Note: Descriptions are shown in the official language in which they were submitted.
I
CA 2898751 2017-04-10
METHODS FOR TREATING A FERROUS METAL SUBSTRATE
FIELD OF THE INVENTION
[0001] The present invention relates to methods for treating a ferrous
metal
substrate, such as cold rolled steel, hot rolled steel, and electrogalvanized
steel. The present
invention also relates to coated ferrous metal substrates. The present
invention also relates
to methods for removing iron from a pretreatment bath when the pretreatment
bath is on the
processing line, both in the presence of an article to be coated by the
pretreatment
composition and when the pretreatment bath is off-shift.
BACKGROUND INFORMATION
[0002] The use of protective coatings on metal substrates for improved
corrosion
resistance and paint adhesion is common. Conventional techniques for coating
such
substrates include techniques that involve pretreating the metal substrate
with a phosphate
conversion coating and chrome-containing rinses. Typical phosphate conversion
coatings
operate in the range of about at least 1,000 parts per million ("ppm") of
phosphate, which
leads to waste treatment issues. The use of such phosphate and/or chromate-
containing
compositions, therefore, imparts environmental and health concerns.
[0003] As a result, chromate-free and/or phosphate-free pretreatment
compositions
have been developed. Such compositions are generally based on chemical
mixtures that in
some way react with the substrate surface and bind to it to form a protective
layer. For
example, pretreatment compositions based on a group IIIB or IVB metal compound
have
recently become more prevalent.
[0004] When processing ferrous metal substrates through a pretreatment
composition based on a group IIIB or IVB metal compound, however, the
concentration of
ferric (Fe'3) iron in a bath of the pretreatment composition increases over
time as more iron
based metal is treated. In particular, soluble (Fe+2) iron from the substrate
becomes
insoluble (Fe+3) through Fe+2 concentration build up, oxidation, and
subsequent reaction
with oxygen and water. The resulting insoluble rust, i.e., hydrated iron (III)
oxide
(Fc203.n.H20) and/or iron (III) oxide-hydroxide (Fe0(OH)), flocculates and the
insoluble
1
11
CA 2898751 2017-04-10
rust particles resist settling out during the mild agitation present while
processing parts. As
a result, the insoluble rust particles can adhere to or deposit on the
substrate and be carried
to subsequent processing steps (particularly when filtration equipment is not
available),
such as a downstream electrocoat bath that is employed to deposit an organic
coating. Such
cross-contamination can detrimentally affect the performance of such
subsequently
electrodeposited coatings.
[0005] As a result, it is conventional practice in the industry to
periodically dilute
the pretreatment bath to reduce soluble iron concentration as a preventative
measure and to
add a replenisher to the pretreatment bath in order to replenish the bath
ingredients and to
regain coating ability. In some instances, the pretreatment bath has to be
removed from the
processing line to perform methods for removing rust therefrom. Alternatively,
the
pretreatment bath must be discharged every one to two weeks and a fresh bath
made up.
Each of these practices is costly due the significant product loss, waste
treatment, and
inconvenience.
[0006] As a result, it would be desirable to provide improved methods for
treating
a ferrous metal substrate and for removing soluble iron that address at least
some of the
foregoing.
SUMMARY OF THE INVENTION
[0007] In certain respects, the present invention is directed to methods
for coating a
ferrous metal substrate.
[0008] In certain respects, the method for coating a ferrous metal
substrate
comprises: (a) contacting the ferrous metal substrate with an aqueous
pretreatment
composition having a pH of 4 to 5.5 and comprising: (a) a Group IIIB and/or
IVB metal
compound; (b) phosphate ions; and (c) water, wherein the Group IIIB and/or IVB
metal
compound is present in the pretreatment composition in an amount of 10 to 500
ppm metal
and the weight ratio of Group IIIB and/or IVB metal to phosphate ions in the
pretreatment
composition is at least 0.8:1; and wherein the phosphate ions are maintained
in a bath of the
pretreatment composition in an amount: (i) sufficient to essentially prevent
the formation
of insoluble rust in the bath; and (ii) insufficient to prevent the deposition
of a Group IIIB
2
I I
CA 2898751 2017-04-10
or IVB metal film having a coverage of at least 10 mg/m2 on the ferrous metal
substrate;
and (iii) resulting in a weight ratio of phosphate to ferric ions of 1 to
1.8:1; and then (b)
contacting the substrate with a coating composition comprising a film-forming
resin to form
a coated metal substrate that exhibits corrosion resistance properties.
[0009] In certain other respects, the method for coating a ferrous metal
substrate
comprises: (a) contacting the ferrous metal substrate with an aqueous
pretreatment
composition having a pH of 4 to 5.5 and comprising: (a) a Group IIIB and/or
IVB metal
compound; (b) phosphate ions; and (c) water, wherein the Group IIIB and/or IVB
metal
compound is present in the pretreatment composition in an amount of 10 to 500
ppm metal
and the weight ratio of Group IIIB and/or IVB metal to phosphate ions in the
pretreatment
composition is at least 0.8:1; and wherein the phosphate ions are maintained
in a bath of the
pretreatment composition in an amount: (i) sufficient to essentially prevent
the formation
of insoluble rust in the bath; and (ii) insufficient to prevent the deposition
of a Group IIIB
or IVB metal film having a coverage of at least 10 mg/m2 on the ferrous metal
substrate;
and (iii) resulting in a weight ratio of phosphate to additional soluble iron
in the ferrous
state in a range of 1.8 to 10:1; and then (b) contacting the substrate with a
coating
composition comprising a film-forming resin to form a coated metal substrate
that exhibits
corrosion resistance properties.
[00010] In certain other respects, the present invention is directed to
methods for
removing iron from a pretreatment bath comprising steps that are performed
when the
pretreatment bath is off-shift.
[00011] In certain respects, the off-shift methods for removing iron from a
pretreatment bath containing a pretreatment composition comprising a Group
IIIB and/or
Group IV metal, comprise: (a) reducing the pH of the pretreatment bath by at
least 0.2; (b)
adding phosphate ions to the pretreatment bath and in (a); and (c) raising the
pH of the
pretreatment bath in (b) by at least 0.2.
[00012] In certain other respects, the off-shift methods for removing iron
from a
pretreatment bath containing a pretreatment composition comprising a Group
IIIB and/or
Group IVB metal, comprise: (a) adding an acid to the pretreatment bath to
reduce the pH
3
I I
CA 2898751 2017-04-10
of the pretreatment composition to below 4.0; (b) adding phosphate ions to the
pretreatment
bath in (a); and (c) raising the pH of the pretreatment bath in (b) to 4.0 and
5.5.
[00013] The present invention is also directed to substrates treated and
coated
thereby.
BRIEF DESCRIPTION OF THE DRAWINGS
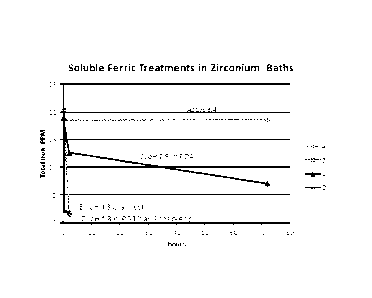
[00014] Figs. 1 and 2 are graphical representations of observed results of
Example 3;
[00015] Fig. 3 is a graphical representation of observed results of Example
4;
[00016] Fig. 4 is a graphical representation of observed results of Example
5; and
1000171 Fig. 5 is a graphical representation of observed results of Example
6.
DETAILED DESCRIPTION OF THE INVENTION
[00018] For purposes of the following detailed description, it is to be
understood that
the invention may assume various alternative variations and step sequences,
except where
expressly specified to the contrary. Moreover, other than in any operating
examples, or
where otherwise indicated, all numbers expressing, for example, quantities of
ingredients
used in the specification and claims are to be understood as being modified in
all instances
by the term "about". Accordingly, unless indicated to the contrary, the
numerical
parameters set forth in the following specification and attached claims are
approximations
that may vary depending upon the desired properties to be obtained by the
present invention.
At the very least, and not as an attempt to limit the application of the
doctrine of equivalents
to the scope of the claims, each numerical parameter should at least be
construed in light of
the number of reported significant digits and by applying ordinary rounding
techniques.
[00019] Notwithstanding that the numerical ranges and parameters setting
forth the
broad scope of the invention are approximations, the numerical values set
forth in the
specific examples are reported as precisely as possible. Any numerical value,
however,
inherently contains certain errors necessarily resulting from the standard
variation found in
their respective testing measurements.
[00020] Also, it should be understood that any numerical range recited
herein is
intended to include all sub-ranges subsumed therein. For example, a range of
"1 to 10" is
intended to include all sub-ranges between (and including) the recited minimum
value of 1
4
I I
CA 2898751 2017-04-10
and the recited maximum value of 10, that is, having a minimum value equal to
or greater
than 1 and a maximum value of equal to or less than 10.
[00021] In this application, the use of the singular includes the plural
and plural
encompasses singular, unless specifically stated otherwise. In addition, in
this application,
the use of "or" means "and/or" unless specifically stated otherwise, even
though "and/or"
may be explicitly used in certain instances.
[00022] In this application, the term "off-shift" means that an article to
be coated by
the pretreatment composition is absent from the pretreatment bath, but does
not mean that
the pretreatment bath is necessarily removed from the process line.
[00023] In this application, the term "total iron" or "total Fe" means the
total amount
of iron in a pretreatment bath, including but not limited to ferric (Fe i2)
iron and ferrous
(Fe+3) iron.
[00024] In this application, unless specifically indicated to the contrary,
when it is
stated that a pretreatment composition is "substantially free" of a particular
component, it
means that the material being discussed is present in the composition, if at
all, as an
incidental impurity. In other words, the material is not intentionally added
to the
composition, but may be present at minor or inconsequential levels, because it
was carried
over as an impurity as part of an intended composition component. Moreover,
when it is
stated that a pretreatment composition is "completely free" of a particular
component it
means that the material being discussed is not present in the composition at
all.
[00025] As previously mentioned, certain embodiments of the present
invention are
directed to methods for treating a ferrous metal substrate. Suitable ferrous
metal substrates
for use in the present invention include those that are often used in the
assembly of
automotive bodies, automotive parts, and other articles, such as small metal
parts, including
fasteners, i.e., nuts, bolts, screws, pins, nails, clips, buttons, and the
like. Specific examples
of suitable ferrous metal substrates include, but are not limited to, cold
rolled steel, hot
rolled steel, steel coated with zinc metal, zinc compounds, or zinc alloys,
such as
electrogalvanized steel, hot-dipped galvanized steel, galvanealed steel, and
steel plated with
zinc alloy. Moreover, the ferrous metal substrate being treating by the
methods of the
present invention may be a cut edge of a substrate that is otherwise treated
and/or coated
I I
CA 2898751 2017-04-10
over the rest of its surface. The metal ferrous substrate coated in accordance
with the
methods of the present invention may be in the form of, for example, a sheet
of metal or a
fabricated part.
[00026] The ferrous metal substrate to be treated in accordance with the
methods of
the present invention may first be cleaned to remove grease, dirt, or other
extraneous matter.
This is often done by employing mild or strong alkaline cleaners, such as are
commercially
available and conventionally used in metal pretreatment processes. Examples of
alkaline
cleaners suitable for use in the present invention include ChemkleenTM 163,
177, 611L, and
490MX, each of which are commercially available from PPG Industries, Inc. Such
cleaners
are often followed and/or preceded by a water rinse.
[00027] As previously indicated, certain embodiments of the present
invention are
directed to methods for treating a metal substrate that comprise contacting
the metal
substrate with a pretreatment composition comprising a group IIIB and/or IVB
metal. As
used herein, the term "pretreatment composition" refers to a composition that
upon contact
with the substrate reacts with and chemically alters the substrate surface and
binds to it to
form a protective layer.
[00028] Often, the pretreatment composition comprises a carrier, often an
aqueous
medium, so that the composition may be in the form of a solution or dispersion
of a group
IIIB and/or IVB metal compound in the carrier. In these embodiments, the
solution or
dispersion may be brought into contact with the substrate by any of a variety
of known
techniques, such as dipping or immersion, spraying, intermittent spraying,
dipping followed
by spraying, spraying followed by dipping, brushing, or roll-coating. In
certain
embodiments, the solution or dispersion when applied to the metal substrate is
at a
temperature ranging from 50 to 150 F (10 to 65 C). The contact time is often
from 2
seconds to five minutes, such as 30 seconds to 2 minutes.
[00029] As used herein, the term "group IIIB and/or IVB metal" refers to an
element
that is in group IIIB or group IVB of the CAS Periodic Table of the Elements
as is shown,
for example, in the Handbook of Chemistry and Physics, 63rd edition (1983).
Where
applicable, the metal itself may be used. In certain embodiments, a group IIIB
and/or IVB
metal compound is used. As used herein, the term "group IIIB and/or IVB metal
6
I I
CA 2898751 2017-04-10
compound" refers to compounds that include at least one element that is in
group IIIB or
group IVB of the CAS Periodic Table of the Elements.
[00030] In certain
embodiments, the group IIIB and/or IVB metal compound used in
the pretreatment composition may be a compound of zirconium, titanium,
hafnium, or a
mixture thereof. Suitable compounds of zirconium include, but are not limited
to,
hexafluorozirconic acid, alkali metal and ammonium salts thereof, ammonium
zirconium
carbonate, zirconium basic carbonate, zirconyl nitrate, zirconium carboxylates
and
zirconium hydroxy carboxylates, such as hydrofluorozirconic acid, zirconium
acetate,
zirconium oxalate, ammonium zirconium glycolate, ammonium zirconium lactate,
ammonium zirconium citrate, and mixtures thereof. Suitable compounds of
titanium
include, but are not limited to, fluorotitanic acid and its salts. A suitable
compound of
hafnium includes, but is not limited to, hafnium nitrate.
[00031] In certain
embodiments, the group IIIB and/or IVB metal compound is
present in a bath of the pretreatment composition in an amount of at least 10
ppm metal,
such as at least 20 ppm metal, at least 30 ppm metal, or, in some cases, at
least 50 ppm
metal (measured as elemental metal). In certain embodiments, the group IIIB
and/or IVB
metal compound is present in the bath of the pretreatment composition in an
amount of no
more than 500 ppm metal, such as no more than 150 ppm metal, or, in some
cases, no more
than 80 ppm metal (measured as elemental metal). The amount of group IIIB
and/or IVB
metal in the pretreatment composition can range between any combination of the
recited
values inclusive of the recited values.
[00032] As
previously indicated, the pretreatment compositions used in certain
embodiments of the methods of the present invention comprise phosphate ions.
In certain
embodiments, the source of phosphate ions is phosphoric acid, such as 75%
phosphoric
acid, although other sources of phosphate ions are contemplated by the present
invention,
such as, for example, monosodium phosphate or disodium phosphate. In certain
other
embodiments, the pretreatment compositions of the methods of the present
invention are
substantially free of phosphate ions.
[00033] As
indicated previously, in certain embodiments of the methods of the
present invention, the phosphate ions are maintained in a bath of the
pretreatment
7
11
CA 2898751 2017-04-10
composition in an amount sufficient to essentially prevent the formation of
insoluble rust in
the bath. As used herein, the term "maintained" means that the amount of
phosphate ions
is regulated and, as necessary, adjusted to essentially prevent the formation
of insoluble
rust. As used herein, the phrase "essentially prevent the formation of
insoluble rust" means
that insoluble rust, i.e., including but not limited to, hydrated iron (III)
oxide (Fe20311H20)
and/or iron (III) oxide-hydroxide (Fe0(OH)), is prevented from forming in the
bath to an
extent that an orange or red-brown appearance indicative of the formation of
such
compounds in the bath is not visible to the naked eye. Rather, in certain
embodiments of
the present invention, the phosphate ions are maintained in the bath in an
amount sufficient
to complex with the soluble iron etched from the surface of the ferrous metal
substrate being
treated to form iron (III) phosphate (FePO4) in the bath, which results in the
bath having a
whitish appearance, rather than an orange or red-brown appearance associated
with the
presence of rust and which results in the formation of an insoluble sludge
that can be
removed from the bath using conventional filtration equipment. Certain
embodiments of
the present invention, therefore, limit the amount of ferric iron (Fe+3) in
the bath (from the
ferrous metal substrate) that is available to become insoluble rust that can
deposit on the
substrate and be carried to subsequent processing equipment, such as a
downstream spray
nozzles, pumps, rinse baths, and electrocoat baths for the deposition of an
organic coating.
As previously indicated, such cross-contamination can detrimentally affect the
performance
of such subsequently deposited coatings.
[00034] In certain embodiments of the methods of the present invention, the
phosphate ions are also maintained in the bath of the pretreatment composition
in an amount
insufficient to prevent the deposition of a Group IIIB or IVB metal film
having a coverage
(total film weight) of at least 10mg/m2, such as at least 100 mg/m2 or, in
some cases, 100
to 500 mg/m2, on the ferrous metal substrate. It has been discovered that
there is,
particularly at the bath pH's used in the present invention, a delicate
balance between the
phosphate ions complexing with the soluble iron etched from the ferrous metal
substrate to
form iron phosphate, as is desired, and complexing with the Group IIIB or IVB
metal
present in the bath, which is not desired because it would prevent the
deposition of a
sufficient Group IIIB or IVB metal film on the ferrous metal substrate.
8
I I
CA 2898751 2017-04-10
1000351 It has been discovered that the presence of 1 to 1.8, such as 1.2
to 1.6 parts
by weight phosphate ions to every 1 part by weight ferric (Fe+3) ions in a
composition is
sufficient to essentially prevent the formation of insoluble rust as described
above while
being insufficient to prevent the deposition of a Group IIIB or IVB metal film
having a
coverage of at least 100 mg/m2, such as at least 10mg/m2, on a ferrous metal
substrate. As
a result, in certain embodiments of the methods of the present invention, the
phosphate ions
are maintained in the bath at a level that results in a weight ratio of
phosphate ions to ferric
ions of 1 to 1.8:1, in some cases 1.2 to 1.6:1. If the weight ratio of
phosphate ions to ferric
ions is less than 1:1, then there may be too little phosphate in the bath to
essentially prevent
the formation of insoluble rust in the bath as described above. If the weight
ratio of
phosphate ions to ferric ions is greater than 1.8:1, then the amount of
phosphate ions may
be sufficient to prevent the deposition of an adequate Group IIIB or IVB metal
film on a
ferrous metal substrate. The ratio of phosphate ions to ferric ions in the
pretreatment
composition can range between any combination of the recited values inclusive
of the
recited values.
[00036] In addition, in certain embodiments of the methods of the present
invention,
the phosphate ions are maintained in the bath at a level that results in a
weight ratio of group
IIIB and/or IVB metal to phosphate ions in the bath of at least 50:1, in some
cases at least
25:1, in some cases at least 12.5:1, in some cases at least 3:1, and in some
cases at least 2:1.
If the weight ratio of group IIIB and/or IVB metal to phosphate ions is less
than 2:1, then
there may be too much phosphate in the bath, thereby negatively impacting on
the ability to
deposit a sufficient Group IIIB or IVB metal film on the ferrous metal
substrate.
[00037] As is apparent, because the pretreatment compositions of the
present
invention comprise, in some cases, 20 to 500 ppm group IIIB and/or IVB metal,
such as 30
to 150 ppm, or, in some cases, 30 to 80 ppm group IIIB and/or IVB metal, in
certain
embodiments of the methods of the present invention, relatively little
phosphate ion is often
present in the bath since the phosphate ions are, in certain embodiments,
maintained in the
bath at a level that results in a weight ratio of group IIIB and/or IVB metal
to phosphate
ions in the bath of at least 2:1, in some cases at least 3:1. As a result, in
certain embodiments,
such a bath comprises no more than 30 ppm, such as 10 to 30 ppm, phosphate
ions. Yet,
9
I
CA 2898751 2017-04-10
the presence of a small level of phosphate ions has been shown to have a
dramatic effect on
useful bath life by preventing the formation of insoluble rust in the
pretreatment bath for up
to months or years in certain embodiments, such as by removing iron from the
pretreatment
bath.
[00038] As discussed above, when processing ferrous metal substrates
through a
pretreatment composition based on a group IIIB or IVB metal compound, the
concentration
of ferric (Fe') iron in a bath of the pretreatment composition increases over
time as more
iron based metal is treated. The result is that such a bath accumulates
insoluble rust that
can deposit on the substrate being treated and be carried to subsequent
processing steps. To
avoid this, such a bath must often be replaced periodically, in some cases
once per week. It
has been surprisingly discovered, however, that the presence of the
aforementioned small
levels of phosphate can prevent the formation of insoluble rust, without
preventing the
formation of an adequate group IIIB and/or IVB metal film, such that the bath
can be
operated for several months, maybe indefinitely, without replacement. That
such a small
level of phosphate could extend bath life to such a significant degree was
surprising and not
anticipated. Moreover, the presence of phosphate ions in such small amount
results in the
formation of a minimal amount of sludge that is more than offset by the
prevention of
insoluble rust, such that waste disposal issues are not a significant concern.
[00039] In certain embodiments, the pretreatment composition also comprises
an
electropositive metal. As used herein, the term "electropositive metal" refers
to metals that
are more electropositive than the metal substrate. This means that, for
purposes of the
present invention, the term "electropositive metal" encompasses metals that
are less easily
oxidized than the metal of the metal substrate that is being treated. As will
be appreciated
by those skilled in the art, the tendency of a metal to be oxidized is called
the oxidation
potential, is expressed in volts, and is measured relative to a standard
hydrogen electrode,
which is arbitrarily assigned an oxidation potential of zero. The oxidation
potential for
several elements is set forth in the table below. An element is less easily
oxidized than
another element if it has a voltage value, E*, in the following table, that is
greater than the
element to which it is being compared.
I
CA 2898751 2017-04-10
Element Half-cell reaction Voltage, E*
Potassium Krf + e ¨> K -2.93
Calcium Ca' + 2e ¨> Ca -2.87
Sodium Na + + e ¨> Na -2.71
Magnesium Mg2+ + 2e --> Mg -2.37
Aluminum Al3+ + 3e ¨> Al -1.66
Zinc Zn" + 2e Zn -0.76
Iron Fe" + 2e ¨> Fe -0.44
Nickel Ni" + 2e ¨> Ni -0.25
Tin Sn' + 2e ¨> Sn -0.14
Lead Pb' + 2e ¨> Pb -0.13
Hydrogen 2H+ + 2e --> H2 -0.00
Copper Cu" + 2e ¨> Cu 0.34
Mercury Hg2' + 2e ¨> 2Hg 0.79
Silver Ag+ + e ¨> Ag 0.80
Gold Au3+ + 3e ¨> Au 1.50
[00040] Thus, as will be apparent, when the metal substrate comprises a
ferrous
metal, as is the case in the present invention, suitable electropositive
metals for inclusion in
the pretreatment composition include, for example, nickel, tin, copper,
silver, and gold, as
well mixtures thereof.
[00041] In certain embodiments, the source of electropositive metal in the
pretreatment composition is a water soluble metal salt. In certain embodiments
of the
present invention, the water soluble metal salt is a water soluble copper
compound. Specific
examples of water soluble copper compounds, which are suitable for use in the
present
invention include, but are not limited to, copper cyanide, copper potassium
cyanide, copper
sulfate, copper nitrate, copper pyrophosphate, copper thiocyanate, disodium
copper
ethylenediaminetetraacetate tetrahydrate, copper bromide, copper oxide, copper
hydroxide,
copper chloride, copper fluoride, copper gluconate, copper citrate, copper
lauroyl
sarcosinate, copper formate, copper acetate, copper propionate, copper
butyrate, copper
lactate, copper oxalate, copper phytate, copper tartarate, copper malate,
copper succinate,
11
CA 2898751 2017-04-10
copper malonate, copper maleate, copper benzoate, copper salicylate, copper
aspartate,
copper glutamate, copper fumarate, copper glycerophosphate, sodium copper
chlorophyllin,
copper fluorosilicate, copper fluoroborate and copper iodate, as well as
copper salts of
carboxylic acids in the homologous series formic acid to decanoic acid, copper
salts of
polybasic acids in the series oxalic acid to suberic acid, and copper salts of
hydroxycarboxylic acids, including glycolic, lactic, tartaric, malic and
citric acids.
[00042] When
copper ions supplied from such a water-soluble copper compound are
precipitated as an impurity in the form of copper sulfate, copper oxide, etc.,
it may be
preferable to add a complexing agent that suppresses the precipitation of
copper ions, thus
stabilizing them as a copper complex in the solution.
[00043] In certain
embodiments, the copper compound is added as a copper complex
salt such as K3Cu(CN)4 or Cu-EDTA, which can be present stably in the
composition on its
own, but it is also possible to form a copper complex that can be present
stably in the
composition by combining a complexing agent with a compound that is
difficultly soluble
on its own. Examples thereof include a copper cyanide complex formed by a
combination
of CuCN and KCN or a combination of CuSCN and KSCN or KCN, and a Cu-EDTA
complex formed by a combination of CuSO4 and EDTA.2Na.
[00044] With
regard to the complexing agent, a compound that can form a complex
with copper ions can be used; examples thereof include polyphosphates, such as
sodium
tripolyphosphate and hexametaphosphoric acid; aminocarboxylic acids, such as
ethylenediaminetetraacetic acid, hydroxyethylethylenediaminetriacetic acid,
and
nitrilotriacetic acid; hydroxycarboxylic acids, such as tartaric acid, citric
acid, gluconic acid,
and salts thereof; aminoalcohols, such as triethanolamine; sulfur compounds,
such as
thioglycolic acid and thiourea, and phosphonic acids, such as
nitrilotrimethylenephosphonic
acid, ethylenediaminetetra(methylenephosphonic acid) and
hydroxyethylidenediphosphonic acid.
[00045] In certain
embodiments, the electropositive metal, such as copper, is
included in the pretreatment compositions in an amount of at least 1 ppm, such
as at least 5
ppm, or in some cases, at least 10 ppm of total metal (measured as elemental
metal). In
certain embodiments, the electropositive metal is included in such
pretreatment
12
I I
CA 2898751 2017-04-10
compositions in an amount of no more than 500 ppm, such as no more than 100
ppm, or in
some cases, no more than 50 ppm of total metal (measured as elemental metal).
The amount
of electropositive metal in the pretreatment composition can range between any
combination of the recited values inclusive of the recited values.
[00046] As indicated, the operating pH of the pretreatment composition used
in the
methods of the present invention ranges from 4.0 to 5.5, in some cases, 4.0 to
5.0, 4.5 to
5.5, or, in yet other cases, 4.5 to 5Ø The pH of the pretreatment
composition may be
adjusted using, for example, any acid or base as is necessary.
[00047] In addition to the previously described components, the
pretreatment
compositions used in the methods of the present invention may comprise any of
a variety
of additional optional components. For example, in certain embodiments, the
pretreatment
compositions used in the methods of the present invention comprises a
polyhydroxy
functional cyclic compound as is described in United States Patent No.
6,805,756 at col. 3,
line 9 to col. 4, line 32. In other embodiments, however, the pretreatment
compositions
used in the methods of the present invention are substantially free, or, in
some cases,
completely free, of any such polyhydroxy functional cyclic compound.
[00048] In certain embodiments, the pretreatment compositions used in the
methods
of the present invention comprise an oxidizer-accelerator, such as those
described in United
States Patent No. 6,805,756 at col. 4, line 52 to col. 5, line 13, and United
States Patent No.
6,193,815 at col. 4, line 62 to col. 5, line 39. By contrast, in other
embodiments, the
pretreatment compositions are substantially free, or, in some cases,
completely free, of any
such an oxidizer-accelerator.
[00049] In certain embodiments, the pretreatment composition comprises an
organic
film forming resin, such as the reaction product of an alkanolamine and an
epoxy-functional
material containing at least two epoxy groups, such as those disclosed in
United States
Patent No. 5,653,823; a resin containing beta hydroxy ester, imide, or sulfide
functionality,
incorporated by using dimethylolpropionie acid, phthalimide, or
mercaptoglycerine as an
additional reactant in the preparation of the resin; the reaction product is
that of the
diglycidyl ether of Bisphenol A (commercially available from Shell Chemical
Company as
EPON 880), dimethylol propionic acid, and diethanolamine in a 0.6 to 5.0:0.05
to 5.5:1
13
I I
CA 2898751 2017-04-10
mole ratio; water soluble and water dispersible polyacrylic acids as disclosed
in United
States Patent Nos. 3,912,548 and 5,328,525; phenol formaldehyde resins as
described in
United States Patent Nos. 5,662,746; water soluble polyamides such as those
disclosed in
WO 95/33869; copolymers of maleic or acrylic acid with allyl ether as
described in
Canadian patent application 2,087,352; and water soluble and dispersible
resins including
epoxy resins, aminoplasts, phenol-formaldehyde resins, tannins, and polyvinyl
phenols as
discussed in United States Patent No. 5,449,415. By contrast, in other
embodiments, the
pretreatment compositions are substantially free, or, in some cases,
completely free, of any
organic film-forming resin, such as one or more of those described above.
[00050] In certain embodiments, the pretreatment compositions used in the
methods
of the present invention comprise fluoride ion, such as is described in United
States Patent
No. 6,805,756 at col. 6, lines 7-23. In certain embodiments, the fluoride ion
is introduced
into the composition through the Group IIIB and/or IVB metal compound. In
certain
embodiments, the pretreatment compositions are substantially free, or, in some
cases,
completely free, of any fluoride ion introduced to the pretreatment
composition from a
source other than through the Group IIIB and/or IVB metal compound.
[00051] In certain embodiments, the pretreatment compositions used in the
methods
of the present invention comprise a polysaccharide, such as is described in
United States
Patent No. 6,805,756 at col. 6, lines 53-64 and International Application WO
2005/001158
at page 3, lines 17-23. By contrast, in other embodiments, the pretreatment
compositions
are substantially free, or, in some cases, completely free, of any such
polysaccharide.
[00052] In certain embodiments, the pretreatment compositions used in the
methods
of the present invention comprise a phosphate acid ester, a water-soluble
polyethylene
glycol ester of a fatty acid, and/or nitric acid, such as is described in
United States Patent
No. 5,139,586 at col. 6, lines 31-63. By contrast, in other embodiments, the
pretreatment
compositions are substantially free, or, in some cases, completely free, of a
phosphate acid
ester, a water-soluble polyethylene glycol ester of a fatty acid, and/or
nitric acid.
[00053] In certain embodiments, the pretreatment compositions used in the
methods
of the present invention comprise vanadium and/or cerium ions, such as is
described in
United States Patent No. 4,992,115 at col. 2, line 47 to col. 3, line 29 and
United States
14
11
CA 2898751 2017-04-10
Patent Application Publication No. 2007/0068602. By contrast, in other
embodiments, the
pretreatment compositions are substantially free, or, in some cases,
completely free, of
vanadium and/or cerium ions.
[00054] In certain embodiments, the pretreatment compositions used in the
methods
of the present invention comprise a phosphorous acid, hypophosphorous acid
and/or salts
thereof, such as is described in United States Patent No. 5,728,233 at col. 4,
lines 24-37.
By contrast, in other embodiments, the pretreatment compositions are
substantially free, or,
in some cases, completely free, of phosphorous acid, hypophosphorous acid
and/or salts
thereof
[00055] In certain embodiments, the pretreatment compositions used in the
methods
of the present invention comprise a Group IIA metal, such as is described in
United States
Patent No. 5,380,374 at col. 3, lines 25-33, and/or a Group IA metal, such as
is described
in United States Patent No. 5,441,580 at col. 2, line 66 to col. 3, line 4. By
contrast, in other
embodiments, the pretreatment compositions are substantially free, or, in some
cases,
completely free, of any Group IIA metal and/or any Group lA metal.
[00056] In certain embodiments, the pretreatment compositions used in the
methods
of the present invention comprise a molybdenum compound, such as is described
in UK
Patent Application GB 2 259 920 A. By contrast, in other embodiments, the
pretreatment
compositions are substantially free, or, in some cases, completely free, of
any molybdenum
compound.
[00057] In certain embodiments, the pretreatment compositions used in the
methods
of the present invention comprise one or more ions of metals selected from the
group
consisting of scandium, yttrium, lanthanum, praseodymium, neodymium, samarium,
europium, gadolinium, terbium, dysprosium, holmium, erbium, thulium,
ytterbium, and
lutetium, such as is described in United States Patent No. 5,104,577 at col.
2, line 60 to col.
3, line 26. By contrast, in other embodiments, the pretreatment compositions
are
substantially free, or, in some cases, completely free, of any ions of metals
selected from
the group consisting of scandium, yttrium, lanthanum, praseodymium, neodymium,
samarium, europium, gadolinium, terbium, dysprosium, holmium, erbium, thulium,
ytterbium, and lutetium.
I I
CA 2898751 2017-04-10
[00058] The pretreatment composition may optionally contain other
materials, such
as nonionic surfactants and auxiliaries conventionally used in the art of
pretreatment. In an
aqueous medium, water dispersible organic solvents, for example, alcohols with
up to about
8 carbon atoms, such as methanol, isopropanol, and the like, may be present;
or glycol ethers
such as the monoalkyl ethers of ethylene glycol, diethylene glycol, or
propylene glycol, and
the like. When present, water dispersible organic solvents are typically used
in amounts up
to about ten percent by volume, based on the total volume of aqueous medium.
[00059] Other optional materials include surfactants that function as
defoamers or
substrate wetting agents.
[00060] In certain embodiments, the pretreatment composition also comprises
a
filler, such as a siliceous filler. Non-limiting examples of suitable fillers
include silica,
mica, montmorillonite, kaolinite, asbestos, talc, diatomaceous earth,
vermiculite, natural
and synthetic zeolites, cement, calcium silicate, aluminum silicate, sodium
aluminum
silicate, aluminum polysilicate, alumina silica gels, and glass particles. In
addition to the
siliceous fillers other finely divided particulate substantially water-
insoluble fillers may also
be employed. Examples of such optional fillers include carbon black, charcoal,
graphite,
titanium oxide, iron oxide, copper oxide, zinc oxide, antimony oxide,
zirconia, magnesia,
alumina, molybdenum disulfide, zinc sulfide, barium sulfate, strontium
sulfate, calcium
carbonate, and magnesium carbonate. By contrast, in other embodiments, the
pretreatment
compositions are substantially free, or, in some cases, completely free, of
any such filler.
[00061] In certain embodiments, the pretreatment composition is
substantially or, in
some cases, completely free of chromate and/or heavy metal phosphate, such as
zinc
phosphate. As used herein, the term "substantially free" when used in
reference to the
absence of chromate and/or heavy metal phosphate in the pretreatment
composition, means
that these substances are not present in the composition to such an extent
that they cause a
burden on the environment. As used herein, the term "completely free", when
used with
reference to the absence of a heavy metal phosphate and/or chromate, means
that there is
no heavy metal phosphate and/or chromate in the composition at all.
[00062] As will be appreciated, in certain embodiments, the pretreatment
composition utilized in the methods of the present invention consists
essentially of or, in
16
I
CA 2898751 2017-04-10
some cases, consists of: (a) a Group IIIB and/or IVB metal compound, such as a
zirconium
compound; (b) a source of phosphate ions, such as phosphoric acid; and (c)
water. In certain
other embodiments, the pretreatment composition utilized in the methods of the
present
invention consists essentially of or, in some cases, consists of: (a) a Group
IIIB and/or IVB
metal compound, such as a zirconium compound; and (c) water. In certain
embodiments,
such pretreatment compositions include fluoride ions introduced to the
pretreatment
composition through the Group IIIB and/or IVB metal compound. As used herein,
the
phrase "consists essentially of" means that the composition does not include
any other
components that would materially affect the basic and novel characteristic(s)
of the
invention. For the purposes of the present invention, this means that the
pretreatment
composition does not include any components that would materially affect the
pretreatment
composition's ability to be successfully employed in the methods of the
present invention.
[00063] In certain embodiments, the film coverage (total film weight) of
the residue
of the pretreatment coating composition is at least 10 milligrams per square
meter (mg/m2),
such as 100 to 500 mg/m2, or, in some cases at least 50 mg/m2. The thickness
of the
pretreatment coating can vary, but it is generally very thin, often having a
thickness of less
than 1 micrometer, in some cases it is from 1 to 500 nanometers, and, in yet
other cases, it
is 10 to 300 nanometers, such as 20 to 100 nanometers.
[000641 In certain embodiments, the off-shift method is used to remove
soluble iron
from the pretreatment bath such that the pretreatment bath, at the completion
of the off-shift
method, is substantially free of iron, thereby essentially preventing the
formation of
insoluble rust in the operating bath of the pretreatment composition. As used
herein, the
term "substantially free," when used in reference to iron in the operating
bath of the
pretreatment composition, means that the total iron is present in an amount of
less than 10
ppm. As described herein, in certain embodiments, the bath of the pretreatment
composition
is substantially free of phosphate ions when the bath is operating, such as in
pretreatment
systems in which the presence of phosphate in the pretreatment bath may
adversely affect
the deposition of the pretreatment composition on the substrate. In such
embodiments, the
off-shift method of removing iron from the pretreatment bath may be
particularly useful for
such pretreatment systems that are substantially free of phosphate ions as a
method of
17
I I
CA 2898751 2017-04-10
essentially preventing the formation of insoluble rust in the pretreatment
bath. Additionally,
as described herein, in certain other embodiments, the bath of the
pretreatment composition
comprises phosphate ions as a method of essentially preventing the formation
of insoluble
rust in the pretreatment bath. In such embodiments, the off-shift method of
removing iron
from the pretreatment bath may be particularly useful as an additional or
supplemental
method of essentially preventing the formation of insoluble rust in the
pretreatment bath.
[00065] As previously indicated, in certain embodiments, the pretreatment
bath has
an operating pH of greater than 4.0, such as between 4.2 and 5.5, preferably
between 4.5
and 5.0, and most preferably 4.8. In certain embodiments, a first step of the
off-shift method
of removing iron from the pretreatment bath comprises reducing the pH of the
pretreatment
bath by at least 0.2, such as by at least 0.5 or at least 1.0, such that the
pH of the pretreatment
bath is reduced to between 1.0 and 3.8, and preferably between 2.5 and 3.3. In
certain
embodiments, the pH of the pretreatment bath is reduced by the addition of an
acid to the
pretreatment bath, including as non-limiting examples, a Group IVB fluro metal
acid such
as hexafluorozirconic acid and hexafluorotitanic acid, phosphoric acid,
sulfuric acid,
sulfamic acid, nitric acid, and mixtures thereof.
[00066] In certain embodiments of the off-shift method of removing iron
from the
pretreatment bath, the first step of reducing the pH of the pretreatment bath
is accomplished
by adding a sufficient amount of an acid to the pretreatment bath to reduce
the pH as
discussed above.
[00067] In certain embodiments of the off-shift method of removing iron
from the
pretreatment bath, a second step comprises adding phosphate ions to the
pretreatment bath.
In certain embodiments, the sources of phosphate ions may be alkali metal and
ammonium
orthophosphates present as either the monohydrogen or dihydrogen type,
including as
examples monosodium phosphate, disodium phosphate, and mixtures thereof. In
certain
embodiments, Zircobond Additive P, a monosodium phosphate solution
commercially
available from PPG Industries, Inc., Euclid, Ohio, is used as the source of
the phosphate
ions.
[00068] In certain embodiments of the off-shift method of removing iron
from the
pretreatment bath, a third step comprises adding an oxidizing agent to the
pretreatment bath.
18
CA 2898751 2017-04-10
In such embodiments, the oxidizing agent is a peroxide compound, air, sodium
nitrite,
sodium bromate, and mixtures thereof In a preferred embodiment, the peroxide
compound
is hydrogen peroxide.
[00069] In certain embodiments of the off-shift method of removing iron
from the
pretreatment bath, the source of the phosphate ions and the oxidizing agent
are each added
in amounts that are sufficient to result in a pretreatment bath that is
substantially free of
iron.
[00070] In certain embodiments of the off-shift method of removing iron
from the
pretreatment bath, a fourth step comprises raising the pH of the pretreatment
bath by at least
0.2. In embodiments, the pH is raised to above 4.0, such as 4.2 to 5.2, 4.5 to
5.0, and 4.8.
In certain embodiments, the pH is raised by adding a sufficient amount of an
alkaline
composition to the pretreatment bath, including as non-limiting examples
caustic soda,
caustic potash, and sodium hydroxide. In embodiments, the alkaline composition
is
Chemfil Buffer, a commercial product available from PPG Industries, Inc.,
Euclid, Ohio,
can be used in a quantity sufficient to achieve the desired operating pH.
[00071] In certain embodiments of the off-shift method of the present
invention, the
phosphate ions are added to the pretreatment bath in an amount sufficient to
complex with
the soluble iron etched from the surface of the ferrous metal substrate being
treated to form
iron (III) phosphate (FePO4) in the bath, which results in the bath having a
whitish
appearance, rather than an orange or red-brown appearance associated with the
presence of
rust and which results in the formation of an insoluble sludge that can be
removed from the
bath using conventional filtration equipment. In certain embodiments of the
off-shift
method of the present invention, a fifth step comprises filtering the
pretreatment bath using
such conventional filtration equipment to remove solid matter from the
pretreatment bath,
i.e., iron phosphate, iron oxides, iron hydroxides, or any other insoluble
sludge that forms
in the pretreatment bath. In certain embodiments, the step of filtering may
immediately
follow raising the pH of the pretreatment bath by at least 0.2. In certain
other embodiments,
the step of filtering may follow an equilibration period during which this
insoluble sludge
settles to the bottom of the pretreatment bath, such as 1 to 10 hours after
raising the pH of
the pretreatment bath.
19
I I
CA 2898751 2017-04-10
[00072] The off-shift method of the present invention, therefore, removes
soluble
iron in the bath (from the ferrous metal substrate) that is available to
become insoluble rust
that can deposit on the substrate and be carried to subsequent processing
equipment, such
as a downstream spray nozzles, pumps, rinse baths, and electrocoat baths for
the deposition
of an organic coating. As previously indicated, such cross-contamination can
detrimentally
affect the performance of such subsequently deposited coatings. It has been
surprisingly
discovered, however, that lowering the pH of the pretreatment bath below the
operating pH
and then adding the aforementioned small levels of phosphate and optionally
oxidant, can
essentially remove iron in the bath, thereby preventing the formation of
insoluble rust in the
pretreatment bath, without preventing the formation of an adequate group IIIB
and/or IVB
metal film after the bath pH is raised to operating levels, such that the bath
can be operated
for several months, maybe indefinitely, without replacement. That such steps
could extend
bath life to such a significant degree was surprising and not anticipated.
[00073] Following contact with the pretreatment solution, the substrate may
be rinsed
with water and dried.
[00074] In certain embodiments of the methods of the present invention,
after the
substrate is contacted with the pretreatment composition, it is then contacted
with a coating
composition comprising a film-forming resin. Any suitable technique may be
used to
contact the substrate with such a coating composition, including, for example,
brushing,
dipping, flow coating, spraying and the like. In certain embodiments, however,
as described
in more detail below, such contacting comprises an electrocoating step wherein
an
electrodepositable composition is deposited onto the metal substrate by
electrodeposition.
[00075] As used herein, the term "film-forming resin" refers to resins that
can form
a self-supporting continuous film on at least a horizontal surface of a
substrate upon removal
of any diluents or carriers present in the composition or upon curing at
ambient or elevated
temperature. Conventional film-forming resins that may be used include,
without
limitation, those typically used in automotive OEM coating compositions,
automotive
refinish coating compositions, industrial coating compositions, architectural
coating
compositions, coil coating compositions, and aerospace coating compositions,
among
others.
I I
CA 2898751 2017-04-10
[00076] In certain embodiments, the coating composition comprises a
thermosetting
film-forming resin. As used herein, the term "thermosetting" refers to resins
that "set"
irreversibly upon curing or crosslinking, wherein the polymer chains of the
polymeric
components are joined together by covalent bonds. This property is usually
associated with
a cross-linking reaction of the composition constituents often induced, for
example, by heat
or radiation. Curing or crosslinking reactions also may be carried out under
ambient
conditions. Once cured or crosslinked, a thermosetting resin will not melt
upon the
application of heat and is insoluble in solvents. In other embodiments, the
coating
composition comprises a thermoplastic film-forming resin. As used herein, the
term
"thermoplastic" refers to resins that comprise polymeric components that are
not joined by
covalent bonds and thereby can undergo liquid flow upon heating and are
soluble in
solvents.
[00077] As previously indicated, in certain embodiments, the substrate is
contacted
with a coating composition comprising a film-forming resin by an
electrocoating step
wherein an electrodepositable composition is deposited onto the metal
substrate by
electrodeposition. In the process of electrodeposition, the metal substrate
being treated,
serving as an electrode, and an electrically conductive counter electrode are
placed in
contact with an ionic, electrodepositable composition. Upon passage of an
electric current
between the electrode and counter electrode while they are in contact with the
electrodepositable composition, an adherent film of the electrodepositable
composition will
deposit in a substantially continuous manner on the metal substrate.
[00078] Electrodeposition is usually carried out at a constant voltage in
the range of
from 1 volt to several thousand volts, typically between 50 and 500 volts.
Current density
is usually between 1.0 ampere and 15 amperes per square foot (10.8 to 161.5
amperes per
square meter) and tends to decrease quickly during the electrodeposition
process, indicating
formation of a continuous self-insulating film.
[00079] The electrodepositable composition utilized in certain embodiments
of the
present invention often comprises a resinous phase dispersed in an aqueous
medium
wherein the resinous phase comprises: (a) an active hydrogen group-containing
ionic
21
CA 2898751 2017-04-10
electrodepositable resin, and (b) a curing agent having functional groups
reactive with the
active hydrogen groups of (a).
[00080] In certain embodiments, the electrodepositable compositions
utilized in
certain embodiments of the present invention contain, as a main film-forming
polymer, an
active hydrogen-containing ionic, often cationic, electrodepositable resin. A
wide variety
of electrodepositable film-forming resins are known and can be used in the
present
invention so long as the polymers are "water dispersible," i.e., adapted to be
solubilized,
dispersed or emulsified in water. The water dispersible polymer is ionic in
nature, that is,
the polymer will contain anionic functional groups to impart a negative charge
or, as is often
preferred, cationic functional groups to impart a positive charge.
1000811 Examples of film-forming resins suitable for use in anionic
electrodepositable compositions are base-solubilized, carboxylic acid
containing polymers,
such as the reaction product or adduct of a drying oil or semi-drying fatty
acid ester with a
dicarboxylic acid or anhydride; and the reaction product of a fatty acid
ester, unsaturated
acid or anhydride and any additional unsaturated modifying materials which are
further
reacted with polyol. Also suitable are the at least partially neutralized
interpolymers of
hydroxy-alkyl esters of unsaturated carboxylic acids, unsaturated carboxylic
acid and at
least one other ethylenically unsaturated monomer. Still another suitable
electrodepositable
film-forming resin comprises an alkyd-aminoplast vehicle, i.e., a vehicle
containing an
alkyd resin and an amine-aldehyde resin. Yet another anionic
electrodepositable resin
composition comprises mixed esters of a resinous polyol, such as is described
in United
States Patent No. 3,749,657 at col. 9, lines 1 to 75 and col. 10, lines 1 to
13. Other acid
functional polymers can also be used, such as phosphatized polyepoxide or
phosphatized
acrylic polymers as are known to those skilled in the art.
[00082] As aforementioned, it is often desirable that the active hydrogen-
containing
ionic electrodepositable resin (a) is cationic and capable of deposition on a
cathode.
Examples of such cationic film-forming resins include amine salt group-
containing resins,
such as the acid-solubilized reaction products of polyepoxides and primary or
secondary
amines, such as those described in United States Patent Nos. 3,663,389;
3,984,299;
3,947,338; and 3,947,339. Often, these amine salt group-containing resins are
used in
22
I I
CA 2898751 2017-04-10
combination with a blocked isocyanate curing agent. The isocyanate can be
fully blocked,
as described in United States Patent No. 3,984,299, or the isocyanate can be
partially
blocked and reacted with the resin backbone, such as is described in United
States Patent
No. 3,947,338. Also, one-component compositions as described in United States
Patent
No. 4,134,866 and DE-OS No. 2,707,405 can be used as the film-forming resin.
Besides
the epoxy-amine reaction products, film-forming resins can also be selected
from cationic
acrylic resins, such as those described in United States Patent Nos. 3,455,806
and 3,928,157.
[00083] Besides amine salt group-containing resins, quaternary ammonium
salt
group-containing resins can also be employed, such as those formed from
reacting an
organic polyepoxide with a tertiary amine salt as described in United States
Patent Nos.
3,962,165; 3,975,346; and 4,001,101. Examples of other cationic resins are
ternary
sulfonium salt group-containing resins and quaternary phosphonium salt-group
containing
resins, such as those described in United States Patent Nos. 3,793,278 and
3,984,922,
respectively. Also, film-forming resins which cure via transesterification,
such as described
in European Application No. 12463 can be used. Further, cationic compositions
prepared
from Mannich bases, such as described in United States Patent No. 4,134,932,
can be used.
[00084] In certain embodiments, the resins present in the
electrodepositable
composition are positively charged resins which contain primary and/or
secondary amine
groups, such as described in United States Patent Nos. 3,663,389; 3,947,339;
and 4,116,900.
In United States Patent No. 3,947,339, a polyketimine derivative of a
polyamine, such as
diethylenetriamine or triethylenetetraamine, is reacted with a polyepoxide.
When the
reaction product is neutralized with acid and dispersed in water, free primary
amine groups
are generated. Also, equivalent products are formed when polyepoxide is
reacted with
excess polyamines, such as diethylenetriamine and triethylenetetraamine, and
the excess
polyamine vacuum stripped from the reaction mixture, as described in United
States Patent
Nos. 3,663,389 and 4,116,900.
[00085] In certain embodiments, the active hydrogen-containing ionic
electrodepositable resin is present in the electrodepositable composition in
an amount of 1
to 60 percent by weight, such as 5 to 25 percent by weight, based on total
weight of the
electrodeposition bath.
23
I I
CA 2898751 2017-04-10
[00086] As indicated, the resinous phase of the electrodepositable
composition often
further comprises a curing agent adapted to react with the active hydrogen
groups of the
ionic electrodepositable resin. For example, both blocked organic
polyisocyanate and
aminoplast curing agents are suitable for use in the present invention,
although blocked
isocyanates are often preferred for cathodic electrodeposition.
[00087] Aminoplast resins, which are often the preferred curing agent for
anionic
electrodeposition, are the condensation products of amines or amides with
aldehydes.
Examples of suitable amine or amides are melamine, benzoguanamine, urea and
similar
compounds. Generally, the aldehyde employed is formaldehyde, although products
can be
made from other aldehydes, such as acetaldehyde and furfural. The condensation
products
contain methylol groups or similar alkylol groups depending on the particular
aldehyde
employed. Often, these methylol groups are etherified by reaction with an
alcohol, such as
a monohydric alcohol containing from 1 to 4 carbon atoms, such as methanol,
ethanol,
isopropanol, and n-butanol. Aminoplast resins are commercially available from
American
Cyanamid Co. under the trademark CYMEL and from Monsanto Chemical Co. under
the
trademark RESIMENE.
[00088] The aminoplast curing agents are often utilized in conjunction with
the active
hydrogen containing anionic electrodepositable resin in amounts ranging from 5
percent to
60 percent by weight, such as from 20 percent to 40 percent by weight, the
percentages
based on the total weight of the resin solids in the electrodepositable
composition.
[00089] As indicated, blocked organic polyisocyanates are often used as the
curing
agent in cathodic electrodeposition compositions. The polyisocyanates can be
fully blocked
as described in United States Patent No. 3,984,299 at col. 1, lines 1 to 68,
col. 2, and col. 3,
lines 1 to 15, or partially blocked and reacted with the polymer backbone as
described in
United States Patent No. 3,947,338 at col. 2, lines 65 to 68, col. 3, and col.
4 lines 1 to 30.
By "blocked" is meant that the isocyanate groups have been reacted with a
compound so
that the resultant blocked isocyanate group is stable to active hydrogens at
ambient
temperature but reactive with active hydrogens in the film forming polymer at
elevated
temperatures usually between 90 C and 200 C.
24
I I
CA 2898751 2017-04-10
[00090] Suitable polyisocyanates include aromatic and aliphatic
polyisocyanates,
including cycloaliphatic polyisocyanates and representative examples include
diphenylmethane-4,4'-diisocyanate (MDI), 2,4- or 2,6-toluene diisocyanate
(TDI),
including mixtures thereof, p-phenylene diisocyanate, tetramethylene and
hexamethylene
diisocyanates, dicyclohexylmethane-4,4'-diisocyanate, isophorone diisocyanate,
mixtures
of phenylmethane-4,4'-diisocyanate and polymethylene polyphenylisocyanate.
Higher
polyisocyanates, such as triisocyanates can be used. An example would include
triphenylmethane-4,4',4"-triisocyanate. Isocyanate ( )-prepolymers with
polyols such as
ncopentyl glycol and trimethylolpropane and with polymeric polyols such as
polycaprolactone diols and triols (NCO/OH equivalent ratio greater than 1) can
also be used.
[00091] The polyisocyanate curing agents are typically utilized in
conjunction with
the active hydrogen containing cationic electrodepositable rcsin in amounts
ranging from 5
percent to 60 percent by weight, such as from 20 percent to 50 percent by
weight, the
percentages based on the total weight of the resin solids of the
electrodepositable
composition.
[00092] In certain embodiments, the coating composition comprising a film-
forming
resin also comprises yttrium. In certain embodiments, yttrium is present in
such
compositions in an amount from 10 to 10,000 ppm, such as not more than 5,000
ppm, and,
in some cases, not more than 1,000 ppm, of total yttrium (measured as
elemental yttrium).
[00093] Both soluble and insoluble yttrium compounds may serve as the
source of
yttrium. Examples of yttrium sources suitable for use in lead-free
electrodepositable
coating compositions are soluble organic and inorganic yttrium salts such as
yttrium acetate,
yttrium chloride, yttrium formate, yttrium carbonate, yttrium sulfamate,
yttrium lactate and
yttrium nitrate. When the yttrium is to be added to an electrocoat bath as an
aqueous
solution, yttrium nitrate, a readily available yttrium compound, is a
preferred yttrium source.
Other yttrium compounds suitable for use in electrodepositable compositions
are organic
and inorganic yttrium compounds such as yttrium oxide, yttrium bromide,
yttrium
hydroxide, yttrium molybdate, yttrium sulfate, yttrium silicate, and yttrium
oxalate.
Organoyttrium complexes and yttrium metal can also be used. When the yttrium
is to be
I
CA 2898751 2017-04-10
incorporated into an electrocoat bath as a component in the pigment paste,
yttrium oxide is
often the preferred source of yttrium.
[00094] The electrodepositable compositions described herein are in the
form of an
aqueous dispersion. The term "dispersion" is believed to be a two-phase
transparent,
translucent or opaque resinous system in which the resin is in the dispersed
phase and the
water is in the continuous phase. The average particle size of the resinous
phase is generally
less than 1.0 and usually less than 0.5 microns, often less than 0.15 micron.
[00095] The concentration of the resinous phase in the aqueous medium is
often at
least 1 percent by weight, such as from 2 to 60 percent by weight, based on
total weight of
the aqueous dispersion. When such compositions are in the form of resin
concentrates, they
generally have a resin solids content of 20 to 60 percent by weight based on
weight of the
aqueous dispersion.
[00096] The electrodepositable compositions described herein are often
supplied as
two components: (1) a clear resin feed, which includes generally the active
hydrogen-containing ionic electrodepositable resin, i.e., the main film-
forming polymer, the
curing agent, and any additional water-dispersible, non-pigmented components;
and (2) a
pigment paste, which generally includes one or more colorants (described
below), a
water-dispersible grind resin which can be the same or different from the main-
film forming
polymer, and, optionally, additives such as wetting or dispersing aids.
[00097] In certain embodiments, the two component electrodepositable
composition
is embodied in the form of an electrodeposition bath, as is well known to
those skilled in
the art, wherein components (1) and (2) are dispersed in an aqueous medium
which
comprises water and, usually, coalescing solvents. An advantage of the methods
of the
present invention, as indicated earlier, is that such baths can be prevented
from being
contaminated with rust, even in the absence of filtration equipment.
[00098] As aforementioned, besides water, the aqueous medium may contain a
coalescing solvent. Useful coalescing solvents are often hydrocarbons,
alcohols, esters,
ethers and ketones. The preferred coalescing solvents are often alcohols,
polyols and
ketones. Specific coalescing solvents include isopropanol, butanol, 2-
ethylhexanol,
isophorone, 2-methoxypentanone, ethylene and propylene glycol and the
monoethyl
26
I
CA 2898751 2017-04-10
monobutyl and monohexyl ethers of ethylene glycol. The amount of coalescing
solvent is
generally between 0.01 and 25 percent, such as from 0.05 to 5 percent by
weight based on
total weight of the aqueous medium.
[00099] In addition, a colorant and, if desired, various additives such as
surfactants,
wetting agents or catalyst can be included in the coating composition
comprising a film-
forming resin. As used herein, the term "colorant" means any substance that
imparts color
and/or other opacity and/or other visual effect to the composition. The
colorant can be
added to the composition in any suitable form, such as discrete particles,
dispersions,
solutions and/or flakes. A single colorant or a mixture of two or more
colorants can be
used.
[000100] Example colorants include pigments, dyes and tints, such as those
used in
the paint industry and/or listed in the Dry Color Manufacturers Association
(DCMA), as
well as special effect compositions. A colorant may include, for example, a
finely divided
solid powder that is insoluble but wettable under the conditions of use. A
colorant can be
organic or inorganic and can be agglomerated or non-agglomerated. Colorants
can be
incorporated by use of a grind vehicle, such as an acrylic grind vehicle, the
use of which
will be familiar to one skilled in the art.
[000101] Example pigments and/or pigment compositions include, but are not
limited
to, carbazole dioxazine crude pigment, azo, monoazo, disazo, naphthol AS, salt
type (lakes),
benzimidazolone, condensation, metal complex, isoindolinone, isoindoline and
polycyclic
phthalocyanine, quinacridone, perylene, perinone, diketopyrrolo pyrrole,
thioindigo,
anthraquinone, indanthrone, anthrapyrimidine, flavanthrone, pyranthrone,
anthanthrone,
dioxazine, triarylcarbonium, quinophthalone pigments, diketo pyrrolo pyrrole
red
("DPPBO red"), titanium dioxide, carbon black and mixtures thereof The terms
"pigment"
and "colored filler" can be used interchangeably.
[000102] Example dyes include, but are not limited to, those that are
solvent and/or
aqueous based such as pthalo green or blue, iron oxide, bismuth vanadate,
anthraquinone,
perylene, aluminum and quinacridone.
[000103] Example tints include, but are not limited to, pigments dispersed
in
water-based or water miscible carriers such as AQUA-CHEM 896 commercially
available
27
I I
CA 2898751 2017-04-10
from Degussa, Inc., CHARISMA COLORANTS and MAXITONER INDUSTRIAL
COLORANTS commercially available from Accurate Dispersions division of Eastman
Chemical, Inc.
[000104] As noted above, the colorant can be in the form of a dispersion
including,
but not limited to, a nanoparticle dispersion. Nanoparticle dispersions can
include one or
more highly dispersed nanoparticle colorants and/or colorant particles that
produce a
desired visible color and/or opacity and/or visual effect. Nanoparticle
dispersions can
include colorants such as pigments or dyes having a particle size of less than
150 nm, such
as less than 70 nm, or less than 30 nm. Nanoparticles can be produced by
milling stock
organic or inorganic pigments with grinding media having a particle size of
less than 0.5
mm. Example nanoparticle dispersions and methods for making them are
identified in U.S.
Patent No. 6,875,800 B2. Nanoparticle dispersions can also be produced by
crystallization,
precipitation, gas phase condensation, and chemical attrition (i.e., partial
dissolution). In
order to minimize re-agglomeration of nanoparticles within the coating, a
dispersion of
resin-coated nanoparticles can be used. As used herein, a "dispersion of resin-
coated
nanoparticles" refers to a continuous phase in which is dispersed discreet
"composite
microparticles" that comprise a nanoparticle and a resin coating on the
nanoparticle.
Example dispersions of resin-coated nanoparticles and methods for making them
are
identified in United States Patent Application Publication 2005-0287348 Al,
filed June 24,
2004, U.S. Provisional Application No. 60/482,167 filed June 24, 2003, and
United States
Patent Application Serial No. 11/337,062, filed January 20, 2006.
10001051 Example special effect compositions that may be used include
pigments
and/or compositions that produce one or more appearance effects such as
reflectance,
pearlescence, metallic sheen, phosphorescence, fluorescence, photochromism,
photosensitivity, thermochromism, goniochromism and/or color-change.
Additional
special effect compositions can provide other perceptible properties, such as
opacity or
texture. In certain embodiments, special effect compositions can produce a
color shift, such
that the color of the coating changes when the coating is viewed at different
angles.
Example color effect compositions are identified in U.S. Patent No. 6,894,086.
Additional
color effect compositions can include transparent coated mica and/or synthetic
mica, coated
28
I I
CA 2898751 2017-04-10
silica, coated alumina, a transparent liquid crystal pigment, a liquid crystal
coating, and/or
any composition wherein interference results from a refractive index
differential within the
material and not because of the refractive index differential between the
surface of the
material and the air.
[000106] In certain
embodiments, a photosensitive composition and/or photochromic
composition, which reversibly alters its color when exposed to one or more
light sources,
can be used. Photochromic and/or photosensitive compositions can be activated
by
exposure to radiation of a specified wavelength. When the composition becomes
excited,
the molecular structure is changed and the altered structure exhibits a new
color that is
different from the original color of the composition. When the exposure to
radiation is
removed, the photochromic and/or photosensitive composition can return to a
state of rest,
in which the original color of the composition returns. In certain
embodiments, the
photochromic and/or photosensitive composition can be colorless in a non-
excited state and
exhibit a color in an excited state. Full color-change can appear within
milliseconds to
several minutes, such as from 20 seconds to 60 seconds. Example photochromic
and/or
photosensitive compositions include photochromic dyes.
[000107] In certain
embodiments, the photosensitive composition and/or
photochromic composition can be associated with and/or at least partially
bound to, such as
by covalent bonding, a polymer and/or polymeric materials of a polymerizable
component.
In contrast to some coatings in which the photosensitive composition may
migrate out of
the coating and crystallize into the substrate, the photosensitive composition
and/or
photochromic composition associated with and/or at least partially bound to a
polymer
and/or polymerizable component in accordance with certain embodiments of the
present
invention, have minimal migration out of the coating. Example
photosensitive
compositions and/or photochromic compositions and methods for making them are
identified in U.S. Application Serial No. 10/892,919 filed July 16, 2004.
[000108] In
general, the colorant can be present in the coating composition in any
amount sufficient to impart the desired visual and/or color effect. The
colorant may
comprise from 1 to 65 weight percent, such as from 3 to 40 weight percent or 5
to 35 weight
percent, with weight percent based on the total weight of the composition.
29
I I
CA 2898751 2017-04-10
[000109] After deposition, the coating is often heated to cure the
deposited
composition. The heating or curing operation is often carried out at a
temperature in the
range of from 120 to 250 C, such as from 120 to 190 C, for a period of time
ranging from
to 60 minutes. In certain embodiments, the thickness of the resultant film is
from 10 to
50 microns.
[000110] As will be appreciated by the foregoing description, certain
embodiments of
the present invention are also directed to methods for preventing rust
contamination of
coating equipment even in the absence of filtration equipment in a process
wherein a ferrous
metal substrate is being coated. In certain embodiments, such methods comprise
utilizing
a pretreatment composition having a pH of 4 to 5.5 and comprising, or, in some
cases,
consisting essentially of: (a) a Group IIIB and/or IVB metal compound; (b)
phosphate ions;
and (c) water. In such embodiments of the methods of the present invention,
the phosphate
ions are maintained in a bath of the pretreatment composition in an amount:
(i) sufficient to
essentially prevent the formation of insoluble rust in the bath; and (ii)
insufficient to prevent
the deposition of a Group IIIB and/or IVB metal film having a coverage of at
least 10 mg/ft'
on the ferrous metal substrate. In certain other embodiments, such methods
comprise an
off-shift method of removing iron from a pretreatment bath comprising a Group
IIIB and/or
Group IVB metal that, in certain embodiments, is substantially free of
phosphate ions during
operation, and in certain other embodiments, comprises phosphate ions. The off-
shift
method comprises the steps of: (a) reducing the pH of the pretreatment bath by
at least 0.2;
(b) adding phosphate ions to the pretreatment bath in (a); (c) adding an
oxidizing agent to
the pretreatment bath in (b); and (d) raising the pH of the pretreatment bath
in (c) by at least
0.2. In such off-shift methods of removing iron from the pretreatment bath,
insoluble rust
may be essentially removed from the pretreatment bath. In certain embodiments,
the off-
shift method further comprises the step of filtering the pretreatment bath
using filtration
equipment.
[000111] As will also be appreciated, the present invention is also
directed to methods
for coating a ferrous metal substrate. In certain embodiments, these methods
comprise: (a)
contacting the ferrous metal substrate with an aqueous pretreatment
composition having a
pH of 4 to 5.5 and comprising or, in some cases, consisting essentially of:
(i) a Group IIIB
I I
CA 2898751 2017-04-10
and/or IVB metal compound; (ii) phosphate ions; and (ii) water, wherein the
phosphate ions
are maintained in a bath of the pretreatment composition in an amount
sufficient to
essentially prevent the formation of insoluble rust in the bath; and then (b)
contacting the
substrate with a coating composition comprising a film-forming resin to form a
coated metal
substrate that exhibits corrosion resistance properties. In certain other
embodiments, such
methods comprise: (a) removing iron from a pretreatment bath when the
pretreatment bath
is off-shift ; and then (b) contacting the ferrous metal substrate with an
aqueous pretreatment
composition having a pH of 4 to 5.5 and comprising, or in some cases,
consisting essentially
of: (i) a Group IIIB and/or Group IVB metal; and (ii) water; wherein the
pretreatment
composition is, in certain embodiments, substantially free of phosphate ions;
and then (c)
contacting the substrate with a coating composition comprising a film-forming
resin to form
a coated metal substrate that exhibits corrosion resistance properties. In
such methods, the
step of removing iron from the pretreatment bath when the pretreatment bath is
off-shift
comprises, or in some cases, consists essentially of: (a) reducing the pH of
the pretreatment
bath by at least 0.2; (b) adding phosphate ions to the pretreatment bath in
(a); (c) adding an
oxidizing agent to the pretreatment bath in (b); and (d) raising the pH of the
pretreatment
bath in (c) by at least 0.2. As used herein, the term "corrosion resistance
properties" refers
to the measurement of corrosion prevention on a metal substrate utilizing the
test described
in ASTM B117 (Salt Spray Test). In this test, the coated substrate is scribed
with a knife
to expose the bare metal substrate according to ASTM D1654-92. The scribed
substrate is
placed into a test chamber where an aqueous salt solution is continuously
misted onto the
substrate. The chamber is maintained at a constant temperature. The coated
substrate is
exposed to the salt spray environment for a specified period of time, such as
250, 500 or
1000 hours. After exposure, the coated substrate is removed from the test
chamber and
evaluated for corrosion along the scribe. Corrosion is measured by "scribe
creep", which
is defined as the total distance the corrosion has traveled across the scribe
measured in
millimeters. When it is stated that a substrate "exhibits corrosion resistance
properties" it
means that the scribe creep exhibited by the ferrous metal substrate is no
more than 3
millimeters after testing in accordance with ASTM B117 for 500 hours in a salt
spray
environment in the case where the substrate is coated with a polyester powder
paint
31
I I
CA 2898751 2017-04-10
commercially available from PPG Industries, Inc. as PCT79111, according to the
manufacturer's instructions.
[000112] Illustrating the invention are the following examples that arc not
to be
considered as limiting the invention to their details. All parts and
percentages in the
examples, as well as throughout the specification, are by weight unless
otherwise indicated.
EXAMPLES
Example 1
[000113] In one experiment, five clean steel panels were placed in a water
solution of
a pH of about 1.8 ¨ 2.4 containing fluorozirconic acid and phosphoric acid
(for 90 ppm Zr
and 10 ppm PO4-3). After building ferrous concentration to approximately 30
ppm, the
panels were removed from the clear solution and divided into one gallon (3.78
liters)
portions.
[000114] The first gallon was subdivided further into 700 ml portions to
which (75%
by wgt.) phosphoric acid was added to yield a series of baths with phosphate
ions at 10, 25,
50, 75 and 100 ppm. The same series of phosphate levels was repeated with
Zirconium at
125, 150 and 200 ppm.
[000115] The pH in all sample baths was adjusted 5Ø The baths containing
30 ppm
of ferrous iron and various amounts of zirconium and phosphate ions were
allowed to stand
in a quiescent state for two days. After two days, the appearance of the
individual baths
was noted. The results summarized in Table 1.0 below demonstrate that, in this
example, a
zirconium bath containing 30ppm of total iron will converted from a brown to a
white
appearance in the presence of between 25 and 50 ppm of phosphate ion. The
brown
appearance is indicative of the formation of an iron oxide or an iron
oxyhydroxide.
[000116] The matrix of results showed that all of the 10 ppm PO4' baths
developed
rust colored water and mostly brown precipitate to the same degree; i.e.,
without regard to
the Zr level. The next lightest colored ones were all the 25 ppm PO4-3 baths
which also had
lighter colored precipitates. All the 50 ppm PO4-3 baths were nearly color-
free with
crystalline like precipitates that were barely noticeable off-white. The 75
and 100 ppm PO4-
32
I I
CA 2898751 2017-04-10
. ,
3 baths were all color-free with white crystalline precipitate. This white
precipitate was
ferric phosphate, possibly with insignificant amounts of zirconium compounds.
[000117] This example shows that a phosphate to ferric weight
ratio of at least 1:1,
such as at least 1.2:1, such as 1 to 1.8:1, is sufficient to essentially
prevent the formation of
insoluble rust in a pretreatment bath comprising a group IIIB and/or IVB metal
when the
bath is used to treat a ferrous metal substrate.
Table 1.0
Zirconium, Phosphate, Precipitate Total Iron,
pH
PPm PPm Appearance ppm
90 10 Brown 30 5.0
90 25 Brown 30 5.0
90 50 White 30 5.0
90 75 White 30 5.0
90 100 White 30 5.0
125 10 Brown 30 5.0
125 25 Brown 30 5.0
125 50 White 30 5.0
125 75 White 30 5.0
125 100 White 30 5.0
150 10 Brown 30 5.0
150 25 Brown 30 5.0
150 50 White 30 5.0
150 75 White 30 5.0
150 100 White 30 5.0
200 10 Brown 30 5.0
200 25 Brown 30 5.0
200 50 White 30 5.0
200 75 White 30 5.0
200 100 White 30 5.0
33
: ,
I I
CA 2898751 2017-04-10
Example 2
[000118] Steel panels were cleaned using a conventional alkaline-based
cleaner,
rinsed twice in city water, treated in baths containing zirconium in a range
of 10 ¨ 150 ppm
and phosphate in a range of 10 -100 ppm, and then subsequently rinsed in city
water. The
treated steel panels were painted with either P590 cationic epoxy
electrodeposited coating
or PCT79111 triglycidyl isocyanurate-polyester powder coating, both of which
being
commercially available from a PPG Industries Inc. Corrosion performance was
determined
by exposing the zirconium treated and painted panels to a neutral salt-spray,
according to
ASTM B117, for the times indicated in Table 2Ø Acceptable performance for
the cationic
epoxy electrodeposited coating at 1000 hours of neutral salt-spray exposure in
this test was
4.0 - 5.0mm of 1/2 width scribe loss. Acceptable performance for the TGIC-
polyester
powder paint at 500 hours of neutral salt-spray exposure is 2.0 - 3.0mm of 1/2
width scribe
loss. The results below demonstrate the acceptable corrosion performance can
be obtained
when phosphate ions are added to the zirconium treatment bath. As shown in
Example 1.0,
at a low concentration of phosphate ion, the treatment bath turned brown,
indicating the
presence of iron oxide or iron oxyhydroxide.
Table 2.0
% Width Scribe Loss,
Aged
Experiment mm
PO4 Zr pH Fe, ppm Bath
P590 PCT79111
color
1128 hrs 500 hrs
1 10 10 5.0 10 9.0 Na Brown
2 10 150 5.0 10 3.7 1.75 Brown
3 55 80 5.0 10 2.9 2.8 White
4 100 80 5.0 10 4.4 2.7 White
100 150 5.0 10 3.1 2.35 White
Example 3
[000119] A pretreatment solution was prepared to which increasing amounts
of
hexafluorozirconic acid were added. Prior to coating cold rolled steel panels,
the bath pH
was adjusted to 4.7. Panels from ACT Labs (Hillsdale, MI) were first spray
cleaned in an
alkaline cleaner (PPG Industries Chemkleen 611L, at 2% and 140-150 F) and
rinsed twice
34
I I
CA 2898751 2017-04-10
before entering the pretreatment zone. The zirconium bath was sprayed onto the
panels for
60 seconds at 9 psi. They were then rinsed with city water and finally with a
deionized
water halo prior to an infrared drying step.
10001201 Panel samples were obtained at 0, 10, 15, 20, 50, and 80 ppm
zirconium bath
levels. Sections of each were analyzed via XPS (X-Ray photoelectron
spectroscopy) for
determination of layer thickness of zirconium in the coatings. The depth of
the zirconium
layer was determined to be the nanometer at which the profile crossed back
down to the
10% atomic percent level. The resulting table of depths was graphed vs. the
zirconium bath
concentration as illustrated in Fig. 1.
[000121] Using panels from the same series, an anionic acrylic electrocoat,
commercially available from PPG Industries, Inc. as Powercron 395 was applied
to three
panels at each level prior to corrosion testing per ASTM B117 and D1654-92.
Results are
illustrated in Fig. 2. The results confirm that a good degree of corrosion
protection is
reached that coincides with the attainment of a minimum thickness, i.e., from
a bath with
20 ppm zirconium.
Example 4
[0001221 In practice, baths heavily contaminated with rust are opaque
brownish red
and are preceded by the appearance of translucent orange solutions, indicating
the initial
conversion to insoluble ferric complexes. In one experiment, ten gallons of a
low pH bath
(-2.7) containing 100 ppm zirconium was sprayed with steel panels for several
hours until
the total iron reached 50 ppm. Ferrous iron was approximately 40 ppm. Though
the bath
contained ten ppm of soluble ferric ions, it was clear and colorless. A large
sample was
divided into portions to which increasing levels of phosphate were added to
determine the
level that would prevent the initial discoloration of the bath after raising
the pH to 5. For
the control sample with no phosphate, the level of ferric increased to 24 ppm
just before the
bath began turning color. Results of this experiment are shown in Table 3Ø
I
CA 2898751 2017-04-10
Table 3.0
Initial pH = 5, clear bath, ferric available ¨ 24 ppm
Pat ppm Bath pH next day Bath color next day Precipitate color
0 3.94 light orange Brown-orange
3.98 light orange Brown-orange
4.04 slightly orange Orange
4.15 slightly orange Orange
4.24 slightly orange Orange
4.38 slightly orange Light orange
4.48 slightly orange Light orange
4.54 light yellow Orange-white
4,54 very light yellow White, orange tint
[000123] With increased PO4 level, the color change took longer and was not
as
intense as the zero phosphate control. In addition, the p11 dropped down to
the levels shown
in the table after overnight storage, indicating the completion of the
oxidation and
precipitation steps. The pH decrease was smaller as more phosphate was used.
After a
certain level of phosphate, the pH remained constant ¨ indicating an excess
beyond the
amount needed for the ferric. Over a couple days, the precipitate quality was
evident, as
described in Table 3Ø Without enough phosphate in the system, the
precipitate developed
as a flocculent brown oxide, resulting in a substantial decrease in pH. With
enough
phosphate, the precipitate was white with a density that promoted removal of
the iron before
it could be carried downstream.
[000124] Zirconium levels were also checked to determine the effect of any
excess
phosphate. Fig. 3 shows that although some zirconium was depleted from the
system, the
loss was not substantial. As the phosphate converts the soluble ferric complex
to an
insoluble ferric phosphate, the point of equivalent addition of phosphate to
ferric can be
seen by the plateau of the pH. This occurred at approximately 35-40 ppm of
phosphate for
the 24 ppm of ferric.
36
CA 2898751 2017-04-10
[000125] Thus, in working bath above, just 25-35 ppm of phosphate per 24
ppm of
ferric would be enough to inhibit the development of a reddish brown bath with
only minor
depletion of the zirconium. Bath life for this example would be significantly
longer than
that typically seen in competitive industrial baths based on a group IIIB
and/or IVB metal
but which do not include phosphate ions. The phosphate to ferric ratio is in
the range of 1:1
to 1.8:1 on a weight basis. Higher ratios could begin to deplete too much
zirconium.
Example 5
[000126] A concentrate containing iron was obtained by hanging clean steel
panels
over two days into a solution of hexafluorozirconic acid in deionized water
that contained
no phosphate. The final ferrous level was approximately 900 ppm and ferric was
33 ppm.
The concentrate was then diluted in city water to provide approximately 20 ppm
ferrous and
3 ppm ferric. Varying amounts of phosphoric acid were added followed by enough
hydrogen peroxide to convert all the ferrous to ferric. The pH was then
adjusted to 4.7 for
each bath. After standing quiescent over one day, the baths were analyzed for
phosphate
and zirconium. The results are plotted in Fig. 4. As is apparent,
approximately 30 ppm
phosphate would be enough to remove the 20 ppm ferric while maintaining most
of the
original 65 ppm of zirconium in solution.
Example 6
[000127] Example 6 was carried out to demonstrate that ferric iron (Fe+3)
can be
removed from the pretreatment bath off-shift.
[000128] A stock solution was prepared from 3 liters of city water and 1.2
g
fluorozirconic acid solution (45%). The stock solution had a target of 85 ppm
Zr. To this,
0.38 ml of ferric sulfate (50% solution) was added for a target solution
having 20 ppm ferric
ion. The stock solution had a pH of 2.9.
[000129] The stock solution was split into Baths A-D, each containing 900m1
of the
stock solution. As described in more detail below, a Hach meter was used in
this Example
(and in Examples 6 and 7) to measure ferrous iron (Fe+2) and total iron
concentrations at
various time points. Where it was desired to obtain the concentration of
ferric iron (Fe+3)
in a particular bath, ferric iron concentration was calculated as the
difference between total
37
I I
CA 2898751 2017-04-10
iron concentration and ferrous iron concentration. In Example 6, none of Baths
A-D
contained any ferrous iron (Fe') at any time point measured.
[000130] Bath A served as the control to which the ferric iron (Fe+3) and
total iron
concentrations (ppm) of baths B, C and D (treated as described below) were
compared.
[000131] 0.1g of Chemfil Buffer, an alkaline solution commercially
available from
PPG Industries, Inc., was added as a source of alkalinity to control Bath A
for a resulting
pH of 3.4. As illustrated in Figure 5, the ferric iron (Fe+3) concentration
(ppm) in Bath A
was about 18.6 ppm for the duration of the 72 hr experiment. There was a
barely visible
rust colored precipitate that formed in Bath A. These data confirm that ferric
iron (Fe') is
fairly stable at a pH range of about 3.4.
[000132] 0.5g of Chemfil Buffer was added to the 900m1 stock solution of
Bath B to
raise the pH of the bath to 4.8, which was within the standard operating range
for a bath
containing the pretreatment composition described herein. As illustrated in
Figure 5, the
ferric iron (Fe+3) concentration in Bath B decreased from an initial
concentration of about
21 ppm to a concentration of about 2 ppm by 2 hr after raising the pH of the
pretreatment
bath to 4.8. These data indicate that most of the soluble ferric iron was
converted to rust or
ferric oxide, which is insoluble in the pretreatment composition. A rusty
precipitate was
visible in the Bath B by 2 hr after raising the pH.
[000133] 0.09g of monosodium phosphate solution, provided as Zircobond
Additive
P, available from PPG Industries, Inc., Euclid, OH (45% by weight) was added
to the 900m1
stock solution of Bath C. Bath C contained 14ppm of phosphate and had a pH of
2.9 that
was steady over the 72 hour duration of the experiment. As illustrated in
Figure 5, the ferric
iron (Fe-13) concentration in Bath C decreased from about 18 ppm to about 12
ppm in the
first 2 hours of the experiment, and then continued to gradually decrease over
the duration
of the 72 hr experiment to a final concentration of 7 ppm. A white precipitate
was visible in
Bath C within the first hours of the experiment, and by the end of the
experiment, a slightly
tan precipitate formed, indicating that removal of ferric iron was gradual and
incomplete
when the pH was below the normal operating level.
[000134] 0.09g of monosodium phosphate solution, provided as Zircobond
Additive
P, (45% by weight) was added to the 900 ml stock solution of Bath D. Bath D
contained
38
,
CA 2898751 2017-04-10
34ppm of phosphate. As illustrated in Figure 5, the ferric iron (Fe+3)
concentration of Bath
D was 20ppm. 0.5g Chemfil Buffer was added to Bath D to raise the pH to 4.75
and the
bath immediately became cloudy. After allowing the crystals to settle, a bath
sample was
filtered through a five micron syringe filter and this filtrate was checked
for total iron. The
ferric iron concentration of Bath D was 2 ppm and two hours later (at the
conclusion of the
experiment) was 1.9 ppm. The bath was clear with a small white precipitate.
[000135] The data from Example 6 demonstrate that the addition of phosphate
to the
pretreatment bath removes a large portion of the ferric iron at low pH and
substantially all
of the ferric in a shorter time after the pH is raised back to the operating
range. These data
confirm that ferric iron may be removed from the pretreatment bath when the
bath is
off-shift.
Example 7
[000136] The data illustrated in Figure 5 and described in Example 6
demonstrated
that ferric iron was removed from the pretreatment bath by adding phosphate to
the
pretreatment bath at a low pH. However, in practice, pretreatment baths that
have been
used to treat substrate often contain ferrous iron that has to be converted to
ferric iron in
order to be removed from the pretreatment bath. Example 7 and the data
illustrated in Table
4 and described herein demonstrate that the addition of an oxidizing agent to
the
pretreatment bath improves removal of iron that was initially in the ferrous
state.
[000137] A stock solution was prepared from 3 liters of city water and 1.2
g
fluorozirconic acid solution (45%). The stock solution had a target of 85 ppm
Zr. To this,
0.32g of ferrous sulfate heptahydrate was added for a target solution having
20 ppm ferric
ion (Fe'2) and 23 ppm total iron. The stock solution had a pH of 3.1.
[000138] The stock solution was split into Baths E-G, each containing 900m1
of the
stock solution. Bath E served as the control to which the ferrous iron (Fe+2)
and total iron
concentrations (ppm) of baths F and G (treated as described below) were
compared. Using
a Hach meter, ferrous iron and total iron concentrations were monitored in
each Bath at
periodic intervals over the 44 hour duration of the experiments
[000139] Bath E served as a control. Bath E had an initial pH of 3.1. A few
drops of
Chemfil Buffer were added to the bath to increase the pH to 3.5, which, as
illustrated in
39
I I
CA 2898751 2017-04-10
Table 4, remained steady for the duration of the experiment. Also as
illustrated in Table 4,
the total iron concentration (ppm) in Bath E dropped from 22.8ppm initially to
22.1ppm at
the end of the 44hr experiment. Ferrous iron (Fe+2) concentration was
initially 19.8ppm
and dropped to 15.7ppm at the end of the 44hr experiment. The bath remained
clear for the
duration of the experiment, with no red color forming. These data indicate
that all of the
iron in the bath remained in solution as ferrous iron, with only a minor
conversion of the
ferrous iron to ferric iron. These data demonstrate that at low pH (i.e., a pH
lower than
operating pH) there is only minimal conversion of ferrous iron to ferric iron.
[000140] 0.093g of monosodium phosphate (45% solution) was added to Bath F
to
yield a solution that had 43ppm phosphate and a ratio of PO4:total iron of
about 1.8:1. 0.5g
Chemfil Buffer was then added to the bath to yield a pH of 4.7. The pH of Bath
F decreased
slightly over the duration of the experiment, and was 4.38 at 44hr. As
illustrated in Table
4, the total iron concentration in Bath E dropped from 22.8ppm initially to
18.5ppm by
30min and 14.7ppm at the end of the 44hr experiment. Ferrous iron
concentration was
initially 19.8ppm, decreased to 17.2ppm by 30min, and was 12.4ppm at the end
of the 44
hr experiment. Some white precipitate, indicating the formation of ferric
phosphate, formed
in the bath over the duration of the experiment. These data indicate that the
addition of
phosphate, followed by increasing the 1,11 to between 4.38 and 4.7, removed
only some of
the soluble iron as ferric phosphate because, though not intending to be
limited by theory,
oxidation of ferrous by increasing pH alone was relatively slow and limited by
pH related
equilibria.
[000141] As illustrated in Table 4, Bath G initially had a pH of 3.0, a
total iron
concentration of 22.8ppm and a ferrous iron concentration of 19,8ppm. 0.1g of
monosodium
phosphate (45% solution) was added to Bath G immediately prior to adding 0.32g
hydrogen
peroxide (3% wt. solution). By 15min after adding the hydrogen peroxide, the
total iron
concentration was decreased to 10.2ppm, the ferrous iron concentration was
decreased to
0.4ppm, and pH was 2.6. Some white precipitate formed in the bath, indicating
that the
iron-phosphate complex was partially completed. Next, the pH of the bath was
increased
to 4.7 by adding 0.6g Chemfil Buffer, and 15 minutes later (i.e., 46 minutes
after the start
of the experiment), nearly all of the iron was removed, with the total iron
concentration
I I
CA 2898751 2017-04-10
. .
being 5ppm and the ferrous iron concentration being 0.1ppm. At the conclusion
of the
experiment (i.e., 44hr after the start), the pH of the bath was 4.6, the total
iron concentration
was 0.24ppm, and the ferrous iron concentration was 0.02ppm. These data
demonstrated
that the addition of phosphate and hydrogen peroxide to the bath significantly
improved the
removal of iron from the bath at operating pH.
Table 4.0
Total iron
Time Elapsed Ferrous
Bath pH (PPm) Notes
(hr:min) (Fe+2), PPm
E 3.1 19.8 22.8
E 0:00 3.5 18.3 23.2 Added
alkaline
buffer
E 44:00 3.54 15.7 22.1 Clear
bath
F 3.12 19.8 218 +Zircobond ADDP
F 0:00 4.7 19.8 22.8 Added alkaline
buffer
F 0:30 4.7 17.2 18.5
F 43:40 4.38 12.4 14.7 White
precipitate in
a trace amount
G 0:00 3.10 19.8 22.8
Zircobond ADDP +
hydrogen peroxide
G 0:15 2.6 0.4 10.2
Added alkaline
G 0:31 4.7 Not Not
buffer; bath turned
measured measured
opaque white
G 0:46 4.7 0.1 5.0
G 44:00 4.62 0.02 0.24 White
precipitate
formed
Example 8
[000142] In this Example, an operating pretreatment bath was made by adding
3.60g
hexafluorozirconic acid to 3 liters of water to yield a solution having 240ppm
zirconium.
An amount of Chemfil Buffer sufficient to raise the pH of the solution to 4.5
was added.
0.31g of ferrous sulfate heptahydrate was added to obtain 20ppm ferrous iron.
In order to
prevent the formation of rust particles, approximately 14 drops of
hexafluorozirconic acid
41
i
CA 2898751 2017-04-10
were immediately added to decrease the pH to 3.3. The bath was clear. Using a
Hach
meter, total iron concentration was measured to be 23.2ppm and ferrous iron
was 19.5ppm.
[000143] Phosphate was added to the bath on an approximate 1:1 molar ratio
(or 1.8:1
by weight) to the total iron that was to be precipitated. For this bath,
41.5ppm phosphate
from 0.175 g of a phosphoric acid solution (75% by wt.) was added for an
excess of about
8-9 ppm. After mixing for lmin, 1.27 g of hydrogen peroxide solution (3% by
wt.) was then
added based on a 1:1 molar ratio to ferrous iron (with a slight excess). The
ferrous iron was
converted to ferric iron in less than 1 min.
[000144] In order to precipitate all of the ferric iron as ferric
phosphate, the bath pH
was raised slowly to 4.75 by adding Chemfil Buffer drop-wise. If raised too
quickly, some
insoluble ferric oxide, as rust, could form instead of ferric phosphate. As
the pH increased,
a white cloudiness developed in the bath that eventually became a flocculent
that
completely settled within 10 minutes to yield a clear bath. This final
solution contained
0.2ppm total iron, with no detectable ferrous iron. The residual phosphate was
8.5ppm,
which was consistent with the mass balance calculation.
[000145] It will be appreciated by those skilled in the art that changes
could be made
to the embodiments described above without departing from the broad inventive
concept
thereof. It is understood, therefore, that this invention is not limited to
the particular
embodiments disclosed, but it is intended to cover modifications which are
within the spirit
and scope of the invention, as defined by the appended claims.
42