Note: Descriptions are shown in the official language in which they were submitted.
CA 02838501 2013-12-05
WO 2013/004534
PCT/EP2012/062224
1
COMPOSITION CONSISTANT EN UN OXYDE MIXTE DE ZIRCONIUM ET
DE CERIUM A REDUCTIBILITE ELEVEE, PROCEDE DE PREPARATION ET
UTILISATION DANS LE DOMAINE DE LA CATALYSE
La présente invention concerne une composition consistant en un oxyde
mixte de zirconium et de cérium, à réductibilité élevée, son procédé de
préparation et son utilisation dans le domaine de la catalyse.
On utilise à l'heure actuelle pour le traitement des gaz d'échappement
des moteurs à combustion interne (catalyse postcombustion automobile) des
catalyseurs dits multifonctionnels. Par multifonctionnels, on entend les
catalyseurs capables d'opérer non seulement l'oxydation en particulier du
monoxyde de carbone et des hydrocarbures présents dans les gaz
d'échappement mais également la réduction notamment des oxydes d'azote
également présents dans ces gaz (catalyseurs "trois voies"). Les produits à
base d'oxyde de cérium, d'oxyde de zirconium et éventuellement d'un ou
plusieurs oxydes d'autres terres rares apparaissent aujourd'hui comme des
constituants particulièrement importants et intéressants rentrant dans la
composition de ce type de catalyseurs. Pour être efficaces, ces constituants
doivent présenter une surface spécifique importante même après avoir été
soumis à des températures élevées, par exemple d'au moins 900 C.
Une autre qualité requise pour ces constituants de catalyseurs est la
réductibilité. On entend par réductibilité, ici et pour le reste de la
description, la
capacité du catalyseur à se réduire en atmosphère réductrice et à se réoxyder
en atmosphère oxydante. La réductibilité peut se mesurer notamment par la
quantité d'oxygène mobile ou d'oxygène labile par unité de masse du matériau
et pour une gamme de température donnée. Cette réductibilité et, par
conséquent, l'efficacité du catalyseur, sont maximales à une température qui
est actuellement assez élevée pour les catalyseurs à base des produits
précités. Or, il existe un besoin en catalyseurs dont les performances soient
suffisantes dans des gammes de température plus faibles.
Dans l'état actuel de la technique, il apparaît que les deux
caractéristiques mentionnées plus haut sont souvent difficiles à concilier,
c'est
à dire qu'une réductibilité élevée à plus basse température a pour
contrepartie
une surface spécifique plutôt faible.
CA 02838501 2013-12-05
WO 2013/004534
PCT/EP2012/062224
2
L'objet de l'invention est de fournir une composition de ce type qui
présente en combinaison une surface spécifique élevée et une bonne
réductibilité à température plus basse.
Dans ce but, la composition de l'invention consiste essentiellement en un
oxyde mixte de zirconium et de cérium, ayant une teneur en oxyde de
zirconium d'au moins 45% en masse, et elle est caractérisée en ce qu'elle
présente après calcination à 1000 C, 4 heures une surface spécifique d'au
moins 25 m2/g et une quantité d'oxygène mobile entre 200 C et 400 C d'au
moins 0,5 ml 02/g.
D'autres caractéristiques, détails et avantages de l'invention apparaîtront
encore plus complètement à la lecture de la description qui va suivre faite en
référence aux dessins annexés et dans lesquels :
- la figure 1 est un schéma d'un réacteur utilisé pour la mise en oeuvre du
procédé de préparation de la composition de l'invention;
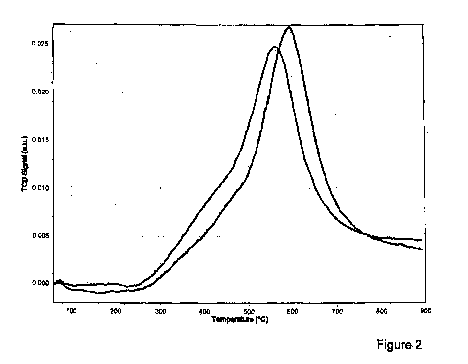
- la figure 2 représente les courbes obtenues par une mesure de
réductibilité par réduction programmée en température d'une composition
selon l'invention et d'un produit comparatif.
Pour la suite de la description, on entend par surface spécifique, la
surface spécifique B.E.T. déterminée par adsorption d'azote conformément à
la norme ASTM D 3663-78 établie à partir de la méthode BRUNAUER -
EMMETT- TELLER décrite dans le périodique "The Journal of the American
Chemical Society, 60, 309 (1938)".
En outre, les calcinations et notamment les calcinations à l'issue
desquelles sont données les valeurs de surface sont des calcinations sous air
à un palier de température sur la durée indiquée sauf indication contraire.
Les teneurs ou quantités sont données en masse d'oxyde par rapport à
l'ensemble de la composition sauf indication contraire. L'oxyde de cérium est
sous forme d'oxyde cérique.
On précise pour la suite de la description que, sauf indication contraire,
dans les fourchettes de valeurs qui sont données, les valeurs aux bornes sont
incluses.
La quantité d'oxygène mobile ou labile correspond à la moitié de la
quantité en mole d'hydrogène consommée par réduction de l'oxygène de la
composition pour former de l'eau et mesurée entre différentes bornes de
température, entre 200 C et 450 C ou bien entre 200 et 400 C. Cette mesure
est faite par réduction programmée en température sur un appareil
AUTOCHEM II 2920 avec un réacteur en silice. On utilise l'hydrogène comme
gaz réducteur à 10% en volume dans l'argon avec un débit de 30 mL/mn. Le
CA 02838501 2013-12-05
WO 2013/004534
PCT/EP2012/062224
3
protocole expérimental consiste à peser 200 mg de l'échantillon dans un
récipient préalablement taré. L'échantillon est ensuite introduit dans une
cellule en quartz contenant dans le fond de la laine de quartz. L'échantillon
est
enfin recouvert de laine de quartz, positionné dans le four de l'appareil de
mesure et un thermocouple est placé au coeur de l'échantillon. On détecte un
signal avec un détecteur de conductivité thermique. La consommation de
l'hydrogène est calculée à partir de la surface manquante du signal
d'hydrogène entre 200 C et 450 C ou encore entre 200 C et 400 C.
La température maximale de réductibilité (température à laquelle le
captage de l'hydrogène est maximal et où, en d'autres termes, la réduction du
cérium IV en cérium III est aussi maximale et qui correspond à une labilité
maximale en 02 de la composition) est mesurée en effectuant une réduction
en température programmée, telle que décrite ci-dessus. Cette méthode
permet de mesurer la consommation d'hydrogène d'une composition selon
l'invention en fonction de la température et d'en déduire la température à
laquelle le taux de réduction du cérium est maximum.
La mesure de réductibilité est faite par réduction programmée en
température sur un échantillon qui a été préalablement calciné 4 heures à
1000 C sous air. La montée en température se fait de 50 C à 900 C à raison
de 10 C/mn. Le captage de l'hydrogène est calculé à partir de la surface
manquante du signal d'hydrogène de la ligne de base à la température
ambiante à la ligne de base à 900 C.
La température maximale de réductibilité se traduit par un pic sur la
courbe obtenue par la méthode de réduction en température programmée qui
a été décrite. Il faut noter toutefois que dans certains cas une telle courbe
peut
comporter deux pics.
Les compositions selon l'invention se caractérisent tout d'abord par la
nature de leurs constituants.
Ces compositions consistent essentiellement en un oxyde mixte ou un
mélange d'oxydes de zirconium et de cérium.
La teneur en oxyde de zirconium est d'au moins 45%. Elle peut être plus
particulièrement au moins 60% et encore particulièrement au moins 70%.
Cette valeur peut être plus particulièrement d'au plus 95% et encore plus
particulièrement d'au plus 90%.
Selon une variante de l'invention les compositions de l'invention peuvent
peuvent contenir en outre un ou plusieurs éléments additionnels qui peuvent
être choisis dans le groupe comprenant le fer, le cobalt, le strontium, le
cuivre
et le manganèse. Ce ou ces éléments additionnels sont présents
CA 02838501 2013-12-05
WO 2013/004534
PCT/EP2012/062224
4
généralement sous forme d'oxyde. Dans le cas de cette variante les
compositions de l'invention consistent alors essentiellement en un oxyde mixte
ou un mélange d'oxydes de zirconium et de cérium et d'un ou plusieurs
éléments additionnels précités. La quantité d'élément additionnel est
généralement d'au plus 10%, elle peut être plus particulièrement comprise
entre 2% et 8%.
Par consistent essentiellement on entend que les compositions
peuvent comporter d'autres éléments sous forme de traces ou d'impuretés,
comme l'hafnium notamment, mais qu'elles ne comportent pas d'autres
éléments susceptibles notamment d'avoir une influence sur leur surface
spécifique et/ou leurs propriétés de réductibilité. En particulier, les
compositions de l'invention ne contiennent pas de terres rares autres que le
cérium.
Les compositions de l'invention ont pour caractéristique de présenter une
quantité d'oxygène mobile importante dans une gamme de température
relativement basse. Cette quantité exprimée en ml d'oxygène par gramme de
composition est d'au moins 0,5 ml 02/g entre 200 C et 400 C. Cette quantité
peut être notamment d'au moins 0,6 ml 02/g. Des quantités jusqu'à environ au
moins 1 ml 02/g peuvent être atteintes.
Dans une gamme de température un peu plus large, c'est-à-dire entre
200 C et 450 C, les compositions de l'invention présentent une quantité
d'oxygène mobile d'au moins 0,9 ml 02/g plus particulièrement d'au moins 1 ml
02/g. Des quantités jusqu'à environ au moins 1,5 ml 02/g peuvent être
atteintes.
Les compositions de l'invention ont pour autre caractéristique le fait de
présenter après calcination à 1000 C pendant 4 heures une température
maximale de réductibilité d'au plus 580 C, plus particulièrement d'au plus
570 C. Cette température maximale de réductibilité peut être notamment d'au
moins 530 C.
Les compositions de l'invention présentent aussi des caractéristiques
particulières de surface spécifique. En effet, tout en présentant de bonnes
propriétés de réductibilité à basse température, elles offrent en outre des
surfaces spécifiques élevées même à hautes températures.
Ainsi, elles présentent après calcination à 1000 C, 4 heures une surface
spécifique d'au moins 25 m2/g, plus particulièrement d'au moins 30 m2/g et
encore plus particulièrement d'au moins 35 m2/g. Dans ces mêmes conditions
de calcination des surfaces spécifiques jusqu'à une valeur d'environ 45 m2/g
peuvent être obtenues.
CA 02838501 2013-12-05
WO 2013/004534
PCT/EP2012/062224
Par ailleurs, après calcination à 1100 C, 4 heures, ces compositions
présentent une surface spécifique d'au moins 8 m2/g, plus particulièrement
d'au moins 10 m2/g et encore plus particulièrement d'au moins 12 m2/g. Dans
ces mêmes conditions de calcination des surfaces spécifiques jusqu'à une
5 valeur d'environ 20 m2/g peuvent être obtenues.
Une autre caractéristique intéressante des compositions de l'invention
est qu'elles peuvent se présenter sous forme de particules désagglomérables.
Ainsi, par un simple traitement par ultrasons, ces particules peuvent
présenter,
après un tel traitement et quelle que soit la taille des particules au départ,
un
diamètre moyen (d50) d'au plus 10 pm, plus particulièrement d'au plus 8 pm et
encore plus particulièrement d'au plus 6 pm.
Les valeurs de granulométrie données ici et pour le reste de la
description sont mesurées au moyen d'un granulométre laser de type Malvern
Mastersizer 2000 (module HydroG) sur un échantillon de particules dispersées
dans une solution à 1g/I d'hexaméthylphosphate (HMP) et soumis aux
ultrasons (120 W) pendant 5 minutes.
Les compositions de l'invention peuvent se présenter sous la forme d'une
solution solide pure de l'oxyde de cérium et de l'oxyde de zirconium. On
entend par là que le cérium est présent totalement en solution solide dans
l'oxyde de zirconium. Les diagrammes en diffraction RX de ces compositions
révèlent en particulier, au sein de ces dernières, l'existence d'une phase
unique clairement identifiable et correspondant à celle d'un oxyde de
zirconium
cristallisé dans le système quadratique, traduisant ainsi l'incorporation du
cérium dans le réseau cristallin de l'oxyde de zirconium, et donc l'obtention
d'une solution solide vraie. Il est à noter que les compositions de
l'invention
peuvent présenter cette caractéristique de solution solide même après
calcination à température élevée, par exemple au moins 1000 C, 4 heures.
Le procédé de préparation des compositions de l'invention va maintenant
être décrit. Ce procédé peut être mis en oeuvre selon deux variantes en
fonction du type de réacteur utilisé.
Selon une première variante, le procédé est caractérisé en ce qu'il
comprend les étapes suivantes :
- (a) on forme un mélange liquide comprenant des composés du cérium, du
zirconium et, éventuellement, de l'élément additionnel;
- (b) on fait réagir en continu dans un réacteur ledit mélange avec un composé
basique, le temps de séjour du milieu réactionnel dans la zone de mélange du
réacteur étant d'au plus 100 millisecondes ce par quoi on obtient un précipité
à
la sortie du réacteur;
CA 02838501 2013-12-05
WO 2013/004534
PCT/EP2012/062224
6
- (c) on chauffe en milieu aqueux ledit précipité, le milieu étant maintenu
à un
pH d'au moins 5;
- (d) on ajoute au précipité obtenu à l'étape précédente un additif, choisi
parmi
les tensioactifs anioniques, les tensioactifs non ioniques, les polyéthylène-
glycols, les acides carboxyliques et leurs sels et les tensioactifs du type
éthoxylats d'alcools gras carboxyméthylés;
- (e) on calcine le précipité ainsi obtenu.
Selon une seconde variante, le procédé de l'invention est caractérisé en
ce qu'il comprend les étapes suivantes :
- (a') on forme un mélange liquide comprenant des composés du cérium, du
zirconium et, éventuellement, de l'élément additionnel;
- (b') on fait réagir en continu dans un réacteur centrifuge ledit mélange
avec
un composé basique, le temps de séjour du milieu réactionnel dans la zone de
mélange du réacteur étant d'au plus 10 secondes ce par quoi on obtient un
précipité à la sortie du réacteur;
- (c') on chauffe en milieu aqueux ledit précipité, le milieu étant
maintenu à un
pH d'au moins 5;
- (d') on ajoute au précipité obtenu à l'étape précédente un additif,
choisi parmi
les tensioactifs anioniques, les tensioactifs non ioniques, les polyéthylène-
glycols, les acides carboxyliques et leurs sels et les tensioactifs du type
éthoxylats d'alcools gras carboxyméthylés;
- (e') on calcine le précipité ainsi obtenu.
La différence entre les deux variantes de procédé se situe aux étapes (b)
et (b'). Les autres étapes de procédé sont identiques pour les deux variantes.
De ce fait, la description qui va être faite ci-dessous pour les étapes (a),
(c), (d)
et (e) de la première variante s'applique de même aux étapes (a'), (c'), (d')
et
(e') de la seconde variante.
La première étape (a) du procédé consiste donc à préparer un mélange
en milieu liquide des composés des éléments constitutifs de la composition,
c'est à dire du cérium, du zirconium et, éventuellement, de l'élément
additionnel.
Le mélange se fait généralement dans un milieu liquide qui est l'eau de
préférence.
Les composés sont de préférence des composés solubles. Ce peut être
notamment des sels de zirconium et de cérium. Dans le cas de la préparation
de compositions comprenant un ou plusieurs éléments additionnels du type
mentionné plus haut, le mélange de départ comprendra en outre un composé
de cet ou ces éléments additionnels.
CA 02838501 2013-12-05
WO 2013/004534
PCT/EP2012/062224
7
Ces composés peuvent être choisis parmi les nitrates, les sulfates, les
acétates, les chlorures, le nitrate céri-ammoniacal.
A titre d'exemples, on peut ainsi citer le sulfate de zirconium, le nitrate de
zirconyle ou le chlorure de zirconyle. Le nitrate de zirconyle est utilisé le
plus
généralement. On peut citer aussi notamment les sels de cérium IV tels que
nitrates ou le nitrate céri-ammoniacal par exemple, qui conviennent ici
particulièrement bien. De préférence, on utilise du nitrate cérique. Il est
avantageux d'utiliser des sels de pureté d'au moins 99,5% et plus
particulièrement d'au moins 99,9%. Une solution aqueuse de nitrate cérique
peut par exemple être obtenue par réaction de l'acide nitrique sur un oxyde
cérique hydraté préparé d'une manière classique par réaction d'une solution
d'un sel céreux, par exemple le nitrate céreux, et d'une solution d'ammoniaque
en présence d'eau oxygénée. On peut également, de préférence, utiliser une
solution de nitrate cérique obtenue selon le procédé d'oxydation
électrolytique
d'une solution de nitrate céreux tel que décrit dans le document FR-A- 2 570
087, et qui constitue ici une matière première intéressante.
On notera ici que les solutions aqueuses de sels de cérium et de sels de
zirconyle peuvent présenter une certaine acidité libre initiale qui peut être
ajustée par l'addition d'une base ou d'un acide. Il est cependant autant
possible de mettre en oeuvre une solution initiale de sels de zirconium et de
cérium présentant effectivement une certaine acidité libre comme mentionné
ci-dessus, que des solutions qui auront été préalablement neutralisées de
façon plus ou moins poussée. Cette neutralisation peut se faire par addition
d'un composé basique au mélange précité de manière à limiter cette acidité.
Ce composé basique peut être par exemple une solution d'ammoniaque ou
encore d'hydroxydes d'alcalins (sodium, potassium,...), mais de préférence
une solution d'ammoniaque.
On notera enfin que lorsque le mélange de départ contient du cérium
sous forme III, il est préférable de faire intervenir dans le cours du procédé
un
agent oxydant, par exemple de l'eau oxygénée. Cet agent oxydant peut être
utilisé en étant ajouté au milieu réactionnel lors de l'étape (a), lors de
l'étape
(b) ou encore au début de l'étape (c).
Le mélange peut être indifféremment obtenu soit à partir de composés
initialement à l'état solide que l'on introduira par la suite dans un pied de
cuve
d'eau par exemple, soit encore directement à partir de solutions de ces
composés puis mélange, dans un ordre quelconque, desdites solutions.
Dans la deuxième étape (b) du procédé, on met en présence ledit
mélange avec un composé basique pour les faire réagir. On peut utiliser
CA 02838501 2013-12-05
WO 2013/004534
PCT/EP2012/062224
8
comme base ou composé basique les produits du type hydroxyde. On peut
citer les hydroxydes d'alcalins ou d'alcalino-terreux. On peut aussi utiliser
les
amines secondaires, tertiaires ou quaternaires. Toutefois, les amines et
l'ammoniaque peuvent être préférés dans la mesure où ils diminuent les
risques de pollution par les cations alcalins ou alcalino terreux. On peut
aussi
mentionner l'urée. Le composé basique peut être plus particulièrement utilisé
sous forme d'une solution.
La réaction entre le mélange de départ et le composé basique se fait en
continu dans un réacteur. Cette réaction se fait donc en introduisant en
continu
les réactifs et en soutirant en continu aussi le produit de la réaction.
La réaction doit se faire dans des conditions telles que le temps de séjour
du milieu réactionnel dans la zone de mélange du réacteur est d'au plus 100
millisecondes. On entend par zone de mélange du réacteur la partie du
réacteur dans laquelle sont mis en présence le mélange de départ précité et le
composé basique pour que la réaction ait lieu. Ce temps de séjour peut être
plus particulièrement d'au plus 50 millisecondes, de préférence il peut être
d'au plus 20 millisecondes. Ce temps de séjour peut être par exemple compris
entre 10 et 20 millisecondes.
On réalise de préférence l'étape (b) en utilisant un excès
stoechiométrique de composé basique pour s'assurer d'un rendement maximal
de précipitation.
La réaction se fait de préférence sous forte agitation, par exemple dans
des conditions telles que le milieu réactionnel est en régime turbulent.
La réaction se fait généralement à température ambiante.
On peut utiliser un réacteur de type mélangeur rapide.
Le mélangeur rapide peut être en particulier choisi parmi les mélangeurs (ou
tubes) en T ou en Y symétriques, les mélangeurs (ou tubes) en T ou en Y
asymétriques, les mélangeurs à jets tangentiels, les mélangeurs Hartridge-
Roughton, les mélangeurs vortex.
Les mélangeurs (ou tubes) en T ou en Y symétriques sont généralement
constitués de deux tubes opposés (tubes en T) ou formant un angle inférieur à
180 (tubes en Y), de même diamètre, déchargeant dans un tube central dont
le diamètre est identique ou supérieur à celui des deux tubes précédents. Ils
sont dits symétriques car les deux tubes d'injection des réactifs
présentent
le même diamètre et le même angle par rapport au tube central, le dispositif
étant caractérisé par un axe de symétrie. De préférence, le tube central
présente un diamètre deux fois plus élevés environ que le diamètre des tubes
CA 02838501 2013-12-05
WO 2013/004534
PCT/EP2012/062224
9
opposés ; de même la vitesse de fluide dans le tube central est de préférence
égale à la moitié de celle dans les tubes opposés.
On préfère cependant employer, en particulier lorsque les deux fluides à
introduire ne présentent pas le même débit, un mélangeur (ou tube) en T ou
en Y asymétriques plutôt qu'un mélangeur (ou tube) en T ou en Y symétrique.
Dans les dispositifs asymétriques, un des fluides (le fluide de plus faible
débit
en général) est injecté dans le tube central au moyen d'un tube latéral de
diamètre plus faible. Ce dernier forme avec le tube central un angle de 900 en
général (tube en T) ; cet angle peut être différent de 90 (tube en Y),
donnant
des systèmes à co-courant (par exemple angle de 45 ) ou à contre-courant
(par exemple angle de 135 ) par rapport à l'autre courant.
De manière avantageuse, on utilise dans le procédé selon la présente
invention un mélangeur à jets tangentiels, par exemple un mélangeur
Hartridge-Roughton.
La figure 1 est un schéma qui représente un mélangeur de ce type. Ce
mélangeur 1 comprend une chambre 2 ayant au moins deux admissions
tangentielles 3 et 4 par lesquelles entrent séparément (mais en même temps)
les réactifs, c'est-à-dire ici le mélange formé à l'étape (a) et le composé
basique, ainsi qu'une sortie axiale 5 par laquelle sort le milieu réactionnel
et
ce, de préférence, vers un(e) réacteur (cuve) disposé(e) en série après ledit
mélangeur. Les deux admissions tangentielles sont de préférence situées
symétriquement et de manière opposée par rapport à l'axe central de la
chambre 2.
La chambre 2 du mélangeur à jets tangentiels, Hartridge-Roughton utilisé
présente généralement une section circulaire et est de préférence de forme
cylindrique.
Chaque tube d'admission tangentielle peut présenter une hauteur interne
(a) en section de 0,5 à 80 mm.
Cette hauteur interne (a) peut être comprise entre 0,5 et 10 mm, en
particulier entre 1 et 9 mm, par exemple entre 2 et 7 mm. Cependant,
notamment à l'échelle industrielle, elle est de préférence comprise entre 10
et
80 mm, en particulier entre 20 et 60 mm, par exemple entre 30 et 50 mm.
Le diamètre interne de la chambre 2 du mélangeur à jets tangentiels,
Hartridge-Roughton employé peut être compris entre 3a et 6a, en particulier
entre 3a et 5a, par exemple égal à 4a; le diamètre interne du tube de sortie
axiale 5 peut être compris entre la et 3a, en particulier entre 1,5a et 2,5a,
par
exemple égal à 2a.
CA 02838501 2013-12-05
WO 2013/004534 PCT/EP2012/062224
La hauteur de la chambre 2 du mélangeur peut être comprise entre la et
3a en particulier entre 1,5 et 2,5a, par exemple égal à 2a.
La réaction conduite à l'étape (b) du procédé conduit à la formation d'un
précipité qui est évacué du réacteur et récupéré pour la mise en oeuvre de
5 l'étape (c).
Dans le cas de la seconde variante, l'étape (b') est mise en oeuvre dans
un réacteur de type centrifuge. Par réacteur de ce type on entend les
réacteurs rotatifs utilisant la force centrifuge.
On peut citer comme exemples de ce type de réacteurs, les mélangeurs
10 ou réacteurs rotor-stators, les réacteurs à disque tournant (sliding
surface
reactor), dans lesquels les réactifs sont injectés sous cisaillement élevé
dans
un espace confiné entre le fond du réacteur et un disque tournant à vitesse
élevée, ou encore les réacteurs dans lesquels la force centrifuge permet aux
liquides de se mélanger intimement en films minces. Dans cette catégorie
figurent le Spinning Disc Reactor (SDR) ou le Rotating Packed Bed reactor
(RPB), décrit dans la demande de brevet US 2010/0028236 Al. Le réacteur
décrit dans cette demande de brevet comporte une structure poreuse ou
garnissage, en céramique, en mousse métallique ou en matière plastique, de
forme cylindrique et qui tourne à vitesse élevée autour d'un axe longitudinal.
Les réactifs sont injectés dans cette structure et se mélangent sous l'effet
de
fortes forces de cisaillement du fait des forces centrifuges pouvant atteindre
plusieurs centaines de g créées par le mouvement de rotation de la structure.
Le mélange des liquides dans des veines ou des films très fins permet
d'atteindre ainsi des tailles nanométriques.
Le procédé selon la seconde variante de l'invention peut donc être mis
en oeuvre en introduisant dans la structure poreuse précitée le mélange formé
à l'étape (a').
Les réactifs ainsi introduits peuvent être soumis à une accélération d'au
moins 10 g, plus particulièrement d'au moins 100 g et qui peut être comprise
par exemple entre 100 g et 300 g.
Compte tenu de leur conception, ces réacteurs peuvent être utilisés avec
des temps de séjour du milieu réactionnel dans leur zone de mélange (au
même sens que donné plus haut pour la première variante) plus élevés que
pour la première variante de procédé, c'est-à-dire jusqu'à plusieurs secondes
et en général d'au plus 10 s. De préférence, ce temps de séjour peut être d'au
plus 1 s, plus particulièrement d'au plus 20 ms et encore plus
particulièrement
d'au plus 10 ms.
CA 02838501 2013-12-05
WO 2013/004534
PCT/EP2012/062224
11
Comme pour la variante précédente on réalise de préférence l'étape (b')
en utilisant un excès stoechiométrique de composé basique et cette étape se
fait généralement à température ambiante.
A l'issue de l'étape (b') le précipité obtenu est évacué du réacteur et
récupéré pour la mise en oeuvre de l'étape suivante.
L'étape (c) ou (c') est une étape de chauffage du précipité en milieu
aqueux.
Ce chauffage peut être réalisé directement sur le milieu réactionnel
obtenu après réaction avec le composé basique ou sur une suspension
obtenue après séparation du précipité du milieu réactionnel, lavage éventuel
et
remise dans l'eau du précipité. La température à laquelle est chauffé le
milieu
est d'au moins 90 C et encore plus particulièrement d'au moins 100 C. Elle
peut être comprise entre 100 C et 200 C. L'opération de chauffage peut être
conduite en introduisant le milieu liquide dans une enceinte close (réacteur
fermé du type autoclave). Dans les conditions de températures données ci-
dessus, et en milieu aqueux, on peut préciser, à titre illustratif, que la
pression
dans le réacteur fermé peut varier entre une valeur supérieure à 1 Bar (105
Pa) et 165 Bar (1,65. 107 Pa), de préférence entre 5 Bar (5. 105 Pa) et 165
Bar
(1,65. 107 Pa). On peut aussi effectuer le chauffage dans un réacteur ouvert
pour les températures voisines de 100 C.
Le chauffage peut être conduit soit sous air, soit sous atmosphère de gaz
inerte, de préférence l'azote.
La durée du chauffage peut varier dans de larges limites, par exemple
entre 1 minute et 2 heures, ces valeurs étant données à titre tout à fait
indicatif.
Le milieu soumis au chauffage présente un pH d'au moins 5. De
préférence, ce pH est basique, c'est à dire qu'il est supérieur à 7 et, plus
particulièrement, d'au moins 8.
Il est possible de faire plusieurs chauffages. Ainsi, on peut remettre en
suspension dans l'eau, le précipité obtenu après l'étape de chauffage et
éventuellement un lavage puis effectuer un autre chauffage du milieu ainsi
obtenu. Cet autre chauffage se fait dans les mêmes conditions que celles qui
ont été décrites pour le premier.
L'étape suivante du procédé peut se faire selon deux variantes.
Selon une première variante, on ajoute au milieu réactionnel issu de
l'étape précédente un additif qui est choisi parmi les tensioactifs
anioniques,
les tensioactifs non ioniques, les polyéthylène-glycols, les acides
CA 02838501 2013-12-05
WO 2013/004534
PCT/EP2012/062224
12
carboxyliques et leurs sels et les tensioactifs du type éthoxylats d'alcools
gras
carboxyméthylés.
En ce qui concerne cet additif on pourra se référer à l'enseignement de la
demande WO-98/45212 et utiliser les tensioactifs décrits dans ce document.
On peut mentionner comme tensioactifs du type anionique les
éthoxycarboxylates, les acides gras éthoxylés ou propoxylés, notamment ceux
de la marque ALKAMULS , les sarcosinates de formule
R-C(0)N(CH3)CH2000-, les bétaînes de formule RR'NH-CH3-000-, R et R'
étant des groupes alkyles ou alkylaryles, les esters phosphates, notamment
ceux de la marque RHODAFAC , les sulfates comme les sulfates d'alcool, les
sulfates d'éther alcool et les éthoxylats d'alcanolamide sulfatés, les
sulfonates
comme les sulfosuccinates, les alkyl benzène ou alkyl naphtalène sulfonates.
Comme tensioactif non ionique on peut mentionner les tensioactifs
acétyléniques, les alcools gras éthoxylés ou propoxylés, par exemple ceux des
marques RHODASURF ou ANTAROX , les alcanolamides, les oxydes
d'amine, les alcanolamides éthoxylés, les amines éthoxylées ou propoxylées à
longues chaînes, par exemple ceux de la marque RHODAMEEN , les
copolymères oxyde d'éthylène/oxide de propylène, les dérivés du sorbitan,
l'éthylène glycol, le propylène glycol, le glycérol, les esters polyglyceryle
et
leurs dérivés éthoxylés, les alkylamines, les alkylimidazolines, les huiles
éthoxylées et les alkylphénols éthoxylés ou propoxylés, notamment ceux de la
marque IGEPAL . On peut citer aussi en particulier les produits cités dans
WO-98/45212 sous les marques IGEPAL , DOWANOL , RHODAMOX et
ALKAMIDE .
En ce qui concerne les acides carboxyliques, on peut utiliser notamment
les acides mono- ou dicarboxyliques aliphatiques et parmi ceux-ci plus
particulièrement les acides saturés. On peut utiliser aussi des acides gras et
plus particulièrement les acides gras saturés. On peut citer ainsi notamment
les acides formique, acétique, proprionique, butyrique, isobutyrique,
valérique,
caproïque, caprylique, caprique, laurique, myristique, palmitique, stéarique,
hydroxystéarique, éthy1-2-hexanoïque et béhénique. Comme acides
dicarboxyliques, on peut mentionner les acides oxalique, malonique,
succinique, glutarique, adipique, pimélique, subérique, azélaïque et
sébacique.
Les sels des acides carboxyliques peuvent aussi être utilisés, notamment
les sels ammoniacaux.
A titre d'exemple, on peut citer plus particulièrement l'acide laurique et le
laurate d'ammonium.
CA 02838501 2013-12-05
WO 2013/004534
PCT/EP2012/062224
13
Enfin, il est possible d'utiliser un tensioactif qui est choisi parmi ceux du
type éthoxylats d'alcools gras carboxyméthylés.
Par produit du type éthoxylats d'alcool gras carboxyméthylés on entend
les produits constitués d'alcools gras éthoxylés ou propoxylés comportant en
bout de chaîne un groupement CH2-000H.
Ces produits peuvent répondre à la formule :
R1-0-(CR2R3-CR4R5-0)n-CH2-COOH
dans laquelle R1 désigne une chaîne carbonée, saturée ou insaturée,
dont la longueur est généralement d'au plus 22 atomes de carbone, de
préférence d'au moins 12 atomes de carbone; R2, R3, R4 et R5 peuvent être
identiques et représenter l'hydrogène ou encore R2 peut représenter un
groupe CH3 et R3, R4 et R5 représentent l'hydrogène; n est un nombre entier
non nul pouvant aller jusqu'à 50 et plus particulièrement compris entre Set
15,
ces valeurs étant incluses. On notera qu'un tensio-actif peut être constitué
d'un mélange de produits de la formule ci-dessus pour lesquels R1 peut être
saturé et insaturé respectivement ou encore des produits comportant à la fois
des groupements -CH2-CH2-0- et -C(CH3)-CH2-0-.
Une autre variante consiste à séparer d'abord le précipité issu de l'étape
(c) puis à ajouter l'additif tensioactif à ce précipité.
La quantité de tensio-actif utilisée, exprimée en pourcentage en poids
d'additif par rapport au poids de la composition calculé en oxyde, est
généralement comprise entre 5% et 100% plus particulièrement entre 15% et
60%.
Après l'addition du tensio-actif, on maintient le mélange obtenu de
préférence sous agitation pendant une durée qui peut être d'environ une
heure. On sépare ensuite éventuellement le précipité du milieu liquide par
tout
moyen connu.
Le précipité séparé peut éventuellement être lavé, notamment par de
l'eau ammoniaquée.
Dans une dernière étape du procédé selon l'invention, le précipité
récupéré, éventuellement séché, est ensuite calciné. Cette calcination permet
de développer la cristallinité du produit formé et elle peut être également
ajustée et/ou choisie en fonction de la température d'utilisation ultérieure
réservée à la composition selon l'invention, et ceci en tenant compte du fait
que la surface spécifique du produit est d'autant plus faible que la
température
de calcination mise en oeuvre est plus élevée. Une telle calcination est
généralement opérée sous air, mais une calcination menée par exemple sous
CA 02838501 2013-12-05
WO 2013/004534
PCT/EP2012/062224
14
gaz inerte ou sous atmosphère contrôlée (oxydante ou réductrice) n'est bien
évidemment pas exclue.
En pratique, on limite généralement la température de calcination à un
intervalle de valeurs comprises entre 300 et 900 C sur une durée qui peut être
par exemple comprise entre 1 heure et 10 heures.
Selon une autre variante du procédé de l'invention, les éléments
additionnels fer, cobalt, strontium, cuivre et manganèse peuvent ne pas être
ajoutés lors de la préparation de la composition telle qu'elle a été décrite
plus
haut mais ils peuvent être apportés en utilisant la méthode d'imprégnation.
Dans ce cas la composition issue de la calcination d'oxyde mixte de zirconium
et de cérium est imprégnée par une solution d'un sel d'élément additionnel
puis soumise à une autre calcination dans les mêmes conditions que celles
données plus haut.
Le produit issu de la calcination se présente sous la forme d'une poudre
et, si nécessaire, il peut être désaggloméré ou broyé en fonction de la taille
souhaitée pour les particules constituant cette poudre.
Les compositions de l'invention peuvent aussi éventuellement être mises
en forme pour se présenter sous forme de granulés, billes, cylindres ou nids
d'abeille de dimensions variables.
Les compositions de l'invention peuvent être utilisées comme catalyseurs
ou supports de catalyseur. Ainsi, l'invention concerne aussi des systèmes
catalytiques comprenant les compositions de l'invention. Pour de tels
systèmes, ces compositions peuvent ainsi être appliquées sur tout support
utilisé habituellement dans le domaine de la catalyse, c'est à dire notamment
des matériaux inertes thermiquement. Ce support peut être choisi parmi
l'alumine, l'oxyde de titane, l'oxyde de cérium, l'oxyde de zirconium, la
silice,
les spinelles, les zéolites, les silicates, les phosphates de silicoaluminium
cristallins, les phosphates d'aluminium cristallins.
Les compositions peuvent aussi être utilisées dans des systèmes
catalytiques comprenant un revêtement (wash coat) à propriétés catalytiques
et à base de ces compositions, sur un substrat du type par exemple monolithe
métallique par exemple FerCralloy, ou en céramique, par exemple en
cordiérite, en carbure de silicium, en titanate d'alumine ou en mullite. Le
revêtement peut comporter lui aussi un matériau inerte thermiquement du type
de ceux mentionnés plus haut. Ce revêtement est obtenu par mélange de la
composition avec ce matériau de manière à former une suspension qui peut
être ensuite déposée sur le substrat.
CA 02838501 2013-12-05
WO 2013/004534
PCT/EP2012/062224
Ces systèmes catalytiques et plus particulièrement les compositions de
l'invention peuvent trouver de très nombreuses applications. Ils sont ainsi
particulièrement bien adaptés à, et donc utilisable dans la catalyse de
diverses
réactions telles que, par exemple, la déshydratation, l'hydrosulfuration,
5 l'hydrodénitrification, la désulfuration, l'hydrodésulfuration, la
déshydrohalogénation, le reformage, le reformage à la vapeur, le craquage,
l'hydrocraquage, l'hydrogénation, la déshydrogénation, l'isomérisation, la
dismutation, l'oxychloration, la déshydrocyclisation d'hydrocarbures ou autres
composés organiques, les réactions d'oxydation et/ou de réduction, la réaction
10 de Claus, l'oxydation de gaz issus de sources stationnaires ainsi que le
traitement des gaz d'échappement des moteurs à combustion interne, la
démétallation, la méthanation, la shift conversion, l'oxydation catalytique
des
suies émises par les moteurs à combustion interne comme les moteurs diesel
ou essence fonctionnant en régime pauvre.
15 Les systèmes catalytiques et les compositions de l'invention peuvent
être
utilisés comme pièges à NOx ou encore dans un procédé SCR, c'est-à-dire un
procédé de réduction des NOx dans lequel cette réduction est effectuée par de
l'ammoniac ou un précurseur de l'ammoniac comme l'urée.
Dans le cas de ces utilisations en catalyse, les compositions de
l'invention sont employées généralement en combinaison avec des métaux
précieux, elles jouent ainsi le rôle de support pour ces métaux. La nature de
ces métaux et les techniques d'incorporation de ceux-ci dans les compositions
supports sont bien connues de l'homme du métier. Par exemple, les métaux
peuvent être le platine, le rhodium, le palladium ou l'iridium, ils peuvent
notamment être incorporés aux compositions par imprégnation.
Parmi les utilisations citées, le traitement des gaz d'échappement des
moteurs à combustion interne (catalyse post combustion automobile) constitue
une application particulièrement intéressante. De ce fait, l'invention
concerne
aussi un procédé de traitement des gaz d'échappement des moteurs à
combustion interne qui est caractérisé en ce qu'on utilise à titre de
catalyseur
un système catalytique tel que décrit ci-dessus ou une composition selon
l'invention et telle que décrite précédemment.
Des exemples vont maintenant être donnés.
EXEMPLE 1
Cet exemple concerne une composition à 80% de zirconium et 20% de
cérium, ces proportions étant exprimées en pourcentages massiques des
oxydes Zr02 et Ce02.
CA 02838501 2013-12-05
WO 2013/004534
PCT/EP2012/062224
16
Dans un bécher agité, on introduit la quantité nécessaire de nitrate de
cérium et de nitrate de zirconium. On complète ensuite avec de l'eau distillée
de façon à obtenir 1 litre d'une solution de nitrates à 120 g/I (exprimé ici
et
pour l'ensemble des exemples en équivalent d'oxyde).
Dans un autre bécher agité, on introduit une solution d'ammoniaque (10
mol/i) et on complète ensuite avec de l'eau distillée de façon à obtenir un
volume total de 1 litre et un excès stoechiométrique en ammoniaque de 40%
par rapport aux cations à précipiter.
On maintient sous agitation constante les deux solutions préparées
précédemment et on les introduit en continu dans un mélangeur rapide
Hartridge-Roughton du type de celui de la figure 1 et de hauteur d'entrée (a)
de 2 mm. Le pH en sortie du mélangeur est de 9,25. Le débit de chacun des
réactifs est de 301/h et le temps de séjour de 12 ms.
La suspension de précipité ainsi obtenue est placée dans un autoclave
en acier inoxydable équipé d'un mobile d'agitation. La température du milieu
est portée à 150 C pendant 2h sous agitation.
On ajoute à la suspension ainsi obtenue 33 grammes d'acide laurique. La
suspension est maintenue sous agitation pendant 1 heure.
La suspension est alors filtrée sur Büchner et le précipité filtré est ensuite
lavé à l'eau ammoniaquée.
Le produit obtenu est ensuite porté à 700 C pendant 4 heures en palier
puis désaggloméré dans un mortier.
EXEMPLE 2 COMPARATIF
Cet exemple concerne la même composition que celle de l'exemple 1.
On part des mêmes réactifs et on prépare 1 litre d'une solution de
nitrates de cérium et de zirconium.
Dans un réacteur agité, on introduit une solution d'ammoniaque (10 mol/l)
et on complète ensuite avec de l'eau distillée de façon à obtenir un volume
total de 1 litre et un excès stoechiométrique en ammoniaque de 40% par
rapport aux nitrates à précipiter.
La solution de nitrates est introduite dans le réacteur sous agitation
constante en 1 heure. On procède ensuite après la précipitation de la même
manière que dans l'exemple 1.
On donne dans les tableaux 1 et 2 ci-dessous, les caractéristiques des
produits obtenus dans les exemples.
CA 02838501 2013-12-05
WO 2013/004534 PCT/EP2012/062224
17
Tableau 1
Surface spécifique SBET (m2/g) après calcination
4h à
90000 1000 C 1100 C
1 56 39 14
2 comparatif 47 33 11
Tableau 2
Température Quantité d'02
Quantité d'02
maximale de
labile entre 200 et labile entre 200
réductibilité ( C) 400 C (mL/g) et
450 C (mL/g)
1000 C(2) 1000 C(2) 1000 C(2)
1 559 0,7 1,2
2 comparatif 597 0,4 0,8
(2) Cette température est celle à laquelle a été préalablement soumise,
pendant 4h, la composition avant la mesure de réductibilité.
Il est à noter que les produits des exemples 1 et 2 se présentent sous la
forme d'une solution solide après calcination 4 heures à 900 C ou à 1000 C.
La figure 2 donne les courbes obtenues en mettant en oeuvre la mesure
de réductibilité décrite plus haut. La température figure en abscisse et la
valeur
du signal mesuré est donnée en ordonnée. La température maximale de
réductibilité est celle qui correspond à la hauteur maximale du pic de la
courbe. La figure donne les courbes obtenues pour les compositions des
exemples 1 (courbe avec le pic le plus à gauche de la figure) et 2 comparatif
(courbe avec le pic le plus à droite)