Note: Descriptions are shown in the official language in which they were submitted.
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
ANTI-ANGIOGENESIS THERAPY FOR THE TREATMENT OF
PREVIOUSLY TREATED BREAST CANCER
RELATED APPLICATION
This application claims priority to and the benefit of United States
Provisional
Application Serial No. 61/266,343, filed December 3, 2009 and United States
Provisional
Application Serial No. 61/234,28 1, filed August 15, 2009, the specifications
of which are
incorporated herein in their entirety.
FIELD OF THE INVENTION
This invention relates in general to treatment of human diseases and
pathological
conditions. More specifically, the invention relates to anti-angiogenesis
therapy, either alone
or in combination with other anti-cancer therapies, for the treatment of
previously treated
breast cancer.
BACKGROUND
Cancer remains to be one of the most deadly threats to human health. In the
U.S., cancer
affects nearly 1.3 million new patients each year, and is the second leading
cause of death
after heart disease, accounting for approximately 1 in 4 deaths. It is also
predicted that cancer
may surpass cardiovascular diseases as the number one cause of death within 5
years. Solid
tumors are responsible for most of those deaths. Although there have been
significant
advances in the medical treatment of certain cancers, the overall 5-year
survival rate for all
cancers has improved only by about 10% in the past 20 years. Cancers, or
malignant tumors,
metastasize and grow rapidly in an uncontrolled manner, making timely
detection and
treatment extremely difficult.
Breast cancer is a disease that kills many women each year in the United
States.
According to the American Cancer Society, approximately 40,000 will die from
the disease in
2008. Over 180,000 new cases of breast cancer are diagnosed annually, and it
is estimated
that one in eight women will develop breast cancer. These numbers indicate
that breast cancer
is one of the most dangerous diseases facing women today. Metastatic breast
cancer is
generally incurable with only a few patients achieving long-term survival
after standard
chemotherapy. Greenberg et al., J. Clin. Oncol. 14:2197-2205 (1996).
Knowledge of the basic biology of breast cancer has expanded exponentially
over the
1
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
last three decades with some having an impact on therapy. A multinational,
open-label phase
II trial of 222 women with HER2 overexpressing metastatic breast cancer found
a response
rate of 15% with six confirmed complete responses using a recombinant
humanized
monoclonal antibody (trastuzumab, also known as Herceptin , Genentech, South
San
Francisco) directed against HER2 (Cobleigh et al., Proc. Am. Soc. Clin. Oncol.
17:97 (1998)).
A randomized phase III trial evaluated the safety and efficacy of adding
Herceptin to first-line
chemotherapy with either paclitaxel or the combination of doxorubicin plus
cyclophosphamide. Overall response rate and time to progression significantly
improved with
the addition of Herceptin to chemotherapy compared to chemotherapy alone
(Slamon et al.,
Proc. Am. Soc. Clin. Oncol. 17:98 (1998)). The addition of Herceptin prolonged
overall
survival (Norton et al., Proc. Am. Soc. Clin. Oncol. 18:127a (1999)).
Though trastuzumab is the first novel, biologically-based therapeutic agent
approved for
the treatment of a subpopulation of breast cancer patients having HER2
overexpressing
cancers, several other approaches have shown promise and have entered the
clinic. However,
there are estimates that 75 percent of women will newly diagnosed metastatic
breast cancer
are HER2-negative. Compounds which inhibit angiogenesis have generated
particular interest
for reaching additional breast cancer populations and have been and are the
subject of clinical
trials both in the US and abroad.
Since cancer is still one of the most deadly threats, additional cancer
treatments for
patients are needed. The invention addresses these and other needs, as will be
apparent upon
review of the following disclosure.
SUMMARY
Uses of an anti-VEGF antibody for effectively treating breast cancer patients
with
previously treated metastic breast cancer are provided. In particular, the
invention provides
data from a randomized phase III clinical trial of bevacizumab (AVASTIN ) in
combination
with chemotherapy regimes in subjects with previously treated metastic breast
cancer in
human subjects. Such chemotherapy regimes include taxane therapy (e.g.,
paclitaxel q3wk or
weekly, paclitaxel protein-bound particles (e.g., Abraxane ) or docetaxel),
gemcitabine,
vinorelbine or capecitabine therapy. The success of the trial shows that
adding anti-VEGF
antibody to chemotherapy provides statistically significant and clinically
meaningful benefits
as a second line therapy for previously treated breast cancer patients.
2
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
The results obtained in clinical studies of the use of bevacizumab in human
subjects with
metastatic breast cancer show that the efficacy, as evaluated by progression
free survival
(PFS) was positive especially when compared to PFS data for chemotherapeutic
agents alone.
Subjects in the clinical trials who received bevacizumab in combination with
chemotherapy
(taxane therapy, capecitabine, gemcitabine or vinorelbine) had an increase in
progression free
survival compared to subjects treated with chemotherapy alone. The difference
was
significantly significant.
Accordingly, the invention provides a method of treating a patient diagnosed
with
previously treated metastatic breast cancer, comprising subjecting the patient
to a treatment
regimen combining a chemotherapy with the administration of an effective
amount of an anti-
VEGF antibody. The treatment regimen combining the chemotherapy with the
administration
of the anti-VEGF effectively extends the progression free survival (PFS) of
the patient.
In certain embodiments, the PFS is extended about 0.5 months, 1 month, 1.2
months, 2
months, 2.1 months, 2.2 months, 2.8 months, 3 months, etc. In one embodiment,
the PFS is
extended about 2.1 months. In one embodiment, the PFS is extended about 2.2
months. In
one embodiment, the PFS is extended about 2.8 months.
Any chemotherapeutic agent exhibiting anticancer activity can be used
according to the
invention. In certain embodiments, the chemotherapeutic agent is selected from
the group
consisting of alkylating agents, antimetabolites, folic acid analogs,
pyrimidine analogs, purine
analogs and related inhibitors, vinca alkaloids, epipodopyyllotoxins,
antibiotics, L-
Asparaginase, topoisomerase inhibitor, interferons, platinum cooridnation
complexes,
anthracenedione substituted urea, methyl hydrazine derivatives, adrenocortical
suppressant,
adrenocorticosteroides, progestins, estrogens, antiestrogen, androgens,
antiandrogen, and
gonadotropin-releasing hormone analog. In certain embodiments, the
chemotherapeutic agent
is for example, capecitabine, taxane, paclitaxel, docetaxel, paclitaxel
protein-bound particles
(e.g., Abraxane ), gemcitabine, vinorelbine or combinations thereof. In
certain
embodiments, the chemotherapeutic agent is for example, capecitabine, taxane,
paclitaxel,
docetaxel, paclitaxel protein-bound particles (e.g., Abraxane ), gemcitabine,
or combinations
thereof. In certain embodiments, the chemotherapeutic agent is for example,
capecitabine,
taxane, paclitaxel, docetaxel, paclitaxel protein-bound particles (e.g.,
Abraxane ), or
combinations thereof. Two or more chemotherapeutic agents can be used in a
cocktail to be
administered in combination with administration of the anti-VEGF antibody.
3
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
Clinical benefits of the treatments according to the invention can be measured
by, for
example, duration of progression free survival (PFS), time to treatment
failure, objective
response rate and duration of response.
Accordingly, the invention features a method of instructing a human subject
with
previously treated, e.g., breast, cancer by providing instructions to receive
treatment with an
anti-VEGF antibody so as to increase progression free survival of the subject,
to decrease the
subject's risk of cancer recurrence or to increase the subject's likelihood of
survival. In some
embodiments the method further comprises providing instructions to receive
treatment with at
least one chemotherapeutic agent. The treatment with the anti-VEGF antibody
may be
concurrent with or sequential to the treatment with the chemotherapeutic
agent. In certain
embodiments the subject is treated as instructed by the method of instructing.
The invention also provides a promotional method, comprising promoting the
administration of an anti-VEGF antibody for treatment of previously treated,
e.g., breast,
cancer in a human subject. In some embodiments, the method further comprises
promoting
the administration of at least one chemotherapeutic agent. Administration of
the anti-VEGF
antibody may be concurrent with or sequential to administration of the
chemotherapeutic
agent. Promotion may be conducted by any means available. In some embodiments,
the
promotion is by a package insert accompanying a commercial formulation of the
anti-VEGF
antibody. The promotion may also be by a package insert accompanying a
commercial
formulation of the chemotherapeutic agent. Promotion may be by written or oral
communication to a physician or health care provider. In some embodiments, the
promotion
is by a package insert where the package inset provides instructions to
receive therapy with
anti-VEGF antibody. In some embodiments, the promotion is followed by the
treatment of
the subject with the anti-VEGF antibody with or without the chemotherapeutic
agent.
The invention provides a business method, comprising marketing an anti-VEGF
antibody for treatment of previously treated, e.g., breast, cancer in a human
subject so as to
increase progression free survival, or decrease the subject's likelihood of
cancer recurrence or
increase the subject's likelihood of survival. In some embodiments the method
further
comprises marketing a chemotherapeutic agent for use in combination with the
anti-VEGF
antibody. In some embodiments the marketing is followed by treatment of the
subject with
the anti-VEGF antibody with or without the chemotherapeutic agent.
Also provided is a business method, comprising marketing a chemotherapeutic
agent
in combination with an anti-VEGF antibody for treatment of previously treated,
e.g., breast,
4
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
cancer in a human subject so as to increase progression free survival, or
decrease the subject's
likelihood of cancer recurrence or increase the subject's likelihood of
survival. In some
embodiments, the marketing is followed by treatment of the subject with the
combination of
the chemotherapeutic agent and the anti-VEGF antibody.
In each of the methods of the invention the anti-VEGF antibody may be
substituted with
a VEGF specific antagonist, e.g., a VEGF receptor molecule or chimeric VEGF
receptor
molecule as described below. In certain embodiments of the methods of the
invention the
anti-VEGF antibody is bevacizumab. The anti-VEGF antibody, or antigen-binding
fragment
thereof, can be a monoclonal antibody, a chimeric antibody, a fully human
antibody, or a
humanized antibody. Exemplary antibodies useful in the methods of the
invention include
bevacizumab (AVASTIN ), a G6 antibody, a B20 antibody, and fragments thereof.
In
certain embodiments, the anti-VEGF antibody has a heavy chain variable region
comprising
the following amino acid sequence:
EVQLVESGGG LVQPGGSLRL SCAASGYTFT NYGMNWVRQA PGKGLEWVGW
INTYTGEPTY AADFKRRFTF SLDTSKSTAY LQMNSLRAED TAVYYCAKYP
HYYGSSHWYF DVWGQGTLVT VSS (SEQ ID No. 1)
and a light chain variable region comprising the following amino acid
sequence:
DIQMTQSPSS LSASVGDRVT ITCSASQDIS NYLNWYQQKP GKAPKVLIYF
TSSLHSGVPS RFSGSGSGTD FTLTISSLQP EDFATYYCQQ YSTVPWTFGQ
GTKVEIKR (SEQ ID No. 2).
The antibody, or antigen-binding fragment thereof, can also be an antibody
that lacks
an Fc portion, an F(ab')2, an Fab, or an Fv structure.
In one embodiment, the treatment is a combination of a VEGF-specific
antagonist,
e.g., anti-VEGF antibody, and at least one chemotherapeutic agent.
Each of the methods of the invention may be practiced in relation to the
treatment of
cancers including, but not limited to, carcinoma, lymphoma, blastoma, sarcoma,
and
leukemia. More particular examples of such cancers include breast cancer,
squamous cell
cancer, small-cell lung cancer, non-small cell lung cancer, adenocarcinoma of
the lung,
squamous carcinoma of the lung, cancer of the peritoneum, hepatocellular
cancer,
gastrointestinal cancer, pancreatic cancer, glioblastoma, cervical cancer,
ovarian cancer, liver
5
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
cancer, bladder cancer, hepatoma, colon cancer, colorectal cancer, endometrial
or uterine
carcinoma, salivary gland carcinoma, kidney cancer, liver cancer, prostate
cancer, renal
cancer, vulval cancer, thyroid cancer, hepatic carcinoma, gastric cancer,
melanoma, and
various types of head and neck cancer. In some embodiments, the subject has
HER2-negative
metastic previously treated breast cancer.
Each of the above aspects can further include monitoring the subject for
recurrence of
the cancer. Monitoring can be accomplished, for example, by evaluating
progression free
survival (PFS) or overall survival (OS) or objective response rate (ORR). In
one embodiment,
the PFS or the OS or the ORR is evaluated after initiation of treatment.
Depending on the type and severity of the disease, preferred dosages for the
anti-VEGF
antibody, e.g., bevacizumab, are described herein and can range from about 1
g/kg to about 50
mg/kg, most preferably from about 5 mg/kg to about 15 mg/kg, including but not
limited to 5
mg/kg, 7.5 mg/kg, 10 mg/kg or 15 mg/kg. The frequency of administration will
vary
depending on the type and severity of the disease. For repeated
administrations over several
days or longer, depending on the condition, the treatment is sustained until
the cancer is
treated or the desired therapeutic effect is achieved, as measured by the
methods described
herein or known in the art. In one example, the anti-VEGF antibody of the
invention is
administered once every week, every two weeks, or every three weeks, at a dose
range from
about 5 mg/kg to about 15 mg/kg, including but not limited to 5 mg/kg, 7.5
mg/kg, 10 mg/kg
or 15 mg/kg. However, other dosage regimens may be useful. The progress of the
therapy of
the invention is easily monitored by conventional techniques and assays.
In additional embodiments of each of the above aspects, the VEGF-specific
antagonist,
e.g., anti-VEGF antibody is administered locally or systemically (e.g., orally
or
intravenously). In other embodiments, one aspect of the treatment is with the
VEGF-specific
antagonist in extended treatment phase or maintenance therapy, as assessed by
the clinician or
described herein.
In other embodiments, treatment with the VEGF-specific antagonist for
previously
treated metastatic breast cancer is in combination with an additional anti-
cancer therapy,
including but not limited to, surgery, radiation therapy, chemotherapy,
differentiating therapy,
biotherapy, immune therapy, an angiogenesis inhibitor, a cytotoxic agent and
an anti-
proliferative compound. Treatment with the VEGF-specific antagonist can also
include any
combination of the above types of therapeutic regimens. In some embodiments,
the
chemotherapeutic agent and the VEGF-specific antagonist are administered
concurrently.
6
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
In the embodiments which include an additional anti-cancer therapy, the
subject can be
further treated with the additional anti-cancer therapy before, during (e.g.,
simultaneously), or
after administration of the VEGF-specific antagonist. In one embodiment, the
VEGF-specific
antagonist, administered either alone or with an anti-cancer therapy, can be
administered as
maintenance therapy.
Other features and advantages of the invention will be apparent from the
following
Detailed Description, the drawings, and the claims.
BRIEF DESCRIPTION OF THE DRAWINGS
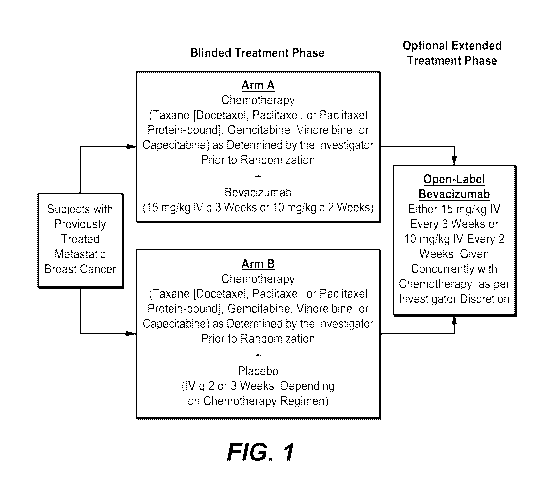
Figure 1 depicts the study design for the previously treated metastatic breast
cancer
trial using bevacizumab (Arm A) or placebo (Arm B) with various
chemotherapies.
Figure 2 depicts the primary endpoint analysis of PFS of the study in Figure
1.
Figure 3 depicts the cohort-specific analyses of PFS of the study in Figure 1.
Figure 4 depicts the objective response rate of the study in Figure 1.
DETAILED DESCRIPTION
1. DEFINITIONS
The term "VEGF" or "VEGF-A" is used to refer to the 165-amino acid human
vascular
endothelial cell growth factor and related 121-, 145-, 189-, and 206- amino
acid human
vascular endothelial cell growth factors, as described by, e.g., Leung et al.
Science, 246:1306
(1989), and Houck et al. Mol. Endocrin., 5:1806 (1991), together with the
naturally occurring
allelic and processed forms thereof. VEGF-A is part of a gene family including
VEGF-B,
VEGF-C, VEGF-D, VEGF-E, VEGF-F, and P1GF. VEGF-A primarily binds to two high
affinity receptor tyrosine kinases, VEGFR-1 (Flt- 1) and VEGFR-2 (Flk-1/KDR),
the latter
being the major transmitter of vascular endothelial cell mitogenic signals of
VEGF-A.
Additionally, neuropilin-1 has been identified as a receptor for heparin-
binding VEGF-A
isoforms, and may play a role in vascular development. The term "VEGF" or
"VEGF-A" also
refers to VEGFs from non-human species such as mouse, rat, or primate.
Sometimes the
VEGF from a specific species is indicated by terms such as hVEGF for human
VEGF or
mVEGF for murine VEGF. The term "VEGF" is also used to refer to truncated
forms or
fragments of the polypeptide comprising amino acids 8 to 109 or 1 to 109 of
the 165-amino
acid human vascular endothelial cell growth factor. Reference to any such
forms of VEGF
7
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
maybe identified in the application, e.g., by "VEGF (8-109)," "VEGF (1-109)"
or
"VEGF165." The amino acid positions for a "truncated" native VEGF are numbered
as
indicated in the native VEGF sequence. For example, amino acid position 17
(methionine) in
truncated native VEGF is also position 17 (methionine) in native VEGF. The
truncated native
VEGF has binding affinity for the KDR and Flt-1 receptors comparable to native
VEGF.
An "anti-VEGF antibody" is an antibody that binds to VEGF with sufficient
affinity and
specificity. The antibody selected will normally have a binding affinity for
VEGF, for
example, the antibody may bind hVEGF with a Kd value of between 100 nM-1 pM.
Antibody
affinities may be determined by a surface plasmon resonance based assay (such
as the
BlAcore assay as described in PCT Application Publication No. W02005/012359);
enzyme-
linked immunoabsorbent assay (ELISA); and competition assays (e.g. RIA's), for
example. In
certain embodiments, the anti-VEGF antibody of the invention can be used as a
therapeutic
agent in targeting and interfering with diseases or conditions wherein the
VEGF activity is
involved. Also, the antibody may be subjected to other biological activity
assays, e.g., in
order to evaluate its effectiveness as a therapeutic. Such assays are known in
the art and
depend on the target antigen and intended use for the antibody. Examples
include, but are not
limited to, the HUVEC inhibition assay; tumor cell growth inhibition assays
(as described in
WO 89/06692, for example). An anti-VEGF antibody will usually not bind to
other VEGF
homologues such as VEGF-B or VEGF-C, nor other growth factors such as P1GF,
PDGF or
bFGF.
A "VEGF antagonist" refers to a molecule capable of neutralizing, blocking,
inhibiting,
abrogating, reducing or interfering with VEGF activities including its binding
to one or more
VEGF receptors. VEGF antagonists include anti-VEGF antibodies and antigen-
binding
fragments thereof, receptor molecules and derivatives which bind specifically
to VEGF
thereby sequestering its binding to one or more receptors, anti-VEGF receptor
antibodies and
VEGF receptor antagonists such as small molecule inhibitors of the VEGFR
tyrosine kinases.
A "native sequence" polypeptide comprises a polypeptide having the same amino
acid
sequence as a polypeptide derived from nature. Thus, a native sequence
polypeptide can have
the amino acid sequence of naturally-occurring polypeptide from any mammal.
Such native
sequence polypeptide can be isolated from nature or can be produced by
recombinant or
synthetic means. The term "native sequence" polypeptide specifically
encompasses naturally-
occurring truncated or secreted forms of the polypeptide (e.g., an
extracellular domain
8
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
sequence), naturally-occurring variant forms (e.g., alternatively spliced
forms) and naturally-
occurring allelic variants of the polypeptide.
A polypeptide "variant" means a biologically active polypeptide having at
least about 80%
amino acid sequence identity with the native sequence polypeptide. Such
variants include, for
instance, polypeptides wherein one or more amino acid residues are added, or
deleted, at the N-
or C-terminus of the polypeptide. Ordinarily, a variant will have at least
about 80% amino acid
sequence identity, more preferably at least about 90% amino acid sequence
identity, and even
more preferably at least about 95% amino acid sequence identity with the
native sequence
polypeptide.
The term "antibody" is used in the broadest sense and includes monoclonal
antibodies
(including full length or intact monoclonal antibodies), polyclonal
antibodies, multivalent
antibodies, multispecific antibodies (e.g., bispecific antibodies), and
antibody fragments (see
below) so long as they exhibit the desired biological activity.
Throughout the specification and claims, the numbering of the residues in an
immunoglobulin heavy chain is that of the EU index as in Kabat et al.,
Sequences of Proteins
of Immunological Interest, 5th Ed. Public Health Service, National Institutes
of Health,
Bethesda, Md. (1991), expressly incorporated herein by reference. The "EU
index as in
Kabat" refers to the residue numbering of the human IgGI EU antibody.
The "Kd" or "Kd value" according to this invention is in one embodiment
measured
by a radiolabeled VEGF binding assay (RIA) performed with the Fab version of
the antibody
and a VEGF molecule as described by the following assay that measures solution
binding
affinity of Fabs for VEGF by equilibrating Fab with a minimal concentration of
(125I)-labeled
VEGF(109) in the presence of a titration series of unlabeled VEGF, then
capturing bound
VEGF with an anti-Fab antibody-coated plate (Chen, et al., (1999) J. Mol Biol
293:865-881).
In one example, to establish conditions for the assay, microtiter plates
(Dynex) are coated
overnight with 5 ug/ml of a capturing anti-Fab antibody (Cappel Labs) in 50 mM
sodium
carbonate (pH 9.6), and subsequently blocked with 2% (w/v) bovine serum
albumin in PBS
for two to five hours at room temperature (approximately 23 C). In a non-
adsorbant plate
(Nunc #269620), 100 pM or 26 pM [125I]VEGF(109) are mixed with serial
dilutions of a Fab
of interest, e.g., Fab-12 (Presta et al., (1997) Cancer Res. 57:4593-4599).
The Fab of interest
is then incubated overnight; however, the incubation may continue for 65 hours
to insure that
equilibrium is reached. Thereafter, the mixtures are transferred to the
capture plate for
9
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
incubation at room temperature for one hour. The solution is then removed and
the plate
washed eight times with 0.1% Tween-20 in PBS. When the plates had dried, 150
ul/well of
scintillant (MicroScint-20; Packard) is added, and the plates are counted on a
Topcount
gamma counter (Packard) for ten minutes. Concentrations of each Fab that give
less than or
equal to 20% of maximal binding are chosen for use in competitive binding
assays.
According to another embodiment the Kd or Kd value is measured by using
surface plasmon
resonance assays using a BlAcoreTM-2000 or a BlAcoreTM-3000 (BlAcore, Inc.,
Piscataway,
NJ) at 25 C with immobilized hVEGF (8-109) CM5 chips at -10 response units
(RU).
Briefly, carboxymethylated dextran biosensor chips (CM5, BlAcore Inc.) are
activated with
N-ethyl-N'- (3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC) and N-
hydroxysuccinimide (NHS) according to the supplier's instructions. Human VEGF
is diluted
with 10mM sodium acetate, pH 4.8, into 5ug/ml (-0.2uM) before injection at a
flow rate of
5u1/minute to achieve approximately 10 response units (RU) of coupled protein.
Following the
injection of human VEGF, 1M ethanolamine is injected to block unreacted
groups. For
kinetics measurements, two-fold serial dilutions of Fab (0.78 nM to 500 nM)
are injected in
PBS with 0.05% Tween 20 (PBST) at 25 C at a flow rate of approximately
25u1/min.
Association rates (k n) and dissociation rates (k ff) are calculated using a
simple one-to-one
Langmuir binding model (BIAcore Evaluation Software version 3.2) by
simultaneous fitting
the association and dissociation sensorgram. The equilibrium dissociation
constant (Kd) was
calculated as the ratio k ff/k n. See, e.g., Chen, Y., et al., (1999)J. Mol
Biol 293:865-881. If
the on-rate exceeds 106 M_1 S_1 by the surface plasmon resonance assay above,
then the on-rate
is can be determined by using a fluorescent quenching technique that measures
the increase or
decrease in fluorescence emission intensity (excitation = 295 nm; emission =
340 nm, 16 nm
band-pass) at 25 C of a 20nM anti-VEGF antibody (Fab form) in PBS, pH 7.2, in
the presence
of increasing concentrations of human VEGF short form (8-109) or mouse VEGF as
measured
in a spectrometer, such as a stop-flow equipped spectrophometer (Aviv
Instruments) or a
8000-series SLM-Aminco spectrophotometer (ThermoSpectronic) with a stirred
cuvette. The
"Kd" or "Kd value" according to this invention in one embodiment is measured
by techniques
known in the art.
A "blocking" antibody or an antibody "antagonist" is one which inhibits or
reduces
biological activity of the antigen it binds. For example, a VEGF-specific
antagonist antibody
binds VEGF and inhibits the ability of VEGF to induce vascular endothelial
cell proliferation
or to induce vascular permeability. Preferred blocking antibodies or
antagonist antibodies
completely inhibit the biological activity of the antigen.
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
Unless indicated otherwise, the expression "multivalent antibody" is used
throughout
this specification to denote an antibody comprising three or more antigen
binding sites. For
example, the multivalent antibody is engineered to have the three or more
antigen binding
sites and is generally not a native sequence IgM or IgA antibody.
"Antibody fragments" comprise only a portion of an intact antibody, generally
including
an antigen binding site of the intact antibody and thus retaining the ability
to bind antigen.
Examples of antibody fragments encompassed by the present definition include:
(i) the Fab
fragment, having VL, CL, VH and CH1 domains; (ii) the Fab' fragment, which is
a Fab
fragment having one or more cysteine residues at the C-terminus of the CH1
domain; (iii) the
Fd fragment having VH and CH1 domains; (iv) the Fd' fragment having VH and CH1
domains and one or more cysteine residues at the C-terminus of the CH1 domain;
(v) the Fv
fragment having the VL and VH domains of a single arm of an antibody; (vi) the
dAb
fragment (Ward et at., Nature 341, 544-546 (1989)) which consists of a VH
domain; (vii)
isolated CDR regions; (viii) F(ab')2 fragments, a bivalent fragment including
two Fab'
fragments linked by a disulphide bridge at the hinge region; (ix) single chain
antibody
molecules (e.g. single chain Fv; scFv) (Bird et at., Science 242:423-426
(1988); and Huston et
at., PNAS (USA) 85:5879-5883 (1988)); (x) "diabodies" with two antigen binding
sites,
comprising a heavy chain variable domain (VH) connected to a light chain
variable domain
(VL) in the same polypeptide chain (see, e.g., EP 404,097; WO 93/11161; and
Hollinger et
at., Proc. Natl. Acad. Sci. USA, 90:6444-6448 (1993)); (xi) "linear
antibodies" comprising a
pair of tandem Fd segments (VH-CHI-VH-CH1) which, together with complementary
light
chain polypeptides, form a pair of antigen binding regions (Zapata et at.
Protein Eng.
8(10):1057-1062 (1995); and US Patent No. 5,641,870).
The term "monoclonal antibody" as used herein refers to an antibody obtained
from a
population of substantially homogeneous antibodies, i.e., the individual
antibodies comprising
the population are identical except for possible naturally occurring mutations
that may be
present in minor amounts. Monoclonal antibodies are highly specific, being
directed against a
single antigen. Furthermore, in contrast to polyclonal antibody preparations
that typically
include different antibodies directed against different determinants
(epitopes), each
monoclonal antibody is directed against a single determinant on the antigen.
The modifier
"monoclonal" is not to be construed as requiring production of the antibody by
any particular
method. For example, the monoclonal antibodies to be used in accordance with
the invention
may be made by the hybridoma method first described by Kohler et at., Nature
256:495
11
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
(1975), or may be made by recombinant DNA methods (see, e.g., U.S. Patent No.
4,816,567).
The "monoclonal antibodies" may also be isolated from phage antibody libraries
using the
techniques described in Clackson et at., Nature 352:624-628 (1991) or Marks et
at., J. Mol.
Biol. 222:581-597 (1991), for example.
An "Fv" fragment is an antibody fragment which contains a complete antigen
recognition and binding site. This region consists of a dimer of one heavy and
one light chain
variable domain in tight association, which can be covalent in nature, for
example in scFv. It
is in this configuration that the three CDRs of each variable domain interact
to define an
antigen binding site on the surface of the VH-VL dimer. Collectively, the six
CDRs or a subset
thereof confer antigen binding specificity to the antibody. However, even a
single variable
domain (or half of an Fv comprising only three CDRs specific for an antigen)
has the ability to
recognize and bind antigen, although usually at a lower affinity than the
entire binding site.
As used herein, "antibody variable domain" refers to the portions of the light
and
heavy chains of antibody molecules that include amino acid sequences of
Complementarity
Determining Regions (CDRs; ie., CDR1, CDR2, and CDR3), and Framework Regions
(FRs).
VH refers to the variable domain of the heavy chain. VL refers to the variable
domain of the
light chain. According to the methods used in this invention, the amino acid
positions
assigned to CDRs and FRs may be defined according to Kabat (Sequences of
Proteins of
Immunological Interest (National Institutes of Health, Bethesda, Md., 1987 and
1991)).
Amino acid numbering of antibodies or antigen binding fragments is also
according to that of
Kabat.
As used herein, the term "Complementarity Determining Regions" (CDRs; i.e.,
CDR1,
CDR2, and CDR3) refers to the amino acid residues of an antibody variable
domain the
presence of which are necessary for antigen binding. Each variable domain
typically has three
CDR regions identified as CDR1, CDR2 and CDR3. Each complementarity
determining
region may comprise amino acid residues from a "complementarity determining
region" as
defined by Kabat (i.e. about residues 24-34 (L1), 50-56 (L2) and 89-97 (L3) in
the light chain
variable domain and 31-35 (H1), 50-65 (H2) and 95-102 (H3) in the heavy chain
variable
domain; Kabat et at., Sequences of Proteins of Immunological Interest, 5th Ed.
Public Health
Service, National Institutes of Health, Bethesda, MD. (1991)) and/or those
residues from a
"hypervariable loop" (i.e. about residues 26-32 (L1), 50-52 (L2) and 91-96
(L3) in the light
chain variable domain and 26-32 (H1), 53-55 (H2) and 96-101 (H3) in the heavy
chain
variable domain; Chothia and Lesk J. Mol. Biol. 196:901-917 (1987)). In some
instances, a
complementarity determining region can include amino acids from both a CDR
region defined
12
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
according to Kabat and a hypervariable loop. For example, the CDRH1 of the
heavy chain of
antibody 4D5 includes amino acids 26 to 35.
"Framework regions" (hereinafter FR) are those variable domain residues other
than
the CDR residues. Each variable domain typically has four FRs identified as
FRl, FR2, FR3
and FR4. If the CDRs are defined according to Kabat, the light chain FR
residues are
positioned at about residues 1-23 (LCFR1), 35-49 (LCFR2), 57-88 (LCFR3), and
98-107
(LCFR4) and the heavy chain FR residues are positioned about at residues 1-30
(HCFR1), 36-
49 (HCFR2), 66-94 (HCFR3), and 103-113 (HCFR4) in the heavy chain residues. If
the CDRs
comprise amino acid residues from hypervariable loops, the light chain FR
residues are
positioned about at residues 1-25 (LCFR1), 33-49 (LCFR2), 53-90 (LCFR3), and
97-107
(LCFR4) in the light chain and the heavy chain FR residues are positioned
about at residues 1-
25 (HCFR1), 33-52 (HCFR2), 56-95 (HCFR3), and 102-113 (HCFR4) in the heavy
chain
residues. In some instances, when the CDR comprises amino acids from both a
CDR as
defined by Kabat and those of a hypervariable loop, the FR residues will be
adjusted
accordingly. For example, when CDRH1 includes amino acids H26-H35, the heavy
chain
FR1 residues are at positions 1-25 and the FR2 residues are at positions 36-
49.
The "Fab" fragment contains a variable and constant domain of the light chain
and a
variable domain and the first constant domain (CH1) of the heavy chain.
F(ab')2 antibody
fragments comprise a pair of Fab fragments which are generally covalently
linked near their
carboxy termini by hinge cysteines between them. Other chemical couplings of
antibody
fragments are also known in the art.
"Single-chain Fv" or "scFv" antibody fragments comprise the VH and VL domains
of
antibody, wherein these domains are present in a single polypeptide chain.
Generally the Fv
polypeptide further comprises a polypeptide linker between the VH and VL
domains, which
enables the scFv to form the desired structure for antigen binding. For a
review of scFv, see
Pluckthun in The Pharmacology of Monoclonal Antibodies, Vol 113, Rosenburg and
Moore
eds. Springer-Verlag, New York, pp. 269-315 (1994).
The term "diabodies" refers to small antibody fragments with two antigen-
binding
sites, which fragments comprise a heavy chain variable domain (VH) connected
to a light
chain variable domain (VL) in the same polypeptide chain (VH and VL). By using
a linker that
is too short to allow pairing between the two domains on the same chain, the
domains are
forced to pair with the complementary domains of another chain and create two
antigen-
binding sites. Diabodies are described more fully in, for example, EP 404,097;
WO 93/11161;
and Hollinger et al., Proc. Natl. Acad. Sci. USA, 90:6444-6448 (1993).
13
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
The expression "linear antibodies" refers to the antibodies described in
Zapata et al.,
Protein Eng., 8(10):1057-1062 (1995). Briefly, these antibodies comprise a
pair of tandem Fd
segments (VH-CHI-VH-CH1) which, together with complementary light chain
polypeptides,
form a pair of antigen binding regions. Linear antibodies can be bispecific or
monospecific.
The monoclonal antibodies herein specifically include "chimeric" antibodies
(immunoglobulins) in which a portion of the heavy and/or light chain is
identical with or
homologous to corresponding sequences in antibodies derived from a particular
species or
belonging to a particular antibody class or subclass, while the remainder of
the chain(s) is
identical with or homologous to corresponding sequences in antibodies derived
from another
species or belonging to another antibody class or subclass, as well as
fragments of such
antibodies, so long as they exhibit the desired biological activity (U.S.
Patent No. 4,816,567;
and Morrison et al., Proc. Natl. Acad. Sci. USA 81:6851-6855 (1984)).
"Humanized" forms of non-human (e.g., murine) antibodies are chimeric
antibodies
which contain minimal sequence derived from non-human immunoglobulin. For the
most
part, humanized antibodies are human immunoglobulins (recipient antibody) in
which
residues from a hypervariable region of the recipient are replaced by residues
from a
hypervariable region of a non-human species (donor antibody) such as mouse,
rat, rabbit or
nonhuman primate having the desired specificity, affinity, and capacity. In
some instances, Fv
framework region (FR) residues of the human immunoglobulin are replaced by
corresponding
non-human residues. Furthermore, humanized antibodies may comprise residues
which are
not found in the recipient antibody or in the donor antibody. These
modifications are made to
further refine antibody performance. In general, the humanized antibody will
comprise
substantially all of at least one, and typically two, variable domains, in
which all or
substantially all of the hypervariable loops correspond to those of a non-
human
immunoglobulin and all or substantially all of the FR regions are those of a
human
immunoglobulin sequence. The humanized antibody optionally also will comprise
at least a
portion of an immunoglobulin constant region (Fc), typically that of a human
immunoglobulin. For further details, see Jones et at., Nature 321:522-525
(1986); Riechmann
et al., Nature 332:323-329 (1988); and Presta, Curr. Op. Struct. Biol. 2:593-
596 (1992).
A "human antibody" is one which possesses an amino acid sequence which
corresponds to that of an antibody produced by a human and/or has been made
using any of
the techniques for making human antibodies as disclosed herein. This
definition of a human
antibody specifically excludes a humanized antibody comprising non-human
antigen-binding
residues. Human antibodies can be produced using various techniques known in
the art. In
14
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
one embodiment, the human antibody is selected from a phage library, where
that phage
library expresses human antibodies (Vaughan et al. Nature Biotechnology 14:309-
314 (1996):
Sheets et al. Proc. Natl. Acad. Sci. 95:6157-6162 (1998)); Hoogenboom and
Winter, J. Mol.
Biol., 227:381 (1991); Marks et al., J. Mol. Biol., 222:581 (1991)). Human
antibodies can
also be made by introducing human immunoglobulin loci into transgenic animals,
e.g., mice in
which the endogenous immunoglobulin genes have been partially or completely
inactivated.
Upon challenge, human antibody production is observed, which closely resembles
that seen in
humans in all respects, including gene rearrangement, assembly, and antibody
repertoire. This
approach is described, for example, in U.S. Pat. Nos. 5,545,807; 5,545,806;
5,569,825;
5,625,126; 5,633,425; 5,661,016, and in the following scientific publications:
Marks et al.,
Bio/Technology 10: 779-783 (1992); Lonberg et al., Nature 368: 856-859 (1994);
Morrison,
Nature 368:812-13 (1994); Fishwild et al., Nature Biotechnology 14: 845-51
(1996);
Neuberger, Nature Biotechnology 14: 826 (1996); Lonberg and Huszar, Intern.
Rev. Immunol.
13:65-93 (1995). Alternatively, the human antibody may be prepared via
immortalization of
human B lymphocytes producing an antibody directed against a target antigen
(such B
lymphocytes may be recovered from an individual or may have been immunized in
vitro).
See, e.g., Cole et al., Monoclonal Antibodies and Cancer Therapy, Alan R.
Liss, p. 77 (1985);
Boerner et al., J. Immunol., 147 (1):86-95 (1991); and U.S. Pat. No.
5,750,373.
An "affinity matured" antibody is one with one or more alterations in one or
more
CDRs thereof which result an improvement in the affinity of the antibody for
antigen,
compared to a parent antibody which does not possess those alteration(s).
Preferred affinity
matured antibodies will have nanomolar or even picomolar affinities for the
target antigen.
Affinity matured antibodies are produced by procedures known in the art. Marks
et al.
Bio/Technology 10:779-783 (1992) describes affinity maturation by VH and VL
domain
shuffling. Random mutagenesis of CDR and/or framework residues is described
by: Barbas et
al. Proc Nat. Acad. Sci, USA 91:3809-3813 (1994); Schier et al. Gene 169:147-
155 (1995);
Yelton et al. J. Immunol. 155:1994-2004 (1995); Jackson et al., J. Immunol.
154(7):3310-9
(1995); and Hawkins et al., J. Mol. Biol. 226:889-896 (1992).
A "functional antigen binding site" of an antibody is one which is capable of
binding a
target antigen. The antigen binding affinity of the antigen binding site is
not necessarily as
strong as the parent antibody from which the antigen binding site is derived,
but the ability to
bind antigen must be measurable using any one of a variety of methods known
for evaluating
antibody binding to an antigen. Moreover, the antigen binding affinity of each
of the antigen
binding sites of a multivalent antibody herein need not be quantitatively the
same. For the
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
multimeric antibodies herein, the number of functional antigen binding sites
can be evaluated
using ultracentrifugation analysis as described in Example 2 of U.S. Patent
Application
Publication No. 20050186208. According to this method of analysis, different
ratios of target
antigen to multimeric antibody are combined and the average molecular weight
of the
complexes is calculated assuming differing numbers of functional binding
sites. These
theoretical values are compared to the actual experimental values obtained in
order to evaluate
the number of functional binding sites.
An antibody having a "biological characteristic" of a designated antibody is
one which
possesses one or more of the biological characteristics of that antibody which
distinguish it
from other antibodies that bind to the same antigen.
In order to screen for antibodies which bind to an epitope on an antigen bound
by an
antibody of interest, a routine cross-blocking assay such as that described in
Antibodies, A
Laboratory Manual, Cold Spring Harbor Laboratory, Ed Harlow and David Lane
(1988), can
be performed.
A "species-dependent antibody" is one which has a stronger binding affinity
for an
antigen from a first mammalian species than it has for a homologue of that
antigen from a
second mammalian species. Normally, the species-dependent antibody "binds
specifically" to
a human antigen (e.g., has a binding affinity (Kd) value of no more than about
1 x 10.7 M, or
no more than about 1 x 10-8 M or no more than about 1 x 10-9 M) but has a
binding affinity for
a homologue of the antigen from a second nonhuman mammalian species which is
at least
about 50 fold, or at least about 500 fold, or at least about 1000 fold, weaker
than its binding
affinity for the human antigen. The species-dependent antibody can be any of
the various
types of antibodies as defined above, but typically is a humanized or human
antibody.
As used herein, "antibody mutant" or "antibody variant" refers to an amino
acid
sequence variant of the species-dependent antibody wherein one or more of the
amino acid
residues of the species-dependent antibody have been modified. Such mutants
necessarily
have less than 100% sequence identity or similarity with the species-dependent
antibody. In
one embodiment, the antibody mutant will have an amino acid sequence having at
least 75%
amino acid sequence identity or similarity with the amino acid sequence of
either the heavy or
light chain variable domain of the species-dependent antibody, more preferably
at least 80%,
more preferably at least 85%, more preferably at least 90%, and most
preferably at least 95%.
Identity or similarity with respect to this sequence is defined herein as the
percentage of amino
acid residues in the candidate sequence that are identical (i.e same residue)
or similar (i.e.
amino acid residue from the same group based on common side-chain properties,
see below)
16
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
with the species-dependent antibody residues, after aligning the sequences and
introducing
gaps, if necessary, to achieve the maximum percent sequence identity. None of
N-terminal,
C-terminal, or internal extensions, deletions, or insertions into the antibody
sequence outside
of the variable domain shall be construed as affecting sequence identity or
similarity.
To increase the half-life of the antibodies or polypeptide containing the
amino acid
sequences of this invention, one can attach a salvage receptor binding epitope
to the antibody
(especially an antibody fragment), as described, e.g., in US Patent 5,739,277.
For example, a
nucleic acid molecule encoding the salvage receptor binding epitope can be
linked in frame to
a nucleic acid encoding a polypeptide sequence of this invention so that the
fusion protein
expressed by the engineered nucleic acid molecule comprises the salvage
receptor binding
epitope and a polypeptide sequence of this invention. As used herein, the term
"salvage
receptor binding epitope" refers to an epitope of the Fc region of an IgG
molecule (e.g., IgGi,
IgG2, IgG3, or IgG4) that is responsible for increasing the in vivo serum half-
life of the IgG
molecule (e.g., Ghetie et al., Ann. Rev. Immunol. 18:739-766 (2000), Table 1).
Antibodies
with substitutions in an Fc region thereof and increased serum half-lives are
also described in
W000/42072, WO 02/060919; Shields et al., J. Biol. Chem. 276:6591-6604 (2001);
Hinton,
J. Biol. Chem. 279:6213-6216 (2004)). In another embodiment, the serum half-
life can also
be increased, for example, by attaching other polypeptide sequences. For
example, antibodies
or other polypeptides useful in the methods of the invention can be attached
to serum albumin
or a portion of serum albumin that binds to the FcRn receptor or a serum
albumin binding
peptide so that serum albumin binds to the antibody or polypeptide, e.g., such
polypeptide
sequences are disclosed in WOO 1/45746. In one embodiment, the serum albumin
peptide to
be attached comprises an amino acid sequence of DICLPRWGCLW. In another
embodiment,
the half-life of a Fab is increased by these methods. See also, Dennis et al.
J. Biol. Chem.
277:35035-35043 (2002) for serum albumin binding peptide sequences.
A "chimeric VEGF receptor protein" is a VEGF receptor molecule having amino
acid
sequences derived from at least two different proteins, at least one of which
is as VEGF
receptor protein. In certain embodiments, the chimeric VEGF receptor protein
is capable of
binding to and inhibiting the biological activity of VEGF.
An "isolated" antibody is one that has been identified and separated and/or
recovered
from a component of its natural environment. Contaminant components of its
natural
environment are materials that would interfere with diagnostic or therapeutic
uses for the
antibody, and may include enzymes, hormones, and other proteinaceous or
nonproteinaceous
solutes. In certain embodiments, the antibody will be purified (1) to greater
than 95% by
17
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
weight of antibody as determined by the Lowry method, and most preferably more
than 99%
by weight, (2) to a degree sufficient to obtain at least 15 residues of N-
terminal or internal
amino acid sequence by use of a spinning cup sequenator, or (3) to homogeneity
by SDS-
PAGE under reducing or nonreducing conditions using Coomassie blue or, silver
stain.
Isolated antibody includes the antibody in situ within recombinant cells since
at least one
component of the antibody's natural environment will not be present.
Ordinarily, however,
isolated antibody will be prepared by at least one purification step.
By "fragment" is meant a portion of a polypeptide or nucleic acid molecule
that
contains, preferably, at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%,
95%, or
more of the entire length of the reference nucleic acid molecule or
polypeptide. A fragment
may contain 10, 20, 30, 40, 50, 60, 70, 80, 90, or 100, 200, 300, 400, 500,
600, or more
nucleotides or 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 120, 140, 160, 180,
190, 200 amino
acids or more.
An "anti-angiogenesis agent" or "angiogenesis inhibitor" refers to a small
molecular
weight substance, a polynucleotide, a polypeptide, an isolated protein, a
recombinant protein,
an antibody, or conjugates or fusion proteins thereof, that inhibits
angiogenesis,
vasculogenesis, or undesirable vascular permeability, either directly or
indirectly. It should be
understood that the anti-angiogenesis agent includes those agents that bind
and block the
angiogenic activity of the angiogenic factor or its receptor. For example, an
anti-angiogenesis
agent is an antibody or other antagonist to an angiogenic agent as defined
throughout the
specification or known in the art, e.g., but are not limited to, antibodies to
VEGF-A or to the
VEGF-A receptor (e.g., KDR receptor and/or Flt-1 receptor), VEGF-trap, anti-
PDGFR
inhibitors such as GleevecTM (Imatinib Mesylate). Anti-angiogensis agents also
include native
angiogenesis inhibitors, e.g., angiostatin, endostatin, etc. See, e.g.,
Klagsbrun and D'Amore,
Annu. Rev. Physiol., 53:217-39 (1991); Streit and Detmar, Oncogene, 22:3172-
3179 (2003)
(e.g., Table 3 listing anti-angiogenic therapy in malignant melanoma); Ferrara
& Alitalo,
Nature Medicine 5:1359-1364 (1999); Tonini et al., Oncogene, 22:6549-6556
(2003) (e.g.,
Table 2 listing known antiangiogenic factors); and Sato. Int. J. Clin. Oncol.,
8:200-206 (2003)
(e.g., Table 1 lists anti-angiogenic agents used in clinical trials).
A "maintenance" dose herein refers to one or more doses of a therapeutic agent
administered to the patient over or after a treatment period. Usually, the
maintenance doses
are administered at spaced treatment intervals, such as approximately every
week,
approximately every 2 weeks, approximately every 3 weeks, or approximately
every 4 weeks.
18
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
"Survival" refers to the patient remaining alive, and includes progression
free survival
(PFS) and overall survival (OS). Survival can be estimated by the Kaplan-Meier
method, and
any differences in survival are computed using the stratified log-rank test.
"Progression free survival (PFS)" refers to the time from treatment (or
randomization)
to first disease progression or death. For example it is the time that the
patient remains alive,
without return of the cancer, e.g., for a defined period of time such as about
0.5 months, 1
month, 2 months, 2.1 months, 2.2. months, 2.8 months, 3 months, etc., from
initiation of
treatment or from initial diagnosis. In one embodiment, the PFS is extended
about 2.1
months. In one aspect of the invention, PFS can be assessed by Response
Evaluation Criteria
in Solid Tumors (RECIST).
"Overall survival" refers to the patient remaining alive for a defined period
of time,
such as about 1 year, about 2 years, about 3 years, about 4 years, about 5
years, about 10
years, etc., from initiation of treatment or from initial diagnosis.
By "extending survival" or "increasing the likelihood of survival" is meant
increasing
PFS and/or OS in a treated patient relative to an untreated patient (i.e.
relative to a patient not
treated with a VEGF-specific antagonist, e.g., a VEGF antibody), or relative
to a control
treatment protocol, such as treatment only with the chemotherapeutic agent,
such as those use
in the care for breast cancer. Survival is monitored for at least about one
month, two months,
four months, six months, nine months, or at least about 1 year, or at least
about 2 years, or at
least about 3 years, or at least about 4 years, or at least about 5 years, or
at least about 10
years, etc., following the initiation of treatment or following the initial
diagnosis.
Hazard ratio (HR) is a statistical definition for rates of events. For the
purpose of the
invention, hazard ratio is defined as representing the probability of an event
in the
experimental arm divided by the probability of an event in the control arm at
any specific
point in time. "Hazard ratio" in progression free survival analysis is a
summary of the
difference between two progression free survival curves, representing the
reduction in the risk
of death on treatment compared to control, over a period of follow-up.
The term "concurrently" is used herein to refer to administration of two or
more
therapeutic agents, where at least part of the administration overlaps in
time. Accordingly,
concurrent administration includes a dosing regimen when the administration of
one or more
agent(s) continues after discontinuing the administration of one or more other
agent(s).
By "maintenance therapy" is meant a therapeutic regimen that is given to
reduce the
likelihood of disease recurrence or progression. Maintenance therapy can be
provided for any
length of time, including extended time periods up to the life-span of the
subject.
19
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
Maintenance therapy can be provided after initial therapy or in conjunction
with initial or
additional therapies. Dosages used for maintenance therapy can vary and can
include
diminished dosages as compared to dosages used for other types of therapy.
The terms "cancer" and "cancerous" refer to or describe the physiological
condition in
mammals that is typically characterized by unregulated cell growth. Included
in this
definition are benign and malignant cancers as well as dormant tumors or
micrometastatses.
Examples of cancer include but are not limited to, carcinoma, lymphoma,
blastoma, sarcoma,
and leukemia. More particular examples of such cancers include breast cancer,
squamous cell
cancer, lung cancer (including small-cell lung cancer, non-small cell lung
cancer,
adenocarcinoma of the lung, and squamous carcinoma of the lung), cancer of the
peritoneum,
hepatocellular cancer, gastric or stomach cancer (including gastrointestinal
cancer), pancreatic
cancer, glioblastoma, cervical cancer, ovarian cancer, liver cancer, bladder
cancer, hepatoma,
colon cancer, colorectal cancer, endometrial or uterine carcinoma, salivary
gland carcinoma,
kidney or renal cancer, liver cancer, prostate cancer, vulval cancer, thyroid
cancer, hepatic
carcinoma and various types of head and neck cancer, as well as B-cell
lymphoma (including
low grade/follicular non-Hodgkin's lymphoma (NHL); small lymphocytic (SL) NHL;
intermediate grade/follicular NHL; intermediate grade diffuse NHL; high grade
immunoblastic NHL; high grade lymphoblastic NHL; high grade small non-cleaved
cell NHL;
bulky disease NHL; mantle cell lymphoma; AIDS-related lymphoma; and
Waldenstrom's
Macroglobulinemia); chronic lymphocytic leukemia (CLL); acute lymphoblastic
leukemia
(ALL); Hairy cell leukemia; chronic myeloblastic leukemia; and post-transplant
lymphoproliferative disorder (PTLD), as well as abnormal vascular
proliferation associated
with phakomatoses, edema (such as that associated with brain tumors), and
Meigs' syndrome.
By "metastasis" is meant the spread of cancer from its primary site to other
places in
the body. Cancer cells can break away from a primary tumor, penetrate into
lymphatic and
blood vessels, circulate through the bloodstream, and grow in a distant focus
(metastasize) in
normal tissues elsewhere in the body. Metastasis can be local or distant.
Metastasis is a
sequential process, contingent on tumor cells breaking off from the primary
tumor, traveling
through the bloodstream, and stopping at a distant site. At the new site, the
cells establish a
blood supply and can grow to form a life-threatening mass. Both stimulatory
and inhibitory
molecular pathways within the tumor cell regulate this behavior, and
interactions between the
tumor cell and host cells in the distant site are also significant.
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
By "subject" is meant a mammal, including, but not limited to, a human or non-
human
mammal, such as a bovine, equine, canine, ovine, or feline. Preferably, the
subject is a
human. Patients are also subjects herein.
For the methods of the invention, the term "instructing" a subject means
providing
directions for applicable therapy, medication, treatment, treatment regimens,
and the like, by
any means, but preferably in writing, such as in the form of package inserts
or other written
promotional material.
For the methods of the invention, the term "promoting" means offering,
advertising,
selling, or describing a particular drug, combination of drugs, or treatment
modality, by any
means, including writing, such as in the form of package inserts. Promoting
herein refers to
promotion of a therapeutic agent, such as a VEGF antagonist, e.g., anti-VEGF
antibody or
chemotherapeutic agent, for an indication, such as breast cancer treatment,
where such
promoting is authorized by the Food and Drug Administration (FDA) as having
been
demonstrated to be associated with statistically significant therapeutic
efficacy and acceptable
safety in a population of subjects
The term "marketing" is used herein to describe the promotion, selling or
distribution
of a product (e.g., drug). Marketing specifically includes packaging,
advertising, and any
business activity with the purpose of commercializing a product.
A "population" of subjects refers to a group of subjects with cancer, such as
in a
clinical trial, or as seen by oncologists following FDA approval for a
particular indication,
such as breast cancer therapy.
The term "anti-cancer therapy" refers to a therapy useful in treating cancer.
Examples
of anti-cancer therapeutic agents include, but are limited to, e.g., surgery,
chemotherapeutic
agents, growth inhibitory agents, cytotoxic agents, agents used in radiation
therapy, anti-
angiogenesis agents, apoptotic agents, anti-tubulin agents, and other agents
to treat cancer,
such as anti-HER-2 antibodies, anti-CD20 antibodies, an epidermal growth
factor receptor
(EGFR) antagonist (e.g., a tyrosine kinase inhibitor), HERI/EGFR inhibitor
(e.g., erlotinib
(Tarceva ), platelet derived growth factor inhibitors (e.g., GleevecTM
(Imatinib Mesylate)), a
COX-2 inhibitor (e.g., celecoxib), interferons, cytokines, antagonists (e.g.,
neutralizing
antibodies, small molecule inhibitors, etc.) that bind to one or more of the
following targets
ErbB2, ErbB3, ErbB4, PDGFR-beta, B1yS, APRIL, BCMA or VEGF receptor(s), VEGF,
TRAIL/Apo2, and other bioactive and organic chemical agents, etc. Combinations
thereof are
also included in the invention.
21
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
The term "cytotoxic agent" as used herein refers to a substance that inhibits
or prevents
the function of cells and/or causes destruction of cells. The term is intended
to include
radioactive isotopes (e.g. At211, 1131, I125 Y90 Re186 Re188 Sm153 Bi212, P32
and radioactive
isotopes of Lu), chemotherapeutic agents, and toxins such as small molecule
toxins or
enzymatically active toxins of bacterial, fungal, plant or animal origin,
including fragments
and/or variants thereof.
A "chemotherapeutic agent" is a chemical compound useful in the treatment of
cancer.
Examples of chemotherapeutic agents include is a chemical compound useful in
the treatment
of cancer. Examples of chemotherapeutic agents include alkylating agents such
as thiotepa
and CYTOXAN cyclosphosphamide; alkyl sulfonates such as busulfan, improsulfan
and
piposulfan; aziridines such as benzodopa, carboquone, meturedopa, and uredopa;
ethylenimines and methylamelamines including altretamine, triethylenemelamine,
trietylenephosphoramide, triethiylenethiophosphoramide and
trimethylolomelamine;
acetogenins (especially bullatacin and bullatacinone); a camptothecin
(including the synthetic
analogue topotecan); bryostatin; callystatin; CC-1065 (including its
adozelesin, carzelesin and
bizelesin synthetic analogues); cryptophycins (particularly cryptophycin 1 and
cryptophycin
8); dolastatin; duocarmycin (including the synthetic analogues, KW-2189 and
CB1-TM1);
eleutherobin; pancratistatin; a sarcodictyin; spongistatin; nitrogen mustards
such as
chlorambucil, chlornaphazine, cholophosphamide, estramustine, ifosfamide,
mechlorethamine, mechlorethamine oxide hydrochloride, melphalan, novembichin,
phenesterine, prednimustine, trofosfamide, uracil mustard; nitrosureas such as
carmustine,
chlorozotocin, fotemustine, lomustine, nimustine, and ranimnustine;
antibiotics such as the
enediyne antibiotics (e. g., calicheamicin, especially calicheamicin gammalI
and
calicheamicin omegall (see, e.g., Agnew, Chem Intl. Ed. Engl., 33: 183-186
(1994));
dynemicin, including dynemicin A; bisphosphonates, such as clodronate; an
esperamicin; as
well as neocarzinostatin chromophore and related chromoprotein enediyne
antiobiotic
chromophores), aclacinomysins, actinomycin, authramycin, azaserine,
bleomycins,
cactinomycin, carabicin, carminomycin, carzinophilin, chromomycinis,
dactinomycin,
daunorubicin, detorubicin, 6-diazo-5-oxo-L-norleucine, ADRIAMYCIN doxorubicin
(including morpholino-doxorubicin, cyanomorpholino-doxorubicin, 2-pyrrolino-
doxorubicin
and deoxydoxorubicin), epirubicin, esorubicin, idarubicin, marcellomycin,
mitomycins such
as mitomycin C, mycophenolic acid, nogalamycin, olivomycins, peplomycin,
potfiromycin,
puromycin, quelamycin, rodorubicin, streptonigrin, streptozocin, tubercidin,
ubenimex,
zinostatin, zorubicin; anti-metabolites such as methotrexate and 5-
fluorouracil (5-FU); folic
22
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
acid analogues such as denopterin, methotrexate, pteropterin, trimetrexate;
purine analogs
such as fludarabine, 6-mercaptopurine, thiamiprine, thioguanine; pyrimidine
analogs such as
ancitabine, azacitidine, 6-azauridine, carmofur, cytarabine, dideoxyuridine,
doxifluridine,
enocitabine, floxuridine; androgens such as calusterone, dromostanolone
propionate,
epitiostanol, mepitiostane, testolactone; anti-adrenals such as
aminoglutethimide, mitotane,
trilostane; folic acid replenisher such as frolinic acid; aceglatone;
aldophosphamide glycoside;
aminolevulinic acid; eniluracil; amsacrine; bestrabucil; bisantrene;
edatraxate; defofamine;
demecolcine; diaziquone; elfornithine; elliptinium acetate; an epothilone;
etoglucid; gallium
nitrate; hydroxyurea; lentinan; lonidainine; maytansinoids such as maytansine
and
ansamitocins; mitoguazone; mitoxantrone; mopidanmol; nitraerine; pentostatin;
phenamet;
pirarubicin; losoxantrone; podophyllinic acid; 2- ethylhydrazide;
procarbazine; PSK
polysaccharide complex (JHS Natural Products, Eugene, OR); razoxane; rhizoxin;
sizofiran;
spirogermanium; tenuazonic acid; triaziquone; 2,2',2"-trichlorotriethylamine;
trichothecenes
(especially T-2 toxin, verracurin A, roridin A and anguidine); urethan;
vindesine; dacarbazine;
mannomustine; mitobronitol; mitolactol; pipobroman; gacytosine; arabinoside
("Ara-C");
cyclophosphamide; thiotepa; taxoids, e.g., TAXOL paclitaxel (Bristol- Myers
Squibb
Oncology, Princeton, N.J.), ABRAXANE Cremophor-free, albumin-engineered
nanoparticle
formulation of paclitaxel (American Pharmaceutical Partners, Schaumberg,
Illinois), and
TAXOTERE doxetaxel (Rhone- Poulenc Rorer, Antony, France); chloranbucil;
GEMZAR
gemcitabine; 6-thioguanine; mercaptopurine; methotrexate; platinum analogs
such as
cisplatin, oxaliplatin and carboplatin; vinblastine; platinum; etoposide (VP-
16); ifosfamide;
mitoxantrone; vincristine; NAVELBINE vinorelbine; novantrone; teniposide;
edatrexate;
daunomycin; aminopterin; xeloda; ibandronate; irinotecan (Camptosar, CPT-11)
(including
the treatment regimen of irinotecan with 5-FU and leucovorin); topoisomerase
inhibitor RFS
2000; difluorometlhylornithine (DMFO); retinoids such as retinoic acid;
capecitabine;
combretastatin; leucovorin (LV); oxaliplatin, including the oxaliplatin
treatment regimen
(FOLFOX); lapatinib (Tykerb ); inhibitors of PKC-alpha, Raf, H-Ras, EGFR
(e.g., erlotinib
(Tarceva )) and VEGF-A that reduce cell proliferation and pharmaceutically
acceptable salts,
acids or derivatives of any of the above.
Also included in this definition are anti-hormonal agents that act to regulate
or inhibit
hormone action on tumors such as anti-estrogens and selective estrogen
receptor modulators
(SERMs), including, for example, tamoxifen (including NOLVADEX tamoxifen),
raloxifene, droloxifene, 4-hydroxytamoxifen, trioxifene, keoxifene, LY117018,
onapristone,
and FARESTON= toremifene; aromatase inhibitors that inhibit the enzyme
aromatase, which
23
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
regulates estrogen production in the adrenal glands, such as, for example,
4(5)-imidazoles,
aminoglutethimide, MEGASE megestrol acetate, AROMASIN exemestane,
formestanie,
fadrozole, RIVISOR vorozole, FEMARA letrozole, and ARIMIDEX anastrozole;
and
anti-androgens such as flutamide, nilutamide, bicalutamide, leuprolide, and
goserelin; as well
as troxacitabine (a 1,3-dioxolane nucleoside cytosine analog); antisense
oligonucleotides,
particularly those which inhibit expression of genes in signaling pathways
implicated in
abherant cell proliferation, such as, for example, PKC-alpha, Ralf and H-Ras;
ribozymes such
as a VEGF expression inhibitor (e.g., ANGIOZYME ribozyme) and a HER2
expression
inhibitor; vaccines such as gene therapy vaccines, for example, ALLOVECTIN
vaccine,
LEUVECTIN vaccine, and VAXID vaccine; PROLEUKIN rIL-2; LURTOTECAN
topoisomerase 1 inhibitor; ABARELIX rmRH; and pharmaceutically acceptable
salts, acids
or derivatives of any of the above.
The term "cytokine" is a generic term for proteins released by one cell
population which
act on another cell as intercellular mediators. Examples of such cytokines are
lymphokines,
monokines, and traditional polypeptide hormones. Included among the cytokines
are growth
hormone such as human growth hormone, N-methionyl human growth hormone, and
bovine
growth hormone; parathyroid hormone; thyroxine; insulin; proinsulin; relaxin;
prorelaxin;
glycoprotein hormones such as follicle stimulating hormone (FSH), thyroid
stimulating
hormone (TSH), and luteinizing hormone (LH); epidermal growth factor; hepatic
growth
factor; fibroblast growth factor; prolactin; placental lactogen; tumor
necrosis factor-alpha and
-beta; mullerian-inhibiting substance; mouse gonadotropin-associated peptide;
inhibin;
activin; vascular endothelial growth factor; integrin; thrombopoietin (TPO);
nerve growth
factors such as NGF-alpha; platelet-growth factor; transforming growth factors
(TGFs) such
as TGF-alpha and TGF-beta; insulin-like growth factor-I and -II;
erythropoietin (EPO);
osteoinductive factors; interferons such as interferon-alpha, -beta and -gamma
colony
stimulating factors (CSFs) such as macrophage-CSF (M-CSF); granulocyte-
macrophage-CSF
(GM-CSF); and granulocyte-CSF (G-CSF); interleukins (ILs) such as IL-1, IL-
lalpha, IL-2,
IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-l0, IL-11, IL-12; a tumor
necrosis factor such as
TNF-alpha or TNF-beta; and other polypeptide factors including LIF and kit
ligand (KL). As
used herein, the term cytokine includes proteins from natural sources or from
recombinant cell
culture and biologically active equivalents of the native sequence cytokines.
A "growth inhibitory agent" when used herein refers to a compound or
composition
which inhibits growth of a cell in vitro and/or in vivo. Thus, the growth
inhibitory agent may be
24
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
one which significantly reduces the percentage of cells in S phase. Examples
of growth
inhibitory agents include agents that block cell cycle progression (at a place
other than S phase),
such as agents that induce G1 arrest and M-phase arrest. Classical M-phase
blockers include the
vincas (vincristine and vinblastine), TAXOL , and topo II inhibitors such as
doxorubicin,
epirubicin, daunorubicin, etoposide, and bleomycin. Those agents that arrest
G1 also spill over
into S-phase arrest, for example, DNA alkylating agents such as tamoxifen,
prednisone,
dacarbazine, mechlorethamine, cisplatin, methotrexate, 5-fluorouracil, and ara-
C. Further
information can be found in The Molecular Basis of Cancer, Mendelsohn and
Israel, eds.,
Chapter 1, entitled "Cell cycle regulation, oncogenes, and antineoplastic
drugs" by Murakami et
al. (WB Saunders: Philadelphia, 1995), especially p. 13.
The term "prodrug" as used in this application refers to a precursor or
derivative form
of a pharmaceutically active substance that is less cytotoxic to tumor cells
compared to the
parent drug and is capable of being enzymatically activated or converted into
the more active
parent form. See, e.g., Wilman, "Prodrugs in Cancer Chemotherapy" Biochemical
Society
Transactions, 14, pp. 375-382, 615th Meeting Belfast (1986) and Stella et al.,
"Prodrugs: A
Chemical Approach to Targeted Drug Delivery," Directed Drug Delivery,
Borchardt et al.,
(ed.), pp. 247-267, Humana Press (1985). The prodrugs of this invention
include, but are not
limited to, phosphate-containing prodrugs, thiophosphate-containing prodrugs,
sulfate-
containing prodrugs, peptide-containing prodrugs, D-amino acid-modified
prodrugs,
glycosylated prodrugs, (3-lactam-containing prodrugs, optionally substituted
phenoxyacetamide-containing prodrugs or optionally substituted phenylacetamide-
containing
prodrugs, 5-fluorocytosine and other 5-fluorouridine prodrugs which can be
converted into the
more active cytotoxic free drug. Examples of cytotoxic drugs that can be
derivatized into a
prodrug form for use in this invention include, but are not limited to, those
chemotherapeutic
agents described above.
By "radiation therapy" is meant the use of directed gamma rays or beta rays to
induce
sufficient damage to a cell so as to limit its ability to function normally or
to destroy the cell
altogether. It will be appreciated that there will be many ways known in the
art to determine
the dosage and duration of treatment. Typical treatments are given as a one
time
administration and typical dosages range from 10 to 200 units (Grays) per day.
By "reduce or inhibit" is meant the ability to prevent/delay or to cause an
overall
decrease preferably of 20% or greater, more preferably of 50% or greater, and
most preferably
of 75%, 85%, 90%, 95%, or greater. Reduce or inhibit can refer to the symptoms
of the
disorder being treated, the presence or size of metastases or micrometastases,
the size of the
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
primary tumor, the presence or the size of the dormant tumor, or the size or
number of the
blood vessels in angiogenic disorders.
The term "intravenous infusion" refers to introduction of a drug into the vein
of an animal
or human patient over a period of time greater than approximately 5 minutes,
preferably between
approximately 30 to 90 minutes, although, according to the invention,
intravenous infusion is
alternatively administered for 10 hours or less.
The term "intravenous bolus" or "intravenous push" refers to drug
administration into a
vein of an animal or human such that the body receives the drug in
approximately 15 minutes or
less, preferably 5 minutes or less.
The term "subcutaneous administration" refers to introduction of a drug under
the skin of
an animal or human patient, preferable within a pocket between the skin and
underlying tissue,
by relatively slow, sustained delivery from a drug receptacle. The pocket may
be created by
pinching or drawing the skin up and away from underlying tissue.
The term "subcutaneous infusion" refers to introduction of a drug under the
skin of an
animal or human patient, preferably within a pocket between the skin and
underlying tissue, by
relatively slow, sustained delivery from a drug receptacle for a period of
time including, but not
limited to, 30 minutes or less, or 90 minutes or less. Optionally, the
infusion may be made by
subcutaneous implantation of a drug delivery pump implanted under the skin of
the animal or
human patient, wherein the pump delivers a predetermined amount of drug for a
predetermined
period of time, such as 30 minutes, 90 minutes, or a time period spanning the
length of the
treatment regimen.
The term "subcutaneous bolus" refers to drug administration beneath the skin
of an
animal or human patient, where bolus drug delivery is preferably less than
approximately 15
minutes, more preferably less than 5 minutes, and most preferably less than 60
seconds.
Administration is preferably within a pocket between the skin and underlying
tissue, where the
pocket is created, for example, by pinching or drawing the skin up and away
from underlying
tissue.
A "disorder" is any condition that would benefit from treatment with the
antibody. This
includes chronic and acute disorders or diseases including those pathological
conditions which
predispose the mammal to the disorder in question. Non-limiting examples of
disorders to be
treated herein include cancer; benign and malignant tumors; leukemias and
lymphoid
malignancies; neuronal, glial, astrocytal, hypothalamic and other glandular,
macrophagal,
epithelial, stromal and blastocoelic disorders; and inflammatory, angiogenic
and immunologic
disorders.
26
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
The term "therapeutically effective amount" refers to an amount of a drug
effective to
treat a disease or disorder in a mammal. In the case of cancer, the
therapeutically effective
amount of the drug may reduce the number of cancer cells; reduce the tumor
size; inhibit (i.e.,
slow to some extent and preferably stop) cancer cell infiltration into
peripheral organs; inhibit
(i.e., slow to some extent and preferably stop) tumor metastasis; inhibit, to
some extent, tumor
growth; and/or relieve to some extent one or more of the symptoms associated
with the
disorder. To the extent the drug may prevent growth and/or kill existing
cancer cells, it may
be cytostatic and/or cytotoxic. For cancer therapy, efficacy in vivo can, for
example, be
measured by assessing the duration of survival, duration of progression free
survival (PFS),
the response rates (RR), duration of response, and/or quality of life.
"Treatment" refers to both therapeutic treatment and prophylactic or
preventative
measures. Those in need of treatment include those already with the disorder
as well as those
in which the disorder is to be prevented.
The word "label" when used herein refers to a detectable compound or
composition
which is conjugated directly or indirectly to the polypeptide. The label may
be itself be
detectable (e.g., radioisotope labels or fluorescent labels) or, in the case
of an enzymatic label,
may catalyze chemical alteration of a substrate compound or composition which
is detectable.
II. ANTI-VEGF ANTIBODIES AND ANTAGONISTS
(i) VEGF Antigen
The VEGF antigen to be used for production of antibodies may be, e.g., the
VEGF165
molecule as well as other isoforms of VEGF or a fragment thereof containing
the desired
epitope. Other forms of VEGF useful for generating anti-VEGF antibodies of the
invention
will be apparent to those skilled in the art.
Human VEGF was obtained by first screening a cDNA library prepared from human
cells, using bovine VEGF cDNA as a hybridization probe. Leung et at. (1989)
Science,
246:1306. One cDNA identified thereby encodes a 165-amino acid protein having
greater
than 95% homology to bovine VEGF; this 165-amino acid protein is typically
referred to as
human VEGF (hVEGF) or VEGF165. The mitogenic activity of human VEGF was
confirmed
by expressing the human VEGF cDNA in mammalian host cells. Media conditioned
by cells
27
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
transfected with the human VEGF cDNA promoted the proliferation of capillary
endothelial
cells, whereas control cells did not. Leung et at. (1989) Science, supra.
Although a vascular endothelial cell growth factor could be isolated and
purified from
natural sources for subsequent therapeutic use, the relatively low
concentrations of the protein
in follicular cells and the high cost, both in terms of effort and expense, of
recovering VEGF
proved commercially unavailing. Accordingly, further efforts were undertaken
to clone and
express VEGF via recombinant DNA techniques. (See, e.g., Ferrara, Laboratory
Investigation
72:615-618 (1995), and the references cited therein).
VEGF is expressed in a variety of tissues as multiple homodimeric forms (121,
145, 165,
189, and 206 amino acids per monomer) resulting from alternative RNA splicing.
VEGF121 is
a soluble mitogen that does not bind heparin; the longer forms of VEGF bind
heparin with
progressively higher affinity. The heparin-binding forms of VEGF can be
cleaved in the
carboxy terminus by plasmin to release a diffusible form(s) of VEGF. Amino
acid sequencing
of the carboxy terminal peptide identified after plasmin cleavage is Argiio-
Alaiii. Amino
terminal "core" protein, VEGF (1-110) isolated as a homodimer, binds
neutralizing
monoclonal antibodies (such as the antibodies referred to as 4.6.1 and
3.2E3.1.1) and soluble
forms of VEGF receptors with similar affinity compared to the intact VEGF165
homodimer.
Several molecules structurally related to VEGF have also been identified
recently,
including placenta growth factor (PIGF), VEGF-B, VEGF-C, VEGF-D and VEGF-E.
Ferrara
and Davis-Smyth (1987) Endocr. Rev., supra; Ogawa et al. J. Biological Chem.
273:31273-
31281(1998); Meyer et at. EMBO J., 18:363-374(1999). A receptor tyrosine
kinase, Flt-4
(VEGFR-3), has been identified as the receptor for VEGF-C and VEGF-D. Joukov
et al.
EMBO. J. 15:1751(1996); Lee et al. Proc. Natl. Acad. Sci. USA 93:1988-
1992(1996); Achen
et al. (1998) Proc. Natl. Acad. Sci. USA 95:548-553. VEGF-C has been shown to
be involved
in the regulation of lymphatic angiogenesis. Jeltsch et al. Science 276:1423-
1425(1997).
(ii) Anti- VEGF Antibodies
Anti-VEGF antibodies that are useful in the methods of the invention include
any
antibody, or antigen binding fragment thereof, that bind with sufficient
affinity and specificity
to VEGF and can reduce or inhibit the biological activity of VEGF. An anti-
VEGF antibody
will usually not bind to other VEGF homologues such as VEGF-B or VEGF-C, nor
other
growth factors such as P1GF, PDGF, or bFGF.
In certain embodiments of the invention, the anti-VEGF antibodies include, but
are not
limited to, a monoclonal antibody that binds to the same epitope as the
monoclonal anti-VEGF
28
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
antibody A4.6.1 produced by hybridoma ATCC HB 10709; a recombinant humanized
anti-
VEGF monoclonal antibody generated according to Presta et al. (1997) Cancer
Res. 57:4593-
4599. In one embodiment, the anti-VEGF antibody is "Bevacizumab (BV)", also
known as
"rhuMAb VEGF" or "AVASTIN ". It comprises mutated human IgGi framework regions
and antigen-binding complementarity-determining regions from the murine anti-
hVEGF
monoclonal antibody A.4.6.1 that blocks binding of human VEGF to its
receptors.
Approximately 93% of the amino acid sequence of bevacizumab, including most of
the
framework regions, is derived from human IgG 1, and about 7% of the sequence
is derived
from the murine antibody A4.6. 1.
Bevacizumab and other humanized anti-VEGF antibodies are further described in
U.S.
Pat. No. 6,884,879 issued Feb. 26, 2005. Additional antibodies include the G6
or B20 series
antibodies (e.g., G6-31, B20-4.1), as described in PCT Publication No.
W02005/012359, PCT
Publication No. W02005/044853, and US Patent Application Publication US2009-
0142343,
the content of these patent applications are expressly incorporated herein by
reference. For
additional antibodies see U.S. Pat. Nos. 7,060,269, 6,582,959, 6,703,020;
6,054,297;
W098/45332; WO 96/30046; W094/10202; EP 0666868B1; U.S. Patent Application
Publication Nos. 2006009360, 20050186208, 20030206899, 20030190317,
20030203409, and
20050112126; and Popkov et al., Journal of Immunological Methods 288:149-164
(2004).
Other antibodies include those that bind to a functional epitope on human VEGF
comprising
of residues F17, M18, D19, Y21, Y25, Q89,191, K101, E103, and C104 or,
alternatively,
comprising residues F17, Y21, Q22, Y25, D63, 183 and Q89.
In one embodiment of the invention, the anti-VEGF antibody has a heavy chain
variable
region comprising the following amino acid sequence:
EVQLVESGGG LVQPGGSLRL SCAASGYTFT NYGMNWVRQA PGKGLEWVGW
INTYTGEPTY AADFKRRFTF SLDTSKSTAY LQMNSLRAED TAVYYCAKYP
HYYGSSHWYF DVWGQGTLVT VSS (SEQ ID No. 1)
and a light chain variable region comprising the following amino acid
sequence:
DIQMTQSPSS LSASVGDRVT ITCSASQDIS NYLNWYQQKP GKAPKVLIYF
TSSLHSGVPS RFSGSGSGTD FTLTISSLQP EDFATYYCQQ YSTVPWTFGQ
GTKVEIKR (SEQ ID No. 2).
29
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
A "G6 series antibody" according to this invention, is an anti-VEGF antibody
that is
derived from a sequence of a G6 antibody or G6-derived antibody according to
any one of
Figures 7, 24-26, and 34-35 of PCT Publication No. W02005/012359, the entire
disclosure of
which is expressly incorporated herein by reference. See also PCT Publication
No.
W02005/044853, the entire disclosure of which is expressly incorporated herein
by reference.
In one embodiment, the G6 series antibody binds to a functional epitope on
human VEGF
comprising residues F17, Y21, Q22, Y25, D63, 183 and Q89.
A "B20 series antibody" according to this invention is an anti-VEGF antibody
that is
derived from a sequence of the B20 antibody or a B20-derived antibody
according to any one
of Figures 27-29 of PCT Publication No. W02005/012359, the entire disclosure
of which is
expressly incorporated herein by reference. See also PCT Publication No.
W02005/044853,
and US Patent Application Publication US2009-0142343, the content of these
patent
applications are expressly incorporated herein by reference. In one
embodiment, the B20
series antibody binds to a functional epitope on human VEGF comprising
residues F17, M18,
D19, Y21, Y25, Q89,191, KlOl, E103, and C104.
A "functional epitope" according to this invention refers to amino acid
residues of an
antigen that contribute energetically to the binding of an antibody. Mutation
of any one of the
energetically contributing residues of the antigen (for example, mutation of
wild-type VEGF
by alanine or homolog mutation) will disrupt the binding of the antibody such
that the relative
affinity ratio (IC50mutant VEGF/IC50wild-type VEGF) of the antibody will be
greater than 5
(see Example 2 of WO2005/012359). In one embodiment, the relative affinity
ratio is
determined by a solution binding phage displaying ELISA. Briefly, 96-well
Maxisorp
immunoplates (NUNC) are coated overnight at 4 C with an Fab form of the
antibody to be
tested at a concentration of 2ug/ml in PBS, and blocked with PBS, 0.5% BSA,
and 0.05%
Tween20 (PBT) for 2h at room temperature. Serial dilutions of phage displaying
hVEGF
alanine point mutants (residues 8-109 form) or wild type hVEGF (8-109) in PBT
are first
incubated on the Fab-coated plates for 15 min at room temperature, and the
plates are washed
with PBS, 0.05% Tween20 (PBST). The bound phage is detected with an anti-M13
monoclonal antibody horseradish peroxidase (Amersham Pharmacia) conjugate
diluted 1:5000
in PBT, developed with 3,3', 5,5'-tetramethylbenzidine (TMB, Kirkegaard &
Perry Labs,
Gaithersburg, MD) substrate for approximately 5 min, quenched with 1.0 M
H3PO4, and read
spectrophotometrically at 450 nm. The ratio of IC50 values (IC50,ala/IC50,wt)
represents the
fold of reduction in binding affinity (the relative binding affinity).
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
(iii) VEGF receptor molecules
Two VEGF receptors have been identified, Flt-1 (also called VEGFR-1) and KDR
(also
called VEGFR-2). Shibuya et al. (1990) Oncogene 8:519-527; de Vries et al.
(1992) Science
255:989-991; Terman et al. (1992) Biochem. Biophys. Res. Commun. 187:1579-
1586. The
specificity of each receptor for each VEGF family member varies but VEGF-A
binds to both
Flt-1 and KDR. Neuropilin-1 has been shown to be a selective VEGF receptor,
able to bind
the heparin-binding VEGF isoforms (Soker et al. (1998) Cell 92:735-45). Both
Flt-I and KDR
belong to the family of receptor tyrosine kinases (RTKs). The RTKs comprise a
large family
of transmembrane receptors with diverse biological activities. At present, at
least nineteen
(19) distinct RTK subfamilies have been identified. The receptor tyrosine
kinase (RTK)
family includes receptors that are crucial for the growth and differentiation
of a variety of cell
types (Yarden and Ullrich (1988) Ann. Rev. Biochem. 57:433-478; Ullrich and
Schlessinger
(1990) Cell 61:243-254). The intrinsic function of RTKs is activated upon
ligand binding,
which results in phosphorylation of the receptor and multiple cellular
substrates, and
subsequently in a variety of cellular responses (Ullrich & Schlessinger (1990)
Cell 61:203-
212). Thus, receptor tyrosine kinase mediated signal transduction is initiated
by extracellular
interaction with a specific growth factor (ligand), typically followed by
receptor dimerization,
stimulation of the intrinsic protein tyrosine kinase activity and receptor
trans-phosphorylation.
Binding sites are thereby created for intracellular signal transduction
molecules and lead to the
formation of complexes with a spectrum of cytoplasmic signaling molecules that
facilitate the
appropriate cellular response. (e.g., cell division, differentiation,
metabolic effects, changes in
the extracellular micro environment) see, Schlessinger and Ullrich (1992)
Neuron 9:1-20.
Structurally, both Flt-1 and KDR have seven immunoglobulin-like domains in the
extracellular domain, a single transmembrane region, and a consensus tyrosine
kinase
sequence which is interrupted by a kinase-insert domain. Matthews et al.
(1991) Proc. Natl.
Acad. Sci. USA 88:9026-9030; Terman et al. (1991) Oncogene 6:1677-1683.
VEGF receptor molecules, or fragments thereof, that specifically bind to VEGF
can be
used in the methods of the invention to bind to and sequester the VEGF
protein, thereby
preventing it from signaling. In certain embodiments, the VEGF receptor
molecule, or VEGF
binding fragment thereof, is a soluble form, such as sFlt-1. A soluble form of
the receptor
exerts an inhibitory effect on the biological activity of the VEGF protein by
binding to VEGF,
thereby preventing it from binding to its natural receptors present on the
surface of target
cells. Also included are VEGF receptor fusion proteins, examples of which are
described
below.
31
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
A chimeric VEGF receptor protein is a receptor molecule having amino acid
sequences
derived from at least two different proteins, at least one of which is a VEGF
receptor protein
(e.g., the flt-1 or KDR receptor), that is capable of binding to and
inhibiting the biological
activity of VEGF. In certain embodiments, the chimeric VEGF receptor proteins
of the
invention consist of amino acid sequences derived from only two different VEGF
receptor
molecules; however, amino acid sequences comprising one, two, three, four,
five, six, or all
seven Ig-like domains from the extracellular ligand-binding region of the flt-
1 and/or KDR
receptor can be linked to amino acid sequences from other unrelated proteins,
for example,
immunoglobulin sequences. Other amino acid sequences to which Ig-like domains
are
combined will be readily apparent to those of ordinary skill in the art.
Examples of chimeric
VEGF receptor proteins include, e.g., soluble Flt-1/Fc, KDR/Fc, or FLt-
1/KDR/Fc (also
known as VEGF Trap). (See for example PCT Application Publication No.
W097/44453)
A soluble VEGF receptor protein or chimeric VEGF receptor proteins of the
invention
includes VEGF receptor proteins which are not fixed to the surface of cells
via a
transmembrane domain. As such, soluble forms of the VEGF receptor, including
chimeric
receptor proteins, while capable of binding to and inactivating VEGF, do not
comprise a
transmembrane domain and thus generally do not become associated with the cell
membrane
of cells in which the molecule is expressed.
III. THERAPEUTIC USES OF ANTI-VEGF ANTIBODIES
The invention encompasses antiangiogenic therapy, a novel cancer treatment
strategy
aimed at inhibiting the development of tumor blood vessels required for
providing nutrients to
support tumor growth. Because angiogenesis is involved in both primary tumor
growth and
metastasis, the antiangiogenic treatment provided by the invention is capable
of inhibiting the
neoplastic growth of tumor at the primary site as well as preventing
metastasis of tumors at
the secondary sites, therefore allowing attack of the tumors by other
therapeutics.
Specifically, the invention provides a method of treating a patient diagnosed
with
previously treated metastatic breast cancer, comprising subjecting the patient
to a treatment
regimen combining a chemotherapy with the administration of an effective
amount of an anti-
VEGF antibody.
Combination Therapies
The invention features the use of a combination of at least one VEGF-specific
antagonist with one or more additional anti-cancer therapies. Examples of anti-
cancer
therapies include, without limitation, surgery, radiation therapy
(radiotherapy), biotherapy,
32
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
immunotherapy, chemotherapy, or a combination of these therapies. In addition,
cytotoxic
agents, anti-angiogenic and anti-proliferative agents can be used in
combination with the
VEGF-specific antagonist.
In certain aspects, the invention provides a method of treating previously
treated breast
cancer, by administering effective amounts of an anti-VEGF antibody and one or
more
chemotherapeutic agents to a patient susceptible to, or diagnosed with,
previously treated
metastatic cancer. A variety of chemotherapeutic agents may be used in the
combined
treatment methods of the invention. An exemplary and non-limiting list of
chemotherapeutic
agents contemplated is provided herein under "Definition", or described
herein.
In one example, the invention features the use of a VEGF-specific antagonist
with one
or more chemotherapeutic agents (e.g., a cocktail) or any combination thereof.
The combined
administration includes simultaneous administration, using separate
formulations or a single
pharmaceutical formulation, and consecutive administration in either order,
wherein
preferably there is a time period while both (or all) active agents
simultaneously exert their
biological activities. Preparation and dosing schedules for such
chemotherapeutic agents may
be used according to manufacturers' instructions or as determined empirically
by the skilled
practitioner. Preparation and dosing schedules for chemotherapy are also
described in
Chemotherapy Service Ed., M. C. Perry, Williams & Wilkins, Baltimore, Md.
(1992). The
chemotherapeutic agent may precede, or follow administration of the VEGF-
specific
antagonist or may be given simultaneously therewith.
In some other aspects, other therapeutic agents useful for combination tumor
therapy
with the antibody of the invention include antagonist of other factors that
are involved in
tumor growth, such as but not limited to EGFR, ErbB2 (also known as Her2)
ErbB3, ErbB4,
or TNF. Sometimes, it may be beneficial to also administer one or more
cytokines to the
patient. In one embodiment, the VEGF antibody is co-administered with a growth
inhibitory
agent. For example, the growth inhibitory agent may be administered first,
followed by the
VEGF antibody. However, simultaneous administration or administration of the
VEGF
antibody first is also contemplated. Suitable dosages for the growth
inhibitory agent are those
presently used and may be lowered due to the combined action (synergy) of the
growth
inhibitory agent and anti-VEGF antibody.
The formulation herein may also contain more than one active compound as
necessary
for the particular indication being treated, preferably those with
complementary activities that
do not adversely affect each other. For example, it may be desirable to
further provide
antibodies which bind to EGFR, VEGF (e.g. an antibody which binds a different
epitope on
33
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
VEGF), VEGFR, or ErbB2 (e.g., Herceptin ) in the one formulation.
Alternatively, or in
addition, the composition may comprise a cytotoxic agent, cytokine, growth
inhibitory agent
and/or small molecule VEGFR antagonist. Such molecules are suitably present in
combination in amounts that are effective for the purpose intended.
In certain aspects, other therapeutic agents useful for combination cancer
therapy with
the antibody of the invention include other anti-angiogenic agents. Many anti-
angiogenic
agents have been identified and are known in the arts, including those listed
by Carmeliet and
Jain (2000). In one embodiment, the anti-VEGF antibody of the invention is
used in
combination with another VEGF antagonist or a VEGF receptor antagonist such as
VEGF
variants, soluble VEGF receptor fragments, aptamers capable of blocking VEGF
or VEGFR,
neutralizing anti-VEGFR antibodies, low molecule weight inhibitors of VEGFR
tyrosine
kinases and any combinations thereof. Alternatively, or in addition, two or
more anti-VEGF
antibodies may be co-administered to the patient.
For the prevention or treatment of disease, the appropriate dosage of VEGF-
specific
antagonist will depend on the type of disease to be treated, as defined above,
the severity and
course of the disease, whether the VEGF-specific antagonist is administered
for preventive or
therapeutic purposes, previous therapy, the patient's clinical history and
response to the
VEGF-specific antagonist, and the discretion of the attending physician. The
VEGF-specific
antagonist is suitably administered to the patient at one time or over a
series of treatments. In
a combination therapy regimen, the VEGF-specific antagonist and the one or
more anti-cancer
therapeutic agent of the invention are administered in a therapeutically
effective or synergistic
amount. As used herein, a therapeutically effective amount is such that co-
administration of a
VEGF-specific antagonist and one or more other therapeutic agents, or
administration of a
composition of the invention, results in reduction or inhibition of the cancer
as described
above. A therapeutically synergistic amount is that amount of a VEGF-specific
antagonist
and one or more other therapeutic agents necessary to synergistically or
significantly reduce or
eliminate conditions or symptoms associated with a particular disease.
The VEGF-specific antagonist and the one or more other therapeutic agents can
be
administered simultaneously or sequentially in an amount and for a time
sufficient to reduce
or eliminate the occurrence or recurrence of a tumor, a dormant tumor, or a
micrometastases.
The VEGF-specific antagonist and the one or more other therapeutic agents can
be
administered as maintenance therapy to prevent or reduce the likelihood of
recurrence of the
tumor.
34
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
As will be understood by those of ordinary skill in the art, the appropriate
doses of
chemotherapeutic agents or other anti-cancer agents will be generally around
those already
employed in clinical therapies, e.g., where the chemotherapeutics are
administered alone or in
combination with other chemotherapeutics. Variation in dosage will likely
occur depending on
the condition being treated. The physician administering treatment will be
able to determine
the appropriate dose for the individual subject.
In addition to the above therapeutic regimes, the patient may be subjected to
radiation
therapy.
In certain embodiments, the administered VEGF antibody is an intact, naked
antibody.
However, the VEGF antibody may be conjugated with a cytotoxic agent. In
certain
embodiments, the conjugated antibody and/or antigen to which it is bound
is/are internalized
by the cell, resulting in increased therapeutic efficacy of the conjugate in
killing the cancer
cell to which it binds. In one embodiment, the cytotoxic agent targets or
interferes with
nucleic acid in the cancer cell. Examples of such cytotoxic agents include
maytansinoids,
calicheamicins, ribonucleases and DNA endonucleases.
The invention also features a method of instructing a human subject with
previously
treated breast cancer by providing instructions to receive treatment with an
anti-VEGF
antibody so as to increase the time for progression free survival, to decrease
the subject's risk
of cancer recurrence or to increase the subject's likelihood of survival. In
some embodiments
the method further comprises providing instructions to receive treatment with
at least one
chemotherapeutic agent. The treatment with the anti-VEGF antibody may be
concurrent with
or sequential to the treatment with the chemotherapeutic agent. In certain
embodiments the
subject is treated as instructed by the method of instructing. Treatment of
breast cancer by
administration of an anti-VEGF antibody with or without chemotherapy may be
continued
until cancer recurrence or death.
The invention further provides a promotional method, comprising promoting the
administration of an anti-VEGF antibody for treatment of previously treated
breast cancer in a
human subject. In some embodiments the method further comprises promoting the
administration of at least one chemotherapeutic agent. Administration of the
anti-VEGF
antibody may be concurrent with or sequential to administration of the
chemotherapeutic
agent. Promotion may be conducted by any means available. In some embodiments
the
promotion is by a package insert accompanying a commercial formulation of the
anti-VEGF
antibody. The promotion may also be by a package insert accompanying a
commercial
formulation of the chemotherapeutic agent. Promotion may be by written or oral
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
communication to a physician or health care provider. In some embodiments the
promotion is
by a package insert where the package inset provides instructions to receive
breast cancer
therapy with anti-VEGF antibody. In a further embodiment, the package insert
include some
or all of the results under Example 1. In some embodiments the promotion is
followed by the
treatment of the subject with the anti-VEGF antibody with or without the
chemotherapeutic
agent.
The invention provides a business method, comprising marketing an anti-VEGF
antibody for treatment of previously treated breast cancer in a human subject
so as to increase
the subject's time for progression free survival, to decrease the subject's
likelihood of cancer
recurrence or increase the subject's likelihood of survival. In some
embodiments the method
further comprises marketing a chemotherapeutic agent for use in combination
with the anti-
VEGF antibody. In some embodiments the marketing is followed by treatment of
the subject
with the anti-VEGF antibody with or without the chemotherapeutic agent.
Also provided is a business method, comprising marketing a chemotherapeutic
agent
in combination with an anti-VEGF antibody for treatment of previously treated
breast cancer
in a human subject so as to increase the subject's time for progression free
survival, to
decrease the subject's likelihood of cancer recurrence or increase the
subject's likelihood of
survival. In some embodiments the marketing is followed by treatment of the
subject with the
combination of the chemotherapeutic agent and the anti-VEGF antibody.
IV DOSAGES, AND DURATION
The VEGF-specific antagonist composition will be formulated, dosed, and
administered in a fashion consistent with good medical practice. Factors for
consideration in
this context include the particular disorder being treated, the particular
subject being treated,
the clinical condition of the individual patient, the cause of the disorder,
the site of delivery of
the agent, the method of administration, the scheduling of administration, and
other factors
known to medical practitioners. The "therapeutically effective amount" of the
VEGF-specific
antagonist to be administered will be governed by such considerations, and is
the minimum
amount necessary to prevent, ameliorate, or treat, or stabilize, the cancer;
to increase the time
until progression (duration of progression free survival) or to treat or
prevent the occurrence
or recurrence of a tumor, a dormant tumor, or a micrometastases of previously
treated cancer.
The VEGF-specific antagonist need not be, but is optionally, formulated with
one or more
agents currently used to prevent or treat cancer or a risk of developing a
cancer. The effective
amount of such other agents depends on the amount of VEGF-specific antagonist
present in
36
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
the formulation, the type of disorder or treatment, and other factors
discussed above. These
are generally used in the same dosages and with administration routes as used
hereinbefore or
about from 1 to 99% of the heretofore employed dosages.
Depending on the type and severity of the disease, about 1 g/kg to 100 mg/kg
(e.g.,
0.1-20 mg/kg) of VEGF-specific antagonist is an initial candidate dosage for
administration to
the patient, whether, for example, by one or more separate administrations, or
by continuous
infusion. A typical daily dosage might range from about 1 g/kg to about 100
mg/kg or more,
depending on the factors mentioned above. Particularly desirable dosages
include, for
example, 5 mg/kg, 7.5 mg/kg, 10 mg/kg, and 15 mg/kg. For repeated
administrations over
several days or longer, depending on the condition, the treatment is sustained
until the cancer
is treated, as measured by the methods described above or known in the art.
However, other
dosage regimens may be useful. In one example, if the VEGF-specific antagonist
is an
antibody, the antibody of the invention is administered once every week, every
two weeks, or
every three weeks, at a dose range from about 5 mg/kg to about 15 mg/kg,
including but not
limited to 5 mg/kg, 7.5 mg/kg, 10 mg/kg or 15 mg/kg. The progress of the
therapy of the
invention is easily monitored by conventional techniques and assays. In other
embodiments,
such dosing regimen is used in combination with a chemotherapy regimen as the
second line
therapy for treating previously treated metastatic breast cancer. Further
information about
suitable dosages is provided in the Example below.
The duration of therapy will continue for as long as medically indicated or
until a
desired therapeutic effect (e.g., those described herein) is achieved.
The VEGF-specific antagonists of the invention are administered to a subject,
e.g., a
human patient, in accord with known methods, such as intravenous
administration as a bolus
or by continuous infusion over a period of time, by intramuscular,
intraperitoneal,
intracerobrospinal, subcutaneous, intra-articular, intrasynovial, intrathecal,
oral, topical, or
inhalation routes. Local administration is particularly desired if extensive
side effects or
toxicity is associated with VEGF antagonism. An ex vivo strategy can also be
used for
therapeutic applications. Ex vivo strategies involve transfecting or
transducing cells obtained
from the subject with a polynucleotide encoding a VEGF antagonist. The
transfected or
transduced cells are then returned to the subject. The cells can be any of a
wide range of types
including, without limitation, hematopoietic cells (e.g., bone marrow cells,
macrophages,
monocytes, dendritic cells, T cells, or B cells), fibroblasts, epithelial
cells, endothelial cells,
keratinocytes, or muscle cells.
37
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
For example, if the VEGF-specific antagonist is an antibody, the antibody is
administered by any suitable means, including parenteral, subcutaneous,
intraperitoneal,
intrapulmonary, and intranasal, and, if desired for local immunosuppressive
treatment,
intralesional administration. Parenteral infusions include intramuscular,
intravenous,
intraarterial, intraperitoneal, or subcutaneous administration. In addition,
the antibody is
suitably administered by pulse infusion, particularly with declining doses of
the antibody.
Preferably the dosing is given by injections, most preferably intravenous or
subcutaneous
injections, depending in part on whether the administration is brief or
chronic.
In another example, the VEGF-specific antagonist compound is administered
locally,
e.g., by direct injections, when the disorder or location of the tumor
permits, and the injections
can be repeated periodically. The VEGF-specific antagonist can also be
delivered
systemically to the subject or directly to the tumor cells, e.g., to a tumor
or a tumor bed
following surgical excision of the tumor, in order to prevent or reduce local
recurrence or
metastasis, for example of a dormant tumor or micrometastases.
Alternatively, an inhibitory nucleic acid molecule or polynucleotide
containing a
nucleic acid sequence encoding a VEGF-specific antagonist can be delivered to
the
appropriate cells in the subject. In certain embodiments, the nucleic acid can
be directed to
the tumor itself.
The nucleic acid can be introduced into the cells by any means appropriate for
the
vector employed. Many such methods are well known in the art (Sambrook et al.,
supra, and
Watson et al., Recombinant DNA, Chapter 12, 2d edition, Scientific American
Books, 1992).
Examples of methods of gene delivery include liposome mediated transfection,
electroporation, calcium phosphate/DEAE dextran methods, gene gun, and
microinjection.
V. PHARMACEUTICAL FORMULATIONS
Therapeutic formulations of the antibodies used in accordance with the
invention are
prepared for storage by mixing an antibody having the desired degree of purity
with optional
pharmaceutically acceptable carriers, excipients or stabilizers (Remington's
Pharmaceutical
Sciences 16th edition, Osol, A. Ed. (1980)), in the form of lyophilized
formulations or
aqueous solutions. Acceptable carriers, excipients, or stabilizers are
nontoxic to recipients at
the dosages and concentrations employed, and include buffers such as
phosphate, citrate, and
other organic acids; antioxidants including ascorbic acid and methionine;
preservatives (such
as octadecyldimethylbenzyl ammonium chloride; hexamethonium chloride;
benzalkonium
chloride, benzethonium chloride; phenol, butyl or benzyl alcohol; alkyl
parabens such as
38
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
methyl or propyl paraben; catechol; resorcinol; cyclohexanol; 3-pentanol; and
m-cresol); low
molecular weight (less than about 10 residues) polypeptides; proteins, such as
serum albumin,
gelatin, or immunoglobulins; hydrophilic polymers such as
polyvinylpyrrolidone; amino acids
such as glycine, glutamine, asparagine, histidine, arginine, or lysine;
monosaccharides,
disaccharides, and other carbohydrates including glucose, mannose, or
dextrins; chelating
agents such as EDTA; sugars such as sucrose, mannitol, trehalose or sorbitol;
salt-forming
counter-ions such as sodium; metal complexes (e.g. Zn-protein complexes);
and/or non-ionic
surfactants such as TWEENTM, PLURONICSTM or polyethylene glycol (PEG).
Examples of
lyophilized anti-VEGF antibody formulations are described in WO 97/04801,
expressly
incorporated herein be reference.
Optionally, the formulation contains a pharmaceutically acceptable salt,
typically, e.g.,
sodium chloride, and often at about physiological concentrations. Optionally,
the
formulations of the invention can contain a pharmaceutically acceptable
preservative. In some
embodiments the preservative concentration ranges from 0.1 to 2.0%, typically
v/v. Suitable
preservatives include those known in the pharmaceutical arts. Benzyl alcohol,
phenol, m-
cresol, methylparaben, and propylparaben are examples of preservatives.
Optionally, the
formulations of the invention can include a pharmaceutically acceptable
surfactant at a
concentration of 0.005 to 0.02%.
In one embodiment, bevacizumab is supplied for therapeutic uses in 100 mg and
400 mg
preservative-free, single-use vials to deliver 4 ml or 16 ml of bevacizumab
(25 mg/ml). The
100 mg product is formulated in 240 mg a, a-trehalose dehydrate, 23.2 mg
sodium phosphate
(monobasic, monohydrate), 4.8 mg sodium phosphate (dibasic, anhydrous), 1.6 mg
polysorbate 20, and Water for Injection, USP. The 400 mg product is formulated
in 960 mg
a, a-trehalose dehydrate, 92.8 mg sodium phosphate (monobasic, monohydrate),
19.2 mg
sodium phosphate (dibasic, anhydrous), 6.4 mg polysorbate 20, and Water for
Injection, USP.
See also the label for bevacizumab. Bevacizumab is currently available
commercially in
certain countries.
The formulation herein may also contain more than one active compound as
necessary
for the particular indication being treated, preferably those with
complementary activities that
do not adversely affect each other. For example, it may be desirable to
further provide
antibodies which bind to EGFR, VEGF (e.g. an antibody which binds a different
epitope on
VEGF), VEGFR, or ErbB2 (e.g., Herceptin ) in the one formulation.
Alternatively, or in
addition, the composition may comprise a cytotoxic agent, cytokine, growth
inhibitory agent
39
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
and/or VEGFR antagonist (e.g., small molecule inhibitor, a polypeptide, etc.).
Such
molecules are suitably present in combination in amounts that are effective
for the purpose
intended.
The active ingredients may also be entrapped in microcapsules prepared, for
example, by
coacervation techniques or by interfacial polymerization, for example,
hydroxymethylcellulose or gelatin-microcapsules and poly-(methylmethacylate)
microcapsules, respectively, in colloidal drug delivery systems (for example,
liposomes,
albumin microspheres, microemulsions, nano-particles and nanocapsules) or in
macroemulsions. Such techniques are disclosed in Remington's Pharmaceutical
Sciences 16th
edition, Osol, A. Ed. (1980).
Sustained-release preparations may be prepared. Suitable examples of sustained-
release preparations include semipermeable matrices of solid hydrophobic
polymers
containing the antibody, which matrices are in the form of shaped articles,
e.g., films, or
microcapsule. Examples of sustained-release matrices include polyesters,
hydrogels (for
example, poly(2-hydroxyethyl-methacrylate), or poly(vinylalcohol)),
polylactides (U.S. Pat.
No. 3,773,919), copolymers of L-glutamic acid and y ethyl-L-glutamate, non-
degradable
ethylene-vinyl acetate, degradable lactic acid-glycolic acid copolymers such
as the LUPRON
DEPOTTM (injectable microspheres composed of lactic acid-glycolic acid
copolymer and
leuprolide acetate), and poly-D-(-)-3-hydroxybutyric acid. While polymers such
as ethylene-
vinyl acetate and lactic acid-glycolic acid enable release of molecules for
over 100 days,
certain hydrogels release proteins for shorter time periods. When encapsulated
antibodies
remain in the body for a long time, they may denature or aggregate as a result
of exposure to
moisture at 37 C, resulting in a loss of biological activity and possible
changes in
immunogenicity. Rational strategies can be devised for stabilization depending
on the
mechanism involved. For example, if the aggregation mechanism is discovered to
be
intermolecular S-S bond formation through thio-disulfide interchange,
stabilization may be
achieved by modifying sulfhydryl residues, lyophilizing from acidic solutions,
controlling
moisture content, using appropriate additives, and developing specific polymer
matrix
compositions.
The formulations to be used for in vivo administration may be sterile. This is
readily
accomplished by filtration through sterile filtration membranes.
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
VI EFFICACY OF THE TREATMENT
The main advantage of the treatment of the invention is the ability of
producing marked
anti-cancer effects in a human patient without causing significant toxicities
or adverse effects,
so that the patient benefited from the treatment overall. The efficacy of the
treatment of the
invention can be measured by various endpoints commonly used in evaluating
cancer
treatments, including but not limited to, tumor regression, tumor weight or
size shrinkage,
time to progression, duration of survival, progression free survival, overall
response rate,
duration of response, and quality of life. Because the anti-angiogenic agents
of the invention
target the tumor vasculature and not necessarily the neoplastic cells
themselves, they represent
a unique class of anticancer drugs, and therefore may require unique measures
and definitions
of clinical responses to drugs. For example, tumor shrinkage of greater than
50% in a 2-
dimensional analysis is the standard cut-off for declaring a response.
However, the anti-
VEGF antibody of the invention may cause inhibition of metastatic spread
without shrinkage
of the primary tumor, or may simply exert a tumouristatic effect. Accordingly,
novel
approaches to determining efficacy of an anti-angiogenic therapy can be
optionally employed,
including for example, measurement of plasma or urinary markers of
angiogenesis and
measurement of response through radiological imaging.
In another embodiment, the invention provides methods for increasing
progression free
survival of a human patient susceptible to or diagnosed with a previously
treated cancer.
Time to disease progression is defined as the time from administration of the
drug until
disease progression or death. In one embodiment, the combination treatment of
the invention
using anti-VEGF antibody and one or more chemotherapeutic agents significantly
increases
progression free survival by at least about 0.5 months, 1 month, 2 months, 2.1
months, 2.2
months, 2.8 months or more. In one embodiment, the combination treatment of
the invention
using anti-VEGF antibody and one or more chemotherapeutic agents significantly
increases
progression free survival by about 1 to about 5 months, when compared to a
treatment with
chemotherapy alone. In one embodiment, the PFS median in months is 7.2 in the
patients
treated with bevacizumab and compared to 5.1 months in the therapy without
bevacizumab,
with a HR of 0.775, p-value (log-rank) 0.0072. In another embodiment, the PFS
in months is
found in Figure 3.
In yet another embodiment, the treatment of the invention significantly
increases
response rate in a group of human patients susceptible to or diagnosed with a
previously
treated cancer who are treated with various therapeutics. Response rate is
defined as the
percentage of treated patients who responded to the treatment. In one aspect,
the combination
41
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
treatment of the invention using anti-VEGF antibody and one or more
chemotherapeutic
agents significantly increases response rate in the treated patient group
compared to the group
treated with chemotherapy alone.
In one aspect, the invention provides methods for increasing duration of
response in a
human patient or a group of human patients susceptible to or diagnosed with a
cancer.
Duration of response is defined as the time from the initial response to
disease progression.
In one embodiment, the invention can be used for increasing the duration of
survival of a
human patient susceptible to or diagnosed with a cancer.
VII Antibody Production
(i) Polyclonal antibodies
Polyclonal antibodies are preferably raised in animals by multiple
subcutaneous (sc) or
intraperitoneal (ip) injections of the relevant antigen and an adjuvant. It
may be useful to
conjugate the relevant antigen to a protein that is immunogenic in the species
to be
immunized, e.g., keyhole limpet hemocyanin, serum albumin, bovine
thyroglobulin, or
soybean trypsin inhibitor using a bifunctional or derivatizing agent, for
example,
maleimidobenzoyl sulfosuccinimide ester (conjugation through cysteine
residues), N-
hydroxysuccinimide (through lysine residues), glutaraldehyde, succinic
anhydride, SOC12, or
RIN=C=NR, where R and RI are different alkyl groups.
Animals are immunized against the antigen, immunogenic conjugates, or
derivatives by
combining, e.g., 100 g or 5 g of the protein or conjugate (for rabbits or
mice, respectively)
with 3 volumes of Freund's complete adjuvant and injecting the solution
intradermally at
multiple sites. One month later the animals are boosted with 1/5 to 1/10 the
original amount
of peptide or conjugate in Freund's complete adjuvant by subcutaneous
injection at multiple
sites. Seven to 14 days later the animals are bled and the serum is assayed
for antibody titer.
Animals are boosted until the titer plateaus. Preferably, the animal is
boosted with the
conjugate of the same antigen, but conjugated to a different protein and/or
through a different
cross-linking reagent. Conjugates also can be made in recombinant cell culture
as protein
fusions. Also, aggregating agents such as alum are suitably used to enhance
the immune
response.
42
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
(ii) Monoclonal antibodies
Various methods for making monoclonal antibodies herein are available in the
art. For
example, the monoclonal antibodies may be made using the hybridoma method
first described
by Kohler et at., Nature, 256:495 (1975), or by recombinant DNA methods (U.S.
Patent No.
4,816,567).
In the hybridoma method, a mouse or other appropriate host animal, such as a
hamster or
macaque monkey, is immunized as hereinabove described to elicit lymphocytes
that produce
or are capable of producing antibodies that will specifically bind to the
protein used for
immunization. Alternatively, lymphocytes may be immunized in vitro.
Lymphocytes then are
fused with myeloma cells using a suitable fusing agent, such as polyethylene
glycol, to form a
hybridoma cell (Goding, Monoclonal Antibodies: Principles and Practice, pp.59-
103
(Academic Press, 1986)).
The hybridoma cells thus prepared are seeded and grown in a suitable culture
medium
that preferably contains one or more substances that inhibit the growth or
survival of the
unfused, parental myeloma cells. For example, if the parental myeloma cells
lack the enzyme
hypoxanthine guanine phosphoribosyl transferase (HGPRT or HPRT), the culture
medium for
the hybridomas typically will include hypoxanthine, aminopterin, and thymidine
(HAT
medium), which substances prevent the growth of HGPRT-deficient cells.
Preferred myeloma cells are those that fuse efficiently, support stable high-
level
production of antibody by the selected antibody-producing cells, and are
sensitive to a
medium such as HAT medium. Among these, examples of myeloma cell lines are
murine
myeloma lines, such as those derived from MOPC-21 and MPC-11 mouse tumors
available
from the Salk Institute Cell Distribution Center, San Diego, California USA,
and SP-2 or X63-
Ag8-653 cells available from the American Type Culture Collection, Rockville,
Maryland
USA. Human myeloma and mouse-human heteromyeloma cell lines also have been
described
for the production of human monoclonal antibodies (Kozbor, J. Immunol.,
133:3001 (1984);
Brodeur et at., Monoclonal Antibody Production Techniques and Applications,
pp. 51-63
(Marcel Dekker, Inc., New York, 1987)).
Culture medium in which hybridoma cells are growing is assayed for production
of
monoclonal antibodies directed against the antigen. Preferably, the binding
specificity of
monoclonal antibodies produced by hybridoma cells is determined by
immunoprecipitation or
43
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
by an in vitro binding assay, such as radioimmunoassay (RIA) or enzyme-linked
immunoabsorbent assay (ELISA).
After hybridoma cells are identified that produce antibodies of the desired
specificity,
affinity, and/or activity, the clones may be subcloned by limiting dilution
procedures and
grown by standard methods (Goding, Monoclonal Antibodies: Principles and
Practice, pp.59-
103 (Academic Press, 1986)). Suitable culture media for this purpose include,
for example,
D-MEM or RPMI-1640 medium. In addition, the hybridoma cells maybe grown in
vivo as
ascites tumors in an animal.
The monoclonal antibodies secreted by the subclones are suitably separated
from the
culture medium, ascites fluid, or serum by conventional immunoglobulin
purification
procedures such as, for example, protein A-Sepharose, hydroxylapatite
chromatography, gel
electrophoresis, dialysis, or affinity chromatography.
DNA encoding the monoclonal antibodies is readily isolated and sequenced using
conventional procedures (e.g., by using oligonucleotide probes that are
capable of binding
specifically to genes encoding the heavy and light chains of the monoclonal
antibodies). The
hybridoma cells serve as a source of such DNA. Once isolated, the DNA may be
placed into
expression vectors, which are then transfected into host cells such as E. coli
cells, simian COS
cells, Chinese hamster ovary (CHO) cells, or myeloma cells that do not
otherwise produce
immunoglobulin protein, to obtain the synthesis of monoclonal antibodies in
the recombinant
host cells. Recombinant production of antibodies will be described in more
detail below.
In a further embodiment, antibodies or antibody fragments can be isolated from
antibody
phage libraries generated using the techniques described in McCafferty et al.,
Nature,
348:552-554 (1990). Clackson et al., Nature, 352:624-628 (1991) and Marks et
al., J. Mol.
Biol., 222:581-597 (1991) describe the isolation of murine and human
antibodies,
respectively, using phage libraries. Subsequent publications describe the
production of high
affinity (nM range) human antibodies by chain shuffling (Marks et al.,
Bio/Technology,
10:779-783 (1992)), as well as combinatorial infection and in vivo
recombination as a strategy
for constructing very large phage libraries (Waterhouse et al., Nuc. Acids.
Res., 21:2265-2266
(1993)). Thus, these techniques are viable alternatives to traditional
monoclonal antibody
hybridoma techniques for isolation of monoclonal antibodies.
The DNA also may be modified, for example, by substituting the coding sequence
for
human heavy- and light-chain constant domains in place of the homologous
murine sequences
44
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
(U.S. Patent No. 4,816,567; Morrison, et al., Proc. Natl Acad. Sci. USA,
81:6851 (1984)), or
by covalently joining to the immunoglobulin coding sequence all or part of the
coding
sequence for a non-immunoglobulin polypeptide.
Typically such non-immunoglobulin polypeptides are substituted for the
constant
domains of an antibody, or they are substituted for the variable domains of
one antigen-
combining site of an antibody to create a chimeric bivalent antibody
comprising one antigen-
combining site having specificity for an antigen and another antigen-combining
site having
specificity for a different antigen.
(iii) Humanized and human antibodies
A humanized antibody has one or more amino acid residues introduced into it
from a
source which is non-human. These non-human amino acid residues are often
referred to as
"import" residues, which are typically taken from an "import" variable domain.
Humanization
can be essentially performed following the method of Winter and co-workers
(Jones et at.,
Nature, 321:522-525 (1986); Riechmann et at., Nature, 332:323-327 (1988);
Verhoeyen et at.,
Science, 239:1534-1536 (1988)), by substituting rodent CDRs or CDR sequences
for the
corresponding sequences of a human antibody. Accordingly, such "humanized"
antibodies are
chimeric antibodies (U.S. Patent No. 4,816,567) wherein substantially less
than an intact
human variable domain has been substituted by the corresponding sequence from
a non-
human species. In practice, humanized antibodies are typically human
antibodies in which
some CDR residues and possibly some FR residues are substituted by residues
from analogous
sites in rodent antibodies.
The choice of human variable domains, both light and heavy, to be used in
making the
humanized antibodies is very important to reduce antigenicity. According to
the so-called
"best-fit" method, the sequence of the variable domain of a rodent antibody is
screened against
the entire library of known human variable-domain sequences. The human
sequence which is
closest to that of the rodent is then accepted as the human framework (FR) for
the humanized
antibody (Sims et at., J. Immunol., 151:2296 (1993); Chothia et at., J. Mol.
Biol., 196:901
(1987)). Another method uses a particular framework derived from the consensus
sequence of
all human antibodies of a particular subgroup of light or heavy chains. The
same framework
may be used for several different humanized antibodies (Carter et at., Proc.
Natl. Acad. Sci.
USA, 89:4285 (1992); Presta et al., J. Immnol., 151:2623 (1993)).
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
It is further important that antibodies be humanized with retention of high
affinity for the
antigen and other favorable biological properties. To achieve this goal,
according to one
embodiment, humanized antibodies are prepared by a process of analysis of the
parental
sequences and various conceptual humanized products using three-dimensional
models of the
parental and humanized sequences. Three-dimensional immunoglobulin models are
commonly available and are familiar to those skilled in the art. Computer
programs are
available which illustrate and display probable three-dimensional
conformational structures of
selected candidate immunoglobulin sequences. Inspection of these displays
permits analysis
of the likely role of the residues in the functioning of the candidate
immunoglobulin sequence,
i.e., the analysis of residues that influence the ability of the candidate
immunoglobulin to bind
its antigen. In this way, FR residues can be selected and combined from the
recipient and
import sequences so that the desired antibody characteristic, such as
increased affinity for the
target antigen(s), is achieved. In general, the CDR residues are directly and
most substantially
involved in influencing antigen binding.
Humanized anti-VEGF antibodies and affinity matured variants thereof are
described in,
for example, U.S. Pat. No. 6,884,879 issued February 26, 2005.
It is now possible to produce transgenic animals (e.g., mice) that are
capable, upon
immunization, of producing a full repertoire of human antibodies in the
absence of
endogenous immunoglobulin production. For example, it has been described that
the
homozygous deletion of the antibody heavy-chain joining region (JH) gene in
chimeric and
germ-line mutant mice results in complete inhibition of endogenous antibody
production.
Transfer of the human germ-line immunoglobulin gene array in such germ-line
mutant mice
will result in the production of human antibodies upon antigen challenge. See,
e.g.,
Jakobovits et at., Proc. Natl. Acad. Sci. USA, 90:2551 (1993); Jakobovits et
at., Nature,
362:255-258 (1993); Bruggermann et at., Year in Immuno., 7:33 (1993); and
Duchosal et at.
Nature 355:258 (1992).
Alternatively, phage display technology (McCafferty et at., Nature 348:552-553
(1990))
can be used to produce human antibodies and antibody fragments in vitro, from
immunoglobulin variable (V) domain gene repertoires from unimmunized donors.
According
to this technique, antibody V domain genes are cloned in-frame into either a
major or minor
coat protein gene of a filamentous bacteriophage, such as M13 or fd, and
displayed as
functional antibody fragments on the surface of the phage particle. Because
the filamentous
particle contains a single-stranded DNA copy of the phage genome, selections
based on the
46
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
functional properties of the antibody also result in selection of the gene
encoding the antibody
exhibiting those properties. Thus, the phage mimics some of the properties of
the B-cell.
Phage display can be performed in a variety of formats; for their review see,
e.g., Johnson,
Kevin S. and Chiswell, David J., Current Opinion in Structural Biology 3:564-
571 (1993).
Several sources of V-gene segments can be used for phage display. Clackson et
at., Nature,
352:624-628 (1991) isolated a diverse array of anti-oxazolone antibodies from
a small random
combinatorial library of V genes derived from the spleens of immunized mice. A
repertoire of
V genes from unimmunized human donors can be constructed and antibodies to a
diverse
array of antigens (including self-antigens) can be isolated essentially
following the techniques
described by Marks et at., J. Mol. Biol. 222:581-597 (1991), or Griffith et
at., EMBO J.
12:725-734 (1993). See, also, U.S. Patent Nos. 5,565,332 and 5,573,905.
As discussed above, human antibodies may also be generated by in vitro
activated B
cells (see U.S. Patents 5,567,610 and 5,229,275).
Human monoclonal anti-VEGF antibodies are described in U.S. Patent No.
5,730,977,
issued March 24, 1998.
(iv) Antibody fragments
Various techniques have been developed for the production of antibody
fragments.
Traditionally, these fragments were derived via proteolytic digestion of
intact antibodies (see,
e.g., Morimoto et at. , Journal of Biochemical and Biophysical Methods 24:107-
117 (1992)
and Brennan et at., Science, 229:81 (1985)). However, these fragments can now
be produced
directly by recombinant host cells. For example, the antibody fragments can be
isolated from
the antibody phage libraries discussed above. Alternatively, Fab'-SH fragments
can be
directly recovered from E. coli and chemically coupled to form F(ab')2
fragments (Carter et
at., Bio/Technology 10:163-167 (1992)). According to another approach, F(ab')2
fragments
can be isolated directly from recombinant host cell culture. Other techniques
for the
production of antibody fragments will be apparent to the skilled practitioner.
In other
embodiments, the antibody of choice is a single chain Fv fragment (scFv). See
WO 93/16185.
(vi) Other amino acid sequence modifications
Amino acid sequence modification(s) of the antibodies described herein are
contemplated. For example, it may be desirable to improve the binding affinity
and/or other
biological properties of the antibody. Amino acid sequence variants of the
antibody are
prepared by introducing appropriate nucleotide changes into the antibody
nucleic acid, or by
47
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
peptide synthesis. Such modifications include, for example, deletions from,
and/or insertions
into and/or substitutions of, residues within the amino acid sequences of the
antibody. Any
combination of deletion, insertion, and substitution is made to arrive at the
final construct,
provided that the final construct possesses the desired characteristics. The
amino acid changes
also may alter post-translational processes of the antibody, such as changing
the number or
position of glycosylation sites.
A useful method for identification of certain residues or regions of the
antibody that
are preferred locations for mutagenesis is called "alanine scanning
mutagenesis" as described
by Cunningham and Wells Science, 244:1081-1085 (1989). Here, a residue or
group of target
residues are identified (e.g., charged residues such as arg, asp, his, lys,
and glu) and replaced
by a neutral or negatively charged amino acid (most preferably alanine or
polyalanine) to
affect the interaction of the amino acids with antigen. Those amino acid
locations
demonstrating functional sensitivity to the substitutions then are refined by
introducing further
or other variants at, or for, the sites of substitution. Thus, while the site
for introducing an
amino acid sequence variation is predetermined, the nature of the mutation per
se need not be
predetermined. For example, to analyze the performance of a mutation at a
given site, ala
scanning or random mutagenesis is conducted at the target codon or region and
the expressed
antibody variants are screened for the desired activity.
Amino acid sequence insertions include amino- and/or carboxyl-terminal fusions
ranging in length from one residue to polypeptides containing a hundred or
more residues, as
well as intrasequence insertions of single or multiple amino acid residues.
Examples of
terminal insertions include antibody with an N-terminal methionyl residue or
the antibody
fused to a cytotoxic polypeptide. Other insertional variants of the antibody
molecule include
the fusion to the N- or C-terminus of the antibody to an enzyme (e.g. for
ADEPT) or a
polypeptide which increases the serum half-life of the antibody.
Another type of variant is an amino acid substitution variant. These variants
have at
least one amino acid residue in the antibody molecule replaced by a different
residue. The
sites of greatest interest for substitutional mutagenesis include the
hypervariable regions, but
FR alterations are also contemplated.
Substantial modifications in the biological properties of the antibody are
accomplished
by selecting substitutions that differ significantly in their effect on
maintaining (a) the
structure of the polypeptide backbone in the area of the substitution, for
example, as a sheet or
helical conformation, (b) the charge or hydrophobicity of the molecule at the
target site, or (c)
the bulk of the side chain. Amino acids may be grouped according to
similarities in the
48
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
properties of their side chains (in A. L. Lehninger, in Biochemistry, second
ed., pp. 73-75,
Worth Publishers, New York (1975)):
(1) non-polar: Ala (A), Val (V), Leu (L), Ile (I), Pro (P), Phe (F), Trp (W),
Met (M)
(2) uncharged polar: Gly (G), Ser (S), Thr (T), Cys (C), Tyr (Y), Asn (N), Gln
(Q)
(3) acidic: Asp (D), Glu (E)
(4) basic: Lys (K), Arg (R), His(H)
Alternatively, naturally occurring residues may be divided into groups based
on common
side-chain properties:
(1) hydrophobic: Norleucine, Met, Ala, Val, Leu, Ile;
(2) neutral hydrophilic: Cys, Ser, Thr, Asn, Gln;
(3) acidic: Asp, Glu;
(4) basic: His, Lys, Arg;
(5) residues that influence chain orientation: Gly, Pro;
(6) aromatic: Trp, Tyr, Phe.
Non-conservative substitutions will entail exchanging a member of one of these
classes
for another class.
Any cysteine residue not involved in maintaining the proper conformation of
the
antibody also may be substituted, generally with serine, to improve the
oxidative stability of
the molecule and prevent aberrant crosslinking. Conversely, cysteine bond(s)
may be added
to the antibody to improve its stability (particularly where the antibody is
an antibody
fragment such as an Fv fragment).
An example of a substitutional variant involves substituting one or more
hypervariable
region residues of a parent antibody (e.g. a humanized or human antibody).
Generally, the
resulting variant(s) selected for further development will have improved
biological properties
relative to the parent antibody from which they are generated. A convenient
way for
generating such substitutional variants involves affinity maturation using
phage display.
Briefly, several hypervariable region sites (e.g. 6-7 sites) are mutated to
generate all possible
amino substitutions at each site. The antibody variants thus generated are
displayed in a
monovalent fashion from filamentous phage particles as fusions to the gene III
product of
M13 packaged within each particle. The phage-displayed variants are then
screened for their
49
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
biological activity (e.g. binding affinity) as herein disclosed. In order to
identify candidate
hypervariable region sites for modification, alanine scanning mutagenesis can
be performed to
identify hypervariable region residues contributing significantly to antigen
binding.
Alternatively, or additionally, it may be beneficial to analyze a crystal
structure of the antigen-
antibody complex to identify contact points between the antibody and human
VEGF. Such
contact residues and neighboring residues are candidates for substitution
according to the
techniques elaborated herein. Once such variants are generated, the panel of
variants is
subjected to screening as described herein and antibodies with superior
properties in one or
more relevant assays may be selected for further development.
Another type of amino acid variant of the antibody alters the original
glycosylation
pattern of the antibody. By altering is meant deleting one or more
carbohydrate moieties
found in the antibody, and/or adding one or more glycosylation sites that are
not present in the
antibody.
Glycosylation of antibodies is typically either N-linked or O-linked. N-linked
refers to
the attachment of the carbohydrate moiety to the side chain of an asparagine
residue. The
tripeptide sequences asparagine-X-serine and asparagine-X-threonine, where X
is any amino
acid except proline, are the recognition sequences for enzymatic attachment of
the
carbohydrate moiety to the asparagine side chain. Thus, the presence of either
of these
tripeptide sequences in a polypeptide creates a potential glycosylation site.
O-linked
glycosylation refers to the attachment of one of the sugars N-
aceylgalactosamine, galactose, or
xylose to a hydroxyamino acid, most commonly serine or threonine, although 5-
hydroxyproline or 5-hydroxylysine may also be used.
Addition of glycosylation sites to the antibody is conveniently accomplished
by altering
the amino acid sequence such that it contains one or more of the above-
described tripeptide
sequences (for N-linked glycosylation sites). The alteration may also be made
by the addition
of, or substitution by, one or more serine or threonine residues to the
sequence of the original
antibody (for O-linked glycosylation sites).
Where the antibody comprises an Fc region, the carbohydrate attached thereto
may be
altered. For example, antibodies with a mature carbohydrate structure that
lacks fucose
attached to an Fc region of the antibody are described in US Pat Appl No US
2003/0157108
Al, Presta, L. See also US 2004/0093621 Al (Kyowa Hakko Kogyo Co., Ltd).
Antibodies
with a bisecting N-acetylglucosamine (G1cNAc) in the carbohydrate attached to
an Fc region
of the antibody are referenced in WO03/0l 1878, Jean-Mairet et at. and US
Patent No.
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
6,602,684, Umana et at. Antibodies with at least one galactose residue in the
oligosaccharide
attached to an Fc region of the antibody are reported in W097/30087, Patel et
at. See, also,
W098/58964 (Raju, S.) and W099/22764 (Raju, S.) concerning antibodies with
altered
carbohydrate attached to the Fc region thereof.
It may be desirable to modify the antibody of the invention with respect to
effector
function, e.g. so as to enhance antigen-dependent cell-mediated cyotoxicity
(ADCC) and/or
complement dependent cytotoxicity (CDC) of the antibody. This may be achieved
by
introducing one or more amino acid substitutions in an Fc region of the
antibody.
Alternatively or additionally, cysteine residue(s) may be introduced in the Fc
region, thereby
allowing interchain disulfide bond formation in this region. The homodimeric
antibody thus
generated may have improved internalization capability and/or increased
complement-
mediated cell killing and antibody-dependent cellular cytotoxicity (ADCC). See
Caron et at.,
J. Exp Med. 176:1191-1195 (1992) and Shopes, B. J. Immunol. 148:2918-2922
(1992).
Homodimeric antibodies with enhanced anti-tumor activity may also be prepared
using
heterobifunctional cross-linkers as described in Wolff et at. Cancer Research
53:2560-2565
(1993). Alternatively, an antibody can be engineered which has dual Fc regions
and may
thereby have enhanced complement lysis and ADCC capabilities. See Stevenson et
at. Anti-
Cancer Drug Design 3:219-230 (1989).
W000/42072 (Presta, L.) describes antibodies with improved ADCC function in
the
presence of human effector cells, where the antibodies comprise amino acid
substitutions in
the Fc region thereof. Preferably, the antibody with improved ADCC comprises
substitutions
at positions 298, 333, and/or 334 of the Fc region (Eu numbering of residues).
Preferably the
altered Fc region is a human IgGI Fc region comprising or consisting of
substitutions at one,
two or three of these positions. Such substitutions are optionally combined
with
substitution(s) which increase C l q binding and/or CDC.
Antibodies with altered Clq binding and/or complement dependent cytotoxicity
(CDC)
are described in W099/51642, US Patent No. 6,194,551B1, US Patent No.
6,242,195B1, US
Patent No. 6,528,624B1 and US Patent No. 6,538,124 (Idusogie et al.). The
antibodies
comprise an amino acid substitution at one or more of amino acid positions
270, 322, 326,
327, 329, 313, 333 and/or 334 of the Fc region thereof (Eu numbering of
residues).
To increase the serum half life of the antibody, one may incorporate a salvage
receptor
binding epitope into the antibody (especially an antibody fragment) as
described in US Patent
5,739,277, for example. As used herein, the term "salvage receptor binding
epitope" refers to
51
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
an epitope of the Fc region of an IgG molecule (e.g., IgG1, IgG2, IgG3, or
IgG4) that is
responsible for increasing the in vivo serum half-life of the IgG molecule.
Antibodies with improved binding to the neonatal Fc receptor (FcRn), and
increased
half-lives, are described in W000/42072 (Presta, L.) and US2005/0014934A1
(Hinton et al.).
These antibodies comprise an Fc region with one or more substitutions therein
which improve
binding of the Fc region to FcRn. For example, the Fc region may have
substitutions at one or
more of positions 238, 250, 256, 265, 272, 286, 303, 305, 307, 311, 312, 314,
317, 340, 356,
360, 362, 376, 378, 380, 382, 413, 424, 428 or 434 (Eu numbering of residues).
In one
embodiment, the Fc region-comprising antibody variant with improved FcRn
binding
comprises amino acid substitutions at one, two or three of positions 307, 380
and 434 of the
Fc region thereof (Eu numbering of residues). In one embodiment, the antibody
has 307/434
mutations.
Engineered antibodies with three or more (preferably four) functional antigen
binding
sites are also contemplated (US Appln No. US2002/0004587 Al, Miller et al.).
Nucleic acid molecules encoding amino acid sequence variants of the antibody
are
prepared by a variety of methods known in the art. These methods include, but
are not limited
to, isolation from a natural source (in the case of naturally occurring amino
acid sequence
variants) or preparation by oligonucleotide-mediated (or site-directed)
mutagenesis, PCR
mutagenesis, and cassette mutagenesis of an earlier prepared variant or a non-
variant version
of the antibody.
(v) Immunoconjugates
The invention also pertains to immunoconjugates comprising the antibody
described
herein conjugated to a cytotoxic agent such as a chemotherapeutic agent, toxin
(e.g. an
enzymatically active toxin of bacterial, fungal, plant or animal origin, or
fragments thereof),
or a radioactive isotope (i.e., a radio conjugate).
Chemotherapeutic agents useful in the generation of such immunoconjugates have
been
described above. Enzymatically active toxins and fragments thereof which can
be used
include diphtheria A chain, nonbinding active fragments of diphtheria toxin,
exotoxin A chain
(from Pseudomonas aeruginosa), ricin A chain, abrin A chain, modeccin A chain,
alpha-
sarcin, Aleurites fordii proteins, dianthin proteins, Phytolaca americans
proteins (PAPI,
PAPII, and PAP-S), momordica charantia inhibitor, curcin, crotin, sapaonaria
officinalis
52
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
inhibitor, gelonin, mitogellin, restrictocin, phenomycin, enomycin and the
tricothecenes. A
variety of radionuclides are available for the production of radioconjugate
antibodies.
Examples include 212Bi, 1311, 131In, 90Y and 186Re.
Conjugates of the antibody and cytotoxic agent are made using a variety of
bifunctional
protein coupling agents such as N-succinimidyl-3-(2-pyridyldithiol) propionate
(SPDP),
iminothiolane (IT), bifunctional derivatives of imidoesters (such as dimethyl
adipimidate
HCL), active esters (such as disuccinimidyl suberate), aldehydes (such as
glutareldehyde), bis-
azido compounds (such as bis (p-azidobenzoyl) hexanediamine), bis-diazonium
derivatives
(such as bis-(p-diazoniumbenzoyl)-ethylenediamine), diisocyanates (such as
tolyene 2,6-
diisocyanate), and bis-active fluorine compounds (such as 1,5-difluoro-2,4-
dinitrobenzene).
For example, a ricin immunotoxin can be prepared as described in Vitetta et
at. Science 238:
1098 (1987). Carbon-14-labeled 1-isothiocyanatobenzyl-3-methyldiethylene
triaminepentaacetic acid (MX-DTPA) is an exemplary chelating agent for
conjugation of
radionucleotide to the antibody. See W094/11026.
In another embodiment, the antibody may be conjugated to a "receptor" (such
streptavidin) for utilization in tumor pretargeting wherein the antibody-
receptor conjugate is
administered to the patient, followed by removal of unbound conjugate from the
circulation
using a clearing agent and then administration of a "ligand" (e.g. avidin)
which is conjugated
to a cytotoxic agent (e.g. a radionucleotide).
(vi) Immunoliposomes
The antibody disclosed herein may also be formulated as immunoliposomes.
Liposomes containing the antibody are prepared by methods known in the art,
such as
described in Epstein et at., Proc. Natl. Acad. Sci. USA, 82:3688 (1985); Hwang
et at., Proc.
Natl Acad. Sci. USA, 77:4030 (1980); and U.S. Pat. Nos. 4,485,045 and
4,544,545.
Liposomes with enhanced circulation time are disclosed in U.S. Patent No.
5,013,556.
Particularly useful liposomes can be generated by the reverse phase
evaporation method
with a lipid composition comprising phosphatidylcholine, cholesterol and PEG-
derivatized
phosphatidylethanolamine (PEG-PE). Liposomes are extruded through filters of
defined pore
size to yield liposomes with the desired diameter. Fab' fragments of the
antibody of the
invention can be conjugated to the liposomes as described in Martin et at. J.
Biol. Chem. 257:
286-288 (1982) via a disulfide interchange reaction. A chemotherapeutic agent
is optionally
contained within the liposome. See Gabizon et at. J. National Cancer Inst.
81(19)1484 (1989)
53
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
VIII. ARTICLES OF MANUFACTURE AND KITS
In another embodiment of the invention, an article of manufacture containing
materials
useful for the treatment of the disorders described above is provided. The
article of
manufacture comprises a container, a label and a package insert. Suitable
containers include,
for example, bottles, vials, syringes, etc. The containers may be formed from
a variety of
materials such as glass or plastic. The container holds a composition which is
effective for
treating the condition and may have a sterile access port (for example the
container may be an
intravenous solution bag or a vial having a stopper pierceable by a hypodermic
injection
needle). At least one active agent in the composition is an anti-VEGF
antibody. The label on,
or associated with, the container indicates that the composition is used for
treating the
condition of choice. The article of manufacture may further comprise a second
container
comprising a pharmaceutically-acceptable buffer, such as phosphate-buffered
saline, Ringer's
solution and dextrose solution. It may further include other materials
desirable from
a commercial and user standpoint, including other buffers, diluents, filters,
needles, and
syringes. In addition, the article of manufacture comprises a package inserts
with instructions
for use, including for example instructing the user of the composition to
administer the anti-
VEGF antibody composition and a chemotherapeutic agent to the patient with
previously
treated breast cancer and optionally HER2 negative. In certain embodiments,
the patient has
metastatic cancer. In some embodiments, the patient has previously treated
metastatic breast
cancer and is HER2 negative. The package insert may optionally contain some or
all of the
results found in Example 1.
The VEGF-specific antagonist can be packaged alone or in combination with
other
anti-cancer therapeutic compounds as a kit. The kit can include optional
components that aid
in the administration of the unit dose to patients, such as vials for
reconstituting powder forms,
syringes for injection, customized IV delivery systems, inhalers, etc.
Additionally, the unit
dose kit can contain instructions for preparation and administration of the
compositions. In
certain embodiments, the instructions comprises instructions for use,
including for example
instructing the user of the composition to administer the anti-VEGF antibody
composition and
a chemotherapeutic agent to the patient with previously treated breast cancer
and optionally
HER2 negative. In certain embodiments, the patient has metastatic cancer. In
some
embodiments, the patient has previously treated metastatic breast cancer and
is HER2
negative. The instructions may optionally contain some or all of the results
found in
Example 1. The kit may be manufactured as a single use unit dose for one
patient, multiple
54
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
uses for a particular patient (at a constant dose or in which the individual
compounds may
vary in potency as therapy progresses); or the kit may contain multiple doses
suitable for
administration to multiple patients ("bulk packaging"). The kit components may
be
assembled in cartons, blister packs, bottles, tubes, and the like.
Deposit of Materials
The following hybridoma cell line has been deposited under the provisions of
the
Budapest Treaty with the American Type Culture Collection (ATCC), Manassas,
VA, USA:
Antibody Designation ATCC No. Deposit Date
A4.6.1 ATCC HB-10709 March 29, 1991
The following example is intended merely to illustrate the practice of the
invention and
is not provided by way of limitation. The disclosures of all patent and
scientific literatures
cited herein are expressly incorporated in their entirety by reference.
EXAMPLE
Example 1. Bevacizumab in Combination with Chemotherapy Regimens in Subjects
with Previously Treated Metastic Breast Cancer
Metastatic breast cancer (MBC) is an incurable disease, with the majority of
patients
succumbing to their disease within 2 year of diagnosis. See, Greenberg, et
al., 1996, J. Clin.
Oncol. 14:2197-205. Approximately 60% of patients with advanced stage disease
present
with a local recurrence and 40% present with distant metastasis after adjuvant
therapy. Only
10% of patients present with metastatic disease at initial diagnosis. See,
Ryberg et al., 2001
Ann. Oncol. 12:81-7.
The treatment algorithm for patients with MBC is based on several factors that
include
clinical, pathologic, and histologic characteristics such as human epidermal
growth factor-2
(HER2) amplification, hormone receptor status, prior response to and/or
failure of hormonal
agents, number and specific sites of metastatic disease, and treatment history
in both the
metastatic and adjuvant settings. Numerous cytotoxic chemotherapy agents have
shown
activity in MBC, including anthracyclines, taxanes, gemcitabine, capecitabine,
and
vinorelbine. The response rates and progression-free intervals seen with these
agents vary,
depending on the extent and type of prior therapy and the extent of metastatic
diseases. In
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
general, anthracyline-based combination therapy and taxanes (paclitaxel and
docetaxel) have
shown the greatest activity in the metastatic setting, with response rates of
40-50% and
median progression-free survival (PFS) of approximately 6-9 months in patients
without prior
exposure to agents in the adjuvant setting and without prior exposure to
chemotherapy for the
treatment of metastatic disease. See, Hamilton and Hortobagyi 2005 J. Clin.
Oncol. 23:1760-
75. As expected, patients who progress after or during the first treatment
have a shorter
progression-free interval (4-6 months) and survival (8-12 months). Thus, there
is a need to
find additional treatments that can be incorporated into regimens that will
extend both the
progression-free interval and survival of patients with recurrent disease.
In the second-line treatment setting for MBC, many agents have demonstrated
activity,
including anti-tubulin drugs (taxanes, vinorelbine) and anti-metabolites
(capecitabine,
gemcitabine). Compared with mitomycin and vinblastine, docetaxel was the first
single agent
to significantly increase the time to disease progression (4.4 vs. 2.5 months;
p=0.001) and
survival (11.4 vs. 8.7 months; p=0.0097) in a large Phase III study of MBC
patients who had
received prior anthracyline therapy. See, Nabholtz et al., 1999 J. Clin.
Oncol. 17:1413-24.
The addition of capecitabine to docetaxel compared with docetaxel alone led to
further
improvements in time to progression (6.1 vs. 4.2 months; hazard ratio =0.652)
and survival
(14.5 vs. 11.5 months; hazard ratio=0.775) in patients who had previously
received or were
not candidates for anthracyclines; however, there was a substantial increase
in toxicity with
this combination. See, O'Shaughnessy et al., 2002 J Clin. Oncol. 20:2812-23.
Additional
studies with either single or combination agents have failed to demonstrate
clear survival
advantages. See, Hamilton and Hortobagyi 2005 J. Clin. Oncol. 23:1760-75.
Based on the
experience with multiple agents used alone or in combination, the current
treatment paradigm
is sequential single-agent therapy with the choice of a specific agent(s)
determined by a
number of factors, including prior therapy, treatment-free interval, toxicity
profile, and patient
preference.
Despite the availability of these multiple agents, additional treatments for
MBC
patients in second-line setting are needed. New treatments directed at
delaying disease
progression while avoiding systemic toxicity would represent a significant
advance in the
treatment of these patients.
A Phase III study (RIBBON 2) of Avastin (bevacizumab) in combination with
chemotherapy increased the time women with metastatic HER2-negative breast
cancer whose
initial chemotherapy had stopped working lived without the disease worsening
(progression-
free survival or PFS), compared to chemotherapy alone. The doctors treating
the women in
56
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
the study chose the type of chemotherapy used in combination with Avastin and
the
chemotherapies were assessed together in the primary endpoint analysis.
Adverse events were
consistent with those previously reported for Avastin, and no new Avastin
safety signals were
observed in the study.
Ribbon 2 was an international, multicenter, randomized, double-blind, placebo-
controlled clinical study that enrolled 684 patients with metastatic HER2-
negative breast
cancer who had previously received chemotherapy for their metastatic disease.
See Study
Conduct in Table 1 and Patient Characteristics in Table 2. The trial evaluated
the addition of
either Avastin (bevacizumab) or placebo to an investigator's choice of
chemotherapy. The
following chemotherapy regimens were used in the study: taxanes: paclitaxel,
protein-bound
paclitaxel or docetaxel; gemcitabine; capecitabine; or vinorelbine.
Table 1: Study Conduct
Chemo Cohort Taxanes Gemcitabine Capecitabine Vinorelbine
Enrolled, n (%) 304 (44.4) 160 (23.4) 144 (21.1) 76 (11.1)
Bevacizumab 201 108 97 53
(BV)
Placebo (PL) 103 52 47 23
Eligibility _ in the study included the following:
Ages Eligible for Study: 18 Years and older
Genders Eligible for Study: Both
Accepts Healthy Volunteers: No
Inclusion Criteria in the study included the following:
Signed Informed Consent Form
>18 years of age
Histologically confirmed carcinoma of the breast with measurable or non-
measurable
metastatic disease that has progressed (patients with a history of brain
metastasis are eligible
for study participation [U.S. only], as long as their brain metastases have
been treated and they
57
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
have no evidence of progression or hemorrhage after treatment and no ongoing
requirement
for dexamethasone)
Progression of disease during or following administration of one (non-
investigational)
chemotherapy regimen administered in the first-line setting
ECOG performance status of 0 or 1
For women of childbearing potential, use of an effective means of non-hormonal
contraception
Life expectancy >3 months
Willingness and capacity to comply with study and follow-up procedures
Exclusion Criteria in the study included the following:
Prior hormonal therapy only as treatment for metastatic disease without
chemotherapy.
Patients must have received chemotherapy for their metastatic disease in the
first-line setting.
Hormone therapy alone is not allowed.
For subjects who have received prior anthracycline-based therapy,
documentation of
left ventricular ejection fraction < 50% by either multiple gated acquisition
(MUGA) or
echocardiogram (ECHO)
Treatment with more than one prior cytotoxic regimen for MBC
HER2-positive status (patients who have unknown HER2 status, and for whom
determination of HER2 status is not possible, are eligible for this study)
Unknown ER and PR status
Radiation therapy other than for palliation or brain metastasis, biologic
therapy, or
chemotherapy for MBC within 21 days prior to Day 0
Prior therapy with bevacizumab or other VEGF pathway-targeted therapy
Untreated brain metastasis
Inadequately controlled hypertension
Unstable angina
New York Heart Association Grade II or greater CHF
History of myocardial infarction within 6 months prior to Day 0 (the day of
the first
bevacizumab/placebo infusion)
58
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
History of stroke or transient ischemic attack within 6 months prior to Day 0
Clinically significant peripheral vascular disease
Evidence of bleeding diathesis or coagulopathy
Major surgical procedure, open biopsy, or significant traumatic injury within
28 days
prior to Day 0; anticipation of need for major elective surgical procedure
during the study
Minor surgical procedures, fine-needle aspirations, or core biopsies within 7
days prior
to Day 0
History of abdominal fistula, gastrointestinal perforation, or intra-abdominal
abscess
within 6 months prior to Day 0
Serious, non-healing wound, ulcer, or bone fracture
History of anaphylactic reaction to monoclonal antibody therapy not controlled
with
treatment premedication
History of other malignancies within 5 years of Day 0, except for tumors with
a
negligible risk for metastasis or death, such as adequately controlled basal
cell carcinoma or
squamous cell carcinoma of the skin or carcinoma in situ of the cervix
inadequate organ function
Pregnancy (positive serum pregnancy test) or lactation
Any other diseases, metabolic dysfunction, physical examination finding, or
clinical
laboratory finding giving reasonable suspicion of a disease or condition that
contraindicates
the use of an investigational drug or that may affect the interpretation of
the results or renders
the subject at high risk from treatment complications
Bevacizumab (5mg/kg weekly equivalent)
15 mg/kg IV q 3 weeks; or
10 mg/kg IV q 2 weeks
Plus
Chemotherapy
Taxane
Paclitaxel (e.g,. Taxol ): 90 mg/m2 IV every week for 3 weeks followed by 1
week of rest; or
59
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
Paclitaxel (e.g., Taxol ): 175 mg/m2 IV every 3 weeks; or
Paclitaxel protein-bound particles (Abraxane ): 260 mg/m2 IV every 3 weeks;
or
Docetaxel (Taxotere ): 75-100 mg/m2 IV every 3 weeks; or
Gemcitabine (Gemzar ): 1250 mg/m2 IV on Days 1 and 8 of each 3-week cycle; or
Vinorelbine (Navelbine ): 30 mg/m2 IV every week; or
Capecitabine (Xeloda ): 1000mg/m2 orally twice daily on Days 1-14 of each 3-
week
cycle
In the study, PFS was defined as the time from randomization to disease
progression or
death as assessed by the treating physicians in the study (investigator-
assessed). Secondary
endpoints included objective response rate, duration of response, one-year
survival rate,
overall survival, PFS assessment by chemotherapy type and safety. The results
indicated a
prolongation in PFS, the primary endpoint in a pooled cohort of patients
receiving avastin +
chemotherapy (HR = 0.775; p-value (log-rank) = 0.0072). The median PFS (95%
Cl) was 7.2
months (6.5, 7.6) in the bevacizumab/chemotherapy arm (Arm A of Figure 1,
n=459) verses
5.1 months (4.1, 6.0) in the placebo/chemotherapy arm (Arm B of Figure 1
n=225). See
Table 3 Primary Efficacy Analysis of PFS and Figure 2 (Primary Endpoint of
PFS) and
Figure 3 (Cohort-Specific Analyses of PFS). PFS results were generally
consistent across
chemotherapy cohorts with the exception of the small vinorelbine sub-group.
Other sub-
groups (age, triple negative, etc.) were generally consistent with the primary
PFS results. The
observed improvement in overall PFS was supported by the secondary efficacy
endpoint of
ORR. See Figure 4. See Table 4 for Interim Efficacy Analysis of OS. No new
bevacizumab
safety signals were observed in the study. See, e.g., Table 5, Safety Summary,
Safety
Population, Table 6 Selected AEs, Safety Population and Table 7 Safety Summary
by
Chemotherapy Cohort, Safety Population. Bevacizumab was beneficial to patients
as a
second-line treatment.
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
Table 2 Patient Characteristics, ITT Population
Chemo/PL Chemo/BV
(n=225) (n=459)
Age, yr
Median 55.0 55.0
Mean (range) 55 (23-90) 55.6 (25-86)
>65, % 19.6 22.0
ECOG PS 1, % 50.9 50.2
No. metastatic sites
Mean (range) 2.5 (0-6) 2.5 (0-6)
> 3 sites, % 47.1 44.5
Bone only disease, % 9.8 6.8
Visceral disease, % 25.3 26.8
HR positive, % 73.3 71.7
Triple negative, % 20.9 24.4
Interval from MBC diagnosis
to first PD
70.7 72.5
>6 mo, %
HER2-negative, % 85.3 83.9
-unknown, % 13.8 14.6
Measurable disease, % 79.6 78.9
BV=bevacizumab, HR=hormone receptor, PD=disease progression, PL=placebo.
Table 3 Primary Efficacy Analysis of PFS, ITT Population
Chemo/PL Chemo/BV
(n=225) (n=459)
No. of events, n (%) 184 (81.8) 372 (81.0)
Earliest contributing event, n (%)
Progressive disease 170 (75.6) 341 (74.3)
Death 14 (6.2) 31(6.8)
PFS (months)
Median 5.1 7.2
(95% CI) (4.1-6.0) (6.5-7.6)
Stratified analysis
HR (95% CI) 0.78 (0.64-0.93)
p-value (log-rank) 0.0072
61
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
Table 4 Interim Efficacy Analysis of OS*, ITT Population
Chemo/PL Chemo/BV
(n=225) (n=459)
No. of deaths, n (%) 109 (48.4) 206 (44.9)
OS (mo)
Median 16.4 18.0
(95% Cl) (14.6-20.2) (17.1-20.2)
Stratified analysis
HR (95% Cl) 0.90 (0.71-1.14)
p-value (log-rank) 0.3741
1- r survival rate (%) 66.2 69.5
*This is the interim analysis at 57% information (315 events).
Table 5 Safety Summary, Safety Population
Chemo/PL Chemo/BV
n (%) (n=221) (n=458)
Selected AEs* (_>Grade 3) 50 (22.6) 163 (35.6)
SAE 39 (17.6) 112 (24.5)
AE leading to 16(7.2) 61 (13.3)
discontinuation/death
AE leading to death 5(2.3) 6(l.3)
AE=adverse event, BV=bevacizumab, PL=placebo, SAE=serious adverse event.
* AEs previously shown to be associated with BV
Table 6 Selected AEs (? Grade 3), Safety Population
Chemo/PL
n (%) (n=221) Chemo/BV (n=458)
Neutropenia 32 (14.5) 81 (17.7)
Hypertension l (O.5) 41(9.0)
Sensory neuropathy 13 (5.9) 30 (6.6)
Proteinuria l (O.5) 15 (3.3)
Febrile neutropenia 6 (2.7) 10 (2.2)
Bleeding events 0 (0) 8(l.7)
Left ventricular systolic
dysfunction 0 (0) 4 (0.9)
ATE 3(l.4) 3 (0.7)
GI perforation 0 (0) 3 (0.7)
Wound dehiscence 0 (0) 3 (0.7)
RPLS 0 (0) 0 (0)
ATE=arterial thrombotic event, Gl=gastrointestinal, RPLS=reversible posterior
leukoencephalopathy syndrome.
62
CA 02771086 2012-02-13
WO 2011/022264 PCT/US2010/045147
Table 7 Safety Summary by Chemotherapy Cohort, Safety Population
Tax/PL Gem/PL Gem/BV
n (%) (n=101) Tax/BV (n=200) (n=52) (n=108)
Selected AE 29 (28.7) 79 (39.5) 5 (9.6) 34 (31.5)
(Grade >3)
SAE 12 (11.9) 53 (26.5) 8(15.4) 25 (23.1)
AE leading to
BV/PL 4(4.0) 12(6.0) 4(7.7) 6(5.6)
discontinuation
or death
AE leading to 0(0) 3 (1.5) 2(3.8) 2(1.9)
death
Cape/PL Vin/PL Vin/BV
n (%) (n=46) Cape/BV (n=97) (n=22) (n=53)
Selected AE 2(4.3) 20 (20.6) 14 (63.6) 30 (56.6)
(Grade >3)
SAE 12 (26.1) 18 (18.6) 7(31.8) 16 (30.2)
AE leading to
BV/PL 4 (8.7) 6 (6.2) 1 (4.5) 4 (7.5)
discontinuation
or death
AE leading to 2(4.3) 1 (1.0) 1(4.5) 0(0)
death
AE=adverse event, SAE=serious adverse event, Tax=taxane, Gem=gemcitabine,
Cape=capecitabine, Vin=vinorelbine.
63