Note: Descriptions are shown in the official language in which they were submitted.
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
COATED AND CURED PROPPANTS
RELATED APPLICATIONS
[1] This application claims benefit of priority of U.S. Application Serial
No.
13/626,055 entitled "Coated and Cured Proppants" and filed on September 25,
2012 which is
a continuation-in-part of U.S. Patent Application Serial No. 13/099,893
entitled "Coated and
Cured Proppants" and filed on May 3, 2011 and also a continuation-in-part of
U.S. Patent
Application Serial No. 13/188,530 entitled "Coated and Cured Proppants" and
filed on July 22,
2011. The contents of these co-pending applications are hereby incorporated by
reference.
FIELD OF INVENTION
[2] The invention relates to a method for the production of coated
proppants, and also
to the proppants obtained according to this method, to the uses thereof and to
methods which use
the proppants.
DACKGROIJND OF THE INVENTION
[3] Well fracturing is an often used technique to increase the efficiency
and
productivity of oil and gas wells. Overly simplified, the process involves the
introduction of a
fracturing fluid into the well and the use of fluid pressure to fracture and
crack the well strata.
The cracks allow the oil and gas to flow more freely from the strata and
thereby increase
production rates in an efficient manner.
[4] There are many detailed techniques involved in well fracturing, but one
of the
most important is the use of a solid "proppant" to keep the strata cracks open
as oil, gas, water and
other fluids found in well flow through those cracks. The proppant is carried
into the well with the
fracturing fluid which itself may contain a variety of viscosity enhancers,
gelation agents,
surfactants, etc. These additives also enhance the ability of the fracturing
fluid to carry proppant
to the desired strata depth and location. The fracturing fluid for a
particular well may or may not
use the same formulation for each depth in the strata.
1/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
[5] Proppants can be made of virtually any generally solid particle that
has a
sufficiently high crush strength to prop open cracks in a rock strata at great
depths and
temperatures of about 35 C and higher. Sand and ceramic proppants have proved
to be
especially suitable for commercial use.
[6] A proppant that is flushed from the well is said to have a high "flow
back." Flow
back is undesirable. In addition to closure of the cracks, the flushed
proppants are abrasive and
can damage or clog the tubular goods used to complete the well, valves and
pipelines in
downstream processing facilities.
[7] One type of synthetic resin coatings can be used to impart a degree of
adhesion to
the proppant so that flow back is substantially reduced or eliminated. Such
resins can include
phenol resin, epoxy resin, polyurethane-phenol resin, furane resin, etc. See
published US Patent
Application Nos. 2002/0048676,2003/0131998, 2003/0224165, 2005/0019574,
2007/0161515
and 2008/0230223 as well as US Patent Nos. 4,920,192; 5,048,608; 5,199,491;
6,406,789;
6,632,527; 7,624,802; and published international application WO 2010/049467,
the disclosures
of which are herein incorporated by reference.
[8] With some coatings, the synthetic coating is not completely cured when
the
proppant is introduced into the well. The coated, partially-cured proppants
are free flowing, but
the coating resin is still slightly reactive. The final cure is intended to
occur in situ in the strata
fracture at the elevated closure pressures and temperatures found "down hole."
[9] Such partially cured coatings can also exhibit a number of performance
issues
ranging from:
= A lack of storage stability if stored in a hot environment. This type
situation could
result in a completion of the curing process while in storage making the
coated proppant
incapable of bonding when placed in the fracture.
= Leaching chemicals out of the partially cured coating that could
interfere with the
viscosity profile of the fluid used to carry the proppant into the fracture or
the chemical
breaker system that is relied on to reduce the frac fluid viscosity after
completion of the
fracturing treatment.
= Erosion of the partially cured coating when the coated proppant is
handled
pneumatically in order to place in the field storage bins at the well site.
2/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
= Premature curing in the fracture due to extended exposure to the elevated
temperatures found downhole but before the cracks heal and begin to force the
proppant grains into contact with each other.
[10] A second type of synthetic coating is described as being pre-cured or
tempered. In
this case the coating is essentially cured during the manufacturing process.
This type of coating
will strengthen the substrate particle so that it can withstand a higher
stress level before grain
failure. Such a pre-cured coating with also exhibit the following traits: (1)
Excellent storage
stability; (2) Minimal chemicals that can be leached out of the coating to
interfere with carrier
fluid viscosity or breaker systems; and (3) A coating that is resilient to the
abrasion of pneumatic
handling.
[11] The main limitation of a pre-cured coating is that it cannot create
significant
particle to particle bonding when placed in the fracture and temperature and
closure pressure are
applied. This means that a pre-cured coated particle can do little to prevent
proppant flowback
after the well is opened up to start the clean-up process or to produce the
well. Such pre-cured
products can also exhibit reductions in bonding capability and/or strength if
exposed to elevated
temperatures during handling or storage.
[12] Proppants based on polyurethane chemistries have a number of potential
advantages over phenol resin systems. Most notably, the reaction rates used to
make
polyurethane coatings are generally faster than phenol resins, cure at lower
temperatures and do
not have gaseous emissions that require specialized recovery equipment. The
coating step with
polyurethanes can be carried out at temperatures of about 10 C to about 250
C although
temperatures of less than about 110 C are preferred to minimize emissions
during the coating
process as well as energy use. Polyurethane coatings can also be performed
without the use of
solvents, whereas many of the known methods, as a rule, require organic
solvents for the resinous
coating. The components in polyurethane systems are also generally easier to
use and pose lower
environmental issues. These factors could reduce the cost to make coated
proppants. [13]
Previously described polyurethanes have not, however, achieved widespread
adoption due to their performance in the hot, wet, high pressured environment
encountered in the
fracture. The stability of the coating to this environment, the ability of the
coating to prevent
particle failure (e.g., by crushing) and to develop strong particle-to-
particle bonds, have
contributed to poor flowback control and less than desirable fracture
conductivity.
3/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
[14] Low temperature wells pose certain problems for coated proppants.
Prior to the
present invention, the only option that was available to the oil and gas well
industry for
controlling flowback in a well having a low formation temperature, e.g., <140
F (60 C) was to
use a partially cured, phenolic-coated proppant in combination with a type of
plasticizer called a
bond "activator". Without the bond activator, the phenolic coating cured too
slowly to generate
sufficient bond strength in a reasonable amount of time. The activator
plasticizer softens the
coating so that the coating can gain some adhesion properties when the coated
proppant solids are
pushed into contact due to the closure stress from the closing of the
fractured strata cracks. This
adhesion will never result in a substantial measurable unconfined compressive
strength but can
result in a somewhat consolidated sample. The activator would be used at
concentrations ranging
from 5-20 gallons/ 1000 gallons of fracturing fluid (known as "frac" fluid).
While the activator
can help the phenolic coated proppant to function (to some degree) in low
temperature
applications it does have the following issues:
= The activator loadings add substantial cost to the treatment.
= The activator chemistry can create problems with frac fluid rheology and
breakers
systems.
= The use of an activator does not result in a significant measureable
particle to
particle bond strength.
= The activator is another factor in trying to quantify the effects of a
fracturing
treatment on the environment.
= The phenolic coating also has environmental issues because of the
components
that can be leached out of the coating (formaldehyde, phenol and
hexamethylenetetramine)
SUMMARY OF THE INVENTION
[15] It would be desirable to develop a coated proppant that combined
adequate crush
resistance, resistance to dusting when handled pneumatically and the limited
leaching of
chemicals that is exhibited by pre-cured coatings with the ability to create
the particle-to-particle
bonds that resist proppant flowback which are exhibited by a partially cured
proppant coating.
Even more desirably, this bonding ability is relatively unaffected by
prolonged exposure to
4/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
elevated temperatures in the fracture so that the coated proppants continue to
bond as stress is
applied when the fracture heals and forces the coated proppants together.
[16] It would also be desirable to have a coated proppant that retained its
conductivity
under the conditions prevailing within an actively producing well field
stratum.
[17] It would further be desirable to have a coating that not only
exhibited all of these
properties but which had desirable fracture conductivity and a short
production cycle.
[18] It would be especially preferred if a proppant could be substantially
covered with
a fast cure coating that could be produced to a free flowing state in a short
period, e.g., less than
about four minutes, while also exhibiting good crush resistance, resistance to
hot water coating
loss and low dust generation during pneumatic conveyance.
[19] It would also be desirable to have a coating that would not require
the use of an
activator in order to generate measurable bond strength at low temperature,
that would eliminate
the need for an added activator which might affect interactions between the
frac fluid and a
breaker, and avoid environmental effects from the use of an activator and
components that can
come out of a phenolic coating on the proppant solids.
[20] These and other objectives of the invention that will become apparent
from the
description herein can be accomplished by a coating and coating process that
comprises the step
of: coating a proppant solid with a substantially homogeneous coating mixture
that comprises (i)
at least one isocyanate-functional component having at least 2 isocyanate
groups, and (ii) at least
one curing agent comprising a monofunctional alcohol, amine or amide, in an
amount sufficient
and under conditions sufficient to substantially cure said proppant coating
and form free flowing,
coated proppants in a period of time of less than about four minutes to form a
free-flowing,
substantially cured, coated proppant.
[21] A coated, free flowing proppant according to the invention comprises a
solid
proppant core particle that is substantially covered with a coating that
comprises the reaction
product of a coating mixture that comprises at least one isocyanate component
and a curing agent
to form a substantially fully cured proppant coating that is capable of
forming particle-to-particle
bonds at elevated temperature and pressure, such as those found downhole in an
oil or gas well.
[22] The coating process of the present invention results one or more
layers of cured
polyurethane around a solid proppant core that is substantially cured and
crosslinked quickly to
produce a coated proppant product that acts like it has a hybrid coating,
i.e., one that acts like a
pre-cured coating in its resists dissolution of the coating under the rigorous
combination of high
5/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
heat, agitation, abrasion and water found downhole in an oil or gas well;
exhibits good crush
resistance and fracture conductivity; and has a tough coating that exhibits
low levels of dust or
fines generation during pneumatic conveyance as well as in use downhole but
also exhibits traits
of a partially cured coating in its ability to form particle-particle bonds
with similarly coated
proppants at downhole conditions. In addition, the coating process has a high
production rate due
to its low cycle time for the coating/curing process, low emission level and a
low overall
production cost and does not lose bonding capability if exposed to elevated
temperatures during
handling or storage.
BRIEF DESCRIPTION OF THE FIGURES
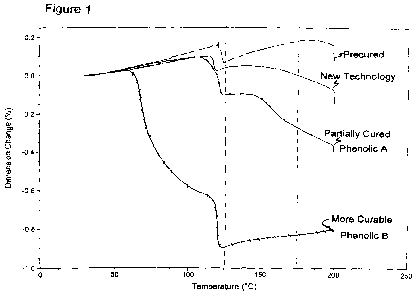
[23] Figure 1 is a TMA plot of Dimension Change at various Temperatures in
a TMA
test of pre-cured, partially cured Phenolic A, partially cured Phenolic B that
is more curable than
Phenolic A, and the coating of the present invention as discussed in Example
6.
[24] Figure 2 is a bar chart of crush test results of various coated
proppants tested in
Example 6.
[25] Figure 3 is a chart of Unconfined Compressive Strength for the coated
proppants
of Example 6.
[26] Figure 4 charts the fracture conductivity of three coated proppants
used in
Example 6.
[27] Figure 5 depicts the results of coating loss tests in simulated well
down-hole
conditions.
[28] Figures 6 and 7 illustrate the results of comparative conductivity
tests in a
simulated low temperature well using a proppant according to the present
invention and a prior
art proppant coated with a phenolic resin.
[29] Figure 8 shows the results of high temperature tests for unconfined
compressive
strength under high temperature well conditions and either with or without a
three hour preheat
exposure.
DETAILED DESCRIPTION OF THE INVENTION
[30] The coating formulation of the present invention includes a
substantially
homogeneous mixture of a curable coating formulation that comprises: (a) at
least one
6/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
isocyanate-functional reactant having at least 2 isocyanate groups, and (ii)
at least one curing
agent comprising a monofunctional alcohol, amine or amide. The coating
formulation may
further comprise one or more curing agents in the form of amine reactants,
metal catalysts,
and/or polyol-functional reactants. The components are used in an amount
sufficient and
under conditions that are also sufficient to substantially cure the proppant
coating and form
free flowing, coated proppants in a fairly short period of time. The coated
proppant thus
exhibits the good handling and low dust characteristics of a pre-cured product
but also
exhibits in-strata consolidation characteristics and flow-back resistance that
are like a partially
cured product.
[31] The coating process of the present invention applies one or more
layers of a
curable coating formulation around a solid proppant core that is quickly and
substantially
cured to resist dissolution of the coating under the rigorous combination of
high heat,
agitation, abrasion and water that are found downhole in a well. Preferably,
the cured coating
exhibits a sufficient resistance to a 10 day autoclave test or 10 day
conductivity test so that the
coating resists loss by dissolution in hot water of less than 25 wt%, more
preferably less than
15 wt%, and even more preferably a loss of less than 5 wt%. The substantially
cured coating
of the invention thus resists dissolution in the fractured stratum while also
exhibiting
sufficient particle- to-particle reaction bond strength to resist flow back
and sufficiently high
crush strength to maintain conductivity of the fractures.
[32] A preferred testing method for the effectiveness of a proppant is
described in ISO
13503-5:2006(E) "Procedures for measuring the long term conductivity of
proppants", the
disclosure of which is herein incorporated by reference. ISO 13503-5:2006
provides standard
testing procedures for evaluating proppants used in hydraulic fracturing and
gravel packing
operations. ISO 13503-5:2006 provides a consistent methodology for testing
performed on
hydraulic fracturing and/or gravel packing proppants. The "proppants"
mentioned henceforth in
this part of ISO 13503-5:2006 refer to sand, ceramic media, resin-coated
proppants, gravel
packing media, and other materials used for hydraulic fracturing and gravel-
packing operations.
ISO 13503-5:2006 is not applicable for use in obtaining absolute values of
proppant pack
conductivities under downhole reservoir conditions, but it does serve as a
consistent method by
which such downhole conditions can be simulated and compared in a laboratory
setting.
[33] The present invention is particularly directed to a proppant coating
technology
that exhibits decidedly different traits when kept dry (as in a storage bin)
as opposed to when the
7/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
coated proppant is added to the fracturing fluid and pumped into the fracture.
One way to
characterize this difference is by analyzing the test results from a TMA
(thermal mechanical
analyzer). In a dry state, the preferred coating will show a Tg softening
point that is well above
any possible storage temperature (>75 C). This assures that the coated
product can be safely
stored as long as it is kept relatively dry. However, when water is added to
the TMA test sample,
the resulting Tg is now measured at a level that is < 50 C. At this Tg, the
coated sand will have
the necessary properties to promote adhesion at low temperature applications
once the fracture
has closed and the resulting differential stress is placed on the proppants.
Since the coating's
ability to bond is not related to a chemical reaction rate, once the fracture
has closed thereby
exerting a closure stress on the proppant pack, there is no need for an
extended shut-in period,
e.g., 18 ¨ 24 hours, before opening the well up to cleanup and production.
THE ISO CYANATE COMPONENT
[34] The isocyanate-functional component for the present invention
comprises an
isocyanate-functional component with at least 2 reactive isocyanate groups.
Other isocyanate-
containing compounds may be used, if desired. Examples of suitable isocyanate
with at least 2
isocyanate groups an aliphatic or an aromatic isocyanate with at least 2
isocyanate groups (e.g. a
diisocyanate, triisocyanate or tetraisocyanate), or an oligomer or a polymer
thereof can
preferably be used. These isocyanates with at least 2 isocyanate groups can
also be carbocyclic
or heterocyclic and/or contain one or more heterocyclic groups.
[35] The isocyanate-functional component with at least 2 isocyanate groups
is
preferably a compound or oligomer of compounds of the formula (III) or a
compound of the
formula (IV):
8/51
CA 02889928 2015-04-29
WO 2014/052459
PCT/US2013/061688
(R2)q
A R1 NCO'
I
(III)
(R2)q (R2)(4
(OCN - R1 A R3 A Rl NCO)
r s
(IV)
9/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
[36] In the formulas (III) and (IV), A is each, independently, an aryl,
heteroaryl,
cycloalkyl or heterocycloalkyl. Preferably, A is each, independently, an aryl
or cycloalkyl.
More preferably A is each, independently, an aryl which is preferably phenyl,
naphthyl or
anthracenyl, and most preferably phenyl. Still more preferably A is a phenyl.
[37] The above mentioned heteroaryl is preferably a heteroaryl with 5 or 6
ring atoms,
of which 1, 2 or 3 ring atoms are each, independently, an oxygen, sulfur or
nitrogen atom and the
other ring atoms are carbon atoms. More preferably the heteroaryl is selected
among pyridinyl,
thienyl, furyl, pyrrolyl, imidazolyl, pyrazolyl, pyrazinyl, pyrimidinyl,
pyridazinyl, oxazolyl,
isoxazolyl or furazanyl.
[38] The above mentioned cycloalkyl is preferably a C340-cycloalkyl, more
preferably
a C5_7-cycloalkyl.
[39] The above mentioned heterocycloalkyl is preferably a heterocycloalkyl
with 3 to
ring atoms (more preferably with 5 to 7 ring atoms), of which one or more
(e.g. 1, 2 or 3) ring
atoms are each, independently, an oxygen, sulfur or nitrogen atom and the
other ring atoms are
carbon atoms. More preferably the heterocycloalkyl is selected from among
tetrahydrofuranyl,
piperidinyl, piperazinyl, aziridinyl, acetidinyl, pyrrolidinyl,
imidazolidinyl, morpholinyl,
pyrazolidinyl, tetrahydrothienyl, octahydroquinolinyl, octahydroisoquinolinyl,
oxazolidinyl or
isoxazolidinyl. Still more preferably, the heterocycloalkyl is selected from
among
tetrahydrofuranyl, piperidinyl, piperazinyl, pyrrolidinyl, imidazolidinyl,
morpholinyl,
pyrazolidinyl, tetrahydrothienyl, oxazolidinyl or isoxazolidinyl.
[40]1 i
In the formulas (III) and (IV), each R s, independently, a covalent bond or C1-
4-
alkylene (e.g. methylene, ethylene, propylene or butylene). Preferably each R2
is hydrogen or a
covalent bond.
[41] In the formulas (III) and (IV), each R2 is each, independently,
hydrogen, a
halogen (e.g. F, Cl, Br or I), a C1_4-alkyl (e.g. methyl, ethyl, propyl or
butyl) or C14-alkyoxy (e.g.
methoxy, ethoxy, propoxy or butoxy). Preferably, each R2 is, independently,
hydrogen or a C1-4-
alkyl. More preferably each R2 is hydrogen or methyl.
[42] In the formula (IV), R3 is a covalent bond, a C1_4-alkylene (e.g.
methylene,
ethylene, propylene or butylene) or a group ¨(CH2)R31-0-(CH2)R32-, wherein R31
and R32 are
each, independently, 0, 1, 2 or 3. Preferably, R3 is a -CH2- group or an -0-
group.
[43] In the formula (III), p is equal to 2, 3 or 4, preferably 2 or 3, more
preferably 2.
10/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
[44] In the formulas (III) and (IV), each q is, independently, an integer from
0 to 4,
preferably 0, 1 or 2. When q is equal to 0, the corresponding group A has no
substituent R2, but
has hydrogen atoms instead of R2.
[45] In the formula (IV), each r and s are, independently, 0, 1, 2, 3 or 4,
wherein the
sum of r and s is equal to 2, 3 or 4. Preferably, each r and s are,
independently, 0, 1 or 2, wherein
the sum of r and s is equal to 2. More preferably, r is equal to 1 and s is
equal to 1.
[46] Examples of the isocyanate with at least 2 isocyanate groups are:
toluol-2,4-
diisocyanate; toluol-2,6-diisocyanate; 1,5-naphthalindiisocyanate; cumo1-2,4-
diisocyanate; 4-
methoxy- 1,3-phenyldiisocyanate; 4-chloro-1,3-phenyldiisocyanate;
diphenylmethane-4,4-
diisocyanate; diphenylmethane-2,4-diisocyanate; diphenylmethane-2,2-
diisocyanate; 4-bromo-
1,3-phenyldiisocyanate; 4-ethoxy-1,3-phenyl-diisocyanate; 2,4' -diisocyanate
diphenylether; 5,6-
dimethy1-1,3-phenyl-diisocyanate; methylenediphenyl diisocyanate (including
2,2'-MDI, 2,4' -
MDI and 4,4"-MDI); 4,4-diisocyanato-diphenylether; 4,6-dimethy1-1,3-
phenyldiisocyanate;
9,10-anthracene-diisocyanate; 2,4,6-toluol triisocyanate; 2,4,4'-
triisocyanatodiphenylether; 1,4-
tetramethylene diisocyanate; 1,6-hexamethylene diisocyanate; 1,10-
decamethylene-diisocyanate;
1,3-cyclohexylene diisocyanate; 4,4' -methylene-bis-(cyclohexylisocyanate);
xylol diisocyanate;
1-isocyanato-3-methyl-isocyanate-3,5,5-trimethylcyclohexane (isophorone
diisocyanate); 1-3-
bis(isocyanato- 1-methylethyl) benzol (m-TMXDI); 1,4-bis(isocyanato-1-
methylethyl) benzol (p-
TMXDI); oligomers or polymers of the above mentioned isocyanate compounds; or
mixtures of
two or more of the above mentioned isocyanate compounds or oligomers or
polymers thereof.
[47] Particularly preferred isocyanates with at least 2 isocyanate groups
are toluol
diisocyanate, methylenediphenyl diisocyanate, diphenylmethane diisocyanate, an
oligomer based
on toluol diisocyanate, an oligomer based on methylenediphenyl diisocyanate
(poly-MDI) or an
oligomer based on diphenylmethane diisocyanate and polymers thereof.
CURING AGENTS
[48] The coatings of the invention can be cured with at least one of a
variety of curing
agents, including reactive, non-reactive (e.g., "catalysts") and partially
reactive agents.
Generally, preferred curing agents are selected from amine-based curing
agents, hydroxyl-
functional curing agents, polyols, and/or metal-based catalysts. Particularly
preferred curing
agents are one or more monofunctional alcohols, amines and/or amides. The
amine-based curing
11/51
CA 02889928 2015-04-29
WO 2014/052459
PCT/US2013/061688
agents may also be used as a mixture of a fast-acting first curing agent and a
second, latent
curing agent. Either of these first and/or second amine-based curing agents
may be reactive,
nonreactive or partially reactive.
[49] Suitable single amine-based curing agent or a mixture of amine-based
curing
agents can include, but are not limited to, ethylene diamine; hexamethylene
diamine; 1-methyl-
2,6-cyclohex yl diamine; 2,2,4- and 2,4,4-trimethy1-1,6-hexanediamine; 4,4'-
bis-(sec-
butylamino)-dicyclohexylmethane and derivatives thereof; 1,4-bis-(sec-
butylamino)-
cyclohexane; 1,2-bis-(sec-butylamino)-cyclohexane; 4,4'-dic yclohex ylmethane
diamine; 1,4-
cyclohexane-bis- (methylamine); 1,3-c yclohexane-bis- (methylamine), isomers,
and mixtures
thereof; diethylene glycol bis-(aminopropyl)ether; 2-methylpentamethylene-
diamine;
diaminocyclohexane, isomers, and mixtures thereof; diethylene triamine;
triethylene tetramine;
tetraethylene pentamine; propylene diamine; 1,3-diaminopropane; dimethylamino
propylamine;
diethylamino propylamine; imido-bis-(propylamine); monoethanolamine,
diethanolamine;
triethanolamine; monoisopropanolamine, diisopropanolamine; isophoronediamine;
4,4'-
methylenebis-(2-chloroaniline); 3,5-dimethylthio-2,4-toluenediamine; 3,5-
dimethylthio-2,6-
toluenediamine; 3,5-diethylthio-2,4-toluenediamine; 3,5-diethylthio-2,6-
toluenediamine; 4,4'-
bis-(sec-butylamino)-benzene; and derivatives thereof; 1,4-bis-(sec-
butylamino)-benzene; 1,2-
bis-(sec-butylamino)-benzene; N,N1-dialkylamino-diphenylmethane;
trimethyleneglycol-ci-p-
aminobenzoate; polytetramethyleneoxide-di-p-aminobenzoate; 4,4'-methylenebis-
(3-chloro-2,6-
diethyleneaniline);4,4'-methylenebis-(2,6-diethylaniline); meta-
phenylenediamine;
paraphenylenediamine; N,N'-diisopropyl-isophoronediamine; polyoxypropylene
diamine;
propylene oxide-based triamine; 3,3'-dimethy1-4,4'-ciaminocyclohexylmethane;
and mixtures
thereof. In one embodiment, the amine-terminated curing agent is 4,4'-bis-(sec-
butylamino)-
dicyclohexylmethane. Preferred amine-based curing agents for use with the
present invention
include triethylenediamine; bis(2-dimethylaminoethyl)ether;
tetramethylethylenediamine;
pentamethyldiethylenetriamine; 1,3,5-tris (3-(dimethylamino)prop y1)-
hexahydro-s-triazine and
other tertiary amine products of alkyleneamines.
[50] Additionally, other catalysts that promote the reaction of isocyanates
with
hydroxyls and amines that are known by the industry can be used in the present
invention, e.g.,
transition metal co-catalysts of Groups III or IV used for polyurethane foams.
A particularly
preferred metal co-catalyst is a tin complex such as stannous 2-ethylhexanoate
or an organotin
compound, such as dibutyltin dilaurate and tin-containing salts.
12/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
[51] Also preferred are catalysts that promote isocyanate trimerization
over other
reaction mechanisms. See, e.g., US Patent No. 5,264,572 (cesium fluoride or
tetraalkylammonium fluoride), US Patent No. 3,817,939 (organic carbonate
salt), and US Patent
No. 6,127,308 (lithium salts, lithium hydroxide, allophane catalysts such as
tin-2-ethylhexanoate
or tin octoate, and organic compounds containing at least one hydroxyl group),
the disclosures of
which are herein incorporated by reference.
[52] The amine-based curing agent may have a molecular weight of about 64
or
greater. In one embodiment, the molecular weight of the amine-curing agent is
about 2000 or
less. In addition, any of the amine-terminated moieties listed above for use
as the isocyanate-
reactive component to form the prepolymer may be used as curing agents to
react with the
prep olymers.
[53] Of the list above, the saturated amine-based curing agents suitable
for use with the
present invention include, but are not limited to, ethylene diamine;
hexamethylene diamine; 1-
methy1-2,6-c yclohex yl diamine; 2,2,4- and 2,4,4-trimethy1-1,6-hexanediamine;
4,4'-bis-(sec-
butylamino)-dicyclohexylmethane;1,4-bis-(sec-butylamino)-cyclohexane;1,2-bis-
(sec-
butylamino-cyclohexane; derivatives of 4,4'-bis-(sec-butylamino)-
dicyclohexylmethane; 4,4'-
dic yclohex ylmethane diamine; 1,4-c yclohexane-bis- (methylamine); 1,3-c yc
lohexane-bis-
(methylamine); diethylene glycol bis-(aminopropyl) ether; 2-
methylpentamethylene-diamine;
diaminocyclohexane; diethylene triamine; triethylene tetramine; tetraethylene
pentamine;
propylene diamine; dipropylene triamine; 1,3-diaminopropane; dimethylamino
propylamine;
diethylamino prop ylamine; imido-bis- (propylamine); monoethanolamine,
diethanolamine;
triethanolamine; monoisopropanolamine, diisopropanolamine;
triisopropanolamine;
isophoronediamine; N,N'-diisopropylisophorone diamine and mixtures thereof.
[54] In one embodiment, the curative used with the prepolymer include 3,5-
dimethylthio-2,4-toluenediamine,3,5-dimethyl-thio-2,6-toluenediamine, 4,4'-
bis-(sec-
butylamino)-diphenylmethane, N,N'-diisopropyl-isophorone diamine;
polyoxypropylene
diamine; propylene oxide-based triamine; 3,3'-dimethy1-4,4'-
diaminocyclohexylmethane; and
mixtures thereof.
[55] Because unhindered primary diamines result in a rapid reaction between
the
isocyanate groups and the amine groups, in certain instances, a hindered
secondary diamine may
be more suitable for use in the prepolymer. Without being bound to any
particular theory, it is
believed that an amine with a high level of stearic hindrance, e.g., a
tertiary butyl group on the
13/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
nitrogen atom, has a slower reaction rate than an amine with no hindrance or a
low level of
hindrance. For example, 4,4'-bis-(sec-butylamino)-dicyclohexylmethane
(CLEARLINK 1000
from Huntsman Corporation in The Woodlands, Texas) may be suitable for use in
combination
with an isocyanate to form the polyurea prepolymer. In addition, N,N'-
diisopropyl-isophorone
diamine, also available from Huntsman Corporation, under the tradename
JEFFLINK , may be
used as the secondary diamine curing agent.
[56] In addition, a trifunctional curing agent can be used to help improve
cross-linking
and, thus, to further improve the chemical and/or abrasion resistance of the
coating. In one
embodiment, a triol such as trimethylolpropane or a tetraol such as N,N,N',N'-
tetrakis (2-
hydroxylpropyl)ethylenediamine may be added to the formulations.
[57] The curing agents of the present invention can be added to the coating
formulation with the polyol component, the amine-reactive polyol component,
any of the
additives (e.g., coloring agents) or added simultaneously as any of these
components or pre-
coated on the proppant. Preferably, the curing agent is mixed with or co-
applied to the solid
proppant core as the first isocyanate and any other reactants are mixed so
that the curing process
has begun by the time the coating formulation is applied to the surface of the
solid proppant core.
It is also possible to premix the isocyanate and polyol together immediately
before entry into the
mixer. This probably would give a slightly more uniform distribution of the
chemicals in the
coating. Alternately, it would be possible to premix the polyol and curing
agent before they are
added to the isocyanate.
[58] Most preferably, the isocyanate, polyol, combination of (a) polyol and
(b) curing
agent or each individually are continuously added to solid proppant in a
moving mixer at a rate
that is not substantially greater than the rate of the crosslinking reaction
between and among the
ingredients. The specific rate will depend on the size of the mixer, the type
of mixer, and whether
batch or continuous production is desired. The goal is to substantially
completely coat the
proppant solid with a coating that becomes cured in the mixer and is
discharged as a free-
flowing, discrete particulates. The amperage draw rate of the mixer can be
used as a guide in
tumbling-type mixers because the build-up of an uncured, tacky coating on the
proppant solids
will increase the load on the mixer motor which can be monitored by a simple
amp meter.
Adding the reaction components at a rate that is consistent with the reaction
rate of the curing
process avoids substantial increases in amperage allows the coating process,
avoids stalling the
motor or interrupting the coating process, and maximizes the productivity of
the equipment used
14/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
to perform the coating/curing process. In a preferred process using laboratory
scale equipment, a
few seconds after beginning to add the polyol, the isocyanate is added at a
controlled rate over a
relatively short period, e.g., about a minute.
HYDROXY-FUNCTIONAL CURING AGENTS
[59] The proppant coating of the invention may also be cured alone or with
other
curing agents with a single hydroxy-terminated curing agent (i.e., a monol
such as C1-C20
alcohols such as ethanol, isopropyl, butanol, or stearyl alcohol) or a mixture
of hydroxy-
terminated curing agents. The appropriate use of such a monol capping agent or
chain terminator
can help to control the impact of internal, unreacted ¨NCO groups that can
have adverse
properties on the final coating. Indeed, the use of monol within the range
from about 1 equivalent
wt% to about 30 equivalent wt% relative to the weight of any added polyhydroxy
compounds
can bring into the coating certain properties that are not related to the
original isocyanate or
polyhydroxy component, such as enhanced or decreased hydrophobicity, corrosion
resistance,
viscosity modification in fracturing fluid, reduce the frictional drag of
production fluids once in
the fracture, ion exchange and antimicrobial effects.
[60] Suitable hydroxy-terminated curing agents include, but are not limited
to, ethanol,
ethylene glycol; diethylene glycol; polyethylene glycol; propylene glycol; 2-
methy1-1,3-
propanediol; 2,-methyl-1,4-butanediol; dipropylene glycol; polypropylene
glycol; 1,2-
butanediol; 1,3-butanediol; 1,4-butanediol; 2,3-butanediol; 2,3-dimethy1-2,3-
butanediol;
trimethylolpropane; cyclohexyldimethylol; triisopropanolamine; N,N,N'N'-tetra-
(2-
hydroxypropy1)-ethylene diamine; diethylene glycol bis-(aminopropyl)ether; 1,5-
pentanediol; 1,6-
hexanediol; 1,3-bis-(2-hydroxyethoxy)cyclohexane; 1,4-cyclohexyldimethylol;
1,3-bis-[2-(2-
hydroxyethoxy)ethoxy]cyclohexane; 1,3-his- { 2- [2-(2-
hydroxyethoxy)ethoxy]ethoxyl
cyclohexane; polytetramethylene ether glycol, preferably having a molecular
weight ranging
from about 250 to about 3900; resorcinol-di-(beta-hydroxyethyl)ether and its
derivatives;
hydroquinone-di-(beta-hydroxyethyl)ether and its derivatives; 1,3-bis-(2-
hydroxyethoxy)benzene; 1,3-bis-[2-(2-hydroxyethoxy)ethoxy]benzene; 1,3-bis- 1
24242-
hydroxyethoxy)ethoxylethoxylbenzene; N,N-bis(beta.-hydroxypropyl)aniline; 2-
propano1-1,1'-
phenylaminobis; and mixtures thereof.
15/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
[61] The hydroxy-terminated curing agent may have a molecular weight of at
least
about 50. In one embodiment, the molecular weight of the hydroxy-terminated
curing agent is
about 2000 or less. In yet another embodiment, the hydroxy-terminated curing
agent has a
molecular weight of about 250 to about 3900. It should be understood that
molecular weight, as
used herein, is the absolute weight average molecular weight and would be
understood as such
by one of ordinary skill in the art.
[62] The saturated hydroxy-terminated curing agents, included in the list
above, are
preferred when making a light stable composition. Those saturated hydroxy-
terminated curing
agents include, but are not limited to, ethylene glycol; diethylene glycol;
polyethylene glycol;
propylene glycol; 2-methyl-1,3-propanediol; 2,-methyl-1,4-butanediol;
dipropylene glycol;
polypropylene glycol; 1,2-butanediol; 1,3-butanediol; 1,4-butanediol; 2,3-
butanediol; 2,3-
dimethy1-2,3-butanediol; trimethylolpropane; cyclohexyldimethylol;
triisopropanolamine;
N,N,N,Nt-tetra-(2-hydroxypropy1)-ethylene diamine; diethylene glycol bis-
(aminopropyl)ether;
1,5-pentanediol; 1,6-hexanediol; 1,3-bis-(2-hydroxyethoxy)cyclohexane; 1,4-
cyclohexyldimethylol; 1,3-bis42-(2-hydroxyethoxy)ethoxylcyclohexane; 1,3-bis-{
24242-
hydroxyethoxy)ethoxylethoxy}cyclohexane; polytetramethylene ether glycol
having molecular
weight ranging from about 250 to about 3900; and mixtures thereof.
[63] The amount of curing agent that is added to the coating will generally
fall within
the range from about 0.01 wt% to about 95 wt% of the complete coating
formulation.
POLYOL CURING AGENTS
[64] A polyol component can be added to the coating formulation. The polyol
component may or may not have reactive amine functionality and can comprise
oxides,
polyesters, polyamides, polyurethane, epoxy, silicone or polysiloxane, or
vinyl backbones which
react to become an integral part of the resulting coating on the proppant
core.
[65] A useful polyurethane coating is a phenolic polyurethane made with a
phenolic
polyol according to a patent application that was filed with the German Patent
Office under no.
DE 10 2010 051 817.4 on November 19, 2010 and entitled "Proppant Coating
Technology", the
disclosure of which is herein incorporated by reference. This patent is
summarized below in the
context of the process of the present invention.
16/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
[66] Another polyol component for the present process comprises a
phenol resin that
comprises a condensation product of a phenol and an aldehyde, such as
formaldehyde. The
phenol resin is preferably a resole or novolak phenol resin and more
preferably a benzyl ether
resin.
[67]
The resole-type phenol resin can be obtained, for example, by condensation of
phenol or of one or more compounds of the following formula (I), with
aldehydes, preferably
formaldehyde, under basic conditions.
OH
I. (R)p
(I)
[68] In the formula (I):
[69] "R" is in each case, independently, a hydrogen atom, a halogen atom,
C1_16-alkyl
(preferably C1_12-alkyl, more preferably C1_6-alkyl, and still more preferably
methyl, ethyl, propyl or butyl) or ¨OH;
[70] "p" is an integer from 0 to 4, preferably 0, 1, 2 or 3, and more
preferably 1 or 2.
Those in the art will understand that when p is 0, the compound of formula (I)
is
phenol.
[71]
Novolak-type phenol resin for the present invention comprises the condensation
product of phenol or of one or more compounds of the formula (I) defined
above, with
aldehydes, preferably formaldehyde, under acidic conditions.
[72]
In another preferred embodiment, the phenol resin is a benzyl ether resin of
the
general formula (II):
¨ ¨ ¨ ¨
OH OH OH
R R R R
I I I I
HC ___
0 0 C 0 C 0 _________ C 0 H H H H H
A D A D A D
B B B
¨ m1 ¨ ¨ n ¨
m2
(II)
17/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
[73] In the formula (II):
[74] A, B and D each are, independently, a hydrogen atom, a halogen atom, a C1-
16-
hydrocarbon residue, -(C1_16-alkylene)-0H, -OH, an -0-(C1_16-hydrocarbon
residue), phenyl, -(C1_6-alkylene)-phenyl, or -(C1_6-alkylene)-phenylene-OH;
[75] The halogen atom is F, Cl, Br or I;
[76] The C1-16-hydrocarbon-residue is preferably C1_16-alkyl, C2_16-alkenyl
or C2_16-
alkinyl, more preferably C1_12-alkyl, C2_12-alkenyl or C2_12-alkinyl, still
more
preferably Ci_6-alkyl, C2-6-alkenyl or C2_6-alkinyl, and still more preferably
C1-4-
alkyl, C2_4-alkenyl or C2_4-alkinyl, and still more preferably C1_12-alkyl,
and still
more preferably C1_6-alkyl, and still more preferably methyl, ethyl, propyl or
butyl, and most preferably methyl;
[77] The residue -(C1_16-alkylene)-OH is preferably -(C1_12-alkylene)-0H, more
preferably -(C1_6-alkylene)-0H, and still more preferably -(C1_4-alkylene)-0H,
and most preferably a methylol group (-CH2-0H);
[78] The -0- (C1_16-hydrocarbon)-residue is preferably C1_16-alkoxy, more
preferably C1-12-
alkoxy, and still more preferably C1_6-alkoxy, and still more preferably C1-4-
alkoxy, and still more preferably -0-CH3, -0-CH2CH3, -0-(CH2)2CH3 or -0-
(CH2)3CH3;
[79] The residue -(C1_6-alkylene)-phenyl is preferably -(C1_4-alkylene)-
phenyl, and
more preferably -CH2-phenyl;
[80] The residue -(C1_6-alkylene)-phenylene-OH is preferably -(C1_4-alkylene)-
phenylene-
OH, and more preferably -CH2-phenylene-OH;
[81] R is a hydrogen atom of a Ci_6-hydrocarbon residue (e.g. linear or
branched C1-6-
alkyl). R is particularly preferred as a hydrogen atom. This is the case, for
example, when formaldehyde is used as aldehyde component in a condensation
reaction with phenols in order to produce the benzyl ether resin of the
formula
(II);
[82] m1 and m2 are each, independently, 0 or 1.
[83] n is an integer from 0 to 100, preferably an integer from 1 to 50,
more preferably
from 2 to 10, and still more preferably from 2 to 5; and
[84]
wherein the sum of n, m1 and m2 is at least 2.
18/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
[85] In a still further embodiment, the polyol component is a phenol resin
with
monomer units based on cardol and/or cardanol. Cardol and cardanol are
produced from cashew
nut oil which is obtained from the seeds of the cashew nut tree. Cashew nut
oil consists of about
90% anacardic acid and about 10% cardol. By heat treatment in an acid
environment, a mixture
of cardol and cardanol is obtained by decarboxylation of the anacardic acid.
Cardol and cardanol
have the structures shown below:
OH OH
___________________________ C15H31_n
C15 H31 _n
HO
n=0,246 n=0,2,4,6
Cardanol Cardol
[86] As shown in the illustration above, the hydrocarbon residue (¨Ci5H31,)
in cardol
and/or in cardanol can have one (n=2), two (n=4) or three (n=6) double bonds.
Cardol
specifically refers to compound CAS-No. 57486-25-6 and cardanol specifically
to compound
CAS-No. 37330-39-5.
[87] Cardol and cardanol can each be used alone or at any particular mixing
ratio in
the phenol resin. Decarboxylated cashew nut oil can also be used.
[88] Cardol and/or cardanol can be condensed into the above described
phenol resins,
for example, into the resole- or novolak-type phenol resins. For this purpose,
cardol and/or
cardanol can be condensed e.g. with phenol or with one or more of the above
defined compounds
of the formula (I), and also with aldehydes, preferably formaldehyde.
[89] The amount of cardol and/or cardanol which is condensed in the phenol
resin is
not particularly restricted and preferably is from about 1 wt% to about 99
wt%, more preferably
about 5 wt% to about 60 wt%, and still more preferably about 10 wt% to about
30 wt%, relative
to 100 wt% of the amount of phenolic starting products used in the phenol
resin.
[90] In another embodiment, the polyol component is a phenol resin obtained by
condensation of cardol and/or cardanol with aldehydes, preferably
formaldehyde.
[91] A phenol resin which contains monomer units based on cardol and/or
cardanol as
described above, or which can be obtained by condensation of cardol and/or
cardanol with
19/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
aldehydes, has a particularly low viscosity and can thus preferably be
employed with a low
addition or without addition of reactive thinners. Moreover, this kind of long-
chain, substituted
phenol resin is comparatively hydrophobic, which results in a favorable shelf
life of the coated
proppants obtained by the method according to the present invention. In
addition, a phenol resin
of this kind is also advantageous because cardol and cardanol are renewable
raw materials.
[92] Apart from the phenol resin, the polyol component can still contain other
compounds containing hydroxyl groups. The other compounds containing hydroxyl
groups can
be selected from the compounds containing hydroxyl groups that are known to be
useful for
making polyurethanes, e.g., hydroxy-functional polyethers, hydroxy-functional
polyesters,
alcohols or glycols. One preferred compound containing hydroxyl groups is, for
instance, castor
oil. Compounds containing hydroxyl groups such as alcohols or glycols, in
particular cardol
and/or cardanol, can be used as reactive thinners.
[93] The amount of the other compounds containing hydroxyl groups depends
on the
desired properties of the proppant coating and can suitably be selected by the
person skilled in
the art. Typical amounts of compounds containing hydroxyl groups are in the
range of between
about 10 wt% and about 80 wt%, preferably from about 20 wt% to about 70 wt%,
relative to 100
wt% of the polyol component.
[94] The process of the present invention is particularly useful when the
proppants are
coated with a condensation reaction product that has been made with an excess
of isocyanate
component with respect to the polyol or curing agent component. In step (a)
therefore, 100 parts
by weight of the polyol component is used with about 100 to about 600,
preferably about 210 to
about 530, more preferably about 220 to about 420, and still more preferably
about 230 to about
400 parts by weight of the isocyanate base value. Ratios of iso:polyol from
about 10:90 to as low
as 100:0 are usable depending on the equipment, conditions and production rate
provided that the
coating and reaction are completed during the coating process. The preferred
range of iso:polyol
is generally within the range from about 10:90 to about 90:10.
[95] The isocyanate base value defines the amount of the isocyanate
component which
is equivalent to 100 parts by weight of the polyol component. The NCO-content
(%) of the
isocyanate component is defined herein according to DIN ISO 53185. To
determine the OH-
content (%) of the polyol component, first the so-called OH-number is
determined in mg KOH/g
according to DIN ISO 53240 and this value is divided by 33, in order to
determine the OH-
content. Thus, in step (a) an excess of NCO-groups in the isocyanate component
of between
20/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
about 100 and about 500%, preferably about 110 to about 430%, more preferably
about 120% to
about 320%, and still more preferably about 130% to about 300%, relative to
the OH-groups in
the polyol component is used.
[96] Moreover, in step (a) one or more additives can be mixed with the
proppant, the
polyol component and the isocyanate component. These additives are not
particularly restricted
and can be selected from the additives known in the specific field of coated
proppants. Provided
that one of these additives has hydroxyl, amine or amide functional groups, it
should be
considered as a different reactive group-containing compound, as described
above in connection
with the polyol component. If one of the additives has isocyanate groups, it
should be
considered as a different isocyanate-group-containing compound. Additives with
hydroxyl
groups and isocyanate groups can be simultaneously considered as different
hydroxyl-group-
containing compounds and as different isocyanate-group-containing compounds.
REACTIVE AMINES OR AMIDES
[97] The coating formulation of the present invention also optionally
includes a
reactive amine or reactive amide component, preferably an amine-terminated
compound or an
amide. The coating formulation can, however, be made effectively and with good
properties in
the absence or substantial absence of a reactive amine component apart from
the reactive polyol
and isocyanate components. The reactive amine component can enhance crosslink
density within
the coating and, depending on component selection, can provide additional
characteristics of
benefit to the cured coating. Reactive amine components for use in the present
invention include
C1-C40 amine-terminated, amine-containing, or amide compounds such as
monoamines (e.g.,
butyl amine), amides (e.g., fatty acid amides, stearyl amides), diamines,
triamines, amine-
terminated glycols such as the amine-terminated polyalkylene glycols sold
commercially under
the trade name JEFFAMINE from Huntsman Performance Products in The Woodlands,
Texas.
The use of amides can be particularly useful for enhancing flow and
hydrophobic properties as
well as the antimicrobial properties of the coatings.
[98] Suitable diamines include primary, secondary and higher polyamines and
amine-
terminated compounds. Suitable compounds include, but are not limited to,
ethylene diamine;
propylenediamine; butanediamine; hexamethylenediamine; 1,2-diaminopropane; 1,4-
diaminobutane; 1,3-diaminopentane; 1,6-diaminohexane; 2,5-diamino-2,5-
dimethlhexane; 2,2,4-
21/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
and/or 2,4,4-trimeth y1-1,6-diaminohex ane; 1,11-diaminoundecane; 1,12-
diaminododecane; 1,3-
and/or 1,4-cyclohexane diamine; 1-amino-3,3,5-trimethy1-5-aminomethyl-
cyclohexane; 2,4-
and/or 2,6-hexahydrotoluylene diamine; 2,4' and/or 4,4'-diaminodicyclohexyl
methane and 3,3'-
dialky1-4,4'-diamino-dicyclohexyl methanes such as 3,3'-dimethy1-4,4-diamino-
dicyclohexyl
methane and 3,3'-diethyl-4,4'-diaminodicyclohexyl methane; aromatic polyamines
such as 2,4-
and/or 2,6-diaminotoluene and 2,6-diaminotoluene and 2,4' and/or 4,4'-
diaminodiphenyl
methane; and polyoxyalkylene polyamines (also referred to herein as amine
terminated
polyethers).
[99] Mixtures of polyamines may also be employed in preparing aspartic
esters, which
is a secondary amine derived from a primary polyamine and a dialkyl maleic or
fumaric acid
ester, for use in the invention. Representative examples of useful maleic acid
esters include
dimethyl maleate, diethyl maleate, dibutyl maleate, dioctyl maleate, mixtures
thereof and
homologs thereof.
[100] Suitable triamines and higher multifunctional polyamines for use in
the present
coating include diethylene triamine, triethylenetetramine, and higher homologs
of this series.
[101] JEFFAMINE diamines include the D, ED, and EDR series products. The D
signifies a diamine, ED signifies a diamine with a predominately polyethylene
glycol (PEG)
backbone, and EDR designates a highly reactive, PEG based diamine.
[102] JEFFAMINE D series products are amine terminated polypropylene
glycols with
the following representative structure:
H2,,h,õ..õ,.."-.....N
N N H2
0 X
C Fi3 CH3
JEFFAMINE x MW
D-230 -2.5 230
D-400 -6.1 430
D-2000 -33 2,000
D-4000 (XTJ-510) -68 4,000
[103] JEFFAMINE EDR-148 (XTJ-504) and JEFFAMINE EDR-176 (XTJ-590) amines
are much more reactive than the other JEFFAMINE diamines and triamines. They
are
represented by the following structure:
22/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
H-2
0 0
2
LH3 H3 CH3
JEFFAMINE y x z MW
HK-511 2.0 -1.2 220
ED-600 (XTJ-500) -9.0 -3,6 600
ED-900 (XTJ-501) -12.5 -6.0 900
ED-2003 (XTJ-502) -39 -6.0 2,000
[104] JEFFAMINE T series products are triamines prepared by reaction of
propylene
oxide (PO) with a triol intiator followed by amination of the terminal
hydroxyl groups. They are
exemplified by the following structure:
CH 3
_AO
H2C7nµ y NH2
H2 N H2
X
CH 3 H 3
Moles PO
JEFFAMINE R n (x+y+z) WV*
T-403 C2H5 1 5-6 440
T-3000 (XTJ-509) H 0 50 3000
T-5000 H 0 85 5000
[105] The SD Series and ST Series products consist of secondary amine
versions of the
JEFFAMINE core products. The SD signifies a secondary diamine and ST signifies
a secondary
trimine. The amine end-groups are reacted with a ketone (e.g. acetone) and
reduced to create
hindered secondary amine end groups represented by the following terminal
structure:
CH3 CH3
One reactive hydrogen on each end group provides for more selective reactivity
and
makes these secondary di- and triamines useful for intermediate synthesis and
intrinsically
slower reactivity compared with the primary JEFFAMINE amines.
23/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
JEFFAMINEe Base Product MW*
SD-231 (XI-J-584) 0-230 315
SD-401 (XTJ-585) 0-400 515
SD-2001 (XTJ-576) 0-2000 2050
ST-404 (XTJ-586) T-403 565
[106] See also U.S. Patent Nos. 6,093,496; 6,306,964; 5,721,315; 7,012,043;
and
Publication U.S. Patent Application No. 2007/0208156 the disclosures of which
are hereby
incorporated by reference.
[107] Additionally, the amine containing compound can be monofunctional as
primary
amines and amides, each capable of incorporating desireable properties into
the coating, e.g.,
hydrophobic characteristics, better flow properties and antimicrobial
properties.
ADDITIVES
[108] The proppant coating compositions of the invention may also include
various
additives. For example, the coatings of the invention may also include
pigments, tints, dyes, and
fillers in an amount to provide visible coloration in the coatings. Other
materials include, but are
not limited to, reaction enhancers or catalysts, crosslinking agents, optical
brighteners, propylene
carbonates, coloring agents, fluorescent agents, whitening agents, UV
absorbers, hindered amine
light stabilizers, defoaming agents, processing aids, mica, talc, nano-
fillers, silane coupling
agents, antislip agents, water affinity or repulsion components, impact
modifiers, water-activated
catalysts, viscosifiers, flowaids, anticaking agents, wetting agents,
toughening agents such as one
or more block copolymers, and components that act to remove at least some
portion of the heavy
metals and/or undesirable solutes found in subterranean groundwater. See,
copending US patent
application serial number 13/224726 filed on 1 September 2011 entitled "Dual
Function
Proppants", the disclosure of which is herein incorporated by reference. The
additives are
preferably present in an amount of about 15 weight percent or less. In one
embodiment, the
additive is present in a non-zero amount of about 5 percent or less by weight
of the coating
composition. Especially preferred are amorphous silica (e.g., silica flour,
fumed silica and silica
dispersions) and silica alternatives (such as those used in sandblasting as an
alternative to silica
or organofunctional silane like the DYNASYLAN fluids from Evonik Degussa
Corporation in
Chester, PA) that act as anticaking agents or dispersions that are applied to
the outer surfaces of
the coated proppant solid to prevent the formation of agglomerates during
packing and shipping.
24/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
Applied amounts of the amorphous silica are generally within the range from
about 0.001 wt% to
about 1 wt% based on the dry proppant weight.
[109] Other additives can include, for example, solvents, softeners,
surface-active
agents, molecular sieves for removing the reaction water, thinners and/or
adhesion agents can be
used. Silanes are a particularly preferred type of adhesion agent that
improves the affinity of the
coating resin for the surface of the proppant. Silanes can be mixed in as
additives in step (a), but
can also be converted chemically with reactive constituents of the polyol
component or of the
isocyanate component. Functional silanes such as amino-silanes, epoxy-, aryl-
or vinyl silanes
are commercially available and, as described above, can be used as additives
or can be converted
with the reactive constituents of the polyol component or of the isocyanate
component. In
particular, amino-silanes and epoxy-silanes can be easily converted with the
isocyanate
component.
[110] An optional, additional additive is a contaminant removal component
that will
remove, sequester, chelate or otherwise clean at least one contaminant,
especially dissolved or
otherwise ionic forms of heavy metals and naturally occurring radioactive
materials (NORMS),
from subterranean water or hydrocarbon deposits within a fractured stratum
while also propping
open cracks in said fractured stratum. Preferably, the contaminant removal
component is
associated with the proppant solid as a chemically distinct solid that is
introduced together with
the proppant solid as: (a) an insoluble solid secured to the outer or inner
surface of the proppant
solid with a coating formulation that binds the solids together, (b) as a
solid lodged within pores
of the proppant solid or (c) as a chemical compound or moiety that is mixed
into or integrated
with a coating or the structure of the proppant solid. See copending US patent
application serial
number 13/224726 filed on 2 September 2011 entitled "Dual Function Proppants"
the disclosure
of which is herein incorporated by reference. Additional added functionality
can also be in the
form of fracturing fluid breakers, de-emulsifiers, and bactericides.
[111] The added functionality of an auxiliary particle to the proppant may
also be in the
form of an ion exchange resin that is pretreated or which itself constitutes a
dissolvable solid for
the slow release of corrosion or scale inhibitors. Such slow release materials
could prove
beneficial and advantageous to the overall operation and maintenance of the
well.
25/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
PROPPANT CORE SOLIDS
[112] The proppants can be virtually any small solid with an adequate crush
strength
and lack of chemical reactivity. Suitable examples include sand, ceramic
particles (for instance,
aluminum oxide, silicon dioxide, titanium dioxide, zinc oxide, zirconium
dioxide, cerium
dioxide, manganese dioxide, iron oxide, calcium oxide, magnesium oxide, or
bauxite), or also
other granular materials.
[113] Proppant sands are a preferred type of proppant for the present
invention. Sand is
mainly used in the hydraulic fracturing process of natural gas and oil wells
to increase their
productivity of valuable natural resources. Proppant sand is monocrystalline
with a high silica
content of at least 80 wt%, and more typically greater than about 97 wt%
silica.
[114] American Petroleum Institute specifications place the following
limitations on
sieve distribution for proppants suitable for use in a fracture:
= At least 90% of material must fall between the two mesh sizes,
= No more than 10% of the material may be coarser than the largest mesh
size,
= No more than 0.1% of the material may be coarser than the next largest
mesh size [e.g.
for 20/40, up to 10% of the proppant may be between 16 and 20 mesh, but no
more than
0.1% can exceed 16 mesh], and
= No more than 1% of material is permitted to fall onto the pan.
[115] According to bulk density, proppant is divided into: low-density,
medium density,
high-density. According to the anti-crushing strength, proppant is divided
into 52Mpa, 69Mpa,
86Mpa and 103Mpa four series. Specifications of proppant sand are generally:
12-18 mesh, 12-
20 mesh, 16-20 mesh, 16-30 mesh, 20-40 mesh between 30-50 mesh, 40-60 mesh, 40-
70 mesh
and smaller. The proppants to be coated preferably have an average particle
size within the range
from about 50 [t. m and about 3000 [t. m, and more preferably within the range
from about 100 [t. m
to about 2000 [t.m.
COATING METHOD
[116] The method for the production of coated proppants according to the
present
invention can be implemented without the use of solvents. Accordingly, the
mixture obtained in
step (a) in one embodiment of the method is solvent-free, or is essentially
solvent-free. The
26/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
mixture is essentially solvent-free, if it contains less than 20 wt%,
preferably less than 10 wt%,
more preferably less than 5 wt%, and still more preferably less than 3 wt%,
and most preferably
less than 1 wt% of solvent, relative to the total mass of components of the
mixture.
[117] Preferably, the method is implemented without the use of organic
solvents. In
this case, the mixture obtained in step (a) is free of organic solvents, or is
essentially free of
organic solvents. The mixture is essentially free of organic solvents, if it
contains less than 20
wt%, preferably less than 10 wt%, more preferably less than 5 wt%, and still
more preferably
less than 3 wt%, and most preferably less than 1 wt% of solvent, relative to
the total mass of
components of the mixture.
[118] In step (a) the proppant is preferably heated to an elevated
temperature and then
contacted with the coating components. Preferably, the proppant is heated to a
temperature
within the range of about 50 C to about 150 C to accelerate cros slinking
reactions in the applied
coating.
[119] The temperature of the coating process is not particularly restricted
outside of
practical concerns for safety and component integrity. The preferred
conditions for the
coating/curing step of the present invention are generally at conditions
within the range of about
50 to about 175 C, more preferably at a temperature within the range from
about 75 C to about
150 C, and most preferably at a temperature within the range from about 80 C
to about 135 C.
This temperature avoids a number of emissions issues, reduces the amount of
energy consumed
in the coating process and also reduces the cooling time for the coated
proppants for further
handling and packaging.
[120] The mixer used for the coating process is not particularly restricted
and can be
selected from among the mixers known in the specific field. For example, a pug
mill mixer,
agitation mixer, drum mixer, plate-type mixer, tubular mixer, trough mixer or
conical mixer can
be used. The easiest way is mixing in a rotating drum. As continuous mixer, a
worm gear can,
for example, be used.
[121] Mixing can be carried out on a continuous or discontinuous basis. In
suitable
mixers it is possible, for example, to add adhesion agents, isocyanate, amine
and optional
ingredients continuously to the heated proppants. For example, isocyanate
components, amine
reactant and optional additives can be mixed with the proppant solids in a
continuous mixer
(such as a worm gear) in one or more steps to make one or more layers of cured
coating.
27/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
[122] Preferably, the proppant, isocyanate component, curing agent, amine
reactant,
polyol, and optional additives are mixed homogeneously. Thus, the isocyanate
component and
amine reactant are distributed uniformly on the surface of the proppants. The
coating ingredients
are preferably kept in motion throughout the entire mixing process.
[123] It is also possible to arrange several mixers in parallel, series, or
serially in several
runs in one mixer.
[124] Importantly, the time, temperature, chemistry and reaction rate of
the
coating/curing process can be combined in proportions that will affect
performance
characteristics of the resulting cured coating. Preferably, an isocyanate-
containing component is
used in an amount within the range from about 100 wt% to about 400 wt% based
on a reactive
polyol-containing component in the curable coating mixture. Lower proportions
of excess
isocyanate can be used to move the curing process towards substantially
complete reaction of all
of the ¨NCO groups within the applied proppant coating by the time the product
is discharged as
a free-flowing solid. The lower amount of isocyanate-containing component tend
to add more
thermoplastic properties to the coating for better performance in low
temperature applications.
Preferably, the proppant coating is cured to an amount less than about 10 wt%
of reactive ¨NCO
groups based on the originally applied weight of the proppant coating. The
most preferred low
temperature proppants according to the invention contain have a weight ratio
of isocyanate-
functional component that is within the range from about 100-175 wt% of the
polyol-functional
component with a low coating loss under simulated downhole testing conditions.
[125] Having more unreacted ¨NCO groups can be useful to develop more
thermoset
characteristics in the coating thereby making the proppant better suited for
high temperature
applications. In such a case, a higher amount of isocyanate is used. The
preferred high
temperature product contains about 200-400% by weight of isocyanate-functional
component to
polyol-functional component and exhibits a coating loss of less than about 2%
in simulated
downhole testing conditions.
[126] The coating is preferably performed at the same time as the curing of
the coating
on the proppant. In the present invention, the coated proppant becomes free-
flowing at a time of
less than 5 minutes, preferably within the range of 1-4 minutes, more
preferably within the range
of 1-3 minutes, and most preferably within the range of 1-2.5 minutes to form
a coated,
substantially cured, free-flowing, coated proppant. This short cycle time
combines with the
relatively moderate coating temperatures to form a coating/curing process that
provides lower
28/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
energy costs, smaller equipment, reduced emissions from the process and the
associated
scrubbing equipment, and overall increased production for the coating
facility.
[127] The coating material may be applied in more than one layer. In this
case, the
coating process is repeated as necessary (e.g. 1-5 times, 2-4 times or 2-3
times) to obtain the
desired coating thickness. A typical size range for coated proppant is
typically within the range
of about 16 to about 100 mesh.
[128] The amount of coating resin, that is, of the polyurethane resin
applied to a
proppant, is preferably between about 0.5 and about 10 wt%, more preferably
between about 1%
and about 5 wt%, resin relative to the mass of the proppant as 100 wt%. With
the method
according to the present invention proppants can be coated at temperatures
between about 10 C
and about 150 C and preferably in a solvent-free manner. The coating process
requires a
comparatively little equipment and if necessary can also be carried out near
the sand or ceramic
substrate source, near the geographically location of the producing field or
at/near the well itself.
[129] The coated proppants can additionally be treated with surface-active
agents,
anticaking agents, or auxiliaries, such as talcum powder or stearate or other
processing aids such
as fine amorphous silica to improve pourability, wettability (even to the
extent that a water
wetting surfactant can be eliminated), dispersability, reduced static charge,
dusting tendencies
and storage properties of the coated product.
[130] If desired, the coated proppants can be baked or heated for a period
of time
sufficient to further enhance the ultimate performance of the coated particles
and further react
the available isocyanate, hydroxyl and reactive amine groups that might remain
in the coated
proppant. Such a post-coating cure may occur even if additional contact time
with a catalyst is
used after a first coating layer or between layers. Typically, the post-
coating cure step is
performed like a baking step at a temperature within the range from about 1000
- 200 C for a
time of about 1 minute to 4 hours, preferably the temperature is about 125 -
200 C for -1-30
minutes.
[131] Even more preferably, the coated proppant is cured for a time and
under
conditions sufficient to produce a coated proppant that exhibits a loss of
coating of less than 25
wt%, preferably less than 15 wt%, and even more preferably less than 5 wt%
when tested
according to simulated downhole conditions under ISO 13503-5:2006(E). Even
more preferably,
the coated proppant exhibits the low dust and handling characteristics of a
pre-cured proppant
(see API RP 60) but also exhibits a crush test result at 10,000 psi of less
than 2%, more
29/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
preferably less than 1.5%, and especially less than 1%. The coated proppants
of the invention
preferably also have an unconfined compressive strength of greater than 100
psi and more
preferably more than 500 psi with a fracture conductivity at a given closure
stress that is
substantially equal to, or greater than, the conductivity of a phenolic
coating used in the same
product application range.
I TSING THE COATED PROPPANTS
[132] The invention also includes the use of the coated proppants in
conjunction with a
fracturing liquid to increase the production of petroleum or natural gas.
Techniques for
fracturing an unconsolidated formation that include injection of consolidating
fluids are also well
known in the art. See U.S. Patent No. 6,732,800 the disclosure of which is
herein incorporated by
reference. Generally speaking, a fluid is injected through the wellbore into
the formation at a
pressure less than the fracturing pressure of the formation. The volume of
consolidating fluid to
be injected into the formation is a function of the formation pore volume to
be treated and the
ability of the consolidating fluid to penetrate the formation and can be
readily determined by one
of ordinary skill in the art. As a guideline, the formation volume to be
treated relates to the height
of the desired treated zone and the desired depth of penetration, and the
depth of penetration is
preferably at least about 30 cm radially into the formation. Please note that
since the
consolidation fluid is injected through the perforations, the treated zone
actually stems from the
aligned perforations.
[133] Before consolidating the formation, according to a preferred
embodiment, an acid
treatment is performed by injection of an acidic fluid. As it is well known in
the art, this acidic
treatment typically includes several stages such as an acid preflush, one or
more stages of acid
injection and an overflush.
[134] After the perforation and the consolidation, the final step is the
fracturing step.
Although a resin treatment alone may have been sufficient in preventing early
sand production
the resin reduces the permeability of the formation around the wellbore. The
primary purpose of
the fracture treatment is to connect the wellbore to the formation and in
doing so by pass any
damage and act as a filter allowing the production of hydrocarbons while
holding back formation
material. The high surface area associated with a fracture makes it a very
effective filter, for
30/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
example, a 13.7 m fracture length with 25 cm height has a surface area of 368
m2, compared to
the open hole flow area for a gravel pack of 3.2 m2 with the same zone height.
[135] Techniques for hydraulically fracturing a subterranean formation will
be known to
persons of ordinary skill in the art, and will involve pumping the fracturing
fluid into the
borehole and out into the surrounding formation. The fluid pressure is above
the minimum in situ
rock stress, thus creating or extending fractures in the formation. In order
to maintain the
fractures formed in the formation after the release of the fluid pressure, the
fracturing fluid
carries a proppant whose purpose is to prevent the fracturing from closing
after pumping has
been completed.
[136] The fracturing liquid is not particularly restricted and can be
selected from among
the frac liquids known in the specific field. Suitable fracturing liquids are
described, for
example, in WC Lyons, GJ Plisga, Standard Handbook Of Petroleum And Natural
Gas
Engineering, Gulf Professional Publishing (2005). The fracturing liquid can
be, for example,
water gelled with polymers, an oil-in-water emulsion gelled with polymers, or
a water-in-oil
emulsion gelled with polymers. In one preferred embodiment, the fracturing
liquid comprises
the following constituents in the indicated proportions: 1000 1 water, 20 kg
potassium chloride,
0.120 kg sodium acetate, 3.6 kg guar gum (water-soluble polymer), sodium
hydroxide (as
needed) to adjust a pH-value from 9 to 11, 0.120 kg sodium thiosulfate, 0.180
kg ammonium
persulfate and optionally a crosslinker such as sodium borate or a combination
of sodium borate
and boric acid to enhance viscosity.
[137] In addition, the invention relates to a method for the production of
petroleum or
natural gas which comprises the injection of the coated proppant into the
fractured stratum with
the fracturing liquid, i.e., the injection of a fracturing liquid which
contains the coated proppant,
into a petroleum- or natural gas-bearing rock layer, and/or its introduction
into a fracture in the
rock layer bearing petroleum or natural gas. The method is not particularly
restricted and can be
implemented in the manner known in the specific field.
[138] Suitable proppants include, but are not limited to, sand, bauxite,
glass beads, and
ceramic beads and resin-coated versions of each. The proppant will typically
exhibit a size
within the range from about 8 to about 100 U.S. Standard Mesh in size.
Mixtures of suitable
proppants can be used. The concentration of proppant in the fracturing fluid
can be any
concentration known in the art, and will typically be in the range of about
0.5 to about 20 pounds
of proppant added per gallon of clean fluid.
31/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
[139] The fracturing fluid can contain an added proppant-retention agent,
e.g. a fibrous
material, a curable resin coated on the proppant, platelets, deformable
particles, or a sticky
proppant coating to trap proppant particles in the fracture and prevent their
production through
the wellbore. Fibers, in concentration that preferably ranges from about 0.1%
to about 5.0% by
weight of proppant, for example selected from natural organic fibers,
synthetic organic fibers,
glass fibers, carbon fibers, ceramic fibers, inorganic fibers, metal fibers
and mixtures thereof, in
combination with curable resin-coated proppants are particularly preferred.
The proppant-
retention agent is intended to keep proppant solids in the fracture, and the
proppant and
proppant-retention agent keep formation particles from being produced.
EXAMPLES
[140] Conductivity testing was performed at simulated downhole conditions
using the
method and procedures found in ISO 13503-5:2006. In such tests, a closure
stress is applied
across a test unit for 50 hours to allow the proppant sample bed to reach a
semi-steady state
condition. Initially the pack is allowed something around 16 hours to
stabilize at 1000 psi
closure stress and the test temperature before elevating the closure stress on
the proppant. As the
fluid is forced through the proppant bed, the pack width, differential
pressure, pressure drop,
temperature and flow rates are measured at each stress. Proppant pack
permeability and
conductivity are then calculated.
[141] Multiple flow rates are used to verify the performance of the
transducers, and to
determine Darcy flow regime at each stress; an average of the data at these
flow rates is reported.
The test fluid is 2 wt% potassium chloride substitute solution filtered to
311m absolute. The
initial conductivity, permeability and width is measured and compared to the
final conductivity,
permeability and width after each stress period. Stress is applied and
maintained using an Isco
260D. Stress is applied at 100 psi/minute.
[142] Width of the proppant pack is determined by assembling the
conductivity cell
with the Ohio sandstone wafers and shims without the sample proppants. The
distance between
the width bars that are attached to each end of the conductivity cells are
measured at each of the
four corners and recorded. The cells are then assembled with the proppant
samples. The
measurements are made again at the beginning and ending of each stress period.
Width is
32/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
determined by subtracting the average of the zero from the average of each of
the stress width
values. Conductivity is calculated using Darcy's equation.
Conductivity; kWf =26.78 Q/(AP)
Permeability; k=321.41.0/[(AP)Wf]
wherein:
k is the proppant pack permeability, expressed in Darcy's;
kWf is the proppant pack conductivity, expressed in millidarcy-feet
II is the viscosity of the test liquid at test temperature, expressed
in centipoises;
Q is the flow rate, expressed in cubic centimeters per minute;
AP is the differential pressure, expressed in psi;
Wf is proppant pack width, expressed in inches.
[143] Sieve analysis is performed using the procedure found in ISO 13503-2
"Measurements of proppants used in hydraulic fracturing and gravel pack
operations" Standard
US mesh screens are used to separate the sample by size. Not more than 0.1%
should be greater
than the first specified sieve and not more than 1% should be retained in the
pan. There should
be at least 90% retained in the specified screens.
[144] To determine the magnitude of coating loss during the conductivity
test, samples
of the proppant pack are taken, dried in an oven and weighed. They are then
subjected to a
temperature of 960 C for 2.5 hours. At the end of this period the samples are
cooled and
weighed again. The difference between the sample weight after drying but
before being
subjected to the furnace compared to the sample weight after the time in the
furnace, equates to
the coating weight. Comparing this number to the same test performed on a
sample of the coated
material before being subjected to the conductivity test, will equate to the
coating weight lost due
to the long term exposure to the conditions of the conductivity tests.
[145] The procedure used in an autoclave test would be as follows:
[146] The autoclave test utilizes what amounts to a pressure cooker to
subject the coated
sands to a hot wet environment that is above the boiling temperature of water.
Approximately 20
g of sample is placed in a jar along with 150 ml of distilled water. The lids
are placed on sample
jars but not tightened. The samples are placed in the autoclave and the
chamber is sealed. Heat is
applied until the autoclave temperature reaches 250-265 F (121 -129 C). The
samples are
maintained under these conditions for the ten day period. At the end of the
test period the
autoclave is cooled down, opened and the sample jars removed. Each sample is
washed with
33/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
distilled water and then placed in an oven to dry. The dried samples are then
put through a
standard test for determination of coating loss. This result is compared to
the results of a coating
test that was run on the original sample. The difference in coating weight
before and after the
autoclave test, quantifies the amount of coating that was dissolved by the
exposure to a hot water
environment.
EXAMPLE 1:
[147] Ten pounds of Minnesota 40/70 fracturing sand is heated to 200 F
in a laboratory
mixer at which point the following components are added in the sequence and
timing as given
below in Tables 1 and 2. The weight ratio of the poly-MDI to phenolic polyol
in this example is
75/25.
Table 1
WEIGHT (grams) COMPONENT
4540 Minnesota sand
4.5 silane coupling agent
2.3 50% red iron oxide in castor oil
6.9 1,3,5-tris(3-(dimethylamino)propy1)-hexahydro-s-triazine
(JEFFCAT TR90)
34.2 Phenolic Polyol comprising 48% phenolic resin, 28% cashew nut
oil, 24% castor oil
102.2 Poly-MDI (32% NCO content)
2.5 Wetting agent
34/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
Table 2
TIME (minutes: seconds) ADDITION/COMMENT
0:00 Sand is at 200 F
0:00 4.5 gms silane coupling agent is added over 30 secs
0:00 JEFFCAT TR90 is added over 20 secs
0:00 Blend of red iron oxide and Phenolic Polyol is added
over 60
seconds
0:10 Poly-MDI is added over 60 seconds
2:00 Product is free flowing
3:30 Wetting agent is added over 5 seconds
4:00 Product is discharged at 180 F
[148] In this and the other examples presented herein, it was noticed that
the JEFFCAT
TR90 catalyst increased the reaction rate sufficiently that the amperage on
the associated mixer
was not exceeded as the coating reactants were metered into the proppant
solids in the moving
mixer. This suggests that the coating became cured at a rate that was
consistent with the feed rate
so that the liquid viscosity did not increase the electrical load on the
mixer. This same method of
controlled, metered addition would also apply for other formulations and
chemistries under the
present invention in order to keep the contents reacting at a rate that does
not tax the load on the
mixing equipment.
[149] The resin coated sand from the example above tested at 2.75% coating
level from
the mixer. When subjected to a three day 250 F autoclave test, the coating
level was measured
again at 2.34% reflecting the good resistance to hot water removal of the
coating.
EXAMPLE 2:
[150] Ten pounds of Genoa 40/70 fracturing sand is heated to 204 F in a
laboratory
mixer at which point the following components are added in the sequence and
timing as given
below in Tables 3 and 4. The weight ratio of the poly MDI to phenolic polyol
in this example is
75/25.
35/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
Table 3
WEIGHT (grams) COMPONENT
4540 Genoa sand
4.5 A1100 silane coupling agent
2.3 50% red iron oxide in castor oil
6.9 JEFFCAT TR90
18.2 Phenolic Polyol comprised of 48% phenolic resin, 28%
cashew
nut oil, 24% castor oil
54.5 Poly-MDI (32% NCO content)
2.3 Wetting agent
Table 4
TIME (minutes: seconds) ADDITION/COMMENT
0:00 Sand is at 204 F
0:00 4.5 gms A1100 is added over 10 secs
0:00 JEFFCAT TR90 is added over 10 secs
0:00 blend of red iron oxide and Phenolic Polyol is added
over 30
seconds
0:10 polyMDI is added over 30 seconds
2:00 Product is free-flowing
3:30 Wetting agent is added over 5 seconds
4:00 Product is discharged at 182 F
[151] The resin coated sand from the example above tested as having 1.48%
coating
from the mixer. When subjected to a three day 250 F autoclave test, the
coating level was
measured again at 1.43% reflecting the good resistance to hot water removal of
the coating.
EXAMPLE 3:
[152] Ten pounds of Minnesota fracturing sand is heated to 200 F in a
laboratory mixer
at which point the following components are added in the sequence and timing
as given below in
Tables 5 and 6. The weight ratio of the poly MDI to phenolic polyol in this
example is 92/8.
36/51
CA 02889928 2015-04-29
WO 2014/052459
PCT/US2013/061688
Table 5
WEIGHT (grams) COMPONENTS:
4540 Minnesota sand
4.5 A1100 silane coupling agent
2.3 50% red iron oxide in castor oil
6.9 JEFFCAT TR 90
12 Phenolic Polyol comprised of 48% phenolic resin, 28%
cashew
nut oil, 24% castor oil
135 Poly-MDI (32% NCO content)
2.5 Wetting agent
Table 6
TIME (minutes: seconds) ADDITION/COMMENT
0:00 Sand is at 202 F
0:00 A1100 is added over 20 secs
0:00 JEFFCAT TR90 is added over 20 secs
0:00 Blend of red iron oxide and Phenolic Polyol is added over 60
seconds
0:10 Poly-MDI is added over 60 seconds
2:00 Product is free flowing
3:30 Wetting agent is added over 5 seconds
4:00 Product is discharged at 170 F
[153] The resin coated sand from example 3 tested at 2.80% coating level
from the
mixer. When subjected to a three day 250 F autoclave test, the coating level
was measured
again at 2.56% reflecting the good resistance to hot water removal of the
coating.
EXAMPLE 4:
[154] One kg of 40/70 Minnesota fracturing sand is heated to 210 F in a
laboratory
mixer at which point the following components are added in the sequence and
timing as given
below in Tables 7 and 8. The weight ratio of the poly-MDI to the aminated
polyalkyleneglycol
(JEFFAMINE D230) is 63/37.
37/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
Table 7
WEIGHT (grams) COMPONENTS
1000 Minnesota sand
1 A1100 silane coupling agent
20 Poly-MDI (32% NCO content)
12 JEFFAMINE D230
Table 8
TIME (minutes: seconds) ADDITION/COMMENT
0:00 Sand is at 210 F
0:00 A1100 is added over 10 secs
0:10 Poly-MDI is added over 30 secs
0:50 JEFFAMINE D230 is added over 10 secs
2:00 Product is free flowing
4:00 Product is discharged at 140 F
[155] The resin coated sand from the example above tested at 2.90% coating
level from
the mixer. When subjected to a three day 250 F autoclave test, the coating
level was measured
again at 2.83% reflecting the good resistance to hot water removal of the
coating.
EXAMPLE 5:
[156] One kg of 40/70 Minnesota fracturing sand is heated to 210 F in a
laboratory
mixer at which point the following components are added in the sequence and
timing as given
below in Tables 9 and 10. The weight ratio of the poly-MDI to the aminated
polyalkyleneglycol
(JEFFAMINE D230 from Huntsman Corporation) is 63/37.
Table 9
WEIGHT (grams) COMPONENTS
1000 Minnesota sand
1 A1100 silane coupling agent
20 Poly-MDI (32% NCO content)
12 Aminatedpolyalkyleneglycol
0.6 Triethylenediamine
38/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
Table 10
TIME (minutes: seconds) ADDITION/COMMENT
0:00 Sand is at 210 F
0:00 A1100 is added over 10 secs
0:10 Poly-MDI is added over 30 secs
0:50 Preblended Aminated polyalkyleneglycol and
Triethylenediamine
are added over 10 s
2:00 Product is free flowing
4:00 Product is discharged at 145 F
[157] The resin coated sand from the example above tested at 2.84% coating
level from
the mixer. When subjected to a three day 250 F autoclave test, the coating
level was measured
again at 2.63% reflecting the good resistance to hot water removal of the
coating.
EXAMPLE 6:
[158] In this example, a series of test results were performed to
demonstrate the
properties of proppant coatings that include completely reacted (pre-cured)
and partially cured
phenolic coatings as compared to the coating of the present invention ("new
technology
coating"). The graph in Figure 1 illustrates the TMA results for (a) Pre-cured
phenolic coated
sand, (b) New Technology coated sand using the formulation of Example 1, (c) a
partially cured,
phenolic-coated sand (also identified as Phenolic A) and (d) a somewhat more
curable, phenolic-
coated sand (also identified as Phenolic B)
[159] The ThermoMechanical Analyzer (TMA) as supplied by TA Instruments is
a
device that accurately imposes a small force (i.e., a load) onto a sample
which is then subjected
to a desired temperature ramp over a defined time. During this increasing
temperature period,
the force is held constant. The probe which imposes the force is connected to
a sophisticated
micrometer that is capable of measuring fractions of a micron change in the
position of the
probe. Any change in the position of the probe can be interpreted to reflect
an expansion or
contraction of the sample that is brought about by the temperature change(s).
In many
applications, the sample merely expands as it is being heated (for instance
raw sand) thereby
39/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
creating a database that refers to the coefficient of thermal expansion. The
TMA has the ability
to run the samples in a variety of environments.
[160] In general, a pre-cured, phenolic-coated sand will be characterized
by a plot that
is essential flat (parallel to the X axis) or has a positive slope. This
response is indicative of a
coating that is essentially reacted in which there is little to no remaining
reactivity that remains in
the coating.
[161] If, however, a more curable or partially cured phenolic coating is
tested, the TMA
plot will exhibit a negative slope as early as 80 C to 100 C, but more often
after about 125 C to
about 175 C. This type of plot is characteristic of a coating that has
retained a level of reactivity
even after completing the manufacturing process. The more negative the slope
and the lower the
temperature in which the slope turns negative, the more reactivity that has
been left in the
coating.
[162] As shown in Figure 1, the top curve is labeled "Pre-cured" and is
indicative of the
response of a phenolic coating that is no longer reactive. The two lower
curves are labeled
"Partially Cured Phenolic A" and "More Curable Phenolic B." These curves
represent the TMA
results from two levels of partially cured coatings. The second curve labeled
"New Technology"
shows a response that is similar to the pre-cured coating curve but actually
shows properties that
fall between a pre-cured coating and the less reactive partially cured
coating. The shape of the
New Technology curve indicates that the New Technology coating would exhibit
some
properties that are similar to a pre-cured coating and others that may be
similar to the partially
cured coatings.
[163] The plot of "Crush Results" in Figure 2 illustrates the comparable
strength of sand
coated with pre-cured phenolic coating, two partially cured coatings (labeled
A and B) and the
New technology coating. Historically, the pre-cured phenolic coated sand would
show a lower
crush percentage (in the ISO test procedure) than partially cured coated sand.
These crush test
results follow this trend with the pre-cured coating sand having a crush of
2.05% and the two
partially coatings (Phenolic coatings A and B) having crushes of 3.93% and
4.95% respectively.
It is important to notice that the coating having the more remaining
reactivity (Phenolic B) has
the highest crush value. The New Technology coating actually tested out having
the lowest crush
value (0.69%). So in the crush evaluation The New Technology coating performed
like a
superior, pre-cured coated sand.
40/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
[164] The plot entitled "Unconfined Compressive Strength" in Figure 3
represents a
strength measurement of the particle to particle bonds of a coated proppant
sand. Historically, a
pre-cured coated phenolic sand possess little if any ability to form particle
to particle bonds of
any measureable strength. In this test the coating labeled Phenolic A
exhibited a bond strength
UCS of 449 psi. The coating labeled Phenolic B had a UCS of 155 psi. Since the
TMA indicated
that Phenolic B was a more reactive coating than Phenolic A, one would expect
that the UCS
results should be reversed. That would be true if the coated sands had the
same resin level (LOT).
However the plot entitled "Coated Sand Loss On Ignition" in Figure 5, shows
that Phenolic A
actually has a 3.97% phenolic coating while phenolic coating B has a 2.84%
resin coating. This
could be one explanation for the unexpected UCS results.
[165] In Figure 3, the pre-cured phenolic coating sand showed only a weak
bonding
capability with a measurement of 7 psi. This level of bonding would indicate
that the pre-cured
phenolic coating is not capable of forming particle-to-particle bonds that
would consolidate the
proppant or be effective in controlling proppant flowback.
[166] The New Technology coating exhibited the TMA appearance of a pre-
cured
coated sand in Figure 1, it yielded the highest bond strength (UCS= 576 psi)
of any sample
tested. See Figure 3. These dual results are new and unexpected for a coated
proppant.
[167] The plot entitled "Fracture Conductivity @ 4000 psi" in Figure 4
reveals a data
point from a long term conductivity test. Presented on the plot are the
conductivity numbers for
the two partially cured phenolic coated sands and a sand coated with the New
Technology.
Historically, the conductivity test results for a partially cured phenolic
coating will meet or
exceed that of a pre-cured coating. The plot shows that the New Technology
coating has a
conductivity similar to the partially cured coating of Phenolic A and superior
to partially cured
coating of Phenolic B. This is in spite of the fact the Phenolic A has a
significantly higher
coating level than the New technology and Phenolic B is marginally higher than
the New
Technology coating (see Figure 5).
[168] In summary, The New Technology coated sand exhibits the thermal
properties of
a pre-cured phenolic coated sand and crush resistance superior to a pre-cured
phenolic coating. It
also shows a bonding capability superior to the partially cured phenolic
coated sand and a
comparable if not superior fracture conductivity (when measured at 4,000 psi
in a long term
conductivity test). This would seem to indicate that the new technology
contains traits and
41/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
performance properties from both type coating and would best be described as a
"hybrid" coating
technology.
EXAMPLE 7
[169] A proppant coating formulation for Example 7 was prepared with an
Iso:Polyol
ratio of 0.65 equivalent weight at a process temperature of 198 F (92 C) and
made from the
curable coating ingredients shown in Table 11:
Table 11
INGREDIENT WEIGHT (LBS.)
Sand 1000.00
Dynasylan AMEO (Silane)* 1.00
Red 2B in Castor Oil 1.00
Dow 801X Polyol 19.34
Dow ISO (XUS17557.00) 20.11
Dabco T-12 (DBTDL) 0.17
5i02.0H 2-12
Chemicals 43.62- 53.62
[170] * Dynasylan AMEO from Evonik Degussa Corporation in Chester, PA is a
bifunctional silane possessing a reactive primary amino group and hydrolyzable
ethoxysilyl
groups. The dual nature of its reactivity is represented by its manufacturer
to allow Dynasylan
AMEO to bind chemically to both inorganic materials (e.g. glass, metals,
fillers) and organic
polymers (e.g. thermosets, thermoplastics, elastomers) thus functioning as an
adhesion promoter,
crosslinker, and/or surface modifier.
[171] Table 12 shows the timing and duration for the order of addition of
the
components making up the curable coating mixture of the invention.
Table 12
COMPONENT START TIME (s) DURATION (s) END TIME (s)
Silane 0 3 3
Color 5 10 15
Polyol & catalyst 20 60 80
Isocyanate 30 60 90
Additive 120 20 140
Discharge 180
42/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
[172] What results from the above coating and curing process is a
substantially cured
and coated proppant having handling characteristics like a pre-cured, resin-
coated proppant but
with the ability to form interparticle bonds under downhole conditions like a
curable, resin-
coated proppant. The resulting product is then further contacted with a finely
divided anticaking
agent, like an amorphous silica or silica substitute in dry form or as a
dispersion.
[173] The preferred anticaking agents are either a dry form of very small
amorphous
silica or a dispersion of nanometer-sized fumed silica. The following Table 13
summarizes the
differences between the additives:
Table 13
ANTICAKING % SURFACE PARTICLE STARTING LBS /
AGENT SOLIDS AREA SIZE
CONC. WT. % 1000 LBS
Colloidal silica 30 Medium - 50-100 nm 0.300 10.00
High
1st Dispersion (aq) 15 Low <20 nm 0.075 5.00
of fumed silica
2nd Dispersion (aq) 20 Medium <20 nm 0.075 3.75
of fumed silica
3rd Dispersion (aq) 30 Low <20 nm 0.075 2.50
of fumed silica
[174] Tests of unconfined compressive strength with a conventional proppant
tester at
125 F (52 C), 24 hour shut-in, 1000 psi with a 2 wt% KC1 solution and without
use of a bond
activator plasticizer show that a 16/30 size of coated proppant sand according
to the invention
exhibits an unconfined compressive strength of 100 psi, and a 20/40 size blend
of coated sand
exhibits an unconfined compressive strength of 92 psi. Comparative tests
against similarly sized
proppants that use a partially-cured phenolic resin coating and 1.5 wt% of a
bond activator
plasticizer show no unconfined compressive strength under the same conditions.
In other words,
the coated proppant of the invention forms a shaped sample exhibiting
interparticle bonding of
92-100 UCS while the phenolic resin proppant remains loose particulates that
show no
interparticle bond strength even though an activator was added to promote such
bonds.
[175] Some time is necessary before adequate interparticle bonds are
developed when
using the present invention. The bonds do not form instantaneously in low
temperature wells at
100-125 F (38 -52 C). Generally, at least about 5 hours is desirable with at
least 12 hours is
useful for most low temperature wells with proppants according to the present
invention. This is
referred to in the industry as the "shut-in" time in which the proppant is
subjected to downhole
43/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
conditions that reflect healing and crack closure of the fractured field
strata which exert
compressive stresses on the coated proppants within the fracture cracks.
[176] Figures 6 and 7 show the results of comparative conductivity tests
reflected in
Table 14 below that compare the proppants of Example 7 against two proppants
with partially
cured phenolic coatings. Figures 6 and 7 show that the 16/30 proppant of the
invention exhibits a
79% greater conductivity at 2000 psi and a 32% higher conductivity at 4000 psi
than the prior art
phenolic proppant coating. Figure 7 shows similar results with the 20/40
proppant with a 29%
higher conductivity at 2000 psi and 13% greater at 4000 psi.
Table 14- Conductivity (md-ft)
CLOSURE STRESS (psi)
SAMPLE 1K 2K 4K 6K
20/40 Invention (Low Temp Cured) 4703 3574 2117 1192
20/40 Competitor A 3615 2773 1877 1317
16/30 Competitor B 6014 4826 2970 1735
16/30 Invention (Low Temp Cured) 13563 8640 3945 1701
[177] Hot water leaching tests show that the proppant coatings of the
present invention
show that the coating is highly resistant to leaching on components and
unreacted materials.
Indeed, the test water after the test was classifiable as safe to the limits
of tap water for drinking.
This contrasts with many phenolic coatings that can leach phenols and
formaldehyde after
prolonged exposure to hot water.
EXAMPLE 8
[178] High temperature wells present other performance issues for coated
proppants and
the formation of a consolidated pack yet there is a continued need for
proppant coatings that can
produce interparticle bonds despite extended exposure to an elevated
temperature of at least 200
F (93 C) for a period of time, e.g., at least about two hours or longer,
without interparticle
contact due to closure stress while also reducing the generation of loose
fine, resisting cyclic
stress and being compatible with frac fluids, breakers and environmental
concerns. The coating
of this example is specifically directed to a coating that is well suited to
higher temperature
wells.
44/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
[179] A 30/50 sand coating according to the invention was prepared having
the
ingredients in the curable mixture as shown in Table 15 in amounts sufficient
to form a 1.75 wt%
coating on the underlying sand core solid. The sand was pre-heated to 210 F
(99 C).
Table 15
INGREDIENTS AMOUNT (GMS)
Sand 2000.00
Polyol 7.90
Dynasylan AMEO 2.00
Distilled Water 0.00
ISO 23.70
Chembetain CAS Surfactant 3.00
Dabco TMR 0.21
Dabco T-12 0.10
Black colorant in Castor Oil 2.00
NanoArc AL-2125 0.00
Green colorant in Castor Oil 0.00
Chemical 38.92
Total 2038.92
[180] The ingredients of the curable mixture were added at the times and
for the
durations shown in Table 16.
Table 16
INGREDIENT START (S) DURATION (S) END (S)
Silane 0 5 5
Colorants in Oil 5 5 10
Catalyst + Polyol 20 60 80
ISO 30 60 90
CAS 95 5 100
Discharge 180
45/51
CA 02889928 2015-04-29
WO 2014/052459 PCT/US2013/061688
[181] The coated sand of Example 8 and two prior art proppants with
partially cured
phenolic coatings were then subjected to an unconfined compressive strength
test using the
equipment and materials described in Example 7 but operated at 250 F (121
C). In one set of
conditions, the proppants were subjected to a three hour preheating that would
be characteristic
of the exposure times and temperatures found in a high temperature well. A
comparison set of
tests was performed without the preheating to gauge the ability of the
proppant to resist the
effects of extended exposure to heat before closure stress was applied. The
results are shown in
Figure 8.
[182] Inspection of Figure 8 will show that both prior art coated proppants
(Competitive
Product 1 and Competitive Product 2) exhibit good UCS when there was no
preheating and
substantially diminished UCS when preheating was experienced. In contract, the
coated proppant
of the invention experienced consistent performance that was the same as, or
better than, the
prior art proppant products.
[183] Once those skilled in the art are taught the invention, many
variations and
modifications are possible without departing from the inventive concepts
disclosed herein. The
invention, therefore, is not to be restricted except in the spirit of the
appended claims
46/51