Note: Descriptions are shown in the official language in which they were submitted.
CA 02792724 2012-10-19
CHELATING COMPOUNDS AND IMMOBILIZED lETHERED CHELATORS
This application is a continuation-in-part of U.S. Patent Application Serial
No.
13/052,477, filed 21 March 2011 which is a divisional of U.S. Patent
Application Serial No.
12/104,066, filed 16 April 2008 which is now issued U.S. Patent 7,932,326, the
entire
disclosures of which are incorporated herein by reference.
TECHNICAL FIELD AND INDUSTRIAL APPLICABILITY OF THE INVENTION
[0001] The present invention relates generally to the chemical field and, more
particularly, to novel chelating agents, useful intermediates for synthesizing
those chelating
agents, the immobilization of those agents on a solid support resin, and the
use of those
chelating resins to remove metal ions from aqueous solutions.
BACKGROUND OF THE INVENTION
[0002] A chelator or chelating agent is a polydentate ligand that bonds to
more than
one coordination site of a metal ion. Chelating agents have long been known in
the art to be
useful in chemical analysis, in environmental remediation and in medicine. In
chelation
therapy, a chelating agent is employed to bind a poisonous metal agent such as
mercury,
arsenic, iron, lead or aluminum in order to displace the ion from biological
ligands such as
proteins and convert the metal ion into a less toxic form that can be excreted
without further
interaction with the body.
[0003] The present invention relates to (1) novel chelating agents or
compounds, (2)
novel immobilized, tethered chelators comprising the novel chelating compounds
linked to
immobilized supports and (3) methods of employing the novel compounds and
chelators to
remove trivalent and tetravalent metal ion such as Al3+ and Pu4+ from aqueous
systems in
situ, in vivo and in vitro.
[0004] There have been previous studies of tripodal, trihydroxamic acids.
Most of
these ligands are based on tripodal platforms of tris(2-aminethyl)amine (tren)
(Matsumoto et
al., Chem. Commun. 2001, 978-979; Matsumoto et al., Inorg. Chem., 2001, 40:
190-191;
1
CA 02792724 2012-10-19
Matsumoto et al., Inorg. Chem. 2004, 43: 8538-8546; Ng et al., Inorg. Chem.
1989, 28:
2062-2066), tris(3-aminopropyl)amine (Matsumoto et al., Eur. J. Inorg. Chem.
2001, 2481-
2484); or nitrilotriacetic acid (nta) (Lee et al, J. Med. Chem. 1985, 28: 317-
323; Hara et al.,
Inorg. Chem. 2000, 39: 5074-5082). These studies teach that such ligands form
Fe3+
complexes with binding constants in the range of 1028 to 1033, so long as
there are five or six
atoms connecting the bridgehead atom of the platform and the first atom of the
hydroxamate
functional group on the sidearm (Matsumoto et al., Eur. J. Inorg. Chem. 2001,
2481-2484;
Matsumoto et al., Inorg. Chem. 2001, 40: 190-191; Ng et al., Inorg. Chem.
1989, 28: 2062-
2066). These ligands include amide functional groups in the sidearms, and the
iron
complexes appear to be stabilized by intramolecular hydrogen bonding between
the amide
functional groups (Matsumoto et al., Inorg. Chem. 2001, 40:190-191).
[0005] The common feature of all the above ligands is that the bridgehead atom
is a
tertiary nitrogen. To attach these ligands to a solid support via this
nitrogen would require
the formation of a quaternary ammonium group. This is expected to have an
adverse effect
on the chelating ability of the ligand. It will introduce a permanent positive
charge on the
ligand, resulting in electrostatic repulsion of the target metal ion. In some
cases, it will also
require a change in the conformation of the metal complex.
[0006] A few tripodal tris(hydroxamate) ligands have been prepared in which
the
bridgehead atom is a carbon, rather than a nitrogen. These ligands are built
on tripodal bases
of either 1,1,1-tris(hydroxymethypethane (Motekaitis et al., Inorg. Chem.
1991, 30: 1554-
1556) or 1,1,1-tris(hydroxymethyl)propane (D ayan et al., Inorg. Chem. 1993,
32: 1467-
1475). Hydroxamate groups were added to these tripodal bases through ether
linkages.
These studies teach that one needs 4 or 5 atoms between the bridgehead carbon
and the first
atom of the hydroxamate functional group for strong metal binding. The Fe3+
complexes of
these ligands have binding constants of 1026 to 1028. However, it is not
possible to link these
ligands to a polymeric support through the quaternary carbon bridgehead atom.
[0007] The current invention is based in the use of hydroxyalkylaminomethanes,
especially the common buffer tris (1,1,1-tris(hydroxymethyl)aminomethane), as
the tripodal
base. The use of hydroxylalkylaminomethanes allows us to construct tripodal
chelating
functional groups that will mimic the high metal binding affinities of the
ligands already in
2
CA 02792724 2012-10-19
the literature, but it also provides a free amine group that can be used to
easily attach the
ligands to a variety of solid supports.
[0008] In issued U.S. Patent 7,932,326, all of the hydroxamate ligands are
derived
from tris[tris(hydroxymethyl)aminomethane]. The oxygen atoms of tris are
alkylated with
alkyl groups of various lengths terminating in hydroxamic acids. The amine of
the tris is
linked to a polymer support via sulfonamide, carboxamide or urea groups
amongst others, to
give the resin supported ligands. This document relates to an extension of
that work.
SUMMARY OF THE INVENTION
[0009] In accordance with the purposes of the present invention as described
herein,
novel di- and tripodal compounds are disclosed for use as chelating agents.
Such compounds
include, but are not limited to, novel tripodal trihydroxamate chelating
agents having a
tris(hydroxylalky)aminomethane platform, such chelating agents bonded to a
polymeric
resin, useful intermediates for making such chelating agents and to a method
of removing a
trivalent metal such as aluminum from a solution using such chelating agents.
[0010] In the following description there is shown and described several
different
embodiments of the invention, simply by way of illustration of some of the
modes best suited
to carry out the invention. As it will be realized, the invention is capable
of other different
embodiments and its several details are capable of modification in various,
obvious aspects
all without departing from the invention. Accordingly, the drawings and
descriptions will be
regarded as illustrative in nature and not as restrictive.
BRIEF DESCRIPTION OF THE DRAWINGS
[0011] The accompanying drawings, incorporated herein and forming a part of
the
specification, illustrate several aspects of the present invention and
together with the
description serve to explain certain principles of the invention. In the
drawings:
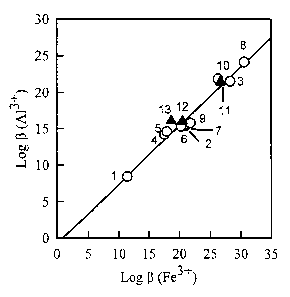
[0012] Figure 1 is a linear free energy relationship showing the correlation
between
the binding affinities of Fe3+ and Al3+ with hydroxamate ligands. Each data
point represents
a ligand, with the log f3 value for Fe3+ as the x-coordinate and the log r3
value for Al3+ as the
y-coordinate. The open symbols represent reference compounds described in the
literature.
3
CA 02792724 2012-10-19
The data points are: 1-3 represent the 1:1, 1:2, and 1:3 complexes with
acetohydroxamic
acid. Points 4 - 7 represent a series of linear dihydroxamates, in which the
hydroxamate
groups are separated by 4, 5, 6, or 7 methylene groups. Points 8 and 9 are the
binding
constants of the desferrioxamine (DFO) complex and the protonated complex of
DFO. Point
is mesitylenetrihydroxamic acid. The filled triangles represent compounds from
the
current invention. Point 11 represents the complexes of Ligand 1, point 12
represents the
protonated complexes of Ligand 1, and point 13 represents the complexes of
Ligand 7.
[0013] Figure 2 is a graph demonstrating the binding of A13' to 50 mg Resin 1
in
which the concentration of free A13+ remaining in solution after the addition
of 50 mcg Al at
time 0 to either 100 ml or 5 ml of 4-morpholineethanesulfonic acid (MES)
buffer at pH 5 has
been determined by electrothermal atomic absorption spectroscopy (ETAAS).
[0014] Figure 3 is a spectrophotometric assay showing the binding of Al3+ to
Resin 1
following addition of 22.8 mg of Resin 1 to 3 ml of 0.15 mM Al-ferron at pH 5.
Spectra
show the decrease in the absorbance of Al-ferron at 364 nm and the increase in
the
absorbance of free ferron at 434 nm. Spectrum 10 shows the reference spectrum
for 0.15
mM ferron.
[0015] Figure 4 is a graph illustrating the binding of A134 to Resin 1
following the
sequential addition of six aliquots of 100 mcg of Al to 50 mg of Resin 1
suspended in 100 ml
of pH 5 MES buffer. The first aliquot of Al was added at time = 0. Five
subsequent
additions were made at 12 hr intervals at the time indicated by the arrows on
the graph. The
free Al concentration was determined by ETAAS; and
[0016] Figure 5 is a graph illustrating the binding of A134 to Resin 1
following the
addition of 250 mg of Resin 1 to 0.5 ml of 0.23M calcium gluconate containing
¨ 9000 ng
Al/ml. The free Al concentration was determined by ETAAS.
[0017] Figure 6 is a plot of the fraction of Al remaining in solution as a
function of
time during the extraction of A134 by resin 9. Three solutions were extracted:
a 0.1 M MES
buffer at pH 6.06, which had been spiked with 6.3 ppm Al; a solution of 0.46 M
gluconate
containing 3.4 ppm Al, which had been adjusted to pH 6.4 by the addition of
tetramethylammonium hydroxide; and a commercial sample of 0.23 M
calcium(gluconate)2,
which contained 5.9 ppm Al and had a pH of 6.07. In each experiment,
approximately 240
4
CA 02792724 2012-10-19
mg of resin 9 was added to 10 ml of solution. The mixtures were stirred by a
magnetic
overhead stirrer during the extraction. At periodic times, a 100 uL aliquot
was removed
from the sample and analyzed by inductively coupled plasma-mass spectrometry
to
determine the Al concentration.
[0018] Figure 7 is a plot of the fraction of Al remaining in a solution of
commercial
calcium(gluconate)2 during the extraction of Al3+ by resin 9 (filled and open
circles) and by
the commercial chelating resin Chelex (filled triangles). In each experiment,
approximately
240 mg of resin was added to 10 ml of solution. The mixtures were stirred by a
magnetic
overhead stirrer during the extraction. At periodic times, a 100 uL aliquot
was removed
from the sample and analyzed by inductively coupled plasma-mass spectrometry
to
determine the Al concentration.
[0019] Figures 8a-8c illustrate three different embodiments of cartridges that
may be
filled with the immobilized chelating agents described herein.
[0020] Figure 8d is a partially cross sectional view of the cartridge
illustrated in
Figure 8a.
[0021] Figures 9a-9c illustrate three different embodiments, the first two of
a vessel
holding a flow-through packet containing the immobilized chelating agents
described herein,
and the third holding free immobilized chelating agents as described herein.
[0022] Figure 10 is a plot of percent removal of Al versus flow rate using the
device
illustrated in Figure 8a filled with resin 9.
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS OF THE INVENTION
[0023] The present invention relates generally to novel chelating compounds
having a
general formula of
Ri¨N¨R3
R2
[0024] wherein It' = hydrogen or tosylate, R2 = hydrogen, methyl, ethyl, n-
propyl or
isopropyl and 5
CA 02792724 2012-10-19
, ,
and R3=
a.)
0, pH
µC-N,
( \CH2/) x (Rc4H2):ji
C-N
0õ d y =R4
OH
(CH2) z =R4
wherein x, y, and z vary independently from 2 to 4, and R4 = hydrogen or CI-
C)0 straight or
branched alkyl;
b.)
HO
N-CI,
(CH2)x / R4 OH 0 µ ii
1 ,(CH2)-N-C\
0õ 0 y R4
H
1
cCH2); N¨C=R4
wherein x, y, and z vary independently from 2 to 4 and R4 = hydrogen or C1-C10
straight or
branched alkyl;
C)
\C0N(R4)0H
\ \
0) c0 / CON(R4)0H
0 CON(R4)0H
wherein R4 = hydrogen or C1-C10 straight or branched alkyl;
6
CA 02792724 2012-10-19
d.)
--CON(R4)0H
CON(R4)0H
o)C0 0---/
ON /OTh CON(R4)0H
wherein R4 = hydrogen or C1-C10 straight or branched alkyl;
e.)
0
0 (CR52)-1---N/ OH
/ NH / n R4
0 0 (CR52)_1/ n ,OH0
0 0 R4
0 \ 5(CR 2) n R4
wherein n = 2 or 3, R5 = hydrogen or methyl, and R4 = hydrogen or C1-C10
straight or
branched alkyl;
f.) C¨N OH
sR4
R4
C-14
8 OH
-0
0 OH
C¨N
sR4
7
CA 02792724 2012-10-19
wherein R4 = hydrogen or C1-C10 straight or branched alkyl;
g) 0 OOH 0 OH /A
0 OOH R4
'R4
wherein R4 = hydrogen or C1-C10 straight or branched alkyl; and
OH
h.) CLN 0 \R4
0\ )1 N 0 OH
R4
wherein R5 = hydrogen or methyl and R4 = hydrogen or C1-C10 straight or
branched alkyl.
[0025] The novel compounds of the present invention are particularly
useful as
chelators or chelating agents. One preferred use of the free ligands would be
in vivo
chelation therapy to remove metal ions such as Fe3+ and Al3+ from the body.
[0026] The compounds include an amine functional group that allows the
ligands to
be easily linked to an insoluble matrix via a sulfonamide linkage, an amide
linkage or a urea
linkage to provide immobilized, tethered chelators. Typically, the insoluble
matrix
comprises a resin support. The resin support may take the form of a macro-
porous
polystyrene such as commercially available under the trademark XAD-4 sold by
Rohm and
Haas. Other polymer resins useful in the present invention include but are not
limited to,
polyacrylate, sepharose and silica gel.
10027] The overall process of adding a chelating compound of the
present invention
to a polystyrene resin via a sulfonamide bond is shown in Scheme 1, where NR2H-
Ligand in
8
CA 02792724 2012-10-19
,
this and subsequent schemes refers to, the free amine form (R1 = H; R2 =
hydrogen, methyl,
ethyl, n-propyl or isopropyl) of any of the free ligands represented by R3 = a
through h.
, o
CI ES-Lagani \\___14H,Ligand
INSOLUBLE ,/=\ CO INSOLUBLE __(--=-\4. a - INSOLUBLE
- MATRIX ,,__J '' IRE Ts? MATRIX \ /7 II
MATRIX n__/
0 . 0
macroporous modified resin resin linker
ligand
polystyrene resin,
e.g. XAD-4
[0028] Scheme 1
[0029] The overall process of adding a chelating compound of the
present invention
to a resin support by means of an amide linkage is shown in Scheme 2.
0 0
Et3N, THT, DCC
INSOLUBLE ¨7-.= INSOLUBLE i 1
C /
/P. --- \
MATRIX MATRIX \ ITH2-
Dgard
\ / \ 0H
\ NH=ligand
\¨
[0030] Scheme 2
[0031] The overall process of adding a chelating compound of the
present invention
to a resin support by means of a urea linkage is shown in Scheme 3.
0 0
1
Insoluble Matrix 410 Insoluble Matrix, 4.
I, NH2 NH)0¨N
7 0 \
0
N-0-'-
0 /
' 2
Et3N, THF 0
r I
Insoluble Matrix, .
)- ,
HR2N¨Ligand
N NR--Ligand
H
9
CA 02792724 2012-10-19
[0032] Scheme 3
[0033] For certain applications it may be desirable to elongate the linker
by adding
polyethylene glycol units between the resin support and the ligand in order to
increase the
rate of metal binding to the resin-hound ligand. These elongated linkers are
added using
commercially available amine capped polyethylene glycols of variable length,
with the use of
a urea functional group to covalently bind the ligand and linker moieties.
[0034] The elongation process is illustrated in Scheme 4 using the linker
3-oxa-1,5-
pentanediamine as a specific example.
0 0
II 11 Et3N, THEa-
Insoluble Matrix 11 Insoluble Matrix/
11
0 H2Nas-'-'N H2
0 0
0 Et3N, THF
Insoluble Matrix; 11
0 0 H HR2N¨Ligand
0
0
2
0
Insoluble Matrix] Ito H 0 NR¨Ligand
0
[0035] Scheme 4
[0036] Other commercially available amine-capped polyethyleneglycols
include the
compounds
10
CA 02792724 2012-10-19
CH3
0 .1"-\\ 0 NH2 ) 2 n= 10-12 n
CH3NH2
which give chelating resins with the structures shown below
0
Insoluble Matrix, = S¨NH 0 0
NH NR2¨Ligand
0
Insoluble Matrix, 4. 0 H CH3 0
0 CH3 n=10-12 NH NR2¨Ligand
[0037] The immobilized, tethered chelators of the present invention
comprise the
chelating compounds identified above bound to a resin support through an
appropriate
linkage. The immobilized, tethered chelators of the present invention may be
generally
described as having the following formula:
R6-N-R3
R2
wherein R6 =
11
CA 02792724 2012-10-19
\ 0
Insoluble 9 Insoluble
Matrix 0 Matrix
9
Insoluble Insoluble CH2¨NH¨
Matrix ¨CH2¨NH Matrix
Insoluble 0
Matrix
0
0
!I
Insoluble
Matrix 0 HNH
Insoluble \ 9 H CH3 0
Matrix
0 µ.., r,u
1 13 fl
n = 10-12
R2 = hydrogen, methyl, ethyl; n-propyl or isopropyl and
R3 =
a.)
¨Nõ 4 N
/C orc1-1H2),._ OH
(\CH2)x ¨
Y sR4
?I pH
(CH2)z
R4
wherein x, y, and z vary independently from 2 to 4 and R4 = hydrogen or Cl-C10
straight or
branched alkyl;
12
CA 02792724 2012-10-19
b.)
HO ;0
(CH2)x OHO
r(CH2)-N-C
0 y R4
OH n
/;-
CH2)--N¨Cs
z R-
wherein x, y, and z vary independently from 2 to 4, and R4 = hydrogen or C1-
C10 straight or
branched alkyl;
c.) õO/ \CON(R4)0H
\CON(R4)0H
OD( 0/
0
\ /CON(R4)0H
wherein R4 = hydrogen or C1-C10 straight or branched alkyl;
d.)
--CON(R4)0H
CON(R4)0H
OX0 0-7
0\
/0-\CON(R4)0H
wherein R4 = hydrogen or C1-C10 straight or branched alkyl;
13
CA 02792724 2012-10-19
, '
e.) 5 0
0 /(CR 2)__ILn NPH\
fiLNIFI R4
0 0 i(CIR-2)n-..fiN,OH c 0
,C0............õõ)---NH
\
0 0 R4
\ ,NH OH
II \ 51L N:
0 (CR-2)n se
wherein n = 2 or 3, R5 = hydrogen or methyl, and R4 = hydrogen or CI-C10
straight or
branched alkyl;
0 pH
f.) 0 8-N , FrA
R4
9¨N
---E-30 * 0 OH
0
0 0 OH
841,
R4
wherein R4 = hydrogen or C1-C10 straight or branched alkyl;
g) OOH
. C-14,
0H4 ogirf ,ip
OOH
%R4
14
CA 02792724 2012-10-19
wherein R4 = hydrogen or C1-C10 straight or branched alkyl; and
h.) O OH
rN \R4
0
R5 0 OH
0\ ) N
R4
wherein R5 = hydrogen or methyl and R4 = hydrogen or C1-C10 straight or
branched alkyl.
[0038] PREPARATION OF COMPOUNDS OF THIS INVENTION
[0039] Selected chelating agents and chelating resins from this invention are
listed in
Table 1.
Table 1. Partial list of chelating agents and chelating resins included in
this invention
Ri-N-R3
R2
15
CA 02792724 2012-10-19
, . . .
Free Ligands (R1 = Tosyl, 12-=H)
Ligand IC x y z R2 R5
Ligand 1 a 2 2 2 H
Ligand 2 a, 3 3 3 H
Ligand".,. a 4 4 4 H
Ligand 4 '.: 4 4 2 H
Ligand 5 c H
Ligand 6 d H
Ligand 7 b Methyl
Ligand a 3 3 2 H
Ligand 9 a
Resins (R6 = polystyrenesulfonate, R4=H)
Resin 1 a 2 2 2 H
Resin 2 h Methyl
[0040] Example 1
[0041] Synthesis of Ligand 1
[0042] The overall synthesis of Ligand 1 is shown in Scheme 5. The
aminotriol (1,
Tris buffer) was reacted with acrylonitrile in the presence of a catalytic
amount of base to
give the trinitrile (Intermediate 1) (Newkome, G.R. and X. Lin, Symmetrical,
four-
directional, poly(ether-amide) cascade polymers. Macromolecules, 1991, 24(6):
1443-1444).
Reaction of Intermediate in refluxing methanolic HC1 gave the tris(methyl
ester)
(Intermediate 2). Reaction of Intermediate 2 with tosyl chloride gave the
sulfonamide tris
ester (Intermediate 3). This ester was converted to the trihydroxamic acid
(Ligand 1) by
reaction with 0-trimethylsilyi hydroxylamine (NH,OTMS) in methanol.
16
CA 02792724 2012-10-19
dioxane, KOH (cat), H2N_e00
CNI oN Me0H, HCI,
reflux
H2N OH
1 OH
33% 1.25 eq per OH
\ /C N
55%
Intermediate 1
//CO2Me
/CO2Me
Et3N, THF
0 0
H2NOCOMe
g¨IF\i¨e ---¨0O2Me
=0 so201
8 0
CO2Me
Intermediate 3 CO2Me
Intermediate 2
,CONHOH
NH2OTMS Me0H KOH
AcOH Me0H
s-N
\-0 'CONHOH
Ligand 1 CONHOH
[0043] Scheme 5
[0044] Synthesis of Intermediate
1
[0045] To a stirred solution of
tris(hydroxymethyl)aminomethane (50.0 g, 412.0
mmol) and KOH (2.3 g, 4.5% of the weight of alcohol) in 1,4-dioxane (150 mL)
was added
acrylonitrile (71.17 g, 1342.4 mmol) drop wise over a period of lh, after
which a clear
solution was obtained. After stirring at room temperature for 24 h, the
mixture was made
acidic (¨ pH = 2) by the addition of dil. HC1. After extraction with CH2C12 (3
x 100 mL) the
combined organic layers were dried over sodium sulfate and evaporated to give
tris[(cyanoethoxy)methyl]aminomethane (Intermediate 1), 43.2 g (33.5%). IR
(neat) 3588,
3368, 2251 cm-3, IHNMR (CDC13) 6 3.68 (t, J=6.0 Hz, 6H), 3.44 (s, 61-I), 2.61
(t, J=6.0 Hz),
1.68 (br s, 2H); 13C NMR (CDC13) 6 118.2, 72.7, 65.9, 56.3, 19.0; HRMS (El,
MH') calcd
for C13H211\1403: 281.16147, found: 281.16138. (Newkome, G.R. and X. Lin,
Symmetrical,
four-directional, poly(ether-amide) cascade polymers. Macromolecules, 1991.
24(6): p.
1443-1444.
[0046] Synthesis of Intermediate
2
17
CA 02792724 2012-10-19
[0047] Dry HC1 gas was passed through a solution of intermediate 1 (52.6 g,
187.0
mmol) in dry methanol (150 mL) until the solution was saturated with HC1. The
mixture
was refluxed overnight. After the solution was cooled, NH4C1 was removed by
filtration,
and the filtrate was concentrated to give a gum. The gum was taken up in THF,
filtered, and
the filtrate was concentrated to get the tris ester (Intermediate 2) 37.0 g
(55.0%). IR (neat)
3394, 1735 cm-1; 11-1 NMR (CDC13) 63.69 (t,2Jh_h=6.31 Hz, 6H), 13C NMR (CDC13)
5 172.4,
69.0, 67.1, 59.6, 52.0, 34.7; HRMS (El, MH+) calcd for Cl6H30N09: 380.19217,
found:
380.19205. (Nierengarten, J.F.; Habicher, T.; Kessinger, R.; Cardullo, F.,
Diuederich, F.;
Gramlich, V.; Gisselbrecht, J.P.; Boudon, D.; Gross, M., Macrocylization on
the fullerene
core. Direct regio- and diasterioselective multi-functionalization of
[60]fullerene, and
synthesis offullerene-dendrimer derivatives. Helv. Chim. Acta, 1997, 80: 2238-
2276).
[0048] Synthesis of Intermediate 3
100491 To a stirred solution of tosyl chloride (10.0 g, 52.4 mmol) and the
tris ester
(Intermediate 2) (19.90 g, 52.4 mmol) in CH2C12 was added NEt3 (6.37 g, 62.9
mmol) and
the mixture was heated at reflux overnight. The solvent was removed in vacuo,
and the
residue was redissolved in CH2C12 (200 mL) and washed with water (3 x 100 mL).
The
organic layer was dried over Na2SO4 and concentrated to give a gum. Column
chromatography using silica gel with 50% ethyl acetate in hexane yielded a
gummy solid of
Intermediate 3 (20.4 g, 73%), which later crystallized on storing at room
temperature.
Finally it was characterized by X-ray crystallography. IR (neat) 3610, 3287,
1736 cm-1; 114
NMR (CDC13) 5 7.78, 7.76, 7.27, 7.24 (s each, 4H), 3.68 (s, 9H), 3.51 (s, 6H),
3.51 (t, 24-h=
6.5 Hz, 6H), 2.41 (t, 23h_h= 6.5 Hz, 6H) 2.41 (s, 3H); 13C NMR (CDC13) 5
172.1, 142.8, 140.5,
129.2, 127.0, 69.9, 66.7, 62.4, 51.9, 34.7, 21.6; BERMS (El, MH+) calcd for
C23H36N0NS:
534.20095, found: 534.20093.
[0050] Synthesis of Ligand 1
[0051] To a stirred solution of the tris(ester) (Intermediate 3) (8.23 g, 15.4
mmol) in
methanol (100 mL) was added NH2OTMS (9.74 g, 92.5 mmol) followed by KOH (2.60
g,
46.0 mmol). After 6 h at room temperature, the reaction mixture was treated
with 20 g of
prewashed Amberlyst-15 and swirled for 1 h. The resin was filtered off and the
filtrate was
evaporated to give a gum. Recrystallization from acetone:hexane (1:1) yielded
the tris
18
CA 02792724 2012-10-19
hydroxamate (Ligand 1), 5.02 g, (61%) which was characterized by X-ray
crystallography.
IR (neat) 3184, 1631 cm-1; 1H NMR (CDC13) ö 7.76, 7.73, 7.42, 7.39 (s each,
4H), 3.46 (t, J =
5.8 Hz, 6H), 3.40 (s, 6H), 3.31 (s, 3H, Me0H), 2.40 (s, 3H), 2.30 (t, J = 5.8
Hz, 6H); 13C
NMR (CDC13) 8 171.1, 145.0, 139.0, 130.1, 127.0, 69.2, 67.0, 63.0, 49.2
(CH3OH), 33.3,
21.0 HRMS (El, MH ) calcd for C20H33N4O1S: 537.18677, found: 537.18665.
[0052] Example 2.
[0053] Synthesis of Ligand 2
[0054] The overall synthesis of Ligand 2 is shown in Scheme 6. The
trimethyl
orthoester of 4-iodo-1-butyric acid (3), in which the vulnerable sp2 carbon
has been
protected, is known to alkylate alkoxides (Srivastava, R. P., Hajda, J.
Stereospecific synthesis
of ether phospholipids. Preparation of 1-0-(3'-carboxypropyl)-glycero-3-
phosphoserine
from glyceric acid. Tetrahedron Lett. 1991, 32, 6525-6528) (Method A). Thus
treatment of
the BOC-protected trio! (2) with sodium hydride and the trimethyl ortho ester
(3) in DMF,
followed by deprotection with anhydrous methanolic HC1 gives the triester
(Intermediate 4).
Alternatively, reductive alkylation of the trimethylsilylated trio! (BSA,
reflux) with 3-
cyanopropionaldehyde (Iwanami, K., Kentaro Y., Takeshi, 0. An Efficient and
Convenient
Method for the Direct Conversion of Alkyl Silyl Ethers into Corresponding
Alkyl Ethers
Catalyzed by Iron (III) Chloride. Synthesis 2005, 2669-2672) (Method B),
followed by
treatment with anhydrous HC1 in refluxing methanol should also yield
intermediate 4.
Intermediate 4 is tosylated to give Intermediate 5, which is then converted to
the
corresponding hydroxamic acid (Ligand 2) by treatment with 0-
(trimethylsilyphydroxylamine).
CO2Me
1, R = H HO¨KOH OMeOMe 3
/
11?.flec(319 1 NaH, OMe
t Method A 0¨v--0 CO2Me
2, R = Boc RHN OH 2. HC1, Me0H
H2NI \-0_TCO2Me
1 FeC13, Et3S1H, H(0=)CCH2CH2CN Intermediate 4
Method B
2 Me0H, HO
r CO2Me (- --1"CONHOH
'¨
0
0 0
Et3N, THE / NH2OTMS Me0H KOH
0 CONHOH
= 0CO2Me
AcOH Me0H =g4 11
0
0 0\ F-CONHOH
0 0\ ,-002Me
19
O Intermediate 5
Ligand 2
CA 02792724 2012-10-19
[0055] Scheme 6
[0056] Example 3.
[0057] Synthesis of Ligand 3.
[0058] The overall synthesis of Ligand 3 is shown in Scheme 7. Treatment
of the
BOC-protected triol (2) with sodium hydride and the trimethyl ortho ester or 5-
iodo-1-
pentanoic acid (4) in DMF, followed by deprotection with anhydrous methanolic
HCI gives
the triester (Intermediate 6). Alternatively, reductive alkylation of the
trimethylsilylated triol
(Bis silylacetamide, reflux) with 4-cyanobutryoaldehyde (Iwanami, K., Kentaro
Y., Takeshi,
0.. An Efficient and Convenient Method for the Direct Conversion of Alkyl
Silyl Ethers into
Corresponding Alkyl Ethers Catalyzed by Iron (III) Chloride. Synthesis 2005,
2669-2672)
(Method B), followed by treatment with anhydrous HC1 in refluxing methanol
should also
yield intermediate 6. Intermediate 6 is tosylated to give Intermediate 7,
which is then
converted to the corresponding hydroxamic acid (Ligand 3) by treatment with 0-
(trimethylsilyl)hydroxylamine).
CO2Me
OMe
/\/\/ CO2Me
HO¨OH 4
1. NaH, I OMe
Ve6c1?1 r Method A
2, R = Boc RHNI \¨OH 2. HCI, Me0H
H2NI
CO2Me
1. FeCI3, Et3S1H, H(0=)CCH2CH2CH2CN
Method B Intermediate 6
2. Me0H, HCI
CO Me CONHOH
(;_0/\/.\/
CONHOH
Et3N, THF 0
9/\/\/co2Me NH2OTMS, Me0H, KOH ik 0
S N
,-N
AcOH Me0H " H
SO2CI oo\/v\CO2Me
\¨ \,/\/\CONHOH
Intermediate 7 Ligand 3
[0059] Scheme 7
20
CA 02792724 2012-10-19
10060] Example 4
[0061] Synthesis of Ligand 4. The binding constants for Ligand 1 (see below)
indicate that two arms of the ligand bind to metal ions very strongly, but
that steric hindrance
weakens the binding of the third arm. In the heteropodal Ligand 4, the length
of two of the
ligand arms have been extended to relieve this internal strain. The synthesis
of ligand 4 is
shown in Scheme 8. To prepare heteropodal trihydroxamic ligands, two of the
hydroxyls on
the aminotriol (tris) are first blocked by a protecting group. The aminotriol
(1) is converted
to the known cyclic acetal (5) using a published 2 step, 1 pot procedure (Ooi,
H., Ishibashi,
N., Iwabuchi, U., Ishihara, J., Hatakeyama, S.. A concise Route to (+)-
Lactacystin. J. Org.
Chem. 2004, 69, 7765-7768). Alternatively, the diol can be protected as the
benzylidene
(6a) (Balakumar, V., A highly regio- and chemoselective reductive cleavage of
benzylidene
acetals with EtA1C12-Et3SiH, Synlet, 2004, 647-650; Low, J.N., B.F. Milne, J.-
N. Ross, and
J.L. Wardell, Derivatives of 1V,N'-bis[2-Hydroxy-1,1-
bis(hydroxymethyl)ethyl]ethanediamide. Journal of the Brazilian Chemical
Society, 2002,
13: 207-217) using similar reaction conditions, which results in additional
options for
deprotection later in the synthetic sequence. Addition of the remaining free
alcohol to
acrylonitrile yields the mononitrile (7) (Newkome, G. R., Lin, X. Symmetrical,
four-
directional, poly(ether-amide) cascade polymers. Macromolecules. 1991, 24,
1443-1444).
Simultaneous deprotection of the acetal and methanolysis of the nitrile with a
refluxing
solution of methanolic HC1 yields the monoester-diol (8). The diol is then
alkylated by the
addition of the trimethyl ortho ester of 5-iodopentanoic acid to form the
triester (Intermediate
8). An additional complication in the alkylation of the monoester diol is base-
catalyzed beta
elimination of the alkoxy group beta to the ester. Intermediate 8 is tosylated
to give
Intermediate 9, which is converted to the corresponding trihydroxamate (Ligand
4) by the
addition of 0-(trimethylsilyphydroxylamine.
21
CA 02792724 2012-10-19
,
/ON
HOThr-OH
B20, DMF then
HO-v-Ov NaOH, DMSO
0
¨KO
Me2C(OMe)2, pTSA
CN õ...-...-::-.'
H20-0H
HNI \¨Of\
H,N 0K
HO-v-0 ''_
R
1
13o c 5
7
H"-0
BOG
6a, R = H
6b. R = OMe
OMe
iCO2Me
HCI, Me0H
r CO2Me
1 NaH,
0
,.....,..)<OMe
4
r--\_102Me
_ \O--v¨OH
I
OMe
OThr
_,.
2. HCI, Me0H
H2N7\¨OH
H2 NI \--0\ _...
02Me
8
Intermediate 8
(CO2Me
rCONHOH
0
\CD
Et3N,THF
0
/¨\_/CO2Me
p Fici----\_ JCONHOH
I
t.
40.
NH2OTMS, Me0H, KOH
, = S¨N
H
AcOH Me0H
H P SO2CI
0
0\ j--\
0
\___I-7LONHOH
CO2Me
Intermediate 9
Ligand 4
[0062]
Scheme 8
[0063]
Example 5.
[0064]
Synthesis of Ligand 5. The synthesis of the heteropodal Ligand 5 is described
in Scheme 9. The extension of one arm of the ligand is achieved by the
reaction of the acetal
protected aminotriol (5) with chloroacetic acid, followed by selective
reduction of the
carboxylic acid and deprotection of the cyclic acetal to give the unsymmetric
triol (9).
Adding the triol to acrylonitrile gives the trinitrile Intermediate 10, and
methanolysis of the
nitrile gives the tris(ester) (Intermediate 11).
This compound is tosylated to give
Intermediate 12. The addition of 0-(trimethylsilyl)hydroxylamine to
Intermediate 12 gives
the heteropodal Ligand 5.
22
CA 02792724 2012-10-19
=
HO
HO
HO¨v¨Oy NaH, CICH2CO2H BH3 THF
HN/ \-0/ \
Eoc 5 HN1 \-0/\
Boc
Boc
HO
0 CN
HCI, H20, Me0H 'o OHNaOH, DMSO
OO CN
H2N/ \--OH CN
9 H2N CN
/ \ Intermediate 10
0 CO2Me
HCI, Me0H, reflux - Ox0 CO2Me /¨\ Et3N,
THF
SO2C'
H2N 0 CO2Me
Intermediate 11
/\ / \
/\./.0 CO2Me /\,0
CONHOH
0 0
II 2e 0/ \CO M NH2OTMS, Me0H, KOH II
0/ \CONHOH
S ¨N =S¨N
8 H AcOH Me0H 8 H
0 CO2Me 0
CONHOH
\
Ligand 5
Intermediate 12
= [0065] Scheme 9
[0066] Example 6.
[0067] Synthesis of Ligand 6
[0068] The synthesis of Ligand 6 is shown in Scheme 10. The
diol (8) from Scheme
8 is reprotected at the amine with a Boc group to give (10). The remaining
hydroxyls are
reductively alkylated with aldehyde (11) followed by methanolysis to yield the
triester
(Intermediate 13). The aldehyde (11) is easily prepared in two steps from
glycol. Although
synthesis of the ester (12) would provide a more direct approach, the reaction
would be
complicated by competing, rapid lactonization to lactone (13). Intermediate 13
is tosylated to
give intermediate 14. This compound is treated with 0-
(trimethylsilyl)hydroxylamine to
give the heteropodal trihydroxamic acid Ligand 6.
23
CA 02792724 2012-10-19
, .
(CO2Me
(CO2Me 0 0---\ 11 /---\
CO2Me
1, FeCI3, Et3SiH \\--/ CN . 0¨v-0 0--/
0¨v¨OH 2. HCI, Me0H, reflux H2N/ \--0 0¨\ \-
-/ CO2Me
RC OH
8, R = H Intermediate
13
10, R = Boc
0
1. Glycol (10 equiv.), NaH HO 0¨\ PCC, CH2Cl2 0 0¨\
HO 0¨\ CD)
CICH2CN
\¨/ CO2Me CN --,-
' \-1 ON
11 12
13
('CO2Me
0
KCO2Me /---\ CO2Me
0¨v¨O 0¨/ Et3N, THF ip ___.(c_ /¨\
ICO2Me NH2OTMS, Me0H. KOH
. = s-N 0 0_, ).--
ll H AcOH Me0H
H2N/ \-0 0--\ * SO2CI 0 0 0¨\
\¨/ CO2Me \¨/ CO2Me
Intermediate 13
Intermediate 14
(CONHOH
0
o______ /--\ CONHOH
11 i¨N 0 0¨/
\--/ CONHOH
Ligand 6
[0069] Scheme 10
[0070] Example 7.
[0071] Synthesis of Ligand 7. The overall synthesis of Ligand 7
is shown in Scheme
11.
24
CA 02792724 2012-10-19
/ \ /CO2Me
OH dioxane, KOH (cat) 0 CN Me0H, HCI, reflux 0/
H2N¨& H2N -CCH3
OH --;"---CN 1.25 eq per OH 0 CN 55 A 0
\ /
33%
Intermediate 15 CO2Me
Intermediate 16
/CO2Me
Et3N, THF 0= 0/
g1CH3
SO2C! I I \-0
0
Intermediate 17 CO2Me
,CONHOH
NH2OTMS Me0H KOH 0 CH3 /
= 4_,1 \co
AcOH Me0H
0
Ligand 7 CONHOH
[0072] Scheme 11
[0073] Synthesis of Intermediate 15
[0074] To a stirred solution of 2-amino-2-methy1-1.3-propanediol (50.0
g, 475.5
mmol) and KOH (1.0 g, 2% of the weight of diol) in 1,4-dioxane (100 mL) was
added
acrylonitrile (56.7 g, 1070.0 mmol) dropwise over a period of 1 h, after which
a clear
solution was obtained. After stirring at room temperature for 24 h, 200 mL of
CH2Cl2 was
added to the mixture. The mixture was extracted with water and the organic
layer was dried
over sodium sulfate. The solvent was evaporated to yield a thick oil.
Distillation under
reduced pressure (160f 'C/ 10 mm Hg) yielded Intermediate 15, 39.5 g (39%). IR
(neat)
3517, 3360, 2250 cm-1; 1H NMR (CDC13) 8 3.68 (t, J=6.1 Hz, 4H), 3.33 (Abq, Av
= 14.2 Hz,
J=8.5 Hz, 4H), 2.60 (t, J=6.1 Hz, 4H), 1.44 (br s, 2H), 1.06(s, 3H; 13C NMR
(CDC13) 6 118.2,
76.5, 65.8, 52.8, 22.6, 19.0 HR.MS (El, MI-1 ) calcd for C10H18N302:
212.14002, found:
212.13989.
[0075] Synthesis of Intermediate 16
25
CA 02792724 2012-10-19
[0076] Dry HCI gas was passed through a solution of Intermediate 16
(39.48 g, 186.1
mmol) in dry methanol (150 mL) until the solution was saturated with HC1. The
mixture was
refluxed overnight. After cooling, NH4C1 was removed by filtration, and the
filtrate was
concentrated to give a gum. The gum was redissolved in THF, filtered, and the
filtrate was
concentrated to get the diester (Intermediate 16) 30.0 g (57.0%). IR (neat)
3409, 1727 cm-1;
'H NMR (CDC13) 8 3.79 (t, J=6.1 Hz, 2H), 3.70 (2, 6H), 3.62 (s, 4H). 2.64 (t,
J=6.1 Hz, 411),
1.42 (s, 3H); 13C NMR (CDC13) 8 172.6, 71.7, 67.2, 58.0, 52.1, 34.8, 18.4;
HRMS (EL mir)
calcd for Cl2H24N06: 278.16047, found: 278.16037.
[0077] Synthesis of Intermediate 17
[0078] To a stirred solution of tosyl chloride (10.0 g, 52.4 mmol) and
Intermediate 16
(14.54 g, 52.4 mmol) in CH2C12 was added NEt3 (6.37 g, 62.9 mmol) and the
mixture was
heated at reflux overnight. The solvent was removed in vacuo, and the residue
was
redissolved in CH2C12 (200 mL) and washed with water (3 x 100 mL). The organic
layer
was dried over Na2SO4 and concentrated to give a gum. Column chromatography
using
silica gel with 40% ethyl acetate in hexane yielded a gummy solid of
Intermediate 17, (16.9
g, 74%). IR (neat) 3604, 1736, 1735 cm-1; IFINMR (CDC13) 8 7.82, 7.55, 7.29,
7.26 (s each,
4H), 3.70 (s, 6H), 3.65 (t, J=6.2 Hz, 4H), 3.33 (Abq, Av = 47.0 Hz, J = 9.1Hz,
4H), 2.53 (t, J
= 6.2 Hz, 411), 2.41 (s, 311), 1.10 (s, 3H); 13C NMR (CDCI3) 8 172.2, 143.1,
140.7, 129.6,
127.0, 73.7, 66.8, 58.9, 51.9, 34.9, 21.7, 18.2; HRMS (El, MH+) calcd for
Ci9H30N08S:
432.16931, found: 432.16922.
[0079] Synthesis of Ligand 7
[0080] To a stirred solution of the ester (Intermediate 17) (3.61 g, 8.3
mmol) in
methanol (50 mL) was added NH2OTMs (3.52 g, 33.4 mmol) followed by KOH (0.94
g, 16.7
mmol). After 6 h at room temperature, the mixture was treated with 7.0 g of
prewashed
Amberlyst-15 and swirled for 1 h. The resin was filtered off and the filtrate
was evaporated
to give a gum. Recrystallization from CH2C12:ether (1:1) yielded Ligand 7,
2.46 g, (68%).
IR (neat) 3233, 1633 cm-1; NMR (CDC13) 8 7.76, 7.68, 7.42, 7.39 (s each, 411),
3.54 (m,
411), 3.35 (s, 411), 3.30 (Abq, Av 22.6 Hz, J = 10.0 Hz, 4H), 2.40 (s, 311),
2.34 (t, J 5.8
Hz, 611), 1.08 (s, 3H); 13C NMR (CDC13) 6 171.1, 145.0, 139.0, 130.2, 127.0,
73.5, 67.0,
26
CA 02792724 2012-10-19
59.5, 49.3, 21.0, 18.8; HRMS (El, MH ) calcd for C17H28N308S: 434.15983,
found:
434.15970.
[0081] Example 8
[0082] Synthesis of Resin 1. Resin 1 was prepared by synthesizing the
chelating
functional group (R3= a) of Ligand 1 on the surface of a polystyrene resin.
The synthesis of
Resin 1 is shown in Scheme 12. Macro-porous polystyrene beads (14, Amberlite
XAD-4)
were reacted with chlorosulfonic acid to give the polymeric sulfonyl chloride
(15) (Emerson,
D. W., Emerson, R.R., Joshi, S.C., Sorensen, E. M., Turek, J.E. Polymer-bound
sulfonylhydrazine functionality. Preparation, characterization, and reactions
of
copoly(styrene-divinylbenzenesulfonylhydrazine). J. Org. Chem. 1979, 44: 4634-
4640; Hu, J.
-B., Zhao, G., Ding, Z. -D. Enantioselective reduction of ketones catalyzed by
polymer-
supported sulfonamide using NaBH4/Me3SiC1 (or BF3*OEt2) as reducing agent.
Angewandte Chemie, International Edition 2001, 40: 1109-1111). The procedures
in
Scheme 5 were used to prepare the methyl ester of the free amine form of
ligand 1
(Intermediate 2). Addition of Intermediate 2 to the sulfonyl chloride form of
the resin (15)
gave the sulfonamide triester (Intermediate 18). The ester functional groups
were converted
to hydroxamic acids by reaction with 0-trimethylsily1 hydroxylamine in
methanol to give
Resin 1. The successful conversion of the esters to hydroxamic acids was
judged from the
IR spectra. The number of ligand molecules on the surface of the resin was
calculated from
the S and N combustion analysis of the resin to be 0.3 mmoles ligand per gram
of resin.
27
CA 02792724 2012-10-19
41 Cii= ISO3H 4<¨¨S02C1 Et3N, THF \ 0 H r---CO2Me
XAD-4 15 0 0
14 H2N-0/¨0O2Me
Intermediate 18
Intermediate 2
f"-CONHOH
NH2OTMS MeOH, KOH 0 H _C ZCONHOH¨
AcOH, Me0H 8
_-CONHOH
Resin 1
[0083] Scheme 12
[0084] Synthesis of Sulfonyl Chloride Resin:
[0085] To 35 g of macroporous styrene-divinylbenzene copolymer (20-60
mesh, avg.
pore diameter: 40A, Amberlite XAD-4) in 100 mL of 1,2-dichloroethane was added
160 g
(1.37 mol) of technical grade chlorosulfonic acid with occasional swirling.
The mixture was
kept at room temperature for 12 h. The product was filtered using a glass frit
and was
washed successively with two portions of dichloromethane (DCM), two portions
of DCM-
THF mixture, two portions of THF, and a final wash with DCM. The vacuum dried
polymer
was ready to use and was stored under argon at low temperature. IR (neat)
3521, 1369, 1171
cm ; Anal. Found: C, 57.17; H, 5.50; S, 10.23; Cl, 8.49; calculated loading S,
3.22 mmol/g,
Cl, 2.39 mmol/g.
[0086] Synthesis of Intermediate 18
[0087] To polymeric sulfonyl chloride (15) (2.0 g, 5.0 mmol) in THF (50
mL) was
added a solution of the tris ester of the free ligand (Intermediate 2) (7.58
g, 20.0 mmol) in
THF (30 mL) followed by triethylamine (2.0 g, 20.0 mmol) and the mixture was
swirled for
four days at room temperature. The polymer was then filtered off and washed
successively
with THF, water, THF, DCM and dried in vacuo. IR (neat) 3494, 1732, 1169 cm-1;
Anal.
Found: C, 58.96; H, 6.81; S, 8.22; N, 2.66; calculated loading: S, 2.57
mmol/g, N, 1.90
mmol/g.
[0088] Synthesis of Resin 1
28
CA 02792724 2012-10-19
[0089] To the resin-bound triester
(Intermediate 18) (1.7 g, 4.25 mmol) in methanol
(40 mL) was added NH2OTMs (4.02 g, 38.2 mmol) dropwise with stirring at room
temperature. KOH (2.14 g, 38.2 mmol) was added and the mixture was swirled for
12 h.
The product, resin 1, was filtered off and washed successively with methanol,
water, and
methanol. The resin was then swirled with dil. acetic acid for an hour and
washed
successively with methanol, water, THF, and dried over pump. IR (neat) 3479,
1644, 1173
cm-1; Anal. Found: C, 54.65 H, 5.57; S, 9.51; N, 1.59; Loading: S, 2.97
mmol/g, N, 1.14
mmol/g.
[0090] Example 9
[0091] Synthesis of Resin 2. The
overall synthesis of Resin 2 is shown in Scheme 13.
* CISO3H
so2c1 Et3N, THF
\ 0
H
XAD-4 14
15 H2N-4---CH3 /O r¨N-0O2Me
0
0002Me
¨"\---0O2Me
Intermediate 19
Intermediate 16
NH2OTMS Me0H, KOH
H_C
r---CONHOH
AcOH, Me0H
/
CH 30
L'¨\.--CONHOH
Resin 2
[0092] Scheme 13.
[0093] Synthesis of Intermediate
19.
The synthesis of the chlorosulfonated
polystyrene resin (15) is described in Scheme 12. To this polymeric sulfonyl
chloride (5.2 g,
13.0 mmol) in THF (80 mL) was added a solution containing Intermediate 16 from
Scheme
11, (14.4 g, 52.0 mmol) in THF (50 mL) followed by triethylamine (5.26 g, 52.0
mmol). The
suspension was swirled for four days at room temperature. The product,
Intermediate 19,
was then filtered off and washed successively with THF, water, THF, DCM and
dried over
pump. IR (neat) 3492, 1735, 1170 cm-1; Anal. Found: C, 60.28; H, 7.04; S,
8.42; N, 2.61;
calculated loading: S, 2.63 mmol/g, N, 1.86 mmol/g.
29
CA 02792724 2012-10-19
[0094] Synthesis of Resin 2.
[0095] To the resin-bound diester (Intermediate 19) (5.27 g, 13.17 mmol) in
methanol
(60 mL) was added NH2OTMs (11.08g, 105.3 mmol) dropwise with stirring at room
temperature to give Resin 2. KOH (2.95 g, 52.6 mmol) was added and the mixture
was
swirled for 12 h. The resin was filtered off and washed successively with
methanol, water,
and methanol. The resin was then swirled with dilute acetic acid for an hour
and washed
successively with methanol, water, THF, and dried over pump. IR (neat) 3468,
1643, 1176
cm-I; Anal. Found: C, 53.09 H, 5.59; S, 8.82; N, 1.95; Loading: S, 2.75
mmol/g, N, 1.39
mmol/g.
[0096] Example 10
[0097] Ligands immobilized via an amide linkage
[0098] The triester intermediate of each ligand containing a free amine group
(R1 = H)
is coupled to a resin bearing a carboxylic acid using a coupling reagent such
as
dicyclohexylcarbodiimide (DCC) or N-ethyl-N"-(3-dimethylaminopropyl)
carbodiimide
(EDC) with a tertiary amine base in TI-IF solution. The esters are then
converted to the
hydroxamic acid as described above for the sulfonamide linked system. This
process is
described in Scheme 14 using Intermediate 2 as an example.
30
CA 02792724 2012-10-19
z_./CO2Me _/CO2Me
0
Et3N, THF, DCC
H2N-"e0--¨0O2Me Matrix
Insoluble =cu
0 o
Insoluble
C ¨OH
CO2Me Matrix CO2Me
Intermediate 20
Intermediate 2
/...._/CONHOH
NH2OTMS, Me0H, KOH
Insoluble 4/lik9 1-1Ã.9
matrix 0--¨CONHOH
AcOH, Me0H C¨N_ '-0
CONHOH
Resin 3
[0099] Scheme 14
[00100] Example 11
[00101] Ligands immobilized via a urea linker
[00102] The amine of a triester intermediate is coupled to a resin bearing
an amine via
a urethane linkage using a reagent such as N,N-disuccinimidyl carbonate (16),
carbonyl
diimidazole or triphosgene with a tertiary amine base in THF solution. The
esters are then
converted to the hydroxamic acid as described above for the sulfonamide linked
system.
This process is shown in Scheme 15 using Intermediate 2 as an example.
31
CA 02792724 2012-10-19
,
, . .
0
Insoluble Matrix 0I
' Insoluble Matrix, O S
NH2
NH).0-N
7 0
0
N- 0--r-'-'
0 12
16
/CO2Me
Et3N, THE
0 /
Insoluble Matrix, it
CO Me 2 NAN-6
/ H H
0 CO2Me
\
H2N_e?0,,
Intermediate 21 \
CO2Me
CO2Me
0
\
\
CO2Me
Intermediate 2
/CONHOH
NH2OTMS Me0H KOH0
/
> Insoluble MatrixiI =
AcOH Me0H0-\___CONHOH
NA N--6
H H 0
\
Resin 4 \
CONHOH
[00103] Scheme 15
[00104] Example 12
[00105] Extension of the Linker Group. In Paragraph [0032]
and [0036] we showed
three options for longer linkers that might be used to connect the chelating
agent to the
polymer resin. These linkers insert polyethylene glycol units between the
aromatic ring of
the resin and the amine group attached to the bridgehead carbon of the
chelating agent.
[00106] The invention includes the use of three amine capped
polyethylene glycol
(PEG) based linkers, 3-oxa-1,5-diaminopentane (17), 4,7,10-trioxa-1,13-
tridecanediamine
(18), and the 2-aminopropane capped polyethylene glycol with 10-12 PEG units
(19), all of
which are commercially available in bulk. A representative attachment scheme
using
Intermediate 2 and the 2 PEG unit diamine (17) is shown in Scheme 16, along
with the
structures of the two other PEG linkers (18,19).
32
CA 02792724 2012-10-19
,
. ,
Et3N THF0 Insoluble Matrix' = II
Insoluble Matrix 4. SO2CI . 1 S_ N'C)NH
11 H
0
15 H2N7-..õ..Ø.,...---.,NH2 20
17
0 0
0
,
Insoluble Matrix' 4.
"
/0H 0
21 0
N-0
\ 2
16 -
/CO2Me
0 /
Et3N, THE 0 0
Insoluble Matrix' =
' /CO2Me 1 8 H H H 0 CO2Me
/ \
\
Intermediate 22 CO2Me
H2N-60
CO2Me
0
\\
Intermediate 2 CO2Me
CONHOH
//
0
NH2OTMS Me0H KOH 0 0
' Insoluble Matrix; 0 ¨ N '---'''' N j---- N-C-0-\_CONHOH
H H 0
AcOH Me0H 0 H \
\CONHOH
Resin 5
Alternative Linkers
CH3
..(-0 NH2 )2H2N )J,0c),N H2
0 '
n
18 n = 10-12CH3
[00107] Scheme 16
19
[00108] Each amine capped polyethylene glycol linker is attached to the
activated resin
(15) by using an excess of the diamine to ensure complete capping. The
sulfonamide linked
tether (20) is activated as the N-hydroxysuccinimide (NHS) with N,N'-
disuccinimidyl
carbonate (16) (Takeda, K., Y. Akagi, A. Saiki, T. Tsukahara, and H. Ogura,
Studies on
activating methods of functional groups. Part X. Convenient methods for
syntheses of active
carbamates, ureas, and nitroureas using N,N'-disuccinimido carbonate (DSC).
Tetrahedron
Letters, 1983, 24: 4569-4572.), followed by washing to remove the excess
carbonate and the
33
CA 02792724 2012-10-19
N-hydroxysuccinimide byproduct from the resin to produce (21). Reaction of the
activated
urethane with the amino-triester (Intermediate 2) provides the resin capped
product
Inteimediate 22 which is expected to be stable to hydroxylamine and aqueous
conditions.
Final conversion to the trihydroxamate (Resin 5) is accomplished with 0-
trimethylsily1
hydroxylamine in methanol.
[00109] Example 13. Binding of Metal Ions by the Free Ligands
[00110] The acid dissociation constants for the trihydroxamate Ligand 1 and
the
dihydroxamate Ligand 7 have been determined by potentiometric titration of the
free ligands
in 0.1 M KNO3 at 25 C. The overall ligand protonation constants for Ligand 1
are log 13011 =
10.26, log 13012 = 19.68, and log 13013 = 28.15. The overall ligand
protonation constants for
Ligand 7 are log Pon = 9.80 and log 13m: = 18.49. These protonation constants
have been
used in the calculations of the metal chelate stability constants described
below.
[00111] The binding of A134-, Fe3+, an a series of divalent metal ions to
Ligand 1 has
been evaluated by potentiometric titration in 0.1 M KNO3 at 25 C. For most of
the metal
ions, two complexes were detected. In one complex, all three of the
hydroxamate groups
were coordinated to the central metal one. The stability of these complexes is
described by
the overall binding constant
[ML] (3)
A = {M][L]
where L refers to the fully deprotonated. "nonionic form of ligand 1, and
charges on the
species have been omitted for clarity.
[00112] The potentiometric analysis also detected a protonated metal chelate,
designated as MHL. The position of the 112 and-to-metal charge transfer band
in the visible
spectrum of the MI-IL complex of Fe3-1 indicated that in the MHL complexes,
two of the
hydroxamate groups are coordinated to the metal ion, while the third
hydroxamate group is
protonated and not bound to the metal ion. The stability of the MHL complexes
is described
by the overall binding constant
34
CA 02792724 2012-10-19
=
[MHL]
All = [M1{1-1-11] (4)
[00113] The calculated binding constants for the complexes of Ligand 1 are
listed in
Table 2. The binding constant for Al3+ is log 1311 0= 21.44. Ligands with only
two
hydroxamates have binding constants of about log f3110 ¨ 15 (Evers, A.,
Hancock, R.D.,
Martell, A.E., Motekaitis, R.J.., Metal ion recognition in ligands with
negatively charged
oxygen donor groups. Complexation of Fe(111), Ga(II1), In (III), Al(II1), and
other highly
charged metal ions, Inorg. Chem. 1989, 28: 2189-2195). The larger value of log
p,,0 for
Ligand 1 confirms that all three hydroxamate groups of the ligand are bound to
the Ar'.
Table 2: Binding constants for metal complexes of the trihydroxamate ligand,
Ligand 1.
Fe3+ Al3+ Cu2+ Ni24 Zr124 Mn2+ Ca2'
Log 1311, 27.60 26.27 23.61 19.10 19.13 17.06 13.34
Log 13,10 23.78 21.44 10.73 10.13 8.95 3.71
[00114] The data in Table 2 confirm that Ligand 1 shows very high
selectivity for the
binding of trivalent metal ions such as Al3+ and Fe3+ in preference to the
binding of Ca'''.
This is a critical property, as it allows this ligand to bind trivalent metal
ions in the presence
of very high concentrations of Ca2+.
[00115] Ligand 1 showed good selectivity for Al3+ and Fe3' in comparison
to the
divalent transition metal ions Cu2+, Ni2+, Zn2+, and Mn2 . However, the
binding affinities for
these metal ions were still appreciable, especially for the binding of Cu2+.
Thus it is not
claimed that the invention can remove Al' and/or Fe3+ from pharmaceutical
solutions
without also removing significant amounts of Cu2+ and Zn2+. The proposed
process for
reducing Al3+ in total parenteral nutrition (TPN) solutions involves the
removal of Al'' from
the calcium gluconate and sodium phosphate component solutions, rather than
treating the
35
CA 02792724 2012-10-19
final TPN solution. Treating the final TPN solution with the invention is
likely to remove a
large percentage of the essential ions Cu2+ and Zn2"-.
[001161 Example 14. Metal Binding by Ligand 7
[001171 The dihydroxamate Ligand 7 forms 1:1 complexes with all the metal ions
studied in which both hydroxamate groups are coordinated to the metal ion. The
stability of
these complexes is characterized by the values of log 13110 shown in Table 3.
The 1:1
complex of Al3+, Zn2+ and Mn2+ hydrolyze to form the mixed-ligand hydroxo
complexes
ML(OH). characterized by the overall binding constant
[ML(OH)][H] (5)
/811-1[M][L]
Speciation calculations based on the stability constants in Table 3 indicated
that the Al
complex of Ligand 7 existed as a mixture of the ML and ML(OH) complexes over
the pH
range of 3 to 7. If an immobilized form of Ligand 7 (Resin 2) is used to
remove Al3+ from
solutions within this pH range, the formation of the ML(OH) complex will
stabilize the
immobilized Al and facilitate removal of Al3+ from the solution.
Table 3. Binding Constants for Metal Complexes of Ligand 7
Al3+ Cu2+ Ni2+ Zn2+ Mn2+
Pilo 16.07 13.97 9.02 9.18 7.15
P11-1 11.06 0.35 -0.1
[00118]
[00119] Example 15. Binding of Al to Resin 1
[00120] The compounds and compositions of the present invention are useful in
a
method of removing a trivalent metal ion such as A13+ from an aqueous
solution. This is
performed by treating the aqueous solution with an effective amount of the
compound or
composition of the present invention. In the most preferred embodiment, the
invention
36
CA 02792724 2012-10-19
consists of a resin to which the chelating agent is attached by a covalent
bond to form a
chelating resin.
1001211 In one method of use, the resin is stirred in a solution. After the
metal ions
from the solution bind to the resin, the metal-depleted solution and the metal-
laden resin are
separated by filtration or decantation.
[00122] In a second method of use, the resin is packed in a column, and the
metal-
containing solution is passed through the column. The metal ions are retained
on the
column, while the metal-depleted solution exits from the outlet of the column.
1001231 In one possible application, the invention would be used to reduce the
amount
of Al3+ contained in total parenteral nutrition solutions, particularly for
TPN solutions given
to neonates. The binding constants shown in Tables 1 and 2 indicate that
treatment of the
final TPN solution with the invention is likely to remove essential metal ions
such as Fe3+,
Cu2+ and Zn2+ in addition to A13+. Thus the strongly preferred process is to
use the invention
to remove the Al3+ from small volume parenteral (SVP) solutions that are used
in the
preparation of TPN solutions.
1001241 The primary "culprit" SVP solutions, which are contaminated with
aluminum
thereby contributing aluminum to the final TPN admixture and therefore to the
patient, are
calcium gluconate and sodium phosphate (Driscoll, M. and D.F. Driscoll, Am. J.
Health-
Syst. Pharm. 2005, 62: 312-315). It should be appreciated that removal of Al3+
from these
solutions is difficult because the anions of these salts, gluconate and
phosphate, respectively,
are themselves strong Al-binding agents (R.J. Motekaitis and A.E. Martell,
Inorg. Chem.
1984, 23: 18-23; K. Atkari, T. Kiss, R. Bertani, and R.B. Martin, Inorg. Chem.
1996, 35:
7089-7094). Thus the invention must compete against high concentrations of
these anions in
order to remove Al3+ from the solution.
[00125] In one possible application, the compositions of the present invention
are
loaded into a flow-through filter device such as illustrated in Figures 8a-8d
and described in
greater detail below. As the SVP solution flows through the device, the
aluminum is
extracted from the solution. The device is provided in-line between the
container of the SVP
culprit solution and the TPN bag being prepared by the automated TPN
compounder. The
device has on its outlet side a membrane filter with a pore size small enough
to sterilize the
37
CA 02792724 2012-10-19
solution by filtration, retain the resin in the device and block release of
large particles from
the device. A screen on the inlet side contains the resin. Leur lock or
similar connectors on
the inlet and outlet sides enable easy connection to standard i.v. fluid
administration sets.
[00126] In another medical application, the compounds and compositions of the
present invention are utilized to ensure that aluminum is not inadvertently
included in the
dialysis solution used in peritoneal dialysis or hemodialysis. Another example
is home
peritoneal dialysis, where tap water is used to prepare the dialysate. If the
tap water contains
significant aluminum, which might have been introduced during the water
treatment process,
or might enter in the raw water, and which is not adequately removed during
the water
treatment process, the aluminum could enter the patient. In addition the
compounds and
compositions of the present invention could be used on a bulk scale in
industry to remove
aluminum from solutions such as the solutions that go into SVP containers or
any material or
process that is contaminated with aluminum, such as the guanine nucleotide-
binding
regulatory component (G/F) of adenylate cyclase, with which aluminum binds to
activate
adenylate cyclase. The following experimental data support the utility of the
claimed
compounds and compositions.
[00127] To demonstrate the ability of Resin I to bind Al, 50 mg of the resin
was
suspended in 100 ml of a buffered (0.10 M 4-morpholineethanesulfonic acid)
aqueous
solution at pH 5, and 25 mcg Al was added, as an acidic solution of aluminum
chloride. The
free Al3+ concentration in the sample solution was measured as a function of
time by
ETAAS. The results are shown in Figure 2. The 50 mg of resin removed 94% of
the Al3+
from the solution after 94 hours and 97.4% of the Al'' from the solution after
287 hours (see
Fig. 2 and Table 5). The removal of the A13+ followed first order kinetics,
with a half-life of
10.5 hrs.
[00128] The Al3+ removal experiment was repeated by adding 25 mcg of Ar and 50
mg of Resin 1 to a smaller volume of only 5 ml of MES buffer at pH 5. The
results are
shown in Figure 2. Under these conditions, 98% of the Al3+ was removed from
the solution
within 12 hr and 99.9% after 24 hours.
[00129] The removal of A131- from MES buffer was also followed by a
spectrophotometric assay in which the weaker chelating agent 7-iodo-8-
hydroxyquinoline-5-
38
CA 02792724 2012-10-19
sulfonic acid (ferron) was used as an indicator for free A13. The data are
shown in Figure 3.
The starting solution contains a 1:1 ratio of 150 microMolar A13" and ferron
in a total volume
of 3.0 ml, and the initial spectrum shows the peak at 360 nm indicative of the
Al-ferron
complex. A total of 25 mg of Resin 1 was added to the solution, and the
removal of A13"
from the solution was monitored by the loss of the absorbance of the Al-ferron
complex at
360 nm and the corresponding increase in the absorbance of free ferron at 440
nm. Based on
a comparison to the final absorbance to that of a standard solution of free
ferron, it is
estimated that the resin removed approximately 80% of the A134. The rate of Al
removal
corresponds to a half-life of approximately 90 min. The smaller percentage of
Al removed
reflected the competition for A13 from the ferron. These data were used to
estimate an
equilibrium constant for the binding of Al3+ to the Resin 1 as described
below.
[00130] To determine the capacity of Resin 1 to bind Al, sequential aliquots
of 100
mcg of A134 were added at 8 hr intervals to 50 mg of the resin suspended in
100 ml of MES
buffer at pH 5. The total amount of Al added to the solution was 6,000 ng/ml.
The
concentration of free Al remaining in the solution was followed by ETAAS. The
results are
shown in Figure 4. Because of the slow rate of Al removal in dilute solutions,
the
concentration of A134- accumulates to a total of approximately 4,000 ng/ml
after the addition
of the final aliquot of A134. However, after 200 hrs the resin removed about
85% of the
added Al3+, reducing the free Al concentration to about 1,000 ng/ml. This
indicates that the
binding capacity of Resin 1 is at least 10,000 mcg A134 per gram of resin.
1001311 The binding affinity of Resin 1 has been evaluated from four different
types of
experiments and the results are summarized in Table 5. Binding constants for
the
immobilized ligand were calculated using the speciation program HySS. A
multicomponent
equilibrium model was constructed for each reaction solution, in which the
binding constant
of Resin 1 was the only unknown binding constant. This constant was then
adjusted
manually until the HySS speciation model results matched the experimentally
determined
value for the percentage of Al bound to the resin. In all the calculations of
the binding
constants for the resin, the protonation constants for the immobilized ligand
are assumed to
be the same as those of the free ligand so that the resulting equilibrium
constant can be
expressed as a value of log13110, rather than a pH-dependent effective binding
constant.
39
CA 02792724 2012-10-19
[00132] The binding affinity of Resin 1 was determined from the final Al
concentrations shown in Figure 2 for the removal of A13 from MES buffer. In
these
calculations, the only competitive binding agent was hydroxide ion. The
calculations used
hydrolysis constants for the Al3+ for 0.1 M ionic strength taken from Mesmer
and Baes (The
Hydrolysis of Cation, Wiley, New York, 1976). The values of 13110 for the
immobilized
ligand of Resin 1 are listed in Table 5.
[00133] The aluminum binding constant for Resin 1 has been calculated from the
spectrophotometric data shown in Figure 3. In addition to A13" hydrolysis
constants, these
calculations included the Al binding constants of ferron from Martell and
Smith (Critical
Stability Constants, Vol 3, Plenum, New York, 1979). The binding constant for
Resin 1 is
listed in Table 5.
[00134] The Al binding constant for Resin 1 has been determined by competition
against the well-known hexadentate chelating agent 1,10- diaza-4,7-di
oxadecane-1,1,10,10-
tetraacetic acid (EGTA). A known amount of Resin 1 was allowed to equilibrate
in a pH 5.7
solution containing both EGTA and A13 . After equilibration, the concentration
of Al bound
in the solution to EGTA was determined by ETAAS. Protonation constants and the
Al-
binding constant for EGTA were taken from Martell and Smith (Critical
Stability Constants,
vol 1, Plenum, New York, 1974). The binding constant for Resin 1 is listed in
Table 5.
[00135] Aluminum binding constants for the Resin 1 were also measured by
competition against gluconic acid. A 50 mg aliquot of the resin was added to 1
ml of a
commercial solution of 0.23 M Ca(gluconate)2. Analysis by ETAAS showed that
the
untreated solution contained 115 mcMolar Al3+ as a contaminant. No other Al
was added to
the solution. Resin 1 removed approximately 10% of the Al from this solution.
Based on the
known binding constants for Al-gluconate, HySS was used to calculate the
binding constant
for the immobilized ligand on Resin 1. The binding constant for Resin 1 is
listed in Table 5.
[00136] Competition experiments versus gluconate were repeated using samples
in
which the commercial Ca(gluconate)2 solution was diluted with pH 5 MES buffer.
These
solutions contained gluconate concentrations of 0.215 M, 0.1 M, and 0.046 M
gluconic acid.
The results are listed in Table 5.
40
CA 02792724 2012-10-19
[00137] To determine the ability of Resin 1 to bind Al at ratios of mg
resin per ml of
Ca(gluconate) that more closely model conditions one expects in a filtration
device, another
competition experiment versus gluconate was conducted using samples in which
250 mg of
Resin 1 was added to 0.50 ml of the commercial Ca(gluconate)2 solution. The
initial Al
concentration was 9130 ng/ml. The concentration of free Al remaining in the
solution was
followed by ETAAS. The results are shown in Figure 5.
Table 5: Summary of Experiments to Calculate the Al binding constant (f3i10)
for Resin 1
Solution Total Volume pH mg resin Total Al % Al Log plio
(m1) (microMolar)removed
0.1 MMES 100 5.0 50 9.26 97.4 18.9
0.1 MMES 5 5.0 50 185 99.9 18.2
0.15 mM ferron 3 5 25 150 78 19.2
mM EGTA 1 5.7 50 14.9 58 18.8
0.23 M Ca(glu)2 1 5.3 50 115 10 18.5
0.108 M Ca(glu)2 1 5.2 50 53.8 18 18.5
0.05 M Ca(glu)2 1 5.4 50 25.0 44 18.7
0.023 M Ca(glu)2 1 5.3 50 11.5 72 18.9
0.23 M Ca(glu)2 0.5 4.3 250 338 40 19.0
[00138] The overall average binding constant for the immobilized ligand of
Resin 1
from the data in Table 5 is log 13110 = 18.7 + 0.3. This value is about 2.7
log units less than
the binding constant for free Ligand 1. Unfavorable steric interactions with
the resin may be
a factor, particularly when large, bulky ligands are bonded to the resin (M.
Feng, L. van der
41
CA 02792724 2012-10-19
Does, and A. Bantjes, J. Appl. Polymer Sci., 1995, 56: 1231-1237). The
invention includes
resins with longer groups linking the ligand to the polymer. It is anticipated
this elongation
of the linker will result in better agreement between the binding constants of
the free and
immobilized ligands.
[00139] It is significant
that the binding constants calculated from competition with the
gluconate solutions are in good agreement with the other values in Table 5.
This confirms
that the presence of high concentrations of Ca2+ in the gluconate solutions
has essentially no
impact on the ability of the resin to remove Al from gluconate solutions.
[00140] EXPANDED DETAILED
DESCRIPTION OF THE PREFERRED EMBODIMENTS OF
THE INVENTION
[00141] The present invention
relates to generally novel chelating agents having
general formula of
R1-N-R2
R3
[00142] wherein R1 =
hydrogen, sulfonamide, urea, carbamate, carboxamide, aryl or
alkyl, R2 = hydrogen, methyl, ethyl, n-propyl, isopropyl or similar alkyl
substituent
[00143] and R3 =
a)
(CH2)x-0 0 R4 / R4 N-OH
(CH2)z-0 0(CH2)y-OM'NLOH
o HO N-R4
[00144] wherein x, y, and z
vary independently from 1 to 4, X = CH2 and 0, and R4 ¨
hydrogen, methyl, ethyl, n-propyl, isopropyl or similar alkyl substituent.
42
CA 02792724 2012-10-19
[00145] [Note: when x=y=z=1, this is the TRIS platform]
b)
0 pH
>\¨N
µR4
(CH2)x-0 0
(CH2)y-01-- N'R4
(CH2)z-0 0H
0 OH
[00146] wherein x, y, and z vary independently from 1 to 4, and R4 =
hydrogen,
methyl, ethyl, n-propyl, isopropyl or similar alkyl substituent.
[00147] Note: when x=y=z=1, this is the TRIS platform]
c)
Ho, 0
(CH2)x-0 0
(CH2)y-ON--\
(CH2)z-0 HO
0 \
[00148] wherein x and y vary independently from 1 to 4, X = CH2 and 0, and R4
=
hydrogen, methyl, ethyl, n-propyl, isopropyl or similar alkyl substituent.
43
CA 02792724 2012-10-19
d) pH
(CH2)x-0 0R4
(CH2)y ON N
(CH2)z-0 HO )
pH
0 \
[00149] wherein x and y vary independently from 1 to 4, X CH, and 0, and
R4 =
hydrogen, methyl, ethyl, n-propyl, isopropyl or similar alkyl substituent.
e)
(CH2)x-0¨(CH2)y¨CON(OH)¨CH2(CF12)zCONR4OH
c R5
(CH2)x-0¨(CH2)y¨CON(OH)¨CH2(CH2)zCONR4OH
[00150] wherein x varies from 1-4, y varies from 1-2, and z varies
independently from
2 to 8, R4 = hydrogen, methyl, ethyl, n-propyl, isopropyl or similar alkyl
substituent, and R5
= hydrogen, methyl, ethyl, n-propyl, isopropyl or similar alkyl substituent.
(CH2)x-0¨(CH2)y¨CON(OH)¨CH2(CH2)zCH2N(COR6)0H
c R5
(CH2)x-0¨(CH2)y¨CON(OH)¨CH2(CH2)zCH2N(COR6)0H
[00151] wherein x varies from 1-4, y varies from 1-2, and z varies
independently from
2 to 8, R5 = hydrogen, methyl, ethyl, n-propyl, isopropyl or similar alkyl
substituent, and R6
= hydrogen, methyl, ethyl, n-propyl, isopropyl or similar alkyl substituent,or
Ph or similar
aryl substituent
[00152] The present invention also includes generally novel tripodal
triesters as
precursors or useful intermediates to chelating ligands having general formula
of
44
CA 02792724 2012-10-19
R1-N-R2
R7
[00153] wherein R' = hydrogen,
sulfonamide, urea, carboxamide or benzyl, R2 --
hydrogen, methyl, ethyl, n-propyl, isopropyl or similar alkyl substituent and
R7 =
g)
OR8
(CH2)x--o
(CH2)y-00R8
(CH2)z-0 0
OR8
[00154] wherein x, y, and z vary
independently from 1 to 4, X = CH2 and 0, and R8 ¨
hydrogen, methyl, ethyl, n-propyl, isopropyl or similar alkyl substituent and
Ph or similar
aryl substituent.
h)
0
(CH2)x-0 0
(CH2)z-0(CE12)y¨OLOR8
0 OR8
[00155] wherein x, y, and z vary
independently from 1 to 4, X = CH2 and 0, and R8 =
hydrogen, methyl, ethyl, n-propyl, isopropyl or similar alkyl substituent and
Ph or similar
aryl substituent.
45
CA 02792724 2012-10-19
[00156] The novel compounds of the present invention are
particularly useful as
chelators or chelating agents. One preferred use of the free ligands would be
in vivo
chelation therapy to remove metal ions such as Fe3+ and A13+ from the body.
[00157] The compounds include an amine functional group that allows
the ligands to
be easily linked to an insoluble matrix via a sulfonamide linkage, an amine
linkage, an amide
linkage, or a urea linkage to provide immobilized, tethered chelators.
Typically, the
insoluble matrix comprises a resin support. The resin support may take the
form of a macro-
porous polystyrene such as commercially available under the trademark XAD-4
sold by
Rohm and Haas. Other polymer resins useful in the present invention include
but are not
limited to, polyacrylate, sepharose and silica gel.
[00158] The overall process of adding a chelating compound of the
present invention
to a polystyrene resin via a sulfonamide bond, where NR2H-Ligand in this and
subsequent
schemes refers to the free amine form (le = H; R2 = hydrogen, methyl, ethyl, n-
propyl or
isopropyl) of any of the free ligands represented by R3 = a through f is shown
in scheme 17.
Scheme 17
CISO3H 0 Et3N, THF
Insoluble Matrix Insoluble Matrix
, g CI
8 RHN-ligand
macroporous polystyren resin
modified resin
e.g. XAD-4
0 H
Insoluble Matrix, 411 g¨N¨Ligand
0
linker ligand
resin
[00159] An alternative process of adding a chelating compound of the
present
invention to a polystyrene resin via a sulfonamide bond, where NR2H-Ligand-
prcursor in this
and subsequent schemes refers to the free amine form (1t1 = H; R2 = hydrogen,
methyl, ethyl,
n-propyl or isopropyl) of any of the triesters represented by R3 = g through h
is shown in
Scheme 18.
[00160] Scheme 18
46
CA 02792724 2012-10-19
O CISO3H 0 Et3N,
THF
Insoluble Matrixl S¨CI ' Insoluble Matrix;
411 g CI
8 8 RHN-
ligand precursor
macroporous polystyren resin
modified resin
e.g. XAD-4
O H NH2OR', Me0H, Me0Na
Insoluble Matrix; g¨N¨Ligand precursor
ii
O R' = H, TMS
0 H
Insoluble Matrixi 4r ¨N¨Ligand
6
linker ligand
resin
CO2Me
(CH2)x-0
RHN (CH2)y
(CH2)z-0
0 CO2Me
S-CI
8
CO2Me
a it 0, ri (cH2)y-0,CO2Me (CH2)x-0
CONHOH
8 (cH2)z¨o
0R (CH2)x-0
CO2Me
N (CH2)y_cy----v=CONHOH
6 (cH2)z--0\
CONHOH
[00161] The overall process of adding a chelating compound of the
present invention
to a resin support by means of a urea linkage is shown in Scheme 19.
[00162] Scheme 19
47
CA 02792724 2012-10-19
'
, , ,
, '
Et3N, THF
0
Insoluble Matrix! .
NH2 0=0=N-ligand precursor
k Insoluble Matrix! ii
N A NH-ligand precursor
H
modified resin
NI-120R', Me0H, Me0Na
0
, Insoluble Matrix l . 1
NANH-ligand
R' = H, TMS
H
Insoluble (-1
Matrix = New
/1CO2Me
/CO2Me itif lik
(CH2)x-0
COCl2, CH2Cl2
(CH2)x-0 /
NH2
0CO2Me
CO2Me
,
H2N ( (CH2)y (CH2)z-0
aq NaHCO ' 3 0/
/0, ,N (CH2)z-0(CH2)y-0
Et3N, THF
\
\
\
\
CO2Me
CO2Me
/ /CO2Me
(?' (CH2)x-0
NH2OR', Me0H, Me0Na
NH--"N c (CH2)y-0
CO,Me
H
R' = H, TMS
(CH2)z-0
\
CONHOH
\
CO2Me Giv<¨
//
ICI' (CH2)x-0
NI-1--I''-N c (CH2)y-0--- CONHOH
H
(CH2)z-0
\
\
CONHOH
1001631 An alternative process of
adding a chelating compound of the present
invention to a resin support by means of a urea linkage is shown in Scheme 20.
1001641 Scheme 20
48
CA 02792724 2012-10-19
, .
,
COCl2, CH2C1 2 1
Insoluble Matrix . , Insoluble Matrix, =
NH2 aq NaHCO3 N=C=0
modified resin
Et3N, THF i 0
NH2OR', Me0H, Me0Na
- Insoluble Matrix, 411 A
,,
RHN-ligand precursor N NR-ligand
precursor
H
R' = H, TMS
0
Insoluble Matrix; .
NANR-ligand
H
CO2Me
/-1
Insoluble ri
(c H2)x-o
Matrix = '40.- RHN c
(CH2)y _cy\-0O2Me
(CH2)z-0
\--\
e= II COCl2, CH2Cl2 . it 11
CO2Me )
NH2 aq NaHCO3
NCO Et3N, THF
CO2Me
/
/
(cH2)x-0
CO2Me NH2OR', Me0H, Me0Na
it .0 N HN c (CH2)y-0
.
R R' = H, TMS
(CH2)z-0
\ CONHOH
\ /
CO2Me (ex¨ /
If? (CH2)x ¨0
NH --"IN (CH2)y_0ONHOH
R
(CH2)z-0
\
\
CONHOH
[00165] The overall process of adding a chelating compound of the
present invention
to a resin support by means of an amine linkage is shown in Scheme 21.
[00166] Scheme 21
49
CA 02792724 2012-10-19
N-methylpyrrolidone
Nal1
Insoluble Matrix! 1 -
Insoluble Matrix, II
CI RHN-ligand precursor
N¨Ligand precursor
modified resin
NH2OR, Me0H, Me0Na Insoluble Matrix 441
N¨Ligand
R' = H, TMS
CO2Me
(CH2)x-0
CO2Me
RHN (CH2)y-0
(CH2)z-0
CO2Me
h(CH2)x-0CI N-methylpyrrolidonCe 2Me
RN c (CH2)y-0 CO2Me
Nal
Insoluble
(cH2)z-0
Matrix
CO2Me
CONHOH
NH2OR', Me0H, Me0Na (;)_(_)
(cHox-0
R ' = H, TMS RN (CH2)y¨OCONHOH
(CH2)z-0
CONHOH
1001671 The overall process of adding a chelating compound of
the present invention
to a resin support by means of an amide linkage is shown in Scheme 22.
50
CA 02792724 2012-10-19
[00168] Scheme 22
Insoluble Matrix'
0 Et3N, THE
Insoluble Matrixl
0
Cl RHN-ligand precursor
4111
N¨Ligand precursor
modified resin
NH2OR', Me0H, Me0Na
' Insoluble Matrix,' 111
R' = H, TMS
N¨Ligand
CO2Me
Insoluble rA
Matrix w
(CH2)x--0
_(:)CO2Me
RHN (CH2)y
(CH2)z-0
CO2Me
CO2Me =
0 (CH2)x-0
" THF, Et3N
RN -c (CH2)y-07
CO2Me Cl
(CH2)z-0
CO2Me
/CONHOH
NH2OR', Me0H, Me0Na
0
/
R' = H, TMS =
RN (cH2)x-0(CH2)y-0
CONHOH
(CH2)z-0
CONHOH
[00169] For certain applications it may
be desirable to elongate the linker by adding
polyethylene glycol units between the resin support and the ligand in order to
increase the
rate of metal binding to the resin-bound ligand. These elongated linkers are
added using
commercially available amine capped polyethylene glycols of variable length,
with the use of
a urea functional group to covalently bind the ligand and linker moieties.
51
CA 02792724 2012-10-19
41H2N
N H2
NH2 N=C=0
CO2Me
(CH2)x-0
CO2Me
,N
(CH2)y-0
/c
0/ ' (CH2)z-0
0
NH
CO2Me
2
Insoluble
CO2Me
Matrix
w
4. 0
(CH2)x-0
(CH2)y_oCO2Me
H H
(CH2)z-0
CO2Me
CO2NHOH
a II
(cH2)x¨o/
(CH2)y-0
CO2NHOH
H H
(CH2)z-0
CO2NHOH
1001701
Other commercially available amine-capped polyethyleneglycols include the
compound
H2 )0 \
2
[00171]
which gives chelating resins with the structures shown below
9
Insoluble Matrix l= 1¨NH
O
0ONH
NR2¨Ligand
0
or
52
CA 02792724 2012-10-19
0 0
Insoluble Matrix, 111
[00172] The immobilized, tethered chelators of the present invention comprise
the
chelating compounds identified above bound to a resin support through an
appropriate
linkage. The immobilized, tethered chelators of the present invention may be
generally
described as having the following formula:
R6¨N¨R3
R2
[00173] wherein R6 =
0 0
Insoluble g_ Insoluble
Matrix Matrix
0
0
!I
Insoluble(¨)_ Insoluble
Matrix / CH2 NH Matrix )¨CH2¨NH-
_
0
Insoluble
Matrix
0
0II 0
Insoluble
MatrixNH--0,,8 H
Insoluble c¨)_c?H JCH3 9
Matrix S¨N 0
8
CH3
n = 10-12
[00174] R2= hydrogen, methyl, ethyl; n-propyl or isopropyl and
[00175] R3 =
53
CA 02792724 2012-10-19
a)
R4
N-OH
(C1-12)X-0 R4
(C1-12))/-0 'OH
(CH2)z-0 0
()
Ho'N-R4
[00176] wherein x, y, and z vary independently from 1 to 4, X = CH2
and 0, and R4 =
hydrogen, methyl, ethyl, n-propyl, isopropyl or similar alkyl substituent.
b)
0 ,OH
R4
(CH2)x-0 0
(CH2))/-01--- N-R4
(CH2)z¨O\ OH
)7-N)R4
0 OH
[00177] wherein x, y, and z vary independently from 1 to 4, and R4 =
hydrogen,
methyl, ethyl, n-propyl, isopropyl or similar alkyl substituent
c) HO, 0
R4--N1
(CH2)z-0 HdpH
NX
0
[00178] wherein x and y vary independently from 1 to 4, X = CH2 and 0,
and R4 =
hydrogen, methyl, ethyl, n-propyl, isopropyl or similar alkyl substituent.
54
CA 02792724 2012-10-19
d)
0 pH
R4
(CH2)x-0 0
(CI-12)y -0/N-N
(CH2)z-0 HO /OH 1
0
[00179] wherein x and y vary independently from 1
to 4, X = CH2 and 0, and R4 =
hydrogen, methyl, ethyl, n-propyl, isopropyl or similar alkyl substituent.
e)
(CH2)x-0--(CH2)y-CON(OH)-CH2(CH2),CONR4OH
R5
(CH2)x-0-(CH2)y-CON(OH)-CH2(CH2),CONR4OH
[00180] wherein x varies from 1-4, y varies from 1-
2, and z varies independently from
2 to 8, R4 = hydrogen, methyl, ethyl, n-propyl, isopropyl or similar alkyl
substituent, and R5
= hydrogen, methyl, ethyl, n-propyl, isopropyl or similar alkyl substituent.
(CH2)x--0¨(cH2)y¨CoN(0H)¨CH2(CF12),CH2N(COR6)0H
c R5
(CH2)x--0¨(CH2)y¨CoN(oH)¨cH2(CH2),CH2N(COR6)0H
[00181] wherein x varies from 1-4, y varies from 1-
2, and z varies independently from
2 to 8, R5 = hydrogen, methyl, ethyl, n-propyl, isopropyl or similar alkyl
substituent, and R6
= hydrogen, methyl, ethyl, n-propyl, isopropyl or similar alkyl substituent,or
Ph or similar
aryl substituent
[00182] Example 16
55
CA 02792724 2012-10-19
, . .,
[00183] General Synthesis of Tripodal ligands.
[00184] A general approach to the amino triol scaffolds (24a-d to 27a-d)
employs the
chemistry of nitroalkanes. Starting with 2-nitroethanol (23a), 3-nitropropanol
(23b), 4-
nitrobutanol (23c) or 5-nitropentanol (23d), two identical alkenol chains can
be added with 1
carbon atom (24a-d), 2 carbon atoms (25a-d), 3 carbon atoms (26a-d), or 4
carbon atoms
(27a-d). In all cases, reduction of the nitro group with hydrogen over Ti
Raney nickel will
yield the amino compounds. Protection of the nitroalkenols (23a-d) with a TBS
group
(series B) will yield amino triols with one chain differentiated for further
reaction.
[00185] Scheme 24
0 (cH2)x¨OR
(cHox¨OR ,,,,,,, 1. Pd(0), --',--K,J
1. Na2CO3, H20, CH20 02N(CH2)x¨OR
, H2N c (cHoy¨OH
H2N (CH2)y OH ,
A R = H 2.T1 Raney Nickel
2. T1 Raney Nickel, (CH2)z¨OH
(CH2)z¨OH B R = SiMe2t-Bu H2, Me0H
H2, Me0H
23a x = 1
24ax= y=z=1 1. t-BuOK
THF 23b x = 2 's= 1. '-'- CHO 27ax=1,y=z=4
24bx= 2,y=z= 1
23c x = 3 s=,, 2. NaBH4 27bx=2,y=z=4
24c x= 3,y=z= 1 o 23d x= 4
24dx= 4,y =z= 1 µ-3. T1 Raney Nickel 27c
x = 3, y = z = 4
2. Ti Raney Nickel '1-42, Me0H 27dx=y=z=4
H2, Me0H
(CH2)x¨OR
(CH2)x¨OR
H2N c (CH2)y¨OH
H2N
(CH2)y¨OH 26ax=1,y=z=3
25a x = 1, y = z = 2 (0H2)z¨OH (CH2)z¨OH 26b x = 2, y
= z = 3
25bx=y=z= 2 26cx=y=z= 3
25cx=3,y=z=2 AR=H
26d x= 4, y =z= 3
25d x= 4, y = z = 2 B R = SiMe2t-Bu
[00186] The tripodal ligands with different length side arms can be
prepared by the
reactions of aminomethyl triol scaffolds (24-27)] with acrylonitrile then
HC1/Me0H (28-31),
or ethyl diazoacetate (36-39). The resulting esters (28-31 and 36-39) are
converted to the
hydroxamates (32-35 and 40-43) with hydroxylamine or TMSONE12.
[00187] Scheme 25
56
CA 02792724 2012-10-19
,
COX
/
/
(CH2)x-OH^-,- (CH2)x-0
X = OMe TMSONH2 or
1. KOH, dioxane, ON ,--,,,COX
NH2OH.HCI
H2N (CH2)y¨OH 2. HCl, Me0H, reflux ' H2N
(CH2)y-0 Me0H
(CH2)z¨OH (CH2)z-0
X = NHOH ' NaOH
\
Series A X= OMe28a-d to 31a-d
\
23a-d to 27a-d X= NHOH,32a-d to35a-
d COX
[00188] Scheme 26
/----cox
(cH2)x---OH (cH2)x--0
x = OEt TMSONH2 or
1. Rh2(0Ac)4, N2CHCO2Et
NH2OH.HCI
H2N c (CH2)y¨OH ' H2N c (CH2)y¨OCOX
Me0H
(CH2)z¨OH(CH2)z-0
X = NHOH * NaOH
Series A
23a-d to 27a-d X= OEt, 36a-d to 39a-
d
X= NHOH, 40a-d to 43a-d
[00189] Example 17
[00190] Synthesis of ligand 8
[00191] Scheme 27
1.(Boc)20, Me0H' BocHN
1.CF3002H, 0H2012,
H2N OH it, 20 h, 68%
C)."--0O2Et it, 73%
.
,
0-"---CO2Et
OH 2. Rh2(0Ac)4,
2. TsCI, Et3N,
ethyl diazoacetate, 0
26c OH CH2Cl2, it, 6 h, 73% \--0O2Et
CH2Cl2, 40 C, 15 h, 91%
Intermediate 23
TsHN Cr¨''CO2Et NH2OTMS, TsHN
C)---CONHOH
`-' CO2Et Me0H, it, 30 h 'µC)----CONHOH
0 80% 0
\--0O2Et \---CONHOH
' Intermediate 24
Ligand 8
[00192] Boc- protection of the amino group of amino triol 26c
followed by rhodium
acetate catalyzed alkylation of hydroxyl groups with ethyl diazoacetate gave
intermediate
23. Boc- deprotection and reprotection of the amine by a tosyl group proceeded
smoothly to
give intermediate 24. Reaction of tri-ester (Intermediate 24) with 0-
trimethylsily1
57
CA 02792724 2012-10-19
hydroxylamine produced the ligand 8. The ligand 8 is a colorless solid and was
purified by
recrystalization from ethanol/isopropanol mixture (1:1) and its structure was
determined by
x-ray crystallography.
100193] To a stirred solution of aminotriol 26c (1.65g, 8.06 mmol) in dry Me0H
(33
mL) was added (Boc)20 (1.86g, 8.5 mmol) and the mixture was stirred at room
temperature
for 20 h. The solvent was evaporated under reduced pressure and the residue
was purified by
column chromatography (Si02, CH2C12/Me0H gradient) to give the product (1.681
g, 68%)
which was crystallized from C112C12 as white crystals: IR (neat) 3453, 3400,
3307, 3228,
2947, 2872, 1689 cm-1; 'H NMR (Me0D) 8 (ppm) 3.43 (t, J= 6.6 Hz, 6H), 1.57-
1.51 (m,
6H), 1.42-1.35 (m, 6H), 1.32 (s, 911); 13C (Me0D) 8 (ppm) 156.5, 79.3, 63.4,
57.9, 32.4,
28.8, 27.5.
[00194] To a stirred solution of Boc protected amino triol (0.6 g, 1.96 mmol)
and
Rh2(0Ac)4 (0.043 g, 0.098 mmol) in CH2C12 (6 mL) was added a solution of ethyl
diazoacetate (1.05 ml, 9.98 mmol) in CH2C12 (50 ml) over a period of 1 h
(syringe pump).
After the addition was complete, the mixture was stirred for 5 h at room
temperature. The
solvent was removed under reduced pressure and the crude product was purified
by column
chromatography (Si02, hexane/Et0Ac gradient) to give intermediate 23 (0.8 g,
73%): IR
(neat) 3356, 2952, 2875, 1749, 1729, 1640 cm-1, 1FINMR (CDC13) 8 (ppm) 4.20
(q, J¨ 7.1
Hz, 6H), 4.05 (s, 611), 3.49 (t, J= 6.3 Hz, 6H), 1.63-1.57 (m, 12H), 1.39 (s,
911), 1.27 (t, J-
7.1 Hz, 911); 13C (CDC13) 5 (ppm) 170.6, 154.3 78.7 72.1, 68.5, 60.9, 56.9,
31.8, 28.6, 23.7,
14.4; HRMS (FAB) calcd for C271-149N011Na [M+Nar: 586.32037. Found: 586.31950.
[00195] Intermediate 23 (1.147 g, 2.03 mmol) was in a 1:1 mixture of CF3CO2H
(2.3
mL, 31 mmol) and CH2C12 (2.3 mL) and the resulting solution was stirred at
room
temperature. The reaction was monitored by TLC (50% Et0Ac in hexane). When the
reaction was complete, the solvent was evaporated under reduced pressure. The
residue was
dissolved in CH2C12 (10 mL) and a saturated solution of Na2CO3 was added
dropwise while
cautiously shaking the flask until CO2 evolution ceased. The layers were
separated and the
aqueous layer was extracted with twice more with CH2C12 (2x20 mL). The
combined organic
layers were dried over Na2SO4 and evaporated under reduced pressure to give
the crude
58
CA 02792724 2012-10-19
product, which was purified by column chromatography (Si02, CH2C1,/Me0H
gradient) to
give the free amine as a colorless oil (0.683 g, 73%): IR (neat) 3436, 2945,
2864, 1744, 1634,
cm-1; 1H NMR (CDC13) 8 (ppm) 4.19 (q, J= 7.1 Hz, 6H), 4.05 (s, 6H), 3.52 (t,
J= 5.7 Hz,
6H), 1.65-1.54 (m, 12H), 1.24 (t, J= 7.1 Hz, 9H); 13C (CDC13) 8 (ppm) 170.6,
72.1, 68.5,
61.0, 54.7, 35.2, 23.7, 14.3.
[00196] To the stirred solution of the free amine (0.68 g, 1.46 mmol) and
tosyl chloride
(0.66 g, 3.5 mmol) in CH2C12 (13 ml) was added Et3N (0.40 mL, 2.87 mmol) and
the mixture
was heated at 40 C for 17 h. The solvent was evaporated under reduced
pressure and the
residue was dissolved in CH2C12 (25 ml) and washed with water (2x25 mL). The
aqueous
layer was extracted with CH2Cl2(2x25 mL) and the combined organic layers was
dried over
Na2SO4 and concentrated under reduced pressure. The resiude was purified by
column
chromatography (Si02, hexane/Et0Ac gradient) to give intermediate 24 as a
gummy solid
(0.826 g, 91%): IR (neat) 3278, 2948, 2877, 1747 cm-1, 1H NMR (CDC13) 8 (ppm)
7.68 (d, J
= 8.2 Hz, 2H), 7.17 (d, J= 8.2 Hz, 2H), 4.10 (q, J= 7.1 Hz, 6H), 3.88 (s, 6H),
3.24 (t, J= 5.9
Hz, 6H), 2.31 (s, 3H), 1.48-1.38 (m, 12H), 1.18 (t, J= 7.1 Hz, 9H); 13C
(CDC13) 8 (ppm)
170.3, 142.6, 140.7, 129.3, 126.7, 72.4, 68.1, 61.9, 61.0, 32.7, 23.2, 21.3,
14.1; HRMS
(FAB) calcd for C29H47N011SNa [M+Nar: 640.27673. Found: 640.27700
[00197] To a solution of the triester (intermediate 24) (0.54 g, 1.46 mmol) in
Me0H (6
mL) was added NH2OTMS (1.00 mL, 8.17 mmol) and the mixture was stirred at room
temperature for 30 h. The solvent was evaporated under reduced pressure to
give a foamy
solid. Recrystallization from a 1:1 mixture of Et0H and iPrOH yielded ligand 8
as a white
crystalline solid (0.55 g, 80%): IR (neat) 3256, 2953, 2872, 1642 cm "1; 1H
NMR (D20) 8
(ppm) 7.81 (d, J= 8.3 Hz, 2H), 7.44 (d, J= 8.3 Hz, 2H), 3.99 (s, 6 H), 3.35
(t, J= 5.9 Hz,
611), 2.43 (s, 3H), 1.52 (m, 12H); 13C (Me0D) 8 (ppm) 169.2, 144.4, 142.7,
130.7, 128.0,
72.9, 70.1, 63.0, 33.7, 24.2, 21.5; HRMS (FAB) calcd for C23H39N4011S [M+H]:
579.23358.
Found: 579.23320. The product structure was confirmed X-ray crystallography.
[00198] Example 18
[00199] Synthesis of Ligand 9
[00200] Scheme 28
59
CA 02792724 2012-10-19
K2CO3, H20, 02N OH T-1 Raney Nickel,, H2NOH
02N OH CH20 (2 eq.) H2, Et0H, 45
psi
23c 30 C, 3 h, 84% OH OH rt, 20 h, 95 %
OH OH
intermediate 25 interemediate 26
1. Me0H, dry HCI
acrylonitrile, dioxane H2N reflux, 15 h, 74
%TsHN CO2Me
KOH (cat.), rt, 24 h, 49 % 0 2. TsCI, Et3N,
CH2Cl2, 40 C, 15 h
75%CN \--0O2Me
intermediate 27 intermediate 28
NH2OTMS
Me0H, KOH (cat.), 0 0¨\--CONHOH
amberlyst-15,
rt, 7 h, 88 A)
CONHOH ligand 9
[00201] The base catalyzed addition of two equivalents of
formaldehyde to 3-
nitropropanol (Griesser, H.; Ohrlein, R.; Ehrler, R.; Jager, V. Synthesis
1999, 77, 236) 23c
gave intermediate 25. The nitro group was reduced to an amine by hydrogenation
using T-1
Raney nickel as a catalyst and hydrogen at 45 psi. The amino triol
intermediate 26 was
alkylated with acrylonitrile to give trinitrile. Treatment with HCI in
methanol at reflux gave
the aminotriester, which protected by tosylation to give intermediate 28.
Reaction of
intermediate 28 with 0-trimethylsily1 hydroxylamine produced the ligand 9.
[00202] To a solution of 3-nitropropanol (6.70 g, 63.7 mmol)
(Griesser, H.; Ohrlein,
R.; Ehrler, R.; Jager, V. Synthesis 1999, 77, 236 ) in H20 (7 mL) were added
37 weight %
formaldehyde solution (10 mL, 128 mmol) and solid K2CO3.3/2H20 (21.0 g, 127
mmol) and
the mixture was stirred at room temperature for 1 h (reaction was exothermic)
and then at 30
C for lh. The reaction was monitored by TLC (10% Me0H in CHC13). 20% aqueous
HC1
solution was added drop wise with stirring until the effervescence of CO2
ceased. The
resulting mixture was washed with CH2C12 (2x30 mL) to remove some impurities
and the
aqueous layer was evaporated under reduced pressure. The residue was
triturated with hot
Et0H (3 x 50 mL) and the Et0H was filtered and evaporated under reduced
pressure to yield
intermediate 25 as a thick oil (8.85 g, 84%). IR (neat) 3378, 2949, 2888 cm-1;
11-1 NMR
60
CA 02792724 2012-10-19
(D20) 8 (ppm) 4.05 (ABq, A6 = 22.2 Hz, J = 12.4 Hz, 4H) 3.73 (t, J = 6.6 Hz,
2H), 2.26 (t, J
= 6.6 Hz, 2H); 13C NMR (D20) 6 (ppm) 93.9, 62.1, 57.0, 33.3.
[00203] Freshly prepared T-1 Raney nickel (3.6 g) was transferred to a Parr
hydrogenation flask as a slurry in absolute Et0H (45 mL). Intermediate 25
(4.80 g, 29.1
mmol) was dissolved in absolute Et0H (45 mL) and transferred to the
hydrogenation flask.
The resulting mixture was shaken on a Parr hydrogenator under 45 psi of H2 at
room
temperature for 20 h. The flask was removed from the hydrogenator and flushed
with argon
for 20 min. The catalyst (pyrophoric when dry) was then filtered through a
short pad of
celite, which was never allowed to dry. The celite was washed with Et0H (3 x
30 mL). The
Et0H was evaporated to give intermediate 26 a viscous brown gel (traces of
Et0H was
always present) (3.7 g, 95%) which was used in the next step without further
purification. IR
(neat) 2931, 2875 cm-1; 111 NMR (D20) 5 (ppm) 3.72 (t, J= 7.2 Hz, 2H), 3.47
(s, 4H), 1.65
(t, J = 7.2 Hz, 2H); 13C NMR (D20) 8 (ppm) 64.7, 56.9, 54.8, 35.2
[00204] To a stirred solution of intermediate 26 (3.65 g, 27.0 mmol) and KOH
pellets
(0.4 g) in 1,4-dioxane (13 mL) was added acrylonitrile (6.3 mL, 94 mmol) drop
wise over a
period of 1 h. Once the addition was complete, the mixture was stirred at room
temperature
for 24 h. The solvent was evaporated under reduced pressure to yield thick
liquid residue
which was dissolved in CH2C12 (50 mL) and washed with H20 (50 mL). The aqueous
layer
was re-extracted with additional portions of CH2C12 (2x30 mL). The combined
CH2C12
fractions were dried over Na2SO4 and evaporated under reduced pressure and the
residue was
purified by column chromatography (SiO2, 5% Et0H in Et0Ac) to give
intermediate 27 as a
thick brown oil (3.89 g, 49%): IR (neat) 3371, 3307, 2873, 2245 cm-1; 11-1 NMR
(CDC13) 5
(ppm) 3.68 (t, J = 6.0 Hz, 4H), 3.65 (t, J = 6.1 Hz, 2H), 3.62 (t, J = 6.2 Hz,
2H), 3.41 (ABq,
A8 = 19.5 Hz, J- 8.7 Hz, 4H), 2.60 (t, J = 6.0 Hz, 4H), 2.59 (t, J = 6.0 Hz,
2H), 1.72 (t, J=
6.1 Hz, 2H); '3C NMR (CDC13) 8 (ppm) 118.2, 75.1, 67.5, 66.0, 65.8, 54.7,
34.6, 19.1, 19.0;
HRMS (FAB) C,41-123N403 [M+Hr calcd 295.17703, found 295.17720
[00205] Dry HC1 gas was bubbled into a solution of intermediate 27 (1.43 g,
4.86
mmol) in Me0H (12 ml) until it was saturated. The resulting mixture was heated
at reflux for
h. Saturated Na2CO3 was added drop wise while stirring the mixture until CO2
61
CA 02792724 2012-10-19
effervescence ceased. The solution was extracted with Et0Ac (3x30 mL). The
combined
organic layers were washed with brine, dried over Na2SO4 and evaporated to
obtain the the
amino triester as a liquid (1.41 g, 74%), which was used in the next step
without further
purification. IR (neat) 2951, 2875, 1732 cm-I; NMR (CDC13) 8 (ppm) 3.74- 3.63
(m,
15H), 3.53 (t, J= 6.5 Hz, 2H), 3.26 (ABq, AS = 13.8 Hz, J= 8.9 Hz, 4H), 2.55
(t, J= 6.3 Hz,
6H), 2.03 (br s, 2H), 1.62 (t, J= 6.4 Hz, 2H); 13C NMR (CDC13) 6 (ppm) 172.3,
172.2, 75.1,
67.4, 66.8, 66.3, 54.7, 51.8, 51.7, 35.1, 35.0, 34.5; HRMS (FAB) Ci7H32N09
[M+1-11+ calcd
394.20770, found 394.20860
[00206] To a stirred solution of the amino triester (1.40 g, 3.56 mmol)
and TsC1 (1.60
g, 8.40 mmol) in CH2C12 (32 mL) was slowly added Et3N (1.0 mL, 7.2 mmol) and
the
resulting mixture was heated at reflux for 20 h. The solvent was evaporated
under reduced
pressure and the residue was dissolved in CH2C12 (50 mL), washed with water
(2x25 mL),
dried over Na2SO4 and concentrated under reduced pressure. The crude product
was purified
by column chromatography (Si02, hexanes/Et0Ac gradient) to give intermediate
28 as a
thick oil (1.42 g, 74%): IR (neat) 3287, 2953, 2876, 1732 cm-1; 111 NMR
(CDC13) 6 (ppm)
7.73 (d, J= 8.3 Hz, 2H), 7.24 (d, J= 8.3 Hz, 2H), 5.58 (s, 1H), 3.67 (s, 3H),
3.64 (s, 6H),
3.58 (t, J= 6.3 Hz, 2H), 3.50-3.36 (m, 10H), 2.52 (t, J= 6.3 Hz, 2H), 2.39 (t,
J= 6.3 Hz,
4H), 2.38 (s, 3H), 1.88 (t, J= 6.0 Hz, 2H); 1-3C NMR (CDC13) 8 (ppm) 172.1,
172.0, 142.8,
140.6, 129.3, 126.9, 72.1, 66.9, 66.5, 66.2, 61.6, 51.8, 51.7, 34.8, 34.6,
32.6, 21.5; HRMS
(FAB) C24H38N011S [M+Hr calcd 548.21655, found 548.21570
[00207] To a solution of intermediate 28 (0.5 g, 0.91 mmol) in dry Me0H
(5.5 mL)
was added NH2OTMS (0.67 mL, 5.48 mmol) and the resulting solution was stirred
at room
temperature. The reaction was monitored by TLC (50% Et0Ac in hexanes). No
reaction was
observed after 3 h. KOH (0.3 g) was added and stirring was continued for an
additional 45
min. Amberlyst-15 (2.6 g, washed with dry Me0H) was added to the reaction
mixture and
stirring was continued for 1 h. The mixture was filtered and the filtrate was
evaporated under
reduced pressure to obtain ligand 9 as a solid (0.44 g, 88%): IR (neat) 3215,
2876, 1638 cm-1;
11-1 NMR (D20) 8 (ppm) 7.80 (d, J= 8.2 Hz, 2H), 7.47 (d, J= 8.2 Hz, 2H), 3.64
(t, J= 6.0
Hz, 2H), 3.48-3.52 (m, 6H), 3.39 (s, 4H), 2.44 (s, 3H), 2.40 (t, J= 6.0 Hz,
2H), 2.32 (t, J=
62
CA 02792724 2012-10-19
5.8 Hz, 4H), 1.85 (t, J = 6.7 Hz, 2H); 13C NMR (Me0D) 8 (ppm) 171.0, 170.9,
144.4, 141.9,
123.5, 127.9, 72.5, 67.6, 67.3, 62.5, 54.9, 34.4, 33.3, 21.5; HRMS (FAB) C211-
135N4011S
[M+Hf calcd 551.20227, found 551.20260
[00208] Example 19
[00209] General Synthesis of Macrocyclic Ligands.
[00210] A general approach to macrocyclic ligands begins with mono
protected
aminotriols (series B, 24ad-27ad). The free hydroxyls are alkylated with t-
butyl acrylate
under basic conditions. The hydroxyl protecting is removed and the hydroxyl is
alkylated
methyl acrylate. Finally, the amine is protected with a Cbz group to give
compounds 44a-d to
47a-d. The t-butyl groups are removed selectively with TFA and the resulting
diacids are
converted into the acid chlorides using oxalyl chloride. Macrocycles are
prepared by
reaction of diacid chlorides with benzyl protected hydroxylamines under high
dilution
conditions. The remaining methyl is reacted with 0-benzyl hydroxylamine and
then benzyl
groups are removed via hydrogenolysis to yield the hydroxamic acids 50a-d to
53a-d.
[00211] Scheme 29.
CO2Me
(CH2)x¨OR 1. KOH, dioxane, CO2t-Bu (CH2)x-0
H2N ( (CH2)y¨OH 2. HF.Py, CH2C12 CbzNH (CH2)y¨OCO2t-Bu
(CH2)z¨OH 3. KOH, dioxane, (CH2)z-0
Series B 4. CbzCI CO2Me
24a-d to 27a-d 44a-d to 47a-d CO2t-Bu
CONHOH
1. TFA0
(CH2)x
2. DMF, (C0C 3. BnONH2 ¨012)2, CH3CN
NH2 c (CH2)y
high dilution 4. H2, Pd.0
OBn OBn 2OH, Hd )
H¨N
50a-d to 53a-d
AX=CH2,BX=0 0
X-- 48, X = CH2
49, X = 0
[00212] Example 20
[00213] General Synthesis of Tetrahydroxamic Acids Type A
63
CA 02792724 2012-10-19
[00214] Tetrahydroxamates, designed to mimic the structure of DF0-13, can
be
prepared from protected amino diacids. The amino diol scaffold for the diacids
are prepared
using chemistry similar to that for the aminotriols (para 00199). Starting
with nitroalkanes
(54), two identical alkenol chains can be added with 1 carbon atom (55), 2
carbon atoms
(56), 3 carbon atoms (57), or 4 carbon atoms (58). In all cases, reduction of
the nitro group
with hydrogen over Ti Raney nickel will yield the amino compounds.
[00215] Scheme 30
0 (CH2)4-0H
(CH2)¨OH 1. Pd(0),
1. Na2003, H20, CH20 02N R H2N ( R
H2N R 2. Ti Raney nickel 2. Ti Raney Nickel,
H2(CH2)4-0H
(CH2)-0H H2, Me0H 54 R = Me, Et,etc
55 1. t-BuOK 58
THF 1. CHO
2, NaBH4
2. T1 Raney Nickel 3. Ti Raney Nickel,
H2, Me0H H2, Me0H
(CH2)2-0H (CH2)3-0H
H2N R
H2N R
(CH2)2-0H
56 57 (CH2)3-0H
The diesters with different length side arms can be prepared by the reactions
of aminodiol
scaffolds (55-58)] with acrylonitrile then HCl/Me0H (59-62) or diazoacetate
(67-70). The
resulting esters (59-62 and 67-70) are converted to the acids (63-66 and 71-
74) by hydrolysis
with lithium hydroxide.
Scheme 31
64
CA 02792724 2012-10-19
COR'
(CH2)x-OH(CH2)x-0 59-62 R' = OMe
H2N R H2N 1. KOH, dioxane, CN R
LiOH
(CH2)x¨OH 2. HCI, Me0H, reflux (CH2)x-0 63-66 R = OH
55-58 COR'
(CH2)x-OH (CH2)x-0 67-70 R' = OEt
H2N R 1. Rh2(0Ac)4, N2CHCO2Et"*" H2N R
LiOH
(CH2)x¨OH (CH2)x-0 71-74 R' = OH -
= ---
55-58 \¨COR'
For series A, the acids are coupled with a protected monoacyl dihydroxylamine
(78). The
oximes are made by oxidizing diols to dialdehydes and condensing with 0 benzyl
(or other
protected) hydroxylamine to give an oxime. Reduction of the oxime with sodium
cyanoborohydride under acidic conditions give the protected dihydroxylamine
which is
monoacylated with one equivalent acetic anhydride.
Scheme 32
1. PCC, Celite,
CH2Cl2, rt, 2h, HCI.Me0H (2N),
HOCH2(CH2),CH2OH BnON=CH2(CH2),CH2N-0Bn
75 2. BnONH2.HCI, 76 Me0H, rt,
3 h,
Py, Et0H, NaBH3CN,
ref lux, 4 h
Ac20 (1 eq.), DMAP,
BnONHCH2(CH2)zCH2NHOBn
BnONHCH2(CH2)zCH2N(Ac)0Bn
77 Py, CH2Cl2, rt, 2 h 78
The acids are coupled with two equivalents of protected monoacyl
dihydroxylamine (78)
using dicyclohexyl carbodiimide (DCC) and then the benzyl groups are removed
via
hydrogenolysis to give the tetrahydroxamic acids 79
Scheme 33
65
CA 02792724 2012-10-19
(CH2)x-0¨(CH2)y¨COOH
1. DCC, HOBt,
R
Py, DMAP,
(CH2)z-0¨(CH2)y¨COOH CH2Cl2, 0 C to it,
63-66 or 71-74 15h
2. H2, Pd/C (10%),
BnONHCH2(CH2),CH2N(Ao)0Bn Me0H, rt, 1 h
78 (CH2)x-
0¨(CH2)y¨CONH(OH)CH2(CH2),C1-12NH(Ac)OH
R
(CH2)z-0¨(CH2)y¨CONH(OH)CH2(CH2),CH2NH(Ac)OH
79
[00216] Example 21
[00217] (Type A) Tetrahydroxamate Ligand 10
[00218] PCC oxidation of 1,9-nonanediol gave nonanedial, which
was reacted with 0-
benzylhydroxylamine hydrochloride and pyridine in refluxing ethanol to produce
the
dioxime intermediate 29 Sodium cyanoborohydride reduction of dioxime gave
benzyl
protected bis-hydroxylamine intermediate 30. Acetylation of one of the
hydroxylamine
nitrogen using one equivalent of acetic anhydride led to the formation of
intermediate 31.
[00219] Scheme 34
1. PCC, Celite,
CH2Cl2, rt, 2h,
NaBH3CN,
66%
HCI.Me0H (2N),
HO OH
r N
Bn0 µ0Bn Me0H, rt, 3 h,
1,9-nonanediol L. B nONH2.HCI,
76%
Py, Et0H, intermediate 29
reflux at 79 C, 4 h
67%
Ov
Ac20 (1 eq.), DMAP,
N, ,N
BnO,N OBn Py, CH2Cl2, it, 2 h,
30 % BnO µ0Bn
intermediate 30
intermediate 31
66
CA 02792724 2012-10-19
=
[00220] The dicarboxylic acid (intermediate 32) was prepared (Scheme 35) by
the base
catalyzed hydrolysis of intermediate 17 (R. A. Yokel, W. R. Harris, C. D.
Spilling and C.-G.
Zhan (2011) US Patent 7,932,326).
[00221] Scheme 35
i) Li0H.H20,
Aq. THF (50%), 000 to rt
TsHNi ii) 2N HC1 (to pH 1) TsHN7
intermediate 17 intermediate 32
[00222] Finally, the two partners 32 and 31 were coupled using DCC (Scheme
36) and
deprotected by hydrogenation with H2 over 10% Pd on C to obtain ligand 10.
[00223] Scheme 36
1, DCC, HOBt,
Py, DMAP, 0
TsNN/ CH2C12, 000 to it' OH
intermediate 32 15 h 7 OH
CD Tsl-INKcy¨-CONN,OH
2. H2, Pd/C (10 /o), OH 7
BnO ,N \OBn Me0H, it, 1 h Ligand 10
intermediate 31
[00224] To a solution of PCC (32.33 g, 150.0 mmol) and with a suspension of
celite
(11.0 g) in CH2C12 (300 mL) was added 1,9-nonanediol (10.0 g, 62.4 mmol). This
solution
was stirred at room temperature and the reaction was monitored by TLC (1:1
hexanes/Et0Ac). After 2 h the reaction was complete. The reaction mixture was
passed
through a short column of silica and celite, which was washed with CH2C12
(5x50 mL). The
solvent evaporated under reduced pressure and the residue was purified by
column
chromatography (Si02 hexanes/Et0Ac gradient) to give the dialdehyde (A. Ozane,
L
Pouysegu, D. Depernet, B. Francois, S. Quideau Organic Letter 2003, 5, 2903-
2906) as a
colorless liquid (6.4 g, 66%): 1H NMR (CDC13) 8 (ppm) 9.73 (t, J= 1.8 Hz, 2H),
2.40 (td, J
67
CA 02792724 2012-10-19
= 7.3, 1.8 Hz, 4H), 1.58 (m, 4H), 1.32 (br s, 6H); 13C NMR (CDC13) 5 (ppm)
202.8, 43.9,
29.2, 29.0, 22Ø
[00225] To a solution of nonanedial (5.60 g, 35.8 mmol) and 0-
benzylhydroxylamine
hydrochloride (15.0 g, 94.0 mmol) in Et0H (120 mL) was added pyridine (7.6 mL,
94 mmol)
drop wise. The resulting solution was heated at reflux for 4 h. The solvent
was evaporated
under reduced pressure and the residue was dissolved in CH2C12 (200 mL) and
washed with
water (3x100 mL). The aqueous layer was re-extracted with CH2C12 (2x100 mL).
The
combined organic layers were dried over Na2SO4, evaporated under reduced
pressure, and
the residue was purified by column chromatography (Si02 hexanes) to give
intermediate 29
as a colorless liquid (8.01 g, 61%) as a 1.5:1 mixture of geometric isomers.
IR (neat) 3030,
2925, 2856 cm-1; 1H NMR (CDC13) 5 (ppm) 7.46 (t, J 6.2 Hz, 0.6H), 7.39-7.31
(m, 5H),
6.69 (t, J= 5.4 Hz, 0.4H), 5.13 (s, 0.8 H), 5.08 (s, 1.2H), 2.39 (app q,
0.8H), 2.20 (app q,
1.2H), 1.48 (m 2H), 1.32 ( m, 3H); 13C NMR (CDC13) 8 (ppm) 152.6, 151.7,
138.3, 137.8,
128.5 (x2), 128.3, 128.0, 127.9, 127.8, 75.8, 75.6, 29.6, 29.3, 29.1, 29.0,
26.7, 26.2, 25.9;
HRMS (FAB) C23H31N202 [M+H] calcd 367.23856, found 367.23830. One of the
isomers
was isolated and characterized completely. 11-1 NMR (CDC13) 5 (ppm) 7.39 (t,
J= 6.2 Hz,
2H), 7.31-7.24 (m, 10H), 5.00 (s, 4H), 2.13 (app q, 4H), 1.41 (quin, J= 6.8
Hz, 4H), 1.25 (m,
6H); 13C NMR (CDC13) 8 (ppm) 151.7, 137.9, 128.5, 128.4, 128.0, 127.9, 75.6,
29.6, 29.1,
29.0, 26.7.
[00226] To a solution of intermediate 29 (0.11 g, 0.3 mmol) and NaCNBH3 (0.042
g,
0.66 mmol) in Me0H (2 mL) was added 2N HC1 in Me0H drop wise until the
solution pH
was between 3 and 4. The resulting mixture was stirred for 3 h at room
temperature. The
solvent was evaporated under reduced pressure and the solid residue was
dissolved in water
(2 mL) and 6 N KOH solution was added drop wise to adjust the solution pH to
>9. The
aqueous solution was extracted with CH2C12 (3 x10 ml), and combined organic
extracts were
washed with brine (20 mL), dried over Na2SO4 and evaporated under reduced
pressure. The
residue was purified by column chromatography (Si02hexanes/Et0Ac gradient) to
give the
intermediate 30 as a colorless liquid (0.083 g, 76%): IR (neat) 3028, 2924,
2852, 1453, 1363
cm-1; 111 NMR (CDC13) 5 (ppm) 7.30-7.38 (m, 10H), 4.73 (s, 4H), 2.94 (t, J=
7.0 Hz, 4H),
68
CA 02792724 2012-10-19
1.52 (app quin, J=6.7 Hz, 4H), 1.30 (br s, 10H); 13C NMR (CDC13) 8 (ppm)
138.2, 128.5
(x2), 127.9, 76.3, 52.3, 29.6, 27.4, 27.3.
[00227] To a solution of intermediate 30 (0.40 g, 1.08 mmol) in CH2C12 (15 mL)
was
added DMAP (0.13 g, 1.06 mmol), pyridine (0.18 mL, 2.22 mmol), and Ac20 (0.102
mL,
1.08 mmol) sequentially and the resulting mixture was stirred at room
temperature for 2 h.
The mixture was diluted with CH2C12 (20 mL) was added and washed with
saturated
NaHCO3 (20 mL). Organic layer was dried over Na2SO4 and evaporated under
reduced
pressure and the solid residue was purified by column chromatography (Si02,
hexanes/Et0Ac gradient) to give intermediate 31 as a colorless liquid (0.120
g, 30%): IR
(neat) 3031, 2927, 2854, 1660 cm-1; 1H NMR (CDC13) 8 (ppm) 7.31-7.39 (m, 10H),
5.56 (br
s, 1H), 4.82 (s, 2H), 4.71 (s, 2H), 3.63 (t, J= 6.9 Hz, 2H), 2.93 (t, J= 7.1
Hz, 2H), 2.10 (s,
3H), 1.64 (m, 2H), 1.50 (m, 211), 1.29 (br s, 10H); 13C NMR (CDC13) 5 (ppm)
154.2, 138.2,
134.7, 129.3, 129.0, 128.9, 128.5 (x2), 127.9, 76.4, 76.3, 52.3, 29.6, 29.4,
27.5, 27.3, 27.0,
26.9, 20.7; HRMS (FAB) C25H37N203 [M+H] calcd 413.28040, found 413.2776
[00228] To a solution of intermediate 17 (3.00 g, 6.96 mmol) in aqueous THF
(1:1, 30
ml) at 0 C, was added Li0H.H20 (1.17 g, 27.9 mmol) and the resulting mixture
was stirred
for 1 h. After 1 h, the flask was allowed to warm to room temperature while
stirring was
continued. When all starting material was consumed (TLC 1:1 Et0Ac/hexanes),
the reaction
was quenched with 2N HC1 and solution pH was adjusted to 1. The mixture was
filtered (to
remove LiC1) and extracted with CH2C12 (3x50 mL). The combined organic
extracts were
washed with brine, dried over Na2SO4 and evaporated under reduced pressure to
give the di-
acid as a white powder (2.67 g, 95%) which was used in the next step without
further
purification. IR (neat) 3300, 3051, 2925, 2875, 1697 cm -1; 111 NMR (D20) 6
(ppm) 7.84 (d,
J= 8.3 Hz, 2H), 7.45 (d, J= 8.3 Hz, 2H), 3.60-3.53 (m, 411), 3.37 (ABq, A5 =
22.8 Hz, J=
Hz, 4H), 2.53 (t, J= 6.0 Hz, 4H), 2.44 (s, 3H), 1.14 (s, 311); 13C NMR (Me0D)
8 (ppm)
175.6, 144.3, 142.5, 130.5, 128.0, 74.8, 68.0, 60.1, 35.7, 21.5, 19.4; HRMS
(FAB)
C17H26N08S [M+Hr calcd 404.13790, found 404.13880.
69
CA 02792724 2012-10-19
[00229] Ligand 10. To a solution of intermediate 31(0.40 g, 0.97 mmol) and
DMAP
(0.16 g, 1.31 mmol) in CH2C12 (10 mL) and pyridine (0.11 mL, 1.4 mmol) was
added a
solution of intermediate 32 (0.18 g, 0.45 mmol) and HOBt (0.14 g, 1.03 mmol)
in CH2C12
(10 mL) and the mixture was cooled to 0 C. DCC (0.2 g, 0.97 mmol) was added
and the
mixture was stirred for 1 h at 0 C, then it was allowed to warm to room
temperature and was
stirred for an additional 15 h. The solvent was evaporated under reduced
pressure and the
residue was purified by column chromatography (Si02, hexanes/Et0Ac gradient)
to give the
tetrabenzyl tetrahydroxamate as a thick colorless oil (0.342 g, 65%): IR
(neat) 3033, 2930,
2856, 1650, 1601 cm-1; 1H NMR (CDC13) 5 (ppm) 7.75 (d, J= 8.2 Hz, 2H), 7.45-
7.33 (m,
20H), 7.17 (d, J= 8.2 Hz, 2H), 5.74 (s, 1H), 4.78 (s, 4H), 4.76 (s, 4H), 3.63-
3.59 (m, 12H),
3.30 (ABq, M = 52.5 Hz, J= 9.2 Hz, 4H), 2.66-2.55 (m, 4H), 2.25 (s, 3H), 2.04
(s, 6H), 1.58
(m, 8H), 1.23 (br s, 20H), 1.08 (s, 3H); 13C NMR (CDC13) 8 (ppm) 172.2, 172.0,
142.4,
140.8, 134.4, 134.3, 129.9, 129.3, 129.2, 129.0, 128.7, 128.6, 128.5, 127.0,
126.7, 126.2,
77.6, 76.2, 76.0, 73.7, 66.7, 58.6, 45.2, 32.5, 29.2, 29.0, 26.7, 26.5, 21.3,
20.4, 18.1; HRMS
(FAB) C67H94N5012S [M+H] calcd 1192.66199, found 1192.66110
[00230] To a solution of tetrabenzyl tetrahydroxamate (0.34 g, 0.28 mmol) in
Me0H
(11 mL) was added 10% Pd/C (0.074 g). Flask was then evacuated and flushed
with H2 from
two balloons and the mixture was stirred under H2 at room temperature until
the starting
material was consumed ('TLC, 70% Et0Ac in hexanes). The reaction flask was
purged with
Ar and the mixture was filtered. The filtrate was evaporated under reduced
pressure to give
ligand 10 as a foamy solid (0.22 g, 91%): IR (neat) 3360, 3060, 2928, 2857,
1611 cm-1; 11-1
NMR (Me0D) 5 (ppm) 7.68 (d, J= 8.2 Hz, 2H), 7.23 (d, J= 8.2 Hz, 2H), 3.52-3.46
(m,
12H), 3.27-3.17 (m, 4H overlapping with Me0H peak), 2.58 (t, J= 6.0 Hz, 4H),
2.31 (s, 3H),
1.99 (s, 6H), 1.51 (br s, 8H), 1.21 (br s, 20H), 1.00 (s, 3H); 13C NMR (Me0D)
8 (ppm)
173.6, 173.5, 144.2, 142.6, 130.5, 128.0, 75.0, 68.0, 60.1, 48.9, 33.9, 30.6,
30.4, 27.8, 21.6,
20.3, 19.5; HRMS (FAB) C39H70N50/2S [M+H] calcd 832.4742, found 832.4725
[00231] Example 22
[00232] Tetrahydroxamate ligand 11
70
CA 02792724 2012-10-19
, =
[00233] Ligand 11 is prepared from amino diester intermediate 17.
The amine is
protected with Cbz to give intermediate 33. Hydrolysis with lithium hydroxide
yields diacid
intermediate 34 which is coupled with intermediate 31 using DCC. Global
deprotection of
the Cbz and benzyl protecting groups gives ligand 11.
[00234] Scheme 37
0 ----0O2Me
H2N/ CbzHNK0
intermediate 17 intermediate 33
i) Li0H.H20,
DCC, HOBt, Py, DMAP,
Aq. THF (50%), 0 C to it
CH2Cl2, 0 C to it, 15 h
ii) 2N HCI (to pH 1) CbzHNK0-------0O2H
(2.2 eq.) 0y
intermediate 34 H
BnO" N \OBn
intermediate 31
OH
H2, Pd/C (10%), 0 -OH
Me0H, rt, 1 h X 7
N .0H
OH 7
Ligand 11
[00235] To a stirred solution of intermediate 17 (R. A. Yokel, W. R.
Harris, C. D.
Spilling and C.-G. Zhan (2011) US Patent 7,932,326) (2.0 g, 7.2 mmol) in THF
(57 mL) was
added 10% aqueous Na2CO3 (57.0 mL, 53.7 mmol). After 15 minutes at room
temperature
CbzCl (1.13 mL, 7.93 mmol) was added and stirring was continued for an
additional 5 h. The
layers were separated and the aq. layer was extracted with Et0Ac (2x30 mL).
The combined
organic extracts were washed with water and brine, dried over Na2SO4 and
evaporated under
reduced pressure. The residue was purified by column
chromatography (SiO2,
hexane/Et0Ac gradient) to give intermediate 33 as a colorless oil (1.9 g,
65%): IR (neat)
3366, 2951, 2875, 1731 cm-1; 11-1 NIVIR (300 MHz, CDC13) 8 (ppm) 7.33-7.31 (m,
5H), 5.28
(s, 1H), 5.01 (s, 2H), 3.67 (t, J= 6.2 Hz, 4H), 3.64 (s, 6H), 3.47 (ABq, A8 =
42.9 Hz, J= 9.0
71
CA 02792724 2012-10-19
Hz, 4H), 2.53 (t, J= 6.2 Hz, 411), 1.29 (s, 311); 13C NMR (300 MHz, CDC13) 8
(ppm) 172.0,
155.1, 137.4, 128.5, 128.0, 72.9, 66.8, 66.1, 55.7, 51.7, 34.8, 19.8; FIRMS
(FAB) C20H30N08
[M+H] calcd 412.19714, found 412.19750
[00236] To a solution of intermediate 33 (7.40 g, 18.0 mmol) in 50%
aqueous THF (75
mL) was added LiOH (1.68 g, 70.1 mmol) at 0 C and the resulting mixture was
stirred for 1
h. After 1 h, the flask was allowed to warm to room temperature with continued
stirring.
Once the starting material was consumed (TLC 1:1 Et0Ac/hexanes), the reaction
was
quenched with 2N HC1 to adjust the solution pH to 2. The mixture was extracted
with
CH2C12 (3x100 mL) and the combined extracts were washed with brine, dried over
Na2SO4,
and evaporated under reduced pressure to give intermediate 34 as a colorless
gel (6.8 g,
quant) which was used in the next step without further purification. IR (neat)
3033, 2939,
2878, 1709 cm -1; 11-1 NMR (300 MHz, CDC13) 8 (ppm) 10.28 (br s, 211), 7.35-
7.29 (m, 511),
5.34 (br s, 1H), 5.04 (s, 211), 3.68 (t, J 6.2 Hz, 4H), 3.49 (ABq, A8 = 39.6
Hz, J= 8.9 Hz,
4H), 2.57 (t, J= 6.2 Hz, 4H), 1.31 (s, 3H); 13C NMR (300 MHz, CDC13) 8 (ppm)
177.3,
155.3, 136.5, 128.5 (x2), 128.3, 128.1, 128.0, 77.3, 72.8, 66.4, 55.7, 34.6,
19.0; FIRMS
(FAB) Ci8H26N08 [M+Hr calcd 384.1658, found 384.1648
[00237] To a solution of intermediate 31(1.1 g, 2.66 mmol) and DMAP (0.44
g, 3.60
mmol) in CH2C12 (50 mL) and pyridine (0.29 mL 3.58 mmol), was added
intermediate 34
(0.47 g, 1.22 mmol) and HOBt (0.37 g, 2.71 mmol) in CH2C12 (50 mL). The
mixture was
cooled to 0 C and DCC (0.58 g, 2.81 mmol) was added. The mixture was stirred
for 1 h at 0
C, then allowed to warm to room temperature and stirred for additional 15 h.
The solvent
was concentrated under reduced pressure and the solids were removed by
filtration (DCC
urea) and washed with CH2C12. The filtrate was evaporated under reduced
pressure and the
residue was purified by column chromatography (SiO2' Et0Ac/hexanes gradient)
to give the
Cbz tetrabenzyl tetrahydroxylamine as a white solid product (0.80 g, 56%): IR
(neat) 3029,
2930, 2856, 1719, 1649 cm-1; NMR (300 MHz, CDC13) 8 (ppm) 7.36-7.25 (m, 5H),
5.49
(s, 1H), 5.00 (s, 211), 4.79 (s, 414), 4.78 (s, 4H), 3.71 (t, J= 6.2 Hz, 4H),
3.60-3.55 (m, 8H),
3.49 (ABq, A5 = 45.8 Hz, J= 9.1 Hz, 4H), 2.64 (t, J= 5.9 Hz, 4H), 2.07 (s,
6H), 1.60-1.58
72
CA 02792724 2012-10-19
, .
(m, 811), 1.32 (s, 3H), 1.24 (s, 20H); 13C NMR (300 MHz, CDC13) 6 (ppm) 172.4,
172.2,
155.1, 136.8, 134.5, 129.1 (x2), 128.9, 128.8, 128.7, 128.4, 127.9, 127.8,
77.4, 76.3, 76.2,
73.2, 67.1, 65.9, 55.7, 45.3, 33.9, 32.7, 29.4, 29.3, 29.2, 29.1, 26.8, 26.7,
26.6, 20.5, 19.0;
HRMS (FAB) C68H94N5012 [M+H] calcd 1172.68994, found 1172.68620.
[00238] To a solution of Cbz tetrabenzyl tetrahydroxylamine (0.50 g, 0.43
mmol) in
Me0H (16 mL) was added 10% Pd/C (0.11 g) under argon. The flask was evacuated
and
flushed with 112 (balloons) and then mixture was stirred under H2 at room
temperature until
the starting material was consumed (TLC analysis 70% Et0Ac in hexanes). The
reaction
mixture was then flushed with Ar and filtered. The filtrate was evaporated
under reduced
pressure to obtain ligand 11 as a sticky solid (0.29 g, quant): IR (neat) 3133
(broad), 2927,
2854, 1607 cm-1; 111 NMR (300 MHz, Me0D) 8 (ppm) 3.52 (t, J= 6.1 Hz, 4H), 3.37
(t, J =-
7.1 Hz, 4H), 3.35 (t, J= 7.1 Hz, 4H), 3.18 (ABq, M = 26.4 Hz, J= 9.6 Hz, 4H),
2.53 (t, J-
6.1 Hz, 4H), 1.85 (s, 6H), 1.38 (m, 8H), 1.09 (m, 20H), 0.92 (s, 3H); 13C NMR
(300 MHz,
Me0D) 8 (ppm) 173.5, 173.4, 74.9, 68.3, 56.5, 48.9, 34.8, 33.8, 30.7, 30.4,
27.8, 26.8, 26.2,
20.4 (x2); HRMS (FAB) C32H64N5010 [M+11]+ calcd 678.46533, found 678.46620
[00239] Example 23
[00240] General Synthesis of Tetrahydroxamic Acids Type B
[00241] The synthesis of type B tetrahydroxamate ligands begins with selective
reduction of oc,co diacid mono esters 80 (Z > 4) using borane in THF to give
the hydroxy
esters, which are reoxidized to give the ester aldehydes 81. The aldehydes are
condensed
with 0-benzyl (or other protected) hydroxylamine to give oximes 82. Reduction
with
sodium cyanoborohydride under acidic conditions will yield the hydroxylamine
esters 83.
[00242] Scheme 38
73
CA 02792724 2012-10-19
, ,
1. BH3.THF
BnONH2.HCI,t,
THF, -18 C to r
Py, Et0H,
4 h, 75 %
,
HO2C(CH2)zCO2Me
- 0=CH(CH2)zCO2Me
80 2. PCC, silica, CH2C12,
at 4 h,,
rt, 2 h 86 %
81 89%
NaBH3CN,
BnON=CH(CH2),CO2Me
BnONHCH2(CH2),CO2Me
2 N HCI. Me0H, ,
82 Me0H, rt, 97%
83
The hydroxylamine esters 83 are condensed with diacids (63-66 or 71-74) to
benzyl
protected dihydroxamic acids 84. Reaction of the ester with 0-trimethylsily1
hydroxylamine
in Me0H and removal of the benzyl groups via hydrogenolysis will yield the
type B
tetrahydroxamates 85.
Scheme 39
(CH2)x¨o¨(cH2)y¨CooH
R DCC, HOBt,
(CH2)x-0¨(CH2)y¨CONH(OBn)-
CH2(CH2)zCO2Me
(CH2)z-0¨(CH2)y¨COOH
c - c R
Py, DMAP, CH2-I2'
(63-66 or 71-74) 0 C to rt, 82
% (CH2)z-0¨(CH2)y¨CONH(OBn)
CH2(CH2)zCO2Me
BnONHCH2(CH2),CO2Me
84
83
1. TMSONH2, Me0H, (CH2)x-
0¨(CH2)y¨CONH(OH)CH2(CH2),CONHOH
rt, 20 h
R
85
2. H2, PcI/C (10%), ''. C
(CH2)z-0¨(CH2)y¨CONH(OH)CH2(CH2)zCONHOH
Me0H, rt
[00243] Example 24
[00244] (Series B) Tetrahydroxamte Ligand 12
[00245] The acid group of monomethyl azelate was
selectively reduced with BH3 to
give an alcohol (Scheme 40), which was reoxidized with PCC to form the
aldehyde
intermediate 35. The aldehyde was reacted with 0-benzylhydroxylamine
hydrochloride in
74
CA 02792724 2012-10-19
the refluxing ethanol to obtain the oxime ester intermediate 36, which was
reduced to the
benzyl protected hydroxylamine ester intermediate 37.
[00246] Scheme 40
1. BH3.THF
0 0 THF, -18 C to rt, BnONH2.HCI,
4 h, 75 % 0 0 Py, Et0H,
HO OMe 2. PCC, silica, CH2Cl2, OMe ref lux at
, 4 h,
Monomethyl Azelate rt, 2 h 86 % intermediate 35 89%
BnO,N
0 NaBH3CN, 0
OMe 2 N HC1. r BnO, N Me0H, OMe
intermediate 36 Me0H, it, 97% intermediate 37
[00247] DCC mediated coupling of dicarboxylic acid intermediate 17 with
intermediate 37 produced the benzyl protected hydroxamic acid intermediate 38
(Scheme
41), which was purified by chromatography. The ester groups were then
converted to the
hydroxamic acids by reaction with 0-trimethylsily1 hydroxylamine. The benzyl
protected
hydroxamic acid groups were deprotected using hydrogenolysis to give ligand
12.
[00248] Scheme 41
OBn 0
TsHNK0--------0O2H I DCC, HOBt, 7 OMe
intermediate 32 0 Py, DMAP, CH2Ci2 TsHN/ Me,
BnO,N 0 C to it, 82 % OBn 70
OMe
intermediate 38
intermediate 37
OH 0
OCON OH
1. TMSONH2, Me0H, 7H
it, 20 h
TsH N-OH
2. H2, Pd/C (10%), OH 0
Me0H, it ligand12
[00249] To a solution of intermediate 35 (Kai, K.; Takeuchi, J.;
Kataoka, T.;
Yokoyama, M.; Watanabe, N. Tetrahedron 2008, 64, 6760 ) (3.40 g, 18.2 mmol)
and 0-
75
CA 02792724 2012-10-19
benzylhydroxylamine hydrochloride (3.79 g, 23.7 mmol) in Et0H (73 mL) was
added
pyridine (3.84 mL, 47.4 mmol) drop wise. The resulting solution was heated at
reflux for 3 h.
The solvent was evaporated under reduced pressure and the residue was
triturated with
Et0Ac (5x20 mL). The Et0Ac fractions were combined and filtered, then
evaporated under
reduced pressure. The residue was purified by column chromatography (Si02
hexanes/Et0Ac gradient) to give intermediate 36 as a thick colorless liquid
(4.7 g, 89%) as
the mixture of two geometric isomers: IR (neat) 3027, 2929, 2856, 1736, cm-1;
11-1 NMR
(CDC13) 8 (ppm) 7.44 (t, J = 6.2 Hz, 0.6H), 7.37-7.27 (m, 5H), 6.67 (t, J =
5.5 Hz, 0.4H),
5.11 (s, 0.8H), 5.06 (s, 1.2H), 3.67 (s, 3H), 2.37 (m, 0.6H), 2.31 (m, 2H),
2.17 (m, 1.4H),
1.62 (m, 2H), 1.47 (m, 2H), 1.31 (m, 6H); '3C NMR (CDC13) 8 (ppm) 174.4,
152.6, 151.7,
138.3, 137.8, 128.5 (x 2), 128.4, 128.0, 127.9 (x 2), 75.8, 75.6, 51.6, 34.2,
29.6, 29.3, 29.1,
29.0, 26.7, 26.3, 25.9, 25Ø
1002501 One of the isomers was isolated and characterized, but
isomerized soon on
standing: 'H NMR (CDC13) 8 (ppm) 7.44 (t, J= 6.2 Hz, 1H), 7.37-7.27 (m, 5H),
5.06 (s, 2H),
3.67 (s, 3H), 2.30 (t, J= 7.4Hz, 2H), 2.17 (app q, J= 6.4 Hz, 2H), 1.62 (m,
2H), 1.47 (m,
2H), 1.31 (br s, 6H).
[00251] To a solution of intermediate 36 (4.70 g, 16.1 mmol) and NaCNBH3
(1.12 g,
17.8 mmol) in Me0H (100 mL) was added 2N HC1 in Me0H drop wise at room
temperature
until the solution pH (checked by universal indicator) was adjusted to 3-4,
then the solution
was stirred for 3 h at room temperature. The solvent was evaporated under
reduced pressure
to give solid residue, which was dissolved in water (100 mL). 6 N KOH solution
was added
drop wise to adjust the solution the pH to >9. The aqueous solution was
extracted with
CH2C12 (3x100 mL). The combined organic layers were washed with brine, dried
over
Na2SO4 and evaporated under reduced pressure. The residue was purified by
column
chromatography (Si02 2% Et0Ac in hexanes) to give the hydroxylamine
intermediate 37 as
a colorless liquid (4.6 g, quant): IR (neat) 3022, 2928, 2854, 1736 cm-1; NMR
(CDC13) 8
(ppm) 7.36-7.27 (m, 5H), 4.71 (s, 2H), 3.67 (s, 3H), 2.92 (t, J = 7.0 Hz, 2H),
2.30 (t, J = 7.4
Hz, 2H), 1.62 (m, 2H), 1.50 (m, 2H), 1.30 (br s, 8H); 13C NMR (CDC13) 8 (ppm)
174.5,
76
CA 02792724 2012-10-19
138.1, 128.5, 127.9, 76.3, 52.3, 51.6, 34.2, 29.4, 29.3, 29.2, 27.4, 27.2,
25.1; HRMS (FAB)
C17H28NO3 [M+H]+ calcd 294.20691, found 294.20750
[00252] To a solution of intermediate 37 (0.10 g, 0.34 mmol) and DMAP (0.06 g,
0.49
mmol) in CH2C12 (4 mL) and pyridine (0.04 mL, 0.49 mmol) was added
intermediate 32
(0.06 g, 0.15 mmol) and HOBt (0.05 g, 0.37 mmol) in CH2C12 (4 mL). The mixture
was
cooled to 0 C and DCC (0.07 g, 0.34 mmol) was added. The mixture was stirred
for 1 h at 0
C, then it allowed to warm to room temperature and stirred for additional 20
h. The solvent
was evaporated under reduced pressure and the liquid residue was purified by
column
chromatography (Si02 hexanes/EtoAc gradient) to give intermediate 38 as a
thick colorless
oil (0.142 g, 60%): IR (neat) 3269, 3028, 2930, 2857, 1734, 1653 crn-1; 111
NMR (CDC13) 8
(ppm) 7.77 (d, J= 8.2 Hz, 2H), 7.41-7.36 (m, 10H), 7.22 (d, J= 8.2 Hz, 2H),
5.65 (s, 1H),
4.82 (s, 4H), 3.66-3.63 (m, 14H), 3.34 (ABq, A8 = 87 Hz, J= 9.2 Hz, 4H), 2.67-
2.57 (m,
4H), 2.38 (s, 3H), 2.28 (t, J= 7.5 Hz, 4H), 1.62-1.57 (m, 8H), 1.28 (m, 16H),
1.09 (s, 3H);
13C NMR (CDC13) 8 (ppm) 174.4, 172.5, 142.8, 141.0, 134.6, 129.5, 129.3,
129.1, 128.9,
127.0, 76.5, 74.0, 76.1, 58.9, 51.6, 45.6, 34.2, 34.8, 29.3, 29.2, 26.8, 25.0,
21.6, 18.3; HRMS
(FAB) C511176N3012S [M+H] calcd 954.51495, found 954.51690.
[00253] To a solution of intermediate 38 (2.03 g, 2.13 mmol) in dry Me0H (45
mL)
was added KOH (0.80 g, 14.3 mmol), NH2OTMS (1.22 mL, 9.38 mmol) and the
resulting
solution was stirred at room temperature for 15 h. Amberlyst-15 (6 g, washed
with dry
Me0H) was added to the reaction mixture and stirred for additional 1 h. The
mixture was
filtered and the filtrate was evaporated under reduced pressure to obtain the
dibenzyl
tetrhydroxamate as a white foamy solid (2.01 g, quant.) which was used without
further
purification in the next step: IR (neat): 3258, 2927, 2857, 1632, 1454, 1110
cm-1; 1H NMR
(Me0D) 8 (ppm) 7.63 (d, J= 8.2 Hz, 2H), 7.15 (d, J= 8.2 Hz, 2H), 4.76 (s, 4H),
3.55 (app t,
J= 5.9 Hz, 4H), 3.42 (app t, J= 5.8 Hz, 4H), 3.23-3.12 (ABq, 4H over laps with
Me0D),
2.47 (app t, J= 5.8 Hz, 4H), 2.24 (s, 3H), 2.13 (t, J= 7.4 Hz, 1.4H), 1.94 (t,
J= 7.3 Hz,
2.3H), 1.48 (m, 811), 1.17 (m, 16H), 0.95 (s, 3H); HRMS (FAB) C49H74N5012S
[M+H] calcd
956.50543, found 956.50500
77
CA 02792724 2012-10-19
,
[00254] To a solution of dibenzyl tetrahydroxamate (2.0 g, 2.1 mmol) in Me0H
(100
mL) under argon was added 10% Pd/C (0.22 g). The flask was then evacuated,
flushed with
H2 (balloons) and the mixture was stirred under H2 at room temperature for 3h.
The reaction
mixture was filtered and the filtrate was evaporated under reduced pressure to
give ligand 12
as a foamy solid (1.3 g, 81%): IR (neat) 3500-2600 (broad), 2927, 2856, 1613
cm-1; 1H NMR
(Me0D) 8 7.58 (d, J= 8.0 Hz, 2H), 7.14 (d, J= 8.0 Hz, 2H), 3.44-3.36 (m, 8 H),
3.16-3.09
(m, 4H, overlaps with CD30D), 2.47 (t, J = 5.7 Hz, 4H), 2.21 (s, 3H), 1.89 (t,
J= 7.2 Hz,
4H), 1.41 (br. S, 8H), 1.12 (br. S, 16H), 0.89 (s, 3H); 13C NMR (Me0D) 8
173.6, 173.1,
144.3, 142.5, 130.5, 128.0, 74.9, 68.0, 60.1, 52.1, 34.9, 33.8, 30.3, 30.1,
27.8, 27.7, 26.8,
26.1, 21.6, 19.4; HRMS (FAB) C35H62N5012S [M+Hr calcd 776.41150, found
776.41130.
[00255] Example 25
[00256] Preparation of Urea-linked Tris Hydroxamic Acid Resin 6
[00257] Scheme 42
78
CA 02792724 2012-10-19
.
,
0 /
0 /
--CD
Method A
¨0
/
/
j
H2N¨fo
0 0
Triphosgene
).
N
=
tO
,
0
0C
CH2Cl2, aq NaHCO3
0 0
0 0 0
0
0
intermediate 2
intermediate 39
NCO Triphosgene
NH2
Method B
j 4--/
intermediate 41
0 OH
0 /
\--NH
\--0
0/
0/
HNt 0 0
HN
, tO 0 4-7 1) NH2OTMS
HN¨i 0 10H-OH
N
HN¨
31r
4---/ 0 0
C)v
H
2) aq. AcOH
Resin 6
,I,N,OH
Y0
0
0
(Method A:1.04 meq/g)
intermediate 40
(Method B: 0.54 meq/g)
(Method A:1.23 meq/g)
(Method B: 1.23 meq/g)
1002581
Method A ¨ Amine resin and ruus Isocyanate Route
[00259]
To 12.26 g (32.3 mmol) of the free amine intermediate 2 in 130 mL of CH2C12
was added 130 rnL of saturated' NaHCO3. The stirred mixture was treated
PORTION WISE
with 3.2 g (10.8 mmol) of triphosgene. This reaction is fast and results in a
lot of gas
formation. After stirring for 15 min, the organic phase was separated. The
aqueous layer
was extracted twice with CH2C12, combined organic extracts dried and
concentrated to afford
intermediate 39 (12.2 g, 93%) as a clear, dark oil: IR(ATR) 2955, 2878, 2245
(NCO), 1733
(CO), 1437; 111 NMR (CDC13, 300 MHz) 8 2.52 (t, 3H, 6.3 Hz), 3.39 (s, 6H),
3.62 (s, 9H),
3.68 (t, 3H, 6.3 Hz); 13C NMR (CDC13, 75 MHz) 8 34.7, 51.6, 63.7, 67.0, 71.1,
127.2 (CNO),
171.8.
79
CA 02792724 2012-10-19
[00260] To 2 g (4.4 mmol) of aminomethyl resin (Aldrich 564095; Macroporous 30-
60
mesh, 2.2 meq/g) was added 10 mL of CH2C12 followed by 2.3 mL of
diisopropylethyl amine
(DIEA). The mixture was treated with 5.3 g (13.2 mmol) of intermediate 39 and
allowed to
agitate at rt overnight. The resin was subsequently filtered and washed three
times each with
CH2C12, Me0H, H20, saturated NaHCO3, H20, Me0H and Et20. After filtration the
resin
was dried under reduced pressure overnight at rt to afford 3.11 g of a light
tan resin
intermediate 40 IR(ATR) 3382 (br), 3023, 2924, 1737 (C=0), 1680; Elemental
Analysis: C,
74.31; H, 7.53; N, 3.44; EA shows loading of 1.23 meq/g based on N analysis.
[00261] A suspension of 2.74 g (3.37 mmol) of urea-triester resin intermediate
40 in
17 mL of Me0H was prepared in the 60 mL peptide reactor. The mixture was
treated with
3.02 mL of NH2OTMS followed by 1.34 g of KOH in 10 mL of Me0H. The mixture was
allowed to agitate by rocking overnight. The mixture was filtered and washed
three times
each with Me0H, H20, Me0H and H20. The resin was treated with 10% aq. acetic
acid and
allowed to agitate for one hour. The resin was filtered, washed three times
each with Me0H,
H20, Et20 and dried under reduced pressure to give 2.85 g of the trihydroxamic
acid resin 6
IR(ATR) 3205, 2919, 1636, 1550. Elemental Analysis: C, 66.02; H, 7.08; N,
7.28; EA shows
loading of 1.04 meq/g based on N analysis.
[00262] Method B ¨ Isocyanate resin and TRIS Amine Route
[00263] To 2 g (4.4 mmol) of aminomethyl resin (Aldrich 564095; macroporous 30-
60
mesh, 2.2 meq/g) was added 10 mL of CH2C12 followed by 770 uL of DIEA and 1.3
g (4.4
mmol) of triphosgene. The mixture was allowed to rock in a peptide reactor for
15 min and
subsequently filtered and washed with CH2C12. The resin was suspended in 10 mL
CH2C12,
treated with 2.31 mL of DIEA and 5.0 g (13.2 mmol) of aminetriester
intermediate 2 and
allowed to rock overnight. Filtration followed by washing three times each
with CH2C12,
Me0H, H20, satr NaHCO3, H20, Me0H and Et20 afforded intermediate 40. The
product
was dried in vacuo at rt overnight to give 2.35 g of an off-white resin:
IR(ATR) 3300 (br,
weak), 3025, 2923, 2260 (very weak, residual isocyanate), 1738, 1679, 1601.
Elemental
Analysis: C, 81.06; H, 7.62; N, 3.41; EA shows loading of 1.23 meq/g based on
N analysis.
[00264] A suspension of 2.2 g (2.7 mmol) of urea-triester resin intermediate
40 in 10
mL of Me0H was prepared in the 60 mL peptide reactor. The mixture was treated
with 1.2
80
CA 02792724 2012-10-19
mL of NH2OTMS followed by 0.56 g of KOH in 8 mL of Me0H. The mixture was
allowed
to rock overnight. The mixture was filtered and washed three times each with
Me0H and
H20. The resin was treated with 10% aq. acetic acid and allowed to rock for
one hour. The
resin was filtered, washed three times each with H20, Me0H, Et20 and dried
under reduced
pressure to give 2.2 g of the trihydroxamic acid resin 6: IR(ATR) 3311, 3023,
2920, 1651,
1600. Elemental Analysis: C, 80.46; H, 7.51; N, 3.75; Elemental analysis shows
loading of
0.54 meq/g based on N analysis.
1002651 Example 26
1002661 Preparation of Polyalkoxy Tether-linked Tris Hydroxamic Acid Resin 7
81
CA 02792724 2012-10-19
,
0-1-0\---\
0
1) CH2Cl2, TEA, triphosgene NH
p. (a___ j HN- NH2
4-1NH2 2) CH2Cl2, TEA, 0
0(0}-120H200H20H20H2NH2)2
intermediate 42(0.95 meq/g)
\
070 o
/ 0-1¨
ro o
d---\__o 0 /
o/
\ Y-0 HN NH HN HN---\0____\
...z.- 4 ¨/ o -i o o \
intermediate 39 0
0-
,
0
intermediate 43 (0.71 meq/g)
o
0 0 o \--NHpH
1) NH2OTMS 0i
2) aq. AcOH
HN---"No\
HN-i
NH , 0 0 e 0
HN-OH
Resin 7 (0.70 meq/g) rENII,OH
0
[00267] To 5 g (11 mmol) of aminomethyl resin (Sigma-Aldrich cat#
564095; 2.2
meq/g) in 25 mL CH2C12 in the 60 mL peptide reactor was added 1.53 mL of
triethylamine.
The mixture was treated portionwise with 3.26 g (11 mmol) of triphosgene.
After 15 min the
mixture was filtered and washed three times with CH2C12. The resin was
suspended in 25
mL CH2C12 and treated with 4.6 mL FLA and 7.23 nth of 4,7,10-trioxa-1,13-
tridecanediamine. After rocking for two days, the mixture was filtered and
washed three
times each with CH2C12, Me0H, H20, Me0H, Et20 and allowed to dry under a
stream of N2
for 1h. The resin was dried overnight in vacuo at rt to afford 6.177 g of an
off-white resin
82
CA 02792724 2012-10-19
intermediate 42. IR (ATR) 3560 (br, weak), 2920, 2880, 1655. Elemental
Analysis: C,
80.14; H, 8.18; N, 4.01; EA shows loading of 0.95 meq/g based on N analysis.
[00268] To 6.14 g of resin intermediate 42 (5.8 mmol) was added 30 mL of
CH2C12
followed by 2.3 mL of diisopropylethylamine (DIEA). The mixture was treated
with 5.3 g of
isocyanate intermediate 39 and allowed to agitate by rocking for 2 days at rt.
The resin was
filtered and washed three times each with CH2C12, Me0H, H20, satr NaHCO3, H20,
Me0H
and Et20. After filtration the resin was dried in vacuo overnight at rt to
afford 5.5 g of a light
tan resin intermediate 43: IR(ATR) 3322 (br), 3023, 2920, 2865, 1736 (C=0),
1685, 1655.
Elemental Analysis: C, 78.52; H, 7.84; N, 3.99; EA shows loading of 0.71 meq/g
based on N
analysis.
[00269] A suspension of 5.5 g of triester resin intermediate 43 (3.8 mmol) in
15 mL
of Me0H was prepared in a 60 mL peptide reactor. The mixture was treated with
4.2 mL of
NH2OTMS followed by a solution of 1.92 g of KOH in 20 mL of Me0H. The mixture
was
allowed to rock overnight at rt. The mixture was subsequently filtered and
washed three
times each with Me0H and H20. The resin was treated with 10% aq. acetic acid
and
allowed to rock for one hour. The resin was filtered, washed three times each
with H20,
Me0H, Et20 and dried in vacuo at rt to give 5.96 g of the trihydroxamic acid
resin 7:
IR(ATR) 3200, 2915, 1670, 1650, 1552. Elemental Analysis: C, 76.24; H, 7.77;
N, 4.68; EA
shows loading of 0.67 meq/g based on N analysis.
[00270] Example 27
[00271] A urea linked unsymmetrical resin 8
[00272] Scheme 44
83
CA 02792724 2012-10-19
H2NO CO2Me L.A.)C12, CH2C12 OCN-0
CO2Me Q/NH2
\O--\
N---0O2Me aq NaHCO3
iPr2NEt, CH2C12
--0O2Me CO2Me
intermediate 44 intermediate 45 CO2Me
H H H H
0 0
?v CO2Me 11TMS-ONH2 r.1
CONHOH
0 0
CO2Me Me0H, KOH CONHOH
intermediate 46 OC 2Me resin 8 \--
CONHOH
[00273] To a vigorously stirred solution of amine triester
intermediate 44 (12.0 g, 30.5
mmol) in CH2C12 (100 mL) and saturated NaHCO3 (100 mL) was added triphosgene
(3.08 g,
36.0 mmol) in small portions. Once the addition was complete, the mixture was
stirred for an
additional 15 min., then the layers were separated and the aqueous layer was
extracted with
CH2C12 (2x100 mL). The combined organic extracts were dried over Na2SO4,
evaporated
under reduced pressure, and the residue was purified by column chromatography
(Si02,
hexanes/Et0Ac gradient) to give the isocyanate intermediate 45 as a colorless
oil (5.14 g,
41%): IR (neat) 2957, 2874, 2244, 1733 cm-1; 1H N1VIR (300 MHz, CDC13) 8 3.70
(t, J= 6.3
Hz, 4H), 3.64 (s, 9H), 3.63 (t, J= 6.3 Hz, 2H), 3.50 (t, J= 6.3 Hz, 2H),
3.39(s, 4H), 2.54 (t,
J= 6.2 Hz, 4H), 2.52 (t, J = 6.2 Hz, 2H), 1.72 (t, J- 6.4 Hz, 2H); 13C NMR
(300 MHz,
CDC13) 6 172.1, 171.9, 126.6, 73.4, 66.8, 66.7, 66.2, 62.7, 51.8, 51.7, 34.9,
34.8, 33.8;
HRMS (FAB) calcd for CI8H301\1010 [M + Hr: 420.1870. Found 420.1885.
[00274] To a suspension of aminomethyl resin (Aldrich 564095,
macroporous 30-60
mesh) (1.84 g, 2.2 meq/g, 4.05 mmol of -NH2), in CH2C12 (15 mL) was added
diisopropylethyl amine (2.12 ml, 12.2 mmol) followed by the isocyanate
intermediate 45
(5.10 g, 12.2 mmol). The suspension was shaken using orbital shaker overnight
at room
temperature. The resin was filtered and washed 3 times each with CH2C12, Me0H,
H20,
saturated NaHCO3, H20, Me0H and Et20. It was dried under reduced pressure to
obtain pale
yellow resin intermediate 46 (2.86 g). Wt. added to the resin = 1.02 g; IR
(AIR): 3393,
3026, 2912, 2869, 1736 (CO), 1673 cm-1; Elemental analysis C = 75.50%, H =
7.59%, N =
84
CA 02792724 2012-10-19
3.35%, loading = 0.87 meq/g (based on %N); %C indicates 76% conversion of
available NH2
groups of the resin.
[00275] A suspension of the urea triester resin intermediate 46 (2.68 g,
1.14 meq/g -
maximum loading, 3.06 mmol) in Me0H (20 mL) was shaken for 15 minutes. A
solution of
KOH (1.54 g, 27.59 mmol) in Me0H (5 mL) was added followed by NH2OTMS (3.37
mL,
27.6 mmol) and the mixture was shaken for 20 h using orbital shaker. The resin
was filtered
and washed 3 times each with Me0H, H20. The resin was then suspended in 10%
aqueous
AcOH (20 mL) and for shaken for 30 minutes. The resin was filtered and washed
3 times
with 10 % aqueous CH3CO2H, H20, Me0H and Et20 and dried under reduced pressure
to
give a light yellow colored resin 8 (2.7 g). Wt. gained by the resin = 0.02 g;
IR (A'TR): 3221,
3025, 2920, 1641 (C=0 hydroxamate, sharp), 1551 cm-i; Elemental analysis C =
71.48%, H
= 7.36%, N = 4.65%, loading = 0.58 meq/g (based on %N). ,
[00276] Example 28
[00277] Scheme 45
0 /
ON
0
HO
orj
OH acrylonitrile
Me0H, HCI 0
H N-0 i
H2N 2
KOH, H tO 0
46% 2 acetonitrile
OH 1.,CN
C) 0
TRIS
CN .r0,,
intermediate 1 intermediate 2
0 / 0
o OH
,-0
),--Nil-1
Method A
/
0 1) NH2OTMS
o/
clCa HNtO 0 2) aq. AcOH
t
Nal, NMP0 0 Method B LõJLN-OH
1) NH2OH, Me0H, Me0Na C)
H
=,..,1y0.., 2) aq. AcOH H
intermediate 47 (0.51 meq/g) 8 OH
Resin 9 0
Method A (0.46 meq/g)
Method B (0.49 meq/g)
[00278] Tris[2-(cyanoethoxy)methyllmethylamine (intermediate 1) by an
improved method. A stirred suspension of tris-(hydroxymethyl)aminomethane
(127.0 g) in
acetonitrile (500 mL) in a 2L round bottom flask equipped with an overhead
stirrer was
85
CA 02792724 2012-10-19
treated with KOH (5.0 g). Acrylonitrile (207 mL) was added to the stirred
suspension over a
few minutes. After lh, an exotherm was observed to 36 C. After 3h, the
mixture was a
homogeneous, slightly orange solution and showed complete conversion based on
111 NMR
analysis. After a total of 4 h, the mixture was concentrated under reduced
pressure to afford
intermediate 1 (237.8 g, 89%) as a light tan oil. IR(neat) 3504, 3288, 2857,
2250(CN); r1D23
= 1.4687 1H NMR (CDC13, 300 MHz) M.45 (brs, 2H), 2.53 (t, 6.0 Hz, 6H), 3.34
(s, 6H),
3.59 (t, 6.0 Hz, 6H); 13C NMR (CDC13, 75 MHz) 618.8, 56.0, 65.7, 72.5, 188.1.
This
compound has been previously prepared, however the yield appears to be
improved with
acetonitrile solvent.
[00279] A 10 mL oven dried peptide reactor was charged with Merrifield resin
(2.0 g)
(Marcroporous, 100-200 mesh, 150-75 urn, 1.2 mmol/g, SigmaAldrich 564087, Lot
#05629MC, washed and dried) and anhydrous NaI (400 mg). The mixture was
suspended in
a solution of intermediate 2 (2.93 g) in of anhydrous NMP (12 mL) and agitated
for 7 days at
rt. The mixture was sampled at regular intervals and the reaction progress
monitored by IR
(ATR). After a total of 7 days the reaction was complete. The product was
collected by
filtration and washed sequentially three times each with DMA, H20, Me0H and
Et20 to give
a light, tan resin intermediate 47: IR(ATR) 3026, 2921, 1739 (C=0), 1602;
Elemental
Analysis: C, 83.03; H, 7.59; N, 0.72; EA shows loading of 0.51 meq/g (53%
conversion of
benzylchloride groups). The resin was kept in the peptide reactor and used
directly in the
next step.
[00280] Preparation of Trishydroxamic Acid Resin 9 using Method A. A
suspension of the ester resin intermediate 47 in anhydrous Me0H (5 mL) was
treated with
NH2OTMS (2.42 mL), followed by a solution of KOH (1.11 g) in anhydrous Me0H (5
mL).
The mixture was agitated overnight, then the resin was washed three times each
with Me0H
and H20, treated with 10% aq. AcOH and agitated for lh. The mixture was
filtered and
treated a 2nd time with 10% AcOH for 30 min. The resin was filtered, washed
three times
each with Me0H, H20, Et20 and dried under reduced pressure to give of the
trihydroxamic
acid resin 9 (1.84 g): IR(ATR) 3210, 3026, 2921, 1652, 1602, 1493; Elemental
Analysis: C,
80.33; H, 7.46; N, 2.55; EA shows loading of 0.46 meq/g.
86
CA 02792724 2012-10-19
, , , .,
[00281] Preparation of Trishydroxamic Acid Resin 9 using Method B. A
mixture
of 900 mg of hydroxylamine hydrochloride in 30 mL of 0.5_M sodium methoxide in
methanol was stirred at rt for 15 mm. The mixture was filtered to remove NaC1
salts and
added directly to 2.0 g of triester resin intermediate 47 (1.24 mmol) in a
flask equipped with
an overhead stirrer, heating mantle and kept under nitrogen. The stirred
mixture was heated
to 45 C and allowed to stir for 3 days. After filtration the resin was washed
three times each
with methanol, 10% aq. acetic acid, H20, methanol, and ethyl ether. The resin
was dried in
vacuo overnight to afford an amber resin 9: IR(ATR) 3210, 2921, 1652, 1602.
Elemental
Analysis: C, 78.95; H, 7.43; N, 2.74; EA shows loading of 0.49 meq/g based on
N
analysis.
[00282] Example 29
[00283] Resin linked tetra hydroxamic acid
[00284] Scheme 46
87
CA 02792724 2012-10-19
0 0 THP-ONH2 THPO.,N
0
OMe Et0H, Py I OMe
intermediate 35 intermediate 48
NaBH3CN, Me0H THPO,N 0 OMe DCC,
DMAP, CH2Cl2
HCI intermediate 49
CbzHN/\--0-----CO2H
intermediate 34
OTHP
0 --CON CO2Me 1) Me0H, Pd.C, H2
CbZHN000NCOMe 2) COCl2,
CH2Cl2, aq NaHCO3
OTHP
intermediate 50
OTHP CO2Me (ar---NH2
OC~OCOCO2Me CH2Cl2,
1Pr2NEt
OTHP
intermediate 51
OTHP
0 0 CO2Me 1) TMS-
ONH2, Me0H, KOH
NNOCONH OTHP CO2Me 2) Me0H,
Ts0H
intermediate 52
OH
0 CONHOH
ar N OH CONHOH siH
resin 10
1002851 To a solution of aldehyde intermediate 35 (Kai, K.;
Takeuchi, J.; Kataoka, T.;
Yokoyama, M.; Watanabe, N. Tetrahedron 2008, 64, 6760 ) (2.89 g, 15.5 mmol)
and 0-
(tetrahydro-2H- pyran-2-yl)hydroxylamine (2.0 g, 17.1 mmol) in Et0H (62 mL) at
room
temperature was added pyridine (1.88 mL, 23.3 mmol) drop wise. The resulting
solution was
heated at reflux for 4 h. Solvent was evaporated under reduced pressure and
the residue was
dissolved in CH2C12 (100 mL) and washed with water (2x100 mL). The aqueous
layer was
88
CA 02792724 2012-10-19
re-extracted with CH2C12 (2x100 mL). The combined organic layers were dried
over Na2SO4
and evaporated reduced pressure. The crude product was purified by column
chromatography
(SiO2hexanes) to give the oxime intermediate 48 as a colorless liquid (3.70 g,
84%) and as a
mixture of geometric isomers: IR (neat) 2933, 2856, 1736 cm-1; 'H NMR (CDC13)
8 (ppm)
7.42 (t, J= 6.3 Hz, 0.4H), 7.71 (t, J= 5.5 Hz, 0.611), 5.19 (m, 1.2H), 5.15
(m, 0.8H), 3.89-
3.84 (m, 1H), 3.62 (s, 3H), 3.59-3.53 (m, 1H), 2.35 (m, 1.5H), 2.26 (m, 2H),
2.18 (m, 0.5H),
1.90-1.40 (m, 10H), 1.29-1.26 (m, 6H); 13C NMR (CDC13) 8 (ppm) 174.2, 153.5,
152.9,
100.6, 100.5, 63.2, 63.1, 51.4, 34.0, 29.5, 29.1, 29.0, 28.9 (x 3), 26.6,
26.1, 25.8, 25.2(x 2),
24.9, 20.1, 20.0; HRMS (FAB) C15H28N04 [M+Hr calcd 286.20184, found 286.20110
[00286] To a solution of intermediate 48 (3.60 g, 12.31 mmol) and NaCNBH3
(0.95 g,
15.1 mmol) in Me0H (100 mL) was added 2N HC1 in Me0H drop wise until the
solution pH
was adjusted to 4 (pH was never allowed to go down from 4). The mixture was
stirred for 3 h
at rt. The solvent was evaporated under reduced pressure to give solid residue
which was
dissolved in water (100 mL) and then 6 N KOH solution was added drop wise to
adjust the
solution pH to >9. The aqueous mixture was extracted with CH2C12 (3x100 mL).
The organic
layers were combined and washed with brine, dried over Na2SO4 and evaporated
under
reduced pressure. The crude product was purified by column chromatography
(SiO2
hexanes/Et0Ac gradient) to give intermediate 49 as a colorless liquid (2.80 g,
78%): IR
(ATR, neat) 2931, 2854, 1736 cm-1; 111 NMR (CDC13) 8 (ppm) 5.53 (s, 1H), 4.73-
4.71 (m,
1H), 3.89-3.82 (m, 1H), 3.59 (s, 3H), 3.53-3.46 (m, 111), 2.96-2.82 (m, 2H),
2.23 (t, J= 7.4
Hz, 211), 1.75-1.39 (m, 10H), 1.25 (m, 8H); 13C NMR (CDC13) 5 (ppm) 174.2,
101.4, 63.1,
52.2, 51.4, 34.0, 29.3, 29.2, 29.1, 29.0, 27.2, 27.1, 25.3, 24.9, 20.2; HRMS
(FAB) C15H30N04
[M+Hr calcd 288.21747, found 288.21780
[00287] To a stirred solution of intermediate 49 (2.30 g, 8.00 mmol) and DMAP
(1.33
g, 10.9 mmol) in CH2C12 (70 mL) and pyridine (0.88 mL, 10.9 mmol) was added
intermediate 34 (1.39 g, 3.63 mmol) in CH2C12 and the mixture was cooled to 0
C. DCC
(1.65 g, 8.00 mmol) was added and the mixture was stirred for 1 h at 0 C. The
mixture was
then allowed to warm to room temperature and was stirred for additional 15 h.
The solvent
89
CA 02792724 2012-10-19
=
was concentrated under reduced pressure, filtered and washed with CH2C12. The
filtrate was
evaporated under reduced pressure and the residue was purified by column
chromatography
(Si02hexanes/Et0Ac gradient) to give intermediate 50 as a colorless oil (2.50
g, 76%): IR
(neat) 3329, 2930, 2856, 1732, 1655 cm-1; 1H NMR (CDC13) 8 (ppm) 7.33-7.24 (m,
511),
5.51 (s, 1H), 4.99 (s, 2H), 4.87 (s, 2H), 3.92-3.87 (m, 2H), 3.78-3.68 (m,
6H), 3.61 (s, 6H),
3.58-3.37 (m, 8H), 2.65-2.60 (m, 4H), 2.25 (t, J= 7.5 Hz, 4H), 1.76-1.54 (m,
20H), 1.29 (s,
3H), 1.24 (br s, 1611); 13C NMR (CDC13) 8 (ppm) 174.1, 172.7, 155.1, 136.7,
136.6, 128.3 (x
2), 127.8, 127.7, 104.3, 77.4, 73.2, 73.1, 67.9, 67.0, 65.8, 63.4, 55.7, 55.6,
51.3, 50.1, 48.6,
48.1, 35.5, 33.9, 32.9, 32.5, 30.6, 29.0 (x 2), 28.9, 28.8 (x 2), 26.6, 26.5,
26.0, 25.6, 25.1,
19.8, 19.1; HRMS (FAB) C48H79N30i4Na [M+Na] calcd 944.5460, found 944.5488.
[00288] To a solution of intermediate 50 (2.50 g, 2.70 mmol) in Me0H (200 mL)
under argon was added 10% Pd/C (0.275 g). The flask was evacuated then flushed
with 112
(balloons) and resulting the mixture was stirred under 112 at room temperature
until TLC
analysis (75% Et0Ac in hexanes) indicated that the starting material was
consumed. The
reaction flask was then purged with argon and filtered. The filtrate was
evaporated under
reduced pressure to give the amine as thick oil (2.1 g, 100%) which was used
directly in the
next step. To a stirred biphasic mixture of the amine (2.1 g, 2.7 mmol) in
CH2C12 (30 mL)
and saturated NaHCO3 (30 mL) was added triphosgene (0.26 g, 0.89 mmol) in
small
portions. After 20 min after the addition of triphosgene the layers were
separated and the
aqueous layer was washed with CH2C12 (2x30 mL). The combined organic layers
were dried
over Na2SO4 and evaporated under reduced pressure to give the isocyanate
intermediate 51
as a thick colorless oil product (1.7 g, 78 %) which was found to be pure
enough to use in the
next step without further purification. IR (neat) 2932, 2856, 2241, 1736, 1655
crn-1; 1H NMR
(CDC13) 8 (ppm) 4.89 (br s, 211), 3.96-3.89 (m, 2H), 3.81-3.72 (m, 6H), 3.62
(s, 6H), 3.60-
3.53 (m, 4H), 3.41-3.32 (m, 4H), 2.68- 2.61 (m, 4H), 2.26 (t, J= 7.4 Hz, 4H),
1.78-1.57 (m,
20H), 1.26 ( br s, 16H), 1.18 (s, 3H); 13C NMR (CDC13) 8 (ppm) 174.3, 172.7,
126.8, 104.5,
77.4, 75.3, 75.2, 75.1, 68.2, 67.3, 63.6, 61.2, 61.1, 61.0, 55.0, 51.5, 50.2,
48.3, 35.7, 34.1,
33.1, 32.6, 30.8, 29.5, 29.2, 29.1, 26.9, 26.8, 26.2, 25.5, 25.4, 22.5, 19.8;
HRMS (FAB)
C411-171N3013Na [M+Nar calcd 836.48846, found 836.48570.
90
CA 02792724 2012-10-19
[00289] To a suspension of the aminomethyl resin (1.1 g, 2.2 meq/g, 2.42 mmol
of ¨
N112, Aldrich 564095, macroporous 30-60 mesh) in CH2C12 (17 mL) and
diisopropyl ethyl
amine (2.66 mL, 15.3 mmol) was added the isocyanate intermediate 51(5.0 g,
6.10 mmol),
and the resulting mixture was shaken on orbital shaker for 24 h at room
temperature. The
resin was filtered and washed 3 times each with CH2CH2, Me0H, H20, saturated
NaHCO3,
H20, Me0H and Et20. It was then dried under reduced pressure to obtain light
colored resin
intermediate 52 (1.51 g). Wt. added to the resin = 0.41 g. IR (ATR): 3377,
3022, 2924,
2848, 1734 (C=0 ester), 1655 (C=0 hydroxamate cm-1. Elemental analysis C
81.23%, H
7.96%, N 3.67%. loading = 0.28 meq/g (based on %N). Change in %C indicates 43%
conversion of available NH2 groups of the resin.
[00290] To a suspension of the resin intermediate 52 (1.51 g, 0.30 meq/g, 0.45
mmol)
in Me0H (10 mL) was added KOH (0.15 g, 2.7 mmol) and NH2OTMS (0.33 mL, 2.7
mmol)
and the mixture was shaken for 20 h on an orbital shaker. The resin was
isolated by filtration
and washed 3 times with Me0H. The resin was re-suspended in a solution of
Ts0H.H20
(0.52 g, 2.7 mmol) in Me0H (10 mL) and shaken for for 3 h. The resin was
isolated by
filtration and washed 3 times each with Me0H, H20, Me0H and Et20 and dried
under
reduced pressure to give the product as pale yellow colored resin 10 (1.73 g).
Wt. gained by
the resin = 0.22 g. IR (ATR): 3382 (br), 3500-2500 (br), 3024, 2922, 2854,
1646 (C=0
hydroxamate) cm'. Elemental analysis C 75.74%, H 7.51%, N 3.68%. Loading =
0.19 meq/g
(based on %N).
[00291] Example 30
[00292] Binding of Al to resin 9
[00293] The compounds and compositions of the present invention are useful in
a
method of removing a trivalent metal ion such as Al3+ from an aqueous
solution. This is
accomplished by treating the aqueous solution with the invention, which
consists of a resin to
which the chelating agent is attached by a covalent bond to form a chelating
resin.
91
CA 02792724 2012-10-19
[00294] To demonstrate the ability of resin 9 to bind A13 , a 240 mg portion
of the resin
was suspended in three different aqueous solutions: (1) an aqueous solution
buffered at pH
6.06 by 0.1 M MES (4-morpholineethanesulfonic acid); (2) a solution of
gluconate that had
been adjusted to pH 6.42 by the addition of tetramethylammonium hydroxide; and
(3) a
commercial sample of calcium(gluconate)2, which had a pH of 6.07. Each
solution was
stirred by a magnetic overhead stirrer to keep the resin suspended in solution
with significant
mechanical damage as might result from use of a stir bar.
, [00295] At periodic times, the stirring was stopped for 1-2 minutes to let
the resin
settle, and a 100 uL aliquot was removed from the sample solution. Five I, of
concentrated,
metal-free nitric acid was added immediately to each aliquot removed to
stabilize the Al3+ in
solution. Samples were collected and subsequently analyzed by inductively
coupled plasma-
mass spectrometry to determine the Al concentration. Figure 6 shows plots of
the fraction of
original Al concentration remaining in the extracted solutions as a function
of the extraction
time.
[00296] The pH of the sample solutions was measured before and immediately
after
the completion of the extraction experiment. The addition of the resin and the
extraction
produce essentially no change in the pH of any of the solutions tested.
[00297] In the extraction of Al3+ from the MES buffer, the only competition
to binding
to the chelating resin is the hydrolysis of Al3+ to a mixture of Al-hydroxide
complexes.
Figure 6 shows that the concentration of Al3+ decreases to ¨0. In the case of
both gluconate
and calcium gluconate solutions, chelation of Al3+ by gluconate is competitive
with the
binding of Al3+ to the resin. Nevertheless, resin 9 removes approximately 90%
of the Al
from both of these solutions.
[00298] Figure 7 shows the extraction of three solutions of commercial
calcium(gluconate)2. The figure includes data from two duplicate extractions
of the
calcium(gluconate)2 by 240 mg portions of resin 9. The resin removes ¨90% of
the total Al.
The rate of Al removal from the solution can be fit to a single-exponential
function to give an
apparent first-order rate constant for Al removal of 4.2 hr-1, which
corresponds to a half-life
for Al removal of only 10 minutes. The solid line in Figure 7 is the least
squares fit of one of
the two data sets to the single-exponential function.
92
CA 02792724 2012-10-19
1002991 For the purpose of comparison, another aliquot of the same commercial
calcium(gluconate)2 solution was extracted with the commercial chelating resin
Chelex .
This resin consists of polystyrene beads to which the chelating agent
iminodiacetic acid has
been linked by covalent bonds. This resin is Widely used in a variety of
applications to
remove metal ions from solution. Figure 7 shows that the Chelex resin is not
able to remove
any significant fraction of Al3+ from the calcium(gluconate)2 solution. This
poor extraction
reflects the avidity with which gluconate binds Al3+, and demonstrates a
strong chelating
agent is required to compete with gluconate to remove Al3+ from the solution.
[00300] The Al-binding constants for gluconate have been reported (R.J.
Motekaitis
and A.E. Martell, Inorg. Chem. 1984, 23: 18-23). Given these binding constants
for Al-
gluconate, the percentage of Al remaining in a gluconate solution in
equilibrium with resin-9
can be used to estimate the effective Al binding constant of the
trihydroxamate chelating
agent covalently bound to the resin. The removal of 90% of the Al corresponds
to an Al3+-
resin binding constant of log K = 20.6.
[00301] The immobilized chelating agents described in this document may be
used in a
number of difference devices. Figures 8a-8d disclose three different
embodiments of
cartridge 10 filled with any of the immobilized chelating agents described in
this document.
Each cartridge 10 contains a sealed body 12 having a first Luer Lock fitting
14 at the inlet
end and a second Luer Lock fitting 16 at the outlet end. The resin 18
including the
immobilized chelating agent is held in the body 12. A membrane 20 covers the
outlet 22
thereby preventing any particles that might be released from the resin 18 from
exiting the
cartridge 10. A mesh, sieve, frit or membrane 24 covers the inlet 26 and
functions to
maintain the resin 18 in position in the body 12 of the cartridge 10.
1003021 The body 12 and resin 18 contained therein may have a cylindrical
shape. The
Figure 8a and 8d embodiment has a relatively intermediate length and diameter.
The Figure
8b embodiment has a relatively long length and relatively narrow diameter. The
Figure 8c
embodiment has a relatively short length and a relatively large diameter.
[00303] Each of the embodiments has a different diameter and length which
affects the
flow characteristic of any fluid passing through the cartridge 10 and the
resin 18 holding the
93
CA 02792724 2012-10-19
immobilized chelating agent. The embodiment chosen for use will depend upon
the
application.
[00304] All of the embodiments 8a-8c provide a straight flow path from the
inlet 26 to
the outlet 22. While not illustrated, it should be appreciated that the
cartridge 10 may be
substantially any shape including non-linear, arcuate, even coiled. Further,
while the
illustrated cartridges 10 are all symmetrical it should be appreciated that
nonsymmetrical
shapes could be provided. In addition, while all the illustrated cartridges 10
are cylindrical in
cross section, substantially any other shape of cross section may be provided
including but
not limited to frustoconical, helical, square, hexagon and T-shaped.
[00305] The cartridge 10 may be used with a continuous flow of solution
passing
through the cartridge or in a stop-flow mode where solution is introduced into
the cartridge,
stopped for a time to allow the chelating agent to act, and then expelled from
the cartridge.
[00306] Figures 9a and 9b both show vessels 50 that hold a packet 52
containing resin
beads 54 holding the immobilized chelating agent. The packet 52 is made from a
semi-
permeable membrane similar, for example, to the paper used in the production
of tea bags.
[00307] In the figure 9a embodiment, the vessel 50 is opened and a solution to
be
treated and a packet 52 containing resin beads 54 are both introduced into the
vessel. The
vessel 50 is closed and the solution is agitated by magnetic stirrer (not
shown) or other means
to facilitate interaction and contact between the solution which freely passes
through the
packet 52 and the chelating agent that is immobilized on the resin beads 54
sealed in the
packet.
[00308] In the embodiment illustrated in Figure 9b, the vessel 50 includes an
inlet 56
and an outlet 58. The packet 52 is placed in the open vessel 50 and the lid 60
of the vessel
50 is then closed. A pump (not shown) is then actuated to pump solution
through the vessel
by means of the inlet 56 and outlet 58. Solution flow may be continuous or
stop-flow mode
in the manner described above. Agitation of solution within the vessel 50 may
also be
provided. Depending on the application, the vessel 50 may be a sterile vessel.
[00309] In an alternative approach, the resin beads 54 holding the chelating
agent may
be placed directly in the vessel 50 without a packet (see Figure 9c).
Membranes may be
provided over the inlet and outlet to insure the resin beads and any particles
they might
94
CA 02792724 2012-10-19
release are maintained in the vessel 50. Lure Lock fittings may also be
provided at the inlet
and outlet if desired.
[00310] Figure 10 is a plot showing the mean percentage of aluminum removed
from a
10% calcium gluconate injection USP as a function of flow rate using the
device illustrated
in Figure 8a and the resin and immobilized chelating agent of resin 9. The
device had an
internal cavity diameter of 6 mm and a length of 20 mm. It held approximately
245 mg of
resin and immobilized chelating agent at a loading rate of 0.55 mmoles/gram
resin.
[00311] As noted above, the chelating agents described in this document are
particularly useful in removing aluminum from solutions including calcium
gluconate. It
should be appreciated, however, that they are also very useful in a multitude
of other
applications involving the separation of trivalent metal ions from a solution.
Such
applications include but are not limited to: (1) removal of Al from dialysis
fluids and
biological products such as albumin and other pharmaceutical solutions in
which it is a
contaminant, such as phosphates; (2) treatment of metal overload of any
trivalent hard or
intermediate acid, according to the HSAB theory (for example iron from blood
transfusions
in beta-thalassemia); (3) as a complexing agent for MR1 contrast enhancement
(for example
with gadolinium); (4) as a treatment for poisoning with tri- and tetravalent
metal ions,
including radioactive elements that workers may be inadvertently exposed to
and are
potential chemical warfare agents (dirty bomb components) (for example
americium, cerium,
and plutonium); (5) extraction of metal ions from solution either to isolate
the pure metal or
as a pre-concentration step prior to some sort of elemental analysis ; (6)
environmental
remediation by complexing and removing toxic metals from contaminated water
and/or soils;
and (7) use as a complexing agent for radionuclides of metals such as Ga or In
for use as
diagnostic imaging agents or as therapeutic radiopharmaceuticals.
[00312] The invention has been described herein with reference to certain
preferred
embodiments. However, as obvious variations will become apparent to those
skilled in the
art, the invention is not to be considered as limited thereto.
95