Note: Descriptions are shown in the official language in which they were submitted.
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
Energy Recovery Ventilation Sulfonated Block
Copolymer Laminate Membrane
Field of the Invention
The present disclosure relates to a membrane for use in an energy recovery
ventilation core unit. In particular, the present disclosure relates to a
membrane made up of a
microporous substrate laminated with a sulfonated block copolymer having at
least at least
two polymer end blocks that contain little or no sulfonic acid or sulfonate
functionality and at
least one polymer interior block which contains an effective amount of
sulfonic acid or
sulfonate functionality. The present disclosure further relates to an energy
recovery
ventilation unit having a core which employs such membranes.
Background of the Invention
It is well known that heating and cooling systems are employed for temperature
control of buildings and various housing. Often fresh air will be ushered from
outside of the
building or house while exhaust air from within will be returned outdoors.
Generally, a large
amount of energy is expended in such cooling and heating systems. One way to
conserve the
cost for this expenditure of energy is by exchanging some of the heat and
moisture between
the air streams as they are entering and exiting the structure.
Accordingly such systems for exchanging the heat and humidity of the air
streams
have come to be known as energy recovery ventilation (ERV) systems. ERV
involves the
sensible and latent heat exchange of exhaust inside air with fresh outdoor
air. The basis for
such exchange is that the exhaust air flow and the intake airflow will possess
different water
vapor pressures and will furthermore be at different temperatures. For
example, in summer if
the intake airflow is warm and humid, energy is recovered by exchanging both
the sensible
heat and the latent heat with the cool and low humidity exhaust air.
Alternatively, in winter,
.. if the outdoor air is cold and dry, energy is recovered by exchanging both
the dry cold air
with the warmer, more humid exhaust air.
ERV systems are usually employed in conjunction with a heating and/or cooling
system, and are made up of a device having an ERV core unit. The core unit is
generally
comprised of various stacked membranes separated by some type of barrier. The
intake and
- 1 -
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
exhaust air streams are transported to the core unit and made to pass by one
another without
intermixture on each side of the stacked plates.
Sensible heat exchange is generally simpler to accomplish since a thin layer
barrier
may transfer heat rather easily. On the other hand, latent heat transfer is
affected by the
change in humidity between the air streams. Accordingly, what is needed
therefore is a
system that allows for both the efficient exchange of both sensible and latent
heat of the
various intake and exhaust air flows.
Summary of the Invention
Latent heat is effected in large proportion by the change in humidity of the
incoming
and exiting air. Accordingly, latent heat transfer becomes in large degree a
function of the
ERV membrane's ability to transport water vapor between the two aiflow
streams.
What has been found and disclosed herein is an ERV system for the improved
sensible and latent heat exchange between inflow and outflow air streams. This
has been
achieved by a membrane disclosed herein made up of a microporous substrate
with a
laminate having a sulfonated block copolymer. The sulfonated block copolymer
has high
water vapor transport rates, thus facilitating efficient latent heat exchange.
In some embodiments, disclosed herein is a laminate membrane for a core in an
energy recovery system for the exchange of heat and moisture between air
streams passing
through the system, the membrane including:
a fibrous microporous support substrate,
a sulfonated block copolymer having at least one end block A and at least one
interior
block B wherein each A block contains essentially no sulfonic acid or
sulfonate ester
functional groups and each B block is a polymer block containing from about 10
to about
100 mol percent sulfonic acid or sulfonate ester functional groups based on
the number of
monomer units,
wherein the sulfonated block copolymer is laminated on the microporous support
substrate,
Furthermore, the membrane can include a spacer element interposed between
itself
and a second membrane, the spacer and the membrane forming a layer in an ERV
core. In
further embodiments, the core has a plurality of said layers stacked one upon
the other.
- 2 -
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
In other embodiments, the microporous substrate is a fibrous woven or non-
woven
material. In further embodiments, the microporous substrate is selected from
the group
consisting of carbon, fiberglass, polyester, polyethylene, polyethylene
terephthalate,
cellulose, cellulose nitrate, cellulose acetate, nylon,
polytetrafluoroethylene.
In some embodiments, the sulfonated block copolymer is heat laminated, solvent
laminated or adhesive laminated onto the microporous support substrate.
Furthermore, the
sulfonated block copolymer laminate can be blended with additional
hydrogenated and non-
hydrogenated thermoplastic elastomeric styrenic block copolymers.
In other embodiments, each A block comprises one or more segments selected
from
polymerized (i) para-substituted styrene monomers, (ii) ethylene, (iii) alpha
olefins of 3 to 18
carbon atoms; (iv) 1,3-cyclodiene monomers, (v) monomers of conjugated dienes
having a
vinyl content less than 35 mol percent prior to hydrogenation, (vi) acrylic
esters, (vii)
methacrylic esters, and (viii) mixtures thereof.
In other embodiments, each B block comprises segments of one or more vinyl
aromatic monomers selected from polymerized (i) unsubstituted styrene
monomers, (ii)
ortho-substituted styrene monomers, (iii) meta-substituted styrene monomers,
(iv) alpha-
m ethyl styrene, (v) 1,1-di ph enyl ethyl en e , (vi) 1 ,2-diph enyl ethyl en
e and (vii) mixtures thereof.
In other embodiments, the sulfonated block copolymer has the general
configuration
A-B-A, A-B-A-B-A, (A-B-A)õX, (A-B)11X, A-D-B-D-A, A-B-D-B-A, (A-D-B)õ,X, (A-B-
D)X or mixtures thereof, where n is an integer from 2 to about 30, and X is a
coupling agent
residue and wherein each D block is a polymer block resistant to sulfonation
and the plurality
of A blocks , B blocks, or D blocks are the same or different.
In other embodiments, each D block is selected from the group consisting of
(i) a
polymerized or copolymerized conjugated diene selected from isoprene, 1,3-
butadiene
having a vinyl content prior to hydrogenation of between 20 and 80 mol
percent, (ii) a
polymerized acrylate monomer, (iii) a silicon polymer, (iv) polymerized
isobutylene and (v)
mixtures thereof, wherein any segments containing polymerized 1,3-butadiene or
isoprene
are subsequently hydrogenated
- 3 -
In still other embodiments, disclosed herein is an energy recovery system
having a core
unit permitting heat and moisture exchange between at least two air streams,
the core unit
including:
a plurality of spacer elements arranged in a stacked configuration, the spacer
elements
forming air passageways configured for the flow of at least two independent
air streams
therethrough,
a plurality of laminated membranes with the spacer interposed therebetween,
the
membranes being comprised of a laminated microporous fibrous substrate
support, the laminate
layer comprising a sulfonated block copolymer, the sulfonated block copolymer
having at least
one end block A and at least one interior block B wherein each A block
contains essentially no
sulfonic acid or sulfonate ester functional groups and each B block is a
polymer block containing
from about 10 to about 100 mol percent sulfonic acid or sulfonate ester
functional groups based
on the number of monomer units of the B block.
In further embodiments, the passageways are made up of two sets of passageways
with a
first set arranged in a first direction and a second set arranged in a second
direction different from
said first direction thereby enabling the at least two independent air streams
to have a cross-flow
pattern.
In further embodiments, the spacer element is metal, fiberglass or plastic.
Additionally, the
microporous substrate can be fibrous woven or non-woven material. Furthermore,
the microporous
substrate is selected from the group consisting of carbon, fiberglass,
polyester, polyethylene,
polyethylene terephthalate, cellulose, cellulose nitrate, cellulose acetate,
nylon,
polytetrafluoroethylene. In other embodiments, the sulfonated block copolymer
is heat laminated,
solvent laminated or adhesive laminated on the microporous support substrate.
In other embodiments, there is provided a laminate membrane for a core in an
energy
recovery system for the exchange of heat and moisture between air streams
passing
through said system, said membrane comprising: a fibrous microporous support
substrate, a film
comprising a sulfonated block copolymer having at least one end block A and at
least one interior
block B wherein each A block contains essentially no sulfonic acid or
sulfonate ester functional
groups and each B block is a polymer block containing from about 10 to about
100 mol percent
sulfonic acid or sulfonate ester functional groups based on the number of
monomer units, wherein
-4-
CA 2810409 2018-09-14
the film is heat laminated, solvent laminated or adhesive laminated on the
microporous support
substrate.
In other embodiments, there is provided an energy recovery system having a
core unit
permitting heat and moisture exchange between at least two air streams, said
core unit comprising:
a plurality of spacer elements arranged in a stacked configuration, said
spacer elements forming
air passageways configured for the flow of at least two independent air
streams therethrough, a
plurality of laminated membranes with said spacer elements interposed
therebetween, said
membranes comprising a laminate layer on_a fibrous microporous substrate
support, said laminate
layer comprising a heat laminated, or adhesive laminated film of a sulfonated
block copolymer,
said sulfonated block copolymer having at least one end block A and at least
one interior block B,
wherein each A block contains essentially no sulfonic acid or sulfonate ester
functional groups and
each B block is a polymer block containing from about 10 to about 100 mol
percent sulfonic acid
or sulfonate ester functional groups based on the number of monomer units of
the B block.
Brief Description of the Drawings
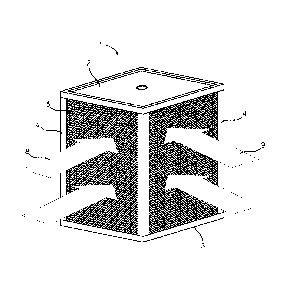
Fig. 1 illustrates a perspective view of an ERV core unit.
Fig. 2 illustrates an air exchange membrane with spacer element.
Fig. 3 illustrates spacer elements in a transverse configuration.
Fig. 4 illustrates an arrangement of stacked spacer elements.
Fig. 5 illustrates spacers having elongate ribs.
-4a-
CA 2810409 2018-09-14
81633953
Fig. 6 illustrates spacers made up of plates.
Fig. 7 illustrates a roller assembly.
Detailed Description of the Invention
A detailed description of embodiments of the present invention is disclosed
herein;
however, it is to be understood that the disclosed embodiments arc merely
exemplary, and
that the invention may be embodied in various and alternative forms of the
disclosed
embodiments. Therefore, specific structural and functional details which are
addressed in the
embodiments disclosed herein are not to be interpreted as limiting, but merely
as a basis for
the claims and as a representative basis for teaching one skilled in the art
to variously employ
the present invention.
In the event of conflict between this specification and publications, patent
applications, and
patents referenced herein, the present specification, including definitions,
is intended to control.
Unless specifically stated otherwise, all technical terms used herein have the
meaning
as commonly understood by those skilled in the art.
Moreover, unless specifically stated otherwise, the following expressions as
used
herein are understood to have the following meanings.
Unless specifically stated otherwise, the expression "coated" or "coating"
means the
application or bonding of a polymer in solution or liquid form to a substrate
or other material.
In contrast to "coated," unless specifically stated otherwise, the expression
"lamination" means the application or bonding of a cast polymer membrane or
polymer film
to a substrate or other material.
Disclosed herein is an improved ERV system for the exchange of sensible and
latent
heat between an intake airflow stream and an exhaust airflow stream. ERV
systems employ
a core unit having a stack of multiple moisture permeable membranes separated
by spacers.
Both the intake airflow streams as well as the exiting airflow streams are
transported to the
ERV core unit. In the core unit, the air streams are separated by the
membranes in the core
unit as they flow past one another. In this way heat and moisture are
exchanged between the
two air streams.
- 5 -
CA 2810409 2018-01-25
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
Energy Recovery Ventilation Core Units
One embodiment of an ERV core unit 1 is shown in Fig. 1. As shown therein the
unit
has a housing made up of a top cover 2 and a bottom cover 3 with side supports
4. Within
the housing is held exchange element 5 made up of a plurality of air exchange
membranes
separated by plurality spacer elements. In the embodiment shown, the fresh
intake air flow
is shown by arrow 8 and furthermore the exhaust air flow is shown by arrow 9.
Referring now to Fig. 2, illustrated is air exchange membranes 6 with spacer
elements
7 interposed therebetween. Spacer elements 7 are configured to provide
channels for airflow
between membranes 6. The size of such channels may be such as to provide an
air gap from
about 5 to 30 mm. In the embodiment shown in Fig. 2, this is done by forming
the spacers 7
into a ridged formation. The crests thus form longitudinal apertures along the
length of the
spacers in one direction, thus permitting flow both above and below the spacer
depending on
the formation of the ridges. Such spacers may be made up of fiberglass,
aluminum or plastic.
Other materials may similarly be used which provide strength and maintain the
membranes
separated for permitting and directing airflow. The material should be such
that both air and
moisture is not permitted to pass through the spacers themselves.
In the embodiment shown in Fig. 3, the spacers are ridged and arranged in a
transverse configuration. In this example, one set of spacers 7a are arranged
so that the air
channels are aligned in one longitudinal direction, while a second set of
spacers 7b are
arranged such that the air channels are aligned in a second longitudinal
direction, each set
being stacked in alternating fashion. Accordingly, fresh intake air 8 may be
passed through
the spacer air channels in one direction, while the exhaust air 9 is passed
through the spacer
air channels in a second direction. Furthermore, as membranes are arranged on
either side of
the spacer elements 7, heat and moisture can be exchanged through the membrane
without
intermixture of the different air streams. While Fig. 3 illustrates the
spacers 7a and 7b
disassembled, Fig. 4 shows this arrangement of spacers stacked one on top of
the other as
they would be in a core unit (with membranes between each).
An example of ERV units have cores with ridged spacers are sold for example by
Innergytech in their enthalpy heat exchanger units. Additionally, for example
US 6,536,514
discloses a ridged spacer with a moisture permeable membranes.
- 6 -
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
While ridged spacers are illustrated in Figs. 2-4, it will be understood that
other types
of spacers maybe used. For example, as shown in Fig 5, instead of being
ridged, the spacers
may be made up of singular elongate ribs 10 adhered to the surface of
membranes 11, which
are which extend along the length of the core unit, with each successive layer
alternating to
form a cross flow pattern. In still other examples, as shown in Figs. 6, the
spacers may be
made up of a plate 12 which can be laid in between membranes. Such plates may
have an
outer frame 13, with longitudinal partitions 14 spaced a distance from one
another. Such
plates may be made up of fiberglass, aluminum or plastic. A membrane 6 is
placed between
plates 12, with plates 12 being stacked in an alternating cross flow pattern.
For example in
Fig. 6, the top plate extends in one direction while the lower plate extends
in a second
direction, which is perpendicular in the illustrative example. The partitions
14 form air
channels 15 which direct the air streams flowing through the ERV unit.
Apertures are placed
in the side of the outer frame 13 to allow air flow in to the air channels 15.
Spacers such as that shown in Figs. 2-6, may have configurations other than
longitudinal, for example, they may be diagonal or jagged, or have other
shapes. By
adjusting the shape of the partitions the residence time of the air streams in
the ERV core can
be increased thereby improving heat and moisture exchange. Furthermore, such
spacers can
be stacked such that the intake and exhaust air streams have cross flows in
any direction, for
example transversely, right angles, or any non-parallel configuration.
Further, parallel
configurations can be employed as well if the gases can be prevented from
being intermixed
as they enter or exit the core.
Energy Recovery Ventilation Membranes
The spacers are provided in order to allow membrane surfaces to contact the
intake
and exhaust air flow streams in a non-parallel direction one on each side of a
membrane
without intermixture thereof. The membranes allow transfer of moisture between
the air
streams, and thus allowing sensible heat exchange. Accordingly, the ability to
more
efficiently transfer moisture greatly affects the effectiveness and efficiency
of the ERV unit.
As disclosed herein, it has been found a surprisingly improved ERV membrane
having a
polymer film laminated to a substrate for improved water vapor transport
between the intake
and exhaust gas streams
- 7 -
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
a. Membrane substrate
The substrate is employed with the membrane to provide mechanical strength
while
additionally facilitating water vapor transport. Accordingly, it should be
made of a porous
material to allow moisture to pass through with as little resistance as
possible while also
providing structural integrity. The porous substrate may be those known and
used in the art,
many of which are commercially available.
Accordingly, the substrate to be used with the membrane disclosed herein
includes
porous cellulosic fibrous materials. Microporous films may also be used.
Materials include,
for example, fabrics, polymeric films and fibers, and cellulosic materials
(such as paper).
The substrate may be composed of natural and/or synthetic fibers. Fabrics
include wovens,
non-wovens, knits and cross-laid fabrics.
Further, the substrate may be composed of filaments, glass yams, fiberglass,
non-
corroding metal fibers (such as nickel fibers), as well as well as carbon
fibers. Synthetic
fibers include polyolefms, polyethylene, polypropylene, and polyesters.
Exemplary substrates also include polyvinylidene fluoride,
polytetrafluoroethylene,
nylon, polyethersulfone, polypropylene, polyamide, cellulose, cellulose
nitrate, cellulose
acetate, cellulose nitrate/acetate po I ytetrafluoroethyl ene, Polyethylene
Terephth al ate (PET),
and Polyether ether ketone (PEEK).
Additives or coatings (other than coating of the sulfonated polymer) may be
added to
.. the substrate to improve other properties. Such additives should not
interfere with the
effectiveness and efficiency of the ERV unit, or introduce any harmful
components into the
air streams. One type of additives is flame retardants which may be employed
to inhibit or
prevent fire or the spread of fire. For example, non-halogen flame retardants
may be
employed as well as phosphorus containing compounds. Halogen flame retardants
may
.. include bromine containing retardants. Other useful flame retardants known
in the art may
be used.
Biocides may also be applied, including fungicides, microbicides and
bactericides, for
preventing growth of molds, mildew, fungus, bacteria, viruses, and parasites
as well as other
biological organisms that may be harmful to humans or reduce the efficiency of
the ERV
unit.
- 8 -
81633953
Other additive; may be added to the substrate to increase its strength,
porosity and
life, or reduce odor such as anti-oxidants, silica, alumina and zeolites.
b. Membrane Polymer
The membrane to be used in the ERV core disclosed herein is a polymeric film
layer
laminated onto a porous substrate. As disclosed herein, the polymeric film is
composed of or
includes a sulfonated block copolymer. It has been surprisingly found that the
when the
sulfonated Nock copolymer disclosed herein is laminated onto a porous
substrate, water
vapor transport, and thus latent heat exchange, is significantly improved.
In some
embodiments, the sulfonated block copolymers which are used in the polymer
film layer
include sulfonated block copolymers as described in US 2007/0021569 to Willis
et al.
Furthermore, the sulfonated block copolymers which include the sulfonated
block
copolymers as described in US 2007/0021569 may be prepared according to the
process
of WO 2008/089332 to Dada et al.
1. Sulfonated Block Copolymers
The block copolymers needed to prepare the sulfonated block copolymers may be
made by a number of different processes, including anionic polymerization,
moderated
anionic polymerization, cationic polymerization, Ziegler-Natta polymerization,
and living
chain or stable free radical polymerization. Anionic polymerization is
described below in
more detail, and in the referenced documents. Moderated anionic polymerization
processes
for making styrenic block copolymers are disclosed, for example, in US
6,391,981,
US 6,455,651 and US 6,492,469. Cationic polymerization processes for preparing
block
copolymers are disclosed, for example, in US 6,515,083 and US 4,946,899.
Living Ziegler-Natta polymerization processes that can be used to make block
copolymers were recently reviewed by G.W. Coates, P.D. Hustad, and S. Reinartz
in Angew.
Chem. Int. Ed., 41, 2236-2257 (2002); a subsequent publication by H. Zhang and
K. Nomura
(J. Am. Chem. Soc., Comm., 2005) describe living Ziegler-Natta techniques for
making
styrenic block copolymers specifically. The extensive work in the field of
nitroxidc mediated
- 9 -
CA 2810409 2018-01-25
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
living radical polymerization chemistry has been reviewed; see C.J. Hawker,
A.W. Bosman,
and E. Harth, Chem. Rev., 101(12), 3661-3688 (2001). As outlined in this
review, styrenic
block copolymers can be synthesized by living or stable free radical
techniques. Nitroxide
mediated polymerization methods are preferred living chain or stable free
radical
polymerization processes when preparing the precursor polymers.
2. Polymer Structure
One aspect of the disclosure relates to the polymer structure of the
sulfonated block
copolymers. In one embodiment, the neutralized block copolymers have at least
two polymer
end or outer blocks A and at least one saturated polymer interior block B
wherein each A
block is a polymer block which is resistant to sulfonation and each B block is
a polymer
block which is susceptible to sul fon ati on .
Preferred block copolymer structures have the general configuration A-B-A, (A-
B)n(A), (A-B-A)n, (A-B-A)nX, (A-B)nX, A-B-D-B-A, A-D-B-D-A, (A-D-B)n(A), (A-B-
D)n(A), (A-B-D)nX, (A-D-B)nX or mixtures thereof, where n is an integer from 2
to about
30, X is coupling agent residue and A, B and D are as defined hereinafter.
[00001] Most preferred structures are linear structures such as A-B-A, (A-
B)2X, A-B-D-
B-A, (A-B-D)2X, A-D-B-D-A, and (A-D-B)2X and radial structures such as (A-B)nX
and
(A-D-B)nX where n is 3 to 6. Such block copolymers are typically made via
anionic
polymerization, stable free radical polymerization, cationic polymerization or
Ziegler-Natta
polymerization. Preferably, the block copolymers are made via anionic
polymerization. It
will be understood by those skilled in the art that in any polymerization, the
polymer mixture
will include a certain amount of A-B diblock copolymer, in addition to any
linear and/or
radial polymers. The respective amounts have not been found to be detrimental.
The A blocks are one or more segments selected from polymerized (i) para-
substituted styrene monomers, (ii) ethylene, (iii) alpha olefins of 3 to 18
carbon atoms; (iv)
1,3-cyclodiene monomers, (v) monomers of conjugated dienes having a vinyl
content less
than 35 mol percent prior to hydrogenation, (vi) acrylic esters, (vii)
methacrylic esters, and
(viii) mixtures thereof. If the A segments are polymers of 1,3-cyclodiene or
conjugated
- 10 -
81633953
dicnes, the segments will be hydrogenated subsequent to polymerization of the
block
copolymer and before sulfonation of the block copolymer.
The para-substituted styrene monomers arc selected from para-methylstyrene,
para-
ethylstyrene, para-n-propylstyrene, paru-iso-propylstyrene, para-n-
butylstyrene, par-see-
butylstyrene, para-iso-butylstyrene, para-t-butylstyrene, isomers of partt-
decylstyrene,
isomers of para-dodecylstyrene and mixtures of the above monomers. Preferred
pars-
substituted styrene monomers are para-t-butylstyrene and para-methylstyrene,
with para-t-
butylstyrene being most preferred. Monomers may be mixtures of monomers,
depending on
the particular source. It is desired that the overall purity of the para-
substituted styrene
monomers be at least 90%-wt,, preferably at least 95%-wt., and even more
preferably at least
98%-vet, of the desired pars-substituted styrene monomer.
When the A blocks are polymer segments of ethylene, it may be useful to
polymerize
ethylene via a Ziegler-Natta process, as taught in the references in the
review article by G.W.
Coates et al, as cited above, which disclosure is herein incorporated by
reference. It is
preferred to make the ethylene blocks using anionic polymerization techniques
as taught in
US 3,450,795. The block molecular weight for such ethylene blocks will
typically be between
about 1,000 and about 60,000.
When the A blocks are polymers of alpha olefins of 3 to 18 carbon atoms, such
polymers are prepared by via a Ziegler-Natta process, as taught in the
references in the
above-cited review article by G.W. Coates et al. Preferably, the alpha-olefins
arc propylene,
butylene, hexene or octene, with propylene being most preferred. The block
molecular
weight for each of such alpha-olefin blocks typically is between about 1,000
and about
60,000.
When the A blocks are hydrogenated polymers of 1,3-cyclodiene monomers, such
monomers are selected fmm the group consisting of 1,3-cyclohexadiene, 1,3-
cycloheptadiene
and 1,3-cyclooetadiene. Preferably, the cyclodiene monomer is 1,3-
cyclohexadiene.
Polymerization of such cyclodiene monomers is disclosed in US 6,699,941.
It will be necessary to hydrogenate the A blocks when using cyclodiene
monomers since
non-hydrogenated polymerized cyclodiene blocks are
- 11 -
CA 2810409 2018-01-25
81633953
susceptible to sulfonation. Accordingly, after synthesis of the A block with
1,3-cyclodiene
monomers, the block copolymer will be hydrogenated.
When the A blocks are hydrogenated polymers of conjugated acyclic dimes having
a
vinyl content less than 35 mol percent prior to hydrogenation, it is preferred
that the
conjugated diene is 1,3-butadiene. It is necessary that the vinyl content of
the polymer prior
to hydrogenation be less than 35 mol percent, preferably less than 30 mol
percent. In certain
embodiments, the vinyl content of the polymer prior to hydrogenation will be
less than 25
mol percent, even more preferably less than 20 mol percent, and even less than
15 mol
percent with one of the more advantageous vinyl contents of the polymer prior
to
hydrogenation being less than 10 mol percent. In this way, the A blocks will
have a
crystalline structure, similar to that of polyethylene. Such A block
structures are disclosed in
US 3,670,054 and in US 4,107,236.
The A blocks may also be polymer segments of acrylic esters or methacrylic
esters.
Such polymer blocks may be made according to the methods
disclosed in US 6,767,976. Specific examples of the methacrylic
ester include esters of a primary alcohol and methacrylic acid, such as methyl
methacrylate,
ethyl methacrylate, propyl methacrylate, n-butyl methacrylate, isobutyl
methacrylate, hexyi
methacrylate, 2-ethylhexyl methacrylate, dodecyl methacrylate, lauryl
methacrylate,
methoxyethyl methacrylate,
dimethylaminoethyl methacrylate, d iethy I aminoethyl
methacrylate, glycidyl methacrylate, trimethoxysilylpropyl methacrylate,
trifluoromethyl
methacrylate, trifluoroethyl methacrylate; esters of a secondary alcohol and
methacrylic acid,
such as isopropyl methacrylate, cyclohexyl methacrylate and isobornyl
mctliacrylate; and
esters of a tertiary alcohol and methacrylic acid, such as tert-butyl
methacrylate. Specific
examples of the acrylic ester include esters of a primary alcohol and acrylic
acid, such as
methyl acrylate, ethyl acrylate, propyl acrylate, n-butyl acrylate, isobutyl
acrylate, hexyl
acrylate, 2-ethylhexyl acrylate, dodecyl acrylate, lauryl acrylate,
methoxyethyl acrylate,
dimethylaminoethyl acrylate, diethylamino ethyl acrylate,
glycidyl acrylate,
trimethoxysilylpropyl acrylate, trifluoromethyl acrylate, trifluoroethyl
acrylate; esters of a
secondary alcohol and acrylic acid, such as isopropyl acrylate, cyclohexyl
acrylatc and
- 12 -
CA 2810409 2018-01-25
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
isobomyl acrylate; and esters of a tertiary alcohol and acrylic acid, such as
tert-butyl acrylate.
If necessary, as raw material or raw materials, one or more of other anionic
polymerizable
monomers may be used together with the (meth)acrylic ester. Examples of the
anionic
polymerizable monomer that can be optionally used include methacrylic or
acrylic monomers
such as trimethylsilyl methacrylate, N-,N-dimethylmethacrylamide, N,N-
diisopropylmethacrylamide, N,N-diethylmethacrylamide, N,N-
methylethylmethacrylamide,
N,N-di-tert-butylmethacrylamide, trimethylsilyl acrylate, N,N-
dimethylacrylamide, N,N-di-
isopropylacrylamide, N,N-methylethylacrylamide and N,N-di-tert-
butylacrylamide.
Moreover, there may be used a multifunctional anionic polymerizable monomer
having in
the molecule thereof two or more methacrylic or acrylic structures, such as
methacrylic ester
structures or acrylic ester structures (for example, ethylene glycol
diacrylate, ethylene glycol
dimethacrylate, 1,4-butanediol diacrylate, 1,4-butanediol dimethacrylate, 1,6-
hexanediol
diacrylate, 1,6-hexane diol dimethacrylate,
trimethylolpropane triacrylate and
trimethylolpropane trimethacrylate).
In the polymerization processes used to make the acrylic or methacrylic ester
polymer
blocks, only one of the monomers, for example, the (meth)acrylic ester may be
used, or two
or more thereof may be used in combination. When two or more of the monomers
are used in
combination, any copolymerization form selected from random, block, tapered
block and the
like copolymerization forms may be effected by selecting conditions such as a
combination
of the monomers and the timing of adding the monomers to the polymerization
system (for
example, simultaneous addition of two or more monomers, or separate additions
at intervals
of a given time).
The A blocks may also contain up to 15 mol percent of the vinyl aromatic
monomers
such as those present in the B blocks which are addressed in more detail in
the following. In
some embodiments, the A blocks may contain up to 10 mol percent, preferably
they will
contain only up to 5 mol percent, and particularly preferably only up to 2 mol
percent of the
vinyl aromatic monomers as mentioned for the B blocks. However, in the most
preferred
embodiments, the A blocks will contain no vinyl monomers as present in the B
blocks. The
sulfonation level in the A blocks may be from 0 up to 15 mol percent of the
total monomers
in the A block. It will be understood by those skilled in the art that
suitable ranges include
- 13 -
81633953
any combination of the specified mol percents even if the specific combination
and range is
not listed herewith.
The B blocks, in each case, comprises segments of one or more polymerized
vinyl
aromatic monomers selected from unsubstituted styrene monomer, ortho-
substituted styrene
monomers, meta-substituted styrene monomers, alpha-methylstyrene monomer, 1,1-
diphenylethylene monomer, 1,2-diphenylethylene monomer, and mixtures thereof.
In
addition to the monomers and polymers mentioned above, the B blocks may also
comprise a
partially or completely hydrogenated copolymer of such monomer(s) with a
conjugated diene
selected from 1,3-butadiene, isoprene and mixtures thereof, having a vinyl
content of
between 20 and 80 mol percent. These copolymers with partially or completely
hydrogenated
dienes may be random copolymers, tapered copolymers, block copolymers or
controlled
distribution copolymers. In one preferred embodiment, the B blocks are
selectively partially
or completely hydrogenated and comprise a copolymer of conjugated dienes and
the vinyl
aromatic monomers noted in this paragraph. In another preferred embodiment,
the B blocks
arc unsubstitutcd styrene monomer blocks which are saturated by virtue of the
nature of the
monomer and do not require the added process step of hydrogenation. The B
blocks having a
controlled distribution structure are disclosed in US 7,169,848.
US 7,169,848 also discloses the preparation of sulfonated block
copolymers. The B blocks comprising a styrene block are described herein. In a
preferred
embodiment, the B blocks are made up of unsubstituted styrene and will not
require a
separate hydrogenation step.
In another aspect of the present disclosure, the block copolymer includes at
least one
impact modifier block D having a glass transition temperature less than 20 C.
In one
embodiment, the impact modifier block D comprises a hydrogenated polymer or
copolymer
of a conjugated diene selected from isoprene, 1,3-butadiene and mixtures
thereof the
butadiene portion of the polymer block having a vinyl content prior to
hydrogenation of
between 20 and 80 mol percent and the polymer block having a number average
molecular
weight of between 1,000 and 50,000. In another embodiment, the impact modifier
block D
comprises an acrylate or silicone polymer having a number average molecular
weight of
- 14 -
CA 2810409 2018-01-25
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
1,000 to 50,000. In still another embodiment, the impact modifier block D
block is a polymer
block of isobutylene having a number average molecular weight of 1,000 to
50,000.
Each A block independently has a number average molecular weight between about
1,000 and about 60,000 and each B block independently has a number average
molecular
weight between about 10,000 and about 300,000. Preferably each A block has a
number
average molecular weight of between 2,000 and 50,000, more preferably between
3,000 and
40,000 and even more preferably between 3,000 and 30,000. Preferably each B
block has a
number average molecular weight of between 15,000 and 250,000, more preferably
between
20,000 and 200,000, and even more preferably between 30,000 and 100,000. It
will be
understood by those skilled in the art that suitable ranges include any
combination of the
specified number average molecular weights even if the specific combination
and range is
not listed herewith. These molecular weights are most accurately determined by
light
scattering measurements, and are expressed as number average molecular weight.
Preferably,
the sulfonated polymers have from about 8 mol percent to about 80 mol percent,
preferably
from about 10 to about 60 mol percent A blocks, more preferably more than 15
mol percent
A blocks and even more preferably from about 20 to about 50 mol percent A
blocks.
The relative amount of vinyl aromatic monomers which are unsubstituted styrene
monomer, ortho-substituted styrene monomer, meta-substituted styrene monomer,
alpha-
methylstyrene monomer, 1,1-diphenylethylene monomer, and 1,2-diphenylethylene
monomer in the sulfonated block copolymer is from about 5 to about 90 mol
percent,
preferably from about 5 to about 85 mol percent. In alternative embodiments,
the amount is
from about 10 to about 80 mol percent, preferably from about 10 to about 75
mol percent,
more preferably from about 15 to about 75 mol percent, with the most preferred
being from
about 25 to about 70 mol percent. It will be understood by those skilled in
the art that suitable
ranges include any combination of the specified mol percents even if the
specific
combination is not listed herewith.
In a preferred embodiment, the mol percent of vinyl aromatic monomers which
are
unsubstituted styrene monomer, ortho-substituted styrene monomer, meta-
substituted styrene
monomer, alpha-methylstyrene monomer, 1,1-diphenylethylene monomer, and 1,2-
diphenylethylene monomer in each B block is from about 10 to about 100 mol
percent,
- 15 -
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
preferably from about 25 to about 100 mol percent, more preferably from about
50 to about
100 mol percent, even more preferably from about 75 to about 100 mol percent
and most
preferably 100 mol percent. It will be understood by those skilled in the art
that suitable
ranges include any combination of the specified mol percents even if the
specific
combination and range is not listed herewith.
Typical levels of sulfonation are such that each B block contains one or more
sulfonic
functional groups. Preferred levels of sulfonation are 10 to 100 mol percent
based on the mol
percent of vinyl aromatic monomers which are unsubstituted styrene monomer,
ortho-
substituted styrene monomer, meta-substituted styrene monomer, alpha-
methylstyrene
monomer, 1,1-diphenylethylene monomer, and 1,2-diphenylethylene monomer in
each B
block, more preferably about 20 to 95 mol percent and even more preferably
about 30 to 90
mol percent. It will be understood by those skilled in the art that suitable
ranges of
sulfonation include any combination of the specified mol percents even if the
specific
combination and range is not listed herewith. The level of sulfonation is
determined by
titration of a dry polymer sample, which has been re-dissolved in
tetrahydrofuran with a
standardized solution of NaOH in a mixed alcohol and water solvent.
3. Overall Anionic Process to Prepare Polymers
The anionic polymerization process comprises polymerizing the suitable
monomers
in solution with a lithium initiator. The solvent used as the polymerization
vehicle may be
any hydrocarbon that does not react with the living anionic chain end of the
forming
polymer, is easily handled in commercial polymerization units, and offers the
appropriate
solubility characteristics for the product polymer. For example, non-polar
aliphatic
hydrocarbons, which are generally lacking in ionizable hydrogen atoms make
particularly
suitable solvents. Frequently used are cyclic alkanes, such as cyclopentane,
cyclohexane,
cycloheptane, and cyclooctane, all of which are relatively non-polar. Other
suitable solvents
will be known to those skilled in the art and can be selected to perform
effectively in a given
set of process conditions, with polymerization temperature being one of the
major factors
taken into consideration.
- 16 -
81633953
Starting materials for preparing the block copolymers of the present
disclosure
include the initial monomers noted above. Other important starting materials
for anionic
copolymerizations include one or more polymerization initiators, in the
present disclosure
suitable initiators include, for example, alkyl lithium compounds such as s-
butyllithium, n-
butyllithium, t-butyllithium, arnyllithium and the like and other organo
lithium compounds
including di-initiators such as the di-sec-butyl lithium adduct of m-
diisopropenyl benzene.
Other such di-initiators are disclosed in US 6,492,469.
Of the various polymerization initiators, s-butyllithium is
preferred. The initiator can be used in the polymerization mixture (including
monomers and
solvent) in an amount calculated on the basis of one initiator molecule per
desired polymer
chain. The lithium initiator process is well known and is described in, for
example, US
4,039,593 and US Re. 27,145.
Polymerization conditions to prepare the block copolymers of the present
disclosure
are typically similar to those used for anionic polymerizations in general.
The polymerization
is preferably carried out at a temperature of from about -30 C to about 150 C,
more
preferably about 10 C to about 100 C, and most preferably, in view of
industrial limitations,
from about 30 C to about 90 C. The polymerization is carried out in an inert
atmosphere,
preferably under nitrogen, and may also be accomplished under pressure within
the range of
from about 0.5 to about 10 bars. This copolymerization generally requires less
than about 12
hours, and can be accomplished in from about 5 minutes to about 5 hours,
depending upon
the temperature, the concentration of the monomer components, and the
molecular weight of
the polymer that is desired. When two or more of the monomers are used in
combination, any
copolymerization form selected from random, block, tapered block, controlled
distribution
block, and the like copolymerization forms may be utilized.
It will be understood by those skilled in the art that the anionic
polymerization
process may be moderated by the addition of a Lewis acid, such as an aluminum
alkyl, a
magnesium alkyl, a zinc alkyl or combinations thereof. The effects of the
added Lewis acid
on the polymerization process are
- 17 -
CA 2810409 2018-01-25
81633953
1) to lower thc viscosity of the living polymer solution allowing for a
process that
operates at higher polymer concentrations and thus uses less solvent,
2) to enhance the thermal stability of the living polymer chain end which
permits
polymerization at higher temperatures and again, reduces the viscosity of the
polymer solution allowing for the use of less solvent, and
3) to slow the rate of reaction which permits polymerization at higher
temperatures
while using the same technology for removing the heat of reaction as had been
used in the standard anionic polymerization process.
The processing benefits of using Lewis acids to moderate anionic
polymerization
techniques have been disclosed in US 6,391,981, US 6,455,651
and US 6,492,469. Related information is disclosed in US 6,444,767 and
US 6,686,423. The polymer made by such a moderated,
anionic polymerization process can have the same structure as one prepared
using the conventional anionic polymerization process and as such,
this process can be useful in making the polymers of the present
disclosure. For Lewis acid moderated, anionic polymerization processes,
reaction
temperatures between 100 C and 150 C are preferred as at these temperatures it
is possible
to take advantage of conducting the reaction at very high polymer
concentrations. While a
stoichiometric excess of the Lewis acid may be used, in most instances there
is not sufficient
benefit in improved processing to justify the additional cost of the excess
Lewis acid. It is
preferred to use from about 0.1 to about 1 mole of Lewis acid per mole of
living, anionic
chain ends to achieve an improvement in process performance with the
moderated, anionic
polymerization technique.
Preparation of radial (branched) polymers requires a post-polymerization step
called
"coupling". In the above radial formulas n is an integer of from 3 to about
30, preferably
from about 3 to about 15, and more preferably from 3 to 6, and X is the
remnant or residue of
a coupling agent. A variety of coupling agents is known in the art and can be
used in
preparing the block copolymers. These include, for example, dihaloalkanes,
silicon halides,
siloxanes, multifunctional epoxides, silica compounds, esters of monohydric
alcohols with
carboxylic acids, (e.g. methylbenzoate and dimethyl adipate) and epoxidized
oils. Star-
- 18 -
CA 2810409 2018-01-25
81633953
shaped polymers are prepared with polyaIkenyl coupling agents as disclosed in,
for example,
US 3,985,830, US 4,391,949 and US 4,444,953; as well as CA 716,645.
Suitable polyalkenyl coupling agents include divinylbenzene, and preferably
m-divinylbenzene. Preferred are tetra-alkoxysilanes such as
tetra-methoxysilane (TMOS) and tetra-ethoxysilane (TEOS), tri-alkoxysilanes
such
as methyltrimethoxysilane (MTMS), aliphatic diesters such as dimethyl adipate
and diethyl
adipate, and diglycidyl aromatic epoxy compounds such as.diglycidyl ethers
deriving from
the reaction of his-phenol A and epichlorohydrin.
Linear polymers may also be prepared by a post-polymerization "coupling" step.
However, unlike radial polymers, "n" in the above formulas is the integer 2,
and X is the
remnant or residue of a coupling agent.
4. Process to Prepare Hydrogenated Block Copolymers
As noted, in some cases - i.e., (1) when there is a dime in the B interior
blocks, (2)
when the A block is a polymer of a 1,3-cyclodiene, (3) when there is an impact
modifier
block D and (4) when the A block is a polymer of a conjugated diene having a
vinyl content
of less than 35 mol percent - it is necessary to selectively hydrogenate the
block copolymer to
remove any ethylenic unsaturation prior to sulfonation. Hydrogenation
generally improves
thermal stability, ultraviolet light stability, oxidative stability, and,
therefore, weatherability
of the final polymer, and reduces the risk of sulfonating the A block or the D
block.
Hydrogenation can be carried out via any of the several hydrogenation or
selective
hydrogenation processes known in the prior art. Such hydrogenation has been
accomplished
using methods such as those taught in, for example, US 3,595,942,
US 3,634,549, US 3,670,054, US 3,700,633, and US Re. 27,145.
These methods operate to hydrogenate polymers containing ethylenic
unsaturation and are based upon operation of a suitable catalyst. Such a
catalyst, or catalyst
precursor, preferably comprises a Group 8 to 10 metal such as nickel or cobalt
which is
combined with a suitable reducing agent such as an aluminum alkyl or hydride
of a metal
selected from Groups 1, 2 and 13 of the Periodic Table of the Elements,
particularly lithium,
magnesium or aluminum. This preparation can be accomplished in a suitable
solvent or
- 19 -
CA 2810409 2018-01-25
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
diluent at a temperature from about 20 C to about 80 C. Other catalysts that
are useful
include titanium based catalyst systems.
Hydrogenation can be carried out under such conditions that at least about 90
percent
of the conjugated diene double bonds are reduced, and between zero and 10
percent of the
arene double bonds are reduced. Preferred ranges are at least about 95 percent
of the
conjugated diene double bonds reduced, and more preferably about 98 percent of
the
conjugated diene double bonds are reduced.
Once the hydrogenation is complete, it is preferable to oxidize and extract
the catalyst
by stirring the polymer solution with a relatively large amount of aqueous
acid (preferably 1
to 30 percent by weight acid), at a volume ratio of about 0.5 parts aqueous
acid to 1 part
polymer solution. The nature of the acid is not critical. Suitable acids
include phosphoric
acid, sulfuric acid and organic acids. This stirring is continued at about 50
C for from about
30 to about 60 minutes while sparging with a mixture of oxygen and nitrogen.
Care must be
exercised in this step to avoid that an explosive mixture of oxygen and
hydrocarbons is
formed.
5. Process to Make Sulfonated Polymers
According to the multiple embodiments disclosed herein, the above prepared
block
copolymers are sulfonated to obtain a sulfonated polymer product that is in
solution and in
micellar form. In this micellar form, the sulfonated block copolymer can be
neutralized prior
to casting a membrane, and at the same time, the risk of gelling and/or
precipitation of the
sulfonated block copolymer while in solution is reduced.
Without being bound by any particular theory, it is the present belief that
the micelle
structure of the sulfonated block copolymer can be described as having a core
comprising the
sulfonated block or blocks having a substantial amount of spent sulfonating
agent residues
which is surrounded by the sulfonation resistant block or blocks which, in
turn, are swollen
by an organic non-halogenated aliphatic solvent. As will be further described
in more detail
below, the sulfonated blocks are highly polar due to the presence of sulfonic
acid and/or
sulfonate ester functional groups. Accordingly, such sulfonated blocks are
sequestered into a
core, while the outer sulfonation resistant blocks form a shell which is
solvated by a non-
- 20 -
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
halogenated aliphatic solvent. In addition to forming discrete micelles, there
may also be
formation of polymer aggregates. Without being bound by any particular theory,
polymer
aggregates can be described as discrete or non-discrete structures resulting
from association
of polymer chains in ways other than the description provided for micelles,
and/or loosely
aggregated groups of two or more discrete micelles. Accordingly, the solvated
sulfonated
block copolymer in micellar form may include discrete micelles and/or
aggregates of
micelles, with such solution optionally including aggregated polymer chains
having
structures other than the micelle structure.
Micelles can be formed as a result of the sulfonation process, or
alternatively, the
block copolymer may arrange in a micelle structure prior to sulfonation.
In some embodiments, for the formation of micelles, the sulfonation processes
as
described in WO 2008/089332 may be employed. The methods are useful for
preparing
sulfonated styrenic block copolymers as described in US 2007/021569.
After polymerization, the polymer can be sulfonated using a sulfonation
reagent such
as an acyl sulfate in at least one non-halogenated aliphatic solvent. In some
embodiments, the
precursor polymer can be sulfonated after being isolated, washed, and dried
from the reaction
mixture resulting from the production of the precursor polymer. In some other
embodiments,
the precursor polymer can be sulfonated without being isolated from the
reaction mixture
resulting from the production of the precursor polymer.
fi) Solvent
The organic solvent is preferably a non-halogenated aliphatic solvent and
contains a
first non-halogenated aliphatic solvent which serves to solvate one or more of
the sulfonation
resistant blocks or non-sulfonated blocks of the copolymer. The first non-
halogenated
aliphatic solvent may include substituted or unsubstituted cyclic aliphatic
hydrocarbons
having from about 5 to 10 carbons. Non-limiting examples include cyclohexane,
methyl cyclohexane, cyclopentane, cycloheptane, cyclooctane and mixtures
thereof. The most
preferable solvents are cyclohexane, cyclopentane and methylcyclohexane. The
first solvent
may also be the same solvent used as the polymerization vehicle for anionic
polymerization
of the polymer blocks.
-21-
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
In some embodiments, the block copolymer may be in micellar form prior to
sulfonation even in the case of using only a first solvent. The addition of a
second non-
halogenated aliphatic solvent to a solution of the precursor polymer in the
first non-
halogenated aliphatic solvent can result in or assist the "pre-formation" of
polymer micelles
and/or other polymer aggregates. The second non-halogenated solvent, on the
other hand, is
preferably chosen such that it is miscible with the first solvent, but is a
poor solvent for the
sulfonation susceptible block of the precursor polymer in the process
temperature range and
also does not impede the sulfonation reaction. In other words, preferably, the
sulfonation
susceptible block of the precursor polymer is substantially insoluble in the
second non-
halogenated solvent in the process temperature range. In the case where the
sulfonation
susceptible block of the precursor polymer is polystyrene, suitable solvents
which are poor
solvents for polystyrene and can be used as the second non-halogenated solvent
include
linear and branched aliphatic hydrocarbons of up to about 12 carbons, for
example, hexane,
heptane, octane, 2-ethyl hexane, isooctane, nonane, decane, paraffinic oils,
mixed paraffinic
solvents, and the like. One preferred example of the second non- halogenated
aliphatic
solvent is n-heptane.
The pre-formed polymer micelles and/or other polymer aggregates allow the
sulfonation of the polymer to proceed essentially without disabling gelling at
considerably
higher concentration than can be achieved without the addition of the second
solvent. In
addition, this approach can substantially improve the utility of more polar
acyl sulfates, such
as C3 acyl sulfate (propionyl sulfate), in terms of polymer sulfonation
conversion rate and
minimization of by-products. In other words, this approach may improve the
utility of more
polar sulfonation reagents. Such acyl sulfates are further described below.
(ii) Polymer Concentration
In accordance with some embodiments, high levels of styrene sulfonation can be
achieved in a manner that is substantially free of polymer precipitation and
free of disabling
gelling in the reaction mixture, the reaction product, or both, by maintaining
the precursor
polymer concentration below a limiting concentration of the precursor polymer,
at least
during the early stages of sulfonation. It will be understood by those skilled
in the art that
- 22 -
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
minor amounts of polymers may deposit on surfaces as a result of localized
solvent
evaporation in the course of processing in a mixture that is substantially
free of polymer
precipitation. For example, in accordance with some embodiments, a mixture is
considered to
be substantially free of polymer precipitation when no more than 5% of the
polymer in the
mixture has precipitated.
The polymer concentration at which the sulfonation can be conducted depends
upon
the composition of the starting polymer, since the limiting concentration
below which
polymer gelling is non-disabling or negligible depends upon the polymer
composition. As
stated above, the limiting concentration may also be dependent on other
factors such as the
identity of the solvent or the solvent mixture used and the desired degree of
sulfonation.
Generally, the polymer concentration falls within the range of from about 1%-
wt. to about
30%-wt., alternatively from about 1%-wt. to about 20%-wt., alternatively from
about 1%-wt.
to about 15%-wt., alternatively from about 1%-wt. to about 12%-wt., or
alternatively from
about 1%-wt. to about 10%-wt., based on the total weight of a reaction mixture
that is
preferably substantially free of halogenated solvents. It will be understood
by those skilled in
the art that suitable ranges include any combination of the specified mol
percents even if the
specific combination and range is not listed herewith.
In accordance with some embodiments of the presently described technology, the
initial concentration of the precursor block polymer or mixture of precursor
block polymers
should be maintained below the limiting concentration of the precursor
polymer(s),
alternatively in the range of from about 0.1%-wt. to a concentration that is
below the limiting
concentration of the precursor polymer(s), alternatively from about 0.5%-wt.
to a
concentration that is below the limiting concentration of the precursor
polymer(s),
alternatively from about 1.0%-wt. to a concentration that is about 0.1%-wt.
below the
limiting concentration of the precursor polymer(s), alternatively from about
2.0%-wt. to a
concentration that is about 0.1%-wt. below the limiting concentration of the
precursor
polymer(s),alternatively from about 3.0%-wt. to a concentration that is about
0.1%-wt. below
the limiting concentration of the precursor polymer(s), alternatively from
about 5.0%-wt. to a
concentration that is about 0.1%-wt. below the limiting concentration of the
precursor
polymer(s), based on the total weight of the reaction mixture. It will be
understood by those
-23 -
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
skilled in the art that suitable ranges include any combination of the
specified mol percents
even if the specific combination and range is not listed herewith.
At least in some embodiments, maintaining the polymer concentration below the
limiting concentration can result in reaction mixtures with reduced
concentrations of by-
product carboxylic acid relative to the higher concentration conditions that
lead to gelling.
It will be understood by those skilled in the art, however, that during the
production
of the sulfonated polymer in some embodiments of the present technology,
especially in a
semi-batch or continuous production process, the total concentration of the
polymer(s) in the
reaction mixture may be above the limiting concentration of the precursor
polymer.
(iii) Sulfonation agent
According to multiple embodiments, acyl sulfate may be used for sulfonating
the
polymerized block copolymer. The acyl group preferably is derived from a C2 to
C8,
alternatively C3 to Cg, alternatively C3 to C5, linear, branched, or cyclic
carboxylic acid,
anhydride, or acid chloride, or mixtures thereof. Preferably, these compounds
do not contain
non-aromatic carbon-carbon double bonds, hydroxyl groups, or any other
functionality that is
reactive with acyl sulfate or decomposes readily under sulfonation reaction
conditions. For
example, acyl groups that have aliphatic quaternary carbons in the alpha-
position from the
carbonyl functionality (e.g., acyl sulfate derived from trimethylacetic
anhydride) appear to
decompose readily during polymer sulfonation reaction, and preferably should
be avoided in
the presently described technology. Also included in the scope of useful acyl
groups for the
generation of acyl sulfate in the present technology are those derived from
aromatic
carboxylic acids, anhydrides, and acid chlorides such as benzoic and phthalic
anhydride.
More preferably, the acyl group is selected from the group of acetyl,
propionyl, n-butyryl,
and isobutyryl. Even more preferably, the acyl group is isobutyryl. It has
been discovered
that isobutyryl sulfate can afford high degrees of polymer sulfonation and
relatively minimal
by-product formation.
The formation of acyl sulfate from a carboxylic anhydride and sulfuric acid
can be
represented by the following reaction:
- 24 -
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
0 0 0 0 0
õKR 0
+ HO¨S-01-1 ¨*P.
0 0¨S¨OH OH
0
0
Acyl sulfates are subject to slow decomposition during the course of
sulfonation reactions
forming alpha-sulfonated carboxylic acids of the following formula:
0
R'
*LOH
SO3H
In one embodiment of the presently described technology, the acyl sulfate
reagent is
obtained from a carboxylic anhydride and sulfuric acid in a reaction that is
conducted in a
separate "pre-generation" reaction prior to addition to a solution of polymer
in a non-
halogenated aliphatic solvent. The pre-generation reaction can be conducted
with or without
a solvent. When a solvent is used to pre-generate the acyl sulfate, the
solvent is preferably
non-halogenated. Alternatively, the acyl sulfate reagent can be obtained in an
in-situ reaction
within a solution of the polymer in a non-halogenated aliphatic solvent. In
accordance with
this embodiment of the present technology, the molar ratio of anhydride to
sulfuric acid can
be from about 0.8 to about 2, and preferably from about 1.0 to about 1.4. The
sulfuric acid
used in this preferred method preferably has a concentration of about 93% to
about 100% and
more preferably has a concentration of about 95% to about 100%, by weight. It
will be
understood by those skilled in the art that oleum may be used as an
alternative to sulfuric
acid in an in-situ reaction to generate acyl sulfate, provided that the oleum
strength is
sufficiently low so as to avoid or minimize unintended charring of the
reaction mixture.
In another embodiment of the present technology, the acyl sulfate reagent can
be
obtained from a carboxylic anhydride and oleum in a reaction that is conducted
in a separate
"pre- generation" reaction prior to addition to a solution of polymer in
aliphatic solvent,
wherein the oleum strength is in the range of from about 1% to about 60% free
sulfur
-25 -
CA 02810409 2013-03-04
WO 2012/050860 PC T/
U S2011/053599
trioxide, alternatively from about 1% to about 46% free sulfur trioxide,
alternatively from
about 10% to about 46% free sulfur trioxide, and wherein the molar ratio of
anhydride to
sulfuric acid present in the oleum is from about 0.9 to about 1.2.
Additionally, the acyl sulfate reagent can be prepared from a carboxylic
anhydride via
reaction with any combination of sulfuric acid, oleum, or sulfur trioxide.
Further, the acyl
sulfate reagent can be prepared from a carboxylic acid via reaction with
chlorosulfonic acid,
oleum, sulfur trioxide, or any combination thereof. Moreover, the acyl sulfate
reagent can
also be prepared from a carboxylic acid chloride via reaction with sulfuric
acid.
Alternatively, the acyl sulfate may be prepared from any combination of
carboxylic acid,
anhydride, and/or acid chloride.
The sulfonation of polymer styrenic repeat units with the acyl sulfate can be
represented by the following reaction:
0
+ Ro--OH ROH
0
so3H
The acyl sulfate reagent that may be used relative to the moles of sulfonation
susceptible monomer repeat units present in the polymer solution in amounts
ranging from
very low levels for lightly sulfonated polymer products to high levels for
heavily sulfonated
polymer products. The molar amount of the acyl sulfate can be defined as the
theoretical
amount of the acyl sulfate that can be generated from a given method, the
amount being
dictated by the limiting reagent in the reaction. The molar ratio of acyl
sulfate to styrene
.. repeat units (i.e., sulfonation susceptible units) in accordance with some
embodiments of the
present technology may range from about 0.1 to about 2.0, alternatively from
about 0.2 to
about 1.3, alternatively from about 0.3 to about 1Ø
In accordance with at least some embodiments of the presently described
technology,
the degree of sulfonation of the vinyl aromatic monomers susceptible to
sulfonation in the
.. block polymers is greater than about 0.4 milliequivalents (meq) sulfonic
acid per gram
sulfonated polymer (0.4 meq/g), alternatively greater than about 0.6 meq
sulfonic acid per
- 26 -
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
gram sulfonated polymer (0.6 meq/g), alternatively greater than about 0.8 meq
sulfonic acid
per gram sulfonated polymer (0.8 meq/g), alternatively greater than about 1.0
meq sulfonic
acid per gram sulfonated polymer (1.0 meq/g), alternatively greater than about
1.4 meq
sulfonic acid per gram sulfonated polymer (1.4 meq/g). For example, after the
precursor
polymers described above are sulfonated in accordance with the methods of the
presently
described technology, the typical levels of sulfonation are where each B block
contains one
or more sulfonic functional groups. Preferred levels of sulfonation are from
about 10 to about
100 mol percent, alternatively from about 20 to 95 mol percent, alternatively
from about 30
to 90 mol percent, and alternatively from about 40 to about 70 mol percent,
based on the mol
percent of sulfonation susceptible vinyl aromatic monomers in each B block,
which can be,
for example, unsubstituted styrene monomer, ortho-substituted styrene monomer,
meta-
substituted styrene monomer, alp ha-methylstyrene monomer, 1,1-diphenyl
ethylene
monomer, 1,2-diphenyl ethylene monomer, a derivative thereof, or a mixture
thereof. It will
be understood by those skilled in the art that suitable ranges of sulfonation
level include any
combination of the specified mol percents even if the specific combination and
range is not
listed herewith.
The level or degree of sulfonation of a sulfonated polymer can be measured by
NMR
and/or titration methods as known to people skilled in the art, and/or a
method using two
separate titrations as described in the Examples below and may be appreciated
by people
skilled in the art. For example, a resulting solution from the methods of the
present
technology can be analyzed by 1H-NMR at about 60 C ( 20 C). The percentage
styrene
sulfonation can be calculated from the integration of aromatic signals in the
1H-NMR
spectrum. For another example, the reaction product can be analyzed by two
separate
titrations (the "two-titration method") to determine the levels of styrenic
polymer sulfonic
acid, sulfuric acid, and non-polymeric by-product sulfonic acid (e.g. 2-sulfo-
alkylcarboxylic
acid), and then to calculate the degree of styrene sulfonation based on mass
balance.
Alternatively, the level of sulfonation can be determined by titration of a
dry polymer
sample, which has been re-dissolved in tetrahydrofuran with a standardized
solution of
NaOH in a mixture of alcohol and water. In the latter case, rigorous removal
of by-product
acids are preferably ensured.
-27 -
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
Although embodiments for sulfonating polymers are described above in the
context
of acyl sulfate reagents, the utility of other sulfonation reagents are also
contemplated. For
example, the use of those sulfonation reagents derived from
complexing/reacting sulfur
trioxides and phosphate esters such as triethylphosphate has been demonstrated
in the present
technology. The chemistry of such sulfonation reagents is known in the art to
afford aromatic
sulfonation with significant degrees of sulfonic acid alkyl ester
incorporation. As such, the
resultant sulfonated polymers likely contain both sulfonic acid and sulfonic
acid alkyl ester
groups. Other contemplated sulfonation reagents include, but are not limited
to, those derived
from the reaction or complexation of sulfur trioxide with phosphous pentoxide,
polyphophoric acid, 1,4-dioxane, triethylamine, etc.
(iv) Reaction conditions
The sulfonation reaction between the acyl sulfates and sulfonation susceptible
block
copolymers such as aromatic-containing polymers (e.g., styrenic block
copolymers) can be
conducted at a reaction temperature in the range of from about 20 C to about
150 C,
alternatively from about 20 C to about 100 C, alternatively from about 20 C to
about 80 C,
alternatively from about 30 C to about 70 C, alternatively from about 40 C to
about 60 C
(e.g., at about 50 C). The reaction time can be in the range of from
approximately less than 1
minute to approximately 24 hours or longer, dependent on the temperature of
the reaction. In
some preferred acyl sulfate embodiments that utilize in-situ reaction of
carboxylic anhydride
and sulfuric acid, the initial temperature of the reaction mixture can be
about the same as the
intended sulfonation reaction temperature. Alternatively, the initial
temperature may be lower
than the intended subsequent sulfonation reaction temperature. In a preferred
embodiment,
the acyl sulfate can be generated in-situ at about 20 C to about 40 C (e.g.,
at about 30 C) for
.. about 0.5 to about 2 hours, alternatively about 1 to about 1.5 hours, and
then the reaction
mixture can be heated to about 40 C to about 60 C to expedite the completion
of the
reaction.
Although not required, an optional reaction quenching step can be conducted
through
the addition of a quenching agent, which can be, for example, water or
hydroxyl-containing
-28-
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
compounds such as methanol, ethanol, or isopropanol. Typically in such a step,
an amount of
the quenching agent at least sufficient to react with residual unreacted acyl
sulfate may be
added.
In some embodiments of the presently described technology, the sulfonation of
the
aromatic-containing polymer in a non-halogenated aliphatic solvent can be
carried out by
contacting the aromatic-containing polymer with a sulfonation reagent in a
batch reaction or
a semi-batch reaction. In some other embodiments of the present technology,
the sulfonation
can be carried out in a continuous reaction, which can be enabled, for
example, through the
use of a continuous stirred tank reactor or a series of two or more continuous
stirred tank
reactors.
As a result of sulfonation, the micelle cores contain sulfonation susceptible
blocks
having sulfonic acid and/or sulfonate ester functionality which are surrounded
by an outer
shell containing sulfonation resistant blocks of the block copolymer. The
driving force for
this phase segregation (causing the micelle formation) in solution has been
attributed to the
considerable difference in polarity between the sulfonated block(s) and the
non-sulfonated
blocks of the sulfonated block copolymer. The latter blocks are freely
solvable by a non-
halogenated aliphatic solvent, for example the first solvent disclosed above.
On the other
hand, the sulfonated polymer block(s) may arrange to concentrate in the core
of micelle.
Once the sulfonation reaction is completed, the block copolymers can be cast
directly
into an article form (e.g., membrane) without the necessity of isolating the
block copolymer.
In this particular embodiment the polymeric film (e.g., membrane) can be
submerged in
water and will retain its form (solid) while in the water. In other words, the
block copolymer
will not dissolve in water or disperse in water.
(v) Additional components
Further, the copolymers disclosed herein can be compounded with other
components
not adversely affecting the copolymer properties or the membrane formed from
the
sulfonated block copolymer. Further, the disclosed block copolymers may be
blended with a
large variety of other polymers, including olefin polymers, styrene polymers,
tackifying
resins, hydrophilic polymers and engineering thermoplastic resins, with
polymer liquids such
- 29 -
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
ionic liquids, natural oils, fragrances, and with fillers such as nanoclays,
carbon nanotubes,
fullerenes, and traditional fillers such as talcs, silica and the like.
In addition, the sulfonated polymers of the present invention may be blended
with
conventional styrene/diene and hydrogenated styrene/diene block copolymers,
such as the
styrene block copolymers available from Kraton Polymers LLC. These styrene
block
copolymers include linear hydrogenated and non-hydrogenated S-B-S, S-I-S, S-EB-
S, S-EP-
S block copolymers. Also included are radial block copolymers based on styrene
along with
isoprene and/or butadiene and selectively hydrogenated radial block
copolymers.
Additionally, the styrene block copolymers S-B-S, S-I-S, S-EB-S, S-EP-S may be
functionalized, for example with a monocarboxylic or polycarboxylic acid
compound, such
as maleic acid or a derivative such as maleic anhydride. The preferred acid
compounds are
unsaturated monocarboxylic and polycarboxylic-containing acids (C1-Cio) with
preferably at
least one olefinic unsaturation, and anhydrides, salts, esters, ethers and
other substituted
derivatives from such acids. Examples of such materials include maleic acid,
fumaric acid,
itaconic acid, citraconic acid, acrylic acid, acrylic polyethers, acrylic
anhydride, methacrylic
acid, crotonic acid, isocrotonic acid, mesaconic acid, angelic acid, maleic
anhydride, itaconic
anhydride and citraconi c anhydride. The preferred monomers for fun cti on al
i zing styreni c
block copolymers are maleic anhydride, maleic acid, fumaric acid and their
derivatives.
These functionalized styrenic block copolymers (F-SBC) may be blended with the
sulfonated
block copolymer (S-SBC) in a ratio (F-SBC/S-SBC) of 20/80 to 80/20, more
preferably from
30/70 to 70/30 or most preferably 60/40 to 40/60. Additionally, other acid
functionalities
may be used as well as known as the art.
Olefin polymers include, for example, ethylene homopolymers, ethylene/alpha-
olefin
copolymers, propylene homopolymers, propylene/alpha-olefin copolymers, high
impact
polypropylene, butylene homopolymers, butylene/alpha olefin copolymers, and
other alpha
olefin copolymers or interpolymers. Representative polyolefins include, for
example, but are
not limited to, substantially linear ethylene polymers, homogeneously branched
linear
ethylene polymers, heterogeneously branched linear ethylene polymers,
including linear low
density polyethylene (LLDPE), ultra or very low density polyethylene (ULDPE or
VLDPE),
medium density polyethylene (MDPE), high density polyethylene (HDPE) and high
pressure
- 30 -
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
low density polyethylene (LDPE). Other polymers included hereunder are
ethylene/acrylic
acid (EEA) copolymers, ethylene/methacrylic acid (EMAA) ionomers,
ethylene/vinyl acetate
(EVA) copolymers, ethylene/vinyl alcohol (EVOH) copolymers, ethylene/cyclic
olefin
copolymers, polypropylene homopolymers and copolymers, propylene/styrene
copolymers,
ethylene/propylene copolymers, polybutylene, ethylene carbon monoxide
interpolymers (for
example, ethylene/carbon monoxide (ECO) copolymer, ethylene/acrylic
acid/carbon
monoxide terpolymer and the like). Still other polymers included hereunder are
polyvinyl
chloride (PVC) and blends of PVC with other
materials.
Styrene polymers include, for example, crystal polystyrene, high impact
polystyrene,
medium impact polystyrene, styrene/acrylonitrile copolymers,
styrene/acrylonitrile/butadiene
(ABS) polymers, syndiotactic polystyrene, sulfonated polystyrene and
styrene/olefin
copolymers. Representative styrene/olefin copolymers are substantially random
ethylene/styrene copolymers, preferably containing at least 20, more
preferably equal to or
greater than 25 weight percent copolymerized styrene monomer.
Exemplary materials that could be used as additional components would include,
without limitation:
1) pigments, antioxidants, stabilizers, surfactants, and fl ow promoters;
2) particulates, fillers and oils; and
3) solvents and other materials added to enhance processability and
handling of
the composition.
With regard to the pigments, antioxidants, stabilizers, surfactants, and flow
promoters, these components, when utilized in compositions with the sulfonated
block
copolymers of the present invention may be included in amounts up to and
including 10%,
i.e., from 0 to 10%, based on the total weight of the composition. When any
one or more of
these components are present, they may be present in an amount from about
0.001 to about
5%, and even more preferably from about 0.001 to about 1%.
With regard to particulates, fillers and oils, such components may be present
in an amount up
to and including 50%, from 0 to 50%, based on the total weight of the
composition. When
any one or more of these components arc present, they may be present in an
amount from
about 5 to about 50%, preferably from about 7 to about 50%.
-31-
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
C. Lamination
As discussed above, the sulfonated block copolymer as disclosed herein is
formed
into a film for laminating onto the porous substrate. It was surprisingly
found that laminating
the block copolymer onto the substrate produced significantly improved MVTR
compared to
direct solution coating; the MVTR of the laminates was nearly equivalent to
that of the pure
polymer film of equal thickness. As MVTR is correlated to sensible heat
transfer, improved
MVTR is indicative of improved ERV unit efficiency. Accordingly ERV core units
employing such membranes will show improved efficiency and effectiveness.
Lamination may be conducted as known in the art. Generally, the sulfonated
block
copolymer will be formed into a film, optionally with other components,
(hereinafter
"polymeric film") and joined with the porous substrate to form the laminate
membrane.
Multiple methods may be employed for lamination of the polymeric film for
attachment or
bonding of the polymeric film to the porous substrate. For example, lamination
can be
conducted by cold lamination or heat (thermal) lamination. Additionally, sonic
bonding may
be employed for lamination.
Heat lamination is carried out by contacting the polymeric film with the
porous
substrate under temperature and pressure thereby forming a bond between the
two. The
lamination can take place in a vessel such as an oven or other machine or
apparatus which
enables pressing of the polymeric film and porous substrate together.
Generally the
temperature ranges from 95 to 450 F, and the pressure can have a range from
100 to 7,000
psi. Residence time, or time subjected to the increased temperature and
pressure can be from
seconds to 10 minutes. Thereafter, the membrane can be cooled at room
temperature and
pressure to produce the finalized membrane. Various types of laminating
assemblies known
in the art can be employed to contact the polymeric film and substrate under
heat and
25 pressure. Adhesives may also be used in heat lamination processes.
Further, heat activated
adhesives may be employed.
One type of heat lamination utilizes a press. A press can have two flat metal
platens
that are each individually heated and contain thermometers for temperature
validation. In
addition, the two heated metal platens can be drawn together under adjustable
pressure. A
30 film of the sulfonated block copolymer disclosed herein can be placed on
a substrate to be
-32 -
CA 02810409 2013-03-04
55201-1
placed between the metal platens thus forming a two ply membrane. Two pieces
of metal
foil can be placed around this two ply arrangement and then put into the press
under pressure.
The typical operating conditions may include temperatures from 150-450oF,
pressure from
100-7000 psi and residence time of approximately one to two minutes.
Temperature,
pressure and time may be varied to achieve desired laminate bonding.
Besides laminating by means of a press, roller assembles may also be used.
Such a
roller assembly is shown in Fig. 7. As indicated a sulfonated polymer film 20
is fed past sub-
rollers 22a and between top roller 23 and bottom roller 24. A substrate 21 is
also fed past
sub-rollers 22b and between top roller 23 and bottom roller 24. The polymer
film 20 and
substrate 21 are pinched between the top roller 23 and bottom roller 24 thus
bonding the
polymer film 20 to the substrate 21 and forming laminate membrane 25. Either
the top roller
23, bottom roller 24, or both rollers, may be heated sufficient for bonding
the polymer film to
the substrate. Additionally, the rollers 23 and 24 can be separated or pushed
together to
adjust the pressure at which the lamination occurs. Temperatures may be from
150-450oF,
pressures from 100-7000 psi. Furthermore, as the substrate and polymer films
are fed into
the rollers, this allows for large quantities of laminated substrate to
produced. Line speed
may vary depending on temperature and pressure and the desired quality and
quantity of the
laminate membrane.
Accordingly, commercial processes make use of such roller
assemblies for preparing large quantities of laminate product.
An additional method of lamination is termed solvent bonding. This can be done
under heat or room temperature. In this type of lamination, an organic solvent
is applied to
the sulfonated block copolymer film. The portion of the sulfonated polymer
film contacted
with the solvent accordingly softens. The film is then pressed onto the
substrate thereby
forming a bond between the portions softened by the organic solvent and the
substrate.
Organic solvents may be used which have the effect of solvating portions of
the polymeric
film. Such organic solvents include alcohols, alkyls, ketones, acetates,
ethers, and aromatic
solvents, such as toluene, and benzene.
Another method for lamination is cold lamination, which generally utilizes wet
or dry
adhesives. While labeled cold, such temperature includes room temperatures.
Adhesive
lamination is carried out by applying adhesive to one side of the polymeric
film and then
- 33 -
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
contacting it with a porous substrate. However, the adhesive should be applied
to cover a
particular surface area of the substrate and polymeric film so as to minimize
blockages which
may prevent vapor from passing through. Adhesives may have the effect of
blocking certain
pores of the substrate, thereby reducing efficiency. Accordingly, with
adhesive lamination, it
is preferable to minimize the area covered by the adhesive. The adhesive
employed can be
for example urethane or latex based.
In either cold or heated lamination, laboratory or commercial scale lamination
assemblies may also be used for contacting and bonding the polymeric film and
substrate to
form a laminate. There are many possible configurations for commercial scale
lamination,
including rollers, presses, the most prevalent in the art being the use of
dual, heated pinch
rollers.
When contacting the polymeric film with the porous substrate, measures can be
taken
to ensure that the film will be flat against the porous substrate. For example
a lamination
apparatus with rollers can be applied across the surfaces to flatten and
remove bubbles.
After lamination samples can be analyzed using Scanning Electron Microscopy
(SEM) to view the quality of the lamination.
Illustrative Embodiments
The following examples are intended to be illustrative only, and are not
intended to
be, nor should they be construed as, limiting the scope of the present
invention in any way.
a. Materials and Methods
Degree of Sulfonation: The degree of sulfonation as described herein and as
determined by titration was measured by the following potentiometric titration
procedure.
The sulfonation reaction product solution was analyzed by two separate
titrations (the "two-
titration method") to determine the levels of styrenic polymer sulfonic acid,
sulfuric acid, and
non-polymeric by-product sulfonic acid (2-sulfoisobutyric acid). For each
titration, an aliquot
of about five (5) grams of the reaction product solution was dissolved in
about 100 mL of
tetrahydrofuran and about 2 mL of water and about 2 mL of methanol were added.
In the first
titration, the solution was titrated potentiometrically with 0.1 N
cyclohexylamine in methanol
to afford two endpoints; the first endpoint corresponded to all sulfonic acid
groups in the
-34 -
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
sample plus the first acidic proton of sulfuric acid, and the second endpoint
corresponded to
the second acidic proton of sulfuric acid. In the second titration, the
solution was titrated
potentiometrically with 0.14 N sodium hydroxide in about 3.5:1 methanol :water
to afford
three endpoints: The first endpoint corresponded to all sulfonic acid groups
in the sample
plus the first and second acidic proton of sulfuric acid; the second endpoint
corresponded to
the carboxylic acid of 2-sulfoisobutyric acid; and the third endpoint
corresponded to
isobutyric acid.
The selective detection the of the second acidic proton of sulfuric acid in
the first
titration, together with the selective detection of the carboxylic acid of 2-
sulfoisobutyric acid
in the second titration, allowed for the calculation of acid component
concentrations.
The degree of sulfonation as described herein and as determined by 1H-NMR was
measured using the following procedure. About two (2) grams of non-neutralized
sulfonated
polymer product solution was treated with several drops of methanol and the
solvent was
stripped off by drying in a 50 C vacuum oven for approximately 0.5 hours. A 30
mg sample
.. of the dried polymer was dissolved in about 0.75 mL of tetrahydrofuran-d8
(THF-d8), to
which was then added with a partial drop of concentrated H2SO4 to shift
interfering labile
proton signals down fi el d away from aromatic proton signals in subsequent
NMR analysis.
The resulting solution was analyzed by 1H-NMR at about 60 C. The percentage
styrene
sulfonation was calculated from the integration of 1H-NMR signal at about 7.6
part per
million (ppm), which corresponded to one-half of the aromatic protons on
sulfonated styrene
units; the signals corresponding to the other half of such aromatic protons
were overlapped
with the signals corresponding to non-sulfonated styrene aromatic protons and
tert-butyl
styrene aromatic protons.
The ion exchange capacity as described herein was determined by the
potentiometric
titration method described above and was reported as milliequivalents of
sulfonic acid
functionality per gram of sulfonated block copolymer.
MVTR: A number of test methods are known for the determination of water vapor
transmission (MVTR) through materials. In the examples disclosed herein, a
modified
ASTM Method E96-80-B is employed, which is also referred to as "the upright
water cup
method."
-35 -
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
This method includes placing a sample of the membrane tightly onto the mouth
of a
test cup that is partially filled with water. Air of known relative humidity
and temperature
(the test is isothermal) is moved across the cup, thereby coming in contact
with the
membrane on the mouth of the cup. As air passes over the membrane, moisture
vapor is
drawn through the membrane sample.
MVTR was measured using membrane samples that were cut using a die of 13/16
inch diameter. These circular samples were then mounted inside glass vials
using a rubber
gasket to ensure a good, leak-free seal. The samples vials were filled with
deionized water to
the point of leaving an approximate 0.25 inch gap (headspace) between the
water level and
the inner membrane surface. Temperature and Relative Humidity was controlled
by using a
Tenney Environmental Chamber. The Chamber additional had 10 volt internal
computer
fans to facilitate air flow.
Water-filled sample vials were initially weighed on an analytical balance
accurate to
0.1mg. Over an eight hour period, the samples were weighed on two hour
intervals and the
MVTR was calculated as an average in the units of g,/m2 x day. Typical
standard deviations
ranged from about 20 to 100 g/m2 x day. The rate calculations were done by a
least squares
line fit method revealing R values greater than 0.98.
As will be understood in complex measurements, there may be some small
fluctuations of the absolute magnitude of the MVTR measured in this manner.
Accordingly,
in order to account for this the data in Table 1 is also presented as a
percentage compared to a
control. In this case the control is a uniform neat 0.5 mil thickness film of
SBC-1. As a
standard operation procedure, a control was run with each set of measurements.
b. Experiments
Preparation of Sulfonated Block Copolymer SBC-1
A pentablock copolymer having the configuration A-D-B-D-A was prepared by
sequential anionic polymerization where the A blocks are polymer blocks of
para-tert-
butylstyrene (ptBS), the D blocks were comprised of polymer blocks of
hydrogenated
isoprene (Ip), and the B blocks werere comprised of polymer blocks of
unsubstituted styrene
(S). Anionic polymerization of the t-butylstyrene in cyclohexane was
inititated using sec-
- 36 -
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
butyllithium affording an A block having a molecular weight of 15,000 g/mol.
Isoprene
monomers were then added to afford a second block with a molecular weight of
9,000 g/mol
(ptBS-Ip-Li). Subsequently, styrene monomer was added to the living (ptBS-Ip-
Li) diblock
copolymer solution and was polymerized to obtain a living triblock copolymer
(ptBS-Ip-S-
Li). The polymer styrene block was comprised only of polystyrene having a
molecular
weight of 28,000 g/mol. To this solution was added another aliquot of isoprene
monomer
resulting in an isoprene block having a molecular weight of 11,000 g/mol.
Accordingly, this
afforded a living tetrablock copolymer structure (ptBS-Ip-S-Ip-Li). A second
aliquot of
para-tert butyl styrene monomer was added, and polymerization thereof was
terminated by
adding methanol to obtain a ptBS block having a molecular weight of about
14,000 g/mol.
The ptBS-Ip-S-Ip-ptBS was then hydrogenated using a standard
Co2+/triethylaluminum
method to remove the C=C unsaturation in the isoprene portion of the
pentablock. The block
polymer was then sulfonated directly (without further treatment, not
oxidizing, washing, nor
"finishing") using an i-butyric anhydride/sulfuric acid reagent. The
hydrogenated block
copolymer solution was diluted to about 10% solids by the addition of heptane
(roughly an
equal volume of heptane per volume of block copolymer solution). Sufficient i-
butyric
anhydride and sulfuric acid (1/1 (mol/mol)) were added to afford 2.0 meq of
sulfonated
polystyrene functionality per g of block copolymer. The sulfonation reaction
was terminated
by the addition of ethanol (2 mol ethanol/mol of i-butyric anhydride) The
resulting polymer
.. was found, by potentiometric titration, to have an "Ion Exchange Capacity
(IEC)" of 2.0 meq
of ¨S03H/g of polymer. The solution of sulfonated polymer had a solids level
of about 10%
wt/wt in a mixture of heptane, eyclohexane, and ethyl i-butyrate.
Preparation of Laminate Membranes
After preparation of films formed from SBC-1, the films were then laminated to
a
substrate. Heat Lamination, designated as "heat bonding" in the examples
below, was
accomplished by using a press. The press has two flat metal platens that are
each
individually heated and contain thermometers for temperature validation. In
addition, the
two heated metal platens can be drawn together under adjustable pressure.
Thus, in all the
heat-laminated examples, a 5 inch x 5 inch square of 0.5mil thick polymer film
was layered
on to a 4 inch by 4 inch square of substrate. Two pieces of metal foil were
put outside this
-37 -
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
two ply arrangement. This entire sandwich was then put into a press and placed
under
pressure. The operating conditions included a temperatures within the 320-350
F range,
pressure in the 1000-5000 pound pressure range and heating time of
approximately one to
two minutes.
Since the polymeric film sample was slightly larger than the 4"x4" square of
substrate, after lamination the unbonded edge was used for a manual peel test
to determine
the bond strength. In all the reported examples, the excess unbonded membrane
would tear
first, leaving the bond to the substrate intact. Samples were also observed
using Scanning
Electron Microscopy (SEM) to view the quality of the lamination. In all the
reported cases,
the laminates were essentially defect-free and the polymeric film had the
appearance of flow
(ingress) within the fabric of the substrates.
For examples designated as "solvent bonding" the polymer film was applied by
means of an organic solvent. In this case an organic solvent was applied to
the polymer film.
This has the effect of causing a portion of the film which was contacted with
the solvent to
soften. The film is then pressed to the substrate. With such pressure, the
portions softened
by the organic solvent facilitate the bonding of the film to the substrate.
Preparation of Comparative Solvent Cast Membranes
Generally, coating involves application of the polymer to the substrate in
solution or
liquid form. In the examples herein designated as "Solution coating", the
following
procedure was used. The mass and dimension of the substrate was first
determined in order
to calculate a coat weight (expressed as grams per square meter ¨ gsm). Next,
this substrate
was placed onto a glass plate and a 3wt% solution of the sulfonated block
copolymer
(2.0IEC) was poured on top of the substrate followed by the removal of excess
solution with
a glass rod. The solvent cast membrane was allowed to dry completely then the
added mass
of the polymer was determined to complete the coat weight calculation.
Although not used
in these examples, solution coating of the sulfonated block copolymer
solutions could be
introduced by simple dip coating or spray coating.
- 38 -
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
4.0 Samples for Comparative Data
Table 1 ¨ Comparative MVTR
Experiment Substrate Polymer Membrane Membrane Control % of
No. formation
MVTR MVTR Control*
PET Scrim SB C-1 Solution 320 2200 15
Coated
Comp 1
PET Scrim SB C-1 Solution 334 2200 15
Coated
Comp 2
PET Scrim SB C-1 Laminated 2270 2600 87
(solvent
Exp 1 bonding)
PET Scrim SB C-1 Laminated 2400 2600 92
(heat bonding)
Exp 2
PET Scrim SBC-1 Laminated 2400
2600 96
laminated on a (heat bonding)
commercially
Exp 3 lamination line
Carbon Fiber SBC-1 Laminated 2500 2600 96
Veil (heat bonding)
Exp 4
Fiber Glass SB C-1 Laminated 2500 2600 96
Exp 5 Scrim (heat bonding)
Nylon Veil SBC-1 Laminated 2500 2600 95
Exp 6 (heat bonding)
Comparative commercially available membranes
Experiment Membrane Membrane
Control % of
No.
MVTR MVTR Control*
Comp 3 Innergytech Coated cellulosic
Paper 1780 2030 88
Comp 4 Mitsubishi Lossnay cellulosic
Paper 1640 2230 74
MVTR measurement at 25 C and 50% Relative Humidity
Table 1 compares the use of coated membranes to laminated membranes with
respect
to MVTR. Membranes with higher MVTR when used in ERV core units will promote
more
effective latent heat transfer and thus great ERV effectiveness and
efficiency.
In particular Comp 1 and Comp 2 demonstrate MVTR with respect to copolymer
SBC-1 solution coated onto a substrate, in this case PET scrim. Exps 1-6
demonstrate
- 39 -
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
MVTR with respect to copolymer SBC-1 laminated onto various substrates
including PET
Scrim, Carbon Fiber Veil, Fiber Glass Scrim and Nylong Veil. The heated
laminate was
virtually colorless in all cases except the carbon fiber veil which is
naturally black in color.
Furthermore, Exps 1-6 laminated membranes surprisingly and unexpectedly
produced
significantly improved MVTR as compared Comp 1 and 2 coated membranes. This
can
further be seen in that both Comp 1 and 2 employ a PET Scrim, and Exp 1 and 2
also utilize
PET scrim. Thus all conditions are the same ¨ the same substrate and the same
SBC-1
polymer is used, except however, Comp 1 and 2 are solution coated whereas Exp
1 and 2 are
laminated. Thus it can be seen that SBC-1 laminated onto substrate produces
exceptional
MVTR for use in ERV units.
Moreover Table 1 also shows comparative results for membranes from several
well
known commercially available ERV units, namely experiment numbers Comp 3 and
4.
These results demonstrate a substantial advantage of the laminated SBC-1
membranes as
compared to the comparative commercial membranes. In particular, Exps 3-6
demonstrate a
MVTR of above 96%. Generally MVTR's above 90% are advantageous as compared to
conventional membranes, and MVTR's above 95% are exceptional.
Accordingly, Table 1 demonstrates the unexpected and significant advantages
obtained by the membranes according to the disclosure herein using SBC-1
laminates. As
MVTR performance correlates to latent heat transfer efficiency, such membranes
would
provide significantly improved and exceptional MVTR for use in ERV units.
- 40 -
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
Table 2¨ Commercial Roller lamination MVTR measurement
Exp Substrate Polymer Membrane Membrane Control % of
No. Formation MVTR MVTR Control
Exp Apex RCO8 Nylon SBC-1 Laminated 2310 2250 103
7 Woven Veil (heat
bonding)'
Exp Apex XA91A PET S B C -1 Laminated 2370 2250 105
8 Woven Veil (heat
bonding)'
Exp 0.8 Non-woven PET SBC-1 Thermal 2300 2250 102
9 Scrim lamination
on
commercial
line'
Exp Trilaminate (Non- S B C -1 Thermal 2200 2700 81
woven PET 0.8 oz lamination
outer layers and on
MD9200 film inner commercial
layer) line'
MVTR measurement at 25 C and 50% Relative Humidity
1 Conditions for heat bonding lamination using a commercial thermal
calendaring lamination line:
260 C, 450 psi, 8 yards per minute line speed
5 Table 2 illustrates membranes laminated on commercial roller lamination
lines and
the respective MVTR results. The "control" in Table 2 is a 0.5 mil film of SBC-
1 which
shows very good MVTR values. As illustrated in Table 2, by comparing the
experimental
results of the control to the laminated membranes, it can be seen that the
MVTR values are
not significantly affected with the bonding of a substrate.
Accordingly, with SBC-1
10 laminated to a substrate, MVTR values can be reached which are close to
those of the SBC-1
polymer alone.
Exp. 7-9 illustrates thermal lamination (commercial line provided by 3M Powell
Company) MVTR values arc obtained which are equal to or slightly better than
the control.
The values higher than the controls may be due to the compression (thinning)
of the SBC-1
polymer layer during the process that runs the laminate under pressure and
temperature
between calendar rolls.
-41-
CA 02810409 2013-03-04
WO 2012/050860 PCT/US2011/053599
Exp. 10 illustrates the production of a trilaminate consisting of non-woven
PET
scrim for outer layers and a 0.5 mil SBC-1 film in the center. The extra
thickness of the
additional PET layer creates greater resistance to moisture vapor transport
hence the lower
MVTR performance.
- 42 -