Note: Descriptions are shown in the official language in which they were submitted.
CA 02862143 2014-06-27
. .
1
METHOD FOR PRODUCING 6-CHLORODIBENZO[D,9[1,3,2]DIOXAPHOSPHEPIN
BACKGROUND OF THE INVENTION
The present invention relates to a process for preparing 6-chloro-
dibenzo[df][1,3,2]dioxaphosphepin (2,2'-biphenylphosphomonochloridite).
PRIOR ART
Organic diphosphite compounds have found extremely widespread use, for example
as chelat-
ing ligands in homogeneous catalysis and also as flame retardants, UV
stabilizers, etc. Particu-
lar rhodium complexes comprising chelating diphosphite compounds have been
found to be
useful as catalysts for the hydroformylation of olefins, since they firstly
have a high catalytic ac-
tivity and secondly lead to predominantly linear aldehydes which are preferred
for many applica-
tions. Organic diphosphite compounds are also suitable as ligands for
transition metal complex
catalysts for hydrocyanation, hydrogenation, carbonylation, hydroacylation,
hydroamidation,
hydroesterification, hydrosilylation, hydroboration, alcoholysis,
isomerization, allylic alkylation or
hydroalkylation.
Such diphosphite compounds, their preparation and their use as ligands in a
hydroformylation
process are described, for example, in EP 0 214 622 A2, EP 0 285 136 A2, US
4,668,651,
US 4,748,261, US 4,769,498, US 4,885,401, US 5,235,113, US 5,391,801, US
5,663,403,
US 5,728,861, US 6,172,267 and DE 103 60 771 Al.
Organic diphosphites of the general formula (4) are usually prepared by a
process which com-
prises the following steps:
a) reaction of a compound of the formula (1) (= first aromatic diol) with
phosphorus trichloride
to give the phosphochloridite (2)
CA 02862143 2014-06-27
2
Rxn Rx
1. OH 'V0
PCI3 \
OH ¨
-2 HCI
4
* o/P CIIk
Rxn
Rxn
(1) (2)
b) reaction of the phosphochloridite (2) with a compound of the formula (3)
(= second aro-
matic diol) to give the chelating diphosphite (4)
R<,
le fen
o
\
RxnAla /P ¨O
OH 0/
lir 0/ RY
\ Yni = 111 m
2 o P¨CI + R
. lik - 2 HCI lirt RYm
HO 0
/
Rxn (3) 0 ¨P
\
0
(2) Rxn =
SI
(4) Rxn
The groups of the organic diphosphites which are derived from the first
aromatic diol (1) will
hereinafter also be referred to as "side wings".
Phosphochloridites of the formula (2) and especially 6-
chlorodibenzo[d,f][1,3,2]dioxaphosphepin
are thus important intermediates in the preparation of ligands for many
homogeneous transition
metal catalysts. There is therefore a continual need for processes which allow
preparation of
these compounds with very effective utilization of the starting materials in
very high yields and
good purity. Owing to the toxicity and corrosivity of PCI3, there is a need
for processes which
make possible a very low excess of the PCI3 used in the synthesis in order to
keep the streams
to be handled in the synthesis, recycling and disposal as small as possible.
In the preparation of phosphomonochloridites and chelating diphosphites,
hydrogen halide is
obtained in the condensation reaction of the alcohols or phenols used with
PCI3 and this has to
CA 02862143 2014-06-27
3
be removed from the reaction mixture. One possibility is neutralization with a
base, with nitrogen
bases frequently being used. In this procedure, an at least stoichiometric
amount of base based
on the hydrogen halide liberated has to be used. The base is frequently also
used in excess.
Catalytic activity of the hydrohalic acid salts of the nitrogen bases in the
condensation reactions
has not hitherto been described.
WO 2003/062171 and WO 2003/062251 describe a process for the removal of acids
from reac-
tion mixtures by means of an auxiliary base which with the acid forms a salt
which is liquid at
temperatures at which the desired product is not significantly decomposed
during the removal of
the liquid salt and the salt of the auxiliary base with the desired product or
the solution of the
desired product in a suitable solvent forms two immiscible liquid phases. In
other words, the
acid salts of the auxiliary base behave like ionic liquids which are
essentially immiscible with the
actual reaction solvent. Preferred auxiliary bases of this type are 1-
methylimidazole, 1-n-
butylimidazole, 2-methylpyridine and 2-ethylpyridine. The processes described
in
WO 2003/062171 and WO 2003/062251 are suitable, inter alia, for
phosphorylation reactions
such as the above-described synthesis of phosphomonochloridites and the
reaction thereof with
an aromatic diol to give a chelating diphosphite compound. According to the
teaching of
WO 2003/062171 and WO 2003/062251, the nitrogen base is used in an at least
stoichiometric
amount based on hydrogen halide liberated.
CN 101684130A describes a process for preparing chelating phosphites, in which
the phos-
phomonochloridite forming the side wings is introduced as a solution in
dichloromethane into the
reaction and the aromatic diol which bridges the two phosphorus atoms is
introduced as a solu-
tion in triethylamine or a triethylamine/dichloromethane mixture. To prepare
the phosphochlorid-
ite, the document teaches reacting the aromatic diol with from 1 to 10
equivalents of PCI3 and
distilling off excess PCI3. The use of an acid salt of a nitrogen base as
catalyst is not described.
In the specific examples, 2,2'-biphenol is reacted with PCI3 at 100 C in the
absence of organic
solvent. The reaction thus occurs below the melting point of 2,2'-biphenol,
i.e. as a suspension
of the diol in a large excess of PCI3 (molar ratio of diol: PCI3 = 1:6.1). In
addition, this document
indicates that when the unpurified phosphochloridite obtained in this way is
used in a solvent
other than dichloromethane, e.g. toluene, only turbid suspensions are obtained
because of the
impurities present. However, the inventors of the present invention were able
to demonstrate
the contrary for the inventive process described below.
WO 2010/052090 and WO 2010/052091 describe processes for preparing
6-chlorodibenzo[d,f][1,3,2]-dioxaphosphepin, in which 2,2'-dihydroxybiphenyl
is added as a melt
CA 02862143 2014-06-27
4
or as a suspension in an inert solvent to an excess of phosphorus trichloride
under inert gas
while stirring and the gases formed are discharged from the reaction mixture
and neutralized.
This process has the disadvantage that the PCI3 has to be used in a large
molar excess over
the diol. Thus, according to the general teaching of these documents, a from 2-
to 25-fold ex-
cess is used, while an approximately 12-fold excess is used in the examples.
These documents
do not teach carrying out the reaction in the presence of a catalyst.
WO 2008/124468 describes a calixarene-bisphosphite composition for use as
ligand in a transi-
tion metal complex catalyst. In example 1(a), the preparation of 2,2'-
biphenylphosphomonochloridite is described. Here, 3.7 equivalents of PCI3 are
added at room
temperature to one equivalent of o,o'-biphenyldiol and the suspension obtained
is subsequently
heated until the evolution of HCI abates and subsequently distilled in a high
vacuum, with the
desired phosphochloridite being obtained in a 78% yield. A significant
disadvantage of this pro-
cedure is that the reaction is virtually uncontrollable after mixing the
reactants and is thus prob-
lematical from a safety point of view. It is less the thermal safety which is
a problem, since this
reaction is endothermic, but rather the risk that on the production scale the
HCI discharging sys-
tem can fail, for example due to an excessively high reaction rate or
blockage, which can lead to
a pressure buildup with the associated consequences. Furthermore, a greater
capital invest-
ment is necessary since the HCI scrubber has to be made larger in order to
cope with sudden
larger amounts of HCI per unit time.
WO 2010/042313 describes, inter alia, a process for preparing
phosphomonochloridites by re-
acting PCI3 with an aromatic diol in a slurry which additionally comprises an
organic solvent and
less than 5 mol /0, based on the aromatic diol, of a nitrogen base.
Specifically, in example 1 of
this document 2,2'-dihydroxybiphenyl is reacted as a suspension in toluene
with PCI3 in the
presence of a catalytic amount of pyridine at 0 C. It is a critical feature of
this process that a
substantial part of the aromatic diol used is insoluble in the organic
solvent. Thus, a large molar
excess of PCI3 over the diol is always available for the actual reaction in
the organic phase,
even though the overall molar excess of PCI3 is lower. However, this document
also indicates
that undesirable by-products are formed at an excess of PCI3 which is too low.
WO 2009/120210 and the US patent US 2009/0247790 having the same priority have
a disclo-
sure content comparable to that of WO 2010/042313. They describe a process for
preparing
phosphomonochloridites, in which the reaction of PCI3 with an aromatic diol is
carried out in a
solution comprising less than 5 mol% of a nitrogen base, based on mol of
aromatic diol, with
HCI formed being driven off from the reaction solution and the reaction being
carried out under
CA 02862143 2014-06-27
essential isothermal conditions. For the reaction, PCI3 is initially charged
in a reaction zone and
a solution or suspension of the diol in an organic solvent is fed into the
reaction zone.
It is an object of the present invention to provide a simple, effective and
safe process for prepar-
ing phosphochloridites. It should make the preparation of the
phosphochloridites with very effec-
tive utilization of the starting materials in very high yields and good purity
possible. The phos-
phochloridite compound obtained should preferably have a purity which allows
it to be used as
intermediate for the preparation of chelating phosphites without complicated
intermediate purifi-
cation.
It has now surprisingly been found that this object is achieved by a process
for preparing phos-
phochloridites, which comprises reacting an aromatic diol with PCI3 in the
presence of a catalytic
amount of an acid salt of a nitrogen base and in the absence of external
organic solvents. It is
also surprising that only very small excesses of PCI3 are necessary when the
PCI3 is added to a
melt of 2,2'-dihydroxybiphenyl, i.e. under process conditions under which the
2,2'-
dihydroxybiphenyl is always present in a relatively high excess over PCI3
until shortly before the
end of the reaction. Furthermore, it is surprising that when the PCI3 is
introduced into the gas
space above the surface of the melt of 2,2'-dihydroxybiphenyl, virtually no
loss of PCI3 due to
vaporization and entrainment in the offgas stream occurs despite the
significantly lower boiling
point of PCI3 compared to the melting point of 2,2'-dihydroxybiphenyl.
SUMMARY OF THE INVENTION
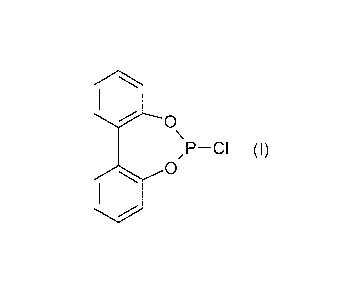
The invention provides a process for preparing 6-
chlorodibenzo[d,f][1,3,2]dioxaphosphepin (I)
SO
,P--CI (I)
SO
which comprises reacting 2,2'-dihydroxybiphenyl (Al)
CA 02862143 2014-06-27
6
OH HO
(Al)
with PCI3 in the presence of a catalytic amount of an acid salt of a nitrogen
base, wherein the
reaction is carried out in the absence of external organic solvents.
DESCRIPTION OF THE INVENTION
For the purposes of the invention, the expression "external organic solvent"
refers to compo-
nents which act as solvent and are different from the starting materials and
catalysts used for
preparing the phosphochloridites of the general formula (I) and the reaction
products formed.
The product of the 6-chlorodibenzo[d,f][1,3,2]dioxaphosphepin synthesis is
soluble in toluene to
give a clear solution (at 40 C as 90% solution and at 20 C as 50% solution)
and can be used
without a complicated work-up to prepare organic diphosphites. To avoid
misunderstandings, it
should be pointed out that the production of a toluene solution of (I) serves
merely to provide (I)
for subsequent reactions and does not represent a work-up or purification
step.
The process of the invention has the following advantages:
It allows the preparation of 6-chlorodibenzo[d,f][1,3,2]dioxaphosphepin (I) in
high yields
and with good selectivities.
The chlorodibenzo[d,f][1,3,2]dioxaphosphepin is obtained in high purity.
Compared to the processes known from the prior art, the process requires a
very small
excess of PCI3 for the synthesis of the phosphochloridite. Owing to the
toxicity and the
corrosivity of PCI3, this is particularly advantageous since the PCI3 streams
to be handled
in the synthesis, recycling and disposal can be kept small.
Since no volatile organic solvents are used, the problem of combustion of the
vapors of
these solvents before the HCI formed in the condensation reaction is separated
off by
scrubbing with a base does not arise.
The acid salts of nitrogen bases, especially N-methylimidazolium
hydrochloride, which are
used as catalyst are suitable for the reaction of PCI3 with aromatic diols
which have a cer-
tain residual water content (up to about 0.3% by weight, based on the total
weight of the
CA 02862143 2014-06-27
7
diol used). A lower outlay is therefore required for drying of the diol and
its storage and
use under anhydrous conditions.
Both the intermediate (A3) and the undesirable by-product (A4) are found only
in traces of
<2% in the end product in the preparation of the phosphochloridites (1) as a
result of addi-
tion of the PC13 to the initially charged diol (Al).
R' R2 R3
,
R5 RR
* R4C)
OH
p-O
R4 0- R5 R5
R3 'R R7 R8
R2
(A3)
R2
R7 R2 R3 1 R3
R8 R8
Ri
1101 0 411 R4 1.1 R4
0-P=so
p-O R5
R4 0 R8 110 R5 el
le R6
R3 R7
R6 R8
R7
R2 R1
(A4)
According to the invention, the reaction of the diol (Al) with PC13 is carried
out in the presence
of a catalytic amount of an acid salt of a nitrogen base.
The acid salt is preferably derived from a nitrogen base selected from among
in each case un-
substituted or substituted imidazoles, pyridines, 1H-pyrazoles, 1-pyrazolines,
3-pyrazolines,
imidazolines, thiazoles, oxazoles, 1,2,4-triazoles and 1,2,3-triazoles.
The acid salt is particularly preferably derived from a nitrogen base selected
from among in
each case unsubstituted or substituted imidazoles and pyridines.
Particularly preferred nitrogen bases are 3-chloropyridine, 4-
dimethylaminopyridine,
2-methylpyridine (a-picoline), 3-methylpyridine (13-picoline), 4-
methylpyridine (y¨picoline),
2-ethylpyridine, 2-ethyl-6-methylpyridine, quinoline, isoquinoline, 1-
methylimidazole,
CA 02862143 2014-06-27
=
8
1,2-dimethylimidazole, 1-(n-butyl)imidazole, 1,4,5-trimethylimidazole, 1,4-
dimethylimidazole,
imidazole, 2-methylimidazole, 1-butyl-2-methylimidazole, 4-methylimidazole, 1-
(n-
pentyl)imidazole, 1-(n-hexyl)imidazole, 1-(n-octyl)imidazole, 1-(2'-
aminoethyl)imidazole, 2-ethyl-
4-methylimidazole, 2-ethylimidazole, 1-(2'-cyanoethyl)imidazole and
benzotriazole.
In particular, the acid salt is derived from a nitrogen base selected from
among 1-(C1-C4-
alkyl)imidazoles, 2-(Ci-C4-alkyl)pyridines, 3-(Ci-C4-alkyl)pyridines and 4-(Ci-
C4-alkyl)pyridines.
The acid salt is especially derived from a nitrogen base selected from among
1-methylimidazole, 1-(n-butyl)imidazole, 2-methylpyridine and 2-ethylpyridine.
Acids with which the nitrogen bases can form salts are, for example, hydrogen
chloride (NCI),
hydrogen bromide (HBr), sulfuric acid (H2SO4, to form sulfates or
hydrogensulfates), methyl-
sulfuric acid (HO(S02)0CH3), ethylsulfuric acid (HO(S02)0C2H5), phosphoric
acid (H3PO4, to
form phosphates, hydrogenphosphates or dihydrogenphosphates), p-
toluenesulfonic acid, ben-
zenesulfonic acid, benzoic acid, 2,4,6-trimethylbenzoic acid, mandelic acid,
methanesulfonic
acid, ethanesulfonic acid or trifluoromethanesulfonic acid. Preferred acids
with which the nitro-
gen bases can form salts are, for example, hydrogen chloride, p-
toluenesulfonic acid, me-
thanesulfonic acid, 2,4,6-trimethylbenzoic acid and trifluoromethanesulfonic
acid. Particular
preference is given to hydrogen chloride.
Especially, N-methylimidazolium hydrochloride is used as acid salt of a
nitrogen base in the
process of the invention.
The amount of acid salt of the nitrogen base used is preferably from 0.01 to 5
mol%, particularly
preferably from 0.05 to 2 mol%, in particular from 0.1 to 1 mol%, based on the
molar amount of
diol (Al).
In the process of the invention, the reaction of the diol (Al) with PCI3 is
carried out essentially
without addition of free nitrogen bases. The preparation of the
phosphochloridites of the general
formula (I) is thus carried out according to the process of the invention not
as described in WO
03/062171 and WO 03/062251. That is to say, in the process of the invention,
the hydrogen
chloride liberated in the reaction is not separated off by means of an
auxiliary base which with
hydrogen chloride forms a salt which is liquid at temperatures at which the
phosphochloridites of
the general formula (I) are not significantly decomposed and the
phosphochloridites of the gen-
eral formula (I) or a solution thereof in a suitable solvent forms two
immiscible liquid phases.
r.
CA 02862143 2014-06-27
9
The molar ratio of the gradually added PCI3 to the amount of diol (Al) used is
more than 1:1,
preferably at least 1.1:1, in particular at least 1.2:1, at the end of the
reaction.
The molar ratio of the gradually added PCI3 to the amount of diol (Al) used is
preferably not
more than 2.5:1, particularly preferably not more than 2:1, in particular not
more than 1.8:1, es-
pecially not more than 1.6:1, more especially not more than 1.4:1, at the end
of the reaction.
As mentioned above, the process of the invention makes it possible to prepare
6-chlorodibenzo[d,f][1,3,2]dioxaphosphepin (I) using only a small excess of
PCI3.
The reaction is preferably carried out at a temperature in the range from 20
to 250 C, particular-
ly preferably from 50 to 200 C.
In a specific embodiment of the process of the invention for preparing the
phosphochloridites of
the general formula (I), the diol (Al) is used as a melt for the reaction.
To provide a melt, the diol (Al) is heated to a temperature above the melting
point so that it
goes over into a liquid state. If the diol (Al) is used as a technical-grade
compound comprising
impurities which lower the melting point, the melting point can also be below
that of the pure
compound. Pure 2,2'-dihydroxybiphenyl melts at from 108 to 110 C.
In a specific embodiment of the process of the invention, the PCI3 is added to
a melt of the diol
(Al).
The PCI3 is preferably introduced into the space above the melt of the diol
(Al). This can be
achieved using a conventional addition device whose outlet opening ends above
the melt. The
PCI3 can be introduced in the form of individual droplets or as a jet. The
amount fed in can be
regulated by means of a conventional metering device, e.g. a valve, metering
pump, etc. The
reaction can thus be carried out in a metering-controlled manner. At least
those surfaces which
come into contact with the PCI3 are made of a corrosion-resistant material
such as glass, Teflon,
enamels, etc.
The boiling point of PCI3 (76.1 C at 1013 mbar) is below the melting point of
the diol (Al) (108-
110 C). The reaction is therefore preferably carried out using one of the
following measures:
,
CA 02862143 2014-06-27
- addition of the PCI3 in sufficiently small amounts per time interval,
- use of a cooling device, e.g. a reflux condenser, in order to separate
off vaporized PCI3 as
condensate and recirculate it to the reaction zone.
In general, the reaction is carried out at ambient pressure (1013 mbar), but
higher or lower
pressures can also be used.
In a specific embodiment, the reaction is carried out in the presence of a gas
which is inert un-
der the reaction conditions. Suitable inert gases of this type are, for
example, nitrogen, argon or
helium. In a useful embodiment, the liquid reaction medium is blanketed with
an inert gas. In a
further useful embodiment, a stream of inert gas is passed through the liquid
reaction medium.
The stream of inert gas passed through the liquid reaction medium can at the
same time serve
to strip the reaction medium in order to remove the HCI formed more
effectively. In a preferred
embodiment, an offgas stream is taken from the reaction zone and subjected to
scrubbing to
remove the HCI comprised therein. Suitable washing media are water and aqueous
alkaline
washing media.
If the reaction zone is connected to a cooling device, e.g. a reflux
condenser, in order to avoid
PCI3 losses by vaporization, the offgas stream, optionally together with at
least one inert gas, is
preferably also firstly passed through the cooling device and only then the
offgas scrubber.
The reaction is preferably carried out until at least 95% by weight of the
dial (Al), particularly
preferably at least 98% by weight of the diol (Al), has been converted into
6-chlorodibenzo[d,f][1,3,2]dioxaphosphepin (I).
If the reaction mixture still comprises unreacted PCI3 after the conclusion of
the reaction, this
can be separated off by conventional methods, preferably by distillation. To
separate off the
PCI3 by distillation, it is possible to employ one of the following measures:
increasing the temperature of the reaction mixture,
- applying a reduced pressure,
introducing a stream of inert gas into the reaction mixture,
a combination of at least two of these measures.
The phosphochloridites obtained by the process of the invention are
particularly advantageous
for preparing organic diphosphites.
CA 02862143 2014-06-27
11
The invention further provides a process for preparing organic diphosphites of
the general for-
mula (II)
R2
1
1 10 0 R II R4 4111
0--R
p¨O
0
40 0 R4 it Ri
R3 R2
(II)
where
R1, R2, R3 and R4 are each, independently of one another, hydrogen, Ci-C12-
alkyl, Cl-C12-
alkoxy, C3-C12-cycloalkyl, C3-C12-heterocycloalkyl, C6-C20-aryl, chlorine,
bromine, hydroxy,
acyl or alkoxycarbonyl,
where two adjacent radicals R, to R4 together with the carbon atom of the
benzene ring to
which they are bound can also form a fused ring system with a further benzene
ring,
where C1-C12-alkyl and C1-C12-alkoxy can each be unsubstituted or substituted
by one or
more identical or different radicals Ra selected from among C3-C12-cycloalkyl,
C3-C12-
heterocycloalkyl, C5-C20-aryl, fluorine, chlorine, cyano, acyl and
alkoxycarbonyl,
where C3-C12-cycloalkyl and C3-C12-heterocycloalkyl can each be unsubstituted
or substi-
tuted by one or more identical or different radicals Rb selected from among C1-
C12-alkyl,
C1-C12-alkoxy, C3-C12-cycloalkyl, C3-C12-heterocycloalkyl, C6-C20-aryl,
fluorine, chlorine,
bromine, cyano, formyl, acyl and alkoxycarbonyl,
where C6-C20-aryl can in each case be unsubstituted or substituted by one or
more identi-
cal or different radicals Rc selected from among C1-C12-alkyl, C1-C12-alkoxy,
C3-C12-
cycloalkyl, C3-C12-heterocycloalkyl, C6-C20-aryl, fluorine, chlorine, bromine,
cyano, formyl,
acyl and alkoxycarbonyl,
CA 02862143 2014-06-27
12
which comprises
a) reacting 2,2'-dihydroxybiphenyl (Al)
OH HO
11 II (Al)
with PCI3 in the presence of a catalytic amount of an acid salt of a nitrogen
base and in
the absence of an external organic solvent to give
6-chlorodibenzo[d,f][1,3,2]dioxaphosphepin (I),
b) reacting the 6-chlorodibenzo[d,f][1,3,2]dioxaphosphepin (I) with a diol
of the general for-
mula (A2)
R2 R3
R1 II R4
(A2)
OH
HO
R4 410 Ri
R3 R2
to give the organic diphosphite (II).
For the purposes of the invention, halogen is fluorine, chlorine, bromine or
iodine, preferably
fluorine, chlorine or bromine.
In the following, the expression "C1-C12-alkyl" comprises straight-chain and
branched C1-C12-
alkyl groups. Preference is given to straight-chain or branched C1-C8-alkyl
groups and very par-
ticularly preferably C1-C6-alkyl groups. Examples of C1-C12-alkyl groups are,
in particular, me-
thyl, ethyl, propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, n-
pentyl, 2-pentyl,
2-methylbutyl, 3-methylbutyl, 1,2-dimethylpropyl, 1,1-dimethylpropyl, 2,2-
dimethylpropyl, 1-
ethylpropyl, n-hexyl, 2-hexyl, 2-methylpentyl, 3-methylpentyl, 4-methylpentyl,
1,2-dimethylbutyl,
1,3-dimethylbutyl, 2,3-dimethylbutyl, 1,1-dimethylbutyl, 2,2-dimethylbutyl,
3,3-dimethylbutyl,
1,1,2-trimethylpropyl, 1,2,2-trimethylpropyl, 1-ethylbutyl, 2-ethylbutyl, 1-
ethyl-2-methylpropyl, n-
4
CA 02862143 2014-06-27
13
heptyl, 2-heptyl, 3-heptyl, 2-ethylpentyl, 1-propylbutyl, n-octyl, 2-
ethylhexyl, 2-propylheptyl,
nonyl, decyl.
The above explanations of the expression "C1-C12-alkyl" also apply to the
alkyl groups in Cl-C12-
alkoxy.
Substituted Ci-C12-alkyl groups and substituted Cl-C12-alkoxy groups can,
depending on their
chain length, have one or more (e.g. 1, 2, 3, 4 or 5) substituents Ra. The
substituents Ra are
preferably selected independently from among C3-C12-cycloalkyl, C3-C12-
heterocycloalkyl, 06-
020-aryl, fluorine, chlorine, bromine, cyano, formyl, acyl and alkoxycarbonyl.
For the purposes of the present invention, the expression "alkylene" refers to
straight-chain or
branched alkanediyl groups having preferably from 1 to 6 carbon atoms. These
include meth-
ylene (-CH2-), ethylene (-0H2-0H2-), n-Propylene (-CH2-CH2-0H2-), isopropylene
(-CH2-
CH(CH3)-), etc.
For the purposes of the present invention, the expression "03-C12-cycloalkyl"
comprises mono-
cyclic, bicyclic or tricyclic hydrocarbon radicals having from 3 to 12, in
particular from 5 to 12,
carbon atoms. These include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl,
cycloheptyl, cy-
clooctyl, cyclododecyl, cyclopentadecyl, norbornyl, bicyclo[2.2.2]octyl or
adamantyl.
For the purposes of the present invention, the expression "C3-C12-
heterocycloalkyl" comprises
nonaromatic, saturated or partially unsaturated cycloaliphatic groups having
from 3 to 12, in
particular from 5 to 12, carbon atoms. C3-012-Heterocycloalkyl groups
preferably have from 4 to
8, particularly preferably 5 or 6, ring atoms. In contrast to the cycloalkyl
groups, 1, 2, 3 or 4 of
the ring carbons in the heterocycloalkyl groups are replaced by heteroatoms or
heteroatom-
comprising groups. The heteroatoms or heteroatom-comprising groups are
preferably selected
from among -0-, -S-, -C(=0)- and -S(=0)2-. Examples of C3-C12-heterocycloalkyl
groups are
tetrahydrothiophenyl, tetrahydrofuranyl, tetrahydropyranyl and dioxanyl.
Substituted C3-C12-cycloalkyl groups and substituted C3-C12-heterocycloalkyl
groups can, de-
pending on their ring size, have one or more (e.g. 1, 2, 3, 4 or 5)
substituents Rb. The substitu-
ents Rb are preferably selected independently from among C1-C12-alkyl, C1-012-
alkoxy, C3-C12-
cycloalkyl, 03-C12-heterocycloalkyl, C6-C20-aryl, fluorine, chlorine, bromine,
cyano, formyl, acyl
and alkoxycarbonyl. Substituted C3-C12-cycloalkyl groups preferably bear one
or more, e.g. 1, 2,
CA 02862143 2014-06-27
14
3, 4 or 5, C1-C6-alkyl groups. Substituted C3-C12-heterocycloalkyl groups
preferably bear one or
more, e.g. 1, 2, 3, 4 or 5, Cl-C6-alkyl groups.
Examples of substituted C3-C12-cycloalkyl groups are 2- and 3-
methylcyclopentyl, 2- and 3-
ethylcyclopentyl, 2-, 3- and 4-methylcyclohexyl, 2-, 3- and 4-ethylcyclohexyl,
2-, 3- and 4-
propylcyclohexyl, 2-, 3- and 4-isopropylcyclohexyl, 2-, 3- and 4-
butylcyclohexyl, 2-, 3- and 4-
sec-butylcyclohexyl, 2-, 3- and 4-tert-butylcyclohexyl, 2-, 3- and 4-
methylcycloheptyl, 2-, 3- and
4-ethylcycloheptyl, 2-, 3- and 4-propylcycloheptyl, 2-, 3- and 4-
isopropylcycloheptyl, 2-, 3- and
4-butylcycloheptyl, 2-, 3- and 4-sec-butylcycloheptyl, 2-, 3- and 4-tert-
butylcycloheptyl, 2-, 3-, 4-
and 5-methylcyclooctyl, 2-, 3-, 4- and 5-ethylcyclooctyl, 2-, 3-, 4- and 5-
propylcyclooctyl.
For the purposes of the present invention, the expression "C6-C20-aryl"
comprises monocyclic or
polycyclic aromatic hydrocarbon radicals. These have from 6 to 20 ring atoms,
in particular from
6 to 14 ring atoms. Aryl is preferably phenyl, naphthyl, indenyl, fluorenyl,
anthracenyl, phenan-
threnyl, naphthacenyl, chrysenyl, pyrenyl, coronenyl, perylenyl, etc. In
particular, aryl is phenyl
or naphthyl.
Substituted C6-C20-aryl groups can, depending on their ring size, have one or
more (e.g. 1, 2, 3,
4 or 5) substituents Rc. The substituents Rc are preferably selected
independently from among
C1-C12-alkyl, C1-C12-alkoxy, C3-C12-cycloalkyl, C3-C12-heterocycloalkyl, C6-
C20-aryl, fluorine, chlo-
rine, bromine, cyano, nitro, formyl, acyl and alkoxycarbonyl.
Substituted Cs-Cm-aryl is preferably substituted phenyl or substituted
naphthyl. Substituted C6-
C20-aryl groups preferably bear one or more, e.g. 1, 2, 3, 4 or 5,
substituents selected from
among C1-C6-alkyl groups, Ci-C6-alkoxy groups, chlorine and bromine.
For the purposes of the present invention, the term "acyl" refers to alkanoyl
or aroyl groups
which generally have from 2 to 11, preferably from 2 to 8, carbon atoms. They
include, for
example, acetyl, propanoyl, butanoyl, pentanoyl, hexanoyl, heptanoyl-, 2-
ethylhexanoyl, 2-
propylheptanoyl, pivaloyl, benzoyl or naphthoyl.
For the purposes of the present invention, the term carboxylate preferably
refers to a derivative
of a carboxylic acid function, in particular a carboxylic ester function or a
carboxamide function.
Such functions include, for example, esters with C1-C4-alkanols such as
methanol, ethanol, n-
propanol, isopropanol, n-butanol, sec-butanol and tert-butanol. They also
include the primary
amides and their N-alkyl and N,N-dialkyl derivatives.
CA 02862143 2015-11-24
Fused ring systems can be aromatic, hydroaromatic and cyclic compounds joined
by fusion
(fused). Fused ring systems comprise two, three or more than three rings.
Depending on the
way in which they are joined, a distinction is made among fused ring systems
between ortho-
fusion, i.e. each ring shares an edge or two atoms with each adjacent ring,
and pen-fusion in
which a carbon atom belongs to more than two rings. Among fused ring systems,
preference is
given to ortho-fused ring systems.
As regards step a), what has been said above for the preparation of
phosphochloridites of the
general formula (I) is fully referenced.
Step b)
In the organic diphosphites of the general formula (II), preference is given
to the radicals R3 and
R4 together forming a fused-on benzene ring and R1 and R2 each being hydrogen,
i.e. the group
of the formula
R2 R3
R1 11 R4
R4 it R1
R3 R2
is
44/
410
The diols of the general formula (A2)
CA 02862143 2014-06-27
16
R2 R3
R1 lik R4
(A2)
OH
HO
R4 . Ri
R3 R2
are preferably selected from among 3,3',5,5'-tetramethy1-1,1'-biphenyl-2,2'-
diol, 3131,5,51-
tetraethy1-1,11-bipheny1-2,2'-diol, 3,3',5,5'-tetra-n-propy1-1,1-bipheny1-2,2'-
diol, 3,3'-dimethy1-5,5'-
dichloro-1,1'-bipheny1-2,2'-diol, 3,3'-diethyl-5,5'-dibromo-1,11-bipheny1-2,2'-
diol, 3,3'-dimethy1-
5,5'-diethy1-1,1'-biphenyl-2,2'-diol, 3,3'-dimethy1-5,5'-di-n-propy1-1,1'-
biphenyl-2,2'-diol, 3,31,5,51-
tetraisopropy1-1,11-bipheny1-2,2'-diol, 3,3',5,5'-tetra-n-buty1-1,1'-bipheny1-
2,2'-diol, 3,31,5,51-
tetraisobuty1-1,1'-bipheny1-2,21-diol, 3,31,5,51-tetra-sec-buty1-1,1-bipheny1-
2,2'-diol, 3,31,5,51-
tetra(1,1-dimethylethyl)-1,11-bipheny1-2,2'-diol, 3,3'-di(1,1-dimethylethyl)-
5,5'-di-n-amy1-1,1-
bipheny1-2,2'-diol, 3,3',5,5'-tetrakis(1,1-dimethylpropy1)-1111-biphenyl-2,2'-
diol, 3,3'-di(1,1-
dimethylethyl)-5,5'-bis(1,1-dimethylpropy1)-1,11-biphenyl-2,2'-diol, 3,3'-
di(1,1-dimethylethyl)-5,5'-
di-n-hexy1-1,11-biphenyl-2,2'-diol, 3,3'-di(1,1-dimethylethyl)-5,5'-di-2-hexy1-
1,1-biphenyl-2,2'-diol,
3,3'-di(1,1-dimethylethyl)-5,5'-di-3-hexy1-1,1'-biphenyl-2,2"-diol, 3,3'-
di(1,1-dimethylethyl)-5,5'-di-
n-hepty1-1,11-biphenyl-2,2"-diol, 3,3'-di(1,1-dimethylethyl)-5,5'-di-2-heptyl-
1,1-biphenyl-2,2'-diol,
3,3'-di(1,1-dimethylethyl)-5,5'-di-3-hepty1-1,11-biphenyl-2,2'-diol, 3,3'-
di(1,1-dimethylethyl)-5,5'-di-
4-hepty1-1,11-biphenyl-2,2'-diol, 3,3'-di(1,1-dimethylethyl)-5,5'-di-n-octy1-
1,11-biphenyl-2,2'-diol,
3,3'-di(1,1-dimethylethyl)-5,5'-di-2-octy1-1,1'-biphenyl-2,2'-diol, 5,5'-di-3-
octy1-1,1'-bipheny1-2,2'-
diol, 3,3'-di(1,1-dimethylethyl)-5,5'-di-4-octy1-1,1'-biphenyl-2,2'-diol, 3,3'-
di(1,1-dimethylethyl)-
5,5'-bis(1,1,3,3-tetramethylbuty1)-1,1'-biphenyl-2,2'-diol, 3,3',5,5'-
tetrakis(1,1,3,3-tetra-
methylbuty1)-1,1'-bipheny1-2,2'-diol, 3,3'-di(1,1-dimethylethy1)-5,5',6,6'-
tetramethy1-1,11-bipheny1-
2,2'-diol, 3,3'-di(1,1-dimethylethyl)-5,5'-diphenyl-1,1'-biphenyl-2,2'-diol,
3,3'-di(1,1-dimethylethyl)-
5,5'-bis(2,4,6,-trimethylpheny1)-1,1'-biphenyl-2,2'-diol, 3,3'-di(1,1-
dimethylethyl)-5,5'-dimethoxy-
1,1'-biphenyl-2,2'-diol, 3,3'-di(1,1-dimethylethyl)-5,5'-diethoxy-1,1'-
bipheny1-2,2'-diol, 3,3'-di(1,1-
dimethylethyl)-5,5'-di-n-propoxy-1,11-biphenyl-2,2'-diol, 3,3'-di(1,1-
dimethylethyl)-5,5'-di-
isopropoxy-1,1-bipheny1-2,2'-diol, 3,3'-di(1,1-dimethylethyl)-5,5'-di-n-butoxy-
1,1'-bipheny1-2,Z-
dial, 3,3'-di(1,1-dimethylethyl)-5,5'-di-sec-butoxy-1,11-bipheny1-2,2'-diol,
3,3'-di(1,1-
dimethylethyl)-5,5'-diisobutoxy-1,11-bipheny1-2,2'-diol, 3,3'-di(1,1-
dimethylethyl)-5,5'-di-tert-
butoxy-1,1-bipheny1-2,2'-diol and 1,1'-binaphthaliny1-2,2'-diol.
CA 02862143 2014-06-27
17
The diol (A2) is particularly preferably 3,3',5,5'-tetra(1,1-dimethylethyI)-
1,11-biphenyl-2,2'-diol.
That is to say, particular preference is given to the radicals R1 and R3 in
the organic diphos-
phites of the general formula (II) all being tert-butyl and R2 and R4 all
being hydrogen.
The 6-chlorodibenzo[d,f][1,3,2]dioxaphosphepin (I) prepared by the process of
the invention is
particularly useful for preparing the following organic diphosphites (II):
(CH3)3C C(CH3)3
(0H3)3c 0(0H3)3
0 0
0-1,13 P-0
oI
0
110
(CH3)3C CH3 H3C C(CH3)3
411
(CH3)3C C(CH3)3
0 0
la 0-7 ¨o.
oI
0
= 411
CA 02862143 2014-06-27
=
18
CH30 OCH3
41.
(CH3)3C C(CH)3
0 0
0-7 P-0 =
oI
0
411
(CH3)3C C(CH3)3
CH30 OCH3
0 0
0-7 -o.
0 0
=
410
111
0 0
si 0¨Fr P-0 410
o
0
411 441
In particular, the organic diphosphite of the formula (II) is 6,6'4[3,3',5,5'-
tetrakis(1,1-
dimethylethyl)-1,11-biphenyl]-2,2'-diyl]bis(oxy)]bisdibenzo [d,f] [1,3,2]-
dioxaphosphepin.
Step b) of the preparation of the diphosphites (II) can in principle be
carried out by means of
known phosphorus halide-alcohol condensation reactions, as described, for
example, in
CA 02862143 2015-11-24
19
EP 0 214 622 A2, US 4,668,651, US 4,748,261, US 4,769,498, US 4,885,401, US
5,235,113,
US 5,391,801, US 5,663,403, US 5,728,861, US 6,172,267, WO 2003/062171 and
WO 2003/062251.
The reaction in step b) is preferably carried out in the presence of a base.
Suitable bases are generally, for example, alkali metal hydroxides, alkaline
earth metal hydrox-
ides, NO3, alkali metal carbonates, alkaline earth metal carbonates, alkali
metal hydrogencar-
bonates, alkaline earth metal hydrogencarbonates, tertiary amines and basic
ion-exchange res-
ins, etc. These include, for example, NaOH, KOH, Ca(OH)2, triethylamine,
tripropylamine, tribu-
tylamine, etc. Preference is given to tertiary amines and more particualrly
triethylamine.
In a specific embodiment, the reaction in step b) is carried out by means of a
process as de-
scribed in WO 2003/062171 and WO 2003/062251. Here, step b) is then carried
out in the pres-
ence of a base selected from among bases which with the hydrohalic acid formed
in the respec-
tive reaction step form a salt which is liquid at temperatures at which the
reaction product of the
respective reaction step is not significantly decomposed during the removal of
the liquid salt and
the salt forms two immiscible liquid phases with the reaction medium of the
respective reaction
step.
Suitable bases of this type are described in WO 2003/062171 and WO
2003/062251. Prefer-
ence is given to using a base selected from among 1-methylimidazole, 1-n-
butylimidazole, 2-
methylpyridine and 2-ethylpyridine in step b).
In the last-named process variant, the major part of the acid salts formed
from HCI and base in
the condensation reaction in step b) can advantageously be removed by simple
phase separa-
tion.
The organic diphosphites (II) are advantageous as ligands for catalysts for
hydroformylation,
hydrocyanation or hydrogenation.
The invention will be illustrated below with the aid of the following,
nonlimiting example.
Examples
Example 1:
CA 02862143 2014-06-27
Synthesis of 6-chlorodibenzo[d,f][1,3,2]clioxaphosphepin using
methylimidazolium hydrochloride
as catalyst
2,2'-Dihydroxybiphenyl (931.1 g, 5.0 mol) and 1-methylimidazoliumhydrochloride
(0.9 g,
7.6 mmol) were placed under nitrogen in a 2000 ml double-walled reactor and,
after melting of
the 2,2'-dihydroxybiphenyl, heated to an internal temperature of 142 C. The
introduction of PCI3
(861.2 g, 6.263 mol) was then commenced with stirring, ensuring that the PCI3
did not get onto
the hot reactor wall. The rate of introduction was regulated so that the
attached HCI scrubbing
tower could completely absorb the HC1formed. A total of three hours were
required for the in-
troduction of the PCI3. After the introduction of the PCI3, the mixture was
stirred at 140 C for
another three hours to give a fluid yellow reaction mixture. The reactor was
subsequently evac-
uated over a period of 40 minutes to a final vacuum of 16 mbar in order to
remove the excess
PCI3. The last residues of PCI3 were removed by stirring under reduced
pressure at 140 C/16
mbar and the mixture was subsequently cooled to 65 C. After admission of
nitrogen, toluene
(139.2 g) was added and the 90% strength solution (1390 g) of the product
obtained in this way
was drained into a screw-cap bottle and closed under argon. According to 31P-
NMR, the product
had a purity of 98.7%.