Note: Descriptions are shown in the official language in which they were submitted.
CHEMICAL INDUCERS OF FETAL HEMOGLOBIN
Background
1. Field of the Invention
The present invention is directed to methods and compositions for the
treatment of the
beta-globin blood disorders sickle cell disease and beta-thalassemia and, in
particular,
compositions and methods comprising ambroxol, benserazide, desloratadine,
resveratrol, NSC-
95397, and/or MS-275 (entinostat).
2. Background of the Invention
Inherited disorders of production of the beta-chain of adult hemoglobin A
(beta-
thalassemia) or mutations affecting the structure of the beta-globin chain
(sickle cell disease) are
the most common genetic diseases in the world, afflicting millions of
individuals worldwide, and
are designated by WHO as a global health burden. Disorders of hemoglobin
synthesis include
deficiencies of globin synthesis such as thalassemia syndromes and structural
abnormalities of
globin such as sickle cell syndromes and syndromes associated with unstable
hemoglobins.
The beta hemoglobin disorders are characterized by hemolytic anemia, shortened
red
blood cell lifespan, which reduces oxygen transport throughout the body and
multi-organ
damage. Pharmacological augmentation of fetal hemoglobin (beta-globin chain)
production, to
replace the defective or missing beta-globin chains, is a promising
therapeutic approach.
Fetal globin (also known as gamma globin) normally combines with alpha globin
chains
prenatally to form fetal hemoglobin (HbF). Fetal globin is replaced by beta
globin after birth,
which then combines with alpha globin to form adult hemoglobin A. Fetal globin
performs the
same function as beta globin, and can combine with the alpha chains to
generate a healthy form
of hemoglobin thereby reducing high concentrations of unmatched alpha globin
chains.
30
1
CA 2922013 2017-10-03
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
The various types of beta-thalassemias are syndromes resulting from mutations
which
produce a deficiency of globin chains. In beta-thalassemia. the unmatched
alpha globin chains
aggregate inside red blood cells (RBCs) and their progenitors, causing the
premature destruction
of RBCs and RBC progenitors, which results in anemia, transfusion-dependence,
iron overload,
organ failure, and early death.
In sickle cell disease (SCD), one amino acid substitution in the beta globin
chain results
in the generation of sickling hemoglobin (HbS), which allows polymerization
with repeated
cycles of deoxygenation. Polymerization results in "sickling" of RBCs. The
sickled RBCs
undergo hemolysis, while adhesive sickled RBCs occlude the microcirculation,
provoking
widespread tissue ischemia and organ infarction. The natural history of SCD is
marked by
painful crises and acute chest syndrome, and eventual potentially life-
threatening sequelae,
including renal insufficiency, retinitis, osteonecrosis, osteomyelitis,
aplastic crises, functional
asplenism, stroke, priapism, and severe pulmonary hypertension.
Many efforts to stimulate HbF production have accordingly been undertaken, but
pharmacologic reactivation of high-level HbF expression with non-toxic and
tolerable
therapeutic agents that are orally available (for worldwide therapeutic
application) has been an
elusive therapeutic goal for many years. In sickle cell disease, average HbF
levels in adult
patients are 5-7%; but levels of HbF greater than 15-20% in 70% or more of red
blood cells
expressing HbF (F-cells) are typically required to ameliorate most of the
clinical complications.
One HbF stimulant therapeutic, hydroxyurea (HU), is FDA-approved for treatment
of sickle cell
disease and benefits approximately 40% of subjects, with most benefit
occurring those who
attain absolute HbF levels greater than 0.5 g/dl or 20%. HbF levels achieved
are often not
sufficiently high to completely ameliorate all complications. Additional
therapeutics, especially
non-cytotoxic agents which can be used in combinations with HU, could provide
additional
.. benefit. There are no therapeutic agents approved for the beta-thalassemia
syndromes.
Fetal hemoglobin (HbF, gamma2a1pha2) produced by the gamma-globin gene (HBG),
is the
major human hemoglobin in utero, and is replaced by beta-globin expression in
infancy, even
when the beta-globin genes are mutated. Fetal globin when present in even
small amounts
prevents sickling. In beta-thalassemia, increasing synthesis of fetal globin
chains reduces the
globin chain imbalance, caused by excess of alpha globin and deficiency of
beta globin, and
consequent anemia. Fetal globin is the major modulator of disease severity and
a generally-
2
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
accepted target for drugs to ameliorate these diseases. A major unmet medical
need exists for
high-potency HbF inducers, particularly for patients with lower baseline
levels of HbF, and for
the many who do not have a robust response to HU alone. A combination
chemotherapy
approach is needed to increase response rates in sickle disease. Baseline HbF
expression is
influenced by genetic modifiers. The 3 most influential quantitative trait
loci (QTLs) are
responsible for 50% of variability in HbF levels. Polymorphisms in these QTLs
are associated
with higher baseline HbF levels. One QTL, BCL-11A, is a repressor of fetal
globin synthesis.
The inventions have been identified to suppress the expression of this
repressor at the mRNA
and protein levels. Several of these drug candidates act, in part, through
suppression of
repressors of fetal globin (Bcl-11A, LSD-1, HDAC-3, HDAC-2, and KLF-1).
The thalassemia syndromes are a heterogeneous group of disorders all
characterized by a
lack of or a decreased synthesis of the globin chains of HbA. Deficiencies of
alpha-globin
expression are referred to as alpha-thalassemias and deficiencies of beta-
globin. beta-
thalassemias. The hemolytic consequences of deficient globin chain synthesis
result from
.. decreased synthesis of one chain and also an excess of the complementary
chain. Free chains
tend to aggregate into insoluble inclusions within erythrocytes causing
premature destruction of
maturing erythrocytes and their precursors, ineffective erythropoiesis, and
the hemolysis of
mature red blood cells. The underlying defects of hemoglobin synthesis have
been elucidated
over the years and largely reside in the nucleic acid sequences which express
or control the
expression of alpha or beta-globin protein.
A small number of therapeutic agents of different chemical classes can induce
HbF
experimentally, with only a few are orally-active or currently in clinical
testing. Three general
classes of therapeutic agents have been shown to induce HbF significantly in
subjects with sickle
cell disease and beta-thalassemia, include: cytotoxic chemotherapeutic agents
(such as
Hydroxyurea (HU), 5-azacytidine, and decitabine), erythropoietin (EPO)
preparations, and
short chain fatty acids (SCFAs) and derivatives (SCFADs), which include some
HDAC
inhibitors. Additionally, there are a variety of small molecules have been
shown to effect
hemoglobin or fetal globin expression. Early experiments demonstrated that
acetate
(CH3COOH), propionate (CH3CH2COOH), butyrate (CH3CH2CH2COOH) and isobutyrate
(CH3CH(CH3)COOH) all induced hemoglobin synthesis in cultured Friend leukemia
cells (E.
Takahashi et al., Gann 66:577-80, 1977).
3
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
Additional studies showed that polar compounds, such as acid amides, and fatty
acids
could stimulate the expression of both fetal and adult globin genes in murine
erythroleukemia
cells (U. Nudel et al., Proc. Natl. Acad. Sci. USA 74:1100-4, 1977).
Hydroxyurea
(H2NCONHOH), another relatively small molecule, was found to stimulate globin
expression
(N.L. Letvin et al., N. Engl. J. Med. 310:869-73, 1984). Stimulation, however,
did not appear to
be very specific to fetal globin (S. Charache et al., Blood 69:109-16, 1987).
Hydroxyurea (HU) is
also a well-known carcinogen making its widespread and long term use as a
pharmaceutical
impractical. One of the major breakthroughs in the treatment of
hemoglobinopathies was made
when it was discovered that butyric acid (butanoic acid; CH3CH2CH2COOH)
accurately and
specifically stimulated transcription of the human fetal (gamma) globin gene
(G.A. Partington et
al., EMBO J. 3:2787-92, 1984). Some of these have shown proof-of-principle,
but, except for
HU, has required parenteral administration or large doses, which were not
suitable for broad
application.
While three SCFA agents have been reported to induce gamma-globin expression
and to
increase hemoglobin levels in subjects with beta-thalassemia, rendering some
beta-thalassemia
subjects transfusion- independent, these prior generations of SCFAs, including
arginine butyrate
(AB) and sodium phenylbutyrate (SPB), have limited utility as a therapeutic
agent in vivo, as
they are either rapidly metabolized, required intravenous (IV) infusions, or
required large doses
which were difficult for subjects to tolerate lone-term. Furthermore, these
first generation SCFAs
are also known to inhibit erythroid cell proliferation, and therefore require
titration and
intermittent dosing, complicating their use in conditions of anemia where
compensatory
erythroid cell proliferation is desirable. There, thus remains an unmet
clinical need for a
therapeutic agent that induces gamma-globin gene expression and does not
inhibit erythroid cell
proliferation (i.e., is not cytotoxic), and which is more applicable for wide
application in these
genetic diseases.
Summary of the Invention
The present invention relates to agents that function as fetal globin-inducing
agents, for
the treatment of beta-globin disorders, such as sickle cell anemia and beta-
thalassemias, with
higher efficacy than prior generation agents in predictive in vivo models and
which do not have
the limitations of prior generation candidates for long term use.
Lead candidates were investigated in in vivo non-primate models. While some
identified
4
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
compounds are already FDA-approved for other medical indications, their safety
profiles are
benign and they are not known to increase fetal-globin expression.
Accordingly, subjects in
need of increased fetal-globin expression can be selected and administered the
compounds
identified herein for treatment of beta-globin diseases, such as Sickle Cell
Disease and the
diverse variations of beta-thalassemia syndromes.
The disclosure herein provides novel methods and compositions for increasing
the amount
of fetal hemoglobin in a subject with a blood disorder, including beta-
thalassemias and sickle
cell disease. Fetal hemoglobin (HbF: a1pha2, gamma2) is an endogenous type of
hemoglobin
which is present in all humans, but is normally suppressed in infancy to
levels below 2%.
Decades of biochemical, clinical, and epidemiologic research have shown that
any increase in
HbF and F-cell levels reduce the severity of sickle cell disease, or alleviate
the anemia of beta-
thalassemia. It is well-established that fetal globin (gamma-globin) chains
interfere with the
polymerization of sickle hemoglobin, preventing many pathologic consequences
of sickling, and
also that adequate levels of fetal hemoglobin (also referred to as hemoglobin
F or HbF), correlate
with mild or benign courses in sickle cell disease (SCD).
Accordingly, pharmacologic augmentation of fetal hemoglobin (HbF, gamma-
globin)
production replaces diminished beta-globin chains in the beta-thalassemias and
inhibits HbS
polymerization in sickle cell disease. Despite long-term efforts, regulatory
approval has been
obtained for only one chemotherapeutic agent. Phannacologic reactivation of
high-level HbF
expression with non-cytotoxic, tolerable therapeutics is still an unmet
medical need for this
global health burden. To investigate potential therapeutic libraries for
unrecognized HbF
inducers, a high-throughput screening (HTS) program was developed to
interrogate diverse
chemical libraries, including a library of FDA-approved and clinical stage
drugs. Ushila this
assay, unexpected new and highly potent HbF-inducing drugs were identified,
some of which are
already in clinical use for other medical indications and have established
safety profiles.
The invention overcomes the problems and disadvantages associated with current
strategies and designs, and provides new compositions, methods, and aids for
the treatment and
prevention of blood disorders. In particular, the present invention generally
relates to increasing
the percentage of fetal hemoglobin (HbF or gamma-globin) in the blood of a
subject without
decreasing proliferation of cells, the method comprising administering to the
subject a
composition comprising at least one of, or any combination of HbF-inducing
drugs, which
5
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
include, ambroxol, benserazide, desloratadine, resveratrol, NSC-95397, or MS-
275.
In some embodiments, the composition comprises benserazide or desloratadine or
MS-
275, or any pharmaceutically acceptable salt, polymorph or ester thereof.
One embodiment of the invention is directed to compositions that comprise one
or more
compounds which stimulate the proliferation of hemoglobin-producing cells, the
expression of
hemoglobin or the expression of embryonic or fetal globin in mammalian cells.
Chemical
compounds, or HbF-inducing agents include, ambroxol, benserazide,
desloratadine, resveratrol,
NSC-95397. or MS-275.
Another embodiment of the invention is directed to compositions comprising at
least one
or any combination of HbF-inducing agents selected from the group of ambroxol,
benserazide,
desloratadine, resveratrol, NSC-95397, or MS-275, in particular benserazide,
desloratadine or
MS-275 that stimulate the proliferation of hemoglobin producing and other
types of cells, the
expression of hemoglobin or the expression of embryonic or fetal globin in
mammalian cells, but
do not decrease or otherwise adversely affect cell viability. Such HbF-
inducing agents include
benserazide, desloratadine or MS-275. Cell viability may be assayed by DNA
fragmentation
assays or cell division assays, or by measuring the amount of nucleic acid or
protein synthesis
which occurred in treated cells as compared to untreated cells. Cells tested
may be normal
healthy cells, subject cells to be treated or cells in tissue culture.
Another embodiment of the invention is directed to methods for the treatment
of blood
disorders. Compositions containing an effective amount of one or more agents
selected from, at
least one or any combination of HbF-inducing agents selected from the group
including,
ambroxol, benserazide, desloratadine, resveratrol, NSC-95397, or MS-275, in
particular
benserazide, desloratadine or MS-275 which stimulate the proliferation of
hemoglobin producing
cells or the expression of embryonic or fetal globin from cells are
administered to patients.
Patients may be any mammal such as a human. Administration may be by
parenteral or
nonparenteral means, but is preferably oral or intravenous. Treatment may be
for short periods of
time, e.g., pulsed or continuous throughout the lifetime of the patient.
Another embodiment of the invention is directed to methods for the treatment
of blood
disorders comprising the administration of compositions containing
therapeutically effective
amounts of an HbF-inducing agent selected from at least one or any combination
of ambroxol,
benserazide, desloratadine, resveratrol, NSC-95397, or MS-275, in particular
benserazide.
6
desloratadine or MS-275 which increases the proportion or number of
reticulocytes that express
embryonic or fetal globin and the amount of embryonic or fetal globin
expressed per cell.
Another embodiment of the invention is directed to methods for regulating the
expression
of a globin gene such as an embryonic or fetal globin gene or an at least
partially functional
pseudo-globin gene in mammalian cells. In some embodiments, treated cells,
e.g., ex vivo, or
products expressed from these cells, can be harvested and introduced or
reintroduced to a subject
to treat or prevent a blood disorder.
Another embodiment of the invention is directed to methods for regulating the
proliferation of hemoglobin expressing cells. Cells in culture or in patients
are exposed to
compositions of HbF-inducing agents selected from the group including, at
least one or any
combination of ambroxol, benserazide, desloratadine, resveratrol, NSC-95397,
or MS-275, in
particular benserazide, desloratadine or MS-275 and induced to proliferate..
Another
embodiment of the invention is directed to methods for the prevention of the
clinical
manifestations of the hemoglobinopathy or beta thalassemic disorders.
Compositions
containing an effective amount of agents as disclosed herein, e.g., at least
one or any
combination of ambroxol, benserazide, desloratadine, resveratrol, NSC-95397,
or MS-275, in
particular benserazide, desloratadine or MS-275 which stimulate the
proliferation of hemoglobin
producing cells or the expression of embryonic or fetal globin are
administered to patients
suspected of having a blood disorder. The subject may be any mammal such as a
human and is
preferably an adolescent, child, or infant. Administration may be by any route
including
parenteral and nonparenteral routes, but is preferably oral or intravenous.
Treatment may be for
short periods of time or continuous throughout the lifetime of the patient.
Other embodiments and advantages of the invention are set forth in part in the
description, which follows, and in part, may be obvious from this description,
or may be learned
from the practice of the invention.
In accordance with an aspect of the present invention, there is provided
benserazide for
use as a fetal hemoglobin inducing agent in the treatment of a blood disorder
wherein the agent
increases the total hemoglobin or the hematocrit value of the blood.
In accordance with another aspect of the present invention, the blood disorder
is one or
more of a sickle-cell syndrome, sickle cell disease, HbSS, HbSC, HbS/beta-
thalassemia, HbS-
Arab, a beta-thalassemia syndrome, beta-zero-thalassemia, beta plus
thalassemia, thalassemia
7
CA 2922013 2017-10-12
major, thalassemia intermedia, HbE/beta-thalassemia, delta/beta-thalassemia,
or a globin gene
mutation disease.
In accordance with another aspect of the present invention, the agent is
formulated to
increase the percentage of fetal hemoglobin in the blood of a patient or
proportion of fetal
hemoglobin in relation to non-fetal hemoglobin.
In accordance with another aspect of the present invention, the formulation
comprises an
amount of agent that, 15 minutes or more after administration to the patient,
produces a blood
concentration of at least 1 nM.
In accordance with another aspect of the present invention, the formulation
comprises an
amount of agent that, 15 minutes or more after administration to the patient,
produces a blood
concentration in the patient of at least 0.2 p.M.
In accordance with another aspect of the present invention, the amount of
fetal
hemoglobin in the blood increases by two percent or more within 28 days.
In accordance with another aspect of the present invention, the agent is a
salt, an acid, an
ester, an amine, an aldehyde, a ketone, an amide, an alcohol, a carbonyl, an
amine, an alkene, or
a polymorph.
In accordance with another aspect of the present invention, the agent at
ambient
temperature in use, is a liquid, a suspension, a powder or a gel.
In accordance with another aspect of the present invention, the agent is
formulated for
.. administration to the patient orally, by injection, by suppository or by a
transdermal patch.
In accordance with another aspect of the present invention, the agent is
formulated for
administration as a single dose, continuously, in pulsed doses or by
intermittent or non-
continuous doses.
In accordance with another aspect of the present invention, the agent further
comprises a
second fetal hemoglobin inducing agent or an erythropoietic agent.
In accordance with another aspect of the present invention, there is provided
a
pharmaceutical composition comprising benserazide and a pharmaceutically
acceptable carrier
formulated for oral, injectable, transdermal or rectal administration to a
patient for treatment of a
blood disorder.
In accordance with another aspect of the present invention, the composition is
used at a
therapeutically effective dose for increasing the percentage of fetal
hemoglobin or proportion of
7a
CA 2922013 2017-10-12
fetal hemoglobin in relation to non-fetal hemoglobin in the blood of a patient
for treating or
preventing a blood disorder.
In accordance with another aspect of the present invention, the blood disorder
is one or
more of a sickle-cell syndrome, sickle cell disease, MSS, HbSC, HbS/beta-
thalassemia, HbS-
.. OArab, a beta-thalassemia syndrome, beta-zero-thalassemia, beta plus
thalassemia, thalassemia
major, thalassemia intermedia, HbE/beta-thalassemia, delta/beta-thalassemia,
or a globin gene
mutation disease.
In accordance with another aspect of the present invention, thc composition is
formulated
for administration as a liquid, a suspension, a powder or a gel.
In accordance with another aspect of the present invention, the composition is
formulated
for administration as a single dose, continuously, in pulsed doses or by
intermittent or non-
continuous doses.
In accordance with another aspect of the present invention, there is provided
a benzamide
histone deacetylase inhibitor for use as a fetal hemoglobin inducing agent in
the treatment of a
blood disorder wherein said agent increases the total hemoglobin or hematocrit
value of the
blood.
In accordance with another aspect of the present invention, the benzamide
histone
deacetylase inhibitor is entinostat (MS-275).
In accordance with another aspect of the present invention, the blood disorder
is one or
more of a sickle-cell syndrome, sickle cell disease, HbSS, HbSC, HbS/beta-
thalassemia, HbS-
OArab, a beta-thalassemia syndrome, beta-zero-thalassemia, beta plus
thalassemia, thalassemia
major, thalassemia intermedia, HbE/beta-thalassemia, delta/beta-thalassemia or
a globin gene
mutation disease.
In accordance with another aspect of the present invention, the agent is
formulated to
increase the percentage of fetal hemoglobin in the blood of a patient or
proportion of fetal
hemoglobin in relation to non-fetal hemoglobin.
In accordance with another aspect of the present invention, the formulation
comprises an
amount of agent that, 15 minutes or more after administration to the patient,
produces a blood
concentration of at least 1 nM.
In accordance with another aspect of the present invention, the formulation
comprises an
amount of agent that, 15 minutes or more after administration to the patient,
produces a blood
7b
CA 2922013 2017-10-12
concentration in the patient of at least 0.2 M.
In accordance with another aspect of the present invention, the amount of
fetal
hemoglobin in the blood increases by two percent or more within 28 days.
In accordance with another aspect of the present invention, the agent is a
salt, an acid, an
ester, an amine, an aldehyde, a ketone, an amide, an alcohol, a carbonyl, an
amine, an alkene, or
a polymorph.
In accordance with another aspect of the present invention, the agent at
ambient
temperature in use, is a liquid, a suspension, a powder or a gel.
In accordance with another aspect of the present invention, the agent is
formulated for
administration to a patient orally, by injection, by suppository or by a
transdermal patch.
In accordance with another aspect of the present invention, the agent is
formulated for
administration as a single dose, continuously, in pulsed doses or by
intermittent or non-
continuous doses.
In accordance with another aspect of the present invention, the agent further
comprises a
.. second fetal hemoglobin inducing agent or an crythropoietic agent.
In accordance with another aspect of the present invention, there is provided
a
pharmaceutical composition comprising a benzamide histonc deacetylase
inhibitor and a
pharmaceutically acceptable carrier formulated for oral, injectable,
transdermal or rectal
administration to a patient for treatment of a blood disorder.
In accordance with another aspect of the present invention, the benzamidc
histone
deacetylase inhibitor of the pharmaceutical composition is entinostat (MS-
275).
In accordance with another aspect of the present invention, the composition is
used in a
therapeutically effective dose for increasing the percentage of fetal
hemoglobin or proportion of
fetal hemoglobin in relation to non-fetal hemoglobin in the blood of a patient
for treating or
preventing a blood disorder.
In accordance with another aspect of the present invention, the blood disorder
is one or
more of a sickle-cell syndrome, sickle cell disease, HbSS, HbSC, HbS/beta-
thalassemia, HbS-
OArab, a beta-thalassemia syndrome, beta-zero-thalassemia, beta plus
thalassemia, thalassemia
major, thalassemia intermedia, HbE/beta-thalassemia, delta/beta-thalassemia,
or a globin gene
mutation disease.
In accordance with another aspect of the present invention, the composition is
formulated
7c
CA 2922013 2017-10-12
for administration as a liquid, a suspension, a powder or a gel.
In accordance with another aspect of the present invention, the composition is
formulated
for administration as a single dose, continuously, in pulsed doses or by
intermittent or non-
continuous doses.
In accordance with another aspect of the present invention, there is provided
a
pharmaceutical composition comprising one or more of ambroxol, desloratadine,
resveratrol or
NSC-95397, and a pharmaceutically acceptable carrier formulated for oral,
injectable,
transdermal or rectal administration to a patient for treatment of a blood
disorder.
In accordance with another aspect of the present invention, the blood disorder
is one or
more of a sickle-cell syndrome, sickle cell disease, HbSS, HbSC, HbS/beta-
thalassemia, HbS-
OArab, a beta-thalassemia syndrome, beta-zero-thalassemia, beta plus
thalassemia, thalassemia
major, thalassemia intermedia, HbE/beta-thalassemia, delta/beta-thalassemia or
a globin gene
mutation disease.
In accordance with another aspect of the present invention, the pharmaceutical
composition is used in a therapeutically effective dose for inducing fetal
hemoglobin in a patient
to treat or prevent a blood disorder.
Description of the Drawings
Figure 1A is a schematic showing an illustration of the high throughput
screening assay (HTS)
with the cell construct comprising a locus control region (LCR) linked to the
gamma-globin gene
promoter and enhanced GFP (EGFP).
Figure 1B depicts a small panel of approved therapeutics were found to induce
7-globin
expression from the library of compounds and US and EMA approved medicinal
products tested.
7d
CA 2922013 2017-10-12
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
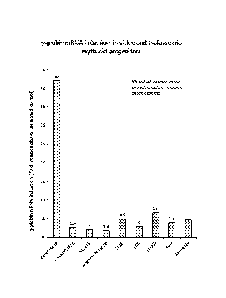
Figure 2 shows a table of the fold increase fetal globin mRNA in erythroid
progenitors in
response to benserazide and MS-275 and other compounds identified in the high-
throughput screen.
Figure 3 shows gamma-globin mRNA expression in the blood of baboons
administered fetal
hemoglobin inducing agent benserazide. Benserazide was compared to 2 other
candidates (i)
.. desloratadine (DLT), which was administered orally (0.5 mg/kg/dose), three
times a week over two
weeks, and (ii) MS-275, which was administered orally three times a week for
two weeks at 0.2
mg/kg/dose. Doses of Benserazide are shown in the bars above the graph, with
Benserazide administered
at 1 mg/kg (Open squares) and 2 mg/kg (dark squares). Benserazide resulted in
up to 33-fold induction of
gamma-globin mRNA in vivo (Figure 3A) and an increase in total hemoglobin (Hb)
(by 1.5g/dL)
(Figure 3B).
Figure 4 shows the mean induction of fetal globin with Benserazide (BEN) and
Desloratidine
(DLT) as compared to other inducers in the Anaemic Baboon model. Benserazide
induced fetal
globin mRNA by a mean of 27-fold; Desloratidine (DLT) induced a 11-fold
increase above
baseline.
.. Figure 5 shows the mean increase of induction of F-Reticulocytes during and
after
administration of Benserazide (BEN) in the anemic baboon model.
Figure 6 shows the effect of benserazide compared to a known Hb inducer
(Hydoxyurea (HU) in
a transgenic mice model comprising the entire human non-alpha (gamma delta
beta-globin) gene
locus. Benserazide induced a mean increase in total hemoglobin (Hb) at 5 weeks
(Figure 6A)
and induced an absolute 13% mean increase in the number of F-cells (from 0.1%
to 9% (mice
#1), 0.4% to 18% (mice #2), and 0.13% to 12% (mice #3); and 10 to 33-fold
increase in mean
fluorescence intensity (MFI) (Figure 6B).
Figure 7. The structures of the Compounds named in this application.
Figure 8. Various derivative forms of MS-275 as structures I, II and III.
Figure 9 depicts a comparison of the magnitude of fetal globin mRNA induction
over baseline
levels in anemic baboons by new drugs compared to hydroxyurea (HU) (Figure
9A), and
proportions of new red blood cells with fetal globin protein, F-reticulocytes,
(Figure 9B).
Figures 10A-F depict Benserazide reduction of binding of two repressors, LSD1
and HDAC3, to
the fetal globin gene promoter in erythroid progenitors from sources as
indicated.
Figure 11 depicts the suppression of repressor proteins BCL11A and KLF1 in
different erythroid
progenitors treated with new drugs as compared with untreated control levels
by Western blot
analysis.
8
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
Description of the Invention
The present invention is directed to compounds, compositions and methods that
increase
gamma-globin expression in vivo, and in particular, compounds, compositions
and methods for
the treatment of gamma-globin disorders such as, but not limited to Sickle
Cell anemia and beta-
thalassemia. In particular, the present invention generally relates to
increasing the percentage of
fetal hemoglobin (HbF or gamma-globin) in the blood of a subject, the method
comprising
administering to the subject a composition comprising at least one of, or any
combination of
ambroxol, benserazide, desloratadine, resveratrol, NSC-95397, or MS-275. In
some
embodiments, the composition comprises benserazide or desloratadine or MS-275,
or any
pharmaceutically acceptable salt, polymorph or ester thereof.
For convenience, certain terms employed herein, in the specification, examples
and
appended claims are collected here. Unless stated otherwise, or implicit from
context, the
following terms and phrases include the meanings provided below. Unless
explicitly stated
otherwise, or apparent from context, the terms and phrases below do not
exclude the meaning
that the term or phrase has acquired in the art to which it pertains. The
definitions are provided to
aid in describing particular embodiments, and are not intended to limit the
claimed invention,
because the scope of the invention is limited only by the claims. Unless
otherwise defined, all
technical and scientific terms used herein have the same meaning as commonly
understood by
one of ordinary skill in the art to which this invention belongs.
The term "pharmaceutically acceptable excipient", as used herein, refers to
carriers and
vehicles that are compatible with the active ingredient (for example, a
compound of the
invention) of a pharmaceutical composition of the invention (and preferably
capable of
stabilizing it) and not deleterious to the subject to be treated. For example,
solubilizing agents
that form specific, more soluble complexes with the compounds of the invention
can be utilized
as pharmaceutical excipients for delivery of the compounds. Suitable carriers
and vehicles are
known to those of extraordinary skill in the art. The term "excipient" as used
herein will
encompass all such carriers, adjuvants, diluents, solvents, or other inactive
additives. Suitable
pharmaceutically acceptable excipients include, but are not limited to, water,
salt solutions,
alcohol, vegetable oils, polyethylene glycols, gelatin, lactose, amylose,
magnesium stearate, talc,
silicic acid, viscous paraffin, perfume oil, fatty acid monoglycerides and
diglycerides,
petroethral fatty acid esters, hydroxymethyl-cellulose, polyvinylpyrrolidone.
etc. The
9
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
pharmaceutical compositions of the invention can also be sterilized and, if
desired, mixed with
auxiliary agents, e.g., lubricants, preservatives, stabilizers, wetting
agents, emulsifiers, salts for
influencing osmotic pressure, buffers, colorings, flavorings and/or aromatic
substances and the
like, which do not deleteriously react with the active compounds of the
invention.
Thus, as used herein, the term "pharmaceutically acceptable salt." is a salt
formed from
an acid and a basic group of a compound of the invention. Illustrative salts
include, but are not
limited, to sulfate, citrate, acetate, oxalate, chloride, bromide, iodide,
nitrate, bisulfate,
phosphate, acid phosphate, isonicotinate, lactate, salicylate, acid citrate,
tartrate, oleate, tannate,
pantothenate, bitartrate, ascorbate, succinate, maleate, gentisinate,
fumarate, gluconate,
glucaronate, saccharate, formate, benzoate, glutamate, methanesulfonate,
ethanesulfonate,
benzenesulfonate, p-toluenesulfonate, and pamoate salts.
The term "pharmaceutically acceptable salt" also refers to a salt prepared
from a
compound of the invention having an acidic functional group, such as a
carboxylic acid
functional group, and a pharmaceutically acceptable inorganic or organic base.
Suitable bases
include, but are not limited to, hydroxides of alkali metals such as sodium,
potassium, and
lithium; hydroxides of alkaline earth metal such as calcium and magnesium;
hydroxides of other
metals, such as aluminum and zinc; ammonia, and organic amines, such as
unsubstituted or
hydroxy-substituted mono-, di-, or trialkylamines; dicyclohexylamine; tributyl
amine; pyridine;
N-methyl, N-ethylamine; diethylamine; triethylamine; mono-, his-, or tris-(2-
hydroxy-lower
alkyl amines), such as mono-, his-, or tris-(2-hydroxyethyl)amine, 2-hydroxy-
tert-butylamine, or
tris-(hydroxymethyl)methylamine, N,N,-di-lower alkyl-N-(hydroxy lower alkyl)-
amines, such
as N,N- dimethyl-N-(2-hydroxyethyl)amine, or tri-(2-hydroxyethyl)amine; N-
methyl-D-
glucamine; and amino acids such as arginine, lysine, and the like. Other
pharmaceutically
acceptable salts are described in the Handbook of Pharmaceutical Salts.
Properties, Selection,
and Use (P. Heinrich Stahl and C. Wermuth, Eds., Verlag Helvetica Chica Acta,
Zurich,
Switzerland (2002)).
As used herein, a "prodrug" refers to compounds that can be converted via some
chemical
or physiological process (e.g., enzymatic processes and metabolic hydrolysis).
The prodrug
compound often offers advantages of solubility, or delayed release in an
organism. The term
"prodrug" is also meant to include any covalently bonded carriers, which
release the active
compound in vivo when such prodrug is administered to a subject. Prodrugs of
an active
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
compound may be prepared by modifying functional groups present in the active
compound in
such a way that the modifications are cleaved, either in routine manipulation
or in vivo, to the
parent active compound. Prodrugs include compounds wherein a hydroxy, amino or
mercapto
group is bonded to any group that, when the prodrug of the active compound is
administered to a
subject, cleaves to form a free hydroxy, free amino or free mercapto group,
respectively.
Examples of prodrugs include, but are not limited to, acetate, formate and
benzoate derivatives
of an alcohol or acetamide, formamide and benzamide derivatives of an amine
functional group
in the active compound and the like.
The term "subject" is used interchangeably herein with "patient" and refers to
a
vertebrate, preferably a mammal, more preferably a primate, still more
preferably a human.
Mammals include, without limitation, humans, primates, wild animals, rodents,
feral animals,
farm animals, sports animals, and pets. Primates include chimpanzees,
cynomologous monkeys,
spider monkeys, and macaques, e.g., Rhesus. Rodents include mice, rats,
woodchucks, ferrets,
rabbits and hamsters. Domestic and game animals include cows, horses, pigs,
deer, bison,
buffalo, feline species, e.g., domestic cat, canine species, e.g., dog, fox,
wolf, avian species, e.g.,
chicken. emu, ostrich, and fish, e.g., trout, catfish and salmon. Patient or
subject includes any
subset of the foregoing, e.g., all of the above, but excluding one or more
groups or species such
as humans, primates or rodents. In certain embodiments of the aspects
described herein, the
subject is a mammal, e.g., a primate, e.g., a human. A subject can be male or
female.
The term "therapeutically effective amount" as used herein refers to an amount
sufficient
to affect a beneficial or desired clinical result upon treatment.
Alternatively, a "therapeutically
effective amount" is an amount of a compound of this invention sufficient to
confer a therapeutic
or prophylactic effect on the treated subject a hemoglobinopathy and/or
thalassemia.
Therapeutically effective amounts will vary, as recognized by those skilled in
the art, depending
on the specific disease treated, the route of administration, the excipient
selected, and the
possibility of combination therapy. Generally, a therapeutically effective
amount can vary with
the subject's history, age, condition, sex, as well as the severity and type
of the medical condition
in the subject, and administration of other pharmaceutically active agents.
The terms "increased" ,"increase" or "enhance" or "activate" are all used
herein to
generally mean an increase by a statistically significant amount; for the
avoidance of any doubt,
the terms "increased". "increase" or "enhance" or "activate" means an increase
of at least 2% as
11
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
compared to a reference level
The term "statistically significant" or "significantly" refers to statistical
significance and
generally means a two standard deviation (2 SD) below normal, or lower,
concentration of the
marker. The term refers to statistical evidence that there is a difference. It
is defined as the
probability of making a decision to reject the null hypothesis when the null
hypothesis is actually
true. The decision is often made using the p-value.
The term "substantially" as used herein means a proportion of at least about
60%, or
preferably at least about 70% or at least about 80%, or at least about 90%, at
least about 95%, at
least about 97% or at least about 99% or more or any integer between 70% and
100%.
As used herein the term "comprising" or "comprises" is used in reference to
compositions, methods, and respective component(s) thereof, that are essential
to the invention,
yet open to the inclusion of unspecified elements, whether essential or not.
As used herein the term "consisting essentially of" refers to those elements
required for a
given embodiment. The term permits the presence of additional elements that do
not materially
affect the basic and novel or functional characteristic(s) of that embodiment
of the invention.
The term "consisting of" refers to compositions, methods, and respective
components
thereof as described herein, which are exclusive of any element not recited in
that description of
the embodiment.
As used in this specification and the appended claims, the singular forms "a,"
"an," and
"the" include plural references unless the context clearly dictates otherwise.
Thus for example,
references to "the method" includes one or more methods, and/or steps of the
type described
herein and/or which will become apparent to those persons skilled in the art
upon reading this
disclosure and so forth.
Other than in the operating examples, or where otherwise indicated, all
numbers
expressing quantities of ingredients or reaction conditions used herein should
be understood as
modified in all instances by the term "about." The term "about" when used in
connection with
percentages can mean 1%.
In this application and the claims, the use of the singular includes the
plural unless
specifically stated otherwise. In addition, use of "or" means "and/or" unless
stated otherwise.
Moreover, the use of the term "including", as well as other forms, such as
"includes" and
"included", is not limiting. Also, terms such as "element" or "component"
encompass both
12
CA 02922013 2016-02-19
WO 2015/026939
PCT/US2014/051887
elements and components comprising one unit and elements and components that
comprise more
than one unit unless specifically stated otherwise.
HbF-inducing agent
In particular, the present invention generally relates to increasing the
percentage of fetal
hemoglobin (HbF or gamma-globin) in the blood of a subject, the method
comprising
administering to the subject a composition comprising at least one of, or any
combination of
HbF-inducing agents, selected from the group comprising; ambroxol,
benserazide, desloratadine,
resveratrol, NSC-95397, or MS-275.
In some embodiments, an HbF-inducing agent is benserazide or desloratadine or
MS-275,
or any pharmaceutically acceptable salt, polymorph or ester thereof.
In certain embodiments, the amount of fetal globin in the blood of the subject
increases
with administration of an HbF-inducing agent as disclosed herein to the
subject. In some
embodiments, the number of F-cells in the blood of the subject increases with
administration of
one or more HbF-inducing agent as disclosed herein. In certain embodiments,
the number of F-
reticulocytes in the blood of the subject increases on administration of an
HbF-inducing agent as
disclosed herein. In some embodiments, the amount of total fetal hemoglobin in
the blood of the
subject increases. In certain embodiments, the amount of total hemoglobin in
the blood of the
subject increases. In some embodiments, hematocrit increases. In certain
embodiments, red
blood cell production increases.
In some embodiments, a composition comprising at least one HbF-inducing agent
as
disclosed herein is administered in an effective amount to increase the
expression of gamma-
globin in the blood by a statistically significant increase as compared to in
the absence of a
compound. In some embodiments, a composition comprising at least one HbF-
inducing agent as
disclosed herein is administered to increase the expression of gamma-globin in
the blood by a
statistically significant increase as compared to in the presence of a control
agent
such as, for example, hydroxyurea.
In some embodiments, a composition comprising at least one HbF-inducing agent
as
disclosed herein, for example, any one, or any combination of ambroxol,
benserazide,
desloratadine, resveratrol, NSC-95397, or MS-275, or in particular,
benserazide, desloratadine,
or MS-275 is administered in an effective amount to increase the level of
gamma-goblin
expression in blood by at least about 2%, as compared to either in the absence
of the
13
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
composition, or as compared to a positive control agent, such ST20.
In some embodiments, a composition comprising at least one HbF-inducing agent
as
disclosed herein, for example, any one, or any combination of ambroxol,
benserazide,
desloratadine, resveratrol, NSC-95397, or MS-275, or in particular,
benserazide, desloratadine,
or MS-275 is administered in an effective amount to increase the total
hemoglobin in the blood
above the baseline.
In some embodiments, administration of one or more HbF-inducing agent is a
pulsed
administration. In certain embodiments, a pulsed administration comprises
administering one or
more HbF-inducing agent for about 8 weeks, followed by not administering an
HbF-inducing
agent for about 4 weeks. In some embodiments, the pulsed administration
comprises
administering at least one HbF-inducing agent for about 6 weeks, followed by
not administering
an HbF-inducing agent for about 2 weeks. In certain embodiments, the pulsed
administration
comprises administering at least one HbF-inducing agent for about 4 weeks,
followed by not
administering an HbF-inducing agent for about 2 weeks. In some embodiments,
the pulsed
administration comprises administering at least one HbF-inducing agent for
about 4 days a week
or for about 2 weeks or 4 weeks, followed by not administering an HbF-inducing
agent for about
1 week or 2 weeks. In some embodiments, pulsed administration comprises pulses
of
administering at least one HbF-inducing agent for about 1 day, about 2 days,
about 3 days, about
4 days, about 5 days, about 6 days, about 7 days, about 10 days, about 2
weeks, about 3 weeks,
about 4 weeks, about 5 weeks. about 6 weeks, about 7 weeks, about 8 weeks,
about 2 months,
about 3 months, about 4 months, about 5 months, about 6 months, about 9
months, about 12
months. In certain embodiments, pulsed administration comprises intervals of
not administering
an HbF-inducing agent of about 1 day, about 2 days, about 3 days, about 4
days, about 5 days,
about 6 days, about 7 days, about 10 days, about 2 weeks, about 3 weeks, about
4 weeks, about 5
weeks, about 6 weeks, about 7 weeks, about 8 weeks, about 2 months, about 3
months, about 4
months, about 5 months, about 6 months, about 9 months, about 12 months. In
some
embodiments, administration is continuous. In certain embodiments,
administration is for the
lifetime of the subject.
In some embodiments, a subject is a mammal. In certain embodiments, a mammal
is an
animal. In some embodiments, an animal is a horse or a dog. In certain
embodiments, the
mammal is a human. In some embodiments, the human is a child. In certain
embodiments, a
14
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
human is under the age of 18. In some embodiments, a human is under the age of
10. In some
embodiments, a human is under the age of 2.
Further provided herein are methods for increasing the percentage of fetal
hemoglobin in
the blood of a subject diagnosed with a beta-thalassemia comprising
administering to the subject
a composition comprising at least one HbF-inducing agent or a pharmaceutically
acceptable salt,
or ester thereof, wherein after the administration the percentage of fetal
hemoglobin in the blood
of the subject increases by a statistically significant amount as compared to
the absence of
administration of the HbF-inducing agent, or a control agent, such as, for
example, ST20.
In some embodiments, provided herein are methods for increasing the percentage
of fetal
hemoglobin in the blood of a subject diagnosed with a beta-thalassemia
comprising
administering to the subject a composition comprising one or more HbF-inducing
agents, as the
free acid, a pharmaceutically acceptable salt, or ester thereof.
In some embodiments, the percentage of fetal hemoglobin in the blood of a
subject
increases after one week of administering as compared to baseline. In other
embodiments the
percentage of fetal hemoglobin in the blood of a subject increases after two
weeks of
administering as compared to baseline. In certain embodiments, the percentage
of fetal
hemoglobin in the blood of a subject increases after four weeks of
administering as compared to
baseline. In some embodiments, the percentage of fetal hemoglobin in the blood
of a subject
increases after one day of administering as compared to baseline. In certain
embodiments the
percentage of fetal hemoglobin in the blood of a subject increases after 3
days of administering
as compared to baseline.
In some embodiments, the methods for increasing the percentage of fetal
hemoglobin
further comprises administering to the subject at least one other therapeutic
agent with at least
one HbF-inducing agent, wherein the therapeutic agent can be selected from the
group consisting
of; hydroxyurea, decitabine, an HDAC inhibitor, sodium 2,2 dimethylbutyrate,
ST20 or any
combination thereof.
Benserazide
Benserazide (also called SERAZIDErm or Ro 4-4602) is a peripherally-acting
aromatic
L-amino acid decarboxylase (AAAD) or DOPA decarboxylase inhibitor, which is
unable to cross
the blood-brain barrier (Figure 7). Benserazide has a systematic (IUPAC) name
(RS)-2-amino-
3-hydroxy-N'-(2,3,4- trihydroxybenzyl)propanehydrazide, and CAS number 14919-
77-8 and
formula: C10H15N305, and Molecular mass of 257.243 g/mol.
Desloratadine
Desloratadine, known as 8-chloro-6,11-dihydro-11-(4-piperidylidene)-
5H-benzo[5,6]cyclohepta[1,2-b-]pyridine (Figure 7). Desloratadine is currently
marketed as
CLARINEXTM in the United States. CLAPJNEXTM is prescribed as an antihistamine
for
prevention or treatment of allergenic reactions, which may result in symptoms
such as sneezing,
itchy eyes and hives U.S. Pat. No. 4,659,716, by reference discloses
descarbonylethoxyloratadine
(also known as Desloratadine), which possesses antihistaminic properties with
substantially no
sedative properties. The 4,659,716 patent describes a process for the
preparation of
Desloratadine by dissolving loratadine in water and basifying with dilute
solution of potassium
carbonate to obtain a pink-colored oil. The organic material is extracted with
chloroform, washed
with water and triturated with hexane. Desloratadine is obtained by
recrystallisation of the
extracted organic material with large volume of hexane after charcolisation.
U.S. Pat. No. 6,506,767 (hereinafter 767) , discloses two polymorphic forms of
desloratadine, labeled Forms I and II. The XRPD peaks and the FTIR spectrum
for the forms are
also disclosed in the '767 patent. According to this patent '767 patent,
discloses certain alcoholic
solvents, e.g., hexanol and methanol produce 100% polymorph form 1, but
others, e. g., 3-
methyl-1 -butanol and cyclohexanol produce significant amounts of form 2.
Chlorinated solvents,
e. g., dichloromethane produce form 1 substantially free of form 2. Ether
solvents such as
dioxane produced form 1 substantially free of form 2 but other alkane ethers,
e.g., di-isopropyl
ether produced form 1 with significant amounts of form 2 and di-n-butyl ether
favored formation
of form 2. Ketones such as methyl isobutyl ketone produced crystalline
polymorph form 1
essentially free of form 2 but methyl butyl ketone produced 8:1 ratio of form
1 to form 2. Use of
methyl isobutyl ketone is preferred to produce crystalline polymorph form 1
essentially free of
form 2. Only ethyl acetate and di-n-butyl ether were found to produce
crystalline polymorph
form 2 substantially free of form 1. Use of di-n-butyl ether is preferred for
producing crystalline
form 2 substantially free of form 1. According to this patent the polymorph
form obtained from
U.S. Pat. No. 4,659,716 is a mixture of form I and form II.
Teva International Patent Application No. W02004/080461, claims a
pharmaceutical
composition of desloratadine comprising of a mixture of crystalline
Desloratadine of form I and
II in a weight to weight ratio of about 25% to about 75% of either form to the
other and a
16
CA 2922013 2017-10-03
pharmaceutically acceptable excipient.
Desloratadine or its pharmaceutically acceptable salts thereof can be used in
the methods
and compositions as disclosed herein, and are well known in the art and is
disclosed and can be
manufactured as taught in the following U.S. Patent Applications,
2010/0129310; 2010/0216831;
2010/0069402; 2010/0022576; 2010/0021542; 2008/0118555; 2007/0244144;
2007/0135472;
2007/0060756; 2007/0053974; 2007/0014855; 2007/0004671; 2006/0276495;
2006/0223841;
2006/0154948; 2006/0100435.
MS-275
MS-275 is also called MS-27-275 or N-(2-arninopheny1)-44N-(pyridin-3-
yl-methoxycarbonyparninomethylThenzarnide (Figure 7). MS-275 is also commonly
known in
the art as Entinostat, and is an orally bioavailable, highly selective, class
I histone deacetylase
(HDAC) inhibitor with a long half-life that allows for weekly or every- other-
week dosing.
Entinostat is currently being investigated in multiple phase 2 clinical
studies in cancer in
combination with other agents.
MS-275 is a synthetic benzamide derivative that has been shown to inhibit
cellular
histone deacetylase activity and to block growth in a variety of human tumor
cell lines (A. Saito,
etal., 1999, Proc. Natl. Acad. Sci. USA, A synthetic inhibitor of histone
deacetylase, MS-27-
275, with marked in vivo antitumor activity against human tumors, 96:4592-7).
The chemical
structure of MS-275 (structure I in Figure 8). MS-275 is chemically
synthesized using methods
known in the art. One such method is described in T. Suzuki et al., 1999, J.
Med. Chem.,
Synthesis and histone deacetylase inhibitory activity of new benzamide
derivatives, 42:3001-3.
Additional information relating to synthesis of MS-275 and related compounds
is found in
Japanese Unexamined Patent Publication Hei No. 10-152462. MS-275 is also
available from
various sources. One such source is Nihon Schering K.K. Another source is the
National Cancer
Institute (MS-275 is NSC No. 706995). Two benzamide derivatives closely
related to MS-275
(structures II and III in Figure 8). MS-275 can have derivatives as shown as
structures I, II and
III, in U.S. Patent 6,841,565. A polymorph of MS-275 is disclosed in GB patent
GB0907347.9
entitled N-(2-aminopheny1)-4-[N-(pyridine-3-YL)-methoxycarbonyl-aminomethyl]-
benzamide
(MS-275) polymorph B.
Treatment of diseases
In one embodiment, the invention relates to compositions for the treatment and
17
CA 2922013 2017-10-03
prevention of blood disorders such as anemia, thalassemia, and sickle cell
disease. Compositions
stimulate the specific expression of a gamma-globin protein, without
inhibiting cell proliferation,
and can increase the development of hemoglobin-expressing or other myeloid
cells.
Blood Disorders
In another embodiment, the invention relates to methods and medical aids which
utilize
these compositions to treat blood disorders and/or to ameliorate symptoms
associated with blood
disorders. The term "blood disorders" as used herein includes
hemoglobinopathies and
thalassemias. Blood disorders include disorders that can be treated,
prevented, or otherwise
ameliorated by the administration of a compound of the invention. Treatable
blood disorders
also include syndromes such as hemoglobin C, D and E disease, hemoglobin
lepore disease, and
HbH and HbS diseases. Treatment ameliorates one or more symptoms associated
with the
disorder. Compositions provided to the subject may include any combination of
the proteins or
chemical compounds of the invention or known to those of ordinary skill in the
art.
Administration of the composition may be short term, continuous or sporadic as
necessary.
Patients with a suspected or diagnosed with a blood disorder may only require
composition
treatment for short periods of time or until symptoms have abated or have been
effectively
eliminated.
Another embodiment of the invention is directed to methods for the treatment
of patients
with blood disorders comprising the administration of one or more compositions
of the
invention. Compositions to be administered contain a therapeutically effective
amount of a
chemical compound. A therapeutically effective amount is that amount which has
a beneficial
effect to the subject by alleviating one or more symptoms of the disorder or
by simply reducing
premature mortality. For example, a beneficial effect may be a decrease in
pain on an annual or
daily basis, a decrease in duration, frequency or intensity of pain crises, an
increased hematocrit,
an improved erythropoiesis with decreased hemolysis, decrease in fatigue or an
increased
endurance or stamina or walking ability to walk a certain distance without
shortness of breath, or
increased strength. Preferably, a therapeutic amount is that amount of
chemical compound or
agent that stimulates or enhances the expression of non-adult globin such as
embryonic or fetal
18
CA 2922013 2017-10-03
CA 02922013 2016-02-19
WO 2015/026939
PCT/US2014/051887
globin (HbF), or the proliferation of embryonic, fetal globin expressing
cells. In some
embodiments, a therapeutic amount is that amount of chemical compound or agent
as disclosed
herein that increases the percentage of expression of non-adult globin such as
embryonic or fetal
globin (HbF), or the proliferation of embryonic, fetal or adult globin
expressing cells.
Administration
In some embodiments, compositions comprising at least one or any combination
of
ambroxol, benserazide, desloratadine, resveratrol, NSC-95397, or MS-275, in
particular
benserazide, desloratadine or MS-275 can be directly or indirectly
administered to the patient.
Indirect administration can also be performed, for example, by administering
the composition to
cells ex vivo and subsequently introducing the treated cells to the patient.
The cells may be
obtained from the subject to be treated or from a genetically related or
unrelated patient. Related
patients offer some advantage by lowering the immunogenic response to the
cells to be
introduced. For example, using techniques of antigen matching, immunologically
compatible
donors can be identified and utilized.
Direct administration of compositions comprising at least one or any
combination of
ambroxol, benserazide, desloratadine, resveratrol, N SC-95397, or MS-275, in
particular
benserazide, desloratadine or MS-275 can also be by oral, parenteral,
sublingual, rectal such as
suppository or enteral administration, or by pulmonary absorption or topical
application.
Parenteral administration may be by intravenous injection, subcutaneous
injection. In some
embodiments, a composition comprising at least one or any combination of
ambroxol,
benserazide, desloratadine, resveratrol, NSC-95397, or MS-275, in particular
benserazide,
desloratadine or MS-275 can be administered by transdermal transfusion such as
with a dermal
or cutaneous patch, by direct contact with, for example, bone marrow through
an incision or
some other artificial opening into the body. Compositions may also be
administered to the nasal
passages as a spray. Arteries of the nasal area provide a rapid and efficient
access to the
bloodstream and immediate access to the pulmonary system. Access to the
gastrointestinal tract,
which can also rapidly introduce substances to the blood stream, can be gained
using oral,
enema, or injectable forms of administration. Compositions may be administered
as a bolus
injection or spray, or administered sequentially over time (episodically) such
as every two, four.
six or eight hours, every day (QD) or every other day (QOD), or over longer
periods of time such
as weeks to months. Compositions may also be administered in a timed-release
fashion such as
19
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
by using slow-release resins and other timed or delayed release materials and
devices.
Orally active compositions comprising at least one or any combination of
ambroxol,
benserazide, desloratadine, resveratrol, NSC-95397, idarubicin or MS-275, in
particular
benserazide, desloratadine or MS-275 are more preferred as oral administration
is usually the
safest, most convenient and economical mode of drug delivery. Oral
administration is usually
disadvantageous because compositions are poorly absorbed through the
gastrointestinal lining.
Compounds which are poorly absorbed tend to be highly polar. Consequently,
compounds which
are effective, as described herein, may be made orally bioavailable by
reducing or eliminating
their polarity. This can often be accomplished by formulating a composition
with a
complimentary reagent which neutralizes its polarity, or by modifying the
compound with a
neutralizing chemical group. Oral bioavailability is also a problem because
drugs are exposed to
the extremes of gastric pH and gastric enzymes. These problems can be overcome
in a similar
manner by modifying the molecular structure to withstand very low pH
conditions and resist the
enzymes of the gastric mucosa such as by neutralizing an ionic group, by
covalently bonding an
ionic interaction, or by stabilizing or removing a disulfide bond or other
relatively labile bond.
Prophylactic treatments involve administration of a composition of the
invention to a
subject having a confirmed or suspected blood disorder without having any
overt symptoms. For
example, otherwise healthy patients who have been genetically screened and
determined to be at
high risk for the future development of a blood disorder may be administered
compositions of
the invention prophylactically. Administration can begin at birth and
continue, if necessary, for
life. Both prophylactic and therapeutic uses are readily acceptable because
these compounds are
generally safe and non-toxic.
Individual pulses of an HbF-inducing agent as disclosed herein can be
delivered to the
patient continuously over a period of several hours, such as about 2, 4, 6, 8,
10, 12, 14 or 16
hours, or several days, such as 2, 3, 4, 5, 6, or 7 days, preferably from
about 1 hour to about 24
hours and more preferably from about 3 hours to about 9 hours. Alternatively,
periodic doses
can be administered in a single bolus or a small number of injections of the
composition over a
short period of time, typically less than 1 or 2 hours. For example, arginine
butyrate has been
administered over a period of 4 days with infusions for about 8 hours per day
or overnight,
followed by a period of 7 days of no treatment. This has been shown to be an
effective regimen
for many thalassemic disorders. Fetal hemoglobin levels rise substantially and
there is a
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
significant rise in the number of both adult and fetal hemoglobin expressing
cells. In certain
instances, a substantial rise in HbF means that there are positive
consequences that raise the
patient's standard of living such as, for example, increased activity or
mobility, fewer side-
effects, fewer hospital stays or visits to the physician, or fewer
transfusions. For instance, HbF
levels above 20% are generally considered to be sufficient to eliminate
symptoms associated
with sickle cell disease.
With pulse therapy, in vivo levels of an HbF-inducing agent thereof can drop
below that
level required for effective continuous treatment. Pulsed administration can
reduce the amount
of a composition comprising an HbF-inducing agent thereof administered to the
patient per dose,
and/or per total treatment regimen with an increased effectiveness. Pulsed
administration can
provide a saving in time, effort and expense and a lower effective dose can
lessen the number
and severity of complications that can be experienced by a subject. As such,
pulsing can be more
effective than continuous administration of the same composition.
In some embodiments, individual pulses can be delivered to a subject
continuously over a
period of several hours, such as about 2, 4, 6, 8, 10, 12. 14 or 16 hours, or
several days, such as
2, 3, 4, 5, 6, or 7 days, or from about 1 hour to about 24 hours or from about
3 hours to about 9
hours. Alternatively, periodic doses can be administered in a single bolus or
a small number of
injections of the composition comprising an HbF-inducing agent thereof over a
short period of
time, for example, less than 1 or 2 hours. For example, arginine butyrate can
be administered
.. over a period of 4 days with infusions for about 8 hours per day or
overnight, followed by a
period of 7 days of no treatment.
The interval between pulses or the interval of no delivery can be greater than
24 hours or
can be greater than 48 hours, and can be for even longer such as for 3, 4, 5,
6, 7, 8, 9 or 10 days,
two, three or four weeks or even longer. The interval between pulses can be
determined by one
of ordinary skill in the art, for example, as demonstrated herein in the
Examples, by measuring
the gamma-globin expression level in the blood in the subject after
administration of the pulse
dose, and administering a pulse when the mRNA gamma-globin level reaches a
certain pre-
defined low threshold limit. Such pre-defined low threshold limits can be
determined by one of
ordinary skill in the art, and can be, for example, about baseline level, or
about 100% or about
200% above baseline level mRNA gamma-globin expression (e.g., mRNA gamma-
globin
expression without administration of an HbF-inducing agent). Alternatively, in
some
21
embodiments, the interval between pulses can be calculated by administering
another dose of a
composition comprising an HbF-inducing agent, and when the active component of
the
composition is no longer detectable in the patient prior to delivery of the
next pulse.
Alternatively, intervals can also be calculated from the in vivo half-life of
the composition.
The interval between pulses can also be determined by one of ordinary skill in
the art, for
example, as demonstrated herein in the Examples, by measuring the percent
increase in absolute
hemoglobin (see Fig 3), percent F-reticulocytes (see Figure 5), percent
increase in F-cells (see
Figure 6), in the blood in the subject after administration of the pulse dose,
and administering a
pulse when the mRNA gamma-globin or total hemoglobin (Hb) level increases by,
for example
lo about a 1.0 or about 0.5 g/dL increase in total hemoglobin or by 0.3-
fold increase in fetal globin
mRNA above levels prior to treatment.
In some embodiments, the number of pulses in a single therapeutic regimen can
be as
little as two, but can be from about 5 to 10, 10 to 20, 15 to 30 or more.
In some embodiments, a subject can receive one or more compositions comprising
an
HbF-inducing agent for life according to the methods of this invention, for
example, where the
subject has a permanent or incurable blood disorder, e.g., an inherited blood
disorder.
Compositions can be administered by most any means, and can be delivered to
the subject as an
oral formulation, or injection (e.g. intravenous, subcutaneous, intra-
arterial), infusion or
instillation. Various methods and apparatus for pulsing compositions by
infusion or other forms
of delivery to the patient are disclosed in U.S. Pat. Nos. 4,747,825;
4,723,958; 4,948,592;
4,965,251 and 5,403,590.
In one embodiment, a composition comprising an HbF-inducing agent thereof can
be
administered to a subject for about 2, or about 3, or about 4, or about five
days, or more than five
days, and then a subsequently administered after an appropriate interval for
an additional period
of time, for example, for about 2, or about 3, or about 4, or about five days,
or more than five
days. Cycles of treatment may occur in immediate succession or with an
interval of no treatment
between cycles.
In some embodiments, a composition comprising an HbF-inducing agent can be
administered to a subject before a chemotherapeutic treatment, or radiation
treatment is
administered to the subject. In alternative embodiments, a composition
comprising an HbF-
inducing agent can be co-administered to a subject In some embodiments, a
composition
22
CA 2922013 2017-10-03
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
comprising an HbF-inducing agent can be co-administered with a pharmaceutical
composition
comprising an comprising one or more addition agents. The pharmaceutical
compositions can be
provided by pulsed administration. For example, a composition comprising an
HbF-inducing
agent can be administered to a subject, followed by a chemotherapeutic
treatment, or radiation
treatment after an interval of time has passed, and this order of
administration the same or similar
time interval can be repeated, for example, at least 1, 2, 3, 4, 5, 6, 7, 8,
9, or 10 or more times.
Method for Increasing Percent HbF and/or Total Hemoglobin
Provided herein are methods and compositions for increasing the percentage of
fetal
hemoglobin in the blood of a subject comprising administering to a subject a
composition
comprising an HbF-inducing agent as disclosed herein as the free acid, a
pharmaceutically
acceptable salt, or ester thereof. Further provided herein are methods and
compositions for
increasing total hemoglobin in the blood of a subject, comprising
administering to the subject a
composition comprising an HbF-inducing agent as disclosed herein as the free
acid, a
pharmaceutically acceptable salt, or ester thereof. Also provided herein are
methods and
.. compositions for increasing total hemoglobin, hematocrit, and red blood
cells in a subject,
comprising administering to said subject an HbF-inducing agent as disclosed
herein as the free
acid, a pharmaceutically acceptable salt, or ester thereof. Further provided
herein are methods
and compositions for increasing total hemoglobin, hematocrit, or red blood
cells, or a
combination thereof, comprising administering to said subject an HbF-inducing
agent as
disclosed herein.
In some embodiments, administering an HbF-inducing agent as disclosed herein
does not
suppress erythropoiesis at concentrations associated with biologic activity.
In certain
embodiments, administering an HbF-inducing agent as disclosed herein
stimulates cell
proliferation. In some embodiments, administering an HbF-inducing agent as
disclosed herein
inhibits apoptosis of erythroid progenitors. In further embodiments,
administering an HbF-
inducing agent as disclosed herein stimulates erythroid cell proliferation and
survival. In some
embodiments, administering an HbF-inducing agent as disclosed herein
stimulates erythroid cell
proliferation. In certain embodiments, administering an HbF-inducing agent as
disclosed herein
stimulates erythroid cell survival. In some embodiments, administering an HbF-
inducing agent
as disclosed herein stimulates red blood cell production. In certain
embodiments, administering
an HbF-inducing agent as disclosed herein leads to a longer survival of
sickled blood cells.
23
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
In some embodiments, administering an HbF-inducing agent as disclosed herein
stimulates erythropoiesis. In certain embodiments, administering an HbF-
inducing agent as
disclosed herein induces expression of the fetal globin gene promoter. In some
embodiments,
administering an HbF-inducing agent as disclosed herein increases fetal globin
levels. In certain
embodiments, administering an HbF-inducing agent as disclosed herein increases
RBC
production. In some instances, increased RBC production is assayed by
reticulocytes, total
hemoglobin (Hgb), and hematocrit (Hct).
In certain embodiments, administering an HbF-inducing agent as disclosed
herein
increases the amount of fetal globin in the blood of the subject. In some
embodiments,
.. administering an HbF-inducing agent as disclosed herein increases the
amount of fetal
hemoglobin in the blood of the subject. In certain embodiments, administering
an HbF-inducing
agent as disclosed herein increases the amount of total hemoglobin in the
blood of the subject. In
some embodiments, administering an HbF-inducing agent as disclosed herein
increases the
percentage of reticulocytes in the blood of the subject. In certain
embodiments, administering an
HbF-inducing agent as disclosed herein increases the number of reticulocytes
in the blood of the
subject. In some embodiments, administering an HbF-inducing agent as disclosed
herein
increases hematocrit.
In contrast to ST20 or hydroxyurea, an HbF-inducing agent as disclosed herein
is
effective at increasing percent HbF at a total daily dose which is below the
maximum tolerated
dose. In some instances, administering an HbF-inducing agent as disclosed
herein does not
necessitate the careful dose titration currently required for treatment with
ST20.
In addition, the total daily dose of an HbF-inducing agent as disclosed herein
which is
effective in increasing the percentage of HbF is significantly lower than the
dose required for
other SCFAD like arginine butyrate. In some embodiments, a subject can be
administered an
HbF-inducing agent as disclosed herein with other agents, including but not
limited to 2,2-
dimethylbutyrate is administered as sodium 2,2-dimethylbutyrate. 2,2-
Dimethylbutyrate
includes, but is not limited to, 2,2-dimethylbutyric acid, sodium 2,2-
dimethylbutyrate, potassium
2,2-dimethylbutyrate, magnesium 2,2-dimethylbutyrate, calcium 2,2-
dimethylbutyrate, arginine
2,2-dimethylbutyrate, lysine 2,2-dimethylbutyrate, choline 2,2-
dimethylbutyrate, methyl 2.2-
dimethylbutyrate (2,2-dimethylbutyric acid methyl ester), ethyl 2,2-
dimethylbutyrate, propyl
2,2-dimethylbutyrate, or isopropyl 2,2-dimethylbutyrate.
24
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
In certain embodiments, the subject has been diagnosed with a blood disorder.
In some
embodiments, the blood disorder is sickle cell disease. In other embodiments,
the blood disorder
is a beta thalassemia. In certain embodiments, the beta-thalassemia is beta
thalassemia
intermedia. In some embodiments, the beta thalassemia is beta-thalassemia
major. In certain
embodiments, the beta thalassemia is beta thalassemia minor (beta-thalassemia
trait). In some
embodiments, the beta thalassemia is HbE beta-thalassemia. In certain
embodiments, the beta
thalassemia is HbS beta-thalassemia.
In certain embodiments, a subject is administered a composition comprising an
HbF-
inducing agent as disclosed herein, daily. In further embodiments,
administration is continuous.
In some embodiments, the administration of a composition comprising an HbF-
inducing agent as
disclosed herein is by pulsed administration. In certain embodiments, pulsed
administration
comprises administering an HbF-inducing agent pulse for about 1 day. about 2
days, about 3
days, about 4 days, about 5 days, about 6 days, about 7 days, about 10 days,
about 2 weeks,
about 3 weeks, about 4 weeks. about 5 weeks, about 6 weeks, about 7 weeks,
about 8 weeks,
about 2 months. about 3 months, about 4 months, about 5 months, about 6
months, about 9
months, about 12 months. in some embodiments, pulsed administration comprises
intervals of
not administering an HbF-inducing agent for about 1 day, about 2 days, about 3
days, about 4
days, about 5 days, about 6 days, about 7 days, about 10 days, about 2 weeks,
about 3 weeks,
about 4 weeks, about 5 weeks. about 6 weeks, about 7 weeks, about 8 weeks,
about 2 months,
about 3 months, about 4 months, about 5 months, about 6 months, about 9
months, about 12
months. In certain embodiments, administration is for the lifetime of the
subject.
In some embodiments, a composition comprising an HbF-inducing agent is
administered
every other day. In certain embodiments, the pulsed administration comprises
administering a
composition comprising an HbF-inducing agent for about 5 days per week. In
some
embodiments, the pulsed administration comprises administering a composition
comprising an
HbF-inducing agent for about 5 days, followed by not administering an HbF-
inducing agent for
about 2 days. In certain embodiments, the pulsed administration comprises
administering an
HbF-inducing agent for about 2 weeks, followed by not administering an HbF-
inducing agent
for about 1 week. In some embodiments, the pulsed administration comprises
administering an
HbF-inducing agent for about 2 weeks, followed by not administering an HbF-
inducing agent for
about 2 weeks. In certain embodiments, the pulsed administration comprises
administering an
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
HbF-inducing agent for about 4 weeks, followed by not administering an HbF-
inducing agent for
about 1 week. In some embodiments, the pulsed administration comprises
administering an
HbF-inducing agent for about 4 weeks, followed by not administering an HbF-
inducing agent for
about 2 weeks. In further embodiments, the pulsed administration comprises
administering an
HbF-inducing agent for about 6 weeks, followed by not administering an HbF-
inducing agent
for about 2 weeks. In certain embodiments, the pulsed administration comprises
administering
an HbF-inducing agent for about 8 weeks, followed by not administering an HbF-
inducing agent
for about 2 weeks. In some embodiments, the pulsed administration comprises
administering an
HbF-inducing agent for about 8 weeks, followed by not administering an HbF-
inducing agent for
about 4 weeks.
In some instances, administering a composition comprising an HbF-inducing
agent to a
subject with one genotype of beta thalassemia is more effective in raising
percent HbF than
administering DMB (ST20) to a subject with a different genotype of beta-
thalassemia. Further
provided herein are methods and compositions comprising diagnosing a beta-
thalassemia
genotype of a patient, determining a treatment plan considering the beta-
thalassemia genotype,
and optionally increasing the percentage of fetal hemoglobin in the blood of
the patient,
comprising administering to the patient an HbF-inducing agent as the free
acid, a
pharmaceutically acceptable salt, or ester thereof.
Pharmaceutical Compositions
In some embodiments, a pharmaceutical composition comprising an HbF-inducing
agent
administered according to a method of the invention are administered orally in
effective dosages,
depending upon the weight, body surface area, and condition of the subject
being treated. In
some instances, variations occur depending upon the species of the subject
being treated and its
individual response to said medicament, as well as on the type of
pharmaceutical formulation
chosen and the time period and interval at which such administration is
carried out. In some
embodiments, the administration of the pharmaceutical composition comprising
an
HbF-inducing agent according to a method of the invention is carried out in
single or multiple
doses. For example, the composition can be administered in a wide variety of
different dosage
forms, i.e., it may be combined with various pharmaceutically acceptable inert
carriers in the
form of tablets, dragees, capsules, lozenges, troches, hard candies, aqueous
suspensions, elixirs,
syrups, and the like. Such carriers include solid diluents or fillers, sterile
aqueous media and
26
CA 02922013 2016-02-19
WO 2015/026939
PCT/US2014/051887
various non-toxic organic solvents, etc. Moreover, oral pharmaceutical
compositions can be
suitably sweetened and/or flavored.
In certain embodiments, pharmaceutical compositions comprising an HbF-inducing
agent
are suitable for oral administration. Suitable pharmaceutical compositions for
oral
administration can be in the form of capsules, tablets, pills, lozenges,
cachets, dragees, powders,
granules; or as a solution or a suspension in an aqueous or non-aqueous
liquid; or as an oil-in-
water or water-in-oil liquid emulsion; or as an elixir or syrup; and the like;
each containing a
predetermined amount of a compound of the present invention as an active
ingredient. When
intended for oral administration in a solid dosage form (i.e., as capsules,
tablets, pills and the
like), the pharmaceutical compositions of the invention will typically
comprise a compound of
the present invention as the active ingredient and one or more
pharmaceutically-acceptable
carriers, such as sodium citrate or dicalcium phosphate. Optionally or
alternatively, such solid
dosage forms may also comprise: filters or extenders, such as starches,
microcrystalline
cellulose, lactose, sucrose, glucose, mannitol, and/or silicic acid; binders,
such as
carboxymethylcellulose, alginates, gelatin, polyvinylpyrrolidone, sucrose
and/or acacia;
humectants, such as glycerol; disintegrating agents, such as agar-agar,
calcium carbonate, potato
or tapioca starch, alginic acid, certain silicates, and/or sodium carbonate;
solution retarding
agents, such as paraffin; absorption accelerators, such as quaternary ammonium
compounds:
wetting agents, such as cetyl alcohol and/or glycerol monostearate;
absorbents, such as kaolin
and/or bentonite clay; lubricants, such as talc, calcium stearate, magnesium
stearate, solid
polyethylene glycols, sodium lauryl sulfate, and/or mixtures thereof; coloring
agents; and
buffering agents.
Solid compositions of a similar type may also be employed as fillers in
gelatin capsules;
preferred materials in this connection also include lactose or milk sugar as
well as high
molecular weight polyethylene glycols. When aqueous suspensions and/or elixirs
are desired for
oral administration, the active ingredient may be combined with various
sweetening or flavoring
agents, coloring matter or dyes, and, if so desired, emulsifying and/or
suspending agents as well,
together with such diluents as water, ethanol, propylene glycol, glycerin and
various like
combinations thereof.
Release agents, wetting agents, coating agents, sweetening, flavoring and
perfuming
agents, preservatives and antioxidants can also be present in the
pharmaceutical compositions of
27
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
the invention. Examples of pharmaceutically-acceptable antioxidants include:
water-soluble
antioxidants, such as ascorbic acid, cysteine hydrochloride, sodium bisulfate,
sodium
metabi sulfate sodium sulfite and the like; oil-soluble antioxidants, such as
ascorbyl palmitate,
butylated hydroxyanisole (BHA), butylated hydroxytoluene (BHT), lecithin,
propyl gallate,
alpha-tocopherol, and the like; and metal-chelating agents, such as citric
acid, ethylenediamine
tetraacetic acid (EDTA), sorbitol, tartaric acid, phosphoric acid, and the
like. Coating agents for
tablets, capsules, pills and like, include those used for enteric coatings,
such as cellulose acetate
phthalate (CAP), polyvinyl acetate phthalate (PVAP), hydroxypropyl
methylcellulose phthalate,
methacrylic acid, methacrylic acid ester copolymers, cellulose acetate
trimellitate (CAT),
carboxymethyl ethyl cellulose (CMEC), hydroxypropyl methyl cellulose acetate
succinate
(HPMCAS), and the like.
In addition, the pharmaceutical compositions of the present invention may
optionally
contain opacifying agents and may be formulated so that they release the
active ingredient only,
or preferentially, in a certain portion of the gastrointestinal tract,
optionally, in a delayed manner.
Examples of embedding compositions which can be used include polymeric
substances and
waxes. The active ingredient can also be in micro-encapsulated form, if
appropriate, with one or
more of the above-described excipients.
If desired, pharmaceutical compositions of the present invention may also be
formulated
to provide slow or controlled release of the active ingredient using, by way
of example,
hydroxypropyl methyl cellulose in varying proportions; or other polymer
matrices, liposomes
and/or microspheres. Sustained release compositions can be formulated
including those wherein
the active component is derivatized with differentially degradable coatings,
e.g., by
microencapsulation, multiple coatings, etc.
It will be appreciated that the actual preferred amounts of active compounds
used in a
given therapy will vary according to the particular compositions formulated.
Optimal
administration rates for a given protocol of administration can be readily
ascertained by those
skilled in the art using conventional dosage determination tests conducted
with regard to the
foregoing guidelines.
It will also be understood that normal, conventionally known precautions will
be taken
.. regarding the administration of the compounds of the invention generally to
ensure their efficacy
under normal use circumstances. Especially when employed for treatment of
humans and
28
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
animals in vivo. the practitioner should take all sensible precautions to
avoid conventionally
known contradictions and toxic effects.
The composition, shape, and type of dosage forms of the invention will
typically vary
depending on their use. This aspect of the invention will be readily apparent
to those skilled in
the art. See, e.g., Remington's Pharmaceutical Sciences (1990) 18th ed., Mack
Publishing,
Eastern Pa.
In certain embodiments, the pharmaceutical compositions of the invention are
packaged
in a unit dosage form. The term "unit dosage form" or "unit dose" refers to a
physically discrete
unit suitable for dosing a patient, i.e., each unit containing a predetermined
quantity of active
agent calculated to produce the desired therapeutic effect either alone or in
combination with one
or more additional units. For example, such unit dosage forms may be capsules,
tablets, pills, and
the like. Unit doses can also be prepared to contain any useful amount of an
active ingredient
(e.g., an HbF-inducing agent). For example, a unit dose can comprise 10 mg, 20
mg, 30 mg, 40
mg, 50 mg, 60 mg, 70 mg, 80 mg, 90 mg, 100 mg, 110 mg, 120 mg, 130 mg, 140 mg,
150 mg,
160 mg, 170 mg, 180 mg, 190 mg, 200 mg, 210 m2, 220 mg. 230 mg, 240 mg, 250
mg. 260 mg,
270 mg, 280 mg, 290 mg, 300 mg, 310 mg, 320 mg, 330 mg, 340 mg, 350 mg, 360
mg. 370 mg,
380 mg, 390 mg, 400 mg, 410 mg, 420 mg, 430 mg, 440 mg. 450 mg, 460 mg, 470
mg, 480 mg,
490 mg, 500 mg, 510 mg, 520 mg, 530 mg, 540 mg, 550 mg. 560 mg, 570 mg, 580
mg, 590 mg,
600 mg, 625 mg, 650 mg, 675 mg, 700 mg, 750 mg, 800 mg. 850 mg, 900 mg, 950
mg, 1000
mg, or more of an HbF-inducing agent per unit dose. Milligrams per dose can
refer to either the
free acid form of an HbF-inducing agent, or an HbF-inducing agent in a salt or
ester form.
Administrations can be repeated on consecutive or non-consecutive days. Thus,
daily
administrations can be performed for 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13,
14, 15, 16, 17, 18, 19,
20, 25, 30, 35, 40, 45, 50, 55, 60, or more consecutive days. For example,
administration of 10
mg/kg of an HbF-inducing agent is performed twice a day (at a total daily dose
of 20 mg/kg) for
14 consecutive days. Alternatively, administration may occur for multiple
days, but on non-
consecutive days separated by 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 or more days. For
example,
administration of 15 mg/kg of an HbF-inducing agent is performed on every
other day following
therapy onset. In another instance, administration of an HbF-inducing agent is
performed for 5
days per week. Such dosing regimens can be tailored to an individual patient,
based on any
number of clinically relevant parameters including, but not limited to
toxicity, tolerance, side-
29
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
effects, effectiveness, etc.
Combination Therapy
In certain embodiments, the pharmaceutical composition is administered alone
or in
combination with other known compositions for treating blood disorders in a
subject, e.g., a
mammal. In some embodiments, mammals include cats, dogs, pigs, horses, cows,
rats, mice,
monkeys, chimpanzees, baboons, and humans. In specific embodiments, the mammal
is a
human. In some embodiments, the human is a child. In certain embodiments the
human is under
the age of 18. In some embodiments, the human is under the age of 10. In some
embodiments,
the human is under the age of 2. In one embodiment, the subject is suffering
from a blood
disorder. In another embodiment, the subject is at risk of suffering from a
blood disorder.
The language "in combination with" a known composition is intended to include
simultaneous administration of the composition of the invention and the known
composition,
administration of the composition of the invention first, followed by the
known composition and
administration of the known composition first, followed by the composition of
the invention.
Any of the compositions known in the art for treating blood disorders can be
used in the methods
of the invention.
In some embodiments, in addition to the use of an HbF-inducing agent for the
treatment
of blood disorders, concomitant administration of other pharmaceutical and
nutraceutical
compounds occurs. For example, persons suffering from sickle cell disease are
given an HbF-
inducing agent other agents as disclosed herein), folic acid supplements (for
blood cell
production), opioids or analgesics (for pain management), and/or antibiotics
(for treating
secondary infections). In further embodiments, administration of an HbF-
inducing agent for the
treatment of blood disorders is combined with the administration of natural or
synthetic
erythropoietin. In certain instances, concomitant treatment with an HbF-
inducing agent and a
.. second agent occurs at the same time, or on different regimen schedules. In
some embodiments
an HbF-inducing agent is an orally bio-available compound that is active at
well tolerated doses.
Administration of the compositions comprising HbF-inducing agents as described
herein
may be by oral, parenteral, sublingual, rectal, or enteral administration, or
pulmonary absorption
or topical application. Compositions can be directly or indirectly
administered to the patient.
Indirect administration is performed, for example, by administering the
composition to cells ex
vivo and subsequently introducing the treated cells to the subject, e.g.,
human patient.
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
Alternatively, the cells may be obtained from the patient to be treated or
from a genetically
related or unrelated patient. Related patients offer some advantage by
lowering the
immunogenic response to the cells to be introduced. For example, using
techniques of antigen
matching, immunologically compatible donors can be identified and utilized.
The compositions comprising HbF-inducing agents can be purchased commercially
and
prepared as a mixed composition using techniques well-known to those of
ordinary skill in the
art. Direct administration of a composition comprising HbF-inducing agents to
a subject can be
by oral, parenteral, sublingual, rectal such as suppository or enteral
administration, or by
pulmonary absorption or topical application. Parenteral administration may be
by intravenous
(IV) injection. subcutaneous (s.c.) injection, intramuscular (i.m.) injection,
intra-arterial
injection, intrathecal (i.t.) injection, intra-peritoneal (i.p.) injection, or
direct injection or other
administration to the subject.
Alternatively, pharmaceutical compositions comprising HbF-inducing agents
and/or salts
thereof can be added to the culture medium of cells ex vivo. In addition to
the active compound,
.. such compositions comprising HbF-inducing agents can contain
pharmaceutically-acceptable
carriers and other ingredients known to facilitate administration and/or
enhance uptake (e.g.,
saline, dimethyl sulfoxide, lipid, polymer, affinity-based cell specific-
targeting systems). In some
embodiments, a composition comprising HbF-inducing agents and/or salts thereof
can be
incorporated in a gel, sponge, or other permeable matrix (e.g., formed as
pellets or a disk) and
placed in proximity to the endothelium for sustained, local release. In some
embodiments, a
composition comprising HbF-inducing agents and/or salts thereof can be
administered in a single
dose or in multiple doses which are administered at different times.
Pharmaceutical compositions comprising HbF-inducing agents and/or salts
thereof can be
administered by any known route. By way of example, a composition c comprising
HbF-
inducing agents and/or salts thereof can be administered by a mucosal,
pulmonary, topical, or
other localized or systemic route (e.g., enteral and parenteral). The phrases
"parenteral
administration" and "administered parenterally" as used herein means modes of
administration
other than enteral and topical administration, usually by injection, and
includes, without
limitation, intravenous, intramuscular, intra-arterial. intrathecal,
intraventricular, intracapsular,
intraorbital, intracardiac, intradermal, intraperitoneal, transtracheal,
subcutaneous, subcuticular,
intraarticular. sub capsular, subarachnoid, intraspinal, intracerebro spinal,
and intrasternal
31
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
injection, infusion and other injection or infusion techniques, without
limitation. The phrases
"systemic administration," "administered systemically", "peripheral
administration" and
"administered peripherally" as used herein mean the administration of the
agents as disclosed
herein such that it enters the animal's system and, thus, is subject to
metabolism and other like
processes, for example, subcutaneous administration.
The phrase "pharmaceutically acceptable" is employed herein to refer to those
compounds, materials, compositions, and/or dosage forms which are, within the
scope of sound
medical judgment, suitable for use in contact with the tissues of human beings
and animals
without excessive toxicity, irritation, allergic response, or other problem or
complication,
commensurate with a reasonable benefit/risk ratio.
The phrase "pharmaceutically acceptable carrier" as used herein means a
pharmaceutically acceptable material, composition or vehicle, such as a liquid
or solid filler,
diluent, excipient, solvent or encapsulating material, involved in carrying or
transporting the
subject agents from one organ, or portion of the body, to another organ, or
portion of the body.
Each carrier must be "acceptable" in the sense of being compatible with the
other ingredients of
the formulation, for example the carrier does not decrease the impact of the
agent on the
treatment. In other words, a carrier is pharmaceutically inert.
Suitable choices in amounts and timing of doses, formulation, and routes of
administration of a composition comprising HbF-inducing agents and/or salts
thereof can be
made with the goals of achieving a favorable response in the subject with a
blood disorder, e.g.,
thalassemia, aplastic anemia and hemoglobinopathy and avoiding undue toxicity
or other harm
thereto (i.e., safety). Therefore, "effective" refers to such choices that
involve routine
manipulation of conditions to achieve a desired effect.
A bolus of the formulation of a composition comprising HbF-inducing agents
and/or salts
thereof administered to an individual over a short time once a day is a
convenient dosing
schedule. Alternatively, the effective daily dose can be divided into multiple
doses for purposes
of administration, for example, two to twelve doses per day. Dosage levels of
active ingredients
in a pharmaceutical composition comprising HbF-inducing agents and/or salts
thereof can also
be varied so as to achieve a transient or sustained concentration of the
compound or derivative
thereof in an individual, especially in and around the blood circulation and
to result in the desired
therapeutic response or protection. But it is also within the skill of the art
to start doses at levels
32
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
lower than required to achieve the desired therapeutic effect and to gradually
increase the dosage
until the desired effect is achieved.
In some embodiments, the amount of a composition comprising HbF-inducing
agents
and/or salts thereof can be administered is dependent upon factors known to a
person skilled in
the art such as bioactivity and bioavailability of the compound (e.g., half-
life in the body,
stability, and metabolism); chemical properties of the compound (e.g.,
molecular weight,
hydrophobicity, and solubility); route and scheduling of administration, and
the like. It will also
be understood that the specific dose level to be achieved for any particular
individual can depend
on a variety of factors, including age, gender, health, medical history,
weight, combination with
one or more other drugs, and severity of disease.
Production of compounds comprising HbF-inducing agents and/or salts thereof
according
to present regulations are regulated for good laboratory practices (GLP) and
good manufacturing
practices (GMP) by governmental regulatory agencies for pharmaceuticals (e.g.,
U.S. Food and
Drug Administration). This requires accurate and complete record keeping, as
well as
monitoring of QA/QC. Oversight of patient protocols by agencies and
institutional panels is also
envisioned to ensure that informed consent is obtained; safety, bioactivity,
appropriate dosage,
and efficacy of products are studied in phases; results are statistically
significant; and ethical
guidelines are followed. Similar oversight of protocols using animal models,
as well as the use of
toxic chemicals, and compliance with regulations is required.
Dosages, formulations, dosage volumes, regimens, and methods for analyzing
results
aimed at increasing the proliferation of blood cells, and increasing absolute
neutrophil count
(ANC) can vary. Thus, minimum and maximum effective dosages vary depending on
the method
of administration. Increase in ANC in a subject can occur within a specific
dosage range, which
varies depending on, for example, the race, sex, gender, age, and overall
health of the subject
receiving the dosage, the route of administration, whether a composition
comprising HbF-
inducing agents and/or salts thereof is administered in conjunction with other
molecules, and the
specific regimen of administration of the composition comprising HbF-inducing
agents and/or
salts thereof. For example, in general, nasal administration requires a
smaller dosage than oral,
enteral, rectal, or vaginal administration.
In an alternative embodiment, for oral and/or enteral formulations of a
composition
comprising HbF-inducing agents and/or salts thereof, tablets can be formulated
in accordance
33
with conventional procedures employing solid carriers well-known in the art.
Capsules employed
for oral formulations to be used with the methods of the present invention can
be made from any
pharmaceutically acceptable material, such as gelatin or cellulose
derivatives. Sustained release
oral delivery systems and/or enteric coatings for orally administered dosage
forms are also
contemplated, such as those described in U.S. Pat. No. 4,704,295, "Enteric
Film-Coating
Compositions," issued Nov. 3, 1987; U.S. Pat. No.4, 556,552, "Enteric Film-
Coating
Compositions," issued Dec. 3, 1985; U.S. Pat. No. 4,309,404, ''Sustained
Release Pharmaceutical
Compositions," issued Jan. 5, 1982; and U.S. Pat. No. 4,309,406, "Sustained
Release
Pharmaceutical Compositions," issued Jan. 5, 1982. Examples of solid carriers
include starch,
sugar, bentonite, silica, and other commonly used carriers. Further non-
limiting examples of
carriers and diluents which can be used in the formulations of the present
invention include
saline, syrup, dextrose, and water.
Enteric Coated Formulation
In some embodiments, oral formulations of a composition comprising HbF-
inducing
agents and/or salts thereof can be in the form of a tablet formulation, for
example, comprising
HbF-inducing agents and/or salts thereof with an enteric polymer casing. An
example of such a
preparation can be found in W02005/021002. The active material in the core can
be present in a
micronized or solubilized form. In addition to active materials the core can
contain additives
conventional to the art of compressed tablets. Appropriate additives in such a
tablet can
comprise diluents such as anhydrous lactose, lactose monohydrate, calcium
carbonate,
magnesium carbonate, dicalcium phosphate or mixtures thereof; binders such as
microcrystalline
cellulose, hydroxypropylmethylcellulose, hydroxypropyl-cellulose,
polyvinylpyrrolidone, pre-
gelatinised starch or gum acacia or mixtures thereof; disintegrants such as
microcrystalline
cellulose (fulfilling both binder and disintegrant functions) cross-linked
polyvinylpyrrolidone,
sodium starch glycollate, croscarmellose sodium or mixtures thereof;
lubricants, such as
magnesium stearate or stearic acid, glidants or flow aids, such as colloidal
silica, talc or starch,
and stabilizers such as desiccating amorphous silica, coloring agents, flavors
etc. In some
embodiments, a tablet comprises lactose as diluent. When a binder is present,
it is preferably
hydroxypropylmethyl cellulose. In some embodiments, a tablet comprises
magnesium stearate
as lubricant. In some embodiments, a tablet comprises croscarmellose sodium as
disintegrant, or
can comprise a microcrystalline cellulose.
34
CA 2922013 2017-10-03
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
In some embodiments, a diluent can be present in a range of 10 - 80% by weight
of the
core. The lubricant can be present in a range of 0.25- 2% by weight of the
core. The
di sintegrant can be present in a range of 1 - 10% by weight of the core.
Microcrystalline
cellulose, if present, can be present in a range of 10- 80% by weight of the
core.
In some embodiments, the active ingredient, e.g., HbF-inducing agents and/or a
salt
thereof comprises between 10 and 50% of the weight of the core, more
preferably between 15
and 35% of the weight of the core (calculated as free base equivalent). The
core can contain any
therapeutically suitable dosage level of the active ingredient e.g., HbF-
inducing agents and/or a
salts thereof, but preferably contains up to 150 mg as free base of the active
ingredient. In some
embodiments, the core contains 20, 30, 40, 50, 60, 80 or 100 mg as free base
of the active
ingredient. The active ingredient e.g., HbF-inducing agents and/or a salts
thereof can be present
as the free base, or as any pharmaceutically acceptable salt. If the active
ingredient e.g., HbF-
inducing agents is present as a salt, the weight is adjusted such that the
tablet contains the desired
amount of active ingredient, calculated as free base of the salt. In some
embodiments, the active
ingredient e.g., HbF-inducing agents is present as a hydrochloride salt.
In some embodiments, the core can be made from a compacted mixture of its
components. The components can be directly compressed, or can be granulated
before
compression. Such granules can be formed by a conventional granulating process
as known in
the art. In an alternative embodiment, the granules can be individually coated
with an enteric
casing, and then enclosed in a standard capsule casing.
In some embodiments, the core can be surrounded by a casing which comprises an
enteric polymer. Examples of enteric polymers are cellulose acetate phthalate,
cellulose acetate
succinate, methylcellulose phthalate, ethylhydroxycellulose phthalate,
polyvinylacetate pthalate,
polyvinylbutyrate acetate, vinyl acetate-maleic anhydride copolymer, styrene-
maleic mono-ester
copolymer, methyl acrylate- methacrylic acid copolymer or methacrylate-
methacrylic acid-octyl
acrylate copolymer. These can be used either alone or in combination, or
together with other
polymers than those mentioned above. The casing can also include insoluble
substances which
are neither decomposed nor solubilized in living bodies, such as alkyl
cellulose derivatives such
as ethyl cellulose, cross-linked polymers such as styrene-divinylbenzene
copolymer,
polysaccharides having hydroxyl groups such as dextran, cellulose derivatives
which are treated
with bifunctional crosslinking agents such as epichlorohydrin, dichlorohydrin
or 1, 2-, 3, 4-
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
diepoxybutane. The casing can also include starch and/or dextrin.
In some embodiments, an enteric coating materials are the commercially
available
EUDRAGITO enteric polymers such as EUDRAGITO L, EUDRAGIT Sand EUDRAGITO
NE, used alone or with a plasticizer. Such coatings are normally applied using
a liquid medium,
and the nature of the plasticizer depends upon whether the medium is aqueous
or non-aqueous.
Plasticizers for use with aqueous medium include propylene glycol, triethyl
citrate, acetyl triethyl
citrate or CITROFLEXO or CITROFLEXO A2. Non- aqueous plasticizers include
these, and
also diethyl and dibutyl phthalate and dibutyl sebacate. A preferred
plasticizer is Triethyl citrate.
The quantity of plasticizer included will be apparent to those skilled in the
art.
In some embodiments, a casing can also include an anti-tack agent such as
talc, silica or
glyceryl monostearate. In some embodiments, an anti-tack agent is glyceryl
monostearate.
Typically, the casing can include around 5 - 25 wt% plasticizer and up to
around 50 wt % of anti-
tack agent, preferably 1-10 wt% of anti-tack agent.
If desired, a surfactant can be included to aid with forming an aqueous
suspension of the
polymer. Many examples of possible surfactants are known to the person skilled
in the art.
Preferred examples of surfactants are polysorbate 80, polysorbate 20, or
sodium lauryl sulphate.
If present, a surfactant can form 0.1- 10% of the casing, preferably 0.2- 5%
and particularly
preferably 0.5- 2%.
In one embodiment, there is a seal coat included between the core and the
enteric coating.
A seal coat is a coating material which can be used to protect the enteric
casing from possible
chemical attack by any alkaline ingredients in the core. The seal coat can
also provide a
smoother surface, thereby allowing easier attachment of the enteric casing. A
person skilled in
the art would be aware of suitable coatings. Preferably the seal coat is made
of an OPADRYO
coating, and particularly preferably it is OPADRY White OY-S-28876.
In one embodiment, the pharmaceutically active ingredient is HbF-inducing
agents or a
salt thereof. In some embodiments, an example of an enteric-coated formulation
as described in
W02005/021002, comprises varying amounts of HbF-inducing agents. In that
example, lactose
monohydrate, microcrystalline cellulose, the active ingredient, the
hydroxypropyl methyl
cellulose and half of the croscarmellose sodium were screened into a 10 Litre
Fielder high-shear
blender (any suitable high shear blender could be used) and blended for 5
minutes at 300 rpm
with the chopper off. The mixture was then granulated by the addition of about
750 ml water
36
whilst continuing to blend. The granules were dried in a Glatt 3/5 fluid bed
drier, screened by
Comil into a PharmatecTM 5 Liter bin blender and then blended with any lactose
anhydrous given
in the formula plus the remainder of the croscarmellose sodium over 5 minutes
at 20 rpm.
Magnesium stearate was screened into the blender and the mixing process
continued for a further
1 minute at 10 rpm. The lubricated mix was compressed using a Riva Piccolla
rotary tablet press
fitted with 9.5mm round normal convex punches (any suitable tablet press could
be used). The
sealcoat, and subsequently the enteric coat, are applied by spraying of an
aqueous suspension of
the coat ingredients in a Manesty 10 coater using parameters for the coating
process as
recommended by the manufacturers of the coating polymers (again, any suitable
coater could be
used). Other enteric-coated preparations of this sort can be prepared by one
skilled in the art,
using these materials or their equivalents.
Other formulations and routes of administration
In alternative embodiments, the compositions as disclosed herein is by an
infusion pump
(to infuse, for example, the compositions as disclosed herein into the
subject's circulatory
system) is generally used intravenously, although subcutaneous, arterial, and
epidural infusions
are occasionally used. Injectable forms of administration are sometimes
preferred for maximal
effect. When long-term administration by injection is necessary, medi-ports,
in-dwelling
catheters, or automatic pumping mechanisms are also preferred, wherein direct
and immediate
access is provided to the arteries in and around the heart and other major
organs and organ
systems.
In some embodiments, compositions as disclosed herein comprising HbF-inducing
agents
and/or salts thereof can be administered to a specific site may be by
transdermal transfusion,
such as with a transdermal patch, by direct contact to the cells or tissue, if
accessible, such as a
skin tumor, or by administration to an internal site through an incision or
some other artificial
opening into the body. Alternatively, in some embodiments, compositions as
disclosed herein
comprising HbF-inducing agents and/or salts thereof can also be administered
to the nasal
passages as a spray. Diseases localized to the head and brain area are
treatable in this fashion, as
arteries of the nasal area provide a rapid and efficient access to the upper
areas of the head.
Sprays also provide immediate access to the pulmonary system and are the
preferable methods
for administering compositions to these areas. Access to the gastrointestinal
tract is gained using
oral, enema, or injectable forms of administration. For example,
administration of the
37
CA 2922013 2017-10-03
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
compositions as disclosed herein comprising HbF-inducing agents and/or salts
thereof to a
subject is preferably oral. As a result, the subject can undergo
administration of a composition
comprising HbF- inducing agents and/or salts at home.
As indicated above, orally active compositions comprising HbF-inducing agents
and/or
salts thereof are preferred for at least a portion of the cycle of therapy, as
oral administration is
usually the safest, most convenient, and economical mode of drug delivery.
Consequently,
compositions as disclosed herein comprising HbF-inducing agents and/or salts
thereof can be
modified to increase their oral bioavailable by reducing or eliminating their
polarity. This can
often be accomplished by formulating a composition with a complimentary
reagent that
neutralizes its polarity, or by modifying the compound with a neutralizing
chemical group. Oral
bioavailability is also a problem, because drugs are exposed to the extremes
of gastric pH and
gastric enzymes. Accordingly, problems associated with oral bioavailability
can be overcome by
modifying the molecular structure to be able to withstand very low pH
conditions and resist the
enzymes of the gastric mucosa such as by neutralizing an ionic group, by
covalently bonding an
ionic interaction, or by stabilizing or removing a disulfide bond or other
relatively labile bond.
In some embodiments, the compositions as disclosed herein comprising HbF-
inducing
agents and/or salts thereof can be used in combination with other agents to
maximize the effect
of the compositions administered in an additive or synergistic manner.
Accordingly,
compositions as disclosed herein comprising HbF-inducing agents and/or salts
thereof can also
comprise proteinaceous agents such as growth factors and/or cytokines. Such
proteinaceous
agents may also be aminated, glycosylated, acylated, neutralized,
phosphorylated, or otherwise
derivatized to form compositions that are more suitable for the method of
administration to the
patient or for increased stability during shipping or storage. Cytokines that
are useful to be
included in the compositions comprising HbF-inducing agents and/or salts
thereof include, but
are not limited to, granulocyte/macrophage colony stimulating factor (GM-CSF),
stem cell factor
(SCF erythropoietin (EPO), steel factor, activin, inhibin, the bone
morphogenic proteins (BMPs),
retinoic acid or retinoic acid derivatives such as retinol.
Compositions as disclosed herein comprising HbF-inducing agents and/or salts
thereof
can be physiologically stable at therapeutically effective concentrations.
Physiological stable
compounds of HbF-inducing agents or salts thereof not break down or otherwise
become
ineffective upon administration to a subject or prior to having a desired
effect. Compounds of
38
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
HbF-inducing agents that are structurally resistant to catabolism, and, thus,
physiologically
stable, or coupled by electrostatic or covalent bonds to specific reagents to
increase physiological
stability. Such reagents include amino acids such as arginine, glycine,
alanine, asparagine,
glutamine, histidine, or lysine, nucleic acids including nucleosides or
nucleotides, or substituents
.. such as carbohydrates, saccharides and polysaccharides, lipids, fatty
acids, proteins, or protein
fragments. Useful coupling partners include, for example, glycol, such as
polyethylene glycol,
glucose, glycerol, glycerin, and other related substances.
Physiological stability of a composition comprising an HbF-inducing agent
and/or salts
thereof can be measured from a number of parameters such as the half-life of
the an HbF-
inducing agent compound or salts thereof, or the half-life of active metabolic
products derived
from the an HbF-inducing agent compound or salts thereof. In some embodiments,
compositions comprising an HbF-inducing agent and/or salts thereof have in
vivo half-lives of
greater than about fifteen minutes, greater than about one hour, greater than
about two hours, and
greater than about four hours, eight hours, twelve hours, or longer. A
compound of an HbF-
inducing agent or its salts is stable using this criteria, however,
physiological stability can also be
measured by observing the duration of biological effects on the patient.
Clinical symptoms that
are important from the patient's perspective include a reduced frequency or
duration, or
elimination of the need for transfusions or chelation therapy. Preferably, a
stable composition
comprising an HbF-inducing agent and/or salts thereof has an in vivo half-life
of greater than
about 15 minutes, a serum half-life of greater than about 15 minutes, or a
biological effect which
continues for greater than 15 minutes after treatment has been terminated or
the serum level of
the compound has decreased by more than half. Preferably, compositions as
disclosed herein
comprising an HbF-inducing agent and/or salts thereof are also not
significantly biotransformed,
degraded, or excreted by catabolic processes associated with metabolism.
Although there may
be some biotransformation, degradation, or excretion, these functions are not
significant, if the
composition is able to exert its desired effect.
In some embodiments, compositions as disclosed herein comprising an HbF-
inducing
agent and/or salts thereof are also safe at effective dosages. Safe
compositions are compositions
that are not substantially toxic (e.g. cytotoxic or myelotoxic), or mutagenic
at required dosages,
do not cause adverse reactions or side effects, and are well-tolerated.
Although side effects may
occur, compositions are substantially safe if the benefits achieved from their
use outweigh
39
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
disadvantages that may be attributable to side effects. Unwanted side effects
include nausea,
vomiting, hepatic or renal damage or failure, hypersensitivity, allergic
reactions, cardiovascular
problems, gastrointestinal disturbances, seizures, and other central nervous
system difficulties,
fever, bleeding or hemorrhaging, serum abnormalities, and respiratory
difficulties. Preferably,
compositions useful for treating blood disorders preferably do not
substantially affect the
viability of a blood cell such as a normal mammalian blood cell. Normal cell
viability or the
viability of blood cell, e.g., hematopoietic cell can be determined from
analyzing the effects of
the composition on one or more biological processes of the blood or
hematopoietic cell. Useful
combination therapies will be understood and appreciated by those of skill in
the art. Potential
advantages of such combination therapies include the ability to use less of
each of the individual
active ingredients to minimize toxic side effects, synergistic improvements in
efficacy, improved
ease of administration or use, and/or reduced overall expense of compound
preparation or
formulation.
In some embodiments, the composition comprising an HbF-inducing agent and/or
salts
thereof can be administered to an adult, an adolescent, a child, a neonate, an
infant or in utero. In
some embodiments, a composition comprising an HbF-inducing agent and/or salts
thereof can be
administered to a subject via a continuous infusion throughout the cycle of
therapy.
Alternatively, a composition comprising an HbF-inducing agent and/or salts
thereof can be
administered to a the subject over a single span of a few to several hours per
day every day
throughout the first period of the cycle of therapy. Alternatively, in some
embodiments a
composition comprising an HbF-inducing agent and/or salts thereof can be
administered to a
subject in a single parenteral bolus, or orally, daily for several days
throughout the treatment
regimen or cycle, or weekly.
In some embodiments, a composition comprising an HbF-inducing agent and/or
salts
.. thereof can be prepared in solution as a dispersion, mixture, liquid,
spray, capsule, or as a dry
solid such as a powder or pill, as appropriate or desired. Solid forms may be
processed into
tablets or capsules or mixed or dissolved with a liquid such as water,
alcohol. saline or other salt
solutions, glycerol, saccharides or polysaccharide, oil, or a relatively inert
solid or liquid.
Liquids, pills, capsules or tablets administered orally may also include
flavoring agents to
increase palatability. Additionally, in some embodiments, a composition
comprising an HbF-
inducing agent and/or salts thereof can further comprise agents to increase
shelf-life, such as
CA 02922013 2016-02-19
WO 2015/026939
PCT/US2014/051887
preservatives, anti-oxidants, and other components necessary and suitable for
manufacture and
distribution of the composition. Compositions comprising an HbF-inducing agent
and/or salts
thereof can further comprise a pharmaceutically acceptable carrier or
excipient. Carriers are
chemical or multi-chemical compounds that do not significantly alter or affect
the active
ingredients of the compositions. Examples include water, alcohols such as
glycerol and
polyethylene glycol, glycerin, oils, salts such as sodium, potassium,
magnesium, and ammonium,
fatty acids, saccharides, or polysaccharides. Carriers may be single
substances or chemical or
physical combinations of these substances.
Administration Therapy
In some embodiments, a composition comprising an HbF-inducing agent and/or
salts
thereof can contain chemicals that are substantially non-toxic. Substantially
non-toxic means
that the composition, although possibly possessing some degree of toxicity, is
not harmful to the
long-term health of the patient. Although the active component of the
composition may not be
toxic at the required levels, there may also be problems associated with
administering the
necessary volume or amount of the final form of the composition to the
patient. For example, if
composition comprising an HbF-inducing agent contains a salt, although the
active ingredient
may be at a concentration that is safe and effective, there can be a harmful
build-up of sodium,
potassium, or another ion. With a reduced requirement for the composition or
at least the active
component of that composition, the likelihood of such problems can be reduced
or even
eliminated. Consequently, although patients may suffer minor or short term
detrimental side-
effects, the advantages of taking the composition outweigh the negative
consequences.
Doses of administration
The amount of an HbF-inducing agent that can be combined with a carrier
material to
produce a single dosage form will generally be that amount of the compound
that produces a
therapeutic effect. Generally out of one hundred percent, this amount will
range from about
0.01% to 99% of the compound, preferably from about 5% to about 70%, most
preferably from
10% to about 30%. The data obtained from the cell culture assays and animal
studies can be
used in formulating a range of dosage for use in humans. The dosage of such
compounds lies
preferably within a range of circulating concentrations that include the ED50
(the dose
.. therapeutically effective in 50% of the population) with little or no
toxicity. The dosage may
vary within this range depending upon the dosage form employed and the route
of administration
41
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
utilized.
The therapeutically effective dose of an HbF-inducing agent can be estimated
initially
from cell culture assays, for example, one can measure the percent increase in
mRNA gamma-
globin in the blood on administration, as disclosed herein. A dose may be
formulated in animal
models to achieve a circulating plasma concentration range that includes the
IC50 (i.e., the
concentration of the therapeutic which achieves a half-maximal inhibition of
symptoms) as
determined in cell culture. Levels in plasma may be measured, for example, by
high
performance liquid chromatography. The effects of any particular dosage can be
monitored by a
suitable bioassay.
The dosage of an HbF-inducing agent may be determined by a physician and
adjusted, as
necessary, to suit observed effects of the treatment. Generally, the
compositions are
administered so that an HbF-inducing agent or a prodrug thereof is given at a
dose from l jig/kg
to 150 mg/kg, 1 p g /kg to 100 mg/kg, 1 pg /kg to 50 mg/kg, 1 pg /kg to 20
mg/kg, 1 pg /kg to 10
mg/kg, 1 pg /kg to 1 mg/kg, 100 jig /kg to 100 mg/kg, 100 jig /kg to 50 mg/kg,
100 [tg /kg to 20
mg/kg, 100 p g /kg to 10 mg/kg, 100 p.g/kg to 1 mg/kg, 1 mg/kg to 100 mg/kg, 1
mg/kg to 50
mg/kg, 1 mg/kg to 20 mg/kg, 1 mg/kg to 10 mg/kg, 10 mg/kg to 100 mg/kg, 10
mg/kg to 50
mg/kg, or 10 mg/kg to 20 mg/kg. It is to be understood that ranges given here
include all
intermediate ranges. for example, the range 1 mg/kg to 10 mg/kg includes 1
mg/kg to 2 mg/kg, 1
mg/kg to 3 mg/kg, 1 mg/kg to 4 mg/kg, 1 mg/kg to 5 mg/kg, 1 m2/kg to 6 mg/kg,
1 mg/kg to 7
mg/kg, 1 mg/kg to 8 mg/kg, 1 mg/kg to 9 mg/kg, 2 mg/kg to 10 mg/kg, 3 mg/kg to
10 mg/kg, 4
mg/kg to 10 mg/kg, 5 mg/kg to 10 mg/kg, 6 mg/kg to 10 mg/kg, 7 mg/kg to 10
mg/kg, 8 mg/kg
to 10 mg/kg, 9 mg/kg to 10 mg/kg, and the like. It is to be further understood
that the ranges
intermediate to the given above are also within the scope of this invention,
for example, in the
range 1 mg/kg to 10 mg/kg, dose ranges such as 2 mg/kg to 8 mg/kg, 3 mg/kg to
7 mg/kg, 4
mg/kg to 6 mg/kg, and the like.
In some embodiments, the compositions comprising an HbF-inducing agent are
administered at a dosage so that an HbF-inducing agent or a metabolite thereof
has an in vivo,
e.g., serum or blood, concentration of less than 500 nM, less than 400 nM,
less than 300 nM, less
than 250 nM, less than 200 nM, less than 150 nM, less than 100 nM, less than
50 nM, less than
25 nM, less than 20, nM, less than 10 nM, less than 5 nM, less than 1 nM, less
than 0.5 nM, less
than 0.1 nM, less than 0.05 nM, less than 0.01 nM, less than 0.005 nM, or less
than 0.001 nM
42
after 15 mins, 30 mills, 1 hr, 1.5 hrs, 2 hrs, 2.5 hrs, 3 hrs, 4 hrs, 5 hrs, 6
hrs, 7 hrs, 8 hrs, 9 hrs, 10
hrs, 11 hrs, 12 hrs or more of time of administration. In a preferred
embodiment the blood
concentration is at least 200nM after 15 minutes or more.
With respect to duration and frequency of treatment, it is typical for skilled
clinicians to
monitor subjects in order to determine when the treatment is providing
therapeutic benefit, and to
deteimine whether to increase or decrease dosage, increase or decrease
administration frequency,
discontinue treatment, resume treatment or make other alteration to treatment
regimen. The
dosing schedule can vary from once a week to daily depending on a number of
clinical factors,
such as the subject's sensitivity to an HbF-inducing agent acid. The desired
dose can be
administered every day or every third, fourth, fifth, or sixth day. The
desired dose can be
administered at one time or divided into subdoses, e.g., 2-4 subdoses and
administered over a
period of time, e.g., at appropriate intervals through the day or other
appropriate schedule. Such
sub-doses can be administered as unit dosage forms. In some embodiments of the
aspects
described herein, administration is chronic, e.g., one or more doses daily
over a period of weeks
or months. Examples of dosing schedules are administration daily, twice daily,
three times daily
or four or more times daily over a period of 1 week, 2 weeks, 3 weeks, 4
weeks, 1 month, 2
months, 3 months, 4 months, 5 months, or 6 months or more.
An HbF-inducing agent or a prodrug thereof can be administrated to a subject
in
combination with one or more pharmaceutically active agents. Exemplary
pharmaceutically
active compound include, but are not limited to, those found in Harrison's
Principles of Internal
Medicine, 13th Edition, Eds. T.R. Harrison et al. McGraw-Hill N.Y., NY;
Physician's Desk
Reference, 50th Edition, 1997, Oradell New Jersey, Medical Economics Co.;
Pharmacological
Basis of Therapeutics, 8th Edition, Goodman and Gilman, 1990; United States
Pharmacopeia,
The National Formulary, USP XII NF XVII, 1990; current edition of Goodman and
Gilman's
The Pharmacological Basis of Therapeutics; and current edition of The Merck
Index.
The following examples illustrate embodiments of the invention, but should not
be
viewed as limiting the scope of the invention.
Examples
Dual luciferase reporter assay
A counter-screening assay to determine the gamma-globin-specificity of hits,
using an
assay which measures gamma-globin gene promoter induction relative to beta-
globin gene
43
CA 2922013 2017-10-12
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
promoter induction. This assay is fluorescence-based, and consists of a dual-
luciferase reporter
construct containing the LCR and beta-globin promoter linked to renilla
luciferase and the A-
gamma-gl obin promoter linked to firefly luciferase ( LCR-beta-pr-Rluc-Agamma-
pr-Fluc
cassette), stably-transfected into GM979 cells.
The orientation of the promoters and uLCR allowed detection only of strong,
specific
inducers of the gamma-globin gene promoter.
Erythroid progenitor cultures
Fetal globin-inducing activity in human erythroid progenitors was assessed.
Human
erythroid progenitors were cultured from peripheral blood samples, exposed to
the test
compounds at varying concentrations and for varying durations and analyzed for
globin chain
mRNA ratios. Erythroid progenitors were cultured from cord blood CD34+ cells.
Briefly,
CD34+ cells in the human cord blood were separated using a Ficoll-paque
density gradient.
CD34+ cells were cultured in H4230 medium containing 2mM L-glutamine, 1%
Methylcellulose
in Iscove's Medium, 30% Fetal Bovine Serum, 1% Bovine Serum Albumin and 10-4 M
beta-
mercaptoethanol. Methylcellulose H4230 medium was supplemented with EPO (0.5
U/ml) and
IL-3 (20 ng/ml) to support BFU-e growth. Cells were cultured in 35 x 100mm
mini-dishes and
incubated in a humidified atmosphere containing 5% CO2, at 37 C. Different
concentrations of
the test compounds were added at the time the cultures were established. Each
compound was
tested in three different cultures. BFU-e colonies grown in mini-dishes were
counted on day 14
and harvested for mRNA analysis.
mRNA analysis by real-time PCR
On day 14. RNA was extracted from cultured erythroid cells, and relative
quantification
PCR was performed. Briefly, cDNA was generated from equal amounts of total RNA
extracted
using The PerfectPure RNA Purification Kit (5 Prime Inc Gaithersburg, MD).
Real-time PCR
was performed using an ABI 7500 Real-Time PCR system (Applied Biosystems,
Foster City,
CA). Levels of globin mRNA were calculated by the delta-delta Ct method.
Isolated total RNA
was used as a template for cDNA synthesis and real-time PCR was performed
using appropriate
primer sets. GAPDH levels were used for standardization. Western blotting
nuclear extract of
K562 cells and 14-day-old BFU-e were analyzed by electrophoresis using 5-24%
gradient SDS-
.. polyacrylamide mini-gels (BIORAD Laboratories. Hercules CA). Proteins in
the gels were
transferred to Immobilon-P membranes. Blots were then incubated with BCL-11A
polyclonal
44
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
(Novus Biologicals, Littleton Co) or mouse monoclonal (Santa Cruz
Biotechnology, Santa Cruz
CA) antibodies, after washing blots were incubated with anti-rabbit IgG-
horseradish peroxidase
secondary antibody (ECL, Little Chalfont Buckinghamshire UK). BCL-11A bands
were detected
on the X-ray film using Western Lightning Reagents (Perking Elmer Inc.,
Waltham, MA).
Studies in non-human primates
Studies to evaluate pharmacokinetic properties and gamma-globin induction were
performed in juvenile baboons (Papio hamadryas anubis). Briefly, animals were
chronically
phlebotomized on a daily basis to achieve stable anemia, maintaining a total
hemoglobin level of
7.0 to 7.5 g/dl. Candidate compounds were administered intravenously or orally
once daily in
single doses for pharmacokinetic studies, or once daily in single doses, 4-5
days per week for 4-5
weeks, for pharmacodynamic studies. The compound Desloratadine was
administered
intravenously to a baboon at doses of 50 or 200 mg/kg once daily, 5 days per
week for 4 weeks,
to evaluate gamma-globin gene expression. MS-275 was administered
intravenously at a dose of
10 mg/kg once daily. 4-5 days per week for four weeks, to assess gamma-globin
gene expression
in baboon 5002. Levels of gamma-globin mRNA expression and globin chain
synthesis were
assessed in baboons before and during treatment with test compounds. A washout
period
between administrations of different compounds in the same baboon was
provided. MS-275 and
desloratidine both induced fetal globin mRNA and total hemoglobin levels
increased following
MS-275 administration.
EXAMPLE 1: Benserazide is identified as a novel HbF inducing agent using a
High throughput
Screening Assay (HTS). Benserazide was discovered in a novel high-throughput
screening
(HTS) program which interrogated 3 chemical libraries of compounds and
included a library of
medicinal products which are already approved in USA or in the European Union
(by the
European Medicines Agency) for other medical indications. Prior to the HTS,
the potential of
Benserazide to induce HbF was unknown. Using a gamma-globin gene promoter
linked to GFP
(Figure 1), the HTS assay was adapted from low throughput to a robotic high-
throughput
screening system, and screening was performed over a 3¨log concentration
range, on the diverse
chemical libraries. The screening assay was developed from a cell-based
reporter, stably
transfected with a construct containing the 1.4-kilobase (kb) KpnI-BglII
fragment of the human
locus control region (LCR) linked to the gamma-globin gene promoter and the
enhanced green
fluorescent protein (EGFP) reporter gene (Figure 1B). Because EGFP messenger
RNA (mRNA)
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
is very stable, positive changes averaged 1.2- to 2-fold, and weak inducers
are not detectable in
this system. Two-fold or higher induction over control indicates strong
inducers of gamma-
globin gene activity. The HTS assay was developed in 96-well format on a Tecan
SpectraFluor
Plus, incorporating multiple positive and negative control wells in each
plate. Optimal
.. fluorescence measurements were identified. Analysis of two sets of 10,000
compounds was
performed, testing over a 3-log range of concentrations. From the library of
compounds and US
and EMA approved medicinal products tested, a small panel of approved
therapeutics were
found to induce y-globin expression (Figure 1B). The activity of the potential
candidate
compounds identified using the HTS was validated in a secondary assay using
dual luciferases
linked to the fetal and adult globin promoters. The secondary assay confirmed
that Benserazide
induces only the gamma globin promoter and not general globin promoters. The
racemate form
of Benserazide was selected for further analysis and was referred to as PB-04.
EXAMPLE 2: Benserazide induces a mean 42-fold induction of Fetal globin (gamma-
globin)
mRNA in erythroid progenitors from sickle cell and beta thalassemia patients.
Erythroid
progenitors were isolated from cord blood and from human patients with beta
thalassaemia or
sickle cell disease and CD34+ cells were enriched. CD34+ erythroid progenitors
were cultured
for a first phase to eliminate white cells and then in a second phase to
expand and differentiate
the erythroid cells in the presence of erythropoietin. This method produces
95% erythroid cells.
mRNA was extracted after 12 days and real-time RT-PCR was performed for
expression levels
of gamma-globin mRNA. Briefly, cDNA was generated from equal amounts of total
RNA
extracted using the PerfectPure RNA purification kit (5 Prime Inc.
Gaithersburg, MD) or RNA
STAT-60 isolation reagent (Teltest, Friendswood TX). Real-time PCR was
performed using an
ABI 7500 Real-Time PCR system (Applied Biosystems, Foster City, CA). Levels of
gamma-
globin mRNA were calculated by the AACt method. GAPDH and 18S levels were used
for
standardization. Benserazide caused a mean 42-fold increase in fetal globin
(gamma-globin)
mRNA in human erythroid progenitors (Figure 2), which is significantly higher
than the
magnitude of induction by other known fetal globin inducing agents, which only
caused a 2-6.5
fold increase in gamma-globin mRNA.
EXAMPLE 3: In vivo results: Anemic Baboon Study. Benserazide and Desloratidine
(DLT)
induce a 33-fold and 11-fold increase in fetal Globin Induction respectively,
and increase
erythroid cell production in anemic baboons. The effect of Benserazide to
induce fetal globin
46
was assessed in vivo in a baboon model of anemia. This baboon model (Pace et
al., 2002.
Short-chain fatty acid derivatives induce fetal globin expression and
erythropoiesis in vivo.
Blood. 100(13): 4640-4648) requires that anemia be present for induction of
gamma-globin to be
detected. One juvenile baboon (Papio hamadryas anubis) was chronically
phlebotomized (by 3.7
to 5 mL/kg/day) to achieve and maintain stable anemia with total hemoglobin of
7.0 to 7.5 g/dl, a
level necessary for the modulation of globin genes in this species. The
animals were
supplemented with iron dextran and folic acid to support active
erythropoiesis. The phlebotomy
regimen effectively exchanged the animals' total blood volume every 10 to 20
days. Assays of
gamma-globin mRNA expression were performed with the approval of the
corresponding animal
research institutions. The drug candidate was administered based on doses
adjusted for more
rapid metabolism in the baboon, but projected as equivalent to doses
previously used or studied
in humans as follows: Benserazide was administered orally, at 1 mg/kg for 4
days/week and 2
mg/kg for two weeks. A washout period was provided between administrations of
different
compounds in the same baboon. Levels of gamma-globin mRNA expression, total
hemoglobin,
and % F-cells were assessed before and during treatment with test compounds.
Benserazide was
compared to 2 other candidates (i) desloratadine (DLT), which was administered
orally (0.5
mg/kg/dose), three times a week over two weeks, and (ii) MS-275, which was
administered
orally three times a week for two weeks at 0.2 mg/kg/dose. Doses of
Benserazide are shown in
the bars above the graph, with Benscrazide administered at 1 mg/kg (Open
squares) and 2 mg/kg
(dark squares). The time course is consistent with the time required for new
red blood cells to
develop and enter the circulation. A lag follows each time-off the drug. Gamma
globin mRNA
was analyzed by RT-PCR prior to drug administration, 3 times/week during and
following drug
administration, and change from baseline was assessed. Changes in gamma-globin
mRNA is
shown in Figure 3A. Benserazide resulted in up to 33-fold induction of gamma-
globin mRNA in
vivo. Benscrazide also caused an increase in total hemoglobin (Hb) (by
1.5g/dL) (Figure 3B)
despite the daily phlebotomy, indicating that Benserazide has an independent
effect on
erythropoiesis, which is also beneficial in the treatment of patients with
beta-thalassemia and
sickle cell disease. Furthermore, both Benserazide (BEN) and Desloratidine
(DLT) resulted in a
mean increase in fetal globin mRNA in vivo in the Anaemic Baboon model, with
Benserazide
inducing a mean 27-fold increase, and Desloratidine (DLT) inducing a 11-fold
increase above
baseline (Figure 4), both of which arc significantly higher than other known
inducers of fetal
47
CA 2922013 2017-10-12
globin (Hydroxyurea (HU) and ST20 (dimethylbutyrate)), which induced only a
1.5-3.5-fold
increase of fetal globin mRNA above the baseline.
EXAMPLE 4: In vivo results: Anemic Baboon Study. Benserazide results in a 15%
increase the
number of F-retieulocytes in Anemic Baboons. F-reticulocytes of Benserazide
treated anemic
baboons were analyzed using flow cytometry. The peripheral blood of the
baboons was analyzed
using Flow cytometry to detect HbF protein and proportions of cells expressing
fetal globin
protein as previously described (Pace et al., 2002. Short-chain fatty acid
derivatives induce fetal
globin expression and erythropoiesis in vivo. Blood. 100(13): 4640-4648).
Briefly, cells were
washed with PBS containing 0.1% BSA, and fixed. After washing twice, the cells
were
permeabilized in PBS with 0.1% Triton-X100 and incubated. Cells were thcn
washed,
resuspended in PBS containing 0.1 % BSA. PerCP isotype labeled and unstained
cells were used
as controls. Thiazole orange was used to determine proportions and populations
of reticulocytes.
A custom-synthesized PerCP mouse anti-human antibody that detects baboon F-
cells was used to
label fetal globin-containing cells. Samples were incubated in the dark at
room temperature for
30 min, washed several times with PBS containing 0.1 % BSA and analyzed by
flow cytometry
on a FACScalibur (Becton Dickinson, Franklin Lakes, NJ). Figure 5 shows that
Benserazide
increases F-reticulocytes by 15% following the 2 mg/kg dose. The time course
of response is
consistent with the time required for new red blood cells to mature and enter
the circulation.
EXAMPLE 5: In vivo results: Transgenic Murine Study. Benserazide induces human
-y-globin
gene expression in a mouse model. The effect of Benserazide was compared to a
known Hb
inducer (Hydoxyurea (HU) in a transgenic mice model comprising the entire
human non-alpha
(gamma delta beta-globin) gene locus. Mice transgenic for the human beta-
globin gene locus
including the locus control region (LCR) were previously described (Pace ct
al., 2002.
Short-chain fatty acid derivatives induce fetal globin expression and
erythropoiesis in vivo.
Blood. 100(13): 4640-4648). Mice were treated with either (i) hydroxyurea (HU)
- administered
at 100 mg/kg/dose once daily for 5 days/week, or (ii) Benserazide -
administered at 2
mg/kg,/dose, 3 times per week for 5 weeks or (iii) water in the same volume
(100 microliters) as
the drug candidates as a negative control. Dosing was performed by
intraperitoneal injection (i.p)
to ensure consistent drug delivery. Blood was sampled for globin mRNA and for
F-cell
quantitation and fluorescent intensity of F-cells by flow cytometry. Total
hemoglobin (Hb) was
assayed on a Horiba ABX60 at baseline, 2 and 5 weeks (sampling was limited due
to constraints
48
CA 2922013 2017-10-12
on amount of blood that can be withdrawn safely from mice). Benserazide
induced a mean
increase in total hemoglobin (Hb) at 5 weeks, which was significantly better
as compared to mice
administered hydroxyurea (HI]), (the only FDA approved drug for fetal globin
induction)
(Figure 6A). Importantly, Benserazide induced an
10
48a
CA 2922013 2017-10-12
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
absolute 13% mean increase in the number of F-cells (from 0.1% to 9% (mice
#1), 0.4% to 18%
(mice #2), and 0.13% to 12% (mice #3); and 10 to 33-fold increase in mean
fluorescence
intensity (MFI) (Figure 6B). Responses were observed within one week, and
continually
increased in the Benserazide treated mice, whereas the percentage of F-cells
declined in the
Hydroxyurea-treated mice.
Figure 9 depicts a comparison of the magnitude of fetal globin mRNA induction
over
baseline levels in anemic baboons by new drugs compared to hydroxyurea (HU)
(Figure 9A),
and proportions of new red blood cells with fetal globin protein, F-
reticulocytes, (Figure 9B).
Reponses are significantly higher than those induced by the only US and EMA-
approved drug,
hydroxyurea. F-reticulocytes are 3 to 5 times higher with the new drugs than
induced by
hydroxyurea.
Figures 10A-F depict Benserazide reduction of binding of two repressors, LSD1
and
HDAC3, to the fetal globin gene promoter in erythroid progenitors from sources
as indicated.
Figure 11 depicts the suppression of repressor proteins BCLI1A and KLF1 in
different
erythroid progenitors treated with new drugs as compared with untreated
control levels by
Western blot analysis.
Prior Experience in Development of Fetal Globin-Inducers and Selection of
Higher Potency
Candidates
Three lead drug candidates are expected to have higher clinical activity than
prior-
generation HbF-inducers. This expectation is based on their activity in
multiple assays shown to
be clinically predictive for prior-generation drugs. The first-generation
inducer, Arginine
Butyrate, induced beta-globin expression 3-fold in sickle cell patients from a
mean baseline HbF
of 7% to mean 21% and significantly increased HbF and total Hb levels (by a
mean 2.8 g/d1) in
subjects with beta-thalassemia and rendered some beta-thalassemia patients
transfusion-
independent for greater than 7 years. However, arginine butyrate (AB) is
rapidly-metabolized,
requires intravenous (IV) infusions which are difficult long-term, and is also
a broad HDAC
inhibitor, suppressing cell proliferation. Experience with this therapy
demonstrated the
importance of preserving erythroid cell proliferation for optimal HbF
induction. This
fundamental principle is important for successful application of any fetal
globin induction
therapy. Next-generations of oral derivatives have rapidly induced HbF in
short Phase I trials,
49
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
(compared to the 6 months required for HU effects); SDMB increased HbF by a
mean of 9%
(range 5-21%) in beta-thalassemia within 8 weeks. Yet, higher potency inducers
are likely
needed for the most severe patients. Inducers of fetal globin structurally-
unrelated compounds
which actively induce the gamma-globin genes in vitro and in vivo. From a
library of 13,000
structurally unrelated compounds tested against a putative receptor site, 2
candidates, RB16 and
RB7 have 5- to 7-fold higher potency and favorable PK profiles in primates.
RB7 is particularly
novel, in that it displaces a repressor complex containing HDAC3 and recruits
EKLF to the
promoter. Yet all such new chemical entities must pass daunting, lengthy, and
costly toxicology
and manufacturing hurdles, and may have unknown safety issues.
The system utilizes a stably-transfected construct containing the human LCR
linked to
the gamma-globin gene promoter and the enhanced green fluorescent protein
(EGFP) reporter
gene, and was adapted from low-throughput to high-throughput application. The
HTS assay also
allowed an estimate of the cytotoxicity of the tested compounds. Because EGFP
mRNA has long
stability, positive changes average 1.2- to 2-fold; weak inducers are not
detectable in this system;
and 2-fold or higher induction indicates very strong inducers of beta-globin
promoter activity.
Two campaigns screening 10,000 compounds were performed. From the "FDA-
approved
compound" library, a number of hits were identified, of which a few showed
higher potency than
butyrates in secondary, confirmatory assays. Several were eliminated as
hemoglobinopathy
therapeutics because of cytotoxicity (e.g., Idarubicin) or the need for
parenteral administration,
(but these were nonetheless validated in confirmatory assays as potent HbF-
inducers). Follow-on
confirmatory assays included a dual-luciferase reporter assay that analyzes
specificity by
comparative induction of the gamma- vs beta-globin promoters, globin mRNA
analyses by RT-
PCR, and initial erythroid cultures from normal subjects with low baseline HbF
Bfu-e; these
published assays are validated for their correlation with subsequent clinical
activity in humans,
with induction in Bfu-e having greater correlation than reporter assays.
Significantly, the HTS assay identified 10 drugs, already FDA-approved by the
U.S. or
Canadian drug authorities for other indications (and confirmed in secondary
assays) as potent
inducers of gamma-globin gene expression (active at 2-3 logs lower
concentration than butyrate,
and generating higher induction of gamma-globin mRNA in human erythroid
progenitors). These
drugs hits are 3-8 times more potent than an oral SCFAD HbF inducer currently
in clinical trials
(SDMB). Three drugs were selected with high potency in specifically inducing
the gamma-
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
globin promoter. One (MS-275) also suppresses a repressor of gamma-globin
transcription,
BCL-11A. Two of the 3 are approved in young children, which is important as
organ damage in
these diseases begins in childhood.
One issue which confounds prediction of clinical efficacy in the beta-globin
disease
population, however, is patient variability in HbF genetic modifier profiles
and resulting baseline
HbF levels. Such variables can only be studied in erythroid cells cultured
from beta-
hemoglobinopathy patients. Analysis of the effects of the 3 lead drug
candidates in genotyped
hemoglobinopathy patient erythroid progenitor cultures are used for final
selection of the lead
compound for clinical development. This selected therapeutic can then
immediately be evaluated
in Phase 2 clinical trials in patients with b-globin diseases, without need
for the costly (and "high
risk") preclinical toxicology, mutagenicity, and manufacturing development
necessary for a new
chemical entity.
The targeted therapeutic action is induction of high-level expression of fetal
globin,
without concomitant erythroid cell cytostasis or cytotoxicity. Two clinically-
approved
therapeutics (desloratidine and benserazide) and a third extensively
clinically-studied
therapeutic, MS-275, were selected for evaluation in patients' erythroid
progenitors. The agents
have shown 5-fold gamma-globin induction in erythroid cells cultured from
normal subjects,
compared to untreated controls from the same subjects, and each had greater
effects than induced
by HU or butyrate. Because hemoglobinopathy patients (as opposed to cultures
from normal
subjects) have a wide range of basal HbF levels which may affect the magnitude
of responses to
therapeutics. These agents augment HbF expression to the greatest degree in
erythroid
progenitors cultured from patients with lower baseline HbF levels.
The 3 preferred oral therapeutics include:
= Desloratidine (DLT): A drug requiring prescription for treatment of
allergic symptoms,
with a benign safety profile in extended use. In baboon studies, this agent
induced HbF within 4
days of administration to a higher degree than almost all other test agents
within the same
animal..
= Benserazide (BEN): Approved in a combination formulation to enhance the
PK profile
of L-dopa, and reported to have no clinical effects itself. No adverse effects
of this therapeutic
or drug interactions have been identified. A proprietary extended-release
formulation is
developed. This drug has never been approved for any use except in combination
with other
51
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
drugs.
MS-275, an oral. class I-specific HDAC inhibitor. Its primary adverse effect
is fatigue,
but it is administered infrequently, once every 2 weeks, and is therefore
worthwhile to evaluate.
It does not have adverse cardiac side effects of many pan-HDAC inhibitors. MS-
275 suppresses
expression of BCL-11A, a transcriptional repressor which down-regulates beta-
globin
transcriptional activity. As (pan-)HDAC inhibitors inhibit erythroid cell
proliferation, MS-275
would be given in an intermittent or pulsed regimen in patients. Of the 3
leading candidates,
BEN is a preferred pharmaceutical agent as it has slight pro- erythropoietic
activity. DLT is
another preferred agent as stable GMP formulations for extended release are
available.
Erythroid progenitor cultures
Three drugs/compounds are investigated for their efficacy in up-regulating HbF
synthesis
in erythroid progenitor culture experiments (Figure 2). Progenitor cultures in
patients were
predictive of subsequent HbF response rates in patients to prior-generation
inducers which have
been less effective in the baboon than the newly discovered agents. Peripheral
blood is
collected from (de-identified) sickle cell patients given a unique patient
identifier number
(UPIN), and in whom alpha- and beta-globin genotypes, and the status of the 3
genetic modifiers
(QTL) are documented. Erythroid progenitors are cultured +/- the 3 candidate
drugs and vehicle
controls, and +/- HU, at the optimal concentrations established. Two drugs
which have shown
activity in clinical trials (Arginine butyrate and hydroxyurea) are used as
positive controls for
comparison with the candidate drugs. The gamma-globin induction assays are
performed on
erythroid progenitors cultured from 3 groups of at least 15 genotyped sickle
cell patients,
stratified for baseline HbF in 3 ranges: low (less than 2%), average HbF (3-
8%), and the typical
range in HU-responsive adults (8-11%). HbF levels are obtained on the
submitted de-identified
samples by HPLC. Approximately 8 samples with baseline HbF levels greater than
15% may
also be analyzed. Analyses is performed as previously described on cultured
progenitors
(Boosalis. 2001; 2011) for: (i) gamma-globin mRNA, by quantitative RT-PCR;
(ii) F-cells by
flow cytometry; (iii) HbF by reverse-phase HPLC; and (iv) erythroid colony
size (mean
cells/colony) and Bfu-e number/ per CD34 or mononuclear cells cultured.
The magnitude of positive changes in these parameters, in absolute terms and
as a percent
of baseline, and the proportion of subjects' cultures in which a positive
change occurs, are
compared to the subjects' untreated control progenitors, and +/- addition of
HU. Descriptive
52
CA 02922013 2016-02-19
WO 2015/026939 PCT/US2014/051887
statistics are used to assess differences between treated and untreated
control progenitors from
the same subjects. 1) highest magnitude of HbF induction above baseline in low
and moderate
basal HbF patient groups; and, 2) the highest proportion of positive
responses, defined as at least
a 5% absolute increase compared to untreated control. Paired-tests and
Fisher's exact test
(GraphPad Prism V software) are used to determine if significant differences
are produced in%
F-cells, HbF, and gamma-globin mRNA parameters, compared to levels in the same
subjects'
vehicle-treated control cultures. Whether addition of HU with test drugs
results in higher
induction compared to cultures treated with HU alone, or with test drugs
alone, are statistically
evaluated; t-tests and Wilcoxon tests for paired differences and other tests
as indicated are used.
Positive changes in greater than 35% of cultures are based on positive changes
in 75% of
progenitors cultured from normal subjects; however, the magnitude of change in
hemoglobinopathy patients' progenitors is much more likely significant. All
drug candidates
should increase HbF expression in greater than or equal to 60% of subjects'
colonies, based on
response rates in cells cultured from normal subjects. As growth of erythroid
cells is inhibited
by HU, and by HDAC inhibitors to a lesser degree, HU (positive control) are
utilized at low
nanomolar concentrations. If colony growth is severely suppressed by HU, it
may be added
transiently. Selection is based on the agent that produces the greatest
magnitude of HbF
induction compared to untreated controls in cultures from the low and moderate
baseline HbF
levels. If there is no significant difference between the magnitude of HbF
responses, or the
response rates with the different drugs, selection of the clinical candidate
are based on effects on
erythroid proliferation and side-effect profile (favoring DLT or Benserazide),
and the most rapid
regulatory path (DLT).
Formulation of the drug Desloratidine may include higher dose requirements
than
required for allergy treatment. Benserazide may include extended-release
preparations based on
its published pharmacokinetics. Benserazide is currently formulated
commercially in
combination with L-dopa. Because of its solubility profile, Desloratidine is
prepared in a
proprietary medicinal formulation as a solution or tablet. Validated assays
for the analytical work
are established for the required formulation development for all 3.
Formulation developments
include pH solubility and pH stability profiles; selection of buffer and
buffer strength; selection
of formulation type-solution, tablet, immediate or extended release; selection
of co-solvents and
complexing agents, (e.g., for extended release, Benserazide); selection of
preservatives based on
53
pH; selection of sweeteners and flavoring agents (from a panel of 15 taste-
masking agents);
composition of final formulation (active pharmaceutical ingredient [API],
excipients,
preservatives); dissolution of any extended release formulation; establishment
of specifications
for future batches, including sterility testing; accelerated stability study.
Other embodiments and uses of the invention will be apparent to those skilled
in the art
from consideration of the specification and practice of the invention
disclosed herein. The term
comprising, where ever used, is intended to include the terms consisting and
consisting
essentially of Furthermore, the terms comprising, including, containing and
the like are not
intended to be limiting. It is intended that the specification and examples be
considered
exemplary only with the true scope and spirit of the invention indicated by
the following claims.
54
CA 2922013 2017-10-03