Note: Descriptions are shown in the official language in which they were submitted.
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
PROCESS AND HIGH SURFACE AREA ELECTRODES FOR THE
ELECTROCHEMICAL REDUCTION OF CARBON DIOXIDE
FIELD
[0001] The present disclosure generally relates to the field of
electrochemical reactions, and more particularly to methods and/or
systems for electrochemical reduction of carbon dioxide using high
surface area electrodes.
BACKGROUND
[0002] The combustion of fossil fuels in activities such as electricity
generation, transportation, and manufacturing produces billions of
tons of carbon dioxide annually. Research since the 1970s indicates
increasing concentrations of carbon dioxide in the atmosphere may
is be responsible for altering the Earth's climate, changing the pH of
the ocean and other potentially damaging effects. Countries around
the world, including the United States, are seeking ways to mitigate
emissions of carbon dioxide.
[0003] A mechanism for mitigating emissions is to convert carbon
dioxide into economically valuable materials such as fuels and
industrial chemicals. If the carbon dioxide is converted using energy
from renewable sources, both mitigation of carbon dioxide emissions
and conversion of renewable energy into a chemical form that can be
stored for later use may be possible.
SUMMARY OF THE PREFERRED EMBODIMENTS
[0004] The present invention is directed to using high surface area
electrodes and particular electrolyte solutions to produce single
carbon (Cl) chemicals, including formic acid, and multi-carbon (C2+)
based chemicals (i.e., chemicals with two or more carbon atoms in
i
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
the compound). The present invention includes the process, system,
and various components thereof.
[0005] It is to be understood that both the foregoing general
description and the following detailed description are exemplary and
explanatory only and are not necessarily restrictive of the disclosure
as claimed. The accompanying drawings, which are incorporated in
and constitute a part of the specification, illustrate an embodiment
of the disclosure and together with the general description, serve to
io explain the principles of the disclosure.
BRIEF DESCRIPTION OF THE DRAWINGS
[0006] The numerous advantages of the present disclosure may be
better understood by those skilled in the art by reference to the
is accompanying figures in which:
FIG. 1 is a flow diagram of a preferred electrolyzer system for
the reduction of carbon dioxide in accordance with an embodiment of
the present disclosure;
FIG. 2 is a flow diagram of a preferred electrochemical
20 acidification system;
FIG. 3 is a flow diagram of another preferred system for the
electrochemical reduction of carbon dioxide;
FIG. 4 is a flow diagram of another preferred electrochemical
acidification system incorporating bipolar membranes;
25 FIG. 5 is flow diagram of another preferred electrochemical
electrolyzer system incorporating an ion exchange compartment for
the reduction of carbon dioxide; and
FIG. 6 is a flow diagram of a nano-filtration system in
accordance with an embodiment of the present disclosure;
2
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
FIG. 7 is a chart illustrating cumulative yield of formate over
time in accordance with an embodiment described with reference to
Example 1 of the present disclosure;
FIG. 8 is a chart illustrating cumulative yield of formate over
time in accordance with an embodiment described with reference to
Example 2 of the present disclosure;
FIG. 9 is a chart illustrating cumulative yield of formate over
time in accordance with an embodiment described with reference to
Example 3 of the present disclosure;
io FIG. 10 is a chart illustrating cumulative yield of formate over
time in accordance with an embodiment described with reference to
Example 4 of the present disclosure;
FIG. 11 is a chart illustrating cumulative formate yield versus
time in accordance with an embodiment described with reference to
is Example 9 of the present disclosure;
FIG. 12 is a chart illustrating formate concentration versus
time in accordance with an embodiment described with reference to
Example 9 of the present disclosure;
FIG. 13 is a chart illustrating cumulative formate yield versus
20 time in accordance with an embodiment described with reference to
Example 10 of the present disclosure;
FIG. 14 is a chart illustrating formate concentration versus
time in accordance with an embodiment described with reference to
Example 10 of the present disclosure;
25 FIG. 15 is a chart illustrating operating cell voltage versus time
in accordance with an embodiment described with reference to
Example 11 of the present disclosure;
FIG. 16 is a chart illustrating catholyte formate concentration
versus time in accordance with an embodiment described with
30 reference to Example 11 of the present disclosure;
3
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
FIG. 17 is a chart illustrating formate current efficiency versus
time in accordance with an embodiment described with reference to
Example 11 of the present disclosure;
FIG. 18 is a chart illustrating catholyte pH versus time in
accordance with an embodiment described with reference to Example
11 of the present disclosure;
FIG. 19 is a chart illustrating formate current efficiency versus
time in accordance with an embodiment described with reference to
Example 12 of the present disclosure;
FIG. 20 is a chart illustrating catholyte formate concentration
versus time in accordance with an embodiment described with
reference to Example 12 of the present disclosure; and
FIG. 21 is a chart illustrating catholyte pH versus time in
accordance with an embodiment described with reference to Example
12 of the present disclosure.
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
[0007] Reference will now be made in detail to the presently
preferred embodiments of the present disclosure, examples of which
are illustrated in the accompanying drawings.
[0008] In accordance with some embodiments of the present
disclosure, an electrochemical system is provided that converts
carbon dioxide to organic products including formate and formic acid.
Use of a cathode comprising a high surface area three dimensional
material, an acidic anolyte, and a catholyte comprising bicarbonate
facilitates the process.
[0009] Before any embodiments of the invention are explained in
detail, it is to be understood that the embodiments described below
do not limit the scope of the claims that follow. Also, it is to be
understood that the phraseology and terminology used herein is for
4
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
the purpose of description and should not be regarded as limiting.
The use of terms such as "including," "comprising," or "having" and
variations thereof herein are generally meant to encompass the item
listed thereafter and equivalents thereof as well as additional items.
Further, unless otherwise noted, technical terms may be used
according to conventional usage.
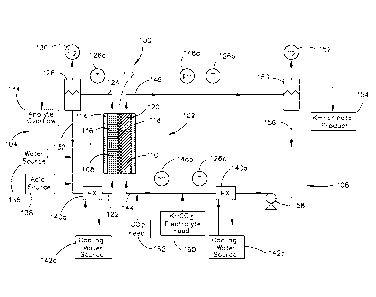
[0010] Referring to FIG. 1, a flow diagram of an electrolyzer system
100 is shown in accordance with an embodiment of the present
invention. The electrolyzer system 100 may be utilized for the
electrochemical reduction of carbon dioxide to organic products or
organic product intermediates. Preferably, the electrolyzer system
100 reduces carbon dioxide to an alkali metal formate, such as
potassium formate. The electrolyzer system 100 generally includes
is an electrolyzer 102, an anolyte recycle loop 104, and a catholyte
recycle loop 106. The electrolyzer system 100 may include as process
feeds/inputs carbon dioxide, a catholyte comprising bicarbonate
(preferably potassium bicarbonate, but other bicarbonate-based
compounds are contemplated instead of or in addition to potassium
bicarbonate), and an acidic anolyte (preferably sulfuric acid, but may
include other acids, instead of, or in addition to sulfuric acid). The
product of the electrolyzer system 100 is generally an alkali metal
formate, such as potassium formate, and may include excess
catholyte, carbon dioxide, hydrogen, oxygen, and/or other unreacted
process inputs.
[0011] The electrolyzer 102 generally includes an anode compartment
108 and a cathode compartment 110, and may further include a
cation exchange membrane 112 to separate the anode compartment
108 from the cathode compartment 110. The anode compartment
108 includes an anode 114 suitable to oxidize water. In a preferred
implementation, the anode 114 is a titanium anode having an anode
5
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
electrocatalyst coating which faces the cation exchange membrane
112. For instance, the anode 114 may include an anode mesh screen
116 that includes a folded expanded titanium screen with an anode
electrocatalyst coating. The anode mesh screen 116 may provide
spacing and contact pressure between the anode 114 and the cation
exchange membrane 112. The anode 114 may also include one or
more electrical current connection posts (not shown) on a backside of
the anode 114.
io [0012] The cathode compartment 110 generally includes a cathode
118 mounted within the cathode compartment 110. The cathode 118
preferably includes a metal electrode with an active electrocatalyst
layer on a front surface of the cathode 118 facing the cation
exchange membrane 112, and may include one or more electrical
is current conduction posts (not shown) on a backside of the cathode
118. The cathode 118 preferably includes a high surface area
cathode structure 120. The high surface area cathode structure 120
may be mounted between the cation exchange membrane 112 and
the cathode 118 for conducting electrical current into the high
20 surface area cathode structure 120. The interface between the high
surface area cathode structure 120 and the cation exchange
membrane 112 may include an insulator screen (not shown), such as a
thin expanded plastic mesh insulator screen to minimize direct
contact between the high surface area cathode structure 120 and the
25 cation exchange membrane 112.
[0013] The anode compartment 108 generally includes an anode feed
stream 122 that includes a dilute acid anolyte solution. The anode
feed stream 122 may enter a bottom of the anode compartment 108
30 to flow by a face of the anode 114 and through the anode mesh
screen 116. The reaction in the anode compartment 108 may include
deriving oxygen (02, i.e., gaseous oxygen) and hydrogen ions (W) or
6
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
protons from the oxidation of water at an applied current and voltage
potential. The hydrogen ions or protons are generally available for
the reactions within the cathode compartment 110 via the cation
exchange membrane 112. The gaseous oxygen and other liquids
leaving the anode compartment 108 of the electrolyzer 102 leave as
anode exit stream 124. The anode exit stream 124 may be monitored
by a temperature sensor 126a and may flow to an anolyte disengager
128 suitable for separating the oxygen from the anode exit stream
124. The anolyte disengager 128 may process the anode exit stream
io 124 into an oxygen stream 130, an anolyte recycle stream 132, and an
anolyte overflow stream 134. The oxygen stream 130 may be vented
from the anolyte disengager 128. The anolyte stream 132 may be
combined with water (preferably deionized water) from a water
source 136 and with acid (preferably sulfuric acid) from an acid
is source 138. The water source 136 and the acid source 138 in the
anolyte recycle loop 104 may maintain anolyte acid strength and
volume for the anode feed stream 122. The temperature of the
anode feed stream 122 may be regulated by a heat exchanger 140a
coupled with a cooling water source 142a prior to entering the anode
20 compartment 108 of the electrolyzer 102.
[0014] The cathode compartment 110 generally includes a cathode
feed stream 144 that includes carbon dioxide and a catholyte. In a
preferred implementation, the catholyte is a bicarbonate compound,
25 such as potassium bicarbonate (KHCO3), which is saturated with
carbon dioxide. The cathode feed stream 144 may enter a bottom of
the cathode compartment 110 to flow by a face of the cathode 118
and through the high surface area cathode structure 120. The
reaction in the cathode compartment 110 may reduce carbon dioxide
30 to formate at an applied current and voltage potential. The reaction
products and any unreacted materials (e.g., excess catholyte
solution) may exit the cathode compartment 110 as cathode exit
7
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
stream 146. The cathode exit stream 146 may be monitored by a pH
sensor 148a and a temperature sensor 126b and may flow to a
catholyte disengager 150 suitable for separating gaseous components
(e.g., hydrogen) from the cathode exit stream 146. The catholyte
disengager 150 may process the cathode exit stream 146 into a
hydrogen stream 152, a product stream 154, and a catholyte recycle
stream 156. The hydrogen stream 152 may be vented from the
catholyte disengager 150. The product stream 154 preferably
includes an alkali metal formate (such as potassium formate where
io the electrolyte includes potassium bicarbonate) and may include
excess catholyte. The catholyte stream 156 may be processed by a
catholyte recirculation pump 158 and a heat exchanger 140b coupled
with a cooling water source 142b. A temperature sensor 126c may
monitor the catholyte stream 156 downstream from the heat
is exchanger 140b having cooling water source 142b. A fresh catholyte
electrolyte feed 160 may be metered into the catholyte stream 156,
where the fresh catholyte electrolyte feed 160 may adjust the pH of
the cathode feed stream 144 into the cathode compartment 110 of
the electrolyzer 102, which may control final product overflow rate
20 and establish the formate product concentration. The pH may be
monitored by pH sensor 148b. A carbon dioxide stream 162 may be
metered into the cathode feed stream 144 downstream from the
catholyte electrolyte feed 160 prior to entering the cathode
compartment 110 of the electrolyzer 102. Preferably, the carbon
25 dioxide saturates the catholyte entering the cathode compartment.
[0015] When using an acidic anolyte, where protons are passed
through the membrane into the cathode compartment, the pH of the
electrolyzer 102 may be controlled or maintained through use of an
30 alkali metal bicarbonate and/or carbonate in combination with water
to control the pH of the catholyte. By controlling the pH of the
8
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
catholyte at an optimum value, the cell may more efficiently convert
carbon dioxide into Cl and C2 products with a higher conversion rate
than if a non-optimum pH value was maintained or if no pH control
mechanism was employed. In a preferred process, the catholyte is
constantly recirculated to maintain an adequate and uniform carbon
dioxide concentration at cathode surfaces coated with an
electrocatalyst. A fresh catholyte feed stream may be used to
control the pH of the catholyte and to control the product
concentration in the product overflow stream. The mass flow rate of
io the catholyte feed to the cathode compartment (e.g., mass flow of
potassium bicarbonate) is preferably balanced with the introduction
of protons into the catholyte and with the formation of hydroxide
from the inefficient byproduct reaction of water splitting at the
cathode. The
concentration of the potassium bicarbonate is
is important, since it provides volume to the catholyte, which will
dilute the product in the catholyte.
[0016] For pH control of the catholyte, potassium bicarbonate is
preferred, in a concentration range of 5 to 600 gm/L, or more
20 preferably in the 10 to 500 gm/L range. If the feed concentration of
bicarbonate to the catholyte is fixed, a separate feed of water may
be employed into the catholyte to control final product
concentration. In another implementation, potassium carbonate may
be used as a feed for pH control. Potassium carbonate has a much
25 higher solubility in water than potassium bicarbonate, and is
preferably used in a concentration range of 5 to 1,500 gm/L.
[0017] Referring now to FIG. 2, a block diagram of an electrochemical
acidification system 200 is shown in accordance with an embodiment
30 of the present invention. The electrochemical acidification system
200 may be utilized to acidify the product stream 154 from the
electrolyzer system 100. Preferably,
the electrochemical
9
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
acidification system 200 acidifies an alkali metal formate, such as
potassium formate, to form an organic acid, such as formic acid, and
co-produce an alkali metal hydroxide, such as potassium hydroxide.
The electrochemical acidification system 200 generally includes an
electrochemical acidification unit 202, an anolyte recycle loop 204,
and a catholyte recycle loop 206. The electrochemical acidification
system 200 may include as process feeds/inputs the product stream
154 from the electrolyzer system 100 (which preferably includes an
alkali metal formate), water in each of the anolyte recycle loop 204
io and the catholyte recycle loop 206, and an acidic anolyte (preferably
sulfuric acid, but may include other acids, instead of, or in addition
to sulfuric acid). The product of the electrochemical acidification
system 200 is generally an organic acid, such as formic acid, and an
alkali metal hydroxide, and may include residual alkali metal
is formate, bicarbonate catholyte, carbon dioxide, hydrogen, oxygen,
and/or other unreacted process inputs.
[0018] The electrochemical acidification unit 202 is preferably a
three-compartment electrochemical acidification unit or cell. The
20 electrochemical acidification unit 202 generally includes an anode
compartment 208, a cathode compartment 210, and a central ion
exchange compartment 212 bounded by cation exchange membranes
214a and 214b on each side. The anode compartment 208 includes an
anode 216 suitable to oxidize water. In a preferred implementation,
25 the anode 216 is a titanium anode having an anode electrocatalyst
coating which faces the cation exchange membrane 214a. The
cathode compartment 210 includes a cathode 218 suitable to reduce
water and to generate an alkali metal hydroxide. In a preferred
implementation, hydrogen ions (W) or protons are generated in the
30 anode compartment 208 when a potential and current are applied to
the electrochemical acidification unit 202. The hydrogen ions (1-1+) or
protons pass through the cation exchange membrane 214a into the
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
central ion exchange compartment 212. The product stream 154
from the electrolyzer system 100 is preferably introduced to the
electrochemical acidification unit 202 via the central ion exchange
compartment 212, where the hydrogen ions (F1+) or protons displace
the alkali metal ions (e.g., potassium ions) in the product stream 154
to acidify the stream and produce a product stream 260 including an
organic acid product, preferably formic acid. The displaced alkali
metal ions may pass through the cation exchange membrane 214b to
the cathode compartment 210 to combine with hydroxide ions (OH)
io formed from water reduction at the cathode 218 to form an alkali
metal hydroxide, preferably potassium hydroxide.
[0019] The central ion exchange compartment 212 may include a
plastic mesh spacer (not shown) to maintain the dimensional space in
is the central ion exchange compartment 212 between the cation
exchange membranes 214a and 214b. In an embodiment, a cation ion
exchange material 220 is included in the central ion exchange
compartment 212 between the cation exchange membranes 214a and
214b. The cation ion exchange material 220 may include an ion
20 exchange resin in the form of beads, fibers, and the like. It is
contemplated that the cation ion exchange material 220 may increase
electrolyte conductivity in the ion exchange compartment solution,
and may reduce the potential effects of carbon dioxide gas on the
cell voltage as bubbles are formed and pass through the central ion
25 exchange compartment 212.
[0020] The anode compartment 208 generally includes an anode feed
stream 222 that includes an acid anolyte solution (preferably a
sulfuric acid solution). The gaseous oxygen and other liquids leaving
30 the anode compartment 208 of the electrochemical acidification unit
202 leave as anode exit stream 224. The anode exit stream 224 may
be monitored by a temperature sensor 226a and may flow to an
11
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
anolyte disengager 228 suitable for separating the oxygen from the
anode exit stream 224. The anolyte disengager 228 may process the
anode exit stream 224 into an oxygen stream 230, an anolyte recycle
stream 232, and an anolyte overflow stream 234. The oxygen stream
230 may be vented from the anolyte disengager 228. The anolyte
stream 232 may be combined with water (preferably deionized water)
from a water source 236 and with acid (preferably sulfuric acid) from
an acid source 238. The water source 236 and the acid source 238 in
the anolyte recycle loop 204 may maintain anolyte acid strength and
volume for the anode feed stream 222. The temperature of the
anode feed stream 222 may be regulated by a heat exchanger 240a
coupled with a cooling water source 242a prior to entering the anode
compartment 208 of the electrochemical acidification unit 202.
[0021] The cathode compartment 210 generally includes a catholyte
feed stream 244 that includes water and may include an alkali metal
hydroxide that circulates through the catholyte recycle loop 206.
The reaction products, which may include the alkali metal hydroxide
and hydrogen gas, may exit the cathode compartment 210 as cathode
exit stream 246. The cathode exit stream 246 may be monitored by a
temperature sensor 226b and may flow to a catholyte disengager 248
suitable for separating gaseous components (e.g., hydrogen) from the
cathode exit stream 246. The catholyte disengager 248 may process
the cathode exit stream 246 into a hydrogen stream 250, a catholyte
stream 252, and a catholyte overflow stream 254, which may include
KOH. The hydrogen stream 250 may be vented from the catholyte
disengager 248. The catholyte stream 252 preferably includes an
alkali metal hydroxide (such as potassium hydroxide where the
product steam 154 includes potassium formate). The catholyte
stream 252 may be processed by a catholyte recirculation pump 256
and a heat exchanger 240b coupled with a cooling water source 242b.
12
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
A temperature sensor 226c may monitor the catholyte stream 252
downstream from the heat exchanger 240b. The catholyte stream
252 may be combined with water (preferably deionized water) from a
water source 258, where the water may be metered to control the
concentration of the alkali metal hydroxide in the catholyte feed
stream 244 entering the cathode compartment 210.
[0022] Referring now to FIG. 3, a flow diagram of a preferred system
300 for the electrochemical reduction of carbon dioxide to an organic
io acid product is shown. The system 300
may incorporate the
electrolyzer system 100 (described with reference to FIG. 1) and the
electrochemical acidification system 200 (described with reference to
FIG. 2), and preferably includes a potassium hydroxide recycle loop
302 suitable for the production of potassium bicarbonate from
is potassium hydroxide and carbon dioxide. The system 300 may also
incorporate carbon dioxide processing components for the separation
(e.g., gas separation units 304a, 304b, 304c, 304d) and recovery of
carbon dioxide from process streams.
20 [0023] The
system 300 generally includes carbon dioxide, an alkali
metal hydroxide (preferably potassium hydroxide), an acid
(preferably sulfuric acid), and water (preferably deionized water) as
process inputs and generally includes an organic acid (preferably
formic acid), oxygen gas, and hydrogen gas as process outputs. The
25 organic acid may undergo additional processing to provide a desired
form and concentration. Such processing may include evaporation,
distillation, or another suitable physical separation/concentration
process.
30 [0024] The
chemistry of the reduction of carbon dioxide in the system
300 may be as follows.
13
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
[0025] Hydrogen atoms are adsorbed at the electrode from the
reduction of water as shown in equation (1).
1-1+ + e- 4 Had (1)
[0026] Carbon dioxide is reduced at the cathode surface with the
adsorbed hydrogen atom to form formate, which is adsorbed on the
surface as in equation (2).
CO2 + Had 4 HCOOad (2)
[0027] The adsorbed formate on the surface then reacts with another
adsorbed hydrogen atom to form formic acid that is then released
into the solution as in equation (3)
HCOOad + Had 4 HCOOH (3)
[0028] The competing reaction at the cathode is the reduction of
water where hydrogen gas is formed as well as hydroxide ions as in
equation (4).
2H20 + 2e- 4 H2 + 20H- (4)
[0029] The anode reaction is the oxidation of water into oxygen and
hydrogen ions as shown in equation (5).
2H20 4 4W + 4e- + 02 (5)
[0030] High Surface Area Cathode
[0031] As described with reference to FIG. 1, the cathode 118
preferably includes a high surface area cathode structure 120. The
high surface area cathode structure 120 preferably includes a void
volume ranging from 30% to 98%. The specific surface area of the
high surface area cathode structure 120 is preferably from 2 cm2/cm3
to 500 cm2/cm3 or higher. The surface area also can be defined as
total area in comparison to the current distributor/ conductor back
plate, with a preferred range of 2x to 1000x or more.
14
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
[0032] The cathode 118 preferably includes electroless indium on tin
(Sn) coated copper woven mesh, copper screen, copper fiber as well
as bronze and other are copper-tin alloys, nickel and stainless steels.
The metals may be precoated with other metals, such as to
adequately form a suitable base for the application of the indium and
other preferred cathode coatings. The cathode may also include
Indium-Cu intermetallics formed on the surfaces of copper fiber,
woven mesh, copper foam or copper screen. The intermetallics are
generally harder than the soft indium metal, and may provide
io desirable mechanical properties in addition to usable catalytic
properties. The cathode may also include, but is not limited to
coatings and/or metal structures containing Pb, Sn, Hg, -IL In, Bi, and
Cd, their alloys, and combinations thereof. Metals including Ti, Nb,
Cr, Mo, Ag, Cd, Hg, -IL An, and Pb as well as Cr-Ni-Mo steel alloys
is among many others may be incorporated. The cathode 118 may
include a single or multi-layered electrode coating, such that the
electrocatalyst coating on the cathode substrate includes one or more
layers of metals and alloys. A preferred electrocatalyst coating on
the cathode includes a tin coating on a high surface area copper
20 substrate with a top layer/coating of indium. The indium coating
coverage preferably ranges from 5% to 100% as indium.
[0033] In the use of indium alloys on the exposed catalytic surfaces of
the electrode, the indium composition preferably ranges from 5% to
25 99% as indium in alloys with other metals, including Sn, Pb, Hg, -IL Bi,
Cu, and Cd and their mixed alloys and combinations thereof. It is
also contemplated to include Au, Ag, Zn, and Pd into the coating in
percentages ranging from 1% to 95%.
30 [0034] Additionally, metal oxides may be used or prepared as
electrocatalysts on the surfaces of the base cathode structure. For
example, lead oxide can be prepared as an electrocatalyst on the
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
surfaces of the base cathode structure. The metal oxide coating
could be formed by a thermal oxidation method or by electro-
deposition followed by chemical or thermal oxidation.
[0035] Additionally, the cathode base structure can also be gradated
or graduated, such that the density of the cathode can be varied in
the vertical or horizontal directions in terms of density, void volume,
or specific surface area (e.g., varying fiber sizes). The cathode
structure may also consist of two or more different electrocatalyst
io compositions that are either mixed or located in separate regions of
the cathode structure in the catholyte compartment.
[0036] During normal operation of the electrolyzer 102, the
performance of the system may decrease with regard to formate
is yield which may result from catalyst loss or over-coating of the
catalyst with impurities, such as other metals that may be plated
onto the cathode 118. The surfaces of the cathode 118 may be
renewed by the periodic addition of indium salts or a mix of
indium/tin salts in situ during operation of the electrolyzer 102.
20 Depending on the composition of the cathode 118, it is contemplated
that other or additional metal salts may be added in situ including
salts of Ag, Au, Mo, Cd, Sn, and other suitable metals, singly or in
combination. The electrolyzer 102 may be operated at full rate
during operation, or temporarily operated at a lower current density
25 with or without any carbon dioxide addition during the injection of
the metal salts. The conditions under which to renew the cathode
surface with the addition of these salts may differ depending on
desired renewal results. The use of an occasional brief current
reversal during electrochemical cell operation may also be employed
30 to potentially renew the cathode surfaces.
16
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
[0037] In particular embodiments, the electrolyzer 102 is operated at
pressures exceeding atmospheric pressure, which may result in higher
current efficiency and permit operation of the electrolyzer 102 at
higher current densities than when operating the electrolyzer 102 at
or below atmospheric pressure.
[0038] In preparing cathode materials for the production of organic
chemicals, the addition of metal salts that can reduce on the surfaces
of the cathode structure can be also used, such as the addition of Ag,
Au, Mo, Cd, Sn, and other suitable metals. Such addition of metal
salts may provide a catalytic surface that may be otherwise difficult
to prepare directly during cathode fabrication or for renewal of the
catalytic surfaces.
[0039] A preferred method for preparing the high surface area
cathode structure 120 is using an electroless plating solution which
may include an indium salt, at least one complexing agent, a
reducing agent, a pH modifier, and a surfactant. The preferred
procedure for forming an electroless indium coating on the high
surface area cathode may include combining in stirred deionized
water the following materials: Trisodium citrate dihydrate (100g/L),
EDTA-disodium salt (15g/L), sodium acetate (10g/L), InCl3
(anhydrous, 10g/L), and Thiodiglycolic acid (0.3g/L, e.g., 3 mL of
100mg/mL solution). A pre-mixed stock deposition solution that has
been stirred (preferably for multiple hours, e.g., overnight) may also
be used. The procedure also includes heating the mixture to about
40 C. The procedure also includes adding 40 mL TiCl3 (20 wt. % in 2%
HU) per liter [0.05 mM] and adding 7M ammonia in methanol until
the pH of the mixture is approximately 7 (-15 mL ammonia solution
per liter) at which point ammonium hydroxide (28% ammonia
solution) is used to adjust the pH to between approximately 9.0 and
9.2. The procedure then includes heating the mixture to about 60 C.
17
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
If the pH drops, adjust the pH to approximately 9.0 with ammonium
hydroxide solution. The procedure then includes heating the mixture
to about 75 C, where deposition may begin at about 65 C. The
procedure includes holding the mixture at 75 C for about one hour.
[0040] A preferred procedure for the metallic coating of copper
substrates may include rinsing bare copper substrates in acetone to
clean the copper surface (e.g., removing residual oils or grease that
may be present on the copper surface) and then rinsing the acetone-
treated copper substrates in deionized water. The procedure also
includes immersing the bare copper substrates in a 10% sulfuric acid
bath for approximately 5 minutes, and then rinsing with deionized
water. The procedure also includes depositing approximately 25 pm
of tin on the copper surface. The deposition may be done using a
is commercial electroless tinning bath (Caswell, Inc.) operated at 60 C
for 15 minutes. Following tin deposition, parts are rinsed thoroughly
in deionized water. The procedure
also includes depositing
approximately 1 pm of indium on the tinned copper surface. The
deposition may be done using an electroless bath operated at 90 C
for 60 minutes. Following indium
deposition, parts are rinsed
thoroughly in deionized water. The procedure may also include
treated the copper/tin/indium electrode in a 5 wt% nitric acid bath
for 5 minutes. Such treatment may improve electrode stability as
compared to an untreated copper/tin/indium electrode. In another
implementation, the electroless tin plated copper substrate may be
dipped into molten indium for coating.
[0041] In particular implementations, cathode substrates may be
treated with catalytic materials for carbon dioxide reduction. Four
example treatments are presented by the following.
18
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
[0042] A first treatment may include coating a conductive substrate
(e.g., vitreous carbon or metal) in a conductive sol-gel containing
sufficient catalyst material to yield a high active surface area. The
conductive component of the sol-gel may be catalytically active.
After coating the substrate with the catalytic sol-gel, the sol-gel is
allowed to undergo a high degree of polymerization/cross-linking.
The combined substrate/sol-gel structure may then be pyrolized at
high temperature to convert organic material to amorphous (and
potentially conductive) carbon. The pyrolized structure may also be
io subjected to chemical treatments that selectively remove the organic
material or the silica phase, leading to a high catalyst content
coating.
[0043] The second treatment may include binding relatively small
is particles (e.g., micron or nanometer scale) to a substrate using a
binding agent such as amines, thiols, or other suitable binding agent.
The binding agent is preferably conductive to pass current between
the substrate and catalyst particles. The catalyst particles preferably
include conjugated organic molecules, such as diphenybenzene. If
20 the substrate is also made of catalyst material the binding agent may
have symmetrical binding groups, otherwise binding agents with two
different binding groups may be utilized.
[0044] The third treatment may include coating a substrate in a slurry
25 containing catalyst material (which may be in salt form) and a binding
agent. The slurry may also contain a conductive additive, such as
carbon black, carbon nanotubes, or other suitable conductive
additive. The slurry coating may then be dried to form a conformal
coating over the substrate. The substrate and dried slurry coating
30 may be heated in order to fuse the various constituent materials into
a mechanically robust, conductive, and catalytic material. In a
19
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
particular implementation, the heating of the substrate and dried
slurry coating occurs in a reducing environment.
[0045] The fourth treatment may include coating a substrate with
semiconducting metal chalcogenides by applying a precursor to the
substrate, removing solvent, and baking the substrate to convert the
precursor material to a monolithic semiconducting metal
chalcogenide coating. The coating materials may include, but are
not limited to, Na4SnS4, Na4Sn2S6, K4SnTe4, Na3AsS3, (NH4)4Sh2S6,
io (NH4)3A5S3, and (NH4)2M0S4.
[0046] Other coating and electrocatalyst preparation techniques
include applying thermal oxides onto a substrate, forming an
intermetallic with a substrate, and applying semiconductor materials
is on a substrate. In an embodiment, the thermal oxidation of various
metal salts painted onto various metal and ceramic substrates is
preferred for forming high surface area materials suitable for the
electrochemical reduction of carbon dioxide. The thermal oxidation
may be similar to that used for forming electrocatalysts on titanium
20 for use as anode materials in electrochemical chlorine cells, such as
iridium oxide and ruthenium oxide. In another embodiment, indium
is electroplated onto a copper foil, then the copper foil is heated to
40 C above the melting point of indium, until indium is melted on the
foil surface, and forming a golden intermetallic with copper, and
25 then cooled. The formation of the intermetallic can be done in air or
under an inert gas atmosphere (e.g., argon or helium) or under a full
or partial vacuum. The electroplated material preferably provides
approximately 50% Faradaic conversion efficiency, and may be
utilized as a coating on planar metal back plates and also on copper
30 fibers. An intermetallic may also be formed with tin-plated copper
substrates. In a further embodiment, a semiconductor material may
be applied to a substrate by gaseous deposition, sputtering, or other
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
suitable application methods. The substrate is preferably a metallic
substrate. The semiconductor materials may be doped to P-type or
N-type as desired.
[0047] In the four treatments and other coating techniques described
above, certain measures may be taken to improve the quality
(mechanical, electrical, etc.) of the bond between the substrate and
catalyst. Such measures may involve creating functional groups on
the substrate surface that can undergo chemical bonding with the
io catalyst or a binding agent, or the creation of geometrical features in
the substrate surface that facilitate bonding with an applied catalyst
coating.
[0048] The substrate for the high surface area cathodes described
is herein may include RVC materials, such as carbon and graphite,
metal foams, woven metals, metal wools made from fibers, sintered
powder metal films and plates, metal and ceramic beads, pellets,
ceramic and metal column and trickle bed packing materials, metal
and inorganic powder forms, metal fibers and wools, or other suitable
20 substrate materials. The specific surface area of the physical forms
preferably include a specific surface area between approximately 2
and 2,000 cm2/cm3 or greater.
[0049] The electrode or high surface area structure of an electrode
25 may incorporate alloys as fibers or wools, and may be coated with
various compounds, and subsequently fired in air or in a reducing
atmosphere oven, to form stable oxides on the surfaces which are
electrocatalytic in the reduction of carbon dioxide. Other cathode
materials may include metallic glasses and amorphous metals.
[0050] Referring now to FIG. 4, a particular implementation of the
acid acidification system 200 of FIG. 2 is shown utilizing bipolar
membranes in an electrochemical acidification unit 402. By utilizing
21
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
bipolar membranes in electrochemical acidification unit 402, the
alkali metal formate (e.g., potassium formate) may be acidified in
addition to recovering potassium hydroxide. The use of the bipolar
membranes may reduce the voltage required for the acidification of
the alkali metal formate and may reduce the number of actual
anodes and cathodes needed for the electrochemical stack. The
bipolar membranes preferably consist of a cation membrane and an
anion membrane that have been bonded together, and function by
splitting water at the two membrane interface, forming hydrogen (H+)
io ions from the cation membrane and hydroxide ions (OH) from the
anion membrane.
[0051] Referring now to FIG. 5, an alternative embodiment of the
electrochemical system 100 of FIG. 1 is shown. The electrolyzer 502
is in FIG. 5 includes an ion exchange compartment 504 in addition to an
anode 506 compartment and a cathode compartment 508. This ion
exchange compartment 504 functions similarly as the acid
acidification compartment 212 in electrochemical acidification unit
202 as shown in FIG. 2. The alkali metal formate product (e.g.,
20 potassium formate) and unreacted KHCO3 from the cathode
compartment is passed through the ion exchange compartment 504 to
provide a formic acid product with CO2 and some residual KHCO3.
The hydrogen ions (W) passing through the adjacent membrane 510a
on the anode compartment side displace the alkali metal ions (e.g.,
25 10 in the stream passing through the central ion exchange
compartment 504 so that the alkali metal formate is acidified and the
alkali metal ions and remaining hydrogen ions pass through the
adjoining membrane 510b on the cathode compartment 508 and into
the catholyte. This will allow operation of the catholyte at higher pH
30 conditions if required for obtaining high Faradaic current efficiencies
with the cathodes selected for the process.
22
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
[0052] In an indium-based cathode system, the preferred catholytes
include alkali metal bicarbonates, carbonates, sulfates, phosphates,
and the like. Other preferred catholytes include borates, ammonium,
and hydroxides. Other catholytes may include chlorides, bromides,
and other organic and inorganic salts. Non-aqueous electrolytes, such
as propylene carbonate, methanesulfonic acid, methanol, and other
ionic conducting liquids may be used, which may be in an aqueous
mixture, or as a non-aqueous mixture in the catholyte. The
io introduction of micro bubbles of carbon dioxide into the catholyte
stream may improve carbon dioxide transfer to the cathode surfaces.
[0053] Referring now to FIG. 6, a nano-filtration system may be
utilized between the electrolyzer system 100, as shown in FIG. 1, and
is the electrochemical acidification system 200, as shown in FIG. 2. The
nano-filtration system is preferably utilized to separate alkali metal
formate (e.g., potassium formate) from bicarbonate leaving the
electrolyzer system 100 (e.g., stream 154) to reduce the amount of
bicarbonate entering the electrochemical acidification unit 202. The
20 nano-filtration system preferably uses a nano-filtration
filter/membrane under pressure for selective separation of the
bicarbonate from the alkali metal formate. The nano-filtration
filter/membrane separates monovalent anions (e.g., formate) from
divalent anions (e.g., carbonate) using a high pressure pump and
25 suitable selected membranes for the separation. When utilizing the
nano-filtration system as a separation tool between the electrolyzer
system 100 and the electrochemical acidification system 200, the
bicarbonate in the formate/bicarbonate product (e.g., stream 154) is
preferably converted to carbonate in order to efficiently separate the
30 formate from the carbonate with the nano-filtration
filter/membrane. The nano-filtration system may include a mixer,
such as a mixing tank, to mix the formate/bicarbonate product
23
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
stream with a potassium hydroxide (KOH) stream. The mixer may
promote the conversion of potassium bicarbonate to potassium
carbonate to facilitate the separation of the formate from the
carbonate. A high pressure pump then sends the potassium
formate/carbonate stream into a nano-filtration unit which includes
the nano-filtration filter/membrane. The nano-filtration unit
produces a low-carbonate-containing potassium formate permeate
stream which is then sent to the electrochemical acidification system
200 as shown in FIG. 2 as stream 154, to enter the electrochemical
io acidification unit 202. The potassium carbonate containing reject
stream leaving the nano-filtration unit is preferably sent to the
KHCO3 block of FIG. 3, where the potassium carbonate is mixed with
KOH and CO2 for conversion to potassium bicarbonate. The potassium
bicarbonate is preferably utilized as a feed to the cathode
is compartment of the electrolyzer 102 of the electrolyzer system 100.
The nano-filtration separation system may consist of multiple units
connected in a series flow configuration to increase the total
separation efficiency of the carbonate from formate separation. The
system may also utilize recycle streams to recycle an output stream
20 from one unit to the input of another unit to maintain flow and
pressures as well as to increase the recovery of the formate.
[0054] Depending on the chemistry of the electrochemical systems
described herein, the pH of the catholyte preferably ranges from 3 to
25 12. The desired pH of the catholyte may be a function of the
catholyte operating conditions and the catalysts used in the cathode
compartment, such that there is limited or no corrosion at the
electrochemical cell.
30 [0055] Preferable catholyte cross sectional area flow rates may
include a range of 2 to 3,000 gpm/ft2 or more ( 0.0076 to 11.36
24
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
m3/m2), with a flow velocity range of 0.002 to 20 ft/sec (0.0006 to
6.1 m/sec).
[0056] A homogenous heterocyclic catalyst is preferably utilized in
the catholyte. The homogenous heterocyclic catalyst may include,
for example, one or more of 4-hydroxy pyridine, adenine, a
heterocyclic amine containing sulfur, a heterocyclic amine containing
oxygen, an azole, a benzimidazole, a bipyridine, furan, an imidazole,
an imidazole related species with at least one five-member ring, an
indole, a lutidine, methylimidazole, an oxazole, phenanthroline,
pterin, pteridine, a pyridine, a pyridine related species with at least
one six-member ring, pyrrole, quinoline, or a thiazole, and mixtures
thereof.
[0057] Preferred anolytes for the system include alkali metal
hydroxides, such as KOH, NaOH, Li0H; ammonium hydroxide;
inorganic acids such as sulfuric, phosphoric, and the like; organic
acids such as methanesulfonic acid; non-aqueous and aqueous
solutions; alkali halide salts, such as the chlorides, bromides, and
iodine types such as NaCl, NaBr, LiBr, and Nal; and acid halides such
as HCl, HBr and HI. The acid halides and alkali halide salts will
produce for example chlorine, bromine, or iodine as a halide gas or as
dissolved aqueous products from the anolyte compartment. Methanol
or other hydrocarbon non-aqueous liquids can also be used, and
would form some oxidized organic products from the anolyte.
Selection of the anolyte would be determined by the process
chemistry product and requirements for lowering the overall
operating cell voltage. For example, the formation of bromine at the
anode requires a significantly lower anode voltage potential than
chlorine formation, and iodine is even lower than that of bromine.
This allows for a significant power cost savings in the operation of
both of the electrochemical units when bromine is generated in the
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
anolyte. The formation of a halogen, such as bromine, in the anolyte
may then be used in an external reaction to produce other
compounds, such as reactions with alkanes to form bromoethane,
which may then be converted to an alcohol, such as ethanol, or an
alkene, such as ethylene, and the halogen acid byproduct from the
reaction can be recycled back to the electrochemical cell anolyte.
[0058] Operation of the electrolyzer catholyte at a higher operating
pressure may allow more carbon dioxide to dissolve in the aqueous
io electrolyte than at lower pressures (e.g., ambient pressures).
Electrochemical cells may operate at pressures up to about 20 to 30
psig in multi-cell stack designs, although with modifications, they
could operate at up to 100 psig. The electrolyzer anolyte may also be
operated in the same pressure range to minimize the pressure
is differential on the membrane separating the two electrode
compartments. Special electrochemical designs are required to
operate electrochemical units at higher operating pressures up to
about 60 to 100 atmospheres or greater, which is in the liquid CO2
and supercritical CO2 operating range.
[0059] In a particular implementation, a portion of the catholyte
recycle stream may be separately pressurized using a flow restriction
with backpressure or using a pump, with CO2 injection, such that the
pressurized stream is then injected into the catholyte compartment
of the electrolyzer. Such a configuration may increase the amount of
dissolved CO2 in the aqueous solution to improve the conversion yield.
[0060] Catholyte and anolyte operating temperatures preferably
range from -10 to 95 C, more preferably 5 to 60 C. The minimum
operating temperature will be limited to the electrolytes used and
their freezing points. In general, the lower the temperature, the
higher the solubility of CO2 in the aqueous solution phase of the
26
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
electrolyte, and would help in obtaining higher conversion and
current efficiencies. A
consideration for lower operating
temperatures is that the operating electrolyzer cell voltages may be
higher, so an optimization may be required to produce the chemicals
at the lowest operating cost.
[0061] The electrochemical cell design may include a zero gap, flow-
through design with a recirculating catholyte electrolyte with various
high surface area cathode materials. Other designs include: flooded
io co-current packed and trickle bed designs with the various high
surface area cathode materials, bipolar stack cell designs, and high
pressure cell designs.
[0062] Anodes for use in the electrochemical system may depend on
is various system conditions. For acidic anolytes and to oxidize water
to generate oxygen and hydrogen ions, the anode may include a
coating, with preferred electrocatalytic coatings including precious
metal oxides, such as ruthenium and iridium oxides, as well as
platinum, rhodium, and gold and their combinations as metals and
20 oxides deposited on valve metal substrates, such as titanium,
tantalum, zirconium, and niobium. For other
anolytes, such as
alkaline or hydroxide electrolytes, the anode made include carbon,
cobalt oxides, stainless steels, nickel, and their alloys and
combinations which may be stable as anodes suitable under alkaline
25 conditions.
[0063] As described herein, the electrochemical system may employ a
membrane positioned between the anode compartment and the
cathode compartment. Cation ion exchange type membranes are
30 preferred, especially those that have a high rejection efficiency to
anions, for example perfluorinated sulfonic acid based ion exchange
membranes such as DuPont Nafion brand unreinforced types N117
27
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
and N120 series, more preferred PTFE fiber reinforced N324 and N424
types, and similar related membranes manufactured by Japanese
companies under the supplier trade names such as Flemion . Other
multi-layer perfluorinated ion exchange membranes used in the chlor
alkali industry have a bilayer construction of a sulfonic acid based
membrane layer bonded to a carboxylic acid based membrane layer,
which efficiently operates with an anolyte and catholyte above a pH
of about 2 or higher. These membranes have a much higher anion
rejection efficiency. These are sold by DuPont under their Nafion
io trademark as the N900 series, such as the N90209, N966, N982, and
the 2000 series, such as the N2010, N2020, and N2030 and all of their
types and subtypes. Hydrocarbon based membranes, which are made
from various cation ion exchange materials can also be used if the
anion rejection is not as critical, such as those sold by Sybron under
is their trade name lonac , AGC Engineering (Asahi Glass) under their
Selemion trade name, and Tokuyama Soda, among others available
on the market.
[0064] Example Electrolyzer Design
20 [0065] The electrolyzer design used in laboratory examples may
incorporate various thickness high surface area cathode structures
using added spacer frames and also provide the physical contact
pressure for the electrical contact to the cathode current conductor
backplate.
[0066] An electrochemical bench scale cell with an electrode
projected area of about 108 cm2 was used for much of the bench
scale test examples. The electrochemical cell was constructed
consisting of two electrode compartments machined from 1.0 inch
(2.54 cm) thick natural polypropylene. The outside dimensions of the
anode and cathode compartments were 8 inches (20.32 cm) by 5
inches (12.70 cm) with an internal machined recess of 0.375 inches
28
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
(0.9525 cm) deep and 3.0 inches (7.62 cm) wide by 6 inches (15.24
cm) tall with a flat gasket sealing area face being 1.0 inches (2.52
cm) wide. Two holes were drilled equispaced in the recess area to
accept two electrode conductor posts that pass though the
compartment thickness, and having two 0.25 inch (0.635 cm) drilled
and tapped holes to accept a plastic fitting that passes through 0.25
inch (0.635 cm) conductor posts and seals around it to not allow
liquids from the electrode compartment to escape to the outside.
The electrode frames were drilled with an upper and lower flow
io distribution hole with 0.25 inch pipe threaded holes with plastic
fittings installed to the outside of the cell frames at the top and
bottom of the cells to provide flow into and out of the cell frame,
and twelve 0.125 inch (0.3175 cm) holes were drilled through a 45
degree bevel at the edge of the recess area to the upper and lower
is flow distribution holes to provide an equal flow distribution across
the surface of the flat electrodes and through the thickness of the
high surface area electrodes of the compartments.
[0067] For the anode compartment cell frames, an anode with a
20 thickness of 0.060 inch (0.1524 cm) and 2.875 inch (7.3025 cm) width
and 5.875 inch (14.9225 cm) length with two 0.25 inch (0.635 cm)
titanium diameter conductor posts welded on the backside were
fitted through the two holes drilled in the electrode compartment
recess area. The positioning depth of the anode in the recess depth
25 was adjusted by adding plastic spacers behind the anode, and the
edges of the anode to the cell frame recess were sealed using a
medical grade epoxy. The electrocatalyst coating on the anode was a
Water Star WS-32, an iridium oxide based coating on a 0.060 inch
(0.1524 cm) thick titanium substrate, suitable for oxygen evolution in
30 acids. In addition, the anode compartment also employed an anode
folded screen (folded three times) that was placed between the
anode and the membrane, which was a 0.010 inch (0.0254 cm) thick
29
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
titanium expanded metal material from DeNora North America
(EC626), with an iridium oxide based oxygen evolution coating, and
used to provide a zero gap anode configuration (anode in contact
with membrane), and to provide pressure against the membrane from
the anode side which also had contact pressure from the cathode
side.
[0068] For the cathode compartment cell frames, 316L stainless steel
cathodes with a thickness of 0.080 inch (0.2032 cm) and 2.875 inch
io (7.3025 cm) width and 5.875 inch (14.9225 cm) length with two 0.25
inch (0.635 cm) diameter 316L SS conductor posts welded on the
backside were fitted through the two holes drilled in the electrode
compartment recess area. The positioning depth of the cathode in
the recess depth was adjusted by adding plastic spacers behind the
is cathode, and the edges of the cathode to the cell frame recess were
sealed using a fast cure medical grade epoxy.
[0069] A copper bar was connected between the two anode posts and
the cathode posts to distribute the current to the electrode back
20 plate. The cell was assembled and compressed using 0.25 inch (0.635
cm) bolts and nuts with a compression force of about 60 in-lbs force.
Neoprene elastomer gaskets (0.0625 inch (0.159 cm) thick) were used
as the sealing gaskets between the cell frames, frame spacers, and
the membranes.
[0070] EXAMPLE 1
[0071] The above cell was assembled with a 0.010 inch (0.0254 cm)
thickness indium foil mounted on the 316L SS back conductor plate
using a conductive silver epoxy. A multi-layered high surface area
cathode, comprising an electrolessly applied indium layer of about 1
micron thickness that was deposited on a previously applied layer of
electroless tin with a thickness of about 25 micron thickness onto a
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
woven copper fiber substrate. The base copper fiber structure was a
copper woven mesh obtained from an on-line internet supplier,
PestMall.com (Anteater Pest Control Inc.). The copper fiber
dimensions in the woven mesh had a thickness of 0.0025 inches
(0.00635 cm) and width of 0.010 inches (0.0254 cm). The prepared
high surface area cathode material was folded into a pad that was
1.25 inches (3.175 cm) thick and 6 inches (15.24 cm) high and 3
inches (7.62 cm) wide, which filled the cathode compartment
dimensions and exceeded the adjusted compartment thickness
io (adding spacer) which was 0.875 inches (2.225 cm) by about 0.25
inches (0.635 cm). The prepared cathode had a calculated surface
area of about 3,171 cm2, for an area about 31 times the flat cathode
plate area, with a 91% void volume, and specific surface area of 12.3
cm2/cm3. The cathode pad was compressible, and provided the
is spring force to make contact with the cathode plate and the
membrane. Two layers of a very thin (0.002 inches thick) plastic
screen with large 0.125 inch (0.3175 cm) holes were installed
between the cathode mesh and the Neon 324 membrane.
Neoprene gaskets (0.0625 inch (0.159 cm) thick) were used as the
20 sealing gaskets between the cell frames and the membranes. The
electrocatalyst coating on the anode in the anolyte compartment was
a Water Star WS-32, an iridium oxide based coating, suitable for
oxygen evolution in acids. In addition, the anode compartment also
employed a three-folded screen that was placed between the anode
25 and the membrane, which was a 0.010 inch (0.0254 cm) thick
titanium expanded metal material from DeNora North America
(EC626), with an iridium oxide based oxygen evolution coating, and
used to provide a zero gap anode configuration (anode in contact
with membrane), and to provide pressure against the membrane from
30 the anode side which also had contact pressure from the cathode
side.
31
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
[0072] The cell assembly was tightened down with stainless steel
bolts, and mounted into the cell station, which has the same
configuration as shown in FIG. 1 with a catholyte disengager, a
centrifugal catholyte circulation pump, inlet cell pH and outlet cell
pH sensors, a temperature sensor on the outlet solution stream. A 5
micron stainless steel frit filter was used to sparge carbon dioxide
into the solution into the catholyte disengager volume to provide
dissolved carbon dioxide into the recirculation stream back to the
catholyte cell inlet.
[0073] The anolyte used was a dilute 5% by volume sulfuric acid
solution, made from reagent grade 98% sulfuric acid and deionized
water.
[0074] In this test run, the system was operated with a catholyte
composition containing 0.4 molar potassium sulfate aqueous with 2
gm/L of potassium bicarbonate added, which was sparged with
carbon dioxide to an ending pH of 6.60.
[0075] Operating Conditions:
Batch Catholyte Recirculation Run
Anolyte Solution: 0.92 M H2504
Catholyte Solution: 0.4 M K2504, 0.14mM KHCO3
Catholyte flow rate: 2.5 LPM
Catholyte flow velocity: 0.08 ft/sec
Applied cell current: 6 amps (6,000 mA)
Catholyte pH range: 5.5 - 6.6, controlled by periodic additions of
potassium bicarbonate to the catholyte solution recirculation loop.
Catholyte pH declines with time, and is controlled by the addition of
potassium bicarbonate.
[0076] Results:
32
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
Cell voltage range: 3.39 - 3.55 volts (slightly lower voltage when the
catholyte pH drops)
Run time: 6 hours
Formate Faradaic yield: Steady between 32 - 35%, calculated taking
samples periodically. See FIG. 7.
Final formate concentration: 9,845 ppm
[0077] EXAMPLE 2
[0078] The same cell as in Example 1 was used with the same
cathode, which was only rinsed with water while in the
electrochemical cell after the run was completed and then used for
this run.
[0079] In this test run, the system was operated with a catholyte
composition containing 0.375 molar potassium sulfate aqueous with
40 gm/L of potassium bicarbonate added, which was sparged with
carbon dioxide to an ending pH of 7.05.
[0080] Operating Conditions:
Batch Catholyte Recirculation Run
Anolyte Solution: 0.92 M H2504
Catholyte Solution: 0.4 M K2504, 0.4 M KHCO3
Catholyte flow rate: 2.5 LPM
Catholyte flow velocity: 0.08 ft/sec
Applied cell current: 6 amps (6,000 mA)
Catholyte pH range: Dropping from 7.5 to 6.75 linearly with time
during the run.
[0081] Results:
Cell voltage range: 3.40 - 3.45 volts
Run time: 5.5 hours
33
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
Formate Faradaic yield: Steady at 52% and slowly declining with time
to 44% as the catholyte pH dropped. See FIG. 8.
Final formate concentration: 13,078 ppm
[0082] EXAMPLE 3
[0083] The same cell as in Examples 1 and 2 was used with the same
cathode, which was only rinsed with water while in the
electrochemical cell after the run was completed and then used for
io this run.
[0084] In this test run, the system was operated with a catholyte
composition containing 0.200 molar potassium sulfate aqueous with
40 gm/L of potassium bicarbonate added, which was sparged with
is carbon dioxide to an ending pH of 7.10.
[0085] Operating Conditions:
Batch Catholyte Recirculation Run
Anolyte Solution: 0.92 M H2504
20 Catholyte Solution: 0.2 M K2504, 0.4 M KHCO3
Catholyte flow rate: 2.5 LPM
Catholyte flow velocity: 0.08 ft/sec
Applied cell current: 9 amps (9,000 mA)
Catholyte pH range: Dropping from 7.5 to 6.65 linearly with time
25 during the run, and then additional solid KHCO3 was added to the
catholyte loop in 10 gm increments at the 210, 252, and 290 minute
time marks which brought the pH back up to about a pH of 7 for the
last part of the run.
30 [0086] Results:
Cell voltage range: 3.98 - 3.80 volts
Run time: 6.2 hours
34
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
Formate Faradaic yield: 75% declining to 60% at a pH of 6.65, and
then increasing to 75% upon the addition of solid potassium
bicarbonate to the catholyte to the catholyte loop in 10 gm
increments at the 210, 252, and 290 minute time marks and slowly
declining down with time 68% as the catholyte pH dropped to 6.90.
See FIG. 9.
Final formate concentration: 31,809 ppm.
[0087] EXAMPLE 4
[0088] The same cell as in Examples 1, 2, and 3 was used with the
same cathode, which was only rinsed with water while in the
electrochemical cell after the run was completed and then used for
this run.
[0089] In this test run, the system was operated with a catholyte
composition containing 1.40 molar potassium bicarbonate (120 gm/L
KHCO3), which was sparged with carbon dioxide to an ending pH of
7.8.
[0090] Operating Conditions:
Batch Catholyte Recirculation Run
Anolyte Solution: 0.92 M H2504
Catholyte Solution: 1.4 M KHCO3
Catholyte flow rate: 2.6 LPM
Catholyte flow velocity: 0.09 ft/sec
Applied cell current: 11 amps (11,000 mA)
Catholyte pH range: Dropping from around 7.8 linearly with time
during the run to a final pH of 7.48
[0091] Results:
Cell voltage range: 3.98 - 3.82 volts
Run time: 6 hours
CA 02883127 2015-02-25
WO 2014/042781 PCT/US2013/053554
Formate Faradaic yield: 63% and settling down to about 54 - 55%. See
FIG. 10.
Final formate concentration: 29,987 ppm.
[0092] Prophetic EXAMPLE 5
[0093] This example contemplates separation of product potassium
formate from potassium carbonate! bicarbonate supporting
electrolyte by membrane nano-filtration (NF) (FIG. 10). The test
would involve two commercial NF membranes. The feed solution
io would comprise 1.2M KHCO3 + 0.6M K-formate and its pH would be
adjusted to 7, 9, and 11 for three separate runs (for each
membrane).
[0094] All NF tests would be performed in GE-Osmonic Sepa
is permeator (active membrane area of 0.0137 m2) at applied pressure
of 40 bar (580 psig) and 50 C. During each run 3 liters of feed solution
would be passed through and the permeate would be collected into a
measuring cylinder (to determine volume) and the elapsed time
recorded. The permeate would later be analyzed for total carbonate
20 (HCO3 - + C032-) and formate. From such data, the permeability (in
L/m2 h bar) and solute rejections (in %) would be calculated as
follows:
volume collected (L)
Permeability -
membrane area(m2)x elapsed time(h)
[S]Feed¨[S]Permeate
25 % Rejection - ________________ x 100
[S]Feed
Where [S] denotes molar concentration of solute that could be either
formate or total carbonate.
30 [0095] Expected results are summarized below:
GE-Desal DK membrane
Feed pH %Rejection Permeabiility
Total L/m2 h bar
carbonate Formate
36
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
7 11.4 2.2 1.72
9 30.3 -9.7 1.07
11 81.8 -46.3 0.36
Dow-Filmtec NF270 membrane
Feed pH %Rejection Permeabiility
Total L/m2 h bar
carbonate Formate
7 11.0 2.6 1.91
9 29.5 -5.4 1.20
11 80.1 -43.8 0.44
[0096] Prophetic EXAMPLE 6
[0097] A single permeation test could be performed with DK
membrane, using a formate-enriched Feed solution comprising 1.2M
KHCO3 + 1.2M K-formate. The test could be done at pH 11 and all
other conditions would be as in the above Example 1.
[0098] Such a test would likely give 79.9% and -33.8% rejection for
total carbonate and formate, respectively. The permeability would
be 0.32 L/m2 h bar.
[0099] EXAMPLE 7
[00100] The same cell as in Examples 1, 2, and 3 was used,
except for using 701 gm of tin shot (0.3 - 0.6 mm diameter) media
with an electroless plated indium coating as the cathode. The
cathode compartment thickness was 0.875 inches.
[00101] In this test run, the system was operated with a
catholyte composition containing 1.40 molar potassium bicarbonate
(120 gm/L KHCO3), which was sparged with carbon dioxide to an
ending pH of 8.0
[00102] The cell was operated in a batch condition with no
overflow for the first 7.3 hrs, and then a 1.40 molar potassium
bicarbonate feed was introduced into the catholyte at a rate of about
37
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
1.4 mL/min, with the overflow collected and measured, and a sample
of the loop was collected for formate concentration analysis.
[00103] Operating Conditions:
Batch Catholyte Recirculation Run
Anolyte Solution: 0.92 M H2504
Catholyte Solution: 1.4 M KHCO3
Catholyte flow rate: 3.2 LPM
Applied cell current: 6 amps (6,000 mA)
io Catholyte pH range: Dropping slowly from around a pH of 8 linearly
with time during the run to a final pH of 7.50
[00104] Results:
Cell voltage range: 3.98 - 3.82 volts
is Run time: Batch mode: 7.3 hours
Feed and product overflow: 7.3 hours to end of run at 47 hours.
[00105] The formate Faradaic efficiency was between 42% and
52% during the batch run period where the formate concentration
20 went up to 10,490 ppm. During the feed and overflow period, the
periodic calculated efficiencies varied between 32% and 49%. The
average conversion efficiency was about 44%. The formate
concentration varied between 10,490 and 48,000 ppm during the feed
and overflow period. The cell voltage began at around 4.05 volts,
25 ending up at 3.80 volts.
[00106] EXAMPLE 8
[00107] Electrolyses were performed using a 3-compartment
glass cell of roughly 80 mL total volume. The cell was constructed to
30 be gas tight with Teflon bushings. The compartments were separated
by 2 glass frits. A 3-electrode assembly was employed. One
compartment housed the working electrode and the reference
electrode (Accumet silver/silver chloride) which contained the
38
CA 02883127 2015-02-25
WO 2014/042781 PCT/US2013/053554
aqueous electrolyte and catalyst as stated. The center compartment
also contained the electrolyte and catalyst solution as stated. The
third compartment was filled with 0.5 molar K2SO4 aqueous
electrolyte solution sparged with CO2 with a pH of about 4.5 and
housed the counter electrode (TELPRO (Stafford, TX) - Mixed Metal
Oxide Electrode). The working electrode compartment was purged
with carbon dioxide during the experiment. The solutions were
measured by ion chromatography for formic acid, analyzing the
solution before (a blank) and after electrolysis. The tests were
conducted under potentiometric conditions using a 6 channel Arbin
Instruments MSTAT, operating at -1.46 or -1.90 volts vs. an SCE
reference electrode for about 1.5 hrs.
Cathode Experiment Formate Formate Applied Current Time
Evaluated Designation Produced Yield % Potential (ma)
(hrs)
(PPrn) (volts)
Electroplated DK80 1,818 75.8 -1.9 50 1.5
indium on tin foil
Electroplated DK82 1,956 64.0 -1.9 58.5 1.5
indium on tin foil
Untreated tin DK80 1,260 54.3 -1.9 44.5 1.5
foil
Electroplated DK83 1,887 31.7 -1.9 123 1.5
indium on
copper foil
Tin foil DK80 604 18.0 -1.9 54.8 1.5
(untreated)
Copper screen DK79 1,813 30.6 -1.46 97.9 1.5
with electroless
indium coating
Copper screen DK78 1,387 43.9 -1.46 63.6 1.5
with electroless
indium annealed
at 200 C
[00108] EXAMPLE 9
[00109] The same cell as in Examples 1, 2, and 3 was used,
except for using 890.5 gm of tin shot (3 mm diameter) media and
39
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
with a tin foil coating as the cathode. The cathode compartment
thickness was 1.25 inches and the system was operated in a batch
mode with no feed input. Carbon dioxide was sparged to saturate the
solution in the catholyte disengager.
[00110] Packed Tin Bed Cathode Detail:
Weight: 890.5 gm tin shot
Tin shot: 3 mm average size
Total compartment volume: 369 cm3
Calculated tin bead surface area: 4,498 cm2
io Calculated packed bed cathode specific surface area: 12.2 cm2/cm3
Calculated packed bed void volume: 34.6%
[00111] In this test run, the system was operated with a
catholyte composition containing 1.40 molar potassium bicarbonate
is (120 gm/L KHCO3), which was sparged with CO2 to an ending pH of
about 8.0
[00112] The cell was operated in a batch condition with no
overflow and a sample of the catholyte loop was collected for
20 formate concentration analysis periodically.
[00113] Operating Conditions:
Batch Catholyte Recirculation Run
Anolyte Solution: 0.92 M H2504
25 Catholyte Solution: 1.4 M KHCO3
Catholyte flow rate: 3.0 LPM (upflow)
Catholyte flow velocity: 0.068 ft/sec
Applied cell current: 6 amps (6,000 mA)
Catholyte pH range: Increasing slowly from around a pH of 7.62
30 linearly with time during the run to a final pH of 7.73
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
[00114] Results:
Cell voltage range: Started at 3.84 volts, and slowly declined to 3.42
volts
Run time: Batch mode, 19 hours
[00115] The formate Faradaic efficiency started at about 65%
and declined after 10 hours to 36% and to about 18.3% after 19 hours.
The final formate concentration ended up at 20,500 ppm at the end
of the 19 hour run. See Figures 11 and 12.
io
[00116] EXAMPLE 10
[00117] The same cell as in Examples 1, 2, and 3 was used,
except for using 805 gm of indium coated tin shot (3 mm diameter)
media and with a 0.010 inch (0.0254 cm) thickness indium foil
is mounted on the 316L SS back conductor plate using a conductive
silver epoxy as the cathode. The cathode compartment thickness was
1.25 inches and the system was operated in a batch mode with no
feed input. Carbon dioxide was sparged to saturate the solution in
the catholyte disengager. The tin shot was electrolessly plated with
20 indium in the same method as used in Examples 1 - 4 on the tin-
coated copper mesh. The indium coating was estimated to be about
0.5- 1.0 microns in thickness.
[00118] Indium-Coated Tin Shot Packed Bed Cathode Detail:
25 Weight: 890.5 gm, indium coating on tin shot
Indium coated tin shot: 3 mm average size
Total compartment volume: 369 cm3
Calculated tin bead surface area: 4498 cm2
Packed bed cathode specific surface area: 12.2 cm2/cm3
30 Packed bed void volume: 34.6%
41
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
[00119] In this test run, the system was operated with a
catholyte composition containing 1.40 molar potassium bicarbonate
(120 gm/L KHCO3), which was sparged with CO2 to an ending pH of
about 8.0
[00120] The cell was operated in a batch condition with no
overflow and a sample of the catholyte loop was collected for
formate concentration analysis periodically.
[00121] Operating Conditions:
Batch Catholyte Recirculation Run
Anolyte Solution: 0.92 M H2504
Catholyte Solution: 1.4 M KHCO3
Catholyte flow rate: 3.0 LPM (upflow)
is Catholyte flow velocity: 0.068 ft/sec
Applied cell current: 6 amps (6,000 mA)
Catholyte pH range: Decreased slowly from around a pH of 7.86
linearly with time during the run to a final pH of 5.51
[00122] Results:
Cell voltage range: Started at 3.68 volts, and slowly declined to 3.18
volts
Run time: Batch mode, 24 hours
[00123] The formate Faradaic efficiency started at about 100%
and varied between 60% to 85%, ending at about 60% after 24 hours.
The final formate concentration ended up at about 60,000 ppm at the
end of the 24 hour run. Dilution error of the samples at the high
formate concentrations may have provided the variability seen in the
yield numbers. See Figures 13 and 14.
42
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
[00124] EXAMPLE 11
[00125] The same cell as in Examples 1, 2, and 3 was used with
a newly prepared indium on tin electrocatalyst coating on a copper
mesh cathode. The prepared cathode had calculated surface areas of
about 3,171 cm2, for an area about 31 times the flat cathode plate
area, with a 91% void volume, and specific surface area of 12.3
cm2/cm3.
[00126] In this test run, the system was operated with a
io catholyte composition containing 1.40 M potassium bicarbonate (120
gm/L KHCO3), which was sparged with CO2 to an ending pH of 7.8
before being used.
[00127] The cells were operated in a recirculating batch mode
is for the first 8 hours of operation to get the catholyte formate ion
concentration up to about 20,000 ppm, and then a fresh feed of 1.4 M
potassium bicarbonate was metered into the catholyte at a feed rate
of about 1.2 mL/min. The overflow volume was collected and volume
measured, and the overflow and catholyte loop sample were sampled
20 and analyzed for formate by ion chromatography.
[00128] Operating Conditions:
Cathode: Electroless indium on tin on a copper mesh substrate
Continuous Feed with Catholyte Recirculation Run - 11.5 days
25 Anolyte Solution: 0.92 M H2504
Catholyte Solution: 1.4 M KHCO3
Catholyte flow rate: 3.2 LPM
Catholyte flow velocity: 0.09 ft/sec
Applied cell current: 6 amps (6,000 mA)
[00129] Results:
43
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
Cell voltage versus time: FIG. 15 illustrates results of cell voltage
versus time, displaying a stable operating voltage of about 3.45
volts over the 11.5 days after the initial start-up.
Continuous Run time: 11.5 days
Formate Concentration Versus Time: FIG. 16 shows results of the
formate concentration versus time.
Formate Faradaic yield: FIG. 17 illustrates the calculated formate
current efficiency versus time measuring the formate yield
from the collected samples.
Final formate concentration: About 28,000 ppm.
Catholyte pH: FIG. 18 illustrates the catholyte pH change over the
11.5 days, which slowly declined from a pH of 7.8 to a pH
value of 7.5. The feed rate was not changed during the run,
but could have been slowly increased or decreased to maintain
a constant catholyte pH in any optimum operating pH range.
[00130] EXAMPLE 12
[00131] The same cell as in Examples 1, 2, and 3 was used with
a newly prepared indium on tin electrocatalyst coating on a copper
mesh cathode. The prepared cathode had calculated surface areas of
about 3,171 cm2, for an area about 31 times the flat cathode plate
area, with a 91% void volume, and specific surface area of 12.3
cm2/cm3.
[00132] In this test run, the system was operated with a
catholyte composition containing 1.40 M potassium bicarbonate (120
gm/L KHCO3), which was sparged with CO2 to an ending pH of 7.8
before being used.
[00133] The cells were operated in a recirculating batch mode
for the first 8 hours of operation to get the catholyte formate ion
44
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
concentration up to about 20,000 ppm, and then a fresh feed of 1.4 M
potassium bicarbonate was metered into the catholyte at a feed rate
of about 1.2 mL/min. The overflow volume was collected and volume
measured, and the overflow and catholyte loop sample were sampled
and analyzed for formate by ion chromatography.
[00134] Operating Conditions:
Cathode: Electroless indium on tin on a copper mesh substrate
Continuous Feed with Catholyte Recirculation Run - 21 days
Anolyte Solution: 0.92 M H2504
Catholyte Solution: 1.4 M KHCO3
Catholyte flow rate: 3.2 LPM
Catholyte flow velocity: 0.09 ft/sec
[00135] Applied cell current: 6 amps (6,000 mA)
[00136] Results:
Cell voltage versus time: The cell showed a higher operating voltage
of about 4.40 volts, higher than all of our other cells, because
of an inadequate electrical contact pressure of the cathode
against the indium foil conductor back plate. The cell
maintained operation for an extended run.
Continuous Run time: 21 days
[00137] Formate Faradaic yield: FIG. 19 illustrates calculated
formate current efficiency versus time measuring the formate yield
from the collected samples. The formate Faradaic current efficiency
declined down into the 20% range after 16 days.
[00138] Formate Concentration Versus Time: FIG. 20 illustrates
results of the formate concentration versus time. On day 21, 0.5 gm
of indium (III) carbonate was added to the catholyte while the cell
CA 02883127 2015-02-25
WO 2014/042781
PCT/US2013/053554
was still operating at the 6 ampere operating rate. The formate
concentration in the catholyte operating loop was 11,330 ppm before
the indium addition, which increased to 13,400 ppm after 8 hours,
and increased to 14,100 ppm after 16 hours when the unit was shut
down after 21 days of operation.
[00139] Catholyte pH: FIG. 21 illustrates the catholyte pH
change over the continuous operation period, which operated in the
7.6 to 7.7 pH range except for an outlier data point near day 16 when
io the feed pump had stopped pumping. The feed rate was not changed
during the run, but could have been increased or decreased to
maintain a constant pH operation in an optimum range.
[00140] It is believed that the present disclosure and many of its
is attendant advantages will be understood by the foregoing
description, and it will be apparent that various changes may be
made in the form, construction and arrangement of the components
thereof without departing from the scope and spirit of the disclosure
or without sacrificing all of its material advantages. The form herein
20 before described being merely an explanatory embodiment thereof, it
is the intention of the following claims to encompass and include such
changes.
46