Note: Descriptions are shown in the official language in which they were submitted.
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
1
Combination of an EGFR T790M Inhibitor and an EGFR Inhibitor for the Treatment
of Non-Small Cell Lung Cancer
Field of the Invention
This invention relates to a method of treating non-small cell lung cancer by
administering a combination of an EGFR T790M inhibitor in combination with a
low-dose
amount of a panHER inhibitor. This invention also relates to a method of
treating non-
small cell lung cancer by administering a combination of an irreversible EGFR
T790M
inhibitor in combination with an EGFR inhibitor.
Background
Non-small cell lung cancer is the leading cause of cancer death worldwide,
with
an estimated 1.4 million new cases diagnosed each year. In lung
adenocarcinoma,
which is the most common form of non-small cell lung cancer, patients
harboring
mutations in the epidermal growth factor receptor (EGFR) constitute between 10-
30 %
of the overall population. It is this segment of patients for whom EGFR
inhibitors such as
erlotinib or gefitinib can be most effective (Paez et al. Science 2004; Lynch
et al. NEJM
2004; Pao et al, PNAS 2004). The most common activating mutations associated
with
good response to these inhibitors are deletions within exon 19 (e.g. E746-
A750) and
point mutations in the activation loop (exon 21, in particular, L858R).
Additional somatic
mutations identified to date but to a lesser extent include point mutations:
G719S,
G719C, G719A, L861 and small insertions in Exon 20 (Shigematsu et al JNCI
2005;
Fukuoka et al. JCO 2003; Kris et al JAMA 2003 and Shepherd et al NEJM 2004).
While these agents can be effective treatments for the EGFR mutant sub-
population, the majority of patients who initially respond develop resistance.
The
primary mechanism of resistance, observed in approximately 50 % of patients,
is due to
a second mutation (T790M) which occurs at the gatekeeper threonine residue
(Kosaka
et al CCR 2006; Balak et al CCR 2006 and Engelman et al Science 2007).
Improved therapies for the treatment of non-small cell lung cancer comprise a
large unmet medical need and the identification of novel combination regimens
are
required to improve treatment outcome.
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
2
Summary of the Invention
Each of the embodiments described below can be combined with any other
embodiment described herein not inconsistent with the embodiment with which it
is
combined. Furthermore, each of the embodiments described herein envisions
within its
scope pharmaceutically acceptable salts of the compounds described herein.
Accordingly, the phrase or a pharmaceutically acceptable salt thereof" is
implicit in the
description of all compounds described herein.
Some embodiments described herein relate to a method of treating non-small
cell
lung cancer comprising administering to a patient in need thereof an effective
amount of
an irreversible EGFR T790M inhibitor in combination with an effective amount
of an
EGFR inhibitor.
In further embodiments of the method of the present invention, the
irreversible
EGFR T790M inhibitor is 1-{(3R,4R)-3-[({5-chloro-2-[(1-methyl-1H-pyrazol-4-
yl)am ino]-
7H-pyrrolo[2,3-d]pyrim idin-4-ylloxy)methyI]-4-methoxypyrrolidin-1-yllprop-2-
en-1-one, or
a pharmaceutically acceptable salt thereof.
In some embodiments of the method of the present invention, the irreversible
EGFR T790M inhibitor is N-methyl-N-[trans-3-({2-[(1-methyl-1H-pyrazol-4-
yl)amino]-5-
(pyridin-2-yI)-7H-pyrrolo[2,3-d]pyrimidin-4-ylloxy)cyclobutyl]prop-2-enamide,
or a
pharmaceutically acceptable salt thereof.
In certain embodiments of the method of the present invention, the
irreversible
EGFR T790M inhibitor is N-[trans-3-({5-chloro-2-[(1,3-dimethyl-1H-pyrazol-4-
y1)amino]-
7H-pyrrolo[2,3-d]pyrim ino)cyclobutyI]-N-methylprop-2-enamide, or
a
pharmaceutically acceptable salt thereof.
In further embodiments of the method of the present invention, the
irreversible
EGFR T790M inhibitor is 1-{(3R,4R)-3-[({5-chloro-2-[(3-methoxy-1-methyl-1H-
pyrazol-4-
yl)am ino]-7H-pyrrolo[2,3-d]pyrimidin-4-ylloxy)methyI]-4-methoxypyrrolidin-1-
yllprop-2-
en-1-one, or a pharmaceutically acceptable salt thereof.
In embodiments of the method of the present invention, the EGFR inhibitor is
selected from the group consisting of gefitinib, erlotinib, icotinib,
vandetanib, lapatinib,
neratinib, afatinib, pelitinib, dacomitinib, canertinib, cetuximab and
panitumumab, or a
pharmaceutically acceptable salt thereof.
In further embodiments of the method of the present invention, the EGFR
inhibitor is selected from the group consisting of gefitinib, erlotinib,
icotinib, vandetanib,
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
3
lapatinib, neratinib, afatinib, pelitinib, dacomitinib, and canertinib, or a
pharmaceutically
acceptable salt thereof.
In some embodiments of the method of the present invention, the EGFR inhibitor
is selected from the group consisting of gefitinib, erlotinib, afatinib, and
dacomitinib, or a
pharmaceutically acceptable salt thereof.
In certain embodiments of the method of the present invention, the EGFR
inhibitor is gefitinib, or a pharmaceutically acceptable salt thereof.
In some embodiments of the method of the present invention, the EGFR inhibitor
is erlotinib, or a pharmaceutically acceptable salt thereof.
In embodiments of the method of the present invention, the EGFR inhibitor is
afatinib, or a pharmaceutically acceptable salt thereof.
In some embodiments of the method of the present invention, the EGFR inhibitor
is dacomitinib, or a pharmaceutically acceptable salt thereof.
In further embodiments of the method of the present invention, the EGFR
inhibitor is a reversible EGFR inhibitor.
In further embodiments of the method of the present invention, the reversible
EGFR inhibitor is selected from the group consisting of gefitinib, erlotinib,
icotinib,
vandetanib, and lapatinib, or a pharmaceutically acceptable salt thereof.
In embodiments of the method of the present invention, the reversible EGFR
inhibitor is gefitinib, or a pharmaceutically acceptable salt thereof.
In further embodiments of the method of the present invention, the reversible
EGFR inhibitor is erlotinib, or a pharmaceutically acceptable salt thereof.
In additional embodiments of the method of the present invention, the EGFR
inhibitor is an irreversible EGFR inhibitor.
In embodiments of the method of the present invention, the irreversible EGFR
inhibitor is selected from the group consisting of neratinib, afatinib,
pelitinib, dacomitinib,
and canertinib, or a pharmaceutically acceptable salt thereof.
In further embodiments of the method of the present invention, the
irreversible
EGFR inhibitor is afatinib, or a pharmaceutically acceptable salt thereof.
In some embodiments of the method of the present invention, the irreversible
EGFR inhibitor is dacomitinib, or a pharmaceutically acceptable salt thereof.
Some embodiments of the present invention relate to a method of treating non-
small cell lung cancer comprising administering to a patient in need thereof
an effective
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
4
amount of an EGFR T790M inhibitor in combination with a panHER inhibitor,
wherein
the panHER inhibitor is administered according to a non-standard clinical
dosing
regimen.
In further embodiments of the method of the present invention, the non-
standard
clinical dosing regimen is a non-standard clinical dose or a non-standard
dosing
schedule.
In some embodiments of the method of the present invention, the non-standard
clinical dosing regimen is a low-dose amount of the panHER inhibitor.
In embodiments of the method of the present invention, the non-standard
clinical
dosing regimen is an intermittent dosing regimen.
In further embodiments of the method of the present invention, the EGFR T790M
inhibitor is selected from the group consisting of Go6976, PKC412, AP26113,
HM61713,
VVZ4002, CO-1686 and TAS-2913, or a pharmaceutically acceptable salt thereof.
In further embodiments of the method of the present invention, the EGFR T790M
inhibitor is 1-{(3R,4R)-3-[({5-chloro-2-[(1-methyl-1H-pyrazol-4-yl)amino]-7H-
pyrrolo[2,3-
d]pyrim idin-4-ylloxy)methyI]-4-methoxypyrrolidin-1-yllprop-2-en-1-one, or a
pharmaceutically acceptable salt thereof.
In embodiments of the method of the present invention, the EGFR T790M
inhibitor is N-methyl-N-[trans-3-({2-[(1-methyl-1H-pyrazol-4-yl)amino]-5-
(pyridin-2-y1)-7H-
pyrrolo[2,3-d]pyrimidin-4-ylloxy)cyclobutyl]prop-2-enamide, or a
pharmaceutically
acceptable salt thereof.
In additional embodiments of the method of the present invention, the EGFR
T790M inhibitor is Nqtrans-3-({5-chloro-2-[(1,3-dimethyl-1H-pyrazol-4-
yl)amino]-7H-
pyrrolo[2,3-d]pyrimidin-4-yllamino)cyclobuty1]-N-methylprop-2-enamide, or a
pharmaceutically acceptable salt thereof.
In further embodiments of the method of the present invention, the EGFR T790M
inhibitor is 1-{(3R,4R)-3-[({5-chloro-2-[(3-methoxy-1-methyl-1H-pyrazol-4-
yl)amino]-7H-
pyrrolo[2,3-d]pyrim idin-4-ylloxy)methyI]-4-methoxypyrrolidin-1-yllprop-2-en-1-
one, or a
pharmaceutically acceptable salt thereof.
In some embodiments of the method of the present invention, the panHER
inhibitor is selected from the group consisting of lapatinib, neratinib,
afatinib, pelitinib,
dacomitinib, and canertinib, or a pharmaceutically acceptable salt thereof.
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
In certain embodiments of the method of the present invention, the panHER
inhibitor is selected from the group consisting of lapatinib, neratinib,
afatinib, pelitinib,
dacomitinib, and canertinib, or a pharmaceutically acceptable salt thereof.
In further embodiments of the method of the present invention, the panHER
5 inhibitor is afatinib, or a pharmaceutically acceptable salt thereof.
In embodiments of the method of the present invention, the panHER inhibitor is
dacomitinib, or a pharmaceutically acceptable salt thereof.
In further embodiments of the method of the present invention, the panHER
inhibitor is an irreversible EGFR inhibitor.
In certain embodiments of the method of the present invention, the
irreversible
panHER inhibitor is selected from the group consisting of neratinib, afatinib,
pelitinib,
dacomitinib, and canertinib, or a pharmaceutically acceptable salt thereof.
In embodiments of the method of the present invention, the irreversible panHER
inhibitor is afatinib, or a pharmaceutically acceptable salt thereof.
In some embodiments of the method of the present invention, the irreversible
panHER inhibitor is dacomitinib, or a pharmaceutically acceptable salt
thereof.
Certain embodiments of the present invention relate to a method of treating
non-
small cell lung cancer comprising administering to a patient in need thereof a
synergistic
amount of an EGFR T790M inhibitor in combination with an EGFR inhibitor.
In some embodiments of the method of the present invention, the EGFR T790M
inhibitor is selected from the group consisting of Go6976, PKC412, AP26113,
HM61713,
VVZ4002, CO-1686 and TAS-2913, or a pharmaceutically acceptable salt thereof.
In further embodiments of the method of the present invention, the EGFR T790M
inhibitor is 1-{(3R,4R)-3-[({5-chloro-2-[(1-methyl-1H-pyrazol-4-yl)am ino]-7H-
pyrrolo[2,3-
d]pyrimidin-4-ylloxy)methy1]-4-methoxypyrrolidin-1-yllprop-2-en-1-one, or a
pharmaceutically acceptable salt thereof.
In embodiments of the method of the present invention, the EGFR T790M
inhibitor is N-methyl-N-[trans-3-({2-[(1-methyl-1H-pyrazol-4-yl)amino]-5-
(pyridin-2-y1)-7H-
pyrrolo[2,3-d]pyrimidin-4-ylloxy)cyclobutyl]prop-2-enam ide, or a
pharmaceutically
acceptable salt thereof.
In certain embodiments of the method of the present invention, the EGFR T790M
inhibitor is N-[trans-3-({5-chloro-2-[(1,3-dimethyl-1H-pyrazol-4-y1)amino]-7H-
pyrrolo[2,3-
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
6
d]pyrimidin-4-yllamino)cyclobuty1]-N-methylprop-2-enamide, or a
pharmaceutically
acceptable salt thereof.
In some embodiments of the method of the present invention, the EGFR T790M
inhibitor is 1-{(3R,4R)-3-[({5-chloro-2-[(3-methoxy-1-methyl-1H-pyrazol-4-
yl)am ino]-7H-
pyrrolo[2,3-d]pyrimidin-4-ylloxy)methy1]-4-methoxypyrrolidin-1-yllprop-2-en-1-
one, or a
pharmaceutically acceptable salt thereof.
In further embodiments of the method of the present invention, the EGFR
inhibitor is selected from the group consisting of gefitinib, erlotinib,
icotinib, vandetanib,
lapatinib, neratinib, afatinib, pelitinib, dacomitinib, canertinib, cetuximab
and
panitumumab, or a pharmaceutically acceptable salt thereof.
In some embodiments of the method of the present invention, the EGFR inhibitor
is selected from the group consisting of gefitinib, erlotinib, icotinib,
vandetanib, lapatinib,
neratinib, afatinib, pelitinib, dacomitinib, and canertinib, or a
pharmaceutically
acceptable salt thereof.
In certain embodiments of the method of the present invention, the EGFR
inhibitor is selected from the group consisting of gefitinib, erlotinib,
afatinib, and
dacomitinib, or a pharmaceutically acceptable salt thereof.
In some embodiments of the method of the present invention, the EGFR inhibitor
is gefitinib, or a pharmaceutically acceptable salt thereof.
In embodiments of the method of the present invention, the EGFR inhibitor is
erlotinib, or a pharmaceutically acceptable salt thereof.
In some embodiments of the method of the present invention, the EGFR inhibitor
is a panHER inhibitor.
In further embodiments of the method of the present invention, the panHER
inhibitor is selected from the group consisting of lapatinib, neratinib,
afatinib, pelitinib,
dacomitinib, and canertinib, or a pharmaceutically acceptable salt thereof.
In embodiments of the method of the present invention, the panHER inhibitor is
selected from the group consisting of lapatinib, neratinib, afatinib,
pelitinib, dacomitinib,
and canertinib, or a pharmaceutically acceptable salt thereof.
In some embodiments of the method of the present invention, the panHER
inhibitor is afatinib, or a pharmaceutically acceptable salt thereof.
In additional embodiments of the method of the present invention, the panHER
inhibitor is dacomitinib, or a pharmaceutically acceptable salt thereof.
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
7
In additional embodiments of the method of the present invention, the panHER
inhibitor is an irreversible EGFR inhibitor.
In some embodiments of the method of the present invention, the irreversible
panHER inhibitor is selected from the group consisting of neratinib, afatinib,
pelitinib,
dacomitinib, and canertinib, or a pharmaceutically acceptable salt thereof.
In embodiments of the method of the present invention, the irreversible panHER
inhibitor is afatinib, or a pharmaceutically acceptable salt thereof.
In certain embodiments of the method of the present invention, the
irreversible
panHER inhibitor is dacomitinib, or a pharmaceutically acceptable salt
thereof.
Some embodiments of the present invention relate to a synergistic combination
of
(a) an EGFR T790M inhibitor; and (b) an EGFR inhibitor, wherein component (a)
and
component (b) are synergistic.
In some embodiments of the combination of the present invention, the EGFR
T790M inhibitor is selected from the group consisting of Go6976, PKC412,
AP26113,
HM61713, VVZ4002, CO-1686 and TAS-2913, or a pharmaceutically acceptable salt
thereof.
In further embodiments of the combination of the present invention, the EGFR
T790M inhibitor is 1-{(3R,4R)-3-[({5-chloro-2-[(1-methyl-1H-pyrazol-4-
y1)amino]-7H-
pyrrolo[2,3-d]pyrim idin-4-ylloxy)methyI]-4-methoxypyrrolidin-1-yllprop-2-en-1-
one, or a
pharmaceutically acceptable salt thereof.
In embodiments of the combination of the present invention, the EGFR T790M
inhibitor is N-methyl-N-[trans-3-({2-[(1-methyl-1H-pyrazol-4-yl)amino]-5-
(pyridin-2-y1)-7H-
pyrrolo[2,3-d]pyrimidin-4-ylloxy)cyclobutyl]prop-2-enam ide, or a
pharmaceutically
acceptable salt thereof.
In additional embodiments of the combination of the present invention, the
EGFR
T790M inhibitor is Nqtrans-3-({5-chloro-2-[(1,3-dimethyl-1H-pyrazol-4-
yl)amino]-7H-
pyrrolo[2,3-d]pyrimidin-4-yllamino)cyclobuty1]-N-methylprop-2-enamide, or a
pharmaceutically acceptable salt thereof.
In some embodiments of the combination of the present invention, the EGFR
T790M inhibitor is 1-{(3R,4R)-3-[({5-chloro-2-[(3-methoxy-1-methyl-1H-pyrazol-
4-
yl)am ino]-7H-pyrrolo[2,3-d]pyrimidin-4-ylloxy)methyI]-4-methoxypyrrolidin-1-
yllprop-2-
en-1-one, or a pharmaceutically acceptable salt thereof.
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
8
In some embodiments of the combination of the present invention, the EGFR
inhibitor is selected from the group consisting of gefitinib, erlotinib,
icotinib, vandetanib,
lapatinib, neratinib, afatinib, pelitinib, dacomitinib, canertinib, cetuximab
and
panitumumab, or a pharmaceutically acceptable salt thereof.
In embodiments of the combination of the present invention, the EGFR inhibitor
is
selected from the group consisting of gefitinib, erlotinib, icotinib,
vandetanib, lapatinib,
neratinib, afatinib, pelitinib, dacomitinib, and canertinib, or a
pharmaceutically
acceptable salt thereof.
In embodiments of the combination of the present invention, the EGFR inhibitor
is
selected from the group consisting of gefitinib, erlotinib, afatinib, and
dacomitinib, or a
pharmaceutically acceptable salt thereof.
In certain embodiments of the combination of the present invention, the EGFR
inhibitor is gefitinib, or a pharmaceutically acceptable salt thereof.
In further embodiments of the combination of the present invention, the EGFR
inhibitor is erlotinib, or a pharmaceutically acceptable salt thereof.
In some embodiments of the combination of the present invention, the EGFR
inhibitor is a panHER inhibitor.
In further embodiments of the combination of the present invention, the panHER
inhibitor is selected from the group consisting of lapatinib, neratinib,
afatinib, pelitinib,
dacomitinib, and canertinib, or a pharmaceutically acceptable salt thereof.
In additional embodiments of the combination of the present invention, the
panHER inhibitor is selected from the group consisting of lapatinib,
neratinib, afatinib,
pelitinib, dacomitinib, and canertinib, or a pharmaceutically acceptable salt
thereof.
In embodiments of the combination of the present invention, the panHER
inhibitor
is afatinib, or a pharmaceutically acceptable salt thereof.
In some embodiments of the combination of the present invention, the panHER
inhibitor is dacomitinib, or a pharmaceutically acceptable salt thereof.
In embodiments of the combination of the present invention, the panHER
inhibitor
is an irreversible EGFR inhibitor.
In additional embodiments of the combination of the present invention, the
irreversible panHER inhibitor is selected from the group consisting of
neratinib, afatinib,
pelitinib, dacomitinib, and canertinib, or a pharmaceutically acceptable salt
thereof.
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
9
In some embodiments of the combination of the present invention, the
irreversible
panHER inhibitor is afatinib, or a pharmaceutically acceptable salt thereof.
In embodiments of the combination of the present invention, the irreversible
panHER inhibitor is dacomitinib, or a pharmaceutically acceptable salt
thereof.
Brief Description of the Drawings
Figure 1 shows that Sanger sequencing identified a C>T EGFR T790M mutation
in RPC9 clone 3 and clone 6, with the percentages shown representing castPCR
quantified EGFR T790M alleles with respect to the total EGFR alleles.
Figure 2 shows dose response curves in cell viability assays for PC9 and RPC9
clones 3 and 6 that were treated with various concentration of dacomitinib
(Figure 2A) or
erlotinib (Figure 2B).
Figure 3 shows dose response curves in an RPC9 clone 6 cell viability assay.
Figure 3A shows dose response curves of Compound A ("compd A") and dacomitinib
("daco") alone and in combination. Figure 3B shows dose response curves of
Compound A and erlotinib ("erlo") alone and in combination. Figure 3C shows
the
percent of inhibition at selected concentrations of Compound A as a single
agent and in
combination with dacomitinib and erlotinib.
Figure 4 shows dose response curves in an RPC9 clone 6 cell viability assay.
Figure 4A shows dose response curves of Compound B ("compd B") and dacomitinib
("daco") alone and in combination. Figure 4B shows dose response curves of
Compound B and erlotinib ("erlo") alone and in combination. Figure 4C shows
the
percent of inhibition at selected concentrations of Compound B as a single
agent and in
combination with dacomitinib and erlotinib.
Figure 5 shows dose response curves in an RPC9 clone 6 cell viability assay.
Figure 5A shows dose response curves of Compound A ("compd A") alone and in
combination with dacomitinib ("daco"). Figure 5B shows dose response curves of
Compound A alone and in combination with gefitinib ("gefi"). Figure 5C shows
dose
response curves of Compound A alone and in combination with afatinib ("afat").
Figure
5D shows the percent of inhibition at selected concentrations of Compound A as
a
single agent and in combination with dacomitinib, gefitinib and afatinib.
Figure 6 shows dose response curves in an RPC9 clone 6 cell viability assay.
Figure 6A shows dose response curves of Compound B ("compd B") alone and in
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
combination with dacomitinib ("daco"). Figure 6B shows dose response curves of
Compound B alone and in combination with gefitinib ("gefi"). Figure 6C shows
dose
response curves of Compound B alone and in combination with afatinib ("afat").
Figure
4D shows the percent of inhibition at selected concentrations of Compound B as
a
5 single agent and in combination with dacomitinib, gefitinib and afatinib.
Figure 7 shows a Western immunoblot of the phosphorylation levels of EGFR,
AKT, and ERK in RPC9 clone 6 cells. GAPDH was included as a protein loading
control.
Figure 7A shows the RPC9 clone 6 cells treated with DMSO, dacomitinib,
Compound A
or a combination of dacomitinib + Compound A ("Compd A"). Figure 7B shows the
10 RPC9 clone 6 cells treated with DMSO, erlotinib, Compound A or a
combination of
erlotinib + Compound A ("Compd A").
Figure 8 shows the densitometry results on the bands of the Western immunoblot
(Figure 7A) of the RPC9 clone 6 cells treated with DMSO, dacomitinib, Compound
A or
a combination of dacomitinib ("Daco") + Compound A ("Compd A"). Inhibition of
pEGFR
Y1068 (Figure 8A), pAKT S473 (Figure 8B), and pERK T202/Y204 (Figure 8C) was
determined by comparison to the DMSO control.
Figure 9 shows the densitometry results on the bands of the Western immunoblot
(Figure 7B) of the RPC9 clone 6 cells treated with DMSO, erlotinib, Compound A
or a
combination of erlotinib ("Erlo") + Compound A ("Compd A"). Inhibition of
pEGFR
Y1068 (Figure 9A), pAKT S473 (Figure 9B), and pERK T202/Y204 (Figure 9C) was
determined by comparison to the DMSO control.
Figure 10 shows a Western immunoblot of the phosphorylation levels of EGFR,
AKT, and ERK in RPC9 clone 6 cells. GAPDH was included as a protein loading
control.
Figure 10A shows the RPC9 clone 6 cells treated with DMSO, dacomitinib,
Compound B
or a combination of dacomitinib + Compound B ("Compd B"). Figure 10B shows the
RPC9 clone 6 cells treated with DMSO, erlotinib, Compound B or a combination
of
erlotinib + Compound B ("Compd B").
Figure 11 shows the densitometry results on the bands of the Western
immunoblot (Figure 10A) of the RPC9 clone 6 cells treated with DMSO,
dacomitinib,
Compound B or a combination of dacomitinib ("Daco") + Compound B ("Compd B").
Inhibition of pEGFR Y1068 (Figure 11A), pAKT S473 (Figure 11B), and pERK
T202N204 (Figure 11C) was determined by comparison to the DMSO control.
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
11
Figure 12 shows the densitometry results on the bands of the Western
immunoblot (Figure 10B) of the RPC9 clone 6 cells treated with DMSO,
erlotinib,
Compound B or a combination of erlotinib ("Erlo") + Compound B ("Compd B").
Inhibition of pEGFR Y1068 (Figure 12A), pAKT S473 (Figure 12B), and pERK
T202N204 (Figure 12C) was determined by comparison to the DMSO control.
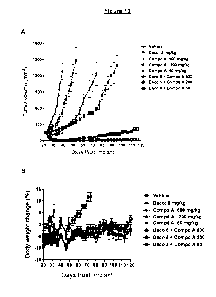
Figure 13 graphs the results of the xenograft model with RPC9 clone 6 tumor
bearing SCID mice, which were randomized, daily and orally treated with
vehicle,
dacomitinib, Compound A, or dacomitinib ("Daco") + Compound A ("Compd A").
Figure
13A graphs the tumor volumes, which were measured 3 times per week and graphed
with mean and standard error of the mean. Figure 13B graphs the body weight of
each
group, which was recorded daily and percentage changes were graphed with mean
and
standard error of the mean.
Figure 14 graphs the results of the xenograft model with RPC9 clone 6 tumor
bearing SCID mice, which were randomized, daily and orally treated with
vehicle,
dacomitinib, Compound B, or dacomitinib ("Daco") + Compound B (Compd B").
Figure
14A graphs the tumor volumes, which were measured 3 times per week and graphed
with mean and standard error of the mean. Figure 14B graphs the body weight of
each
group, which was recorded daily and percentage changes were graphed with mean
and
standard error of the mean.
Figure 15 graphs the results of the xenograft model with RPC9 clone 6 tumor
bearing SCID mice, which were randomized, daily and orally treated with
vehicle,
dacomitinib, Compound B, or dacomitinib ("Daco") + Compound B ("Compd B").
Figure
15A graphs the tumor volumes of single agent treatment groups. Figure 15B
graphs the
tumor volumes of combination treatment groups.
Figure 16 graphs the results of the xenograft model with RPC9 clone 6 tumor
bearing SCID mice, which were randomized, daily and orally treated with
vehicle,
erlotinib, Compound A, or erlotinib ("Erlo") + Compound A (Compd A"). Figure
16A
graphs the tumor volumes, which were measured 3 times per week and graphed
with
mean and standard error of the mean. Figure 16B graphs the body weight of each
group, which was recorded daily and percentage changes were graphed with mean
and
standard error of the mean.
Detailed Description of the Invention
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
12
The members of the human epidermal growth factor receptor/epidermal growth
factor receptor (HER/EGFR) family of receptors include EGFR/HER-1,
HER2/neu/erbB-
2, HER3/erbB-3 and HER4/erbB-4.
EGFR inhibitors effectively inhibit the common activating mutations (L858R and
delE746-A750) of EGFR. The common activating mutations are also referred to as
single mutants or single mutant forms. Examples of EGFR inhibitors include
gefitinib,
erlotinib, icotinib, vandetanib, lapatinib, neratinib, afatinib, pelitinib,
dacomitinib and
canertinib. Monoclonal antibody inhibitors of EGFR, such as cetuximab and
panitumumab, are also EGFR inhibitors, as defined in the present invention.
Inhibitors of EGFR may be reversible or irreversible inhibitors. Reversible
inhibitors of the tyrosine kinase domain of the EFGR molecule attach to and
periodically
detach from the receptor. Gefitinib, erlotinib, icotinib, vandetanib and
lapatinib are
examples of reversible EGFR inhibitors. Irreversible inhibitors of the
tyrosine kinase
domain of the EFGR molecule bind to EGFR irreversibly. Neratinib, afatinib,
pelitinib,
dacomitinib and canertinib are examples of irreversible EGFR inhibitors.
EGFR inhibitors are inhibitors of at least one member of the HER family.
Gefitinib, erlotinib, icotinib and vandetanib are selective EGFR/HER-1
tyrosine kinase
inhibitors (TKI). Cetuximab and panitumumab are monoclonal antibodies specific
to
EGFR/HER-1.
A pan-HER inhibitor is an agent that block multiple members of the HER family.
Lapatinib, neratinib, afatinib, pelitinib, dacomitinib and canertinib are
examples of pan-
HER inhibitors. Lapatinib, neratinib, afatinib and pelitinib inhibit the EGFR
and HER2
members of the HER family. Dacomitinib and canertinib inhibit the EGFR, HER2,
and
HER4 members of the HER family.
EGFR T790M inhibitors effectively inhibit the common activating mutations
(L858R and delE746-A750) and the gatekeeper mutation (T790M). The EGFR T790M
inhibitors of the present invention preferentially inhibit the double mutant
forms of EGFR
(L858R/T790M and delE746-A750/T790M) over the single mutants (L858R and
delE746-A750). Examples of EGFR T790M inhibitors include Go6976, PKC412,
AP26113, HM61713, VVZ4002, CO-1686 and TAS-2913.
Inhibitors of EGFR T790M may be reversible or irreversible inhibitors. Go6976,
PKC412 and AP26113 are examples of reversible EGFR T790M inhibitors. HM61713,
VVZ4002, CO-1686 and TAS-2913 are examples irreversible EGFR T790M inhibitors.
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
13
EGFR T790M inhibitors of the present invention also include 1-{(3R,4R)-3-[({5-
chloro-2-[(1-methyl-1H-pyrazol-4-yl)amino]-7H-pyrrolo[2,3-d]pyrimidin-4-
ylloxy)methyl]-
4-methoxypyrrolidin-1-yllprop-2-en-1-one ("Compound A"), N-methyl-N-[trans-3-
({2-[(1-
methy1-1H-pyrazol-4-y1)am ino]-5-(pyridin-2-y1)-7H-pyrrolo[2,3-d]pyrim idin-4-
ylloxy)cyclobutyl]prop-2-enamide ("Compound B"), Nqtrans-3-({5-chloro-2-[(1,3-
dimethyl-1H-pyrazol-4-yl)amino]-7H-pyrrolo[2,3-d]pyrimidin-4-
yllamino)cyclobutyl]-N-
methylprop-2-enamide ("Compound C"); and 1-{(3R,4R)-34({5-chloro-2-[(3-methoxy-
1-
methyl-1H-pyrazol-4-y1)amino]-7H-pyrrolo[2,3-d]pyrimidin-4-ylloxy)methyl]-4-
methoxypyrrolidin-1-yllprop-2-en-1-one ("Compound D"), or a pharmaceutically
acceptable salt thereof. Compound A, Compound B, Compound C and Compound D are
examples of irreversible EGFR T790M inhibitors.
The following abbreviations may be used herein: Ac (acetyl); APCI (atomic
pressure chemical ionization); Boc (tert-butoxycarbonyl); Boc20 (di-tert-butyl
dicarbonate); BrettPhos Palladacycle (chloro[2-(dicyclohexylphosphino)-3,6-
dimethoxy-
2',4', 6'-triisopropy1-1,1'-biphenyl][2-(2-aminoethyl)phenyl]palladium(II));
DCC (1,3-
dicyclohexylcarbodiimide); DCM (dichloromethane); Deoxo-Fluor (bis(2-
methoxyethyl)aminosulfur trifluoride); DIAD (diisopropyl azodicarboxylate);
DIEA
(diisopropylethylamine); DIPEA (N,N-diisopropylethylamine); DMAP (4-
dimethylaminopyridine); DMEM (Dulbecco's modified Eagle's medium); DMF
(dimethylformamide); DMSO (dimethylsulphoxide); DPPA (diphenyl
phosphorazidate);
EGTA GEthylenebis(oxyethylenenitrilo)]tetraacetic acid); eq (equivalent); Et
(ethyl);
Et0H (ethanol); Et0Ac (ethyl acetate); Et20 (diethyl ether); FBS (fetal bovine
serum);
HATU (2-(7-aza-1H-benzotriazole-1-y1)-1,1,3,3-tetramethyluronium
hexafluorophosphate); HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic
acid);
HMDS (bis(trimethylsilyl)amine, which is also known as hexamethyldisilazane or
hexamethyldisiloxane); HOAc (acetic acid); HPLC (high-performance liquid
chromatography); iPr (isopropyl); iPrMgC1(isopropylmagnesium chloride); iPrOH
(isopropyl alcohol); KHMDS (potassium bis(trimethylsilyl)amide); LAH (lithium
aluminum
hydride); LCMS (liquid chromatography-mass spectrometry); LiHMDS (lithium
bis(trimethylsilyl)amide); Me (methyl); Me0H (methanol); MeCN (acetonitrile);
MTBE
(methyl tert-butyl ether); N (normal); N/A (not available); NaHMDS (sodium
bis(trimethylsilyl)amide); N/D (not determined); NIS (N-iodosuccinimide); NMM
(N-
methylmorpholine); NMR (nuclear magnetic resonance); Pd2(dba)3
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
14
(tris(dibenzylideneacetone)dipalladium(0)); PG (protecting group); Ph
(phenyl);
Ph1(0Ac)2 (odobenzene diacetate); PMSF (phenylmethylsulfonyl fluoride); psi
(pounds
per square inch); Rf (retention factor); RPM! (Roswell Park Memorial
Institute); rt (room
temperature); sat. (saturated); SCX (strong cation exchange); SEM (2-
(trimethylsilyl)ethoxymethyl); SEM-CI (2-(trimethylsilyl)ethoxymethyl
chloride); SFC
(supercritical fluid chromatography); TBAF (tetrabutylammonium fluoride);
TBDPS (tert-
butyldiphenylsilyl); TBS (tert-butyldimethylsilyl); t-BuXPhos Palladacycle
(chloro[2-(di-
tert-butylphosphino)-2',4',6'-triisopropy1-1,1'-biphenyl][2-(2-
aminoethyl)phenylApalladium(11); TFA (trifluoroacetate); THF
(tetrahydrofuran); TLC
(thin layer chromatography); toluene (methylbenzene); tosyl (p-
toluenesulfonyl); and
Xantphos (4,5-bis(diphenylphosphino)-9,9-dimethylxanthene).
Some embodiments relate to the pharmaceutically acceptable salts of the
compounds described herein. Pharmaceutically acceptable salts of the compounds
described herein include the acid addition and base addition salts thereof.
Some embodiments also relate to the pharmaceutically acceptable acid addition
salts of the compounds described herein. Suitable acid addition salts are
formed from
acids which form non-toxic salts. Non-limiting examples of suitable acid
addition salts,
i.e., salts containing pharmacologically acceptable anions, include, but are
not limited to,
the acetate, acid citrate, adipate, aspartate, benzoate, besylate,
bicarbonate/carbonate,
bisulphate/sulphate, bitartrate,borate, camsylate, citrate, cyclamate,
edisylate, esylate,
ethanesulfonate, formate, fumarate, gluceptate, gluconate, glucuronate,
hexafluorophosphate, hibenzate, hydrochloride/chloride, hydrobromide/bromide,
hydroiodide/iodide, isethionate, lactate, malate, maleate, malonate, mesylate,
methanesulfonate, methylsulphate, naphthylate, 2-napsylate, nicotinate,
nitrate, orotate,
oxalate, palmitate, pamoate, phosphate/hydrogen phosphate/dihydrogen
phosphate,
pyroglutamate, saccharate, stearate, succinate, tannate, tartrate, p-
toluenesulfonate,
tosylate, trifluoroacetate and xinofoate salts.
Additional embodiments relate to base addition salts of the compounds
described
herein. Suitable base addition salts are formed from bases which form non-
toxic salts.
Non-limiting examples of suitable base salts include the aluminium, arginine,
benzathine, calcium, choline, diethylamine, diolamine, glycine, lysine,
magnesium,
meglumine, olamine, potassium, sodium, tromethamine and zinc salts.
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
The compounds described herein that are basic in nature are capable of forming
a wide variety of salts with various inorganic and organic acids. The acids
that may be
used to prepare pharmaceutically acceptable acid addition salts of such basic
compounds described herein are those that form non-toxic acid addition salts,
e.g., salts
5 containing pharmacologically acceptable anions, such as the
hydrochloride,
hydrobromide, hydroiodide, nitrate, sulfate, bisulfate, phosphate, acid
phosphate,
isonicotinate, acetate, lactate, sal icylate, citrate, acid citrate, tartrate,
pantothenate,
bitartrate, ascorbate, succinate, maleate, gentisinate, fumarate, gluconate,
glucuronate,
saccharate, formate, benzoate, glutamate, methanesulfonate, ethanesulfonate,
10 benzenesulfonate, p-toluenesulfonate and pamoate [i.e., 1,1'-methylene-
bis-(2-hydroxy-
3-naphthoate)] salts. The compounds described herein that include a basic
moiety,
such as an amino group, may form pharmaceutically acceptable salts with
various
amino acids, in addition to the acids mentioned above.
The chemical bases that may be used as reagents to prepare pharmaceutically
15 acceptable base salts of those compounds of the compounds described
herein that are
acidic in nature are those that form non-toxic base salts with such compounds.
Such
non-toxic base salts include, but are not limited to those derived from such
pharmacologically acceptable cations such as alkali metal cations (e.g.,
potassium and
sodium) and alkaline earth metal cations (e.g., calcium and magnesium),
ammonium or
water-soluble amine addition salts such as N-methylglucamine-(meglumine), and
the
lower alkanolammonium and other base salts of pharmaceutically acceptable
organic
amines.
The compounds of the embodiments described herein include all stereoisomers
(e.g., cis and trans isomers) and all optical isomers of compounds described
herein
(e.g., R and S enantiomers), as well as racemic, diastereomeric and other
mixtures of
such isomers. While all stereoisomers are encompassed within the scope of our
claims,
one skilled in the art will recognize that particular stereoisomers may be
preferred.
In some embodiments, the compounds described herein can exist in several
tautomeric forms, including the enol and imine form, and the keto and enamine
form and
geometric isomers and mixtures thereof. All such tautomeric forms are included
within
the scope of the present embodiments. Tautomers exist as mixtures of a
tautomeric set
in solution. In solid form, usually one tautomer predominates. Even though one
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
16
tautomer may be described, the present embodiments includes all tautomers of
the
present compounds.
The present embodiments also include atropisomers of the compounds described
herein. Atropisomers refer to compounds that can be separated into
rotationally
restricted isomers.
Hem isalts of acids and bases may also be formed, for example, hem isulphate
and hemicalcium salts.
For a review on suitable salts, see Handbook of Pharmaceutical Salts:
Properties,
Selection, and Use by Stahl and Wermuth (Wiley-VCH, 2002). Methods for making
pharmaceutically acceptable salts of compounds described herein are known to
one of
skill in the art.
The term "solvate" is used herein to describe a molecular complex comprising a
compound described herein and one or more pharmaceutically acceptable solvent
molecules, for example, ethanol.
The compounds described herein may also exist in unsolvated and solvated
forms. Accordingly, some embodiments relate to the hydrates and solvates of
the
compounds described herein.
Compounds described herein containing one or more asymmetric carbon atoms
can exist as two or more stereoisomers. Where a compound described herein
contains
an alkenyl or alkenylene group, geometric cis/trans (or Z/E) isomers are
possible.
Where structural isomers are interconvertible via a low energy barrier,
tautomeric
isomerism (tautomerism) can occur. This can take the form of proton
tautomerism in
compounds described herein containing, for example, an imino, keto, or oxime
group, or
so-called valence tautomerism in compounds which contain an aromatic moiety. A
single compound may exhibit more than one type of isomerism.
Included within the scope of the present embodiments are all stereoisomers,
geometric isomers and tautomeric forms of the compounds described herein,
including
compounds exhibiting more than one type of isomerism, and mixtures of one or
more
thereof. Also included are acid addition or base salts wherein the counterion
is optically
active, for example, d-lactate or 1-lysine, or racemic, for example, dl-
tartrate or dl-
arginine.
Cis/trans isomers may be separated by conventional techniques well known to
those skilled in the art, for example, chromatography and fractional
crystallisation.
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
17
Conventional techniques for the preparation/isolation of individual
enantiomers
include chiral synthesis from a suitable optically pure precursor or
resolution of the
racemate (or the racemate of a salt or derivative) using, for example, chiral
high
pressure liquid chromatography (HPLC).
Alternatively, the racemate (or a racemic precursor) may be reacted with a
suitable optically active compound, for example, an alcohol, or, in the case
where a
compound described herein contains an acidic or basic moiety, a base or acid
such as
1-phenylethylamine or tartaric acid. The resulting diastereomeric mixture may
be
separated by chromatography and/or fractional crystallization and one or both
of the
diastereoisomers converted to the corresponding pure enantiomer(s) by means
well
known to a skilled person.
The term "treating", as used herein, unless otherwise indicated, means
reversing,
alleviating, inhibiting the progress of, or preventing the disorder or
condition to which
such term applies, or one or more symptoms of such disorder or condition. The
term
"treatment", as used herein, unless otherwise indicated, refers to the act of
treating as
"treating" is defined immediately above.
A patient to be treated according to this invention includes any warm-blooded
animal, such as, but not limited to human, monkey or other lower-order
primate, horse,
dog, rabbit, guinea pig, or mouse. For example, the patient is human. Those
skilled in
the medical art are readily able to identify individual patients who are
afflicted with non-
small cell lung cancer and who are in need of treatment.
The term "additive" means that the result of the combination of the two
compounds
or targeted agents is the sum of each agent individually. The terms "synergy"
or
"synergistic" are used to mean that the result of the combination of the two
agents is more
than the sum of each agent together. A "synergistic amount" is an amount of
the
combination of the two agents that result in a synergistic effect.
Determining a synergistic interaction between one or two components, the
optimum
range for the effect and absolute dose ranges of each component for the effect
may be
definitively measured by administration of the components over different w/w
ratio ranges
and doses to patients in need of treatment. For humans, the complexity and
cost of
carrying out clinical studies on patients renders impractical the use of this
form of testing
as a primary model for synergy. However, the observation of synergy in in
vitro models or
in vivo models can be predictive of the effect in humans and other species and
in vitro
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
18
models or in vivo models exist, as described herein, to measure a synergistic
effect and
the results of such studies can also be used to predict effective dose and
plasma
concentration ratio ranges and the absolute doses and plasma concertrations
required in
humans and other species by the application of pharmacokinetic/pharmacodynamic
methods.
In an embodiment, the method of the invention is related to a method of
treating
non-small cell lung cancer comprising administering to a patient in need
thereof an
effective amount of an EGFR T790M inhibitor in combination with a panHER
inhibitor,
where the panHER inhibitor is administered according to a non-standard
clinical dosing
regimen, in amounts sufficient to achieve synergistic effects. In this
embodiment, the
method of the invention is related to a synergistic combination of targeted
therapeutic
agents, specifically an EGFR T790M inhibitor and a panHER inhibitor.
In an embodiment, the method of the invention is related to a method of
treating
non-small cell lung cancer comprising administering to a patient in need
thereof an
effective amount of an EGFR T790M inhibitor in combination with a low-dose
amount of
a panHER inhibitor, in amounts sufficient to achieve synergistic effects. In
this
embodiment, the method of the invention is related to a synergistic
combination of
targeted therapeutic agents, specifically an EGFR T790M inhibitor and a panHER
inhibitor.
In another embodiment, the method of the invention is related to a method of
treating non-small cell lung cancer comprising administering to a patient in
need thereof
an effective amount of an irreversible EGFR T790M inhibitor in combination
with an
effective amount of an EGFR inhibitor, in amounts sufficient to achieve
synergistic
effects. In this embodiment, the method of the invention is related to a
synergistic
combination of targeted therapeutic agents, specifically an irreversible EGFR
T790M
inhibitor and an EGFR inhibitor.
As used herein, an "effective" amount refers to an amount of a substance,
agent,
compound, or composition that is sufficient to prevent or inhibit the growth
of tumor cells or
the progression of cancer metastasis in the combination of the present
invention.
Therapeutic or pharmacological effectiveness of the doses and administration
regimens
may also be characterized as the ability to induce, enhance, maintain or
prolong remission
in patients experiencing specific tumors.
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
19
A "non-standard clinical dosing regimen," as used herein, refers to a regimen
for
administering a substance, agent, compound, or composition, which effectively
inhibits
the single mutant forms (L858R and delE746-A750) of EGFR, but which is
different than
the amount or dose typically used in a clinical setting. A "non-standard
clinical dosing
regimen," includes a "non-standard clinical dose" or a "non-standard dosing
schedule".
A "low-dose amount", as used herein, refers to an amount or dose of a
substance, agent, compound, or composition, which effectively inhibits the
single mutant
forms (L858R and delE746-A750) of EGFR, but which is an amount or dose lower
than
the amount or dose typically used in a clinical setting.
Those skilled in the art will be able to determine, according to known
methods,
the appropriate amount or dosage of each compound, as used in the combination
of the
present invention, to administer to a patient, taking into account factors
such as age,
weight, general health, the compound administered, the route of
administration, the
nature and advancement of the non-small cell lung cancer requiring treatment,
and the
presence of other medications.
The practice of the method of this invention may be accomplished through
various
administration regimens. The compounds of the combination of the present
invention can
be administered intermittently, concurrently or sequentially. Repetition of
the
administration regimens may be conducted as necessary to achieve the desired
reduction
or diminution of cancer cells. In an embodiment, the compounds of the
combination of the
present invention can be administered in an intermittent dosing regimen.
Administration of the compounds of the combination of the present invention
can
be effected by any method that enables delivery of the compounds to the site
of action.
These methods include oral routes, intraduodenal routes, parenteral injection
(including
intravenous, subcutaneous, intramuscular, intravascular or infusion), topical,
and rectal
administration.
The compounds of the method or combination of the present invention may be
formulated prior to administration. The formulation will preferably be adapted
to the
particular mode of administration. These compounds may be formulated with
pharmaceutically acceptable carriers as known in the art and administered in a
wide
variety of dosage forms as known in the art. In making the pharmaceutical
compositions
of the present invention, the active ingredient will usually be mixed with a
pharmaceutically acceptable carrier, or diluted by a carrier or enclosed
within a carrier.
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
Such carriers include, but are not limited to, solid diluents or fillers,
excipients, sterile
aqueous media and various non-toxic organic solvents. Dosage unit forms or
pharmaceutical compositions include tablets, capsules, such as gelatin
capsules, pills,
powders, granules, aqueous and nonaqueous oral solutions and suspensions,
lozenges,
5 troches, hard candies, sprays, creams, salves, suppositories, jellies,
gels, pastes,
lotions, ointments, injectable solutions, elixirs, syrups, and parenteral
solutions
packaged in containers adapted for subdivision into individual doses.
Parenteral formulations include pharmaceutically acceptable aqueous or
nonaqueous solutions, dispersion, suspensions, emulsions, and sterile powders
for the
10 preparation thereof. Examples of carriers include water, ethanol,
polyols (propylene
glycol, polyethylene glycol), vegetable oils, and injectable organic esters
such as ethyl
oleate. Fluidity can be maintained by the use of a coating such as lecithin, a
surfactant,
or maintaining appropriate particle size. Exemplary parenteral administration
forms
include solutions or suspensions of the compounds of the invention in sterile
aqueous
15 solutions, for example, aqueous propylene glycol or dextrose solutions.
Such dosage
forms can be suitably buffered, if desired.
Additionally, lubricating agents such as magnesium stearate, sodium lauryl
sulfate and talc are often useful for tableting purposes. Solid compositions
of a similar
type may also be employed in soft and hard filled gelatin capsules. Preferred
materials,
20 therefor, include lactose or milk sugar and high molecular weight
polyethylene glycols.
When aqueous suspensions or elixirs are desired for oral administration the
active
compound therein may be combined with various sweetening or flavoring agents,
coloring matters or dyes and, if desired, emulsifying agents or suspending
agents,
together with diluents such as water, ethanol, propylene glycol, glycerin, or
combinations
thereof.
Methods of preparing various pharmaceutical compositions with a specific
amount of active compound are known, or will be apparent, to those skilled in
this art.
For examples, see Remington's Pharmaceutical Sciences, Mack Publishing
Company,
Easter, Pa., 15th Edition (1975).
The invention also relates to a kit comprising the therapeutic agents of the
combination of the present invention and written instructions for
administration of the
therapeutic agents. In one embodiment, the written instructions elaborate and
qualify
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
21
the modes of administration of the therapeutic agents, for example, for
simultaneous or
sequential administration of the therapeutic agents of the present invention.
The examples and preparations provided below further illustrate and exemplify
the compounds described herein and methods of preparing such compounds. The
scope of the embodiments described herein is not limited in any way by the
following
examples and preparations. In the following examples, molecules with a single
chiral
center, unless otherwise noted, exist as a racemic mixture. Those molecules
with two
or more chiral centers, unless otherwise noted, exist as a racemic mixture of
diastereomers. Single enantiomers/diastereomers may be obtained by methods
known
to those skilled in the art.
In the examples shown, salt forms were occasionally isolated as a consequence
of the mobile phase additives during HPLC based chromatographic purification.
In these
cases, salts such as formate, trifluorooacetate and acetate were isolated and
tested
without further processing. It will be recognized that one of ordinary skill
in the art will be
able to realize the free base form by standard methodology (such as using ion
exchange
columns, or performing simple basic extractions using a mild aqueous base).
In general, the compounds described herein may be prepared by processes
known in the chemical arts, particularly in light of the description contained
herein.
Certain processes for the manufacture of the compounds described herein are
provided
as further features of the embodiments and are illustrated in the reaction
schemes
provided below and in the experimental section.
Examples
Example 1: Preparation of 1-{(3R14R)-345-chloro-2-(1-methy1-1H-pyrazol-4-
ylamino)-7H-pyrrolo[213-dlpyrimidin-4-yloxymethyll-4-methoxy-pyrrolidin-1-
vIlpropenone trifluoroacetate (also known as "1-{(3R,4R)-3-[({5-chloro-2-[(1-
methyl-1H-pyrazol-4-yl)aminol-7H-pyrrolor2,3-dlpyrimidin-4-ylloxy)methyll-4-
methoxypyrrolidin-1-yllprop-2-en-1 -one trifluoroacetate" and "1 -((3R,41R)-3-
(((5-
chloro-2-((1 -methyl-1 H-pyrazol-4-ynamino)-7H-pyrrolo[213-dlpyri midi n-4-
viloxylmethyl)-4-methoxypyrrol idi n-1 -yl)prop-2-en-1 -one trifluoroacetate")
(trifluoroacetate salt of "Compound A")
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
22
HN
CI
N
HN N 0
0
N-N
H3C
H30 0H20 TFA
Step 1: Preparation of (3S,4R)-1-benzy1-4-methoxy-pyrrolidine-3-carboxylic
acid
methyl ester
H C 0
3 `o-4do CH3
To a solution of (E)-3-methoxy-acrylic acid methyl ester (50 g, 430.6 mmol) in
2-
Me-THF (600 mL) and TFA (6.7 mL) at 0 C was added N-(methoxymethyl)-N-
(trimethylsilylmethyl)-benzylamine (204 g, 2 eq) dropwise. After addition,
reaction was
allowed to warm to rt and stirred for 2 hrs. Reaction was transferred to a
separatory
funnel and washed with sat. NaHCO3, sat. NaCI, then dried over Na2504 and the
solvent removed to leave the crude racemic product as a yellow oil which was
purified
on 5i02 (10% ¨35 % Et0Ac/heptane) to give the racemic trans product as a
yellow oil
(82.7 g). Enantiomer separation by chiral-SFC (Chiralpak AD-H 4.6 x 250 mm
column 4
% Me0H w/0.1 % diethylamine, 140 bar, 3.0 mL/min) gave the desired single
isomer
product which was verified by comparison with a known standard (34 g, 31.7 %
yield).
Specific rotation [a]027 = +23.8 (C=1.3, Me0H). 1H NMR (400 MHz, DMSO-d6) 6
ppm
2.55 - 2.63 (m, 2 H) 2.69 (dd, J=9.95, 6.42 Hz, 1 H) 2.82 - 2.88 (m, 1 H) 2.90
- 2.96 (m,
1 H) 3.23 (s, 3 H) 3.51 -3.63 (m, 2 H) 3.66 (s, 3 H) 4.07 - 4.12 (m, 1 H) 7.22
- 7.39 (m, 5
H). m/z (APCI+) for (C14H19NO3) 250.0 (M+H)+.
Step 2: Preparation of (3S,4R)-4-methoxy-pyrrolidine-1,3-dicarboxylic acid 1 -
tert-
butyl ester 3-methyl ester
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
23
HO 0
3 j0-4
0 0
H CCH
3 CH3 3
A solution of (3S,4R)-1-benzy1-4-methoxy-pyrrolidine-3-carboxylic acid methyl
ester (35 g, 140.4 mmol) in ethanol (500 mL) was purged with nitrogen and then
Pd(OH)2 (2 g, 0.1 eq) was added and the mixture stirred overnight under an
atmosphere
of hydrogen gas at approximately 15 psi (via hydrogen balloon). The reaction
was then
filtered through Celite and di-tert-butyldicarbonate (30.9 g, 1 eq) was added
to the
resulting filtrate slowly with stirring. After one hr the reaction was
concentrated and the
crude material was purified through a short silica column eluting with 10
(:)/0
Et0Ac/heptane for 2 volumes then 1:1 Et0Ac/heptane until the product was
completely
eluted. Product fractions were combined and concentrated to give the title
compound
as a clear oil, (35.81 g, 98% yield). 1H NMR (400 MHz, DMSO-d6) 6 ppm 1.39 (s,
9 H)
3.17 (br. s., 1 H) 3.23 - 3.28 (m, 4 H) 3.35 - 3.53 (m, 3 H) 3.65 (s, 3 H)
4.06 (d, J=4.78
Hz, 1 H). m/z (APC1+) for product minus Boc (C7H13NO3) 160.1 (M+H)+. Specific
Rotation: [a]D= -12.5 degrees (C=0.87, Me0H).
Step 3: Preparation of (3R,4R)-3-hydroxymethy1-4-methoxy-pyrrolidine-1-
carboxylic acid tert-butyl ester
HO¨%
CCCH3
0 0
H C CHCH
3 3 3
Lithium borohydride (12.7 g, 4 eq) was added portionwise to a solution of
(3S,4R)-4-methoxy-pyrrolidine-1,3-dicarboxylic acid 1-tert-butyl ester 3-
methyl ester
(35.81 g, 138.1 mmol) in THF (600 mL), then the reaction was heated to 60 C
for 4 hrs.
The reaction was quenched with water at 0 C and extracted with Et0Ac. The
organic
layer was washed with sat. NaCl and dried over Na2504. The solvent was removed
and
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
24
the residue was purified through a plug of Si02 (3:1 Et0Ac/heptane) to yield
the title
compound as a clear oil (29.35 g, 92 (:)/0 yield). 1H NMR (400 MHz, chloroform-
d) 6 ppm
1.46 (s, 9 H) 2.37 -2.47 (m, 1 H) 3.19 (dd, J=11.08, 5.29 Hz, 1 H) 3.33 (d,
J=4.03 Hz, 4
H) 3.50 - 3.66 (m, 4H) 3.77 - 3.83 (m, 1 H). m/z (APCI+) for product minus Boc
(C6H13NO2) 132.2 (M+H)+. Specific Rotation: [a]D= +9.3 degrees (C=0.86, Me0H).
Step 4: Preparation of (3R,4R)-3-15-chloro-2-(1-methy1-1H-pyrazol-4-ylamino)-
7H-pyrrolof2,3-d1pyrimidin-4-yloxymethy11-4-methoxy-pyrrolidine-1-carboxylic
acid tett-
butyl ester
HN
N Cl
HN N 0
0
LON-1( CH3
N-NH
-
H3C-0
H3C CH3 3
Method A: (using microwave heating)
To a solution 2,4,5-trichloro-7H-pyrrolo[2,3-d]pyrimidine (904 mg, 4.1 mmol)
and
(3R,4R)-3-hydroxymethy1-4-methoxy-pyrrolidine-1-carboxylic acid tert-butyl
ester (940
mg, 4.1 mmol) in 1,4-dioxane (15 mL) in a microwave vial was added potassium
tert-
pentoxide (25% w/w in toluene, 1.6 mL, 3.5 mmol). The resulting solution was
stirred at
ambient temperature for 15 min. LCMS showed a quantitative formation of
(3R,4R)-3-
(2,5-dichloro-7H-pyrrolo[2,3-d]pyrimidin-4-yloxymethyl)-4-methoxy-pyrrolidine-
1-
carboxylic acid tert-butyl ester. To this resulting reaction solution was
added 1-methyl-
1H-pyrazol-4-ylamine (474 mg, 4.9 mmol) and t-BuXPhos palladacycle (110 mg,
0.04
mol eq). The reaction mixture was stirred and heated to 100 C using microwave
at
normal absorption level for 45 min. The reaction mixture was filtered through
Celite and
the filtrate was evaporated to give a dark color residue. The crude material
was purified
via flash chromatography eluting with a gradient of 0 (:)/0 - 100 (:)/0 Et0Ac
in heptanes to
give the title compound (1.78 g, 76% yield). 1H NMR (400 MHz, DMSO-d6) 6 ppm
11.50 (br. s., 1 H) 9.06 (s, 1 H) 7.85 (s, 1 H) 7.52 (s, 1 H) 7.05 (d, J=2.27
Hz, 1 H) 4.30 -
4.53 (m, 2 H) 3.86 - 3.96 (m, 1 H) 3.80 (s, 3 H) 3.55 - 3.68 (m, 1 H) 3.43 -
3.53 (m, 1 H)
3.24 - 3.31 (m, 3 H) 2.71 (br. s., 1 H) 1.39 (br. s., 9 H). m/z (APCI+) for
product minus
Boc; C16H20CIN702 378.1 (M+H)+ with Cl isotope pattern.
Method B: using thermal heating
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
To a solution 2,4,5-trichloro-7H-pyrrolo[2,3-d]pyrimidine (9.28 g, 41.7 mmol)
and
(3R,4R)-3-hydroxymethyl-4-methoxy-pyrrolidine-1-carboxylic acid tert-butyl
ester (9.65
g, 41.7 mmol) in 1,4-dioxane (100 mL) in a round bottom flask was added
potassium
tert-pentoxide (25 (:)/0 w/w in toluene, 80 mL, 167 mmol). The resulting
reaction solution
5 was stirred at ambient temperature for 30 min. LCMS showed a quantitative
formation
of (3R,4R)-3-(2,5-dichloro-7H-pyrrolo[2,3-c]pyrimidin-4-yloxymethyl)-4-methoxy-
pyrrolidine-1-carboxylic acid tert-butyl ester. To the resulting reaction
solution was
added 1-methyl-1H-pyrazol-4-ylamine (4.86 g, 50.1 mmol) and t-BuXPhos
palladacycle
(1.1 g, 1.67 mmol, 0.04 mol eq). The reaction mixture was stirred and heated
to 90 C
10 in an oil bath for 1 hr. The reaction mixture was then filtered through
Celite and the
filtrate was evaporated to remove the volatiles to give a dark gum that was
then
dissolved in ethyl acetate (300 mL) and filtered through a silica gel plug.
The filtrate was
evaporated and the residue was purified via flash chromatography eluting with
a
gradient of 0 (:)/0 - 100% Et0Ac in heptanes to give the title compound (12.4
g, 62 (:)/0
15 yield). 1H NMR (400 MHz, DMSO-d6) 6 ppm 11.51 (br. s., 1 H) 9.07 (s, 1
H) 7.86 (s, 1
H) 7.52 (s, 1 H) 7.06 (d, J=2.20 Hz, 1 H) 4.31 - 4.54 (m, 2 H) 3.92 (br. s., 1
H) 3.80 (s, 3
H) 3.55 - 3.68 (m, 1 H) 3.44 - 3.55 (m, 1 H) 3.30 (d, J=18.34 Hz, 3 H) 2.72
(br. s., 1 H)
1.39 (br. s., 9 H). m/z (APCI+) for C21H28CIN704 378.2 (M+H)+ with Cl isotope
pattern.
Step 5: Preparation of f5-chloro-44(3R,4R)-4-methoxy-pyrrolidin-3-ylmethoxy)-
20 7H-pyrrolo[2,3-dlpyrimidin-2-y11-(1-methyl-1H-pyrazol-4-y1)-amine
trifluoroacetate
HN
N Cl
HN N 0
CNN H NH
1-130'
TFA
To a solution of (3R,4R)-3-[5-chloro-2-(1-methyl-1H-pyrazol-4-ylamino)-7H-
pyrrolo[2,3-d]pyrimidin-4-yloxymethyl]-4-methoxy-pyrrolidine-1-carboxylic acid
tert-butyl
ester (12.40 g, 26 mmol) in DCM (60 mL) at 0 C was added TFA (10.1 mL, 208
mmol)
25 and the resulting solution was stirred at ambient temperature for 2.5
hrs. The volatiles
were removed and to the residue was added ethyl ether (150 mL). The resulting
suspension was stirred for 2 hrs then filtered to afford a light pink solid.
This was
washed with ethyl ether (30 mL) and dried to give the title compound (15.69 g,
quant) as
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
26
a TFA salt. 1H NMR (400 MHz, DMSO-d6) 6 ppm 11.56 (br. s., 1 H) 9.09 (s, 3 H)
7.85
(s, 1 H) 7.54 (s, 1 H) 7.09 (d, J=2.32 Hz, 1 H) 4.48 (d, J=6.48 Hz, 2 H) 4.11
(br. s., 1 H)
3.81 (s, 3 H) 3.46 -3.60 (m, 1 H) 3.35 -3.45 (m, 2 H) 3.32 (s, 3 H) 3.15 (dq,
J=12.01,
6.02 Hz, 1 H) 2.88 (m, J=6.42, 6.42 Hz, 1 H). m/z (APCI+) for parent molecule
C16H20CIN702 378.2 (M+H)+ with Cl isotope pattern.
Step 6: Preparation of 14(3R,4R)-345-chloro-2-(1-methy1-1H-pyrazol-4-ylamino)-
7H-pyrrolof2,3-dipyrimidin-4-yloxymethyll-4-methoxy-pyrrolidin-1-yllpropenone
trifluoroacetate
HN
N Cl
HN N 0
/NN
HO 30 H2C
TFA
A mixture of [5-chloro-44(3R,4R)-4-methoxy-pyrrolidin-3-ylmethoxy)-7H-
pyrrolo[2,3-d]pyrimidin-2-y1]-(1-methy1-1H-pyrazol-4-y1)-amine (15.0 g (2 TFA
salt)), 24.7
mmol), ethyl acetate (200 mL) and saturated aqueous NaHCO3 (100 mL) was
stirred at
0 C for 10 min. Acryloyl chloride (2.3 mL, 29 mmol, 1.1 mol eq) was added
dropwise
and the resulting mixture was stirred at ambient temperature for 30 min. Ethyl
acetate
(150 mL) was added and the organic layer was separated. The aqueous layer was
extracted with ethyl acetate (150 mL) and the combined organic layers were
dried over
Na2504 and evaporated to give a solid that was purified by SFC (ZymorSPHER HAP
5p
21.2 x 150 mm column eluting with 35% Et0H in CO2 at 120 bar, flow 64 mL/min)
to
give the title compound as an off white solid (8.3 g, 78% yield). 1H NMR (400
MHz,
DMSO-d6) 6 ppm 11.51 (s, 1 H) 9.07 (s, 1 H) 7.86 (s, 1 H) 7.52 (s, 1 H) 7.05
(s, 1 H)
6.59 (ddd, J=16.75, 10.27, 1.34 Hz, 1 H) 6.14 (dd, J=16.75, 2.32 Hz, 1 H) 5.68
(dt,
J=10.27, 2.32 Hz, 1 H) 4.44 (d, J=6.24 Hz, 2 H) 3.82 -4.09 (m, 2 H) 3.80 (s, 3
H) 3.57 -
3.76 (m, 2 H) 3.47 - 3.54 (m, 1 H) 3.31 (d, J=4.65 Hz, 3 H) 2.67 - 2.92 (m, 1
H). m/z
(APCI+) for parent molecule C19H22CIN703 431.9 (M+H)+ with Cl isotope pattern.
Alternate Example 1: Preparation of 1-{(3R14R)-3-[({5-chloro-2-[(1-methy1-1 H-
Pvrazol-4-vnaminol-7H-pyrrolo[2,3-d]pyrimidin-4-v1}oxv)methyll-4-
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
27
methoxypyrrolidin-1-yllprop-2-en-1-one (also known as, "1-((3R,4R)-3-(((5-
chloro-
2-((1-methyl-1H-pyrazol-4-ynamino)-7H-pyrrolo[213-dipyrimidin-4-ynoxy)methyl)-
4-
methoxypyrrol idi n-1 -yl)prop-2-en-1 -one") ("Compound A")
HN
CI
N
HN N 0
0
N-N
H3
0 s
H3C H2C
Step 1: Preparation of methyl (3,4-trans)-1-benzy1-4-methoxypyrrolidine-3-
carboxylate
0 ,CH3
H3C-0 0
Under a nitrogen atmosphere with magnetic stirring, methyl trans-3-
methoxyacrylate (500 mL, 540 g, 4.65 mol) and benzyl
methoxymethyltrimethylsilylamine (595 mL, 552.1 g, 2.3 mol) were mixed. To
this
mixture was added TFA (2.7 mL, 4.14 g, 36.3 mmol) which resulted in an
exotherm to
approximately 95 C in 30 seconds. The resulting mixture was then heated at
reflux for 1
hr (note: at the beginning the reflux temperature was approximately at 104 C
and after
1 hr it had dropped to approximately at 90 C). Three batches of this scale
plus another
batch using 325 mL benzyl methoxymethyltrimethylsilylamine compound were
performed. Two of these batches were combined and poured into 2 N HCI (5 L).
The
mixture was extracted with Et0Ac (3 L and 2 L). With cooling on ice, the
aqueous layer
was brought to pH 9 by adding 50 % NaOH (aq). The aqueous layer was extracted
with
Et0Ac (2.5 L, 1.5 L and 1 L). The combined organic layers were washed with
brine (3 L)
and dried over Na2504. The same workup was done for the remaining batches. All
organic layers were filtered and the filtrate was concentrated in vacuo to
give crude title
compound (racemic-trans, 1640 g). After purification in batches by bulb-to-
bulb
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
28
distillation (0.1 mbar, 100 C ¨145 C) the title compound was isolated as a
yellow oil
(69 % overall yield).
Step 2: Preparation of methyl (3,4-trans)-4-methoxypyrrolidine-3-carboxylate
0 OH3
H30¨O,,
Methyl (3,4-trans)-1-benzy1-4-methoxypyrrolidine-3-carboxylate (463.3 g, 1858
mmol) was dissolved in iPrOH (2 L). To this solution was added 20 % Pd(OH)2/C
(50 g,
37 % moist, Aldrich) and the mixture was stirred vigorously. A pressure of
11.8 bar H2
was applied and refilling was done several times until 1H-NMR showed complete
conversion. The mixture was filtered through Celite and the Celite was rinsed
with
iPrOH. The filtrate was concentrated in vacuo to give the title compound as a
dark
yellow liquid (266 g, 90 % yield). Used as is in next step.
Step 3: Preparation of (3R,4S)-3-methoxy-4-(methoxycarbonyl)pyrrolidinium
(2R,3R)-2,3-bis(benzyloxy)-3-carboxypropanoate
0 CH3
0 0
0'
=
N+ OLJ:C)
H H 0 OH
To a warm solution of 0,0-dibenzoyl-L-tartaric acid (1 kg, 2.79 mol) in
ethanol (5
L) was added a solution of methyl (3,4-trans)-4-methoxypyrrolidine-3-
carboxylate (480
g, 2.84 mole) in ethanol (1 L). The clear solution was seeded and allowed to
crystallize
overnight. The resulting solid was isolated and washed with ethanol. This
enriched
material was recrystallized 5 times from ethanol (2 times 5 L, 4 L, 3.5 L and
3 L) to
afford 216 g (15% yield) of the salt with an enantiomeric excess of 98% (Rt
12.18 min
using condition below).
Chiral purity determination:
Sample preparation: 5 mg salt was mixed with DCM (1.5 mL) and 2 N NaOH (0.2
mL). The DCM layer was dried and analysed by gas chromatography.
Column: Agilent Cyclosil B; 30m x 250 cm x 0.25 cm
Temp: 90 C (0 min) to 5 C/min to 180 C (4 min). Total run time 22 min.
Inj temp.: 250 C
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
29
Detector: 250 C; FID
Inj vol.: 1.0 pL
Split ratio: 25:1
Column flow: 2.2 mL/min (H2)
Step 4: Preparation of 1-tert-butyl 3-methyl (3S,4R)-4-methoxypyrrolidine-1,3-
dicarboxylate
0 CH3
c?
H3C-R ¨
d
N
0 0
H.,C*CH3
- CH3
A mixture of (3R,4S)-3-methoxy-4-(methoxycarbonyl)pyrrolidinium (2R,3R)-2,3-
bis(benzyloxy)-3-carboxypropanoate (216 g, 420 mmol) in DCM and saturated
NaHCO3
was stirred mechanically, and Boc20 (119 g, 546 mmol) was added portionwise.
The
mixture was stirred overnight at rt and the layers were separated. The aqueous
phase
was extracted with DCM and the combined organic layers washed with brine,
dried and
concentrated. This afforded 138 g of the crude title product mixed with 33 %
Boc20 (85
% yield). Used directly in next step.
Step 5: Preparation of tert-butyl (3R,4R)-3-(hydroxymethyl)-4-
methoxypyrrolidine-
1-carboxylate
H3c-o, i0H
N
0 0
H.,C*CH3
- CH3
1-Tert-butyl 3-methyl (3S,4R)-4-methoxypyrrolidine-1,3-dicarboxylate (138 g,
containing Boc20, approx. 2:1, - 0.35 mol) was dissolved in 2 L THF. The
solution was
mechanically stirred and cooled to -78 C. A solution of lithium aluminium
hydride (2.4
M in THF, 200 mL, 0.5 mol) was added dropwise over 30 min, keeping the
temperature
below -70 C. The temperature was then allowed to rise to -30 C. A saturated
solution
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
of sodium potassium tartrate tetrahydrate (aqueous, 100 mL) was added slowly
to
quench the reaction. The solid material was filtered off over a bed of Na2SO4.
The filtrate
was concentrated in vacuo to give the title compound (66 g, 0.28 mol, - 80 A
yield) as
an almost colorless syrup. 1H-NMR (300 MHz, CDCI3): ppm 3.80 (q, J= 5.1 Hz,
1H),
5 3.62 (d, J=6.2 Hz, 2H), 3.56 (m, 1H), 3.52 (d, J=7.4 Hz, 1H), 3.40-3.30
(m, 1H), 3.36 (s,
3H), 2.42 (m, 1H), 1.46 (s, 9H). m/z (GCMS) for Ci FI21 NO4 231.2 (M)+. m/z
(APCI+)
for Ci iF121 NO4132.0 (M+H)+. Specific rotation: [a]D = +11.6 degrees (c 0.77,
Me0H).
Chiral purity determination method: Chiralpak AD-H 21.2 x 250 mm 5u column
eluted
with mobile phase of 12 A MeOH: 88 A CO2 at 35 C and held to 120 bar. Flow
rate of
10 62 mUmin. Rt at 3.36 min.
Step 6: Preparation of tert-butyl (3R,4R)-3-f({5-chloro-2-[(1-methyl-1H-
pyrazol-4-
yl)am ino]-7H-pyrrolo[2,3-d]pyrimidin-4-yl}oxy)methyI]-4-methoxypyrrolidine-1-
carboxylate
CI
HN N 0
0
CH3cH3
H3C-0
H3C' CH3
15
To a solution 2,4,5-trichloro-7H-pyrrolo[2,3-d]pyrimidine (9.28 g, 41.7 mmol)
and
tert-butyl (3R,4R)-3-(hydroxymethyl)-4-methoxypyrrolidine-1-carboxylate 9.65
g, 41.7
mmol) in 1,4-dioxane (100 mL) in a round bottom flask was added potassium tert-
pentoxide (25 A w/w in toluene, 80 mL, 167 mmol). The resulting reaction
solution was
stirred at ambient temperature for 30 min. LCMS showed a quantitative
formation of
20 intermediate tert-butyl (3R,4R)-3-{[(2,5-dichloro-7H-pyrrolo[2,3-
d]pyrimidin-4-
yl)oxy]methy1}-4-methoxypyrrolidine-1-carboxylate. To the above reaction
solution was
added 1-methyl-1H-pyrazol-4-ylamine (4.86 g, 50.1 mmol), t-BuXPhos
palladacycle (1.1
g, 1.67 mmol, 0.04 mol eq) and the reaction mixture was stirred and heated to
90 C in
an oil bath for 1 hr. LCMS indicated the reaction was complete. The reaction
mixture
25 was filtered through Celite, and the Celite washed with ethyl acetate
(200 mL). The
combined filtrates were evaporated to remove the volatiles to give a dark
color residue.
This residue was dissolved in ethyl acetate (300 mL) and filtered through a
silica gel
plug. The filtrate was evaporated and the residue was purified via flash
chromatography
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
31
eluting with a gradient of 0 - 100 (:)/0 Et0Ac in heptanes to give the title
compound (12.4
g, 62% yield). 1H NMR (400 MHz, DMSO-d6) 6 ppm 11.51 (br. s., 1 H) 9.07 (s, 1
H)
7.86 (s, 1 H) 7.52 (s, 1 H) 7.06 (d, J=2.20 Hz, 1 H) 4.31 - 4.54 (m, 2 H) 3.92
(br. s., 1 H)
3.80 (s, 3 H) 3.55 - 3.68 (m, 1 H) 3.44 - 3.55 (m, 1 H) 3.30 (d, J=18.34 Hz, 3
H) 2.72 (br.
s., 1 H) 1.39 (br. s., 9 H). m/z (APCI+) for C21H28CIN704 378.2 (M+H)+ with Cl
isotope
pattern. Optical rotation: [a]d= -8.3 degrees (c=0.24, Me0H).
Step 7: Preparation of (3R,4R)-3-f({5-chloro-2-f(1-methyl-1H-pyrazol-4-
yl)aminol-
7H-pyrrolo[2,3-d]pyrimidin-4-yl}oxy)methyl]-4-methoxypyrrolidinium
trifluoroacetate
HN 0
N CI
0
HN N 0
N-N =
H3C-0'
H3d
To a solution of tert-butyl (3R,4R)-3-[({5-chloro-2-[(1-methyl-1H-pyrazol-4-
yl)am ino]-7H-pyrrolo[2,3-d]pyrimidin-4-yl}oxy)methyI]-4-methoxypyrrolidine-1-
carboxylate (12.40 g, 26 mmol) in DCM (60 mL) in a water bath was added TFA
(10.1
mL, 208 mmol) and the resulting solution was stirred at ambient temperature
for 2.5 hrs.
The volatiles were removed to give a residue to which was added ethyl ether
(150 mL).
The resulting suspension was stirred for 2 hrs and the light pink solid was
collected by
filtration, washed with ethyl ether (30 mL) and dried to give the title
product (15.69 g,
100% yield) 1H NMR (400 MHz, DMSO-d6) 6 ppm 11.56 (br. s., 1 H) 9.09 (s, 3 H)
7.85
(s, 1 H) 7.54 (s, 1 H) 7.09 (d, J=2.32 Hz, 1 H) 4.48 (d, J=6.48 Hz, 2 H) 4.11
(br. s., 1 H)
3.81 (s,3 H) 3.46 -3.60 (m, 1 H) 3.35 -3.45 (m, 2 H) 3.32 (s,3 H) 3.15 (dq,
J=12.01,
6.02 Hz, 1 H) 2.88 (m, J=6.42, 6.42 Hz, 1 H). m/z (APCI+) for parent molecule
C16H20CIN702 378.2 (M+H)+ with Cl isotope pattern. Optical rotation: [a]d= -
4.1 degrees
(c=0.24, Me0H).
Step 8: Preparation of 14(3R,4R)-345-chloro-2-(1-methyl-1H-pyrazol-4-ylamino)-
7H-pyrrolof2,3-dlpyrim idin-4-yloxymethy11-4-methoxy-pyrrolidin-1-yllprop2-en-
1-one
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
32
HN
CI
N
HN N 0
0
N-N
H3C-0
H3C H2C
A mixture of (3R,4R)-3-[({5-chloro-2-[(1-methyl-1H-pyrazol-4-yl)am ino]-7H-
pyrrolo[2,3-d]pyrim idin-4-ylloxy)methyI]-4-methoxypyrrolidinium
trifluoroacetate (15.0 g
24.7 mmol), ethyl acetate (200 mL) and saturated aqueous NaHCO3 (100 mL) was
stirred at 0 C for 10 min. Acryloyl chloride (2.3 mL, 29 mmol, 1.1 mol eq)
was added
dropwise and the resulting mixture was stirred at ambient temperature for 30
min. Ethyl
acetate (150 mL) was added and the organic layer was separated; the aqueous
layer
was extracted with ethyl acetate (150 mL) and the combined organic layers were
dried
over Na2SO4 and evaporated to give a solid, which was purified via flash
chromatography eluting with a gradient of 0 - 50 (:)/0 ethanol in ethyl
acetate to give a
white solid. This solid was then recrystallized from ethanol (10 mL of ethanol
for 1 g of
crude) with light heating using heat gun and seeded with crystal seeds. Upon
cooling
the white crystals were collected by filtration and washed with ethanol (3 mL
of ethanol
for 1 g of crude) to give the title compound (7.47 g, 70 (:)/0) as a white
solid. 1H NMR (400
MHz, DMSO-d6) 6 ppm 11.50 (br. s., 1 H) 9.06 (s, 1 H) 7.85 (s, 1 H) 7.51 (s,1
H) 7.04
(d, J=2.32 Hz, 1 H) 6.58 (ddd, J=16.78, 10.30, 1.16 Hz, 1 H) 6.13 (dd,
J=16.81, 2.38 Hz,
1 H) 5.67 (dt, J=10.33, 2.23 Hz, 1 H) 4.43 (d, J=6.24 Hz, 2 H) 3.95 -4.05 (m,
1 H) 3.68-
3.85 (m, 4 H) 3.56 - 3.66 (m, 2 H) 3.44 - 3.53 (m, 1 H) 3.30 (d, J=4.65 Hz, 3
H) 2.68 -
2.90 (m, 1 H). m/z (APCI+) for parent molecule C19H22CIN703 432.1 (M+H)+ with
Cl
isotope pattern. Chiral purity determination: Whelk-01 (R,R) 4.6 x 250 mm
column 30 (:)/0
Et0H at 140 bar, 3mUmin. Rt= -8.8 min, Peak 1, >99% ee. Optical rotation:
[a]D22 =
-3.1 degrees (c 0.14, Et0H). Elemental analysis: Theoretical: C, 52.84; H,
5.13; Cl,
8.21; N, 22.70;. Found: C, 52.45; H, 5.38; Cl, 7.91; N, 22.02.
Example 2: Preparation of N-methyl-N-ftrans-3-({24(1-methyl-1H-pyrazol-4-
vnaminol-5-(pyridin-2-v1)-7H-Pyrrol0F2,3-dlpyrimidin-4-v1}oxv)cyclobutyllprop-
2-
enamide ("Compound B")
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
33
HN N,
N /
HN N Q
0
N¨N
\CH3 /N--1
H30
CH
Step 1: Preparation of 2,4-dichloro-5-iodo-7H-pyrrolo[2,3-c]pyrimidine
HN
N
,
-N CI
To a solution of 2,4-dichloro-7H-pyrrolo[2,3-d]pyrimidine, (50.0 g, 266 mmol,
1.00
equiv) in DMF (266 mL, 1.0 M) was added N-iodosuccinimide (62.8 g, 279 mmol,
1.05
equiv) at a rate such that the internal temperature was maintained below 50
C. The
reaction mixture was stirred vigorously and cooled in an ambient temperature
bath for
1.5 hrs. The reaction mixture was diluted with ice water (1.5 L), and the
resulting
precipitate was isolated by filtration. The precipitate was washed with ice
water (2 x 500
mL) and dried in vacuo at 45 C for 36 hrs to give the title compound (81.5 g,
98 %
yield) as an off-white solid. 1H NMR (400 MHz, DMSO-d6) 6 ppm 13.09 (br. s., 1
H),
7.95 (s, 1 H). m/z (APCI+) for C6H2C12IN3 313.9 (M+H)+.
Step 2: Preparation of 2,4-dichloro-5-iodo-74[2-(trimethylsilypethoxy]methy1}-
7H-
Pvrrolof2,3-dlpyrimidine
CH,
H3
I
H 3C
N
Cl N Cl
To a cooled (0 C) solution of 2,4-dichloro-5-iodo-7H-pyrrolo[2,3-d]pyrimidine
(81.4 g, 259 mmol, 1.00 equiv) and diisopropylethylamine (105 mL, 596 mmol,
2.30
equiv) in THF (600 mL) was added 2-(trimethylsilyl)ethoxymethyl chloride (59.5
mL, 337
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
34
mmol, 1.30 equiv) in a dropwise manner over 5 min. The reaction mixture was
allowed
to stir at 0 C for 3 hrs. At that time the reaction mixture was filtered, and
the filtrate was
concentrated in vacuo. The resulting thick oil was diluted with Et0Ac (400
mL), washed
sequentially with sat. aqueous NH4CI (2 x 200 mL) and brine (2 x 200 mL),
dried over
Na2SO4 and concentrated in vacuo. The resulting material was dissolved in a
minimum
volume of DCM (50 mL) and diluted with heptane (200 mL). This solution was
concentrated to a total volume of 150 mL which facilitated the trituration of
the title
compound. This mixture was filtered to give the title compound (84.1 g) and a
filtrate
that was concentrated and further purified via flash chromatography eluting
with a
gradient of 0 - 15 (:)/0 Et0Ac in heptane to provide an additional portion of
the title
compound (26.5 g). These two portions were combined to give the desired
product
(110.6 g) as an off-white solid. 1H NMR (400 MHz, CDCI3) 6 ppm 7.50 (s, 1 H),
5.57 (s,
2 H), 3.61 (t, J=8.0 Hz, 2 H), 0.95 (t, J=8.0 Hz, 2 H), 0.01 (s, 9 H). m/z
(APCI+) for
C12H16C121N30Si 444.0 (M+H)+.
Step 3: Preparation of 2,4-dichloro-5-(pyridin-2-y1)-7-{f2-
(trimethylsilypethoxylmethy11-7H-pyrrolof2,3-dipyrimidine
CH,
H I
3
H3C
N,
N
CI'N CI
To a cooled (-78 C) solution of 2,4-dichloro-5-iodo-7-{[2-
(trimethylsilypethoxy]methy11-7H-pyrrolo[2,3-d]pyrimidine (30.0 g, 68.0 mmol,
1.00
equiv) in THF (350 mL) was added a solution of i-PrMgCI (47.3 mL, 94.6 mmol,
1.40
equiv, 1.00 M THF) in a dropwise manner over 4 min. The reaction mixture was
stirred
at -78 C for 2 h and then treated with a freshly prepared solution of ZnBr2
(24.7 g, 110
mmol, 1.62 equiv, dried at 130 C) in THF (100 mL) in a dropwise manner over
15 min.
The mixture was stirred at -78 C for an additional 1 hr then warmed to
ambient
temperature and stirred for an additional 0.5 hrs. At this time the reaction
mixture was
treated with Pd(PPh3)4 (3.94 g, 3.38 mmol, 0.05 equiv) and 2-iodopyridine
(10.8 mL, 101
mol, 1.50 equiv) and heated to 65 C for 10 hrs. Upon cooling to ambient
temperature,
the reaction mixture was concentrated to a volume of -200 mL, diluted with
water (600
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
mL), sat. aqueous sodium potassium tartrate (100 mL) and Et0Ac (400 mL). The
layers
were separated and the aqueous layer was extracted with Et0Ac (4 x 300 mL).
The
combined organics were washed with brine (300 mL), dried (Na2SO4), and
concentrated
in vacuo. The resulting oil was purified via flash chromatography eluting with
a gradient
5 of 0 ¨ 30 (:)/0 Et0Ac in heptane to provide the title compound (23.5 g,
87 (:)/0 yield) as an
oil that converted on standing to a light tan solid. 1H NMR (400 MHz, CDCI3) 6
ppm
8.76 - 8.63 (m, 1 H), 7.83 - 7.77 (m, 1 H), 7.74 (s, 1 H), 7.67 (d, J=7.8 Hz,
1 H), 7.35 -
7.29 (m, 1 H), 5.68 (s, 2 H), 3.61 (dd, J=7.6, 8.9 Hz, 2 H), 1.03 - 0.92 (m, 2
H), -0.01 (s,
9 H). m/z (APCI+) for C17H20C12N40Si 395.1 (M+H)+.
10 Step 4: Preparation of tert-butyl (trans-3-{ftert-
butyl(dimethyl)silylloxylcyclobuty1)-
carbamate
HO OH
3
H3C
CH3
0 CH3
HN 0
oxCH3
H3C CH3
To a cooled (0 C) solution of tert-butyl (cis-3-hydroxycyclobutyl)carbamate
(62.0
g, 330 mmol, 1.00 equiv) and 4-nitrobenzoic acid (60.8 g, 360 mmol, 1.10
equiv) in THF
15 (1.0 L) was sequentially treated with PPh3 (130 g, 490 mol, 1.48 equiv)
and diethyl
azodicarboxylate (86.3 g, 490 mmol, 1.48 equiv). After the addition the
reaction mixture
was refluxed for 4 days, cooled to ambient temperature, and concentrated in
vacuo. The
residue was crystallized from i-PrOH to give a white solid (63 g).
To a solution of the above obtained 4-nitro benzoate ester (63 g) in Me0H (1.0
L)
20 and H20 (200 mL) was added K2CO3 (51.6 g, 370 mmol). The resulting
mixture was
refluxed for 2 hrs, cooled to ambient temperature, and filtered. The filtrate
was
concentrated in vacuo, and partitioned between Et0Ac and aqueous 10 (:)/0
Na2CO3.
The resulting organic layer was washed with brine and concentrated to afford a
white
solid (31.0 g).
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
36
To a solution of the above obtained alcohol (75.0 g, 400 mmol, 1.00 equiv) in
pyridine (1.0 L) was added TBSCI (91.0 g, 600 mmol, 1.50 equiv). The reaction
mixture
was stirred at ambient temperature for 2 hrs and then concentrated in vacuo.
The
resulting residue was purified via flash chromatography eluting with a
gradient of 0 - 10
(:)/0 Et0Ac in petroleum ether to provide the title compound (111 g, 92 (:)/0
yield) as a white
solid. 1H NMR (400 MHz, DMSO-d6) 6 ppm 7.13 (d, 1 H) 4.38 - 4.47 (m, 1 H) 3.90
(br.
s., 1 H) 2.00 - 2.16 (m, 4 H) 1.36 (s, 9 H) 0.81 -0.89 (m, 9 H) -0.01 -0.01
(m, 6 H). m/z
(APCI+) for C10H23NOSi 202.1 (M-Boc+H)+.
Step 5: Preparation of tert-butyl (trans-3-{[tert-
butyl(dimethyl)silyl]oxy}cyclobuty1)-
methylcarbamate
H3C a-13
H3C
\ CH3
0 CH3
H3CNy0
oxcH3
H3c cH3
To a solution of tert-butyl (trans-3-{[tert-
butyl(dimethyl)silyl]oxylcyclobutyl)carbamate (111 g, 370 mmol, 1.00 equiv) in
THF (1.0
L) was added NaH (60 (:)/0 dispersion in oil, 22.2 g, 550 mmol, 1.50 equiv) in
portions.
After the addition, the reaction mixture was stirred for an additional 0.5
hrs, cooled (0
C), and treated with methyl iodide (38.2 mL, 615 mmol, 1.66 equiv) in a
dropwise
manner. After an additional 5 hrs at ambient temperature, the reaction mixture
concentrated in vacuo, and the resulting residue was purified via flash
chromatography
eluting with 10 (:)/0 Et0Ac in petroleum ether to provide the title compound
(97.5 g, 84 (:)/0
yield) as an oil. 1H NMR (400 MHz, DMSO-d6) 6 ppm 4.64 (br. s., 1 H) 4.29 -
4.37 (m, 1
H) 2.73 (s, 3 H) 2.31 -2.43 (m, 2 H) 1.97 -2.08 (m, 2 H) 1.37 (s, 9 H) 0.86
(s, 9 H) -0.01
- 0.05 (m, 6 H). m/z (APCI+) for C11H25NOSi 216.2 (M-Boc+H)+.
Step 6: Preparation of tert-butyl (trans-3-hydroxycyclobutyl)methylcarbamate
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
37
OH
H3C -.0
oxCH3
H3C CH3
To a solution of tert-butyl (trans-3-{[tert-
butyl(dimethyl)silyl]oxylcyclobuty1)-
methylcarbamate (195 g, 600 mmol, 1.00 equiv) in THF (1.0 L) was added TBAF
(930
mL, 930 mmol, 1.55 equiv, 1 M in THF). The reaction mixture was stirred at
ambient
temperature for 3 hrs and then concentrated in vacuo. The resulting residue
was
partitioned between Et0Ac (1.0 L) and sat. aqueous NH4CI (500 mL). The
resulting
organic layer was concentrated in vacuo, and the resulting residue was
purified via flash
chromatography eluting with 10 (:)/0 Et0Ac in petroleum ether to provide the
title
compound (88 g, 76 (:)/0 yield) as a white solid. 1H NMR (400 MHz, CDCI3) 6
ppm 4.78
(s, 1 H), 4.41-4.38 (m, 1 H), 2.82 (s, 3 H), 2.41-2.38 (m, 2 H), 2.23-2.20 (m,
2 H), 1.47(s,
9 H). m/z (ESI+) for C10H18NO3 146.1 (M-tBu+H)+.
Step 7: Preparation of tert-butyl methylftrans-3-f(2-[(1-methyl-1H-pyrazol-4-
y1)amino]-5-(pyridin-2-y1)-7-{[2-(trimethylsilypethoxy]methyll-7H-pyrrolo[2,3-
d]pyrimidin-
4-y1)oxylcyclobutyl}carbamate
H3 ,
C CH
Si
H3C
0--"N
N /
HN N 0
N¨N
H3C ,N 0
HC
0 CH3
)(CH3
H3C
To a cooled solution of tert-butyl (trans-3-hydroxycyclobutyl)methylcarbamate
(12.9 g, 64.0 mmol, 1.15 equiv) in THF (100 mL) was added potassium
bis(trimethylsilyl)amide (12.4 g, 62.3 mmol, 1.12 equiv) in three portions.
After the
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
38
addition, the alkoxide solution was allowed to warm to ambient temperature
over 0.5 hr.
A separate flask was charged with 2,4-dichloro-5-(pyridin-2-y1)-7-{[2-
(trimethylsilypethoxy]methy11-7H-pyrrolo[2,3-d]pyrimidine (22.0 g, 55.6 mmol,
1.00
equiv) and THF (300 mL) and cooled (0 C). This solution was treated with the
alkoxide
solution via cannula over 5 min, and then stirred for an additional 0.5 hr at
0 C. The
reaction mixture was then diluted with brine (100 mL), water (200 mL), and
Et0Ac (600
mL). The layers were separated and the aqueous layer was extracted with Et0Ac
(4 x
200 mL). The combined organic layer was then washed with brine (200 mL), dried
(Na2SO4), and concentrated in vacuo. The resulting viscous oil (31.0 g) was
used
without further purification in the next step.
To a solution of the above obtained oil (31.0 g) in 1,4-dioxane (300 mL) was
added 1-methyl-1H-pyrazol-4-amine (7.57 g, 77.9 mmol, 1.40 equiv), Pd2dba3
(2.68 g,
2.78 mmol, 0.05 equiv), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (3.25
g, 5.56
mmol, 0.10 equiv), and C52CO3 (45.8 g, 139 mmol, 2.5 equiv). The reaction
mixture
was then sparged with a stream of nitrogen gas for 20 min and heated at 105 C
for 10
hrs with vigorous stirring. Upon cooling to ambient temperature, the reaction
mixture
was diluted with Et0Ac (500 mL), filtered through Celite, and concentrated in
vacuo.
The resulting residue was purified via flash chromatography eluting with a
gradient of 0
- 80 (:)/0 Et0Ac in heptane to provide the title compound (25.6 g, 74 (:)/0
yield) as an
orange foam. 1H NMR (400 MHz, DMSO-d6) 6 ppm 9.17 (s, 1 H), 8.56 (d, J=4.3 Hz,
1
H), 8.14 (d, J=7.9 Hz, 1 H), 7.93 (br. s., 1 H), 7.87 -7.79 (m, 1 H), 7.69 (s,
1 H), 7.55 (s,
1 H), 7.23 (dd, J=5.0, 7.2 Hz, 1 H), 5.57 (br. s., 2 H), 5.47 (br. s., 1 H),
4.84 - 4.66 (m, 1
H), 3.82 (s, 3 H), 3.63 - 3.53 (m, 2 H), 2.84 (s, 3 H), 2.75 - 2.62 (m, 2 H),
2.47 - 2.34 (m,
2 H), 1.38 (s, 9 H), 0.86 (t, J=8.0 Hz, 2 H), -0.11 (s, 9 H). m/z (APCI+) for
C31H44N804Si
621.3 (M +H)+.
Step 8: Preparation of 3-chloro-N-methyl-N-{trans-3-[(2-[(1-methyl-1H-pyrazol-
4-
y1)amino]-5-(pyridin-2-y1)-7-{[2-(trimethylsilypethoxy]methy1}-7H-pyrrolo[2,3-
d]pyrimidin-
4-yl)oxylcyclobutyllpropanamide
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
39
H3õ
CCH3
Si
H3C
0"--\N \ N,
N
HN N 0
N-N
H3C
H3C
CI
To a cooled (0 C) solution of tert-butyl methyl{trans-3-[(2-[(1-methyl-1H-
pyrazol-
4-y1)amino]-5-(pyridin-2-y1)-7-{[2-(trimethylsilypethoxy]methyll-7H-
pyrrolo[2,3-
d]pyrimidin-4-y1)oxy]cyclobutyllcarbamate (25.0 g, 40.3 mmol, 1.00 equiv) in
MeCN (600
mL) was added TFA (70.0 mL, 914 mmol, 22.7 equiv) in a dropwise manner. The
reaction mixture was stirred at 0 C and then allowed to warm to ambient
temperature
overnight. The reaction mixture was then cooled (0 C), adjusted to pH = 8
with
aqueous NaOH, and the phases separated. The aqueous phase was extracted with
Et0Ac (3 x 300 mL), and the combined organic phases were washed with brine (2
x 200
mL), dried (Na2SO4), and concentrated in vacuo. The resulting residue was
partially
purified by via flash chromatography eluting with a gradient of 2 ¨ 7 % Me0H
in DCM to
provide 4-{[trans-3-(methylamino)cyclobutyl]oxyl-N-(1-methyl-1H-pyrazol-4-y1)-
5-pyridin-
2-y1-7-{[2-(trimethylsilypethoxy]methy11-7H-pyrrolo[2,3-d]pyrimidin-2-amine as
a yellow
gum, which was used directly in the next step.
To a solution of 4-{[trans-3-(methylamino)cyclobutyl]oxyl-N-(1-methyl-1H-
pyrazol-
4-y1)-5-pyridin-2-y1-7-{[2-(trimethylsilypethoxy]methy11-7H-pyrrolo[2,3-
d]pyrimidin-2-
amine and DIPEA (16.0 mL, 91.9 mmol, 1.61 equiv) in DCM (500 mL) was added 3-
chloropropionyl chloride (8.25 mL, 86.4 mmol, 1.51 equiv) in a dropwise manner
and
then stirred at ambient temperature for 1 hr. The reaction mixture was then
washed with
H20 (2 x 100 mL) and brine (100 mL), dried (Na2SO4), and concentrated in
vacuo. The
resulting gum was triturated with MTBE (250 mL), and the solid was filtered
and dried in
vacuo to give the title compound (24.0 g, 68.9 % yield) as a yellow solid. 1H
NMR (400
MHz, DMSO-d6) 6 ppm 9.10 -9.25 (m, 1 H) 8.51 - 8.61 (m, 1 H) 8.10 -8.25 (m, 1
H)
7.92 -8.05 (m, 1 H) 7.82 - 7.91 (m, 1 H) 7.67 -7.76 (m, 1 H) 7.47 - 7.60 (m, 1
H) 7.17 -
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
7.30 (m, 1 H) 5.55 - 5.64 (m, 2 H) 5.40 - 5.54 (m, 1 H) 4.63 - 5.31 (m, 1 H)
3.83 (s, 3 H)
3.74 - 3.81 (m, 2 H) 3.53 - 3.66 (m, 2 H) 2.92 - 3.08 (m, 3 H) 2.81 - 2.89 (m,
2 H) 2.58 -
2.79 (m, 2 H) 2.29 -2.46 (m, 2 H) 0.75 - 0.93 (m, 2 H) -0.10 (s, 9 H). m/z
(APCI+) for
C29H39CIN803Si 611.2 (M +H)+.
5 Step 9: Preparation of N-methyl-N-ftrans-3-({2-f(1-methyl-1H-pyrazol-4-
yl)aminol-
5-(Pvridin-2-v1)-7H-pyrrolo[2,3-dlpyrimidin-4-ylloxy)cyclobutyllprop-2-enamide
HN N,
N /
HN N q
0
N¨N
\CH3
H3C CH
To a solution of 3-chloro-N-methyl-N-{trans-3-[(2-[(1-methyl-1H-pyrazol-4-
yl)am ino]-5-(pyridin-2-y1)-7-{[2-(trimethylsilypethoxy]methy11-7H-pyrrolo[2,3-
d]pyrim idin-
10 4-yl)oxy]cyclobutyllpropanamide (24.0 g, 39.2 mmol, 1.00 equiv) in
DCM/iPrOH (9:1,
250 mL) was added an HCI solution (250 mL, 4 M in 1,4-dioxane). The reaction
mixture
was stirred at ambient temperature overnight, concentrated in vacuo to afford
3-chloro-
N-[trans-3-({7-(hydroxymethyl)-2-[(1-methyl-1H-pyrazol-4-y1)amino]-5-pyridin-2-
y1-7H-
pyrrolo[2,3-d]pyrim idin-4-ylloxy)cyclobutyI]-N-methylpropanam ide that was
used in the
15 next step without purification.
The intermediate from the previous step was dissolved in 1,4-dioxane (80 mL)
and concentrated aqueous NH4OH (50 mL). The reaction mixture was stirred at
ambient
temperature for 3 hrs and then concentrated in vacuo. The residue was
triturated with
MTBE (100 mL), and the solid filtered and dried in vacuo to give 3-chloro-N-
methyl-N-
20 [trans-3-({2-[(1-methyl-1H-pyrazol-4-yl)amino]-5-pyridin-2-y1-7H-
pyrrolo[2,3-d]pyrimidin-
4-ylloxy)cyclobutyl]propanamide as a yellow solid that was used in the next
step without
further purification.
To a solution of 3-chloro-N-methyl-N-[trans-3-({2-[(1-methyl-1H-pyrazol-4-
yl)am ino]-5-pyridin-2-y1-7H-pyrrolo[2,3-d]pyrim idin-4-
ylloxy)cyclobutyl]propanamide in
25 Et0H (400 mL) was added K2CO3 (21.4 g, 155 mmol, 3.95 equiv) and the
reaction
mixture was stirred at ambient temperature overnight. The reaction mixture was
filtered
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
41
to remove inorganic salts, concentrated in vacuo, and dissolved in Et0Ac (100
mL).
MTBE (200 mL) was added to precipitate the crude product, which was collected
by
filtration. The filtrate was concentrated and purified via flash
chromatography eluting
with a gradient of 2 - 7 (:)/0 Me0H in DCM to provide an additional portion of
the crude
product. The combined crude material was purified via reverse phase
chromatography
using a YMC-Actus Triact C18 (150 mm x 30 mm x 5 m) column eluting with a
gradient
of 5% MeCN in H20 (0.225 (:)/0 HCOOH) to 25 (:)/0 MeCN in H20 (0.225 (:)/0
HCOOH) to
give the formate salt of the title compound (7.14 g, 37 (:)/0 over 3 steps) as
a yellow solid.
To a solution of the formate salt of the title compound (5.14 g) in H20 (200
mL)
was added sat. aqueous NaHCO3 (100 mL) and Et0Ac (200 mL) sequentially. The
layers were separated, and the aqueous layer was extracted with Et0Ac (8 x 100
mL).
The combined organics were dried (Na2SO4) and concentrated in vacuo to give
the title
compound as an amorphous solid. A portion of this amorphous solid (-3 g) was
dissolved in a minimum volume of Et0H/Et0Ac (-1:1, -120 mL), concentrated in
vacuo
to a volume of -10 mL, and diluted with Et0Ac (40 mL). This solution was
seeded with
-5 mg of crystalline product and allowed to stir at ambient temperature
overnight. The
crystallization flask was cooled (0 C) for 1 hr to promote further
crystallization. The
product was collected by filtration and dried in vacuo to give the title
compound (2.63 g)
as a white crystalline material containing Et0Ac (0.038 equiv) and Et0H (0.03
equiv).
mp = 204.9 C. 1H NMR (400 MHz, DMSO-d6, 30 C) 6 ppm 11.65 (s, 1 H), 8.93 (s,
1
H), 8.54 (d, J=3.9 Hz, 1 H), 8.14 (d, J=8.1 Hz, 1 H), 7.86 (s, 1 H), 7.82 (t,
J=7.3 Hz, 1 H),
7.53 (s, 1 H), 7.52 (s, 1 H), 7.20 (ddd, J=1.0, 4.9, 7.4 Hz, 1 H), 6.71 (br.
s., 1 H), 6.08
(br. s., 1 H), 5.66 (br. s., 1 H), 5.53 (br. s., 1 H), 5.31 -4.79 (m, 1 H),
3.82 (s, 3 H), 3.15 -
2.93 (m, 3 H), 2.76 (br. s., 2 H), 2.46 (br. s., 2 H). 1H NMR (400 MHz, DMSO-
d6, 80 C)
8 ppm 11.41 (br. s., 1 H), 8.59 (s, 1 H), 8.55 - 8.52 (m, 1 H), 8.13 (d, J=8.1
Hz, 1 H),
7.84 (s, 1 H), 7.80 (dt, J=1.9, 7.7 Hz, 1 H), 7.55 (s, 1 H), 7.50 (s, 1 H),
7.18 (ddd, J=1.0,
4.8, 7.4 Hz, 1 H), 6.66 (dd, J=10.6, 16.8 Hz, 1 H), 6.05 (dd, J=2.3, 16.8 Hz,
1 H), 5.63
(dd, J=2.3, 10.5 Hz, 1 H), 5.59 -5.53 (m, 1 H), 5.00 (t, J=8.0 Hz, 1 H), 3.82
(s, 3 H),
3.04 (s, 3 H), 2.82 - 2.71 (m, 2 H), 2.55 - 2.45 (m, 2 H). m/z (APCI+) for
C23H24N802
445.2 (M +H)+. Elemental analysis: found C, 61.96; H, 5.50; N, 24.93.
C23H24N802 +
0.038 Et0Ac + 0.030 equiv Et0H requires C, 62.06; H, 5.49; N, 24.95.
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
42
Example 3: N-ftrans-3-({5-chloro-24(1,3-dimethyl-1H-pyrazol-4-ynaminol-7H-
Pyrrolo[213-dipyrimidin-4-yl}amino)cyclobutyll-N-methylprop-2-enamide
("Compound C")
Fi
CI
N
HN N
N¨N 0
CH3
H3C
CH2
Step 1: Preparation of tert-butyl methyl{cis-341-methyl-1-
(trimethylsilypethoxylcyclobutyllcarbamate
Fi3c ,cH3
H3CsicH3
Fi3c
cH3
H3c
o cH3
To a solution of tert-butyl (cis-3-hydroxycyclobutyl)carbamate (10.5 g, 56
mmol,
1.00 equiv) in pyridine (150 mL) was added TBSCI (12.7 g, 84 mmol, 1.50
equiv). After
addition, the mixture was stirred at ambient temperature for 3 hrs. TLC
(petroleum ether
/ Et0Ac = 3/1) showed the starting material was completely consumed. The
reaction
mixture was concentrated, and the residue was extracted with Et0Ac (3 x 100
mL). The
combined organic layers were concentrated to give crude tert-butyl {cis-341-
methyl-1-
(trimethylsilypethoxy]cyclobutyllcarbamate as an oil, which was used for the
next step
without further purification.
Crude tert-butyl {cis-341-methyl-1-(trimethylsilypethoxy]cyclobutyllcarbamate
was diluted in THF (300 mL) and treated with NaH (60 %, 3.36 g, 84 mmol, 1.50
equiv)
in portions and stirred at rt for 30 min. The mixture was cooled to 0 C and
treated with
methyl iodide (23.9 g, 168 mmol, 3.0 equiv) in a dropwise manner. After
addition, the
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
43
reaction mixture was stirred at ambient temperature for 5 hrs. TLC (petroleum
ether /
Et0Ac = 10/1) indicated that the starting material had been consumed
completely. The
reaction mixture was concentrated and purified via silica gel chromatography
(petroleum
ether! Et0Ac = 10/1) to give the title compound (18 g, 100%) as an oil.
Step 2: Preparation of tert-butyl (cis-3-hydroxycyclobutyl)methylcarbamate
OH
CH3
H3C-
0 CH3
To a solution of tert-butyl methyl{cis-341-methyl-1-
(trimethylsilypethoxy]cyclobutyllcarbamate (18 g, 56 mmol, 1.0 equiv) in THF
(200 mL)
was added TBAF (22.0 g, 84 mmol, 1.50 equiv) in portions. After addition, the
mixture
was stirred at ambient temperature for 3 hrs. TLC (petroleum ether: Et0Ac =
2:1)
showed the starting material was completely consumed. The reaction mixture was
concentrated, and the residue was purified by column chromatography (petroleum
ether:
Et0Ac = 2:1) to afford the title compound (8.9 g, 79 % yield) as a white
solid.
Step 3: Preparation of tert-butyl (trans-3-aminocyclobutyl)methylcarbamate
NH2
cH3
H3C-
CH3
To a vigorously stirred cooled (-30 C) solution of tert-butyl (cis-3-
hydroxycyclobutyl)methylcarbamate (18.0 g, 0.089 mol, 1.0 equiv) and
triethylamine (37
mL, 0.267 mol, 3.0 equiv) in DCM (300 mL) was added MsCI (14.1 g, 0.123 mol,
1.38
equiv) in a dropwise manner over 30 min. The reaction mixture was then allowed
to
warm to ambient temperature and stirred for 1 hr. The reaction mixture was
diluted with
water (100 mL) and DCM (200 mL). The organic phase was separated, washed with
water (2 x 100 mL), sat. aqueous NR4C1(3 x 100 mL) and brine (100 mL), dried
over
anhydrous Na2504, and concentrated to give crude cis-3-Rtert-
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
44
butoxycarbonyl)(methyl)amino]cyclobutyl methanesulfonate as a yellow solid,
which was
used in the next step without further purification.
The above obtained crude cis-3-Rtert-butoxycarbonyl)(methyl)amino]cyclobutyl
methanesulfonate was dissolved in DMF (250 mL) and treated with NaN3 (28.77 g,
0.44
mol, 5 equiv). The resulting mixture was then heated to 70 C and stirred
overnight.
After cooling, water (1500 mL) and Et0Ac (300 mL) were added to the reaction
mixture.
The phases were separated and the aqueous layer was extracted with Et0Ac (3 x
300
mL). The combined organic phases were washed with sat. aqueous NaHCO3 (2 x 100
mL), water (2 x 200 mL) and brine (100 mL), dried over anhydrous Na2SO4, and
evaporated to give a crude tert-butyl (trans-3-
azidocyclobutyl)methylcarbamate, which
was used directly in the next step.
To a mixture of the above crude tert-butyl (trans-3-
azidocyclobutyl)methylcarbamate and Pd/C (2.5 g) in Me0H (100 mL) under a
hydrogen
atmosphere was added saturated NH3 in Me0H (200 mL) via syringe. The resulting
mixture was stirred at ambient temperature for 36 hrs. The reaction mixture
was filtered,
and the filtrate was concentrated under reduced pressure. This crude material
was
purified by column chromatography with Et0Ac/petroleum ether from 1/10 to 1/1
to
afford the title compound (13.6 g, 76.4 % yield over 3 steps) as yellow
liquid.
Step 4: Preparation of 2,4,5-trichloro-7-{f2-(trimethylsilypethoxylmethy11-7H-
pyrrolof2,3-dlpyrimidine
cH3
H3C
CI
N
CI I ,
CI
To a solution of 2,4-dichloro-7-{[2-(trimethylsilypethoxy]methy11-7H-
pyrrolo[2,3-
d]pyrimidine (100.0 g, 316 mmol, 1.0 equiv), as prepared in Example 2, step 1,
in DMF
(1800 mL) was added N-chlorosuccinimide (44.5 g, 332 mmol, 1.05 equiv) at
ambient
temperature. The resulting mixture was then stirred at 80 C for 3 hrs. The
reaction
mixture was then cooled to ambient temperature and poured into ice water (3
L). The
white precipitate formed was collected and dried in vacuo to give the title
compound
(99.7 g, 90 % yield) as a gray solid. 1H NMR (400 MHz, CDCI3) 6 ppm = 7.35 (s,
1H),
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
5.58 (s, 2H), 3.60 - 3.49 (m, 2H), 1.00 - 0.89 (m, 2H), -0.02 (s, 9H). m/z
(APCI+) for
C12H16C13N30Si 352.0 (M+H)+.
Step 5: Preparation of 1,3-dimethy1-1H-pyrazol-4-amine
NH2
N-N
CH3
5 A solution of 1,3-dimethy1-1H-pyrazol-4-amine hydrochloride (800 mg) in
Me0H
(15 mL) was treated with hydroxide resin (Bio Rad AG 1-X2 resin, catolog #143-
1255)
until pH - 8 was obtained. The mixture was stirred for 15 min. The resin was
filtered off
and washed with several portions of Me0H. The filtrate was concentrated under
reduced pressure to give the title compound (615.3 mg, 94 (:)/0 yield). This
material was
10 used without further purification. 1H NMR (400 MHz, DMSO-d6) 6 ppm =
6.89 (s, 1H)
3.57 (s, 3H) 3.54 (br. s., 2H) 1.96 (s, 3H). m/z (APCI+) for C5H9N3 112.1
(M+H)+.
Step 6: Preparation of tert-butyl {trans-3-[(2,5-dichloro-74[2-
(trimethylsilypethoxylmethy11-7H-pyrrolof2,3-dipyrimidin-4-
vl)aminolcyclobutyllmethylcarbamate
H3C CH
/ 3
H3C
CI
N
I
CI N' NH
CH3
H3CNC5(
15 0 H3C CH3
A mixture of tert-butyl (trans-3-am inocyclobutypmethylcarbamate (1020 mg,
5.1mmol, 1.2 equiv), 2,4,5-trichloro-7-{[2-(trimethylsilypethoxy]methy11-7H-
pyrrolo[2,3-
d]pyrimidine (1500 mg, 4.253 mmol, 1.0 equiv) and DIPEA (2.12 mL, 12.8 mmol,
3.0
equiv) in MeCN (21.0 mL, 0.2M) was heated at 80 C for 5.5 hrs. The reaction
mixture
20 was diluted with water and extracted with Et0Ac. The organic layer was
dried over
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
46
Na2SO4 and concentrated. The residue was purified via flash chromatography (10
to 30
(:)/0 Et0Ac in heptane) to give the tile compound (2.22 g, 100% yield) as a
clear gum. 1H
NMR (400 MHz, DMSO-d6) 6 ppm = 7.54 (s, 1H) 7.05 (d, J=5.99 Hz, 1H) 5.40 (s,
2H)
4.74 (br. s., 1H) 4.41 - 4.54 (m, 1H) 3.45 - 3.54 (m, 2H) 2.83 (s, 3H) 2.52 -
2.63 (m, 2H)
2.28 - 2.42 (m, 2H) 1.40 (s, 9H) 0.78 - 0.89 (m, 2H) -0.08 (s, 9H). m/z
(APCI+) for
C22H35Cl2N503Si 516.2 (M+H)+.
Step 7: Preparation of tert-butyl ftrans-3-f(5-chloro-2-[(1,3-dimethyl-1H-
pyrazol-4-
y1)amino]-7-{[2-(trimethylsilypethoxy]methyl}-7H-pyrrolo[2,3-d]pyrimidin-4-
y1)aminolcyclobutyl}methylcarbamate
H3c\ /cH3
H3C
CI
N
HN/\ NH
N-N
CH3H3
H3C
0 FI3C CH3
A flask containing 1,3-dimethy1-1H-pyrazol-4-amine (567 mg, 5.10 mmol, 1.2
equiv) and tert-butyl grans-3-[(2,5-dichloro-7-{[2-
(trimethylsilyl)ethoxy]nethy1}-7H-
pyrrolo[2,3-d]pyrimidin-4-0amino]cyclobutylynethylcarbarnate
(2197 mg, 4.253 mmol, 1.00 equiv) was charged with Pd2(dba)3 (393 mg, 0.425
mmol,
0.1 equiv), Xantphos (259 mg, 0.425 mmol, 0.1 equiv) and C52CO3 (4160 mg, 12.8
mmol, 3.0 equiv). 1,4-Dioxane (42 mL, 0.1 M) was added and the mixture was
heated to
105 C for 18 hrs. The reaction was cooled tort and filtered through a pad of
Celite.
The filtrate was concentrated, and the residue was purified via flash
chromatography (40
-60 (:)/0 Et0Ac in heptane) to give the title compound (1950 mg, 78 (:)/0
yield) as a foam.
1H NMR (400 MHz, DMSO-d6) 6 ppm = 8.03 (s, 1H) 7.87 (s, 1H) 7.10 (s, 1H) 6.33
(d,
J=6.24 Hz, 1H) 5.35 (s, 2H) 4.67 (br. s., 1H) 4.55 (br. s., 1H) 3.68 - 3.75
(m, 3H) 3.45 -
3.53 (m, 2H) 2.85 (s, 3H) 2.52 -2.60 (m, 2H) 2.29 -2.38 (m, 2H) 2.12 (s, 3H)
1.40 (s,
9H) 0.78 -0.88 (m, 2H) -0.10 (s, 9H). m/z (APCI+) for C27H43CIN803Si 591.3
(M+H)+.
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
47
Step 8: Preparation of N-ftrans-3-({5-chloro-2-f(1,3-dimethyl-1H-pyrazol-4-
yl)aminol-7H-
Pyrrolo[2,3-dlpyrimidin-4-yllamino)cyclobutyll-N-methylprop-2-enamide
CI
N
HN N NH
1
0
N¨N
CH3
H3C L
CH2
To a cooled (0 C) solution of tert-butyl {trans-3-[(5-chloro-2-[(1,3-dimethyl-
1 H-
pyrazol-4-y1)amino]-7-{[2-(trimethylsilypethoxy]methyll-7H-pyrrolo[2,3-
c]pyrimidin-4-
y1)amino]cyclobutyllmethylcarbamate (1950 mg, 3.30 mmol, 1.0 equiv) in DCM (42
mL)
was added TFA (31 mL). The reaction mixture was allowed to come to rt and was
stirred
for an additional 16 hrs. The reaction mixture was then diluted with toluene
(30 mL) and
concentrated to dryness. The resulting crude residue was dissolved in 1,4-
dioxane (20
mL) and concentrated aqueous NH4OH (20 mL) and stirred at ambient temperature
for 3
hrs. The reaction mixture was then evaporated to dryness. The resulting crude
solid
was partitioned between Et0Ac (140 mL) and sat. aqueous Na2CO3 (140 mL) and
treated with acryloyl chloride (0.420 mL, 5.19 mmol, 1.58 equiv) with vigorous
stirring for
1 hr. At this time the layers were separated and the aqueous layer was
extracted with
Et0Ac (50 mL). The combined organic layers were dried (Na2504), diluted with
toluene
(30 mL), and evaporated to dryness. The resulting solid was purified by SFC
using a
ZymorSpher HAP 150 x 21.2 mm column with 20 ¨ 40 % Et0H @4 %/min, 140 bar, 55
mL/min to afford the title compound (950 mg, 69 % yield) as a grey powder. 1H
NMR
(400 MHz, DMSO-d6, 26 C) 6 ppm = 11.16 (br. s., 1H), 7.80 (br. s., 1H), 7.77
(s, 1H),
6.88 (d, J=2.4 Hz, 1H), 6.74 (dd, J=10.5, 16.7 Hz, 1H), 6.32 (br. s., 1H),
6.07 (d, J=15.9
Hz, 1H), 5.66 (d, J=10.4 Hz, 1H), 5.30 -4.75 (m, 1H), 4.59 (br. s., 1H), 3.71
(s, 3H), 3.15
- 2.88 (m, 3H), 2.62 (br. s., 2H), 2.40 (br. s., 2H), 2.08 (s, 3H). 1H NMR
(400 MHz,
DMSO-d6, 80 C) 6 ppm = 10.93 (br. s., 1H), 7.73 (s, 1H), 7.41 (br. s., 1H),
6.82 (d,
J=2.2 Hz, 1H), 6.68 (dd, J=10.5, 16.9 Hz, 1H), 6.16 (d, J=5.9 Hz, 1H), 6.05
(dd, J=2.4,
16.8 Hz, 1H), 5.63 (dd, J=2.4, 10.5 Hz, 1H), 4.94 (t, J=8.0 Hz, 1H), 4.60 (dd,
J=4.0, 8.7
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
48
Hz, 1H), 3.72 (s, 3H), 3.04 (s, 3H), 2.72 -2.57 (m, 2H), 2.41 (ddd, J=4.5,
8.9, 13.6 Hz,
2H), 2.10 (s, 3H). m/z (APCI+) for C19H23CIN803 415.1 (M+H)+.
Example 4: Preparation of 1-{(3R,4R)-3-[({5-chloro-2-[(3-methoxy-1-methyl-1 H-
Pvrazol-4-ynaminol-7H-pyrrolor2,3-dlpyrimidin-4-ylloxy)methyll-4-
methoxypyrrolidin-1-yllprop-2-en-1-one ("Compound 0")
HN
H30\ HN N 0
0¨OH
3
0( N "%õ,
N -
CH3
CH2
Prepared in a manner analogous to Example 1, substituting 1-methyl-1H-pyrazol-
4-am ine with 3-methoxy-1-methyl-1H-pyrazol-4-amine and other non-critical
substitutions.
Experimental Procedures for Key Intermediates
Preparation I. Preparation of tert-butyl-3-(hydroxymethyl)-4-
(methoxymethyl)pyrrolidine-1-carboxylate
HO
0CHrC)3
¨C)\ ,CH3
0 iC
H3C H3
Step 1: Preparation of ethyl (2E)-4-{[tert-butyl(dimethyl)silyl]oxy}but-2-
enoate
0
H3C\
H3C
>rH \C
H3C 3 OCH3
cH
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
49
DIEA (2.75 mL, 16.6 mmol) and LiCI (5.54 g, 129 mmol) were added to a solution
of tert-butyldimethylsilyloxy acetaldehyde (3.22 g, 18.5 mmol) and
diethylmethylphosphonoacetate (4.66 g, 22.2 mmol) in CH3CN (40 mL) and the
mixture
was stirred at rt for 24 hrs. The mixture was quenched with water (50 mL) and
extracted
with Et0Ac (50 mL). The organic layer was dried over MgSO4 and concentrated.
The
residue was purified via flash chromatography eluting with 25 % Et0Ac/heptane
to give
the title compound as a colorless oil (3.27 g, 72 % yield). 1H NMR (400 MHz,
chloroform-d) 6 ppm 6.91 (dt, J=15.42, 3.49 Hz, 1 H) 6.01 (dt, J=15.61, 2.27
Hz, 1 H)
4.25 (dd, J=3.27, 2.27 Hz, 2 H) 4.12 (q, J=7.22 Hz, 2 H) 1.21 (t, J=7.18 Hz, 3
H) 0.84 (s,
9 H) 0.00 (s, 6 H).
Step 2: Preparation of trans-ethyl-1-benzy1-4-
({ftertbutyl(dimethyl)silylloxylmethyl)
pyrrolidine-3-carboxylate
H3C0OH3
H3C 1-"CH3
H3C-\ 0\
0 __________________________________________
N
401
To a solution of ethyl (2E)-4-{[tert-butyl(dimethyl)silyl]oxylbut-2-enoate
(3.27 g,
13.4 mmol) and N-benzy1-1-methoxy-N-((trimethylsilyl)methyl)methanamine (4.14
g,
17.5 mmol) in CH2Cl2 (30 mL) was added TFA (0.280 mL, 3.64 mmol) at 0 C. The
reaction was stirred at rt overnight. The mixture was quenched with water (50
mL) and
extracted with Et0Ac (two x 50 mL). The combined organic layers were dried
over
Mg504 and concentrated. The residue was purified via flash chromatography
eluting
with 20 % Et0Ac/heptane to give the title compound as a pale yellow oil (2.61
g, 53 %
yield). 1H NMR (400 MHz, chloroform-d) 6 ppm 7.08 - 7.41 (m, 5H), 4.10 (q, J =
7.13 Hz,
2H), 3.42 - 3.73 (m, 4H), 2.37 - 2.90 (m, 6H), 1.22 (t, J = 7.05 Hz, 3H), 0.84
(s, 9H), 0.00
(d, J = 1.26 Hz, 6H).
Step 3: Preparation of trans-l-tert-butyl 3-ethyl-4-ffltert-
butyl(dimethyl)silylloxylmethyl) pyrrolidine-1,3-dicarboxylate
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
H3CN/CH3
H3CsC-CH3
/ CH3
H3C¨\ ____41)0
0
0 0
H3C-4¨CH3
CH3
To a solution of trans-ethyl-1-benzy1-4-ffltert-
butyl(dimethyl)silyl]oxylmethyl)
pyrrolidine-3-carboxylate (trans mixture) (3.25 g, 8.61 mmol) in Et0H (40 mL)
was
added Pd(OH)2 (300 mg) and Boc20 (1.90 g, 8.61 mmol). The mixture was stirred
5 under H2 (50 psi, 50 C) overnight. The mixture was filtered through
Celite and the
filtrate was concentrated. The residue was purified via flash chromatography
eluting
with 5 % -10 % Et0Ac/heptane to give the title compound as a colorless oil
(3.08 g, 92
% yield). 1H NMR (400 MHz, chloroform-d) 6 ppm 4.13 -4.25 (m, 2 H) 3.65 (m, 5
H)
3.14 - 3.29 (m, 1 H) 2.84 - 3.00 (m, 1 H) 2.47 - 2.70 (m, 1 H) 1.46 (s, 9 H)
1.27 (td,
10 J=7.11, 2.64 Hz, 3 H) 0.85 -0.92 (m, 9 H) 0.05 (s,6 H).
Step 4: Preparation of trans-tert-buty1-3-({ftert-
butyl(dimethyl)silylloxylmethyl)-4-
(hydroxymethyl)pyrrolidine-1-carboxylate
H3C JF13
H3C1-:CH3
0/ CH3
N
0 0
H3C+CH3
CH3
LiBH4 (911 mg, 39.7 mmol) was added to a solution of trans-l-tert-butyl 3-
ethyl-4-
15 (fitert-butyl(dimethyl)silyl]oxylmethyl)pyrrolidine-1,3-dicarboxylate
(3.08 g, 7.95 mmol) in
THF (25 mL). The mixture was heated to reflux for 3 hrs. The reaction mixture
was
cooled to rt, then quenched with water (15 mL) and stirred at rt for 1 hr. The
mixture
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
51
was diluted with water (60 mL) and extracted with ethyl acetate (two x 80 mL).
The
combined organic layers were washed with brine, dried over Na2SO4 and
concentrated
in vacuo to give a colorless oil. The crude product was purified via flash
chromatography
eluting with 30 % Et0Ac/heptane to give the title compound as a colorless oil
(2.34 g, 86
% yield). 1H NMR (400 MHz, chloroform-d) 6 ppm 3.73 (m, 1H), 3.61 (m, 2H),
3.52 (m,
2H), 3.45 (m, 1H), 2.90 - 3.09 (m, 2H), 2.04 - 2.32 (m, 2H), 1.46 (s, 9H),
0.92 (s, 9H),
0.10 (d, J = 1.01 Hz, 6H).
Step 5: Preparation of trans-tert-butyl-3-ffltert-
butyl(dimethyl)silyl]oxy}methyl)-4-
(methoxymethyl)pyrrolidine-1-carboxylate
H3C JH3
H3CsCCH3
0 CH3
H3C/
N
0 0
H3C+CH3
CH3
Tetrabutylammonium iodide (0.110 g, 0.28 mmol), 50% aqueous NaOH (20 mL)
and dimethyl sulfate (0.325 mL, 3.41 mmol) were added to a solution of trans-
tert-buty1-
3-(fftert-butyl(dimethyl)silyl]oxy}methyl)-4-(hydroxymethyl)pyrrolidine-1-
carboxylate
(0.982 g, 2.84 mmol) in CH2Cl2 (20 mL). The reaction was stirred at rt
overnight. TLC
showed some starting material remaining so additional dimethyl sulfate (0.150
mL) was
added to the reaction mixture and stirred at rt for 3 hrs. Aqueous NH3OH (30
mL) was
added to the reaction mixture and stirred at rt for 1 hr. The mixture was
diluted with
water (20 mL) and extracted with CH2Cl2 (two x 30 mL). The organic layer was
dried
over Mg504 and concentrated. The residue was purified via flash chromatography
eluting with 10% Et0Ac/heptane to give the title compound as a colorless oil
(451 mg,
44 % yield). 1H NMR (400 MHz, chloroform-d) 6 ppm 3.60 -3.70 (m, 1H), 3.55
(br. s.,
2H), 3.37 - 3.48 (m, 1H), 3.34 (m, 4H), 3.05 - 3.23 (m, 2H), 2.22 - 2.40 (m,
1H), 2.07 -
2.21 (m, 1H), 1.43 - 1.49 (m, 9H), 0.89 (s, 9H), 0.05 (s, 6H).
Step 6: Preparation of trans-tert-butyl-3-(hydroxymethyl)-4-
(methoxymethyl)pyrrolidine-1-carboxylate
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
52
H
HC O
00
H3C+CH3
CH3
TBAF (1.0 M in THF, 2.45 mL, 2.45 mmol) was added to a solution of trans-tert-
buty1-3-(fftert-butyl(dimethyl)silyl]oxylmethyl)-4-(methoxymethyl)pyrrolidine-
1-
carboxylate (290 mg, 0.81 mmol) in THF (5 mL). The mixture was stirred at rt
for 1 hr.
The mixture was quenched with water and extracted with Et0Ac. The organic
layer was
dried over MgSO4 and concentrated. The crude product was used without
purification in
subsequent steps.
Preparation 2. Preparation of tert-butvl (trans-3-
aminocyclobutvnmethvIcarbamate
H C
3
CH3
0
H2N
Step 1: Preparation of 3-methylidenecyclobutanecarboxylic acid
HO
)=0
H2C
To a solution of 3-methylidenecyclobutanecarbonitrile (110 g, 1.18 mol) in
ethanol
(500 mL) and water (500 mL) was added potassium hydroxide (264 g, 4.7 mol) and
the
resulting mixture was refluxed overnight. The ethanol was removed under
reduced
pressure, and then the solution was cooled to below 10 C and acidified with
concentrated HCI to pH 1. The mixture was extracted with Et0Ac (two x 500 mL)
and
the combined organic extracts were dried over anhydrous sodium sulfate and
concentrated under vacuum to afford compound the title compound (132 g, 100 %
yield)
as yellow oil.
Step 2: Preparation of tert-butyl (3-methylidenecyclobutyl)carbamate
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
53
CH3
H 0+CH
CH3 3
0
H2C
To a solution of 3-methylidenecyclobutanecarboxylic acid (132 g, 1.17 mol) and
Et3N (178 g, 1.76 mol) in tert-butyl alcohol (1 L) was added dropwise DPPA
(574 g, 1.41
mol) and the resulting mixture was refluxed overnight. The mixture was then
quenched
with water (100 mL). After removal of the tert-butyl alcohol, the residue was
treated with
sat. NH4CI (500 mL), and the resulting solid precipitate was collected, washed
with sat.
NH4CI and sat. NaHCO3 to give the title compound (165 g, 77 % yield) as a
white solid.
Step 3: Preparation of tert-butyl (3-oxocyclobutyl)carbamate
H 0,/CH3
N C H 3
CH3
0
0
To a solution of tert-butyl (3-methylidenecyclobutyl)carbamate (165 g, 0.91
mol)
in CH2Cl2 (1000 mL) and Me0H (1000 mL) was bubbled 03 at -78 C until the
solution
turned blue. TLC (petroleum ether: Et0Ac = 10:1) showed that the starting
material was
consumed completely. Nitrogen gas was then bubbled through the reaction to
remove
excess 03, and then the mixture was quenched with Me25 (200 mL) and stirred
for an
hour. The solution was concentrated to give a residue, which was washed with
sat.
NaHCO3 and water to yield the title compound (118 g, 70 % yield) as a white
solid.
Step 4: Preparation of tert-butyl (cis-3-hydroxycyclobutyl)carbamate
OH3
H
\1,H3c' 1Ni¨CH3
HO
To a solution of tert-butyl (3-oxocyclobutyl)carbamate (100 g, 54 mmol) in THF
(2000 mL) at -72 C was added dropwise a solution of lithium trisec-
butylhidridoborate
(648 mL, 1 M) in THF over 1.5 hrs. The resulting solution was allowed to warm
up to rt
and stirred for another 1 hr. TLC (petroleum ether: Et0Ac = 2:1) showed that
the
starting material was consumed completely. The reaction was quenched with
NH4CI
aqueous. Water (1000 mL) and Et0Ac (2000 mL) were added to the mixture. The
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
54
organic layer was separated, dried over MgSO4 and concentrated to give crude
material,
which was purified by column chromatography with petroleum ether: Et0Ac from
10:1 to
1:2 to afford the title compound (62 g, 61 % yield) as a white solid.
Step 5: Preparation of tert-butyl {cis-3-[1-methyl-1-
(trimethylsilypethoxylcyclobutyllcarbamate
OH3
H 0,(
7
H C CH3
HO OH)3 I 0 3
H3 C¨Si+0
/
H3C CH3
To a solution of tert-butyl (cis-3-hydroxycyclobutyl)carbamate (62 g, 0.33
mol) in
pyridine (1 L) was added TBSCI (159 g, 1.056 mol). After addition, the mixture
was
stirred at ambient temperature overnight. TLC (petroleum ether: Et0Ac = 2:1)
showed
the starting material was consumed completely. The reaction was then
concentrated
and diluted with Et0Ac (1 L), and the organic layer was separated and washed
with
water (three x 300 mL) and brine (200 mL), dried over Mg504, filtered and
concentrated
to dryness to give crude title compound (108 g), which was used for the next
step
directly without further purification.
Step 6: Preparation of tert-butyl methyl{cis-341-methy1-1-
(trimethylsilyDethoxylcyclobutyllcarbamate
H,C CH3
0
CH3
tN
H C
HC CH3 I __________________________________ I 0 3
3 \
H3 C¨Si+0
/
H3C CH3
To a solution of crude tert-butyl {cis-341-methyl-1-
(trimethylsilypethoxy]cyclobutyllcarbamate (108 g) in THF (1 L) was added NaH
(60 %
in oil, 39.6 g, 0.99 mol) in portions and the resulting mixture was stirred at
rt for 30 min.
The mixture was then cooled to 0 C and iodomethane (140.58 g, 0.99 mol) was
added
dropwise. After addition, the mixture was stirred from 0 C to rt overnight.
The mixture
was quenched with sat. NH4CI, and water was added (200 mL), and extracted with
Et0Ac. The organic layer was washed with brine, dried over Na2504, then
evaporated
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
to give crude product which was purified via silica gel chromatography to give
the title
compound (68.9 g, 87 % yield) as an oil.
Step 7: Preparation of tert-butyl (cis-3-hydroxycyclobutyl)methylcarbamate
HO OH
3\ ----Z
/ -CH3
r
HO
5 To a solution of tert-butyl methyl{cis-341-methy1-1-
(trimethylsilypethoxy]cyclobutyllcarbamate (68.9 g, 0.217 mol) in pyridine
(800 mL) was
added TBAF (62 g, 0.24 mol) in portions. After addition, the mixture was
stirred at rt for
2 hrs. The mixture was evaporated to dryness, and the residue was dissolved in
1000
mL of ethyl acetate and washed with conc. NH4CI (three x 200 mL). The organic
layer
10 was dried over Na2504, filtered and concentrated to give the crude
product, which was
purified by column chromatography with Et0Ac/petroleum ether from 1/20 to 1/5
to
afford the title compound (26.3 g, 60 % yield) as a white solid.
Step 8: Preparation of cis-3-Rtert-butoxycarbonyl)(methyl)aminolcyclobutyl
methanesulfonate
HO CH
3 \N0 3
C 3H
0 ______________________________________ r ,H3C
0.0
H3C;S-0
Triethylamine (4.14 mL, 29.79 mmol) was added into the solution of tert-butyl
(cis-3-hydroxycyclobutyl)methylcarbamate (2.0 g, 9.93 mmol) in CH2Cl2 (30 mL)
and the
resulting mixture was cooled to -30 C upon vigorous stirring. Mesyl chloride
(1.36 g,
11.91 mmol) was added dropwise over a ten minute period. The mixture was then
allowed to warm to rt and stirred for an hour until TLC analysis (Me0H/CH2C12
= 1/15)
showed the reaction was complete. The reaction mixture was then washed with
water
(two x 10 mL), aq. NH4CI (10 mL), brine (10 mL), dried over anhydrous Na2504
and
concentrated to give the title compound (2.5 g, 91 % yield) as yellow solid,
which was
used for next step directly.
Step 9: Preparation of tert-butyl (trans-3-azidocyclobutyl)methylcarbamate
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
56
HO OH3
3 X oz
/ ¨CH3
,H3c
_______________________________ S.
1\ls
Cis-3-Rtert-butoxycarbonyl)(methyl)amino]cyclobutyl methanesulfonate (2.5 g,
8.94 mmol) was dissolved in DMF (25 mL) and NaN3 (2.84 g, 43.69 mmol) was
added.
The resulting mixture was then heated to 70 C and stirred overnight. After
cooling,
water (150 mL) was added and the mixture was extracted with Et0Ac (three x 50
mL).
The combined organic phases were washed with water (three x 20 mL) and brine
(20
mL), dried over anhydrous Na2SO4, then concentrated in vacuo to give the title
compound (1.8 g, 89 % yield) as a yellow liquid, which was used without
further
purification.
Step 10: Preparation of tert-butyl (trans-3-am inocyclobutyl)methylcarbamate
H3C\ CH3
\01-13C
H2N
To the mixture of tert-butyl (trans-3-azidocyclobutyl)methylcarbamate (1.8 g,
7.95
mmol) and Pd/C (200 mg) in Me0H (5 mL) under hydrogen atmosphere (hydrogen
balloon) was added NH3(g)/Me0H (saturated, 50 mL) via syringe. The resulting
mixture
was stirred at rt for three hours until TLC analysis (Et0Ac:petroleum ether =
1:2)
showed the reaction was complete. Pd/C was filtered off and the resulting
solution was
concentrated and dried in vacuum to afford crude title compound (1.6 g), which
was
used for the next steps without further purification.
25
CA 02904797 2015-09-09
WO 2014/140989 PCT/1B2014/059401
57
Table 1
Example
No. LRMS
(Scheme) Structure and Compound Name m/z 1H NMR
H:2 1H NMR (400 MHz,
CI DMSO-d6) 6 ppm
N 11.51 (s, 1 H) 9.07
(s, 1 H) 7.86 (s, 1 H)
HN N 0
0 7.52 (s, 1 H) 7.05 (s,
1
14.4:0N
---1 H) 6.59 (ddd,
(Scheme F) N-N
J=16.75, 10.27, 1.34
/ H3C-0 Hz, 1 H) 6.14 (dd,
H3C H2C 431.9 J=16.75, 2.32 Hz, 1
TFA salt of=TFA H) 5.68 (dt, J=10.27,
Compound
2.32 Hz, 1 H) 4.44
A 1-{(3R,4R)-3-[({5-chloro-2-[(1- (d, J=6.24 Hz, 2 H)
methyl-1H-pyrazol-4-y1)amino]- 3.82 -4.09 (m, 2 H)
7H-pyrrolo[2,3-d]pyrimidin-4- 3.80 (s, 3 H) 3.57 -
ylloxy)methy1]-4- 3.76 (m, 2 H) 3.47 -
methoxypyrrolidin-1-yllprop-2-en- 3.54 (m, 1 H) 3.31
1-one trifluoroacetate (d, J=4.65 Hz, 3 H)
2.67 - 2.92 (m, 1 H).
1H NMR (400 MHz,
DMSO-d6) 6 ppm
H:2._ 11.50 (br. s., 1 H)
N
CI 9.06 (s, 1 H) 7.85 (s,
,k , 1 H) 7.51 (s, 1 H)
HN N 0 7.04 (d, J=2.32 Hz, 1
11.44:0N 0
---H) 6.58 (ddd,
J=16.78, 10.30, 1.16
H3C
Alternate 1 N-N Hz, 1 H) 6.13 (dd,
/ -0
H3C H2C 432.1 J=16.81, 2.38 Hz, 1
Compound H) 5.67 (dt, J=10.33,
A 2.23 Hz, 1 H) 4.43
1-{(3R,4R)-3-[({5-chloro-2-[(1-
(d, J=6.24 Hz, 2 H)
methyl-1H-pyrazol-4-y1)amino]-
3.95 -4.05 (m, 1 H)
7H-pyrrolo[2,3-d]pyrimidin-4-
3.68-3.85 (m, 4 H)
ylloxy)methyI]-4-
3.56 - 3.66 (m, 2 H)
methoxypyrrolidin-1-yllprop-2-en-
3.44 - 3.53 (m, 1 H)
1-one
3.30 (d, J=4.65 Hz, 3
H) 2.68 - 2.90 (m, 1
H).
CA 02904797 2015-09-09
WO 2014/140989 PCT/1B2014/059401
58
Example
No. LRMS
(Scheme) Structure and Compound Name m/z 1H NMR
1H NMR (700 MHz,
HN \ N, DMSO) 6 ppm 11.68
N \ / (br. s., 1 H) 8.96 (s,
A 1 H) 8.55 (d, J=3.96
HN N Q Hz, 1 H) 8.15 (d,
1 J=7.70 Hz, 1 H) 7.75
- 7.94 (m, 2 H) 7.54
2 '7
(d, J=8.36 Hz, 2 H)
(Scheme I) N-N q 0
\ 7.21 (dd, J=6.93,
cH3 /N---1
Compound H3c \---.. -CH2 445'1 5.17 Hz, 1 H)
6.57-
6.92 (m, 1 H) 5.92 -
B 6.22 (m, 1 H) 5.61 -
N-methyl-N-[trans-3-({2-[(1- 5.82 (m, 1 H) 5.54
methyl-1H-pyrazol-4-y1)amino]-5- (br. s., 1 H) 5.25 (br.
(pyridin-2-yI)-7H-pyrrolo[2,3- s.,1 H) 3.83 (s, 3 H)
d]pyrimidin-4-
2.97 -3.15 (m, 3 H)
ylloxy)cyclobutyl]prop-2-enamide 2.76 (br. s., 2 H)
2.34 -2.49 (m, 2 H)
HIT....._ 1H NMR (400 MHz,
ci DMSO-d6) 6 ppm
N 11.16 (br. s., 1 H)
A , 7.77 (s, 2 H) 6.88 (s,
HN N NH 1 H) 6.74 (dd,
i
J=16.63, 10.52 Hz, 1
3 H C--.....,
3 \ H) 6.30 (d, J=6.11
(Scheme B) N-N 0 Hz, 1 H)6.07 (d,
. ,N.---
-z----
CH, H3C \
415.1 J=15.89 Hz, 1 H)
-
Compound 5.66 (d, J=10.03 Hz,
.CH2
C 1 H) 4.81 - 5.19 (m,
1 H) 4.59 (br. s., 1
N-[trans-3-({5-chloro-2-[(1,3- H) 3.71 (s, 3 H) 2.93
dimethy1-1H-pyrazol-4-y1)amino]- -3.15 (m, 3 H) 2.62
7H-pyrrolo[2,3-d]pyrimidin-4- (br. s., 2 H) 2.39(br.
yllamino)cyclobutyI]-N- s., 2 H) 2.08 (s, 3 H)
methylprop-2-enamide
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
59
Example
No. LRMS
(Scheme) Structure and Compound Name m/z 1H NMR
1H NMR (400 MHz,
DMSO-d6) 6 ppm
11.43 (br. s., 1 H)
N 7.80(s, 1 H)
7.56 (d,
I J=2.27 Hz, 1 H)
6.91
HN N 0
H C (s, 1 H) 6.49
(dd,
3 3
=,õ O-CH J=16.80,
10.23 Hz, 1
/ H) 6.04 (dd,
4 N-N J=16.67, 2.27
Hz, 1
(Scheme F) CH3 H) 5.58 (dt,
J=10.29,
Compound 462.1 1.80 Hz, 1 H)
4.21 -
4.39 (m, 2 H) 3.89 -
o CH2
3.99 (m, 1 H) 3.79 -
3.87 (m, 1 H) 3.64 -
1-{(3R,4R)-3-[({5-chloro-2-[(3- 3.74 (m, 3 H)
3.62
methoxy-1-methyl-1H-pyrazol-4- (d, J=5.05 Hz, 1
H)
yl)amino]-7H-pyrrolo[2,3- 3.55 - 3.60 (m,
3 H)
c]pyrimidin-4-ylloxy)methy1]-4- 3.45 - 3.54 (m,
1 H)
methoxypyrrolidin-1-yllprop-2-en- 3.29 -3.44 (m, 1
H)
1-one 3.20 (d, J=4.55
Hz, 3
H) 2.59 -2.77 (m, 1
H)
Biological Examples
Example 5: pEGFR Y1068 ELISA Assay
In order to profile the effect of EGFR T790M inhibitors in cells with
different
EGFR mutation status, inhibition of phosphorylation of EGFR at Tyr1068 was
determined in cells with wildtype EGFR and EGFR double mutants (L858R+T790M,
EGFR delE746-A750 +T790M). Phosphorylation of EGFR at Y1068 was measured by
PathScan Phospho-EGF Receptor (Try1068) Sandwich ELISA kit (#7240, Cell
Signaling Technology , Danvers, MA). The PathScan Phospho-EGF Receptor
(Tyr1068) Sandwich ELISA Kit is a solid phase sandwich enzyme-linked
immunosorbent
assay (ELISA) that detects endogenous levels of phospho-EGF Receptor (Tyr1068)
protein. The following cell lines were evaluated in this assay: A549 (EGFR
wildtype,
endogenous), NCI-H1975 (EGFR L858R+T790M, endogenous),
NIH3T3/EGFR_wildtype, NIH3T3/EGFR L858R+T790M and PC9-DRH (EGFR delE746-
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
A750 +T790M). NIH/3T3 parental, A549, and NCI-H1975 cells were purchased from
the
American Type Culture Collection (Manassas, VA). All cells were cultured
according to
ATCC recommendations. A549 cells were grown in RPM! media (Invitrogen,
Carlsbad)
supplemented with 10 % FBS (Sigma, St Louis, MO), and with 1 % Penn/Strep
5 (Invitrogen). NCI-H1975 cells were grown in RPM! (Invitrogen)
supplemented with 10%
FBS (Sigma), and with 1 % Penn/Strep (Invitrogen). NIH/3T3 cells were grown in
DMEM (Invitrogen) supplemented with 10 % newborn calf serum (Invitrogen), and
NIH3T3/EGFR mutant cells were grown in complete media with 5 pg/mL puromycin
(Invitrogen). PC9-DRH cells were generated and cultured as described in
Example 6.
10 Plasm ids (pLPCX) with various EGFR constructs were made by GenScript
(Piscataway,
NJ), and stable pools of NIH/3T3 cells expressing these constructs were made
at Pfizer
La Jolla. Cells were plated in complete culture media (50 pL/well) on the
bottom of clear
tissue culture treated microtiter plates (#3595, Corning Inc, Corning, NY) and
allowed to
adhere overnight at 37 C, 5 % CO2. Cells were seeded at the following
concentrations:
15 (A549: 40,000/well, NCI-H1975: 40,000/well, NIH3T3: 20,000/well, PC9-
DRH:
50,000/well). The following day, compound dilution plates were prepared in 96
well
clear V-bottom 0.5 mL polypropylene block plates (#3956, Corning, Inc). All
cell lines
were not evaluated for each compound. Each compound evaluated was prepared as
a
DMSO stock solution (10 mM). Compounds were tested in duplicate on each plate,
with
20 an 11-point serial dilution curve (1:3 dilution). Compound treatment (50
pL) was added
from the compound dilution plate to the cell plate. The highest compound
concentration
was 1 or 10 pM (final), with a 0.3 % final DMSO (#D-5879, Sigma)
concentration. Plates
were then incubated for 2 hrs at 37 C, 5 % CO2. For NIH3T3/wildtype assay,
cells
were serum starved for 24 hrs prior to compound treatment; cells were treated
in serum-
25 free media as described and then stimulated for 10 min with EGF (100
ng/mL,
Calbiochem/EMD Chemicals, Gibbstown, NJ). For A549/wildtype assay, cells were
plated in full-serum (10 %) media for 24 hrs prior to compound treatment;
cells were
treated in full serum media as described and then stimulated for 10 min with
EGF (40
ng/mL/starvation media, Invitrogen). Immediately prior to the end of the
incubation, ice-
30 cold lysis buffer was prepared (lx Cell Lysis Buffer (#9803, Cell
Signaling Technology),
1 mM sodium orthovanadate (Na3VO4, #96508, Sigma), 1 mM phenylmethanesulfonyl
fluoride (PMSF, 52332, CalBiochem/EMD Chemicals), complete Mini EDTA-free
Protease Inhibitor Cocktail Tablet (1 tablet/10 mL, #11836170001, Roche,
Indianapolis,
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
61
IN), and PhosSTOP Phosphatase Inhibitor Cocktail Tablet (1 tablet/10 mL,
#04906837001, Roche) in pure water. At the end of 2 hrs, media was flicked off
and
cells were washed once with ice-cold 1 mM Na3VO4 in PBS (100 L/well,
Invitrogen).
The wash was then flicked off and ice-cold lysis buffer was added to the cells
(50
L/well). The plate was shaken for 20-30 min at 4 C to completely lyse the
cells.
Sample diluent (50 L/well) was added to the ELISA plate, and the lysate (50
4) was
diluted into the sample diluent in each well of the ELISA plate. Plates were
sealed and
incubated overnight at 4 C with shaking. The next day, wells were washed four
times
with lx Wash Buffer; plates were taped on lint-free paper after the final wash
prior to
adding Add Detection Antibody (green, 100 L/well) to each well and incubating
for 1 hr
at 37 C. After incubation, wells were washed as described. HRP-Linked
secondary
antibody (red, 100 L/well) was added to each well and incubated for 30 min at
37 C.
After incubation, the wells were washed as described. TMB Substrate (100
L/well) was
added to each well and the plate incubated for 10 minutes at 37 C or 30
minutes at
room temperature maximum. Stop Solution (100 L/well) was added to each well
at the
end of the incubation and plates were shaken gently for a few seconds.
Absorbance
was read at 450 nm within 30 min after addition of Stop Solution on a
PerkinElmer
EnVision Excite Multilabel Reader Method for Absorbance or on a Molecular
Devices
SpectraMax384 Reader for absorbance. Data were analyzed using a four-parameter
fit
in Microsoft Excel.
The results of the pEGFR Y1068 ELISA assays for the compounds tested are
listed in Table 2. The pEGFR ELISA IC50 data shown in Table 2 for T790M_L858R
is
for 3T3 cell lines, unless otherwise indicated.
Table 2
pEGFRY1068
ELISA3T3T790M_ pEGFRY1068 pEGFRY1068
Example L858R PC9-DRH ELISAA549
Number IC50 (nM) IC50 (nM) IC50 (nM)
1 7 N/D >4,287
Alternate 1 15
2 6(H1975) N/D 1650
3 27 14 4286
4 12 6 >10,000
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
62
Example 6: Generation and Characterization of RPC9 and PC9-DRH Cells
Step 1: Generation of RPC9 cells from PC9 cells
In parental PC9 cells, the EGFR delE746-A750 mutant allele is amplified and no
wild-type EGFR allele can be detected. Parental PC9 cells were utilized in the
generation of the RPC9 cells. PC9 cells were cultured at 37 C with 5 % CO2 in
RPM!
1640 medium supplemented with 10% heat inactivated FBS. To generate EGFR
inhibitor resistant cell lines, PC9 cells were initially treated with 0.5 nM
dacomitinib.
Once cells grew up to 90 % of confluence, they were split and the drug
concentration
was escalated by two-fold. After six weeks of such treatment, PC9 cells could
grow in 2
nM dacomitinib. Single cell clones were generated and ten were selected for
futher
characterization. Those resistant cells were maintained in growth medium
containing 2
pM erlotinib, and were named RPC9 for Resistant PC9.
Step 2: CastPCR analysis
Genomic DNA was extracted from clones of RPC9 cells using Qiagen DNA mini
kit following manufacture's recommendations and subjected to castPCR (primer
sets:
Hs00000106_wt and Hs00000105) following the protocol from the manufacturer
(ABI).
Data were analyzed via ABI mutation detector software.
Step 3: Cell viability IC50 determination
3000 RPC9 cells per well were seeded in 90 pL of growth medium in duplicate
wells of a 96 well plate (Corning). 24 hours later, cells were treated with
dacomitinib or
erlotinib in an 11 point titration of 3-fold dilution in 10 pL growth medium.
The highest
final concentration was 10 pM. After 72 hours of treatment, cells were
analyzed via
CTG assay (Promega) following manufacture's instructions.
Step 4: Characterization of RPC9 cells harboring EGFR T790M mutation
In the 10 clones that were generated in Step 1, Sanger sequencing identified
the
C>T mutation in EGFR exon20, which corresponds to the clinically relevant
T790M
mutation. Sequences of representative clones, RPC9 clone 3 and clone 6, are
shown in
Figure 1. Further validation via castPCR showed there were 10.2 % and 11.9 %
of
EGFR alleles harboring the EGFR T790M mutation in RPC9 clones 3 and 6,
respectively (Figure 1). PC9 cells are very sensitive to dacomitinib (Figure
2A). Even
the lowest concentration (0.17 nM) of dacomitinib inhibited 96% of PC9 cell
viability
(Figure 2A). IC50 could not be calculated according to the dose response
curve. RPC9
clones 3 and 6 were more resistant with IC5os of 73 and 64 nM (Figure 2A).
When
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
63
RPC9 clones 3 and 6 were treated with erlotinib, RPC9 clones 3 and 6 showed
more
than 200 fold increase in IC50 compared with the PC9 cells in the cell
viability assay
(Figure 2B).
Accordingly, RPC9 cells which harbor EGFR T790M mutation and are resistant to
EGFR inhibitors such as dacomitinib and erlotinib were generated. The RPC9
cells
contain a mixture of both single mutant (EGFR delE746-A750) and double mutant
(EGFR delE746-A750 and T790M) EGFR alleles, since the EGFR T790M allele
constitutes about 10 % of the total EGFR alleles in RPC9 cells.
Step 5: Generation and Characterization of PC9-DRH cells
In addition to RPC9 cells, PC9-DRH cells (DRH = dacomitinib resistant high
T790M) were also generated. The RPC9 cell pool resistant to 2 nM dacomitinib
was
further challenged with increasing concentrations of dacomitinib from 2 nM to
2 M in 8
weeks, as described in Step 1. PC9-DRH cells were maintained in growth medium,
as
described in Step 1, containing 2 M dacomitinib. PC9-DRH cells were analyzed,
as
described in Steps 2, 3, and 4. PC9-DRH cells contain 70 % of their EGFR
alleles as
double mutant EGFR delE746-A750 and T790M. Similar to RPC9 cells, PC9-DRH
cells
are resistant to dacomitinib (IC50 = 1,651 nM), erlotinib (IC50 >10,000 nM),
and gefitinib
(IC50 >10,000 nM). When used in the pEGFR Y1068 ELISA assay as described in
Example 5, the 2 pM dacomitinib was removed from growth medium and cells were
allowed to grow for 36 hours prior to use in the ELISA assay.
Example 7: RPC9 Cell Viability utilizing EGFR T790M inhibitors alone or in
combination with dacomitinib or erlotinib
3000 RPC9 cells, as prepared in Example 6, per well were seeded in 90 pL of
growth medium into duplicate wells of a 96 well plate (Corning). 24 hours
later, cells
were treated with one of the EGFR T790M inhibitors, Compound A, Compound B,
Compound C or Compound D in an 11 point titration of three-fold dilution with
or without
either 4 nM dacomitinib or 300 nM erlotinib in 10 pL growth medium. The
highest final
concentration was 10 pM of Compound A, Compound B, Compound C or Compound D.
After 72 hours of treatment, cells were analyzed via CTG assay (Promega)
following
manufacture's instruction.
The free plasma concentration at steady-state from the standard clinical
dosing
regimen of dacomitinib and erlotinib are 4 nM and 300 nM, respectively. At
those
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
64
concentrations, dacomitinib and erlotinib completely inhibited parental PC9
cell viability
(Figure 2A and 2B). Neither drug significantly inhibited RPC9 cell viability
at the same
concentrations (Figure 2A and 2B).
The inhibition of viability in RPC9 clone 6 cells was potentiated by a
combination
of Compound A with either dacomitinib or erlotinib (Figures 3A and 3B). The
viability
IC50 of Compound A was 17 nM when combined with 4 nM dacomitinib and 15 nM
when
combined with 300 nM erlotinib (Table 3). The viability IC50s for Compound A
in
combination decrease over 11 fold compared with that of Compound A treatment
alone.
Similarly, when RPC9 clone 6 cells were treated with Compound B, dacomitinib
and
erlotinib also sensitized RPC9 clone 6 to Compound B (Figures 4A and 4B). The
IC50 of
Compound B was 4 nM in combination with dacomitinib and 5 nM in combination
with
erlotinib (Table 3). The viability IC50s decreased by 9.5 fold and 7.6 fold
compared with
that of Compound B treatment alone. Importantly, the projected human exposure
for
Compound A is 190 nM at which concentration Compound A alone inhibited cell
viability
about 40 %. When combined with dacomitinib or erlotinib, the same
concentration of
Compound A achieved maximal inhibition (83 %) (Figure 3A and 3B). Similarly
for
Compound B, the projected human exposure is 90 nM at which concentration,
Compound B alone achieved 64 % inhibition. The combinations further
potentiated the
inhibition up to 84 % (Figure 4A and 4B). Thus, a combination with dacomitinib
or
erlotinib enhances the viability effect for Compound A and Compound B. In
addition to
Compound A and Compound B, Compound C and Compound D were synergistic with
the clinically relevant concentrations of dacomitinib and erlotinib (Table 3).
Table 3. Viability IC50 of EGFR T790M inhibitors alone or in combination with
dacomitinib or erlotinib in RPC9 clone 6.
IC50 (nM)
in combination with in combination
Compound single agent dacomitinib with erlotinib
dacomitinib 67
erlotinib 6433
Compound A 199 17 15
Compound B 38 4 5
Compound C 234 19 16
Compound D 441 30 21
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
In conclusion, compounds which specifically target the single mutant form of
EGFR, such as dacomitinib and erlotinib, used at their clinically relevant
concentrations
potentiated compounds which preferentially inhibit the double mutant form of
EGFR,
such as Compound A, Compound B, Compound C and Compound D, in clinically
5 relevant models that harbor both double mutant and single mutant forms of
EGFR.
Example 8: RPC9 Clone 6 Cell Viability utilizing EGFR T790M inhibitors alone
or in
combination with dacomitinib, qefitinib, or afatinib
Using the method of Example 7, cells were treated with one of the EGFR T790M
10 inhibitors, Compound A or Compound B, with or without either 4 nM
dacomitinib, 20 nM
gefitinib or 20 nM afatinib.
The free plasma concentration at steady-state from the standard clinical
dosing
regimen of dacomitinib is 4 nM. The free plasma concentration at steady-state
from the
standard clinical dosing regimen of gefitinib and afatinib is 20 nM.
15 The inhibition of viability in RPC9 clone 6 cells was potentiated by a
combination
of Compound A with either dacomitinib, gefitinib or afatinib (Figures 5A, 5B
and 5C).
Similarly, when RPC9 clone 6 cells were treated with Compound B, dacomitinib,
gefitinib
or afatinib also sensitized RPC9 clone 6 to Compound B (Figures 6A, 6B and
6C). Thus,
each of Compound A and Compound B were synergistic with the clinically
relevant
20 concentrations of dacomitinib, gefitinib and afatinib (Figures 5 and 6).
Similar to the
discussion in example 7, the viability IC50s of Compound A decreased 19 fold
and 14
fold when combined with gefitinib and afatinib, respectively. The IC50s of
Compound B
decreased 10 fold and 8 fold when combined with gefitinib and afatinib,
respectively.
In conclusion, compounds which specifically target the single mutant form of
25 EGFR, such as dacomitinib, gefitinib or afatinib, used at their
clinically relevant
concentrations potentiated compounds which preferentially inhibit the double
mutant
form of EGFR, such as Compound A and Compound B, in clinically relevant models
that
harbor both double mutant and single mutant forms of EGFR.
30 Example 9: EGFR T790M inhibitors in combination with dacomitinib or
erlotinib in
Allele Mixture Models
Methodology
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
66
Cell Culture: RPC9 clone 6 cells were generated and subcloned as described in
Example 6. Cells were cultured in RPM! with 10% FBS and were maintained under
selective pressure (2 nM dacomitinib). For experiments, selective pressure was
removed, cells plated onto 10 cm dishes and incubated overnight (37 C, 5 %
CO2) to
achieve 70-80 % confluence for treatments.
Treatments: Dacomitinib, erlotinib, Compound A, and Compound B were
dissolved in 100 % DMSO. Dacomitinib (4 nM) and erlotinib (300 nM) were used
at their
free plasma exposure from standard clinical dosing regimen. Compound A and
Compound B were used at a range starting below the target modulation IC50
value and
up to the predicted clinical free plasma exposure for each compound,
respectively.
Cells were treated with dacomitinib or erlotinib and/or a titration of
Compound A or
Compound B, or with control (DMSO). Treatment was applied for 6 hours; at the
end of
the incubation period, cell pellets were collected and frozen until ready for
analysis.
Immunoblotting: Cell pellets were treated with lysis buffer (150 mM NaCI, 1.5
mM
MgC12, 50 mM HEPES, 10% glycerol, 1 mM EGTA, 1 % Triton X-100, 0.5% NP-40)
supplemented with 1 mM Na3VO4, 1 mM PMSF, 1 mM NaF, 1 mM p-glycerophosphate,
protease inhibitor cocktail (Roche, Indianapolis, IN), and phosphatase
inhibitor cocktail
(Roche). Protein concentration of cell lysates was determined using the BCA
Protein
Assay (Pierce/Thermo Fisher Scientific, Rockford, IL) per the manufacturer's
instructions. Protein (10 pg) was resolved by SDS-PAGE and transferred onto
nitrocellulose membranes (Bio-Rad CriterionTM System, Hercules, CA). Blots
were
probed with primary antibodies to detect proteins of interest. EGFR, pEGFR
Y1068,
AKT, pAKT S473, ERK, and pERK T202/204 antibodies were purchased from Cell
Signaling Technology, Inc (Danvers, MA). GAPDH antibody was purchased from
Santa
Cruz Biotechnology (Santa Cruz, CA). After incubation with secondary
antibodies,
membranes were visualized by chemiluminescence (Pierce/Thermo Fisher
Scientific)
and densitometry was performed on the FluorChem Q Imaging System
(ProteinSimple,
Santa Clara, CA).
Results
Cells treated with both dacomitinib and Compound A exhibited a decrease in
pEGFR signaling that is consistent with a greater effect of combination versus
single
agent therapy at the two lowest doses (77 % inhibition with dacomitinib, 24 %
inhibition
with 10 nM Compound A versus 91 % with dacomitinib + 10 nM Compound A; 21%
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
67
inhibition with 30 nM Compound A versus 99 % with dacomitinib + 30 nM Compound
A)
(Figure 7A, 8A). These combinations exhibited greater than additive inhibition
of pERK
signaling at the lowest dose (34 % inhibition with dacomitinib, 27 %
inhibition with 10 nM
Compound A versus 100 % with dacomitinib + 10 nM Compound A; and an additive
effect at the next highest dose of (34 % inhibition with dacomitinib, 64 %
inhibition with
30 nM Compound A versus 99 % with dacomitinib + 30 nM Compound A; (Figure 7A,
8C). Higher doses with single agent therapy led to greater inhibition of pEGFR
and
pERK signaling; additivity calculations were constrained by the maximal
inhibition
observed in the assay. In contrast, inhibition of pAKT appeared to be additive
at the
lowest concentration of (37 % inhibition with dacomitinib, 22 % inhibition
with 10 nM
Compound A versus 64 % with dacomitinib + 10 nM Compound A; but achieved no
greater than 60-65 % inhibition even at the higher doses (Figure 7A, 8B).
Cells treated with both erlotinib and Compound A exhibited a decrease in pEGFR
signaling greater than the additive effect of single agent therapy at the
three lowest
concentrations of Compound A (61 % inhibition with erlotinib, 41 % inhibition
with 10 nM
Compound A versus 86 % inhibition with erlotinib + 10 nM Compound A; 27 %
inhibition
with 30 nM Compound A versus 91 % inhibition with erlotinib + 30 nM Compound
A; 16
% inhibition with 100 nM Compound A versus 95 % inhibition with erlotinib +
100 nM
Compound A) (Figure 7B, 9A). These treatments exhibited greater than additive
inhibition of pERK signaling at the two lowest doses (31 % inhibition with
erlotinib, 38 %
inhibition with 10 nM Compound A (versus 100 % with erlotinib + 10 nM Compound
A;
54 % inhibition with 30 nM Compound A versus 100 % with erlotinib + 30 nM
Compound
A; Figure 7B, 9C). Higher doses with single agent therapy led to greater
inhibition of
pEGFR and pERK signaling; additivity calculations were constrained by the
maximal
inhibition observed in the assay. In contrast, inhibition of pAKT appeared to
be greater
than additive at the lowest concentration of Compound A (18 % inhibition with
erlotinib, 3
% inhibition with 10 nM Compound A versus 48 % with erlotinib + 10 nM Compound
A;
Figure 7B, 9B) and additive at the next highest dose (18 % inhibition with
erlotinib, 31 %
inhibition with 30 nM Compound A versus 49 % with erlotinib + 30 nM Compound
A;
(Figure 7B, 9B), but achieved no greater than 50 % inhibition even at the
higher doses.
Cells treated with both dacomitinib and Compound B exhibited a decrease in
pEGFR signaling greater than the additive effect of single agent therapy at
the two
lowest doses (54 % inhibition with dacomitinib, 46 % inhibition with 3 nM
Compound B
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
68
versus 81 % with dacomitinib + 3 nM Compound B; 17 % inhibition with 10 nM
Compound B versus 90 % with dacomitinib + 10 nM Compound B) and additive at
the
next highest dose (54 % inhibition with dacomitinib, 33 % inhibition with 30
nM
Compound B versus 90% with dacomitinib + 30 nM Compound B) (Figure 10A, 11A).
These same cells exhibited an additive inhibition of pERK signaling at the
lowest dose
(57 % inhibition with dacomitinib, 55 % inhibition with 3 nM Compound B versus
100 %
with dacomitinib + 3 nM Compound B; (Figure 10A, 11C). Higher doses with
single
agent therapy led to greater inhibition of pEGFR and pERK signaling;
additivity
calculations were constrained by the maximal inhibition observed in the assay.
In
contrast, inhibition of pAKT achieved no greater than 72 % inhibition even at
the higher
doses, and did not achieve partial or greater than additive results (Figure
10A, 11B).
Cells treated with both erlotinib and Compound B exhibited a decrease in pEGFR
signaling greater than the additive effect of single agent therapy at all
concentrations of
Compound B (12 % inhibition with erlotinib, 2 % inhibition with 3 nM Compound
B
versus 74% with erlotinib + 3 nM Compound B; 8 % inhibition with 10 nM
Compound B
versus 66% inhibition with erlotinib + 10 nM Compound B; 22 % inhibition with
30 nM
Compound B versus 82 % with erlotinib + 30 nM Compound B; 60 % inhibition with
100
nM Compound B versus 84 % with erlotinib + 100 nM Compound B) (Figure 10B,
12A).
These treatments exhibited greater than additive inhibition of pERK signaling
at the
lowest dose of Compound B (41 % inhibition with erlotinib, 39 % inhibition
with 3 nM
Compound B versus 99 % with erlotinib + 3 nM Compound B; Figure 10B, 12C).
Higher
doses with single agent therapy led to greater inhibition of pEGFR and pERK
signaling;
additivity calculations were constrained by the maximal inhibition observed in
the assay.
In contrast, inhibition of pAKT appeared to be greater than additive at the
lowest
concentration of Compound B (29 % inhibition with erlotinib, 15 % inhibition
with 3 nM
Compound B versus 54 % with erlotinib + 3 nM Compound B; but achieved no
greater
than 62 % inhibition even at the higher doses (Figure 10B, 12B).
Example 10: EGFR T790M inhibitors in combination with dacomitinib or erlotinib
in the RPC9 clone 6 (dacomitinib and erlotinib resistant) Xenoqraft Model
Background:
RPC9 clone 6 cells were generated from parental PC9 cells as described in
Example 6. Parental PC9 cells contain EGFR delE746-A750 and are sensitive to
the
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
69
treatments of dacomitinib and erlotinib. RPC9 clone 6 cell line was one of the
selected
resistant clones generated by dose-escalation treatment with dacomitinib
("daco").
RPC9 clone 6 cells contain approximately 10 % EGFR delE746-A750 and T790M and
90 % EGFR delE746-A750 alleles. Therefore, in the in vitro assays, RPC9 clone
6 was
resistant to dacomitinib/erlotinib single agent treatments due to the EGFR
delE746-A750
and T790M allele as well as resistant to Compound A ("compd A") and Compound B
("compd B") single agent treatments due to the EGFR delE746-A750 allele.
Combination of Compound A or Compound B and clinically relevant concentrations
of
dacomitinib or erlotinib generated a synergistic effect on cell viability via
synergistic
inhibition of EGFR signal pathway. Therefore, the in vivo animal studies were
performed to evaluate whether combination of Compound A or Compound B with
dacomitinib or erlotinib would generate a synergistic anti-tumor effect in the
RPC9 clone
6 xenograft model.
Methods:
Four- to six-week-old SCID beige female mice were obtained from Charles River
lab and maintained in pressurized ventilated caging at the Pfizer La Jolla
animal facility.
All studies were approved by Pfizer Institutional Animal Care and Use
Committees.
Tumors were established by subcutaneously injecting 5x106 RPC9 clone 6 cells
suspended 1:1 (v/v) with reconstituted basement membrane (Matrigel, BD
Biosciences).
For tumor growth inhibition (TGI) studies, mice with established tumors of -
300 mm3
were selected and randomized, then treated with EGFR T790M inhibitors as
single
agent or in combination with dacomitinib or erlotinib using the indicated
doses and
regimens. Tumor dimensions were measured with vernier calipers and tumor
volumes
were calculated using the formula of 7/6 x larger diameter x (smaller
diameter)2. Tumor
growth inhibition percentage (TGI %) was calculated as 100 x (1-AT/AC). Tumor
regression percentage was calculated as 100 x (1-AT/starting tumor size).
Compound A was formulated in spray dried dispersion suspension in 0.5 %
Methoce1/20mM Tris Buffer at pH 7.4. Compound B was formulated in in-situ
lactate
salt solution with 0.5 % Methocel. Dacomitinib was formulated in 0.1 M lactic
acid
solution at pH 4.5. Erlotinib was formulated in 40 % Captisol . All drugs were
formulated and dosed at the concentration of 10 m L/kg. Tumor bearing mice
were orally
and daily administrated with indicated treatments; body weight and health
observation
were recorded daily.
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
Results:
Study I: Combination of Compound A and dacomitinib
In this study, RPC9 clone 6 tumor-bearing mice were randomized and treated
with either single agent of Compound A or dacomitinib, or Compound A in
combination
5 with dacomitinib. Dacomitinib at 5 mg/kg gave an average unbound drug
concentration
of 4 nM in mouse plasma, which matches the average clinical exposure from the
clinical
dose of 45 mg/kg/day. The body weight change percentages were plotted in
Figure
13B, and indicated that all dose groups of this study were well tolerated with
body
weight loss less than 10 %. Compound A was dosed at 500 mg/kg, 200 mg/kg, and
50
10 mg/kg as single agent or in combination with dacomitinib at 5 mg/kg as
indicated in the
Figure 13A. At study day 39, when the tumor sizes of vehicle group reached
average
1200 mm3, single agent treatment of dacomitinib generated a tumor growth
stasis and
single agent treatments of compound A generated a dose-dependent tumor growth
inhibition as illustrated in Figure 13A and Table 4. Combination of all dose
ranges of
15 Compound A with dacomitinib generated complete tumor regression as
illustrated in
Table 4.
To further assess the combination effects on the tumor regression, mice in the
single and combination treatment groups of Compound A at 200 mg/kg and 50
mg/kg
continued to receive treatments until the study day 61. The tumor growth
inhibition and
20 tumor regression were calculated and illustrated in Table 4. Results
indicate that (1)
combination of compound A with dacomitinib generated complete tumor regression
at
both dose ranges, (2) tumors in the single agent treatment groups of compound
A at
both 200 mg/kg and 50 mg/kg progressed in a dose-dependent manner which mimics
in
vitro resistance probably driven by EGFR delE746-A750 allele, and (3) tumors
in the
25 single treatment group of dacomitinib also progressed which mimics the
in vitro
resistance probably driven by EGFR delE746-A750 and T790M allele in this RPC9
clone
6 xenograft model.
The treatment period was further extended to assess the in vivo resistance by
single agent treatment and combination treatment. The single treatment group
of
30 dacomitinib continued to progress and was terminated at day 74 when
tumor size
reached above 1200 mm3. Similarly, the single treatment group of compound A at
200
mg/kg continued to progress and was terminated at day 95 when tumor size
reached
above 1400 mm3. Therefore, tumors in the single treatment groups of either
dacomitinib
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
71
or compound A continued to progress and demonstrated the in vivo resistance
similar to
the in vitro characteristics. Combination of dacomitinib and compound A at 50
mg/kg
group was able to achieve 100 % TGI, furthermore, combination of dacomitinib
and
compound A at 200 mg/kg maintained tumor regression until the end of study at
day 120
as shown in Figure 13A and Table 4.
Table 4: Tumor Growth Inhibition and Regression in Study I.
Study day Day 39 Day 61
Day 120
TGI Regression TGI Regression TGI Regression
daco_5 mg/kg 96% 45% NAb
compd A_500 mg/kg 7% NA*
compd A_200 mg/kg 91% 65% NAc
compd A_50 mg/kg 68% NAa
compd A_500 mg/kg +
daco_5 mg/kg 100% NA*
compd A_200 mg/kg +
daco_5 mg/kg 100% 100% 100%
compd A_50 mg/kg +
daco_5 mg/kg 100% 100% 80%
*: this study group was terminated at day 39, since there were no tumors
detectable for combination arm, animals were used for safety end points.
a: this study group was terminated at day 53, mean tumor volume was
above 1200 mm3.
b: this study group was terminated at day 74, mean tumor volume was
above 1200 mm3.
C: this study group was terminated at day 95, mean tumor volume was
above 1400 mm3.
Study II: Combination of Compound B and dacomitinib
In this study, RPC9 clone 6 tumor-bearing mice were randomized and treated
with either single agent of Compound B or dacomitinib, or Compound B in
combination
with dacomitinib. Dacomitinib was dosed at 5 mg/kg and 1.5 mg/kg. Compound B
was
dosed at 50 mg/kg, 15 mg/kg, and 5 mg/kg as indicated in the Figure 14A. The
body
weight change percentages were plotted in Figure 14B, and indicated that all
dose
groups of this study were well tolerated with body weight loss less than 10 %.
At study
day 36, when the tumor sizes of vehicle group reached average 1000 mm3, single
agent
treatments of dacomitinib generated a dose-dependent tumor growth inhibition
as
illustrated in Figure 15A. Single agent treatments of Compound B at 5 mg/kg
and 15
mg/kg were not significantly effective due to the extreme low dosages, while
single
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
72
agent treatment of Compound B at 50 mg/kg gave a 47 % T G I as shown in Figure
15A.
Combination of Compound B and dacomitinib generated dose-dependent tumor
regression as shown in Figure 15B. The tumor growth inhibition and regression
were
calculated and illustrated in Table 5.
Table 5: Tumor Growth Inhibition and Regression in Study II.
Study day Day 36
TGI Regression
Dacomitinib_5 mg/kg 97%
Dacomitinib_1.5 mg/kg 80%
Compound 13_50 mg/kg 47%
Compound B_15 mg/kg 2%
Compound B_5 mg/kg 18%
Compound 13_50 mg/kg +
Dacomitinib_5 mg/kg 92%
Compound B_15 mg/kg +
Dacomitinib_5 mg/kg 72%
Compound B_5 mg/kg +
Dacomitinib_5 mg/kg 52%
Compound 13_50 mg/kg +
Dacomitinib_1.5 mg/kg 31%
Study III: Combination of compound A and erlotinib
In this study, RPC9 clone 6 tumor-bearing mice were randomized and treated
with either single agent of Compound A or erlotinib, or Compound A in
combination with
erlotinib. Erlotinib at 25 mg/kg gave an average unbound drug concentration of
300 nM
in mouse plasma, which matches the average clinical exposure. The body weight
change percentages were plotted in Figure 16B, and indicated that all dose
groups of
this study were well tolerated with body weight loss less than 10 %. Compound
A was
dosed at 400 mg/kg, 200 mg/kg, and 50 mg/kg as single agent or in combination
with
erlotinib at 25 mg/kg as indicated in the Figure 16A. At study day 45, when
the tumor
sizes of vehicle group reached above 1500 mm3, single agent treatment of
erlotinib
generated Si % tumor growth inhibition and single agent treatments of compound
A
generated a dose-dependent tumor growth inhibition as illustrated in Figure
16A and
Table 6. Combination of all dose ranges of compound A with erlotinib generated
tumor
regression as illustrated in Table 6.
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
73
To further assess the combination effects on the tumor regression, mice in the
single and combination treatment groups of Compound A at 200 mg/kg and 50
mg/kg
continued to receive treatments until the study day 73. The tumor growth
inhibition and
tumor regression were calculated and illustrated in Table 6. Similar to the
results of
Study I in combination with dacomitinib, the results from this study also
indicate that
combination of Compound A with erlotinib generated tumor regression at both
dose
ranges, and tumors in the single agent treatment groups of compound A or
erlotinib
continued to progress indicating the in vivo resistance driven by either del
or del/T790M
allele.
Therefore, in conclusion, combination of compound A with either dacomitinib or
erlotinib achieved synergistic antitumor activity to induce tumor regression
in the
resistant RPC9 clone 6 xenograft model.
Table 6: Tumor Growth Inhibition and Regression in Study III.
Study day Day 45 Day 73
TGI Regression TGI
Regression
Erlotinib_25 mg/kg 51% NAa
Compound A_400 mg/kg 39% NA*
Compound A_200 mg/kg 94% 76%
Compound A_50 mg/kg 63% NAb
Compound A_400 mg/kg +
Erlotinib_25 mg/kg 92% NA*
Compound A_200 mg/kg
+ Erlotinib_25 mg/kg 80% 75%
Compound A_50 mg/kg +
Erlotinib_25 mg/kg 44% 35%
*: these groups were terminated at day 45, since there were no tumors
detectable
for combination arm, animals were used for safety end points.
a: this group was terminated at day 52, mean tumor volume was above 1500
M M3 .
b: this group was terminated at day 55, mean tumor volume was above 1500
M M3 .
Conclusions:
RPC9 clone 6 xenograft model which harbors both EGFR delE746-A750 and
EGFR delE746-A750/T790M alleles exhibited tumor progression when treated with
compound A, compound B, dacomitinib, or erlotinib as single agent treatment.
The
model showed complete regression when treated with high doses of compound A
with
clinical relevant dose of dacomitinib or erlotinib as combination therapy, as
well as
CA 02904797 2015-09-09
WO 2014/140989
PCT/1B2014/059401
74
demonstrated dose-dependent tumor regression when treated with low doses of
compound B in combination with dacomitinib. Therefore, current preclinical
animal
studies have successfully demonstrated that the combination strategy of EGFR
T790M
selective inhibitors with dacomitinib or erlotinib is a mechanism based and
potentially
clinically translatable strategy to develop EGFR T790M clinical candidates in
NSCLC
patients with both primary and acquired EGFR mutations.