Note: Descriptions are shown in the official language in which they were submitted.
ABEXINOSTAT SALT, ASSOCIATED CRYSTALLINE FORM,
PREPARATION METHOD THEREOF
AND THE PHARMACEUTICAL COMPOSITIONS CONTAINING SAME
The present invention relates to N-hydroxy-4- {2-[3-(N,N-
dimethylaminomethyl)-
benzofuran-2-ylcarbonylaminolethoxy 1 benzamide tosylate, or a solvate
thereof.
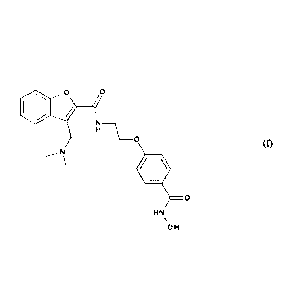
Alternatively, the subject-matter of the invention relates to a tosylate salt
of abexinostate
of formula (I):
H
_____________________________________ 0
11)-
=0
Htst\
OH
More particularly, the invention is directed to the salt of formula (II):
H3C SO3H
H
\ _____________________________________ 0
(II)
=0
HN
OH
The present invention relates also to the crystalline form I of N-hydroxy-4-
{243-(N,N-
dimethy laminomethyl)benzofuran-2-y lc arbony lamino] ethoxy 1 benzami de
tosylate, to the
preparation method thereof and also to the pharmaceutical compositions
containing same.
N-hydroxy-4- {2- [3-(N,N-dimethylaminomethyl)benzofuran-2-
ylcarbonylaminolethoxy 1-
benzamide, also known as abexinostate, is a histone deacetylase (HDAC)
inhibitor
- 1 -
Date Recue/Date Received 2020-09-10
CA 02903369 2015-09-01
described in patent application W02004/092115. It allows inhibition of cell
growth and
induces apoptosis in cultured tumour cells in vitro, and it inhibits tumour
growth in vivo
in xenograft models (Buggy et al., Mol. Cancer Ther 2006 5(5) 1309). In view
of its
pharmacological profile, abexinostate is intended for use in the treatment of
cancer.
From the industrial point of view it is imperative to be able to synthesize
the compound
with excellent purity, especially in a perfectly reproducible form, having
valuable
characteristics of dissolution, filtration, drying, ease of formulation and
stability allowing
its prolonged storage without particular requirements for temperature, light,
humidity or
oxygen levels.
Patent application W02004/092115 describes two different routes for obtaining
abexinostate. In both cases, 3-methyl-benzofuran-2-carboxylic acid is used as
starting
material, but functionalisation of this central nucleus by the
dimethylaminomethyl group
in the 3-position is carried out at different stages in the synthesis process,
namely before
or after coupling of the benzofuran-2-carboxylic acid derivative with methyl 4-
(2-
aminoethoxy)benzoate. Obtaining abexinostate hydrochloride is specifically
described in
the W02004/092115 application. However, using this salt on an industrial scale
is
delicate because of its hygroscopic properties.
The present invention describes a process for obtaining abexinostate tosylate
(abexinostate 4-methylbenzenesulfonate) in a well-defined, perfectly
reproducible
crystalline form, and presenting a very good stability compatible with the
industrial
constraints of preparation (especially drying) and storage of pharmaceutical
compositions.
The crystalline form I of abexinostate tosylate is characterised by an powder
diffraction
pattern X having the following diffraction lines (Bragg's angle 2 theta,
expressed in
degrees +0.2 ): 6.50; 9.94; 11.35; 12.33; 14.08; 18.95; 21.08; 27.05. Even
more
particularly, the crystalline form I of abexinostate tosylate is characterised
by the
following diffraction lines: 6.50; 9.94; 11.35; 12.33; 14.08; 18.95; 19.61;
19.96; 21.08;
22.82; 23.61; 27.05.
More specifically, crystalline form I of abexinostate tosylate is
characterised by the
powder diffraction pattern X hereinbelow, measured using a F'ANalytical X1Pert
Pro
- 2 -
CA 02903369 2015-09-01
MPD diffractometer with an X'Celerator detector and e#ressed in terms of line
position
(Bragg's angle 2 theta, expressed in degrees 0.2 ) and interplanar distance d
(expressed
in A):
Angle 2-theta Interplanar
Line no.
(degrees) distance (A)
1 6.50 13.581
2 9.94 8.894
3 11.35 7.789
4 12.33 7.173
14.08 6.285
6 18.95 4.683
7 19.61 4.526
8 19.96 4.449
9 21.08 4.215
22.82 3.897
11 23.61 3.768
12 27.05 3.296
Furthermore, the crystalline form I of abexinostate tosylate has been
characterised by
5 Raman spectroscopy. Significant peaks were observed at the following
positions: 940 cm-
, 1088 cm-I, 1132 cm', 1242 cm-I, 1360 cm-I, 1608 cm1
.
Alternatively, the crystalline form I of abexinostate tosylate may be
characterised by the
powder diffraction pattern X which includes the 12 significant lines presented
previously
and also by a Raman spectrum having a significant peak at the position 1608
cm*
10 Finally, the crystalline form I of abexinostate tosylate has also been
characterised by
solid-state NMR spectroscopy. Significant peaks were observed at 121.2 ppm,
122.1
ppm, 123.5 ppm, 126.0 ppm, 126.8 ppm, 128.2 ppm, 128.9 ppm, 143.4 ppm, 144.6
ppm,
153.8 ppm, 159 ppm, 161.2 ppm and 162.1 ppm.
More specifically, the 13C CP/MAS (Cross Polarization Magic Angle Spinning)
spectra
have the following peaks (expressed in ppm 0.2 ppm):
- 3 -
CA 02903369 2015-09-01
Chemical shift Chemical shift
Peak no. Peak no.
(PPI11) (PPm)
1 162.1 10 126.0
2 161.2 11 123.5
3 159.0 12 122.1
4 153.8 13 121.3
144.6 14 65.9
6 143.4 15 50.6
7 128.9 16 46.9
8 128.2 17 45.0
9 126.8 18 21.9
The invention relates also to a preparation method of crystalline form I of
abexinostate
tosylate, characterised in that abexinostate is crystallised from a polar
medium in the
presence of para-toluenesulphonic acid. Preferably, the polar medium is
composed of one
or more solvents selected from water, alcohols, ketones and esters, it being
understood
5 that:
- "alcohols" means the C1-C6 alcohols such as methanol, ethanol, propanol,
isopropanol, butanol, isobutanol, pentanol, 2-pentanol, 3-pentanol,
isopentanol,
hexanol,
- "ketones" means the C3-C6 ketones such as acetone, methyl ethyl ketone, 2-
pentanone, 3-pentanone, 3-methyl-2-butanone, 2-hexanone, 3-hexanone, ethyl
isopropyl ketone, methyl isopropyl ketone, 2,2-dimethy1-3-butanone,
- "esters" means the C3-C8 esters such as ethyl formate, isopropyl formate,
ethyl
acetate, propyl acetate, isopropyl acetate, butyl acetate, isobutyl acetate,
tert-butyl
acetate, pentyl acetate, isopentyl acetate, hexyl acetate.
Preferred alcohols are ethanol and isopropanol. Among the preferred solvents
preference
will also be given to acetone and methyl ethyl ketone among the ketones and to
ethyl
acetate among the esters.
Alternatively, the polar medium is a binary mixture, one constituent of which
is water.
Even more preferably, the polar medium is a binary mixture selected among:
acetone/water, ethanol/water, isopropanol/water, and methyl ethyl
ketone/water.
- 4 -
CA 02903369 2015-09-01
In the crystallisation process according to the invention, abexinostate (free
base) obtained
by any process may be used.
The invention relates also to another preparation method of the crystalline
form I of
abexinostate tosylate, in which process the crystallisation is seeded using a
very small
amount of the crystalline form I of abexinostate tosylate.
In this second crystallisation process according to the invention,
abexinostate (free base)
obtained by any process may also be used.
Obtaining the crystalline form I of abexinostate tosylate has the advantage of
making it
possible to prepare pharmaceutical formulations having a consistent and
reproducible
composition, presenting good characteristics of dissolution and stability,
which is
especially advantageous when the formulations are intended for oral
administration. More
specifically, use of the crystalline form I of abexinostate tosylate is
especially valuable in
an industrial context in view of its low hygroscopicity.
The crystalline form I of abexinostate tosylate is intended for the treatment
of cancer,
more particularly for the treatment of a carcinoma, a tumour, a neoplasm, a
lymphoma, a
melanoma, a glioma, a sarcoma, or a blastoma.
The invention relates also to the pharmaceutical compositions comprising, as
active
ingredient, a tosylate salt of abexinostate, even more particularly the
crystalline form I of
abexinostate tosylate, together with one or more appropriate, non-toxic, inert
excipients.
Among the pharmaceutical compositions according to the invention those that
are suitable
for oral, parenteral (intravenous or subcutaneous) or nasal administration,
tablets or
dragees, granules, sublingual tablets, capsules, lozenges, suppositories,
creams,
ointments, dermal gels, injectable preparations, drinkable suspensions and
chewing gums
can be more particularly mentioned.
Preference is given to pharmaceutical compositions administered orally.
The useful dosage varies according to the sex, age and weight of the patient,
the
administration route, the nature of the cancer and any associated treatments;
and the
- 5 -
CA 02903369 2015-09-01
useful dosage ranges from 20 mg to 480 mg of N-hydroxy-4-{2-[3-(N,N-
d m ethyl am i nom ethyl)benzofuran-2-ylcarbonylam ino]ethoxyl benzamide
per day
expressed in terms of the free base.
The Examples below illustrate the invention but do not limit it in any way.
Example I: Process for obtaining the crystalline form I of abexinostate
tosylate
1.66 kg of abexinostate (free base) are placed in 9.48 kg of a mixture of
isopropanol/water (50/50 weight/weight) at ambient temperature. The para-
toluenesulphonic acid monohydrate (0.83 kg) is added in 2.36 kg of water at
ambient
temperature. The mixture is then heated at 75 C for 30 minutes before being
cooled to
to 0 C. When crystallisation is complete, the suspension is filtered at 20
C. After drying, the
crystalline form I of abexinostate tosylate is obtained with a yield of about
85 % and a
purity greater than 99 %. The solid was characterised by the powder
diffraction pattern X,
Raman spectrum and NMR spectrum as set out in the following Examples 3-6.
Example 2: Process for ohtaining the crystalline form I of abexinostate
tosylate
(seeding)
33.9 kg of abexinostate (free base) are placed in 170 kg of a mixture of
isopropanol/water
(45.6/54.4 weight/weight) at ambient temperature. A solution composed of para-
toluenesulphonic acid monohydrate (17.06 kg) in water (24.1 kg) is added. The
medium
is then heated at 70-75 C, cooled, and seeded with 1.935 kg of crystalline
form I of
abexinostate tosylate. The suspension is then filtered at 20 C. After drying,
the crystalline
form I of abexinostate tosylate is obtained with a yield of about 86 % and a
purity greater
than 99 %. The solid was characterised by the powder diffraction pattern X,
Raman
spectrum, and NMR spectrum as set out in the following Examples 3-6.
- 6 -
CA 02903369 2015-09-01
Example 3: The crystalline form I of abexinostate tosylate (powder diffraction
pattern X)
Recording of the data was carried out using a PANalytical X'Pert Pro MPD
diffractometer
with an X'Celerator detector under the following conditions:
- Voltage 45 kV, current 40 mA,
- Mounting: theta/theta,
- Anode: copper,
- K alpha-1 wavelength: 1.54060 A,
- K alpha-2 wavelength: 1.54443 A,
- K alpha-2/K alpha-1 ratio: 0.5,
- Measurement mode: continuous from 3 to 55 (Bragg's angle 2 theta) in
increments
of 0.017 ,
- Measurement time per step: 35.53 s.
The powder diffraction pattern X of the form 1 of abexinostate tosylate
obtained
according to the process of Example 1 or 2 is expressed in terms of line
position (Bragg's
angle 2 theta, expressed in degrees 0.2 ), interplanar distance (expressed in
A) and
relative intensity (expressed as a percentage relative to the most intense
line). The
significant lines have been collated in the following table:
Relative
Angle 2-theta Interplanar
Line no. intensity
(degrees) distance (A)
(%)
1 6.50 13.581 75.6
2 9.94 8.894 58.4
3 11.35 7.789 19.1
4 12.33 7.173 23.7
5 14.08 6.285 33.1
6 18.95 4.683 100
7 19.61 4.526 53.9
8 19.96 4.449 50.9
9 21.08 4.215 93.5
10 22.82 3.897 28.5
11 23.61 3.768 32.6
12 27.05 3.296 16.0
- 7 -
CA 02903369 2015-09-01
Example 4: Crystalline form I of abexinostate tosylate (crystal unit cell)
A saturated solution of abexinostate tosylate in 2,2,2-trifluoroethanol is
prepared by
stirring a suspension for 24 hours at ambient temperature, followed by
filtration. 1 mL of
the resulting solution is then poured into a 1.8-mL HPLC vial, to which 0.25
mL of water
is added. The solution is maintained at ambient temperature for 75 minutes.
After
centrifuging and then drying, the solid is isolated for analysis. From among
the crystals
obtained a crystal of sufficient quality is taken for single-crystal X-ray
diffraction
analysis.
The crystalline structure of the above single crystal was determined using a
Bruker Kappa
CCD diffractometer equipped with an FR590 generator having a molybdenum
anticathode (XMoKa 1 = 0.7093 A) with an angular range from 2 to 27.5 in
terms of 0.
The following parameters were established:
- crystal unit cell: triclinic
- unit cell parameters: a = 10.467 A, b = 14.631 A, c = 20.159 A, a = 73.971 .
13 = 79.040 , = 72.683
- space group: P -1
- number of molecules in the unit cell: 4
- volume of the unit cell: Vurnt cell = 2813.0 A3
- density: d = 1.345 g/cm3.
Example 5: Crystalline form I of abexinostate tosylate (Raman spectrum)
The form I of abexinostate tosylate was characterised by Raman spectroscopy.
The
spectra were recorded in diffuse reflectance mode (Raman Station 400,
PerkinElmer)
using a 785 nm laser. The signal was recorded by a CCD detector. The
wavelength shift
depends on the material and is characteristic of that material, which allows
analysis of the
chemical composition and of the molecular arrangement of the sample studied.
The
spectra were acquired with maximum power (100 % laser capacity), a spot size
of
100 i.tm, twenty exposures of 2 seconds and a spectral resolution of 2 cm-1.
The spectral
range explored ranges from 0 to 3278 cm-1.
- 8 -
CA 02903369 2015-09-01
Significant peaks were observed at the following positions: 940 cm-1, 1088 cm-
1,
1132 cm', 1242 cm11, 1360 cm', 1608 cm-1.
Example 6: Crystalline form I of abexinostate tosylate (solid NMR spectrum)
The form I of abexinostate tosylate was also characterised by solid-state NMR
spectroscopy. The 13C NMR spectra were recorded at ambient temperature using a
Bruker
SB Avance spectrometer with a 4-mm CP/MAS SB VTN type probe under the
following
conditions:
- Frequency: 125.76 MHz,
- Spectral width: 40 kHz,
- Magic angle spinning rate of sample: 10 kHz,
- Pulse sequence: CP (Cross Polarization) with SPINAL64 decoupling (decoupling
power: 80 kHz),
- Repetition delay: 10 s,
- Acquisition time: 35 ms,
- Contact time: 4 ms,
- Number of scans: 4096.
An apodisation function ("5 Hz line broadening") is applied to the collected
signal before
the Fourier transform. The spectra thereby obtained were referenced relative
to a sample
of adamantane (the highest-frequency peak of adamantane has a chemical shift
of
38.48 ppm).
The peaks observed have been collated in the following table (expressed in ppm
+
0.2 ppm):
Chemical shift Chemical shift
Peak no. Peak no.
(ppm) (ppm)
1 162.1 10 126.0
2 161.2 11 123.5
3 159.0 12 122.1
4 153.8 13 121.3
5 144.6 14 65.9
6 143.4 15 50.6
7 128.9 16 46.9
8 128.2 17 45.0
9 126.8 18 21.9
- 9 -
CA 02903369 2015-09-01
Example 7: Pharmaceutical composition
Formula for the preparation of 1000 tablets each containing 100 mg of
abexinostate
(expressed in terms of the base equivalent):
Abexinostate tosylate ........................................ 143.4 g
Lactose monohydrate ................................. 213.1 g
Magnesium stearate ........................................... 2.5 g
Maize starch ................................................. 75 g
Maltodextrin ................................................. 50 g
Anhydrous colloidal silica ................................... 1 g
....................................................... Sodium
carboxymethylcellulose 15 g
Example 8: Hygroscopicity
Hygroscopicity of the form I of abexinostate tosylate was assessed using
gravimetric
water vapor adsorption (DVS ¨ Dynamic Vapor Sorption). A sample of 5 to 10 mg
of the
.. drug substance, accurately weighed, was placed into a DVS sample pan
working at 25 C
under controlled humidity. The mass variation was recorded whilst drying under
0 per
cent RH (relative humidity) and during two subsequent cycles of increasing and
decreasing linear variations of relative humidity in the range 0-90 per cent
RH at a rate of
10 per cent per hour. The relative humidity was maintained constant when it
reached
either 0 or 90 per cent RH until the mass variation was less than 0.002 per
cent per minute
within a limit of time of 15 h.
An increase in weight lower than 0.5% was detected by DVS analysis when a
sample was
exposed to relative humidities included between 0% to 90% at 25 C.
- 10-