Note: Descriptions are shown in the official language in which they were submitted.
CA 02767675 2012-01-09
WO 2011/011276 PCT/US2010/042255
TITLE
METHODS FOR SYNTHESIZING POLYETHER DIOLS AND
POLYESTER DIOLS
5J
CROSS- REF ERENQ E TO RELATED APPLICATIONS
This application claims the benefit of provisional U .S. Application
Serial No. 61/227518,
FIELD OF THE INVENTION
The invention relates to methods for synthesizing polyether dials
and polyester dials. The methods provide reduced color as compared to
such polymers made using conventional methods.
5 BACKGROUND
Polytrimethylene ether glycol (hereinafter also referred to as
"P ") produced from the acid rata yzed polycondensation ofI 3-
propanediol (hereinafter also referred to as ''PDO) can have quality
problems, in particular the color of the polymer may not be acceptable to
the industry. The raw material PDO and the polymerization process
contihions and stability of the polymer are responsible for discoloration to
some extent.
Various pre-polymerization treatment methods are disclosed in the
prior art to remove color precursors present in the P. Attempts have
also been made to reduce the color of clyt; methylene ether glycols past.
polymerization. For example: Sunkara et al. describes a process for
reducing color in P03G by contacting P036 with an adsorbent and then
separating the P036 from the adsorbent (U S. Patent 7,294,746).
Pre- or post-polymerization methods may undesireably add
additional steps, time, and expense to production processes. Attempts
have also been made to alter reaction conditions to control product color
during polymerization. For exampe, U.S, Patent Application Publication
No. 2005/272911 discloses methods of controlling color formation by
_11_
CA 02767675 2012-01-09
WO 2011/011276 PCT/US2010/042255
carrying out the dehydration-condensation reaction in the presence of a
catalyst composed of an acid and a base:
There exists a need for improvred and convenient methods to
reduce color of P03G.
BRIEF DESCRIPTION OF THE FIGURES
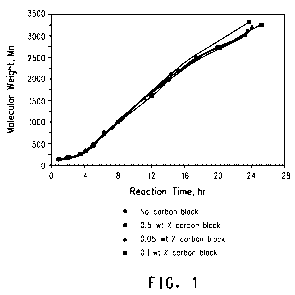
Figure 1 illustrates the molecular wai ;lit development of 1,3-propanedioi
polymerization with and without carbon black addition.
Figure 2 illustrates P030 product color development as a function of
molecular weight with and without carbon black during polymerization.
SUMMARY OF THE INVENTION
One aspect of the present invention is a process comprising:
is contacting reactants with a catalyst and carbon black to form a
reaction product, wherein the reactannts comprise at least one selected
from the group consisting of: a diol of formula 0H( H2) OH where n is an
integer of 2 or greater; or a polyol thereof; and a diacid of formula
HOOC(CH2) C OH where z is an integer of 4 or greater, or a polymer
thereof,
Another aspect of the present invention is a process comprising
contacting reactants with a catalyst and carbon black to form a reaction
product, wherein the reactants comprise a dial of formula OH(CH_2),,OH
where n is an integer greater than or equal to 2 or a polyol thereof; and
a diacid of formula H 0C(CH2)wCO0H where z is an integer greater than
or equal to 4 or a polymer thereof; and wherein the reac'.on product is a
polyester dial.
A further aspect of the present invention is a process comprising
process comprising contacting reactants with a catalyst and carton black
to form a reaction product:. wherein the reactants comprise a diol of
formula 0H(CH2) OH where n is an integer greater than or equal to 3 or a
polyol thereof; or a viol of formula H t C CH ),COOH where z is greater
than or equal to or a polyol thereof; and wherein the reaction product is
a polyaather dial.
-2-
CA 02767675 2012-01-09
WO 2011/011276 PCT/US2010/042255
DETAILED DES. RIPTION
Unless otherwise stated, all percentages, parts, ratios, etc_ are by
height. Further, when an amount: concentration or other value or
parameter is given as either a range, preferred range or a list of upper
preferable values and lower preferable values, this is to be understood as
specifically disclosing all ranges formed from any pair of any upper range
limit or preferred value and any lower range l:Ã .i or preferred value.,
regardless of whether ranges are separately disclosed_
Processes disclosed herein employ carbon black. Carbon black is
an adsorbent, and athough it is present during reactions in the processes
described herein, it is not a "reactant" as the term is used herein. The
term "adsorbent" refers to materials that commonly are used to remove
relatively small amounts of undesired components, Whether such removal
is by the process of adsorption or absorption, As used herein, :`carbon
black' refers to carbon black, activated carbon, or charcoal. Activated
carbon is available commercially in different forms such as powder,
granular, and shaped products. The preferred form is powdered activated
carbon, Various brands of carbon may be used, including, but not limited
to, Merit America G60, NORIT RO 0.8, Calgon PWA, BL, and WPH, and
Ceca ACTICARBONE ENO. Also suitable are Darco KB-G or Darco S-51
(Norit), or ADP Carbon' (CalgoÃa Carbon), Suitable forms of carbon black
also include those having a particle size range of about 2.7 micron to
about 130 micron. Other forms will be known to those skilled in the art,
Other adsorbents suitable for the processes disclosed herein are
commercially available from various sources and in many forms and
include alumina, silica, diatomaceous earth, montmorillonite clays. Fuller's
earth, kaolin minerals and derivatives thereof.
"Color" and " color bodies" refer to visible color that can be
quantified by the use of a spectroeolori titer in the range of visible light,
using wavelengths of approximately 400 to 800 nm, and by comparison
with pure water. Color precursors in P00 are not visible in this range, but
contribute color during and after polymerization.
-3-
CA 02767675 2012-01-09
WO 2011/011276 PCT/US2010/042255
Provided herein is a process of producing polymeric reaction
product in the presence of carbon black. The processs comprnses
polycondensing reactants comprising 1, -propanediiol, poly-1. -
propanÃdiol or a mixture thereof in the presence of acid polycondensation
catalyst and carbon black to form a reaction product. In some
embodiments, the process further comprises separating the reaction
product from the carbon black. In some embodiments, the reactants
further comprise a co monomer dial.
In some embodiments, the reaction product has a molecular weight
greater than about 500 or a molecular weight of about 500 to about 5000.
In some embodiments, the reaction product has an APHA color of less
than about 250 or less than about 50.
In some embodiments, the reaction product comprises
polytrimethylene ether glycol. In some embodiments, the polytrin thylene
ether glycol is contacted with a monocarboxylic acid to form a dicarboxylic
acid ester of polytrimethyiene ether glycol.
In accordance with the present invention, it has been found that
carbon black reduces polymer color when present during polymerization
(Figure 2, Examples). In preferred embodiments, the carbon black has a
desirable effect on polymer color without substantially affecting polymer
molecular weight development (Figure 1, Examples). At the same
reaction temperature and acid concentration, for a given polymer
molecular weight, polymer color decreases with an increase in amount of
carbon black addition. Also, in situ removal of color species may allow a
polymerization process to be operated at a higher temperature and higher
catalyst concentrations facihtating produd:on of a certain molecular weight.
product in a shorter polymerization time pe6od,
In one embodiment, a process comprises contacting reactants with
a catalyst and carbon black to form a reaction product, wherein said
reactants comprise at least one of;
(a) a dial of formula H(CH2) H where n is an integer greater than or
equal to 2, or a polyol thereof; or
(b)a diacid of formula HOOC(CH2) COOH where z is an integer greater
than or equal to 4, or a polymer thereof.
-4-
CA 02767675 2012-01-09
WO 2011/011276 PCT/US2010/042255
Also provided is a process comprising contacting reactants with a
catalyst and carbon black to form a polyester diol reaction product wherein
the reactants comprise both
(a) a dial of formula OH(CH2)ROH where n is an integer greater than or
equal to 2 or a polyol thereof; and
(b) ,a diacid of formula HOO (CH2) COON where z is an integer greater
than or equal to 4 or a polymer thereof.
Further provided is a process comprising contacting reactant with a
catalyst and carbon black to form a polyether dial reaction product wherein
the reactants comprise a diol of formula OH(I2)SOH where n is an
integer greater than or equal to 3 or polyols thereof; or a dial of formula
HOOC( H2)COOH where z is an integer greater t; hare or equal to 6 or
polyols thereof.
Also disclosed is a process comprising contacting reactants with a
catalyst and carbon black to form a reaction product wherein the reactants
comprise a dial of formula OH(CH ),OH where n is an integer greater t' an
or equal to 2, or polyols thereof; and wherein said diol is 1,3-propane dial.
In another aspect, the reactants further comprise a comonomer Biol. In
one embodiment, the reaction product comprises polytrimethyylene ether
glycol.
In some embodiments, the carbon black is about 0.05 to about 5
weight percent based on the total weight of the reactants. In some
embodiments, the process includes separating the reaction product from
the carbon black by, for example, filtration.
In some embodiments, the catalyst for the processes comprises a
titanium catalyst or an acid catalyst. In some embodiments, the reaction
products of the processes have an APHA color of less than about 2g;
less than about 100, less than about 50, less than about 40, or less than
about 30 Also provided is a process comprising polycondensing reactants
comprising 1,3-propanediol, poly 1,3-propanediol or a mixture thereof, in
the presence of acid and carbon black. In one embodiment, the reaction
product comprises polytrimethylene ether glycol. In some embodiments,
the I,3-pr'opanediol, the poly-1,3-propanediol or mixtures thereof comprise
-5-
CA 02767675 2012-01-09
WO 2011/011276 PCT/US2010/042255
bio-derived I3-propanediol._ In some aspects the acid comprises sulfuric
acid. In further embodiments the reactants comprise comonomer diol and
the comonomer diol can, in some embodiments, be ethylene glycol.
In some embodiments, the process further comprises contacting
the polytrimethyl ne ether glycol with a monocars)oxylic acid to form a
dicarboxylic acid ester of polytrimethylene ether glycol. In some aspects,
the monoc rboxylic acid is 2-ethylhexanoic acid.
In some embodiments, the molecular weight of the reaction product
is greater than about 500. In some preferred embodiments, the molecular
weight is from about 500 to about 5000. In some embodiments, the
product has an APHA color of less than about 250, less than about 100,
less than about 50, less than about 40 or less than about 30,
The processes disclosed herein can, in some embodiments., be
used to make polytrimethylene ether glycol,
In the processes disclosed herein, carbon black may be added at
any time dur3og the polycondensatÃon reaction, Depending on the reaction
conditions and the time of addition, the reactants present during the
polycondensation in fi~w presence of carbon black can include monomer
dials or polyols thereof, or diacids or polymers thereof. In one example,
the reactants comprise P00 monomer, poly-1, 3-propanediol. or mixtures
thereof. Poly-I;3-propanedlol includes oligomers of P00 including PDO diner
and PDO trimer.
The processes disclosed herein can be used to produce reaction
products from reactants comprising at least one of a diol of formula
QH(CHw)00H where n is an integer greater than or equal to 2, or a polyol
thereof; or a diacid of formula HO0C%CH;_1,C OH where z is an integer
greater than or equal to 4, or a polymer thereof, The reactants can
include both a diol (or a polyol thereof) and a diacid (or a polymer therof)
such as, for example, when the reaction product is a polyester dial.
Reaction products may be homopolymers or copolymers.
Polyester diol reaction products can be prepared using known
methods from aliphatic, cycloaliphatic or aromatic dicarboxylic or
polycarboxylic acids or anhydrides thereof (for example, succinic, glutaric,
adipic, pimelic, subenc, azelaic, sebacic, nonanedicarboxylic,
-6-
CA 02767675 2012-01-09
WO 2011/011276 PCT/US2010/042255
decanedicrboxylic, terephthalic, isophthalic, o-phthalic,
tetra hydrophthalic, h x hydrop thafÃc or trimelH tic acid) as well as acid
anhydrides (such as o-phthalic, trimellÃtic or succinic acid anhydride or a
mixture thereof) and dihydric alcohols such as, for example,ethanediol.,
diethylene, triethylene, tetraethyl ne glycol, `I ,2wpropaned 1, dipropylene,
tripropylene, tetrapropylene glycol, 1,3-propan diol, 1;4-hutanediol, 1, -
butanediol', 2,3-butanediol, 1, -pentanediol, 1.S.hexanedlol, 2.2YdÃr ethyl-
I ,3-propanedÃol, 1,4-dÃfydroxycyclohexane, I ,4_dimethylolcyclohexane,
1, -octanediol, 1,10-decanediol, 1.12-doc ecanediol or mixtures thereof.
Dols suitable for the processes disclosed herein include aliphatic
diols, for example, ethylenediol, Il ,6-hexanediol; 1, 7-heptanediol _ 1,8-
octanediol, 1,9-nonanediol. 1,10-decanedÃol, 1;12-dodecanedÃol,
3,3,4,4,5,5-hexafluro-1, -pentan diol, 2,2,3,3,4:4,5,5-octafluoro-1, -
hexanedlol, 3.3,4,4,5, ,6, ,7,7,8,8; ,9,10 l0-hexadecafluoro-1,12-
dbdecanediol, cycloaliphatic dials, for example, 1,4-c clohexanediol, 1, -
cyclohexanedirethanol and isosorhide, polyhydroxy compounds, for
example, glycerol, trimethylolpropane, and pentaerythritol. Other suitable
dials include 2-methyl-1,3-propanedÃol, 2,2-dimethyl-1,3-propanediol, 2,2-
di ethyl- 1, 3-propaneiol, 2-ethyl-2-(hydroxyme hyl)-1,3-propanediol, 1,6-
hexa ed ol, 1,8-octanediol, 1;10-decanediol, isosorbide,and mixtures
thereof. In some embodiments, preferred dials are 1.3-propanediol and
ethylene glycol.
Catalysts suitable for the production of polyester dials include
organic and inorganic compounds of titanium, lanthanum, tin, antimony,
zirconium, manganese. zinc, phosphorus and mixtures thereof. Titanium
catalysts such as tetraisopropyl titanate and tetrabutyl titanate are
preferred and can be added in an amount of at least about 25 ppm and up
to about 1000 ppm titanium by weight, based on the weight of the polymer.
The processes disclosed herein can be used to produce polyether
dial reaction products. For example the processes can be used to
produce reaction products from reactants comprising at least one of a dial
of formula OH(CH2)õ H where n is an integer greater than or equal to 3,
or a polyol' thereof, or a dial of formula H(CH~>) OH where nis an integer
greater than or equal to 6, or a polyol thereof. Diols of formula
-7-
CA 02767675 2012-01-09
WO 2011/011276 PCT/US2010/042255
OH(CH ),,OH where n is 2, 4, or 5 may not be preferred, as they may
cyclize.
In one embodiments the reaction product comprises P03 G.
Methods of making PO from 1,-propanediol are described in the art,
for example, in U.S. Application Publication Nos. 20020007043 and
20020010374. As shown in the Examples herein, polyether diols such as
PO can be produced by polycondensing P00 using an acid catalyst.
Suitable catalysts, for processes to produce poiyether dials include those
acids with a plea less than about 4, preferably with a pKa less than about
2, and include inorganic acids, organic sulfonic acids, heteropolyacids,
perfluoro-alkyl sulfonic acids and mixtures thereof Also suitable are metal
salts of acids with a plea less than about 4, including metal sulfonates,
metal trifluoroacetates, metal triftates, and mixtures thereof including
mixtures of the salts with their conjugate acids, Specific examples of
catalysts include sulfuric acid, fluorosulfonic acid, phosphorous acid, p-
toluenesulfonic acid, benzenesulfonic acid, phosphotungstic acid,
phosphorolybdic acid, trifluoromethanesulfonic acid, 1,1,2,2
tetrafluoroethanesulfonic acid, I .1 1,2, ,;3-hexafluoropropanesulfonic acid,
bismuth triflate, yttrium tr;flate, ytterbium triflate, neodymium triflate,
lanthanum triflate, scandium triflate, zirconium triflate. A preferred
catalyst
for P03 is sulfuric acid. Other suitable catalysts include superacids and
N.A': iON solid catalysts (E.I. DuPont de Nernours & Co).
A particularly preferred source of PDO is via a fermentation process
using a renewable biological source, As an illustrative example of a
starting material from a renewable source, biochemical routes to PDO
have been described that utilize feedstocks produced from biological and
renewable resources such as corn feed stock. For example, bacterial
strains able to convert glycerol into 1,3-ropanediol are found in the
species / !ebsle /ca, Citrobacter, Clostridium, and Lactobacillus, The
tec;hIn:que is disclosed in several publications: including US 633362,
U85686276 and U 58210 2. U 821 92 discloses, inter atria, a process
for the biological production of PDO from glycerol using recombinant
organisms. The process incorporates E. co/i bacteria, transformed with a
heterologous pdu diol dehydratase gene, having specificity for I ,2-
-s-
CA 02767675 2012-01-09
WO 2011/011276 PCT/US2010/042255
propaaned()l. The transformed E. fcoti is grown in the presence of glycerol
as a carbon source and PDO is isolated from the growth media. Since
both bactena and yeasts can convert glucose (e.g., corn sugar) or other
carbohydrates to glycerol, the processes disclosed in these publications
provide a rapid, inexpensive and environmentally responsible source of
P00 monomer.
The biologically-derived P00, such as produced by the processes
described and referenced above, contains carbon from the atmospheric
carbon dioxide incorporated by plants, which compose the feedstock for
the production of the P00, In this way, the biologically-derived P00
preferred for use in the context of the present invention contains only
renewable carbon, and not fossil fuel-based or petroleum-based carbon,
The polymers based thereon utilizing the biological ly-den ved P0,
therefore, have less impact on the environment as the P00 used does not
deplete diminishing fossil fuels and, upon degradation, releases carbon
back to the atmosphere for use by plants once again, Thus, the
compositions of the present invention can be characterized as more
natural and having less environmental impact than similar compositions
comprising petroleum based dials.
Preferably the P00 used as a reactant or as a component of the
reactants in the processes disclosed herein has a purity of greater than
about 99%, and more preferably greater than about 99.9%, by weight as
determined by gas chromatographic analysis, Particularly preferred is
purified P00 as discloser in US7088 8, US7084311 and
US 00500999 Al
In one embodiment the product of the process is P3. Product
P03G can be P03G homo- or co-polymer. For example, the P0Ã can be
polymerized with other diols ("comonomer diols') to make copolymer. The
PDO copolymers useful in the process can contain up to 50 percent by
weight (preferably 20 percent by weight or less) of comonomer diols in
addition to the 1,8-propanediol and/or its oligomers. A preferred
comonomer dial is ethylene glycol. Other coronorer dials that are
suitable for use in the process include aliphatic diols, for example,
ethylenediol, 1,6-hexane iol, 1,7-heptanediol, 1,8-octane iol, 1,9-
-9-
CA 02767675 2012-01-09
WO 2011/011276 PCT/US2010/042255
nonanediol, 1,10-decanedÃol, 1,1 2-dod ne iol,, 3,,3 4,4, , -hexa uro-
3,5- entanedÃol., 2:2:3 3,4,4,5,5 o>-Ctay~iSuoFo-1,$-hexane iotl,
3,3 4 4,5 ,6,6,7,7, ,5,g,9;10,10-hrexadecafluoro-1,12-dodecanediol,
cycloalÃphatic di.c ls, for example, 1,4-cyclohexanediol, 1,4-
cyc ohexanedimethanol and isosorbide, polyhydroxy compounds, for
example, glycerol, trimethyÃo pÃo ane, and entaer'ythrntol. Other suitable
comonomer diols are selected from the group consisting of 2-methyl-1 3-
propanediol, 2,24methyl-1,3-propanedÃol, 2,2-dietÃhyÃ-1,3-proparne rol, 2-
ethyl-2-(hydroxymethyÃ)-1,3-propanediol, 1,6-hexanediol, 1,8-octanÃedÃol,
1,1Ã3-decanediol, isosorbide, and mixtures thereof. Thermal stabilizers,
antioxidants and coloring materials may be added to the polymerization
mixture or to the polymer if desired.
In one embodiment, a process comprises causing reactants to
polymerize in the presence of carbon black. For a given reaction
temperature and catalyst concentration, product APIA color values for a
polymer of a given molecular weight or molecular weight range are
reduced as compared to the color values for the product polymerized
without the presence of carbon black. It will be appreciated that preferred
color values or preferred reductions may vary depending on the desired
molecular weight or the desired end use of the product. However, armed
with this disclosure, one of skill in the art will be able to adjust the
process
conditions to achieve the desired effect on the color of the product.
It is desired that reaction i the presence carbon black results in
polymer with an APHA color of less than about 100, and, more preferably,
less than 50. Preferably, the APIA color is less than about 40, more
preferably, less than 30, So, in certain embodiments, the APHA color is
about 30 to about 100 APHA. APHA color values are a measure of color
as defined in A. TM-D-1209 (see Test Method 1, below).
The molecular weight of the product polymer is typically within the
range of about 250 to about 5000. Preferably, the molecular weight is
about 500 to about 4000. In some embodiments, the product polymer has
a molecular weight of about 250 to about 2250. In some embodiments the
product polymer has a molecular weight of about 1i 000 to 2250.
-10-
CA 02767675 2012-01-09
WO 2011/011276 PCT/US2010/042255
The amount of carbon black used depends on factors including the
process conditions such as reaction volume, contact time and
temperature. Carbon black can be added at any time during the reaction,
but is preferably added at. the beginning of the reaction. It. can be
premixed with reactant or catalyst before addition into the reactor. The
amount added may be based on the weight of the monomer or polymer
phase at the time of addition. For example, if the reactants comprise PDO
and comonomer, the amount will be based on the total weight of PDO and
comonomer initially added. For continuous operations, it should be based
on the total weight of reactants in the reactor,
About 0.05 to about 5 weight percent carbon black may be
employed, and about 0.1 to about 1 weight percent carbon black is
preferred. It is preferred that the amount added is sufficient to reduce
color, and preferably the amount added is sufficient to reduce color to less
than 100 ARIA or more preferably to less than 50 APHA,
The contacting of the reactants with carbon black is carved out
under conditions suitable for polymerization. The contacting occurs in the
presence of acid and preferably at a temperature of about 120 to . 0 C,
preferably 150 to 180'C. The reaction is conducted for a period of about
3 to 50 hours, and preferably about 3 to about 15 hours.
Suitable processes for removal of the carbon black such as filtration
are well known to those skilled in the art. Other filter media can be used
and will be well known to those skilled in the art, the requirements being a
fineness of filter sufficient to retain the carbon black and inert to the
glycol.
A batch process can be used, wherein carbon black is added into
the reactor at any stage of reaction, and, after a period of time, separated
out by suitable means, for example, by filtration, cent,.ifugation, etc, The
process of the invention may also be conducted in a continuous or semi-
continuous fashion. For example, the reactants may be mixed with carbon
black and be pumped from a storage tank into a reactor. Carbon black
can be added into the reactor at any stage of reaction, The feed rate is
adjusted for the kind, amount, and prior use of carbon black in the bed and
the color level of the feedstock so that the carbon black is present in the
reactor sufficiently long to give a product with the desired color reduction.
-11-
CA 02767675 2012-01-09
WO 2011/011276 PCT/US2010/042255
Other variations will be recognized by those skilled in the art. Although it
is contemplated that the process described herein can be used in
conjunction with methods known in the art wherein the raw materials are
pretreated to remove color (such as, for example, in U.S. Patent
6,238,948) or methods wherein the polymer products are post-treated to
remove color (such as, for example, in U.S. Patent 75294,746) it is also
believed that use of the process described herein eliminate or diminish the
necessity of such pretreatment steps and still produce polymer of desired
low APIA color.in some embodiments, the product has desired ;PHA.
color at the end of the polymerization, and in other embodiments, the
product achieves desired APHA color after further purification.The
processes disclosed herein can be used for the decolorization of P03G
prepared by polymerization of PDO prepared from petrochemical sources,
such as the process using acrolein, and for P03G prepared by
polymerization of PDO prepared by biochemical routes.
In accordance with a further embodiment of the present invention, a
product comprises (i) carbon black, and (ii) P03G wherein the P 03G has
an APHA color of less than about 250, In certain embodiments, the.APH.A
color is less than about 100, less than about 50, less than about 40, or
less than about 30. Also, the product may contain about 0.05 to about 5
weight percent of carbon black or preferably about 0.1 to about 1 weight
percent of carbon black.
In one embodiment, the process forms P03G and further
comprises esterification of the product P0 3G by reaction with a
monocarboxylic acid and/or equivalent, as described in copending U.S;
Application Publication No. 20080108845. By "monocarbo ylic acid
equivalent" is meant compounds that perform substantially like
monocarboxylic acids in reaction with polymeric glycols and dials; as
would be generally recognized by a person of ordinary skill in the relevant
art. Monocarboxyxlic acid equivalents for the purpose of the present
invention include, for example, esters of monocarboxylic acids, and ester-
forming derivatives such as acid halides (e.g., acid chlorides) and
anhydrides. Preferably, a monocarboxylic acid is used having the formula
R--COOH, wherein R is a substituted or unsubstituted aromatic, aliphatic
-12-
CA 02767675 2012-01-09
WO 2011/011276 PCT/US2010/042255
or cycloaliphatic organic moiety containing from 6 to 40 carbon atoms.
Mixtures of different monocarboxylic acids and/or equivalents are also
suitable.
The monocarboxylic acid (or equivalent) can contain any
substituent groups or combinations thereof (such as functional groups like
amide, amine, carbonyl, halsde, hydroxyl, etc.) so long as the substituent
groups do not interfere with the esteriflcation reaction or adversely affect
the roperties of the resulting ester product.
Suitable monocarboxylic acids and their derivatives include lauric,
myristic, palmitic, stearic, arachidic, benzoic, caprylic, palmitic, erucic,
palmitoleic, pentadecanoic, heptadecanoic, nonadecanoic, linoleic,
arachidoniiic, oleic, valeric,roic, capric and 2-ethylhexanoic acids, and
mixtures thereof. In a preferred embodiment, the monocarboxylic acid is
2-ethyihexancic acid. In some embodiments, the dic-arboxylic acid esters
produced by the processes provided herein, in particular the bis-2-
ethylhexanoate esters will have uses as functional fluids, for example, as
lubricants.
For preparation of the carboxylic acid esters, the P 03G can be
contacted, preferably in the presence of an inert gas, with the
monocarboxyllc acid(s) at temperatures ranging from about 100 C to
about 275 G, from about 1 0 C to 250'C, and most preferably at about
120"G, The process can be carried out at atmospheric pressure or under
vacuum. During the contacting water is formed and can be removed in the
inert gas stream or under vacuum to drive the reaction to completion,
To facilitate the reaction of PO with carboxylic acid an
ester/canon catalyst is generally used, preferably an acid catalyst.
Examples of suitable acid catalysts n,',ude but are not limited to sulfuric
acid, hydrochloric acid, phosphoric acid, hydriodic acid. Other suitable
catalysts include heterogeneous catalysts such as zeolites,
heteropolyacid, amberlyst,. and ion exchange resin. A particularly
preferred acid catalyst is sulfuric acid. The amount of catalyst used in the
contacting of P03G with monocarbaxylic acid can be from about 0.01 wt
% to about 10 wt % of the reaction mixture, preferably from 0.1 wt % to
-13-
CA 02767675 2012-01-09
WO 2011/011276 PCT/US2010/042255
about 5 wE %, and more preferably from about . wt. % to about 2 *1 %,
of the reaction mixture.
Any ratio of monocarbcxylic acid, or derivatives thereof, to glycol
hydroxyl groups can be used, The preferred ratio of acid to hydroxyl
groups is from about 31 to about 12, where the ratio can be adjusted to
shift the ratio of monoester to diester in the product. Generally to favor
production of diesters sightly more than a 11 ratio is used. To favor
production of monoesters, a 0-5-1 ratio or less of monocarboxylic acid to
hydroxyl is used.
A preferred process comprises pol'lycondensing 1,3-propanediol in
the presence of carbon black to polytrinethylene ether glycol using an
acid catalyst (as described herein), then subsequently adding
monocarboxylic acid and carrying out the esterifcation to form a
dicarboxylic acid ester of P0 3G, It is preferred that the contacting of
P03G with a monocaà boxylic acid is carried out without first isolating and
purifying the P03 G.
The polycondensation reaction is continued until desired molecular
weight is reached, and then the monocarboxylic acid is subsequently
added to the reaction mixture. The reaction is continued while the water
byproduct is removed.. At this stage both esterification and etherification
reactions occur simultaneously. Thus, in a preferred process, the acid
catalyst used for polycondensation of diolis also used for esterification
without adding addW.Wonal catalyst. However, it is contemplated that
additional': catalyst can be added at the esterification stage.
In an alternative procedure, the esterification reaction can be
carried out on purified P03G by addition of an ster fication catalyst and
monocarboxylic acid followed by heating and removal of water,
Regardless of which esterification procedure is followed, after the
esterification step any by products are removed, and then the catalyst
residues remaining from polycondensation and/or esterification are
removed in order to obtain an ester product that is stable, ;particularly at
high temperatures. This may be accomplished by hydrolysis of the crude
ester product by treatment with water at from about 80T to about OO'C
for a time sufficient to hydrolyze any residual acid esters derived from the
-14-
CA 02767675 2012-01-09
WO 2011/011276 PCT/US2010/042255
catalyst without impacting significantly the carboxylic acid esters. The time
required can vary from about I to about 8 hours. If the hydrolysis is carried
out under pressure, high r temperatures and correspondingly shorter
times are possible. At this point the product may contain theaters,
monoesters, or a combination of theaters and monoesters, and small
amounts of acid catalyst, unreacted carboxylic acid and dial depending on
the reaction conditions, However, dicarboxylic acid esters are preferred;
and processes which produce dicarboxylrc acid esters are preferred.
The hydrolyzed polymer is further purified to remove water, acid
catalyst and unreacted carboxylic acid by the known conventional
techniques such as water washings, base neutralization, filtration and/or
distillation. Unreacted dial and acid catalyst can, for example., be removed
by washing with deionized water. Unreacted carboxylic acid also can
removed, for example, by washing with deionied water or aqueous base
solutions, or by vacuum stripping) If desired, the product can be
fractionated further toÃsolate lbw molecular weight esters by a fractional
distillation under reduced po essure.
EXAMPLES
Materials, Equipment, and Test Methods
The bio-derived PDO used in the Examples herein is commercially
available from E.I. DuPont de Nemours & Co. as DuPont Tate & Lyle Bic-
PD rr" For Examples 2, 3, and 4, carbon black (Nor it Carbon) was
obtained from Univar (product name Dar o .) G-60). For examples 6, and
7, carbon black was type ADP carbon (Calgon Carbon).
Test Method 1. Color Measurement and APHA Values.
A Hunterlab Color Quest XE Spe trocolorimet r (Reston, Va.) was
used to measure the polymer color resulting from the absence or presence
of carbon black treatment. Color numbers of the polymer are measured
as APHA values (Platinum-Cobalt System) according to ATM D-1209.
-15-
CA 02767675 2012-01-09
WO 2011/011276 PCT/US2010/042255
The polymer molecular weights were calculated from their hydroxyl
numbers obtained from NMR or were determined using a previously
generated standard curve based on polymer viscosity.
C :arative Example A: Control Pc:=Ãymer:zatio
12 kg of bio-based PDO monomer was added to a 20L glass
reactor equipped with a condenser and an agitator, purged with N2 at the
rate 5L/min. The reactant was heated up to 170 C with agitation speed of
250 rpm. When the reactant temperature reached 170 C, 187.5 g of
sulfuric acid was added into the reactor. The time of sulfuric acid addition
was set as reaction starting point. Polymerization proceeded at 170 C.
The reaction volatiles were condensed in the condenser and the polymer
product was accumulated in the reactor. Polymer samples were taken
periodically for color and molecular weight analysis. The number average
molecular weight of polymer was determined by N MR and the product
color was determined using a Hunter Lab Color quest XE machine and
expressed as APHA index. Molecular weight development is shown in
Figure 1 and product color is shown in Figure 2.
Example 0,05 weight -percent of Carbon Black
The equipment and polymerization procedures were the same as in
Comparative Example A except for carbon black addition. 0.05 weight
percent of carbon black (D f o G-60, Univar) on the basis of bÃo-based
PLO was added together with the monomer at the beginning of the
polymerization. Carbon black was mixed with monomer under agitation
when the reactor temperature was increased to 170'C. 187.5 g of sulfuric
acid was added at 170 C and the polymerization occurred in the present of
carbon black, Product molecular weight and color were measured after
carbon black removal by filtration at ambient temperature using a syringe
filter. The product color was measured by visual comparison of the
samples with a series of standard samples determined using a Hunter Lab
Color quest XE machine and expressed as APHA index. The molecular
weight and color developments are shown in Figures1 and 2 respectively.
-16-
CA 02767675 2012-01-09
WO 2011/011276 PCT/US2010/042255
Examle 2: g.1 wei ht percent of Carbon Black
The equipment and polymerization procedures were the same as in
Example 1 except for amount. of carbon black addition. 0,1 weight percent
of carbon black on the basis of bio-based PDO was added together with
the monomer at the beginning of the polymerization, The molecular
weight and color developments are shown in Figures 1 and 2 respectively
Exam le 3: 0.5 wei. ht percent of Carbon Black
The equipment and polymerization procedures were the same as in
Example 1 excent for amount. of carbon black addition, 0.5 weight percent
of carbon black on the basis of biowbased PDO was added together with
the monomer at the beginning of the polymerization, The molecular
weight and color developments are shown in Figures 1 and 2 respectively.
Comparative Ex m .le B: Control poi m rization
900 g of bio-based PDO monomer:. 11.5g of 0.98 percent purity
sulfuric acid, and 6.1g of 10 weight percent sodium carbonate solution in
dernineralized water (for color control) were added to a 1 L glass reactor
equipped with a condenser and an agitator, purged with N2 at the rate of
35L/min. The reactant was heated up to 170'C with agitation speed of
120 rpm. The time the heat was turned on was set as the reaction starting
point. Polymerization proceeded at 1 70C. The reaction volathles were
condensed in the condenser and polymer product was accumulated in the
reactor. The polymer samples were taken periodically for molecular
weight analysis, using a viscometr. The total reaction time is 18 hours.
The number average molecular weight of polymer was determined from its
viscosity, which is calibrated based on NMR measurements. The product
color was determined using Hunter Lab Color quest ?E machine and
expressed as APHA index. The molecular weight and color of final crude
polymer are shown in Table 1.
Example 0. +ei tit percent of Carbon Black, added at reaction times of
2 and 5 hours
-17-
CA 02767675 2012-01-09
WO 2011/011276 PCT/US2010/042255
900 g of biro-based P00 monomer and 11.5g of 0.98 percent purity
sulfuric acid were added to a 1 L glass reactor equipped with a condenser
and an agitator, purged with N2 at the rate of 35L/min, The reactant was
heated up to 170' wA,th agitation speed of 120 rpm. The time the heat
was turned on was sat as the reaction starting point, Polymerization
proceeded at 170 C. A mixture of 2 g of carbon black in about 10 g bio-
P00 is added into the reaction at reaction times of 2 and 5 hours. The
reaction volatiles were condensed in the condenser and polymer product
was accumulated in the reactor, The polymer samples were taken
periodically for molecular weight analysis, using a viscometer, Total
reaction time is 25 hours. The number average molecular weight of
polymer was determined from its viscosity. The product color was
measured by visual comparison of the samples with a series of standard
samples determined using a Hunter Lab Color quest XE machine and
expressed as APHA index. The molecular weight and color of final crude
polymer are shown in Table 1.
Example 5., 0,5 !eight percent of Carbon Black, added at reaction time of
4 hours
900 g of bio-based P00 monomer and 11.5g of 0.98 percent purity
sulfuric acid were added to a IL glass reactor equipped with a condenser
and an agitator, purged with N2 at the rate of ;'i>> 'rlmin. The reactant was
heated up to 170`with agitation speed of 120 rpm. The time the heat
was turned on was set as the reaction starting point. Polymerization
proceeded at 170 C. A mixture of 4 g of carbon black in about 10 g bio-
PDO is added into the reaction at reaction time of 4 hours. The reaction
volatiles were condensed in the condenser and polymer product was
accumulated in the reactor. The polymer samples were taken periodically
for molecular weight analysis, using a viscometer. Total reaction time is
25 hours. The number average molecular weight of polymer was
determined from its viscosity. The product color was measured by visual
comparison of the samples with a series of standard samples determined
using a Hunter Lab Color quest XE machine and expressed as APHA
-18-
CA 02767675 2012-01-09
WO 2011/011276 PCT/US2010/042255
index. The molecular weight and color of final crude polymer are shown in
Table 1
Table 1. Result summary
----------------------- ------------------------------------------ ------------
-------------------------------------------------------------------------------
--- --------------------------------
rumple Heat/Reacton Viscosity (Cp) M based on oior
time (hr) viscosity(g/moi) (APHA)
Conip 8 18 7,246 3,244 --500
4
,444 4,836 200
--------------- ---------------------------------------------------------------
---------------------------------------------------------- --------------------
----------
Exam (PROPHETIQ .EsterÃfi tion fP0 G
PDO is polymerized to form P030 homopolymer in the presence of
carbon black as described in other Examples. When the reaction product
reaches a MW of about 300 (or a viscosity of 150 cP), -ethylhexanoic
acid is added to the reaction mixture to esterify the P030 hor,
mopolyr er,
The amount of 2-ethylhexanoic acid addled is about 60 % of the original
PLO charged into the reactor. No addl=tE anal acid catalyst is added. The
temperature is reduced to 120'C,. and the reaction is carried out for about
6 to 7 additional hours with no changes in the pressure. The resulting
ester product is tested for color as described and is analyzed using proton
NMR and IR for MW and % esterification respectively. It is preferred that
the color will be below about 200 APHA and that the % estenfcation will
be at least 80%. The reaction product is then purified by rreutrali ing the
acid and removing the impurities from the product using methods known in
the art, for example as in Pat, Publication 20080108845.
-19-