Note: Descriptions are shown in the official language in which they were submitted.
AZAINDOLE COMPOUNDS, SYNTHESIS THEREOF,
AND METHODS OF USING THE SAME
Background
[0001] This application claims priority to Indian Provisional Patent
Application No. 31 96/
CHE/2013, filed July 17, 2013, entitled "Azaindole Compounds, Synthesis
Thereof, and Methods
of Using the Same," and also claims priority to a second and updated
provisional application
having the same serial number (3196/CHE/2013) and a filing date of April 30,
2014.
[0002] Tuberculosis (TB) continues to cause considerable morbidity and
mortality worldwide,
despite having an effective and economical quadruple drug therapy regimen, put
in place 40 years
ago (Raviglione, M. et al. Lancet 379, 1902-1913 (2012); World Health
Organization. Global
Tuberculosis Report (2012)). It is gratifying to see US Food and Drug
Administration (FDA)'s
recent accelerated approval of Janssen's Sirturo (bedaquiline) for multidrug-
resistant tuberculosis
(MDR-TB), putting an end to four-decade-long lull for a new TB drug with novel
mechanism of
action (Cohn, J. Science 339, 130-131 (2013)). However, the impact of Sirturo
on disease
landscape and patient's lives needs to be seen; in the context of associated
safety risks and the
burden of post marketing studies.
[0003] The nitro-benzothiazinones (BTZs) and related compounds are known to
inhibit
decaprenylphosphoryl-3-D-ribose2 '-epimerase 1 (DprE 1) involved in the
conversion of
decaprenylphosphoryl-P-D-ribose (DPR) to decaprenylphosphoryl-P-D-
arabinofuranose (DP A), a
precursor of mycobacterial cell wall arabinan (Trefzer, C. et al. J. Am. Chem.
Soc. 132,
13663-13665). This reaction is catalysed by a heteromeric enzyme decaprenyl-
phospho-ribose 2'-
epimerase (DprE), which occurs via a sequential oxidation-reduction mechanism
involving an
intermediate ( decaprenylphosphory1-2-keto-P-D-erythro-pentofuranose, DPX).
This enzyme is
composed of two proteins encoded by the dprE 1 and dprE2 genes. DprE 1 enzyme
is the FAD-
containing oxidoreductase, while DprE2 is the NADH-dependent reductase
(Mikusova, K. et al. J.
Bacteriol. 187, 8020-8025 (2005); Makarov, V. et al. Science 324, 801-804
(2009)).
[0004] The identification of BTZ043 as a covalent inhibitor of DprEl with
potent
antimycobacterial activity confirms the validity of this target for a novel TB
therapy (Science 324,
801-804 (2009)). However, it remains to be understood whether non-nitro
inhibitors of
1
Date Recue/Date Received 2021-01-18
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
DprEl will lead to efficacy in vivo? Additionally, is nanomolar cellular
activity essential for in
vivo efficacy? Greater understanding in relation to these aspects of DprE1
inhibition will
significantly influence future TB drug discovery efforts directed at this
target. Thus, a need
exists in the art for additional compounds that target DprEl.
2
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
Sunimary of the Invention
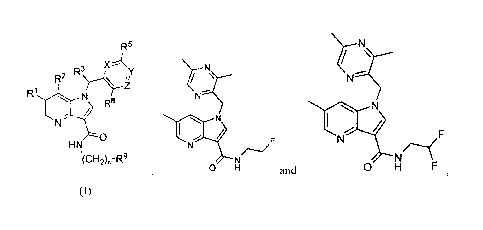
100051 In some aspects, the invention provides, at least in part, a
compound of formula (I):
R5
R2 R3µ Y
8
HN
µ(CH2)n-R9
(I)
wherein
RI is selected from hydrogen, fluorine, bromine, -OCH3 and methyl;
R2 is hydrogen or methyl;
R3 is hydrogen or methyl;
X is N or CR4;
R4 is selected from hydrogen, fluorine and ¨OCH3;
R5 is selected from hydrogen, fluorine, -CF3 and -CN;
Y is N or CR6;
R6 is hydrogen or methyl;
Z is N or CR7;
R7 is selected from hydrogen, fluorine, -OCH3, -OCHF2, -OCH2CF3 and ¨N(CH3)2;
R8 is selected from hydrogen, fluorine, methyl and -OCH3;
n is 1 or 2;
R9 is selected from fluorine, cyclopropyl, -OCH3, -OH, -0CF3, -CHF2, -CH(F)CH3
and ¨
CH(OH)CH3, or a pharmaceutically acceptable salt thereof.
100061 In some aspects, the invention provides, at least in part, a
pharmaceutical composition
comprising a compound of formula (I), or a pharmaceutically acceptable salt
thereof, and a
pharmaceutically acceptable carrier or diluent.
[0007] In some aspects, the invention provides a compound of formula (I) or
a
pharmaceutically acceptable salt therof, for use in the treatment of
tuberculosis or a
Mycobacterium infection.
3
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
[0008] In some aspects, the invention provides a compound of formula (I)
for use in the
manufacture of a medicament for the treatment of tuberculosis or a
Mycobacterium infection.
[0009] In some aspects, the invention provides a method of treating
tuberculosis or a
Mycobacterium infection comprising administering to a subject in need thereof
a therapeutically
effective amount of a compound of formula (I) or a pharmaceutically acceptable
salt thereof.
[0010] In some aspects, the invention provides a pharmaceutical composition
comprising a
compound of formula (I) or a pharmaceutically acceptable salt therof, for use
in the treatment of
tuberculosis or a Mycobacterium infection.
[0011] In some aspects, the invention provides a compound of formula (I) or
a
pharmaceutically acceptable salt therof, for use in the inhibition of DprEl .
100121 In some aspects, the invention provides a compound of formula (I)
for use in the
manufacture of a medicament for inhibition of DprEl.
[00131 In some aspects, the invention provides a method of inhibiting DprE
1 comprising
administering to a therapeutically effective amount of a compound of formula
(I) or a
pharmaceutically acceptable salt thereof
[00141 In some aspects, the invention provides a pharmaceutical composition
comprising a
compound of formula (I) or a pharmaceutically acceptable salt thereof, for
inhibiting DprEl.
4
Brief Descriptions of the Drawings and Tables
100151 Figure 1 illustrates the in vitro cell cidality and in vivo efficacy
of (a) kinetic of cell
cidality for compound 3, (b) kinetic of cell cidality for compound 4, (c)
acute efficacy in mouse
TB model (d) chronic efficacy in mouse TB model.
100161 Figure 2 illustrates the intracellular efficacy of compounds 3 and 4
in THP1 model.
100171 Figure 3 illustrates (a) Time vs concentration profiles of compound
3 and 4 in mice
following oral administration at 100mg/kg (with ABT), (b) Time vs
concentration profiles of
compound 3 and 4 in rat following oral administration at 30mWkg (c) Time vs
concentration
profiles of compound 3 and 17 in rat following IV bolus at 0.5 and 2 mg/kg
respectively.
100181 Figure 4 illustrates (a) Time vs concentration profile of compound 3
in mice
following multiple oral administration at 30 & 100 mg/kg in chronically
infected mice (with
ABT), (b) Time vs concentration profile of compound 4 in mice following
multiple oral
administration at 30 & 100 mg/kg in chronically infected mice (with ABT), (c)
ELF PK of
compound 3 in healthy mice at 100 mg/kg, (d) ELF PK of compound 4 in healthy
mice at 100
mg/kg.
100191 Figure 5 illustrates (a) Time vs. concentration profiles of 1,4-
azaindole compounds
following multiple oral administration at 100 mg/kg in chronically Mtb
infected rats, (b)
Summary of efficacy of 1,4-azaindole compounds in Chronic TB infection model
in Wistar Rats
following 6 days a week po dosing for four weeks. The net logl 0 cfu
reduction/left lobe of the
lungs was obtained by subtracting lung bacterial counts from the vehicle
treated controls. All
compounds exhibited statistically significant effect vs untreated controls
(*p<0.05).
100201 Figure 6 illustrates Synthetic Scheme 1 for the synthesis of
Intermediates 3-9.
100211 Figure 7 illustrates Synthetic Scheme 2 for the synthesis of
Intermediates 11-15.
10022 I Figure 8 illustrates Synthetic Scheme 3 for the synthesis of
Intermediates 17-21.
100231 Figure 9 illustrates Synthetic Scheme 4 for the synthesis of
Intermediates 23-25.
100241 Figure 10 illustrates Synthetic Scheme 5 for the synthesis of
Intermediates 27-30.
100251 Figure II illustrates Synthetic Scheme 6 for the synthesis of
Intermediates 32-33.
100261 Figure 12 illustrates Synthetic Scheme 7 for the synthesis of
Intermediates 35-37.
100271 Figure 13 illustrates Synthetic Scheme 8 for the synthesis of
Intermediates 39-44.
100281 Figure 14 illustrates Synthetic Scheme 9 for the synthesis of
Intermediates 41-47.
Date Recue/Date Received 2020-10-02
[0029] Figure 15 illustrates Synthetic Scheme 10 for the synthesis of
Intermediates 48a and
48b-50.
[0030] Figure 16 illustrates the synthesis of compounds in (a)-(d) Examples
1-4.
[0031] Figure 17 illustrates the synthesis of compounds in (a)-(d) Examples
5-8.
[0032] Figure 18 illustrates the synthesis of compounds in (a)-(d) Examples
9-12.
[0033] Figure 19 illustrates the synthesis of compounds in (a)-(d) Examples
13-16.
[0034] Figure 20 illustrates the synthesis of compounds in (a)-(d) Examples
17-20.
[0035] Figure 21 illustrates the synthesis of compounds in (a)-(d) Examples
21-24.
[0036] Figure 22 illustrates the synthesis of compounds in Example 25.
[0037] Figure 23 illustrates the synthesis of compounds in (a)-(d) Examples
26-29.
[0038] Figure 24 illustrates the synthesis of compounds in (a)-(c) Examples
30-32.
[0039] Table 2, Pathogen specificity.
[0040] Table 3, Activity against drug sensitive and drug resistant Mtb.
6
Date Recue/Date Received 2020-10-02
CA 02918487 2016-01-15
WO 2015/009525 PCT1US2014/046100
Detailed Description of the Invention
00411 Compounds
100421 In some aspects, the invention provides compounds of Formula (I),
wherein
[0043] In some aspects, the invention provides, at least in part, a
compound of formula (I):
R5
n3 X
R2 rµ
R8
I /
¨0
HN
(I)
wherein
R1 is selected from hydrogen, fluorine, bromine, -OCH3 and methyl;
R2 is hydrogen or methyl;
R3 is hydrogen or methyl;
X is N or CR4;
R4 is selected from hydrogen, fluorine and ¨OCH3;
R5 is selected from hydrogen, fluorine, -CF3 and -CN;
Y is N or CR6;
R6 is hydrogen or methyl;
Z is N or CR7;
7 i R s selected from hydrogen, fluorine, -OCH3, -OCHF2, -OCH2CF3 and
¨N(CH3)2;
R8 is selected from hydrogen, fluorine, methyl and ¨OCH3;
n is 1 or 2;
R9 is selected from fluorine, cyclopropyl, -OCH3, -OH, CHF2,
-CH(F)CH3 and ¨
CH(OH)CH3, or a pharmaceutically acceptable salt thereof.
[0044] In some aspects, RI and R2 are each hydrogen.
100451 In some aspects, RI is hydrogen and R2 is methyl.
[0046] In some aspects, RI is selected from fluorine, bromine and methyl
and R2 is
hydrogen.
100471 In some aspects, RI, R2 and R3 are each hydrogen.
7
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
100481 In some aspects, RI is methyl and R2 and R3 are each hydrogen.
[0049] In some aspects, n is 1 and R9 is cyclopropyl.
[0050] In some aspects, n is 1 and R9 is -CH(F)CH3.
[0051] In some aspects, n is I and R9 is -CHF2.
100521 In some aspects, n is I and R9 is -CH(OH)CH3.
100531 In some aspects, n is 2 and R9 is fluorine.
100541 In some aspects, n is 2 and R9 is -0Me.
[0055] In some aspects, n is 2 and R9 is ¨OH.
[0056] In some aspects, n is 2 and R9 is -0CF3.
100571 In some aspects, X is N, R5 is hydrogen, Y is N, Z is CR7, R7 is
¨0Me and R8 is
methyl.
[0058] In some aspects, X is CR4, R4 is hydrogen, R5 is selected from
fluorine, -CN and
¨CF3. Y is CR6, R6 is hydrogen, Z is N and R8 is ¨0Me.
[0059] In some aspects, RI is hydrogen, R2 is hydrogen, R3 is hydrogen, X
is N, R5 is
hydrogen, Y is N, Z is CR7, R7 is ¨0Me and R8 is methyl.
100601 In some aspects, RI is methyl, R2 is hydrogen, R3 is hydrogen, X is
N, R5 is hydrogen,
Y is N, Z is CR7, R7 is ¨0Me and R8 is methyl.
[00611 In some aspects, RI is fluorine, R2 is hydrogen, R3 is hydrogen, X
is N, R5 is
hydrogen, Y is N, Z is CR7, R7 is ¨0Me and R8 is methyl.
[0062] In some aspects, RI is bromine, R2 is hydrogen, R3 is hydrogen, X is
N, R5 is
hydrogen, Y is N, Z is CR7, R7 is ¨0Me and R8 is methyl.
[0063] In some aspects, RI is hydrogen, R2 is hydrogen, R3 is methyl, X is
CR4, R4 is
fluorine, R5 is hydrogen, Y is CR6, R6 is hydrogen, Z is CR7, R7 is hydrogen,
R8 is fluorine, n is 2
and R9 is fluorine.
[0064] In some aspects, RI is hydrogen, R2 is methyl, R3 is hydrogen, X is
CR4, R4 is -0Me,
R5 is hydrogen, Y is CR6, R6 is hydrogen, Z is CR7, R7 is fluorine, R8 is
hydroge, n is 1 and R9 is
cyclopropyl.
[0065] In some aspects, RI is hydrogen, R2 is hydrogen, R3 is hydrogen, X
is N, R5 is
hydrogen, Y is N, Z is CR7, R7 is ¨0Me, R8 is methyl, n is 2 and R9 is
fluorine.
[0066] In some aspects, RI is hydrogen, R2 is hydrogen, R3 is hydrogen, X
is N, R5 is
hydrogen, Y is N, Z is CR7, R7 is ¨0Me, R8 is methyl. n is 1 and R9 is
cyclopropyl.
8
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
100671 In some aspects, RI is hydrogen, R2 is hydrogen, R3 is hydrogen, X is
CR4, R4 is -0Me,
R5 is hydrogen, Y is CR6, R6 is hydrogen, Z is CR7, R7 is fluorine, R8 is
fluorine, n is 2 and R9 is
fluorine.
100681 In some aspects, R1 is hydrogen, R2 is hydrogen, R3 is hydrogen, X
is CR4, R4 is
hydrogen, R5 is hydrogen, Y is CR6, R6 is hydrogen, Z is N, R8 is -0Me, n is 2
and R9 is
-0Me.
100691 In some aspects. RI is hydrogen, R2 is hydrogen, R3 is hydrogen, X
is N, R5 is
hydrogen, Y is N, Z is CR7, R7 is -0Me, R8 is methyl, n is 2 and R9 is -0Me.
100701 In some aspects, RI is methyl, R2 is hydrogen, R3 is hydrogen, X is
N, R5 is hydrogen,
Y is N, Z is CR7, R7 is -0Me, R8 is methyl, n is 2 and R9 is fluorine.
100711 In some aspects, RI is hydrogen, R2 is hydrogen, R3 is hydrogen, X
is N, R5 is
hydrogen, Y is N, Z is CR7, R7 is -0Me, R8 is methyl, n is 2 and R9 is -OH.
100721 In some aspects, RI is methyl, R2 is hydrogen, R3 is hydrogen, X is
N, R5 is hydrogen,
Y is N, Z is CR7, R7 is -0Me, R8 is methyl, n is 1 and R9 is cyclopropyl.
100731 In some aspects, RI is methyl, R2 is hydrogen, R3 is hydrogen, X is
N, R5 is hydrogen,
Y is N, Z is CR7, R7 is -0Me, R8 is methyl, n is 2 and R9 is -0Me.
100741 In some aspects, R is hydrogen, R2 is hydrogen, R3 is hydrogen, X is
N, R5 is
hydrogen, Y is CR6, R6 is hydrogen, Z is CR7 and R7 is -OCH2CF3, R8 is methyl,
n is 2 and R9 is
fluorine.
100751 In some aspects, RI is hydrogen, R2 is hydrogen, R3 is hydrogen, X
is CR4, R4 is
hydrogen, R5 is -CF3. Y is CR6, R6 is hydrogen, Z is N, R8 is -0Me, n is 2 and
R9 is fluorine.
100761 In some aspects, RI is hydrogen, R2 is hydrogen, R3 is hydrogen, X
is CR4, R4 is
hydrogen, R5 is -CN, Y is CR6, R6 is hydrogen, Z is N, R8 is -0Me, n is 2 and
R9 is fluorine.
[00771 In some aspects, RI is methyl, R2 is hydrogen, R3 is hydrogen, X is
CR4, R4 is
hydrogen, R5 is fluorine, Y is CR6, R6 is hydrogen, Z is N, R8 is -0Me, n is 2
and R9 is fluorine.
100781 In some aspects, RI is methyl, R2 is hydrogen, R3 is hydrogen, X is
N, R5 is hydrogen,
Y is N, Z is CR7, R7 is -0Me, R8 is methyl, n is 1 and R9 is CH(F)CH3.
100791 In some aspects, RI is methyl, R2 is hydrogen, R3 is hydrogen, X is
N, R5 is hydrogen,
Y is N, Z is CR7, R7 is -0Me, R8 is methyl, n is 2 and R9 is -OH.
100801 In some aspects, RI is hydrogen, R2 is hydrogen, R3 is hydrogen, X
is CR4, R4 is
hydrogen, R5 is -fluorine, Y is CR6, R6 is methyl, Z is N. R8 is -0Me, n is 2
and R9 is fluorine.
9
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
[0081] In some aspects, RI is fluorine, R2 is hydrogen, R3 is hydrogen, X
is N, R5 is
hydrogen, Y is N, Z is CR7, R7 is -0Me, R8 is methyl, n is 2 and R9 is
fluorine.
[0082] In some aspects, RI is bromine, R2 is hydrogen, R3 is hydrogen, X is
N, R5 is
hydrogen, Y is N, Z is CR7, R7 is -0Me, R8 is methyl, n is 2 and R9 is
fluorine.
[0083] In some aspects, RI is methyl, R2 is hydrogen, R3 is hydrogen, X is
N, R5 is hydrogen,
Y is CR6, R6 is hydrogen, Z is CR7, R7 is -OCH2CF3, R8 is methyl, n is 2 and
R9 is fluorine.
[0084] In some aspects, RI is methyl, R2 is hydrogen, R3 is hydrogen, X is
CR4, R4 is
hydrogen, R5 is -CF3. Y is CR6, R6 is hydrogen, Z is N, R8 is -0Me, n is 1 and
R9 is CH(F)CH3.
[0085] In some aspects, RI is hydrogen, R2 is hydrogen, R3 is hydrogen, X
is N, R5 is
hydrogen, Y is N, Z is CR7, R7 is -0Me, R8 is methyl, n is 2 and R9 is -0CF3.
[0086] In some aspects, RI is methyl, R2 is hydrogen, R3 is hydrogen, X is
CR4, R4 is
hydrogen, R5 is fluorine, Y is CR6, R6 is hydrogen, Z is N, R8 is -0Me, n is 1
and R9 is -
CH(F)CH3.
[0087] In some aspects, RI is hydrogen, R2 is hydrogen, R3 is hydrogen, X
is N, R5 is
hydrogen, Y is N, Z is CR7, R7 is -0Me, R8 is methyl, n is 1 and R9 is
cyclopropyl.
[0088] In some aspects, RI is methyl, R2 is hydrogen, R3 is hydrogen, X is
CR4, R4 is
hydrogen, R5 is fluorine, Y is CR6, R6 is methyl, Z is N, R8 is methyl, n is 2
and R9 is fluorine.
[0089] In some aspects, RI is methyl, R2 is hydrogen, R3 is hydrogen, X is
N, R5 is hydrogen,
Y is CR6, R6 is hydrogen, Z is CR7, R7 is -OCH2CF3, R8 is methyl, n is 2 and
R9 is -OH.
[0090] In some aspects, RI is methyl, R2 is hydrogen, R3 is hydrogen, X is
N, R5 is hydrogen,
Y is N, Z is CR7, R7 is -0Me, R8 is methyl, n is I and R9 is -CH(OH)CH3.
[0091] In some aspects, RI is methyl, R2 is hydrogen, R3 is hydrogen, X is
N, R5 is hydrogen,
Y is N, Z is CR7, R7 is -N(CH3)2, R8 is methyl, n is 2 and R9 is fluorine.
100921 In some aspects, RI is methyl, R2 is hydrogen, R3 is hydrogen. X is
N, R5 is hydrogen,
Y is N, Z is CR7, R7 is -0Me, R8 is methyl, n is 1 and R9 is CHF2.
[0093] In some aspects, RI is hydrogen, R2 is hydrogen, R3 is hydrogen, X
is CR4, R4 is
hydrogen, R5 is hydrogen, Y is CR6, R6 is methyl, Z is CR7, R7 is hydrogen, R8
is methyl, n is 2
and R9 is fluorine.
100941 In some aspects, RI is hydrogen, R2 is hydrogen, R3 is hydrogen, X
is CR4, R4 is
fluorine, R5 is hydrogen, Y is CR6, R6 is hydrogen, Z is CR7, R7 is hydrogen,
R8 is -0Me, n is 1
and R9 is cyclopropyl.
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
10095) In
some aspects, RI is methyl, R2 is hydrogen, R3 is hydrogen, X is N, R5 is
hydrogen,
Y is N, Z is CR7, R7 is -OCH3, R8 is methyl, n is 1 and R9 is -CHF2-
[0096] In
some aspects, RI is methyl, R2 is hydrogen, R3 is hydrogen, X is N, R5 is
hydrogen,
Y is N, Z is CR7, R7 is -N(CH3)2, R8 is methyl, n is 2 and R9 is -OH.
100971 In
some aspects, RI is methyl, R2 is hydrogen, R3 is hydrogen, X is N, R5 is
hydrogen,
Y is N, Z is CR7, R7 is -OCHF2, R8 is methyl, n is 2 and R9 is -OH.
100981 In
some aspects, RI is -OCH3, R2 is hydrogen, R3 is hydrogen, X is N, R5 is
hydrogen, Y is N, Z is CR7, R7 is -OCH3, R8 is methyl, n is 2 and R9 is -OH.
100991 In
some aspects, RI is -OCH3, R2 is hydrogen, R3 is hydrogen, X is N, R5 is
hydrogen, Y is N, Z is CR7, R7 is -OCH3, R8 is methyl, n is 1 and R9 is -CHF2.
1001001 In some aspects, RI is -OCH3, R2 is hydrogen, R3 is hydrogen, X is N,
R5 is
hydrogen, Y is N, Z is CR7, R7 is -OCH3, R8 is methyl, n is 2 and R9 is -OH.
In some
aspects, RI is -OCH3, R2 is hydrogen, R3 is hydrogen, X is N, R5 is hydrogen,
Y is N, Z is CR7,
R7 is -N(CH3)2, R8 is methyl, n is 2 and R9 is F.
101001 In
some aspects, RI is -OCH3, R2 is hydrogen, R3 is hydrogen, X is N, R5 is
hydrogen, Y is N, Z is CR7, R7 is -N(CH3)2, R8 is methyl, n is 1 and R9 is -
CHF2.
101011 In
some aspects, RI is -OCH3, R2 is hydrogen, R3 is hydrogen, X is N, R5 is
hydrogen, Y is N, Z is CR7, R7 is -N(CH3)2, R8 is methyl, n is 2 and R9 is -
OH.
101021 In
some aspects, RI is -OCH3, R2 is hydrogen, R3 is hydrogen, X is N, R5 is
hydrogen, Y is N, Z is CR7, R7 is -OCHF2, R8 is methyl, n is 2 and R9 is F.
[01031 In
some aspects, RI is -OCH3, R2 is hydrogen, R3 is hydrogen, X is N, R5 is
hydrogen, Y is N, Z is CR7, R7 is -OCHF2, R8 is methyl, n is 1 and R9 is -
CHF2.
(01041 In
some aspects, RI is -OCH3, R2 is hydrogen, R3 is hydrogen, X is N, R5 is
hydrogen, Y is N, Z is CR7, R7 is -OCHF2, R8 is methyl, n is 2 and R9 is -OH.
101051 In
some aspects, RI is -OCH3, R2 is hydrogen, R3 is hydrogen, X is N, R5 is
hydrogen, Y is N, Z is CR7, R7 is -OCHF2, R8 is methyl, n is 2 and R9 is F.
101061 In
some aspects, RI is methyl, R2 is hydrogen, R3 is hydrogen, Xis N, R5 is
hydrogen,
Y is CR6, R6 is methyl, Z is N, R8 is methyl, n is 2 and R9 is F.
101071 In
some aspects, RI is methyl, R2 is hydrogen, R3 is hydrogen, X is N, R5 is
hydrogen,
N. is CR6, R6 is methyl, Z is N, R8 is methyl, n is 1 and R9 is CHF2.
11
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
[0108] In some aspects, the compounds of formula (I) include the compounds
in Table 1, or
pharmaceutically acceptable salts thereof
[0109] Table 1
Compound
Compound Mtb MIC ( M)
NO.
N
I /
1 12.5
o
1-(l -(2,6-difluorophenyl)ethyl)-N-(2-fluoroethyl)-1H-
pyrrolo[3,2-b] pyridine-3-carboxamide
0
CY.ts. =
/
2 tsr F 6.25
o
N-(Cyclopropylmethyl)-1-(5-fluoro-2-methoxybenzy1)-7-
methyl-lH-pyrrolo[3,2-b]pyridine-3-carboxamide
\o
NA"--
3CTIII 1.56
0
N-(2-fluoroethyl)-1-((6-methoxy-5-methylpyrimidin-4-
yl)methyl)-1H-pyrrolo[3,2-b]pyridine-3-carboxamide
12
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
\o
4 14/
<0.781
0 11
N-(cyclopropylmethyl)-146-methoxy-5-methylpyrimidin-
4-yOmethyl)-1H-pyrrolo[3,2-b]pyridine-3-carboxamide
¨0
N
F
<0.66
0
1-(2,3-difluoro-6-methoxybenzy1)-N-(2-fluoroethyl)-1H-
pyrrolo[3,2-b]pyridine-3-carboxamide
N
6
1.3
0¨.
1-((5-fluoro-2-methoxypyridin-3-ypmethyl)-N-(2-
methoxyethyl)-1H-pyrrolo[3,2-b]pyridine-3-carboxamide
7
/ <1.33
1-1
1-((6-methoxy-5-methylpyrim idin-4-yl)methyl)-N-(2-
methoxycthyl)-1H-pyrrolo[3,2-b]pyridine-3-carbox am ide
13
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
LA_O
8 I <0.53
N-(2-fluoroethyl)-14(6-methoxy-5-methylpyrimidin-4-
yl)methyl)-6-methyl-1H-pyrrolo[3,2-Npyridine-3-
carboxamide
0
9 13.9
OH
N-(2-hydroxyethyl)-1-((6-methoxy-5-methylpyrimidin-4-
y1)methy1)-1H-pyrrolo[3,2-b]pyridine-3-carboxamide
0
I 1.56
N
0 H
N-(cyclopropylmethyl)-1-((6-methoxy-5-methylpyrimidin-
4-yl)methyl)-6-methyl-1H-pyrrolo[3,2-b]pyridine-3-
carboxamide
\o
N
11
0.781
0 Fl
14(6-methoxy-5-methylpyrimidin-4-yl)methyl)-N-(2-
I methoxyethyl)-6-meth y1-1H-pyrrolo[3,2-b]pyridine-3-
carboxamide
14
CA 02918487 2016-01-15
WO 2015/009525 PCT1US2014/046100
F ______________________________________________________________________
Ft-F
0
(-- ----
N--
12 N 6.25
F
N
N
0 H
N-(2-fluoroethyl)-14(3-methyl-4-(2,2,2-
trifluoroethoxy)pyridin-2-yl)methyl)-1H-pyrrolo[3,2-
___________ 1)] pyridine-3 -carboxamide
¨0
rx....., N F
Isr H F F
13 N <0.53
0 \.......\
F
N-(2-fluoroethyl)-1-((2-methoxy-5-
(trifluoromethyl)pyridin-3-yl)methyl)-1H-pyrrolo[3,2-
b]pyridine-3-carboxamide _ __________________________________
N
CN 1 \ \
--Cc-
14 I / F 3.12
N
fr----/-
N
0 H
1-((5-cyano-2-methoxypyridin-3-yOmethyl)-N-(2-
fluoroethyl)-1H-pyrrolo[3,2-Npyridine-3-carboxamide
--cs
/ N\
I /
N
15 H
N 1.56
F
1((5-fluoro-2-methox ypyridin-3 -yl)methyl)-N-(2-
fl uoroethyl)-6-methy1-1H-pyrrolo[3,2-b]pyridine-3-
carbox amide
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
\o
16 N 0.156
N
N =
0 H
(S)-N-(2-fluoropropy1)-14(6-methoxy-5-methylpyrimidin-
4-yOmethyl)-6-methyl-IH-pyrrolo[3,2-Npyridine-3-
carboxamide
o
17 1.56
OH
0 H
N-(2-hydroxyethyl)-1-((6-methoxy-5-methyl pyrim i d i n-4-
yl)methyl)-6-methy1-1H-pyrmlo[3,2-b]pyridine-3-
___________ carboxam ide
N\
1 N
18
,F 0.781
N
1-((5-fluoro-2-methoxy-6-methylpyridin-3-yl)methyl)-N-
___________ (2-fluoroethyl)-1H-pyrrolo[3,2-b]pyridine-3-carboxamide
rts\'/N
F
19 6.25
6-fl uoro-N-(2-fluoroethyl)-14(6-methoxy-5-
methylpyrimidin-4-yOmethyl)-1H-pyrrolo[3,2-Npyridine-
___________ 3-carbox amide
16
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
0
I
20 o 0.781
6-bromo-N-(2-fluoroethyl)- 1 4(6-methoxy-5-
methylpyrimidin-4-Amethyp- 1 H-pyrrolo[3,2-b]pyridine-
3-earboxamide
F
21 1.56
F
0 11
N-(2-fluoroethyl)-6-methyl- 1-((3 -methy1-4-(2,2,2-
trifluoroethoxy)pyridin-2-yl)meth y1)- 1 H-pyrrolo[3,2-
___________ b]pyridine-3-carboxamide
F
I
22 r%1 <0.391
o Ls/
(S)-N-(2-fluoropropy1)- 1 -((2-methoxy-5-
(trifluoromethyppyridi n-3-yl)methyl)-6-methyl- 1 H-
pyrrolo[3,2-b]pyridine-3-carboxamide
17
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
/
N
23
F 3.12
0 H
14(6-methoxy-5-methylpyrimidin-4-yl)methyl)-N-(2-
(trifluoromethoxy) ethyl)-1H-pyrrolo[3,2-b]pyridine-3-
carboxamide
N I
F '
24 <0.391
rs-1
N
0 H
(S)-14(5-fluoro-2-methoxypyridin-3-yl)methyl)-N-(2-
fluoropropy1)-6-methy1-1H-pyrrolo[3,2-b]pyri cline-3-
carboxamide
no--
1
25 25
2-cyclopropyl-N-(14(6-methoxy-5-methylpyrimidin-4-
yptincthyl)-1H-pyrrolo[3,2-13]pyridin-3-yDacetamide
rcc--
N
26 3.12
0
14(5- fluoro-2,6-dimethylpyridin-3 -yl)methyl)-N-(2-
fluoroethyl )-6-methyl -1H-pyrrolo[3,2-b]pyridine-3-
carboxamide
18
CA 02918487 2016-01-15
WO 2015/009525
PCT/US2014/046100
0
N-
N
I
27 3.12
o
OH
N-(2-hydroxyethyl)-6-methy1-14(3-methyl-4-(2,2,2-
trifluoroethoxy) pyridin-2-yOmethyl)-1H-pyrrolo[3,2-
________ pyridine-3-carboxamide
\o
28 6.25
OH
N
O H
(R)-N-(2-hydroxypropy1)-14(6-methoxy-5-
methylpyrimidin-4-yl)methyl)-6-methyl-IH-pyrrolo[3,2-
b]pyridine-3-carboxamide
NI-
(
29 1.56
N F
0 H
1-06-(dimethylamino)-5-methylpyrimidin-4-yOmethyl)-N-
(2-fluoroethyl) -6-methyl- I H-pyrrolo[3,2-b]pyridine-3-
carboxamide
\o
N
30 N
1Y,N i F <0.391
OH
N-(2,2-di fluoroethyl)-1-((6-methoxy-5-methylpyrimidin-4-
ypmethyl)-6-methyl-IH-pyrrolo[3,2-bjpyridine-3-
carboxamide
19
CA 02918487 2016-01-15
WO 2015/009525
PCT/US2014/046100
-N
31 tkr F 1.56
0 H
1-(2,4-dimethylbenzy1)-N-(2-fluoroethyl)-1 H-pyrrolo[3,2-
b]pyridine-3-carboxamide
r\--o
32 /
1.56
0 H
N-(cyclopropylmethyl)-1-(2-fluoro-6-methoxybenzy1)-1H-
pyrrolo[3,2-b]pyridine-3-carboxamide
33 tsi/
<0.39
H F
N-(2,2-difluoroethyl)-1-((6-methoxy-5-methylpyrimidin-4-
yl)methyl)-6-methyl-IH-pyrrolo[3,2-b]pyridine-3-
carboxamide
\
NA\
N
34 I / <0.39
OH
0 H
14(6-(dimethylamino)-5-methylpyrimidin-4-yl)methyl)-N-
(2-hydroxyethyl)-6-methyl-IH-pyrrolo[3,2-b]pyridine-3-
carboxamide
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
Fj:0
35 I
OH <0.39 /
0 H
1-06-(difluoromethoxy)-5-methylpyrimidin-4-yl)methyl)-
N-(2-hydroxyethyl)-6-methyl-1H-pyrrolo[3,2-b]pyridine-
3-carboxamide
\o
oI
N
36 <0.39
,F
0 H
N-(2-fluoroethyl)-6-methoxy-14(6-methoxy-5-
methylpyrimidin-4-yl)methyl)-1H-pyrrolo[3,2-Npyridine-
3-earboxamide
oI
N
37 / <0.39
0 H F
N-(2,2-difluoroethyl)-6-methoxy-14(6-methoxy-5-
methylpyrimidin-4-yl)methyl)-1H-pyrrolo[3,2-b]pyridine-
3-carboxamide
21
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
oI
N
38 / 0.78
OH
0 H
N-(2-hydroxyethyl)-6-methoxy-14(6-methoxy-5-
methylpyrimidin-4-yl)methyl)-1H-pyrrolo[3,2-b]pyridine-
3-carboxamide
14---
6 ,
39 I / <0.39
0 H
1-06-(dimethylamino)-5-methylpyrimidin-4-yl)methyl)-N-
(2-fluoroethyl)-6-methoxy-1H-pyrrolo[3,2-b]pyridine-3-
___________ carboxamide
40 <0.39
N/
0 H F
N-(2,2-difluoroethyl)-1-06-(dimethylamino)-5-
methylpyrimidin-4-yOmethyl)-6-methoxy-IH-pyrrolo[3,2-
b]pyridine-3-carboxamide
22
CA 02918487 2016-01-15
WO 2015/009525
PCT/US2014/046100
\N'
0 N
41 / <039
OH
O H
1-06-(dimethylamino)-5-methylpyrimidin-4-yl)methyl)-N-
(2-hydroxyethyl)-6-methoxy-1H-pyrrolo[3,2-b]pyridine-3-
earboxamide
N
I
42 O(N <0.39
O H
1-06-(difluoromethoxy)-5-methylpyrimidin-4-yOmethyl)-
N-(2-fluoroethyl)-6-methoxy-1H-pyrrolo[3,2-b]pyridine-3-
carboxamide
F--(0
0
43 N <0.39
/
= H F
N-(2,2-di fluoroethyl)-1-06-(di tluoromethoxy)-5-
methylpyrimidin-4-yl)methyl)-6-methoxy-IH-pyrrolo [3,2-
b]pyridine-3-carboxamide
23
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
F--(0
44 o =
Ni 0.78
OH
0 H
1-06-(difluoromethoxy)-5-methylpyrimidin-4-yl)methyl)-
N-(2-hydroxyethyl)-6-methoxy-IH-pyrrolo[3,2-b]pyridine-
3-carboxamide
).--
45 / <0.39
0 H
14(3,5-dimethylpyrazin-2-yl)methyl)-N-(2-fluoroethyl)-6-
methyl-1H-pyrrolo[3,2-b]pyridine-3-carboxamide
I 1\1/
46 <0.39
0 H F
N-(2,2-difluoroethyl)-1-((3,5-dimethylpyrazin-2-
yl)methyl)-6-methyl-1H-pyrrolo[3,2-b]pyridine-3-
carboxamide
101101 Pharmaceutical Compositions
10111] In some aspects, the invention provides a pharmaceutical composition
comprising a
compound of formula (1), or a pharmaceutically acceptable salt thereof, and a
pharmaceutically
acceptable diluent or carrier.
01121 The language "pharmaceutically acceptable" includes compounds,
materials,
compositions, and/or dosage forms which are, within the scope of sound medical
judgment,
suitable for use in contact with the tissues of human beings and animals
without excessive
24
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
toxicity, irritation, allergic response, or other problem or complication,
commensurate with a
reasonable benefit/risk ratio.
101131 The
compounds of formula (I) may form stable pharmaceutically acceptable acid or
base salts, and in such cases administration of a compound as a salt may be
appropriate.
Examples of acid addition salts include acetate, adipate, ascorbate, benzoate,
benzenesulfonate,
bicarbonate, bisulfate, butyrate, camphorate, camphorsulfonate, choline,
citrate, cyclohexyl
sulfamate, diethylenediamine, ethanesulfonate, fumarate, glutamate, glycolate,
hemisulfate,
2-hydroxyethylsulfonate, heptanoate, hexanoate, hydrochloride, hydrobromide,
hydroiodide,
hydroxymaleate, lactate, malate, maleate, methanesulfonate, meglumine, 2-
naphthalenesulfonate,
nitrate, oxalate, pamoate, persulfate, phenylacetate, phosphate, diphosphate,
picrate, pivalate,
propionate, quinate, salicylate, stearate, succinatc, sulfamate, sulfanilate,
sulfate, tartrate, tosylate
(p-toluenesulfonate), trifluoroacetate, and undecanoate.
Examples of base salts include
ammonium salts; alkali metal salts such as sodium, lithium and potassium
salts; alkaline earth
metal salts such as aluminum, calcium and magnesium salts; salts with organic
bases such as
dicyclohexylamine salts and N-methyl-D-glucamine; and salts with amino acids
such as arginine,
lysine, omithine, and so forth. Also, basic nitrogen-containing groups may be
quatemized with
such agents as: lower alkyl halides, such as methyl, ethyl, propyl, and butyl
halides; dialkyl
sulfates such as dimethyl, diethyl, dibutyl; diamyl sulfates; long chain
halides such as decyl,
lauryl, myristyl and stearyl halides; arylalkyl halides such as benzyl bromide
and others.
Non-toxic physiologically acceptable salts are preferred, although other salts
may be useful, such
as in isolating or purifying the product.
101141 The
salts may be formed by conventional means, such as by reacting the free base
form of the product with one or more equivalents of the appropriate acid in a
solvent or medium
in which the salt is insoluble, or in a solvent such as water, which is
removed in vacuo or by
freeze drying or by exchanging the anions of an existing salt for another
anion on a suitable
ion-exchange resin.
101151 The
compositions of the invention may be in a form suitable for oral use (for
example
as tablets, lozenges, hard or soft capsules, aqueous or oily suspensions,
emulsions, dispersible
powders or granules, syrups or elixirs), for topical use (for example as
creams, ointments, gels,
or aqueous or oily solutions or suspensions), for administration by inhalation
(for example as a
finely divided powder or a liquid aerosol), for administration by insufflation
(for example as a
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
finely divided powder) or for parenteral administration (for example as a
sterile aqueous or oily
solution for intravenous, subcutaneous, intramuscular or intramuscular dosing
or as a suppository
for rectal dosing).
101161 The compositions of the invention may be obtained by conventional
procedures using
conventional pharmaceutical excipients well known in the art. Thus,
compositions intended for
oral use may contain, for example, one or more coloring, sweetening, flavoring
and/or
preservative agents.
101171 Suitable pharmaceutically acceptable excipients for a tablet
formulation include, for
example, inert diluents such as lactose, sodium carbonate, calcium phosphate
or calcium
carbonate; granulating and disintegrating agents such as corn starch or
algenic acid; binding
agents such as starch; lubricating agents such as magnesium stearate, stearic
acid or talc;
preservative agents such as ethyl or propyl p-hydroxybenzoate; and anti-
oxidants, such as
ascorbic acid. Tablet formulations may be uncoated or coated either to modify
their
disintegration and the subsequent absorption of the active ingredient within
the gastrointestinal
tract, or to improve their stability and/or appearance, in either case, using
conventional coating
agents and procedures well known in the art.
101181 Compositions for oral use may be in the form of hard gelatin
capsules in which the
active ingredient is mixed with an inert solid diluent, for example, calcium
carbonate, calcium
phosphate or kaolin, or as soft gelatin capsules in which the active
ingredient is mixed with water
or an oil such as peanut oil, liquid paraffin, or olive oil.
[01191 Aqueous suspensions generally contain the active ingredient in
finely powdered form
or in the form of nano or micronized particles together with one or more
suspending agents, such
as sodium carboxymethylcellulose, methylcellulose,
hydroxypropylmethylcellulose, sodium
alginate, polyvinyl-pyrrolidone, gum tragacanth and gum acacia; dispersing or
wetting agents
such as lecithin or condensation products of an alkylene oxide with fatty
acids (for example
polyoxethylene stearate), or condensation products of ethylene oxide with long
chain aliphatic
alcohols, for example heptadecaethyleneoxycetanol, or condensation products of
ethylene oxide
with partial esters derived from fatty acids and a hexitol such as
polyoxyethylene sorbitol
monooleate, or condensation products of ethylene oxide with long chain
aliphatic alcohols, for
example heptadecaethyleneoxycetanol, or condensation products of ethylene
oxide with partial
esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol
monooleate, or
26
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
condensation products of ethylene oxide with partial esters derived from fatty
acids and hexitol
anhydrides, for example polyethylene sorbitan monooleate. The aqueous
suspensions may also
contain one or more preservatives such as ethyl or propyl p-hydroxybenzoate;
anti-oxidants such
as ascorbic acid); coloring agents; flavoring agents; and/or sweetening agents
such as sucrose,
saccharine or aspartame.
[01201 Oily suspensions may be formulated by suspending the active
ingredient in a
vegetable oil such as arachis oil, olive oil, sesame oil or coconut oil or in
a mineral oil such as
liquid paraffin. The oily suspensions may also contain a thickening agent such
as beeswax, hard
paraffin or cetyl alcohol. Sweetening agents such as those set out above, and
flavoring agents
may be added to provide a palatable oral preparation. These compositions may
be preserved by
the addition of an anti-oxidant such as ascorbic acid.
[0121] Dispersible powders and granules suitable for preparation of an
aqueous suspension
by the addition of water generally contain the active ingredient together with
a dispersing or
wetting agent, suspending agent and one or more preservatives. Suitable
dispersing or wetting
agents and suspending agents are exemplified by those already mentioned above.
Additional
excipients such as sweetening, flavoring and coloring agents, may also be
present.
[0122] The pharmaceutical compositions of the invention may also be in the
form of oil-in-
water emulsions. The oily phase may be a vegetable oil, such as olive oil or
arachis oil, or a
mineral oil, such as for example liquid paraffin or a mixture of any of these.
Suitable
emulsifying agents may be, for example, naturally-occurring gums such as gum
acacia or gum
tragacanth, naturally-occurring phosphatides such as soya bean, lecithin, an
esters or partial
esters derived from fatty acids and hexitol anhydrides (for example sorbitan
monooleate) and
condensation products of the said partial esters with ethylene oxide such as
polyoxyethylene
sorbitan monooleate. The emulsions may also contain sweetening, flavoring and
preservative
agents.
[0123] Syrups and elixirs may be formulated with sweetening agents such as
glycerol,
propylene glycol, sorbitol, aspartame or sucrose, and may also contain a
demulcent, preservative,
flavoring and/or coloring agent.
101241 The pharmaceutical compositions may also be in the form of a sterile
injectable
aqueous or oily suspension, which may be formulated according to known
procedures using one
or more of the appropriate dispersing or wetting agents and suspending agents,
which have been
27
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
mentioned above. A sterile injectable preparation may also be a sterile
injectable solution or
suspension in a non-toxic parenterally-acceptable diluent or solvent, for
example a solution in
1,3-butanediol.
[0125] Compositions for administration by inhalation may be in the form of
a conventional
pressurized aerosol arranged to dispense the active ingredient either as an
aerosol containing
finely divided solid or liquid droplets. Conventional aerosol propellants such
as volatile
fluorinated hydrocarbons or hydrocarbons may be used and the aerosol device is
conveniently
arranged to dispense a metered quantity of' active ingredient.
[0126] For further information on formulation the reader is referred to
Chapter 25.2 in
Volume 5 of Comprehensive Medicinal Chemistry (Corwin Hansch; Chairman of
Editorial
Board), Pergamon Press 1990.
101271 The amount of active ingredient that is combined with one or more
excipients to
produce a single dosage form will necessarily vary depending upon the host
treated and the
particular route of administration. For further information on Routes of
Administration and
Dosage Regimes the reader is referred to Chapter 25.3 in Volume 5 of
Comprehensive Medicinal
Chemistry (Corwin Hansch; Chairman of Editorial Board), Pergamon Press 1990.
[0128] Methods of Use
[0129] In some aspects, the invention provides a compound of formula (I) or
a
pharmaceutically acceptable salt therof, for use in the treatment of
tuberculosis or a
Mycobacterium infection.
[0130] In some aspects, the invention provides a compound of formula (I) in
the manufacture
of a medicament for use in the treatment of tuberculosis or a Mycobacterium
infection.
[0131] In some aspects, the invention provides a method of treating
tuberculosis or a
Mycobacterium infection comprising administering to a subject in need thereof
a therapeutically
effective amount of a compound of formula (I) or a pharmaceutically acceptable
salt thereof.
[0132] The language "therapeutically effective amount" includes an amount
of the co-
crystals described herein that will elicit the biological or medical response
of a subject, for
example, the reduction or inhibition of enzyme or protein activity related to
a Mycobacterium
infection or tuberculosis, amelioration of symptoms of a Mycobacterium
infection or
tuberculosis, or the slowing or delaying of progression of a Mycobacterium
infection or
tuberculosis. In some embodiments, the language "therapeutically effective
amount" includes
28
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
the amount of a co-crystal described herein, that when administered to a
subject, is effective to at
least partially alleviate, inhibit, and/or ameliorate a Mycobacterium
infection or tuberculosis,
and/or reduce or inhibit the bacterial growth, replication or bacterial load
of Mycobacterium in
a subject.
(01331 The term "subject" includes warm blooded mammals, for example,
primates, cows,
sheep, dogs, cats, rabbits, rats, voles, seals and mice. In some embodiments,
the subject is a
primate, for example, a human. In some embodiments, the subject is suffering
from a
Mycobacterium infection or tuberculosis. In some embodiments, the subject is
in need of
treatment (e.g., the subject would benefit biologically or medically from
treatment).
[0134] The language "inhibit," "inhibition" or "inhibiting" includes a
decrease in the
baseline activity of a biological activity or process.
101351 The language "treat," "treating" and "treatment" includes the
reduction or inhibition
of enzyme or protein activity related to a Mycobacterium infection or
tuberculosis in a subject,
amelioration of one or more symptoms of a Mycobacterium infection or
tuberculosis in a
subject, or the slowing or delaying of progression of a Mycobacterium
infection or tuberculosis
in a subject. The language "treat," "treating" and "treatment" also includes
the reduction or
inhibition of the bacterial growth, replication or a reduction or inhibition
of the bacterial load of
Mycobacterium in a subject.
101361 The language "Mycobacterium infection" includes infections caused by
one or more
of the species of the Mycobacterium tuberculosis complex, e.g., Mycobacterium
tuberculosis,
Mycobacterium bovis, Mycobacterium africanum, Mycobacterium canetti,
Mycobacterium
caprae, Mycobacterium microti or Mycobacterium pinnipedii. In some
embodiments, the
Mycobacterium infection is a Mycobacterium tuberculosis infection.
[0137] The term "tuberculosis" refers to the disease caused by an infection
in a subject of
one or more species of the Mycobacterium tuberculosis complex. The term -
tuberculosis"
includes latent tuberculosis (LTBI), non-drug resistant tuberculosis, multiple
drug resistant
tuberculosis (MDR-TB) and extensively drug resistant tuberculosis (XRD-TB).
The language
"latent tuberculosis" includes an infection of a subject caused by one or more
species of
Mycobacterium tuberculosis complex but where the subject does not necessarily
exhibit
symptoms a tuberculosis disease. The language "non-drug resistant
tuberculosis" includes
tuberculosis caused by an infection by one or more species of the
Mycobacterium tuberculosis
29
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
complex that exhibits no antibacterial resistance to standard tuberculosis
therapy. The language
"multiple drug resistant tuberculosis (MDR-TB)" includes tuberculosis caused
by an infection by
of one or more species of the Mycobacterium tuberculosis complex that is
resistant to rifampicin
and isoniazid. The language "extensively drug resistant tuberculosis (XRD-TB)"
includes
tuberculosis caused by an infection by one or more species of the
Mycobacterium tuberculosis
complex that is resistant to rifampicin and isoniazid, as well as any member
of the quinolone
family, and is also resistant to at least one of kanamycin, capreomycin and
amikacin. In some
embodiments, the tuberculosis infection is acute. In some embodiments, the
tuberculosis
infection is chronic.
[0138] In some aspects, the invention provides a compound of formula (I) or
a
pharmaceutically acceptable salt therof, for inhibiting DprEl.
[0139) In some aspects, the invention provides a compound of formula (I) in
the manufacture
of a medicament for use in inhibiting DprEl.
101401 In some aspects, the invention provides a method of inhibiting DprE
1 comprising
contacting a cell with a therapeutically effective amount of a compound of
formula (I) or a
pharmaceutically acceptable salt thereof.
101411 Combinations
101421 The compounds described herein may be applied as a sole therapy or
may involve one
or more other substances and/or treatments. Such co-treatment may be achieved
by way of the
simultaneous, sequential or separate administration of the individual
components of the
treatment. Where the administration is sequential or separate, the delay in
administering the
second component should not be such as to lose the beneficial effect of the
combination.
Suitable classes and substances include one or more antibacterial agents
useful in the treatment
of Mycobacterium infections and/or tuberculosis, such as, for example,
rifampicin, isoniazid,
pyrizinamide, ethambutol, quinolones (e.g., ciprofloxacin, levofloxacin,
moxifloxacin and
gatifloxacin), aminoglycosides (e.g., streptomycin, kanamycin, and amikacin),
polypeptides
(e.g., capreomycin, viomycin and enviomycin), rifabutin, clarithromycin,
linezolid,
thioacetazone, thioridazine, arginine, vitamin D and R207910.
Examples
[01431 All anhydrous solvents, reagent grade solvents for chromatography
and starting
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
materials were purchased from either Sigma Aldrich Chemical Co. or Fisher
Scientific. Water
was distilled and purified through a Milli-Q water system (Millipore Corp.,
Bedford, MA).
General methods of purification of compounds involved the use of silica
cartridges purchased
from Grace Purification systems. The reactions were monitored by TLC on
precoated Merck 60
F254 silica gel plates and visualized using UV light (254 nm). All compounds
were analyzed for
purity by HPLC and characterized by 11-1 NMR using Bruker 300 MHz NMR and/or
Bruker 400
MHz NMR spectrometers. Chemical shifts are reported in ppm (8) relative to the
residual
solvent peak in the corresponding spectra; chloroform 7.26, methanol 8 3.31,
DMSO-d6 8 3.33
and coupling constants (J) are reported in hertz (Hz) (where s = singlet, bs =
broad singlet, d =
doublet, dd = double doublet, bd = broad doublet, ddd = double doublet of
doublet, t = triplet, tt
¨ triple triplet, q = quartet, m = multiplet) and analyzed using ACD NMR data
processing
software. Mass spectra values are reported as m/z. All reactions were
conducted under Nitrogen
unless otherwise noted. Solvents were removed in vacuo on a rotary evaporator.
[0144] Abbreviations: NMP = N-methyl Pyrrolidine; HC1 = hydrochloric acid;
DMF = N,N-
dimethylformamide; NaH = sodium hydride. El = electrospray ionization; HRMS =
high
resolution mass spectrometry.
101451 Figure 6 shows Synthetic Scheme 1 for the synthesis of Intermediates
3-9.
101461 Intermediate 3: To a stirred suspension of NaH (60%) in dry DMF,
diethyl malonate
was added drop wise over a period of 30 min. To this mixture substituted 2-
chloro-3-
nitropyridine was added portion wise over a period of 1 h and stirred the
contents at r.t for 90
min. The contents were heated to 80 C over a period of 30 min and maintained
for 1 h. DMF
was evaporated from the reaction mixture under vacuum and the residue was
diluted with water.
The reaction mixture pH was adjusted to 5-6 range and extracted with ethyl
acetate. The organic
layer was washed with water, brine, dried over anhydrous Na2SO4 and
concentrated by rotary
evaporation to get an orange oily liquid. The compound was used as such in
next step without
further purification.
[0147] Intermediate 4: To a stirred solution of intermediate 3 in DMSO:H20
was added
LiC1 and the reaction mixture was stirred at 80 C for 16 h. Then the reaction
mixture was
poured in to water and extracted with ethyl acetate. The combine organic layer
was washed with
31
water, brine, dried over sodium sulfate, and concentrated at reduced pressure.
The crude product
was purified by silica gel column chromatography using ethyl acetate in hexane
to afford
intermediate 4.
[0148] Intermediate 5: To a stirred solution of intermediate 4 in DMF, was
added DMF-
DMA and the reaction mixture was stirred at 80 C for 16 h. After completion of
reaction the
reaction mixture was poured in to ice-water and extracted with ethyl acetate.
The combined
organic layer was washed with water, brine, dried over sodium sulfate, and
concentrated at
reduced pressure to yield intermediate 5. The crude material was taken for
next step without
purification.
[0149] Intermediate 6: To the stirred solution of intermediate 5 in acetic
acid was added Fe-
powder at once and the mixture was stirred at 60 C for 2 h. Then the reaction
mixture was diluted
with methanol and filter through CeliteTM. The filtrate was concentrated under
reduced pressure.
The crude product was purified by silica gel column chromatography using ethyl
acetate to
afford intermediate 6 as a solid.
[0150] Intermediate 7: To a stirred solution of intermediate 6 and K2CO3 in
DMF was
added aryl halide and the reaction mixture was stirred at RT for 16 h. The
resulting mixture was
poured in to water and extracted with dichloromethane. The combined organic
layer was washed
with water, brine, dried over sodium sulfate, and concentrated at reduced
pressure to yield
intermediate 7 as a solid or liquid.
OR
101511 Intermediate 6 and K2CO3 was taken in dry DMF under nitrogen
atmosphere. To this
aryl halide was added. The resulting reaction was stirred at 80 C for 3 h. DMF
was evaporated to
dryness, diluted with water and extracted with DCM. Purification was done on
CombiflashTM
system to get intermediate 7 as a solid or liquid.
OR
101521 Intermediate 6 was dissolved in DMF under nitrogen atmosphere. To
this at 0 C
NaH was added. After 5 min aryl halide was added. The resulting reaction was
stirred at rt for 6
h. Reaction was poured on ice water, added ethyl acetate. Organic layer was
separated and
32
Date Recue/Date Received 2020-10-02
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
washed with brine and concentrated. Purification was done on combiflash system
to obtain
intermediate 7 as a solid or liquid.
[0153] Intermediate 8: To a solution of intermediate 7 in ethanol, was
added lithium
hydroxide in water. The reaction mixture was stirred for 5 h at RT. The
solvent was evaporated
under the reduced pressure to yield intermediate 8 as a off-white solid.
OR
[0154] To a solution of intermediate 7 in methanol was added lithium
hydroxide in water.
The reaction mixture was stirred at 60 C for 3 h. The solvent was evaporated
under the reduced
pressure, nutralized with acetic acid to yield intermediate 8 as a off-white
solid.
[0155] Figure 7 shows Synthetic Scheme 2 for the synthesis of Intermediates
11-15.
[0156] Intermediate 11: To a solution of 2-chloro-5-
(trifluoromethyl)pyridine (5 g, 27.60
mmol) in methanol was added sodium methoxide (2.98 g, 55.20 mmol) at 0 C. The
reaction
mixture was stirred for 6 h at RT. Then the solvent was removed under vacuum.
The resulting
mixture was poured into water (100 mL) added water extracted with ethyl
acetate (2 x 50 mL).
The combined organic layer was washed with water, brine, dried over sodium
sulfate, and
concentrated at reduced pressure to yield 2-methoxy-5-
(trifluoromethyl)pyridine (11) as a pale
yellow liquid (4 g, 82.1%).
[0157] Intermediate 12: To a stirred solution of 2-methoxy 5-
(trifluoromethyl) pyridine (4
g, 22.58 mmol) was dissolved in acetonitrile (50 ml) was added NBS (6 g, 33.87
mmol) portion
wise at 00C. The reaction mixture was stirred for overnight at RT. The solvent
was removed
under vacuum, quenched with water (100 mL) and extracted with ethyl acetate.
The combined
organic layer was washed with water, brine, dried over sodium sulfate, and
concentrated at
reduced pressure to yield 3-bromo-2-methoxy-5-(trifluoromethyl)pyridine (12)
as a pale yellow
liquid (2 g, 34.7%).
[0158] Intermediate 13: To a stirred solution of 3-bromo-2-methoxy-5-
(trifluoromethyl)pyridine (23) (1.2 g, 4.68 mmol) dissolved in mixture of
methanol (10 ml)/
toluene (10 ml) was added tri ethyl amine (1 ml,
7.65 mmol), [1,1'-
33
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
Bis(diphenylphosphino)ferrocene]palladium(II) dichloride ( 70 mg, 0.102 mmol).
The reaction
mixture was carbonylated under CO [5 kg] at 80 C for 12 h. Then the solvent
was filtered
through celite and solvent was concentrated on vacuum, get crude. The crude
compound was
purified by using silica gel chromatography eluting 30% ethyl acetate in
hexaen to afford methyl
2-methoxy-5-(trifluoromethypnicotinate (13) as a liquid (0.5 g, 45.4%).
[01591 Intermediate 14: To a solution of methyl 2-methoxy-5-
(trifluoromethypnicotinate
(13) (0.5 g, 2.12 mmol) dissolved in DCM was added DIBAL-H (6.38 ml, 1M in
Toluene) at
0 C. The reaction mixture was stirred for 2h at RT. Then the reaction mixture
was quenched
with saturated NH4C1 solution and extracted with ethyl acetate (2 x 25 mL).
The combined
organic layer was washed with water, brine, dried over sodium sulfate, and
concentrated at
reduced pressure to yield (2-methoxy-5-(trifluoromethyppyridin-3-ypmethanol
(14) as a pale
yellow liquid (0.4 g, 90%).
101601 intermediate 15: To a solution of 2-methoxy-5-
(trifluoromethyl)pyridin-3-
yl)methanol (14) (0.4 g, 1.93 mmol) dissolved in DCM was added thionyl
chloride (0.38 ml,
3.86 mmol). The reaction mixture was stirred for 2 h at RT. The solvent was
evaporated under
the reduced pressure and the reaction mixture was poured in to water (50 mL)
and extracted with
DCM (2 x 50 mL). The combined organic layer was washed with water, brine,
dried over
sodium sulfate, and concentrated at reduced pressure to yield 3-(chloromethyl)-
2-methoxy-5-
(trifluoromethyl)pyridine (15) as a liquid. (Yield 0.3 g, 69.76%)
(0161j Figure 8 shows Synthetic Scheme 3 for the syntheis of Intermediates
17-21.
101621 Intermediate 17: To a solution of 2,6-dichloro-5-fluoronicotinic
acid (16) (5 g, 23.8
mmol) in methanol (50m1) was added thionyl chloride in dropwise (5.66 g,
47.62mmo1) at 0 C
and 2 drops of DMF [vigourous bubbling was observed]. The mixture was stirred
at room
temperature for 3h. To this methanol was added and stirred the reaction
mixture for 2 h at RT.
The reaction mixture is concentrated under reduced pressure to and the mixture
was poured in to
ice cold water (20 mL) and extracted with dichloromethane (2 x 50 mL). The
combined organic
layer was washed with water, brine and solvent was evaporated under reduced
pressure to yield
methyl 2,6-dichloro-5-fluoronicotinate (17) (5g, 93.8%).
34
CA 02918487 2016-01-15
WO 2015/009525 PCT1US2014/046100
[0163] Intermediate 18: A mixture of methyl 2,6-dichloro-5-fluoronicotinate
(17) ( 3.5 g
15.62 mmol), trimethylborox in (1.96 g, 15.62 mmol), 1,1'-
Bis(diphenylphosphino)ferrocene-
palladium(Mdichloride dichloromethane (1.276 g, 1.562 mmol) and cesium
carbonate 15.27 g,
46.86 mmol) was heated at 110 C overnight. The mixture is cooled to room
temperature,
diluted with water, and extracted with Et0Ac. The combined organic phase is
washed with
water followed by brine, then dried over anhydrous sodium sulfate and
concentrated under
reduced pressure. The residue is purified by flash silica gel chromatography
using a gradient of
0-30% Et0Aciheptanes to provide methyl 2-chloro-5-fluoro-6-methylnicotinate
(18) 0.8 g as a
colorless solid (0.8 g 25 %).
[0164] Intermediate 19: To a stirred solution of methyl 2-chloro-5-
fluoro-6-
methylnicotinate (18) (0.8 g, 3.92 mmol,) in THF (35 mL) was added sodium
methanolate (0.42
g, 7.85 mmol) at 0 C. The mixture was stirred at 60 C for 6 hours and cooled
to room
temperature. Then the mixture was poured into water and extracted with
dichloromethane (2 x
50 mL). The combined organic layer was washed with water, brine and solvent
was evaporated
under reduced pressure. The residue was purified by column chromatography on
silica gel
eluting with 10% ethyl acetate in hexane to give methyl 5-fluoro-2-methoxy-6-
methylnicotinate
(19) (250 mg, 32%) as a white solid.
[0165] Intermediate 20: To a solution of methyl 5-fluoro-2-methoxy-6-
methylnicotinate
(19) (0.25 g, mmol) in MDC was added DIBAL-H in dropwise (2.5 ml, 1M in
Toluene) at 0 C.
The reaction mixture was stirred for 2h at RT. Reaction was quenched with
saturated
ammonium chloride solution and extracted with DCM (2 x 50 ml). The combined
organic layer
was washed with water, brine and solvent was evaporated under reduced pressure
to yield (5-
fluoro-2-methoxy-6-methylpyridin-3-yl)methanol (20) as a liquid (0.2 g 90%).
101661 Intermediate 21: To a solution of (5-fluoro-2-methoxy-6-
methylpyridin-3-
yl)methanol (20) (0.2 g, 1.16 mmol) in DCM was added thionyl chloride (0.278
g, 2.33 mmol).
The reaction mixture was stirred for 2 h at RT. The solvent was evaporated
under the reduced
pressure and the reaction mixture was poured in to water (50 mL) and extracted
with DCM (2 x
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
50 mL). The combined organic layer was washed with water, brine, dried over
sodium sulfate,
and concentrated at reduced pressure to yield to afford 3-(chloromethyl)-5-
fluoro-2-methoxy-6-
methylpyridine (21) as a pale yellow liquid (0.2 g) 90%.
[0167] Figure 9 shows Synthetic Scheme 4, for the synthesis of
Intermediates 23-25.
[0168] Intermediate 23: To a solution of ethyl 6-chloro-5-methylpyrimidine-
4-carboxylate
(35) (0.9 g, 4.48 mmol) dissolved in dioxane(20 ml) were added dimethyl amine
in THF (1M,
22.43 ml, 22.43 mmol) and diisopropyl ethylamine(3 ml, 22.43 mmol). The
reaction mixture
was heated to 80 OC for 16 h. Then the mixture is cooled to room temperature,
diluted with
water, and extracted with Et0Ac (2 x 50 mL). The combined organic layer was
washed with
water, brine, dried over sodium sulfate, and concentrated at reduced pressure
to yield ethyl 6-
(dimethylamino)-5-methylpyrimidine-4-carboxylate (23) as a light yellow
liquid; 0.4 g (43%).
1(11691 Intermediate 24: To a stirred solution of ethyl 6-(dimethylamino)-5-
methylpyrimidine-4-carboxylate (23) (0.4 g, 1.91 mmol) in Me0H (10 mL) was
added in
portionwise NaBH4 (0.14 g, 3.82 mmol) at 0 C and the mixture was stirred for
16 h at RT. The
solvent was evaporated under the reduced pressure and the reaction mixture was
poured in to
water (20 mL) and extracted with ethyl acetate (3 x 30 mL). The combined
organic layer was
washed with water, brine, dried over sodium sulfate, and concentrated at
reduced pressure to
yield (6-(dimethylamino)-5-methylpyrimidin-4-yOmethanol (24); 0.Ig , (31.3%).
[0170] Intermediate 25: To a solution of (6-(dimethylamino)-5-
methylpyrimidin-4-
yl)methanol (24) (0.1 g, 0.60 mmol) in DCM was added thionyl chloride (0.14 g,
1.1961 mmol).
The reaction mixture was stirred for 4 h at RT. The solvent was removed under
reduced pressure
to afford 6-(Chloromethyl)-N,N,5-trimethylpyrimidin-4-amine (25) as a white
solid; 0.1 g
90.9%).
[01711 Figure 10 shows Synthetic Scheme 5, for the synthesis of
intermediates 27-30.
[0172] Intermediate 27: 2-Chloro-6-trifluoro methyl pyridine (1.0 g,
5.5mmol) in THF
solution (10 ml) was added slowly to a cold (at -78 C) solution of LDA (4.58
ml, 8.2 mmol) in
dry THF (15 ml). The resulting mixture was stirred for 4 h at -78 OC, before
the addition of
36
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
methyl iodide (0.705 ml, 0.0082 mol) in THF (4 m1). Stirring was continued for
4 h at -75 C,
before quenching with water (10 ml) at the same temperature, and further
addition of water (15
ml) at 0 C after 2h. The crude product was extracted with ethyl acetate,
washed with brine
solution, and the combined organic layer was dried over Na2SO4, and solvent
removed in
vacuum to afford a crude solid, which was purified by flash column
chromatography on silica
(ethyl acetate/ petrpleum ether (0-10%) to afford 27 as a off- white solid;
(0.6g, 33%).
101731 Intermediate 28: In a tiny cave apparatus, 2-chloro-3-methyl-6-
(trifluoro
methyppyridine (27) (0.5 g, 2.5 mmol) was dissolved in Me0H (10 ml) and added
TEA (0.5
m1,3.7 mmol) which was degassed for 15 minutes with nitrogen before adding
Pd(dppt)C12.DCM complex (0.061 g, 0.075 mmol). Tiny cave was filled with CO
gas (75Psi, 5
kg) and heated at 75 C for 16 h. The mixture was filtered through celite and
washed with
methanol. Combined filtrate was concentrated and the crude was purified flash
column
chromatography on silica (ethyl acetate/ hexane (0-10%) to afford methyl 3-
methy1-6-
(trifluoromethyppicolinate (28): off- white solid; (280 mg, 49.9%).
[0174] Intermediate 29: To a solution of methyl 3-methyl-6-(trifluoro
methyl)picolinate
(0.28 g) in DCM (10 ml) at -78 C was added DIBAL-H (1.92 m1,1.91 mmol) under
nitrogen.
The reaction mixture was allowed to reach rt and stirred for 1 h. The RM was
quenched with
ammonium chloride solution and extracted with ethyl acetate (3 30 mL). The
combined organic
layer was washed with brine, dried over Na2SO4 and concentrated to give 3-
methy1-6-
(trifluoromethyl)pyridin-2-yl)methanol (29). The crude alcohol was taken
directly for next step
(180 mg, 72%).
[0175] Intermediate 30: To a solution of (3-methyl-6-
(trifluoromethyl)pyridin-2-
yl)methanol (29) (0.18g,0.94 mmol) in DCM (5m1) was added thionyl chloride
(0.112 g, 0.95
mmol) and stirred at rt for 2 h. After completion of the reaction, mixture was
concentrated and
washed with hexane to afford 2-(chloromethyl)-3-methyl-6-
(trifluoromethyl)pyridine (30). The
crude solid was taken directly for the next step (180 mg, 91.8%).
[0176] Figure 11 shows Synthetic Scheme 6, for the synthesis of
Intermediates 32-33.
37
CA 02918487 2016-01-15
WO 2015/009525 PCT1US2014/046100
101771 Intermediate 32: To a stirred solution of 1-(2,4-
dimethylphenyl)ethan- 1 -one (1.0 g,
0.0067 mol) in Me0H (10 ml) was added NaBH4 (0.77 g, 0Ø020 mol) portion-wise
at 0 C.
The resultant mixture was stirred at RT for 1 h. After completion of the
reaction, the mixture
was quenched with ice cold water (2.5 ml) and solvent was concentrated under
reduced pressure.
The reaction mixture was diluted with water and extracted with Et0Ac (3 x 15
m1). The
combined organic layer was washed with water, brine, dried over sodium
sulfate, and
concentrated at reduced pressure. The crude mixture was just passed through a
pad of silica gel
and washed with 3:1 hexanes: Et0Ac to afford (0.9 g, 88.8%) to give the title
compound 32 as a
white solid.
[0178] Intermediate 33: To a stirred solution of 1-(2,4-
dimethylphenyl)ethan-l-ol (32) (0.9
g, 0.006 mol) in CH2C12 (10 ml) was added phosphorus tribromide (0.85 ml,
0.009 mol) at room
temperature. Stirring was continued for another 2 h at the same temperature.
The mixture was
quenched with water, and was extracted with CH2Cl2 (3x 15 ml). The combined
organic layer
was washed with water, brine, dried over sodium sulfate, and concentrated at
reduced pressure to
give the title product (33) as a light orange solid (900 mg, 70.8% yield).
101791 Figure 12 shows Synthetic Scheme 7, for the synthesis of
Intermediates 35-37.
101801 Intermediate 35: A solution of 2,5-dibromo-3-methylpyridine (1) 3 g,
12.1 mmol) in
methanol (20 ml) was added sodium methoxide (2M, 20 mL) and refluxed at 100 C
for 2h. The
reaction mixture was poured on ice water and neutralized with aqueous
hydrochloric acid (1M)
and extracted with dichloromethane(2 x 15m1). The combined organic layer was
washed with
water, brine, dried over sodium sulfate, and concentrated at reduced pressure
to give 5-Bromo-2-
metlioxy-3-methyl-pyridine (35), which was used without further purification
(1.9 g, 77.8%).
[0181] Intermediate 36: To a solution of 5-bromo-2-methoxy-3-methyl-
pyridine (1.0 g, 5.0
mmol) in DMF (10 ml) was added CuCN (0.534 g, 6.0 mmol) and the resulting
mixture is heated
at reflux for 28 h. After cooling to room temperature the mixture is diluted
with Et0Ac and
washed with 10% ammonia solution followed by water and brine solution. The
organic layer
was separated, dried over MgSO4 and evaporated under reduced pressure. The
residue was
purified by flash chromatography (5% Et0Ac/Hexane) to give 5-cyano-2-methoxy-3-
methyl-
38
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
pyridine (36) (0.68 g, 93%).
[0182] intermediate 37: To a solution of methyl 5-eyano-2-methoxy-3-methyl-
pyridine (36)
(0.25g, 1.7 mmol) in CC14 (10 mL) was added N-bromosuccinimide (346 mg, 1.7
mmol) and
2',2-azobisisobutyronitrile (13.0 mg, 0.085 mmol). The reaction mixture was
stirred at 80 C for
1 h. The reaction mixture was filtered and the filtrate was concentrated to
afford 5-
(bromomethyl)-6-methoxy nicotinonitrile (37) as a yellow solid; (180 mg,
46.9%).
101831 Figure 13 shows Synthetic Scheme 8, for the synthesis of
Intermediates 39-44.
f01841 Intermediate 39: To a solution of 38 (300 g, 2.94 mol) and ethyl
propionate (429.4 g,
2.94 mol) in 1.8 L of anhydrous Et0H was added Na0Et (300 g, 4.41 mol) at room
temperature.
The mixture was stirred overnight. After cooling, the mixture was adjusted to
pH = 7 with 6N
HCI. The mixture was concentrated in vacuum. The residue was diluted with
water, and then
extracted with Et0Ac. The combined Et0Ac layers were dried over Na2SO4 and
concentrated in
vacuo to furnish the product 39 (400 g, 67%) as a red liquid without further
purification for the
next step. 1H NMR: (400 MHz, CDC13) 8 ppm 4.103-4.386 (m, 5H); 1.246-1.439 (m,
9H).
[0185] Intermediate 40: A mixture of 39 (300 g, 1.485 mol), formimidamide
acetate (225 g,
2.12 mol) and Na0Et (160 g, 2.36 mol) in Et0H (2000 mL) was heated to reflux
for 12 hours.
After cooling, the mixture was adjusted to pH = 7 with 6N HC1. The mixture was
concentrated
in vacuum. The residue was diluted with water, and then extracted with DCM.
The combined
DCM layers were washed with water, brine and concentrated in vacuo. The crude
product was
purified by chromatography on silica gel (PE: Et0Ac =1:1--pure Et0Ac) to
afford the product as
a white solid (80 g, 30% yield). ES+MS m/z: 183.0 (M+1). 1H NMR: (400 MHz,
CDC13) 8 ppm
8.126 (s,1H); 4.345-4.399 (m,2H); 2.242 (s, 3H); 1.337-1.373 (t, 3H).
[0186] Intermediate 41: A solution of 40 (80 g, 0.44 mol) in P0C13 (800 g)
was heated to
reflux for 4 hours. After cooling, the excess POC13 was removed under reduced
pressure to give
41(88 g crude, 100% yield) as black oil. ES+MS m/z: 201.0 (M+1). 11-1 NMR:
(400 MHz,
CDC13) 8 ppm 8.954 (s,1H); 4.492-4.545 (m,2H); 2.591 (s, 3H); 1.454-1.490 (t,
3H).
39
CA 02918487 2016-01-15
WO 2015/009525 PCT1US2014/046100
[0187]
Intermediate 42: To a solution of 41(88 g, 0.4 mol) in CH3OH (IL) was added
CH3ONa (40 g, 0.74 mol) at room temperature and the mixture was stirred at
room temperature
overnight. The mixture was adjusted to pH = 7 with 6N HC1. The mixture was
concentrated in
vacuum. The residue was diluted with water, and then extracted with Et0Ac. The
combined
Et0Ac layers were dried over anhydrous Na2SO4 and concentrated in vacuum. The
crude
product was purified by chromatography on silica gel (PE: Et0Ac =5:1) to
afford 42 (10 g, 14 %
yield) as a yellow oil. ES+MS m/z: 197.0 (M+1). NMR:
(400 MHz, CDC13) 8 ppm 8.626 (s, 1
H), 4.357-4.410 (m, 2 H), 3.969 (s, 3 H), 2.279 (s, 3 H),1.344-1.380 (t,3 H).
101881
Intermediate 43: To solution of 42 (10 g, 0.057 mol) in CH3OH (100 mL) was
added
NaBH.4 (10 g, 0.29 mol) at 0 C and the resulting reaction was allowed warm to
room temperature
and stirred for 2 h. The mixture was concentrated in vacuum and partitioned
between water and
Et0Ac. The water layer was extracted with Et0Ac. The combined Et0Ac layers
were dried
over Na2SO4, filtered and concentrated in vacuum to afford 43 (7.5 g, 85 %
yield) as a white
solid. ES+MS m/z: 155.0 (M+1). NMR:
(400 MHz, CDC13) 8 ppm 8.629 (s, 1 H), 4.641 (s, 2
H), 4.007 (s, 3 H), 2.028 (s, 3 H).
101891
Intermediate 44: To solution of 43 (7.5 g, 48.7 mmol) in DCM (150 mL) was
added
S0C12 (75 g, 0.64 mol) at 0 C. The resulting reaction was allowed warm to room
temperature
and stirred for 2 h. TLC showed the starting material was consumed. The
mixture was
concentrated in vacuum to provide 44 (8.3 g, 99 % yield) as a yellow
solid._ES+MS m/z: 173.0
(M+1). NMR:
(400 MHz, CDC13) 8 ppm 8.877 (s, 1 H), 4.992 (s, 2 H), 4.207 (s, 3 H), 2.317
(s, 3 H).
101901 Figure 14 shows Synthetic Scheme 9, for the synthesis of
Intermediates 41-47.
[01911
Intermediate 41: A solution of ethyl 6-hydroxy-5-methylpyrimidine-4-
carboxylate
40 (100 g, 549 mmol) in P0CI3 (1000 ml) was heated to reflux for 5 h at 100 C
in sealed tube.
After cooling to RT, the excess POC13 was removed under reduced pressure, then
quenched with
ice water and extracted with Et0Ac. The combined organic phase is washed with
water, brine
and concentrated under reduced pressure to afford ethyl 6-chloro-5-
methylpyrimidine-4-
carboxylate 41 as a black liquid. Yield: 80 g (72%). This material was used as
such for next step
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
without purification.
[0192] Intermediate 45: To a solution of ethyl 6-chloro-5-methylpyrimidine-
4-carboxylate
(41) (80 g, 398.0 mmol)dissolved in dioxane (800 ml) were added dimethyl amine
in THF (2M,
600 ml, 1196.0 mmol) and diisopropyl ethylamine(330 ml, 1990.0 mmol). The
reaction mixture
was heated to 80 C for 16 h. Then the mixture is cooled to room temperature,
diluted with
water, and extracted with Et0Ac (2 x 50 mL). The combined organic layer was
washed with
water, brine, dried over sodium sulfate, and concentrated at reduced pressure
to yield ethyl 6-
(dimethylamino)-5-methylpyrimidine-4-carboxylate (45) as a light yellow
liquid; yield: 60 g
(72%).
[0193] Intermediate 46: To a stirred solution of ethyl 6-(dimethylamino)-5-
methylpyrimidine-4-carboxylate (45) (60.0 g, 287.0 mmol) in Et0H (600 mL) was
added in
portionwise NaBH4 (21.82 g, 574.0 mmol) at 0 C and the mixture was stirred for
16 h at RT.
The solvent was evaporated under the reduced pressure and the reaction mixture
was poured in
to water (200 mL) and extracted with ethyl acetate (3 x 100 mL). The combined
organic layer
was washed with water, brine, dried over sodium sulfate and concentrated at
reduced pressure to
yield (6-(dimethylamino)-5-methylpyrimidin-4-yl)methanol (46); 47 g, (97%).
[0194] Intermediate 4 7: To a solution of (6-(dimethylamino)-5-
methylpyrimidin-4-
yl)methanol (46) (40 g, 239 mmol) in DCM was added thionyl chloride (35 ml,
478 mmol). The
reaction mixture was stirred for 4 h at RT. The solvent was removed under
reduced pressure to
afford 6-(Chloromethyl)-N,N,5-trimethylpyrimidin-4-amine (47) as a brown
solid; Yield: 40 g
(90.9%); IF1 NMR (4001\4Hz ,DMSO-d6) 8 = 8.69 (s, 1H), 4.86 (s. 2H), 3.27 (s,
61-1), 2.35 (s,
3H).
[0195] Figure 15 shows Synthetic Scheme 10, for the synthesis of
Intermediates 48a and
48b-50.
[01961 Intermediate 486: In a round bottom flask (5 litre) was taken Ethyl
6-hydroxy-5-
methylpyrimidine-4-carboxylate 40 (85 g, 466 mmol), chlorodifluoroacetic acid
sodium salt
(106.7 g, 699 mmol), sodium carbonate (98.9 g, 933 mmol), acetonitrile (1500
ml) and DMF
41
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
(425 m1). The reaction mixture was heated to 90 C for 16 h. The Progress of
the reaction was
monitored by LCMS. The reaction mixture was cooled to room temperature and
then neutralized
with saturated ammonium chloride. The solvent was removed under vacuum and
extracted with
ethyl acetate. The combined organic layer was washed with water, brine and
concentrated under
reduced pressure. The crude compound was purified through silica gel
chromatography eluting
(3-4% ethyl acetate) to afford ethyl 6-(difluoromethoxy)-5-methylpyrimidine-4-
carboxylate 48b
as a pale yellow liquid. Yield: 14 g (13%).
[0197] Intermediate 49: To a stirred solution of 6-(difluoromethoxy)-5-
methylpyrimidine-4-
carboxylate 48b (14 g, 60.30 mmol) in ethanol (200 mL), NaBH4 (4.58 g, 120.59
mmol) was
added at 0 C and the mixture was stirred for 16 h at RT. Then the solvent was
evaporated under
the reduced pressure and the reaction mixture was poured in to water and
extracted with ethyl
acetate. The organic layer was washed with water brine and concentrated under
reduced
pressure to afford (6-(difluoromethoxy)-5-methylpyrimidin-4-yl)methanol 39 as
a yellow solid.
Yield: 8.4 g (73.3%).
[0198] Intermediate 50: To a solution of (6-(difluoromethoxy)-5-
methylpyrimidin-4-
yl)methanol 49 (15 g, 78.94 mmol) dissolved in DCM (150 ml) added thionyl
chloride (8.59 ml,
118.42 mmol). The reaction mixture was stirred for 4 h at RT. The solvent was
removed under
vacuum pump to afford 4-(chloromethyl)-6-(difluoromethoxy)-5-methylpyrimidine
50 as a
brown solid. Yield: 14 g (85%); I H NMR (400 MHz, DMSO-d6) 8 = 8.74 (s, 1H),
7.77 (t., 1H, J
= 95.4 Hz), 4.81 (s, 2H), 2.24 (s, 3H).
[0199] Example I: 1-(1-(2,6-difluorophenyt)ethyl)-N-(2-fluoroethyl)-1H-
pyrrolo[3,2-b]
pyridine-3-c a rbox a mide
QI\t_p
z F
[0200] See Figure 16(a). 1-(1-(2,6-difluorophenyl)ethyl)-1H-pyrrolo[3,2-
b]pyridine-3-
42
CA 02918487 2016-01-15
WO 2015/009525 PCT1US2014/046100
carboxylic acid (0.090 g, 0.30 mmol),2-fluoroethanamine (0.019 g, 0.30 mmol)
and
triethylamine (0.166 mL, 1.19 mmol) was taken in DCM (15 mL) under N2 and
stirred. After 5
min. 1-Propanephosphonic acid cyclic anhydride (0.379 g, 1.19 mmol) was added.
The resulting
reaction was stirred at rt for 40 min. LCMS analysis showed formation of
required product.
Reaction was diluted with DCM and water. DCM layer was extracted and washed
with brine
and dried over sodium sulphate and concentrated. Purification was done on
Waters RP system to
get product 1-(1-(2,6-difluorophenyl)ethyl)-N-(2-fluoroethyl)-1H-pyrrolo
[3,2-b] pyridine-3-
carboxamide (0.040 g, 38.7 %) as a solid. ES+MS m/z: 348.40. 1H NMR (300 MHz,
DMSO-d6)
8 ppm 2.05 (d, J=6.97 Hz, 3 H) 3.67 (q, J=5.21 Hz, 1 H) 3.71 - 3.82 (m, 1 H)
4.49 (t, J=4.90 Hz,
1 H) 4.65 (t, J=4.99 Hz, 1 H) 6.22 (q, J=7.16 Hz, 1 H) 7.13 (t, J=8.57 Hz, 2
H) 7.29 (dd, J=8.48,
4.71 Hz, 1 H) 7.34 - 7.52 (m, 1 H) 7.80 (d, J=8.29 Hz, 1 H) 8.41 (s, 1 H) 8.50
(d, J=4.71 Hz, 1
H) 8.92 (t, J=5.75 Hz, 1 H).
[02011 E.vantple 2: \-(C3, clopropylmethyl)-1-(5-fluoro-2-methoxybenzy1)-7-
methyl-1H-
p rrolo13,2-hip ridine-3-carboxamide
-0
N
I /
0 \......õ(7
[0202] See Figure 16(b). To a stirred solution of 1-(5-fluoro-2-
methoxybenzy1)-7-methyl-
1H-pyrrolo[3,2-b]pyridine-3-carboxylic acid (0.22 g, 0.000699 moles) in
dichloromethane (10
mL) were added 2-fluoroethan- 1 -amine (0.06 g, 0.000836 moles), triethylamine
(0.29 ml, 0.002
moles) and T3P (1.32 ml, 0.002 moles) and the mixture was stirred for 16 h at
room temperature.
The reaction mixture was poured in to water and extracted with
dichloromethane. The combined
organic layer was washed with water, brine and the solvent was evaporated
under reduced
pressure. The crude was purified by flash column chromatography using 50%
ethyl acetate in
hexane to afford N-(cyclopropylmethyl)-1-(5-fluoro-2-methoxybenzy1)-7-methyl-
1H-
pyrrolo[3,2-b]pyridine-3-carboxamide as a off-white solid. Yield-24%. ES+MS
m/z: 368. 11-1
NMR (400 MHz, DMS0-4) 8 ppm: 0.22-0.24 (m, 2H), 0.48-0.50 (m, 2H), 1.05-1.09
(m, 1H),
2.44 (s, 3H), 3.28 (t, 2H, J = 6.2 Hz), 3.86 (s, 3H), 5.63 (s, 2H), 5.97-6.00
(m, 1H), 7.03-7.04 (m,
43
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
1H), 7.12-7.14 (m, 2H), 8.22 (s, 1H), 8.37 (d, 1H, J = 4.8 Hz), 9.01 (t, 1H,
J= 5.6 Hz).
102031 Example 3: N-(2 -flu oroethyl)-1-((6-methoxy-5-methylpyrimidin-4-
yl)methyl)-
ridinc-3-carboxamide
o
)-1=1//
0 H
10204] See Figure 16(c). 1-((6-methoxy-5-methylpyrimidin-4-yl)methyl)-1H-
pyrrolo[3,2-
11pyridine-3-carboxylic acid (250 mg, 0.84 mmol) was taken in a 100 ml single
necked flask
equipped with an air condenser connected to nitrogen source. DCM (10 mL) was
added to get a
suspension. Triethyl amine (1.162 mL, 8.38 mmol) was added to get a clear
solution. I-
Propanephosphonic acid cyclic anhydride (1.497 mL, 2.51 mmol) was added
followed by the
addition of 2-fluoroethanamine hydrochloride (83 mg, 0.84 mmol). The reaction
mass was
stirred at RT for overnight, suspension was observed. Mier completion of the
reaction, diluted
with DCM, added water, and separated the DCM layer washed with brine solution.
The DCM
layer was dried over sodium sulphate, evaporated and purified the compound by
column
chromatography. Yield - 52%. ES+MS m/z: 344(M+1). 1H NMR (300 MHz, DMSO-d6)
ppm
2.25 (s, 20 H) 3.67 (d, J=5.46 Hz, 7 H) 3.77 (d, J=5.46 Hz, 7 H) 4.50 (t,
J=4.90 Hz, 7 H) 4.66 (t,
J=4.99 Hz, 7 H) 5.69 (s, 13 H) 7.26 (dd, J=8.29, 4.71 Hz, 7 H) 7.94 (d, J=8.48
11z, 7 H) 8.28 (s,
7 H) 8.41 (s, 6 H) 8.49 (d, J=4.52 Hz, 7 H) 8.95 (t, J=5.84 Hz, 7 H).
102051 Example 4: N-(cyclopropylmethyl)-1-((6-methoxy-5-methylpyrimidin-4-
yl)methyl)-1H-pyrrolo[3,2-W. ridine-3-carboxamide
44
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
\o
0 H
102061 See Figure 16(b). 14(6-methoxy-5-methylpyrimidin-4-yl)methyl)-1H-
pyrrolo[3,2-
b]pyridine-3-carboxylic acid (200 mg, 0.67 mmol) was taken in DCM (10 mL).
Added 1-
Propanephosphonic acid cyclic anhydride (427 mg, 1.34 mmol) followed by the
addition of
Triethyl amine (339 mg, 3.35 mmol) and cyclopropylmethanamine (95 mg, 1.34
mmol). The
reaction mass was stirred at RT for overnight. After the completion of the
reaction added water
and extracted with DCM. The organic layer was washed with water and brine
slution. The
organic layer was separated, dried over sodium sulphate. Evaporated the
organic layer to get the
residue, which was purified by column chromatography to get the pure compound.
Yield- 74%.
ES+MS 352.38(M+1). 1H NMR (300 MHz, DMSO-d6) 6 ppm 0.02 (q, J=4.58 Hz, 2
H)
0.18 -0.31 (m, 2 H) 0.83 (t, J=6.88 Hz, 1 H) 2.00 (s, 3 H) 2.98 -3.11 (m, 3 H)
3.69 (s, 3 H) 5.43
(s, 2 H) 7.00 (dd, J=8.29, 4.71 Hz, 1 H) 7.68 (dd, J=8.29, 1.13 Hz, 1 H) 7.98
(s, 1 H) 8.17 (s, 1
H) 8.24 (dd, J=4.71, 1.13 Hz, 1 H) 8.55 (t, J=5.75 Hz, 1 H).
102071 Example 5: 1-(2,3-difluoro-6-methoxybenzyl)-N-(2-fluoroethyl)-1H-
pyrrolo[3,2-
hi ridine-3-carboxamide
-0
I NI/ F F
lµr
0
[02081 See Figure 17(a). In a 50mL round-bottomed flask N-(2-fluoroethyl)-
1H-pyrrolo[3,2-
b]pyridine-3-carboxamide (0.1 g, 0.48 mmol) was DMF (10 mL) to give a
colourless suspension.
the reaction mixture was cooled to 0 C and potassium carbonate (0.200 g, 1.45
mmol) and 2-
(bromomethy1)-3,4-difluoro- 1 -methoxybenzene (0.114 g, 0.48 mmol) was added
then the RM
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
was stirred at 80 C for 4h. The reaction was monitored by LCMS. DMF was
concentrated under
vacuao, added water and extracted with DCM. The combined organic layer was
washed with
brine, dried over sodium sulphate, filtered and evaporated to give crude
product. The crude
material was purified on reverse phase preparative HPLC system to get 1-(2,3-
difluoro-6-
methoxybenzy1)-N-(2-fluoroethyl)-1H-pyrrolo[3,2-b]pyridine-3-carboxamide
(0.060 g, 34.2 %).
ES+MS m/z: 364(M+1) H NMR (DMSO-d6, 300 MHZ): 8 ppm 8.90 (br. s., 1 H), 8.50
(d,
J=4.3 Hz, 1 H), 8.02- 8.15 (m, 2 H), 7.30- 7.52 (m, 2 H), 6.92 (d, J=8.7 Hz, 1
H), 5.53 (s, 2 H),
4.63 (t, J=4.4 Hz, 1 H), 4.47 (t, J=4.8 Hz, 1 H),3.86 (s, 3 H), 3.74 (d, J=5.3
Hz, 1 H), 3.65 (d,
J=5.3 Hz, 1 H).
102091 Example 6: 14(5-fluoro-2-niethox
1)methyl)-N-(2-methoxyethyl)-1H-
pyrrolo[3,2-14yridine-3-carboxaniide
F4..õ"c
0 H
[0210] See Figure 17(b). 1-((5-
fluoro-2-methoxypyridin-3-yOmethyl)-1H-pyrrolo[3,2-
b]pyridine-3-carboxylic acid (100 mg, 0.33 mmol) was taken in dichloromethane
(15 mL) to get
a suspension. -Methyl amine (230 mL, 1.66 mmol) was added to get clear
solution. Added 1-
Propanephosphonic acid cyclic anhydride (198 mL, 0.66 mmol) and stirred at RI
for 5 minutes.
2-methoxyethanamine (74.8 mg, 1.00 mmol) was added and stirred the reaction
mass at RI for
2hr. After the completion of the reaction diluted the reaction mass with DCM,
washed with
water, brine solution. The DCM layer was separated, dried over sodium sulphate
and evaporated
to get the crude compound. The compound was purified by silica gel
chromatography using
methanol and dichloromethane as eluent. Yield- 63%. ES+MS m/z: 359.1(M+1).
NMR (500
MHz, DMSO-d6) 8 ppm 3.44 - 3.52 (m, 2 H) 3.56 (q, J=5.46 Hz, 2 H) 3.89 (s, 3
II) 5.47 (s, 2 H)
7.31 (dd, J=8.20, 4.73 Hz, 1 H) 7.46 (dd, J=8.20, 2.84 Hz, 1 H) 8.09 - 8.22
(m, 2 H) 8.31 (s, 1 H)
8.51 (dd, J=4.73, 0.95 Hz, 1 H) 8.85 (t, J=5.52 Hz, 1 H).
[02111 Example 7: 1((6-methoxy-5-methylpyrimidin-4-yl)meth et11
1)-
46
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
1H-pyrrolo[3,2-blpyridine-3-carboxamide
0
NN-k.
(r:kql
0 -
0 H
[0212] See Figure 17(c). 1((6-methoxy-5-methylpyrimidin-4-yOmethyl)-1 H-
pyrrolo[3,2-
b]pyridine-3-carboxylic acid (200mg, 0.67 mmol) was taken in DCM (10 mL) to
get a
suspension. Triethyl amine (0.929 mL, 6.70 mmol) was added to get a clear
solution. 1-
Propanephosphonic acid cyclic anhydride (1.197 mL, 2.01 mmol) was added and
stirred at RT
for 5 minutes. 2-methoxyethanamine (151 mg, 2.01 mmol) was added and stirred
at RT for
overnight. After the completion of the reaction, diluted the reaction mass
with DCM, washed
with water, brine solution and then evaporated to get the crude compound. The
compound was
purified by silica gel chromatography. Yield-90%. ES+MS
356.2(M+1). H NMR (300
MHz, DMSO-d6) 8 ppm 2.24 (s, 3 H) 3.30 (s, 3 H) 3.43 - 3.63 (m, 4 H) 3.93 (s,
3 H) 5.68 (s, 2
H) 7.24 (dd, J=8.29, 4.71 Hz, 1 H) 7.92 (dd, .1=8.38, 1.22 Hz, 1 H) 8.25 (s, 1
H) 8.41 (s, 1 H)
8.48 (dd, J=4.71, 1.13 Hz, 1 H) 8.85 (t, J=5.65 Hz, 1 H).
[0213] Example 8: N-(2-fluoroethyI)-1-((6-methoxy-5-methylp:µ
1)meth), 1)-6-
methyl-1H-pyrrolo13,2-bI pyridine-3-carboxamide
Ic-
N
0 H
102141 See Figure 17(d). 14(6-
methoxy-5-methylpyrimidin-4-yl)methyl)-6-methyl-1H-
pyrrolo[3,2-b]pyridine-3-carboxylic acid (0.190 gm, 0.61 mmol) and 2-
fluoroethanamine (0.077
g, 1.22 mmol), TEA (0.254 mL, 1.83 mmol) was added. After 3 mint -
Propanephosphonic acid
cyclic anhydride (0.484 g, 1.52 mmol) was added. The resulting reaction
mixture was stirred at
rt for 50 min. LCMS analysis confirmed the formation of required product.
Reaction was diluted
47
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
with DCM and water.DCM layer was extracted and washed with brine and dried
over sodium
sulphate and concentrated. Purification was performed on Waters RP system to
get product N-
(2-fluoroethyl)-14(6-methoxy-5-methylpyrimidin-4-yl)methyl)-6-methyl-1 H-
pyrrolo [3,2-
b]pyridine-3-carboxamide (0.090 g, 41.4 %). ES+MS m/z: 358.36. H NMR (300 MHz,
DMSO-d6) 8 PPm 2.17 - 2.30 (3, 3 H) 2.40 (s, 3 H) 3.59 - 3.73 (m, 1 H) 3.73 -
3.83 (m, 1 H) 3.94
(s, 3 H) 4.50 (t. J=4.99 Hz, 1 H) 4.66 (t, J=4.99 Hz, 1 H) 5.64 (s, 2 H) 7.76
(s, 1 H) 8.15 (s, 1 H)
8.35 (s, 1 H) 8.42 (s. 1 H) 8.87 (t, J=5.84 Hz, 1 H).
102151 Example 9: N-(2-hydroxyethyl)-14(6-methoxy-5-methylpyrimidin-4-
yOmethyl)-
1 rrolo13,2-hipyridine-3-carboxamide
\
./\
0 L...\
OH
102161 See Figure 18(a). 14(6-methoxy-5-methylpyrimidin-4-yl)methyl)-1H-
pyrrolo[3,2-
b]pyridine-3-carboxylic acid (1 g, 3.35 mmol) was taken in DCM (20 mL) to get
a suspension.
Triethyl amine (1.394 mL, 10.06 mmol) was added followed by the addition of 1-
Propanephosphonic acid cyclic anhydride (3.991 mL, 6.70 mmol). The reaction
mass was stirred
at RT for 5 minutes. Ethanol amine (6.02 mL, 10.06 mmol) was added and stirred
at RT for 2h.
After completion of the reaction, diluted with DCM and then washed with water
and brine
solution. The organic layer was separated, dried, evaporated and the crude
compound was
purified by silica gel chromatography. Yield-45%. ES+MS miz: 342(M+1). 11-1
NMR (300
MHz, DMSO-d6) 8 ppm 2.24 (s, 3 11) 3.37 - 3.64 (m, 4 H) 3.94 (s, 3 H) 4.80 (t,
J=4.99 Hz, 1 H)
5.67 (s, 2 H) 7.24 (dd, J=8.29, 4.52 Hz, 1 H) 7.92 (d, J=8.10 Hz, 1 H) 8.23
(s, 1 H) 8.41 (s, 1 H)
8.47 (d, J=4.52 Hz, 1 H) 8.85 (t, J=5.37 Hz, 1 H).
102171 Example 10: N-(cyclopropylmethy1)-14(6-methoxy-5-meth:s 1pyrimidin-4-
:s1)meth1)-6-mettil-1H-pyrrolor3,2-blp.),ridine-3-carboxamide
48
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
0
OH
102181 See Figure 18(b). 1-((6-
methoxy-5-methylpyrimidin-4-yl)methyl)-6-methyl-1H-
pyrrolo[3,2-b]pyridine-3-carboxylic acid (0.100 g, 0.32 mmol)and
cyclopropylmethanamine
(0.046 g, 0.64 mmol), TEA (0.134 mL, 0.96 mmol) was added. After
3 minl-
Propanephosphonic acid cyclic anhydride (0.255 g, 0.80 mmol) was added. The
resulting
reaction mixture was stirred at rt for 50 min. LCMS analysis showed formation
of required
product. Reaction was diluted with DCM and water. DCM layer was extracted and
washed with
brine and dried over sodium sulphate and concentrated. Purification was done
on Waters RP
system to get product N-(cyclopropylmethyl)-14(6-methoxy-5-methylpyrimidin-4-
yOmethyl)-6-
methyl-1H-pyrrolo[3,2-b]pyridine-3-carboxamide (0.045 g, 38.5 %). ES+MS m/z:
366.44. ill
NMR (300 MHz, DMSO-d6) 8 ppm 0.21 -0.33 (m, 2H) 0.41 -0.56 (m, 2 H) 1.08 (m,
J=6.97 Hz,
1 H) 2.24 (s, 3 H) 2.40 (s, 3 H) 3.26 (d., 2 H) 3.94 (s, 3 H) 5.63 (s, 2 H)
7.75 (s, 1 H) 8.11 (s, 1
H) 8.35 (s, 1 H) 8.42 (s, 1H) 8.74 (t, J=5.65 Hz, 1 H).
[02191 Example 11: 14(6- methoxy-5-methylpyrimid Dmethyl)-N-(2-
methoxyethyl)-6-meth I- Ill-py rrolo[3,2-b]pyridine-3-carboxa in ide
\o
0 -...
0 H
(02201 See Figure 18(c). 14(6-
methoxy-5-methylpyrimidin-4-yOmethyl)-6-methyl-11-1-
pyrrolo[3,2-b]pyridine-3-carboxylic acid (0.100 g, 0.32 mmol)and 2-
methoxyethanamine (0.048
g, 0.64 mmol), TEA (0.134 mL, 0.96 mmol) was added. After 3 min, 1-
Propanephosphonic acid
cyclic anhydride (0.255 g, 0.80 mmol) was added. The resulting reaction
mixture was stirred at
rt for 50 min. LCMS analysis showed formation of required product. Reaction
was diluted with
49
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
DCM and water. DCM layer was extracted and washed with brine and dried over
sodium
sulphate and concentrated. Purification was done on Waters RP system to get
product 1-((6-
methoxy-5-methylpyrimidin-4-yl)methyl)-N-(2-methoxyethyl)-6-methyl-IH-
pyrrolo[3,2-
b]pyridine-3-carboxamide (0.045 g, 38.0 %). ES+MS m/z: 370.21. iff NMR (300
MHz,
DMSO-d6) 8 ppm 2.17 - 2.28 (s , 3H) 2.40 (s, 3 H) 3.30 (s, 13 H) 3.50 (d,
J=4.52 Hz, 2 H) 3.55
(t, J=5.18 Hz, 2 H) 3.94 (s, 3 H) 5.63 (s, 2 H) 7.74 (s, 1 H) 8.12 (s, 1 H)
8.34 (s, 1 H) 8.42(s, 1
H) 8.78 (t, J=5.46 Hz, 1H).
102211 Example 12: N-(2-fluoroethyl)-1-((3-methyl-4-(2,2,2-
trifluoroethoxy)pyridin-2-
yl)methyl)-1H-pyrrolo[3,2-131pyridine-3-carboxamide
F
Fr,
t-Nk
0 H
102221 See Figure 18(d). 1 -((3-methyl-4-(2,2,2-trifl uoroethoxy)pyridin-
2-yl)methyl)-1 H-
pyrrolo[3,2-b]pyridine-3-carboxyl i c acid (100 mg, 0.27 mmol) was taken in
DCM (10 mL) to get
a clear solution. Triethyl amine (0.190 mL, 1.37 mmol) was added followed by
the addition of
1-Propanephosphonic acid cyclic anhydride solution in ethyl acetate (0.326 mL,
0.55 mmol) and
stirred the reaction mass for 5 minutes. 2-Fluoroethylamine hydrochloride
(54.5 mg, 0.55 mmol)
was added and stirred the reaction mass for 3h. After completion of the
reaction, diluted with
DCM and then washed with water and brine solution. The organic layer was
separated, dried,
evaporated and the crude compound was purified by silica gel chromatography
using methanol
and dichloromethane as eluent. Yield-49%. ES+MS miz: 411(M+1). H NMR (300 MHz,
DMSO-d6) 8 ppm 2.25 (s, 3 H) 3.67 (q, J=5.21 Hz, 1 H) 3.76 (q, J=5.21 Hz, 1 H)
4.50 (t, J=4.99
Hz, 1 H) 4.66 (t, J=4.90 Hz, 1 H) 4.90 (q, J=8.85 Hz, 2 H) 5.68 (s, 2 H) 7.06
(d, J-5.84 Hz, 1 H)
7.24 (dd, J=8.38, 4.62 Hz, 1 H) 7.93 (d, J=7.35 Hz, 1 H) 8.17 (d, J=5.65 Hz, 1
H) 8.24 (s, 1 H)
8.48 (d, J=3.77 Hz, 1 H) 8.94 (t, J=5.75 Hz, 1 H).
[02231 Example 13: N-(2-fluoroetkl)-1 -((2-nictho -5-(trilluorometh 1)p
ridin-3-
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
yl)methyl)-1H-pyrrolo[3,2-b] pyridine-3-earboxamide
/
I F F
O
[0224j See Figure 19(a). To a stirred solution of 1-((2-methoxy-5-
(trifluoromethyl)pyridin-
3-yl)methyl)-1H-pyrrolo[3,2-b]pyridine-3-carboxylic acid (60 mg, 0.177 mmol)
in
dichloromethane (5 mL) were added 2-fluoroethan-1 -amine hydrochloride (26 mg,
0.26 mmol),
triethylamine (0.053 g, 0.531 mmol) and T3P (0.33 g, 0.531 mmol) and reaction
mixture was
stirred for 16 h at room temperature. Then the reaction mixture was poured in
to water and
extracted with dichloromethane. The reaction mixture was poured into water and
extracted with
dichloromethane . The combined organic layer was washed with water,brine and
solvent was
evaporated under reduced pressure. The crude product was purified by silica
gel column
chromatography using silica gel column chromatography to afford N-(2-
fluoroethyl)-14(2-
methoxy-5-(trifluoromethyl)pyridin-3-y1)methyl)-1H-pyrrolo[3,2-b]pyridine-3-
carboxamide as a
white solid. Yield: 15 mg. (22.3%). ES+MS m/z: 397. 1H NMR (400 MHz, DMSO-d6):
8 ppm
3.66-3.70 (m, 2H),3.72-3.76 (m, 2H), 4.00 (s, 3H), 4.50-4.52 (m, 2H), 4.62-
4.64 (m, 2H), 5.76 (s,
2H), 7.30-7.33 (m, 1H), 7.92 (s, 1H), 8.11 (d, 1H, J = 8.3 Hz), 8.32 (s, I H),
8.50-8.52 (m, 1H),
8.66-8.67 (m, 1H), 8.94 (t, 11-1, J = 5.6 Hz).
102251 EA ample 1-1: I -((5-cyano-2-methox ridin-
3-yl)methy1)-N-(2-fluoroethyl)-1H-
p, rrolo[3.2-blp ridine-3-carboxamide
7/
irS
Ni 0\
0
102261 See Figure 19(b). To a solution of I 4(5-cyano-2-methoxy pyridin-3-
y1) methyl)-1H-
51
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
pyrrolo[3,2-b] pyridine-3-carboxylic acid (0.10 gm,0.32 mmol) in DCM, were
added TEA
(0Ø98 g, 0.135 m1,0.97 mmol), 2-fluoroethan-1 -amine hydrochloride (0.095 g,
0.97 mmol) and
T3P (0.308g, 0.97 mmol). The reaction mixture was stirred at RT for 12 h.
Water was added to
the reaction mixture and extracted with DCM. The combined organic layer was
washed with
water, brine, dried over sodium sulfate, and concentrated at reduced pressure.
The crude product
was purified by flash column chromatography to afford 145-yano-2-
methoxypyridin-3-
yl)methyl)-N-(2-fluoroethyl)-1H-pyrrolo[3,2-b]pyridine-3-carboxamide- the
product as a solid
(17 mg,14.9%). ES+MS m/z: 354. 11-1 NMR (400 MHz, DMSO-d6): 8 8.95 (t, J =
5.80 Hz, 1H),
8.66 (d, J = 2.04 Hz, 1H), 8.51 (d, J = 4.08 Hz, 1H), 8.32 (s, 1H), 8.11 (d, J
= 8.40 Hz, 1H), 7.93
(d, J = 1.92 Hz, 1H), 7.30-7.33 (m, 1H), 5.49 (s, 2H), 5.30 (t, J = 5.00 Hz,
1H), 4.51 (t, J = 4.96
Hz, 1H), 3.91 (s, 3H), 3.72-3.76 (m, 1H), 3.66-3.70 (m, 1H).
[0227] Example 15: 1-((5-fluoro-2-methox py ridin-3-yl)methyl)-N-(2-
fluoroeth:s,1)-6-
methyl-1H-pyrrolo13,2-blpyridine-3-carboxamide
-00
rF
I /
0
[0228] See Figure 19(c). 1-((5-fluoro-2-methoxypyridin-3-yl)methyl)-6-
methyl-1H-
pyrrolo[3,2-b]pyridine-3-carboxylic acid (60 mg, 0.19 mmol)and 2-
fluoroethanamine (21.60 mg,
0.34 mmol), TEA (0.080 ml,, 0.57 mmol) was added. After 3 minl-
Propanephosphonic acid
cyclic anhydride (151 mg, 0.48 mmol) was added. The resulting reaction mixture
was stirred at
rt for 50 min. LCMS analysis showed formation of required product. Reaction
was diluted with
DCM and water.DCM layer was extracted and washed with brine and dried over
sodium
sulphate and concentrated. Purification was done on Waters RP system to get
product 14(5-
fluoro-2-methoxypyridi n-3-yOmethyl)-N-(2-fluoroethyl)-6-methyl-1H-pyrro lo[3
,2-b]pyridine-3-
carboxamide (25.00 mg, 36.5 %). ES+MS m/z: 361.33. 11-1 NMR (300 MHz, DMSO-d6)
6 PPm
2.44 (s, 3 H) 3.65 (d, J=5.27 Hz, 1 H) 3.75 (d, J=5.27 Hz, 1 H) 3.90 (s, 3 H)
4.49 (t, J=4.99 Hz,
1H) 4.64 (t, J=4.99 Hz, 1 H) 5.42 (s, 2 H) 7.36 (dd, J=8.29, 3.01 Hz, 1 H)
7.93 (s, 11-1) 8.12 (d,
J=3.01 Hz, 1 H) 8.23 (s, 1H) 8.38 (s, 5 H) 8.88 (t, J=5.65 Hz, 1 H).
52
CA 02918487 2016-01-15
WO 2015/01)9525 PCTIUS2014/046100
102291 Example 16: (S)-N-(2-fluoroprop31)-14(6-methoxy-5-methylpyrimidin-4-
yl)methyl)-6-meth 1-111-pyrrolo13,2-b1p3 ridine-3-carboxamide
N
I
N
OH r
[0230] See Figure 19(d). 1-((6-methoxy-5-methylpyrimidin-4-yOmethyl)-6-
methyl-1H-
pyrrolo[3,2-b]pyridine-3-carboxylic acid (100 mg, 0.32 mmol) was taken in
dichloromethane (10
mL) to get a suspension. Added triethyl amine (0.133 mL, 0.96 mmol) followed
by the addition
of 1-Propanephosphonic acid cyclic anhydride (0.381 mL, 0.64 mmol). The
reaction mass was
stirred at RT for 5 minutes. (R)-2-fluoropropan- 1-amine (49.4 mg, 0.64 mmol)
was added and
stirred at RT for overnight. After the completion of the reaction, diluted the
reaction mass with
DCM, washed ith water, and brine solution. The organic layer was dried over
sodium sulphate,
evaporated and the crude compound was purified by silica gel chromatography.
Yield-75%.
ES+MS m/z: 373.2 (M+1). 11-1 NMR (300 MHz, DMSO-d6) 8 ppm 1.25 - 1.42 (m, 4 H)
2.24 (s, 3
H) 2.40 (s, 3 H) 3.42 - 3.81 (m, 2 H) 3.94 (s, 3 H) 4.65 - 4.85 (m, 1 H) 4.93
(td, J=6.50, 3.39 Hz,
1 H) 5.64 (s, 2 H) 7.76 (s, 1 H) 8.16 (s, 1 H) 8.29 - 8.47 (m, 2 H) 8.91 (t,
J=6.03 Hz, 1 H).
102311 Example 17: N-(2-hydroxyethyl)-14(6-methoxy-5-methylpyrimidin-4-
.N1)methyl)-6-methyl-1H-pyrrolo[3,2-131pyridine-3-carboxamide
0 H
[0232] See Figure 20(a). 1-((6-methoxy-5-methylpyrimidin-4-yl)methyl)-6-
methyl-1H-
pyrrolo[3,2-b]pyridine-3-carboxylic acid (75 mg, 0.24 mmol) was taken in
dichloromethane (10
mL) to get a suspension. Added tricthyl amine (0.0669 mL, 0.48 mmol) followed
by the
53
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
addition of 1-Propanephosphonic acid cyclic anhydride (0.286 mL, 0.48 mmol).
The reaction
mass was stirred at RT for 5 minutes. Ethanol amine (0.029 mL, 0.48 mmol) was
added and
stirred at RT for overnight. After the completion of the reaction, diluted the
reaction mass with
DCM, washed with water and brine solution. The DCM layer was dried over sodium
sulphate,
evaporated to get the crude compound which was purified by column
chromatography. Yield-
52.7%. ES+MS m/z: 356.4(M+1). NMR (300 MHz, DMSO-d6) 8 ppm 2.23 (s, 3 H) 2.39
(s, 3
H) 3.39 - 3.65 (m, 4 H) 3.93 (s, 3 H) 4.84 (t, J=5.09 Hz, 1 H) 5.63 (s, 2 H)
7.74 (s, 1 H) 8.12 (s, 1
H) 8.33 (s, 1 H) 8.41 (s, 1 H) 8.80 (t, J=5.65 Hz, 1 H).
102331 Example 18: 1((5-fluoro-2-methoxy-6-meth .. ridin-3-1)methyl)-N-(2-
fluoroethyl)-1 H-pyrrolo[3,2-131pyridine-3-carboxamide
N\
F
0 H
102341 See Figure 20(b). To the stirred solution of methyl 1-((5-fluoro-2-
methoxy-6-
methylpyridin-3-yl)methyl)-1H-pyrrolo[3,2-b]pyridine-3-carboxylic acid (0.15
g,0.47 mmol) in
dichloromethane (10 mL), were added 2-fluoroethan- 1-amine hydrochloride (71
mg, 0.71
mmol), triethylamine (0.142 g, 1.41 mmol) and T3P (0.9 g, 1.41 mmol) and the
mixture was
stirred for 16 h at room temperature. Then the reaction mixture was poured
into water and
extracted with dichloromethane . The combined organic layer was washed with
water, brine,
dried over sodium sulfate, and concentrated at reduced pressure. The residue
was purified by
silica gel column chromatography to yield 14(5-fluoro-2-metboxy-6-
methylpyridin-3-
yl)methyl)-N-(2-fluoroethyl)-1H-pyrrolo[3,2-b]pyridine-3-carboxamide as a
white solid. Yield:
30 mg (18%). ES+MS tn/z: 361(M+1). 1H NMR (400 MHz, DMSO-do): 8 ppm 2.31 (d,
3H, J =
2.9 Hz), 3.64-3.68 (m, 2H), 3.71-3.75 (m, 2H), 3.85 (s, 3H), 4.49-4.51 (m,
2H), 4.61-4.63 (m,
2H), 5.41 (s, 2H), 7.28-7.32 (m, 1H), 7.45 (d, 1H, = 9.0 Hz), 8.08-8.11 (m,
1H), 8.29 (s, 1H),
8.49-8.50(m, 1H), 8.92 (t, 1H, J = 5.8 Hz).
102351 Example 19: 6-fluoro-N-(2-fluoroethyl)-1-((6-methoxy-5-
methylpyrimidin-4-
54
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
yl)methyl)-1H-pyrrolo[3,2-b]pyridine-3-carboxamide
F N.7:j
0 v...1
[0236] See Figure 20(c). 6-
fluoro-1-((o-methoxy-5-methylpyrimidin-4-yl)methyl)-1H-
pyrrolo[3,2-b]pyridine-3-carboxylic acid (20 mg, 0.06 mmol) and 2-
fluoroethanamine (7.18 mg,
0.11 mmol), TEA (0.026 mL, 0.19 mmol) was added. After 3 mm, 1-
Propanephosphonic acid
cyclic anhydride (50.3 mg, 0.16 mmol) was added. The resulting reaction
mixture was stirred at
rt for 50 min. LCMS analysis showed formation of required product. Reaction
was diluted with
DCM and water.DCM layer was extracted and washed with brine and dried over
sodium
sulphate and concentrated. Purification was done on Waters RP system to get
product 6-fluoro-
N-(2-fluoroethyl)-1-((6-methoxy-5-methylpyrimidin-4-yl)methyl)-1H-pyrrolo[3,2-
b]pyridine-3-
carboxamide (20.00 mg, 88 %). ES+MS rn/z: 362. NMR (300 MHz, DMSO-d6) 6 ppm
2.24
(s, 3 H) 3.57 - 3.70 (q, 1 H) 3.70 - 3.80 (q, 1 H) 3.94 (s, 3 H) 4.43 - 4.58
(t, 1 H) 4.66 (t, J=4.99
Hz, 1 H) 5.68 (s, 2 H) 8.03 (dd, J=9.89, 2.54 Hz, 1 H) 8.29 (s, 6 H) 8.40 (s,
1 H) 8.53 (t, J=2.07
Hz, 1 H) 8.71 (t, J=5.84 Hz, 1 H).
[0237] Example 20: 6-bromo-N-(2-fluoroethyl)-14(6-methoxy-5-methylpyrimidin-
4-
yOmethyly1H-p)rrolo13,2-bipyridine-3-carboxamide
Br NJ' r_<
0-
I N,
0
[02381 See Figure 20(d). To a
stirred solution of 6-bromo-14(6-methoxy-5-
methylpyrimidin-4-yOmethyl)-1H-pyrrolo[3,2-b]pyridine-3-carboxylic acid (0.13
g, 0.34 mmol)
in dichloromethane (10 mL), were added 2-fluoroethan-1-amine (0.06 g, 0.68
mmol),
triethylamine (0.1 g, 1.02 mmol) and T3P (0.32 g, 1.02 mmol) and the mixture
was stirred at RT
for 16 h. Then the reaction mixture was poured into water and extracted with
dichloromethane.
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
The organic layer was washed with brine and concentrated. The crude product
was purified by
silica gel column chromatography and subsequent PREP purification to afford 6-
bromo-N-(2-
fluoroethyl)-14(6-metho xy-5-methylpyrimidin-4-yl)methyl)-1H-pyrrolo [3 ,2-
b]pyridine-3 -
carboxamide as a white solid. Yield: 25 mg (17%). ES+MS m/z: 424.2(M+1). 11-1
NMR (400
MHz, DMS046): 8 ppm 2.24 (s, 3H), 3.68 (bs, 1H), 3.75 (bs, 1H), 4.51 (bs, 1H),
4.64 (bs, 1H),
5.70 (s, 2H), 8.28 (s, 1H), 8.39 (d. 1H. J = 10.9 Hz), 8.58 (s, 1H), 8.64 (bs,
2H)
102391 Example 21: N-(2-fluoroethyl)-6-methyl-1-43- methy1-4-(2,2,2-
tri fl uo roethoxy)pyridin-2-) l) niethyl)-1H-pyrrolo 13,2-hi pyridine-3-
carboxantide
F
Fir
0
i=rk
0 H
102401 See Figure 21(a). 6-methyl- I -43-methy1-4-(2,2,2-
trifluoroethoxy)pyridin-2-
yOmethyl)-1H-pyrrolo[3,2-b] pyridine-3-carboxylic acid (111 mg, 0.29 mmol) was
taken
dichloromethane (10 mL). Triethyl amine (0.204 mL, 1.46 mmol) was added to get
a clear
solution. I -Propanephosphonic acid cyclic anhydride (0.348 mL, 0.59 mmol) was
added and
stirred at RI for 5 minutes. Added 2-Fluoroethylamine hydrochloride (87 mg,
0.88 mmol) and
stirred at RT for overnight. After the completion of the reaction, diluted the
reaction mass with
dichloromethanc, washed with water, brine solution. The organic layer was
separated, dried over
sodium sulphate, evaporated to get the crude compound. The compound was
purified by silica
gel chromatography using methanol and dichloromethane as eluent. Yield- 44.3%.
ES+MS nilz:
425.2(M+1). 1H NMR (300 MHz, DMSO-d6) 8 ppm 2.24 (s, 3 H) 2.39 (s, 3 H) 3.66
(q, J=5.21
Hz, 1 H) 3.75 (q, J=5.15 Hz, 1 H) 4.49 (t, J=4.99 Hz, 1 H) 4.65 (t, J---4.99
Hz, 1 H) 4.90 (q,
J=8.85 Hz, 2 H) 5.63 (s, 2 H) 7.07 (d, J=5.65 Hz, 1 H) 7.76 (s, 1 H) 8.11 (s,
1 H) 8.17 (d, J=5.65
Hz, 1 H) 8.34 (s, 1 H) 8.88 (t, J=5.84 Hz, 1 H).
102411 Example 22: (S)-N-(2-fluoropropy1)-1-02-methoxy-5-
(trifluoromethyl)pyridin-3-
Orneth1)-6-meth1-1H-pyrrolo[3,2-b1p ridine-3-carboxamide
56
CA 02918487 2016-01-15
WO 2015/009525 PCTIUS2014/046100
F
F
N --N
r-c---
I 0\
'..
NI-1__H
0 N\......7
:.-F
102421 See Figure 21(b). In a 25mL thermal vial was charged with 1-02-
methoxy-5-
(trifluoromethyppyridin-3-yOmethyl)-6-methyl-IFI-pyrrolo[3,2-b]pyridine-3-
carboxylic acid
(100 mg, 0.27 mmol) and HATU (125 mg, 0.33 mmol) were taken in NMP (4 mL) and
stirred
for 10min at rt. Then (S)-2-fluoropropan- 1 -amine hydrochloride (37.3 mg,
0.33 mmol) and
triethyl amine (0.114 mL, 0.82 mmol) were added and stirred for lh at RT. The
LCMS showed
completion of the reaction. The reaction mixture was poured into water and
extracted with
chloroform. The organic layer was dried and concentrated and crude was
submitted to reverse
phase purification. The pure fractions were concentrated to get (S)-N-(2-
fluoropropy1)-14(2-
methoxy-5-(trifluoromethyl)pyridin-3-yl)methyl)-6-methyl-1H-pyrrolop ,2-b]pyri
di ne-3-
carboxamide (82 mg, 70.6 %) a solid. ES+MS miz: 425(M+1). 1H NMR (300 MHz,
DMSO-d6)
S ppm 1.15 - 1.44 (m, 3 H) 2.44 (s, 3 H) 3.39 - 3.79 (m, 2 H) 3.98 (s, 3 H)
4.75 (td, J=6.50, 3.39
Hz, 1 H) 4.92 (td, J=6.50, 3.20 Hz, 1 H) 5.48 (s, 2 H) 7.81 (d, J=2.26 Hz, 1
H) 7.91 - 8.03 (m, 1
H) 8.25 (s, 1 H) 8.34 - 8.42 (m, 1 H) 8.53 - 8.62 (m, 1 H) 8.90 (t, J=5.93 Hz,
1 H).
102431 Example 23: l-((6-niethox3-5-Inethylpyrimidin-4-1)meth1)-N-(2-
(tritluoromethox),) ethyl)-1H-pyrrolo13,2-blpyridine-3-carboxamide
0---
/ ir-----'
/ /
0--IF_F
N F
0 H
102441 See Figure 21(c). 1 -((6-methoxy-5-methylpyrimidin-4-yl)methyl)-1H-
pyrrolo [3,2-
b]pyridine-3-carboxylic acid (40 mg, 0.13 mmol) was taken in a 50m1 single
necked flask
equipped with an air condenser connected to nitrogen source. NMP (3 ml, 31.17
mmol) was
added to get a solution. Triethyl amine (0.056 ml, 0.40 mmol) was added
followed by the
57
1
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
addition of 2-(trifluoromethoxy)ethanamine hydrochloride (44.4 mg, 0.27 mmol).
The reaction
mass was stirred at RT for 5 minutes. HATU (61.2 mg, 0.16 mmol) was added and
stirred at RT
for 30 min. After the completion of the reaction, few drops of methanol was
added and clear
solution was subjected for reverse phase HPLC purification to get 1-((6-
methoxy-5-
methylpyrimidin-4-yl)methyl)-N-(2-(trifluoromethoxy)ethyl)-1H-pyrrolo[3,2-
b]pyridine-3-
carboxamide (15.00 mg, 27.3 %). ES+MS m/z: 410 (M+1). 1H NMR (300 MHz, DMSO-
d6)
ppm 2.25 (s, 3 H) 3.68 - 3.80 (m, 2 H) 3.93 (s, 3 H) 4.24 (t, J=5.27 Hz, 2 H)
5.69 (s, 2 H) 7.26
(dd, J=8.29, 4.71 Hz, 1 H) 7.94 (d, 3=7.35 Hz, 1 H) 8.28 (s, 1 H) 8.38 - 8.54
(m, 2 H) 8.96 (s, 1
H).
[0245] Example 24: (S)-14(5-fluoro-2-methoxypyridin-3-yl)methyl)-N-(2-
fluoropropyl)-
6-methyl-IH-pyrrolo[3,2-blpyridine-3-carboxamide
--N I
F
0 H -
[0246] See Figure 21(d). 14(5-fluoro-2-methoxypyridin-3-yl)methyl)-6-methyl-IH-
pyrrolo[3,2-b]pyridine-3-carboxylic acid (147 mg, 0.47 mmol) was taken in a
50m1 single
necked flask equipped with an air condenser connected to nitrogen source. NMP
(3 ml, 31.17
mmol) was added to get a suspension. HATU (213 mg, 0.56 mmol) was added
followed by the
addition of (S)-2-fluoropropan-l-amine (71.9 mg, 0.93 mmol). The reaction mass
was stirred at
RT for 5 minutes. Triethyl amine (0.195 ml, 1.40 mmol) was added and stirred
at RT for 10
minutes.
[0247] After the completion of the reaction added water, and extracted with
ethyl acetate.
The organic layer was dried over sodium sulphate, evaporated to get the crude
compound. The
crude compound was purified by Gilson prep HPLC to get (S)-14(5-fluoro-2-
methoxypyridin-3-
yl)methyl)-N-(2-fluoropropy1)-6-methyl-1H-pyrrolo[3,2-b]pyridine-3-carboxamide
(110 mg,
63.0 %). ES+MS m/z: 375 (M+1). 1H NMR (300 MHz, DMSO-d6) 8 ppm 1.24 - 1.37 (m,
3 H)
2.44 (s, 3 H) 3.47 (s, 1 H) 3.64 (br. s., 1 H) 3.75 (s, 1 H) 3.90 (s, 3 H)
4.76 (br. s., 1 H) 4.90 (br.
s., 1 H) 5.42 (s, 2 H) 7.38 (s, 1 II) 7.93 (s, I H) 8.12 (d, 3=3.01Hz, 1 H)
8.23 (s, 1 H) 8.38 (s, 1
58
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
H) 8.90 (s, 1 H).
[0248] Example 25: 2-cyclopropyl-N-(14(6-methoxy-5-methylpyrimidin-4-
yl)methyl)-
1 H-pyrrolo [3,2-b] pyridin-3-yl)acetamide
r_5_1(s1
0
25N
[0249] See Figure 22.
102501 3-Nitro-1H-pyrrolo[3,2-blpyridine (25b): To a solution of compound-1
(5 g, 0.042
moles) in con.H2S0.4 (50 mL), con.HNO3 (3 mL, 0.063 mole) was added at -10 C.
At this
temperature the reaction mixture was stirred for 5 h. Then the mixture was
poured into ice cold
water (100 mL), neutralized with aq. NaOH (10%) and extracted with
ethylacctate (2 x 100 mL).
The combined organic layer was washed with brine and the solvent was
evaporated under
reduced pressure to yield 3-Nitro-1H-pyrrolo[3,2-b]pyridine (25b) 3 g (43.4%).
102511 1((6-methoxy-5-meth rimidin-1-1)inet113,1)-3-nitro-111-
pyrrolo[3,2-
him r idin c (25c): To a stirred suspension of 3-Nitro-1H-pyrrolo[3,2-
b]pyridine (25b) (0.5 g,
3.04 mmol) and K2CO3 (0.5 g, 9.12 mmol) in DMF (10 mL) was added 4-
(chloromethyl)-6-
methoxy-5-methylpyrimidine (0.7 g, 6.09 mmol) and the resulting mixture was
stirred for 16 h at
room temperature. Then the reaction mixture was poured in to water (50 mL) and
extracted with
dichloromethane (2 x 50 mL). The combined organic layer was washed with brine
and the
solvent was evaporated under reduced pressure to yield 1-((6-methoxy-5-
methylpyrimidin-4-
yl)methyl)-3-nitro-1H-pyrrolo[3,2-b]pyridine (25c) [300 mg (33%)] as a pale
yellow solid.
[0252] 1-((6-methoxy-5-methylpyrimidin-4-yl)methyl)-1H-pyrrolo13,2-b1p:4,
ri ci i n-3-
amine (25d): To a solution of 1-((6-methoxy-5-methylpyrimidin-4-yOmethyl)-3-
nitro-1H-
pyrrolo[3,2-b]pyridine (0.15 g, 0.58 mmol) in ethanol (5 mL) was added Pd/C
(0.03 g). The
reaction mixture was hydrogenated under balloon pressure for 16 h at RT. The
solvent was
59
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
filtered and evaporated under the reduced pressure to yield 14(6-methoxy-5-
methylpyrimidin-4-
yOmethyl)-1H-pyrrolo[3,2-b]pyridin-3-amine (25d 0.1 g (74%).
102531 2-cyc lop ropyl-N-(14(6-methoxy-5-methylpyrimidin-4-yl)methyl)-111-
pyrrolo[3,2-blpyridin-3-yl)acetamide: To a stirred solution of 1-((6-methoxy-5-
methylpyrimidin-4-yOmethyl)-1H-pyrrolo[3,2-b]pyridin-3-amine (25d) (0.1 g,
0.37 mmol) in
dichloromethane (10 mL) were added triethylamine (0.15 mL, 1.11 mmol), T3P
(0.35 g, 1.11
mmol) and 2-cyclopropylacetic acid (0.037 g, 0.37 mmol and the mixture was
stirred for 16 h at
room temperature. Then the reaction mixture was poured into water (20 mL) and
extracted with
dichloromethane (2 x 50 mL). The combined organic layer was washed with brine
and the
solvent was evaporated under reduced pressure. The crude product was purified
by silica gel
column chromatography using 50% cthylacetate in hexane to afford 2-cyclopropyl-
N-(14(6-
methoxy-5-methylpyrimidin-4-yl)methyl)-1H-pyrrolo[3,2-b]pyridin-3-yl)acetamide
as off-white
solid; yield: 25 mg (19%). ES+MS rn/z: 352 (M+1). 11-1 NMR (400 MHz, DMSO-d6):
8 ppm
2.25 (s,3H), 3.67-3.69 (m, 1H), 3.73-3.76 (m, 1H), 4.50-4.52 (m, I H), 4.62-
4.64 (m, 1H), 5.49 (s,
2H), 6.94-6.96 (m, 3H), 7.27-7.30 (m, 1H), 8.08 (d, J = 8.2 Hz), 8.45 (s, 1H),
8.50 (d, J = 3.88
Hz), 8.92 (t, J = 5.6 Hz).
102541 Example 26: 1-((5-fluoro-2,6-dimethylpyridin-3-yl)methyl)-N-(2-
fluoroethyl)-6-
methyl -1H-pyrrolo[3,2-b1pyridine-3-carboxamide
Arc-
IN;
'N
0
[0255i See Figure 23(a). In a 25mL thermal vial was charged with 145-fluoro-
2,6-
dimethylpyridin-3-yl)methyl)-6-methyl-IH-pyrrolo[3,2-b]pyridine-3-carboxylic
acid (100 mg,
0.32 mmol) and HATU (146 mg, 0.38 mmol) were taken in NMP (4 mL) and stirred
for 10min at
rt. Then 2-fluoroethanamine (20.13 mg, 0.32 mmol) and Triethyl amine (133 mL,
0.96 mmol)
were added and stirred for lh at rt. The LCMS showed completion of the
reaction. The reaction
mixture was poured into water and extracted with chloroform. The organic layer
was dried and
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
concentrated and crude was submitted to reverse phase purification. The pure
fractions were
concentrated to get 1-((5-fluoro-2,6-dimethylpyridin-3-yOmethyl)-N-(2-
fluoroethyl)-6-methyl-
IH-pyrrolo[3,2-b]pyridine-3-carboxamide (75 mg, 65.6 %) as a solid. ES+MS m/z:
359(M+1).
11-1 NMR (300 MHz, DMSO-d6) 8 ppm 2.37 (d, J=2.64 Hz, 3 H) 2.42 (s, 6 H) 3.66
(d, J=5.46 Hz,
1 H) 3.75 (d, J=5.27 Hz, 1 I-1) 4.49 (t, J=4.90 Hz, 1 H) 4.65 (t, J=4.99 Hz, 1
H) 5.52 (s, 2 H) 6.85
(d, J=10.17 Hz, 1 H) 7.86 (s, 1 H) 8.15 (s, 1 H) 8.34 - 8.46 (m, 1 H) 8.89 (t,
J=5.84 Hz, 1 H).
102561 Example 27: N-(2-hydroxyethyl)-6-methy1-1-((3-methyl-4-(2,2,2-
trifluoroethoxy)
pyridin-2-yl)methyl)-1H-pyrrolo13,2-131pyridine-3-earboxamide
7.3 /
0
5r
0
OH
[02571 See Figure 23(b). 6-methy1-1-43-methyl-4-(2,2,2-
trifluoroethoxy)pyridin-2-
yOmethyl)-1H-pyrrolo[3,2-b]pyridine-3-carboxylic acid (55 mg, 0.14 mmol) and
NMP (13.95
I, 0.14 mmol) was taken in a 50m1 single necked flask equipped with an air
condenser
connected to nitrogen source. HATU (66.2 mg, 0.17 mmol) was added to get a
suspension.
Added ethanol amine (17.50 1, 0.29 mmol) followed by the addition of triethyl
amine (60.6 I,
0.43 mmol). The reaction mass was stirred at RT for 5 minutes. LCMS showed
completion of
the reaction. The crude compound was purified by Gilson Prep HPLC to get pure
N-(2-
hydroxyethyl)-6-methy1-1-03-methy1-4-(2,2,2-trifluoroethoxy)pyridin-2-
yOmethyl)-1H-
pyrrolo[3,2-b]pyridine-3-carboxamide (15.00 mg, 24.49 %). ES+MS m/z: 423
(M+1). 11-1 NMR
(300 MHz, DMSO-d6) 8 ppm 2.23 (s, 3 H) 2.39 (s, 3 H) 3.45 (br. s., 2H) 3.53
(br. s., 2 H) 4.83
(s, 1 H) 4.90 (d, J=9.04 Hz, 2 H) 5.62 (s, 2 H) 7.06 (d,J=5.65 Hz, 1 H) 7.75
(s, 1 H) 8.07 (s, 1 H)
8.17 (d, J=5.09 Hz, 1 H) 8.32 (s, 1H) 8.79 (s, 1 H).
102581 Example 28: (R)-N-(2-hydroxypropy1)-14(6-methoxy-5-meth I pyrimidin-
4-
yl)methyl) -6-methyl-1H-pyrrolo[3,2-14yridine-3-carboxamide
61
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
\
OH
N
0 H
102591 See Figure 23(c). 14(6-methoxy-5-methylpyrimidin-4-ypmethyl)-6-
methyl-1H-
pyrrolo[3,2-b]pyridine-3-carboxylic acid (75 mg, 0.24 mmol) was taken in a
thermal reactor.
DCM (5 mL) was added to get a suspension. Added triethyl amine (0.067 mL, 0.48
mmol)
followed by the addition of 1-Propanephosphonic acid cyclic anhydride (0.286
mL, 0.48 mmol).
(R)-1-aminopropan-2-ol (36.1 mg, 0.48 mmol) was added and stirred at RT for
ON. After
completion of the reaction, reaction mixture was concentrated and dissolved in
DCM:Me0H.
This crude compound was subjected for reverse phase purification to obtain (R)-
N-(2-
hydroxypropy1)-14(6-methoxy-5-methylpyrimidi n-4-yl)methyl)-6-methyl-1 H-
pyrrolo [3,2-
b]pyridine-3-carboxamide (35.0 mg, 39.5 %) as a off-white solid. ES+MS miz:
370 (M+1). 1H
NMR (300 MHz, DMSO-d6) & ppm 1.10 (d, J=5.84 Hz, 3 H) 2.24 (s, 3 H) 2.39 (s, 3
H) 3.16 -
3.29 (m, 1 H) 3.37 - 3.48 (m, 1 H) 3.77 (br. s., 1H) 3.93 (s, 3 H) 4.85 (d,
J=4.33 Hz, 1 H) 5.63 (s,
2 H) 7.74 (s, 1 H) 8.12 (s, 1 H) 8.33 (s, 1 H) 8.41 (s, 1 H) 8.82 (br. s., 1
H).
102601 Example 29: 1-(0-(dimethy1amino)-5-meth3.1p ri tuidin-4-3,1)methyl)-
N-(2-
fluoroethyl) -6-methy1-1H-pyrrolo13,2-h pyridine-3-carbo mide
--N
0 H
102611 See Figure 23(d). 1-06-(dimethylamino)-5-methylpyrimidin-4-
yl)methyl)-6-methyl-
IH-pyrrolo[3,2-b]pyridine-3-carboxylic acid (75 mg, 0.23 mmol) was taken in a
thermal reactor.
DCM (5 mL) was added to get a suspension. Triethyl amine (0.064 mL, 0.46 mmol)
was added
followed by the addition of 1-Propanephosphonic acid cyclic anhydride (0.274
mL, 0.46 mmol).
2-fluoroethanamine hydrochloride (22.94 mg, 0.23 mmol) was added and stirred
at RT for ON.
62
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
After the completion of the reaction, reaction mixture was concentrated and
dissolved in
DCM:Me0H. This crude compound was subjected for reverse phase purification.
The final
compound obtianed 14(6-(dimethylamino)-5-methylpyrimidin-4-yl)methyl)-N-(2-
fluoroethyl)-6-
methyl-1H-pyrrolo[3,2-b]pyridine-3-carboxamide (12.00 mg, 14.05 %) as a white
solid.
ES+MS rn/z: 371 (M+1). 1H NMR (300 MHz, DMSO-d6) 8 ppm 2.27 (s, 3 H) 2.40 (s,
3 H) 2.95
(s, 6 H) 3.66 (d, J=5.46 Hz, 1 H) 3.75 (d, J=5.27 Hz, 1 H) 4.49 (t, J=4.99
Hz,! H) 4.65 (t, J=4.99
Hz, 1 H) 5.54 (s, 2 H) 7.75 (s, 1 H) 8.13 (s, 1 H) 8.22 (s, 1 H) 8.34 (s, 1 H)
8.88 (t, J=5.84 Hz, 1
H).
102621 Example 30: \ -(2,2-difluoroethyl)-14(6-methoxy-5-meth I pµ rimidin-
4-
1)meth 1)-6-meth:s1-1H-pyrrolo13,2-131pyridine-3-earboxamide
Ic-
N
0 H F
102631 See Figure 24(a). 1-((6-methoxy-5-methylpyrimidin-4-yl)methyl)-6-
methyl-11-1-
pyrrolo[3,2-b]pyridine-3-carboxylic acid (100 mg, 0.32 mmol) was taken in a
thermal reactor.
DCM (3 mL) was added to get a suspension. Triethyl amine (0.089 mL, 0.64 mmol)
was added
followed by the addition of 1-Propanephosphonic acid cyclic anhydride (0.381
mL, 0.64 mmol).
2,2-difluoroethanamine (26.0 mg, 0.32 mmol) was added and stirred at RT for
ON. After the
completion of the reaction, reaction mixture was concentrated and dissolved in
DCM:Me0H.
This crude compound was subjected for reverse phase purification to obtain N-
(2,2-
di fluoroethyl )-1-((6-methoxy-5-methylpyrimidin-4-yl)methyl)-6-methyl-1H-
pyrrolo [3,2-
b]pyridine-3-carboxamide (20.00 mg, 16.64 %) as a off white solid. ES+MS m/z:
376 (M+1).
NMR (300 MHz, DMSO-d6) 8 ppm 2.24 (s, 4 H) 2.40 (s, 3 H) 3.77 - 3.89 (m, 2 H)
3.93 (s, 4
H) 5.65 (s, 2 H) 7.77 (s, 1 H) 8.20 (s, 1 H) 8.40 (s,1 H) 8.36 (s, 1 H) 8.91
(br. s., 1 H).
[0264] Example 31: 1-(2,4-dimethlbenzy1)-N-(2-fluoroethyl)-1H-pyrro1o[3,2-
b]pyridine-3-earhoxamide
63
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
-
0 H
[0265] See Figure 24(b). 1-(2,4-dimethylbenzy1)-1H-pyrrolo [3 ,2-b]pyridine-
3-carboxyl ic
acid (200 mg, 0.71 mmol) was taken in a 100m1 single necked flask equipped
with an air
condenser connected to nitrogen source. Added CH2C12 (10 mL) to get a clear
solution.
Triethylamine (5 mL, 35.87 mmol) was added followed by the addition of 1-
Propylphosphonic
acid cyclic anhydride (2 mL, 1.43 mmol) and 2-Fluoroethylamine hydrochloride
(142 mg, 1.43
mmol). The reaction mass was stirred at RT for overnight. This crude compound
was subjected
for reverse phase purification to obtain 1-(2,4-dimethylbenzy1)-N-(2-
fluoroethyl)-1H-
pyrrolo[3,2-b]pyridine-3-carboxamide (50.00 mg, 22 %) as a off white solid.
ES+MS Ink: 326
(M+1). 1H NMR (300 MHz, DMSO-d6) 8 ppm 2.21 (s, 3 H) 2.24 (s, 3 H) 3.57 - 3.86
(m, 2 H)
4.50 (t, J=4.99 Hz, 1 H) 4.66 (t, J=4.99 Hz, 1 H) 5.41 - 5.60 (m, 2 H) 6.72
(d, J=7.72 Hz, 1 H)
6.94 (d, J=7.54 Hz, 1 H) 7.05 (s, 1 H) 7.28 (dd, J=8.29, 4.71 Hz, 1 H) 7.98
(dd, J=8.38, 1.04 Hz,
1 H) 8.13 (s, 1 H) 8.51 (dd, J=4.62, 1.04 Hz, 1 H) 8.94 (t, J=5.84 Hz, 1 H).
[0266] Example 32: N-(cyclopropylmethyl)-1-(2-fluoro-6-methoxybenzy1)-1H-
pyrrolo[3,2-blpyridine-3-carboxamide
F
0 H
[0267] See Figure 24(c). In a 50 mL round-bottomed flask 1-(2-fluoro-6-
methoxybenzy1)-
1H-pyrrolo[3,2-b]pyridine-3-carboxylic acid (.130 g. 0.43 mmol),
cyclopropylmethanamine
(0.040 g, 0.56 mmol) and TEA (0.181 mL, 1.30 mmol) was taken DCM (10 mL) under
N2. To
this 1-Propanephosphonic acid cyclic anhydride (0.317 g, 1.00 mmol) was added.
The resulting
reaction was stirred at RT for 50 min. LCMS analysis showed formation of
required product.
Reaction was diluted with DCM and water. DCM layer was extracted and washed
with brine
64
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
and dried over sodium sulphate and concentrated. Purification was done on
Waters RP system to
get product N-(cyclopropylmethyl)-1-(2-fluoro-6-methoxyben7y1)-1H-pyrrolo[3,2-
b]pyridine-3-
carboxamide (0.060 g, 39.2 %). ES+MS m/z: 354 (M+1). NMR
(300 MHz, DMSO-d6) 8
ppm 0.24 (q, J=4.90 Hz, 2 H) 0.38 - 0.50 (m, 2 H) 1.03 (t, J=7.06 Hz, 1H) 3.25
(t, .1=6.22 Hz, 2
H) 3.88 (s, 3 H) 5.47 (s, 2 H) 6.83 - 6.99 (m, 2 H) 7.26 - 7.46 (m, 2 H) 8.02
(s, 1H) 8.07 (d,
J=8.10 Hz, 1 H) 8.49 (d, J=3.96 Hz, 1 H) 8.75 (t, J=5.65 Hz, 1 H).
[02681 Example 33: N-(2,2-difluoroethyl)-14(6-methoxy-5-methylpyrimidin-4-
y1)methyl)-6-methyl-1H-pyrrolo[3,2-blpyridine-3-carboxamide
[02691 ES+MS m/z: 376
[02701 11-1 NMR (300 MHz, DMSO-d6) 8 ppm 2.18 - 2.31 (m, 3 H) 2.40(s, 3H)
3.84 - 3.97
(m, 5 H) 5.62 - 5.70 (m, 2 H) 6.01- 6.22 (tt,1 H) 7.78 (s, 1 H) 8.20 (s, 1 H)
8.38-8.41 (d, J=14.51
Hz, 2 H) 8.87 - 8.96 (m, 1 H).
[02711 Example 34: 1-06-(dimethylamino)-5-meth timid i 11-4- I ) methyl)-
N-(2-
hydroxy ethyl)-6-methy1-1H-pyrrolo[3,2- hi py ridin e-3-carbox a mid e
[02721 ES+MS m/z: 369
[02731 NMR
(300 MHz, DMSO-d6) 8 ppm 2.27 (s, 3 H) 2.40 (s, 3 H) 2.95 (s, 6 H) 3.41 -
3.58 (m, 4 H) 4.84 (t, J=4.99 Hz, 1 H) 5.53 (s, 2 H) 7.73 (s, 1 H) 8.10 (s, 1
H) 8.22 (s, 1 H) 8.33
(s, 1 H) 8.80 (t, J=5.46 Hz, 1 H).
[02741 Example 35: 1-1(6-(Difluoromethoxy)-5-methylpyrimidin-4-1)methyl)-N-
(2-
hydroxyethyl)-6-meth:s 1-1 H-py rrolo[3,2-blpyridine-3-carboxam id c
[02751 ES+MS / 392
[02761 NMR
(400MHz, DMSO-d6) 8 ppm 2.32 (s, 3H), 2.40 (s, 3H), 3.46 (d, J=5.6 Hz,
2H), 3.56 (d, J=5.1 Hz, 2H), 5.75 (s, 2H), 4.83 (brs, 1H), 8.02 - 7.53 (m,
2H), 8.21 - 8.04 (m,
I H), 8.35 (s, 1H), 8.52 (s, 1H), 8.81 (br s, 1H).
102771 Example 36: N-(2-fluoroethyl)-6-methoxy-14(6-methoxy-5-
methylpyrimidin-4-
) I )nictl 1)- 1 H-p) rrolo13,2-131pyridine-3-carboxamide
102781 ES+MS miz: 374
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
[0279] 11-1 NMR (300 MHz, DMSO-d6) 6 ppm 2.14 - 2.32 (m, 3 H) 3.64 - 3.86
(m, 5 H)
3.94 (s, 3 H) 4.49 (t, J-4.99 Hz, 1 H) 4.65 (t, J=4.90 Hz, 1 H) 5.64 (s, 2 H)
7.63 (d, J=2.45 Hz, 1
H) 8.07 (s, 1 H) 8.26 (d, J=2.45 Hz, 1 H) 8.43 (s, 1 H) 8.76 (t, J=5.93 Hz, 1
H).
[02801 Example 37: N-(2,2-difluoroethyl)-6-methoxy-14(6-methoxy-5-
methylpyrimidin-
4-yl)methyl)-1H-pyrrolo[3,2-blpyridine-3-carboxamide
[02811 ES+MS m/z: 392
[0282] NMR (300 MHz, DMSO-d6) 6 ppm 2.18 - 2.31 (m, 3 H) 3.77 - 3.97 (m, 8
H) 5.62
- 5.69 (in, 2 H) 6.00-6.38(tt, 1H) 7.64 (d, J=2.45 Hz, 1 H) 8.11 (s, 1 H) 8.27
(d, J=2.45 Hz, 1 H)
8.43 (s, 1 H) 8.74 - 8.84 (m, 1 H).
[0283] Example 38: N-(2-hydroxyethyl)-6-methoxy-1-((6-methoxy-5-
methylpyrinddin-
4-y1)methyl)-1H-pyrrolo[3,2-blpyridine-3-carboxamide
[0284] ES+MS miz: 372
102851 11-1 NMR (300 MHz, DMSO-d6) 6 ppm 2.23 (s, 3 H) 3.31 - 3.43 (m, 4 H)
3.81 (s, 3
H) 3.94 (s, 3 H) 4.83 (t, J=4.99 Hz, 1 H) 5.63 (s, 2 H) 7.61 (d, J=2.45 Hz, 1
H) 8.03 (s, 1 H) 8.25
(d, J=2.45 Hz, 1 H) 8.43 (s, 1 H) 8.64 - 8.74 (m, 1 H).
102861 Example 39: 1-(6-(Dimethylamino)-5-methylpyrimidin--t-yl)methyD-N-
(2-
flu o roethyl)-6-methoxy-1H-pyrrolo[3,2-b1 pyridine-3-carbox a m icie
[02871 ES+MS Ink: 387
(0288) iff NMR (400MHz, DMSO-d6) 8 ppm 2.28 (s, 3H), 2.96 (s, 6H), 3.67 (q,
J=5.2 Hz,
1H), 3.74 (q, J=5.4 Hz, 1H), 3.83 (s, 3H), 4.51 (t, J=5.0 Hz, 1H), 4.63 (t,
J=5.0 Hz, 1H), 5.55 (s,
2H), 7.62 (d, J=2.4 Hz, 1H), 8.05 (s, 1H), 8.27 - 8.25 (m, 2H), 8.77 (t, J=6.0
Hz, 1H).
(0289) Example 40: N-(2,2-Difluoroethyl)-1-((6-(d i met h la mino)-5-meth
ylpyrimidin-4-
yl)methyl)-6-methoxy-1H-pyrrolo[3,2-b] pyridine-3-carho a nude
102901 ES+MS miz: 405
102911 iff NMR (400MHz, DMSO-d6) 8 ppm 2.27 (s, 3H), 2.95 (s, 6H), 4.00 -
3.84 (m, 5H),
5.55 (s, 2H), 6.32 - 6.04 (m, 1H), 7.62 (d, J=2.3 Hz, 1H), 8.08 (s, 1H), 8.27 -
8.24 (m, 2H), 8.79
(t, J=6.0 Hz, 1H).
66
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
102921 Example 41: 1((6-(Dimethylamino)-5-methylpyrimidin-4- I) methyll)-N-
(2-
hydroxyethyl)-6-methoxy-1H-pyrrolo[3,2-131 pyridine-3-carbo = a m i de
[0293] ES+MS m/z: 385
102941 NMR
(400MHz, DMSO-d6) 8 ppm 2.28 (s, 3H), 2.96 (s, 6H), 3.46 - 3.43 (m, 2H),
3.55-3.54 (m, 2H), 3.82 (s, 3H), 4.82 (br. s., I H), 5.54 (s, 2H), 7.60 (d,
J=2.4 Hz, 1H), 8.01 (s,
1H), 8.37 - 8.17 (m, 21-1), 8.69 (t, J=5.7 Hz, 1H).
102951 Example 42: 14(6-(Difluoromethoxy)-5-methylpyrimidin-4-yOmethyD-N-(2-
tluoroethyl)-6-methoxy-1H-pyrrolo13,2-b1pyridine-3-carboxamide
102961 ES+MS m/z: 410
10297] 11-1 NMR (400MHz, DMSO-d6) 8 ppm 2.31 (s, 3H), 3.68-3.67 (m, 1H),
3.77 - 3.71
(m, 1H), 3.82 (s, 3H), 4.52 (t, J=4.9 Hz, 1H), 4.64 (t, J=4.9 Hz, 1H), 5.75
(s, 2H), 7.99-7.63 (m,
2H), 8.09 (s, 1H), 8.27 (d, J=2.2 Hz, 1H), 8.54 (s, 1H), 8.78 (t, J=5.7 Hz,
IH).
102981 Example 43: N-(2,2-Difluoroethy1)-1-((6-(difluoromethoxv)-5-
methv1p'rimidin-
4-yI)methyI)-6-methoxy-1H-pyrro1o[3,2-bpyridine-3-carhoxa mide
102991 m/z: 428
103001 H NMR
(400MHz, DMSO-d6) 8 ppm 2.32 (s, 3H), 3.90 - 3.82 (m, 5H), 5.76 (s, 211),
6.34 - 6.05 (m, 1H), 7.99-7.63 (m, 2H), 8.13 (s, 1H), 8.29 (d, J=2.4 Hz, 1H),
8.81 (t, J=6.2 Hz,
1H), 8.54 (s, 1H), 8.81 (t, J=6.2 Hz, 1 H).
[03011 Example 44: 1-06-(difluoromethox )-5-methylpyrimidin-4-yl)methyl)-N-
(2-
hydroxyethyl)-6-methoxy-1H-pyrrolo13,2-blp rid i ne-3-earbo am id e
[03021 ES+MS m/z: 408
103031 NMR
(400MHz, DMSO-d6) 8 ppm 2.30 (s, 3H), 3.46 - 3.42 (m, 2H), 3.56 - 3.52
(m, 2H), 3.82 (s, 3H) 4.81 (t, J=5.0 Hz. 1H), 5.73 (s, 2 H), 7.97-7.61 (m,
2H), 8.04 (s, 1H), 8.25
(d, J=2.3 Hz, 1H), 8.53 (s, 1H), 8.69 (t, J=5.6 Hz, 1H).
103041 Example 45: 14(3,5-di metb:µ 1pyrazin-2-yl)methyD-N-(2-fluorocth yI)-
6-meth yl-
1 H-pyrrolo[3,2-b1pyridine-3-carbo x a mide
[03051 ES+MS m/z: 342
67
[0306] 1H NMR (300 MHz, DMSO-d6) 6 ppm 2.40 (d, J=0.94 Hz, 6 H) 2.58 (s, 4
H) 3.63 -
3.70 (m, 1 H) 3.75 (q, J=5.53 Hz, 1 H) 4.49 (t, 1=4.99 Hz, 1 H) 4.65 (t,
J=5.18 Hz, 1 H) 5.67 (s,
2 H) 7.75 - 7.79 (m, 1 H) 8.12 - 8.17 (m, 2 H) 8.33 - 8.37 (m, 1 H) 8.87 (t,
1=5.93 Hz, 1 H).
[0307] Examgle 46: N-(2,2-difluoroethyl)-1-((3,5-dimethylpyrazin-2-
y1)methyl)-6-
methyl-1H-pyrrolo [3,2-b] pyridine-3-carboxamide
[0308] ES=MS m/z: 360
[0309] 1H NMR (300 MHz, DMSO-d6) 6 ppm 2.39 (s, 6 H) 2.58 (s, 3 H) 3.76 -
3.95 (m, 2
H) 5.68 (s, 2 H) 5.98 - 6.05 (m, 1 H) 6.15 - 6.22 (m, 1 H) 6.35 - 6.40 (m, 1H)
7.78 (s, 1 H) 8.14
(s, 1 H) 8.19 (s, 1 H) 8.36 (s, 1 H) 8.85 - 8.97 (m, 1 H).
[0310] Minimal inhibitory concentration (NBC) and minimal bactericidal
concentration
(MBC)
10311] Mycobacterium tuberculosis (Mtb) H37Rv ATCC 27294 used for MIC
detetmination
was grown as reported in Jayaram et. al. (2003). The inoculum used for all the
experiments was
derived from a single seed lot that had been maintained at -70 C. Briefly, Mtb
was grown in
roller bottles at 37 C for 7 to 10 days in Middlebrook 7H9 broth supplemented
with 0.2%
glycerol, 0.05% fweenTm 80 (Sigma), and 10% albumin dextrose catalase (Difco
Laboratories,
Detroit, Mich.); referred to as 7H9 broth in the remainder of the document.
The cells were
harvested by centrifugation, washed twice in 7H9 broth, and resuspended in
fresh 7H9 broth.
Aliquots of 0.5 ml were dispensed, and the seed-lot suspensions were stored at
-70 C. After 24
hours at -70 C, one vial was thawed and plated for colony forming unit (CFU)
enumeration. All
test compound stocks and dilutions were prepared in DMSO.
[0312] Mtb MICs of test compounds were determined in 7H9 broth by a
standard
microdilution method (Balganesh et. al. 2010) with some modifications.
Briefly, 1 1 of serial
two-fold dilutions of test compound were put in 384 well plate, with the final
concentrations
ranging from 100 M- 0.19 M. Control wells included media and culture controls.
40 1 (3-7 x
105 CFUlml) of the bacterial culture was added to all the wells except the
media control wells.
The plates were packed in gas permeable polythene bags and incubated at 37 C
for 5 days.
Following this incubation period, 8 1 of a freshly prepared 1:1 mixture of
Resazurin (0.02% in
water), and 10% Tween 80 was added to all the wells. The plates were re-
incubated for an
68
Date Recue/Date Received 2020-10-02
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
additional 24 hours at 37 C and the colour conversion of all wells recorded. A
blue colour in the
well was interpreted as no growth, and a pink colour was scored as growth.
Minimum Inhibitory
Concentration (MIC) was defined as the lowest drug concentration which
prevented the colour
change from blue to pink. Absorbance at 575nm & 610nm was monitored and their
ratio
calculated. The least concentration which yielded 80% inhibition was
considered as MIC.
Isoniazid is used as reference drug for this assay.
(0313] Aliquots from sample wells (MIC and higher) from the MIC plates were
diluted 1:10
and plated on 7H10 agar plates. Plates were incubated at 37 C for 3-4 weeks,
CFU was
enumerated. The least compound concentration that resulted in a reduction of
two logioCFU
from the start CFU was considered as MBC.
103141 MIC for drug sensitive and single drug resistant M. tuberculosis
isolates
10315] This assay was set up using the same protocol as above, however the
incubation
period was extended to 2-3 weeks. Cell growth was monitored by
turbidometrically and the least
concentration which showed no growth was identified as MIC. With the single
drug resistant
strains, the respective resistance marker drug was included as positive
control.
[0316] Method for MIC determination for other bacteria (Gram positives &
Gram
negatives):
103171 MIC values for different bacterial strains (Staphylococcus aureus
ARC517,
Streptococcus pneumoniae ARC548, Haemophilus influenzae ARC446, Haemophilus
influenzae
ARC158, Escherichia colt ARC523, Escherichia coli ARC524, Pseudomonas
aeruginosa
ARC545, P. aeruginosa ARC546, Klebsiella pneumoniae ARC1865, Mycobacterium
smegmatis
(Msm) ATCC607, Msm mc2155 and Candida albicans ARC526 were determined
according to
Clinical Laboratory Standards Institute (CLSI) guidelines (National Committee
for Clinical
Laboratory Standards. 2009) using 384 well format in cation adjusted Muller
Hinton broth
media. Media control, culture control and appropriate reference drug controls
were included.
Growth is monitored by checking absorbance at 600nm. Minimum inhibitory
concentration
(MIC) was taken as the concentration that resulted in a growth inhibition of?
80%.
[0318] Killing kinetics in 7119 Broth and human THP-1 Macrophages
69
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
103191 The killing kinetics assay in 7H9 broth was performed in a 200 ILL
volume using 96-
well plates with Middlebrook 7H9 medium. Serial two-fold dilutions of
compounds were made
in DMSO separately, with the concentrations ranging from 128 to 0.25 mg/L.
From each of
these dilutions, 44 was added respective wells in a 96-well plate which
contained
approximately 3 X 107 CFU/mL of Mtb H37Rv. The plates were incubated at 37 C
and on days
0, 3, 7, 10, 14 aliquots were diluted in Middlebrook 7H9 broth and plated on
Middlebrook 7H11
agar plates. Bacterial colonies were enumerated after 21-28 days. Data were
expressed as the
logioCFU for each drug treatment.
[03201 Intracellular efficacy of 1,4-azaindoles in THP-1 macrophages
[03211 THP-1 cells (ATCC) were cultured in 75cm2 flask to confluence using
RPM! 1640
with 10% fetal calf serum (Sigma, St. Louis, Mo.) supplemented with 2mM L-
glutamine. The
cells were grown in a 37 C incubator with 5% CO2 and 95% air till they reach a
density of
500,000 cells/mL. From the culture, cells at a density of 1-2 x 105 cells/mL
were infected with
M. tuberculosis H37Rv at a multiplicity of infection (MOI) of 1:10
(macrophage: bacteria) for 2
hours at 37 C (batch infection). After 2 hours, the cells were washed twice
with pre-warmed
phosphate buffered saline to remove extracellular bacteria and then
resuspended in complete
RPMI1640. Phorbol myristate acetate (Sigma) at 40 nM concentration was used to
differentiate
the cells to macrophage and were allowed adhere to 96-well plate for 24 hours
at 37 C. After 24
hours, varying concentrations of the test compounds are added to the
monolayers and incubated
for 7 days. The macrophage monolayers were periodically observed under a
microscope to
monitor adverse changes in the cell morphology due to drug toxicity. At the
start of drug
treatment and at 7 days post-treatment, the monolayers were gently washed and
lyscd with
0.04% SDS and plated on Middlebrook 7H11 agar plates. Bacterial colonies were
enumerated
after 21-28 days. Data were expressed as the logioCFU for each drug treatment.
103221 Antimicrobial Activity against hypoxia induced non-replicating
persistent (NRP)
Mtb cells
103231 M. tuberculosis H37Rv cultures were adapted to hypoxic conditions as
described in
Wayne and Hayes (1996) with minor modifications. Briefly, Mtb cells were grown
in Dubos
Tween broth in McCartney bottles with a magnetic bead using a defined head-
space ratio (HSR)
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
of 0.5. Methylene blue was added as a redox indicator (final concentration of
1.5 g/mL) to all
bottles to monitor oxygen depletion. The MacCartney bottles were placed on a
magnetic stirrer
set at 180rpm, inside a 37 C incubator. The methylene blue indicator started
to fade by day 8
and completely decolorized by 12 days. The antimicrobial activity of various
compounds against
NRP Mtb cells was determined in 96-well microtiter plates using a 14-day old
hypoxia adapted
culture as described above under the MIC determination section. The entire
assay was
performed in a hypoxic chamber (DuPoy) by exposing hypoxic cells to varying
concentrations of
compounds for 7 days at 37 C. An anaerobic indicator strip was placed inside
the chamber to
visually confirm the removal of oxygen during the entire process. Bacterial
enumeration was
performed on Middlebrook 7H11 agar plates. Isoniazid and nigericin were used
as controls in
the assay. Isoniazid showed no reduction in the bacterial CFU even at 10 g/mL
concentration
indicating a strict NRP state. Data are expressed as the logioCFU for each
drug treatment.
103241 A549 cytotoxicity
[0325] The in vitro cytotoxicity of compounds were measured against A549
human lung
carcinoma cells as describedin Eakin et. al (2012). Briefly, A549 cells (ATCC)
were grown in
RPMI medium (GIBCO-BRL) containing 10% heat-inactivated fetal bovine serum
(GIBCO-
BRL) and 1mM L-glutamine (GIBCO-BRL) at a density of 1,000 cells/well. After
incubation
of the cells with compound in a CO2 atmosphere at 37 C for 72 hours, cell
viability was
determined following addition of 10 M of resazurin solution (Sigma), by
measuring
fluorescence (excitation at 535 nm, emission at 590 mn) using a fluorimeter.
The concentration
at which growth is inhibited by 50% is taken as IC50 value.
103261 Mutant generation, Resistance frequency & Whole genome sequencing &
analysis
[0327] Generation of resistant mutant strains and resistance frequency
103281 Spontaneous resistant mutants were raised against compound 31 & 32
using a single
step selection method. Briefly, a mid-logarithmic phase culture of Mtb H37Rv
was centrifuged
and concentrated 100-fold to achieve a bacterial number of ¨1010 CFU/mL.
Varying dilutions of
the bacterial culture were plated onto compound containing plates
(concentration corresponding
to 4X, 8X and 16 X MIC conc. Appropriate dilutions of the bacterial culture
were also plated on
71
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
drug-free Middlebrook 7H11 agar to enumerate the bacterial numbers in the
culture. Plates were
incubated for 4 weeks at 37 C and the CFUs in drug-free plates were
enumerated. The drug-
containing plates were incubated for up to 6 weeks at 37 C to confirm the
final number of
spontaneously resistant colonies. The spontaneous rate of resistance was
calculated by dividing
the number of colonies on drug -containing plates (at a given concentration)
divided by the total
number of viable bacteria estimated on drug-free plates. Resistant colonies
were randomly
picked from the drug containing plates and gown in complete 7H9 broth to
determine their level
of resistance against the specific, compound as well as, other standard TB
drugs with different
mechanisms of action.
103291 Whole genome sequencing
[03301 Total DNA for whole genome sequencing was extracted from resistant
Mtb cells
using standard Phenol-chloroform method. Yield was quantitated on a Qubit 2.0
fluorometer
using the dsDNA broad range assay kit (Life Technologies, Grand Island, NY).
Library
generation was carried out using the Nextera XT DNA sample preparation kit and
Nextera XT
index primers (IIlumina, San Diego, CA). The recommended procedure was
followed with the
following exceptions; a high initial starting concentration of DNA was used
and the library
normalization step at the end was omitted in favour of qPCR library
quantification. qPCR was
performed on a BioRad CFX96 cycler using the Kapa BioSytems (Woburm, MA)
Library
quantification kit (KK4824). Libraries where diluted to a standard
concentration of 4 nM and
2.5 p.1 of each sample (8-12 samples depending) were combined and denatured
with IN NaOH
(final concentration 0.1N NaOH) for 5 minutes. Sufficient sample was diluted
to 600 I to
provide a multiplexed sample of 15-20 pmol. Samples were sequenced on an
IIlumina MiSeq
V2 instrument as 2X150 paired-end single index reads. All sequencing was
targeted at ¨50-fold
coverage.
103311 Assembly and analysis of sequence reads was performed off-instrument
using
CLCBio Genomics Workbench v 6.0 (Cambridge, MA). Fastq files were processed
and
analyzed as follows; duplicate sequence reads were removed and remaining reads
were trimmed
for quality and minimum length (50 bp). Reads were then de novo assembled
under high
stringency (fraction length = 0.9, similarity fraction = 0.99) using default
mismatch/insertion/deletion costs. Detection of SNPs / indels in mutant
isolates was
72
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
accomplished by mapping the processed reads to a reference parent assembly
using the same
assembly conditions. Quality based SNPs were detected at a minimum frequency
of 80% using
default criteria. Relevant SNPS / indels were verified by BLAST comparison of
the region
against the de novo assembly to help eliminate possible errors due to the
directed mapping
assembly.
[03321 Pharmacokinetics (PK) of Azaindole compounds:
[03331 PK of azaindoles compounds was performed in mice (healthy and
infected) and rats.
Mice were pretreated with 100 mg/kg ABT two hours prior to the compound
administration. PK
data from healthy mice was used to design the dosing regimen for the efficacy
study while,
information from infected mice was used for the PK-PD analysis.
[03341 BALB/c mice or Wistar rats were administered test compounds 3, 4, 8
and 17 in
separate groups, via oral gavage. All oral administration was performed as
suspensions in 0.5 %
HPMC, and 0.1% Tween 80. In a separate groups test compound 3 (0.5 mg/kg) and
17 (2
mg/kg) were administered intravenously as a solution (20 % v/v DMA in
phosphate buffered
saline). All blood samples were collected via sapheneous vein into Microvette
CB300
(Starstedt, Germany) tubes coated with Lithium-Heparin, and plasma was
prepared from the
collected blood by centrifugation.
103351 Single mouse infected POPK: Compounds and reference drugs were
formulated in
0.5 % HPMC (hydroxypropyl methyl cellulose) and 0.1% Tween80 suspensions.
BALB/c mice
(3 mice/group) were administered via oral gavage at 50, 100 and 200 mg/kg.
Pharmacokinetics
was performed on infected mice on 24th day of dosing (Rennard, 1986). Blood
samples were
collected from each mouse at 0.5, 1.5, 3, 5, 7 and 24 hours post compound
administration.
About 30 pt blood samples were collected by serial sampling from all groups
via sapheneous
vein into Microvette CB3008 (Starstedt, Germany) tubes coated with Lithium-
Heparin and
plasma (10 L) was prepared following centrifugation. Plasma samples were
stored at ¨ 20 C
until analysis using LC-MS/MS.
103361 Epithelial lining (luid (ELF) PK: ELF PK was performed in healthy
mice (three mice
/group) as described previously (Solapure et. al, 2013) after administration
of a single oral dose
73
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
of 100 mg/kg compound formulated in 0.5 % HPMC, and 0.1% Tween 80 suspensions.
After
0.5, 1.5, 3, 5, 7,17 and 24 h of dosing, mice were anesthetized using
isoflourane and blood was
collected through Retro-Orbital Plexus puncture. Broncho-Alveolar Lavage (BAL)
was
performed after tracheotomy using 0.7 mL of ice-cold PBS. Urea estimation kit,
DIUR-500
(Bio-assay Systems, U.S.A) was used for urea estimation in plasma and BAL
samples. Volume
of ELF was calculated after normalizing the urea concentration in BAL with
that of plasma as
described in Marry et. at, 2011. Plasma and BAL samples were stored at ¨ 20 C
until analysis
using LC-MS/MS.
[03371 Plasma and BAL sample analysis: One mg/mL stock solution of each
compound was
prepared in Dimethylsulfoxide (DMSO) and diluted two-fold with acetonitrile. A
sixteen point
calibration curve was utilized for each analyte, and the standard curves
ranged from 0.001 to 40
g/mL. Plasma/BAL samples were precipitated by adding chilled acetonitrile
(1:10 v/v)
containing carbamazepine as internal standard (250 ng/mL). Samples were
vortexed, and
centrifuged at 4000 rpm for 30 min at 10 C. The resulting supernatant was
mixed with mobile
phase (50% acetonitrile in water with 0.1% formic acid). 10 j.tL of sample was
injected on to a
liquid chromatographic system (Waters-ACQUTY UPLC) coupled to triple
quadrupole mass
spectrometer (Waters-ACQUTY-TQD; MS/MS). Samples were acquired in positive ion
mode
and detected by multiple reaction monitoring (MRM). Concentrations of the
analyte were
determined from a standard curve obtained by plotting known concentrations of
the analyte
against peak area ratios (analyte/internal standard peak response).
103381 Healthy and infected PK data Analysis: PK analysis of the plasma
concentration-time
relationships were performed with WinNonlin Phoenix Software (version 6.2;
Pharsight, USA).
A Non-compartmental analysis program, model 200, was used to calculate PK
parameters. The
maximum concentration of drug in plasma (Cmax), time to Cmax (Tmax),
elimination half-life (t%),
and AUC from time zero to infinity (AUCo_co) were estimated. AUC was computed
using
trapezoidal rule (linear up and log down) and AUC0_. value was considered only
when AUC
extrapolated was not more than 20% of original value. Minimum of three sample
points in the
terminal slope were used to estimate to calculate half-life.
74
CA 02918487 2016-01-15
WO 2015/009525 PCTIUS2014/046100
103391 Analysis of ELF PK
103401 Volume of ELF in the BAL samples was calculated as volume of BAI,
multiplied by
the ratio of urea concentrations in BAL and plasma as described (Solapure et.
al, 2013; Marry et.
al, 2011) The compound concentration in ELF was calculated by multiplying
concentration in
BAL samples by the ratio of BAL volume to the ELF volume. AUCO-a) in plasma
and ELF
were calculated by Non-compartmental analysis WinNonlin Phoenix Software
(version 6.2;
Pharsight, USA). Free plasma AUC was calculated after multiplying
concentrations at each
time point by the free fraction in plasma. Lung ELF penetration ratio was
calculated as a ratio of
AUC0.. in ELF to free AUC0,0 in free plasma/total plasma during the same time
interval. This
ratio, measured in healthy mice after single dose administration, was assumed
to remain constant
during multiple dose efficacy study in the infected mice. Sparse sample
analysis in WinNonlin
was used for estimating standard error (SE) associated with AUC estimate.
103411 In vivo efficacy studies
[03421 Mycobacterium tuberculosis infection inoculums: Mtb H37Rv (ATCC
27294),
sensitive to all the standard antimycobacterial agents, was grown as mentioned
above. After 7-
days, cells were harvested by centrifugation, washed twice in 7H9 broth and re-
suspended in
fresh 7H9 broth. One mL aliquots were dispensed and stored at -70 C. The
frozen stocks were
thawed on the day of animal infection and used as inoculums.
[0343] Ethics statement and Animals: All animal experiment protocols and
usage was
approved by Institutional Animal Ethics Committee (IAEC), registered with the
Committee for
the Purpose of Control and Supervision (CPCSEA), Government of India. Male
BALB/c mice
were purchased from RCC Hyderabad, and Rats from Bioneeds, Bangalore, India.
Mice and rats
(6-8 weeks) 8 were randomly assigned into groups of three or four per cage,
and were kept for
one week acclimatization before initiating the study. Animals were housed
under standard
conditions with a 12 hr day-night cycle. Feed (Nutrilab ) and water were given
ad libitum.
Infected mice were maintained in individually ventilated cages (Allentown
Technologies, USA)
in bio-safety level 3 (BSL-3) facility. All procedures including dosing and
blood sampling for
pharrnacokinetics on infected mice were performed under strict bio-
containment.
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
[03441 Aerosol Infection: Mice and rats were infected with M. tuberculosis
via inhalation
procedure using modified Madison aerosol equipment (Jayaram et. al. 2003).
Acute infection
model was established by high dose aerosol infection that instilled ¨104
CFU/lung in mice and
the drug treatment started three days post infection (Schroeder et. al, 2003;
Jayaram et al. 2003).
In contrast, the chronic infection model (mice and rats) (Schroeder et. al,
2003; Jayaram et al.
2003; Kumar et al. 2014) was developed with low dose Mtb aerosol infection,
that delivered
¨50-100 bacilli/lung and the drug treatment started 28 days post infection.
Bacterial numbers
present in the lungs at the beginning of drug treatment was determined. At the
end of the
treatment, mice was euthanized, lungs were aseptically removed, and
homogenized in 3.0 mL gel
saline using Wheaton Teflon-Glass tissue grinders. Lung homogenates were
serially diluted in
10-fold steps and plated onto Middlebrook 7H11 agar plates supplemented with
10% ADC.
Plates were incubated at 370C with 5% a)) for 3 weeks to obtain isolated
colonies.
[03451 In Vivo Dose-Response Studies in mice: Infected mice were pre-
treated with daily
oral doses of 100 mg/kg of Aminobenzotriazole (ABT) two hours prior to
compound
administration, to block P450 metabolism enzymes. Azaindole compounds 3 & 4
were
formulated in 0.5% (w/v) HPMC and 0.1% Tween 80 (Sigma chemical co. USA)
suspensions
and delivered by oral gavages. In acute mice model, animals were dosed with
50, 100 mg/kg of
compound 3 & 4 and with lsoniazid at 3 mg/kg as a positive control. In chronic
model 30 and
100 mg/kg doses of compound 3 & 4 were used. Rifampicin at 10 mg/kg was used
as reference
drug control. Two separate vehicle control groups with and without ABT were
used to rule out
any adverse effect of ABT on Mtb infection. All drugs and test compounds were
administered
orally for four weeks, on a 6/7 day per week dosing format. Forty eight hours
after completion
of dosing period, animals were euthanized with CO,, lungs were aseptically
removed and CFU
enumerated following plating as described above.
[03461 In Vivo Dose-Response Studies in rats: Azaindole compounds 8, 17, 30
& 34 were
formulated in 0.5% (w'v) HPMC and 0.1% Tween 80 (Sigma chemical co. USA)
suspensions
and delivered by oral gavages. In chronic rat model, animals were dosed with
30, 100 mg/kg of
compounds 8, 17, 30 & 34. Rifampicin at 10 mg/kg was used as reference drug
control. All
drugs and test compounds were administered orally for four weeks, on a 6/7 day
per week dosing
76
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
format. Forty eight hours after completion of dosing period, animals were
euthanized with CO2,
lungs were aseptically removed and CFU enumerated following plating as
described above.
[0347] Statistical Analysis: The colony counts obtained from plating were
transformed to
Log10 (X+1), where x equals the total number of viable bacilli present in a
given sample. Prism
software version 4 (Graph Pad Software, Inc., San Diego, California) was used
for plotting
pharmacodynamic effects. Dunnet's multiple-comparison test was used to
differentiate
statistical differences in lung CFU in treated versus untreated mice.
103481 Solubility Assay
103491 Solubility of the compounds in 0.1M phosphate buffer, pH 7.4 was
determined as
described. Glyburide was used as QC standard in the assay. Briefly, compounds
were diluted in
ACN/water (40:60) to desired concentration, samples were dried using Genevac
for 4 hrs and
subsequently 800 I of buffer was added. Compound containing plates were
stirred for 24 hrs at
25 C on Eppendorf 'Thermomix R at 750rpm. Finally, compound concentration was
estimated
using UV and MS analysis.
103501 Plasma protein binding assay
103511 Protein binding is measured using the equilibrium dialysis
technique. Compound is
added to 10% plasma giving a concentration of 20 M and dialysed with isotonic
buffer for 18
hours at 37 C. The plasma and buffer solutions are analysed using generic
LCUVMS and the
first apparent binding constant for the compound derived. Thc binding constant
is then used to
determine the % free in 100% plasma.
[03521 Metabolic stability assay (mouse/human microsomal
10353j luM compound was incubated with lmg/mL of microsomes (Pooled HLM/MLM
with 20mg/m1 protein conc.) at 37 C in 166 L of buffer (100mM phosphate
buffer, pH-7.4)
containing 2mM NADPH solution. 20 L of incubation mix was quenched with 4
volumes
chilled acetonitrile at different time points i.e. 0, 2, 5, 10, 20 and 30min
in a fresh 96 well plate.
The quench plate was centrifuged at 4000rpm for 15 min. 304 of supernatant was
diluted to
300 L with 50% acetonitrile in water and substrate depletion was analyzed
using LC-MS/MS.
77
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
103541 Metabolic stability assay (rat/human hepatocyte Clint)
103551 Viability of cryopreserved hepatocytes was determined using trypan
blue and the cell
conc. was adjusted to 106 cells per mL with buffer (ICHB buffer). 1 M compound
(in
Acetonitrile; 0.01% DMSO) was incubated with 5004 of hepatocyte cells (1
million cells per
mL) in a NUNC plate. Reaction was stopped at different time points (0, 5, 15,
30, 60, 90 and
120 min) by addition of 3 volumes of chilled acetonitrile to 1004 of reaction
mixture and
centrifuged at 4 C for 15 min. Supernatants were analyzed using LC-MS/MS for
substrate
depletion.
103561 logD
[03571 Octanol-water partition coefficient (Log D) based on the shake-flask
principle has
been measured as follows. The aqueous solution used was 10 mM sodium phosphate
buffer pH
7.4. 20 I.LL of 10mM compound dissolved in DMSO was taken in glass vial plate.
DMSO was
removed using GeneVac. 435 tL of octanol was added using Tomtec, stirred for 5
min to
dissolve. Further mixing was done by inversion for 5 h at 25 C, subsequently
centrifuged for 30
min at 3000 RPM. LC/UV/APPI/MS quantitation of both aqueous and octanol layers
was carried
out. Log D value was determined according to the following equation.
LogD = Log(lOctanol/Octanol inj volume'
Buffer/Buffer inj volume
[03581 The method has been validated for log D ranging from -2 to 5Ø
103591 hERG assay
103601 Compounds were tested on voltage-gated ion channels using the medium-
throughput
electrophysiology IonWorksTM device. Detailed methods regarding the running of
lonWorksTM
have been published (Schroeder 2013). For carrying out the experiment, a boat
in the "Cells"
position of the IonWorksTM instrument was loaded with the cell suspension, and
a 96-well PBS
destination plate was placed in the "Plate 1" position. A 384-well
PatchPlatcTM was placed in the
IonWorksTM plenum and held in position using the plenum clamp. From this point
the
experiment progress is automated and ultimately reports a non-cumulative
concentration-effect
curve for test compound.
78
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
Summary
[0361] The disclosed compounds, although cidal for Mtb and Mycobacterium
smegmatis
(Msm), did not show activity against broad-spectrum pathogens, thus suggesting
excellent target
pathogen specificity (Table 2, Figure 25). The compounds in the series retain
MIC for drug
sensitive and single drug resistant clinical isolates of Mtb (Table 3, Figure
26), suggesting their
potential for drug sensitive and MDR TB therapy. The compounds exhibited time
dependent kill
kinetics against replicating Mtb, with ¨ 4 logIO reductions in colony forming
units (CFU) by day
at a concentration of 1-4 fold of MIC (Figure 1). The compounds were also
active on
intracellular Mtb, with ¨ 1 logl 0 reduction in CFU at concentrations 1- to 4-
fold greater than the
MIC in THP1 cells infected with Mtb (Figure 2). In addition to their potent
activity on
replicating bacteria, a subset of molecules in the series show moderate
activity against non-
replicating Mtb under hypoxic conditions (cidality measured as HBC in Wayne
model)
represented by compound 3 (Table 4). The compound in the series were found to
be non-
cytotoxic on A549 human lung adenocarcinoma epithelial cell line post 72 hours
treatment
(MMIC >100 M, Table 4). We have also observed >95% THP-1 macrophage viability
following 7 days of compound exposure at maximum of 32 111µ4 (Table 4).
[0362] Figure 25 shows Table 2, Pathogen specificity.
[0363] Figure 26 shows Table 3, Activity against drug sensitive and drug
resistant Mtb.
103641 Table 4. Microbiological properties of Compounds
MMIC
Compound Mtb MIC Mtb MBC Mtb HBC
(A549)
No (11M) (11M) (PM) (1AM)
3 1.56-3.12 1.56-3.12 50 >100
4 0.39-1.56 0.78-1.56 >100 >100
8 <0.39 <0.39 100 >100
17 1.56-3.12 0.78-1.56 >100 >100
103651 Spontaneous resistant mutants with reduced susceptibility to 1,4-
azaindoles arose at a
frequency of 2.9 x 1 e at 8x MIC concentration (Table 5). Whole gertome
sequencing of the
resistant Mtb mutants revealed a single nucleotide change in dprE 1 (Rv 3790),
resulting in an
amino acid substitution at 314 position (Tyr His)
with no significant secondary target
79
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
observed. While the compounds in the series were cross-resistant to mutant
strain (Tyr314His),
resistance was not observed for reference drugs including BTZ043. The
cystein387 DprEl
mutations (Cys --0 Ser, Cys --4 Gly) that impart resistance to BTZ043
(Makarov, V. et al.
Science, 324, 801-804 (2009), did not show cross resistance to 1,4-azaindoles
(Table 6).
Furthermore, target specificity was re-confirmed by MIC modulation on over-
expression of
DprEl, as also seen for BTZ043 (Table 6).
103661 Table 5. Resistance frequency
Mtb H37Rv Mtb H37Rv Mtb H37Rv MMIC (A549)
MIC (01) MBC (IIM) HBC (pM) (12M)
Compound 31 1.56-3.12 3.12-6.25 12.5-25 >82
Compound 32 1.56-3.12 1.56-6.25 >200 >100
Mtb DprEl OE Mtb DprE1 Mtb DprE1 C387G Mtb DprEl Y314H
MIC( M) C387S MIC ( 1.1M) MIC( M)
Compound 31 25 0.78 0.78 25
Compound 32 50 0.39 0.39 200
103671 Table 6. Cross-resistance within series and reference compounds
Mtb Mtb Mtb DprE1 NItb DprEl Mtb DprEl
Values in M H37Rv DprEl OE C3875 C387G Y314H
Compound 3 3.12 50 3.12 0.78 >100
Compound 4 0.39 /5 0.39 0.39 >100
Compound 8 0.39 6.25 0.39 0.39 >200
Compound 17 1.56 100 0.39 0.78 >200
BTZ043 0.003 50 >0.1 >0.1 0.0015-0.003
Isoniazid 0.06 0.03 0.06 0.06 0.06
Ethambutol 2 2 2 4 2.0-4.0
Rifampicin 0.01 0.003 0.003 0.003 0.006
TMC207 0.6 0.15 0.3 0.15 0.15-0.3
1
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
Moxifloxacin 0.125 0.06 0.06 0.06 0.06
Ofloxacin 1 0.5 0.5 0.5 0.5
D-cycloserine 8 8 8 8 8.0-16.0
Clofazimine 0.125 0.06 0.03 0.03 0.125
103681 The compounds in the series were profiled for in vitro drug
metabolism and
pharmacokinetics (DMPK) properties, representative compounds are shown in
Table 7. The
dried DMSO solubility for compounds 3,4 and 8 was lower than compound 17; the
improved
solubility may be attributed to hydroxyethyl amide side chain. The protein
binding (% free)
values for compounds 3-4, 8 and 17 was between 5% and 22%. The predicted
clearance for
compounds 3-4, 8 and 17 ranged from 4 to 18% of liver blood flow (%LBF),
estimated by using
human microsomes, human hepatocytes and rat hepatocytes. In contrast, the
predicted clearance
was higher from mouse microsomes (Table 8), suggesting species specific
clearance
mechanisms. The permeability measured by Caco-2 assay suggested that these
compounds are
highly permeable with no significant efflux observed. The compounds in the
series did not show
inhibition of CYP enzymes at 50 NI (Table 7), suggesting their potential for
combination
therapy. In vitro safety profiling of compounds 3-4, 8 and 17 against a panel
of human targets
and cardiac channels revealed no major safety liabilities associated with this
series (Table 7).
103691 Table 7. DMPK and Safety properties of compounds
Compound 3 4 8 17
logD 2.1 3.0 2.6 1.8
Solubility (IM) 8a 5a 124
Human CLpred microsomes (%
10.4 15.6 10.1 16.1
LBF)
Human CLpred hepatocytes (%
6.0 4.3 4.3 9.2
LBF)
Rat Ci-pred hepatocytes (% 13
13 14.9 17.9
LBF)
Human PPB (% free) 9.8 5 5 22
Caco-2 A-B/B-A (1E-6.cm/s) 25/17 38/24 33/17 11/30
81
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
CYPb inhibition (pM) >50 >50 >50 >50
hERG ( M) >33 >33 >33 >33
Secondary pharmacology hits
No significant hits
IC50 GIM)
[a]: Kinetic solubility in test media >100 M, [b]: CYP1A2, CYP2C9, CYP2C19,
CYP2D6,
CYP3A4.
103701 Table 8. High intrinsic
clearance for mouse microsomes
Compound 3 4 8 17
Mo micrsomal Clint 104 1 80 108 25
(il/min/mg)
103711 Based on in vitro properties, compounds 3-4, 8 and 17 were profiled
for in vivo PK in
mouse and rat to assess oral exposures. The PK exposures in mouse were
measured in the
presence of aminobenzotriazole (ABT), a Pan-inhibitor of CYP isoforms was used
to block
mouse specific clearance. Significant oral exposures were observed for
compounds 3-4, 8 and
17 in both rat and mouse (Figure 3, Table 9). In the rat, a good correlation
was observed
between in vitro and in vivo clearance with 92% bioavailability for compound
17 (Table 9). The
in vivo efficacy of two representative compounds (3 & 4) was assessed in
BALB/c mice in
"acute" and "chronic TB models" (Jayaram et al. 2003; Marry et al. 2011; Kumar
et at. 2014). In
the acute model, treatment was started 3 days post infection, whereas
treatment was started on
day 28 in the chronic model. Four weeks of treatment of compounds 3 & 4
reduced the bacterial
burden in the lungs by >1.5 log10 CFU and statistically significant dose
dependent efficacy was
observed (Figure 1). The oral exposures of compound 3 & 4, assessed from
infected animals
showed AUCs ranging from 200-700 M.h and concentrations were maintained above
the MIC
for ¨10h after each dose (%T> MIC of ¨10 hrs), resulting into efficacy in
chronic mouse model
(Figure 4; Table 10). Interestingly, the levels of compounds 3 & 4 measured in
healthy mouse
lung epithelial lining fluid (ELF PK) was comparable to free plasma levels for
both the
compounds (Figure 4, Table 11), demonstrating significant exposures at the
target site. Thus, a
good correlation was observed between plasma and/or ELF levels and
pharmacodynamic effect.
In the acute and chronic mouse model, the animals tolerated the administered
doses for one
month, and no adverse effects were observed in terms of body weight and gross
pathology.
82
CA 02918487 2016-01-15
WO 2015/009525 PCT1US2014/046100
[03721 Table 9. Phannacokinetic parameters of compounds in healthy BALB/c
mice and
Wistar rats following single dose administration (data are mean S.D., n=3
unless otherwise
stated).
Compound 3 4 8i7b _________________
Dose (mg/kg) 100 100 30 50
AUCc,..
521.50 19.60 346.73 42.75 64.19 420.30 23.5
Mouse POPK ( M.h)
(+ABT) Tõ.õ (h) 6.33 1.15 4.67 1.15 2 1.33 0.58
C.õ( M) 66.01 11.17 44.12 13.0 13.36 74.79
15.61
Ti 2 (h) 1.91 0.11 3.63 1.46 4.15 1.780.10
Dose (mg/kg) 0.5 2
CL
15.50 3.21 27.35
(111/min/kg)
Võ(1.11cg) 1.05 0.28 ND ND 7.34
Rat 1VPK
AUCo-t
1.60 0.29 3.46
( M.h)
T1 2 (h) 2.21+2.51 2.06
Coll (p1M) 1.76 0.42 2.64
Dose (mg/kg) 30 30 30 20
AUCO.,õ
101.38 22.86 29.29 59.82
(.thel.h)
Rat POPle (h) 2.0 3.0 4.5 0.5
C1.õ(1.1.M) 15.01 2.35 3.69 17.84
T1.2 (h) 2.01 3.64 3.58 2.74
96% ND ND 100%
ND: not determined; 'compound 5 (n=2); b compound 6 for Rat MI( (n=2); for all
compounds (n=2)
103731 Table 10. ELF penetration ratio of 1,4-aaindoles in healthy BAL13/c
mice following
oral single dose administration (data are mean S.D., n=3 unless otherwise
stated).
Compound 3 4
Matrix ELF Plasma /Plasma ELF Plasma fPlasma
83
CA 02918487 2016-01-15
WO 2015/009525
PCT/US2014/046100
Dose (mg/kg) 100 100
74.0 63.84+ 4.85 39.92 38.09 1.52
Cmax (PM)
3.77 7.37 0.56 13.71 5.86 0.07
712.85 573.271 43.46 410.37 406.81 12.97
AUC0_0, (h* M)
176.16 78.76 5.96 91.52 44.56 2.78
ELF penetration
ratio (based on
7.29-26.10 12.4-50.9
free plasma
AUC)*
ELF penetration
ratio (based on
0.60-1.97 0.52-1.51
total plasma
AUC)
*calculated based on Hu PPB % free; ELF penetration ratio calculated at 95%
confidence
interval
103741 Table 11. Pharmacokinetic parameters of 1,4-azaindoles in infected
BALB/c mice
following multiple oral doses (data are mean S.D., n=3 unless otherwise
stated).
Compound 3 4
Dose 50 50
100 100
(me/kg)
Mouse
C. (j1M) 17.79 5.61 41.28 10.28 38.91 15.16 72.62 8.31
infected
AUCo_. 106.08 332.15 199.62 529.03
POPK from
( M*h) 3.04 152.51 101.21 85.07
acute
T. (h) 4.33 1.15 3.83 2.84 4.50 + 2.78 5.67 2.31
efficacy
1112(h) 2.38 0.19 3.02 1 1.99 2.23 0.35 2.02 0.09
(+ABT)
% fT>MIC 23 41 39 50
% T>MIC 58 66 75 83
Mouse Dose 30 30
1001 100b
infected (mg/kg)
POPK from C.. (j.1M) 32.78 71.24 17.36 1.76 43.17
84
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
chronic 14.41
efficacy AUCc,.. 251.94 226.98 +
695.76 772.33
(+ABT) ( M*h) 64.01 28.87
Tmax (h) 2.00 0.87 3 5.67 2.31 5
Tu2 (h) 4.46 0.16 4.4 3.85 0.54 4.21
%f T>MIC 29 63 29 100
% T>MIC 92 100 100 100
a compound 3 (n=2); b compound 4 (n=1)
[0375] Table 12. Pharmacokinetic parameters (Mean SD) of 1,4-azaindole
compounds
following multiple oral dose (100 mg/kg) administration in Mtb infected male
Wistar rats.
Compound 8 17 30 34
Dose (mg/kg) 100
Cmax (11M) 11.9 3.0 46.1 7.7 3.4 2.0 31.5 3.0
Tmax (h) 1.5 0.6 5.3 1.2 3.3 2.3 2.0 0.0
AliCo_. ( M*h) 98.4 31.9 986.4 274.3 23.3 11.7 166.1 = 43.9
T112 (h) 7.2 1.3 3.8 0.6 3.3 1.1 4.6 0.0
CA 02918487 2016-01-15
WO 2015/009525 PCT/US2014/046100
REFERENCES
Jayaram et. al. Antimicrob. Agents Chemother. 47, 2118-2124 (2003).
Balganesh et. al. Antimicrob. Agents Chemother. 54, 5167-5172 (2010).
National Committee for Clinical Laboratory Standards. 2009. Volume 29, Number
2.
National Committee for Clinical Laboratory Standards, Wayne, PA.
Wayne and Hayes Infect. Immun. 64, 2062-2069 (1996).
Eakin et. al, Antimicrob. Agents Chemother 56, 1240-1246 (2012).
Reddy et. Eur. I Pharm. Sci. 47, 444-450 (2012).
Louie et. al. Antimicrob. Agents Chemother 53, 3325-30 (2009).
Rennard, S.I. et. al. J. App!. PhysioL 60, 532-538 (1986).
Solapure et. al, Antimicrob. Agents Chemother. 57,2506-2510 (2013).
Marry et. al, Antimicrob. Agents Chemother. 55, 1237-1247 (2011).
Schroeder et. al, J. BiomoL Screen. 8, 50-64 (2003).
Marry et al. Antimicrob. Agents Chemother. 55, 1237-1247 (2011).
Kumar et aL Tuberculosis. (2014).
86
Table 2
ze. )
MI C ( M)
H37Rv ATCC Msm Msm Eco ARC
Hin ARC Hin ARC Pae ARC Pae ARC Kpn ARC Sau
ARC Spn ARC Spn ARC Cal ARC
Compounds Eco toIC
et- 27294 ATCC mc2155 523
446 158 545 546 1865 517 548 546 527
n.)
0
cji1/41 Compound 3 2 6.25 2 >200 >200
>200 >200 >200 >200 >200 >200 >200 >200 >200
Compound 4 <0.39 <0.39 <0.39 >200 >200 >200
>200 >200 >200 - >200 >200 >200 >300 >200
Compound 8 <0.39 <0.39 <0.39 >200 >200
>200 >200 >200 >200 >200 >200 >200 >200 >200
Compound 17 2 3.125 2 >200 >200 >200
>200 >200 >200 >200 >200 >200 >200 >200
Msm ATCC Mycobacterium smegmatis 607
Msm mc2155 Mycobacterium smeg ma tis ma 155
Eco ARC523 Escherichia colt ARC523
Eco toIC Escherichia coil tolCstr ain (to/C mutant)
op
HinARC 446 Haemophilus influenza e ARC446
HinARC 158 Haemophilus inlluenzae ARC1S8 (arra mutant)
PaeARC 545 Pseudomontz aeruginosaARC545
PaeARC 546 Pseudomonasoeruginosa ARC546(MexA8OXY mutant)
KpnARC 1865 Klebstella pneumoricre ARC1865
SauARC 517 Staphylococcus CROCUS ARC517
SpnARC 548 Streptococcus pneurnon i.e ARC548
SpnARC546 Streptococcus pn eumon toe ARC548
CalARC 527 Candida a I bicons ARC527
o
0, Table 3
e
p3
m
.ci
C
6
a Af. tuberculosis Sensitive
strains I StrR I InhR I FtifR
x MIC(04)
I H371tv ATCC Beijing DKU- DKU- DKU- DKU- ATCC ATCC ATCC
Rif
Compound
27294 Erdman (E-47/94) Harlingen CDC1551 TAI14149 SA161 5A310
6570
O 76
97A 211 220 35811 35820 35822 Res
o.
n.) Compound 3 3.125 3.125 1.56 1.56 1.56
1.56 3.125 1.56 3.125 3.125 3.125 1.56
1.56 3.125 3.125 3.125 3.125
o Compound 4 0.39 0.39 0.78
0.39 0.78 0.78 0.78 0.39 0.78 0.39
0.39 0.78 0.78 0.39 0.39 0.39 0.78
.1c%P.: - .
Compound 8 0.78 0.39 0.39 0.78 0.39
1.56 0.78 0.39 0.78 0.78 0.39 0.78 0.78 0.39
0.39 0.39 0.39
4
0 Compound 17 1.56 1.56 1.56 0.78 0.78
1.56 0.78 0.78 1.56 0.78 0.78 0.39 0.78 0.78
0.78 0.78 0.78
n.) .
Streptomycin 0.34 0.34 0.17
0.17 0.34 0.34 0.34 0.34 0.17 0.17 0.17 0.68 0.34 >5 >5 0.17 0.17
Isonlazid , 0.24 0.24 0.24 0.24 0.12
0.12 0.12 _ 0.12 ' 0.12 0.12 0.12 , 0.12 0.12 '
0.12 0.12 >29 0.12
Rlfampidn 0.015
0.007 0.003 . 0.003 0.003 . 0.003 0.003
0.003 0.003 0.003 0.003 _ 0.003 0.003 0.003 , 0.003 0.003 _ >4.8
StrR- streptomycin resistance
InhR- isoniazid resistance
RifR - rifarnpicin resistance
00
00