Note: Descriptions are shown in the official language in which they were submitted.
CA 02762224 2011-11-16
WO 2011/143753 PCT/CA2011/000579
TITLE RECOVERY AND PURIFICATION OF HYDROXY
FATTY ACIDS FROM SOURCE OILS
FIELD OF THE INVENTION
This invention relates to processes for recovery, purification and enrichment
of hydroxy fatty acids from source oils.
BACKGROUND
Oil recovered from castor seed (Ricinus communis L.) is an important raw
material in many industrial processes and/or syntheses due to its high content
(i.e., in
the range of 80% - 90%) of the hydroxy fatty acid (HFA) ricinoleic acid (Eq
1).
O
OH
OH Eq I
The highly reactive hydroxy groups can be engaged in various chemical
reactions and result in diverse end products such as lubricants, coatings and
pharmaceuticals. For example, after recovery from castor oil, ricinoleic acid
may be
further converted to sebacic acid and capryl alcohol. Sebacic acid can be
polymerized
with hexamethylene diisocyanate to produce nylon-6,10. Capryl alcohol can be
used
in the production of plasticizers. Dehydration of ricinoleic acid, which
occurs at the
hydroxyl group, produces conjugated double bond structures. Accordingly,
ricinoleic
acids can be used as semi-drying oils.
However, castor plants and their seeds contain ricin and ricinine that are
extremely toxic to many organisms including mammals, avian species and marine
life. Consequently, there are significant safety concerns associated with the
harvesting
and processing of castor seed crops to produce oil.
CA 02762224 2011-11-16
WO 2011/143753 2 PCT/CA2011/000579
One strategy to overcome the disadvantages of working with castor seed and
castor oil has been to genetically modify other types of oil-seed plants to
produce
ricinoleic acid. However, it has been found that in comparison to castor seed,
such
genetically transformed plants typically produce very low levels of ricinoleic
acid. As
a result, considerable efforts and expense are required to recover, purify and
enrich
ricinoleic acid and other HFA from source oils produced from genetically
modified
plants.
It is possible to recover the hydroxy fatty acids lesquerolic acid (14-hydroxy-
11-eicosenoic acid) and auricolic acid (14-hydroxy-11,17-eicosadienoic acid)
from
Lesquerella fendleri and Lesquerella gordonii oils by a process incorporating
low-
temperature crystallization. Lesquerella oils are first hydrolyzed for 3 hours
then
acidified to obtain free fatty acids (FFA). The FFA are then extracted with
hexane,
washed with phosphate buffer and dried to recover the FFA. The FFA are then
dissolved in hexane and chilled to -25 C overnight to allow crystallization
and
separation of the HFA. Finally, HFA are filtered, washed and dried. Although
this
method enriched lesquerolic acid and auricolic acid from 55-59% to 85-99% with
94% yield, the process required large amounts of solvents, long processing
times and
carefully controlled processing temperatures (i.e. -25 C).
Another method to isolate HFA from source oils is based on salt solubility
fractionation. Potassium salts of ricinoleic acid are isolated from castor oil
by their
different solubilities in different solvent systems at certain temperatures.
However, in
addition to the complexity of the process, the method is not effective for the
separation of ricinoleic acid from oleic acid and linoleic acid.
Other strategies assessed separation and recovery of HFA from source oils
based on urea fractionation of the fatty acids. However, it was found that
this
approach is more useful for the separation and recovery of polyunsaturated
fatty acids
(PUFA) rather than HFA.
Other methods for recovery of HFA from source oils incorporate liquid-liquid
extraction steps. These approaches are based on the polarities of FFA/FAME and
their
CA 02762224 2011-11-16
WO 2011/143753 3 PCT/CA2011/000579
solubilities in bi-phase solvent systems. However, these processes are complex
and
require large amounts of solvents and long processing times.
SUMMARY OF THE INVENTION
The exemplary embodiments of the present invention relate to processes for
recovering, purifying and enriching hydroxy fatty acids from source oils. The
processes generally comprise the steps of. (a) methylating a source oil to
form a blend
of hydroxy fatty acid methyl esters and non-hydroxy fatty acid methyl esters;
(b)
separating the hydroxy fatty acid methyl esters from the non-hydroxy fatty
acid
methyl esters with an organic solvent mixture comprising methylpentane/hexane
and
a short-chain alcohol; and (c) separately recovering the hydroxy fatty acid
methyl
esters and the non-hydroxy fatty acid methyl esters.
Some exemplary embodiments relate to a process for the recovery,
purification and enrichment of hydroxy fatty acids exemplified by ricinoleic
acid (12-
hydroxy-9-cis-octadecenoic acid), densipolic acid (12-hydroxy-cis-9,15-
octadecadienoic acid), lesquerolic acid (14-hydroxy-cis-II-eicosenoic acid),
and
auricolic acid (14-hydroxy-cis-11,17-eicosadienoic acid), and the like.
Some exemplary embodiments of the present invention relate to processes for
recovery and processing of non-hydroxy fatty acid methyl esters into biodiesel
fuels
and/or lubricants.
BRIEF DESCRIPTION OF THE DRAWINGS
The present invention will be described in conjunction with reference to the
following drawing, in which:
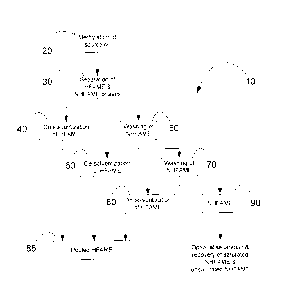
Fig. I is a schematic flowchart showing an exemplary process of the present
invention for separation and recovery of hydroxy fatty acids from a source
oil;
Fig. 2 is a schematic flowchart showing another exemplary process of the
present invention for separation and recovery of hydroxy fatty acids from a
source oil;
and
CA 02762224 2011-11-16
WO 2011/143753 4 PCT/CA2011/000579
Fig. 3 is a schematic flowchart showing an exemplary process for separating
non-hydroxy fatty acid methyl esters into saturated non-hydroxy fatty acid
methyl
esters and unsaturated non-hydroxy fatty acid methyl esters.
DETAILED DESCRIPTION OF THE INVENTION
Unless otherwise defined, all technical and scientific terms used herein have
the same meaning as commonly understood by one of ordinary skill in the art to
which this invention belongs. In order that the invention herein described may
be fully
understood, the following acronyms, terms and definitions are provided herein.
As used herein, the term "depleted" means lessened in quantity or content.
As used herein, the term "enriched" means increased in quantity or content.
As used herein, the term "selective" means to take by preference so as to
increase the percentage of the selected object(s), item(s) or thing(s) in the
selected
portion.
As used herein, FFA means free fatty acids.
As used herein, FAME means fatty acid methyl esters.
As used herein, HFA means hydroxy fatty acids.
As used herein, HFAME means hydroxy fatty acid methyl esters.
As used herein, NHFA means non-hydroxy fatty acids.
As used herein, NHFAME means non-hydroxy fatty acid methyl esters.
The exemplary embodiments of the present invention relate to processes for
the recovery of HFA from source oils. Some aspects relate to purification of
the
recovered HFA. Some aspects relate to processes for the concentration of
and/or
enrichment of purified HFA. Source oils are derived from plant materials
containing
HFA. Exemplary plant materials include seeds, nuts, stems, leaves, tubers and
the
like. The plant materials may be harvested or otherwise recovered from plants
that are
naturally occurring and/or hybridized and/or genetically modified and/or
genetically
CA 02762224 2011-11-16
WO 2011/143753 5 PCT/CA2011/000579
engineered. Suitable plants are exemplified by those producing high-oil
content grain
or seeds or nuts that comprise HFA, including but not limited to castor bean,
camelina, legumes, palms, Lesquerella sp., genetically modified Brassica sp.,
genetically modified Brassicacea sp., genetically modified camelina,
genetically
modified Lesquerella sp., genetically modified maize, genetically modified
legumes,
and genetically modified palm. Particularly suitable are non-food-use host
plants that
have been genetically engineered with a gene sequence coding for expression of
one
or more HFA (i.e., ricinoleic acid and/or densipolic acid and/or lesquerolic
acid
and/or auricolic acid) operably linked to a prornotor. Surprisingly, it has
been found
that the processes of the present invention are useful for recovering HFAME
from
seed or nut oils from genetically engineered plants wherein the source oils
contain
less than 6% HFAME by weight, and then purifying and concentrating the HFAME
from the source oils to about 68% w/w and greater.
Exemplary hydroxy fatty acids that can be recovered and/or purified and/or
enriched by the processes of the present invention include ricinoleic acid (12-
hydroxy-9-octadecadienoic acid), densipolic acid (12-hydroxy-9,15-
octadecadienoic
acid), lesquerolic acid (14-hydroxy-cis-II-eicosenoic acid), and auricolic
acid (14-
hydroxy-11,17-eicosadienoic acid), among others.
Certain exemplary embodiments of the present invention relate to processes
for concurrent or sequential recovery of HFA and NHFA from source oils. Some
aspects relate to separate purification of the recovered HFA and NHFA. Some
aspects
relate to separate concentration and/or enrichment of the purified HFA and
NHFA.
Suitable source oils are derived from plant materials containing HFA and NHFA,
for
example derived from seeds or nuts produced by plants. The plants may be
naturally
occurring, hybridized, genetically modified, and/or genetically engineered.
Suitable
plants are exemplified by those producing high-oil content grain or seed or
nuts that
comprise HFA, including but not limited to castor bean, camelina, Lesquerella
sp.,
genetically modified Brassica sp., genetically modified Brassicacea sp.,
genetically
modified camelina, genetically modified Lesquerella sp., genetically modified
maize,
genetically modified legumes, and genetically modified palm. Particularly
suitable are
non-food-use host plants that have been genetically engineered with a gene
sequence
coding for expression of ricinoleic acid. Surprisingly, it has been found that
the
CA 02762224 2011-11-16
WO 2011/143753 6 PCT/CA2011/000579
processes of the present invention are useful for recovering HFA from seed or
nut
materials containing less than 6% HFA by weight, then purifying and enriching
the
HFAME to about 68% w/w and greater, while concurrently recovering, purifying
and
enriching the NHFAME to about 90% w/w and greater.
Certain exemplary embodiments of the present invention relate to processes
for concentrating and/or purifying HFAME from source oils comprising HFA
(Figs. 1
and 2). A selected source oil is methylated 20 by intermixing with
KOH/methanol
mixture to obtain FAME. FAME are then mixed with 80% methanol and a non-polar
solvent exemplified by methylpentane/hexane. The resulting mixture 30 is
allowed to
separate into a NHFAME-containing upper phase and a HFAME-containing lower
phase. HFAME are recovered by desolventization of the lower phase 40. The
upper
NHFAME phase is washed with a C1_3 alcohol exemplified by 80% methanol 50 and
then allowed to separate into a second NHFAME-containing upper phase and a
second HFAME-containing lower phase. Additional HFAME are recovered by
desolventization of the second lower phase 60. The second upper NHFAME phase
is
extracted a third time with 80% methanol 70 and then allowed to separate into
a third
NHFAME-containing upper phase and a third HFAME-containing lower phase.
Additional HFAME are recovered by desolventization of the third lower phase
80.
The recovered HFAME from each extraction can be pooled 85. The enriched HFAME
contains ricinoleic acid and/or densipolic acid and/or lesquerolic acid and/or
auricolic
acid and the like. It is optional to continue washing the third NHFAME-
containing
upper phase 90 if necessary to separate additional HFAME that can subsequently
be
recovered by desolventization 100 and pooling 105 with the HFAME previously
recovered by the process shown in Fig. 2.
The washed NHFAME-containing phases 90, 110 comprise a blend of
saturated NHFAME and unsaturated NHFAME. If so desired, the saturated NHFAME
and unsaturated NHFAME can be separated and recovered from the NHFAME phases
by a process 200 comprising complexation with a urea-organic solvent mixture
220 to
create a saturated NHFAME enriched solids fraction and an unsaturated NHFAME
enriched liquid fraction 210 (Fig. 3). The solvent of choice for use in this
step of the
process is a C 1-3 alcohol with or without water at a weight ratio of at least
about 2:1
solvent to urea, preferably about 3:1 to 10:1, most preferably about 4:1 to
5:1. Urea
CA 02762224 2011-11-16
WO 2011/143753 7 PCT/CA2011/000579
should be employed at a weight ratio of at least 1:1 urea to fatty acid,
preferably at a
weight ratio of about 2:1 to 5:1, most preferably about 3:1. A weight ratio of
less than
about 1:1 tends to result in incomplete complexation of the fatty acids while
a weight
ratio in excess of about 5:1 increases processing cost without a concomitant
increase
in yield or separation efficiency.
Separation of the enriched saturated fatty acid-enriched NHFAME solids
fraction 230 and unsaturated fatty acid-enriched NHFAME liquid fraction 240
from
the blend 210 can be achieved by any of the well-known solid-liquid separation
techniques. Suitable processes and systems include specifically, but not
exclusively,
decantation, countercurrent decantation, gravity sedimentation, filtration,
expression,
centrifugation and combinations thereof. The spent urea-solvent solution 250
is
recovered from the unsaturated-NHFAME enriched liquid fraction 240 after which
the unsaturated-NHFAME can be intermixed with fresh urea-organic solvent
mixture
220 to further precipitate saturated fatty acid-enriched solids which can then
be
recovered 250 from the remaining unsaturated NHFAME enriched liquid fraction
260. The spent urea-solvent solution 290 is recovered from the unsaturated-
NHFAME
enriched liquid fraction 280 after which the unsaturated-NHFAME 300 are washed
with water acidified to a pH of about 3-4 310. After separation of the waste
water
stream 330, the purified unsaturated NHFAME 320 can be used to produce if so
desired, a cold-tolerant biodiesel fuel.
The following examples are provided to more fully describe the invention and
are presented for non-limiting illustrative purposes.
EXAMPLES
EXAMPLE 1: Purification qfHFAME using castor oil as the source oil
Castor oil was dried under vacuum for 30 min at 100 C and then cooled to
60 C. Dried KOH (2% w/w of oil weight) was dissolved in methanol (20% w/w of
oil weight) in a beaker. Once the oil was cooled to 60 C, the KOH/methanol
mixture
was added to the oil. The mixture was mixed in the rotary evaporator for 4 h.
Then,
the mixture was transferred into a separatory funnel for phase separation. The
top
layer comprised castor oil methyl esters. The bottom layer was recovered and
suitably
CA 02762224 2011-11-16
WO 2011/143753 8 PCT/CA2011/000579
discarded. The top layer was transferred to a beaker and heated to 60 C under
nitrogen after which, a soap analysis was then done on the heated top layer.
Then,
Trysil S615 was added to the heated top layer (1% w/w Trysil S615 per 1000 ppm
of
soap) followed by mixing for 15 min before filtering to recover and separate
the
residual soap from the castor oil methyl esters. The castor oil methyl ester
fraction,
comprising HFAME and NHFAME, was then dried under vacuum and stored for
subsequent use as the starting material in an exemplary embodiment of the
HFAME
purification process of the present invention.
Separation and purification of HFAME from castor oil methyl ester fraction
was carried out as follows. In a reparatory funnel, lOg castor oil methyl
esters were
mixed with a mixture containing 200g of 80% methanol and 50 g of
methylpentane/hexane mixture to obtain a ratio of 1:20:5 (w/w/w; methyl esters
: 80%
methanol : methylpentane/hexane). The mixture was shaken vigorously for 30 s
and
then allowed to separate into two layers. NHFAME separated into the top layer
while
the HFAME separated into the bottom layer. The layers were separated and
desolventized to obtain enriched NHFAME and HFAME fractions. Ten L of each
fraction were diluted with 4mL hexane and individually passed through a gas
chromatograph (Agilent model 6890N) equipped with a DB-23 column (0.25 mm X
30 M, 0.25 m thick) and a flame ionization detector. The fatty acid
composition
(FAC) of castor oil, enriched HFAME and NHFAME are listed in Table 1.
CA 02762224 2011-11-16
WO 2011/143753 9 PCT/CA2011/000579
Table 1: Fatty acid compositions of castor oil methyl esters, HFAME and
NHFAME.
FAME Castor oil HFAME NHFAME
16:0 1.37 - 2.44
18:0 1.46 - 2.70
18:1-9 3.73 0.20 6.72
18:1-11 0.60 - 1.11
18:2 5.15 0.44 9.39
18:3 0.57 - 0.86
20:0 0.11 - 0.19
20:1-11 0.45 - 0.66
18:1-OH 86.18 99.37 75.13
Total HFA 86.18 99.37 75.13
Total FAME 99.61 100.00 99.19
These data show that the process increased the HFAME concentration in the
recovered and enriched fraction from 86.18% to 99.37%.
EXAMPLE 2: Purification of HFA ME from oil crushed from genetically
engineered soy plants
An exemplary process used for recovering, purifying and enriching HFA from
soy oil is illustrated in Fig 1. Seeds were harvested from genetically
engineered
soybean plants that were provided with a castor bean oleate hydroxylase gene
sequence coding for a non-native HFA, using the pMS737.4 vector. As a
consequence
of the introduced oleate hydroxylase gene sequence, the genetically modified
soybean
plants produced ricinoleic acid (i.e. a non-native HFA in soy). The seeds were
crushed
to produce soybean oil comprising the non-native HFA (i.e., HF-soy oil). The
HF-soy
oil was dried under vacuum for 30 min at 100" C and then cooled to 60 C.
Dried
KOH (2% w/w of oil weight) was dissolved in methanol (20% w/w of oil weight)
in a
beaker. Once the oil was cooled to 60 C, the KOH/methanol mixture was added
to
the oil. The mixture was mixed in the rotary evaporator for 4 h. Then, the
mixture was
transferred into a reparatory funnel for phase separation. The top layer
comprised soy
oil methyl esters. The bottom layer was recovered and suitably discarded. The
top
CA 02762224 2011-11-16
WO 2011/143753 10 PCT/CA2011/000579
layer was transferred to a beaker and heated to 60 C under nitrogen after
which, a
soap analysis was then done on the heated top layer. Then, Trysil S615 was
added to
the heated top layer (I% w/w Trysil S615 per 1000 ppm of soap) followed by
mixing
for 15 min before filtering to recover and separate the residual soap from the
soy oil
methyl esters. The soy oil methyl ester fraction, comprising a blend of HFAME
and
NHFAME, was then dried under vacuum and stored for subsequent use as the
starting
material in an exemplary embodiment of the HFAME purification process of the
present invention.
The soy oil methyl ester fraction comprising the blend of HFAME and
NHFAME, was mixed together with 80% methanol and 100% hexane/methylpentane
in the ratio of 1:5:5 (w/w/w). This first mixture was stirred for about 1-2
hat ambient
room temperature under nitrogen. The first mixture was then transferred to a
separatory funnel and allowed to rest for a period of time to allow phase
separation to
occur (referred to as the first extraction). The first bottom phase comprising
the
HFAME and methanol was recovered from the reparatory funnel and then
desolventized by rotary evaporation. The first top phase comprising the NHFAME
and hexane/methylpentane was recovered and then mixed together with 80%
methanol and 100% hexane/methypentane in the ratio of 1:5:5 (w/w/w). This
second
mixture was stirred for about 1-2 h at ambient room temperature under
nitrogen. The
second mixture was then transferred to a separatory funnel and allowed to rest
for a
period of time to allow phase separation to occur (referred to as the second
extraction). The second bottom phase comprising the HFAME and methanol was
recovered from the reparatory funnel and then desolventized by rotary
evaporation.
The second top phase comprising NHFAME was recovered and then mixed together
with 80% methanol and 100% methylpentane/hexane in the ratio of 1:5:10
(w/w/w).
This third mixture was stirred for about 1-2 h at ambient room temperature
under
nitrogen. The third mixture was then transferred to a separatory funnel and
allowed to
rest for a period of time to allow phase separation to occur (referred to as
the third
extraction). The third bottom phase comprising HFAME and methanol was
recovered
from the separatory funnel and then desolventized by rotary evaporation. The
third
top phase comprising NHFAME was recovered and then desolventized by rotary
evaporation. The desolventized NHFAME were suitable for further processing to
produce cold-tolerant biodiesel and/or lubricants. The fatty acid composition
of
CA 02762224 2011-11-16
WO 2011/143753 11 PCT/CA2011/000579
HFAME from each of the extractions and of the NHFAME were analyzed and
quantified by diluting a 10 L aliquot of each extraction with 4mL hexane and
then
passing it through a gas chromatograph (Agilent model 6890N) equipped with a
DB-
23 column (0.25 mm X 30 M, 0.25 m thick) and a flame ionization detector. The
data showing the fatty acid compositions of HF-soy oil, purified HFAME and
NHFAME fractions are listed in Table 2.
Table 2: Fatty acid compositions of HF-soy oil, HFAME and NHFAME fractions
FAME HF-soy oil HFAME-1 * HFAME-2** HFAME-3*** NHFAME
% (w/w) % (w/w) % (w/w) % (w/w) % (w/w)
16:0 8.27 1.77 2.13 1.82 8.56
18:0 4.55 0.45 0.58 0.50 4.75
18:1-9 31.76 5.86 7.29 6.29 33.06
18:1-11 1.55 - - - 1.62
18:2 42.37 13.18 15.96 14.22 43.74
18:3 4.48 2.19 2.63 2.40 4.57
20:0 0.44 0.43 - - 0.47
20:1-11 0.30 - - - 0.31
18:1-OH 5.40 65.82 61.93 65.50 2.47
18:2-OH 0.44 7.47 6.23 6.11 2.47
Total HFAME 5.84 73.29 68.15 71.61 2.47
Total FAME 99.56 97.17 96.74 96.85 99.54
% yield (w/w) 2.83 2.35 1.15 93.42
* 1St extraction
**2^d extraction
` d extraction
***3
The data show that although the total HFAME content of HF-soy oil was less
than 6% (w/w), in each of the three washings and extractions of methylated HF-
soy
oil fatty acids with a solvent mixture of methanol and hexane, the % HFAME
content
in the recovered and enriched product was over 68%, while the total yield of
NHFAME was 93.42% (w/w).
CA 02762224 2011-11-16
WO 2011/143753 12 PCT/CA2011/000579
EXAMPLES 3-5: Enrichment of HFAME using model systems as source oils
Plant systems may be genetically engineered to enable their production of
C18:1-OH from C18:1-9. In addition, C18:2 may also be derived from C18:1-9. It
is
likely that the synthesis and production of these three fatty acids is
interelated, and
may also vary considerably in different types of plants. Furthermore, the
polarities of
C18:1-9 and C18:2 may be similar to C18:1-OH and thereby, affect separation
and
recovery of C18:1-OH from source oils. Therefore, model systems comprising
different mixtures (i.e., formulae) of C18:1, C18:2 and C18:1-OH were prepared
and
used to assess the efficiencies of exemplary embodiments of the present
process for
separation, recovery, purification and enrichment of C18:1-OH.
EXAMPLE 3:
Five model mixtures of fatty acid methyl esters were prepared to assess the
effects of variable linoleic acid (C18:2) and hydroxylated oleic acid (C18:1-
OH)
levels on the separation and purification of C18:1-OH with an exemplary
process of
the present invention. The model mixtures comprised methyl esters of palmitic
acid
(C16:0), stearic acid (C18:0), oleic acid (C18:1-9; C18:1-11), linoleic acid
(C18:2),
linolenic acid (C18:3), arachidic acid (C20:0), eicosenoic acid (C20:1-11),
and
ricinoleic acid (C18:1-OH). Methyl esters of palmitic acid, stearic acid,
oleic acid,
linoleic acid, linolenic acid, arachidic acid, and eicosenoic acid were
purchased from
Nu-Chek Prep Inc. (Elysian, MN, US). Ricinoleic acid methyl ester was
separated and
recovered from castor oil following the process described in Example 1. The
purity
and concentration of the resulting C18:1-OH HFAME were determined by GC
analysis. Twenty-gram quantities of each formula were prepared and stored in a
freezer until required. The five model mixtures i.e. formulae 1-5 were
prepared for
this study as shown in Table 3. For this study, the oleic acid content of each
formula
was constant while the levels of linoleic acid and ricinoleic acid were
varied.
CA 02762224 2011-11-16
WO 2011/143753 11 PCT/CA2011/000579
Table 3:
FAME (g) Formula I Formula 2 Formula 3 Formula 4 Formula 5
C16:0 1.71 1.71 1.71 1.71 1.71
C18:0 0.90 0.90 0.91 0.91 0.92
C 18:1-9 6.41 6.42 6.43 6.41 6.42
C18:1-11 0.34 0.33 0.36 0.33 0.32
C18:2 8.60 8.21 7.60 7.21 6.61
C18:3 0.95 1.06 0.97 0.92 0.94
C20:0 0.10 0.10 0.10 0.11 0.10
C20:1-11 0.07 0.07 0.06 0.07 0.06
C18:1-OH 1.01 1.41 2.02 2.42 3.01
Total 20.10 20.21 20.16 20.09 20.10
FAME
The HFAME and NHFAME components of each formula were fractionated as
follows. Approximately I Og of model FAME were mixed with 80% methanol and
hexane/methylpentane at a ratio of 1:5:5 (w/w/w) in a separatory funnel. The
mixture
was shaken vigorously for 30 s. The mixture was then allowed to separate into
two
clear layers before each phase was recovered and desolventized. Two
fractionations
(replicates) were carried out and each fractionation was done with 10 g of
sample.
The fatty acid compositions in the samples were analyzed using GC as described
in
the previous examples. The weight yields of HFAME were determined
gravimetrically and the percentage yields were calculated. The study was
repeated a
second time, the data were averaged and are shown in Table 4.
CA 02762224 2011-11-16
WO 2011/143753 PCT/CA2011/000579
l~ oo M N o0
Q o N M N N , , r
O -a a)
00 O \O M
Lt. p M
oo 0, I M Q:
C 00 M V O~-
V1 V> O r M O
\ ~t N N ~ ~ N O
(y --~ O ~n O a ~h
C
Li, U 01 M M M \O In
00
00 cN O O
V-) CC 1 t N M N o0
Q o M oo N r~ N N
M Lt ,--~ O d O C~ N M
cd r. _
Li., M N M M O c- M M O
k v~ [~ oo O ~n M 00
00
00 M --O O
O W
C C M M '-" O
0a) \ 00 M M O\ i i M
N
U cd
O p N
00 M M
- ^ k 00 4 C- - ~n O O
C
U
W
M M 00
p Q _ M O I
'z~
c CZ
O '^ 00 M O O M
u
U
w - x -
o o c~ r o ;Q ai
CC 00 0o c)
00 - N o
H
CA 02762224 2011-11-16
WO 2011/143753 15 PCT/CA2011/000579
The data in Table 4 show that when C18:1-OH was present in the model oil
mixture at
a very low level i.e., 3.82% of the total FAC, the process recovered and
enriched a fraction
that comprised over 70% (Formula 1). As the C18:1-OH concentration was
successively
increased from 3.82% in the Formula I mixture to 11.94% in the Formula 5
mixture, the
concentration of C18:1-OH in their corresponding HFAME fractions increased
from 70.14%
to 84.75%, thereby demonstrating the separation, recovery and enrichment of
HFA from
various source oils by this process.
EXAMPLE 4
Five model mixtures of fatty acid methyl esters were prepared to assess the
effects of
variable oleic acid (C18:1) and ricinoleic acid (C18:1-OH) levels on the
separation and
purification of hydroxy fatty acids with an exemplary process of the present
invention. The
model mixtures comprised methyl esters of palmitic acid (C16:0), stearic acid
(C18:0), oleic
acid (C18:1-9; C18:1-11), linoleic acid (C18:2), linolenic acid (C18:3),
arachidic acid
(C20:0), eicosenoic acid (C20:1-11), and ricinoleic acid (C18:1-OH). Methyl
esters of
palmitic acid, stearic acid, oleic acid, linoleic acid, linolenic acid,
arachidic acid, and
eicosenoic acid were purchased from Nu-Chek Prep Inc. (Elysian, MN, US).
Ricinoleic acid
methyl ester was separated and recovered from castor oil following the process
described in
Example 1. The purity and concentration of the resulting C18:1-OH HFAME were
determined by GC analysis. Twenty-gram quantities of each formula were
prepared and
stored in a freezer until required. The five model mixtures i.e. formulae 6-10
were prepared
for this study as shown in Table 5. For this study, the linoleic acid content
of each formula
was constant while the levels of oleic acid and ricinoleic acid were varied.
The HFAME and NHFAME components of each formula were fractionated, enriched
and analyzed as described in Example 3. The weight yields of HFAME were
determined
gravimetrically and the percentage yields were calculated. The study was
repeated a second
time, the data were averaged and are shown in Table 6.
CA 02762224 2011-11-16
WO 2011/143753 16 PCT/CA2011/000579
Table 5:
FAME (g) Formula 6 Formula 7 Formula 8 Formula 9 Formula 10
C16:0 1.71 1.71 1.74 1.71 1.71
C18:0 0.90 0.91 0.91 0.91 0.91
C 18:1-9 6.41 6.01 5.42 5.02 4.41
C18:1-11 0.34 0.33 0.32 0.35 0.34
C 18:2 8.60 8.61 8.63 8.61 8.61
C 18:3 0.95 0.92 0.92 0.93 1.01
C20:0 0.10 0.11 0.11 0.10 0.11
C20:1-11 0.07 0.072 0.06 0.06 0.06
C18:1-01-1 1.01 1.41 2.00 2.41 3.02
Total FAME 20.10 20.07 20.12 20.09 20.17
CA 02762224 2011-11-16
WO 2011/143753 PCT/CA2011/000579
N N O N N O.
Q o N M .-. 00 i i
O Ls - O O [~ ~t
C~ M
00
O O O~ ~p M [~ O N
[~. Cl N M [~ N M
M N N M 7T
o M t N N N i i N
~ ~ o .-~ O M O ~ ONC
O
O ~ O z. ~ O N oo N ~ ~ ~' C
N o0 00 V-) M
.?C 00 N O O
w c
00 N p 00
00 [y o N O N O Cl 00
C N
O O O~ `O O N 00 z
oc o0 00 Lr) c
C . r o0 ~ N =-~ ~ ~ O O ~
^
N
`-~"~ 00 N \O N M
O o O 'C 00 M N i i
N
U O
M Q)
w [~ 00 N 00 lzt to N
N kn
'~ 00 o o -r
>, o
a)
O w \O M M 00 `O 'l=
o w N o o M
U N
cd
U O s" 00 M M 00 N
in m
O Y. .--~ M ~' O O M
O o
U
Q)
W O O " N M O O O
~O \0 00 00 00 O
O Q . 00 N o
H
CA 02762224 2011-11-16
WO 2011/143753 18 PCT/CA2011/000579
The data in Table 6 show that when C 18:1-OH was present in the model oil
mixture at
a very low level i.e., 3.82% of the total FAC, the process recovered and
enriched a fraction
that comprised over 70% (Formula 6). As the C18:1-OH concentration was
successively
increased from 3.82% in the Formula 6 mixture to 12.63% in the Formula 10
mixture, the
concentration of C 18:1-OH in their corresponding HFAME fractions increased
from 70.14%
to 79.94%, thereby demonstrating the separation, recovery and enrichment of
HFA from
various source oils by this process.
EXAMPLE 5
Five model mixtures of fatty acid methyl esters were prepared to assess the
effects of
variable linoleic acid (C 18:2) and oleic acid (C 18:1) levels on the
separation and purification
of hydroxy fatty acids with an exemplary process of the present invention. The
model
mixtures comprised methyl esters of palmitic acid (C 16:0), stearic acid (C
18:0), oleic acid
(C18:1-9; C18:1-11), linoleic acid (C18:2), linolenic acid (C18:3), arachidic
acid (C20:0),
eicosenoic acid (C20:1-11), and ricinoleic acid (C18:1-OH). Methyl esters of
palmitic acid,
stearic acid, oleic acid, linoleic acid, linolenic acid, arachidic acid, and
eicosenoic acid were
purchased from Nu-Chek Prep Inc. (Elysian, MN, US). Ricinoleic acid methy
ester was
separated and recovered from castor oil following the process described in
Example 1. The
purity and concentration of the resulting C18:1-O11 HFAME were determined by
GC
analysis. Twenty-gram quantities of each formula were prepared and stored in a
freezer until
required. The five model mixtures i.e. formulae 11-15 were prepared for this
study as shown
in Table 7. For this study, the levels of oleic acid and linoleic acid were
varied, while the
levels of hydroxylated oleic acid were constant across the five formulae.
The HFAME and NHFAME components of each formula were fractionated, enriched
and analyzed as described in Example 3. The weight yields of HFAME were
determined
gravimetrically and the percentage yields were calculated. The study was
repeated a second
time, the data were averaged and are shown in Table 8.
CA 02762224 2011-11-16
WO 2011/143753 19 PCT/CA2011/000579
Table 7:
FAME (g) Formula 11 Formula 12 Formula 13 Formula 14 Formula 15
C16:0 1.71 1.71 1.70 1.70 1.70
C18:0 0.90 0.91 0.90 0.91 0.91
C 18:1-9 6.42 6.01 5.42 5.00 4.41
C18:1-11 0.33 0.33 0.32 0.32 0.49
C 18:2 8.21 8.61 9.22 9.60 10.21
C18:3 1.06 0.92 0.93 0.93 0.93
C20:0 0.10 0.11 0.10 0.10 0.10
C20:1-11 0.07 0.07 0.07 0.06 0.06
C18:1-OH 1.41 1.41 1.41 1.41 1.41
Total FAME 20.21 20.07 20.08 20.03 20.22
CA 02762224 2011-11-16
WO 2011/143753 PCT/CA2011/000579
w
N C N O N O
00
Q v~ o N oo
ri
cN
oc a, v, N o
oo N ri ~r o o iri
N
o0
l~ M M ~n O M ~ M
M p~ O~ r [~ I ~p
O ~O O N N
00
O O
00 M N
k oo ~t N O O
w
V~ d M N O 00
\ M 00 DD i i DD
00
Liz. N O O N N
O N
~" p p sue. O 01
(~ O r1 \O M o0 I M 00
U oo r O O tIi
00 N `O N M
o O `O 00 M N i i
N O O _ N N
N N
O S
~ ^
C) c~ O
9 M
:"O
N w ~: `n M If )
oo o O ~t
' 1 -
- 8 0 d\ M M O M V}-
00 V' M M V-)
- (y .-O ~D O N N
cd N
O' O ~" 'C I 00 In ' M M
w d ' ' ' In M M
Y p
U
N
00 O O ; N M O ; O v
oo .-. N O
u. -- - N o
cz
H
CA 02762224 2011-11-16
WO 2011/143753 21 PCT/CA2011/000579
The data in Table 8 show that when C18:1-OH was present in the model oil
mixture at a very low level i.e., 5.36% of the total FAC, the process
recovered and
enriched a fraction that comprised over 70% (Formula 11). Increasing the C18:2
concentration and decreasing the C18:1 concentration while keeping the C18:1-
OH
concentration constant resulted in declines in the amounts of C18:1-OH
recovered.
However, even in Formula 15, the recovered and purified HFA fraction comprised
over 65% C18:1-OH thereby demonstrating the separation, recovery and
enrichment
of HFA from various source oils by this process.
EXAMPLE 6: Purification of HFAME from lesquerella oil
An exemplary process used for recovering, purifying and enriching HFAME
from lesquerella oil is illustrated in Fig 2. The lesquerella oil was dried
under vacuum
for 30 min at 100 C and then cooled to 60 C. Dried KOH (1-2% w/w of oil
weight)
was dissolved in methanol (20% w/w of oil weight) in a beaker. Once the oil
was
cooled to 60 C, the KOH/methanol mixture was added to the oil. The mixture
was
mixed in the rotary evaporator for 4 h. Then, the mixture was transferred into
a
separatory funnel for phase separation. The top layer comprised lesquerella
oil methyl
esters. The bottom layer was recovered and suitably discarded. The top layer
was
transferred to a beaker and heated to 60 C under nitrogen after which, a soap
analysis
was then done on the heated top layer. Then, Trysil 300 was added to the
heated top
layer (1% w/w Trysil 300 per 1000 ppm of soap) followed by mixing for 15 min
before filtering to recover and separate the residual soap from the
lesquerella oil
methyl esters. The lesquerella oil methyl ester fraction, comprising a blend
of
HFAME and NHFAME, was then dried under vacuum and stored for subsequent use
as the starting material in an exemplary embodiment of the HFAME purification
process of the present invention.
The lesquerella oil methyl ester fraction comprising the blend of HFAME and
NHFAME was mixed together with methanol (80%-90% methanol with remaining
being water) and 100% hexane/methylpentane in the ratio of 1:5:5 (w/w/w). This
first
mixture was stirred for about 1-2 h at ambient room temperature under
nitrogen. The
first mixture was then transferred to a separatory funnel and allowed to rest
for a
CA 02762224 2011-11-16
WO 2011/143753 22 PCT/CA2011/000579
period of time to allow phase separation to occur (referred to as the first
extraction).
The first bottom phase comprising the HFAME and methanol was recovered from
the
separatory funnel and then desolventized by rotary evaporation. The first top
phase
comprising the NHFAME and hexane/methylpentane was recovered and then mixed
together with methanol (80%-90% methanol with remaining being water) and 100%
hexane in the ratio of 1:5:5 (w/w/w). This second mixture was stirred for
about 1-2 h
at ambient room temperature under nitrogen. The second mixture was then
transferred
to a separatory funnel and allowed to rest for a period of time to allow phase
separation to occur (referred to as the second extraction). The second bottom
phase
comprising the HFAME and methanol was recovered from the separatory funnel and
then desolventized by rotary evaporation. The second top phase comprising
NHFAME was recovered and then mixed together with methanol (80%-90%
methanol with remaining being water) and 100% methylpentane/hexane in the
ratio of
1:5:10 (w/w/w). This third mixture was stirred for about 1-2 h at ambient room
temperature under nitrogen. The third mixture was then transferred to a
separatory
funnel and allowed to rest for a period of time to allow phase separation to
occur
(referred to as the third extraction). The third bottom phase comprising HFAME
and
methanol was recovered from the separatory funnel and then desolventized by
rotary
evaporation. The third top phase comprising NHFAME was recovered and then
mixed
together with methanol (80%-90% methanol with remaining being water) and 100%
methylpentane/hexane in the ratio of 1:5:10 (w/w/w). This fourth mixture was
stirred
for about 1-2 h at ambient room temperature under nitrogen. The fourth mixture
was
the transferred to a separatory funnel and allowed to rest for a period of
time to allow
phase separation to occur (referred to as the fourth extraction). The fourth
bottom
phase comprising HFAME and methanol was recovered from the separatory funnel
and then desolventized by rotary evaporation. The fourth top phase comprising
NHFAME was recovered and then desolventized by rotary evaporation. The
desolventized NIIFAME were suitable for further processing to produce cold-
tolerant
biodiesel and/or lubricants. The fatty acid composition of HFAME from each of
the
extractions and of the NHFAME were analyzed and quantified by diluting a 10 L
aliquot of each extraction with 4mL hexane and then passing it through a gas
chromatograph (Agilent model 6890N) equipped with a DB-23 column (0.25 mm X
30 M, 0.25 m thick) and a flame ionization detector. The data showing the
fatty acid
CA 02762224 2011-11-16
WO 2011/143753 23 PCT/CA2011/000579
compositions of lesquerella oil, purified HFAME and NHFAME fractions are
listed in
Table 9 (fractionation using 80% methanol) and Table 10 (fractionation using
90%
methanol).
Table 9: Fatty acid compositions of lesquerella oil, HFAME and NHFAME
fractions
resulted from fractionation using 80% methanol).
FAME Lesquerella HFAME-la HFAME-2b HFAME-3 HFAME-4d NHFAME
oil % area % area % area % area % area % area
16:0 1.29 0.13 0.05 1.49
16:1 1.00 0.15 0.06 0.98
18:0 1.89 2.40
18:1-9 13.42 1.28 1.33 0.96 1.09 17.26
18:1-11 2.02 0.19 0.20 0.16 2.39
18:2 7.45 1.07 1.14 0.86 0.95 9.29
18:3 12.50 2.72 2.92 2.26 2.45 15.49
20:0 0.17 0.22
20:1-11 0.89 1.16
18:1-OH 0.54 1.49 1.32 1.29 1.16 0.35
20:1-OH 55.94 86.43 86.81 88.36 87.96 46.80
20:2-OH 2.90 6.63 6.24 6.27 6.01 2.16
Total 59.38 94.54 94.37 95.93 95.13 49.32
HFAME
Total 100.01 99.81 100.24 100 99.89 99.99
FAME
yield
yie 6.83 6.95 4.26 4.50 77.55
a 1 st extraction
b 2d extraction
3rd extraction
d 4 i extraction
The data show that total HFAME contents of all four HFAME fractions are
above 94% when fractionation is carried out using 80% methanol. After four
successive fractionations, 36% of HFAME was recovered from the original
lesquerella oil. By combining the four HFAME fractions, we can obtain a HFAME
fraction containing 95% total HFAME with a yield of 22.54%.
CA 02762224 2011-11-16
WO 2011/143753 24 PCT/CA2011/000579
Table 10: Fatty acid compositions of lesquerella oil, HFAME and NHFAME
fractions
resulted from fractionation using 90% methanol).
FAME Lesquerella HFAME-1 a HFAME-2b HFAME-3c HFAME-4d NHFAME
oil % area % area % area % area % area % area
16:0 1.29 0.25 0.38 0.35 0.47 2.49
16:1 1.00 0.23 0.35 0.33 0.45 1.54
18:0 1.89 0.26 0.41 0.36 0.49 4.23
18:1-9 13.42 2.67 4.17 3.82 5.19 29.16
18:1-11 2.02 0.37 0.58 0.54 0.73 4.02
18:2 7.45 1.96 3.00 2.86 3.88 14.84
18:3 12.50 4.39 6.49 6.40 8.61 23.13
20:0 0.17 0.40
20:1-11 0.89 0.19 0.16 0.23 2.07
18:1-OH 0.54 1.00 0.76 0.68 0.52 0.22
20:1-OH 55.94 83.54 79.18 80.41 75.75 17.41
20:2-OH 2.90 5.33 4.40 4.18 3.51 0.61
Total 59.38 89.88 84.34 85.27 79.77 18.13
HFAME
Total 100.01 100.00 99.91 100.09 99.83 100.12
FAME
% yield 25.77 21.17 7.34 5.58 40.14
(w/w)
a 1st extraction
b 2nd extraction
3`d extraction
d 4d' extraction
The data show that total HFAME content of all four HFAME fractions are
above 79% when fractionation is carried out using 90% methanol. After four
successive fractionations, 87.02% of HFAME was recovered from the original
lesquerella oil. By combining the four HFAME fractions, we can obtain a HFAME
fraction containing 86% total HFAME with a yield of 59.86%.
HFAME fraction obtained from 90% methanol fractionation had lower
HFAME concentration (86%) compared to the one obtained from 80% methanol
CA 02762224 2011-11-16
WO 2011/143753 25 PCT/CA2011/000579
fractionation (95%); however yield of HFAME fraction obtained from 90%
methanol
fractionation was higher (59.86%) than the one obtained from 80% methanol
fractionation (22.54%). Depends on the HFAME concentration needed, different
percentage methanol can be used in the fractionation.
EXAMPLE 7: Purification of HFAME fronm genetically engineered camelina
oil
The exemplary process used for recovering, purifying and enriching HFA
from genetically engineered camelina oil is illustrated in Fig 1. The seeds
were
pressed to produce camelina oil comprising the non-native HFA (i.e., HF-
camelina
oil). The pressed camelina cake was then extracted with methylpentane/hexane
to
obtain residue oil. The pressed and solvent extracted oil were combined to
obtain
crude HF-camelina oil. The crude HF-camelina oil was dried under vacuum for 30
min at 100 C and then cooled to 60 C. Dried KOH (1-2% w/w of oil weight) was
dissolved in anhydrous methanol (20% w/w of oil weight) in a beaker. Once the
oil
was cooled to 60 C, the KOH/methanol mixture was added to the oil. The
mixture
was mixed in the rotary evaporator for 4 h. Then, the mixture was transferred
into a
separatory funnel for phase separation. The top layer comprised camelina oil
methyl
esters. The bottom layer was recovered and suitably discarded. The top layer
was
transferred to a beaker and heated to 60 C under nitrogen after which, a soap
analysis
was then done on the heated top layer. Then, Trysil 300 was added to the
heated top
layer (1% w/w Trysil 300 per 1000 ppm of soap) followed by mixing for 15 min
before filtering to recover and separate the residual soap from the camelina
oil methyl
esters. The camelina oil methyl ester fraction, comprising a blend of HFAME
and
NHFAME, was then distillated at 220 C under vacuum (0.1-0.2 mmHg).The
distilled
camelina methyl esters were water washed and stored for subsequent use as the
starting material in an exemplary embodiment of the HFAME purification process
of
the present invention.
The camelina oil methyl ester fraction comprising the blend of HFAME and
NHFAME, was mixed together with methanol (70-90% with the remaining being
water) and 100% hexane/methylpentane in the ratio of 1:5:5 (w/w/w). This first
mixture was stirred for about 1-2 h at ambient room temperature under
nitrogen. The
CA 02762224 2011-11-16
WO 2011/143753 26 PCT/CA2011/000579
first mixture was then transferred to a separatory funnel and allowed to rest
for a
period of time to allow phase separation to occur (referred to as the first
extraction).
The first bottom phase comprising the HFAME and methanol was recovered from
the
reparatory funnel and then desolventized by rotary evaporation. The first top
phase
comprising the NHFAME and hexane/methylpentane was recovered and then mixed
together with methanol and 100% hexane in the ratio of 1:5:5 (w/w/w). This
second
mixture was stirred for about 1-2 h at ambient room temperature under
nitrogen. The
second mixture was then transferred to a separatory funnel and allowed to rest
for a
period of time to allow phase separation to occur (referred to as the second
extraction). The second bottom phase comprising the HFAME and methanol was
recovered from the separatory funnel and then desolventized by rotary
evaporation.
The second top phase comprising NHFAME was recovered and then mixed together
with methanol and 100% methylpentane/hexane in the ratio of 1:5:10 (w/w/w).
This
third mixture was stirred for about 1-2 h at ambient room temperature under
nitrogen.
The third mixture was then transferred to a separatory funnel and allowed to
rest for a
period of time to allow phase separation to occur (referred to as the third
extraction).
The third bottom phase comprising HFAME and methanol was recovered from the
separatory funnel and then desolventized by rotary evaporation. The third top
phase
comprising NHFAME was recovered and then desolventized by rotary evaporation.
The desolventized NHFAME were suitable for further processing to produce cold-
tolerant biodiesel and/or lubricants. The fatty acid composition of HFAME from
each
of the extractions and of the NHFAME were analyzed and quantified by diluting
a 10
L aliquot of each extraction with 4mL hexane and then passing it through a gas
chromatograph (Agilent model 6890N) equipped with a DB-23 column (0.25 mm X
30 M, 0.25 m thick) and a flame ionization detector. The data showing the
fatty acid
compositions of HF-camelina oil, purified HFAME and NHFAME fractions are
listed
in Table 11.
CA 02762224 2011-11-16
WO 2011/143753 27 PCT/CA2011/000579
Table 11: Fatty acid compositions of HF-camelina oil, distilled camelina
methyl
esters, HFAME and NHFAME fractions resulted from 80% methanol fractionation.
HF- distilled
FAME camelina camelina HFAME-1 a HFAME-2b HFAME-3` NHFAME
oil %area methyl esters % area % area % area % area
% area
16:0 6.59 6.63 1.22 1.73 1.68 7.02
18:0 4.90 5.30 0.52 0.81 0.74 5.70
18:1-9 30.60 30.44 5.11 7.41 7.10 32.42
18:1-11 1.31 0.93 0.19 0.27 0.26 1.11
18:2 10.58 10.77 2.91 4.06 4.01 11.41
18:3 18.23 18.41 7.66 10.37 10.51 19.26
20:0 1.30 1.36 0.25 0.10 0.11 1.46
20:1-11 14.37 14.54 1.32 2.07 1.90 15.77
20:2 0.49 0.45 0.10 0.10 0.49
20:3 0.32 0.35 0.07 0.10 0.10 0.37
22:1 0.83 0.72 0.23 0.17 0.14 0.72
18:1-OH 3.86 3.77 30.10 28.95 30.18 1.55
18:2-OH 4.67 4.48 46.53 39.74 38.74 1.36
20:1-OH 0.46 0.45 1.79 2.02 2.32 0.22
20:2-011 0.19 0.18 1.04 1.08 1.20
Total 9.18 8.88 79.46 71.79 72.44 3.13
HFAME
Total 98.70 98.78 98.94 98.98 99.09 98.86
FAME
% yield 3.13 2.67 1.30 92.90
(w/w)
a 1 s` extraction
b 2 a extraction
'3 d extraction
The data show that the total HFAME content of each of the three HFAME
fraction was increased from 9.18% to greater than 70%. By combining the three
fractions, a HFAME fraction was obtained with a yield of 7.1% and HFAME
concentration of 75.29%.
CA 02762224 2011-11-16
WO 2011/143753 28 PCT/CA2011/000579
While particular exemplary embodiments of the present invention have been
described in the foregoing, it is to be understood that other embodiments are
possible
within the scope of the present invention and are intended to be included
herein. In
view of numerous changes and variations that will be apparent to persons
skilled in
the art, the scope of the present invention is to be considered limited solely
by the
appended claims.