Note: Descriptions are shown in the official language in which they were submitted.
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
FENOFIBRATE FORMULATION
BACKGROUND
1. Cross-Reference to Related Applications
[0001] This is a Continuation-in-Part of Application No. 13/531,955, filed
June 25,
2012. The entire disclosure of the prior application is hereby incorporated by
reference in its entirety.
2. Field
[0002] This invention relates generally to immediate release fenofibrate
dosage
forms.
3. Description of Related Art
[0003] Fenofibrate is an active principle which is very poorly soluble in
water, and
the absorption of fenofibrate in the digestive tract is limited. An increase
in its
solubility leads to better digestive absorption. Various approaches have been
explored
in order to increase the solubility of fenofibrate, including micronization of
the active
principle, addition of a surfactant, and comicronization of fenofibrate with a
surfactant.
[0004] Fenofibrate is freely soluble in methanol and acetonitrile, and
insoluble in
water. Having no ionizable group, fenofibrate solubility is not influenced by
changes
in medium pH. However, the aqueous solubility of Fenofibrate can be improved
in the
presence of surfactants. As the concentration of the surfactant sodium lauryl
sulfate,
for example, increases from 0.0 M to 0.1 M, fenofibrate solubility increases
from 0.8
mg/L to 910.8 mg/L.
- 1 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
[0005] There are various lipid regulating agents, such as atorvastatin,
cerivastatin,
ezetimibe, fluvastatin, lovastatin, pitavastatin, pravastatin, probucol,
rosuvastatin,
simvastatin and fibrates. The fibrates are a group of drugs which are known as
hypolipidaemic agents. They include benzafibrate, clofibrate, ciprofibrate,
fenofibrate
and gemfibrozil. The fibrates have the beneficial effect of lowering
cholesterol levels
in the blood and hence reducing the risk of coronary heart disease.
[0006] Fenofibrate is a fibric acid derivative that has been marketed since
the mid
1970's (1998 in the United States) as a lipid regulating drug. The chemical
name of
fenofibrate is 244-(4-chlorobenzoyl) phenoxy]-2-methyl-propionic acid,
1¨methyl
ester. It has a molecular formula C20H2104C1 and a molecular weight of 360.83.
The
melting point of fenofibrate is 79 C to 82 C. Fenofibrate is a white solid
that is stable
under ordinary conditions. Fenofibrate is absorbed as fenofibric acid, which
is
responsible for the pharmacological activity.
[0007] Fenofibrate has an extremely low solubility in water of around 6
micrograms/ml. This can adversely affect absorption of drugs of the drug
substance in
vivo, leading to poor bioavailability. Consequently higher amounts of the drug
substance are required to achieve the desired blood levels. The poor
solubility of the
fenofibrate also restricts the options available for formulating the drug
substance.
[0008] The standards for bioavailability and/or bioequivalence depend on
several
natural log transformed parameters associated with the rate and extent of
absorption.
Specifically, the rate and extent of absorption is measured by the parameters
AUCL,
AUCI, and Cmax. The parameter AUCL is the area under the plasma concentration-
time curve from time zero to time t, where t is the last time point with
measurable
concentration for individual formulation. The parameter AUCI is the area under
the
- 2 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
plasma concentration-time curve from time zero to time infinity. Additionally,
Cmax,
sometimes referred to as Cpeak, is the maximum plasma concentration of the
drug.
For two products to be bioequivalent, the 90% confidence interval of the
relative
mean Cmax, AUCL, and AUCI of the test product to reference product should be
within 80% to 125%.
[0009] Many methods have been used to enhance dissolution rates of poorly
water-
soluble or insoluble drugs in general, and fenofibrate in particular. Such
methods
include micronization of fenofibrate, microcrystallization of fenofibrate,
preparation
solid fenofibrate dispersions, and coprecipitation of fenofibrate with inert,
water-
soluble compounds as carriers. Other methods include grinding fenofibrate with
an
inert water-insoluble compound, so that fenofibrate is adsorbed onto the inert
compound.
[0010] European Patent EP 256933 teaches fenofibrate granules which contain
micronized fenofibrate. The crystalline fenofibrate particles are less than 50
microns
in size. The micronized fenofibrate may be granulated with various types of
binder
polymers, such as polyvinylpyrrolidone, methacrylic polymers, cellulose
derivatives,
and polyethylene glycols, where an organic solvent is used for the
granulation.
[0011] European Patent EP 330532 teaches improving the bioavailability of
fenofibrate by comicronizing fenofibrate with a solid wetting agent or
surfactant, such
as sodium lauryl sulfate. The comicronizate is then granulated by wet
granulation in
order to improve the flow capacities of the powder and to facilitate filling
into gelatin
capsules. The comicronizate may be granulated with excipients such as lactose,
starch, polyvinyl pyrrolidone and/or magnesium stearate. A formulation of the
composition described in EP 330532 was compared to the formulation described
in
- 3 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
patent EP 256933, discussed above, and found to show a statistically
significant
increase in bioavailability vs. the formulation of EP 256933. Specifically, 67
mg
fenofibrate in the formulation of EP 330532 gave the same absorption in vivo
as 100
mg fenofibrate of the formulation of EP 256933. Therefore, the process
described in
EP 330532 led to a new dosage form in which the active ingredient, co-
micronized
with a solid surfactant, was able to show improved dissolution, and thus
increased
bioavailability, which makes it possible, for a given level of effectiveness,
to decrease
the daily dose of the medicament.
[0012] U.S. Patent Number 4,895,726 teaches to improve fenofibrate
bioavailability
using a composition containing a comicronized mixture of particles of
fenofibrate and
a solid surfactant. The co-micronization was carried out in an accelerated air-
jet mill
until the powder obtained has a mean particle size is less than 15 microns.
The
powder was mixed with lactose and starch and granulated. The dried granules
were
mixed with polyvinylpyrrolidone and magnesium stearate and filled in gelatin
capsules. U.S. Patent Number 4,895,726 teaches that there is no statistically
significant difference between the in vivo bioavailability of 200 mg of co-
micronized
fenofibrate according to the invention of U.S. Patent Number 4,895,726 and 300
mg
of non-micronized fenofibrate. In other words, this patent proved that co-
micronized
fenofibrate at a 200 mg dose is bioequivalent to a 300 mg dose of a non-
micronized
fenofibrate formulation according to EP 330532.
[0013] U.S. Patent Number 4,800,079 describes a granular medicine based on
fenofibrate, each granule comprising an inert core, a layer based on
fenofibrate, and a
protective layer. The medicine is characterized in that the fenofibrate is
present in the
form of crystalline microparticles having a size of less than 30 microns, and
- 4 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
preferably less than 10 microns. The layer based on fenofibrate includes a
binder
selected from the group consisting of methacrylic polymers,
polyvinylpyrrolidone,
cellulose derivatives, and polyethylene glycols.
[0014] U.S. Patent Number 7,101,574 describes a pharmaceutical composition in
the
form of granules, containing neutral microgranules, supporting a layer of a
composition comprising micronized fenofibrate, a surfactant and, a cellulose
binding
cellulose derivative, preferably hydroxypropylmethylcellulose (HPMC), as a
solubilizing adjuvant. The cellulose derivative represents less than 20 wt. %
of the
composition, while the fenofibrate is present in an amount greater than or
equal to
60% by weight of the pharmaceutical composition. The pharmaceutical
composition
reported in U.S. Patent Number 7,101,574 describes the fenofibrate:HPMC mass
ratio
as being between 5:1 and 15:1. The formulation disclosed in U.S. Patent Number
7,101,574 provides enhanced bioavailability of the active principle. U.S.
Patent
Number 7,101,574 compares the in vivo release profile of gelatin capsules
containing
the disclosed granules to gelatin capsules containing an equivalent dose of
the
formulation of EP 330532, marketed under the trade name Lipanthyl 200M. The
maximum plasma concentration (Cmax) of the disclosed formulation under fasting
conditions was reported to be 4.71 microgram/mL, compared to a Cmax of 2.35
microgram/mL attained with the formulation of EP 330532.
[0015] This present disclosure provides a formulation having a higher
bioavailability
than commercially available products containing fenofibrate, including ANTARA
Capsules, sold by Lupin Laboratories and containing 130 mg of fenofibrate. The
ANTARA Capsules are made according to the teachings of U.S. Patent Number
7,101,574 and/or U.S. Patent Number 7,863,331.
- 5 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
[0016] In various embodiments, the formulation of the present disclosure
comprises
Fenofibrate or a related drug, HPMC, a surfactant, such as sodium lauryl
sulfate, an
inert support, such as sugar spheres (20-45 mesh or 850-355 microns) and other
excipients. In various embodiments, the inert supports may have a size of 35-
45 mesh
or 500-355 microns). In other embodiments, the inert supports may have a size
of 20-
25 mesh or 850-710 microns). The fenofibrate to HPMC weight ratio is between
about 3.5: 1 and about 4.5:1, and the amount of sodium lauryl sulfate is
between about
0.3% and about 10% by weight. The fenofibrate formulation and method of
manufacturing disclosed herein provides an improvement in Cmax, compared to
ANTARA capsules. In various embodiments, the fenofibrate formulation and
method of manufacturing disclosed herein provides an improvement in Cmax
without
significantly affecting values for AUCL and/or AUCI, when compared to ANTARA
capsules. Various embodiments disclosed herein may afford an improvement in
Cmax and an increase in AUCL and/or AUCI, compared to ANTARA capsules.
SUMMARY
[0017] A summary of various embodiments is presented herein. Some
simplifications
and omissions may be made in the following summary, which is intended to
highlight
and introduce some aspects of the various exemplary embodiments, but not to
limit
the scope of the invention. Detailed descriptions of a preferred exemplary
embodiment adequate to allow those of ordinary skill in the art to make and
use the
inventive concepts will follow in later sections. Various embodiments
disclosed
herein relate to a dosage form comprising an effective amount of fenofibric
acid,
pharmaceutically acceptable salts thereof, pharmaceutically acceptable esters
thereof,
- 6 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
or prodrugs thereof. In various embodiments, the active agent is fenofibrate,
an ester
of fenofibric acid which is hydrolyzed to fenofibric acid in vivo. Fenofibrate
is a
prodrug of fenofibric acid.
[0018] Various embodiments disclosed herein relate to a fenofibrate
formulation
comprising a dosage form containing a plurality of beads or particles. The
beads or
particles collectively comprise a pharmaceutical composition containing a drug
selected from the group consisting of fenofibric acid, pharmaceutically
acceptable
salts thereof, pharmaceutically acceptable esters thereof, and prodrugs
thereof; from
0.3% to 10% by weight of the beads or particles of a surfactant; and from
about 5% to
about 15% by weight of the beads or particles of a water soluble or water
dispersible
cellulosic binder. The beads or particles may comprise from about 20% to about
60%
by weight of the drug. In various embodiments, the beads or particles may each
comprise an inert core, with the pharmaceutical composition being coated on
the inert
core.
[0019] In various embodiments, the mass ratio of the drug to the binder in the
dosage
form is between about 3.5:1 and 4.5:1, preferably between about 3.8:1 and
about
4.4:1, more preferably between about 3.9:1 and about 4.35:1. In some
embodiments,
the dosage form produces a first Cmax in vivo that is between about 10% and
about
50% higher, preferably 20% to 45% higher, than a comparative Cmax produced by
a
comparative dosage form comprising the drug and the binder in a ratio of
between 5:1
and 15:1, where the comparative dosage form further comprises 1% to 10% by
weight
of the surfactant. The dosage form disclosed herein and the comparative dosage
form
each contain equivalent amounts of the drug. Suitable examples of comparative
dosage forms include the dosage forms described in the Examples of U.S. Patent
No.
- 7 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
7,863,331 and the Examples of U.S. Patent No. 7,101,574.
[0020] In certain embodiments, where the pharmaceutical composition comprises
about 45% to about 55% by weight of said drug, based on the weight of the
beads or
particles, the dosage form produces a Cmax in vivo that is between about 10%
and
about 30% higher than a comparative Cmax from the comparative dosage form. In
other embodiments, where the pharmaceutical composition comprises about 25% to
about 35% by weight of the drug, based on the weight of the beads or
particles, the
dosage form produces a Cmax in vivo that is between about 35% and about 50%
higher than a comparative Cmax from the comparative dosage form.
[0021] In various embodiments, the dosage form comprises a plurality of first
beads
or first particles; and a plurality of second beads or second particles. Each
of the first
beads or first particles comprises from about 45% to about 55% by weight of
the
fenofibrate drug, from 0.3% to 10% by weight of a surfactant; and from about
5% to
about 15% by weight of a water soluble or water dispersible cellulosic binder.
The
mass ratio of the drug to the binder in said first beads or first particles is
between
about 3.5:1 and 4.5:1. Each of the second beads or second particles comprises
from
about 25% to about 35% by weight of the fenofibrate drug, from 0.3% to 10% by
weight of a surfactant; and from about 5% to about 15% by weight of a water
soluble
or water dispersible cellulosic binder. The mass ratio of the drug to the
binder in the
second beads or second particles is between about 3.5:1 and about 4.5:1. In
various
embodiments, the dosage form comprising a combination of first beads or first
particles and second beads or second particles produces a Cmax in vivo that is
greater
than a Cmax produced by a dosage form containing only the first beads, and
less than
a Cmax produced by a dosage form containing only the second beads.
- 8 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
[0022] The dosage form comprising a combination of first beads or first
particles and
second beads or second particles contains from 20% to 80% by weight of the
first
beads or first particles, preferably between 30% and 70% by weight of the
first beads
or first particles, more preferably between 40% and 60% by weight of the first
beads
or first particles, based on the total weight of all beads or particles. The
precise ratio
of the first beads or first particles to the second beads or second particles
may be
adjusted to control Cmax. For example, a dosage form comprising 30% by weight
of
the first beads and 70% of the second beads will exhibit a higher Cmax than a
dosage
form comprising 70% by weight of the first beads and 30% of the second beads.
[0023] Various embodiments disclosed herein relate to a dosage form comprising
a
plurality of beads or particles, where the beads or particles comprise a
pharmaceutical
composition containing from about 20% to about 55% by weight of a drug
selected
from the group consisting of fenofibric acid, pharmaceutically acceptable
salts
thereof, pharmaceutically acceptable esters thereof, and prodrugs thereof;
from 0.3%
to 10% by weight of said beads or particles of a surfactant; and from about 5%
to
about 15% by weight of a water soluble or water dispersible cellulosic binder.
The
mass ratio of the drug to the binder in the dosage form is between about 3.5:1
and
4.5:1.
[0024] According to various embodiments described herein, the surfactant used
in the
pharmaceutical compositions disclosed herein may be anionic surfactants,
nonionic
surfactants, or cationic surfactants, preferably anionic surfactants. A
preferred
surfactant is sodium lauryl sulfate.
[0025] The standards for bioavailability depend on several natural log
transformed
parameters associated with the rate and extent of absorption. Specifically,
- 9 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
bioavailability depends on the parameters Cmax, AUCL, and AUCI. the 90%
confidence interval of the relative mean Cmax, AUCL, and AUCI of the test
product
to reference product should be within 80% to 125%.
[0026] Various embodiments disclosed herein relate to a dosage form which is
more
bioavailable than ANTARA@ capsules having an equivalent amount of fenofibrate,
and therefore is not bioequivalent to ANTARA@ capsules having an equivalent
amount of fenofibrate. Specifically, the dosage forms disclosed herein exhibit
a higher
Cmax than ANTARA@ capsules having an equivalent amount of fenofibrate. The
disclosed dosage form comprises a defined amount of fenofibrate, which may be
between 40 and 200 mg fenofibrate, preferably between 40 and 160 mg micronized
fenofibrate. In various embodiments, the disclosed composition has a high
bioavailability, with a Cmax which is between 10% and 50% higher, preferably
between 20% and 45% higher, than ANTARA@ capsules having an equivalent
amount of fenofibrate. In various embodiments, ratio of AUCL or AUCI of the
disclosed composition to ANTARA@ capsules having an equivalent amount of
fenofibrate, falls with the range of 80% to 125%.
[0027] In various embodiments disclosed herein, the disclosed compositions
have an
AUCL or an AUCI value which is greater than the corresponding AUCL or AUCI
value for ANTARA@ capsules having an equivalent amount of fenofibrate, as well
as
a Cmax which is higher than the corresponding Cmax for ANTARA@ capsules. More
specifically, the disclosed compositions may have an AUCL or an AUCI value
which
is at least 10% greater, preferably 10% to 50% greater, more preferably 10% to
30%
greater, than the corresponding AUCL or AUCI value for ANTARA@ capsules.
[0028] Various dosage forms disclosed herein also have a higher
bioavailability than
- 10-
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
the dosage forms described in the Examples of U.S. Patent No. 7,863,331 and
the
Examples of U.S. Patent No. 7,101,574.
[0029] Fenofibrate dosage forms disclosed herein may be used to reduce
cholesterol
levels in patients at risk of cardiovascular disease. The fenofibrate dosage
forms
disclosed herein reduce both low-density lipoprotein (LDL) and very low
density
lipoprotein (VLDL) levels, as well as increasing high-density lipoprotein
(HDL)
levels and reducing triglycerides level. The fenofibrate dosage forms
disclosed herein
may be used alone, or in conjunction with statins in the treatment of
hypercholesterolemia and hypertriglyceridemia.
BRIEF DESCRIPTION OF THE DRAWINGS
[0030] In order to better understand various exemplary embodiments, reference
is
made to the accompanying drawings, wherein:
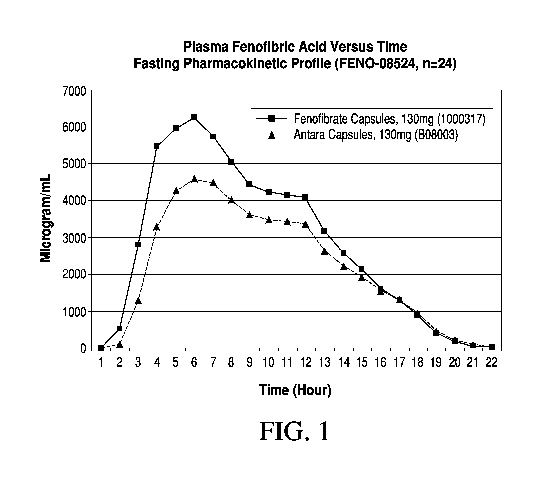
[0031] FIG. 1 shows the concentration of fenofibrate in plasma over time upon
administration of 130 mg ANTARA capsules and administration of 130 mg
capsules according to Example 4.
[0032] FIG. 2 and FIG. 3 show the concentration of fenofibrate in plasma over
time
upon administration of 130 mg ANTARA capsules and administration of 130 mg
capsules according to Example 4.
[0033] FIG. 4 shows the concentration of fenofibrate in plasma over time upon
administration of 130 mg ANTARA capsules and administration of 130 mg capsules
according to Example 6.
[0034] Fig. 5 shows the concentration of fenofibrate in plasma over time upon
administration of 130 mg ANTARA capsules and administration of 130 mg
- 11-
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
capsules according to Example 7.
[0035] FIG. 6 and FIG. 7 show the concentration of fenofibrate in plasma over
time
upon administration of 130 mg ANTARA capsules and administration of 130 mg
capsules according to Example 8.
DETAILED DESCRIPTION
1. Dosage Forms Which Are Bioequivalent to ANTARA Capsules
[0036] Certain embodiments disclosed herein relate to dosage forms comprising
a
combination of high bioavailability beads and low bioavailability beads. In
certain
embodiments, the high bioavailability fenofibrate-containing beads contain
micronized fenofibrate, a surfactant, and a binder which is water-soluble or
water-
dispersible. Suitable binders include hydroxypropylmethylcellulose (HPMC);
hydroxypropyl cellulose; hydroxyethyl cellulose; c arb oxymethylcellulo se ;
povidone
and chitosan, with HPMC being a preferred binder. A suitable HPMC binder is
commercially available under the trade name Pharmacoat 603, from Harke Group.
[0037] In certain embodiments, the low bioavailability fenofibrate-containing
beads
contain micronized fenofibrate, and a binder which is water-soluble or water-
dispersible. Suitable binders include hydroxypropylmethylcellulose (HPMC);
hydroxypropyl cellulose; hydroxyethyl cellulose; c arb oxymethylcellulo se ;
povidone
and chitosan, with HPMC being a preferred binder.
[0038] In various embodiments, the high bioavailability fenofibrate-containing
beads
contain a surfactant in an amount of between about 0.3% by weight and about
10% by
weight, preferably between about 0.5% by weight and about 5% by weight, more
preferably between about 0.5% by weight and about 3% by weight. The low
- 12-
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
bioavailability fenofibrate-containing beads contain a surfactant in an amount
of
between about 0% by weight and about 0.25% by weight, preferably between about
0% by weight and about 0.05% by weight, more preferably 0% by weight.
[0039] The low bioavailability fenofibrate-containing beads (slow beads) and
the high
bioavailability fenofibrate-containing beads (fast beads) may be co-
administered in a
single dosage form. The low bioavailability fenofibrate-containing beads and
the high
bioavailability fenofibrate-containing beads may be combined and filled into a
hard
gelatin shell to form a capsule.
[0040] Alternatively, they may be combined with a water-soluble or water-
dispersible
binder, and compressed along with tableting excipients to form an immediate-
release
solid oral dosage form such as a tablet. Such compressed tablets may include a
combination of slow beads and fast beads mixed together and combined with the
binder and excipients prior to compression. As an alternative, slow beads may
be
mixed with the binder and excipients prior to a first compression step to form
a first
layer containing slow beads; and then the fast beads may be combined with the
binder
and excipients prior to a second compression step. In the second compression
step, the
formulation of fast beads and binder is deposited on the first layer, and the
formulation of fast beads is compressed to form a bilayer tablet. If desired,
suitable
colorants may be added to either or both of the slow bead formulation and the
fast
bead formulation so that the layers of the bilayer tablet are visually
distinguishable.
Alternatively, the formulation of fast beads may be compressed initially to
form the
first layer, with the second layer containing the formulation of slow beads.
[0041] In various embodiments, the fast beads and the slow beads may be
combined,
and then blended with suitable excipients, including a binder, a water-soluble
or
- 13 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
water-dispersible filler, a disintegrant and/or a lubricant. The resulting
mixture may
be compressed into multiple mini-tablets, which may then be then encapsulated
in a
suitable size two piece hard gelatin capsule shell.
[0042] In other embodiments, the fast beads may be blended with suitable
excipients,
and then compressed into multiple mini-tablets. Similarly, the slow beads may
be
blended with suitable excipients, and then compressed into multiple mini-
tablets.
Mini-tablets containing the fast beads may be mixed with mini-tablets
containing the
slow beads, and the resulting admixture may then be then encapsulated in a
suitable
size two piece hard gelatin capsule shell. In various embodiments, the mini-
tablets
containing the fast beads and the mini-tablets containing the slow beads
contain equal
amounts of fenofibrate, and are combined in a predetermined ratio. In various
embodiments, a capsule containing mini-tablets containing fast beads and mini-
tablets
containing slow beads contains from 50% to 80% of mini-tablets containing fast
beads and from 20% to 50% of mini-tablets containing the slow beads.
[0043] In various embodiments, the dosage of fenofibrate may take the form of
multiple tablets to be co-administered. In various embodiments, a first tablet
may
contain fenofibrate in the form of fast beads only (a fast tablet), while a
second tablet
may contain fenofibrate in the form of slow beads only (a slow tablet). With
this
approach, one slow tablet may be co-administered with at least one fast
tablet,
preferably from one to three fast tablets.
[0044] In various embodiments, one tablet containing fast beads or a
combination of
slow and fast beads may be prepared as disclosed herein, and then combined
with a
granulated powder of fenofibrate and encapsulated in a suitable size two piece
hard
gelatin capsule shell. Alternatively, a combination of fast beads and
granulated
- 14-
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
powder of fenofibrate can be encapsulated to achieve the desired drug release
profile.
[0045] In various embodiments, the slow and fast beads can also be
manufactured by
extrusion spheronization technology. As an alternative to slow and fast beads,
the
dosage forms disclosed herein may be manufactured from granules manufactured
by
spray drying techniques. For example, a slurry of fenofibrate, a cellulosic
binder, and
a surfactant in an amount of between about 0.3% by weight and about 10% by
weight,
based on solids content, may be spray dried to form fast granules. A slurry of
fenofibrate, a cellulosic binder, and a surfactant in an amount of between
about 0% by
weight and about 0.25% by weight, based on solids content, may be spray dried
to
form slow granules. The fast and slow granules may be used as substitutes for
fast and
slow beads.
[0046] In various embodiments, the fenofibrate beads or granules can be used
for
manufacturing combination pharmaceutical products. In some embodiments, the
combination products may contain fenofibrate and a second drug, such as a
statin,
niacin, or metformin. The combination products may be manufactured by applying
a
fenofibrate suspension onto a core material, where the core material contains
at least
one pharmaceutical active, such as a statin, niacin, or metformin.
[0047] In some embodiments, the slow and fast beads may be combined and filled
into a unit dose sealed pouch. The contents of the pouch may be dispersed in a
liquid
such as juice or water, and the patient may drink the resulting dispersion.
[0048] In various embodiments, the ratio of the fast and slow beads (fast
beads:slow
beads) in the dosage forms disclosed herein is between 50:50 and 90:10,
preferably
between 60:40 and 90:10, most preferably between about 75:25 and 80:20. The
beads
disclosed herein may be prepared by spraying the drug layer suspension onto
inert
- 15 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
cores, preferably inert cores having a 20 to 50 mesh particle size, i.e., 300
microns to
850 microns. In some embodiments, the inert cores may have a mesh size of 20
to 25,
i.e., from 710 to 850 microns. In other embodiments, the inert cores may have
a mesh
size of 35 to 45, i.e., from 355 to 500 microns. In further embodiments, the
fast beads
may be made from inert cores having a mesh size of between 20 mesh and 25
mesh,
while the slow beads may be made from inert cores having a mesh size of
between 35
mesh and 45 mesh. In other embodiments, the fast beads may be made from 35 to
45
mesh cores, while the slow beads may be made from 20 to 25 mesh cores.
[0049] In one embodiment, an HPMC binder is solubilized in water or a polar
organic
solvent. Micronized fenofibrate is added to the binder solution to form a drug
suspension. The surfactant is added to the drug suspension. Optionally, an
antifoaming agent is incorporated into the drug suspension. Suitable
antifoaming
agents include silicones, such as dimethicone. Suitable solvents include Class
3
solvents, i.e., solvents of low toxic potential. Preferred Class 3 solvents
include polar
solvents suitable for dissolving or dispersing HPMC, such as water, Acetone,
Anisole,
1-Butanol, 2-Butanol, 3-Methyl- 1-butanol, Methyl ethyl ketone, Methyl
isobutyl
ketone, 2-Methyl-l-propanol, Dimethyl sulfoxide, Ethanol, 1-Pentanol, 1-
Propanol,
and 2-Propanol, and mixtures thereof. The resulting drug suspension is
homogenized,
and then sprayed onto the sugar spheres. In various embodiments, the drug
suspension
is homogenized for a minimum of 8 hours, preferably 8 to 48 hours, more
preferably
8 to 24 hours, most preferably 8 to 10 hours, prior to spraying onto the inert
cores.
[0050] In various embodiments, the beads disclosed herein may be prepared by
spraying the drug layer suspension onto inert cores made from insoluble inert
materials, such as silicon dioxide, calcium phosphate dihydrate, dicalcium
phosphate,
- 16-
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
calcium sulfate dihydrate, microcrystalline cellulose, cellulose derivatives,
calcium
carbonate, dibasic calcium phosphate anhydrous, dibasic calcium phosphate
monohydrate, tribasic calcium phosphate, magnesium carbonate, magnesium oxide
and activated carbon. In various embodiments, the beads disclosed herein may
be
prepared by spraying the drug layer suspension onto soluble cores, such as
sugar
spheres, more particularly, spheres of sugars selected from the group
consisting of
like dextrose, lactose, anhydrous lactose, spray-dried lactose, lactose
monohydrate,
mannitol, starches, sorbitol, and sucrose. Other materials which may be used
as inert
cores include insoluble inert plastic materials, such as spherical or nearly
spherical
core beads of polyvinylchloride or polystyrene. Mixtures of these core
materials may
be used. In certain embodiments, low bioavailability fenofibrate-containing
beads
(slow beads) may be prepared using a different core material from high
bioavailability
fenofibrate-containing beads (fast beads).
[0051] In various embodiments, the drug suspension is sprayed onto the inert
cores
contains a surfactant. Suitable surfactants include anionic surfactants,
nonionic
surfactants, cationic surfactants, or mixtures thereof. Preferably, the
surfactants are
anionic surfactants. Suitable anionic surfactants include sodium oleate,
sodium
dodecyl sulfate, sodium diethylhexyl sulfosuccinate, sodium dimethylhexyl
sulfosuccinate, sodium di-2-ethylacetate, sodium 2-ethylhexyl sulfate, sodium
lauryl
sulfate; sodium undecane-3- sulfate, sodium ethylphenylundecanoate, and
carboxylate
soaps. Preferred anionic surfactants include C8 to C24 sulfate monoester
surfactants.
More preferred anionic surfactants include sodium 2-ethylhexyl sulfate, sodium
lauryl
sulfate; and sodium undecane-3-sulfate. Suitable cationic surfactants include
benzalkonium halide salts. Suitable nonionic surfactants include C8-C28
ethoxylated
- 17 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
alcohols, mono-, di-, and trimesters of glycerol, and Polysorbate 80. In
various
embodiments, low bioavailability fenofibrate-containing beads (slow beads) may
be
prepared using a different surfactant from high bioavailability fenofibrate-
containing
beads (fast beads). In various embodiments, the slow beads include from 0% to
about
0.25% by weight of a surfactant, preferably from 0% to about 0.05% by weight
of a
surfactant, more preferably 0% by weight of a surfactant. In various
embodiments, the
fast beads include from about 0.3% to about 10% by weight of a surfactant,
preferably
from 0.3% to about 5% by weight of a surfactant, more preferably from about
0.5% to
about 2% by weight of a surfactant.
[0052] In various embodiments, the slow beads contain between about 30% and
about
80% micronized fenofibrate, relative to the total weight of the slow beads.
Preferably,
the slow beads contain between about 40% and about 59% micronized fenofibrate,
more preferably between about 45% and about 55% micronized fenofibrate. The
beads may contain between about 30% and about 59% micronized fenofibrate,
relative to the total weight of the slow beads. Preferably, the fast beads
contain
between about 40% and about 59% micronized fenofibrate, more preferably
between
about 45% and about 55% micronized fenofibrate.
[0053] The fast beads disclosed herein contain fenofibrate and a binder in a
ratio of
fenofibrate:binder of from about 1:1 to less than 5:1, preferably from about
2:1 to
about 4.5:1, more preferably from about 3.5:1 to about 4.5:1. In various
embodiments,
the binder is HPMC. In various embodiments, the fast beads disclosed herein
contain
fenofibrate and HPMC in a ratio of fenofibrate:HPMC of about 4:1. In some
embodiments, the fast beads additionally comprise from 0.3% to about 10% by
weight
of sodium lauryl sulfate, preferably from 0.5% to about 2% by weight of sodium
- 18 -
CA 02878011 2014-12-23
WO 2014/003810
PCT/US2012/061486
lauryl sulfate.
[0054] The slow beads disclosed herein contain fenofibrate and a binder in a
ratio of
fenofibrate:binder of from greater than 5:1 to about 15:1, preferably from
about 6:1 to
about 12:1, more preferably from about 7:1 to about 9:1. In various
embodiments, the
binder is HPMC, and the slow beads contain fenofibrate and HPMC in a ratio of
fenofibrate:HPMC of about 8:1. In some embodiments, the slow beads
additionally
comprise from 0% to about 0.25% by weight of sodium lauryl sulfate, and are
preferably free of sodium lauryl sulfate.
[0055] Fenofibrate, which is a prodrug of fenofibric acid, may be used as a
micronized fenofibrate powder. The fenofibrate powder may be fenofibrate Form
I as
disclosed in U.S. Patent Publication 2009/0149533; fenofibrate Form II as
disclosed
in U.S. Patent Publication 2009/0149533; amorphous fenofibrate; hydrates or
solvates
of fenofibrate, or a mixture thereof. Fenofibrate may be partially or
completely
replaced with fenofibric acid; pharmaceutically acceptable salts of fenofibric
acid; Cl
to C5 esters or prodrugs of fenofibric acid, or a mixture thereof.
[0056] In various embodiments, the fast and slow beads are made using
micronized
fenofibrate with a weight-average particle diameter (D50) of between 1 and 15
microns, preferably between 4 and 10 microns. Preferably, the fast and slow
beads are
made using micronized fenofibrate where at least 99% of the fenofibrate
particles
have a particle diameter of less than 50 microns.
2. Dosage
Forms Which Are More Bioavailable than ANTARA Capsules
[0057] Various embodiments disclosed herein provide fenofibrate dosage forms
comprising a faster (higher bioavailability) formulation than commercially
available
- 19-
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
products containing fenofibrate, including ANTARA Capsules, sold by Lupin
Laboratories and containing 130 mg of fenofibrate. Various embodiments of the
formulation disclosed herein comprise fenofibric acid, a salt thereof, a
derivative
thereof, or a prodrug thereof. A suitable formulation includes fenofibrate, an
ester
prodrug of fenofibric acid which undergoes hydrolysis in vivo.
[0058] Various embodiments of the dosage form disclosed herein include beads
or
particles containing a drug, which may be fenofibric acid or a salt or ester
thereof;
from 0.3% to 10% by weight of the beads or particles of a surfactant; and from
about 5% to about 15% by weight of the beads or particles of a water soluble
or water
dispersible cellulosic binder. In various embodiments, the mass ratio of the
drug to the
binder in said dosage form is between about 3.5:1 and 4.5:1, preferably
between about
3.8 and about 4.4, more preferably between about 3.9 and about 4.35.
[0059] In various embodiments, a formulation comprising fenofibric acid or a
salt or
ester thereof; from 0.3% to 10% by weight of a surfactant; and from about 5%
to
about 15% by weight of a water soluble or water dispersible cellulosic binder
may be
converted into particles or granules by conventional methods. Suitable methods
include spray-drying a solution of fenofibrate, a surfactant, and a water
soluble or
water dispersible cellulosic binder to form solid granules. In certain
embodiments, a
solution of fenofibrate, a surfactant, and a water soluble or water
dispersible cellulosic
binder may be sprayed onto inert cores to form solid granules.
[0060] In various embodiments, the formulation comprises beads or granules
made
from fenofibrate, HPMC, sodium lauryl sulfate, and inert cores. The beads or
granules
disclosed herein may be prepared by spraying a drug layer suspension
comprising a
solvent, fenofibrate, HPMC, and sodium lauryl sulfate onto inert cores,
preferably
- 20 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
inert cores having a 20 to 50 mesh particle size, i.e., 300 microns to 850
microns. In
some embodiments, the inert cores may have a mesh size of 20 to 25, i.e., from
710 to
850 microns. In other embodiments, the inert cores may have a mesh size of 35
to 45,
i.e., from 355 to 500 microns. In further embodiments, the beads or granules
may
comprise a first population of beads from inert cores having a mesh size of
between
20 mesh and 25 mesh, and a second population of beads from inert cores having
a
mesh size of between 35 mesh and 45 mesh.
[0061] In certain embodiments, an HPMC binder is solubilized in water or a
polar
organic solvent. Micronized fenofibrate is added to the binder solution to
form a drug
suspension. The surfactant is added to the drug suspension. Optionally, an
antifoaming agent is incorporated into the drug suspension. Suitable
antifoaming
agents include silicones, such as dimethicone. Suitable solvents include polar
solvents
suitable for dissolving or dispersing HPMC, such as water, Acetone, Anisole, 1-
Butanol, 2-Butanol, 3-Methyl- 1 -butanol, Methyl ethyl ketone, Methyl isobutyl
ketone, 2-Methyl-l-propanol, Dimethyl sulfoxide, Ethanol, 1-Pentanol, 1-
Propanol,
and 2-Propanol, and mixtures thereof. The resulting drug suspension is
homogenized,
and then sprayed onto the sugar spheres. In various embodiments, the drug
suspension
is homogenized for a minimum of 8 hours, preferably 8 to 48 hours, more
preferably
8 to 24 hours, most preferably 8 to 10 hours, prior to spraying onto the inert
cores.
[0062] In various embodiments, the beads disclosed herein may be prepared by
spraying the drug layer suspension onto inert cores made from insoluble inert
materials, such as silicon dioxide, calcium phosphate dihydrate, dicalcium
phosphate,
calcium sulfate dihydrate, microcrystalline cellulose, cellulose derivatives,
calcium
carbonate, dibasic calcium phosphate anhydrous, dibasic calcium phosphate
- 21 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
monohydrate, tribasic calcium phosphate, magnesium carbonate, magnesium oxide
and activated carbon. In various embodiments, the beads disclosed herein may
be
prepared by spraying the drug layer suspension onto soluble cores, such as
sugar
spheres, more particularly, spheres of sugars selected from the group
consisting of
like dextrose, lactose, anhydrous lactose, spray-dried lactose, lactose
monohydrate,
mannitol, starches, sorbitol, and sucrose. Other materials which may be used
as inert
cores include insoluble inert plastic materials, such as spherical or nearly
spherical
core beads of polyvinylchloride or polystyrene. Mixtures of these core
materials may
be used.
[0063] In various embodiments, the inert cores may be sugar spheres (35-45
mesh or
355 to 500 microns). The fenofibrate to HPMC weight ratio is between 3.5:1 and
4.5:1, and the surfactant is used in an amount of between about 0.3 wt. % and
about
wt. %. In various embodiments, the surfactant is an anionic surfactant, such
as
sodium lauryl sulfate (SLS). In various embodiments, the amount of sodium
lauryl
sulfate is 0.3% to 10% w/w, preferably 0.4 % to 5% w/w, more preferably 0.5%
to
2% w/w. The aqueous drug suspension containing fenofibrate, HPMC, sodium
lauryl
sulfate and simethicone was mixed for a minimum of 8 hours in a Ross Mixer
before
spraying on to the fluidized sugar sphere substrates in a ROTOR granulator.
The
fenofibrate formulation and method of manufacturing of this present invention
provides an improvement in Cmax by 1.2 times than that of the commercial
formulation (ANTARNO) and/ or an AUC improvement of at least 1.4 times than
that of the commercial formulation ANTARA capsules when dosed in the fasted
state.
[0064] In various embodiments, the formulation comprises Fenofibrate, HPMC,
- 22 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
sodium lauryl sulfate, sugar spheres (35-45 mesh or 355-500 microns) and other
excipients. The fenofibrate to HPMC weight ratio may be about 4 0.2 to about
1, and
the amount of sodium lauryl sulfate may be about 0.5 to about 2% w/w. The
fenofibrate formulation and method of manufacturing of this present invention
provides a value of Cmax which is 10% to 50%, preferably 20% to 45%, higher
than
that of the commercial formulation (ANTARA ) and/ or a value of AUC which is
10% to 60%, preferably 15% to 50%, more preferably 20 to 45%, higher than that
of
ANTARA capsules when dosed in the fasted state. In this present invention,
the
absorption of fenofibrate in fasted healthy volunteers is significantly
(P<0.05)
enhanced when compared to the commercial formulation such as ANTARA 130 mg
capsules.
[0065] An embodiment of the invention is directed to a fenofibrate,
composition
wherein the pharmacokinetic profile of the composition resulted in higher
bioavailability than ANTARA Capsules, 130mg, in particular as defined by Cmax
and AUC guidelines given by the U.S. Food and Drug Administration. The
increase in
bioavailability may permit a reduction in total dosage for some patients. The
improved bioavailability may allow administration of smaller doses of
fenofibrate to
achieve equivalent pharmacokinetics profiles.
3. Examples
[0066] Various embodiments will be described in the following non-limiting
examples. In the following examples, sugar spheres were used as the base
substrate
onto which the drug suspension was sprayed. The drug suspension was sprayed
onto
the sugar spheres in a fluidized bed fitted with a ROTOR Insert. The sugar
spheres
-23 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
had a 20-25 mesh particle size distribution, providing a uniform surface area
for drug
layering.
[0067] The micronized fenofibrate used in the following examples had a weight
average particle size D50 of between 5 and 7 microns, with at least 99% of
particles
having a particle size of <50 microns (D99); at least 90% of particles having
a particle
size of <15 microns (D90); and no more than 10% of particles having a particle
size
of <1 micron (D10).
[0068] Purified water was selected as a solvent for preparation of the drug
suspension, as it provides a suitable medium for dissolving the hypromellose
binder
and suspending the micronized fenofibrate drug substance.
[0069] Hypromellose (Pharmacoat 603) was used as a binder in the drug
suspension, as it aids in adhering the drug to the sugar sphere substrate
during
processing. Sodium lauryl sulfate (SLS) was used as a surfactant in preparing
the fast
beads. Sodium lauryl sulfate is a commonly used excipient in solid oral dosage
forms
to enhance wetting and improve drug dissolution rate. This excipient is
employed to
enhance the aqueous wettability of fenofibrate in the drug layering suspension
and to
enhance the drug release from the high bioavailability, or fast, drug layered
beads.
Sodium lauryl sulfate was not used in the slow beads in the following
examples,
resulting in reduced drug release from the slow beads.
[0070] Simethicone, an antifoaming agent, was incorporated in the drug
suspension to
minimize the potential to generate foam during preparation of the drug
layering
suspension.
[0071] In some examples, approximately 0.1% w/w micronized talc was blended
with
the Fenofibrate Intermediate Beads, Type B prior to encapsulation to dissipate
static
- 24 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
charge and ensure efficient filling during encapsulation.
[0072] In various examples discussed below, formulations disclosed herein were
administered to healthy adult volunteers in bioequivalence studies under
fasting and
fed conditions. The bioavailability achieved with the formulations disclosed
herein is
comparable to the bioavailability achieved with the administration of Lupin' s
ANTARA@ capsules, where the ANTARA@ capsules contain a single population of
granules having a defined concentration of fenofibrate. The formulations
disclosed
herein include two populations of fenofibrate beads, including fast beads
which have
a higher bioavailability than the beads in ANTARA@ capsules, and slow beads
which
have a lower bioavailability than the beads in ANTARA@ capsules.
[0073] Manufacturing Process: The manufacture of Fenofibrate Intermediate
Beads or
Pellets or Particles involves: Drug Suspension Manufacturing Rotor Drug
Layering Fluid Bed Drying Screening Via MeshFinal Blending. The
manufacturing process was shown to provide Fenofibrate Intermediate Beads with
acceptable assay and content uniformity characteristics.
[0074] Manufacture of Fenofibrate Capsules (Micronized) proceeds via
encapsulation
of Fenofibrate Intermediate Beads. In certain embodiments disclosed herein,
capsules
are prepared from a homogeneous population of Fenofibrate Intermediate Beads.
These beads may comprise fenofibrate; from 0.3% to 10% by weight of a
surfactant;
and from about 5% to about 15% by weight of a water soluble or water
dispersible
cellulosic binder, where the mass ratio of fenofibrate to the binder s between
about
3.5:1 and 4.5:1. The homogeneous population of Fenofibrate Intermediate Beads
is
supplied to a capsule filing station, which is used to prepare dosage forms by
encapsulating the population of beads in appropriately sized capsule shells,
preferably
-25 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
gelatin capsule shells.
[0075] In some embodiments, dosage forms are prepared from a heterogeneous
population of Fenofibrate Intermediate Beads. The heterogeneous population of
Fenofibrate Intermediate Beads may be prepared by combining at least two types
of
fenofibrate beads having different release properties to form a blend. The
blend may
be prepared first, and then filled into capsule shells using a single capsule
filling
station. Alternatively, capsules may be prepared using two or more capsule
filling
stations. Filling station I may be used to fill a first type of Fenofibrate
Intermediate
Beads into a capsule shell, and Filling station II may then be used to
encapsulate a
second type of Fenofibrate Intermediate Beads into the capsule shell, where
the first
and second types of beads are different. The manufacturing process was shown
to
provide a finished product with acceptable assay and content uniformity
characteristics.
[0076] Drug Layer Suspension Manufacturing: The drug layer suspension in this
invention is manufactured using a high speed homogenizer mixer (Ross Model HSM
105, attached with a rotor/stator mixing blade used for the large scale
manufacturing).
During experimentation, several different types of mixer/disperser mixing head
attachments were evaluated: slotted rotor/stator disperser, saw tooth
disperser, slotted
stator, and a fine screen stator with slotted disperser. Table 1 summarizes
the
formulation compositions and the drug release characteristics from these
experiments.
All formulations processed well and their drug release characteristics were
similar,
indicating that mixer type has no impact on formulation performance. Based on
this
evaluation, the rotor/stator configuration was chosen for the manufacture of
the drug
layering suspension. In addition, the process requires the suspension to be
mixed with
- 26 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
a homogenizer mixer for a minimum of 8 hours prior to drug layering, with
continuous agitation of the suspension maintained throughout the drug layering
process. The homogenization time minimum of 8 hours can be further reduced,
based
on processing efficiency.
Table 1: Fenofibrate Capsules USP, 130 mg
Evaluation of Homogenizer Mixing Head
Fine screen
Slotted Rotor/
Saw tooth Slotted Stator
stator
Stator
w/dispersator
X07- X07-
X07-047- X07-047-
Part I % % 047- % 047- %
58A4 58A5
58A6 58A7
Sugar Spheres (#35 -
279.5 62.1 279.5 62.1 279.5 62.
279.5 62.1
#45) 1
Part II
Fenofibrate . 28
130.0 28.9 130.0 28.9 130.0 130.0 28.9
(Micronized) 9
Sodium Lauryl 0 .
2
9.0 2.00 9.0 2.00 9.0 9.0 2.00
Sulfate 0
Pharmacoat 603
31.5 7.0 31.5 7.0 31.5 7.0 31.5 7.0
(Hypromellose)
Purified Water* (682.0) (682.0) (682.0) (682.0)
Total 450.0 450.0 450.0 450.0
Dissolution condition: 1000 mL purified water, 0.01M sodium lauryl
ANTARA 130mg sulfate,
USP Apparatus 2, at 75 rpm
0.01
Time M 0.01M SLS 0.01 M SLS 0.01 M SLS
0.01 M SLS
SLS
min 21% 19% 21% 21% 19%
min 24% 19% 21% 21% 18%
min 24% 19% 21% 21% 18%
min 24% 18% 21% 21% 18%
min 24% 18% 21% 21% 18%
60 min 24% 18% 21% 21% 18%
*Purified water removed during processing
[0077] Description of Rotor Drug Layering Process: The fluidization pattern in
the
rotor processor can be best characterized as a spiraling helix. Three factors
act on the
- 27 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
beads or pellets or particles (materials) to produce this flow pattern. The
rotating disk
of the ROTOR insert provides centrifugal force which forces the rotating
materials
toward the wall of the processing chamber at the periphery of the rotor
insert, while
conditioned upward airflow through the rotor gap develops a vertical force
causing
the materials to become fluidized. The fluidization air pushes the moving
materials
upward into the expansion chamber until gravity overcomes the upward air
velocity
and the material falls toward the center of the disk where there is little air
movement.
The drug layer suspension is sprayed tangentially onto the rotating particles,
while
heated process air causes the applied drug layer suspension to dry before the
particles
move again into the spraying zone. This cyclical process is repeated many
hundreds
of times until the appropriate quantity of solids are applied to the rotating
core
substrate (material).
[0078] The efficiency of the drug layering process is dependent on the
relationship
between particle movement within the processor, drug layering suspension spray
rate
and the rate of solvent evaporation. The movement of the particles during
rotor drug
layering process is dependent on rotor speed and air flow. Rotor speed is
considered a
critical parameter since it can affect the integrity of the beads. Slow speeds
can lead to
product agglomeration while excessive speeds can cause attrition. Rotor speed
is
adjusted to maintain proper particle movement as the weight of the batch
increases
during drug layering process. Once proper movement of the particle bed is
established, the deposition of drug layering solids onto the core substrate is
controlled
by the rate at which drug layering suspension is applied to core beads or
pellets, and
the rate at which solvent is removed from the system. The example of
appropriate
process parameters (inlet air temperature, product temperature, air flow,
spray rate,
- 28 -
CA 02878011 2014-12-23
WO 2014/003810
PCT/US2012/061486
nozzle atomizing air pressure, nozzle size and Rotor speed) utilized for
manufacturing
the beads in large scale batches utilizing FL-Multi-60 with a Rotogranulator
Insert are
summarized in the following Table 2.
Table 2. Rotor drug layering Process Parameters for manufacturing the beads or
pellets or
particles using a 30" rotor inserted with a FL-Multi-60 Fluid Bed Dryer. Batch
Size: 50 kg
Example 4 Example 6 Example
7
R&D- R&D- R&D-I R&D- R&D-
11976 11975 2052 12133
12128
Inlet Temperature ( C) 50-65 54-65 53-64 52-67 56-
69
Product Temperature ( C) 31-34 30-35 30-32 34-32 36-
36
Air Flow (CFM) 509-672 507-671 503-660 503-674 499-
652
Rotor Speed (rpm) 100-125 100-125 100-125 99-125 100-
125
Rotor Gap 4.0-6.0 4.0-6.0 4.0-6.0 4.0-6.5 4.0-
6.0
Atomization Air Pressure
55 55 55 55 55
(psi)
Spray Rate (g/min) 107-267 100-267 100-267 87-287 93-
273
Spray Nozzle Tip Size
1.2 1.2 1.2 1.2 1.2
(mm)
Example 1
[0079] Two formulations of fenofibrate containing beads were prepared. Each
formulation was prepared by spraying a fenofibrate suspension onto sugar
spheres
having a size of 35 to 45 mesh in a fluidized bed. In one formulation
(designated X07-
047-58A1), the drug suspension included fenofibrate and HPMC in a ratio of
4:1,
while in the other formulation (designated X07-047-62A1), the drug suspension
included fenofibrate and HPMC in a ratio of 2.4: lwere evaluated. The amounts
of the
various ingredients are set forth in Table 3 below. The bead formulations are
prepared
in a fluidized bed by spraying a water-based suspension of micronized
fenofibrate,
HPMC, and sodium lauryl sulfate onto the sugar spheres.
- 29 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
Table 3: Fenofibrate Capsules USP, 130 mg
(Compositions containing 7% w/w or 12% w/w Pharmacoat 603 as a binder)
X07-047-58A1 X07-047-62A1
(7% Pharmacoat (12% Pharmacoat
603) 603)
Part! mg % mg %
Sugar Spheres (35-45 mesh) 279.5 62.1 257.0 57.1
Part II
Fenofibrate Jet Milled (Micronized) 130.0 28.9 130.0 28.9
Sodium Lauryl Sulfate 9.0 2.0 9.0 2.0
Pharmacoat 603 (HPMC, Hypromellose) 31.5 7.0 54.0 12.0
Purified Water* (682.0) (956.0)
Total 450.0 450.0
*Removed during processing and does not contribute to the dry weight.
Table 4. Dissolution of Fenofibrate Capsules USP
Dissolution condition: 1000 mL purified water, 0.01M Sodium Lauryl Sulfate
(SLS),
USP Apparatus 2, at 75 rpm
ANTARA X07-047-58A1 X07-047-62A1
Time 0.01 M SLS 0.01 M SLS 0.01 M SLS
min 21% 21% 20%
min 24% 21% 20%
min 24% 21% 20%
min 24% 21% 19%
min 24% 21% 19%
60 min 24% 21% 19%
[0080] The formulation with 7% HPMC and the branded product ANTARA yielded
similar drug release characteristics in 1000 mL purified water containing 0.01
M
sodium lauryl sulfate, USP Apparatus 2, at 75 rpm, as seen in Table 4.
Example 2
[0081] The effect of a nonionic surfactant or wetting agent on the formulation
was
studied by evaluating the in vitro drug release characteristics of capsules
containing
beads. In these formulations, the drug to binder (Fenofibrate:HPMC) ratio was
kept
- 30 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
constant at 4:1, as seen in Table 5 below. The X07-047-64A1 was formulated
without
any surfactant, whereas the X07-047-65A1 was formulated with 2% Polysorbate
80.
These formulations were manufactured by mixing micronized fenofibrate, HPMC,
and Polysorbate 80 in water to form a drug suspension, and spraying the drug
layer
suspension onto the 35-45 mesh sugar spheres in a fluidized bed dryer inserted
with a
ROTOR.
Table 5: Fenofibrate Capsules USP, 130 mg ¨
Compositions
Effect of a non-ionic surfactant, Polysorbate 80
X07-047-64A1 X07-047-65A1
7% 7%
Pharmacoat,
Pharmacoat, 2%
Polysorbate
No Surfactant 80
Part! mg % mg %
Sugar Spheres (35-45 mesh) 288.5 64.1 279.5 62.1
Part II
Fenofibrate Jet Milled
130.0 28.9 130.0 28.9
(Micronized)
Polysorbate 80 0.0 0.0 9.0 2.0
Pharmacoat 603 (Hypromellose) 31.5 7.0 31.5 7.0
Purified Water (682.0) (682.0)
Total 450.0 450.0
- 31 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
Table 6. Dissolution of Fenofibrate Capsules USP
Dissolution condition: 1000 mL purified water, 0.01 M sodium lauryl
sulfate,
USP Apparatus 2, at 75 rpm
ANTARA X07-047-64A1 X07-047-
65A1
Time 0.01 M SLS 0.01 M SLS 0.01 M SLS
10 min 21% 21% 20%
15 min 24% 21% 20%
20 min 24% 21% 20%
30 min 24% 20% 20%
40 min 24% 20% 20%
60 min 24% 20% 20%
[0082] The formulations with 7% HPMC yielded similar drug release
characteristics
in 1000 mL purified water containing 0.01 M sodium lauryl sulfate, USP
Apparatus 2,
at 75 rpm, to the drug release characteristics of the branded product ANTARA ,
as
seen in Table 6. This result was seen regardless of the presence or absence of
a
nonionic surfactant.
Example 3¨Fenofibrate dosage forms which are more bioavailable than
ANTARA capsules
[0083] A formulation containing fenofibrate and HPMC in a ratio of 4:1 was
evaluated. The formulation additionally contained 2%, by weight of the
formulation,
of the anionic surfactant SLS, as seen in Table 7. A drug suspension
containing
micronized fenofibrate, HPMC (hypromellose, Pharmacoat 603), sodium lauryl
sulfate, and the antifoaming agent simethicone, a mixture of
polydimethylsiloxane
and hydrated silica gel, was prepared in purified water, and sprayed onto 35-
45 mesh
sugar spheres in a large scale fluid bed dryer (FL-M-60) equipped with a rotor
granulator insert to produce the drug layered intermediate beads. Simethicone
was
- 32-
CA 02878011 2014-12-23
WO 2014/003810
PCT/US2012/061486
incorporated to the drug layering suspension at a low level (0.044 %w/w) to
minimize
foaming during preparation. Following drug layering, the dried beads were
blended
with micronized talc, screened to remove agglomerates, and machine
encapsulated
into the two piece hard gelatin capsules (size#OEL). Table 7 presents a
summary of
the composition.
Table 7: Fenofibrate Capsules USP, 130 mg (Lot 1000317)
Composition
(Contains Fenofibrate Beads: 2% SLS, 4:1 Drug:HPMC)
Part I (Drug Layer Suspension) mg/g %
Fenofibrate Micronized Intermediate 130.0 28.87
Hypromellose 2910 (Pharmacoat 603) 31.5 7.0
Sodium Lauryl Sulfate (Empicol LX/N) 9.0 2.0
Simethicone 0.198 0.044
Purified Water, USP (342.0)
Part II
Sugar Spheres (35/45 mesh) 279.297 62.035
Total 450.0
Talc, Micronized 0.225 0.05
Total 450.22
[0084] The in vitro drug release of the above formulation was evaluated in a
1000 mL
purified water containing 0.05 M SLS using USP Apparatus II at 75 rpm. Table 8
below summarizes the dissolution characteristics.
Table 8: Fenofibrate Capsules USP, 130 mg (Lot 1000317)
Dissolution Condition: USP Apparatus II, 75 rpm, 1000mL, 0.05 M SLS
Product 10min
15min 20min 30min 40min 60min
ANTARA Capsules, 130
54% 74% 84% 93% 97% 100%
mg; B080033
Fenofibrate Capsules,
54% 69% 78% 92% 99% 101%
130mg #1000317
[0085] As seen in Table 8 above, the drug release rate of the formulation set
forth in
Table 7 (2% SLS, 4:1 Drug:HPMC) is comparable to the drug release rate of the
branded product ANTARA . Both formulations release 54% of the incorporated
- 33 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
fenofibrate in 10 minutes; 92% to 93% of the incorporated fenofibrate in 30
minutes;
and substantially all of the incorporated fenofibrate in 60 minutes.
[0086] The formulation of Table 7 was assessed in a pilot bioequivalence
study, and
compared to ANTARA Capsules. Both the formulation of Table 7 and the
ANTARA Capsules contained 130 mg fenofibrate. The pilot bioequivalence study
was an open-label, single-dose, randomized, two-period, two-treatment
crossover
study, using 24 normal healthy subjects. A summary of the pharmacokinetic data
is
presented in Table 9. Figure 1 presents the pharmacokinetic profile of the
formulations in Table 7.
Table 9: Fenofibrate Capsules USP, 130 mg (Lot 1000317)
Initial Pilot Bioequivalence Study Results - Fasting (n=24) Conditions, Study
(FENO-
08254)
Fenofibric Acid ¨ AUCL Fenofibric Acid ¨ CPEAK
Tpeak
Mean Intra Intra
Lot # Ratio 90% Mean Ratio 90%
(ng=hr/ (MIT) CI ng/mL (MIT) CI subject
subject (hours)
104.6
128.1-
1000317 141825 1.10 1
6549.0 1.41 19.3 3.45
54.9
115.5
B08003-
127830 4750.0 4.13
ANTARA
[0087] The FDA has provided guidance for bioequivalence between a branded
product and a generic equivalent. In general, bioequivalence depends on
several
natural log transformed parameters associated with the rate and extent of
absorption.
Specifically, the entire 90% confidence interval for the ratio of the test to
reference
area under the curve from zero to the last detectable concentration, AUCL,
must fall
between 80 and 125% of the corresponding AUCL of the branded product for
therapeutic equivalence. Additionally, the entire 90% confidence interval for
the ratio
of the test to reference maximum plasma concentration, Cmax, must also fall
between
- 34 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
80 and 125% of the corresponding Cmax of the branded product for therapeutic
equivalence to be declared.
[0088] The results of Table 9 indicate that the formulation of Table 7
exhibited a
similar extent of absorption (AUCL) compared to ANTARA capsules, but showed
elevated drug concentration in the plasma. The 90% confidence interval for the
AUCL is 104.6-115.5%, which falls within the FDA's desired confidence interval
ratio of 80%-125%.
[0089] However, the Cmax parameter for the formulation of Table 7 is 138% of
the
corresponding value for the branded product, with a 90% confidence interval of
128.1%-155%. This result falls outside the FDA's desired confidence interval
ratio of
80%-125%. Accordingly, the formulation of Table 7 exhibits higher
bioavailability
under fasting conditions than the reference product ANTARA . The beads
prepared
in this formulation may be combined with slow beads, and used as fast beads in
a
formulation which is bioequivalent to ANTARA capsules. Alternatively, the
beads
prepared in this formulation may be used as the only beads in a formulation
which has
a greater bioavailability than ANTARA capsules. As seen in the results of
Table 9, a
130 mg fenofibrate capsule containing only beads prepared according to Example
3
would show a moderate increase in AUCL of about 10%, when compared to the
branded product ANTARA . Further, a 130 mg fenofibrate capsule containing only
beads prepared according to Example 3 would show a pronounced increase in Cmax
of about 40%, when compared to the branded product ANTARA .
- 35 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
Example 4¨Fenofibrate dosage forms which are more bioavailable than
ANTARA capsules
[0090] Two different formulations containing 0.5% w/w sodium lauryl sulfate as
a
surfactant or 2% w/w sodium lauryl sulfate were manufactured. Both
formulations
contained fenofibrate and HPMC in a 4:1 ratio, as seen in Table 10. A drug
suspension containing micronized fenofibrate, with a mean particle size of 10
microns, HPMC (hypromellose, Pharmacoat 603), sodium lauryl sulfate, and the
antifoaming agent simethicone, a mixture of polydimethylsiloxane and hydrated
silica
gel, was prepared in purified water, and sprayed onto 20-25 mesh sugar spheres
in a
large scale fluid bed dryer (FL-M-60) equipped with a rotor granulator insert
to
produce the drug layered intermediate beads. A ratio of 4:1 fenofibrate to
HPMC was
used in the following formulations. Following the drug layering, the dried
beads were
blended with micronized talc, screened to remove the agglomerates and machine
encapsulated using two piece hard gelatin capsules (size#OEL).
- 36 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
Table 10: Fenofibrate Capsules USP, 130 mg, #1000374
Fenofibrate Beads (520.0 mg/gram), Lot#R&D-I1976, 2% SLS, 4:1 Drug:Pharmacoat
603
Fenofibrate Capsules USP, 130 mg, #1000375
Fenofibrate Beads (520.0 mg/gram), Lot#R&D-I1975, 0.5% SLS, 4:1
Drug:Pharmacoat 603
Fenofibrate Capsules, 130mg
Fenofibrate Capsules, 130 mg
#1000374 #1000375
Fenofibrate Fenofibrate
Intermediate Fenofibrate Intermediate Fenofibrate
R&D-I1976, Capsules, 130 mg, R&D-I1975, Capsules, 130
mg,
2% SLS, #1000374 0.5% SLS,
#1000375
4:1 Drug:HPMC 4:1 Drug:HPMC
Part I-Drug Layer mg/ mg/
mg/g % % mg/g % %
Suspension capsule capsule
Fenofibrate
520.0 52.0 130.0 51.95 520.0 52.0 130.0 51.95
Micronized
Pharmacoat 603
126.0 12.6 31.5 12.6 126.0 12.6 31.5 12.6
(Hypromellose)
Sodium Lauryl Sulfate 20.0 2.0 5.0 2.0 5.0 0.5 1.25
0.5
Simethicone 0.22 0.022 0.055 0.022 0.22 0.022
0.055 0.022
Purified Water* (2735.0) (2735.0)
Part II
Sugar Spheres
333.78 33.378 83.445 33.348 348.78 34.878 87.195 34.85
(20/25 mesh)
Total 1000.0 100 250.0 1000.0 100 250.0
Talc, micronized 0.225 0.0899
0.225 0.0899
Total Fill Weight 250.225 250.225
*Removed during processing
[0091] The in vitro drug release of the above formulation was evaluated in a
1000 mL
purified water containing 0.05 M sodium lauryl sulfate using USP Apparatus II
at 75
rpm. Table 11 summarizes the dissolution characteristics.
[0092] As seen in Table 11 below, the drug release rate of the formulations
set forth
in Table 10 is faster than the drug release rate of the branded product ANTARA
.
Formulation #1000374, containing 2% sodium lauryl sulfate, releases 64% of the
incorporated fenofibrate in 10 minutes; and substantially all of the
incorporated
fenofibrate in 30 minutes. ANTARAO, in contrast, releases 54% of the
incorporated
- 37 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
fenofibrate in 10 minutes; and 93% of the incorporated fenofibrate in 30
minutes.
Formulation #1000375, containing 0.5% sodium lauryl sulfate, is more closely
comparable to ANTARA@ capsules, releasing 61% of the incorporated fenofibrate
in
minutes; 94% of the incorporated fenofibrate in 30 minutes; and substantially
all of
the incorporated fenofibrate in 60 minutes.
Table 11: Fenofibrate Capsules USP, 130mg
Dissolution Condition: USP Apparatus II, 75 rpm, 1000 mL, 0.05 M SLS
Product 10min 15min 20min 30min 40min 60min
ANTARA Capsules, 130 mg; B08003 54% 74% 84% 93%
97% 100%
Fenofibrate Capsules, 13 Omg
64% 85% 94% 100% 102% 103%
#1000374
Fenofibrate Capsules, 13 Omg
61% 77% 86% 94% 98% 102%
#1000375
[0093] The above formulations were assessed against the reference listed drug
product in two separate pilot bioequivalence studies versus ANTARA@ 130 mg
Capsules (B08003), under fasting conditions in open-label, single-dose,
randomized,
two-period, two-treatment crossover studies initiated with 28 normal healthy
adult
subjects each. The methodology was similar to the methodology used in Example
3.
Summaries of the pharmacokinetic data from these studies are presented in
Tables 12
and 13. Figures 2 and 3 present the pharmacokinetic profile of the
formulations in
Table 10.
- 38 -
CA 02878011 2014-12-23
WO 2014/003810
PCT/US2012/061486
Table 12: Fenofibrate Capsules USP, 130 mg
Pilot Bioequivalence Study Results - Fasting (n=28) Conditions Study (FEN0-
0968)
(Formulation containing drug layered beads manufactured
with 2% sodium lauryl sulfate, #20-#25 Sugar Spheres, Drug:Pharmacoat 603,
4:1)
Fenofibric Acid ¨ AUCL Fenofibric Acid ¨ CPEAK Tpea
k
Intra Intra
Mean Rati Rati
90 subje Mean 90 subje
Lot # (ng=hr % o % o (hour
% ct ng/m % ct
Brand / CV (MIT CV (MIT s)
CI % L CI %
mL) ) )
CV CV
105 119
10003 12505 27
4 ' '
9 5428. 40
109 - 8%
74 7 3
35' 1 28 - 16% (2-'6)
'
113 138
B0800 11484 26. 4191. 31. 5.0
3 9 9 0 4 (2-6)
Table 13: Fenofibrate Capsules USP, 130 mg
Pilot Bioequivalence Study Results - Fasting (n=25) Conditions Study (FEN0-
0969)
(Formulation containing drug layered beads manufactured with 0.5% sodium
lauryl sulfate, #20-
#25 Sugar Spheres, Drug:Pharmacoat 603, 4:1)
Fenofibric Acid ¨ AUCL Fenofibric
Acid ¨ CPEAK Tpeak
Mean Intra Intra
Lot # % Ratio 90%
subject Mean % Ratio 90%
(ng=hr/
subject (hours)
Brand CV (MIT) CI ng/mL CV (MIT) CI
mL) % CV % CV
1000375 123828 35.8 1.12 107- 111- 4.4117
9% 4969 28.5 1.22 135 21%
(2-9)
4.5
B08003 110477 33.6 4099 34.8
(2-10)
[0094] The results of Table 12 indicate that Formulation #1000374, containing
2%
sodium lauryl sulfate, exhibited a similar extent of absorption (AUCL)
compared to
ANTARA capsules, but showed elevated drug concentration in the plasma. The
90% confidence interval for the AUCL is 105-113%, which falls within the FDA's
desired confidence interval ratio of 80%-125%. However, the Cmax parameter for
Formulation #1000374 has a 90% confidence interval of 119-138%. This result
falls
outside the FDA's desired confidence interval ratio of 80%-125%.
- 39 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
[0095] The results of Table 13 indicate that the Formulation #1000375,
containing
0.5% sodium lauryl sulfate, also exhibited elevated drug concentration in the
plasma,
compared to ANTARA@ capsules. The 90% confidence interval for the Cmax
parameter for Formulation #1000375 has a 90% confidence interval of 111-135%,
falling outside the FDA's desired confidence interval ratio of 80%-125%.
[0096] The pharmacokinetic results presented in the above table show that
these
modified formulations (containing 20-25 mesh sugar spheres as inert cores and
0.5 to
2% sodium lauryl sulfate) showed higher bioavailability than the reference
product
ANTARA .
[0097] The beads prepared in the formulations of Example 4 are not suitable
for use
as the only beads in a formulation which is intended to be bioequivalent to
ANTARA@ capsules. However, the beads prepared in this formulation may be
combined with slow beads, and used as fast beads in a formulation which is
bioequivalent to ANTARA@ capsules. Alternatively, the beads prepared in this
formulation may be used as the only beads in a formulation which has a greater
bioavailability than ANTARA@ capsules.
Example 5
[0098] On the basis of the bioavailability study results of Examples 3 and 4,
sodium
lauryl sulfate was removed from the formulation and the effect of three
different
binder concentrations of Pharmacoat 603 (4.3% w/w, 6.5% w/w and 13.0% w/w) on
the drug release was evaluated. A drug suspension containing micronized
fenofibrate,
HPMC (hypromellose, Pharmacoat 603), and the antifoaming agent simethicone was
prepared in purified water, and sprayed onto 20-25 mesh sugar spheres in a
fluid bed
dryer ( GPCG 3) equipped with a rotor granulator insert to produce the drug
layered
- 40 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
intermediate beads. The amount of fenofibrate was held constant at 130
mg/capsule,
while the ratio of fenofibrate was varied between 4:1 and 12:1, as seen in
Table 14.
Table 14: Fenofibrate Capsules USP, 130 mg
(Formulation composition without sodium lauryl sulfate)
X07-047- X07-047-82A1 X07-047-83A1
81A1 8:1, Drug:
12:1, Drug:
4:1 Drug: Pharmacoat
Pharmacoat
Pharmacoat 603 ratio, 603 ratio,
603 ratio,
6.5%Pharmaco 4.3%Pharmaco
13%Pharmaco at at 603
at 603 603
Part! mg % mg % mg %
Sugar Spheres (#20 - #25) 87.5 35.0 103.75 41.5
109.2 43.7
Part II
Fenofibrate (Micronized) 130.0 52.0 130.0 52.0
130.0 52.0
Pharmacoat 603 (Hypromellose) 32.5 13.0 16.25 6.5 10.8
4.3
Purified Water* (682.0) (682.0) (682.0)
Total 250.0 250.0 250.0
*Removed during processing
[0099] The in vitro drug release of the formulations of Table 14 was evaluated
in a
1000 mL purified water containing 0.025 M sodium lauryl sulfate using USP
Apparatus II at 75 rpm. Table 15 summarizes the drug release characteristics,
as seen
in Table 15.
- 41 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
Table 15: Fenofibrate Capsules USP, 130mg
Dissolution Condition: USP Apparatus II, 75 rpm, 1000mL, 0.025 M SLS
Drug: %
Product HPMC HPMC 10min 15min 20min 30min 40min 60min
.
ratio
ANTARA@
Capsules, 130 mg; 35%
52% 62% 74% 83% 92%
B08003
Fenofibrate
Capsules, 130mg 4:1
13%w/w 72% 80% 95% 97% 98% 99%
#X07-047-81A1
Fenofibrate
Capsules, 130mg 8:1
6.5%w/w 51% 68% 77% 85% 89% 94%
#X07-047-82A1
Fenofibrate
Capsules, 130mg 12:1
4.3%w/w 18% 28% 38% 49% 57% 66%
#X07-047-83A1
[00100] The data presented in the above shows that higher concentrations of
HPMC
yielded more rapid drug release in formulations containing no sodium lauryl
sulfate.
Example 6¨Fenofibrate dosage forms which are less bioavailable than
ANTARA capsules
[00101] Formulation X07-047-82A1 containing 6.5% w/w Pharmacoat 603 with no
sodium lauryl sulfate was manufactured in a large scale equipment as lot
1000442
using Size#OEL capsules shell and evaluated in a bioequivalence study versus
ANTARA@ capsules. Table 16 describes this formulation.
- 42 -
CA 02878011 2014-12-23
WO 2014/003810
PCT/US2012/061486
Table 16: Fenofibrate Capsules, 130 mg, #1000442
(Contains drug layered beads manufactured without sodium lauryl sulfate (SLS),
#20-
#25 Sugar Spheres, 8:1 Drug:Pharmacoat 603)
Fenofibrate Intermediate
Fenofibrate Capsules USP,
Beads, R&D-I 2052,
130 mg,
8:1 Drug:Pharmacoat
#1000442
603
Part I-A (Drug Layer
mg/g % mg/capsule %
Suspension)
Fenofibrate Micronized 520.0 52.0 130.0
51.95324
Hypromellose 2910
65.0 6.5 16.25 6.5
(Pharmacoat 603)
Simethicone 0.22 0.022 0.055 0.02198
Purified Water* (2735.0)
Part-IB
Sugar Spheres (20/25 mesh) 414.78 41.478 103.695 41.4407
Total 1000.0 100 250.0
Talc, micronized 0.225 0.08991
Total Fill Weight 250.225 100.0
*Purified removed during processing
[00102] The in vitro drug release of the above formulation was evaluated in a
1000 mL
purified water containing 0.05 M sodium lauryl sulfate using USP Apparatus II
at 75
rpm. Table 17 below summarizes the drug release characteristics.
Table 17: Fenofibrate Capsules USP, 130mg
Dissolution (USP Apparatus II, 75 rpm, 1000mL, 0.05 M SLS)
Product 10min 15min 20min 30min
40min 60min
ANTARA@ Capsules, 130
54% 74% 84% 93% 97% 100%
mg; B08003
Fenofibrate Capsules,
14% 23% 35% 62% 78% 88%
130mg #1000442
[00103] As seen in Table 17, the capsules of Table 16 release the drug more
slowly
than the comparative ANTARA@ capsules. After 10 minutes, the comparative
ANTARA@ capsules released 54% of the drug, while the capsules of Table 14
released only14% of the drug. Additionally, the capsules of Table 14 were
assessed
in a bioequivalence study versus ANTARA@ Capsules, 130 mg in an open-label,
- 43 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
single-dose, randomized, two-period, two-treatment crossover study using 32
normal
healthy subjects (27 completed the study). The methodology was similar to the
methodology used in Example 3. A summary of the pharmacokinetic data from this
study is presented in Table 18. Also, a plot showing the pharmacokinetic
profile of the
capsules of Table 16 is shown in Figure 4.
Table 18: Fenofibrate Capsules USP, 130 mg (Lot 1000442)
Pilot Bioequivalence Study Results - Fasting (n=27) Conditions
Study (FEN0-09288)
Fenofibric Acid ¨ AUCL Fenofibric Acid ¨ CPEAK
Tpeak*
M ean Intra Intra
Lot # % Ratio 90% subject Mean % Ratio 90% subject
(L) ng=hr/ (hours)
Brand CV (MIT) CI % ng/mL CV (MIT) CI %
m
CV CV
5.0 (3-
1000442 76734 40.0 0.56 13% 2263.4 31.8 0.49 18.0%
59 53 10)
B08003 134952 34.9 4748.7 36.8
4.0(2 -
6)
* Median values presented (range of values)
[00104] The capsules of Table 16 exhibited poor bioavailability under fasting
conditions due to a low AUCL and Cmax. The results of Table 18 indicate that
the
capsules of Table 16, containing fenofibrate and HPMC in a ratio of 8:1 and no
sodium lauryl sulfate, exhibited low absorption (AUCL) compared to ANTARA
capsules. The capsules of Table 16 showed between 53% and 59% of the
absorption
(AUCL) observed with the branded product, with a confidence interval of 90%.
Additionally, the Cmax parameter for capsules of Table 16 has a 90% confidence
interval of 45% to 53% when compared to the branded product. This result falls
outside the FDA's desired confidence interval ratio of 80%-125%, and indicates
that
the capsules of Table 16 are not bioequivalent to ANTARA capsules.
- 44 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
Example 7¨Fenofibrate dosage forms which are bioequivalent to ANTARA
capsules
[00105] Three alternative intermediate bead formulations were scaled-up. The
first
group of beads, Fenofibrate Intermediate Beads, Type A, Lot#R&D-I2133, contain
0.5% sodium lauryl sulfate. The second group of beads, Fenofibrate
Intermediate
Beads, Type B, Lot#R&D-I2134, contain 2% sodium lauryl sulfate. The third
group
of beads, Fenofibrate Intermediate Beads, Type C, Lot# R&D-I2128, contains no
sodium lauryl sulfate.
[00106] Based on pharmacokinetic analysis, two capsule formulations containing
different ratios of the above beads were machine encapsulated in size#OEL and
dosed
in a bioequivalence study. The first capsule formulation, lot #1000529,
contained an
80:20 ratio of Type A beads and Type C beads, and the second capsule
formulation,
lot #1000530, contained a 75:25 ratio of Type B and Type C beads. Tables 19,
20, and
21 summarize these two capsule formulations and drug release characteristics.
- 45 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
Table 19: Fenofibrate Capsules USP, 130 mg, #1000529
(Contains 80% w/w Fenofibrate Intermediate Beads, Type A (0.5%SLS), Lot# R&D-
I12133 and
20% w/w Fenofibrate Intermediate Beads, Type C (No SLS), Lot# R&D-I12128)
Fenofibrate
Fenofibrate Fenofibrate
Intermediate 80%w/w 20%w/w
Intermediate Beads Capsules USP,
Beads Type C,
Type A, 0.5%SLS, 130 mg,
No SLS, R&D- Type A Type C
R&D-I2133 #1000529
12128, Beads Beads
Drug:HPMC, 4:1 80:20
Drug:HPMC, 8:1
mg/g % mg/g % Mg mg mg %
Part I-A (Drug
Layer Suspension)
Fenofibrate
520.0 52.0 520.0 52.0 104.0 26.0 130.0 51.95
Micronized
Hypromellose 2910
126.0 12.6 65.0 6.5 25.2 3.25 28.45 11.37
(Pharmacoat 603)
Sodium Lauryl
5.0 0.5 1.0 1.0
0.4
Sulfate
Simethicone
0.22 0.022 0.22 0.022 0.044 0.011 0.055 0.022
Purified Water* (2735.0) (2735.0)
Part-IB
Sugar Spheres
348.78 34.878 414.78 41.48 69.756 20.739 90.495 36.17
(20/25 mesh)
Total 1000.0 100 1000.0 100 200.0 50.0
250.0
Talc, micronized 0.225 0.225
0.09
Total Capsule Fill
250.225
Wt.
*Removed during processing
- 46 -
CA 02878011 2014-12-23
WO 2014/003810
PCT/US2012/061486
Table 20: Fenofibrate Capsules USP, 130 mg, #1000530
(Contains 75% w/w Fenofibrate Intermediate Beads, Type B (2%SLS), Lot#R&D-
I2134 and
25% w/w Fenofibrate Intermediate Beads, Type C (No SLS), Lot# R&D-I2128)
Fenofibrate Fenofibrate 75%w/w 25%w/w Fenofibrate
Intermediate Intermediate Type B
Type C Capsules USP,
Beads Type B, Beads Type C, Beads Beads 130 mg,
2%SLS, R&D-I2128, No #1000530
Drug:Pharmacoat, SLS
75:25
4:1, R&D-I2134 Drug:HPMC, 8:1
mg/g % mg/g % mg mg mg %
Part I (Drug
Layer Suspension)
Fenofibrate
520.0 52.0 520.0 52.0 97.5 32.5 130.0 51.95
Micronized
Hypromellose 2910
126.0 12.6 65.0 6.5
23.625 4.0625 27.6875 11.07
(Pharmacoat 603)
Sodium Lauryl
20.0 2.0 NA NA 3.75 3.75
1.5
Sulfate
Simethicone
0.22 0.022 0.22 0.022 0.04125 0.01375 0.055 0.22
Purified Water* (2735.0) (2735.0) (2735.0)
Part II
Sugar Spheres
333.78 33.38 414.78 41.478 62.58375 25.92375 88.5075 35.37
(20/25 mesh)
Total 1000.0 1000.0 1000.0 100 187.5 62.5 250.0
Talc, micronized 0.225 0.225
0.09
Total Capsule
250.225
Fill Wt, mg
*Removed during processing
[00107] The drug release characteristics of the above two capsule formulations
are
evaluated using USP Apparatus II, 75 rpm, 1000mL, 0.05 M SLS, and are
summarized in Table 21. As seen in Table 21, the dissolution rate of the
capsule
formulations of Tables 19 and 20 is slower than the dissolution rate of ANTARA
capsules containing an equivalent amount of fenofibrate, i.e., 130 mg.
Additionally,
the drug release rate of the capsule formulations of Tables 19 and 20 is
slower than a
capsule prepared using the formulation of Table 14, containing fenofibrate and
HPMC
in a ratio of 8:1, with no sodium lauryl sulfate. The dissolution rate of the
capsule
formulations of Tables 19 and 20 is also substantially slower than a capsule
prepared
- 47 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
using Type B beads alone, where the Type B beads contain fenofibrate and HPMC
in
a ratio of 4.1:1, with 2% sodium lauryl sulfate.
Table 21: Fenofibrate Capsules USP, 130mg
Dissolution Condition:USP Apparatus II, 75 rpm, 1000mL, 0.05 M SLS
Product 10min 15min 20min 30min 40min 60min
ANTARA Capsules, 130 mg;
49% 73% 84% 94% 97% 100%
B08017
Fenofibrate Intermediate Beads,
63% 78% 84% 90% 93% 97%
R&D 12052, Type C Beads
Fenofibrate Intermediate Beads,
92% 98% 100% 100% 100% 100%
R&D 12134, Type B Beads
Fenofibrate Capsule, 130 mg, Lot.
33% 50% 64% 79% 88% 96%
1000529
Fenofibrate Capsule, 130 mg, Lot.
45% 60% 68% 78% 85% 93%
1000530
[00108] The capsules of Tables 19 and 20 were assessed in a bioequivalence
study
versus ANTARA Capsules, 130 mg, in an open-label, single-dose, randomized,
two-period, two-treatment crossover study using 21 normal healthy subjects.
The
methodology was similar to the methodology used in Example 3. A summary of the
pharmacokinetic data from this study is presented in Table 22. A plot showing
pharmacokinetic profile, specifically plasma levels, is shown in Figure 5.
- 48 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
Table 22: Fenofibrate Capsules USP, 130 mg
Pilot Bioequivalence Study Results - Fasting (n=21) Conditions; Study (FENO-
1018)
Treatment A: Lot 1000529 contains 80% Fast Beads (0.5% SLS) and 20% Slow Beads
(No SLS);
Dose: 1 x 130mg
Treatment B: Lot 1000530 contains 75% Fast Beads (2%SLS) and 25% Slow Beads
(No SLS);
Dose: lx 130 mg
Treatment C: ANTARA Capsules, Lot: B08017 ; Dose: 1 x 130 mg; Oscient/Lupin
Mean Mean 90% CI
Treatment A= Treatment C=
Lot 1000529 Lot B08017
AUCI (ng.hr/mL) 122189.62 124010.37 94-106
AUCL(ng.hr/mL) 113485.24 117167.45 92-105
CPEAK (ng/mL) 4498.10 4720.35 88-108
TPEAK (Hour)* 3.5 (2-10) 4 (2-12)
Treatment B= Treatment C=
Lot 1000530 Lot B08017
AUCI (ng.hr/mL) 116320.92 124010.37 90-102
AUCL(ng.hr/mL) 108823.93 117167.45 89-101
CPEAK (ng/mL) 4656.90 4720.35 90-109
TPEAK (Hour)* 4.0(2.0-24) 4 (2-12)
* Median values, with range in parentheses.
[00109] The pharmacokinetic results presented in Table 22 demonstrate that the
formulations of Tables 19 and 20 were each bioequivalent to ANTARA .
Formulation #1000529, containing 80% Type A beads and 20% Type C beads,
exhibited an extent of absorption (AUCL) which was between 92% and 105% of the
absorption observed with ANTARA capsules, within a confidence interval of
90%.
Formulation #1000530, containing 75% Type B beads and 25% Type C beads,
exhibited an extent of absorption (AUCL) which was between 89% and 101% of the
absorption observed with ANTARA capsules, within a confidence interval of
90%.
This result falls within the FDA's desired confidence interval ratio of 80%-
125%.
[00110] Additionally, the Cmax parameter for Formulation #1000529 was 88% to
108% of the Cmax observed with ANTARA capsules, within a confidence interval
- 49 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
of 90%; and the Cmax parameter for Formulation #1000530 was 90% to 109% of the
Cmax observed with ANTARA@ capsules, within a confidence interval of 90%. This
result falls within the FDA's desired confidence interval ratio of 80%-125%.
The
results of Table 22 indicate that the Formulation #1000529 and Formulation
#1000530 are each bioequivalent to ANTARA@ capsules.
Example 8¨Fenofibrate dosage forms which are bioequivalent to ANTARA@
capsules
[00111] A large scale batch, Formulation#1000596 was manufactured using the
Formulation composition of 1000530 (shown in Table 20). This capsule contains
two
different types of Fenofibrate Intermediate Beads, one containing no
surfactant
(Fenofibrate Intermediate Beads, Type C, 520 mg/g) and the other containing
sodium
lauryl sulfate as a surfactant (Fenofibrate Intermediate Beads, Type B). Each
capsule
contains 25%w/w Fenofibrate Intermediate Beads Type C (520 mg/g) and 75%w/w
Fenofibrate Intermediate Beads Type B which corresponds to theoretical fill
weights
of 62.5 mg and 187.725 mg, respectively. The actual fill weight of each bead
is
adjusted based on the potency factor assigned to each bead prior to
encapsulation.
Each bead type was filled into the capsule shell using a separate dosing
station during
encapsulation.
[00112] The resulting Capsules USP, 130 mg (Lot. 100596) were dosed in a
bioequivalence study versus the reference listed drug, ANTARA@ Capsules, 130
mg.
The study was a single dose, open label, randomized, 2-period, 2-treatment
crossover
of the test and reference products administered under fasting and post-
prandial
conditions. Statistical analyses of the data revealed that the 90% confidence
intervals
were within the acceptable bioequivalent range of 80% and 125% for the natural
log
- 50 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
transformed parameters AUCL, AUCI, and CPEAK for fenofibric acid. This study
demonstrates that Capsules of Lot. 100596, 130 mg are bioequivalent to ANTARA
Capsules, 130 mg, following a single, oral 130 mg (1 x 130mg capsule) dose
administered under fasting and post-prandial conditions. A summary of the
pharmacokinetic data from this study is presented in Table 23 and Table 24.
Figure 6
and Figure 7 display the pharmacokinetic profile, specifically plasma
concentration of
fenofibrate over time, for Capsules of Lot. 100596, 130 mg, which are
bioequivalent
to ANTARA Capsules, 130 mg.
Table 23: Fenofibrate Capsules USP, 130 mg (Lot#1000596)
Bioequivalence Study Results - Fasting (n=29) Conditions; Study (FENO-10202)
(Formulation 1000596 contains 75% Fast Beads (2% SLS Beads) and 25% Slow Beads
(No SLS))
Fenofibric Acid ¨ AUCL Fenofibric Acid ¨ CPEAK
Tpeak
M ean Intra Intra
Lot # % Ratio 90% subject Mean % Ratio 90% subject
(ng=hr/
(hours)
L) CV (MIT) CI % ng/mL CV (MIT) CI %
m
CV CV
92- 99- 4.2
1000596 121033 36.5 0.97 11' 5% 4617 35.0 1.08 117 19%
102 (2-10)
4.5
B08017 123562 34.4 4321 39.4
(2-10)
Table 24: Fenofibrate Capsules USP, 130 mg (Lot#1000596)
Bioequivalence Study Results - Fed (Post-Prandial) (n=31) Conditions; Study
(FENO-10203)
(Formulation 1000596 contains 75% Fast Beads (2% SLS Beads) and 25% Slow Beads
(No SLS))
Fenofibric Acid ¨ AUCL Fenofibric Acid ¨ CPEAK
Tpeak
M ean Intra Intra
Lot # % Ratio
90% subject Mean % Ratio 90% subject
(ng=hr/
(hours)
L) CV (MIT) CI % ng/mL CV
(MIT) CI %
m
CV CV
94- 96-
5.7
1000596 140411 31.6 0.977 22% 8300 17.8 1.00 10%
100 ' 104
(3-12)
6.1
B08017 145211 32.0 8381 20.6
(2-12)
- 51 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
4. Comparative Example
[00113] The Comparative Example presented herein is drawn from U.S. Patent
Number 7,863,331.
[00114] Example 4 of U.S. Patent Number 7,863,331 presents a formulation
comprising inert cores coated with a layer of fenofibrate, HPMC, and SLS, as
presented in Table 25 below.
Table 25: Fenofibrate Capsules USP, Prepared according to Example 4 of
U.S. Patent Number 7,863,331
FORMULA PERCENTAGE BY MASS
Neutral cores 16.44
Micronized fenofibrate 63.69
Ilydroxypropylmethyl cellulose 3.0 12.04
Viscosity cP
Sodium lauryl sulfate 3.25
Dimethicone 0.25
Simethicone 0.03
Talc 0.63
Outer layer
Ilydroxypropylmethyl cellulose 6.0 2.57
Viscosity cP
Talc 1.1
[00115] Example 6 of U.S. Patent Number 7,863,331 presents bioavailability
data for a
130 mg fenofibrate capsule prepared according to the formulation of Table 25.
The
130 mg capsule has a Cmax in the fasted state of 4375 ng/mL. These results are
compared to the results obtained with various exemplary embodiments described
herein in Table 26.
- 52-
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
Table 26: Fenofibrate Capsules USP, 130 mg
Bioequivalence Data
Fenofibric Acid¨AUCL
Fenofibric Acid¨Cmax
Lot #
Mean (ng=hr/mL) Mean (ng/mL)
B08017 (ANTARAIO)a 123455 4321
Example 6, U.S. 7,863,331 114853 4375
1000317b 141825 6549
1000374' 125054 5428.7
1000375' 123828 4969
a. See Table 23.
b. See Table 9; capsules containing beads with 28.9% fenofibrate, as disclosed
herein.
c. See Table 12; capsules containing beads with 52% fenofibrate, as disclosed
herein.
d. See Table 13; capsules containing beads with 52% fenofibrate, as disclosed
herein.
[00116] Thus, Table 26 compares results obtained with the prior art products
to
capsules containing fenofibrate beads or granules disclosed herein. The
granules
disclosed herein comprise fenofibrate; from 0.5% to 2% by weight of a
surfactant; and
from about 5% to about 15% by weight of a water soluble or water dispersible
cellulosic binder. The mass ratio of the drug to the binder in the dosage
forms
disclosed herein and listed in Table 26 (Lot #1000317, Lot # 1000374, and Lot
#1000375) is about 4:1 to about 4.15. The prior art granules described in
Example 6
of U.S. Patent Number 7,863,331 comprise fenofibrate; 3.25% by weight of a
surfactant; and about 12% by weight of a water soluble or water dispersible
cellulosic
binder. The mass ratio of the drug to the binder in the dosage form described
in
Example 6 of U.S. Patent Number 7,863,331 is 5.29. As seen in Table 26,
reducing
the mass ratio of the drug to the binder from 5.29 to about 4.1 to about 4.15
significantly increases bioavailability, seen as an increase in Cmax.
- 53 -
CA 02878011 2014-12-23
WO 2014/003810 PCT/US2012/061486
[00117] Although the various exemplary embodiments have been described in
detail
with particular reference to certain exemplary aspects thereof, it should be
understood
that the invention is capable of other embodiments and its details are capable
of
modifications in various obvious respects. As is readily apparent to those
skilled in
the art, variations and modifications can be affected while remaining within
the spirit
and scope of the invention. Accordingly, the foregoing disclosure,
description, and
figures are for illustrative purposes only and do not in any way limit the
invention,
which is defined only by the claims.
- 54 -