Note: Descriptions are shown in the official language in which they were submitted.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries / Version 08-02-2011
HYDROXYALKYL BENZYL PYRAZOLES, AND USE THEREOF FOR THE TREATMENT OF
HYPERPROLIFERATIVE AND ANGIOGENIC DISEASES
The present application relates to novel 1-[3-(hydroxyalkyl)benzyl]-1H-
pyrazole derivatives, to
processes for preparation thereof, to use thereof for treatment and/or
prevention of diseases and to
use thereof for production of medicaments for treatment and/or prevention of
diseases, more
particularly for treatment and/or prevention of hyperproliferative and
angiogenic diseases and
those diseases which arise from metabolic adaptation to hypoxic states. Such
treatments can be
effected in the form of monotherapy or else in combination with other
medicaments or further
therapeutic measures.
Cancers are the consequence of uncontrolled cell growth of a wide variety of
different tissues. In
many cases the new cells penetrate into existing tissue (invasive growth), or
they metastasize into
remote organs. Cancers occur in a wide variety of different organs and often
progress in a manner
specific to the tissue. The term "cancer" as a generic term therefore
describes a large group of
defined diseases of different organs, tissue and cell types.
In 2002, 4.4 million people worldwide were diagnosed with tumours of the
breast, intestine,
ovaries, lung or prostate. In the same year, approx. 2.5 million deaths were
assumed to be a
consequence of these diseases (Globocan 2002 Report). In the USA alone, in
2005, more than 1.25
million new cases and more than 500 000 deaths were predicted from cancers.
The majority of
these new cases relate to cancers of the intestine (- 100 000), lung ('- 170
000), breast (-S 210 000)
and prostate (- 230 000). A further increase in cancers of approx. 15% over
the next 10 years is
expected (American Cancer Society, Cancer Facts and Figures 2005).
Some tumours at early stages can be removed by surgical and radiotherapy
measures. Metastasized
tumours can generally only be treated palliatively by chemotherapeutics. The
aim here is to
achieve the optimum combination of an improvement in the quality of life and
prolonging of life.
Chemotherapies are often composed of combinations of cytotoxic medicaments.
The majority of
these substances have bonding to tubulin as their mechanism of action, or they
are compounds
which interact with the formation and processing of nucleic acids. As of
recently, these also
include enzyme inhibitors which interfere with epigenetic DNA modification or
cell cycle
progression (e.g. histone deacetylase inhibitors, aurora kinase inhibitors).
Since such therapies are
toxic, there has recently been an increasing focus on targeted therapies in
which specific processes
in the cell are blocked without a high level of toxic stress. These especially
include inhibitors of
kinases which inhibit the phosphorylation of receptors and signal transmission
molecules. One
example thereof is imatinib, which is used very successfully for treatment of
chronic myeloid
leukaemia (CML) and gastrointestinal stromal tumours (GIST). Further examples
are substances
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-2-
which block EGFR kinase and HER2, such as erlotinib, and VEGFR kinase
inhibitors, such as
sorafenib and sunitinib, which are used for kidney cell carcinomas, liver
carcinomas and advanced
stages of GIST.
With an antibody directed against VEGF, it has been possible to prolong the
life expectancy of
colorectal carcinoma patients. Bevacizumab inhibits the growth of blood
vessels, which is an
obstacle to rapid expansion of a tumour, since it requires connection to the
blood vessel system for
continuously functioning supply and disposal.
One stimulus for angiogenesis is hypoxia, which occurs time and again with
solid tumours, since
blood supply is inadequate because of the unregulated growth. If there is a
lack of oxygen, cells
switch their metabolism from oxidative phosphorylation to glycolysis, so as to
stabilize the ATP
level in the cell. This process is controlled by a transcription factor which
is upregulated
depending on the oxygen content in the cell. This transcription factor, called
"hypoxia-induced
factor" (HIF), is normally removed posttranslationally by rapid degradation
and prevented from
being transported into the cell nucleus. This is accomplished by the
hydroxylation of two proline
units in the oxygen-degradable domain (ODD) and one asparagine unit in the
vicinity of the C
terminus by the enzymes prolyl dehydrogenase and FIH ("factor inhibiting
HIF"). After the
modification of the proline units, HIF can be degraded with mediation by the
Hippel-Lindau
protein (part of a ubiquitin-E3-ligase complex) via the proteasome apparatus
(Maxwell, Wiesener
et al., 1999). In the event of oxygen deficiency, the degradation does not
take place and the protein
is upregulated and leads to transcription or to blockage of the transcription
of numerous (more than
100) other proteins (Semenza and Wang, 1992; Wang and Semenza, 1995).
The transcription factor HIF is formed by the regulated a-subunit and a
constitutively present (--
subunit (ARNT, aryl hydrocarbon receptor nuclear translocator). There are
three different species
of the a-subunit, 1a, 2a and 3a, the latter being assumed to be a suppressor
if anything (Makino,
Cao et al., 2001). The HIF subunits are bHLH (basic helix loop helix) proteins
which dimerize via
their HLH and PAS (Per-Amt-Sim) domains, which starts their transactivation
activity (Jiang, Rue
et al., 1996).
In the most important tumour entities, overexpression of the HIFIa protein is
correlated with
increasing blood vessel density and enhanced VEGF expression (Hirota and
Semenza, 2006). At
the same time, glucose metabolism is moved towards glycolysis, and the Krebs
cycle is reduced in
favour of the production of cell units. This also implies a change in lipid
metabolism. Such
changes appear to guarantee the survival of the tumours. If, on the other
hand, the activity of HIF
is now inhibited, the development of tumours could consequently be suppressed.
This has already
been observed in various experimental models (Chen, Zhao et al., 2003;
Stoeltzing, McCarty et al.,
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-3-
2004; Li, Lin et al., 2005; Mizukami, Jo et al., 2005; Li, Shi et al., 2006).
Specific inhibitors of the
HIF-controlled metabolism should therefore be suitable as tumour therapeutics.
WO 2004/089303-A2 describes diary]-substituted pyrazoles as mGluR5 modulators
for treatment
of psychiatric disorders. WO 2010/072352-Al and WO 2010/085584-Al disclose 3-
phenyl-5-(IH-
pyrazol-4-yl)-1,2,4-oxadiazole derivatives as sphingosine-l-phosphate agonists
for treatment of
autoimmune and vascular disorders.
WO 2005/030121-A2 and WO 2007/065010-A2 describe the usability of particular
pyrazole
derivatives for inhibition of the expression of HIF and HIF-regulated genes in
tumour cells.
WO 2008/141731-A2 discloses heteroaryl-substituted N-benzylpyrazoles as
inhibitors of the HIF
regulation pathway for treatment of cancers. However, it has been found that
many of these
compounds do not have sufficient inhibitory activity or else, on the basis of
their pharmacokinetic
properties in animal models, are expected to have such a long half-life (>48
h) in the human body
that significant substance accumulation is probable after repeated once-daily
administration.
It was therefore an object of the present invention to discover and provide
novel compounds which
firstly act as potent inhibitors of the transactivating action of the
transcription factor HIF and
secondly have a pharmacokinetic profile which allows repeated once-daily
administration without
simultaneous occurrence of clinically relevant accumulation. Such properties
could also lead
overall to a broadening of the clinical employability of these HIF inhibitors
and more particularly
facilitate the combinability thereof with other active ingredients, for
example conventional tumour
chemotherapeutics.
This object is achieved by the inventive compounds described hereinafter. In
structural terms, this
novel group of N-benzylpyrazole derivatives features a hydroxyalkyl
substituent in the 3 position
of the benzyl head group, which surprisingly leads to an improved profile of
properties of the
compounds.
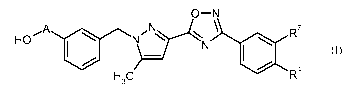
The present invention relates specifically to compounds of the general formula
(I)
O-N
R7
HOPN N I \
H 3 C / R4 (1)
in which
A represents a group of the formula
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-4-
R3A R3B
R1A R18
*
* q* or * * in which
R2A R2B
* denotes the point of attachment of the hydroxyl group,
* * denotes the point of attachment of the phenyl ring,
RIA and RIB each independently of one another represent hydrogen, deuterium,
methyl,
hydroxymethyl or trifluoromethyl
or
are joined to one another and, together with the carbon atom to which they are
attached, form a cyclopropane-1,1-diyl, cyclobutane-1,l-diyl, oxetane-3,3-diyl
or
tetrahydro-2H-pyran-4,4-diyl ring,
R2A and R2B each independently of one another represent hydrogen, deuterium,
methyl or
trifluoromethyl,
and
R3A and R3B each independently of one another represent hydrogen, fluorine,
methyl,
hydroxymethyl or trifluoromethyl
or
are joined to one another and, together with the carbon atom to which they are
attached, form a cyclopropane-1,l-diyl, cyclobutane-1,1-diyl, oxetane-3,3-diyl
or
tetrahydro-2H-pyran-4,4-diyl ring,
R4 represents trifluoromethoxy, trifluoromethylsulphanyl,
trifluoromethylsulphonyl,
pentafluorosulphanyl, trimethylsilyl or a group of the formula
# R6
5A` 5B
R R in which
# denotes the point of attachment of the phenyl ring,
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-5-
R5A and R5B each independently of one another represent hydrogen, fluorine,
methyl, ethyl,
n-propyl or isopropyl
or
are joined to one another and, together with the carbon atom to which they are
attached, form a cyclopropane-1,l-diyl, cyclobutane-1,1-diyl, cyclopentane-1,1-
diyl, cyclohexane-1,1-diyl, oxetane-3,3-diyl or tetrahydro-2H-pyran-4,4-diyl
ring
and
R6 represents hydrogen, fluorine, methyl, trifluoromethyl, methoxymethyl or
ethoxymethyl,
and
R7 represents hydrogen, fluorine or methyl,
and the salts, solvates and solvates of the salts thereof.
In the context of the present invention, preference is given to compounds of
the formula (I) in
which
A represents a group of the formula
Hoc, CH3 *~**
* ** * ** * H3C CH3
F F H3C CH3
* * * * ** or * * *
1AX X R2A R2B R2A R2B R2A Rte
in which
* denotes the point of attachment of the hydroxyl group,
** denotes the point of attachment of the phenyl ring,
and
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-6-
R2A and R2B both represent hydrogen or deuterium,
R4 represents trifluoromethyl, trifluoromethoxy, trifluoromethylsulphanyl,
pentafluorosulphanyl, trimethylsilyl or a group of the formula
R6
R5A R5B in which
# denotes the point of attachment of the phenyl ring,
R5A and R5B both represent methyl or are joined to one another and, together
with the
carbon atom to which they are attached, form a cyclopropane-1,l-diyl or
tetrahydro-2H-pyran-4,4-diyl ring
and
R6 represents fluorine, methyl or trifluoromethyl,
and
R7 represents hydrogen or methyl,
and the salts, solvates and solvates of the salts thereof.
Particular preference is given in the context of the present invention to
compounds of the formula
(I) in which
A represents a group of the formula
H, C CH3 F F
* ** * X A
** H3C CH3 *`/`**
H3C CH3
or
D D
in which
* denotes the point of attachment of the hydroxyl group,
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-7-
and
* * denotes the point of attachment of the phenyl ring,
R4 represents trifluoromethoxy, trifluoromethylsulphanyl or a group of the
formula
F #X CH3 # X C F3 #C F3
or
H3C CH3 , H3C CH3 H3C CH3
in which
# denotes the point of attachment of the phenyl ring,
and
R7 represents hydrogen,
and the salts, solvates and solvates of the salts thereof.
In a further aspect, the present invention also relates to certain prodrugs of
compounds of the
formula (I). In general, the term "prodrugs" refers here to covalent
derivatives of the compounds of
the formula (I), which may themselves be biologically active or inactive, but
are converted while
present in the body, for example by a metabolic or hydrolytic route, into
compounds of the formula
M.
Accordingly, the present invention also relates to compounds of the formula (I-
PD)
O-N
RPD O~q PN __~ \ R'
N
H3C R4 (I-PD)
in which
A, R4 and R7 each have the meanings given above
and
RPD represents a prodrug group of the formula
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-8-
R9A O
HO OII I
HOP"I ## or R9B.N # in which
$
R
# denotes the point of attachment of the oxygen atom,
R8 represents hydrogen or (C i-C4)-alkyl,
and
R9A and R9B each independently of one another represent hydrogen or methyl,
and the salts, solvates and solvates of the salts thereof.
In the context of the present invention, preference is given to compounds of
the formula (I-PD) in
which
RPD represents a prodrug group of the formula
0
CH
HO 0 13 0
H N
H O B ~P H3C N # or z #
~## $
R
in which
# denotes the point of attachment of the oxygen atom,
and
R8 represents methyl, isopropyl, isobutyl or sec-butyl,
and the salts, solvates and solvates of the salts thereof.
The compounds of the formula (I-PD) are prodrugs of the compounds of the
formula (I) with a
good solubility in aqueous or other physiologically compatible media; they
additionally offer the
possibility of salt formation with appropriate bases or acids, which can lead
to a further increase in
solubility. Accordingly, the compounds of the formula (I-PD) and their salts
are in particular also
suitable for intravenous administration forms. This could also open up
additional therapeutic fields
of use for these compounds.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-9-
Compounds according to the invention are thus the compounds of the formulae
(I) and (I-PD) and
the salts, solvates and solvates of the salts thereof, the compounds,
encompassed by the formulae
(I) and (I-PD), of the formulae specified hereinafter and the salts, solvates
and solvates of the salts
thereof, and the compounds encompassed by the formulae (I) and (I-PD) and
specified hereinafter
as working examples and the salts, solvates and solvates of the salts thereof,
to the extent that the
compounds encompassed by the formulae (I) and (I-PD) and specified hereinafter
are not already
salts, solvates and solvates of the salts.
Depending on their structure, the compounds according to the invention may
exist in different
stereoisomeric forms, i.e. in the form of configurational isomers or if
appropriate also as
conformational isomers (enantiomers and/or diastereomers, including those in
the case of
atropisomers). The present invention therefore encompasses the enantiomers and
diastereomers
and the respective mixtures thereof. The stereoisomerically uniform
constituents can be isolated
from such mixtures of enantiomers and/or diastereomers in a known manner;
chromatography
processes are preferably used for this, in particular HPLC chromatography on
an achiral or chiral
phase.
Where the compounds according to the invention can occur in tautomeric forms,
the present
invention encompasses all the tautomeric forms.
The present invention also encompasses all suitable isotopic variants of the
compounds according
to the invention. An isotopic variant of a compound according to the invention
is understood here
to mean a compound in which at least one atom within the compound according to
the invention
has been exchanged for another atom of the same atomic number, but with a
different atomic mass
than the atomic mass which usually or predominantly occurs in nature. Examples
of isotopes
which can be incorporated into an inventive compound are those of hydrogen,
carbon, nitrogen,
oxygen, phosphorus, sulphur, fluorine, chlorine, bromine and iodine, such as
2H (deuterium), 3H
(tritium), 13Cs 14Cs 15N> 17Q> 180e 32P, 33Pe 33S, 34S, 35S, 36S, 18F s 36C1>
82 Br, 1231e 1241, 1291 and 1311.
Particular isotopic variants of an inventive compound, especially those in
which one or more
radioactive isotopes have been incorporated, may be beneficial, for example,
for the examination
of the mechanism of action or of the active ingredient distribution in the
body; due to
comparatively easy preparability and detectability, especially compounds
labelled with 3H or 14C
isotopes are suitable for this purpose. In addition, the incorporation of
isotopes, for example of
deuterium, can lead to particular therapeutic benefits as a consequence of
greater metabolic
stability of the compound, for example to an extension of the half-life in the
body or to a reduction
in the active dose required; such modifications of the inventive compounds may
therefore in some
cases also constitute a preferred embodiment of the present invention.
Isotopic variants of the
inventive compounds can be prepared by generally customary processes known to
those skilled in
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-10-
the art, for example by the methods described below and the procedures
reported in the working
examples, by using corresponding isotopic modifications of the particular
reagents and/or starting
compounds therein.
Preferred salts in the context of the present invention are physiologically
compatible salts of the
inventive compounds. Also encompassed are salts which are not themselves
suitable for
pharmaceutical applications but can be used, for example, for isolation or
purification of the
compounds according to the invention.
Physiologically acceptable salts of the compounds according to the invention
include acid addition
salts of mineral acids, carboxylic acids and sulphonic acids, for example
salts of hydrochloric acid,
hydrobromic acid, sulphuric acid, phosphoric acid, methanesulphonic acid,
ethanesulphonic acid,
toluenesulphonic acid, benzenesulphonic acid, naphthalenedisulphonic acid,
acetic acid,
trifluoroacetic acid, propionic acid, lactic acid, tartaric acid, malic acid,
citric acid, fumaric acid,
maleic acid and benzoic acid.
Physiologically acceptable salts of the compounds according to the invention
also include salts of
conventional bases, by way of example and with preference alkali metal salts
(e.g. sodium and
potassium salts), alkaline earth metal salts (e.g. calcium and magnesium
salts) and ammonium salts
derived from ammonia or organic amines having 1 to 16 carbon atoms, by way of
example and
with preference ethylamine, diethylamine, triethylamine,
ethyldiisopropylamine,
monoethanolamine, diethanolamine, triethanolamine, dicyclohexylamine,
dimethylaminoethanol,
procaine, dibenzylamine, N-methylmorpholine, arginine, lysine, ethylenediamine
and N-
methylpiperidine.
In the context of the invention, solvates refer to those forms of the
compounds according to the
invention which, in the solid or liquid state, form a complex by coordination
with solvent
molecules. Hydrates are a specific form of the solvates in which the
coordination is with water.
Preferred solvates in the context of the present invention are hydrates.
In the context of the present invention, the substituents, unless specified
otherwise, are each
defined as follows:
In the context of the invention, (C1-C4 -a) lkyl is a straight-chain or
branched alkyl radical having 1
to 4 carbon atoms. Preferred examples include: methyl, ethyl, n-propyl,
isopropyl, n-butyl,
isobutyl, sec-butyl, tert-butyl.
In the context of the present invention, all radicals which occur more than
once are each defined
independently of one another. If radicals in the compounds according to the
invention are
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-11-
substituted, the radicals may be mono- or polysubstituted, unless specified
otherwise. Substitution
by one or by two or three identical or different substituents is preferred.
Substitution by one or by
two identical or different substituents is particularly preferred. Very
particular preference is given
to substitution by one substituent.
The individual radical definitions specified in the respective combinations or
preferred
combinations of radicals are, independently of the respective combinations of
the radicals
specified, also replaced as desired by radical definitions of other
combinations.
Very particular preference is given to combinations of two or more of the
abovementioned
preferred ranges.
The present invention further provides a process for preparing the compounds
of the formulae (I)
and (I-PD), characterized in that initially an N'-hydroxyamidine of the
formula (II)
HOB
N
R7
\
H2N I
/ R4 (II)
in which R4 and R7 have the meanings given above,
is condensed with a pyrazolecarboxylic acid of the formula (III)
O
HNC OH
H3C (III)
to give a 1,2,4-oxadiazole derivative of the formula (IV)
O-N
R
HNC N
- I /
H3C R4 (IV)
in which R4 and R7 have the meanings given above,
the compound (IV) is then reacted in the presence of a base with a compound of
the formula (V)
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-12-
R10O'-A X
(V)
in which A has the meaning given above,
X represents a leaving group such as, for example, chlorine, bromine, iodine,
mesylate,
triflate or tosylate
and
R10 represents hydrogen or represents a customary hydroxyl protective group
such as, for
example, acetyl, tetrahydropyranyl, trimethylsilyl, triisopropylsilyl, tert-
butyldimethylsilyl
or tert-butyl(diphenyl)silyl,
to give a compound of the formula (VI)
O-N
R10 O" pN R'
N
Icr,
H3C R4 (VI)
in which A, R4, R' and R10 each have the meanings given above,
the hydroxyl protective group R10 is then - if present - removed by customary
methods and the
resulting compound of the formula (I)
O-N
A , \ R7 Nlzz~
HOB PN N
H3C R4 (1)
in which A, R4 and R7 have the meanings given above,
is finally, if desired, converted with a compound of the formula (VII)
RPD-OH (VII),
or an activated form of this compound in which RPD has the meaning given above
by known methods into the prodrug compound of the formula (I-PD)
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-13-
O-N
A ', \ R7
RPDOPN N I /
H3C R a (I-PD)
in which A, RPD, R4 and R7 each have the meanings given above,
and the resulting compounds of the formula (I) or (I-PD) are optionally
separated into their
enantiomers and/or diastereomers and/or converted using the appropriate (i)
solvents and/or (ii)
bases or acids into the solvates, salts and/or solvates of the salts thereof.
The condensation reaction (II) + (III) (IV) is preferably carried out with the
aid of a
carbodiimide such as N'-(3-dimethylaminopropyl)-N-ethylcarbodiimide (EDC), in
combination
with 1-hydroxy-lH-benzotriazole (HOBt) as active ester component, or with the
aid of a phosgene
derivative such as 1,1'-carbonyldiimidazole (CDI) in a high-boiling dipolar-
aprotic solvent such as,
for example, N,N-dimethylformamide or dimethyl sulphoxide.
The initial coupling step of this reaction is generally carried out in a
temperature range of from
0 C to +50 C; the cyclization to the 1,2,4-oxadiazole is then accomplished by
subsequent heating
of the reaction mixture at temperatures of from +100 C to +150 C. The reaction
can be performed
at atmospheric, elevated or reduced pressure (for example from 0.5 to 5 bar);
in general, the
reaction is carried out at atmospheric pressure.
Inert solvents for the process step (IV) + (V) -> (VI) are, for example,
halogenated hydrocarbons
such as dichloromethane, trichloromethane, carbon tetrachloride,
trichloroethylene or
chlorobenzene, ethers such as diethyl ether, diisopropyl ether, methyl tert-
butyl ether,
tetrahydrofuran, 1,4-dioxane, 1,2-dimethoxyethane or bis(2-methoxyethyl)
ether, hydrocarbons
such as benzene, toluene, xylene, pentane, hexane, cyclohexane or mineral oil
fractions, or dipolar
aprotic solvents such as acetone, methyl ethyl ketone, ethyl acetate,
acetonitrile, NN-
dimethylformamide (DMF), N,N-dimethylacetamide (DMA), dimethyl sulphoxide
(DMSO), NN'-
dimethylpropyleneurea (DMPU), N-methylpyrrolidinone (NMP) or pyridine. It is
equally possible
to use mixtures of the solvents mentioned. Preference is given to using
tetrahydrofuran or 1,4-
dioxane.
Suitable bases for the reaction (IV) + (V) -+ (VI) are customary inorganic or
organic bases. These
preferably include alkali metal hydroxides, for example lithium hydroxide,
sodium hydroxide or
potassium hydroxide, alkali metal alkoxides such as sodium methoxide or
potassium methoxide,
sodium ethoxide or potassium ethoxide or sodium or potassium tert-butoxide,
alkali metal hydrides
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-14-
such as sodium hydride or potassium hydride, or amides such as sodium amide,
lithium
bis(trimethylsilyl)amide or potassium bis(trimethylsilyl)amide or lithium
diisopropylamide.
Preference is given to using potassium tert-butoxide. If appropriate, the
addition of an alkylation
catalyst, for example lithium bromide, sodium iodide, potassium iodide, tetra-
n-butylammonium
bromide or benzyltriethylammonium chloride, may be advantageous.
The reaction is generally carried out in a temperature range of from -20 C to
+100 C, preferably at
from 0 C to +60 C. The reaction can be performed at atmospheric, elevated or
reduced pressure
(for example in the range from 0.5 to 5 bar); in general, the reaction is
carried out at atmospheric
pressure.
Customary methods for removing a hydroxyl protective group R10 include, for
example, in the case
of ester derivatives the basic or acidic hydrolysis or the reaction with
organometallic compounds
(such as, for example, methylmagnesium bromide or ethylmagnesium bromide), in
the case of
tetrahydropyranyl ethers the acidic hydrolysis and in the case of silyl ethers
likewise the hydrolysis
or the treatment with fluorides (such as, for example, potassium fluoride or
tetra-n-
butylammonium fluoride) [cf., for example, T.W. Greene and P.G.M. Wuts,
Protective Groups in
Organic Synthesis, Wiley, New York, 1999].
Activated forms of the compound (VII) which are suitable for the introduction
of the prodrug
group RPD [transformation (I) (I-PD)] are, for example, corresponding
chlorides or anhydrides,
including mixed anhydrides, or else certain ester or amide derivatives. Any
other hydroxyl or
amino groups present in the RPD radical are appropriately present here in
temporarily protected
form and are then released again at the end of the reaction sequence by
customary methods. In this
process step, the protective group used for a hydroxyl function is preferably
benzyl, which is
removed again by hydrogenolysis, and the preferred amino protective group is
tert-
butoxycarbonyl, which can be cleaved off by treatment with a strong acid such
as hydrochloric
acid or trifluoroacetic acid [cf., for example, M. Bodanszky and A. Bodanszky,
The Practice of
Peptide Synthesis, Springer-Verlag, Berlin, 1984; M. Bodanszky, Principles of
Peptide Synthesis,
Springer-Verlag, Berlin, 1993; T.W. Greene and P.G.M. Wuts, Protective Groups
in Organic
Synthesis, Wiley, New York, 1999; see also Reaction Schemes 8-10 below].
Alternatively, compounds of the formula (I) according to the invention can
also be prepared by
initially reacting, in the presence of a base, the compound of the formula
(IV) described above
with a compound of the formula (VIII)
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-15-
Z X
(VIII)
in which X has the meaning given above
and
Z generally represents a substituent or a functional group which allows the
introduction or
construction of the hydroxyalkyl group HO-A-, defined above, by subsequent
chemical
transformations,
to give a compound of the formula (IX)
O-N
pN
Z '
R
N
H3C R4 (IX)
in which R4, R7 and Z have the meanings given above,
and then converting these by transformations of group Z, carried out in
accordance with methods
known from the literature, into compounds of the formula (I).
The reaction (IV) + (VIII) -+ (IX) is carried out under analogous reaction
conditions with regard to
solvent, base and temperature, as described above for the process step (IV) +
(V) - (VI).
Examples of substituents or functional groups Z in formula (VIII) which allow
the introduction or
construction of the hydroxyalkyl group HO-A- defined above are firstly halides
such as bromide or
iodide and secondly radicals containing a carbonyl group, such as
alkoxycarbonyl,
alkoxycarbonylalkyl, hydroxycarbonyl, hydroxycarbonylalkyl, formyl,
alkylcarbonyl or
alkylcarbonylalkyl. The conversions in question are carried out by known
methods familiar to the
person skilled in the art and include, for example, reactions such as the
reduction with complex
metal hydrides, the 1,2- addition of organometallic compounds (for example
Grignard compounds)
to carbonyl compounds, the hydroxylation, C-alkylation and C-acylation and
also the introduction
and removal of temporary protective groups [see also the Reaction Schemes 1 -
7 below and the
preparation, described in detail in the Experimental Part, of the Working
Examples].
The compounds of the formulae (II), (III), (V), (VII) and (VIII) are
commercially available or
described as such in the literature, or they can be prepared in a way obvious
to the person skilled in
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-16-
the art, in analogy to methods published in the literature. Numerous detailed
instructions and
literature information for the preparation of the starting materials are also
to be found in the
experimental part in the section on the preparation of the starting compounds
and intermediates.
The preparation of the compounds according to the invention can be illustrated
in an exemplary
manner by the reaction schemes below:
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-17-
Scheme I
0 HO _N
thlN OH + R7
H4q a
H3C R~
CD I
Or
EDUHOBt
O 0-N
MeO or + VN N 1 I R 7
qC R
KOtB u
0 0-N
7
MeO ~N R
H3 Rt
\ 1 IH+
2 eq. MeMgBr
\'1
0 -N
,N ~ 1 R7
H3C CH3 0 -N HO
HO
C
-11-b~l V-N R7 R
~CRt
KOtBu
H3C CH3 O -N R7 HN~ H0 ` 0Ms + _~-N Ra
H3C
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-18-
Scheme 2
ON
p"N__, RAcO OMs + HN
H3C R
KOtBu
O-N
AcO N~N~ "N\ I \ R'
/ R4
H3C
EtMgBr
O-N
HO N~
R4
H3C
KOtBu
ON
N
HO I \ Br + HPNN
/ 4
/ H3C R
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-19-
Scheme 3
O-N
~
HO N~N N
H3C R
R4
1. 'PrMgCI
2. [:O
O-N
NON R7
H3C / R4
1. iPrMgCI
2. O O
O
O-N
HO N~N~ R7
N
H3C R4
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-20-
Scheme 4
O-N
EtO I \ Br HN,N R7
O
H3C R4
KO'Bu
O-N
EtO \ N.N \ \ R7
H3C R4
1 eq. MeMgBr
O-N
H3C
~N N\ N ~ ~R7
H3C R4
LiAIH4
O-N
HO \ pN R7
CH3 / H3C R4
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-21-
Scheme 5
O-N
,N \ 7
EtOOC D N N I \ R
R
H3C / 4
1.NaH
2. (EtO)2CO
COOEt O-N
N 7
EtOOC P N R
H3C R
1.NaH
2. Mel
COOEt O-N
EtOOC I N, N \ \ \ R7
H3C
H3C R
NaBH4
HO
O-N
HO N,N~ R7
H3C _ N
H3C R4
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-22-
Scheme 6
F F I CHs Cu/Sn F F CH UAIH4
x + I -> EtOOC 3
EtOOC Br
F F F F
HO CHs NBS I HO Br
/ AIBN
J F O-N
HO HN,N\ R' KOtBu
Br + - N I .
H3C R4
F F O-N
HOJ N,N N \ \ R'
H3C / R4
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-23-
Scheme 7
McOOC Br BrCH2CH2Br Br LIAID4
McOOC
LiHMDS
HO Br TIPSOTf ,p Br 1. BuLi
s TIPS --
D D D D I/ 2. DMF
H
O LiAIH4 p Ms20
TIS 0 -> TIPSY OH
D D D D I/ Et3N
TIW O I OMs
D O-N
,N \ R7 1. KO1Bu
TIPS~O OMs + HN N
D D I / H C / R4 2. TBAF
3
0-N
HO I N,N \ R7
D D
H3C R4
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-24-
Scheme 8
O-N
H0-~ A PN N I R
H3C R4
(RO)2P(=O)CI
or I base
[(RO)2P(=0)]20
O-N
RO~IIII A PN R'
ROP-( ` N I \
H3C R4
H2, Pd/C
or
Me3SiBr
0 O-N
H0 jII ,A ~N~ 11-1 R'
HO'P-O N I \
H3C R4
[R = phenyl or benzy]].
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-25-
Scheme 9
0 -N
HO "A N `N. tiN 1 Pi
P-N B2 2
OI +
0 H3C P4
1. Tetrazole
2. mCPBA
0 -N
7
CCO 01,0 N ` P
PO -0 'A N~ N ~'-a ),:~, F3C _ P¾
3
H2, Pd/C
0 0 -N
P7
H O:P -0 f A Np
HO N
H 3 C P4
[cf. Y. Watanabe et al., Tetrahedron Lett. 1990, 31 (2), 255-256.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-26-
Scheme 10
0 0 0 -N
A ~N ` 1 R7
tBu0~N 0 + H0~ I N N
~ a
Rs 0 H3C R
base
I0 0-N
tBuO,rN_''xO,A I:rH3 N,N~ R7
0 R$ -
C Ra
HCI
0 O -N
H2N O,A N.N~ ~N a R 7
Rg __ -
Ra
The inventive compounds have valuable pharmacological properties and can be
used for
prevention and treatment of diseases in humans and animals.
The compounds according to the invention are highly potent inhibitors of the
regulatory HIF
pathway. In addition, the compounds according to the invention have
advantageous
pharmacokinetic properties with regard to their distrubution volume and/or
their clearance and the
half-life derived therefrom, rendering them suitable for repeated once-daily
administration.
On the basis of their profile of action, the inventive compounds are
especially suitable for
treatment of hyperproliferative diseases in humans and in mammals in general.
The compounds
can inhibit, block, reduce or lower cell proliferation and cell division, and
secondly increase
apoptosis.
The hyperproliferative diseases for the treatment of which the compounds
according to the
invention can be employed include, inter alia, psoriasis, keloids, formation
of scars and other
proliferative diseases of the skin, benign diseases, such as benign prostate
hyperplasia (BPH), and
in particular the group of tumour diseases. In the context of the present
invention, these are
understood to mean especially the following diseases, but without any
limitation thereto:
mammary carcinomas and mammary tumours (ductal and lobular forms, also in
situ), tumours of
the respiratory tract (parvicellular and non-parvicellular carcinoma,
bronchial carcinoma), cerebral
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-27-
tumours (e.g. of the brain stem and of the hypothalamus, astrocytoma,
medulloblastoma,
ependymoma and neuro-ectodermal and pineal tumours), tumours of the digestive
organs
(oesophagus, stomach, gall bladder, small intestine, large intestine, rectum),
liver tumours (inter
alia hepatocellular carcinoma, cholangiocellular carcinoma and mixed
hepatocellular and
cholangiocellular carcinoma), tumours of the head and neck region (larynx,
hypopharynx,
nasopharynx, oropharynx, lips and oral cavity), skin tumours (squamous
epithelial carcinoma,
Kaposi sarcoma, malignant melanoma, Merkel cell skin cancer and non-
melanomatous skin
cancer), tumours of soft tissue (inter alia soft tissue sarcomas,
osteosarcomas, malignant fibrous
histiocytomas, lymphosarcomas and rhabdomyosarcomas), tumours of the eyes
(inter alia
intraocular melanoma and retinoblastoma), tumours of the endocrine and
exocrine glands (e.g.
thyroid and parathyroid glands, pancreas and salivary gland), tumours of the
urinary tract (tumours
of the bladder, penis, kidney, renal pelvis and ureter) and tumours of the
reproductive organs
(carcinomas of the endometrium, cervix, ovary, vagina, vulva and uterus in
women and carcinomas
of the prostate and testicles in men). These also include proliferative blood
diseases in solid form
and as circulating blood cells, such as lymphomas, leukaemias and
myeloproliferative diseases, for
example acute myeloid, acute lymphoblastic, chronic lymphocytic, chronic
myelogenic and hair
cell leukaemia, and AIDS-correlated lymphomas, Hodgkin's lymphomas, non-
Hodgkin's
lymphomas, cutaneous T cell lymphomas, Burkitt's lymphomas and lymphomas in
the central
nervous system.
These well-described diseases in humans can also occur with a comparable
aetiology in other
mammals and can be treated there with the compounds of the present invention.
In the context of this invention, the term "treatment" or "treat" is used in
the conventional sense
and means attending to, caring for and nursing a patient with the aim of
combating, reducing,
attenuating or alleviating a disease or health abnormality, and improving the
quality of life
impaired by this disease, as, for example, in the event of a cancer.
The inventive compounds act as modulators of the HIF regulation pathway and
are therefore also
suitable for treatment of diseases associated with a harmful expression of the
HIF transcription
factor. This applies especially to the transcription factors HIF-la and HIF-
2a. The term "harmful
expression of HIF" here means abnormal physiological presence of HIF protein.
This can be
caused by excessive synthesis of the protein (mRNA- or translation-related),
by reduced
degradation or by inadequate counter-regulation in the functioning of the
transcription factor.
HIF-la and HIF-2a regulate more than 100 genes. This applies to proteins which
play a role in
angiogenesis and are therefore directly relevant to tumours, and also those
which influence
glucose, amino acid and lipid metabolism, and cell migration, metastasis and
DNA repair, or
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-28-
improve the survival of tumour cells by suppressing apoptosis. Others act more
indirectly via
inhibition of the immune reaction and upregulation of angiogenic factors in
inflammation cells.
HIF also plays an important role in stem cells, and here especially tumour
stem cells, which are
reported to have elevated HIF levels. The inhibition of the HIF regulation
pathway by the
compounds of the present invention thus also has a therapeutic influence on
tumour stem cells,
which do not have a high proliferation rate and therefore are affected only
inadequately by
cytotoxic substances (cf. Semenza, 2007; Weidemann and Johnson, 2008).
Changes in cell metabolism by HIF are not exclusive to tumours, but also occur
in other hypoxic
pathophysiological processes, whether chronic or transient. HIF inhibitors -
such as the compounds
of the present invention - are therapeutically helpful in those connections in
which, for example,
additional damage arises from adaptation of cells to hypoxic situations, since
damaged cells can
cause further damage if they do not function as intended. One example of this
is the formation of
epileptic foci in partly destroyed tissue following strokes. A similar
situation is found in the case
of cardiovascular diseases if ischaemic processes occur in the heart or in the
brain as a
consequence of thromboembolic events, inflammations, wounds, intoxications or
other causes.
These can lead to damage such as a locally retarded action potential, which in
turn can bring about
arrhythmias or chronic heart failure. In transient form, for example as a
result of apnoea, there may
under certain circumstances be essential hypertension, which can lead to known
sequelae, for
example stroke and cardiac infarction.
Inhibition of the HIF regulation pathway such as is achieved by the compounds
according to the
invention can therefore also be helpful for diseases such as heart failure,
arrhythmia, cardiac
infarction, apnoea-induced hypertension, pulmonary hypertension, transplant
ischaemia,
reperfusion damage, stroke and macular degeneration, as well as for recovery
of nerve function
after traumatic damage or severance.
Since HIF is one of the factors which control the transition from an
epithelial to a mesenchymal
cell type, which is important especially for the lung and kidney, the
inventive compounds can also
be used to prevent or control fibroses of the lung and kidney associated with
HIF.
Further diseases for the treatment of which the compounds according to the
invention can be used
are inflammatory joint diseases, such as various forms of arthritis, and
inflammatory intestinal
diseases, such as, for example, Crohn's disease.
Chuvash polycythaemia is mediated by HIF-2a activity during erythropoiesis, in
the spleen among
other organs. The inventive compounds, as inhibitors of the HIF regulation
pathway, are therefore
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-29-
also suitable here for suppressing excessive erythrocyte formation and hence
for alleviating the
effects of this disease.
The compounds of the present invention can also be used for treatment of
diseases associated with
excessive or abnormal angiogenesis. These include, inter alia, diabetic
retinopathy, ischaemic
retinal vein occlusion and retinopathy in premature babies (cf. Aiello et al.,
1994; Peer et al.,
1995), age-related macular degeneration (AMD; cf. Lopex et al., 1996),
neovascular glaucoma,
psoriasis, retrolental fibroplasia, angiofibroma, inflammation, rheumatic
arthritis (RA), restenosis,
in-stent restenosis and restenosis following vessel implantation.
Increased blood supply is additionally associated with cancerous, neoplastic
tissue and leads here
to accelerated tumour growth. Moreover, the growth of new blood and lymph
vessels facilitates the
formation of metastases and hence the spread of the tumour. New lymph and
blood vessels are also
harmful to allografts in immunoprivileged tissues, such as the eye, which, for
example, increases
susceptibility to rejection reactions. Compounds of the present invention can
therefore also be used
for therapy of one of the aforementioned diseases, for example by inhibition
of the growth of or a
reduction in the number of blood vessels. This can be achieved via inhibition
of endothelial cell
proliferation or other mechanisms for preventing or attenuating the formation
of vessels and via a
reduction of neoplastic cells by apoptosis.
In the case of obesity, HIF-1 a becomes enriched in the adipose tissue,
resulting in a HIF-mediated
shift in the catabolism in the direction of glycolysis, such that an increased
amount of glucose as
an energy carrier is consumed. This leads at the same time to reduced lipid
metabolism and hence
to storage of lipids in the tissue. The inventive substances are therefore
also suitable for treatment
of HIF-la-mediated enrichment of lipids in the tissue, especially in the case
of obesity.
The present invention further provides for the use of the compounds according
to the invention for
the treatment and/or prophylaxis of disorders, in particular the disorders
mentioned above.
The present invention further provides for the use of the compounds according
to the invention for
producing a medicament for treatment and/or prophylaxis of disorders, in
particular the disorders
mentioned above.
The present invention furthermore provides the use of the compounds according
to the invention in
a method for treatment and/or prevention of diseases, in particular the
abovementioned diseases.
The present invention further provides a method for treatment and/or
prophylaxis of disorders, in
particular the disorders mentioned above, using an effective amount of at
least one of the
compounds according to the invention.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-30-
The inventive compounds can be used alone or, if required, in combination with
one or more other
pharmacologically active substances, provided that this combination does not
lead to undesirable
and unacceptable side effects. The present invention furthermore therefore
provides medicaments
containing at least one of the compounds according to the invention and one or
more further active
compounds, in particular for treatment and/or prevention of the abovementioned
diseases.
For example, the compounds of the present invention can be combined with known
antihyperproliferative, cytostatic or cytotoxic substances for treatment of
cancer diseases. The
combination of the compounds according to the invention with other substances
customary for
cancer therapy or also with radiotherapy is therefore indicated in particular,
since hypoxic regions
of a tumour respond only weakly to the conventional therapies mentioned,
whereas the compounds
of the present invention display their activity there in particular.
Suitable active compounds in the combination which may be mentioned by way of
example are:
aldesleukin, alendronic acid, alfaferone, alitretinoin, allopurinol, aloprim,
aloxi, altretamine,
aminoglutethimide, amifostine, amrubicin, amsacrine, anastrozole, anzmet,
aranesp, arglabin,
arsenic trioxide, aromasin, 5-azacytidine, azathioprine, BCG or tice-BCG,
bestatin, betamethasone
acetate, betamethasone sodium phosphate, bexarotene, bleomycin sulphate,
broxuridine,
bortezomib, busulfan, calcitonin, campath, capecitabine, carboplatin, casodex,
cefesone,
celmoleukin, cerubidin, chlorambucil, cisplatin, cladribin, clodronic acid,
cyclophosphamide,
cytarabine, dacarbazine, dactinomycin, daunoxome, decadron, decadron
phosphate, delestrogen,
denileukin diftitox, depomedrol, deslorelin, dexrazoxane, diethylstilbestrol,
diflucan, docetaxel,
doxifluridine, doxorubicin, dronabinol, DW-166HC, eligard, elitek, ellence,
emend, epirubicin,
epoetin-alfa, epogen, eptaplatin, ergamisol, estrace, estradiol, estramustine
sodium phosphate,
ethinylestradiol, ethyol, etidronic acid, etopophos, etoposide, fadrozole,
farstone, filgrastim,
finasteride, fligrastim, floxuridine, fluconazole, fludarabin, 5-
fluorodeoxyuridine monophosphate,
5-fluorouracil (5-FU), fluoxymesterone, flutamide, formestane, fosteabine,
fotemustine,
fulvestrant, gammagard, gemcitabine, gemtuzumab, gleevec, gliadel, goserelin,
granisetron
hydrochloride, histrelin, hycamtin, hydrocortone, erythro-hydroxynonyladenine,
hydroxyurea,
ibritumomab tiuxetan, idarubicin, ifosfamide, interferon-alpha, interferon-
alpha-2, interferon-
alpha-2a, interferon-alpha-2(3, interferon-alpha-nl, interferon-alpha-n3,
interferon-beta, interferon-
gamma-la, interleukin-2, intron A, iressa, irinotecan, kytril, lentinan
sulphate, letrozole,
leucovorin, leuprolide, leuprolide acetate, levamisole, levofolic acid calcium
salt, levothroid,
levoxyl, lomustine, lonidamine, marinol, mechlorethamine, mecobalamin,
medroxyprogesterone
acetate, megestrol acetate, melphalan, menest, 6-mercaptopurine, mesna,
methotrexate, metvix,
miltefosine, minocycline, mitomycin C, mitotane, mitoxantrone, modrenal,
myocet, nedaplatin,
neulasta, neumega, neupogen, nilutamide, nolvadex, NSC-631570, OCT-43,
octreotide,
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-31-
ondansetron hydrochloride, orapred, oxaliplatin, paclitaxel, pediapred,
pegaspargase, pegasys,
pentostatin, picibanil, pilocarpine hydrochloride, pirarubicin, plicamycin,
porfimer sodium,
prednimustine, prednisolone, prednisone, premarin, procarbazine, procrit,
raltitrexed, rebif,
rhenium-186 etidronate, rituximab, roferon-A, romurtide, salagen, sandostatin,
sargramostim,
semustine, sizofiran, sobuzoxane, solu-medrol, streptozocin, strontium-89
chloride, synthroid,
tamoxifen, tamsulosin, tasonermin, tastolactone, taxoter, teceleukin,
temozolomide, teniposide,
testosterone propionate, testred, thioguanine, thiotepa, thyrotropin,
tiludronic acid, topotecan,
toremifen, tositumomab, tastuzumab, teosulfan, tretinoin, trexall,
trimethylmelamine, trimetrexate,
triptorelin acetate, triptorelin pamoate, UFT, uridine, valrubicin,
vesnarinone, vinblastine,
vincristine, vindesine, vinorelbine, virulizin, zinecard, zinostatin-
stimalamer, zofran; ABI-007,
acolbifen, actimmune, affinitak, aminopterin, arzoxifen, asoprisnil,
atamestane, atrasentan, avastin,
CCI-779, CDC-501, celebrex, cetuximab, crisnatol, cyproterone acetate,
decitabine, DN-101,
doxorubicin-MTC, dSLIM, dutasteride, edotecarin, eflornithine, exatecan,
fenretinide, histamine
dihydrochloride, histrelin hydrogel implant, holmium-166 DOTMP, ibandronic
acid, interferon-
gamma, intron-PEG, ixabepilone, keyhole limpet hemocyanine, L-651582,
lanreotide, lasofoxifen,
libra, lonafarnib, miproxifen, minodronate, MS-209, liposomal MTP-PE, MX-6,
nafarelin,
nemorubicin, neovastat, nolatrexed, oblimersen, onko-TCS, osidem, paclitaxel
polyglutamate,
pamidronate disodium, PN-401, QS-21, quazepam, R-1549, raloxifen, ranpirnas,
regorafenib, 13-
cis-retic acid, satraplatin, seocalcitol, Sorafenib, T-138067, tarceva,
taxoprexin, thymosin-alpha-1,
tiazofurin, tipifarnib, tirapazamine, TLK-286, toremifen, transMID-107R,
valspodar, vapreotide,
vatalanib, verteporfin, vinflunin, Z-100, zoledronic acid and combinations of
these.
In a preferred embodiment, the compounds of the present invention can be
combined with
antihyperproliferative agents, which can be, by way of example - without this
list being conclusive:
aminoglutethimide, L-asparaginase, azathioprine, 5-azacytidine, bleomycin,
busulfan,
camptothecin, carboplatin, carmustine, chlorambucil, cisplatin, colaspase,
cyclophosphamide,
cytarabine, dacarbazine, dactinomycin, daunorubicin, diethylstilbestrol, 21,2'-
difluorodeoxycytidine, docetaxel, doxorubicin (adriamycin), epirubicin,
epothilone and its
derivatives, erythro-hydroxynonyladenine, ethinylestradiol, etoposide,
fludarabin phosphate, 5-
fluorodeoxyuridine, 5-fluorodeoxyuridine monophosphate, 5-fluorouracil,
fluoxymesterone,
flutamide, hexamethylmelamine, hydroxyurea, hydroxyprogesterone caproate,
idarubicin,
ifosfamide, interferon, irinotecan, leucovorin, lomustine, mechlorethamine,
medroxyprogesterone
acetate, megestrol acetate, melphalan, 6-mercaptopurine, mesna, methotrexate,
mitomycin C,
mitotane, mitoxantrone, paclitaxel, pentostatin, N-phosphonoacetyl L-aspartate
(PALA),
plicamycin, prednisolone, prednisone, procarbazine, raloxifen, semustine,
streptozocin, tamoxifen,
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-32-
teniposide, testosterone propionate, thioguanine, thiotepa, topotecan,
trimethylmelamine, uridine,
vinblastine, vincristine, vindesine and vinorelbine.
The inventive compounds can also be combined in a very promising manner with
biological
therapeutics, such as antibodies (for example avastin, rituxan, erbitux,
herceptin) and recombinant
proteins, which additively or synergistically enhance the effects of
inhibition of the HIF signal
pathway transmission.
Inhibitors of the HIF regulation pathway, such as the inventive compounds, can
also achieve
positive effects in combination with other therapies directed against
angiogenesis, for example
with avastin, axitinib, recentin, regorafenib, sorafenib or sunitinib.
Combinations with inhibitors of
the proteasome and of mTOR and antihormones and steroidal metabolic enzyme
inhibitors are
particularly suitable because of their favourable profile of side effects.
Generally, the following aims can be pursued with the combination of compounds
of the present
invention with other cytostatically or cytotoxically active agents:
= improved efficacy in slowing the growth of a tumour, in reducing its size or
even in the
complete elimination thereof, compared with treatment with an individual
active ingredient;
= the possibility of using the chemotherapeutics used in a lower dosage than
in the case of
monotherapy;
= the possibility of a more tolerable therapy with fewer side effects compared
with individual
administration;
= the possibility of treatment of a broader spectrum of tumours;
= the achievement of a higher rate of response to the therapy;
= a longer survival time of the patient compared with present-day standard
therapy.
In addition, the inventive compounds can also be used in conjunction with
radiotherapy and/or
surgical intervention.
The present invention further provides medicaments which comprise at least one
compound
according to the invention, typically together with one or more inert,
nontoxic, pharmaceutically
suitable auxiliaries, and for the use thereof for the aforementioned purposes.
The compounds according to the invention may act systemically and/or locally.
For this purpose,
they can be administered in a suitable manner, for example by the oral,
parenteral, pulmonal, nasal,
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-33-
sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctival, otic
route, or as an implant or
stent.
The compounds according to the invention can be administered in administration
forms suitable
for these administration routes.
Suitable administration forms for oral administration are those which work
according to the prior
art, which release the compounds according to the invention rapidly and/or in
a modified manner
and which contain the compounds according to the invention in crystalline
and/or amorphized
and/or dissolved form, for example tablets (uncoated or coated tablets, for
example with gastric
juice-resistant or retarded-dissolution or insoluble coatings which control
the release of the
inventive compound), tablets or films/oblates which disintegrate rapidly in
the oral cavity,
films/lyophilizates or capsules (for example hard or soft gelatin capsules),
sugar-coated tablets,
granules, pellets, powders, emulsions, suspensions, aerosols or solutions.
Parenteral administration can bypass an absorption step (e.g. intravenously,
intraarterially,
intracardially, intraspinally or intralumbally) or include an absorption (e.g.
intramuscularly,
subcutaneously, intracutaneously, percutaneously or intraperitoneally).
Suitable administration
forms for parenteral administration include injection and infusion
formulations in the form of
solutions, suspensions, emulsions, lyophilizates or sterile powders.
For the other administration routes, suitable examples are inhalable
medicament forms (including
powder inhalers, nebulizers), nasal drops, solutions or sprays, tablets,
films/oblates or capsules for
lingual, sublingual or buccal administration, suppositories, ear or eye
preparations, vaginal
capsules, aqueous suspensions (lotions, shaking mixtures), lipophilic
suspensions, ointments,
creams, transdermal therapeutic systems (e.g. patches), milk, pastes, foams,
sprinkling powders,
implants or stents.
Oral and parenteral administration are preferred, especially oral and
intravenous administration.
The compounds according to the invention can be converted to the
administration forms
mentioned. This can be done in a manner known per se, by mixing with inert,
nontoxic,
pharmaceutically suitable excipients. These excipients include carriers (for
example
microcrystalline cellulose, lactose, mannitol), solvents (e.g. liquid
polyethylene glycols),
emulsifiers and dispersing or wetting agents (for example sodium
dodecylsulphate,
polyoxysorbitan oleate), binders (for example polyvinylpyrrolidone), synthetic
and natural
polymers (for example albumin), stabilizers (e.g. antioxidants, for example
ascorbic acid), dyes
(e.g. inorganic pigments, for example iron oxides) and flavour and/or odour
correctants.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-34-
In general, it has been found to be advantageous in the case of parenteral
administration to
administer amounts of about 0.00 1 to 1 mg/kg, preferably about 0.01 to 0.5
mg/kg, of body weight
to achieve effective results. In the case of oral administration, the dosage
is about 0.01 to 100
mg/kg, preferably about 0.01 to 20 mg/kg and most preferably 0.1 to 10 mg/kg
of body weight.
It may nevertheless be necessary where appropriate to deviate from the stated
amounts, specifically
as a function of the body weight, route of administration, individual response
to the active
ingredient, nature of the preparation and time or interval over which
administration takes place.
For instance, in some cases, less than the aforementioned minimum amount may
be sufficient, while
in other cases the upper limit mentioned must be exceeded. In the case of
administration of greater
amounts, it may be advisable to divide them into several individual doses over
the day.
The working examples which follow illustrate the invention. The invention is
not limited to the
examples.
The percentages in the tests and examples which follow are, unless indicated
otherwise,
percentages by weight; parts are parts by weight. Solvent ratios, dilution
ratios and concentration
figures for liquid/liquid solutions are each based on volume.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-35-
A. Examples
Abbreviations and acronyms:
abs. absolute
Ac acetyl
AIBN 2,2'-azobis(isobutyronitrile)
aq. aqueous
Ex. Example
Bu butyl
approx. approximately
CDI 1,1'-carbonyldiimidazole
Cl chemical ionization (in MS)
d doublet (in NMR)
d day(s)
DAST diethylaminosulphur tri fluoride
TLC thin layer chromatography
DCI direct chemical ionization (in MS)
dd doublet of doublets (in NMR)
DMAP 4-N,N-dimethylaminopyridine
DME 1,2-dimethoxyethane
DMF N,N-dimethylformamide
DMSO dimethyl sulphoxide
dt doublet of triplets (in NMR)
EDC N'-(3-dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride
ee enantiomeric excess
El electron impact ionization (in MS)
eq. equivalent(s)
ESI electrospray ionization (in MS)
Et ethyl
GC gas chromatography
h hour(s)
HOBt 1-hydroxy-IH-benzotriazole hydrate
HPLC high-pressure, high-performance liquid chromatography
`Pr isopropyl
LC-MS liquid chromatography-coupled mass spectrometry
LiHMDS lithium hexamethyldisilazide
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-36-
lit. literature (reference)
m multiplet (in NMR)
mCPBA meta-chloroperbenzoic acid
Me methyl
min minute(s)
MPLC medium pressure liquid chromatography (over silica gel; also called
"flash chromatography")
Ms methanesulphonyl (mesyl)
MS mass spectrometry
NBS N-bromosuccinimide
NMP N-methyl-2-pyrrolidinone
NMR nuclear magnetic resonance spectroscopy
Pd/C palladium on activated carbon
PEG polyethylene glycol
Pr propyl
quart quartet (in NMR)
quint quintet (in NMR)
Rf retention index (in TLC)
RT room temperature
Rt retention time (in HPLC)
s singlet (in NMR)
sept septet (in NMR)
t triplet (in NMR)
TBAF tetra-n-butylammonium fluoride
`Bu tert-butyl
Tf trifluoromethylsulphonyl (triflyl)
TFA trifluoroacetic acid
THE tetrahydrofuran
TIPS triisopropylsilyl
UV ultraviolet spectrometry
v/v ratio by volume (of a solution)
tog. together
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-37-
HPLC, LC/MS and GC/MS methods:
Method I (LC/MS):
MS instrument type: Micromass ZQ; apparatus type HPLC: HP 1100 series; UV DAD;
column:
Phenomenex Gemini 3 , 30 mm x 3.00 mm; mobile phase A: 1 1 of water + 0.5 ml
of 50% strength
formic acid, mobile phase B: 1 1 of acetonitrile + 0.5 ml of 50% strength
formic acid; gradient: 0.0
min 90% A --> 2.5 min 30% A -+ 3.0 min 5% A 4.5 min 5% A; flow rate: 0.0 min 1
ml/min
2.5 min/3.0 min/4.5 min 2 ml/min; oven: 50 C; UV detection: 210 nm
Method 2 (LC/MS):
Instrument: Micromass Quattro Micro MS with HPLC Agilent Series 1100; column:
Thermo
Hypersil GOLD 3 , 20 mm x 4 mm; mobile phase A: 1 1 of water + 0.5 ml of 50%
strength formic
acid, mobile phase B: 1 1 of acetonitrile + 0.5 ml of 50% strength formic
acid; gradient: 0.0 min
100% A -> 3.0 min 10% A -* 4.0 min 10% A -* 4.01 min 100% A (flow rate 2.5
ml/min) 5.00
min 100% A; oven: 50 C; flow rate: 2 ml/min; UV detection: 210 nm
Method 3 (LC/MS):
MS instrument type: Micromass ZQ; apparatus type HPLC: Waters Alliance 2795;
column:
Phenomenex Synergi 2.5p MAX-RP 100A Mercury 20 mm x 4 mm; mobile phase A: 1 1
of water
+ 0.5 ml of 50% strength formic acid, mobile phase B: 1 1 of acetonitrile +
0.5 ml of 50% strength
formic acid; gradient: 0.0 min 90% A -> 0.1 min 90% A -> 3.0 min 5% A -3 4.0
min 5% A -
4.01 min 90% A; flow rate: 2 ml/min; oven: 50 C; UV detection: 210 nm
Method 4 (LC/MS):
Instrument: Micromass Quattro Premier with Waters UPLC Acquity; column: Thermo
Hypersil
GOLD 1.9 , 50 mm x 1 mm; mobile phase A: 1 1 of water + 0.5 ml of 50% strength
formic acid,
mobile phase B: 1 I of acetonitrile + 0.5 ml of 50% strength formic acid;
gradient: 0.0 min 90% A
-* 0.1 min 90% A -* 1.5 min 10% A -> 2.2 min 10% A; flow rate: 0.33 ml/min;
oven: 50 C; UV
detection: 210 nm
Method 5 (LC/MS):
Instrument: Micromass Quattro LCZ with HPLC Agilent series 1100; column:
Phenomenex
Synergi 2.5.x MAX-RP 100A Mercury 20 mm x 4 mm; mobile phase A: 1 I of water +
0.5 ml of
50% strength formic acid, mobile phase B: 1 I of acetonitrile + 0.5 ml of 50%
strength formic acid;
gradient: 0.0 min 90% A 0.1 min 90% A --> 3.0 min 5% A -* 4.0 min 5% A -> 4.1
min 90% A;
flow rate: 2 ml/min; oven: 50 C; UV detection: 208-400 nm.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-38-
Method 6 (LC/MS):
Instrument: Waters Acquity SQD UPLC System; column: Waters Acquity UPLC HSS T3
1.8p 50
mm x 1 mm; mobile phase A: 1 I of water + 0.25 ml of 99% strength formic acid,
mobile phase B:
1 1 of acetonitrile + 0.25 ml of 99% formic acid; gradient: 0.0 min 90% A ->
1.2 min 5% A -* 2.0
min 5% A; flow rate: 0.40 ml/min; oven: 50 C; UV detection: 210-400 nm.
Method 7 (LC/MS):
MS instrument type: Waters ZQ; apparatus type HPLC: Agilent 1100 Series; UV
DAD; column:
Thermo Hypersil GOLD 3 , 20 mm x 4 mm; mobile phase A: 1 1 of water + 0.5 ml
of 50%
strength formic acid, mobile phase B: 1 1 of acetonitrile + 0.5 ml of 50%
strength formic acid;
gradient: 0.0 min 100% A 3.0 min 10% A - 4.0 min 10% A 4.1 min 100% A (flow
rate 2.5
ml/min); oven: 55 C; flow rate: 2 ml/min; UV detection: 210 nm
Method 8 (LC/MS):
MS instrument type: Micromass ZQ; apparatus type HPLC: HP 1100 series; UV DAD;
column:
Phenomenex Gemini 3 , 30 mm x 3.00 mm; mobile phase A: 1 1 of water + 0.5 ml
of 50% strength
formic acid, mobile phase B: 1 1 of acetonitrile + 0.5 ml of 50% strength
formic acid; gradient: 0.0
min 90% A -3 2.5 min 30% A -> 3.0 min 5% A -3 4.5 min 5% A; flow rate: 0.0 min
I ml/min -*
2.5 min/3.0 min/4.5 min 2 ml/min; oven: 50 C; UV detection: 210 nm
Method 9 (LC/MS):
Instrument: Micromass GCT, GC 6890; column: Restek RTX-35, 15 m x 200 m x
0.33 m;
constant flow rate with helium: 0.88 ml/min; oven: 70 C; inlet: 250 C;
gradient: 70 C, 30 C/min
- 310 C (maintained for 3 min).
Method 10 (analytical HPLC
Instrument: HP 1100 with DAD detection; column: Kromasil 100 RP-18, 60 mm x
2.1 mm, 3.5
m; mobile phase A: 5 ml of perchloric acid (70% strength) / I of water, mobile
phase B:
acetonitrile; gradient: 0 min 2% B -> 0.5 min 2% B -+ 4.5 min 90% B -> 6.5 min
90% B -* 6.7
min 2% B -* 7.5 min 2% B; flow rate: 0.75 ml/min; column temperature: 30 C; UV
detection: 210
nm
Method 11 (preparative HPLC)
Column: GROM-SIL 120 ODS-4 HE, 10 m, 250 mm x 30 mm; mobile phase and
gradient
programme: acetonitrile/0.1% aq. formic acid 10:90 (0-3 min),
acetonitrile/0.1% aq. formic acid
10:90 -> 95:5 (3-27 min), acetonitrile/0.1% aq. formic acid 95:5 (27-34 min),
acetonitrile/0.1%
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-39-
aq. formic acid 10:90 (34-38 min); flow rate: 50 ml/min, temperature: 22 C; UV
detection: 254
nm
Method 12 (preparative HPLC
Column: Daiso C18 Bio Spring Column, 10 m, 300 mm x 100 mm; mobile phase and
gradient
programme: water/methanol 80:20 (0-6 min), water/methanol 80:20 -> 20:80 (6-60
min),
water/methanol 20:80 (60-95 min), water/methanol 10:90 (95-105 min),
water/methanol 80:20
(105-113 min); flow rate: 250 ml/min, temperature: 25 C; UV detection: 240 nm
Method 13 (preparative HPLC)
Column: Reprosil-Pur C18, 10 m, 250 mm x 30 mm; mobile phase and gradient
programme:
acetonitrile/0.1% aq. formic acid 10:90 (0-3 min), acetonitrile/0.1% aq.
formic acid 10:90 -* 95:5
(3-27 min), acetonitrile/0.1% aq. formic acid 95:5 (27-34 min),
acetonitrile/0.1% aq. formic acid
10:90 (34-38 min); flow rate: 50 ml/min, temperature: 22 C; UV detection: 254
nm
Method 14 (preparative HPLCZ
Column: YMC-ODS-AQ, C18, 10 gm, 30 mm x 250 mm; mobile phase and gradient
programme:
methanol/0.1% aq. TFA 50:50 (0:00-4:25 min) -> 70:30 (4:25-4:50 min) -> 90:10
(4:50-11:50
min) -> 100:0 (11:50-12:00 min) -> 100:0 (12:00-14:50 min); flow rate: 50
ml/min, temperature:
22 C; UV detection: 210 nm
Method 15 (preparative HPLC
Column: YMC-ODS-AQ, C18, 10 m, 30 mm x 250 mm; mobile phase and gradient
programme:
methanol/0.1% aq. TFA 60:40 (0:00-4:25 min) -> 80:20 (4:25-4:50 min) -+ 100:0
(4:50-11:50
min) -> 100:0 (11:50-14:50 min); flow rate: 50 ml/min, temperature: 22 C; UV
detection: 210 nm
Method 16 (preparative HPLC):
Column: Sunfire C18 OBD 5 m, 20 mm x 250 mm; mobile phase: water/methanol/1%
aq. TFA
20:75:5 (0:00-7:00 min); flow rate: 25 ml/min, temperature: 40 C; UV
detection: 210 nm
Method 17 (preparative HPLC):
Column: Reprosil C18, 10 gm, 250 mm x 30 mm; mobile phase and gradient
programme:
acetonitrile/0.1% aq. ammonia 20:80 (0-3 min), acetonitrile/0.1% aq. ammonia
20:80 - 98:2 (3-
min), acetonitrile/0.1% aq. ammonia 98:2 (35-40 min); flow rate: 50 ml/min,
temperature:
22 C; UV detection: 210 nm
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-40-
Method 18 (preparative HPLC
Column: Sunfire C18 OBD 5 pm, 20 mm x 250 mm; mobile phase:
water/acetonitrile/1% aq. TFA
24:70:06 (0:00-14:00 min); flow rate: 25 ml/min, temperature: 40 C; UV
detection: 210 nm
Method 19 (LC/MS):
Instrument: Thermo DFS, Trace GC Ultra; column: Restek RTX-35, 15 m x 200 m x
0.33 pm;
constant flow rate with helium: 1.20 ml/min; oven: 60 C; inlet: 220 C;
gradient: 60 C, 30 C/min
-> 300 C (maintained for 3.33 min).
For all the reactants or reagents for which the preparation is not described
explicitly in the
following, they were obtained commercially from generally accessible sources.
For all the other
reactants or reagents for which the preparation likewise is not described in
the following and
which were not commercially obtainable or were obtained from sources which are
not generally
accessible, reference is made to the published literature in which their
preparation is described.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-41-
Starting materials and intermediates:
Example 1A
N'-Hydroxy-4-(1,1,1-trifluoro-2-methylpropan-2-yl)benzenecarboximidamide
HOB
N
H 2 N F
F
F
H3C CH3
Step 1: 2-(4-Bromophenyl)-1,1,1-trifluoropropan-2-ol
Br
/ OH
H3C F
F F
A suspension of dichloro(dimethyl)titanium in a heptane/dichloromethane
mixture was first
prepared as follows: 100 m] (100 mmol) of a 1 M solution of titanium
tetrachloride in
dichloromethane were cooled to -30 C, 100 ml (100 mmol) of a 1 M solution of
dimethylzinc in
heptane were added dropwise and the mixture was subsequently stirred at -30 C
for 30 min. This
suspension was then cooled to -40 C and a solution of 10 g (39.5 mmol) of 1-(4-
bromophenyl)-
2,2,2-trifluoroethanone in 50 ml of dichloromethane was added. The mixture was
subsequently
stirred at -40 C for 5 min, the temperature was then allowed to come to RT and
the mixture was
stirred at RT for a further 2 h. 50 ml of water were slowly added dropwise,
while cooling with ice,
and the mixture was then diluted with a further 300 ml of water. It was
extracted twice with
dichloromethane, the combined dichloromethane phases were washed once with
water, dried over
anhydrous magnesium sulphate and filtered and the solvent was removed on a
rotary evaporator.
The residue was purified by column chromatography over silica gel (mobile
phase:
cyclohexane/ethyl acetate 85:15). 10.5 g (100% of theory) of the title
compound were obtained
which, according to 'H NMR, still contained residues of solvent.
'H NMR (400 MHz, CDC13, S/ppm): 7.52 (d, 2H), 7.47 (d, 2H), 1.76 (s, 3H).
LC/MS (Method 1, ESlpos): R, = 2.27 min; m/z = 268 [M+H]+;
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-42-
Step 2: 2-(4-Bromophenyl)-1,1,1-trifluoropropan-2-yl methanesulphonate
Br
O-S-CH3
H3C F
F F
3.12 g (78.05 mmol, 60% strength in mineral oil) of sodium hydride were
initially introduced into
45 ml of THE under argon and a solution of 10.5 g (39.03 mmol) of the compound
obtained in
Example IA / step 1 in 20 ml of THE was added dropwise at RT. After the
mixture had been
stirred at RT for 1 h and at 40 C for 30 min, a solution of 8.94 g (78.05
mmol) of
methanesulphonyl chloride in 45 ml of THE was added dropwise and the reaction
mixture was
stirred at 40 C for a further 60 min. 50 ml of water were then slowly added
dropwise to the
mixture and the mixture was diluted with saturated aqueous sodium bicarbonate
solution and
extracted twice with ethyl acetate. The combined ethyl acetate phases were
dried over anhydrous
magnesium sulphate and filtered and the solvent was removed on a rotary
evaporator. The residue
was stirred in hexane and the solid obtained was filtered off and dried in
vacuo. This gave 12.4 g
(92% of theory) of the title compound.
1H NMR (400 MHz, CDC13, 8/ppm): 7.58 (d, 2H), 7.43 (d, 2H), 3.16 (s, 3H), 2.28
(s, 3H).
LC/MS (Method 2, ESlpos): R, = 2.32 min, m/z = 364 [M+NH4]+
Step 3: 1-Bromo-4-(1,1,1-trifluoro-2-methylpropan-2-yl)benzene
Br
F
F
H3C CH3
12.4 g (35.72 mmol) of the compound obtained in Example lA / step 2 were
initially introduced
into 250 ml of dichloromethane and the mixture was cooled to 0 C. 35.7 ml
(71.44 mmol) of a 2
M solution of trimethylaluminium were then slowly added dropwise at 0 C,
while stirring, and the
mixture was then allowed to come to RT and was subsequently stirred at RT for
a further 1.5 h.
120 ml of a saturated aqueous sodium bicarbonate solution were slowly added
dropwise to the
mixture, followed by 40 ml of a saturated aqueous sodium chloride solution.
The mixture was
filtered through kieselguhr and the kieselguhr was washed twice with
dichloromethane. The
combined dichloromethane phases were washed once with saturated aqueous sodium
chloride
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
r= .
-43-
solution and dried over anhydrous magnesium sulphate and the solvent was
removed on a rotary
evaporator. This gave 8.69 g (87% of theory) of the title compound in a purity
of 95%.
'H NMR (400 MHz, CDC13, S/ppm): 7.49 (d, 2H), 7.33 (d, 2H), 1.55 (s, 6H).
LC/MS (Method 3, ESlpos): R, = 2.54 min, no ionization
GC/MS (Method 9, ESIpos): R, = 3.48 min, m/z = 266 [M]+.
Step 4: 4-(I,I,I-Trifluoro-2-methylpropan-2-yl)benzenecarbonitrile
NC
F
F
H3C CH3
3.34 g (12.50 mmol) of the compound obtained in Example IA / step 3 were
initially introduced
into 2.5 ml of degassed DMF under argon, 881 mg (7.50 mmol) of zinc cyanide
and 867 mg (0.75
mmol) of tetrakis(triphenylphosphine)palladium(0) were added and the mixture
was stirred at 80 C
overnight. After cooling to RT, the reaction mixture was diluted with ethyl
acetate and solid
constituents were filtered off. The filtrate was washed twice with 2 N aqueous
ammonia solution
and once with saturated aqueous sodium chloride solution, dried over anhydrous
magnesium
sulphate and freed from the solvent on a rotary evaporator. The residue was
purified by column
chromatography over silica gel (mobile phase: cyclohexane/ethyl acetate
85:15). This gave 2.08 g
(78% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 7.68 (d, 2H), 7.62 (d, 2H), 1.60 (s, 6H).
GC/MS (Method 9, ESIpos): R, = 3.83 min, m/z = 213 [M]+.
Step 5: N'-Hydroxy-4-(1,1,1-trifluoro-2-methylpropan-2-
yl)benzenecarboximidamide
HOB
N
H 2 N F
F
F
H 3 C CH3
A mixture of 2.40 g (11.26 mmol) of the compound from Example IA / step 4,
1.72 g (24.77
mmol) of hydroxylamine hydrochloride and 3.45 ml (24.77 mmol) of triethylamine
in 60 ml of
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-44-
ethanol was stirred under reflux for I h. After cooling to RT, the solvent was
removed on a rotary
evaporator. Ethyl acetate was added to the residue and the solid present was
filtered off. The ethyl
acetate solution was washed successively with water and saturated aqueous
sodium chloride
solution, dried over anhydrous magnesium sulphate and filtered. After removal
of the solvent, the
oil obtained was triturated with petroleum ether. After the resulting solid
had been filtered off with
suction and dried under a high vacuum, 2.65 g (96% of theory) of the title
compound were
obtained.
'H NMR (400 MHz, CDC13, S/ppm): 8.0 (s, broad, 1H), 7.62 (d, 2H), 7.52 (d,
2H), 4.88 (s, broad,
2H), 1.60 (s, 6H).
LC/MS (Method 2, ESlpos): Rt = 1.34 min; m/z = 247 [M+H]+;
Example 2A
4-(2-F luoropropan-2-yl)-N'-hydroxybenzenec arboxi midamide
HOB
N
H2N
F
H3C CH3
Step I. 4-(2-Fluoropropan-2-yl)benzenecarbonitrile
NC \
/ F
H 3 C CH3
At a temperature of 0 C, 1.20 g (7.44 mmol) of diethylaminosulphur trifluoride
(DAST) were
added to a solution of 1.00 g (6.20 mmol) of 4-(2-hydroxypropan-2-yl)benzene
carbonitrile
[obtained from 4-(propan-2-yl)benzene carbonitrile according to J.L. Tucker et
al., Synth. Comm.
2006, 36 (15), 2145-2155] in 20 ml of dichloromethane. The reaction mixture
was stirred at RT for
2 h and then diluted with water and extracted with dichloromethane. The
organic phase was
washed with water, dried over anhydrous magnesium sulphate and filtered. After
removal of the
solvent on a rotary evaporator, the residue was purified by means of MPLC
(silica gel, mobile
phase: cyclohexane/ethyl acetate 95:5). This gave 675 mg (67% of theory) of
the title compound.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-45-
'H NMR (400 MHz, CDCI3i 6/ppm): 7.57 (d, 2H), 7.48 (d, 2H), 1.72 (s, 3H), 1.68
(s, 3H).
LC/MS (Method 2, ESlpos): R, = 2.12 min; m/z = 163 [M+H]+;
Step 4-(2-Fluoropropan-2-yl)-N'-hydroxybenzenecarboximidamide
HO.N
HzN
F
H3C CH3
By the process described under Example IA / step 5, 756 mg (93% of theory) of
the title
compound were obtained from 675 mg (4.14 mmol) of the compound from Example 2A
/ step 1.
'H NMR (400 MHz, CDCI3, S/ppm): 7.62 (d, 2H), 7.41 (d, 2H), 4.89 (s, broad,
2H), 1.72 (s, 3H),
1.68 (s, 3H).
LC/MS (Method 2, ESIpos): R, = 1.04 min; m/z = 197 [M+H]+;
Example 3A
N'-Hydroxy-4-[(trifluoromethyl)sulphonyl]benzenecarboximidamide
HONI N
HzN \ F
F
~S\ F
0 0
By the process described under Example IA /step 5, 4.60 g (19.56 mmol) of 4-
[(trifluoromethyl)sulphonyl]benzenecarbonitrile [W. Su, Tetrahedron. Lett.
1994, 35 (28), 4955-
4958] gave 5.08 g (97% of theory) of the title compound.
'H NMR (400 MHz, DMSO-d6, S/ppm): 10.26 (s, 1H), 8.13 (dd, 4H), 6.12 (s, 2H).
LC/MS (Method 2, ESIpos): R, = 1.57 min; m/z = 269 [M+H]+;
Example 4A
4-(3 -Fluorooxetan-3 -yl)-N'-hydroxybenzenecarboximidamide
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-
46-HO.N
H 2 N
F
0
Step 1: 4-(3-Hydroxyoxetan-3-yl)benzenecarbonitrile
NC COH
0
11 ml (21.8 mmol) of a 2 M solution of isopropylmagnesium chloride in diethyl
ether were added
dropwise to a solution of 5.0 g (21.8 mmol) of 4-iodobenzonitrile in 100 ml of
anhydrous THE at
-40 C under inert conditions. After the mixture had been stirred at the same
temperature for 1.5 h,
it was cooled down to -78 C and was slowly added to a solution, likewise
cooled to -78 C, of 2.95
g (32.7 mmol, 80% in dichloromethane) of 3-oxooxetane [G. Wuitschik et al.,
Angew. Chem. Int.
Ed. Engl. 2006, 45 (46), 7736-7739] in 100 ml of anhydrous THE with the aid of
a cannula. When
the addition had ended, the reaction mixture was stirred first at -78 C for 10
min, then at 0 C for
2 h and finally at RT for 30 min. A few ml of saturated aqueous ammonium
chloride solution were
then added. The solvent was then largely removed on a rotary evaporator. The
residue obtained
was diluted with 200 ml of water and extracted three times with approx. 200 ml
of ethyl acetate
each time. The combined organic extracts were washed successively with water
and saturated
sodium chloride solution. After drying over anhydrous magnesium sulphate, the
mixture was
filtered and the solvent was removed on a rotary evaporator. The crude product
obtained was
purified by crystallization from cyclohexane/ethyl acetate 10:1. This gave
2.42 g (63% of theory)
of the title compound.
'H NMR (400 MHz, DMSO-d6, 6/ppm): 7.88 (d, 2H), 7.80 (d, 2H), 6.63 (s, 1H),
4.79 (d, 2H), 4.65
(d, 2H).
HPLC (Method 10): R, = 3.09 min
MS (DCI, NH3): m/z = 193 [M+NH4]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-47-
Step 2: 4-(3-Fluorooxetan-3-yl)benzenecarbonitrile
NC TIII:1;x; F
O
A solution of 662 mg (4.11 mmol) of diethylaminosulphur trifluoride (DAST) in
5 ml of
dichloromethane was added dropwise to a suspension of 600 mg (3.43 mmol) of
the compound
from Example 4A / step 1 in 55 ml of dichloromethane at -78 C under inert
conditions. After 30
min at -78 C, the reaction mixture was warmed very rapidly to -20 C with the
aid of an ice/water
bath. After approx. 30 seconds, 20 m] of I M sodium hydroxide solution were
added and the
mixture was allowed to warm to RT. After dilution with 150 ml of water, the
mixture was
extracted three times with approx. 50 ml of diethyl ether each time. The
combined organic extracts
were dried over anhydrous magnesium sulphate. After filtration, the solvent
was removed on a
rotary evaporator. The crude product was purified by means of MPLC (silica
gel, mobile phase:
cyclohexane/ethyl acetate 8:1). This gave 495 mg (82% of theory) of the title
compound.
'H NMR (400 MHz, CDC13, S/ppm): 7.76 (d, 2H), 7.73 (d, 2H), 5.15 (dd, 2H),
4.81 (dd, 2H).
LC/MS (Method 2, ESlpos): R, = 1.59 min; m/z = 178 [M+H]+;
Step 3: 4-(3-Fluorooxetan-3-yl)-N'-hydroxybenzenecarboximidamide
HORN
H2N
F
O
By the process described under Example IA / step 5, 470 mg (86% of theory) of
the title
compound were obtained from 450 mg (2.54 mmol) of the compound from Example 4A
/ step 2.
'H NMR (400 MHz, DMSO-d6, S/ppm): 9.71 (s, 1H), 7.77 (d, 2H), 7.54 (d, 2H),
5.87 (broad s,
2H), 4.97 (dd, 2H), 4.91 (dd, 2H).
HPLC (Method 10): R, = 2.64 min
MS (DCI, NH3): m/z = 211 [M+H]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
- 48 -
LC/MS (Method 2, ESlpos): Rt = 0.80 min; m/z = 211 [M+H]+;
Example 5A
4-(4-Fluorotetrahydro-2H-pyran-4-yl)-N'-hydroxybenzenecarboximidamide
HOB
N
H 2 N
F
O
Step 1: 4-(4-Hydroxytetrahydro-2H-pyran-4-yl)benzenecarbonitrile
NC
h C 0H
O
By the process described under Example 4A / step 1, 25.0 g (109 mmol) of 4-
iodobenzonitrile
were reacted with 16.4 g (164 mmol) of tetrahydro-4H-pyran-4-one to give 7.56
g (34% of theory)
of the title compound.
'H NMR (400 MHz, DMSO-d6, S/ppm): 7.80 (d, 2H), 7.70 (d, 2H), 5.30 (s, IH),
3.81-3.70 (m,
4H), 2.02-1.94 (m, 2H), 1.51-1.48 (m, 2H).
HPLC (Method 10): Rt = 3.35 min
MS (DCI, NH3): m/z = 204 [M+H]+, 221 [M+NH4]+-
Step 2: 4-(4-Fluorotetrahydro-2H-pyran-4-yl)benzenecarbonitrile
NC
hFF
0
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-49-
By the process described under Example 4A / step 2, 6.5 g (31.98 mmol) of the
compound from
Example 5A / step I were reacted to give 3.73 g (57% of theory) of the title
compound.
'H NMR (400 MHz, CDC13, S/ppm): 7.68 (d, 2H), 7.50 (d, 2H), 3.98-3.83 (m, 4H),
2.23-2.05 (m,
2H), 1.91-1.85 (m, 2H).
HPLC (Method 10): R, = 4.04 min
MS (DC I, NH3): m/z = 223 [M+NH4]+
Step 3: 4-(4-Fluorotetrahydro-2H-pyran-4-yl)-N'-hydroxybenzenecarboximidamide
HONI N
H2N I \
/ F
O
By the process described under Example IA / step 5, 3.57 g (88% of theory) of
the title compound
were obtained from 3.5 g (17.05 mmol) of the compound from Example 5A / step
2.
'H NMR (500 MHz, DMSO-d6, S/ppm): 9.64 (s, IH), 7.70 (d, 2H), 7.44 (d, 2H),
5.81 (s, 2H), 3.88-
3.83 (m, 2H), 3.73-3.67 (m, 2H), 2.23-2.06 (m, 2H), 1.87-1.81 (m, 2H).
HPLC (Method 10): R, = 3.06 min
MS (DCI, NH3): m/z = 239 [M+H]+.
LC/MS (Method 4, ESIpos): R, = 0.40 min; m/z = 239 [M+H]+;
Example 6A
4-Cyclohexyl-N'-hydroxybenzenecarboximidamide
HO.
N
H2N
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
r = ~.
-50-
Analogously to the process described under Example IA / step 5, 240 mg (1.29
mmol) of 4-
cyclohexylbenzenecarbonitrile [E. Riguet et al., J. Organomet. Chem. 2001, 624
(1-2), 376-379]
gave 252 mg (89% of theory) of the title compound.
'H NMR (400 MHz, DMSO-d6, S/ppm): 9.51 (s, 1H), 7.56 (d, 2H), 7.20 (d, 2H),
5.72 (s, broad,
2H), 2.52-2.48 (m, 1H), 1.81-1.74 (m, 4H), 1.73-1.67 (m, 1H), 1.45-1.31 (m,
4H), 1.28-1.19 (m,
I H).
LC/MS (Method 5, ESIpos): R, = 1.24 min, m/z = 219 [M+H]+.
Example 7A
N'-Hydroxy-4-(pentafluoro-k6-sulphanyl)benzenecarb oximi damide
HO.
N
H2N
F
/ IMF
FIF
F
Analogously to the process described under Example IA / step 5, 5.00 g (21.8
mmol) of 4-
(pentafluoro-X6-sulphanyl)benzenecarbonitrile [P.J. Crowley et al., Chimia
2004, 58 (3), 138-142]
gave 5.50 g (96% of theory) of the title compound.
'H NMR (400 MHz, DMSO-d6, 6/ppm): 9.99 (s, IH), 7.94-7.85 (m, 4H), 6.00 (s,
2H).
LC/MS (Method 2, ESlpos): R, = 1.49 min, m/z = 263 [M+H]+.
Example 8A
N'-Hydroxy-4-[(trifluoromethyl)sulphanyl]benzenecarboximidamide
HORN
H2N F F
S" 'F
Analogously to the process described under Example lA / step 5, 5.20 g (25.6
mmol) of 4-
[(trifluoromethyl)sulphanyl]benzenecarbonitrile gave 5.98 g (97% of theory) of
the title
compound.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-51-
'H NMR (400 MHz, DMSO-d6, S/ppm): 9.90 (s, I H), 7.80 (d, 2H), 7.72 (d, 2H),
5.94 (s, 2H).
LC/MS (Method 2, ESlpos): R, = 1.42 min, m/z = 237 [M+H]+.
Example 9A
N'-Hydroxy-4-(trimethylsilyl)benzenecarboximidamide
HOB
N
H2N I
S'CH3
H 3 C CH3
Analogously to the process described under Example IA / step 5, 2.10 g (11.9
mmol) of 4-
(trimethylsilyl)benzenecarbonitrile [P. di Raddo et al., J. Chem. Soc. Chem.
Commun. 1984, (3),
159-160] gave 2.20 g (91% pure, 80% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 7.95 (s, broad, 1H), 7.60 (d, 2H), 7.55 (d,
2H), 4.86 (s, broad,
2H), 0.27 (s, 9H).
LC/MS (Method 6, ESlpos): R, = 0.75 min, m/z = 209 [M+H]+.
Example 1OA
4-(1-Fluorocyclobutyl)-N'-hydroxybenzenecarboximidamide
HONI N
H2N
j"'~ I F
Step 1: 4-(1-Hydroxycyclobutyl)benzenecarbonitrile
NC
\ 1 OH
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-52-
Analogously to the process described under Example 4A / step 1, 9.47 g (83% of
theory) of the
title compound were obtained from 15.00 g (65.5 mmol) of 4-iodobenzonitrile,
34.4 ml (68.8
mmol) of isopropylmagnesium chloride solution (2 M in diethyl ether) and 7.4
ml (98.2 mmol) of
cyclobutanone. The purification of the product was carried out by means of
MPLC (silica gel,
mobile phase: cyclohexane/ethyl acetate 10:1 -> 4:1).
'H NMR (400 MHz, CDCI3, S/ppm): 7.67 (d, 2H), 7.62 (d, 2H), 2.58-2.51 (m, 2H),
2.44-2.37 (m,
2H), 2.23-2.04 (m, 2H), 1.83-1.72 (m, IH).
HPLC (Method 10): Rt = 3.47 min
MS (DCI, NH3): m/z = 191 [M+NH4]+
Step 2: 4-(1-Fluorocyclobutyl)benzenecarbonitrile
NC
1 F
Analogously to the process described under Example 4A / step 2, 1.39 g (69% of
theory) of the
title compound were obtained from 2.00 g (11.5 mmol) of the compound from
Example 1OA / step
1 and 1.8 ml (13.9 mmol) of diethylaminosulphur trifluoride (DAST). The
purification of the
product was carried out by means of MPLC (silica gel, mobile phase:
cyclohexane/ethyl acetate
10:1 -> 5:1).
'H NMR (400 MHz, CDC13, 6/ppm): 7.69 (d, 2H), 7.57 (d, 2H), 2.78-2.62 (m, 2H),
2.58-2.48 (m,
2H), 2.20-2.09 (m, 1H), 1.87-1.75 (m, 1H).
GC/MS (Method 9, EIpos): Rt = 4.71 min, m/z = 155 [M-HF]+.
Step 3: 4-(1-Fluorocyclobutyl)-N'-hydroxybenzenecarboximidamide
H0.
N
H2N
F
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-53-
Analogously to the process described under Example IA / step 5, 1.16 g of the
title compound
(78% of theory) were obtained starting from 1.25 g (7.13 mmol) of the compound
from Example
IOA / step 2.
'H NMR (400 MHz, CDC13, 6/ppm): 7.67 (d, 2H), 7.50 (d, 2H), 4.87 (s, broad,
2H), 2.72-2.52 (m,
5H), 2.16-2.05 (m, 1H), 1.82-1.71 (m, 1H).
HPLC (Method 10): Rt = 3.17 min
MS (DCI, NH3): m/z = 209 [M+H]+.
Example 11A
N'-Hydroxy-4-(tetrahydro-2H-pyran-4-yl)benzenecarboximidamide
HOB
N
H2N
O
Step 1: 4-(Tetrahydro-2H-pyran-4-yl)benzonitrile
NC
O
186 mg (0.594 mmol) of nickel(II) iodide, 90 mg (0.594 mmol) of trans-2-
aminocyclohexanol
hydrochloride and 3.63 g (19.8 mmol) of sodium hexamethyldisilazide were added
to a solution of
2.91 g (19.8 mmol) of 4-cyanophenylboronic acid [M. Nishimura et al.,
Tetrahedron 2002, 58 (29),
5779-5788] in 20 ml of isopropanol. The suspension obtained in this way was
stirred at RT under
an argon atmosphere for 5 min. 2.1 g (9.90 mmol) of 4-iodotetrahydropyran
[Heuberger et al., J.
Chem. Soc. 1952, 910] were then added. After the reaction mixture had been
stirred at a
temperature of 75 C for 15 h, it was cooled to RT and largely freed from
inorganic salts with
dichloromethane by filtration over approx. 50 g of silica gel. The crude
product was purified by
MPLC (silica gel, mobile phase: dichloromethane). This gave 986 mg (53% of
theory) of the title
compound.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-54-
'H NMR (400 MHz, CDC13, 8/ppm): 7.60 (d, 2H), 7.32 (d, 2H), 4.12-4.07 (m, 2H),
3.56-3.50 (m,
2H), 2.87-2.79 (m, IH), 1.86-1.73 (m, 4H).
GC/MS (Method 9, Elpos): Rt = 5.97 min, m/z = 187 [M]+.
Step 2: N'-Hydroxy-4-(tetrahydro-2H-pyran-4-yl)benzenecarboximidamide
HO.
N
H2N
O
Analogously to the process described under Example IA / step 5, 480 mg (2.56
mmol) of the
compound from Example I IA / step 1 were reacted to give 525 mg (93% of
theory) of the title
compound.
'H NMR (400 MHz, CDC13, 6/ppm): 7.58 (d, 2H), 7.26 (d, 2H), 6.79 (broad, IH),
4.82 (s, broad,
2H), 4.11-4.05 (m, 2H), 3.57-3.50 (m, 2H), 2.83-2.74 (m, IH), 1.87-1.73 (m,
4H).
LC/MS (Method 2, ESIpos): R, = 0.92 min; m/z = 221 [M+H]+;
Example 12A
N'-Hydroxy-4-isobutylbenzenecarboximidamide
HOB
N
H 2 N CH3
CH3
Step 1: 4-Isobutylbenzenenitrile
NC \ CH3
CH3
Under inert, oxygen-free conditions, a mixture of 5.0 g (23.5 mmol) of 1-bromo-
4-
isobutylbenzene, 3.14 g (26.7 mmol) of zinc cyanide, 963 mg (2.35 mmol) of
dicyclohexyl-(2',6'-
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-55-
dimethoxybiphenyl-2-yl)phosphane and 1.08 g (1.17 mmol) of
tris(dibenzylidenacetone)dipalladium in 230 ml of DMF/water (99:1) was heated
at 120 C for lh.
After cooling to RT, the mixture was diluted with approx. 1000 ml of water and
extracted three
times with approx. 150 ml of ethyl acetate each time. The combined organic
extracts were washed
successively with water and saturated sodium chloride solution. After drying
over anhydrous
magnesium sulphate, the mixture was filtered and the filtrate was freed from
the solvent on a
rotary evaporator. The residue obtained was purified by means of filtration
with suction through
silica gel using 10:1 cyclohexane/ethyl acetate as mobile phase. This gave
3.04 g (81% of theory)
of the title compound.
1H NMR (400 MHz, CDC13, S/ppm): 7.56 (d, 2H), 7.23 (d, 2H), 2.53 (d, 2H), 1.94-
1.83 (m, 1H),
0.90 (d, 6H).
GC/MS (Method 9, Elpos): R, = 4.05 min, m/z = 159 [M]+.
Step 2: N'-Hydroxy-4-isobutylbenzenecarboximidamide
HOB
N
HZN I CH3
CH3
Analogously to the process described under Example IA / step 5, 3.03 g (19.0
mmol) of the
compound from Example 12A / step 1 were reacted to give 3.39 g (93% of theory)
of the title
compound.
'H NMR (400 MHz, DMSO-d6, S/ppm): 9.53 (s, IH), 7.57 (d, 2H), 7.14 (d, 2H),
5.74 (broad, 2H),
2.46 (d, 2H), 1.89-1.79 (m, 1H), 0.87 (d, 6H).
LC/MS (Method 6, ESlpos): Rt = 0.68 min, m/z = 193 [M+H]+
Example 13A
N'-Hydroxy-4-isopropylbenzenecarboximidamide
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-56-
HORN
H2N
CH3
CH3
Analogously to the process described under Example IA / step 5, 5.00 g (34.4
mmol) of 4-
isopropylbenzenenitrile gave 4.65 g (94% pure, 71% of theory) of the title
compound.
'H NMR (400 MHz, DMSO-d6, 6/ppm): 9.53 (s, 1H), 7.58 (d, 2H), 7.23 (d, 2H),
5.74 (s, broad,
2H), 2.89 (sept, 1H), 1.20 (d, 6H).
LC/MS (Method 4, ESlpos): Rt = 0.64 min, m/z = 179 [M+H]+.
Example 14A
N'-Hydroxy-4-[I -(methoxymethyl)cyclobutyl]benzenecarboximidamide
HO.N
H2N
eCH3
Step 1: Ethyl 1-(4-bromophenyl)cyclobutanecarboxylate
Br
\ I /\
O CH3
45 ml (45.2 mmol) of a 1 M solution of lithium hexamethyldisilazide in THE
were added to a
solution of 10.0 g (41.1 mmol) of 4-bromophenylacetic acid ethyl ester in 250
ml of anhydrous
THE at 0 C. After 15 min, 5.4 ml (53.5 mmol) of 1,3-dibromopropane were added.
The reaction
mixture was allowed to warm to RT and was subsequently stirred at this
temperature for 1 h. It was
then cooled again to 0 C and a further 45 ml (45.2 mmol) of lithium
hexamethyldisilazide solution
(1 M in THF) were added. Thereafter, the mixture was warmed again to RT. After
1 h, the reaction
was ended by addition of approx. 10 ml of saturated aqueous ammonium chloride
solution. The
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-57-
THE was largely removed on a rotary evaporator. The residue was diluted with
water and extracted
with ethyl acetate. The organic extract was washed successively with water and
saturated sodium
chloride solution. After drying over anhydrous magnesium sulphate, the mixture
was filtered and
the filtrate was freed from the solvent on a rotary evaporator. The crude
product obtained in this
way was coarsely purified by means of filtration with suction over approx. 300
g of silica gel with
cyclohexane/ethyl acetate 3:1 as the mobile phase. 7.1 g (44% of theory,
purity of 73%) of the title
compound were obtained, this being reacted further in this form.
'H NMR (400 MHz, CDC13, S/ppm): 7.44 (d, 2H), 7.17 (d, 2H), 4.10 (quart, 2H),
2.85-2.79 (m,
2H), 2.49-2.41 (m, 2H), 2.10-1.98 (m, IH), 1.91-1.81 (m, 1H), 1.18 (t, 3H).
MS (DCI, NH3): m/z = 300/302 [M+NH4]+
LC/MS (Method 2, ESlpos): Rt = 2.70 min, m/z = 283/285 [M+H]+.
Step 2: [1-(4-Bromophenyl)cyclobutyl]methanol
Br
OH
7.20 g (25.4 mmol) of the compound from Example 14A / step 1 were dissolved in
150 ml of
anhydrous THF, and 25 ml (25 mmol) of a I M solution of lithium aluminium
hydride in THE
were added dropwise at 0 C. When the addition had ended, the ice/water bath
was removed and
stirring was continued at RT. After 1 h, the reaction was ended by - initially
cautious - addition of
approx. 450 ml of saturated aqueous ammonium chloride solution. It was then
extracted with ethyl
acetate. After drying of the organic extract over anhydrous magnesium sulphate
and subsequent
filtration, the solvent was removed on a rotary evaporator. 6.04 g (88% of
theory, 90% pure) of the
title compound were obtained.
'H NMR (400 MHz, CDC13, S/ppm): 7.43 (d, 2H), 7.02 (d, 2H), 3.72 (d, 2H), 2.33-
2.20 (m, 4H),
2.13-2.01 (m, IH), 1.93-1.83 (m, 1H).
MS (DCI, NH3): m/z = 258/260 [M+NH4]+
GC/MS (Method 9, ESlpos): R, = 5.77 min, m/z = 240/242 [M]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-58-
Step 3: 1-Bromo-4-[I-(methoxymethyl)cyclobutyl]benzene
Br
/CH3
,or
1.28 g (31.9 mmol) of a 60% strength suspension of sodium hydride in mineral
oil were added to a
solution of 7.0 g (29.0 mmol) of the compound from Example 14A / step 2 in 120
ml of anhydrous
DMF at approx. 5 C. After the mixture had been stirred at this temperature for
1 h, 2.2 ml
(34.8 mmol) of methyl iodide were added. The reaction mixture was allowed to
warm to RT and
stirring was continued for 15 h. The reaction mixture was then concentrated to
a volume of approx.
20 ml on a rotary evaporator. Approx. 500 ml of water were added and the
mixture was extracted
three times with approx. 200 ml of diethyl ether each time. The combined
organic extracts were
washed with saturated sodium chloride solution and dried over anhydrous
magnesium sulphate.
After filtration and removal of the solvent on a rotary evaporator, the crude
product obtained was
purified by means of filtration with suction over approx. 200 g of silica gel
with cyclohexane/ethyl
acetate 50:1 as the mobile phase. This gave 4.92 g (66% of theory) of the
title compound.
'H NMR (400 MHz, CDC13, 6/ppm): 7.41 (d, 2H), 7.04 (d, 2H), 3.48 (s, 2H), 3.27
(s, 3H), 2.32-
2.22 (m, 4H), 2.12-2.00 (m, 1H), 1.90-1.80 (m, 1H).
MS (DCI, NH3): m/z = 272/274 [M+NH4]+
GC/MS (Method 9, ESlpos): R, = 5.25 min, m/z = 254/256 [M]+.
Step 4: 4-[ 1-(Methoxymethyl)cyclobutyl]benzonitrile
NC
1
CH3
Analogously to the process described under Example 12A / step 1, 1.82 g (48%
of theory) of the
title compound were obtained from 4.80 g (18.8 mmol) of the compound from
Example 14A / step
3.
'H NMR (400 MHz, CDC13, S/ppm): 7.58 (d, 2H), 7.24 (d, 2H), 3.52 (s, 2H), 3.26
(s, 3H), 2.34-
2.24 (m, 4H), 2.16-2.03 (m, 1 H), 1.92-1.83 (m, I H).
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-59-
LC/MS (Method 4, ESlpos): Ry = 1.22 min; m/z = 202 [M+H]+;
Step 5: N-Hydroxy-4-[1-(methoxymethyl)cyclobutyl]benzenecarboximidamide
HORN
O/CH3
H2N Or
Analogously to the process described under Example IA / step 5, 2.04 g (96% of
theory) of the
title compound were obtained from 1.82 g (9.04 mmol) of the compound from
Example 14A / step
4.
'H NMR (400 MHz, CDC13, S/ppm): 7.55 (d, 2H), 7.20 (d, 2H), 7.10 (broad, IH),
4.83 (broad,
2H), 3.51 (s, 2H), 3.27 (s, 3H), 2.36-2.25 (m, 4H), 2.12-2.01 (m, 1H), 1.90-
1.81 (m, 1H).
LC/MS (Method 6, ESIpos): Ry = 0.61 min; m/z = 235 [M+H]+;
Example 15A
N-Hydroxy-4-[ 1-(trifluoromethyl)cyclopropyl]benzenecarboximidamide
HOB
N
H2N F F
F
Step 1: 1-Bromo-4-[I-(trifluoromethyl)cyclopropyl]benzene
Br
F F
F
Activated zinc bromide on montmorillonite was first prepared as follows: 7.0 g
(31.1 mmol) of
zinc bromide were initially charged in a 1 litre flask in 225 ml of methanol,
and 28.2 g of K 10
montmorillonite were added. Subsequently, the suspension was stirred at RT for
I h. Then the
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-60-
mixture was concentrated to dryness on a rotary evaporator. The remaining fine
powder was
heated to bath temperature 200 C in a sand bath under gentle vacuum (approx.
500 mbar) for 1 h
and then allowed to cool under argon.
The title compound was then prepared as follows: 49.63 g (267 mmol) of 1-
phenyl-l-
(trifluoromethyl)cyclopropane were initially charged in 1.25 litres of
pentane, and the activated
zinc bromide on montmorillonite obtained above was added. Then the reaction
vessel was wrapped
with aluminium foil on the outside, in order to reduce the incidence of light.
137 ml (2.67 mol) of
bromine were slowly added dropwise while stirring. Subsequently, the reaction
mixture was stirred
in the dark at RT for 16 h. Then, while cooling with ice, 1 litre of saturated
aqueous sodium
sulphite solution was added dropwise. The solids were filtered off with
suction and washed twice
with pentane. After phase separation, the filtrate was extracted twice more
with 1 litre each time of
pentane. The combined organic extracts were dried over anhydrous sodium
sulphate, filtered and
freed from the solvent on a rotary evaporator under only gentle vacuum. This
gave 77.18 g (92%
pure, 100% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 7.47 (d, 2H), 7.33 (s, 2H), 1.37-1.34 (m, 2H),
1.03-0.98 (m,
2H).
GC/MS (Method 9, ESlpos): Rt = 3.43 min, m/z = 264/266 [M]+.
Step 2: 4-[1-(Trifluoromethyl)cyclopropyl]benzonitrile
NC
F F
F
A solution of 75.0 g (283 mmol) of the compound from Example 15A / Step 1 in a
mixture of 990
ml of DMF and 10 m] of water was freed of oxygen by repeated application of a
gentle vacuum
and venting with argon. Then 37.87 g (322 mmol) of zinc cyanide and 32.69 g
(28.3 mmol) of
tetrakis(triphenylphosphine)palladium(0) were added. The reaction mixture was
subsequently
heated to 120 C for 5 h. After cooling to RT, insolubles were filtered off and
the residue was
washed with a little DMF. The filtrate was subsequently freed from the solvent
on a rotary
evaporator. The resulting crude product was dissolved in 1.5 litres of ethyl
acetate and the mixture
was washed twice with 500 ml each time of saturated ammonium chloride solution
and once with
500 ml of saturated sodium chloride solution. After drying the organic phase
over anhydrous
magnesium sulphate, the mixture was filtered and the filtrate was concentrated
on a rotary
evaporator. The oil obtained was purified by means of suction filtration
through 175 g of silica gel
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-61-
with 40:1 cyclohexane/ethyl acetate as the eluent. This gave, after
concentration of the product
fractions and drying under high vacuum, 49.7 g (83% of theory) of the title
compound.
'H NMR (400 MHz, CDC13i 6/ppm): 7.65 (d, 2H), 7.57 (d, 2H), 1.46-1.42 (m, 2H),
1.09-1.03 (m,
2H).
GC/MS (Method 9, ESlpos): R, = 3.79 min, m/z = 211 [M]+.
&p 3: N-Hydroxy-4-[1-(trifluoromethyl)cyclopropyl]benzenecarboximidamide
HOB
N
H2N F F
F
14.48 g (208 mmol) of hydroxylammonium chloride and 29 ml (208 mmol) of
triethylamine were
added to a solution of 20.0 g (94.7 mmol) of the compound from Example 15A /
Step 2 in 500 ml
of ethanol. The reaction mixture was heated under reflux for 2 h.
Subsequently, about half of the
solvent was removed on a rotary evaporator. 1.5 litres of water were added to
the remaining
mixture, and the resulting suspension was stirred at RT for 20 min. Then the
solid was filtered off
with suction, washed with a little cold water and dried under high vacuum. For
further purification,
it was stirred with a mixture of 120 ml of pentane and 30 ml of
dichloromethane. This gave, after
the solid had again been filtered off with suction and dried, 15.79 g (68% of
theory) of the title
compound.
'H NMR (400 MHz, DMSO-d6, 6/ppm): 9.68 (s, 1H), 7.67 (d, 2H), 7.46 (d, 2H),
5.83 (s, broad,
2H), 1.36-1.32 (m, 2H), 1.15-1.11 (m, 2H).
LC/MS (Method 4, ESlpos): Rt = 0.80 min, m/z = 245 [M+H]+.
Example 16A
3 -Fluoro-N'-hydroxy-4-(trifluoromethoxy)benzenecarbox imidamide
HORN
F
H 2 N I N:ZZ F F
0 XF
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-62-
Analogously to the process described under Example IA / step 5, 5.0 g (23.9
mmol) of 3-fluoro-4-
(trifluoromethoxy)benzonitrile gave 5.7 g (99% of theory) of the title
compound.
'H NMR (400 MHz, CDC13, S/ppm): 7.53-7.49 (dd, 1H), 7.45-7.41 (m, IH), 7.37-
7.31 (t, IH), 4.87
(s, broad, 2H).
LC/MS (Method 6, ESlpos): R, = 0.74 min; m/z = 239 [M+H]+;
Example 17A
N'-Hydroxy-3-methyl-4-(tri fluoromethoxy)benzenecarboximidamide
HORN
CH3
H 2 N F F
LO X F
Step 3-Methyl-4-(trifluoromethoxy)benzonitrile
NC / CH3
O F
Analogously to the process described under Example 12A / step 1, 5.00 g (19.6
mmol) of 3-
methyl-4-(trifluoromethoxy)bromobenzene gave 3.55 g (90% pure, 81% of theory)
of the title
compound.
'H NMR (400 MHz, CDC13, S/ppm): 7.59 (d, IH), 7.53 (dd, 1H), 7.31 (d, 1H),
2.36 (s, 3H).
GC/MS (Method 9, DCI / NH3): R, = 2.58 min, m/z = 201 [M]+.
Step 2: N'-Hydroxy-3-methyl-4-(trifluoromethoxy)benzenecarboximidamide
HORN
CH3
H 2 N F F
0 XF
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-63-
Analogously to the process described under Example IA / step 5, 2.36 g (87%
pure, 70% of
theory) of the title compound were obtained from 2.50 g (12.4 mmol) of the
compound from
Example 17A / step 1.
LC/MS (Method 6, ESlpos): Rt = 0.67 min, m/z = 235 [M+H]+.
Example 18A
4-[l -(Ethoxymethyl)cyclobutyl]-N-hydroxybenzenecarboximidamide
HORN
H 2 N OCH3
Step 1: 1-Bromo-4-[I-(ethoxymethyl)cyclobutyl]benzene
Br
O CH3
Analogously to the process described under Example 14A / step 3, 1.28 g (77%
of theory) of the
title compound were obtained from 1.50 g (6.22 mmol) of the compound from
Example 14A / step
2 and 557 tl (7.46 mmol) of bromoethane.
'H NMR (400 MHz, CDC13, S/ppm): 7.40 (d, 2H), 7.05 (d, 2H), 3.51 (s, 2H), 3.38
(quart, 2H),
2.29-2.24 (m, 4H), 2.11-2.00 (m, I H), 1.89-1.79 (m, 1 H), 1.10 (t, 3H).
GC/MS (Method 9, ESlpos): R, = 5.37 min, m/z = 268/270 [M]+.
Step 2: 4-[ 1-(Ethoxymethyl)cyclobutyl]benzonitrile
NC
0 CH3
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-64-
Analogously to the process described under Example 12A / step 1, 0.64 g (69%
of theory) of the
title compound were obtained from 1.15 g (4.29 mmol) of the compound from
Example 18A / step
1.
'H NMR (400 MHz, CDC13, S/ppm): 7.59 (d, 2H), 7.27 (d, 2H), 3.54 (s, 2H), 3.37
(quart, 2H),
2.31-2.27 (m, 4H), 2.16-2.03 (m, IH), 1.91-1.82 (m, 1H), 1.08 (t, 3H).
LC/MS (Method 6, ESlpos): R, = 1.18 min, m/z = 216 [M+H].
Step 3: 4-[I-(Ethoxymethyl)cyclobutyl]-N'-hydroxybenzenecarboximidamide
HOB
N
H2N / OCH3
Analogously to the process described under Example IA / step 5, 725 mg (90%
pure, 90% of
theory) of the title compound were obtained from 635 mg (2.95 mmol) of the
compound from
Example 18A / step 2.
'H NMR (400 MHz, DMSO-d6, S/ppm): 9.55 (s, 1H), 7.58 (d, 2H), 7.14 (d, 2H),
5.79 (broad, 2H),
3.50 (s, 2H), 3.35 (quart, 2H), 2.23-2.20 (m, 4H), 2.08-1.97 (m, 1H), 1.82-
1.73 (m, 1H), 1.03 (t,
3H).
LC/MS (Method 6, ESlpos): R, = 0.67 min, m/z = 249 [M+H]+.
Example 19A
N'-Hydroxy- 4-[] -(methoxymethyl)cyclopentyl]benzenecarboximidamide
HONI N
H2N /
0 "CH3
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-65-
Step 1: Ethyl 1-(4-bromophenyl)cyclopentanecarboxylate
Br
O
OCH3
Analogously to the process described under Example 14A / step 1, 75.0 g (308
mmol) of ethyl (4-
bromophenyl)acetate and 47.5 ml (401 mmol) of 1,4-dibromobutane gave 100 g
(98% of theory,
about 90% pure) of the title compound which was reacted further in this form.
GC/MS (Method 9, Elpos): R, = 6.22 min, m/z = 296/298 [M]+.
Step 2: [1-(4-Bromophenyl)cyclopentyl]methanol
Br
I
OH
100.0 g (336 mmol) of the compound from Example 19A / step I were dissolved in
1800 ml of
anhydrous THF, and 337 ml (337 mmol) of a 1 M solution of lithium aluminium
hydride in THE
were added dropwise at 0 C. When the addition had ended, the ice/water bath
was removed and
stirring was continued at RT. After 1 h, the reaction was ended by - initially
cautious - addition of
approx. 100 ml of saturated aqueous ammonium chloride solution. Most of the
THE was then
removed on a rotary evaporator. The residue obtained was divided between 1.5
litres each of 2 M
hydrochloric acid and ethyl acetate. After phase separation, the organic
extract was dried over
anhydrous magnesium sulphate. After filtration, the solvent was removed on a
rotary evaporator.
The residue obtained was chromatographed on 1.5 kg of silica gel using
cyclohexane/ethyl acetate
95:5 -> 80:20 as mobile phase. This gave 45.7 g (53% of theory) of the title
compound.
'H NMR (400 MHz, DMSO-d6, 6/ppm): 7.44 (d, 2H), 7.23 (d, 2H), 4.62 (t, IH),
3.33 (d, 2H), 1.97-
1.90 (m, 2H), 1.72-1.56 (m, 6H).
MS (DC I, NH3): m/z = 272/274 [M+NH4]+
GC/MS (Method 9, Elpos): R, = 6.34 min, m/z = 254/256 [M]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-66-
Step 3: 1-Bromo-4-[I-(methoxymethyl)cyclopentyl]benzene
Br
Ia6 /CH3
Analogously to the process described under Example 14A / step 3, 9.52 g (90%
of theory) of the
title compound were obtained from 10.0 g (39.1 mmol) of the compound from
Example 19A / step
2 and 2.9 ml (43.1 mmol) of iodomethane.
'H NMR (400 MHz, CDC13, S/ppm): 7.40 (d, 2H), 7.19 (d, 2H), 3.33 (s, 2H), 3.22
(s, 3H), 2.00-
1.94 (m, 2H), 1.87-1.79 (m, 2H), 1.76-1.66 (m, 4H).
GC/MS (Method 9, Elpos): R, = 5.78 min, m/z = 268/270 [M]+.
Step 4: 4-[ 1-(Methoxymethyl)cyclopentyl]benzonitrile
NC
/CH3
Analogously to the process described under Example 12A / step 1, 2.21 g (55%
of theory) of the
title compound were obtained from 5.00 g (18.6 mmol) of the compound from
Example 19A / step
3.
'H NMR (400 MHz, CDC13, S/ppm): 7.58 (d, 2H), 7.42 (d, 2H), 3.35 (s, 2H), 3.22
(s, 3H), 2.05-
1.99 (m, 2H), 1.88-1.70 (m, 6H).
LC/MS (Method 4, ESIpos): R, = 1.31 min, m/z = 216 [M+H]+.
Step 5. N'-Hydroxy- 4-[1-(methoxymethyl)cyclopentyl]benzenecarboximidamide
HORN
H2N
0 oeCH3
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-67-
Analogously to the process described under Example IA / step 5, 2.32 g (93% of
theory) of the
title compound were obtained from 2.17 g (10.1 mmol) of the compound from
Example 19A / step
4.
'H NMR (400 MHz, DMSO-d6, S/ppm): 9.53 (s, IH), 7.56 (d, 2H), 7.29 (d, 2H),
5.73 (broad, 2H),
3.32 (s, 2H), 3.12 (s, 3H), 1.97-1.90 (m, 2H), 1.82-1.75 (m, 2H), 1.73-1.58
(m, 4H).
LC/MS (Method 6, ESlpos): Rt = 0.69 min, m/z = 249 [M+H]+.
Example 20A
3-Fluoro-N'-hydroxy-4-[ 1-(methoxymethyl)cyclobutyl]benzenecarboximidamide
HOB
N
F
HZN /
O/C'H3
Step 1: Ethyl (4-bromo-2-fluorophenyl)acetate
Br / F O
O C H 3
100 pl of concentrated sulphuric acid were added to a solution of 50.0 g
(0.215 mol) of 4-bromo-2-
fluorophenylacetic acid in 500 ml of ethanol, and the mixture was then heated
at reflux for 16 h.
After cooling to RT, most of the solvent was removed on a rotary evaporator.
The residue obtained
was taken up in 500 ml of ethyl acetate and washed with 250 ml of saturated
aqueous sodium
bicarbonate solution. After drying over anhydrous magnesium sulphate, the
mixture was filtered
and the filtrate was evaporated to dryness. 53.2 g (90% of theory, 95% pure)
of the title compound
were obtained.
GC/MS (Method 9, Elpos): R, = 4.74 min, m/z = 260/262 [M]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-68-
Step 2: Ethyl I -(4-bromo-2-fluorophenyl)cyclobutanecarboxylate
Br F
O
OCH3
Analogously to the process described under Example 14A / step 1, 67.6 g (75%
pure, 82% of
theory) of the title compound, which was reacted further in this form, were
obtained from 53.0 g
(0.202 mol) of the compound from Example 20A / step 1 and 26.8 ml (0.264 mol)
of 1,3-
dibromopropane.
LC/MS (Method 6, ESlpos): Rt = 1.31 min, m/z = 301/303 [M+H]+.
Step 3: [1-(4-Bromo-2-fluorophenyl)cyclobutyl]methanol
Br F
I
OH
Analogously to the process described under Example 14A / step 2, 23.9 g (85%
pure, 50% of
theory) of the title compound were prepared from 67.6 g (0.150 mol) of the
compound from
Example 20A / step 2. Here, in deviation from the procedure described under
Example 14A / step
2, it was necessary during work-up to add, after addition of the saturated
ammonium chloride
solution, as much more 2 M hydrochloric acid until a clear aqueous phase was
obtained.
'H NMR (400 MHz, DMSO-d6, 6/ppm): 7.38 (dd, 1H), 7.32 (dd, 1H), 7.02 (t, 1H),
4.80 (t, IH),
3.57 (d, 2H), 2.24-2.19 (m, 4H), 2.07-1.95 (m, 1H), 1.83-1.74 (m, 1H).
Step 4: 4-Bromo-2-fluoro-l-[1-(methoxymethyl)cyclobutyl]benzene
Br F
1
0 -ICH3
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-69-
Analogously to the process described under Example 14A / step 3, 12.8 g (49.4
mmol) of the
compound from Example 20A / step 3 and 3.7 ml (59.3 mmol) of methyl iodide
were converted
into 12.6 g (93% of theory) of the title compound.
'H NMR (400 MHz, DMSO-d6, S/ppm): 7.39 (dd, 1H), 7.33 (dd, 1H), 7.06 (t, IH),
3.53 (s, 2H),
3.18 (s, 3H), 2.32-2.17 (m, 4H), 2.12-2.01 (m, 1H), 1.85-1.75 (m, IH).
GC/MS (Method 9, Elpos): R, = 5.04 min, m/z = 272/274 [M]+.
Step 5: 3-Fluoro-4-[I-(methoxymethyl)cyclobutyl]benzonitrile
NC F
I O -ICH3
Analogously to the process described under Example 12A / step 1, 7.01 g (63%
of theory) of the
title compound were obtained from 12.60 g (46.1 mmol) of the compound from
Example 20A /
step 4.
'H NMR (400 MHz, DMSO-d6, S/ppm): 7.72 (dd, 1H), 7.62 (dd, 1H), 7.30 (t, IH),
3.59 (s, 2H),
3.18 (s, 3H), 2.36-2.20 (m, 4H), 2.15-2.03 (m, 1H), 1.86-1.77 (m, 1H).
Step 6: 3-Fluoro-N'-hydroxy-4-[ I-
(methoxymethyl)cyclobutyl]benzenecarboximidamide
HORN
F
HZN
OI-ICH3
Analogously to the process described under Example IA / step 5, 2.04 g (90%
pure, 86% of
theory) of the title compound were obtained from 5.00 g (22.8 mmol) of the
compound from
Example 20A / step 5.
'H NMR (400 MHz, DMSO-d6, S/ppm): 9.69 (s, IH), 7.43 (dd, I H), 7.33 (dd, I
H), 7.10 (t, I H),
5.83 (s, broad, 2H), 3.55 (s, 2H), 3.18 (s, 3H), 2.34-2.19 (m, 4H), 2.12-2.01
(m, 1H), 1.84-1.77 (m,
1 H).
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-70-
LC/MS (Method 6, ESIpos): R, = 0.70 min, m/z = 253 [M+H]+.
Example 21A
3 -Fluoro-N'-hydroxy-4-(1,1,1-trifluoro-2-methylpropan-2-
yl)benzenecarboximidamide
HOB
N
F
H2N F F
F
H3C CH3
Step 1: 1-[4-Bromo-2-fluoro-3-(trimethylsilyl)phenyl]-2,2,2-trifluoroethanone
CH3
H3C--Si,CH3
Br F
F F
F
Under argon and at a bath temperature of -20 C, 78 ml (125 mmol) of a 1.6 M
solution of n-
butyllithium in hexane were slowly added dropwise to a solution of 17.60 g
(124 mmol) of 2,2,6,6-
tetramethylpiperidine in 110 ml of THF. After 30 min of stirring at -20 C, the
mixture was cooled
further to a bath temperature of -70 C, and a solution of 28.0 g (113 mmol) of
(2-brom-6-
fluorophenyl)(trimethyl)silane [obtained from I -bromo-3-fluorobenzene and
chloro(trimethyl)silane according to S. Lulinski et al., J. Org. Chem. 2003,
68 (24), 9384-9388] in
30 ml THE was added. After I h of stirring at a bath temperature of -70 C,
17.70 g (124.6 mmol)
of ethyl trifluoroacetate were added dropwise at -70 C. The mixture was then
allowed to warm
slowly to RT and stirred at RT for another hour. Saturated aqueous ammonium
chloride solution
was then added and the mixture was extracted twice with ethyl acetate. The
combined ethyl acetate
phases were washed once with sodium chloride solution, dried over magnesium
sulphate, filtered
and concentrated. This gave 42.0 g (82% pure, 89% of theory) of the title
compound.
GC/MS (Method 9, Elpos): R, = 3.92 min, m/z = 342/344 [M]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
t '.
-71-
Step 2: 1-(4-Bromo-2-fluorophenyl)-2,2,2-trifluoroethanone
Br F
F F
F
O
At RT, 120 ml (120 mmol) of a 1 M solution of tetra-n-butylammonium fluoride
in THE were
added to a solution of 42.0 g (100 mmol, purity 82%) of the compound from
Example 21A / step 1
in 140 ml of THF. After 30 min of stirring at RT, the mixture was diluted with
ethyl acetate and
washed once with water. The aqueous phase was reextracted once with ethyl
acetate. The
combined organic phases were then washed once with saturated sodium chloride
solution, dried
over magnesium sulphate, filtered and concentrated. The residue obtained was
purified by means
of flash chromatography (silica gel, mobile phase: cyclohexane -->
cyclohexane/ethyl acetate 95:5).
Removal of the solvent gave 18.90 g (92% pure, 64% of theory) of the title
compound.
'H NMR (400 MHz, CDC13, 8/ppm): 7.78 (t, 1 H), 7.49 (dd, 1 H), 7.45 (dd, 1 H).
GC/MS (Method 9, Elpos): R, = 2.63 min, m/z = 270/272 [M]+.
Step 3: 2-(4-Bromo-2-fluorophenyl)-1,1,1-trifluoropropan-2-ol
Br F
F F
F
H3C OH
A suspension of dichloro(dimethyl)titanium in a heptane/dichloromethane
mixture was first
prepared as follows: 160 ml (160 mmol) of a 1 M solution of titanium
tetrachloride in
dichloromethane were cooled to -30 C, 160 ml (160 mmol) of a I M solution of
dimethylzinc in
heptane were then added dropwise and the mixture was subsequently stirred at -
30 C for 30 min.
The suspension was then cooled to -40 C, and a solution of 19.4 g (65.9 mmol,
purity 92%) of the
compound from Example 21A / step 2 in 80 ml of dichloromethane was added. The
mixture was
stirred at -40 C for another 5 min, the bath temperature was then allowed to
come to RT and the
mixture was stirred at RT for a further 2 h. 80 ml of water were then slowly
added dropwise, while
cooling with ice, and the mixture was then diluted with a further 250 ml of
water. The mixture was
extracted twice with in each case 250 ml of dichloromethane, the combined
dichloromethane
phases were washed once with 350 ml of water, dried over anhydrous magnesium
sulphate and
filtered and the solvent was removed on a rotary evaporator. This gave 23.7 g
(> 100% of theory)
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-72-
of a residue which comprised the title compound in a purity of 92% according
to 'H NMR and was
reacted further in this form.
'H NMR (400 MHz, CDC13, S/ppm): 7.52 (t, 1 H), 7.34 (dd, I H), 7.29 (dd, I H),
3.06-2.99 (m, 1 H),
1.86 (s, 3H).
LC/MS (Method 6, ESlpos): Rt = 1.08 min, m/z = 331/333 [M-H+HCO2H]-.
GC/MS (Method 19, Elpos): Rt = 3.61 min, m/z = 286/288 [M]+.
Step 4: 2-(4-Bromo-2-fluorophenyl)-1,1,1-trifluoropropan-2-ylmethanesulphonate
Br F
O
O-S-CH 3
I I
H3C FO
F F
At RT, a solution of 23.7 g (75.9 mmol, purity 92%) of the compound from
Example 21A / step 3
in 40 ml of THE was added dropwise to a suspension of 6.08 g of sodium hydride
(60% in mineral
oil, 152 mmol) in 90 ml of THF. After I h of stirring at RT and a further 30
min at 40 C, a solution
of 11.8 ml (151.91 mmol) of methanesulphonyl chloride in 90 ml THE was added
dropwise, and
the mixture was then stirred at 40 C for 1 h. 100 ml of water were then slowly
added dropwise.
The reaction mixture was then diluted with saturated aqueous sodium
bicarbonate solution and
extracted twice with ethyl acetate. The combined organic phases were dried
over magnesium
sulphate, filtered and concentrated. The residue obtained in this manner was
triturated with
pentane. The solid was filtered off, washed once with pentane and air-dried.
This gave 25.6 g (92%
of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 7.42 (t, 1H), 7.37 (dd, 1H), 7.32 (dd, 1H),
3.19 (s, 3H), 2.33
(s, 3H).
LC/MS (Method 4, ESlpos): Rt = 1.34 min, m/z = 382/384 [M+NH4]+.
Step 5: 4-Bromo-2-fluoro-l-(1,1,1-trifluoro-2-methylpropan-2-yl)benzene
Br F
F F
F
H3C CH3
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-73-
At 0 C, 70 ml (140 mmol) of a 2 M solution of trimethylaluminium in heptane
were added slowly
with stirring to a solution of 25.6 g (70.1 mmol) of the compound from Example
21A / step 4 in
480 ml of dichloromethane. The bath temperature was allowed to come to RT and
the mixture was
stirred at RT for another 1 h. 230 ml of a saturated aqueous sodium
bicarbonate solution and 75 ml
of a saturated aqueous sodium chloride solution were then added slowly. The
mixture was filtered
through kieselguhr and the filter residue was washed twice with
dichloromethane. The filtrate was
combined with the wash solution and washed once with saturated aqueous sodium
chloride
solution, dried over magnesium sulphate, filtered and concentrated. This gave
18.77 g (94% of
theory) of the title compound.
'H NMR (400 MHz, CDC13, 8/ppm): 7.32-7.24 (m, 3H), 1.63 (s, 6H).
GC/MS (Method 9, Elpos): R, = 2.99 min, m/z = 283/285 [M]+.
Step 6: 3-Fluoro-4-(1,1,1-trifluoro-2-methylpropan-2-yl)benzonitrile
NC F
F F
F
H3C CH3
Under argon, 2.47 g (21 mmol) of zinc cyanide and 2.43 g (2.10 mmol) of
tetrakis(triphenylphosphine)palladium(0) were added to a solution of 10.0 g
(35.1 mmol) of the
compound from Example 21A / step 5 in 50 ml of DMF. The mixture was stirred at
80 C
overnight. After cooling to RT, the solid present was filtered off and washed
with a little DMF.
The filtrate and the wash solution were combined and concentrated. The residue
was purified by
means of column chromatography (silica gel, mobile phase: cyclohexane/ethyl
acetate 95:5). This
gave 5.40 g (67% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 7.57 (t, IH), 7.45 (dd, 1H), 7.38 (dd, 1H),
1.67 (s, 6H).
GC/MS (Method 9, Elpos): Rt = 3.22 min, m/z = 231 [M]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-74-
Step 7: 3-Fluoro-N'-hydroxy-4-(1,1,1-trifluoro-2-methylpropan-2-
yl)benzenecarboximidamide
HOB
N
F
HzN I F F
\ F
H3C CH3
Analogously to the process described under Example IA / step 5, 5.81 g (94% of
theory) of the
title compound were obtained from 5.40 g (23.36 mmol) of the compound from
Example 2lA /
step 6.
'H NMR (400 MHz, CDC13, 6/ppm): 7.46 (t, 1H), 7.41-7.31 (m, 2H), 4.84 (s,
broad, 2H), 1.65 (s,
6H).
LC/MS (Method 6, ESlpos): Rt = 0.77 min, m/z = 265 [M+H]+.
Example 22A
5-(5-Methyl-1 H-pyrazol-3-yl)-3-[4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazole
O-N
PloIN H
N F
F
H 3 C O F
Method A:
At RT, 23.3 g (0.121 mol) of EDC, 16.4 g (0.121 mol) of HOBt and 26.7 g (0.121
mol) of N'-
hydroxy-4-(trifluoromethoxy)benzenecarboximidamide were added successively to
a solution of
15.3g (0.121 mol) of 5-methyl-lH-pyrazole-3-carboxylic acid in 600 ml of
anhydrous DMF. The
mixture was stirred first at RT for 2 h and then at 140 C for 5 h. After
cooling, the mixture was
diluted with 2 litres of water and extracted three times with I litre of ethyl
acetate each time. The
combined organic extracts were washed successively with water and saturated
sodium chloride
solution. After drying over anhydrous magnesium sulphate, the mixture was
filtered and the
solvent was removed on a rotary evaporator. The crude product obtained was
purified by means of
filtration with suction over a suction filter filled with silica gel (mobile
phase: cyclohexane/ethyl
acetate 5:1 -> 1:1). The product fractions were combined and the solvent was
removed on a rotary
evaporator to such an extent that the product just started to precipitate out.
The precipitation was
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-75-
brought to completion at RT. By filtration and further concentration of the
mother liquor, two
fractions of solid were obtained, which were combined and dried under a high
vacuum. A total of
19.7 g (52% of theory) of the title compound was thus obtained.
Method B:
To a suspension of 20.25 g (125 mmol) of 1,1'-carbonyldiimidazole (CDI) in 75
ml of anhydrous
DMF was added dropwise, at RT within 15 min, a solution of 15.0 g (119 mmol)
of 5-methyl-lH-
pyrazole-3-carboxylic acid in 75 ml of anhydrous DMF. After the mixture had
been stirred at RT
for 1 h 45 min, 26.19 g (119 mmol) of N'-hydroxy-4-
(trifluoromethoxy)benzenecarboximid amide
were added. Subsequently, the reaction mixture was heated to 110 C for 4 h.
After cooling, the
majority of the solvent was removed on a rotary evaporator. 800 ml of water
were added to the
residue, and it was stirred for a few minutes. The undissolved product was
filtered off with suction
and washed with diethyl ether. The ether phase was removed from the filtrate
and the aqueous
phase was extracted once more with diethyl ether. 25 ml of methanol were added
to the combined
ether extracts, and the product which had been filtered off with suction
beforehand was suspended
therein. After stirring for a few minutes, the mixture was filtered off with
suction again. The
residue was dried under high vacuum and gave a first fraction of the title
compound (8.79 g). The
same amount of water was added to the filtrate, and it was extracted by
shaking. After phase
separation, the organic phase was washed with saturated sodium chloride
solution, dried over
anhydrous magnesium sulphate and finally freed from the solvent on a rotary
evaporator. After the
residue had been dried under high vacuum, a second fraction of the title
compound was obtained in
this way (24.43 g). A total of 33.32 g (90% of theory) of the title compound
was thus obtained.
'H NMR (400 MHz, CDC13, 6/ppm): 10.64 (broad, 1H), 8.23 (d, 2H), 7.34 (d, 2H),
6.82 (s, 1H),
2.46 (s, 3H).
HPLC (Method 10): R, = 4.72 min
MS (DC I, NH3): m/z = 311 [M+H]+.
LC/MS (Method 4, ESIpos): R, = 1.28 min; m/z = 311 [M+H]+;
Example 23A
5-(5-Methyl- 1 H-pyrazol-3-yl)-3-[4-(1,1,1-trifluoro-2-methylpropan-2-
yl)phenyl]-1,2,4-oxadiazole
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-76-
O-N
Pi
HN - N F F
H3C F
H3C CH3
Analogously to the process described under Example 22A (Method A), 7.57 g
(60.0 mmol) of 5-
methyl-lH-pyrazole-3-carboxylic acid and 14.8 g (60.0 mmol) of the compound
from Example 1A
gave 14.7 g (73% of theory) of the title compound. Here, after the reaction
had ended, for work-up
the product was precipitated by adding the reaction mixture to ice-water with
stirring, filtered off
with suction, subsequently washed with water and dried under high vacuum.
'H NMR (400 MHz, DMSO-d6, S/ppm): 11.80 (s, broad, 1H), 8.17 (d, 2H), 7.63 (d,
2H), 6.83 (s,
IH), 2.46 (s, 3H), 1.63 (s, 6H).
LC/MS (Method 4, ESlpos): Rt = 1.34 min, m/z = 337 [M+H]+.
Example 24A
3-[4-(2-Fluoropropan-2-yl)phenyl]-5-(5-methyl-1 H-pyrazol-3-yl)-1,2,4-
oxadiazole
O-N PNH N
/ F
H3C
H3C CH3
Analogously to the process described under Example 22A (Method A), 3.15 g
(25.0 mmol) of 5-
methyl-lH-pyrazole-3-carboxylic acid and 4.91 g (25.0 mmol) of the compound
from Example 2A
gave 5.0 g (68% of theory) of the title compound. Here, after the reaction had
ended, for work-up
the product was precipitated by adding the reaction mixture to ice-water with
stirring, filtered off
with suction, subsequently washed with water and dried under high vacuum.
'H NMR (400 MHz, DMSO-d6, 6/ppm): 13.54 (s, broad, IH), 8.08 (d, 2H), 7.62 (d,
2H), 6.81 (s,
1H), 2.33 (s, 3H), 1.72 (s, 3H), 1.68 (s, 3H).
LC/MS (Method 2, ESIpos): Rt = 2.19 min, m/z = 287 [M+H]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-77-
Example 25A
5-(5-Methyl-1 H-pyrazol-3-yl)-3-{4-[(trifluoromethyl)sulphonyl]phenyl }-1,2,4-
oxadiazole
O-N PHN - N F F
H C S" 'F
3 // \\
O O
Analogously to the process described under Example 22A (Method A), 2.37 g
(18.7 mmol) of 5-
methyl-IH-pyrazole-3-carboxylic acid and 5.03 g (18.7 mmol) of the compound
from Example 3A
gave 3.4 g (51 % of theory) of the title compound.
'H NMR (400 MHz, DMSO-d6, 8/ppm): 13.62 (s, broad, 1H), 8.49 (d, 2H), 8.38 (d,
2H), 6.83 (s,
1H), 2.34 (s, 3H).
LC/MS (Method 4, ESlpos): R, = 1.25 min, m/z = 359 [M+H]+.
Example 26A
5 -(5-Methyl-I H-pyrazol-3 -yl)-3 -[4-(trimethyls ilyl)phenyl] -1,2,4-
oxadiazol e
O-N
PPN
HN N
H C Si" CH3
3
H3C CH3
Analogously to the process described under Example 22A (Method A), 2.52 g
(20.0 mmol) of 5-
methyl-lH-pyrazole-3-carboxylic acid and 4.63 g (20.0 mmol, 90% pure) of the
compound from
Example 9A gave 4.67 g (78% of theory) of the title compound. Here, after the
reaction had ended,
for work-up the product was precipitated by adding the reaction mixture to ice-
water with stirring,
filtered off with suction, subsequently washed with water and dried under high
vacuum.
'H NMR (400 MHz, CDC13, 6/ppm): 11.3 (s, broad, I H), 8.12 (d, 2H), 7.63 (d,
2H), 6.81 (s, I H),
2.43 (s, 3H), 0.31 (s, 9H).
LC/MS (Method 3, ESIpos): Rt = 2.39 min, m/z = 299 [M+H]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-78-
Example 27A
5-(5-Methyl-1 H-pyrazol-3-yl)-3-[4-(trifluoromethyl)phenyl]-1,2,4-oxadiazole
O-N
ppN
HN N
I F
H 3 C
F F
Analogously to the process described under Example 22A (Method A), 2.40 g
(19.0 mmol) of 5-
methyl-lH-pyrazole-3-carboxylic acid and 3.88 g (19.0 mmol) of N'-hydroxy-4-
(trifluoromethyl)benzenecarboximidamide gave 3.95 g (71% of theory) of the
title compound.
Here, after the reaction had ended, for work-up the product was precipitated
by adding the reaction
mixture to 500 ml of ice-water with stirring, filtered off with suction,
subsequently washed with
water and dried under high vacuum.
'H NMR (400 MHz, CDC13, 6/ppm): 10.52 (broad, 1H), 8.32 (d, 2H), 7.77 (d, 2H),
6.82 (s, 1H),
2.63 (s, 3H).
LC/MS (Method 6, ESlpos): R, = 1.11 min, m/z = 295 [M+H]+.
Example 28A
5-(5-Methyl-1 H-pyrazol-3-yl)-3-[4-(tetrahydro-2H-pyran-4-yl)phenyl]-1,2,4-
oxadiazole
O-N
PN
HN
H3C
O
Analogously to the process described under Example 22A (Method A), from 469 mg
(3.72 mmol)
of 5-methyl-lH-pyrazole-3-carboxylic acid and 820 mg (3.72 mmol) of the
compound from
Example 11A, 450 mg of the title compound were obtained after extraction of
the crude product by
stirring in acetonitrile, and a further 97 mg of the title compound were
obtained after purification
of the mother liquor by preparative HPLC (Method 11) (yield 47 % of theory in
total).
'H NMR (400 MHz, DMSO-d6, S/ppm): 13.52 (s, 1H), 8.01 (d, 2H), 7.49 (d, 2H),
6.79 (s, 1H),
3.99-3.95 (m, 2H), 3.49-3.42 (m, 2H), 2.92-2.84 (m, 1H), 2.34 (s, 3H), 1.77-
1.65 (m, 4H).
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-79-
LC/MS (Method 6, ESlpos): Rt = 0.98 min, m/z = 311 [M+H]+.
Example 29A
3-[3-Fluoro-4-(trifluoromethoxy)phenyl]-5-(5-methyl-I H-pyrazol-3-yl)-1,2,4-
oxadiazole
O-N
Pi N \ F
HN- I F F
H 3 C / OXF
Analogously to the process described under Example 22A (Method A), 2.0 g (15.9
mmol) of 5-
methyl-IH-pyrazole-3-carboxylic acid and 3.78 g (15.9 mmol) of the compound
from Example
16A gave 3.15 g (92% pure, 56% of theory) of the title compound. In this case
the product was
obtained not by purification by chromatography but by washing the crude
product with water and
pentane and subsequent drying under reduced pressure.
'H NMR (400 MHz, CDC13, S/ppm): 12.0-9.5 (broad, 1H), 8.10-7.97 (m, 2H), 7.46-
7.41 (t, IH),
6.81 (s, 1H), 2.47 (s, 3H).
LC/MS (Method 6, ESlpos): R, = 1.16 min, m/z = 329 [M+H]+.
Example 30A
5-(5-Methyl-1 H-pyrazol-3-yl)-3-{4-[ 1-(trifluoromethyl)cyclopropyl]phenyl }-
1,2,4-oxadiazole
O-N PHN - N F F
H3C F
Method A:
Analogously to the process described under Example 22A (Method A), 1.19 g
(9.42 mmol) of 5-
methyl-IH-pyrazole-3-carboxylic acid and 2.30 g (9.42 mmol) of the compound
from Example
15A gave 1.05 g (62% of theory) of the title compound. Here, the purification
of the crude product
was carried out by means of preparative HPLC (Method 13).
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-80-
Method B:
To a suspension of 6.75 g (41.6 mmol) of 1,1'-carbonyldiimidazole (CDI) in 25
ml of anhydrous
DMF was added dropwise, at RT within 15 min, a solution of 5.0 g (36.6 mmol)
of 5-methyl-lH-
pyrazole-3-carboxylic acid in 25 ml of anhydrous DMF. After the mixture had
been stirred at RT
for 1 h 45 min, 9.68 g (39.6 mmol) of the compound from Example 15A were
added.
Subsequently, the reaction mixture was heated to 110 C for 2.5 h. After
cooling to RT, 800 ml of
water were slowly added with vigorous stirring, causing the precipitation of
the product. The solid
was filtered off with suction and washed with a little cold water. The crude
product, which was
still moist, was recrystallized from a boiling mixture of 300 ml of ethanol
and 350 ml of water.
This gave 11.03 g (83% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 11.04 (s, broad, 1H), 8.16 (d, 2H), 7.60 (d,
2H), 6.82 (s, 1H),
2.45 (s, 3H), 1.43-1.40 (m, 2H), 1.11-1.07 (m, 2H).
LC/MS (Method 6, ESIpos): R, = 1.14 min, m/z = 335 [M+H]+.
Example 31A
3-{3-Fluoro-4-[1-(methoxymethyl)cyclobutyl]phenyl}-5-(5-methyl-IH-pyrazol-3-
yl)-1,2,4-
oxadiazole
O-N PHNC N\ N F
//'++ ~CH3
H3C O
Analogously to the process described under Example 22A (Method A), 2.50 g
(19.8 mmol) of 5-
methyl-IH-pyrazole-3-carboxylic acid and 5.0 g (19.8 mmol) of the compound
from Example 20A
gave 2.07 g (30% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 10.80 (broad, I H), 7.91 (dd, 1 H), 7.80 (dd,
1 H), 7.24 (t, 1 H),
6.80 (s, 1H), 3.67 (s, 2H), 3.29 (s, 3H), 2.45 (s, 3H), 2.48-2.31 (m, 4H),
2.19-2.07 (m, IH), 1.95-
1.86 (m, I H).
LC/MS (Method 4, ESIpos): R, = 1.34 min, m/z = 343 [M+H]+.
Example 32A
3-{4-[ I-(Ethoxymethyl)cyclobutyl]phenyl }-5-(5-methyl-1 H-pyrazol-3-yl)-1,2,4-
oxadiazole
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-81-
O-N
..IN
HN N
H3C OCH3
Analogously to the process described under Example 22A (Method A), 368 mg
(2.92 mmol) of 5-
methyl-1H-pyrazole-3-carboxylic acid and 725 mg (2.92 mmol) of the compound
from Example
18A gave 608 mg (60% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 11.13 (broad, IH), 8.10 (d, 2H), 7.30 (d, 2H),
6.81 (s, IH),
3.58 (s, 2H), 3.39 (quart, 2H), 2.45 (s, 3H), 2.41-2.29 (m, 4H), 2.15-2.04 (m,
1H), 1.92-1.83 (m,
1H), 1.10 (t, 3H).
LC/MS (Method 2, ESlpos): Rt = 2.52 min, m/z = 339 [M+H]+.
Example 33A
3-{4-[I-(Methoxymethyl)cyclopentyl]phenyl}-5-(5-methyl-iH-pyrazol-3-yl)-1,2,4-
oxadiazole
O-N PNH N
H C OlCH3
3
Analogously to the process described under Example 22A (Method A), 1.18 g
(9.34 mmol) of 5-
methyl-IH-pyrazole-3-carboxylic acid and 2.32 g (9.34 mmol) of the compound
from Example
19A gave 1.31 g (41% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 10.82 (broad, 1H), 8.10 (d, 2H), 7.46 (d, 2H),
6.80 (s, 1H),
3.41 (s, 2H), 3.23 (s, 3H), 2.43 (s, 3H), 2.07-2.00 (m, 2H), 1.97-1.89 (m,
2H), 1.78-1.71 (m, 4H).
LC/MS (Method 6, ESlpos): R, = 1.19 min, m/z = 339 [M+H]+.
Example 34A
5-(5-Methyl-I H-pyrazol-3-yl)-3-[3-methyl-4-(trifluoromethoxy)phenyl]-1,2,4-
oxadiazole
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-82-
O-N
CH3
pN \ /
HN I F F
H 3 C O " _ F
Analogously to the process described under Example 22A (Method A), 1.50 g
(11.9 mmol) of 5-
methyl-lH-pyrazole-3-carboxylic acid and 2.79 g (11.9 mmol) of the compound
from Example
17A gave 1.81 g (94% pure, 44% of theory) of the title compound.
'H NMR (400 MHz, DMSO-d6, S/ppm): 13.56 (broad, IH), 8.12 (s, I H), 8.02 (d, I
H), 7.54 (d,
I H), 6.81 (s, I H), 2.39 (s, 3H), 2.35 (s, 3H).
LC/MS (Method 6, ESlpos): R, = 1.17 min, m/z = 325 [M+H]+.
Example 35A
3-[4-(3-Fluorooxetan-3-yl)phenyl]-5-(5-methyl-1 H-pyrazol-3-yl)-1,2,4-
oxadiazole
O-N PNH N \
I
F
H3C
O
Analogously to the process described under Example 22A (Method A), 300 mg
(2.38 mmol) of 5-
methyl-IH-pyrazole-3-carboxylic acid and 500 mg (2.38 mmol) of the compound
from Example
4A gave 204 mg (28% of theory) of the title compound. In this case, the crude
product was
purified not by chromatography but by crystallization from ethanol.
'H NMR (400 MHz, CDC13, 6/ppm): 8.27 (d, 2H), 7.72 (d, 2H), 6.81 (s, 1H), 5.05
(dd, 2H), 5.01
(dd, 2H), 2.47 (s, 3H).
LC/MS (Method 2, ESIpos): R, = 1.93 min, m/z = 301 [M+H]+.
Example 36A
5-(5-Methyl-1 H-pyrazol-3-yl)-3-{4-[(trifluoromethyl)sulphanyl]phenyl }-1,2,4-
oxadiazole
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-83-
O-N
HpN
N I F F
H3C / S" _F
Analogously to the process described under Example 22A (Method A), 600 mg
(4.76 mmol) of 5-
methyl-IH-pyrazole-3-carboxylic acid and 1.12 g (4.76 mmol) of the compound
from Example 8A
gave 485 mg (31% of theory) of the title compound. In this case, the crude
product was purified
not by chromatography but by crystallization from ethanol.
'H NMR (400 MHz, CDC13, 8/ppm): 10.60 (broad, 1H), 8.23 (d, 2H), 7.79 (d, 2H),
6.81 (s, IH),
2.47 (s, 3H).
LC/MS (Method 6, ESlpos): R, = 1.17 min, m/z = 327 [M+H]+.
Example 37A
3-(4-Cyclohexylphenyl)-5-(5-methyl-1H-pyrazol-3-yl)-1,2,4-oxadiazole
O-N
PPN
HN ~ N I ~
H Analogously to the process described under Example 22A (Method A), 6.66 g
(52.8 mmol) of 5-
methyl-lH-pyrazole-3-carboxylic acid and 11.54 g (52.8 mmol) of the compound
from Example
6A gave 9.02 g (55% of theory) of the title compound. In this case, the crude
product was purified
not by chromatography but by crystallization from ethanol.
'H NMR (400 MHz, CDC13, 8/ppm): 10.91 (s, broad, IH), 8.09 (d, 2H), 7.33 (d,
2H), 6.81 (s, 1H),
2.61-2.52 (m, IH), 2.45 (s, 3H), 1.95-1.82 (m, 4H), 1.80-1.73 (m, 1H), 1.52-
1.34 (m, 4H), 1.33-
1.21 (m, 1 H).
LC/MS (Method 6, ESlpos): R, = 1.31 min, m/z = 309 [M+H]+.
Example 38A
5-(5-Methyl-I H-pyrazol-3-yl)-3-[4-(pentafluoro-),6-sulphanyl)phenyl]-1,2,4-
oxadiazole
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-84-
O-N
PPN
HN N
F
/ IMF
H 3 C FSNF
F
Analogously to the process described under Example 22A (Method A), 1.58 g
(12.5 mmol) of 5-
methyl-IH-pyrazole-3-carboxylic acid and 3.28 g (12.5 mmol) of the compound
from Example 7A
gave 2.87 g (65% of theory) of the title compound. Here, after the reaction
had ended, for work-up
the product was precipitated by adding the reaction mixture to ice-water with
stirring, filtered off
with suction, subsequently washed with water and dried under high vacuum.
'H NMR (400 MHz, CDC13, S/ppm): 11.0 (s, broad, 1H), 8.29 (d, 2H), 7.89 (d,
2H), 6.82 (s, IH),
2.46 (s, 3H).
LC/MS (Method 6, ESlpos): R, = 1.13 min, m/z = 353 [M+H]+.
Example 39A
3-(4-Isobutylphenyl)-5-(5-methyl-I H-pyrazol-3-yl)-1,2,4-oxadiazole
O-N PHN - N I CH3
H3C CH3
3.19 g (16.7 mmol) of EDC, 2.55 g (16.7 mmol) of HOBt and 3.35 g (17.4 mmol)
of the compound
from Example 12A were added successively to a solution of 2.0 g (15.9 mmol) of
5-methyl-lH-
pyrazole-3-carboxylic acid in 80 ml of anhydrous DMF. The mixture was stirred
at RT for 1 h and
then heated at 140 C for 30 min. After cooling to RT, most of the solvent was
removed on a rotary
evaporator. About 500 ml of water were added to the residue and the mixture
was extracted three
times with about 200 ml of diethyl ether each time. The combined organic
extracts were washed
successively with water and saturated sodium chloride solution. After drying
over anhydrous
magnesium sulphate, the mixture was filtered and the filtrate was evaporated
on a rotary
evaporator. The residue obtained was purified by means of MPLC (silica gel,
mobile phase:
cyclohexane/ethyl acetate 2:1). Final trituration with about 50 ml of pentane
gave 1.7 g (38% of
theory) of the title compound.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-85-
'H NMR (400 MHz, CDC13, S/ppm): 10.84 (broad, IH), 8.08 (d, 2H), 7.27 (d, 2H),
6.81 (s, 1H),
2.54 (d, 2H), 2.44 (s, 3H), 1.97-1.87 (m, IH), 0.93 (d, 6H).
LC/MS (Method 7, ESlpos): Rt = 2.59 min, m/z = 283 [M+H]+
Example 40A
3-{4-[1-(Methoxymethyl)cyclobutyl]phenyl}-5-(5-methyl-IH-pyrazol-3-yl)-l,2,4-
oxadiazole
O-N
N
HN N
CH
3
H3C O
Analogously to the process described under Example 39A, 1.08 g (8.52 mmol) of
5-methyl-lH-
pyrazole-3-carboxylic acid and 2.0 g (8.52 mmol) of the compound from Example
14A gave 1.87 g
(46% of theory) of the title compound. For the MPLC purification of the crude
product, a mobile
phase gradient of cyclohexane/ethyl acetate (5:1 -> 1:1) was used.
'H NMR (400 MHz, CDC13, S/ppm): 11.57 (broad, 1H), 8.10 (d, 2H), 7.30 (d, 2H),
6.81 (s, IH),
3.57 (s, 2H), 3.29 (s, 3H), 2.45 (s, 3H), 2.41-2.28 (m, 4H), 2.15-2.03 (m,
1H), 1.93-1.84 (m, 1H).
LC/MS (Method 4, ESlpos): Rt = 1.28 min, m/z = 325 [M+H]+.
Example 41A
3-(4-Isopropylphenyl)-5-(5-methyl-I H-pyrazol-3-yl)-1,2,4-oxadiazole
O-N
PPN
HN N
113C CH 3
CH3
Analogously to the process described under Example 39A, 2.0 g (15.9 mmol) of 5-
methyl-lH-
pyrazole-3-carboxylic acid and 3.11 g (17.4 mmol) of the compound from Example
13A gave 2.20
g (52% of theory) of the title compound.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-86-
'H NMR (400 MHz, CDC13, 6/ppm): 8.10 (d, 2H), 7.36 (d, 2H), 6.81 (s, 1H), 2.97
(sept, 1H), 2.43
(s, 3H), 1.29 (d, 6H).
LC/MS (Method 7, ESlpos): Rt = 2.42 min, m/z = 269 [M+H]+.
Example 42A
3-(4-tert-Butylphenyl)-5-(5-methyl-IH-pyrazol-3-yl)-1,2,4-oxadiazole
O-N
~N
HN N
I CH3
H3C
INI~
H3C CH3
Analogously to the process described under Example 39A, 2.50 g (19.8 mmol) of
5-methyl-lH-
pyrazole-3-carboxylic acid and 4.19 g (21.8 mmol) of 4-tert-butyl-N'-
hydroxybenzenecarboximidamide were converted into 2.60 g (46% of theory) of
the title
compound.
'H NMR (400 MHz, CDC13, S/ppm): 11.08 (s, broad, 1H), 8.10 (d, 2H), 7.51 (d,
2H), 6.81 (s, IH),
2.46 (s, 3H), 1.37 (s, 9H).
LC/MS (Method 6, ESlpos): Rt = 1.21 min, m/z = 283 [M+H]+.
Example 43A
3-[4-(4-Fluorotetrahydro-2H-pyran-4-yl)phenyl]-5-(5-methyl-lH-pyrazol-3-yl)-
1,2,4-oxadiazole
O-N
PN H
N
/ F
H3C
O
At RT, a solution of 4.30 g (34.1 mmol) of 5-methyl-lH-pyrazole-3-carboxylic
acid in 20 ml of
anhydrous DMF was added dropwise over a period of about 15 min to a suspension
of 5.81 g
(35.8 mmol) of 1,1'-carbonyldiimidazole in 25 ml of anhydrous DMF. After the
mixture had been
stirred at RT for 105 min, 8.12 g (34.1 mmol) of the compound from Example 5A
were added. The
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-87-
reaction mixture was then heated at 110 C for 5 h. After cooling to RT, the
mixture was slowly
stirred into 800 ml of water. The precipitated product was filtered off with
suction and washed
with water. The moist crude product was then recrystallized from 430 ml of
ethanol. The crystal
fraction gave, after filtration and drying, 8.31 g (74% of theory) of the
title compound. A further
fraction was obtained by concentrating the mother liquor (1.69 g, 85% pure,
13% of theory).
1H NMR (400 MHz, CDC13, S/ppm): 10.73 (broad, 1H), 8.20 (d, 2H), 7.52 (d, 2H),
6.81 (s, 1H),
4.00-3.88 (m, 4H), 2.45 (s, 3H), 2.30-2.11 (m, 2H), 1.98-1.91 (m, 2H).
LC/MS (Method 8, ESlpos): R, = 4.24 min, m/z = 329 [M+H]+.
Example 44A
3-[4-(1-Fluorocyclobutyl)phenyl]-5-(5-methyl-1H-pyrazol-3-yl)-1,2,4-oxadiazole
O-N
HN N I \
/ F
H3C
Analogously to the process described under Example 43A, 2.50 g (19.8 mmol) of
5-methyl-lH-
pyrazole-3-carboxylic acid and 4.13 g (19.8 mmol) of the compound from Example
l0A gave 4.41
g (75% of theory) of the title compound. The reaction mixture was added to
water with stirring and
then extracted three times with in each case about 200 ml of ethyl acetate.
The combined organic
extracts were washed with saturated sodium chloride solution and dried over
anhydrous
magnesium sulphate. After filtration and evaporation of the solvent, the crude
product was purified
by crystallization from ethanol.
'H NMR (400 MHz, CDC13, S/ppm): 11.36 (broad, 1H), 8.20 (d, 2H), 7.60 (d, 2H),
6.82 (s, 1H),
2.77-2.54 (m, 4H), 2.47 (s, 3H), 2.19-2.08 (m, 1H), 1.87-1.77 (m, 1H).
LC/MS (Method 6, ESlpos): R, = 1.07 min, m/z = 299 [M+H]+.
Example 45A
3-[3-Fluoro-4-(1,1,1-trifluoro-2-methylpropan-2-yl)phenyl]-5-(5-methyl-1 H-
pyrazol-3-yl)-1,2,4-
oxadiazole
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-88-
O-N
pN , \ F
HN / I F F
H3C F
H3C CH3
Analogously to the process described under Example 39A, 2.71 g (21.5 mmol) of
5-methyl-lH-
pyrazole-3-carboxylic acid and 5.68 g (21.5 mmol) of the compound from Example
21A gave 4.36
g (95% pure, 54% of theory) of the title compound.
'H NMR (400 MHz, CDC13, 6/ppm): 7.94 (d, 1H), 7.88 (d, 1H), 7.57 (t, 1H), 6.81
(s, 1H), 2.45 (s,
3H), 1.69 (s, 6H).
LC/MS (Method 6, ESIpos): Rt = 1.16 min, m/z = 355 [M+H]+.
Example 46A
3-(2-Hydroxypropan-2-yl)benzyl methanesulphonate
H C C H 3 O O
HO O1-1 SNI C H 3
Step 1: 2-[3-(Hydroxymethyl)phenyl]propan-2-ol
H 3 C CH3
HO I \ OH
1.5 ml (3.47 mmol) of a 2.4 M solution of lithium aluminium hydride in THE
were added slowly to
a suspension of 500 mg (2.78 mmol) of 3-(2-hydroxypropan-2-yl)benzoic acid in
10 ml of THF.
The mixture was then heated to a bath temperature of 80 C for 2 h. After
cooling to RT, 50 ml of 1
N hydrochloric acid were added and the mixture was extracted three times with
30 ml of tert-butyl
methyl ether each time. The combined organic phases were dried over sodium
sulphate, filtered
and concentrated. After the residue had been dried in vacuo, 455 mg (97% pure,
99% of theory) of
the title compound were obtained.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-89-
'H NMR (400 MHz, CDC13, 6/ppm): 7.51 (s, 1H), 7.42 (d, 1H), 7.34 (t, 1H), 7.25
(d, 1H), 4.71 (s,
2H), 1.59 (s, 6H).
LC/MS (Method 6, ESlpos): R, = 0.57 min, m/z = 149 [M+H-H20]+.
Step 2: 3-(2-Hydroxypropan-2-yl)benzy] methanesulphonate
H3C CH3 O O
HO O/ CH3
Under argon, 1.1 ml (7.88 mmol) of triethylamine at RT, followed by 1.01 g
(5.78 mmol) of
methanesulphonic anhydride at 0 C, were added to a solution of 873 mg (5.25
mmol) of the
compound from Example 46A / step 1 in 50 ml of dichloromethane. After 1 h of
stirring at RT, the
mixture was washed successively with 100 ml of aqueous ammonium chloride
solution and 100 ml
of sodium chloride solution. The organic phase was dried over magnesium
sulphate, filtered and
concentrated. Drying of the residue under reduced pressure gave 1.13 g (88% of
theory) of the title
compound.
'H NMR (400 MHz, CDC13, S/ppm): 7.56 (s, 1H), 7.51 (d, 1H), 7.39 (t, 1H), 7.32
(d, 1H), 5.25 (s,
2H), 2.94 (s, 3H), 1.59 (s, 6H).
LC/MS (Method 6, ESIpos): R, = 0.74 min, m/z = 227 [M+H-H2O]+.
Example 47A
1-(3-{[(Methylsulphonyl)oxy]methyl}phenyl)cyclopropyl acetate
O O
H3C O O/ CH3
Step 1: 1-[3-({[tert-Butyl(dimethyl)silyl]oxy}methyl)phenyl]cyclopropanol
H3C\ ,CH3
HO O/Si)( CH3
1 H3C CH3
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-90-
Preparation of solution A: 60 ml of methanol and a drop of concentrated
hydrochloric acid were
added to 12.32 g (70.7 mmol) of [(1-ethoxycyclopropyl)oxy](trimethyl)silane,
and the mixture was
stirred at RT overnight. The solvent was then removed at RT and under a
reduced pressure of not
less than 30 mbar on a rotary evaporator. This gave 6.26 g (61.27 mmol) of 1-
ethoxycyclopropanol, which were dissolved in 80 ml of THF. Under argon, this
solution was then
cooled to -70 C, and 30.6 ml (61.27 mmol) of a 2 M solution of ethylmagnesium
chloride in THF
were added. The cooling bath was then removed, and the solution was stirred
without cooling until
an internal temperature of 0 C had been reached.
Preparation of solution B: Under argon and at -40 C, 47.1 ml (61.27 mmol) of a
1.3 M solution of
isopropylmagnesium chloride/lithium chloride complex in THF were added to a
solution of 19.40
g (55.70 mmol) of tert-butyl[(3-iodobenzyl)oxy]dimethylsilane in 280 m] of
THF, and the mixture
was stirred at -40 C for 1 h.
Once the two solutions had been prepared, solution A was added to solution B
at 0 C. The reaction
mixture was then heated under reflux for 1 h. After cooling to RT, saturated
aqueous ammonium
chloride solution was added, and the mixture was extracted twice with tert-
butyl methyl ether. The
combined organic phases were washed once with saturated sodium chloride
solution, dried over
magnesium sulphate, filtered and concentrated. The residue was purified by
means of flash
chromatography (silica gel, mobile phase: cyclohexane - cyclohexane/ethyl
acetate 85:15).
Removal of the solvent gave 9.55 g (60% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 7.32-7.24 (m, 2H), 7.22-7.16 (m, 2H), 4.74 (s,
2H), 2.36 (s,
1 H), 1.26 (dd, 2H), 1.06 (dd, 2H), 0.94 (s, 9H), 0.11 (s, 6H).
MS (DCI, NH3): m/z = 296 [M+NH4]+
Step 2: 1-[3-({ [tert-Butyl(dimethyl)silyl]oxy}methyl)phenyl]cyclopropyl
acetate
0 H3C\ CH3
H3C O oolsl C H 1 H3C CH3
At RT, 21.4 ml (42.87 mmol) of a 2 M solution of ethylmagnesium chloride in
THF, directly
followed by 3.0 ml (42.87 mmol) of acetyl chloride, were added to a solution
of 9.55 g (34.3
mmol) of the compound from Example 47A / step 1 in 100 ml of THF. After 5 min
of stirring at
RT, saturated aqueous ammonium chloride solution was added, and the mixture
was extracted
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-91-
twice with ethyl acetate. The combined organic phases were dried over
magnesium sulphate,
filtered and concentrated. This gave 11.25 g (94% pure, 96% of theory) of the
title compound.
'H NMR (400 MHz, CDC13, 6/ppm): 7.30-7.24 (m, 2H), 7.20-7.13 (m, 2H), 4.72 (s,
2H), 2.04 (s,
3H), 1.31-1.25 (m, 2H), 1.24-1.18 (m, 2H), 0.94 (s, 9H), 0.09 (s, 6H).
MS (DCI, NH3): m/z = 338 [M+NH4]+-
Step 3: 1-[3-(Hydroxymethyl)phenyl]cyclopropyl acetate
O
H 3 C'J~ O OH
At RT, 65.6 ml (65.6 mmol) of a I M solution of tetra-n-butylammonium fluoride
in THE were
added to a solution of 11.25 g (32.82 mmol, purity 94%) of the compound from
Example 47A /
step 2. The mixture was stirred at RT for 30 min and then diluted with ethyl
acetate and washed
once with water. The aqueous phase was reextracted once with ethyl acetate.
The combined
organic phases were washed once with saturated sodium chloride solution, dried
over magnesium
sulphate and concentrated. This gave 8.0 g (80% pure, 95% of theory) of the
title compound.
'H NMR (400 MHz, CDC13, S/ppm): 7.24-7.12 (m, 5H), 4.58 (s, 2H), 1.95 (s, 3H),
1.22-1.17 (m,
2H), 1.16-1.10 (m, 2H).
Step 4: 1-(3-{[(Methylsulphonyl)oxy]methyl}phenyl)cyclopropyl acetate
O O
\l
H3C O I O/ CH3
At 0 C, 2.8 ml (37.24 mmol) of methanesulphonyl chloride were added dropwise
to a solution of
8.0 g (31.03 mmol, purity 80%) of the compound from Example 47A / step 3 and
5.6 ml (40.34
mmol) of triethylamine in 90 ml THF. The mixture was then slowly warmed to RT,
stirred at RT
for another 10 min and then diluted with ethyl acetate. The mixture was washed
once with water,
and the aqueous phase was reextracted once with ethyl acetate. The combined
organic phases were
washed once with saturated sodium chloride solution, dried over magnesium
sulphate and
concentrated. The residue obtained was purified by means of flash
chromatography (silica gel,
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-92-
mobile phase: cyclohexane/ethyl acetate 95:5 -4 70:30). Removal of the solvent
and drying of the
residue under reduced pressure gave 8.45 g (95% pure, 91% of theory) of the
title compound.
'H NMR (400 MHz, CDC13, S/ppm): 7.39-7.27 (m, 4H), 5.22 (s, 2H), 2.90 (s, 3H),
2.06 (s, 3H),
1.35-1.28 (m, 2H), 1.27-1.20 (m, 2H).
LC/MS (Method 4, ESIpos): R, = 1.02 min, m/z = 285 [M+H]+.
Example 48A
3-(2-Hydroxy-2-methylpropyl)benzyl methanesulphonate
00
HO O,,SIII CH3
H3C CH3
Std 1-(3-Bromophenyl)-2-methylpropan-2-ol
HO Br
H 3 C CH3
At 0 C, 55 ml (164 mmol) of a 3 M solution of methylmagnesium chloride in THE
were added
dropwise to a solution of 15.0 g (65.5 mmol) of methyl (3-bromophenyl)acetate
in 600 m] of
anhydrous THE After the addition had ended, stirring was continued at 0 C for
1 h. The ice/water
bath was then removed, and stirring was continued at RT overnight. About 1.2
litres of saturated
aqueous ammonium chloride solution were then added, and the mixture was
extracted three times
with about 200 ml of ethyl acetate each time. The combined organic extracts
were washed with
saturated sodium chloride solution, dried over anhydrous magnesium sulphate,
filtered and finally
freed from the solvent under reduced pressure. The residue obtained was
purified by filtration with
suction through silica gel using the mobile phase cyclohexane/ethyl acetate
10:1 -> 1:1. This gave
8.04 g (53% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 7.41-7.37 (m, 2H), 7.20-7.13 (m, 2H), 2.73 (s,
2H), 1.32 (s,
1 H), 1.23 (s, 6H).
GC/MS (Method 9, Elpos): R, = 4.56 min, m/z = 210/212 [M-H20]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-93-
Step 2. 3-(2-Hydroxy-2-methylpropyl)benzaldehyde
0
HO
H 3 C CH3
At -78 C, 13.7 ml (21.8 mmol) of an n-butyllithium solution (1.6 M in hexane)
were added
dropwise to a solution of 2.50 g (10.9 mmol) of the compound from Example 48A
/ step 1 in 100
ml of anhydrous THF. After the addition had ended, the mixture was stirred at -
78 C for another
30 min, and then, also at -78 C, 2.6 ml (32.8 mmol) of anhydrous N,N-
dimethylformamide were
added. The cooling bath was then removed, and stirring was continued at RT
overnight. About 100
ml of saturated aqueous ammonium chloride solution were then added, and the
mixture was
extracted three times with about 100 ml of ethyl acetate each time. The
combined organic extracts
were washed successively with water and saturated sodium chloride solution,
dried over anhydrous
magnesium sulphate, filtered and finally freed from the solvent under reduced
pressure. The crude
product obtained in this way was purified by means of MPLC (silica gel, mobile
phase:
cyclohexane/ethyl acetate 2:1). This gave 1.15 g (59% of theory) of the title
compound.
'H NMR (400 MHz, CDC13, S/ppm): 10.01 (s, 1H), 7.79-7.74 (m, 2H), 7.53-7.47
(m, 2H), 2.86 (s,
2H), 1.25 (s, 6H).
GC/MS (Method 9, Elpos): R, = 4.76 min, m/z = 160 [M-H201+-
Step 3: 1-[3-(Hydroxymethyl)phenyl]-2-methylpropan-2-ol
HO Y*~ OH
H3C CH3
At 0 C, 6.0 ml (6.00 mmol) of a lithium aluminium hydride solution (1.0 M in
THF) were added
dropwise to a solution of 1.07 g (6.00 mmol) of the compound from Example 48A
/ step 2 in 30 ml
of anhydrous THF. After the addition had ended, stirring was continued at RT
for 1 h. Carefully,
1-2 ml of saturated aqueous ammonium chloride solution and then about 30 ml of
ethyl acetate
were then added. Anhydrous magnesium sulphate was added in the amount required
to take up the
aqueous phase completely. After filtration, the filtrate was freed from the
solvent on a rotary
evaporator and the residue was dried under high vacuum. This gave 1.09 g (100%
of theory) of the
title compound.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-94-
'H NMR (400 MHz, CDC13, S/ppm): 7.31 (t, 1H), 7.25 (dd, 1H), 7.22 (dd, 1H),
7.14 (dd, 1H), 4.69
(s, broad, 2H), 2.78 (s, 2H), 1.79 (broad, IH), 1.41 (s, broad, 1H), 1.23 (s,
6H).
GC/MS (Method 9, Elpos): Rt = 5.00 min, m/z = 162 [M-H201-'-
Step 4: 3-(2-Hydroxy-2-methylpropyl)benzyl methanesulphonate
00
HO O,,SI-I CH3
H3C CH3
At 0 C, 1.12 g (6.41 mmol) of methanesulphonic anhydride were added to a
solution of 1.05 g
(5.83 mmol) of the compound from Example 48A / step 3 and 1.2 ml (8.74 mmol)
of triethylamine
in 60 ml of anhydrous dichloromethane. The mixture was then stirred at RT for
another I h. The
reaction mixture was then transferred into a separating funnel and quickly
washed successively
with semi-saturated aqueous ammonium chloride solution and water. After drying
over anhydrous
magnesium sulphate, the mixture was filtered and the filtrate was freed from
the solvent on a
rotary evaporator. This gave 1.5 g (99% of theory) of the title compound.
MS (DCI, NH3): m/z = 276 [M+NH4]+
Example 49A
Ethyl 2-[3-(bromomethyl)phenyl]-2-methylpropanoate
H 3 C CH3
H3CO
Br
I
Step 1: Ethyl 2-methyl-2-(3-methylphenyl)propanoate
H3C CH3
H3CO CH3
O I
10.0 g (56.1 mmol) of ethyl m-tolylacetate and 15.9 g (112 mmol) of methyl
iodide were initially
charged in 300 ml of abs. THE and cooled to about -70 C. 6.93 g (61.7 mmol) of
potassium tert-
butoxide were added, and the mixture was stirred without further cooling for 1
h. The mixture was
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-95-
then cooled again to about -70 C, another 6.93 g (61.7 mmol) of potassium tert-
butoxide were
added, and the mixture was stirred without cooling for a further hour. The
mixture was then
diluted with ethyl acetate, washed with water and saturated aqueous sodium
chloride solution,
dried over anhydrous magnesium sulphate and filtered. The filtrate was freed
from the solvent on a
rotary evaporator. This gave 9.7 g (76% of theory) of the title compound.
GC/MS (Method 9, Elpos): R, = 4.23 min, m/z = 206 [M]+.
Step 2: Ethyl 2-[3-(bromomethyl)phenyl]-2-methylpropanoate
H3C CH3
H3C~0
Br
(
3.70 g (17.9 mmol) of the compound from Example 49A / step 1 were initially
charged in 55 ml of
acetonitrile, 3.19 g (17.9 mmol) of N-bromosuccinimide and 29 mg (0.18 mmol)
of 2,2'-azobis-2-
methylpropanenitrile (AIBN) were added and the mixture was heated under reflux
overnight. The
mixture was then freed from the solvent on a rotary evaporator, the residue
was suspended in
pentane and the solid was separated off by filtration. The filtrate was freed
from the solvent on a
rotary evaporator. The residue obtained was a yellow oil which still contained
starting material but
was reacted directly without further purification.
Yield: 4.64 g (64% of theory, purity according to GC/MS 71%)
GC/MS (Method 9, Elpos): R, = 5.71 min, m/z = 284/286 [M]+.
Example 50A
3-(1-{[(Triisopropylsilyl)oxy]methyl}cyclopropyl)benzyl methanesulphonate
H3C` CH3
H3C IY 00
>--Si-0
O~S~CH
3
HC
3 H C CH
3 3
/
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-96-
Step 1: Methyl 1-(3-bromophenyl)cyclopropanecarboxylate
.1O Br --~7 H3C
O /
At 0 C, 48 ml (48.0 mmol) of a 1 M solution of lithium hexamethyldisilazide
(LiHMDS) in THF
were added to a solution of 10.0 g (43.6 mmol) of methyl (3-
bromophenyl)acetate in 250 ml of
anhydrous THF. After 15 min at 0 C, 4.9 ml (56.7 mmol) of 1,2-dibromoethane
were added. The
ice/water bath was removed, and the mixture was stirred at RT for another 1 h.
The mixture was
then cooled once more to 0 C, and a further 48 ml (48.0 mmol) of the LiHMDS
solution were
added. After the addition had ended, stirring was continued at RT for 63 h.
About 250 ml of
saturated aqueous ammonium chloride solution were then added, and the reaction
mixture was
extracted three times with about 200 ml of ethyl acetate each time. The
combined organic extracts
were washed successively with water and saturated sodium chloride solution,
dried over anhydrous
magnesium sulphate, filtered and finally freed from the solvent under reduced
pressure. The
residue obtained was purified by means of filtration with suction through
silica gel using 20:1
cyclohexane/ethyl acetate as mobile phase. This gave 6.24 g (56% of theory) of
the title
compound.
'H NMR (400 MHz, CDC13, 6/ppm): 7.50 (m, 1H), 7.39 (m, 1H), 7.27 (m, 1H), 7.19
(m, 1H), 3.63
(s, 3H), 1.62-1.60 (m, 2H), 1.20-1.17 (m, 2H).
GC/MS (Method 9, Elpos): Rt = 5.27 min, m/z = 254/256 [M]+.
Step 2: [1-(3-Bromophenyl)cyclopropyl]methanol
HO Br
At -78 C, 13.7 ml (13.7 mmol) of a 1 M solution of lithium aluminium hydride
in THF were added
to a solution of 3.50 g (13.7 mmol) of the compound from Example 50A / step I
in 70 ml of
anhydrous THF. After 1 h of stirring, about 3 ml of saturated aqueous ammonium
chloride solution
were added and the reaction mixture was allowed to warm to RT. The mixture was
then diluted
with about 80 ml of ethyl acetate, and anhydrous magnesium sulphate was then
added in such an
amount that the aqueous phase was taken up completely. After filtration, the
filtrate was
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-97-
concentrated and the residue was purified by means of MPLC (silica gel; mobile
phase:
cyclohexane --> cyclohexane/ethyl acetate 5:1). This gave 1.37 g (44% of
theory) of the title
compound.
'H NMR (400 MHz, CDC13, S/ppm): 7.52 (s, I H), 7.36 (d, I H), 7.29 (d, I H),
7.18 (t, IH), 3.66 (d,
2H), 1.44 (t, 1H), 0.91-0.84 (m, 4H).
GC/MS (Method 9, Elpos): R, = 5.26 min, m/z = 226/228 [M]+.
Step 3: {[1-(3-Bromophenyl)cyclopropyl]methoxy}(triisopropyl)silane
H
H C3C` /CH3
3 >- Si-0 Br
H3C
H3C CH3 /
At about -50 C, 1.55 ml (6.19 mmol) of triisopropylsilyl triflate were added
to a solution of 1.34 g
(5.90 mmol) of the compound from Example 50A / step 2 and 948 mg (8.85 mmol)
of 2,6-lutidine
in 25 ml of anhydrous dichloromethane. After 30 min, the cooling bath was
removed, and stirring
was continued at RT for 1 h. Approx. 50 m] of water were then added and the
mixture was
extracted three times with about 50 ml of ethyl acetate each time. The
combined organic extracts
were washed with saturated sodium chloride solution, dried over anhydrous
magnesium sulphate,
filtered and finally freed from the solvent under reduced pressure. The
residue obtained was
purified by means of MPLC (silica gel, mobile phase: cyclohexane/ethyl acetate
5:1). This gave
1.93 g (85% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 7.52 (s, 1H), 7.31 (d, 1H), 7.27 (d, 1H), 7.13
(t, 1H), 3.74 (s,
2H), 1.02 (m, 3H), 0.99 (d, 18H), 0.91-0.89 (m, 2H), 0.78-0.75 (m, 2H).
GC/MS (Method 9, Elpos): R, = 6.87 min, m/z = 339/341 [M-'Pr]+.
Step 4: 3-( 1-{ [(Triisopropylsilyl)oxy]methyl } cyclopropyl)benzaldehyde
H H C3CY CH3
3 XSi-O
-
H
H3C
H3C CH3
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-98-
Analogously to the process described under Example 48A / step 2, 1.92 g (5.01
mmol) of the
compound from Example 50A / step 3 and corresponding amounts of n-butyllithium
and N,N-
dimethylformamide gave 1.48 g (88% of theory) of the title compound after MPLC
purification
(silica gel, mobile phase: cyclohexane/ethyl acetate 10:1).
1H NMR (400 MHz, CDC13, S/ppm): 10.00 (s, IH), 7.89 (s, IH), 7.72 (d, I H),
7.65 (d, I H), 7.43 (t,
1H), 3.79 (s, 2H), 1.01 (sept, 3H), 0.98 (d, 18H), 0.96-0.94 (m, 2H), 0.83-
0.81 (m, 2H).
GC/MS (Method 9, Elpos): R, = 7.00 min, m/z = 289 [M-'Pr]+.
Step 5: [3-(1-{[(Triisopropylsilyl)oxy]methyl} cyclopropyl)phenyl]methanol
H H 3CCH3
3 C
Si-O
HC --~7 OH
H3C '1~ CH3 I /
Analogously to the process described under Example 50A / step 2, 1.40 g (4.21
mmol) of the
compound from Example 50A / step 4 and the corresponding amount of lithium
aluminium hydride
gave 1.10 g (78% of theory) of the title compound. Here, chromatographic
purification of the
product was dispensed with.
'H NMR (400 MHz, CDC13, S/ppm): 7.38 (s, 1H), 7.31-7.25 (m, 2H), 7.20 (d, 1H),
4.67 (d, 2H),
3.79 (s, 2H), 1.60 (t, 1H), 1.02 (sept, 3H), 1.00 (d, 18H), 0.93-0.90 (m, 2H),
0.77-0.75 (m, 2H).
GC/MS (Method 9, Elpos): R, = 7.18 min, m/z = 291 [M-'Pr]+.
Step 6: 3-(1-{[(Triisopropylsilyl)oxy]methyl}cyclopropyl)benzyl
methanesulphonate
H H 3C CH3
3C O
Si_O O-S-CH 3
H 3 C
I II
H3C CH3 O
Analogously to the process described under Example 48A / step 4, 820 mg (2.45
mmol) of the
compound from Example 50A / step 5 and corresponding amounts of triethylamine
and
methanesulphonic anhydride gave 1.01 g (100% of theory) of the title compound.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-99-
'H NMR (400 MHz, CDC13, 6/ppm): 7.42 (s, 1H), 7.40 (d, 1H), 7.32 (t, IH), 7.25
(d, 1H), 5.21 (s,
2H), 3.77 (s, 2H), 2.91 (s, 3H), 1.02 (sept, 3H), 0.98 (d, 18H), 0.93-0.91 (m,
2H), 0.79-0.76 (m,
2H).
LC/MS (Method 6, ESlpos): R, = 1.59 min, m/z = 413 [M+H]+.
MS (DCI, NH3): m/z = 430 [M+NH4]+
Example 51A
3-(1-{Dideutero[(triisopropylsilyl)oxy]methyl}cyclopropyl)benzyl
methanesulphonate
H3C CH3
H3C OO
~Si-O
H3C -j N~ O CH3
H 3C)-, CH 3 D D
Step 1: [1-(3-Bromophenyl)cyclopropyl]-1,1-dideuteromethanol
HO 7 Br
D D
Analogously to the process described under Example 50A / step 2, 3.50 g (13.7
mmol) of the
compound from Example 50A / step 1 were reacted with lithium aluminium
deuteride (instead of
lithium aluminium hydride) to give 1.39 g (44% of theory) of the title
compound.
'H NMR (400 MHz, CDC13, S/ppm): 7.51 (s, IH), 7.36 (d, 1H), 7.29 (d, 1H), 7.18
(t, 1H), 1.43 (s,
I H), 0.90-0.86 (m, 4H).
GC/MS (Method 9, Elpos): R, = 5.26 min, m/z = 228/230 [M]+.
Step 2: ({[1-(3-Bromophenyl)cyclopropyl]-1,1-
dideuteromethyl}oxy)(triisopropyl)silane
H
H C3C\ /CH3
3 XSi-O Br
H3C
H3C CH3 D D
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-100-
Analogously to the process described under Example 50A / step 3, 1.38 g (6.02
mmol) of the
compound from Example 51A / step 1 were reacted with triisopropylsilyl
triflate to give 2.03 g
(88% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 7.52 (s, 1 H), 7.31 (d, 1 H), 7.27 (d, 1 H),
7.13 (t, 1 H), 1.03 (m,
3H), 1.00 (d, 18H), 0.91-0.88 (m, 2H), 0.78-0.75 (m, 2H).
GC/MS (Method 9, Elpos): R, = 6.86 min, m/z = 341/343 [M-'Pr]+.
Step 3: 3-(I-([(Triisopropylsilyl)oxyldideuteromethyllcyclopropyl)benzaldehyde
H 3 C O
H 3CX)Si" /CH3
-O H
HC
H3C )", CH3 D D
Analogously to the process described under Example 48A / step 2, 2.01 g (5.23
mmol) of the
compound from Example 51A / step 2 and corresponding amounts of n-butyllithium
and N,N-
dimethylformamide gave 1.75 g (100% of theory) of the title compound.
'HNMR (400 MHz, CDC13, S/ppm): 10.00 (s, IH), 7.89 (s, IH), 7.72 (d, 1H), 7.64
(d, 1H), 7.43 (t,
1H), 1.01 (sept, 3H), 0.98 (d, 18H), 0.97-0.93 (m, 2H), 0.83-0.81 (m, 2H).
GC/MS (Method 9, Elpos): R, = 7.00 min, m/z = 291 [M-'Pr]+.
Step 4: [3 -(1 -{ [(Triisopropylsilyl)oxy]dideuteromethyl
}cyclopropyl)phenyl]methanol
H H 3C` CH3
3C
X)~"Si-O
H3C OH
H 3CCH 3 D D
Analogously to the process described under Example 50A / step 2, 1.74 g (5.21
mmol) of the
compound from Example 51A / step 3 and the corresponding amount of lithium
aluminium hydride
gave 1.45 g (83% of theory) of the title compound after MPLC purification
(silica gel, mobile
phase: cyclohexane/ethyl acetate 10:1).
'H NMR (400 MHz, CDC13, S/ppm): 7.37 (s, 1H), 7.30-7.25 (m, 2H), 7.20 (d, 1H),
4.67 (d, 2H),
1.60 (t, 1H), 1.02 (sept, 3H), 1.00 (d, 18H), 0.93-0.90 (m, 2H), 0.77-0.75 (m,
2H).
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-101-
GC/MS (Method 9, Elpos): Rt = 7.18 min, m/z = 293 [M-'Pr]+.
Step 5: 3-(1-{[(Triisopropylsilyl)oxy]dideuteromethyl}cyclopropyl)benzyl
methanesulphonate
H 3C\ CH3
H 3C IY O
Si-O O-S-CH 3
HH CH D D I
3C 3
Analogously to the process described under Example 48A / step 4, 1.18 g (3.51
mmol) of the
compound from Example 51A / step 4 and corresponding amounts of triethylamine
and
methanesulphonic anhydride gave 1.46 g (100% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 7.42 (s, 1H), 7.40 (d, 1H), 7.32 (t, 1H), 7.25
(d, 1H), 5.21 (s,
2H), 2.91 (s, 3H), 1.02 (sept, 3H), 0.98 (d, 18H), 0.93-0.90 (m, 2H), 0.79-
0.76 (m, 2H).
LC/MS (Method 6, ESlpos): Rt = 1.59 min, m/z = 415 [M+H]+.
MS (DCI, NH3): m/z = 432 [M+NH4]+
Example 52A
2-[3-(Bromomethyl)phenyl]-2,2-difluoroethanol
F F
HO Br
Step 1: Ethyl difluoro(3-methylphenyl)acetate
F F
H3CNl/O CH3 )fy '*--~" O I /
At RT and under argon, 25.0 g (123 mmol) of ethyl bromo(difluoro)acetate and
41.0 g (225 mmol)
of copper bronze (90/10 copper/tin alloy) were added to a solution of 23.35 g
(107 mmol) of 3-
iodotoluene in 110 ml of DMSO. The reaction mixture was then stirred at 50 C
for 16 h. After
cooling to RT, the mixture was introduced into 200 ml of 1 N hydrochloric
acid, and 100 ml of
ethyl acetate were added. Any solids present were filtered off, and the
filtrate was extracted twice
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-102-
with in each case 50 ml of I N hydrochloric acid and 50 ml of ethyl acetate.
The combined organic
phases were washed in each case once with 200 ml of water and 200 ml of
saturated sodium
chloride solution, dried over sodium sulphate, filtered and concentrated. The
residue was purified
by means of column chromatography (silica gel, mobile phase: isohexane/ethyl
acetate 98:2 -->
90:10). Removal of the solvent gave 21.41 g (54% of theory) of the title
compound.
'H NMR (400 MHz, CDC13, 6/ppm): 7.43-7.38 (m, 2H), 7.37-7.21 (m, 2H), 4.30
(quart, 2H), 2.40
(s, 3H), 1.31 (t, 3H).
GC/MS (Method 9, Elpos): R, = 3.72 min, m/z = 214 [M]+.
Step 2: 2,2-Difluoro-2-(3-methylphenyl)ethanol
F F
HO CH3
At RT and under argon, 1.51 g (40 mmol) of sodium borohydride were added
slowly to a solution
of 8.57 g (40.0 mmol) of the compound from Example 52A / step 1 in 70 ml of
ethanol. After 30
min of stirring at RT, 300 ml of tert-butyl methyl ether and 300 ml of 1 N
hydrochloric acid were
added slowly to the reaction mixture. The aqueous phase was then reextracted
once with 200 ml of
tert-butyl methyl ether. The combined organic phases were dried over sodium
sulphate, filtered
and concentrated at RT on a rotary evaporator using a pressure just
sufficiently reduced. This gave
7.17 g (> 100% of theory) of a residue comprising the target compound and also
residual solvent.
'H NMR (400 MHz, CDC13, 6/ppm): 7.35-7.24 (m, 3H), 3.96 (t, 2H), 2.40 (s, 3H).
GC/MS (Method 9, Elpos): R, = 3.32 min, m/z = 172 [M]+.
Step 3: 2-[3-(Bromomethyl)phenyl]-2,2-difluoroethanol
F F
HOJ Br
At RT, 7.47 g (42.0 mmol) of N-bromosuccinimide and 328 mg (2.00 mmol) of 2,2'-
azobis-2-
methylpropanenitrile (AIBN) were added to a solution of 6.88 g (approx. 40
mmol) of the
compound from Example 52A / step 2 in 150 ml of acetonitrile. The mixture was
heated at a bath
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-103-
temperature of 80 C for 6 h. After cooling to RT, the solvent was removed and
the residue was
triturated with a mixture of 100 ml of pentane and 50 ml of ethyl acetate. The
solid was filtered off
with suction and washed twice with 15 ml of the 2:1 mixture of pentane and
ethyl acetate. The
filtrate and the wash solutions were combined, washed in each case once with
200 m] of saturated
aqueous sodium sulphite solution and 200 ml of saturated aqueous sodium
chloride solution, dried
over sodium sulphate, filtered and concentrated. This gave 9.72 g (70% pure,
68% of theory) of the
title compound, which was reacted further in this form.
'H NMR (400 MHz, CDC13, S/ppm): 7.56-7.42 (m, 4H), 4.51 (s, 2H), 3.98 (m, 2H).
GC/MS (Method 9, Elpos): R, = 5.06 min, m/z = 250 [M]+.
Example 53A
Ethyl [3-(bromomethyl)phenyl](difluoro)acetate
F F
H3CO IIZZ~ Y
Br
O I /
4.05 g (22.74 mmol) of N-bromosuccinimide and 178 mg (1.08 mmol) of 2,2'-
azobis-2-
methylpropanenitrile (AIBN) were added to a solution of 4.64 g (21.7 mmol) of
the compound
from Example 52A / step I in 110 ml of acetonitrile. The mixture was heated at
a bath temperature
of 80 C for 2.5 h. After cooling to RT, the solvent was removed and the
residue was triturated with
30 ml of ethyl acetate and 60 ml of pentane. The solid was filtered off and
washed twice with
10 ml of pentane each time. The filtrate was combined with the wash phases,
washed successively
with 100 ml of 10% strength aqueous sodium thiosulphate solution, 100 ml of
water and 100 ml of
saturated sodium chloride solution, dried over sodium sulphate and
concentrated. This gave 5.56 g
(70% pure, 62% of theory) of the title compound, which was reacted further in
this form.
'H NMR (400 MHz, CDC13, S/ppm): 7.64 (s, 1H), 7.58-7.28 (m, 3H), 4.51 (s, 2H),
4.31 (quart,
2H), 1.31 (t, 3H).
GC/MS (Method 9, Elpos): Rt = 5.24 min, m/z = 282 [M]+.
Example 54A
Methyl 3-[(5-methyl-3-{3-[4-(1,1,1-trifluoro-2-methylpropan-2-yl)phenyl]-1,2,4-
oxadiazol-5-yl}-
I H-pyrazol- l -yl)methyl]benzoate
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-104-
0 O-N
H3CII 0 N~ \
N I F F
/
H 3 C F
H3C CH3
Method A:
1.0 g (2.97 mmol) of the compound from Example 23A and 0.82 g (3.56 mmol) of
methyl 3-
bromomethylbenzoate were dissolved in 30 ml of abs. THF, 0.37 g (3.27 mmol) of
potassium tert-
butoxide was added with ice bath cooling and the mixture was stirred at RT
overnight. Water was
then added, and the mixture was extracted three times with about 50 ml of
ethyl acetate each time.
The combined organic extracts were washed with saturated sodium chloride
solution. After drying
over anhydrous magnesium sulphate, the mixture was filtered and the filtrate
was concentrated on
a rotary evaporator. The residue was chromatographed on silica gel (mobile
phase:
cyclohexane/ethyl acetate 4:1). This gave 0.74 g (51 % of theory) of the title
compound.
Method B:
At 0 C, 3.67 g (32.7 mmol) of solid potassium tert-butoxide were added to a
solution of 10.0 g
(29.7 mmol) of the compound from Example 23A and 8.86 g (38.6 mmol) of methyl
3-
bromomethylbenzoate in 300 ml of anhydrous 1,4-dioxane, and the mixture was
then stirred at RT
for 15 h. About 600 ml of water were then added, and the mixture was extracted
three times with
about 250 ml of ethyl acetate each time. The combined organic extracts were
washed with
saturated sodium chloride solution, dried over anhydrous magnesium sulphate,
filtered and finally
freed from the solvent on a rotary evaporator. At RT, the crude product was
triturated in a mixture
of 50 ml of pentane and 5 ml of diisopropyl ether for 30 min. The solid was
filtered off, the filtrate
was discarded and the residue was once more triturated with
pentane/diisopropyl ether as
described. Once more the residue was filtered off and dried under high vacuum,
giving 11.0 g
(72% of theory, 95% pure) of the title compound.
'H NMR (400 MHz, DMSO-d6, 6/ppm): 8.10 (d, 2H), 7.91 (d, 1H), 7.84 (s, 1H),
7.77 (d, 2H), 7.55
(t, 1H), 7.47 (d, 1H), 6.96 (s, 1H), 5.61 (s, 2H), 3.85 (s, 3H), 2.34 (s, 3H),
1.61 (s, 6H).
LC/MS (Method 6, ESlpos): Rt = 1.38 min, m/z = 485 [M+H]+.
Example 55A
2,2,2-Tri fluoro- l -{ 3-[(5-methyl-3-{ 3-[4-(trifluoromethoxy)phenyl]-1,2,4-
oxadiazol-5-yl }-1 H-
pyrazol- l -yl)methyl]phenyl } ethanone
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-105-
0 O-N
F pN
N F F
XFF H3C O X F
At -20 C, 2.6 ml (3.42 mmol) of a 1.3 M solution of isopropylmagnesium
chloride/lithium chloride
complex in THE were added rapidly to a solution of 600 mg (1.14 mmol) of the
compound from
Example 81A in 25 ml of anhydrous THF. After 4 min, the reaction mixture was
cooled to about
-40 C, and 644 pl (4.56 mmol) of trifluoroacetic anhydride were then added in
one portion. The
reaction mixture was stirred at -40 C for 10 min, 1 ml of saturated aqueous
ammonium chloride
solution was then added and the mixture was warmed to RT. Approx. 100 ml of
water were then
added and the mixture was extracted three times with approx. 50 ml of ethyl
acetate each time. The
combined organic extracts were washed with saturated sodium chloride solution,
dried over
anhydrous magnesium sulphate, filtered and finally freed from the solvent on a
rotary evaporator.
The residue obtained was first subjected to coarse purification by means of
MPLC (about 50 g of
silica gel, mobile phase: cyclohexane/ethyl acetate 4:1). After the product
fraction had been
evaporated, the residue was triturated in a mixture of 25 ml of pentane and 1
ml of
dichloromethane. The product was filtered off with suction and then dried
under high vacuum.
This gave 233 mg (39% of theory, 95% pure) of the title compound.
'H NMR (400 MHz, CDC13, 6/ppm): 8.25 (d, 2H), 8.02 (d, 1H), 7.90 (s, 1H), 7.55
(t, IH), 7.50 (d,
1H), 7.33 (d, 2H), 6.86 (s, 1H), 5.53 (s, 2H), 2.32 (s, 3H).
LC/MS (Method 6, ESlpos): Rt = 1.22 min, no ionization.
Example 56A
Methyl 3-[(5-methyl-3-{3-[4-(tetrahydro-2H-pyran-4-yl)phenyl]-1,2,4-oxadiazol-
5-yl}-1H-pyrazol-
1-yl)methyl]benzoate
O O-N
H3C~O N~
N
H3C
O
At 0 C, 70 mg (0.620 mmol) of solid potassium tert-butoxide were added to a
solution of 175 mg
(0.564 mmol) of the compound from Example 28A and 168 mg (0.733 mmol) of
methyl 3-
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
- 106 -
bromomethylbenzoate in 6 ml of anhydrous THF, and the mixture was then stirred
at RT for 4 h.
The reaction was then ended by addition of 2 drops of water, and methanol was
added until all
solid components had just dissolved. This solution was then purified directly
by preparative HPLC
(Methode 11). This gave 208 mg (80% of theory) of the title compound.
'H NMR (400 MHz, CDC13, 8/ppm): 8.15 (d, 2H), 7.98 (d, 1H), 7.89 (s, IH), 7.42
(t, 1H), 7.34 (d,
2H), 7.33 (d, 1H), 6.82 (s, IH), 5.50 (s, 2H), 4.13-4.08 (m, 2H), 3.91 (s,
3H), 3.54 (dt, 2H), 2.87-
2.79 (m, 1H), 2.28 (s, 3H), 1.92-1.78 (m, 4H).
LC/MS (Method 6, ESIpos): R, = 1.24 min, m/z = 459 [M+H]+.
Example 57A
Methyl 3-({3-[3-(4-tert-butylphenyl)-1,2,4-oxadiazol-5-yl]-5-methyl-IH-pyrazol-
l-
yl } methyl)benzoate
O O-N
\
H3C~0 PN N ' ~~
/ CH3
H3C
H3C CH3
At 0 C, 298 mg (2.66 mmol) of solid potassium tert-butoxide were added to a
solution of 500 mg
(1.77 mmol) of the compound from Example 42A and 811 mg (3.54 mmol) of methyl
3-
bromomethylbenzoate in 15 ml of anhydrous THF, and the mixture was then
stirred at RT for 15 h.
About 45 ml of water were then added, and the mixture was extracted three
times with about 20 ml
of ethyl acetate each time. The combined organic extracts were washed with
saturated sodium
chloride solution, dried over anhydrous magnesium sulphate, filtered and
finally freed from the
solvent on a rotary evaporator. MPLC (silica gel, mobile phase:
cyclohexane/ethyl acetate 5:1) of
the crude product gave 493 mg (64% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 8.13 (d, 2H), 7.98 (d, 1H), 7.89 (s, 1H), 7.50
(d, 2H), 7.42 (t,
1H), 7.33 (d, 1H), 6.83 (s, IH), 5.50 (s, 2H), 3.91 (s, 3H), 2.28 (s, 3H),
1.37 (s, 9H).
LC/MS (Method 6, ESIpos): R, = 1.41 min, m/z = 431 [M+H]+.
Example 58A
Methyl 3-{[3-(3-{4-[I-(methoxymethyl)cyclobutyl]phenyl}-1,2,4-oxadiazol-5-yl)-
5-methyl-IH-
pyrazol-1-yl]methyl) benzoate
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-107-
0 O-N
H3C~0
-1 Nz~ -oo pN \ H C / 0I-ICH3
3
Analogously to the process described under Example 57A, 325 mg (1.00 mmol) of
the compound
from Example 40A and methyl 3-bromomethylbenzoate gave 206 mg (44% of theory)
of the title
compound.
'H NMR (400 MHz, CDCI3, 6/ppm): 8.14 (d, 2H), 7.98 (d, I H), 7.90 (s, IH),
7.42 (t, I H), 7.33 (d,
1H), 7.30 (d, 2H), 6.83 (s, IH), 5.50 (s, 2H), 3.91 (s, 3H), 3.55 (s, 2H),
3.28 (s, 3H), 2.43-2.28 (m,
4H), 2.28 (s, 3H), 2.15-2.03 (m, IH), 1.93-1.83 (m, 1H).
LC/MS (Method 6, ESlpos): Rt = 1.38 min, m/z = 473 [M+H]+.
Example 59A
Methyl 3-{[3-(3-{3-fluoro-4-[1-(methoxymethyl)cyclobutyl]phenyl}-1,2,4-
oxadiazol-5-yl)-5-
methyl-I H-pyrazol-l -yl]methyl }benzoate
0 O-N
H3C~0 Ni F
- N
HC / 0"CH3
Analogously to the process described under Example 57A, 350 mg (1.02 mmol) of
the compound
from Example 31A and methyl 3-bromomethylbenzoate gave 233 mg (46% of theory)
of the title
compound.
'H NMR (400 MHz, CDC13, S/ppm): 7.99 (d, IH), 7.93 (dd, 1H), 7.89 (s, IH),
7.83 (dd, 1H), 7.43
(t, 1H), 7.33 (d, IH), 7.23 (d, 1H), 6.83 (s, 1H), 5.51 (s, 2H), 3.91 (s, 3H),
3.67 (s, 2H), 3.28 (s,
3H), 2.48-2.40 (m, 2H), 2.37-2.30 (m, 2H), 2.28 (s, 3H), 2.19-2.07 (m, 1H),
1.94-1.85 (m, IH).
LC/MS (Method 6, ESlpos): Rt = 1.42 min, m/z = 491 [M+H]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-108-
Example 60A
Methyl 3-{ [3-(3-{4-[ I-(methoxymethyl)cyclopentyl]phenyl }-1,2,4-oxadiazol-5-
yl)-5-methyl-1 H-
pyrazol-1-yl]methyl }benzoate
O O-N
H3CN10 PN N
H C 0~CH3
3
Analogously to the process described under Example 57A, 350 mg (1.03 mmol) of
the compound
from Example 33A and methyl 3-bromomethylbenzoate gave 174 mg (35% of theory)
of the title
compound. In this case, the product was isolated not by MPLC but by
preparative HPLC (Method
11).
'H NMR (400 MHz, CDCI3, S/ppm): 8.12 (d, 2H), 7.99 (d, 1H), 7.90 (s, 1H), 7.45
(d, 2H), 7.42 (t,
1H), 7.33 (d, 1H), 6.83 (s, 1H), 5.50 (s, 2H), 3.91 (s, 3H), 3.41 (s, 2H),
3.23 (s, 3H), 2.28 (s, 3H),
2.07-2.00 (m, 2H), 1.96-1.88 (m, 2H), 1.79-1.70 (m, 4H).
LC/MS (Method 6, ESIpos): R, = 1.43 min, m/z = 487 [M+H]+.
Example 61A
Methyl 3-{ [3-(3-{4-[ 1-(ethoxymethyl)cyclobutyl]phenyl }-1,2,4-oxadiazol-5-
yl)-5-methyl-1 H-
pyrazol-1-yl]methyl}benzoate
O O-N
H3CN1 0 PN N \
H3C O CH3
Analogously to the process described under Example 56A, 150 mg (0.443 mmol) of
the compound
from Example 32A and methyl 3-bromomethylbenzoate gave 43 mg (19% of theory)
of the title
compound. In addition, 85 mg (41 % of theory) of the corresponding benzoic
acid were isolated.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-109-
'H NMR (400 MHz, CDC13, 6/ppm): 8.13 (d, 2H), 7.98 (d, 1H), 7.90 (s, 1H), 7.43
(t, 1H), 7.33 (d,
1H), 7.30 (d, 2H), 6.83 (s, IH), 5.50 (s, 2H), 3.91 (s, 3H), 3.57 (s, 2H),
3.38 (quart, 2H), 2.40-2.30
(m, 4H), 2.27 (s, 3H), 2.15-2.03 (m, IH), 1.92-1.83 (m, 1H), 1.09 (t, 3H).
LC/MS (Method 6, ESlpos): R, = 1.45 min, m/z = 487 [M+H]+.
Example 62A
Methyl 3-{ [5-methyl-3-(3-{4-[(trifluoromethyl)sulphanyl]phenyl}-1,2,4-
oxadiazol-5-yl)-1 H-
pyrazol-1-yl]methyl}benzoate
O O-N
H3C~'0 PN ~ X N FF
H3C S F
At 0 C, 112 mg (0.996 mmol) of solid potassium tert-butoxide were added to a
solution of 250 mg
(0.766 mmol) of the compound from Example 36A and 211 mg (0.919 mmol) of
methyl 3-
bromomethylbenzoate in 7.5 ml of anhydrous 1,4-dioxane, and the mixture was
then stirred at RT
for 15 h. About 25 ml of water were then added, and the mixture was extracted
three times with
about 20 ml of ethyl acetate each time. The combined organic extracts were
washed with saturated
sodium chloride solution, dried over anhydrous magnesium sulphate, filtered
and finally freed
from the solvent on a rotary evaporator. MPLC (silica gel, mobile phase:
cyclohexane/ethyl acetate
10:1 -+ 5:1) of the crude product gave 268 mg (74% of theory) of the title
compound.
'H NMR (400 MHz, CDC13, 6/ppm): 8.27 (d, 2H), 7.99 (d, 1H), 7.89 (s, 1H), 7.78
(d, 2H), 7.43 (t,
1H), 7.34 (d, IH), 6.83 (s, 1H), 5.51 (s, 2H), 3.91 (s, 3H), 2.29 (s, 3H).
LC/MS (Method 6, ESlpos): R, = 1.39 min, m/z = 475 [M+H].
Example 63A
Methyl 3 -[(3 - { 3 -[4-(4-fluorotetrahydro-2H-pyran-4-yl)phenyl] -1,2,4-
oxadiazol-5-yl } -5-methyl-1 H-
pyrazol- l -yl)methyl]benzoate
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
- 110 -
0 O-N
H PN N \
1/ F
H 3 C
O
Analogously to the process described under Example 62A, 500 mg (1.52 mmol) of
the compound
from Example 43A and methyl 3-bromomethylbenzoate gave 434 mg (60% of theory)
of the title
compound. In this case, the product was isolated not by MPLC but by
preparative HPLC (Method
11).
'H NMR (400 MHz, CDC13, S/ppm): 8.12 (d, 2H), 7.99 (d, IH), 7.90 (s, IH), 7.52
(d, 2H), 7.42 (t,
IH), 7.34 (d, I H), 6.84 (s, IH), 5.51 (s, 2H), 4.00-3.87 (m, 4H), 3.91 (s,
3H), 2.29 (s, 3H), 2.29-
2.12 (m, 2H), 1.98-1.92 (m, 2H).
LC/MS (Method 6, ESIpos): R, = 1.23 min, m/z = 477 [M+H]+.
Example 64A
Methyl 3-[(5-methyl-3-{ 3-[4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-yl }-
1 H-pyrazol-1-
yl)methyl]benzoate
O O-N
H3C"0 pN__,___l F F
H3C O F
Analogously to the process described under Example 54A (Method A), 300 mg
(0.97 mmol) of the
compound from Example 22A and methyl 3-bromomethylbenzoate gave 430 mg (89% of
theory)
of the title compound.
'H NMR (400 MHz, DMSO-d6, 6/ppm): 8.20 (d, 2H), 7.91 (d, IH), 7.84 (s, 1H),
7.59 (d, 2H), 7.55
(t, IH), 7.48 (d, IH), 6.96 (s, IH), 5.61 (s, 2H), 3.85 (s, 3H), 2.34 (s, 3H).
LC/MS (Method 4, ESIpos): Rt = 1.54 min, m/z = 459 [M+H]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
- 111 -
Example 65A
Methyl 3-[(3-{ 3-[4-(1-fluorocyclobutyl)phenyl]-1,2,4-oxadiazol-5-yl}-5-methyl-
1 H-pyrazol-l -
yl)methyl]benzoate
O O-N
H3CN1 0 PN J \
N
/ F
H3C
Analogously to the process described under Example 54A (Method A), 200 mg
(0.67 mmol) of the
compound from Example 44A and methyl 3-bromomethylbenzoate gave 210 mg (68% of
theory)
of the title compound. In this case, the product was isolated by preparative
HPLC (Method 17).
1H NMR (400 MHz, DMSO-d6, 6/ppm): 8.12 (d, 2H), 7.91 (d, I H), 7.84 (s, 1 H),
7.70 (d, 2H), 7.55
(t, IH), 7.47 (d, IH), 6.96 (s, 1H), 5.61 (s, 2H), 3.85 (s, 3H), 2.64 (t, 2H),
2.59 (t, 2H), 2.35 (s, 3H),
2.07 (m, 1H), 1.77 (m, I H).
LC/MS (Method 6, ESlpos): R, = 1.37 min, m/z = 447 [M+H]+.
Example 66A
1-[3-({ 3-[3-(4-tent-Butylphenyl)-1,2,4-oxadiazol-5-yl]-5-methyl-I H-pyrazol-1-
yl} methyl)phenyl]cyclopropylacetate
O O-N
H CO PN \
3 N
CH3
H3C
H 3 C CH3
At 0 C, 103 mg (0.92 mmol) of potassium tert-butoxide were added to a solution
of 200 mg (0.71
mmol) of the compound from Example 42A and 232 mg (0.78 mmol, purity 95%) of
the compound
from Example 47A in 5 ml of THF. The reaction mixture was then stirred at RT
for 1 h. After
dilution with ethyl acetate, the mixture was washed once with water and the
aqueous phase was
reextracted once with ethyl acetate. The combined organic phases were washed
once with
saturated sodium chloride solution, dried over magnesium sulphate, filtered
and concentrated. This
gave 350 mg (80% pure, 84% of theory) of the title compound.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-112-
'H NMR (400 MHz, CDCl3, 6/ppm): 8.13 (d, 2H), 7.51 (d, 2H), 7.30-7.24 (m, IH),
7.18 (d, 1H),
7.10 (s, 1H), 7.01 (d, IH), 6.81 (s, 1H), 5.44 (s, 2H), 2.26 (s, 3H), 2.01 (s,
3H), 1.36 (s, 9H), 1.31-
1.24 (m, 2H), 1.21-1.15 (m, 2H).
LC/MS (Method 4, ESlpos): Rt = 1.64 min, m/z = 471 [M+H]+.
Example 67A
1-{ 3-[(5-Methyl-3-{3-[4-(tetrahydro-2H-pyran-4-yl)phenyl]-1,2,4-oxadiazol-5-
yl }-1 H-pyrazol- l -
yl)methyl]phenyl}cyclopropyl acetate
0 O-N
H C"k 0 IIZZZ P \
s N
H3C
0
At 0 C, 65 mg (0.58 mmol) of potassium tert-butoxide were added to a solution
of 139 mg (0.45
mmol) of the compound from Example 28A and 147 mg (0.49 mmol, purity 95%) of
the compound
from Example 47A in 3.5 ml of THF. After warming to RT, the mixture was
stirred at RT for a
further hour. After dilution with ethyl acetate, the mixture was washed once
with water and the
aqueous phase was reextracted once with ethyl acetate. The combined organic
phases were washed
once with saturated sodium chloride solution, dried over magnesium sulphate,
filtered and
concentrated. The residue was purified by means of column chromatography
(silica gel, mobile
phase: cyclohexane/ethyl acetate 7:3). Removal of the solvent gave 164 mg (79%
pure, 58% of
theory) of the title compound.
LC/MS (Method 6, ESIpos): Rt = 1.27 min, m/z = 499 [M+H]+.
Example 68A
1-{3-[(5-Methyl-3-{ 3-[4-(trimethylsilyl)phenyl]-1,2,4-oxadiazol-5-yl }-1 H-
pyrazol-l -
yl)methyl]phenyl}cyclopropyl acetate
0 O-N
H C0 N~
3 N
CH
H3C Si
H3C CH3
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-113-
Analogously to the process described in Example 67A, 211 mg (0.71 mmol) of the
compound from
Example 26A and 232 mg (0.78 mmol, purity 95%) of the compound from Example
47A gave 231
mg (90% pure, 60% of theory) of the title compound. Here, the chromatographic
purification of
the product was carried out using the mobile phase cyclohexane/ethyl acetate
4:1.
'H NMR (400 MHz, CDC13, 6/ppm): 8.17 (d, 2H), 7.64 (d, 2H), 7.30-7.26 (m, IH),
7.18 (d, I H),
7.10 (s, 1H), 7.01 (d, 1H), 6.81 (s, 1H), 5.44 (s, 2H), 2.26 (s, 3H), 2.01 (s,
3H), 1.31-1.15 (m, 4H),
0.31 (s, 9H).
LC/MS (Method 6, ESlpos): R, = 1.49 min, m/z = 487 [M+H]+.
Example 69A
1-{ 3-[(5-Methyl-3-{ 3-[4-(1,1,1-trifluoro-2-methylpropan-2-yl)phenyl]-1,2,4-
oxadiazol-5-yl }-1 H-
pyrazol-l-yl)methyl]phenyl}cyclopropyl acetate
O O-N
PN
H N \
3C O I N I F
F
H3C F
H3C CH3
Analogously to the process described in Example 67A, 238 mg (0.71 mmol) of the
compound from
Example 23A and 232 mg (0.78 mmol, purity 95%) of the compound from Example
47A gave 286
mg (88% pure, 68% of theory) of the title compound. Here, the chromatographic
purification of
the product was carried out here using the mobile phase cyclohexane/ethyl
acetate 3:1.
'H NMR (400 MHz, CDC13, S/ppm): 8.20 (d, 2H), 7.62 (d, 2H), 7.30-7.26 (m, IH),
7.21-7.17 (d,
I H), 7.10 (s, IH), 7.01 (d, I H), 6.82 (s, I H), 5.44 (s, 2H), 2.27 (s, 3H),
2.01 (s, 3H), 1.62 (s, 6H),
1.33-1.15 (m, 4H).
LC/MS (Method 6, ESlpos): R, = 1.40 min, m/z = 525 [M+H]+.
Example 70A
1-[3-({ 3-[3-(4-Isopropylphenyl)-1,2,4-oxadiazol-5-yl]-5-methyl-I H-pyrazol- l
-
yl}methyl)phenyl]cyclopropyl acetate
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
- 114 -
0 O-N
'J~ PN
H
3 C O N
C H 3
H3C
CH3
Analogously to the process described in Example 66A, 190 mg (0.71 mmol) of the
compound from
Example 41A and 232 mg (0.78 mmol, purity 95%) of the compound from Example
47A gave
359 mg (69% pure, 76% of theory) of the title compound.
LC/MS (Method 6, ESlpos): Rt = 1.41 min, m/z = 457 [M+H]+.
Example 71A
1-[3-({ 3-[3-(4-Isobutylphenyl)-1,2,4-oxadiazol-5-yl]-5-methyl-1 H-pyrazol- l -
yl}methyl)phenyl]cyclopropyl acetate
O O-N
.-K PN
H
C O 3 N CH
H 3 C C H 3
Analogously to the process described in Example 66A, 200 mg (0.71 mmol) of the
compound from
Example 39A and 232 mg (0.78 mmol, purity 95%) of the compound from Example
47A gave
381 mg (72% pure, 82% of theory) of the title compound.
LC/MS (Method 6, ESIpos): R, = 1.47 min, m/z = 471 [M+H]+.
Example 72A
1-(3-{ [5-Methyl-3-(3-{4-[ 1-(trifluoromethyl)cyclopropyl]phenyl }-1,2,4-
oxadiazol-5-yl)-1 H-
pyrazol-1-yl]methyl } phenyl)cyclopropyl acetate
O O-N
H C~O P 3 I N F F
H3C F
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-115-
Analogously to the process described in Example 66A, 237 mg (0.71 mmol) of the
compound from
Example 30A and 232 mg (0.78 mmol, purity 95%) of the compound from Example
47A gave
408 mg (70% pure, 77% of theory) of the title compound.
LC/MS (Method 6, ESlpos): R, = 1.40 min, m/z = 523 [M+H]+.
Example 73A
1-{3-[(5-Methyl-3-{ 3-[4-(pentafluoro-),6-sulphanyl)phenyl]-1,2,4-oxadiazol-5-
yl}-1 H-pyrazol- l -
yl)methyl]phenyl}cyclopropyl acetate
0 0-N
PN \
H 3C O N \
/ IMF
H3C FS~F
F
Analogously to the process described in Example 66A, 275 mg (0.71 mmol) of the
compound from
Example 38A and 232 mg (0.78 mmol, purity 95%) of the compound from Example
47A gave
423 mg (75% pure, 75% of theory) of the title compound. In this case, the
reaction time was 4 h at
RT.
LC/MS (Method 6, ESlpos): Rt = 1.39 min, m/z = 541 [M+H]+.
Example 74A
1-{3-[(3-{3-[4-(2-Fluoropropan-2-yl)phenyl]-1,2,4-oxadiazol-5-yl}-5-methyl-1 H-
pyrazol- l -
yl)methyl]phenyl}cyclopropyl acetate
0 O-N
PN H
3C 0 \ _ _ ', \
N
F
H3C
"N~
H3C CH3
Analogously to the process described in Example 66A, 223 mg (0.78 mmol) of the
compound from
Example 24A and 255 mg (0.86 mmol, purity 95%) of the compound from Example
47A gave
406 mg (75% pure, 82% of theory) of the title compound. In this case, the
reaction time was 4 h at
RT.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-116-
LC/MS (Method 6, ESlpos): R, = 1.34 min, m/z = 475 [M+H]+.
Example 75A
1-{ 3-[(5-Methyl-3-{ 3-[4-(trifluoromethyl)phenyl]-1,2,4-oxadiazol-5-yl}-1 H-
pyrazol-l -
yl)methyl]phenyl}cyclopropyl acetate
O O-N
'J~ PN
H
3 C O N
F
H3C
F F
Analogously to the process described in Example 66A, 230 mg (0.78 mmol) of the
compound from
Example 27A and 255 mg (0.86 mmol, purity 95%) of the compound from Example
47A gave
375 mg (74% pure, 74% of theory) of the title compound. In this case, the
reaction time was 16 h
at RT.
LC/MS (Method 6, ESlpos): Rt = 1.55 min, m/z = 483 [M+H]+.
Example 76A
1-{3-[(3-{ 3-[4-(4-Fluorotetrahydro-2H-pyran-4-yl)phenyl]-1,2,4-oxadiazol-5-yl
}-5-methyl-1 H-
pyrazol-1-yl)methyl]phenyl}cyclopropyl acetate
O O-N
H3C O PN
N
F
H3C
O
Analogously to the process described in Example 66A, 256 mg (0.78 mmol) of the
compound from
Example 27A and 255 mg (0.86 mmol, purity 95%) of the compound from Example
47A gave
415 mg (65% pure, 67% of theory) of the title compound. In this case, the
reaction time was 16 h
at RT.
LC/MS (Method 4, ESlpos): Rt = 1.44 min, m/z = 517 [M+H]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-117-
Example 77A
1-[3-({3-[3-(4-Cyclohexylphenyl)-1,2,4-oxadiazol-5-yl]-5-methyl-1 H-pyrazol-l -
yl)methyl)phenyl]cyclopropyl acetate
O O-N
H C~O PNN
s N
H3C
Analogously to the process described in Example 66A, 195 mg (0.63 mmol) of the
compound from
Example 37A and 207 mg (0.70 mmol, purity 95%) of the compound from Example
47A gave
318 mg (42% pure, 43% of theory) of the title compound. In this case, the
reaction time was 4 h at
RT.
LC/MS (Method 6, ESlpos): R, = 1.59 min, m/z = 497 [M+H]+.
Example 78A
1-(3-{ [5-Methyl-3-(3-{4-[(trifluoromethyl)sulphanyl]phenyl } -1,2,4-oxadiazol-
5-yl)-1 H-pyrazol- l -
yl]methyl}phenyl)cyclopropyl acetate
O O-N
PN
H
sC O N
X
H3C S F
Analogously to the process described in Example 66A, 254 mg (0.78 mmol) of the
compound from
Example 36A and 255 mg (0.86 mmol, purity 95%) of the compound from Example
47A gave
396 mg (72% pure, 70% of theory) of the title compound. In this case, the
reaction time was 4 h at
RT.
LC/MS (Method 6, ESlpos): R, = 1.25 min, m/z = 441 [M+H]+.
Example 79A
1-{ 3-[(3-{3-[4-(1-Fluorocyclobutyl)phenyl]-1,2,4-oxadiazol-5-yl }-5-methyl-1
H-pyrazol-l -
yl)methyl]phenyl}cyclopropyl acetate
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
- 118 -
0 O-N
N ___ '~
H C~O P \
s N F
H3C
At 0 C, 113 mg (1.01 mmol) of potassium tert-butoxide were added to a solution
of 233 mg (0.78
mmol) of the compound from Example 36A and 255 mg (0.86 mmol, purity 95%) of
the compound
from Example 47A in 5.5 ml of THF. After warming to RT, the mixture was
stirred at RT for a
further 4 h. After dilution with ethyl acetate, the mixture was washed once
with water and the
aqueous phase was reextracted once with ethyl acetate. The combined organic
phases were washed
once with saturated sodium chloride solution, dried over magnesium sulphate,
filtered and
concentrated. The residue was purified by means of preparative HPLC (Method
15). The clean
product-containing fractions were combined, saturated aqueous sodium
bicarbonate solution was
added and the mixture was reconcentrated to a residual volume of aqueous
phase. The mixture was
extracted twice with ethyl acetate. The combined organic phases were dried
over magnesium
sulphate, filtered and concentrated. This gave 187 mg (62% pure, 30% of
theory) of the title
compound.
LC/MS (Method 6, ESlpos): R, = 1.37 min, m/z = 487 [M+H]+.
Example 80A
1-{ 3-[(3-{3-[3-Fluoro-4-(1,1,1-trifluoro-2-methylpropan-2-yl)phenyl]-1,2,4-
oxadiazol-5-yl }-5-
methyl-lH-pyrazol-l-yl)methyl]phenyl}cyclopropyl acetate
0 O-N
H C N~ N \ F
3 _ N I F F
11,511
H3C F
H3C CH3
Analogously to the process described in Example 66A, 200 mg (0.54 mmol, purity
95%) of the
compound from Example 45A and 176 mg (0.59 mmol, purity 96%) of the compound
from
Example 47A gave 192 mg (69% pure, 46% of theory) of the title compound. In
this case, the
reaction time was 4 h at RT. The crude product was purified by means of
preparative HPLC
according to Method 15.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-119-
LC/MS (Method 6, ESlpos): Rt = 1.45 min, m/z = 543 [M+H]+.
Example 81A
5-[ 1-(3-Iodobenzyl)-5-methyl-1 H-pyrazol-3-yl]-3-[4-(trifluoromethoxy)phenyl]-
1,2,4-oxadiazole
O-N
PN
N F F
H3C O F
Analogously to the process described under Example 54A (Method A), 500 mg
(1.61 mmol) of the
compound from Example 22A and 570 mg (1.93 mmol) of 3-iodobenzyl bromide were
reacted to
give 690 mg (78% of theory) of the title compound. Here, the product was
isolated by means of
preparative HPLC (Method 17).
'H NMR (400 MHz, DMSO-d6, 6/ppm): 8.20 (d, 2H), 7.69 (m, IH), 7.63 (s, IH),
7.59 (d, 2H),
7.18 (d, 2H), 6.94 (s, 1H), 5.49 (s, 2H), 2.35 (s, 3H).
LC/MS (Method 2, ESlpos): Rt = 3.05 min, m/z = 527 [M+H]+.
Example 82A
Ethyl {3-[(5-methyl-3-{3-[4-(1,1,1-trifluoro-2-methylpropan-2-yl)phenyl]-1,2,4-
oxadiazol-5-yl}-
1 H-pyrazol- l -yl)methyl]phenyl } acetate
O-N
PN
H3CO
N F F
H3C F
H3C C H 3
Analogously to the process described under Example 54A (Method A), 0.50 g
(1.49 mmol) of the
compound from Example 23A and 0.50 g (1.93 mmol) of ethyl [3-
(bromomethyl)phenyl]acetate
[lit. S. R. Kasibhatla et al., J. Med. Chem. 2000, 43 (8), 1508-1518] were
converted into 0.47 g
(62% of theory) of the title compound. Here, the product was isolated by means
of preparative
HPLC (Method 17).
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-120-
'H NMR (400 MHz, DMSO-d6, S/ppm): 8.09 (d, 2H), 7.77 (d, 2H), 7.33 (t, 1H),
7.21 (d, 1H), 7.10
(d, 2H), 6.94 (s, IH), 5.50 (s, 2H), 4.04 (quart, 2H), 3.65 (s, 2H), 2.33 (s,
3H), 1.61 (s, 6H), 1.14 (t,
3H).
LC/MS (Method 6, ESlpos): R, = 1.43 min, m/z = 513 [M+H]+.
Example 83A
Ethyl {3-[(5-methyl-3-{3-[4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-yl}-1H-
pyrazol-1-
yl)methyl]phenyl } acetate
0-N
Fi3C0 PN
N FF
H3C 0 F
15.5 g (138 mmol) of potassium tert-butoxide were added to a solution of 39.0
g (126 mmol) of the
compound from Example 22A in 1 litre of anhydrous THF. At about 5 C (ice/water
bath), a
solution of 38.8 g (151 mmol) of ethyl [3-(bromomethyl)phenyl]acetate [lit.:
S. R. Kasibhatla et
al., J. Med. Chem. 2000, 43 (8), 1508-1518] in 700 ml of anhydrous THE was
then added
dropwise. After the reaction mixture had been stirred at RT for 40 h, the
mixture was diluted with
1 litre of ethyl acetate. About 100 g of anhydrous magnesium sulphate were
added, and the
heterogeneous mixture was stirred vigorously. The mixture was then filtered
and the filtrate was
freed from the solvent on a rotary evaporator. The crude product was purified
by filtration with
suction through about 2 kg of silica gel (particle size 0.06-0.2 mm) using a
cyclohexane/ethyl
acetate gradient [9:1 (5 litres) - 8:2 (4 litres) - 7:3 (4 litres) -* 6:4 (4
litres) - 1:1 (4 litres)] as
mobile phase. This gave 33.8 g (48% of theory, 87% pure) of the title
compound.
'H NMR (400 MHz, DMSO-d6, S/ppm): 8.20 (d, 2H), 7.60 (d, 2H), 7.33 (t, IH),
7.21 (d, 1H), 7.11
(s, IH), 7.10 (d, IH), 6.93 (s, 1H), 5.49 (s, 2H), 4.05 (quart, 2H), 3.65 (s,
2H), 2.34 (s, 3H), 1.14 (t,
3H).
LC/MS (Method 6, ESlpos): R, = 1.37 min, m/z = 487 [M+H]+.
Example 84A
1-{ 3-[(5-Methyl-3-{ 3-[4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-yl}-1 H-
pyrazol-l -
yl)methyl]phenyl } acetone
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
- 121 -
O-N
H3C pN N IIZZ~ F F
O x
H 3 C O F
At 0 C, 50.5 m] (152 mmol) of methylmagnesium bromide solution (3 M in diethyl
ether) were
added dropwise to a solution of 33.5 g (68.9 mmol) of the compound from
Example 83A in 700 ml
of anhydrous THF. When the addition had ended, the ice/water bath was removed
and stirring was
continued at RT. After 2 h, the reaction was ended by careful addition of 50
ml of saturated
aqueous ammonium chloride solution. The reaction was then diluted with 700 ml
of ethyl acetate,
and 100 g of anhydrous magnesium sulphate were added. After brief stirring,
the mixture was
filtered and the filtrate was freed from the solvent on a rotary evaporator.
The residue obtained was
purified by filtration with suction through 770 g of silica gel (particle size
0.06-0.2 mm) using a
cyclohexane/ethyl acetate gradient [8:2 (8 litres) -> 7:3 (8 litres) -> 6:4 (8
litres) -> 1:1 (4 litres)]
as mobile phase. Evaporation of the solvent gave 22.5 g of a crude product
which, in addition to
the title compound, mainly contained the compound 2-methyl-l-{3-[(5-methyl-3-
{3-[4-
(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-yl}-IH-pyrazol-1-
yl)methyl]phenyl}propan-2-ol (see
Example 43).
7.6 g of this crude product were recrystallized from 61 ml of boiling
diisopropyl ether. The crystals
obtained at RT were filtered off with suction and washed twice with 5 ml of
cold diisopropyl ether
each time. The material obtained in this manner consisted of pure 2-methyl-l-
{3-[(5-methyl-3-{3-
[4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-yl} -1 H-pyrazol-l-
yl)methyl]phenyl} propan-2-ol
(see Example 43). The mother liquor was freed from the solvent on a rotary
evaporator, and the
residue was separated into its components by means of preparative HPLC (Method
12). This gave
540 mg of the title compound and a further 2.85 g of 2-methyl-l-{3-[(5-methyl-
3-{3-[4-
(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-yl}-1H-pyrazol-1-
yl)methyl]phenyl}propan-2-ol (see
Example 43). The 540 mg of the title compound were finally crystallized from 5
ml of diisopropyl
ether. Drying under high vacuum gave 373 mg of the title compound in pure form
(3.5% of theory,
based on 7.6 g of crude product that had been subjected to further
processing).
'H NMR (400 MHz, CDC13, 6/ppm): 8.25 (d, 2H), 7.33 (d, 2H), 7.31 (t, IH), 7.14
(d, I H), 7.07 (d,
IH), 7.01 (s, IH), 6.82 (s, 1H), 5.43 (s, 2H), 3.69 (s, 2H), 2.29 (s, 3H),
2.14 (s, 3H).
LC/MS (Method 6, ESlpos): R, = 1.31 min, m/z = 457 [M+H]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-122-
Example 85A
Ethyl 2-methyl-2-(3-{ [5-methyl-3-(3-{4-[ I-
(trifluoromethyl)cyclopropyl]phenyl}-1,2,4-oxadiazol-
5-yl)-1 H-pyrazol-1-yl]methyl } phenyl)propanoate
H 3 C CH3 O-N
H3CO PN
N F F
O
H 3 C F
At 0 C, 99 mg (0.88 mmol) of potassium tert-butoxide were added to a solution
of 227 mg
(0.68 mmol) of the compound from Example 30A and 300 mg (0.75 mmol, purity
71%) of the
compound from Example 49A in 5 ml of THF. The mixture was allowed to warm to
RT and then
stirred overnight at RT. After addition of ethyl acetate and water, the
aqueous phase was extracted
once with ethyl acetate. The combined organic phases were washed once with
saturated sodium
chloride solution, dried over magnesium sulphate, filtered and concentrated.
The residue was
purified by means of preparative HPLC (Method 15). The clean product-
containing fractions were
combined and concentrated to a residual volume of aqueous phase. After
addition of saturated
aqueous sodium bicarbonate solution, the mixture was extracted twice with
ethyl acetate. The
combined organic phases were dried over magnesium sulphate, filtered and
concentrated. Drying
of the residue under reduced pressure gave 264 mg (71% of theory) of the title
compound.
tH NMR (400 MHz, CDC13, S/ppm): 8.19 (d, 2H), 7.59 (d, 2H), 7.29-7.26 (m, 2H),
7.16 (s, 1H),
7.03-6.98 (m, 1H), 6.81 (s, IH), 5.44 (s, 2H), 4.09 (quart, 2H), 2.26 (s, 3H),
1.52 (s, 6H), 1.42-1.39
(m, 2H), 1.12 (t, 3H), 1.01-1.08 (m, 2H).
LC/MS (Method 6, ESlpos): Rt = 1.49 min, m/z = 539 [M+H]+.
Example 86A
Ethyl 2-methyl-2- { 3-[(5-methyl-3- { 3-[4-(tetrahydro-2H-pyran-4-yl)phenyl]-
1,2,4-oxadi azol-5-yl } -
1 H-pyrazol-1-yl)methyl]phenyl } propanoate
H3C CH3 O-N
H3CO N~ N
O I I
H3C
0
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-123-
58 mg (0.513 mmol) of solid potassium tert-butoxide were added to a solution
of 145 mg (0.467
mmol) of the compound from Example 28A and 173 mg (0.607 mmol) of the compound
from
Example 49A in 5 ml of anhydrous THE at 0 C. After removal of the ice/water
bath, the reaction
mixture was stirred at RT for 16 h. 1 ml of methanol was then added, and the
reaction mixture was
separated directly into its components by means of preparative HPLC (method
11). In this manner,
198 mg (77% of theory, 93% pure) of the title compound were obtained.
'H NMR (400 MHz, CDC13, S/ppm): 8.15 (d, 2H), 7.35 (d, 2H), 7.29-7.24 (m, 2H),
7.15 (s, 1H),
7.00 (d, 1H), 6.80 (s, 1H), 5.44 (s, 2H), 4.12-4.06 (m, 2H), 4.09 (quart, 2H),
3.55 (dt, 2H), 2.87-
2.80 (m, 1H), 2.27 (s, 3H), 1.91-1.78 (m, 4H), 1.53 (s, 6H), 1.14 (t, 3H).
LC/MS (Method 4, ESlpos): R, = 1.52 min, m/z = 515 [M+H]+.
Example 87A
Ethyl 2-methyl-2-{3-[(5-methyl-3-{3-[4-(1,1,1-tri fluoro-2-methylpropan-2-
yl)phenyl]-1,2,4-oxa-
diazol-5-yl }-1 H-pyrazol-1-yl)methyl]phenyl } propanoate
H3C CH3 O-N
H 3 C ,0 PN
11-~
O / F F
H3C F
H3C CH3
Analogously to the process described under Example 85A, 0.50 g (1.49 mmol) of
the compound
from Example 23A and 0.55 g (1.93 mmol) of the compound from Example 49A were
reacted to
give 0.49 g (59% of theory) of the title compound. The purification of the
product was carried out
by means of preparative HPLC according to Method 17.
'H NMR (400 MHz, DMSO-d6, 6/ppm): 8.09 (d, 2H), 7.77 (d, 2H), 7.34 (t, 1H),
7.27 (d, 1H), 7.14
(s, 1H), 7.06 (d, 1H), 6.93 (s, 1H), 5.51 (s, 2H), 4.00 (quart, 2H), 2.32 (s,
3H), 1.61 (s, 6H), 1.47 (s,
6H), 1.03 (t, 3H).
LC/MS (Method 6, ESIpos): R, = 1.50 min, m/z = 541 [M+H]+.
Example 88A
Ethyl 2-methyl-2-{ 3-[(5-methyl-3-{ 3-[4-(trifluoromethoxy)phenyl]-1,2,4-
oxadiazol-5-yl}-1 H-
pyrazol-l-yl)methyl]phenyl}propanoate
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
- 124 -
H3C CH3 O-N PH3CO ___, N- N I F F
O
H3C / O F
Analogously to the process described under Example 85A, 0.14 g (0.44 mmol) of
the compound
from Example 22A and 0.15 g (0.53 mmol) of the compound from Example 49A were
reacted to
give 0.14 g (60% of theory) of the title compound. The purification of the
product was carried out
by means of preparative HPLC according to Method 17.
'H NMR (400 MHz, DMSO-d6, 6/ppm): 8.20 (d, 2H), 7.60 (d, 2H), 7.34 (t, IH),
7.27 (d, 1H), 7.14
(s, 1H), 7.06 (d, 1H), 6.93 (s, IH), 5.51 (s, 2H), 4.00 (quart, 2H), 2.32 (s,
3H), 1.46 (s, 6H), 1.04 (t,
3H).
LC/MS (Method 6, ESlpos): R, = 1.44 min, m/z = 515 [M+H]+.
Example 89A
Ethyl difluoro{3-[(5-methyl-3-{3-[4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-
5-yl}-1H-pyrazol-
1-yl)methyl]phenyl } acetate
F F O-N
H3CO
PNN I'll N F F
H3C O F
At RT, 814 mg (2.11 mmol, purity 76%) of the compound from Example 53A were
added to a
solution of 546 mg (1.76 mmol) of the compound from Example 22A in 20 ml of
THF. At 0 C,
257 mg (2.29 mmol) of potassium tert-butoxide were then added, and the mixture
was stirred at
RT for 20 h. A further 150 mg (1.34 mmol) of potassium tert-butoxide were then
added, and the
mixture was stirred at RT for another 6 h. After dilution with ethyl acetate,
the mixture was
extracted once with water and the aqueous wash phase was reextracted once with
ethyl acetate.
The combined organic phases were washed once with saturated sodium chloride
solution, dried
over magnesium sulphate, filtered and concentrated. The residue was taken up
in methanol and
purified by means of column chromatography (silica gel, mobile phase:
cyclohexane/ethyl acetate
7:3). Removal of the solvent and drying under reduced pressure gave 125 mg of
a product which,
according to 'H NMR, contained the title compound and the corresponding methyl
ester in a total
proportion of about 60% and was used as such for sebsequent reactions.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-125-
LC/MS (Method 6, ESlpos): Rt = 1.43 min, m/z = 523 [M+H]+.
Example 90A
Diethyl {3-[(5-methyl-3-{3-[4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-yl}-
IH-pyrazol-1-
yl)methyl]phenyl } malonate
H3C
0 0
O-N
N N I FF
H3C O F
0.23 g (1.23 mmol) of sodium hydride (60% dispersion in mineral oil) and 0.20
ml (1.64 mmol) of
diethyl carbonate were initially charged in 5.6 ml of abs. THF. With ice bath
cooling, a solution of
0.40 g (0.82 mmol) of the compound from Example 83A in 1.6 ml of abs. THE was
added
dropwise, and the mixture was then heated at reflux for 2 h. The mixture was
then poured into 50
ml of ice water and extracted twice with 50 ml of ethyl acetate each time. The
combined organic
phases were washed with water and saturated sodium chloride solution, dried
over anhydrous
magnesium sulphate and filtered. The filtrate was freed from the solvent on a
rotary evaporator,
and the residue was purified by means of preparative HPLC (Method 17). This
gave 0.29 g (63%
of theory) of the title compound.
'H NMR (400 MHz, DMSO-d6, S/ppm): 8.20 (d, 2H), 7.59 (d, 2H), 7.38 (t, IH),
7.32 (d, 1H), 7.19
(d, 2H), 6.93 (s, 1H), 5.52 (s, 2H), 4.97 (s, IH), 4.11 (m, 4H), 2.33 (s, 3H),
1.13 (t, 6H).
LC/MS (Method 6, ESlpos): Rt = 1.38 min, m/z = 559 [M+H]+.
Example 91A
Diethyl methyl{3-[(5-methyl-3-{3-[4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-
5-yl}-1H-pyrazol-
1 -yl)methyl]phenyl }malonate
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
- 126 -
H3C\
0 0
CH3 O-N
H3C~0
N 1-1 N I FF
H3C O F
With ice bath cooling, 22 mg (0.55 mmol) of sodium hydride (60% dispersion in
mineral oil) were
added to 0.28 g (0.50 mmol) of the compound from Example 90A in 5.0 m] of abs.
DMF, the
mixture was stirred for 5 min and 34 l (0.55 mmol) of methyl iodide were then
added. The
mixture was stirred at RT for 3 h, a little 1 N hydrochloric acid was added
and the product was
purified directly by means of preparative HPLC (Method 17). This gave 0.25 g
(86% of theory) of
the title compound.
'H NMR (400 MHz, DMSO-d6, 6/ppm): 8.20 (d, 2H), 7.59 (d, 2H), 7.38 (t, 1H),
7.29 (d, 1H), 7.17
(d, 1H), 7.12 (s, 1H), 6.93 (s, 1H), 5.52 (s, 2H), 4.11 (m, 4H), 2.32 (s, 3H),
1.73 (s, 3H), 1.11 (t,
6H).
LC/MS (Method 6, ESlpos): Rt = 1.45 min, m/z = 573 [M+H]+.
Example 92A
5-[5-Methyl-l -(3-{2-methyl-2-[(3-oxido-1,5-dihydro-2,4,3-benzodioxaphosphepin-
3-
yl)oxy]propyl } benzyl)-I H-pyrazol-3-yl]-3-[4-(trifluoromethoxy)phenyl]-1,2,4-
oxadiazole
O
O-PLO O-N
PN
N I F F
H3C CH3
H3 / O F
At RT, 684 mg (2.86 mmol) of 3-diethylamino-1,5-dihydro-2,4,3-
benzodioxaphosphepine and 311
mg (4.45 mmol) of tetrazole were added to a solution of 300 mg (0.635 mmol) of
the compound
from Example 43 in 10 ml of dichloromethane. After the mixture had been
stirred at RT for 10
min, first 40 l of water and then, at -40 C, 1.1 g (4.45 mmol) of 70%
strength meta-
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
- 127 -
chloroperbenzoic acid (mCPBA) were added. The mixture was initially stirred at
-40 C for a
further 10 min, and the cooling bath was then removed and stirring was
continued at RT for 16 h.
50 ml of water were then added, and the mixture was extracted three times with
about 25 ml of
ethyl acetate each time. The combined organic extracts were washed
successively with water and
saturated sodium chloride solution, dried over anhydrous magnesium sulphate,
filtered and finally
freed from the solvent on a rotary evaporator. The residue was purified by
means of MPLC (silica
gel, mobile phase: cyclohexane/ethyl acetate 5:1 -> 1:1). After evaporation of
the appropriate
fractions, the product was redissolved in ethyl actetate and washed with
saturated sodium
bicarbonate solution to remove the last traces of meta-chloroperbenzoic acid.
The solution was
once more dried over anhydrous magnesium sulphate, filtered and evaporated,
giving 255 mg
(59% of theory) of the title compound.
1H NMR (400 MHz, CDC13, S/ppm): 8.25 (d, 2H), 7.37-7.31 (m, 4H), 7.29-7.23 (m,
3H), 7.19 (d,
I H), 7.08 (s, I H), 7.05 (d, I H), 6.77 (s, I H), 5.41 (s, 2H), 5.18-4.98 (m,
4H), 3.04 (s, 2H), 2.23 (s,
3H), 1.53 (s, 6H).
LC/MS (Method 4, ESlpos): R, = 1.55 min, m/z = 455 [M-C8H9O4P+H]+, 655 [M+H]+.
Example 93A
Dibenzyl 2-methyl-2-{3-[(5-methyl-3-{ 3-[4-(1,1,1-trifluoro-2-methylpropan-2-
yl)phenyl]-1,2,4-
oxadiazol-5-yl}-I H-pyrazol-1-yl)methyl]phenyl}propyl phosphate
/ \ 0 H C C H 3 O-N
I I
O-PO PN O N I F F
/
H3C F
H3C CH3
With ice bath cooling, 18 mg (0.44 mmol) of sodium hydride (60% dispersion in
mineral oil) were
added to 0.20 g (0.40 mmol) of the compound from Example 60 in 8 ml of abs.
THF, and the
mixture was stirred at this temperature for 5 min. 0.24 g (0.44 mmol) of
tetrabenzyl pyrophosphate
was then added, and the mixture was stirred without further cooling for about
another 4 h. The
mixture was then diluted with ethyl acetate, washed with water and saturated
sodium chloride
solution, dried over anhydrous magnesium sulphate and filtered. The filtrate
was freed from the
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-128-
solvent on a rotary evaporator, and the residue was purified by means of
preparative HPLC
(Method 17). This gave 0.15 g (49% of theory) of the title compound.
'H NMR (400 MHz, DMSO-d6, 6/ppm): 8.08 (d, 2H), 7.76 (d, 2H), 7.37-7.26 (m,
13H), 6.97 (d,
1H), 6.88 (s, 1H), 5.46 (s, 2H), 4.85 (d, 4H), 3.97 (d, 2H), 2.29 (s, 3H),
1.61 (s, 6H), 1.24 (s, 6H).
LC/MS (Method 6, ESlpos): Rt = 1.56 min, m/z = 759 [M+H]+.
Example 94A
Dibenzyl 2-methyl-2-{3-[(5-methyl-3-{ 3-[4-(trifluoromethoxy)phenyl]-1,2,4-
oxadiazol-5-yl }-1 H-
pyrazol- l -yl)methyl]phenyl } propyl phosphate
0 H 3 C CH3 O-N
_ II
O P N I \
H3C O F
I
Analogously to the process described under Example 93A, 0.10 g (0.21 mmol) of
the compound
from Example 61 gave 92 mg (59% of theory) of the title compound.
'H NMR (400 MHz, DMSO-d6, 6/ppm): 8.18 (d, 2H), 7.58 (d, 2H), 7.35-7.25 (m,
13H), 6.98 (d,
1H), 6.88 (s, 1H), 5.46 (s, 2H), 4.85 (d, 4H), 3.96 (d, 2H), 2.30 (s, 3H),
1.24 (s, 6H).
LC/MS (Method 6, ESlpos): Rt = 1.54 min, m/z = 733 [M+H]+.
Example 95A
1-{ 3-[(3-{ 3-[3-Fluoro-4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-yl }-5-
methyl-I H-pyrazol-l -
yl)methyl]phenyl}cyclopropyl acetate
O O-N
H CO PN \ F
s N
H3C 0 F
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-129-
Analogously to the process described in Example 66A, 250 mg (0.762 mmol) of
the compound
from Example 29A and 249 mg (0.838 mmol, purity 96%) of the compound from
Example 47A
gave 411 mg (72% pure, 75% of theory) of the title compound. In this case, the
reaction time was
16hatRT.
LC/MS (Method 6, ESlpos): R, = 1.39 min, m/z = 517 [M+H]+.
Example 96A
5-[5-Methyl- l -(3 -{2-methyl-2-[(3-oxido-1,5-dihydro-2,4,3-
benzodioxaphosphepin-3-
yl)oxy]propyl} benzyl)-1 H-pyrazol-3-yl]-3-[4-
(trifluoromethyl)sulphanyl]phenyl }-1,2,4-oxadiazole
O
O-PLO O-N
O PN
7(yL
N I F F
CH3
H3C / S X F
At RT, 1.10 g (4.61 mmol) of 3-diethylamino-1,5-dihydro-2,4,3-
benzodioxaphosphepine and 502
mg (7.16 mmol) of tetrazole were added to a solution of 500 mg (1.02 mmol) of
the compound
from Example 53 in 16 ml of dichloromethane. After the mixture had been
stirred at RT for 10
min, first 65 l of water and then, at -40 C, 1.77 g (7.16 mmol) of 70%
strength meta-
chloroperbenzoic acid (mCPBA) were added. The mixture was initially stirred at
-40 C for a
further 10 min, and the cooling bath was then removed and stirring was
continued at RT for 16 h.
100 ml of water were then added, and the mixture was extracted three times
with about 50 ml of
ethyl acetate each time. The combined organic extracts were washed
successively with saturated
sodium bicarbonate solution, water and saturated sodium chloride solution,
dried over anhydrous
magnesium sulphate, filtered and finally freed from the solvent on a rotary
evaporator. The residue
was purified by means of MPLC (silica gel, mobile phase: cyclohexane/ethyl
acetate 5:1 -p 1:1).
Evaporation of the appropriate fractions and drying of the residue under high
vacuum gave 618 mg
(85% of theory, 94% pure) of the title compound.
'H NMR (400 MHz, CDC13, 8/ppm): 8.27 (d, 2H), 7.79 (d, 2H), 7.35-7.31 (m, 3H),
7.29-7.23 (m,
2H, partially covered by CHC13 signal), 7.20 (d, 1H), 7.08 (s, IH), 7.05 (d,
1H), 6.78 (s, 1H), 5.41
(s, 2H), 5.17-4.99 (m, 4H), 3.03 (s, 2H), 2.23 (s, 3H), 1.53 (s, 6H).
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-130-
LC/MS (Method 6, ESlpos): Rt = 1.42 min, m/z = 471 [M-C8H9O4P+H]+, 671 [M+H]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-131-
Working examples:
Example 1
{ 3-[(5-Methyl-3-{3-[4-(1,1,1-trifluoro-2-methylpropan-2-yl)phenyl]-1,2,4-
oxadiazol-5-yl}-1 H-
pyrazol-l-yl)methyl]phenyl }methanol
0-N
HO PNN \
I N I F F
H3C / F
H 3 C C H 3
At 0 C, 7.83 mg (0.21 mmol) of lithium aluminium hydride were added to 100 mg
(0.21 mmol) of
the compound from Example 54A in 2 ml of abs. THF, and the mixture was stirred
with ice bath
cooling for another 15 min. Saturated aqueous ammonium chloride solution,
anhydrous
magnesium sulphate and ethyl acetate were than added in succession. The
mixture was then
filtered, and the filtrate was evaporated on a rotary evaporator. The residue
was purified by
preparative HPLC. This gave 60 mg (64% of theory) of the title compound.
'H NMR (400 MHz, DMSO-d6, 8/ppm): 8.09 (d, 2H), 7.77 (d, 2H), 7.32 (t, 1H),
7.24 (d, IH), 7.18
(s, IH), 7.06 (d, IH), 6.93 (s, I H), 5.50 (s, 2H), 5.20 (t, I H), 4.47 (d,
2H), 2.34 (s, 3H), 1.61 (s,
6H).
LC/MS (Method 6, ESlpos): Rt = 1.25 min, m/z = 457 [M+H]+.
Example 2
2,2,2-Tri fluoro-1-{3-[(5-methyl-3-{ 3-[4-(trifluoromethoxy)phenyl]-1,2,4-
oxadiazol-5-yl }-1 H-
pyrazol- l -yl)methyl]phenyl } ethanol (racemate)
F
F F
O-N
PNN
HO lo-I
N F\ F
H3C 0 F
9.5 mg (0.249 mmol) of solid sodium borohydride were added to a solution of
200 mg (0.383
mmol) of the compound from Example 55A in a mixture of 10 ml of 1,4-dioxane
and I ml of
water. After the reaction mixture had been stirred at RT for 1 h, first 2 ml
of 1 M hydrochloric acid
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-132-
and then about 60 ml of water were added. The mixture was extracted three time
with about 60 ml
of ethyl acetate each time. The combined organic extracts were washed with
saturated sodium
chloride solution, dried over anhydrous magnesium sulphate, filtered and
finally freed from the
solvent on a rotary evaporator. The crude product obtained in this manner was
triturated with a
mixture of 25 ml of pentane and 1 ml of dichloromethane. Filtration with
suction and drying of the
residue under high vacuum gave 185 mg (94% of theory, 97% pure) of the title
compound.
'H NMR (400 MHz, CDC13, 6/ppm): 8.24 (d, 2H), 7.43 (d, 1H), 7.39 (t, 1H), 7.34
(s, IH), 7.32 (d,
2H), 7.19 (d, IH), 6.82 (s, 1H), 5.48 (s, 2H), 5.02 (s, 1H), 2.99 (s, broad,
IH), 2.27 (s, 3H).
LC/MS (Method 6, ESlpos): R, = 1.29 min, m/z = 499 [M+H]+.
Example 3
1,1,1-Tri fluoro-2-{3-[(5-methyl-3-{ 3-[4-(tri fluoromethoxy)phenyl]-1,2,4-
oxadiazol-5-yl }-I H-
pyrazol-l-yl)methyl]phenyl}propan-2-ol (racemate)
F F
H3C F O-N
HO PN N F\ /F
H3C O F
At -30 C, 731 l (0.950 mmol) of a 1.3 M solution of isopropylmagnesium
chloride/lithium
chloride complex in THE were added rapidly to a solution of 250 mg (0.475
mmol) of the
compound from Example 81A in 10 ml of anhydrous THF. After 30 min at -30 C,
266 mg (2.37
mmol) of 1,1,1-trifluoroacetone were added, and the cooling bath was removed.
After the reaction
mixture had warmed to RT, about 1 ml of methanol was added. The reaction
mixture was then
dried over anhydrous magnesium sulphate, filtered and finally freed from the
solvent on a rotary
evaporator. The residue obtained was first freed from unpolar impurities by
means of MPLC
(about 50 g of silica gel, mobile phase: cyclohexane/ethyl acetate 4:1).
Further purification of the
product was then carried out by means of preparative HPLC (Method 13). This
gave 49 mg (11%
of theory) of the title compound.
'H NMR (400 MHz, CDC13, 6/ppm): 8.25 (d, 2H), 7.51 (d, 1H), 7.47 (s, 1H), 7.37
(t, 1H), 7.33 (d,
2H), 7.14 (d, 1H), 6.83 (s, 1H), 5.49 (s, 2H), 2.70 (s, broad, IH), 2.28 (s,
3H), 1.77 (s, 3H).
LC/MS (Method 6, ESlpos): Rt = 1.31 min, m/z = 513 [M+H]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-133-
Example 4
2-{3-[(5-Methyl-3-{ 3-[4-(tetrahydro-2H-pyran-4-yl)phenyl]-1,2,4-oxadiazol-5-
yl}-1 H-pyrazol- l -
yl)methyl]phenyl } propan-2-ol
H 3 C CH3 O-N
N)
HO \ \
N
H3C
O
At 0 C, 153 l of (0.459 mmol) of methylmagnesium chloride solution (3 M in
THF) were added
dropwise to a solution of 100 mg (0.218 mmol) of the compound from Example 56A
in 2.2 ml of
anhydrous THF. The ice/water bath was then removed, and stirring was continued
at RT. After 3 h,
about 5 drops of saturated aqueous ammonium chloride solution were added. The
mixture was
diluted with about 10 ml of ethyl acetate, and anhydrous magnesium sulphate
was then added in
such an amount that the aqueous phase was taken up completely. The mixture was
then filtered and
the filtrate was freed from the solvent on a rotary evaporator. The residue
gave, by means of
preparative HPLC (Method 11), 36 mg of the title compound (66% of theory,
based on
conversion), and 45 mg of the starting material were recovered.
'H NMR (400 MHz, DMSO-d6, S/ppm): 8.01 (d, 2H), 7.48 (d, 2H), 7.41 (s, 1H),
7.38 (d, IH), 7.28
(t, I H), 6.97 (d, I H), 6.91 (s, I H), 5.49 (s, 2H), 5.01 (s, I H), 3.99-3.94
(m, 2H), 3.46 (dt, 2H),
2.91-2.83 (m, 1H), 2.34 (s, 3H), 1.78-1.65 (m, 4H), 1.40 (s, 6H).
LC/MS (Method 6, ESlpos): Rt = 1.19 min, m/z = 459 [M+H]+.
Example 5
2-[3-({ 3-[3-(4-tert-Butylphenyl)-1,2,4-oxadiazol-5-yl]-5-methyl-I H-pyrazol-l
-
yl)methyl)phenyl]propan-2-ol
H 3 C CH3 O-N
HO PN
CH3
H3C
H3C CH3
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-134-
Analogously to the process described under Example 4, 100 mg (0.232 mmol) of
the compound
from Example 57A and methylmagnesium chloride gave 66 mg (66% of theory) of
the title
compound.
'H NMR (400 MHz, CDC13, 6/ppm): 8.13 (d, 2H), 7.51 (d, 2H), 7.41 (d, 1H), 7.37
(s, IH), 7.30 (t,
1H), 7.01 (d, 1H), 6.81 (s, 1H), 5.47 (s, 2H), 2.29 (s, 3H), 1.73 (s, 1H),
1.55 (s, 6H), 1.37 (s, 9H).
LC/MS (Method 4, ESlpos): R, = 1.54 min, m/z = 413 [M-H2O+H]+.
Example 6
2-(3-{ [3-(3-{4-[ 1-(Methoxymethyl)cyclobutyl]phenyl }-1,2,4-oxadiazol-5-yl)-5-
methyl-I H-pyrazol-
1-yl]methyl } phenyl)propan-2-ol
H3C CH3 O-N
PN
H
ON
H C O"CH3
3
Analogously to the process described under Example 4, 150 mg (0.317 mmol) of
the compound
from Example 58A and methylmagnesium chloride gave 114 mg (75% of theory) of
the title
compound. Here, the reaction was terminated after 3 h at RT by addition of 100
l of acetic acid.
The reaction mixture was then evaporated to dryness on a rotary evaporator.
The title compound
was isolated from the residue obtained by means of MPLC (silica gel, mobile
phase:
cyclohexane/ethyl acetate 5:1 -> 3:1).
'H NMR (400 MHz, CDC13, 6/ppm): 8.13 (d, 2H), 7.40 (d, 1H), 7.36 (s, IH), 7.30
(t, 1H), 7.29 (d,
2H), 7.01 (d, 1H), 6.80 (s, 1H), 5.47 (s, 2H), 3.56 (s, 2H), 3.28 (s, 3H),
2.43-2.28 (m, 4H), 2.29 (s,
3H), 2.15-2.03 (m, IH), 1.93-1.83 (m, 1H), 1.74 (s, 1H), 1.56 (s, 6H).
LC/MS (Method 4, ESlpos): R, = 1.49 min, m/z = 455 [M-H2O+H]+.
Example 7
2-(3-{ [3-(3-{ 3-Fluoro-4-[ I-(methoxymethyl)cyclobutyl]phenyl }-1,2,4-
oxadiazol-5-yl)-5-methyl-
I H-pyrazol-1-yl]methyl } phenyl)propan-2-ol
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-135-
H3C CH3 O-N
HONA \ \ F
- N
/ H C O~CH3
3
Analogously to the process described under Example 4, 150 mg (0.306 mmol) of
the compound
from Example 59A and methylmagnesium chloride gave 120 mg (80% of theory) of
the title
compound.
'H NMR (400 MHz, CDC13, S/ppm): 7.93 (dd, 1H), 7.82 (dd, 1H), 7.40 (d, IH),
7.37 (s, 1H), 7.30
(t, I H), 7.22 (d, IH), 7.00 (d, I H), 6.81 (s, I H), 5.47 (s, 2H), 3.66 (s,
2H), 3.28 (s, 3H), 2.48-2.40
(m, 2H), 2.37-2.30 (m, 2H), 2.29 (s, 3H), 2.19-2.07 (m, 1H), 1.94-1.85 (m,
1H), 1.73 (s, IH), 1.55
(s, 6H).
LC/MS (Method 6, ESlpos): R, = 1.36 min, m/z = 473 [M-H2O+H]+, 491 [M+H]+.
Example 8
2-(3-{ [3-(3-{4-[ 1-(Methoxymethyl)cyclopentyl]phenyl }-1,2,4-oxadiazol-5-yl)-
5-methyl-1 H-
pyrazol-1-yl]methyl } phenyl)prop an-2-ol
H 3 C CH3 O-N
PN
H
ON
H C OCH3
3C
Analogously to the process described under Example 4, 145 mg (0.298 mmol) of
the compound
from Example 60A and methylmagnesium chloride gave 119 mg (82% of theory) of
the title
compound.
'H NMR (400 MHz, CDC13, 8/ppm): 8.12 (d, 2H), 7.96 (d, 2H), 7.40 (d, IH), 7.37
(s, 1H), 7.30 (t,
IH), 7.01 (d, IH), 6.81 (s, 1H), 5.47 (s, 2H), 3.41 (s, 2H), 3.22 (s, 3H),
2.29 (s, 3H), 2.07-2.00 (m,
2H), 1.96-1.88 (m, 2H), 1.82 (s, 1H), 1.78-1.70 (m, 4H), 1.55 (s, 6H).
LC/MS (Method 6, ESlpos): R, = 1.37 min, m/z = 469 [M-H2O+H]+, 487 [M+H]+.
CA 02798374 2012-11-05
= , BHC 10 1 009 Foreign Countries
-136-
Example 9
2-(3-{ [3-(3-{4-[ 1-(Ethoxymethyl)cyclobutyl]phenyl }-1,2,4-oxadiazol-5-yl)-5-
methyl-1 H-pyrazol-
1-yl]methyl}phenyl)propan-2-ol
H3C CH3 O-N
HO p
N
H3C O CH3
Analogously to the process described under Example 4, 36.5 mg (0.075 mmol) of
the compound
from Example 61A and methylmagnesium chloride gave 30.8 mg (84% of theory) of
the title
compound.
'H NMR (400 MHz, CDC13, S/ppm): 8.13 (d, 2H), 7.41 (d, 1H), 7.36 (s, 1H), 7.30
(t, 1H), 7.29 (d,
2H), 7.01 (d, 1H), 6.81 (s, 1H), 5.47 (s, 2H), 3.57 (s, 2H), 3.38 (quart, 2H),
2.41-2.30 (m, 4H), 2.28
(s, 3H), 2.15-2.03 (m, 1H), 1.92-1.82 (m, IH), 1.76 (s, 1H), 1.55 (s, 6H),
1.10 (t, 3H).
LC/MS (Method 2, ESlpos): R, = 2.83 min, m/z = 469 [M-H2O+H]+, 487 [M+H]+.
Example 10
2-(3-{ [5-Methyl-3-(3-{4-[(trifluoromethyl)sulphanyl]phenyl} -1,2,4-oxadiazol-
5-yl)-I H-pyrazol-l -
yl]methyl } phenyl)propan-2-ol
H 3 C CH3 O-N
HO PN N F\ F
Fi3C / S F
Analogously to the process described under Example 4, 100 mg (0.211 mmol) of
the compound
from Example 62A and methylmagnesium chloride gave 76 mg (77% of theory) of
the title
compound. In this case, the reaction time at RT was 1.5 h.
'H NMR (400 MHz, CDC13, S/ppm): 8.27 (d, 2H), 7.78 (d, 2H), 7.41 (d, 1H), 7.37
(s, 1H), 7.30 (t,
IH), 7.01 (d, IH), 6.82 (s, 1H), 5.47 (s, 2H), 2.29 (s, 3H), 1.74 (s, 1H),
1.55 (s, 6H).
LC/MS (Method 4, ESIpos): R, = 1.53 min, m/z = 457 [M-H2O+H]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-137-
Example 11
2-{ 3-[(3-{3-[4-(4-Fluorotetrahydro-2H-pyran-4-yl)phenyl]-1,2,4-oxadiazol-5-
yl}-5-methyl-1 H-
pyrazol- l -yl)methyl]phenyl }propan-2-ol
H3C CH3 0-N
PN
N \ HO H3C
0
Analogously to the process described under Example 4, 100 mg (0.210 mmol) of
the compound
from Example 63A and methylmagnesium chloride gave 83 mg (83% of theory) of
the title
compound. In this case, the reaction time at RT was 1.5 h.
'H NMR (400 MHz, CDC13, S/ppm): 8.23 (d, 2H), 7.52 (d, 2H), 7.41 (d, IH), 7.37
(s, IH), 7.30 (t,
I H), 7.01 (d, I H), 6.82 (s, I H), 5.47 (s, 2H), 4.00-3.87 (m, 4H), 2.29 (s,
3H), 2.29-2.21 (m, 2H),
1.98-1.91 (m, 2H), 1.76 (s, 1H), 1.55 (s, 6H).
LC/MS (Method 6, ESIpos): Rt = 1.16 min, m/z = 459 [M-H2O+H]+, 477 [M+H]+.
Example 12
2-{ 3-[(3-{ 3-[3-Fluoro-4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-yl} -5-
methyl-1 H-pyrazol- l -
yl)methyl]phenyl } propan-2-ol
H3C CH3 O-N
110 PN \ \ \ F
N F\ F
H3C 0 F
At RT, 58 mg (0.52 mmol) of potassium tert-butoxide, followed by a solution of
147 mg (0.60
mmol) of the compound from Example 46A in 1.5 ml of THF, were added to a
solution of 131 mg
(0.40 mmol) of the compound from Example 29A in 2 ml of THF. The mixture was
stirred at RT
for 1 h. 30 ml of water were then added, and the mixture was extracted three
times with 30 ml of
ethyl acetate each time. The combined organic phases were dried over sodium
sulphate, filtered
and concentrated. The residue was purified by means of preparative HPLC
(Method 14). The clean
product fractions were combined and concentrated to a residual volume of
aqueous phase. The pH
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-138-
was adjusted to 7 by addition of saturated aqueous sodium bicarbonate
solution, and the aqueous
phase was then extracted twice with 30 ml of ethyl acetate each time. The
combined organic
phases were then dried over sodium sulphate, filtered and concentrated. This
gave 191 mg (53% of
theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 8.08 (dd, 1H), 8.06-8.01 (m, 1H), 7.46-7.39
(m, 2H), 7.37 (s,
1 H), 7.30 (t, 1 H), 7.01 (d, 1 H), 6.82 (s, 1 H), 5.47 (s, 2H), 2.29 (s, 3H),
1.56 (s, 6H).
LC/MS (Method 6, ESlpos): R, = 1.33 min, m/z = 477 [M+H]+.
Example 13
2-{ 3-[(3-{3-[4-(2-Fluoropropan-2-yl)phenyl]-1,2,4-oxadiazol-5-yl }-5-methyl-I
H-pyrazol-l -
yl)methyl]phenyl}propan-2-ol
H3C CH3 O-N
HO N~
F
H3C
H3C CH3
Analogously to the process described in Example 12, 114 mg (0.40 mmol) of the
compound from
Example 24A and 147 mg (0.60 mmol) of the compound from Example 46A were used
to obtain
81 mg (43% of theory) of the title compound. Here, purification by preparative
HPLC was carried
out according to Method 15.
'H NMR (400 MHz, CDC13, S/ppm): 8.20 (d, 2H), 7.50 (d, 2H), 7.43-7.38 (d, 1H),
7.36 (s, 1H),
7.30 (t, 1H), 7.03-6.98 (d, IH), 6.82 (s, 1H), 5.47 (s, 2H), 2.29 (s, 3H),
1.75 (s, 3H), 1.70 (s, 3H),
1.55 (s, 6H).
LC/MS (Method 2, ESlpos): Rt = 2.59 min, m/z = 417 [M-H2O+H]+.
Example 14
2-(3-{ [5-Methyl-3-(3-{4-[(trifluoromethyl)sulphonyl]phenyl}-1,2,4-oxadiazol-5-
yl)-I H-pyrazol-l -
yl] methyl } phenyl)propan-2-ol
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-139-
H 3 Q CH3 O-N
HO p \
N F F
H3C USX F
O O
Analogously to the process described in Example 12, 143 mg (0.40 mmol) of the
compound from
Example 25A and 147 mg (0.60 mmol) of the compound from Example 46A were used
to obtain
78 mg (37% of theory, purity 95%) of the title compound. Here, purification by
preparative HPLC
was carried out according to Method 15.
'H NMR (400 MHz, CDCI3, 6/ppm): 8.52 (d, 2H), 8.18 (d, 2H), 7.43-7.36 (m, 2H),
7.31 (t, 1H),
7.01 (d, 1H), 6.84 (s, 1H), 5.48 (s, 2H), 2.31 (s, 3H), 1.56 (s, 6H).
LC/MS (Method 2, ESlpos): R, = 2.63 min, m/z = 489 [M-H2O+H]+.
Example 15
2-(3-{[5-Methyl-3-(3-{4-[1-(trifluoromethyl)cyclopropyl]phenyl}-1,2,4-
oxadiazol-5-yl)-1H-
pyrazol-1-yl]methyl } phenyl)propan-2-ol
H3C CH3 O-N
HO/\ PN
N F F
H3C F
Analogously to the process described in Example 12, 134 mg (0.40 mmol) of the
compound from
Example 30A and 147 mg (0.60 mmol) of the compound from Example 46A were used
to obtain
103 mg (50% of theory, purity 95%) of the title compound. Here, purification
by preparative
HPLC was carried out according to Method 15.
'H NMR (400 MHz, CDC13, S/ppm): 8.18 (d, 2H), 7.58 (d, 2H), 7.39 (d, IH), 7.36
(s, 1H), 7.30 (t,
1H), 7.01 (d, IH), 6.81 (s, 1H), 5.46 (s, 2H), 2.29 (s, 3H), 1.55 (s, 6H),
1.43-1.37 (m, 2H), 1.12-
1.05 (m, 2H).
LC/MS (Method 2, ESIpos): R, = 2.74 min, m/z = 463 [M-H2O+H]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-140-
Example 16
2-{ 3-[(5-Methyl-3-{ 3-[4-(trimethylsilyl)phenyl]-1,2,4-oxadiazol-5-yl }-1 H-
pyrazol-l -
yl)methyl]phenyl} propan-2-ol
H 3 C CH3 O-N
PN
H
O N
HC Si CH 3
3
H3C CH3
Analogously to the process described in Example 12, 120 mg (0.40 mmol) of the
compound from
Example 26A and 147 mg (0.60 mmol) of the compound from Example 46A were used
to obtain
89 mg (46% of theory, purity 92%) of the title compound. Here, purification by
preparative HPLC
was carried out according to Method 15.
'H NMR (400 MHz, CDC13, 6/ppm): 8.17 (d, 2H), 7.64 (d, 2H), 7.40 (d, 1H), 7.36
(s, 1H), 7.30 (t,
1H), 7.00 (d, 1H), 6.82 (s, 1H), 5.47 (s, 2H), 2.28 (s, 3H), 1.55 (s, 6H),
0.31 (s, 9H).
LC/MS (Method 2, ESlpos): Rt = 2.94 min, m/z = 447 [M+H]+.
Example 17
2-{ 3-[(5-Methyl-3-{ 3-[4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-yl }-I H-
pyrazol- l -
yl)methyl]phenyl } propan-2-ol
H 3 C CH3 O-N
.01 N
HO N N F F
H3C O F
Analogously to the process described under Example 4, 150 mg (0.33 mmol) of
the compound
from Example 64A and methylmagnesium bromide solution (1.4 M in THE/toluene)
gave 51 mg
(34% of theory) of the title compound. In this case, the reaction time at RT
was 1 h, and
purification of the crude product by preparative HPLC was carried out
according to Method 17.
'H NMR (400 MHz, DMSO-d6, 6/ppm): 8.20 (d, 2H), 7.59 (d, 2H), 7.41 (s, IH),
7.38 (d, IH), 7.28
(t, 1 H), 6.97 (d, I H), 6.93 (s, 1 H), 5.49 (s, 2H), 5.01 (s, I H), 2.34 (s,
3H), 1.40 (s, 6H).
LC/MS (Method 6, ESIpos): R, = 1.31 min, m/z = 459 [M+H]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-141-
Example 18
2-{3-[(5-Methyl-3-{3-[4-(1,1,1-trifluoro-2-methylpropan-2-yl)phenyl]-1,2,4-
oxadiazol-5-yl }-1 H-
pyrazol-1-yl)methyl]phenyl } propan-2-ol
H3C CH3 O-N
HO PN
N F F
H3C F
H3C CH3
At 0 C, 15.9 ml (47.7 mmol) of methylmagnesium chloride solution (3 M in THF)
were added
dropwise to a solution of 11.0 g (22.7 mmol) of the compound from Example 54A
in 220 ml of
anhydrous THF. The ice/water bath was then removed, and stirring was continued
at RT. After I h,
about 100 ml of saturated aqueous ammonium chloride solution and 1.5 litres of
water were added.
The mixture was extracted three time with about 500 ml of ethyl acetate each
time. The combined
organic extracts were washed with saturated sodium chloride solution, dried
over anhydrous
magnesium sulphate, filtered and finally freed from the solvent on a rotary
evaporator. The crude
product obtained was purified by filtration with suction through about 330 g
of silica gel using the
mobile phase cyclohexane/ethyl acetate 4:1 -> 1:1. After the solvent had been
removed from the
combined product fractions, the product was crystallized from 120 ml of
ethanol/water (2:1)
(boiling point -- RT). This gave, after filtration and drying under high
vacuum, 5.79 g (53% of
theory) of the title compound.
Melting point: 167 C
'H NMR (400 MHz, CDC13, 6/ppm): 8.20 (d, 2H), 7.62 (d, 2H), 7.41 (d, IH), 7.37
(s, IH), 7.31 (t,
1H), 7.01 (d, 1H), 6.83 (s, 1H), 5.47 (s, 2H), 2.29 (s, 3H), 1.75 (s, IH),
1.62 (s, 6H), 1.55 (s, 6H).
LC/MS (Method 6, ESlpos): R, = 1.32 min, m/z = 467 [M-H2O+H]+, 485 [M+H]+.
Example 19
2-{3-[(3-{3-[4-(1-Fluorocyclobutyl)phenyl]-1,2,4-oxadiazol-5-yl}-5-methyl-I H-
pyrazol-l -
yl)methyl] phenyl } propan-2-ol
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-142-
H3 Q CH3 O-N
HO N~ \
N
I~ - I F
H3C
Analogously to the process described under Example 4, 180 mg (0.40 mmol) of
the compound
from Example 65A and methylmagnesium chloride gave 70 mg (40% of theory) of
the title
compound. In this case, the reaction time at RT was 1 h, and purification of
the crude product by
preparative HPLC was carried out according to Method 17.
'H NMR (400 MHz, DMSO-d6, S/ppm): 8.12 (d, 2H), 7.70 (d, 2H), 7.41 (s, 1H),
7.38 (d, 1H), 7.28
(t, 1H), 6.96 (d, 1H), 6.94 (s, IH), 5.49 (s, 2H), 5.03 (s, 1H), 2.64 (t, 2H),
2.59 (t, 2H), 2.35 (s, 3H),
2.07 (m, 1 H), 1.77 (m, 1 H), 1.40 (s, 6H).
LC/MS (Method 6, ESIpos): R, = 1.29 min, m/z = 447 [M+H]+.
Example 20
1-[3-({3-[3-(4-tert-Butylphenyl)-1,2,4-oxadiazol-5-yl]-5-methyl-I H-pyrazol- l
-
yl}methyl)phenyl]cyclopropanol
0-N
HO N",I N 1'1~ V V / CH3
H3C
H3C CH3
250 mg (0.532 mmol) of the compound from Example 66A were dissolved in 5 ml of
anhydrous
THF, and a total of 2.2 m] of ethylmagnesium chloride solution (2 M in THF)
were added
dropwise at RT. The progress of the reaction was monitored by analytical HPLC,
and only the
amount of Grignard reagent required for complete conversion was added. 2 ml of
water were then
added. Once most of the solvent and the water had been removed on a rotary
evaporator, the
resulting residue was purified by preparative HPLC (Method 15). The clean
product fractions were
combined and concentrated to a residual volume of aqueous phase. After
addition of saturated
aqueous sodium bicarbonate solution, the aqueous phase was extracted twice
with 30 ml of ethyl
acetate each time. The combined organic phases were then dried over magnesium
sulphate, filtered
and concentrated. This gave 97 mg (32% of theory) of the title compound.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-143-
'H NMR (400 MHz, CDC13, 6/ppm): 8.13 (d, 2H), 7.50 (d, 2H), 7.29 (d, IH), 7.21-
7.14 (m, 2H),
6.98 (d, 1H), 6.80 (s, 1H), 5.44 (s, 2H), 2.27 (s, 3H), 1.36 (s, 9H), 1.29-
1.23 (m, 3H), 1.01 (dd,
2H).
LC/MS (Method 4, ESlpos): R, = 1.53 min, m/z = 429 [M+H]+.
Example 21
1-{3-[(5-Methyl-3-{ 3-[4-(tetrahydro-2H-pyran-4-yl)phenyl]-1,2,4-oxadiazol-5-
yl }-1 H-pyrazol- l -
yl)methyl]phenyl}cyclopropanol
O-N
HO PN
N
H3C
O
At 0 C, 1.6 ml (3.21 mmol) of ethylmagnesium chloride solution (2 M in THF)
were added slowly
to a solution of 160 mg (0.25 mmol, 79% pure) of the compound from Example 67A
in 5 ml of
THF. The mixture was allowed to warm to RT and stirred at RT for 30 min. After
removal of the
solvent, the residue was purified by means of column chromatography (silica
gel, mobile phase:
cyclohexane/ethyl acetate 6:4). The combined product fractions were
concentrated, and the residue
was re-purified by preparative thin-layer chromatography (silica gel, mobile
phase:
cyclohexane/ethyl acetate 1:1). The product zone was extracted with
dichloromethane/methanol
(95:5). Removal of the solvent gave 15 mg (12% of theory, purity 94%) of the
title compound.
'H NMR (400 MHz, CDC13, 8/ppm): 8.15 (d, 2H), 7.35 (d, 2H), 7.31-7.26 (m, IH),
7.21-7.14 (m,
2H), 6.99 (d, 1H), 6.81 (s, 1H), 5.45 (s, 2H), 4.13-4.06 (m, 2H), 3.55 (td,
2H), 2.89-2.78 (m, 1H),
2.28 (s, 3H), 1.92-1.77 (m, 4H), 1.29-1.24 (m, 2H), 1.04-0.99 (m, 2H).
LC/MS (Method 6, ESlpos): R, = 1.15 min, m/z = 457 [M+H]+.
Example 22
1-{3-[(5-Methyl-3-{3-[4-(trimethylsilyl)phenyl]-1,2,4-oxadiazol-5-yl}-1H-
pyrazol-l-
yl)methyl]phenyl } cyclopropanol
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-144-
O-N
HO N~
N
I
/ Si,CH3
H3C
H3C CH3
At 0 C, 2.4 ml (4.73 mmol) of ethylmagnesium chloride solution (2 M in THF)
were added slowly
to a solution of 230 mg (0.43 mmol, 90% pure) of the compound from Example 68A
in 7.5 m] of
THF. The mixture was allowed to warm to RT and stirred at RT for 30 min. The
reaction was then
once more cooled to 0 C, and 5 ml of water, followed by 5 ml of I M
hydrochloric acid, were
added dropwise. The mixture was diluted further with water and extracted twice
with ethyl acetate.
The combined ethyl acetate phases were washed once with saturated sodium
chloride solution,
dried over magnesium sulphate, filtered and concentrated. The residue was
purified by means of
preparative HPLC (Method 15). The clean product fractions were combined and
concentrated to a
to residual volume of aqueous phase. After addition of saturated aqueous
sodium bicarbonate
solution, the mixture was extracted twice with ethyl acetate, and the combined
organic phases were
dried over magnesium sulphate, filtered and concentrated. Drying of the
residue under reduced
pressure gave 121 mg (58% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 8.17 (d, 2H), 7.64 (d, 2H), 7.30-7.26 (m, 1H),
7.20 (s, IH),
7.16 (d, IH), 6.98 (d, IH), 6.81 (s, IH), 5.45 (s, 2H), 2.28 (s, 3H), 1.29-
1.24 (m, 2H), 1.03-0.99 (m,
2H), 0.31 (s, 9H).
LC/MS (Method 4, ESIpos): R, = 1.59 min, m/z = 445 [M+H]+.
Example 23
1-{ 3-[(5-Methyl-3-{ 3-[4-(1,1,1-trifluoro-2-methylpropan-2-yl)phenyl]-1,2,4-
oxadiazol-5-yl }-1 H-
pyrazol-l-yl)methyl]phenyl}cyclopropanol
O-N
HO PN F F
H3C F
H3C CH3
Analogously to the process described under Example 22, 285 mg (0.48 mmol, 88%
pure) of the
compound from Example 69A and ethylmagnesium chloride solution solution gave
120 mg (52%
of theory) of the title compound.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-145-
'H NMR (400 MHz, CDC13, 6/ppm): 8.19 (d, 2H), 7.62 (d, 2H), 7.28 (t, 1H), 7.20
(s, 1H), 7.16 (d,
I H), 6.99 (d, I H), 6.81 (s, IH), 5.45 (s, 2H), 2.28 (s, 3H), 1.62 (s, 6H),
1.27 (dd, 2H), 1.01 (dd,
2H).
LC/MS (Method 4, ESlpos): R, = 1.49 min, m/z = 483 [M+H]+.
Example 24
1-[3-({ 3-[3-(4-Isopropylphenyl)-1,2,4-oxadiazol-5-yl]-5-methyl-1 H-pyrazol-l -
yl } methyl)phenyl]cyclopropanol
O-N
HO N~
N
CH3
H3C
CH3
Analogously to the process described under Example 22, 359 mg (0.55 mmol, 69%
pure) of the
compound from Example 70A and ethylmagnesium chloride solution gave 126 mg
(55% of theory)
of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 8.12 (d, 2H), 7.34 (d, 2H), 7.29 (d, IH), 7.21-
7.13 (m, 2H),
6.99 (d, IH), 6.80 (s, 1H), 5.44 (s, 2H), 2.97 (sept, 1H), 2.49 (s, IH), 2.27
(s, 3H), 1.31-1.24 (m,
8H), 1.01 (dd, 2H).
LC/MS (Method 6, ESlpos): R, = 1.30 min, m/z = 415 [M+H]+.
Example 25
1-[3-({ 3-[3-(4-Isobutylphenyl)-1,2,4-oxadiazol-5-yl]-5-methyl-I H-pyrazol-l -
yl } methyl)phenyl]cyclopropanol
O-N
PN
H
O N CH3
H3C CH3
Analogously to the process described under Example 22, 380 mg (0.57 mmol, 72%
pure) of the
compound from Example 71A and ethylmagnesium chloride solution gave 132 mg
(54% of theory)
of the title compound.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-146-
'H NMR (400 MHz, CDC13, 8/ppm): 8.10 (d, 2H), 7.31-7.24 (m, 3H), 7.20 (s, 1H),
7.16 (d, 1H),
6.99 (d, IH), 6.80 (s, I H), 5.44 (s, 2H), 2.54 (d, 2H), 2.51 (s, I H), 2.27
(s, 3H), 1.98-1.85 (m, I H),
1.26 (dd, 2H), 1.01 (dd, 2H), 0.93 (d, 6H).
LC/MS (Method 6, ESlpos): R, = 1.37 min, m/z = 429 [M+H]+.
Example 26
1-(3-{ [5-Methyl-3-(3-{4-[ I-(trifluoromethyl)cyclopropyl]phenyl }-1,2,4-
oxadiazol-5-yl)-1 H-
pyrazol-1-yl]methyl } phenyl)cyclopropanol
O-N
HO PN \
N F F
H3C F
Analogously to the process described under Example 22, 400 mg (0.54 mmol, 70%
pure) of the
compound from Example 72A and ethylmagnesium chloride solution gave 118 mg
(46% of theory)
of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 8.18 (d, 2H), 7.58 (d, 2H), 7.31-7.27 (m, 1H),
7.20 (s, 1H),
7.16 (d, 1H), 6.99 (d, 1H), 6.81 (s, IH), 5.45 (s, 2H), 2.42 (s, 1H), 2.28 (s,
3H), 1.40 (dd, 2H), 1.27
(dd, 2H), 1.12-1.05 (m, 2H), 1.01 (dd, 2H).
LC/MS (Method 6, ESIpos): R, = 1.29 min, m/z = 481 [M+H]+.
Example 27
1-{ 3-[(5-Methyl-3-{ 3-[4-(pentafluoro-X6-sulphanyl)phenyl]-1,2,4-oxadiazol-5-
yl }-1 H-pyrazol- l -
yl)methyl]phenyl) cyclopropanol
O-N
HO ~ - N F
Vlo N~
/ IoF
H3C F"I I 'F
t-
Analogously to the process described under Example 22, 422 mg (0.59 mmol, 75%
pure) of the
compound from Example 73A and ethylmagnesium chloride solution gave 107 mg
(35% of theory,
purity 94%) of the title compound.
CA 02798374 2012-11-05
= . BHC 10 1 009 Foreign Countries
-147-
'H NMR (400 MHz, CDC13, 8/ppm): 8.31 (d, 2H), 7.89 (d, 2H), 7.32-7.27 (m, 1H),
7.20 (s, 1H),
7.16 (d, 1H), 6.99 (d, 1H), 6.82 (s, IH), 5.46 (s, 2H), 2.39 (s, broad, 1H),
2.29 (s, 3H), 1.27 (dd,
2H), 1.02 (dd, 2H).
LC/MS (Method 6, ESlpos): R, = 1.27 min, m/z = 499 [M+H]+.
Example 28
1-{ 3-[(3-{3-[4-(2-Fluoropropan-2-yl)phenyl]-1,2,4-oxadiazol-5-yl }-5-methyl-1
H-pyrazol-l -
yl)methyl]phenyl} cyclopropanol
O-N
HO PNN \ \
H3C
H3C CH3
Analogously to the process described under Example 22, 405 mg (0.64 mmol, 75%
pure) of the
compound from Example 74A and ethylmagnesium chloride solution gave 145 mg
(52% of theory)
of the title compound.
'H NMR (400 MHz, CDC13, 5/ppm): 8.19 (d, 2H), 7.50 (d, 2H), 7.29 (d, 1H), 7.20
(s, 1H), 7.16 (d,
1H), 6.99 (d, 1H), 6.81 (s, 1H), 5.45 (s, 2H), 2.42 (s, 1H), 2.28 (s, 3H),
1.75 (s, 3H), 1.69 (s, 3H),
1.26 (dd, 2H), 1.01 (dd, 2H).
LC/MS (Method 6, ESlpos): R, = 1.20 min, m/z = 433 [M+H]+.
Example 29
1-{ 3-[(5-Methyl-3-{ 3-[4-(trifluoromethyl)phenyl]-1,2,4-oxadiazol-5-yl }-1 H-
pyrazol-l -
yl)methyl]phenyl } cyclopropanol
O-N
HO PN \
N
F
H3C
F F
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-148-
Analogously to the process described under Example 22, 375 mg (0.58 mmol, 75%
pure) of the
compound from Example 75A and ethylmagnesium chloride solution gave 148 mg
(58% of theory)
of the title compound.
'H NMR (400 MHz, CDC13, 8/ppm): 8.34 (d, 2H), 7.76 (d, 2H), 7.31-7.27 (m, 1H),
7.21 (s, 1H),
7.16 (d, 1 H), 6.99 (d, 1 H), 6.82 (s, 1 H), 5.45 (s, 2H), 2.39 (s, 1 H), 2.29
(s, 3 H), 1.27 (dd, 2H), 1.02
(dd, 2H).
LC/MS (Method 6, ESlpos): R, = 1.25 min, m/z = 441 [M+H]+.
Example 30
1-{3-[(3-{ 3-[4-(4-Fluorotetrahydro-2H-pyran-4-yl)phenyl]-1,2,4-oxadiazol-5-yl
}-5-methyl-1 H-
pyrazol-1-yl)methyl]phenyl } cyclopropanol
O-N
HO PN__'~,
N
F
H3C
O
Analogously to the process described under Example 22, 415 mg (0.60 mmol, 75%
pure) of the
compound from Example 76A and ethylmagnesium chloride solution gave 141 mg
(49% of theory)
of the title compound. Here, preparative HPLC was carried out according to
Method 14.
'H NMR (400 MHz, CDC13, 8/ppm): 8.22 (d, 2H), 7.52 (d, 2H), 7.31-7.27 (m, 1H),
7.20 (s, 1H),
7.16 (d, 1H), 6.99 (d, 1H), 6.81 (s, 1H), 5.45 (s, 2H), 4.00-3.94 (m, 211),
3.94-3.85 (m, 2H), 2.47 (s,
1H), 2.28 (s, 3H), 2.28-2.10 (m, 2H), 1.99-1.89 (m, 2H), 1.27 (dd, 2H), 1.01
(dd, 2H).
LC/MS (Method 6, ESlpos): R, = 1.15 min, m/z = 475 [M+H]+.
Example 31
1-[3-({ 3-[3-(4-Cyclohexylphenyl)-1,2,4-oxadiazol-5-yl]-5-methyl-1 H-pyrazol-1-
yl } methyl)phenyl]cyclopropanol
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
- 149 -
O-N
.'IN
HO N
N
H3C
Analogously to the process described under Example 22, 318 mg (0.24 mmol, 42%
pure) of the
compound from Example 77A and ethylmagnesium chloride solution gave 50 mg (37%
of theory)
of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 8.11 (d, 2H), 7.33 (d, 2H), 7.30-7.26 (m, IH),
7.19 (s, 1H),
7.16 (d, I H), 6.99 (d, I H), 6.80 (s, I H), 5.44 (s, 2H), 2.60-2.51 (m, IH),
2.43 (s, I H), 2.27 (s, 3H),
1.95-1.82 (m, 4H), 1.77 (d, IH), 1.52-1.34 (m, 4H), 1.32-1.22 (m, 3H), 1.01
(dd, 2H).
LC/MS (Method 6, ESlpos): R, = 1.45 min, m/z = 455 [M+H]+.
Example 32
1-(3-{ [5-Methyl-3-(3-{4-[(trifluoromethyl)sulphanyl]phenyl} -1,2,4-oxadiazol-
5-yl)-1 H-pyrazol-l -
yl] methyl } phenyl)cyclopropanol
O-N
HO N N F F
X
H3C S F
Analogously to the process described under Example 22, 395 mg (0.55 mmol, 72%
pure) of the
compound from Example 78A and ethylmagnesium chloride solution gave 125 mg
(46% of theory)
of the title compound. Here, preparative HPLC was carried out according to
Method 14.
'H NMR (400 MHz, CDC13, 3/ppm): 8.26 (d, 2H), 7.78 (d, 2H), 7.29 (t, 1H), 7.20
(s, IH), 7.17 (d,
I H), 6.99 (d, I H), 6.81 (s, I H), 5.45 (s, 2H), 2.31 (s, I H), 2.28 (s, 3H),
1.27 (dd, 2H), 1.02 (dd,
2H).
LC/MS (Method 6, ESlpos): R, = 1.32 min, m/z = 473 [M+H]+.
Example 33
1-{3-[(3-{3-[4-(I -Fluorocyclobutyl)phenyl]-1,2,4-oxadiazol-5-yl}-5-methyl-IH-
pyrazol-l -
yl)methyl]phenyl } cyclopropanol
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
- 150 -
O-N
HO Vl N N - / F
H3C
Analogously to the process described under Example 22, 185 mg (0.24 mmol, 62%
pure) of the
compound from Example 79A and ethylmagnesium chloride solution gave 86 mg (76%
of theory)
of the title compound. Here, preparative HPLC was carried out according to
Method 14.
'H NMR (400 MHz, CDC13, 6/ppm): 8.23 (d, 2H), 7.59 (d, 2H), 7.31-7.27 (m, 1H),
7.20 (s, 1H),
7.16 (d, 1H), 6.99 (d, 1H), 6.82 (s, 1H), 5.45 (s, 2H), 2.78-2.54 (m, 4H),
2.49 (s, 1H), 2.28 (s, 3H),
2.19-2.07 (m, IH), 1.87-1.74 (m, I H), 1.27 (dd, 2H), 1.01 (dd, 2H).
LC/MS (Method 6, ESlpos): R, = 1.25 min, m/z = 445 [M+H]+.
Example 34
1-{3-[(5-Methyl-3-f 3-[4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-yl}-1 H-
pyrazol- l -
yl)methyl]phenyl } cyclopropanol
O-N
HO PN__ \ \
N F\ F
11511
H3C O F
Analogously to the processes described under Example 66A and Example 22, 70 mg
(0.23 mmol)
of the compound from Example 22A, 102 mg (0.345 mmol) of the compound from
Example 47A
and ethylmagnesium chloride solution gave, in two steps, 12 mg (12% of theory)
of the title
compound. Here, preparative HPLC was carried out according to Method 18.
'H NMR (400 MHz, DMSO-d6, 6/ppm): 8.20 (d, 2H), 7.60 (d, 2H), 7.27 (t, 1H),
7.22 (s, 1H), 7.08
(d, 1H), 6.96 (d, IH), 6.93 (s, IH), 5.93 (s, 1H), 5.48 (s, 2H), 2.34 (s, 3H),
1.09 (dd, 2H), 0.92 (dd,
2H).
LC/MS (Method 7, ESlpos): R, = 2.68 min, m/z = 457 [M+H]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-151-
Example 35
1 -{ 3-[(3-{3-[3-Fluoro-4-(1,1,1-trifluoro-2-methylpropan-2-yl)phenyl]-1,2,4-
oxadiazol-5-yl}-5-
methyl-1 H-pyrazol- l -yl)methyl]phenyl } cyclopropanol
O-N
HO \ N~ F
- N I \ F F
/ / /
H3C F
H3C CH3
Analogously to the process described under Example 22, 192 mg (0.25 mmol,
purity 69%) of the
compound from Example 80A and ethylmagnesium chloride solution gave 70 mg (57%
of theory)
of the title compound. Here, to purify the crude product, a preparative thin-
layer chromatography
was carried out first (silica gel, mobile phase: cyclohexane/ethyl acetate
1:1). The product zone
was extracted with dichloromethane/methanol 95:5. Further purification was
carried out by means
of preparative HPLC (Method 15). When the product fractions, with saturated
aqueous sodium
bicarbonate solution added, were concentrated, the title compound precipitated
as a solid which
was filtered off, washed twice with water and dried under reduced pressure.
'H NMR (400 MHz, CDC13, 6/ppm): 7.96 (dd, 1H), 7.91 (dd, 1H), 7.55 (t, IH),
7.32-7.26 (m, 2H),
7.20 (s, I H), 7.16 (d, I H), 6.99 (d, I H), 6.81 (s, I H), 5.45 (s, 2H), 2.47
(s, IH), 2.28 (s, 3H), 1.69
(s, 6H), 1.27 (dd, 2H), 1.02 (dd, 2H).
LC/MS (Method 4, ESlpos): R, = 1.58 min, m/z = 501 [M+H]+.
Example 36
1-{3-[(5-Methyl-3-{ 3-[4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-yl }-1 H-
pyrazol-l -
yl)methyl]phenyl } cyclobutanol
O-N
PN N F\ F
HO
11
H3C O F
0.10 g (0.19 mmol) of the compound from Example 81 A were dissolved in 3.8 ml
of abs. THF, and
0.22 g of isopropylmagnesium chloride/lithium chloride (14% in THF, 0.21 mmol)
was added
dropwise at -40 C. The mixture was then stirred at about -40 C to -30 C for 1
h. 16 tl (0.21 mmol)
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-152-
of cyclobutanone were then added, and the mixture was stirred without further
cooling for 1 h. The
mixture was then concentrated on a rotary evaporator, the residue was
suspended in ethyl acetate
and anhydrous magnesium sulphate was added. The mixture was filtered and the
filtrate was
concentrated on a rotary evaporator. The residue was purified by means of
preparative HPLC
(Method 17). This gave 18 mg (20% of theory) of the title compound.
'H NMR (400 MHz, DMSO-d6, S/ppm): 8.20 (d, 2H), 7.59 (d, 2H), 7.42 (d, 1H),
7.39 (s, 1H), 7.33
(t, 1H), 7.02 (d, 1H), 6.93 (s, 1H), 5.51 (s, 2H), 5.50 (s, 1H), 2.35 (s, 3H),
2.35 (m, 2H), 2.25 (m,
2H), 1.90 (m, 1 H), 1.62 (m, 1 H).
LC/MS (Method 6, ESlpos): Rt = 1.31 min, m/z = 471 [M+H]+.
Example 37
4-{ 3-[(5-Methyl-3-{ 3-[4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-yl}-1 H-
pyrazol-1-
yl)methyl]phenyl } tetrahydro-2H-pyran-4-ol
O
O-N
HO N~ N F F
H3C O F
100 mg (0.19 mmol) of the compound from Example 81A were dissolved in 3.8 ml
of abs. THF,
and 220 mg of isopropylmagnesium chloride/lithium chloride (14% in THF, 0.21
mmol) were
added dropwise at -40 C. The mixture was then stirred at about -40 C to -30 C
for 0.5 h. A
solution of 21 pl (0.21 mmol) of tetrahydro-2H-pyran-4-one in 0.2 ml of abs.
THF was then added
dropwise, and the mixture was stirred without further cooling for 0.5 h.
Saturated aqueous
ammonium chloride solution and ethyl acetate were then added, and the organic
phase was washed
with water and saturated sodium chloride solution, dried over anhydrous
magnesium sulphate and
filtered. The filtrate was evaporated on a rotary evaporator, and the residue
was purified by means
of preparative HPLC (Method 17). This gave 66 mg (66% of theory) of the title
compound.
'H NMR (400 MHz, DMSO-d6, S/ppm): 8.20 (d, 2H), 7.59 (d, 2H), 7.42 (d, 2H),
7.33 (t, 1H), 7.02
(d, IH), 6.93 (s, IH), 5.51 (s, 2H), 5.05 (s, 1H), 3.76 (t, 2H), 3.70 (dd,
2H), 2.35 (s, 3H), 1.93 (td,
2H), 1.50 (d, 2H).
LC/MS (Method 4, ESlpos): R1 = 1.38 min, m/z = 501 [M+H]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-153-
Example 38
2-{ 3-[(5-Methyl-3-{ 3-[4-(1,1,1-trifluoro-2-methylpropan-2-yl)phenyl]-1,2,4-
oxadiazol-5-yl }-I H-
pyrazo l-I-yl)methyl]phenyl } ethanol
O-N
HO NN_ N
N I ~ F F
H3C / F
H3C CH3
0.10 g (0.20 mmol) of the compound from Example 82A were dissolved in 2.0 ml
of abs. THF, and
0.20 ml (0.20 mmol) of a I M solution of lithium aluminium hydride in THF was
added at 0 C.
The mixture was stirred with ice bath cooling for another 5 min. Saturated
aqueous ammonium
chloride solution, anhydrous magnesium sulphate and ethyl acetate were than
added in succession.
The solid was filtered off, and the filtrate was evaporated on a rotary
evaporator. This gave 72 mg
(77% of theory) of the title compound.
'H NMR (400 MHz, DMSO-d6, S/ppm): 8.09 (d, 2H), 7.77 (d, 2H), 7.27 (t, 1H),
7.16 (d, 1H), 7.09
(s, 1H), 6.99 (d, IH), 6.93 (s, IH), 5.47 (s, 2H), 4.62 (t, IH), 3.58 (quart,
2H), 2.70 (t, 2H), 2.34 (s,
3H), 1.61 (s, 6H).
LC/MS (Method 6, ESlpos): R, = 1.27 min, m/z = 471 [M+H]+.
Example 39
1,1,1-Trifluoro-3-{3-[(5-methyl-3-{3-[4-(trifluoromethoxy)phenyl]-1,2,4-
oxadiazol-5-yl}-1 H-
pyrazol-l-yl)methyl]phenyl}propan-2-ol (racemate)
O-N
HO P
N F F
F F
F H3C
O F
At -20 C, 1.1 ml (1.43 mmol) of a 1.3 M solution of isopropylmagnesium
chloride/lithium chloride
complex in THF were added rapidly to a solution of 250 mg (0.475 mmol) of the
compound from
Example 81A in 10 ml of anhydrous THF. After 2 minutes, 1.8 mg (0.01 mmol) of
copper(I) iodide
were added, and after a further minute 205 l (2.37 mmol) of
trifluoromethyloxirane. The reaction
mixture was then warmed to RT. After 20 min at RT, 100 ml of 25% strength
aqueous ammonia
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-154-
solution were added, and the mixture was extracted three times with about 100
ml of ethyl acetate
each time. The combined organic extracts were washed with saturated sodium
chloride solution,
dried over anhydrous magnesium sulphate, filtered and finally freed from the
solvent on a rotary
evaporator. The residue obtained was first freed from unpolar impurities by
means of MPLC
(about 50 g of silica gel, mobile phase: cyclohexane/ethyl acetate 4:1).
Further purification of the
product was then carried out by means of preparative HPLC (Method 13). This
gave 17 mg (7% of
theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 8.24 (d, 2H), 7.33 (d, 2H), 7.32 (t, 1H), 7.22
(d, IH), 7.12 (s,
IH), 7.08 (d, I H), 6.82 (s, I H), 5.43 (s, 2H), 4.13 (m, I H), 3.02 (dd, I
H), 2.84 (dd, I H), 2.31 (d,
1 H), 2.29 (s, 3H).
LC/MS (Method 6, ESlpos): R, = 1.33 min, m/z = 513 [M+H]+.
Example 40
1-{ 3-[(5-Methyl-3-{ 3-[4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-yl}-1 H-
pyrazol-l -
yl)methyl]phenyl}propan-2-ol (racemate)
O-N
HO PN N F~/F
CH3 /~
HsC O F
At 0 C, 164 l of lithium aluminium hydride solution (1 M in THF) were added
to a solution of
150 mg (0.329 mmol) of the compound from Example 84A in 7.5 ml of anhydrous
THF. The
reaction mixture was stirred at 0 C for 1 h, and the reaction was then ended
by addition of 20 l of
glacial acetic acid. The solvent was removed on a rotary evaporator and the
crude product was
purified by means of MPLC (silica gel, mobile phase: cyclohexane/ethyl acetate
2:1 -+ 1:2). This
gave 122 mg (81% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 8.26 (d, 2H), 7.33 (d, 2H), 7.29 (t, 1H), 7.16
(d, IH), 7.03 (d
and s, zus. 2H), 6.81 (s, I H), 5.44 (s, 2H), 4.03-3.97 (m, IH), 2.77 (dd, I
H), 2.67 (dd, I H), 2.29 (s,
3H), 1.48 (d, IH), 1.22 (d, 3H).
LC/MS (Method 2, ESIpos): R, = 2.68 min, m/z = 459 [M+H]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-155-
Example 41 and Example 42
1-{3-[(5-Methyl-3-{ 3-[4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-yl }-1 H-
pyrazol-l -
yl)methyl]phenyl}propan-2-ol (enantiomers I and 2)
O-N
HO tN)O: H3C O F
100 mg of the racemic compound from Example 40 were dissolved in 6 ml of
isopropanol and, in
portions, separated into the enantiomers by preparative HPLC on a chiral phase
[column:
Daicel Chiralpak AD-H, 5 gm, 250 mm x 20 mm; mobile phase:
isohexane/isopropanol 1:1 (0-10
min); flow rate: 15 ml/min; temperature: 30 C; UV detection: 220 nm]:
Example 41 (enantiomer 1):
10 Yield: 33 mg
Rt = 6.63 min, 99% ee [analytical chiral HPLC: column: Daicel Chiralpak AD-H,
5 gm, 250 mm x
4.6 mm; mobile phase: isohexane/isopropanol/diethylamine 50:49.8:0.2 (0-10
min); flow rate: 1.0
ml/min; temperature: 30 C; UV detection: 220 nm].
Example 42 (enantiomer 2):
15 Yield: 30 mg
Rt = 7.75 min, 98% ee [analytical chiral HPLC: column: Daicel Chiralpak AD-H,
5 gm, 250 mm x
4.6 mm; mobile phase: isohexane/isopropanol/diethylamine 50:49.8:0.2 (0-10
min); flow rate: 1.0
ml/min; temperature: 30 C; UV detection: 220 nm].
'H NMR (400 MHz, CDC13, 6/ppm): 8.25 (d, 2H), 7.33 (d, 2H), 7.29 (t, IH), 7.16
(d, I H), 7.03 (d
and s, zus. 2H), 6.82 (s, I H), 5.44 (s, 2H), 4.03-3.96 (m, 1 H), 2.77 (dd, 1
H), 2.68 (dd, I H), 2.29 (s,
3H), 1.57 (d, 1H), 1.22 (d, 3H).
LC/MS (Method 6, ESlpos): Rt = 1.28 min, m/z = 459 [M+H]+.
Example 43
2-Methyl-l -{ 3-[(5-methyl-3-{ 3-[4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-
5-yl}-1 H-pyrazol-l -
yl)methyl]phenyl } propan-2-ol
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
- 156 -
O-N
HO N,N
N I F F
H 3 C CH3
H3C / O F
At 0 C, 50.5 ml (152 mmol) of methylmagnesium bromide solution (3 M in diethyl
ether) were
added dropwise to a solution of 33.5 g (68.9 mmol) of the compound from
Example 83A in 700 ml
of anhydrous THF. When the addition had ended, the ice/water bath was removed
and stirring was
continued at RT. After 2 h, the reaction was ended by careful addition of 50
ml of saturated
aqueous ammonium chloride solution. The reaction was then diluted with 700 ml
of ethyl acetate,
and 100 g of anhydrous magnesium sulphate were added. After brief stirring,
the mixture was
filtered and the filtrate was freed from the solvent on a rotary evaporator.
The crude product
obtained was purified by filtration with suction through 770 g of silica gel
(particle size 0.06-0.2
mm) using a cyclohexane/ethyl acetate gradient [8:2 (8 litres) -* 7:3 (8
litres) - 6:4 (8 litres)
1:1 (4 litres)] as mobile phase. Evaporation of the solvent gave 22.5 g of a
crude product which
mainly contained the title compound and in addition also the compound 1-{3-[(5-
methyl-3-{3-[4-
(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-yl}-1H-pyrazol-I-
yl)methyl]phenyl}acetone (see
Example 84A). 7.6 g of this crude product were recrystallized from 61 ml of
boiling diisopropyl
ether. The crystals obtained at RT were filtered off with suction, washed
twice with 5 ml of cold
diisopropyl ether each time and dried under high vacuum. This gave 5.19 g (73%
of theory) of the
pure title compound.
'H NMR (400 MHz, CDC13, 6/ppm): 8.25 (d, 2H), 7.33 (d, 2H), 7.29 (t, 1H), 7.17
(d, 1H), 7.05 (d,
1 H), 7.03 (s, I H), 6.81 (s, I H), 5.45 (s, 2H), 2.73 (s, 2H), 2.29 (s, 3H),
1.31 (s, I H), 1.20 (s, 6H).
LC/MS (Method 6, ESlpos): R, = 1.33 min, m/z = 473 [M+H]+.
Example 44
1-[3-({ 3-[3-(4-tert-Butylphenyl)-1,2,4-oxadiazol-5-yl]-5-methyl-I H-pyrazol-l-
yl } methyl)phenyl]-
2-methylpropan-2-ol
O-N
HO PN N
H3C CH3 CH3
H3C
H3C CH3
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-157-
119 mg (1.06 mmol) of solid potassium tert-butoxide were added to a solution
of 200 mg (0.708
mmol) of the compound from Example 42A and 366 mg (1.42 mmol) of the compound
from
Example 48A in 8 ml of anhydrous THE at 0 C. The reaction mixture was then
stirred at RT for 30
min. After addition of about 1 ml of methanol, the reaction mixture was
separated directly into its
components by means of preparative HPLC (Method 13). This gave 242 mg (77% of
theory) of the
title compound.
'H NMR (400 MHz, CDC13, S/ppm): 8.13 (d, 2H), 7.50 (d, 2H), 7.28 (t, 1H), 7.16
(d, IH), 7.05 (d,
1H), 7.02 (s, 1H), 6.80 (s, 1H), 5.45 (s, 2H), 2.73 (s, 2H), 2.28 (s, 3H),
1.36 (s, 9H), 1.29 (s, 1H),
1.19 (s, 6H).
LC/MS (Method 6, ESlpos): R, = 1.41 min, m/z = 445 [M+H]+.
Example 45
1-(3-{ [3-(3-{4-[ I-(Methoxymethyl)cyclobutyl]phenyl }-1,2,4-oxadiazol-5-yl)-5-
methyl-1 H-pyrazol-
1-yl]methyl}phenyl)-2-methylpropan-2-ol
O-N
HO N,~
N
H 3 C CH3
H C OCH3
3
Analogously to the process described in Example 44, 105 mg (0.324 mmol) of the
compound from
Example 40A and 167 mg (0.647 mmol) of the compound from Example 48A were used
to obtain
130 mg (83% of theory) of the title compound.
'H NMR (400 MHz, CDC13, 6/ppm): 8.13 (d, 2H), 7.29 (d, 2H), 7.28 (t, 1H), 7.16
(d, IH), 7.04 (d,
I H), 7.02 (s, I H), 6.81 (s, I H), 5.45 (s, 2H), 3.55 (s, 2H), 3.28 (s, 3H),
2.73 (s, 2H), 2.41-2.29 (m,
4H), 2.27 (s, 3H), 2.15-2.03 (m, 1H), 1.93-1.83 (m, 1H), 1.29 (s, broad, 1H),
1.19 (s, 6H).
LC/MS (Method 6, ESIpos): R, = 1.37 min, m/z = 487 [M+H]+.
Example 46
2-Methyl-l -{3-[(5-methyl-3-{ 3-[4-(trimethylsilyl)phenyl]-1,2,4-oxadiazol-5-
yl }-1 H-pyrazol- l -
yl)methyi]phenyl } propan-2-ol
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
- 158 -
O-N
HO Ni
H3C CH3 CH3
H3C Si
H3C CH3
Analogously to the process described in Example 44, 100 mg (0.335 mmol) of the
compound from
Example 26A and 173 mg (0.670 mmol) of the compound from Example 48A were used
to obtain
112 mg (73% of theory) of the title compound.
'H NMR (400 MHz, CDC13, 6/ppm): 8.17 (d, 2H), 7.64 (d, 2H), 7.28 (t, 1H), 7.17
(d, 1H), 7.05 (d,
I H), 7.03 (s, IH), 6.81 (s, IH), 5.46 (s, 2H), 2.73 (s, 2H), 2.28 (s, 3H),
1.30 (s, I H), 1.19 (s, 6H),
0.31 (s, 9H).
LC/MS (Method 6, ESlpos): R, = 1.46 min, m/z = 461 [M+H]+.
Example 47
2-Methyl-l-(3-{[5-methyl-3-(3-{4-[1-(trifluoromethyl)cyclopropyl]phenyl}-1,2,4-
oxadiazol-5-yl)-
I H-pyrazol-1-yl]methyl} phenyl)propan-2-ol
O-N
HO
Y'' PN
N I F F
H 3 C CH3
H 3 C F
Analogously to the process described in Example 44, 100 mg (0.285 mmol) of the
compound from
Example 30A and 147 mg (0.571 mmol) of the compound from Example 48A were used
to obtain
116 mg (82% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 8.18 (d, 2H), 7.58 (d, 2H), 7.28 (t, 1H), 7.17
(d, 1H), 7.04 (d,
1H), 7.03 (s, 1H), 6.80 (s, IH), 5.45 (s, 2H), 2.74 (s, 2H), 2.28 (s, 3H),
1.42-1.39 (m, 2H), 1.31 (s,
1H), 1.19 (s, 6H), 1.11-1.07 (m, 2H).
LC/MS (Method 6, ESIpos): R, = 1.35 min, m/z = 497 [M+H]+.
Example 48
1-{ 3-[(3-{ 3-[3-Fluoro-4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-yl }-5-
methyl-1 H-pyrazol-l -
yl)methyl]phenyl } -2-methylpropan-2-ol
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-159-
0-5
HO
Y "' PN F
N I F F
H 3 C CH3
H3C O F
Analogously to the process described in Example 44, 100 mg (0.305 mmol) of the
compound from
Example 29A and 157 mg (0.609 mmol) of the compound from Example 48A were used
to obtain
110 mg (74% of theory) of the title compound.
'H NMR (400 MHz, CDC13, 8/ppm): 8.08 (dd, IH), 8.03 (ddd, IH), 7.43 (dd, I H),
7.29 (t, I H),
7.17 (d, IH), 7.04 (d, I H), 7.03 (s, I H), 6.81 (s, I H), 5.45 (s, 2H), 2.73
(s, 2H), 2.29 (s, 3H), 1.31
(s, 1 H), 1.20 (s, 6H).
LC/MS (Method 6, ESlpos): R, = 1.35 min, m/z = 491 [M+H]+.
Example 49
2-Methyl-l-{3-[(5-methyl-3-{3-[4-(trifluoromethyl)phenyl]-1,2,4-oxadiazol-5-
yl}-1H-pyrazol-l-
yl)methyl]phenyl } propan-2-ol
O-N
HO~ N~ \ \ \
N
H 3 C CH3 / - / F
H3C
F F
Analogously to the process described in Example 44, 100 mg (0.340 mmol) of the
compound from
Example 27A and 175 mg (0.680 mmol) of the compound from Example 48A were used
to obtain
126 mg (81 % of theory) of the title compound.
'H NMR (400 MHz, CDC13, 8/ppm): 8.33 (d, 2H), 7.76 (d, 2H), 7.29 (t, 1H), 7.17
(d, IH), 7.05 (d,
1 H), 7.03 (s, I H), 6.82 (s, 1 H), 5.45 (s, 2H), 2.73 (s, 2H), 2.29 (s, 3H),
1.31 (s, I H), 1.20 (s, 6H).
LC/MS (Method 6, ESIpos): R, = 1.31 min, m/z = 457 [M+H]+.
Example 50
1 -{3-[(3-{ 3-[4-(4-Fluorotetrahydro-2H-pyran-4-yl)phenyl]-1,2,4-oxadiazol-5-
yl }-5-methyl-I H-
pyrazol-1-yl)methyl]phenyl } -2-methylpropan-2-ol
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
- 160 -
O-N
HO N N
N
H 3 C CH3 / H 3 C
O
Analogously to the process described in Example 44, 58 mg (0.177 mmol) of the
compound from
Example 43A and 91 mg (0.353 mmol) of the compound from Example 48A were used
to obtain
64 mg (74% of theory) of the title compound.
'H NMR (400 MHz, CDC13, 8/ppm): 8.22 (d, 2H), 7.52 (d, 2H), 7.29 (t, 1H), 7.17
(d, IH), 7.05 (d,
1H), 7.03 (s, 1H), 6.82 (s, 1H), 5.45 (s, 2H), 4.00-3.88 (m, 4H), 2.73 (s,
2H), 2.28 (s, 3H), 2.30-
2.11 (m, 2H), 1.98-1.91 (m, 2H), 1.32 (s, broad, 1H), 1.20 (s, 6H).
LC/MS (Method 6, ESlpos): R, = 1.22 min, m/z = 491 [M+H]+.
Example 51
2-Methyl-l-{3-[(5-methyl-3-{3-[4-(tetrahydro-2H-pyran-4-yl)phenyl]-1,2,4-
oxadiazol-5-yl}-1H-
pyrazol-1-yl)methyl]phenyl } propan-2-ol
O-N
HO PN
N
H 3 C CH3 /
H3C
O
Analogously to the process described in Example 44, 75 mg (0.242 mmol) of the
compound from
Example 28A and 94 mg (0.362 mmol) of the compound from Example 48A were used
to obtain
98 mg (86% of theory) of the title compound.
'H NMR (400 MHz, CDC13, 6/ppm): 8.15 (d, 2H), 7.35 (d, 2H), 7.29 (t, 1H), 7.16
(d, 1H), 7.05 (d,
IH), 7.03 (s, I H), 6.81 (s, I H), 5.45 (s, 2H), 4.13-4.08 (m, 2H), 3.55 (dt,
2H), 2.88-2.80 (m, I H),
2.73 (s, 2H), 2.28 (s, 3H), 1.92-1.78 (m, 4H), 1.22 (s, IH), 1.20 (s, 6H).
LC/MS (Method 6, ESIpos): R, = 1.22 min, m/z = 473 [M+H]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-161-
Example 52
1-{3-[(3-{3-[4-(3-Fluorooxetan-3-yl)phenyl]-1,2,4-oxadiazol-5-yl }-5-methyl-1
H-pyrazol-l -
yl)methyl]phenyl } -2-methylpropan-2-ol
O-N
H O \ PN H3C CH3 F
H3C
0
Analogously to the process described in Example 44, 70 mg (0.233 mmol) of the
compound from
Example 35A and 90 mg (0.350 mmol) of the compound from Example 48A were used
to obtain
76 mg (70% of theory) of the title compound.
'H NMR (400 MHz, CDC13, 6/ppm): 8.29 (d, 2H), 7.71 (d, 2H), 7.29 (t, 1 H),
7.17 (d, 1 H), 7.05 (d,
I H), 7.03 (s, I H), 6.82 (s, I H), 5.46 (s, 2H), 5.06 (dd, 2H), 5.00 (dd,
2H), 2.73 (s, 2H), 2.29 (s,
3H), 1.31 (s, broad, 1H), 1.20 (s, 6H).
LC/MS (Method 6, ESlpos): R, = 1.17 min, m/z = 463 [M+H]`.
Example 53
2-Methyl- l -(3-{ [5-methyl-3-(3-{4-[(trifluoromethyl)sulphanyl]phenyl}-1,2,4-
oxadiazol-5-yl)-1 H-
pyrazol-1-yl]methyl } phenyl)propan-2-ol
0-N
HO~ PNN
H 3 C CH3
N
HsC S F
Analogously to the process described in Example 44, 80 mg (0.245 mmol) of the
compound from
Example 36A and 95 mg (0.368 mmol) of the compound from Example 48A were used
to obtain
87 mg (73% of theory) of the title compound.
'H NMR (400 MHz, CDC13, 6/ppm): 8.27 (d, 2H), 7.78 (d, 2H), 7.29 (t, IH), 7.17
(d, 1H), 7.05 (d,
I H), 7.03 (s, I H), 6.82 (s, I H), 5.45 (s, 2H), 2.73 (s, 2H), 2.29 (s, 3H),
1.29 (s, broad, I H), 1.20 (s,
6H).
LC/MS (Method 6, ESIpos): Rt = 1.38 min, m/z = 489 [M+H]+.
CA 02798374 2012-11-05
= BHC 10 1 009 Foreign Countries
-162-
Example 54
1-(3-{ [3-(3-{3-Fluoro-4-[ 1-(methoxymethyl)cyclobutyl]phenyl }-1,2,4-
oxadiazol-5-yl)-5-methyl-
1 H-pyrazol-1-yl]methyl}phenyl)-2-methylpropan-2-ol
O-N
HO
Y PN F
H 3 C CH3 H TJiCI>-'TOC
Analogously to the process described in Example 44, 100 mg (0.292 mmol) of the
compound from
Example 31A and 151 mg (0.5 84 mmol) of the compound from Example 48A were
used to obtain
58 mg (39% of theory) of the title compound.
' H NMR (400 MHz, CDC13, S/ppm): 7.93 (dd, 1 H), 7.82 (dd, 1 H), 7.29 (t, I
H), 7.24 (t, 1 H), 7.16
(d, I H), 7.05 (d, I H), 7.02 (s, IH), 6.81 (s, I H), 5.45 (s, 2H), 3.67 (s,
2H), 3.28 (s, 3H), 2.73 (s,
2H), 2.48-2.40 (m, 2H), 2.37-2.30 (m, 2H), 2.27 (s, 3H), 2.19-2.06 (m, 1H),
1.94-1.84 (m, IH),
1.29 (s, 1H), 1.19 (s, 6H).
LC/MS (Method 6, ESlpos): R1 = 1.39 min, m/z = 505 [M+H]+.
Example 55
1-(3-{ [3-(3-{4-[ I-(Methoxymethyl)cyclopentyl]phenyl}-1,2,4-oxadiazol-5-yl)-5-
methyl-1 H-
pyrazol-1-yl]methyl}phenyl)-2-methylpropan-2-ol
0-N
HO~ PN ,_ N
N
C
H3C CH3 CH3
H3C
Analogously to the process described in Example 44, 100 mg (0.295 mmol) of the
compound from
Example 33A and 153 mg (0.591 mmol) of the compound from Example 48A were used
to obtain
66 mg (45% of theory) of the title compound.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-163-
'H NMR (400 MHz, CDC13, S/ppm): 8.12 (d, 2H), 7.45 (d, 2H), 7.29 (t, 1 H),
7.17 (d, 1 H), 7.04 (d,
IH), 7.03 (s, 1H), 6.81 (s, IH), 5.45 (s, 2H), 3.41 (s, 2H), 3.23 (s, 3H),
2.73 (s, 2H), 2.28 (s, 3H),
2.07-2.00 (m, 2H), 1.96-1.88 (m, 2H), 1.79-1.71 (m, 4H), 1.30 (s, 1H), 1.19
(s, 6H).
LC/MS (Method 2, ESlpos): R, = 2.82 min, m/z = 501 [M+H]+.
Example 56
2-Methyl- l -{ 3-[(5-methyl-3-{3-[3-methyl-4-(trifluoromethoxy)phenyl]-1,2,4-
oxadiazol-5-yl }-1 H-
pyrazol-1-yl)methyl]phenyl}propan-2-ol
O-N
HO PN N CF F
H 3 C CH3 I / X
H 3 C O F
Analogously to the process described in Example 44, 125 mg (0.385 mmol) of the
compound from
Example 34A and 149 mg (0.577 mmol) of the compound from Example 48A were used
to obtain
60 mg (32% of theory) of the title compound.
'H NMR (400 MHz, CDC13, 8/ppm): 8.14 (d, 1H), 8.06 (dd, 1H), 7.32 (d, IH),
7.29 (t, 1H), 7.17
(d, 1H), 7.05 (d, IH), 7.03 (s, IH), 6.81 (s, 1H), 5.46 (s, 2H), 2.73 (s, 2H),
2.39 (s, 3H), 2.28 (s,
3H), 1.30 (s, IH), 1.19 (s, 6H).
LC/MS (Method 6, ESlpos): R, = 1.37 min, m/z = 487 [M+H]+.
Example 57
2-Methyl-l-{ 3-[(5 -methyl-3-{ 3-[4-(1,1,1-tri fluoro-2-methylpropan-2-
yl)phenyl]-1,2,4-oxadiazol-5-
yl } -1 H-pyrazol-1-yl)methyl]phenyl}propan-2-ol
O-N
HO PNN , X
YN"' N F F
H 3 C CH3
H3C F
H3C CH3
At 0 C, 212 pl of methylmagnesium chloride solution (3 M in diethyl ether)
were added to a
solution of 155 mg (0.302 mmol) of the compound from Example 82A in 3 ml of
anhydrous THF.
The ice/water bath was then removed, and stirring was continued at RT. After
1.5 h, the reaction
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-164-
was ended by addition of 0.5 ml of saturated aqueous ammonium chloride
solution. The reaction
was then diluted with about 9 ml of ethyl acetate, and anhydrous magnesium
sulphate was added.
After brief stirring, the mixture was filtered and the filtrate was freed from
the solvent on a rotary
evaporator. The residue obtained was triturated with 5 ml of pentane. The
solid was filtered off
and dried under high vacuum. This gave 125 mg (83% of theory) of the title
compound.
'H NMR (400 MHz, CDC13, S/ppm): 8.20 (d, 2H), 7.63 (d, 2H), 7.29 (t, IH), 7.16
(d, I H), 7.05 (d,
1H), 7.03 (s, 1H), 6.81 (s, 1H), 5.46 (s, 2H), 2.73 (s, 2H), 2.27 (s, 3H),
1.63 (s, 6H), 1.30 (s, 1H),
1.19 (s, 6H).
LC/MS (Method 6, ESlpos): R, = 1.34 min, m/z = 499 [M+H]+.
Example 58
2-Methyl-2-(3-{ [5-methyl-3-(3-{4-[ 1-(trifluoromethyl)cyclopropyl]phenyl }-
1,2,4-oxadiazol-5-yl)-
I H-pyrazol-l-yl]methyl }phenyl)propan-l -ol
H3C CH3 O-N
H O PN N \ N
N I F F
H3C / F
Under argon and at 0 C, 0.37 ml (0.37 mmol) of lithium aluminium hydride
solution (1 M in THF)
was added dropwise to a solution of 200 mg (0.37 mmol) of the compound from
Example 85A in
3.5 ml of THF. The mixture was stirred at 0 C for another hour, and I ml of
water was then added
dropwise. After warming to RT, the mixture was diluted with ethyl acetate and
the solid present
was filtered off. The solid was washed twice with ethyl acetate, and the
combined organic phases
were dried over magnesium sulphate, filtered and concentrated. The residue was
purified by means
of preparative HPLC (Method 14). Removal of the solvent and drying of the
residue under reduced
pressure gave 17 mg (9% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 8.18 (d, 2H), 7.58 (d, 2H), 7.35-7.27 (m, 3H),
7.22 (s, 1H),
6.99 (d, 1H), 6.81 (s, 1H), 5.45 (s, 2H), 3.59 (s, 2H), 2.29 (s, 3H), 1.40
(dd, 2H), 1.30 (s, 6H), 1.25
(s, 1H), 1.11-1.05 (m, 2H).
LC/MS (Method 6, ESIpos): R, = 1.37 min, m/z = 497 [M+H]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-165-
Example 59
2-Methyl-2-{ 3-[(5 -methyl-3-{ 3-[4-(tetrahydro-2H-pyran-4-yl)phenyl]-1,2,4-
oxadiazol-5-yl }-1 H-
pyrazol- l -yl)methyl]phenyl }propan- l -ol
H3C CH3 O-N
HOJ PN N
H3C
O
At -78 C, 130 l (0.130 mmol) of lithium aluminium hydride solution (1 M in
THF) were added to
a solution of 74 mg (0.129 mmol) of the compound from Example 86A in 3 ml of
anhydrous THF.
The reaction mixture was then stirred first at -78 C for 1 h and then at 0 C
for a further 30 min.
About 9 ml of 1 M aqueous sodium hydroxide solution were then added, and the
mixture was
extracted three times with in each case about 10 ml of ethyl acetate. The
combined organic extracts
were washed successively with water and saturated sodium chloride solution,
dried over anhydrous
magnesium sulphate, filtered and finally freed from the solvent on a rotary
evaporator. The residue
obtained gave, by means of preparative HPLC (Method 11), 42 mg (69% of theory)
of the title
compound.
'H NMR (400 MHz, CDC13, S/ppm): 8.14 (d, 2H), 7.36-7.28 (m, 4H), 7.22 (s, IH),
6.99 (d, 1H),
6.81 (s, 1H), 5.45 (s, 2H), 4.10 (dd, 2H), 3.59 (d, 2H), 3.54 (dt, 2H), 2.88-
2.78 (m, 1H), 2.29 (s,
3H), 1.91-1.78 (m, 4H), 1.30 (s, 6H), 1.24 (t, 1H).
LC/MS (Method 6, ESlpos): Rt = 1.21 min, m/z = 473 [M+H]+.
Example 60
2-Methyl-2-{3-[(5-methyl-3-{ 3-[4-(1,1,1-trifluoro-2-methylpropan-2-yl)phenyl]-
1,2,4-oxadiazol-5-
yl}-1H-pyrazol-1-yl)methyl]phenyl}propan-l-ol
H3C CH3 O-N
HO N
- N I ~ F F
H3C F
H3C CH3
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-166-
Analogously to the process described under Example 59, 80 mg (0.15 mmol) of
the compound
from Example 87A were reacted to give 50 mg (68% of theory) of the title
compound. Here,
purification by preparative HPLC was carried out according to Method 17.
'H NMR (400 MHz, DMSO-d6, S/ppm): 8.09 (d, 2H), 7.77 (d, 2H), 7.32-7.25 (m,
3H), 6.92 (d,
2H), 5.49 (s, 2H), 4.67 (s, broad, 1H), 3.39 (d, 2H), 2.34 (s, 3H), 1.61 (s,
6H), 1.20 (s, 6H).
LC/MS (Method 6, ESlpos): R, = 1.36 min, m/z = 499 [M+H]+.
Example 61
2-Methyl-2-{3-[(5-methyl-3-{3-[4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-
yl }-1 H-pyrazol-l -
yl)methyl]phenyl}propan-l -ol
H3C CH3 O-N
HO PN
N I F F
HsC / O F
Analogously to the process described under Example 59, 80 mg (0.16 mmol) of
the compound
from Example 88A were reacted to give 40 mg (54% of theory) of the title
compound. Here,
purification by preparative HPLC was carried out according to Method 17.
'H NMR (400 MHz, DMSO-d6, S/ppm): 8.20 (d, 2H), 7.59 (d, 2H), 7.32-7.25 (m,
3H), 6.92 (d,
2H), 5.49 (s, 2H), 4.67 (t, 1H), 3.39 (d, 2H), 2.34 (s, 3H), 1.20 (s, 6H).
LC/MS (Method 4, ESIpos): Rt = 1.48 min, m/z = 473 [M+H]+.
Example 62
1,1-Dideutero-2-methyl-2-f 3-[(5-methyl-3-{3-[4-(1,1,1-trifluoro-2-
methylpropan-2-yl)phenyl]-
1,2,4-oxadiazol-5-yl }-1 H-pyrazol-1-yl)methyl]phenyl }propan-l -ol
H 3 C CH3 O-N
HO PN --~ D
D N F F H3C F
H3C CH3
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-167-
Analogously to the process described under Example 59, 300 mg (0.555 mmol) of
the compound
from Example 87A and 555 gl (0.555 mmol) of lithium aluminium deuteride
solution (1 M in
THF) were reacted to give 166 mg (60% of theory) of the title compound. Here,
purification by
preparative HPLC was carried out according to Method 13.
'H NMR (400 MHz, CDC13, 6/ppm): 8.20 (d, 2H), 7.62 (d, 2H), 7.32 (d, 1H), 7.31
(t, 1H), 7.22 (s,
IH), 7.00 (d, 1H), 6.82 (s, 1H), 5.45 (s, 2H), 2.29 (s, 3H), 1.61 (s, 6H),
1.30 (s, 6H), 1.24 (s, broad,
I H).
LC/MS (Method 6, ESlpos): R, = 1.35 min, m/z = 501 [M+H]+.
Example 63
(1-{3-[(5-Methyl-3-{3-[4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-yl}-1H-
pyrazol-l-
yl)methyl]phenyl } cyclopropyl)methanol
O-N
HO N
N F F
H 3 C O F
68 mg (0.604 mmol) of solid potassium tert-butoxide were added to a solution
of 125 mg (0.403
mmol) of the compound from Example 22A and 249 mg (0.604 mmol) of the compound
from
Example 50A in 5 ml of anhydrous THF at 0 C. The reaction mixture was stirred
at RT for 6 days
and then at 40 C for another 2 h. Approx. 50 ml of water were then added and
the mixture was
extracted three times with approx. 50 ml of ethyl acetate each time. The
combined organic extracts
were washed successively with water and saturated sodium chloride solution,
dried over anhydrous
magnesium sulphate, filtered and finally freed from the solvent on a rotary
evaporator. The residue
obtained was redissolved in 5 ml of THF, and 483 pl (0.483 mmol) of tetra-n-
butylammonium
fluoride solution (1 M in THF) were added. After 1 h at RT, the reaction
mixture was diluted with
2 ml of methanol and then separated directly into its components by means of
preparative HPLC
(Method 13). In this manner, 85 mg (45% of theory) of the title compound were
obtained.
'H NMR (400 MHz, CDC13, S/ppm): 8.26 (d, 2H), 7.33 (d, 2H), 7.32-7.25 (m, 2H),
7.22 (s, 1H),
7.00 (d, 1H), 6.81 (s, 1H), 5.44 (s, 2H), 3.66 (s, 2H), 2.30 (s, 3H), 1.39
(broad, 1H), 0.85 (s, 4H).
LC/MS (Method 6, ESIpos): R, = 1.29 min, m/z = 471 [M+H]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-168-
Example 64
(1-{3-[(5-Methyl-3-{3-[4-(1,1,1-trifluoro-2-methylpropan-2-yl)phenyl]-1,2,4-
oxadiazol-5-yl}-1H-
pyrazol-1-yl)methyl]phenyl } cyclopropyl)methanol
O-N
HO N
N F F
H3C F
H3C CH3
Analogously to the process described in Example 63, 109 mg (0.323 mmol) of the
compound from
Example 23A and 200 mg (0.485 mmol) of the compound from Example 50A were used
to obtain,
in two partial steps, 124 mg (77% of theory) of the title compound. Here, in
the first partial step
the reaction mixture was stirred at RT for 16 h; subsequent heating to 40 C
was dispensed with.
'H NMR (400 MHz, CDC13, 6/ppm): 8.20 (d, 2H), 7.62 (d, 2H), 7.32-7.25 (m, 2H),
7.22 (s, IH),
7.00 (d, 1H), 6.82 (s, IH), 5.43 (s, 2H), 3.66 (d, 2H), 2.29 (s, 3H), 1.62 (s,
6H), 1.43 (t, broad, 1H),
0.86 (s, 4H).
LC/MS (Method 4, ESlpos): R, = 1.51 min, m/z = 497 [M+H]+.
Example 65
[ 1-(3-{ [5-Methyl-3-(3-{4-[I-(trifluoromethyl)cyclopropyl]phenyl}-1,2,4-
oxadiazol-5-yl)-] H-
pyrazol-1-yl]methyl}phenyl)cyclopropyl]methanol
O-N
HO N~
N I F F
H3C / F
Analogously to the process described in Example 63, 108 mg (0.323 mmol) of the
compound from
Example 30A and 200 mg (0.485 mmol) of the compound from Example 50A were used
to obtain,
in two partial steps, 141 mg (89% of theory) of the title compound. Here, in
the first partial step
the reaction mixture was stirred at RT for 16 h; subsequent heating to 40 C
was dispensed with.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-169-
'H NMR (400 MHz, CDC13, S/ppm): 8.19 (d, 2H), 7.59 (d, 2H), 7.31-7.25 (m, 2H),
7.22 (s, 1H),
7.00 (d, 1H), 6.82 (s, 1H), 5.43 (s, 2H), 3.66 (s, 2H), 2.29 (s, 3H), 1.46-
1.39 (m, 3H), 1.10-1.07 (m,
2H), 0.85 (s, 4H).
LC/MS (Method 4, ESlpos): Rt = 1.50 min, m/z = 495 [M+H]+.
Example 66
[1-(3-{ [5-Methyl-3-(3-{4-[(trifluoromethyl)sulphanyl]phenyl}-1,2,4-oxadiazol-
5-yl)-1 H-pyrazol-l -
yl] methyl } phenyl)cyclopropyl]methanol
O-N
HO PNN
N F F
H3C S F
Analogously to the process described in Example 63, 105 mg (0.323 mmol) of the
compound from
Example 36A and 200 mg (0.485 mmol) of the compound from Example 50A were used
to obtain,
in two partial steps, 130 mg (83% of theory) of the title compound. Here, in
the first partial step
the reaction mixture was stirred at RT for 16 h; subsequent heating to 40 C
was dispensed with.
'H NMR (400 MHz, CDC13, 6/ppm): 8.27 (d, 2H), 7.78 (d, 2H), 7.31-7.26 (m, 2H),
7.22 (s, 1H),
7.00 (d, 1H), 6.82 (s, 1H), 5.46 (s, 2H), 3.66 (s, 2H), 2.30 (s, 3H), 1.43
(broad, 1H), 0.86 (s, 4H).
LC/MS (Method 4, ESlpos): R, = 1.53 min, m/z = 487 [M+H]+.
Example 67
[1-(3-{ [5-Methyl-3-(3-{4-[(trifluoromethyl)sulphonyl]phenyl}-1,2,4-oxadiazol-
5-yl)-1 H-pyrazol- l -
yl]methyl}phenyl)cyclopropyl]methanol
O-N
HO PN
N F F
H3C ~S\ X F
O O
Analogously to the process described in Example 63, 116 mg (0.323 mmol) of the
compound from
Example 25A and 200 mg (0.485 mmol) of the compound from Example 50A were used
to obtain,
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-170-
in two partial steps, 74 mg (44% of theory) of the title compound. Here, in
the first partial step the
reaction mixture was stirred at RT for 16 h; subsequent heating to 40 C was
dispensed with.
'H NMR (400 MHz, CDC13, S/ppm): 8.52 (d, 2H), 8.18 (d, 2H), 7.32-7.27 (m, 2H),
7.23 (s, 1H),
7.00 (d, 1H), 6.84 (s, 1H), 5.45 (s, 2H), 3.66 (d, 2H), 2.31 (s, 3H), 1.41 (t,
1H), 0.86 (s, 4H).
LC/MS (Method MHZ-QP-GO-1, ESIpos): R, = 1.43 min, m/z = 519 [M+H]+.
Example 68
Dideutero(1-{ 3-[(5-methyl-3-{ 3-[4-(tri fluoromethoxy)phenyl]-1,2,4-oxadiazol-
5-yl } -1 H-pyrazol- l -
yl)methyl]phenyl } cyclopropyl)methanol
O-N
HO PN
N F F
D D
H3C O F
62 mg (0.556 mmol) of solid potassium tert-butoxide were added to a solution
of 115 mg (0.371
mmol) of the compound from Example 22A and 231 mg (0.556 mmol) of the compound
from
Example 51A in 5 ml of anhydrous THF at 0 C. After removal of the ice/water
bath, the reaction
mixture was stirred at RT for 16 h. About 50 ml of water were then added, and
the mixture was
extracted three times with in each case about 50 ml of ethyl acetate. The
combined organic extracts
were washed successively with water and saturated sodium chloride solution,
dried over anhydrous
magnesium sulphate, filtered and finally freed from the solvent on a rotary
evaporator. The residue
obtained was redissolved in 5 ml of THF, and 445 pl (0.445 mmol) of tetra-n-
butylammonium
fluoride solution (1 M in THF) were added. After 1 h at RT, the reaction
mixture was diluted with
2 ml of methanol and then separated directly into its components by means of
preparative HPLC
(Method 13). In this manner, 105 mg (60% of theory) of the title compound were
obtained.
'H NMR (400 MHz, CDC13, S/ppm): 8.25 (d, 2H), 7.33 (d, 2H), 7.30-7.24 (m, 2H),
7.21 (s, 1H),
7.00 (d, IH), 6.82 (s, 1H), 5.43 (s, 2H), 2.30 (s, 3H), 1.39 (broad, 1H), 0.85
(s, 4H).
LC/MS (Method 6, ESlpos): R, = 1.28 min, m/z = 473 [M+H]+.
Example 69
Dideutero(I-{3-[(5-methyl-3-{3-[4-(1,1,1-trifluoro-2-methylpropan-2-yl)phenyl]-
1,2,4-oxadiazol-
5-yl }-1 H-pyrazol-l-yl)methyl]phenyl } cyclopropyl)methanol
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
- 171 -
O-N
HO N
D D N F F
H3C F
H3C CH3
Analogously to the process described in Example 68, 108 mg (0.322 mmol) of the
compound from
Example 23A and 200 mg (0.482 mmol) of the compound from Example 51A were used
to obtain,
in two partial steps, 57 mg (35% of theory) of the title compound. Here, in
the second partial step,
the reaction mixture was stirred at RT for 2 h.
'H NMR (400 MHz, CDC13, S/ppm): 8.20 (d, 2H), 7.62 (d, 2H), 7.31-7.27 (m, 2H),
7.22 (s, 1H),
7.00 (d, IH), 6.82 (s, IH), 5.43 (s, 2H), 2.29 (s, 3H), 1.63 (s, 6H), 1.43
(broad, 1H), 0.83 (s, 4H).
LC/MS (Method 4, ESlpos): R, = 1.51 min, m/z = 499 [M+H]+.
Example 70
Dideutero[1-(3-{[5-methyl-3-(3-{4-[1-(trifluoromethyl)cyclopropyl]phenyl}-
1,2,4-oxadiazol-5-yl)-
I H-pyrazol- l -yl]methyl) phenyl)cyclopropyl]methanol
O-N
HO PN__' N I F F
D D / /
H3C F
Analogously to the process described in Example 68, 107 mg (0.322 mmol) of the
compound from
Example 30A and 200 mg (0.482 mmol) of the compound from Example 51A were used
to obtain,
in two partial steps, 73 mg (45% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 8.19 (d, 2H), 7.59 (d, 2H), 7.31-7.25 (m, 2H),
7.22 (s, 1H),
7.00 (d, 1H), 6.82 (s, 1H), 5.43 (s, 2H), 2.29 (s, 3H), 1.43-1.39 (m, 3H),
1.11-1.07 (m, 2H), 0.85 (s,
4H).
LC/MS (Method 6, ESIpos): R, = 1.31 min, m/z = 497 [M+H]+.
Example 71
Dideutero [ 1-(3 - { [5-methyl-3-(3-{ 4-[(tri fluoromethyl)sulphanyl]phenyl } -
1,2,4-oxadiazol-5-yl)-1 H-
pyrazol-l-yl]methyl } phenyl)cyclopropyl] methanol
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
- 172 -
O-N
HO N,
N ~ F F
D XD I
H3C / S F
Analogously to the process described in Example 68, 105 mg (0.322 mmol) of the
compound from
Example 36A and 200 mg (0.482 mmol) of the compound from Example 51A were used
to obtain,
in two partial steps, 81 mg (52% of theory) of the title compound.
'H NMR (400 MHz, CDC13, 6/ppm): 8.28 (d, 2H), 7.79 (d, 2H), 7.31-7.27 (m, 2H),
7.22 (s, I H),
7.00 (d, 1H), 6.83 (s, IH), 5.44 (s, 2H), 2.29 (s, 3H), 1.45 (broad, IH), 0.84
(s, 4H).
LC/MS (Method 6, ESlpos): R, = 1.34 min, m/z = 489 [M+H]+.
Example 72
Dideutero[ 1-(3-{ [5-methyl-3-(3-{4-[(tri fluoromethyl)sulphanyl]phenyl }-
1,2,4-oxadiazol-5-yl)-I H-
pyrazol-1-yl]methyl} phenyl)cyclopropyl]methanol
O-N
HO PN N
N I F F
D D
HC ~S\ X F
3
O O
Analogously to the process described in Example 68, 115 mg (0.322 mmol) of the
compound from
Example 25A and 200 mg (0.482 mmol) of the compound from Example 51A were used
to obtain,
in two partial steps, 41 mg (25% of theory) of the title compound.
'H NMR (400 MHz, CDC13, 6/ppm): 8.52 (d, 2H), 8.18 (d, 2H), 7.32-7.27 (m, 2H),
7.23 (s, 1H),
7.00 (d, 1H), 6.83 (s, IH), 5.45 (s, 2H), 2.31 (s, 3H), 1.41 (t, 1H), 0.84 (s,
4H).
LC/MS (Method 6, ESlpos): R, = 1.25 min, m/z = 521 [M+H]+.
Example 73
Dideutero(1-{3-[(3-{3-[4-(4-fluorotetrahydro-2H-pyran-4-yl)phenyl]-1,2,4-
oxadiazol-5-yl}-5-
methyl-lH-pyrazol-1-yl)methyl]phenyl}cyclopropyl)methanol
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-173-
O-N
HO PN D D N I/ F
H3C
O
Analogously to the process described in Example 68, 65 mg (0.198 mmol) of the
compound from
Example 43A and 123 mg (0.297 mmol) of the compound from Example 51A were used
to obtain,
in two partial steps, 60 mg (61% of theory) of the title compound.
'H NMR (400 MHz, CDC13, 6/ppm): 8.23 (d, 2H), 7.52 (d, 2H), 7.31-7.26 (m, 2H),
7.21 (s, 1H),
7.00 (d, 1H), 6.82 (s, IH), 5.43 (s, 2H), 4.00-3.87 (m, 4H), 2.29 (s, 3H),
2.30-2.11 (m, 2H), 1.98-
1.91 (m, 2H), 1.41 (s, broad, 1H), 0.84 (s, 4H).
LC/MS (Method 6, ESlpos): R, = 1.18 min, m/z = 491 [M+H]+.
Example 74
2,2-Difluoro-2-{3-[(5-methyl-3-{3-[4-(trimethylsilyl)phenyl]-1,2,4-oxadiazol-5-
yl}-1H-pyrazol-l-
yl)methyl]phenyl} ethanol
F F O-N
HOJ PN
\ N \ \ I / ,CH3
3C
H3C CH3
Under argon and at 0 C, 85 mg (0.75 mmol) of potassium tert-butoxide were
added to a solution of
155 mg (0.52 mmol) of the compound from Example 26A in 4 ml of THF. After 30
min of stirring
at 0 C, 392 mg (1.56 mmol) of the compound from Example 52A in 2 ml of THE
were added, and
the mixture was stirred at RT for 16 h. A further 58 mg (0.52 mmol) of
potassium tert-butoxide
were then added, and the mixture was stirred at RT for another 2 h. 30 ml of
water and 30 ml of
ethyl acetate were then added to the mixture. After phase separation, the
aqueous phase was
extracted once with 30 ml of ethyl acetate. The combined organic phases were
dried over sodium
sulphate, filtered and concentrated. The residue was purified by means of
column chromatography
(silica gel, mobile phase: cyclohexane/ethyl acetate 7:3). After removal of
the solvent, the product
fraction was purified once more by preparative thin-layer chromatography
(silica gel, mobile
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-174-
phase: dichloromethane/methanol 97:3). Removal of the solvent and drying of
the residue under
reduced pressure gave 47 mg (19% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 8.17 (d, 2H), 7.64 (d, 2H), 7.47 (d, 1H), 7.42
(t, 1H), 7.36 (s,
IH), 7.23 (d, I H), 6.84 (s, IH), 5.49 (s, 2H), 3.95 (t, 2H), 2.29 (s, 3H),
0.31 (s, 9H).
LC/MS (Method 6, ESlpos): Rt = 1.37 min, m/z = 469 [M+H]+.
Example 75
2-[3-({ 3-[3-(4-tert-Butylphenyl)-1,2,4-oxadiazol-5-yl]-5-methyl-1 H-pyrazol-l
-yl } methyl)phenyl]-
2,2-difluoroethanol
F F O-N
HOJ PN__'
CH3
H3C
H3C CH3
Under argon and at 0 C, 89 mg (0.80 mmol) of potassium tert-butoxide were
added to a solution of
155 mg (0.55 mmol) of the compound from Example 42A in 3 ml of THF. After 30
min of stirring
at 0 C, 414 mg (1.65 mmol) of the compound from Example 52A were added. The
mixture was
then allowed to warm to RT and stirred at RT for 16 h. 30 ml of water and 30
ml of ethyl acetate
were then added, and the phases were separated. The aqueous phase was
extracted once more with
30 ml of ethyl acetate. The combined organic phases were dried over sodium
sulphate, filtered and
concentrated. The residue was purified by means of column chromatography
(silica gel, mobile
phase: cyclohexane/ethyl acetate 4:1). Removal of the solvent and drying of
the residue under
reduced pressure gave 93 mg (36% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 8.13 (d, 2H), 7.51 (d, 2H), 7.47 (d, 1H), 7.42
(t, IH), 7.36 (s,
1H), 7.23 (d, 1H), 6.83 (s, 1H), 5.49 (s, 2H), 3.95 (dt, 2H), 2.29 (s, 3H),
2.10 (t, IH), 1.36 (s, 9H).
LC/MS (Method 4, ESlpos): Rt = 1.53 min, m/z = 453 [M+H]+.
Example 76
2,2-Difluoro-2-{3-[(5-methyl-3-{3-[4-(1,1,1-trifluoro-2-methylpropan-2-
yl)phenyl]-1,2,4-
oxadiazol-5-yl } -1 H-pyrazol- l -yl)methyl]phenyl } ethanol
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-175-
- F O-N
HOJ ~N
N \ \ \
- N I F F
H 3 C F
H3C CH3
Analogously to the process described in Example 75, 168 mg (0.50 mmol) of the
compound from
Example 23A and 377 mg (1.50 mmol) of the compound from Example 52A were used
to obtain
66 mg (26% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 8.19 (d, 2H), 7.62 (d, 2H), 7.47 (d, 1H), 7.43
(t, 1H), 7.36 (s,
1 H), 7.24 (d, 1 H), 6.83 (s, I H), 5.49 (s, 2H), 3.95 (dt, 2H), 2.29 (s, 3H),
2.10 (t, 1 H), 1.62 (s, 6H).
LC/MS (Method 6, ESlpos): R, = 1.30 min, m/z = 507 [M+H]+.
Example 77
2,2-Difluoro-2-{3-[(3-{3-[4-(3-fluorooxetan-3-yl)phenyl]-1,2,4-oxadiazol-5-yl}-
5-methyl-I H-
pyrazol- l -yl)methyl]phenyl } ethanol
F F O-N
HOJ PN
N I/ F
H3C
0
Analogously to the process described in Example 75, 126 mg (0.42 mmol) of the
compound from
Example 35A and 317 mg (1.26 mmol) of the compound from Example 52A were used
to obtain
38 mg (19% of theory) of the title compound. Here, after chromatography on
silica gel, the product
was repurified by preparative HPLC (Method 16).
'H NMR (400 MHz, CDC13, 6/ppm): 8.28 (d, 2H), 7.71 (d, 2H), 7.46 (d, IH), 7.44
(t, 1H), 7.36 (s,
1H), 7.24 (d, IH), 6.84 (s, IH), 5.50 (s, 2H), 5.15 (dd, 2H), 4.90 (d, 2H),
3.95 (t, 2H), 2.30 (s, 3H).
LC/MS (Method 4, ESlpos): R, = 1.25 min, m/z = 471 [M+H]+.
Example 78
2,2-Difluoro-2-(3-{[5-methyl-3-(3-{4-[I-(trifluoromethyl)cyclopropyl]phenyl}-
1,2,4-oxadiazol-5-
yl)-1 H-pyrazol-1-yl]methyl} phenyl)ethanol
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-176-
F F O-N
HO PN
N I F F
H3C F
Analogously to the process described in Example 75, 167 mg (0.50 mmol) of the
compound from
Example 30A and 377 mg (1.50 mmol) of the compound from Example 52A were used
to obtain
93 mg (36% of theory) of the title compound. Here, the silica gel
chromatography was carried out
using the mobile phase cyclohexane/ethyl acetate 3:1.
'H NMR (400 MHz, CDC13, 6/ppm): 8.18 (d, 2H), 7.59 (d, 2H), 7.46 (d, 1H), 7.44
(t, IH), 7.36 (s,
1H), 7.24 (d, 1H), 6.83 (s, 1H), 5.49 (s, 2H), 3.95 (td, 2H), 2.29 (s, 3H),
2.07 (t, 1H), 1.42-1.38 (m,
2H), 1.11-1.06 (m, 2H).
LC/MS (Method 6, ESlpos): R, = 1.32 min, m/z = 505 [M+H]+.
Example 79
2,2-Difluoro-2-{ 3-[(5-methyl-3-{3-[4-(trifluoromethoxy)phenyl]-1,2,4-
oxadiazol-5-yl }-1 H-
pyrazo l-1-yl)methyl]phenyl -yl)methyljphenyl) ethanol
F F O-N
HO PN
N F F
H 3 C O F
At RT, 38 mg (1.02 mmol) of sodium borohydride were added to a solution of
1.33 g (1.02 mmol,
purity 40%) of the compound from Example 89A in 20 ml of ethanol. After 1 h of
stirring at RT
and addition of another 38 mg (1.02 mmol) of sodium borohydride and 5 ml of
methanol, the
solvent was removed. The residue was triturated with ethyl acetate, solid
components were filtered
off and the filtrate was concentrated. Since, according to LC/MS, the residue
obtained consisted
mainly of the carboxylic acid which corresponds to Example 89A, this residue
was taken up in 20
ml of methanol and 2 ml (27 mmol) of thionyl chloride were added dropwise at 0
C. The mixture
was then stirred at 0 C for 1 h. The mixture was then concentrated, and the
residue was dried
under reduced pressure. This residue was dissolved in 20 ml of ethanol, and
another 38 mg (1.02
mmol) of sodium borohydride were added. After 30 min of stirring at RT, the
mixture was
concentrated. The residue was taken up in ethyl acetate and water and
acidified with 10% strength
citric acid. After phase separation, the aqueous phase was reextracted once
with ethyl acetate. The
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-177-
combined organic phases were dried over magnesium sulphate, filtered and
concentrated. The
residue was purified by means of preparative HPLC (Method 15). Subsequent
column
chromatography on silica gel (mobile phase: dichloromethane/methanol 98.5:1.5)
gave, after
removal of the solvent and drying of the residue under reduced pressure, 248
mg (51% of theory)
of the title compound.
'H NMR (400 MHz, CDC13, 8/ppm): 8.25 (d, 2H), 7.47 (d, 1H), 7.43 (t, 1H), 7.36
(s, 1H), 7.33 (d,
2H), 7.24 (d, IH), 6.83 (s, 1H), 5.49 (s, 2H), 3.95 (td, 2H), 2.30 (s, 3H),
2.19 (t, 1H).
LC/MS (Method 6, ESlpos): R, = 1.25 min, m/z = 481 [M+H]+.
Example 80
2-Methyl-2-{3-[(5-methyl-3-{3-[4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-
yl}-1H-pyrazol-l-
yl)methyl]phenyl } propan-1,3 -diol
HO
C H 3 O-N
HO PN
N F F
H3C O F
With ice bath cooling, 79 mg (2.10 mmol) of sodium borohydride were added to a
solution of 0.12
g (0.21 mmol) of the compound from Example 91 A in 4 ml of abs. THE and 4 ml
of methanol, and
the mixture was stirred at 40 C overnight. A further 79 mg of sodium
borohydride were then
added, and the mixture was stirred at 40 C for another I h. This was followed
by two further
additions of in each case 79 mg of sodium borohydride, where the reaction
mixture was in each
case stirred at 40 C for I h. Saturated aqueous ammonium chloride solution and
water were then
added, and the reaction was extracted with ethyl acetate. The organic phase
was washed with water
and saturated sodium chloride solution, dried over anhydrous magnesium
sulphate and filtered.
The filtrate was freed from the solvent on a rotary evaporator, and the
residue was purified by
means of preparative HPLC (Method 17). This gave 60 mg (59% of theory) of the
title compound.
'H NMR (400 MHz, DMSO-d6, 6/ppm): 8.20 (d, 2H), 7.59 (d, 2H), 7.32 (d, 2H),
7.26 (t, 1H), 6.93
(d, 2H), 5.48 (s, 2H), 3.54 (s, 4H), 2.35 (s, 3H), 1.17 (s, 3H).
LC/MS (Method 2, ESIpos): Rt = 2.46 min, m/z = 489 [M+H]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
- 178-
Example 81
2-Methyl-l -{3-[(5-methyl-3-{3-[4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-
yl}-1 H-pyrazol-l -
yl)methyl]phenyl}propan-2-yl dihydrogenphosphate
OH
I
HO-P
-0 O-N
I PN
N
H 3 C C H 3 H 3 C O X
F
A solution of 197 mg (0.301 mmol) of the compound from Example 92A in 6 ml of
methanol/water 10:1 was hydrogenated in a flow-through hydrogenation apparatus
("H-Cube"
from ThalesNano, Budapest, Hungary) (conditions: 10% Pd/C catalyst, 1 bar, 1
ml/min, 22 C).
After removal of the solvent on a rotary evaporator, the crude product was
purified by means of
preparative HPLC (Method 11). This gave 91 mg (54% of theory) of the title
compound.
'H NMR (400 MHz, CDC13, 8/ppm): 8.19 (d, 2H), 7.30 (d, 2H), 7.20 (t, 1H), 7.19
(s, 1H), 7.09 (d,
1H), 6.96 (d, 1H), 6.78 (s, 1H), 5.43 (s, 2H), 2.89 (s, 2H), 2.26 (s, 3H),
1.41 (s, 6H).
LC/MS (Method 4, ESlpos): R, = 1.29 min, m/z = 455 [M-H3PO4+H]+, 553 [M+H]+.
Example 82
2-Methyl-2-{3-[(5-methyl-3-{ 3-[4-(1,1,1-trifluoro-2-methylpropan-2-yl)phenyl]-
1,2,4-oxadiazol-5-
yl}-1H-pyrazol-1-yl)methyl]phenyl}propyl dihydrogenphosphate
0
H3C CH3 O-N
11
HO-P-0 PN OH N F F
H3C F
H3C CH3
With ice bath cooling, 52 l (0.39 mmol) of bromotrimethylsilane were added to
a solution of
136 mg (0.18 mmol) of the compound from Example 93A in 3.6 ml of
dichloromethane, and the
mixture was stirred at RT overnight. 1 N hydrochloric acid was then added, and
the mixture was
diluted with more dichloromethane. The organic phase was washed with water and
saturated
sodium chloride solution, dried over anhydrous sodium sulphate and filtered.
The filtrate was
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-179-
finally freed from the solvent on a rotary evaporator. This gave 104 mg (95%
of theory) of the title
compound.
'H NMR (400 MHz, DMSO-d6, S/ppm): 8.08 (d, 2H), 7.76 (d, 2H), 7.35-7.24 (m,
3H), 6.92 (d,
2H), 5.49 (s, 2H), 3.83 (d, 2H), 2.33 (s, 3H), 1.60 (s, 6H), 1.25 (s, 6H).
LC/MS (Method 6, ESlpos): R, = 1.13 min, m/z = 579 [M+H]+.
Example 83
2-Methyl-2-{3-[(5-methyl-3-{3-[4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-
yl}-1 H-pyrazol-l -
yl)methyl]phenyl}propyl dihydrogenphosphate
0
11 H3C CH3 O-N
HO-P-0 PN
F
N
O H I / \ F
H3C O F
Analogously to the process described under Example 82, 90 mg (0.12 mmol) of
the compound
from Example 94A gave 58 mg (81% of theory) of the title compound.
'H NMR (400 MHz, DMSO-d6, S/ppm): 8.20 (d, 2H), 7.59 (d, 2H), 7.35-7.28 (m,
3H), 6.94 (d,
IH), 6.93 (s, 1H), 5.50 (s, 2H), 3.84 (d, 2H), 2.35 (s, 3H), 1.26 (s, 6H).
LC/MS (Method 6, ESlpos): R, = 1.09 min, m/z = 553 [M+H]+.
Example 84
2-Methyl-2- { 3-[(5-methyl-3-{ 3-[4-(1,1,1-tri fluoro-2-methylpropan-2-
yl)phenyl]-1,2,4-oxadiazol-5-
yl }-1 H-pyrazol-1-yl)methyl]phenyl }propyl N,N-dimethylglycinate
H3C-N-CH3
H3C CH O-N
p PN
N F F
O I / /
H3C F
H3C CH3
60 mg (0.12 mmol) of the compound from Example 60 and 21 mg (0.13 mmol) of N,N-
dimethylglycyl chloride hydrochloride were initially charged in 2.4 ml of
dichloromethane, 18 pl
(0.13 mmol) of triethylamine were added and the mixture was stirred at RT
overnight. After the
CA 02798374 2012-11-05
= BHC 10 1 009 Foreign Countries
-180-
addition of another 21 mg (0.13 mmol) of N,N-dimethylglycyl chloride
hydrochloride and 18 pl of
triethylamine, the mixture was stirred at RT for another 1 h. The mixture was
then diluted with
ethyl acetate and washed successively with water, saturated sodium bicarbonate
solution and
saturated sodium chloride solution. The organic phase was dried over anhydrous
magnesium
sulphate and filtered. The filtrate was finally freed from the solvent on a
rotary evaporator. This
gave 65 mg (86% of theory) of the title compound.
'H NMR (400 MHz, DMSO-d6, 6/ppm): 8.09 (d, 2H), 7.77 (d, 2H), 7.36-7.29 (m,
3H), 6.98 (d,
1H), 6.93 (s, 1H), 5.50 (s, 2H), 4.11 (s, 2H), 3.05 (s, 2H), 2.34 (s, 3H),
2.10 (s, 6H), 1.61 (s, 6H),
1.29 (s, 6H).
LC/MS (Method 6, ESlpos): R, = 1.12 min, m/z = 584 [M+H]+.
Example 85
(S)-2-Methyl-2-{ 3-[(5-methyl-3-f 3-[4-(1,1,1-trifluoro-2-methylpropan-2-
yl)phenyl]-1,2,4-
oxadiazol-5-yl}-1 H-pyrazol-1-yl)methyl]phenyl}propyl L-valinate
NH2 H3C CH3 O-N
ll_r~ H3C O PN
F F
CH3 O
H 3 C F
H3C CH3
100 mg (0.20 mmol) of the compound from Example 60 were initially charged in 4
ml of
dichloromethane, 73 mg (0.30 mmol) of tert-butyl (4S)-4-isopropyl-2,5-dioxo-
1,3-oxazolidine-3-
carboxylate and 25 mg (0.20 mmol) of 4-N,N-dimethylaminopyridine were added
and the mixture
was stirred at RT overnight. The mixture was then diluted with ethyl acetate
and washed
successively with saturated ammonium chloride solution, water, semi-
concentrated sodium
bicarbonate solution and saturated sodium chloride solution. The organic phase
was dried over
anhydrous magnesium sulphate and filtered, and the filtrate was freed from the
solvent on a rotary
evaporator. At RT, the residue was stirred in 2 ml of a 4 M solution of
hydrogen chloride gas in
dioxane for 30 min. The mixture was then freed from the solvent on a rotary
evaporator.
Purification of the residue by preparative HPLC (Method 17) gave 48 mg (40% of
theory) of the
title compound.
'H NMR (400 MHz, DMSO-d6, 8/ppm): 8.09 (d, 2H), 7.77 (d, 2H), 7.37-7.28 (m,
3H), 6.97 (d,
1 H), 6.93 (s, 1 H), 5.49 (s, 2H), 4.16 (d, 1 H), 4.01 (d, 1 H), 2.98 (d, 1
H), 2.33 (s, 3H), 1.64 (m, 1 H),
1.61 (s, 6H), 1.30 (d, 6H), 0.70 (d, 3H), 0.63 (d, 3H).
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
- 181 -
LC/MS (Method 6, ESlpos): R, = 1.16 min, m/z = 598 [M+H]+.
Example 86 and Example 87
2,2,2-Trifluoro- l -{ 3-[(5-methyl-3-{ 3-[4-(trifluoromethoxy)phenyl]-I,2,4-
oxadiazol-5-yl}-1 H-
pyrazol-l-yl)methyl]phenyl}ethanol (enantiomers 1 and 2)
F
F F
O-N
HO PNN \
N I FF
H 3 C / O \ / F
The racemate from Example 2 was separated chromatographically into its optical
isomers. To this
end, 157 mg (0.316 mmol) of the racemate from Example 2 were dissolved in a
mixture of in each
case 20 ml of isopropanol and isohexane. In portions of in each case 0.5 ml,
this solution was
separated by HPLC on a chiral stationary phase [column material: Daicel
Chiralpak AS-H, 5 m,
250 mm x 20 mm; mobile phase: isohexane/isopropanol 90:10; flow rate: 20
ml/min; temperature:
25 C; UV detection: 230 nm]. The target fractions of the separated enantiomers
were in each case
freed on a rotary evaporator from the solvent and then triturated with a
mixture of 5 ml of pentane
and I ml of diethyl ether at RT. Filtration with suction and drying of the
solid under high vacuum
gave the enantiomers in pure form.
Example 86 (enantiomer 1):
Yield: 48 mg (61 % of theory)
Rt = 4.32 min, >99% ee [analytical chiral HPLC: column: Daicel AS-H, 5 m, 250
mm x 4 mm;
mobile phase: isohexane/isopropanol 60:40; flow rate: 1 ml/min; temperature:
25 C; UV detection:
230 nm].
'H NMR (400 MHz, CDC13, S/ppm): 8.24 (d, 2H), 7.43 (d, 1H), 7.39 (t, 1H), 7.34
(s, 1H), 7.32 (d,
2H), 7.19 (d, 1 H), 6.82 (s, I H), 5.48 (s, 2H), 5.02 (m, 1 H), 2.74 (s,
broad, I H), 2.27 (s, 3H).
LC/MS (Method 6, ESIpos): Rt = 1.27 min, m/z = 499 [M+H]+.
Example 87 (enantiomer 2):
Yield: 41 mg (52% of theory)
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-182-
R, = 4.62 min, 99% ee [analytical chiral HPLC: column: Daicel AS-H, 5 [Lm, 250
mm x 4 mm;
mobile phase: isohexane/isopropanol 60:40; flow rate: 1 ml/min; temperature:
25 C; UV detection:
230 nm].
'H NMR (400 MHz, CDC13, S/ppm): 8.24 (d, 2H), 7.43 (d, 1H), 7.39 (t, IH), 7.35
(s, 1H), 7.32 (d,
2H), 7.19 (d, 1H), 6.82 (s, 1H), 5.48 (s, 2H), 5.02 (m, 1H), 2.77 (s, broad,
1H), 2.27 (s, 3H).
LC/MS (Method 6, ESIpos): Rt = 1.27 min, m/z = 499 [M+H]+.
Example 88 and Example 89
1,1,1-Trifluoro-2-{3-[(5-methyl-3-{ 3-[4-(trifluoromethoxy)phenyl]-1,2,4-
oxadiazol-5-yl }-1 H-
pyrazol-l-yl)methyl]phenyl}propan-2-ol (enantiomers 1 and 2)
F F
H3C F O-N
HO Nz~ PN N F\ F
H3C O F
The racemate from Example 3 was separated chromatographically into its optical
isomers. To this
end, 191 mg (0.373 mmol) of the racemate from Example 3 were dissolved in a
mixture of in each
case 20 ml of isopropanol and isohexane. In portions of in each case 1.0 ml,
this solution was
separated by HPLC on a chiral stationary phase [column material: Daicel
Chiralpak AS-H, 5 m,
250 mm x 20 mm; mobile phase: isohexane/isopropanol 85:15; flow rate: 20
ml/min; temperature:
C; UV detection: 230 nm]. The target fractions of the separated enantiomers
were freed on a
rotary evaporator from the solvent and then triturated with a mixture of 5 ml
of pentane and 1 ml
of diethyl ether at RT. Filtration with suction and drying of the solid under
high vacuum gave the
enantiomers in pure form.
20 Example 88 (enantiomer 1):
Yield: 54 mg (56% of theory)
R, = 5.65 min, >99% ee [analytical chiral HPLC: column: Daicel AS-H, 5 m, 250
mm x 4 mm;
mobile phase: isohexane/isopropanol 60:40; flow rate: 1 ml/min; temperature:
25 C; UV detection:
230 nm].
25 'H NMR (400 MHz, CDC13, 6/ppm): 8.25 (d, 2H), 7.51 (d, 1H), 7.47 (s, IH),
7.38 (t, 1H), 7.33 (d,
2H), 7.14 (d, 1H), 6.83 (s, 1H), 5.49 (s, 2H), 2.58 (s, broad, 1H), 2.28 (s,
3H), 1.77 (s, 3H).
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-183-
LC/MS (Method 6, ESlpos): R, = 1.32 min, m/z = 513 [M+H]+.
Example 89 (enantiomer 2):
Yield: 73 mg (76% of theory)
R, = 6.44 min, >99% ee [analytical chiral HPLC: column: Daicel AS-H, 5 m, 250
mm x 4 mm;
mobile phase: isohexane/isopropanol 60:40; flow rate: 1 ml/min; temperature:
25 C; UV detection:
230 nm].
'H NMR (400 MHz, CDC13, 8/ppm): 8.25 (d, 2H), 7.51 (d, 1H), 7.47 (s, IH), 7.38
(t, 1H), 7.33 (d,
2H), 7.14 (d, IH), 6.83 (s, 1H), 5.49 (s, 2H), 2.58 (s, broad, IH), 2.28 (s,
3H), 1.77 (s, 3H).
LC/MS (Method 6, ESlpos): R, = 1.32 min, m/z = 513 [M+H]+.
Example 90
1-{ 3-[(5-Methyl-3-{ 3-[3-fluoro-4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-5-
yl }-1 H-pyrazol-l -
yl)methyl]phenyl } cyclopropanol
O-N
HO PN N \ FF F
X
H3C O F
Analogously to the process described under Example 22, 410 mg (0.571 mmol,
purity 72%) of the
compound from Example 95A and ethylmagnesium chloride solution gave 100 mg
(37% of theory)
of the title compound. Here, the reaction time at RT was 2 h, and purification
by preparative HPLC
was carried out according to Method 14.
'H NMR (400 MHz, CDC13, 6/ppm): 7.96 (dd, 1H), 7.91 (dd, 1H), 7.55 (t, 1H),
7.32-7.26 (m, 2H),
7.20 (s, 1 H), 7.16 (d, I H), 6.99 (d, I H), 6.81 (s, I H), 5.45 (s, 2H), 2.47
(s, 1 H), 2.28 (s, 3H), 1.69
(s, 6H), 1.27 (dd, 2H), 1.02 (dd, 2H).
LC/MS (Method 6, ESlpos): R, = 1.29 min, m/z = 475 [M+H]+.
Example 91
(1-{ 3-[(3-{ 3-[3-Fluoro-4-(1,1,1-trifluoro-2-methylpropan-2-yl)phenyl]-1,2,4-
oxadiazol-5-yl }-5-
methyl-I H-pyrazol- l -yl)methyl]phenyl } cyclopropyl)methanol
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
- 184 -
O-N
HO pN
F
N F F
H 3 C F
H3C CH3
77 mg (0.687 mmol) of solid potassium tert-butoxide were added to a solution
of 197 mg (0.529
mmol) of the compound from Example 45A and 240 mg (0.582 mmol) of the compound
from
Example 50A in 4 ml of anhydrous THE at 0 C. The reaction mixture was stirred
at RT for 16 h.
793 pl (0.793 mmol) of tetra-n-butylammonium fluoride solution (1 M in THF)
were then added.
After a further 30 min at RT, the mixture was diluted with water and extracted
with ethyl acetate.
After drying of the organic phase over anhydrous magnesium sulphate and
filtration, the solvent
was removed on a rotary evaporator. The crude product obtained was purified by
means of
preparative HPLC (Method 14). The product-containing fractions were combined
and made basic
using saturated sodium bicarbonate solution. All volatile components except
for a small residual
volume of water were then removed on a rotary evaporator. The mixture was
extracted with ethyl
acetate. Drying of the extract over anhydrous magnesium sulphate, filtration
and evaporation gave
76 mg (28% of theory) of the title compound.
'H NMR (400 MHz, CDC13, 6/ppm): 7.97 (d, IH), 7.92 (d, 1H), 7.32-7.25 (m, 3H,
partially
obscured by the CHC13 signal), 7.22 (s, IH), 7.00 (d, IH), 6.81 (s, 1H), 5.44
(s, 2H), 3.66 (s, 2H),
2.29 (s, 3H), 1.69 (s, 6H), 1.57 (broad, IH), 0.85 (s, 4H).
LC/MS (Method 6, ESlpos): Rt = 1.36 min, m/z = 515 [M+H]+.
Example 92
(1-{3-[(3-{3-[4-(4-Fluorotetrahydro-2H-pyran-4-yl)phenyl]-1,2,4-oxadiazol-5-
yl}-5-methyl-1 H-
pyrazol-1-yl)methyl]phenyl}cyclopropyl)methanol
O-N
PN HO
N
F
H3
O
Analogously to the process described in Example 63, 65 mg (0.198 mmol) of the
compound from
Example 43A and 122 mg (0.297 mmol) of the compound from Example 50A were used
to obtain,
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-185-
in two partial steps, 64 mg (66% of theory) of the title compound. Here, in
the first partial step the
reaction mixture was stirred at RT for 16 h; subsequent heating to 40 C was
dispensed with.
'H NMR (400 MHz, CDC13, S/ppm): 8.23 (d, 2H), 7.52 (d, 2H), 7.32-7.25 (m, 2H,
partially
obscured by the CHC13 signal), 7.22 (s, 1H), 7.00 (d, 1H), 6.82 (s, 1H), 5.43
(s, 2H), 4.00-3.87 (m,
4H), 3.65 (s, 2H), 2.29 (s, 3H), 2.27-2.12 (m, 2H), 1.98-1.91 (m, 2H), 1.43
(s, broad, 1H), 0.85 (s,
4H).
LC/MS (Method 6, ESlpos): Rt = 1.18 min, m/z = 489 [M+H]+.
Example 93
2,2-Difluoro-2-[3-({ 3-[3-(4-isopropylphenyl)-1,2,4-oxadiazol-5-yl]-5-methyl-1
H-pyrazol-l -
yl}methyl)phenyl]ethanol
F F O-N
HOJ \ PN
CH3
H3C
CH3
Analogously to the process described in Example 75, 134 mg (0.50 mmol) of the
compound from
Example 41A and 377 mg (1.50 mmol) of the compound from Example 52A were used
to obtain
95 mg (42% of theory) of the title compound.
'H NMR (400 MHz, CDC13, S/ppm): 8.12 (d, 2H), 7.47 (d, 1H), 7.42 (t, IH), 7.36
(s, 1H), 7.35 (d,
2H), 7.23 (d, IH), 6.83 (s, 1H), 5.48 (s, 2H), 3.95 (dt, 2H), 2.98 (sept, 1H),
2.29 (s, 3H), 2.18 (t,
I H), 1.29 (d, 6H).
LC/MS (Method 6, ESlpos): Rt = 1.29 min, m/z = 439 [M+H]+.
Example 94
2,2-Difluoro-2-{3-[(3-{3-[3-fluoro-4-(trifluoromethoxy)phenyl]-1,2,4-oxadiazol-
5-yl}-5-methyl-
I H-pyrazol- l -yl)methyl]phenyl} ethanol
F F O-N
HOJ \ PN__' F
N F F
H3C 0 F
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-186-
Analogously to the process described in Example 75, 164 mg (0.50 mmol) of the
compound from
Example 29A and 377 mg (1.50 mmol) of the compound from Example 52A were used
to obtain
101 mg (41% of theory) of the title compound. Here, the chromatographic
purification on silica gel
was carried out using the mobile phase cyclohexane/ethyl acetate 3:1.
'H NMR (400 MHz, CDC13, S/ppm): 8.08 (dd, IH), 8.03 (d, 1H), 7.49-7.41 (m,
3H), 7.36 (s, 1H),
7.24 (d, IH), 6.83 (s, 1H), 5.50 (s, 2H), 3.95 (dt, 2H), 2.30 (s, 3H), 2.10
(t, 1H).
LC/MS (Method 6, ESlpos): R, = 1.27 min, m/z = 499 [M+H]+.
Example 95
2,2-Difluoro-2-(3-{ [5-methyl-3-(3-{4-[(trifluoromethyl)sulphonyl]phenyl }-
1,2,4-oxadiazol-5-yl)-
1 H-pyrazol-1-yl]methyl}phenyl)ethanol
F F O-N
HOJ PN
N F F
H3C USX X F
O O
Analogously to the process described in Example 75, 179 mg (0.50 mmol) of the
compound from
Example 25A and 377 mg (1.50 mmol) of the compound from Example 52A were used
to obtain
29 mg (10% of theory) of the title compound.
'H NMR (400 MHz, CDC13, 6/ppm): 8.52 (d, 2H), 8.18 (d, 2H), 7.48 (d, 1H), 7.44
(t, 1H), 7.36 (s,
1H), 7.25 (d, IH, partially obscured by the CHC13 signal), 6.83 (s, 1H), 5.50
(s, 2H), 3.96 (t, 2H),
2.31 (s, 3H).
LC/MS (Method 6, ESlpos): Rt = 1.27 min, m/z = 529 [M+H]+.
Example 96
2-Methyl-l-(3-{[5-methyl-3-(3-{4-[(trifluoromethyl)sulphanyl]phenyl}-1,2,4-
oxadiazol-5-yl)-1H-
pyrazol-I-yl]methyl }phenyl)propan-2-yl dihydrogenphosphate
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-187-
OH
I
HO-P-0 O-N
O pN
N F F
H3C C H 3
H3C / S F
28 mg (0.026 mmol) of palladium (10% on activated charcoal) were added to a
solution of 348 mg
(0.519 mmol) of the compound from Example 96A, and the mixture was
hydrogenated in a Parr
apparatus at RT and a hydrogen pressure of I bar. The reaction time was 6 days
in total. During
this time, three more 28-mg-portions of the palladium catalyst were added.
After the reaction had
ended, the catalyst was removed by filtration through a little kieselguhr, and
the filtrate was
evaporated to dryness. The crude product obtained was purified by preparative
HPLC [column: X-
Bridge Shield, 5 m, 100 mm x 19 mm; mobile phase: acetonitrile/water/1% aq.
trifluoroacetic
acid 60:35:5 (0-6 min); flow rate: 25 ml/min; temperature: 25 C; UV detection:
210 nm]. This
gave 131 mg (44% of theory) of the title compound.
'H NMR (400 MHz, DMSO-d6, 6/ppm): 8.22 (d, 2H), 7.93 (d, 2H), 7.28 (t, IH),
7.22 (d, 1H), 7.13
(s, 1H), 7.03 (d, IH), 6.93 (s, IH), 5.48 (s, 2H), 2.92 (s, 2H), 2.33 (s, 3H),
1.31 (s, 6H).
LC/MS (Method 6, ESlpos): R, = 1.17 min, m/z = 471 [M-H3PO4+H]+, 569 [M+H]+.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-188-
B. Assessment of pharmacological activity
The pharmacological activity of the inventive compounds can be demonstrated by
in vitro and in
vivo studies, as known to the person skilled in the art. The usefulness of the
substances according
to the invention can be illustrated by way of example by in vitro (tumour)
cell experiments and in
vivo tumour models such as are described below. The connection between an
inhibition of the HIF
transcription activity and the inhibition of tumour growth is demonstrated by
numerous studies
described in the literature (cf. e.g. Warburg, 1956; Semenza, 2007).
B-1. HIF-luciferase assay
HCT 116 cells were transfected in a stable manner with a plasmid which
contained a luciferase
reporter under the control of an HIF-responsive sequence. These cells were
sown in microtitre
plates [20 000 cells/cavity in RPMI 1640 medium with 10% foetal calf serum
(FCS) and 100 [tg/ml
of hygromycin]. Incubation was carried out overnight under standard conditions
(5% C02, 21%
02, 37 C, moistened). The following morning the cells were incubated with
various concentrations
of the test substances (0-10 gmol/1) in a hypoxia chamber (1% 02). After 24 h,
Bright Glo reagent
(Promega, Wisconsin, USA) was added in accordance with the manufacturer's
instructions, and
after 5 min the luminescence was measured. Cells which were incubated under
normoxia served as
background controls.
The IC50 values from this assay for representative embodiment examples are
listed in the following
table:
Example No. IC50 [nmol/1]
5 0.5
18 0.5
22 0.5
23 0.4
26 0.9
27 0.5
28 0.5
30 0.5
32 0.6
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-189-
Example No. IC50 Inmol/11
46 0.5
53 1.5
57 2.0
60 2.0
62 0.9
72 2.0
79 4.5
86 4.0
89 2.0
90 1.0
91 1.5
The compound 5-[5-methyl-l-(4-methylbenzyl)-1H-pyrazol-3-yl]-3-[4-
(trifluoromethoxy)phenyl]-
1,2,4-oxadiazole, described as Example 1 in WO 2008/141731-A2, which, unlike
the inventive
compounds, does not have a hydroxyalkyl substituent on the benzyl head group,
exhibits an IC50
value in this assay of 30 nmol/l.
B-2. Suppression of HIF target genes in vitro:
Human bronchial carcinoma cells (H460 and A549) were incubated for 16 h with
variable
concentrations of the test substances (1 nM to 10 M) under normoxic
conditions and under a 1 %
oxygen partial pressure (see HIF-luciferase assay). The total RNA was isolated
from the cells and
transcribed into cDNA and the mRNA expression of HIF target genes was analysed
in real-time
PCR. Active test substances already lower the mRNA expression of the HIF
target genes compared
with untreated cells under normoxic conditions, but above all under hypoxic
conditions.
B-3. Human xenograft tumour model:
Human tumour xenograft models in immunodeficient mice were used to assess the
substances. For
this purpose, tumour cells were cultured in vitro and implanted
subcutaneously, or tumour
xenotransplant pieces were transplanted further subcutaneously. The animals
were treated by oral,
subcutaneous or intraperitoneal therapy after the tumour had been established.
The activity of the
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
_190-
test substances was analysed in monotherapy and in combination therapy with
other
pharmacological active substances. In addition, the tumour inhibitory potency
of test substances on
tumours of advanced size (approx. 100 mm2) was characterized. The state of
health of the animals
was checked daily, and the treatments were performed in accordance with animal
welfare
regulations. The tumour area was measured with slide gauges (length L, breadth
B = shorter
dimension). The tumour volume was calculated by the formula (L X B2)/2. The
inhibition in
tumour growth was determined at the end of the study as the T/C ratio of the
tumour areas and
tumour weights and as the TGI value (tumour growth inhibition, calculated from
the formula [1-
(T/C)] x 100) (T = tumour size in the treated group; C = tumour size in the
untreated control
group).
The influence of the test substances on the tumour vessel architecture and the
blood flow within
the tumour was identified with the aid of computer microtomography and
ultrasound microstudies
on treated and untreated tumour-carrying mice.
C. Determination of solubility and stability
The solubility of the progrug compounds according to the invention in aqueous
systems and their
stability to nonspecific hydrolysis and in plasma can be determined by the
following tests:
C-1. Determination of the solubility in aqueous buffer solution pH 6.5:
The test substance was dissolved in DMSO, and aqueous PBS buffer (pH 6.5) was
then added such
that the final DMSO concentration was 1 per cent by volume. The resulting
suspension was shaken
at RT for 24 h. After ultracentrifugation at 224 OOOg for 30 min, the
supernatant was diluted with
DMSO and analysed by HPLC. Quantification was carried out via a two-point
calibration curve of
the test compound in question in DMSO.
The solubilities determined in this manner for some representative working
examples and their
prodrug forms are given below:
Example No. Solubility [mg/1]
43 0.1
81 40
60 <1
82 330
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-191-
Example No. Solubility [mg/1]
61 0.4
83 50
53 0.1
96 20
C-2. Determination of pH-dependent hydrolysis stability:
0.3 mg of the test substance (prodrug) was weighed into a 2 ml HPLC vial, and
0.6 ml of
acetonitrile or acetonitrile/DMSO mixture (with up to 20 per cent by volume of
DMSO) was
added. To dissolve the substance, the sample vessel was placed into an
ultrasound bath for approx.
seconds. Subsequently, 1.0 ml of the particular aqueous buffer solution was
added
(commercially available buffer solutions of pH 2.0, 7.0 and 10.0) and the
sample was treated again
in the ultrasound bath. Over a period of 24 hours at 25 C, 5 l of the sample
solution were
analysed by HPLC every hour for its content of unchanged prodrug or active
ingredient released
10 therefrom by hydrolysis. Quantification was effected via the area
percentages of the corresponding
HPLC peaks.
The stability values for representative prodrug compounds at pH 7.0 (Examples
82 and 84) and pH
6.5 (Example 96) are listed below:
Example 60 % Example 84 % Example 60
Example 82
Time [h] active prodrug active
prodrug
ingredient ingredient
0 100 0 98 2
2 100 0 97 3
4 100 0 98 2
6 100 0 97 3
12 100 0 97 3
24 100 0 95 5
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-192-
% Example 53
% Example 96
Time [h] active
prodrug
ingredient
0 100 0
2 100 0
4 100 0
6 100 0
12 100 0
24 100 0
As a function of pH, the following values were obtained for these examples
after 24 h:
Example 60 % Example 84 % Example 60
Example 82
pH active prodrug active
prodrug
ingredient ingredient
2 99 1 100 0
7 100 0 95 5
100 0 66 34
Example 53
% Example 96
pH active
prodrug
ingredient
2 100 0
6.5 100 0
10 100 0
5 C-3. Determination of plasma stability in vitro:
1 mg of the test substance (prodrug) was weighed into a 2 ml HPLC vial, and
1.5 ml of DMSO and
1 ml of water were added. To dissolve the substance, the sample vessel was
placed into an
ultrasound bath for approx. 10 seconds. 0.5 ml of rat plasma or human plasma
at 37 C was added
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-193-
to 0.5 ml of this solution. The sample was agitated and approx. 10 l were
taken for a first analysis
(time to). Within the period up to 2 hours after commencement of incubation, 4-
-6 further aliquots
were taken for quantification. The sample was kept at 37 C over the test
period. Characterization
and quantification were effected by HPLC.
The stability values in rat plasma are listed below for representative prodrug
examples:
% Example 60 % Example 84 % Example 60
Example 82
Time [h] active prodrug active
prodrug
ingredient ingredient
0 100 0 98 2
0.5 100 0 86 14
1 100 0 77 23
2 100 0 66 34
4 100 0 54 46
24 100 0 34 66
% Example 53
% Example 96
Time [h] active
prodrug
ingredient
0 100 0
0.5 100 0
1 100 0
2 100 0
4 100 0
In human plasma, the following values were obtained for these examples:
Example 60 % Example 84 % Example 60
Example 82
Time [h] active prodrug active
prodrug
ingredient ingredient
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-194-
% Example 60 % Example 84 % Example 60
Example 82
Time [h] active prodrug active
prodrug
ingredient ingredient
0 100 0 98 2
0.5 100 0 98 2
1 100 0 98 2
2 100 0 97 3
4 100 0 97 3
24 100 0 95 5
% Example 53
Example 96
Time [h] active
prodrug
ingredient
0 100 0
0.5 100 0
1 100 0
2 100 0
4 100 0
D. Determination of pharm acokinetic parameters
The pharmacokinetic parameters of the inventive compounds after intravenous or
peroral
administration can be determined as follows:
The substance to be examined was administered to animals (for example mice or
rats)
intravenously as a solution (for example in corresponding plasma with a small
addition of DMSO
or in a PEG/ethanol/water mixture), and peroral administration was effected as
a solution (for
example in Solutol/ethanol/water or PEG/ethanol/water mixtures) or as a
suspension (e.g. in
tylose), in each case via a gavage. After administration of the substance,
blood was taken from the
animals at fixed times. The blood was heparinized, then plasma was obtained
therefrom by
centrifugation. The substance was quantified analytically in the plasma via LC-
MS/MS. From the
plasma concentration/time plots determined in this way, using an internal
standard and with the aid
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-195-
of a validated computer program, the pharmacokinetic parameters, such as AUC
(area under the
concentration/time curve), Cmax (maximum plasma concentration), t1/2 (half
life), Vss (distribution
volume) and CL (clearance), and the absolute and relative bioavailability F
and Ffei (i.v./p.o.
comparison or comparison of suspension to solution after p.o. administration),
were calculated.
To determine the active ingredient release from a prodrug compound, the
prodrug was
administered either intravenously or perorally, as described above, and the
concentrations both of
the prodrug and of the active ingredient released were quantified in the
processed plasma.
D-1. Pharmacokinetic parameters after intravenous administration in rats:
The substance to be examined was administered intravenously to rats, in each
case in amounts
between 0.3 and 1.0 mg/kg as a solution in plasma which contained up to 2%
DMSO. Below, the
kinetic parameters determined in this manner are shown for some exemplary
working examples
[CLplasma = plasma clearance]:
Example No. t12 [h] CLplasma [I/h/kg] Vss 11/kg]
18 7.5 0.7 4.0
23 6.8 1.2 6.6
32 5.5 0.8 4.3
53 6.7 1.1 8.1
57 8.0 1.3 8.6
66 1.4 3.5 4.8
79 7.5 0.9 8.5
90 7.2 0.7 4.7
The compound 5-[5-methyl-l-(4-methylbenzyl)-1H-pyrazol-3-yl]-3-[4-
(trifluoromethoxy)phenyl]-
1,2,4-oxadiazole, described as Example I in WO 2008/141731-A2, which, unlike
the inventive
compounds, does not have a hydroxyalkyl substituent on the benzyl head group,
exhibits the
following data for this parameter after intravenous administration in rats:
t1/2 = 30 h, CLpiasma = 0.4
1/h/kg, Vss = 6.9 1/kg.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-197-
E. Working Examples for pharmaceutical compositions
The compounds according to the invention can be converted to pharmaceutical
formulations as
follows:
Tablet:
Composition:
100 mg of the compound according to the invention, 50 mg of lactose
(monohydrate), 50 mg of
maize starch (native), 10 mg of polyvinylpyrrolidone (PVP 25) (BASF,
Ludwigshafen, Germany)
and 2 mg of magnesium stearate.
Tablet weight 212 mg, diameter 8 mm, radius of curvature 12 mm.
Preparation:
The mixture of the compound according to the invention, lactose and starch is
granulated with a
5% solution (w/w) of the PVP in water. The granules are dried and mixed with
the magnesium
stearate for 5 minutes. This mixture is pressed with a conventional tableting
press (for tablet
dimensions see above). The guide value used for the pressing is a pressing
force of 15 kN.
Suspension which can be administered orally:
Composition:
1000 mg of the inventive compound, 1000 mg of ethanol (96%), 400 mg of
Rhodigel (xanthan
gum from FMC, Pennsylvania, USA) and 99 g of water.
A single dose of 100 mg of the compound according to the invention corresponds
to 10 ml of oral
suspension.
Preparation:
The Rhodigel is suspended in ethanol and the compound according to the
invention is added to the
suspension. The water is added while stirring. The mixture is stirred for
about 6 h until the
swelling of the Rhodigel is complete.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-198-
Solution which can be administered orally:
Composition:
500 mg of the compound according to the invention, 2.5 g of polysorbate and 97
g of polyethylene
glycol 400. A single dose of 100 mg of the compound according to the invention
corresponds to 20
g of oral solution.
Preparation:
The compound according to the invention is suspended in the mixture of
polyethylene glycol and
polysorbate while stirring. The stirring operation is continued until
dissolution of the compound
according to the invention is complete.
i.v. solution:
The compound according to the invention is dissolved in a concentration below
the saturation
solubility in a physiologically acceptable solvent (e.g. isotonic saline,
glucose solution 5% and/or
PEG 400 solution 30%). The solution is subjected to sterile filtration and
dispensed into sterile and
pyrogen-free injection vessels.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
_199-
F. Literature
= Globocan 2002 Report
IARC International Agency for Research on Cancer: Globocan 2002,
http://www-dep.iarc.fr/globocan/downloads.htm
= American Cancer Society, Cancer Facts and Figures 2005
American Cancer Society: Cancer Facts and Figures 2007,
http://www.cancer.org/docroot/STT/content/STT_lx_Cancer Facts_Figures_2007.asp
= Gibbs JB, 2000
Gibbs JB: Mechanism-based target identification and drug discovery in cancer
research,
Science 2000, 287 (5460), 1969-1973.
= Semenza and Wang, 1992
Semenza GL, Wang GL: A nuclear factor induced by hypoxia via de novo protein
synthesis binds
to the human erythropoietin gene enhancer at a site required for
transcriptional activation,
Mol. Cell. Biol. 1992, 12 (12), 5447-5454.
= Wang and Semenza, 1995
Wang GL, Semenza GL: Purification and characterization of hypoxia-inducible
factor 1,
J. Biol. Chem. 1995, 270 (3), 1230-1237.
= Wang, Jiang et al., 1995
Wang GL, Jiang BH, Rue EA, Semenza GL: Hypoxia-inducible factor I is a basic-
helix-loop-
helix-PAS heterodimer regulated by cellular 02 tension, PNAS 1995, 92 (12),
5510-5514.
= Jiang, Rue et al., 1996
Jiang BH, Rue E, Wang GL, Roe R, Semenza GL: Dimerization, DNA binding, and
transactivation
properties of hypoxia-inducible factor 1, J. Biol. Chem. 1996, 271 (30), 17771-
17778.
= Makino, Cao et al., 2001
Makino Y, Cao R, Svensson K, Bertilsson G, Asman M, Tanaka H, Cao Y,
Poellinger L:
Nature 2001, 414 (6863), 550-554.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-200-
Jiang, Semenza et al., 1996
Jiang BH, Semenza GL, Bauer C, Marti HH: Hypoxia-inducible factor I levels
vary exponentially
over a physiologically relevant range of 02 tension, Am. J. Physiol. 1996,
271, 1172-1180).
= Maxwell, Wiesener et al., 1999
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff
CC,
Ratcliffe PJ: The tumour suppressor protein VHL targets hypoxia-inducible
factors for oxygen-
dependent proteolysis, Nature 1999, 399 (6733), 271-275.
= Hirota and Semenza, 2006
Hirota K, Semenza GL: Regulation of angiogenesis by hypoxia-inducible factor
1,
Crit. Rev. Oncol. Hematol. 2006, 59 (1), 15-26.
= Chen, Zhao et al., 2003
Chen J, Zhao S, Nakada K, Kuge Y, Tamaki N, Okada F, Wang J, Shindo M,
Higashino F, Takeda
K, Asaka M, Katoh H, Sugiyama T, Hosokawa M, Kobayashi M: Dominant-negative
hypoxia-
inducible factor-1 alpha reduces tumorigenicity of pancreatic cancer cells
through the suppression
of glucose metabolism, Am. J. Pathol. 2003, 162 (4), 1283-1291.
= Stoeltzing, McCarty et al., 2004
Stoeltzing 0, McCarty MF, Wey JS, Fan F, Liu W, Belcheva A, Bucana CD, Semenza
GL, Ellis
LM: Role of hypoxia-inducible factor-lalpha in gastric cancer cell growth,
angiogenesis, and
vessel maturation, J. Natl. Cancer Inst. 2004, 96 (12), 946-956.
= Li, Lin et al., 2005
Li L, Lin X, Stayer M, Shoemaker A, Semizarov D, Fesik SW, Shen Y: Evaluating
hypoxia-
inducible factor-1 alpha as a cancer therapeutic target via inducible RNA
interference in vivo,
Cancer Res. 2005, 65 (16), 7249-7258.
= Mizukami, Jo et al., 2005
Mizukami Y, Jo WS, Duerr EM, Gala M, Li J, Zhang X, Zimmer MA, Iliopoulos 0,
Zukerberg
LR, Kohgo Y, Lynch MP, Rueda BR, Chung DC: Induction of interleukin-8
preserves the
angiogenic response in HIF-lalpha-deficient colon cancer cells, Nat. Med.
2005, 11 (9), 992-997.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-201 -
= Li, Shi et al., 2006
Li J, Shi M, Cao Y, Yuan W, Pang T, Li B, Sun Z, Chen L, Zhao RC: Knockdown of
hypoxia-
inducible factor-lalpha in breast carcinoma MCF-7 cells results in reduced
tumor growth and
increased sensitivity to methotrexate, Biochem. Biophys. Res. Commun. 2006,
342, 1341-1351).
= Semenza, 2007
Semenza GL: Drug Discov. Today 2007, 12 (19-20), 853-859.
= Weidemann and Johnson, 2008
Weidemann A, Johnson RS: Cell Death and Differentiation 2008, 15, 621-627.
= Aiello et al., 1994
Aiello et al.: New Engl. J. Med. 1994, 331, 1480.
= Peer et al., 1995
Peer et al.: Lab. Invest. 1995, 72, 638.
= Lopez et al., 1996
Lopez et al.: Invest. Ophthalmol. Vis. Sci. 1996, 37, 855.
= Warburg, 1956
Warburg 0: Science 1956, 123 (3191), 309-314.
CA 02798374 2012-11-05
BHC 10 1 009 Foreign Countries
-196-
D-2. Pharmacokinetic parameters after peroral administration in rats:
The substance to be examined was administered perorally to rats, in each case
in amounts between
1 and 3 mg/kg as a solution in Solutol/ethanol/water (40:10:50 or 40:20:40).
Below, the kinetic
parameters determined in this manner are shown for some exemplary working
examples [AUCõ
= dose-standardized exposition (area under the concentration/time curve)]:
Example No. t112 [h] AUCõorm [kg-h/11 F [%]
18 7.9 0.75 54
53 11 0.99 111
57 8.6 0.28 36
The compound 5-[5-methyl-l-(4-methylbenzyl)-IH-pyrazol-3-yl]-3-[4-
(trifluoromethoxy)phenyl]-
1,2,4-oxadiazole, described as Example 1 in WO 2008/141731-A2, which, unlike
the inventive
compounds, does not have a hydroxyalkyl substituent on the benzyl head group,
exhibits the
following data for this parameter after peroral administration in rats: t112 =
29 h, AUCõo,,,, = 1.9
kg=h/l, F = 74%.
D-3. Active compound release in vivo after intravenous prodrug administration
in the rat:
Following intravenous administration of the prodrug from Example 81 (0.35
mg/kg, dissolved in
plasma with 1% DMSO added) to rats, there was a rapid and complete release of
the
corresponding active compound (Example 43): The half-life for the release was
about 2 minutes,
and the dose-standardized exposition (AUCno,,,, value) of the free active
compound following
prodrug administration was 3.1 kg=h/l and was therefore - within the limits of
measurement
accuracy - identical to the value after direct administration of the active
compound (3.0 kg-h/1).