Note: Descriptions are shown in the official language in which they were submitted.
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
CRYSTALLINE FORM OF INDAZOLYL AMIDE DERIVATIVES FOR THE TREATMENT
GLUCOCORTICOID RECEPTOR MEDIATED DISORDERS
Field of the invention
The present invention relates to new solid state forms of a drug, to
pharmaceutical
compositions containing them, to processes for obtaining them and to the use
of the new
solid state forms and compositions containing them in medical treatment.
Background of the invention
In the formulation of drug compositions, it is desirable for the drug
substance to be
in a form in which it can be conveniently handled and processed. This is of
importance, not
only from the point of view of obtaining a commercially viable manufacturing
process, but
also from the point of view of subsequent manufacture of pharmaceutical
formulations
comprising the active compound. In particular, pharmaceutical compositions
which are
formulated for inhaled administration must be in a form which enables
appropriate
processing techniques, such as micronisation, and which enables delivery using
a suitable
delivery device, for example a dry powder inhaler, a metered dose inhaler, a
nebuliser or a
nasal delivery device.
Chemical stability, solid state stability and "shelf life" of the active
ingredients are
also very important factors. The drug substance, and compositions containing
it, should be
capable of being effectively stored over appreciable periods of time, without
exhibiting a
significant change in the active component's physico-chemical characteristics
(e.g. its
chemical composition, density, melting point, hygroscopicity and solubility).
Moreover, it is desirable to be able to provide the drug in a form which is as
chemically pure as possible.
Furthermore, crystalline drug compounds have been shown to provide more
reliable and reproducible plasma concentration profiles following
administration to a
patient.
Moreover, different crystalline forms of a compound may exhibit different
physico-
chemical properties, such as melting point, solubility and hygroscopicity.
Furthermore, different crystalline forms of a compound may exhibit different
pharmacokinetic characteristics, such as total lung exposure, total lung
retention, total
blood exposure, peak plasma exposure and oral bioavailability.
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
2
Additionally, it is desirable for the drug substance to be in a
thermodynamically
stable form in order to prevent or minimize the risk of conversion to another
alternative
form during the manufacturing or formulation process, or during or following
administration to a patient.
Amorphous, or semi-amorphous materials may present significant problems in
this
regard. For example, such materials are typically difficult to handle and to
formulate,
provide for an unreliable solubility, and are often found to be unstable and
chemically
impure.
The skilled person will appreciate that, if a drug can be found in a stable
crystalline
form, the above problems may be solved.
Thus, in the manufacture of commercially viable, and pharmaceutically
acceptable,
drug compositions, it is desirable, wherever possible, to provide the drug in
a substantially
crystalline, and stable, form.
It is to be noted, however, that this goal is not always achievable. Indeed,
typically,
it is not possible to predict, from molecular structure alone, what the
crystallization
behaviour of a compound will be. This can usually only be determined
empirically.
International patent application WO 2008/076048 discloses a number of
compounds, which have been found to be useful as modulators of the
glucocorticoid
receptor, which modulators are of the general formula (I):
D1 R4
1 x 2
R
............,.......õ........ is ,
A¨C(Z) ¨NI Rla 3 N
Rx R (I)
Y NI
W
(wherein R1, Ria, R25 R35 R45 RX5 A,
W, X, Y and Z have meanings given in the description
of WO 2008/076048) and pharmaceutically acceptable salts thereof.
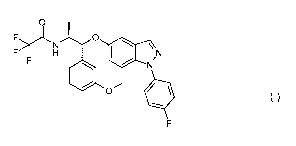
WO 2008/076048 also discloses, as Example 6, the specific compound 2,2,2-
trifluoro-N- [( 1R,2 S)- 1 - [1 -(4-fluorophenyl)indazol-5 -yl] oxy- 1 -(3 -
methoxyphenyl)prop an-
2-yl]acetamide (referred to hereinafter as Compound (I)). The Compound (I)
obtained by
following the procedure described therein is non-crystalline.
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
3
0
F>N0 0
F H \ N
F NI
1.1 0 .
Compound (I)
F
Disclosure of the invention
We have now found the Compound (I) may be prepared in crystalline form,
including a new, thermodynamically stable, crystalline form of Compound (I),
or a
pharmaceutically acceptable salt thereof.
Thus, according to one aspect of the invention, there is provided a
substantially
crystalline form of Compound (I), or a pharmaceutically acceptable salt
thereof
In another aspect of the invention, there is provided a substantially
crystalline form
of Compound (I).
In another aspect of the invention, there is provided Form A of Compound (I).
In one aspect of the invention, Form A of Compound (I) is substantially
crystalline.
In another aspect of the invention, Form A of Compound (I) is crystalline.
In another aspect of the invention, there is provided Form B of Compound (I).
In one aspect of the invention, Form B of Compound (I) is substantially
crystalline.
In another aspect of the invention, Form B of Compound (I) is crystalline.
Forms A and B of Compound (I) are described in further detail hereafter.
In so far as any of the cystalline forms of Compound (I) may exists as
solvate, such
solvates form a part of the present invention. Solvates of Compound (I)
include hydrates
and alcoholates (such as propanol and iso-propanol solvates).
According to a further aspect of the invention, there is provided a
substantially
crystalline anhydrate form of Compound (I). In a still further aspect,
Compound (I) is not
in the form of a salt. In a yet still further aspect, Compound (I) is not in
the form of a
solvate, i.e. it is an "ansolvate". Hence, the term "anhydrate" encompasses
"ansolvate".
We have found that Compound (I) may be obtained in forms that are
substantially
crystalline in nature. When herein reference is made to compounds of the
invention being
crystalline, suitably the degree of crystallinity as determined by X-ray
powder diffraction
data is for example greater than about 60%, such as greater than about 80%,
particularly
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
4
greater than about 90%, more particularly greater than about 95%. In
embodiments of the
invention, the degree of crystallinity as determined by X-ray powder
diffraction data is
greater than about 98%, wherein the % crystallinity refers to the % by weight
of the total
sample mass which is crystalline.
It is stated hereinbefore that Compound (I) may be produced in a crystalline
form
that is an anhydrate. By this we mean that the crystalline form contains less
than 10% of
hydrate form(s) (e.g. a monohydrate) of Compound (I).
In a further aspect of the invention Form A and Form B of Compound (I) are
anhydrate crystalline forms.
Form B of Compound (I)
In another aspect, Form B of Compound (I) is characterised by an X-ray powder
diffraction pattern, measured using a wavelength of X-rays 1.5418 A, with
peaks at 2-
Theta (in degrees) of 9.2, 17.4 and 21.5.
In a further aspect, Form B of Compound (I) is characterised by an X-ray
powder
diffraction pattern, measured using a wavelength of X-rays 1.5418 A, with
peaks at 2-
Theta (in degrees) of 9.2, 11.8, 15.7, 17.4 and 21.5.
In a yet still further aspect, Form B of Compound (I) is characterised by an X-
ray
powder diffraction pattern, measured using a wavelength of X-rays 1.5418 A,
with peaks at
2-Theta (in degrees) as shown in Table 1 hereafter.
In a yet still further aspect, Form B of Compound (I) is characterised by and
hence
the form may be characterized by the X-ray powder diffraction pattern
substantially as
shown in Figure 1, when measured using a wavelength of X-rays 1.5418 A. .
In a further aspect, Form B of Compound (I) is characterized by a differential
scanning calorimetry curve, at a heating rate of 5 C per minute in a closed
aluminium cup
under a nitrogen atmosphere, exhibiting an onset temperature of the melting
endotherm of
about 109 C.
In a further aspect, Form B of Compound (I) is characterized by the
differential
calorimetry curve substantially as shown in Figure 2.
In another aspect, Form B of Compound (I) is characterized by a differential
scanning calorimetry curve, at a heating rate of 5 C per minute in a closed
aluminium cup
under a nitrogen atmosphere, exhibiting an onset temperature of the melting
endotherm of
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
about 109 C and/or an X-ray powder diffraction pattern, measured using a
wavelength of
X-rays 1.5418 A, with peaks at 2-Theta (in degrees) of 9.2, 11.8, 15.7, 17.4
and 21.5.
In another aspect, Form B of Compound (I) is characterized by the differential
calorimetry curve substantially as shown in Figure 2 and/or an X-ray powder
diffraction
5 pattern substantially as shown in Figure 1.
Suitably a crystalline modification of a compound according to the invention
is
substantially free from other crystalline modifications of the compound. For
example in
one embodiment Form B of Compound (I) is substantially free of Form A of
Compound
(I). Suitably, a described crystalline modification of Compound (I) that is
substantially
free from other crystalline modifications of the compound includes less than,
for example,
20%, 15%, 10%, 5%, 3% or particularly, less than 1% by weight of other
crystalline forms
of that compound.
Crystalline anhydrates of Compound (I) may be prepared as described herein by
crystallizing Compound (I) from one or more suitable solvents or mixtures
thereof
Anhydrate may be produced by crystallization from a solvent system which is
substantially
free of water (which may have been dried, and/or may be dried during the
crystallization
process). Solvent may be dried during the crystallization process, for example
by
decreasing the water content of a mixture of the compound to be crystallized
in a suitable
organic solvent / aqueous solvent system (e.g. by increasing the amount of
organic solvent
that is present and/or removal of water by formation of an azeoptrope, with
successive
distillations). However, crystalline anhydrates of Compound (I) may also be
prepared from
water and/or water/alcohol mixtures.
Compounds of the invention that are anhydrates typically contain no more than
2%,
particularly 1%, more particularly 0.5% and more particularly 0.2% (w/w)
water, whether
such water is bound (crystal water or otherwise) or not.
In order to ensure that crystalline forms as described herein are prepared in
the
absence of other crystalline forms, crystallisations may be carried out by
seeding with
nuclei and/or seed crystals of the desired crystalline form in the absence of
nuclei and/or
seed crystals of other crystalline forms.
The skilled person will appreciate that the concentration in solution of the
compound that is to be crystallised, and the solvent system that is used, may
influence
crystallisation temperatures and crystallisation times.
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
6
Different crystalline forms may have different solubility in different organic
solvents at any given temperature. In this respect, above-mentioned, or other,
solvents
may be employed as "antisolvents" (i.e. a solvent in which compounds of the
invention are
poorly soluble, but which is miscible with another solvent, in which compounds
of the
invention are more soluble), and may thus aid the crystallisation process.
As may be appreciated by the skilled person, the crystalline form that is
obtained
depends upon both the kinetics and the thermodynamics of the crystallisation
process.
Under certain thermodynamic conditions (solvent system, temperature, pressure
and
concentration of the compound of the invention), one crystalline form may be
more stable
than another (or indeed any other). However, other crystalline forms that may
have, in
comparison, a relatively low thermodynamic stability, may be kinetically-
favoured. Thus,
in addition, kinetic factors, such as time, impurity profile, agitation, the
presence of seeds,
etc. may also influence which forms appear. Thus, the procedures discussed
herein may be
adapted by the skilled person as appropriate in order to obtain the particular
crystalline
form of Compound (I).
Compounds of the invention may be dried using standard techniques. It will be
appreciated by the skilled person that drying temperature and drying time may
affect the
solid state properties and/or the solid state form of compounds of the
invention. For
example, dehydration may occur at low humidity and/or elevated temperatures
and/or
reduced pressure. Hence, the crystalline anhydrates of compounds of the
invention may
also be formed by dehydration of a hydrate.
Preparation of Crystalline Forms of Compound (I)
According to a further aspect of the invention there is provided a process for
the
production of a compound of the invention which comprises crystallizing
Compound (I)
from a solution, suspension or slurry of Compound (I) with a suitable solvent
system. In
such a process it is important to leave the solution, suspension or slurry
mixed for a
sufficient period of time. The length of time depends on the level of
saturation so that
highly saturated solutions may crystallize within hours or a day or two,
whereas less
saturated solutions may require longer (for example a week or more).
Suitable mixing, for example by stirring, is believed to be important,
possibly since
it creates sites for primary, as well as secondary nucleation, thus speeding
up the
crystallisation process. Once available, the addition of seed crystals (of the
form to be
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
7
crystallised) to the solution, suspension or slurry will speed up the
crystallisation process
since the time for primary nucleation will then be shortened. Thus, a further
process of the
invention provides the production of a compound of the invention which
comprises
crystallising Compound (I) from a solution, suspension or slurry of the
compound with a
suitable solvent using seeds of the relevant compound to initiate and/or
facilitate
crystallisation. Suitable solvents include alcohols (such as ethanol, propanol
and
isopropanol), ethyl acetate, isopropyl acetate, aqueous systems and suitable
mixtures
thereof (for example, water/propanol, water/isopropanol). Other suitable
solvents include
ethereal solvents (such as methyl tert-butyl ether). Antisolvents (such as
heptanes) may
also be used as appropriate. A particular process of the invention comprises
the use of a
two solvent system that favours aggregation of crystals, i.e use of a good
solvent and an
antisolvent, such as the good solvent 1-propanol and the antisolvent n-heptane
or the good
solvent methyl tert-butyl ether and the anti-solvent n-heptane.
Form B of Compound (I) may be prepared by crystallization of Compound (I) in
amorphous form in a suitable solvent system. Thus in one aspect of the
invention,
Compound (I) in amorphous form is suspended or slurried (or partially
dissolved) in a
suitable solvent system and thereafter the suspension or slurry is heated and
then allowed
to cool. In another aspect the suspension or slurry is heated to a sufficient
temperature to
afford dissolution of compound (I) before being allowed to cool. In a further
aspect the
suspension or slurry is heated to at least 75 C (such as at least 80 C for
example about
87 C). In another aspect the solvent system includes any suitable solvent, or
mixture of
solvents, that do not result in the formation of a solvate of Compound (I) at
room
temperature. In another aspect, the solvent system may include those in which
Compound
(I) is only partially (or is at least partially) soluble. In a further aspect,
the solvent system
comprises a two solvent system comprising a good solvent and an antisolvent.
In a still
further aspect, the solvent system comprises an organic solvent that is polar,
e.g. alcohols
(such as lower alkyl alcohols, e.g. a C1_6 alcohol for example 1-propanol or
isopropanol) or
acetates (such as isopropyl acetate) and an alkyl antisolvent such as
heptanes. In a yet
further aspect the solvent system comprises isopropyl acetate and n-heptane.
In a still
further aspect, n-heptane constitutes at least 75% w/w (e.g. at least 85% such
as about
90%) of the total solvent employed in the solvent system. That is, the solvent
system may
contain up to 25% w/w (e.g. up to 15%, or about 10%) of isopropyl acetate.
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
8
The crystallization of Form B of Compound (I) may also be promoted by the
addition of seed crystals of Form B (once available). Thus in one aspect of
the invention
there is provided a crystalline form obtainable by such a seeding process. In
order to obtain
Form B by this process, Compound (I) in amorphous form may be suspended or
slurried
(or at least partially dissolved) in a suitable solvent system (solvent system
V) and
thereafter seeds of Form B (for example 0.2 to 1.5% w/w, e.g. 0.5 to 1.0% w/w,
such as
0.5% w/w) added optionally followed by the addition of an anti-solvent (anti-
solvent W).
In a further aspect of the invention, solvent system V includes any suitable
solvent, or
mixture of solvents, that do not result in the formation of a solvate of
Compound (I). In
another aspect, solvent system V may include those in which Compound (I) is
only
partially (or is at least partially) soluble. In a further aspect, solvent
system V comprises a
two solvent system comprising a good solvent and an antisolvent.
In one embodiment, solvent system V comprises an organic solvent that is
polar,
e.g. alcohols (such as lower alkyl alcohols, e.g. a C1_6 alcohol) and an alkyl
antisolvent
such as heptanes. In a further aspect solvent system V comprises 1-propanol
and n-
heptane. In a further aspect, n-heptane constitutes at least 50% w/w (e.g. at
least 60% such
as about 70%) of the total solvent system employed in solvent system V. That
is, solvent
system V may contain up to 50% w/w (e.g. up to 40%, or about 30%) of 1-
propanol. In a
further aspect, antisolvent W is an alkyl antisolvent such as n-heptane. In a
still further
aspect of the invention, solvent system V is heated and then cooled before the
seed crystals
are added and the mixture is allowed to cool further before antisolvent W is
added. In a
further aspect, solvent system V is heated to at least 55 C (such as at least
60 C for
example about 65 C) then allowed to cool to 40 C to 50 C (such as 45 C to 50 C
for
example about 50 C) then the seed crystals of Form B are added (for example
0.2 to 1.5%
w/w, e.g. 0.5 to 1.0% w/w, such as 0.5% w/w) and the mixture gradually cooled
to at least
room temperature (such as at least 25 C for example at least 15 C such as at
least 8 C).
In a further embodiment of the invention, solvent system V comprises an
organic
solvent that is ethereal and an alkyl antisolvent such as a heptane. In a
further aspect
solvent system V comprises methyl tert-butyl ether and n-heptane. In a further
aspect, n-
heptane constitutes at least 20% w/w (e.g. such as about 30%) of the total
solvent system
employed in solvent system V. That is, solvent system V may contain up to 80%
w/w (e.g.
up to about 70%) of methyl tert-butyl ether. In a further aspect, antisolvent
W is an alkyl
antisolvent such as n-heptane. In a still further aspect of the invention,
solvent system V is
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
9
heated and then cooled before the seed crystals are added. In a further
aspect, solvent
system V is heated to at least 30 C (such as about 35 C) then allowed to cool
to 20 C to
25 C (such as about 21 C) then the seed crystals of Form B added.
In a further embodiment of the invention, the amorphous form of Compound (I)
referred to herein is synthesised via the route described in Example 1, steps
(ii) to (vii).
The terms "suspended" and "slurried" (or "partially dissolved") are well
understood
by the skilled person. For instance to form a suspension or slurry, an excess
of the solid
substance, relative to the solubility in the solvent, is added such that there
is (undissolved)
solid in the solvent system throughout the "suspension" or "slurrying"
procedure.
Crystalline Form B of Compound (I) may also be prepared by seeding a
suspension
of an alternative form of Compound (I) (hereinafter referred to as "Form A")
in a solvent
system with seeds of Form B of Compound (I). The preparation of Form A of
Compound
(I) is described below at Example 4 and Example 5. The characteristic X-ray
powder
diffraction pattern peaks of Form A are tabulated in Table 2 below and the X-
ray powder
diffraction diffractogram shown in Figure 3 below. The characteristic
differential
calorimetry curve of Form A of Compound (I) is shown in Figure 4 below.
There is further provided a crystalline form of Compound (I) obtainable by
such a
(crystallisation) conversion process. The skilled person will understand that
a suspension
process is essentially a "slurrying" process or a process that involves at
least partial (but
not complete) dissolution in a solvent system.
In an aspect of the invention therefore, there is provided the conversion of
one
crystalline form (e.g. one anhydrate form) of Compound (I) to another. In
particular Form
A may be converted to Form B. There is therefore provided a crystalline form
obtainable
by such a (crystallisation) conversion process.
In order to obtain Form B of Compound (I) by this process, Form A may be
suspended or slurried (or at least partially dissolved) in a suitable solvent
system and
thereafter seeds of Form B (for example 1.0 to 2.5% w/w, e.g. 2 % w/w) added.
In one
aspect of the invention, solvent systems employed to obtain Form B of Compound
(I) by
suspension or slurrying (i.e. to achieve the conversion of Form A of Compound
(I) to Form
B) include any suitable solvent, or mixture of solvents, that do not result in
the formation
of a solvate of Compound (I). In another aspect, solvent systems may include
those in
which Compound (I) is only partially (or is at least partially) soluble. In a
further aspect,
the solvent system comprises a two solvent system comprising a good solvent
and a
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
moderate solvent. In a still further aspect, the solvent system comprises an
organic solvent
that is polar, e.g. alcohols (such as lower alkyl alcohols, e.g. a Ci_6
alcohol) and water. In a
yet further aspect the solvent system comprises isopropanol and water. In a
further aspect,
water constitutes at least 60% w/w (e.g. at least 75% such as about 80%) of
the total
5 solvent system employed to obtain Form B. That is, the solvent system may
contain up to
40% w/w (e.g. up to 25%, or about 20%) of isopropanol.
The phase conversion in the solvent system to obtain Form B of Compound (I)
may
take a number of hours or days (e.g. 4 days, see Example 3 hereinafter), but
the length of
time may be reduced depending on for example the temperature of the process
(or it may
10 take longer if performed at lower temperatures) or the concentration of
the solution, etc.
However, the skilled person can easily determine the length of time taken for
conversion to
Form B.
We have found that Form B of Compound (I) has improved physical properties
when compared with other forms of Compound (I) which may have previously been
prepared (for example compared with the amorphous free-base form or with Form
A).
Form B of Compound (I) has, for example, a different hygroscopicity profile
compared to
the amorphous free-base form which may be useful in formulations comprising
the
compounds of the invention. Furthermore, Form B of Compound (I) has a
different
solubility profile and/or dissolution rate compared to Form A (in various
solvents, for
example buffered aqueous systems or propanol/heptane systems) and a different
melting
point compared to Form A which may be useful in the manufacturing process and
in
formulations comprising the compounds of the invention. Furthermore, we have
found that
by employing the crystallisation or conversion processes described herein, it
is possible to
produce Form B of Compound (I) with a high chemical purity.
Moreover, we have found that Form A of Compound (I) is converted to Form B in
the conversion processes described herein, showing that Form B is a
thermodynamically
more stable form of Compound (I), at least at the relevant temperature range,
and may be
particularly advantageous for use as a medicament.
Furthermore, we have found that Form B of Compound (I) exhibits different
pharmacokinetic properties compared with other forms of Compound (I) (such as
Form A).
Form B of Compound (I) exhibits an increased level of total lung exposure
(expressed as
"Area Under the Curve" or AUC) when compared with Form A. Moreover, Form B of
Compound (I) exhibits a reduced level of peak blood level (expressed as Cmax)
when
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
11
compared with Form A. Differences in pharmacokinetic properties may lead to
differences
in the pharmacological efficacy and may provide improved safety margins.
The term "stability" as defined herein includes chemical stability and solid
state
stability.
By "chemical stability", we include that the compound can be stored in an
isolated
solid form, or in the form of a solid formulation in which it may be provided
in admixture
with pharmaceutically acceptable carriers, diluents or adjuvants, under normal
storage
conditions, with an insignificant degree of chemical degradation or
decomposition.
By "solid state stability", we include that the compound can be stored in an
isolated
solid form, or in the form of a solid formulation in which it may be provided
in admixture
with pharmaceutically acceptable carriers, diluents or adjuvants, under normal
storage
conditions, with an insignificant degree of solid state transformation (e.g.
crystallisation,
recrystallisation, loss of crystallinity, solid state phase transition,
hydration, dehydration,
solvatisation or desolvatisation).
Examples of "normal storage conditions" include temperatures of between minus
80 and plus 50 C (preferably between 0 and 40 C and more preferably ambient
temperature, such as between 15 and 30 C), pressures of between 0.1 and 2 bars
(preferably atmospheric pressure), and/or exposure to 460 lux of UV/visible
light, for
prolonged periods (i.e. greater than or equal to six months). Under such
conditions,
compounds of the invention may be found to be less than about 15%, more
preferably less
than about 10%, and especially less than about 5%, chemically
degraded/decomposed, or
solid-state transformed, as appropriate. The skilled person will appreciate
that the above-
mentioned upper and lower limits for temperature and pressure represent
extremes of
normal storage conditions, and that certain combinations of these extremes
will not be
experienced during normal storage (e.g. a temperature of 50 C and a pressure
of 0.1 bar).
The term "normal storage conditions" may also include relative humidities of
between 5 and 95% (preferably 10 to 60%). However, in the case of certain
crystalline
forms according to the invention, changes in conformation or crystal structure
by hydration
and/or dehydration may occur as a result of prolonged exposure to certain
extremes of
relative humidities, at normal temperatures/pressures.
Although compounds of the invention (i.e. the crystalline forms) are
preferably not
in the form of salts, salts that may be mentioned include acid addition salts
and base
addition salts.
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
12
The preparation and characterisation of compounds of the invention are
described
hereinafter. Different crystalline forms of the compounds of the invention may
be readily
characterised using X-ray powder diffraction (XRPD) methods, for example as
described
hereinafter. Standard DSC techniques may also be used.
As may also be appreciated by the skilled person, the crystalline form that is
obtained may be significantly influenced by the synthetic process undertaken
to produce
the compound to be crystallised. In particular factors such as the nature of
reactants, the
nature of the reagents, the nature of the solvents and the nature of the
purification
techniques (if any) utilised in any previous steps (and in particular in the
penultimate
In one aspect of the invention, therefore, there is provided a process for the
preparation of Form B of Compound (I).
Preparation of Compound (I)
In another aspect there is provided a process for the preparation of Compound
(I)
H2N ei\ N 0
el
N
(II) F>r
Li (III)
0 = F
F
F
wherein Ll is an alkoxy or trifluoroacetoxy group. In a further aspect the
acylating agent is
ethyl trifluoracetate. In a still further aspect compound (II) is in the
hydrochloride salt-
form.
The compound of formula (III) is either commercially available or it may be
prepared using well-known chemistry from commercially available starting
materials. The
methods which may be utilized to couple compounds of formula (II) and formula
(III) are
well known in the art. For example, the coupling reaction may be carried out
by mixing
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
13
compounds of formula (II) and (III) in a suitable solvent (e.g. an ethereal
solvent such as
methyl tert-butyl ether) in the presence of a suitable base (e.g. an organic
base such as
triethylamine) at a suitable temperature (e.g. ambient temperature) for a
suitable time (e.g.
24 hours).
In a further aspect, there is provided a process for the preparation of a
compound of
formula (II), which comprises the deprotection of a compound of formula (IV):
1 )
/5
R
S, 0 el
i
N
S (IV)
0 .
F
wherein Rl is alkyl (unsubstituted or substituted by silylalkyl),
dialkylamino, aryl
(unsubstituted or substituted by one or more of halogen, haloalkyl, alkyl or
NO2) or
heteroaryl (unsubstituted or substituted by one or more of halogen, haloalkyl,
alkyl or
NO2). In a still further aspect Rl is aryl (unsubstituted or substituted by
one or more of
halogen, haloalkyl, alkyl or NO2).
The methods which may be utilized to deprotect compounds of formula (IV) are
well known in the art. For example, where Rl is aryl, the deprotection may be
carried out
by mixing a compound of formula (IV) in a suitable solvent (e.g. an organic
solvent such
as acetonitrile) in the presence of a suitable base (e.g. an inorganic base
such as potassium
carbonate) with a suitable deprotecting agent such as a thiol nucelophile
(e.g. thioglycolic
acid) at a suitable temperature (e.g. 60-100 C such as 75 C) for a suitable
time (e.g. 18
hours). Alternatively, the deprotection may be carried out using other well-
known
deprotecting agents, for example strong acids (such as hydrobromic acid or
sulphuric acid),
strong reducing agents (such as ground magnesium or sodium in liquid ammonia
or
sodium naphthalene or tributyl tin hydride) or samarium iodide. Alternative
methods for
carrying out this deprotection are described in standard chemistry texts, for
example
Greene, T.W. & Wuts, P.G.M. (2006), Greene's Protective Groups in Organic
Synthesis,
Wiley-Interscience; or Kocienski, P. (2005), Protecting Groups, Thieme.
In a yet further aspect, there is provided a process for the preparation of a
compound of formula (II), which comprises the deprotection of a compound of
formula
(V):
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
14
o2N 'Jo
& jo
,_!, N :
ki I-1 , 101 \ N
41) N
(V)
F
using methods well-known in the art, for example those described above and in
the
Examples herein.
In a further aspect of the invention there is provided a process for the
preparation of
compounds of formula (IV), which comprises the in-situ formation of a
protected aziridine
(X) from amino-alcohol (VI) followed by in-situ aziridine ring-opening with
hydroxyindazole (XI):
¨ ¨ ¨ ¨
RP Ri ,P
0 CI
S, OH R14 Lo, -,,UH 0
MsCI
0 H E µ, ==
H3N
0 ,
(VII) el õIII) 0- .1
0
(Ix)s-
(VI) - - -
HO RigP I 0
Ry) 0 "N
base6 1\r " N
6 N + el ,
N
_
* 1.1
I' (:)
F
_
(X) (XI) (IV) F
wherein Rl is alkyl (unsubstituted or substituted by silylalkyl),
dialkylamino, aryl
(unsubstituted or substituted by one or more of halogen, haloalkyl, alkyl or
NO2) or
heteroaryl (unsubstituted or substituted by one or more of halogen, haloalkyl,
alkyl or
NO2). In a still further aspect Rl is aryl (unsubstituted or substituted by
one or more of
halogen, haloalkyl, alkyl or NO2).
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
The process to the free-base form of the compound of formula (VI) (ie. not in
salt-
form) is described in WO 2008/076048 (example 6b). The formation of the
hydrochloride
salt of the compound of formula (VI) is described in the Examples herein. The
compound
of formula (VII) is either commercially available or it may be prepared using
well-known
5 chemistry from commercially available starting materials. Processes to
compound of
formula (XI) are described in WO 2008/079073 (example 1) and in the Examples
herein.
The methods which may be utilized to form compounds of formula (IV) are well
known in
the art, for instance those described in the Examples herein.
In a still further aspect of the invention there is provided a process for the
10 preparation of compounds of formula (V), which comprises the in-situ
formation of a
protected aziridine (XV) from amino-alcohol (VI) followed by in-situ aziridine
ring-
opening with hydroxyindazole (XI):
¨ ¨ ¨ ¨
02N 0 o o
i. i.
,P o'N 00 p 0--N ilo p
es,CI msoi
P, L
CI +LOH OH
H3N z ,4P'N -11.
______________________ 1.
(xii) 6 6
S o' el o' lei o'
(VI) ¨ (mil) ¨ (xiv)
¨ ¨ 0
i.
0 40
base 4 .N ,P
p, ,U
6 N :
¨v.
0 ",N
N
40 C) F
_
(XV) (XI) (V) F
The process to the free-base form of the compound of formula (VI) (ie. not in
salt-
form) is described in WO 2008/076048 (example 6b). The formation of the
hydrochloride
salt of the compound of formula (VI) is described in the Examples herein. The
compound
of formula (XII) is commercially available. Processes to compound of formula
(XI) are
described in WO 2008/079073 (example 1) and in the Examples herein.
The methods which may be utilized to form compound of formula (V) are well
known in the art. For example, compound of formula (XIII) may be prepared by
mixing
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
16
compounds of formula (VI) and formula (XII) in a suitable solvent (for example
an organic
solvent such as 2-methyltetrahydrofuran) in the presence of a suitable base
(for example an
organic base such as N-methylmorpholine) at a suitable temperature (e.g.
ambient
temperature) for a suitable time (e.g. 1 hour). Compound of formula (XIV) may
be
prepared by adding methanesulfonyl chloride directly to the reaction mixture
containing
compound of formula (XIII) and a suitable base (for example an organic base
such as N-
methylmorpholine) at a suitable temperature (e.g. 40 C) for a suitable time
(e.g. 16 hours).
Compound of formula (XV) may be prepared by adding a suitable base (such as an
inorganic base, e.g. sodium hydroxide) to a compound of formula (XIV) in a
suitable
solvent (such as an organic solvent e.g. 2-methyltetrahydrofuran) at a
suitable temperature
(e.g. ambient temperature). Alternatively the suitable base (such as an
inorganic base, e.g.
sodium hydroxide) can be added directly to the reaction solution containing
the compound
of formula (XIV) after a method of work-up well-known to those skilled in the
art (for
example washing with an aqueous acidic solution such as aqueous hydrochloric
acid
followed by washing with water). Compound of formula (V) may be prepared by
adding
compound of formula (XI) directly to the reaction mixture containing compound
of
formula (XV) and a suitable base (such as an inorganic base, e.g. sodium
hydroxide) at a
suitable temperature (e.g. 40 C) for a suitable time (e.g. 17 hours).
Further information on the processes of the invention and the products
obtainable
therefrom are described in the Examples herein.
In a further embodiment of the invention there is provided a compound of
formula
(IV):
1 0
R4 0
u
, el : \
H : N
N
S 0 . (IV)
F
wherein Rl is alkyl (unsubstituted or substituted by silylalkyl),
dialkylamino, aryl
(unsubstituted or substituted by one or more of halogen, haloalkyl, alkyl or
NO2) or
heteroaryl (unsubstituted or substituted by one or more of halogen, haloalkyl,
alkyl or
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
17
NO2). In a further aspect there is provided the use of compound (IV), or a
salt thereof, as
defined herein, as a pharmaceutical intermediate.
In a still further aspect Rl is aryl (unsubstituted or substituted by one or
more of
halogen, haloalkyl, alkyl or NO2).
In another embodiment of the invention there is provided a compound of formula
(V). In a further aspect there is provided the use of compound (V), or a salt
thereof, as
defined herein, as a pharmaceutical intermediate.
02N 'Jo
IISNO
\
0 H 0 N
i
I.N
(V)
0 it
F
In a further embodiment of the invention there is provided a compound of
formula
(II) in the hydrochloride salt-form (compound (XVI)). In a further aspect
there is provided
the use of the compound (XVI), as defined herein, as a pharmaceutical
intermediate.
a
0
H3N+ 0\ N
N/
(XVI)
. o,
F
Alkyl groups and moieties are straight or branched chain and comprise, for
example, 1 to 6 (such as 1 to 4) carbon atoms. Examples of alkyl groups are
methyl, ethyl,
n-propyl, iso-propyl or tert-butyl.
Aryl groups and moieties are monocyclic or multicyclic aromatic carbocycles
comprising, for example, 6 to 14 (such as 6 to 10) carbon atoms. Examples of
aryl groups
Dialkylamino means a ¨N(alkyl)2 group in which alkyl is defined as above.
Examples of dialkylamino groups are dimethylamino or diethylamino.
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
18
Haloalkyl means an alkyl group as defined above which is substituted by one or
more halo atoms. Examples of haloalkyl are trifluoromethyl and trifluoroethyl.
Silylalkyl means a ¨Si(alkyl)3 group in which alkyl is defined as above.
Examples
of silylalkyl groups are trimethylsilyl, triethylsilyl and tert-butyl
dimethylsilyl.
We have found that this new synthetic process to Compound (I) has a number of
advantages over the process previously described in WO 2008/076048. The need
for large-
scale chromatography is removed, avoiding significant time, resource
allocation and costs
in purification. The formation of intermediates in-situ without the need for
additional
isolation reduces the need for significant work-up and reaction mixture
manipulations.
Furthermore, the relatively low-yielding copper-promoted coupling step
described in WO
2008/076048 is avoided, providing enhanced selectivity of nitrogen over oxygen
and better
overall reaction control. Furthermore, we have found that this new synthetic
process and
the crystallisation step described herein reliably provide Form B of Compound
(I).
Medical Use
Because of their ability to bind to the glucocorticoid receptor the compounds
of the
invention are useful as anti-inflammatory agents, and can also display
antiallergic,
immunosuppressive and anti-proliferative actions. Thus, compounds of the
invention, or a
pharmaceutically acceptable salt thereof can be used as a medicament for the
treatment or
prophylaxis of one or more of the following pathologic conditions (disease
states) in a
mammal (such as a human):
(0 Lung diseases, which coincide with inflammatory, allergic and/or
proliferative
processes:
chronically obstructive lung diseases of any origin, mainly bronchial asthma,
chronic
obstructive pulmonary disease (COPD)
bronchitis of different origins
Adult respiratory distress syndrome (ARDS), acute respiratory distress
syndrome
Bronchiectases
all forms of restructive lung diseases, mainly allergic alveolitis
all forms of pulmonary edema, mainly toxic pulmonary edema
sarcoidoses and granulomatoses, such as Boeck's disease
(ii) Rheumatic diseases/auto-immune diseases/degenerative joint diseases,
which
coincide with inflammatory, allergic and/or proliferative processes:
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
19
all forms of rheumatic diseases, especially rheumatoid arthritis, acute
rheumatic fever,
polymyalgia rheumatica, collagenoses, Behcet's disease
reactive arthritis
inflammatory soft-tissue diseases of other origins
arthritic symptoms in degenerative joint diseases (arthroses)
traumatic arthritides
collagen diseases of other origins, for example systemic lupus erythematodes,
discoid
lupus erythematosus, sclerodermia, polymyositis, dermatomyositis,
polyarteritis nodosa,
temporal arteritis
Sjogren's syndrome, Still syndrome, Felty's syndrome
Vitiligo
Soft-tissue rheumatism
(iii) Allergies, which coincide with inflammatory, allergic and/or
proliferative
processes:
All forms of allergic reactions, for example Quincke's edema, insect bites,
allergic
reactions to pharmaceutical agents, blood derivatives, contrast media, etc.,
anaphylactic
shock, urticaria, allergic vascular diseases
Allergic vasculitis
inflammatory vasculitis
(iv) Vascular inflammations (vasculitides)
Panarteritis nodosa, temporal arteritis, erythema nodosum
Polyarteris nodosa
Wegner's granulomatosis
Giant-cell arteritis
(v) Nephropathies, which coincide with inflammatory, allergic and/or
proliferative
processes:
nephrotic syndrome
all nephritides, such as, for example, glomerulonephritis
(vi) Liver diseases, which coincide with inflammatory, allergic and/or
proliferative
processes:
acute liver cell decomposition
acute hepatitis of different origins, for example virally-, toxically- or
pharmaceutical agent-
induced
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
chronically aggressive and/or chronically intermittent hepatitis
(vii) Gastrointestinal diseases, which coincide with inflammatory, allergic
and/or
proliferative processes:
regional enteritis (Crohn's disease)
5 Gastritis
Reflux esophagitis
ulcerative colitis
gastroenteritis of other origins, for example native sprue
(viii) Proctological diseases, which coincide with inflammatory, allergic
and/or
10 proliferative processes:
anal eczema
fissures
haemorrhoids
idiopathic pro ctitis
15 (ix) Eye diseases, which coincide with inflammatory, allergic and/or
proliferative
processes:
allergic keratitis, uvenitis iritis
conjunctivitis
blepharitis
20 optic neuritis
chorioiditis
sympathetic ophthalmia
(x) Diseases of the ear-nose-throat area, which coincide with inflammatory,
allergic
and/or proliferative processes:
allergic rhinitis, hay fever
otitis externa, for example caused by contact dermatitis, infection, etc.
otitis media
(xi) Neurological diseases, which coincide with inflammatory, allergic
and/or
proliferative processes:
cerebral edema, mainly tumor-induced cerebral edema
multiple sclerosis
acute encephalomyelitis
different forms of convulsions, for example infantile nodding spasms
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
21
Meningitis
spinal cord injury
Stroke
(xii) Blood diseases, which coincide with inflammatory, allergic and/or
proliferative
processes:
acquired haemolytic anemia
thrombocytopenia such as for example idiopathic thrombocytopenia
M. Hodgkins or Non-Hodgkins lymphomas,
thrombocythemias,
erythrocytoses
(xiii) Tumor diseases, which coincide with inflammatory, allergic and/or
proliferative
processes:
acute lymphatic leukaemia
malignant lymphoma
lymphogranulomatoses
lymphosarcoma
extensive metastases, mainly in breast and prostate cancers
(xiv) Endocrine diseases, which coincide with inflammatory, allergic and/or
proliferative
processes:
endocrine orbitopathy
thyrotoxic crisis
de Quervain's thyroiditis
Hashimoto '5 thyroiditis
Hyperthyroidism
Basedow's disease
Granulomatous thyroiditis
Lymphadenoid goiter
(xv) Transplants, which coincide with inflammatory, allergic and/or
proliferative
processes;
(xvi) Severe shock conditions, which coincide with inflammatory, allergic
and/or
proliferative processes, for example anaphylactic shock
(xvii) Substitution therapy, which coincides with inflammatory, allergic
and/or
proliferative processes, with:
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
22
innate primary suprarenal insufficiency, for example congenital adrenogenital
syndrome
acquired primary suprarenal insufficiency, for example Addison's disease,
autoimmune
adrenalitis, meta-infective, tumors, metastases, etc.
innate secondary suprarenal insufficiency, for example congenital
hypopituitarism
acquired secondary suprarenal insufficiency, for example meta-infective,
tumors, etc.
(xviii) Emesis, which coincides with inflammatory, allergic and/or
proliferative processes:
for example in combination with a 5-HT3-antagonist in cytostatic-agent-induced
vomiting.
(xix) Pains of inflammatory origins, e.g., lumbago
Without prejudice to the foregoing, the compounds of the invention can also be
used to treat disorders such as: diabetes type I (insulin-dependent diabetes),
Guillain-Barre
syndrome, restenoses after percutaneous transluminal angioplasty, Alzheimer's
disease, acute
and chronic pain, arteriosclerosis, reperfusion injury, thermal injury,
multiple organ injury
secondary to trauma, acute purulent meningitis, necrotizing enterocolitis and
syndromes
associated with hemodialysis, leukopheresis, granulocyte transfusion, Conies
Syndrome,
primary and secondary hyperaldosteronism, increased sodium retention,
increased
magnesium and potassium excretion (diuresis), increased water retention,
hypertension
(isolated systolic and combined systolic/diastolic), arrhythmias, myocardial
fibrosis,
myocardial infarction, Bartter's Syndrome, disorders associated with excess
catecholamine
levels, diastolic and systolic congestive heart failure (CHF), peripheral
vascular disease,
diabetic nephropathy, cirrhosis with edema and ascites, oesophageal varicies,
muscle
weakness, increased melanin pigmentation of the skin, weight loss,
hypotension,
hypoglycemia, Cushing's Syndrome, obesity, glucose intolerance, hyperglycemia,
diabetes
mellitus, osteoporosis, polyuria, polydipsia, inflammation, autoimmune
disorders, tissue
rejection associated with organ transplant, malignancies such as leukemias and
lymphomas, rheumatic fever, granulomatous polyarteritis, inhibition of myeloid
cell lines,
immune proliferation/apoptosis, HPA axis suppression and regulation,
hypercortisolemia,
modulation of the Thl/Th2 cytokine balance, chronic kidney disease,
hypercalcemia, acute
adrenal insufficiency, chronic primary adrenal insufficiency, secondary
adrenal
insufficiency, congenital adrenal hyperplasia, Little's syndrome, systemic
inflammation,
inflammatory bowel disease, Wegener's granulomatosis, giant cell arthritis,
osteoarthritis,
angioneurotic edema, tendonitis, bursitis, autoimmune chronic active
hepatitis, hepatitis,
cinhosis, panniculitis, inflamed cysts, pyoderma gangrenosum, eosinophilic
fasciitis,
relapsing polychondritis, sarcoidosis Sweet's disease, type 1 reactive
leprosy, capillary
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
23
hemangiomas, lichen planus, erythema nodosum acne, hirsutism, toxic epidermal
necrolysis, erythema multiform, psychoses, cognitive disorders (such as memory
disturbances) mood disorders (such as depression and bipolar disorder),
anxiety disorders
and personality disorders.
As used herein the term "congestive heart failure" (CHF) or 'congestive heart
disease" refers to a disease state of the cardiovascular system whereby the
heart is unable
to efficiently pump an adequate volume of blood to meet the requirements of
the body's
tissues and organ systems. Typically, CHF is characterized by left ventricular
failure
(systolic dysfunction) and fluid accumulation in the lungs, with the
underlying cause being
attributed to one or more heart or cardiovascular disease states including
coronary artery
disease, myocardial infarction, hypertension, diabetes, valvular heart
disease, and
cardiomyopathy. The term "diastolic congestive heart failure" refers to a
state of CHF
characterized by impairment in the ability of the heart to properly relax and
fill with blood.
Conversely, the term "systolic congestive heart failure" refers to a state of
CHF
characterized by impairment in the ability of the heart to properly contract
and eject blood.
As will be appreciated by one of skill in the art, physiological disorders may
present as a
"chronic" condition, or an "acute" episode. The term "chronic", as used
herein, means a
condition of slow progress and long continuance. As such, a chronic condition
is treated
when it is diagnosed and treatment continued throughout the course of the
disease.
Conversely, the term "acute" means an exacerbated event or attack, of short
course,
followed by a period of remission. Thus, the treatment of physiological
disorders
contemplates both acute events and chronic conditions. In an acute event,
compound is
administered at the onset of symptoms and discontinued when the symptoms
disappear.
In another aspect the present invention provides a compound of the invention,
or a
pharmaceutically acceptable salt thereof, for use in therapy (such as a
therapy described
above).
In yet another aspect the present invention provides the use of a compound of
the
invention, or a pharmaceutically acceptable salt thereof, in the manufacture
of a
medicament for the treatment of a glucocorticoid receptor mediated disease
state (such as a
disease state described above).
In a further aspect the invention provides the use of a compound of the
invention,
or a pharmaceutically acceptable salt thereof, in the manufacture of a
medicament for use
in the treatment of an inflammatory condition (such as an arthritic).
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
24
In a further aspect the invention provides the use of a compound of the
invention,
or a pharmaceutically acceptable salt thereof, in the manufacture of a
medicament for the
treatment of a respiratory condition (for example a lung disease as described
above).
In a still further aspect the invention provides the use of a compound of the
invention, or a pharmaceutically acceptable salt thereof, in the manufacture
of a
medicament for the treatment of asthma.
In another aspect the invention provides the use of a compound of the
invention, or
a pharmaceutically acceptable salt thereof, in the manufacture of a medicament
for the
treatment of COPD.
In another aspect the invention provides the use of a compound of the
invention, or
a pharmaceutically acceptable salt thereof, in the manufacture of a medicament
for the
treatment of allergic rhinitis.
In another aspect the invention provides the use of a compound of the
invention, or
a pharmaceutically acceptable salt thereof, in the manufacture of a medicament
for the
treatment of irritable bowel syndrome.
In another aspect the present invention provides a compound of the invention,
or a
pharmaceutically acceptable salt thereof, for use in treating an inflammatory
condition,
asthma, COPD, allergic rhinitis or irritable bowel syndrome.
In yet another aspect the present invention provides a method of treating a
glucocorticoid receptor mediated disease state (such as a disease state
described above) in
a mammal (such as man), which comprises administering to a mammal in need of
such
treatment an effective amount of a compound of the invention, or a
pharmaceutically
acceptable salt thereof
In another aspect the present invention provides a method of treating an
inflammatory condition (such as an arthritic) in a mammal (such as man), which
comprises
administering to a mammal in need of such treatment an effective amount of a
compound
of the invention, or a pharmaceutically acceptable salt thereof
In another aspect the present invention provides a method of treating a
respiratory
condition (such as a lung disease described above) in a mammal (such as man),
which
comprises administering to a mammal in need of such treatment an effective
amount of a
compound of the invention, or a pharmaceutically acceptable salt thereof
In a still further aspect the invention provides a method of treating asthma
in a
mammal (such as man), which comprises administering to a mammal in need of
such
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
treatment an effective amount of a compound of the invention, or a
pharmaceutically
acceptable salt thereof
In a still further aspect the invention provides a method of treating COPD in
a
mammal (such as man), which comprises administering to a mammal in need of
such
5 treatment an effective amount of a compound of the invention, or a
pharmaceutically
acceptable salt thereof
In a still further aspect the invention provides a method of treating allergic
rhinitis
in a mammal (such as man), which comprises administering to a mammal in need
of such
treatment an effective amount of a compound of the invention, or a
pharmaceutically
10 acceptable salt thereof
In a still further aspect the invention provides a method of treating
irritable bowel
syndrome in a mammal (such as man), which comprises administering to a mammal
in
need of such treatment an effective amount of a compound of the invention, or
a
pharmaceutically acceptable salt thereof.
15 The present invention further provides a method of treating a
glucocorticoid
receptor mediated disease state (such as a disease state described above), an
inflammatory
condition, asthma, COPD, allergic rhinitis and/or irritable bowel syndrome, in
a mammal
(such as man), which comprises administering to a mammal in need of such
treatment an
effective amount of a compound of the invention, or a pharmaceutically
acceptable salt
20 thereof.
In the context of the present specification, the term "therapy" and
"treatment" also
includes prophylaxis and prevention unless there are specific indications to
the contrary.
The terms "therapeutic" and "therapeutically" should be construed accordingly.
In this specification, unless stated otherwise, the terms "inhibitor" and
"antagonist"
25 mean a compound that by any means, partly or completely, blocks the
transduction
pathway leading to the production of a response by the agonist. An agonist may
be a full or
partial agonist.
The term "disorder", unless stated otherwise, means any condition and disease
associated with glucocorticoid receptor activity.
Pharmaceutical composition
In order to use a compound of the invention, or a pharmaceutically acceptable
salt
thereof, for the therapeutic treatment of a mammal, said active ingredient is
normally
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
26
formulated in accordance with standard pharmaceutical practice as a
pharmaceutical
composition.
Therefore in another aspect the present invention provides a pharmaceutical
composition comprising a compound of the invention, or a pharmaceutically
acceptable
salt thereof, (active ingredient) and a pharmaceutically acceptable adjuvant,
diluent or
carrier. One embodiment relates to the use of a pharmaceutical composition
comprising a
compound of the invention, or a pharmaceutically acceptable salt thereof, for
treating a
glucocorticoid receptor mediated disease state (such as a disease state
described above), an
inflammatory condition, asthma and/or COPD.
A further aspect the present invention provides a process for the preparation
of said
composition comprising mixing the active ingredient with a pharmaceutically
acceptable
adjuvant, diluent or carrier. Depending on the mode of administration, the
pharmaceutical
composition can comprise from 0.05 to 99 %w (per cent by weight), for example
from 0.05
to 80 %w, such as from 0.10 to 70 %w (for example from 0.10 to 50 %w), of
active
ingredient, all percentages by weight being based on total composition.
A pharmaceutical composition of the present invention can be administered in a
standard manner for the disease condition that it is desired to treat, for
example by topical
(such as to the lung and/or airways or to the skin), oral, rectal or
parenteral administration.
Thus, a compound of the invention or a pharmaceutically acceptable salt
thereof, may be
formulated into the form of, for example, an aerosol, a powder (for example
dry or
dispersible), a tablet, a capsule, a syrup, a granule, an aqueous or oily
solution or
suspension, an (lipid) emulsion, a suppository, an ointment, a cream, drops,
or a sterile
injectable aqueous or oily solution or suspension.
A suitable pharmaceutical composition of this invention is one suitable for
oral
administration in unit dosage form, for example a tablet or capsule containing
between 0.1
mg and 10 g of active ingredient.
In another aspect a pharmaceutical composition of the invention is one
suitable for
intravenous, subcutaneous, intraarticular or intramuscular injection.
In one embodiment the compounds of the invention or a pharmaceutically
acceptable salt thereof, are administered orally.
In another embodiment the compounds of the invention, or a pharmaceutically
acceptable salt thereof, are administered by inhalation.
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
27
In another embodiment the compounds of the invention, or a pharmaceutically
acceptable salt thereof, are administered nasally.
Inhalation (particularly oral inhalation) is a particularly useful method for
administering a compound of the invention (or a pharmaceutically acceptable
salt thereof)
when treating respiratory diseases such as chronic obstructive pulmonary
disease (COPD)
or asthma. When administered by oral inhalation, a compound of the invention
(or a
pharmaceutically acceptable salt thereof) may be used effectively at a daily
dose in the ,ug
range, for example up to 500 pg, such as from 0.1 to 50 jig, from 0.1 to 40
,g, from 0.1 to
30 ,g, from 0.1 to 20 i.tg or from 0.1 to 10 ,g, of a compound of the
invention (or a
pharmaceutically acceptable salt thereof) as active ingredient.
A pharmaceutical composition of the invention may be administered by oral
inhalation in any suitable form and using any suitable inhaler device.
Suitable inhaler
devices are known to persons skilled in the art and may be manual or breath
actuated. The
pharmaceutical composition may be formulated as a dry powder, as a suspension
(in a
liquid or gas) or as a solution (in a liquid) for administration by oral
inhalation by means of
a suitable inhaler device.
Inhaler devices suitable for pulmonary administration include metered dose
inhalers (MDIs), dry powder inhalers (DPIs), nebulisers and soft mist
inhalers. Multi-
chamber devices may be used to allow for delivery of a compound of the
invention (or a
pharmaceutically acceptable salt thereof) and one or more further active
ingredients (when
present).
A preferred metered dose inhaler device is a pressurised metered dose inhaler
(pMDI).
A pharmaceutical composition for use in a pMDI may be provided in the form of
a
solution or suspension comprising the active ingredient and one or more
excipients, the
excipients including a suitable propellant in which the active ingredient is
dissolved or
dispersed. Suitable propellants are known to persons skilled in the art and
include
hydrocarbon, chlorofluorocarbon and hydrofluoroalkane propellants, or mixtures
of any
such propellants. Examples of propellants are 1,1,1,2,-tetrafluoroethane (HFA
or HFC
134a) and 1,1,1,2,3,3,3-heptafluoropropane (HFA or HFC 227), each of which may
be
used alone or in combination with other propellants and/or other excipients.
A pMDI device contains the pharmaceutical composition in a pressurised
container.
The active ingredient is delivered by actuating a valve of the container of
the pMDI device.
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
28
Actuation may be manual or breath actuated. In a manually actuated pMDI, the
device is
actuated by a user as they inhale, for example by pressing a suitable release
mechanism on
the pMDI device. A breath actuated pMDI device is actuated automatically when
the user
inhales through a mouthpiece of the pMDI. Examples of pMDI devices include for
example Rapihaler0, VannairO, Ventolin0 HFA, Evohaler0, MaxairO, Autohaler0
and
Easi-Breathe .
A metered dose inhaler device (such as a pMDI) may be used in combination with
a spacer device. Suitable spacer devices are well known to persons skilled in
the art and
include Nebuchamber0 or Volumatic0.
A pharmaceutical composition for use in a dry powder inhaler device is
provided in
the form of a dry powder comprising the active ingredient and one or more
excipients, the
excipients typically including a suitable carrier and/or diluent and/or
coating agent. The
active ingredient is provided in an inhalable form and preferably the
particles of the active
ingredient have a mass median aerodynamic diameter of less than about 10 m,
more
preferably of less than about 5 m, for example from 1 to 5 m. Persons skilled
in the art
may measure the mass median aerodynamic diameter using standard techniques
known to
them. Inhalable forms of the active ingredient may be may be prepared by a
variety of
techniques, including spray-drying, freeze-drying and micronisation.
The dry powder composition may take the form of a powder agglomerate or an
ordered mixture.
When the dry powder composition takes the form of an ordered mixture the
mixture
may comprise inhalable particles of the active ingredient formulated with
carrier particles
that aid flow from the dry powder inhaler device into the lung. The particles
of the active
ingredient adhere to the carrier particles to form an ordered (interactive)
powder mixture.
Suitable carrier particles for inclusion in such dry powder compositions are
known, and
include sugars, for example, lactose, glucose, raffinose, melezitose,
lactitol, maltitol,
trehalose, sucrose, mannitol and starch. Suitable carriers are lactose
particles and they may
have a mass median aerodynamic diameter of greater than 90[tm.
When the dry powder composition takes the form of a powder agglomerate the
agglomerate may comprise the active ingredient in the form of microparticles
formulated
with one or more diluents. Suitable diluents include sugars, for example
lactose, mannitol
and sucrose.
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
29
The active ingredient and/or excipients used in powder compositions for
inhalation
may be conditioned before, during or after formulation. Conditioning may be
useful in, for
example, restoring crystallinity and maintaining aerodynamic properties of the
particles.
Conditioning processes are well known and include exposure of particles to
controlled
temperature and humidity/solvent vapour. Examples of conditioning processes
include
those described in W092/018110 and W095/05805.
Dry powder inhaler devices may be single dose, multiple unit dose or multi-
dose
(reservoir) inhalers, and may utilise a dry powder or a powder-containing
capsule.
In single-dose dry powder inhaler devices, individual doses are provided,
usually in
capsules (such as gelatine capsules), and are loaded into the device before
use. Examples
of these devices include Spinhaler , Rotahaler , AeroliserTM, Inhalator and
Eclipse
devices. Multiple unit dose dry powder inhaler devices contain a number of
individually
packaged doses, either as multiple capsules (such as gelatine capsules) or in
blister packs.
Examples of these devices include Diskhaler , Diskus and Aerohaler devices
and
breath-actuated, dry-powder inhaler devices having multiple cavities for
powder arranged
in a disc or ring, such as is disclosed in W02005/002654, W02012/010877, or
W02012/010878. In multi-dose (reservoir) dry powder inhaler devices, the
active
ingredient is stored in a bulk powder reservoir from which individual doses
are metered.
Examples of these devices include Turbuhaler , Easyhaler , Novolizer ,
Clickhaler ,
Spiromax , Airmax and Pulvinal devices.
Nebuliser devices may for example be used to administer the active ingredient
as an
aqueous suspension or, preferably, solution, with or without a suitable pH
and/or tonicity
adjustment, either as a unit-dose or multi-dose formulation. Suitable
nebulisers are well
known to persons skilled in the art and include the eFlow0.
Nasal administration of a compound of the invention (or a pharmaceutically
acceptable salt thereof) may be provided by means of a spray from a suitable
nasal delivery
device, such as a spray pump or an MDI nasal delivery device, for example
Rhinocort
Aqua . Alternatively, the compound of the invention (or a pharmaceutically
acceptable
salt thereof) may be administered nasally as a powder using a suitable DPI
device, for
example Rhinocort0 or Turbuhaler0.
A pharmaceutical composition for use in a spray pump or MDI nasal delivery
device may comprise a compound of the invention (or a pharmaceutically
acceptable salt
thereof) dispersed or preferably dissolved in a suitable aqueous medium. Where
it is
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
desirable to limit the penetration of the active ingredient into the lung and
to retain the
active ingredient in the nasal cavity, it may be necessary to use particles of
the active
ingredient having a mean mass aerodynamic diameter greater than about 10gm,
for
example from 10 gm to 50 gm.
5 Buffers, pharmaceutically-acceptable cosolvents such as polyethylene
glycol,
polypropylene glycol, glycerol or ethanol or complexing agents such as hydroxy-
propyl 13-
cyclodextrin may be used to aid formulation.
The above formulations may be obtained by conventional procedures well known
in the pharmaceutical art. Tablets may be enteric coated by conventional
means, for
10 example to provide a coating of cellulose acetate phthalate.
The invention further relates to combination therapies or compositions wherein
the
compounds of the invention, or a pharmaceutically acceptable salt thereof, or
a
pharmaceutical composition comprising the compounds of the invention, or a
pharmaceutically acceptable salt thereof, is administered concurrently
(possibly in the
15 same composition) or sequentially with one or more agents for the
treatment of any of the
above disease states.
For example, for the treatment of rheumatoid arthritis, osteoarthritis, COPD,
asthma, irritable bowel syndrome or allergic rhinitis a compound of the
invention, or a
pharmaceutically acceptable salt thereof, can be combined with one or more
agents for the
20 treatment of such a condition. Where such a combination is to be
administered by
inhalation, then the one or more agents is selected from the list comprising:
= a PDE4 inhibitor including an inhibitor of the isoform PDE4D;
= a selective 132 adrenoceptor agonist such as metaproterenol,
isoproterenol,
isoprenaline, albuterol, salbutamol, formoterol, salmeterol, terbutaline,
25 orciprenaline, bitolterol mesylate, pirbuterol, indacaterol, olodaterol,
milveterol or
vilanterol;
= a muscarinic receptor antagonist (for example a Ml, M2 or M3 antagonist,
such
as a selective M3 antagonist) such as ipratropium bromide, tiotropium bromide,
oxitropium bromide, pirenzepine, telenzepine, aclidinium bromide or
30 glycopyrronium bromide;
= a steroid (such as budesonide);
= a modulator of chemokine receptor function (such as a CCR1 receptor
antagonist);
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
31
= an inhibitor of p38 kinase function;
= an inhibitor of matrix metalloproteases, most preferably targeting MMP-2,
-9 or
MMP-12; or,
= an inhibitor of neutrophil serine proteases, most preferably neutrophil
elastase or
proteinase 3.
In another embodiment of the invention where such a combination is for the
treatment of COPD, asthma or allergic rhinitis, the compounds of the
invention, or a
pharmaceutically acceptable salt thereof, can be administered by inhalation or
by the oral
route and the other agent, e.g. xanthine (such as aminophylline or
theophylline) can be
administered by inhalation or by the oral route. The compounds of the
invention, or a
pharmaceutically acceptable salt thereof, and the other agent, e.g xanthine
may be
administered together. They may be administered sequencially. Or they may be
administered separately.
Examples
The invention is illustrated, but in no way limited, by the following Examples
and
with reference to the enclosed Figures.
The following abbreviations may be used:
DSC Differential scanning calorimeter
HPLC High-performance liquid chromatography
MTDSC Modulated temperature differential scanning calorimeter
NMR Nuclear magnetic resonance
UV Ultraviolet
XRPD X-Ray powder diffraction
PS80 Polysorbate 80
LOD Loss on drying
LC Liquid chromatography
GC Gas chromatography
General Procedures
X-Ray powder diffraction analysis (XRPD) were performed on samples prepared
according to standard methods, for example those described in Giacovazzo, C.
et at (1995),
Fundamentals of Crystallography, Oxford University Press; Jenkins, R. and
Snyder, R. L.
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
32
(1996), Introduction to X-Ray Powder Diffractometry, John Wiley & Sons, New
York;
Bunn, C. W. (1948), Chemical Crystallography, Clarendon Press, London; or
Klug, H. P.
& Alexander, L. E. (1974), X-ray Diffraction Procedures, John Wiley and Sons,
New
York. X-ray analyses were performed using a Panalytical X'Pert PRO MPD
instrument
with the following parameters:
= CuKc, (1.5418A)
= 45 kV and 40 mA
= 20 20 400
= 4 /min, incr. 0.016
= Rotating Silicon wafer
= Ambient conditions
= Approximately 2 mg of a test sample was placed on the sample holder and
smeared out
on the silicon surface using a flat Teflon bar.
It is known in the art that an X-ray powder diffraction pattern may be
obtained
which has one or more measurement errors depending on measurement conditions
(such as
equipment, sample preparation or machine used). In particular, it is generally
known that
intensities in an X-ray powder diffraction pattern may fluctuate depending on
measurement
conditions and sample preparation. For example, persons skilled in the art of
X-ray powder
diffraction will realise that the relative intensities of the peaks may vary
according to the
orientation of the sample under test and on the type and setting of the
instrument used. The
skilled person will also realise that the position of reflections can be
affected by the precise
height at which the sample sits in the diffractometer and the zero calibration
of the
diffractometer. The surface planarity of the sample may also have a small
effect. Hence a
person skilled in the art will appreciate that the diffraction pattern data
presented herein is
not to be construed as absolute and any crystalline form that provides a
powder diffraction
pattern substantially identical to those disclosed herein fall within the
scope of the present
disclosure. Generally, a measurement error of a diffraction angle in an X-ray
powder
diffraction pattern is about 5% or less, typically plus or minus 0.2 2-theta.
Melting point was determined by Differential Scanning Calorimetry (DSC) using
standard methods, for example those described in Hohne, G. W. H. et al (1996),
Differential Scanning Calorimetry, Springer, Berlin. The calorimetric response
of a test
sample to increasing temperature was investigated using a TA Instruments Q2000
Modulated Temperature Differential Scanning Calorimeter (MTDSC) using a
modulation
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
33
of 0.50 C in intervals of 40 seconds and a ramp rate of 5 C per minute.
Approximately 1
mg of test sample was placed in aluminium cups with lids (no crimping) under a
nitrogen
atmosphere. Where a melting point is quoted, this refers to the onset
temperature of the
melting endotherm.
A person skilled in the art will appreciate that slight variations in the
melting point
measured by DSC may occur as a result of variations in sample purity, sample
preparation
and the measurement conditions (e.g. heating rate). It will be appreciated
that alternative
readings of melting point may be given by other types of equipment or by using
conditions
different to those described hereinafter. Hence the melting point and
endotherm figures
quoted herein are not to be taken as absolute values and such measurement
errors are to be
taken into account when interpreting DSC data. Typically, measurement errors
using DSC
may vary by 0.5 C or less. However, as a skilled person will realise,
melting point can
vary with sample purity and degree of crystallinity of the sample. Even low
levels of
impurities can affect the measured melting point. Therefore, the melting
points disclosed
herein may vary by 5 C from the values quoted herein and reference to a
substance
having a melting point of "about" are to be interpreted as having a value of
5 C from the
values quoted. It is to be understood that references to melting points
disclosed herein
refer to the onset temperature of the melting endotherm. A person skilled in
the art can use
routine optimization/calibration to set up instrumental parameters for a
differential
scanning calorimeter so that data comparable to the data presented herein can
be collected.
A person skilled in the art will further appreciate that slight variations in
the
measurements of solubility given in the examples herein may occur as a result
of sample
purity, polymorph purity, sample preparation and the measuring conditions
(e.g.
temperature, time and degree of agitation). It will be appreciated that
alternative
measurements of solubility may be given by using conditions different to those
described
hereinafter. Hence the measurements of solubility quoted herein are not to be
taken as
absolute values.
It will be further appreciated by a person skilled in the art that slight
variations in
the measurements of hygroscopicity given in the examples herein may occur as a
result of
sample purity, polymorph purity, sample preparation and the measuring
conditions (e.g.
equipment and parameters used). It will be appreciated that alternative
measurements of
hygroscopicity may be given by using conditions different to those described
hereinafter.
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
34
Hence the measurements of hygroscopicity quoted herein are not to be taken as
absolute
values.
Proton (1H) nuclear magnetic resonance (NMR) spectra were acquired using
Varian
(Inova 400 MHz) or Bruker (Avance 500 or DPX 300) spectrometers, at 25 C or
300 K.
Samples were prepared as solutions in a suitable deuterated solvent (d6-DMS0 ¨
d6-
dimethyl sulfoxide, CDC13 ¨ d-chloroform, or d6-acetone), optionally
containing
trimethylsilane (TMS). Sample solutions may also contain an internal standard
(either
maleic acid or 2,3,5,6-tetrachloronitrobenzene) for assay determination and/or
added
trifluoroacetic acid, to move exchangeable proton signals (e.g. from maleic
acid) away
from analyte resonances. Spectral data is reported as a list of chemical
shifts (6, in ppm)
with a description of each signal, using standard abbreviations (s = singlet,
d = doublet, m
= multiplet, t = triplet, q = quartet, br = broad, etc.). Spectra were
referenced relative to
TMS (6 = 0.00 ppm), d5-DMS0 (6 = 2.50 ppm), chloroform (6 = 7.24 ppm) or d5-
acetone
(6 = 2.05 ppm). J-Coupling constants are listed, where measured, in the
descriptions of the
resonances. Slight variation of chemical shifts and J-coupling constants may
occur, as is
well known in the art, as a result of variations in sample preparation, such
as analyte
concentration variations and including or omitting additives (for example NMR
assay
standards or trifluoroacetic acid).
Loss-on-drying analysis was performed using a Mettler Toledo HR83 Moisture
Analyser or Perkin-Elmer TGA7 Thermogravimetric Analyzer.
Large scale reactions were carried out in glass-lined steel reactors fitted
with heat
transfer jackets and serviced with appropriate ancillary equipment. Large
scale preparative
chromatography was performed using a Novasep LC150 preparative HPLC system,
equipped with a dynamic axial compression column, of internal diameter 15 cm.
Standard
laboratory glassware and equipment was used for small scale processes.
Starting materials,
solvents and reagents were purchased commercially and used as supplied.
Liquid chromatography (LC) was performed on reversed phase columns packed
with octadecyl or phenyl bonded silica. Agilent 1100 HPLC instruments equipped
with
UV detectors (k = 230 nm unless stated otherwise) were used. Stationary phase
particle
size, column dimensions, mobile phases (acetonitrile and water, pH adjusted
with
trifluoroacetic acid or ammonium formate/ammonia), gradient timetables, flow
rates and
temperature suitable for the specific analyses were used. Sample solutions
were prepared
at a main analyte concentration of approximately 0.5 mg mL-1 using suitable
diluents.
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
Gas chromatography (GC) was performed using helium as carrier gas on a DB-624
capillary column. Agilent 6890 GC instruments equipped with flame-ionisation
detectors
were used.
Water analysis was performed by coulometric Karl-Fischer titration using a
5 Metrohm 756 KF coulometer.
Summary of XRPD Analysis of Form A and Form B of Compound (I)
Crystals of Compound (I) Form A and Form B obtained as described herein were
analysed by XRPD and the results are tabulated below (RI represents relative
intensity)
10 and are shown in the respective Figures.
Table 1 shows the most significant peaks in the XRPD-diffractogram of
crystalline Form
B of Compound (I). Table 2 shows the most significant peaks in the XRPD-
diffractogram
of crystalline Form A of Compound.
A number of weak and very weak peaks have been omitted. Due to preferred
orientation
effects some of the weak omitted peaks may become more significant.
Table 1 (Form B) Table 2 (Form A)
Position RI Position RI
2-Theta 2-Theta
21.5 vs 18.7 vs
20.6 s 18.1 vs
20.3 s 17.8 vs
19.9 s 16.7 s
17.4 vs 14.9 vs
15.7 vs 13.3 vs
14.4 s 11.9 s
11.8 vs 10.7 s
9.9 s 9.7 vs
9.2 vs 8.4 5
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
36
Abbreviations
vs = very strong; s = strong
Summary of Figures
Figure 1 shows the XRPD-diffractogram of crystalline Form B of Compound (I).
Figure 2 shows the differential scanning calorimetry profile of crystalline
Form B of
Compound (I).
Figure 3 shows the XRPD-diffractogram of crystalline Form A of Compound (I).
Figure 4 shows the differential scanning calorimetry profile of crystalline
Form A of
Compound (I).
Example 1: Preparation of Form B of Compound (I)
(i) 2,2,2-Trifluoro-N-[(1R,2S)-141-(4-fluorophenyl)indazol-5-ylloxy-1-(3-
methoxyphenyl)propan-2-yllacetamide (Form B)
F 0
0
F N
el 0 41100
F
The solution obtained from Example 1, step (ii) was further diluted with 1-
propanol
(17.0 kg) and adjusted to 60 C. n-Heptane (50.4 kg) was charged gradually
whilst
maintaining the temperature at 60 C. The solution was cooled to 50 C,
charged with
seeds of Form B of Compound (I) (50 g) to initiate crystallisation, and
gradually cooled to
8 C. Additional n-heptane (25.5 kg) was charged over 20 minutes before
analysing a
sample of the filtered crystals by XRPD, confirming crystallisation of Form B.
The batch
was filtered in two roughly equal parts, diluting the second part with further
additional n-
heptane (3.5 kg) before filtering. The filter cake was washed with a cold
mixture of 1-
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
37
propanol (7.2 kg) and n-heptane (18.0 kg) and dried under vacuum. Yield 9.15
kg (18.7
mol, 77%). 99.8% Assay (NMR) and 0.2% LOD, both by mass.
11-1NMR (400 MHz, d6-DMS0) 8 9.50 (d, J = 8.5 Hz, 1H), 8.17 (d, J = 0.8 Hz,
1H), 7.73 (m, 2H), 7.69 (d, J = 9.1 Hz, 1H), 7.39 (m, 2H), 7.26 (dd, J = 8.2,
7.7 Hz, 1H),
7.20 (dd, J = 9.1, 2.4 Hz, 1H), 7.13 (d, J = 2.4 Hz, 1H), 6.98 (d, J= 7.7 Hz,
1H), 6.95 (m,
1H), 6.83 (dd, J = 8.2, 2.6 Hz, 1H), 5.27 (d, J = 6.4 Hz, 1H), 4.25 (m, 1H),
3.72 (s, 3H),
1.33 (d, J = 6.8 Hz, 3H).
The XRPD diffractogram of the form of Compound (I) obtained by way of
Example 1 (Form B) is shown in Figure 1 below and is tabulated in Table 1
above.
The DSC profile of the form of Compound (I) obtained by way of Example 1
(Form B) is shown in Figure 2 below. The compound exhibited an onset
temperature of the
melting endotherm of 109 C.
The starting material used in step (i) was prepared as follows.
(ii) 2,2,2-Trifluoro-N-[(1R,2S)-141-(4-fluorophenyl)indazol-5-yl]oxy-1-(3-
methoxyphenyl)propan-2-yl]acetamide
o
FF>1))., 0
-LENI _ el \ N
F N
el 0 104
F
Ethyl trifluoroacetate (14.3 kg, 101 mol) followed by a methyl tert-butyl
ether
(11.2 kg) line wash was charged to a stirring suspension of (1R,25)-1-[1-(4-
fluorophenyl)indazol-5-yl]oxy-1-(3-methoxyphenyl)propan-2-amine hydrochloride
(14.15
kg, 25.1 mol; assay = 75.9% by mass) in methyl tert-butyl ether (35.9 kg) and
triethylamine (7.6 kg, 75 mol). The mixture was stirred at 22 C for 24 hours,
by which
time LC analysis showed that there was 0.8% residual starting material.
Aqueous sodium hydroxide (53 kg, 45 mol) was charged, the mixture stirred for
30
minutes (excess ethyl trifluoroacetate is hydrolysed to trifluoroacetic acid
during this time)
and then allowed to separate into layers before discarding the lower (aqueous)
phase. The
upper (organic) phase was washed successively with aqueous hydrochloric acid
(50 kg, 50
mol) and then water (54 kg), and then screened through a 0.6 [tm filter into a
clean vessel.
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
38
The solution was concentrated by distilling off solvent (31 kg) at < 70 C
(atmospheric pressure). Further solvent (37 kg) was distilled off at < 70 C
under reduced
pressure (900-100 mbar) whilst continuously replacing with 1-propanol (35 kg).
Analysis
showed that there was 11.8 kg (24.2 mol) of 2,2,2-trifluoro-N-[(1R,25)-141-(4-
fluorophenyl)indazol-5-yl]oxy-1-(3-methoxyphenyl)propan-2-yl]acetamide and 7.1
kg of
1-propanol, with < 0.05% methyl tert-butyl ether and < 0.1% water by volume.
The
solution was used directly in the crystallisation step. Yield 96%.
(iii) (1R,2S)-141-(4-Fluorophenyl)indazol-5-yl]oxy-1-(3-methoxyphenyl)propan-2-
amine hydrochloride
ci
H3+1\1: o el ",N
N
el 0 410
F
N-R1S,2R)-2-[1-(4-Fluorophenyl)indazol-5-yl]oxy-2-(3-methoxypheny1)-1-methyl-
ethy1]-4-nitro-benzenesulfonamide (23.3 kg, 35.6 mol; assay = 88.1% by mass)
and
potassium carbonate (19.90 kg, 144 mol; 325-mesh sieved grade) were added to
stirring
acetonitrile (322 kg). After sparging the reaction mixture to remove oxygen
for one hour,
thioglycolic acid (7.10 kg, 77.1 mol) was added before heating to 75 C. After
24 hours,
extra thioglycolic acid (1.75 kg, 19.0 mol) and potassium carbonate (4.92 kg,
35.6 mol)
were charged. After a further 18 hours, LC analysis showed 98.7% conversion of
the 4-
nitro-benzenesulfonamide starting material to the amine product by area.
Water (203.6 kg) was charged and then the mixture concentrated by distilling
off
solvent (126.9 kg) at atmospheric pressure. Methyl tert-butyl ether (165.8 kg)
was
charged, and after allowing the mixture to settle into layers, the lower
(aqueous) phase was
removed. The organic phase was washed successively with 1M sodium hydroxide
(235.6
kg, 225 mol), 1M hydrochloric acid (229.7 kg, 240 mol) and finally saturated
aqueous
sodium chloride (229.6 kg).
After concentrating the solution by distilling off solvent (151.9 kg) at
atmospheric
pressure, it was diluted with methyl tert-butyl ether (328.1 kg), re-
concentrated (327.8 kg
solvent distilled off) and diluted again at 40 C with more methyl tert-butyl
ether (123.9
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
39
kg). (The water content of the resulting mixture was determined as 21 g L-1.)
The slurry
was cooled to 2 C, diluted with further methyl tert-butyl ether (152.2 kg)
and then filtered.
The solids were washed with methyl tert-butyl ether (34.6 kg) and then dried
at 50 C
under vacuum (final LOD analysis 0.2%). Yield 14.3 kg (25.4 mol, 71% by
moles).
75.9% Assay and 2.5% residual methyl tert-butyl ether, both by mass (NMR). The
major
part of the remaining mass is sodium chloride.
1H NMR (400 MHz, d6-DMS0) 6 8.09 (s, 1H), 7.68 - 7.63 (m, 3H), 7.35 - 7.29
(m, 2H), 7.30 (dd, J= 2.4, 9.1 Hz, 1H), 7.27 (t, J = 7.8 Hz, 1H), 7.12 (d, J =
2.3 Hz, 1H),
6.96 -6.92 (m, 2H), 6.84 (dd, J = 2.4, 8.3 Hz, 1H), 5.62 (d, J= 3.1 Hz, 1H),
3.67 (s, 3H),
3.66 (qd, J= 3.1, 6.8 Hz, 1H), 1.17 (d, J= 6.8 Hz, 3H). The three exchangeable
ammonium protons are coalesced with the exchangeable protons of maleic acid
(added to
the NMR sample) and water in the recorded spectrum.
(iv) N-[(1S,2R)-241-(4-Fluorophenyl)indazol-5-yl]oxy-2-(3-methoxypheny1)-1-
methyl-ethyl]-4-nitro-benzenesulfonamide
0-
1 +
, N
0' el
"
0 H I ,N
lei N
0 it
F
A slurry of (1R,2S)-2-amino-1-(3-methoxyphenyl)propan-1-ol hydrochloride
(12.25 kg, 55.9 mol; 99.4% assay by mass) and 4-nitrobenzenesulfonyl chloride
(13.6 kg,
61.4 mol) in 2-methyltetrahydrofuran (260 kg) was heated to 40 C. A mixture
of N-
methylmorpholine (28.4 kg, 280 mol) and 2-methyltetrahydrofuran (23.9 kg) was
added
over 30 minutes, followed by a 2-methyltetrahydrofuran (12.5 kg) line wash.
After stirring
for one hour, LC analysis showed 99.9% conversion by area (k = 254 nm) of the
amine
starting material to N-[(1S,2R)-2-hydroxy-2-(3-methoxypheny1)-1-methyl-ethyl]-
4-nitro-
benzenesulfonamide.
Methanesulfonyl chloride (12.6 kg, 110 mol) was added at 40 C over 10 minutes
followed by a 2-methyltetrahydrofuran (12.5 kg) linewash. The reaction mixture
was
stirred for 16 hours at 40 C to give [(1R,25)-1-(3-methoxypheny1)-2-[(4-
nitrophenyl)sulfonylamino]propyl] methanesulfonate
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
After sequential washing with 5M hydrochloric acid (61.1 kg, 283 mol) and then
water (56.2 kg), 10M Sodium hydroxide (30.4 kg, 223 mol) was added, followed
by a line
wash with water (12.4 kg), to form (2S,3S)-2-(3-methoxypheny1)-3-methy1-1-(4-
nitrophenyl)sulfonyl-aziridine.
5 1-(4-Fluorophenyl)indazol-5-ol (14.32 kg, 61.2 mol; 97.5% assay by mass)
was
added and the reaction mixture stirred for 17 hours at 40 C to form the
subtitle compound.
(LC analysis showed 0.8% of the residual aziridine intermediate by area, k =
254 nm.)
The lower liquid phase was removed after allowing the mixture to settle and
the
upper (organic) phase was then washed sequentially with 5M hydrochloric acid
(60.7 kg,
10 284 mol) and then water (56.5 kg). Solvent (200 kg) was distilled off at
atmospheric
pressure and then toluene (129.2 kg) was added whilst distilling off further
solvent (122.0
kg), matching the rate of toluene addition with the rate of distillation. (GC
analysis of the
solution then showed 5.8% 2-methyltetrahydrofuran by volume.)
The solution was diluted with toluene (109.1 kg), cooled to 50 C, seeded
15 (approximately 2-5 g of the subtitle compound, slurried in toluene) and
further cooled to 0
C. Finally, the solids were filtered off, washed with toluene (50.5 kg) and
dried at 40 C
under vacuum (final LOD analysis 0.6%). Yield 23.30 kg (35.6 mol, 64% by
moles).
88.1% Assay and 12.8% toluene, both by mass (NMR).
11-1NMR (400 MHz, d6-DMS0) 6 8.47 (d, J= 8.3 Hz, 1H), 8.08 (d, J = 8.8 Hz,
20 2H), 8.02 (s, 1H), 7.93 (d, J= 8.9 Hz, 2H), 7.74- 7.68 (m, 2H), 7.60 (d,
J = 9.1 Hz, 1H),
7.40 -7.34 (m, 2H), 7.22 -7.18 (m, 1H)*, 7.08 (dd, J = 2.4, 9.1 Hz, 1H), 6.84
(d, J = 7.7
Hz, 1H), 6.76 - 6.72 (m, 3H), 5.01 (d, J= 4.5 Hz, 1H), 3.72 (dqd, J = 4.5,
6.8, 8.3 Hz, 1H),
3.65 (s, 3H), 1.09 (d, J = 6.8 Hz, 3H). The indicated (*) resonance is
obscured by toluene
signals.
(v) 1-(4-Fluorophenyl)indazol-5-ol
HO
eI \ N
N'
F
A slurry of 5-hydroxy-1H-indazole (12.4 kg, 91.5 mol; assay 99% by mass),
tris(dibenzylideneacetone)dipalladium(0) (1.44 kg, 1.57 mol) and 2-di-tert-
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
41
butylphosphino-2',4',6'-triisopropylbiphenyl (1.35 kg, 3.18 mol) in 2-
methyltetrahydrofuran (42.5 kg) was prepared. Sodium tert-butoxide (17.45 kg,
182 mol)
solution in 2-methyltetrahydrofuran (53.5 kg), 1-chloro-4-fluorobenzene (12.45
kg, 95.4
mol) and then a 2-methyltetrahydrofuran (10.50 kg) line wash were charged
sequentially to
the stirring mixture. The slurry was heated at 73 C for 15 hours (LC analysis
then showed
0.4% residual 5-hydroxy-1H-indazole by area, with k = 222 nm).
Water (73.5 kg), heptane (18.9 kg), 10M hydrochloric acid (8.8 kg, 77 mol) and
a
water (12.3 kg) line wash were then charged after cooling to 50 C. After
phase
separation, the subtitle compound was extracted from the organic phase with
two portions
of aqueous sodium hydroxide (91.5 kg at 0.7 M, 62 mol and then 51.3 kg at 1.0
M, 48
mol). The combined sodium hydroxide extracts were diluted with ethanol (74.6
kg), acetic
acid (4.8 kg, 80 mol) and then, gradually over 45 minutes, a solution of
acetic acid (5.5 kg,
92 mol) in ethanol (19.5 kg). After cooling to -10 C, the solids were
filtered off, washed
with a mixture of water (37.1 kg) and ethanol (19.8 kg) and dried at 40 C
under vacuum
(final LOD analysis 0.3%). Yield 15.40 kg (65.8 mol, 72% by moles). 97.5%
assay by
mass (NMR).
1H NMR (500 MHz, d6-acetone) 6 8.31 (s, 1H), 8.08 (s, 1H), 7.82 -7.78 (m, 2H),
7.68 (d, J = 9.0 Hz, 1H), 7.37 - 7.32 (m, 2H), 7.20 (d, J = 2.0 Hz, 1H), 7.10
(dd, J = 2.0,
9.0 Hz, 1H).
(vi) (1R,2S)-2-Amino-1-(3-methoxyphenyl)propan-l-ol hydrochloride
ci
H 3+ N 0 H
_
lei 0
To a solution of tert-butyl N-R1S)-2-(3-methoxypheny1)-1-methy1-2-oxo-
ethyl]carbamate (24.54 kg, 87.8 mol) in toluene (86 kg) and isopropanol (53.5
kg, 890
mol), aluminium isopropoxide (3.7 kg, 18 mol) was added. The reaction was
heated up to
50 C and stirred for 13 hours to give tert-butyl N-R1S,2R)-2-hydroxy-2-(3-
methoxypheny1)-1-methyl-ethyl]carbamate.
1M Hydrochloric acid (50.6 kg, 50 mol) was added over 1 hour, maintaining the
temperature between 15-25 C, followed by a water rinse (2 kg). After phase
separation,
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
42
the aqueous layer was extracted with two portions of ethyl acetate (34 kg per
portion). The
combined organics were washed with saturated sodium hydrogen carbonate (50.3
kg) and
saturated aqueous sodium chloride (50.3 kg).
The organic solution was concentrated in vacuo to an oil and ethyl acetate
(110 kg)
was charged. The organic solution was concentrated in vacuo to an oil and
ethyl acetate
(110 kg) was charged.
Hydrogen chloride gas (9.7 kg, 266 mol) was charged over 4 hours, maintaining
the
temperature between 0-5 C. The reaction contents were heated to 15 C and
stirred for 12
hours.
Methyl tert-butyl ether (90 kg) was added and the vessel contents cooled to 0
C.
The slurry was stirred for 2 hours at 0 C and filtered under vacuum. The damp
cake was
slurry washed with methyl tert-butyl ether (72 kg), stirring for 30 minutes at
0 C before
filtering under vacuum. The filter cake was washed with methyl tert-butyl
ether (20 kg)
and then dried in a vacuum oven at 50 C. Yield 16.3 kg (74.2 mol, 85% by
moles).
99.1% Assay by mass (NMR).
1H NMR (400 MHz, d6-DMS0) 6 8.09 (s, 3H), 7.28 (t, J= 8.0 Hz, 1H), 6.94 ¨ 6.91
(m, 2H), 6.87 ¨ 6.83 (m, 1H), 6.03 (d, J= 4.2 Hz, 1H), 4.91 (t, J = 3.5 Hz,
1H), 3.75 (s,
3H), 3.38 (qd, J= 3.0, 6.7 Hz, 1H), 0.94 (d, J= 6.7 Hz, 3H).
(Ai) Tert-butyl N-[(1S)-2-(3-methoxypheny1)-1-methyl-2-oxo-ethyl]carbamate
0
>ON
o
To a cooled (0-5 C) solution of N-(tert-butyloxycarbony1)-L-alanine (45.0 kg,
238
mol) in dichloromethane (596.5 kg) was added 1,1-carbonyldiimidazole (52.5 kg,
324 mol)
over 3 hours and the resultant solution maintained at 0-5 C for 30 minutes.
N,0-
Dimethylhydroxylamine hydrochloride (31.5 kg, 323 mol) was added over 1 hour
30
minutes and the resultant solution maintained at 0-5 C for 30 minutes.
After stirring for 14 hours at 15 C, the reaction mixture was washed
sequentially
with two portions of 1M hydrochloric acid (164.5 kg, 163 mol and 166 kg, 164
mol), 10%
aqueous sodium hydrogen carbonate (164.5 kg) and then 20% aqueous sodium
chloride
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
43
solution (199 kg) to give an organic phase solution of tert-butyl N-[(1S)-2-
(methoxy
(methyl)amino)-1-methy1-2-oxo-ethyl]carbamate (606 kg). Half of the solution
(303 kg)
was solvent swapped by distillation into tetrahydrofuran (220 kg).
The solution was cooled to below 10 C and isopropylmagnesium chloride 1.91 M
in tetrahydrofuran (54 kg, about 114 mol) added over 1 hour 20 minutes,
maintaining the
temperature between 10-15 C, followed by a tetrahydrofuran rinse (3 kg). 3-
Methoxy
phenylmagnesium bromide 0.86 M in THF (203 kg, about 202 mol) was gradually
added,
maintaining the temperature between 10-15 C, followed by a tetrahydrofuran
rinse (3 kg).
After warming to 20 C, 20% aqueous acetic acid (101 kg) was added,
maintaining
the temperature below 30 C, followed by a water rinse (5 kg). After phase
separation, the
aqueous layer was back-extracted with ethyl acetate (91 kg). The combined
organic layers
were washed with saturated aqueous sodium hydrogen carbonate solution (101 kg)
and
then saturated aqueous sodium chloride solution (100 kg).
The organic layer was solvent swapped into heptane (80 kg) by distillation,
then
methyl tert-butyl ether (6.3 kg) was charged and the slurry stirred at 20 C
for 6 hours. The
solids were then filtered off, washed with a mixture of heptane (15.75 kg) and
methyl ten'-
butyl ether (4.25 kg), and dried in a 50 C vacuum oven. Yield 26.2 kg (93.8
mol, 79% by
moles).
1H NMR (300 MHz, CDC13) 6 7.55 (m, 2H), 7.38 (t, J=7.8Hz, 1H), 7.13 (m, 1H),
5.61 (m, 1H), 5.27 (m, 1H), 3.85 (s, 3H), 1.39-1.46 (m, 12H).
Example 2: Preparation of seed crystals of Form B of Compound (I)
o
F>1).N JO
F F \
I N
el 0 .4
F
2,2,2-Trifluoro-N-R1R,2S)-1- [1-(4-fluorophenyl)indazol-5 -yl] oxy-1-(3 -
methoxyphenyl)propan-2-yl]acetamide (7.5 g) in amorphous form (prepared as
described
in Example 4 steps (i) to (iv) below but without the crystallization step in
step (i)) was
charged to a solution of 9:1 v:v heptane:isopropyl acetate (75 mL). The
solution was
heated to 87 C which afforded dissolution. The solution was allowed to cool
to 15 C.
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
44
The resulting product was filtered off and dried at 40 C in a vacuum oven to
constant
weight. Yield 6.40 g (85%). XRPD diffractogram consistent with Form B
reference
(Table 1 and Figure 1). DSC profile consistent with Form B reference (Figure
2).
Example 3: Conversion of Form A to Form B of Compound (I)
(i) Conversion of Form A of Compound (I) to Form B of Compound (I)
0
F
NI
el N
0 104
F
A Form B seed of Compound (I) (28.4 g) was charged to a stirring slurry of
Form
A of Compound (I) (1.45 kg) in water (101 L) and 2-propanol (22.5 L). The
slurry was
stirred for 4 days at 20-25 C, by which time XRPD analysis showed conversion
to Form B
was complete. The Form B product was filtered off and dried at 40 C in a
vacuum oven
to constant weight. Yield 1.41 kg (97%). 98.8% Assay (NMR).
lti NMR (400 MHz, d6-DMS0) 6 9.50 (d, J= 8.5 Hz, 1H), 8.18 (d, J= 0.9 Hz,
1H), 7.78 - 7.73 (m, 2H), 7.70 (d, J= 9.1 Hz, 1H), 7.42- 7.35 (m, 2H), 7.27
(t, J= 7.9 Hz,
1H), 7.23 (dd, J= 2.4, 9.1 Hz, 1H), 7.15 (d, J= 2.2 Hz, 1H), 7.01 (d, J= 7.8
Hz, 1H), 7.00
- 6.98 (m, 1H), 6.85 (ddd, J= 0.8, 2.6, 8.3 Hz, 1H), 5.30 (d, J= 6.4 Hz, 1H),
4.28 (dqd, J
= 6.4, 6.8, 8.5 Hz, 1H), 3.74 (s, 3H), 1.36 (d, J= 6.8 Hz, 3H).
Example 4: Preparation of Form A of Compound (I)
(i) 2,2,2-Trifluoro-N-[(1R,2S)-141-(4-fluorophenyl)indazol-5-yl]oxy-1-(3-
methoxyphenyl)propan-2-yl]acetamide (Form A)
0
F
N)
lei N
0 404
F
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
Trifluoroacetic anhydride (1.14 L, 8.14 mol) was charged to a stirring 20 C
suspension of (1 R,2S)-141-(4-fluorophenyl)indazol-5-yl]oxy-1-(3-
methoxyphenyl)propan-
2-amine hydrochloride (2.58 kg, 5.42 mol; 90% assay by mass) in methyl tert-
butyl ether
(9.30 L) and triethylamine (2.80 L, 20.1 mol) over 45 minutes, keeping the
temperature
5 below 40 C. After 5 minutes, LC analysis showed < 0.05% amine starting
material
remaining by area.
The reaction mixture was washed successively with water (9.28 L), 1M
hydrochloric acid (9.28 L, 9.28 mol), 7% aqueous sodium bicarbonate (9.28 L,
8.03 mol)
and water (9.30 L). The organic phase was evaporated at 40-50 C under reduced
pressure
10 (500 mbar), dissolved in 2-propanol (9.30 L) and evaporated again at 40-
50 C, 50-500
mbar.
The residue was dissolved in 2-propanol (18.6 L), screened through a 1 [tm
filter
into a clean vessel, and then water was charged until the solution became
turbid (11.3 L
was required). Two portions of the turbid solution (0.325 and 1.30 L) were
removed and
15 the first stirred with a Form A seed of 2,2,2-trifluoro-N-R1R,25)-141-(4-
fluorophenyl)indazol-5-yl]oxy-1-(3-methoxyphenyl)propan-2-yl]acetamide (1.1 g)
in a
clean flask until a thick slurry was produced, and then the second portion was
added,
continuing to stir until the slurry thickened once again. The main
crystallisation vessel was
heated to about 60 C to re-dissolve some small deposits of amorphous solids
before
20 cooling back to 25 C. The seed slurry was warmed to about 35 C for
better mobility and
then charged to the main crystallisation vessel. The slurry was warmed to 30
C during a 3
hour stir to reduce viscosity. Water (5.70 L) was then charged over 15 minutes
and stirring
continued for 16 hours whilst the temperature was slowly varied in the range
30-38 C.
The 30 C slurry was filtered and washed with a mixture of 2-propanol (2.30 L)
and water
25 (3.50 L). The solids were dried to constant mass at 40 C and reduced
pressure. Yield
2.45 kg (93%). 99.4% Assay (NMR), 0.04% water, 0.1% 2-propanol (all by mass).
1FINMR (400 MHz, d6-DMS0) 6 9.52 (d, J= 8.4 Hz, 1H), 8.18 (s, 1H), 7.77 -
7.72 (m, 2H), 7.70 (d, J = 9.1 Hz, 1H), 7.43 - 7.37 (m, 2H), 7.27 (t, J= 7.9
Hz, 1H), 7.21
(dd, J = 2.4, 9.1 Hz, 1H), 7.14 (d, J = 2.3 Hz, 1H), 6.99 (d, J= 7.8 Hz, 1H),
6.97- 6.95 (m,
30 1H), 6.84 (dd, J= 2.3, 8.1 Hz, 1H), 5.28 (d, J= 6.3 Hz, 1H), 4.26 (dqd,
J = 6.3, 6.8, 8.4
Hz, 1H), 3.73 (s, 3H), 1.34 (d, J = 6.8 Hz, 3H).
The XRPD diffractogram of the form of Compound (I) obtained by way of
Example 4 (Form A of Compound (I)) is shown in Figure 3 below.
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
46
The DSC profile of the form of Compound (I) obtained by way of Example 4
(Form Aof Compound (I)) is shown in Figure 4 below. The compound exhibited an
onset
temperature of the melting endotherm of 83 C.
The (1R,2S)-141-(4-fluorophenyl)indazol-5 -yl] oxy-1-(3 -methoxyphenyl)prop an-
2-
amine hydrochloride starting material was prepared as follows.
(ii) (1R,2S)-141-(4-Fluorophenyl)indazol-5-ylloxy-1-(3-
methoxyphenyl)propan-2-
amine hydrochloride
CI
"
I ,N
N
el 0 410
F
(1R,2S)-1-[1-(4-F luorophenyl)indazol-5 -yl] oxy-1-(3 -methoxyphenyl)prop an-2-
amine solution (37.8 kg, 7.89 mol; 8.2% assay by mass) obtained using the
method of
Example 4 step (iii) below, was evaporated to a thick oil then re-dissolved in
ethyl acetate
(15.5 L). The solution was washed with 1M hydrochloric acid (15.4 L, 15.4 mol)
and then
aqueous sodium chloride (15.5 L; 10% assay by mass). The solution was
evaporated to a
thick oil at 40 C under reduced pressure and re-dissolved in ethanol (6.2 L).
Methyl tert-butyl ether (21.6 L) was charged at 50 C and the solution then
cooled
to 0 C. The crystallised (1R,2S)-141-(4-fluorophenyl)indazol-5-yl]oxy-1-(3-
methoxyphenyl)propan-2-amine hydrochloride was filtered off, washed with four
portions
of methyl tert-butyl ether (6.2 L per portion) and dried at 40 C to constant
mass. Yield
2.98 kg (6.27 mol, 79% by moles). 90.0% Assay by mass (NMR). Contains residual
methyl tert-butyl ether. 99.9% LC purity.
1FINMR (400 MHz, d6-DMS0) 6 8.60 (s, 3H), 8.21 (d, J= 0.9 Hz, 1H), 7.79 -
7.73 (m, 3H), 7.44 - 7.38 (m, 2H), 7.36 (dd, J = 2.4, 9.2 Hz, 1H), 7.33 (t, J=
7.2 Hz, 1H),
7.18 (d, J = 2.3 Hz, 1H), 7.02 - 6.99 (m, 2H), 6.91 - 6.88 (m, 1H), 5.79 (d,
J= 3.0 Hz,
1H), 3.75 (s, 3H), 3.68 (qd, J= 3.0, 6.8 Hz, 1H), 1.22 (d, J= 6.8 Hz, 3H).
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
47
(iii) (1R,2S)-141-(4-Fluorophenyl)indazol-5-yl] oxy-1-(3-
methoxyphenyl)propan-2-
amine
H2N o
. 0, "
1 ,N
el N
0 .
F
Water was removed from a solution of 1-(4-fluoropheny1)-5-iodo-indazole (2.20
kg, 4.40 mol; 67.6% assay by mass) in butyronitrile (8.5 L) by azeotropic
distillation at
atmospheric pressure, removing the aqueous phase of the distillate but
returning the
organic phase to the vessel. (Afterwards, the water content was determined to
be 1.3 g L-1).
Caesium carbonate (4.65 kg, 14.3 mol) and (1R,25)-2-amino-1-(3-
methoxyphenyl)propan-
1-ol hydrochloride (1.17 kg, 5.29 mol; 98.4% assay by mass) were charged and
the
mixture sparged with nitrogen for 60 minutes at 50-60 C. A separately
prepared solution,
which had been sparged with nitrogen for 40 minutes at 80 C, of copper (I)
iodide (0.21
kg, 1.10 mol), N,N-dimethylglycine (0.23 kg, 2.23 mol) and triethylamine (0.31
L, 2.22
mol) in butyronitrile (6.6 L) was then charged. The reaction mixture was
heated at 105 C
for 18 hours.
After cooling to 25 C the reaction mixture was washed with water (two
portions,
each of 11 L) and 1M hydrochloric acid (11 L, 11 mol). The organic layer was
concentrated to a thick oil at 60 C by solvent evaporation under reduced
pressure. Ethyl
acetate (110 was added and then the mixture concentrated at 40 C by solvent
evaporation
under reduced pressure to give crude (1R,2S)-1-E1 -(4-fluorophenyl)indazol-5-
yl]oxy-1-(3-
methoxyphenyl)propan-2-amine hydrochloride. An additional batch of the crude
product
was prepared from further 1-(4-fluoropheny1)-5-iodo-indazole (2.14 kg, 4.38
mol; 69.3%
assay by mass) by the same method and the two batches were dissolved in ethyl
acetate
(21.6 L). The solution was washed with a mixture of 0.01N aqueous disodium
ethylenediaminetetraacetate (disodium EDTA) (21.6 L) and aqueous sodium
chloride (3.6
L; 20% assay by mass) and then diluted with ethyl acetate (1.0 L). The
solution was
washed twice with a mixture of 0.1N aqueous disodium EDTA (21.6 L for each
portion)
and sodium chloride (3.6 L for each portion; 20% assay by mass), then with
aqueous
sodium bicarbonate (21.6 L; 7.0% assay by mass) and then with aqueous sodium
chloride
(21.6 L; 10% assay by mass).
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
48
The solution was chromatographed in portions (0.2 L per cycle, 59 cycles) on
Kromasil 60A 10 gm silica (2.0 kg), eluting with 15:85 v:v ethanol:isohexane
(20 L per
cycle) containing 1% diethylamine. At the end of each cycle, the column was
washed with
1:1 ethanol:ethyl acetate (3 L) and then equilibrated with 15:85 v:v
ethanol:isohexane (4
L); both solutions contained 1% diethylamine. Selected fractions were
concentrated under
reduced pressure to give a solution containing 1.21 kg (3.09 mol) of (1R,2S)-
141-(4-
fluorophenyl)indazol-5-yl]oxy-1-(3-methoxyphenyl)propan-2-amine. Yield 35%,
92.5%
LC purity by area (k = 254 nm).
(iv) 1-(4-Fluoropheny1)-5-iodo-indazole
1 \,
01 N
N
it
F
A mixture of 2-fluoro-5-iodobenzaldehyde (3.70 kg, 14.2 mol; 96.1% assay by
mass) and 4-fluorophenylhydrazine hydrochloride (2.50 kg, 14.2 mol; 92.4%
assay by
mass) in N-methylpyrrolidone (25 L) was stirred for 5 hours at 20 C.
Caesium carbonate (13.89 kg, 42.6 mol) was charged and the mixture stirred at
115
C for 3.5 hours. Water (18.3 L) was charged after adjusting the reaction
temperature to
80 C and once the solids were dissolved, the mixture was allowed to separate
into layers.
The lower layer was discarded.
The upper layer and an N-methylpyrrolidone (2 L) rinse were transferred into
stirring water (11.3 L), maintained at 62 C throughout the addition. After
cooling to 20
C, 1-(4-fluoropheny1)-5-iodo-indazole was filtered off, washed with two
portions of water
(16.0 L and 17.5 L) followed by 2-methylpentane (16.5 L), and suction dried
for around 18
hours at 20 C. Yield 6.28 kg (11.9 mol, 84% by moles). 64% Assay (LC), 20%
water,
both by mass. 98.6% LC purity (k = 254 nm).
1H NMR (400 MHz, d6-DMS0) 6 8.31 (d, J = 0.9 Hz, 1H), 8.28 (dd, J = 0.7, 1.6
Hz, 1H), 7.78 -7.73 (m, 2H), 7.70 (dd, J = 1.6, 8.8 Hz, 1H), 7.61 (ddd, J =
0.7, 0.9, 8.9
Hz, 1H), 7.45 - 7.38 (m, 2H).
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
49
Example 5: Preparation of seed crystals of Form A of Compound (I)
F 0
F>1)11-11 el \,N
F N
0 0 414
F
2,2,2-Trifluoro-N-R1R,2S)-1-[1-(4-fluorophenyl)indazol-5-yl]oxy-1-(3-
methoxyphenyl)propan-2-yl]acetamide in amorphous form (125 mg) (prepared as
described in Example 4 above but without the crystallization step in step (i))
was
dissolved in dichloromethane (3 ml) and 40 1 of this solution was transferred
to an LC-
vial. 80 1 of an organic solvent mixture (ethyl acetate:heptane (1:99%)) was
then added to
the LC-vial and the resulting mixture stirred at 40 C for 7 days. The
resulting crystals were
analysed via XRPD. The XRPD diffractogram was consistent with Form A reference
(Figure 3). The DSC profile was consistent with Form A reference (Figure 4).
Example 6: Alternative Crystallisation Procedure to Form B of Compound (I)
2,2,2-Trifluoro-N-R1R,25)-141-(4-fluorophenypindazol-5 -yl]oxy-1-(3 -
methoxyphenyl)propan-2-yl]acetamide was prepared according to Example 1, steps
(ii) to
(vii) but without the exchange of solvent in step (ii) (ie. without distilling
off methyl ten'-
butyl ether and thereafter replacing with 1-propanol).
Rather, immediately following the filtration step in Example 1, step (ii), the
concentration of the methyl tert-butyl ether solution was adjusted to a 5.5 ml
solution/g of
2,2,2-trifluoro-N-[(1R,25)-1-[1-(4-fluorophenyl)indazol-5-yl]oxy-1-(3-
methoxyphenyl)propan-2-yl]acetamide through addition of further methyl tert-
butyl ether.
At this point 2.2 ml n-heptane/g was added slowly and the solution was heated
to 35 C.
The solution was then cooled to 21 C. The solution was seeded with Form B
crystals. The
formed crystal slurry was stirred overnight. 5 ml n-heptane/g was added and
the crystals
were filtered off after 5 hours stirring. The crystals were washed with a
mixture of 0.4 ml
methyl tert-butyl ether/g and 0.6 ml n-heptane/g. The crystals were dried at
40 C vacuum.
Crystallization yield: 86%. XRPD diffractogram consistent with Form B
reference (Table 1
and Figure 1). DSC profile consistent with Form B reference (Figure 2).
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
Example 7: Solubility of Form B of Compound (I)
Two suspensions of Form B of 2,2,2-trifluoro-N-R1R,2S)-1-[1-(4-
5 fluorophenyl)indazol-5-yl]oxy-1-(3-methoxyphenyl)propan-2-yl]acetamide in
a solution
comprising 1.2 mM citric acid buffer, 0.05% PS80 and 9 mg/g sodium chloride
were
prepared at concentrations of 0.5 mg/g and 0.05 mg/g, and at a pH of 3.8
(adjusting with
sodium hydroxide). The suspensions were left standing for two months at 5 C
then allowed
to warm to room temperature and transferred to 5 mL vials. Each suspension is
centrifuged
10 twice (Sigma 2-16KCH, 8000 rpm, 25 C) and the supernatant transferred to
a new vial
after each centrifuge. The solubility of the supernatant is determined at room
temperature
using HPLC (Agilent Technologies 1100).
Solubility of Form B = 7 g/mL.
15 Example 8: Hygroscopicity of Form B of Compound (I)
The gravimetric response of Form B of 2,2,2-trifluoro-N-R1R,25)- 14144-
fluorophenyl)indazol-5-yl]oxy-1-(3-methoxyphenyl)propan-2-yl]acetamide to
changes in
humidity was measured using a DVS Advantage (Surface Measurement Systems)
20 Gravimetrical Vapour Sorption (GVS) instrument. Approximately 10 mg of
the sample
was evaluated using the following conditions: 120 min under dry conditions
followed by
drying to 90% relative humidity and then at subsequent decreasing levels of
humidity in
steps of 10%.
Humidity uptake of Form B at 80% relative humidity = 0.07%.
Example 9: Pharmacokinetic Properties of Inhaled Form A and Form B of
Compound (I) in Rat Lung and Rat Blood
The level of total lung and blood exposure (expressed as "Area Under the
Curve"
or AUC) and the level of peak blood level (expressed as Cmax) of Form A and
Form B of
Compound (I) when administered via the inhaled route were measured using the
following
protocol.
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
51
1. Inhalation
Exposure by the inhalation route was performed using a nose-only "flow-past"
exposure chamber (Munster AG, Switzerland). Forms A and B of Compound (I) were
administered as a dry powder inhalation (DPI) to the rat (10 min exposure) and
compound
concentration was measured in plasma up to 24 hours after administration.
Calibration of the inhalation system.
A small animal inhalation system (MIVIS) was used to deliver and measure the
inhaled dose. To measure the aerosol concentration a light scattering
instrument was used
(Casella 950 AMS, London, UK). Before exposure the substance correlation
factor was
estimated by taking filter samples (AP40 Millipore, n=2+2). The filters were
positioned in
the inhalation system in the same way as the animals were connected. The
amount of
Compound (I) Forms A and B on filters were analysed by HPLC. The correlation
factor
was used in the dose measurement program where particle concentration and
tidal volume
was used to estimate the inhaled dose. Target concentration on the Casella was
1,5 mg/m3
(Casella no: 034022, range: 0-2000). The calibration was validated by filter
sampling
(2+2) at a flow of 0.25 L/min.
Particle size measurements.
The particle size close to the inhalation side was measured using a Mercer
Seven
Stages Impactor (In-Tox products USA) (n =7+1 filter, flow rate 0.25 L/min).
The
deposition probability of the inhaled particles was considered similar to that
previously
reported for the rat (Raabe OG, Yeh HC, Newton GJ, Phalen RF, Velasques DJ.
Deposition of inhaled monodisperse aerosols in small rodents. In: Walton WH,
editor.
Inhaled Particles IV. New York: Pergamon Press; 1977. p 3-21).
Test system (DPI).
Micronized Compound (I) Forms A and B (30% API, 70% lactose) were pressed in
medium dust containers (pressure = 1.2 Bar) and aerosol generated by a
modified, Wright
Dust Feed (WDF) during 10 minutes by scraping of the substance from the
pressed tablet.
The speed on the WDF (1200 - 1400 rpm) was controlled by a Motomatic II and
the flow
through WDF was 8.0 L/min. The air supply to each animal port was 0.3 L/min
which is
approximately two times the respiratory minute volume for a 210 g rat and
considered
sufficient to cover the oxygen requirement of the animal (Crosfill ML,
Widdicombe JG.
Physical characteristics of the chest and lungs and the work of breathing in
different
CA 02839398 2013-12-13
WO 2013/001294 PCT/GB2012/051503
52
mammalian species. J Physio11961;158(1):1-14). Breathing volume and particle
concentration were monitored during inhalation.
Treatment.
Target lung dose was 50 jig/kg (10 min inhalation). The administration was
performed in the morning. The rats were observed continuously during the
experiment and
up to at least 2 hours after administration.
Calculations of inhaled dose (ID)
Inhaled dose = ((chamber concentration * exposure time * respiratory minute
volume) /
body weight in kg)
Calculations of Body Dose (BD)
Body Dose = Inhaled dose * fraction deposited in body
Calculations of Lung Dose (LD)
Lung Dose = Inhaled dose * fraction deposited in lung
2. Termination
Animals were terminated periodically during 24 hours after the inhalation of
compound.
Anaesthetization was performed with an overdose of pentobarbital given
intraperitoneally
(ip) (60 mg/mL, 10 mL/kg). Animals were weighed and a terminal blood sample
was
taken. The rib cage was opened and the lung and trachea dissected. The
dissected organs
were weighed.
3. Bio analysis
The blood samples were protein precipitated by cold, acidified acetonitrile
containing a
volume marker. After centrifugation the supernatant was diluted to match the
mobile phase
and the extracts were quantified using LC-MS/MS. The lung samples were
prepared for
analysis by first pulverizing in liquid nitrogen and then homogenizing in
Ringer solution
by adaptive focused acoustic energy (Covaris). The homogenates were protein
precipitated
by cold, acidified acetonitrile containing a volume marker and after
centrifugation the
supernatants were diluted to match the mobile phase for analysis by LC-MS/MS
(Agilent
6460 triple quadropole with Agilent 1200 binary pump and a CTC autosampler).
CA 02839398 2013-12-13
WO 2013/001294
PCT/GB2012/051503
53
4. Calculations
Lung and blood samples from the experiment were analysed by LC-MS/MS. The
concentration vs time profiles were analysed by compartmental modelling (2
compartments) using WinNonlin. In this way, parameters were obtained that were
used to
calculate peak areas (AUC) and Cmax.
The values for total lung exposure (lung AUC), peak blood level (blood Cmax)
and total
blood exposure (blood AUC) for Form A and Form B of Compound (I) obtained via
this
method are tabulated in Table 3 below.
Table 3
Form A Form B
Lung AUC (hr*nmol/L) 9110 21289
Blood Cmax (nmol/L) 83 55
Blood AUC (hr*nmol/L) 288 246