Note: Descriptions are shown in the official language in which they were submitted.
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
PURIFICATION OF PROTEINS USING HYDROPHOBIC INTERACTION
CHROMATOGRAPHY
CROSS REFERENCE TO RELATED APPLICATIONS
The present application claims priority to International Application No.
PCT/US2013/031352, filed March 14, 2013, the disclosure of which is
incorporated by reference
herein in its entirety.
FIELD OF INVENTION
The instant invention relates to the field of protein production and
purification, and in
particular to compositions and processes for reducing the levels of
impurities, including process-
related impurities (e.g., host cell proteins and media components) and/or
product-related
substances (e.g., product charge variants, aggregates, and fragments).
BACKGROUND OF THE INVENTION
Hydrophobic interaction chromatography (HIC) is a purification technique that
exploits
the interaction of HIC media with hydrophobic regions present on a protein of
interest, such as an
antibody, and/or those present on an impurity to separate a protein of
interest present in a sample
mixture. HIC is often utilized in either a bind-elute mode, in which the
protein of interest
remains bound to HIC media until eluted during an elution phase, or a flow
through mode, in
which the protein of interest flows through the column while the impurity
binds to the media.
Recently, a chromatographic method termed "weak partitioning mode" has been
described for the purification of proteins (U.S. Patent No. 8,067,182).
According to U.S. Patent
No. 8,067,182, this method allows for the binding of both product and impurity
and is defined by
a intermediate partition coefficient (Kp) for the product. Compared to the
flow-through mode, in
which the Kp for the product is typically low (e.g., <0.1), thereby allowing
the product to flow
through the column while the impurity is bound, and the bind-elute mode, in
which the Kp for
the product is typically high (e.g., >20), thereby allowing the product to
remain bound until
eluted during an elution phase, in the weak portioning mode, the Kp for the
product is in the
range of 0.1-20.
Importantly, U.S. Patent No. 8,067,182 teaches the criticality of this Kp
range.
Specifically, U.S. Patent No. 8,067,182 teaches that Kp values greater than 20
result in a
decreased load challenge at the point of contaminant breakthrough as the
product begins to
compete with the contaminant for binding sites on the media. In addition, U.S.
Patent No.
-1-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
8,067,182 teaches that Kp values greater than 20 result in decreased product
recovery in that the
isocratic wash conditions are not effective at washing the bound product off
the column in a
reasonable number of wash volumes. Accordingly, U.S. Patent No. 8,067,182,
stresses the
criticality of a Kp range to achieve desired purification (see columns 9 and
10).
When applied to HIC, the weak partitioning mode described in U.S. Patent No.
8,067,182
requires an even more narrow Kp range. As set forth in Example 4, weak
partitioning for HIC
required a Kp less than 10. Patentees report that HIC performance deteriorates
with respect to
both contaminant reduction and product recovery at stronger binding
conditions.
SUMMARY OF THE INVENTION
The present invention is directed to methods for purifying a protein of
interest, e.g., an
antibody, from a sample including the protein of interest and at least one
impurity, e.g., an
aggregate, by employing a novel hydrophobic interaction chromatography (HIC)
method. The
present invention is based, at least in part, on the finding that both flow
through and bind-elute
techniques can be combined to achieve greater purification and recovery of a
protein of interest.
Moreover, the present invention is predicated, at least in part, on the
surprising finding that such
methodology can be employed under isocratic wash conditions and at stronger
binding conditions
than previously appreciated, for example, at a Kp greater than 10, so as to
achieve greater
purification and recovery.
In one aspect, the present invention is directed to a method for producing a
preparation
including a protein of interest and having a reduced level of at least one
impurity, said method
comprising: (a) contacting a sample including the protein of interest and at
least one impurity, to
a hydrophobic interaction chromatography (HIC) media, in the presence of a
load buffer such
that (i) a portion of the protein of interest binds to the HIC media and (ii)
a substantial portion of
the at least one impurity binds to the HIC media; (b) collecting a flow
through fraction including
the protein of interest unbound to the HIC media; (c) washing the HIC media
with a wash
buffer that is substantially the same as the load buffer such that a
substantial portion of the
protein of interest bound to the HIC media is released from the media; and
(d) collecting a
wash fraction including the protein of interest released from the HIC media,
wherein each
of the flow through and wash fractions include the protein of interest and
have a reduced level of
the at least one impurity.
In various embodiments, the portion of the protein of interest binds to the
HIC media at a
Kp of greater than 10, 15, 20, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140,
150, 160, 170, 180,
-2-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
190, 200, 220, 250 or 300. For example, in various embodiments, the portion of
the protein of
interest binds to the HIC media at a Kp of greater than 10, the portion of the
protein of interest
binds to the HIC media at a Kp of greater than 20, or the portion of the
protein of interest binds to
the HIC media at a Kp of greater than 100.
In a particular embodiment, the protein of interest is adalimumab.
In one embodiment, a substantial portion of the impurity bound to the HIC
media remains
bound upon washing with the wash buffer. In one embodiment, the flow through
and/or wash
fractions are substantially free of the at least one impurity.
In one embodiment, the at least one impurity is an aggregate of the protein of
interest, for
example, selected from the group consisting of a multimer, a dimer, a trimer,
a tetramer, an
oligomer and other high molecular weight species. In a particular embodiment,
the protein of
interest is adalimumab and the at least one impurity is an aggregate of
adalimumab. For
example, the aggregate may be selected from the group consisting of multimer
1, multimer 2 and
multimer 3.
In another embodiment, the impurity is a process-related impurity or a product-
related
substance. For example, the impurity may be a process-related impurity
selected from the group
consisting of a host cell protein, a host cell nucleic acid, a media
component, and a
chromatographic material. Alternatively, the impurity may be a product-related
substance
selected from the group consisting of a charge variant, an aggregate of the
protein of interest, a
fragment of the protein of interest and a modified protein.
In a particular embodiment the impurity is an acidic or basic variant, for
example, of
adalimumab. In a particular embodiment, the basic variant is a lysine variant
species, for
example, an antibody, or antigen-binding portion thereof, having heavy chains
with either zero,
one or two C-terminal lysines. In another embodiment, the impurity is an
acidic species (AR),
for example, selected from the group consisting of a charge variant, a
structure variant, a
fragmentation variant, a process-related impurity and a product-related
impurity. In a particular
embodiment, the acidic species is AR1 and the charge variant is a deamidation
variant, a
glycation variant, an afucosylation variant, a MGO variant and/ or a citric
acid variant. In
another embodiment, the acidic species is AR1 and the structure variant is a
glycosylation variant
and/ or an acetonation variant. In yet another embodiment, the acidic species
is AR1 and the
fragmentation variant is a Fab fragment variant, a C-terminal truncation
variant or a variant
missing a heavy chain variable domain. In yet a further embodiment, the acidic
species is AR2
and the charge variant comprises a deamidation variant and/ or glycation
variant.
-3-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
In a particular embodiment, the impurity is a fragment such as an Fc or a Fab
fragment.
In another embodiment, the impurity is a modified protein such as a deamidated
protein or
glycosylated protein.
In one embodiment, the protein of interest is an antibody or antigen-binding
fragment
thereof, a soluble protein, a membrane protein, a structural protein, a
ribosomal protein, an
enzyme, a zymogen, an antibody molecule, a cell surface receptor protein, a
transcription
regulatory protein, a translation regulatory protein, a chromatin protein, a
hormone, a cell cycle
regulatory protein, a G protein, a neuroactive peptide, an immunoregulatory
protein, a blood
component protein, an ion gate protein, a heat shock protein, an antibiotic
resistance protein, a
functional fragment of any of the preceding proteins, an epitope-containing
fragment of any of
the preceding proteins, and combinations thereof.
In a particular embodiment, the protein of interest is an antibody or antigen-
binding
fragment thereof such as a humanized antibody or antigen-binding portion
thereof, a human
antibody or antigen-binding portion thereof, a chimeric antibody or antigen-
binding portion
thereof, or a multivalent antibody. In one embodiment, the antibody, or
antigen-binding
fragment thereof, comprises a heavy chain constant region selected from the
group consisting of
IgG 1, IgG2, IgG3, IgG4, IgM, IgA and IgE constant regions. In another
embodiment, the
antibody, or antigen-binding fragment thereof is selected from the group
consisting of a Fab
fragment, a F(ab')2 fragment, a single chain Fv fragment, an SMIP, an
affibody, an avimer, a
nanobody, and a single domain antibody.
In one embodiment, the methods of the invention further include repeating
steps (a)-(d) at
least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 or 20 times using the
flow through fraction,
wash fraction, or combination thereof having a reduced level of the at least
one impurity. In
certain embodiments, the flow through fraction and the wash fraction are
combined.
In one embodiment, the portion of the protein of interest that binds to the
HIC media is at
least about 20%, at least about 30%, at least about 40%, at least about 50%,
at least about 60%, at
least about 70%, at least about 80% or at least about 90% of the protein of
interest in the sample.
Alternatively or in combination, the substantial portion of the protein of
interest released from
the HIC media upon washing with the wash buffer is about at least 20%, at
least about 30%, at
least about 40%, at least about 50%, at least about 60%, at least about 70%,
at least about 80%, at
least about 90% or about 100% of the amount of protein of interest bound to
the HIC media.
Alternatively or in combination, the substantial portion of the at least one
impurity that binds to
the HIC media is at least about 50%, at least about 55%, at least about 60%,
at least about 65%,
-4-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
at least about 70%, at least about 75%, at least about 80%, at least about
85%, at least about 90%,
at least about 95% or about 100% of the at least one impurity in the sample.
In certain embodiments, the accumulative yield of the protein of interest in
the flow
through fraction and/or wash fraction is at least about 35%, at least about
40%, at least about
45%, at least about 50%, at least about 55%, at least about 60%, at least
about 65%, at least about
70%, at least about 75%, at least about 80%, at least about 85%, at least
about 90%, at least about
95%, or about 100%. Alternatively or in combination, the accumulative yield of
the protein of
interest in any one flow through fraction and/or wash fraction is at least
about 4%, at least about
10%, at least about 20%, at least about 30%, at least about 40%, at least
about 50%, at least about
55%, at least about 60%, at least about 65%, at least about 70%, at least
about 75%, at least about
80%, at least about 85, at least about 90%, at least about 95% or about 100%.
Alternatively or in
combination, the reduced level of the at least one impurity of the flow
through fraction and/or
wash fraction is at least about 50%, at least about 55%, at least about 60%,
at least about 65%, at
least about 70%, at least about 75%, at least about 80%, at least about 85%,
at least about 90%, at
least about 95% or about 100% of the at least one impurity in the sample.
In certain embodiments, the accumulative aggregate reduction of the at least
one impurity
in any one flow through fraction and/or wash fraction is at least about 0.1%,
at least about 0.2%,
at least about 0.5%, at least about 1.0%, at least about 2.0%, at least about
3.0%, at least about
4.0%, at least about 5.0%, at least about 10.0%, or at least about 20.0%.
Alternatively or in
combination, the accumulative aggregate reduction of the at least one impurity
in the flow
through fraction and/or wash fraction is at least about 0.1%, at least about
0.2%, at least about
0.5%, at least about 1.0%, at least about 2.0%, at least about 3.0%, at least
about 4.0%, at least
about 5.0%, at least about 10.0%, or at least about 20.0%.
In certain embodiments, the at least one impurity binds to the HIC media at a
Kp of
greater than 250, greater than 300, greater than 400, greater than 500,
greater than 600, greater
than 700, greater than 800, greater than 900, or greater than 1000. In certain
embodiments, the
protein of interest and the at least one impurity have a Kp ratio less than
1:10, 1:9, 1:8, 1:7, 1:6,
1:5, 1:4, 1:3 or 1:2.
In certain embodiments, the Kd for the binding of the protein of interest to
the HIC media
is at least about 0.2, at least about 0.3, at least about 0.4, at least about
0.5, or at least about 0.6.
Alternatively or in combination, the Kd for the binding of the at least one
impurity to the HIC
media is less than or equal to about 0.001, about 0.005, about 0.01, about
0.02, about 0.05, about
0.1, about 0.15 or about 0.2. In particular embodiments, the Kd for the
binding of the protein of
-5-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
interest to the HIC media is less than 50, 45, 40, 35, 30, 25, 20, 15, 10 or 5
times the Kd for the
binding of the at least one impurity to the HIC media.
In certain embodiments, the protein of interest has a Qmax of at least about
20, at least
about 30, at least about 40, at least about 50, at least about 60 or at least
about 100. In certain
embodiments, the at least one impurity has a Qmax of at least about 2, at
least about 5, at least
about 10, at least about 20, at least about 30 or at least about 40.
In certain embodiments, the HIC media comprises at least one hydrophobic
ligand. For
example, the HIC media may be selected from the group consisting of alkyl-,
aryl-ligands, and
combinations thereof. For example, the HIC media may be selected from the
group consisting of
butyl, hexyl, phenyl, octyl, or polypropylene glycol ligands. In a particular
embodiment, the HIC
media is selected from the group consisting of CaptoPhenyl, Phenyl SepharoseTM
6 Fast Flow
with low or high substitution, Phenyl SepharoseTM High Performance, Octyl
SepharoseTM High
Performance, FractogelTM EMD Propyl, FractogelTM EMD Phenyl, Macro-PrepTM
Methyl,
Macro-PrepTM t-Butyl, WP HI-Propyl (C3)TM, ToyopearlTm ether, ToyopearlTm
phenyl,
ToyopearlTm butyl, ToyoScreen PPG, ToyoScreen Phenyl, ToyoScreen Butyl,
ToyoScreen
Hexyl, HiScreen Butyl FF, HiScreen Octyl FF, and Tosoh Hexyl. In one
embodiment, the HIC
media is a column.
In various embodiments, the load buffer and/or wash buffer comprise a salt
selected from
the group consisting of ammonium sulfate, sodium sulfate, sodium chloride,
ammonium chloride,
sodium bromide or a combination thereof. In a particular embodiment, the load
buffer and the
wash buffer include a sulfate salt, a citrate salt, or a combination thereof.
For example, the
sulfate salt may be ammonium sulfate or sodium sulfate. In certain
embodiments, the citrate salt
is sodium citrate. In various embodiments, the load buffer and/or the wash
buffer comprise a
cation selected from the group consisting of Ba2+, Ca2+, Mg2+, Li, Cs, Na, K+,
Rb+, and NH4,
and/or an anion selected from the group consisting of P043-, 5042-, CH3CO3-,
Cl-, Br-, NO3-,
C104-, f, and SCN- or a combination thereof.
In one embodiment, the salt has a concentration of between about 50 mM and
2000 mM.
In a particular embodiment, the load buffer and the wash buffer have a pH
between about 4.0 and
8.5 or between about 5.0 and 7Ø In certain embodiments, the load buffer and
the wash buffer
have a pH of about 4.0, about 4.5, about 5.0, about 5.5, about 6, about 6.5,
about 7.0, about 7.5,
about 8.0, or about 8.5.
In one embodiment, the load buffer and the wash buffer are the same. In one
embodiment, the load buffer and the wash buffer are substantially the same.
For example, the
-6-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
salt concentration and/or the pH of the wash buffer may be within about 20%,
15%, 10% or 5%
of the salt concentration, and/or pH of the loading buffer.
In certain embodiments, about 100 g to about 800 g of the sample are contacted
per one
liter of HIC media. Alternatively or in combination, about 0.2 g to about 120
g of the at least one
impurity is contacted per one liter of HIC media. In certain embodiments, the
sample has a
protein concentration of about 2 mg/ml to about 50 mg/ml. In certain
embodiments, the sample
has a protein of interest concentration of about 2 mg/ml to about 50 mg/ml.
Alternatively or in
combination, the concentration of the at least one impurity in the sample is
about 0.01 to about
5.0 mg/ml.
In various embodiments, the level of the at least one impurity is reduced by
at least 60%,
at least 70%, at least 80%, at least 90%, or at least 95% of the at least one
impurity in the sample.
In one embodiment, the at least one impurity is a host cell protein. For
example, the host
cell protein may be reduced by at least 0.25, at least 0.5, at least 0.75, at
least 1.0, at least 1.25 or
at least 1.5 log reduction fraction.
In one embodiment, the HIC media has a dynamic binding capacity of at least
about 2 g,
at least about 5 g, at least about 10 g, at least about 20 g, at least about
30 g, at least about 40 g, at
least about 50 g, at least about 60 g, at least about 70 g, at least about 90
g, or at least about 100 g
of sample per one liter of media.
In one embodiment, a precursor sample including the protein of interest has
been
subjected to affinity chromatography to generate the sample. Alternatively or
in combination, the
preparation including a protein of interest and having a reduced level of one
impurity is subjected
to affinity chromatography. In such embodiments, affinity chromatography may
be performed
using affinity chromatographic media selected from the group consisting of
Protein A, G, A/G, L
media, and MabSuRe Protein A media.
In one embodiment, a precursor sample including the protein of interest has
been
subjected to ion exchange chromatography to generate the sample. Alternatively
or in
combination, the preparation including a protein of interest and having a
reduced level of one
impurity is subjected to ion exchange chromatography. In such embodiments, ion
exchange
chromatography may be performed using ion exchange chromatography media
selected from the
group consisting of (i) a cation exchange media, for example, comprising
carboxymethyl (CM),
sulfoethyl(SE), sulfopropyl(SP), phosphate(P) or sulfonate(S) ligands, and
(ii) an anion exchange
media, for example, comprising diethylaminoethyl (DEAE), quaternary aminoethyl
(QAE) or
quaternary amine (Q) group ligands.
-7-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
In one embodiment, a precursor sample including the protein of interest has
been
subjected to mixed mode chromatography to generate the sample. Alternatively
or in
combination, the method involves subjecting the preparation including a
protein of interest and
having a reduced level of one impurity to mixed mode chromatography, for
example, using
CaptoAdhere resin.
In one embodiment, a precursor sample including the protein of interest has
been
subjected to a filtration step to generate the sample. Alternatively or in
combination, the method
involves subjecting the preparation including a protein of interest and having
a reduced level of
one impurity to a filtration step, for example, a depth filtration step, a
nanofiltration step, an
ultrafiltration step, and an absolute filtration step, or a combination
thereof.
In one aspect, the present invention is directed to a pharmaceutical
composition including
the preparation produced by any of the foregoing methods.
In another aspect, the present invention is directed to a method for producing
a
preparation including adalimumab and having a reduced level of at least one
aggregate, by
(a) contacting a sample of adalimumab and at least one aggregate, to a HIC
media, in the
presence of a load buffer such that (i) a portion of the adalimumab in the
sample binds to the HIC
media and (ii) a substantial portion of the at least one aggregate binds to
the HIC media; (b)
collecting a flow through fraction of the adalimumab unbound to the HIC media;
(c)
washing the HIC media with a wash buffer that is substantially the same as the
load buffer such
that a substantial portion of the adalimumab bound to the HIC media is
released from the media;
and (d) collecting a wash fraction of the adalimumab released from the HIC
media, wherein each
of the flow through and wash fractions comprise adalimumab and have a reduced
level of the at
least one aggregate.
In one embodiment of the foregoing method, adalimumab binds to the HIC media
at a Kp
of greater than 10, 15, 20, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150,
160, 170, 180, 190,
200, 220, 250 or 300. For example, adalimumab binds to the HIC media at a Kp
of greater than
10. Alternatively, adalimumab binds to the HIC media at a Kp of greater than
20. Alternatively,
adalimumab binds to the HIC media at a Kp of greater than 90.
In a particular embodiment, the aggregate is multimer 1, multimer 2 or
multimer 3.
In various embodiments, the sample includes between 200g and 700g protein per
liter of
HIC media. In certain embodiments, the HIC media is selected from the group
consisting of GE
CaptoPhenyl, Tosoh Hexyl, GE Butyl FF, Butyl, Hexyl, Phenyl, Octyl, GE Butyl
FF, PPG. In
certain embodiments, the load buffer and the wash buffer include ammonium
sulfate, sodium
-8-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
sulfate, sodium citrate, or a combination thereof. Alternatively or in
combination, the pH of the
load buffer and the wash buffer is between 5 and 7. Alternatively or in
combination, the salt
concentration of the load buffer and the wash buffer is between about 150 mM
and 1000 mM.
In another aspect, the present invention provides a pharmaceutical composition
comprising a low-aggregate composition and a pharmaceutical acceptable
carrier.
In one aspect, the present invention provides a pharmaceutical composition
comprising a
preparation of adalimumab produced by the foregoing methods and a
pharmaceutically
acceptable carrier. In another aspect, the present invention provides a
pharmaceutical
composition comprising a low-aggregate composition of adalimumab and a
pharmaceutically
acceptable carrier. For example, the composition may include less than 5%, 4%,
3%, 2.5%, 2.4%,
2.3%, 2.2%, 2.1%, 2%, 1.5%, 1.4%, 1.3%, 1.2%, 1.1%, 1%, 0.5%, 0.1% of
aggregates, e.g.,
MM1, MM2 and MM3. Alternatively, the composition may include less than 2%,
1.5%, 1.4%,
1.3%, 1.2%, 1.1%, 1%, 0.5%, 0.1% of aggregates, e.g.,MM1, MM2 and MM3.
Alternatively,
the composition may include less than 1%, 0.5%, 0.1% of aggregates, e.g.,MM1,
MM2 and
MM3.
BRIEF DESCRIPTIONS OF THE DRAWINGS
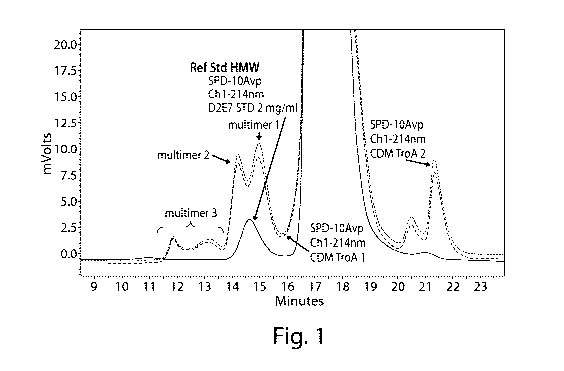
Figure 1 depicts a size exclusion chromatography (SEC) chromatogram used to
determine the molecular weight distribution of a sample of adalimumab. In
combination with
multi-angle light scattering (MALS) analysis (data not shown), the apparent
molecular weight of
each peak was determined and identified as a multimer or the reference
standard as indicated.
Multimer 1 (MM1), Multimer 2 (MM2) and Multimer 3 (MM3) were identified as
depicted.
Figures 2A-2B depicts schematic chromatograms for two modes of chromatographic
operation: bind-elute mode (Figure 2A) and flow-through mode (Figure 2B). In
the bind-elute
mode, there is strong binding of the protein of interest and the impurity.
Elution conditions are
chosen to selectively elutes the protein of interest. In the flow-through mode
there is weak
binding of the product and strong binding of the impurity.
Figure 3 depicts selection of operating conditions appropriate for an
antibody:media:buffer combination. A sample was loaded at 20 g/L and a linear
gradient elution
was performed over 20 CVs to identify the salt concentration at the monomer
and aggregate
peak. The salt concentration at or near the elution peak of the monomer is the
concentration at
which the monomer is eluted from the HIC media.
-9-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
Figure 4 depicts a process chromatogram for the HIC purification of
Adalimumab,
wherein a GE CaptoPhenyl column was equilibrated at 1.1 M AmSO4 pH 7.0
(Tris/Acetate) for
CVs, Adalimumab was prepared at 1.1 M AmSO4 and loaded to the column at 20 g-
protein/L
of media. The column was then washed with 10 CVs of the equilibration buffer
and a linear
gradient from 1.1 M to 0 M AmSO4 pH 7.0 (Tris/Acetate) over 20 CVs was
performed. See
Example 1.
Figure 5 depicts a process chromatogram for the HIC purification of
Adalimumab,
wherein a GE CaptoPhenyl column was equilibrated with 400 mM NaCit pH 5.6 for
10 CVs,
Adalimumab was prepared at 400 mM NaCit pH 5.6 and then loaded to the column
at 500 g-
protein/L-media. Finally, the column was washed with 7 CVs of the
equilibration buffer. See
Example 1.
Figure 6 depicts results of an experiment wherein a feed stream was serially
diluted to
cover a range of load concentrations from 4 to 15 mg/mL and loaded at 500 g/L
to a CaptoPhenyl
column in 400 mM NaCit pH 5.6. The results indicate the impact that the
concentration of
loaded protein can have on aggregate reduction. See Example 7.
Figure 7 depicts the effect of aggregate load concentration on dynamic binding
capacity
and aggregate clearance. The column is conditioned and loaded at different
sample load
concentrations. The flow-through is fractionated to determine the product
quality at different
times during the load and breakthrough. Using protein mass and product quality
for each of the
collected fractions, the accumulative impurity (e.g., aggregate) can be
calculated. The
accumulative impurity of the preparation is reduced when the concentration of
the aggregate in
the load is reduced, even when the total load is unchanged (e.g., 500 g/L).
See Example 13.
Figures 8A-8C depicts the effect of overall load protein concentration in the
sample. The
column is conditioned and loaded at different sample load concentrations. The
flow through is
fractionated to determine the product quality at different times during the
load and breakthrough
(Figure 8A). Using protein mass and product quality for each of the collected
factions, the
accumulative aggregate impurity can be calculated. The accumulative aggregate
impurity of the
preparation is reduced when the protein concentration of the sample is
reduced. The
Equilibrium Binding Isotherms for both the monomer and aggregate show that for
all of the
loading conditions (Figure 8B and Figure 8C), the monomer was in the non-
linear part of its
binding isotherm (e.g., equilibrium binding capacity is independent of monomer
concentration),
and the aggregate was in or near the linear part of its binding isotherm
(e.g., equilibrium binding
-10-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
capacity is dependent on aggregate concentration). Aggregate dynamic binding
capacity = f(Co,
t). See Example 13.
Figure 9 depicts the modulation of the recovery-yield for a given target
impurity
clearance by diluting the load material to a specific range. See Example 13.
Figure 10 depicts dynamic binding capacity (DBC), conventionally measured at
10%
breakthrough, as greater than the equilibrium binding capacity (EBC), based on
the data
presented in Figure 5. See Example 1.
Figure 11 depicts a dynamic binding capacity of >75 g/L. Following an
isocratic wash
and regeneration step, the remaining protein bound to the resin is <35 g/L.
Figures 12A-12B depicts determination of Apparent Binding Capacity (Figure
12A), and
Actual Binding Capacity (Figure 12B). See Example 12.
Figures 13A-13B depict the results of experiments wherein aliquots of resin
are
incubated with a load covering a range of protein concentrations at room
temperature for 3 hours,
after which the protein solution is then removed, and replaced with
equilibration buffer (Wash
simulation) and incubated at room temperature for 3 hours (repeated, Wash II).
After each
incubation, the concentration of the protein solution is measured and used to
calculated the
amount of protein ((Figure 13A) monomer D2E7, (i.e., Adalimumab), and (Figure
13B)
aggregate D2E7) bound to the resin (g protein / L resin) and plotted against
the concentration of
the protein solution at the end of the incubation (e.g., equilibrium). See
Example 11.
Figures 14A-14B depict the results outlined in Figures 13A-13B, highlighting
the fact
that at initial equilibrium a significant amount of monomer/aggregate is bound
to the resin.
However, after the protein solution is replaced with equilibration buffer (see
arrow), the
monomer desorbs from the resin and back into solution, whereas the aggregate
remains bound.
See Example 11.
Figures 15A-15B depict a determination of the binding monomer and aggregate
D2E7
(based on data provided in Figures 13A-13B) by fitting the experimental
equilibrium binding
data to the Langmuir Isotherm using the equation: q = (qma, X Cequil) (Ka +
Cequil); where q =
amount of protein bound to resin [=1 g/L-resin; qmax = maximum amount of
protein bound to
resin [=] g/L-resin; Cequil = solution concentration of protein [=1 g/L-soln;
and Kd = equilibrium
dissociation constant. See Example 11.
Figure 16 depicts the comparison of Apparent and Actual bound protein under
flow
conditions (partial partitioning) as a function of salt concentration. Binding
of the protein of
-11-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
interest is significant >10 g/L. The majority (>65%) of this monomer bound
during the load
desorbs during the isocratic wash (i.e., reversibly bound). The mass balance
of the impurity
demonstrates irreversible binding. See Example 12.
DETAILED DESCRIPTION OF THE INVENTION
The present invention is directed to methods for purifying a protein of
interest, e.g., an
antibody, from a sample comprising the protein of interest and at least one
impurity, e.g., an
aggregate, by employing a novel hydrophobic interaction chromatography (HIC)
method. The
present invention is based, at least in part, on the finding that both flow
through and bind-elute
techniques can be combined to achieve greater purification and recovery of a
protein of interest,
e.g., an antibody. Moreover, the present invention is predicated, at least in
part, on the surprising
finding that such methodology can be employed under isocratic wash conditions
and at stronger
binding conditions than previously appreciated, for example, at a Kp greater
than 10 or at a Kp
greater than 20, so as to achieve greater purification and recovery.
In one aspect, the present invention provides a method for producing a
preparation
including a protein of interest, e.g., an antibody such as adalimumab, and
having a reduced level
of at least one impurity, e.g., an aggregate, by (a) contacting a sample
including the protein of
interest and at least one impurity, to a hydrophobic interaction
chromatography media, in the
presence of a load buffer such that (i) a portion of the protein of interest
binds to the hydrophobic
interaction chromatography (HIC) media and (ii) a substantial portion of the
at least one impurity
binds to the HIC media; (b) collecting a flow through fraction including the
protein of interest
unbound to the HIC media; (c) washing the HIC media with a wash buffer that is
substantially
the same as the load buffer such that a substantial portion of the protein of
interest bound to the
HIC media is released from the media; and (d) collecting a wash fraction
including the protein of
interest released from the HIC media, wherein each of the flow through and
wash fractions
include the protein of interest and have a reduced level of the at least one
impurity. In a
particular embodiment, the portion of the protein of interest binds to the HIC
media at a Kp of
greater than 10. In another embodiment, the portion of the protein of interest
binds to the HIC
media at a Kp of greater than 20. In another embodiment, the portion of the
protein of interest
binds to the HIC media at a Kp of greater than 100.
In certain embodiments, the purification strategies of the present invention
may include
one or more chromatography and/or filtration steps to achieve a desired degree
of purification
prior to exposure of the sample comprising the protein of interest, e.g., an
antibody such as
-12-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
adalimumab, to the HIC media. For example, in certain embodiments, such pre-
HIC
chromatography step(s) can include one or more steps of chromatography and/ or
filtration. In
one embodiment the chromatography is ion exchange chromatography, mixed mode
chromatography, and/or affinity chromatography. In another embodiment, the
filtration step is
depth filtration, nanofiltration, ultrafiltration and/ or absolute filtration.
In certain embodiments,
the purification strategies of the present invention may include one or more
additional
chromatography and/or filtration steps after the HIC purification step. For
example, in certain
embodiments, such post-HIC chromatography step(s) can include one or more
steps of
chromatography and/ or filtration. In one embodiment, the chromatography is
ion exchange
chromatography, mixed mode chromatography, and/or affinity chromatography. In
another
embodiment, the filtration step is depth filtration, nanofiltration,
ultrafiltration and/ or absolute
filtration.
In addition, in certain embodiments, the present invention is directed toward
pharmaceutical compositions comprising one or more proteins of interest
purified by methods
described herein. In a particular embodiment, the present invention is
directed to a
pharmaceutical composition comprising adalimumab and having a reduced level of
aggregates.
DEFINITIONS
Unless otherwise defined herein, scientific and technical terms used in
connection with
the present invention shall have the meanings that are commonly understood by
those of
ordinary skill in the art. The meaning and scope of the terms should be clear,
however, in the
event of any latent ambiguity, definitions provided herein take precedent over
any dictionary or
extrinsic definition. Further, unless otherwise required by context, singular
terms, for example,
those characterized by "a" or "an", shall include pluralities, e.g., one or
more impurities. In this
application, the use of "or" means "and/or", unless stated otherwise.
Furthermore, the use of the
term "including," as well as other forms of the term, such as "includes" and
"included", is not
limiting. Also, terms such as "element" or "component" encompass both elements
and
components comprising one unit and elements and components that comprise more
than one unit
unless specifically stated otherwise.
As used herein, the term "sample", refers to a liquid composition including
the protein of
interest and one or more impurities. In a particular embodiment, the sample is
a "clarified
harvest", referring to a liquid material containing a protein of interest, for
example, an antibody
of interest such as adalimumab, that has been extracted from cell culture, for
example, a
- 13-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
fermentation bioreactor, after undergoing centrifugation to remove large solid
particles and
subsequent filtration to remove finer solid particles and impurities from the
material.
In various embodiments, the sample may be partially purified. For example, the
sample
may have already been subjected to any of a variety of art recognized
purification techniques,
such as chromatography, e.g., ion exchange chromatography, mixed mode
chromatography,
and/or affinity chromatography, or filtration, e.g., depth filtration,
nanofiltration, ultrafiltration
and/ or absolute filtration.
The term "precursor sample", as used herein refers to a liquid composition
containing the
protein of interest and, optionally, one or more impurities, either derived
from the clarified
harvest, or a partially purified intermediate sample that is subject to a
purification or treatment
step prior to being subjected to HIC. Impurities in a precursor sample may be
derived from the
production, purification or treatment of the protein of interest prior to
subjecting the resulting
sample to HIC.
The term "protein of interest", as used herein refers to a target protein
present in a sample,
purification of which is desired. In various embodiment, the protein of
interest is an antibody or
antigen-binding fragment thereof, a soluble protein, a membrane protein, a
structural protein, a
ribosomal protein, an enzyme, a zymogen, an antibody molecule, a cell surface
receptor protein,
a transcription regulatory protein, a translation regulatory protein, a
chromatin protein, a
hormone, a cell cycle regulatory protein, a G protein, a neuroactive peptide,
an
immunoregulatory protein, a blood component protein, an ion gate protein, a
heat shock protein,
an antibiotic resistance protein, a functional fragment of any of the
preceding proteins, an
epitope-containing fragment of any of the preceding proteins, and combinations
thereof. In a
particular embodiment, the protein of interest is a monomer.
In a particular embodiment, the protein of interest is an antibody, or an
antigen binding
portion thereof. The term "antibody" includes an immunoglobulin molecule
comprised of four
polypeptide chains, two heavy (H) chains and two light (L) chains inter-
connected by disulfide
bonds. Each heavy chain is comprised of a heavy chain variable region
(abbreviated herein as
HCVR or VH) and a heavy chain constant region (CH). The heavy chain constant
region is
comprised of three domains, CH1, CH2 and CH3. Each light chain is comprised of
a light chain
variable region (abbreviated herein as LCVR or VL) and a light chain constant
region. The light
chain constant region is comprised of one domain, CL. The VH and VL regions
can be further
subdivided into regions of hypervariability, termed complementarity
determining regions
(CDRs), interspersed with regions that are more conserved, termed framework
regions (FR).
-14-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
Each VH and VL is composed of three CDRs and four FRs, arranged from amino-
terminus to
carboxy-terminus in the following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4.
The term
"antibody", as used herein, also includes alternative antibody and antibody-
like structures, such
as, but not limited to, dual variable domain antibodies (DVD-Ig).
The term "antigen-binding portion" of an antibody (or "antibody portion")
includes
fragments of an antibody that retain the ability to specifically bind to an
antigen (e.g., hIL-12,
hTNFa, or hIL-18). It has been shown that the antigen-binding function of an
antibody can be
performed by fragments of a full-length antibody. Examples of binding
fragments encompassed
within the term "antigen-binding portion" of an antibody include (i) a Fab
fragment, a
monovalent fragment comprising the VL, VH, CL and CH1 domains; (ii) a F(ab')2
fragment, a
bivalent fragment comprising two Fab fragments linked by a disulfide bridge at
the hinge region;
(iii) a Fd fragment comprising the VH and CH1 domains; (iv) a Fv fragment
comprising the VL
and VH domains of a single arm of an antibody, (v) a dAb fragment (Ward et
al., (1989) Nature
341:544-546, the entire teaching of which is incorporated herein by
reference), which comprises
a VH domain; and (vi) an isolated complementarity determining region (CDR).
Furthermore,
although the two domains of the Fv fragment, VL and VH, are coded for by
separate genes, they
can be joined, using recombinant methods, by a synthetic linker that enables
them to be made as
a single protein chain in which the VL and VH regions pair to form monovalent
molecules
(known as single chain Fv (scFv); see, e.g., Bird et al. (1988) Science
242:423-426; and Huston
et al. (1988) Proc. Natl. Acad. Sci. USA 85:5879-5883, the entire teachings of
which are
incorporated herein by reference). Such single chain antibodies are also
intended to be
encompassed within the term "antigen-binding portion" of an antibody. Other
forms of single
chain antibodies, such as diabodies are also encompassed. Diabodies are
bivalent, bispecific
antibodies in which VH and VL domains are expressed on a single polypeptide
chain, but using a
linker that is too short to allow for pairing between the two domains on the
same chain, thereby
forcing the domains to pair with complementary domains of another chain and
creating two
antigen binding sites (see, e.g., Holliger, P., et al. (1993) Proc. Natl.
Acad. Sci. USA 90:6444-
6448; Poljak, R. J., et al. (1994) Structure 2:1121-1123, the entire teachings
of which are
incorporated herein by reference). Still further, an antibody may be part of a
larger
immunoadhesion molecule, formed by covalent or non-covalent association of the
antibody with
one or more other proteins or peptides. Examples of such immunoadhesion
molecules include
use of the streptavidin core region to make a tetrameric scFv molecule
(Kipriyanov, S. M., et al.
(1995) Human Antibodies and Hybridomas 6:93-101, the entire teaching of which
is
incorporated herein by reference) and use of a cysteine residue, a marker
peptide and a C-
-15-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
terminal polyhistidine tag to make bivalent and biotinylated scFv molecules
(Kipriyanov, S. M.,
et al. (1994) Mol. Immunol. 31:1047-1058, the entire teaching of which is
incorporated herein by
reference). Antibody portions, such as Fab and F(ab')2 fragments, can be
prepared from whole
antibodies using conventional techniques, such as papain or pepsin digestion,
respectively, of
whole antibodies. Moreover, antibodies, antibody portions and immunoadhesion
molecules can
be obtained using standard recombinant DNA techniques, as described herein. In
one aspect, the
antigen binding portions are complete domains or pairs of complete domains.
The term "human antibody" includes antibodies having variable and constant
regions
corresponding to human germline immunoglobulin sequences as described by Kabat
et al. (See
Kabat, et al. (1991) Sequences of proteins of Immunological Interest, Fifth
Edition, U.S.
Department of Health and Human Services, NIH Publication No. 91-3242). The
human
antibodies of the invention may include amino acid residues not encoded by
human germline
immunoglobulin sequences (e.g., mutations introduced by random or site-
specific mutagenesis in
vitro or by somatic mutation in vivo), e.g., in the CDRs and in particular
CDR3. The mutations
can be introduced using the "selective mutagenesis approach." The human
antibody can have at
least one position replaced with an amino acid residue, e.g., an activity
enhancing amino acid
residue which is not encoded by the human germline immunoglobulin sequence.
The human
antibody can have up to twenty positions replaced with amino acid residues
which are not part of
the human germline immunoglobulin sequence. In other embodiments, up to ten,
up to five, up
to three or up to two positions are replaced. In one embodiment, these
replacements are within
the CDR regions. However, the term "human antibody", as used herein, is not
intended to
include antibodies in which CDR sequences derived from the germline of another
mammalian
species, such as a mouse, have been grafted onto human framework sequences.
The phrase "recombinant human antibody" includes human antibodies that are
prepared,
expressed, created or isolated by recombinant means, such as antibodies
expressed using a
recombinant expression vector transfected into a host cell, antibodies
isolated from a
recombinant, combinatorial human antibody library, antibodies isolated from an
animal (e.g., a
mouse) that is transgenic for human immunoglobulin genes (see, e.g., Taylor,
L. D., et al. (1992)
Nucl. Acids Res. 20:6287-6295, the entire teaching of which is incorporated
herein by reference)
or antibodies prepared, expressed, created or isolated by any other means that
involves splicing
of human immunoglobulin gene sequences to other DNA sequences. Such
recombinant human
antibodies have variable and constant regions derived from human germline
immunoglobulin
sequences (see, Kabat, E. A., et al. (1991) Sequences of Proteins of
Immunological Interest, Fifth
Edition, U.S. Department of Health and Human Services, NIH Publication No. 91-
3242). In
-16-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
certain embodiments, however, such recombinant human antibodies are subjected
to in vitro
mutagenesis (or, when an animal transgenic for human Ig sequences is used, in
vivo somatic
mutagenesis) and thus the amino acid sequences of the VH and VL regions of the
recombinant
antibodies are sequences that, while derived from and related to human
germline VH and VL
sequences, may not naturally exist within the human antibody germline
repertoire in vivo. In
certain embodiments, however, such recombinant antibodies are the result of
selective
mutagenesis approach or back-mutation or both.
An "isolated antibody" includes an antibody that is substantially free of
other antibodies
having different antigenic specificities (e.g., an isolated antibody that
specifically binds hTNFa is
substantially free of antibodies that specifically bind antigens other than
hTNFa). An isolated
antibody that specifically binds hTNFa may bind TNFa molecules from other
species.
Moreover, an isolated antibody may be substantially free of other cellular
material and/or
chemicals. A suitable anti-TNFa antibody is Adalimumab (AbbVie).
As used herein, the term "adalimumab," also known by its trade name HUMIRA
(AbbVie) refers to a human IgGi antibody that binds human tumor necrosis
factor a (TNFa). In
general, the heavy chain constant domain 2 (CH2) of the adalimumab IgG-Fc
region is
glycosylated through covalent attachment of oligosaccharide at asparagine 297
(Asn-297). The
light chain variable region of adalimumab is provided herein as SEQ ID NO:1,
and the heavy
chain variable region of adalimumab is provided herein as SEQ ID NO:2.
Adalimumab
comprises a light chain variable region comprising a CDR1 of SEQ ID NO:7, a
CDR2 of SEQ ID
NO:5, and a CDR3 of SEQ ID NO:3. Adalimumab comprises a heavy chain variable
region
comprising a CDR1 of SEQ ID NO:8, a CDR2 of SEQ ID NO:6 and CDR3 of SEQ ID
NO:4.
The nucleic acid sequence of the light chain variable region is set forth in
SEQ ID NO:9. The
nucleic acid sequence of the heavy chain variable region is set forth in SEQ
ID NO:10. The full
length amino acid sequence of the light chain is set forth as SEQ ID NO:11 and
the full length
amino acid sequence of the heavy chain is set forth as SEQ ID NO:12.
Adalimumab is
described in U.S. Patent Nos. 6,090,382; 6,258,562; 6,509,015; 7,223,394;
7,541,031; 7,588,761;
7,863,426; 7,919,264; 8,197,813; 8,206,714; 8,216,583; 8,420,081; 8,092,998;
8,093,045;
8,187,836; 8,372,400; 8,034,906; 8,436,149; 8,231,876; 8,414,894; 8,372,401,
the entire
contents of each which are expressly incorporated herein by reference in their
entireties.
Adalimumab is also described in the "Highlights of Prescribing Information"
for HUMIRA
(adalimumab) Injection (Revised Jan. 2008) the contents of which are hereby
incorporated herein
by reference.
-17-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
In one embodiment, adalimumab dissociates from human TNFa with a Kd of 1x10-8
M or
less and a Koff rate constant of 1x10-3 s-1 or less, both determined by
surface plasmon resonance,
and neutralizes human TNFa cytotoxicity in a standard in vitro L929 assay with
an IC50 of 1x10-
7
M or less. In another embodiment, adalimumab dissociates from human TNFa with
a Koff of
5x10-4 s-1 or less, or even more preferably, with a Koff of 1x10-4 s-1 or
less. In still another
embodiment, adalimumab neutralizes human TNFa cytotoxicity in a standard in
vitro L929 assay
with an IC50 of 1x10-8 M or less, an IC50 of 1x10-9 M or less or an IC50 of
1x10-1 M or less.
The term "Koff', as used herein, is intended to refer to the off rate constant
for dissociation of an
antibody from the antibody/antigen complex.
The term "impurity", as used herein refers to any foreign or objectionable
molecule,
including a biological macromolecule such as a DNA, an RNA, or a protein other
than the
protein of interest being purified. Exemplary impurities include, for example,
protein variants,
such as aggregates, high molecular weight species, low molecular weight
species and fragments,
and deamidated species; host cell proteins; proteins that are part of an
absorbent used for affinity
chromatography (e.g. Protein A); endotoxins; and viruses.
The methods of the invention serve to generate a preparation comprising a
protein of
interest and having a reduced level of impurity. As used herein a "reduced
level of impurity"
refers to a composition comprising reduced levels of an impurity as compared
to the levels of the
impurity in the sample prior to purification by the methods of the present
invention. In another
embodiment, the methods of the invention generate a preparation comprising a
protein of interest
and having a reduced level of total impurity. As used herein a "reduced level
of total impurity"
refers to a composition comprising reduced levels of total impurity as
compared to the levels of
the impurity in the sample prior to purification by the methods of the present
invention. In one
embodiment, a preparation having a reduced level of total impurity is free of
impurities or
substantially free of impurities.
The present invention is further directed to low impurity compositions and
methods of
generating the same, for example, low impurity compositions of adalimumab. The
term "low
impurity composition," as used herein, refers to a composition comprising a
protein of interest,
wherein the composition contains less than about 15% total impurities. For
example, a low
impurity composition may contain about 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%,
1.5%, 1.4%,
1.3%, 1.2%, 1.1%, 1%, 0.5%, or less total impurities. In a particular
embodiment, a low impurity
composition comprises about 5%, 4%, 3%, 2.5%, 2.4%, 2.3%, 2.2%, 2.1%, 2%,
1.5%, 1.4%,
1.3%, 1.2%, 1.1%, 1%, 0.5%, 0.1%, or less total impurities.
-18-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
The term "non-low impurity composition," as used herein, refers to a
composition
comprising a protein of interest, which contains more than about 15% total
impurity. For
example, a non-low impurity composition may contain about 15%, 16%, 17%, 18%,
19%, 20%,
21%, 22%, 23%, 24%, 25%, or more total impurities.
In one embodiment, a low impurity composition has improved biological and
functional
properties, including increased efficacy in the treatment or prevention of a
disorder in a subject,
e.g., a disorder in which TNFa activity is detrimental, as compared to a non-
low impurity
composition. In one embodiment, the low impurity composition comprises an anti-
TNFa
antibody, or antigen-binding portion thereof, such as adalimumab or a fragment
thereof. For
example, in one embodiment, a low impurity composition comprising an antibody,
or antigen-
binding portion thereof, exhibits increased cartilage penetration, decreased
bone erosion, and/or
reduced cartilage destruction, as compared to a non-low impurity composition
comprising the
same antibody or antigen binding portion thereof, when administered to a
subject suffering from
a disorder in which TNFa activity is detrimental.
As used herein, the term "increased cartilage penetration" refers to increased
penetration
of cartilage in vivo by a low impurity composition as compared to a non-low
impurity
composition comprising the same antibody or antigen binding portion thereof.
As used herein, the term "reduced cartilage destruction" refers to measurable
decrease in
destruction of cartilage tissue in vivo by a low impurity composition as
compared to a non-low
impurity composition comprising the same antibody or antigen binding portion
thereof. As used
herein, the term "decreased bone erosion" refers to measurable decrease, in
vivo, of the erosion of
bone tissue by a low impurity composition as compared to a non-low aggregate
composition
comprising the same antibody or antigen binding portion thereof. For example,
an in vivo model
of a disease or disorder in which TNFa activity is detrimental, e.g., a mouse
model of arthritis,
can be used to measure cartilage penetration, bone erosion, and/or cartilage
destruction by a
composition comprising an anti-TNFa antibody or antigen binding portion
thereof. One non-
limiting example of an art-recognized mouse model of arthritis is the human
TNF transgenic 197
mouse model of arthritis (TNF-Tg197) (see Keffer, J. et al., EMBO J (1991)
10:4025-4031 for
further description of the TNF-Tg197 model of arthritis).
In another embodiment, a low impurity composition comprising an antibody, or
antigen-
binding portion thereof, exhibits increased protection against the development
of arthritic scores
and/or histopathology scores as compared to a non-low impurity composition
when administered
to an animal model of arthritis, e.g., the TNF-Tg197 model of arthritis. As
used herein, "arthritic
-19-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
scores" refer to signs and symptoms of arthritis in an animal model of
arthritis. As used herein,
"histopathology scores" refer to radiologic damage involving cartilage and
bone as well as local
inflammation.
In another embodiment, a low impurity composition comprising an antibody, or
antigen-
binding portion thereof, exhibits reduced synovial proliferation, reduced cell
infiltration, reduced
chondrocyte death, and/or reduced proteoglycan loss as compared to a non-low
impurity
composition. In another embodiment, a low impurity composition comprising an
anti-TNFa
antibody, or antigen-binding portion thereof, exhibits increased TNFa affinity
as compared to a
non-low impurity composition.
As used herein, the term "a disorder in which TNFa activity is detrimental" is
intended to
include diseases and other disorders in which the presence of TNFa in a
subject suffering from
the disorder has been shown to be or is suspected of being either responsible
for the
pathophysiology of the disorder or a factor that contributes to a worsening of
the disorder.
Accordingly, a disorder in which TNFa activity is detrimental is a disorder in
which inhibition of
TNFa activity is expected to alleviate the symptoms and/or progression of the
disorder. Such
disorders may be evidenced, for example, by an increase in the concentration
of TNFa in a
biological fluid of a subject suffering from the disorder (e.g., an increase
in the concentration of
TNFa in serum, plasma, or synovial fluid of the subject), which can be
detected, for example,
using an anti-TNFa antibody as described above. There are numerous examples of
disorders in
which TNFa activity is detrimental. In one embodiment, the disorder in which
TNFa activity is
detrimental is an autoimmune disorder. In one embodiment, the autoimmune
disorder is selected
from the group consisting of rheumatoid arthritis, juvenile idiopathic
arthritis, rheumatoid
spondylitis, ankylosing spondylitis, psoriasis, osteoarthritis, gouty
arthritis, an allergy, multiple
sclerosis, psoriatic arthritis, autoimmune diabetes, autoimmune uveitis,
nephrotic syndrome,
juvenile rheumatoid arthritis, Crohn's disease, ulcerative colitis, active
axial spondyloarthritis
(active axSpA) and non-radiographic axial spondyloarthritis (nr-axSpA).
Disorders in which
TNFa activity is detrimental are set forth in U.S. Patent No. 6,090,382 and
also in the Humira
Prescribing Information, the contents of each of which are hereby incorporated
herein by
reference. The use of TNFa antibodies and antibody portions obtained using
methods of the
invention for the treatment of specific disorders is discussed in further
detail below.
In a particular embodiment, the impurity is a process-related impurity. As
used herein,
the term "process-related impurity," refers to impurities that are present in
a composition
comprising a protein of interest but are not derived from the protein itself.
Process-related
impurities include, but are not limited to, host cell proteins (HCPs), host
cell nucleic acids,
-20-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
chromatographic materials, and media components.
A "low process-related impurity
composition," as used herein, refers to a composition comprising reduced
levels of process-
related impurities as compared to a composition wherein the impurities were
not reduced. For
example, a low process-related impurity composition may contain about 10%, 9%,
8%, 7%, 6%,
5%, 4%, 3%, 2%, 1%, 0.5%, or less process-related impurities. In one
embodiment, a low
process-related impurity composition is free of process-related impurities or
is substantially free
of process-related impurities.
In one embodiment, the impurity is a host cell protein. The term "host cell
protein"
(HCP), as used herein, is intended to refer to non-protein of interest
proteinaceous impurities
derived from host cells, for example, host cells used to produce the protein
of interest.
In one embodiment, the impurity is a host cell nucleic acid. The term "host
cell nucleic
acids", as used herein, is intended to refer to nucleic acids derived from
host cells, for example,
host cells used to produce the protein of interest.
In a particular embodiment, the impurity is a product-related substance. As
used herein,
the term "product-related substance" refers to variants of the protein of
interest formed during
manufacturing and/or storage of the protein of interest. Specific examples of
product-related
substances include degradants of the protein, truncated forms of the protein,
high molecular
weight species, low molecular weight species, fragments of the protein,
modified forms of the
protein, including deamidated, isomerized, mismatched S-S linked, oxidized or
altered conjugate
forms (e.g., glycosylation, phosphorylation), aggregates including dimers and
higher multiples of
the protein of interest, and charge variants.
In a particular embodiment, the impurity is an aggregate. As used herein, the
term
"aggregate" refers to agglomeration or oligomerization of two or more
individual molecules of
the protein of interest to form, for example, dimers, trimers, tetramers,
oligomers and other high
molecular weight species. Protein aggregates can be soluble or insoluble. In a
particular
embodiment, the aggregate is a multimer of adalimumab. In a particular
embodiment, the
aggregate is a dimer of adalimumab. In another embodiment, the aggregate is a
trimer of
adalimumab. In another embodiment, the aggregate is a tetramer of adalimumab.
In certain embodiments, the sample can comprise more than one type of
aggregate. For
example, but not by way of limitation, the nature of the aggregates and total
aggregate
composition can be identified based on chromatographic residence time. For
example, Figure 1
depicts a size exclusion chromatography (SEC) chromatogram used to determine
the molecular
weight distribution of a sample of adalimumab.
-21-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
As set forth therein, the total aggregate species associated with the
expression of
adalimumab can be divided into multimer 1 (MM1), multimer 2 (MM2) and multimer
3 (MM3).
In various embodiments, the methods of the present invention serve to reduce
the levels of one of
MM1, MM2 or MM3. In another embodiment, the methods of the present invention
serve to
reduce the levels of MM1 and MM2. In another embodiment, the methods of the
present
invention serve to reduce the levels of MM1 and MM3. In another embodiment,
the methods of
the present invention serve to reduce the levels of MM2 and MM3. In yet
another embodiment,
the methods of the present invention serve to reduce the levels of MM1, MM2
and MM3.
In one embodiment, the methods of the invention generate a preparation
comprising a
protein of interest and having a reduced level of aggregate. As used herein, a
"reduced level of
aggregate" refers to a composition comprising reduced levels of an aggregate
as compared to the
levels of the aggregate in the sample prior to purification by the methods of
the present invention.
In one embodiment, a preparation having a reduced level of aggregate is free
of the aggregate or
substantially free of the aggregate. In another embodiment, the methods of the
invention
generate a preparation comprising a protein of interest and having a reduced
level of total
aggregate. As used herein a "reduced level of total aggregate" refers to a
composition
comprising reduced levels of total aggregate as compared to the levels of the
impurity in the
sample prior to purification by the methods of the present invention. In one
embodiment, a
preparation having a reduced level of total aggregate is free of aggregates or
substantially free of
the aggregates.
The present invention is further directed to low aggregate compositions and
methods of
generating the same, for example, low aggregate compositions of adalimumab.
The term "low
aggregate composition," as used herein, refers to a composition comprising a
protein of interest,
wherein the composition contains less than about 15% total aggregates. For
example, a low
aggregate composition may contain about 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%,
1.5%, 1.4%,
1.3%, 1.2%, 1.1%, 1%, 0.5%, or less total aggregates. In a particular
embodiment, a low
aggregate composition comprises about 5%, 4%, 3%, 2.5%, 2.4%, 2.3%, 2.2%,
2.1%, 2%, 1.5%,
1.4%, 1.3%, 1.2%, 1.1%, 1%, 0.5%, 0.1%, or less total aggregates.
In one embodiment, a low aggregate composition of adalimumab can comprise
about
10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2.5%, 2.4%, 2.3%, 2.2%, 2.1%, 2%, 1.5%, 1.4%,
1.3%,
1.2%, 1.1%, 1%, 0.5%, 0.1%, or less of MM1, or 0.0% of MM1. In another
embodiment, a low
aggregate composition of adalimumab can comprise about 10%, 9%, 8%, 7%, 6%,
5%, 4%, 3%,
2.5%, 2.4%, 2.3%, 2.2%, 2.1%, 2%, 1.5%, 1.4%, 1.3%, 1.2%, 1.1%, 1%, 0.5%,
0.1%, or less of
MM2, or 0.0% of MM2. In another embodiment, a low aggregate composition of
adalimumab
-22-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
can comprise about 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2.5%, 2.4%, 2.3%, 2.2%,
2.1%, 2%,
1.5%, 1.4%, 1.3%, 1.2%, 1.1%, 1%, 0.5%, 0.1%, or less of MM3, or 0.0% of MM3.
The term "non-low aggregate composition," as used herein, refers to a
composition
comprising a protein of interest, which contains more than about 15% total
aggregates. For
example, a non-low aggregate composition may contain about 15%, 16%, 17%, 18%,
19%, 20%,
21%, 22%, 23%, 24%, 25%, or more total aggregates . In one embodiment, a non-
low aggregate
composition of adalimumab can comprise about 15%, 16%, 17%, 18%, 19%, 20%,
21%, 22%,
23%, 24%, 25%, or more of MM1. In another embodiment, a non-low aggregate
composition or
adalimumab can comprise about 15%, 16%, 17%, 18%, 19%, 20%, 21%, 22%, 23%,
24%, 25%,
or more of MM2. In another embodiment, a non-low aggregate composition or
adalimumab can
comprise about 15%, 16%, 17%, 18%, 19%, 20%, 21%, 22%, 23%, 24%, 25%, or more
of MM3.
In one embodiment, a low aggregate composition has improved biological and
functional
properties, including increased efficacy in the treatment or prevention of a
disorder in a subject,
e.g., a disorder in which TNFa activity is detrimental, as compared to a non-
low aggregate
composition. In one embodiment, the low aggregate composition comprises an
anti-TNFa
antibody, or antigen-binding portion thereof, such as adalimumab or a fragment
thereof. For
example, in one embodiment, a low aggregate composition comprising an
antibody, or antigen-
binding portion thereof, exhibits increased cartilage penetration, decreased
bone erosion, and/or
reduced cartilage destruction, as compared to a non-low aggregate composition
comprising the
same antibody or antigen binding portion thereof, when administered to a
subject suffering from
a disorder in which TNFa activity is detrimental.
In another embodiment, a low aggregate composition comprising an antibody, or
antigen-
binding portion thereof, exhibits increased protection against the development
of arthritic scores
and/or histopathology scores as compared to a non-low aggregate composition
when
administered to an animal model of arthritis, e.g., the TNF-Tg197 model of
arthritis, as described
above.
In another embodiment, a low aggregate composition comprising an antibody, or
antigen-
binding portion thereof, exhibits reduced synovial proliferation, reduced cell
infiltration, reduced
chondrocyte death, and/or reduced proteoglycan loss as compared to a non-low
aggregate
composition. In another embodiment, a low aggregate composition comprising an
anti-TNFa
antibody, or antigen-binding portion thereof, exhibits increased TNFa affinity
as compared to a
non-low aggregate composition.
-23-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
In a particular embodiment, the impurity is a fragment of the protein of
interest. The term
"fragment" as used herein refers to any truncated form of a protein of
interest, resulting from, for
example, dissociation of a peptide chain, or enzymatic and/or chemical
modifications.
In a particular embodiment, the impurity is a charge variant. The term "charge
variant",
as used herein, refers to the full complement of product variants including,
but not limited to
acidic species, and basic species (e.g., Lys variants). In certain
embodiments, such variants can
include product aggregates and/or product fragments, to the extent that such
aggregation and/or
fragmentation results in a product charge variation as seen in an analytical
technique used for that
purpose.
As used herein, the terms "acidic species," "acidic region," and "AR," refer
to the
variants of a protein, e.g., an antibody or antigen-binding portion thereof,
which are characterized
by an overall acidic charge. For example, in monoclonal antibody (mAb)
preparations, such
acidic species can be detected by various methods, such as ion exchange, for
example, WCX-10
HPLC (a weak cation exchange chromatography), or IEF (isoelectric focusing).
Acidic species
of an antibody may include charge variants, structure variants, and/or
fragmentation variants.
Exemplary charge variants include, but are not limited to, deamidation
variants, afucosylation
variants, methylglyoxal (MGO) variants, glycation variants, and citric acid
variants. Exemplary
structure variants include, but are not limited to, glycosylation variants and
acetonation variants.
Exemplary fragmentation variants include any truncated protein species from
the target molecule
due to dissociation of peptide chain, enzymatic and/or chemical modifications,
including, but not
limited to, Fc and Fab fragments, fragments missing a Fab, fragments missing a
heavy chain
variable domain, C-terminal truncation variants, variants with excision of N-
terminal Asp in the
light chain, and variants having N-terminal truncation of the light chain.
Other acidic species
variants include variants containing unpaired disulfides, host cell proteins,
and host nucleic acids,
chromatographic materials, and media components.
In certain embodiments, a protein composition can comprise more than one type
of acidic
species variant. For example, but not by way of limitation, the total acidic
species can be divided
based on chromatographic residence time. For example, the total acidic species
associated with
the expression of adalimumab can be divided into a first acidic species region
(AR1) and a
second acidic species region (AR2).
AR1 can comprise, for example, charge variants such as deamidation variants,
MGO
modified species, glycation variants, and citric acid variants, structural
variants such as
glycosylation variants and acetonation variants, and/or fragmentation
variants. Other acidic
-24-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
variants such as host cells and unknown species may also be present. In
another embodiment,
AR2 can comprise, for example, charge variants such as glycation variants and
deamidation
variants. Other acidic variants such as host cells and unknown species may
also be present.
With respect, in particular, to adalimumab (and antibodies sharing certain
structural
characteristics of adalimumab, e.g., one or more CDR and/or heavy and light
chain variable
regions of adalimumab), AR1 charge variants can comprise, but are not limited
to, deamidation
variants, glycation variants, afucosylation variants, MGO variants or citric
acid variants. In one
embodiment, deamidation variants result from deamidation occurring at
asparagine residues
comprising Asn393 and Asn329 and at glutamine residues comprising G1n3 and
G1n6. In
another embodiment, the glycation variants result from glycation occurring at
Lys98 and Lys151.
AR1 structure variants can comprise, but are not limited to, glycosylation
variants or acetonation
variants. AR1 fragmentation variants can comprise Fc and Fab fragments,
fragments missing a
Fab, fragments missing a heavy chain variable domain, C-terminal truncation
variants, variants
with excision of N-terminal Asp in the light chain, and variants having N-
terminal truncation of
the light chain. AR2 charge variants can comprise, but are not limited to,
deamidation variants or
glycation variants, wherein the deamidation variants can result from
deamidation occurring at
asparagine residues comprising Asn393 and Asn329 and at glutamine residues
comprising G1n3
and G1n6, and the glycation variants can result from glycation occurring at
Lys98 and Lys151.
Acidic species may also include process-related impurities.
The acidic species may be the result of product preparation (referred to
herein as
"preparation-derived acidic species"), or the result of storage (referred to
herein as "storage-
derived acidic species"). Preparation-derived acidic species are acidic
species that are formed
during the preparation (upstream and/or downstream processing) of the protein,
e.g., the antibody
or antigen-binding portion thereof. For example, preparation-derived acidic
species can be
formed during cell culture ("cell culture-derived acidic species"). Storage-
derived acidic species
are acidic species that are not present in the population of proteins directly
after preparation, but
are formed while the sample is being stored. The type and amount of storage-
derived acidic
species can vary based on the formulation of the sample. Formation of storage-
derived acidic
species can be partially or completely inhibited when the preparation is
stored under particular
conditions. For example, an aqueous formulation can be stored at a particular
temperature to
partially or completely inhibit AR formation. For example, formation or
storage-derived AR can
be partially inhibited in an aqueous formulation stored at between about 2 C
and 8 C, and
completely inhibited when stored at -80 C. In addition, a low AR composition
can be
lyophilized to partially or completely inhibit the formation of storage-
derived AR.
-25-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
The term "low acidic species composition," as used herein, refers to a
composition
comprising an antibody or antigen binding portion thereof, wherein the
composition contains less
than about 15% acidic species. For example, a low acidic species composition
may contain
about 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1.5%, 1.4%, 1.3%, 1.2%, 1.1%, 1%,
0.5%, or
less acidic species. In one embodiment, a low acidic species composition can
comprise about
10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2.5%, 2.4%, 2.3%, 2.2%, 2.1%, 2%, 1.5%, 1.4%,
1.3%,
1.2%, 1.1%, 1%, 0.5%, 0.1%, or less of AR1, or 0.0% of AR1. In another
embodiment, a low
acidic species composition can comprise about 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%,
2.5%, 2.4%,
2.3%, 2.2%, 2.1%, 2%, 1.5%, 1.4%, 1.3%, 1.2%, 1.1%, 1%, 0.5%, 0.1%, or less of
AR2, or 0.0%
of AR2. In a preferred embodiment, a low acidic species composition comprises
about 5%, 4%,
3%, 2.5%, 2.4%, 2.3%, 2.2%, 2.1%, 2%, 1.5%, 1.4%, 1.3%, 1.2%, 1.1%, 1%, 0.5%,
0.1%, or less
acidic species. In one embodiment, a low acidic species composition comprises
about 0.1% or
less AR1 and about 3% or less AR2. In another preferred embodiment, a low
acidic species
composition comprises about 1% or 0.1% or less AR1. In still another preferred
embodiment, a
low acidic species composition comprises about 3% or less AR2. In another
preferred
embodiment, the low AR composition comprises about 1.4% or less AR. For
example, in one
embodiment, the composition comprises about 1.4% AR2 and about 0.0% AR1.
The term "non-low acidic species composition," as used herein, refers to a
composition
comprising an antibody or antigen binding portion thereof, which contains more
than about 15%
acidic species. For example, a non-low acidic species composition may contain
about 15%, 16%,
17%, 18%, 19%, 20%, 21%, 22%, 23%, 24%, 25%, or more acidic species. In one
embodiment,
a non-low acidic species composition can comprise about 15%, 16%, 17%, 18%,
19%, 20%,
21%, 22%, 23%, 24%, 25%, or more of AR1. In another embodiment, a non-low
acidic species
composition can comprise about 15%, 16%, 17%, 18%, 19%, 20%, 21%, 22%, 23%,
24%, 25%,
or more of AR2.
In one embodiment, a low AR composition has improved biological and functional
properties, including increased efficacy in the treatment or prevention of a
disorder in a subject,
e.g., a disorder in which TNFa activity is detrimental, as compared to a non-
low acidic species
composition. In one embodiment, the low AR composition comprises an anti-TNFa
antibody, or
antigen-binding portion thereof, such as adalimumab or a fragment thereof. For
example, in one
embodiment, a low AR composition comprising an antibody, or antigen-binding
portion thereof,
exhibits increased cartilage penetration, decreased bone erosion, and/or
reduced cartilage
destruction, as compared to a non-low acidic species composition comprising
the same antibody
-26-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
or antigen binding portion thereof, when administered to a subject suffering
from a disorder in
which TNFa activity is detrimental.
In another embodiment, a low AR composition comprising an antibody, or antigen-
binding portion thereof, exhibits increased protection against the development
of arthritic scores
and/or histopathology scores as compared to a non-low acidic species
composition when
administered to an animal model of arthritis, e.g., the TNF-Tg197 model of
arthritis, as described
above.
In another embodiment, a low AR composition comprising an antibody, or antigen-
binding portion thereof, exhibits reduced synovial proliferation, reduced cell
infiltration, reduced
chondrocyte death, and/or reduced proteoglycan loss as compared to a non-low
acidic species
composition. In another embodiment, a low AR composition comprising an anti-
TNFa antibody,
or antigen-binding portion thereof, exhibits increased TNFa affinity as
compared to a non-low
acidic species composition.
In another embodiment, the impurity is a lysine variant species. As used
herein, the term
"lysine variant species" refers to an antibody, or antigen-binding portion
thereof, comprising
heavy chains with either zero, one or two C-terminal lysines. For example, the
"Lys 0" variant
comprises an antibody, or antigen-binding portion thereof, with heavy chains
that do not
comprise a C-terminal lysine. The "Lys 1" variant comprises an antibody, or
antigen-binding
portion thereof, with one heavy chain that comprises a C-terminal lysine. The
"Lys 2" variant
comprises an antibody with both heavy chains comprising a C-terminal lysine.
Lysine variants
can be detected, for example, by weak cation exchange chromatography (WCX) of
the
expression product of a host cell expressing the antibody, or antigen-binding
portion thereof.
With respect specifically to adalimumab, three main basic lysine variant
species have been
identified, i.e., Lys 0, Lys 1, and Lys 2.
The term "load buffer", as used herein refers to a salt solution passed
through the HIC
media upon contacting the sample with the HIC media. In certain embodiments,
the load buffer
is passed through the HIC media simultaneously or substantially simultaneously
with passage of
the sample through the HIC media. In certain embodiments, the load buffer is
combined with the
sample prior to passage through the HIC media.
The term "flow through fraction", as used herein refers to the liquid that
passes through
without binding the hydrophobic column upon contacting the sample with the HIC
media during
the load cycle. According to the methods of the present invention, the flow
through fraction
includes protein of interest that does not bind to the HIC media. The flow
through fraction may
-27-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
also include load buffer that passes through the HIC media during the load
cycle and/or a portion
of the impurity that does not bind to the HIC media.
The term "wash buffer", as used herein refers to a salt solution passed
through the HIC
media during the wash cycle.
The term "wash fraction", as used herein refers to the liquid eluted from the
column upon
washing the HIC media with the wash buffer. According to the methods of the
present invention,
the wash fraction includes protein of interest that is released from the HIC
media upon exposure
to the wash buffer. The wash fraction may also include wash buffer that passes
through the HIC
media during the wash cycle and/or a portion of the impurity that does not
bind to the HIC
media.
The term "isocratic", as used herein, refers to wash and load conditions which
are
identical or vary only slightly in terms of, for example, the nature of the
buffer, the salt
concentration, the pH, and the temperature. In particular embodiments, the
wash and load
conditions are substantially the same, for example, the salt concentration
and/or the pH of the
wash buffer are identical to or are adjusted to within about 20%, about 15%,
about 10%, or about
5% of the salt concentration, and/or pH of the loading buffer. In a particular
embodiment, the
wash and load conditions are identical.
The term "load challenge", as used herein refers to the total mass of sample
(e.g., protein
of interest and at least one protein) loaded onto the column in chromatography
applications or
applied to the resin in batch binding, measured in units of mass of product
per unit volume of
resin.
As used herein, the term "dynamic binding capacity" refers to the amount of
total protein
that binds to the HIC media upon breakthrough of 10% of the total protein
load.
As used herein, the term "apparent binding capacity" refers to the amount of
protein of
interest that binds to the HIC media upon breakthrough of 10% of the total
protein load, in
reversible HIC binding applications.
As used herein, the term "actual binding capacity" refers to the amount of
protein of
interest that remains bound to the chromatographic media under isocratic wash
conditions.
As used herein, the term "equilibrium binding capacity" refers to the maximum
amount of
total protein that can be bound under certain conditions.
As used herein, the term "partition coefficient" (Kp) refers to the
equilibrium ratio of the
concentration of protein of interest adsorbed to the HIC media to the
concentration of protein of
-28-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
interest in the solution comprising the unbound protein of interest, under
specified conditions of
pH and solution composition. The partition coefficient Kp corresponds to the
slope of the protein
of interest adsorption isotherm at very low solution concentrations. Kp can be
calculated from
the Qmax (maximum capacity of the HIC media for the protein of interest) and
Kd (dissociation
constant for the HIC media-protein of interest interaction) as follows: Kp =
Q/C = Qmax/Kd.
PROTEIN PURIFICATION
Protein Purification Generally
The present invention provides a method for producing a preparation including
a protein
of interest, e.g., an antibody, and having a reduced level of at least one
impurity, e.g., an
aggregate, by contacting a sample including the protein of interest and at
least one impurity, to a
hydrophobic interaction chromatography media.
In certain embodiments, the compositions of the present invention include, but
are not
limited to, a preparation comprising a protein of interest having a reduced
level of at least one
impurity. For example, but not by way of limitation, the present invention is
directed to
preparations of adalimumab having a reduced level of at least one impurity,
for example,
aggregate. Such preparations having a reduced level of at least one impurity
address the need for
improved product characteristics, including, but not limited to, product
stability, product safety
and product efficacy. In further embodiments, compositions of the present
invention include
pharmaceutical compositions comprising the preparation produced by the methods
of the
invention (e.g., protein of interest having a reduced level of the at least on
impurity) and a
pharmaceutically acceptable carrier.
In certain embodiments, the purification process of the invention begins at
the separation
step when the protein of interest has been produced using production methods
described above
and/or by alternative production methods conventional in the art. Once a
clarified solution or
sample comprising the protein of interest has been obtained, separation of the
protein of interest
from at least one impurity, such as process-related impurities, e.g., other
proteins produced by the
cell, as well as any product-related substances, e.g., charge variants and/or
size variants
(aggregates and fragments), can be performed using a HIC separation step, or a
combination of a
HIC separation step and one or more purification techniques, including
filtration and/or affinity,
ion exchange, and/or mixed mode chromatographic step(s), as outlined herein.
-29-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
Primary Recovery
In certain embodiments, the initial steps of the purification methods of the
present
invention involve the clarification and primary recovery of protein of
interest, for example,
antibody, following production. In certain embodiments, the primary recovery
will include one
or more centrifugation steps to separate the protein of interest from cells
and cell debris.
Centrifugation of the protein containing composition can be run at, for
example, but not by way
of limitation, 7,000 x g to approximately 12,750 x g. In the context of large
scale purification,
such centrifugation can occur on-line with a flow rate set to achieve, for
example, but not by way
of limitation, a turbidity level of 150 NTU in the resulting supernatant. Such
supernatant can
then be collected for further purification, or in-line filtered through one or
more depth filters for
further clarification of the sample.
In certain embodiments, the primary recovery will include the use of one or
more depth
filtration steps to clarify the sample and thereby aid in purifying the
protein of interest in the
present invention. In other embodiments, the primary recovery will include the
use of one or
more depth filtration steps post centrifugation to further clarify the sample.
Non-limiting
examples of depth filters that can be used in the context of the instant
invention include the
Millistak+ XOHC, FOHC, DOHC, A 1HC, B1HC depth filters (EMD Millipore), CunoTM
model
30/60ZA, 60/90 ZA, VR05, VR07, delipid depth filters (3M Corp.). A 0.2 i.tm
filter such as
Sartorius' s 0.45/0.2lim SartoporeTM bi-layer or Millipore' s Express SHR or
SHC filter cartridges
typically follows the depth filters.
In certain embodiments, the primary recovery process can also be a point at
which to
reduce or inactivate viruses that can be present in the sample. For example,
any one or more of a
variety of methods of viral reduction/inactivation can be used during the
primary recovery phase
of purification including heat inactivation (pasteurization), pH inactivation,
solvent/detergent
treatment, UV and 7-ray irradiation and the addition of certain chemical
inactivating agents such
as 13-propio1actone or e.g., copper phenanthroline as in U.S. Pat. No.
4,534,972. In certain
embodiments of the present invention, the sample is exposed to detergent viral
inactivation
during the primary recovery phase. In other embodiments, the sample may be
exposed to low pH
inactivation during the primary recovery phase.
In those embodiments where viral reduction/inactivation is employed, the
sample can be
adjusted, as needed, for further purification steps. For example, following
low pH viral
inactivation, the pH of the sample is typically adjusted to a more neutral pH,
e.g., from about 4.5
-30-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
to about 8.5, prior to continuing the purification process. Additionally, the
mixture may be
diluted with water for injection (WFI) to obtain a desired conductivity.
Hydrophobic Interaction Chromatography
The instant invention features methods for producing a preparation comprising
a protein
of interest (e.g., the anti-TNFa antibody adalimumab, or a fragment thereof)
having a reduced
level of at least one impurity, for example, aggregate, from a sample
comprising the protein of
interest and at least one impurity by contacting the sample with HIC media.
According to the present invention, HIC purification of a protein of interest
comprises
reversible binding of the protein of interest and binding of one or more
impurities through
hydrophobic interaction with hydrophobic moieties attached to a solid matrix
support (e.g.,
agarose). The hydrophobic interaction between molecules results from the
tendency of a polar
environment to exclude non-polar (i.e., hydrophobic) molecules. HIC relies on
this principle of
hydrophobicity of molecules (i.e., the tendency of a given protein to bind
adsorptively to
hydrophobic sites on a hydrophobic adsorbent body) to separate biomolecules
based on their
relative strength of interaction with the hydrophobic moieties (see, e.g.,
U.S. 4,000,098 and U.S.
3,917,527 which are herein incorporated by reference in their entirety). An
advantage of this
separation technique is its non-denaturing characteristics and the stabilizing
effects of salt
solutions used during loading, washing and or eluting.
Hydrophobic interaction chromatography employs the hydrophobic properties of
molecules (e.g., proteins, polypeptides, lipids) to achieve separation of even
closely-related
molecules. Hydrophobic groups on the molecules interact with hydrophobic
groups of the media
or the membrane. In certain embodiments, the more hydrophobic a molecule is,
the stronger it
will interact with the column or the membrane. Thus, HIC steps, such as those
disclosed herein,
can be used to remove a variety of impurities, for example, process-related
impurities (e.g.,
DNA) as well as product-related species (e.g., high and low molecular weight
product-related
species, such as protein aggregates and fragments).
In one aspect, the present invention provides a method for producing a
preparation
including a protein of interest, e.g., an antibody, and having a reduced level
of at least one
impurity, e.g., an aggregate, by (a) contacting a sample including the protein
of interest and at
least one impurity, to a hydrophobic interaction chromatography media, in the
presence of a load
buffer such that (i) a portion of the protein of interest binds to the
hydrophobic interaction
chromatography (HIC) media and (ii) a substantial portion of the at least one
impurity binds to
the HIC media; (b) collecting a flow through fraction including the protein of
interest unbound to
-31-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
the HIC media; (c) washing the HIC media with a wash buffer that is
substantially the same as
the load buffer such that a substantial portion of the protein of interest
bound to the HIC media is
released from the media; and (d) collecting a wash fraction including the
protein of interest
released from the HIC media, wherein each of the flow through and wash
fractions include the
protein of interest and have a reduced level of the at least one impurity. In
a particular
embodiment, the portion of the protein of interest binds to the HIC media at a
Kp of greater than
10. In a particular embodiment, the portion of the protein of interest binds
to the HIC media at a
Kp of greater than 20. In a particular embodiment, the portion of the protein
of interest binds to
the HIC media at a Kp of greater than 100.
In another aspect, the present invention provides a method for producing a
preparation
including a protein of interest, e.g., an antibody, and having a reduced level
of at least one
impurity, e.g., an aggregate, by (a) contacting a sample including the
protein of interest and
at least one impurity, to a HIC media, in the presence of a load buffer such
that (i) a portion of
the protein of interest binds to the HIC media and (ii) a substantial portion
of the at least one
impurity binds to the HIC media; collecting a flow through fraction including
the protein of
interest unbound to the HIC media; (c) washing the HIC media with a wash
buffer that is
substantially the same as the load buffer such that a substantial portion of
the protein of interest
bound to the HIC media is released from the media; and (d) collecting a wash
fraction including
the protein of interest released from the HIC media, wherein either (i) the
substantial portion of
the at least one impurity binds to the HIC media at a Kp greater than 200
and/or (ii) the protein of
interest and the at least on impurity have a Kp ratio less than 1:7; and
wherein each of the flow
through and wash fractions include the protein of interest and have a reduced
level of the at least
one impurity.
In yet another aspect, the present invention provides a method for producing a
preparation
including a protein of interest, e.g., an antibody, and having a reduced level
of at least one
impurity, e.g., an aggregate, by (a) contacting a sample including the protein
of interest and at
least one impurity, to a HIC media, in the presence of a load buffer such that
(i) a portion of the
protein of interest binds to the HIC media, and (ii) a substantial portion of
the at least one
impurity binds to the HIC media; (b) collecting a flow through fraction
including the protein of
interest unbound to the HIC media; (c) washing the HIC media with a wash
buffer that is
substantially the same as the load buffer such that a substantial portion of
the protein of interest
bound to the HIC media is released from the media; and (d) collecting a wash
fraction including
the protein of interest released from the HIC media, wherein the Kd for the
binding of the protein
of interest to the HIC media is less than 60, 50, 45, 40, 35, 30, 25, 20, 15,
10, 5 or 2 times the Kd
-32-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
for the binding of the at least one impurity to the HIC media, and wherein
each of the flow
through and wash fractions include the protein of interest and have a reduced
level of the at least
one impurity.
According to the present invention, a portion of the protein of interest
reversibly binds to
the HIC media while a portion of the protein of interest flows through to form
a flow through
fraction which has a reduced level of impurity. The portion of the protein of
interest that binds to
the HIC media binds reversibly in that the bound protein of interest may be
released therefrom
under isocratic conditions, for example, by use of a wash buffer that is
substantially the same as
the load buffer. In contrast, a substantial portion of the at least impurity
in the sample binds the
HIC media upon loading and a substantial portion thereof remains bound upon
washing the HIC
media with the wash buffer.
The present invention is based, at least in part, on the finding that such
reversible binding
can be achieved at relatively high binding strength. For example, contrary to
the teachings of
U.S. Patent No. 8,067,182 which teaches weak partitioning binding of a
product, i.e., at a Kp of
between 0.1 and 20 and less than 10 for HIC, according to the methods of the
present invention,
the protein of interest may bind at higher Kp levels so as to achieve higher
purification and
greater recovery of the protein of interest. For example, in a particular
embodiment, the protein
of interest binds to the HIC media at a Kp of greater than 10, 15, 20, 50, 60,
70, 80, 90, 100, 110,
120, 130, 140, 150, 160, 170, 180, 190, 200, 220, 250, 300, 400 or 500. In one
embodiment, the
protein of interest binds to the HIC media at a Kp greater than 10. In another
embodiment, the
protein of interest binds to the HIC media at a Kp greater than 20. In another
embodiment, the
protein of interest binds to the HIC media at a Kp of greater than 90.
According to the present invention, the impurity binds at a higher strength
and, thus to a
greater degree to the HIC media, thereby allowing for selective release of the
bound protein of
interest upon wash. For example, in particular embodiments, the at least one
impurity binds to
the HIC media at a Kp of greater than 200, greater than 250, greater than 300,
greater than 400,
greater than 500, greater than 600, greater than 700, greater than 800,
greater than 900, greater
than 1000, or greater than 2000. In a specific embodiment, the at least one
impurity binds to the
HIC media at a Kp of greater than 600.
In a further embodiment, the protein of interest and the at least one impurity
have a Kp
ratio less than 1:10, less than 1:9, less than 1:8, less than 1:7, less than
1:6, less than 1:5, less than
1:4, less than 1:3 or less than 1:2. In a specific embodiment, the protein of
interest and the at
least one impurity have a Kp ratio less than 1:7.
-33-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
The relative strength of binding may also be assessed by determining Kd, the
dissociation
constant for the media-protein of interest interaction, or the media-impurity
interaction. In one
embodiment of the invention, the Kd for the binding of the protein of interest
to the HIC media is
at least about 0.2, at least about 0.3, at least about 0.4, at least about
0.5, at least about 0.6, at
least about 0.7, or at least about 0.8. In a preferred embodiment, the Kd for
the binding of the
protein of interest to the HIC media is at least about 0.4.
In another embodiment of the invention, the Kd of the binding of the at least
one impurity
to the HIC media is less than or equal to about 0.001, about 0.005, about
0.01, about 0.02, about
0.05, about 0.1, about 0.15, about 0.2, about 0.3, about 0.4, about 0.5, or
about 1Ø In a
particular embodiment the Kd for the at least one impurity is less than or
equal to about 0.01.
In another embodiment of the invention, the Kd for the binding of the protein
of interest to
the HIC media is less than 60, 50, 45, 40, 35, 30, 25, 20, 15, 10, 5 or 2
times the Kd for the
binding of the at least one impurity to the HIC media.
The relative binding capacity of the protein of interest may also be assessed
by
determining Qmax, the maximum capacity of the media for the protein of
interest, or for the at
least one impurity. In one embodiment of the invention, the protein of
interest has a Qmax of at
least about 20, at least about 30, at least about 40, at least about 50, at
least about 60, at least
about 100, at least about 250, or at least about 500. In a preferred
embodiment, the protein of
interest has a Qmax of at least about 40.
In another embodiment, the at least one impurity has a Qmax of at least about
2, at least
about 5, at least about 10, at least about 20, at least about 30, at least
about 40, at least about 50,
at least about 75, or at least about 100. In a preferred embodiment, the at
least one impurity has a
Qmax of at least about 5.
In performing the HIC separation, the sample is contacted with the HIC media,
e.g., using
a batch purification technique or using a column or membrane chromatography or
monolithic
material (referred to as HIC media or resin). For example, in the context of
chromatographic
separation, a chromatographic apparatus, commonly cylindrical in shape, is
employed to contain
the chromatographic support media (e.g., HIC media) prepared in an appropriate
buffer solution.
Once the chromatographic material is added to the chromatographic apparatus, a
sample
containing the protein of interest, e.g., an antibody, and the protein of
interest is contacted to the
chromatographic material in the presence of a loading buffer to allow binding
of a portion of the
protein of interest and a substantial portion of the impurity to the HIC
media. A portion of the
protein of interest in the sample binds to the HIC media while a portion of
the protein interest
-34-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
flows through, forming a flow through fraction having a reduced level of
impurity which is
collected.
In one embodiment, the portion of the protein of interest that binds to the
HIC media is at
least about 20%, at least about 30%, at least about 40%, at least about 50%,
at least about 60%, at
least about 70%, at least about 80%, at least about 90%, at least about 95% or
at least about 98%
of the amount of the protein of interest in the sample.
Alternatively or in combination, the substantial portion of the at least one
impurity that
binds to the HIC media is at least about 60%, at least about 65%, at least
about 70%, at least
about 75%, at least about 80%, at least about 85%, at least about 90%, at
least about 95%, at least
about 98% or about 100% of the level of the at least one impurity in the
sample.
Alternatively or in combination, the portion of the protein of interest that
flows through
without binding to the HIC media is at least about 20%, at least about 30%, at
least about 40%, at
least about 50%, at least about 60%, at least about 70%, at least about 80%,
at least about 90%, at
least about 95% or at least about 98% of the amount of the protein of interest
in the sample.
The media is then subjected to a wash buffer, thereby allowing for a portion
of the bound
protein of interest to release from the HIC media in a wash fraction which is
collected, while a
substantial portion of the impurity remains bound to the HIC media. After
loading, the column
can be regenerated with water and cleaned with caustic solution to remove the
bound impurities
before next use.
In one embodiment, the substantial portion of the protein of interest released
from the
HIC media upon washing with the wash buffer is at least about at least about
50%, at least about
60%, at least about 70%, at least about 80%, at least about 90%, at least
about 95%, at least about
98%, or about 100% of the amount of protein of interest bound to the HIC
media.
Alternatively or in combination, the substantial portion of the impurity that
remains
bound to the HIC media is at least about 50%, at least about 60%, at least
about 65%, at least
about 70%, at least about 75%, at least about 80%, at least about 85%, at
least about 90%, at least
about 95%, at least about 98% or about 100% of the impurity bound to the HIC
media during the
load cycle.
In order to achieve the desired reversible binding of the protein of interest
and the
comparable strong binding of the at least one impurity, appropriate selection
of resin, buffer,
concentration, pH and sample load is required. Techniques to identify optimal
conditions for
achieving such desired binding profile are set forth in the Examples below.
-35-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
Hydrophobic interactions are strongest at high salt concentration (and hence
the ionic
strength of the anion and cation components). Adsorption of the protein of
interest to a HIC
column is favored by high salt concentrations, but the actual concentrations
can vary over a wide
range depending on the nature of the protein of interest, salt type and the
particular HIC ligand
chosen. In various embodiments, the salt concentration may be in the range of,
for example,
about 50 mM to about 5000 mM, about 100 mM to about 4000 mM, about 1000 mM to
about
4000 mM, about 50 mM to about 2000 mM, depending, in part, on the salt type
and HIC
adsorbent. In one embodiment the salt concentration is about 50 mM, about 55
mM, about 60
mM, about 65 mM, about 70 mM, about 75 mM, about 80 mM, about 85mM, about
90mM,
about 100mM, about 200mM, about 300 mM, about 400 mM, about 500 mM, about 600
mM,
about 700 mM, about 800 mM, about 900 mM, about 1000 mM, about 1200 mM, about
1400
mM, about 1600 mM, about 1800 mM or about 2000 mM.
Various ions can be arranged in a so-called soluphobic series depending on
whether they
promote hydrophobic interactions (salting-out effects) or disrupt the
structure of water
(chaotropic effect) and lead to the weakening of the hydrophobic interaction.
Cations are ranked
in terms of increasing salting out effect as Ba2+; Ca2+; Mg2+; Li + ; Cs + ;
Na + ; K+ ; Rb+ ; NH4,
while anions may be ranked in terms of increasing chaotropic effect as PO43-;
SO42-; CH3CO3- ;
C1- ; Br- ; NO3- ; C104- ; f; SCN-.
In certain embodiments, the anionic part of the salt is chosen from among
sulfate, citrate,
chloride, or a mixture thereof. In certain embodiments, the cationic part of
the salt is chosen
from among ammonium, sodium, potassium, or a mixture thereof. In general, Na,
K+ or NH4+
sulfates effectively promote ligand-protein interaction in HIC. Salts may be
formulated that
influence the strength of the interaction as given by the following
relationship: (NH4)2SO4 >
Na2SO4 > NaC1 > NH4C1 > NaBr > NaSCN. In general, salt concentrations of
between about
0.75 and about 2 M ammonium sulfate or between about 1 and 4 M NaC1 are
useful. In another
embodiment, the load buffer and the wash buffer comprise a salt of the
Hofmeister series or
lyotropic series of salts.
In one embodiment, the load buffer and the wash buffer comprise a sulfate
salt, a citrate
salt, or a combination thereof. In another embodiment, the sulfate salt in
ammonium sulfate. In
another embodiment, the sulfate salt is a sodium sulfate. In yet another
embodiment, the citrate
salt is sodium citrate. In certain embodiments, the load and/or wash buffer
may be comprised of
at least 2 salts.
-36-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
In certain embodiments, the HIC adsorbent material is composed of a
chromatographic
backbone with pendant hydrophobic interaction ligands. For example, but not by
way of
limitation, the HIC media can be composed of convective membrane media with
pendent
hydrophobic interaction ligands, convective monolithic media with pendent
hydrophobic
interaction ligands, and/or convective filter media with embedded media
containing the pendant
hydrophobic interaction ligands.
In certain embodiments, the HIC adsorbent material can comprise a base matrix
(e.g.,
derivatives of cellulose, polystyrene, synthetic poly amino acids, synthetic
polyacrylamide gels,
cross-linked dextran, cross-linked agarose, synthetic copolymer material or
even a glass surface)
to which hydrophobic ligands (e.g., alkyl, aryl and combinations thereof) are
coupled or
covalently attached using difunctional linking groups such as ¨NH-, -S-, -000-
, etc. The
hydrophobic ligand may be terminated in a hydrogen but can also terminate in a
functional group
such as, for example, NH2, SO3H, PO4H2, SH, imidazoles, phenolic groups or non-
ionic radicals
such as OH and CONH2. In one embodiment, the HIC media comprises at least one
hydrophobic
ligand. In another embodiment, the hydrophobic ligand is selected from the
group consisting of
butyl, hexyl, phenyl, octyl, or polypropylene glycol ligands.
One, non-limiting, example of a suitable HIC media comprises an agarose media
or a
membrane functionalized with phenyl groups (e.g., a Phenyl SepharoseTM from GE
Healthcare or
a Phenyl Membrane from Sartorius). Many HIC medias are available commercially.
Examples
include, but are not limited to, Tosoh Hexyl, CaptoPhenyl, Phenyl SepharoseTM
6 Fast Flow with
low or high substitution, Phenyl SepharoseTM High Performance, Octyl
SepharoseTM High
Performance (GE Healthcare); FractogelTM EMD Propyl or FractogelTM EMD Phenyl
(E. Merck,
Germany); Macro-PrepTM Methyl or Macro-PrepTM t-Butyl columns (Bio-Rad,
California); WP
HI-Propyl (C3)TM (J. T. Baker, New Jersey); ToyopearlTm ether, phenyl or butyl
(TosoHaas, PA);
ToyoScreen PPG, ToyoScreen Phenyl, ToyoScreen Butyl, and ToyoScreen Hexyl are
a rigid
methacrylic polymer bead. GE HiScreen Butyl FF and HiScreen Octyl FF are high
flow agarose
based beads.
In one embodiment, the HIC media has a dynamic binding capacity of at least
about 2 g,
at least about 5 g, at least about 10 g, at least about 20 g, at least about
30 g, at least about 40 g, at
least about 50 g, at least about 60 g, at least about 70 g, at least about 80
g, at least about 90 g, at
least about 100 g, or at least about 200 g of sample per one liter of media.
-37-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
Because the pH selected for any particular purification process must be
compatible with
protein stability and activity, particular pH conditions may be specific for
each application. A
high or low pH may serve to weaken hydrophobic interactions and retention of
proteins changes.
The pH of the HIC purification process is dependent, in part, on the pH of the
buffers
used to load, equilibrate and or wash the chromatographic resin or media.
Accordingly, in one embodiment, the pH of any of the buffers is between about
4.0 and
8.5. In a further embodiment, the pH of any of the buffers is between about
5.0 and 7Ø In one
embodiment, the pH of any of the buffers may be about 4.0, about 4.5, about
5.0, about 5.5,
about 6.0, about 6.5, about 7.0, about 7.5, about 8.0, or about 8.5. In a
preferred embodiment the
pH of any of the buffers is 5Ø In a related embodiment the pH of any of the
buffers is 5.6. In
yet another embodiment, the pH of any of the buffers is 7Ø
In certain embodiments, the load challenge of the sample comprising the
protein of
interest and at least one impurity is adjusted to a total protein load to the
column of between
about 50 and 1000 g/L, or between about 250 and 700 g/L, or between about 350
and 500 g/L of
HIC media. In certain embodiments, the protein concentration of the load
challenge is adjusted
to a total protein concentration of about 0.5 and 50 g/L, or between about 1
and 20 g/L, or
between about 3 and 10 g/L. In one embodiment the load challenge is about 50
g, about 100 g,
about 150 g, about 200 g, about 250 g, about 300 g, about 350 g, about 400 g,
about 450 g, about
500 g, about 550 g, about 600 g, about 650 g, about 700 g, about 750 g, about
800 g, about 850 g,
about 900 g, about 950 g, or about 1000 g of sample per one liter of HIC
media. In a particular
embodiment, the load challenge of the sample is 200 g/L. In another
embodiment, the load
challenge is 350 g/L. In yet another embodiment, load challenge of the sample
is 500 g/L. In yet
another embodiment, the load challenge of the sample is 700 g/L.
In another embodiment, the load challenge for the impurity alone is about 0.1
g, about 0.2
g, about 0.3 g, about 0.4 g, about 0.5 g, about 0.6 g, about 0.7 g, about 0.8
g, about 0.9 g, about
1.0 g, about 1.5 g, about 2.0 g, about 2.5 g, about 3.0 g, about 3.5 g, about
4.0 g, about 4.5 g, or
about 5.0 g of the at least one impurity per one liter of HIC media.
In certain embodiments, impurity (e.g., aggregate) concentration is measured
and used as
a parameter for controlling impurity clearance in the present invention. For
example, but not by
way of limitation, the data presented in the Examples below, demonstrates that
impurity
concentration influences the impurity reduction by hydrophobic interaction
chromatography.
Thus, in certain embodiments, the at least one impurity concentration is
adjusted from about 0.5
to 0.1 g/L, to about 0.1 to 0.05 g/L or to below 0.05 g/L. In another
embodiment, the at least one
-38-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
impurity contacting the HIC media has a concentration of about 0.01, about
0.02, about 0.03,
about 0.04, about 0.05, about 0.06, about 0.07, about 0.08, about 0.09, about
0.1, about 0.15,
about 0.2, about 0.3, about 0.4, about 0.5, about 0.6, about 0.7, about 0.8,
about 0.9, about 1.0,
about 2.0, about 3.0, about 4.0 or about 5.0 g/L.
In certain embodiments, protein of interest concentration (e.g., antibody
monomer) is
measured and used as a parameter for controlling impurity (e.g., aggregate)
clearance in the
present invention. For example, but not by way of limitation, the data
presented in the Examples
below demonstrates that control of the concentration of the protein of
interest can be used to
achieve improved impurity clearance. Thus, in certain embodiments, the protein
of interest
concentration is adjusted from about 15 to 8 g/L, to about 8 to 4 g/L or to
below 4 g/L. In
another embodiment, the protein of interest contacting the HIC media has a
concentration of less
than about 1, about 2, about 3, about 4, about 5, about 6, about 7, about 8,
about 9, about 10,
about 15, about 20, about 25, about 30, about 35, about 40 about 45, about 50,
or about 55 g/L.
In certain embodiments, protein of interest (e.g., antibody monomer) and
impurity (e.g.,
aggregate) concentration is measured and used as a parameter for controlling
impurity clearance
in the present invention. For example, but not by way of limitation, the data
presented in the
Examples demonstrates that control of the protein of interest and monomer
concentrations within
certain ranges can be used to achieve improved impurity clearance. Thus, in
certain
embodiments, the protein of interest concentration is adjusted from about 20
to 15 g/L, about 15
to 8 g/L or to below 4 g/L and the impurity concentration is adjusted to 0.5
to 0.1 g/L, about 0.1
to 0.05 g/L or to below 0.05 g/L to achieve impurity reduction in the present
invention. In
another embodiment, the protein of interest contacting the HIC media has a
concentration of
about 1, about 2, about 3, about 4, about 5, about 6, about 7, about 8, about
9, about 10, about 15,
about 20, about 25, about 30, about 35, about 40 about 45, or about 55 g/L and
the at least one
impurity contacting the HIC media has a concentration of less than about 0.01,
about 0.02, about
0.03, about 0.04, about 0.05, about 0.06, about 0.07, about 0.08, about 0.09,
about 0.1, about
0.15, about 0.2, about 0.3, about 0.4, about 0.5, about 0.6, about 0.7, about
0.8, about 0.9, or
about 1.0 g/L.
In another embodiment, the sample contacting the HIC media has a concentration
of
about 1, about 2, about 3, about 4, about 5, about 6, about 7, about 8, about
9, about 10, about 15,
about 20, about 25, about 30, about 35, about 40 about 45, or about 55 g/L.
In one embodiment, the at least one impurity is an aggregate of the protein of
interest, for
example, selected from the group consisting of a dimer, a trimer, a tetramer,
an oligomer and
-39-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
other high molecular weight species. In a particular embodiment, the protein
of interest is
adalimumab and the at least one impurity is an aggregate of adalimumab. For
example, the
aggregate may be selected from the group consisting of multimer 1, multimer 2
and multimer 3.
In another embodiment, the impurity is a process-related impurity or a product-
related
substance. For example, the impurity may be a process-related impurity
selected from the group
consisting of a host cell protein, a host cell nucleic acid, a media
component, and a
chromatographic material. Alternatively, the impurity may be a product-related
substance
selected from the group consisting of a charge variant, an aggregate of the
protein of interest, a
fragment of the protein of interest and a modified protein.
In a particular embodiment the impurity is an acidic or basic variant, for
example, of
adalimumab. In a particular embodiment, the basic variant is a lysine variant
species, for
example, an antibody, or antigen-binding portion thereof, having heavy chains
with either zero,
one or two C-terminal lysines. In another embodiment, the impurity is an
acidic species (AR),
for example, selected from the group consisting of a charge variant, a
structure variant, a
fragmentation variant, a process-related impurity and a product-related
impurity. In a particular
embodiment, the acidic species is AR1 and the charge variant is a deamidation
variant, a
glycation variant, an afucosylation variant, a MGO variant and/ or a citric
acid variant. In
another embodiment, the acidic species is AR1 and the structure variant is a
glycosylation variant
and/ or an acetonation variant. In yet another embodiment, the acidic species
is AR1 and the
fragmentation variant is a Fab fragment variant, a C-terminal truncation
variant or a variant
missing a heavy chain variable domain. In yet a further embodiment, the acidic
species is AR2
and the charge variant comprises a deamidation variant and/ or glycation
variant.
In a particular embodiment, the impurity is a fragment such as an Fc or a Fab
fragment.
In another embodiment, the impurity is a modified protein such as a deamidated
protein or
glycosylated protein.
In certain embodiments, HIC chromatographic fractions are collected during the
load
and/or wash cycles and are combined after appropriate analysis to provide a
protein preparation
that contains the reduced level of impurities. In certain embodiments, the
flow through fraction
is combined with certain wash fractions to improve the yield of the process
while still achieving
the desired, e.g., reduced level of impurities in the preparation.
-40-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
Additionally, the flow through or wash fractions, or combination thereof may
be
contacted with HIC media again to further purify the sample. In various
embodiments, the
method may be repeated at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15 or 20 times.
In certain embodiments, spectroscopy methods such as UV, NIR, FTIR,
Fluorescence,
Raman may be used to monitor levels of impurities such as aggregates and low
molecular weight
variants (e.g., fragments of the protein of interest) in an on-line, at-line
or in-line mode, which
can then be used to control the level of aggregates in the pooled material
collected from the HIC
methods of the present invention. In certain embodiments, on-line, at-line or
in-line monitoring
methods can be used either on the wash line of the chromatography step or in
the collection
vessel, to enable achievement of the desired product quality/recovery. In
certain embodiments,
the UV signal can be used as a surrogate to achieve an appropriate product
quality/recovery,
wherein the UV signal can be processed appropriately, including, but not
limited to, such
processing techniques as integration, differentiation, moving average, such
that normal process
variability can be addressed and the target product quality can be achieved.
In certain
embodiments, such measurements can be combined with in-line dilution methods
such that ion
concentration/conductivity of the load/wash can be controlled by feedback,
thereby facilitating
product quality control.
In one embodiment, the reduced level of the at least one impurity of the flow
through
fractions and/ or the wash fractions is at least about 60%, at least about
65%, at least about 70%,
at least about 75%, at least about 80%, at least about 85%, at least about
90%, at least about 95%,
at least about 98% or about 100% of the amount of the at least on impurity,
e.g., aggregate or
host cell protein in the sample.
In another embodiment, the impurity is a host cell protein and is reduced by
at least 0.25,
at least 0,5, at least 0.75, at least 1.0, at least 1.25, at least 1.5, at
least 1.75, at least 2.0, or at least
5.0 LFR.
In another embodiment, the accumulative aggregate reduction of the at least
one impurity
in any one flow through fraction and/ or wash fraction collected during the
preparation is at least
about 0.1%, at least about 0.2%, at least about 0,5%, at least about 1.0%, at
least about 2.0%, at
least about 3.0%, at least about 4.0%, at least about 5.0%, at least about
10%, at least about 20%,
or at least about 50%.
In another embodiment, the accumulative aggregate reduction of the at least
one impurity
in the flow through fraction and the wash fractions is at least about 0.1%, at
least about 0.2%, at
-41-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
least about 0.5%, at least about 1.0%, at least about 2.0%, at least about
3.0%, at least about
4.0%, at least about 5.0%, at least about 10.0%, or at least about 20.0%.
In another embodiment, the accumulative yield of the protein of interest in
the flow
through fraction and in the wash fraction is at least about 35%, at least
about 40%, at least about
45%, at least about 50%, at least about 55%, at least about 60%, at least
about 65%, at least about
70%, at least about 75%, at least about 80%, at least about 85%, at least
about 90%, at least about
95%, or about 100%.
In yet another embodiment, the accumulative yield of the protein of interest
in any one
flow through fraction or wash fraction is at least about 4%, at least about
10%, at least about
20%, at least about 30%, at least about 40%, at least about 50%, at least
about 55%, at least about
60%, at least about 65%, at least about 70%, at least about 75%, at least
about 80%, at least about
85, at least about 90%, at least about 95% or about 100%.
Complementary Purification Techniques
In certain embodiments, a combination of HIC and at least one of AEX (anion
exchange
chromatography) and CEX (cation exchange chromatography) and MM (mixed-mode
chromatography) methods can be used to prepare preparations of protein of
interest having a
reduced level of impurity, including certain embodiments where one technology
is used in a
complementary/ supplementary manner with another technology. In certain
embodiments, such a
combination can be performed such that certain sub-species are removed
predominantly by a
particularly technology, such that the combination provides the desired final
composition/product
quality. In certain embodiments, such combinations include the use of
additional intervening
chromatography, filtration, pH adjustment, UF/DF
(ultrafiltration/diafiltration) steps so as to
achieve the desired product quality, ion concentration, and/or viral
reduction.
Affinity Chromatography
In certain embodiments, a precursor sample is subjected to affinity
chromatography to
purify the protein of interest, prior to the methods of the present invention.
Alternatively or in
addition, the wash and/or flow through fractions generated by the methods of
the present
invention can be subjected to affinity chromatography to further purify the
protein of interest. As
noted above, certain embodiments of the present invention will employ one or
more affinity
chromatography steps prior to the HIC purification step, while others will
employ an affinity
chromatography step after or both before and after the HIC purification step.
In certain
embodiments, the affinity chromatography media is a Protein A, G, A/G, or L
media, although
alternative affinity chromatography medias are known in the art. There are a
variety of
-42-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
commercial sources for Protein A media. Suitable medias include, but are not
limited to,
MabSelect SuReTM, MabSelect SuRe LX, MabSelect, MabSelect Xtra, rProtein A
Sepharose
from GE Healthcare, ProSep HC, ProSep Ultra, and ProSep Ultra Plus from EMD
Millipore,
MapCapture from Life Technologies.
In certain embodiments, the Protein A column can be equilibrated with a
suitable buffer
prior to sample loading. Following the loading of the column, the column can
be washed one or
multiple times using a suitable sets of buffers. The Protein A column can then
be eluted using an
appropriate elution buffer. The eluate can be monitored using techniques well
known to those
skilled in the art. The eluate fractions of interest can be collected and then
prepared for further
processing.
The Protein A eluate may be subject to a viral inactivation step either by
detergent or low
pH, provided this step is not performed prior to the Protein A capture
operation. A proper
detergent concentration or pH and time can be selected to obtain desired viral
inactivation results.
After viral inactivation, the Protein A eluate is usually pH and/or
conductivity adjusted for
subsequent purification steps.
The Protein A eluate may be subjected to filtration through a depth filter to
remove
turbidity and/or various impurities from the antibody of interest prior to
additional
chromatographic polishing steps. Examples of depth filters include, but are
not limited to,
Millistak+ XOHC, FOHC, DOHC, A 1HC, and B1HC Pod filters (EMD Millipore), or
Zeta Plus
30ZA/60ZA, 60ZA/90ZA, delipid, VR07, and VRO5 filters (3M). The Protein A
eluate pool may
need to be conditioned to proper pH and conductivity to obtain desired
impurity removal and
product recovery from the depth filtration step.
Ion Exchange Chromatography
In certain embodiments, a precursor sample is subjected to ion exchange
chromatography
to purify the protein of interest, prior to the methods of the present
invention. Alternatively or in
addition, the wash and/or flow through fractions generated by the methods of
the present
invention can be subjected to ion exchange chromatography to further purify
the protein of
interest. As noted above, certain embodiments of the present invention will
employ one or more
ion exchange chromatography steps prior to the HIC purification step, while
others will employ
an ion exchange chromatography step after or both before and after the HIC
purification step.
As used herein, ion exchange separations includes any method by which two
substances
are separated based on the difference in their respective ionic charges,
either on the protein of
interest and/or chromatographic material as a whole or locally on specific
regions of the protein
-43-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
of interest and/or chromatographic material, and thus can employ either
cationic exchange
material or anionic exchange material.
The use of a cationic exchange material versus an anionic exchange material is
based on
the local charges of the protein of interest in a given solution. Therefore,
it is within the scope of
this invention to employ an anionic exchange step prior to the use of a HIC
step, or a cationic
exchange step prior to the use of an HIC step. Furthermore, it is within the
scope of this
invention to employ only a cationic exchange step, only an anionic exchange
step, or any serial
combination of the two either prior to or subsequent to the HIC step.
In performing the separation, the sample containing the protein of interest
(e.g., an
antibody or antigen-binding fragment thereof) can be contacted with the ion
exchange material
by using any of a variety of techniques, e.g., using a batch purification
technique or a
chromatographic technique, as described above in connection with HIC.
Ion exchange chromatography separates molecules based on differences between
the local
charges of the proteins of interest and the local charges of the
chromatographic material. A
packed ion-exchange chromatography column or an ion-exchange membrane device
can be
operated in a bind-elute mode, a flow-through, or a hybrid mode. After washing
the column or
the membrane device with the equilibration buffer or another buffer with
different pH and/or
conductivity, the product recovery is achieved by increasing the ionic
strength (i.e., conductivity)
of the elution buffer to compete with the solute for the charged sites of the
ion exchange matrix.
Changing the pH and thereby altering the charge of the solute is another way
to achieve elution
of the solute. The change in conductivity or pH may be gradual (gradient
elution) or stepwise
(step elution). The column is then regenerated before next use.
Anionic or cationic substituents may be attached to matrices in order to form
anionic or
cationic supports for chromatography. Non-limiting examples of anionic
exchange substituents
include diethylaminoethyl (DEAE), quaternary aminoethyl (QAE) and quaternary
amine (Q)
groups. Cationic substituents include carboxymethyl (CM), sulfoethyl (SE),
sulfopropyl (SP),
phosphate (P) and sulfonate (S). Cellulose ion exchange medias such as DE23TM,
DE32TM,
DE52TM, CM-23Tm, CM-32Tm, and CM-52Tm are available from Whatman Ltd.
Maidstone, Kent,
U.K. SEPHADEVD-based and -locross-linked ion exchangers are also known. For
example,
DEAE-, QAE-, CM-, and SP- SEPHADEX and DEAE-, Q-, CM-and S-SEPHAROSE and
SEPHAROSE Fast Flow, and CaptoTM S are all available from GE Healthcare.
Further, both
DEAE and CM derivitized ethylene glycol-methacrylate copolymer such as
TOYOPEARLTm
DEAE-6505 or M and TOYOPEARLTm CM-6505 or M are available from Toso Haas Co.,
-44-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
Philadelphia, PA, or Nuvia S and UNOSphereTM S from BioRad, Hercules, CA,
Eshmuno S
from EMD Millipore, Billerica, CA.
Mixed Mode Chromatography
In certain embodiments, a precursor sample is subjected to mixed mode
chromatography
to purify the protein of interest, prior to the HIC methods of the present
invention. Alternatively
or in addition, the wash and/or flow through fractions generated by the
methods of the present
invention can be subjected to mixed mode chromatography to further purify the
protein of
interest. As noted above, certain embodiments of the present invention will
employ one or more
mixed mode chromatography steps prior to the HIC purification step, while
others will employ a
mixed mode chromatography step after or both before and after the HIC
purification step.
Mixed mode chromatography is chromatography that utilizes a mixed mode media,
such
as, but not limited to CaptoAdhere available from GE Healthcare. Such a media
comprises a
mixed mode chromatography ligand. In certain embodiments, such a ligand refers
to a ligand
that is capable of providing at least two different, but co-operative, sites
which interact with the
substance to be bound. One of these sites gives an attractive type of charge-
charge interaction
between the ligand and the protein of interest. The other site typically gives
electron acceptor-
donor interaction and/or hydrophobic and/or hydrophilic interactions. Electron
donor-acceptor
interactions include interactions such as hydrogen-bonding, 7E-7E, cation- it,
charge transfer,
dipole-dipole, induced dipole etc. The mixed mode functionality can give a
different selectivity
compared to traditional anion exchangers. For example, CaptoAdhere is designed
for post-
Protein A purification of monoclonal antibodies, where removal of leached
Protein A,
aggregates, host cell proteins, nucleic acids and viruses from monoclonal
antibodies is performed
in flow-through mode (the antibodies pass directly through the column while
the contaminants
are adsorbed). Mixed mode chromatography ligands are also known as
"multimodal"
chromatography ligands.
In certain embodiments, the mixed mode chromatography media is comprised of
mixed
mode ligands coupled to an organic or inorganic support, sometimes denoted a
base matrix,
directly or via a spacer. The support may be in the form of particles, such as
essentially spherical
particles, a monolith, filter, membrane, surface, capillaries, etc. In certain
embodiments, the
support is prepared from a native polymer, such as cross-linked carbohydrate
material, such as
agarose, agar, cellulose, dextran, chitosan, konjac, carrageenan, gellan,
alginate etc. To obtain
high adsorption capacities, the support can be porous, and ligands are then
coupled to the
external surfaces as well as to the pore surfaces. Such native polymer
supports can be prepared
-45-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
according to standard methods, such as inverse suspension gelation (S Hjerten:
Biochim Biophys
Acta 79(2), 393-398 (1964). Alternatively, the support can be prepared from a
synthetic
polymer, such as cross-linked synthetic polymers, e.g. styrene or styrene
derivatives,
divinylbenzene, acrylamides, acrylate esters, methacrylate esters, vinyl
esters, vinyl amides etc.
Such synthetic polymers can be produced according to standard methods, see
e.g. "Styrene based
polymer supports developed by suspension polymerization" (R Arshady: Chimica e
L'Industria
70(9), 70-75 (1988)). Porous native or synthetic polymer supports are also
available from
commercial sources, such as Amersham Biosciences, Uppsala, Sweden.
Viral Filtration
In certain embodiments, a precursor sample is subjected to viral filtration to
purify the
protein of interest, prior to the HIC methods of the present invention.
Alternatively or in
addition, the wash and/or flow through fractions generated by the methods of
the present
invention can be subjected to viral filtration to further purify the protein
of interest. As noted
above, certain embodiments of the present invention will employ one or more
viral filtration
steps prior to the HIC purification step, while others will employ viral
filtration after or both
before and after the HIC purification step.
Viral filtration is a dedicated viral reduction step in the entire
purification process. This
step is usually performed as a post chromatographic polishing step. Viral
reduction can be
achieved via the use of suitable filters including, but not limited to,
Planova 2ONTM, 50 N or
BioEx from Asahi Kasei Pharma, ViresolveTM filters from EMD Millipore,
ViroSart CPV from
Sartorius, or Ultipor DV20 or DV5OTM filter from Pall Corporation. It will be
apparent to one of
ordinary skill in the art to select a suitable filter to obtain desired
filtration performance.
Ultrafiltration/Diafiltration
In certain embodiments, a precursor sample is subjected to ultrafiltration
and/or
diafiltration to purify the protein of interest, prior to the HIC methods of
the present invention.
Alternatively or in addition, the wash and/or flow through fractions generated
by the methods of
the present invention can be subjected to ultrafiltration and/or diafiltration
to further purify the
protein of interest. As noted above, certain embodiments of the present
invention will employ
one or more ultrafiltration and/or diafiltration steps prior to the HIC
purification step, while
others will employ ultrafiltration and/or diafiltration after or both before
and after the HIC
purification step.
Ultrafiltration is described in detail in: Microfiltration and
Ultrafiltration: Principles and
Applications, L. Zeman and A. Zydney (Marcel Dekker, Inc., New York, N.Y.,
1996); and in:
-46-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
Ultrafiltration Handbook, Munir Cheryan (Technomic Publishing, 1986; ISBN No.
87762-456-
9). A preferred filtration process is Tangential Flow Filtration as described
in the Millipore
catalogue entitled "Pharmaceutical Process Filtration Catalogue" pp. 177-202
(Bedford, Mass.,
1995/96). Ultrafiltration is generally considered to mean filtration using
filters with a pore size
of smaller than 0.1 pm. By employing filters having such small pore size, the
volume of the
sample can be reduced through permeation of the sample buffer through the
filter while
antibodies are retained behind the filter.
Diafiltration is a method of using ultrafilters to remove and exchange salts,
sugars, and
non-aqueous solvents, to separate free from bound species, to remove low
molecular-weight
material, and/or to cause the rapid change of ionic and/or pH environments.
Microsolutes are
removed most efficiently by adding solvent to the solution being ultrafiltered
at a rate
approximately equal to the ultratfiltration rate. This washes microspecies
from the solution at a
constant volume, effectively purifying the retained antibody. In certain
embodiments of the
present invention, a diafiltration step is employed to exchange the various
buffers used in
connection with the instant invention, optionally prior to further
chromatography or other
purification steps, as well as to remove impurities from the antibody
preparations.
Exemplary Purification Strategies
In certain embodiments, primary recovery can proceed by sequentially employing
pH
reduction, centrifugation, and filtration steps to remove cells and cell
debris (including HCPs)
from the production bioreactor harvest.
Additionally, the HIC methodology as described herein is utilized to further
purify the
protein of interest. As set forth herein, such methods involve (a) contacting
a sample including
the protein of interest and at least one impurity, to a hydrophobic
interaction chromatography
(HIC) media, in the presence of a load buffer such that (i) a portion of the
protein of interest
binds to the HIC media, for example, at a Kp of at least 10, 20 or 100 and
(ii) a substantial
portion of the at least one impurity binds to the HIC media; (b) collecting a
flow through fraction
including the protein of interest unbound to the HIC media; (c) washing the
HIC media with a
wash buffer that is substantially the same as the load buffer such that a
substantial portion of the
protein of interest bound to the HIC media is released from the media; and (d)
collecting a wash
fraction including the protein of interest released from the HIC media,
wherein each of the flow
through and wash fractions include the protein of interest and have a reduced
level of the at least
one impurity.
-47-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
Examples of buffers that can be used in the context of both the loading and
wash steps of
the present invention include, but are not limited to, the following: about
0.1 M to about 0.6 M
sodium citrate (NaCit), pH 5.6; or about 0.5 M to about 1.1 M ammonium sulfate
(AmSO4), pH
7.0 as well as buffers substantially the same, in that any differences result
in insubstantial
changes to the binding of impurities, but do not substantially affect the
ability to wash and
release antibody product. Such buffers can span a range of varying
"hydrophobicities" based on
the rationales discussed in above.
In certain embodiments, the HIC media employed in the HIC step is CaptoPhenyl
(GE)
resin. In certain embodiments, the CaptoPhenyl (GE) resin is buffer exchanged
into 0.4 M
sodium citrate (NaCit), pH 5.6, and then distributed in 100 L aliquots into
microcentrifuge
tubes. Each tube is then challenged with 2 mL of antibody produce source
material, e.g., a
partially purified cell culture harvest sample, in 0.4 M NaCit, pH 5.6, at a
range of concentrations
from 0.5-15.0 mg/mL and incubated for 3 hours at room temperature with mixing.
The resin is
allowed to settle and the supernatant removed and replaced with 1 mL of fresh
0.4 M NaCit, pH
5.6, buffer and incubated for 2 hours at room temperature with mixing. This
step was repeated
one more time.
In alternative embodiments, the CaptoPhenyl (GE) HIC resin can be packed in
1.0 cm x
10.0 cm (OmniFit) columns. Antibody product HIC-load can be prepared by
diluting the source
material, e.g., a partially purified cell culture harvest sample, with a 1.2 M
stock solution of
sodium citrate (NaCit), pH 5.6, to final concentration in the range of 0.3 to
0.5 M NaCit, pH 5.6.
CaptoPhenyl columns can then be equilibrated with 7 column volumes (CVs) of a
NaCit buffer,
pH 5.6, corresponding to the load concentration. The antibody product solution
can then be
loaded to the column in the range of 200-500 g/L, after which the column is
washed with 20 CVs
of the wash buffer. The column can then be regenerated (3 CVs f 25 mM sodium
phosphate/20
% (v/v) isopropyl alcohol, pH 6.5), cleaned in place (3 CVs 1M NaOH, 60 min
hold), and stored
(5 CVs of 25 mM sodium phosphate/20 % (v/v) isopropyl alcohol, pH 6.5). The
released
solution from the column can be fractionated during the entire run and used to
monitor the
breakthrough of both the protein of interest, e.g., antibody product monomer,
as well as
impurities, e.g., aggregates and host cell protein (HCP).
Such HIC purification steps can be preceded by affinity chromatography, for
example,
but not limited to, the use of Protein A-base affinity chromatography. There
are several
commercial sources for Protein A media. One suitable media is MabSelectTM from
GE
Healthcare. An example of a suitable column packed with MabSelectTM is a
column about 1.0
cm diameter x about 21.6 cm long (-17 mL bed volume). This size column can be
used for
-48-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
bench scale. This can be compared with other columns used for scale ups. For
example, a 20 cm
x 21 cm column whose bed volume is about 6.6 L can be used for commercial
production.
Regardless of the column, the column can be packed using a suitable media such
as
MabSelectTM.
In certain aspects, the Protein A column can be equilibrated with a suitable
buffer prior to
sample loading. An example of a suitable buffer is a Tris/NaC1 buffer, pH of
about 6 to 8, and in
certain embodiments about 7.2. A specific example of suitable conditions is 25
mM Tris, 100
mM NaC1, pH 7.2. Following this equilibration, the sample can be loaded onto
the column.
Following the loading of the column, the column can be washed one or multiple
times using, e.g.,
the equilibrating buffer. Other washes including washes employing different
buffers can be used
before eluting the column. For example, the column can be washed using one or
more column
volumes of 20 mM citric acid/sodium citrate, 0.5 M NaC1 at pH of about 6Ø
This wash can
optionally be followed by one or more washes using the equilibrating buffer.
The Protein A
column can then be eluted using an appropriate elution buffer. An example of a
suitable elution
buffer is an acetic acid/NaC1 buffer, pH around 3.5. Suitable conditions are,
e.g., 0.1 M acetic
acid, pH 3.5. The eluate can be monitored using techniques well known to those
skilled in the art.
For example, the absorbance at 0D280 can be followed. Column eluate can be
collected starting
with an initial deflection of about 0.5 AU to a reading of about 0.5 AU at the
trailing edge of the
elution peak. The elution fraction(s) of interest can then be prepared for
further processing. For
example, the collected sample can be titrated to a pH of about 5.0 using Tris
(e.g., 1.0 M) at a pH
of about 10. Optionally, this titrated sample can be filtered and further
processed.
In certain embodiments, the HIC purification step can also be preceded by an
ion
exchange chromatography step. The ion exchange purification step can occur
before, after, or in
place of an affinity chromatography step. In certain embodiments, where a
Protein A step
precedes the ion exchange step, a Protein A eluate is purified using a cation
exchange column. In
certain embodiments, the equilibrating buffer used in the cation exchange
column is a buffer
having a pH of about 5Ø An example of a suitable buffer is about 210 mM
sodium acetate, pH
5Ø Following equilibration, the column is loaded with sample prepared from
HIC purification
step above. The column is packed with a cation exchange media, such as CM
SepharoseTM Fast
Flow from GE Healthcare. The column is then washed using the equilibrating
buffer. The
column is next subjected to an elution step using a buffer having a greater
ionic strength as
compared to the equilibrating or wash buffer. For example, a suitable elution
buffer can be about
790 mM sodium acetate, pH 5Ø The antibodies will be eluted and can be
monitored using a UV
spectrophotometer set at OD280,.. In a particular example, elution collection
can be from upside
-49-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
3 0D280õõ to downside 8 OD280õõ. It should be understood that one skilled in
the art may vary
the conditions and yet still be within the scope of the invention.
In certain embodiments where a Protein A step precedes an ion exchange step, a
Protein
A eluate is purified using an anion exchange column. A non-limiting example of
a suitable
column for this step is a 60 cm diameter x 30 cm long column whose bed volume
is about 85 L.
The column is packed with an anion exchange media, such as Q SepharoseTM Fast
Flow from GE
Healthcare. The column can be equilibrated using about seven column volumes of
an
appropriate buffer such as Tris/sodium chloride. An example of suitable
conditions is 25 mM
Tris, 50 mM sodium chloride at pH 8Ø A skilled artisan may vary the
conditions but still be
within the scope of the present invention. The column is loaded with the
collected sample from
the HIC purification step outlined above. In another aspect, the column is
loaded from the eluate
collected during cation exchange. Following the loading of the column, the
column is washed
with the equilibration buffer (e.g., the Tris/sodium chloride buffer). The
flow-through
comprising the antibodies can be monitored using a UV spectrophotometer at
OD280,.. This
anion exchange step reduces process related impurities such as nucleic acids
like DNA, and host
cell proteins. The separation occurs due to the fact that the antibodies of
interest do not
substantially interact with nor bind to the solid phase of the column, e.g.,
to the Q SepharoseTM,
but many impurities do interact with and bind to the column' s solid phase.
The anion exchange
can be performed at about 12 C.
In certain embodiments, the cation exchange or anion exchange eluate,
depending on
which ion exchange step is employed, or employed first, is next filtered
using, e.g., a 16 inch
CunoTM delipid filter. This filtration, using the delipid filter, can be
followed by, e.g., a 30-inch
0.45/0.2 i.tm SartoporeTM bi-layer filter cartridge. The ion exchange elution
buffer can be used to
flush the residual volume remaining in the filters and prepared for
ultrafiltration/diafiltration.
In order to accomplish the ultratfiltration/diafiltration step, the filtration
media is prepared
in a suitable buffer, e.g., 20 mM sodium phosphate, pH 7Ø A salt such as
sodium chloride can
be added to increase the ionic strength, e.g., 100 mM sodium chloride.
This
ultrafiltration/diafiltration step serves to concentrate the anti-IL-12, anti-
TNFa, or anti-IL-18
antibodies, remove the sodium acetate and adjust the pH. Commercial filters
are available to
effectuate this step. For example, Millipore manufactures a 30 kD molecular
weight cut-off
(MWCO) cellulose ultrafilter membrane cassette. This filtration procedure can
be conducted at
or around room temperature.
-50-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
In certain embodiments, the sample from the capture filtration step above is
subjected to a
second ion exchange separation step. In certain embodiments, this second ion
exchange
separation will involve separation based on the opposite charge of the first
ion exchange
separation. For example, if an anion exchange step is employed after HIC
purification, the
second ion exchange chromatographic step may be a cation exchange step.
Conversely, if the
HIC purification step was followed by a cation exchange step, that step would
be followed by an
anion exchange step. In certain embodiments the first ion exchange eluate can
be subjected
directly to the second ion exchange chromatographic step where the first ion
exchange eluate is
adjusted to the appropriate buffer conditions. Suitable anionic and cationic
separation materials
and conditions are described above.
In certain embodiments, a mixed mode chromatography step will precede the HIC
chromatography step, thereby forming a mixed mode chromatography sample that
can be
exposed to the HIC media in the HIC chromatography step. Examples of mixed
mode medias
include, but are not limited to: CaptoAdhere (GE Healthcare), PPA-HyperCel
(Pall Life
Sciences), and HEA-HyperCel (Pall Life Sciences). In certain embodiments, the
mixed mode
chromatography step is a CaptoAdhere chromatography step. In certain
embodiments, the mixed
mode chromatography sample is further subject to a filtration step. Filters
well known to those
skilled in the art can be used in this embodiment. In one aspect, the
filtration step is a
nanofiltration step. In certain embodiments, a depth filtration step follows a
filtration step.
In certain embodiments of the invention, the wash and/or flow through
fractions from the
hydrophobic chromatography step are subjected to filtration for the removal of
viral particles,
including intact viruses, if present. A non-limiting example of a suitable
filter is the Ultipor
DV5OTM filter from Pall Corporation. Other viral filters can be used in this
filtration step and are
well known to those skilled in the art. The HIC eluate is passed through a pre-
wetted filter of
about 0.1 i.tm and a 2 x 30-inch Ultipor DVSOTM filter train at around 34
psig. In certain
embodiments, following the filtration process, the filter is washed using,
e.g., the HIC wash
buffer in order to remove any antibodies retained in the filter housing. The
filtrate can be stored
in a pre-sterilized container at around 12 C.
In a certain embodiments, the filtrate from the above is again subjected to
ultrafiltration/diafiltration. This step is important if a practitioner's end
point is to use the
antibody in a, e.g., pharmaceutical formulation. This process, if employed,
can facilitate the
concentration of antibody, removal of buffering salts previously used and
replace it with a
particular formulation buffer. In certain embodiments, continuous
diafiltration with multiple
volumes, e.g., two volumes, of a formulation buffer is performed. A non-
limiting example of a
-51-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
suitable formulation buffer is 5 mM methionine, 2% mannitol, 0.5% sucrose, pH
5.9 buffer (no
Tween). Upon completion of this diavolume exchange the antibodies are
concentrated. Once a
predetermined concentration of antibody has been achieved, then a practitioner
can calculate the
amount of 10% Tween that should be added to arrive at a final Tween
concentration of about
0.005% (v/v).
Certain embodiments of the present invention will include further purification
steps.
Examples of additional purification procedures which can be performed prior
to, during, or
following the ion exchange chromatography method include ethanol
precipitation, isoelectric
focusing, reverse phase HPLC, chromatography on silica, chromatography on
heparin
SepharoseTM, further anion exchange chromatography and/or further cation
exchange
chromatography, chromatofocusing, SDS-PAGE, ammonium sulfate precipitation,
hydroxylapatite chromatography, gel electrophoresis, dialysis, and affinity
chromatography (e.g.,
using protein G, an antibody, a specific substrate, ligand or antigen as the
capture reagent).
In certain embodiments the unbound flow through and wash fractions can be
further
fractionated and a combination of fractions providing a target protein of
interest purity can be
pooled.
In certain embodiments the protein concentration can be adjusted to achieve a
differential
partitioning behavior between the protein of interest and the impurities such
that the purity and/or
yield can be further improved.
In certain embodiments the loading can be performed at different protein
concentrations
during the loading operation to improve the product quality/yield of any
particular purification
step.
In certain embodiments the column temperature, can be independently varied to
improve
the separation efficiency and/or yield of any particular purification step.
In certain embodiments, the loading and washing buffers can be different or
composed of
mixtures of chemicals, while achieving similar "hydrophobic interaction"
behavior such that the
above novel separation can be effected.
In certain embodiments, the loading and washing buffers can be different, in
terms of
ionic strength or pH, while remaining substantially the same in function in
terms of the washout
of the protein of interest achieved during the wash step.
-52-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
In certain embodiments, the loading & washing steps can be controlled by in-
line, at-line
or off-line measurement of the impurity levels, either in the column effluent,
or the collected pool
or both, so as to achieve the protein of interest quality and/or yield.
In certain embodiments, the loading concentration can be dynamically
controlled by in-
line or batch or continuous dilutions with buffers or other solutions to
achieve the partitioning
necessary to improve the separation efficiency and/or yield.
In certain embodiments, additives such as amino acids, sugars, PEG, etc can be
added to
the load or wash steps to modulate the partitioning behavior to achieve the
separation efficiency
and/or yield.
In certain embodiments, the separation can be performed on any type of HIC
media such
as membranes, monoliths or depth filters that have hydrophobic interaction
characteristics.
Mixed mode media can also be employed to enable this method, provided the same
functionality is achieved by appropriately adjusting the column loading and/or
washing
conditions.
METHODS OF ASSAYING SAMPLE PURITY
Assaying Aggregates
In certain embodiments, the levels of product-related substances, such as
aggregates, in
either the initial sample or the flow through and/or wash fractions following
the HIC steps of the
present invention are analyzed. For example, but not by way of limitation, the
aggregates present
in the Adalimumab process samples can be quantified according to the following
methods.
Aggregates may be measured using a size exclusion chromatographic (SEC) method
whereby molecules are separated based on size and/or molecular weight such
that larger
molecules elute earlier from the column. For example, but not by way of
limitation, a SEC
columns useful for the detection of aggregates include: TSK-gel G3000SWxL, 5
i.tm, 125 A, 7.8
X 300 mm column (Tosoh Bioscience), TSK-gel Super 5W3000, 4 i.tm, 250 A, 4.6 X
300 mm
column (Tosoh Bioscience), or Zorbax GF450 column (Agilent Technologies). A
further
example of an SEC column for analysis of monomers and aggregates is the
MAbPacTM SEC-1
(Thermo Scientific) column which may be used under non-denaturing conditions,
in both high-
and low-salt mobile phases, and with volatile eluents.
In certain embodiments, the
aforementioned columns are used along with an Agilent or a Shimazhu HPLC
system. In a
-53-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
particular embodiment of SEC, aggregates may be quantified using a Zorbax
GF450 column on
an Agilent HPLC system.
In certain embodiments, sample injections are made under isocratic elution
conditions
using a mobile phase consisting of, for example, 100 mM sodium sulfate and 100
mM sodium
phosphate at pH 6.8, and detected with UV absorbance at 214 nm. In certain
embodiments, the
mobile phase will consist of 1X PBS at pH 7.4, and elution profile detected
with UV absorbance
at 280 nm.
The elution profile may be further analyzed using multiangle laser light-
scattering
(MALS), to determine the apparent molecular weight of each peak, and allow
identification as a
dimer, tetramer, or other high molecular weight species (Figure 1). The
elution profile may also
be further analyzed using sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-
PAGE). For example, the fraction is mixed with either a non-reducing or
reducing denaturing
sample buffer, treated for two minutes at 98 C in an Eppendorf Thermomixer
Confort, then
loaded in a 5% polyacrylamide tris-HCL gel alongside pre-stained broad range
molecular weight
markers. Electrophoresis is performed using a buffer comprising 0.3% (w/v)
Tris, 1.44% (w/v)
glycine and 0.1% SDS, pH 8.3. Separation is performed at a constant current of
100 V and at
maximally 50 mA for about 1 hour, followed by staining of the gel. In another
embodiment, the
aggregates may be analyzed and the molecular weight determined using high
performance-size
exclusion chromatography followed by native electrospray ionization time-of-
flight mass
spectrometry (ESI-TOF MS). Further methods for assaying levels of aggregates
are provided in
the Examples below.
Assaying Host Cell Protein
The present invention also provides methods for determining the residual
levels of host
cell protein (HCP) concentration in the initial sample or the flow through
and/or wash fractions
following the HIC steps of the present invention. As described above, HCPs are
desirably
excluded from the final preparation. Exemplary HCPs include proteins
originating from the
source of the protein of interest production. Failure to identify and
sufficiently remove HCPs
from the target protein of interest may lead to reduced efficacy and/or
adverse subject reactions
when administered in a therapeutic setting.
As used herein, the term "HCP ELISA" refers to an ELISA where the antibody
used in
the assay is specific to the HCPs produced from cells, e.g., CHO cells, used
to generate the
protein of interest. The antibody may be produced according to conventional
methods known to
those of skill in the art. For example, the antibody may be produced using
HCPs obtained by
-54-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
sham production and purification runs, i.e., the same cell line used to
produce the protein of
interest is used, but the cell line is not transfected with antibody DNA. In
an exemplary
embodiment, the antibody is produced using HPCs similar to those expressed in
the cell
expression system of choice, i.e., the cell expression system used to produce
the protein of
interest.
Generally, HCP ELISA comprises sandwiching a liquid sample comprising HCPs
between two layers of antibodies, i.e., a first antibody and a second
antibody. The sample is
incubated during which time the HCPs in the sample are captured by the first
antibody, for
example, but not limited to goat anti-CHO, affinity purified (Cygnus). A
labeled second
antibody, or blend of antibodies, specific to the HCPs produced from the cells
used to generate
the antibody, e.g., anti-CHO HCP Biotinylated, is added, and binds to the HCPs
within the
sample. In certain embodiments, the first and second antibodies are polyclonal
antibodies. In
certain embodiments, the first and second antibodies are blends of polyclonal
antibodies raised
against HCPs. The amount of HCP contained in the sample is determined using
the appropriate
test based on the label of the second antibody.
HCP ELISA may be used for determining the level of HCPs in preparation or
fraction,
such as a wash fraction or a flow-through obtained using the process described
above. The
present invention also provides a preparation comprising a protein of
interest, wherein the
composition has no detectable level of HCPs as determined by an HCP Enzyme
Linked
Immunosorbent Assay ("ELISA"). In one embodiment, the protein of interest is
adalimumab.
Assaying Charge and Size Variants
In certain embodiments, the levels of product-related substances, such as
acidic species
and other charge variants, in the chromatographic samples produced using the
techniques
described herein are analyzed. For example, but not by way of limitation, the
acidic species and
other charge variants present in the Adalimumab process samples can be
quantified according to
the following methods. Cation exchange chromatography was performed on a
Dionex ProPac
WCX-10, Analytical column 4 mm x 250 mm (Dionex, CA). An Agilent 1200 HPLC
system was
used as the HPLC. The mobile phases used were 10mM Sodium Phosphate dibasic pH
7.5
(Mobile phase A) and 10mM Sodium Phosphate dibasic, 500 mM Sodium Chloride pH
5.5
(Mobile phase B). A binary gradient (94% A, 6% B: 0-20 min; 84% A, 16% B: 20-
22 min; 0%
A, 100%B: 22-28 min; 94% A, 6% B: 28-34 min) was used with detection at 280
nm.
In certain embodiments, the levels of aggregates, monomer, and fragments in
the
chromatographic samples produced using the techniques described herein are
analyzed. In
-55-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
certain embodiments, the aggregates, monomer, and fragments are measured using
a size
exclusion chromatographic (SEC) method for each molecule. For example, but not
by way of
limitation, a TSK-gel G3000SWxL, 5 i.tm, 125 A, 7.8 X 300 mm column (Tosoh
Bioscience) can
be used in connection with certain embodiments, while a TSK-gel Super 5W3000,
4 i.tm, 250 A,
4.6 X 300 mm column (Tosoh Bioscience) can be used in alternative embodiments.
In certain
embodiments, the aforementioned columns are used along with an Agilent or a
Shimazhu HPLC
system. In certain embodiments, sample injections are made under isocratic
elution conditions
using a mobile phase consisting of, for example, 100 mM sodium sulfate and 100
mM sodium
phosphate at pH 6.8, and detected with UV absorbance at 214 nm. In certain
embodiments, the
mobile phase will consist of 1X PBS at pH 7.4, and elution profile detected
with UV absorbance
at 280 nm. In certain embodiments, quantification is based on the relative
area of detected peaks.
ANTIBODY GENERATION
Antibodies to be purified by the methods of the present invention can be
generated by a
variety of techniques, including immunization of an animal with the antigen of
interest followed
by conventional monoclonal antibody methodologies e.g., the standard somatic
cell hybridization
technique of Kohler and Milstein (1975) Nature 256: 495. Although somatic cell
hybridization
procedures are preferred, in principle, other techniques for producing
monoclonal antibody can
be employed e.g., viral or oncogenic transformation of B lymphocytes.
In certain embodiments, the animal system for preparing hybridomas is the
murine
system. Hybridoma production is a well-established procedure. Immunization
protocols and
techniques for isolation of immunized splenocytes for fusion are known in the
art. Fusion
partners (e.g., murine myeloma cells) and fusion procedures are also known.
An antibody can be, in certain embodiments, a human, a chimeric, or a
humanized
antibody. Humanized antibodies of the present disclosure can be prepared based
on the sequence
of a non-human monoclonal antibody prepared as described above. DNA encoding
the heavy
and light chain immunoglobulins can be obtained from the non-human hybridoma
of interest and
engineered to contain non-murine (e.g., human) immunoglobulin sequences using
standard
molecular biology techniques. For example, to create a chimeric antibody,
murine variable
regions can be linked to human constant regions using methods known in the art
(see e.g., U.S.
Patent No. 4,816,567 to Cabilly et al.). To create a humanized antibody,
murine CDR regions
can be inserted into a human framework using methods known in the art (see
e.g., U.S. Patent
No. 5,225,539 to Winter, and U.S. Patent Nos. 5,530,101; 5,585,089; 5,693,762
and 6,180,370 to
Queen et al.).
-56-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
Human monoclonal antibodies can be generated using transgenic or
transchromosomic
mice carrying parts of the human immune system rather than the mouse system.
These
transgenic and transchromosomic mice include mice referred to herein as the
HuMAb Mouse
(Medarex, Inc.), KM Mouse (Medarex, Inc.), and XenoMouse@ (Amgen).
Moreover, alternative transchromosomic animal systems expressing human
immunoglobulin genes are available in the art and can be used to raise
antibodies of the
disclosure. For example, mice carrying both a human heavy chain
transchromosome and a
human light chain tranchromosome, referred to as "TC mice" can be used; such
mice are
described in Tomizuka et al. (2000) Proc. Natl. Acad. Sci. USA 97:722-727.
Furthermore, cows
carrying human heavy and light chain transchromosomes have been described in
the art (e.g.,
Kuroiwa et al. (2002) Nature Biotechnology 20:889-894 and PCT application No.
WO
2002/092812) and can be used to raise the antibodies of this disclosure.
In certain embodiments, the antibodies of this disclosure are recombinant
human
antibodies, which can be isolated by screening of a recombinant combinatorial
antibody library,
e.g., a scFv phage display library, prepared using human VL and VH cDNAs
prepared from
mRNA derived from human lymphocytes. Methodologies for preparing and screening
such
libraries are known in the art. In addition to commercially available kits for
generating phage
display libraries (e.g., the Pharmacia Recombinant Phage Antibody System,
catalog no. 27-9400-
01; and the Stratagene SurfZAPrTh4 phage display kit, catalog no. 240612, the
entire teachings of
which are incorporated herein), examples of methods and reagents particularly
amenable for use
in generating and screening antibody display libraries can be found in, e.g.,
Ladner et al. U.S.
Patent No. 5,223,409; Kang et al. PCT Publication No. WO 92/18619; Dower et
al. PCT
Publication No. WO 91/17271; Winter et al. PCT Publication No. WO 92/20791;
Markland et al.
PCT Publication No. WO 92/15679; Breitling et al. PCT Publication No. WO
93/01288;
McCafferty et al. PCT Publication No. WO 92/01047; Garrard et al. PCT
Publication No. WO
92/09690; Fuchs et al. (1991) Bio/Technology 9:1370-1372; Hay et al. (1992)
Hum Antibod
Hybridomas 3:81-85; Huse et al. (1989) Science 246:1275-1281; McCafferty et
al., Nature
(1990) 348:552-554; Griffiths et al. (1993) EMBO J 12:725-734; Hawkins et al.
(1992) J Mol
Biol 226:889-896; Clackson et al. (1991) Nature 352:624-628; Gram et al.
(1992) PNAS
89:3576-3580; Garrard et al. (1991) Bio/Technology 9:1373-1377; Hoogenboom et
al. (1991)
Nuc Acid Res 19:4133-4137; and Barbas et al. (1991) PNAS 88:7978-7982; the
entire teachings
of which are incorporated herein.
Human monoclonal antibodies of this disclosure can also be prepared using SCID
mice
into which human immune cells have been reconstituted such that a human
antibody response
-57-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
can be generated upon immunization. Such mice are described in, for example,
U.S. Patent Nos.
5,476,996 and 5,698,767 to Wilson et al.
The antibodies or antigen-binding portions thereof can be altered wherein the
constant
region of the antibody is modified to reduce at least one constant region-
mediated biological
effector function relative to an unmodified antibody. To modify an antibody of
the invention
such that it exhibits reduced binding to the Fc receptor, the immunoglobulin
constant region
segment of the antibody can be mutated at particular regions necessary for Fc
receptor (FcR)
interactions (see, e.g., Canfield and Morrison (1991) J. Exp. Med. 173:1483-
1491; and Lund et
al. (1991) J. of Immunol. 147:2657-2662, the entire teachings of which are
incorporated herein).
Reduction in FcR binding ability of the antibody may also reduce other
effector functions which
rely on FcR interactions, such as opsonization and phagocytosis and antigen-
dependent cellular
cytotoxicity.
ANTIBODY PRODUCTION
To express an antibody of the invention, DNAs encoding partial or full-length
light and
heavy chains are inserted into one or more expression vector such that the
genes are operatively
linked to transcriptional and translational control sequences. (See, e.g.,
U.S. Pat. No. 6,914,128,
the entire teaching of which is incorporated herein by reference.) In this
context, the term
"operatively linked" is intended to mean that an antibody gene is ligated into
a vector such that
transcriptional and translational control sequences within the vector serve
their intended function
of regulating the transcription and translation of the antibody gene. The
expression vector and
expression control sequences are chosen to be compatible with the expression
host cell used. The
antibody light chain gene and the antibody heavy chain gene can be inserted
into a separate
vector or, more typically, both genes are inserted into the same expression
vector. The antibody
genes are inserted into an expression vector by standard methods (e.g.,
ligation of complementary
restriction sites on the antibody gene fragment and vector, or blunt end
ligation if no restriction
sites are present). Prior to insertion of the antibody or antibody-related
light or heavy chain
sequences, the expression vector may already carry antibody constant region
sequences.
Additionally or alternatively, the recombinant expression vector can encode a
signal peptide that
facilitates secretion of the antibody chain from a host cell. The antibody
chain gene can be
cloned into the vector such that the signal peptide is linked in-frame to the
amino terminus of the
antibody chain gene. The signal peptide can be an immunoglobulin signal
peptide or a
heterologous signal peptide (i.e., a signal peptide from a non-immunoglobulin
protein).
-58-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
In addition to the antibody chain genes, a recombinant expression vector of
the invention
can carry one or more regulatory sequence that controls the expression of the
antibody chain
genes in a host cell. The term "regulatory sequence" is intended to include
promoters, enhancers
and other expression control elements (e.g., polyadenylation signals) that
control the transcription
or translation of the antibody chain genes. Such regulatory sequences are
described, e.g., in
Goeddel; Gene Expression Technology: Methods in Enzymology 185, Academic
Press, San
Diego, CA (1990), the entire teaching of which is incorporated herein by
reference. It will be
appreciated by those skilled in the art that the design of the expression
vector, including the
selection of regulatory sequences may depend on such factors as the choice of
the host cell to be
transformed, the level of expression of protein desired, etc. Suitable
regulatory sequences for
mammalian host cell expression include viral elements that direct high levels
of protein
expression in mammalian cells, such as promoters and/or enhancers derived from
cytomegalovirus (CMV) (such as the CMV promoter/enhancer), Simian Virus 40
(5V40) (such
as the 5V40 promoter/enhancer), adenovirus, (e.g., the adenovirus major late
promoter
(AdMLP)) and polyoma. For further description of viral regulatory elements,
and sequences
thereof, see, e.g., U.S. Patent No. 5,168,062 by Stinski, U.S. Patent No.
4,510,245 by Bell et al.
and U.S. Patent No. 4,968,615 by Schaffner et al., the entire teachings of
which are incorporated
herein by reference.
In addition to the antibody chain genes and regulatory sequences, a
recombinant
expression vector of the invention may carry one or more additional sequences,
such as a
sequence that regulates replication of the vector in host cells (e.g., origins
of replication) and/or a
selectable marker gene. The selectable marker gene facilitates selection of
host cells into which
the vector has been introduced (see e.g., U.S. Patents Nos. 4,399,216,
4,634,665 and 5,179,017,
all by Axel et al., the entire teachings of which are incorporated herein by
reference). For
example, typically the selectable marker gene confers resistance to drugs,
such as G418,
hygromycin or methotrexate, on a host cell into which the vector has been
introduced. Suitable
selectable marker genes include the dihydrofolate reductase (DHFR) gene (for
use in dhfr- host
cells with methotrexate selection/amplification) and the neo gene (for G418
selection).
An antibody of the invention can be prepared by recombinant expression of
immunoglobulin light and heavy chain genes in a host cell. To express an
antibody
recombinantly, a host cell is transfected with one or more recombinant
expression vectors
carrying DNA fragments encoding the immunoglobulin light and heavy chains of
the antibody
such that the light and heavy chains are expressed in the host cell and
secreted into the medium in
which the host cells are cultured, from which medium the antibodies can be
recovered. Standard
-59-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
recombinant DNA methodologies are used to obtain antibody heavy and light
chain genes,
incorporate these genes into recombinant expression vectors and introduce the
vectors into host
cells, such as those described in Sambrook, Fritsch and Maniatis (eds),
Molecular Cloning; A
Laboratory Manual, Second Edition, Cold Spring Harbor, N.Y., (1989), Ausubel
et al. (eds.)
Current Protocols in Molecular Biology, Greene Publishing Associates, (1989)
and in U.S. Patent
Nos. 4,816,397 & 6,914,128, the entire teachings of which are incorporated
herein.
For expression of the light and heavy chains, the expression vector(s)
encoding the heavy
and light chains is (are) transfected into a host cell by standard techniques.
The various forms of
the term "transfection" are intended to encompass a wide variety of techniques
commonly used
for the introduction of exogenous DNA into a prokaryotic or eukaryotic host
cell, e.g.,
electroporation, calcium-phosphate precipitation, DEAE-dextran transfection
and the like.
Although it is theoretically possible to express the antibodies of the
invention in either
prokaryotic or eukaryotic host cells, expression of antibodies in eukaryotic
cells, such as
mammalian host cells, is suitable because such eukaryotic cells, and in
particular mammalian
cells, are more likely than prokaryotic cells to assemble and secrete a
properly folded and
immunologically active antibody. Prokaryotic expression of antibody genes has
been reported to
be ineffective for production of high yields of active antibody (Boss and Wood
(1985)
Immunology Today 6:12-13, the entire teaching of which is incorporated herein
by reference).
Suitable host cells for cloning or expressing the DNA in the vectors herein
are the
prokaryote, yeast, or higher eukaryote cells described above. Suitable
prokaryotes for this
purpose include eubacteria, such as Gram-negative or Gram-positive organisms,
e.g.,
Enterobacteriaceae such as Escherichia, e.g., E. coli, Enterobacter, Erwinia,
Klebsiella, Proteus,
Salmonella, e.g., Salmonella typhimurium, Serratia, e.g., Serratia marcescans,
and Shigella, as
well as Bacilli such as B. subtilis and B. licheniformis (e.g., B.
licheniformis 41P disclosed in
DD 266,710 published Apr. 12, 1989), Pseudomonas such as P. aeruginosa, and
Streptomyces.
One suitable E. coli cloning host is E. coli 294 (ATCC 31,446), although other
strains such as E.
coli B, E. coli X1776 (ATCC 31,537), and E. coli W3110 (ATCC 27,325) are
suitable. These
examples are illustrative rather than limiting.
In addition to prokaryotes, eukaryotic microbes such as filamentous fungi or
yeast are
suitable cloning or expression hosts for polypeptide encoding vectors.
Saccharomyces
cerevisiae, or common baker's yeast, is the most commonly used among lower
eukaryotic host
microorganisms. However, a number of other genera, species, and strains are
commonly
available and useful herein, such as Schizosaccharomyces pombe; Kluyveromyces
hosts such as,
e.g., K. lactis, K. fragilis (ATCC 12,424), K. bulgaricus (ATCC 16,045), K.
wickeramii (ATCC
-60-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
24,178), K. waltii (ATCC 56,500), K. drosophilarum (ATCC 36,906), K.
thermotolerans, and K.
marxianus; yarrowia (EP 402,226); Pichia pastoris (EP 183,070); Candida;
Trichoderma reesia
(EP 244,234); Neurospora crassa; Schwanniomyces such as Schwanniomyces
occidentalis; and
filamentous fungi such as, e.g., Neurospora, Penicillium, Tolypocladium, and
Aspergillus hosts
such as A. nidulans and A. niger.
Suitable host cells for the expression of glycosylated antibodies are derived
from
multicellular organisms. Examples of invertebrate cells include plant and
insect cells. Numerous
baculoviral strains and variants and corresponding permissive insect host
cells from hosts such as
Spodoptera frugiperda (caterpillar), Aedes aegypti (mosquito), Aedes
albopictus (mosquito),
Drosophila melanogaster (fruitfly), and Bombyx mori have been identified. A
variety of viral
strains for transfection are publicly available, e.g., the L-1 variant of
Autographa californica NPV
and the Bm-5 strain of Bombyx mori NPV, and such viruses may be used as the
virus herein
according to the present invention, particularly for transfection of
Spodoptera frugiperda cells.
Plant cell cultures of cotton, corn, potato, soybean, petunia, tomato, and
tobacco can also be
utilized as hosts.
Suitable mammalian host cells for expressing the recombinant antibodies of the
invention
include Chinese Hamster Ovary (CHO cells) (including dhfr- CHO cells,
described in Urlaub and
Chasin, (1980) PNAS USA 77:4216-4220, used with a DHFR selectable marker,
e.g., as
described in Kaufman and Sharp (1982) Mol. Biol. 159:601-621, the entire
teachings of which
are incorporated herein by reference), NSO myeloma cells, COS cells and 5P2
cells. When
recombinant expression vectors encoding antibody genes are introduced into
mammalian host
cells, the antibodies are produced by culturing the host cells for a period of
time sufficient to
allow for expression of the antibody in the host cells or secretion of the
antibody into the culture
medium in which the host cells are grown. Other examples of useful mammalian
host cell lines
are monkey kidney CV1 line transformed by 5V40 (COS-7, ATCC CRL 1651); human
embryonic kidney line (293 or 293 cells subcloned for growth in suspension
culture, Graham et
al., J. Gen Virol. 36:59 (1977)); baby hamster kidney cells (BHK, ATCC CCL
10); Chinese
hamster ovary cells/-DHFR (CHO, Urlaub et al., Proc. Natl. Acad. Sci. USA
77:4216 (1980));
mouse sertoli cells (TM4, Mather, Biol. Reprod. 23:243-251 (1980)); monkey
kidney cells (CV1
ATCC CCL 70); African green monkey kidney cells (VERO-76, ATCC CRL-1587);
human
cervical carcinoma cells (HELA, ATCC CCL 2); canine kidney cells (MDCK, ATCC
CCL 34);
buffalo rat liver cells (BRL 3A, ATCC CRL 1442); human lung cells (W138, ATCC
CCL 75);
human liver cells (Hep G2, HB 8065); mouse mammary tumor (MMT 060562, ATCC
CCL51);
TRI cells (Mather et al., Annals N.Y. Acad. Sci. 383:44-68 (1982)); MRC 5
cells; F54 cells; and
-61-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
a human hepatoma line (Hep G2), the entire teachings of which are incorporated
herein by
reference.
Host cells are transformed with the above-described expression or cloning
vectors for
antibody production and cultured in conventional nutrient media modified as
appropriate for
inducing promoters, selecting transformants, or amplifying the genes encoding
the desired
sequences.
The host cells used to produce an antibody may be cultured in a variety of
media.
Commercially available media such as Ham's F 1 OTm (Sigma), Minimal Essential
MediumTM
((MEM), (Sigma), RPMI-1640 (Sigma), and Dulbecco's Modified Eagle's MediumTM
((DMEM),
Sigma) are suitable for culturing the host cells. In addition, any of the
media described in Ham et
al., Meth. Enz. 58:44 (1979), Barnes et al., Anal. Biochem. 102:255 (1980),
U.S. Pat. Nos.
4,767,704; 4,657,866; 4,927,762; 4,560,655; or 5,122,469; WO 90/03430; WO
87/00195; or U.S.
Pat. No. Re. 30,985 may be used as culture media for the host cells, the
entire teachings of which
are incorporated herein by reference. Any of these media may be supplemented
as necessary
with hormones and/or other growth factors (such as insulin, transferrin, or
epidermal growth
factor), salts (such as sodium chloride, calcium, magnesium, and phosphate),
buffers (such as
HEPES), nucleotides (such as adenosine and thymidine), antibiotics (such as
gentamycin drug),
trace elements (defined as inorganic compounds usually present at final
concentrations in the
micromolar range), and glucose or an equivalent energy source. Any other
necessary
supplements may also be included at appropriate concentrations that would be
known to those
skilled in the art. The culture conditions, such as temperature, pH, and the
like, are those
previously used with the host cell selected for expression, and will be
apparent to the ordinarily
skilled artisan.
Host cells can also be used to produce portions of intact antibodies, such as
Fab fragments
or scFv molecules. It is understood that variations on the above procedure are
within the scope
of the present invention. For example, in certain embodiments it may be
desirable to transfect a
host cell with DNA encoding either the light chain or the heavy chain (but not
both) of an
antibody of this invention. Recombinant DNA technology may also be used to
remove some or
all of the DNA encoding either or both of the light and heavy chains that is
not necessary for
binding to the antigen to which the putative antibody of interest binds. The
molecules expressed
from such truncated DNA molecules are also encompassed by the antibodies of
the invention. In
addition, bifunctional antibodies may be produced in which one heavy and one
light chain are an
antibody of the invention and the other heavy and light chain are specific for
an antigen other
than the one to which the putative antibody of interest binds, depending on
the specificity of the
-62-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
antibody of the invention, by crosslinking an antibody of the invention to a
second antibody by
standard chemical crosslinking methods.
In a suitable system for recombinant expression of an antibody of the
invention, a
recombinant expression vector encoding both the antibody heavy chain and the
antibody light
chain is introduced into dhfr-CHO cells by calcium phosphate-mediated
transfection. Within the
recombinant expression vector, the antibody heavy and light chain genes are
each operatively
linked to CMV enhancer/AdMLP promoter regulatory elements to drive high levels
of
transcription of the genes. The recombinant expression vector also carries a
DHFR gene, which
allows for selection of CHO cells that have been transfected with the vector
using methotrexate
selection/amplification. The selected transformant host cells are cultured to
allow for expression
of the antibody heavy and light chains and intact antibody is recovered from
the culture medium.
Standard molecular biology techniques are used to prepare the recombinant
expression vector,
transfect the host cells, select for transformants, culture the host cells and
recover the antibody
from the culture medium.
When using recombinant techniques, the antibody can be produced
intracellularly, in the
periplasmic space, or directly secreted into the medium. In one aspect, if the
antibody is
produced intracellularly, as a first step, the particulate debris, either host
cells or lysed cells (e.g.,
resulting from homogenization), can be removed, e.g., by centrifugation or
ultrafiltration. Where
the antibody is secreted into the medium, supernatants from such expression
systems can be first
concentrated using a commercially available protein concentration filter,
e.g., an AmiconTM or
Millipore PelliconTM ultrafiltration unit.
Prior to the process of the invention, procedures for purification of
antibodies from cell
debris initially depend on the site of expression of the antibody. Some
antibodies can be secreted
directly from the cell into the surrounding growth media; others are made
intracellularly. For the
latter antibodies, the first step of a purification process typically
involves: lysis of the cell, which
can be done by a variety of methods, including mechanical shear, osmotic
shock, or enzymatic
treatments. Such disruption releases the entire contents of the cell into the
homogenate, and in
addition produces subcellular fragments that are difficult to remove due to
their small size.
These are generally removed by differential centrifugation or by filtration.
Where the antibody is
secreted, supernatants from such expression systems are generally first
concentrated using a
commercially available protein concentration filter, e.g., an AmiconTM or
Millipore PelliconTM
ultrafiltration unit. Where the antibody is secreted into the medium, the
recombinant host cells
can also be separated from the cell culture medium, e.g., by tangential flow
filtration. Antibodies
-63-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
can be further recovered from the culture medium using the antibody
purification methods of the
invention.
METHODS OF TREATMENT USING THE Low IMPURITY COMPOSITIONS OF THE INVENTION
The low impurity compositions, for example, low aggregate compositions, of the
invention may be used to treat any disorder in a subject for which the
therapeutic protein of
interest (e.g., an antibody or an antigen binding portion thereof) comprised
in the composition is
appropriate for treating.
A "disorder" is any condition that would benefit from treatment with the
protein of
interest. This includes chronic and acute disorders or diseases including
those pathological
conditions which predispose the subject to the disorder in question. In the
case of an anti-TNFa
antibody, or antigen binding portion thereof, such as adalimumab, a
therapeutically effective
amount of the low impurity composition may be administered to treat a disorder
in which TNFa
activity is detrimental.
A disorder in which TNFa activity is detrimental includes a disorder in which
inhibition
of TNFa activity is expected to alleviate the symptoms and/or progression of
the disorder. Such
disorders may be evidenced, for example, by an increase in the concentration
of TNFa in a
biological fluid of a subject suffering from the disorder (e.g., an increase
in the concentration of
TNFa in serum, plasma, synovial fluid, etc. of the subject), which can be
detected, for example,
using an anti-TNFa antibody.
TNFa has been implicated in the pathophysiology of a wide variety of a TNFa-
related
disorders including sepsis, infections, autoimmune diseases, transplant
rejection and graft-versus-
host disease (see e.g., Moeller, A., et al. (1990) Cytokine 2:162-169; U.S.
Patent No. 5,231,024
to Moeller et al.; European Patent Publication No. 260 610 B1 by Moeller, A.,
et al.Vasilli, P.
(1992) Annu. Rev. Immunol. 10:411-452; Tracey, K.J. and Cerami, A. (1994)
Annu. Rev. Med.
45:491-503). Accordingly, the low impurity compositions of the invention may
be used to treat
an autoimmune disease, such as rheumatoid arthritis, juvenile idiopathic
arthritis, or psoriatic
arthritis, an intestinal disorder, such as Crohn's disease or ulcerative
colitis, a
spondyloarthropathy, such as ankylosing spondylitis, or a skin disorder, such
as psoriasis.
Disorders in which TNFa activity is detrimental are well known in the art and
described
in detail in U.S. Patent No. 8,231,876 and U.S. Patent No. 6,090,382, the
entire contents of each
of which are expressly incorporated herein by reference. In one embodiment, "a
disorder in
which TNFa activity is detrimental" includes sepsis (including septic shock,
endotoxic shock,
-64-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
gram negative sepsis and toxic shock syndrome), autoimmune diseases (including
rheumatoid
arthritis, rheumatoid spondylitis, osteoarthritis and gouty arthritis,
allergy, multiple sclerosis,
autoimmune diabetes, autoimmune uveitis, nephrotic syndrome, multisystem
autoimmune
diseases, lupus (including systemic lupus, lupus nephritis and lupus
cerebritis), Crohn's disease
and autoimmune hearing loss), infectious diseases (including malaria,
meningitis, acquired
immune deficiency syndrome (AIDS), influenza and cachexia secondary to
infection), allograft
rejection and graft versus host disease, malignancy, pulmonary disorders
(including adult
respiratory distress syndrome (ARDS), shock lung, chronic pulmonary
inflammatory disease,
pulmonary sarcoidosis, pulmonary fibrosis, silicosis, idiopathic interstitial
lung disease and
chronic obstructive airway disorders (COPD), such as asthma), intestinal
disorders (including
inflammatory bowel disorders, idiopathic inflammatory bowel disease, Crohn's
disease and
Crohn's disease-related disorders (including fistulas in the bladder, vagina,
and skin; bowel
obstructions; abscesses; nutritional deficiencies; complications from
corticosteroid use;
inflammation of the joints; erythem nodosum; pyoderma gangrenosum; lesions of
the eye,
Crohn's related arthralgias, fistulizing Crohn's indeterminant colitis and
pouchitis), cardiac
disorders (including ischemia of the heart, heart insufficiency, restenosis,
congestive heart
failure, coronary artery disease, angina pectoris, myocardial infarction,
cardiovascular tissue
damage caused by cardiac arrest, cardiovascular tissue damage caused by
cardiac bypass,
cardiogenic shock, and hypertension, atherosclerosis, cardiomyopathy, coronary
artery spasm,
coronary artery disease, valvular disease, arrhythmias, and cardiomyopathies),
spondyloarthropathies (including ankylosing spondylitis, psoriatic
arthritis/spondylitis,
enteropathic arthritis, reactive arthritis or Reiter's syndrome, and
undifferentiated
spondyloarthropathies), metabolic disorders (including obesity and diabetes,
including type 1
diabetes mellitus, type 2 diabetes mellitus, diabetic neuropathy, peripheral
neuropathy, diabetic
retinopathy, diabetic ulcerations, retinopathy ulcerations and diabetic
macrovasculopathy),
anemia, pain (including acute and chronic pains, such as neuropathic pain and
post-operative
pain, chronic lower back pain, cluster headaches, herpes neuralgia, phantom
limb pain, central
pain, dental pain, opioid-resistant pain, visceral pain, surgical pain, bone
injury pain, pain during
labor and delivery, pain resulting from burns, including sunburn, post partum
pain, migraine,
angina pain, and genitourinary tract-related pain including cystitis), hepatic
disorders (including
hepatitis, alcoholic hepatitis, viral hepatitis, alcoholic cirrhosis, al
antitypsin deficiency,
autoimmune cirrhosis, cryptogenic cirrhosis, fulminant hepatitis, hepatitis B
and C, and
steatohepatitis, cystic fibrosis, primary biliary cirrhosis, sclerosing
cholangitis and biliary
obstruction), skin and nail disorders (including psoriasis (including chronic
plaque psoriasis,
guttate psoriasis, inverse psoriasis, pustular psoriasis and other psoriasis
disorders), pemphigus
-65-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
vulgaris, scleroderma, atopic dermatitis (eczema), sarcoidosis, erythema
nodosum, hidradenitis
suppurative, lichen planus, Sweet' s syndrome, scleroderma and vitiligo),
vasculitides (including
Behcet' s disease), and other disorders, such as juvenile rheumatoid arthritis
(JRA),
endometriosis, prostatitis, choroidal neovascularization, sciatica, Sjogren's
syndrome, uveitis,
wet macular degeneration, osteoporosis and osteoarthritis.
As used herein, the term "subject" is intended to include living organisms,
e.g.,
prokaryotes and eukaryotes. Examples of subjects include mammals, e.g.,
humans, dogs, cows,
horses, pigs, sheep, goats, cats, mice, rabbits, rats, and transgenic non-
human animals. In specific
embodiments of the invention, the subject is a human.
As used herein, the term "treatment" or "treat" refers to both therapeutic
treatment and
prophylactic or preventative measures. Those in need of treatment include
those already with the
disorder, as well as those in which the disorder is to be prevented.
In one embodiment, the invention provides a method of administering a low
impurity
composition comprising an anti-TNFa antibody, or antigen binding portion
thereof, to a subject
such that TNFa activity is inhibited or a disorder in which TNFa activity is
detrimental is treated.
In one embodiment, the TNFa is human TNFa and the subject is a human subject.
In one
embodiment, the anti-TNFa antibody is adalimumab, also referred to as HUMIRA .
The low impurity compositions can be administered by a variety of methods
known in the
art. Exemplary routes/modes of administration include subcutaneous injection,
intravenous
injection or infusion. In certain aspects, a low impurity compositions may be
orally administered.
As will be appreciated by the skilled artisan, the route and/or mode of
administration will vary
depending upon the desired results.
Dosage regimens may be adjusted to provide the optimum desired response (e.g.,
a
therapeutic or prophylactic response). For example, a single bolus may be
administered, several
divided doses may be administered over time or the dose may be proportionally
reduced or
increased as indicated by the exigencies of the therapeutic situation. In
certain embodiments it is
especially advantageous to formulate parenteral compositions in dosage unit
form for ease of
administration and uniformity of dosage. Dosage unit form as used herein
refers to physically
discrete units suited as unitary dosages for the mammalian subjects to be
treated; each unit
comprising a predetermined quantity of active compound calculated to produce
the desired
therapeutic effect in association with the required pharmaceutical carrier.
The specification for
the dosage unit forms of the invention are dictated by and directly dependent
on (a) the unique
characteristics of the active compound and the particular therapeutic or
prophylactic effect to be
-66-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
achieved, and (b) the limitations inherent in the art of compounding such an
active compound for
the treatment of sensitivity in individuals.
An exemplary, non-limiting range for a therapeutically or prophylactically
effective
amount of a low impurity composition of the invention is 0.01-20 mg/kg, or 1-
10 mg/kg, or 0.3-1
mg/kg. With respect to low impurity compositions comprising an anti-TNFa
antibody, or
antigen-binding portion thereof, such as adalimumab, an exemplary dose is 40
mg every other
week. In some embodiments, in particular for treatment of ulcerative colitis
or Crohn's disease,
an exemplary dose includes an initial dose (Day 1) of 160 mg (e.g., four 40 mg
injections in one
day or two 40 mg injections per day for two consecutive days), a second dose
two weeks later of
80 mg, and a maintenance dose of 40 mg every other week beginning two weeks
later.
Alternatively, for psoriasis for example, a dosage can include an 80 mg
initial dose followed by
40 mg every other week starting one week after the initial dose.
It is to be noted that dosage values may vary with the type and severity of
the condition to
be alleviated. It is to be further understood that for any particular subject,
specific dosage
regimens should be adjusted over time according to the individual need and the
professional
judgment of the person administering or supervising the administration of the
compositions, and
that dosage ranges set forth herein are exemplary only and are not intended to
limit the scope or
practice of the claimed composition.
PHARMACEUTICAL FORMULATIONS CONTAINING THE Low IMPURITY COMPOSITIONS OF THE
INVENTION
The present invention further provides preparations and formulations
comprising low
impurity compositions, for example, low aggregate compositions, of the
invention. It should be
understood that any of the proteins of interest, such as antibodies and
antibody fragments
described herein, including proteins of interest having any one or more of the
structural and
functional features described in detail throughout the application, may be
formulated or prepared
as described below. When various formulations are described in this section as
including a
protein of interest, such as an antibody, it is understood that such a protein
of interest may be a
protein having any one or more of the characteristics of the proteins of
interest described herein.
In one embodiment, the antibody is an anti-TNFa antibody, or antigen-binding
portion thereof.
In certain embodiments, the low impurity compositions, for example, low
aggregate
compositions, of the invention may be formulated with a pharmaceutically
acceptable carrier as
pharmaceutical (therapeutic) compositions, and may be administered by a
variety of methods
known in the art. As will be appreciated by the skilled artisan, the route
and/or mode of
-67-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
administration will vary depending upon the desired results. The term
"pharmaceutically
acceptable carrier" means one or more non-toxic materials that do not
interfere with the
effectiveness of the biological activity of the active ingredients. Such
preparations may routinely
contain salts, buffering agents, preservatives, compatible carriers, and
optionally other
therapeutic agents. Such pharmaceutically acceptable preparations may also
routinely contain
compatible solid or liquid fillers, diluents or encapsulating substances which
are suitable for
administration into a human. The term "carrier" denotes an organic or
inorganic ingredient,
natural or synthetic, with which the active ingredient is combined to
facilitate the application.
The components of the pharmaceutical compositions also are capable of being co-
mingled with
the proteins of interest (e.g., antibodies) of the present invention, and with
each other, in a
manner such that there is no interaction which would substantially impair the
desired
pharmaceutical efficacy.
The low impurity compositions, for example, low aggregate compositions, of the
invention are present in a form known in the art and acceptable for
therapeutic uses. In one
embodiment, a formulation of the low impurity compositions, for example, low
aggregate
compositions, of the invention is a liquid formulation. In another embodiment,
a formulation of
the low impurity compositions, for example, low aggregate compositions, of the
invention is a
lyophilized formulation. In a further embodiment, a formulation of the low
impurity
compositions, for example, low aggregate compositions, of the invention is a
reconstituted liquid
formulation. In one embodiment, a formulation of the low impurity
compositions, for example,
low aggregate compositions, of the invention is a stable liquid formulation.
In one embodiment,
a liquid formulation of the low impurity compositions, for example, low
aggregate compositions,
of the invention is an aqueous formulation. In another embodiment, the liquid
formulation is
non-aqueous. In a specific embodiment, a liquid formulation of the low
impurity compositions,
for example, low aggregate compositions, of the invention is an aqueous
formulation wherein the
aqueous carrier is distilled water.
The formulations of the low impurity compositions, for example, low aggregate
compositions, of the invention comprise a protein of interest (e.g., an
antibody) in a concentration
resulting in a w/v appropriate for a desired dose. The protein of interest may
be present in the
formulation at a concentration of about lmg/m1 to about 500mg/ml, e.g., at a
concentration of at
least 1 mg/ml, at least 5 mg/ml, at least 10 mg/ml, at least 15 mg/ml, at
least 20 mg/ml, at least
25 mg/ml, at least 30 mg/ml, at least 35 mg/ml, at least 40 mg/ml, at least 45
mg/ml, at least 50
mg/ml, at least 55 mg/ml, at least 60 mg/ml, at least 65 mg/ml, at least 70
mg/ml, at least 75
mg/ml, at least 80 mg/ml, at least 85 mg/ml, at least 90 mg/ml, at least 95
mg/ml, at least 100
-68-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
mg/ml, at least 105 mg/ml, at least 110 mg/ml, at least 115 mg/ml, at least
120 mg/ml, at least
125 mg/ml, at least 130 mg/ml, at least 135 mg/ml, at least 140 mg/ml, at
least 150 mg/ml, at
least 200 mg/ml, at least 250 mg/ml, or at least 300 mg/ml.
In a specific embodiment, a formulation of the low impurity compositions, for
example,
low aggregate compositions, of the invention comprises at least about 100
mg/ml, at least about
125 mg/ml, at least 130 mg/ml, or at least about 150 mg/ml of protein of
interest (e.g., an
antibody) of the invention.
In one embodiment, the concentration of protein of interest (e.g., antibody),
which is
included in the formulation of the invention, is between about 1 mg/ml and
about 25 mg/ml,
between about 1 mg/ml and about 200 mg/ml, between about 25 mg/ml and about
200 mg/ml,
between about 50 mg/ml and about 200 mg/ml, between about 75 mg/ml and about
200 mg/ml,
between about 100 mg/ml and about 200 mg/ml, between about 125 mg/ml and about
200
mg/ml, between about 150 mg/ml and about 200 mg/ml, between about 25 mg/ml and
about 150
mg/ml, between about 50 mg/ml and about 150 mg/ml, between about 75 mg/ml and
about 150
mg/ml, between about 100 mg/ml and about 150 mg/ml, between about 125 mg/ml
and about
150 mg/ml, between about 25 mg/ml and about 125 mg/ml, between about 50 mg/ml
and about
125 mg/ml, between about 75 mg/ml and about 125 mg/ml, between about 100 mg/ml
and about
125 mg/ml, between about 25 mg/ml and about 100 mg/ml, between about 50 mg/ml
and about
100 mg/ml, between about 75 mg/ml and about 100 mg/ml, between about 25 mg/ml
and about
75 mg/ml, between about 50 mg/ml and about 75 mg/ml, or between about 25 mg/ml
and about
50 mg/ml.
In a specific embodiment, a formulation of the low impurity compositions, for
example,
low aggregate compositions, of the invention comprises between about 90 mg/ml
and about 110
mg/ml or between about 100 mg/ml and about 210 mg/ml of a protein of interest
(e.g., an
antibody).
The formulations of the low impurity compositions, for example, low aggregate
compositions, of the invention comprising a protein of interest (e.g., an
antibody) may further
comprise one or more active compounds as necessary for the particular
indication being treated,
typically those with complementary activities that do not adversely affect
each other. Such
additional active compounds are suitably present in combination in amounts
that are effective for
the purpose intended.
The formulations of the low impurity compositions, for example, low aggregate
compositions, of the invention may be prepared for storage by mixing the
protein of interest (e.g.,
-69-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
antibody) having the desired degree of purity with optional physiologically
acceptable carriers,
excipients or stabilizers, including, but not limited to buffering agents,
saccharides, salts,
surfactants, solubilizers, polyols, diluents, binders, stabilizers, salts,
lipophilic solvents, amino
acids, chelators, preservatives, or the like (Goodman and Gilman's The
Pharmacological Basis of
Therapeutics, 12th edition, L. Brunton, et al. and Remington's Pharmaceutical
Sciences, 16th
edition, Osol, A. Ed. (1999)), in the form of lyophilized formulations or
aqueous solutions at a
desired final concentration. Acceptable carriers, excipients, or stabilizers
are nontoxic to
recipients at the dosages and concentrations employed, and include buffers
such as histidine,
phosphate, citrate, glycine, acetate and other organic acids; antioxidants
including ascorbic acid
and methionine; preservatives (such as octadecyldimethylbenzyl ammonium
chloride;
hexamethonium chloride; benzalkonium chloride, benzethonium chloride; phenol,
butyl or
benzyl alcohol; alkyl parabens such as methyl or propyl paraben; catechol;
resorcinol;
cyclohexanol; 3-pentanol; and m-cresol); low molecular weight (less than about
10 residues)
polypeptide; proteins, such as serum albumin, gelatin, or immunoglobulins;
hydrophilic polymers
such as polyvinylpyrolidone; amino acids such as glycine, glutamine,
asparagine, histidine,
arginine, or lysine; monosaccharides, disaccharides, and other carbohydrates
including trehalose,
glucose, mannose, or dextrins; chelating agents such as EDTA; sugars such as
sucrose, mannitol,
trehalose or sorbitol; salt-forming counter-ions such as sodium; metal
complexes (e.g.,
Zn-protein complexes); and/or non-ionic surfactants such as TWEEN, polysorbate
80,
PLURONICSTM or polyethylene glycol (PEG).
The buffering agent may be histidine, citrate, phosphate, glycine, or acetate.
The
saccharide excipient may be trehalose, sucrose, mannitol, maltose or
raffinose. The surfactant
may be polysorbate 20, polysorbate 40, polysorbate 80, or Pluronic F68. The
salt may be NaC1,
KC1, MgC12, or CaC12
The formulations of the low impurity compositions, for example, low aggregate
compositions, of the invention may include a buffering or pH adjusting agent
to provide
improved pH control. A formulation of the invention may have a pH of between
about 3.0 and
about 9.0, between about 4.0 and about 8.0, between about 5.0 and about 8.0,
between about 5.0
and about 7.0, between about 5.0 and about 6.5, between about 5.5 and about
8.0, between about
5.5 and about 7.0, or between about 5.5 and about 6.5. In a further
embodiment, a formulation of
the invention has a pH of about 3.0, about 3.5, about 4.0, about 4.5, about
5.0, about 5.1, about
5.2, about 5.3, about 5.4, about 5.5, about 5.6, about 5.7, about 5.8, about
5.9, about 6.0, about
6.1, about 6.2, about 6.3, about 6.4, about 6.5, about 6.6, about 6.7, about
6.8, about 6.9, about
7.0, about 7.5, about 8.0, about 8.5, or about 9Ø In a specific embodiment,
a formulation of the
-70-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
invention has a pH of about 6Ø One of skill in the art understands that the
pH of a formulation
generally should not be equal to the isoelectric point of the particular
protein of interest (e.g.,
antibody) to be used in the formulation.
Typically, the buffering agent is a salt prepared from an organic or inorganic
acid or base.
Representative buffering agents include, but are not limited to, organic acid
salts such as salts of
citric acid, ascorbic acid, gluconic acid, carbonic acid, tartaric acid,
succinic acid, acetic acid, or
phthalic acid; Tris, tromethamine hydrochloride, or phosphate buffers. In
addition, amino acid
components can also function in a buffering capacity. Representative amino
acid components
which may be utilized in the formulations of the invention as buffering agents
include, but are
not limited to, glycine and histidine. In certain embodiments, the buffering
agent is chosen from
histidine, citrate, phosphate, glycine, and acetate. In a specific embodiment,
the buffering agent
is histidine. In another specific embodiment, the buffering agent is citrate.
In yet another
specific embodiment, the buffering agent is glycine. The purity of the
buffering agent should be
at least 98%, or at least 99%, or at least 99.5%. As used herein, the term
"purity" in the context
of histidine and glycine refers to chemical purity of histidine or glycine as
understood in the art,
e.g., as described in The Merck Index, 13th ed., O'Neil et al. ed. (Merck &
Co., 2001).
Buffering agents are typically used at concentrations between about 1 mM and
about 200
mM or any range or value therein, depending on the desired ionic strength and
the buffering
capacity required. The usual concentrations of conventional buffering agents
employed in
parenteral formulations can be found in: Pharmaceutical Dosage Form:
Parenteral Medications,
Volume 1, 2nd Edition, Chapter 5, p. 194, De Luca and Boylan, "Formulation of
Small Volume
Parenterals", Table 5: Commonly used additives in Parenteral Products. In one
embodiment, the
buffering agent is at a concentration of about 1 mM, or of about 5 mM, or of
about 10 mM, or of
about 15 mM, or of about 20 mM, or of about 25 mM, or of about 30 mM, or of
about 35 mM, or
of about 40 mM, or of about 45 mM, or of about 50 mM, or of about 60 mM, or of
about 70 mM,
or of about 80 mM, or of about 90 mM, or of about 100 mM. In one embodiment,
the buffering
agent is at a concentration of 1 mM, or of 5 mM, or of 10 mM, or of 15 mM, or
of 20 mM, or of
25 mM, or of 30 mM, or of 35 mM, or of 40 mM, or of 45 mM, or of 50 mM, or of
60 mM, or of
70 mM, or of 80 mM, or of 90 mM, or of 100 mM. In a specific embodiment, the
buffering
agent is at a concentration of between about 5 mM and about 50 mM. In another
specific
embodiment, the buffering agent is at a concentration of between 5 mM and 20
mM.
In certain embodiments, the formulation of the low impurity compositions, for
example,
low aggregate compositions, of the invention comprises histidine as a
buffering agent. In one
embodiment the histidine is present in the formulation of the invention at a
concentration of at
-71-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
least about 1 mM, at least about 5 mM, at least about 10 mM, at least about 20
mM, at least about
30 mM, at least about 40 mM, at least about 50 mM, at least about 75 mM, at
least about 100
mM, at least about 150 mM, or at least about 200 mM histidine. In another
embodiment, a
formulation of the invention comprises between about 1 mM and about 200 mM,
between about
1 mM and about 150 mM, between about 1 mM and about 100 mM, between about 1 mM
and
about 75 mM, between about 10 mM and about 200 mM, between about 10 mM and
about 150
mM, between about 10 mM and about 100 mM, between about 10 mM and about 75 mM,
between about 10 mM and about 50 mM, between about 10 mM and about 40 mM,
between
about 10 mM and about 30 mM, between about 20 mM and about 75 mM, between
about 20 mM
and about 50 mM, between about 20 mM and about 40 mM, or between about 20 mM
and about
30 mM histidine. In a further embodiment, the formulation comprises about 1
mM, about 5 mM,
about 10 mM, about 20 mM, about 25 mM, about 30 mM, about 35 mM, about 40 mM,
about 45
mM, about 50 mM, about 60 mM, about 70 mM, about 80 mM, about 90 mM, about 100
mM,
about 150 mM, or about 200 mM histidine. In a specific embodiment, a
formulation may
comprise about 10 mM, about 25 mM, or no histidine.
The formulations of the low impurity compositions, for example, low aggregate
compositions, of the invention may comprise a carbohydrate excipient.
Carbohydrate excipients
can act, e.g., as viscosity enhancing agents, stabilizers, bulking agents,
solubilizing agents, and/or
the like. Carbohydrate excipients are generally present at between about 1% to
about 99% by
weight or volume, e.g., between about 0.1% to about 20%, between about 0.1% to
about 15%,
between about 0.1% to about 5%õ between about 1% to about 20%, between about
5% to about
15%, between about 8% to about 10%, between about 10% and about 15%, between
about 15%
and about 20%, between 0.1% to 20%, between 5% to 15%, between 8% to 10%,
between 10%
and 15%, between 15% and 20%, between about 0.1% to about 5%, between about 5%
to about
10%, or between about 15% to about 20%. In still other specific embodiments,
the carbohydrate
excipient is present at 1%, or at 1.5%, or at 2%, or at 2.5%, or at 3%, or at
4%, or at 5%, or at
10%, or at 15%, or at 20%.
Carbohydrate excipients suitable for use in the formulations of the invention
include, but
are not limited to, monosaccharides such as fructose, maltose, galactose,
glucose, D-mannose,
sorbose, and the like; disaccharides, such as lactose, sucrose, trehalose,
cellobiose, and the like;
polysaccharides, such as raffinose, melezitose, maltodextrins, dextrans,
starches, and the like;
and alditols, such as mannitol, xylitol, maltitol, lactitol, xylitol sorbitol
(glucitol) and the like. In
one embodiment, the carbohydrate excipients for use in the present invention
are chosen from,
sucrose, trehalose, lactose, mannitol, and raffinose. In a specific
embodiment, the carbohydrate
-72-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
excipient is trehalose. In another specific embodiment, the carbohydrate
excipient is mannitol.
In yet another specific embodiment, the carbohydrate excipient is sucrose. In
still another
specific embodiment, the carbohydrate excipient is raffinose. The purity of
the carbohydrate
excipient should be at least 98%, or at least 99%, or at least 99.5%.
In a specific embodiment, the formulations of the low impurity compositions,
for
example, low aggregate compositions, of the invention may comprise trehalose.
In one
embodiment, a formulation of the invention comprises at least about 1%, at
least about 2%, at
least about 4%, at least about 8%, at least about 20%, at least about 30%, or
at least about 40%
trehalose. In another embodiment, a formulation of the invention comprises
between about 1%
and about 40%, between about 1% and about 30%, between about 1% and about 20%,
between
about 2% and about 40%, between about 2% and about 30%, between about 2% and
about 20%,
between about 4% and about 40%, between about 4% and about 30%, or between
about 4% and
about 20% trehalose. In a further embodiment, a formulation of the invention
comprises about
1%, about 2%, about 4%, about 6%, about 8%, about 15%, about 20%, about 30%,
or about 40%
trehalose. In a specific embodiment, a formulation of the invention comprises
about 4%, about
6% or about 15% trehalose.
In certain embodiments, a formulation of the low impurity compositions, for
example,
low aggregate compositions, of the invention comprises an excipient. In a
specific embodiment,
a formulation of the invention comprises at least one excipient chosen from:
sugar, salt,
surfactant, amino acid, polyol, chelating agent, emulsifier and preservative.
In one embodiment,
a formulation of the invention comprises a salt, e.g., a salt selected from:
NaC1, KC1, CaC12, and
MgC12. In a specific embodiment, the formulation comprises NaCl.
A formulation of the low impurity compositions, for example, low aggregate
compositions, of the invention may comprise at least about 10 mM, at least
about 25 mM, at least
about 50 mM, at least about 75 mM, at least about 80 mM, at least about 100
mM, at least about
125 mM, at least about 150 mM, at least about 175 mM, at least about 200 mM,
or at least about
300 mM sodium chloride (NaC1). In a further embodiment, the formulation may
comprise
between about 10 mM and about 300 mM, between about 10 mM and about 200 mM,
between
about 10 mM and about 175 mM, between about 10 mM and about 150 mM, between
about 25
mM and about 300 mM, between about 25 mM and about 200 mM, between about 25 mM
and
about 175 mM, between about 25 mM and about 150 mM, between about 50 mM and
about 300
mM, between about 50 mM and about 200 mM, between about 50 mM and about 175
mM,
between about 50 mM and about 150 mM, between about 75 mM and about 300 mM,
between
about 75 mM and about 200 mM, between about 75 mM and about 175 mM, between
about 75
-73-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
mM and about 150 mM, between about 100 mM and about 300 mM, between about 100
mM and
about 200 mM, between about 100 mM and about 175 mM, or between about 100 mM
and about
150 mM sodium chloride. In a further embodiment, the formulation may comprise
about 10 mM,
about 25 mM, about 50 mM, about 75 mM, about 80 mM, about 100 mM, about 125
mM, about
150 mM, about 175 mM, about 200 mM, or about 300 mM sodium chloride.
A formulation of the low impurity compositions, for example, low aggregate
compositions, of the invention may also comprise an amino acid, e.g., lysine,
arginine, glycine,
histidine or an amino acid salt. The formulation may comprise at least about
1mM, at least about
10mM, at least about 25 mM, at least about 50 mM, at least about 100 mM, at
least about 150
mM, at least about 200 mM, at least about 250 mM, at least about 300 mM, at
least about 350
mM, or at least about 400 mM of an amino acid. In another embodiment, the
formulation may
comprise between about 1 mM and about 100 mM, between about 10 mM and about
150 mM,
between about 25 mM and about 250 mM, between about 25 mM and about 300 mM,
between
about 25 mM and about 350 mM, between about 25 mM and about 400 mM, between
about 50
mM and about 250 mM, between about 50 mM and about 300 mM, between about 50 mM
and
about 350 mM, between about 50 mM and about 400 mM, between about 100 mM and
about 250
mM, between about 100 mM and about 300 mM, between about 100 mM and about 400
mM,
between about 150 mM and about 250 mM, between about 150 mM and about 300 mM,
or
between about 150 mM and about 400 mM of an amino acid. In a further
embodiment, a
formulation of the invention comprises about 1 mM, 1.6 mM, 25 mM, about 50 mM,
about 100
mM, about 150 mM, about 200 mM, about 250 mM, about 300 mM, about 350 mM, or
about
400 mM of an amino acid.
The formulations of the low impurity compositions, for example, low aggregate
compositions, of the invention may further comprise a surfactant. The term
"surfactant" as used
herein refers to organic substances having amphipathic structures; namely,
they are composed of
groups of opposing solubility tendencies, typically an oil-soluble hydrocarbon
chain and a water-
soluble ionic group. Surfactants can be classified, depending on the charge of
the surface-active
moiety, into anionic, cationic, and nonionic surfactants. Surfactants are
often used as wetting,
emulsifying, solubilizing, and dispersing agents for various pharmaceutical
compositions and
preparations of biological materials. Pharmaceutically acceptable surfactants
like polysorbates
(e.g., polysorbates 20 or 80); polyoxamers (e.g., poloxamer 188); Triton;
sodium octyl glycoside;
lauryl-, myristyl-, linoleyl-, or stearyl-sulfobetaine; lauryl-, myristyl-,
linoleyl- or stearyl-
sarcosine; linoleyl-, myristyl-, or cetyl-betaine; lauroamidopropyl-,
cocamidopropyl-,
linoleamidopropyl-, myristamidopropyl-, palmidopropyl-, or isostearamidopropyl-
betaine (e.g.,
-74-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
lauroamidopropyl); myristamidopropyl-, palmidopropyl-, or isostearamidopropyl-
dimethylamine;
sodium methyl cocoyl-, or disodium methyl oleyl-taurate; and the MONAQUATM
series (Mona
Industries, Inc., Paterson, N.J.), polyethyl glycol, polypropyl glycol, and
copolymers of ethylene
and propylene glycol (e.g., PLURONICSTM, PF68, etc.), can optionally be added
to the
formulations of the invention to reduce aggregation. In one embodiment, a
formulation of the
invention comprises Polysorbate 20, Polysorbate 40, Polysorbate 60, or
Polysorbate 80.
Surfactants are particularly useful if a pump or plastic container is used to
administer the
formulation. The presence of a pharmaceutically acceptable surfactant
mitigates the propensity
for the protein to aggregate. The formulations may comprise a polysorbate
which is at a
concentration ranging from between about 0.001% to about 1%, or about 0.001%
to about 0.1%,
or about 0.01% to about 0.1%. In other specific embodiments, the formulations
of the invention
comprise a polysorbate which is at a concentration of 0.001%, or 0.002%, or
0.003%, or 0.004%,
or 0.005%, or 0.006%, or 0.007%, or 0.008%, or 0.009%, or 0.01%, or 0.015%, or
0.02%.
The formulations of the low impurity compositions, for example, low aggregate
compositions, of the invention may optionally further comprise other common
excipients and/or
additives including, but not limited to, diluents, binders, stabilizers,
lipophilic solvents,
preservatives, adjuvants, or the like. Pharmaceutically acceptable excipients
and/or additives
may be used in the formulations of the invention. Commonly used
excipients/additives, such as
pharmaceutically acceptable chelators (for example, but not limited to, EDTA,
DTPA or EGTA)
can optionally be added to the formulations of the invention to reduce
aggregation. These
additives are particularly useful if a pump or plastic container is used to
administer the
formulation.
Preservatives, such as phenol, m-cresol, p-cresol, o-cresol, chlorocresol,
benzyl alcohol,
phenylmercuric nitrite, phenoxyethanol, formaldehyde, chlorobutanol, magnesium
chloride (for
example, but not limited to, hexahydrate), alkylparaben (methyl, ethyl,
propyl, butyl and the
like), benzalkonium chloride, benzethonium chloride, sodium dehydroacetate and
thimerosal, or
mixtures thereof can optionally be added to the formulations of the invention
at any suitable
concentration such as between about 0.001% to about 5%, or any range or value
therein. The
concentration of preservative used in the formulations of the invention is a
concentration
sufficient to yield a microbial effect. Such concentrations are dependent on
the preservative
selected and are readily determined by the skilled artisan.
Other contemplated excipients/additives, which may be utilized in the
formulations of the
invention include, for example, flavoring agents, antimicrobial agents,
sweeteners, antioxidants,
antistatic agents, lipids such as phospholipids or fatty acids, steroids such
as cholesterol, protein
-75-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
excipients such as serum albumin (human serum albumin (HSA), recombinant human
albumin
(rHA), gelatin, casein, salt-forming counterions such as sodium and the like.
These and
additional known pharmaceutical excipients and/or additives suitable for use
in the formulations
of the invention are known in the art, e.g., as listed in "Remington: The
Science & Practice of
Pharmacy", 21st ed., Lippincott Williams & Wilkins, (2005), and in the
"Physician's Desk
Reference", 60th ed., Medical Economics, Montvale, N.J. (2005).
Pharmaceutically acceptable
carriers can be routinely selected that are suitable for the mode of
administration, solubility
and/or stability of protein of interest (e.g., an antibody), as well known
those in the art or as
described herein.
In one embodiment, the low impurity compositions, for example, low aggregate
compositions, of the invention are formulated with the same or similar
excipients and buffers as
are present in the commercial adalimumab (HUMIRA ) formulation, as described
in the
"Highlights of Prescribing Information" for HUMIRA (adalimumab) Injection
(Revised Jan.
2008) the contents of which are hereby incorporated herein by reference. For
example, each
prefilled syringe of HUMIRA , which is administered subcutaneously, delivers
0.8 mL (40 mg)
of drug product to the subject. Each 0.8 mL of HUMIRA contains 40 mg
adalimumab, 4.93 mg
sodium chloride, 0.69 mg monobasic sodium phosphate dihydrate, 1.22 mg dibasic
sodium
phosphate dihydrate, 0.24 mg sodium citrate, 1.04 mg citric acid monohydrate,
9.6 mg mannitol,
0.8 mg polysorbate 80, and water for Injection, USP. Sodium hydroxide is added
as necessary to
adjust pH.
It will be understood by one skilled in the art that the formulations of the
low impurity
compositions, for example, low aggregate compositions, of the invention may be
isotonic with
human blood, wherein the formulations of the invention have essentially the
same osmotic
pressure as human blood. Such isotonic formulations will generally have an
osmotic pressure
from about 250 mOSm to about 350 mOSm. Isotonicity can be measured by, for
example, using
a vapor pressure or ice-freezing type osmometer. Tonicity of a formulation is
adjusted by the use
of tonicity modifiers. "Tonicity modifiers" are those pharmaceutically
acceptable inert
substances that can be added to the formulation to provide an isotonicity of
the formulation.
Tonicity modifiers suitable for this invention include, but are not limited
to, saccharides, salts
and amino acids.
In certain embodiments, the formulations of the low impurity compositions, for
example,
low aggregate compositions, of the invention have an osmotic pressure from
about 100 mOSm to
about 1200 mOSm, or from about 200 mOSm to about 1000 mOSm, or from about 200
mOSm to
about 800 mOSm, or from about 200 mOSm to about 600 mOSm, or from about 250
mOSm to
-76-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
about 500 mOSm, or from about 250 mOSm to about 400 mOSm, or from about 250
mOSm to
about 350 mOSm.
The concentration of any one component or any combination of various
components, of
the formulations of the low impurity compositions, for example, low aggregate
compositions, of
the invention is adjusted to achieve the desired tonicity of the final
formulation. For example, the
ratio of the carbohydrate excipient to protein of interest (e.g., antibody)
may be adjusted
according to methods known in the art (e.g., U.S. Patent No. 6,685,940). In
certain
embodiments, the molar ratio of the carbohydrate excipient to protein of
interest (e.g., antibody)
may be from about 100 moles to about 1000 moles of carbohydrate excipient to
about 1 mole of
protein of interest, or from about 200 moles to about 6000 moles of
carbohydrate excipient to
about 1 mole of protein of interest, or from about 100 moles to about 510
moles of carbohydrate
excipient to about 1 mole of protein of interest, or from about 100 moles to
about 600 moles of
carbohydrate excipient to about 1 mole of protein of interest.
The desired isotonicity of the final formulation may also be achieved by
adjusting the salt
concentration of the formulations. Pharmaceutically acceptable salts and those
suitable for this
invention as tonicity modifiers include, but are not limited to, sodium
chloride, sodium succinate,
sodium sulfate, potassium chloride, magnesium chloride, magnesium sulfate, and
calcium
chloride. In specific embodiments, formulations of the invention comprise
NaC1, MgC12, and/or
CaC12. In one embodiment, concentration of NaC1 is between about 75 mM and
about 150 mM.
In another embodiment, concentration of MgC12 is between about 1 mM and about
100 mM.
Pharmaceutically acceptable amino acids including those suitable for this
invention as tonicity
modifiers include, but are not limited to, proline, alanine, L-arginine,
asparagine, L-aspartic acid,
glycine, serine, lysine, and histidine.
In one embodiment the formulations of the low impurity compositions, for
example, low
aggregate compositions, of the invention are pyrogen-free formulations which
are substantially
free of endotoxins and/or related pyrogenic substances. Endotoxins include
toxins that are
confined inside a microorganism and are released only when the microorganisms
are broken
down or die. Pyrogenic substances also include fever-inducing, thermostable
substances
(glycoproteins) from the outer membrane of bacteria and other microorganisms.
Both of these
substances can cause fever, hypotension and shock if administered to humans.
Due to the
potential harmful effects, even low amounts of endotoxins must be removed from
intravenously
administered pharmaceutical drug solutions. The Food & Drug Administration
("FDA") has set
an upper limit of 5 endotoxin units (EU) per dose per kilogram body weight in
a single one hour
period for intravenous drug applications (The United States Pharmacopeial
Convention,
-77-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
Pharmacopeial Forum 26 (1):223 (2000)). When therapeutic proteins are
administered in
amounts of several hundred or thousand milligrams per kilogram body weight, as
can be the case
with proteins of interest (e.g., antibodies), even trace amounts of harmful
and dangerous
endotoxin must be removed. In certain specific embodiments, the endotoxin and
pyrogen levels
in the composition are less then 10 EU/mg, or less then 5 EU/mg, or less then
1 EU/mg, or less
then 0.1 EU/mg, or less then 0.01 EU/mg, or less then 0.001 EU/mg.
When used for in vivo administration, the formulations of the low impurity
compositions,
for example, low aggregate compositions, of the invention should be sterile.
The formulations of
the invention may be sterilized by various sterilization methods, including
sterile filtration,
radiation, etc. In one embodiment, the protein of interest (e.g., antibody)
formulation is filter-
sterilized with a presterilized 0.22-micron filter. Sterile compositions for
injection can be
formulated according to conventional pharmaceutical practice as described in
"Remington: The
Science & Practice of Pharmacy", 21st ed., Lippincott Williams & Wilkins,
(2005). Formulations
comprising proteins of interest (e.g., antibodies), such as those disclosed
herein, ordinarily will
be stored in lyophilized form or in solution. It is contemplated that sterile
compositions
comprising proteins of interest (e.g., antibodies) are placed into a container
having a sterile
access port, for example, an intravenous solution bag or vial having an
adapter that allows
retrieval of the formulation, such as a stopper pierceable by a hypodermic
injection needle. In
one embodiment, a composition of the invention is provided as a pre-filled
syringe.
In one embodiment, a formulation of the low impurity compositions, for
example, low
aggregate compositions, of the invention is a lyophilized formulation. The
term "lyophilized" or
"freeze-dried" includes a state of a substance that has been subjected to a
drying procedure such
as lyophilization, where at least 50% of moisture has been removed.
The phrase "bulking agent" includes a compound that is pharmaceutically
acceptable and
that adds bulk to a lyo cake. Bulking agents known to the art include, for
example,
carbohydrates, including simple sugars such as dextrose, ribose, fructose and
the like, alcohol
sugars such as mannitol, inositol and sorbitol, disaccharides including
trehalose, sucrose and
lactose, naturally occurring polymers such as starch, dextrans, chitosan,
hyaluronate, proteins
(e.g., gelatin and serum albumin), glycogen, and synthetic monomers and
polymers.
A "lyoprotectant" is a molecule which, when combined with a protein of
interest (such as
an antibody of the invention), significantly prevents or reduces chemical
and/or physical
instability of the protein upon lyophilization and subsequent storage.
Lyoprotectants include, but
are not limited to, sugars and their corresponding sugar alcohols; an amino
acid such as
-78-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
monosodium glutamate or histidine; a methylamine such as betaine; a lyotropic
salt such as
magnesium sulfate; a polyol such as trihydric or higher molecular weight sugar
alcohols, e.g.,
glycerin, dextran, erythritol, glycerol, arabitol, xylitol, sorbitol, and
mannitol; propylene glycol;
polyethylene glycol; PLURONICSTM; and combinations thereof. Additional
examples of
lyoprotectants include, but are not limited to, glycerin and gelatin, and the
sugars mellibiose,
melezitose, raffinose, mannotriose and stachyose. Examples of reducing sugars
include, but are
not limited to, glucose, maltose, lactose, maltulose, iso-maltulose and
lactulose. Examples of
non-reducing sugars include, but are not limited to, non-reducing glycosides
of polyhydroxy
compounds selected from sugar alcohols and other straight chain polyalcohols.
Examples of
sugar alcohols include, but are not limited to, monoglycosides, compounds
obtained by reduction
of disaccharides such as lactose, maltose, lactulose and maltulose. The
glycosidic side group can
be either glucosidic or galactosidic. Additional examples of sugar alcohols
include, but are not
limited to, glucitol, maltitol, lactitol and iso-maltulose. In specific
embodiments, trehalose or
sucrose is used as a lyoprotectant.
The lyoprotectant is added to the pre-lyophilized formulation in a
"lyoprotecting amount"
which means that, following lyophilization of the protein in the presence of
the lyoprotecting
amount of the lyoprotectant, the protein essentially retains its physical and
chemical stability and
integrity upon lyophilization and storage.
In one embodiment, the molar ratio of a lyoprotectant (e.g., trehalose) and
protein of
interest (e.g., antibody) molecules of a formulation of the invention is at
least about 10, at least
about 50, at least about 100, at least about 200, or at least about 300. In
another embodiment, the
molar ratio of a lyoprotectant (e.g., trehalose) and protein of interest
molecules of a formulation
of the invention is about 1, is about 2, is about 5, is about 10, about 50,
about 100, about 200, or
about 300.
A "reconstituted" formulation is one which has been prepared by dissolving a
lyophilized
protein of interest (e.g., antibody) formulation in a diluent such that the
protein of interest is
dispersed in the reconstituted formulation. The reconstituted formulation is
suitable for
administration (e.g., parenteral administration) to a patient to be treated
with the protein of
interest and, in certain embodiments of the invention, may be one which is
suitable for
intravenous administration.
The "diluent" of interest herein is one which is pharmaceutically acceptable
(safe and
non-toxic for administration to a human) and is useful for the preparation of
a liquid formulation,
such as a formulation reconstituted after lyophilization. In some embodiments,
diluents include,
-79-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
but are not limited to, sterile water, bacteriostatic water for injection
(BWFI), a pH buffered
solution (e.g., phosphate-buffered saline), sterile saline solution, Ringer's
solution or dextrose
solution. In an alternative embodiment, diluents can include aqueous solutions
of salts and/or
buffers.
In certain embodiments, a formulation of the low impurity compositions, for
example,
low aggregate compositions, of the invention is a lyophilized formulation
comprising a protein of
interest (e.g., antibody) of the invention, wherein at least about 90%, at
least about 95%, at least
about 97%, at least about 98%, or at least about 99% of said protein of
interest may be recovered
from a vial upon shaking said vial for 4 hours at a speed of 400 shakes per
minute wherein the
vial is filled to half of its volume with the formulation. In another
embodiment, a formulation of
the invention is a lyophilized formulation comprising a protein of interest of
the invention,
wherein at least about 90%, at least about 95%, at least about 97%, at least
about 98%, or at least
about 99% of the protein of interest may be recovered from a vial upon
subjecting the
formulation to three freeze/thaw cycles wherein the vial is filled to half of
its volume with said
formulation. In a further embodiment, a formulation of the invention is a
lyophilized formulation
comprising a protein of interest of the invention, wherein at least about 90%,
at least about 95%,
at least about 97%, at least about 98%, or at least about 99% of the protein
of interest may be
recovered by reconstituting a lyophilized cake generated from said
formulation.
In one embodiment, a reconstituted liquid formulation may comprise a protein
of interest
(e.g., antibody) at the same concentration as the pre-lyophilized liquid
formulation.
In another embodiment, a reconstituted liquid formulation may comprise a
protein of
interest (e.g., antibody) at a higher concentration than the pre-lyophilized
liquid formulation, e.g.,
.about 2 fold, about 3 fold, about 4 fold, about 5 fold, about 6 fold, about 7
fold, about 8 fold,
about 9 fold, or about 10 fold higher concentration of a protein of interest
than the pre-
lyophilized liquid formulation.
In yet another embodiment, a reconstituted liquid formulation may comprise a
protein of
interest (e.g., antibody) of the invention at a lower concentration than the
pre-lyophilized liquid
formulation, e.g., about 2 fold, about 3 fold, about 4 fold, about 5 fold,
about 6 fold, about 7 fold,
about 8 fold, about 9 fold or about 10 fold lower concentration of a protein
of interest than the
pre-lyophilized liquid formulation.
The pharmaceutical formulations of the low impurity compositions, for example,
low
aggregate compositions, of the invention are typically stable formulations,
e.g., stable at room
temperature.
-80-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
The terms "stability" and "stable" as used herein in the context of a
formulation
comprising a protein of interest (e.g., an antibody) of the invention refer to
the resistance of the
protein of interest in the formulation to aggregation, degradation or
fragmentation under given
manufacture, preparation, transportation and storage conditions. The "stable"
formulations of the
invention retain biological activity under given manufacture, preparation,
transportation and
storage conditions. The stability of the protein of interest can be assessed
by degrees of
aggregation, degradation or fragmentation, as measured by HPSEC, static light
scattering (SLS),
Fourier Transform Infrared Spectroscopy (FTIR), circular dichroism (CD), urea
unfolding
techniques, intrinsic tryptophan fluorescence, differential scanning
calorimetry, and/or ANS
binding techniques, compared to a reference formulation. For example, a
reference formulation
may be a reference standard frozen at -70 C consisting of 10 mg/ml of a
protein of interest of the
invention in PBS.
Therapeutic formulations of the low impurity compositions, for example, low
aggregate
compositions, of the invention may be formulated for a particular dosage.
Dosage regimens may
be adjusted to provide the optimum desired response (e.g., a therapeutic
response). For example,
a single bolus may be administered, several divided doses may be administered
over time or the
dose may be proportionally reduced or increased as indicated by the exigencies
of the therapeutic
situation. It is especially advantageous to formulate parenteral compositions
in dosage unit form
for ease of administration and uniformity of dosage. Dosage unit form as used
herein refers to
physically discrete units suited as unitary dosages for the subjects to be
treated; each unit
contains a predetermined quantity of active compound calculated to produce the
desired
therapeutic effect in association with the required pharmaceutical carrier.
The specification for
the dosage unit forms of the invention are dictated by and directly dependent
on (a) the unique
characteristics of the protein of interest (e.g., antibody)and the particular
therapeutic effect to be
achieved, and (b) the limitations inherent in the art of compounding such a
protein of interest for
the treatment of sensitivity in individuals.
Therapeutic compositions of the low impurity compositions, for example, low
aggregate
compositions, of the invention can be formulated for particular routes of
administration, such as
oral, nasal, pulmonary, topical (including buccal and sublingual), rectal,
vaginal and/or parenteral
administration. The formulations may conveniently be presented in unit dosage
form and may be
prepared by any methods known in the art of pharmacy. The amount of active
ingredient which
can be combined with a carrier material to produce a single dosage form will
vary depending
upon the subject being treated, and the particular mode of administration. The
amount of active
ingredient which can be combined with a carrier material to produce a single
dosage form will
-81-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
generally be that amount of the composition which produces a therapeutic
effect. By way of
example, in certain embodiments, the proteins of interest (including fragments
of the protein of
interest) are formulated for intravenous administration. In certain other
embodiments, the
proteins of interest (e.g., antibodies), including fragments of the proteins
of interest (e.g.,
antibody fragments) are formulated for local delivery to the cardiovascular
system, for example,
via catheter, stent, wire, intramyocardial delivery, intrapericardial
delivery, or intraendocardial
delivery.
Formulations of the low impurity compositions, for example, low aggregate
compositions, of the invention which are suitable for topical or transdermal
administration
include powders, sprays, ointments, pastes, creams, lotions, gels, solutions,
patches and inhalants.
The active compound may be mixed under sterile conditions with a
pharmaceutically acceptable
carrier, and with any preservatives, buffers, or propellants which may be
required (US Patent No.
7,378,110; 7,258,873; 7,135,180; 7,923,029; and US Publication No.
20040042972).
The phrases "parenteral administration" and "administered parenterally" as
used herein
means modes of administration other than enteral and topical administration,
usually by injection,
and includes, without limitation, intravenous, intramuscular, intraarterial,
intrathecal,
intracapsular, intraorbital, intracardiac, intradermal, intraperitoneal,
transtracheal, subcutaneous,
subcuticular, intraarticular, subcapsular, subarachnoid, intraspinal, epidural
and intrasternal
injection and infusion.
Actual dosage levels of the active ingredients in the pharmaceutical
compositions of the
low impurity compositions, for example, low aggregate compositions, of the
invention may be
varied so as to obtain an amount of the active ingredient which is effective
to achieve the desired
therapeutic response for a particular patient, composition, and mode of
administration, without
being toxic to the patient. The selected dosage level will depend upon a
variety of
pharmacokinetic factors including the activity of the particular compositions
of the present
invention employed, or the ester, salt or amide thereof, the route of
administration, the time of
administration, the rate of excretion of the particular compound being
employed, the duration of
the treatment, other drugs, compounds and/or materials used in combination
with the particular
compositions employed, the age, sex, weight, condition, general health and
prior medical history
of the patient being treated, and like factors well known in the medical arts.
In certain embodiments, the proteins of interest (e.g., antibodies) of the
invention can be
formulated to ensure proper distribution in vivo. For example, the blood-brain
barrier (BBB)
excludes many highly hydrophilic compounds. To ensure that the therapeutic
compounds of the
-82-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
invention can cross the BBB (if desired), they can be formulated, for example,
in liposomes. For
methods of manufacturing liposomes, see, e.g., U.S. Pat. Nos. 4,522,811;
5,374,548; 5,399,331.
The liposomes may comprise one or more moieties which are selectively
transported into specific
cells or organs, thus enhance targeted drug delivery (see, e.g., V. V. Ranade
(1989) J. Clin.
Pharmacol. 29:685). Exemplary targeting moieties include folate or biotin
(see, e.g., U.S. Pat.
No. 5,416,016); mannosides (Umezawa et al., (1988) Biochem. Biophys. Res.
Commun.
153:1038); antibodies (P. G. Bloeman et al. (1995) FEBS Lett. 357:140; M.
Owais et al. (1995)
Antimicrob. Agents Chemother. 39:180); surfactant Protein A receptor (Briscoe
et al. (1995) Am.
J. Physiol. 1233:134), different species of which may comprise the
formulations of the invention,
as well as components of the invented molecules; p120 (Schreier et al. (1994)
J. Biol. Chem.
269:9090); see also K. Keinanen; M. L. Laukkanen (1994) FEBS Lett. 346:123; J.
J. Killion; I. J.
Fidler (1994) Immunomethods 4:273. In one embodiment of the invention, the
therapeutic
compounds of the invention are formulated in liposomes; in another embodiment,
the liposomes
include a targeting moiety. In another embodiment, the therapeutic compounds
in the liposomes
are delivered by bolus injection to a site proximal to the desired area. When
administered in this
manner, the composition must be fluid to the extent that easy syringability
exists. It must be
stable under the conditions of manufacture and storage and may be preserved
against the
contaminating action of microorganisms such as bacteria and fungi.
Additionally or
alternatively, the proteins of interest (e.g., antibodies) of the invention
may be delivered locally
to the brain to mitigate the risk that the blood brain barrier slows effective
delivery.
In certain embodiments, the low impurity compositions, for example, low
aggregate
compositions, of the invention may be administered with medical devices known
in the art. For
example, in certain embodiments a protein of interest (e.g., antibody) or a
fragment of protein of
interest (e.g., antibody fragment) is administered locally via a catheter,
stent, wire, or the like.
For example, in one embodiment, a therapeutic composition of the invention can
be administered
with a needleless hypodermic injection device, such as the devices disclosed
in U.S. Pat. Nos.
5,399,163; 5,383,851; 5,312,335; 5,064,413; 4,941,880; 4,790,824; 4,596,556.
Examples of
well-known implants and modules useful in the present invention include: U.S.
Pat. No.
4,487,603, which discloses an implantable micro-infusion pump for dispensing
medication at a
controlled rate; U.S. Pat. No. 4,486,194, which discloses a therapeutic device
for administering
medicants through the skin; U.S. Pat. No. 4,447,233, which discloses a
medication infusion pump
for delivering medication at a precise infusion rate; U.S. Pat. No. 4,447,224,
which discloses a
variable flow implantable infusion apparatus for continuous drug delivery;
U.S. Pat. No.
4,439,196, which discloses an osmotic drug delivery system having multi-
chamber
-83-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
compartments; and U.S. Pat. No. 4,475,196, which discloses an osmotic drug
delivery system.
Many other such implants, delivery systems, and modules are known to those
skilled in the art.
The efficient dosages and the dosage regimens for the reduced level of at
least one
impurity compositions of the invention depend on the disease or condition to
be treated and can
be determined by the persons skilled in the art. One of ordinary skill in the
art would be able to
determine such amounts based on such factors as the subject's size, the
severity of the subject's
symptoms, and the particular composition or route of administration selected.
ALTERNATIVE FORMULATIONS CONTAINING THE Low IMPURITY COMPOSITIONS OF THE
INVENTION
Alternative Aqueous Formulations
The invention also provides a low impurity composition, for example a low
aggregate
composition, formulated as an aqueous formulation comprising a protein of
interest and water, as
described in U.S. Patent No. 8,420,081, the contents of which are hereby
incorporated by
reference. In these aqueous formulations, the protein of interest is stable
without the need for
additional agents. This aqueous formulation has a number of advantages over
conventional
formulations in the art, including stability of the protein of interest in
water without the
requirement for additional excipients, increased concentrations of protein of
interest without the
need for additional excipients to maintain solubility of the protein of
interest, and low osmolality.
These also have advantageous storage properties, as the proteins of interest
in the formulation
remain stable during storage, e.g., stored as a liquid form for more than 3
months at 7 C or
freeze/thaw conditions, even at high protein of interest concentrations and
repeated freeze/thaw
processing steps. In one embodiment, formulations described herein include
high concentrations
of proteins of interest such that the aqueous formulation does not show
significant opalescence,
aggregation, or precipitation.
In one embodiment, an aqueous low impurity composition comprising a protein of
interest, e.g., an antibody, an anti-TNFa antibody or antigen biding portion
thereof, and water is
provided, wherein the formulation has certain characteristics, such as, but
not limited to, low
conductivity, e.g., a conductivity of less than about 2.5 mS/cm, a protein of
interest concentration
of at least about 10 [t.g/mL, an osmolality of no more than about 30
mOsmol/kg, and/or the
protein of interest has a molecular weight (Mw) greater than about 47 kDa. In
one embodiment,
the formulation has improved stability, such as, but not limited to, stability
in a liquid form for an
extended time (e.g., at least about 3 months or at least about 12 months) or
stability through at
-84-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
least one freeze/thaw cycle (if not more freeze/thaw cycles). In one
embodiment, the formulation
is stable for at least about 3 months in a form selected from the group
consisting of frozen,
lyophilized, or spray-dried.
In one embodiment, the formulation has a low conductivity, including, for
example, a
conductivity of less than about 2.5 mS/cm, a conductivity of less than about 2
mS/cm, a
conductivity of less than about 1.5 mS/cm, a conductivity of less than about 1
mS/cm, or a
conductivity of less than about 0.5 mS/cm.
In another embodiment, low impurity compositions included in the formulation
have a
given concentration, including, for example, a concentration of at least about
1 mg/mL, at least
about 10 mg/mL, at least about 50 mg/mL, at least about 100 mg/mL, at least
about 150 mg/mL,
at least about 200 mg/mL, or greater than about 200 mg/mL. In another
embodiment, the
formulation of the invention has an osmolality of no more than about 15
mOsmol/kg.
The aqueous formulations described herein do not rely on standard excipients,
e.g., a
tonicity modifier, a stabilizing agent, a surfactant, an anti-oxidant, a
cryoprotectant, a bulking
agent, a lyroprotectant, a basic component, and an acidic component. In other
embodiments of
the invention, the formulation contains water, one or more proteins of
interest, and no ionic
excipients (e.g., salts, free amino acids).
In certain embodiments, the aqueous formulation as described herein comprise a
low
impurity composition comprising a protein of interest concentration of at
least 50 mg/mL and
water, wherein the formulation has an osmolality of no more than 30 mOsmol/kg.
Lower limits
of osmolality of the aqueous formulation are also encompassed by the
invention. In one
embodiment the osmolality of the aqueous formulation is no more than 15
mOsmol/kg. The
aqueous formulation of the invention may have an osmolality of less than 30
mOsmol/kg, and
also have a high protein of interest concentration, e.g., the concentration of
the protein of interest
is at least 100 mg/mL, and may be as much as 200 mg/mL or greater. Ranges
intermediate to the
above recited concentrations and osmolality units are also intended to be part
of this invention. In
addition, ranges of values using a combination of any of the above recited
values as upper and/or
lower limits are intended to be included.
The concentration of the aqueous formulation as described herein is not
limited by the
protein of interest size and the formulation may include any size range of
proteins. Included
within the scope of the invention is an aqueous formulation comprising at
least 40 mg/mL and as
much as 200 mg/mL or more of a protein of interest, for example, 40 mg/mL, 65
mg/mL, 130
mg/mL, or 195 mg/ml, which may range in size from 5 kDa to 150 kDa or more. In
one
-85-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
embodiment, the protein of interest in the formulation of the invention is at
least about 15 kD in
size, at least about 20 kD in size; at least about 47 kD in size; at least
about 60 kD in size; at least
about 80 kD in size; at least about 100 kD in size; at least about 120 kD in
size; at least about 140
kD in size; at least about 160 kD in size; or greater than about 160 kD in
size. Ranges
intermediate to the above recited sizes are also intended to be part of this
invention. In addition,
ranges of values using a combination of any of the above recited values as
upper and/or lower
limits are intended to be included.
The aqueous formulation as described herein may be characterized by the
hydrodynamic
diameter (Dh) of the proteins of interest in solution. The hydrodynamic
diameter of the protein of
interest in solution may be measured using dynamic light scattering (DLS),
which is an
established analytical method for determining the Dh of proteins. Typical
values for monoclonal
antibodies, e.g., IgG, are about 10 nm. Low-ionic formulations may be
characterized in that the
Dh of the proteins of interest are notably lower than protein of interest
formulations comprising
ionic excipients. It has been discovered that the Dh values of antibodies in
aqueous formulations
made using the disfiltration/ultrafilteration (DF/UF) process, as described in
U.S. Patent No.
8,420,081, using pure water as an exchange medium, are notably lower than the
Dh of antibodies
in conventional formulations independent of protein concentration. In one
embodiment,
antibodies in the aqueous formulation as described herein have a Dh of less
than 4 nm, or less
than 3 nm.
In one embodiment, the Dh of the protein of interest in the aqueous
formulation is smaller
relative to the Dh of the same protein of interest in a buffered solution,
irrespective of protein of
interest concentration. Thus, in certain embodiments, a protein of interest in
an aqueous
formulation made in accordance with the methods described herein, will have a
Dh which is at
least 25% less than the Dh of the protein of interest in a buffered solution
at the same given
concentration. Examples of buffered solutions include, but are not limited to
phosphate buffered
saline (PBS). In certain embodiments, proteins of interest in the aqueous
formulation of the
invention have a Dh that is at least 50% less than the Dh of the protein of
interest in PBS in at the
given concentration; at least 60% less than the Dh of the protein of interest
in PBS at the given
concentration; at least 70% less than the Dh of the protein of interest in PBS
at the given
concentration; or more than 70% less than the Dh of the protein of interest in
PBS at the given
concentration. Ranges intermediate to the above recited percentages are also
intended to be part
of this invention, e.g., about 55%, 56%, 57%, 64%, 68%, and so forth. In
addition, ranges of
values using a combination of any of the above recited values as upper and/or
lower limits are
intended to be included, e.g., about 50% to about 80%.
-86-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
In one aspect, the aqueous formulation includes the protein of interest at a
dosage of
about 0.01 mg/kg-10 mg/kg. In another aspect, the dosages of the protein of
interest include
approximately 1 mg/kg administered every other week, or approximately 0.3
mg/kg administered
weekly. A skilled practitioner can ascertain the proper dosage and regime for
administering to a
subject.
Alternative Solid Unit Formulations
The invention also provides a low impurity composition of the invention
formulated as a
stable composition of a protein of interest, e.g., an antibody, or antigen
binding portion thereof,
and a stabilizer, referred to herein as solid units, as described in Attorney
Docket No. 117813-
31001, the contents of which are hereby incorporated by reference herein.
Specifically, it has been discovered that despite having a high proportion of
sugar, the
solid units comprising the low impurity compositions of the invention maintain
structural rigidity
and resist changes in shape and/or volume when stored under ambient
conditions, e.g., room
temperature and humidity, for extended periods of time (e.g., the solid units
comprising the low
impurity compositions of the invention do not require storage in a sealed
container) and maintain
long-term physical and chemical stability of the protein of interest without
significant
degradation and/or aggregate formation. Moreover, despite having a high
proportion of sugar,
the solid units comprising the low impurity compositions of the invention
remain free-flowing
when stored under ambient conditions, e.g., room temperature and humidity, for
extended
periods of time, and yet are easily dissolved in an aqueous solvent, e.g.,
water (e.g., the solid
units require minimal mixing when contacted with a solvent for
reconstitution). Furthermore, the
solid units comprising the low impurity compositions of the invention may be
prepared directly
in a device for patient use. These properties, when compared to existing
techniques which
require a vial containing a lyophilized protein of interest provided as a cake
(which may not
stabilize a protein of interest for extended periods of time), a separate vial
for a diluent, one or
more sterile syringes, and several manipulation steps, thus provides
alternative approaches for
reconstitution since the solid units comprising the low impurity compositions
of the invention
may be provided, e.g., in a dual chambered cartridge, to make reconstitution
invisible during
patient delivery. Furthermore, the solid units comprising the low impurity
compositions of the
invention are versatile in that they can be readily and easily adapted for
numerous modes of
administration, such as parenteral and oral administration.
As used herein, the term "solid unit," refers to a composition which is
suitable for
pharmaceutical administration and comprises a protein of interest, e.g., an
antibody or peptide,
-87-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
and a stabilizer, e.g., a sugar. The solid unit comprising the low impurity
compositions of the
invention has a structural rigidity and resistance to changes in shape and/or
volume. In one
embodiment, the solid unit comprising the low impurity compositions of the
invention is
obtained by freeze-drying a pharmaceutical formulation of a therapeutic
protein of interest . The
solid unit comprising the low impurity compositions of the invention may be
any shape, e.g.,
geometric shape, including, but not limited to, a sphere, a cube, a pyramid, a
hemisphere, a
cylinder, a teardrop, and so forth, including irregularly shaped units. In one
embodiment, the
solid unit has a volume ranging from about 1 1 to about 20 1. In another
embodiment, the
solid unit is not obtained using spray drying techniques, e.g., the solid unit
is not a powder or
granule.
As used herein, the phrase "a plurality of solid units" refers to a collection
or population
of solid units comprising the low impurity compositions of the invention,
wherein the collection
comprises two or more solid units having a substantially uniform shape, e.g.,
sphere, and/or
volume distribution. A substantially uniform size distribution is intended to
mean that the
individual shapes and/or volumes of the solid units comprising the low
impurity compositions of
the invention are substantially similar and not greater than a 10% standard
deviation in volume.
For example, a plurality of solid units which are spherical in shape would
include a collection of
solid units having no greater than 10% standard deviation from an average
volume of the
spheres. In one embodiment, the plurality of solid units is free-flowing.
KITS AND ARTICLES OF MANUFACTURE COMPRISING THE Low IMPURITY COMPOSITIONS OF
THE
INVENTION
Also within the scope of the present invention are kits comprising the low
impurity
compositions of the invention and instructions for use. The term "kit" as used
herein refers to a
packaged product comprising components with which to administer the protein of
interest (e.g.,
antibody, or antigen-binding portion thereof)), of the invention for treatment
of a disease or
disorder. The kit may comprise a box or container that holds the components of
the kit. The box
or container is affixed with a label or a Food and Drug Administration
approved protocol. The
box or container holds components of the invention which may be contained
within plastic,
polyethylene, polypropylene, ethylene, or propylene vessels. The vessels can
be capped-tubes or
bottles. The kit can also include instructions for administering a protein of
interest (e.g., an
antibody) of the invention.
-88-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
The kit can further contain one more additional reagents, such as an
immunosuppressive
reagent, a cytotoxic agent or a radiotoxic agent or one or more additional
proteins of interest of
the invention (e.g., an antibody having a complementary activity which binds
to an epitope in the
TNFa antigen distinct from a first anti-TNFa antibody). Kits typically include
a label indicating
the intended use of the contents of the kit. The term label includes any
writing, or recorded
material supplied on or with the kit, or which otherwise accompanies the kit.
The invention also provides a pharmaceutical pack or kit comprising one or
more
containers filled with a liquid formulation or lyophilized formulation of a
protein of interest (e.g.,
an antibody or antibody fragment thereof) of the invention. In one embodiment,
a container
filled with a liquid formulation of the invention is a pre-filled syringe. In
a specific embodiment,
the formulations of the invention are formulated in single dose vials as a
sterile liquid. For
example, the formulations may be supplied in 3 cc USP Type I borosilicate
amber vials (West
Pharmaceutical Services - Part No. 6800-0675) with a target volume of 1.2 mL.
Optionally
associated with such container(s) can be a notice in the form prescribed by a
governmental
agency regulating the manufacture, use or sale of pharmaceuticals or
biological products, which
notice reflects approval by the agency of manufacture, use or sale for human
administration.
In one embodiment, a container filled with a liquid formulation of the
invention is a pre-
filled syringe. Any pre-filled syringe known to one of skill in the art may be
used in combination
with a liquid formulation of the invention. Pre-filled syringes that may be
used are described in,
for example, but not limited to, PCT Publications W005032627, W008094984,
W09945985,
W003077976, US Patents US6792743, U55607400, U55893842, U57081107, U57041087,
U55989227, U56807797, U56142976, U55899889, U57699811, U57540382, U57998120,
U57645267, and US Patent Publication No. US20050075611. Pre-filled syringes
may be made
of various materials. In one embodiment a pre-filled syringe is a glass
syringe. In another
embodiment a pre-filled syringe is a plastic syringe. One of skill in the art
understands that the
nature and/or quality of the materials used for manufacturing the syringe may
influence the
stability of a protein formulation stored in the syringe. For example, it is
understood that silicon
based lubricants deposited on the inside surface of the syringe chamber may
affect particle
formation in the protein formulation. In one embodiment, a pre-filled syringe
comprises a
silicone based lubricant. In one embodiment, a pre-filled syringe comprises
baked on silicone.
In another embodiment, a pre-filled syringe is free from silicone based
lubricants. One of skill in
the art also understands that small amounts of contaminating elements leaching
into the
formulation from the syringe barrel, syringe tip cap, plunger or stopper may
also influence
stability of the formulation. For example, it is understood that tungsten
introduced during the
-89-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
manufacturing process may adversely affect formulation stability. In one
embodiment, a pre-
filled syringe may comprise tungsten at a level above 500 ppb. In another
embodiment, a pre-
filled syringe is a low tungsten syringe. In another embodiment, a pre-filled
syringe may
comprise tungsten at a level between about 500 ppb and about 10 ppb, between
about 400 ppb
and about 10 ppb, between about 300 ppb and about 10 ppb, between about 200
ppb and about 10
ppb, between about 100 ppb and about 10 ppb, between about 50 ppb and about 10
ppb, between
about 25 ppb and about 10 ppb.
In certain embodiments, kits comprising proteins of interest (e.g.,
antibodies) of the
invention are also provided that are useful for various purposes, e.g.,
research and diagnostic
including for purification or immunoprecipitation of protein of interest from
cells, detection of
the protein of interest in vitro or in vivo. For isolation and purification of
a protein of interest, the
kit may contain an antibody coupled to beads (e.g., sepharose beads). Kits may
be provided
which contain the antibodies for detection and quantitation of a protein of
interest in vitro, e.g., in
an ELISA or a Western blot. As with the article of manufacture, the kit
comprises a container
and a label or package insert on or associated with the container. The
container holds a
composition comprising at least one protein of interest (e.g., antibody) of
the invention.
Additional containers may be included that contain, e.g., diluents and
buffers, control proteins of
interest (e.g., antibodies). The label or package insert may provide a
description of the
composition as well as instructions for the intended in vitro or diagnostic
use.
The present invention also encompasses a finished packaged and labeled
pharmaceutical
product. This article of manufacture includes the appropriate unit dosage form
in an appropriate
vessel or container such as a glass vial, pre-filled syringe or other
container that is hermetically
sealed. In one embodiment, the unit dosage form is provided as a sterile
particulate free solution
comprising a protein of interest (e.g., an antibody) that is suitable for
parenteral administration.
In another embodiment, the unit dosage form is provided as a sterile
lyophilized powder
comprising a protein of interest (e.g., an antibody) that is suitable for
reconstitution.
In one embodiment, the unit dosage form is suitable for intravenous,
intramuscular,
intranasal, oral, topical or subcutaneous delivery. Thus, the invention
encompasses sterile
solutions suitable for each delivery route. The invention further encompasses
sterile lyophilized
powders that are suitable for reconstitution.
As with any pharmaceutical product, the packaging material and container are
designed to
protect the stability of the product during storage and shipment. Further, the
products of the
invention include instructions for use or other informational material that
advise the physician,
-90-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
technician or patient on how to appropriately prevent or treat the disease or
disorder in question,
as well as how and how frequently to administer the pharmaceutical. In other
words, the article
of manufacture includes instruction means indicating or suggesting a dosing
regimen including,
but not limited to, actual doses, monitoring procedures, and other monitoring
information.
Specifically, the invention provides an article of manufacture comprising
packaging
material, such as a box, bottle, tube, vial, container, pre-filled syringe,
sprayer, insufflator,
intravenous (i.v.) bag, envelope and the like; and at least one unit dosage
form of a
pharmaceutical agent contained within said packaging material, wherein said
pharmaceutical
agent comprises a liquid formulation containing a protein of interest (e.g.,
an antibody). The
packaging material includes instruction means which indicate how that said
protein of interest
(e.g., antibody) can be used to prevent, treat and/or manage one or more
symptoms associated
with a disease or disorder.
The present invention is further illustrated by the following examples which
should not
be construed as limiting in any way. The contents of all cited references,
including literature
references, issued patents, and published patent applications, as cited
throughout this application
are hereby expressly incorporated herein by reference. It should further be
understood that the
contents of all the figures and tables attached hereto are expressly
incorporated herein by
reference. The entire contents of the following applications are also
expressly incorporated
herein by reference: U.S. Provisional Patent Application 61/XXX,XXX, entitled
"STABLE
SOLID PROTEIN COMPOSITIONS AND METHODS OF MAKING SAME", Attorney Docket
Number 117813-31001; U.S. Provisional Patent Application 61/XXX,XXX, entitled
"LOW
ACIDIC SPECIES COMPOSITIONS AND METHODS FOR PRODUCING THE SAME
USING DISPLACEMENT CHROMATOGRAPHY", Attorney Docket Number 117813-73602,
filed on even date herewith; U.S. Provisional Patent Application 61/XXX,XXX,
entitled
"MUTATED ANTI-TNFa ANTIBODIES AND METHODS OF THEIR USE", Attorney Docket
Number 117813-73802, filed on even date herewith; U.S. Provisional Patent
Application
61/XXX,XXX, entitled "LOW ACIDIC SPECIES COMPOSITIONS AND METHODS FOR
PRODUCING THE SAME", Attorney Docket Number 117813-73901, filed on even date
herewith; and U.S. Provisional Patent Application 61/XXX,XXX, entitled
"MODULATED
LYSINE VARIANT SPECIES AND METHODS FOR PRODUCING AND USING THE
SAME", Attorney Docket Number 117813-74101, filed on even date herewith.
-91-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
EXAMPLES
GENERAL MATERIALS AND METHODS
Except where noted, the materials and methods described in connection with the
instant
example were also employed in Examples 1-3, below.
Chromatography Method
Pre-packed media columns were used in the following experiments, except where
specified. The column was equilibrated in a buffer system with appropriate pH
and conductivity.
The column load was prepared from Protein A affinity chromatography eluates or
concentrated
CEX chromatography elutes by buffer exchange (if the eluates were with
different buffer
components from the mixed mode target buffer system) or addition of the stock
solutions and/or
water to obtain the target pH and conductivity as specified (if the eluates
were with the same
buffer components as the mixed mode target buffer system). The prepared load
material was
filtered and loaded on the column according to the target load amount (g
protein/L media) as
specified followed by washing with the equilibration buffer or buffer similar
to equilibration
buffer with volumes as specified. The column Flow Through/Wash were collected
as fractions or
as a pool. HIC column was cleaned with 20% Isopropyl Alcohol solution. 1M NaOH
solution
was used for column cleaning.
Buffer Preparation Method
Buffers were prepared targeting a specific salt concentration in a buffered
system, and
titrating to a specific pH with the conjugate acid or base. For example, an
800 mM Ammonium
Sulfate (AmSO4) pH 7.0 solution was made by dissolving AmSO4 salt in a 20 mM
Tris-Acetate
buffered solution, titrating with acetate, and subsequently bringing up to
volume with water to
achieve the desired Am504 concentration. Load samples were prepared targeting
a specific salt
concentration by addition of concentrated salt solution in a buffered system,
and titrating to a
specific pH with the conjugate acid or base. For example, an 800 mM Am504 pH
7.0 load was
made by mixing the load in a 1:1 ratio with a 1600 mM Am504 pH 7.0 stock
buffer in a 40 mM
Tris-Acetate, and subsequently titrating with Tris or acetate to achieve a
final pH 7Ø
-92-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
Size Exclusion Chromatography
The molecular weight distribution of collected samples were quantified
according to the
following methods. Size exclusion chromatography (SEC) was performed using a
TSK-gel
G3000SWxL, 5[tm, 125 A, 7.8 X 300mm column (Tosoh Bioscience) on an HP Agilent
HPLC
system. Injections were made under isocratic elution conditions using a mobile
phase of 200 mM
sodium sulfate, 100 mM sodium phosphate, pH 6.8, and detected with absorbance
at 214 nm.
Quantification is based on the relative area of detected peaks.
Host Cell Protein (HCP) ELISA
HCP assay is based on process specific antigen based ELISA. Sample dilutions
were
applied to achieve readings within the calibration range. The limit of
quantitation of the assay is
0.625 ng/mL.
UV spectroscopy A280
UV A280 was used to determine protein concentrations for the samples post
protein A
elution. The assay was performed on an Agilent UV Spectrophotometer following
the method
.The protein concentration was determined using Beer-Lambert' s Law, A = cic,
where A is
Absorbance, c is the extinction coefficient, 1 is the path length, and c is
the concentration. The
absorbance was taken at 280 nm, the path length was 1 cm, and the extinction
coefficients were
1.39 for Adalimumab, 1.38 for mAb B, and 1.43 for mAb C.
EXAMPLE 1: DETERMINING OPERATING CONDITIONS APPROPRIATE FOR AN MAB: MEDIA:
BUFFER COMBINATION
The demonstration of the current invention for a specific antibody & media is
provided in
this example, and consists of: 1) Choosing a salt concentration that allows
product and
impurities to bind at a given pH; 2) Loading a small amount of protein to the
column and then
performing a linear gradient elution by decreasing the salt concentration.; 3)
Determining salt
concentration range in which the protein elutes from the HIC media.
In this example, adalimumab and GE CaptoPhenyl were chosen. The column was
equilibrated at 1.1 M Am504 pH 7.0 (Tris/Acetate) for 10 CVs. Adalimumab was
prepared at
1.1 M Am504 and loaded to the column at 20 g-protein/L of resin. The column
was washed with
CVs of the equilibration buffer. A linear gradient from 1.1M to OM Am504 pH
7.0
(Tris/Acetate) over 20CVs was performed. The process chromatogram is shown in
Figure 4.
-93-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
This process can be repeated for any given mAb-media combination for a given
buffer
system. Table 1 shows the DOE parameters determined using the method described
above for
adlimumab in AmSO4 pH 7.0 (Tris/acetate) for 3 different HIC adsorbents.
Table 1: Example Experimental Design Scope determined from LGE with different
resins
Adlimumab - Ammonium Sulfate pH 7.0 (Tris/Acetate)
Resin Buffer Concentration Range
Tosoh Hexyl 250 - 750 mM
GE CaptoPhenyl 300 - 650 mM
GE Butyl FF 800 - 950 mM
In practicing the current invention, the aggregate reduction desired can be
achieved by
appropriate pooling of the load and wash fractions. By collecting and
subsequently determining
the product quality of each fraction throughout the load and wash, the
accumulative aggregate
reduction and accumulative yield can be calculated using the weighted averages
up to a given
fraction. Additionally, the instantaneous yield can be estimated by comparing
the protein
recovered against the total protein loaded to the column at a given fraction.
Sample calculations
are shown below:
Sample Calculation A: Accumulative Yield up to a given fraction
Accumulated Protein Mass Recovered up to Fraction
Accumulative Yield = ___________________________________________
Total Mass Protein Load
Sample Calculation B: Accumulative Aggregate Reduction up to a given fraction
Accumulative Aggregate Reduction =
Accumulated Aggregate Mass Recovered up to Fraction
...Load Agg% ________________________________________________
Accumulated Total Protein Mass Recovered up to Fraction
Sample Calculation C: Instantaneous Yield up to a given fraction
Accumulated Protein Mass Recovered up to Fraction
Instantaneous Yield = __________________________________________
Total Protein Mass Loaded to Column at Fraction
-94-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
The demonstration of the current invention for a specific antibody & resin is
provided in
this example, and consists of
1. For a given salt concentration and optionally pH and HIC media.
2. Loading the HIC media in excess of the dynamic binding capacity for the
product for
the given condition.
3. Washing the column with a buffer containing a similar salt concentration
and
optionally pH used for the equilibration and loading steps.
4. Collecting fractions throughout the loading and wash steps and subsequently
determining the product quality profile (e.g. Aggregate, HCP etc.)
In this example, adalimumab and GE CaptoPhenyl were chosen. The experiment was
performed at 400 mM sodium citrate (NaCit) pH 5.6. The column was equilibrated
with 400 mM
NaCit pH 5.6 for 10 CVs. Adalimumab was prepared at 400 mM NaCit pH 5.6 and
loaded to the
column at 500 g-protein/L-resin. The column was washed with 7 CVs of the
equilibration buffer.
The process chromatogram is shown in Figure 5. Fractions were collected and
analyzed for
product quality and the accumulative yield and accumulative aggregate
reduction calculated,
shown in Table 2. From this example, it is clear to one skilled in the art to
determine a run
condition which delivers a targeted product quality and/or step yield.
This general approach is used to evaluate the performance for a given
operating condition
for any resin/mAb/buffer combination.
Table 2: Accumulative Yield and Aggregate Reduction from Figure 5
Fraction Load Accumulative Recovery Accumulative AAgg
D1 8 g/L 0% 0.82%
D2 45 g/L 4% 0.77%
D3 82 g/L 12% 0.71%
D4 119 g/L 19% 0.67%
D5 156 g/L 26% 0.62%
D6 193 g/L 33% 0.56%
D7 231 g/L 41% 0.51%
El 268 g/L 48% 0.47%
E2 305 g/L 55% 0.43%
E3 342 g/L 62% 0.40%
-95-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
E4 379 g/L 70% 0.37%
E5 416 g/L 77% 0.34%
E6 454 g/L 84% 0.32%
E7 491 g/L 91% 0.29%
F1 500 g/L 93% 0.29%
F2 WASH 99% 0.28%
F3 WASH 100% 0.28%
F4 WASH 101% 0.29%
F5 WASH 101% 0.29%
EXAMPLE 2: DEMONSTRATION OF AGGREGATE REDUCTION WITH HIC RESINS
This data set is compiled to demonstrate the aggregate reduction achieved with
six
different HIC adsorbents. Each resin was evaluated with a 500 g/L load of
adalimumab at a
NaCit concentration near, and slightly higher than, the peak elution
concentration determined
from the process outlined in Example 1. Table 3 outlines the results from
these experiments.
Table 3: Effect of HIC Resins on Aggregate Reduction of Adalimumab
HIC Resin NaCit, pH 5.6 AAgg Yield
400 mM 1.5% 99.8%
Butyl
450 mM 1.2% 85.7%
240 mM 1.2% 93.9%
Hexyl
300 mM 1.1% 100.9%
400 mM 1.5% 96.5%
Phenyl
450 mM 1.2% 90.7%
350 mM 0.4% 98.5%
Octyl
400 mM 0.1% 103.3%
550 mM 1.2% 88.1%
GE Butyl FF
600 mM 1.7% 83.0%
450 mM 0.2% 97.5%
PPG
600 mM 1.0% 38.1%
EXAMPLE 3: DEMONSTRATION OF AGGREGATE REDUCTION WITH OTHER ANTIBODIES, MAB
B AND MAB C
Aggregate reduction technology of the current invention has been demonstrated
with
multiple antibodies using HIC adsorbents. Antibodies have different
hydrophobic properties,
-96-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
leading to interaction behavior on a HIC column that differs from one antibody
to another.
Therefore the impact of salt type and concentration is different for each
antibody.
Table 4 and Table 5, presented below, provide the data obtained for mAb B and
mAB C.
The data clearly demonstrates that the aggregate reduction technology is
effective for alternatives
to adalimumab.
Table 4: Aggregate reduction for mAb B, pI - 9.1
HIC Resin AmSO4, pH 5.0 AAgg Yield
370 mM 0.8% 100%
Hexyl
710 mM 0.6% 93%
340 mM 0.6% 95%
Phenyl
790 mM 0.5% 95%
840 mM 0.6% 99%
Butyl
1000 mM 0.6% 96%
Table 5: Aggregate reduction for mAb C, pI - 7.0
HIC Resin AmSO4, pH 5.0 AAgg Yield
80 mM 5.0% 89.0%
Hexyl
330 mM 4.5% 99.8%
130 mM 3.5% 92.8%
Phenyl
480 mM 2.9% 92.8%
690 mM 5.2% 93.5%
Butyl
880 mM 5.4% 87.9%
EXAMPLE 4 : DEMONSTRATION OF AGGREGATE REDUCTION WITH DIFFERENT SALT
CONCENTRATIONS - ADALIMUMAB
Ion concentration is a key variable in the performance of hydrophobic
interaction
chromatography. For every combination of antibody/resin/pH there is a range of
ion
concentrations that provide aggregate reduction; the strategy outlined in
Example 1. can be
followed to determine the aggregate reduction and the corresponding recovery
for each salt
concentration.
-97-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
Table 6, below, shows the effect of salt concentration on aggregate reduction
and step
yield. In this example CaptoPhenyl and adalimumab were chosen, and evaluated
at a loading of
200-500 g/L in NaCit pH 5.6 at the concentration specified. The data
demonstrates that the
aggregate reduction can be effectively achieved over a range of salt
concentrations, and that the
salt concentration and column loading can be balanced to achieve a desired
step yield and final
product quality
Table 6: Effect of Ion Concentration on Aggregate Reduction
NaCit pH 5.6 Load Yield AAgg
200 g/L 92% 0.59%
300 mM 350 g/L 96% 0.33%
500 g/L 97% 0.24%
200 g/L 90% 0.76%
400 mM 350 g/L 94% 0.43%
500 g/L 96% 0.35%
200 g/L 85% 1.09%
500 mM 350 g/L 91% 0.97%
500 g/L 94% 0.86%
EXAMPLE 5: DEMONSTRATION OF AGGREGATE REDUCTION WITH DIFFERENT BUFFER
SYSTEMS WITH ADALIMUMAB
In addition to the salt concentration, the salt anion and cation types are key
variables in
hydrophobic interaction chromatography. The invention has been demonstrated
with ammonium
sulfate, sodium sulfate, and sodium citrate. As one skilled in the art would
appreciate the optimal
salt concentration and optionally pH are different for each salt type and was
derived by using the
strategy outlined in Example 1. Table 7 shows the data of aggregate reduction
and corresponding
recovery for the different anion/cation types and different HIC adsorbents.
-98-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
Table 7: Effect of Anion/Cation Type Aggregate Reduction
Resin Buffer System Load Yield AAgg
630 mM AmSO4 pH 7.0 300 g/L 95% 2.1 %
300 mM AmSO4 pH 7.0 300 g/L 99% 1.1 %
425 mM Na504 pH 7.0 300 g/L 95% 1.9 %
CaptoPhenyl
240 mM Na504 pH 7.0 300 g/L 101`)/0 1.1 %
500 mM NaCit pH 5.6 350 g/L 91% 1.0 %
300 mM NaCit pH 5.6 350 g/L 96% 0.2 %
725 mM Am504 pH 7.0 300 g/L 94% 1.7 %
275 mM Am504 pH 7.0 300 g/L 103% 0.9 %
460 mM Na504 pH 7.0 300 g/L 97% 0.7 %
Tosoh Hexyl
180 mM Na504 pH 7.0 300 g/L 101% 0.6 %
440 mM NaCit pH 5.6 300 g/L 87% 0.5 %
150 mM NaCit pH 5.6 300 g/L 97% 0.5 %
800 mM Am504 pH 7.0 300 g/L 100% 0.7 %
1000 mM Am504 pH 7.0 300 g/L 94% 1.6 %
750 mM Na504 pH 7.0 300 g/L 96% 1.8 %
Butyl FF
700 mM Na504 pH 7.0 300 g/L 101`)/0 1.7 %
700 mM NaCit pH 5.6 300 g/L 98% 1.6 %
600 mM NaCit pH 5.6 300 g/L 95% 1.5 %
EXAMPLE 6: DEMONSTRATION OF AGGREGATE REDUCTION WITH DIFFERENT LOADING
Furthermore, the strategy outlined in Example 1. to reduce aggregates through
careful
control of ion concentration, ion type, HIC adsorbent, and pH can be applied
to various ranges of
protein loading. Aggregate reduction for a range of protein loadings (e.g. 250-
700 g/L) for
CaptoPhenyl using a 400 mM NaCit pH 5.6 buffer is shown in Table 8, displaying
a robust
aggregate reduction across an expansive loading range.
-99-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
Table 8: Impact of Column loading
Load Yield AAgg AAgg/LoadAgg
250 g/L 95% 0.29 % 87%
500 g/L 100% 0.25 % 77%
700 g/L 100% 0.21 % 65%
EXAMPLE 7: DEMONSTRATION OF AGGREGATE REDUCTION WITH DIFFERENT LOAD
CONCENTRATION - ADALIMUMAB
In addition to the strategy outlined in Example 6. to reduce aggregates
through careful
control of ion concentration, ion type, and HIC adsorbent, it has been shown
that the
concentration of the load protein can have an effect on aggregate reduction.
In this example, a
feed stream was serial diluted to cover a range of load concentrations from 4
to 15 mg/mL and
loaded at 500 g/L to a CaptoPhenyl column in 400mM NaCit pH 5.6. The effect of
decreasing
the concentration of the load protein is shown in Figure 6.
EXAMPLE 8: DEMONSTRATION OF HCP REDUCTION IN ADDITION To AGGREGATE
REDUCTION
HIC chromatography can also be effective in reducing host cell protein (HCP)
levels. In
the present invention, it has been demonstrated that HCP levels can be
effectively reduced under
operating conditions selected for aggregate reduction.
Table 9 shows HCP removal achieved along with aggregate reduction. The data
clearly
shows that other process related substances/impurities can be achieved using
the current
invention on the HIC adsorbents, and hence functions as an effective polishing
step in the large
scale purification of monoclonal antibodies.
-100-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
Table 9: HCP Removal during HIC Chromatography
HCP
NaCit pH 5.6 Load Yield AAgg
Load Pool
200 g/L 92% 0.59% NA
350 g/L 96% 0.33% 1398 ng/mg 150 ng/mg
300 mM
500 g/L 97% 0.24% 348 ng/mg
200 g/L 99% 0.34% 38 ng/mg 5 ng/mg
200 g/L 90% 0.76% 104 ng/mg
350 g/L 94% 0.43% 1599 ng/mg 148 ng/mg
400 mM
500 g/L 96% 0.35% 350 ng/mg
350 g/L 97% 0.35% 38 ng/mg 6 ng/mg
200 g/L 85% 1.09% 169 ng/mg
350 g/L 91% 0.97% 1528 ng/mg
203 ng/mg
500 mM
500 g/L 94% 0.86% 301 ng/mg
500 g/L 87% 0.35% 38 ng/mg 11 ng/mg
EXAMPLE 9: DEMONSTRATION OF IMPACT OF DYNAMIC AND EQUILIBRIUM BINDING
In the HIC-based separation strategies described herein, the measured dynamic
binding
capacity (DBC), which is conventionally measured at 10% breakthrough, was
found to be greater
than the amount of protein that remained bound after washing the column (a.k.a
equilibrium
binding capacity, EBC) with a buffer with similar pH and salt concentration to
the equilibration
and load conditions. For example, but not by way of limitation, Figure 10
shows an example of
the DBC and EBC for the data presented in Figure 5. In addition, Table 10
shows effect of salt
type, concentration, and HIC resin on DBC and EBC values for Adalimumab.
-101-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
Table 10: Comparison of DBC and EBC values for Adilmumab
Resin Buffer System AAgg DBC EBC
630 mM AmSO4 pH 7.0 2.1 % 27 g/L 16 g/L
300 mM AmSO4 pH 7.0 1.1 % 6 g/L 4 g/L
CaptoPhenyl
425 mM Na504 pH 7.0 1.9 % 22 g/L 15 g/L
240 mM Na504 pH 7.0 1.1 % 6 g/L 4 g/L
1000 mM Am504 pH 7.0 1.6 % 17 g/L 11 g/L
800 mM Am504 pH 7.0 0.7 % 4 g/L 4 g/L
750 mM Na504 pH 7.0 1.8 % 29 g/L 13 g/L
Butyl FF
700 mM Na504 pH 7.0 1.7 % 22 g/L 11 g/L
700 mM NaCit pH 5.6 1.6 % 39 g/L 24 g/L
600 mM NaCit pH 5.6 1.5% 17 g/L 11 g/L
EXAMPLE HIC 10: COMBINATIONS OF HIC WITH ALTERNATIVE SEPARATION STRATEGIES
The methods described herein for reducing aggregates using HIC can be used as
an
independent operation or in combination with other process steps that provide
additional
aggregate reduction or those providing additional complementary and
supplementary
purification. Data for specific separation strategies is provided in Tables 11
and 12. For
example, but not by way of limitation, the following process combinations can
be used:
1. Affinity HIC
2. Affinity AEX HIC
3. Affinity Mixed Mode HIC
-102-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
Table 11: Aggregate reduction with different source materials
HCP LRF
Load Source Buffer Condition Load Yield AAgg (log
reduction
fraction
400 mM NaCit pH 5.6 500 g/L 96% 1.49% NA
ProteinA Eluate
450 mM NaCit pH 5.6 500 g/L 91% 1.22% NA
300 mM NaCit pH 5.6 200 g/L 92% 0.59% 1.0
ProteinA / 400 mM NaCit pH 5.6 350 g/L 94% 0.43% 1.0
AEX FTW
500 mM NaCit pH 5.6 500 g/L 94% 0.86% 0.7
300 mM NaCit pH 5.6 200 g/L 99% 0.34% 0.8
ProteinA /
Mixed Mode FTW 400 mM NaCit pH 5.6 350 g/L 97% 0.35% 0.8
500 mM NaCit pH 5.6 500 g/L 87% 0.35% 0.5
Table 12: Complete Process Train with Protein A Capture ¨ Aggregate, HMW and
HCP
reduction
Process Yield (%) %HMWHCP LRF
reduction
Clarified Harvest 97.00% n/a n/a
Prt-A Eluate Pool 89.60% n/a 1.87
Viral Inactivated
99.70% 0.07 0.39
Filtrate
MM FT pool 91.90% 0.83 1.63
HIC FT-pool 98.50% 0.23 0.46
VF(FT) Filtrate 96.10% No reduction 0.1
BDS (FT) 103.80% No reduction 0.13
EXAMPLE 11: HYBRID HIC BINDING MECHANISM
By estimating the partitioning coefficient Kp, it can be demonstrated that
certain strategies
described in the instant application do not fall under the category of "Weak-
Partitioning (WP)" or
"Flow-Through Overload (FT)" modes as those are described in the art, e.g.,
US2007/0060741.
For example, Figures 13A-13B depict the results of experiments wherein
aliquots of resin are
incubated with a load covering a range of protein concentrations at room
temperature for 3 hours,
after which the protein solution is then removed, and replaced with
equilibration buffer (Wash
simulation) and incubated at room temperature for 3 hours (repeated, Wash II).
After each
incubation, the concentration of the protein solution is measured and used to
calculated the
amount of protein ((A) monomer D2E7, a.ka. Adalimumab, and (B) aggregate D2E7)
bound to
the resin (g protein / L resin) and plotted against the concentration of the
protein solution at the
-103-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
end of the incubation (e.g. equilibrium). Figures 14A-14B depict the
results outlined in
Figures 13A-13B, highlighting the fact that at initial equilibrium a
significant amount of
monomer/aggregate is bound to the resin. However, after the protein solution
is replaced with
equilibration buffer (see arrow), the monomer de-sorbs from the resin and back
into solution,
whereas the aggregate remains bound.
Figures 15A-15B depict a determination of the binding monomer and aggregate
D2E7
(based on data provided in Figures 13A-13B) by fitting the experimental
equilibrium binding
data to the Langmuir Isotherm using the equation: q = (qmax X Cequil) (Ka +
Cequil); where q =
amount of protein bound to resin [=] g/L-resin; qmax = maximum amount of
protein bound to
resin [=] g/L-resin; Cequil = solution concentration of protein [=] g/L-soln;
and Kd = equilibrium
dissociation constant.
By fitting the experimental data, the qmax and Kd for the monomer and the
aggregates can
be calculated.
Species Q.õ [mg I mL] Kd [mgI mL]
Monomer 41.9 0.47
Aggregate 6.0 0.01
Significantly, qmax for both monomer/aggregate and the Kd values (i.e.
strength of
binding) are similar to those of strong hydrophobic interactions, therefore it
is not expected for
this interaction to be "reversible." In addition, by calculating Kp where:
Q Qmax
K =
Species Q.[mg I mL] Kd [mgI mL] C Kd
Monomer 41.9 0.47 90
Aggregate 6.0 0.01 600
it is apparent that the instant technique does not fall within the category of
flow-through (where
Kp < 1) or weak portioning (where Kp = 1-10), but rather fall within the
category of bind-elute
(where Kp > 10).
-104-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
EXAMPLE 12: DETERMINATION OF BINDING CAPACITY AT SATURATION
The following protocol exemplifies the determination of (i) apparent binding
capacity,
i.e., the binding capacity at saturation (when outlet protein conentration
equals inlet protein
concentration) under flow conditions and (ii) the actual binding capacity,
i.e., the amount of
protein that remains bound after an isocratic wash.
A column packed with resin containing a hydrophobic interaction ligand was
equilibrated
with a buffer at a given salt concentration and pH. A protein load in the same
buffer condition as
the equilibration solution was loaded to the column until the protein breaks
through the column,
and the protein concentration at the effluent of the column was equal to the
protein concentration
at the inlet of the column (i.e., saturated). The column was then washed with
the equilibration
solution until the protein concentration at the effluent was effectively zero.
The remaining
protein bound to the column was then eluted with a buffer condition that will
cause the protein to
desorb from the resin.
Taking into account the void volume of the column and chromatography system,
one can
calculate the amount of protein bound to the column at the saturation point by
integrating the area
above the breakthrough curve at the effluent of the column (Figures 12A-12B).
After the
isocratic wash, one can calculate the protein that remained bound to the resin
by integrating the
area under the curve of the elution peak.
The differences between these two values is the 'reversible' binding capacity,
which is
significant when compared to the binding capacity observed at the saturation
point (e.g.,
"apparent binding capacity"). This difference is also a function of the salt
concentration, which
is shown in Figure 16. Figure 16 is a comparison of apparent and actual bound
protein under
flow conditions. Binding of the antibody during loading is significant (>10
g/1). The majority
(>65 %) of the antibody monomer bound during load desorbs during the isocratic
wash (i.e.,
reversibly bound). The mass balance of the impurity demonstrates irreversible
binding.
EXAMPLE 13: DETERMINATION OF BINDING CAPACITY AT SATURATION
A column was conditioned and loaded, as described in Example 12, at different
inlet
protein concentrations. In these experiments, the flow-through fractionated to
determine the
product quality at different times during the loading and breakthrough. Using
the protein mass
-105-
CA 02899310 2015-07-24
WO 2014/143185 PCT/US2013/065797
and product quality for each of the fractions, the accumulative impurity
(e.g., aggregate)
breakthrough can be calculated using the weighted average:
(Sum of total aggregate mass up to fraction,)
A rcu t ti % Aggre.qates ¨ _________________________________________
(Sum of total. pr otein mass zip tofractio.0
This calculation can be plotted for each successive fraction (Figure 8A) and
used to
compare different loading conditions. The Equilibrium Binding Isotherms for
both the monomer
and aggregate show that for all of the loading conditions (Figures 8B-8C), the
monomer was in
the non-linear part of its binding isotherm (e.g., equilibrium binding
capacity is independent of
monomer concentration), and the aggregate was in or near the linear part of
its binding isotherm
(e.g., equilibrium binding capacity is dependent on aggregate concentration).
The clearance of
the aggregate improves by decreasing the overall load protein concentration,
even though this
results in the resin having a lower binding capacity for the aggregate.
In Figure 7, material from a single source was serially diluted to three
different protein
concentrations and subsequently loaded to a column to 500 g/L. This data
clearly demonstrates
that diluting the load material resulted in a better aggregate clearance, even
though the same
amount of impurity was loaded in each case. This is non-intuitive, especially
when considering
that diluting the load protein concentration results in a lower overall
binding capacity for the
impurity as the impurity is in the linear range of the equilibrium binding
isotherm and therefore
the binding capacity decreases linearly with concentration.
Figure 9 is a re-plot of the same data as in Figure 7 to demonstrate that, for
a given target
impurity clearance, the recovery-yield can be modulated by diluting the load
material to a
specific range.
-106-