Note: Descriptions are shown in the official language in which they were submitted.
CA 02901353 2015-08-14
WO 2014/113022 PCT/US2013/022213
DCC MEDIATED COUPLING FOR HALOFENATE MANUFACTURE
[0001] Ester and amide derivatives of (-)-4-chloro-a-(3-
trifluoromethylphenoxy)phenylacetic
acid (I) ((-)-halofenic acid), including (-)-halofenate (II), are chiral
compounds and are useful in
ameliorating a variety of physiological conditions, including conditions
associated with blood
lipid deposition, Type II diabetes, hyperlipidema and hyperuricemia (see,
e.g., U.S. Pat. Nos.
7,199,259 and 6,262,118 which are incorporated herein by reference in their
entireties).
lei rs 1.1
F3C 0 F31/4, 0 0
0 0 OH
ISI N
0 H)..
CI (I) CI n
[0002] Halofenic acid and its ester and amide derivatives contain a single
chiral center alpha
to the carbonyl carbon atom, and therefore exist in two enantiomeric forms.
The administration
of halofenate (i.e., a racemic mixture of the two enantiomers of 2-
acetamidoethyl (4-
chlorophenyl) (3-trifluoromethylphenoxy) acetate) results in lowering plasma
glucose,
triglycerides and serum uric acid. However, this racemic mixture also results
in various adverse
effects including nausea, gastrointestinal ulcers, and gastrointestinal
bleeding. Other side effects
that have been reported with racemic halofenate include potential adverse drug-
drug interactions,
including difficulties controlling anticoagulation with CoumadinTM. It has
been determined that
the (-)-enantiomer of halofenic acid is about twenty-fold less active in its
ability to inhibit
cytochrome P450 2C9 compared to the (+)-enantiomer. It is therefore more
desirable and
advantageous to administer the (-)-enantiomer of halofenate instead of racemic
halofenate.
[0003] Various synthetic routes for preparing (-)-halofenic acid
derivatives, such as (-)-
halofenate, have been reported in literature. However, these derivatives are
often difficult to
prepare in high yields and high enantiomeric purity using known synthetic
methods. Therefore,
there is a need for a process for preparing a-(phenoxy)phenylacetic acid and
derivatives thereof,
such as (-)-halofenate, with high yields and high enantiomeric purity.
1
CA 02901353 2015-08-14
WO 2014/113022 PCT/US2013/022213
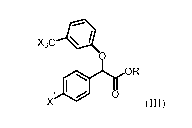
[0004] One aspect provides for a process for the preparation of a compound
of the formula
(III) or a salt thereof:
X3C *
=
R
X (III)
wherein:
R is selected from the group consisting of arylCi_6alkyl-, (Ci_6alkY1)2NC 1_6
alkyl-, C1 _
6 alkyl-NHC1_6alkyl- ,C1_6alkylC(0)NHCi_6alkyl-, ary1C(0)NHC1_6alkyl-,
Ci_6alkyl-
NHC(0)NHCi_6alkyl-, aryloxyCi_6alkyl- and Ci_6alkylNHC(0)NHphenyl-; and
X and X are each independently a halogen;
the process comprising:
contacting a compound of the formula (Ia)
X3C *
=
OH
, 1101 0
X (Ia)
wherein X and X' are each independently a halogen;
with a compound of formula (Ib)
ROH (lb)
wherein R is defined above; and
a coupling agent;
in an aprotic solvent under conditions sufficient to form the compound of
formula (III).
[0005] In one embodiment, the compound of formula (III) obtained from the
crude reaction
mixture (i.e. obtained before recrystallization) may be allowed to remain in
the crude reaction
mixture without requiring workup for an extended period of time, wherein the
compound of the
formula (III) does not undergo epimerization or racemization. For example, the
compound of the
formula (III) may remain in the crude reaction mixture for at least 1.5 hours,
2 hours, 5 hours, 10
hours, 1 5 hours, 1 7 hours or more than 24 hours without any measurable
racemization. In
2
CA 02901353 2015-08-14
WO 2014/113022 PCT/US2013/022213
another embodiment, the compound of the formula (III) may remain in the crude
reaction
mixture at about 0 C, 10 C, 15 C, 20 C or at about room temperature for
the above cited
period of time without any measurable racemization.
[0006] In another embodiment, R is Ci_6alkylC(0)NHCi_6a1ky1 or
ary1C(0)NHC1_6alkyl, X is
F and X is Cl, and the coupling agent is N,N'-dicyclohexylcarbodiimide. In
another embodiment,
R is CH3CONHCH2CH2- and X is F and X' is Cl. In another embodiment, the
compound of the
formula (Ib) is N-acetylethanolamine. In another embodiment, no base is used.
In another
embodiment, the compound of formula (III) is the compound of formula (II):
rs
0
1$1 0
CI (II).
[0007] In another embodiment, the aprotic solvent is selected from the
group consisting of
toluene, xylenes, cyclohexane, diisopropylether, isopropyl acetate, THF,
hexanes, MTBE, and
combinations thereof. In one variation of the process, the aprotic solvent is
toluene. In another
embodiment, the mole ratio of N,N'-dicyclohexylcarbodiimide to the compound of
formula (Ia)
is 1.05:1 to 1.15:1, and the mole ratio of the compound of formula (Ib) to the
compound of
formula (Ia) is 1.02:1 to 1.7:1. In one variation of the process, the mole
ratio of N,N'-
dicyclohexylcarbodiimide to the compound of formula (Ia) is 1.1:1, and the
mole ratio of the
compound of formula (Ib) to the compound of formula (Ia) is 1.5:1. In one
embodiment, the
process is performed using 4-(N,N-dirnethylamino)pyridine (DMAP). In another
embodiment,
no DMAP is used. In another embodiment, the process is performed with no N-
hydroxy based
agent typically used in DCC coupling reactions to avoid racemization (such as,
e.g., 1-
hydroxybenzotriazole (HOBt), N-hydroxysuccinimide (HOSu), or N-hydroxy-5-
norbornene-
endo-2,3-dicarboximide (BONB)).
[0008] In one embodiment, the process further comprises: contacting a
compound of formula
(Ia) with a compound of formula (Ib); cooling the resulting solution at about
0 C; and contacting
a solution of N,N'-dicyclohexylcarbodiimide with the solution comprising the
compounds of
formulae (Ia) and (lb) for a sufficient period of time to form the compound of
formula (III). For
example, one process comprises contacting a solution comprising N-
acetylethanolamine in
toluene with a solution of the compound of formula (Ia) in toluene; cooling
the resulting solution
3
CA 02901353 2015-08-14
WO 2014/113022 PCT/US2013/022213
at about 0 C; and contacting a solution of N,N'-dicyclohexylcarbodiimide in
toluene with the
solution comprising N-acetylethanolamine and the compound of formula (Ia) for
a sufficient
period of time to form the compound of formula (III).
[0009] In one embodiment, the process further comprises isolating
dicyclohexylurea (DCU)
precipitate as a by-product from the filtrate and washing the precipitate with
an aprotic solvent
such as toluene. In another embodiment, the process further comprises adding
an aprotic solvent,
such as cyclohexane, to the filtrate, washing the filtrate with water and
azeotropically distilling
water from the filtrate, and isolating the compound of the formula (III) by
crystallization.
[0010] In certain embodiments, the compound of formula (III) is obtained
with a yield of
about 80% or greater before recrystallization. In certain embodiments, the
compound of
formula (III) is obtained with an enantiomeric excess of about 98% or greater
before
recrystallization. In various embodiments, the compound of formula (III) has
an enantiomeric
excess of about 98%, about 99%, about 99.5%, about 99.9%, or greater before
recrystallization.
In certain embodiments, the compound of formula (III) is obtained with a
chemical purity of
about 98% or greater before recrystallization. In some embodiments, the
compound of formula
(III) has a DCU level of less than 1.0% before recrystallization, less than
about 0.9%, less than
about 0.7%, less than about 0.5%, less than about 0.3% or less than about
0.1%.
[001 11 In one embodiment, the process provides for the preparation of (-)-
halofenate (II) or a
salt thereof:
0
F3s,r . 0 0
40
N
H 1 0 CI ).
CI (II)
the process comprising: contacting a solution of (-)-4-chloro-phenyl-(3-
trifluoromethyl-
phenoxy)-acetic acid in an aprotic solvent with N-acetylethanolamine;
contacting the resulting
solution mixture with a solution of N,N'-dicyclohexylcarbodiimide in an
aprotic solvent to form
(-)-halofenate. In one aspect of the process, the aprotic solvent is selected
from the group
consisting of toluene, xylenes, cyclohexane, di-isopropyl ether, isopropyl
acetate, THF, hexanes
and MTBE or combinations thereof. In another aspect, the aprotic solvent is
toluene. In another
aspect of the process, the contacting the resulting solution mixture with a
solution of N,N'-
4
CA 02901353 2015-08-14
WO 2014/113022 PCT/US2013/022213
dicyclohexylcarbodiimide in an aprotic solvent s performed at about 0 C. In
another aspect, the
process further comprises filtering the DCU by-product from the solution.
[0012] In certain embodiments, the (-)-halofenate is obtained with a yield
of about 80% or
greater. In certain embodiments of the process, the (-)-halofenate is obtained
with an
enantiomeric excess of about 98% or greater before purification, that is,
before any purification
step (e.g. recrystallization) is performed (that is, as the solid is first
obtained from the reaction
mixture). In various embodiments, the (-)-halofenate has an enantiomeric
excess of about 98%,
about 99%, about 99.5%, about 99.9%, or greater before purification. In
certain embodiments of
the process, the (-)-halofenate is obtained with a chemical purity of about
98% or greater before
purification. In some embodiments of the process, the (-)-halofenate has a DCU
level of less
than 1.0%, about 0.5% about 0.3% or about 0.1% before purification.
[0013] In another aspect, the process further comprises purifying (e.g.
recrystallizing) the
compound of formula (III) (e.g. the compound of formula (II), (-)-halofenate).
Recrystallization
may be performed in a variety of solvents, for example using diisopropylether,
cyclohexane, or a
mixture of toluene and cyclohexane. Recrystallization may be used to improve
the enantiomeric
excess or chemical purity of the desired product. For example, in some
embodiments the
compound of formula (III) has an enantiomeric excess of about 99.9% or greater
after
recrystallization. In some embodiments, the compound of formula (III) has a
chemical purity of
about 99% or greater after recrystallization. In some embodiments, the
compound of formula
(III) has a DCU level of less than about 0.5%, less than about 0.3%, or less
than about 0.1%.
Other aspects of the current disclosure are described below.
[0014] Unless defined otherwise, all technical and scientific terms used
herein have the same
meaning as commonly understood by one of ordinary skill in the art. It must be
noted that as
used herein and in the appended claims, the singular forms "a", and, and the
include plural
referents unless the context clearly dictates otherwise.
[0015] "Alkyl" refers to straight, branched, or cyclic aliphatic
hydrocarbons chain groups of
one to ten carbon atoms (Ci_io alkyl), one to six carbon atoms (Ci_6 alkyl) or
one to four carbon
atoms (C1_4 alkyl). Exemplary alkyl groups include, but are not limited to,
methyl, ethyl, n-
propyl, 2-propyl, tert-butyl, pentyl and the like.
[0016] "Aryl" refers to a monovalent monocyclic or bicyclic aromatic
hydrocarbon moiety of
6 to 10 carbon ring atoms. Unless indicated otherwise, an aryl group can be
substituted with one
CA 02901353 2015-08-14
WO 2014/113022 PCT/US2013/022213
or more substituents, such as one, two or three substituents; or one or two
substituents selected
from alkyl, haloalkyl, nitro and halo. More specifically the term aryl
includes, but is not limited
to, phenyl, 1-naphthyl, and 2-naphthyl, and the like, each of which is
optionally substituted with
one or more substituent(s) noted above.
[0017] "Chiral" or "chiral center" refers to a carbon atom having four
different substituents.
However, the ultimate criterion of chirality is non-superimposability of
mirror images.
[0018] The terms "CPTA" and "halofenic acid" are used interchangeably
herein and refer to
(4-chlorophenyl)(3-trifluoromethylphenoxy)acetic acid or 4-chloro-a-(3-
trifluoromethylphenoxy)phenylacetic acid. The (-) optical isomer of CPTA has
an R
configuration at the chiral center, and the (+) optical isomer of CPTA has an
S configuration at
the chiral center.
[0019] "Enantiomeric mixture" refers to a chiral compound having a mixture
of enantiomers,
including a racemic mixture.
[0020] "Enantiomerically enriched" refers to a composition where one
enantiomer is present
in a higher amount than the other enantiomer.
[0021] "Enantiomeric excess" or "% e.e." refers to the amount of difference
between the first
enantiomer and the second enantiomer. Enantiomeric excess is defined by the
equation: % e.e. =
(% of the first enantiomer)-(% of the second enantiomer). Thus, if a
composition comprises 98%
of the first enantiomer and 2% of the second enantiomer, the enantiomeric
excess of the first
enantiomer is 98%-2% or 96%.
[0022] The terms "halogen", "halide" and "halo" are used interchangeably
herein and refer to
F, Cl, Br and I. In one aspect, halogen refers to F and Cl.
[0023] "Haloalkyl" refers to alkyl group as defined herein in which one or
more hydrogen
atoms have been replaced with halogen(s), including perhaloalkyls, such as
trifluoromethyl.
[0024] "Halofenate" refers to 2-acetamidoethyl 4-chlorophenyl-(3-
trifluoromethyl-
phenoxy)acetate (i.e., 4-chloro-a-(3-(trifluoromethyl)phenoxy)benzeneacetic
acid, 2-
(acetylamino)ethyl ester or (4-chlorophenyl)(3-trifluoromethylphenoxy)acetic
acid), 2-
(acetylamino)ethyl ester).
[0025] Unless otherwise stated, the term "phenyl" refers to an optionally
substituted phenyl
group. Suitable phenyl substituents are same as those described in the
definition of "aryl."
Similarly, the term "phenoxy" refers to a group of the formula -0Ara, wherein
Ara is phenyl as
6
CA 02901353 2015-08-14
WO 2014/113022 PCT/US2013/022213
defined herein. Thus, the term "ct-(phenoxy)phenylacetic acid" refers to
acetic acid that is
substituted on the 2-position with an optionally substituted phenyl and
optionally substituted
phenoxy moieties.
[0026] As used herein, the term "treating", "contacting" or "reacting"
refers to adding or
mixing two or more reagents under appropriate conditions to produce the
indicated and/or the
desired product. It should be appreciated that the reaction which produces the
indicated and/or
the desired product may not necessarily result directly from the combination
of two reagents
which were initially added, i.e., there may be one or more intermediates which
are produced in
the mixture which ultimately leads to the formation of the indicated and/or
the desired product.
[0027] Many organic compounds exist in optically active forms, i.e., they
have the ability to
rotate the plane of plane-polarized light. In describing an optically active
compound, the
prefixes R and S are used to denote the absolute configuration of the molecule
about its chiral
center(s). The prefixes "d" and "1" or (+) and (-) are employed to designate
the sign of rotation
of plane-polarized light by the compound, with (-) or (1) meaning that the
compound is
"levorotatory" and with (+) or (d) is meaning that the compound is
"dextrorotatory". There is no
correlation between nomenclature for the absolute stereochemistry and for the
rotation of an
enantiomer. For a given chemical structure, these compounds, called
"stereoisomers," are
identical except that they are mirror images of one another. A specific
stereoisomer can also be
referred to as an "enantiomer," and a mixture of such isomers is often called
an "enantiomeric" or
"racemic" mixture. See, e.g., Streitwiesser, A. & Heathcock, C. H.,
INTRODUCTION TO
ORGANIC CHEMISTRY, 2nd Edition, Chapter 7 (MacMillan Publishing Co., U.S.A.
1981).
[0028] The terms "substantially free of its (+)-stereoisomer,"
"substantially free of its (+)-
enantiomer," are used interchangeably herein and mean that the compositions
contain a
substantially greater proportion of the (-)-isomer in relation to the (+)-
isomer. In one
embodiment, the term "substantially free of its (+) stereoisomer" means that
the composition is at
least 90% by weight of the (-)-isomer and 10% by weight or less of the (+)-
isomer. In another
embodiment, the term "substantially free of its (+)-stereoisomer" means that
the composition
contains at least 99% by weight of the (-)-isomer and 1% by weight or less of
the (+)-isomer. In
another embodiment, the term "substantially free of its (+)-stereoisomer"
means that the
composition contains greater than 99%, 99.5%, 99.8% or 99.9% by weight of the
(-)-isomer.
These percentages are based upon the total amount of isomers in the
composition.
7
CA 02901353 2015-08-14
WO 2014/113022 PCT/US2013/022213
[00291 In one embodiment, the present application discloses a condensation
reaction of (-)-
CPTA with N-acetylethanolamine to provide (-)-halofenate in high chemical
yields, high chiral
purity and provides a favorable by-product profile. In one aspect of the
present application, the
condensation reaction to provide (-)-halofenate may be performed in a single
step.
[0030] Coupling reactions using propylphosphonic anhydride (T3P) under a
variety of
conditions did not provide the desired ester in high yields. It is known that
the by-product of
T3P can be removed by simple washing with water. Ratios of reagents to (-)-
CPTA and
different amount of solvents were studied. Reactions were monitored by LC-MS
and HPLC.
Chiral HPLC methods were used to measure chiral purity (which may be expressed
as
enantiomeric excess). The coupling reaction could be driven to completion
using T3P under
optimized conditions, such as increasing the reaction stoichiommetry,
increasing the
concentration of the reactants and elevated reaction temperatures. However,
upon completion of
the reaction, epimerization of the chiral center invariably resulted under the
reaction conditions.
Quenching the reaction at 0 C still resulted in a 50-50 racemic mixture.
Bases, such as
triethylamine, N,N-diethylisopropylamine and N-methylmorpholine, that have
been used in the
coupling reaction was thought to be the cause of the racemization. However, in
the absence of a
base, these coupling reactions did not proceed to completion.
[0031] Various other coupling reagents were investigated in the coupling
reaction. Base is
needed for the reactions when phosphonium (BOP, PyBOP and PyA0P), aminium
(HBTU,
TBTU, HATU, TATU and HCTU), uronium (TSTU, TNTU, TOTU, TPTU and TDBTU) and
the miscellaneous coupling reagents (CIB, CIP, TCFH and DEPBT) were used.
Carbodiimides
(DCC, DIC and EDC) and imidazolium (CDI) mediated coupling reactions preceded
without
base. Under various conditions attempted, the CDI coupling reactions did not
go to completion.
The name of the specific coupling agents are as follows:
BOP Benzotriazole-1-yl-oxy-tris-(dimethylamino)-phosphonium
hexafluorophosphate
PyBOP Benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate
PyAOP (7-Azabenzotriazol-1-yloxy)tripyrrolidinophosphonium
hexafluorophosphate
HBTU 0-Benzotriazole-N,N,N',N'-tetramethyl-uronium-hexafluoro-phosphate
TBTU 2-(1H-Benzotriazole-1-y1)-1,1,3,3-tetramethyluronium
tetrafluoroborate
HATU 2-(1H-7-Azabenzotriazol-1-y1)--1,1,3,3-tetramethyl uronium
hexafluorophosphate
Methanaminium
TATU 0-(7-Azabenzotriazole-1-y1)-N,N,N,N1-tetramethyluronium
tetrafluoroborate
8
CA 02901353 2015-08-14
WO 2014/113022 PCT/US2013/022213
HCTU 2-(6-Chloro-1H-benzotriazole-1-y1)-1,1,3,3-tetramethylaminium
hexafluorophosphate
TSTU N,N,N',N'-Tetramethy1-0-(N-succinimidyeuronium tetrafluoroborate
TNTU 0-(5-Norbornene-2,3-dicarboximido)-N,N,N',N'-tetramethyluronium
tetrafluoroborate
TOTU 0-[(Ethoxycarbonyl)cyanomethylenamino1-N,N,N',N'-tetramethyluronium
tetrafluoroborate
TPTU 0-(2-0xo-1(2H)pyridy1)-N,N,N',N'-tetramethyluronium tetrafluoroborate
TDBTU N,N,1\11,N1-Tetramethyl-0-(3,4-dihydro-4-oxo-1,2,3-benzotriazin-3-
yl)uranium
tetrafluoroborate
CIB 2-Chloro-1,3-dimethylimidazolidinium tetrafluoroborate
CIP 2-Chloro-1,3-dimethylimidazolidinium hexafluorophosphate
TCFH N,N,N',N'-Tetramethylchloroformamidinium-hexafluorophosphate
DEPBT 3-(Diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one
DCC N,N'-dicyclohexylcarbodiimide
DIC N,N'-diisopropylcarbodiimide
EDC 1-Ethy1-3-(3-dimethylaminopropyl) carbodiimide
CDI Carbodiimidazole
[0032] Typically, the coupling reactions using DCC require a base such as 4-
(N,N-
dimethylamino)pyridine (DMAP). However, under certain conditions, the coupling
reaction of
(-)-halofenic acid with N-acetylethanolamine and DCC resulted in complete
conversion to (-)-
halofenate without the use of DMAP.
[0033] Representative chiral purities (% e.e.) of the coupling products (as
crude reaction
mixtures) using DCC, DIC and EDC (with and without DMAP) were obtained as
follows:
% e.e. % e.e.
Coupling Agent/Conditions
With DMAP No DMAP
DCC (0 C) 99% 98%
DIC (room temp.) Not performed 96%
EDC (room temp.) Not performed 80%
[0034] In the DCC coupling reaction using a catalytic amount of DMAP, upon
the
completion of the reaction, the resulting crude reaction mixture was stored
over a period of time.
Storage of the crude reaction mixture resulted in slow the epimerization of
the (-)-halofenate.
However, when no DMAP was used in the coupling reaction, there was no further
epimerization
after the reaction completion. It was noted that the reaction with DMAP was
slightly faster than
the reaction where DMAP was absent. Upon the completion of the coupling
reaction, it was
9
CA 02901353 2015-08-14
WO 2014/113022
PCT/US2013/022213
observed that the reaction that was performed without DMAP gave slightly lower
chiral purity
(98 % e.e.) when compared to the coupling reaction performed with DMAP, which
afforded a
chiral purity of 99 % e.e. However, one advantage of performing the reaction
without DMAP
was that no further epimerization of the resulting product occurring after the
reaction was
completed such that immediate work-up was not required. Accordingly, DCC may
be used in
the absence of a base in the ester formation step for the synthesis of (-)-
halofenate.
[0035] We
also studied the effect of the ratio of the coupling reagent to (-)-CPTA on
the
reaction. A scale-up reaction of 0.1 mol scale of a DCC mediated (-)-CPTA
coupling reaction
was carried out using the conditions described above. An LC-MS analytical
method was
developed to determine the residual DCU, a by-product of DCC, in the crude and
final product.
It was determined that DCU was present at 0.8% in the crude product, and was
reduced to 0.48%
in the final isolated product after a single recrystallization from
diisopropylether.
Examples
Starting materials and reagents:
(-)-CPTA, Cilag, Lot# 07B2213 (>99% e.e.)
N-Acetylethanolamine, Cilag, waxy solid, Lot# 07C4811
N-Acetylethanolamine, TCI, viscous liquid, catalog number: A0075, Lot# FI01
DCC, Aldrich, 99%, Lot# 13896KMV
DCC Coupling Reactions:
(-)-CPTA DCC N-Acetylethanolamine DMAP Toluene DMA
3.3g 2.27g 2.1 g 2.5 mg 5 + 5 mL 2 mL
1.0 eq 1.1 eq 2 eq O.2%
[0036] DCC in toluene (5 mL) was added to a solution containing (-)-CPTA
and N-
acetylethanolamine in toluene (5 mL) and DMA (2 mL) via a syringe pump over 20
min at 0 C,
and the resulting mixture was stirred at 0 C for 3 hours.
Analytical 3 hours (0 C) 1 day, rt 2 day,
rt
Yield (halofenate) > 99% > 99% > 99%
Chiral Purity ((-)-halofenate) 99% e.e. 97% e.e. 92.6%
e.e.
CA 02901353 2015-08-14
WO 2014/113022
PCT/US2013/022213
[0037] The above representative result demonstrates that the coupling
reaction using DCC
and DMAP provides the desired coupling product. Epimerization of the chiral
center occurred
slowly over time while the product remained in the reaction mixture. Crude
solid from
toluene/cyclohexane: 3.45 g, 94.8% e.e. Crystallization from diisopropylether:
2.4 g (recovery
yield 70% from crude solid), 96.4% e.e.
[0038] When the coupling reaction was performed with dimethylacetamide
(DMA), the
coupling product was driven to completion, and the product was obtained in
high chemical yield,
high chiral purity. However, it was determined that the coupling reaction may
also be performed
without the presence of DMA as a solvent, co-solvent or as an additive to the
reaction, and the
resulting product was obtained in similar chemical yield and chiral purity.
DCC Coupling Reaction without DMA:
(-)-CPTA DCC N-Acetylethanolamine DMAP Toluene
3.3 g 2.27 g 2.1 g 2.5 mg 5 + 5 mL
1.0 eq 1.1 eq 2 eq O.2%
[0039] DDC in toluene (5 mL) was added to the solution via a syringe pump
in 20 min at 0
C and the mixture was stirred at 0 C for 3 hours.
Analytical 30 min (0 C)
Yield (halofenate) > 99%
Chiral Purity ((-)-halofenate) 99% e.e.
[0040] DCC coupling reaction without DMAP
(-)-CPTA DCC N-Acetylethanolamine Base
Toluene
3.3g 2.27g 2.1 g None 7 + 5
mL
1.0 eq 1.1 eq 2 eq None
[0041] DCC in toluene (5 mL) was added to the solution via a syringe pump
in 20 min at 0
C and the mixture was stirred at 0 C for 3 hours and at room temperature.
Analytical 30 min (0 C) 1.5 h (0 C) 17 h (rt) 4 days
Yield (halofenate) 98.9% > 99% > 99% Not
performed
Chiral Purity
> 98% e.e. Not performed 98% e.e. 98%
e.e.
((-)-halofenate)
11
CA 02901353 2015-08-14
WO 2014/113022 PCT/US2013/022213
DCC with lower ratio of reagents:
(-)-CPTA DCC N-Acetylethanolamine Toluene
3.3g 2.17g 1.55g 15 mL + 5 mL
1.0 eq 1.05 eq 1.5 eq
[0042] DCC in toluene (5 mL) was added to the solution via a syringe pump
in 20 min at 0
C and the mixture was stirred at 0 C for 3 hours and at room temperature.
Analytical 1.5 h (0 C)
Yield Halofenate > 99%
Chiral Purity ((-)-halofenate) > 98% e.e.
[0043] DCC using N-Acetylethanolamine from TCI
(-)-CPTA DCC N-Acetylethanolamine
Toluene
3.3g 2.17g 1.55 g 15 mL + 5 mL
1.0 eq 1.05 eq 1.5 eq
[0044] DDC in toluene (5 mL) was added to the solution via a syringe pump
in 20 min at 0
C and the mixture was stirred at 0 C for 3 hours and at room temperature.
Analytical 1.5 h (0 C)
Yield (halofenate) > 99%
Process Summary:
[0045] Scheme 1 illustrates a general method of preparing compounds of
formula (III).
Scheme 1
X3C * X3C *
=
=
OH ROH _____________________________________ )1.= OR
x' 1101 0 x' 1101 0
12
CA 02901353 2015-08-14
WO 2014/113022 PCT/US2013/022213
[0046] In one embodiment, the process for the preparation of the compound
of the formula
(III), such as (R)-2-acetamidoethyl 2-(4-chloropheny1)-2-(3-
(trifluoromethyl)phenoxy)acetate,
may be performed by initially dissolving DCC in an aprotic solvent, such as
toluene. Separately,
a solution of ROH, such as N-acetylethanolamine, is dissolved in an aprotic
solvent, such as
toluene, and the N-acetylethanolamine solution is mixed with a solution of a
compound of the
formula (Ia) wherein X and X' are as defined herein, such as (-)-CPTA, in an
aprotic solvent,
such as toluene. Alternatively, a solution of a compound of the formula Ia,
such as (-)-4-chloro-
phenyl-(3-trifluoromethyl-phenoxy)-acetic acid is prepared in an aprotic
solvent, such as toluene,
and the compound of the formula ROH, wherein R is as defined herein, is then
added to the
mixture. The solution of DCC is then added to the solution comprising the
compound of the
formula (Ia) with the compound of the formula ROH.
[0047] In one aspect, the addition of the solution of DCC to the solution
comprising the
compound of formula (Ia) with ROH is performed below room temperature, such as
below about
15 C, below 10 C, below 5 C or about 0 C for about 1 hour, 2 hours, 3
hours, 5 hours or more.
In one variation, the addition of the DCC solution is performed at about 0 C
for about 1.5 hours
or until all of the DCC solution is added.
[0048] In another aspect, the resulting solution or suspension is stirred
at below room
temperature, such as about 10 C or 0 C, for at least about 3 hours, 5 hours,
7 hours, about 12
hours or more, until the reaction is determined to be complete. Reaction
completion may be
monitored by chromatographic methods, such as by TLC, HPLC or other
spectroscopic methods.
The precipitated DCU by-product may be removed from the solution using
standard methods,
such as filtration over Whatman paper, filtration on a Buchner funnel,
optionally with silica gel
and/or celite to remove the DCU. Depending on the solvent or solvent mixtures
that are used in
the process, more than one filtration steps to remove DCU may require as the
DCU precipitates
out over time. In one variation, the DCU cake is washed with a solvent, such
as toluene. To the
filtrate contain the product is added an organic solvent, such as cyclohexane,
and the resulting
solution is washed with water, and water is separated from the organic
solution. Residual water
is then removed from the organic solution by drying, such as the use of drying
agent (sodium
sulfate, magnesium sulfate etc.) and or by azeotropic distillation of the
cyclohexane-toluene
solution.
13
CA 02901353 2015-08-14
WO 2014/113022 PCT/US2013/022213
[0049] A solid compound of formula (III), such as (R)-2-acetamidoethyl 2-(4-
chloropheny1)-
2-(3-(trifluoromethyl)phenoxy)acetate, may be obtained from solution. In one
variation, to the
reaction solution is added an organic solvent, such as cyclohexane, and the
resulting mixture is
heated to above room temperature, such as about 35 C, 37 C or about 40 C.
Optionally, the
resulting solution is seeded with crystals of the desired product, such as
crystals of (R)-2-
acetamidoethyl 2-(4-chloropheny1)-2-(3-(trifluoromethyl)phenoxy)acetate, and
mixed at the
same temperature for at least about 1 hour, 2 hours, 3 hours or about 4 hours
or more, and the
mixture is allowed to cool slowly to room temperature or below room
temperature, such for at
least about 1 hour, 2 hours, 3 hours or about 4 hours or more, and the
resulting crystals are
filtered and washed with a cold (about 10 C or colder) solvent such as
cyclohexane. The
crystalline product may be dried to afford the desired product.
[0050] Different reaction work up conditions, crystallization conditions
and isolation
conditions using various modifications of the processes as described herein
allow the isolation of
(-)-halofenate in greater than about 80% yield, greater than about 85% yield,
greater than about
90% yield, greater than about 95% yield and greater than about 97% yield. In
various aspects,
the above described processes allow the isolation of (-)-halofenate at about
98% e.e., about 99%
e.e., about 99.5% e.e., about 99.9% e.e. or greater. In various aspects, the
above described
processes allow the isolated (-)-halofenate to have a DCU level of about 1%,
about 0.5%, about
0.4%, about 0.3%, about 0.2%, about 0.1%, about 0.05% or about 0.01% or less.
In certain
aspects, these e.e. and chemical purity levels are obtained before further
purification (e.g.
recrystallization) of the compound. In one aspect of the process described
herein, the product
obtained, (-)-halofenate, is substantially free of its (+)-stereoisomer.
[0051] It was determined that, depending on the reaction conditions and the
solvent or
solvent mixtures employed for each isolation step, DCU may be precipitated out
of the solution
at different rates over time, and upon precipitation, DCU may be isolated from
the desired
solution containing the product by a filtration process.
[0052] DCC 0.1 mol of CPTA reaction, using N-acetylethanolamine from TCI
Preparation of (R)-2-acetamidoethyl 2-(4-chloropheny1)-2-(3-
(trifluoromethyl)phenoxy)acetate
14
CA 02901353 2015-08-14
WO 2014/113022 PCT/US2013/022213
0
Fõ 0 0
1
OS-NÄ
H 01 0
CI II
_
[0053] DCC (22.7 g, 110 mmol) was dissolved in toluene (50 mL) at 20 C. To
a 500 mL of
round-bottom flask were added N-acetylethanolamine (15.5 g, 150 mml), toluene
(150 mL) and
(-)-4-chloro-phenyl)-(3-trifluoromethyl-phenoxy)-acetic acid (33.1 g, 100
mmol). The mixture
was stirred at 20 C until a clear solution was formed. The solution was
cooled with an ice-water
bath. To the solution was added DCC in toluene solution dropwise at 0 C in 90
min. The
resulted suspension was stirred at 0 C for 3 hours and then 20 C over night.
Dicyclohexylurea
(DCU, obtained as by product) was filtered off and washed with toluene (25 mL)
two times. To
the filtrate (containing the desired product, (-)-halofenate) was added
cyclohexane (100 mL).
The solution was washed with water (100 mL) three times. The residual water
was removed by
azeotropic distillation with cyclohexane and toluene (about 180 mL). To the
resulting solution
was added cyclohexane (200 mL) at 40 C and seeded with (-)-halofenate (33
mg). The resulting
suspension was stirred at 40 C for 2 hours, then cooled to 10 C over 1 hour
and stirred for an
additional 1 hour. The precipitated crystals were filtered and washed with
cold (<10 C)
cyclohexane. The wet product was dried under vacuum over night to yield the
title compound as
a white solid. Yield: 35.4 g (80.5 %), % e.e. 99.5 %; chemical purity 98.6%
(DCU 0.80 %).
[0054] The dry solid (35 g) was recrystallized from diisopropylether (350
mL) to give the
title compound as a white crystalline solid. Yield: 28.5 g (81.4 %), e.e. 99.9
%; chemical purity
99.3 % (DCU 0.48%).
[0055] While the foregoing description describes specific embodiments,
those with ordinary
skill in the art will appreciate that various modifications and alternatives
can be developed.
Accordingly, the particular embodiments and examples described above are meant
to be
illustrative only, and not to limit the scope of the invention, which is to be
given the full breadth
of the appended claims, and any and all equivalents thereof.