Note: Descriptions are shown in the official language in which they were submitted.
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
PATENT COOPERATION TREATY APPLICATION
ZERO VALENT IRON/ IRON OXIDE MINERAL/FERROUS IRON COMPOSITE FOR
TREAMENT OF A CONTAMINATED FLUID
INVENTOR:
Yongheng Huang
Citizen of the People's Republic of China
Filed Via EFS on September 20, 2010
CA 02787032 2016-01-18
WO 20-11/O35263 PCl/US211101049528
ZERO VALENT IRON/ IRON OXIDE MINERAL/FERROUS IRON COMPOSITE FOR
TREAMENT OF A CONTAMINATED FLUID
FIELD OF THE INVENTION
[0003] The present invention relates to remediation of contaminated fluids.
BACKGROUND
[0004] It is common to treat various sources of liquids in order to remove
contaminants.
Examples of sources of liquids for treatment include surface water, ground
water, and industrial
waste streams. Industrial waste stream refers to liquid streams of various
industrial processes.
An industrial waste stream may be produced at any stage of a process. The
waste stream may be
wastewater, which herein refers to a primarily water-based liquid stream.
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
[0005] Wastewater treatment is one of the most important and challenging
environmental
problems associated with coal-based power generation. Using wet scrubbers to
clean flue gas is
becoming more popular worldwide in the electrical power industry. In the
coming years,
hundreds of wet scrubbers will be installed in the US alone. While wet
scrubbers can greatly
reduce air pollution, toxic metals in the resulting wastewater present a major
environmental
problem. The industry prepares to invest billions of dollars in the next
decade to meet the ever
more stringent environmental regulations; unfortunately, a cost-effective and
reliable technology
capable of treating such complicated wastewater is still not available.
[0006] The compositions of FGD wastewaters vary greatly, depending not
only on the
types of coal and limestone used but also on the types of scrubber and
processes used.
Pretreatment method and management practices also affect wastewater
characteristics.
According to a recent survey by EPRI (2006), untreated raw FGD wastewater
could have TSS in
¨10,000 mg/L but after settlement, it falls to ¨10 mg/L; the pH typically
falls in 5.8-7.3; sulfate
is in the range of 1,000-6,000 mg/L; nitrate-N at level of 50 mg/L is not
uncommon; chloride,
alkalinity and acidity vary from hundreds to thousands ppm; selenium exists in
various forms,
ranging from dozens of ppb to over 5 ppm, among which, selenate could account
for more than
half of total Se; arsenic ranges from a few ppb to hundreds of ppb; mercury
ranges from below 1
ppb to hundreds of ppb; and boron can be as high as hundreds of ppm.
[0007] It is desirable, for example, to remove selenium from wastewater.
Treatment of
selanate-Se in wastewater is often considered to be one of the most difficult
in toxic metal
treatments. Selenium is a naturally occurring chemical element in rocks, soils
and natural
waters. Although Se is an essential micronutrient for plants and animals, it
can be toxic at
elevated levels and some of Se species may be carcinogenic. The hexavalent
selenium is stable
in oxic environments and exists as the selenate (5e042-) anion, which is
weakly sorbed by
mineral materials and generally soluble. Tetravalent Se is the stable valence
state under mildly
reducing or anoxic condition (0.26 V < Eh < 0.55 V at pH 7). It exists as the
selenite (Se032-)
anion, which tends to be bound onto mineral surfaces (e.g., Fe and Mn oxides).
Selenate and
3
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
selenite are more toxic due to their high bioavailability than elemental
selenium or metallic
seleni des.
[0008] Further, for example, it is desirable to remove mercury from
wastewater. In
particular, the future EPA guideline for total mercury is < 12 part per
trillion (ppt) or ng/L.
Metal sulfide chemistry is well understood and has been used in various ways
in water treatment
system to achieve reduction of dissolved toxic metals from water. For example,
organosulfide
has been used as a water treatment reagent to precipitate Hg and other toxic
metals in water
industry. Iron sulfide materials (FeS or FeS2 ores) have been used as
adsorbent for toxic metals
removal. Conventional sulfide-based toxic metal removal technology has not
been able to
achieve the desired mercury removal level in many applications. For example,
direct application
of organosulfide has been found unable to achieve Hg removal below 12 ppt in
the treated
effluent that is required by the new federal or local EPA guideline.
[0009] A biological treatment system, ABMet, has been patented and is being
marketed
by GE Water.
[0010] However, there remains a need for a cost-effective and reliable
treatment process
for removing a contaminant from an aqueous fluid.
SUMMARY
[0011] The present inventor has discovered a novel composition, system, and
process for
treating an aqueous fluid so as to reduce the concentration of a contaminant.
The composition,
system, and process are robust, flexible, and based on cost-effective
materials. The present
inventor has developed a chemical treatment process that can cost-effectively
treat all major
pollutants in the flue gas desulfurization (FGD) wastewater in a single
process.
[0012] The present inventor developed a fluidized reacting system using a
hybrid
reactive solid/secondary reagent reactor that can cost-effectively remove many
toxic metals from
wastewater. The system and process are effective to treat an aqueous
suspension. The system
uses a reactive solid and a secondary reagent as reactive agents to rapidly
reduce selenate to
become insoluble selenium species, which are then adsorbed or precipitated
along with various
4
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
of other toxic metals (such as As and Hg, if present) in wastewater onto the
iron oxide sludge.
The system is particularly effective for removing selenate-Se.
[0013] According to various embodiments, a composition, system, and process
involve a
composite for removing a contaminant from a fluid stream, where the composite
comprises zero
valent iron, an iron oxide mineral, and ferrous iron, wherein the ferrous iron
is disposed so as to
facilitate maintenance of the iron oxide mineral, and wherein the composite is
active for
removing the contaminant from the fluid stream.
[0014] The present process is effective for removing almost all toxic
metals in an
aqueous suspension; in addition, it can remove oxyanion pollutants and
metalloids. More
particularly, contaminants removable by the present system and process are:
most toxic metals
such as arsenic, mercury, selenium, cobalt, lead, cadmium, chromium, silver,
zinc, nickel,
molybdenum, and the like; metalloid pollutants such as boron and the like;
many oxyanion
pollutants, such as nitrate, bromate, iodate, and periodate, and the like, and
combinations thereof.
[0015] The present system and process use common, non-toxic, and
inexpensive
chemicals. The present chemical treatment system costs much less to construct
and operate than
biological treatment systems, which tend to be more complex.
[0016] The present system and process are versatile and flexible. The
present system and
process are more robust and manageable than a biological process when exposed
to toxic
chemicals or any disturbances and changes in wastewater quality and quantity.
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
BRIEF DESCRIPTION OF DRAWINGS
[0017] The foregoing summary as well as the following detailed description
will be
better understood when read in conjunction with the appended drawings. It
should be
understood, however, that the present invention is not limited to the precise
arrangements and
instrumentalities shown herein. The components in the drawings are not
necessarily to scale,
emphasis instead being placed upon clearly illustrating the principles of the
present invention.
Moreover, in the drawings, like reference numerals designate corresponding
parts throughout the
several views.
[0018] The invention may take physical form in certain parts and
arrangement of parts.
For a more complete understanding of the present invention, and the advantages
thereof,
reference is now made to the following descriptions taken in conjunction with
the accompanying
drawings, in which:
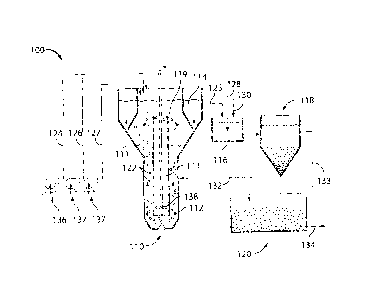
[0019] Figure 1 is a schematic illustrating a single-stage fluidized bed
reactor;
[0020] Figure 2 is a flow chart illustrating a three-stage reaction system;
and
[0021] Figure 3 is a schematic illustrating a single-stage fluidized bed
ZVI/Fe0x/Fe(II);
[0022] Figure 4 is a flow-chart of a hybrid ZVI/Fe0x/Fe(II) prototype
treatment system
incorporating a sulfide generator;
[0023] Figure 5 is a schematic illustrating treatment of groundwater;
[0024] Figures 6A, 6B, 6C, and 7 are SEM micrographs of ZVI/Fe0x/Fe(II)
particles;
[0025] Figure 8 shows a cartoon of formation of particles with and without
Fe2+;
[0026] Figure 9 illustrates an iron corrosion model of ZVI/Fe0x/Fe(II)
particles;
[0027] Figure 10 is schematic of batch testing of ZVI/Fe0x/Fe(II)
particles.
[0028] Figure 11 shows data illustrating removal of selenate-Se from FGD
wastewater by
a treatment system containing ZVI/Fe0x/Fe(II) particles; and
[0029] Figure 12 shows data illustrating removal of total mercury over time
from FGD
wastewater by a treatment system containing ZVI/Fe0x/Fe(II) particles.
6
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
DETAILED DESCRIPTION
[0030] In the present description and claims, it will be understood that
all numbers are
approximate within convention as understood by one of ordinary skill in the
art. That is, for
example, 9 is to be understood as about 9, and likewise for other numbers.
[0031] The present inventor has discovered a novel system for treating
wastewater. It
will be understood that wastewater is illustrative of an aqueous fluid. For
example, the present
inventor contemplates treating oil refinery waste. Further, the present
inventor contemplates
treating wetlands. The aqueous fluid may be a suspension.
[0032] Experiments have demonstrated the system operable for removal of
selenium
present as selenate. It will be understood that selenium is illustrative of a
toxic material. Other
common toxic materials are contemplated. For example, the present inventor
contemplates
removing metals such as arsenic, mercury, cobalt, lead, cadmium, chromium,
silver, zinc, nickel,
molybdenum, and the like; metalloid pollutants such as boron and the like; and
many oxyanion
pollutants, such as nitrate, bromate, iodate, and periodate, and the like. It
will be understood that
toxic materials are illustrative of contaminants.
[0033] According to some embodiments, a reactor system includes a reactive
solid. The
reactive solid may include zero valent iron. It will be understood that iron
is illustrative of a
reactive solid. The present inventor believes that iron is particularly
practical. However, the
present inventor contemplates other treatment materials. For example,
according to some
embodiments, the reactive solid includes zinc. It will be understood that a
reactive system may
include the reactive solid in zero valent form.
[0034] According to some embodiments, the reactive solid includes a base
material and a
supplementary material. Zero valent iron is illustrative of a base material.
The supplementary
material may include an iron oxide mineral. The iron oxide mineral may be
magnetite. The
supplementary material may assist the functionality of the base material. The
reactive solid may
be in the form of a plurality of particles. A reactive solid particle may
include a core and a shell.
The core may include primarily the base material. The shell may include
primarily the
7
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
supplementary material. The shell may be continuous. Alternatively, the shell
may be
discontinuous. The shell may include a plurality of particles of the
supplementary material.
[0035] According to some embodiments, a reactor system includes a secondary
solid.
The secondary solid may assist the functionality of the reactive solid. The
secondary solid may
be in the form of particles. Thus, the reactor system may include a plurality
of reactive solid
particles and a second plurality of secondary solid particles. The secondary
solid may be in
equilibrium with the reactive solid. The secondary solid may include the same
material as the
supplementary material. Thus, when the supplementary material includes
magnetite, the
secondary solid may include magnetite.
[0036] According to some embodiments, a reactor system includes an additive
solid. The
additive solid may include a material promoting mercury removal. The material
may be an iron
sulfide. An iron sulfide may be selected from among FeS, FeS2, and
combinations thereof The
iron sulfide may be pyrite.
[0037] According to some embodiments, a reactor a system further includes a
secondary
reagent. The secondary reagent may include ferrous iron. The ferrous iron may
be present as
ferrous ion. It will be understood that ferrous iron is illustrative of a
secondary reagent.
Aluminum ion, A13 , may substitute for (e.g. added as aluminum sulfate) for
ferrous iron.
[0038] The present inventor believes that ferrous iron acts as a
passivation reversal agent
for zero valent iron. Thus, the secondary reagent may be adapted to act as a
passivation reversal
agent. Thus, according to some embodiments, the reactive system further
includes a passivation
reversal agent suitable for a zero valent reactive solid as may be
advantageous. Passivation is
generally the process of rendering an active material, for example zero valent
zinc, inactive. The
mechanism of action is complex. While not wishing to be limited by theory, the
present inventor
believes that passivation is partially caused by corrosion of iron in a water
environment. The
present inventor believes that ferrous iron acts to cause conversion of iron
corrosion product on
the surface of the zero valent iron to magnetite. The present inventor
believes that boron and
dissolved silica in the wastewater further contributes to passivation of zero
valent iron and that
8
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
ferrous iron facilitates removal of the boron and dissolved silica from the
zero valent iron
reactive system.
[0039] According to some embodiments, a sufficient amount of magnetite is
produced so
as to optimize removal of toxic materials by a reaction system including zero
valent iron.
According to some embodiments, the process uses a highly reactive mixture of
zerovalent iron
(Fe ), iron oxide minerals (Fe0x), and ferrous iron (Fen) to react with,
absorb, and precipitate
various toxic metals and metalloids from wastewater, forming chemically inert
and well
crystallized magnetite (Fe304) particles that can be separated from water and
disposed with
encapsulated pollutants. Thus, according to some embodiments, the process
produces removable
solids. According to some embodiments, the removable solids contain
encapsulated toxic
material. According to some embodiments, the encapsulated toxic material is
solid. According
to some embodiments, the removable solids contain toxic material encapsulated
in magnetite.
[0040] According to some embodiments, a reactor system further includes an
additive.
The additive may be in the form of an additive solid. The additive solid may
include iron
sulfide. Alternatively, or in combination, the additive may be in the form of
an additive reagent
in solution. The additive reagent may include sulfide ion. The additive
reagent may be adapted
to promote the removal of mercury.
[0041] It will be understood that mercury is illustrative of toxic metals
whose removal
may be improved with the addition of a sulfur-containing species, such a
sulfide ion or iron
sulfide. In particular, a sulfur-containing additive may be adapted to promote
the removal of
mercury, lead, copper, cadmium, zinc, and the like. It will be understood that
improvement with
a sulfur-containing additive is illustrative of improvement of removal of a
contaminant with
addition of an additive, as disclosed further herein.
[0042] According to some embodiments, referring to Figure 1, reactor 110
includes
internal settling zone 114 in communication with a reactive zone 111. The
reactor is illustrated
in schematic in FIG. 1. According to some embodiments, reactive zone 111 is
maintained near
neutral pH. According to some embodiments, internal settling zone 114 uses
gravitational forces
to separate solids from liquids. According to some embodiments, mostly liquids
remain in
9
WO 2011/035263 PCT/US2010/049528
settling zone 114. According to some embodiments, internal settling zone 114
is towards the top
of reactor 110 (FIG. 1). According to some embodiments, communication with
reactive zone 111
is via inlet 115 at the bottom of the internal settling zone 114. According to
some embodiments,
effluent 125 is removed from the top region of internal settling zone 114.
[0043] According to some embodiments, the effluent is very clear. It
will be understood
that a clear effluent is illustrative of an effluent substantially free of
removable solids. As disclosed
herein, removable solids may contain magnetite. Magnetite is known to be
black. Settling observed
in previous experiment over time shows clearer separation of black material
and clear fluid over
time. The present inventor believes that settling for a separating method is
particularly efficient.
However, other suitable separating methods are contemplated.
[0044] Still referring to Figure 1, according to some embodiments,
reactive zone 111 includes
central conduit 113. Central conduit 113 improves mixing. For example,
according to some
embodiments, Central conduit 113 promotes convective motion.
[0045] Still referring to Figure 1, thus, according to some embodiments,
reactor system
100 operates in part as fluidized bed reactor 110 that employs motorized
stirrer 138 in conjunction
with central flow conduit 113 to create circular flow 119 within reactor 110
and provide an
adequate mixing between reactive solids 122 and wastewater 124. Internal
settling zone 114 was
created to allow solid-liquid separation and return of the solid into
fluidized zone 112. It will be
understood that as used herein the terms "fluidized bed reactor" is defined to
refer to reactor that
provide a flow of reactive solids within the reactor so as to provide adequate
mixing between
reactive solids and wastewater. According to some embodiments, the reactor
includes a stirrer and
operates similarly to a stirred tank reactor. According to some embodiments,
flow is created by a
conventional method known to one of ordinary skill in the art for creating
flow in a fluidized bed
reactor and the reactor operates with a conventional fluidized bed.
3828002
CA 2787032 2020-03-19
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
[0046] FIG. 1 is a schematic illustrating an embodiment of the system and
process.
Single-stage fluidized-bed system 100 includes fluidized reactive zone 112, an
internal
solid/liquid separating zone 114, an aerating basin 116, final settling basin
118, and optional
sand filtration bed 120.
[0047] Still referring to FIG. 1, fluidized zone 112 is the main reactive
space where
reactive solid 122, in the form of particles, is completely mixed with
wastewater 124 and
secondary reagent 126 and where various physical-chemical processes
responsible for toxic
metal removal occur.
[0048] Still referring to FIG. 1, internal settling zone 114 is to allow
particles to separate
from water and be retained in fluidized zone 112. For high density particles,
an internal settling
zone with a short hydraulic retention time is sufficient for complete
solid/liquid separation. This
eliminates the need of a large external clarifier and a sludge recycling
system.
[0049] Still referring to FIG. 1, aeration basin 116 has two purposes: (1)
to eliminate
residual secondary reagent in effluent 125 from fluidized zone 112; and (2) to
increase the
dissolved oxygen level. For a single-stage reactor, effluent from fluidized
reactive zone will
always contain certain amount of secondary reagent. Oxidation of secondary
reagent will
consume alkalinity and therefore will lower the pH. To accelerate oxidation of
secondary
reagent, aeration basin 116 should maintain a pH of above 7Ø Chemicals such
as Ca(OH)2,
NaOH, and Na2CO3 could be used for pH control.
[0050] Still referring to FIG. 1, final settling tank 118 is to remove
flocculent formed in
aeration basin 116. The floc (fluffy) settled to the bottom can be returned as
returned sludge 132
to fluidized zone 112 and transformed by secondary reagent 126 into dense
particulate matter.
[0051] Still referring to FIG. 1, upon final settling, sand filtration bed
120 may be used to
further polish the intermediate treated water133 before discharge as treated
water 134.
[0052] Still referring to FIG. 1, the post-FBR (fluidized bed reactor)
stages (aeration-
settling-filtration) may not be needed under certain operation conditions.
[0053] Still referring to FIG. 1, shown also are wastewater pump 136,
reagent pumps
137, auxiliary reagent 127 (e.g. HC1), air 128, and pH control chemical 130.
11
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
[0054] Referring now to FIG. 2, several fluidized-bed reactors 210 can be
combined to
form a multi-stage treatment system 200. It is recommended that each stage
maintain its own
reactive solid. That is, the solids are separated in each stage. In order to
achieve a separate solid
system, each stage may have its own internal solid-liquid separation
structure.
[0055] Still referring to FIG. 2, depending on operating conditions in FBRs
240, 242,
244, wastewater 224 characteristics, and discharge 234 standards, the post FBR
treatments
(aeration 216 + final clarifier 218 + sand filtration 220) may not be needed.
[0056] Although a multi-stage system is more complex and may result in a
higher initial
construction cost, a multi-stage fluidized-bed reactor system has several
major advantages.
[0057] A multi-stage system can achieve higher removal efficiency than a
single-stage
system under comparable conditions. Further, the FGD wastewater may contain
certain
chemicals (e.g., phosphate and dissolved silica) that may be detrimental to
the high reactivity of
the reactive solids. A multi-stage system can intercept and transform these
harmful chemicals in
the first stage and thus reducing the exposure of the subsequent stages to the
negative impact of
these detrimental chemicals. As such, a multi-stage configuration is more
stable and robust.
[0058] A multi-stage configuration facilitates the control of nitrate
reduction, for
example in an iron-based system. In a single stage system, because the
presence of dissolved
oxygen carried in raw wastewater, it tends to be difficult to operate the
system in a rigorous
anaerobic environment. In a multi-stage system, stage 1 can remove virtually
all dissolved
oxygen; as a result, the subsequent stages can be operated under rigorous
anaerobic environment.
[0059] A multi-stage system allows flexible control of different chemical
conditions in
each individual reacting basin. The chemical conditions in each reactive basin
can be controlled
by adjusting the pumping rate of supplemental chemicals and turning aeration
on or off. A
multi-stage system can be operated in a mode of multiple feeding points. Each
stage may be
operated under different pH and dissolved oxygen condition.
[0060] A multi-stage system will lower chemical consumption. In a single-
stage
complete-mixed system, secondary reagent in the reactor is desirably
maintained at a relatively
high concentration in order to maintain high reactivity of reactive solids. As
a result, the residual
12
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
secondary reagent in the effluent will be high. This means that more secondary
reagent will be
wasted and more NaOH (or lime) consumption will be required just to neutralize
and precipitate
the residual secondary reagent in the effluent. As a result, more solid sludge
will be produced
and waste disposal cost will increase. In a multi-stage system, residual
secondary reagent from
stage 1 can still be used in stage 2. In this case, secondary reagent can be
added in a way that
conforms to its actual consumption rate in each stage. As a result, it is
possible to control
residual secondary reagent in the effluent in the final stage to be much lower
than the one in a
single stage system.
[0061] Referring to FIG. 3, according to some embodiments, in the system
and process
illustrated by FIG. 1, the reactive solid 323 includes zero valent iron (ZVI)
and iron oxide
mineral (FeOx), and the secondary reagent is Fe2'. Thus, referring to FIG. 3,
single-stage
fluidized-bed ZVI/FeOx/Fe(II) system 300 includes a fluidized reactive zone
312, an internal
solid/liquid separating zone 314, an aerating basin 316, a final settling
basin 318, and an optional
sand filtration bed 320.
[0062] Still referring to FIG. 3, fluidized zone 312 is the main reactive
space where ZVI
and FeOx reactive solids are completely mixed with wastewater 324 and
dissolved Fe2 326 and
where various physical-chemical processes responsible for toxic metal removal
occur.
[0063] Still referring to FIG. 3, internal settling zone 114 is to allow
ZVI and FeOx to
separate from water and be retained in fluidized zone 112. Because of high
density of fully or
partially crystallized FeOx particles, an internal settling zone with a short
hydraulic retention
time would be suffice for complete solid/liquid separation. This eliminates
the need of a large
external clarifier 318 and a sludge 332 recycling system.
[0064] Still referring to FIG. 3, aeration basin 330 has two purposes: (1)
to eliminate
residual dissolved Fe2- in the effluent from fluidized zone; and (2) to
increase dissolved oxygen
level. For single-stage reactor 310, effluent from fluidized reactive zone 312
will always contain
certain amount of dissolved Fe2-. Oxidation of Fe2' will consume alkalinity
and therefore will
lower the pH. To accelerate oxidation of dissolved Fe2', aeration basin 316
should maintain a
pH of above 7Ø Chemicals such as Ca(OH)2, NaOH, and Na2CO3 could be used for
pH control.
13
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
[0065] Still referring to FIG. 3, final settling tank 318 is to remove
iron oxide flocculent
formed in aeration basin 316. The ferric oxide floc (fluffy) settled to the
bottom can be returned
as returned sludge 332 to the fluidized zone 312 and transformed by Fe2+ into
dense particulate
matter.
[0066] Still referring to FIG. 3, upon final settling, sand filtration bed
320 may be used to
further polish the treated water before discharge.
[0067] Still referring to FIG. 3, reactive solid 323 may initially be zero
valent iron, with
the iron oxide mineral formed in situ. The iron oxide mineral may coat the
zero valent iron.
Reactive solid 323 may be in the form of particles.
[0068] Still referring to FIG. 3, shown also are wastewater pump 336,
reagent pumps
337, auxiliary reagent 327 (e.g. HC1), air 328, and pH control chemical 330.
[0069] Referring to FIG. 4, a reactor system may include a standalone
sulfide generator.
The standalone sulfide generator may produce small amount of sulfide ions
before introducing
into the reactor. The sulfide ions may contribute to precipitating toxic
metals. The sulfide
generator could be a packed-bed filter column filled with a powder (mixed with
sand if necessary
to improve its porosity and hydraulic conductivity). The powder may be a
sulfide generating
material. Fore example the powder may be FeS or FeS2. A low concentration acid
may be
flowed through the column to dissolve the powder and steadily and gradually
release a stream of
acid leachate rich of sulfide ions to add into the reactor. Addition of
sulfide ions to the reactor is
particularly useful for removal of mercury, lead, copper, cadmium, zinc and
the like from a
liquid stream.
[0070] According to some embodiments, a method of treating an aqueous
fluid
incorporates a chemical process to generate inorganic sulfide ions and
introduce the sulfide ions
into water treatment process that can result in rapid precipitation and
significantly improved
removal efficiency of dissolved toxic metal including mercury and many other
toxic metals of
major environmental concern. The composition, system, and process
incorporating sulfide
generation may be used in conjunction with the Hybrid Zerovalent
Iron/Fe0x/Fe(II) water
treatment system disclosure herein. Sulfide generation may use a sulfide
generator. The sulfide
14
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
generator could be a standalone toxic metal treatment system or a subsystem
that can be
incorporated into other water chemical treatment processes such as the Hybrid
Zerovalent
Iron/Fe0x/Fe(II) water treatment system.
[0071] Most of dissolved toxic metal ions (e.g., Hg and Pb) can bind with
sulfide ions to
form metal sulfide that are extremely low in solubility. A filter cartridge
filled with FeS as
reactive material may be employed as a sulfide generator. When low
concentration of acid (e.g.,
0.005 M HC1) flows through the FeS filter, acid could gradually dissolve FeS
to become Fe2+ and
S2- (<0.0025M). Because H2S has high solubility in water (about 3.8 g/L or
0.11 M H2S at 20
C), the small concentration of S2- will remain dissolved in water and
therefore no H2S gas
bubble will be formed, which could minimize the danger posed by toxic H2S gas.
FeS acid-
leaching solution may be introduced into a treatment reactor where the
dissolved sulfide ion
could bind with various toxic metal ions and precipitate and mineralize
together with other solid
phase (various iron oxides in the Hybrid Zerovalent Iron/Fe0x/Fe(II) water
treatment system).
For most applications in which toxic metals are present in low or sub-ppm
level, addition of low
ppm level of sulfide would be sufficient to precipitate all of the concerned
toxic metals. The
residual S2- may be readily precipitated by the dissolved Fe2 (accompanied
with S2-) and other
non toxic metals present in the water, and therefore pose no threat in the
treated effluent.
[0072] Referring to Figure 5, according to some embodiments, the present
zero valent
iron (ZVI) is used to build a permeable reactive barrier for remediation of
groundwater. Figure 5
shows bedrock 512, permeable zone 514, contaminated plume 516, toxic materials
of 518 (e.g.
chlorinated organics, heavy metals), permeable reactive barrier 520, heavy
metals retained 522,
organics degraded 524, and remediated groundwater 530.
[0073] Referring again to Figure 3, iron-based system 300 can be operated
under various
controlled conditions as needed.
[0074] According to some embodiments, an iron-based technique employs a
mixture of
zerovalent iron (ZVI or Fe ) and iron oxide minerals (FeOx), and Fe(II)
species to react with,
adsorb, precipitate, and remove various toxic metals, metalloids and other
pollutants from the
contaminated wastewater. According to some embodiments, an iron-based physical-
chemical
WO 2011/035263 PCT/US2010/049528
treatment process that employs a hybrid Zerovalent Iron/Fe0x/Fe(11) Reactor to
treat toxic metal-
contaminated wastewater. For example, according to some embodiments, the
present system and
process involve a hybrid Zerovalent Iron/Fe0x/Fe(II) reactor for removing
toxic metals in
wastewater. According to some embodiments, the process employs a fluidized bed
system and
uses a reactive mixture of Fe , Fe" and FeOx to absorb, precipitate, and react
with various toxic
metals, metalloids and other pollutants for wastewater decontamination.
According to some
embodiments, toxic metals are encapsulated within iron oxide crystalline
(mainly magnetite
powder) that are chemically inert and physically dense for easier solid-liquid
separation and final
disposal.
[0075] While not wishing to be limited by theory, the present inventor
believes that the
following are contributing mechanisms for the present system and process when
it is iron based: a)
using the reducing power of Fe and Fe') to reduce various contaminants in
oxidized forms to become
insoluble or non-toxic species; b) using high adsorption capacity of iron
oxide surface for metals to
remove various dissolved toxic metal species from wastewater; and c) promoting
mineralization of iron
oxides and growth of certain iron oxide crystals so that surface-adsorbed or
precipitated toxic metals
and other pollutants could be incorporated into the iron oxides and remain
encapsulated in a stabilized
form for final disposal.
[0076] The present technology provides a practical and cost-effective
method to remove
dissolved silica from a liquid stream. Embodiments of the present composite
solid, reactor,
activation process (also termed pretreatment process), use of the activation
process for making the
composite solid, and use of the composite solid for treating a liquid stream
to remove toxic metals
have been previously described by the present inventor. Removal of dissolved
silica was
discovered by the present inventor in the course of laboratory testing of
removal of toxic metals
from flue gas desulfurization water using the present composite solid. In
turn, it will be understood
that in the description with respect to dissolved silica below,
16
3828002
CA 2787032 2020-03-19
WO 2011/035263 PCT/US2010/049528
dissolved silica is illustrative of a contaminant.
[0077] The technology promotes precipitation of dissolved silica from
the liquid stream. The
technology has an advantage of economy, through low operating costs of using
inexpensive materials.
Further, the present technology has the advantage environmental benefit, by
reducing the amount of
solid waste produced by silica removal and the energy consumption of the
silica removal process. Still
further, the technology has the advantage of effective operation at neutral pH
and ambient temperature,
increasing efficiency.
[0078] The present technology involves using a composite for silica
removal. The
composite solid may include zero-valent iron and an activating material. The
activating material
may be adapted to overcome the tendency of zero-valent iron to passivate in
solution. Thus, the
activating material may act as a promoter. That is the composite solid has
increased activity for
silica removal as compared to zero valent iron. Alternatively or in
combination, the activating
material may be adapted to electronically mediate an electrochemical reaction
between the zero -
valent iron and dissolved silica so as to facilitate precipitation of
dissolved silica. Thus, the
activating material may be semi-conducting. The composite solid may include a
plurality of
composite particles. The composite particle may have a zero-valent iron core
and a layer of
activating material. The layer may coat the surface of the zero-valent iron
core. The layer may be
in the form of a reactive film. The activating material may be present as one
or more iron minerals.
For example, the activating material may include magnetite. The activating
material may be well
crystallized. The present composite solid is reactive for silica removal.
[0079] The present composite solid may be produced by an activation
process. The
activation process may involve oxidizing a portion of a zero-valent iron
precursor so as to form
an intermediate material and exposing the intermediate portion to dissolved
ferrous ion so to
form the activating material. The ferrous ion may adsorb onto the intermediate
material. The
ferrous ion may covert the intermediate material into a layer of the
activating material. The zero-
valent iron precursor may be provided as a plurality of particles, such as in
a powder. The
intermediate material may include an iron corrosion material. The iron
corrosion material may
17
3828002
CA 2787032 2020-03-19
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
contain one or more of ferric oxide and amorphous mixed valent ferric-ferrous
(oxy)hydroxides.
When the zero-valent iron precursor is a particle, the intermediate material
may form as an
intermediate layer over a zero-valent core. Exposing such an intermediate
layer to ferrous ion
transforms the intermediate layer into a layer of activating material. This
activating may include
maintaining the zero-valent iron precursor in an oxidizing environment. The
oxidizing
environment may be a solution may contain an oxidant. Suitable oxidants
include dissolved
oxygen, nitrate, nitrite, selenate, hypochlorite, hydrogen peroxide, iodate,
periodate, bromate,
and the like. Oxidant is consumed in the activation process when a portion of
the zero-valent
iron is oxidized to form activating material. When the oxidant is dissolved
oxygen, the dissolved
oxygen may be provided through aeration. When the oxidant is nitrate, nitrite,
or selenate, the
oxidant may be provided as a dissolved salt.
[0080] Alternatively, or in combination the composite is produce in situ as
part of the
silica removal process.
[0081] The present composite solid may be used in a silica removal process.
The silica
removal process may involve contacting an influent stream with a plurality of
the composite
particles so as to produce an effluent stream, where the effluent stream is
reduced in dissolved
silica with respect to the influent stream. The silica removal process may
utilize a reactive
system that includes a reaction zone including a fluidized bed reactor and a
plurality of the
composite particles in fluidized bed in the reactor. The contacting may occur
in the reaction
zone. The reactive system may include a plurality of reaction zones. For
example, the reactive
system may be a multi-stage reactor system. The reduction in the concentration
of dissolved
silica of the effluent stream with respect to the influent stream may be
greater than 70 %. For
example, the reduction may be at last 80 %. For example the reduction may be
at least 90%.
When the reactive system is a multi-stage reactor system, the first reactor
stage may be primarily
for removal of dissolved silica from a liquids stream and one or more later
stages for other
treatment of the liquid stream. For example, later stages may remove toxic
materials.
[0082] The fluidized bed reactor may include an internal settling zone. The
internal
settling zone may help to retain a high concentration of iron corrosion
products. Thus, the
18
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
settling zone may facilitate the maintenance of the activating material in the
composite. The
internal settling zone may further provide extra large surface area to
facilitate adsorption,
polymerization, and precipitation of dissolved silica.
[0083] The reactive system may further include ferrous iron as ion in
solution. The
ferrous iron may be adsorbed on the surface of the composite solid. While not
wishing to be
limited by theory, it appears that a continuous corrosion reaction of zero-
valent iron may play a
role in promoting rapid polymerization of dissolved silica. Addition of
external ferrous ion
(Fe2+) may play a role in inducing formation of a magnetite coating on zero-
valent iron and
maintain high reactivity of zero-valent iron at near neutral pH.
Alternatively, or in combination,
addition of external ferrous ion may play a role as to allow oxidation and
precipitation of ferrous
ion at near netural pH to directly contribute to removal of dissolved silica.
[0084] The removal process may include sustaining an iron corrosion
reaction.
Sustaining the iron corrosion reaction may be accomplished by providing
ferrous ion to the
reaction zone and maintaining the reaction zone in an oxidizing environment.
Addition of
ferrous ion in the presence of an oxidant in situ facilitates formation of
activating material in situ.
The oxidizing environment may be a solution may contain an oxidant. Suitable
oxidants include
dissolved oxygen, nitrate, nitrite, selenate, hyprchlorite, hydrogen peroxide,
iodate, periodate,
bromate, and the like, and combinations thereof. Oxidant is consumed in the
iron corrosion
reaction. When the oxidant is dissolved oxygen, the dissolved oxygen may be
provided through
aeration. When the oxidant is nitrate, nitrite, or selenate, the oxidant may
be provided as a
dissolved salt. A process for treating a liquid stream in a multi-stage
reactor system may include
sustaining an iron corrosion reaction in the first reaction zone.
[0085] High concentrations of FeO, maintained in the reactor may contribute
to the
removal of silica by providing large surface area with surface charge
conditions conducive to
polymerization of dissolved silica. FeO x may be present as magnetite (Fe304).
Maghemite (y-
Fe2O3) may also be present in the reactor. The presence of maghemite may
improve removal of
dissolved silica. The maghemite may be present as particles of maghemite.
Maghemite has been
observed when zero valent iron is aerated to promote iron corrosion in the
present of dissolved
19
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
Fe2 and in the absence of nitrate, selenate, or other oxidants. The present
inventor believes that
the maghemite is produced by oxidation of magnetite. Thus, according to some
embodiments, a
chemical system as disclosed herein comprises an additive, where the additive
comprises
maghemite.
[0086] Colloidal or precipitated silica floc when retained in the reactor
may also
contribute to polymerization and precipitation of dissolved silica.
[0087] It will be understood that aspects of the embodiments described
herein may be
used singly or in combination. According to some embodiments, a treatment
system for treating
a fluid stream, comprises a chemical reactor system comprising a fluidized bed
reactor
comprising a reactive zone. The chemical reactor system may further comprise
an internal
settling zone in communication with the reactive zone. The internal settling
zone may be located
in the top region of the chemical reactor system. The internal settling zone
may comprise an
opening at the bottom of the internal settling zone adapted for the
communication with the
reactive zone. The internal settling zone may comprise an outlet adapted for
removal of effluent
from the internal settling zone. The reactive zone may comprise a conduit. The
conduit may be
central with respect to the reactive zone. The treatment system may be a multi-
stage system
comprising an additional reactor system. The treatment system may further
comprise vessel
comprising a sulfide ion generator. The reactive zone may comprise a reactive
solid and a
secondary reagent. The reactive solid may comprise iron. The secondary reagent
may comprise
ferrous iron. The reactive solid may further comprise an iron oxide mineral.
The iron oxide
mineral may comprise magnetite. The treatment system may further comprise an
additive
reagent. The additive reagent may comprise sulfide ion. The treatment system
may further
comprise an additive solid. The additive solid may comprise an iron sulfide
compound. The
fluid stream may comprise a waste steam. The fluid stream may comprise a toxic
material. The
toxic material may be selected from the group consisting of selenium, arsenic,
mercury,
aluminum, antimony, beryllium, thallium, chromium, cobalt, lead, cadmium,
silver, zinc, nickel,
molybdenum, nitrates, bromates, iodates, periodates, and borates.
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
[0088] According to some embodiments, a process for treating a fluid stream
comprises
feeding the fluid stream to a treatment system according to any one of the
above-described
embodiments. The process may further comprise removing a toxic material from
the fluid
stream. The removing may comprise: a) at least one of reacting, adsorbing, and
precipitating the
toxic material from the fluid stream so as to form removable solids in treated
effluent; and b)
separating the removable solids from the fluid stream. The removable solids
may comprise at
least a portion of the toxic material encapsulated in the removable solids.
[0089] According to some embodiments, a process for treating wastewater
comprising a
toxic material, comprising exposing the wastewater to a reactive material
system so as to remove
toxic material from the wastewater, wherein the reactive material system
comprises zero valent
iron particles and ferrous iron, wherein the exposing comprises: a) at least
one of reacting,
adsorbing, and precipitating the toxic material from the wastewater so as to
form removable
solids in treated wastewater, wherein the removable solids comprise at least a
portion of the toxic
material encapsulated in at least a portion of an iron mineral derived from
the reactive material
system; and b) separating the removable solids from the treated wastewater.
The removable
solids may further comprise precipitated sulfide.
[0090] According to some embodiments, a new and improved fluidized bed
apparatus for
wastewater treatment comprises a fluidized bed, a fluidized reactive zone, an
internal solid/liquid
separating zone in fluid communication with said reactive zone, an aerating
basin, and a settling
basin. The apparatus may further comprise control and metering systems for
monitoring and
manipulating chemical processes within said reactor. The apparatus may further
comprise a sand
filtration bed. The apparatus may further comprise a central conduit in the
fluidized bed reactor
to promote convective fluid flow enhancing mixing. The apparatus may further
comprise a
motorized stirrer in conjunction with said central conduit configured so fluid
flow within the
conduit is down and flow within the fluidized bed reactor outside the conduit
is up. The
apparatus may further comprise at least one additional fluidized bed apparatus
configured as
stages in series with said first apparatus. The apparatus may further comprise
control and
metering systems for monitoring and manipulating chemical processes run within
said reactors.
21
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
According to some embodiments, the chemical process conditions within
different stages are
varied to optimize results. According to some embodiments, the first stage is
optimized for silica
removal. The apparatus may further comprise a sulfide ion generator in fluid
communication
with the fluidized reactive zone. The fluidized reactive zone comprises a
composition
comprising zero valent iron, iron oxide mineral, and ferrous iron. The
fluidized reactive zone
may further comprise sulfide ion. Alternatively or in combination, the
fluidized reactive zone
may further comprise an iron sulfide compound.
[0091] According to some embodiments, a composition for treating a fluid
stream
comprises zero valent iron, iron oxide mineral, and ferrous iron.
[0092] According to some embodiments, a chemical system for treating a
fluid stream
comprises zero valent iron, iron oxide mineral, ferrous iron, and an additive.
The additive may
comprise an additive reagent. The additive reagent may comprise ionic sulfide.
Alternatively or
in combination, the additive may comprise an additive solid. The additive
solid may comprise
an iron sulfide compound. Alternatively or in combination, the additive solid
may comprise
maghemite. The additive solid may be present as particle comprising the
additive solid, where
the additive solid particles are distinct from the composite. The composite
may be present as
particles of the composite. The chemical system may comprise composite
particles each
comprising a core and a layer layered on the core, where the cores comprise
the zero valent iron,
and the layers comprise a first portion of the iron oxide mineral. The
chemical system may
further comprise secondary particles comprising a second portion of the iron
oxide mineral.
[0093] According to some embodiments, a composite comprises zero-valent
iron and a
predetermined activating material selected so as to increase the activity of
the composite for
removal of a contaminant. The contaminant may be a toxic material. The toxic
material may be
selected from the group consisting of selenium, arsenic, mercury, aluminum,
antimony,
beryllium, thallium, chromium, cobalt, lead, cadmium, silver, zinc, nickel,
molybdenum, nitrates,
bromates, iodates, periodates, and borates. Alternatively or in combination,
the contaminant may
be silica. The activating material may be adapted to mediate an
electrochemical reaction
between the zero-valent iron and the contaminant so as to facilitate
precipitation of the
22
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
contaminant. The activating material may be selected from the group consisting
of zero-valent
iron promoters, semi-conductors, and combinations thereof. The activating
material may
comprise an iron mineral. The iron oxide mineral may comprise magnetite. The
composite may
comprise a particle, having a core comprising zero-valent iron and a layer
over the core, wherein
the layer comprises the activating material. The composite particle may
further comprise a
second layer over the first layer. The second layer may comprise a plurality
of fingers extending
from the first layer. The second layer may comprise a non-activating material.
The non-
activating material may comprise lepidocrocite.
[0094] According to some embodiments, a reactor system for removing a
contaminant
from a liquid stream comprises a fluidized bed reactor configured for
increasing the efficiency of
removal of the contaminant from the liquid stream. The contaminant may be a
toxic material.
The toxic material may be selected from the group consisting of selenium,
arsenic, mercury,
aluminum, antimony, beryllium, thallium, chromium, cobalt, lead, cadmium,
silver, zinc, nickel,
molybdenum, nitrates, bromates, iodates, periodates, and borates.
Alternatively, or in
combination, the contaminant may be silica. The fluidized bed reactor may
comprise an internal
settling zone. Alternatively, or in combination, the fluidized bed reactor may
comprise a central
conduit.
[0095] According to some embodiments, a composite is made by a method
comprising:
a) oxidizing a portion of zero valent iron so as to produce an intermediate
material; and b)
exposing the intermediate material to ferrous ion so as to produce a composite
comprising the
remaining zero valent iron and an activating material. Step (b) may comprise
transforming the
intermediate material into the activating material. Step (a) may comprise
providing a dissolved
oxidant. The dissolved oxidant may be selected from the group consisting of
oxygen, nitrate,
nitrite, selanate, hypochlorite, hydrogen peroxide, iodate, periodate,
bromate, and the like, and
combinations thereof The intermediate material may comprise an iron corrosion
product. The
activating material may be adapted to electronically mediate an
electrochemical reaction between
the zero-valent iron and the contaminant so as to facilitate precipitation of
the contaminant. The
activating material may be selected from the group consisting of zero-valent
iron promoters,
23
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
semi-conductors, and combinations thereof. The activating material may
comprise an iron
mineral. The iron oxide mineral may comprise magnetite. The activating
material may increase
the activity of the composite for removal of a contaminant in comparison with
zero valent iron.
The contaminant may comprise a toxic material. The toxic material may be
selected from the
group consisting of selenium, arsenic, mercury, aluminum, antimony, beryllium,
thallium,
chromium, cobalt, lead, cadmium, silver, zinc, nickel, molybdenum, nitrates,
bromates, iodates,
periodates, and borates. The contaminant may comprise dissolved silica. The
composite may
comprise a particle, having a core comprising the zero-valent iron and a layer
over the core,
wherein the layer comprises the activating material.
[0096] According to some embodiments, a process for activating zero valent
iron for
removing a contaminant from a liquid stream, comprises a) oxidizing a portion
of the zero valent
iron so as to produce an intermediate material; and b) exposing the
intermediate material to
ferrous ion so as to produce a composite comprising the remaining zero valent
iron and an
activating material. The contaminant may be a toxic material. The toxic
material may be
selected from the group consisting of selenium, arsenic, mercury, aluminum,
antimony,
beryllium, thallium, chromium, cobalt, lead, cadmium, silver, zinc, nickel,
molybdenum, nitrates,
bromates, iodates, periodates, and borates. Alternatively, or in combination,
the contaminant
may be silica. Step (b) may comprise transforming the intermediate material
into the activating
material. Step (a) may comprise providing a dissolved oxidant. The dissolved
oxidant may be
selected from the group consisting of oxygen, nitrate, nitrite, selenate,
hypochlorite, hydrogen
peroxide, iodate, periodate, bromate, and the like, and combinations thereof.
The intermediate
material may comprise an iron corrosion product. The activating material may
be adapted to
electronically mediate an electrochemical reaction between the zero-valent
iron and the
contaminant so as to facilitate precipitation of the contaminant. The
activating material may be
selected from the group consisting of zero-valent iron promoters, semi-
conductors, and
combinations thereof. The activating material may comprise an iron mineral.
The iron oxide
mineral may comprise magnetite.
24
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
[0097] According to some embodiments, a process for removing a contaminant
from an
influent stream comprises contacting the influent stream with a composite
comprising zero-
valent iron and an activating material under removal-promoting conditions so
as to produce an
effluent stream reduced in concentration of contaminant with respect to the
influent stream. The
contaminant may be a toxic material. The toxic material may be selected from
the group
consisting of selenium, arsenic, mercury, aluminum, antimony, beryllium,
thallium, chromium,
cobalt, lead, cadmium, silver, zinc, nickel, molybdenum, nitrates, bromates,
iodates, periodates,
and borates. Alternatively, or in combination, the contaminant may be silica.
The reduction in
contaminant concentration may be greater than 70 %. The reduction in
contaminant
concentration may be greater than 80 %. The reduction in contaminant
concentration may be
greater than 90 %. The activating material may be adapted to electronically
mediate an
electrochemical reaction between the zero-valent iron and the contaminant so
as to facilitate
precipitation of the contaminant. The activating material may be selected from
the group
consisting of zero-valent iron promoters, semi-conductors, and combinations
thereof. The
activating material may comprise an iron mineral. The iron oxide mineral may
comprise
magnetite. The removal-promoting conditions comprise substantially neutral pH.
The pH may
be between 6 and 8. The pH may be between 7 and 8. The removal-promoting
conditions may
comprise ambient temperature.
[0098] According to some embodiments, a composite for removing a
contaminant from a
fluid stream, comprising zero valent iron, an iron oxide mineral, and ferrous
iron, wherein the
ferrous iron is disposed so as to facilitate maintenance of the iron oxide
mineral, and wherein the
composite is active for removing the contaminant from the fluid stream. The
contaminant
maycomprise a toxic material. The toxic material is selected from the group
consisting of
selenium, arsenic, mercury, aluminum, antimony, beryllium, thallium, chromium,
cobalt, lead,
cadmium, silver, zinc, nickel, molybdenum, nitrates, bromates, iodates,
periodates, and borates.
Alternatively, or in combination, the toxic material may comprises a
phosphate. The
contaminant may comprise dissolved silica. The iron oxide mineral may comprise
a zero valent
iron promoter with respect to removal of the contaminant from the fluid
stream. The iron oxide
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
mineral may comprise a conducting material. The iron oxide mineral may
comprise Fe(H),
Fe(111), and oxygen. The iron oxide mineral may comprise Fe(H), Fe(M), oxygen,
and
hydrogen. The iron oxide mineral may comprise magnetite. The zero valent iron
and iron oxide
mineral may together comprise a reactive solid, where the reactive solid is
suspended in a
solution, and where the ferrous iron is selected from the group consisting of
ferrous iron
dissolved in the solution and ferrous iron bound to the surface of the iron
oxide mineral. The
fluid stream may be at near neutral pH. The composite may be made by a method
comprising
activating the zero valent iron, wherein the activating comprises adding
ferrous ion and an
oxidant to a solution in which the zero valent iron is suspended, where the
adding allows
formation of the iron oxide mineral. The adding may comprise pre-treating the
zero valent iron
outside the presence of the fluid stream containing the contaminant. The
adding may comprise
activating the zero valent iron in situ in the presences of the fluid
containing the contaminant.
According to some embodiments, a chemical system comprises a composite
according to any
one of the above-described embodiments and a solution, where the composite is
disposed in the
solution, and where the chemical system further comprising an additive
disposed in the solution.
The additive may be selected from the group consisting of maghemite particles,
dissolved ionic
sulfide, iron sulfide particles, and combinations thereof. According to some
embodiments, a
treatment system for treating a fluid stream comprises a chemical system
according to any of the
above-described embodiments and a reactor, wherein the reactor comprises a
reactive zone
containing the chemical system. The reactor may further comprise a settling
zone in
communication with the reactive zone. Alternatively, or in combination, the
reactor may further
comprises a central conduit adapted so as to circulate the chemical system
within the reactive
zone. The treatment system may comprise a second reactor such that the
treatment system
comprises a multi-stage system. The first reactor may be optimized for removal
of dissolved
silica and the second reactor may optimized for removal of the contaminant,
wherein the
contaminant comprises a toxic material. The treatment system may further
comprising a sulfide
generator in communication with the reactor. According to some embodiments, a
process
comprises contacting a composite according to any of the above-described
embodiments with a
26
CA 02787032 2016-01-18
WO 2011/035263 PCT/U S20101049528
fluid stream in a reaction zone under removal-promoting conditions so as to
remove a portion of
the contaminant from the aqueous stream so as to produce an effluent. The
removal-promoting
conditions may comprise near neutral pH. The pH may be between 6 and 8. The
removal-
promoting conditions may comprise ambient temperature. The contaminant may
comprise
removing dissolved silica so as to produce the effluent and the process may
comprise removing a
toxic material from the effluent. The may comprises providing a concentration
of dissolved
ferrous iron in the reaction zone selected so as to optimize activity of the
composite for removing
the contaminant.
[0099] The scope of the claims should not be limited by specific embodiments
and examples
provided in the disclosure, but should be given the broadest interpretation
consistent with the
disclosure as a whole.
EXAMPLES
[00100] Example 1. Iron corrosion model for activity of Hybrid
ZVI/Fe0x/Fe(11)
material.
[00101] While not wishing to be limited to theory, the present inventor
proposes the
following findings. Passivation of Fe is caused by ferric oxides or amorphous
ferrous
oxyhydroxides. Ferric oxides or amorphous ferrous oxyhydroxides arc formed
under most
natural or engineered environments. A magnetite coating on Fe can maintain
high Fe'
reactivity. Adding dissolved Fe2 can promote transformation of ferric oxides
to magnetite under
the right chemical environments. In this way. Fe reactivity can be sustained.
[00102] While not wishing to be limited by theory, the present inventor
proposes a semi-
conducting corrosion model. Referring to Figures 6 and 7, SEM micrographs of a
corrosion
coating on zero valent iron show a) an outer layer dominated by lepidoprocite,
b) middle layer
including both magnetite and lepidocrocite, and c) an inner layer dominated by
magnetite. In
27
WO 2011/035263 PCT/US2010/049528
the presence of certain strong oxidants such as dissolved oxygen,
hypochlorite, hydrogen peroxide,
nitrite, iodate, periodate, nitrate and the like in an aqueous solution with
near neutral or weak
alkaline pH, that is for example pH 6-10, iron corrosion in such aqueous
chemical environment
tends to develop a ferric oxide coating (e.g., lepidocrocite) as part of its
corrosion products.
Referring to Figure 8, source iron grain 810 include Fe(0) 812, sa-Fe2O3 814,
and Fe304 816. Without
Fe2+, iron grains 812 would be coated by lepidocrocite 820 (including 7-Fe0OH)
from Fe(0)-nitrate
(or selanate) reaction, forming undesirable particles 818. With Fe2+,
lepidocricite 826 (including 7-
Fe0OH) would be rapidly converted into magnetite 824 (including Fe304),
followed by rapid
reduction of selenate, forming desirable particles 822. Referring to Figure 9,
while not wishing to
be limited by theory, aspects of a model include the following. Some reaction
sites are located at
the bottom of pores. Electronic properties of iron oxides are: magnetite is an
excellent
semiconductor, in which electrons can move almost freely; whereas
lepidocrocite is an electron
barrier. Reactivity of Fe involves a balance between the oxidizing power of a
compound and the
electron transfer resistance of the yielded iron corrosion coating.
[00103]
[00104] It will be understood that a corrosion coating can result on zero
valent iron in an
oxidizing environment.
[00105] It will be understood that lepidocrocite is illustrative of other
passivating iron oxide
materials, as described herein. It will be understood that passivating ferric
oxides include lepidocrocite,
maghemite, hematite, and other non-conducting ferric oxides.
[00106] It will be understood that magnetite is illustrative of iron
oxide minerals. The iron oxide
mineral may be non-stoichiometric. The iron oxide mineral may be a conductive.
As used herein
conductive includes both metal-like and semi-conductive. The iron oxide
mineral may be a defect iron
oxide mineral. For example, magnetite is known to have a defect structure
where atoms can be missing
and charge compensated for. Magnetite has a spinel structure with
semiconducting properties. While
not wishing to be limited by theory, the present inventors believe
28
3828002
CA 2787032 2020-03-19
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
that the spinet structure and/or semi-conducting properties facilitate the
ability of magnetite to
activate zero valent iron for removal of contaminants from a fluid stream.
[00107] According to some embodiments, an iron oxide mineral is formed by
transformation of a passivating ferric oxide. Alternatively, or in
combination, iron oxide mineral
is formed by transformation of zero valent iron.
[00108] It will be understood that as part of a chemical system for removal
of a
contaminant from an aqueous stream ferrous iron, as Fe(II), may be present as
Fe2+ (ferrous ion)
in solution, surface bound Fe2+, or Fe(II) incorporated into reactive solids.
[00109] The present inventor contemplates that one possible role of Fe2+ is
that surface
bound Fe2+ facilitates one or more of formation and maintenance of the iron
oxide mineral. The
surface bound Fe2+may facilitate conversion of ferric oxide to magnetite.
Surface bound Fe(II)
species could be labile. For example, a surface bound Fe(II) species may
undergo one or more
of the following: exchange with one or more of Fe2+in solution and Fe(II) in
the iron oxide
mineral, change valence state, and be oxidized. As a surface bound Fe(II)
species undergoes a
labile process it may be replenished so as to maintain the concentration of
surface bound Fe(II).
[00110] It will be understood that formation of the hybrid ZVI/Fe0x/Fe(II)
via a corrosion
coating of passivating ferric oxide is illustrative of formation of the hybrid
ZVI/Fe0x/Fe(H).
Alternatively, using a nitrate pretreatment process as described, for example,
hybrid
ZVI/Fe0x/Fe(II) forms directly upon adding ferrous iron and nitrate to a zero
valent iron
suspended in a solution.
[00111] It will be understood that the finger structure shown in Figure 9
is illustrative of a
porous structure. The porous structure may be of a corrosion coating.
Alternatively, or in
combination, the porous structure may be of a passivating ferric oxide.
[00112] It will be understood that the core/shell structure shown in Figure
8 is a simplified
schematic illustration of a hybrid ZVI/Fe0x/Fe(II) material. The iron oxide
mineral may be
interpenetrated with one or more of the zero valent iron and a passivating
ferric oxide. Thus, the
hybrid ZVI/Fe0x/Fe(II) material may include an interpenetrating network. This
is illustrated in
29
WO 2011/035263 PCT/US2010/049528
Figure 6, in which an outer layer was dominated by lepocrocite, an inner layer
was dominated
magnetite, and a middle layer included both lepidocrocite and magnetite.
[00113] It will be understood that a porous passivating ferric oxide may
partially cover the
iron oxide mineral in a composite, with the pores of the porous passivating
ferric oxide allowing
Fe(II) in solution to diffuse to the surface of the iron oxide mineral so as
to become surface bound
Fe(II).
[00114] It will be understood that an advantage of the hybrid
ZVI/Fe0x/Fe(II), is a sustainably
high level of activity. Therefore, in comparison to zero valent iron, the
hybrid ZVI/Fe0x/Fe(II)
improves both the activity and the lifetime.
[00115] It will be understood that the hybrid ZVI/Fe0x/Fe(II), as
described in this example is
illustrative of a reactive solid, as disclosed herein.
[00116] Example 2. Experimental Results of Using a Hybrid ZVI/FethdFe(II)
Reactive System
to Treat FGD Wastewater
[00117] The present system and process are a result of laboratory
research conducted by the
present inventor to develop a cost-effective method for removing toxic metals
in the flue gas
desulfurization wastewater generated from wet scrubbers of coal-fired steam
electric power plants.
Although developed specifically for treating the FGD wastewater with selenium
as the main target
contaminant, this chemical reactive system is suitable for general application
of removing a wide
spectrum of toxic metals in industrial wastewater, tail water of mining
operations, and
contaminated groundwater, and like contaminated aqueous streams containing
like contaminants.
[00118] According to various experimental embodiments, as shown herein, a
single stage may
achieve 90% selenate removal from synthetic wastewater within 4 hr reaction
time. A three-stage
system, in comparison, may achieve a 96% removal rate from synthetic
wastewater. The synthetic
wastewater did not contain dissolved silica. As disclosed herein, when the
aqueous stream to be treated
contains dissolved silica, the present inventor contemplates removal of the
3828002
CA 2787032 2020-03-19
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
dissolved silica in one or more stages before removal of other contaminants
such as toxic
materials.
[00119] The present inventor believes that some exemplary novel aspects are
as follows.
A first aspect is discovery of the role of externally-added Fe2+ in sustaining
the reactivity of Fe
with respect to selenate reduction. Externally-added Fe2+ may convert less
reactive ferric oxide
coating on Fe particles into a highly reactive and electron-conducting mixed-
valent Fe304 oxide
coatings and therefore rejuvenate the passivated Fe surface. A second aspect
is discovery that
surface-bound Fe(II) on magnetite (Fe304) particles can rapidly reduce
selenate to insoluble
elemental Se and be removed from the liquid phase. The present inventors have
found, based on
extensive tests, that surface-bound Fe(II) on magnetite (or in combination the
reactive Fe(II) in
non-stoichiometric magnetite) could rapidly reduce selenate to selenite and
then elemental
selenium. Selenite appears to be an intermediate product and will finally be
further converted to
elemental Se. A third aspect is discovery that the chemical conditions that
promote the
formation of magnetite (Fe304) as a reaction product from the oxidations of
Fe(0) and surface-
bound Fe(II) (coupled with reductions of dissolved oxygen, nitrate, and
selenate in the water). A
fourth aspect is development of a circulating bed system (e.g. a fluidized bed
and/or stirred
system) with an internal settling zone and a central conduit that can (a)
retain high concentration
of Fe304 solid particles and therefore offer abundant reactive surface area
that can host surface
bound Fe(II)-selenate redox reaction; (b) offer an effective mixing condition
so that Fe , Fe:304
and s.b.Fe(II) can achieve their respective roles in removing toxic metals;
(c) avoid excess
diffusion of oxygen from air into the reactive system so that less Fe and
Fe(II) are wasted. A
fifth aspect is development of a multiple-stage fluidized bed system that will
(a) achieve better
toxic metal removal efficiency than a single stirred-tank reactor; (b)
mitigate the inhibitive effect
of certain impurities in water such as dissolved silica on iron corrosion
reaction through the use
of first stage so as to maintain high toxic metal removal efficiency in the
following stages; (c)
control nitrate reduction efficiency to a level of desire; (d) reduce
consumption of ferrous salt
and Fe ; (e) reduce or completely eliminate residual dissolved Fe2'.
[00120] Bench Scale Tests
31
CA 02787032 2016-01-18
WO 2011/035263 PCT/US2010/049528
[00121] Single Stage Reactor
[00122] Three bench-scale fluidized-bed reactors were fabricated and
operated.
[00123] Reactortil had an internal settling zone (the compartment on the
left side) in
which it allows reactive solid to separate from the water and be retained
within the fluidized
zone. Reactor#2 is identical to Reactor/41 Reactor#1 and #2 both had an
operating capacity of
7.2 L and had an internal settling zone (0.5 L) within the reactors.
[00124] Reactortt3 is an integral system that has an internal settling zone
(far left), an
aeration basin (near left), and a second settling basin (right) within the
reactor. Reactor#3 had an
operating capacity of 10 L. It had a built-in aeration basin (0.6 L) and a
built-in final settling
TM
basin (Figures 5A and 5B). Peristaltic pumps (Masterflex pumps, Cole-Farmer,
Illinois) were
used to pump in wastewater and the needed chemical reagents. A small aquarium
air pump
(purchased from Wal-Mart) as used to provide aeration. A motorized stirrer
(max. 27 watt,
adjustable rpm 100-2000, three-blade propeller stirrer) was used to provided
mixing condition.
[00125] Zcrovalent iron powder used in the tests was obtained from Hcpure
Technology
Inc., including H200+ and HC15 (see Batch Test results for more details).
Other reagents used
in the operation include HC1, FeCl2, and NaOH.
[00126] Startup
[00127] Contrary to what many experts in ZVI technology believed, fresh ZVI
is not
necessarily more effective for chemical reduction of selenate. Batch test
results (Appendix B
and Appendix C) confirmed that ZVI grains coated with magnetite could achieve
a much higher
reaction rate than ZVI grains of a relative fresh surface with little or very
thin iron rusts. To
improve performance of a ZVI system, a unique start-up process is employed to
coat the ZVI
powder surface with a more reactive and passivation-resistant, chemically-
stable magnetite
coating. When a reactor was started with using fresh ZVI powder, it took some
time under
carefully controlled chemical environment to coat ZVI with a magnetite layer.
[00128] Several factors are desirably considered in order to have a rapid
and successful
start-up for a treatment system. First, the physical chemical properties of
iron, most important
the size distribution of iron particles, are considered. Both reductions of
selenate by ZVI and by
32
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
surface bound Fe(II) (s.b.Fe(I1)) on magnetite are surface-mediated
heterogeneous reactions;
therefore, increasing solid-liquid interfacial area would increase overall
reaction rate. Fine ZVI
powders could provide larger surface area and therefore achieve higher
selenate reduction under
comparable conditions. This was confirmed in batch tests. The continuous flow
reactor tests
were successfully started up five times. It appears that finer iron particles
(dominant size: <45
um in diameter) may be started up faster than larger particles (dominant size:
45-150 gm in
diameter). The chemical purity of ZVI powder was found to not a major factor.
In batch and
continuous-flow tests, various purities and composition of ZVI powder were
used. No major
differences were observed among the different iron sources with respect to
reaction mechanism
and rate for selenate reduction. Over time, the zerovalent iron grains may all
be coated with a
magnetite coating and in the present of dissolved Fe2' , they all achieve high
reactivity for
selenate reduction.
[00129] Generation of a magnetite coating on a ZVI particle is helpful to
the success of
the system. Appropriate aqueous chemical conditions must be maintained for the
purpose. Iron
corrosion could produce various iron oxides under different chemical
conditions. Our batch and
continuous flow reactor tests show that in order to generate magnetite from
iron corrosion
reaction, three conditions must be met: a pH of 6.5 to 7.5; adequate dissolved
Fe2+ that can form
s.b.Fe(II); and appropriate species and concentration of oxidants. Oxidants
can be certain
oxyanions such as selenate, nitrate, nitrite, iodate (103-) and periodate (104-
) in the wastewater.
Oxidation of ZVI by these oxidants tends to form ferric oxides (most likely
lepidocrocite, y-
Fe0OH). The small quantity of ferric oxides can be transformed to magnetite in
the presence of
surface-adsorbed Fe(II). Dissolved oxygen can also serve as an oxidant to
generate magnetite
(Huang et al. 2006). Low-intensity aeration in the early stage could
accelerate the magnetite-
coating process. High-intensity aeration should be avoided because it could
form large quantity
of ferric oxides even in the presence of dissolved Fe2+ and moreover, it will
waste ZVI. Our
experiences from five successful start-ups using simulated FGD wastewater
indicates that in
general the system will take about one to two weeks for the fresh ZVI to
mature; over time, the
system will gradually improve before reaching a state of high performance.
33
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
[00130] As an alternative (and recommended) start-up procedure, we used
nitrate solution
(add 30 mg/L nitrate-N in tap water, operating HRT = 12 hr) instead of
simulated FGD
wastewater to feed the system. Nitrate would be completely reduced and in the
presence of
adequate dissolved Fe2+, a high quality (better crystallized and less
amorphous, containing less
ferric oxides or ferrous hydroxides) magnetite coating can be formed on ZVI
particles. Start-up
with nitrate solution would take only two days.
[00131] A general start up procedure and exemplary controlled parameters
included one or
more of the following:
1) Selection of ZVI sources. Finer iron powder (< 50 gm) is preferred. Low
iron purity and
rusty surface in general are not a problem.
2) Add 80-100 g/L ZVI powder in the fluidized zone. Turn on mixing equipment.
3) Start-up with FGD wastewater
= Feed FGD wastewater at a rate equivalent to HRT=12 hrs. The exact
compositions of
raw FGD wastewater may vary widely, but in general contains high concentration
of
C1, sulfate, and relative high concentration of nitrate.
= Feed FeCl2 solution (0.1 M FeC12 in 0.005 M HC1 solution) at a rate
equivalent to 1.5
m mole Fe2+ per 1 L wastewater
= Feed HC1 at a rate to control the pH in the fluidized zone at 6.8-7.2.
= If the FGD wastewater contains limited concentration of nitrate (e.g.,
below 10 mg/L
nitrate-N), then a low intensity aeration in the fluidized bed should be
provided to
accelerate the formation of a magnetite coating.
[00132] Start-up with nitrate solution:
= Feed nitrate solution (30 ing/L nitrate-N) at a rate equivalent to HRT=12
hrs.
= Feed FeCl2 solution (0.1 M FeC12 in 0.005 M HC1 solution) at a rate
equivalent to 1.5
m mole Fe2 per 1 L wastewater
= Adjust HC1 solution (0.1 M HO) feeding rate to control the pH in the
fluidized zone
at 7.0-7.5.
34
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
[00133] Normal Operation
[00134] Once started up successfully, the system requires only low-level
maintenance
effort. Routine operations and maintenances include one or more of:
(a) Monitor the quality of wastewater entering the system. The most important
parameters
include: pH, alkalinity, acidity, total suspended solid (TSS). Of course,
toxic constituents
in the raw wastewater should be monitored.
(b) Monitor the pH in the fluidized reactive zone. Performance of the system
depends
mostly on pH. For a single-stage system, pH in the reactive zone should be
maintained
within 6.5 to 7.5. Both HC1 and FeC12 can be used to control the system.
(c) Monitor the pH in the aeration basin. Dissolved Fe2 can be oxidized more
rapidly at pH
> 7Ø Formation and settling properties of ferric oxide flocculent depends
also on pH.
Therefore, it is recommended that aeration basin be operated at pH 7.5-8Ø
(d) Monitor the performance of settling tank and sand filtration bed. The
maintenance
requirements arc no different from those unit processes in typical water or
wastewater
treatment plants. Most importantly, the settled sludge should be discharged or
returned at
an appropriate rate to avoid excessive build-up of the reactor.
(e) Excess solid discharge and disposal.
If the raw wastewater contains relative high suspended solids, a pre-settling
basin may be
needed to reduce TSS in wastewater before entering the system. This can avoid
accumulation of
inert TSS in the fluidized reactive zone that might dilute the effective ZVI/
FeOx solid
concentration.
[00135] For a single-stage reactor, the concentration of total solid in the
fluidized zone
could be maintained between 80 and 120 g/L. Assuming that 30 mg Fe27L be
converted to
Fe304 and the reactor is operated at HRT = 4 hours (based on test results), we
estimate that it will
add 0.25 g/L FeOx solid per day and therefore will take 160 days for the
reactor to increase its
solid from 80 g/L to 120 g/L. This estimate conforms to the fact that during a
three-month
continuous flow test (hydraulic retention time varies between 3 to 12 hours),
we discharge no
solid from the fluidized bed reactor.
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
[00136] ZVI/ FeO x reactive solids are considered mature when the surface
of ZVI grains is
covered with well crystallized magnetite (dark black color after dry) and a
significant presence of
discrete magnetite crystalline (may be aggregated into a larger particle due
to its strong magnetic
property). Unlike typical ZVI powder, matured ZVI/Fea, reactive solids will
not cement easily
when settled at the bottle. Therefore, the reactor could be stopped for weeks
with no risk of iron
powder cementation. That is, the reactor can be stopped and restarted very
flexibly without a
need to vacate the ZVI/ FeO x mixture from the reactor.
[00137] Results
[00138] Results of testing are described in Appendix A and Appendix D. The
results
demonstrate that a single-stage reactive system alone can effectively remove
high concentration
of selenate within a relatively short reaction time. A multiple-stage system
can further improve
the overall performance. Since for most FGD wastewater, Se(VI) concentration
will be lower
than 5 mg/L used in this test (most typically, 1-2 mg/L), the present inventor
estimates that an
HRT of less than 4 hours would be sufficient for most applications. Moreover,
the reactor is
operated at near neutral pH.
[00139] Multi-Stage Reactor
[00140] The start-up procedure and normal operation requirements described
for a single-
stage system can be similarly applied for a multistage system. Again, it is
desirable that nitrate
solution be used for rapid start-up. Nitrate solution was also found to be
very effective in
rejuvenating a fouled system in which the system was accidentally acidified
(pH dropped to
below 4.0) for a few hours, which might permanently damage iron oxide
reactivity and resulted
in extremely poor performance even after returning to normal operation
conditions.
[00141] In this test, Reactor#1, #2, and #3 was combined in sequence to
form a three-stage
FBRs treatment system. This system was a 24-liter three-stage ZVI/FeO/Fe(II)
fluidized-bed
reactor system. Initial testing on the three-stage system is described in
Appendix A and
Appendix D.
36
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
[00142] Continuous flow tests were conducted for six months on the bench-
scale (24 liter)
three state fluidized bed system based on the ZVI/Fe0x/Fe(II) technique with
high-strength raw
FGD wastewater.
[00143] The system was demonstrated during a 6 month testing period to be a
complete
success, as shown in Table 1.
Table 1.
Major Concentration in Concentration after Removal
Pollutants FGD wastewater treatment Efficiency
dissolved Se
Selenium 7.8 mg/L Sc042--Se > 98%
<0.15 mg/L
dissolved Hg
Mercury 335 g/L dissolved Hg > 99.9%
<0.2 g/L
400 g/L dissolved dissolved As
Arsenic >99.9%
As(III) and As(V) <0.2 g/L,
nitrate-N
Nitrate 26 mg/L nitrate-N > 80%
<5.0 mg/L
200-600 mgiL projected to be
Boron **
dissolved B >70%
[00144] Notes: * The original raw FGD wastewater contains only less than
0.6 1..ig/L total
dissolved As. To evaluate Arsenic treatment effectiveness, 400 p.g/L arsenite-
As and arsenate-As
was added. ** Removal of dissolved boron in the system is still being tested
and needs to be
further verified.
[00145] Laboratory Tests
[00146] Extensive laboratory tests have been conducted to understand the
treatment
conditions and mechanisms.
[00147] Settling of reactive solid (black) from fluid (clear) has been
observed by the
present inventor.
37
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
[00148] This inventor has conducted extensive batch tests (Appendix B,
Appendix C, and
Appendix D) in addition to the continuous flow tests (Appendix A and Appendix
D) to
investigate the fundamental chemistry and application issues in the
complicated reactive system
that comprised of Fe , dissolved Fe2+, various FeOx in different forms and
compositions,
dissolved oxygen, simulated FGD wastewater or real FGD wastewater with a very
complex
matrix of constituents. Laboratory experiments and their results are described
in details and
discussed in depth in the Appendix A, Appendix B, Appendix C, and Appendix D.
[00149] Those Appendices include Figure Captions as follows, renumbering
sequentially.
First, for continuous test the figures captions are as follows.
[00150] Findings from these tests are summarized as below:
1) In a rigorous anaerobic condition, selenate (at ppm level concentration)
cannot be
effective reduced by pure Fe (with fresh surface that contains negligible
iron oxides).
Only negligible selenate could be reduced. That is, reactivity of Fe will be
naturally
passivated by the presence of selenate. This explains why previous
investigators
failed to achieve a sustainable removal when using Fe to reduce selenate.
Se042- + 2 Fe + 2 H20 4 Se ,1, + 2 Fe0ON + 2 OH- (eq. 1)
Lepidocrocite (y-Fe0OH) forms a passive coating on the surface of Fe particle
and
therefore inhibits further reaction between Fe and selenate.
2) In the presence of dissolved oxygen, selenate could be reduced by Fe at a
modest
rate; however, to sustain the desired selenate-Fe reaction, much of Fe will
be
wastefully consumed by dissolved oxygen as a result. The implication is: An
excessive aerated Fe system might be able to remove selenate, but the process
is
economically infeasible due to wasteful consumption of Fe by oxygen and
generation of large quantity of iron oxide sludge.
3) Reduction of selenate could be greatly accelerated in the presence of
dissolved Fe2 at
circum-neutral pH environment. The reaction rate increases as dissolved Fe2'
increases. A presence of 0.3 mM dissolved Fe2' will be adequate. At near
neutral pH
and anaerobic environment, the reaction will form magnetite as their product.
38
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
Se042- + 2 Fe + Fe2+ 4 Se(0)1, + Fe304 (eq. 2)
In this reaction, the direct role of Fe2 might be to facilitating the
conversion of
passive Fe0OH to reactive Fe304 and therefore, greatly accelerating the
reaction.
4) Selenate could be rapidly reduced by s.b.Fe(II) on activated magnetite
surface at near
neutral or weak acidic pH in the absence of Fe .
Fe304
Fe2+ (aqueous) > s.b.Fe(") + 2 I-1+ (eq. 3)
Se042- + 9 s.b.Fe(II) 4 Se(0),1, + 3 Fe304 + 2 OH- (eq. 4)
Unlike Fe2+ in the equation 2, Fe(II) here serves as a reductant and directly
contributes one electron to the reduction of selenate.
5) Nitrate, which is often present at tens of ppm level in the FGD wastewater,
will not
inhibit selenate reduction by Fe . Indeed, nitrate was found to slightly
accelerate
selenate reduction by Fe . In contrast, reduction of nitrate by Fe will be
inhibited by
the presence of selenate. In a rigorous anaerobic environment, reduction of
nitrate by
Fe can occur only after selenate is completely reduced in the system.
6) Both reductions of nitrate and selenate by Fe will consume certain amount
of Fe2+.
Nitrate reduction consumes 0.75 mM Fe(II)/1.0 mM nitrate; selenate reduction
consume approximately 1.0 mM Fe(II)/1.0 mM selenate.
7) The complex matrix of constituents in the FGD wastewater may affect
selenate
reduction rate in the Fe /Fe0x/Fe(II) system. Sulfate will slow down the
reaction rate
several folds. Chloride at a concentration below 800 mg/L does not affect the
reaction rate. Even with the interference of high concentrations of chloride
and
sulfate, the overall reaction rate still remains reasonably fast.
8) Source of Fe . The mechanisms of Fe -selenate reaction will not be altered
by the use
of difference Fe0 sources. Tests with different purities of Fe show that Fe
purity
has no apparent relationship with the achievable reaction rate. There is no
obvious
advantage from the use of high pure (>99%), little rusted, electrolytic iron
powder
(Fisher Scientific) over low-grade (95%), industrial iron filings. The size of
iron
power however does matter. Fine iron powder will provide more reactive surface
39
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
than coarse iron powder. Fine iron powder may also mature faster and ease
start-up
of the system.
[00151] Pilot Scale Tests (Prophetic)
The success of the laboratory-scale prototype has paved the road for
constructing a pilot-
scale system and conducting extended field demonstrations to further evaluate,
develop and
refine the technology.
[00152] The present inventor contemplates a pilot-scale treatment system
based on a
proved laboratory-scale prototype and conduct long-term field tests to further
develop the
technique and finalize its design for commercialization.
[00153] The pilot scale test may involve one or more steps, such as: design
and construct a
pilot treatment system based on the laboratory prototype; conduct on-site long-
term
demonstrations in conjunction with further laboratory mechanistic study;
collaborate closely with
industry and other stakeholders to further refine the system to meet the
industrial needs and
environmental goals. Contemplated pilot scale tests are further described in
Appendix D.
[00154] The present inventor contemplates an integral treatment system that
can treat
FGD wastewater at a flow rate of 2 to 5 gallon per minute, which represents
about 1% of
wastewater expected from a 1,000 megawatt power plant. The pilot system may be
mounted on
a trailer that is adapted to be hauled to different test sites.
[00155] Example 5 below describes field experiments that are a realization
of on site
bench-scale continuous-flow treatment demonstration tests. An exemplary pilot
scale prototype
is contemplated capable of treating 1-3 gpm.
[00156] Industrial Operation (Prophetic)
[00157] Based on the bench scale test described above, the present inventor
estimates that
for treating a 500 gpm FGD waste stream from a 1,000 megawatt, a iron-based
system will
consume per year: 200 to 400 ton of iron chemical (est. bulk price: $1,000 to
$2,000/ton); 200 to
400 tons of concentrated HCl; 50-200 kilowatt electric power consumption.
Further, the present
inventor estimates that for treating a 500 gpm FGD waste stream from a 1,000
megawatt coal-
fired facility, a iron-based (e.g. hybrid ZVI/Fe0x/Fe(II)) treatment system
will generate per year
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
: 300 to 800 tons of iron oxide (weight in dry mass; laden with toxic metals),
solid waste to be
disposed.
[00158]
Example 3. Sulfide Generation for Enhancing Toxic Metal Removal in
Hybrid Zero-Valent Iron/Fe0x/Fe(II) Water Treatment System
[00159] This
example demonstrate use of sulfide generation to provide sulfide ions to
further improve the heavy metal removal capability of the Hybrid Zerovalent
Iron/Fe0x/Fe(II)
water treatment system described in Example 2. The Hybrid Zerovalent
Iron/Fe0x/Fe(II) water
treatment system was demonstrated in Example 2 to remove selenium from
industrial wastewater
(represented by flue gas desulfurization wastewater) by chemically
transforming highly soluble
selenate-selenium to insoluble elemental or selenide-selenium. The treatment
system was also
found to be effective in removing significant percentages of most toxic metals
and metalloids of
major environmental concern. Despite the great success in selenium removal,
the Hybrid ZVI
process may have difficulty in meeting the future EPA guideline for total
mercury < 12 ppt
without further process improvement.
[00160] A
bench-scale prototype Hybrid Zerovalent Iron/Fe0x/Fe(II) treatment system
was developed and demonstrated through a continuous-flow field test for
treating real FGD
wastewater. The removal efficiency for selenate-selenium and total mercury
(dissolved Hg2+
varied from about 2 ppb to 60 ppb in raw FGD wastewater) was found to be about
99.8% and
99.99%, respectively, with total Se < 10 ppb and total Hg < 5 ppt in the
treated effluent. The
prototype also achieved over 97% removal for many other toxic metals including
arsenic, lead,
chromium, cadmium, vanadium and nickel. Despite the high success of field
test, the removal
mechanism of the treatment system for toxic metals such as Hg (other than Se)
was not
adequately understood.
Therefore, there is no guarantee that the Hybrid Zerovalent
Iron/Fe0x/Fe(II) treatment system could achieve similar efficiency for Hg and
other metals
when treating wastewater of different constituent matrix.
[00161] The
bench scale test was followed up with the field test to conduct additional
laboratory tests (both batch and continuous-flow) to verify the treatment
effectiveness of both
conventional ZVI and the hydrid ZVI/Fe0x/Fe(II) for Hg removal. We found that
both
41
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
conventional and hybrid ZVI could ensure 90% removal of dissolved Hg2 removal
in a simpler
water matrix (simulated wastewater spiked with Hg2'). For example, when using
the prototype
reactor to treat a Hg-spiked tap water (supplied from groundwater, have
various concentration of
Ca2+, Mg2+, Nat, C1, 5042-, carbonate and dissolved silica etc ), 12 hr
treatment could reduce
Hg2+ from 150 ppb to about 10 to 25 ppb. Extending reaction time from 2 hrs to
24 hrs will only
marginally improve Hg removal. The 90% removal of Hg was not acceptable to the
industry.
Similar results were observed when treating DI water spiked with Hg2t.
Separate batch tests
with various combinations of water quality and constituents confirmed that
high removal Hg by
ZVI process is not guaranteed.
[00162] The extraordinary high Hg removal observed in the field
demonstration may be
attributed to certain constituents in the real FGD wastewater. This was
confirmed from
controlled batch test that compared Hg removal from real FGD wastewater with
synthetic
(composition known) wastewater. In comparable batch tests, a ZVI reactive
system could reduce
dissolved Hg2' from 153 ppb to below 0.5 ppb when treating real FGD
wastewater: in contrast, it
only reduced Hg2+ from 150 ppb to about 20 ppb when synthetic wastewater was
used. A
number of factors (pH, nitrate, selenate, and Cl-, sulfate, dissolved silica
etc) were screened.
Two most likely constituents in the real FGD water were identified that may be
responsible for
enhancing Hg2+ removal: one is iodate (or periodate), another is phosphate.
The iodate (or
periodate-- the two could not be differentiated with the IC analysis) are
present in the FGD
wastewater at ppm level. Phosphate is also existed in low ppm level. In a
continuous-flow test,
when the synthetic wastewater (spiked with 150 ppb Hg2P) is spiked with 5 ppm
iodate and 5
ppm phosphate, we observed significant improvement of Hg removal: the
dissolved Hg in the
treated effluent was lowered from > 15 ppb without iodate/phosphate additive
to about 0.7 ppb
with iodate/phosphate additive.
[00163] The presence of both iodate and phosphate in the raw FGD water
during the field
demonstration may be the main contributing factor for achieving a 99.99% Hg
reduction. From
our previous batch tests, we demonstrate that iodate or periodate could be
rapidly reduced to
iodide in a zero-valent iron system. Therefore, the true effective constituent
that enhances Hg
42
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
removal could be iodide through formation of mercury iodide minerals. More
tests are now
undergoing to further investigate Hg removal mechanism in the presence of
extremely complex
constituent matrix in real FGD wastewater. Other factors such as trace amount
of various metal
ions (e.g., A13+) could contribute to enhanced Hg removal through complex co-
precipitation
process in the presence of high concentration of Fe0x in the Hybrid ZVI
reactors.
[00164] Potential Solutions to Improve Hg Removal of Hybrid ZVI System are
as follows.
Solution 1 is Adding trace amount of Iodate/Periodate/iodide into the reactor
to improve Hg
removal in the ZVI process. Solution 2 is adding trace amount of phosphate
into the reactor to
improve Hg removal in the ZVI process. Solution 3 is adding sulfide
constituents into the
reactor to improve Hg removal in the ZVI process. Solution 1 and 2 can be
easily accomplished
by using soluble iodide or phosphate salts. Solution 3 can be accomplished by
the use of
organosulfide as additive to the ZVI reactor. A standalone Sulfide Generator
is an Alternative
Solution.
[00165] Removing toxic metal through sulfide-metal chemistry is desirable
in
consideration of the characteristics of the Hybrid ZVI reactor. First, the
hybrid ZVI reactor
could provide an anaerobic and neural pH environment where sulfide ion could
play a dedicated
role in precipitating mercury and other toxic metals. Although sulfide could
be precipitated by
ferrous iron, most of toxic metal sulfide has a much lower solubility than
that of FeS. For
example, FeS has a solubility constant of Ksp=8x10-19; in comparison, HgS has
a solubility
constant of 2x10-53 and CuS of 8x1037. As such in the co-presences of these
metal ions, sulfide
could first be used to form less soluble precipitate like HgS. Once formed,
tracer amounts of
mental sulfides could be assimilated and encapsulated in the bulk of Fe0x,
which will drive the
continued reduction of Hg and other toxic metals through the treatment trains.
[00166] To exploit the metal sulfide chemistry with the Hybrid
ZVI/Fe0x/Fe(II) system,
different methods were tried.
[00167] First method. Additive solid.
[00168] This method involves adding FeS (or FeS2) into the reactor as part
of reactive
solid to provide adsorption and precipitation sites for mercury.
43
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
[00169] This approach was evaluated in continuous flow reactor tests. We
found that
addition of 30 g of FeS (and FeS2 in a second test) into the mixture of 500 g
ZVI/Fe304/Fe(II)
only improve mercury removal slightly compared to the ones without adding FeS.
The less-
than-expected removal improvement is probably attributable to the fact that
the ZVI reactor is
operated at near neutral pH and thus the dissolution of FeS is negligible. In
addition, in the
presence of substantial dissolved Fe(II) and continued precipitation of Fe(II)
to form FeOx, any
reactive FeS surface suitable for Hg2+ adsorption and precipitation might be
quickly occupied by
fresh Fe(II) precipitation. Therefore, unless we operated the reactor under
acidified condition
(e.g., pH < 4), adding FeS in a solid powder form will not be able to
significantly improve Hg (or
other toxic metals) removal. For a multi-stage Hybrid ZVI/Fe0x/Fe(II) reactor,
it is feasible that
the first stage reactor could be operated under acidic condition (feeding
adequate HO) that the
added HC1 could be consumed to dissolve both Fe0 and FeS and produce Fe2 and
S2-. The
produced Fe2' and S2 could be used in the second (and subsequent) stage
reactors where the
operating condition could resemble that of a typical Hybrid ZVI/Fe0x/Fe(II)
reactor. The
disadvantage is that this modification will consume more ZVI and produce
excessive H2S that
may pose a safety danger or result in odor problem.
[00170] Second method. Additive reagent.
[00171] As an alternative to adding FeS to promote toxic metal sulfide
precipitation, the
reactive system can include a standalone sulfide generator (Figure 4) to
produce small amount of
sulfide ions before introducing into the reactor to precipitate toxic metals.
[00172] A sulfide generator could be a packed-bed filter column filled with
FeS (or FeS2)
powder (mixed with sand if necessary to improve its porosity and hydraulic
conductivity). A
low concentration acid is flowed through the column to dissolve FeS and
steadily and gradually
release a stream of acid leachate rich of sulfide ions to add into the
reactor. We found that 0.005
M HC1 is sufficient to dissolve FeS.
[00173] In-situ generating sulfide is easier than using Na2S salt to supply
sulfide. Na2S is
highly reactive, dangerous to handle, and highly unstable in atmosphere
environment (react with
44
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
moisture and oxygen). In contrast, FeS is relatively stable under typical
environment. The
gradual dissolution of FeS by low concentration of acid can be relatively
safely handled.
[00174] Figure 4 show a flow-chart of the hybrid ZVI/Fe0x/Fe(II) prototype
treatment
system incorporated a sulfide generator to improve mercury removal. Referring
to Figure 4,
toxic metals were removed as wastewater cascaded through four reactors in
series. Sulfide was
introduced in to Reactor 1. Fe2+ was added to Reactor 1, 2, and 3. Lime was
added to Aerating
Basin.
[00175] Preliminary results
[00176] Experimental Set-up: hybrid ZVI/Fe0x/Fe(II) in two stages (R1 and
R2), each 6.0
L effective reactive volume; Sulfide generator: 1 in internal diameter x 8 in
height glass column,
filled with 20 g FeS mixed with 75 mL silica sand (grain diameter 0.25-0.42
mm). Sulfide
leachate is introduced into R1 .
[00177] Operating conditions: Wastewater feed solution: simulated
wastewater made of
tap water spiked with 200 ppb Hg2'; Flow rate: 16.7 mL/min (or 1 liter per
hour); equivalent
reaction time = 6 hr for each stage reactor (12 hr in total); Sulfur generator
feed: 5 mM HO;
flow rate: 0.3 mL/min; estimated S2- (including H2S and HS) in the leachate =
80 mg/L.
Equivalent dose per liter wastewater = 1.5 mg/L; Fe2 feed: 0.5 mM.
[00178] Results:
[00179] When sulfide generator was operated to add 1.5 mg S2- per 1 liter
wastewater,
Hg2+ concentration in effluent of R1 was below detection limit (0.1 ppb) of
AAS-hydride
generation method. That is, 99.95% Hg removal could be achieved in a single
stage within 6 hr
reaction time. Based on our preliminary test, it appears that such high
removal was achieved
almost instantly in the reactor. A reaction time of 6 hr is not essential.
Note that the actual Hg
concentration in the effluent might be substantially lower than 0.1 ppb.
[00180] In the absence of sulfide generator, dissolved Hg concentration was
about 20 ppb
in the effluent of R1 and >10 ppb in the effluent of R2. That is, the hybrid
ZVI/Fe0x/Fe(II) can
only remove about 90% dissolved mercury. The poor additional Hg removal
suggests that
extending reaction time and stages would not significantly improve Hg removal.
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
[00181] A small amount of sulfide (in this test, 1.5 mg/L) is sufficient
for greatly
improving Hg removal. The presence of significant concentration of Fe2 does
not impede the
function of sulfide. The small amount of sulfide does not interfere with
reactivity of ZVI in term
of selenate reduction.
[00182] During the test, there is no noticeable H2S bad smell in the R1 .
The added sulfide
is fully consumed (or fixed) in the reactor.
[00183] Example 4. Treatment of fluid streams containing dissolved silica.
[00184] A bench scale prototype system with an effective volume of 20
liters was built.
Laboratory and field continuous flow tests were conducted for four months. The
system treated
40 liters water of high dissolved silica. Both artificially composited water
and real industrial
water were tested. The results demonstrated that the present reactive system
could efficiently
reduce dissolved silica in water from 230 mg/L (as SiO2) to below 10 mg/L. It
was observed that
iron corrosion products accounted for up to 80% of 200 g/L of reactive solid
in the reactor. The
reactor operated at substantially neutral pH. Conditions included ambient
temperature and
atmospheric pressure. The process produced limited solid waste.
[00185] Removal efficiency
[00186] A single stage reactor demonstrated high removal efficiency. In
particular, over
90% of dissolved silica was removed. In a field demonstration for treating
flue gas
desulfurization wastewater, a single stage reactive system, with one reactor,
consistently reduced
dissolved silica from about 70 mg/L as SiO2 to below 4.0 mg/L within 6 hours.
In treating
artificially composited water, the single stage reactor can reduce dissolved
silica from about 250
mg/L to below 10 mg/L.
[00187] In a two stage reactive system, with the first reactor the same as
in the one stage
system, in the field demonstration for treating flue gas desulfurization
wastewater, after passing a
second reactor, dissolved silica in the wastewater was further reduced to
below 1.0 mg/L.
[00188] Materials consumption
[00189] Removal of dissolved silica will consume only about 0.5 mg zero
valent iron and
0.3 mg ferrous iron for each 1 mg of dissolved silica.
46
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
[00190] pH
[00191] Removal was achieved in experiments between pH 7 and 8. Therefore
the
process will require no significant pH adjustment to the water of most
industrial applications.
This avoids the use of chemicals for increasing pH in pretreatment of a liquid
stream before
silica removal. Further, it avoids non-neutral pH driven precipitation of Ca
and Mg ions that
account for much of excessive waste solids when they are present in treated
water.
[00192] Temperature
[00193] The experiments giving high removal efficiency were conducted at
ambient
temperature. Ambient temperature is typically 22 C, but it will be understood
by one of
ordinary skill in the art that ambient temperature may be within a range near
that typical value.
[00194] Energy
[00195] The experiments used a motorized stirrer to provide adequate (not
intensive)
mixing between the composite solids and water.
[00196] Liquid Stream Composition
[00197] The process was effective for removing dissolved silica from
various water
qualities and compositional matrices. For example, high total dissolved salts
(including Na' ,
Ca2f, Mg2f, Cr, SO4-, and HCO3- ions) up to 20,0000 mg/L was found to barely
affect the
overall removal efficiency of the system in experiments. Organic matters (such
as sugar and
acetate) in the water up to 2,000 mg/L did not affect the dissolved silica
removed by the process.
[00198] Field Testing within a Waste Treatment Process
[00199] The high efficiency and reliability of waste treatment process
incorporating
dissolved silica removal was demonstrated in a five-week field test conducted
with a multi-stage
four reactor, 30-liter prototype system. The prototype accepted raw FGD
wastewater, reduced
all major pollutants of concern and produced a high-quality effluent. Reactor
1 alone removed
over 95% of dissolved silica, from about 70 ppm to below 5 ppm. Reduction of
dissolved silica
by Reactor 1 aided the function of the other reactors. The multi-stage
prototype consistently
reduced total Selenium, which existed mainly as selenate ion, from about 3,000
ug/L to < 7
ug/L. Total mercury was reduced from about 50 ug/L to <0.005 !AWL. Nitrate was
reduced
47
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
from about 25 mg/L to < 0.2 mg/L. in addition, Arsenic, Lead, Cadmium,
Chromium and
Vanadium were all reduced to sub-ppb level.
[00200] The waste treatment process uses inexpensive chemicals and produces
limited
amount of solid waste. The expendable chemical cost for treating 1 m3 of the
FGD wastewater is
estimated to be less than $0.5. Leaching tests (following the USEPA TCLP
method) was
conducted to determine the toxicity of the resultant solid waste. The leachate
was found to
contain < 0.1 mg/L of total Se, < 0.2 g/L of total Hg and < 0.1 ug/L of total
As, all of which are
well below the regulatory limits. The preliminary results suggest that the
solid waste could be
treated as non-hazardous waste.
[00201] Example 5. Field Demonstration of a Hybrid ZVI/Fe0x Reactive System
for
Treating the FGD Wastewater
[00202] Overview
[00203] This example illustrates that the present technology is adapted to
help industries
to meet stringent effluent regulations for toxic metals.
[00204] The wet scrubber is becoming more popular as an effective
technology for flue
gas desulfurization in coal-fired electric power industry. While wet scrubbers
can significantly
reduce air pollution, wet scrubbers produce waste liquid streams that are
laden with various toxic
metals including mercury and selenium of various forms.
[00205] The field demonstration described in this example illustrates that
the present
technology provides a high-performing, cost-effective, and reliable technology
that is capable of
treating flue gas desulfurization (FGD) wastewater to comply with rigorous
discharge
regulations on toxic metals. For example, the results met a desired reduction
level for selenium
and mercury of: total Se < 50 ppb and total Hg < 12 ppt, respectively.
[00206] The field demonstration permitted evaluation of the effectiveness
of an exemplary
Hybrid ZVI/Fe0x chemical treatment process for removing toxic metals in the
wastewater
generated from the flue gas desulfurization processes of coal-fired power
plants. The main target
pollutants in the field demonstration were dissolved selenium (Se) and mercury
(Hg) in the FGD
wastewater. Further, the field demonstration permitted evaluations of removal
of other
48
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
contaminants of concern including various trace toxic metals such as arsenic
(As), lead (Pb),
cadmium (Cd), chromium (Cr), nickel (Ni), vanadium (V), and zinc (Zn);
nutrients such as
nitrate and phosphate; and boron (B).
[00207] General apparatus, materials, and methods
[00208] The field demonstration described in this example used a Hybrid
ZVI/Fe0x
treatment system, exemplary of the treatment system shown in Figure 3. The
integrated Hybrid
ZVI/Fe0x treatment system employed the reactivity of elemental iron to create
a highly reactive
solid mixture of zero-valent iron particles and a special type of iron oxide
for chemical
transformation and mineralization of most toxic metals in water. The hybrid
system was
particularly effective for removing hexavalent selenium. This process employed
a special
mechanism to reverse the loss of chemical reactivity of zero-valent iron
powder due to the
formation of passive corrosion coatings on the zero-valent iron surface. The
process featured a
reactor design adapted to promote and direct the reactive power of the iron
corrosion process
toward cleaning up various harmful constituents in impaired water. The system
was designed to
minimize wasteful consumption of zero-valent iron power and thus significantly
reduce waste
sludge production.
[00209] The treatment system included reactor units and post-treatment
units. A four-
stage continuously stirred tank reactor (CSTR) reactive unit (similarly to
Figures 2, with four
stages, rather than three), with sequential CSTR stages termed R1, R2, R3, and
R4, was used in
this field test. Dissolved Se, Hg and other toxic metals and contaminants were
transformed and
removed in the reactors. The post-treatment consists of aeration + final
clarification + rapid sand
filtration, which was used to remove the residual dissolved iron and the
suspended solids.
[00210] The combined effective volume of four reactors is 30 liters. The
effective
volumes of R1-R4 are 9.0, 9.0, 6.0 and 6.0 liters, respectively. The influent
(FGD wastewater)
and chemical reagent solutions were delivered by peristaltic pumps (Masterflex
pumps, Cole-
Palmer). The mixing in each reactor was provided by an overhead motorized
stirrer. Aeration
was provided by a small aeration pump (purchased from a Wal-Mart store, for
household
aquarium use).
49
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
[00211] Three main chemicals used are zero-valent iron, Reagent B, and
Reagent C
= ZVI: The zero-valent iron powder used in this test was consisted of
various sizes (5-50
microns) and shapes of fine particles (Figure 4). The surface of ZVI powder is
covered
with rust. The purity of Fe is about 95%, with impurities consisted of about
3.5-4.5%
carbon, max.1.5% silicon, max. 2.5% oxygen. The specific surface area of iron
powder
(BET surface) was measured as 1.5 m2/g.
= Reagent B to the reactor: Surface regeneration solution is an acidified
FeSO4 solution (75
mM Fe2 and 3 mM HC1).
= Reagent C to the aereating basin: Solution of 150 mM NaHCO3 and 150 mM
Na2CO3.
[00212] The field test lasted for five weeks. The first week was the start-
up period, during
which the treatment system was optimized and stabilized. At the beginning of
the start-up, 400 g
fresh ZVI was added to each reactor. To ease and accelerate the start-up at
the field site, the iron
powders had been pre-conditioned for one week in to modify their surface
composition and
enhance their surface reactivity. The partially started-up reactors were sent
to field site for use.
After the treatment system was re-assembled the first-week's effort involved
mainly adjusting
flow rates of Reagent B and C to optimize system performance. The flow rate of
Reagent B was
adjusted between 0.1 and 0.4 L/d. Flow rate of Reagent C was adjusted between
0.2 and 0.6 L/d.
[00213] After the start-up (Week 1), the treatment system was operated
without major
accidents or problems. In Week 2, the main outlet of Reactor 1 was clogged,
resulting in
overflow of unknown amount of the reactive solids from Reactor 1 into Reactor
2. The outlet
was cleaned up and the tubing was replaced to restore normal effluent flow.
Since the accident
caused no significant changes in the overall system performance, no additional
measurement was
taken to compensate the loss of reactive solid in reactor 1. The accident in
the second week
inevitably complicated efforts to estimate ZVI consumption rates in Reactor 1
and 2. A power
outage of lasting unknown period might also occur during the weekend of second
week. The
treatment system was operated under more normal conditions during the final
three three weeks.
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
[00214] Throughout five week test period, raw FGD wastewater was fed at a
constant rate
of 30 liter/day (or 1.25 liter/hr). The corresponding hydraulic retention time
was about 24 hr.
During Week 2 to Week 5, reagent B was pumped at an equal flow rate of 0.3
L/day into each of
Reactor 1, 2 and 3. Reagent B was used to maintain reactivity of zero-valent
iron and to produce
secondary, highly reactive species for removal of toxic metals. Reactor 4 did
not receive
Reagent B. Reagent C was pumped at a flow rate of 0.5 L/day into the aeration
basin. Reagent
C was used to neutralize and precipitate the residual dissolved Fe2+ in the
effluent from reactors.
[00215] The prototype system was used to treat raw FGD wastewater that was
pretreated
only with settling in an equalization tank. A 250 liter tank was used as a
feeding tank to store
raw FGD wastewater for use of one week. In total, five tanks of wastewater
were used. Raw
FGD wastewater has an initial pH of 6.7. The pH was slightly increased to
about 7.1 to 7.3 by
adding NaHCO1 at an amount of 0.06 g/L. The wastewater was highly brackish,
containing
about 20 g/L total dissolved salts.
[00216] Temperature was not controlled during the test. The operating
temperature
appeared to mirror the ambient temperature, which varied from standard room
temperature when
the windows were closed to outdoor temperature when the windows were open,
which was as
low as 40 F in the early morning. .
[00217] Influent and effluent samples were taken twice a week on Monday and
Thursday
and submitted for outside analysis of toxic metals. The results from the EPA-
certified outside
laboratory were used to evaluate system performance in Se and Hg removal.
Additional water
samples were collected daily during workdays and transferred to the present
inventor's
laboratory for supplementary analysis. These results were mainly used to
monitor the status of
the system and adjust its operation. The present inventor's laboratory also
analyzed and
characterized iron oxide samples.
[00218] Results
[00219] Removal of contaminants
51
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
[00220] The performance for removal of contaminants was evaluated. Table 2
provides
summary results for removal of selected contaminants in treating high-strength
raw FGD
wastewater.
Table 2:
Influent Removal
Pollutants Effluent
(as total metal) Efficiency
Selenium 1910 2950 ppb Total Se < 7 ppb >99.8%
Total Hg < 0.005
Mercury 22 to 61 ppb >99.99%
ppb
Arsenic 6.4 to 10.6 ppb Total As < 0.3 ppb >97%
Cadmium 45 to 73 ppb Total Cd < 0.3 ppb >99%
Chromium 25 to 55 ppb Total Cr < 0.6 ppb >98%
Nickel 231 to 266 ppb Total Ni < 7.0 ppb >97%
Lead 3.3 ppb Total Pb < 0.08 ppb >97%
Zinc 901 to 1350 ppb Total Zn < 2.0 ppb >99.8%
Vanadium 17 to 23 ppb Total V <0.15 ppb >99.8%
Nitrate-N < 0.2
Nitrate 30 ppm Nitrate-N >99%
ppm
[00221] Removal of specific contaminants is described below.
[00222] Selenium. The treatment system was proven to be capable of
effectively
removing dissolved selenium in form of selenate at ppm level (Figure 11).
Removal of selenate
is considered the main technical challenge for FGD wastewater treatment.
During the entire test
period, total selenium in the final effluent had never been higher than 50
ppb. In fact, total
selenium in the final effluent was consistently below 10 ppb once the system
was successfully
started-up. The effluent from Reactor 2 contained less then 25 ppb, which
means that over 99%
selenate-Se had been removed by the first two stages. For selenium removal,
stages 3 and 4
52
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
appeared to be redundant, which means that the treatment time of 24 hr could
be significantly
shortened. The results demonstrate that the technology can meet the targeted
treatment standard
(total Se <50ppb) anticipated by the industry and governments.
[00223] Mercury. The treatment system achieved a remarkable Hg removal
efficiency,
consistently reducing Hg from tens of parts per billion to below 0.01 ppb.
During the entire test
period (including the start-up stage), total Hg in the effluent was never
above 0.005 ppb (Figure
12). The treatment could meet the most stringent wastewater discharge standard
for Hg (i.e.,
0.012 ppb). Analysis indicated that total Hg was reduced to below 0.1 ppb in
the effluent of
Reactor 1, which means that over 99.9% total Hg was removed in the first
stage. The results
suggest that the reaction time for reducing total Hg to below 0.0012 ppb could
be significantly
less than 24 hr.
[00224] Various other toxic metals. The results confirmed that this
treatment system
could effectively remove a broad spectrum of toxic metals including Arsenic,
Cadmium,
Chromium, Nickel, Lead, Zinc, and Vanadium. The treatment system consistently
removed over
97% of these metals.
[00225] Copper. Dissolve Cu2 (or Cu') is known to easily react with Fe and
be reduced
to Cu (solid). Previous laboratory investigation had confirmed that dissolved
Cu can be easily
removed by a zero-valent iron reactive system. According to outside analysis,
however, Cu was
the only metal that the system not only did not remove, but actually increased
after treatment.
This abnormality might be most likely caused by the corrosion of a copper
weight block that was
attached to the influent end of the reagent tubing to ensure that the intake
reached to the bottom
of Reagent C tank. Copper appeared to have corroded in alkaline (Na2CO3)
conditions, released
significant amount of dissolved cupric ions and resulted in increased level of
Cu in the final
effluent.
[00226] Nitrate. The ZVI reactors consistently removed over 99% of nitrate
during the
test. Nitrate-N was reduced from about 25 mg/L to below 0.2 mg/L. Most nitrate
(>99%) had
been removed by Reactor 3. The nitrate concentration in the effluent is well
below 10 mg/L as
N, which is the Maximum Contaminant Level for drinking water. It appears that
most of nitrate
53
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
was converted to ammonium. NH4 -N concentration increased from negligible to
about 20 mg/L
in the final effluent. As a result of this transformation, break-point
chlorination would be
desirable as a post-treatment process to oxidize ammonium to nitrogen gas to
complete the
removal of nitrogen nutrient for the FGD wastewater. Break-point chlorination
is a mature and
cost-effective technology that has been widely used in industry to remove low
level ammonium
in water/wastewater.
[00227] Dissolved silica. Dissolved silica was removed very effectively by
the system.
Real-time on site analysis by the present inventor confirmed that Reactor 1
alone removed over
95% of dissolved silica, from about 70 ppm to below 5 ppm. The increase of
dissolved silica
after Reactor 1 could be caused by dissolution of silica sand in filtration
bed or redissolution of
polymerized silica in Reactor 3 and 4.
[00228] Boron. Boron existed mainly as borate. Based on outside analysis,
no significant
amount of borate was removed during the treatment. However, previous
laboratory tests
suggested that the treatment system could achieve a much improved boron
removal under certain
conditions. For example, increasing operating pH in the reactor to near 8.0
was found to achieve
a much better borate removal.
[00229] Total dissolved solids. The system didn't reduce or increase total
dissolved solids
in any significant scale. Ca2+ and Mg2- ions in the influent passed the
treatment system without
much change. Limited removal of Ca2+ and Mg2- are desirable because it means
that Ca2+ and
Mg2- will not contribute to excessive solid waste production. There is an
obvious increase in
Na + as NaHCO3 and Na2CO3 are added during the treatment.
[00230] Other impurities. Fluoride and Bromide ions are present at a level
of about 10
mg/L in the influent. In the effluent, F- concentration appears to be reduced
to below 5 mg/L.
Phosphate in the influent was below ppm level and not detected in the treated
effluent. In a
ZVI/Fe0x system, phosphate is expected to be completely precipitated and
removed from the
solution. I- was not present in the influent, but was detected at a level of a
few mg/L in the
treated effluent. It was likely that iodate (I03-) and/or periodate (104-)
ions were present in the
54
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
influent. Previous laboratory tests confirmed that 103- and 104- could be
converted to I- by the
treatment system.
[00231] Chemicals consumed
[00232] Based on the field test results, for treating one cubic meter of
high strength FGD
wastewater, the system will consume: 150-250 g Fe , which costs about $1.5/kg;
200-300 g iron
salt, which costs about $0.2/kg; and < 50 g CaO (lime). The total expendable
chemical cost is
projected to be less than $0.5 per 1 m3 wastewater. For treating a 500 gpm FGD
waste stream,
the projected expendable chemical cost will be less than $500,000 per year.
[00233] Solid waste produced
[00234] Production of solid waste can be calculated by applying principle
of mass
balance. Based on the amount of chemicals added into the system and the
changes of total
dissolved solids in the water, it can be estimated that the system will
produce 0.5-1.0 kg waste
solid per 1 m3 wastewater treated.
[00235] The solid waste was mainly composed of magnetite and polymerized
silica. X-
ray diffraction spectra of spent solid particles from the four reactors were
obtained. The analyses
showed that the main compositions of the solids are magnetite (Fe304)
crystalline. TEM and
EDS micrographs of the reactive solids collected in R1 at the end of test were
obtained. The
analyses showed that the solids mainly consist of magnetite crystalline (P2)
and polymerized
silica (P1). Several other forms of iron minerals like hematite, maghemite and
lepidocrocite may
also be present. The well crystallized magnetite and ferric oxides in general
are chemically
stable.
[00236] Leaching tests following the USEPA TCLP method was conducted to
determine
the toxicity of the resultant solid waste. The leachate was found to contain <
0.1 mg/L of total
Se, < 0.2 ug/L of total Hg and < 0.1 ug/L of total As, all of which are well
below the regulatory
limits. TCLP hazardous limits are 1.0 mg/L for total selenium, 0.2 mg/L for
total Hg, and 5.0
mg/L for total As. Concentration of other toxic metals (lead, zinc, etc.) in
the leachate has not
been analyzed. The preliminary results suggest that the solid waste could be
treated as non-
hazardous waste.
CA 02787032 2012-07-10
WO 2011/035263 PCT/US2010/049528
[00237] Speciation of Se was analyzed in the solid waste by the present
inventor's
laboratory. It was found that elemental selenium accounts for about 60% and
selenide for about
40% of total selenium in the solid waste. Thus, results demonstrate that
soluble selenate was
removed from liquid phase through chemical reduction by ZVI to become
insoluble elemental
selenium and FeSe.
[00238] The present inventor contemplates that solid wastes may come from
several
sources. A first source is iron oxides formed through a corrosion reaction of
ZVI. The corrosion
reaction may involve one or more of reduction of nitrate, reduction of
dissolved oxygen (carried
over in the influent or aeration through open liquid surface in the reactors),
reduction of water,
and reduction of other oxyanions such as iodate. A second source is
polymerization and
precipitation of dissolved silica (possibly in association with FeOx). A third
source is iron
oxides formed through precipitation and oxidation of externally added Fe2'. A
fourth source is
CaCO3 precipitate formed when Na2CO3 (or CaO) is used to provide alkalinity
and maintain pH.
[00239] Discussion
[00240] Most of nitrate and selenate reduction had been removed in the
first and second
reactor. Most of toxic metals could have been removed in Reactor 1. In this
field test, Reactor 3
and 4 appeared to operate in an idle mode, receiving negligible pollutants
from upstream. It
could be inferred from this result that hydraulic retention time could be
significantly shortened in
future tests; e.g., from 24 hrs to 12 hrs. Reagent B added into Reactor 3 was
wasted. By the
present inventor's estimate, consumption of Reagent B could be halved. In
fact, during the start-
up stage Reagent B was once provided at a rate of about 0.15 L/d per reactor
for two days; the
results showed that the system still achieved well acceptable performance.
[00241] The system was operated at a rather conservative mode due to the
lack of in-situ
monitoring measure. The strategy was also used to reduce the maintenance need
and improve
flexibility and adaptability of the system in handling variable wastewater
qualities. Under
operation with in-situ, real-time, monitoring and automation, consumption of
chemicals and
other operating controls could be further optimized.
56
CA 02787032 2016-01-18
WO 2011/035263 PCT/1iS2010/049528
[002421 The example illustrates that the present technology offers many
competitive
advantages to industry. In particular, simplicity, reliability and efficiency
are advantages of the
present technology. More particularly, eight advantages of the present process
for removing a
contaminant from an aqueous stream are simplicity, versatility, robustness,
low initial capital
cost, low operating cost, limited maintenance, limited sludge production, and
minimization of
risky byproducts. With respect to simplicity, the present process requires no
complicated and
expensive pretreatments or post-treatments, and it accepts raw wastewater and
produces
dischargeable effluent in a single integral process. With respect to
versatility, the present process
removes most toxic metals and metalloids from various industrial waste
streams. With respect to
robustness, the present process is less susceptible to temperature variation
and water quality
disturbance and is suitable for treating water with high salts and dissolved
organic matter. With
respect to low initial capital cost, the present process does not require
expensive equipment.
With respect to low operating cost, the present process uses common,
inexpensive, nontoxic
substances (zero-valent iron and iron salts). For example, the expendable
material operating cost
will be less than $0.5 per cubic meter for treating highly polluted and
complicated FGD
wastewater. With respect to limited maintenance, the present process
facilitates process
monitoring and adjustment with standard sensors and operational controls. With
respect to
limited sludge production, the present process operates at near-neutral pH,
which reduces
chemical consumption and limits sludge production. With respect to
minimization of risky
byproducts, the present process involves little chance of forming extremely
toxic organic
mercury (or selenium) compounds.
57