Note: Descriptions are shown in the official language in which they were submitted.
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
AUTOPHAGY INDUCER AND INIHBITOR COMBINATION THERAPY FOR THE
TREATMENT OF NEOPLASMS
FIELD OF THE INVENTION
The RNAi constructs and combination therapy disclosed herein relate to the
treatment of neoplasms.
BACKGROUND OF INVENTION
Aberrant activation of the class I phosphatidylinositol 3-kinase (PI3K)/Akt
pathway has been widely implicated in a variety of cancers. This is not only
as a result of
abnormal activities of various upstream growth factors and their receptors,
but also through
direct alterations of the PI3K and Akt isoforms, and more frequently,
inactivation of the
tumor suppressor phosphatase and tensin homolog (PTEN), a phospholipid
phosphatase that
negates the activity of PI3K. The three Akt isoforms represent attractive
cancer therapeutic
targets (Samuels, Y., and K. Ericson, (2006), Oncogenic PI3K and its role in
cancer. Curr
Opin Oncol. 18:77-82; Stambolic, V., and J.R. Woodgett, (2006), Functional
distinctions of
protein kinase B/Akt isoforms defined by their influence on cell migration.
Trends Cell Biol.)
Genetic ablations of the 3 Akt genes in mice have revealed both distinct and
overlapping
functions of each isoform in normal physiology (Chen, W.S., P.Z. Xu, K.
Gottlob, M.L.
Chen, K. Sokol, T. Shiyanova, I. Roninson, W. Weng, R. Suzuki, K. Tobe, T.
Kadowaki, and
N. Hay, (2001), Growth retardation and increased apoptosis in mice with
homozygous
disruption of the Akt1 gene. Genes Dev. 15:2203-8; Cho, H., J. Mu, J.K. Kim,
J.L.
Thorvaldsen, Q. Chu, E.B. Crenshaw, 3rd, K.H. Kaestner, M.S. Bartolomei, G.I.
Shulman,
and M.J. Birnbaum, (2001), Insulin resistance and a diabetes mellitus-like
syndrome in mice
lacking the protein kinase Akt2 (PKB beta). Science. 292:1728-31; Cho, H.,
J.L.
Thorvaldsen, Q. Chu, F. Feng, and M.J. Birnbaum, (2001), Aktl/PKBalpha is
required for
normal growth but dispensable for maintenance of glucose homeostasis in mice.
J Biol Chem.
276:38349-52. Epub 2001 Aug 31; Easton, R.M., H. Cho, K. Roovers, D.W.
Shineman, M.
Mizrahi, M.S. Forman, V.M. Lee, M. Szabolcs, R. de Jong, T. Oltersdorf, T.
Ludwig, A.
Efstratiadis, and M.J. Birnbaum, (2005), Role for Akt3/protein kinase Bgamma
in attainment
of normal brain size. Mol Cell Biol. 25:1869-78; Peng, X.D., P.Z. Xu, M.L.
Chen, A. Hahn-
Windgassen, J. Skeen, J. Jacobs, D. Sundararajan, W.S. Chen, S.E. Crawford,
K.G. Coleman,
and N. Hay, (2003), Dwarfism, impaired skin development, skeletal muscle
atrophy, delayed
1
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
bone development, and impeded adipogenesis in mice lacking Aktl and Akt2.
Genes Dev.
17:1352-65; Tschopp, 0., Z.Z. Yang, D. Brodbeck, B.A. Dummler, M. Hemmings-
Mieszczak, T. Watanabe, T. Michaelis, J. Frahm, and B.A. Hemmings, (2005),
Essential role
of protein kinase B gamma (PKB gamma/Akt3) in postnatal brain development but
not in
glucose homeostasis. Development. 132:2943-54. Epub 2005 Jun 1; Yang, Z.Z., 0.
Tschopp,
N. Di-Poi, E. Bruder, A. Baudry, B. Dummler, W. Wahli, and B.A. Hemmings,
(2005),
Dosage-dependent effects of Aka /protein kinase Balpha (PKBalpha) and
Akt3/PKBgamma
on thymus, skin, and cardiovascular and nervous system development in mice.
Mol Cell Biol.
25:10407-18) and tumor initiation (Chen, M.L., P.Z. Xu, X.D. Peng, W.S. Chen,
G. Guzman,
X. Yang, A. Di Cristofano, P.P. Pandolfi, and N. Hay, (2006), The deficiency
of Aktl is
sufficient to suppress tumor development in Pten+/- mice. Genes Dev. 20:1569-
74; Ju, X., S.
Katiyar, C. Wang, M. Liu, X. Jiao, S. Li, J. Zhou, J. Turner, M.P. Lisanti,
R.G. Russell, S.C.
Mueller, J. Ojeifo, W.S. Chen, N. Hay, and R.G. Pestell, (2007), Aktl governs
breast cancer
progression in vivo. Proc Natl Acad Sci U S A. 104:7438-43; Maroulakou, I.G.,
W. Oemler,
S.P. Naber, and P.N. Tsichlis, (2007), Aktl ablation inhibits, whereas Akt2
ablation
accelerates, the development of mammary adenocarcinomas in mouse mammary tumor
virus
(MMTV)-ErbB2/neu and MMTV-polyoma middle T transgenic mice. Cancer Res. 67:167-
77; Skeen, J.E., P.T. Bhaskar, C.C. Chen, W.S. Chen, X.D. Peng, V. Nogueira,
A. Hahn-
Windgassen, H. Kiyokawa, and N. Hay, (2006), Ala deficiency impairs normal
cell
proliferation and suppresses oncogenesis in a p53-independent and mTORC1-
dependent
manner. Cancer Cell. 10:269-80). The relative contribution of the three Akt
isoforms in
maintaining human tumor growth, however, remains elusive.
Human cancers usually co-express two or all three Akt isoforms, and
amplification or hyperactivation of each isoform has been documented in
different types of
cancers (Altomare, D.A., and J.R. Testa, (2005), Perturbations of the AKT
signaling pathway
in human cancer. Oncogene. 24:7455-64; Stahl, J.M., A. Sharma, M. Cheung, M.
Zimmerman, J.Q. Cheng, M.W. Bosenberg, M. Kester, L. Sandirasegarane, and G.P.
Robertson, (2004), Deregulated Akt3 activity promotes development of malignant
melanoma.
Cancer Res. 64:7002-10). Mounting evidence suggests that Akt isoforms may be
differentially regulated depending on the external stimuli and the tissue
studied, and may
regulate distinct aspects of cellular processes in a cell and tissue-specific
manner (Dufour, G.,
M.J. Demers, D. Gagne, A.B. Dydensborg, I.C. Teller, V. Bouchard, I. Degongre,
J.F.
Beaulieu, J.Q. Cheng, N. Fujita, T. Tsuruo, K. Vallee, and P.H. Vachon,
(2004), Human
intestinal epithelial cell survival and anoikis. Differentiation state-
distinct regulation and
2
CA 02821378 2013-06-12
WO 2012/087336 PCT/US2010/062096
roles of protein kinase B/Alct isoforms. J Biol Chem. 279:44113-22. Epub 2004
Aug 6; Irie,
H.Y., R.V. Pearline, D. Grueneberg, M. Hsia, P. Ravichandran, N. Kothari, S.
Natesan, and
J.S. Brugge, (2005), Distinct roles of Akt 1 and Alct2 in regulating cell
migration and
epithelial-mesenchymal transition. J Cell Biol. 171:1023-34; Kim, D., S. Kim,
H. Koh, S.O.
Yoon, A.S. Chung, K.S. Cho, and J. Chung, (2001), Alct/PKB promotes cancer
cell invasion
via increased motility and metalloproteinase production. FASEB J. 15:1953-62;
Samuels, Y.,
L.A. Diaz, Jr., 0. Schmidt-Kittler, J.M. Cummins, L. Delong, I. Cheong, C.
Rago, D.L. Huso,
C. Lengauer, K.W. Kinzler, B. Vogelstein, and V.E. Velculescu. 2005. Mutant
PIK3CA
promotes cell growth and invasion of human cancer cells. Cancer Cell. 7:561-
73; Tanno, S.,
S. Tanno, Y. Mitsuuchi, D.A. Altomare, G.H. Xiao, and J.R. Testa, (2001), AKT
activation
up-regulates insulin-like growth factor I receptor expressi6n and promotes
invasiveness of
human pancreatic cancer cells. Cancer Res. 61:589-93; Yoeli-Lerner, M., G.K.
Yiu, I.
Rabinovitz, P. Erhardt, S. Jauliac, and A. Toker, (2005), Akt blocks breast
cancer cell
motility and invasion through the transcription factor NFAT. Mol Cell. 20:539-
50).
Akt is well known for its anti-apoptotic activity, leading to its depiction as
a
survival kinase (Amaravadi, R., and C.B. Thompson, (2005), The survival
kinases Akt and
Pim as potential pharmacological targets. J Clin Invest. 115:2618-24).
However, inhibiting
components of the PI3K/Akt pathway often does not induce substantial apoptosis
without
additional pro-apoptotic insults. This is exemplified in a recent study, where
a dual
PI3K/mTOR inhibitor that efficiently inhibited phosphorylation of Akt, blocked
proliferation
of glioma xenografts without the induction of apoptosis (Fan, Q.W., Z.A.
Knight, D.D.
Goldenberg, W. Yu, K.E. Mostov, D. Stokoe, K.M. Shokat, and W.A. Weiss,
(2006), A dual
PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell.
9:341-9).
In a recent study, alkylating agents were given in combination with
,
chloroquine (WO 2006/078774). No results were presented on the combination of
kinase
inhibitors with chloroquine.
BRIEF SUMMARY OF INVENTION
The subject matter disclosed herein relates to agents and methods of treating
neoplasms with an agent that is a kinase inhibitor and is also an inducer of
autophagy in
combination with an agent that is an inhibitor of autophagy.
DESCRIPTION OF THE FIGURES
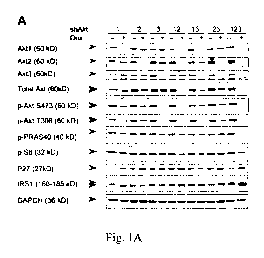
Figure 1 shows inducible (knockdown) KD of Akt isoforms and their effect on
3
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
xenograft tumor growth. (A) Immunoblot analysis of Akt isoforms and various
downstream
proteins in stable PC3 clones expressing the inducible shRNA constructs. Each
clone was
induced to express the respective shRNA(s) with 1 gg/m1 Dox grown under 10%
FBS for 7
days. Double arrowheads indicate slight differences in the mobility of the 3
Akt isoforms
detected by total and phospho-Akt antibodies, and the mobility shift of IRS1.
Figure 1B
shows the effect of Akt KD on xenograft tumor growth. Representative
experiments showing
the growth of PC3 xenograft tumors containing the various shRNAs treated with
vehicle
control (-Dox, filled circles) or Dox (+Dox, open circles) (see details in
Table 3). Error bars
represent SEM. *, P <0.05; **, P < 0.005.
Figure 2 depicts that Akt KD resulted in cell cycle delay and elevated
autophagy without substantial apoptosis. (A) Histological analysis of PC3-
shAkt123 tumors
treated with Dox or vehicle control for 5, 15, or 21 d as indicated. Tumor
tissues were
analyzed by IHC using antibodies specific for Ki-67 or by the TUNEL assay.
Pathologist's
scoring of the signal intensity for each sample is indicated in parentheses.
Bars, 100 gm. (B
and C) Effect of triple-Akt KD on cell cycle progression under serum
starvation (ss)
compared with cells grown under 10% FBS. Cells containing shRNAs targeting
EGFP or all
three Akt isoforms were pretreated for 2 d with or without Dox in medium
containing 10%
FBS and changed to 0% (B) or 0.5% (C) FBS. Cell cycle profiles were analyzed
at the
indicated time points after serum withdrawal. Error bars represent SEM (n=3).
The
percentage of change in each cell cycle phase with Dox versus without Dox
treatment is also
shown.
Figure 3 depicts autophagy induced in PC3 and U87MG cells by Akt KD. (A)
EM images of PC3 (ad) and U87MG (e-g) cells grown in the absence (a and f) or
presence
(b-e and g) of Dox-induced Akt 123 KD for 5 d. Arrows, degradative
autolysosomes. Double
arrows, initial AVs. Arrowhead, phagophore isolation membrane. M,
mitochondrion in an
AV. Asterisks, glycogen particle clusters. Bars: (a, b, f, and g) 0.5 gm: (c
and d) 200 nm; (e)
1 gm. (B) Quantification of the number of AVs per unit cytoplasmic area of 4.5
gm2 (n? 64)
and the percentage of cytoplasmic area occupied by AV in randomly sampled
cytoplasmic
areas (n = 5 areas of >200 11m2) of PC3 and U87MG cells with and without Dox-
induced
shAkt 123 expression. Error bars represent SEM. (e) Dox-induced Akt silencing
caused
degeneration in PC3 and U87MG tumors. (C) (a) PC3 tumors expressing the
control EGFP
shRNA after 15 d of Dox treatment. The tumor cells contain large nuclei and
nucleoli, some
lipid droplets (asterisks), and are connected by cell junctions (arrowheads).
(b-d) PC3 tumors
expressing shAkt 123 after 15 (b and c) or 10 d (d) of Dox treatment. (b)
Cells and nuclei in
4
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
these tumors often appear shrunken. Arrows, AVs. E, eosinophil. (c) Two AVs
(arrows)
found among dilated RER cisternae in a degenerating tumor cell. (d) Ultrathin
cryosection
with immunogold labeling of human LAMP!. Label occurs on lysosomes (arrow) and
AVs
(top inset). Some of the tumor cells also contain human LAMPl-positive dense
bodies with a
shape reminiscent of microautophagy (bottom inset; de Waal, E.J., H. Vreeling-
Sindelarova,
J.P. Schellens, J.M. Houtkooper, and J. James, (1986), Quantitative changes in
the lysosomal
vacuolar system of rat hepatocytes during short-term starvation. A
morphometric analysis
with special reference to macro- and microautophagy. Cell Tissue Res. 243:641-
8). The
tumor cells have widened nuclear envelope and ER cisterns (asterisks), which
contain small
cytoplasmic islands (arrowheads). (e) U87MG tumor after 5 d of vehicle
treatment. (f-h)
U87MG-shAkt 123 tumor after 5 d of Dox treatment. Arrows, AVs. (h) In some
tumor
samples, cells with glycogen clusters (asterisks) and glycogen-containing AVs
occur. Bars:
(a-c) 2 gm: (e and 0 1 gm: (g) 0.5 gm: (d and h) 200 nm.
Figure 4 depicts lysosomotropic agents accelerated cell death in combination
with Akt KD. (A) CQ treatment caused accumulation of GFP-LC3 dots in Dox-
treated PC3-
shAkt 123 cells. PC3-shAkt 123 cells stably expressing GFP-LC3 were pretreated
with or
without 1 jig/ml Dox for 6 d and treated with or without 10 gIVI CQ. GFP
fluorescence was
imaged after 1 d of CQ treatment. Arrowheads point to representative GFP dots
or clumps.
Bar, 10 gm. (B) Effect of shAkt123 and 10 gM CQ on LC3 processing, PARP
cleavage, and
total Akt in PC3-shAkt 123 cells treated with or without Dox or CQ. The ratio
of LC3-11 to
LC3-1 and cleaved (Cl) to hill-length (FL) PARP was quantified from
immunoblots of cell
lysates made at days 1 and 2 of CQ treatment. Immunoblots of day 2 samples are
shown.
Molecular masses are indicated in kilodaltons parenthetically next to each
protein. Data are
representative of three independent experiments. (C) CQ promoted cell death in
PC3 cells
induced to express shAkt123, whereas 3-MA pretreatment delayed this effect.
PC3-shAkt123
cells were preincubated with 1 jig/ml Dox for 3 d to induce shRNA expression
before cells
were seeded into fresh medium containing 10 gM CQ or 2.5 nM Ba with or without
Dox. 1
mM 3-MA was added with Dox, both during and after the pretreatment. Cell
viability was
determined at days 2, 3, and 4 under 0.5% (e) or 0% (D) FBS (cells treated
with Ba alone
under 0% FBS were followed for 2 and 3 d only). The percentage of the annexin
V-positive
PI-negative population was determined at days 2, 3, and 4 under 0.5% FBS.
Caspase-3/7
activity was determined at days 2 and 3 under 0% FBS and expressed as relative
fluorescence
units (RFU, in thousands) normalized to the same number of cells. Error bars
represent SD of
three independent experiments.
5
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
Figure 5 depicts CQ accelerated cell death in combination with 111-5. (A) PC3
cells were treated with DMSO or 0.5 gM 111-5 in the presence or absence of 10
gM CQ under
0.5% FBS. Cell viability was determined by PI exclusion at days 2, 3, and 5.
Annexin V
staining was analyzed at days 2 and 3 and broken down into PI+ or PI-
populations. (B) Time
course of cell viability in PC3 cells treated with 0.5 (III-5-0.5) or 20 gM
(III-5-20) 111-5 with
or without 10 gM CQ or 3 mM 3-MA. PC3 cells pretreated with 111-5 for 24 h
under 1 %
FBS were split into medium containing 0.5% FBS in the presence or absence of
CQ. 3-MA
was added immediately before 111-5, 24 h before CQ addition. Cell viability
was determined
by PI exclusion at the indicated time points after CQ addition. Error bars
represent SEM (n =
3). LC3-11 to LC3-1 ratios were determined from quantitation of immunoblots.
(C) CQ
dramatically increased the size and number of MDC+ vacuoles in PC3 cells
treated with III-
5, whereas 3-MA suppressed this effect. Cells were cultured in medium
containing 0.5% FBS
and treated with DMSO, 0.5 gM 111-5, 10 gM CQ, and 5 mM 3-MA, alone or in
combinations as indicated. MDC staining at 48 h is shown. Bar, 10 gm.
Figure 6 depicts CQ accelerated cell death in combination with 11-4. (A) PC3
cells were treated with DMSO or 4 1.1M 11-4 in the presence or absence of 10
gM CQ under
0.5% FBS. Cell viability was determined by PI exclusion over the course of 10
d. Error bars
represent SEM. Representative data from one of three independent experiments
are shown.
(B) Immunoblot analysis of cell lysates collected at the indicated time points
from the
experiment shown in A. Arrowheads indicate the positions for LC3-1 and -II,
CathD 43, and
CathD 28. Quantifications of the indicated markers are shown in C. CathD 43,
the 43-50-kD
forms of cathepsin D precursors. CathD 28, the 28-kD cathepsin D heavy chain.
Figure 7 depicts accumulation of AVOs preceded plasma membrane rupture
and correlated with the appearance of apoptotic and anucleated cells with Alct
inhibitor
("Alcti"), in this example compound 11-4, and CQ treatment. (A) PC3 cells
treated with
DMSO, 10 gM 11-4, 10 gM CQ, or both under 5% FBS were followed for 3 d using
time-
lapse microscopy. Representative images of the cells at the indicated time
points are shown.
White arrowheads indicate the fusion between two adjacent cells before plasma
membrane
rupture in cells treated with both agents. Bar, 10 gm. (B) PC3 cells treated
with the indicated
agents were stained with AO and analyzed by multispectral imaging flow
cytometry. (left)
Brightfield (BF), nuclei (green), vacuoles (red), and green/red composite
images of three
representative cells with each treatment are shown. Bars, 10 gm. (middle)
plotting AO green
intensity versus AO green bright detail area revealed three distinct
populations: R2
anucleated cells, R3 apoptotic cells, and R4 live cells. (right) AO red
intensity for R4 is
6
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
plotted on the histogram with an arbitrary gate (R5) drawn to include events
with the
brightest AO red intensity. R2, R3, and R4 histograms are overlaid in the Akti
(II-4) + CQ
plot only. (C) Statistics for each population shown in B. *, percentage of
total single cells; **,
mean fluorescence intensity of R4 live cells; ***, percentage of R4 live
cells.
Figure 8 depicts Akt inhibition induces mitochondrial superoxide and cellular
ROS production, which is augmented by CQ. (A) PC3 cells cultured in 0.5% FBS
were
treated with DMSO, 3 JIM 11-4, 10 gM CQ, or both, stained with MitoSOX red
dye, and
examined by fluorescence microscopy. Images at 24 h are shown. Bar, 10 gm. (B)
PC3 cells
treated as in A were stained with the Image-iT LIVE green ROS Detection kit
and examined
by fluorescence microscopy at 24 h. Bright field (BF) images of cells are also
shown. Bar, 10
gm. (C) Quantification of MitoSOX red and ROS green fluorescence intensities
by flow
cytometry at 24 h. Cells were treated as in A and B. Error bars represent SEM
(n = 3).
Figure 9 depicts CQ selectively accelerated cell death in Akti-treated PTEN-
null cells in vitro and enhanced the antitumor efficacy of Akt KD in vivo. (A)
PTEN-/- (-/-)
MEFs were more sensitive than isogenic PTEN+/+ (+/+) counterparts to the
combined
treatment with 11-4 and CQ. MEFs were treated with 5 gM each of 11-4 and CQ
under 1 %
FBS, and cell viability was determined at days 0, 2, and 3 by PI exclusion.
Error bars
represent SEM (n = 3). (B) Mean tumor volumes of PC3 xenograft tumors treated
daily with
vehicle (Veh), Dox only, CQ only, or both Dox and CQ over a 28-d period. The
vehicle and
vehicle + CQ groups were followed for up to 18 d before terminated because of
weight loss
from the tumor burdens. Error bars represent SEM (n = 10 tumors in each
cohort). (C)
Scatterplot of the tumor volumes in the Dox only and Dox + CQ groups on day 28
(P = 0.05).
Horizontal bars indicate mean tumor volumes. Numbers of tumors with complete
remission
(CR, dashed line) are indicated for each group. (D) Individual tumor growth
plotted as a
percentage of tumor volume change compared with day 0 for the Dox only and Dox
+ CQ
cohorts shown in A. Dashed lines indicate -100% change from the starting tumor
volumes,
i.e., complete tumor regression. Numbers of tumors with smaller ( O% change)
or larger
(>0% change) than the starting tumor volumes on day 28 are indicated.
Figure 10 depicts increased AV accumulation and apoptosis in PC3 tumor
with combined Akt123 KD and CQ treatment. (A) (a) EM images of PC3-shAkt123
tumors
treated for 5 d with CQ only. Arrows, dense AVs and lysosomes; N, nucleolus.
(b) Dox only.
Arrows, AVs with a less dense appearance than in a. (c and d) Both Dox and CQ.
(c)
Numerous dense and enlarged AVs (arrows) accumulate in tumor cells. An
apoptotic cell
(Ap) is partially surrounded by a macrophage (M). T, tumor cell. (d) Apoptotic
nuclei (Ap)
7
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
among the AV-loaded (arrows) tumor cells. Insets, enlarged images of AVs (a-c)
and
abnormal mitochondria (*) in each tumor. Bars: (a-c) 2 gm: (d) 1 gm. (B)
Quantification of
the percentage of cytoplasmic area occupied by AVs in randomly sampled
cytoplasmic areas
(n = 6 areas of >80 gm2). (C) Percentage of apoptotic nuclei among randomly
sampled tumor
cell nuclei (n = 3-4 sets of 100 tumor cell nuclei). (B and C) Error bars
represent SEM; *, P <
0.0005 compared with the other three groups.
Figure 11 depicts Akt knockdown by shRNA induces autophagy gene
expression. PC3 cells were induced to express shRNA to indicated Akt isoforms
for 72 hours
by Doxycycline, RNA was extracted from both Dox treated (Dox+) or untreated
control
(Dox-) cells. Microarray analysis was carried out using Affymetrix chips.
Ratios of the
expression levels of each autophagy gene from Dox+ and Dox- samples are shown.
Data are
mean values from 3 independent experiments.
Figure 12 depicts Akt inhibitors that induce autophagy gene expression. PC3
cells were treated with DMSO vehicle control or various Akt inhibitors,
including 1-(1-(4-(5-
hydroxy-6-methyl-3-phenylpyrazin-2-yl)benzyl)piperidin-4-y1)-1H-benzo[d] im
idazol-2(3H)-
one (II-
1), 1 -(1-(4-(6-hydroxy-5-isobuty1-3-phenylpyrazin-2-yl)benzyl)piperidin-4-y1)-
1H-
benzo[d] imidazol-2 (3H)-one (II-2), 1-
(1 -(4 -(7-pheny1-1H-im idazo [4,5-g] quinoxal in-6-
yObenzyppiperidin-4-y1)-1H-benzo [d] imidazol-2(3H)-one (II-4), (S)-2-(4-
chloropheny1)-1 -
(4 -((5R,7R)-7-hydroxy-5-methy1-6,7-dihydro-5H-cyclopenta[d] pyrim idin-4-
yl)piperazin-1-
y1)-3-(isopropylamino)propan-1-one (III-4), and (S)-1-(3-(N-(4-(3-
phenyl isoxazol-5-
yOthiazol-2-y1)thiophene-2-carboxamido)propyl)piperidine-2-carboxamide (IV-1),
at 1 or 5
OM for 6 or 24 hours. RNA was extracted from the cells. Microarray analysis
was carried out
using Affymetrix chips. Expression levels of each autophagy gene are
normalized to the
DMSO controls. Data are mean values from 3 independent experiments.
Figure 13 depicts various mTOR, PI3K and Akt inhibitors alone induce
increased autophagic vacuole accumulation as measured by side scatter (SSC) in
a flow
cytometer. Inhibitors include 3-pheny1-2-(4-((4-(5-(pyridin-2-y1)-111-1,2,4-
triazol-3-
yOpiperidin-1-y1)methyl)pheny1)-1,6-naphthyridin-5(6H)-one (II-3), benzyl 2-(4-
(3-
ethylureido)pheny1)-4-(1,4-oxazepan-4-y1)-51-1-pyrrolo [3,4 -d] pyrim idine-
6(7H)-carboxylate
(III-1); 1 -ethy1-3 -(4 -(4-morphol ino-7-(pyrimidin-2-y1)-5,6,7,8-
tetrahydropyrido [3,4-
d] pyrimidin-2 -yl)phenyl)urea
(III-2), (R)-1-(4-((2 -(2-am inopyrim idin-5-y1)-7-methy1-4-
morphol inoth ieno[3,2-d] pyrimidin-6-yl)methyl)piperazin-l-y1)-2-
hydroxypropan-1 -one (III-
3), 4 -
(2 -(1H-indazol-4-y1)-6-((4-(methylsul fonyl)p iperazin-l-yl)methyl)th ieno
[3,2-
d]pyrimidin-4-yl)morpholine (III-6), (S)-N-(4-(benzo [d] [1,3] dioxo1-5-
ypthiazol-2-y1)-N-(3-
8
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
(2-formylpiperidin- 1 -yl)propyl)thiophene-2-carboxamide (IV-2) and Rapamycin.
Figure 14 depicts CQ (10 M) increases the number and size of autophagic
vacuoles when combined with the various mTOR, PI3K, and Akt inhibitors.
Figure 15 depicts various mTOR, PI3K and Akt inhibitors alone induce
increased autophagic vacuole accumulation as measured by the red to green
fluorescent ratio
after acridine orange staining.
Figure 16 depicts CQ increases the accumulation of autophagic vacuoles when
combined with the various mTOR, PI3K and Akt inhibitors as measured by the red
to green
fluorescent ratio after acridine orange staining.
Figure 17 depicts Akt inhibitor 111-4 and PI3K inhibitor 111-6 both synergize
with CQ to accelerate cell death.
Figure 18 depicts relative levels of Akt isoforms in cancer cell lines and
inducible knockdown of Akt isoforms in U87MG cells and xenograft tumors. (A)
Relative
expression levels of each Akt isoform in tumor cell lines. Akt proteins in
total cell lysates
from each cell line were analyzed by Western blot analysis using isoform-
specific antibodies
and quantified using recombinant proteins of each isoform loaded on the same
gel as
standards. Data are representative of two independent experiments. (B) Western
blot
validation of Akt KD in U87MG cells expressing the inducible shRNA constructs.
Stable
pools ofU87MG cells containing the indicated constructs targeting the
respective Akt
isoforms were induced to express shRNA with 1 mg/ml Dox for 3 days. Total cell
lysates
were analyzed using the indicated antibodies. GAPDH levels were determined as
a loading
control. Molecular weights are indicated in lcDa parenthetically next to each
protein. (C-F)
U87 MG xenograft tumors containing the indicated shRNAs were treated with
vehicle control
or Dox. Each cohort consisted of ten mice. (G) Dox has no effect on the growth
of wild-type
PC3 cells. (H) PC3 tumor growth retardation induced by a second shAlctl
construct. Error
bars represent SEM. *P < 0.05; **p < 0.005.
Figure 19 depicts the effect of shGFP and Akt isoform KDs on cell cycle
progression, apoptosis and autophagy. (A) Steady-state cell cycle profiles of
PC3 stable
clones containing the indicated shRNAs with or without Dox treatment (1 ilg/m1
for 96
hours). Error bars represent standard deviations of 2 independent experiments.
(B) Cell cycle
profiles in (A) expressed as the percentage of change in each phase with Dox
treatment
compared to the same clone without Dox treatment. Data are representative of
at least 2
independent experiments with at least 0.5 x 106 cells analyzed for each
condition. (C) EM
quantification of the number of AVs per unit cytoplasmic area of 4.5 t1n2 (n =
130) of PC3
9
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
and U87MG cells showing no significant difference in AV numbers with or
without Dox-
induced shGFP expression. Error bars represent SEM. (D) Increased punctate LC3
immunofluorescence staining in PC3 and U87MG cells after 5 days of Dox-induced
shAlct123
expression compared to untreated controls. Bar, 10 n. (E) Percentage of PC3-
shAkt123 cells
with GFP-LC3 dots under the indicated treatments. PC3-shAkt123 cells stably
expressing
GFP-LC3 were pretreated as in Fig. 4 and the percentage of cells with >5
punctate GFP dots
visible was determined. Note that although CQ treatment alone caused
perinuclear GFP-LC3
clumps in the cells, these are morphologically different from the widespread
cytoplasmic
GFP-LC3 dots in Dox treated cells. Error bars represent standard deviation
from 2 random
fields of at least 15 cells each. (F) CQ treatment causes accumulation of MDC-
labeled
vacuoles in Dox-treated PC3 cells expressing shAkt123. PC3-shAkt123 cells were
incubated
in the presence or absence of 1 g/m1 Dox, with or without 10 M CQ, for 5
days before
labeling with MDC. Scale bar: 10 m. (G) Flow cytometry analysis of acridine
orange-
stained PC3-shAkt123 cells treated with 1 g/m1 Dox, 10 M CQ or both. FL1-H
indicates
intensity of green fluorescence of the nucleus. FL3-H indicates intensity of
red fluorescence
of the AVOs. Percentage of cells with high FL3-H/FL1-H ratio, characteristic
of cells With
high levels of AVOs (Paglin et al., 2001), is indicated on the histogram. The
data shown are
representative of three independent experiments.
Figure 20. Effect of LAMP2, Atg7, protease inhibitors, cathepsin D siRNA
and pepstatin A on PC3 cell viability in combination with PI-103 or Alcti-1/2.
(A)
Immunoblot showing KD of LAMP2 by siRNA oligos a-d and their pool compared to
a non-
targeting control oligo (siCtr1). (B) Effect of LAMP2 siRNA oligos on PC3 cell
viability.
PC3 cells were transfected with 80 nM of siRNA under 0.5% FBS, PI-103 (0.5 M)
or
DMSO was added 2 days post-transfection and cell viability (PI exclusion) was
analyzed 4
days after PI-103 addition. (C) Atg7 immunoblot of PC3 cells transfected with
Atg7 siRNA
pool (Santa Cruz) or a non-targeting control oligo. (D) Effect of Atg7 siRNA
oligos on PC3
cell viability under the different treatments. PC3 cells were transfected with
20 nM of siRNA
under 0.5% FBS, Akti-112 (5 M) or DMSO was added with or without CQ (10 M) 2
days
post-transfection and cell viability (PI exclusion) was analyzed 2 and 3 days
after compound
addition. *, P <0.05 between the two conditions. (E) PC3 cells treated with
the indicated
concentrations of zVAD.fmk added together with DMSO, 10 M CQ, 3 M Alcti-1/2,
or both
Alcti-1/2 and CQ. Cell viability was determined at day 3 by PI exclusion. *,
P< 0.05; **, P<
0.001 between the two conditions. (F) PC3 cells treated with the indicated
concentrations of
zFA.fink added together with DMSO, 10 M CQ, 3 M Akti-1/2, or both Akti-1/2
and CQ.
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
Cell viability was determined at day 3 by PI exclusion. *, P< 0.05; **, P<
0.001 between the
two conditions. (F) PC3 cells treated with the indicated concentrations of
zFA.fmk added
together with DMSO, 10 M CQ, 3 M Akti-1/2, or both Akti-1/2 and CQ. Cell
viability
was determined at day 3 by PI exclusion. *, P< 0.05 compared to either Akti-
1/2 alone or
zFA.fmk alone treated cells. (G-H) PC3 cells pre-treated with the indicated
concentrations of
CA-074-Me (G) or ALLN (H) for 2 hours prior to addition of DMSO, 10 M CQ, 3
WI
Akti-1/2, or both Akti-1/2 and CQ. Cell viability was determined at day 3 by
PI exclusion. *,
P <0.05 compared to either Akti-1/2 alone or the protease inhibitors alone
treated cells. (I)
Effect of cathepsin D siRNA oligos on PC3 cell viability under the different
treatments. PC3
cells were transfected with 10 nM of cathepsin D siRNA pool (Santa Cruz) or a
non-targeting
control under 0.5% FBS, Akti-1/2 (5 M) or DMSO was added with or without CQ
(10 M)
2 days post-transfection and cell viability (PI exclusion) was analyzed 2 days
after compound
addition. Cathepsin D knockdown was confirmed by immunoblot analysis shown at
the upper
right corner. (J) Effect of pepstatin A on PC3 cell viability under the
different treatments.
PC3 cells were treated with DMSO, 5 M Akti-1/2, 10 04 CQ or both under 0.5%
FBS with
or without 200 M pepstatin A. Cell viability (PI exclusion) was analyzed 2
days after
compound addition. *, P < 0.05 between the two conditions. (B, D-J) Error bars
represent
SEM (n
= 3). Molecular weights are indicated in kDa parenthetically next to each
protein
for immunoblots in A, C & I.
Figure 21. CQ promoted mitochondrial membrane depolarization and cellular
ROS accumulation in combination with Akti-l/2 (A) CQ enhanced Akti-1/2-induced
mitochondria depolarization. PC3 cells cultured in 0.5% FBS were treated with
DMSO, 3
M Akti-1/2, 10 M CQ, or both and stained with the MitoPT dye (Immunochemistry
Technologies, LLC) at various time points. Images at 48h are shown. Healthy
mitochondria
show punctate red stains of JC- laggregates (MitoPT-R), while cells with
depolarized
mitochondria show diffuse green stains of JC-1 monomers (MitoPT -G). Merged
images
between red and green channels are also shown (MitoPT -M). Scale bar: 20 m.
(B)
Percentage of cells showing red punctate stains and diffuse green stains from
treatments
described in (A). >86 cells from two random fields of fluorescent images of
each treatment
were counted. Error bars represent standard deviations between the two fields.
(C) 3-MA
reduced ROS signals generated by CQ, Akti and Akti + CQ. PC3 cells cultured in
0.5% FBS
were pre-treated with 3-MA overnight, then treated with DMSO, 5 M Akti-1/2,
10 /VI CQ,
or both for 48 hours and stained with the Image-iT LIVE Green ROS Detection
Kit. Total
fluorescence intensity from all cells was collected using the Isocyte
(Blueshift
11
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
Biotechnologies), a laser scanning imager. Green fluorescence intensity is
collected with a
488 nm laser and a 510-540 nm band pass filter, and normalized to cell number
determined
by Hoechst staining using a 405 nm laser and a 430-480 nm band pass filter.
Error bars,
standard deviation between two independent experiments. (D) Representative
images of the
green ROS signals and bright field (BF) taken under a Nikon TE300 inverted
microscope.
Akti-1/2 alone first induced a homogeneous increase in ROS level, but by 48
hours the
fluorescence became significantly reduced and localized to perinuclear and
cytoplasmic
vacuoles resembling autolysosomes. Although CQ alone had little effect on ROS
levels,
combination with Akti-1/2 caused a prolonged increase in vacuolar
fluorescence, as well as
an increased population of cells with persistent homogeneous fluorescence,
many of which
showing morphological signs of apoptosis within 48 hours. Arrowheads point to
representative cells exhibiting vacuolar fluorescence. Asterisks indicate
representative cells
with homogeneous green fluorescence and also exhibiting apoptotic morphology.
Scale bar:
gm.
15 Figure
22. NAC rescued cell death induced by Akti + CQ. (A) NAC reduced
MitoSOX Red signal induced by the various treatments. PC3 cells were either
pretreated for
1 day with 5 mM NAC then washed off (NACpr) and treated with the indicated
agents, or
pretreated for 1 hour with 5 mM NAC and incubated with the indicated agents in
the
continuous presence of NAC (NAC). MitoSOX signals were determined 24 hours
after Akti-
20 1/2
addition by Flow Cytometry. 5 gM Akti-1/2 and 10 gM CQ were used. Continuous
NAC
treatment is required for MitoSOX Red signal reduction at this time point. (B)
Viability (PI
exclusion) of cells treated as in (A) was determined 4 days after Akti-1/2
addition by Flow
Cytometry. NAC pretreatment showed a small, insignificant decrease in cell
death in the
Akti + CQ group, and significant rescue is seen with continuous NAC treatment.
(A and B)
Error bars represent SEM (n = 3). (C) PC3 cells stably expressing GFP-LC3 were
treated as
in (A) and analyzed by immunoblots at 48 hours after Akti-1/2 addition. I3-
actin and GAPDH
were used as loading controls. Quantifications of p62 and cleaved GFP levels
normalized to
GAPDH, and LC3-II to LC3-I ratios are shown on the right. Akti caused a
reduction in p62
levels, increased LC3-I turnover (both endogenous and GFP-LC3-I) and
concomitant LC3-II
and cleaved GFP accumulation. CQ increased the level of p62 both with and
without Akti,
consistent with its blocking of p62 degradation in the autolysosomes. CQ also
induced LC3-II
and cleaved GFP accumulation due to its blocking of their degradation in the
autolysosomes.
NAC treatment counteracted all these effects induced by Akti with or without
CQ but did not
affect Akti's ability to inhibit pAkt or pS6. NACpr showed some but weaker
effects than
12
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
continuous NAC treatment. (D) Time lapse fluorescence images of PC3 cells
stably
expressing GFP-LC3 treated with DMSO, 3 p.M Akti-1/2 in the presence or
absence of 5 mM
NAC (added 1 hour prior to Akt-1/2) at 2, 11 and 23 hours after Akti-1/2
addition.
Arrowheads point to representative cells with visible GFP-LC3 dots. Scale bar:
50 Rm.
Figure 23 depicts the pHUSH vector system used to make shRNAs
specifically targeting Akt isoforms. The pHUSH vector system comprises an
shRNA
expression shuttle plasmid (pShuttle-H1) and a viral vector backbone (pHUSH-
GW;
GW=Gateway) that contains a TetR-IRES-Puro cassette to enable Tet-regulated
shRNA
expression. The Akt shRNA vectors were constructed by (1) designing and
cloning shRNA
sequences into pShuttle-H1 (2) transferring the Hl-shRNA cassette into pHUSH-
GW by a
Gateway (Invitrogen) recombination reaction and (3) packaging the completed HI-
pHUSH
plasmid as a retrovirus. For each shRNA, a 19bp siRNA sequence was designed
using an
appropriate algorithm against the coding sequence of an Akt gene(s). The shRNA
sequence
was converted into an shRNA hairpin sequence, and then the corresponding
double-stranded
DNA oligo was synthesized and cloned into pShuttle-H1 as shown. The
effectiveness of
each shRNA in pShuttle-H1 vector was verified by transient transfection into
cells and the
degree of knockdown of each Akt isoform examined by western blots. The
validated HI-
shRNA cassette was then transferred into the pHUSH-GW vector and packaged as a
retrovirus (Table 1 summarizes the validated sequences used). Cells stably
expressing each
shRNA were generated by retroviral infection with single or combination of
shRNA-
containing viruses. For single Akt isoform knockdowns, cells were infected
with one
retroviral vector encoding an shRNA construct singly targeting each Akt
isoform (constructs
252 & 253 for Akt 1, 254 & 255 for Akt2, and 259 & 260 for Akt3) and stable
clones were
selected using 5 mg/ml puromycin. For dual Aka and Akt2 knockdown, a single
shRNA
targeting both Akt 1 and 2 simultaneously (construct 256 & 257) was used. Dual
Akt2 and 3
(constructs 255 and 261), or triple Alctl, 2 and 3 (constructs 257 and 261)
knockdowns were
achieved by co-infecting the cells with two retroviral vectors containing
different antibiotic
selection markers (puromycin and hygromycin), each encoding one single shRNA,
and stable
clones were selected using 5 mg/ml puromycin and 300 mg/ml hygromycin. For
dual Akt 1
and 3 knockdown, either a single shRNA targeting both Aka and 3 (construct
258), or co-
infection with two shRNA vectors (constructs 253 and 261) was employed (Table
2). All
shRNAs shown in Table 2 have been validated in cultured cells. The efficiency
and tumor
inhibitory effect of shRNAs validated in xenograft models are summarized in
Table 3.
Figure 24 depicts a model of the mechanism of cell death induced by the
13
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
combination of chloroquine with Akt inhibition. Akt inhibition alone (by
shRNA, specific
Akt inhibitors or class I PI3K inhibitors) can activate autophagy through
multiple
mechanisms, including decreased mTORC1 activity downstream of Akt, (Corradetti
MN,
Guan KL. Upstream of the mammalian target of rapamycin: do all roads pass
through
mTOR? Oncogene (2006); 25:6347-60), increased activity of Fox() proteins (Zhao
J, Brault
JJ, Schild A, Goldberg AL. Coordinate activation of autophagy and the
proteasome pathway
by fox0 transcription factor, Autophagy (2008); 4:378-80), and decreased
glucose and
energy metabolism. We also observed an accumulation of abnormal mitochondria
and signs
of ER stress (unpublished data) in cells with Akt knockdown or inhibition,
both of which can
induce autophagy. (Yorimitsu T, Klionslcy DJ. Endoplasmic reticulum stress: a
new pathway
to induce autophagy. Autophagy (2007); 3:160-2). Abnormal mitochondria can
generate a
ROS signal, resulting in elevated autophagic removal of the damaged
mitochondria and
attenuation of the oxidative stress. 3-methyladenine (3-MA) and other non-
selective pan-
PI3K inhibitors that inhibit class III PI3K, such as wortmannin or LY294002,
block the
induction of autophagy. Impaired autolysosomal degradation caused by CQ (or
other
inhibitors of lysosomal enzyme activity) can result in aggregation of
deleterious ROS
generators that further amplify the ROS damage. Multiple downstream events can
lead to
both apoptosis-like and non-apoptotic cell death. It is unclear whether the
autophagic
response induced by Akt inhibition alone can eventually lead to cell death
directly in some
cells, or require additional insults.
Figure 25A-E depict AV accumulation in PC3 cells treated with Akt inhibitor,
CQ and their combination. (Panels A-C): PC3 cells grown under 0.5% FBS were
treated with
(A) DMSO control, (B) 10 M CQ, and (C) 5 12M Akti-1/2 for 1 day. Both CQ and
Akti-1/2
alone induced accumulation of AVs (arrows). (Panels D-E): Combined treatment
of Akti-1,2
and CQ resulted in accumulation of larger AVs and the appearance apoptotic
nuclei. (D)
After 1 day of treatment, many cells contained very large AVs (arrows),
distended ER
cisterns (arrowheads), and appear largely vacuolated (small arrows). (E) After
2 days of
treatment, apoptosis became apparent in a large proportion of the cells
(asterisk). Scale bars,
2 ttm.
Figures 26A-B depicts measured ED50 for CQ alone, the AKTi 111-4 alone,
and their combination, in multiple cell lines. Figure 26A depicts the compound
ratio work
table for CQ and the AKTi. Figure 26B depicts the CI (combination index) by
using bar
graphs for data obtained by CellTiter-Glo assay at day 4 of treatment with
10%serum. The
X-axis shows the cell line and the Y-axis shows the concentration in pM. The
combination
14
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
of the AKTi with CQ significantly lowers the ED50 for both the AKTi and CQ.
Figures 27A-B depicts data for the PC3 (PTEN-, p53- and Al) cell line.
Figure 27A depicts the CI values at ED50, ED75 and ED90 when the AKTi 111-4
and CQ are
combined at varying ratios. The data show combination ratios of III-4:CQ in
the ranges of
5:1 to 1:800, respetively, with the lowest ED50, ED75 and ED90 values in the
ratios of about
1:1.5 to about 1:200, with alternative ratios in the range of about 1:3 to
about 1:50, with
additionally alternative ratios of about 1:12 to about 1:50, with particular
ratio of about 1:25,
respectively. 27B depicts growth inhibition curves of the AKTi 111-4 alone, CQ
alone, and
the AKTi and CQ combination dosed in a 1:25 ratio (about the EC50 ratio). The
x-axis
indicates 111-4 concentration in 1.1M and the y-axis indicates the % of
control. The data
indicate that the combination inhibits growth in the cell line at lower
concentrations of 111-4
than either 111-4 alone or CQ alone.
Figures 28A-B depicts data for the MDA-361.1 (P12-K mut (E545K), Her2+,
HR+ and Luminal) cell line. Figure 28A depicts the CI values at ED50, ED75 and
ED90
when the AKTi 111-4 and CQ are combined at varying ratios. The data show
combination
ratios of III-4:CQ in the ranges of 5:1 to 1:800, respetively, with the lowest
ED50, ED75 and
ED90 values in the ratios of about 1:1.5 to about 1:200, with alternative
ratios in the range of
about 1:3 to about 1:25, with particular ratio of about 1:12.5, respectively.
Figure 28B
depicts growth inhibition curves of the AKTi 111-4 alone, CQ alone, and the
AKTi and CQ
combination dosed in a 1:12.5 ratio (about the EC50 ratio). The x-axis
indicates 111-4
concentration in tiM and the y-axis indicates the % of control. The data
indicate that the
combination inhibits growth in the cell line at lower concentrations of 111-4
than either 111-4
alone or CQ alone.
Figures 29A-B depicts data for the MDA-MB-231 (Kras, Braf, p53 mut,
Triple- and Basal) cell line. Figure 29A depicts the CI values at ED50, ED75
and ED90
when the AKTi 111-4 and CQ are combined at varying ratios. The data show
combination
ratios of III-4:CQ in the ranges of 5:1 to 1:800, respetively, with the lowest
ED50, ED75 and
ED90 values in the ratios of about 2.5:1 to about 1:1:3, with particular ratio
of about 1.25:1,
respectively. Figure 29B depicts growth inhibition curves of the AKTi 111-4
alone, CQ alone,
and the AKTi and CQ combination dosed in a 1.25:1 ratio (about the EC50
ratio). The x-axis
indicates 111-4 concentration in jiM and the y-axis indicates the % of
control. The data
indicate that the combination inhibits growth in the cell line at lower
concentrations of 111-4
than either 111-4 alone or CQ alone.
Figures 30A-B depicts data for the U87MG (PTEN-, PI3K mut (1391M)) cell
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
line. Figure 30A depicts the CI values at ED50, ED75 and ED90 when the AKTi
111-4 and
CQ are combined at varying ratios. The data show combination ratios of III-
4:CQ in the
ranges of 5:1 to 1:800, respetively, with the lowest ED50, ED75 and ED90
values in the
ratios of about 2.5:1 to about 1:25, with alternative ratios in the range of
about 1.25:1 to about
1:3, with a particular ratio of about 1:1.5, respectively. Figure 30B depicts
growth inhibition
curves of the AKTi 111-4 alone, CQ alone, and the AKTi and CQ combination
dosed in a
1:1.5 ratio (about the EC5 0 ratio with minimum CI). The x-axis indicates 111-
4 concentration
in M and the y-axis indicates the % of control. The data indicate that the
combination
inhibits growth in the cell line at lower concentrations of 111-4 than either
111-4 alone or CQ
alone.
Figures 3 1A-C depicts data for the Panc-1 (Akt2 amp, Kras mut, p53 mut) cell
line. Figure 3 lA depicts the CI values at ED50, ED75 and ED90 when the AKTi
111-4 and
CQ are combined at varying ratios. The data show combination ratios of III-
4:CQ in the
ranges of 5:1 to 1:800, respetively, with the lowest ED50, ED75 and ED90
values in the
ratios of about 2.5:1 to about 1:1.3, with a particular ratio of about 1.25:1,
respectively.
Figure 3 1B depicts growth inhibition curves of the AKTi 111-4 alone, CQ
alone, and the
AKTi and CQ combination dosed in a 1.28:1 ratio (about the EC50 ratio with
minimum CI).
The x-axis indicates 111-4 concentration in pM and the y-axis indicates the %
of control. The
data indicate that the combination inhibits growth in the cell line at lower
concentrations of
111-4 than either 111-4 alone or CQ alone. Figure 3 IC depicts growth
inhibition curves of the
AKTi 111-4 alone, CQ alone, and the AKTi and CQ combination dosed in a 1:1.56
ratio
(about the EC1 0 ratio with minimum CI). The x-axis indicates 111-4
concentration in M and
the y-axis indicates the % of control. The data indicate that the combination
inhibits growth
in the cell line at lower concentrations of 111-4 than either 111-4 alone or
CQ alone.
Figures 32A-B and 34A-B depict data showing correlation between
Autophagy induction and apoptosis induction when each compound is combined
with CQ at
24 hrs after the given compound treatment measured by Acridine Orange staining
in 53 7MEL
melanoma cell line and SKBR3 Breast cancer cell line, respectively. Control
was DMSO.
Compounds tested were 1 -
( 1 -(4-(7-phenyl- 1 H-imidazo [4,5 -dquinoxal in-6-
(pyrid in-2-y1)- 1 H- 1 ,2,4-triazol-3-yl)piperidin- 1 -yl)methyl)phenyI)- 1
,6-n aphthyridin-5 (6f1)-
one (II-3), (S)-
2-(4-chloropheny1)- 1 -(44(5R,7R)-7-hydroxy-5-methy1-6,7-dihydro-5H-
cyclopenta[d]pyrim idin-4-yl)p iperazin- 1 -y1)-3 -(isopropylamino)propan- 1 -
one (III-4), 4-(2-
( 1 H-indazol-4-y1)-6-04-(methylsulfonyppiperazin- 1 -yl)methyl)thieno [3 ,2-
d]pyrim idin-4-
1 6
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
yl)morpholine (III-6), (R)-N-(2,3-dihydroxypropoxy)-3,4-difluoro-2-
(2-fluoro-4-
iodophenylamino)benzamide (VII), (S)-1-ethy1-3-(4-(4-(3-methylmorpholino)-7-
(pyrimidin-
2-y1)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-2-yOphenyOurea (III-2a) and
rapamycin.
Figures 33A-G and 35A-G show dose response curve of apoptosis induction in
537MEL melanoma and SKBR3 Breast cancer cells, respectively, treated with the
indicated
compound alone or with 10 M CQ, measured by Annezin V (AnnV) and Prop idium
iodide
(PI) staining.
In above Figures, a positive correlation is shown between autophagy induction
and apoptosis induction when the compound is combined with CQ. In the case of
the
compound that does not induce autophagy (VII), no synergistic apoptosis
induction is shown
when combined with CQ.
DETAILED DESCRIPTION OF THE INVENTION
DEFINITIONS
The term "neoplasms" encompasses "cancer" and "cancerous," which refer to
or describe the physiological condition in mammals that is typically
characterized by
unregulated cell growth. A "tumor" comprises one or more cancerous cells.
Examples of
cancer include, but are not limited to, carcinoma, lymphoma, blastoma,
sarcoma, and
leukemia or lymphoid malignancies. More particular examples of such cancers
include
squamous cell cancer (e.g., epithelial squamous cell cancer), lung cancer
including small- cell
lung cancer, non-small cell lung cancer ("NSCLC"), adenocarcinoma of the lung
and
squamous carcinoma of the lung, cancer of the peritoneum, hepatocellular
cancer, gastric or
stomach cancer including gastrointestinal cancer, pancreatic cancer,
glioblastoma, cervical
cancer, ovarian cancer, liver cancer, bladder cancer, hepatoma, breast cancer,
colon cancer,
rectal cancer, colorectal cancer, endometrial or uterine carcinoma, salivary
gland carcinoma,
kidney or renal cancer, prostate cancer, vulval cancer, thyroid cancer,
hepatic carcinoma, anal
carcinoma, penile carcinoma, as well as head and neck cancer. In one
embodiment, the
neoplasm is other than a glycolysis dependent cancer. In another embodiment,
the neoplasm
is prostate, breast, glioma or pancreatic cancer. In another embodiment, the
neoplasm is
prostate, breast or ovarian cancer. In another embodiment, the neoplasm
comprises PTEN or
PI3K mutations. In another embodiment, the neoplasm is resistant to inhibitors
of the Akt
kinase pathway.
The term "alkyl" as used herein refers to a saturated linear or branched-chain
17
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
monovalent hydrocarbon radical of one to twelve carbon atoms, wherein the
alkyl radical
may be optionally substituted independently with one or more substituents
described below.
Examples of alkyl groups include, but are not limited to, methyl (Me, -CH3),
ethyl (Et, -
CH2CH3), 1-propyl (n-Pr, n-propyl, -CH2CH2CH3), 2-propyl (i-Pr, i-propyl, -
CH(CH3)2), 1-
butyl (n-Bu, n-butyl, -CH2CH2CH2CH3), 2-methyl- 1-propyl (i-Bu, i-butyl, -
CH2CH(CH3)2),
2-butyl (s-Bu, s-butyl, -CH(CH3)CH2CH3), 2-methyl-2-propyl (t-Bu, t-butyl, -
C(CH3)3), 1-
pentyl (n-pentyl, -CH2CH2CH2CH2CH3), 2-pentyl (-CH(CH3)CH2CH2CH3), 3-pentyl (-
CH(CH2CH3)2), 2-methyl-2-butyl (-C(CH3)2CH2CH3), 3-methyl-2-butyl
(-
CH(CH3)CH(CH3)2), 3-methyl-l-butyl (-CH2CH2CH(CH3)2), 2-methyl- I -butyl (-
CH2CH(CH3)CH2CH3), 1-hexyl (-CH2CH2CH2CH2CH2CH3), 2-hexyl (-
CH(CH3)CH2CH2CH2CH3), 3-hexyl (-CH(CH2CH3)(CH2CH2CH3)), 2-methyl-2-pentyl
C(CH3)2CH2CH2CH3), 3-methyl-2-pentyl (-CH(CH3)CH(CH3)CH2CH3), 4-methyl-2-
pentyl
CH(CH3)CH2CH(CH3)2), 3-methyl-3-pentyl (-C(CH3)(CH2CH3)2), 2-methyl-3-pentyl (-
CH(CH2CH3)CH(CH3)2), 2,3-dimethy1-2-butyl (-C(CH3)2C11(CH3)2), 3,3-dimethy1-2-
butyl (-
CH(CH3)C(CH3)3, 1-heptyl, 1-octyl, and the like.
The term "alkenyl" refers to linear or branched-chain monovalent hydrocarbon
radical of two to twelve carbon atoms with at least one site of unsaturation,
i.e., a carbon-
carbon, sp2 double bond, wherein the alkenyl radical may be optionally
substituted
independently with one or more substituents described herein, and includes
radicals having
"cis" and "trans" orientations, or alternatively, "E" and "Z" orientations.
Examples include,
but are not limited to, ethylenyl or vinyl (-CH=CH2), allyl (-CH2CH=CH2), and
the like.
The term "allcynyl" refers to a linear or branched monovalent hydrocarbon
radical of two to twelve carbon atoms with at least one site of unsaturation,
i.e., a carbon-
carbon, sp triple bond, wherein the alkynyl radical may be optionally
substituted
independently with one or more substituents described herein. Examples
include, but are not
limited to, ethynyl propynyl (propargyl, -CH2C-CH), and the like.
The terms "carbocycle", "carbocyclyl", "carbocyclic ring" and "cycloalkyl"
refer to a monovalent non-aromatic, saturated or partially unsaturated ring
having 3 to 12
carbon atoms as a monocyclic ring or 7 to 12 carbon atoms as a bicyclic ring.
Bicyclic
carbocycles having 7 to 12 atoms can be arranged, for example, as a bicyclo
[4,5], [5,5], [5,6]
or [6,6] system, and bicyclic carbocycles having 9 or 10 ring atoms can be
arranged as a
bicyclo [5,6] or [6,6] system, or as bridged systems such as
bicyclo[2.2.1]heptane,
bicyclo[2.2.2]octane and bicyclo[3.2.2]nonane. Examples of monocyclic
carbocycles
18
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, 1-
cyclopent-1-enyl, 1-
cyc lopent-2-enyl, 1-cyclopent-3-enyl, cyclohexyl, 1-cyclohex-1-enyl, 1-
cyclohex-2-enyl, 1-
cyc lohex-3 -enyl, cyclohexadienyl, cycloheptyl, cyclooctyl, cyclononyl,
cyclodecyl,
cycloundecyl, cyclododecyl, and the like.
"Aryl" or "aromatic" means a monovalent aromatic hydrocarbon radical of 6-
20 carbon atoms derived by the removal of one hydrogen atom from a single
carbon atom of
a parent aromatic ring system. Some aryl groups are represented in the
exemplary structures
as "Ar". Aryl includes bicyclic radicals comprising an aromatic ring fused to
a saturated,
partially unsaturated ring, or aromatic carbocyclic or heterocyclic ring.
Typical aryl groups
include, but are not limited to, radicals derived from benzene (phenyl),
substituted benzenes,
naphthalene, anthracene, biphenyl, indenyl, indanyl, 1,2-dihydronapthalene,
1,2,3,4-
tetrahydronapthyl, and the like. Aryl groups are optionally substituted
independently with
one or more substituents described herein.
The terms "heterocycle," "hetercycly1" and "heterocyclic ring" are used
interchangeably herein and refer to a saturated, a partially unsaturated
(i.e., having one or
more double and/or triple bonds within the ring) or aromatic carbocyclic
radical of 3 to 20
ring atoms in which at least one ring atom is a heteroatom selected from
nitrogen, oxygen and
sulfur, the remaining ring atoms being C, where one or more ring atoms is
optionally
substituted independently with one or more substituents described below. A
heterocycle may
be a monocycle having 3 to 7 ring members (2 to 6 carbon atoms and 1 to 4
heteroatoms
selected from N, 0, P, and S) or a bicycle having 7 to 10 ring members (4 to 9
carbon atoms
and 1 to 6 heteroatoms selected from N, 0, P, and S), for example: a bicyclo
[4,5], [5,5],
[5,6], or [6,6] system. Heterocycles are described in Paquette, Leo A.;
"Principles of Modern
Heterocyclic Chemistry" (W.A. Benjamin, New York, 1968), particularly Chapters
1, 3, 4, 6,
7, and 9; "The Chemistry of Heterocyclic Compounds, A series of Monographs"
(John Wiley
& Sons, New York, 1950 to present), in particular Volumes 13, 14, 16, 19, and
28; and J.
Am. Chem. Soc. (1960) 82:5566. The term "heterocycle" includes
heterocycloalkoxy.
"Heterocycly1" also includes radicals where heterocycle radicals are fused
with a saturated,
partially unsaturated ring, or aromatic carbocyclic or heterocyclic ring.
Examples of
heterocyclic rings include, but are not limited to, pyrrolidinyl,
tetrahydrofuranyl,
dihydrofuranyl, tetrahydrothienyl, tetrahydropyranyl, dihydropyranyl,
tetrahydrothiopyranyl,
piperidino, morpholino, thiomorpholino, thioxanyl, piperazinyl,
homopiperazinyl, azetidinyl,
oxetanyl, thietanyl, homopiperidinyl, oxepanyl, thiepanyl, oxazepinyl,
diazepinyl,
thiazepinyl, 2-pyrrolinyl, 3-pyrrolinyl, indolinyl, 2H-pyranyl, 4H-pyranyl,
dioxanyl, 1,3-
19
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
dioxolanyl, pyrazolinyl, dithianyl, dithiolanyl, dihydropyranyl,
dihydrothienyl,
dihydrofuranyl, pyrazolidinylimidazolinyl, imidazolidinyl, 3-azabicyco [3
.1.0]hexanyl, 3-
azabicyclo[4.1.0]heptanyl, azabicyclo[2.2.2]hexanyl, 3H-indoly1 quinolizinyl
and N-pyridyl
ureas. Spiro moieties are also included within the scope of this definition.
Examples of a
heterocyclic group wherein 2 ring carbon atoms are substituted with oxo (-----
0) moieties are
pyrimidinonyl and 1,1-dioxo-thiomorpholinyl. The heterocycle groups herein are
optionally
substituted independently with one or more substituents described herein.
The term "heteroaryl" or "heteroaromatic" refers to a monovalent aromatic
radical of 5-, 6-, or 7-membered rings, and includes fused ring systems (at
least one of which
is aromatic) of 5-20 atoms, containing one or more heteroatoms independently
selected from
nitrogen, oxygen, and sulfur. Examples of heteroaryl groups are pyridinyl
(including, for
example, 2-hydroxypyridinyl), imidazolyl, imidazopyridinyl, pyrimidinyl
(including, for
example, 4-hydroxypyrimidinyl), pyrazolyl, triazolyl, pyrazinyl, tetrazolyl,
furyl, thienyl,
isoxazolyl, thiazolyl, oxazolyl, isothiazolyl, pyrrolyl, quinolinyl,
isoquinolinyl, indolyl,
benzimidazolyl, benzofuranyl, cinnolinyl, indazolyl, indolizinyl,
phthalazinyl, pyridazinyl,
triazinyl, isoindolyl, pteridinyl, purinyl, oxadiazolyl, triazolyl,
thiadiazolyl, thiadiazolyl,
furazanyl, benzofurazanyl, benzothiophenyl, benzothiazolyl, benzoxazolyl,
quinazolinyl,
quinoxalinyl, naphthyridinyl, and furopyridinyl. Heteroaryl groups are
optionally substituted
independently with one or more substituents described herein.
The heterocycle or heteroaryl groups may be carbon (carbon-linked), nitrogen
(nitrogen-linked) or oxygen (oxygen-linked) attached where such is possible.
By way of
example and not limitation, carbon bonded heterocycles or heteroaryls are
bonded at position
2, 3, 4, 5, or 6 of a pyridine, position 3, 4, 5, or 6 of a pyridazine,
position 2, 4, 5, or 6 of a
pyrimidine, position 2, 3, 5, or 6 of a pyrazine, position 2, 3, 4, or 5 of a
furan,
tetrahydrofuran, thiofuran, thiophene, pyrrole or tetrahydropyrrole, position
2, 4, or 5 of an
oxazole, imidazole or thiazole, position 3, 4, or 5 of an isoxazole, pyrazole,
or isothiazole,
position 2 or 3 of an aziridine, position 2, 3, or 4 of an azetidine, position
2, 3, 4, 5, 6, 7, or 8
of a quinoline or position 1, 3, 4, 5, 6, 7, or 8 of an isoquinoline.
The terms "treat" and "treatment" refer to both therapeutic treatment and
prophylactic or preventative measures, which prevent or slow down (lessen) an
undesired
physiological change or disorder, such as the growth, development or spread of
cancer. In
one embodiment, the term "treat" and "treatment" refer to therapeutic
treatment, which or
slows down (lessen) an undesired physiological change or disorder, such as the
growth,
development or spread of cancer. For purposes of this invention, beneficial or
desired clinical
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
results include, but are not limited to, alleviation of symptoms, diminishment
of extent of
disease, stabilized (i.e., not worsening) state of disease, delay or slowing
of disease
progression, amelioration or palliation of the disease state, and remission
(whether partial or
total), whether detectable or undetectable. "Treatment" can also mean
prolonging survival as
compared to expected survival if not receiving treatment. Those in need of
treatment include
those already with the condition or disorder as well as those prone to have
the condition or
disorder or those in which the condition or disorder is to be prevented.
As used herein, "glycolysis dependent cancer" is meant to refer to cancer that
is characterized by cancer cells that rely on glucose metabolism for
essentially all of their
energy needs excluding energy that may be obtained by autophagy. Cancer cells
of
glycolysis dependent cancer may be capable of some level of non-glycolytic
metabolism but
such level does not prevent the cancer cells from undergoing cell death by
apoptosis or
autophagy in the absence of a glucose energy source. There are numerous
methods of
determining whether or not a cancer is dependent upon glycolysis. Samples of
tumors can be
excised and examined in vitro by any one of several well known assays to
determine if the
cells are dependent on glycolysis. Such methods can determine whether or not
the cells
utilize aerobic or anaerobic glycolysis. FDG-PETscan technology uses high
levels of glucose
uptake as a marker for detection. The cancer cells that take up the detectable
glucose
derivative 18-fluoro-2-deoxyglucose can be located on a computer image of the
patient's
anatomy. Those cancers which can be detected by FDG-PETscan technology have a
high
likelihood of being dependent on glycolysis.
The phrase "therapeutically effective amount" means an amount of a
compound of the present invention that (i) treats the particular disease,
condition, or disorder,
(ii) attenuates, ameliorates, or eliminates one or more symptoms of the
particular disease,
condition, or disorder, or (iii) prevents or delays the onset of one or more
symptoms of the
particular disease, condition, or disorder described herein. In the case of
cancer, the
therapeutically effective amount of the drug may reduce the number of cancer
cells; reduce
the tumor size; inhibit (i.e., slow to some extent and/or stop) cancer cell
infiltration into
peripheral organs; inhibit (i.e., slow to some extent and/or stop) tumor
metastasis; inhibit, to
some extent, tumor growth; and/or relieve to some extent one or more of the
symptoms
associated with the cancer. To the extent the drug may prevent growth and/or
kill existing
cancer cells, it may be cytostatic and/or cytotoxic. For cancer therapy,
efficacy can be
measured, for example, by assessing the time to disease progression (UP)
and/or
determining the response rate (RR).
21
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
The term "mammal" includes, but is not limited to, humans, mice, rats, guinea
pigs, monkeys, dogs, cats, horses, cows, pigs, and sheep, and poultry.
The phrase "pharmaceutically acceptable salt" as used herein, refers to
pharmaceutically acceptable organic or inorganic salts of a compound of the
invention.
Exemplary salts include, but are not limited, to sulfate, citrate, acetate,
oxalate, chloride,
bromide, iodide, nitrate, bisulfate, phosphate, acid phosphate, isonicotinate,
lactate,
salicylate, acid citrate, tartrate, oleate, tannate, pantothenate, bitartrate,
ascorbate,
succinate, maleate, gentisinate, fumarate, gluconate, glucuronate, saccharate,
formate,
benzoate, glutamate, methanesulfonate "mesylate", ethanesulfonate,
benzenesulfonate, p-
toluenesulfonate, and pamoate (i.e., 1,1'-methylene-bis -(2-hydroxy-3-
naphthoate)) salts.
A pharmaceutically acceptable salt may involve the inclusion of another
molecule such as
an acetate ion, a succinate ion or other counter ion. The counter ion may be
any organic or
inorganic moiety that stabilizes the charge on the parent compound.
Furthermore, a
pharmaceutically acceptable salt may have more than one charged atom in its
structure.
Instances where multiple charged atoms are part of the pharmaceutically
acceptable salt
can have multiple counter ions. Hence, a pharmaceutically acceptable salt can
have one or
more charged atoms and/or one or more counter ion.
If the compound of the invention is a base, the desired pharmaceutically
acceptable salt may be prepared by any suitable method available in the art,
for example,
treatment of the free base with an inorganic acid, such as hydrochloric acid,
hydrobromic
acid, sulfuric acid, nitric acid, methanesulfonic acid, phosphoric acid and
the like, or with
an organic acid, such as acetic acid, maleic acid, succinic acid, mandelic
acid, fumaric
acid, malonic acid, pyruvic acid, oxalic acid, glycolic acid, salicylic acid,
a pyranosidyl
acid, such as glucuronic acid or galacturonic acid, an alpha hydroxy acid,
such as citric
acid or tartaric acid, an amino acid, such as aspartic acid or glutamic acid,
an aromatic
acid, such as benzoic acid or cinnamic acid, a sulfonic acid, such as p-
toluenesulfonic acid
or ethanesulfonic acid, or the like. Acids which are generally considered
suitable for the
formation of pharmaceutically useful or acceptable salts from basic
pharmaceutical
compounds are discussed, for example, by P. Stahl et al, Camille G. (eds.)
Handbook of
Pharmaceutical Salts. Properties, Selection and Use. (2002) Zurich: Wiley-VCH;
S. Berge
et al, Journal of Pharmaceutical Sciences (1977) 66(1) 119; P. Gould,
International J. of
Pharmaceutics (1986) 33 201 217; Anderson et al, The Practice of Medicinal
Chemistry
(1996), Academic Press, New York; Remington's Pharmaceutical Sciences, 18th
ed.,
(1995) Mack Publishing Co., Easton PA; and in The Orange Book (Food & Drug
22
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
Administration, Washington, D.C. on their website). These disclosures are
incorporated
herein by reference thereto.
If the compound of the invention is an acid, the desired pharmaceutically
acceptable salt may be prepared by any suitable method, for example, treatment
of the free
acid with an inorganic or organic base, such as an amine (primary, secondary
or tertiary),
an alkali metal hydroxide or alkaline earth metal hydroxide, or the like.
Illustrative
examples of suitable salts include, but are not limited to, organic salts
derived from amino
acids, such as glycine and arginine, ammonia, primary, secondary, and tertiary
amines, and
cyclic amines, such as piperidine, morpholine and piperazine, and inorganic
salts derived
from sodium, calcium, potassium, magnesium, manganese, iron, copper, zinc,
aluminum
and lithium.
The phrase "pharmaceutically acceptable" indicates that the substance or
composition is compatible chemically and/or toxicologically, with the other
ingredients
comprising a formulation, and/or the mammal being treated therewith.
The phrase "substantially corresponding to" means that a sequence has
inconsequential variations from the known or target sequence. In one example,
a sequence
has about 80% homology with a known or target sequence. In another example, a
sequence has 85% homology. In another example, a sequence has 90, 91, 92, 93,
94, 95,
96, 97, 98, 99 or above % homology. Methods are known in the art for
determining %
homology.
A "solvate" refers to a physical association or complex of one or more solvent
molecules and a compound of the invention. The compounds of the invention may
exist in
unsolvated as well as solvated forms. Examples of solvents that form solvates
include, but
are not limited to, water, isopropanol, ethanol, methanol, DMSO, ethyl
acetate, acetic acid,
and ethanolamine. The term "hydrate" refers to the complex where the solvent
molecule is
water. This physical association involves varying degrees of ionic and
covalent bonding,
including hydrogen bonding. In certain instances the solvate will be capable
of isolation,
for example when one or more solvent molecules are incorporated in the crystal
lattice of
the crystalline solid. Preparation of solvates is generally known, for
example, M. Caira et
al, J. Pharmaceutical Sci., 93(3), 601 611 (2004). Similar preparations of
solvates,
hemisolvate, hydrates and the like are described by E. C. van Tonder et al,
AAPS
PharmSciTech., 5(1), article 12 (2004); and A. L. Bingham et al, Chem.
Commun., 603
604 (2001). A typical, non-limiting, process involves dissolving the inventive
compound
in desired amounts of the desired solvent (organic or water or mixtures
thereof) at a higher
23
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
than ambient temperature, and cooling the solution at a rate sufficient to
form crystals
which are then isolated by standard methods. Analytical techniques such as,
for example
I.R. spectroscopy, show the presence of the solvent (or water) in the crystals
as a solvate
(or hydrate).
The term "synergistic" as used herein refers to a therapeutic combination
which is more effective than the additive effects of the two or more single
agents. A
determination of a synergistic ,interaction between a kinase inhibitor that
induces
autophagy and one or more inhibitor of autophagy may be based on the results
obtained
from the assays described herein. A synergistic effect may be attained when
the active
ingredients are: (1) co-formulated and administered or delivered
simultaneously in a
combined, unit dosage formulation; (2) delivered by alternation or in parallel
as separate
formulations; or (3) by some other regimen. When delivered in alternation
therapy, a
synergistic effect may be attained when the compounds are administered or
delivered
sequentially, e.g., by different injections in separate syringes. In general,
during
alternation therapy, an effective dosage of each active ingredient is
administered
sequentially, i.e., serially, whereas in combination therapy, effective
dosages of two or
more active ingredients are administered together. The combinations provided
herein have
been evaluated, and the data can be analyzed utilizing a standard program for
quantifying
synergism, additivism, and antagonism. An example of a program used for
calculating
synergism is that described by Chou and Talalay, in "New Avenues in
Developmental
Cancer Chemotherapy," Academic Press, 1987, Chapter 2.
As used herein, "autophagy inhibitor" is meant to refer to composition which
decreases the level of autophagy in a cell undergoing autophagy in its
presence compared to
the level of autophagy in a cell undergoing autophagy in its absence.
Autophagy is a catabolic
process of bulk lysosomal degradation and recycling of cytoplasmic material
and organelles,
characterized by the appearance of autophagic vacuoles in the cytoplasm,
leading to self-
digestion of cytoplasmic organelles and other constituents in the lysosomal
compartments.
While autophagy may be capable of ultimate cell killing when allowed to reach
its limit,
autophagy can provide a temporary survival mechanism for cells under stress
conditions, but
can also make cells vulnerable to several forms of cell death under specific
circumstances.
Inhibiting autophagy can either promote or inhibit cell death depending on the
conditions and
agents used (Amaravadi, R.K., D. Yu, J.J. Lum, T. Bui, M.A. Christophorou,
G.I. Evan, A.
Thomas-Tikhonenko, and C.B. Thompson, (2007), Autophagy inhibition enhances
therapy-
induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 117:326-
336;
24
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
Kroemer, G., and M. Jaattela. 2005. Lysosomes and autophagy in cell death
control. Nat Rev
Cancer. 5:886-97; Levine, B., and J. Yuan, (2005), Autophagy in cell death: an
innocent
convict. J Clin Invest. 115:2679-2688; Lockshin, R.A., and Z. Zakeri, (2004),
Apoptosis,
autophagy, and more. Int J Biochem Cell Biol. 36:2405-19). Autophagy is a
catabolic process
that has distinct phases. These include, induction, sequestration, fusion and
degradation
phases. Inhibitors of autophagy can inhibit one or more of the phases. In an
embodiment,
autophagy inhibitors inhibit the later stages of autophagy. In one example,
autophagy
inhibitors inhibit the sequestration, fusion and degradation phases of
autophagy. In one
example, autophagy inhibitors inhibit the fusion and degradation phases of
autophagy. In one
example, autophagy inhibitors inhibit the degradation phase of autophagy.
Useful inhibitors
of autophagy include siRNA; antisense RNA; agents that inhibit the expression
or function of
LAMP2, LAMP1 or an autophagy (Atg) gene (e.g., Atg 1 , Atg4, Atg8, Atg5, Atg7
or Atg12);
3-methyladenine; lysosomotropic agents, which can also be antiparasitic, such
as
chloroquine, hydroxychloroquine or suramin, a vacuolar proton -ATPase
inhibitor, such as
Bafilomycin Al, an agent acting on the circulatory system, such as Amiodarone
or
Perhexilene, a cytotoxic agent, such as Vinblastine, an agent influencing
lipid metabolism, an
antibiotic, such as monensin, or a hormone, such as, Glucagon or estradiol,
lysosomotropic
agents, such as ammonium chloride, cAMP or methylamine, ATPase inhibitors,
protease
inhibitors, lysosomal protease inhibitors such as cathepsin inhibitors and
cathepsin
knockdown, as well as LAMP knockdown, e.g. LAMP1 and LAMP2. In another
embodiment, modulators of lysosomal activity can be combined with kinase
inhibitors that
induce autophagy to provide a combination therapy for neoplasms. Lysosomes are
organelles
that contain digestive enzymes (acid hydrolases). Such enzymes include lipase,
which digests
lipids, carbohydrases, which digest carbohydrates (e.g., sugars), proteases,
which digest
proteins, nucleases, which digest nucleic acids, and phosphoric acid
monoesters. In one
example, the modulator acts to inhibit lysosomal activity. In an embodiment,
the combination
provides a synergistic effect.
KINASE INHIBITORS
There are hundreds of kinases, but not all kinase inhibitors also induce
autophagy. For example, inhibitors of the bRaf and MEK kinase do not induce
autophagy.
In one example, the MEK inhibitor (R)-N-(2,3-dihydroxypropoxy)-3,4-difluoro-2-
(2-fluoro-
4-iodophenylamino)benzamide (VII) does not induce an increase in autophagy. In
another
example, CQ does not induce apoptosis in cancer cells (for example, melanoma
and breast
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
cancers) when combined with compound VII. Described herein are assays to
determine
whether a kinase inhibitor also induces autophagy. Inhibitors of kinases that
induce
autophagy include inhibitors of Akt (eg. Akt-1, Akt-2 and Akt-3), PI3K, mTOR,
PDK1 and
p70S6K. The Akt kinase inhibitor can be a pan-Akt inhibitor, an allosteric Akt
inhibitor or a
selective inhibitor of Akt-1, Akt-2 or Akt-3.
In one embodiment, the Akt kinase inhibitor is a compound of Formula I:
A
R1 N R5
çJLJ
R201 o
and tautomers, resolved enantiomers, resolved diastereomers, solvates, and
salts thereof,
wherein,
R1 is H, Me, Et and CF3;
R2 is H or Me; R5 is H or Me;
A is:
R6 R7
(CRCRd)n
(CHOrn
(CRaRb)p
F>r
wherein G is phenyl optionally substituted by one to four R9 groups or a 5-6
membered
heteroaryl optionally substituted by a halogen;
R6 and R7 are independently H, OCH3, (C3-C6 cycloalky1)-(CH2), (C3-C6
cycloalkyl)-
(CH2CH2), V4CH2)13.1 wherein V is a 5-6 membered heteroaryl, W-(CH2)1_2
wherein W is
phenyl optionally substituted with F, Cl, Br, I, OMe, CF3 or Me, C3-C6-
cycloalkyl optionally
substituted with C1-C3 alkyl or 0(C1-C3 alkyl), hydroxy-(C3-C6-cycloalkyl),
fluoro-(C3-C6-
cycloalkyl), CH(CH3)CH(OH)phenyl, 4-6 membered heterocycle optionally
substituted with
F, OH, C1-C3 alkyl, cyclopropylmethyl or C(=0)(C1-C3 alkyl), or C1-C6-alkyl
optionally
substituted with one or more groups independently selected from OH, oxo, 0(Ci-
C6-alkyl),
CN, F, NH2, NH(C1-C6-alkyl), N(C1-C6-alky1)2, cyclopropyl, phenyl, imidazolyl,
piperidinyl,
26
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
pyrrolidinyl, morpholinyl, tetrahydrofuranyl, oxetanyl or tetrahydropyranyl,
or R6 and R7
together with the nitrogen to which they are attached form a 4-7 membered
heterocyclic ring
optionally substituted with one or more groups independently selected from OH,
halogen,
oxo, CF3, CH2CF3, CH2CH2OH, 0(C1-C3 alkyl), C(=0)CH3, NH2, NHMe, N(Me)2,
S(0)2CH3, cyclopropylmethyl and C1-C3 alkyl;
Ra and Rb are H, or Ra is H, and Rb and R6 together with the atoms to which
they are attached
form a 5-6 membered heterocyclic ring having one or two ring nitrogen atoms;
Rc and Rd are H or Me, or Rc and Rd together with the atom to which they are
attached from a
cyclopropyl ring;
R8 is H, Me, F or OH, or R8 and R6 together with the atoms to which they are
attached form a
5-6 membered heterocyclic ring having one or two ring nitrogen atoms;
each R9 is independently halogen, C1-C6-alkyl, C3-C6-cycloalkyl, 0-(C1-C6-
alkyl), CF3,
OCF3, S(C1-C6-alkyl), CN, OCH2-phenyl, CH20-phenyl, NH2, NH-(C1-C6-alkyl), N-
(C1-C6-
alky1)2, piperidine, pyrrolidine, CH2F, CHF2, OCH2F, OCHF2, OH, S02(C1-C6-
alkyl),
C(0)NH2, C (0)NH(C 1 -C6-alkyl), and C(0)N(C 1 -C6-alky1)2 ;
RI is H or Me; and
m, n and p are independently 0 or 1.
Another embodiment includes Akt inhibitors of Formula I, wherein RI is
methyl; R2, R5 and R1 are H; G is phenyl optionally substituted with 1-3 R9;
R9 is halogen, C1-C3
alkyl, NC, CF3, OCF3 OCH3 or OCH2Phenyl; Rc and Rd are H or methyl; m, n and p
are 0 or 1;
and R8 is H or methyl.
Another embodiment includes Akt inhibitors of Formula I, including the
compounds:
27
CA 02821378 2013-06-12
WO 2012/087336 PCT/US2010/062096
I
* 9
NH F NH
NH HN NH
0 1
0
0 F lai 0 0
0 110 N F3C *I (N) Br
10 CN) CI IW N
CI ( ) N N ( ) Me0 N
N (N)
1 IN
,:LAtNI
eCLN eCLN
)a-LN
I
N I
- N I
- N I
,: N
HO HOs HO HO
HO
(01
Y r-NJ L\
HN r
F
NH NH2 N NH
F 0 io 0 0 F fal 0 0
(N) F io N
F3C. W' (N) Br CN) Br 1101 N Br W
0 (N) N CN)
N N
.LAN
.ra)*N1
,a--'Llµl eLAL;1 I
aAN
I I N I
HO - N
HO HO HO Ha
r0.1
Y NH2 H
N
NH I
NH
NH
0
r&I 40 Cs
0 401 0 F io 0 1 N (10 (
F3C-0 lir (N) CI (N) CI N NN) ( )
I IN F ( )
1 N
1 IN I IN
a, a, eCLN
I WN1
- N
- N
Ho HO
HO HO Ha
NH2 (A YF3c,
1 nO
NH NH 0 NH Y
01. 0
0 F rap o o 0 NH
CI IWP rN 0
L ) CI 11 CN) CI 1W- 1 (NJ CI N I. (N)IN)
N N N -*INI Br 1, )
,z N erLI ;I
HO - N 4 N 'N
Ho- HO HO HO N
HO
28
CA 02821378 2013-06-12
WO 2012/087336 PCT/US2010/062096
nO
\l/
NH
N
...24H z,NH NHH
_
(
CI ) CI (N) ci\ S (N) Brf
(N) CI 10
S N
4 IN ( ) CI p ( )
N N N N
N
MI
e
eCLN 4)\XI
e(LN
N Ck'N
I 1 Ls'a
1 ,)
HO HO HO HO
HO
rNH ),
HN NJ NJ NI
0
B Nr ) CI ( ) CI . (N; CI * CN; CI
N N CI (N)
1 N N
001 I LCI'''N
I I
I
N - N
...: N
HO HO HO Ho HO
'
PREPARATION OF FORMULA I COMPOUNDS
Compounds of Formula I may be prepared according to methods described in
U.S. Patent Appl. Ser. No. 11/773,949, filed July 5, 2007, entitled
"Hydroxylated and
Methoxylated Pyrimidyl Cyclopentanes as AKT Protein Kinase Inhibitors," which
is incorporated
by reference herein, for all purposes.
Compounds of Formula I may be prepared singly or as compound libraries
comprising at least 2, for example 5 to 1,000 compounds, or 10 to 100
compounds. Libraries of
compounds of Formula I may be prepared by a combinatorial 'split and mix'
approach or by
multiple parallel syntheses using either solution phase or solid phase
chemistry.
For illustrative purposes, Schemes 1-4 show a general method for preparing the
compounds of Formula I as well as key intermediates. Those skilled in the art
will appreciate that
other synthetic routes may be used. Although specific starting materials and
reagents are
depicted in the Schemes and discussed below, other starting materials and
reagents can be easily
substituted to provide a variety of derivatives and/or reaction conditions. In
addition, many of the
compounds prepared by the methods described below can be further modified in
light of this
disclosure using conventional chemistry well known to those skilled in the
art.
29
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
Me00C) 1NH2 HSX ,õ111, ?H
Reduction H I
Chlorination
__________________________________________ _
0:11) H2N N
1 2 3 4
y oc y oc
N
C ) N
C )
I
Oxidation y --- - o
5"---?"--
1
SNAr N N
. NJ-DR -.5R---, Hydrolysis N -....õ
N N N
oI OAc OAc OH
6 7 8
H Ry,..J0
N N
HCI C) 1. Acylation C Nj
2. HCI
----....
N
111-1-1-)R rag R6-..N-R7
N N I
OH OHR = (CR`Rd)n
\
9 10 (CH2)m
..õõ(CFORtip-1--,,s
G R8
Scheme 1
Scheme 1 shows a method of preparing compound 10 of Formula I wherein R1 is
H, R2 is OH and R5 is H. Formation of pyrimidine 2 can be accomplished by the
reaction of the
5 keto ester 1 with thiourea in the presence of a base such as KOH in an
appropriate solvent, such
as ethanol. After reduction of the mercapto group of compound 2 under standard
reducing
conditions (e.g., Raney Ni and NH4OH) to provide compound 3, the
hydroxypyrimidine 3 can be
chlorinated under standard conditions (e.g., POC13 in DIEA/DCE) to provide
compound 4.
Compound 4 is then oxidized under standard conditions (e.g., MCPBA in an
appropriate solvent
such as CHC13) to give the pyrimidine-oxide 5. Treatment of the pyrimidine-
oxide with acetic
anhydride gives the rearrangement product 6. Compound 7 is obtained by
reacting compound 6
with an appropriately substituted piperidine under standard SNAr reaction
conditions to provide
compound 7. Compound 7 is hydrolyzed to provide compound 8, which is then
deprotected to
yield the intermediate 9. Acylation of the piperazinyl cyclopenta[d]pyrimidine
9 with an
appropriated amino acid in the presence of a coupling reagent such as HBTU,
followed by
deprotection if necessary, gives compound 10 of Formula I.
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
s
03 0 COOEt H2N A NH2
11 12 13 14
(+)-pulegone
OH ,.. OH ,... CI ...
_ CI Acetic
oxidation N ---... '''..
anhydride,
A
N--1----). :15 reduction N -1*---...b chlorination
______õ.._ u........... ..... ----..- k ...... -...- Q...
_.
HS
0-
15 16 17 18
Boc yoc 1. HCI R yO
N. Acylation N
C DI LiOH CN ) 3. HCI
,
N ,.... N ... N
N--L-.): N -A-.): N -1---*
CI ..z. OAc OH OH
N----1):: __ 20 21 22
OAc 1 .HCI R .__.,,
Boc Boc -1"
19 ri N 2 Acylation
LiOH
3. HCI N
; N )
="*..--- N
N--
0Ac OH OH
23 24 25
1 NaH
Met
R ..f0
Boc 1. HCI
14 2 Acylation.. N j
R= R6õN R7
3. HCI ...r
I ; ) N _
N -
\
k
N" OMe:: N--1------r:
N---
, (CRaRb)p¨ki ---1.--)
G OMe
R8
26 27
Scheme 2
Scheme 2 shows a method of preparing compounds 22, 25 and 27 of Formula I
wherein RI, R2 and R5 are methyl. According to Scheme 2, bromination of (+)-
pulegone 11 with
bromine gives the dibromide 12. The treatment of the dibromide 12 with a base
such as sodium
ethoxide provides the pulegenate 13. Ozonolysis of the pulegenate 13 gives the
ketoester 14.
Treatment of the keto ester 14 with thiourea in the presence of a base such as
KOH in ethanol,
followed by reduction of the mercapto group under standard conditions (e.g.
Raney Ni catalyst in
ammonia) affords the hydroxypyrimidine 16. Chlorination of the
hydroxypyrimidine 16 under
standard conditions (e.g., POC13) provides the 4-chloropyrimidine 17. The
oxidation of the 4-
chloropyrimidine 17 with an oxidizing agent such as MCPBA or hydrogen peroxide
provides the
N-oxide 18. Rearrangement of the N-oxide 18 with acetic anhydride yields the
intermediate 19.
Compound 19 is reacted with the desired piperazine according to the procedure
described in
31
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
Scheme 1 to provide compound 20 where R5 is H and 23 where R5 is Me. Compounds
20 and 23
are subjected to chiral separation using RPLC with chiral stationary and then
hydrolyzed upon
treatment with a base such as lithium hydroxide to provide compounds 21 and
24, respectively.
After deprotection, compounds 21 and 24 are then reacted with the appropriate
amino acid to
provide compounds 22 and 25, respectively.
Alternatively, the 7-hydroxy group of compound 24 may be alkylated with
alkylation reagent such as alkyl halide in the presence of a base such as NaH
or KOH to provide
compound 26 where R2 is Me. After deprotection, compound 26 is then reacted
with the
appropriate amino acid to provide compound 27.
14 N H4 OAc
o,...--
NH..6õ..,... ecr3 Halogenation
o 1-11,1 0 N
64
63
Boc
N
Ha I Boc
Cl oc
N R5 B
N
C 1
H
1---- --1---
N R5 Ac20a
---
Oxidation .
eal . N R5
N
eaNI eal
N
65 N li
66 67
Boc Boc Boa
N N
N
CN1 B' Hydrolysis C N1 R5 Oxida C tion N1 Rs Asymmetric
i
69 Reduction
N
N
Ac0 HO 0
68
Boc
N Boc R y-O
R y-0
C 1 OR CN12. Acyla 1'HCI
N Ire tIon L
N Rs 3. Functionalisation _.õ. N .,._
R
___L. O CN1
N
________________________________________ . N R5
-61 Lai :c-:1.---3
1 N
N A N
N
HO H6
RS
H6
72 73 HO
71 74
R = R 5,_ _. R7
N
1
(C R`R ' ),,
\ R5= H, Me, Et, CF3
(C H2),,,
10 G
R5
Scheme 3
Scheme 3 shows an alternative method of preparing compounds 73 and 74.
According to Scheme 3, amination of 14 using an ammonia synthon gives 63.
Pyrimidine
15 formation using, for example, ammonium formate in the presence of
formamide at 50 C-250 C
and/or at high pressure gives the bicyclic unit 64. Activation of 64 using,
for example, POC13 or
SOC12 gives the activated pyrimidine 65. Displacement of this leaving group,
using a suitable
protected/substituted piperidine at 0 C to 150 C gives the piperidine 66.
Oxidation, using, for
example, m-chloroperoxybenzoic acid ("MCPBA" or "m-CPBA") or Oxone at -20 C
to 50 C
32
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
gives the N-oxide 67. Treatment with an acylating agent (eg. acetic anhydride)
followed by
heating (40 C to 200 C) causes rearrangement to give 68. Hydrolysis, using,
for example LiOH
or NaOH at 0 C to 50 C gives the alcohol 69. Oxidation, using for example,
Swern conditions,
Mn04 or pyridine-S03 complex at appropriate temperatures gives the ketone 70.
Asymmetric
reduction using, for example, a catalytic chiral catalyst in the presence of
hydrogen, the CBS
catalyst or a borohydride reducing agent in the presence of a chiral ligand
gives rise to either the
(R) or the (S) stereochemistry at the alcohol 71 or 72. Alternatively, a non-
chiral reducing agent
could be used (eg. H2, Pd/C), allowing the methyl group on the cyclopentane
unit to provide
facial selectivity and ultimately diastereoselectivity. If the reduction
gives a lower
diastereoselctivity, the diastereomers could be separated by (for example)
chromatography,
crystallization or derivitization. Finally deprotection of the Boc-group,
using, for example, acid
at 0 C to 50 C, acylation using an appropriately functionalized amino acid and
final
functionalization of the amine of this amino acid (eg. removal of any
protecting group, alkylation,
reductive amination or acylation to introduce new substituents) gives rise to
the final compounds
73 and 74.
R'X
Acylation õ oc NB Lewis Add
Saponification
s/)
HO2C 00 X Irrl'
Boc 0 S
(1) (2) (3) (4)
0.y0H
Boc
R. N
(5)
Scheme 4
Introduction of a chiral auxiliary (e.g. Evans oxazolidinone, etc.) to
compound
(1) may be accomplished by standard acylation procedures to give the conjugate
(2). For
example, treatment of the acid with an activating agent (e.g. COC12) or mixed
anhydride
formation (e.g. 2,2-dimethylpropanoyl chloride) in the presence of an amine
base at -20 C to
100 C followed by treatment with the appropriate chiral auxiliary (X) gives
compound (2).
The stereochemistry and choice of the chiral auxiliary may determine the
stereochemistry of
the newly created chiral center and the diastereoselectivity. Treatment of
compound (2) with
a Lewis acid (eg. TiCI4) at low temperature (e.g. -20 C to -100 C) and an
amine base (e.g.
Hunig's base) followed by the use of an appropriately substituted imminium ion
precursor (3)
at low temperature then gives rise to compound (4). The temperature, Lewis
acid and chiral
33
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
auxiliary may all be expected to influence the diastereoselectivity of the
addition adduct.
Finally, saponification under mild conditions (e.g. Li0H/H20 at -10 C to 30 C)
gives rise to
the desired acid (5).
In another embodiment, the kinase inhibitor is an Akt inhibitor of the
following formula:
R5
Rsa
A
NR =
0
(Ni
Ria N R3
,L117)1
R2 R2a 5
stereoisomers, tautomers or pharmaceutically acceptable salts thereof,
wherein:
G is phenyl optionally substituted with one to three Ra groups or a 5-6
membered
heteroaryl optionally substituted by a halogen;
R1 and RI a are independently selected from H, Me, CF3,
CHF2 or CH2F;
R2 is H, F or ¨OH;
R2a is H;
R3 is H;
R4 is H, or C1-C4 alkyl optionally substituted with F, -OH or -0(C1-C3 alkyl);
R5 and R5a are independently selected from H and CI-CI alkyl, or R5 and R5a
together with
the atom to which they are attached form a 5-6 membered cycloalkyl or 5-6
membered
heterocycle, wherein the heterocycle has an oxygen heteroatom;
each Ra is independently halogen, C1-C6-alkyl, C3-C6-cycloalkyl, -0-(C1-C6-
alkyl), CF3,
-0CF3, S(CI-C6-alkyl), CN, -OCH2-phenyl, NH2, -NO2, -N-(Ci-C6-alky1)2,
piperidine, pyrrolidine, CH2F, CHF2, -OCH2F, -OCHF2, -OH, -S02(Ci-C6-alkyl),
C(0)NH2,
C(0)NH(Ci-C6-alkyl), and C(0)N(C1-C6-alky1)2; and
j is 1 or 2.
Another embodiment includes Akt inhibitor compounds, including:
34
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
CL rAl r--1
rY
,NH 4, N.,
F =NNH 4, N..., 4, NH 4, NH
F 0 Sr'Cf 0
IP N F3C * 0 0
0 *
N ,. N 5 N 5 N
S N
F3C ( ) ( ) -
N CN) V ( ) Br ( ) Br
1 IN N N (N)
e(j'N
)aAN
,CLN LCLi N
eCLN
HO HO
Ho HO HO
F
F
7-1
YF c-INH CH ANH A NH
4, N
\ 0 0
0 rill 0 r& ra
40 S
0 F 0
CI (N) CI 5 N CI W.- (N) CI 11" (N) F3C 1WA (N) F3C N
(N) (N)
1 IN N N N
00I
eL)*N
erl'N
I
eLAN
I
I .)
Ho - N F Ho HO F
HO
n [-I 1-1A (JO NH
N-..
H`
0 0 0 0 0 0
N
IP (NI CI 40 C CI (101 101 C I (.1 N
CI (SI (N) CI N) N C ) CI (N) (N)
1 IN N N N
001 eCjN
1
erLN
I L el'I'N
1 .) LAN
. )
.:
HO HO Ho HO HO Ho: N
/4- n 7
\ 0H cN.....N__\ n
4. N- CN.....õcF3 ry
4, NH 4. N OH 4, NH
0 0 0
N
40 N 5 No cl 0 C ) v 5 rN) a 0 (NI,
c, r ) ci CN) N N Is. ) Br 5 N
N (N)
4 1.'1%1
LLAN eCL N
erLN
001 e(L. N
- N
.)N
HO HO HO N
Ho HO F .
In one embodiment, the kinase inhibitor is an Akt inhibitor compound of
Formula II:
õ,,.........,,õ.....,,,,,(CR1R2)p ......,,,,.
I II
X
A B
wherein, R1 and R2 are independently hydrogen, C1-05 alkyl, hydroxyl, C1.5
alkoxy or amine;
p is an integer from 1 to 6; A is a 5-14 carbon cyclic, bicyclic or tricyclic
aromatic or
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
heteroaromatic ring, which can be optionally substituted with halogen, OH,
amino,
dialkylamino, monoalkylamino, C1-C6-alkyl or phenyl, which is optionally
substituted with
halogen, OH, C1-C3 alkyl or cyclopropylmethyl; and in one embodiment A has one
of the
following structures:
N 1 N
j I 1 and
- 5
R4 Eµ;5 Ra N R N N R5 N R'
0
wherein D and E are independently ¨CH or N;
wherein R3 and R4 are each independently hydrogen, halogen, OH, amino,
dialkylamino,
monoalkylamino or C1-C6-alkyl, which is optionally substituted with halogen,
OH, C1-C3
alkyl or cyclopropylmethyl;
R5 is a 5 or 6 membered aromatic or heteroaromatic ring optionally substituted
with halogen,
OH, amino, dialkylamino, monoalkylamino or Ci-C6-alkyl, which is optionally
substituted
with halogen, OH, C1-C3 alkyl or cyclopropylmethyl; in one embodiment R5 is
phenyl;
B is an aromatic, heteroaromatic, cyclic or heterocyclic ring having the
formula:
ss4,X
R7
wherein, Q, T, X and Y are each independently selected from the group
consisting of¨CH, -
CH2, CO, N or 0;
Z is -CH, -CH2, C=0, N, 0 or ¨C=C¨;
R6 and R7 are independently selected from the group consisting of hydrogen,
halogen,
carbonyl and a 5 or 6 membered aromatic or heteroaromatic ring optionally
substituted with halogen,
OH, amino, dialkylamino, monoalkylamino or C1-C6-alkyl, which is optionally
substituted with
halogen, OH, C1-C3 alkyl or cyclopropylmethyl; in one embodiment R6 or le is
pyridinyl, or R6 and
R7 aretaken together to form a 5-6 membered aromatic, heteroaromatic, cyclic
or
heterocyclic ring, which can be optionally substituted with halogen, OH,
amino,
dialkylamino, monoalkylamino or C1-C6-alkyl, which is optionally substituted
with halogen,
OH, C1-C3 alkyl or cyclopropylmethyl; in one embodiment, B has one of the
following
structures:
36
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
r\lf N
X N and 3 ===NX
(- NH
\ IDA
Q n
R7
wherein X, Y, Q, R6 and R7 are as described above, and X', Q' and T' are -CH
or N.
In another embodiment, AKT inhibitors include compounds having the
formula:
r N Na
/1(
wherein: a is 0 or 1; b is 0 or 1; m is 0, 1 or 2; n is 0, 1 or 2; p is 0, 1
or 2; r is 0 or 1; s
is 0 or 1;
Q is selected from: --NR7R8,
N
0 N2C,,
and
N N
(R00-3
õ0 (ROO-3
RI is independently selected from (C=0)a0bCi-C6 alkyl, (C=0)a0baryl, C2-
C6alkenyl,
C2-C6 alkynyl, (C=0)a0bheterocyclyl, (C=0)a0bC3-C6cycloalkyl, CO2H, halogen,
CN, OH,
=
ObCi-C6perfluoroalkyl, 0a(C=0)bNR7R8, Nitc(c0)NR7Rs, S(0)n,Ra, S(0)2NR7R8,
NRcS(0)mRa, oxo, CHO, NO2, NRc(C=0)Oble, 0(C=0)0bC1-C6 alkyl, 0(C=0)0bC3-C6
cycloalkyl, 0(C=0)0baryl, and 0(C=0)0b-heterocycle, wherein said alkyl, aryl,
alkenyl,
alkynyl, heterocyclyl, and cycloalkyl are optionally substituted with one or
more substituents
selected from le;
R2 is independently selected from C1-C6 alkyl, aryl, heterocyclyl, CO2H, halo,
CN,
OH and S(0)2NR7R8, wherein said alkyl, aryl and heterocyclyl are optionally
substituted with
one, two or three substituents selected from Rz;
R7 and R8 are independently selected from H, (C=0)0bCI-C10 alkyl, (C=0)0bC3-C8
cycloalkyl, (C=0)0baryl, (C=0)0bheterocyclyl, CI-C10 alkyl, aryl, C2-
C10alkenyl, C2-C10
alkynyl, heterocyclyl, C3-C8 cycloalkyl, SO2Ra and (C=0)NRb2, wherein said
alkyl,
37
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
cycloalkyl, aryl, heterocylyl, alkenyl, and alkynyl is optionally substituted
with one or more
substituents selected from le, or
R7 and R8 canbe taken together with the nitrogen to which they are attached to
form a
monocyclic or bicyclic heterocycle with 5-7 members in each ring and
optionally containing,
in addition to the nitrogen, one or two additional heteroatoms selected from
N, 0 and S, said
monocyclic or bicyclic heterocycle optionally substituted with one or more
substituents
selected from le;
le is selected from: (C=0),0,(Ci-C10) alkyl, Or(C1-C3)perfluoroalkyl, (Co-
C6)alky1ene-S(0),õRa , oxo, OH, halo, CN, (C=0)r0s(C2-Cio) alkenyl,
(C=0)rOs(C2-C10)
alkynyl, (C=0)10,(C3-C6) cycloalkyl, (C=0),0,(Co-C6) alkylene-aryl,
(C=0),Os(Co-C6)
alkylene-heterocyclyl, (C=0)1Os(Co-C6) alkylene-N(Rb)2, C(0)Ra, (Co-
C6)alkylene-CO2Ra,
C(0)H, (Co-C6)alkylene-CO2H, C(0)N(Rb)2, S(0)mRa, and S(0)2N(Rb)2NRc(C=0)0bRa,
0(C=0)0bCi-Cioalkyl, 0(C=0)0bC3-C8cycloalkyl, 0(C=0)0baryl, and O(CO)0b-
heterocycle, wherein said alkyl, alkenyl, alkynyl, cycloalkyl, aryl, and
heterocyclyl are
optionally substituted with up to three substituents selected from Rb, OH, (C1-
C6)alkoxy,
halogen, CO2H, CN, 0(C=0)C1-C6alkyl, oxo, and N(Rb)2;
Ra is (Ci-C6)alkyl, (C3-C6)cycloalkyl, aryl or heterocyclyl; and
Rb is H, (Ci-C6)alkyl, aryl, heterocyclyl, (C3-C6)cycloalkyl, (CO)0C1-C6
alkyl,
(C=0)C1-C6alkyl or S(0)2Ra;
Rc is selected from: H, C1-C6 alkyl, aryl, C2-C6 alkenyl, C2-C6 alkynyl,
heterocyclyl,
C3-C8 cycloalkyl and C1-C6perfluoroalkyl, wherein said alkyl, cycloalkyl,
aryl, heterocylyl,
alkenyl, and alkynyl is optionally substituted with one or more substituents
selected from le;
or a pharmaceutically acceptable salt or a stereoisomer thereof.
In another embodiment, AKT inhibitors include:
,
v ,lkil,. NI, el Na
Q
(R1)n- i -X.t N
I
/
,
wherein a is 0 or 1; b is 0 or 1; m is 0, 1 or 2; n is 0, 1, 2 or 3; p is 0, 1
or 2; r is 0 or 1;
s is 0 or 1; u, v, w and x are independently selected from: CH and N, provided
that only one
of u, v, w and x may be N;
Q is selected from:--NR5R6,
38
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
oNN rjA and
(R)03H (Rz)0_3
z
R1 isindependently selected from (C=0)a0bCi-C6 alkyl, (C=0)a0baryl, C2-C6
alkenyl,
C2-C6 alkynyl, (C=0)a0bheterocyclyl, (C=0)a0bC3-C6cycloalkyl, CO2H, halogen,
CN, OH,
,
ObC1-C6perfluoroalkyl, 0a(C=0)bNR7R8, NRc(c=o)NR7R8 S(0),,Ra, S(0)2NR7R8,
NleS(0)mRa, oxo, CHO, NO2, NRc(C=0)0bRa, 0(C=0)0bC1-C6 alkyl, 0(C=0)0bC3-C6
cycloalkyl, 0(C=0)0baryl, and 0(C=0)0b-heterocycle, wherein said alkyl, aryl,
alkenyl,
alkynyl, heterocyclyl, and cycloalkyl are optionally substituted with one or
more substituents
selected from le;
R2 is independently selected from C1-C6 alkyl, aryl, heterocyclyl, CO2H, halo,
CN,
OH and S(0)2NR7R8, wherein said alkyl, aryl and heterocyclyl are optionally
substituted with
one, two or three substituents selected from le;
R7 and R8 areindependently selected from H, (C=0)0bCi-Cio alkyl, (C=0)0bC3-C8
cycloalkyl, (C=0)0baryl, (C=0)0bheterocyclyl, Ci-C10 alkyl, aryl, C2-C10
alkenyl, C2-C10
alkynyl, heterocyclyl, C3-C8 cycloalkyl, SO2Ra and (C=0)NRb2, wherein said
alkyl,
cycloalkyl, aryl, heterocylyl, alkenyl, and alkynyl is optionally substituted
with one or more
substituents selected from le, or
R7 and R8 canbe taken together with the nitrogen to which they are attached to
form a
monocyclic or bicyclic heterocycle with 5-7 members in each ring and
optionally containing,
in addition to the nitrogen, one or two additional heteroatoms selected from
N, 0 and S, said
monocyclic or bicyclic heterocycle optionally substituted with one or more
substituents
selected from Rz;
Rz is selected from: (C=0)10,(C1-C1o) alkyl, Or(C1-C3)perfluoroalkyl, (Co-
C6)alkylene-S(0)mRa , oxo, OH, halo, CN, (C=0),0,(C2-C10) alkenyl, (C=0),0,(C2-
Ci0)
alkynyl, (C=0),Os(C3-C6) cycloalkyl, (C=0)rOs(C0-C6) alkylene-aryl,
(C=0)1Os(Co-C6)
alkylene-heterocyclyl, (C=0),Os(Co-C6) alkylene-N(Rb)2, C(0)Ra, (Co-
C6)alkylene-CO2Ra,
C(0)H, (Co-C6)alkylene-CO2H, C(0)N(Rb)2, S(0),nRa, and
S(0)2N(Rb)2NRc(C=0)ObRa,
0(C=0)0bCi-Cloalkyl, 0(C=0)0bC3-C8cycloalkyl, 0(C=0)0baryl, and 0(C=0)0b-
heterocycle, wherein said alkyl, alkenyl, alkynyl, cycloalkyl, aryl, and
heterocyclyl are
optionally substituted with up to three substituents selected from Rb, OH, (CI-
C6)alkoxy,
halogen, CO2H, CN, 0(C=0)C1-C6 alkyl, oxo, and N(Rb)2;
39
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
le is (Ci-C6)allcyl, (C3-C6)cycloalkyl, aryl or heterocyclyl; and
Rb is H, (Ci-C6)alkyl, aryl, heterocyclyl, (C3-C6)cycloalkyl, (C=0)0CI-C6
alkyl,
(C=0)C1-C6alkyl or S(0)212.%
It.' is selected from: H, C1-C6 alkyl, aryl, C2-C6 alkenyl, C2-C6 alkynyl,
heterocyclyl,
C3-C8 cycloalkyl and C1-C6 perfluoroalkyl, wherein said alkyl, cycloalkyl,
aryl, heterocylyl,
alkenyl, and alkynyl is optionally substituted with one or more substituents
selected from Rz;
or a pharmaceutically acceptable salt or a stereoisomer thereof.
In another embodiment, AKT inhibitors include:
Na
,U 0 N 0
v - Q
(R),¨.1¨
Vkix .-
N
I ¨(R2)P
/
)
wherein a is 0 or 1; b is 0 or 1; m is 0, 1 or 2; n is 0, 1, 2 or 3; p is 0, 1
or 2; r is 0 or 1;
s is 0 or 1; u, v, and x are independently selected from CH and N; W is a
bond, CH or N;
Q is selected from:--NR5R6,
ON r)µ, and
N---- ='..?\" N\%Y
(R0-3 H (Rz)0-3
õyin z)
RI is independently selected from (C=0)a0bCi-C6 alkyl, (C=0)a0baryl, C2-C6
alkenyl,
C2-C6 alkynyl, (C=0)a0bheterocyclyl, (C=0)a0bC3-C6cycloalkyl, CO2H, halogen,
CN, OH,
ObCi-C6perfluoroalkyl, 0a(C=0)bNR7R8, NRc(C=0)NR7R8, S(0),õRa, S(0)2NR7R8,
NWS(0)õ,Ra, oxo, CHO, NO2, NRc(C=0)0bRa, 0(C=0)0bCi-C6allcyl, 0(C=0)0bC3-C6
cycloalkyl, 0(C=0)0baryl, and 0(C=0)0b-heterocycle, wherein said alkyl, aryl,
alkenyl,
alkynyl, heterocyclyl, and cycloalkyl are optionally substituted with one or
more substituents
selected from Rz;
R2 is independently selected from C1-C6alkyl, aryl, heterocyclyl, CO2H, halo,
CN,
OH and S(0)2NR7R8, wherein said alkyl, aryl and heterocyclyl are optionally
substituted with
one, two or three substituents selected from le;
R7 and R8 areindependently selected from H, (C=0)0bCi-C10 alkyl, (C=0)0bC3-C8
cycloalkyl, (C=0)0baryl, (C=0)0bheterocyclyl, Ci-C10 alkyl, aryl, C2-C10
alkenyl, C2-C10
CA 02821378 2013-06-12
WO 2012/087336 PCT/US2010/062096
alkynyl, heterocyclyl, C3-C8 cycloalkyl, SO2Ra and (C=0)NRb2, wherein said
alkyl,
cycloalkyl, aryl, heterocylyl, alkenyl, and alkynyl is optionally substituted
with one or more
substituents selected from le, or
R7 and R8 canbe taken together with the nitrogen to which they are attached to
form a
monocyclic or bicyclic heterocycle with 5-7 members in each ring and
optionally containing,
in addition to the nitrogen, one or two additional heteroatoms selected from
N, 0 and S, said
monocyclic or bicyclic heterocycle optionally substituted with one or more
substituents
selected from le;
Rz is selected from: (C=0)rOs(Ci-Ci0) alkyl, Or(C1-C3)perfluoroalkyl, (Co-
C6)alkylene-S(0)õ,Ra , oxo, OH, halo, CN, (C=0)1Os(C2-C1o) alkenyl,
(C=0)1Os(C2-Cio)
alkynyl, (C=0),0,(C3-C6) cycloalkyl, (C=0),Os(Co-C6) alkylene-aryl,
(C=0),Os(Co-C6)
allcylene-heterocyclyl, (C43)r0s(Co-C6) alkylene-N(Rb)2, C(0)Ra, (Co-
C6)alkylene-CO2Ra,
C(0)H, (Co-C6)alkylene-CO2H, C(0)N(Rb)2, S(0)Ra, and S(0)2N(Rb)2NRc(C=0)0bRa,
0(C=0)0bCi-C10 alkyl, 0(C=0)0bC3-Cgcycloalkyl, 0(C=0)0baryl, and 0(C=0)0b-
heterocycle, wherein said alkyl, alkenyl, alkynyl, cycloalkyl, aryl, and
heterocyclyl are
optionally substituted with up to three substituents selected from Rb, OH, (Ci-
C6)alkoxy,
halogen, CO2H, CN, 0(C=0)CI-C6alkyl, oxo, and N(Rb)2;
Ra is (C1-C6)alkyl, (C3-C6)cycloalkyl, aryl or heterocyclyl; and
Rb is H, (Ci-C6)allcyl, aryl, heterocyclyl, (C3-C6)cycloalkyl, (C0)0C i-
C6alkyl,
(C=0)C1-C6alkyl or S(0)2Ra;
le is selected from: H, C1-C6 alkyl, aryl, C2-C6 alkenyl, C2-C6 alkynyl,
heterocyclyl,
C3-C8 cycloalkyl and C1-C6perfluoroalkyl, wherein said alkyl, cycloalkyl,
aryl, heterocylyl,
alkenyl, and alkynyl is optionally substituted with one or more substituents
selected from le;
or a pharmaceutically acceptable salt or a stereoisomer thereof.
Exemplary AKT inhibitors include:
HC N N A
X.s; NH II-I
Ho N 0
=
,
41
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
h L 14111
<1. NJ(
N. 0 ,c
= NFI
11-4
,
0
HO k,, (,,,,,N., õA 11-2
i.õ1 " NH
k 0
=
ti,C CH,
and
411
ar
Hk I .,,,, N ¨
/ \ 11-3
_ .
In one embodiment, the kinase inhibitor is an Alct-1 selective inhibitor, and
is
a compound of Formula IV:
0
A-, _________________________________ Q
\
/ ____________________________ N
R17( N ¨1
_.,. I
(CRisRis)p_NKR"
'
wherein, A, B, D and E are independently S, -CH, 0 or N, wherein depending on
A, B, D and
E, the ring shown in formula IV can be aromatic, heteroaromatic, cyclic or
heterocyclic;
p is an integer from 1 to 6;
1115 and 11.16 are independently selected from the group consisting of
hydrogen, halogen, OH,
amino, dialkylamino, monoalkylamino and Ci-C6-alkyl;
Q is a 5-6 membered aromatic or heteroaromatic ring; in one embodiment Q the
following
structure:
G'
i ¨GO10 ,
wherein, G and G' are independently N, S or -C-----C-;
R' and R" are taken together with the N to which they are bound to form a 5, 6
or 7 member
42
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
heterocyclic ring, which can be optionally substituted with halogen, OH,
amino,
diallcylamino, monoallcylamino, C1-C6-alkyl, and, as an example, has the
following structure,
which depending on G' can be heteroaromatic or heterocyclic, which can further
contain the
above-listed substituents:
ss&N'G'
wherein G' is as described above,
J is an unsubstituted or substituted amide;
R17 is a 5-14 membered aromatic or heteroaromatic ring system, which can be
optionally
substituted; in one embodiment R17 has one of the following structures:
R18_ X
and
R20Lç --c
R19
wherein, X and Y, independently, are N, 0, S or -CH;
R185 R19 and R2 are independently selected from the group consisting of
halogen, OH,
amino, dialkylamino, monoalkylamino, Ci-C6-alkyl or phenyl, which is
optionally substituted
with halogen, OH, C1-C3 alkyl or cyclopropylmethyl; or R18 and R19 are taken
together to
form an aromatic, heteroaromatic, cyclic or heterocyclic ring.
Compounds of Formula IV include:
=;-
0 >
N
I
Iv-1
0
and
0
0
s IV-2
0 49
tat,
Another embodiment includes AKT inhibitors such as perifosine having the
formula:
43
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
cH3
II 0.3
0 0
0_
Another embodiment includes AKT inhibitors such as oligonucleotides,
including antisense oligonucleotides having the sequences: 5'
ccagcccccaccagtccact 3', 5'
cgccaaggagatcatgcagc 3', 5' gctgcatgatctecttggcg 3', 5' agatagctggtgacagacag
3', 5'
cgtggagagatcatctgagg 3', 5' tcgaaaaggtcaagtgctac 3', 5' tggtgcagcggcagcggcag
3' and 5'
ggcgcgagcgcgggcctagc 3'.
In one embodiment, the kinase inhibitor is a compound of Formula III. In one
example, compounds of Formula III include P13-k inhibitors. In another
example, compounds
of Formula III include mTOR inhibitors. Compounds of Formula III have the
formula:
NRR"
R8 A7
R9E\D
III
R10
5
wherein, A, B, D and E are independently ¨CH or N;
R8 and R9 are taken together to form a 5 or 6 membered aromatic,
heteroaromatic, cyclic or
heterocyclic ring, which can be optionally substituted. For example, R8 and R9
can be taken
together with the carbons in formula III to which they are attached to form a
9-10 member
bicyclic ring system. Embodiments of the bicyclic ring systems include the
following
structures, wherein ¨1 indicates a bond in the formula III ring:
Ri .s R-ro Rii_N
Nvi"sr
R12 rrrix R12 ',Pr R12 R12 prrrs
R11 and R11 S
R12 rep, ,
wherein R" and R12 are independently selected from the group consisting of
hydrogen,
halogen, OH, amino, dialkylamino, monoalkylamino, C1-C6-alkyl,
C(=0)0-(CR)le)n-W or phenyl, which is optionally substituted with halogen, OH,
C1-C3
alkyl or cyclopropylmethyl, wherein W is C5-12 aryl or heteroaryl, RY and 12!
are
44
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
independently hydrogen, halogen, -OH or C1_6 alkyl; or R11 and R12 are taken
together to
form a 5-14 membered aromatic or heteroaromatic ring. For example, R11 and R12
can be
taken together with the carbons to which they are attached and the ring in
Formula III above
to form a 12-14 member tricyclic ring system, and in one embodiment has the
following
structure:
0 \
/
"Is
R' and R" are taken together with the N to which they are bound to form a 5, 6
or 7 member
heterocyclic ring, which can be optionally substituted with halogen, OH,
amino,
dialkylamino, monoalkylamino, C1-C6-alkyl, having one of the following
structures, which
can further contain the above-listed substituents:
G
-G'
(Gi and
N
wherein, G and G' are independently C, 0 or N;
R1 is an aromatic or heteroaromatic ring, having the structure:
ArX,y
R13,
wherein, X, Y, Z and Z' are independently --CH or N;
R13 is hydrogen, halogen, OH, amino, dialkylamino, monoalkylamino, Ci-C6-alkyl
or -N-
(C=0)-N-R14, wherein R14 is C1-C6-alkyl. An example of R1 is:
/
no 14
" 9
wherein, J is -N-(C=0)-N-, and R14 is C1-C6-alkyl.
An example compound of Formula III includes the P13-k inhibitor:
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
0
0 N
111-5
NI ---
II OH
9
Another embodiment includes mTOR inhibitors having the following formula:
A
N
I
R2 N B-D ;
stereoisomers, tautomers or a pharmaceutically acceptable salt thereof,
wherein:
A is a ring selected from the group consisting of morpholin-4-yl, 3,4-dihydro-
2H-pyran-
4-yl, 3,6-dihydro-2H-pyran-4-yl, tetrahydro-2H-pyran-4-yl, 1,4-oxazepan-4-yl,
piperidin-l-yl,
and is optionally substituted with from 1 to 2 substituents selected from the
group consisting of
-C(0)0Ra,-C(0)NleRb, -NRaRb, -0Ra, -SRa, -S(0)21tc, -S(0)Re, -Re, halogen, -
NO2, -CN and
-N3, wherein Ra and Rb are each independently selected from hydrogen, C1_6
alkyl, Ci_6 haloalkyl,
C2_6 alkenyl and C3_6 cycloalkyl, or Ra and Rb, together with the nitrogen
atom to which each is
attached, are combined to form a 3- to 6- membered ring, and Re is selected
from C1.6 alkyl, Cl
-
6 haloalkyl, C2-6 alkenyl, C3_6 cycloalkyl;
RI and R2 are combined with the atoms to which they are attached to form an
optionally
substituted pyrrolidine, piperidine or homopiperidine ring, wherein the
nitrogen atom of said
pyrrolidine, piperidine or homopiperidine ring is substituted by the group:
E(F)m(G)p_.
.
wherein E is hydrogen, C6_10 aryl, C510 heteroaryl, C3-10 cycloalkyl, C3-10
heterocycloalkyl, Ci_6 alkyl or Ci_6 heteroalkyl; and wherein E is optionally
substituted with 1 to 5
substituents selected from halogen, C1_6 alkyl, -NRdRe, -SRd, -ORd, -C(0)0Rd, -
C(0)NRdRe,
-C(0)Rd, -NRdC(0)Re, -0c(0)R, -NRdC(0)NRdRe, -0C(0)NRdRe, -C(=NOR()NRdRe,
-NRdC(=N-CN)NRdRe, -NRdS(0)2NRdRe, -S(0)2Rd, -S(0)2NRdRe, -Rf, -NO2, -N3, =0, -
CN,
-(CH2)1-4-NRdRe, -(CH2)14-SRd, -(CH2)14-0Rd, -(CH2)1-4-C(0)0Rd, -(CH2)14-
C(0)NRdRe,
-(CH2)14-C(0)Rd, -(CH2)1-4-NRdC(0)Re, -(CH2)1-4-0C(0)Rf, -(CH2)1-4-
NRdC(0)NRdRe, -(CH2)1-
46
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
4-0C(0)NRdRe, -(CH2)1-4-C(=N0Rd)NRdRe, -(CH2)1-4-1=IRdC(=N-CN)NRdRe,
-(CH2)1-4-NRdS(0)2NRdRe, -(CH2)1-4-S(0)2Rd, -(CH2)1-4-S(0)2NRdRe, -(CH2)1-
44\102,
-(CH2)1-4-N3 or -(CH2)1-4-CN; wherein Rd and Re are each independently
selected from hydrogen,
Ci.6 alkyl, C1_6 haloalkyl, C2_6 alkenyl, C2_6 alkynyl, C3_7 cycloalkyl, C3_7
heterocycloalkyl, phenyl
and -(CH2)14-phenyl, or Rd and Re, when attached to the same nitrogen atom are
combined to
form a 3- to 6-membered ring; Rf is selected from C1_6 alkyl, C1_6 haloalkyl,
C2_6 alkenyl,
C2_6 alkynyl, C3_7 cycloalkyl, C3_7 heterocycloalkyl, phenyl and -(CH2)14-
phenyl;
F is a member selected from the group consisting of C1.6 alkylene, C2_6
alkenylene, C2-6
alkynylene and Ci_6 heteroalkylene; wherein F is independently substituted
with from 0 to 3
substituents selected from the group consisting of halogen, -NRgRh, -SRg, -
ORg, -C(0)0Rg,
-C(0)NRgRh, -NRgC(0)W, -0C(0)W, -NRgC(0)NRgRh, -0C(0)NRgRh, NRgS(0)2NRgRh,
-S(0)2R, -S(0)2NRgRh, -R', -NO2, N3, =0, -CN, -(CH2)1-4-NRgRh, -(CH2)1-4-SRg, -
(CH2)14-0Rg,
-(CH2)1-4-C(0)0Rg, -(CHC(0)NRgRh, -(CH2)14-C(0)Rg, -(CH2)1-4-NRgC(0)Rh,
-(CH2)1-4-0C(0)R1, -(CH2)1-4-NRgC(0)NRgRh, -(CH2)1-4-0C(0)NRgRh,
-(CH2)1-4-NRgS(0)2NRgRh, -(CH2)1-4-S(0)2Rg, -(CH2)1-4-5(0)2NRgRh, -(CH2)14-
NO2,
-(CH2)1-4-N3 and -(CH2)1-4-CN; wherein Rg and Rh are each independently
selected from
hydrogen, C1-6 alkyl, C1-6 haloalkyl, C1_6 heteroalkyl, C3_7 cycloalkyl, C3_7
heterocycloalkyl,
phenyl and -(CH2)14-phenyl, and optionally Rg and Rh, when attached to the
same nitrogen atom
are combined to form a 3- to 6-membered ring; W is selected from Ci_6 alkyl,
C1_6 haloalkyl, C3-7
cycloalkyl, C3_7 heterocycloalkyl, phenyl and -(CH2)1.4-phenyl;
G is a member selected from the group consisting of -C(0)-, -0C(0)-, -NHC(0)-,
-NHC(=NOH)-, -S(0)2- and -NHS(0)2-;
m and p are each independently an integer from 0 to 1, wherein if m and p are
both the
integer 0, then E is not C1_6 alkyl or C1_6 heteroalkyl;
wherein pyrrolidine, piperidine or homopiperidine ring formed by combining W
and R2 is
further substituted with from 0 to 5 substituents selected from the group
consisting of halogen,
-NWRk, -SW, -OW, -C(0)0W, -C(0)NRJRk, -NHC(0)W, -0C(0)W, -Rm, -CN and =0,
wherein
It? and Rk are each independently selected from hydrogen, Ci_6 alkyl, C1.6
haloalkyl, C2.6 alkenyl,
C2.6 alkynyl, C3_5 cycloalkyl and C3.5 heterocycloalkyl, and RI and Rk, when
attached to the same
nitrogen atom, are optionally combined to form a 3- to 6- membered ring; and
le is selected from
C1_6 alkyl, C1_6 haloalkyl, C2_6 alkenyl, C2.6 alkynyl, C3_5 cycloalkyl and
C3_5 heterocycloalkyl;
B is selected from the group consisting of phenylene, pyridylene,
pyrimidylene,
pyridazinylene and pyrazinyline and is substituted with from 0 to 4
substituents selected from
47
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
halogen, -CN, -N3, -NO2, -C(0)011n, -NRTIC(0)R , -NlInC(0)NRnR , -OW', -
NRnR
and RP; wherein Rn and R are independently selected from hydrogen and C1-4
alkyl, Ci_
4 haloalkyl, C1_4 heteroalkyl, C3_7 cycloalkyl and C3-7 heterocycloalkyl, or
when attached to the
same nitrogen atom, IV and Ware optionally are combined to form a 3- to 6-
membered ring; RP
is C1-4 alkyl, C1-4 haloalkyl, C3_7 cycloalkyl and C3-7 heterocycloalkyl,
wherein any two
substituents, not including the D group, located on adjacent atoms of B are
optionally combined
to form a 5- to 6-membered carbocyclic, heterocyclic, aryl or heteroaryl ring;
and
D is a member selected from the group consisting of -NR3C(0)NR4R5, -NR4R5,
-C(0)NR4R5, -0C(0)0R4, -0C(0)NR4R5, -NR3C(=N-CN)NR4R5, -NR3C(=N-0R4)NR4R5,
-NR3c(=N_NR4)NR4R5, -NR3C(0)R4, -NR3C(0)0R4, -NR3S(0)2NR4R5 and -NR3S(0)2R4,
wherein R3 is selected from the group consisting of hydrogen, C1_6 alkyl, C1.6
haloalkyl and
C2.6 alkenyl; R4 and R5 are each independently selected from the group
consisting of
hydrogen, C1_6 alkyl, C1_6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-10
cycloalkyl,
C3_10 heterocycloalkyl, C6-10 aryl and C5_10 heteroaryl, and R4 and R5, when
attached to the
same nitrogen atom, are optionally combined to form a 5- to 7- membered
heterocyclic or
heteroaryl ring; and wherein R3, R4 and R5 are further substituted with from 0
to 3
substituents independently selected from the group consisting of halogen, -
NO2, -CN,
-NIVRr, -SRq, -C(0)0Rq, -C(0)NR`V, -NRqC(0)W, -NRqC(0)0Rs, -(CH2)1_4-NR`V,
-- q, -, -2,14- _ -, -H2,14- _ _ .__q__r,
-(CH2)1-4 R (CH SR (CH C(1451 R (C C"NTR R
-(CH2)1-4-NRqC(0)Rr, -(CH2)14-NRqC(0)0Rr, -(CH2)1-4-CN, -(CH2)14-NO2, -S(0)R',
-S(0)2R', =0, and -Rs; wherein Rq and R' is selected from hydrogen, C1_6
alkyl, C1_6 haloalkyl,
C2_6 alkenyl, C2.6 alkynyl, C1_,6 heteroalkyl, C3.7 cycloalkyl, C3.7
heterocycloalkyl, C6_10 aryl,
C5_10 heteroaryl; and Rs, at each occurrence, is independently selected from
C1-6 alkyl. Cl
-
6 haloalkyl, C1_6 heteroalkyl, C3_7 cycloalkyl, C3-7 heterocycloalkyl, C6_10
aryl and C5_
10 heteroaryl; and wherein the D group and a substituent located on an
adjacent atom of the B
ring are optionally combined to form a 5- to 6- membered heterocyclic or
heteroaryl ring.
In certain embodiments:
A is a ring selected from the group consisting of morpholin-4-yl, 3,4-dihydro-
2H-pyran-
4-yl, 3,6-dihydro-2H-pyran-4-yl, tetrahydro-2H-pyran-4-yl, 1,4-oxazepan-4-yl,
piperidin-l-yl,
optionally substituted by Ci-C6 alkyl;
B is selected from the group consisting of phenylene and pyrimidylene;
D is -NR3C(0)NR4R5, -NR4R5, -C(0)NR4R5, -0C(0)0R4, -0C(0)NR4R5, -NR3C(=N-
CN)NR4R5, -NR3C(=N-OR4)NR41e, -NR3C(=N-NR4)NR4R5, -NR3C(0)R4, -NR3C(0)0R4,
48
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
-NR3S(0)2NR4R5 or -NR3S(0)2R4, wherein R3 is hydrogen or C1.6 alkyl; R4 and R5
are each
independently hydrogen, C1.6 alkyl, C1-6 haloalkyl or C3_10 cycloalkyl, or R4
and R5 arecombined
to form a 5- or 6- membered heterocyclic ring;
R1 and R2 are combined with the atoms to which they are attached to form an
substituted
pyrrolidine, piperidine or homopiperidine ring, wherein the nitrogen atom of
said ring is
substituted by the group:
E(F)m(G)p_
4 ,
wherein E is hydrogen, C6 aryl, C5.6 heteroaryl, C1_15 alkyl or C5-6
heterocycloalkyl,; and
wherein E is optionally substituted with 1 to 5 substituents selected from
halogen, C1.6 alkyl,
-NRdRe, -SRd, -ORd, -C(0)0Rd, -C(0)NRdRe, -C(0)Rd, -N1dC(0)Re, -0C(0)Re,
-NRdC(0)NRdRe, -0C(0)NRciRe, _c(=NoRdwRd-e, _
NRdC(=N-CN)NRdRe, -NRdS(0)2NRdRe,
-S(0)2Rd, -S(0)2NRdRe, -Re, -NO2, -N3, =0, -CN, -(CH2)14-NRdRe, -(CH2)14-SRd, -
(CH2)1_4-0Rd,
-(CH2)1-4-C(0)0Rd, -(CH2)1-4-C(0)NRdRe, -(CH2)14-C(0)Rd, -(CH2)14-NRdC(0)Re,
-(CH2)1-4-0C(0)Rf, -(CH2)1_4-NRdC(0)NRdRe, -(CH2)14-0C(0)NRdRe, -(CH2)1-4-
C(=NORd)NRdRe, -(CH2)1_4-
NRdC(=N-CN)NRdRe, -(CH2)14-NRdS(0)2NRdRe, -(CH2)14-S(0)2Rd,
-(CH2)14-S(0)2NRdRe, -(CH2)14-1\102, -(CH2)14-N3 or -(CH2)1_4-CN; wherein Rd
and Re are each
independently selected from hydrogen, C1_6 alkyl, C1_6 haloalkyl, C2_6
alkenyl, C2_6 alkynyl, C3-7
cycloalkyl, C3_7 heterocycloalkyl, phenyl and -(CH2)14-phenyl, or Rd and Re,
when attached to the
same nitrogen atom are combined to form a 3- to 6-membered ring; Re is
selected from C1_6 alkyl,
Ci_6 haloalkyl, C3_7 cycloalkyl, C3.7 heterocycloalkyl, phenyl and -(CH2)14-
phenyl;
F is C1_6 alkylene;
G is -C(0)-, -0C(0)-, -NHC(0)-, -NHC(=NOH)-, -S(0)2- or -NHS(0)2-; and
m and p are independently 0 or 1.
Another embodiment includes mTOR inhibitor compounds, including:
A
R1
11
R2 B-D
stereoisomers, tautomers, and pharmaceutically acceptable salts thereof,
wherein A is a 5- to
8-membered heterocyclic ring having from 1 to 3 heteroatoms independently
selected from
N, 0 and S as ring vertices, and having from 0 to 2 double bonds; wherein the
A ring is
49
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
further substituted with from 0 to 5 RA substituents selected from the group
consisting of
C(0)0Ra,-C(0)NRaltb, -NRaRb, -0C(0)Rc, -0Ra, -SRa, -S(0)2Rc, -S(0)Rc, -Rc, -
(CH2)14-
NRaRb, -(CH2)1-4-NRaC(0)Rc, -(CH2)14-0Ra, -(CH2)14-SRa, -(CH2)14-S(0)2Rc, -
(CH2)14-
S(0)Rc, halogen, -NO2, -CN and -N3, wherein Ra and Rb are each independently
selected
from hydrogen, C1_6 alkyl, C1_6 haloalkyl, C1_6 heteroalkyl, C2.6 alkenyl,
C2_6 alkynyl, C3_
6 cycloalkyl, phenyl and -(CH2)14(phenyl), and optionally Ra and Rb, together
with the
nitrogen atom to which each is attached, are combined to form a 3- to 7-
membered
heterocyclic ring comprising 1 to 2 heteroatoms selected from N, 0 and S; Rc
is selected from
C1.6 alkyl, C1_6 haloalkyl, C1-6 heteroalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-6
cycloalkyl, phenyl
and -(CH2)14 (phenyl); and any two substituents attached to the same atom in
the 5- to 8-
membered heterocyclic ring are optionally combined to form a 3- to 5- membered
carbocyclic or a 3 to 5- membered heterocyclic ring; RI and R2 are combined
with the atoms
to which they are attached to form a 5- to 8- membered monocyclic or bridged
bicyclic
heterocyclic ring comprising -0- as one of the ring vertices; wherein the 5-
to 8- membered
monocyclic or bridged-bicyclic heterocyclic ring formed by combining RI and R2
further
optionally comprises one additional heteroatom selected from the group
consisting of N, 0
and S, and is substituted with from 0 to 5 RR substituents selected from the
group consisting
of halogen, -NRiRk, -SRJ, -OR, -C(0)OR, -C(0)NRJRk, -NHC(0)RJ, -0C(0)W, -
CN,
=0, =S, =N-CN, -(CH2)14-CN, -(CH2)14-ORJ, -(CH2)14 -NRJRk, -C14 alkylene-OR, -
C14
allcylene-Rm, -C24 alkenylene-Rm, -C24 alkynylene-Rm, -C14 alkylene-C1.9
heteroaryl, C24
alkenylene-C1_9 heteroaryl, C24 alkynylene-C1_9 heteroaryl, C14 alkylene-C6.10
aryl, C24
allcynylene-C6_10 aryl and C24 allcynylene-C6_10 aryl, wherein IV and Rk are
each
independently selected from hydrogen, C1_6 alkyl, C1_6 haloalkyl, CI-6
heteroalkyl,
C2-6 alkenyl, C2-6 alkynyl, C3_7 cycloalkyl, C2-6 heterocycloalkyl, phenyl,
Pyridyl and -(CH2)1-
4-(Ph), and R and Rk, when attached to the same nitrogen atom, are optionally
combined to
form a 3- to 6- membered heterocyclic ring comprising 1 to 2 heteroatoms
selected from N, 0
and S; and Rm is selected from C1-6 alkyl, Ci_6 haloalkyl, C1-6 heteroalkyl,
C2_6 alkenyl,
C2-6 alkynyl, C3-7 cycloalkyl, C2-6 heterocycloalkyl and -(CH2)14-(Ph), and
wherein a C3-7
cycloalkyl, C2-6 heterocycloalkyl, C1_9 heteroaryl or C6-10 aryl portion of a
RR substituent is
substituted with from 0 to 3 substituents selected from the group consisting
of F, Cl, Br, I,
-NH(C14 alkyl), -N(diCi4 alkyl), 0(C14 alkyl), Ci_6 alkyl, C1-6 heteroalkyl, -
C(0)0(C14
alkyl), -C(0)NH(Ci4alkyl), -C(0)N(diCi4 alkyl), -NO2, -CN; wherein when RI and
R2 are
combined to form a monocyclic 5- to 8- membered heterocyclic ring then any two
RR
substitutents attached to the same atom or adjacent carbon atoms in said 5- to
8-membered
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
heterocyclic ring are optionally combined to form a 3- to 7- membered
cycloalkyl ring or a 3-
to 7- membered heterocycloalkyl ring comprising 1 to 2 heteroatoms selected
from N, 0 and
S as ring vertices; B is a member selected from the group consisting of
phenylene and 5- to 6-
membered heteroarylene, and is substituted with from 0 to 4 RB substituents
selected from
halogen, -CN, -N3, -NO2, -C(0)OW, -C(0)NWIR , -NR C(0)R , -NR C(0)NRIIR ,
-NWR , -(CH2)14-C(0)01e, -(CH2)1_4-C(0)NWW, -(CH2)14-ORn, -(CH2)1_4-NR R , -
(CH2)1-
4-SR and RP; wherein R and R are independently selected from hydrogen and
C1.6 alkyl,
C1_6 haloalkyl, C1.6 heteroalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-7 cycloalkyl,
C2.6
heterocycloalkyl, phenyl and -(CH2)1.4-(phenyl) or when attached to the same
nitrogen atom,
R and R are optionally are combined to form a 3- to 6- membered heterocyclic
ring
comprising 1 to 2 heteroatoms selected from N, 0 and S; RP is C1_6 alkyl, C1_6
haloalkyl, C1-6
heteroalkyl, C2-6 alkenyl, C2_6 alkynyl, C3_7 cycloalkyl, C2-6
heterocycloalkyl, phenyl and
-(CH2)14-(phenyl), wherein any two substituents, not including the D group,
located on
adjacent atoms of B are optionally combined to form a 5- to 6-membered
carbocyclic,
heterocyclic, aryl or heteroaryl ring; D is a member selected from the group
consisting of
-NR3C(0)NR4R5, -NR4R5, -C(0)NR4R5, -0C(0)0R4, -0C(0)NR4R5, -NR3C(=N-CN)NR4R5,
-NR3C(=N-0R4)NR4R5, -NR3C(=N-NR4)NR4R5, -NR3C(0)R4, -NR3C(0)0R4,
-NR3S(0)2NR4R5, -NR3S(0)2R4, -NR3C(=S)NR4R5 and -S(0)2R4R5, wherein R3 is
selected
from the group consisting of hydrogen, C1_6 alkyl, C1-6 haloalkyl and C2-6
alkenyl; R4 and R5
are each independently selected from the group consisting of hydrogen, C1_6
alkyl, C1-6
haloalkyl, C1-6 alkylamino-C(=0)-, C2-6 alkenyl, C2.6 alkynyl, C3_10
cycloalkyl, C2-
9 heterocycloalkyl, C6-10 aryl and C1_9 heteroaryl, and R4 and R5, when
attached to the same
nitrogen atom, are optionally combined to form a 5- to 7- membered
heterocyclic or 5- to 6-
membered heteroaryl ring comprising 1 to 3 heteroatoms selected from N, 0 and
S; and
wherein R3, R4 and R5 are further substituted with from 0 to 3 RD substituents
independently
selected from the group consisting of halogen, -NO2, -CN, -C(0)OR,
-C(0)NRqW, -NWIC(0)W, -NRqC(0)0W, -(CHNRqW, -(CH2)14-010, -(CH2)1-4-SWI,
-(CH2)14-C(0)0Rq, -(CH2)14-C(0)NRqW, -(CH2)1_4-NWIC(0)W, -(CH2)14-NRqC(0)0Rr,
-(CH2)14-CN, -(CH2)1_4-NO2, -S(0)W, -S(0)2R', -(CH2)14W, =0, and -Rs; wherein
Rq and W
is selected from hydrogen, Ci_6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2_6
alkynyl, Cl
-
6 heteroalkyl, C3_7 cycloalkyl, C2_6 heterocycloalkyl, C6-10 aryl, Ci_9
heteroaryl; and Rs, at each
occurrence, is independently selected from Cis alkyl, C1-6 haloalkyl, C3-7
cycloalkyl, C2-
6 heterocycloalkyl, C6_10 aryl and C1_9 heteroaryl; and wherein the D group
and a substituent
51
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
located on an adjacent atom of the B ring are optionally combined to form a 5-
to 6-
membered heterocyclic or heteroaryl ring optionally substituted with 1 to 2 RD
substituents.
Another embodiment includes mTOR inhibitor compounds, including:
R3
N N
R1 I
N
R2 A3 A.J..
;6k- D
stereoisomers, tautomers, and pharmaceutically acceptable salts thereof,
wherein RI is
selected from the group consisting of 6- to 10- membered aryl, 5- to 9-
membered heteroaryl,
3- to 12- membered heterocycloalkyl, 3- to 12- membered cycloalkyl, wherein RI
is
substituted with from 0 to 5 R substituents selected from the group consisting
of halogen, F,
Cl, Br, I, -NRaRb, -SRa, -0Ra, -C(0)0Ra, -C(0)NRaRb, -C(0)Ra, -NRaC(0)Rb, -
0C(0)Rc,
-NRaC(0)NRaRb, -0C(0)NRaRb, -NRaS(0)2NRaRb, -S(0)2Ra, -S(0)2NRaRb,Rc,-NO2, -
N3,
=0, -CN, Rdl,-Xl-NRaRb, xlsRa,-X1-0Ra, -X1 -C(0)0Ra, -Xl-C(0)NRaRb, -XI-
C(0)Ra,
-Xl-NRaC(0)Rb, -X1-0C(0)Ra, -Xl-NRaC(0)NRaRb, -X1-0C(0)NRaRb,
-Xl-NRaS(0)2NRaRb, -XI-S(0)2Ra, -Xl-S(0)2NRaRb, -XI-NO2, -XI-N3, -XI-CN, and
X' -R';
wherein Ra and Rb are each independently selected from hydrogen, C1_6 alkyl,
C1_6 haloalkyl,
Ci_6 heteroalkyl, C2.6 alkenyl, C2_6 alkynyl, C3_7 cycloalkyl, C2-7
heterocycloalkyl, phenyl and
-(042)14-phenyl, optionally Ra and Rb, when attached to the same nitrogen atom
are
combined to form a 3- to 6-membered heterocyclic ring comprising 1 to 2
heteroatoms
selected from N, 0 and S; Rc is selected from C1_6 alkyl, C1_6 haloalkyl, C2-6
alkenyl,
C2.6 alkynyl, C3_7 cycloalkyl, C2_7 heterocycloalkyl, phenyl and -(CH2)14-
phenyl; XI is
selected from the group consisting of C1_4 alkylene, C2_4 alkenylene and C2_4
alkynylene; and
Rci is selected from the group consisting of phenyl, 2-pyridyl, 3-pyridyl, 4-
pyridyl, 2-
imidazolyl, 2-indolyl, 1-naphthyl, 2-naphthyl, 2-thienyl, 3-thienyl, 2-
pyrrolyl, 2-furanyl and
3-furanyl, and wherein Rcl is substituted with from 0 to 3 substituents
selected from F, Cl, Br,
I, -NRaRb, -SRa, -0Ra, -S(0)2Ra, -S(0)2NRaRb, -NO2, -N3, =0, -CN, pyridyl,
C1.6 alkyl, C2-6
alkenyl, C2_6 alkynyl and C1-6 heteroalkyl; R2 is selected from the group
consisting of
hydrogen, C1_6 alkyl, C2_6 alkenyl, C2_6 alkynyl, C1_6 heteroalkyl, a 6-to 10
membered aryl, 5-
to 10- membered heteroaryl, a 3- to 12- membered heterocycloalkyl, a 3- to 12
membered
cycloalkyl, -L-C6_113 aryl, -L-C1_9 heteroaryl, -L-C3_12 cycloalkyl and -L-
C2_12
heterocycloalkyl, wherein L is selected from C1_6 alkylene, C2_6 alkenylene,
C2_6 alkynylene
and C1_6 heteroalkylene, and wherein R2 is substituted with from 0 to 5 RR2
substituents
52
CA 02821378 2013-06-12
WO 2012/087336 PCT/US2010/062096
selected from the group consisting of halogen, F, Cl, Br, I, -NRdle, -SRd, -
ORd, -C(0)0Rd,
-C(0)NRdRe, -C(0)Rd, -
NRdC(0)Re, -0C(0)R, -NRdC(0)NRdRe, -0C(0)NRdRe,
-NRdS(0)2NRd- e5 -S(0)2R', -S(0)2NRdRe, -Rf, -NO2, -N3, =0, -CN, -X2-
NRciRe, _v_sRd,
-X2-OR', -X2-C(0)OR', -X2-C(0)NeRe, _)(2_c(0)Rd, _x2_NRac(0)Re,_<,2_
A OC(0)Rd,
_x2_NRcic(o)NRd-e, _
K X2-0C(0)NRdRe, -X2_NRds(0)2NRdRe, ...x2_s(0)2Rd, ...x2_
S(0)2NRdRe5 A2-NO2, -X2-N3 and -X2-CN; wherein Rd and Re are each
independently
selected from hydrogen, C1_6 alkyl, C1_6 haloalkyl, C1_6 heteroalkyl, C2_6
alkenyl, C2_6 alkynyl,
C3-7 cycloalkyl, C2-7 heterocycloalkyl, phenyl and -(CH2)14-phenyl, optionally
Rd and Re,
when attached to the same nitrogen atom are combined to form a 3- to 6-
membered
heterocyclic ring comprising 1 to 2 heteroatoms selected from N, 0 and S; Rf
is selected from
C1_6 alkyl, Ci_6 haloalkyl, C2_6 alkenyl, C2_6 alkynyl, C3_7 cycloalkyl, C2_7
heterocycloalkyl,
phenyl and -(CH2)14-phenyl; and X2 is selected from the group consisting of
C14 alkylene,
C24 alkenylene and C24 alkynylene; R3 isa 5- to 12- membered monocyclic or
bridged
heterocycloalkyl ring, wherein the R3 group is substituted with from 0 to 3
RR3 substituents
selected from the group consisting of -C(0)0Rg,-C(0)NRgRh, -NRgRh, -ORg, -SRg,
-S(0)212.1,
-S(0)RI, -R', halogen, F, Cl, Br, I, -NO2, -CN and -N3, wherein Rg and Rh are
each
independently selected from hydrogen, C1-6 alkyl, C1-6 haloalkyl, C1-6
heteroalkyl,
C2_6 alkenyl and C3_6 cycloalkyl, wherein optionally Rg and Rh, together with
the nitrogen
atom to which each is attached, are combined to form a 3- to 6- membered
heterocyclic ring
comprising 1 to 2 heteroatoms selected from N, 0 and S, and R` is selected
from C1_6 alkyl,
C16 haloalkyl, C2_6 alkenyl, C3.6 cycloalkyl; and if R3 is a monocyclic
heterocycloalkyl ring
then any two RR3 groups attached to the same atom of R3 is optionally combined
to form at 3-
to 7- membered carbocyclic or 3-to 7- membered heterocyclic ring comprising 1
to 2 atoms
selected from N, 0 and S as ring vertices; Al, A2, A3 and A4 are each a member
independently selected from N, C(RA) or C(H), wherein at least three of Al,
A2, A3 and A4 is
each independently C(H) or C(RA), wherein RA at each occurrence is
independently selected
from the group consisting of F, Cl, Br, I, -NO2, -CN, C1-4 alkyl, C24 alkenyl,
C24 alkynyl, or
any two RA groups attached to adjacent atoms are optionally combined to form a
C2-6
heterocyclic ring comprising from 1 to 2 heteroatoms selected from N, 0 and S
as ring
vertices, C3_7 cycloalkyl ring, a C1_5 heteroaryl ring comprising from 1 to 4
heteroatoms
selected from N, 0 and S as ring vertices, or phenyl ring; and D is a member
selected from
the group consisting of -NR4C(0)NR5R6, -NR5R6, -C(0)NR5R6, -0C(0)0R5, -
0C(0)NR5R6,
-NR4C(=N-CN)NR5R6, -
NR4C(=N-OR5)NR5R6, -NR4C(=N-NR5)NR5R6, -NR4C(0)R5,
-NR4C(0)0R5, -
NR4S(0)2NR5R6 and -NR4S(0)2R5, wherein R4 is selected from the group
53
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
consisting of hydrogen, C1-6 alkyl, C1-6 haloalkyl and C2-6 alkenyl; R5 and R6
are each
independently selected from the group consisting of hydrogen, C1_6 alkyl, C1_6
haloalkyl, C2-
6 alkenyl, C2_6 allcynyl, C3-113 cycloalkyl, C2_10 heterocycloalkyl, C643 aryl
and C1_9 heteroaryl,
and R5 and R6, when attached to the same nitrogen atom, are optionally
combined to form a
5- to 7- membered heterocyclic or a 5- to 9- membered heteroaryl ring
comprising 1 to 3
heteroatoms selected from N, 0 and S as ring vertices and substituted with 0-3
RD
substituents; and wherein R4, R5 and R6 are further substituted with from 0 to
3 RD
substituents, wherein RD is independently selected from the group consisting
of halogen, F,
Cl, Br, I, -NO2, -CN, -NRJRk, -ORJ, -SRJ, -C(0)0R, -C(0)NRJRk, -NRJC(0)Rk,
-NRJC(0)0Rni, -X3-NRJRk, -X3-OR, -X3-C(0)0R, -X3-C(0)NRjRk,
-X3-NRIC(0)Rk, -X3-NRV(0)ORk, -X3-CN, -X3-NO2, -S(0)1e, -S(0)21e, =0, and -le;
wherein IV and Rk is selected from hydrogen, C1_6 alkyl, C1_6 haloalkyl, C2-6
alkenyl, C2-
6 alkYnYl, C1_6 heteroalkyl, C3-7 cycloalkyl, C3-7 heterocycloalkyl, C6_10
aryl, C1_9 heteroaryl;
and le, at each occurrence, is independently selected from C1_6 alkyl, C1_6
haloalkyl, C3_
7 cycloalkyl, C3_7 heterocycloalkyl, C6_10 aryl and C1-9 heteroaryl; X3 is
selected from the
group consisting of C14 allcylene, C24 alkenylene and C2.4 alkynylene; and
wherein D and a
RA substituent attached to an atom that is adjacent to the atom to which D is
attached are
optionally combined to form an optionally substituted 5- to 6- membered
heterocyclic or
heteroaryl ring substituted with from 0 to 4 RD substituents.
Another embodiment includes mTOR inhibitor compounds, including:
R2 R3
1µ1,/
N
y2I 2
1.1i
A3
'10 D
stereoisomers, tautomers, and pharmaceutically acceptable salts thereof,
wherein Y1
and Y2 is each independently N or C(R1), but Y1 and Y2 are not both N or are
not both C(R1),
wherein R1 is selected from the group consisting of hydrogen, C1-6 alkyl, C24
alkenyl, C2-6
alkynyl, C1.6 heteroalkyl, 6- to 10- membered aryl, 5- to 9- membered
heteroaryl, 3- to 12-
membered heterocycloalkyl, 3- to 12- membered cycloalkyl, wherein R1 is
substituted with
from 0 to 5 RR1 substituents selected from the group consisting of halogen, F,
Cl, Br, I,
-NRaRb, -SRa, -0Ra, -C(0)0Ra, -C(0)NRaRb, -C(0)1e, -NRaC(0)Rb, -0C(0)Rc,
-NRaC(0)NRaRb, -0C(0)NRaRb, -NRaS(0)2NRaRb, -S(0)2Ra, -S(0)2NRaRb, -NO2, -
N3,
=0, -CN, Rd. -xl_NRaRb,3,3 _
NI( XI -0Ra, -Xl-C(0)0Ra, -X1-C(0)NRaRb, -Xl-
C(0)Ra,
54
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
-Xl-NRaC(0)Rb, -X1-0C(0)Ra, -Xl-NRaC(0)NRafth, -XI -0C(0)NRalth,
-Xl-NRaS(0)2NRaRh, -X1-S(0)2Ra, -Xl-S(0)2NRale, -X1-NO2, -X'-N3, -X'-CN, and
Xl-Rel;
wherein Ra and Rh are each independently selected from hydrogen, C1_6 alkyl,
C1_6 haloalkyl,
C1_6 heteroalkyl, C2_6 alkenyl, C2_6 alkynyl, C3_7 cycloalkyl, C2_7
heterocycloalkyl, phenyl and
-(CH2)14-phenyl, optionally Ra and Rh, when attached to the same nitrogen atom
are
combined to form a 3- to 6-membered heterocyclic ring comprising 1 to 2
heteroatoms
selected from N, 0 and S; Re is selected from C1-6 alkyl, C1_6 haloalkyl, C2_6
alkenyl,
C2_6 alkynyl, C3-7 cycloalkyl, C2_7 heterocycloalkyl, phenyl and -(CH2)1_4-
phenyl; Xl is
selected from the group consisting of Ci4 alkylene, C2_4 alkenylene and C2_4
alkynylene; and
lel is selected from the group consisting of phenyl, 2-pyridyl, 3-pyridyl, 4-
pyridyl, 2-
imidazolyl, 2-indolyl, 1-naphthyl, 2-naphthyl, 2-thienyl, 3-thienyl, 2-
pyrrolyl, 2-furanyl and
3-furanyl, and wherein lel is substituted with from 0 to 3 substituents
selected from F, Cl, Br,
I, -NRaRh, -SRa, -0Ra, -S(0)2Ra, -S(0)2NRaRh, -NO2, -N3, =0, -CN, pyridyl,
C1_6 alkyl,
alkenyl, C2_6 alkynyl and C1_6 heteroalkyl; R2 is selected from the group
consisting of
hydrogen, C1-6 alkyl, C2.6 alkenyl, C2.6 alkynyl, Ci_6 heteroalkyl, -L-C6_10
aryl, -L-C1-9
heteroaryl, -L-C3_12 cycloalkyl and -L-C2_12 heterocycloalkyl, wherein L is
selected from C1_6
alkylene, C2-6 alkenylene, C2_6 alkynylene and C1_6 heteroalkylene, and
wherein R2 is
substituted with from 0 to 5 RR2 substituents selected from the group
consisting of halogen,
F, Cl, Br, I, -NRdRe, -SRd, -ORd, -C(0)0Rd, -C(0)NRdRe, -C(0)Rd, -NRdC(0)1e, -
0C(0)Rf,
-NRdC(0)NRdRe, -0C(0)NRdRe, -NRdS(0)2NRdRe, -S(0)2Rd, -S(0)2NRdRe, -Re, -NO2, -
1\13,
=0, -CN, -X2-NRdRe, -X2-SRd, -X2-OR', -X2-C(0)OR', -X2-C(0)NRdRe, -X2-C(0)Rd, -
X2-
NRdC(0)Re, -X2-0C(0)Rd, -X2-NRdC(0)NRdRe, -X2-0C(0)1\IRdRe, -X2-NRdS(0)2NRdRe,
-X2-S(0)2Rd, -X2-S(0)2NRdRe, -X2-NO2, -X2-N3 and -X2-CN; wherein Rd and Re are
each
independently selected from hydrogen, Ci_6 alkyl, C1_6 haloalkyl, C1_6
heteroalkyl,
C2_6 alkenyl, C2_6 alkynyl, C3-7 cycloalkyl, C2_7 heterocycloalkyl, phenyl and
-(CH2)14-phenyl,
optionally Rd and Re, when attached to the same nitrogen atom are combined to
form a 3- to
6-membered heterocyclic ring comprising 1 to 2 heteroatoms selected from N, 0
and S; Rf is
selected from C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2_6 alkynyl, C3-7
cycloalkyl, C2-7
heterocycloalkyl, phenyl and -(CH2)14-phenyl; and X2 is selected from the
group consisting
of C1-4 alkylene, C2_4 alkenylene and C2_4 alkynylene; R3 is a 5-to 12-
membered monocyclic
or bridged heterocycloalkyl ring, wherein the R3 group is substituted with
from 0 to 3 RR3
substituents selected from the group consisting of -C(0)0Rg,-C(0)NRgRh, -
NRgRh, -ORg,
-SRg, -S(0)21V, -S(0)R1, -R', halogen, F, Cl, Br, I, -NO2, -CN and -N3,
wherein Rg and Rh are
each independently selected from hydrogen, C1_6 alkyl, Ci_6 haloalkyl, C1_6
heteroalkyl,
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
C2-6 alkenyl and C3-6 cycloalkyl, wherein optionally Rg and Rh, together with
the nitrogen
atom to which each is attached, are combined to form a 3- to 6- membered
heterocyclic ring
comprising 1 to 2 heteroatoms selected from N, 0 and S, and R' is selected
from C1_6 alkyl,
C1-6 haloalkyl, C2-6 alkenyl, C3_6 cycloalkyl; and when R3 is a monocyclic
heterocycloalkyl
ring then any two RR3 groups attached to the same atom of R3 is optionally
combined to form
at 3- to 7- membered carbocyclic or 3-to 7- membered heterocyclic ring
comprising 1 to 2
atoms selected from N, 0 and S as ring vertices; A1, A2, A3 and A4 are each a
member
independently selected from N, C(RA) or C(H), wherein at least three of AI,
A2, A3 and A4 is
each independently C(H) or C(RA), wherein RA at each occurrence is
independently selected
from the group consisting of F, Cl, Br, I, -NO2, -CN, C14 alkyl, C24 alkenyl,
C24 alkynyl, or
any two RA groups attached to adjacent atoms are optionally combined to form a
C2-6
heterocyclic ring comprising from 1 to 2 heteroatoms selected from N, 0 and S
as ring
vertices, C3_7 cycloalkyl ring, a Ci.5 heteroaryl ring comprising from 1 to 4
heteroatoms
selected from N, 0 and S as ring vertices, or phenyl ring; and D is a member
selected from
the group consisting of ¨NR4C(0)NR5R6, -NR5R6, -C(0)NR5R6, -0C(0)0R5, -
0C(0)NR5R6,
-NR4C(=N-CN)NR5R6, -NR4C(=N-0R5)NR5R6, -NR4C(=N-NR5)NR5R6, -NR4C(0)R5,
-NR4C(0)0R5, -NR4S(0)2NR5R6 and ¨NR4S(0)2R5, wherein R4 is selected from the
group
consisting of hydrogen, C1_6 alkyl, C1_6 haloalkyl and C2_6 alkenyl; R5 and R6
are each
independently selected from the group consisting of hydrogen, C1_6 alkyl, C1-6
haloalkyl, C2-
6 alkenyl, C2_6 alkynyl, C3_10 cycloalkyl, C2_10 heterocycloalkyl, C6_10 aryl
and C1_9 heteroaryl,
and R5 and R6, when attached to the same nitrogen atom, are optionally
combined to form a
5- to 7- membered heterocyclic or a 5- to 9- membered heteroaryl ring
comprising 1 to 3
heteroatoms selected from N, 0 and S as ring vertices and substituted with 0-3
RD
substituents; and wherein R4, R5 and R6 are further substituted with from 0 to
3 RD
sub stituents, wherein RD is independently selected from the group consisting
of halogen, F,
Cl, Br, I, -NO2, -CN, -NRJRk, -SIV, -C(0)OR, -C(0)NRJRk, -NRJC(0)Rk,
-NRiC(0)01r, -X3-NRJRk, -X3-011.J, -X3-C(0)OR, -X3-C(0)NRJRk,
-X3-NRJC(0)Rk, -X3-NRJC(0)ORk, -X3-CN, -X3-NO2, -S(0)1r, -S(0)2Rm, =0, and
¨1r;
wherein R and Rk is selected from hydrogen, C1_6 alkyl, C1_6 haloalkyl, C2_6
alkenyl, C2-
6 alkynyl, C1_6 heteroalkyl, C3_7 cycloalkyl, C3_7 heterocycloalkyl, C6_10
aryl, Ci_9 heteroaryl;
and Rm, at each occurrence, is independently selected from C1_6 alkyl, C1_6
haloalkyl, C3_
7 cycloalkyl, C3_7 heterocycloalkyl, C6_10 aryl and C1_9 heteroaryl; X3 is
selected from the
group consisting of Ci4 alkylene, C24 alkenylene and C24 alkynylene; and
wherein D and a
RA substituent attached to an atom that is adjacent to the atom to which D is
attached are
56
CA 02821378 2013-06-12
WO 2012/087336 PCT/US2010/062096
optionally combined to form an optionally substituted 5- to 6- membered
heterocyclic or
heteroaryl ring substituted with from 0 to 4 RD substituents.
Another embodiment includes mTOR inhibitor compounds, including:
o
( ) o r)
LN
N(N
N io * C yt, N f\l'.- c*N N 0 1
N N'.---` N 0 it
H H H H N N
H H
C) ro) ro)
v-,,, \--,,,
,r_NaNaf-L,N Q v-N
NN0 IsOiN
N
0 N io A0 N 0 8
N N
N 40 ,it X N io yt,
''''. N N.--*-
-'.
H H H H
H H
Si
o
( C
C) o ) o )
N
N
N
CIS--NNiH2N
-N is
0 Cisj"--N N
-N >,-N , N
N * 0õp "µ_ "
,--L
,s, N io 0
N
NA N
N ,--
NH2 H
H H
0 7-0 0
N
H2N 0 0 aYN
aN --NarL
' N
N\_)--N I , )
N 0 NjrN
io 0 0
N
NAN''' io 0
,
NA N - N NH2
H H H H
o
CN) 0
(N) 0
(N)
0
gLN
I , S--NaLN
gLN
N N I
Uri N a 9 CN
N io 0 -.1\rl,rN I
N, di 0
.1. Nrs'N NANr,,F
H H I F ( I - N A --,
H H N N -
H H
57
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
0 0
/A- ,...--*LN r--\ P KN--._N H Olre-r,
1 isir- N ")1 0 0
c NN
6 0
/
N A N -,-
(
l'NN,---
H H
H H N A N''.
H H
0 0
\ \
N-...AN N
1>.__ 1 N' I 4 I
_,.....õ, ,
r N 0 0 0
N A 0 0 0,0 N
N N NA NS 0
NA N
H H H H H H
0 0
0
( ).= (,N) (
N
N.--../1-. N N1-...,. NN
N' I IN
.õ....,. , is
N 0
N-----% 011
NAN .---,..., A J )-
H H N N N NJ
H H H H
0 0
( ) 0
( ) C ).=
\
N11=-=N F N....._AN
I N-3_4\ N ----.AHN
(---4
410 0 cil N 0 0
j - INI (:)
NA N N-N 5 0
\ .11. ....,.. N.K.N.----
..,
N N
H H N N H H
H H
00
(:)
EN)
/-----.) /"---N 7----AN
0 I N 0 I fl 0 I
\-1\r 0S---.1eL''-'N
I
NA N.----.., NAN N NH2
H H H H
HO
Another embodiment includes the mTOR inhibitor, rapamycin:
58
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
140.,
\00=0
I
Y6 1 I
OiO OH .
HO 0 V
µ
Another embodiment includes P13-k inhibitor compounds of the following
formula:
o
( )
N
0 \ N
\ / N 10 R1
R2 ,
or pharmaceutically acceptable salts thereof, wherein:
Ri and R2 are independently selected from hydrogen, halogen, C1_6 alkyl, -
NRdlte, -SRd,
-ORd, -C(0)0Rd, -C(0)NRdRe, -C(0)Rd, -NRdC(0)Re, -0C(0)R, 4pRdC(0)NRdRe,
-0C(0)NRdIte, -C(=NORd)NRdRe, -NRdC(=N-CN)NRdRe, -NRdS(0)2NRdRe, -S(0)2R',
-S(0)2NRdRe, -Re, -NO2, -N3, =0, -CN, -(CH2)14-NRdRe, -(CH2)1-4-SRd, -(CH2)1-4-
0Rd, -(CH2)1-4-
C(0)0Rd, -(CH2)1-4-C(0)NRdRe, -(CH2)1-4-C(0)Rd, -(CH2)1-4-NRdC(0)Re, -(CH2)1.4-
0C(0)Rf,
-(CH2)1-4-NRdC(0)NRdRe, -(CH2)1-4-0C(0)NRdle, -(CH2)1-4-C(=NORd)NRdRe, -(CH2)1-
4-
NRdC(=N-CN)NRdle, -(CH2)1.4-NRdS(0)2NRdRe, -(CH2)14-S(0)2Rd, -(CH2)1.4-
S(0)2NRdlr,
-(CH2)1-4-NO2, -(CH2)1-4-N3 or -(CH2)14-CN; wherein Rd and Re are each
independently selected
from hydrogen, C1-6 alkyl, C1-6 haloalkyl, C2_6 alkenyl, C2_6 alkynyl, C3_7
cycloalkyl, C3-7
heterocycloalkyl, phenyl and -(CH2)14-phenyl, or Rd and Re, when attached to
the same nitrogen
atom are combined to form a 3- to 6-membered ring; Rf is selected from C1_6
alkyl, C1.6 haloalkyl,
C3_7 cycloalkyl, C3_7 heterocycloalkyl, phenyl and -(CH2)14-phenyl; or
Rl and R2 are taken together with the atoms to which they are attached to form
a fused
5- or 6- membered heterocyclyl or heteroaryl ring, optionally substituted by
oxo, halogen, C1-
C3 alkyl or CF3.
59
CA 02821378 2013-06-12
WO 2012/087336 PCT/US2010/062096
Example P13-k inhibitors include the following:
o
( ) o
(N ) o
C) o
CJ
N
N N
jOft=N dIlf=N
3
I
cI,Df.-N
N- I H
\ / N io NH2 , , N io OH N- I ,
\ / N go F \ / N ilk N;sx,
W 00
0
( ) 0
( ) 0
C) 0
C)
N
N
jx=k= N N
N- I cVN 60.1jk*KI
\ / N * NH N- I
/c) \ / N figp NH N- I N- I ".
N Br \ / N . CI
H
=
In one embodiment, the kinase inhibitor is a PI3K kinase inhibitor of Formulas
V and VI:
0
( ) 0
.---- --.
N N
R
S...../k-, N 2
R1--$___t
.----.......-'L
R1
N R3
R2 S NR3
V VI
or stereoisomers, geometric isomers, tautomers, or pharmaceutically acceptable
salts thereof,
where:
R1 is selected from H, F, Cl, Br, I, CN, ¨(CRiaRis)mNRioRi 1,
c ¨14¨K.1
( 5.K )õNR12C(=
_ y)Rio, (c¨K14¨
K15)nNRI2S(0)2R1 , -(cR14R15)moR10,
_(cR14R15)nS(0)2R10, --(CR14R15)nS(0)2NRIOR11, _c(oR10)R11R14, c(=y)R10,
-C(=Y)ORICI, -C(=Y)NRWRI I, ¨C(=Y)NR120R1 , ¨C(=0)NR12S(0)2R1 ,
-
-C( K=0)NR12(c-14-
KI5)mNRI R11, -NO2, NR12q_y)R11, NR12(_-- ( -
WW1,
NRI2c(=y)NRIOR11, _NR12s(0)2R10, _--12
NK SO2NR1OR11, SRI , s(0)2R10, _
S(0)2NRI R1 I , -SC(Y)R' , -SC(Y)0R10, C1¨C12 alkyl, C2¨C8 alkenyl, C2¨C8
alkynyl,
C3¨C12 carbocyclyl, C2¨C20 heterocyclyl, C6¨C20 aryl, and CI¨Cm heteroaryl;
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
R2 is selected from H, F, Cl, Br, I, CN, CF3, -NO2, -C(=Y)R10, -C(Y)0R'
,
q_y)NRioRt 1, _(CRI4R15)n1NRI RI 1, -(CRI4R15),I0R1 ,
(cR14R15)t NR12.,
0)(CR14R15)NR10R115 NR12c(=y)R10, NR12-
Y)0R10
,
_NRI2g_y)NR1OR11, NR12s02R10, ORIo, _oc(_yr io,
K. OC(=Y)OR1 , -OCHONR1 R11,
-0S(0)2(0R10), -0P(=Y)(0R1 )(0R11), -0P(0R10)(0R11), SRI , -S(0)R10, -S(0)2R10
,
-S(0)2NR10R11, -S(0)(0R1 ), -S(0)2(0R10), -SC(=Y)R10, -SC(=Y)ORI ,
-SC(=Y)NRI R11, CI-C12 alkyl, C2-C8 alkenyl, C2-C8 alkynyl, C3-C12
carbocyclyl, C2-C20
heterocyclyl, C6-C20 aryl, and C1-C20 heteroaryl;
R3 is a carbon linked monocyclic heteroaryl, a carbon linked fused bicyclic C3-
C20
heterocyclyl, or a carbon linked fused bicyclic C1-C20 heteroaryl, where the
monocyclic
heteroaryl, fused bicyclic C3-C20 heterocyclyl, and fused bicyclic CI-C20
heteroaryl are
optionally substituted with one or more groups selected from F, Cl, Br, I, -
CN, -NR1 R11,
Rio, _c(o)Rio, NRioco, -)1(11,
N(C(0)RI 1)2, -NRI C(0)NRI R11, -NR12S(0)2R1 ,
-C(=0)0R10, -C(=0)NRIoR11, -1
C12 alkyl and (C1-C12 alkyl)-ORI ;
RII and R12 are independently H, C1-C12 alkyl, C2-C8 alkenyl, C2-C8 alkynyl,
C3-C12 carbocyclyl, C2-C20 heterocyclyl, C6-C20 aryl, or CI-C20 heteroaryl,
or RI and RII together with the nitrogen to which they are attached form a C2-
C20
heterocyclic ring optionally substituted with one or more groups independently
selected from
oxo, (CH2)m0R12, NR12R12, CF3, F, Cl, Br, I, SO2R12, C(0)R12, NRI2C(=Y)R12,
NRI2S(0)2R12, C(= y)NR12- 12,
K. CI-C12 alkyl, C2-C8 alkenyl, C2-C8 alkynyl, C3-C12
carbocyclyl, C2-C20 heterocyclyl, C6-C20 aryl and C1-C20 heteroaryl;
RI4 and R15 are independently selected from H, C1-C12 alkyl, or -(CH2)n-aryl,
or RI4 and R15 together with the atoms to which they are attached form a
saturated or
partially unsaturated C3-C12 carbocyclic ring; where said alkyl, alkenyl,
alkynyl, carbocyclyl,
heterocyclyl, aryl, and heteroaryl, are optionally substituted with one or
more groups
independently selected from F, CI, Br, I, CN, CF3, -NO2, oxo, R10, -C(=Y)R1 , -
C(=Y)0R1 ,
c(_y)NRioRI 1, _(cRiaRis)nNRio-ii
, -(CRI4R15)n0R1 ,
_NRI2g_y)Rio,
Nee-
u( Y)OR11, -
NRi2c(=y)N- io-ii
, -(CRI4R15)niNRI2S02R1 , =NRI2, OR1 ,
-0C(=Y)R1 , -0C(=Y)ORI , -0C(=Y)NR1 R11, -0S(0)2(0R10), -0P(=Y)(0R1 )(0R1 I
),
-0P(OR1 )(0R11), -SRI , -S(0)R1 , -S(0)2R10, -S(0)2
NRtc. io-ii, _
S(0)(0R1 ),
61
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
-S(0)2(oRio), _
SC(=Y)Rio, _sc (=y)oRio, ¨SC(=y)NRioRii, C1¨C12 alkyl, C2¨C8 alkenyl,
C2¨C8 alkynyl, C3¨C12 carbocyclyl, C2¨C20 heterocyclyl, C6¨C20 aryl, and
C1¨C20 heteroaryl;
Y is 0, S, or NR12;
m is 0, 1, 2, 3, 4, 5 or 6; and
n is 1, 2, 3, 4, 5 or 6.
Example P13-k inhibitors include the following:
HO \
N
\I t,...õ,,,,,,,
\ x'IN
N 111-3
H,C A
N N"', and
N
/ ____________________ \ I ,
--- \
NH 111-6
0 "4 0
N
i
H3C - S ,
r:0
0
=
PREPARATION OF FORMULAE V AND VI COMPOUNDS
The Formula V and VI compounds may be synthesized by synthetic routes
that include processes analogous to those well-known in the chemical arts, and
including WO
2006/046031, which is incorporated herein by reference in its entirety, for
all purposes.
Starting materials are generally available from commercial sources such as
Aldrich
Chemicals (Milwaukee, WI) or are readily prepared using methods well known to
those
skilled in the art (e.g., prepared by methods generally described in Louis F.
Fieser and Mary
Fieser, Reagents for Organic Synthesis, v. 1-19, Wiley, N.Y. (1967-1999 ed.),
or Beilsteins
Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin,
including
supplements (also available via the Beilstein online database).
Formulae V and VI compound may be prepared using procedures to prepare
other thiophenes, furans, pyrimidines (US 6608053; US 6492383; US 6232320; US
6187777;
62
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
US 3763156; US 3661908; US 3475429; US 5075305; US 2003/220365; GB 1393161; WO
93/13664); and other heterocycles, which are described in: Comprehensive
Heterocyclic
Chemistry, Editors Katritzky and Rees, Pergamon Press, 1984.
Formulae V and VI compounds may be converted into a pharmaceutically
acceptable salt, and a salt may be converted into the free compound, by
conventional
methods. Examples of pharmaceutically acceptable salts include salts with
inorganic acids
such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulphuric acid,
nitric acid and
phosphoric acid; and organic acids such as methanesulfonic acid,
benzenesulphonic acid,
formic acid, acetic acid, trifluoroacetic acid, propionic acid, oxalic acid,
malonic acid,
succinic acid, fumaric acid, maleic acid, lactic acid, malic acid, tartaric
acid, citric acid,
ethanesulfonic acid, aspartic acid and glutamic acid. The salt may be a
mesylate, a
hydrochloride, a phosphate, a benzenesulphonate or a sulphate. Salts may be
mono-salts or
bis-salts. For example, the mesylate salt may be the mono-mesylate or the bis-
mesylate.
Formulae V and VI compounds and salts may also exist as hydrates or
solvates.
Protection of functional groups (e.g., primary or secondary amine) of
intermediates may be necessary in preparing Formulae V and VI compounds. The
need for
such protection will vary depending on the nature of the remote functionality
and the
conditions of the preparation methods. Suitable amino-protecting groups
include acetyl,
trifluoroacetyl, t-butoxycarbonyl (BOC), benzyloxycarbonyl (CBz) and 9-
fluorenylmethyleneoxycarbonyl (Fmoc). The need for such protection is readily
determined
by one skilled in the art. For a general description of protecting groups and
their use, see T.
W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New
York, 1991.
For illustrative purposes, Schemes 5-11 show general methods for preparing
the compounds of the present invention as well as key intermediates. For a
more detailed
description of the individual reaction steps, see the Examples section below.
Those skilled in
the art will appreciate that other synthetic routes may be used to synthesize
the inventive
compounds. Although specific starting materials and reagents are depicted in
the Schemes
and discussed below, other starting materials and reagents can be easily
substituted to provide
a variety of derivatives and/or reaction conditions. In addition, many of the
compounds
prepared by the methods described below can be further modified in light of
this disclosure
using conventional chemistry well known to those skilled in the art.
63
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
0 Hal
Cs 02R1 ---
R1 R1 NH 1,111-\S 1,,, 5 S N
_________________________________________________________ _11/1
N - -:0 ______ R1 _), N Hal
NH2
R2 R2 H R2
51 53
R2 Hal
R2 IR\ j2 1
O R1C 2R1 --0- NH ---4-
elt.N
R1 ___ e--1X R1 __ el 1
--=0 S N Hal
S N-
S NH2 H
52 54 56
Scheme 5
Scheme 5 shows a general method for preparation of the thienopyrimidine
intermediates 55 and 56 from 2-carboxyester, 3-amino thiophene, and 2-amino, 3-
carboxy
5 ester thiophene reagents, respectively 51 and 52, wherein Hal is Cl, Br,
or I; and RI, R2, and
RI are as defined for Formulae V and VI compounds, or precursors or prodrugs
thereto.
0 0
( )
Hal ( ) N
1 N
c-tz-N H
i
R1 ___________ \
N Hal
N Hal
R2
R2 59
57
0
( ) 0
0
N
R2 Hal Fl R2 N
R1 / I Al __R1 ________________ / I
S N Hal
S N Hal
58
Schemefi
Scheme 6 shows a general method for selectively displacing a 4-halide from
10 bis-halo thienopyrimidine intermediates 57 and 58 with morpholine under
basic conditions in
an organic solvent to prepare 2-halo, 4-morpholino thienopyrimidine compounds
59 and 60
respectively, wherein Hal is Cl, Br, or I; and RI and R2 are as defined for
Formulae V and VI
compounds, or precursors or prodrugs thereto.
64
CA 02821378 2013-06-12
WO 2012/087336 PCT/US2010/062096
0
0
( ) ( )
N
N
0 s
_ _XL
n \ 1 .,..,..L ._ \ I
N Hal base Rl lµr Hal
R2
R2 63
61
0
0 ( )
( ) R2 N
R2 N R1 C(0)Z
H I base
R10 S reL Hal
S N Hal
64
62
Scheme 7
Scheme 7 shows a general method for derivatizing the 6-position of 2-halo, 4-
morpholino, 6-hydrogen thienopyrimidine compounds 61 and 62 where RI is H.
Treating 61
or 62 with a lithiating reagent to remove the 6 position proton, followed by
adding an
acylating reagent R1 C(0)Z where Z is a leaving group, such as halide, NHS
ester,
carboxylate, or dialkylamino, gives 2-halo, 4-morpholino, 6-acyl
thienopyrimidine
compounds 63 and 64, wherein Hal is Cl, Br, or I; and R2 and RI are as
defined for Formulae
V and VI compounds, or precursors or prodrugs thereto. An example of RI0C(0)Z
to prepare
6-formyl compounds (RI = H) is N,N'-dimethylformamide (DMF).
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
0 0
C ) C )
N
N
cf.N (Hy)-B(0R15)2 67 iSj--1=--- N
RI \ I
N Hal Pd catalyst N Hy
R2 R2
65 68
0 0
R2 () ( )
N (Hy)-13(0R15)2 17 R2 N
.._....,...
Pd catalyst R1
S N Hal S N Hy
66 69
Scheme 8
Scheme 8 shows a general method for Suzuki-type coupling of a 2-halo
pyrimidine intermediate (65 and 66) with a monocyclic heteroaryl, fused
bicyclic
heterocyclyl or fused bicyclic heteroaryl boronate acid (R15 = H) or ester
(R15 = alkyl) reagent
67 to prepare the 2-substituted (Hy), 4-morpholino thienopyrimidine compounds
(68 and 69)
of Formulae V and VI wherein Hal is Cl, Br, or I; and R1 and R2 are as defined
for Formulae
V and VI compounds, or precursors or prodrugs thereto. For reviews of the
Suzuki reaction,
see: Miyaura et al. (1995) Chem. Rev. 95:2457-2483; Suzuki, A. (1999) J.
Organomet.
Chem. 576:147-168; Suzuki, A. in Metal-Catalyzed Cross-Coupling Reactions,
Diederich, F.,
Stang, P.J., Eds., VCH, Weinheim, DE (1998), pp 49-97. The palladium catalyst
may be any
that is typically used for Suzuki-type cross-couplings, such as PdC12(PPh3)2,
Pd(PPh3)4,
Pd(OAc)2, PdC12(dppf)-DCM, Pd2(dba)3/Pt-Bu)3 (Owens et al (2003) Bioorganic &
Med.
Chem. Letters 13:4143-4145; Molander et al (2002) Organic Letters 4(11):1867-
1870; US
6448433).
66
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
Br R10R11NH RioRi iN
-H
70 base 71
O 0
71 Ri owl N
N
X2 \ I = __ =
N R3 N R3
R2 R2
72 74
O 0
R2 N 71 R2 N
Ri 13R 11N
X2 / I I
S Nj-- -R3
73 75
Scheme 9
Scheme 9 shows a general method for the synthesis of allcynes 71, which can
be used to prepare alkynylated derivatives of compounds 72 and 73. Propargylic
amines 71
may be prepared by reaction of propargyl bromide 70 with an amine of the
formula
R10,-.K11 NH (wherein RI and R" are independently selected from H, alkyl,
aryl and heteroaryl,
or RI and R" together with the nitrogen to which they are attached form a
heterocyclic ring)
in the presence of an appropriate base (Cs2CO3 or the like). For reviews of
alkynyl amines
and related syntheses see Booker-Milburn, K.I., Comprehensive Organic
Functional Group
Transformations (1995), 2:1039-1074; and Viehe, H.G., (1967) Angew. Chem.,
Int. Ed. Eng.,
6(9):767-778. Alkynes 71 may subsequently be reacted with intermediates 72 (X2
= bromo
or iodo) or 73 (via Sonogashira coupling), to provide compounds 74 and 75,
respectively,
wherein R2 and R3 are as defined for Formulae V and VI compounds, or
precursors or
prodrugs thereto.
67
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
CI RioRiiN
)
= H
RioRi 1NH \ = H
1:214 R14
R
R1 5 15 76 CuCI, base 77
O 0
( ) ( )
N N
;.f...N
77 RioRiiN
4, ....x--.1;:-N
N R3 Ri4
>\R15 = \ I ) N.... R3
R2 R2
72 78
O 0
( ) ( )
R2 N 77 R2 N
.-,1owl N > _______________________________________
X2 / I )\NI ---i.- rµ
S N"... R3 Ria Ri5 S N R3
73 79
Scheme 10
Scheme 10 shows a general method for the synthesis of alkynes 77, which can
be used to prepare alkynylated derivatives of compounds 72 and 73. Gem-dialkyl
propargylic amines 77 may be prepared using methods described by Zaragoza et
al (2004) J.
Med. Chem., 47:2833. According to Scheme 6, gem-dialkyl chloride 76 (R14 and
RI5 are
independently methyl, ethyl or other alkyl group) can be reacted with an amine
of the formula
RioK ¨ii
NH (wherein RI and R1' are independently selected from H, alkyl, aryl and
heteroaryl,
or RI and R" together with the nitrogen to which they are attached form a
heterocyclic ring)
in the presence of CuCI and an appropriate base (e.g. TEA or the like) to
provide the alkyne
77. Alkyne 77 can be reacted with intermediates 72 or 73 (via Sonogashira
coupling) to
provide compounds 78 and 79, respectively, wherein R2 and R3 are as defined
for Formulae
V and VI compounds, or precursors or prodrugs thereto.
68
CA 02821378 2013-06-12
WO 2012/087336 PCT/US2010/062096
LG RiowiN
R1OR11NH
= __ H
R-14) = ___________ H 1;4)
R15 80 heat R15
81
O 0
81
N RiowiN
X2 \ I ¨>\ = I
N R3 R14 Ris N R3
R2 R2
72 82
O 0
C
R2 N 81 R2 E N
R1OR11N
X2 / I __________________________________________ elk N
I
S N R3 R:4)\
R15 S N R3
73 83
Scheme 11
Scheme 11 shows a general scheme for the synthesis of alkynes 81, which can
be used to prepare alkynylated derivatives of compounds 72 and 73. But-3-yn-1 -
amines 81
(wherein R14 and R15 are independently H, alkyl, aryl, heteroaryl, or R14 and
R15 together
with the carbon atom to which they are attached form a carbocyclic or
heterocyclic ring) can
be prepared from reaction of alkynes 80 (LG = tosylate or other leaving group)
with an amine
of the formula RI R1INH (wherein RI and RI are independently selected from H,
alkyl, aryl
and heteroaryl, or RI and R" together with the nitrogen to which they are
attached form a
heterocyclic ring) using the protocol described by Olomucki M. et al (1960)
Ann. Chim.
5:845. Alkynes 81 can subsequently be reacted with intermediates 72 or 73 (via
Sonogashira
coupling), according to the descriptions provided for Schemes 5 and 6 to
provide compounds
82 and 83, respectively, wherein R2 and R3 are as defined for Formulae V and
VI compounds,
or precursors or prodrugs thereto.
A pharmaceutically acceptable salt of a thienopyrimidine compound of
Formula I to VI may be prepared using conventional techniques. Typically the
process
comprises treating the thienopyrimidine of Formula I as defined above with a
suitable acid in
a suitable solvent.
In the process of the invention as defined above, both the amination step and
69
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
the Pd-mediated cross-coupling step take place under conventional conditions.
The palladium
catalyst may be any that is typically used for Suzuki-type cross-couplings,
such as
PdC12(PPh3)2 The reducing agent is typically a borohydride, such as
NaBH(OAc)3, NaBH4 or
NaCNBH4
METHODS OF TREATING NEOPLASMS
An embodiment includes a method of treating a neoplasm in a mammal
comprising, administering a combination of (i) an inhibitor of a kinase,
wherein said inhibitor
induces autophagy, and (ii) an inhibitor of autophagy in an amount effective
to treat said
neoplasm. The inhibitor of a kinase and the inhibitor of autophagy can be
administered
together or separately, at the same time or at different times. In an
embodiment, the inhibitor
of kinase that induces autophagy and said inhibitor of autophagy are present
in synergistically
effective amounts.
In another embodiment, the method of treating a neoplasm in a mammal
comprising, administering a combination of (i) an inhibitor of a kinase,
wherein said inhibitor
induces autophagy, and (ii) an inhibitor of autophagy in an amount effective
to treat said
neoplasm, further comprises administering a protease inhibitor. Protease
inhibitors are well
known in the art. In one embodiment, the protease inhibitor inhibits lysosomal
cysteine
protease activity or aspartic proteases, such as pepstatin A. The inhibitor of
a kinase,
inhibitor of autophagy and the protease inhibitor can be administered singly,
or in any
combination together or separately, at the same time or at different times.
Methods of blocking or reducing relapse tumor growth or a relapse cancer cell
growth are also provided. In certain embodiments of the invention, the subject
was, or is
concurrently undergoing cancer therapy. The administration of the combination
therapy
described herein blocks or reduces relapse tumor growth or relapse cancer cell
growth.
Another embodiment provides, a method of inducing apoptosis in a cancer cell
comprising administering to said cell (i) an inhibitor of a kinase, wherein
said inhibitor
induces autophagy, and (ii) an inhibitor of autophagy in an amount effective
to induce said
apoptosis. In one example, the effective amount of said kinase inhibitor
and/or inhibitor of
autophagy produces a synergistic apoptosis inducing effect. In another
example, the effective
amount of said kinase and/or said inhibitor of autophagy has an ED50, ED75 or
ED90 that is
lower than the ED50, ED75 or ED90 of the kinase inhibitor or inhibitor of
autophagy alone.
In one example, the kinase inhibitor and inhibitor of autophagy are given in
ratios in the
range of about 2:1 to about 1:50, alternatively about 1.25:1 to about 1:12,
alternatively about
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
1:1 to about 1:5. In one example, 111-4 is dosed in combination with CQ in a
ratio of about
1:25, 1:12.5, 1:1.5, or 1.3:1.
When any variable occurs more than one time in any constituent or in Formula
I, II, III, IV, V or VI, its definition on each occurrence is independent of
its definition at
every other occurrence. Also combinations of substituents and/or variables are
permissible
only if such combinations result in allowable valences.
The method of treating a neoplasm described herein can comprise
administering an inhibitor of kinase that induces autophagy wherein the
inhibitor is an RNA
interference (RNAi) construct in combination with an inhibitor of autophagy.
Figure 23
shows that such an RNAi construct can comprise RNA, DNA or DNA that is
transcribed to
RNA. Preferably, the use of an RNAi construct in the present methods in
combination with
an autophagy inhibitor results in a synergistic killing or inhibitory effect
on a neoplasm.
RNA CONSTRUCTS
In another embodiment, the subject matter disclosed herein relates to RNAi
constructs described herein. The RNAi constructs are useful inhibitors of Akt.
As used herein, an RNAi construct includes shRNA, siRNA, DNA directed
shRNA and siRNA, as well as the DNA itself, DNA oligos and vectors described
herein. In
certain embodiments, siRNA or shRNA is transcribed from an RNAi construct
comprising a
nucleic acid sequence substantially corresponding to a target sequence in one
or more Akt
genes. Preferably, the sequence is selected from SEQ ID Nos: 39-48 and
combinations
thereof. When introduced to a cell, these RNAi constructs are capable of
reducing the
expression of one or more Akt proteins. Reducing the expression of a protein
means that the
expression is lower in a cell than it would be if the RNAi construct had not
been introduced.
Methods for detecting levels of expression are described herein or known in
the art. The
RNAi constructs can reduce the expression of AKT isoforms including Aktl,
Akt2, Akt3 and
combinations thereof.
The RNAi constructs can comprise one or more DNA sequences substantially
corresponding to a sequence selected from the group consisting of SEQ ID Nos:
1-18. DNA
sequences can be synthesized and cloned into a shuttle as described herein. In
another
embodiment, an RNAi construct capable of reducing the expression of one or
more Akt
proteins comprises a RNA sequence substantially corresponding to a sequence
selected from
the group consisting of SEQ ID Nos: 19-38 and combinations thereof. In another
embodiment, the RNAi constructs can comprise a sense RNA strand and a
substantially
71
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
complementary antisense RNA strand, wherein the antisense strand comprises one
or more
sequence substantially corresponding to a sequence selected from SEQ ID Nos:
20, 22, 24,
26, 28, 30, 32, 34, 36 and 38, wherein the sense and antisense strands are
annealed as a RNA
duplex. The duplex can comprise a sense strand comprising one or more
sequences
substantially corresponding to a sequence selected from the group consisting
of SEQ ID Nos:
19, 21, 23, 25, 27, 29, 31, 33, 35 and 37. The sense and antisense strands can
be annealed to
form the duplex in the pair combinations that include the following: SEQ ID
Nos: 19:20,
21:22, 23:24, 25:26, 27:28, 29:30, 31:32, 33:34, 35:36 and 37:38 and
combinations that
include more than one of the pairs. The RNAi construct can contain a hairpin
that covalently
links the sense strand and the antisense strand.
In another embodiment, RNAi constructs that are capable of reducing the
expression of one or more Aid proteins are described herein. Non-limiting
examples of such
RNAi constructs include a construct comprising a nucleotide sequence
substantially
corresponding to SEQ ID No: 32, and additionally comprising a sequence
substantially
corresponding to a sequence selected from the group consisting of SEQ ID Nos:
22, 26 and
36. Another non-limiting example includes a nucleotide sequence
substantially
corresponding to SEQ ID No: 31, and additionally a sequence substantially
corresponding to
a sequence selected from the group consisting of SEQ ID Nos: 21, 25 and 35.
Other
combinations which lower expression of target Akt isoforms can be readily
obtained from the
present disclosure.
Another embodiment is directed to an RNAi construct capable of reducing the
expression of an Akt gene, wherein the construct is a substrate for a Dicer.
Yet another
embodiment is directed to an isolated nucleotide or nucleic acid sequence as
described herein.
An RNAi construct as described herein can be prepared by any known method.
(McIntyre,
GJ, and Fanning GC, BMC Biotechnology (2006), 6:1).
PHARMACEUTICAL FORMULATIONS
Pharmaceutical compositions or formulations of the present invention include
combinations of compounds of Formula I to VI, and other compounds described
herein, a
inhibitor of autophagy, and one or more pharmaceutically acceptable carrier,
glidant, diluent,
or excipient.
The compounds of Formula I to VI, and other compounds described herein,
and inhibitors of autophagy of the present invention may exist in unsolvated
as well as
solvated forms with pharmaceutically acceptable solvents such as water,
ethanol, and the like,
72
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
and it is intended that the invention embrace both solvated and unsolvated
forms.
The compounds of Formula I to VI, and other compounds described herein,
and inhibitors of autophagy of the present invention may also exist in
different tautomeric
forms, and all such forms are embraced within the scope of the invention. The
term
"tautomer" or "tautomeric form" refers to structural isomers of different
energies which are
interconvertible via a low energy barrier. For example, proton tautomers (also
known as
prototropic tautomers) include interconversions via migration of a proton,
such as keto-enol
and imine-enamine isomerizations.
Valence tautomers include interconversions by
reorganization of some of the bonding electrons.
Pharmaceutical compositions encompass both the bulk composition and
individual dosage units comprised of more than one (e.g., two)
pharmaceutically active
agents including a Formula Ito VI compound and a inhibitor of autophagy
selected from the
lists of the additional agents described herein, along with any
pharmaceutically inactive
excipients, diluents, carriers, or glidants. The bulk composition and each
individual dosage
unit can contain fixed amounts of the aforesaid pharmaceutically active
agents. The bulk
composition is material that has not yet been formed into individual dosage
units. An
illustrative dosage unit is an oral dosage unit such as tablets, pills,
capsules, and the like.
Similarly, the herein-described method of treating a patient by administering
a
pharmaceutical composition of the present invention is also intended to
encompass the
administration of the bulk composition and individual dosage units.
Pharmaceutical compositions also embrace isotopically-labeled compounds of
the present invention which are identical to those recited herein, but for the
fact that one or
more atoms are replaced by an atom having an atomic mass or mass number
different from
the atomic mass or mass number usually found in nature. All isotopes of any
particular atom
or element as specified are contemplated within the scope of the compounds of
the invention,
and their uses. Exemplary isotopes that can be incorporated into compounds of
the invention
include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur,
fluorine, chlorine
and iodine, such as 2H, 3H, I IC, 13C, 14C, I3N, 15N, 150, 170, 180, 32F, 33F,
35s, I8F, 36C1, 123/
and 1251. Certain isotopically-labeled compounds of the present invention
(e.g., those labeled
with 3H and 14C) are useful in compound and/or substrate tissue distribution
assays. Tritiated
(3H) and carbon-14 (14C) isotopes are useful for their ease of preparation and
detectability.
Further, substitution with heavier isotopes such as deuterium (2H) may afford
certain
therapeutic advantages resulting from greater metabolic stability (e.g.,
increased in vivo half-
life or reduced dosage requirements) and hence may be preferred in some
circumstances.
73
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
Positron emitting isotopes such as 150, 13N, 11C and 18F are useful for
positron emission
tomography (PET) studies to examine substrate receptor occupancy. Isotopically
labeled
compounds of the present invention can generally be prepared by following
procedures
analogous to those disclosed in the Schemes and/or in the Examples herein
below, by
substituting an isotopically labeled reagent for a non-isotopically labeled
reagent.
The compounds of Formula I to VI, and other compounds described herein,
and inhibitors of autophagy are formulated in accordance with standard
pharmaceutical
practice for use in a therapeutic combination for therapeutic treatment
(including prophylactic
treatment) of hyperproliferative disorders in mammals including humans. The
invention
provides a pharmaceutical composition comprising a Formula I to VI compound in
association with one or more pharmaceutically acceptable carrier, glidant,
diluent, or
excipient.
Suitable carriers, diluents and excipients are well known to those skilled in
the
art and include materials such as carbohydrates, waxes, water soluble and/or
swellable
polymers, hydrophilic or hydrophobic materials, gelatin, oils, solvents, water
and the like.
The particular carrier, diluent or excipient used will depend upon the means
and purpose for
which the compound of the present invention is being applied. Solvents are
generally
selected based on solvents recognized by persons skilled in the art as safe
(GRAS) to be
administered to a mammal. In general, safe solvents are non-toxic aqueous
solvents such as
water and other non-toxic solvents that are soluble or miscible in water.
Suitable aqueous
solvents include water, ethanol, propylene glycol, polyethylene glycols (e.g.,
PEG 400, PEG
300), etc. and mixtures thereof. The formulations may also include one or more
buffers,
stabilizing agents, surfactants, wetting agents, lubricating agents,
emulsifiers, suspending
agents, preservatives, antioxidants, opaquing agents, glidants, processing
aids, colorants,
sweeteners, perfuming agents, flavoring agents and other known additives to
provide an
elegant presentation of the drug (i.e., a compound of the present invention or
pharmaceutical
composition thereof) or aid in the manufacturing of the pharmaceutical product
(i.e.,
medicament).
The formulations may be prepared using conventional dissolution and mixing
procedures. For example, the bulk drug substance (i.e., compound of the
present invention or
stabilized form of the compound (e.g., complex with a cyclodextrin derivative
or other known
complexation agent) is dissolved in a suitable solvent in the presence of one
or more of the
excipients described above. The compound of the present invention is typically
formulated
into pharmaceutical dosage forms to provide an easily controllable dosage of
the drug and to
74
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
enable patient compliance with the prescribed regimen.
The pharmaceutical composition (or formulation) for application may be
packaged in a variety of ways depending upon the method used for administering
the drug.
Generally, an article for distribution includes a container having deposited
therein the
pharmaceutical formulation in an appropriate form. Suitable containers are
well known to
those skilled in the art and include materials such as bottles (plastic and
glass), sachets,
ampoules, plastic bags, metal cylinders, and the like. The container may also
include a
tamper-proof assemblage to prevent indiscreet access to the contents of the
package. In
addition, the container has deposited thereon a label that describes the
contents of the
container. The label may also include appropriate warnings.
Pharmaceutical formulations of the compounds of the present invention may
be prepared for various routes and types of administration. For example, a
compound of
Formula I to VI, or another compound described herein, having the desired
degree of purity
may optionally be mixed with pharmaceutically acceptable diluents, carriers,
excipients or
stabilizers (Remington's Pharmaceutical Sciences (1995) 18th edition, Mack
Publ. Co.,
Easton, PA), in the form of a lyophilized formulation, milled powder, or an
aqueous solution.
Formulation may be conducted by mixing at ambient temperature at the
appropriate pH, and
at the desired degree of purity, with physiologically acceptable carriers,
i.e., carriers that are
non-toxic to recipients at the dosages and concentrations employed. The pH of
the
formulation depends mainly on the particular use and the concentration of
compound, but
may range from about 3 to about 8.
The pharmaceutical formulation is preferably sterile. In
particular,
formulations to be used for in vivo administration must be sterile. Such
sterilization is readily
accomplished by filtration through sterile filtration membranes.
The pharmaceutical formulation ordinarily can be stored as a solid
composition, a lyophilized formulation or as an aqueous solution.
The pharmaceutical formulations of the invention will be dosed and
administered in a fashion, i.e., amounts, concentrations, schedules, course,
vehicles and route
of administration, consistent with good medical practice. Factors for
consideration in this
context include the particular disorder being treated, the particular mammal
being treated, the
clinical condition of the individual patient, the cause of the disorder, the
site of delivery of the
agent, the method of administration, the scheduling of administration, and
other factors
known to medical practitioners. The "therapeutically effective amount" of the
compound to
be administered will be governed by such considerations, and is the minimum
amount
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
necessary to prevent, ameliorate, or treat the coagulation factor mediated
disorder. Such
amount is preferably below the amount that is toxic to the host or renders the
host
significantly more susceptible to bleeding.
As a general proposition, the initial pharmaceutically effective amount of the
compound of Formula Ito VI, or another compound described herein, administered
orally or
parenterally per dose will be in the range of about 0.01-100 mg/kg, namely
about 0.1 to 20
mg,/kg of patient body weight per day, with the typical initial range of
compound used being
0.3 to 15 mg/kg/day.
Acceptable diluents, carriers, excipients and stabilizers are nontoxic to
recipients at the dosages and concentrations employed, and include buffers
such as
phosphate, citrate and other organic acids; antioxidants including ascorbic
acid and
methionine; preservatives (such as octadecyldimethylbenzyl ammonium chloride;
hexamethonium chloride; benzalkonium chloride, benzethonium chloride; phenol,
butyl or
benzyl alcohol; alkyl parabens such as methyl or propyl paraben; catechol;
resorcinol;
cyclohexanol; 3-pentanol; and m-cresol); low molecular weight (less than about
10 residues)
polypeptides; proteins, such as serum albumin, gelatin, or immunoglobulins;
hydrophilic
polymers such as polyvinylpyrrolidone; amino acids such as glycine, glutamine,
asparagine,
histidine, arginine, or lysine; monosaccharides, disaccharides and other
carbohydrates
including glucose, mannose, or dextrins; chelating agents such as EDTA; sugars
such as
sucrose, mannitol, trehalose or sorbitol; salt-forming counter-ions such as
sodium; metal
complexes (e.g., Zn-protein complexes); and/or non-ionic surfactants such as
TWEENTm,
PLURONICSTM or polyethylene glycol (PEG). The active pharmaceutical
ingredients may
also be entrapped in microcapsules prepared, for example, by coacervation
techniques or by
interfacial polymerization, for example, hydroxymethylcellulose or gelatin-
microcapsules
and poly-(methylmethacylate) microcapsules, respectively, in colloidal drug
delivery systems
(for example, liposomes, albumin microspheres, microemulsions, nano-particles
and
nanocapsules) or in macroemulsions. Such techniques are disclosed in
Remington's
Pharmaceutical Sciences 18th edition, (1995) Mack Publ. Co., Easton, PA.
Sustained-release preparations of the compounds of Formula Ito VI, and other
compounds described herein, may be prepared. Suitable examples of sustained-
release
preparations include semipermeable matrices of solid hydrophobic polymers
containing a
compound of Formula I, which matrices are in the form of shaped articles,
e.g., films, or
microcapsules. Examples of sustained-release matrices include polyesters,
hydrogels (for
76
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
example, poly(2-hydroxyethyl-methacrylate), or poly(vinyl alcohol)),
polylactides (US
3773919), copolymers of L-glutamic acid and gamma-ethyl-L-glutamate, non-
degradable
ethylene-vinyl acetate, degradable lactic acid-glycolic acid copolymers such
as the LUPRON
DEPOTTm (injectable microspheres composed of lactic acid-glycolic acid
copolymer and
leuprolide acetate) and poly-D (-) 3-hydroxybutyric acid.
The pharmaceutical formulations include those suitable for the administration
routes detailed herein. The formulations may conveniently be presented in unit
dosage form
and may be prepared by any of the methods well known in the art of pharmacy.
Techniques
and formulations generally are found in Remington's Pharmaceutical Sciences
18th Ed. (1995)
Mack Publishing Co., Easton, PA. Such methods include the step of bringing
into association
the active ingredient with the carrier which constitutes one or more accessory
ingredients. In
general the formulations are prepared by uniformly and intimately bringing
into association
the active ingredient with liquid carriers or finely divided solid carriers or
both, and then, if
necessary, shaping the product.
Formulations of compounds of Formula I to VI, and other compounds
described herein, and inhibitors of autophagy suitable for oral administration
may be
prepared as discrete units such as pills, hard or soft e.g., gelatin capsules,
cachets, troches,
lozenges, aqueous or oil suspensions, dispersible powders or granules,
emulsions, syrups or
elixirs each containing a predetermined amount of a compound of Formula Ito
VI, or another
compound described herein, and a inhibitor of autophagy. Such formulations may
be
prepared according to any method known to the art for the manufacture of
pharmaceutical
compositions and such compositions may contain one or more agents including
sweetening
agents, flavoring agents, coloring agents and preserving agents, in order to
provide a
palatable preparation. Compressed tablets may be prepared by compressing in a
suitable
machine the active ingredient in a free-flowing form such as a powder or
granules, optionally
mixed with a binder, lubricant, inert diluent, preservative, surface active or
dispersing agent.
Molded tablets may be made by molding in a suitable machine a mixture of the
powdered
active ingredient moistened with an inert liquid diluent. The tablets may
optionally be coated
or scored and optionally are formulated so as to provide slow or controlled
release of the
active ingredient therefrom.
Tablet excipients of a pharmaceutical formulation of the invention may
include: Filler (or diluent) to increase the bulk volume of the powdered drug
making up the
tablet; Disintegrants to encourage the tablet to break down into small
fragments, ideally
77
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
individual drug particles, when it is ingested and promote the rapid
dissolution and absorption
of drug; Binder to ensure that granules and tablets can be formed with the
required
mechanical strength and hold a tablet together after it has been compressed,
preventing it
from breaking down into its component powders during packaging, shipping and
routine
handling; Glidant to improve the flowability of the powder making up the
tablet during
production; Lubricant to ensure that the tableting powder does not adhere to
the equipment
used to press the tablet during manufacture. They improve the flow of the
powder mixes
through the presses and minimize friction and breakage as the finished tablets
are ejected
from the equipment; Antiadherent with function similar to that of the glidant,
reducing
adhesion between the powder making up the tablet and the machine that is used
to punch out
the shape of the tablet during manufacture; Flavor incorporated into tablets
to give them a
more pleasant taste or to mask an unpleasant one, and Colorant to aid
identification and
patient compliance.
Tablets containing the active ingredient in admixture with non-toxic
pharmaceutically acceptable excipient which are suitable for manufacture of
tablets are
acceptable. These excipients may be, for example, inert diluents, such as
calcium or sodium
carbonate, lactose, calcium or sodium phosphate; granulating and
disintegrating agents, such
as maize starch, or alginic acid; binding agents, such as starch, gelatin or
acacia; and
lubricating agents, such as magnesium stearate, stearic acid or talc. Tablets
may be uncoated
or may be coated by known techniques including microencapsulation to delay
disintegration
and adsorption in the gastrointestinal tract and thereby provide a sustained
action over a
longer period. For example, a time delay material such as glyceryl
monostearate or glyceryl
distearate alone or with a wax may be employed.
For treatment of the eye or other external tissues, e.g., mouth and skin, the
formulations are preferably applied as a topical ointment or cream containing
the active
ingredient(s) in an amount of, for example, 0.075 to 20% w/w. When formulated
in an
ointment, the active ingredients may be employed with either a paraffinic or a
water-miscible
ointment base. Alternatively, the active ingredients may be formulated in a
cream with an
oil-in-water cream base.
If desired, the aqueous phase of the cream base may include a polyhydric
alcohol, i.e., an alcohol having two or more hydroxyl groups such as propylene
glycol, butane
1,3-diol, mannitol, sorbitol, glycerol and polyethylene glycol (including PEG
400) and
mixtures thereof. The topical formulations may desirably include a compound
which
enhances absorption or penetration of the active ingredient through the skin
or other affected
78
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
areas. Examples of such dermal penetration enhancers include dimethyl
sulfoxide and related
analogs.
The oily phase of the emulsions of this invention may be constituted from
known ingredients in a known manner, including a mixture of at least one
emulsifier with a
fat or an oil, or with both a fat and an oil. Preferably, a hydrophilic
emulsifier is included
together with a lipophilic emulsifier which acts as a stabilizer. Together,
the emulsifier(s)
with or without stabilizer(s) make up an emulsifying wax, and the wax together
with the oil
and fat comprise an emulsifying ointment base which forms the oily dispersed
phase of cream
formulations. Emulsifiers and emulsion stabilizers suitable for use in the
formulation of the
invention include Tween 60, Span 80, cetostearyl alcohol, benzyl alcohol,
myristyl
alcohol, glyceryl mono-stearate and sodium lauryl sulfate.
Aqueous suspensions of the pharmaceutical formulations of the invention
contain the active materials in admixture with excipients suitable for the
manufacture of
aqueous suspensions. Such excipients include a suspending agent, such as
sodium
carboxymethylcellulose, croscarme I lose, povidone, methyl cel lu lose,
hydroxypropyl
methylcellulose, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum
acacia, and
dispersing or wetting agents such as a naturally occurring phosphatide (e.g.,
lecithin), a
condensation product of an alkylene oxide with a fatty acid (e.g.,
polyoxyethylene stearate), a
condensation product of ethylene oxide with a long chain aliphatic alcohol
(e.g.,
heptadecaethyleneoxycetanol), a condensation product of ethylene oxide with a
partial ester
derived from a fatty acid and a hexitol anhydride (e.g., polyoxyethylene
sorbitan
monooleate). The aqueous suspension may also contain one or more preservatives
such as
ethyl or n-propyl p-hydroxybenzoate, one or more coloring agents, one or more
flavoring
agents and one or more sweetening agents, such as sucrose or saccharin.
Pharmaceutical compositions may be in the form of a sterile injectable
preparation, such as a sterile injectable aqueous or oleaginous suspension.
This suspension
may be formulated according to the known art using those suitable dispersing
or wetting
agents and suspending agents which have been mentioned above. The sterile
injectable
preparation may be a solution or a suspension in a non-toxic parenterally
acceptable diluent
or solvent, such as a solution in 1,3-butanediol or prepared from a
lyophilized powder.
Among the acceptable vehicles and solvents that may be employed are water,
Ringer's
solution and isotonic sodium chloride solution. In addition, sterile fixed
oils may
conventionally be employed as a solvent or suspending medium. For this purpose
any bland
fixed oil may be employed including synthetic mono- or diglycerides. In
addition, fatty acids
79
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
such as oleic acid may likewise be used in the preparation of injectables.
The amount of active ingredient that may be combined with the carrier
material to produce a single dosage form will vary depending upon the host
treated and the
particular mode of administration. For example, a time-release formulation
intended for oral
administration to humans may contain approximately 1 to 1000 mg of active
material
compounded with an appropriate and convenient amount of carrier material which
may vary
from about 5 to about 95% of the total compositions (weight:weight). The
pharmaceutical
composition can be prepared to provide easily measurable amounts for
administration. For
example, an aqueous solution intended for intravenous infusion may contain
from about 3 to
500 jig of the active ingredient per milliliter of solution in order that
infusion of a suitable
volume at a rate of about 30 mL/hr can occur.
Formulations suitable for parenteral administration include aqueous and non-
aqueous sterile injection solutions which may contain anti-oxidants, buffers,
bacteriostats and
solutes which render the formulation isotonic with the blood of the intended
recipient; and
aqueous and non-aqueous sterile suspensions which may include suspending
agents and
thickening agents.
Formulations suitable for topical administration to the eye also include eye
drops wherein the active ingredient is dissolved or suspended in a suitable
carrier, especially
an aqueous solvent for the active ingredient. The active ingredient is
preferably present in
such formulations in a concentration of about 0.5 to 20% w/w, for example
about 0.5 to 10%
w/w, for example about 1.5% w/w.
Formulations suitable for topical administration in the mouth include lozenges
comprising the active ingredient in a flavored basis, usually sucrose and
acacia or tragacanth;
pastilles comprising the active ingredient in an inert basis such as gelatin
and glycerin, or
sucrose and acacia; and mouthwashes comprising the active ingredient in a
suitable liquid
carrier.
Formulations for rectal administration may be presented as a suppository with
a suitable base comprising for example cocoa butter or a salicylate.
Formulations suitable for intrapulmonary or nasal administration have a
particle size for example in the range of 0.1 to 500 microns (including
particle sizes in a
range between 0.1 and 500 microns in increments microns such as 0.5, 1, 30
microns, 35
microns, etc.), which is administered by rapid inhalation through the nasal
passage or by
inhalation through the mouth so as to reach the alveolar sacs. Suitable
formulations include
aqueous or oily solutions of the active ingredient. Formulations suitable for
aerosol or dry
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
powder administration may be prepared according to conventional methods and
may be
delivered with other therapeutic agents such as compounds heretofore used in
the treatment
or prophylaxis disorders as described below.
Formulations suitable for vaginal administration may be presented as
pessaries, tampons, creams, gels, pastes, foams or spray formulations
containing in addition
to the active ingredient such carriers as are known in the art to be
appropriate.
It is further contemplated that an agent of the invention (e.g., DNA, RNAi,
shRNA, siRNA, kinase inhibitor, chemotherapeutic agent or anti-cancer agent)
can be
introduced to a subject by gene therapy. Gene therapy refers to therapy
performed by the
administration of a nucleic acid to a subject. In gene therapy applications,
genes are
introduced into cells in order to achieve in vivo synthesis of a
therapeutically effective
genetic product, for example for replacement of a defective gene. "Gene
therapy" includes
both conventional gene therapy where a lasting effect is achieved by a single
treatment, and
the administration of gene therapeutic agents, which involves the one time or
repeated
administration of a therapeutically effective DNA or mRNA. Antisense RNAs and
DNAs
can be used as therapeutic agents for blocking the expression of certain genes
in vivo. It has
already been shown that short antisense oligonucleotides can be imported into
cells where
they act as inhibitors, despite their low intracellular concentrations caused
by their restricted
uptake by the cell membrane. (Zamecnik et al., Proc. Natl. Acad. Sci. USA
83:4143-4146
(1986)). The oligonucleotides can be modified to enhance their uptake, e.g. by
substituting
their negatively charged phosphodiester groups by uncharged groups. For
general reviews of
the methods of gene therapy, see, for example, Goldspiel et al. Clinical
Pharmacy 12:488-
505 (1993); Wu and Wu Biotherapy 3:87-95 (1991); Tolstoshev Ann. Rev.
Pharmacol.
Toxicol. 32:573-596 (1993); Mulligan Science 260:926-932 (1993); Morgan and
Anderson
Ann. Rev. Biochem. 62:191-217 (1993); and May TIBTECH 11:155-215 (1993).
Methods
commonly known in the art of recombinant DNA technology which can be used are
described in Ausubel et al. eds. (1993) Current Protocols in Molecular
Biology, John Wiley
& Sons, NY; and Kriegler (1990) Gene Transfer and Expression, A Laboratory
Manual,
Stockton Press, NY.
In one embodiment, the RNAi constructs or DNA for forming the RNA
constructs of the invention are delivered to cell(s) for treatment, and may be
delivered in
combination with inhibitors of autophagy. There are two major approaches to
getting the
DNA/RNA (optionally contained in a vector) into the patient's cells; in vivo
and ex vivo. For
in vivo delivery the DNA/RNA is injected directly into the patient, usually at
the site where
81
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
the DNA/RNA is required. For ex vivo treatment, the patient's cells are
removed, the
DNA/RNA is introduced into these isolated cells and the modified cells are
administered to
the patient either directly or, for example, encapsulated within porous
membranes which are
implanted into the patient (see, e.g., U.S. Patent Nos. 4,892,538 and
5,283,187). There are a
variety of techniques available for introducing nucleic acids into viable
cells. The techniques
vary depending upon whether the oligonucleotide is transferred into cultured
cells in vitro, or
in vivo in the cells of the intended host. Techniques suitable for the
transfer of
oligonucleotides into mammalian cells in vitro include the use of liposomes,
electroporation,
microinjection, cell fusion, DEAE-dextran, the calcium phosphate precipitation
method, etc.
A commonly used vector for ex vivo delivery of the gene is a retroviral
vector.
Example in vivo nucleic acid transfer techniques include transfection with
viral vectors (such as adenovirus, Herpes simplex I virus, or adeno-associated
virus) and
lipid-based systems (useful lipids for lipid-mediated transfer of the gene are
DOTMA, DOPE
and DC-Chol, for example). For review of the currently known gene marking and
gene
therapy protocols see Anderson et al., Science 256:808-813 (1992). See also WO
93/25673
and the references cited therein. Examples of using viral vectors in gene
therapy can be
found in Clowes et al. J. ain. Invest. 93:644-651 (1994); Kiem et al. Blood
83:1467-1473
(1994); Salmons and Gunzberg Human Gene Therapy 4:129-141 (1993); Grossman and
Wilson Curr. Opin. in Genetics and Devel. 3:110-114 (1993); Bout et al. Human
Gene
Therapy 5:3 -10 (1994); Rosenfeld et al. Science 252:431-434 (1991); Rosenfeld
et al. Cell
68:143-155 (1992); Mastrangeli et al. I Glin. Invest. 91:225-234 (1993); and
Walsh et al.
Proc. Soc. Exp. Biol. Med. 204:289-300 (1993).
In some situations it is desirable to provide the nucleic acid source with an
agent that targets the target cells, such as an antibody specific for a cell
surface membrane
protein or the target cell, a ligand for a receptor on the target cell, etc.
Where liposomes are
employed, proteins which bind to a cell surface membrane protein associated
with
endocytosis may be used for targeting and/or to facilitate uptake, e.g. capsid
proteins or
fragments thereof tropic for a particular cell type, antibodies for proteins
which undergo
internalization in cycling, proteins that target intracellular localization
and enhance
intracellular half-life. The technique of receptor-mediated endocytosis is
described, for
example, by Wu et al., I Biol. Chem. 262, 4429-4432 (1987); and Wagner et al.,
Proc. Natl.
Acad Sci. USA 87, 3410-3414 (1990). For review of gene marking and gene
therapy
protocols see Anderson et al., Science 256, 808-813 (1992).
The formulations may be packaged in unit-dose or multi-dose containers, for
82
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
example sealed ampoules and vials, and may be stored in a freeze-dried
(lyophilized)
condition requiring only the addition of the sterile liquid carrier, for
example water, for
injection immediately prior to use. Extemporaneous injection solutions and
suspensions are
prepared from sterile powders, granules and tablets of the kind previously
described.
Preferred unit dosage formulations are those containing a daily dose or unit
daily sub-dose, as
herein above recited, or an appropriate fraction thereof, of the active
ingredient.
The invention further provides veterinary compositions comprising at least
one active ingredient as above defined together with a veterinary carrier
therefore.
Veterinary carriers are materials useful for the purpose of administering the
composition and
may be solid, liquid or gaseous materials which are otherwise inert or
acceptable in the
veterinary art and are compatible with the active ingredient. These veterinary
compositions
may be administered parenterally, orally or by any other desired route.
COMBINATION THERAPY
The combination therapy may be administered as a simultaneous or sequential
regimen. When administered sequentially, the combination may be administered
in two or
more administrations. The combined administration includes coadministration,
using
separate formulations or a single pharmaceutical formulation, and consecutive
administration
in either order, wherein preferably there is a time period while both (or all)
active agents
simultaneously exert their biological activities.
Suitable dosages for any of the above coadministered agents are those
presently used and may be lowered due to the combined action (synergy) of the
newly
identified agent and other inhibitors of autophagy or treatments.
In a particular embodiment of anti-cancer therapy, a compound of Formula I
to VI, or other compounds described herein, or a stereoisomer, geometric
isomer, tautomer,
solvate, metabolite, or pharmaceutically acceptable salt thereof, is combined
with an inhibitor
of autophagy, and further combined with surgical therapy and radiotherapy. The
amounts of
the compound(s) of Formula I to VI, or other compounds described herein, and
the
inhibitor(s) of autophagy, and the relative timings of administration will be
selected in order
to achieve the desired combined therapeutic effect. In an embodiment, the
therapeutic effect
is a synergistic effect.
The compounds of the invention may be administered by any route
appropriate to the condition to be treated. Suitable routes include oral,
parenteral (including
subcutaneous, intramuscular, intravenous, intraarterial, inhalation,
intradermal, intrathecal,
83
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
epidural, and infusion techniques), transdermal, rectal, nasal, topical
(including buccal and
sublingual), vaginal, intraperitoneal, intrapulmonary and intranasal. Topical
administration
can also involve the use of transdermal administration such as transdermal
patches or
iontophoresis devices. Formulation of drugs is discussed in Remington's
Pharmaceutical
Sciences, 18th Ed., (1995) Mack Publishing Co., Easton, PA. Other examples of
drug
formulations can be found in Liberman, H. A. and Lachman, L., Eds.,
Pharmaceutical Dosage
Forms, Marcel Decker, Vol 3, 2n1 Ed., New York, NY. For local
immunosuppressive
treatment, the compounds may be administered by intralesional administration,
including
perfusing or otherwise contacting the graft with the inhibitor before
transplantation. It will be
appreciated that the preferred route may vary with for example the condition
of the recipient.
Where the compound is administered orally, it may be formulated as a pill,
capsule, tablet,
etc. with a pharmaceutically acceptable carrier, glidant, or excipient. Where
the compound is
administered parenterally, it may be formulated with a pharmaceutically
acceptable
parenteral vehicle or diluent, and in a unit dosage injectable form, as
detailed below.
A dose to treat human patients may range from about 10 mg to about 1000 mg
of Formula Ito VI compound. A typical dose may be about 100 mg to about 300 mg
of the
compound. A dose may be administered once a day (QID), twice per day (BID), or
more
frequently, depending on the pharmacokinetic (PK) and pharmacodynamic (PD)
properties,
including absorption, distribution, metabolism, and excretion of the
particular compound. In
addition, toxicity factors may influence the dosage and administration
regimen. When
administered orally, the pill, capsule, or tablet may be ingested daily or
less frequently for a
specified period of time. The regimen may be repeated for a number of cycles
of therapy.
ARTICLES OF MANUFACTURE
Kits of combinations of inhibitors of a kinase that induces autophagy and
inhibitors of autophagy are also provided. In certain embodiments, a kit
includes inhibitors
of a kinase that induce autophagy and inhibitors of autophagy, a
pharmaceutically acceptable
carrier, vehicle, or diluent, and a container. Instructions for use can also
be included.
In another embodiment of the invention, an article of manufacture, or "kit",
containing compounds of Formulae I to VI useful for the treatment of the
diseases and
disorders described above is provided. In one embodiment, the kit comprises a
container
comprising a compound of Formula I, or a stereoisomer, geometric isomer,
tautomer, solvate,
metabolite, or pharmaceutically acceptable salt thereof. The kit may further
comprise a label
or package insert, on or associated with the container. The term "package
insert" is used to
84
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
refer to instructions customarily included in commercial packages of
therapeutic products,
that contain information about the indications, usage, dosage, administration,
contraindications and/or warnings concerning the use of such therapeutic
products. Suitable
containers include, for example, bottles, vials, syringes, blister pack, etc.
The container may
be formed from a variety of materials such as glass or plastic. The container
may hold a
compound of Formula I to VI or a formulation thereof which is effective for
treating the
condition and may have a sterile access port (for example, the container may
be an
intravenous solution bag or a vial having a stopper pierceable by a hypodermic
injection
needle). At least one active agent in the composition is a compound of Formula
Ito VI, or a
compound described herein. The label or package insert indicates that the
composition is
used for treating the condition of choice, such as cancer. In one embodiment,
the label or
package inserts indicates that the composition comprising a compound of
Formula Ito VI, or
a compound described herein, can be used to treat a disorder resulting from
abnormal cell
growth. The label or package insert may also indicate that the composition can
be used to
treat other disorders. Alternatively, or additionally, the article of
manufacture may further
comprise a second container comprising a pharmaceutically acceptable buffer,
such as
bacteriostatic water for injection (BWFI), phosphate-buffered saline, Ringer's
solution and
dextrose solution. It may further include other materials desirable from a
commercial and
user standpoint, including other buffers, diluents, filters, needles, and
syringes.
The kit may further comprise directions for the administration of the
compound of Formula I to VI, or a compound described herein, and, if present,
the second
pharmaceutical formulation. For example, if the kit comprises a first
composition comprising
a compound of Formula I to VI, or a compound described herein, and a second
pharmaceutical formulation, the kit may further comprise directions for the
simultaneous,
sequential or separate administration of the first and second pharmaceutical
compositions to a
patient in need thereof.
In another embodiment, the kits are suitable for the delivery of solid oral
forms of a compound of Formula I to VI, or a compound described herein, such
as tablets or
capsules. Such a kit preferably includes a number of unit dosages. Such kits
can include a
card having the dosages oriented in the order of their intended use. An
example of such a kit
is a "blister pack". Blister packs are well known in the packaging industry
and are widely
used for packaging pharmaceutical unit dosage forms. If desired, a memory aid
can be
provided, for example in the form of numbers, letters, or other markings or
with a calendar
insert, designating the days in the treatment schedule in which the dosages
can be
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
administered.
According to one embodiment, a kit may comprise (a) a first container with a
compound of Formula I to VI, or a compound described herein, contained
therein; and
optionally (b) a second container with a second pharmaceutical formulation
contained
therein, wherein the second pharmaceutical formulation comprises a second
compound with
anti-hyperproliferative activity. Alternatively, or additionally, the kit may
further comprise a
third container comprising a pharmaceutically-acceptable buffer, such as
bacteriostatic water
for injection (BWFI), phosphate-buffered saline, Ringer's solution and
dextrose solution. It
may further include other materials desirable from a commercial and user
standpoint,
including other buffers, diluents, filters, needles, and syringes.
Where the kit comprises a composition of Formula I to VI, or a compound
described herein, and a second therapeutic agent, i.e. the inhibitor of
autophagy, the kit may
comprise a container for containing the separate compositions such as a
divided bottle or a
divided foil packet; however, the separate compositions may also be contained
within a
single, undivided container. Typically, the kit comprises directions for the
administration of
the separate components. The kit form is particularly advantageous when the
separate
components are preferably administered in different dosage forms (e.g., oral
and parenteral),
are administered at different dosage intervals, or when titration of the
individual components
of the combination is desired by the prescribing physician.
KINASE INHIBITION THAT INDUCES AUTOPHAGY
Data provided herein, show that Akt inhibition, and likewise, inhibition of
other certain kinases, does not always induce a clear apoptotic response.
Autophagy is a
readily detectable response to pan-Akt knockdown or inhibition, Akt-isoform
selective
knockdown or inhibition, or small molecule inhibitors of the Akt, PI3K, mTOR,
PDK1 or
p70S6K pathways. Kinase-inhibition-induced autophagy may sensitize tumor cells
to agents
targeting this lysosomal degradation pathway. Indeed, agents that block the
lysosomal
degradation function could precipitate cell death when combined with kinase
inhibitors that
induce autophagy and promote complete tumor remissions in preclinical models.
Inhibiting,
slowing or blocking the autophagic response may be a promising strategy to
increase the
therapeutic efficacy of kinase inhibitors that induce autophagy, e.g., Akt,
PI3K, mTOR,
PDK-1 and p70S6K inhibitors.
Autophagy is a more sensitive response to Akt inhibition, for example, than
apoptosis in cancer cell lines. Numerous reports have documented deregulation
of the
86
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
phosphatidylinositol 3-kinase (PI3K)/Akt pathway in a variety of cancers,
leading not only to
uncontrolled growth and proliferation, but also to resistance to various cell
death stimuli.
(Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007;
129:1261-74; Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr
Opin Oncol
2006; 18:77-82). Thus, targeting, for example Akt, the serine/threonine kinase
at the central
node of this pathway, or other kinases for which inhibition induces autophagy,
may inhibit
both growth and survival of the malignant cells.
Although Akt is believed to play a critical role in protecting cells from
programmed cell death following various pro-apoptotic insults (Manning BD, et
al.;
Downward J. Mechanisms and consequences of activation of protein kinase B/Akt.
Curr Opin
Cell Biol 1998; 10:262-7), it remains to be determined whether apoptosis is a
prevailing
response to inhibiting Akt activity alone. RNA interference techniques that
specifically
knockdown each of the three Akt isoforms as well as specific inhibitors result
in a significant
proportion of cancer cell lines examined do not readily undergo apoptosis even
when all three
Akt isoforms are greatly reduced. (Koseoglu S, Lu Z, Kumar C, Kirschmeier P,
Zou J. AKT1,
AKT2 and AKT3-dependent cell survival is cell line-specific and knockdown of
all three
isoforms selectively induces apoptosis in 20 human tumor cell lines. Cancer
Biol Ther 2007;
6:755-62). This is consistent with the report that only a small portion of
total Akt activity is
required for apoptosis inhibition in mouse embryonic fibroblast (MEF) cells.
(Liu X, Shi Y,
Birnbaum MJ, Ye K, De Jong R, Oltersdorf T, Giranda VL, Luo Y. Quantitative
analysis of
anti-apoptotic function of Akt in Akt 1 and Akt2 double knock-out mouse
embryonic
fibroblast cells under normal and stressed conditions. J Biol Chem 2006;
281:31380-8). The
sensitivity of cancer cells to apoptosis induction upon Akt inhibition is
likely dependent on
both their genetic background and environmental conditions. For example,
although a higher
level of activated Akt may suggest a relative reliance of tumor cells on this
pathway and may
be a slightly better predictor of apoptotic response to Akt inhibition,
resistance to apoptosis is
also observed in cells with Akt activation, including those with loss of
phosphatase and tensin
homolog (PTEN), a tumor suppressor that negatively regulates PI3K/Akt
activity. (Koseoglu
S, et al). It is conceivable that apoptosis can be suppressed by multiple
mechanisms in
advanced cancer cells as a result of their evolution through stringent
selection pressure.
In contrast, although both PTEN-null cell lines PC3 and U87MG are resistant
to apoptosis in response to inducible shRNA knockdown of all three Akt
isoforms
(shAkt123), data show significantly elevated autophagy in both cell lines.
Autophagy
appears to be a more sensitive response to reduced Akt activity caused by
either specific
87
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
shRNA knockdown or selective inhibitors of the pathway in a variety of cell
lines, whether or
not apoptosis is induced in these cells (Degtyarev and Lin, this work and
unpublished data).
Blocking autophagic degradation accelerated cell death in combination with
Akt inhibition. Autophagy has been implicated both as a mechanism of cell
death and as a
cytoprotective process, depending on the circumstances and cellular contexts.
(Scarlatti F,
Granata R, Meijer AJ, Codogno P. Does autophagy have a license to kill
mammalian cells?
2008). However, PC3 cells expressing shAkt123 can survive for a prolonged
period of time
without significant loss of viability, even under reduced serum conditions.
When grown as
xenograft tumors, although continuous expression of shAkt123 could effectively
inhibit
tumor growth initially, most of the tumors failed to regress completely and
eventually
overcame the inhibition and rebounded within 2-3 weeks. These suggest that
autophagy
induced by inhibiting Akt alone does not effectively eliminate cancer cells
under these
conditions.
Because autophagy is a more sensitive response to Akt knockdown or small
molecule inhibitors, blocking effective autophagy could accelerate cell death
in combination
with Akt inhibition. The lysosomotropic agent chloroquine (CQ) significantly
accelerated
death rate in cells either expressing shAkt123 or treated with relatively
specific small
molecule inhibitors of the pathway, PI-103 (a PI3K/mTOR inhibitor that is
1,000x more
potent on class I than class III PI3K) (Knight ZA, Gonzalez B, Feldman ME,
Zunder ER,
Goldenberg DD, Williams 0, Loewith R, Stokoe D, Balla A, Toth B, Balla T,
Weiss WA,
Williams RL, Shokat KM. A pharmacological map of the P13-K family defines a
role for
pl 1 Oalpha in insulin signaling. Cell 2006; 125:733-47) and Akti-1/2 (a
selective dual Akt1,2
inhibitor) (Barnett SF, Defeo-Jones D, Fu S, Hancock PJ, Haskell KM, Jones RE,
Kahana JA,
Kral AM, Leander K, Lee LL, Malinowski J, McAvoy EM, Nahas DD, Robinson RG,
Huber
HE. Identification and characterization of pleckstrin-homology-domain-
dependent and
isoenzyme-specific Akt inhibitors. Biochem J 2005; 385:399-408), both of which
induced
overt autophagy. Similar results were obtained with Bafilomycin Al, an
inhibitor of vacuolar
proton pump (V-H+-ATPase) that impairs lysosomal acidification. (Yamamoto A,
Tagawa Y,
Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin Al prevents
maturation of
autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes
in rat
hepatoma cell line, H-4-II-E cells. Cell Struct Funct 1998; 23:33-42).
Synergistic growth
inhibitory effect of CQ and Akt inhibitors have also been observed in an
expanded panel of
cancer cell lines.
The onset of cell death in cells treated with both CQ and Akt pathway
88
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
inhibitors was preceded by an accumulation of enlarged autolysosome-like
vacuoles.
Although both Akt inhibition and CQ alone induced accumulation of autophagic
vacuoles
(AVs) (Fig. 25 A-C), further analysis revealed that Akt inhibition alone
resulted in increased
production and maturation of cathepsin D and lysosomal activity, whereas CQ
blocked the
maturation of cathepsin D, leading to accumulation of very large and likely
degradation-
defective vacuoles in cells with Akt inhibition. This coincided with
activation of caspase 3
and a dramatic increase in apoptotic nuclei (Fig. 25 D,E). There was an
increase in
mitochondrial depolarization and reactive oxygen species (ROS) generation in
cells treated
with Akt inhibitor alone. Cytoplasmic ROS generation was attenuated within 48
hours with
Akt inhibitor alone, but CQ co-treatment caused a prolonged ROS accumulation
both in the
cytoplasm and in the vacuoles. Cytoplasmic translocation of cathepsin D due to
increased
lysosomal membrane permeability (LMP) and the subsequent degradation of
cytoplasmic
ROS scavenger thioredoxin was previously proposed as a mechanism of ROS
accumulation
and cell death induction by CQ in combination with another autophagy inducer.
(Carew JS,
Nawrocki ST, Kahue CN, Zhang H, Yang C, Chung L, Houghton JA, Huang P, Giles
FJ,
Cleveland JL. Targeting autophagy augments the anticancer activity of the
histone
deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood
2007;
110:313-22). Although LMP may still be a possible downstream event
contributing to cell
death, no significant increase in thioredoxin degradation prior to the onset
of cell death in our
system (Degtyarev and Lin, unpublished data) was observed. In contrast,
cathepsin D
knockdown or inhibition of lysosomal protease activity was not able to protect
cells from
CQ/Akti-1/2¨induced cell death, but rather had a similar effect to CQ in
promoting cell death
when combined with Alcti-1/2. This is consistent with the opposite effects of
CQ and Akti-
1/2 on cathepsin D maturation, and suggests that preserving (an elevated)
lysosomal
degradation activity may be critical for cell survival in the presence of
elevated autophagic
activity induced by Akt inhibition. Taken together, these findings support a
model whereby
limited mitochondrial depolarization caused by Akt inhibition resulted in an
increase in ROS
signal that promoted autophagy (Scherz-Shouval R, Shvets E, Fass E, Shorer H,
Gil L, Elazar
Z. Reactive oxygen species are essential for autophagy and specifically
regulate the activity
of Atg4. Embo J 2007; 26:1749-60), which in turn removed the damaged
mitochondria and
alleviated the oxidative stress. Impairment of autolysosomal digestion caused
by CQ resulted
in aggregation of deleterious oxidative products, which could further amplify
the ROS
damage (Moore MN, Viarengo A, Donkin P, Hawkins AJ. Autophagic and lysosomal
reactions to stress in the hepatopancreas of blue mussels. Aquat Toxicol 2007;
84:80-91).
89
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
Multiple downstream events, including caspase activation and possibly LMP can
lead to both
apoptosis-like and non-apoptotic cell death (Figure 24). Not to be bound by
theory, Figure
24 depicts a model of the mechanism of cell death by Akt inhibition in
combination with an
autophagy inhibitor (chloroquine).
Dosing of a pan-Akt inhibitor will likely be limited by its side effects, most
notably metabolic effects due to inhibition of insulin signaling. (Amaravadi
et al, 2005). Our
data suggest that at least in cancer models like the PTEN-null PC3 xenograft
tumors,
complete elimination of tumor cells may not be achievable with continuous pan-
Akt
knockdown alone. However, combined treatment with CQ significantly increased
the
incidence of complete tumor remission in xenograft models, although CQ alone
had no
significant effect. This suggests that autophagy induction through Akt
inhibition can
sensitize tumors to this relatively non-toxic drug, clinically approved for
other indications.
Inappropriate inhibition of autophagy could result in loss of its tumor
suppression function or may cause up-regulation of alternative survival
pathways. (Levine B.
Cell biology: autophagy and cancer. Nature 2007; 446:745-7; Wang Y, Singh R,
Massey AC,
Kane SS, Kaushik S, Grant T, Xiang Y, Cuervo AM, Czaja MJ. Loss of
macroautophagy
promotes or prevents fibroblast apoptosis depending on the death stimulus. J
Biol Chem
2008; 283:4766-77). Data herein suggest that the accumulation of defective
autolysosomes is
required for CQ's effect. One potential advantage of blocking degradation
while allowing the
autophagic sequestration to occur is that this may result in the formation of
more toxic ROS
generators in defective autolysosomes such as lipofuscin (Terman A, Gustafsson
B, Brunk
UT. The lysosomal-mitochondrial axis theory of postmitotic aging and cell
death. Chem Biol
Interact 2006; 163:29-37; Moore MN, Viarengo A, Donkin P, Hawkins AJ.
Autophagic and
lysosomal reactions to stress in the hepatopancreas of blue mussels. Aquat
Toxicol 2007;
84:80-91), therefore leading to a more rapid cell death induction. In
addition, cells may not
find an easy escape after they are already well engaged in an autophagic
response.
The PI3K/Akt pathway is crucial to many aspects of cell growth and survival
with multiple components targeted by genomic aberrations more frequently than
any other
pathway in human cancer, making it an attractive target for cancer therapy.
Critical questions
underlying the clinical outcomes of Akt inhibitors are the degree of
selectivity between the
three isoforms needed, and the effects on tumor cell growth and survival
expected from
inhibiting these kinases. The recently described allosteric Akt inhibitors
with unprecedented
selectivity towards Akt 1 and Akt2 provide valuable tools to begin addressing
these questions
(Barnett, S.F., M.T. Bilodeau, and C.W. Lindsley, (2005), The Akt/PKB family
of protein
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
kinases: a review of small molecule inhibitors and progress towards target
validation. Curr
Top Med Chem. 5:109-25). However, to date small molecule inhibitors are
limited in the
degree of specificity that can be achieved, and the in vivo efficacy of the
reported compounds
were not evaluated due to poor pharmacological properties. In addition, Akt3
selective
compounds have not been reported.
RNA interference is a powerful method for suppressing gene expression.
Using a Dox- inducible shRNA approach, we are able to achieve specific KD of
each Akt
isoform, both individually and in all possible combinations, to evaluate the
requirement of
each isoform in the maintenance of tumor growth in vivo. Data provided herein
results
suggest that in both Pten- androgen-independent prostate cancer model PC3 and
glioblastoma
model U87MG, Aktl is the most important isoform in maintaining tumor growth.
This is in
concert with the recent report that Akt 1 deficiency can markedly decrease the
incidence of
tumors in Pten+/- mice, both in tissues where Aktl is the predominantly
expressed isoform
and in those where Aktl is not (Chen et al., 2006). However, in the mouse
genetic study
Aktl was ablated prior to the development of tumors in Pten+/- mice, whereas
in the present
study we allowed the tumors to establish before Aktl KD was induced. Thus,
reducing Aktl
activity not only prevents tumors from developing, but also inhibits the
growth of established
tumors with PTEN deficiency in human cancer models.
Additional KD of Akt2 and Akt3 resulted in a more consistent and pronounced
inhibition of tumor growth. This suggests that Akt2 and Akt3 activities can
partially
compensate for the reduced Aktl activity in maintaining tumor growth. This is
consistent
with the more effective inhibition of downstream targets observed with
combined Akt KDs.
Taking together the recent reports of increased invasiveness associated with
inhibiting Aktl
alone that could be counteracted by simultaneous KD of Akt2, highly selective
Aktl
inhibition may not be desirable. The data provided herein suggest that partial
KD of all three
isoforms can be more effective in tumor growth inhibition. A plausible
scenario would be to
inhibit all three Akt isoforms but with different degrees of activity KD, thus
preserving a
crucial level of isoform activity for their normal physiological functions,
while achieving the
maximum inhibitory effect on tumor growth and progression.
One of the most prominent functions of Akt is to mediate cell survival.
Constitutively active Akt has been reported to protect cells from programmed
cell death
following various pro-apoptotic insults . Whether apoptosis is a primary
response to Akt
inhibition is however less clear, especially in cancer cells where apoptosis
is often suppressed
due to various genetic alterations. Previous experiments using small molecule
inhibitors of
91
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
the PI3KJAkt pathway often generate conflicting results that are obscured by
the inevitable
non-specific effects of these compounds. Data provided herein indicate that
under normal
cell culture conditions, specific KD of any or all three isoforms of Akt can
result in cell cycle
delay without promoting significant apoptosis. This is consistent with a
recent report that
only a small portion of total Akt activity is required for apoptosis
inhibition under normal
growth condition (Liu, X., Y. Shi, M.J. Birnbaum, K. Ye, R. De Jong, T.
Oltersdorf, V.L.
Giranda, and Y. Luo, (2006), Quantitative analysis of anti-apoptotic function
of Akt in Aktl
and Akt2 double knock-out mouse embryonic fibroblast cells under normal and
stressed
conditions. J Biol Chem. 281:31380-8). In contrast, significantly increased
autophagy was
observed in both PC3 and U87MG cells with Akt KDs, suggesting that autophagy
is a more
sensitive response to reduced Akt activity. This is further demonstrated by
using relatively
specific inhibitors of the pathway, including a dual PI3K/mTOR inhibitor and a
dual Akt1,2
inhibitor.
Although the molecular mechanisms of Akt inhibition-induced autophagy
remains to be further elucidated, several possibilities exist. First,
inhibiting Akt can lead to
inhibition of mTOR, which is a known inhibitor of autophagy. Interestingly, a
constitutively
active form of Akt was shown to suppress the induction of autophagy by
rapamycin
(Takeuchi, H., Y. Kondo, K. Fujiwara, T. Kanzawa, H. Aoki, G.B. Mills, and S.
Kondo,
(2005), Synergistic augmentation of rapamycin-induced autophagy in malignant
glioma cells
by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res.
65:3336-46),
raising the possibility that the effect of Akt on autophagy may not be
completely mediated
through the raptor-mTOR activity downstream of Akt, or that the effect of
rapamycin may be
mediated at least in part through inhibiting Akt, e.g. through inhibition of
the assembly of
mTORC2 after prolonged treatment (Sarbassov, D.D., S.M. Ali, S. Sengupta, J.H.
Sheen, P.P.
Hsu, A.F. Bagley, A.L. Markhard, and D.M. Sabatini, (2006), Prolonged
rapamycin treatment
inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 22:159-68. Epub 2006 Apr 6).
Second,
it is possible that other signaling outputs of Akt, such as glucose uptake and
metabolism, or
cell cycle regulation, can also contribute to autophagy regulation independent
of mTOR. Of
note, Akt inhibition stabilizes p27kip1, which was recently shown to mediate
autophagy
under growth factor withdrawal (Liang, J., S.H. Shao, Z.X. Xu, B. Hennessy, Z.
Ding, M.
Larrea, S. Kondo, D.J. Dumont, J.U. Gutterman, C.L. Walker, J.M. Slingerland,
and G.B.
Mills, (2007), The energy sensing LKB1-AMPK pathway regulates p27(kipl)
phosphorylation mediating the decision to enter autophagy or apoptosis. Nat
Cell Biol. 9:218-
24). Third, data provided herein indicate that Akt inhibition induces
mitochondria membrane
92
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
depolarization and increased ROS generation. It was recently shown that
starvation
stimulates formation of ROS in the mitochondria, which serves as a signal to
activate
autophagy (Scherz-Shouval, R., E. Shvets, E. Fass, H. Shorer, L. Gil, and Z.
Elazar, (2007),
Reactive oxygen species are essential for autophagy and specifically regulate
the activity of
Atg4. Embo J. 26:1749-60). It is conceivable that Akt inhibition can induce
autophagy via a
similar mechanism, and elevated autophagy in turn recycles these damaged
mitochondria and
prevents the accumulation of ROS to a detrimental level.
Although excessive autophagy may lead to cell killing when allowed to reach
its limit, inhibiting Akt alone is apparently very ineffective in cell killing
even under reduced
serum conditions in the PTEN-null cancer cell lines that we examined. Under
the in vivo
tumor growth conditions, autophagy may be a potential mechanism by which Akt
inhibition
restricts tumor growth, but may also provide temporary relief from the
metabolic and
oxidative stress imposed by Akt inhibition, which may allow resistance to
occur. Indeed,
most tumors treated with Akt KD alone became resistant and rebound after
initial regression
or stasis. Inhibiting autophagy at an early stage may prevent this temporary
protective effect,
but may also counteract the possible tumor inhibitory effect of autophagy.
Blocking
autophagy completion at a late stage might avoid this counteracting effect.
Indeed,
combination of Akt inhibition with lysosomotropic agents resulted in excessive
accumulation
of degradation-defective autolysosome-like vacuoles that cannot be cleared,
resulting in
accelerated cell death. This combination can not only sabotage the ROS
scavenger and self-
renewal functions of autophagy, but also promote the rupture of defective
autolysosomes and
the release of lysosomal contents into the cytosol, further augmenting the
oxidative stress and
mitochondrial damage, leading to eventual cell death. Recently, it was
reported that
inhibition of autophagy that was induced as an adaptive survival response to
therapy could
enhance apoptosis in a Myc-induced mouse model of lymphoma (Amaravadi et al.,
2007).
As reported herein, autophagy induced by Akt/PI3K inhibition can be exploited
using
lysosomotropic agents to promote the remission of PTEN-null human tumor
xenografts.
Since this effect is expected to correlate positively with the degree of
autophagy induced by a
given treatment, creative combination of lysosomotropic agents with agents
that induce
extensive autophagy, such as inhibitors of the Akt pathway, may profoundly
affect their anti-
cancer efficacy. Degenhardt et al. proposed that autophagy inhibition by Akt
overexpression
could lead to necrosis in the center of tumors while the surrounding tumor
cells might
respond with accelerated growth as a result of combined effect of necrosis-
induced
inflammatory response and Akt-stimulated proliferation . In the presence of
Akt inhibition,
93
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
however, accelerated cell death caused by CQ-induced late autophagy inhibition
might enable
completely eliminate tumor cells before they have time to grow back due to
possible
inflammatory response, because tumor cell proliferation is greatly reduced.
Data provided
herein show that cells with PTEN deficiency are more sensitive to this
combination than cells
with intact PTEN, suggesting that a reasonable therapeutic window might be
achieved. CQ
has already found therapeutic efficacy in several diseases and is well
tolerated (Gustafsson,
L.L., 0. Walker, G. Alvan, B. Beermann, F. Estevez, L. Gleisner, B. Lindstrom,
and F.
Sjoqvist, (1983), Disposition of chloroquine in man after single intravenous
and oral doses.
Br J Clin Pharmacol. 15:471-9; Hagihara, N., S. Walbridge, A.W. Olson, E.H.
Oldfield, and
R.J. Youle, (2000), Vascular protection by chloroquine during brain tumor
therapy with Tf-
CRM107. Cancer Res. 60:230-4). Given the lengthening list of anti-cancer
agents reported to
induce autophagy, CQ and other lysosomotropic agents may find promising new
therapeutic
values in cancer therapy.
EXAMPLES
Materials and Methods
Cell Culture and Reagents: The PTEN' - and PTEN'+ MEFs were maintained
as previously described (Sun, H., R. Lesche, D.M. Li, J. Liliental, H. Zhang,
J. Gao, N.
Gavrilova, B. Mueller, X. Liu, and H. Wu, (1999), PTEN modulates cell cycle
progression
and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and
Akt/protein
kinase B signaling pathway. Proc Nall Acad Sci US A. 96:6199-204). The PC3 and
U87MG
cells were maintained at 37 C and 5% CO2 in DMEM/Ham's F-12 (1:1) containing
10%
tetracycline-free fetal bovine serum. 11-4 was from Calbiochem (Akt inhibitor
VIII) (Barnett,
S.F., D. Defeo-Jones, S. Fu, P.J. Hancock, K.M. Haskell, R.E. Jones, J.A.
Kahana, A.M.
Kral, K. Leander, L.L. Lee, J. Malinowski, E.M. McAvoy, D.D. Nahas, R.G.
Robinson, and
H.E. Huber, (2005), Identification and characterization of pleckstrin-homology-
domain-
dependent and isoenzyme-specific Akt inhibitors. Biochem J. 385:399-408). To
inhibit
autophagy, cells were treated with 5-10 jiM chloroquine, 2.5 nM Bafilomycin Al
or 1 mM 3-
MA (all from Sigma) and analyzed at the indicated time points. Image-iT LIVE
Green
Reactive Oxygen Species Detection Kit was purchased from Molecular Probes.
MitoPT
Mitochondria Permeability Transition Detection Kit was purchased from
Immunochemistry
Technologies, LLC.
Inducible shRNA constructs and generation of inducible-shRNA clones: The
pHUSH tetracycline-inducible retrovirus gene transfer vector has been
described elsewhere
94
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
(Gray, D., A.M. Jubb, D. Hogue, P. Dowd, N. Kljavin, S. Yi, W. Bai, G. Frantz,
Z. Zhang, H.
Koeppen, F.J. de Sauvage, and D.P. Davis, (2005), Maternal embryonic leucine
zipper
kinase/murine protein serine-threonine kinase 38 is a promising therapeutic
target for
multiple cancers. Cancer Res. 65:9751-61; Hoeflich, K.P., D.C. Gray, M.T. Eby,
J.Y. Tien,
L. Wong, J. Bower, A. Gogineni, J. Zha, M.J. Cole, H.M. Stern, Li. Murray,
D.P. Davis, and
S. Seshagiri, (2006), Oncogenic BRAF is required for tumor growth and
maintenance in
melanoma models. Cancer Res. 66:999-1006; US 2007/0026002, herein incorporated
by
reference in its entirety). The complementary double-stranded shRNA oligos
were inserted
into this vector system using a shuttle vector followed by a Gateway
recombination reaction
(Invitrogen) as previously described (Grunwald, V., L. DeGraffenried, D.
Russel, W.E.
Friedrichs, R.B. Ray, and M. Hidalgo, (2002), Inhibitors of mTOR reverse
doxorubicin
resistance conferred by PTEN status in prostate cancer cells. Cancer Res.
62:6141-5; See also
Figure 23 herein). The shRNA sequences used in this study are summarized in
Table 2. All
constructs were verified by sequencing. Retrovirus infection was performed as
described
(Gray et al., 2005; Hoeflich et al., 2006). For single Akt isoform knockdowns,
cells were
infected with one retroviral vector encoding an shRNA construct singly
targeting each Akt
isoform (constructs 252 & 253 for Aktl, 254 & 255 for Akt2, and 259 & 260 for
Akt3) and
stable clones were selected using 5 p,g/m1 puromycin. For dual Aktl and Akt2
KD, a single
shRNA targeting both Aktl and 2 simultaneously (construct 256 & 257) was used.
Dual Akt2
and 3 (constructs 255 and 261), or triple Aktl, 2 and 3 (constructs 257 and
261) knockdowns
were achieved by co-infecting the cells with two retroviral vectors containing
different
antibiotic selection markers (puromycin and hygromycin), each encoding one
single shRNA,
and stable clones were selected using 5 g/ml puromycin and 300 pg/ml
hygromycin. For
dual Aktl and 3 KD, either a single shRNA targeting both Aktl and 3 (construct
258), or co-
infection with two shRNA vectors (constructs 253 and 261) were employed.
Western blot analysis, immunofluorescence, IHC and TUNEL assay: For
Western blot analysis, total protein lysates were subjected to SDS-PAGE and
transferred to
nitrocellulose. Antibodies used were: anti-Aktl, anti-Akt2, anti-Akt3, anti-
total-Akt, anti-p-
Akt (Ser473), anti-p-Akt (Thr308), anti-p-S6 (Ser235/236), anti-PARP and anti-
cleaved
caspase-3 (Cell Signaling Technology); anti-p-PRAS40 (Invitrogen); anti-p27'
(Santa Cruz
Biotechnology); anti-LC3 (Novus); anti-LAMP2 and anti-Cathepsin D (BD
Biosciences); and
anti-GAPDH (Advanced Immunochemical Inc.). Primary antibodies were detected
using IR
Dye 800-conjugated (Rockland) and Alexa-Fluoro 680-conjugated (Molecular
Probes)
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
species-selective secondary antibodies. Detection and quantification were
performed using an
Odyssey infrared scanner (LICOR) using the manufacturer's software. For
immunofluorescence staining, cells were fixed in 3% paraformaldehyde and
permeabilized
with 0.01% digitonin in PBS, followed by a rabbit polyclonal anti-LC3 (Abgent)
primary
antibody detected with a cy3-conjugated anti-rabbit secondary antibody
(Jackson
Immunoresearch). For IHC, formalin-fixed, paraffin-embedded specimens were
collected. 5-
p.m-thick paraffin-embedded sections were stained using an anti-Ki-67 (MIB-1,
DakoCytomation) antibody with the Dako ARK kit (Dako Corporation). Tissues
were
counterstained with haematoxylin, dehydrated, and mounted. In all cases,
antigen retrieval
was performed with the Dako Target Retrieval Kit as per manufacturer's
instructions. For
TUNEL assay, formalin-fixed, paraffin-embedded sections were stained using an
in situ cell
death detection kit (POD; Roche Diagnostic) according to the manufacturer's
instructions.
Xenograft study: Six- to 8-week-old female athymic nude nu/nu mice were
purchased from Charles River Laboratories and maintained in Genentech's
conventional
animal facility. Mice were injected in the right flank with 5-7.5 x 106 cells
resuspended in
200 pi Hank's Balanced Salt Solution (Invitrogen). When tumors reached a mean
volume of
100-300 mm3 the mice with similarly-sized tumors were grouped into treatment
cohorts.
Mice received 5% sucrose or 5% sucrose plus 1 mg/ml Dox in drinking water for
control and
KD cohorts, respectively. Amber-colored water bottles were used and were
changed 3 times
per week. CQ is dissolved in 0.9% physiological saline, filter-sterilized and
administered at
45 mg/kg through either intraperitoneal or subcutaneous routes. Tumors were
measured with
calipers and mice weighed twice per week. Mice whose tumors reached 2000 mm3
or lost
more than 20% body weight were euthanized. Between 8-10 mice were used for
each
treatment group. Statistical significance was analyzed using the JMP software
(SAS Institute,
Inc.).
Electron Microscopy: Cells were grown to monolayer in plastic flasks and
fixed in half-strength Karnovsky fixative (2% paraformaldehyde, 2.5%
glutaraldehyde,
0.025% CaC12.2H20 and 0.1 M sodium cacodylate buffer, pH 7.4); tumors were cut
into
small cubes (-1 mm3) and fixed by immersion in the same fixative or in the
fixative used for
immunoelectron microscopy. Cells and tissues were postfixed with 1 % Osat and
1 %
K4Ru(II)(CN)6 or 1.5 % K3Fe(CN)6, dehydrated in ethanol and embedded in Epon.
Ultrathin
sections were stained with uranyl acetate and lead citrate. Numbers of AV were
counted on
systematically sampled cytoplasmic areas of 4.5 p.m2 (n>64 per condition). The
percent AV
96
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
area was measured by means of a square mesh grid laid over > 5 sets of
systematically
sampled micrographs with each set covering a cytoplasmic area > 80 m2. The
average
percent of apoptotic nuclei in tumor tissues was calculated from the number of
apoptotic
nuclei in 3 to 4 sets of 100 systematically counted tumor cell nuclei.
Immunoelectron microscopy. Small tumor blocks were fixed by immersion in
2 % paraformaldehyde, 0.2 % glutaraldehyde in 0.1 M phosphate buffer, pH 7.4
for 5 h at 4
C. After rinsing with PBS, the blocks were embedded in 12 % gelatin,
cryoprotected with
2.3 M sucrose, and frozen in liquid nitrogen. Ultrathin cryosections were cut
at -120 C,
picked up with 1 % methylcellulose, 1.2 M sucrose, thawed and collected on
copper grids.
After washing with PBS containing 0.02 M glycine, sections were incubated with
rabbit anti-
human LAMP1 antibodies (gift of M. Fukuda, (Carlsson, S.R., J. Roth, F.
Piller, and M.
Fukuda. 1988. Isolation and characterization of human lysosomal membrane
glycoproteins,
h-lamp-1 and h-lamp-2. Major sialoglycoproteins carrying
polylactosaminoglycan. J Biol
Chem. 263:18911-9) or with rat monoclonal anti-mouse LAMP-1 antibody ID4B (T.
August,
Developmental Studies Hybridoma Bank, Iowa City, IA), followed by a secondary
rabbit
anti-rat IgG antibody (Dako). These were subsequently labeled with Protein A
conjugated to
10 nm colloidal gold particles, and contrasted with a 1.8 % methylcellulose,
0.6 % uranyl
acetate mixture.
Cell viability and cell cycle analysis. Cell number and viability was measured
using trypan blue exclusion assay using a Vi-Cell Analyzer (Beckman Coulter),
or labeled
with 1 1..tg/m1 PI in PBS/1% BSA followed by cytofluorometric analysis with a
fluorescence-
activated cell sorter (FACS) (Becton Dickinson). FITC-conjugated Annexin V was
used for
the assessment of phosphatidylserine exposure by FACS analysis. Caspase
activation was
analyzed using a Caspase-Glo 3/7 Assay kit (Promega). For cell cycle analysis,
cells were
fixed with drop-wise addition of chilled 70% ethanol, washed with PBS and
resuspended in
staining solution containing 50 gg/m1 PI and 60 units of RNAse A. DNA content
was
analyzed by flow cytometry using the FlowJo and ModFit software (Becton
Dickinson).
Multispectral imaging flow cytometry: Cells treated with various agents were
stained with Acridine Orange and analyzed by the ImageStream system (Amnis
Corporation,
Seattle, WA) using the IDEAS image analysis program. The DNA AOGreen image and
the
vacuolar AO Red image were first compensated into separate channels, and then
the
percentage of apoptotic/anucleate cells (based on AO nuclear morphology and
intensity) and
vacuolated cells (AO Red+) were quantified. Plotting AO Green Intensity vs AO
Green
97
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
bright detail area revealed three distinct populations: R2 anucleated cells
(low AO Green
labeling, higher area due to masking of diffuse cytoplasm); R3 apoptotic cells
(intermediate
to low AO Green, very low AO Green detail area due to presence of small,
bright
condensated nuclear fragments); R4 live cells (intact bright nucleus). AORed
Intensity is
plotted on the second histogram with an arbitrary gate (R5) drawn to include
events with the
brightest AO Red intensity.
Time-lapse video microscopy: Cells cultured in 24-well plates were imaged on
an Olympus IX81 inverted microscope under environmental control (37 C and 5%
CO2) for
3 days. Imaging started 6 hours after the addition of the compounds and was
taken at 1-hour
intervals.
Example 1
Inducible shRNA KD of Akt isoforms inhibited the growth of PTEN-null
human tumor xenografts in a dose- and isoform dependent manner.
To determine the relative contribution of the three Akt isoforms in
maintaining
tumor growth, we used a tet-inducible shRNA KD method using a recently
described
retroviral vector system, a tet-inducible plasmid vector for HI or U6 short
hairpin (Gray et
al., 2005; Hoeflich et al., 2006). We chose the PTEN-null human prostate
cancer cell line
PC3 and the glioma cell line U87MG (Li, J., C. Yen, D. Liaw, K. Podsypanina,
S. Bose, S.I.
Wang, J. Puc, C. Miliaresis, L. Rodgers, R. McCombie, S.H. Bigner, B.C.
Giovanella, M.
Ittmann, B. Tycko, H. Hibshoosh, M.H. Wigler, and R. Parsons, (1997), PTEN, a
putative
protein tyrosine phosphatase gene mutated in human brain, breast, and prostate
cancer.
Science. 275:1943-7). Both lines express all three Akt isoforms; in PC3 cells,
Akt 1 protein is
expressed at approximately two times the level of Akt2, with Akt3 contributing
to <10% of
total Akt, whereas in U87MG cells, all three Akt proteins are expressed at
equivalent levels
(Fig. 18 A). Stable clones of PC3 and U87MG cells were generated harboring
inducible
shRNA constructs targeting all possible single and combined Akt isoforms
(Table 2). Each
Akt-targeting shRNA (shAkt) caused ¨75-99% KD of the corresponding Akt mRNA
and
proteins upon doxycycline (Dox) induction (Fig. 1 A, Fig. 18 B, and Table 3).
Decreased
steady-state phosphorylation of downstream targets PRAS40 and S6, up-
regulation of
p27KIP1, and feedback stabilization of IRS1 were observed to varying degrees
in response to
the KDs, with the strongest effects observed in cells with all three Akt KDs
(Fig. 1 A).
We next examined the effect of Akt KDs on the ability of PC3 cells to
maintain the growth of established tumors in vivo. Dox-induced KD of Akt2
(shAkt2) or
Akt3 (shAkt3) alone did not result in significant inhibition of tumor growth
(Fig. 1 B and
98
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
Table 3). In contrast, two different shRNA constructs targeting Akt 1 (shAktl)
both showed
significant tumor growth inhibition, each in two out of three independent
clones. Tumor
growth retardation or stasis was typically observed in these clones (Fig. 1 B,
Fig. 18 H, and
Table 3). Simultaneous KD of Akt1,2 (shAkt12) or Akt1,3 (shAkt13) also
inhibited tumor
growth, with almost all tumor growth halted and tumor regression observed in
several of the
Dox-treated mice. Interestingly, KD of both Akt2 and Akt3 (shAkt23) also
resulted in
significant tumor growth inhibition, with no tumor volume doubling during the
2 wk of Dox
treatment, suggesting that Aka activity alone is not sufficient to maintain
optimal tumor
growth. Finally, triple-Akt KD (shAktI23) most effectively inhibited tumor
growth, with
consistent tumor regression observed during the first 2 wk of treatment.
Similar results were
observed in U87MG cells, which express similar levels of the three Akt
isoforms. Among the
three single KDs, only shAkt1 showed significant tumor stasis, and tumor
regression was
again observed with triple-Akt KD (Fig. 18, C-F). Thus, KD of Aktl alone can
inhibit tumor
growth in both PC3 and U87MG xenografts, and this Aka dependency is not simply
a total
Akt dose effect. More pronounced tumor growth inhibition and regression,
however, occurs
in tumors with KD of all three Akt isoforms.
Table 1 DNA oligo sequences used in Akt shRNA vectors (pShuttle-H1 and pHUSH-
GW)
Target Construct DNA OLIGO LIGATED INTO PSHU1TLE-H1 (ALL IN 5'-3 SEQ
ID
gene # DIRECTION, THE 19BP TARGET SEQUENCES ARE No.
BOLDED)
Aktl 252 sense 5'- 1
GATCCCCTGACCATGAACGAGTTTGATTCAAGAGA
TCAAACTCGTTCATGGTCATTTTTTGGAAA-3'
Anti- 5'- 2
AGCTTTTCCAAAAAATGACCATGAACGAGTTTGATC
sense
TCTTGAATCAAACTCGTTCATGGTCAGGG-3'
Aka 253 sense 5'- 3
GATCCCCGTGGACCACTGTCATCGAATTCAAGAGA
TTCGATGACAGTGGTCCACTTTTTTGGAAA-3'
99
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
Anti- 5'- 4
AGCTTTTCCAAAAAAGTGGACCACTGTCATCGAATC
sense
TCTTGAATTCGATGACAGTGGTCCACGGG-3'
Akt2 254 sense 5'- 5
GATCCCCCCTGGAGGCCACGGTACTTTTCAAGAGA
AAGTACCGTGGCCTCCAGGTTTTTTGGAAA-3'
Anti- 5'- 6
AGCTTTTCCAAAAAACCTGGAGGCCACGGTACTTTC
sense
TCTTGAAAAGTACCGTGGCCTCCAGGGGG-3'
Akt2 255 sense 5'-GATCCCCTGACTTCGACTATCTCAAATTCAAGAGA 7
TTTGAGATAGTCGAAGTCATTTTTTGGAAA-3'
Anti- 5'- 8
AGCTTTTCCAAAAAATGACTTCGACTATCTCAAATC
sense
TCTTGAATTTGAGATAGTCGAAGTCAGGG-3'
Akt3 259 sense 5'-GATCCCCGAATTGTAGTCCAACTTCATTCAAGAGA 9
TGAAGTTGGACTACAATTCTTTTTTGGAAA-3'
Anti- 5'- 10
AGCTTTTCCAAAAAAGAATTGTAGTCCAACTTCATC
sense
TCTTGAATGAAGTTGGACTACAATTCGGG-3'
Akt3 260,261 sense 5'-GATCCCCGCACTTTTGGGAAAGTTATTTCAAGAGA 11
ATAACTTTCCCAAAAGTGCTTTTTTGGAAA-3'
Anti- 5'- 12
AGCTTTTCCAAAAAAGCACTTTTGGGAAAGTTATTC
100
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
sense TCTTGAAATAACTTTCCCAAAAGTGCGGG-3'
Akt 1 256 sense 5'- 13
GATCCCCGCTACTACGCCATGAAGATTTCAAGAGA
Akt2
ATCTTCATGGCGTAGTAGCTTTTTTGGAAA-3'
Anti- 5'- 14
AGCTMCCAAAAAAGCTACTACGCCATGAAGATTC
sense
TCTTGAAATCTTCATGGCGTAGTAGCGGG-3'
Akt 1 257 sense 5'- 15
GATCCCCAGGTGCTGGAGGACAATGATTCAAGAGA
Akt2
TCATTGTCCTCCAGCACCTTTTTTTGGAAA-3'
Anti- 5'-AGCTTTTCCAAAAAAAGGTGCTGGAGGACAATGA 16
sense TCTCTTGAATCATTGTCCTCCAGCACCTGGG-3'
Akt 1 258 sense 5'- 17
GATCCCCCTACAACCAGGACCATGAGTTCAAGAGA
Akt2
CTCATGGTCCTGGTTGTAGTTTTTTGGAAA-3'
Anti- 5'- 18
AGCTTTTCCAAAAAACTACAACCAGGACCATGAGT
sense
CTCTTGAACTCATGGTCCTGGTTGTAGGGG-3'
101
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
Table 2 Summary of Akt shRNA constructs used and effective targeting sequences
Effective siRNA 19bp Seq
Construct
Target Selection Target sequenceb
core sequence ID
No.
5.-TGACCATGAA
Akt1 252 Pur sense UGACCAUGAACGAGUUUGA 19
CGAGTTTGA
anti-
UCAAACUCGUUCAUGGUCA 20
sense
5'-GTGGACCAC
Akt1 253 Pur sense GUGGACCACUGTCAUCGAA 21
TGTCATCGAA
anti-sense UUCGAUGACAGUGGUCCAC 22
5'-CCTGGAGGC
Akt2 254 Pur sense CCUGGAGGCCACGGUACUU 23
C ACGGTACTT
anti-sense AAGUACCGUGGCCUCCAGG 24
5'-TGACTTCGAC
Akt2 255 Pur sense UGACUUCGACUAUCUCAAA 25
T ATCTCAAA
anti-sense UUUGAGAUAGUCGAAGUCA 26
5.-GAATTGTAGT
Akt3 259 Pur sense GAAUUGUAGUCCAACUUCA 27
C CAACTTCA
anti-sense UGAAGUUGGACUACAAUUC 28
5-GCAC1ITTGG
Akt3 260 Pur sense GCACUUUUGGGAAAGUUAU 29
GAAAGTTAT
anti-sense AUAACUUUCCCAAAAGUGC 30
5-GCAC1ITTGG
Akt3 261 Hyg
GAAAGTTAT sense GCACUUUUGGGAAAGUUAU 31
102
CA 02821378 2013-06-12
WO 2012/087336 PCT/US2010/062096
anti-sense AUAACUUUCCCAAAAGUGC 32
5'-GCTACTACGC
Akt12 256 Pur sense GCUACUACGCCAUGAAGAU 33
CATGAAGAT
anti-sense AUCUUCAUGGCGUAGUAGC 34
5-AGGTGCTGGA
Akt12 257 Pur sense AGGUGCUGGAGGACAAUGA 35
GGACAATGA
antisense AGGUGCUGGAGGACAAUGA 36
5'-CTACAACCAG
Akt13 258 Pur sense CUACAACCAGGACCAUGAG 37
GACCATGAG
anti-sense CUCAUGGUCCUGGUUGTAG 38
253+ Pur + 253 and 261
Akt13 sense 253 and 261 core
sequences
261 Hyg target sequences
anti-sense 253 and 261 core
sequences
255 + Pur + 255 and 261
Akt23 sense 255 and 261 core
sequences
261 Hyg target sequences
anti-sense 255 and 261 core
sequences
257+ Pur + 257 and 261
Akt123 sense 257 and 261 core
sequences
261 Hyg target sequences
anti-sense 257 and 261 core
sequences
a Antibiotic selection used to establish stable expression of the shRNA. Pur,
puromycin; Hyg,
Hygromycin.
b Sense sequence in the target (all in 5'-3' direction).
siRNA duplexes or shRNA hairpins containing these 19bp core sequences should
also be
effective against the indicated genes (both sense and sense sequences are in
5'-3' direction)
103
CA 02821378 2013-06-12
WO 2012/087336 PCT/US2010/062096
Table 3 Summary of Akt knockdown efficiency and effect on xenograft tumor
growth for
various PC3-Akt shRNA clones
Target Construct Clone Treatment % Akti a % Akt2a % Akt3a n Pc Rd %
TGI DRS
b f % TGI
(d14)e (DRS)e
Akt1 252 2 Dox- 100 100 100 8 6 0
Dox+ 21.9 1.1 93.8 10.2 151.0 58.3 8 8 0 -14 >21 -
252 9 Dox- 100 100 100 8 5 0
Dox+ 22.3 2.3 122.7 11.3 120.3 8.6 8 2 1 72 21
81*
252 10 Dox- 100 100 100 8 8 0
Dox+ 11.2 2.6 89.1 13.9 106.8 3.2 8 5 0 52 20 58*
253 17 Dox- 100 100 100 8 7 0
Dox+ 11.6 1.2 80.1 9.7 106.1 4.3 8 2 3 97* 13 97*
253 25 Dox- 100 100 100 8 5 1
Dox+ 13.9 5.0 82.3 1.3 92.9 5.3 8 7 0 26 >21 -
253 29 Dox- 100 100 100 1 10 0
0
Dox+ 5.9 0.1 70.3 5.6 98.0 1.0 1 8 0 56* 5 56*
0
Akt2 255 4 Dox- 100 100 100 8 7 0
Dox+ 88.0 16.5 11.6 2.4 116.4 32.0 8 5 2 -7 >21 -
255 23 Dox- 100 100 100 1 10 0
0
Dox+ 105.0 6.1 10.4 1.3 106.3 8.0 1 9 0 22 >21 -
0
Akt3 260 6 Dox- 100 100 100 8 6 1
Dox+ 101.0 4.0 95.5 10.5 10.0 4.0 8 4 1 45 >21 -
Akt12 257 4 Dox- 100 100 100 8 3 2
Dox+ 5.7 0.9 13.2 1.1 129.6 11.3 8 0 5 137* 14 137*
257 6 Dox- 100 100 100 8 7 0
Dox+ 6.6 0.3 12.3 1.6 128.9 18.3 8 1 4 80* 11 90*
Akt13 253+261 2 Dox- 100 100 100 8 8 0
Dox+ 5.1 2.0 66.0 2.6 18.0 3.7 8 1 3 102* 7 120*
Akt23 255+261 8 Dox- 100 100 100 8 6 0
_
Dox+ 96.8 18.8 6.3 0.3 9.9 2.1 8 0 0 82* 7 93*
Akt123 257+261 10 Dox- 100 100 100 1 8 2
0
Dox+ 1.2 0.3 9.5 0.7 4.5 2.1 1 0 6 116* 6 135*
0
257+261 12 Dox- 100 100 100 8 6 0
Dox+ 9.0 3.6 21.7 2.9 12.7 0.9 8 0 5 116* 7 110*
257+261 12 Dox- 100 100 100 1 5 0
0
Dox+ 9.0 3.6 21.7 2.9 12.7 0.9 1 1 4 106* 10 129*
0
eGFP 310 3 Dox- 100 100 100 8 6 1
Dox+ 93.7 13.2 95.0 6.4 87.3 4.7 8 7 0 -90 >21 -
104
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
a Percentage of message level after 72 hours of Dox treatment compared to
untreated control determined by real-
time quantitative RT-PCR (Taqman). Data represent mean SEM of at least 3
independent experiments.
b Number of tumors analyzed in each cohort.
'Number of tumors progressed by d14, defined by tumor volume > 2 fold of the
initial size at the start of
treatment.
d Number of tumors regressed by d14, defined by tumor volume < 50% of the
initial size at the start of
treatment.
'Percentage of tumor growth inhibition (TGI) at day 14 or the first day when
significant difference was
achieved, calculated as % TGI = % [Vc(dx-d0)-Vt(dx-d0)1/Vc(dx-d0)*100, where
Vc(dx-d0) is the difference in
mean tumor volume of the control cohort (Vc) between the day of analysis (dx)
and the day when treatment
started (d0), and Vt(dx-d0) is the difference in mean tumor volume of the
treated cohort (Vt) between the day of
analysis and the day when treatment started. % TGI > 100 indicates tumor
regression.
f Day to Reach Significance, number of days taken after treatment before
significant difference between the
control and the treatment group is achieved.
*P <0.05 compared to sucrose vehicle treated group, determined by Student's t
test.
105
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
Example 2
Akt KDs induced cell cycle delay without significant apoptosis
Analysis of PC3 tumors with Akt KDs revealed a mild decrease in the
proliferation marker Ki-67 and no significant increase in TUNEL-positive cells
compared
with control tumors (Fig. 2 A). The lack of apoptosis was also observed in PC3
cells cultured
in vitro. Under 10% FBS, a mild increase in GO/G I and a decrease in S phase
was observed
in cells expressing each shAkt construct. Slightly increased accumulation of
cells in the
G2/M phase was also observed in cells expressing shRNA for Aktl alone and any
combinations of two or three Akt isoforms, suggesting a cell cycle delay in
both DNA
replication and mitosis in these cells. However, no significant sub-G1
population was
observed with any of the KDs (Fig. 19, A and B). Additional experiments also
failed to
detect significant caspase activation in response to Akt KDs in both PC3 and
U87MG cells.
To partially mimic the suboptimal growth condition in the in vivo environment,
we starved
the cells of serum in culture and asked whether the cells became more
sensitive to Akt KD.
Indeed, complete serum starvation or reducing serum to 0.5% resulted in
markedly increased
accumulation of cells in the 00/01 phase. However, still no significant sub-01
peak was
observed for at least 2 d in 0% FBS and 5 d in 0.5% FBS (Fig. 2, B and C).
Example 3
Akt KD promoted autophagy in PC3 and U87MG cells
Because Akt has been shown to inhibit autophagy (Arico, S., A. Petiot, C.
Bauvy, P.F. Dubbelhuis, A.J. Meijer, P. Codogno, and E. Ogier-Denis, (2001),
The tumor
suppressor PTEN positively regulates macroautophagy by inhibiting the
phosphatidylinositol
3-kinase/protein kinase B pathway. J Biol Chem. 276:35243-6. Epub 2001 Jul 26;
Degenhardt et al., 2006), we asked whether specific KD of endogenous Akt could
promote
autophagy. Indeed, EM analysis revealed a significantly increased accumulation
of AV s in
both PC3 and U87MG cells induced to express shAkt123 (Fig. 3, A and B; and
Fig. 19 C).
The accumulation of AV and acidic vesicular organelles (AVOs) was further
confirmed by
localization of the autophagosome marker GFP-LC3, staining with an anti-LC3
antibody, and
fluorescent dyes monodansylcadaverine (MDC) and acridine orange (AO; Fig. 4 A,
Fig. 19,
D-G).
We examined xenograft tumors expressing shAkt by EM. The control GFP-
targeting shRNA-expressing PC3 tumors consist of healthy looking cells
connected by cell-
cell junctions (Fig. 3 C, a). In contrast, cells in the shAkt123-expressing
tumors exhibit
106
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
morphological signs of degeneration and loss of cell-cell contact after 10-15
d of Dox
treatment (Fig. 3 C, b). Late AV s positive for human lysosome-associated
membrane protein
(LAMP)1 are found in degenerating tumor cells (Fig. 3 C, b-d). Also, these
cells often
contain swollen mitochondria and dilated RER that are drastically
disorganized, suggesting a
connection between energy metabolism, ER stress, and autophagy. Chromatin
clumping and
fragmentation characteristic of typical apoptosis are rarely observed in the
degenerating
tumor cells; instead, some AV-containing cells exhibit mild pylcnosis typical
of cells
undergoing autophagic degeneration (Fig. 3 C, b and d).
To determine whether AV accumulation occurred in tumor cells before
morphological signs of degeneration, we examined U87MG tumors with either 5 d
or 3 wk of
Akt KD. In tumors expressing shA.kt 123 for 5 d, most cells showed similar
gross
morphology to vehicle-treated controls, but with an approximately two times
increase in the
percent AV area (from 0.78% in the control tumors to 1.53% in Dox-treated
tumors; P <
0.05; Fig. 3 C, e-h). After 3 wk of Akt KD, U87MG tumors show signs of
degeneration in
many cells similar to PC3 tumors treated for 15 d (unpublished data).
Example 4
Lysosomotropic agents accelerated cell death in PC3 cells with Akt KD
Despite the elevated levels of autophagy and mild cell cycle delay, PC3 cells
expressing shAlct123 can survive in culture for many passages under 10% FBS
without
appreciable increase in cell death. Even under reduced serum (0.5% FBS), there
is only
marginal decrease in viability over a prolonged period (unpublished data).
Although the
literature has been controversial on the effect of early stage autophagy
inhibition on cell
survival, blocking autophagy at a late stage has been more consistently shown
to cause
accelerated cell death under autophagy-inducing conditions (Kanzawa, T., I.M.
Germano, T.
Komata, H. Ito, Y. Kondo, and S. Kondo, (2004), Role of autophagy in
temozolomide-
induced cytotoxicity for malignant glioma cells. Cell Death Differ. 11:448-57;
Boya, P., R.A.
Gonzalez-Polo, N. Casares, J.L. Perfettini, P. Dessen, N. Larochette, D.
Metivier, D. Meley,
S. Souquere, T. Yoshimori, G. Pierron, P. Codogno, and G. Kroemer, (2005),
Inhibition of
macroautophagy triggers apoptosis. Mol Cell Biol. 25:1025-40; Gonzalez-Polo,
R.A., P.
Boya, A.L. Pauleau, A. Jalil, N. Larochette, S. Souquere, E.L. Eskelinen, G.
Pierron, P.
Saftig, and G. Kroemer, (2005), The apoptosis/autophagy paradox: autophagic
vacuolization
before apoptotic death. J Cell Sci. 118:3091-102. Epub 2005 Jun 28; Kroemer
and Jaattela,
2005; Yu, L., F. Wan, S. Dutta, S. Welsh, Z. Liu, E. Freundt, E.H. Baehrecke,
and M.
Lenardo, (2006), Autophagic programmed cell death by selective catalase
degradation. Proc
107
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
Nail Acad Sci U S A. 103:4952-7). Therefore, we investigated the effect of
blocking the
completion of autophagy initiated by Akt KD on cell viability. In PC3-shAkt123
cells stably
expressing GFP-LC3, Akt KD resulted in punctate GFP signals (Fig. 4 A and Fig.
19 E) with
a corresponding reduction of the nonlipidated precursor form of the endogenous
LC3 (LC34)
and a slight increase in the lipidated auto phagosome-localized LC3-II, which
is rapidly
turned over in the autolysosomes (Fig. 4 B; Klionsky et al., 2008). The
lysosomotropic agent
chloroquine (CQ), a weak base amine widely used to inhibit the maturation of
autophagosomes into degradative autolysosomes (Boya, P., R.A. Gonzalez-Polo,
N. Casares,
J.L. Perfettini, P. Dessen, N. Larochette, D. Metivier, D. Meley, S. Souquere,
T. Yoshimori,
G. Pierron, P. Codogno, and G. Kroemer, (2005), Inhibition of macroautophagy
triggers
apoptosis. Mol Cell Biol. 25:1025-40; Kroemer and Jaattela, 2005; Lum, J.J.,
D.E. Bauer, M.
Kong, M.H. Harris, C. Li, T. Lindsten, and C.B. Thompson, (2005), Growth
factor regulation
of autophagy and cell survival in the absence of apoptosis. Cell. 120:237-48),
caused the
appearance of small GFP-LC3 clusters in the perinuclear region. The
combination of CQ
with Akt KD resulted in a much stronger accumulation of GFP-LC3 dots as well
as
augmented accumulation of LC3-II in the presence of continued LC34 turnover,
consistent
with a defect in autolysosomal degradation. Similar accumulation of MDc+
vacuoles was
also observed (Fig. 19 F). This was accompanied by an accelerated cell death
in shAkt123-
expressing cells treated with CQ under 0.5% and, more pronouncedly, 0% FBS
(Fig. 4, C and
D). A second lysosomotropic agent, bafilomycin Al (Ba), which inhibits the
vacuolar proton
pump (VH+-ATPase) and prevents the proper acidification of lysosomal
compartments
(Yamamoto, A., Y. Tagawa, T. Yoshimori, Y. Moriyama, R. Masaki, and Y.
Tashiro, (1998),
Bafilomycin Al prevents maturation of autophagic vacuoles by inhibiting fusion
between
autophagosomes and lysosomes in rat hepatoma cell line, H-44I-E cells. Cell
Struct Funct.
23:33-42), also promoted cell death in combination with shAkt 123. Increased
annexin V -
positive population and caspase-3,7 activity was observed in cells treated
with either CQ or
Ba in combination with Akt KD, correlating with an increase in poly-ADP-ribose
polymerase
(PARP) cleavage in these cells (Fig. 4 B). In contrast, pretreatment with 1 mM
3-MA, an
inhibitor of the earliest stage of autophagosome formation, attributed to its
inhibition of class
III PI3K (Seglen, P.O., and P.B. Gordon, (1982), 3-Methyladenine: specific
inhibitor of
autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc
Natl Acad Sci US
A. 79:1889-92; Petiot, A., E. Ogier-Denis, E.F. Blommaart, A.J. Meijer, and P.
Codogno,
(2000), Distinct classes of phosphatidylinositol 3'-kinases are involved in
signaling pathways
that control macroautophagy in HT-29 cells. J Biol Chem. 275:992-8),
suppressed the cell
108
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
death-promoting effect of either CQ or Ba on shAkt expressing cells (Fig. 4, C
and D). This
suggests that the accelerated cell death caused by the lysosomotropic
inhibitors is dependent
on the accumulation of abnormal AVs.
Example 5
CQ accelerated cell death in combination with PI3K and Akt inhibitors
Recently, a phosphatidylinositol ether lipid analogue that inhibits Akt
activation was reported to induce autophagy with radiosensitizing effect
(Fujiwara et al.,
2007). Because phosphatidylinositol ether lipid analogues are known to have
additional
cellular targets (Gills et al., 2006; Memmott et al., 2008), we asked whether
other specific
inhibitors of PI3K-Akt could also induce autophagy and sensitize cells to late
stage
autophagy inhibition. We first used a dual PI3K/mTOR inhibitor, compound 111-5
(PI-103),
which inhibits the class I PI3Ks and mTOR at nanomolar concentrations but is
>1,000-fold
less potent on the class III PI3K (Knight, Z.A., B. Gonzalez, M.E. Feldman,
E.R. Zunder,
D.D. Goldenberg, 0. Williams, R. Loewith, D. Stokoe, A. Balla, B. Toth, T.
Balla, W.A.
Weiss, R.L. Williams, and K.M. Shokat, (2006), A pharmacological map of the
P13-K family
defines a role for p1 1 Oalpha in insulin signaling. Cell. 125:733-47). In
contrast to the broad-
spectrum PI3K inhibitors wortmannin or LY294002, which are equipotent at
inhibiting both
class I and III PI3Ks and inhibit autophagy caused by the latter activity
(Petiot et al., 2000;
Knight et al., 2006), 111-5 is potent at inducing the accumulation of AVs
(Fig. 5, B and C).
Similar to Akt KD, combination with CQ accelerated the death of cells treated
with 111-5
(Fig. 5 A). The markedly increased LC3-II to LC3-1 ratio and the appearance of
enlarged
vacuoles brightly stained by MDC was observed before the detection of overt
cell death (Fig.
5, B and C). As observed with Akt KD, pretreatment with 3-MA reduced both LC3-
II to -I
ratios and the accumulation of MDC+ vacuoles and slowed down the rate of cell
death (Fig. 5,
B and C). In contrast, the cell death-promoting effect of CQ was partially
mimicked by
siRNA KD of LAMP2, a protein previously shown to be required for the
maturation of
autophagosomes into autolysosomes (Fig. 20, A and B); Gonzalez-Polo et al.,
2005).
A similar effect of CQ was also obtained with one of the most selective Akt
inhibitors reported, the dual Akt 1,2 inhibitor compound 11-4 (Akti-1/2)
(Barnett et al., 2005).
Treatment with 11-4 alone effectively inhibited the phosphorylation of Akt on
both Ser473
and Thr308 residues and significantly reduced the phosphorylation of
downstream target S6
without causing significant cell death (Fig. 6, A and B). Cotreatment with CQ
resulted in a
rapid drop in cell viability with complete cell death observed by day 10.
Immunoblot
analysis revealed a significant accumulation of LC3-II within 24 h of 11-4
treatment, which is
109
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
further enhanced upon CQ cotreatment (Fig. 6, B and C).
To follow the kinetics of cell death, we used time-lapse microscopy to image
live cells treated with CQ and 11-4 (Fig. 7 A). CQ treatment alone caused a
mild decrease in
cell division and a gradual accumulation of dark particles in the perinuclear
region. AO
staining indicates that these particles are AVOs whose formation is inhibited
by 3-MA
(unpublished data). Treatment with 11-4 alone resulted in near-complete
inhibition of cell
division without overt cell death. These cells exhibited a flattened
morphology with
accumulation of AVOs that eventually filled the cytoplasm. Cells treated with
both 11-4 and
CQ showed similar accumulation of AVOs, but cell shrinkage and plasma membrane
rupture
was observed within 48 h. On a few occasions, two neighboring cells were found
to form a
membrane junction that expanded into complete fusion between the two cells
before rupture
of the plasma membrane (Fig. 7 A, white arrowheads).
Similar correlation between AVO accumulation and cell death was observed
using multispectral imaging flow cytometry (Fig. 7, B and C). Treatment with
either CQ or
11-4 alone induced AVO accumulation without significant loss of viability,
whereas the
combination of both resulted in a further increase in AVO accumulation in live
cells and a
concomitant increase in cells with condensed apoptotic nuclei and the
appearance of
anucleated population characteristics of necrotic cells.
Example 6
Autophagy inhibition and degradation defective autolysosome accumulation
both contribute to accelerated cell death induced by CG in combination with 11-
4
To investigate whether autophagy inhibition by itself is sufficient to induce
accelerated cell death in combination with Aid inhibition, we used siRNA to KD
Atg7, a
gene involved in the formation of autophagosomes (Ohsumi, Y. 2001. Molecular
dissection
of autophagy: two ubiquitin-like systems.). KD of Atg7 alone did not show a
significant
effect on cell death but induced a small drop in cell viability by day 3 when
combined with
Akti. However, when combined with both CQ and 11-4, Atg7 KD resulted in a
transient
delay of cell death at day 2 (Fig. 20, C and D). Together with the
aforementioned effect of 3-
MA, these data suggest that autophagy inhibition and defective AV accumulation
both
contribute to the accelerated cell death induced by CQ in combination with Akt
inhibition.
Because autophagy is a key function of the lysosomal compartment (Terman,
A., B. Gustafsson, and U.T. Brunk, (2006), The lysosomal-mitochondrial axis
theory of
postmitotic aging and cell death. Chem Biol Interact. 163:29-37), we examined
the lysosomal
marker LAMPI and cathepsin D, the predominant lysosomal aspartic protease, by
110
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
immunoblotting (Fig. 6, B and C). Compound 11-4 alone induced an increase in
LAMP1
levels, consistent with an elevated lysosomal activity. Cathepsin D is
synthesized as a 43-1(1)
preprocathepsin D that is cleaved cotranslationally and glycosylated to form a
46-1(D
procathepsin D, which is targeted to lysosomes yielding an intermediate that
is further
cleaved into a mature enzyme consisting of a 15-kD light chain and a 28-kD
heavy chain.
Using an antibody that detects both the 28-kD and the precursor forms of
cathepsin D, an
increase in the level of the premature forms of cathepsin D at 43-50 kD was
first detected
after 11-4 treatment alone followed by an increase in the 28-kD heavy chain of
the mature
enzyme, again indicating an increased lysosomal activity. CQ caused
accumulation of the
precursor forms at the expense of the 28-kD chain, consistent with an
inhibition of lysosomal
cysteine protease activity required for the processing and maturation of
cathepsin D (Liaudet-
Coopman, E., M. Beaujouin, D. Derocq, M. Garcia, M. Glondu-Lassis, V. Laurent-
Matha, C.
Prebois, H. Rochefort, and F. Vignon, (2006), Cathepsin D: newly discovered
functions of a
long-standing aspartic protease in cancer and apoptosis. Cancer Lett. 237:167-
79). In cells
treated with both 11-4 and CQ, the precursor forms of cathepsin D accumulated
to even higher
levels than either alone, whereas the mature 28-kD chain decreased gradually.
Compound
11-4 treatment also reduced the level of p62, another marker of autophagic
activity that is
degraded in the autolysosomes (Klionsky et al., 2008), whereas CQ blocked p62
degradation
both with and without 11-4 treatment (Fig. 22 C). Collectively, these data
suggest that Akt
inhibition causes an increased production and maturation of the lysosomal
enzymes, whereas
CQ cotreatment impairs the maturation of these enzymes in the final
autolysosomal
compartment, causing accumulation of defective AVOs. The latter is accompanied
by an
increased cleavage of caspase 3 into the active forms within 2 d (Fig. 6 B)
with a
corresponding increase in caspase activity and cleavage of its substrate PARP
(unpublished
data). zVAD.fmk, a pancaspase inhibitor, partially rescued cell death at all
concentrations
tested (Fig. 20 E). Although zVAD.fmk can also inhibit lysosomal cysteine
proteases at
higher concentrations, the latter have been reported to mediate caspase-
independent cell death
(Foghsgaard et al., 2001). Neither of the broad-spectrum cysteine protease
inhibitors
zFA.fmk and N-Acetyl-Leu-Leu-Nle-CHO nor a more specific cathepsin B inhibitor
CA-
074-Me showed significant rescue of cell death induced by 11-4 and CQ.
Instead, the cysteine
protease inhibitors enhanced cell killing in combination with Akti at 10-50 M
concentrations, although they also showed cytotoxicity alone at higher
concentrations (Fig.
20, F-H). These results suggest that cell death induced by 11-4 and CQ is at
least partially
caspase dependent, whereas lysosomal protease activity may be required for the
survival of
111
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
cells under Akt inhibition.
To further ask whether impaired lysosomal degradation can accelerate cell
death in combination with Akt inhibition, we knocked down cathepsin D using
siRNA.
Indeed, this significantly increased cell death when combined with 11-4 and
further enhanced
the cell-killing effect of CQ when both are combined with 11-4 (Fig. 20 I).
Similarly,
pepstatin A, an inhibitor of aspartic proteases including cathepsin D, also
promoted cell death
together with Alcti -112 (Fig. 20 J)
Example 7
CQ augmented Akti-induced mitochondrial superoxide and cellular reactive
oxygen species (ROS) accumulation
Increasing evidence has suggested an intimate relationship between lysosomes
and mitochondria in the execution of programmed cell death (Bursch, W. (2001),
The
autophagosomal-lysosomal compartment in programmed cell death. Cell Death
Differ. 8:569-
81). The autophagosomal-lysosomal compartment in programmed cell death. Cell
Death
Differ. 8:569-81; Terman, A., T. Kurz, B. Gustafsson, and U.T. Brunk, (2006),
Lysosomal
labilization. IUBMB L/ è. e. 58:531-9). Therefore, we examined the effect of
Akt inhibition and
CQ on mitochondrial membrane potential. Consistent with Akt's function in
maintaining
mitochondrial integrity (Parcellier, A., L.A. Tintignac, E. Zhuravleva, and
B.A. Hemmings,
(2007), PKB and the mitochondria: AKTing on apoptosis. Cell Signal. 0:0), Akti-
112 alone
caused a decrease in mitochondrial membrane potential, although significant
numbers of
polarized mitochondria were still present in the majority of cells. Although
CQ alone did not
have a significant effect, cotreatment of CQ and 11-4 caused an almost
complete loss of
mitochondrial potential, preceding the sharp drop in cell viability (Fig. 21,
A and B).
It has recently been reported that mitochondrial ROS is involved in autophagy
induction (Scherz-Shouval et al., 2007). Because mitochondria are the primary
intracellular
source of superoxide (02-) generation, we analyzed 02- production using
MitoSOX red, an
02- indicator that accumulates in the mitochondria as a function of membrane
potential and
fluoresces upon oxidation and subsequent binding to DNA. Compound 11-4 alone
increased
MitoSOX fluorescence within 6 h (Fig. 8, A and C; and not depicted). Most of
the
fluorescence exhibited a mitochondrial localization pattern with a
subpopulation of cells
showing nuclear fluorescence, consistent with increased mitochondrial
permeability in these
cells. Although CQ alone only caused a mild increase in MitoSOX signal, the
combination
with 11-4 resulted in a significant increase in fluorescence intensity with
most cells exhibiting
a strong nuclear staining pattern. The increase in 02 production was followed
by an increase
112
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
in overall cellular ROS levels within 24 h, as measured using a general ROS-
sensitive probe
(Fig. 8, B and C). Interestingly, cytoplasmic ROS signal induced by 11-4 alone
was attenuated
within 48 h, whereas cotreatment with CQ caused a prolonged increase in ROS
levels (Fig.
21 D and not depicted). This is consistent with the notion that limited
mitochondrial
depolarization caused by Akt inhibition induces a transient ROS signal to
increase autophagy,
which in sum removes the damaged mitochondria. Impaired digestion of cellular
components
caused by CQ results in auto lysosomal aggregation of deleterious oxidative
products such as
ceroid/lipofuscin, which can further amplify the ROS damage (Moore et al.,
2(06), leading to
cell death. 3-MA pretreatment reduced ROS levels induced by 11-4 (Fig. 21 C),
suggesting a
class III PI3K dependence similar to starvation-induced ROS production (Scherz-
Shouval et
al., 2007). Treatment with a general ROS scavenger N-acetylcysteine (NAC)
rescued cell
viability in the presence of 11-4 and CQ (Fig. 22, A and B). In addition, NAC
reduced Akti-
induced LC3 and GFP-LC3 lipidation, p62 degradation, and GFP-LC3 puncta
formation (Fig.
SS, C and D), consistent with an essential role of ROS in autophagy induction
(Scherz-
Shouval et al., 2007).
Example 8
CQ selectively accelerated cell death in Akti-treated PTEN-null cells in vitro
and enhanced
the antitumor efficacy of Akt KD in vivo
Because PC3 cells are PTEN null, we explored whether PTEN status might
affect the sensitivity of cells to Akt inhibition alone or in combination with
CQ using
isogenic PTEN' and PTEN' " mouse embryonic fibroblasts (MEFs). The PTEN' -
MEFs
were previously shown to have elevated Akt pathway activity and are more
sensitive to the
anti proliferative effect of mTOR inhibition than PTEN+t+ MEFs (Sun et al.,
1999). As
shown in Fig. 9 A, the PTEN' - MEFs were also significantly more sensitive to
the cell-killing
effect of combined CQ and 11-4 than their PTEN'+ counterparts. This suggests
that PTEN'
cellsmay be more dependent on autophagic degradation for survival upon Akt
inhibition,
raising the possibility that a reasonable therapeutic index may be achievable
by selective
targeting of the malignant PTEN-null tumor cells using this strategy.
To ask whether PTEN-null tumors also rely on autophagic degradation upon
Akt inhibition in vivo, we examined the effect of CQ on the survival of PC3
xenograft tumors
expressing shAkt 123. As shown in Fig. 9 B, intraperitoneal injection of CQ
alone caused a
small but insignificant reduction in tumor growth rate. Akt KD alone resulted
in significant
tumor growth inhibition with an initial tumor stasis, but most tumors failed
to regress
completely, and rebound occurred in 90% of the tumors within 2-3 wk; no
complete
113
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
remission was achieved. In contrast, complete regression was observed in 40%
of the tumors
treated with both Dox and CQ with stasis maintained in another 20% of the
tumors
throughout the study (Fig. 9, C and D). Similar results were obtained with a
subcutaneous
peritumor injection of CQ (unpublished data). EM examination of tumor samples
taken at day
5 revealed a mild increase in the AV area in tumors treated with either Dox or
CQ alone,
whereas a dramatic accumulation of AVs was observed in a tumor treated with
both Dox and
CQ that showed >50% regression. These AVs are larger than those found in the
Dox- or CQ-
alone tumors and contain dense undigested materials, but usually with a single-
membrane
auto lysosomal appearance and stained positive for human LAMP1, consistent
with impaired
degradation after autophagosome-lysosome fusions. This coincides with an
increased number
of tumor nuclei exhibiting apoptotic morphology as well as AV-containing cell
debris with
compromised plasma membrane integrity and abnormal mitochondria (Fig. 10).
Thus, CQ not
only accelerated cell death in combination with Akt inhibition in vitro but
also increased the
incidence of complete tumor remissions in vivo.
Using a Dox-inducible shRNA approach, we specifically knocked down each
Akt isoform, both individually and in all possible combinations, to evaluate
their requirement
in the maintenance of tumor growth. Our results suggest that in the PTEN-null
PC3 and
U87MG cells, Aktl is the most important isoform, whereas Akt2 and Akt3
activities could
partially compensate for the reduced Akt1 activity in maintaining tumor
growth. Taking
together both the potential metabolic side effects of Akt2 inhibition and the
reported increase
in invasiveness associated with inhibiting Akt 1 alone that could be
counteracted by
simultaneous inhibition of Akt2 (Irie et al., 2005), it may be necessary to
inhibit two or all
three Akt isoforms simultaneously to achieve maximum tumor inhibition, but
with different
degrees of inactivation to preserve crucial levels of isoform activities to
reduce side effects.
One of the most prominent functions of Akt is cell survival. Constitutively
active Akt has been reported to protect cells from programmed cell death after
various
proapoptotic insults (Downward, 1998). However, whether apoptosis is a primary
response
to Akt inhibition is less clear, especially in cancer cells where apoptosis is
often suppressed
because of various genetic alterations. Previous experiments using small
molecule inhibitors
of the PI3K-Akt pathway often generate conflicting results that are obscured
by their
nonspecific effects. Our data indicate that specific KD of Akt can cause cell
cycle delay
without promoting significant apoptosis. This is consistent with a recent
study that only a
small portion of total Akt activity is required for apoptosis inhibition (Liu
et al., 2006). In
contrast, we found that autophagy is a more sensitive response to reduced Akt
activity caused
114
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
by either specific shRNA KD or selective inhibitors of the pathway.
Several mechanisms may contribute to autophagy induction by Akt inhibition.
First, inhibiting Akt can lead to mTORC1 inhibition. mTOR is a known inhibitor
of
autophagy. Interestingly, a constitutively active form of Akt suppressed the
induction of
autophagy by rapamycin (Takeuchi et al., 2005), raising the possibility that
the effect of
rapamycin on autophagy may be mediated at least partially through inhibiting
Akt via its
long-term effect on mTORC2 (Sarbassov et al., 2006). Second, other signaling
outputs of
Akt, such as the Fox proteins (Zhao et al., 2008) or glucose metabolism, can
also contribute
to autophagy regulation independently of mTOR. Third, our data indicate that
Akt inhibition
induces increased mitochondrial superoxide and cellular ROS signals that can
activate
autophagy.
Autophagy activation may lead to eventual cell death when allowed to reach
its limit or may sensitize cells to additional death-inducing stimuli either
through eventual
autophagic cell death or switching to a more rapid death program such as
apoptosis. For
example, Akt inhibition may increase radiosensitivity through augmenting
autophagic
response (Fujiwara et al., 2007), whereas calpain-mediated cleavage of Atg5
may switch
autophagy into apotosis (Yousefi et al., 2006). Here we show that inhibiting
Akt alone is
ineffective in cell killing in the PTEN-null cancer cells that we examined,
but cell death can
be accelerated through blocking autolysosomal degradation. Although autophagy
may be a
potential mechanism by which Akt inhibition restricts tumor growth, it may
also provide
temporary relief from the metabolic and oxidative stress imposed by Akt
inhibition.
Inhibiting autophagy at an early stage may prevent this temporary protective
effect but may
also counteract its tumor inhibitory effect while allowing early escape via
alternative survival
mechanisms. Blocking lysosomal function after tumor cells have become
committed and
reliant on autophagic degradation, however, might avoid this counteracting
effect while
amplifying the oxidative damage and cytotoxic effects through accumulation of
deleterious
oxidative aggregates (Seehafer and Pearce, 2006). Indeed, our data suggest
that a compatible
lysosomal degradation capacity is critical for cell survival in the presence
of elevated
autophagic activity induced by Akt inhibition such that inhibiting lysosomal
function with
lysosomotropic agents, cathepsin D KD or lysosomal protease inhibitors, can
all precipitate
cell death in combination with Akt inhibition. Autophagy, lysosomal changes,
and oxidative
stress have been associated with a lengthening list of anticancer treatments,
and
lysosomotropic agents have shown anticancer activity either alone or in
combination with
115
CA 02821378 2013-06-12
WO 2012/087336
PCT/US2010/062096
other therapeutic agents (Shoemaker and Dagher, 1979; Ohta et al., 1998;
Ostenfeld et al.,
2005; Amaravadi et al., 2007; Carew, J.S., S.T. Nawrocki, C.N. Kahue, H.
Zhang, C. Yang,
L. Chung, J.A. Houghton, P. Huang, F.J. Giles, and J.L. Cleveland, (2007),
Targeting
autophagy augments the anticancer activity of the histone deacetylase
inhibitor SAHA to
overcome Bcr-Abl-mediated drug resistance. Blood. 110:313-22; Fujiwara et al.,
2007;
GrothPedersen et al., 2007). Here we report for the first time that autophagy
induced by
Akt/PI3K/mTOR inhibition can also be exploited using lysosomotropic agents,
such as the
well-tolerated drug CQ, to promote the remission of PTEN-null human tumor
xenografts.
Because this effect is expected to correlate positively with the degree of
autophagy induced
by a given treatment, creative combination of these agents with potent
autophagy inducers,
such as inhibitors of the Akt pathway, may profoundly affect their anticancer
efficacy.
Specific reference is made to U.S. provisional application No. 61/103,198,
herein incorporated by reference in its entirety. Specific reference is also
made to U.S.
provisional application No. 61/160,169, herein incorporated by reference in
its entirety.
Having now fully described this invention, it will be understood to those of
ordinary skill in
the art that the same can be performed within a wide and equivalent range of
conditions,
formulations, and other parameters without affecting the scope of the
invention or any
embodiment thereof. All patents, patent applications, and publications cited
herein are fully
incorporated by reference herein in their entirety.
116