Note: Descriptions are shown in the official language in which they were submitted.
CA 02879939 2015-01-23
WO 2014/024026
PCT/1B2013/001722
METHOD FOR ELICITING IN INFANTS AN IMMUNE RESPONSE AGAINST
RSV AND B. PERTUSSIS
CROSS-REFERENCE TO RELATED APPLICATIONS
[001] This application claims benefit of the filing date of United States
Provisional
Application Number 61/679,928, filed 06 August 2012 and United States Non-
Provisional
Application Number 13/835,829, filed 15 March 2013, the disclosures of which
are
incorporated herein by reference in their entirety.
COPYRIGHT NOTIFICATION PURSUANT TO 37 C.F.R. 1.71(E)
[002] A portion of the disclosure of this patent document contains material
which is
subject to copyright protection. The copyright owner has no objection to the
facsimile
reproduction by anyone of the patent document or the patent disclosure, as it
appears in
the Patent and Trademark Office patent file or records, but otherwise reserves
all
copyright rights whatsoever.
BACKGROUND
[003] This disclosure concerns the field of immunology. More particularly this
disclosure relates to compositions and methods for eliciting an immune
response specific
for Respiratory Syncytial Virus (RSV) and Bordetella pertussis (pertussis).
[004] Human Respiratory Syncytial Virus (RSV) is the most common worldwide
cause
of lower respiratory tract infections (LRTI) in infants less than 6 months of
age and
premature babies less than or equal to 35 weeks of gestation.
[005] RSV is well documented as a cause of yearly winter epidemics of acute
LRTI,
including bronchiolitis and pneumonia. The incidence rate of RSV-associated
LRTI in
otherwise healthy children was calculated as 37 per 1000 child-year in the
first two years
of life (45 per 1000 child-year in infants less than 6 months old) and the
risk of
hospitalization as 6 per 1000 child-years ( per 1000 child-years in the first
six months of
life). Incidence is higher in children with cardio-pulmonary disease and in
those born
prematurely, who constitute almost half of RSV-related hospital admissions in
the USA.
Children who experience a more severe LRTI caused by RSV later have an
increased
1
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
incidence of childhood asthma. These studies demonstrate widespread need for
RSV
vaccines.
[006] The bacterium Bordetella pertussis is the causative agent for whooping
cough, a
respiratory disease that can be severe in infants and young children. WHO
estimates
suggest that, in 2008, about 16 million cases of whooping cough occurred
worldwide, and
that 195,000 children died from the disease. Vaccines have been available for
decades, and
global vaccination is estimated (WHO) to have averted about 687,000 deaths in
2008.
[007] The clinical course of the disease is characterised by paroxysms of
rapid coughs
followed by inspiratory effort, often associated with a characteristic
'whooping' sound. In
serious cases, oxygen deprivation can lead to brain damage; however the most
common
complication is secondary pneumonia. Although treatment with antibiotics is
available, by
the time the disease is diagnosed, bacterial toxins have often caused severe
damage.
Prevention of the disease is therefore of great importance, hence developments
in
vaccination are of significant interest. The first generation of vaccines
against B. pertussis
were whole cell vaccines, composed of whole bacteria that have been killed by
heat
treatment, formalin or other means. These were introduced in many countries in
the 1950s
and 1960s and were successful at reducing the incidence of whooping cough.
Subsequent
improvements have included the development of acellular pertussis vaccines
containing
highly purified B. pertussis proteins ¨ usually at least pertussis toxoid (PT;
pertussis toxin
chemically treated or genetically modified to eliminate its toxicity) and
filamentous
haemagglutinin (FHA), often together with the 69kD protein pertactin (PRN),
and in some
cases further including fimbriae types 2 and 3 (FIMs 2 and 3). These acellular
vaccines are
generally far less reactogenic than whole cell vaccines, and have been adopted
for the
vaccination programmes of many countries.
[008] Both RSV and pertussis are characterized in that immunity wanes after
childhood
and the most severe morbidity and mortality result from infections during the
first few
months of life, before the paediatric vaccination is fully realized for these
pathogens.
[009] Although pertussis vaccination is well established, despite various
attempts to
produce a safe and effective RSV vaccine that elicits durable and protective
immune
responses in healthy and at-risk populations, none of the candidates evaluated
to date have
been proven safe and effective as a vaccine for the purpose of preventing RSV
infection
and/or reducing or preventing RSV disease. Accordingly, there is an unmet need
for a
2
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
combination vaccine which confers protection against both RSV and B. pertussis
infection
and associated disease, which pose particular dangers to neonates and young
infants.
BRIEF SUMMARY
[010] The present disclosure relates to vaccination regimens and methods for
protecting
infants against disease caused by respiratory syncytial virus (RSV) and
Bordetella
pertussis (pertussis) by administering to pregnant women at least one
immunogenic
composition comprising a recombinant antigen RSV F protein analog and a
pertussis
antigen. Active immunization of pregnant women with immunogenic composition(s)
containing an F protein analog and a pertussis antigen elicits maternal
antibodies, which
are transferred to the gestational infant via the placenta, thereby protecting
the infant from
RSV disease following birth. Accordingly, the present disclosure also relates
to
vaccination regimens and methods of immunising a pregnant woman to protect her
infant
from disease caused by RSV and pertussis, to the use of a recombinant RSV F
protein
analog and a pertussis antigen in immunizing a pregnant female to protect her
infant from
disease caused by RSV, and to kits useful for immunizing pregnant women to
protect their
infants against RSV and pertussis.
BRIEF DESCRIPTION OF THE DRAWINGS
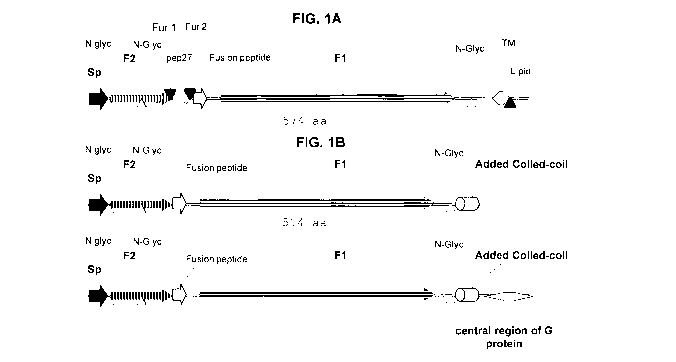
[011] FIG. 1A is a schematic illustration highlighting structural features of
the RSV F
protein. FIG. 1B is a schematic illustration of exemplary RSV Prefusion F
(PreF)
antigens.
[012] FIG. 2 shows the study design for a guinea pig experiment performed in
Example
I.
[013] FIG. 3 shows results from Example 1 following challenge of guinea pig
progeny
with RSV.
[014] FIG. 4 shows the time-course of the neutralizing antibody response in
the guinea
pig model in Example 1.
[015] FIGS. 5A and 5B are graphs illustrating the neutralizing titres and
protection
against RSV challenge infection following immunization with an RSV + pertussis
combination vaccine.
3
CA 02879939 2015-01-23
WO 2014/024026
PCT/1B2013/001722
[016] FIGS. 6A and 6B are graphs illustrating serum antibody titers and
protection
against challenge with Bordetella pertussis following immunization with an RSV
+
pertussis combination vaccine.
DETAILED DESCRIPTION
INTRODUCTION
[017] A particular challenge in the development of a safe and effective
vaccine that
protects infants against diseases caused by Respiratory Syncytial Virus (RSV)
and
Bordetella pertussis (pertussis) is that the highest incidence and severity,
with respect to
morbidity and mortality, is in very young infants. This in itself presents
many challenges.
Young infants, especially those born prematurely, can have an immature immune
system.
There is also a potential risk of interference of maternal antibodies with
vaccination in
very young infants. In the past there has been a problem with enhancement of
RSV
disease with vaccination of young infants against RSV, as well as challenges
arising from
waning of immunity elicited by natural infection and immunization. The present
disclosure concerns methods of protecting young infants, e.g., between birth
and 6 months
of age, from disease caused by both RSV and pertussis by actively immunizing
pregnant
women with an immunogenic composition containing an analog of the RSV F
protein and
a pertussis antigen. The F protein analog and pertussis antigen(s) favorably
elicit
antibodies, which are transferred to the gestational infant via the placenta
resulting in
passive immunological protection of the infant following birth and lasting
through the
critical period for infection and severe disease caused by both RSV and
pertussis.
[018] One aspect of this disclosure concerns a vaccination regimen that
protect infants
(including neonates) against infection or disease caused by both RSV and
pertussis. The
vaccination regimen involves administering to a pregnant female at least one
immunogenic composition capable of boosting (or inducing or eliciting) a
humoral
immune response (e.g., an antibody response) against RSV and pertussis.
Favorably, the
at least one immunogenic composition includes a recombinant RSV antigen
comprising an
F protein analog and a pertussis antigen that is either whole cell pertussis
antigen or
acellular pertussis proteins. Upon administration of the at least one
immunogenic
composition, at least one subset of maternal antibodies specific for RSV and
pertussis
elicited by the immunogenic composition are transferred to the gestational
infant via the
4
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
placents, thereby protecting the infant against infection or disease caused by
both RSV and
pertussis.
[019] Another aspect of this disclosure relates to a method for protecting an
infant
against infection or disease caused by respiratory syncytial virus (RSV) and
pertussis, the
method comprising administering to a pregnant female with a gestational infant
at least
one immunogenic composition comprising a recombinant RSV antigen comprising an
F
protein analog and a pertussis antigen, wherein at least one subset of
maternal antibodies
elicited by the immunogenic composition are transferred to the gestational
infant via the
placenta, thereby protecting the infant against infection or disease caused by
RSV.
[020] In another aspect, the disclosure relates to an immunogenic composition
or
plurality of immunogenic compositions that include a recombinant RSV antigen
comprising an F protein analog and at least one pertussis antigen for use in
protecting an
infant from infection or disease caused by RSV and pertussis, which
immunogenic
composition(s) are formulated for administration to a pregnant female. Upon
administration to a pregnant female, the immunogenic composition(s) are
capable of
boosting a humoral immune response (e.g., inducing, eliciting or augmenting)
at least one
subset of maternal antibodies specific for RSV and pertussis. The maternal
antibodies are
transferred to the gestational infant via the placenta, thereby conferring
protection against
infection and/or disease caused by RSV and pertussis.
[021] In another aspect, this disclosure concerns kits comprising a plurality
of (e.g., 2 or
more) immunogenic compositions formulated for administration to a pregnant
female.
The kit includes (a) a first immunogenic composition comprising an F protein
analog
capable of inducing, eliciting or boosting a humoral immune response specific
for RSV;
and (b) a second immunogenic composition comprising at least one pertussis
antigen
capable of inducing, eliciting or boosting a humoral response specific for B.
pertussis.
The immunogenic compositions are optionally contained in one or more pre-
filled syringe,
for example, in a dual (or multi) chambered syringe. Upon administration to a
pregnant
female, the at least first and second immunogenic compositions of the kit
induce, elicit or
boost at least one subset of RSV-specific antibodies and at least one subset
of pertussis-
specific antibodies, which antibodies are transferred via the placenta to the
gestating infant
carried by the pregnant female. Transfer of maternal antibodies via the
placenta confers
CA 02879939 2015-01-23
WO 2014/024026
PCT/1B2013/001722
protection against infection and/or disease caused by both RSV and pertussis.
[022] In a particular embodiment the vaccination regimen, method, or use or
kit
involves an immunogenic composition (e.g., and administration therof) of an F
protein
analog which is a prefusion F or "PreF" antigen that includes at least one
modification that
stabilizes the prefusion conformation of the F protein. Alternatively, F
analogs stabilized
in the postfusion conformation ("PostF"), or that are labile with respect to
conformation
can be employed in the methods described herein. Generally the F protein
analog, e.g.,
PreF, PostF, etc.) antigen lacks a transmembrane domain, and is soluble, i.e.,
not
membrane bound (for example, to facilitate expression and purification of the
F protein
analog).
[023] In exemplary embodiments, the vaccination regimen, method, use or kit
involves
the administration of an F protein analog that comprises in an N-terminal to C-
terminal
direction: at least a portion or substantially all of an F2 domain and an Fi
domain of an
RSV F protein polypeptide, optionally with a heterologous trimerization
domain. In an
embodiment, there is no furin cleavage site between the F2 domain and the F1
domain. In
certain exemplary embodiments, the F2 domain comprises at least a portion of
an RSV F
protein polypeptide corresponding to amino acids 26-105 of the reference F
protein
precursor polypeptide (Fo) of SEQ ID NO:2 and/or the F1 domain comprises at
least a
portion of an RSV F protein polypeptide corresponding to amino acids 137-516
of the
reference F protein precursor polypeptide (Fo) of SEQ ID NO:2.
[024] For example, in specific embodiments, the F protein analog is selected
from the
group of:
a) a polypeptide comprising a polypeptide selected from the group of SEQ ID
NO:6, SEQ ID NO:8, SEQ ID NO:10, SEQ ID NO:18, SEQ ID NO:20 and
SEQ ID NO:22;
b) a polypeptide encoded by a polynucleotide selected from the group of SEQ
ID NO:5, SEQ ID NO:7, SEQ ID NO:9, SEQ ID NO:17, SEQ ID NO:19
and SEQ ID NO:21, or by a polynucleotide sequence that hybridizes under
stringent conditions over substantially its entire length to a polynucleotide
selected from the group of SEQ ID NO:5, SEQ ID NO:7, SEQ ID NO:9,
SEQ ID NO:17, SEQ ID NO:19 and SEQ ID NO:21, which polypeptide
6
CA 02879939 2015-01-23
WO 2014/024026
PCT/1B2013/001722
comprises an amino acid sequence corresponding at least in part to a
naturally occurring RSV strain;
c) a polypeptide with at least 95% sequence identity to a polypeptide
selected
from the group of SEQ ID NO:6, SEQ ID NO:8, SEQ ID NO:10, SEQ ID
NO:18, SEQ ID NO:20 and SEQ ID NO:22, which polypeptide comprises
an amino acid sequence that does not correspond to a naturally occurring
RSV strain.
[025] Optionally, the F protein analog further comprises a signal peptide.
Optionally,
the F protein analog can further comprise a "tag" or sequence to facilitate
purification,
e.g., a multi-histidine sequence.
[026] In embodiments comprising a heterologous trimerization domain, such a
domain
can comprise a coiled-coil domain, such as an isoleucine zipper, or it can
comprise an
alternative trimerization domain, such as from the bacteriophage T4 fibritin
("foldon"), or
influenza HA.
[027] In certain exemplary embodiments, the F protein analog comprises at
least one
modification selected from:
(i) a modification that alters glycosylation.
(ii) a modification that eliminates at least one non-furin cleavage site;
(iii) a modification that deletes one or more amino acids of the pep27
domain;
and
(iv) a modification that substitutes or adds a hydrophilic amino acid in a
hydrophobic domain of the F protein extracellular domain.
[028] In certain embodiments, the F protein analog comprises a multimer of
polypeptides, for example, a trimer of polypeptides.
[029] With respect to the pertussis antigen, the antigen can be one or more
acellular
pertussis protein(s) or whole cell pertussis antigen. For example, acellular
pertussis
proteins can be selected from the consisting of: pertussis toxoid (PT),
filamentous
haemagglutinin (FHA), pertactin (PRN), fimbrae type 2 (FIM2), fimbrae type 3
(FIM3)
and BrkA. The PT can be chemically toxoided or genetically toxoided (for
example by
one or both of the mutations: R9K and E129G). In certain favorable
embodiments, the
7
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
pertussis antigen comprises a combination of pertussis proteins, for example:
PT and
FHA; PT, FHA and PRN; or PT, FHA, PRN and optionally either or both of FIM2
and
FIM3.
[030] In embodiments that include a whole cell (Pw) pertussis antigen, the Pw
antigen
can have a reduced endotoxin content. A reduced endotoxin content can be
achieved by
chemical extraction of lipo-oligosaccharide (LOS), or by genetic manipulation
of
endotoxin production, for example to induce overexpression or heterologous
expression of
a 3-0-deacylase. In certain favorable embodiments, the Pw antigen comprises B.
pertussis cells comprising at least partially 3-0-deacylated LOS.
[031] In certain embodiments, the F protein analog and/or pertussis antigen
is/are
formulated in an immunogenic composition comprising a pharmaceutically
acceptable
carrier or excipient, such as a buffer. Optionally, the immunogenic
composition also
includes an adjuvant, for example, and adjuvant that includes 3D-MPL, QS21
(e.g., in a
detoxified form), an oil-in-water emulsion (e.g., with or without
immunostimulatory
molecules, such as a-tocopherol), mineral salts, such as aluminium salts
(e.g., aluminium
hydroxide aluminium phosphate), including alum, or combinations thereof.
Preferably, if
the immunogenic composition includes an adjuvant, the adjuvant enhances or
increases a
humoral immune characterized by the production of antibodies, particularly IgG
antibodies of the IgGi subclass. Alternatively the immunogenic composition(s)
are
formulated in the absence of (i.e., without) an adjuvant.
[032] Typically, the immunogenic composition(s) containing the F protein
analog and
the pertussis antigen is administered during the third trimester of pregnancy
(gestation),
although a beneficial effect (especially pregnancies at increased risk of
preterm delivery)
can be obtained prior to the beginning of the third trimester. Thus, although
the F protein
analog can be administered at 26 weeks of gestation (measured from the start
of the last
menstrual period) or later, for example between 26 and 38 weeks of gestation,
or between
28 and 34 weeks of gestation, the F protein analog can also be administered
prior to 26
weeks of gestation.
[033] In certain embodiments of the vaccination regimen, method, use and kit,
the RSV
antigen (recombinant F protein analog) and the pertussis antigen are
coformulated in the
same immunogenic composition. In alternative embodiments, the RSV antigen
8
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
(recombinant F protein analog) and the pertussis antigen are formulated in
different (e.g.,
two different) immunogenic compositions, which can be co-administered (that is
administered at or near the same time, i.e., on the same day) or on different
days, typically
provided that both immunogenic compositions are administered during the third
trimester
of pregnancy, e.g., after 26 weeks of gestation, or after 28 weeks of
gestation, and
typically before 34 weeks of gestation, or before 38 weeks of gestation.
Administration to
the pregnant female can be accomplished by any of a variety of routes, for
example,
intramuscular, cutaneous or intradermal administration routes.
[034] Favorably, the vaccination regimen, method, use, or administration of
the
immunogenic compositions included in the kits elicits an immune response in
the pregnant
female (favorably, a human female), which when passively transferred to her
gestational
infant via the placenta, protects the infant, e.g., from birth to at least
about 6 months of
age. Accordingly, the infant protected by the methods disclosed herein can be
an
immunologically immature infant, such as an infant of less than six months of
age, such as
less than two months of age, for example, less than one month of age, e.g., a
neonate or
newborn. Favorably, at least one subset of antibodies, for example, IgG
antibodies, such
as IgGi antibodies are transferred via the placenta. It is also viewed as
advantageous that
the subset of maternal antibodies includes neutralizing antibodies against
RSV.
[035] In certain embodiments, the transfer of at least one subset of
antibodies via the
placenta can be measured by immunological methods (e.g., ELISA). For example,
in
certain favorable embodiments, RSV-specific antibodies can be detected at a
level at or
greater than 30 mcg/ml in the infant's serum at birth. Similarly, in certain
favorable
embodiments, pertussis-specific antibodies can be detected at a level at or
greater than 10
ELISA units/ml (EU) in the infant's serum at birth. Favorably, the antibodies
specific for
RSV and pertussis are present at birth in the infant's serum at a level that
protects against
(or inhibits) infection and reduces or prevents disease caused by RSV and
pertussis
without impairing the infant's subsequent response to immunization (or
exposure).
[036] Optionally, to ensure continuity of the immune response beyond six
months of
age, the vaccination regimen, method, or use (and administration of the
compositions of
the kit) can involve administering to the infant (delivered of the pregnant
female) one or
more compositions that primes or induces an active immune response against RSV
and/or
9
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
pertussis in the infant. The composition(s) administered to the infant can be
the same or
different from that (or those) administered to the pregnant female. For
example, the
composition administered to the infant can include an RSV antigenic component
that is an
F protein analog or it can include an RSV antigenic component that is a
nucleic acid that
encodes one or more RSV antigens, such as a live attenuated virus, a viral
vector (e.g., an
adenoviral or MVA vector), or a viral replicon particle or other self-
replicating nucleic
acid. The composition can alternatively or additionally include a pertussis
antigenic
component that is an acellular pertussis antigen (Pa, e.g., one or more
pertussis proteins)
or a whole cell pertussis antigen (Pw), as described herein. If both an RSV
antigenic
component and a pertussis antigenic component are administered to the infant,
the RSV
antigenic component and the pertussis antigenic component can be coformulated
in the
same immunogenic composition. Alternatively, the RSV antigenic component and
the
pertussis antigenic component can be formulated in two (or more) different
immunogenic
compositions, which can be co-administered (at the same time or on the same
day) or
which can be administered according to different schedules (e.g., according to
the various
approved and/or recommended pediatric immunization schedules). Optionally, the
composition or compositions administered to the infant can include one or more
additional
antigens of pathogens other than RSV or pertussis (for example, formulated in
vaccines
commonly administered according various pediatric immunization schedules).
Depending
on the formulation, the immunogenic compositions can be administered to the
infant by a
variety of established routes, e.g., intramuscular, cutaneous intradermal
and/or mucosal
(e.g., intranasal).
[037] The vaccination regimens, methods, uses and kits disclosed herein can
reduce the
incidence or severity of infection or disease caused by RSV and pertussis. For
example,
the vaccination regimens, methods, uses and kits can reduce the incidence or
severity of
RSV disease such as LRTI, or by reducing the incidence of severe RSV disease
such as
LRTI. Similarly, the vaccination regimens, methods, uses and kits can reduce
the
incidence or severity of disease (e.g., duration or severity of "whooping"
cough or
pneumonia, or associated symptoms) caused by infection with B. pertussis.
Protecting the
infant favorably includes protecting the infant from severe disease and
hospitalization
caused by RSV and/or pertussis. As such, the methods and uses disclosed herein
can
reduce the incidence of severe disease caused by both RSV and pertussis by 50%
or more,
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
or 60% or more, or 70% or more, as measured by a 50% or more, or 60% or more,
or 70%
or more reduction in the rate of severe LRTI and/or hospitalization and/or
rate of
pneumonia in a cohort of infants of vaccinated mothers compared to infants of
unvaccinated mothers.
TERMS
[038] In order to facilitate review of the various embodiments of this
disclosure, the
following explanations of terms are provided. Additional terms and
explanations can be
provided in the context of this disclosure.
[039] Unless otherwise explained, all technical and scientific terms used
herein have the
same meaning as commonly understood by one of ordinary skill in the art to
which this
disclosure belongs. Definitions of common terms in molecular biology can be
found in
Benjamin Lewin, Genes V, published by Oxford University Press, 1994 (ISBN 0-19-
854287-9); Kendrew et al. (eds.), The Encyclopedia of Molecular Biology,
published by
Blackwell Science Ltd., 1994 (ISBN 0-632-02182-9); and Robert A. Meyers (ed.),
Molecular Biology and Biotechnology: a Comprehensive Desk Reference, published
by
VCH Publishers, Inc., 1995 (ISBN 1-56081-569-8).
[040] The singular terms "a," "an," and "the" include plural referents unless
context
clearly indicates otherwise. Similarly, the word "or" is intended to include
"and" unless
the context clearly indicates otherwise. The term "plurality" refers to two or
more. It is
further to be understood that all base sizes or amino acid sizes, and all
molecular weight or
molecular mass values, given for nucleic acids or polypeptides are
approximate, and are
provided for description. Additionally, numerical limitations given with
respect to
concentrations or levels of a substance, such as an antigen, are intended to
be approximate.
Thus, where a concentration is indicated to be at least (for example) 200 pg,
it is intended
that the concentration be understood to be at least approximately (or "about"
or "¨") 200
pg.
[041] Although methods and materials similar or equivalent to those described
herein
can be used in the practice or testing of this disclosure, suitable methods
and materials are
described below. The term "comprises" means "includes." Thus, unless the
context
requires otherwise, the word "comprises," and variations such as "comprise"
and
11
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
"comprising" will be understood to imply the inclusion of a stated compound or
composition (e.g., nucleic acid, polypeptide, antigen) or step, or group of
compounds or
steps, but not to the exclusion of any other compounds, composition, steps, or
groups
thereof. The abbreviation, "e.g." is derived from the Latin exempli gratia,
and is used
herein to indicate a non-limiting example. Thus, the abbreviation "e.g." is
synonymous
with the term "for example."
[042] The term "F protein" or "Fusion protein" or "F protein polypeptide" or
"Fusion
protein polypeptide" refers to a polypeptide or protein having all or part of
an amino acid
sequence of an RSV Fusion protein polypeptide. Numerous RSV Fusion proteins
have
been described and are known to those of skill in the art. W02008114149 sets
out
exemplary F protein variants (for example, naturally occurring variants).
[043] An "F protein analog" refers to an F protein which includes a
modification that
alters the structure or function of the F protein but which retains the
immunological
properties of the F protein such that an immune response generated against an
F protein
analog will recognize the native F protein. W02010149745, incorporated herein
in its
entirety by reference, sets out exemplary F protein analogs. W02011008974,
incorporated herein in its entirety by reference, also sets out exemplary F
protein analogs.
F protein analogs include for example PreF antigens which include at least one
modification that stabilizes the prefusion conformation of the F protein and
which are
generally soluble, i.e., not membrane bound. F protein analogs also include
post-fusion F
(postF) antigens which are in the post-fusion conformation of the RSV F
protein,
favorably stabilized in such conformation. F analogs further include F protein
in an
intermediate conformation, favorably stabilized in such conformation. Such
alternatives
are also generally soluble.
[044] A "variant" when referring to a nucleic acid or a polypeptide (e.g., an
RSV F or G
protein nucleic acid or polypeptide, or an F analog nucleic acid or
polypeptide) is a nucleic
acid or a polypeptide that differs from a reference nucleic acid or
polypeptide. Usually,
the difference(s) between the variant and the reference nucleic acid or
polypeptide
constitute a proportionally small number of differences as compared to the
referent.
[045] A "domain" of a polypeptide or protein is a structurally defined element
within
the polypeptide or protein. For example, a "trimerization domain" is an amino
acid
12
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
sequence within a polypeptide that promotes assembly of the polypeptide into
trimers. For
example, a trimerization domain can promote assembly into trimers via
associations with
other trimerization domains (of additional polypeptides with the same or a
different amino
acid sequence). The term is also used to refer to a polynucleotide that
encodes such a
peptide or polypeptide.
[046] The terms "native" and "naturally occurring" refer to an element, such
as a
protein, polypeptide or nucleic acid that is present in the same state as it
is in nature. That
is, the element has not been modified artificially. It will be understood,
that in the context
of this disclosure, there are numerous native/naturally occurring variants of
RSV proteins
or polypeptides, e.g., obtained from different naturally occurring strains or
isolates of
RSV. W02008114149, incorporated herein by reference in its entirety, contains
exemplary RSV strains, proteins and polypeptides, see for example Figure 4.
[047] The term "polypeptide" refers to a polymer in which the monomers are
amino
acid residues which are joined together through amide bonds. The terms
"polypeptide" or
"protein" as used herein are intended to encompass any amino acid sequence and
include
modified sequences such as glycoproteins. The term "polypeptide" is
specifically
intended to cover naturally occurring proteins, as well as those which are
recombinantly or
synthetically produced. The term "fragment," in reference to a polypeptide,
refers to a
portion (that is, a subsequence) of a polypeptide. The term "immunogenic
fragment"
refers to all fragments of a polypeptide that retain at least one predominant
immunogenic
epitope of the full-length reference protein or polypeptide. Orientation
within a
polypeptide is generally recited in an N-terminal to C-terminal direction,
defined by the
orientation of the amino and carboxy moieties of individual amino acids.
Polypeptides are
translated from the N or amino-terminus towards the C or carboxy-terminus.
[048] A "signal peptide" is a short amino acid sequence (e.g., approximately
18-25
amino acids in length) that directs newly synthesized secretory or membrane
proteins to
and through membranes, e.g., of the endoplasmic reticulum. Signal peptides are
frequently but not universally located at the N-terminus of a polypeptide, and
are
frequently cleaved off by signal peptidases after the protein has crossed the
membrane.
Signal sequences typically contain three common structural features: an N-
terminal polar
basic region (n-region), a hydrophobic core, and a hydrophilic c-region).
13
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
[049] The terms "polynucleotide" and "nucleic acid sequence" refer to a
polymeric form
of nucleotides at least 10 bases in length. Nucleotides can be
ribonucleotides,
deoxyribonucleotides, or modified forms of either nucleotide. The term
includes single
and double forms of DNA. By "isolated polynucleotide" is meant a
polynucleotide that is
not immediately contiguous with both of the coding sequences with which it is
immediately contiguous (one on the 5' end and one on the 3' end) in the
naturally
occurring genome of the organism from which it is derived. In one embodiment,
a
polynucleotide encodes a polypeptide. The 5' and 3' direction of a nucleic
acid is defined
by reference to the connectivity of individual nucleotide units, and
designated in
accordance with the carbon positions of the deoxyribose (or ribose) sugar
ring. The
informational (coding) content of a polynucleotide sequence is read in a 5' to
3' direction.
[050] A "recombinant" nucleic acid is one that has a sequence that is not
naturally
occurring or has a sequence that is made by an artificial combination of two
otherwise
separated segments of sequence. This artificial combination can be
accomplished by
chemical synthesis or, more commonly, by the artificial manipulation of
isolated segments
of nucleic acids, e.g., by genetic engineering techniques. A "recombinant"
protein is one
that is encoded by a heterologous (e.g., recombinant) nucleic acid, which has
been
introduced into a host cell, such as a bacterial or eukaryotic cell. The
nucleic acid can be
introduced, on an expression vector having signals capable of expressing the
protein
encoded by the introduced nucleic acid or the nucleic acid can be integrated
into the host
cell chromosome.
[051] The term "heterologous" with respect to a nucleic acid, a polypeptide or
another
cellular component, indicates that the component occurs where it is not
normally found in
nature and/or that it originates from a different source or species.
[052] An "antigen" is a compound, composition, or substance that can stimulate
the
production of antibodies and/or a T cell response in a subject, including
compositions that
are injected, absorbed or otherwise introduced intoa subject. The term
"antigen" includes
all related antigenic epitopes. The term "epitope" or "antigenic determinant"
refers to a
site on an antigen to which B and/or T cells respond. The "dominant antigenic
epitopes"
or "dominant epitope" are those epitopes to which a functionally significant
host immune
response, e.g., an antibody response or a T-cell response, is made. Thus, with
respect to a
14
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
protective immune response against a pathogen, the dominant antigenic epitopes
are those
antigenic moieties that when recognized by the host immune system result in
protection
from disease caused by the pathogen. The term "T-cell epitope" refers to an
epitope that
when bound to an appropriate MHC molecule is specifically bound by a T cell
(via a T
cell receptor). A "B-cell epitope" is an epitope that is specifically bound by
an antibody
(or B cell receptor molecule).
[053] An "adjuvant" is an agent that enhances the production of an immune
response in
a non-antigen specific manner. Common adjuvants include suspensions of
minerals
(alum, aluminum hydroxide, aluminum phosphate) onto which antigen is adsorbed;
emulsions, including water-in-oil, and oil-in-water (and variants thereof,
including double
emulsions and reversible emulsions), liposaccharides, lipopolysaccharides,
immunostimulatory nucleic acids (such as CpG oligonucleotides), liposomes,
Toll-like
Receptor agonists (particularly, TLR2, TLR4, TLR7/8 and TLR9 agonists), and
various
combinations of such components.
[054] An "antibody" or "immunoglobulin" is a plasma protein, made up of four
polypeptides that binds specifically to an antigen. An antibody molecule is
made up of
two heavy chain polypeptides and two light chain polypeptides (or multiples
thereof) held
together by disulfide bonds. In humans, antibodies are defined into five
isotypes or
classes: IgG, IgM, IgA, IgD, and IgE. IgG antibodies can be further divided
into four
sublclasses (IgGi, IgG2, IgG3 and IgG4). A "neutralizing" antibody is an
antibody that is
capable of inhibiting the infectivity of a virus. Accordingly, a neutralizing
antibodies
specific for RSV are capable of inhibiting or reducing the infectivity of RSV.
[055] An "immunogenic composition" is a composition of matter suitable for
administration to a human or animal subject (e.g., in an experimental or
clinical setting)
that is capable of eliciting a specific immune response, e.g., against a
pathogen, such as
RSV or Bordetella pertussis). As such, an immunogenic composition includes one
or
more antigens (for example, polypeptide antigens) or antigenic epitopes. An
immunogenic composition can also include one or more additional components
capable of
eliciting or enhancing an immune response, such as an excipient, carrier,
and/or adjuvant.
In certain instances, immunogenic compositions are administered to elicit an
immune
response that protects the subject against symptoms or conditions induced by a
pathogen.
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
In some cases, symptoms or disease caused by a pathogen is prevented (or
reduced or
ameliorated) by inhibiting replication or cellular infection of the pathogen
following
exposure of the subject to the pathogen. In the context of this disclosure,
the term
immunogenic composition will be understood to encompass compositions that are
intended for administration to a subject or population of subjects for the
purpose of
eliciting a protective, preventative or palliative immune response against RSV
or pertussis
(that is, vaccine compositions or vaccines).
[056] An "immune response" is a response of a cell of the immune system, such
as a B
cell, T cell, or monocyte, to a stimulus, such as a pathogen or antigen (e.g.,
formulated as
an immunogenic composition or vaccine). An immune response can be a B cell
response,
which results in the production of specific antibodies, such as antigen
specific neutralizing
antibodies. An immune response can also be a T cell response, such as a CD4+
response
or a CD8+ response. B cell and T cell responses are aspects of a "cellular"
immune
response. An immune response can also be a "humoral" immune response, which is
mediated by antibodies, which can be detected and/or measured, e.g., by an
ELISA assay.
In some cases, the response is specific for a particular antigen (that is, an
"antigen-specific
response"). If the antigen is derived from a pathogen, the antigen-specific
response is a
"pathogen-specific response." A "protective immune response" is an immune
response
that inhibits a detrimental function or activity of a pathogen, reduces
infection by a
pathogen, or decreases symptoms (including death) that result from infection
by the
pathogen. A protective immune response can be measured, for example, by the
inhibition
of viral replication or plaque formation in a plaque reduction assay or
neutralization assay,
or by measuring resistance to pathogen challenge in vivo. Exposure of a
subject to an
immunogenic stimulus, such as a pathogen or antigen (e.g., formulated as an
immunogenic
composition or vaccine), elicits or induces a primary immune response specific
for the
stimulus, that is, the exposure "primes" the immune response. A subsequent
exposure,
e.g., by immunization, to the stimulus can increase or "boost" the magnitude
(or duration,
or both) of the specific immune response. Thus, "boosting" a preexisting
immune
response by administering an immunogenic composition increases the magnitude
of an
antigen (or pathogen) specific response, (e.g., by increasing antibody titre
and/or affinity,
by increasing the frequency of antigen specific B or T cells, by inducing
maturation
effector function, or any combination therof).
16
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
[057] The adjective "pharmaceutically acceptable" indicates that the referent
is suitable
for administration to a subject (e.g., a human or animal subject). Remington's
Pharmaceutical Sciences, by E. W. Martin, Mack Publishing Co., Easton, PA,
15th
Edition (1975), describes compositions and formulations (including diluents)
suitable for
pharmaceutical delivery of therapeutic and/or prophylactic compositions,
including
immunogenic compositions.
[058] The term "reduces" is a relative term, such that an agent reduces a
response or
condition if the response or condition is quantitatively diminished following
administration of the agent, or if it is diminished following administration
of the agent, as
compared to a reference agent. Similarly, the term "protects" does not
necessarily mean
that an agent completely eliminates the risk of an infection or disease caused
by infection,
so long as at least one characteristic of the response or condition is
substantially or
significantly reduced or eliminated. Thus, an immunogenic composition that
protects
against or reduces an infection or a disease, or symptom thereof, can, but
does not
necessarily prevent or eliminate infection or disease in all subjects, so long
as the
incidence or severity of infection or incidence or severity of disease is
measurably
reduced, for example, by at least about 50%, or by at least about 60%, or by
at least about
70%, or by at least about 80%, or by at least about 90% of the infection or
response in the
absence of the agent, or in comparison to a reference agent. In certain
instances, the
reduction is in the incidence of lower respiratory tract infections (LRTI), or
the incidence
of severe LRTI, or hospitalizations due to RSV disease, or in the severity of
disease
caused by RSV. In other instances, the reduction is in the incidence of
pneumonia, or
hospitalization due to disease caused by B. pert ussis.
[059] A "subject" is a living multi-cellular vertebrate organism. In the
context of this
disclosure, the subject can be an experimental subject, such as a non-human
animal, e.g., a
mouse, a cotton rat, guinea pig, cow, or a non-human primate. Alternatively,
the subject
can be a human subject. Similarly, a pregnant female and an infant can be a
non-human
animal or a human female and infant, respectively.
MATERNAL IMMUNIZATION
[060] Lower respiratory tract infection (LRTI) is an infection of any of the
tissues of the
lower respiratory tract with the most severe forms including bronchiolitis and
pneumonia.
17
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
Human Respiratory Syncytial Virus (RSV) is the most common worldwide cause of
lower
respiratory tract infections (LRTI) in infants less than 6 months of age and
premature
babies less than or equal to 35 weeks of gestation. The RSV disease spectrum
includes a
wide array of respiratory symptoms from rhinitis and otitis to pneumonia and
bronchiolitis, the latter two diseases being associated with considerable
morbidity and
mortality.
[061] The bacterium Bordetella pertussis (pertussis) is the causative agent
for whooping
cough, a respiratory disease that can be severe in infants and young children,
with
secondary pneumonia, apnea and respiratory distress and failure being a
serious
complications. Although treatment with antibiotics is available, by the time
the disease is
diagnosed, bacterial toxins can cause severe damage. Both natural infection
with RSV and
immunization with pertussis vaccines result in immunity that wanes over time,
with the
consequence that adolescents and adults can act as reservoirs of these highly
contagious
diseases. This puts neonates at particular risk in the first few months of
life where the
consequences of infection are most severe.
[062] Strategies for protecting vulnerable newborns against pertussis
infection and
disease include "cocooning", i.e., vaccinating adolescents and adults
(including
postpartum women) likely to be in contact with newborns. Vaccination of
pregnant
women (maternal immunization) against pertussis is now recommended in several
countries, whereby anti-pertussis antibodies are transferred placentally to
provide
protection until the infant can be directly vaccinated. Nonetheless, there
remains an unmet
need for a combination vaccine which confers protection against both RSV and
B.
pertussis infection and associated disease, which pose particular dangers to
neonates and
young infants.
[063] A particular challenge in the development of a safe and effective
vaccine that
protects infants against disease caused by RSV and pertussis is that the
highest incidence
and greatest morbidity and mortality is in very young infants. Young infants,
especially
those born prematurely, can have an immature immune system. Physiologically
infants
may be more susceptible to diseases of the lower respiratory tract than older
children.
Thus, protecting young infants from RSV disease, whooping cough and in
particular from
severe LRTI, pneumonia, and respiratory distress and failure is particularly
important.
18
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
There is also a potential risk of interference of antibodies transferred via
the placenta to
the infant ("maternal antibodies") with vaccination of the infant, such that
vaccination in
early infancy may not be sufficiently effective, e.g., to elicit a fully
protective neutralizing
antibody response.
[064] The present disclosure concerns vaccination regimens, methods and uses
of
immunogenic compositions and kits suitable for protecting young infants from
disease
caused by RSV and pertussis by actively immunizing pregnant women with a safe
and
effective immunogenic composition(s) containing an analog of the RSV F protein
and an
acellular or whole cell pertussis antigen. The F protein analog favorably
elicits antibodies
(e.g., neutralizing antibodies), by boosting or increasing the magnitude of
the humoral
response previously primed by natural exposure to (or prior vaccination
against) RSV.
Similarly, the pertussis antigen elicits antibodies, by boosting or increasing
the magnitude
of the humoral response previously primed by natural exposure to, or prior
vaccination
against, pertussis. The antibodies produced in response to the F protein
analog and
pertussis antigen are transferred to the gestational infant via the placenta,
resulting in
passive immunological protection of the infant following birth and lasting
through the
critical period for infection and severe disease caused by RSV and pertussis
(e.g., before
infant vaccination is fully protective). Typically, the passive immunological
protection
=
conferred by this method lasts between birth and at least two months of age,
for example,
up to about 6 months of age, or even longer.
[065] The immunogenic compositions described herein containing an F protein
analog
and/or a pertussis antigen are designed to induce a strong antibody responses
(e.g.,
neutralizing antibodies). Since pregnant mothers have typically been exposed
to RSV one
or more times during their lives, they have an existing primed response to
RSV. The
proportion of the population exposed to RSV infection by adulthood is
essentially 100%.
Pediatric immunization programs designed to protect against and prevent
whooping cough
are widespread. However, despite widespread immunization natural infection
with B.
pertussis is also common. Thus, priming to pertussis is also widespread.
Therefore, the
provision of protection from RSV and pertussis for the infant immediately
after birth and
for the crucial first few months that follow, can be achieved by boosting
these primed
responses as effectively as possible to increase serum antibody responses
(levels) against
RSV and pertussis in the mother, and favorably in respect of particular
antibody
19
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
subclasses (subtypes) such as IgGI that can cross the placenta and provide
protection to
the infant. In one embodiment, immunogenic compositions for use herein do not
include
an adjuvant, or include an adjuvant which favors a strong IgGI response such
as a mineral
salt such as an aluminium or calcium salt, in particular aluminium hydroxide,
aluminium
phosphate or calcium phosphate. Thus in a particular embodiment the F protein
analog for
the methods and uses described herein is favorably formulated with a mineral
salt,
favorably alum. In alternative embodiments, the adjuvant that favors a strong
IgGI
response is an oil-in-water emulsion, or a saponin, such as QS21 (or a
detoxified version
thereof), as will be described in more detail below.
[066] A pregnant female can be a human female, and accordingly, the infant or
gestational infant can be a human infant. For a pregnant human female the
gestational age
of the developing fetus is measured from the start of the last menstrual
period. The
number of weeks post-conception is measured from 14 days after the start of
the last
menstrual period. Thus, when a pregnant human female is said to be 24 weeks
post
conception this will be equal to 26 weeks after the start of her last
menstrual period, or 26
weeks of gestation. When a pregnancy has been achieved by assisted
reproductive
technology, gestational stage of the developing fetus is calculated from two
weeks before
the date of conception.
[067] The term "gestational infant" as used herein means the fetus or
developing fetus
of a pregnant female. The term "gestational age" is used to mean the number of
weeks of
gestation i.e. the number of weeks since the start of the last menstrual
period. Human
gestation is typically about 40 weeks from the start of the last menstrual
period, and may
conveniently be divided into trimesters, with the first trimester extending
from the first
day of the last menstrual period through the 13th week of gestation; the
second trimester
spanning from the 14th through the 27th weeks of gestation, and the third
trimester starting
in the 28th week and extending until birth. Thus, the third trimester starts
at 26 weeks
post-conception and continues through to birth of the infant.
[068] The term "infant" when referring to a human is between 0 and two years
of age It
will be understood that the protection provided by the methods and uses
described herein
can potentially provide protection for an infant into childhood, from aged 2
to 11, or early
childhood for example from ages 2 to 5, or even into adolescence, from aged 12
to 18.
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
However it is during infancy that an individual is most vulnerable to severe
RSV disease
and complications of whooping cough (pertussis) so this is the focus of the
present
disclosure (e.g., from birth to about 6 months of age).
[069] A human infant can be immunologically immature in the first few months
of life,
especially when born prematurely, e.g., before 35 weeks gestation, when the
immune
system may not be sufficiently well developed to mount an immune response
capable of
preventing infection or disease caused by a pathogen in the way that a
developed immune
system would be capable of doing in response to the same pathogen, An
immunologically
immature infant is more likely to succumb to infection and disease caused by a
pathogen
than an infant with a more developed or mature immune system. A human infant
can also
have an increased vulnerability to LRTIs (including pneumonia) during the
first few
months of life for physiological and developmental reasons, for example,
airways are
small and less developed or mature than in children and adults. For these
reasons, when
we refer herein to the first six months of infancy this may be extended for
premature or
pre-term infants according to the amount of time lost in gestational age below
40 weeks or
below 38 weeks or below 35 weeks.
[070] In an alternative embodiment, the pregnant female and its infant are
from any
species such as those described above under "subjects". For a pregnant animal,
such as a
pregnant guinea pig or cow, the time post-conception is measured as the time
since
mating. In humans and in some animals, for example guinea pigs, antibodies
pass from
the mother to the fetus via the placenta. Some antibody isotypes may be
preferentially
transferred through the placenta, for example in humans IgGi antibodies are
the isotype
most efficiently transferred across the placenta. Although subclasses exist in
experimental
animals, such as guinea pigs and mice, the various subclasses do not
necessarily serve the
same function, and a direct correlate between subclasses of humans and animals
cannot
easily be made.
[071] Favorably, protecting the infant by reducing the inhibiting infection
and reducing
incidence or severity of disease caused by RSV and pertussis covers at least
the neonatal
period and very young infancy, for example at least the first several weeks of
life
following birth, such as the first month from birth, or the first two months,
or the first
three months, or the first four months, or the first five months, or the first
six months from
21
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
birth, or longer, e.g., when the infant is a full-term infant delivered at
about 40 weeks of
gestation or later. After the first few months, when the infant is less
vulnerable to the
effects of severe RSV disease and whooping cough, protection against these
infections
may wane. Thus vital protection is provided during the period when it is most
needed. In
the case of a pre-term infant, favorably protection is provided for a longer
period from
birth for example an additional time period at least equaling the time
interval between
birth of the infant and what would have been 35 weeks gestation (i.e., by
about 5 extra
weeks), or 38 weeks gestation (by about 2 extra weeks), or longer depending on
the
gestational age of the infant at birth.
[072] It will be evident that protecting the infant does not necessarily meanl
00%
protection against infection by RSV or by pertussis. Provided that that there
is a reduction
in incidence or severity of infection or disease it will be recognized that
protection is
provided. Protecting the infant favorably includes protecting the infant from
severe
disease and hospitalization caused by RSV and pertussis. As such, the methods
and uses
disclosed herein reduce the incidence or severity of disease caused by RSV and
pertussis
such as LRTI, pneumonia or other symptoms or disease. For example,
administration of
an immunogenic composition containing an F protein analog as disclosed herein
can
reduce the incidence (in a cohort of infants of vaccinated mothers) of LRTI by
at least
about 50%, or at least about 60%, or by 60 to 70 %, or by at least about70%,
or by at least
about 80%, or by at least about 90% compared to infants of unvaccinated
mothers.
Favorably, such administration reduces the severity of LRTI by at least about
50%, or at
least about 60%, or by 60 to 70 %, or by at least about70%, or by at least
about 80%, or by
at least about 90% compared to infected infants of unvaccinated mothers.
Favorably, such
administration reduces the need for hospitalization due to severe RSV disease
in such a
cohort by at least about 50%, or at least about 60%, or by 60 to 70 %, or by
at least
about70%, or by at least about 80%, or by at least about 90% compared to
infected infants
of unvaccinated mothers. Whether there is considered to be a need for
hospitalization due
to severe LRTI, or whether a particular case of LRTI is hospitalized, may vary
from
country to country and therefore severe LRTI as judged according to defined
clinical
symptoms well known in the art may be a better measure than the need for
hospitalization.
With respect to pertussis, administration of an immunogenic composition
containing an
acellular or whole cell pertussis antigen as disclosed herein can reduce the
incidence (in a
22
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
cohort of infants of vaccinated mothers) of severe disease (e.g., pneumonia
and/or
respiratory distress and failure) by at least about 50%, or at least about
60%, or by 60 to 70
%, or by at least about70%, or by at least about 80%, or by at least about 90%
compared to
infants of unvaccinated mothers. Favorably, such administration reduces the
severity of
pneumonia by at least about 50%, or at least about 60%, or by 60 to 70 %, or
by at least
about70%, or by at least about 80%, or by at least about 90% compared to
infected infants
of unvaccinated mothers. Favorably, such administration reduces the need for
hospitalization due to severe complications of pertussis in such a cohort by
at least about
50%, or at least about 60%, or by 60 to 70 %, or by at least about70%, or by
at least about
80%, or by at least about 90% compared to infected infants of unvaccinated
mothers.
[073] Typically, according to the vaccination regimens, methods, uses and
kits, the F
protein analog and pertussis antigen are administered to the pregnant female
during the
third trimester of pregnancy (gestation). The timing of maternal immunization
is designed
to allow generation of maternal antibodies and transfer of the maternal
antibodies to the
fetus. Thus, favorably sufficient time elapses between immunization and birth
to allow
optimum transfer of maternal antibodies across the placenta. Antibody transfer
starts in
humans generally at about 25 weeks of gestation, increasing up 28 weeks and
becoming
and remaining optimal from about 30 weeks of gestation. A minimum of about two
to
four weeks is believed to be needed between maternal immunization as described
herein
and birth to allow effective transfer of maternal antibodies against RSV F
protein and
pertussis antigens (e.g., comprising one or more of pertussis toxoid (PT),
filamentous
haemagglutinin (FHA), pertactin (PRN), fimbrae type 2 (FIM2), fimbrae type 3
(FIM3)
and BrkA, or whole cell pertussis antigen) to the fetus. Thus maternal
immunization can
take place any time after 25 weeks of gestation, for example at or after 25,
26, 27, 28, 29,
30, 31, 32, 33 or 34 weeks of gestation (23, 24, 25, 26, 27, 28, 29, 30, 31,
32, 33, 34, 35 or
36 weeks post-conception), or at or before 38, 37, 36, 35, 34, 33, 32, 31 or
30 weeks of
gestation (36, 35, 34, 33, 32, 31, 29 or 28 weeks post-conception). Favorably
maternal
immunization is carried out between 26 and 38 weeks, such as between 28 and 34
weeks
of gestation.
[074] Favorably maternal immunization is carried out at least two or at least
three or at
least four or at least five or at least six weeks prior to the expected date
of delivery of the
infant. Timing of administration may need to be adjusted in the case of a
pregnant female
23
CA 02879939 2015-01-23
WO 2014/024026
PCT/1B2013/001722
who is at risk of an early delivery, in order to provide sufficient time for
generation of
antibodies and transfer to the fetus.
[075] Favorably, the F protein analog and pertussis antigen(s) or formulation
thereof is
administered to the pregnant female in a single dose, during the period
described.
Maternal immunization against RSV and pertussis, as described herein, can be
considered
as a "booster" for existing maternal immunity against RSV and pertussis that
increases the
immune response against RSV and pertussis that has previously been primed,
e.g., by
natural exposure or vaccination). Thus, it is expected that only a single dose
is required.
If a second dose is administered, this is favorably also within the time
period for
administration for the first dose, favorably with a time gap between the first
and second
doses of for example one to eight weeks or two to six weeks, for example two
weeks or
four weeks or six weeks.
Administration of an F protein analog and a pertussis antigen to a pregnant
female results
in boosting maternal antibody titres, for example, increasing titres of serum
(e.g.,
neutralizing) antibodies, preferably of the Igth subclass. The increased
antibody titre in
the mother results in the passive transfer of RSV-specific and pertussis-
specific antibodies
(e.g., with neutralizing effector function) to the gestating infant across the
placenta via an
active transport mechanism mediated by Fe receptors, e.g., in the
syncytiotrophoblast of
the chorionic villi. Transport across the placenta of RSV- and pertussis-
specific Igth
antibodies resulting from the immunization methods disclosed herein is
expected to be
efficient and result in titres, which in infants born at or near term,
approach, equal or
exceed the titres in maternal circulation. For example, titres of RSV-specific
antibodies
are favorably at levels of at least 30 mcg/mL at birth. Typically, the titres
can be at or
above this level, such as at 40 mcg/mL, 50 mcg/mL, 60 mcg/mL, or even higher,
such as
75 mcg/mL, 80 mcg/mL, 90 mcg/mL, 100 mcg/mL, or even up to 120 mcg/mL or
higher
in healthy infants born at full term gestation.
[0761 Titres of pertussis- (e.g., PT-) specific antibodies are typically
measured by
ELISA in terms of ELISA units/ml (EU), as described, e.g., in Meade et al.,
"Description
and evaluation of serologic assays used in a multicenter trial of acellular
pertussis
vaccines", Pediatrics (1995) 96:570-5. Briefly, for example, microtiter plates
(e.g.,
Immulon 2 , VWR International, West Chester, PA, USA) are coated with standard
quantities of PT, FHA, FIM or PRN. Serial dilutions of serum is incubated for
24
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
approximately 2 h at 28 C and an appropriate dilution of alkaline phosphatase-
conjugated
goat anti-human IgG is added. The reaction is developed and read at 405 nm.
The lower
limit of detection of each specific antibody is determined by multiple
measurements of
serially diluted reference material for each antigen and is set at 1 ELISA
unit (EU) for PT,
FHA and PRN, and 2 EU for FIM. Favorably, following administration of an
immunogenic composition comprising a pertussis antigen as according to the
vaccination
regimen, method, use or as contained in the kits disclosed herein, pertussis-
specific
antibody titres are at levels of at least 10 EU at birth. Typically the titres
can be at or
above this level, such as at 20 EU, 30 EU, 40 EU, 50 EU, 60 EU, 70 EU, 80 EU,
90 EU or
at or above 100 EU. These values can be on an individual basis or on a
population mean
basis. Favorably, the level of antibodies observed at birth is above the
stated thresholds
and persists for several months following birth.
[077] Effector function, e.g., neutralizing capacity (neutralization titre) of
the transferred
antibodies can also be assessed, and provides a measure of functional
attribute of the
antibodies correlated with protection. For example, in the case of RSV, a
specific quantity
of a replication capable RSV virus and a defined dilution of serum are mixed
and
incubated. The virus-serum reaction mixture is then transferred onto host
cells (e.g., HEp-
2 cells) permissive for viral replication and incubated under conditions and
for a period of
time suitable for cell growth and viral replication. Non-neutralized virus is
able to infect
and replicate in the host cells. This leads to the formation of a given number
of plaque
forming units (PFU) on the cell monolayer that can be detected using a
fluorochrome-
tagged anti-RSV antibody. The neutralising titre is determined by calculating
the serum
dilution inducing a specified level of inhibition (e.g., 50% inhibition or 60%
inhibition) in
PFUs compared to a cell monolayer infected with virus alone, without serum.
For
example, the Palivizumab antibody has been shown to have a neutralization
titer (50%
effective concentration [EC50]) expressed as the antibody concentration
required to
reduce detection of RSV antigen by 50% compared with untreated virus-infected
cells of
0.65 mcg per mL (mean 0.75 0.53 mcg per mL; n=69, range 0.07-2.89 mcg per
mL) and
0.28 mcg per mL (mean 0.35 0.23 mcg per mL; n=35, range 0.03-0.88 mcg per
mL)
against clinical RSV A and RSV B isolates, respectively. Thus, in certain
embodiments,
the neutralization titre of antibodies transferred via the placenta to the
gestational infant
can be measured in the infant following birth and has (on a population median
basis) an
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
EC50 of at least about 0.50 mcg/mL (for example, at least about 0.65 mcg/mL),
or greater
for an RSV A strain and an EC50 of at least about 0.3 mcg/mL (for example, at
least about
0.35 mcg/mL), or greater for an RSV B strain. Favorably, the neutralizing
antibody titre
remains above the stated threshold for several weeks to months following
birth.
[078] Toxin-neutralizing effector function of antibodies specific for
pertussis toxin can
also be measured if desired, e.g., in a Chinese Hamster Ovary (CHO) cell
neutralization
assay, for example as described in Gillenius et al., "The standardization of
an assay for
pertussis toxin and antitoxin in microplate culture of Chinese hamster ovary
cells". J. Biol.
Stand. (1985) 13:61-66. However, neutralizing activity in this assay is less
well correlated
with protection.
[079] Optionally, according to the vaccination regimens, methods, uses and
kits
disclosed herein, in order to extend protection against RSV and pertussis
beyond the early
months of life during which the passively transferred maternal antibodies
provide
protection, the infant can be actively immunized to elicit an adaptive immune
response
specific for RSV and/or pertussis. Such active immunization of the infant can
be
accomplished by administering one or more than one composition that contains
an RSV
antigen and/or a pertussis antigen. For example, the (one or more)
composition(s) can
comprise an F protein analog, optionally formulated with an adjuvant to
enhance the
immune response elicited by the antigen. For administration to an infant that
has not been
previously exposed to RSV, the F protein analog can be formulated with an
adjuvant that
elicits immune response that is characterized by the production of T cells
that exhibit a
Thl cytokine profile (or that is characterized by a balance of T cells that
exhibit Thl and
Th2 cytokine profiles).
[080] Alternatively, rather than administering a F protein analog or other
protein subunit
vaccine, the composition that elicits an adaptive immune response to protect
against RSV
can include a live attenuated virus vaccine, or a nucleic acid that encodes
one or more
RSV antigens (such as an F antigen, a G antigen, an N antigen, or a M2
antigen, or
portions thereof). For example, the nucleic acid may be in a vector, such as a
recombinant
viral vector, for example, an adenovirus vector, an adeno-associated virus
vector, an
MVA vector, a measles vector, or the like. Exemplary viral vectors are
disclosed in
W02012 089231, which is incorporated herein for the purpose of illustrating
26
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
immunogenic compositions that contain a viral vector that encodes one or more
RSV
antigens. Alternatively, the nucleic acid can be a self replicating nucleic
acid, such as a
self-replicating RNA, e.g., in the form of a viral replicon, such as an
alphavirus replicon
(e.g., in the form of a virus replicon particle packaged with virus structural
proteins).
Examples of such self-replicating RNA replicons are described in
W02012/103361, which
is incorporated herein for the purpose of disclosing RNA replicons that encode
RSV
proteins and their formulation as immunogenic compositions.
[081] Additionally or alternatively, one or more composition(s) that contain a
pertussis
antigen can be administered to the infant. For example, the composition can
include an
acellular pertussis antigen selected from the group consisting of: pertussis
toxoid (PT),
filamentous haemagglutinin (FHA), pertactin (PRN), fimbrae type 2 (FIM2),
fimbrae type
3 (FIM3) and BrkA, or a combination thereof (e.g., PT and FHA; PT, FHA and
PRN; or
PT, FHA, PRN and either or both of FIM2 and FIM3), for example where the PT is
chemically or is genetically toxoided as described hereinbelow. Alternatively,
the
composition can include a whole cell pertussis antigen as described
hereinbelow.
[082] In certain embodiments, the RSV antigenic component (e.g., recombinant
protein,
such as a F protein analog) and the pertussis antigenic component are
coformulated into a
single immunogenic composition to be administered according to the vaccination
regimen,
method, or use described herein. Alterntively, the RSV antigenic component and
pertussis
antigenic component are formulated in two (or more) different immunogenic
compositions, which can be administered at the same or different times, e.g.,
according to
the various approved and recommended pediatric immunization schedules).
[083] When composition(s) that elicits an adaptive RSV immune response and/or
an
adaptive pertussis immune response is administered to an infant born to a
mother that
received an RSV vaccine as disclosed herein during pregnancy, the composition
can be
administered one or more times. The first administration can be at or near the
time of
birth (e.g., on the day of or the day following birth), or within 1 week of
birth or within
about 2 weeks of birth. Alternatively, the first administration can be at
about 4 weeks
after birth, about 6 weeks after birth, about 2 months after birth, about 3
months after
birth, about 4 months after birth, or later, such as about 6 months after
birth, about 9
months after birth, or about 12 months after birth. For example, in the case
of a
27
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
composition containing a pertussis antigen (e.g., Pa or Pw), it is common to
administer the
vaccine at about 2, 4 and 6 months after birth (followed by additional doses
at 12-18
months and optionally, between 4-7 yrs of age). Thus, in an embodiment, this
disclosure
provides methods for protecting an infant from disease caused by RSV and
pertussis, by
administering one or more compositions that elicits an immune response
specific for RSV
and/or pertussisto an infant born to a female to whom an immunogenic
composition
comprising an F protein analog and a pertussis antigen was administered during
the time
that she was pregnant with the infant.
RSV F PROTEIN ANALOGS
[084] The recombinant RSV antigens disclosed herein, and suitable for use in
the
vaccination regimens, methods, uses and kits, are F protein analogs derived
from (that is,
corresponding immunologically in whole or in part to) the RSV F protein. They
can
include one or more modifications that alter the structure or function of the
F protein but
retain the immunological properties of the F protein such that an immune
response
generated against an F protein analog will recognize the native F protein and
thus
recognize RSV. F protein analogs described herein are useful as immunogens.
[085] Details of the structure of the RSV F protein are provided herein with
reference to
terminology and designations widely accepted in the art, and illustrated
schematically in
FIG. 1A. With reference to the primary amino acid sequence of the F protein
polypeptide
(FIG. 1A), the following terms are utilized to describe structural features of
the F protein
analogs.
[086] The term FO refers to a full-length translated F protein precursor. The
FO
polypeptide can be subdivided into an F2 domain and an Fl domain separated by
an
intervening peptide, designated pep27. During maturation, the FO polypeptide
undergoes
proteolytic cleavage at two furin sites situated between F2 and Fl and
flanking pep27. For
purpose of the ensuing discussion, an F2 domain includes at least a portion,
and as much
as all, of amino acids 1-109, and a soluble portion of an Fl domain includes
at least a
portion, and up to all, of amino acids 137-526 of the F protein. As indicated
above, these
amino acid positions (and all subsequent amino acid positions designated
herein) are given
in reference to the exemplary F protein precursor polypeptide (FO) of SEQ ID
NO:2.
28
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
[087] In nature, the RSV F protein is expressed as a single polypeptide
precursor 574
amino acids in length, designated FO. In vivo, FO oligomerizes in the
endoplasmic
reticulum and is proteolytically processed by a furin protease at two
conserved furin
consensus sequences (furin cleavage sites), RARRio9 (SEQ ID NO:15) and RKRR136
(SEQ ID NO:16) to generate an oligomer consisting of two disulfide-linked
fragments.
The smaller of these fragments is termed F2 and originates from the N-terminal
portion of
the FO precursor. It will be recognized by those of skill in the art that the
abbreviations
FO, Fl and F2 are commonly designated Fo, F1 and F2 in the scientific
literature. The
larger, C-terminal F 1 fragment anchors the F protein in the membrane via a
sequence of
hydrophobic amino acids, which are adjacent to a 24 amino acid cytoplasmic
tail. Three
F2-F1 dimers associate to form a mature F protein, which adopts a metastable
prefusogenic ("prefusion") conformation that is triggered to undergo a
conformational
change upon contact with a target cell membrane. This conformational change
exposes a
hydrophobic sequence, known as the fusion peptide, which associates with the
host cell
membrane and promotes fusion of the membrane of the virus, or an infected
cell, with the
target cell membrane.
[088] The Fl fragment contains at least two heptad repeat domains, designated
HRA
and HRB, and situated in proximity to the fusion peptide and transmembrane
anchor
domains, respectively. In the prefusion conformation, the F2-F1 dimer forms a
globular
head and stalk structure, in which the HRA domains are in a segmented
(extended)
conformation in the globular head. In contrast, the HRB domains form a three-
stranded
coiled coil stalk extending from the head region. During transition from the
prefusion to
the postfusion conformations, the HRA domains collapse and are brought into
proximity
to the HRB domains to form an anti-parallel six helix bundle. In the
postfusion state the
fusion peptide and transmembrane domains are juxtaposed to facilitate membrane
fusion.
[089] Although the conformational description provided above is based on
molecular
modeling of crystallographic data, the structural distinctions between the
prefusion and
postfusion conformations can be monitored without resort to crystallography.
For
example, electron micrography can be used to distinguish between the prefusion
and
postfusion (alternatively designated prefusogenic and fusogenic)
conformations, as
demonstrated by Calder et al., Virology, 271:122-131 (2000) and Morton et al.,
Virology,
311:275-288, which are incorporated herein by reference for the purpose of
their
29
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
technological teachings. The prefusion conformation can also be distinguished
from the
fusogenic (postfusion) conformation by liposome association assays as
described by
Connolly et al., Proc. NatL Acad. Sci. USA, 103:17903-17908 (2006), which is
also
incorporated herein by reference for the purpose of its technological
teachings.
Additionally, prefusion and fusogenic conformations can be distinguished using
antibodies
that specifically recognize conformation epitopes present on one or the other
of the
prefusion or fusogenic form of the RSV F protein, but not on the other form.
Such
conformation epitopes can be due to preferential exposure of an antigenic
determinant on
the surface of the molecule. Alternatively, conformational epitopes can arise
from the
juxtaposition of amino acids that are non-contiguous in the linear
polypeptide.
[090] Typically, the F protein analogs (PreF, PostF, etc.) analogs lack a
transmembrane
domain and cytoplasmic tail, and can also be referred to as an F protein
ectodomain or
soluble F protein ectodomain.
[091] F protein analogs include an F protein polypeptide, which has been
modified to
stabilize the prefusion conformation of the F protein, that is, the
conformation of the
mature assembled F protein prior to fusion with the host cell membrane. These
F protein
analogs are designated "PreF analogs", "PreF" or "PreF antigens", for purpose
of clarity
and simplicity, and are generally soluble. The PreF analogs disclosed herein
are
predicated on the discovery that soluble F protein analogs that have been
modified by the
incorporation of a heterologous trimerization domain exhibit improved
immunogenic
characteristics, and are safe and highly protective when administered to a
subject in vivo.
Exemplary PreF antigens are described in W02010149745, herein incorporated by
reference in its entirety for the purpose of providing examples of PreF
antigens.
[092] F protein analogs also include an F protein polypeptide which has the
conformation of the postfusion F protein and which may be referred to as a
PostF antigen
or postfusion antigen. PostF analogs are described in W02011008974,
incorporated
herein by reference. The PostF antigen contains at least one modification to
alter the
structure or function of the native postfusion F protein.
[093] In a preferred embodiment, the composition for maternal immunization to
protect
an infant against RSV disease includes an RSV PreF analog antigen that is
stabilized in the
prefusion conformation found on the virus prior to cellular adhesion and
fusion. A
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
schematic illustration of exemplary PreF analogs is provided in FIG. 1B. It
will be
understood by those of skill in the art that any RSV F protein can be modified
to stabilize
the prefusion conformation according to the teachings provided herein.
Therefore, to
facilitate understanding of the principles guiding production of PreF (as well
as PostF, and
other conformational) analogs, individual structural components will be
indicated with
reference to an exemplary F protein, the polynucleotide and amino acid
sequence of which
are provided in SEQ ID NOs:1 and 2, respectively. Similarly, where applicable,
G protein
antigens are described in reference to an exemplary G protein, the
polynucleotide and
amino acid sequences of which are provided in SEQ ID NOs:3 and 4,
respectively.
[094] The PreF analogs disclosed herein are designed to stabilize and maintain
the
prefusion conformation of the RSV F protein, such that in a population of
expressed
protein, a substantial portion of the population of expressed protein is in
the prefusogenic
(prefusion) conformation (e.g., as predicted by structural and/or
thermodynamic modeling
or as assessed by one or more of the methods disclosed above). Stabilizing
modifications
are introduced into a native (or synthetic) F protein, such as the exemplary F
protein of
SEQ ID NO:2, such that the major immunogenic epitopes of the prefusion
conformation
of the F protein are maintained following introduction of the PreF analog into
a cellular or
extracellular environment (for example, in vivo, e.g., following
administration to a
subject).
[095] First, a heterologous stabilizing domain can be placed at the C-terminal
end of the
construct in order to replace the membrane anchoring domain of the FO
polypeptide. This
stabilizing domain is predicted to compensate for the HRB instability, helping
to stabilize
the -prefusion conformer. In exemplary embodiments, the heterologous
stabilizing
domain is a protein multimerization domain. One particularly favorable example
of such a
protein multimerization domain is a trimerization domain. Exemplary
trimerization
domains fold into a coiled-coil that promotes assembly into trimers of
multiple
polypeptides having such coiled-coil domains. Examples of trimerization
domains include
trimerization domains from influenza hemagglutinin, SARS spike, HIV gp41,
modified
GCN4, bacteriophage T4 fibritin and ATCase. One favorable example of a
trimerization
domain is an isoleucine zipper. An exemplary isoleucine zipper domain is the
engineered
yeast GCN4 isoleucine variant described by Harbury etal. Science 262:1401-1407
(1993).
The sequence of one suitable isoleucine zipper domain is represented by SEQ ID
NO:11,
31
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
although variants of this sequence that retain the ability to form a coiled-
coil stabilizing
domain are equally suitable. Alternative stabilizing coiled coil trimerization
domains
include: TRAF2 (GENBANK Accession No. Q12933 [gi:23503103]; amino acids 299-
348); Tlu-ombospondin 1 (Accession No. P07996 [gi:135717]; amino acids 291-
314);
Matrilin-4 (Accession No. 095460 [gi:14548117]; amino acids 594-618; CMP
(matrilin-
1) (Accession No. NP 002370 [gi:4505111]; amino acids 463-496; HSF1 (Accession
No.
AAX42211 [gi:61362386]; amino acids 165-191; and Cubilin (Accession No.
NP_001072
[gi:4557503]; amino acids 104-138. It is expected that a suitable
trimerization domain
results in the assembly of a substantial portion of the expressed protein into
trimers. For
example, at least 50% of a recombinant PreF polypeptide having a trimerization
domain
will assemble into a trimer (e.g., as assessed by AFF-MALS). Typically, at
least 60%,
more favorably at least 70%, and most desirably at least about 75% or more of
the
expressed polypeptide exists as a trimer.
[096] Another example of a stabilizing mutation is the addition or
substitution of a
hydrophilic amino acid into a hydrophobic domain of the F protein. Typically,
a charged
amino acid, such as lysine, will be added or substituted for a neutral
residue, such as
leucine, in the hydrophobic region. For example, a hydrophilic amino acid can
be added
to, or substituted for, a hydrophobic or neutral amino acid within the HRB
coiled-coil
domain of the F protein extracellular domain. By way of example, a charged
amino acid
residue, such as lysine, can be substituted for the leucine present at
position 512 the F
protein (relative to the native FO polypeptide; L482K of the exemplary PreF
analog
polypeptide of SEQ ID N0:6). Alternatively, or in addition, a hydrophilic
amino acid can
be added to, or substituted for, a hydrophobic or neutral amino acid within
the HRA
domain of the F protein. For example, one or more charged amino acids, such as
lysine,
can be inserted at or near position 105-106 (e.g., following the amino acid
corresponding
to residue 105 of reference SEQ ID N0:2, such as between amino acids 105 and
106) of
the PreF analog). Optionally, hydrophilic amino acids can be added or
substituted in both
the HRA and FIRB domains. Alternatively, one or more hydrophobic residues can
be
deleted, so long as the overall conformation of the PreF analog is not
adversely impacted.
[097] Secondly, pep27 can be removed. Analysis of a structural model of the
RSV F
protein in the prefusion state suggests that pep27 creates a large
unconstrained loop
between Fl and F2. This loop does not contribute to stabilization of the
prefusion state,
32
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
and is removed following cleavage of the native protein by furin. Thus, pep27
can also be
removed from embodiments that involve a postfusion (or other) conformational
analog.
[098] Third, one or both furin cleavage motifs can be deleted (from between
the F2 and
Fl domains in the native FO protein). One or both furin recognition sites,
located at
positions 105-109 or 106-109 and at positions 133-136 can be eliminated by
deleting or
substituting one or more amino acid of the furin recognition sites, for
example deleting
one or more amino acids or substituting one or more amino acids or a
combination of one
or more substitutions or deletions, or modifying such that the protease is
incapable of
cleaving the PreF (or other F protein analog) polypeptide into its constituent
domains.
Optionally, the intervening pep27 peptide can also be removed or substituted,
e.g., by a
linker peptide. Additionally, or optionally, a non-furin cleavage site (e.g.,
a
metalloproteinase site at positions 112-113) in proximity to the fusion
peptide can be
removed or substituted.
[099] Thus, an F protein analog for use in the methods and uses according to
the
invention can be obtained which is an uncleaved ectodomain having one or more
altered
furin cleavage sites. Such F protein analog polypeptides are produced
recombinantly in a
host cell which secretes them uncleaved at position from amino acid 101 to
161, e.g. not
cleaved at the furin cleavage sites at positions 105-109 and 131-136. In
particular
embodiments, the substitution K131Q, the deletion of the amino acids at
positions 131-
134, or the substitutions K131Q or R133Q or R135Q or R136Q, are used to
inhibit
cleavage at 136/137.
[0100] In an exemplary design, the fusion peptide is not cleaved from F2,
preventing
release from the globular head of the prefusion conformer and accessibility to
nearby
membranes. Interaction between the fusion peptide and the membrane interface
is
predicted to be a major issue in the prefusion state instability. During the
fusion process,
interaction between the fusion peptide and the target membrane results in the
exposure of
the fusion peptide from within the globular head structure, enhancing
instability of the
prefusion state and folding into post-fusion conformer. This conformation
change enables
the process of membrane fusion. Removal of one or both of the furin cleavage
sites is
predicted to prevent membrane accessibility to the N-terminal part of the
fusion peptide,
stabilizing the prefusion state. Thus, in exemplary embodiments disclosed
herein, removal
33
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
of the furin cleavage motifs results in a PreF analog that comprises an intact
fusion
peptide, which is not cleaved by furin during or following processing and
assembly.
[0101] Optionally, at least one non-furin cleavage site can also be removed,
for example
by substitution of one or more amino acids. For example, experimental evidence
suggests
that under conditions conducive to cleavage by certain metalloproteinases, the
F protein
analog can be cleaved in the vicinity of amino acids 110-118 (for example,
with cleavage
occurring between amino acids 112 and 113 of the F protein analog; between a
leucine at
position 142 and glycine at position 143 of the reference F protein
polypeptide of SEQ ID
NO:2). Accordingly, modification of one or more amino acids within this region
can
reduce cleavage of the F protein analog. For example, the leucine at position
112 can be
substituted with a different amino acid, such as isoleucine, glutamine or
tryptophan (as
shown in the exemplary embodiment of SEQ ID NO:20). Alternatively or
additionally,
the glycine at position 113 can be substituted by a serine or alanine. In
further
embodiments the F prtein analogs further contain altered trypsin cleavage
sites, and F
protein analogs are not cleaved by tryp sin at a site between amino acid 101
and 161.
[0102] Optionally, a F protein analog can include one or more modifications
that alters
the glycosylation pattern or status (e.g., by increasing or decreasing the
proportion of
molecules glycosylated at one or more of the glycosylation sites present in a
native F
protein polypeptide). For example, the native F protein polypeptide of SEQ ID
NO:2 is
predicted to be glycosylated at amino acid positions 27, 70 and 500
(corresponding to
positions 27, 70 and 470 of the exemplary PreF analog of SEQ ID NO:6). In an
embodiment, a modification is introduced in the vicinity of the glycosylation
site at amino
acid position 500 (designated N470). For example, the glycosylation site can
be removed
by substituting an amino acid, such as glutamine (Q) in place of the
asparagine at position
500 (of the reference sequence, which corresponds by alignment to position 470
of the
exemplary PreF analog). Favorably, a modification that increases glycosylation
efficiency
at this glycosylation site is introduced. Examples of suitable modifications
include at
positions 500-502, the following amino acid sequences: NGS; NKS; NGT; NKT.
Interestingly, it has been found that modifications of this glycosylation site
that result in
increased glycosylation also result in substantially increased PreF
production. Thus, in
certain embodiments, the PreF analogs have a modified glycosylation site at
the position
corresponding to amino acid 500 of the reference PreF sequence (SEQ ID NO:2),
e.g., at
34
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
position 470 of the PreF analog exemplified by SEQ ID NO:6). Suitable,
modifications
include the sequences: NGS; NKS; NGT; NKT at amino acids corresponding to
positions
500-502 of the reference F protein polypeptide sequence. The amino acid of an
exemplary
embodiment that includes an "NGT" modification is provided in SEQ ID NO:18.
One of
skill in the art can easily determine similar modifications for corresponding
NGS, NKS,
and NKT modifications. Such modifications are favorably combined with any of
the
stabilizing mutations disclosed herein (e.g., a heterologous coiled-coil, such
as an
isoleucine zipper, domain and/or a modification in a hydrophobic region,
and/or removal
of pep27, and/or removal of a furin cleavage site, and/or removal of a non-
furin cleavage
site, and/or removal of a non-furin cleavage site). For example, in one
specific
embodiment, the F protein analog includes a substitution that eliminates a non-
furin
cleavage site and a modification that increases glycosylation. An exemplary
PreF analog
sequence is provided in SEQ ID NO:22 (which exemplary embodiment includes an
"NGT" modification and the substitution of glutamine in the place of leucine
at position
112). For example, in certain exemplary embodiments, the glycosylation
modified PreF
analogs are selected from the group of: a) a polypeptide comprising or
consisting of SEQ
ID NO:22; b) a polypeptide encoded by SEQ ID NO:21 or by a polynucleotide
sequence
that hybridizes under stringent conditions over substantially its entire
length to SEQ ID
NO:21; c) a polypeptide with at least 95% sequence identity to SEQ ID
NO:22.
[0103] More generally, any one of the stabilizing modifications disclosed
herein, e.g.,
addition of a heterologous stabilizing domain, such as a coiled-coil (for
example, an
isoleucine zipper domain), preferably situated at the C-terminal end of the
soluble F
protein analog; modification of a residue, such as leucine to lysine, in the
hydrophobic
HRB domain; removal of pep27; removal of one or both furin cleavage motifs;
removal of
a non-furin cleavage site such as a trypsin cleavage site; and/or modification
of a
glycosylation site can be employed in combination with any one or more (or up
to all-in
any desired combination) of the other stabilizing modifications. For example,
a
heterologous coiled-coil (or other heterologous stabilizing domain) can be
utilized alone or
in combination with any of: a modification in a hydrophobic region, and/or
removal of
pep27, and/or removal of a furin cleavage site, and/or removal of a non-furin
cleavage site,
and/or removal of a non-furin cleavage site. In certain specific embodiments,
the F
protein analog, such as the PreF analog, includes a C-terminal coiled-coil
(isoleucine
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
zipper) domain, a stabilizing substitution in the HRB hydrophobic domain, and
removal of
one or both furin cleavage sites. Such an embodiment includes an intact fusion
peptide
that is not removed by furin cleavage. In one specific embodiment, the F
protein analog
also includes a modified glycosylation site at amino acid position 500.
[0104] The F protein analog for the methods and uses described herein can be
produced
by a method which comprises providing a biological material containing the F
protein
analog (e.g., PreF analog, PostF analog or uncleaved F protein ectodomain,
etc.) and
purifying the analog polypeptide monomers or multimers (e.g., trimers) or a
mixture
thereof from the biological material.
[0105] The F protein analog can be in the form of polypeptide monomers or
trimers, or a
mixture of monomers and trimers which may exist in equilibrium. The presence
of a
single form may provide advantages such as a more predictable immune response
and
better stability.
[0106] Thus, in an embodiment, the F protein analog for use in the invention
is a purified
F protein analog, which may be in the form of monomers or trimers or a mixture
of
monomers and trimers, substantially free of lipids and lipoproteins.
[0107] The F protein polypeptide can be selected from any F protein of an RSV
A or
RSV B strain, or from variants thereof (as defined above). In certain
exemplary
embodiments, the F protein polypeptide is the F protein represented by SEQ ID
NO:2. To
facilitate understanding of this disclosure, all amino acid residue positions,
regardless of
strain, are given with respect to (that is, the amino acid residue position
corresponds to)
the amino acid position of the exemplary F protein. Comparable amino acid
positions of
any other RSV A or B strain can be determined easily by those of ordinary
skill in the art
by aligning the amino acid sequences of the selected RSV strain with that of
the
exemplary sequence using readily available and well-known alignment algorithms
(such
as BLAST, e.g., using default parameters). Numerous additional examples of F
protein
polypeptides from different RSV strains are disclosed in W02008114149 (which
is
incorporated herein by reference for the purpose of providing additional
examples of RSV
F and G protein sequences). Additional variants can arise through genetic
drift, or can be
produced artificially using site directed or random mutagenesis, or by
recombination of
two or more preexisting variants. Such additional variants are also suitable
in the context
36
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
of the F protein analogs utilized in the context of the immunization methods
disclosed
herein.
[0108] In alternative embodiments useful in the methods and uses described
herein the
recombinant RSV protein is an F protein analog as described in W02011008974,
incorporated herein by reference for the purpose of describing additional F
protein
analogs, see for example F protein analogs in Figure 1 of W02011008974 and
also
described in Example 1 of W02011008974.
[0109] In selecting F2 and Fl domains of the F protein, one of skill in the
art will
recognize that it is not strictly necessary to include the entire F2 and/or Fl
domain.
Typically, conformational considerations are of importance when selecting a
subsequence
(or fragment) of the F2 domain. Thus, the F2 domain typically includes a
portion of the
F2 domain that facilitates assembly and stability of the polypeptide. In
certain exemplary
variants, the F2 domain includes amino acids 26-105. However, variants having
minor
modifications in length (by addition, or deletion of one or more amino acids)
are also
possible.
[0110] Typically, at least a subsequence (or fragment) of the Fl domain is
selected and
designed to maintain a stable conformation that includes immunodominant
epitopes of the
F protein. For example, it is generally desirable to select a subsequence of
the Fl
polypeptide domain that includes epitopes recognized by neutralizing
antibodies in the
regions of amino acids 262-275 (palivizumab neutralization) and 423-436
(Centocor's
ch101F MAb). Additionally, desirable to include T cell epitopes, e.g., in the
region of
animo acids 328-355. Most commonly, as a single contiguous portion of the Fl
subunit
(e.g., spanning amino acids 262-436) but epitopes could be retained in a
synthetic
sequence that includes these immunodominant epitopes as discontinuous elements
assembled in a stable conformation. Thus, an F 1 domain polypeptide comprises
at least
about amino acids 262-436 of an RSV F protein polypeptide. In one non-limiting
example
provided herein, the Fl domain comprises amino acids 137 to 516 of a native F
protein
polypeptide. One of skill in the art will recognize that additional shorter
subsequences can
be used at the discretion of the practitioner.
[0111] When selecting a subsequence of the F2 or Fl domain (e.g., as discussed
below
with respect to the G protein component of certain PreF-G analogs), in
addition to
37
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
conformational consideration, it can be desirable to choose sequences (e.g.,
variants,
subsequences, and the like) based on the inclusion of additional immunogenic
epitopes.
For example, additional T cell epitopes can be identified using anchor motifs
or other
methods, such as neural net or polynomial determinations, known in the art,
see, e.g.,
RANKPEP (available on the world wide web at:
mif.dfci.harvard.edu/Tools/rankpep.html); ProPredI (available on the world
wide web at:
imtech.res.iniraghava/propredI/index.html); Bimas (available on the world wide
web at:
www-bimas.dcrt.nih.gov/molbi/hla_bind/index.html); and SYFPEITH (available on
the
world wide web at: syfpeithi.bmi-
heidelberg.com/scripts/MHCServer.d11/home.htm). For
example, algorithms are used to determine the "binding threshold" of peptides,
and to
select those with scores that give them a high probability of MHC or antibody
binding at a
certain affinity. The algorithms are based either on the effects on MHC
binding of a
particular amino acid at a particular position, the effects on antibody
binding of a
particular amino acid at a particular position, or the effects on binding of a
particular
substitution in a motif-containing peptide. Within the context of an
immunogenic peptide,
a "conserved residue" is one which appears in a significantly higher frequency
than would
be expected by random distribution at a particular position in a peptide.
Anchor residues
are conserved residues that provide a contact point with the MHC molecule. T
cell
epitopes identified by such predictive methods can be confirmed by measuring
their
binding to a specific MHC protein and by their ability to stimulate T cells
when presented
in the context of the MHC protein.
[0112] Favorably, the an F protein analog, for example a PreF analogs
(including PreF-G
analogs as discussed below), a Post F analog, or other conformational analog,
include a
signal peptide corresponding to the expression system, for example, a
mammalian or viral
signal peptide, such as an RSV FO native signal sequence (e.g., amino acids 1-
25 of SEQ
ID NO:2 or amino acids 1-25 of SEQ ID NO:6). Typically, the signal peptide is
selected
to be compatible with the cells selected for recombinant expression. For
example, a signal
peptide (such as a baculovirus signal peptide, or the melittin signal peptide,
can be
substituted for expression, in insect cells. Suitable plant signal peptides
are known in the
art, if a plant expression system is preferred. Numerous exemplary signal
peptides are
known in the art, (see, e.g., see Zhang & Henzel, Protein Sc., 13:2819-2824
(2004), which
describes numerous human signal peptides) and are catalogued, e.g., in the
SPdb signal
38
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
peptide database, which includes signal sequences of archaea, prokaryotes and
eukaryotes
(http://proline.bic.nus.edu.sg/spdb/). Optionally, any of the preceding
antigens can include
an additional sequence or tag, such as a His-tag to facilitate purification.
[0113] Optionally, the F protein analog (for example, the PreF or Post F or
other analog)
can include additional immunogenic components. In certain particularly
favorable
embodiments, the F protein analog includes an RSV G protein antigenic
component.
Exemplary chimeric proteins having a PreF and G component include the
following
PreF_V1 (represented by SEQ ID NOs:7 and 8) and PreF _\72 (represented by SEQ
ID
NOs:9 and 10).
[0114] In the PreF-G analogs , an antigenic portion of the G protein (e.g., a
truncated G
protein, such as amino acid residues 149-229) is added at the C-terminal end
of the
construct. Typically, the G protein component is joined to the F protein
component via a
flexible linker sequence. For example, in the exemplary PreF yl design, the G
protein is
joined to the PreF component by a -GGSGGSGGS- linker (SEQ ID NO:14). In the
PreF_V2 design, the linker is shorter. Instead of having the -GGSGGSGGS-
linker (SEQ
ID NO:14), PreF_V2 has 2 glycines (-GG-) for linker.
[0115] Where present, the G protein polypeptide domain can include all or part
of a G
protein selected from any RSV A or RSV B strain. In certain exemplary
embodiments, the
G protein is (or is 95% identical to) the G protein represented by SEQ ID
NO:4.
Additional examples of suitable G protein sequences can be found in
W02008114149
(which is incorporated herein by reference).
[0116] The G protein polypeptide component is selected to include at least a
subsequence
(or fragment) of the G protein that retains the immunodominant T cell
epitope(s), e.g., in
the region of amino acids 183-197, such as fragments of the G protein that
include amino
acids 151-229, 149-229, or 128-229 of a native G protein. In one exemplary
embodiment,
the G protein polypeptide is a subsequence (or fragment) of a native G protein
polypeptide
that includes all or part of amino acid residues 149 to 229 of a native G
protein
polypeptide. One of skill in the art will readily appreciate that longer or
shorter portions
of the G protein can also be used, so long as the portion selected does not
conformationally destabilize or disrupt expression, folding or processing of
the F protein
analog. Optionally, the G protein domain includes an amino acid substitution
at position
39
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
191, which has previously been shown to be involved in reducing and/or
preventing
enhanced disease characterized by eosinophilia associated with formalin
inactivated RSV
vaccines. A thorough description of the attributes of naturally occurring and
substituted
(N191A) G proteins can be found, e.g., in US Patent Publication No.
2005/0042230,
which is incorporated herein by reference.
[0117] Alternatively, the F protein analog can be formulated in an immunogenic
composition that also contains a second polypeptide that includes a G protein
component.
The G protein component typically includes at least amino acids 149-229 of a G
protein.
Although smaller portions of the G protein can be used, such fragments should
include, at
a minimum, the immunological dominant epitope of amino acids 184-198.
Alternatively,
the G protein can include a larger portion of the G protein, such as amino
acids 128-229 or
130-230, optionally as an element of a larger protein, such as a full-length G
protein, or a
chimeric polypeptide.
[0118] For example, with respect to selection of sequences corresponding to
naturally
occurring strains, one or more of the domains can correspond in sequence to an
RSV A or
B strain, such as the common laboratory isolates designated A2 or Long, or any
other
naturally occurring strain or isolate. Numerous strains of RSV have been
isolated to date.
Exemplary strains indicated by GenBank and/or EMBL Accession number can be
found in
W02008114149, which is incorporated herein by reference for the purpose of
disclosing
the nucleic acid and polypeptide sequences of RSV F suitable for use in F
protein analogs
disclosed herein. Additional strains of RSV are likely to be isolated, and are
encompassed
within the genus of RSV. Similarly, the genus of RSV encompasses variants
arising from
naturally occurring (e.g., previously or subsequently identified strains) by
genetic drift,
and/or recombination.
[0119] In addition to such naturally occurring and isolated variants,
engineered variants
that share sequence similarity with the aforementioned sequences can also be
employed in
the context of F protein analogs, including PreF, PostF or other analogs
(including F-G)
analogs. It will be understood by those of skill in the art, that the
similarity between F
protein analog polypeptide (and polynucleotide sequences as described below),
as for
polypeptide (and nucleotide sequences in general), can be expressed in terms
of the
similarity between the sequences, otherwise referred to as sequence identity.
Sequence
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
identity is frequently measured in terms of percentage identity (or
similarity); the higher
the percentage, the more similar are the primary structures of the two
sequences. In
general, the more similar the primary structures of two amino acid (or
polynucleotide)
sequences, the more similar are the higher order structures resulting from
folding and
assembly. Variants of an F protein, polypeptide (and polynucleotide) sequences
typically
have one or a small number of amino acid deletions, additions or substitutions
but will
nonetheless share a very high percentage of their amino acid, and generally
their
polynucleotide sequence. More importantly, the variants retain the structural
and, thus,
conformational attributes of the reference sequences disclosed herein.
[0120] Methods of determining sequence identity are well known in the art, and
are
applicable to F protein analog polypeptides, as well as the nucleic acids that
encode them
(e.g., as decribed below). Various programs and alignment algorithms are
described in:
Smith and Waterman, Adv. AppL Math. 2:482, 1981; Needleman and Wunsch, I Mol.
Biol. 48:443, 1970; Higgins and Sharp, Gene 73:237, 1988; Higgins and Sharp,
CABIOS
5:151, 1989; Corpet et al., Nucleic Acids Research 16:10881, 1988; and Pearson
and
Lipman, Proc. Natl. Acad. Sci. USA 85:2444, 1988. Altschul et al., Nature
Genet.
6:119, 1994, presents a detailed consideration of sequence alignment methods
and
homology calculations. The NCBI Basic Local Alignment Search Tool (BLAST)
(Altschul et al., I MoL Biol. 215:403, 1990) is available from several
sources, including
the National Center for Biotechnology Information (NCBI, Bethesda, MD) and on
the
internet, for use in connection with the sequence analysis programs blastp,
blastn, blastx,
tblastn and tblastx. A description of how to determine sequence identity using
this
program is available on the NCBI website on the internet.
[0121] In some instances, the F protein analog has one or more amino acid
modifications
relative to the amino acid sequence of the naturally occurring strain from
which it is
derived (e.g., in addition to the aforementioned stabilizing modifications).
Such
differences can be an addition, deletion or substitution of one or more amino
acids. A
variant typically differs by no more than about 1%, or 2%, or 5%, or 10%, or
15%, or 20%
of the amino acid residues. For example, a variant F protein analog, e.g.,
PreF or PostF or
other analog polypeptide sequence can include 1, or 2, or up to 5, or up to
about 10, or up
to about 15, or up to about 50, or up to about 100 amino acid differences as
compared to
the relevant portion of a reference F protein sequence (for example, the PreF
analog
41
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
polypeptide sequences of SEQ ID NOs:6, 8, 10, 18, 20 and/or 22. Thus, a
variant in the
context of an RSV F or G protein, or F protein analog, typically shares at
least 80%, or
85%, more commonly, at least about 90% or more, such as 95%, or even 98% or
99%
sequence identity with a reference protein, e.g., in the case of a PreF
analog: the reference
sequences illustrated in SEQ ID NO:2, 4, 6, 8, 10, 18, 20 and/or 22, or any of
the
exemplary PreF analogs disclosed herein. Additional variants included as a
feature of this
disclosure are F protein analogs that include all or part of a nucleotide or
amino acid
sequence selected from the naturally occurring variants disclosed in
W02008114149.
Additional variants can arise through genetic drift, or can be produced
artificially using
site directed or random mutagenesis, or by recombination of two or more
preexisting
variants. Such additional variants are also suitable in the context of the F
protein analog
antigens disclosed herein. For example, the modification can be a substitution
of one or
more amino acids (such as two amino acids, three amino acids, four amino
acids, five
amino acids, up to about ten amino acids, or more) that do not alter the
conformation or
immunogenic epitopes of the resulting F protein analog.
[0122] Alternatively or additionally, the modification can include a deletion
of one or
more amino acids and/or an addition of one or more amino acids. Indeed, if
desired, one
or more of the polypeptide domains can be a synthetic polypeptide that does
not
correspond to any single strain, but includes component subsequences from
multiple
strains, or even from a consensus sequence deduced by aligning multiple
strains of RSV
virus polypeptides. In certain embodiments, one or more of the polypeptide
domains is
modified by the addition of an amino acid sequence that constitutes a tag,
which facilitates
subsequent processing or purification. Such a tag can be an antigenic or
epitope tag, an
enzymatic tag or a polyhistidine tag. Typically the tag is situated at one or
the other end of
the protein, such as at the C-terminus or N-terminus of the antigen or fusion
protein.
[0123] The F protein analogs (and also where applicable, G antigens) disclosed
herein
can be produced using well established procedures for the expression and
purification of
recombinant proteins.
[0124] In brief, recombinant nucleic acids that encode the F protein analogs
are
introduced into host cells by any of a variety of well-known procedures, such
as
electroporation, liposome mediated transfection, Calcium phosphate
precipitation,
42
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
infection, transfection and the like, depending on the selection of vectors
and host cells.
Favorable host cells include prokaryotic (i.e., bacterial) host cells, such as
E. coli, as well
as numerous eukaryotic host cells, including fungal (e.g., yeast, such as
Saccharornyces
cerevisiae and Picchia pastoris) cells, insect cells, plant cells, and
mammalian cells (such
as 3T3, COS, CHO, BHK, HEK 293) or Bowes melanoma cells. Following expression
in
a selected host cell, the recombinant F protein analogs can be isolated and/or
purified
according to procedures well-known in the art. Exemplary expression methods,
as well as
nucleic acids that encode PreF analogs (including PreF-G analogs) are provided
in
W02010149745, which is incorporated herein for the purpose of providing
suitable
methods for the expression and purification of F protein analogs.
PERTUSSIS ANTIGENS
[0125] In the context of the vaccination regimens, methods, uses and kits
disclosed
herein, the at least one B. pertussis antigen can be at least one acellular
pertussis (Pa)
protein selected from the group consisting of: pertussis toxoid (PT),
filamentous
haemagglutinin (FHA), pertactin (PRN), fimbrae type 2 (FIM2), fimbrae type 3
(FIM3).
Such acellular antigens are well known in the art. The antigens are partially
or highly
purified.
[0126] PT can be produced in a variety of ways, for instance by purification
of the toxin
from a culture of B. pertussis followed by chemical detoxification (for
example as
described in W091/12020, incorporated herein by reference), or alternatively
by
purification of a genetically-detoxified analog of PT (for example, as
described in the
following, incorporated herein by reference for the purpose of disclosing
contemplated
genetic modifications of PT: EP306318, EP322533, EP396964, EP322115,
EP275689). In
a particular embodiment, the PT is genetically detoxified. More particularly,
the
genetically-detoxified PT carries one or both of the following substitutions:
R9K and
E129G.
[0127] The pertussis antigenic component can include any 1, 2, 3, 4 or 5 of
the acellular
pertussis antigens PT, FHA, PRN, FIM2 and FIM3, including combinations
thereof. More
particularly, the combinations can include specifically (and without
limitation): PT and
FHA; PT, FHA and PRN; PT, FHA, PRN and FIM2; PT, FHA, PRN and FIM3; and PT,
FHA, PRN, FIM2 and FIM3.
43
CA 02879939 2015-01-23
WO 2014/024026
PCT/1B2013/001722
[0128] In a particular embodiment, PT is used at an amount of 2-5Oug (for
example
exactly or approximately 2.5 or 3.2ug per dose), 5-4Oug (for example exactly
or
approximately 5 or 8ug per dose) or 10-3Oug (for example exactly or
approximately 20 or
25ug per dose).
[0129] In a particular embodiment, FHA is used at an amount of 2-5Oug (for
example
exactly or approximately 2.5 or 34.4ug per dose), 5-4Oug (for example exactly
or
approximately 5 or 8ug per dose) or 10-3Oug (for example exactly or
approximately 20 or
25ug per dose).
[0130] In a particular embodiment, PRN is used at an amount of 0.5-2Oug, 0.8-
15ug (for
example exactly or approximately 0.8 or 1.6ug per dose) or 2-1Oug (for example
exactly
or approximately 2.5 or 3 or 8ug per dose).
[0131] In a particular embodiment, FIM2 and/or FIM3 are used at a total amount
of 0.5-
1Oug (for example exactly or approximately 0.8 or 5ug per dose).
[0132] In a particular embodiment, the pertussis antigenic components include
PT and
FHA at equivalent amounts per dose, being either exactly or approximately 8 or
20 or
25ug. Alternatively, the pertussis antigenic components include PT and FHA at
exactly or
approximately 5 and 2.5ug respectively, or exactly or approximately 3.2 and
34.4ug. In a
further embodiment, the immunogenic composition comprises PT, FHA and PRN at
the
respective exact or approximate amounts per dose: 25:5:8ug; 8:8:2.5ug;
20:20:3ug;
2.5:5:3ug; 5:2.5:2.5ug; or 3.2:34.4:1.6ug.
[0133] Alternatively, or in combination with any of the above-discussed
acellular
pertussis antigens, the pertussis antigenic components can comprise an antigen
derived
from the B. pertussis 'BrkA' antigen (as disclosed in W02005/032584, and Man
et al
(2008), Vaccine, 26(34):4306-4311, incorporated herein by reference).
[0134] In a further embodiment, the at least one Pa antigen can take the form
of an outer
membrane vesicle (OMV) obtained from B. pertussis, as disclosed in Roberts et
al (2008),
Vaccine, 26:4639-4646, incorporated herein by reference. In particular, such
OMV can be
derived from a recombinant B. pertussis strain expressing a lipid A-modifying
enzyme,
such as a 3-0-deacylase, for example PagL (Asensio et al (2011), Vaccine,
29:1649-1656,
44
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
incorporated herein by reference).
[0135] In an alternative embodiment, the at least one B. pertussis antigen is
whole cell
pertussis (Pw) vaccine, such Pw vaccines being well known in the art. Pw can
be
inactivated by several known methods, including mercury-free methods. Such
methods
can include heat (e.g. 55-65 C or 56-60 C, for 5-60 minutes or for 10-30
minutes, e.g.
60 C for 30 minutes), formaldehyde (e.g. 0.1% at 37 , 24 hours),
glutaraldehyde (e.g.
0.05% at room temperature, 10 minutes), acetone-I (e.g. three treatments at
room
temperature) or acetone-II (e.g. three treatments at room temperature and
fourth treatment
at 37 C) inactivation (see for example Gupta et al., 1987, J. Biol. Stand.
15:87; Gupta et
al., 1986, Vaccine, 4:185). Methods of preparing killed, whole cell B.
pertussis (Pw)
suitable for use in the immunogenic compositions of this invention are
disclosed in
W093/24148.
[0136] More particularly, the immunogenic composition of the invention
comprises Pw at
a per-dose amount of (in International Opacity Units, "IOU"): 5-50, 7-40, 9-
35, 11-30, 13-
25, 15-21, or approximately or exactly 20.
[0137] In a particular embodiment of .a. Pw-comprising immunogenic composition
of the
invention, the Pw vaccine component of the composition elicits reduced
reactogenicity.
Reactogenicity (pain, fever, swelling etc) of Pw vaccines is primarily caused
by lipo-
oligosaccharide ('LOS', which is synonymous with lipo-polysaccharide (`LPS')
in the
context of B. pertussis; 'LOS' will be used herein), which is the endotoxin
from the
bacterial outer membrane. The lipid A part of LOS is mainly responsible. In
order to
produce a less reactogenic Pw-containing vaccine (relative to 'traditional' Pw
vaccines
such as produced by the above-discussed inactivation procedures), the
endotoxin can be
genetically or chemically detoxified and/or extracted from the outer membrane.
However,
this must be done in a way which does not substantially impair the
immunogenicity of the
Pw vaccine, as LOS is a potent adjuvant of the immune system.
[0138] In one embodiment, the at least one B. pertussis antigen of the
immunogenic
composition of the invention comprises a 'low reactogenicity' Pw vaccine in
which the
LOS has been genetically or chemically detoxified and/or extracted. For
example, the Pw
vaccine can be subjected to treatment with a mixture of an organic solvent,
such as
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
butanol, and water, as described in W02006/002502 and Dias et al (2012), Human
Vaccines & Immunotherapeutics, 9(2):339-348 which are incorporated herein by
reference
for the purpose of disclosing chemical extraction of LOS.
[0139] In an alternative embodiment, low reactogenicity' is achieved by
deriving the Pw
vaccine from a B. pertussis strain genetically engineered to produce a less
toxic LOS.
W02006/065139 (incorporated herein by reference) discloses genetic 3-0-
deacylation and
detoxification of B. pertussis LOS, resulting in strains comprising at least
partially 3-0-
deacylated LOS. The at least one B. pertussis antigen of the immunogenic
composition of
the invention can therefore be a Pw vaccine derived from a strain of B.
pertussis which has
been engineered to express a lipid A-modifying enzyme, such as a de-0-acylase.
In
particular, such a strain can express PagL as described in W02006/065139, as
well as in
Geurtsen et al (2006), Infection and Immunity, 74(10):5574-5585 and Geurtsen
et al
(2007), Microbes and Infection, 9:1096-1103, all incorporated herein by
reference.
Alternatively or additionally, the strain from which the Pw vaccine is derived
can
naturally, or as a result of engineering: lack the ability to modify its lipid
A phosphate
groups with glucosamine; have a lipid A diglucosamine backbone substituted
with at the
C-3' position with C10-OH or C12-0H; and/or express molecular LOS species that
lack a
terminal heptose. Such a strain, 18-323, is disclosed in Marr et al (2010),
The Journal of
Infectious Diseases, 202(12):1897-1906 (incorporated herein by reference).
IMMUNOGENIC COMPOSITIONS
[0140] Following expression and purification, the F protein analogs are
typically
formulated into immunogenic compositions for administration to a pregnant
female, and
where desired into formulations for administration to the infant following
birth. Such
formulations typically contain a pharmaceutically acceptable carrier or
excipient.
Optionally, additional antigens can also be included in the formulation, such
as another
RSV antigen (e.g., a G protein antigen as described in W02010149745) or a
human
metapneumovirus (hMPV) antigen, a diptheria antigen, a tetanus antigen, or an
influenza
antigen. W02010149743 describes examples of hMPV antigens that can be combined
with RSV antigens, and is incorporated herein by reference for the purpose of
providing
examples of hMPV antigens. Gall S.A. et al. Maternal Immunization with tetanus-
diphtheria-pertussis vaccine: effect on maternal and neonatal serum antibody
levels. Am J
46
CA 02879939 2015-01-23
WO 2014/024026
PCT/1B2013/001722
Obstet Gynecol 2011:204, describes immunization of pregnant mothers with
diphtheria-
tetanus-pertussis (Tdap) vaccine containing pertussis antigens pertactin
(PRN), pertussis
toxin (PT), filamentous hemagglutinin (FHA) and fimbriae (FIM) 2/3 and is
incorporated
herein by reference for the purpose of providing additional details on
maternal
immunization using pertussis antigens.
[0141] Pharmaceutically acceptable carriers and excipients are well known and
can be
selected by those of skill in the art. For example, the carrier or excipient
can favorably
include a buffer. Optionally, the carrier or excipient also contains at least
one component
that stabilizes solubility and/or stability. Examples of
solubilizing/stabilizing agents
include detergents, for example, laurel sarcosine and/or tween. Alternative
solubilizing/stabilizing agents include arginine, and glass forming polyols
(such as
sucrose, trehalose and the like). Numerous pharmaceutically acceptable
carriers and/or
pharmaceutically acceptable excipients are known in the art and are described,
e.g., in
Remington 's Pharmaceutical Sciences, by E. W. Martin, Mack Publishing Co.,
Easton,
PA, 5th Edition (975).
[0142] Accordingly, suitable excipients and carriers can be selected by those
of skill in
the art to produce a formulation suitable for delivery to a subject by a
selected route of
administration.
[0143] Suitable excipients include, without limitation: glycerol, Polyethylene
glycol
(PEG), Sorbitol, Trehalose, N-lauroylsarcosine sodium salt, L ¨proline, Non
detergent
sulfobetaine, Guanidine hydrochloride, Urea, Trimethylamine oxide, KC1, Ca2+,
Mg2+ ,
Mn2+, Zn2+ and other divalent cation related salts, Dithiothreitol,
Dithioerytrol, and 13-
mercaptoethanol. Other excipients can be detergents (including: Tween80,
Tween20,
Triton X-00, NP-40, Empigen BB, Octylglucoside, Lauroyl maltoside, Zwittergent
3-08,
Zwittergent 3-0, Zwittergent 3-2, Zwittergent 3-4, Zwittergent 3-6, CHAPS,
Sodium
deoxycholate, Sodium dodecyl sulphate, Cetyltrimethylammonium bromide).
[0144] In compositions containing an F protein analog and/or a pertussis
antigen for the
vaccination regimens, methods, uses and kits described herein, the composition
is
designed to induce a strong serum (e.g., neutralizing) antibody response.
Mothers have
already been exposed to RSV and pertussis (e.g., by natural infection or
immunization)
and therefore will have an existing primed response, so the goal for providing
protection
47
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
for the infant is to boost this primed response as effectively as possible and
in respect of
the antibody subclasses such as IgGI that can cross the placenta with high
efficiency and
provide protection to the infant. This can be achieved without including an
adjuvant, or by
including an adjuvant that includes only mineral salts such as an aluminium or
calcium
salts, in particular aluminium hydroxide, aluminium phosphate or calcium
phosphate.
Alternatively, this can be achieved by formulating with an oil and water
emulsion
adjuvant, or another adjuvant that enhances the production of antibodies of
the IgGI
subclass. Thus the F protein analog and/or the pertussis antigen for the
vaccination
regimens, methods, uses and kits described herein are favorably formulated
with a mineral
salt, favorably alum (aluminium hydroxide or aluminium phosphate), or with an
oil and
water emulsion adjuvant.
[0145] Any adjuvant is selected to be safe and well tolerated in pregnant
women.
Optionally, the immunogenic compositions also include an adjuvant other than
alum. For
example, adjuvants including one or more of 3D-MPL, squalene (e.g., QS21),
liposomes,
and/or oil and water emulsions are favorably selected, provided that the final
formulation
enhances the production in the pregnant (and typically primed) female of RSV-
specific
antibodies and pertussis antibodies with the desired characteristics (e.g., of
subclass and
neutralizing function).
[0146] In some instances, the adjuvant formulation includes a mineral salt,
such as a
calcium or aluminium (alum) salt, for example calcium phosphate, aluminium
phosphate
or aluminium hydroxide. Where alum is present, either alone, or e.g., in
combination with
3D-MPL, the amount is typically between about 100 jig and lmg, such as from
about
100ug, or about 200ug to about 750ug, such as about 500ug per dose.
[0147] In some embodiments, the adjuvant includes an oil and water emulsion,
e.g., an
oil-in-water emulsion. One example of an oil-in-water emulsion comprises a
metabolisable oil, such as squalene, a tocol such as a tocopherol, e.g., alpha-
tocopherol,
and a surfactant, such as sorbitan trioleate (Span 85TM) or polyoxyethylene
sorbitan
monooleate (Tween 80Tm), in an aqueous carrier. An example of an oil-in-water
emulsion
is MF59. In certain embodiments, the oil-in-water emulsion does not contain
any
additional immunostimulants(s), (in particular it does not contain a non-toxic
lipid A
derivative, such as 3D-MPL, or a saponin, such as QS21). The aqueous carrier
can be, for
48
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
example, phosphate buffered saline. Additionally the oil-in-water emulsion can
contain
span 85 and/or lecithin and/or tricaprylin.
[0148] In another embodiment of the invention the adjuvant composition
comprises an
oil-in-water emulsion and optionally one or more further immunostimulants,
wherein said
oil-in-water emulsion comprises 0.5-10 mg metabolisable oil (suitably
squalene), 0.5-11
mg tocol (suitably a tocopherol, such as alpha-tocopherol) and 0.4-4 mg
emulsifying
agent.
[0149] Other adjuvants that can be used in immunogenic compositions with an F
protein
analog, such as a PreF analog, in the immunogenic compositions for the
vaccination
regimens, methods, uses and kits described here, on their own or in
combination with 3D-
MPL, or another adjuvant described herein, are saponins, such as QS21. Such
adjuvants
are typically not employed (but could be if so desired) with a pertussis
antigen.
[0150] In one embodiment the recombinant F protein analog such as a PreF
antigen for
the methods and uses described herein is formulated with a saponin for example
QS21, in
particular a combination of a preF analog and QS21 is provided.
[0151] In another embodiment the F protein analog such as PreF antigen for the
methods
and uses described herein is formulated with QS21 and 3D-MPL.
[0152] Saponins are taught in: Lacaille-Dubois, M and Wagner H. (1996. A
review of the
biological and pharmacological activities of saponins. Phytomedicine vol 2 pp
363-386).
Saponins are steroid or triterpene glycosides widely distributed in the plant
and marine
animal kingdoms. Saponins are noted for forming colloidal solutions in water
which foam
on shaking, and for precipitating cholesterol. When saponins are near cell
membranes
they create pore-like structures in the membrane which cause the membrane to
burst.
Haemolysis of erythrocytes is an example of this phenomenon, which is a
property of
certain, but not all, saponins.
[0153] Saponins are known as adjuvants in vaccines for systemic
administration. The
adjuvant and haemolytic activity of individual saponins has been extensively
studied in the
art (Lacaille-Dubois and Wagner, supra). For example, Quil A (derived from the
bark of
the South American tree Quillaja Saponaria Molina), and fractions thereof, are
described
49
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
in US 5,057,540 and "Saponins as vaccine adjuvants", Kensil, C. R., Crit Rev
Ther Drug
Carrier Syst, 1996, 12 (1-2):1-55; and EP 0 362 279 Bl. Particulate
structures, termed
Immune Stimulating Complexes (ISCOMS), comprising fractions of Quil A are
haemolytic and have been used in the manufacture of vaccines (Morein, B., EP 0
109 942
Bl; WO 96/11711; WO 96/33739). The haemolytic saponins QS21 and QS17 (HPLC
purified fractions of Quit A) have been described as potent systemic
adjuvants, and the
method of their production is disclosed in US Patent No.5,057,540 and EP 0 362
279 Bl,
which are incorporated herein by reference. Other saponins which have been
used in
systemic vaccination studies include those derived from other plant species
such as
Gypsophila and Saponaria (Bomford et al., Vaccine, 10(9):572-577, 1992).
[0154] QS21 is an Hplc purified non-toxic fraction derived from the bark of
Quillaja
Saponaria Molina. A method for producing QS21 is disclosed in US Patent No.
5,057,540. Non-reactogenic adjuvant formulations containing QS21 are described
in WO
96/33739. The aforementioned references are incorporated by reference herein.
Said
immunologically active saponin, such as QS21, can be used in amounts of
between 1 and
50 g, per human dose of the immunogenic composition. Advantageously QS21 is
used at
a level of about 25 g, for example between 20-30 g, suitably between 21-29 g
or
between 22 -28 g or between 23 -27 g or between 24 -26 g, or 25 g. In another
embodiment, the human dose of the immunogenic composition comprises QS21 at a
level
of about 10 g, for example between 5 and 15 g, suitably between 6 -14 g, for
example
between 7 -13 g or between 8 -12pg or between 9 -11 g, or 10 g. In a further
embodiment, the human dose of the immunogenic composition comprises QS21 at a
level
of about 5 g, for example between 1-9 g, or between 2 -8 g or suitably between
3-7 g or
4 -6 g, or 5 g. Such formulations comprising QS21 and cholesterol have been
shown to
be successful Thl stimulating adjuvants when formulated together with an
antigen. Thus,
for example, PreF polypeptides can favorably be employed in immunogenic
compositions
with an adjuvant comprising a combination of QS21 and cholesterol.
[0155] As indicated above, the adjuvant can include mineral salts such as an
aluminium
or calcium salts, in particular aluminium hydroxide, aluminium phosphate and
calcium
phosphate. Such an adjuvant can also include 3D-MPL For example, an adjuvant
containing 3D-MPL in combination with an aluminium salt (e.g., aluminium
hydroxide or
aluminium phosphate, "alum") is suitable for formulation in an immunogenic
composition
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
containing a F protein analog for administration to a human subject. Such a
formulation is
not typically used with a pertussis antigen, but could be if so desired.
[0156] 3D-MPL is a non-toxic bacterial lipopolysaccharide derivative. An
example of a
suitable non-toxic derivative of lipid A, i.e., monophosphoryl lipid A, or
more particularly
3-Deacylated monophoshoryl lipid A (3D¨MPL). 3D-MPL is sold under the name MPL
by GlaxoSmithKline Biologicals N.A., and is referred throughout the document
as MPL or
3D-MPL. See, for example, US Patent Nos. 4,436,727; 4,877,611; 4,866,034 and
4,912,094. 3D-MPL primarily promotes CD4+ T cell responses with an IFN-y (Thl)
phenotype. 3D-MPL can be produced according to the methods disclosed in
GB2220211
A. Chemically it is a mixture of 3-deacylated monophosphoryl lipid A with 3,
4, 5 or 6
acylated chains. In the compositions of the present invention small particle
3D-MPL can
be used. Small particle 3D-MPL has a particle size such that it can be sterile-
filtered
through a 0.22ttm filter. Such preparations are described in W094/21292.
[0157] A lipopolysaccharide, such as 3D-MPL, can be used at amounts between 1
and
50 g, per human dose of the immunogenic composition. Such 3D-MPL can be used
at a
level of about 25 jig, for example between 20-30 g, suitably between 21-29 g
or between
22 and 28 g or between 23 and 27 g or between 24 and 26 g, or 25 g. In another
embodiment, the human dose of the immunogenic composition comprises 3D-MPL at
a
level of about 10 g, for example between 5 and 151.1g, suitably between 6 and
14 g, for
example between 7 and 13 g or between 8 and 12 g or between 9 and 11 g, or 10
g. In a
further embodiment, the human dose of the immunogenic composition comprises 3D-
MPL
at a level of about 5 g, for example between 1 and 9 g, or between 2 and 8 g
or suitably
between 3 and 7 g or 4 and jig, or 5 g.
[0158] In other embodiments, the lipopolysaccharide can be a 13(1-6)
glucosamine
disaccharide, as described in US Patent No. 6,005,099 and EP Patent No. 0 729
473 Bl.
One of skill in the art would be readily able to produce various
lipopolysaccharides, such
as 3D-MPL, based on the teachings of these references. Nonetheless, each of
these
references is incorporated herein by reference. In addition to the
aforementioned
immunostimulants (that are similar in structure to that of LPS or MPL or 3D-
MPL),
acylated monosaccharide and disaccharide derivatives that are a sub-portion to
the above
structure of MPL are also suitable adjuvants. In other embodiments, the
adjuvant is a
51
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
synthetic derivative of lipid A, some of which are described as TLR-4
agonists, and
include, but are not limited to: 0M174 (2-deoxy-6-o42-deoxy-2-[(R)-3-
dodecanoyloxytetra-decanoylamino]-4-o-phosphono-P-D-glucopyranosy1]-2-[(R)-3-
hydroxytetradecanoylamino]-a-D-glucopyranosyldihydrogenphosphate), (WO
95/14026);
OM 294 DP (3S, 9 R) ¨3-4(R)-dodecanoyloxytetradecanoylamino]-4-oxo-5-aza-9(R)-
[(R)-3-hydroxytetradecanoylamino]decan-1,10-dio1,1,10-
bis(dihydrogenophosphate)
(WO 99/64301 and WO 00/0462); and OM 197 MP-Ac DP ( 3S-, 9R) -3-[(R) -
dodecanoyloxytetradecanoylamino]-4-oxo-5-aza-9-[(R)-3-
hydroxytetradecanoylamino]decan-1,10-dio1,1 -dihydrogenophosphate 10-(6-
aminohexanoate) (WO 01/46127).
[0159] In one specific embodiment, the adjuvant formulation includes 3D-MPL
prepared
in the form of an emulsion, such as an oil-in-water emulsion. In some cases,
the emulsion
has a small particle size of less than 0.2ium in diameter, as disclosed in WO
94/21292. For
example, the particles of 3D-MPL can be small enough to be sterile filtered
through a
0.22micron membrane (as described in European Patent number 0 689 454).
Alternatively, the 3D-MPL can be prepared in a liposomal formulation.
Optionally, the
adjuvant containing 3D-MPL (or a derivative thereof) also includes an
additional
immunostimulatory component.
[0160] Combinations of different adjuvants, such as those mentioned
hereinabove, can
also be used in compositions with F protein analogs such as PreF analogs (and
optionally
also with pertussis antigens if so desired). For example, as already noted,
QS21 can be
formulated together with 3D-MPL. The ratio of QS21 : 3D-MPL will typically be
in the
order of 1 : 10 to 10 : 1; such as 1:5 to 5 : 1, and often substantially 1 :
1. Typically, the
ratio is in the range of 2.5 : 1 to 1 : 1 3D-MPL: QS21. Another combination
adjuvant
formulation includes 3D-MPL and an aluminium salt, such as aluminium
hydroxide.
[0161] Other TLR4 ligands which can be used are alkyl Glucosaminide phosphates
(AGPs) such as those disclosed in WO 98/50399 or US Patent No. 6,303,347
(processes
for preparation of AGPs are also disclosed), suitably RC527 or RC529 or
pharmaceutically acceptable salts of AGPs as disclosed in US Patent No.
6,764,840.
Some AGPs are TLR4 agonists, and some are TLR4 antagonists. Both are thought
to be
useful as adjuvants.
52
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
[0162] Other suitable TLR-4 ligands, capable of causing a signaling response
through
TLR-4 (Sabroe et al, JI 2003 p1630-5) are, for example, lipopolysaccharide
from gram-
negative bacteria and its derivatives, or fragments thereof, in particular a
non-toxic
derivative of LPS (such as 3D-MPL). Other suitable TLR agonists are: heat
shock protein
(HSP) 10, 60, 65, 70, 75 or 90; surfactant Protein A, hyaluronan
oligosaccharides, heparan
sulphate fragments, fibronectin fragments, fibrinogen peptides and b-defensin-
2, and
muramyl dipeptide (MDP). In one embodiment the TLR agonist is HSP 60, 70 or
90.
Other suitable TLR-4 ligands are as described in WO 2003/011223 and in WO
2003/099195, such as compound I, compound II and compound III disclosed on
pages 4-5
of W02003/011223 or on pages 3-4 of W02003/099195 and in particular those
compounds disclosed in W02003/011223 as ER803022, ER803058, ER803732,
ER804053, ER804057, ER804058, ER804059, ER804442, ER804680, and ER804764.
For example, one suitable TLR-4 ligand is ER804057.
[0163] Additional TLR agonists are also useful as adjuvants. The term "TLR
agonist"
refers to an agent that is capable of causing a signaling response through a
TLR signaling
pathway, either as a direct ligand or indirectly through generation of
endogenous or
exogenous ligand. Such natural or synthetic TLR agonists can be used as
alternative or
additional adjuvants. A brief review of the role of TLRs'as adjuvant receptors
is provided
in Kaisho & Akira, Biochimica et Biophysica Acta 1589:1-13, 2002. These
potential
adjuvants include, but are not limited to agonists for TLR2, TLR3, TLR7, TLR8
and
TLR9. Accordingly, in one embodiment, the adjuvant and immunogenic composition
further comprises an adjuvant which is selected from the group consisting of:
a TLR-1
agonist, a TLR-2 agonist, TLR-3 agonist, a TLR-4 agonist, TLR-5 agonist, a TLR-
6
agonist, TLR-7 agonist, a TLR-8 agonist, TLR-9 agonist, or a combination
thereof.
[0164] In one embodiment of the present invention, a TLR agonist is used that
is capable
of causing a signaling response through TLR-1. Suitably, the TLR agonist
capable of
causing a signaling response through TLR-1 is selected from: Tri-acylated
lipopeptides
(LPs); phenol-soluble modulin; Mycobacterium tuberculosis LP; S-(2,3-
bis(palmitoyloxy)-(2-RS)-propy1)-N-palmitoy1-(R)-Cys-(S)-Ser-(S)-Lys(4)-0H,
trihydrochloride (Pam3Cys) LP which mimics the acetylated amino terminus of a
bacterial
lipoprotein and OspA LP from Borrelia burgdorferi.
53
CA 02879939 2015-01-23
WO 2014/024026
PCT/1B2013/001722
[0165] In an alternative embodiment, a TLR agonist is used that is capable of
causing a
signaling response through TLR-2. Suitably, the TLR agonist capable of causing
a
signaling response through TLR-2 is one or more of a lipoprotein, a
peptidoglycan, a
bacterial lipopeptide from M tuberculosis, B burgdorferi or T pallidum;
peptidoglycans
from species including Staphylococcus aureus; lipoteichoic acids, mannuronic
acids,
Neisseria porins, bacterial fimbriae, Yersina virulence factors, CMV virions,
measles
haemagglutinin, and zymosan from yeast.
[0166] In an alternative embodiment, a TLR agonist is used that is capable of
causing a
signaling response through TLR-3. Suitably, the TLR agonist capable of causing
a
signaling response through TLR-3 is double stranded RNA (dsRNA), or
polyinosinic-
polycytidylic acid (Poly IC), a molecular nucleic acid pattern associated with
viral
infection.
[0167] In an alternative embodiment, a TLR agonist is used that is capable of
causing a
signaling response through TLR-5. Suitably, the TLR agonist capable of causing
a
signaling response through TLR-5 is bacterial flagellin.
[0168] In an alternative embodiment, a TLR agonist is used that is capable of
causing a
signaling response through TLR-6. Suitably, the TLR agonist capable of causing
a
signaling response through TLR-6 is mycobacterial lipoprotein, di-acylated LP,
and
phenol-soluble modulin. Additional TLR6 agonists are described in WO
2003/043572.
[0169] In an alternative embodiment, a TLR agonist is used that is capable of
causing a
signaling response through TLR-7. Suitably, the TLR agonist capable of causing
a
signaling response through TLR-7 is a single stranded RNA (ssRNA), loxoribine,
a
guanosine analogue at positions N7 and C8, or an imidazoquinoline compound, or
derivative thereof In one embodiment, the TLR agonist is imiquimod. Further
TLR7
agonists are described in WO 2002/085905.
[0170] In an alternative embodiment, a TLR agonist is used that is capable of
causing a
signaling response through TLR-8. Suitably, the TLR agonist capable of causing
a
signaling response through TLR-8 is a single stranded RNA (ssRNA), an
imidazoquinoline molecule with anti-viral activity, for example resiquimod
(R848);
resiquimod is also capable of recognition by TLR-7. Other TLR-8 agonists which
can be
54
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
used include those described in WO 2004/071459.
[0171] In an alternative embodiment, a TLR agonist is used that is capable of
causing a
signaling response through TLR-9. In one embodiment, the TLR agonist capable
of
causing a signaling response through TLR-9 is HSP90. Alternatively, the TLR
agonist
capable of causing a signaling response through TLR-9 is bacterial or viral
DNA, DNA
containing unmethylated CpG nucleotides, in particular sequence contexts known
as CpG
motifs. CpG-containing oligonucleotides induce a predominantly Thl response.
Such
oligonucleotides are well known and are described, for example, in WO
96/02555, WO
99/33488 and U.S. Patent Nos. 6,008,200 and 5,856,462. Suitably, CpG
nucleotides are
CpG oligonucleotides. Suitable oligonucleotides for use in the immunogenic
compositions of the present invention are CpG containing oligonucleotides,
optionally
containing two or more dinucleotide CpG motifs separated by at least three,
suitably at
least six or more nucleotides. A CpG motif is a Cytosine nucleotide followed
by a
Guanine nucleotide. The CpG oligonucleotides of the present invention are
typically
deoxynucleotides. In a specific embodiment the internucleotide in the
oligonucleotide is
phosphorodithioate, or suitably a phosphorothioate bond, although
phosphodiester and
other internucleotide bonds are within the scope of the invention. Also
included within the
scope of the invention are oligonucleotides with mixed internucleotide
linkages. Methods
for producing phosphorothioate oligonucleotides or phosphorodithioate are
described in
US Patent Nos. 5,666,153, 5,278,302 and WO 95/26204.
[0172] Another class of suitable adjuvants for use in formulations with F
protein analogs
such as PreF analogs (and optionally if desired with pertussis antigens, such
as purified
acellular pertussis proteins) includes OMP-based immunostimulatory
compositions.
OMP-based immunostimulatory compositions are particularly suitable as mucosal
adjuvants, e.g., for intranasal administration. OMP-based immunostimulatory
compositions are a genus of preparations of outer membrane proteins (OMPs,
including
some porins) from Gram-negative bacteria, such as, but not limited to,
Neisseria species
(see, e.g., Lowell et aL, J. Exp. Med. 167:658, 1988; Lowell et al., Science
240:800, 1988;
Lynch et al., Biophys. J. 45:104, 1984; Lowell, in "New Generation Vaccines"
2nd ed.,
Marcel Dekker, Inc., New York, Basil, Hong Kong, page 193, 1997; U.S. Pat. No.
5,726,292; U.S. Pat. No. 4,707,543), which are useful as a carrier or in
compositions for
immunogens, such as bacterial or viral antigens. Some OMP-based
immunostimulatory
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
compositions can be referred to as "Proteosomes," which are hydrophobic and
safe for
human use. Proteosomes have the capability to auto-assemble into vesicle or
vesicle-like
OMP clusters of about 20 nm to about 800 nm, and to noncovalently incorporate,
coordinate, associate (e.g., electrostatically or hydrophobically), or
otherwise cooperate
with protein antigens (Ags), particularly antigens that have a hydrophobic
moiety. Any
preparation method that results in the outer membrane protein component in
vesicular or
vesicle-like form, including multi-molecular membranous structures or molten
globular-
like OMP compositions of one or more OMPs, is included within the definition
of
Proteosome. Proteosomes can be prepared, for example, as described in the art
(see, e.g.,
U.S. Pat. No. 5,726,292 or U.S. Pat. No. 5,985,284). Proteosomes can also
contain an
endogenous lipopolysaccharide or lipooligosaccharide (LPS or LOS,
respectively)
originating from the bacteria used to produce the OMP porins (e.g., Neisseria
species),
which generally will be less than 2% of the total OMP preparation.
[0173] Proteosomes are composed primarily of chemically extracted outer
membrane
proteins (OMPs) from Neisseria menigitidis (mostly porins A and B as well as
class 4
OMP), maintained in solution by detergent (Lowell GH. Proteosomes for Improved
Nasal,
Oral, or Injectable Vaccines. In: Levine MM, Woodrow GC, Kaper JB, Cobon GS,
eds,
New Generation Vaccines. New York: Marcel Dekker, Inc. 1997; 193-206).
Proteosomes
can be formulated with a variety of antigens such as purified or recombinant
proteins
derived from viral sources, including the F protein analogs such as PreF
polypeptides
disclosed herein, e.g., by diafiltration or traditional dialysis processes or
with purified
pertussis antigenic proteins. The gradual removal of detergent allows the
formation of
particulate hydrophobic complexes of approximately 100-200nm in diameter
(Lowell GH.
Proteosomes for Improved Nasal, Oral, or Injectable Vaccines. In: Levine MM,
Woodrow
GC, Kaper JB, Cobon GS, eds, New Generation Vaccines. New York: Marcel Dekker,
Inc. 1997; 193-206).
[0174] "Proteosome: LPS or Protollin" as used herein refers to preparations of
proteosomes admixed, e.g., by the exogenous addition, with at least one kind
of lipo-
polysaccharide to provide an OMP-LPS composition (which can function as an
immunostimulatory composition). Thus, the OMP-LPS composition can be comprised
of
two of the basic components of Protollin, which include (1) an outer membrane
protein
preparation of Proteosomes (e.g., Projuvant) prepared from Gram-negative
bacteria, such
56
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
as Neisseria meningitidis, and (2) a preparation of one or more
liposaccharides. A lipo-
oligosaccharide can be endogenous (e.g., naturally contained with the OMP
Proteosome
preparation), can be admixed or combined with an OMP preparation from an
exogenously
prepared lipo-oligosaccharide (e.g., prepared from a different culture or
microorganism
than the OMP preparation), or can be a combination thereof Such exogenously
added
LPS can be from the same Gram-negative bacterium from which the OMP
preparation was
made or from a different Gram-negative bacterium. Protollin should also be
understood to
optionally include lipids, glycolipids, glycoproteins, small molecules, or the
like, and
combinations thereof The Protollin can be prepared, for example, as described
in U.S.
Patent Application Publication No. 2003/0044425.
[0175] It should be noted that regardless of the adjuvant selected, the
concentration in the
final formulation is calculated to be safe and effective in the target
population of pregnant
females.
[0176] An immunogenic composition for use in the vaccination regimens,
methods, uses
and kits herein typically contains an immunoprotective quantity (or a
fractional dose
thereof) of the antigen, or a quantity which provides passive transfer of
antibodies so as to
be immunoprotective in infants of immunized pregnant females, and can be
prepared by
conventional techniques. Preparation of immunogenic compositions, including
those for
administration to human subjects, is generally described in Pharmaceutical
Biotechnology,
Vol.61 Vaccine Design-the subunit and adjuvant approach, edited by Powell and
Newman, Plenum Press, 1995. New Trends and Developments in Vaccines, edited by
Voller et al., University Park Press, Baltimore, Maryland, U.S.A. 1978.
Encapsulation
within liposomes is described, for example, by Fullerton, U.S. Patent
4,235,877.
Conjugation of proteins to macromolecules is disclosed, for example, by
Likhite, U.S.
Patent 4,372,945 and by Armor et al.,U U.S. Patent 4,474,757.
[0177] Typically, the amount of antigen (e.g., protein) in each dose of the
immunogenic
composition is selected as an amount which induces an immunoprotective
response in the
infant without significant, adverse side effects in the typical subject i.e.
the pregnant
female or the gestating infant. Immunoprotective in this context does not
necessarily
mean completely protective against infection; it means protection against
symptoms or
disease, especially severe disease associated with the virus. The amount of
antigen can
57
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
vary depending upon which specific immunogen is employed. Generally, it is
expected
that each human dose will comprise 1-1000n of each protein or antigen, such as
from
about li_tg to about 100 g, for example, from about 11.1g to about 60i_tg,
such as about 1 g,
about 2 g, about 5 g, about 10 g, about 15tig, about 20tig, about 25[1g, about
30 g,
about 40 g, about 50[Ig, or about 601.1.g. Generally a human dose with be in a
volume of
0.5 ml. Thus the composition for the uses and methods described herein can be
for
example 10[tg or 301,tg or 60mg in a 0.5 ml human dose. The amount utilized in
an
immunogenic composition is selected based on the subject population. An
optimal
amount for a particular composition can be ascertained by standard studies
involving
observation of antibody titres and other responses in subjects. Following an
initial
vaccination, subjects can receive a boost in about 4-12 weeks, or at any time
prior to
delivery of the infant, favorably at least two or at least four weeks prior to
the expected
delivery date.
[0178] Additional formulation details can be found in W02010149745, which is
incorporated herein by reference for the purpose of providing additional
details concerning
formulation of immunogenic compositions comprising F protein analogs such as
PreF
analogs.
[0179] Optionally, the immunogenic compositions containing an F protein analog
such as
a PreF, or PostF analog are formulated with at least one additional antigen of
a pathogenic
organism other than RSV. For example, the pathogenic organism can be a
pathogen of the
respiratory tract (such as a virus that causes a respiratory infection). In
certain cases, the
immunogenic composition contains an antigen derived from a pathogenic virus
other than
RSV, such as a virus that causes an infection of the respiratory tract, such
as influenza or
parainfluenza. Similarly, if desired, a pertussis antigen could also be
formulated with an
antigen of a pathogenic virus other than RSV.
[0180] Coformulation of RSV protein analogs with acellular pertussis antigens
or whole
cell pertussis antigens is described herein, and is one preferred embodiment
of the
vaccination regimens, methods and uses disclosed herein.
[0181] In certain embodiments, immunogenic compositions suitable for the
vaccination
regimens, methods, uses and kits disclosed herein, additionally comprise at
least one
58
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
antigen from at least one pathogenic organism other than RSV and B. pertussis.
In
particular, said at least one pathogenic organism can be selected from the
group consisting
of: Corynebacterium diphtheriae; Clostridium tetani; Hepatitis B virus; Polio
virus;
Haemophilus influenzae type b; N. meningitidis type C; N. meningitidis type Y;
N
meningitidis type A, N meningitidis type W; and N meningitidis type B. More
particularly, said at least one antigen can be selected from the group
consisting of:
diphtheria toxoid (D); tetanus toxoid (T); Hepatitis B surface antigen
(HBsAg);
inactivated polio virus (IPV); capsular saccharide of H influenzae type b
(Hib) conjugated
to a carrier protein; capsular saccharide of N meningitidis type C conjugated
to a carrier
protein (MenC); capsular saccharide of N. meningitidis type Y conjugated to a
carrier
protein (MenY); capsular saccharide of N meningitidis type A conjugated to a
carrier
protein (MenA); capsular saccharide of N. meningitidis type W conjugated to a
carrier
protein (MenW); and an antigen from N meningitidis type B (MenB). In these and
other
embodiments, the additional antigens can be selected to facilitate
administration or reduce
the number of inoculations required to protect a subject against a plurality
of infectious
organisms. For example, the antigen can be derived from any one or more of
influenza,
hepatitis B, diphtheria, tetanus, pertussis, Hemophilus influenza, poliovirus,
Streptococcus or Pneumococcus, among others.
[0182] Combination vaccines containing B. pertussis antigens (Pa or Pw) with D
and T
and various combinations of other antigens such as selected from IPV, HBsAg,
Hib and
conjugated N meningitidis capsular saccharides are well known in the art and
suitable in
the context of the vaccination regimens, methods, uses and kits herein, for
example as
InfanrixTM (such as DTPa-IPV-HBsAg-Hib) and BoostrixTM (such as dTpa)
products. In
this regard, W093/024148, W097/000697 and W098/019702 are incorporated by
reference, as is W002/00249 which discloses a DTPw-HepB-MenAC-Hib composition.
Such combination vaccines are suitable in the context of the vaccination
regimens,
methods, uses and kits disclosed herein.
[0183] Particular immunogenic compositions suitable in the vaccination
regimens,
methods, uses and kits disclosed herein include, in addition to at least one
RSV antigen
and at least one B. pertussis antigen: D and T; D, T and IPV; D, T and HBsAg;
D, T and
Hib; D, T, IPV and HBsAg; D, T, IPV and Hib; D, T, HBsAg and Hib; or D, T,
IPV,
HBsAg and Hib.
59
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
[0184] In a particular embodiment, D is used at the amount per dose of 1-10
International
Units (IU) (for example exactly or approximately 2IU) or 10-40IU (for example
exactly or
approximately 20 or 30IU) or 1-10 Limit of flocculation (Lf units) (for
example exactly or
approximately 2 or 2.5 or 9Lf) or 10-30Lf (for example exactly or
approximately 15 or
25Lf).
[0185] In a particular embodiment, T is used at the amount per dose of 10-30
IU (for
example exactly or approximately 20IU) or 30-50IU (for example exactly or
approximately 40IU) or 1-15Lf (for example exactly or approximately 5 or
10Lf).
[0186] In exemplary embodiments the immunogenic compositions suitable for the
vaccination regimens, methods, uses and kits include, in addition to the at
least one RSV
antigen and at least one B, pertussis antigen, D and T at the respective exact
or
approximate amounts per dose: 30:40IU; 25:10Lf; 20:40IU; 15:5Lf; 2:20IU;
2.5:5Lf;
2:5Lf; 25:10Lf; 9:5Lf. For example, such a composition can comprise (in
addition to the at
least one RSV antigen):
(i) 20-3Oug, for example exactly or approximately 25ug of PT;
(ii) 20-3Oug, for example exactly or approximately 25ug of FHA;
(iii) 1-1Oug, for example exactly or approximately 3 or 8ug of PRN;
(iv) 10-30Lf, for example exactly or approximately 15 or 25Lf of D; and
(v) 1-15Lf, for example exactly or approximately 5 of 10Lf of T.
[0187] By way of another example, such a suitable composition can comprise (in
addition
to the at least one RSV antigen):
(i) 2-1Oug, for example exactly or approximately 2.5 or 8ug of PT;
(ii) 2-1Oug, for example exactly or approximately 5 or 8ug of FHA;
(iii) 0.5-4ug, for example 2-3ug such as exactly or approximately 2.5 or
3ug of
PRN;
(iv) 1-10Lf, for example exactly or approximately 2 or 2.5 or 9Lf of D; and
(v) 1-15Lf, for example exactly or approximately 5 of 10Lf of T.
[0188] Thus, it will be clear that according the vaccination regimens,
methods, uses and
kits disclosed herein, the immunogenic compositions are contemplated for use
in
medicine, and in particular for the prevention or treatment in a human subject
of infection
by, or disease associated with, RSV and B. pertussis. In certain embodiments
containing
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
antigens from other pathogens, such prevention or treatment will extend to the
other
pathogens.
[0189] Maternal immunization as described herein is carried out via a suitable
route for
administration for an RSV vaccine and a pertussis vaccine, including
intramuscular,
intranasal, or cutaneous administration. Favorably, RSV and/or pertussis
maternal
immunization as described herein is carried out cutaneously, which means that
the antigen
is introduced into the dermis and/or epidermis of the skin (e.g.,
intradermally). In a
particular embodiment a recombinant RSV antigen comprising an F protein analog
such as
a PreF antigen or a PostF antigen and/or a pertussis antigen comprising
acellular pertussis
proteins or whole cell pertussis, is delivered to the pregnant female
cutaneously or
intradermally. In a particular embodiment the F protein such as PreF antigen
or postF
antigen is formulated with an adjuvant described herein for example a saponin
such as
QS21, with or without 3D-MPL, for cutaneous or intradermal delivery. In
another
embodiment the F protein analog such as PreF or PostF antigen is formulated
with a
mineral salt such as aluminium hydroxide or aluminium phosphate or calcium
phosphate,
with or without an immunostimulant such as QS21 or 3D-MPL, for cutaneous or
intradermal delivery. Pertussis antigen is typically formulated in combination
with an
aluminium salt and can optionally be administered by a cutaneous or
intradermal route.
Optionally, the F protein analog and pertussis antigen are coformulated, e.g.,
in the
presence of a mineral salt such as aluminium hydroxide or aluminium phosphate
or
calcium phosphate, with or without an immunostimulant such as QS21 or 3D-MPL,
for
cutaneous or intradermal delivery.
[0190] Delivery via the cutaneous route including the intradermal route may
require or
allow a lower dose of antigen than other routes such as intramuscular
delivery. Therefore
also provided is an immunogenic composition for cutaneous or intradermal
delivery
comprising an F protein analog in a low dose e.g. less than the normal
intramuscular dose,
e.g. 50% or less of the normal intramuscular dose, for example 501.1g or less,
or 20 jig or
less, or 10 jig or less or 5 jig or less per human dose. Similarly,
immunogenic
compositions containing pertussis antigens can be formulated at the lower end
of the dose
range (ore at even lower doses) for cutaneous or intradermal administration,
for example,
between 1-10 jig PT, between 1-10 jig FHA, between 0.5-4 1.1.g PRN (with or
without
additional antigenic components). Optionally the immunogenic composition for
61
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
cutaneous or intradermal delivery also comprises an adjuvant e.g. an aluminium
salt or
QS21 or 3D-MPL or a combination thereof.
[0191] Devices for cutaneous administration include short needle devices
(which have a
needle between about 1 and about 2 mm in length) such as those described in US
4,886,499, US5,190,521, US5,328,483, US 5,527,288, US 4,270,537, US 5,015,235,
US
5,141,496, US 5,417,662 and EP1092444. Cutaneous vaccines may also be
administered
by devices which limit the effective penetration length of a needle into the
skin, such as
those described in W099/34850, incorporated herein by reference, and
functional
equivalents thereof. Also suitable are jet injection devices which deliver
liquid vaccines to
the dermis via a liquid jet injector or via a needle which pierces the stratum
corneum and
produces a jet which reaches the dermis. Jet injection devices are described
for example
in US 5,480,381, US 5,599,302, US 5,334,144, US 5,993,412, US 5,649,912, US
5,569,189,US 5,704,911, US 5,383,851, US 5,893,397, US 5,466,220, US
5,339,163,
U55,312,335, US 5,503,627, US 5,064,413, US 5,520, 639, US 4,596,556U5 5
4,790,824,US 4,941,880, US 4,940,460, WO 97/37705 and WO 97/13537.
[0192] Devices for cutaneous administration also include ballistic
powder/particle
delivery devices which use compressed gas to accelerate vaccine in powder form
through
the outer layers of the skin to the dermis. Additionally, conventional
syringes may be used
in the classical mantoux method of cutaneous administration. However, the use
of
conventional syringes requires highly skilled operators and thus devices which
are capable
of accurate delivery without a highly skilled user are preferred. Additional
devices for
cutaneous administration include patches comprising immunogenic compositions
as
described herein. A cutaneous delivery patch will generally comprise a backing
plate
which includes a solid substrate (e.g. occlusive or nonocclusive surgical
dressing). Such
patches deliver the immunogenic composition to the dermis or epidermis via
microprojections which pierce the stratum corneum. Microprojections are
generally
between 10Dm and 2mm, for example 20Dm to 500Dm, 30Dm to lmm, 100 to 200, 200
to 300, 300 to 400, 400 to 500, 500 to 600, 600 to 700, 700, 800, 800 to 900,
100Dm to
400Dm, in particular between about 200Dm and 300Dm or between about 150Dm and
250Dm. Cutaneous delivery patches generally comprise a plurality of
microprojections
for example between 2 and 5000 microneedles for example between 1000 and 2000
microneedles. The microprojections may be of any shape suitable for piercing
the stratum
62
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
corneum,epidermis and/or dermis Microprojections may be shaped as disclosed in
W02000/074765 and W02000/074766 for example. The microprojections may have an
aspect ratio of at least 3:1 (height to diameter at base), at least about 2:1,
or at least about
1:1. One particular shape for the microprojections is a cone with a polygonal
bottom, for
example hexagonal or rhombus-shaped. Other possible microprojection shapes are
shown,
for example, in U.S. Published Patent App. 2004/0087992. In a particular
embodiment,
microprojections have a shape which becomes thicker towards the base. The
number of
microprotrusions in the array is typically at least about 100, at least about
500, at least
about 1000, at least about 1400, at least about 1600, or at least about 2000.
The area
density of microprotrusions, given their small size, may not be particularly
high, but for
example the number of microprotrusions per cm2 may be at least about 50, at
least about
250, at least about 500, at least about 750, at least about 1000, or at least
about 1500. In
one embodiment of the invention the F protein analog is are delivered to the
subject
within 5 hours of placing the patch on the skin of the host, for example,
within 4 hours, 3
hours, 2 hours, 1 hour or 30 minutes. In a particular embodiment , the F
protein analog is
delivered within 20 minutes of placing the patch on the skin, for example
within 30
seconds 1,2, 3,4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18 or 19
minutes.
[0193] The microprojections can be made of any suitable material known to the
skilled
person. In a particular embodiment at least part of the microprojections are
biodegradable,
in particular the tip of the microprojection or the outer most layer of the
microprojection.
In a particular embodiment substantially all the microprojection is
biodegradable. The
term "biodegradable" as used herein means degradable under expected conditions
of in
vivo use (e.g. insertion into skin), irrespective of the mechanism of
biodegradation.
Exemplary mechanisms of biodegradation include disintegration, dispersion,
dissolution,
erosion, hydrolysis, and enzymatic degradation.
[0194] Examples of microprojections comprising antigens are disclosed in
W02008/130587 and W02009/048607. Methods of manufacture of metabolisable
microneedles are disclosed in W02008/130587 and W02010/124255. Coating of
microprojections with antigen can be performed by any method known to the
skilled
person for example by the methods disclosed in W006/055844, W006/055799.
[0195] Suitable delivery devices for cutaneous delivery including intradermal
delivery, in
63
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
the methods and uses described herein include the BD SoluviaTM device which is
a
microneedle device for intradermal administration, the Corium MicroCorTM patch
delivery system, the Georgia Tech microneedle vaccine patch, the Nanopass
microneedle
delivery device and the Debiotech NanojectTM microneedle device. Also provided
is a
cutaneous or intradermal delivery device containing an immunogenic composition
for
RSV maternal immunization as described herein, for example a recombinant F
protein
analog such as PreF antigen, optionally formulated with an adjuvant such as a
mineral salt
e.g. alum, or QS21, or 3D-MPL or a combination thereof.
64
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
EXAMPLES
Example 1 Proof of concept ¨ Maternal immunization in a guinea pig model
[0196] The guinea pig model was selected as placental structure and IgG
transfer is closer
to that of humans than is the case for typical rodent models (reviewed in
Pentsuk and van
der Laan (2009) Birth Defects Research (part B) 86:328-344. The relatively
long
gestational period of the guinea pig (68 days) allows for immunization and
immune
response development during pregnancy. In order to mimic the RSV immune status
of
pregnant women who have been in exposed to RSV throughout their lives and have
a pre-
existing immune response to RSV, female guinea pigs were primed with live RSV
at
either 6 weeks or 10 weeks prior to vaccination.
[0197] Female guinea pigs (N=5/group) were primed intranasally with live RSV
virus
(2.5 x 105 pfu), 6 or 10 weeks prior to vaccination (approximately at the time
of mating or
4 weeks prior to mating). Two groups were left unprimed. Pregnant females were
immunized approximately 6 weeks after the start of gestation with 10 1.1g of
PreF antigen
combined with aluminum hydroxide. One unprimed group of females was injected
with
PBS. Serum samples were collected throughout the priming and gestation period
to
monitor levels of anti-RSV binding and neutralizing antibodies.
[0198] Offspring (7- to 16-day old) were challenged intranasally with live RSV
at 1 x 107
pfu. Four days after challenge, lungs were collected and separated into 7
lobes. Virus was
titrated in 6 of the 7 lobes and total virus particles per gram of lung were
calculated.
[0199] Results are shown in the graph in Figures 2 and 3.
[0200] Similar levels of antibodies were observed on the day of vaccination
whether
guinea pigs had been primed 6 or 10 weeks earlier. Plateau titres were reached
as of 14
days post priming. Neutralising antibody titres do not decline after reaching
a plateau for
at least about 60 days. Thus priming at both time points was equivalent in
this model and
suitable to mimic maternal infection in humans.
[0201] Results from lung viral load in the guinea pig offspring indicate that
offspring
born to primed and vaccinated mothers were protected from RSV challenge, as
compared
to offspring born to unprimed/unvaccinated mothers. In contrast, offspring
born to
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
unprimated/vaccinated mothers were not protected from RSV challenge.
Example 2 Combination Vaccine protects against challenge by RSV
[0202] This example demonstrates protection against RSV elicited by a
combination
vaccine containing RSV and B. Pertussis antigens (PT, FHA and PRN).
Immunogenicity
(neutralizing antibody titers) of two doses of the combined Pa-RSV vaccine was
evaluated
in the Balb/c mouse model, followed by an intranasal RSV challenge to measure
efficacy
of the combination vaccine.
[0203] Groups of BALB/c mice (n=14/group) were immunized intra-muscularly
twice at
a 3-week interval with the formulations displayed in Table 1.
Table 1: Vaccine formulations administered prior to RSV challenge
Vaccine PT (lug) FHA (lug) PRN
(jig) PreF (jig) A1(OH)3(n) Vol N
( 1.)
Pa-RSV w/Alum 6.25 6.25 2 2 50 50 14
Pa-RSV 6.25 6.25 2 2 50 14
Standalone Pa 6.25 6.25 2 50 50 14
Standalone RSV 2 50 50 14
[0204] Sera from
all mice were individually collected on Day 0 (prior to first
immunization), on Day 21 (prior to second immunization) and on Day 35 (2 weeks
after second immunization) and tested for the presence of RSV neutralizing
antibodies
using a plaque reduction assay. Briefly, serial dilutions of each serum were
incubated
with RSV A Long (targeting 100 pfu/well) for 20 mm at 37 C. After incubation,
the
virus-serum mixture was transferred to plates previously seeded with Vero
cells and
emptied of growth medium. On each plate, cells in one column were incubated
with
virus only (100% infectivity) and 2 wells received no virus or serum (cell
controls).
Plates were incubated for 2hrs at 37 C, medium was removed and RSV medium
containing 0.5% CMC (low viscosity carboxymethylcellulose) was added to all
wells.
The plates were incubated for 3 days at 37 C before immunofluorescent
staining.
66
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
[0205] For staining, cell monolayers were washed with PBS and fixed with 1%
paraformaldehyde. RSV-positive cells were detected using a commercial goat
anti-RSV
antiserum followed by a rabbit anti-goat IgG conjugated to FITC. The number of
stained
plaques per well was counted using an automated imaging system. Neutralizing
antibody
titer of each serum was determined as the inverse of the serum dilution
causing 60%
reduction in the number of plaques as compared to the control without serum
(ED60).
Results are illustrated in FIG. 5A.
[0206] The PreF-based vaccine adjuvanted with Al(OH)3 protects mice against an
intranasal RSV challenge and this animal model is therefore useful for
studying the
capability of RSV vaccines to mediate viral clearance in the lungs. The
combination of B.
pertussis (PT, FHA and PRN) and RSV (PreF) antigens in a single vaccine was
then tested
for protective efficacy in the intranasal RSV challenge mouse model. Two weeks
after the
second vaccine dose, mice were challenged by instillation of 50 ill (25 It.L
per nostril) of
live RSV A Long strain (about 1.45x106 pfu/50 IA). Lungs were collected four
days post
challenge for evaluation of lung viral load. Four days after challenge, mice
were
euthanized, the lungs were aseptically harvested and individually weighed and
homogenized. Serial dilutions (8 replicates each) of each lung homogenate were
incubated
with Vero cells and wells containing plaques were identified by
immunofluorescence, 6
days after seeding. The viral titer was determined using the Spearman-Karber
method for
TCID50 calculation and was expressed per gram of lung. The statistical method
employed
is an Analysis of Variance (ANOVA 1) on the log10 values.
[0207] Results are illustrated in FIG. 5B. As expected, 2 lig of PreF combined
with
Al(OH)3 efficiently promoted viral clearance in the lungs compared to mice
vaccinated
with standalone Pa (control group where no protection from RSV challenge is
expected).
Only two animals out of 14 in the PreF group had detectable levels of RSV in
the lungs,
with no RSV detectable in the 12 other animals. Pa-RSV combination vaccine was
equally capable of protecting mice against RSV challenge as shown by only one
out of 14
animals with detectable levels of RSV in the lungs, RSV being undetectable in
the
remaining 13 animals. Overall, animals vaccinated with PreF + Al(OH)3 vaccine
or with
Pa antigens + PreF + Al(OH)3 had significantly lower lung viral titers than
control animals
vaccinated with standalone Pa (P<0.001). In the group vaccinated with Pa
antigens + PreF
in the absence of adjuvant, there was a significant reduction (P<0.001) in
lung viral titers,
67
CA 02879939 2015-01-23
WO 2014/024026
PCT/1B2013/001722
however no animal in this group appeared fully protected from RSV challenge
since virus
was quantifiable in lungs from all animals.
[0208] Using a challenge animal model, we observed that the Pa-RSV combination
vaccine elicited a protective immune response against RSV comparable to that
of RSV
vaccine. This immune response was associated with the production of RSV
neutralizing
antibodies.
Example 3 Combination Vaccine protects against challenge by B. pertussis
[0209] This example demonstrates protection against Bordetella Pertussis
elicited by a
combination vaccine containing RSV and B. Pertussis antigens (PT, FHA and
PRN).
Immunogenicity (neutralizing antibody titers) of two doses of the combined Pa-
RSV
vaccine was evaluated in the Balb/c model, followed by an intranasal challenge
with
infectious B. pertussis to measure efficacy of the combination vaccine.
[0210] Groups of BALB/c mice (n=20/group) were immunized subcutaneously twice
with a 3-week interval with the formulations displayed in Table 2.
Table 1: Vaccine formulations administered prior to B. pertussis challenge
Vaccine PT (gig) FHA (lig) PRN (j.1g)
PreF (jig) Al(OH)3(n) Vol N
( L)
DTPa (1/4 HD) 6.25 6.25 2 125 125 20
Standalone Pa 6.25 6.25 2 50 50 20
Pa-RSV 6.25 6.25 2 2 50 50 20
Standalone RSV 2 50 50 20
[0211] Sera from all mice were individually collected seven days after the
second
immunization (d28 ¨ the day before challenge) and tested for the presence of
anti-PT, -
FHA and -PRN IgG antibodies. In brief, 96-well plates were coated with FHA (2
g/ml),
PT (2 ig/m1) or PRN (6 g/ml) in a carbonate-bicarbonate buffer (50mM) and
incubated
overnight at 4 C. After the saturation step with the PBS-BSA 1% buffer, mouse
sera were
diluted at 1/100 in PBS-BSA 0.2% Tween 0.05% and serially diluted in the wells
from the
plates (12 dilutions, step 1/2). An anti-mouse IgG coupled to the peroxidase
was added
(1/5000 dilution). Colorimetric reaction was observed after the addition of
the peroxidase
68
CA 02879939 2015-01-23
WO 2014/024026 PCT/1B2013/001722
substrate (OPDA), and stopped with HCL 1M before reading by spectrophotometry
(wavelengths: 490-620 nm). For each serum tested and standard added on each
plate, a 4-
parameter logistic curve was fit to the relationship between the OD and the
dilution
(Softmaxpro). This allowed the derivation of each sample titer expressed in
STD titers.
Serological antibody responses specific to Pa antigens (PT, FHA and PRN)
induced by the
vaccines is considered indicative (but not dispositive) of the vaccine ability
to elicit an
antibody responses against the individual antigens found in the Pa vaccine.
FIG. 6A
shows that the DTPa, standalone Pa and Pa-RSV combination promoted PT, FHA and
PRN-specific IgG responses after two immunizations. No antigen-specific
antibodies
were detected in sera from unvaccinated or RSV-vaccinated mice (data not
presented).
Statistical analysis demonstrated equivalence between the anti-PT and anti-FHA
antibody
responses induced by DTPa (Infanrix) and the Pa-RSV combination. The amount of
anti-
PRN specific antibodies induced by the standalone Pa and Pa-RSV combination
vaccines
was also statistically equivalent, demonstrating that the presence of RSV
antigen did not
interfere with the production of anti-pertussis antibody responses.
[0212] To demonstrate protection, one week after the booster, the mice were
challenged
by instillation of 50 1 of bacterial suspension (about 5E6 CFU/50 1) into
the left nostril.
Five mice of each group were euthanized 2 hours, 2 days, 5 days and 8 days
after the
bacterial challenge. The lungs were aseptically harvested and individually
homogenized.
The lung bacterial clearance was measured by counting the colony growth on
Bordet-
Gengou agar plates. Data were plotted according to the mean of number of
colony-
forming unit (CFU ¨ 10g10) per lung in each treatment group for each
collection time. The
statistical method employed is an Analysis of Variance (ANOVA) on the log10
values
with 2 factors (treatment and day) using a heterogeneous variance model.
[0213] In this model, the acellular B. pertussis vaccine (Pa) protects mice
against an
intranasal challenge with the bacteria. This animal model is therefore useful
for studying
the capability of a B. pertussis-based vaccine to mediate bacterial clearance
in the lungs.
The combination of B. pertussis (PT, FHA and PRN) and RSV (Pre-F) antigens in
a single
vaccine was then tested for protective efficacy in the intranasal challenge
mouse model.
Representative results are illustrated in FIG. 6B. As expected, the adjusted
human dose
(one fourth dose of the commercial DTPa vaccine Infanrix) efficiently promoted
bacterial
clearance compared to the unvaccinated mice. Both Pa standalone and Pa-RSV
69
CA 02879939 2015-01-23
WO 2014/024026
PCT/1B2013/001722
combination vaccines were also capable of eliciting a protective immune
response leading
to bacterial elimination. As expected, the standalone Pre-F RSV vaccine was
unable to
protect in this animal model against B. pertussis.
[0214] These results demonstrate in an animal model that the Pa-RSV
combination
vaccine elicited a protective immune response against B. pertussis as well as
against RSV
as demonstrated in Example 2 above. This immune response was associated with
the
production of specific antibodies against the three subunit antigens found in
the acellular
Pa vaccine (PT, FHA and PRN).