Note: Descriptions are shown in the official language in which they were submitted.
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
DESCRIPTION
SOLID PREPARATION
TECHNICAL FIELD OF THE INVENTION
[0001]
s The present invention relates to a solid preparation
showing improved dissolution property of a drug from a
preparation.
BACKGROUND OF INVENTION
[0002]
Hypertension is one of the diseases most frequently found
in adults. According to the 2000 circulatory disease basic
research by the Ministry of Health, Labour and Welfare, the
number of hypertension patients in Japan (those with systolic
blood pressure of not less than 140 mmHg or diastolic blood
pressure of not less than 90 mmHg, or depressor recipients)
reaches about 31 million to 38 million. Hypertension is a
strong risk factor for any circulatory diseases including
cerebrovascular diseases and myocardial infarction. Thus, an
appropriate control of the blood pressure is important for
both improved prognosis of patients, and mitigation of
personal and social burdens.
[0003]
As therapeutic drugs for hypertension, various kinds of
drugs such as hypotensive diuretics, a blockers, R blockers,
angiotensin converting enzyme (ACE) inhibitors, calcium
antagonists, angiotensin II receptor antagonists and the like
have been developed, and many of the patients diagnosed with
hypertension are under treatment with these depressors. For
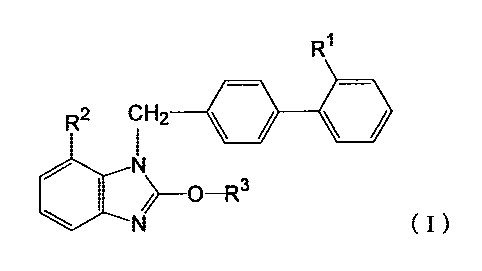
example, a compound represented by the following formula (I):
[0004]
1
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
Ri
R2 CH2
N
_O-R3
N (I)
[0005]
wherein Rl is a monocyclic nitrogen-containing heterocyclic
group having a deprotonatable hydrogen atom, R2 is an
s optionally esterified carboxyl group, and R3 is an optionally
substituted lower alkyl group, or a salt thereof, is known as
an angiotensin II receptor antagonist showing a superior
hypotensive effect and an organ-protective action. JP-B-
2514282 discloses 1-(cyclohexyloxycarbonyloxy)ethyl 2-ethoxy-
1o 1-[[2'-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]benzimidazole-7-
carboxylate (candesartan cilexetil) as examples of the
representative medicament.
[0006]
According to the J-HOME study (The Japan Home vs. Office
15 blood pressure Measurement Evaluation study), about 40% of the
hypertension patients under drug treatment have achieved a
desired blood pressure (outpatient casual blood pressure of
less than 140/90 mmHg), and this means some patients fail to
sufficiently control the blood pressure even with the existing
20 drug treatment. To raise the desired blood pressure achieving
ratio, a more powerful hypotensive treatment is desired.
[0007]
As a drug therapy exerting strong hypotensive effects, a
combination therapy using plural drugs can be mentioned. For
25 example, W001/15674 discloses a combined use of a rennin-
angiotensin inhibitor and other depressor, cholesterol-
lowering drug, diuretic or the like. W002/43807 discloses a
combined use of an angiotensin II receptor antagonist and
other depressor or statin. However, a combined use of plural
30 medicaments for different timings of ingestion may adversely
2
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
affect the drug compliance of patients, where a failure to
control the blood pressure due to forgetfulness is feared. To
more appropriately control the blood pressure, a combination
preparation containing various depressors in one medicament is
strongly desired in clinical practice, since it is an ideal
medicament that exhibits a strong hypotensive effect and
maintains drug compliance of patients.
[0008]
As such combination preparation, a combination
io preparation containing an angiotensin II receptor antagonist
and a calcium antagonist has been suggested. W092/10097
discloses a combination preparation containing an angiotensin
II receptor antagonist and other medicament such as diuretic,
calcium antagonist and the like. JP-A-2006-290899 discloses a
combination preparation containing an imidazolecarboxylate
type angiotensin II receptor antagonist such as olmesartan
medoxomil and the like and a calcium antagonist. US Patent No.
6204281 discloses a combination preparation containing
valsartan, which is an angiotensin II receptor antagonist, and
a 1,4-dihydropyridine compound such as amlodipine and the like,
which is a calcium antagonist, and the like. JP-B-2930252
discloses a combination preparation containing 2-butyl-4-
chloro-l-[(2'-(1H-tetrazol-5-yl)biphenyl-4-
yl)methyl]imidazole-5-carboxylic acid or a pharmaceutically
acceptable salt thereof, each of which is an angiotensin II
receptor antagonist, and diltiazem, which is a calcium
antagonist. Moreover, JP-B-3057471 discloses a combination
preparation containing a benzimidazole derivative, which is an
angiotensin receptor antagonist, and a diuretic or calcium
3o antagonist.
[0009]
A preparation containing a benzimidazole derivative,
which is an angiotensin II receptor antagonist, and a calcium
antagonist is expected to simultaneously show, in clinical
practice, efficacy of the benzimidazole derivative, which is
3
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
an angiotensin II receptor antagonist with a high organ-
protective effect, and efficacy of the calcium antagonist with
a strong hypotensive effect. Moreover, its clinical usefulness
is extremely high, since the action can be enhanced and the
side effects can be reduced depending on the manner of
combination.
[0010]
However, to secure effectiveness and safety of a
pharmaceutical product, not only the effectiveness and safety
io of the active ingredient itself are important but also the
properties of the pharmaceutical preparation, such as the drug
dissolution property in the body and the like, are extremely
important. For example, when dissolution of the drug from the
pharmaceutical preparation is too late, the blood
concentration of the drug does not reach an effective level,
and the expected efficacy may not be sufficiently exhibited.
On the other hand, when dissolution of the drug from the
pharmaceutical preparation is too fast, the blood
concentration of the drug increases rapidly, causing a high
risk of side effects.
[0011]
In other words, pharmaceutical products are required to
ensure the certain level of the drug dissolution, in addition
to the effectiveness and safety. Combination preparations are
required to meet compatibility with various additives and
different conditions required by each active ingredient. Thus,
the development of a preparation meeting all these conditions
is often difficult as compared to a preparation containing a
single active ingredient. In particular, since a benzimidazole
3o derivative, which is an angiotensin II receptor antagonist, is
a poorly soluble compound, the dissolution property of the
preparation may decrease due to the properties of additives
and active ingredient to be combined. When the delayed release
of a drug from the administered combination preparation leads
to decreased drug absorbability, decreased bioavailability,
4
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
i.e., decreased efficacy of active ingredient, and decreased
value of the combination preparation. For a pharmaceutical
preparation to be practicalized, therefore, the composition of
the preparation needs to be adjusted to optimize the
dissolution rate of the active ingredient in the
gastrointestinal tract.
SUMMARY OF THE INVENTION
Problems to be Solved by the Invention
[0012]
It is therefore an object of the present invention to
provide a solid preparation stably containing a benzimidazole
derivative having an angiotensin II receptor antagonistic
action and a calcium antagonist, which is controlled to
optimize dissolution property of these drugs from the
preparation in the gastrointestinal tract.
Means of Solving the Problems
[0013]
The present inventors have conducted intensive studies in
an attempt to solve the above-mentioned problems, and found a
solid preparation comprising (i) a compound represented by the
formula (I) or a salt thereof, (ii) a sugar alcohol, and (iii)
a calcium antagonist, and the preparation shows appropriately
controlled dissolution property in the human gastrointestinal
tract. Specifically, they have taken note of excipients and
found that the dissolution property of a drug from a solid
preparation can be improved by using a highly water-soluble
sugar alcohol, which resulted in the completion of the present
invention.
[0014]
Accordingly, the present invention relates to
[1] a solid preparation comprising (i) a compound represented
by the formula (I):
[0015]
5
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
RI
R2 CH2 1
N
I>_C_R3
N (I)
[0016]
wherein R1 is a monocyclic nitrogen-containing heterocyclic
group having a deprotonatable hydrogen atom, R2 is an
optionally esterified carboxyl group, and R3 is an optionally
substituted lower alkyl group, or a salt thereof, (ii) a sugar
alcohol, and (iii) a calcium antagonist;
[1A] the solid preparation of the above-mentioned [1], wherein
R2 is a carboxyl group optionally esterified by a lower alkyl
io group having a carbon number of 1 to 4, which is optionally
substituted by 1 to 3 substituents selected from a hydroxyl
group, an amino group, a halogen atom, a lower alkanoyloxy
group having a carbon number of 2 to 6, a lower
cycloalkanoyloxy group having a carbon number of 4 to 7, a
carbonyloxy group having a lower alkoxy group having a carbon
number of 1 to 6, a carbonyloxy group having a lower
cycloalkoxy group having a carbon number of 3 to 7, and a
lower alkoxy group having a carbon number of 1 to 4;
[1B] the solid preparation of the above-mentioned [1], wherein
R2 is a 1-(cyclohexyloxycarbonyloxy)ethoxycarbonyl group or a
carboxyl group;
[2] the solid preparation of the above-mentioned [1], [1A] or
[1B], wherein the compound represented by the formula (I) or a
salt thereof is 1-(cyclohexyloxycarbonyloxy) ethyl 2-ethoxy-l-
[[2'-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]benzimidazole-7-
carboxylate, 2-ethoxy-l-[[2'-(1H-tetrazol-5-yl)biphenyl-4-
yl]methyl]-1H-benzimidazole-7-carboxylic acid, 2-ethoxy-l-
[[2'-(4,5-dihydro-5-oxo-1,2,4-oxadiazol-3-yl)biphenyl-4-
yl]methyl]-1H-benzimidazole-7-carboxylic acid or a salt
thereof;
6
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
[3] the solid preparation of the above-mentioned [1], [2],
[1A] or [1B], wherein the compound represented by the formula
(I) or a salt thereof is 1-(cyclohexyloxycarbonyloxy)ethyl 2-
ethoxy-l-[[2'-(1H-tetrazol-5-yl)biphenyl-4-
yl]methyl]benzimidazole-7-carboxylate or a salt thereof;
[4] the solid preparation of the above-mentioned [1], [2],
[1A] or [1B], wherein the compound represented by the formula
(I) or a salt thereof is 2-ethoxy-l-[[2'-(4,5-dihydro-5-oxo-
1,2,4-oxadiazol-3-yl)biphenyl-4-yl]methyl]-1H-benzimidazole-7-
lo carboxylic acid or a salt thereof;
[5] the solid preparation of the above-mentioned [1], [2], [3],
[4], [1A] or [1B], wherein the sugar alcohol is mannitol,
sorbitol or erythritol;
[6] the solid preparation of the above-mentioned [1], [2], [3],
[4], [5], [1A] or [1B], wherein the sugar alcohol is mannitol;
[7] the solid preparation of the above-mentioned [1], [2], [3],
[4], [5], [6], [1A] or [1B], wherein the calcium antagonist is
azelnidipine, amlodipine, aranidipine, efonidipine,
cilnidipine, nicardipine, nisoldipine, nitrendipine,
nifedipine, nilvadipine, barnidipine, felodipine, benidipine,
manidipine or a salt thereof;
[8] the solid preparation of the above-mentioned [1], [2], [3],
[4], [5], [6], [7], [1A] or [1B], wherein the calcium
antagonist is amlodipine or a salt thereof;
[9] the solid preparation of the above-mentioned [1], [2], [3],
[4], [5], [6], [7], [8], [1A] or [1B], further comprising a
polyethylene glycol;
[10] the solid preparation of the above-mentioned [9], wherein
the polyethylene glycol has a molecular weight of 1,000 to
10,000;
[11] a solid preparation comprising (i) 1-
(cyclohexyloxycarbonyloxy)ethyl 2-ethoxy-l-[[2'-(1H-tetrazol-
5-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylate or a
salt thereof, (ii) mannitol, and (iii) amlodipine or a salt
thereof;
7
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
[11A] the solid preparation of the above-mentioned [11],
further comprising a polyethylene glycol;
[11B] the solid preparation of the above-mentioned [11A],
wherein the polyethylene glycol has a molecular weight of
1,000 to 10,000 (preferably 3,000 to 10,000);
[11C] the solid preparation of the above-mentioned [11B],
wherein the content of the polyethylene glycol is 1 to 5 wt%;
[11D] the solid preparation of the above-mentioned [11B],
wherein the content of the polyethylene glycol is 1 to 3 wt%;
to [12] a solid preparation comprising (i) 2-ethoxy-l-[[2'-(4,5-
dihydro-5-oxo-1,2,4-oxadiazol-3-yl)biphenyl-4-yllmethyl]-1H-
benzimidazole-7-carboxylic acid or a salt thereof, (ii)
mannitol, and (iii) amlodipine or a salt thereof;
[12A] the solid preparation of the above-mentioned [12],
is further comprising polyethylene glycol;
[12B] the solid preparation of the above-mentioned [12A],
wherein the polyethylene glycol has a molecular weight of
1,000 to 10,000 (preferably 3,000 to 10,000);
[12C] the solid preparation of the above-mentioned [12B],
20 wherein the content of the polyethylene glycol is 1 to 5 wt%;
[12D] the solid preparation of the above-mentioned [12B],
wherein the content of the polyethylene glycol is 1 to 3 wt%;
[13] the solid preparation of the above-mentioned [1], [2],
[31, [41, [5], [6], [71, [81, [91, [101, [111, [121, [1A],
25 [1B], [11A], [11B], [11C], [11D], [12A], [12B], [12C] or [12D],
which is a tablet;
[14] the solid preparation of the above-mentioned [1], [2],
[3], [4], [5], [6], [7], [8], [9], [10], [11], [12], [1A],
[1B], [11A], [11B], [11C], [11D], [12A], [12B], [12C] or [12D],
30 which is a single-layer tablet;
[15] the solid preparation of the above-mentioned [1], [2],
[31, [41, [5], [6], [7], [81, [91, [101, [111, [121, [13],
[14], [1A], [1B], [11A], [11B], [11C], [11D], [12A], [12B],
[12C] or [12D], which is a prophylactic or therapeutic drug
35 for hypertension, cardiac failure, diabetic nephropathy or
8
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
arteriosclerosis;
[16] the solid preparation of the above-mentioned [1], [2],
[31, [4], [5], [6], [71, [8], [9], [10], [11], [12], [13],
[141, [1A], [1B], [11A], [11B], [11C], [11D], [12A], [12B],
[12C] or [12D], which is a prophylactic or therapeutic drug
for hypertension;
and the like.
EFFECT OF THE INVENTION
[0017]
According to the present invention, a solid preparation
wherein the dissolution of a compound represented by the
above-mentioned formula (I) or a salt thereof, and a calcium
antagonist from the preparation in the gastrointestinal tract
is appropriately controlled, and the stability thereof in the
preparation is finely maintained can be obtained. That is,. the
solid preparation of the present invention is superior in the
dissolution property of a compound represented by the above-
mentioned formula (I) or a salt thereof and a calcium
antagonist from a preparation and the stability thereof.
[0018]
(DETAILED DESCRIPTION OF THE INVENTION)
The solid preparation of the present invention is
explained in detail in the following.
The solid preparation of the present invention is a solid
preparation comprising (i) a compound represented by the
following formula (I):
[0019]
R1
R2 CH2
N
N (I)
[0020]
wherein R1 is a monocyclic nitrogen-containing heterocyclic
9
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
group having a deprotonatable hydrogen atom, R2 is an
optionally esterified carboxyl group, and R3 is an optionally
substituted lower alkyl group, or a salt thereof, (ii) sugar
alcohol, and (iii) a calcium antagonist (hereinafter to be
also referred to as the solid preparation of the present
invention).
[0021]
In the above-mentioned formula (I), as the monocyclic
nitrogen-containing heterocyclic group having a deprotonatable
io hydrogen atom for R1, for example, a tetrazolyl group and a
group represented by the formula
[0022]
N,_
N
H
[0023]
wherein i is -0- or -S- and j is >C=O, >C=S or >S(O)m wherein m
is 0, 1 or 2,
[0024]
and the like can be mentioned. As a preferable group, a
tetrazolyl group, 4,5-dihydro-5-oxo-1,2,4-oxadiazol-3-y1 group
and the like can be mentioned.
4,5-Dihydro-5-oxo-1,2,4-oxadiazol-3-yl group contains 3
tautomers (a', b' and c') represented by the formulas:
[0025]
~O N- ~OH HN ~=O
N/ N N
H
as b' c' and
[0026]
the 4,5-dihydro-5-oxo-1,2,4-oxadiazol-3-yl group includes all
of the above-mentioned a', b' and c'.
[0027]
In the above-mentioned formula (I), examples of the
optionally esterified carboxyl group for R2 include a carboxyl
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
group optionally esterified by a lower alkyl group having a
carbon number of 1 to 4. The aforementioned lower alkyl group
is optionally substituted by 1 to 5 (preferably 1 to 3)
substituent(s) selected from the group consisting of a
hydroxyl group, an amino group, a halogen atom, a lower
alkanoyloxy group having a carbon number of 2 to 6 (e.g.,
acetyloxy group, pivaloyloxy group and the like), a lower
cycloalkanoyloxy group having a carbon number of 4 to 7, a
carbonyloxy group having a lower alkoxy group having a carbon
1o number of 1 to 6 (e.g., methoxycarbonyloxy group,
ethoxycarbonyloxy group and the like), a carbonyloxy group
having a lower cycloalkoxy group having a carbon number of 3
to 7 (e.g., cyclohexyloxycarbonyloxy group and the like), and
a lower alkoxy group having a carbon number of 1 to 4. As a
preferable group, a 1-(cyclohexyloxycarbonyloxy)ethoxycarbonyl
group, a carboxyl group and the like can be mentioned.
[0028]
In the above-mentioned formula (I), as the optionally
substituted lower alkyl group for R3, a lower alkyl group
having a carbon number of 1 to 5, which is optionally
substituted by 1 to 5 (preferably 1 to 3) substituent(s)
selected from the group consisting of a hydroxyl group, an
amino group, a halogen atom and a lower alkoxy group having a
carbon number of 1 to 4, can be mentioned. Preferred is a
lower alkyl group having a carbon number of 2 to 3, and
particularly preferred is an ethyl group.
[0029]
The salt of the compound represented by the above-
identified formula (I) only needs to be a pharmaceutically
3o acceptable salt and, for example, a salt with an inorganic
base, a salt with an organic base, a salt with an inorganic
acid, a salt with an organic acid, a salt with a basic or
acidic amino acid and the like of a compound represented by
the formula (I) can be mentioned. Preferable examples of the
salt with an inorganic base include alkali metal salts such as
11
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
sodium salt, potassium salt and the like; alkaline earth metal
salts such as calcium salt, magnesium salt and the like;
aluminum salt, ammonium salt and the like. Preferable examples
of the salt with an organic base include salts with
trimethylamine, triethylamine, pyridine, picoline,
ethanolamine, diethanolamine, triethanolamine,
dicyclohexylamine, N,N'-dibenzylethylenediamine and the like.
Preferable examples of the salt with an inorganic acid include
salts with hydrochloric acid, hydrobromic acid, nitric acid,
io sulfuric acid, phosphoric acid and the like. Preferable
examples of the salt with an organic acid include salts with
formic acid, acetic acid, trifluoroacetic acid, fumaric acid,
oxalic acid, tartaric acid, maleic acid, citric acid, succinic
acid, malic acid, methanesulfonic acid, benzenesulfonic acid,
p-toluenesulfonic acid and the like. Preferable examples of
the salt with a basic amino acid include salts with arginine,
lysine, ornithine and the like. Preferable examples of the
salt with an acidic amino acid include salts with aspartic
acid, glutamic acid and the like.
[0030]
The compound represented by the above-mentioned formula
(I) or a salt thereof may be a hydrate or a non-hydrate, and a
solvate or a nonsolvate.
Moreover, the compound represented by the above-mentioned
formula (I) or a salt thereof is preferably crystalline, and
the melting point is 100 to 250 C, preferably 120 to 200 C,
particularly 130 to 180 C.
[0031]
As the solid preparation of the present invention, a
compound represented by the above-mentioned formula (I) or a
salt thereof is used. Preferable examples of the compound or a
salt thereof include 1-(cyclohexyloxycarbonyloxy)ethyl 2-
ethoxy-l-[[2'-(1H-tetrazol-5-yl)biphenyl-4-
yl]methyl]benzimidazole-7-carboxylate, 2-ethoxy-l-[[2'-(1H-
tetrazol-5-yl)biphenyl-4-yl]methyl]-1H-benzimidazole-7-
12
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
carboxylic acid, pivaloyloxymethyl 2-ethoxy-l-[[2'-(1H-
tetrazol-5-yl)biphenyl-4-yl]methyl]-1H-benzimidazole-7-
carboxylate, 2-ethoxy-l-[[2'-(4,5-dihydro-5-oxo-1,2,4-
oxadiazol-3-yl)biphenyl-4-yl]methyl]-1H-benzimidazole-7-
carboxylic acid, and salts thereof. Among these, 1-
(cyclohexyloxycarbonyloxy)ethyl 2-ethoxy-l-[[2'-(1H-tetrazol-
5-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylate and a
salt thereof, 2-ethoxy-l-[[2'-(4,5-dihydro-5-oxo-1,2,4-
oxadiazol-3-yl)biphenyl-4-yl]methyl]-1H-benzimidazole-7-
io carboxylic acid and a salt thereof are particularly preferable.
[0032]
In the present invention, the compound represented by the
above-mentioned formula (I) or a salt thereof is contained in
the solid preparation of the present invention in a proportion
of 0.1 to 60 wt%, preferably 1 to 40 wt%, more preferably 3 to
30 wt% based on a free form.
[0033]
As sugar alcohol to be used in the present invention, any
sugar alcohols can be used as long as it simultaneously
establishes the stability of the compound represented by the
above-mentioned formula (I) or a salt thereof in a preparation,
and the dissolution property thereof from a preparation and
can be applied to pharmaceutical products. Examples of the
sugar alcohol to be used in the present invention include
monosaccharide sugar alcohols such as tetritol (e.g.,
erythritol, D-threitol, L-threitol and the like), pentitol
(e.g., D-arabinitol, xylitol and the like), hexitol (e.g., D-
iditol, galactitol (dulcitol), D-glucitol (sorbitol), mannitol
and the like), cyclitol (e.g., inositol and the like) and the
like; disaccharide sugar alcohols such as maltitol, lactitol,
reduced paratinose (isomalt) and the like; oligosaccharide
sugar alcohols such as pentaerythritol, hydrogenated maltose
starch syrup and the like; and the like. Among these,
monosaccharide sugar alcohols are preferable. More preferable
are mannitol, sorbitol and erythritol. Particularly, mannitol
13
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
is preferable, and D-mannitol is especially preferable. The
sugar alcohol may be used alone or two or more kinds thereof
may be used in combination. In addition, the sugar alcohol
simultaneously achieves the stability of a calcium antagonist
in a preparation and dissolution property thereof from the
preparation.
[0034]
In the present invention, the sugar alcohol is contained
in the solid preparation of the present invention in a
io proportion of 15 to 85 wt%, preferably 20 to 80 wt%, more
preferably 25 to 75 wt%.
[0035]
Examples of the calcium antagonist to be used in the
present invention include dihydropyridine compounds such as
azelnidipine, amlodipine, aranidipine, efonidipine,
cilnidipine, nicardipine, nisoldipine, nitrendipine,
nifedipine, nilvadipine, barnidipine, felodipine, benidipine,
manidipine and the like; benzodiazepine compounds such as
diltiazem and the like; and the like. The calcium antagonist
to be used in the present invention also includes salts of the
compounds recited as the aforementioned calcium antagonist.
[0036]
As the calcium antagonist to be used in the present
invention, dihydropyridine compounds, particularly amlodipine
or a salt thereof is preferable. Among these, a salt of
amlodipine is more preferable, and amlodipine besylate is
particularly preferable.
[0037]
In the present invention, a calcium antagonist is
contained in the solid preparation of the present invention in
a proportion of generally 0.05 to 60 wt%, preferably 0.1 to 40
wt%, more preferably 0.5 to 20 wt%, based on a free form.
Specifically, for example, amlodipine is contained in a
proportion of generally 0.05 to 60 wt%, preferably 0.1 to 40
wt%, more preferably 0.5 to 20 wt%, based on a free form.
14
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
[0038]
The solid preparation of the present invention may
further contain an alkylene oxide polymer. Examples of the
alkylene oxide polymer include a polymer of ethylene oxide,
propylene oxide, trimethylene oxide, tetrahydrofuran or the
like (preferably, ethylene oxide). The molecular weight of the
alkylene oxide polymer is preferably 1,000 to 10,000, more
preferably 3,000 to 10,000. The alkylene oxide polymer may be
a copolymer of alkylene oxide, and examples of the copolymer
1o of alkylene oxide include copolymer of two or more kinds of
the above-mentioned alkylene oxide, which has a molecular
weight of 1,000 to 10,000 (preferably 3,000 to 10,000).
The alkylene oxide polymer may be used alone or two or
more kinds thereof may be used in combination.
[0039]
As the alkylene oxide polymer to be used in the present
invention, polyethylene glycol is preferable, polyethylene
glycol having a molecular weight of 1,000 to 10,000 is more
preferable, and polyethylene glycol (e.g., polyethylene glycol
4000, polyethylene glycol 6000, polyethylene glycol 10000)
having a molecular weight of 3,000 to 10,000 is particularly
preferable.
[0040]
In the present invention, the alkylene oxide polymer is
contained in the solid preparation of the present invention in
a proportion of preferably 1 to 5 wt%, more preferably 1 to 3
wt%.
[0041]
A preferable embodiment of the solid preparation of the
present invention includes
a solid preparation wherein the compound represented by the
above-mentioned formula (I) or a salt thereof is 1-
(cyclohexyloxycarbonyloxy)ethyl 2-ethoxy-l-[[2'-(1H-tetrazol-
5-yl)biphenyl-4-yl]methyl]benzimidazole-7-carboxylate
(hereinafter sometimes referred to as "compound A") or a salt
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
thereof, and the calcium antagonist is amlodipine besylate;
a solid preparation wherein the compound represented by the
above-mentioned formula (I) or a salt thereof is 2-ethoxy-l-
[[2'-(4,5-dihydro-5-oxo-1,2,4-oxadiazol-3-yl)biphenyl-4-
yl]methyl]-1H-benzimidazole-7-carboxylic acid (hereinafter
sometimes referred to as "compound B") or a salt thereof and
the calcium antagonist is amlodipine besylate;
and the like.
[0042]
As the solid preparation of the present invention, for
example, a solid preparation suitable for oral administration
such as tablet, granule, fine granule, capsule, pill and the
like can be mentioned, with preference given to tablet and
more preference given to single-layer-tablet. When the solid
preparation of the present invention is a tablet, the form of
the tablet is any of round, oval, oblong and the like. While
the size of the tablet varies depending on the form of the
tablet (round, caplet, oblong etc.), any size is possible as
long as patients can take the tablet with ease. When the solid
preparation of the present invention is a single-layer tablet,
it is easily taken due to its small tablet size.
[0043]
The embodiment of the solid preparation of the present
invention includes
(1) one group granulated preparation
a solid preparation obtained by granulating a mixture
comprising a compound represented by the formula (I) or a salt
thereof, sugar alcohol, and a calcium antagonist (preferably,
a compound represented by the formula (I) or a salt thereof,
sugar alcohol, a calcium antagonist, and a polyethylene
glycol), and compression molding the obtained granulated
material (e.g., one group granulated single-layer tablet);
(2) two group granulated preparation
(a) a solid preparation obtained by mixing and compression
molding the following first part and the second part obtained
16
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
by individual granulation (e.g., two group granulated single-
layer tablet);
(b) a solid preparation obtained by compression molding
(without mixing) the following first part and the second part
obtained by individual granulation (e.g., multi-layer tablet);
(c) a solid preparation obtained by coating one of the
following first part and the second part obtained by
individual granulation with the other (e.g., dry coated
tablet);
first part: a part comprising a compound represented by
the formula-(I) or a salt thereof (preferably, a compound
represented by the formula (I) or a salt thereof, and a
polyethylene glycol)
second part: a part comprising sugar alcohol and a
calcium antagonist
and the like.
The preferable embodiment of the solid preparation of the
present invention is one group granulated preparation (e.g.,
one group granulated single-layer tablet).
[0044]
The solid preparation of the present invention may
contain additives conventionally used in the pharmaceutical
field. Examples of the additive include excipient,
disintegrant, binder, lubricant, pH adjuster, colorant,
surfactant, stabilizer, acidulant, flavor, glidant and the
like. These additives are used in amounts conventionally
employed in the pharmaceutical field. In addition, these
additives may be used in a mixture of two or more kinds
thereof at an appropriate ratio.
[0045]
Examples of the excipient include starches such as corn
starch, potato starch, wheat starch, rice starch, partly
pregelatinized starch, pregelatinized starch, porous starch
and the like, saccharides such as lactose, fructose, glucose,
sucrose and the like, anhydrous calcium phosphate, crystalline
17
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
cellulose, microcrystalline cellulose, Glycyrrhiza uralensis,
sodium hydrogen carbonate, calcium phosphate, calcium sulfate,
calcium carbonate, precipitated calcium carbonate, calcium
silicate and the like.
[0046]
Examples of the disintegrant include amino acid, starch,
corn starch, carboxymethylcellulose, calcium
carboxymethylcellulose, sodium carboxymethyl starch,
carmellose sodium, carmellose calcium, croscarmellose sodium,
1o crospovidone, low-substituted hydroxypropylcellulose,
hydroxypropylstarch and the like.
In the present invention, a disintegrant is contained in
a proportion of preferably 0.1 to 30 wt%, more preferably 1 to
wt%, of the solid preparation of the present invention.
[0047]
Examples of the binder include crystalline cellulose
(e.g., microcrystalline cellulose), hydroxypropylcellulose,
hypromellose, polyvinylpyrrolidone, gelatin, starch, gum
arabic powder, tragacanth, carboxymethylcellulose, sodium
alginate, pullulan, glycerol and the like.
In the present invention, a binder is contained in a
proportion of preferably 0.1 to 40 wt%, more preferably 1 to
10 wt%, of the solid preparation of the present invention.
[0048]
Examples of the lubricant include magnesium stearate,
stearic acid, calcium stearate, talc (purified talc), sucrose
esters of fatty acids, sodium stearyl fumarate and the like.
[0049]
Examples of the pH adjuster include citric acid and a
salt thereof, phosphoric acid and a salt thereof, carbonic
acid and a salt thereof, tartaric acid and a salt thereof,
fumaric acid and a salt thereof, acetic acid and a salt
thereof, amino acid and a salt thereof and the like.
[0050]
Examples of the colorant include food colors such as Food
18
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
Color Yellow No. 5, Food Color Red No. 2, Food Color Blue No.
2 and the like, food lake colors such as Food Yellow No. 4
Aluminum Lake and the like, iron oxide pigments such as red
ferric oxide (colcothar), yellow ferric oxide, triiron
tetraoxide (iron oxide black) and the like; and so on.
[0051]
Examples of the surfactant include sodium lauryl sulfate,
polysorbate 80, polyoxyethylene(160)polyoxypropylene(30)glycol
and the like.
to [0052]
Examples of the stabilizer include tocopherol,
tetrasodium edetate, nicotinamide, cyclodextrins and the like.
[0053]
Examples of the acidulant include ascorbic acid, citric
acid, tartaric acid, malic acid and the like.
[0054]
Examples of the flavor include menthol, peppermint oil,
lemon oil, vanillin and the like.
[0055]
Examples of the glidant include light anhydrous silicic
acid, hydrated silicon dioxide and the like.
[0056]
The solid preparation of the present invention can also
be processed into a film-coated preparation by applying a film
coating such as a coating base, a coating additive and the
like. Examples of the film-coated preparation include a sugar-
coated preparation, sustained-release preparation, enteric
preparation and the like.
[0057]
Preferable examples of the coating base include a sugar
coating base, a water-soluble film coating base, an enteric
film coating base, a sustained-release film coating base and
the like.
[0058]
As the sugar coating base, sucrose is used. Furthermore,
19
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
one or more kinds selected from talc, precipitated calcium
carbonate, gelatin, gum arabic, pullulan, Carnauba wax and the
like may be used in combination.
[0059]
Examples of the water-soluble film coating base include
cellulose polymers such as hydroxypropylcellulose [e.g., NISSO
HPC (grades: L, SL, SL-T, SSL) (trade name); Nippon Soda Co.,
Ltd.], hypromellose [e.g., TC-5 (grades: MW, E, EW, R, RW)
(trade name); Shin-Etsu Chemical Co., Ltd.],
1o hydroxyethylcellulose, methylhydroxyethylcellulose and the
like, synthetic polymers such as polyvinylacetal
diethylaminoacetate, aminoalkylmethacrylate copolymer E
[Eudragit E (trade name); manufactured by ROHM AND HAAS JAPAN
KK.], polyvinylpyrrolidone and the like, polysaccharides such
as pullulan and the like; and so on.
[0060]
Examples of the enteric film coating base include
cellulose polymers such as hypromellose phthalic acid ester,
hydroxypropylmethylcellulose acetate succinate,
carboxymethylethylcellulose, cellulose acetate phthalate and
the like, acrylic acid polymers such as methacrylic acid
copolymer L [Eudragit L (trade name)], methacrylic acid
copolymer LD [Eudragit L-30D55 (trade name); manufactured by
ROHM AND HAAS JAPAN KK.], methacrylic acid copolymer S
[Eudragit S (trade name); manufactured by ROHM AND HAAS JAPAN
KK.] and the like, natural polymers such as shellac and the
like; and so on.
[0061]
Examples of the sustained-release film coating base
include cellulose polymers such as ethylcellulose and the like,
acrylic acid polymers such as aminoalkylmethacrylate copolymer
RS [Eudragit RS (trade name); manufactured by ROHM AND HAAS
JAPAN KK.], ethyl acrylate-methyl methacrylate copolymer
suspension [Eudragit NE (trade name); manufactured by ROHM AND
HAAS JAPAN KK.] and the like; and so on.
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
[0062]
Preferable examples of the coating additives include
light protecting agents such as titanium dioxide and the like,
glidants such as talc and the like, colorants such as red
ferric oxide, yellow ferric oxide and the like, plasticizers
such as polyethylene glycol [e.g., macrogol 6000 (trade name);
manufactured by Sanyo Chemical Industries, Ltd.], triethyl
citrate, castor oil, polysorbates and the like, organic acids
such as citric acid, tartaric acid, malic acid, ascorbic acid
1o and the like; and so on.
[0063]
The solid preparation of the present invention can be
produced by a method known per se (e.g., the method described
in the Japanese Pharmacopoeia 15th Edition, General
Principles).
[0064]
Examples of the method include operations such as mixing,
kneading, granulation, compression molding, film coating and
the like, and an appropriate combination of these operations.
[0065]
Mixing is performed using, for example, a mixer such as a
horizontal cylindrical mixer, a V-type mixer, a tumbler mixer
and the like, and kneading is performed using a rotary vessel-
type kneader such as a ball mill and the like, a fixed vessel-
type kneader such as a screw-type kneader, a Henschel mixer
and the like, a rolling kneader such as a rolling mill, a
taper rolling mill and the like, and the like. Granulation is
performed by a stirring granulation method using a high-speed
stirring granulator, a rolling granulation method using a
container of a pan type, conical drum type, multi-stage
conical drum type, stirring blade-equipped drum type,
vibration type or the like, a fluidized bed granulation and
drying method, a spray drying granulation method, an
extrusion-granulation method, a method using a granulator such
as a Roller-compactor and the like, and the like.
21
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
[0066]
Compression molding is performed, for example, by molding
by an extrusion molding machine, or tableting using a single
punch tableting machine, a rotary tableting machine and the
like.
When compression molding is performed using .a single
stroke tableting machine, a rotary tableting machine and the
like, it is generally preferable to employ a tableting
pressure of 1 to 35 kN/cm2 (preferably 5 to 35 kN/cm2), and
lo further, a taper cutting die to prevent capping.
[0067]
Film coating is performed, for example, by using a pan
coating machine of a horizontal type, an inclination type and
the like, a fluid coating machine of a horizontal rotating-
disc type, an inclined plate type and the like, a fluid
coating machine of a fluidized bed type, a spouted bed type, a
tumbling fluidized bed type and the like, and the like.
[0068]
The solid preparation of the present invention can be
produced, for example, by the following production steps.
(1) When the solid preparation of the present invention is a
one group granulated preparation
A mixture comprising a compound represented by the above-
mentioned formula (I) or a salt thereof, sugar alcohol, and a
calcium antagonist (preferably, a compound represented by the
formula (I) or a salt thereof, sugar alcohol, a calcium
antagonist, and a polyethylene glycol) is granulated, and the
obtained granulated material is compression molded to give the
solid preparation of the present invention (one group
granulated single-layer tablet).
Specifically, a compound represented by the above-
mentioned formula (I) or a salt thereof, a calcium antagonist
and an additive such as excipient, sugar alcohol (e.g., D-
mannitol) and the like are mixed, and the mixture is
granulated while spraying a liquid obtained by dispersing or
22
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
dissolving polyethylene glycol and an additive such as a
binder and the like in a solvent (e.g., water).
An additive such as disintegrant, lubricant and the like
is added to the thus-obtained granulated material and, after
mixing, the mixture is compression molded to give a tablet.
The tablet is coated with a film solution containing a coating
base and the like, whereby the solid preparation of the
present invention (one group granulated single-layer tablet)
is produced.
io [0069]
(2) When the solid preparation of the present invention is a
two group granulated preparation
(a) two group granulated single-layer tablet;
A compound represented by the above-mentioned formula (I)
is or a salt thereof and an additive such as excipient and the
like are mixed, and the mixture is granulated while spraying a
liquid obtained by dispersing or dissolving polyethylene
glycol and an additive such as a binder and the like in a
solvent (e.g., water).
20 On the other hand, a calcium antagonist and an additive
such as excipient, sugar alcohol (e.g., D-mannitol) and the
like are mixed, and the mixture is granulated while spraying a
liquid obtained by dispersing or dissolving an additive such
as a binder and the like in a solvent (e.g., water).
25 An additive such as disintegrant, lubricant and the like
is added to the thus-obtained granulated material containing a
compound represented by the above-mentioned formula (I) or a
salt thereof and the thus-obtained granulated material
containing the calcium antagonist and, after mixing, the
30 mixtures are compression molded to give the solid preparation
of the present invention (two group granulated single-layer
tablet).
(b) multi-layer tablet;
A compound represented by the above-mentioned formula (I)
35 or a salt thereof and an additive such as excipient and the
23
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
like are mixed, and the mixture is granulated while spraying a
liquid obtained by dispersing or dissolving polyethylene
glycol and an additive such as a binder and the like in a
solvent (e.g., water). An additive such as disintegrant,
lubricant and the like is added to the thus-obtained
granulated material to give a mixed granule.
On the other hand, a calcium antagonist and an additive
such as excipient, sugar alcohol (e.g., D-mannitol) and the
like are mixed, and the mixture is granulated while spraying a
io liquid obtained by dispersing or dissolving an additive such
as a binder and the like in a solvent (e.g., water). An
additive such as disintegrant, lubricant and the like is added
to the thus-obtained granulated material to give a mixed
granule.
The thus-obtained mixed granule containing a compound
represented by the above-mentioned formula (I) or a salt
thereof and the thus-obtained mixed granule containing the
calcium antagonist are laid on top of each other and
compression molded to give the solid preparation of the
present invention (multi-layer tablet).
(c) dry coated tablet;
A calcium antagonist and an additive such as excipient,
sugar alcohol (e.g., D-mannitol) and the like are mixed, and
the mixture is granulated while spraying a liquid obtained by
dispersing or dissolving an additive such as a binder and the
like in a solvent (e.g., water). An additive such as
disintegrant, lubricant and the like is added to the obtained
granulated material and, after mixing, the mixture is
compression molded to give a tablet. The tablet is coated with
3o a film solution containing a coating base and the like to give
an inner core tablet.
On the other hand, a compound represented by the above-
mentioned formula (I) or a salt thereof and an additive such
as excipient and the like are mixed, and the mixture is
granulated while spraying a liquid obtained by dispersing or
24
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
dissolving polyethylene glycol and an additive such as a
binder and the like in a solvent (e.g., water). An additive
such as disintegrant, lubricant and the like is added to the
obtained granulated material to give a mixed granule.
s The mixed granule is added as an outer layer to the
above-mentioned inner core tablet and they are compression
molded to give the solid preparation of the present invention
(dry coated tablet).
[0070]
When the solid preparation of the present invention is
granules or fine granules, they can be produced by a method
similar to the above-mentioned methods.
[0071]
When the solid preparation of the present invention is a
capsule, it can be produced by filling the above-mentioned
granules or fine granules in a capsule containing gelatin,
hypromellose and the like. Furthermore, a hard capsule can be
produced by filling a compound represented by the above-
mentioned formula (I) or a salt thereof and a calcium
antagonist together with a sugar alcohol and other excipient
etc. in a capsule containing gelatin, hypromellose and the
like. In addition, a soft capsule can be produced by
encapsulation molding a compound represented by the above-
mentioned formula (I) or a salt thereof and a calcium
antagonist into a given shape with a base containing gelatin
and a plasticizer such as glycerol and the like.
[0072]
The solid preparation of the present invention may have a
punch mark for identification or print on the surface. In
3o addition, it may have a score line for partition.
[0073]
The solid preparation of the present invention is low
toxic and can be safely administered orally or parenterally as
a medicament for a mammal (e.g., human, monkey, cat, swine,
horse, bovine, mouse, rat, guinea pig, dog, rabbit and the
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
like) .
[0074]
A compound represented by the formula (I) or a salt
thereof has a strong angiotensin II receptor antagonistic
action, and therefore, the solid preparation of the present
invention is useful as a prophylactic or therapeutic drug for
(1) a disease developed (or promoted to be developed) by
coarctation or growth of blood vessel or an organ disorder
expressed via angiotensin II receptor, (2) a disease developed
to (or promoted to be developed) by the presence of angiotensin
II, or (3) a disease developed (or promoted to be developed)
by a factor induced by the presence of angiotensin II in the
aforementioned mammals.
[0075]
Examples of the diseases of the above-mentioned (1) to
(3) include hypertension, blood pressure circadian rhythm
abnormality, heart diseases (e.g., cardiac hypertrophy, acute
heart failure, chronic heart failure including cardiac failure,
failure of expansion, cardiac myopathy, angina pectoris,
myocarditis, atrial fibrillation, arrhythmia, tachycardia,
cardiac infraction etc.), cerebrovascular disorders (e.g.,
asymptomatic cerebrovascular disorder, transient cerebral
ischemia, apoplexy, cerebrovascular dementia, hypertensive
encephalopathy, cerebral infarction etc.), cerebral edema,
cerebral circulatory disorder, recurrence and sequela of
cerebrovascular disorders (e.g., neurotic symptom, psychic
symptom, subjective symptom, disorder in daily living
activities etc.), ischemic peripheral circulation disorder,
myocardial ischemia, venous insufficiency, progression of
cardiac insufficiency after myocardial infarction, renal
diseases (e.g., nephritis, glomerulonephritis,
glomerulosclerosis, renal failure, thrombotic vasculopathy,
diabetic nephropathy, complication of dialysis, organ damage
including nephropathy by radiation irradiation etc.),
arteriosclerosis including atherosclerosis (e.g., aneurysm,
26
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
coronary sclerosis, cerebral arteriosclerosis, peripheral
arterial sclerosis etc.), vascular hypertrophy, vascular
hypertrophy or obliteration and organ damages after
intervention (e.g., percutaneous transluminal coronary
angioplasty, stenting, coronary angioscopy, intravascular
ultrasound, dounce thrombolytic therapy etc.), vascular re-
obliteration and restenosis after bypass, polycythemia,
hypertension, organ damage and vascular hypertrophy after
transplantation, rejection after transplantation, ocular
1o diseases (e.g., glaucoma, ocular hypertension etc.),
thrombosis, multiple organ disorder, endothelial dysfunction,
hypertensive tinnitus, other cardiovascular diseases (e.g.,
deep vein thrombosis, obstructive peripheral circulatory
disorder, arteriosclerosis obliterans, thromboangiitis
obliterans, ischemic cerebral circulatory disorder, Raynaud's
disease, Berger disease etc.), metabolic and/or nutritional
disorders (e.g., obesity, hyperlipidemia, hypercholesterolemia,
hyperuricacidemia, hyperkalemia, hypernatremia etc.),
neurodegenerative diseases (e.g., Alzheimer's disease,
Parkinson's disease, amyotrophic lateral sclerosis, AIDS
encephalopathy etc.), central nervous system disorders (e.g.,
damages such as cerebral hemorrhage and cerebral infarction,
and sequela and complication thereof, head injury, spinal
injury, cerebral edema, senile dementia, sensory malfunction,
sensory functional disorder, autonomic nervous system disorder,
autonomic nervous system malfunction, multiple sclerosis etc.),
dementia, defects of memory, disorder of consciousness,
amnesia, anxiety symptom, catatonic symptom, discomfort mental
state, psychopathies (e.g., depression, epilepsy, alcoholism
3o etc.), inflammatory. diseases (e.g., arthritis such as
rheumatoid arthritis, osteoarthritis, rheumatoid myelitis,
periostitis etc.; inflammation after operation or injury;
remission of swelling; pharyngitis; cystitis; pneumonia;
atopic dermatitis; inflammatory intestinal diseases such as
Crohn's disease, ulcerative colitis etc.; meningitis;
27
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
inflammatory ocular disease; inflammatory pulmonary diseases
such as pneumonia, pulmonary silicosis, pulmonary sarcoidosis,
pulmonary tuberculosis etc.), allergic diseases (e.g.,
allergic rhinitis, conjunctivitis, gastrointestinal allergy,
pollinosis, anaphylaxis etc.), chronic obstructive pulmonary
disease, interstitial pneumonia, pneumocytis carinni pneumonia,
collagen diseases (e.g., systemic lupus erythematodes,
scleroderma, polyarteritis etc.), hepatic diseases (e.g.,
hepatitis including chronic hepatitis, hepatic cirrhosis etc.),
io portal hypertension, digestive system disorders (e.g.,
gastritis, gastric ulcer, gastric cancer, gastric disorder
after operation, dyspepsia, esophageal ulcer, pancreatitis,
colon polyp, cholelithiasis, hemorrhoidal disease, varices
ruptures of esophagus and stomach etc.), blood and/or
myelopoietic diseases (e.g., erythrocytosis, vascular purpura,
autoimmune hemolytic anemia, disseminated intravascular
coagulation syndrome, multiple myelopathy etc.), bone diseases
(e.g., fracture, refracture, osteoporosis, osteomalacia, bone
Paget's disease, sclerosing myelitis, rheumatoid arthritis,
joint tissue dysfunction and the like caused by osteoarthritis
of the knee and diseases similar to these), solid tumor,
tumors (e.g., malignant melanoma, malignant lymphoma, cancer
of digestive organs (e.g., stomach, intestine etc.) etc.),
cancer and cachexia following cancer, metastasis cancer,
endocrinopathy (e.g., Addison's disease, Cushing's syndrome,
pheochromocytoma, hyperaldosteronism, primary aldosteronism
etc.), Creutzfeldt-Jakob disease, urinary organ and/or male
genital diseases (e.g., cystitis, prostatic hypertrophy,
prostatic cancer, sex infectious disease etc.), female
3o disorders (e.g., climacteric disorder, gestosis, endometriosis,
hysteromyoma, ovarian disease, breast disease, sex infectious
disease etc.), disease relating to environment and
occupational factors (e.g., radiation hazard, hazard by
ultraviolet, infrared or laser beam, altitude sickness etc.),
respiratory diseases (e.g., cold syndrome, pneumonia, asthma,
28
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
pulmonary hypertension, pulmonary thrombosis and pulmonary
embolism etc.), infectious diseases (e.g., viral infectious
diseases with cytomegalovirus, influenza virus, herpes virus
etc., rickettsiosis, bacterial infectious disease etc.),
toxemias (e.g., sepsis, septic shock, endotoxin shock, Gram-
negative sepsis, toxic shock syndrome etc.),
otorhinolaryngological diseases (e.g., Meniere's syndrome,
tinnitus, dysgeusia, vertigo, disequilibrium, dysphagia etc.),
skin diseases (e.g., keloid, hemangioma, psoriasis etc.),
io intradialytic hypotension, myasthenia gravis, systemic
diseases such as chronic fatigue syndrome and the like, and
the like.
[0076]
The solid preparation of the present invention comprising
a combination of a compound represented by the formula (I) or
a salt thereof and a calcium antagonist is useful as a
prophylactic or therapeutic drug for the above-mentioned
diseases (preferably, a prophylactic or therapeutic drug for
hypertension, cardiac failure, diabetic nephropathy or
arteriosclerosis, more preferably, a prophylactic or
therapeutic drug for hypertension). Moreover, the solid
preparation of the present invention can reduce the doses of a
compound represented by the formula (I) or a salt thereof and
a calcium antagonist as compared to single use thereof.
[0077]
While the dose of a compound represented by the formula
(I) or a salt thereof varies depending on the subject of
administration, administration route, target disease, symptom
and the like, the daily dose for a human adult (body weight 60
3o kg) based on a free form is about 0.05 to 500 mg, preferably
0.1 to 100 mg, more preferably 1 to 100 mg, still more
preferably 2 to 40 mg. For example, the daily dose of compound
A for human adult (body weight 60 kg) is about 1 to 80 mg,
preferably 2 to 32 mg, and the daily dose of compound B for
human adult (body weight 60 kg) is about 1 to 50 mg,
29
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
preferably 10 to 40 mg.
[0078]
While the dose of a calcium antagonist varies depending
on the subject of administration, administration route, target
disease, symptom and the like, the daily dose of, for example,
amlodipine or a salt thereof for a human adult (body weight 60
kg) based on a free form is about 1 to 50 mg, preferably 2.5
to 10 mg.
[0079]
The administration frequency of the solid preparation of
the present invention to the aforementioned mammals is
preferably 1 to 3 times per day, more preferably once per day.
[0080]
Particularly preferable specific examples of the solid
preparation of the present invention include
"a single-layer tablet comprising 8 mg of compound A and 6.93
mg of amlodipine besylate (5 mg as amlodipine) per tablet";
"a single-layer tablet comprising 8 mg of compound A and 3.47
mg of amlodipine besylate (2.5 mg as amlodipine) per tablet";
"a single-layer tablet comprising 4 mg of compound A and 6.93
mg of amlodipine besylate (5 mg as amlodipine) per tablet";
"a single-layer tablet comprising 4 mg of compound A and 3.47
mg of amlodipine besylate (2.5 mg as amlodipine) per tablet";
"a single-layer tablet comprising 40 mg of compound B and 6.93
mg of amlodipine besylate (5 mg as amlodipine) per tablet";
"a single-layer tablet comprising 40 mg of compound B and 3.47
mg of amlodipine besylate (2.5 mg as amlodipine) per tablet";
"a single-layer tablet comprising 20 mg of compound B and 6.93
mg of amlodipine besylate (5 mg as amlodipine) per tablet";
"a single-layer tablet comprising 20 mg of compound B and 3.47
mg of amlodipine besylate (2.5 mg as amlodipine) per tablet";
"a single-layer tablet comprising 10 mg of compound B and 6.93
mg of amlodipine besylate (5 mg as amlodipine) per tablet";
and
"a single-layer tablet comprising 10 mg of compound B and 3.47
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
mg of amlodipine besylate (2.5 mg as amlodipine) per tablet".
[0081]
The solid preparation of the present invention can be
used in combination with one or more different kinds of
medicaments (hereinafter sometimes to be abbreviated as
"concomitant drug"). Examples of the "concomitant drug"
include therapeutic agents for diabetes, therapeutic agents
for diabetic complications, therapeutic agents for
hyperlipidemia, antihypertensive agents, antiobestic agents,
io diuretics, antithrombotic agents and the like.
[0082]
Here, as the therapeutic agent for diabetes, for example,
insulin preparations (e.g., animal insulin preparations
extracted from the pancreas of bovine or swine; human insulin
preparations genetically synthesized using Escherichia coli or
yeast; zinc insulin; protamine zinc insulin; fragment or
derivative of insulin (e.g., INS-1), oral insulin preparation),
insulin sensitizers (e.g., pioglitazone or a salt thereof
(preferably, hydrochloride), rosiglitazone or a salt thereof
(preferably, maleate), Metaglidasen, AMG-131, Balaglitazone,
MBX-2044, Rivoglitazone, Aleglitazar, Chiglitazar,
Lobeglitazone, PLX-204, PN-2034, GFT-505, THR-0921, compounds
described in W02007/013694, W02007/018314, W02008/093639 or
W02008/099794), a-glucosidase inhibitors (e.g., voglibose,
acarbose, miglitol, emiglitate), biguanides (e.g., metformin,
buformin or a salt thereof (e.g., hydrochloride, fumarate,
succinate)), insulin secretagogues (e.g., sulfonylurea (e.g.,
tolbutamide, glibenclamide, gliclazide, chlorpropamide,
tolazamide, acetohexamide, glyclopyramide, glimepiride,
glipizide, glybuzole), repaglinide, nateglinide, mitiglinide
or calcium salt hydrate thereof), dipeptidyl peptidase IV
inhibitors (e.g., Alogliptin or a salt thereof (preferably,
benzoate), Vildagliptin, Sitagliptin, Saxagliptin, BI1356,
GRC8200, MP-513, PF-00734200, PHX1149, SK-0403, ALS2-0426, TA-
6666, TS-021, KRP-104, 2-[[6-[(3R)-3-amino-l-piperidinyl]-3,4-
31
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
dihydro-3-methyl-2,4-dioxo-1(2H)-pyrimidinyl]methyl]-4-
fluorobenzonitrile or a salt thereof), (33 agonists (e.g., N-
5984), GPR40 agonists (e.g., compounds described in
W02004/041266, W02004/106276, W02005/063729, W02005/063725,
W02005/087710, W02005/095338, W02007/013689 or W02008/001931),
GLP-l receptor agonists (e.g., GLP-1, GLP-1MR agent,
Liraglutide, Exenatide, AVE-0010, BIM-51077, Aib(8,35)hGLP-
1(7,37)NH2, CJC-1131, Albiglutide), amylin agonists (e.g.,
pramlintide), phosphotyrosine phosphatase inhibitors (e.g.,
1o sodium vanadate), gluconeogenesis inhibitors (e.g., glycogen
phosphorylase inhibitors, glucose-6-phosphatase inhibitors,
glucagon antagonists, FBPase inhibitors), SGLT2 (sodium-
glucose cotransporter 2) inhibitors (e.g., Depagliflozin,
AVE2268, TS-033, YM543, TA-7284, Remogliflozin, ASP1941),
SGLTl inhibitors, ll(3-hydroxysteroid dehydrogenase inhibitors
(e.g., BVT-3498, INCB-13739), adiponectin or agonist thereof,
IKK inhibitors (e.g., AS-2868), leptin resistance improving
drugs, somatostatin receptor agonists, glucokinase activators
(e.g., Piragliatin, AZD1656, AZD6370, TTP-355, compounds
described in W02006/112549, W02007/028135, W02008/047821,
W02008/050821, W02008/136428 or W02008/156757), GIP (Glucose-
dependent insulinotropic peptide), GPR119 agonists (e.g.,
PSN821), FGF21, FGF analogue and the like can be mentioned.
[0083]
As the therapeutic agent for diabetic complications,
aldose reductase inhibitors (e.g., tolrestat, epalrestat,
zopolrestat, fidarestat, CT-112, ranirestat (AS-3201),
lidorestat), neurotrophic factor and increasing agents thereof
(e.g., NGF, NT-3, BDNF, neurotrophic production/secretion
promoting agent described in W001/14372 (e.g., 4-(4-
chlorophenyl)-2-(2-methyl-l-imidazolyl)-5-[3-(2-
methylphenoxy)propyl]oxazole), compounds described in
W02004/039365), PKC inhibitors (e.g., ruboxistaurin mesylate),
AGE.inhibitors (e.g., ALT946, N-phenacylthiazolium bromide
(ALT766), EXO-226, Pyridorin, pyridoxamine), GABA receptor
32
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
agonists (e.g., gabapentin, pregabalin), serotonin and
norepinephrine reuptake inhibitors (e.g., duloxetine), sodium
channel inhibitors (e.g., lacosamide), active oxygen
scavengers (e.g., thioctic acid), cerebral vasodilators (e.g.,
tiapuride, mexiletine), somatostatin receptor agonists (e.g.,
BIM23190), apoptosis signal regulating kinase-1 (ASK-1)
inhibitors and the like can be mentioned.
[0084]
As the therapeutic agent for hyperlipidemia, HMG-CoA
io reductase inhibitors (e.g., pravastatin, simvastatin,
lovastatin, atorvastatin, fluvastatin, rosuvastatin,
pitavastatin or a salt thereof (e.g., sodium salt, calcium
salt)), squalene synthase inhibitors (e.g., compounds
described in W097/10224, for example, N-[[(3R,5S)-i-(3-
acetoxy-2, 2-dimethylpropyl)-7-chloro-5-(2,3-dimethoxyphenyl)-
2-oxo-1,2,3,5-tetrahydro-4,l-benzoxazepin-3-
yl]acetyl]piperidin-4-acetic acid), fibrate compounds (e.g.,
bezafibrate, clofibrate, simfibrate, clinofibrate), anion
exchange resin (e.g., colestyramine), probucol, nicotinic acid
drugs (e.g., nicomol, niceritrol, niaspan), ethyl icosapentate,
phytosterol (e.g., soysterol, gamma oryzanol (y-oryzanol)),
cholesterol absorption inhibitors (e.g., zechia), CETP
inhibitors (e.g., dalcetrapib, anacetrapib), w-3 fatty acid
preparations (e.g., w-3-acid ethyl esters 90) and the like can
be mentioned.
[0085]
As the antihypertensive agent, angiotensin converting
enzyme inhibitors (e.g., captopril, enalapril, delapril and
the like), angiotensin II antagonists (e.g., candesartan
cilexetil, candesartan, losartan, losartan potassium,
eprosartan, valsartan, telmisartan, irbesartan, tasosartan,
olmesart, olmesartan medoxomil, azilsartan and the like), a
calcium antagonists (e.g., manidipine, nifedipine, amlodipine,
efonidipine, nicardipine, amlodipine, cilnidipine and the
like), R blockers (e.g., metoprolol, atenolol, propranolol,
33
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
carvedilol, pindolol and the like), clonidine and the like can
be mentioned.
[0086]
As the antiobesity agent, monoamine uptake inhibitors
(e.g., phentermine, sibutramine, mazindol, fluoxetine,
tesofensine), serotonin 2C receptor agonists (e.g.,
lorcaserin), serotonin 6 receptor antagonists, histamine H3
receptor, GABA-regulating drugs (e.g., topiramate),
neuropeptide Y antagonists (e.g., velneperit), cannabinoid
1o receptor antagonists (e.g., rimonabant, taranabant),
ghrelinant agonists, ghrelin receptor antagonists, ghrelin
acylation enzyme inhibitors, opioid receptor antagonists (e.g.,
GSK-1521498), orexin receptor antagonists, melanocortin 4
receptor agonists, 11(3-hydroxysteroid dehydrogenase inhibitors
(e.g., AZD-4017), pancreatic lipase inhibitors (e.g., orlistat,
cetilistat), (33 agonists (e.g., N-5984), diacylglycerol
acyltransferase 1 (DGAT1) inhibitors, acetyl CoA carboxylase
(ACC) inhibitors, steaoryl-CoA desaturase inhibitors,
microsomal triglyceride transfer protein inhibitors (e.g., R-
256918), Na-glucose cotransporter inhibitors (e.g., JNJ-
28431754, remogliflozin), NFKB inhibitors (e.g., HE-3286),
PPAR agonists (e.g., GFT-505, DRF-11605), phosphotyrosine
phosphatase inhibitors (e.g., sodium vanadate, Trodusquemin),
GPR119 agonists (e.g., PSN-821), glucokinase activators (e.g.,
AZD-1656), leptin, leptin derivatives (e.g., metreleptin),
CNTF (ciliary neurotrophic factor), BDNF (brain-derived
neurotrophic factor), cholecystokinin agonists, glucagon-like
peptide-1 (GLP-1) preparations (e.g., animal GLP-1
preparations extracted from the pancreas of bovine or swine;
3o human GLP-1 preparations genetically synthesized using
Escherichia coli or yeast; fragment or derivative of GLP-1
(e.g., exenatide, liraglutide)), amylin preparations (e.g.,
pramlintide, AC-2307), neuropeptide Y agonists (e.g., PYY3-36,
derivative of PYY3-36, obinepitide, TM-30339, TM-30335),
oxyntomodulin preparations; FGF21 preparations (e.g., animal
34
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
FGF21 preparations extracted from the pancreas of bovine or
swine; human FGF21 preparations genetically synthesized using
Escherichia coli or yeast; fragment or derivative of FGF21)),
feeding deterrents (e.g., P-57) and the like can be mentioned.
s [0087]
As the diuretic, for example, xanthine derivatives (e.g.,
theobromine sodium salicylate, theobromine calcium salicylate
and the like), thiazide preparations (e.g., ethiazide,
cyclopenthiazide, trichloromethyazide, hydrochlorothiazide,
to hydroflumethiazide, benzylhydrochlorothiazide, penfluthiazide,
poly5thiazide, methyclothiazide and the like), antialdosterone
preparations (e.g., spironolactone, triamterene and the like),
carbonic anhydrase inhibitors (e.g., acetazolamide and the
like), chlorobenzenesulfonamide agents (e.g., chlortalidone,
15 mefruside, indapamide and the like), azosemide, isosorbide,
ethacrynic acid, piretanide, bumetanide, furosemide and the
like can be mentioned.
[0088]
As the antithrombotic agents, for example, heparin (e.g.,
20 heparin sodium, heparin calcium, enoxaparin sodium, dalteparin
sodium), warfarin (e.g., warfarin potassium), anti-thrombin
drugs (e.g., aragatroban, dabigatran), FXa inhibitors (e.g.,
rivaroxaban, apixaban, edoxaban, YM150, compounds described in
W002/06234, W02004/048363, W02005/030740, W02005/058823 or
25 W02005/113504), thrombolytic agents (e.g., urokinase,
tisokinase, alteplase, nateplase, monteplase, pamiteplase),
platelet aggregation inhibitors (e.g., ticlopidine
hydrochloride, clopidogrel, prasugrel, E5555, SHC530348,
cilostazol, ethyl icosapentate, beraprost sodium, sarpogrelate
3o hydrochloride) and the like can be mentioned.
[0089]
When the solid preparation of the present invention and a
concomitant drug are used in combination, the administration
period thereof is not limited, and they may be administered
35 simultaneously or in a staggered manner to the administration
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
subject.
Moreover, the solid preparation of the present invention
and a concomitant drug may be administered as separate
preparations, or as a single preparation containing the solid
preparation of the present invention and a concomitant drug:.
[0090]
The dose of the concomitant drug can be appropriately.
determined based on the dose employed in clinical-situations
of each drug. The mixing ratio of the solid preparation of the
to present invention and a concomitant drug can be appropriately
determined depending on the administration subject,.
administration route, target disease, symptom, combination and
the like. When the subject of administration is human, for
example, a concomitant drug can be used in 0.01 to 100 parts
by weight relative to 1 part by weight of the solid
preparation of the present invention.
Using a concomitant drug in this way, superior effects.of
1) an effect of enhancing the action of the solid preparation
of the present invention or a concomitant drug (synergistic
effect of medicament actions);
2) an effect of reducing the dose of the solid preparation of
the present invention or a concomitant drug (medicament dose-
reducing effect as compared to single administration);
3) an effect of reducing the secondary action of the solid
preparation of the present invention or a concomitant drug;.
and the like can be obtained.
[0091]
The present invention also provides a method of improving
the dissolution property of and a method of stabilizing a.
compound represented by the formula (I) or a salt thereof,
and/or a calcium antagonist, contained in a solid preparation,
which comprises adding sugar alcohol. According to the present
invention, the dissolution property of a compound represented
by the formula (I) or a salt thereof in the solid preparation
are significantly improved.
36
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
EXAMPLES
[0092]
The present invention is explained in more detail in
the following by referring to Examples and Experimental
Examples, which are not to be construed as limitative.
In the following Examples and Experimental Examples, as
the components (additives) other than the active ingredient,
the Japanese Pharmacopoeia 15th Revision, the Japanese
Pharmacopoeia Japanese Pharmaceutical Codex or Japanese
to Pharmaceutical Excipients 2003 compatible products were used.
[0093]
In the following Examples, as a compound represented by
the above-mentioned formula (I) or a salt thereof, compound A
or compound B was used.
[0094]
Example 1
Table 1
Composition per preparation (130 mg)
compound A 8 mg
amlodipine besylate 6.93 mg
(5 mg as amlodipine)
D-mannitol 82.754 mg
microcrystalline cellulose 20 mg
hydroxypropylcellulose 4 mg
macrogol 6000 1.8 mg
croscarmellose sodium 5.6 mg
magnesium stearate 0.9 mg
red ferric oxide 0.016 mg
total 130 mg
[0095]
(1) Hydroxypropylcellulose (720.0 g) and macrogol 6000 (324.0
g) were dissolved in purified water (9000 g) to give binding
solution I. Red ferric oxide (2.880 g) was dispersed in
purified water (2880 g) to give dispersion I. Binding solution
37
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
I was mixed with dispersion I and purified water (720.0 g) to
give binding solution II. Amlodipine besylate (1253 g),
compound A (1449 g), D-mannitol (14880 g) and microcrystalline
cellulose (3600 g) were uniformly mixed in a fluid bed
granulator (FD-S2, POWREX Co., Ltd.), and the mixture was
granulated while spraying binding solution II, and then dried
to give a granule. A part of the obtained granule was milled
with a 1.5 mmcp punching screen in a screening mill(P-3, Showa
Kagakukikai Co., Ltd.) to give a milled granule.
1o (2) To the obtained milled granule (40760 g (2 batches)) were
added croscarmellose sodium (1848 g) and magnesium stearate
(297.0 g), and they were mixed in a tumbler mixer (TM20-0-0,
Suehiro Kakoki Co., Ltd.) to give a mixed granule.
(3) The mixed granule obtained above was tableted by a rotary
tableting machine (AQUARIUS-36K, Kikusui Seisakusho) with a
diameter of 7.0 mm punch (tableting pressure 10 kN, weight per
tablet: 130 mg) to give plain tablets of the composition shown
in Table 1.
[0096]
Example 2
Table 2
Composition per preparation (130 mg)
compound A 8 mg
amlodipine besylate 6.93 mg
(5 mg as amlodipine)
D-mannitol 82.705 mg
microcrystalline cellulose 20 mg
hydroxypropylcellulose 4 mg
macrogol 6000 1.8 mg
croscarmellose sodium 5.6 mg
magnesium stearate 0.9 mg
red ferric oxide 0.065 mg
total 130 mg
[0097]
38
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
(1) Hydroxypropylcellulose (720.0 g) and macrogol 6000 (324.0
g) were dissolved in purified water (9900 g) to give binding
solution I. Red ferric oxide (11.70 g) was dispersed in
purified water (1800 g) to give dispersion I. Binding solution
I was mixed with dispersion I and purified water (540.0 g) to
give binding solution II. Amlodipine besylate (1253 g),
compound A (1449 g), D-mannitol (14870 g) and microcrystalline
cellulose (3600 g) were uniformly mixed in a fluid bed
granulator (FD-S2, POWREX Co., Ltd.), and the mixture was
1o granulated while spraying binding solution II, and then dried
to give a granule. A part of the obtained granule was milled
with a 1.5 mmcp punching screen in a screening mill (P-3, Showa
Kagakukikai Co., Ltd.) to give a milled granule.
(2) To the obtained milled granule (40760 g (2 batches)) were
added croscarmellose sodium (1848 g) and magnesium stearate
(297.0 g), and they were mixed in a tumbler mixer (TM20-0-0,
Suehiro Kakoki Co., Ltd.) to give a mixed granule.
(3) The mixed granule obtained above was tableted by a rotary
tableting machine (AQUARIUS-36K, Kikusui Seisakusho) with a
long diameter of 8.5 mm and short diameter of 5.0 mm punch
(tableting pressure 8 kN, weight per tablet: 130 mg) to give
plain tablets of the composition shown in Table 2.
39
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
[0098]
Example 3
Table 3
Composition per preparation (130 mg)
compound A 8 mg
amlodipine besylate 3.47 mg
(2.5 mg as amlodipine)
D-mannitol 86.165 mg
microcrystalline cellulose 20 mg
hydroxypropylcellulose 4 mg
macrogol 6000 1.8 mg
croscarmellose sodium 5.6 mg
magnesium stearate 0.9 mg
yellow ferric oxide 0.065 mg
total 130 mg
[0099]
(1) Hydroxypropylcellulose (720.0 g) and macrogol 6000 (324.0
g) were dissolved in purified water (9900.g) to give binding
solution I. Yellow ferric oxide (11.70 g) was dispersed in
io purified water (1800 g) to give dispersion I. Binding solution
I was mixed with dispersion I and purified water (540.0 g) to
give binding solution II. Amlodipine besylate (627.7 g),
compound A (1449 g), D-mannitol (15550 g) and microcrystalline
cellulose (3600 g) were uniformly mixed in a fluid bed
granulator (FD-S2, POWREX Co., Ltd.), and the mixture was
granulated while spraying binding solution II, and then dried
to give a granule. A part of the obtained granule was milled
with a 1.5 mmcp punching screen in a screening mill (P-3, Showa
Kagakukikai Co., Ltd.) to give a milled granule.
(2) To the obtained milled granule (40760 g (2 batches)) were
added croscarmellose sodium (1848 g) and magnesium stearate
(297.0 g), and they were mixed in a tumbler mixer (TM20-0-0,
Suehiro Kakoki Co., Ltd.) to give a mixed granule.
(3) The mixed granule obtained above was tableted by a rotary
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
tableting machine (AQUARIUS-36K, Kikusui Seisakusho) with a
long diameter of 8.5 mm and short diameter of 5.0 mm punch
(tableting pressure 8 kN, weight per tablet: 130 mg) to give
plain tablets of the composition shown in Table 3.
[0100]
Example 4
Table 4
Composition per preparation (130 mg)
compound A 4 mg
amlodipine besylate 6.93 mg
(5 mg as amlodipine)
D-mannitol 86.705 mg
microcrystalline cellulose 20 mg
hydroxypropylcellulose 4 mg
macrogol 6000 1.8 mg
croscarmellose sodium 5.6 mg
magnesium stearate 0.9 mg
red ferric oxide 0.065 mg
total 130 mg
[0101]
(1) Hydroxypropylcellulose (720.0 g) and macrogol 6000 (324.0
g) were dissolved in purified water (9900 g) to give binding
solution I. Red ferric oxide (11.70 g) was dispersed in
purified water (1800 g) to give dispersion I. Binding solution
I was mixed with dispersion I and purified water (540.0 g) to
give binding solution II. Amlodipine besylate (1253 g),
compound A (724.3 g), D-mannitol (15600 g) and
microcrystalline cellulose (3600 g) were uniformly mixed in a
fluid bed granulator (FD-S2, POWREX Co., Ltd.), and the
mixture was granulated while spraying binding solution II, and
then dried to give a granule. A part of the obtained granule
was milled with a 1.5 mmcp punching screen in a screening mill
(P-3,. Showa Kagakukikai Co., Ltd.) to give a milled granule.
(2) To the obtained milled granule (40760 g (2 batches)) were
41
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
added croscarmellose sodium (1848 g) and magnesium stearate
(297.0 g), and they were mixed in a tumbler mixer (TM20-0-0,
Suehiro Kakoki Co., Ltd.) to give a mixed granule.
(3) The mixed granule obtained above was tableted by a rotary
tableting machine (AQUARIUS-36K, Kikusui Seisakusho) with a
diameter of 7 mm punch (tableting pressure 10 kN, weight per
tablet: 130 mg) to give plain tablets of the composition shown
in Table 4.
[0102]
1o Example 5
Table 5
Composition per preparation (130 mg)
compound A 4 mg
amlodipine besylate 3.47 mg
(2.5 mg as amlodipine)
D-mannitol 90.165 mg
microcrystalline cellulose 20 mg
hydroxypropylcellulose 4 mg
macrogol 6000 1.8 mg
croscarmellose sodium 5.6 mg
magnesium stearate 0.9 mg
yellow ferric oxide 0.065 mg
total 130 mg
[0103]
(1) Hydroxypropylcellulose (720.0 g) and macrogol 6000 (324.0
g) were dissolved in purified water (9900 g) to give binding
solution I. Yellow ferric oxide (11.70 g) was dispersed in
purified water (1800 g) to give dispersion I. Binding solution
I was mixed with dispersion I and purified water (540.0 g) to
give binding solution II. Amlodipine besylate (627.7 g),
compound A (724.3 g), D-mannitol (16220 g) and
microcrystalline cellulose (3600 g) were uniformly mixed in a
fluid bed granulator (FD-S2, POWREX Co., Ltd.), and the
mixture was granulated while spraying binding solution II, and
42
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
then dried to give a granule. A part of the obtained granule
was milled with a 1.5 mmcp punching screen in a screening mill
(P-3, Showa Kagakukikai Co., Ltd.) to give a milled granule.
(2) To the obtained milled granule (40760 g (2 batches)) were
added croscarmellose sodium (1848 g) and magnesium stearate
(297.0 g), and they were mixed in a tumbler mixer (TM20-0-0,
Suehiro Kakoki Co., Ltd.) to give a mixed granule.
(3) The mixed granule obtained above was tableted by a rotary
tableting machine (AQUARIUS-36K, Kikusui Seisakusho) with a
1o diameter of 7 mm punch (tableting pressure 10 kN, weight per
tablet: 130 mg) to give plain tablets of the composition shown
in Table 5.
[0104]
Example 6
Table 6
Composition per preparation (130 mg)
compound A 8 mg
amlodipine besylate 6.93 mg
(5 mg as amlodipine)
D-mannitol 82.666 mg
microcrystalline cellulose 20 mg
hydroxypropylcellulose 4 mg
macrogol 6000 1.8 mg
croscarmellose sodium 5.6 mg
magnesium stearate 0.9 mg
red ferric oxide 0.104 mg
total 130 mg
[0105]
(1) Hydroxypropylcellulose (720.0 g) and macrogol 6000 (324.0
g) were dissolved in purified water (9900 g) to give binding
solution I. Red ferric oxide (18.72 g) was dispersed in
purified water (1800 g) to give dispersion I. Binding solution
I was mixed with dispersion I and purified water (540.0 g) to
give binding solution II. Amlodipine besylate (1248 g),
43
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
compound A (1446 g), D-mannitol (14870 g) and microcrystalline
cellulose (3600 g) were uniformly mixed in a fluid bed
granulator (FD-S2, POWREX Co., Ltd.), and the mixture was
.granulated while spraying binding solution II, and then dried
to give a granule. A part of the obtained granule was milled
with a 1.5 mmcp punching screen in a screening mill (P-3, Showa
Kagakukikai Co., Ltd.) to give a milled granule.
(2) To the obtained milled granule (20380 g) were added
croscarmellose sodium (924.0 g) and magnesium stearate (148.5
1o g), and they were mixed in a tumbler mixer (TM20-0-0, Suehiro
Kakoki Co., Ltd.) to give a mixed granule.
(3) The mixed granule obtained above was tableted by a rotary
tableting machine (AQUARIUS-36K, Kikusui Seisakusho) with a
long diameter of 8.5 mm and short diameter of 5.0 mm punch
(tableting pressure 8 kN, weight per tablet: 130 mg) to give
plain tablets of the composition shown in Table 6.
[0106]
Example 7
Table 7
Composition per preparation (130 mg)
compound A 8 mg
amlodipine besylate 3.47 mg
(2.5 mg as amlodipine)
D-mannitol 86.126 mg
microcrystalline cellulose 20 mg
hydroxypropylcellulose 4 mg
macrogol 6000 1.8 mg
croscarmellose sodium 5.6 mg
magnesium stearate 0.9 mg
yellow ferric oxide 0.104 mg
total 130 mg
[0107]
(1) Hydroxypropylcellulose (720.0 g) and macrogol 6000 (324.0
g) were dissolved in purified water (9900 g) to give binding
44
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
solution I. Yellow ferric oxide (18.72 g) was dispersed in
purified water (1800 g) to give dispersion I. Binding solution
I was mixed with dispersion I and purified water (540.0 g) to
give binding solution II. Aml.odipine besylate (625.2 g),
compound A (1446 g), D-mannitol (15500 g) and microcrystalline
cellulose (3600 g) were uniformly mixed in a fluid bed
granulator (FD-S2, POWREX Co., Ltd.), and the mixture was
granulated while spraying binding solution II, and then dried
to give a granule. A part of the obtained granule was milled
1o with a 1.5 mmcp punching screen in a screening mill (P-3, Showa
Kagakukikai Co., Ltd.) to give a milled granule.
(2) To the obtained milled granule (20380 g),were added
croscarmellose sodium (924.0 g) and magnesium stearate (148.5
g), and they were mixed in a tumbler mixer (TM20-0-0, Suehiro
Kakoki Co., Ltd.) to give a mixed granule.
(3) The mixed granule obtained above was tableted by a rotary
tableting machine (AQUARIUS-36K, Kikusui Seisakusho) with a
long diameter of 8.5 mm and short diameter of 5.0 mm punch
(tableting pressure 8 kN, weight per tablet: 130 mg) to give
plain tablets of the composition shown in Table 7.
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
[0108]
Example 8
Table 8
Composition per preparation (130 mg)
compound A 8 mg
amlodipine besylate 6.93 mg
(5 mg as amlodipine)
D-mannitol 83.166 mg
microcrystalline cellulose 20 mg
hydroxypropylcellulose 4 mg
macrogol 6000 1.3 mg
croscarmellose sodium 5.6 mg
magnesium stearate 0.9 mg
red ferric oxide 0.104 mg
total 130 mg
[0109]
(1) Hydroxypropylcellulose (144.0 g) was dissolved in purified
water (1980 g) to give binding solution I. Yellow ferric oxide
(3.744 g) was dispersed in purified water (360.0 g) to give
to dispersion I. Binding solution I was mixed with dispersion I
and purified water (108.0 g) to give binding solution II.
Macrogol 6000 (4.680 g) was dissolved in binding solution II
(259.6 g) to give binding solution III. Amlodipine besylate
(24.95 g), compound A (28.80 g), D-mannitol (299.4 g) and
microcrystalline cellulose (72.00 g) were uniformly mixed in a
fluid bed granulator (Lab-1, POWREX Co., Ltd.), and the
mixture was granulated while spraying binding solution III,
and then dried to give a granule. A part of the obtained
granule was sieved with a sieve (16 mesh) to give a milled
granule.
(2) To the obtained milled granule (370.5 g) were added
croscarmellose sodium (16.80 g) and magnesium stearate (2.700
g), and they were mixed in a polyethylene bag (4.9 L) to give
a mixed granule.
46
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
(3) The mixed granule obtained above was tableted by a rotary
tableting machine (Correct 19K, Kikusui Seisakusho) with a
long diameter of 8.5 mm and short diameter of 5.0 mm punch
(tableting pressure 7.5 kN, weight per tablet: 130 mg) to give
plain tablets of the composition shown in Table 8.
[0110]
Example 9
Table 9
1o Composition per preparation (130 mg)
compound A 8 mg
6.93 besylate .93 mg
(5 mg as amlodipine)
D-mannitol 80.566 mg
microcrystalline cellulose 20 mg
hydroxypropylcellulose 4 mg
macrogol 6000 3.9 mg
croscarmellose sodium 5.6 mg
magnesium stearate 0.9 mg
red ferric oxide 0.104 mg
total 130 mg
[0111]
(1) Hydroxypropylcellulose (144.0 g) was dissolved in purified
water (1980 g) to give binding solution I. Red ferric oxide
(3.744 g) was dispersed in purified water (360.0 g) to give
dispersion I. Binding solution I was mixed with dispersion I
and purified water (108.0 g) to give binding solution II.
Macrogol 6000 (14.04 g) was dissolved in binding solution II
(259.6 g) to give binding solution III. Amlodipine besylate
(24.95 g), compound A (28.80 g), D-mannitol (290.0 g) and
microcrystalline cellulose (72.00 g) were uniformly mixed in a
fluid bed granulator (Lab-1, POWREX Co., Ltd.), and the
mixture was granulated while spraying binding solution III,
and then dried to give a granule. A part of the obtained
granule was sieved with a sieve (16 mesh) to give a milled
47
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
granule.
(2) To the obtained milled granule (370.5 g) were added
croscarmellose sodium (16.80 g) and magnesium stearate (2.700
g), and they were mixed in a polyethylene bag (4.9 L) to give
a mixed granule.
(3) The mixed granule obtained above was tableted by a rotary
tableting machine (Correct 19K, Kikusui Seisakusho) with a
long diameter of 8.5 mm and short diameter of 5.0 mm punch
(tableting pressure 7.5 kN, weight per tablet: 130 mg) to give
io plain tablets of the composition shown in Table 9.
[0112]
Example 10
Table 10
Composition per preparation (130 mg)
compound A 8 mg
amlodipine besylate 6.93 mg
(5 mg as amlodipine)
D-mannitol 77.966 mg
microcrystalline cellulose 20 mg
hydroxypropylcellulose 4 mg
macrogol 6000 6.5 mg
croscarmellose sodium 5.6 mg
magnesium stearate 0.9 mg
red ferric oxide 0.104 mg
total 130 mg
[0113]
(1) Hydroxypropylcellulose (144.0 g) was dissolved in purified
water (1980 g) to give binding solution I. Red ferric oxide
(3.744 g) was dispersed in purified water (360.0 g) to give
dispersion I. Binding solution I was mixed with dispersion I
and purified water (108.0 g) to give binding solution II.
Macrogol 6000 (23.40 g) was dissolved in binding solution II
(259.6 g) to give binding solution III. Amlodipine besylate
(24.95 g), compound A (28.80 g), D-mannitol (280.7 g) and
48
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
microcrystalline cellulose (72.00 g) were uniformly mixed in a
fluid bed granulator (Lab-1, POWREX Co., Ltd.), and the
mixture was granulated while spraying binding solution III,
and then dried to give a granule. A part of the obtained
granule was sieved with a sieve (16 mesh) to give a milled
granule.
(2) To the obtained milled granule (370.5 g) were added
croscarmellose sodium (16.80 g) and magnesium stearate (2.700
g), and they were mixed in a polyethylene bag (4.9 L) to give
1o a mixed granule.
(3) The mixed granule obtained above was tableted by a rotary
tableting machine (Correct 19K, Kikusui Seisakusho) with a
long diameter of 8.5 mm and short diameter of 5.0 mm punch
(tableting pressure 7.5 kN, weight per tablet: 130 mg) to give
plain tablets of the composition shown in Table 10.
[0114]
Example 11
Table 11
Composition per preparation (130 mg)
compound A 8 mg
amlodipine besylate 6.93 mg
(5 mg as amlodipine)
D-mannitol 74.066 mg
microcrystalline cellulose 20 mg
hydroxypropylcellulose 4 mg
macrogol 6000 10.4 mg
croscarmellose sodium 5.6 mg
magnesium stearate 0.9 mg
red ferric oxide 0.104 mg
total 130 mg
[0115]
(1) Hydroxypropylcellulose (144.0 g) was dissolved in purified
water (1980 g) to give binding solution I. Red ferric oxide
(3.744 g) was dispersed in purified water (360.0 g) to give
49
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
dispersion I. Binding solution I was mixed with dispersion I
and purified water (108.0 g) to give binding solution II.
Macrogol 6000 (37.44 g) was dissolved in binding solution II
(259.6 g) to give binding solution III. Amlodipine besylate
(24.95 g), compound A (28.80 g), D-mannitol (266.6 g) and
microcrystalline cellulose (72.00 g) were uniformly mixed in a
fluid bed granulator (Lab-1, POWREX Co., Ltd.), and the
mixture was granulated while spraying binding solution III,
and then dried to give a granule. A part of the obtained
io granule was sieved with a sieve (16 mesh) to give a milled
granule.
(2) To the obtained milled granule (370.5 g) were added
croscarmellose sodium (16.80 g) and magnesium stearate (2.700
g), and they were mixed in a polyethylene bag (4.9 L) to give
a mixed granule.
(3) The mixed granule obtained above was tableted by a rotary
tableting machine (Correct 19K, Kikusui Seisakusho) with a
long diameter of 8.5 mm and short diameter of 5.0 mm punch
(tableting pressure 7.5 kN, weight per tablet: 130 mg) to give
plain tablets of the composition shown in Table 11.
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
[0116]
Example 12
Table 12
Composition per preparation (135 mg)
Compound B 40 mg
amlodipine besylate 6.93 mg
(5 mg as amlodipine)
D-mannitol 40.1 mg
microcrystalline cellulose 9.72 mg
hydroxypropylcellulose 5.2 mg
macrogol 6000 4 mg
crospovidone 9.75 mg
microcrystalline cellulose 13 mg
magnesium stearate 1.3 mg
premix I 5 mg
total 135 mg
[0117]
(1) Hydroxypropylcellulose (208.0 g) and macrogol 6000 (160.0
g) were dissolved in purified water (2392.0 g) to give a
binding solution. Amlodipine besylate (277.2 g), compound B
io (1605.0 g), D-mannitol (1599.0 g) and microcrystalline
cellulose (388.8 g) were uniformly mixed in a fluid bed
granulator (FD-5S, POWREX Co., Ltd.), and the mixture was
granulated while spraying the binding solution, and then dried
to. give a granule. A part of the obtained granule was milled
with a 1.5 mmcp punching screen in a screening mill (P-3, Showa
Kagakukikai Co., Ltd.) to give a milled granule.
(2) To the obtained milled granule (3602.0 g) were added
crospovidone (331.5 g), microcrystalline cellulose (442.0 g)
and magnesium stearate (44.201 g), and they were mixed in a
tumbler mixer (TM-15, Suehiro Kakoki co., Ltd.) to give a
mixed granule.
(3) The mixed granule obtained above was tableted by a rotary
tableting machine (Correct 12HUK, Kikusui Seisakusho) with a
51
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
diameter of 7.0 mm punch (tableting pressure 4 kN, weight per
tablet: 130 mg) to give core tablets.
(4) Premix I (240.0 g) was dissolved in purified water (2160.0
g) to give a film coating solution. A film coating was formed
s by uniformly spraying the film coating solution on the core
tablets (3120.0 g) in a film coating machine (DRC-500, POWREX
Co., Ltd.) to give film-coated tablets of the composition
shown in Table 12 (weight per tablet: 135 mg). Here, the
premix I is a premixed powder. The composition of the premix I
1o is shown in Table 12a.
Table 12a
Composition of premix I
ratio by weight
hypromellose 9.0
macrogol 6000 2.0
titanium dioxide 1.0
yellow ferric oxide 0.2
52
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
[0118]
Example 13
Table 13
Composition per preparation (135 mg)
Compound B 20 mg
amlodipine besylate 6.93 mg
(5 mg as amlodipine)
D-mannitol 60.1 mg
microcrystalline cellulose 9.72 mg
hydroxypropylcellulose 5.2 mg
macrogol 6000 4 mg
crospovidone 9.75 mg
microcrystalline cellulose 13 mg
magnesium stearate 1.3 mg
premix I 5 mg
total 135 mg
[0119]
(1) Hydroxypropylcellulose (20.80 g) and macrogol 6000 (16.00
g) were dissolved in purified water (239.2 g) to give a
binding solution. Amlodipine besylate (27.72 g), compound B
1o (80.00 g), D-mannitol (240.4 g) and microcrystalline cellulose
(38.88 g) were uniformly mixed in a fluid bed granulator (Lab-
1, POWREX Co., Ltd.), and the mixture was granulated while
spraying the binding solution, and then dried to give a
granule. A part of the obtained granule was sieved with a
sieve (16 mesh) to give a milled granule.
(2) To the obtained milled granule (370.8 g) were added
crospovidone (34.13 g), microcrystalline cellulose (45.50 g)
and magnesium stearate (4.550 g), and they were mixed in a
polyethylene bag (4.9 L) to give a mixed granule.
(3) The mixed granule obtained above was tableted by a rotary
tableting machine (VEL-5, Kikusui Seisakusho) with a diameter
of 7.0 mm punch (tableting pressure 4 kN, weight per tablet:
130 mg) to give core tablets.
53
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
(4) Premix I (40.00 g) was dissolved in purified water (360.0
g) to give a film coating solution. A film coating was formed
by uniformly spraying the film coating solution on the core
tablets (200.0 g) in a film coating machine (DRC-200, POWREX
Co., Ltd.) to give film-coated tablets of the composition
shown in Table 13 (weight per tablet: 135 mg). Here, the
premix I is a premixed powder. The composition of the premix I
is shown in Table 13a.
to Table 13a
Composition of premix I
ratio by weight
hypromellose 9.0
macrogol 6000 2.0
titanium dioxide 1.0
yellow ferric oxide 0.2
[0120]
Example 14
Table 14
Composition per preparation .(135 mg)
Compound B 20 mg
amlodipine besylate 3.47 mg
(2.5 mg as amlodipine)
D-mannitol 63.56 mg
microcrystalline cellulose 9.72 mg
hydroxypropylcellulose 5.2 mg
macrogol 6000 4 mg
crospovidone 9.75 mg
microcrystalline cellulose 13 mg
magnesium stearate 1.3 mg
premix I 5 mg
total 135 mg
[0121]
54
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
(1) Hydroxypropylcellulose (20.80 g) and macrogol 6000 (16.00
g) were dissolved in purified water (239.2 g) to give a
binding solution. Amlodipine besylate (13.88 g), compound B
(80.00 g), D-mannitol (254.2 g) and microcrystalline cellulose
(38.88 g) were uniformly mixed in a fluid bed granulator (Lab-
1, POWREX Co., Ltd.), and the mixture was granulated while
spraying the binding solution, and then dried to give a
granule. A part of the obtained granule was sieved with a
sieve (16 mesh) to give a milled granule.
1o (2) To the obtained milled granule (370.8 g) were added
crospovidone (34.13 g), microcrystalline cellulose (45.50 g)
and magnesium stearate (4.550 g), and they were mixed in a
polyethylene bag (4.9 L) to give a mixed granule.
(3) The mixed granule obtained above was tableted by a rotary
tableting machine (VEL-5, Kikusui Seisakusho) with a diameter
of 7.0 mm punch (tableting pressure 4 kN, weight per tablet:
130 mg) to give core tablets.
(4) Premix I (40.00 g) was dissolved in purified water (360.0
g) to give a film coating solution. A film coating was formed
by uniformly spraying the film coating solution on the core
tablets (200.0 g) in a film coating machine (DRC-200, POWREX
Co., Ltd.) to give film-coated tablets of the composition
shown in Table 14 (weight per tablet: 135 mg). Here, the
premix I is a premixed powder. The composition of the premix I
is shown in Table 14a.
Table 14a
Composition of premix I
ratio by weight
hypromellose 9.0
macrogol 6000 2.0
titanium dioxide 1.0
yellow ferric oxide 0.2
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
[0122]
Example 15
Table 15
s Composition per preparation (135.154 mg)
Compound B 10 mg
amlodipine besylate 3.465 mg
(5 mg as amlodipine)
D-mannitol 83.535 mg
microcrystalline cellulose 13 mg
hydroxypropylcellulose 3.9 mg
macrogol 6000 1.8 mg
low-substituted 13 mg
hydroxypropylcellulose
magnesium stearate 1.3 mg
hypromellose 3.829 mg
titanium dioxide 0.512 mg
macrogol 6000 0.768 mg
red ferric oxide 0.011 mg
yellow ferric oxide 0.034 mg
total 135.154 mg
[0123]
(1) Hydroxypropylcellulose (35.10 g) and macrogol 6000 (16.20
g) were dissolved in purified water (403.7 g) to give a
binding solution. Amlodipine besylate (10.40 g), compound B
1o (30.00 g), D-mannitol (250.6 g) and microcrystalline cellulose
(39.00 g) were uniformly mixed in a fluid bed granulator (Lab-
1, POWREX Co., Ltd.), and the mixture was granulated while
spraying the binding solution, and then dried to give a
granule. A part of the obtained granule was sieved with a
15 sieve (16 mesh) to give a milled granule.
(2) To the obtained milled granule (289.3 g) were added low-
substituted hydroxypropylcellulose (32.50 g) and magnesium
stearate (3.250 g), and they were mixed in a polyethylene bag
(4.9 L) to give a mixed granule.
20 (3) The mixed granule obtained above was tableted by a rotary
56
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
tableting machine (VEL-5, Kikusui Seisakusho) with a diameter
of 6.5 mm punch (tableting pressure 5 kN, weight per tablet:
130 mg) to give core tablets.
(4) Hypromellose (57.44 g) and macrogol 6000 (11.52 g) were
dissolved in purified water (375.0 g) to give film coating
solution I. Titanium dioxide (7.680 g), red ferric oxide
(0.1650 g) and yellow ferric oxide (0.5100 g) were dispersed
in purified water (330.0 g) to give a dispersion I. Film
coating solution I, dispersion I and purified water (68.25 g)
1o were mixed to give film coating solution II. A film coating
was formed by uniformly spraying film coating solution II on
the core tablets (120.0 g) in.a film coating machine (DRC-200,
POWREX Co., Ltd.) to give film-coated tablets of the
composition shown in Table 15 (weight per tablet: 135.154 mg).
[0124]
Example 16
Table 16
Composition per preparation (135.154 mg)
Compound B 10 mg
amlodipine besylate 6.93 mg
(5 mg as amlodipine)
D-mannitol 80.07 mg
microcrystalline cellulose 13 mg
hydroxypropylcellulose 3.9 mg
macrogol 6000 1.8 mg
low-substituted
hydroxypropylcellulose 13 mg
magnesium stearate 1.3 mg
hypromellose 3.829 mg
titanium dioxide 0.512 mg
macrogol 6000 0.768 mg
red ferric oxide 0.011 mg
yellow ferric oxide 0.034 mg
total 135.154 mg
[0125]
57
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
(1) Hydroxypropylcellulose (35.10 g) and macrogol 6000 (16.20
g) were dissolved in purified water (403.7 g) to give a
binding solution. Amlodipine besylate (20.79 g), compound B
(30.00 g), D-mannitol (240.2 g) and microcrystalline cellulose
s (39.00 g) were uniformly mixed in a fluid bed granulator (Lab-
1, POWREX Co., Ltd.), and the mixture was granulated while
spraying the binding solution, and then dried to give a
granule. A part of the obtained granule was sieved with a
sieve (16 mesh) to give a milled granule.
to (2) To the obtained milled granule (289.3 g) were added low-
substituted hydroxypropylcellulose (32.50 g) and magnesium
stearate (3.250 g),.and they were mixed in a polyethylene bag
(4.9 L) to give a mixed granule.
(3) The mixed granule obtained above was tableted by a rotary
15 tableting machine (VEL-5, Kikusui Seisakusho) with a diameter
of 6.5 mm punch (tableting pressure 5 kN, weight per tablet:
130 mg) to give core tablets.
(4) Hypromellose (57.44 g) and macrogol 6000 (11.52 g) were
dissolved in purified water (375.0 g) to give film coating
20 solution I. Titanium dioxide (7.680 g), red ferric oxide
(0.1650 g) and yellow ferric oxide (0.5100 g) were dispersed
in purified water (330.0 g) to give a dispersion I. Film
coating solution I, dispersion I and purified water (68.25 g)
were mixed to give film coating solution II. A film coating
25 was formed by uniformly spraying film coating solution II on
the core tablets (120.0 g) in a film coating machine (DRC-200,
POWREX Co., Ltd.) to give film-coated tablets of the
composition shown in Table 16 (weight per tablet: 135.154 mg).
58
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
[0126]
Example 17
Table 17
Composition per preparation (135.119 mg)
Compound B 20 mg
amlodipine besylate 3.465 mg
(2.5 mg as amlodipine)
D-mannitol 73.535 mg
microcrystalline cellulose 13 mg
hydroxypropylcellulose 3.9 mg
macrogol 6000 1.8 mg
low-substituted
hydroxypropylcellulose 13 mg
magnesium stearate 1.3 mg
hypromellose 3.829 mg
titanium dioxide 0.512 mg
macrogol 6000 0.768 mg
red ferric oxide 0.01 mg
total 135.119 mg
[0127]
(1) Hydroxypropylcellulose (35.10 g) and macrogol 6000 (16.20
g) were dissolved in purified water (403.7 g) to give a
binding solution. Amlodipine besylate (10.40 g), compound B
io (60.00 g), D-mannitol (220.6 g) and microcrystalline cellulose
(39.00 g) were uniformly mixed in a fluid bed granulator (Lab-
1, POWREX Co., Ltd.), and the mixture was granulated while
spraying the binding solution, and then dried to give a
granule. A part of the obtained granule was sieved with a
is sieve (16 mesh) to give a milled granule.
(2) To the obtained milled granule (289.3 g) were added low-
substituted hydroxypropylcellulose (32.50 g) and magnesium
stearate (3.250 g), and they were mixed in a polyethylene bag
(4.9 L) to give a mixed granule.
20 (3) The mixed granule obtained above was tableted by a rotary
tableting machine (VEL-5, Kikusui Seisakusho) with a diameter
59
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
of 6.5 mm punch (tableting pressure 5 kN, weight per tablet:
130 mg) to give core tablets.
(4) Hypromellose (57.44 g) and macrogol 6000 (11.52 g) were
dissolved in purified water (375.0 g) to give film coating
s solution I. Titanium dioxide (7.680 g) and red ferric oxide
(0.1500 g) were dispersed in purified water (330.0 g) to give
a dispersion I. Film coating solution I, dispersion I and
purified water (68.25 g) were mixed to give film coating
solution II. A film coating was formed by uniformly spraying
io film coating solution II on the core tablets (120.0 g) in a
film coating machine (DRC-200, POWREX Co., Ltd.) to give film-
coated tablets of the composition shown in Table 17 (weight
per tablet: 135.119 mg).
[0128]
15 Example 18
Table 18
Composition per preparation (135.119 mg)
Compound B 20 mg
amlodipine besylate 6.93 mg
(5 mg as amlodipine)
D-mannitol 70.07 mg
microcrystalline cellulose 13 mg
hydroxypropylcellulose 3.9 mg
macrogol 6000 1.8 mg
low-substituted
hydroxypropylcellulose 13 mg
magnesium stearate 1.3 mg
hypromellose 3.829 mg
titanium dioxide 0.512 mg
macrogol 6000 0.768 mg
red ferric oxide 0.01 mg
total 135.119 mg
[0129]
20 (1) Hydroxypropylcellulose (35.10 g) and macrogol 6000 (16.20
g) were dissolved in purified water (403.7 g) to give a
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
binding solution. Amlodipine besylate (20.79 g), compound B
(60.00 g), D-mannitol (210.2 g) and microcrystalline cellulose
(39.00 g) were uniformly mixed in a fluid bed granulator (Lab-
1, POWREX Co., Ltd.), and the mixture was granulated while
s spraying the binding solution, and then dried to give a
granule. A part of the obtained granule was sieved with a
sieve (16 mesh) to give a milled granule.
(2) To the obtained milled granule (289.3 g) were added low-
substituted hydroxypropylcellulose (32.50 g) and magnesium
1o stearate (3.250 g), and they were mixed in a polyethylene bag
(4.9 L) to give amixed granule.
(3) The mixed granule obtained above was tableted by a rotary
tableting machine (VEL-5, Kikusui Seisakusho) with a diameter
of 6.5 mm punch (tableting pressure 5 kN, weight per tablet:
15 130 mg) to give core tablets.
(4) Hypromellose (57.44 g) and macrogol 6000 (11.52 g) were
dissolved in purified water (375.0 g) to give film coating
solution I. Titanium dioxide (7.680 g) and red ferric oxide
(0.1500 g) were dispersed in purified water (330.0 g) to give
20 a dispersion I. Film coating solution I, dispersion I and
purified water (68.25 g) were mixed to give film coating
solution II. A film coating was formed by uniformly spraying
film coating solution II on the core tablets (120.0 g) in a
film coating machine (DRC-200, POWREX Co., Ltd.) to give film-
25 coated tablets of the composition shown in Table 18 (weight
per tablet: 135.119 mg).
61
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
[0130]
Example 19
Table 19
Composition per preparation (135.155 mg)
Compound B 40 mg
amlodipine besylate 3.465 mg
(2.5 mg as amlodipine)
D-mannitol 53.535 mg
microcrystalline cellulose 13 mg
hydroxypropylcellulose 3.9 mg
macrogol 6000 1.8 mg
low-substituted 13 mg
hydroxypropylcellulose
magnesium stearate 1.3 mg
hypromellose 3.829 mg
titanium dioxide 0.512 mg
macrogol 6000 0.768 mg
yellow ferric oxide 0.046 mg
total 135.155 mg
[0131]
(1) Hydroxypropylcellulose (35.10 g) and macrogol 6000 (16.20
g) were dissolved in purified water (403.7 g) to give a
binding solution. Amlodipine besylate (10.40 g), compound B
to (120.0 g), D-mannitol (160.6 g) and microcrystalline cellulose
(39.00 g) were uniformly mixed in a fluid bed granulator (Lab-
1, POWREX Co., Ltd.), and the mixture was granulated while
spraying the binding solution, and then dried to give a
granule. A part of the obtained granule was sieved with a
sieve (16 mesh) to give a milled granule.
(2) To the obtained milled granule (289.3 g) were added low-
substituted hydroxypropylcellulose (32.50 g) and magnesium
stearate (3.250 g), and they were mixed in a polyethylene bag
(4.9 L) to give a mixed granule.
(3) The mixed granule obtained above was tableted by a rotary
tableting machine (VEL-5, Kikusui Seisakusho) with a diameter
62
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
of 6.5 mm punch (tableting pressure 5 kN, weight per tablet:
130 mg) to give core tablets.
(4) Hypromellose (57.44 g) and macrogol 6000 (11.52 g) were
dissolved in purified water (375.0 g) to give film coating
solution I. Titanium dioxide (7.680 g) and yellow ferric oxide
(0.6900 g) were dispersed in purified water (330.0 g) to give
a dispersion I. Film coating solution I, dispersion I and
purified water (68.25 g) were mixed to give film coating
solution II. A film coating was formed by uniformly spraying
io film coating solution II on the core tablets (120.0 g) in a
film coating machine (DRC-200, POWREX Co., Ltd.) to give film-
coated tablets of the composition shown in Table 19 (weight
per tablet: 135.155 mg).
[0132]
Example 20
Table 20
Composition per preparation (135.155 mg)
Compound B 40 mg
amlodipine besylate 6.93 mg
(5 mg as amlodipine)
D-mannitol 50.07 mg
microcrystalline cellulose 13 mg
hydroxypropylcellulose 3.9 mg
macrogol 6000 1.8 mg
low-substituted 13 mg
hydroxypropylcellulose
magnesium stearate 1.3 mg
hypromellose 3.829 mg
titanium dioxide 0.512 mg
macrogol 6000 0.768 mg
yellow ferric oxide 0.046 mg
total 135.155 mg
[0133]
(1) Hydroxypropylcellulose (35.10 g) and macrogol 6000 (16.20
g) were dissolved in purified water (403.7 g) to give a
63
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
binding solution. Amlodipine besylate (20.79 g), compound B
(120.0 g), D-mannitol (150.2 g) and microcrystalline cellulose
(39.00 g) were uniformly mixed in a fluid bed granulator (Lab-
1, POWREX Co., Ltd.), and the mixture was granulated while
spraying the binding solution, and then dried to give a
granule. A part of the obtained granule was sieved with a
sieve (16 mesh) to give a milled granule.
(2) To the obtained milled granule (289.3 g) were added low-
substituted hydroxypropylcellulose (32.50 g) and magnesium
io stearate (3.250 g), and they were mixed in a polyethylene bag
(4.9 L) to give a mixed granule.
(3) The mixed granule obtained above was tableted by a rotary
tableting machine (VEL-5, Kikusui Seisakusho) with a diameter
of 7.0 mm punch (tableting pressure 5 kN, weight per tablet:
130 mg) to give core tablets.
(4) Hypromellose (57.44 g) and macrogol 6000 (11.52 g) were
dissolved in purified water (375.0 g) to give film coating
solution I. Titanium dioxide (7.680 g) and yellow ferric oxide
(0.6900 g) were dispersed in purified water (330.0 g) to give
a dispersion I. Film coating solution I, dispersion I and
purified water (68.25 g) were mixed to give film coating
solution II. A film coating was formed by uniformly spraying
film coating solution II on the core tablets (120.0 g) in a
film coating machine (DRC-200, POWREX Co., Ltd.) to give film-
coated tablets of the composition shown in Table 20 (weight
per tablet: 135.155 mg).
64
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
[0134]
Example 21
Table 21
s Composition per preparation (260 mg)
compound A 8 mg
amlodipine besylate 6.93 mg
(5 mg as amlodipine)
D-mannitol 92.554 mg
microcrystalline cellulose 20 mg
lactose hydrate 89.384 mg
corn starch 20 mg
hypromellose 4 mg
hydroxypropylcellulose 4 mg
macrogol 6000 2.6 mg
croscarmellose sodium 11.2 mg
magnesium stearate 1.3 mg
red ferric oxide 0.032 mg
total 260 mg
[0135]
(1) Hypromellose (720.0 g) was dissolved in purified water
(9000 g) to give binding solution I. Red ferric oxide (2.880
g) was dispersed in purified water (2880 g) to give dispersion
to I. Dispersion I and purified water (720.0 g) were mixed in
binding solution I to give binding solution II. Amlodipine
besylate (1249 g), D-mannitol (16660 g) and microcrystalline
cellulose (3600 g) were uniformly mixed in a fluid bed
granulator (FD-S2, POWREX Co., Ltd.), and the mixture was
15 granulated while spraying binding solution II, and then dried
to give a granule. A part of the obtained granule was milled
with a 1.5 mmcp punching screen in a screening mill (P-3, Showa
Kagakukikai Co., Ltd.) to give milled granule I.
(2) Hydroxypropylcellulose (720.0 g) and macrogol 6000 (468.0
20 g) were dissolved in purified water (9000 g) to give binding
solution III. Red ferric oxide (2.880 g) was dispersed in
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
purified water (2880 g) to give dispersion II. Dispersion II
and purified water (720.0 g) were mixed in binding solution
III to give binding solution IV. Compound A (1436 g), lactose
hydrate (16090 g) and corn starch (3600 g) were uniformly
mixed in a fluid bed granulator (FD-S2, POWREX Co., Ltd.), and
the mixture was granulated while spraying binding solution IV,
and then dried to give a granule. A part of the obtained
granule was milled with a 1.5 mmp punching screen in a
screening mill (P-3, Showa Kagakukikai Co., Ltd.) to give
io milled granule II.
(3) To the obtained milled granule I (20380 g) and milled
granule II (20460 g) were added croscarmellose sodium (1848 g)
and magnesium stearate (214.5 g), and they were mixed in a
tumbler mixer (TM20-0-0, Suehiro Kakoki co., Ltd.) to give a
mixed granule.
(4) The mixed granule obtained above was tableted by a rotary
tableting machine (AQUARIUS-36K, Kikusui Seisakusho) with a
diameter of 8.5 mm punch (tableting pressure 10 kN, weight per
tablet: 260 mg) to give plain tablets of the composition shown
in Table 21.
66
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
[0136]
Example 22
Table 22
Composition per preparation (260 mg)
compound A 8 mg
amlodipine besylate 6.93 mg
(5 mg as amlodipine)
D-mannitol 92.554 mg
microcrystalline cellulose 20 mg
lactose hydrate 89.384 mg
corn starch 20 mg
hypromellose 4 mg
hydroxypropylcellulose 4 mg
macrogol 6000 2.6 mg
croscarmellose sodium 5.6 mg
carmellose calcium 5.6 mg
magnesium stearate 1.3 mg
red ferric oxide 0.032 mg
total 260 mg
[0137]
(1) Hypromellose (720.0 g) was dissolved in purified water
(9000 g) to give binding solution I. Red ferric oxide (2.880
g) was dispersed in purified water (2880 g) to give dispersion
1o I. Binding solution I was mixed with dispersion I and purified
water (720.0 g) to give binding solution II. Amlodipine
besylate (1249 g), D-mannitol (16660 g) and microcrystalline
cellulose (3600 g) were uniformly mixed in a fluid bed
granulator (FD-S2, POWREX Co., Ltd.), and the mixture was
is granulated while spraying binding solution II, and then dried
to give a granule. A part of the obtained granule was milled
with a 1.5 mmcp punching screen in a screening mill (P-3, Showa
Kagakukikai Co., Ltd.) to give milled granule I.
(2) To the obtained milled granule I (20380 g) were added
20 croscarmellose sodium (924.0 g) and magnesium stearate (148.5
67
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
g), and they were mixed in a tumbler mixer (TM-60, Showa
Kagakukikai Co., Ltd.) to give mixed granule I.
(3) Hydroxypropylcellulose (720.0 g) and macrogol 6000 (468.0
g) were dissolved in purified water (9000 g) to give binding
solution III. Red ferric oxide (2.880 g) was dispersed in
purified water (2880 g) to give dispersion II. Dispersion'II
and purified water (720.0 g) were mixed in binding solution
III to give binding solution IV. Compound A (1436 g), lactose
hydrate (16090 g) and corn starch (3600 g) were uniformly
io mixed in a fluid bed granulator (FD-S2, POWREX Co., Ltd.), and
the mixture was granulated while spraying binding solution IV,
and then dried to give a granule. A part of the obtained
granule was milled with a 1.5 mmcp punching screen in a
screening mill (P-3, Showa Kagakukikai Co., Ltd.) to give
milled granule II.
(4) To the obtained milled granule II (20460 g) were added
carmellose calcium (924.0 g) and magnesium stearate (66.00 g),
and they were mixed in a tumbler mixer (TM-60, Showa
Kagakukikai Co., Ltd.) to give mixed granule II.
(5) Mixed granule I (130 mg) and mixed granule II (130 mg)
obtained above were tableted by a rotary tableting machine
(HT-CVX54LS-UW/C&3L, HATA IRON WORKS Co., Ltd.) with a
diameter of 8.5 mm punch (tableting pressure 9 kN, weight per
tablet: 260 mg) to give multi-layer plain tablets of the
composition shown in Table 22.
68
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
[0138]
Example 23
Table 23
Composition per preparation (239 mg)
Compound B 40 mg
lactose hydrate 29.3 mg
corn starch 13 mg
microcrystalline cellulose 13 mg
hydroxypropylcellulose 4 mg
macrogol 6000 4 mg
low-substituted 13 mg
hydroxypropylcellulose
microcrystalline cellulose 13 mg
magnesium stearate 0.7 mg
amlodipine besylate 6.93 mg
(5 mg as amlodipine)
D-mannitol 68.94 mg
microcrystalline cellulose 15.33 mg
hydroxypropylcellulose 3.1 mg
carmellose calcium 5 mg
magnesium stearate 0.7 mg
premix I 9 mg
total 239 mg
[0139]
(1) Hydroxypropylcellulose (155.0 g) was dissolved in purified
water (1395.0 g) to give binding solution I. Amlodipine
besylate (346.7 g), D-mannitol (2447.0 g) and microcrystalline
io cellulose (766.7 g) were uniformly mixed in a fluid bed
granulator (FD-5S, POWREX Co., Ltd.), and the mixture was
granulated while spraying binding solution I, and then dried
to give a granule of an amlodipine besylate layer. A part of
the obtained granule was milled with a 1.5 mmcp punching screen
in a screening mill (P-3, Showa Kagakukikai Co., Ltd.) to give
a milled granule of the amlodipine besylate layer.
(2) Hydroxypropylcellulose (280.1 g) and macrogol 6000 (280.0
69
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
g) were dissolved in purified water (2520.2 g) to give binding
solution II. Compound B (2808.4 g), lactose hydrate (2043.5 g),
corn starch (910.3 g) and microcrystalline cellulose (910.2 g)
were uniformly mixed in a fluid bed granulator (FD-5S, POWREX
Co., Ltd.), and the mixture was granulated while spraying
binding solution II, and then dried to give a granule of the
compound B layer. A part of the obtained granule was milled
with a 1.5 mmcp punching screen in a screening mill (P-3, Showa
Kagakukikai Co., Ltd.) to give a milled granule of the
io compound B layer.
(3) To the milled granule of the obtained amlodipine besylate
layer (3772.0 g) were added carmellose calcium (200.0 g) and
magnesium stearate (28.000 g), and they were mixed in a
tumbler mixer (TM-15, Suehiro Kakoki co., Ltd.) to give a
mixed granule of the amlodipine besylate layer.
(4) To the milled granule of the obtained compound B layer
(3616.1 g) were added low-substituted hydroxypropylcellulose
(455.0 g), microcrystalline cellulose (455.1 g) and magnesium
stearate (24.53 g), and they were mixed in a tumbler mixer
(TM-15, Suehiro Kakoki co., Ltd.) to give a mixed granule of
the compound B layer.
(5) The mixed granule of the amlodipine besylate layer and the
mixed granule of the compound B layer obtained above were
tableted by a rotary tableting machine (HT-Xl2SS-UW&2L, HATA
IRON WORKS Co., Ltd.) with a diameter of 8.0 mm punch
(tableting pressure 7 kN, weight per tablet: 230 mg
(amlodipine besylate layer: 100 mg, compound B layer: 130 mg))
to give multi-layer core tablets.
(6) Premix I (252.0 g) was dissolved in purified water (2268.0
g) to give a film coating solution. A film coating was formed
by uniformly spraying the film coating solution on the core
tablets (3120.0 g) in a film coating machine (DRC-500, POWREX
Co., Ltd.) to give film-coated tablets of the composition
shown in Table 23 (weight per tablet: 239 mg). Here, the
premix I is a premixed powder. The composition of the premix I
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
is shown in Table 23a.
Table 23a
Composition of premix I
ratio by weight
hypromellose 9.0
macrogol 6000 2.0
titanium dioxide 1.0
yellow ferric oxide 0.2
[0140]
Example 24
Table 24
io Composition per preparation (130 mg)
compound A 8 mg
amlodipine besylate 6.93 mg
(5 mg as amlodipine)
D-mannitol 82.666 mg
microcrystalline cellulose 20 mg
hydroxypropylcellulose 4 mg
macrogol 4000 1.8 mg
croscarmellose sodium 5.6 mg
magnesium stearate 0.9 mg
red ferric oxide 0.104 mg
total 130 mg
[0141]
(1) Hydroxypropylcellulose (80.00 g) and macrogol 4000 (36.00
g) were dissolved in purified water (1100 g) to give binding
solution I. Red ferric oxide (2.080 g) was dispersed in
purified water (200.1 g) to give dispersion I. Binding
solution I was mixed with dispersion I and purified water
(60.00 g) to give binding solution II. Amlodipine besylate
(24.95 g), compound A (28.80 g), D-mannitol (297.6 g) and
microcrystalline cellulose (72.00 g) were uniformly mixed in a
71
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
fluid bed granulator (Lab-1, POWREX Co., Ltd.), and the
mixture was granulated while spraying binding solution II
(266.1 g), and then dried to give a granule. A part of the
obtained granule was sieved with a sieve (16 mesh) to give a
milled granule.
(2) To the obtained milled granule (370.5 g) were added
croscarmellose sodium (16.81 g) and magnesium stearate (2.700
g), and they were mixed in a polyethylene bag (4.9 L) to give
a mixed granule.
io (3) The mixed granule obtained above was tableted by a rotary
tableting machine (Correct 19K, Kikusui Seisakusho) with a
long diameter of 8.5 mm and short diameter of 5.0 mm punch
(tableting pressure 8.5 kN, weight per tablet: 130 mg) to give
plain tablets of the composition shown in Table 24.
[0142]
Example 25
Table 25
Composition per preparation (130 mg)
compound A 8 mg
amlodipine besylate 6.93 mg
(5 mg as amlodipine)
D-mannitol 82.666 mg
microcrystalline cellulose 20 mg
hydroxypropylcellulose 4 mg
macrogol 10000 1.8 mg
croscarmellose sodium 5.6 mg
magnesium stearate 0.9 mg
red ferric oxide 0.104 mg
total 130 mg
[0143]
(1) Hydroxypropylcellulose (80.00 g) and macrogol 10000 (36.00
g) were dissolved in purified water (1100 g) to give binding
solution I. Red ferric oxide (2.080 g) was dispersed in
purified water (200.1 g) to give dispersion I. Binding
72
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
solution I was mixed with dispersion I and purified water
(60.10 g) to give binding solution II. Amlodipine besylate
(24.95 g), compound A (28.80 g), D-mannitol (297.6 g) and
microcrystalline cellulose (72.00 g) were uniformly mixed in a
.5 fluid bed granulator (Lab-1, POWREX Co., Ltd.), and the
mixture was granulated while spraying binding solution II
(266.1 g), and then dried to give a granule. A part of the
obtained granule was sieved with a sieve (16 mesh) to give a
milled granule.
1o (2) To the obtained milled granule (370.5 g) were added
croscarmellose sodium (16.80 g) and magnesium stearate (2.710
g), and they were mixed in a polyethylene bag (4.9 L) to give
a mixed granule.
(3) The mixed granule obtained above was tableted by a rotary
15 tableting machine (Correct 19K, Kikusui Seisakusho) with a
long diameter of 8.5 mm and short diameter of 5.0 mm punch
(tableting pressure 8.5 kN, weight per tablet: 130 mg) to give
plain tablets of the composition shown in Table 25.
[0144]
20 Comparative Example 1
Table 26
Composition per preparation (130 mg)
compound A 8 mg
amlodipine besylate 6.93 mg
(5 mg as amlodipine)
lactose hydrate 82.454 mg
corn starch 20 mg
hydroxypropylcellulose 4 mg
macrogol 6000 2.6 mg
carmellose calcium 5.6 mg
magnesium stearate 0.4 mg
red ferric oxide 0.016 mg
total 130 mg
[0145]
73
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
(1) Hydroxypropylcellulose (720.0 g) and macrogol 6000 (468.0
g) were dissolved in purified water (9000 g) to give binding
solution I. Red ferric oxide (2.880 g) was dispersed in
purified water (2880 g) to give dispersion I. Binding solution
I was mixed with dispersion I and purified water (720.0 g) to
give binding solution II. Amlodipine besylate (1253 g),
compound A (1449 g), lactose hydrate (14830 g) and corn starch
(3600 g) were uniformly mixed in a fluid bed granulator (FD-S2,
POWREX Co., Ltd.), and the mixture was granulated while
io spraying binding solution II, and then dried to give a granule.
A part of the obtained granule was milled with a 1.5 mmq
punching screen in a screening mill (P-3, Showa Kagakukikai
Co., Ltd.) to give a milled granule.
(2) To the obtained milled granule (40920 g (2 batches)) were
added carmellose calcium (1848 g) and magnesium stearate
(132.0 g), and they were mixed in a tumbler mixer (TM20-0-0,
Suehiro Kakoki co., Ltd.) to give a mixed granule.
(3) The mixed granule obtained above was tableted by a rotary
tableting machine (AQUARIUS-36K, Kikusui Seisakusho) with a
diameter of 7.0 mm punch (tableting pressure 9 kN, weight per
tablet: 130 mg) to give plain tablets of the composition shown
in Table 26.
[0146]
Experimental Example 1
Dissolution test 1
The dissolution property of the active ingredient
(compound A) from the plain tablets obtained in Examples 1 to
11, 24, 25 and Comparative Example 1 was evaluated by a
dissolution test (1.0(w/w)% polysorbate 20 solution, 900 mL,
37 C, Paddle Method, number of rotation 50 rpm). The
dissolution test was performed according to the Japanese
Pharmacopoeia, 15th Edition, Dissolution Test, Apparatus 2
(Paddle Method). The results are shown in Table 27. Table 27
shows average values, maximum values and minimum values of the
dissolution rate after 15 min from the start of the
74
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
dissolution. Examples 1, 4, 5, 8 to 11, 24, 25 and Comparative
Example 1 show average values, maximum values and minimum
values of the dissolution rates of 6 tablets, and Examples 2,
3, 6 and 7 show average values, maximum values and minimum
values of the dissolution rates of 12 tablets.
[0147]
Table 27
dissolution rate (%) after 15 min
sample average maximum minimum
value value value
Comparative 43 47 39
Example 1
Example 1 56 60 51
Example 2 78 84 64
Example 3 75 81 70
Example 4 68 75 61
Example 5 67 72 64
Example 6 74 82 65
Example 7 74 79 69
Example 8 75 80 71
Example 9 74 78 71
Example 10 62 65 57
Example 11 49 56 39
Example 24 75 83 68
Example 25 82 85 75
[0148]
As shown in Table 27, all of the plain tablets of
so Examples 1 to 11, 24, 25 containing sugar alcohol as an
excipient showed good dissolution property of compound A in
comparison to the sugar alcohol-free plain tablet of
Comparative Example 1.
[0149]
Experimental Example 2
Dissolution test 2
The dissolution property of active ingredient (compound
B) from the film-coated tablet obtained in Example 20 was
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
evaluated by a dissolution test (the Japanese Pharmacopoeia,
15th Edition, 2nd fluid for dissolution test, 900 mL, 37 C,
Paddle Method, number of rotation 50 rpm). The dissolution
test was performed according to the Japanese Pharmacopoeia,
15th Edition, Dissolution Test, Apparatus 2 (Paddle Method).
The results are shown in Table 28. Table 28 shows average
values, maximum values and minimum values of the dissolution
rate of 6 tablets at each time point after 5 to 60 min from
the start of the dissolution.
1o [0150]
Table 28
measurement dissolution rate (%)
time (min average maximum minimum
later) value value value
5 34 30 37
77 74 81
90 88 92
92 90 95
94 92 96
95 93 96
45 96 94 99
60 97 95 101
[01511
As shown in Table 28, the film-coated tablet of Example
20 showed good dissolution property of compound B.
15 [0152]
Experimental Example 3
Stability test
The plain tablets of Examples 1, 4 and 5 were preserved
under conditions of sealed glass bottle and 25 C/60%RH for 12
20 months, the mass of analogues derived from compound A or
amlodipine besylate was measured, and the preservation
stability was evaluated based thereon. The results are shown
in Table 29. The values in the Table 29 show the rates (%) of
the total analogues derived from compound A or amlodipine
76
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
besylate when the content of compound A or amlodipine besylate
(compound A; 8 mg or 4 mg/amlodipine besylate; 6.93 mg or 3.47
mg) is 100%.
[0153]
Table 29
total total
analogue analogue
Sample conditions derived from derived from
compound A amlodipine
besylate
when experiment was 0.21% 0.12%
started
Example 1
25 C/60%RH 12 months
reserved product 0.86% 0.18%
when experiment was 0.49% 0.12%
started
Example 4
25 C/60%RH 12 months
reserved product 1.21% 0.21%
when experiment was 0.48% 0.18%
started
Example 5
25 C/60%RH 12 months
reserved product 1.12% 0.29%
[0154]
As shown in Table 29, the plain tablets of Examples 1, 4
and 5 showed good preservation stability.
INDUSTRIAL APPLICABILITY
io [0155]
The present invention provides a solid preparation
comprising a compound represented by the formula (I) or a salt
thereof, sugar alcohol, and a calcium antagonist, which
appropriately controls dissolution of the compound represented
by the formula (I) or a salt thereof and the calcium
antagonist from the solid preparation in the gastrointestinal
tract, and maintains good stability thereof in the solid
preparation.
77
CA 02760073 2011-10-26
WO 2010/126168 PCT/JP2010/057923
This application is based on patent application Nos.
2009-111381 and 2010-68625 filed in Japan, the contents of
which are incorporated in full herein by this reference.
78