Note: Descriptions are shown in the official language in which they were submitted.
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
HYDROGENATION OF SOLID CARBONACEOUS MATERIALS USING MIXED
CATALYSTS
TECHNICAL FIELD
[0001] This invention relates to systems and processes for pretreating a
carbonaceous
material, for liquefying a carbonaceous material, and for improving efficiency
of
carbonaceous material liquefaction.
BACKGROUND
[0002] Much work has been done over the years on processes for obtaining
liquid and
gaseous products from solid carbonaceous materials such as coal. The known
processes
include both catalytic and non-catalytic reactions. In catalytic processes,
the
hydrocarbonaceous material is typically slurried with a solvent and a
catalyst, and is reacted
in the presence of molecular hydrogen at elevated temperatures and pressures.
[0003] U.S. Patent 5,246,570, for example, describes a coal liquefaction
process in which
a mixture of coal, catalyst, and solvent are rapidly heated to a temperature
of 600-750 F in a
preheater, and then reacted under coal liquefaction conditions in a
liquefaction reaction. U.S.
Patent 5,573,556 describes a process for converting a carbonaceous material to
normally
liquid products comprising heating a slurry that comprises a carbonaceous
material, a
hydrocarbonaceous solvent, and a catalyst precursor to a temperature
sufficient to convert the
catalyst precursor to the corresponding catalyst, and introducing the slurry
into a liquefaction
zone. U.S. Patent 5,783,065 describes a coal liquefaction process comprising
impregnating
coal particles with a catalyst having hydrogenation or hydrogenolysis
activity; introducing
the impregnated coal particles for very short periods into a turbulent flow of
hydrogen
containing gas at a temperature at least about 400 C; and quenching the
temperature of the
products to a temperature significantly less than 400 C.
[0004] Such conventional processes leave much room for improving the liquid
and/or gas
yields of hydroconverted carbonaceous materials, as well as the quality of the
liquid and/or
gas products that are obtained from such processes. Accordingly, a need
remains for
improved systems and processes for hydroconversion of carbonaceous materials,
as well as
improved feed materials for such systems and processes.
1
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
SUMMARY OF THE INVENTION
[0005] The present invention relates to a process for converting solid
carbonaceous
material to a liquid product, comprising maintaining a solid carbonaceous
material in the
presence of at least one active source of zinc and at least one active source
of a second metal
at a reaction temperature of greater than 350 C and at a pressure in the range
of 300 to 5000
psig for a time sufficient to form a liquid product.
[0006] In one aspect, the process comprises preparing a combination of the
solid
carbonaceous material, at least one hydrocarbonaceous liquid, at least one
active source of
zinc and at least one active source of the second metal; and passing the
combination to a
hydroconversion reaction zone and maintaining the solid carbonaceous material
at a reaction
temperature of greater than 350 C and at a pressure in the range of 300 to
5000 psig for a
time sufficient to convert at least a portion of the solid carbonaceous
material to a liquid
product boiling in the temperature range of C5 to 650 C.
[0007] In a further aspect, the step of preparing the combination comprises
preparing a
mixture comprising at least one active source of zinc and at least one active
source of a
second metal; combining the mixture with coal to form catalyst-containing coal
particles;
providing a hydrocarbonaceous liquid to the catalyst-containing coal particles
to prepare the
combination.
[0008] In another aspect, the process of preparing the combination further
comprises
drying the catalyst-containing coal prior to the step of passing the
combination to the
hydroconversion reaction zone.
[0009] In a further aspect, the process further comprises pretreating the
combination at a
pretreatment temperature within the range of 100-350 C and for a time of
between 5 and 600
minutes prior to passing the combination to the hydroconversion reaction zone.
[0010] In a further aspect, before, during or after the step of pretreatment,
at least one
active source of sulfur is added to the solid carbonaceous material in the
preparation of the
combination, wherein the atomic ratio of sulfur to metal components is within
the range of
between 1/1 and 10/1.
[0011] In an aspect, the second metal is iron.
[0012] Several embodiments of the invention, including the above aspects of
the
invention, are described in further detail as follows. Generally, each of
these aspects can be
used in various and specific combinations, and with other aspects and
embodiments unless
otherwise stated herein.
2
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
BRIEF DESCRIPTION OF THE DRAWING
[0013] Fig. 1, Fig. 2, Fig. 3 and Fig. 4 illustrate embodiments of the process
for
converting solid carbonaceous material.
DETAILED DESCRIPTION
[0014] The following terms will be used throughout the specification and will
have the
following meanings unless otherwise indicated.
[0015] The term "catalyst precursor" is used herein to refer to a compound
that is
transformable into a catalyst via chemical reaction with one or more reagents
(such as
sulfiding and/or reducing agents, e.g., hydrogen, such as within a hydrocarbon
medium)
and/or via any other suitable treatment (such as thermal treatment, multi-step
thermal
treatment, pressure treatment, or any combination thereof) whereby the
catalyst precursor at
least partially decomposes into a catalyst.
[0016] The term "active source" is used herein to refer to an atomic,
molecular, complex
or any other form of an element that is a catalyst or a catalyst precursor or
that can be
converted into a catalyst or catalyst precursor. The active source may be in
solution, in slurry
or in particle form. When the active source is deposited on the solid
carbonaceous material,
by, for example, plating, impregnation, coating or washing, a single active
source or a
mixture of active sources may be deposited on individual particles of the
solid carbonaceous
material.
[0017] The term "catalytic material" is used to refer to one or more active
catalysts or
catalyst precursors. The component(s) of catalytic material may be in slurry
or particle form.
In particle form, single or multiple catalysts may be present on individual
particles.
Likewise, when the catalytic material is deposited on the solid carbonaceous
material, by, for
example, plating, impregnation, coating or washing, a single catalyst, or a
mixture of
catalysts or precursors making up the catalytic material may be deposited on
individual
particles of the solid carbonaceous material.
[0018] Unless otherwise specified, coal properties as disclosed herein are on
a dry, ash-
free (daf) basis, wherein ASTM 3173 is used for moisture determination and
ASTM3174 for
ash quantification.
[0019] "d" block elements refer to elements of the Period Table wherein the d
sublevel
of the atom is being filled. Examples include iron, molybdenum, nickel,
manganese,
vanadium, tungsten, cobalt, copper, titanium, chromium, platinum, palladium,
cerium,
zirconium, zinc and tin.
3
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
[0020] Lanthanoid (or lanthanide, or sometimes referred to as rare earths)
elements refer
to the fifteen elements in the Periodic Table with atomic numbers 57 through
71.
[0021] "Oil dispersible" compound means that the compound scatters or
disperses in oil
forming a dispersion. In one embodiment, the oil dispersible compound is oil
soluble which
dissolves upon being mixed with oil.
[0022] For purposes of this disclosure, unless otherwise specified, the
catalyst
composition is defined as the composition of the active source(s) added to the
process,
regardless of the form of the catalytic elements during hydroconversion.
[0023] The present invention relates to the composition and preparation
procedures of a
sulfided zinc-containing catalyst used for hydroconversion of carbonaceous
material
including coal, shale oil, vacuum residuum and bio-fuel stock such as lignin.
The invention
further relates to a hydroconversion process for converting solid carbonaceous
material to a
liquid product in the presence of a catalyst composition comprising zinc. In
embodiments,
the invention further relates to a process for converting a carbonaceous
material, comprising
pretreating a solid carbonaceous material at a pretreatment temperature and in
the presence of
at least one active source of zinc and at least one active source of a second
metal; heating the
pretreated material in the presence of hydrogen to a conversion temperature
which is greater
than the pretreatment temperature; and reacting the heated material for a time
sufficient to
form converted products from the solid carbonaceous material.
Catalyst Formula
[0024] In one embodiment, the catalyst composition as expressed in elemental
form is of
the general formula (RP);(Mt)a(L )b(S )d(CW)e(HX)f(Oy)g(Nz)h. The formula
herein refers to
the catalyst solids, constituting the catalyst slurry in oil. In the equation,
M and L each
represents at least a "d" block element from the Periodic Table such as iron,
molybdenum,
nickel, manganese, vanadium, tungsten, cobalt, copper, titanium, chromium,
platinum,
palladium, cerium, zirconium, zinc and tin. M is different from L. R is
optional, which
represents at least one lanthanoid element from the Periodic Table such as La,
Ce, Nd, etc. In
another embodiment, R is at least an alkali earth metal such as magnesium.
[0025] Also in the equation, p, t, u, v, w, x, y, z representing the total
charge for each of
the components (R, M, L, S, C, H, 0 and N, respectively);
pi+ta+ub+vd+we+xf+yg+zh=O; R
with a subscript i ranging from 0 to 1; M and L with subscripts a and b, with
values of a and
b respectively ranging from 0 to 5, and (0 <= b/a <= 5); S represents sulfur
with the value of
4
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
the subscript d ranging from 0.5(a + b) to 5(a + b); C represents carbon with
subscript e
having a value of 0 to 11(a+b); H is hydrogen with the value of f ranging from
0 to 7(a+b); 0
represents oxygen with the value of g ranging from 0 to 5(a + b); and N
represents nitrogen
with h having a value of 0 to 2(a + b).
[0026] In embodiments, M is iron and L is zinc (or vice versa). In some such
embodiments, the catalyst is of the formula (FezZni_z)a(S)d(C)e(H)j(O)g(N)h,
wherein the zinc
to iron ratio is in the range of 9:1-1:9 (as wt. %). In some such embodiments,
the zinc to iron
ratio is in the range of 1:5 to 5: 1; or in the range of 1: 10 to 1:5.
Pretreatment Process
[0027] In embodiments, the present invention is related to a system and
process for
pretreating a carbonaceous material, for dispersing one or more catalysts or
catalyst
precursors into a carbonaceous material, for enhancing the conversion of a
carbonaceous
material (such as a naturally-occurring solid carbonaceous material, such as
coal) to a liquid
and/or gaseous product, for producing a carbonaceous material of enhanced
reactivity, for
improving efficiency of carbonaceous material (such as coal) liquefaction, as
measured for
example by conversion and liquid yield, and/or for lowering hydrogen
consumption during
liquefaction of carbonaceous material.
[0028] In one embodiment, such pretreating of a carbonaceous material is
performed or
accomplished using reaction conditions (or a combination of conditions, such
as temperature,
pressure, and/or duration of pretreatment) at which substantially no
hydroconversion of the
carbonaceous material occurs (i.e., wherein less than about 20%, less than
about 10% or even
less than about 1% of the carbonaceous material is converted) during the
pretreatment step.
Any suitable process or operating conditions can be utilized to pretreat the
carbonaceous
material. In one embodiment, the pretreatment composition is heated to a
temperature
sufficient to cause one or more catalysts or catalyst precursors to disperse
into the
carbonaceous material, and is maintained, held, and/or kept at this
pretreatment temperature
for a time or duration sufficient to disperse one or more of the catalysts or
catalyst precursors
into the carbonaceous material to a desired degree of dispersion, integration,
and/or
homogeneity. In one embodiment, the pretreatment composition is heated to a
temperature of
about 100-350 C (such as about 150-300 C or even about 180-220 C). In some
such
embodiments, the step of pretreating is conducted at a temperature of about
100-350 C for
about 10-360 minutes.
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
[0029] The pretreatment composition is preferably maintained, kept, and/or
held at the
pretreatment temperature for a time or duration sufficient to cause swelling
of the
carbonaceous material and to allow for dispersion (such as complete dispersion
and/or
homogenous dispersion) of the catalyst or catalyst precursor into the
carbonaceous material.
In one embodiment, for example, the pretreatment composition is maintained,
kept, and/or
held at suitable temperature for a suitable duration to cause the total volume
of voids of the
carbonaceous material (or of each particle of carbonaceous material) to
increase by greater
than about 5%, or about 25% as compared to the carbonaceous material prior to
pretreatment.
In one embodiment, in this regard, the pretreatment composition is maintained
at the
pretreatment temperature for a time of between 5 and 600 minutes, or between
10 and 360
minutes.
[0030] Pretreatment of the carbonaceous material can be performed under any
suitable
atmosphere. In one embodiment, the pretreatment of carbonaceous material
occurs under an
inert atmosphere. In another embodiment, the pretreatment occurs under a
reducing
atmosphere, such as under hydrogen and/or a synthesis gas ("syn-gas")
pressure. In some
embodiments, for example, the pretreatment is performed at a pressure between
atmospheric
pressure and about 500 psig, e.g., a pressure of about 100-450 psig, or about
200-350 psig. In
other embodiments, the pretreatment occurs under a reducing atmosphere at a
pressure
defined by the hydroconversion process, such as at a pressure of about 300-
5000 psig, such as
500-3500 psig, about 1000-3000 psig, or even about 1500-2600 psig. Any
suitable syn-gas
can be used in this regard, such as, for example, a syn-gas that comprises a
1:1 to 2: 1 mixture
of hydrogen with carbon monoxide, and optionally also contains carbon dioxide,
methane,
and/or other components.
[0031] In one embodiment, such pretreatment is performed or accomplished under
conditions sufficient to deposit at least a portion of the catalyst or
catalyst precursor onto the
solid carbonaceous material during pretreatment. In some such embodiments, one
or more
catalysts or catalyst precursors and a liquid contact the solid carbonaceous
material.
[0032] The pretreatment composition comprising the carbonaceous material, one
or more
catalyst or catalyst precursors, and a hydrocarbonaceous liquid can be
prepared in any
suitable manner. In one embodiment, the carbonaceous material, catalyst or
catalyst
precursor, and hydrocarbonaceous liquid are simply mixed to form a
pretreatment
composition, and the pretreatment composition is subjected to pretreatment
conditions. In
another embodiment, the carbonaceous material is contacted with the catalyst
or catalyst
6
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
precursor in the presence of the hydrocarbonaceous liquid, and the
pretreatment composition
is subjected to pretreatment conditions. In another embodiment, the
carbonaceous material is
ground in the presence of the one or more catalysts or catalyst precursors and
the
hydrocarbonaceous liquid, to produce a pretreatment composition in the form of
a slurry; and
the pretreatment composition is subjected to pretreatment conditions. In
another embodiment,
the carbonaceous material is ground in the presence of the hydrocarbonaceous
liquid to
produce a slurry; the one or more catalyst precursors are added to the slurry
to form a
pretreatment composition; and the pretreatment composition is subjected to
pretreatment
conditions. In other embodiment, the catalyst or catalyst precursor is added
at the start of the
pretreatment process. In another embodiment, the catalyst or catalyst
precursor is added at
intervals throughout a pretreatment process. In other embodiments, at least a
portion of the
catalyst or catalyst precursor is deposited on the carbonaceous material
during pretreatment.
[0033] Following pretreatment of the carbonaceous material, the carbonaceous
material
and dispersed catalyst or catalyst precursor, optionally together with the
hydrocarbonaceous
liquid, form an improved feed for a hydroconversion process. Such an improved
feed can be
used for any suitable hydroconversion process to produce a liquid and/or
gaseous product.
Carbonaceous Material
[0034] The carbonaceous material can be any suitable solid carbon containing
material,
such as any naturally occurring solid, or normally solid, carbon containing
material.
Specifically, for example, the carbonaceous material can be coal, such as
anthracite,
bituminous coal, sub-bituminous coal, lignite, or any combination or mixture
thereof. The
carbonaceous material can also be any heteroatom-containing solid carbonaceous
material or
feed, as well as any heavy hydrocarbonaceous feeds, such as, for example,
coal, coke, peat,
shale oil and/or a similar material, such as any solid carbonaceous material
containing a
relatively high ratio of carbon to hydrogen, or combinations or mixtures
thereof. In some
embodiments, at least a portion of the carbonaceous material is in the form of
particles, or
finely divided particles, having any suitable size. For example, at least
about 50 wt% of the
carbonaceous material is in the form of particles having a mean particle
diameter of less than
about 0.5 inches. In embodiments, at least greater than 70 wt% of carbonaceous
material is
in the form of particles having a mean particle diameter in the range of about
0.1 to 0.4
inches. In one embodiment, greater than about 80 wt.% of the carbonaceous
material is in
the form of particles having a mean diameter less than about 0.25 inches. In
another
embodiment, greater than 80 wt% of the carbonaceous material is in the form of
particles
7
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
having a mean diameter in the range of 50 microns to 500 microns, such as 100
microns.
Such particles can be formed in any suitable manner, such as by grinding at
least a portion of
the carbonaceous material. In one embodiment, at least a portion of the
carbonaceous
material is ground in the presence of one or more catalysts or catalyst
precursors and the
hydrocarbonaceous liquid. In another embodiment, at least a portion of the
carbonaceous
material is ground in the presence of the hydrocarbonaceous liquid to form a
slurry, and (such
as subsequently) mixing the slurry with one or more catalysts or catalyst
precursors. In other
embodiments, the carbonaceous material is ground under an inert or a reducing
atmosphere,
such as, for example, hydrogen, nitrogen, helium, argon, syn-gas, or any
combination or
mixture thereof. Any process or equipment may be used to grind the
carbonaceous material,
such as, for example, a hammer mill, a ball mill (such as a wet ball mill, a
conical ball mill, a
rubber roller mill), a rod mill, or a combination thereof.
Hydrocarbonaceous Liquid
[0035] The hydrocarbonaceous liquid can be any suitable liquid (such as
solvent or
diluent) known in the art to be useful for the liquefaction of carbonaceous
materials (such as
solid carbonaceous materials, such as coal). In one embodiment, the
hydrocarbonaceous
liquid is a hydrogen donor solvent, such as any compound(s) which functions as
a hydrogen
donor in hydroconversion conditions. The hydrocarbonaceous liquid can have any
suitable
hydrogen donatability, such as, for example, a hydrogen donatability greater
than about 1.0
wt%, as determined, for example, by NMR.
[0036] In one embodiment, the hydrocarbonaceous liquid comprises a coal-
derived
solvent, or a distillate fraction thereof. In another embodiment, the
hydrocarbonaceous liquid
comprises a hydrogenated aromatic, a naphthenic hydrocarbon, a phenolic
material, or a
similar compound, or a combination or mixture thereof. In another embodiment,
the
hydrocarbonaceous liquid comprises one or more aromatics, such as one or more
alkyl
substituted aromatics. Solvents known to donate hydrogen during liquefaction
include, for
example, the dihydronaphthalenes, the C10 - C12 tetrahydronaphthalenes, the
hexahydrofluorenes, the dihydro-, tetrahydro-, hexahydro- and
octahydrophenanthrenes, the
C12 -C13 acenaphthenes, the tetrahydro-, hexahydro- and decahydropyrenes, the
di-, tetra- and
octahydroanthracenes, and other derivatives of partially saturated aromatic
compounds. They
can be prepared by subjecting a distillate stream from atmospheric
distillation to a
conventional hydrogenation reactor. Particularly effective mixed solvents
include heavy gas
oil fractions (often called vacuum gas oils, or VGO) with an initial boiling
point of about
8
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
343 C. (650 F.) and a final boiling point of about 538 C. (1000 F.). This
stream comprises
aromatics, hydrogenated aromatics, naphthenic hydrocarbons, phenolic
materials, and similar
compounds. If a solvent is used which does not have donatable hydrogen,
hydrogen may be
added from another source.
[0037] The solvent generally boils at a temperature greater than 300 C., such
as, for
example a temperature in the range of 450-900 or 650-850 F. In one embodiment,
the
hydrocarbonaceous liquid is a fluid catalytic cracking (FCC) type process oil
cut that boils at
a temperature of about 500 F or higher (FCC-type process oil (500 F+ cut)). In
another
embodiment, the hydrocarbonaceous liquid is an FCC-type process oil boiling at
a
temperature of about 500 F or less ("FCC-type process oil (500 F- cut)"). In
another
embodiment, the hydrocarbonaceous liquid is a hydrotreated FCC oil. In another
embodiment, the hydrocarbonaceous liquid is tetralin (1,2,3,4
tetrahydronaphthalene). In
another embodiment, the hydrocarbonaceous liquid comprises one or more
compounds that
have an atmospheric boiling point ranging from about 350-850 F.
[0038] Any suitable ratio of hydrocarbonaceous liquid to carbonaceous material
(such as
carbonaceous particles, or even coal particles) can be used in the context of
the present
invention, such as, for example, a ratio in a range of about 1: 10 to about
10: 1, such as 1:6 to
about 6: 1, or a range of about 1:2 to about 2: 1, by weight of the mixture.
In one
embodiment, the ratio of hydrocarbonaceous liquid to carbonaceous material
used in the
pretreatment process is about 0.75:1 to about 1:1.
Catalyst Precursor
[0039] The process for converting a solid carbonaceous material comprises
heating the
carbonaceous material in the presence of a catalyst composition. In
embodiments, the
process for converting a solid carbonaceous material comprises heating a solid
carbonaceous
material in the presence of at least one active source of zinc for a time
sufficient to form a
liquid product from the solid carbonaceous material. In embodiments, the
active source of
zinc is provided to the carbonaceous material is the form of a catalyst
precursor that is
transformable into a catalyst via chemical reaction with one or more reagents
and/or via any
other suitable treatment. The catalyst precursor may be oil soluble, oil
dispersible, water
soluble and/or water dispersible. In embodiments, the process comprises
pretreating the solid
carbonaceous material at a pretreatment temperature and in the presence of at
least one active
source of zinc; heating the pretreated material in the presence of hydrogen to
a conversion
9
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
temperature which is greater than the pretreatment temperature; and reacting
the heated
material for a time sufficient to form a liquid product from the solid
carbonaceous material.
[0040] Suitable catalyst precursors include:
a) zinc metal;
b) zinc containing inorganic compounds, such as the sulfates, nitrates,
carbonates, sulfides, oxysulfides, oxides and hydrated oxides, ammonium
salts and heteropoly acids of zinc;
c) salts of organic acids, such as acyclic and alicyclic aliphatic, carboxylic
acids
containing two or more carbon atoms (non-limiting examples include acetates,
oxylates, citrates);
d) zinc-containing organometallic compounds including chelates such as 1,3-
diketones, ethylene diamine, ethylene diamine tetraacetic acid,,
phthalocyanines, thiocarbamates, phosporothioates, and combinations or
mixtures thereof (non-limiting examples include zinc alkyl dithiocarbamate,
zinc alkyl phosphorodithioate); and/or,
e) zinc salts of organic amines such as aliphatic amines, aromatic amines,,
quaternary ammonium compounds, or combinations or mixtures thereof, and
f) zinc-containing minerals.
[0041] In embodiments, the process for converting a solid carbonaceous
material further
comprises heating the solid carbonaceous material in the presence of at least
one active
source of a second metal. In embodiments, the second metal is selected from
the group
consisting of iron, molybdenum, tungsten, nickel, cobalt, titanium and tin. In
some such
embodiments, the active source of the metal is provided to the carbonaceous
material is the
form of a catalyst precursor that is transformable into a catalyst via
chemical reaction with
one or more reagents and/or via any other suitable treatment. The catalyst
precursor may be
oil soluble, oil dispersible, water soluble and/or water dispersible.
[0042] In embodiments, the catalyst composition comprises for converting the
solid
carbonaceous material further comprises at least one active source of iron.
Suitable catalyst
precursors which provide the active iron source include:
a) iron metal;
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
b) iron containing inorganic compounds, such as the sulfates, nitrates,
carbonates, sulfides, oxysulfides, oxides and hydrated oxides, ammonium
salts and heteropoly acids of iron;
c) salts of organic acids, such as acyclic and alicyclic aliphatic, carboxylic
acids
containing two or more carbon atoms (non-limiting examples include acetates,
oxylates, citrates);
d) iron-containing organometallic compounds including ferrocene, chelates such
as 1,3-diketones, ethylene diamine, ethylene diamine tetraacetic acid,,
phthalocyanines, thiocarbamates, phosphorothioates, and combinations or
mixtures thereof (non-limiting examples include iron alkyl dithiocarbamate,
iron alkyl phosphorodithioate); and/or,
e) iron salts of organic amines such as aliphatic amines, aromatic amines,,
quaternary ammonium compounds, or combinations or mixtures thereof, and
f) iron-containing minerals.
[0043] The catalyst precursor can be formed in any suitable manner prior to
the
hydroconversion process. In one embodiment, for example, one or more catalyst
precursors
are formed by:
a) mixing a hydrocarbonaceous liquid (such as a liquefaction solvent) with an
active source of at least one metal (such as a metal oxide, e.g., iron oxide,
or
other compound containing any suitable metal as discussed herein) to form a
catalyst precursor,
b) combining the catalyst precursor with a carbonaceous material;
c) optionally subjecting the mixture to pretreatment conditions (such as under
hydrogen pressure) in a manner such that one or more catalyst precursors form
in or on the carbonaceous material; and
d) heating the mixture for a time sufficient to form a liquid product
[0044] In embodiments, the catalyst precursors are formed by:
a) mixing a hydrocarbonaceous liquid (such as a liquefaction solvent) with at
least one active source of zinc and with at least one active source of a
second
metal to form a catalyst precursor;
11
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
b) combining the catalyst precursor with a carbonaceous material;
c) optionally subjecting the mixture to pretreatment conditions in a manner
such
that one or more catalyst precursors form in or on the carbonaceous material;
and
d) heating the mixture for a time sufficient to form a liquid product
[0045] In embodiments, the catalyst precursors are formed by
a) mixing a hydrocarbonaceous liquid with an active source of at least one
metal,
b) combining the mixture with a sulfiding agent (such as by passing hydrogen
sulfide through the mixture or adding elemental sulfur to the mixture) in a
manner such that the sulfided metal-containing compound is dispersible,
c) combining the sulfided mixture with a carbonaceous material,
d) optionally subjecting the mixture to pretreatment conditions in a manner
such
that one or more catalyst precursors form in or on the carbonaceous material;
and
e) heating the mixture for a time sufficient to form a liquid product.
[0046] In embodiments, the catalyst precursors are formed by
a) mixing a hydrocarbonaceous liquid with an active source of at least one
metal;
b) combining the catalyst precursor with a carbonaceous material;
c) combining the mixture with a sulfiding agent;
d) optionally subjecting the mixture to pretreatment conditions in a manner
such
that one or more catalyst precursors form in or on the carbonaceous material;
and
e) heating the mixture for a time sufficient to form a liquid product.
[0047] In another embodiment, one or more catalyst precursors are formed by
a) mixing one or more metal containing compounds, a sulfiding agent, and
water,
to form a colloidal suspension,
b) combining the colloidal suspension with a hydrocarbonaceous liquid (such as
a liquefaction solvent) to drive water out of the suspension,
12
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
c) combining the suspension with a carbonaceous material,
d) optionally subjecting the suspension to pretreatment conditions (such as
under
hydrogen pressure), in a manner such that one or more catalyst precursors
form in or on the carbonaceous material; and
e) heating the mixture for a time sufficient to form a liquid product.
[0048] In another embodiment, one or more catalyst precursors are by
a) sulfiding an ammonium containing Group VIB metal compound in an aqueous
phase with hydrogen sulfide, in a substantial absence of hydrocarbon oil, at a
temperature less than about 177 C, to form a presulfided product; and
b) separating ammonia from said presulfided product to form a sulfided
product,
in a manner such that one or more catalyst precursors form in or on the
carbonaceous material.
[0049] In another embodiment, one or more catalyst precursors are formed by a
process
comprising:
a) mixing an active source of zinc and an active source of the second metal
and
water, to form a colloidal suspension or solution;
b) combining the colloidal suspension or solution with a solid carbonaceous
material at conditions sufficient to deposit at least a portion of the zinc
and a
portion of the second metal onto [wherein depositing onto includes depositing
onto the surface of any fractures, pores, or other openings into the internal
volume of the solid carbonaceous material] the solid carbonaceous material;
c) combining the solid carbonaceous material having the active sources of the
metals deposited thereon with a hydrocarbonaceous liquid (such as a
liquefaction solvent); and
d) optionally subjecting the suspension to pretreatment conditions (such as
under
hydrogen pressure), in a manner such that one or more catalyst precursors
form in or on the carbonaceous material; and
e) heating the mixture for a time sufficient to form a liquid product.
[0050] In some such embodiments, the process further comprises combining the
colloidal
suspension or solution and an active source of sulfur with the solid
carbonaceous material.
13
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
[0051] Any suitable amount of the catalytic materials can be used to
hydroconvert the
carbonaceous material in the context of the present invention. In one
embodiment, the
mixture of catalyst precursor, carbonaceous material, and hydrocarbonaceous
liquid
comprises about 25-10000 ppm (such as about 50-9000 ppm, about 100-8000 ppm,
about
250-5000, about 500-3000 ppm, or even about 1000-2000 ppm) of one or more
catalyst or
catalyst precursor by weight, based on the total weight of the mixture. In
embodiments, the
metal content of the catalyst or catalyst precursor refers to added metal, and
does not include
metal which is native to the carbonaceous material, or metal which is eroded
from processing
equipment.
[0052] The catalytic materials can be used in the context of the present
invention in any
suitable form, such as, but not limited to, particulate form, impregnated
within a
carbonaceous material, dispersed in the hydrogen donor solvent, and/or soluble
in the
hydrogen donor solvent. Additionally, the catalytic materials may be used in
processes
employing fixed, moving, and ebullated beds as well as slurry reactors.
[0053] The catalyst precursor(s) can be transformed into a catalyst by thermal
decomposition, such as prior to or during liquefaction, without the addition
of additional
reactants. In other embodiments, following pretreatment, one or more
additional reactants can
be added to the pretreated carbonaceous material mixture (such as prior to or
during the
liquefaction process), to transform the dispersed catalyst precursor into a
catalyst. Any
suitable reactants can be used in this regard, such as for example any
suitable sulfiding or
reducing agents.
Sulfiding Agent Component
[0054] In embodiments, the catalyst composition further comprises at least one
active
source of sulfur. In those embodiments in which catalyst precursors are
utilized, one or more
sulfur compounds can be added subsequent to the pretreating step to activate
the catalyst
precursor to its corresponding sulfided active catalyst. The one or more
sulfur compounds
can be introduced at any point of the system, following pretreatment. In one
embodiment,
one or more sulfur compounds are introduced into the pretreatment zone
following the
performance of the pretreatment process and before the pretreatment
composition is delivered
to the liquefaction zone. In another embodiment, one or more sulfur compounds
are
introduced into the liquefaction zone.
14
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
[0055] In one embodiment, the catalyst is prepared using a sulfiding agent in
the form of
a solution which, under prevailing conditions, is decomposable into hydrogen
sulfide. Such a
sulfiding agent can be used in any suitable amount in preparing the catalyst,
such as in an
amount in excess of the stoichiometric amount required to form the catalyst.
In one
embodiment, the sulfiding agent is present in a sulfur to zinc mole ratio of
at least 3 to 1.
Additionally, any suitable sulfiding agent (such as described above with
respect to the
catalyst precursor) can be used.
[0056] In one embodiment, the sulfiding agent is an aqueous ammonium sulfide.
Such a
sulfiding agent can be prepared in any suitable manner, such as from hydrogen
sulfide and
ammonia. This synthesized ammonium sulfide is readily soluble in water and can
easily be
stored in aqueous solution in tanks prior to use.
[0057] Suitable sulfiding agents include, for example, any sulfur compound
that is in a
readily releasable form, such as, for example, hydrogen sulfide, ammonium
sulfide,
dimethyldisulfide, ammonium sulfate, carbon disulfide, elemental sulfur, and
sulfur-
containing hydrocarbons. Elemental sulfur is preferred in some embodiments,
because of its
low toxicity, low cost, and ease of handling. Additional sulfiding agents
include, for example,
ammonium sulfide, ammonium polysulfide, ammonium thiosulfate, sodium
thiosulfate,
thiourea, dimethyl sulfide, tertiary butyl polysulfide, tertiary nonyl
polysulfide, and mixtures
thereof. In another embodiment, the sulfiding agent is selected from the group
consisting of
alkali- and/or alkaline earth metal sulfides, alkali-and/or alkaline earth
metal hydrogen
sulfides, and mixtures thereof.
[0058] The sulfiding agent can be added in any suitable form. In one
embodiment,
elemental sulfur is added to the carbonaceous material mixture in the form of
a sublimed
powder or as a concentrated dispersion (such as a commercial flower of
sulfur). Allotropic
forms of elemental sulfur, such as orthorhomic and monoclinic sulfur, are also
suitable for
use herein. In one embodiment, the one or more sulfur compounds are in the
form of a
sublimed powder (flowers of sulfur), a molten sulfur, a sulfur vapor, or a
combination or
mixture thereof.
[0059] The sulfiding agent can be used in any suitable concentration. In one
embodiment,
a concentration of sulfur is introduced such that the atomic ratio of sulfur
to metal in the
catalyst precursor is in the range of from about 1: 1 to about 10: 1, such as
from about 2:1 to
about 8:1, about 2:1 to about 7:1, about 2:1 to about 6:1, about 2:1 to about
9:1, about 2:1 to
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
about 8:1, about 2:1 to 7:1, about 3:1 to about 9:1, about 3:1 to about 8:1,
about 3:1 to about
7:1 or even about 3:1 to about 6:1.
Catalyst
[0060] The catalyst contains an active catalytic component in elemental or
compound
form. Examples include finely divided particles, salts, or compounds of the
transition
elements, particularly Groups IV-B, V-B, VI-B or Group VIII of the Periodic
Table of the
Elements, as shown in Handbook of Chemistry and Physics, 45th Edition,
Chemical Rubber
Company, 1964. In embodiments, alkaline earth elements, such as magnesium, may
be
included. In embodiments, lanthanoid (or lanthanide, or sometimes referred to
as rare earths)
elements refer to the fifteen elements in the Periodic Table with atomic
numbers 57 through
71, may be included.
[0061] The catalyst includes any zinc-containing material that is suitable for
use in a
hydroconversion process for a carbonaceous material (such as coal) when
subjected to and/or
when experiencing suitable catalyzing reaction conditions. The catalyst
further comprises
any suitable metal, such as, for example, a metal selected from the group
consisting of Group
IIB metals, Group IIIB metals, Group IVA metals, Group IVB metals, Group VB
metals,
Group VIB metals, Group VIIB metals, Group VIII metals, or a combination or
mixture
thereof, such as in combination with one or more of oxygen, sulfur, nitrogen,
and
phosphorous. In embodiments, a second metal is selected from the group
consisting of Fe,
Mo, W, Co, Ni, Cu, Ti and Sri.
[0062] In embodiments, the sulfided zinc-containing catalyst can be ZnS-FeS,
ZnS-
MoS2, ZnS-WS2, ZnS-CoS, ZnS-NiS, ZnS-CuS, ZnS-TiS2, ZnS-SnS and any of their
combinations and mixtures, for example ZnS-MoS2-TiS2. In the catalyst system,
zinc can be
the rich phase or serve as dopant.
[0063] The amount of zinc that is provided as a catalyst component of the
catalyst is
sufficient to catalyze the conversion of the solid carbonaceous material to
liquid
hydrocarbons; likewise, the amount of the second metal that is provided as a
catalyst
component is sufficient to catalyze the conversion of the solid carbonaceous
material. In
embodiments, zinc is present in the catalyst in an amount of 10 ppm to 10 wt%,
based on dry,
ash free coal. In some such embodiments, zinc is present in the catalyst in
the amount of 0.1
wt% to 5 wt%. An exemplary quantity of zinc, as metal, present in the catalyst
is in the
amount of 0.5 wt% to 2.5 wt%.
16
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
[0064] In embodiments, the second metal in the catalyst is present in an
amount of 10
ppm to 10 wt%, based on dry, ash free coal. In some such embodiments, the
second metal is
present in the catalyst in the amount of 0.1 wt% to 5 wt%. An exemplary
quantity of the
second metal, expressed as a metal, is in the amount of 0.5 wt% to 2.5 wt%. In
some such
embodiments, the second metal in the catalyst is iron. As such, iron is
present in the catalyst
in an amount of 10 ppm to 10 wt%, based on dry, ash free coal. In some such
embodiments,
iron is present in the catalyst in the amount of 0.1 wt% to 5 wt%. An
exemplary quantity of
iron, as metal, present in the catalyst is in the amount of 0.5 wt% to 2.5
wt%. In
embodiments, the molecular ratio between zinc and other metals in combination
can be
between 0.1 to 1 and 10 to 1.
[0065] In embodiments, the catalytic materials are added as finely divided
particulate
metal solids, their oxides, sulfides, etc., e.g., FeSX ; waste fines from
metal refining processes,
e.g., iron, molybdenum, and nickel; crushed spent catalysts, e.g., spent fluid
catalytic
cracking fines, hydroprocessing fines, recovered coal ash, and solid coal
liquefaction
residues. In embodiments, the zinc and the second metal are added as separate
particulate
solids. In other embodiments, the catalyst composition comprises particles
that are richer in
zinc and leaner in the amount of the second metal, or particles that are
richer in the second
metal and leaner in the amount of zinc. In another embodiment, zinc and other
metals can
form bi-metallic compounds as a catalyst precursor rather than being added to
the feed
separately. As an example, ZnXFe(i_X)OOH is prepared by titrating a FeSO4 and
ZnSO4
mixture solution with NH3H2O, followed by oxidizing in flowing air at elevated
temperatures. ZnXFe(i_X)OOH can be pre-sulfided to ZnxFe(i_X)S before mixing
with the feed.
[0066] In embodiments, at least a portion of the catalyst particles are
attached to,
adsorbed onto, absorbed by, supported on or intimately associated with at
least a portion of
the solid carbonaceous material during conversion of the carbonaceous
material. In
embodiments, at least a portion of the catalyst, or catalyst precursor, is
deposited on the solid
carbonaceous material before or during pretreatment, using an aqueous or an
organic liquid to
carry the catalyst or catalyst precursor to the carbonaceous material. In
embodiments, at least
a portion of the catalyst, or catalyst precursor, is deposited on the solid
carbonaceous material
during the step of heating the material to conversion temperature, or during
the conversion
process.
[0067] In an embodiment, the catalyst is prepared using a catalyst precursor
comprising a
metal that comprises a water-soluble zinc component, such as zinc nitrate,
zinc sulfate,
17
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
zinc acetate, zinc chloride, or a mixture thereof. In another embodiment, the
catalyst is
prepared using a catalyst precursor comprising an metal that comprises a zinc
compound
which is at least partly in the solid state, e.g., a water-insoluble zinc
compound such as zinc
carbonate, zinc hydroxide, zinc phosphate, zinc phosphite, zinc formate, zinc
sulfide, zinc
molybdate, zinc tungstate, zinc oxide, zinc alloys such as zinc-molybdenum or
zinc-iron
alloys, or a mixture thereof. In another embodiment, the catalyst is prepared
using a catalyst
precursor comprising a metal that comprises a water-soluble zinc sulfate
solution which
optionally also includes a second promoter metal compound, such as an iron
component in
the solute state selected from iron acetate, chloride, formate, nitrate,
sulfate, or a mixture
thereof. In one embodiment, the catalyst is prepared using a catalyst
precursor that comprises
a metal comprising a zinc sulfate aqueous solution.
[0068] In embodiments, at least a portion of the catalyst particles is
dispersed as particles
separate from the carbonaceous material during the pretreatment step, during
the step of
heating the carbonaceous material to a conversion temperature, or during the
conversion
process.
[0069] In embodiments, the catalyst is dissolved or otherwise suspended in the
liquid
phase, e.g., as fine particles, emulsified droplets, etc. The dispersed
catalyst can be added to
the coal before contact with the hydrocarbonaceous liquid, it can be added to
the
hydrocarbonaceous liquid before contact with the coal, or it can be added to
the coal-liquid
slurry. In some such embodiments, the dispersed catalyst is added in the form
of an
oil/aqueous solution emulsion of a water-soluble compound of the catalyst
hydrogenation
component. The water soluble salt of the catalytic metal can be essentially
any water soluble
salt of metal catalysts. The nitrate or acetate may be the most convenient
form of some
metals. Non- limiting active sources of zinc include zinc nitrate and zinc
acetate. Non-
limiting sources of iron are iron nitrate or iron acetate. In embodiments,
organometallic
complexes such as ferrocene are also employed as sources of iron. For
molybdenum,
tungsten or vanadium, a complex salt such as an alkali metal or ammonium
molybdate,
tungstate, or vanadate may be preferable. Mixtures of two or more metal salts
can also be
used. Particular salts are ammonium heptamolybdate tetrahydrate
[(NH4)6Mo7O24=4H20],
nickel dinitrate hexahydrate [Ni(N03)2.6H20], and sodium tungstate dihydrate
[NaWO4=2H20]. Any convenient process can be used to emulsify the salt solution
in the
hydrocarbon medium. The dispersed dissolution catalyst can also be an oil-
soluble
compound containing a catalytic metal, for example, ferrocene, phosphomolybdic
acid,
18
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
naphthenates of molybdenum, chromium, and vanadium, etc. Suitable oil-soluble
compounds
can be converted to dissolution catalysts in situ.
[0070] In embodiments, the particulate catalyst comprises zinc and a second
metal as an
unsupported catalyst, meaning that the components of the catalyst are not
associated with or
supported on inorganic carriers such as silica, alumina, magnesia, carbon,
etc. In other
embodiments, at least a portion of the metal components of the catalyst
composition are
associated with or supported on at least one inorganic carrier or binder. The
binder material
can comprise any materials that are conventionally utilized as binders in
hydroprocessing
catalysts. Suitable binder material includes, for example, silica, alumina
such as (pseudo)
boehmite, silica-alumina compounds, gibbsite, titania, zirconia, cationic
clays or anionic
clays such as saponite, bentonite, kaoline, sepiolite or hydrotalcite, or
combinations or
mixtures thereof. In one embodiment, one or more binder materials are selected
from silica,
colloidal silica doped with aluminum, silica-alumina, alumina, titanium,
zirconia, or a
mixture thereof. In another embodiment, the binder material comprises a
refractory oxide
material having at least 50 wt.% of titania, on an oxide basis. Any suitable
alumina binder
can be used in the catalyst preparation process. In one embodiment, the
alumina binder has a
surface area ranging from 100 to 400 m2/g, with a pore volume ranging from 0.5
to 1.5 m/g
measured by nitrogen adsorption. Similarly, any suitable titania binder can be
used in the
catalyst preparation process. In one embodiment, the titania of the binder has
an average
particle size of less than 50 microns (such as less than about 5 microns)
and/or greater than
0.005 microns. In another embodiment, the titania of the binder has a BET
surface area of 10
to 700 m2/g.
[0071] In some embodiments, the binder material is a binder that has undergone
peptization. In another embodiment, precursors of the binder materials are
used in the
preparation of the catalyst, wherein the precursor is converted into an
effective or functional
binder during the catalyst preparation process. Suitable binder material
precursors, in this
regard, include alkali metal aluminates (to obtain an alumina binder), water
glass (to obtain a
silica binder), a mixture of alkali metal aluminates and water glass (to
obtain a silica alumina
binder), a mixture of sources of a di-, tri-, and/or tetravalent metal such as
a mixture of water-
soluble salts of magnesium, aluminum and/or silicon (to prepare a cationic
clay and/or
anionic clay), chlorohydrol, aluminum sulfate, or a combination or mixture
thereof. In the
case of supported catalysts, the weight ratio of metal components (ie. zinc
and the second
metal components) to support components is in the range of 10:1 to 1:10.
19
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
[0072] In embodiments, at least a portion of the catalyst particles comprises
additional
components, such as catalyst promoters. Such promoters are selected from the
group
consisting of a non-noble Group VIII metal (such as Ni, Co, Fe), a Group VIB
metal (such as
Cr), a Group IVB metal (such as Ti), a Group IIB metal (such as Zn), a Group
IB metal (such
as Cu) and combinations and mixtures thereof.
[0073] During the conversion process, during which time the solid carbonaceous
material
contacted with the active sources of the catalyst composition and optionally
pretreated at a
temperature in the range of 100-350 C and then heated to conversion
temperature for
conversion of the carbonaceous material to liquid materials, the active
sources of the catalyst
are converted to their active forms. The conversion process is facilitated by
the addition of
sulfur to the catalyst.
[0074] Properly sulfided zinc species such as ZnS and zinc alkyl
dithiocarbamate, zinc
alkyl phosphorodithioate and sulfided metallic species such as MoS2, ammonium
tetrathiomolybdate, NiS, CoS, WS2, SnS, TiS2, CuS, FeS, Fe2S3, molt' alkyl
dithiocarbamate,
iron alkyl dithiocarbamate, titanium alkyl dithiocarbamate, iron alkyl
phosphorodithioate, can
be used directly as catalyst precursors without pre-sulfiding. For a non-
sulfided metal
precursor, including zinc-based zinc metal, zinc oxide, zinc acetate, zinc
nitrate, zinc sulfate
and other zinc salts, zinc minerals and zinc organo compounds; iron-based iron
metal, iron
oxide, ferrous sulfate, ferric nitrate and other iron salts, red mud and other
iron minerals,
ferrocene and other iron organo compounds, molybdenum-based, tungsten-based,
nickel-
based, cobalt-based, titanium-based, copper-based or tin-based metal, oxide,
salts, minerals
and organo compounds, etc., elemental sulfur or other sulfiding agent such as
DMDS, H2S,
CS2, and (NH4)2S can be used to pre-sulfide the catalyst precursor to form
metal sulfides or
the sulfiding agent is added directly during the hydroconversion run to
properly sulfide the
catalyst at the atomic ration of (S/(Zn+other metal))=1/1 to 10/1.
Alternatively, one or more
sulfur compounds can be added during, or subsequent to the pretreating step to
activate the
catalyst or catalyst precursor to its corresponding sulfided active catalyst.
The one or more
sulfur compounds can be introduced at any point of the system. Any suitable
amount of the
one or more sulfur compounds can be used in the context of the present
invention. In one
embodiment, one or more sulfur compounds are introduced into the pretreatment
zone
following the performance of the pretreatment process and before the
pretreatment
composition is delivered to the conversion zone. In another embodiment, one or
more sulfur
compounds are introduced into the conversion (i.e. liquefaction) zone. In one
embodiment, a
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
concentration of sulfur is introduced such that the atomic ration of sulfur to
metal in the
catalyst is from about 2:1 to about 10:1.
[0075] Any suitable sulfur compound may be used in this regard. In one
embodiment,
the sulfiding agent is hydrogen sulfide (H2S). In one embodiment, the
sulfiding agent is in the
form of a solution that under prevailing conditions is decomposable into
hydrogen sulfide,
present in an amount in excess of the stoichiometric amount required to form
the catalyst. In
another embodiment, the sulfiding agent is selected from the group of ammonium
sulfide,
ammonium polysulfide ((NH4)2sX), ammonium thiosulfate ((NH4)2s203), sodium
thiosulfate
(Na2s203), thiourea (CSN2H4), carbon disulfide (CS2), dimethyl disulfide
(DMDS), dimethyl
sulfide (DMS), tertiarybutyl polysulfide (PSTB), tertiarynonyl polysulfide
(PSTN), and
mixtures thereof. In another embodiment, the sulfiding agent is selected from
elemental
sulfur and sulfur containing hydrocarbons. In another embodiment, the
sulfiding agent is
selected from alkali- and/or alkaline earth metal sulfides, alkali- and/or
alkaline earth metal
hydrogen sulfides, and mixtures thereof. The use of sulfiding agents
containing alkali- and/or
alkaline earth metals may require an additional separation process step to
remove the alkali-
and/or alkaline earth metals from the spent catalyst.
[0076] Elemental sulfur may be added to the pretreatment composition in the
form of a
sublimed powder or as a concentrated dispersion (such as a commercial flower
of sulfur).
Allotropic forms of elemental sulfur, such as orthorhombic and monoclinic
sulfur, as also
suitable for use herein. In one embodiment, the one or more sulfur compounds
are in the
form of a sublimed power (flowers of sulfur), a molten sulfur, a sulfur vapor,
or a
combination or mixture thereof.
Other Additives
[0077] Any additional additives can be utilized during or subsequent to the
pretreating
step, such as, to enhance or facilitate the pretreatment process (such as by
enhancing,
facilitating, and/or enhancing dispersion of the catalyst or catalyst
precursor into the
carbonaceous material) and/or to enhance or facilitate hydroconversion of the
pretreated
carbonaceous material.
[0078] Any suitable surfactant can be utilized in the context of the
invention, such as
to improve dispersion, metal surface area, morphology, and/or other
characteristics of the
catalyst or catalyst precursor. Suitable surfactants include, for example, any
anionic
surfactant, zwitterionic surfactant, amphoteric surfactant, nonionic
surfactant, cationic
21
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
surfactant, or combination or mixture thereof. Suitable non-ionic surfactants
include, for
example, polyoxyethylenesorbitan monolaurate, polyoxyethylenated alkyphenols,
polyoxyethylenated alkyphenol ethoxylates, and the like. Suitable cationic
surfactants
include, for example, quartemary long-chain organic amine salts, quarternary
polyethoxylated long-chain organic amine salts, and the like, such as water-
soluble
cationic amines (e.g., cetyl trimethyl ammonium bromide, cetyl trimethyl
ammonium
chloride, dodecyl trimethyl ammonium amine, nonyl trimethyl ammonium chloride,
dodecyl phenol quaternary amine soaps, or combinations or mixtures thereof).
Suitable
anionic surfactants such as sodium succinate compounds, include, for example,
dioctyl
sodium sulfosuccinate or sodium bis(2-ethylhexyl)sulfosuccinate). Suitable
surfactants
can also comprise solvent materials having a high surface tension property,
such as
ethylene carbonate; benzophenone; benzyl cyanide; nitrobenzene; 2-
phenylethanol; 1,3-
propanediol; 1,4-butanediol; 1,5-pentanediol; diethyleneglycol;
triethyleneglycol;
glycerol; dimethyl sulfoxide; N-methyl formamide; N-methyl pyrrolidone; and
combinations and mixtures thereof. Suitable surfactants also include those
surfactants
having a high surface tension, such as N-methyl pyrrolidone. Other examples of
surfactants include acetonitrile, acetone, ethyl acetate, hexane, diethyl
ether, methanol,
ethanol, acetyl acetone, diethylcarbonate, chloroform, methylene chloride,
diethyl ketone,
and combination and mixtures thereof. In another embodiment, the surfactant
comprises
a nitrogen- or phosphorous-containing organic additive having a carbosulfide
phase with
enhanced catalytic activities. The amount of the N-containing / P-containing
organic
additive to be added generally depends on the desired activity of the final
catalyst
composition.
[00791 In another embodiment, the surfactant is an ammonium or phosphonium of
the
formula R1R2R3R4Q+, wherein Q is nitrogen or phosphorous, wherein at least one
of R1,
R2, R3, R4 is an aryl or alkyl group having 8-36 carbon atoms (e.g., C10H21,
C16H33,
C18H37, or a combination thereof), and wherein the remainder of R1, R2, R3, R4
is selected
from the group consisting of hydrogen, an alkyl group having 1-5 carbon atoms,
or a
combination thereof. Suitable such examples of surfactants include:
cetyltrimethylammonium, cetyltrimethylphosphonium,
octadecyltrimethylphosphonium,
cetylpyridinium, myristyltrimethylammonium, decyltrimethylammonium,
dodecyltrimethylammonium, dimethyldidbdecylammonium, or a combination or
mixture
22
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
thereof. The compound from which the above ammonium or phosphonium ion is
derived may be, for example, a hydroxide, halide, silicate, or combination or
mixture
thereof.
[00801 In one embodiment, the surfactant comprises a nitrogen-containing
organic
additive, such as aromatic amines, a cyclic aliphatic amines, a polycyclic
aliphatic
amines, or a combination or mixture thereof. In another embodiment, the
surfactant
comprises a nitrogen-containing organic additive is selected from compounds
containing
at least one primary, secondary, and/or tertiary amine group (such as
hexamethylenediamine, monoethanolamine, diethanolamine, triethanolamine, N,N-
dimethyl-N'-ethylethylenediamine, or a combination or mixture thereof); amino
alcohols
(such as, for example, 2 (2-amino ethyl amino)ethanol, 2 (2-aminoethoxy, or a
combination or mixture thereof) ethanol, 2-amino-l-butanol, 4-amino-l-butanol,
2,2-
diethoxyethylamine, 4,4-diethoxybutylamine, 6-amino-l-hexanol, 2-amino-1,3-
propanediol, 3-amino-1,2-propanediol, 3-amino-l-propanol, or a combination or
mixture
thereof); and amino alkoxy-silanes (such as, for example, 3-glycidoxypropyl)
trimethoxysilane, 3-(2-aminoethylamino) propyltrimethoxysilane, 3-
aminopropyl)trimethoxy-silane, or a combination or mixture thereof).
[00811 In another embodiment, the surfactant is an organic carboxylic acid
surfactant
or stabilizer. In one embodiment, for example, the surfactant is citric acid.
In another
embodiment, the surfactant is pentadecanoic acid, decanoic acid, or other
similar long
chain acids. In yet another embodiment, the surfactant is alginic acid.
[00821 The optional additives can be utilized at any suitable point prior to
or after the
pretreatment process and/or hydroconversion process. In one embodiment, one or
more
additives are combined with one or more of the carbonaceous material,
hydrocarbonaceous liquid, and one or more catalysts or catalyst precursors
prior to
pretreatment. In another embodiment, the additive(s) are combined with the
carbonaceous material, hydrocarbonaceous liquid, and catalysts or catalyst
precursors
during the pretreatment process. In another embodiment, the additive(s) are
combined
with the pretreated carbonaceous material following pretreatment and before
hydroconversion. In yet another embodiment, the additive(s) are combined with
the
pretreated carbonaceous material following during hydroconversion.
23
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
[0083] The additive(s) can be utilized in any suitable concentration. In one
embodiment, for example, the additive(s) are utilized in a concentration of
about 0.001 to
wt.% of the total pretreatment mixture. In another embodiment, the additive(s)
are
utilized in a concentration of about 0.005 to 3 wt.% of the total pretreatment
mixture. In
another embodiment, the additive(s) are utilized in a concentration of about
0.01 to 2
wt.% of the total pretreatment mixture. If the additive(s) are solely added to
the
hydroconversion feedstock, the amount to be added ranges from 0.001 to 0.05
wt. %
(such as about 0.005-0.01 wt.%) of the feed, or in any suitable concentration,
such as
described, for example, in Acta Petrolei Sinica, Vol. 19, Issue 4, pp. 36-44,
ISSN
10018719 and in Khimiya I Tekhnologiya Topilv I Masel, Issue 3, Year 1997, pp.
20-21,
ISSN 00231169, the contents of which are incorporated herein by reference in
their
entirety.
Mixing
[0084] Any suitable process or system can be used to combine and/or mix the
carbonaceous material with the hydrocarbonaceous liquid and the catalysts or
catalyst
precursors. In some embodiments, any suitable mixer is used to simultaneously,
successively,
and/or sequentially mix the carbonaceous material, hydrocarbonaceous liquid,
and the
catalyst or catalyst precursors in a manner suitable to form a homogenous or
heterogeneous
mixture (or slurry), as desired. In other embodiments, a mixer is utilized in
conjunction with
any suitable grinder (such as a hammer mill, a ball mill, a rod mill, or a
combination thereof,
or the like), such that at least a portion of the carbonaceous material is
ground, optionally in
the presence of the hydrocarbonaceous liquid and/or the one or more catalysts
or catalyst
precursors and mixed to form a homogenous or heterogeneous slurry, as desired.
In some
embodiments, the mixer and/or grinder comprises a gas delivery system for
providing an inert
or a reducing atmosphere (such as, for example, hydrogen, nitrogen, helium,
argon, syn-gas,
or any combination or mixture thereof) during mixing and/or grinding of the
carbonaceous
material, the hydrocarbonaceous liquid, and/or the catalyst or catalyst
precursors. In some
embodiments, the mixer and/or grinder are situated upstream of the
pretreatment system. In
other embodiments, the mixer and/or grinder form a portion of the pretreatment
system. In
embodiments, the catalyst precursor used in this process can be mixed directly
to ground coal
or other carbonaceous materials before feeding into the reactor, or added into
coal during coal
solvent grinding. The catalyst can be dissolved and sprayed onto coal or
impregnated onto
24
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
coal by incipient wetness using methanol/ethanol or water as
dissolving/wetting agent. The
catalyst can also be dispersed or soluble in the solvent that is then mixed
with coal.
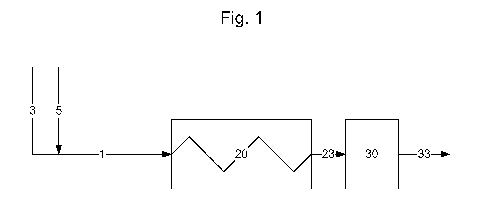
[0085] An embodiment of the invention is illustrated in Fig. 1. Coal feed 3,
with at least
50 wt% of the coal particles having a mean particle diameter of less than 0.5
inches, is
combined with catalytic material 5, comprising an active source of zinc and an
active source
of iron in a molar ratio of zinc to iron within the range of between 0.1/1 to
10/1, and the
combination 1 is passed to preheat furnace 20 for heating to a reaction
temperature in the
range of between 350 C and 500 C. The heated combination of coal and the
catalytic
material 23 leaving the preheat furnace is then passed to reaction zone 30 for
conversion of at
least a portion of the coal to liquid product 33.
[0086] Considering an exemplary process of the invention illustrated in Fig.
2, coal feed
103, with at least 50 wt% of the coal particles having a mean particle
diameter of less than
0.5 inches, is combined with catalytic material 105, comprising an active
source of zinc and
an active source of iron in the molar ratio of zinc to iron within the range
of between 0.1/1 to
10/1, and the combination 101 is passed to pretreatment zone 110 for
maintaining the
combination at a pretreatment temperature within the range of 100-350 C and
for a time of
between 5 and 600 minutes. Following pretreatment, the combination 113 is
passed to
preheat furnace 120 for heating to a reaction temperature in the range of
between 350 C and
500 C. The heated combination of coal and the catalytic material 123 leaving
the preheat
furnace is then passed to reaction zone 130 for conversion of at least a
portion of the coal to
liquid product 133.
[0087] Considering an exemplary process of the invention illustrated in Fig.
3, coal feed
203, with at least 80 wt% of the coal particles having a mean particle
diameter in the range of
50 microns to 500 microns, is passed to pretreatment zone 210. In a particular
exemplary
process, coal is supplied to pretreatment zone as a powder. In another
exemplary process,
coal is supplied as a slurry in a hydrocarbonaceous liquid, such as a coal
derived distillate
fraction.
[0088] A catalytic material 205, comprising an active source of zinc and an
active source
of iron in the molar ratio of zinc to iron within the range of between 3/1 and
1/3, is combined
with the coal particles in the pretreatment zone. In an embodiment, the zinc
is supplied to the
pretreatment zone as an aqueous solution or slurry of a zinc salt such as zinc
nitrate, zinc
chloride, zinc sulfate, zinc acetate, zinc sulfide, zinc oxide or zinc
carbonate. Iron is supplied
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
to the pretreatment zone as an aqueous solution or slurry of an iron salt such
as iron nitrate,
iron chloride, iron sulfate, iron acetate, iron sulfide, iron oxide or iron
carbonate. In another
embodiment, zinc and iron are added as organometallic compounds contained in a
liquid such
as a coal derived distillate fraction. Exemplary organometallic compounds
include zinc alkyl
dithiocarbamate and ferrocene. An active source of sulfur 207 is added to the
pretreatment
zone to supply a sulfur to catalytic metal atomic ratio within the range of
between 2/1 and
6/1. Hydrogen or a hydrogen containing gas 209 is further supplied to the
pretreatment zone
to maintain a pressure within the pretreatment zone within a range of between
atmospheric
pressure and 500 psig. In another embodiment, hydrogen or a hydrogen
containing gas is
supplied to the pretreatment zone to maintain a pressure within the
pretreatment zone within a
range of between 500 psig and 3500 psig. The materials in the pretreatment
zone are
maintained at a pretreatment temperature within the range of 180-220 C and for
a time of
between 5 and 600 minutes. Following pretreatment, the combination 213 is
passed to
preheat furnace 220 for heating to a reaction temperature in the range of
between 350 C and
500 C. The heated combination of coal and the catalytic material 223 leaving
the preheat
furnace is then passed to reaction zone 230 for conversion of at least a
portion of the coal to
liquid product 233.
[0089] Considering an exemplary process of the invention illustrated in Fig.
4, coal feed
303, with at least 50 wt% of the coal particles having a mean particle
diameter of less than
0.5 inches is passed to pretreatment zone 310. A catalytic material 307,
comprising an active
source of zinc and a catalytic material comprising an active source of iron
309 in the molar
ratio of zinc to iron within the range of between 0.1/1 to 10/1, are combined
with the coal
particles in the pretreatment zone, and the combination is maintained at a
pretreatment
temperature within the range of 100-350 C and for a time of between 5 and 600
minutes.
Following pretreatment, the combination 313 is passed to preheat furnace 320
for heating to a
reaction temperature in the range of between 350 C and 500 C. The heated
combination of
coal and the catalytic material 323 leaving the preheat furnace is then passed
to reaction zone
330 for conversion of at least a portion of the coal to liquid product 333.
Hydroconversion
[0090] The carbonaceous material is subjected to any suitable hydroconversion
and/or
liquefaction conditions to produce a product-enriched hydrocarbonaceous
material
comprising any desired liquid and/or gaseous products. The carbonaceous
material (such as
coal) is introduced into at least one hydroconversion zone wherein the
pretreated
26
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
carbonaceous material encounters suitable temperature, pressure, and additives
(such as
sulfur-containing compounds) to at least partially or substantially activate
the catalyst or
catalyst precursor of the pretreated carbonaceous material, and generate
liquid and/or gaseous
products. In one embodiment, for example, greater than about 50 wt.%, such as
about 55
wt.%, about 60 wt.%, about 65 wt.%, about 70 wt.%, about 75 wt.%, about 80
wt.%, about 85
wt.%, about 90 wt.%, about 95 wt.%, about 96 wt.%, about 97 wt.%, about 98
wt.%, or even
about 99 wt.% of the catalyst or catalyst precursor of the pretreated
carbonaceous material
becomes active catalyst, such that it possesses and/or exhibits
hydroconverting activity.
[0091] Suitable hydroconverting temperatures include, but are not limited to,
temperatures greater than about 350 C, such as greater than about 375 C, about
400 C, about
425 C, about 450 C, about 475 C, about 500 C. In some such embodiments, the
step of
hydroconverting the heated material is conducted at a temperature in the range
of between
350 C and 500 C. In some such embodiments, the heated material is reacted in
the
hydroconversion step for a time of at least 10 minutes.
[0092] Suitable hydroconverting pressures include, but are not limited to,
within the
range of 300-5000 psig (such as within the range of about 300-4800 psig, about
300-4600
psig, about 300-4400 psig, about 300-4200 psig, about 400-4000 psig, about 500-
3500 psig,
1000-3000 psig, 1200-2800 psig, 1400-2600 psig, or even about 1500-2600 psig)
of any
suitable gas such as a hydrogen containing gas (such as a hydrogen/methane
mixture, or a
hydrogen/carbon dioxide/water mixture) atmosphere and/or a syn-gas atmosphere.
In one
embodiment, in this regard, the pretreated carbonaceous material is suitable
for low or lower
pressure hydroconversion (such as a hydroconversion pressure less than about
2000 psig,
such as less than about 1800 psig, or even less than about 1600 psig).
Specifically, for
example, hydroconversion of the pretreated carbonaceous material can yield at
least about
10% higher (such as at least about 20%, about 40%, about 60%, about 80%, about
100%,
about 150%, about 200%, about 300%, or even at least about 400% higher liquid
product
yield at a hydroconversion pressure less than about 2000 psig (such as less
than about 1800
psig, or even less than about 1600 psig) than the same carbonaceous material
that has not
been pretreated. In another embodiment, hydroconversion of the pretreated
carbonaceous
material consumes about 10% less (such as about 20%, about 30%, about 40%,
about 50%,
about 60%, about 70%, about 80%, about 90%, or even about 100% less) hydrogen,
as
compared to the same carbonaceous material that has not been pretreated.
27
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
[0093] In embodiments, the hydroconversion of the solid carbonaceous material
is
accomplished by heating the solid carbonaceous material for a time sufficient
to form a liquid
product. In some such embodiments, the solid carbonaceous material is heating
in the
presence of at least one active source of zinc and at least one active source
of a second metal.
In some such embodiments, the solid carbonaceous material is heated at a
reaction
temperature of greater than 350 C and at a pressure in the range of 300 to
5000 psig. In some
such embodiments, the solid carbonaceous material is heated at a reaction
temperature in the
range of between 350 C and 500 C. In some embodiments, the solid carbonaceous
material
is heated for a time within the range of 5 minutes to 600 minutes.
[0094] In one embodiment, hydroconversion and/or liquefaction of the
carbonaceous
material occurs in a single reactor. In another embodiment, hydroconversion
and/or
liquefaction of the carbonaceous material occurs in two or more (such as a
plurality) of zones
or reactors for hydroconversion which may be arranged in any suitable manner
(such as in
parallel, or in series such that, for example, the temperature in each reactor
in series is
progressively higher and/or there is a commensurate increase in the hydrogen
partial pressure
in each downstream reactor). Preferably, hydroconversion and/or liquefaction
of the
pretreated carbonaceous material occurs in a reactor or zone that is separate
and/or distinct
from the pretreatment reactor or zone.
[0095] In one embodiment, hydroconversion and/or liquefaction of the
carbonaceous
material produces a liquid yield greater than about 50%, about 55%, about 60%,
about 65%,
about 70%, about 75%, about 80%, about 85%, about 87%, about 90%, about 95%,
or even
greater than about 99%. In an embodiment, pretreatment of the carbonaceous
material results
in a liquid yield that is at least about 10% higher (such as at least about
15%, about 20%,
about 25%, about 30%, about 35%, or even at least about 40% higher) than the
liquid product
yield of a similar carbonaceous material that is not pretreated prior to
hydroconversion. In
another embodiment, hydroconversion and/or liquefaction of the pretreated
carbonaceous
material produces a total conversion (such as of coal) greater than about 80%,
about 85%,
about 90%, about 95%, or even 99%.
[0096] In some embodiments, pretreatment of the carbonaceous material results
in a total
conversion (such as of coal) that is at least about 5% higher (such as at
least about 10%,
about 12%, about 14%, about 16%, about 18%, or even at least about 20% higher)
than the
conversion of a similar carbonaceous material that is not pretreated prior to
hydroconversion.
In other embodiments, hydroconversion and/or liquefaction of the pretreated
carbonaceous
28
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
material produces less than about 10% (such as less than about 8%, about 6%,
about 4%,
about 3%, about 2%, or even less than about 1%) of Ci-C3 gases.
Separation of Hydroconversion Products
[0097] The effluent from the hydroconversion zone can be fed into any suitable
one or
more separation zones. In one embodiment, the effluent is fed into a first
separation zone
wherein lighter products such as gases, naptha, and distillate are removed via
overhead lines.
Such a first separation zone can be run at a substantially atmospheric
pressure. A bottoms, or
high boiling, fraction of the effluent from the first separation zone can
optionally be recycled
to the hydroconversion reaction zone. All or some of the remaining effluent of
the first
separation zone can be passed to a second separation zone wherein it is
fractionated into a gas
oil fraction and a bottoms fraction. The bottoms fraction of the second
separation zone can be
passed to a third separation zone. A portion of the gas oil can be recycled to
the
hydroconversion zone. In this regard, any suitable high pressure, medium
pressure, and low
pressure separators can be used in the context of the present invention.
Recovery of Catalyst or Catalyst Precursor
[0098] The one or more separation systems or zones can be followed by one or
more
catalyst and/or metal recovery systems or zones in which at least a portion
(such as one or
more metals) of the catalyst and/or catalyst precursor is recovered from one
or more portions
or fractions of the hydroconverted carbonaceous material. In one embodiment,
metal from a
metal-containing catalyst and/or metal-containing catalyst precursor is
recovered in the
recovery system from a solids fraction (such as a residual solids fraction) of
the
hydroconverted carbonaceous material that was separated and/or collected in
the separation
system (and which may include ash).
[0099] The recovery system can be operated at any suitable temperature, such
as at a
temperature of about 1200-1900 C, such as about 1300-1800 C, or even 1400-1700
C. In
one embodiment, the recovery system provides an atmosphere of air that is
suitable to cause
spent catalysts (such as molybdenum sulfides) to be oxidized and sublimated to
Mo03, in the
case where the metal is molybdenum, such as described in U.S. Pat. App. Ser.
No.
60/015,096, filed December 19, 2007, the contents of which are incorporated by
reference in
their entirety. The treated spent catalyst, catalyst precursor, and/or
recovered metal can be
collected and passed from the catalyst recovery zone to a catalyst or catalyst
precursor
preparation zone.
29
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
Catalyst or Catalyst Precursor Preparation
[0100] The one or more recovery systems can be followed by one or more
catalyst or
catalyst precursor preparation systems, in which at least a portion of the
catalyst or catalyst
precursor (such as metal of the catalyst precursor) recovered in the recovery
system is reacted
to form a catalyst or catalyst precursor (such as the same catalyst or
catalyst precursor that
was originally used to pretreat the carbonaceous material).
[0101] In one embodiment, for example, a recovered metal of the catalyst or
catalyst
precursor (such as MoO3) is reacted with a sulfur compound (such as ammonium
sulfide) to
form ammonium tetrathiomolybdate catalyst precursor. The resulting formed
catalyst or
catalyst precursor can then be delivered, optionally in combination with new
or fresh catalyst
precursor, into the pretreatment system and/or the hydroconversion system.
Characterization of the Catalyst
[0102] In embodiments, a catalyst which is active for converting at least a
portion of the
solid carbonaceous material to a liquid product, having the formula
(RP)i(Mt)a(L )b(S )d(CW)e(HX)f(Oy)g(NZ)h and having improved morphology and
dispersion
characteristics, can be characterized using techniques known in the art,
including elemental
analysis, Surface Area analysis (BET), Particle Size analysis (PSA), Powder X-
ray
Diffraction (PXRD), Scanning Electron Microscopy (SEM), Energy Dispersive X-
ray
Analysis (EDS), and other methods. In one method, electron microscopy is used
to
complement the x-ray diffraction study. In another method, the surface area of
the catalyst
is determined using the BET method. In yet another method, scanning tunneling
microscopy
(STM) and density functional theory (DFT) can be used to characterize the
catalyst.
[0103] The catalyst of the formula (RP)i(Mt)a(L )b(S )d(CW)e(HX)J(Oy)g(Nz)h is
characterized as giving excellent conversion rates in the upgrades of coal
depending on the
configuration of the upgrade process and the concentration of the catalyst
used. In one
embodiment, the slurry catalyst provides conversion rates of at least 70% in
one embodiment,
at least 75% in a second embodiment, at least 80% in a third embodiment, and
at least 90% in
a fourth embodiment. In one embodiment of a coal upgrade system employing the
catalyst of
the formula (RP);(Mt)a(L )b(S )d(CW)e(HX)f(Oy)g(Nz)h, at least 98 wt. % of
coal feed is
converted to lighter products. In a second embodiment, at least 98.5% of coal
feed is
converted to lighter products. In a third embodiment, the conversion rate is
at least 99%. In
a fourth embodiment, the conversion rate is at least 95%. In a fifth
embodiment, the
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
conversion rate is at least 80%. As used herein, conversion rate refers to the
conversion of
coal feedstock to less than 1200 F (650 C) boiling point materials.
[0104] In one embodiment, the catalyst has a pore volume in the range of from
0.05 to
5.0 mug as determined by nitrogen adsorption. In a second embodiment, the pore
volume is
in the range of from 0.1 to 4.0 ml/g, such as from 0.1 to 3.0 ml/g or from 0.1
to 2.0 ml/g.
[0105] In embodiments, the catalyst has a surface area of at least 5 m2/g, or
at least
10m2/g, or at least 50 m2/g, or greater than 100 m2/g, or greater than 200
m2/g , as determined
via the B.E.T. method. In embodiments, the catalyst is characterized by
aggregates of
crystallites of 10 to 20 angstrom, for an overall surface area greater than
100 m2/g.
[0106] In embodiments, the catalyst has a particle size ranging from nanometer
to
micrometer ( m) size dimensions. Exemplary suspended catalyst particles have a
median
particle size of 0.0005 to 1000 microns, or a median particle size of 0.001 to
500 microns, or
a median particle size of 0.005 to 100 microns, or a median a particle size of
0.05 to 50
microns. In embodiments, the catalyst in the form of a suspension that is
characterized by a
median particle size of 30 nm to 6000 nm. In embodiments, the catalyst has an
average
particle size in the range of 0.3 to 20 m.
[0107] In embodiments, the catalyst comprises catalyst particles of molecular
dimensions
and/or extremely small particles that are colloidal in size (i.e., less than 1
micrometer or less
than 0.1 micrometer or in the range of 0.1 to 0.001 micrometer). In some
embodiments, the
catalyst is dispersed on the coal surface in 1 to 100 nanometer particles by
impregnation of
the catalyst precursor on the coal. In some embodiments, the catalyst forms a
slurry catalyst,
in a hydrocarbon diluent, having "clusters" of the colloidal particles, with
the clusters having
an average particle size in the range of 1 - 100 micrometers.
[0108] As is further illustrated in the following examples, the systems and
processes
described herein can be used to achieve optimization and efficiency in the
production of any
desired proportions (or yield percentages) of liquid and/or gas products
having a variety of
desired properties. Specifically, a full range of hydroconversion products can
be
accomplished under a variety of hydroconversion conditions (such as at low
hydrogen
pressure and/or with short duration) through selection of any of a variety of
combinations of
hydrocarbonaceous liquid, catalysts and/or catalyst precursors, as well as
pretreatment and
hydroconversion conditions. In this manner, the systems and process offers
tremendous
flexibility to a user in being able to achieve desired hydroconversion
products from any solid
carbonaceous material using any of a variety of different combinations of
hydrocarbonaceous
31
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
liquid, catalyst, and/or catalyst precursor, as well as pretreatment and
hydroconversion
conditions.
EXAMPLE S
Example 1
[0109] Run 1 - A solution of a mixed catalyst precursor iron nitrate
(Fe(N03)3.9H20)
and zinc nitrate (Zn(N03)2 6H20) dissolved in methanol was prepared. A sample
of
moisture-free coal feed (i.e. less than 1 % by weight water) having a particle
size of less than
100 mesh was impregnated to incipient wetness with the solution, at a solution
to coal weight
ratio of 1 to 1, to yield an iron to coal loading on a dry, ash-free basis of
1% iron and a zinc to
coal loading on a dry, ash-free basis of 1 wt% zinc. The catalyst impregnated
coal was then
dried under nitrogen at 105 C for up to 24 hr for to remove the methanol.
[0110] The dried catalyst impregnated coal was mixed with an FCC-type process
oil
(500 F+ cut) hydrocarbonaceous liquid, at a hydrocarbonaceous liquid to coal
ratio of 1.6 to
1. Elemental sulfur was added to sulfide the iron and zinc, at a sulfur to
iron molar ratio of 2
to 1 and a sulfur to zinc ratio of 2 to 1.
[0111] The mixture was then heated quickly in a vessel to 200 C, and held at
200 C for 2
hours, while the hydrogen partial pressure within the vessel increased from
about 100 psia to
about 1000 psia. The mixture was then further heated to 430 C, and then held
at 430 C for 3
hours under a hydrogen partial pressure of 2500 psia.
[0112] After 3 hours the reaction vessel containing the sulfided solvent and
coal mixture,
hydrogen and any reaction products was quenched to room temperature. Product
gases (CO,
C02, C1, C2 and C3) were vented through a wet test meter to determine the gas
yield. Solids,
primarily unconverted coal, ash and catalyst sulfide were separated from
liquid products
(C4+) by filtration. Coal conversion was determined as follows:
Coal conversion = (solids recovered - (ash in coal + recovered catalyst))/coal
feed
By subtracting the solvent added at the beginning of the run, oil yield was
determined based
on coal (dry, ash-free basis). Product yields are tabulated as Run 1 in Table
I.
[0113] Run 2 - Example 1 was repeated using iron sulfate (FeSO4.7H20) and zinc
nitrate
(Zn(N03)2.6H20) mixed catalyst precursor at an iron to coal loading on a dry,
ash-free basis
of 1 wt% iron, and a zinc to coal loading on a dry, ash-free basis of 1 wt%
zinc.. Elemental
32
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
sulfur was added to sulfide the iron and zinc, at a sulfur to iron molar ratio
of 2 to 1 and a
sulfur to zinc ratio of 2 to 1. Product yields are tabulated as Run 2 in Table
I.
[0114] Run 3 - Example 1 was repeated using zinc nitrate (Zn(NO3)2.6H2O) as a
portion
of the mixed catalyst precursor, to yield an zinc to coal loading on a dry,
ash-free (dry, ash-
free) basis of I% zinc. Ferrocene was further added into the solvent coal
mixture at a iron to
coal loading on a dry, ash-free basis of 3% iron.
[0115] Product yields are tabulated as Run 3 in Table I. The results set forth
is Table II
show excellent coal conversation and liquid yield for runs 1, 2 and 3 of
Example 1.
[0116] Detailed product distributions for runs 1, 2 and 3 in Table I are
illustrative of
distributions used to calculate the coal conversion. The liquid product
distribution was
determined by subtracting the simulated distillation result (or boiling curve)
of the
hydro carbonaceous liquid from the simulated distillation result (or boiling
curve) of the
mixture of hydrocarbonaceous liquid and liquid product.
Example 2
[0117] Run 4 - Run 1 was repeated. Product yields tabulated for Run 4 in Table
I
indicate excellent coal conversions and liquid yield for Run 4.
Example 3
[0118] Run 5 - Run 1 was repeated using a iron nitrate (Fe(NO3)3.9H2O) and
zinc nitrate
(Zn(NO3)2.6H2O) mixed catalyst precursor at an iron to coal loading on a dry,
ash-free basis
of 1 wt% iron, and a zinc to coal loading on a dry, ash-free basis of 1 wt%
zinc. No
elemental sulfur was added. Product yields tabulated for Run 5 in Table I
indicate relatively
poor coal conversation and liquid yield.
Example 4
[0119] Run 6 - Run 1 was repeated with iron sulfate (FeSO4.7H2O) dissolved in
methanol as the only catalyst precursor at an iron to coal loading on a dry,
ash-free basis of
1 % iron. Elemental sulfur was added to the solvent and coal mixture at a
sulfur to iron molar
ratio of 2 to 1 to sulfide the iron catalyst. Product yields tabulated as Run
6 in Table I
indicate moderate coal conversion and liquid yield.
33
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
Example 5
[0120] Run 7 - Run 4 was repeated with zinc nitrate (Zn(N03)2.6H2O) dissolved
in
methanol as the only catalyst precursor at a zinc to coal loading on a dry,
ash-free basis of I%
zinc. Elemental sulfur was added to the solvent and coal mixture at a sulfur
to zinc molar
ratio of 2 to 1. The reaction temperature was 430 C rather than 440 C. Product
yields
tabulated as Run 7 in Table I indicate poor coal conversion and poor liquid
yield.
Example 6
[0121] Run 8 - Iron nitrate (Fe(N03)3.9H2O) and zinc nitrate (Zn(N03)2.6H2O)
were
dissolved in de-ionized water. Coal was then dispersed into the aqueous
solution, which was
then titrated drop-wide by base solution of NaOH. Following the titration, the
mixture was
aged at 40 C for 5 hours under a flowing air purge. After aging, the mixture
was centrifuged
and triple rinsed with a large quantity of water to remove excess base. The
resulting mixture
was dried under nitrogen atmosphere at 105 C.
[0122] The reaction mixture was prepared with an FCC-type process oil (500 F+
cut)
hydrocarbonaceous liquid, at a hydrocarbonaceous liquid to coal ratio of 1.6
to 1. Elemental
sulfur (sulfur to iron atomic ratio=2 to 1 plus sulfur to zinc atomic ratio=2
to 1) was added to
sulfide the active metals in-situ.
[0123] The mixture was then heated quickly in a vessel to 200 C, and held at
200 C for 2
hours, while the hydrogen partial pressure within the vessel increased from
about 100 psia to
about 1000 psia. The mixture was then further heated to 430 C, and then held
at 430 C for 3
hours under a hydrogen partial pressure of 2500 psia. Product yields tabulated
for Run 5 in
Table I indicate relatively poor coal conversation and liquid yield. Product
yields tabulated
for Run 8 in Table I indicate relatively poor coal conversation and liquid
yield.
[0124] Run 9 - Run 6 was repeated, but with sulfur being omitted from the
catalyst
preparation. Product yields are tabulated as Run 9 in Table I. Product yields
tabulated for
runs 8 and 9 in Table I indicate a poor coal conversion and liquid yield.
Example 7
[0125] Run 10 - Run 8 was repeated by dissolving iron nitrate (Fe(N03)3.9H20 )
alone in
de-ionized water, with coal at an iron to coal loading on a dry, ash-free coal
basis of I% iron.
Product yields tabulated as Run 10 in Table I indicate moderate coal
conversion and liquid
yield.
34
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
Example 8
[0126] Run 11 - Iron sulfide (FeS) was dispersed in methanol and the
dispersion mixed
with coal, having a particle size of less than 100 mesh, at a solution to coal
weight ratio of 1
to 1, to yield an iron to coal loading on a dry, ash-free (daf) basis of 1%
iron. The mixture
was then dried under nitrogen at 105 C for up to 24 hr for to remove the
methanol.
[0127] The dried catalyst impregnated coal was mixed with an FCC-type process
oil
(500 F+ cut) hydrocarbonaceious liquid, at a hydrocarbonaceous liquid to coal
ratio of 1.6 to
1. Elemental sulfur was added to the solvent and coal mixture at a sulfur to
iron molar ratio
of 2 to 1 to sulfide the iron catalyst.
[0128] The mixture was then heated quickly in a vessel to 200 C, and held at
200 C for 2
hours, while the hydrogen partial pressure within the vessel increased from
about 100 psia to
about 1000 psia. The mixture was then further heated to 440 C, and then held
at 440 C for 3
hours under a hydrogen partial pressure of 2500 psia. Product yields are
tabulated as Run 11
in Table I.
[0129] Run 12 - Run 11 was repeated using a mixture of iron sulfide and zinc
sulfide at a
iron to coal loading of 0.56% iron and a zinc to coal loading of 1% zinc. The
results set forth
in Table I indicate poor coal conversion and liquid yield for both Run 11 and
Run 12.
Example 9
[0130] Run 13 - A sample of moisture-free coal feed (i.e. less than 1 % by
weight water)
having a particle size of less than 100 mesh was mixed with an FCC-type
process oil (500 F+
cut) as solvent, at a solvent to coal ratio of 1.6 to 1. Iron (III)
dimethyldithiocarbamate and
zinc diethyldithiocarbamante was blended in the solvent coal mixture to yield
an iron to coal
loading on a dry, ash-free (daf) basis of 1% iron and a zinc to coal loading
on a dry, ash-free
(daf) basis of 1% zinc. No additional sulfur was added.
[0131] The mixture was then heated quickly in a vessel to 200 C, and held at
200 C for 2
hours, while the hydrogen partial pressure within the vessel increased from
about 100 psia to
about 1000 psia. The mixture was then further heated to 430 C, and then held
at 430 C for 3
hours under a hydrogen partial pressure of 2500 psia.
[0132] The results set forth in Table I show excellent coal conversions and
liquid yield
for Run 13.
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
Example 10
[0133] Run 14 - A solution of the mixed catalyst precursor iron nitrate
(Fe(N03)3.9H20 )
and zinc nitrate (Zn(N03)2.6H20) dissolved in methanol was prepared. A portion
of coal
representing 20% of the total coal sample (moisture-free coal feed (i.e. less
than 1 % by
weight water) having a particle size of less than 100 mesh) was selected. The
coal portion
was impregnated to incipient wetness with the solution, at a solution to coal
weight ratio of 1
to 1. The catalyst impregnated coal portion was then dried under nitrogen at
105 C for up to
24 hr for to remove the methanol. The remaining 80% of the total coal sample
was mixed
with the dried portion, to yield an iron to coal loading on a dry, ash-free
(daf) basis of I%
iron and a zinc to coal loading on a dry, ash-free (daf) basis of I% zinc.
[0134] The dried coal sample was mixed with an FCC-type process oil (500 F+
cut) as
solvent, at a solvent to coal ratio of 1.6 to 1. Elemental sulfur was added to
sulfide the iron
and zinc, at a sulfur to iron molar ratio of 2 to 1 and a sulfur to zinc ratio
of 2 to 1.
[0135] The mixture was then heated quickly in a vessel to 200 C, and held at
200 C for 2
hours, while the hydrogen partial pressure within the vessel increased from
about 100 psia to
about 1000 psia. The mixture was then further heated to 440 C, and then held
at 440 C for 3
hours under a hydrogen partial pressure of 2500 psia.
[0136] Product yields set forth in Table I indicate good coal conversion and
liquid yield.
Example 11
[0137] Run 15 - Moisture-free coal feed (i.e. less than 1 % by weight water)
having a
particle size of less than 100 mesh was dispersed in I% dioctyl sulfosaccinate
sodium
aqueous solution, following by addition of iron nitrate (Fe(N03)3.9H20 ) and
zinc nitrate
(Zn(N03)2.6H20) catalyst precursors, to yield an iron to coal loading on a
dry, ash-free
(daf) basis of 1% iron and a zinc to coal loading on a dry, ash-free (daf)
basis of 1% zinc.
The catalyst impregnated coal portion was then dried under nitrogen at 105 C
for up to 24 hr
for to remove the water.
[0138] The dried coal sample was mixed with an FCC-type process oil (500 F+
cut) as
solvent, at a solvent to coal ratio of 1.6 to 1. Elemental sulfur was added to
sulfide the iron
and zinc, at a sulfur to iron molar ratio of 2 to 1 and a sulfur to zinc ratio
of 2 to 1.
[0139] The mixture was then heated quickly in a vessel to 200 C, and held at
200 C for 2
hours, while the hydrogen partial pressure within the vessel increased from
about 100 psia to
36
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
about 1000 psia. The mixture was then further heated to 440 C, and then held
at 440 C for 3
hours under a hydrogen partial pressure of 2500 psia.
[0140] Product yields set forth in Table I indicate good coal conversion and
liquid yield.
Example 12
[0141] Run 16 - A solution of a mixed catalyst precursor iron nitrate
(Fe(N03)3.9H20)
and zinc nitrate (Zn(N03)2.6H20) dissolved in de-ionized water was prepared. A
sample of
moisture-free coal feed (i.e. less than 1 % by weight water) having a particle
size of less than
100 mesh was dispersed in the aqueous solution, which was treated by NH4OH
drop-wise.
Following the titration, the mixture was aged at 40 C for 5 hours in flowing
air. After aging,
the mixture was centrifuged and triple rinsed with a large quantity of water
to remove excess
base. The resultant mixture was dried under nitrogen atmosphere at 105 C.
[0142] The dried catalyst impregnated coal was mixed with an FCC-type process
oil
(500 F+ cut) as solvent, at a solvent to coal ratio of 1.6 to 1. Elemental
sulfur was added to
sulfide the iron and zinc, at a sulfur to iron molar ratio of 2 to 1 and a
sulfur to zinc ratio of 2
to 1.
[0143] The mixture was then heated quickly in a vessel to 200 C, and held at
200 C for 2
hours, while the hydrogen partial pressure within the vessel increased from
about 100 psia to
about 1000 psia. The mixture was then further heated to 440 C, and then held
at 440 C for 3
hours under a hydrogen partial pressure of 2500 psia.
[0144] Product yields set forth in Table I indicate good coal conversion and
liquid yield.
Example 13
[0145] Run 17 - Run 1 was repeated using ammonium sulfate ((NH4)2SO4) and
ammonium nitrate (NH4NO3) dissolved in methanol in place of a catalyst
precursor at an NH3
to coal loading on a dry, ash-free basis of 1 wt% NH3 from ammonium sulfate,
and an NH3 to
coal loading on a dry, ash-free basis of 1 wt% NH3 from ammonium nitrate.
Elemental sulfur
was added to the solvent and coal mixture to provide 1.14% by weight of sulfur
on the coal.
The results set forth in Table I indicate that this comparative run without
catalyst had poor
coal conversion and low liquid yield.
37
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
Table I
Example Run Liquid Coal Gas Yield (%)
Yield (%) Conversion (%)
1 1 78.0 97.5 14.4
1 2 77.4 97.2 18.3
1 3 77.9 97.4 14.5
2 4 73.5 96.4 14.0
3 5 57.2 93.5 15.0
4 6 66.6 90.7 19.1
7 58.0 90.2 17.5
6 8 55.0 93.0 -----
6 9 56.2 92.3 20.0
7 10 62.5 94.4 19.6
8 11 47.6 92.3 20.9
8 12 44.7 91.1 20.0
9 13 72.7 96.0 14.6
14 67.5 94.3 18.5
11 15 73.8 97.0 14.9
12 16 71.5 93.9 12.6
13 17 33.7 68.0 17.6
38
CA 02781416 2012-05-18
WO 2011/066270 PCT/US2010/057773
Table II
Run 1 2 3
Coal conversion, (%) dry, ash-free basis 97.2 97.5 97.4
Yield (% dry, ash-free coal)
CI-C3 9.2 5.7 6.8
Total gas 18.3 14.4 14.5
C4-350 F 4.8 4.6 6.4
350-650 F 40.4 40.5 35.7
650-850 F 24.6 24.7' 27.8
C4-850 F 69.8 69.8 69.9
850-1000 F 5.9 6.2 6.4
>1000 F 1.7 2.0 1.6
H2O 8.2 11.8 9.0
H2 consumption 6.7 6.7 4.0
C4-850 F liquid bbl/ton dry, ash-free coal 4.9 4.9 4.9
H2 consumption scf/bbl 5432 5432 3239
Hexane insoluble (%dry, ash-free coal) 21.2 19.4 19.5
MCR (% dry, ash-free coal) 4.2 3.0 3.4
Detailed carbon elemental mass balance
C feed from coal 26.40 26.76 26.51
C in the product liquid 22.03 22.16 22.33
C in the product gas 2.71 3.35 2.4
C in the product solid 1.47 1.32 1.29
C out 26.21 26.83 26.01
39