Note: Descriptions are shown in the official language in which they were submitted.
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
COMPOSITIONS AND METHODS FOR THE TREATMENT OF
METABOLIC AND BODY WEIGHT RELATED DISORDERS
Cross-Reference to Related Applications
This international application claims benefit of priority under 35 U.S.C. 120
of pending
application U.S. Serial No. 14/270,130, filed May 5, 2014, which claims
benefit of priority under 35
U.S.C. 120 of pending application U.S. Serial No. 14/052,074, filed October
11,2013, which claims
benefit of priority under 35 U.S.C. 120 of pending application U.S. Serial
No. 14/013,918, filed
August 29, 2013, the entirety of all of which are hereby incorporated by
reference.
Federal Funding Legend
The present invention utilized federal funding from the National Institutes of
Health Grant
GM-63115 and Department of Defense Grant No. DAMD17-03-1-0228. The United
States
Government has certain rights in the invention.
BACKGROUND OF THE INVENTION
Field of the Invention
The present invention generally relates to the fields of medicine and
molecular biology of
metabolic disorders. In
particular aspects, the field of the invention relates to particular
compositions for the treatment of disorders, such as obesity partly due to
upregulation of
thermogenesis. In certain aspects, the compositions comprise fatostatin A and
its analogs or
derivatives.
Description of the Related Art
Metabolic syndrome covers many cardiovascular risk factors including
hypertension,
dyslipidaemia, obesity, type 2 diabetes, pancreatic [3¨cell dysfunction, and
atherosclerosis. A diet
varying in fat or carbohydrate contents contributes to energy metabolism of
animals including
humans. Long chain fatty acids are major source of energy and important
components of the lipids
that comprise the cellular membranes. They are derived from food and
synthesized de novo from
acetyl-CoA. Cholesterol is also derived from food and synthesized from acetyl-
CoA. The conversion
of carbohydrates into acylglycerides through de novo fatty acid and
cholesterol synthesis involves at
least 12 and 23 enzymatic reactions, respectively. Expression levels of the
genes encoding these
enzymes are controlled by three transcription factors, designated sterol
regulatory element-binding
proteins (SREBPs), SREBP-la, -1c and SREBP-2. These membrane-bound proteins
are members
of a class of the basic helix-loop-helix leucin zipper family of transcription
factors. Unlike other leucin
zipper members of transcription factors, SREBPs are synthesized as an ER-
membrane-bound
precursor, which needs to be proteolytically released by two proteases bound
to the Golgi
membrane, Site-1 and Site-2 proteases, in order to activate transcription of
target genes in the
nucleus.
1
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
The proteolytic activation of SREBPs is tightly regulated by sterols through
the interaction
with SREBP cleavage-activating protein (SCAP), an ER-membrane-bound escort
protein of
SREBPs. When sterols accumulate in the ER membranes, the SCAP/SREBP complex
fails to exit
the ER to the Golgi, and thereby the proteolytic processing of SREBPs is
suppressed. SREBPs are
key lipogenic transcription factors that govern the homeostasis of fat
metabolism.
The prior art is deficient in the novel compositions and methods useful for
the treatment of a
variety of metabolic disorders. The present invention fulfills this
longstanding need and desire in the
art.
SUMMARY OF THE INVENTION
The present invention teaches compounds, formulations and pharmaceutical
compositions
thereof and uses thereof for treating a metabolic disorder, such as, but not
limited to, a disease
related to cell hyperproliferation, e.g., a cancer, or a weight-related
condition.
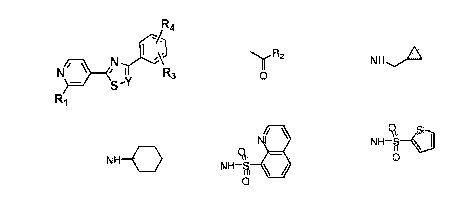
The present invention is directed to a compound having the general structure:
A-B-C
The A ring is a pyridine or a substituted pyridine, a piperidine or a
substituted piperidine, a
pyrrolidine or a substituted pyrrolidine, a thiazole or a substituted
thiazole, a phenyl ring or a
substituted phenyl ring. The B ring is a thioazole or a substituted thioazole,
a piperazine or a
substituted piperazine, a phenyl ring or a substituted phenyl ring. The C ring
is a phenyl ring or a
substituted phenyl ring, a pyridine or a substituted pyridine, a thioazole or
a substituted thioazole.
The present invention also is directed to a compound having the chemical
structure
S \
R2-N\
The R1 substituents are H, halogen, -OH, -0-C1-3alkoxy, -0C(0)R3; R3 is C1-C3
alkyl or aryl, -OCH2-
C(0)0R4; R4 is H or Ci-C3 alkyl, -NHR5; R5 is H, C1-C4 alkyl,
alkylcyclopropane, benzyl, -NHC(0)C--
C3 amide, -NHC(0)0-R6 carbamate; R6 is tert-butyl or benzyl, ¨NH-S02-R7
sulfonamide and R7 is
alkyl or aryl. The R2 substituents may be alkyl or R50C(0)- and Rg is C3-
05alkyl or aryl.
The present invention is directed further still to a compound having the
chemical structure
R2
jJRi
The R1 substituents are H, halogen, -OH, -0-C1-3alkoxy, -0C(0)R3; R3 is C1-C3
alkyl or aryl, -OCH2-
C(0)0R4; R4 is H or Ci-C3 alkyl, -NHR5; R5 is H, C1-C4 alkyl,
alkylcyclopropane, benzyl, -NHC(0)C--
C3 amide, -NHC(0)0-R6 carbamate; R6 is tert-butyl or benzyl, ¨NH-S02-R7
sulfonamide and R7 is
alkyl or aryl. The R2 substituents may be alkyl or R50C(0)- and R3 is C3-
05alkyl or aryl.
The present invention is directed further still to a compound having the
chemical structure
2
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
KN=-.)
The R9 is H, halogen, -OH, -0-01-03 alkoxy, -0C(0)Rii; R11 is C1-C3 alkyl or
aryl, -OCH2-C(0)0R12;
R12 is H or C1-C3 alkyl, -NHR13; R13 is H, 01-04 alkyl, alkylcyclopropane,
benzyl, -NH0(0)01-3amide,
-NH0(0)0-R14carbamate; R14 is tert-butyl or benzyl, ¨NH-S02-R15 sulfonamide;
R15 is alkyl or aryl or
¨S02-NH-R16sulfonamide and R16 is alkyl or aryl. The R10 is nitrogen or
methylene. n is 0 or 1 and
when n is 1, Z is -C=0. A may have the structure
CF3
')-CH_ R
101i7
or CI
wherein R17 is H or 01-03 alkyl
group.
The present invention is directed to the compounds described herein formulated
as a
pharmaceutical composition in a pharmaceutically acceptable excipient or as a
formulation
comprising a food, an animal feed material or a medicine. The present
invention is also directed to
a kit comprising the compounds or combination thereof as described herein or a
pharmaceutical
composition thereof or other formulation thereof and a container housing the
compound.
The present invention is directed further still to a compound that is N-(4-(2-
(2-propylpyridin-
4-yl)thiazol-4-yl)phenyl)methanesulfonamide, tert-butyl-2-(4-(4-
bromophenyl)thiazol-2-y1) pyrrolidine-
1-carboxylate, benzyl 2-(4-(4-bromophenyl)thiazol-2-yl)pyrrolidine-1-
carboxylate, 4-(4-bromophenyl)
-2-(pyrrolidin-2-yl)thiazole, 4-(4-bromopheny1)-2-(1-propylpyrrolidin-2-
yl)thiazole, tert-butyl 3-(4-(4-
bromophenyl)thiazol-2-yl)piperidine-1-carboxylate, benzyl 3-(4-(4-
bromophenyl)thiazol-2-y1)
piperidine-1-carboxylate, 3-(4-(4-bromophenyl)thiazol-2-y1)-1-
propylpiperidine, benzyl 4-(4-(4-bromo
phenyl)thiazol-2-yl)piperidine-1-carboxylate, benzyl (R)-2-(4-(4-
(methylsulfonamido)phenyl)thiazol-2-
yl)pyrrolidine-1-carboxylate,
benzyl 3-(4-(4-(methylsulfonamido)phenyl)th iazol-2-yl)piperidine-1-
carboxylate, benzyl 4-(4-(4-(methylsulfonamido)phenyl)thiazol-2-yl)piperidine-
1-carboxylate, 4-(3-
(pyridin-2-y1)-[1,2,4]triazolo[4,3-b]pyridazin-6-y1)-N-tosylbenzenamine, (4-(5-
chloro-2-methylphenyl)
piperazin-1-y1)(4-(tosylamino)phenyl)
methanone, 4-(44(1-methy1-1H-benzo[d]imidazole-2-y1)
methyl )piperazin-1-y1)-N-tozylbenzenamine, 3-
chloro-4-methyl-N-(6-(4-(3-(trifluoromethyl)
benzyl)piperazin-1-yl)pyridin-3-yl)benzenesulfonamide, 4-
chloro-N-(4-(4-((1-methy1-1H-benzo[d]
imidazol-2-yl)methyl)piperazin-1-yl)phenyl)benzenesulfonamide, (Z)-4-(3-cyano-
3-(4-(2,4-dimethyl
phenyl)thiazol-2-yl)ally1)-N-(thiazol-2-y1)benzenesulfonamide, N-(3-(H-
imidazo[1,2-a]pyridine-2-y1)
phenyl)-4-methyl-2-phenylthiazole-5-carboxamide, N-(3-
(benzo[d]thiazol-2-yl)phenyl)
isonicotinamide, 3-(4-chloropheny1)-4,5-dihydro-1-pheny1-5-(2-phenylthiazol-4-
y1)-1H-pyrazole, N-(4-
(6-methylbenzo[d]thiazol-2-yl)pheny1)-2-(N-m-tolylmethyl
sulfonamido)acetamide, N-(4-(6-methyl
benzo[d]th iazol-2-yl)ph eny1)-2-(N-p-tolylmethyl sulfonam
ido)acetamide; a pharmaceutically
acceptable salt; a stereoisomer thereof; and any combination thereof.
3
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
The present invention is directed further still to a method for treating a
metabolic disorder in
an animal, comprising the step of administering to the individual a
therapeutically effective amount
of at least one compound having the chemical structure A-B-C or as
specifically described herein or
a pharmaceutically acceptable salt or a stereoisomer thereof or a combination
thereof. In preferred
aspects the metabolic disorder is a cancer or a weight-related disorder. The
present invention is
directed to a related method further comprising the step of providing a second
therapy. The second
therapy comprises dietary therapy, physical therapy, behavior therapy,
surgery, drug therapy, and a
combination thereof. In some aspects, the metabolic disorder is diabetes and
the additional therapy
comprises dietary therapy, physical therapy, and drug therapy.
The present invention is directed further still to a method for treating a
cell hyperproliferative
disease in a patient in need thereof. The method comprises the step of
administering to the patient
a therapeutically effective amount of at least one compound described herein,
or a pharmaceutically
acceptable salt or a stereoisomer thereof or a combination thereof.
The present invention is directed further still to a related method for
reducing body weight in
an animal in need thereof. The method comprises administering to the patient a
therapeutically
effective amount of at least one compound described herein, or a
pharmaceutically acceptable salt
or a stereoisomer thereof or a combination thereof.
The present invention is directed further still to a method for reducing body
weight in an
animal in need thereof. The method comprises administering to the animal, in a
pharmaceutically
acceptable medium, a therapeutically effective amount of a compound having the
structure: A-B-C
or as specifically described herein or a pharmaceutically acceptable salt or a
stereoisomer thereof
or a combination thereof. The A ring may be a pyridine or a substituted
pyridine, a piperidine or a
substituted piperidine, a pyrrolidine or a substituted pyrrolidine, a thiazole
or a substituted thiazole, a
phenyl ring or a substituted phenyl ring. The B ring may be a thioazole or a
substituted thioazole, a
piperazine or a substituted piperazine, a phenyl ring or a substituted phenyl
ring. The C ring may be
a phenyl ring or a substituted phenyl ring, a pyridine or a substituted
pyridine, a thioazole or a
substituted thioazole.
The present invention also is directed to a method for increasing
thermogenesis without
reducing lean body mass during weight loss in an animal, comprising the step
of administering to
the animal, in a pharmaceutically acceptable medium, a therapeutically
effective amount of a
compound having the structure:
R4
N-</N1 I A R3
)=/ µSY
or a pharmaceutically acceptable salt or a stereoisomer thereof or a
combination thereof. The R1
substituents may be H, Et, OMe or n-propyl; Y is CH or .
The R2 substituents may be OH,
4
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
OMe, or NH-i-Pr. The R3 substituents may be H, F, or Cl. The R4 substituents
are H, Me, Cl, Br, F,
OH, OBz, OCH2COOMe, OCH2COOH, NH2, NH-i-Pr, NHCOMe, NHSO2Me, NHBn,
N
¨0 NH-9I IP: NH ¨ -0
NH
OMe, NHBoc, , NHTs, , or .
The compound may be a
pharmaceutically acceptable salt or a stereoisomer thereof or a combination
thereof.
The foregoing has outlined rather broadly the features and technical
advantages of the
present invention in order that the detailed description of the invention that
follows may be better
understood. Additional features and advantages of the invention will be
described hereinafter,
which form the subject of the claims of the invention. It should be
appreciated by those skilled in the
art that the conception and specific embodiment disclosed may be readily
utilized as a basis for
modifying or designing other structures for carrying out the same purposes of
the present invention.
It should also be realized by those skilled in the art that such equivalent
constructions do not depart
from the spirit and scope of the invention as set forth in the appended
claims. The novel features
which are believed to be characteristic of the invention, both as to its
organization and method of
operation, together with further objects and advantages will be better
understood from the following
description when considered in connection with the accompanying figures. It is
to be expressly
understood, however, that each of the figures is provided for the purpose of
illustration and
description only and is not intended as a definition of the limits of the
present invention.
BRIEF DESCRIPTION OF THE DRAWINGS
For a more complete understanding of the present invention, reference is now
made to the
following descriptions taken in conjunction with the accompanying drawings.
FIGS. 1A-1B show confirmation of the microarray results by RT-PCR. DU145 cells
were
treated with DMSO (lane 1) and 5 ILIM fatostatin A (lane 2) for 6 hrs (FIG.
1A). Total RNA was then
extracted and subjected to RT-PCR. (FIG. 1B) A summary of the RT-PCR and
microarray data.
ACL, ATP citrate lyase; HMG CoAR, 3-hydroxy-3-methyl-glutaryl-00A reductase;
LDLR, low-density
lipoprotein receptor, MVD, mevalonate pyrophosphate decarboxylase; SCD,
stearyl-CoA
desaturase; INSIG1, insulin-induced gene 1; GAPDH, glyceraldehyde-3-phosphate
dehydrogenase.
FIGS. 2A-2C show that fatostatin A suppresses the ability of endogenous SREBPs
to
activate a reporter gene. HEK293 cells were co-transfected with an SRE-1-
driven luciferase reporter
(pSRE-Luc) (FIG. 2A) and a 13-gal reporter controlled under an actin promoter
(FIG. 2B). The
transfected cells were treated by varied concentrations of fatostatin A or
DMSO alone in a medium
containing lipid-free serum. After 20-hr incubation, luciferase activity was
measured, and the data
were normalized by p-galactosidase activity. In FIG. 2C HEK293 cells were
transfected with pCMV-
SREBP-1 c (1-436) and pSRE-Luc, and the transfected cells were treated with or
without 20 mM
5
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
fatostatin A in a medium containing lipid-free serum. Each value represents
the average of three
independent experiments.
FIGS. 3A-3H show effects of fatostatin A on SREBP-1 and -2. DU145 cells were
treated
with DMSO alone or fatostatin A (1 and 5 'LIM) for 6 hrs. The levels of
precursor and mature forms of
SREBP-1 (FIG. 3A) or SREBP-2 (FIG. 3B) were examined by western blots. Western
blots of actin
are shown in lower panel as a loading control. (FIGS. 3C-3H) Localization of
SREBP-1 was
examined by immunostaining. Cells were treated with DMSO alone (FIGS. 3C-3E)
or 5 'LIM of
fatostatin A (FIGS. 3F-3H) and then stained with DAPI (FIG. 3C and FIG. 3F) or
anti-SREBP-1 (FIG.
3D and FIG. 3G).
FIGS. 4A-4G show inhibition of the insulin-induced adipogenesis by siRNA
knockdown of
the SREBP-1. Two stably transfected clones of 3T3-L1 cells in which the
expression of SREBP-1
were knocked down were established and induced to differentiate into
adipocytes. The knockdown
cells were not differentiated (FIG. 4D and FIG. 4F), whereas 3T3-L1 cells
transfected with an empty
vector (neo) were mostly differentiated into adipocytes (FIG. 4B). FIGS. 4A,
4C and 4E show the
cells without the insulin induction. FIG. 4G is a Western blot analysis of the
clones indicating the
successful knockdown of SREBP-1.
FIGS. 5A-5B demonstrate siRNA knockdown of SREBP-1 blocks the serum-
independent
growth of DU145 prostate cancer cell. In FIG. 5A two stably transfected clones
of DU145 cells in
which the expression of SREBP-1 were knocked down were established and grown
in an MEM
medium containing no serum, 2% fetal bovine serum (FBS), 2% fat-free fetal
bovine serum, or
lpg/mL of IGF1 for three days. The growth rates were measured by WST-1 assays.
The knockdown
cells failed to grow in the MEM medium containing no serum, 2% fat-free FBS,
or 1pg/mL of IGF1
but exhibited as much growth as control cells in the presence of serum. The
experiments were
performed in triplicate. FIG. 5B are Western blots showing the extents of
SREBP-1 knockdown in
clones 1 and 2.
FIGS. 6A-6G demonstrate effects of fatostatin A on mice after
fasting/refeeding fat free diet.
Mice were injected with 30 mg/kg of fatostatin A intraperitoneally daily for
the entire experiments
starting one day before fasting for 48 hrs followed by feeding fat free diet
for another 48 hrs. Loss of
body weight (FIG. 6A) and food intake (FIG. 6B) were determined at the end of
the 48-hr feeding.
FIG. 6C shows serum constituents of the treated and control mice. FIG. 6D is a
representative
western blot for SREBP-1 of the liver extracts from 2 different mice from the
control and fatostatin A
treated groups. The loaded amounts of proteins were normalized. FIG. 6E is a
representative
western blot showing FAS expression (top panel) and a coomassie stained gel
for loading control
(bottom) of liver extracts. FIGS. 6F-6G illustrate activities of FAS and ACC
in liver extracts. Data are
means + SD (n=5); *P <0.05.
FIGS. 7A-7E show effects of two-week treatment of fatostatin A on mice. 5-6
month old
mice were injected daily for two weeks either with 30 mg/kg fatostatin A or
10% DMSO. FIG. 7A
shows body weight before and after the treatment with fatostatin A and FIG. 7B
shows the amounts
of weight loss after treatment. FIG. 7C show the serum levels of glucose,
cholesterol, and
triglycerides (TG) in the treated and control mice. FIG. 7D shows FAS activity
in liver extracts. FIG.
6
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
7E is a representative western blot analysis of the liver extracts of three
mice each for control and
treated groups. The loaded amounts of proteins were normalized. Data are means
+ SD (n=5); *P
<0.05.
FIGS. 8A-8C show effect of fatostatin A on body weight and food consumption.
Two groups
of ob/ob male mice (n=5) were daily injected intraperitoneally with fatostatin
A or 10% DMSO in PBS
to control groups for four weeks. Mice were fed normal chow and on first day
of the experiment and
every day thereafter the weight of the mice and the amount of the food
consumed were measured.
FIG. 8A is a picture of representative control and fatostatin A treated mice.
FIG. 8B shows the
weight of each mouse within each group was measured daily. The average and
variance of the
weights are shown. In FIG. 8C food intake was measured every day and was
expressed as
cumulative food intake per mouse over the 28 days period.
FIGS. 9A-9H demonstrate serum constituents of control and fatostatin A treated
ob/ob mice.
Blood was collected from tail veins of overnight fasted mice, and serum was
collected after
separation from cells. Constituents were determined as described below. The
data are shown as
mean SD, n = 5 mice in each group.
FIGS. 10A-10D show effect of fatostatin on liver and adipose tissue of ob/ob
mice. FIG.
10A shows livers of fatostatin A treated mice (left) and controls (right).
FIG. 10B shows histological
analyses of frozen sections of livers of the control and fatostatin A ob/ob
mice stained with Oil Red-
0 to detect lipid droplets and counter-stained with Mayer's hematoxylin.
Livers of three different
mice treated with fatostatin A, showing a dramatic decrease in red-stained
droplets (top) and
controls showing an abundance of red-stained lipid droplets compared to the
treated mice (bottom).
FIG. 10C shows epididymal fat pads isolated from fatostatin A treated ob/ob
mice (left) and controls
(right). FIG. 100 shows the average weight of livers and epididymal fat pads
isolated from ob/ob
controls and fatostatin A treated mice. The data are shown as mean SD, n = 5
mice in each group
(*p< 0.05).
FIGS. 11A-11B shows triglycerides (FIG. 11A) and cholesterol (FIG. 11B) levels
in livers of
controls and fatostatin A treated ob/ob mice. Lipids were extracted from
livers and triglycerides and
cholesterol were quantified as described below. The data are shown as mean
SD, n = 5 mice in
each group (*P = 0.0004; tP= 0.03).
FIGS. 12A-12D demonstrate that fatostatin A reduces the expression levels and
activities of
lipogenic enzymes. FIG. 12A shows the activity of acetyl-CoA carboxylase (ACC)
and FIG.12B
shows fatty acid synthase measured in liver extracts of ob/ob mice as
described below. FIG. 12C is
a Western Blot analysis of liver crude extracts, from three individual ob/ob
mice, were separated by
4-12% NuPAGE MES gels and probed with different antibodies and detected with
ECL. FIG.12D
shows the ratio of the intensity of the specific bands, of different lipogenic
enzymes from fatostatin A
against control mice after normalization to actin. The data are shown as mean
SD, n = 5 mice in
each group (tP= 0.005; $P= 0.002; *P < 0.05)
FIG. 13 shows transcript levels of liver lipogenic enzymes in control relative
to fatostatin A
ob/ob mice. mRNA levels for each gene was normalized to actin. RNA was
isolated from control
7
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
and fatostatin A treated mice (n=5) and measured by real-time quantitative
RT¨PCR. *P < 0.05 vs.
control.
FIGS. 14A-14F illustrates exemplary compounds 1-66 of the present invention.
FIG. 15 demonstrates a standard luciferase reporter gene assay with exemplary
analogues
2-18.
FIG. 16 shows a standard luciferase reporter gene assay with exemplary
analogues 19-34.
FIG. 17 provides a standard luciferase reporter gene assay with exemplary
analogues 35-
44.
FIGS. 18A-18D show that fatostatin blocks the activation of SREBP. Suppression
by
fatostatin of the ability of endogenous SREBPs to activate a luciferase
reporter gene in a medium
containing lipid-free serum. CHO-K1 cells were transfected with an SRE-1-
driven luciferase reporter
(pSRE-Luc) (FIG. 18A). The transfected cells were treated by varied
concentrations of fatostatin in a
medium containing lipid-free serum. Effect of fatostatin on CHO-K1 cells co-
transfected with pCMV-
SREBP-1c(1-436) and pSRE-Luc in a medium containing lipid-free serum (FIG.
18B). PLAP-BP2 in
transfected CHO-K1 cells remains membrane-bound unless it is cleaved by S1P in
the Golgi and
secreted into the culture medium (left). Treatment with fatostatin (20 pM) or
sterols (10 pg/mL
cholesterol and 1 g/mL 25-hydroxycholesterol) affect cleavage of PLAP-BP2
compared to Et0H
controls (FIG. 18C). Western blot analysis of CHO-K1 cells treated with
fatostatin. P and N denote
the uncleaved membrane precursor and cleaved nuclear forms of SREBP-2,
respectively (FIG.
18D).
FIGS. 19A-19B show that fatostatin blocks the translocation of SREBPs from the
ER to the
Golgi. FIG. 19A is a Western blot analysis showing effects of brefeldin A on
CHO-K1 cells treated
with Et0H alone, sterols (10 ig/m1 cholesterol and 1 pg/m1 25-
hydroxycholesterol), or 20 luM
fatostatin. FIG. 19B is a Western blot analysis with anti-SCAP IgG-9D5 of
cells grown in the
absence or presence of 20 pM fatostatin or sterols (10 pg/mL cholesterol and 1
pg/mL 25-
hydroxycholesterol). Numbers on the right denote the number of N-linked sugar
chains present on
protease-protected SCAP fragments.
FIGS. 20A-20D show the structures of dansyl fatostatin, fatostatin-polyproline
linker-biotin
conjugate and polyproline linker-biotin conjugate and cells treated with the
same. FIG. 20A
illustrates how the polyproline linker was inserted for better projection of
the fatostatin molecule
(Sato et al., 2007). In FIG. 20B CHO-K1 cells treated with dansyl fatostatin
and ER-tracker red
showing localization of dansyl fatostatin in the ER. Scale bar = 10 pm. FIG.
20C illustrates the
interaction of fatostatin with SCAP, shown by western blot analysis with anti-
SCAP, anti-SREBP-1,
anti-SREBP-2, and anti-ATF6 antibodies of proteins bound to Neutravidine-
agarose beads
saturated with biotinylated fatostatin in CHO-K1 membrane extract. FIG. 20D
shows that for the
competition assay, membrane extracts were preincubated with Et0H alone,
cholesterol, or
fatostatin.
FIGS. 21 show effects of fatostatin on liver and adipose tissue of ob/ob mice.
Sections of
the livers of fatostatin-treated and control mice showing red-stained lipid
droplets.
8
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
FIG. 22 demonstrates a western blot analysis of CHO-K1 cells treated with
fatostatin. P and
N denote the uncleaved membrane precursor and cleaved nuclear forms of SREBP-
1, respectively.
FIG. 23 provides an exemplary synthetic scheme of fatostatin, dansyl
fatostatin and
fatostatin-polyproline linker-biotin.
FIG. 24 shows suppression by fatostatin analogues of the ability of endogenous
SREBPs to
activate a luciferase reporter gene in a medium containing lipid-free serum.
CHO-K1 cells were
transfected with pSRE-Luc. The transfected cells were treated by varied
concentrations of fatostatin,
dansyl fatostatin or isopropylamine derivative in a medium containing lipid-
free serum.
FIGS. 25A-25D show that fatostatin reduces the expression levels and
activities of lipogenic
enzymes. Activity of acetyl-CoA carboxylase (ACC) (FIG. 25A) and fatty acid
synthase (FAS) (FIG.
25B) were measured in liver extracts of ob/ob mice. Western Blot analysis of
liver crude extracts
was performed (FIG. 25C). Ratio of the intensity of the specific bands, of
different lipogenic
enzymes from fatostatin against control mice after normalization to actin are
shown (FIG. 25D). The
data are shown as mean SD, n = 5 mice in each group (tP= 0.005; $P= 0.002;
*P < 0.05)
FIG. 26 provides a synthetic scheme of compound 53.
FIG. 27 provides a synthetic scheme of compound 19 and compound 17.
FIG. 28 shows a standard SREBP activation assay with exemplary analogues 45-55
and
19.
FIG. 29 shows a standard SREBP activation assay with exemplary analogues 56-
61and 19.
FIG. 30 shows a standard SREBP activation assay with exemplary analogues 62-
66.
FIG. 31 shows an Inhibitory Concentration (sub 50) data of exemplary compound
53.
FIG. 32 shows an Inhibitory Concentration (sub 50) data of exemplary compounds
58 and
61.
FIGS. 33A-33B show that compound 19 inhibited the growth of breast cancer cell
line SUM
159. Cells were seeded onto 96 well plates at a density of 10000 cells/well in
100u1 medium
containing 2% charcoal stripped serum. After 24 hours, compound 19 was added
to the cells at the
indicated concentrations for another 48 hours. Cell viability was determined
using WST-1 assay.
FIG. 33A: shows the effect of different concentrations of compound 19 on cell
growth as evident
from the changes in the absorbance at A450nm. FIG. 33B: Expression levels of
lipogenic genes
were significantly downregulated by 10 pM treatment of HePG2 cells as
determined by RT PCR
analysis (black bar); values are depicted as means + SD; *P<0.05
FIGS. 34A-34C show that compound 19 inhibited the growth of human liver cancer
cell line
HePG2. Cells were seeded onto 16 well plates at a density of 100000 cells/well
in medium
containing 2% charcoal stripped serum. After 24 hours, compound 19 was added
to the cells at the
indicated concentrations for another48 hours. FIG. 34A shows photographs of
control and treated
HePG2 cells with 25, 50 and 100 pM compound 19. FIG. 34B shows a
representative Western blot
analysis showing that the treated cells (T) had a decreased level of the
mature and active form of
SREBP-1 and higher levels of the precursor compared with the untreated
controls. FIG. 34C shows
expression levels of lipogenic genes were significantly downregulated by 10 pM
treatment of HePG2
9
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
cells and did not affect INSIG2 which is not known to be regulated by SREBP,
as determined by RT
PCR analysis. Values are depicted as means + SD; *P<0.05.
FIGS. 35A-35F show that compound 19 inhibited the growth of human acute
lymphoblastic
leukemia cell line MOLT-4 and human multiple myeloma cell line RPMI8226.
10,000 MOLT-4 cells
(FIGS. 35A-35C) and 20,000 RPMI8226 cells (FIGS. 35D-35F) were seeded onto 96
well plate in
RPM! 1640 medium containing 5% FBS or fat-free charcoal treated serum (FF-FBS)
for 24 hours at
37 C. MOLT-4 and RPMI8226 cells were treated for additional 48 hours with
different concentrations
of compound 19 (None control without DMSO, Vehicle with DMSO, 1, 2, 5, 10 and
20 pM in DMSO;
RPMI8226 cells also were treated with 3 pM in DMSO). At the end of 48 hours
cells were subjected
to MTT assay to determine viability. Values are depicted as means + SD;
*P<0.05.
FIGS. 36A-36F show that compound 17 inhibited the growth of human acute
lymphoblastic
leukemia cell line MOLT-4 and human multiple myeloma cell line RPM18226.
20,000 MOLT-4 cells
(FIGS. 36A-36C) and 20,000 RPMI8226 cells (FIGS. 36D-36F) cells were seeded
onto 96 well
plates in RPM! 1640 Medium containing 5% Fetal bovine serum (FBS) or 5% fat
free charcoal
treated serum (FF-FBS) for 24 hours at 37 C. Cells then were treated for
additional 48 hours with
different concentrations of compound 17 (None control without DMSO, Vehicle
with DMSO, 1, 2, 3,
5, 10 and 20 pM in DMSO). At the end of the 48 hours treatment cells were
subjected to MTT assay
to determine viability. Values are depicted as means + SD (n=3); **P < 0.001;
***P< 0.0001.
Figures 37A-37D: Body weight and composition (fat and lean) in SD rats after
three weeks
of feeding RD and HFHC Diets. Male SD rats 6-7 weeks old with initial body
weight of about 193 +
7.0 gram/rat were fed regular diet (RD) or high fat high carbohydrate diets
for three weeks. Body
weight (Figure 37B) fat weight (Figure 37C) and lean weight (Figure 37D)
content was measured
using ECHO MRI method15. (*P< 0.05)
Figures 38A-38B show cumulative body weight gain (Figure 38A) and food intake
(Figure
38B) after eight weeks of treatment with compound 19. SD rats were fed HFHC
diet for three
weeks before the start of treatment with different doses of compound 19 via
oral gavage or
cottonseed oil vehicle for an additional eight weeks. Body weight and food
intake was measured
and cumulative values were calculated. (Values are means+ S.E.M; *P<0.05;
treated compared to
control vehicle group.
Figure 39A-39E show body composition fat and lean mass determine by ECHO MRI
method. Figure 39A shows total body weight in grams after administration of
compound 19. Figure
39B shows total body fat in grams after administration of compound 19. Figure
39C shows the
percentage of fat per animal after administration of compound 19. Figure 39D
shows total lean
weight in gram after administration of compound 19. Figure 39E shows percent
lean weight per
gram after administration of compound 19. Rats from different experimental
groups were subjected
to body composition analysis every two weeks. Percent of fat and lean mass was
calculated from
the weights of fat and mass relative to body weight. The labels in Figure 39B-
39E correspond to the
same groups as in Figure 39A. Asterisks indicate significant differences
between treated and
control groups (*P < 0.05). Data are means + S.E.M
of n=15).
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
Figures 40A-40D shows the effect of compound 19 on livers of SD rats fed HFHC.
Figure
40A: Representative livers of treated (2.5 and 10 mg/kg) and control rats (n=5
from each group).
Figure 40B: Oil Red 0 staining of frozen sections of livers. Oil droplets are
shown in red. Bars are
indicating 200 micron. Figure 40C: levels of TG and cholesterol in liver
tissues from control and
treated animals; Figure 40D: Fold changes of gene expression controlled by
SREBP-1 and 2 in
treated rats compared to controls measured by real time PCR. ACC: acetyl-CoA
carboxylase; ACL:
ATP citrate-Iyase; SOD: steryl-CoA desaturase MVD Mevalonate decarboxylase;
LDLR; LDL
receptor; INSIG-1: insulin induced gene 1; Means are + SE; *P < 0.05.
Figure 41 shows fold changes in gene expression of UCP2 in treated rats with
compound
19 compared to controls. Total RNA was extracted from liver tissues of control
and compound 19
treated rats with the indicated doses and level of UCP mRNA was determined
using real-time PCR.
DETAILED DESCRIPTION OF THE INVENTION
General Embodiments of the Invention
Metabolic disorders are treated and/or prevented with compounds of the present
invention.
For example, dysregulated biosynthesis of fatty acids and cholesterols and
excessive intake of
dietary fat are correlated with a number of medical complications including at
least obesity,
diabetes, hypertension, and cardiovascular diseases, and in certain aspects
these conditions are
treated and/or prevented with a compound of the invention. Epidemiological
evidence indicates that
metabolic diseases including obesity also promote the development of an
aggressive form of
cancers, including but not limited to prostate cancer.
Upon fat depletion, sterol regulatory element binding proteins (SREBPs) are
proteolytically
released from the membrane and translocated into the nucleus, where they
activate the
transcription of the genes involved in cholesterol and fatty acid
biosynthesis. The present invention
identifies a synthetic small molecule previously known to block both
adipogenesis and cancer cell
growth as a selective inhibitor of the SREBP activation and also provides
analogs and derivatives of
that molecule. The drug-like molecule fatostatin A impairs the proteolytic
activation of SREBPs,
thereby reducing the transcription of their responsive genes in cells. In
mice, fatostatin A blocks the
activation of SREBP-1 in the liver, reduces body weight, lowers the levels of
blood cholesterol and
glucose, and down-regulates lipogenic enzymes. Fatty acid synthase and acetyl-
CoA carboxylase
activities and their expression levels were decreased in the liver of the
treated mice. Fatostatin A
serves as a tool to understand cellular pathways and provides a consensus
molecule as at least
starting point for pharmacological intervention of metabolic diseases, in
certain aspects.
Small molecules that modulate metabolism-related phenotypes serve as tools for
dissecting
the complex associations, in specific embodiments. Fatostatin A causes two
distinct phenotypes in
cultured mammalian cells: complete inhibition of the insulin-induced
adipogenesis of 3T3-L1 mouse
fibroblast cells; and selective repression of the serum-independent insulin-
like growth factor 1
(IGF1)-dependent growth of DU145 human prostate cancer cells.
11
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
In certain aspects of the invention, fatostatin A selectively blocks the
activation of SREBPs,
a key lipogenic transcription factor that activates specific genes involved in
cholesterol and fatty acid
synthesis. The identification of fatostatin A as an inhibitor of SREBPs is
consistent with its anti-
adipogenic property, and indicates a role of SREBPs in the IGF1-dependent
growth of prostate
cancer. The present invention concerns fatostatin A, preferably analog and
derivative compounds
thereof, that blocks the activation of at least SREBP-1, for example as shown
in experimental mice.
Administration of fatostatin A into obese ob/ob mice led to weight loss and
marked reduction of
visceral fat. Moreover, expression of uncoupling protein 1, uncoupling protein
2 and uncoupling
protein 3 is increased and thermogenesis was increased without reducing lean
body mass during
weight loss
The present invention concerns treatment and/or prevention of at least one
symptom of a
metabolic disorder. The metabolic disorder may be of any kind, so long as one
of its symptoms is
improved or prevented with a compound of the present invention. In particular,
though, the
metabolic disease is from one or more inborn errors of metabolism (which may
be referred to as
genetic disorders), such as inherited traits that are due to a defective
metabolic enzyme (for
example one having one or more mutations or disorders that involve mutations
in regulatory
proteins and in transport mechanisms).
Generally, metabolic disorders may be defined as disorders that affect energy
production in
a cell. Although most metabolic disorders are genetic, some may be acquired as
a result of one or
more factors, including diet, toxins, infections, and so forth. Genetic
metabolic disorders may be
caused by genetic defects that result in missing or improperly constructed
enzymes necessary for
some step in the metabolic process of the cell. The largest categories of
metabolic disorders
include the following: 1) glycogen storage diseases (also referred to as
glycogenosis or dextrinosis),
which include disorders that affect carbohydrate metabolism; 2) fatty
oxidation disorders, which
affect fat metabolism and metabolism of fat components; and 3) mitochondrial
disorders, which
affect mitochondria. Examples of glycogen storage diseases (GSD) include at
least GSD type I
(glucose-6-phosphatase deficiency; von Gierke's disease); GSD type ll (acid
maltase deficiency;
Pompe's disease); GSD type III (glycogen debrancher deficiency; Con's disease
or Forbes
disease); GSD type IV (glycogen branching enzyme deficiency; Andersen
disease); GSD type V
(muscle glycogen phosphorylase deficiency; McArdle disease); GSD type VI
(liver phosphorylase
deficiency, Hers's disease); GSD type VII (muscle phosphofructokinase
deficiency; Tarui's disease);
GSD type IX (phosphorylase kinase deficiency); and GSD type XI (glucose
transporter deficiency;
Fanconi-Bickel disease).
Fatty acid metabolism deficiencies may be described as fatty oxidation
disorders or as lipid
storage disorders, in certain embodiments. They may involve one or more inborn
errors of
metabolism that result from enzyme deficiencies that affect the body's ability
to oxidize fatty acids
for the production of energy within muscles, liver, and other cell types, for
example. Examples of
fatty acid metabolism deficiencies include at least coenzyme A dehydrogenase
deficiencies; other
coenzyme A enzyme deficiencies; carnitine-related disorders; or lipid storage
disorders. Examples
of coenzyme A dehydrogenase deficiencies include at least very long-chain acyl-
coenzyme A
12
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
dehydrogenase deficiency (VLCAD); long-chain 3-hydroxyacyl-coenzyme A
dehydrogenase
deficiency (LCHAD); medium-chain acyl-coenzyme A dehydrogenase deficiency
(MCAD); short-
chain acyl-coenzyme A dehydrogenase deficiency (SCAD); and short chain L-3-
hydroxyacyl-coA
dehydrogenase deficiency (SCHAD). Examples of other coenzyme A enzyme
deficiencies include
at least 2,4 Dienoyl-CoA reductase deficiency; 3-hydroxy-3-methylglutaryl-00A
lyase deficiency;
and malonyl-CoA decarboxylase deficiency. Examples of carnitine-related
deficiencies include at
least primary carnitine deficiency; carnitine-acylcarnitine translocase
deficiency; carnitine
palmitoyltransferase I deficiency (CPT); and carnitine palmitoyltransferase ll
deficiency (CPT).
Examples of lipid storage diseases include acid lipase diseases; Wolman
disease; cholesteryl ester
storage disease; Gaucher disease; Niemann-Pick disease; Fabry disease;
Farber's disease;
gangliosidoses; Krabbe disease; and metachromatic leukodystrophy. Other fatty
acid metabolism
disorders include at least mitochondrial trifunctional protein deficiency;
electron transfer flavoprotein
(ETF) dehydrogenase deficiency (GAII & MADD); Tangier disease; and acute fatty
liver of
pregnancy.
Examples of mitochondrial diseases include at least progressive external
ophthalmoplegia (PEO); Diabetes mellitus and deafness (DAD); Leber hereditary
optic neuropathy
(LHON) Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like
syndrome (MELAS);
Myoclonic epilepsy and ragged-red fibers (MERRF); Leigh syndrome; subacute
sclerosing
encephalopathy; Neuropathy, ataxia, retinitis pigmentosa, and ptosis (NARP);
Kearns-Sayre
syndrome (KSS); Myoneurogenic gastrointestinal encephalopathy (MNGIE). In
particular aspects of
the invention, the metabolic disorder is, or has as one of its complications,
one or more of the
following: obesity, hyperlipemia, diabetes, fatty liver, hypertension, and
cardiovascular disease.
The present invention concerns treatment of a disease related to cell
hyperproliferation. In
a particular example cell hyperproliferation may be caused by a cancer or
other neoplastic disease
or disorder. Without being limiting, the hyperproliferative disease may be
cancer of the breast,
respiratory tract, brain, reproductive organs, prostate, digestive tract,
urinary tract, eye, liver, skin,
head and neck, thyroid, parathyroid, lymphoma, sarcoma, melanoma, leukemia,
multiple myeloma,
or a distant metastasis of a solid tumor.
General Compounds of the Invention
Generally, the present invention provides compound, or pharmaceutically
acceptable salts
and stereoisomers thereof, having the general formula:
A-B-C
where A, B, and C can be the same or different and each may be a 5-, 6-, or 7-
membered
ring or a fused bicyclic ring system, the ring being a heterocyclic ring or
non-heterocyclic ring, a
substituted ring or non-substituted ring, A, B and C are either directly
connected or connected
through an intervening atom chain or linker and said atom chain or linker is a
saturated carbon chain
or an unsaturated carbon chain with or without additional functional groups.
Preferably, the A ring is a 6-membered heterocyclic ring with one heteroatom.
The A ring
may be substituted. In preferred embodiments, the ring is a pyridine ring;
more preferably, the
nitrogen atom of the pyridine ring is in the 4-position or in the 2-position
realtive to the position of the
13
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
B ring. Most preferably, the pyridine ring is substituted with a n-propyl
group on a carbon positioned
alpha to the nitrogen heteroatom. In other preferred embodiments, there is a 1-
5 atom side chain,
more preferably a 1-5 carbon side chain, on the A ring. In other illustrative
and non-limiting
embodiments, the A ring may be phenyl, pyrrole, thiophene, furan, pyrimidine,
isoquinoline,
quinoline, benzofuran, indole, oxazole, naphthyl, piperidine, pyrrolidine,
imidazole, imidazol[1,2-
a]pyridine, benzoimidazole, thiazole, or benzothiazole, for example.
In one aspect of the invention, the A ring is a piperidine ring. Preferably,
the nitrogen atom
of the piperidine ring is in the 4-position relative to the position of the B
ring. In other related aspect,
nitrogen atom of the piperidine ring is in the 3-position relative to the
position of the B ring. The
nitrogen atom of the piperidine may be further substituted and wherein the
substitutions are selected
from the group consisting of alkyl, sulfoxide, sulfone, alkyl or aryl
sulfonate, sulfonic acid, and any
combination thereof. For example, the substitution may be a propyl group, a
tert-butyloxycarbonyl
(BOC) or a benzyloxycarbonyl group.
In further aspect of the invention, the A ring is a pyrrolidine ring;
preferably, the nitrogen
atom of the pyrrolidine ring is in the 2-position relative to the position of
the B ring. The nitrogen
atom of the pyrrolidine may be further substituted and wherein the
substitutions are selected from
the group consisting of alkyl, sulfoxide, sulfone, alkyl or aryl sulfonate,
sulfonic acid, and any
combination thereof. For example, the substitution may be a propyl group, a
tert-butyloxycarbonyl
(BOO) or a benzyloxycarbonyl group.
Preferably, the B ring is a 5-membered ring with at least two heteroatoms. The
B ring may
be substituted. In preferred embodiments, the B ring is a thiazole ring. In
other illustrative and non-
limiting embodiments, the B ring may be oxazole, imidazol, isooxazole,
imidazole, thiphene, furan,
pyrimidine, pyrazole, isothiazole, thiazolopyridazine, aryl, or pyrazole, for
example. The B ring may
further be a 6-membered ring with two heteroatoms. For example, the B ring is
a piperazine ring.
Preferably, the C ring is a 6-membered ring, most preferably a phenyl ring.
The C ring may be
substituted. In preferred embodiments, the C ring is methyl substituted. In
other illustrative and
non-limiting embodiments, the C ring may be phenyl, pyridine, pyrrole,
thiophene, furan, pyrimidine,
isoquinoline, quinoline, benzofuran, indole, oxazole, or naphthyl, for
example.
Exemplary compounds are provided in FIGS. 14A-14F and Tables 3 and 4.
Referring to
compound 1, the n-propyl substituted pyridine ring corresponds to the A ring
of the general formula,
the 2,4- substituted thiazole ring corresponds to the B ring of the general
formula, and the methyl
substituted phenyl ring corresponds to the C ring of the general formula. It
should be understood
that substitutions are permissible at any position in any of the A, B, and C
rings and any
substitutions may be the same or different from any other substitutions. Non-
limiting examples of
the substitutions, in addition to those shown in FIGS. 14A-14F, include the
following groups: H (i.e.,
unsubstituted); hydroxy; C1_10 alkyl; C2_10 alkenyl; C2_10 alkynyl; C3_6
cycloalkyl; aryl; heteroaryl;
wherein said alkyl, alkenyl, alkynyl, cycloalkyl, aryl and heteroaryl groups
are optionally substituted
with 1-5 groups selected from the group consisting of hydroxy, -(C=0)Ra; -
(C=0)0Ra, -(C=0)H, -
(C=0)0H, 0(CH2),COORa wherein n=1-10 and wherein Ra is a C1_10 alkyl, 02_10
alkenyl, C2_10
alkynyl, or 03-6 cycloalkyl, aryl, heteroaryl, fluoro, chloro, bromo, iodo,
cyano, carboxy, amino, mono-
14
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
substituted amino and di-substituted amino, mono-substituted amido and di-
substituted amido and
any combination thereof; -(C=0)Ra; -(C=0)0Ra, -(C0)H; -(C=0)0H; -0(CH2)nCOORa
wherein n=1-
and wherein Ra is a Ci_10 alkyl, C2_10 alkenyl, C2_1(3 alkynyl, or C3_6
cycloalkyl, aryl or heteroaryl
fluoro, chloro, bromo, iodo; cyano; carboxy; amino; amido, mono- and di-
substituted amino having
5 a
substitution selected from the group consisting of 01_10 alkyl, C2_10 alkenyl,
C2_10 alkynyl, 03-6
cycloalkyl, aryl, heteroaryl, sulfoxides, sulfones, sulfonates, alkyl
sulfonates, sulfonic acids and any
combination thereof; wherein said alkyl, alkenyl, alkynyl, cycloalkyl, aryl
and heteroaryl are
optionally substituted with 1-5 groups selected from the group consisting of
hydroxy, -(C=0)Ra, -
(C=0)0R0, -(C=0)H, -(C=0)0H, -0(CH2),COORa wherein n=1-10 and wherein Ra is a
C1_10 alkyl,
10 C2_10
alkenyl, C2_10 alkynyl, or C3_6 cycloalkyl, aryl and heteroaryl; fluoro;
chloro; bromo; iodo; cyano;
carboxy; amino; mono- and di- substituted amino with one or more of C1_10
alkyl, C2_10 alkenyl, C2_10
alkynyl groups, and any combination thereof; and, mono- and di- substituted
amido having a
substitution selected from the group consisting of Ci_10 alkyl, C2_10 alkenyl,
C2_10 alkynyl, C3-6
cycloalkyl, aryl, heteroaryl, sulfoxides, sulfones, sulfonates, alkyl
sulfonates, sulfonic acids,
sulfonates, alkyl sulfonates, sulfonic acids and any combination thereof;
wherein said alkyl, alkenyl,
alkynyl, cycloalkyl, aryl and heteroaryl are optionally substituted with 1-5
groups selected from the
group consisting of hydroxyl; -(C=0)Ra; -(C=0)0Ra, -(C0)H; -(C=0)0H, -
0(CH2),COORa wherein
n=1-10 and wherein Ra is a Ci_10 alkyl, 02_10 alkenyl, 02_10 alkynyl, or 03_6
cycloalkyl, aryl or
heteroaryl; fluoro; chloro; bromo; iodo; cyano; carboxy; amino; mono- and di-
substituted amino with
one or more of C1_10 alkyl, 02_10 alkenyl, C2_10 alkynyl groups, and any
combination thereof.
In the present invention, there is a compound, or pharmaceutically acceptable
salt or
stereoisomer thereof, having the general formula: A-B-C, wherein A, B, and C
are the same or
different and wherein each comprises a 5-, 6-, or 7- membered ring or a fused
bicyclic ring system,
the ring being a heterocyclic ring or non-heterocyclic ring, a substituted
ring, or non-substituted ring,
wherein A, B and C are either directly connected or connected through an
intervening atom chain or
linker and wherein said atom chain or linker is a saturated carbon chain or an
unsaturated carbon
chain with or without additional functional groups, wherein any one, any two,
or all three of A, B,
and C are unsubstituted or have one or more substitutions, and wherein any
substitution may be the
same or different from any other substitution, and wherein the substitutions
are consisting of: a)
hydroxy, b) 01-10 alkyl, c) C2-10 alkenyl, d) 02-10 alkynyl, e) 03-6
cycloalkyl, f) aryl, g) heteroaryl
wherein said substitutions in b), c), d), e), f), and/or g) are optionally
further substituted with 1-5
groups consisting of: 1) hydroxy, 2) -(C=0)R9, 3) - (C=0)0Ra, 4) - (C=0)H, 5) -
(C=0)0H, 6) -
0(CH2),COORa wherein n=1-10, 7) halo, 8) cyano, 9) carboxy, 10) amino,11) mono-
substituted
amino, 12) di-substituted amino, 13) amido, 14) mono-substituted amido; 15) di-
substituted amido,
and any combination thereof, wherein in 2), 3), or 6) Ra is a 01-10 alkyl, 02-
10 alkenyl, 02-10
alkynyl, 03-6 cycloalkyl, aryl, or heteroaryl, h) -(C=0)Ra, i) -(C=0)0Ra, j) -
(C0)H, k) -(C=0)0H; I) -
0(CH2)nCOOR9 wherein n=1-10, wherein in h), i), or l), Ra is a Ci_10 alkyl,
02_10 alkenyl, C2_10 alkynyl,
C3_6 cycloalkyl, aryl or heteroaryl, m) halo, n) cyano, o) carboxy, p) amino,
q) mono-substituted
amino, r) di-substituted amino, s) amido, t) mono-substituted amido, and u) di-
substituted amido
wherein one or more of said mono-substituted amino, di-substituted amino, mono-
substituted
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
amido, and di-substituted amido have a substitution selected from the group
consisting of C1-10
alkyl, 02-10 alkenyl, C2-10 alkynyl, C3-6 cycloalkyl, aryl, heteroaryl,
sulfoxide, sulfone, sulfonate,
alkyl sulfonate, sulfonic acid, and any combination thereof, wherein in u)
said alkyl, alkenyl, alkynyl,
cycloalkyl, aryl or heteroaryl are optionally furthersubstituted with 1-5
groups selected from the
group consisting of: i) hydroxy, ii) -(C=0)Ra, iii) -(C=0)0Ra, iv) -(C0)H, v) -
(C=0)0H, vi) -
0(CH2),COOR0 wherein n=1-10, wherein in ii), iii), or vi) Ra is a C1_10 alkyl,
C2_10 alkenyl, C2_10
alkynyl, C3_6 cycloalkyl, aryl or heteroaryl, vii) halo, viii) cyano, ix)
carboxy, x) amino, xi) mono-
substituted amino, xii) di-substituted amino, xiii) amido; xiv) mono-
substituted amido, xv) di-
substituted amido, and any combination thereof.
Exemplary Compounds, Compositions, Formulations and Methods of Use
In one embodiment of the present invention there is provided a compound having
the
chemical structure A-B-C wherein A is a pyridine or a substituted pyridine, a
piperidine or a
substituted piperidine, a pyrrolidine or a substituted pyrrolidine, a thiazole
or a substituted thiazole, a
phenyl ring or a substituted phenyl ring; B is a thioazole or a substituted
thioazole, a piperazine or a
substituted piperazine, a phenyl ring or a substituted phenyl ring; and C is a
phenyl ring or a
substituted phenyl ring, a pyridine or a substituted pyridine, a thioazole or
a substituted thioazole.
In one preferred aspect the chemical structure is:
S
R2¨N)
wherein R1 is H, halogen, -OH, -0-C1-3 alkoxy, -0C(0)R3; R3 is C1-C3 alkyl or
aryl, -OCH2-
C(0)0R4; R4 is H or Ci-C3 alkyl, -NHR5; R5 is H, C1-C4 alkyl,
alkylcyclopropane, benzyl, -NHC(0)C1-
03 amide, -NHC(0)0-R6 carbamate; R6 is tert-butyl or fluorenylmethyl or ¨NH-
S02-R7 sulfonamide;
R7 is alkyl or aryl, R2 is alkyl or R800(0)- and R8 is 03-05 alkyl or aryl.
Particularly, the halogen may
be bromine.
In another preferred aspect the chemical structure is:
R2 s
wherein R1 is H, halogen, -OH, -0-01-3 alkoxy, -0C(0)R3; R3 is 01-03 alkyl or
aryl, -OCH2-
C(0)0R4; R4 is H or C1-C3 alkyl, -NHR5; R5 is H, C1-C4 alkyl,
alkylcyclopropane, benzyl, -NHC(0)C1-
C3 amide, -NHC(0)0-R6 carbamate; R6 is tert-butyl or fluorenylmethyl or ¨NH-
S02-R7 sulfonamide;
R7 is alkyl or aryl, R2 is alkyl or R800(0)- and RB is 03-05 alkyl or aryl.
Particularly, the halogen may
be bromine.
In yet another preferred aspect the chemical structure is:
16
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
wherein R9 is H, halogen, -OH, -0-C1-C3 alkoxy, -0C(0)Rii; R11 is C1-C3 alkyl
or aryl, -
OCH2-C(0)0R12, R12 is H or 01-03 alkyl, -NHR13, R13 is H, C1-C4 alkyl,
alkylcyclopropane, benzyl, -
NHC(0)C1-3amide, -NHC(0)0-Ri4carbamate; R14 is tert-butyl or
fluorenylmethyl, ¨NH-S02-R15
sulfonamide; R15 is alkyl or aryl or ¨S02-NH-R16 sulfonamide; R16 is alkyl or
aryl, R10 is nitrogen or
methylene, n is 0 or 1 and when n is 1, Z is -0=0; and A is
CF3
62¨
CH = Ri7
or CI , wherein R17 is H or 01-03 alkyl
group.
In yet other preferred aspects the chemical structure is:
N N-S02-CH3
00
s 0 µ-'"3
CH3
H3C
Ci
1:10 N
N.N
0
NH
104
17
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
S
N 111(0
0
111. s 141(0
N
0
CN
S
9. 401
HN-S
S--x(
In a related embodiment there is provided pharmaceutical composition
comprising the
compound as described supra and a pharmaceutically acceptable excipient. In
another related
embodiment there is provided the compound as described supra formulated as a
food, an animal
feed material or a medicine. In yet another related embodiment there if
provided a kit comprising
the compound as described supra and a container housing the compound. The
container can be
any appropriate container adapted to store a drug known in the art and
commercially available.
In yet another embodiment of the present invention, preferred compounds are N-
(4-(2-(2-
propylpyridin-4-yl)thiazol-4-yl)phenyl)methanesulfonamide, tert-butyl 2-(4-(4-
bromophenyl)thiazol-2-
yl)pyrrolidine-1-carboxylate, benzyl 2-(4-(4-bromophenyl)thiazol-2-
yl)pyrrolidine-1-carboxylate, 4-(4-
bromopheny1)-2-(pyrrolidin-2-yl)thiazole, 4-(4-bromophenyI)-2-(1-
propylpyrrolidin-2-yl)thiazole, tert-
butyl 3-(4-(4-bromophenyl)thiazol-2-yl)piperidine-1-carboxylate, benzyl 3-(4-
(4-bromophenyl)thiazol-
2-yl)piperidine-1-carboxylate, 3-(4-(4-bromophenyl)thiazol-2-y1)-1-
propylpiperidine, benzyl 44444-
bromophenyl)thiazol-2-yl)pi peridine-1-carboxylate,
benzyl (R)-2-(4-(4-(methylsulfonamido)
phenyl)thiazol-2-yl)pyrrolidine-1-carboxylate, benzyl 3-(4-(4-
(methylsulfonamido)phenyl)thiazol-2-y1)
piperidine-1-carboxylate, benzyl 4-(4-
(4-(methylsulfonamido)phenyl)thiazol-2-yl)piperidine-1-
carboxylate, 4-(3-(pyridin-2-y1)[1,2,4]triazolo[4,3-b]pyridazin-6-y1)-N-
tosylbenzenamine, (4-(5-chloro-
2-methylphenyl) piperazin-1-yI)(4-(tosylamino)phenyl)methanone, 4-(4-((1-
methy1-1H-benzo[d]
imidazole-2-yl)methyl)piperazin-1-y1)-N-tozylbenzenamine, 3-
chloro-4-methyl-N-(6-(4-(3-
(trifluoromethyl)benzyl)piperazin-1-yl)pyridin-3-yl)benzenesulfonamide, 4-
chloro-N-(4-(4-((1-methyl-
1H-benzo[d]imidazol-2-yl)methyl )p iperazin-1-yl)p henyl)benzenesulfonam id e,
(Z)-4-(3-cyano-3-(4-
(2,4-dimethylphenyl)thiazol-2-yl)ally1)-N-(thiazol-2-y1) benzene sulfonamide,
N-(3-(H-imidazo[1,2-a]
pyridine-2-yl)pheny1)-4-methyl-2-phenylth iazole-5-carboxamide, N-(3-
(benzo[d]thiazol-2-y1)
18
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
phenyl)isonicotinamide, 3-(4-
chlorophenyI)-4,5-d ihydro-1-pheny1-5-(2-phenylthiazol-4-y1)-1 H-
pyrazole, N-(4-(6-methylbenzo[d]thiazol-2-yl)pheny1)-2-(N-m-
tolylmethylsulfonamido)acetamide, N-
(4-(6-methylbenzo[d]thiazol-2-yl)pheny1)-2-(N-p-
tolylmethylsulfonamido)acetamide; a
pharmaceutically acceptable salt; a stereoisomer thereof; or any combination
thereof.
In yet another embodiment of the present invention there is provided a method
for treating a
metabolic disorder in an animal, comprising the step of administering to the
animal a therapeutically
effective amount of at least one compound as described supra, or a
pharmaceutically acceptable
salt or a stereoisomer thereof or a combination thereof.
Further to this embodiment the method comprises the step of providing a second
therapy to
the animal. In this further embodiment the second therapy comprises dietary
therapy, physical
therapy, behavior therapy, surgery, drug therapy, chemotherapy, or a
combination thereof.
Particularly the second therapy may be a lifestyle modification, an
antihyperglycemic agent, insulin,
glucagon-like peptide (GLP), a dipeptidyl peptidase-4 inhibitor, a
thiazolidinedione, a lipid lowering
compound or a combination of two or more thereof.
In both embodiments the metabolic disorder is a weight related condition, a
disease related
to cell hyperproliferation, hyperlipemia, diabetes or complications thereof,
fatty liver, hypertension,
or a cardiovascular disease. Particularly, the metabolic disorder may be
obesity, hypertension,
arteriosclerosis, asthma, hyperlipidemia, hyperinsulinemia, non-alcoholic
fatty liver and type 2
diabetes caused by insulin resistance. In an aspect of these embodiments the
metabolic disorder is
a weight-related disorder where the therapeutically effective amount of the
compound increases the
expression of uncoupling protein 1, uncoupling protein 2 or uncoupling protein
3. Also in this aspect
the therapeutically effective amount of the compound increases thermogenesis
without reducing
lean body mass during weight loss in the animal. In another aspect the disease
related to cell
hyperproliferation is a cancer. Particularly, the cancer is a cancer of the
breast, respiratory tract,
brain, reproductive organs, prostate, digestive tract, urinary tract, eye,
liver, skin, head and neck,
thyroid, parathyroid, lymphoma, sarcoma, melanoma, leukemia or a distant
metastasis of a solid
tumor.
In yet another embodiment there is provided a method for treating a cell
hyperproliferative
disease in a patient in need thereof, comprising the step of administering to
the patient a
therapeutically effective amount of at least one compound as described supra,
or a
pharmaceutically acceptable salt or a stereoisomer thereof or a combination
thereof. Particularly,
the cell hyperproliferative disease may be a cancer as described supra.
In a related embodiment, there is provided a method for treating a cancer in a
patient in
need thereof, comprising the step of administering to the patient, a
therapeutically effective amount
of one or more pharmaceutical compositions as described supra. The cancer may
be as described
herein.
In yet another embodiment there is provided a method for reducing body weight
in an
animal in need thereof, comprising the step of administering to the animal, in
a pharmaceutically
acceptable medium, a therapeutically effective amount of one or more of the
compounds as
described supra.
19
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
In yet another embodiment of the present invention, there is provided a method
for
increasing thermogenesis without reducing lean body mass during weight loss in
an animal,
comprising the step of administering to the animal, in a pharmaceutically
acceptable medium, a
therapeutically effective amount of a compound having the structure
R4
,r0
N24 R3
S
wherein the R1 substituents are H, Et, OMe or n-propyl; Y is CH or 0
; R2 is OH, OMe, or NH-
i-Pr; R3 is H, F, or 01; R4 is H, Me, CI, Br, F, OH, OBz, OCH2000Me, OCH2000H,
NH2, NH-i-Pr,
NH.A --
NH__0; NH¨S
N
NHCOMe, NHSO2Me, NHBn, , OMe, NHBoc, , NHTs, 0
or NH¨ I
0 .
or a pharmaceutically acceptable salt or a stereoisomer thereof or a
combination thereof. Using the
method of the present invention, total body fat in the animal is reduced
following administration of
said compound.
In all these embodiments and aspects thereof a person having ordinary skill in
this art could
readily determine a useful dose of a compound of the present invention
depending upon the
metabolic disorder, such as, but not limited to, a disease related to cell
hyperproliferation or a
weight-related disorder, to be treated or the outcome desired. Typically, the
compound is
administered in a dose of from about 1 mg/kg to about 100 mg/kg. The compound
may be
administered in a composition which is in the form of a food, an animal feed
material, or a medicine.
In a preferred aspect, body weight is reduced in the animal by an increase in
uncoupled
thermogenesis with or without a concurrent reduction in lean body mass.
Typically, the compound
increases the expression of uncoupling protein. Representative examples of
uncoupling proteins
include uncoupling protein 1, uncoupling protein 2 and uncoupling protein 3.
The methods of the
present invention would be useful in a variety of situations including, but
not limited to, where the
animal suffers from a weight-related condition selected from the group
consisting of obesity,
hypertension, arteriosclerosis, athsma, hyperlipidemia, hyperinsulinemia, non-
alcoholic fatty liver
and type 2 diabetes caused by insulin resistance. Moreover, a person having
ordinary skill in this
art would readily recognize the utility of providing to the animal a second
therapy including, but not
limited to, a lifestyle modification, an antihyperglycemic agent, insulin,
glucagon-like peptide (GLP),
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
a dipeptidyl peptidase-4 inhibitor, a thiazolidinedione, a lipid lowering
compound, and a combination
of two or more thereof.
As used herein the specification, "a" or "an" may mean one or more. As used
herein in the
claim(s), when used in conjunction with the word "comprising", the words "a"
or "an" may mean one
or more than one. As used herein "another" may mean at least a second or more.
In specific
embodiments, aspects of the invention may "consist essentially of" or "consist
of" one or more
sequences of the invention, for example. Some embodiments of the invention may
consist of or
consist essentially of one or more elements, method steps, and/or methods of
the invention. It is
contemplated that any method or composition described herein can be
implemented with respect to
any other method or composition described herein.
The term "animal" as used herein refers to a mammal, preferably a human,
patient, subject,
or individual that receives or has administered to one or more of the
compounds, compositions or
formulations described herein.
The term "alkyl" as used herein refers to a substituting univalent group
derived by
conceptual removal of one hydrogen atom from a straight or branched-chain
acyclic saturated
hydrocarbon (i.e., -CH3, -CH2CH3, -CH2CH2CH3, -CH(CH3)2, -CH2CH2CH2CH3, -
CH2CH(CH3)2, -
C(CH3)3, etc.).
The term "alkenyl" as used herein refers to a substituting univalent group
derived by
conceptual removal of one hydrogen atom from a straight or branched-chain
acyclic unsaturated
hydrocarbon containing at least one carbon-carbon double bond (i.e., -CH=CH2, -
CH=CHCH3, -
C=C(CH3)2, -CH2CH=CH2, etc.).
The term "alkynyl" as used herein refers to a substituting univalent group
derived by
conceptual removal of one hydrogen atom from a straight or branched-chain
acyclic unsaturated
hydrocarbon containing at least one carbon-carbon triple bond (i.e., -CECH, -
CECCH3, -
CECCH(CH3)2, -CH2CECH, etc.).
The term "aryloxy" as used herein refers to an aryl group with a bridging
oxygen atom, such
as phenoxy (-006H5), or benzoxy (-0CH2C6H5). "Arylamino means an aryl group
with a bridging
amine function such as -NHCH2C6H5. "Arylamido" means an aryl group with a
bridging amide group
such as ¨(C=0)NHCH2C6H5.
The term "alkylidene" as used herein refers to a substituting bivalent group
derived from a
straight or branched-chain acyclic saturated hydrocarbon by conceptual removal
of two hydrogen
atoms from the same carbon atom (i.e., =CH2, =CHCH3, =C(CH3)2, etc.).
The term "cycloalkyl" as used herein refers to a substituting univalent group
derived by
conceptual removal of one hydrogen atom from a saturated monocyclic
hydrocarbon (i.e.,
cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, or cycloheptyl).
The term "aryl" as used herein refers to a substituting univalent group
derived by conceptual
removal of one hydrogen atom from a monocyclic or bicyclic aromatic
hydrocarbon. Examples of
aryl groups are phenyl, indenyl, and naphthyl.
The term "heteroaryl" as used herein refers to a substituting univalent group
derived by the
conceptual removal of one hydrogen atom from a monocyclic or bicyclic aromatic
ring system
21
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
containing 1, 2, 3, or 4 heteroatoms selected from N, 0, or S. Examples of
heteroaryl groups
include, but are not limited to, pyrrolyl, fury!, thienyl, imidazolyl,
pyrazolyl, oxazolyl, isoxazolyl,
thiazolyl, pyridyl, pyrimidinyl, pyrazinyl, benzimidazolyl, indolyl, and
purinyl. Heteraryl substituents
can be attached at a carbon atom or through the heteroatom. Examples of
moncyclic heteroaryl
groups include pyrrolyl, fury!, thienyl, pyrazolyl, oxazolyl, isoxazolyl,
thiazolyl, and pyridyl. Examples
of bicyclic heteroaryl groups include pyrimidinyl, pyrazinyl, benzimidazolyl,
indolyl, and purinyl.
Individual rings may have 5 or 6 atoms. Thus, this includes a 4-membered
moncyclic heteroaryl
group and a 5-memebered monocylcic heteroaryl group. It also includes a
bicyclic heteroaryl group
having one 5-membered ring and one 6-membered ring, and a bicyclic heteroaryl
group having two
6-membered rings.
The term "halo" includes iodo, bromo, chloro and fluoro.
The term "substituted" shall be deemed to include multiple degrees of
substitution by a
substitutent. A substitution occurs where a valence on a chemical group or
moiety is satisfied by an
atome or functional group other than hydrogen. In cases of multiple
substitutions, the substituted
compound can be independently substituted by one or more of the disclosed or
claimed substituent
moieties, singly or plurally. By independently substituted, it is meant that
the (two or more)
substituents can be the same or different.
The term "pharmaceutically acceptable salt" refers herein to a salt of a
compound that
possesses the desired pharmacological activity of the parent compound. Such
salts include: (1) acid
addition salts, formed with inorganic acids such as hydrochloric acid,
hydrobromic acid, sulfuric acid,
nitric acid, phosphoric acid, and the like; or formed with organic acids such
as acetic acid, propionic
acid, hexanoic acid, cyclopentanepropionic acid, glycolic acid, pyruvic acid,
lactic acid, malonic acid,
succinic acid, malic acid, maleic acid, fumaric acid, tartaric acid, citric
acid, benzoic acid, 3-(4-
hydroxybenzoyl)benzoic acid, cinnamic acid, mandelic acid, methanesulfonic
acid, ethanesulfonic
acid, 1,2-ethane-disulfonic acid, 2-hydroxyethanesulfonic acid,
benzenesulfonic acid, 4-
chlorobenzenesulfonic acid, 2-napthalenesulfonic acid, 4-toluenesulfonic acid,
camphorsulfonic
acid, 4-methylbicyclo[2.2.2]-oct-2-ene-1-carboxylic acid, glucoheptonic acid,
3-phenylpropionic acid,
trimethylacetic acid, tertiary butylacetic acid, lauryl sulfuric acid,
gluconic acid, glutamic acid,
hydroxynapthoic acid, salicylic acid, stearic acid, muconic acid, and the
like; or (2) salts formed
when an acidic proton present in the parent compound either is replaced by a
metal ion, e.g., an
alkali metal ion, an alkaline earth ion, or an aluminum ion; or coordinates
with an organic base such
as ethanolamine, diethanolamine, triethanolamine, tromethamine, N-
methylglucamine, and the like.
The term "stereoisomer" means an isomeric molecule whose atomic connectivity
is the
same as one or more other molecules but whose atomic arrangement in space is
different. This
definition includes enantiomers, diastereomers, cis-isomers, trans-isomers,
conformational isomers.
The term "unsubstituted" means all that valences on a chemical group or moiety
are
satisfied by hydrogen.
The term "saturated carbon chain" as used herein refers to a straight or
branched-chain
acyclic saturated hydrocarbon (i.e., -CH3, -CH2-, -CH2CH3, -CH2CH2- -
CH2CH2CH3, -CH2CH2CH2, -
CH2CH2CH2CH3, -CH2CH2CH2CH3-,-CH2CH(CH3)2, -C(CF13)3, etc.).
22
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
The term "unsaturated carbon chain" as used herein refers to a straight or
branched-chain
acyclic unsaturated hydrocarbon containing at least one carbon-carbon double
bond (i.e., -CH=CH2,
-CH=CH-, -CH=CHCH3, -C=C(CH3)2, -CH2CH=CH2, -CH2CH=CH- etc.) or at least one
carbon-
carbon triple bond (i.e., -CECH, -CEO-, -CECCH3, -CECCH-, -CECCH(CH3)2, -
CH2CECH, etc.).
The present invention also includes protected derivatives of compounds
disclosed herein.
For example, when compounds of the present invention contain groups such as
hydroxyl or
carbonyl, these groups can be protected with a suitable protecting group. A
list of suitable
protective groups can be found in T. W. Greene, Protective Groups in Organic
Synthesis, John
Wiley & Sons, Inc. 1981, the disclosure of which is incorporated herein by
reference in its entirety.
The protected derivatives of compounds of the present invention can be
prepared by methods well
known in the art.
The compounds of the present invention may have asymmetric centers, chiral
axes, and
chiral planes, and occur as racemates, racemic mixtures, and as individual
diastereomers, with all
possible isomers and mixtures thereof, including optical isomers, being
included in the present
invention. In addition, the compounds disclosed herein may exist as tautomers
and both tautomeric
forms are intended to be encompassed by the scope of the invention, even
though only one
tautomeric structure may be depicted.
In a specific embodiment of the invention, a composition of the invention
targets one or
more members of a sterol regulatory element binding protein (SREBP) pathway.
The pathway
relates to the proteolytic release of a membrane-bound transcription factor,
SREBP, in specific
aspects, which facilitates transport from the cytoplasm to the nucleus. There,
SREBP binds
elements referred to as the sterol regulatory elements (SREs) present in
regulatory regions of the
genes that encode enzymes associated with production of lipids. Upon binding
of the SREBP to
DNA, transcription of the target gene is modulated, such as upregulated.
Pharmaceutical Preparations
Pharmaceutical compositions of the present invention comprise an effective
amount of one
or more compositions of the invention (and additional agent, where
appropriate) dissolved or
dispersed in a pharmaceutically acceptable carrier. The phrases
"pharmaceutical or
pharmacologically acceptable" refers to molecular entities and compositions
that do not produce an
adverse, allergic or other untoward reaction when administered to an animal,
such as, for example,
a human, as appropriate. The preparation of a pharmaceutical composition that
contains at least
one fatostatin A analog or derivative or additional active ingredient will be
known to those of skill in
the art in light of the present disclosure, as exemplified by Remington's
Pharmaceutical Sciences,
18th Ed. Mack Printing Company, 1990. Moreover, for animal (e.g., human)
administration, it will be
understood that preparations should meet sterility, pyrogenicity, general
safety and purity standards
as required by FDA Office of Biological Standards.
As used herein, "pharmaceutically acceptable carrier includes any and all
solvents,
dispersion media, coatings, surfactants, antioxidants, preservatives (e.g.,
antibacterial agents,
antifungal agents), isotonic agents, absorption delaying agents, salts,
preservatives, drugs, drug
23
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
stabilizers, gels, binders, excipients, disintegration agents, lubricants,
sweetening agents, flavoring
agents, dyes, such like materials and combinations thereof, as would be known
to one of ordinary
skill in the art (see, for example, Remington's Pharmaceutical Sciences, 18th
Ed. Mack Printing
Company, 1990, pp. 1289-1329). Except insofar as any conventional carrier is
incompatible with
the active ingredient, its use in the pharmaceutical compositions is
contemplated.
The fatostatin A analog or derivative may comprise different types of carriers
depending on
whether it is to be administered in solid, liquid or aerosol form, and whether
it need to be sterile for
such routes of administration as injection. The present invention can be
administered intravenously,
intradermally, transdermally, intrathecally,
intraarterially, intraperitoneally, intranasally,
intravaginally, intrarectally, topically, intramuscularly, subcutaneously,
mucosally, orally, topically,
locally, inhalation, e.g., aerosol inhalation, injection, infusion, continuous
infusion, localized
perfusion bathing target cells directly, via a catheter, via a lavage, in
cremes, in lipid compositions,
e.g., liposomes, or by other method or any combination of the forgoing as
would be known to one of
ordinary skill in the art (see, for example, Remington's Pharmaceutical
Sciences, 18th Ed. Mack
Printing Company, 1990).
The fatostatin A analog or derivative may be formulated into a composition in
a free base,
neutral or salt form. Pharmaceutically acceptable salts, include the acid
addition salts, e.g., those
formed with the free amino groups of a proteinaceous composition, or which are
formed with
inorganic acids such as for example, hydrochloric or phosphoric acids, or such
organic acids as
acetic, oxalic, tartaric or mandelic acid. Salts formed with the free carboxyl
groups can also be
derived from inorganic bases such as for example, sodium, potassium, ammonium,
calcium or ferric
hydroxides; or such organic bases as isopropylamine, trimethylamine, histidine
or procaine. Upon
formulation, solutions will be administered in a manner compatible with the
dosage formulation and
in such amount as is therapeutically effective. The formulations are easily
administered in a variety
of dosage forms such as formulated for parenteral administrations such as
injectable solutions, or
aerosols for delivery to the lungs, or formulated for alimentary
administrations such as drug release
capsules and the like.
Further in accordance with the present invention, the composition of the
present invention
suitable for administration is provided in a pharmaceutically acceptable
carrier with or without an
inert diluent. The carrier should be assimilable and includes liquid, semi-
solid, i.e., pastes, or solid
carriers. Except insofar as any conventional media, agent, diluent or carrier
is detrimental to the
recipient or to the therapeutic effectiveness of a the composition contained
therein, its use in
administrable composition for use in practicing the methods of the present
invention is appropriate.
Examples of carriers or diluents include fats, oils, water, saline solutions,
lipids, liposomes, resins,
binders, fillers and the like, or combinations thereof. The composition may
also comprise various
antioxidants to retard oxidation of one or more component. Additionally, the
prevention of the action
of microorganisms can be brought about by preservatives such as various
antibacterial and
antifungal agents, including but not limited to parabens, e.g.,
methylparabens, and propylparabens,
chlorobutanol, phenol, sorbic acid, thimerosal or combinations thereof. In
accordance with the
present invention, the composition is combined with the carrier in any
convenient and practical
24
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
manner, i.e., by solution, suspension, emulsification, admixture,
encapsulation, absorption and the
like. Such procedures are routine for those skilled in the art.
In a specific embodiment of the present invention, the composition is combined
or mixed
thoroughly with a semi-solid or solid carrier. The mixing can be carried out
in any convenient
manner such as grinding. Stabilizing agents can be also added in the mixing
process in order to
protect the composition from loss of therapeutic activity, i.e., denaturation
in the stomach.
Examples of stabilizers for use in an composition include buffers, amino acids
such as glycine and
lysine, carbohydrates such as dextrose, mannose, galactose, fructose, lactose,
sucrose, maltose,
sorbitol, mannitol, etc.
In further embodiments, the present invention may concern the use of a
pharmaceutical lipid
vehicle compositions that include fatostatin A analog or derivative, one or
more lipids, and an
aqueous solvent. As used herein, the term "lipid" will be defined to include
any of a broad range of
substances that is characteristically insoluble in water and extractable with
an organic solvent. This
broad class of compounds are well known to those of skill in the art, and as
the term "lipid" is used
herein, it is not limited to any particular structure. Examples include
compounds which contain long-
chain aliphatic hydrocarbons and their derivatives. A lipid may be naturally
occurring or synthetic
(i.e., designed or produced by man). However, a lipid is usually a biological
substance. Biological
lipids are well known in the art, and include for example, neutral fats,
phospholipids,
phosphoglycerides, steroids, terpenes, lysolipids, glycosphingolipids,
glycolipids, sulphatides, lipids
with ether and ester-linked fatty acids and polymerizable lipids, and
combinations thereof.
One of ordinary skill in the art would be familiar with the range of
techniques that can be
employed for dispersing a composition in a lipid vehicle. For example, the
fatostatin A analog or
derivative may be dispersed in a solution containing a lipid, dissolved with a
lipid, emulsified with a
lipid, mixed with a lipid, combined with a lipid, covalently bonded to a
lipid, contained as a
suspension in a lipid, contained or complexed with a micelle or liposome, or
otherwise associated
with a lipid or lipid structure by any means known to those of ordinary skill
in the art. The dispersion
may or may not result in the formation of liposomes.
The actual dosage amount of a composition of the present invention
administered to an
animal patient can be determined by physical and physiological factors such as
body weight,
severity of condition, the type of disease being treated, previous or
concurrent therapeutic
interventions, idiopathy of the patient and on the route of administration.
Depending upon the
dosage and the route of administration, the number of administrations of a
preferred dosage and/or
an effective amount may vary according to the response of the subject.
In certain embodiments, pharmaceutical compositions may comprise, for example,
at least
about 0.1% of an active compound. In other embodiments, the an active compound
may comprise
between about 2% to about 75% of the weight of the unit, or between about 25%
to about 60%, for
example, and any range derivable therein. Naturally, the amount of active
compound(s) in each
therapeutically useful composition may be prepared is such a way that a
suitable dosage will be
obtained in any given unit dose of the compound. Factors such as solubility,
bioavailability,
biological half-life, route of administration, product shelf life, as well as
other pharmacological
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
considerations will be contemplated by one skilled in the art of preparing
such pharmaceutical
formulations, and as such, a variety of dosages and treatment regimens may be
desirable.
In other non-limiting examples, a dose may also comprise from about 1
microgram/kg/body
weight, about 5 microgram/kg/body weight, about 10 microgram/kg/body weight,
about 50
microgram/kg/body weight, about 100 microgram/kg/body weight, about 200
microgram/kg/body
weight, about 350 microgram/kg/body weight, about 500 microgram/kg/body
weight, about 1
milligram/kg/body weight, about 5 milligram/kg/body weight, about 10
milligram/kg/body weight,
about 50 milligram/kg/body weight, about 100 milligram/kg/body weight, about
200
milligram/kg/body weight, about 350 milligram/kg/body weight, about 500
milligram/kg/body weight,
to about 1000 mg/kg/body weight or more per administration, and any range
derivable therein. In
non-limiting examples of a derivable range from the numbers listed herein, a
range of about 5
mg/kg/body weight to about 100 mg/kg/body weight, about 5 microgram/kg/body
weight to about
500 milligram/kg/body weight, etc., can be administered, based on the numbers
described above.
Alimentary Compositions and Formulations
The fatostatin A analog or derivative is formulated to be administered via an
alimentary
route. Alimentary routes include all possible routes of administration in
which the composition is in
direct contact with the alimentary tract. Specifically, the pharmaceutical
compositions disclosed
herein may be administered orally, buccally, rectally, or sublingually. As
such, these compositions
may be formulated with an inert diluent or with an assimilable edible carrier,
or they may be
enclosed in hard- or soft- shell gelatin capsule, or they may be compressed
into tablets, or they may
be incorporated directly with the food of the diet.
The active compounds may be incorporated with excipients and used in the form
of
ingestible tablets, buccal tables, troches, capsules, elixirs, suspensions,
syrups, wafers, and the like
(Mathiowitz etal., 1997; Hwang etal., 1998; U.S. Pat. Nos. 5,641,515;
5,580,579; and 5,792,451).
The tablets, troches, pills, capsules and the like may also contain the
following: a binder, such as,
for example, gum tragacanth, acacia, cornstarch, gelatin or combinations
thereof; an excipient, such
as, for example, dicalcium phosphate, mannitol, lactose, starch, magnesium
stearate, sodium
saccharine, cellulose, magnesium carbonate or combinations thereof; a
disintegrating agent, such
as, for example, corn starch, potato starch, alginic acid or combinations
thereof; a lubricant, such
as, for example, magnesium stearate; a sweetening agent, such as, for example,
sucrose, lactose,
saccharin or combinations thereof; a flavoring agent, such as, for example
peppermint, oil of
wintergreen, cherry flavoring, orange flavoring, etc. When the dosage unit
form is a capsule, it may
contain, in addition to materials of the above type, a liquid carrier. Various
other materials may be
present as coatings or to otherwise modify the physical form of the dosage
unit. For instance,
tablets, pills, or capsules may be coated with shellac, sugar, or both. When
the dosage form is a
capsule, it may contain, in addition to materials of the above type, carriers
such as a liquid carrier.
Gelatin capsules, tablets, or pills may be enterically coated. Enteric
coatings prevent denaturation of
the composition in the stomach or upper bowel where the pH is acidic. See,
e.g., U.S. Pat. No.
5,629,001. Upon reaching the small intestines, the basic pH therein dissolves
the coating and
26
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
permits the composition to be released and absorbed by specialized cells,
e.g., epithelial
enterocytes and Peyer's patch M cells. A syrup of elixir may contain the
active compound sucrose
as a sweetening agent methyl and propylparabens as preservatives, a dye and
flavoring, such as
cherry or orange flavor. Of course, any material used in preparing any dosage
unit form should be
pharmaceutically pure and substantially non-toxic in the amounts employed. In
addition, the active
compounds may be incorporated into sustained-release preparation and
formulations.
For oral administration the compositions of the present invention may
alternatively be
incorporated with one or more excipients in the form of a mouthwash,
dentifrice, buccal tablet, oral
spray, or sublingual orally- administered formulation. For example, a
mouthwash may be prepared
incorporating the active ingredient in the required amount in an appropriate
solvent, such as a
sodium borate solution (Dobell's Solution). Alternatively, the active
ingredient may be incorporated
into an oral solution such as one containing sodium borate, glycerin and
potassium bicarbonate, or
dispersed in a dentifrice, or added in a therapeutically-effective amount to a
composition that may
include water, binders, abrasives, flavoring agents, foaming agents, and
humectants. Alternatively
the compositions may be fashioned into a tablet or solution form that may be
placed under the
tongue or otherwise dissolved in the mouth.
Additional formulations which are suitable for other modes of alimentary
administration
include suppositories. Suppositories are solid dosage forms of various weights
and shapes, usually
medicated, for insertion into the rectum. After insertion, suppositories
soften, melt or dissolve in the
cavity fluids. In general, for suppositories, traditional carriers may
include, for example, polyalkylene
glycols, triglycerides or combinations thereof. In certain embodiments,
suppositories may be formed
from mixtures containing, for example, the active ingredient in the range of
about 0.5% to about
10%, and preferably about 1% to about 2%.
Parenteral Compositions and Formulations
The fatostatin A analog or derivative may be administered via a parenteral
route. As used
herein, the term "parenteral" includes routes that bypass the alimentary
tract. Specifically, the
pharmaceutical compositions disclosed herein may be administered for example,
but not limited to
intravenously, intradermally, intramuscularly, intraarterially, intrathecally,
subcutaneous, or
intraperitoneally U.S. Pat. Nos. 6,537,514, 6,613,308, 5,466,468, 5,543,158;
5,641,515; and
5,399,363.
Solutions of the active compounds as free base or pharmacologically acceptable
salts may
be prepared in water suitably mixed with a surfactant, such as
hydroxypropylcellulose. Dispersions
may also be prepared in glycerol, liquid polyethylene glycols, and mixtures
thereof and in oils.
Under ordinary conditions of storage and use, these preparations contain a
preservative to prevent
the growth of microorganisms. The pharmaceutical forms suitable for injectable
use include sterile
aqueous solutions or dispersions and sterile powders for the extemporaneous
preparation of sterile
injectable solutions or dispersions (U.S. Patent 5,466,468). In all cases the
form must be sterile and
must be fluid to the extent that easy injectability exists. It must be stable
under the conditions of
manufacture and storage and must be preserved against the contaminating action
of
27
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
microorganisms, such as bacteria and fungi. The carrier can be a solvent or
dispersion medium
containing, for example, water, ethanol, polyol (i.e., glycerol, propylene
glycol, and liquid
polyethylene glycol, and the like), suitable mixtures thereof, and/or
vegetable oils. Proper fluidity
may be maintained, for example, by the use of a coating, such as lecithin, by
the maintenance of the
required particle size in the case of dispersion and by the use of
surfactants. The prevention of the
action of microorganisms can be brought about by various antibacterial and
antifungal agents, for
example, parabens, chlorobutanol, phenol, sorbic acid, thimerosal, and the
like. In many cases, it
will be preferable to include isotonic agents, for example, sugars or sodium
chloride. Prolonged
absorption of the injectable compositions can be brought about by the use in
the compositions of
agents delaying absorption, for example, aluminum monostearate and gelatin.
For parenteral administration in an aqueous solution, for example, the
solution should be
suitably buffered if necessary and the liquid diluent first rendered isotonic
with sufficient saline or
glucose. These particular aqueous solutions are especially suitable for
intravenous, intramuscular,
subcutaneous, and intraperitoneal administration. In this connection, sterile
aqueous media that
can be employed will be known to those of skill in the art in light of the
present disclosure. For
example, one dosage may be dissolved in 1 ml of isotonic NaCI solution and
either added to 1000
ml of hypodermoclysis fluid or injected at the proposed site of infusion, (see
for example,
"Remington's Pharmaceutical Sciences" 15th Edition, pages 1035-1038 and 1570-
1580). Some
variation in dosage will necessarily occur depending on the condition of the
subject being treated.
The person responsible for administration will, in any event, determine the
appropriate dose for the
individual subject.
Sterile injectable solutions are prepared by incorporating the active
compounds in the
required amount in the appropriate solvent with various of the other
ingredients enumerated above,
as required, followed by filtered sterilization. Generally, dispersions are
prepared by incorporating
the various sterilized active ingredients into a sterile vehicle which
contains the basic dispersion
medium and the required other ingredients from those enumerated above. In the
case of sterile
powders for the preparation of sterile injectable solutions, the preferred
methods of preparation are
vacuum-drying and freeze-drying techniques which yield a powder of the active
ingredient plus any
additional desired ingredient from a previously sterile-filtered solution
thereof. A powdered
composition is combined with a liquid carrier such as, e.g., water or a saline
solution, with or without
a stabilizing agent.
The active compound fatostatin A analog or derivative may be formulated for
administration
via various miscellaneous routes, for example, topical (i.e., transdermal)
administration, mucosa!
administration (intranasal, vaginal, etc.) and/or inhalation. Pharmaceutical
compositions for topical
administration may include the active compound formulated for a medicated
application such as an
ointment, paste, cream or powder. Ointments include all oleaginous,
adsorption, emulsion and
water-solubly based compositions for topical application, while creams and
lotions are those
compositions that include an emulsion base only. Topically administered
medications may contain
a penetration enhancer to facilitate adsorption of the active ingredients
through the skin. Suitable
penetration enhancers include glycerin, alcohols, alkyl methyl sulfoxides,
pyrrolidones and
28
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
luarocapram. Possible bases for compositions for topical application include
polyethylene glycol,
lanolin, cold cream and petrolatum as well as any other suitable absorption,
emulsion or water-
soluble ointment base. Topical preparations may also include emulsifiers,
gelling agents, and
antimicrobial preservatives as necessary to preserve the active ingredient and
provide for a
homogenous mixture. Transdermal administration of the present invention may
also comprise the
use of a "patch". For example, the patch may supply one or more active
substances at a
predetermined rate and in a continuous manner over a period of time.
The pharmaceutical compositions may be delivered by eye drops, intranasal
sprays,
inhalation, and/or other aerosol delivery vehicles. Methods for delivering
compositions directly to
the lungs via nasal aerosol sprays has been described e.g., in U.S. Pat. Nos.
5,756,353 and
5,804,212. Likewise, the delivery of drugs using intranasal microparticle
resins and
lysophosphatidyl-glycerol compounds (U.S. Pat. No. 5,725,871) are also well-
known in the
pharmaceutical arts. Transmucosal drug delivery in the form of a
polytetrafluoroetheylene support
matrix is described in U.S. Pat. No. 5,780,045.
The term aerosol refers to a colloidal system of finely divided solid of
liquid particles
dispersed in a liquefied or pressurized gas propellant. The typical aerosol of
the present invention
for inhalation will consist of a suspension of active ingredients in liquid
propellant or a mixture of
liquid propellant and a suitable solvent. Suitable propellants include
hydrocarbons and hydrocarbon
ethers.
Combination Therapy
In order to increase the effectiveness of a composition of the invention, an
additional
therapy may be delivered to an individual having a metabolic disorder. For
example, an individual
that is obese may be administered a composition of the invention in addition
to another therapy for
obesity. Additional obesity therapies include dietary therapy, physical
therapy (exercise), drug
therapy, surgery, and behavioral therapy, for example. Exemplary drug
therapies include, for
example, Xenical Orlistat , Phentermine, and Sibutramine (Meridia ). Exemplary
surgeries include
liposuction and gastric bypass, for example.
For individuals with diabetes, for example, exemplary additional compounds for
therapy
include one or more of the following: Actos (pioglitizone); ACTOSPlus Met;
Amaryl (glimepiride);
Avandaryl (Avandia + Glimiperide); Avandia (rosiglitazone); Avandamet
(rosiglitazone maleate and
metformin hydrochloride); Byettap; Duetact (pioglitazone HCI and glimepiride);
Galvus (Vildagliptin);
Glipizide (Sulfonlyurea); Glucophage (metformin); Glimepiride; Glucovance
(glyburide/metformin);
Glucotrol XL (glipizide extended release); Glyburide; Glyset (miglitol)
glucosidase inhibitor; Januvia
(sitagliptin phosphate); Metaglip (glipizide+metformin); Metformin ¨
biguanide; Prandin (repaglinide);
Precose (acarbose); Rezulin (troglitazone); Starlix (nateglinide). Other
therapies for diabetes
include an improvement in diet and exercise.
29
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
Exemplary Measurement of Metabolic Disorder Treatments
In particular aspects of the invention, an individual is given one or more
compositions of the
present invention and the individual is assessed for an improvement in at
least one symptom of the
metabolic disorder. For example, in particular embodiments when the metabolic
disorder is obesity,
an improvement in obesity may be determined during and/or following treatment
with one or more
compositions of the invention. An improvement in obesity may be measured by
any standard
means, but in particular aspects the improvement in obesity is measured by
weight measurement,
body mass index (BMI) measurement, and/or body part size measurement (such as
waist
measurement), for example. Exemplary methods for calculating BMI includes
dividing a person's
body weight in kilograms by their height in meters squared (weight [kg] height
[m]2). A BMI of 30 or
more is considered obese and a BMI between 25 to 29.9 is considered
overweight. In other aspects
of the invention, an individual with diabetes is tested for an improvement
following administration to
the individual of the therapy of the invention. In one specific embodiment,
the monitoring of
diabetes occurs by blood test. For example, the blood test may measure the
chemical A1C. The
higher the blood sugar, the higher the A1C level will be. In some cases,
cholesterol (including HDL
and/or [DL cholesterol) and/or triglycerides are measured, such as by standard
means in the art. In
specific cases, a fasting lipoprotein profile is performed, such as by
standard means in the art.
Kits of the Invention
Any of the compositions described herein may be comprised in a kit. In a non-
limiting
example, the kit comprises a composition suitable for treatment and/or
prevention of one or more
metabolic disorders. In other embodiments of the invention, the kit comprises
one or more
apparatuses to obtain a sample from an individual. Such an apparatus may be
one or more of a
swab, such as a cotton swab, toothpick, scalpel, spatula, syringe, and so
forth, for example. In
another embodiment, an additional compound is provided in the kit, such as an
additional
compound for treatment and/or prevention of a metabolic disorder. Any
compositions that are
provided in the kits may be packaged either in aqueous media or in lyophilized
form, for example.
The container means of the kits will generally include at least one vial, test
tube, flask, bottle,
syringe or other container means, into which a component may be placed, and
preferably, suitably
aliquoted. Where there are more than one component in the kit, the kit also
will generally contain a
second, third or other additional container into which the additional
components may be separately
placed.
The following examples are included to demonstrate preferred embodiments of
the
invention. It should be appreciated by those of skill in the art that the
techniques disclosed in the
examples which follow represent techniques discovered by the inventor to
function well in the
practice of the invention, and thus can be considered to constitute preferred
modes for its practice.
However, those of skill in the art should, in light of the present disclosure,
appreciate that many
changes can be made in the specific embodiments which are disclosed and still
obtain a like or
similar result without departing from the spirit and scope of the invention.
30
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
EXAMPLE 1
Fatostatin A reduces the expression of SREBP-responsive genes
Gene expression profile comparison of the drug-treated and untreated cells
might reveal
specific molecular pathways affected by fatostatin A. DU145 cells were treated
with fatostatin A or
DMSO alone, and extracted mRNA samples were analyzed by Affimetrix DNA
microarrays mapping
33,000 genes (Table 1).
Table 1. Genes known or likely to be controled by SREBPs were regulated by
Fatostatin A
showed in Microarray result
Genes known to be controlled by SREBP
Gene Code Decreased fold Name of genes
NM_000527.2 0.574349 low density lipoprotein receptor (LDLR)
NM000859.1 0.5 3-hydroxy-3-methylglutaryl-Coenzyme A
reductase
(HMG CoA R)
NM 0002130.1 0.353553 3-hydroxy-3-methylglutaryl-Coenzyme A
synthase 1
(HMG CoA S)
NM_001096.1 0.574349 ATP citrate lyase
NM_000664.1 0.574349 acetyl-Coenzyme A carboxylase alpha
NM_005063.1 0.574349 stearoyl-CoA desaturase (SCD)
NM_002004.1 0.659754 farnesyl pyrophosphate syntlietase
AK000162 0.535887 acetyl-CoA synthetase
NM_000431.1 0.5 me valonate kinase (MVK)
NM002461.1 0.329877 mevalonate decarboxylase (MVD)
NM 003129.2 0.5 squalene epoxidase
Genes relevant to sterol/fat synthesis
Gene Code Decreased fold
NM_022977.1 0.707107 fatty-acid-Coenzyme A ligase long chain 4
NM_004457.2 0.707107 fatty-acid-Coenzyme A ligase long chain 3
NM005931.1 0.659754 fatty acid desaturase 1
N M0019312 0.659754 dihydrolipoamide S-acetyltransferase
AF167438 0.659754 Homo sapiens androgen-regulated short-chain
dehydrogenaseneductase 1
NM006579.1 0.615572 emopamil-bindingprotein (sterol isomerase)
D63807.1 0.615572 lanosterol synthase
BC000408.1 0.574349 acetyl-Coenzyme A acetyltransferase 2
NM_004462.1 0.535887 famesyl-diphosphate famesyltransferase 1
D85181.1 0.5 sterol-05-desaturase
NM_016371.1 0.466517 hydroxysteroid (17-beta) dehydrogenase 7
NM 005542.1 0.329877 Insulin induced gene 1 (INSIG1)
31
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
The results showed that 55% of the genes downregulated (< 0.7 fold) by
fatostatin A are
those known or likely to be controlled by sterol regulatory element binding
protein (SREBP),
including LDL receptor, HMG-CoA reductase, and fatty acid synthase (Horton et
al., 2003). The
downregulation of the representative SREBP-responsive genes were confirmed by
RT-PCR
experiments (FIGS. 1A-16). These results indicated that fatostatin A is a
selective inhibitor of the
SREBP pathway.
To show that fatostatin A impairs the function of SREBPs, the ability of
endogenous
SREBPs to activate transcription of an SREBP-responsive reporter gene was
measured in the
presence or absence of fatostatin A in HEK293 cells (FIGS. 2A-26). Fatostatin
A decreased in a
concentration dependent manner the activation of the reporter gene in which
the expression of
luciferase is controlled by three repeats of sterol regulatory elements. In
contrast, fatostatin A failed
to impair the ability of an exogenously expressed mature form of SREBP-1
(amino acids 1-500) to
activate the reporter gene activity (FIG. 2C). These results indicate that
fatostatin A selectively
blocks the activation process of SREBPs in cells.
EXAMPLE 2
Fatostatin A blocks the proteolytic activation of SREBPS
To examine whether fatostatin A affects the proteolytic activation of SREBPs,
whole cell
lysates of DU145 cells treated with fatostatin A were analyzed by western
blots with an antibody
against the NH2 terminus of SREBP-1 (FIG. 3A). The treatment of fatostatin A
decreased the
amounts of the 68 KDa mature form of SREBP-1 in a dose-dependent manner, while
the amounts
of the 125 KDa precursor form increased. Similar results were obtained for
SREBP-2 with an
antibody against its COOH terminus (FIG. 3B). These results indicate that
fatostatin A directly or
indirectly impairs the proteolytic activation of both SREBP isoforms.
The inhibition of the proteolytic activation of SREBPs would impair the
nuclear translocation
of SREBPs. Effects of fatostatin A on the subcellular localization of SREBP-1
were analyzed by
immunofluoresence microscopy with an antibody against the NH2 terminus of
SREBP-1. When cells
were treated with DMSO alone, SREBP-1 was localized almost exclusively in the
nucleus in a
serum-free (fat free) medium (FIGS. 3C-3E). In contrast, when the cells were
incubated with
fatostatin A, the immunofluorescence of SREBP-1 decreased in the nucleus and
reciprocally
increased outside of the nucleus (FIGS. 3F-3H), indicating that fatostatin A
inhibits the nuclear
localization of SREBP-1.
EXAMPLE 3
Validation of the Fatostatin phenotypes by knocking down SREBP-1
Fatostatin A causes two phenotypes in cultured cells: (i) inhibition of the
insulin-induced
adipogenesis of 3T3-L1 cells and (ii) repression of the serum-independent
growth of DU145
prostate cancer cells. The first phenotype is in complete agreement with the
conclusion that
fatostatin A is a blocker of SREBP-1 because of the known role of SREBP-1 in
lipogenesis
(Tontonoz et a/., 1993). To confirm that under the cell-culture condition, the
expression of SREBP-1
32
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
in 3T3-L1 cells was silenced by transfecting an expression vector of a small
interfering RNA (siRNA)
specific for SREBP-1 (FIG. 4G), and the effects of the silencing on the
insulin-induced adipogenesis
were examined. As expected, the knockdown of the SREBP-1 expression completely
blocked the oil
droplet formation of 3T3-L1 cells (FIGS. 4D-4F, clones 1 and 2), whereas the
control cells
transfected with an empty vector (neo; FIG. 4A) showed as much fat
accumulation as the parental
3T3-L1 cells (FIG. 4B). These results indicate that the fatostatin A-induced
phenotype in 3T3-L1
cells is mediated by the inhibition of SREBP-1.
To test whether the inhibition of SREBP-1 by fatostatin A mediates the
repression of serum-
independent growth of DU145 cells, the expression of SREBP-1 in DU145 cells
was silenced
similarly by transfecting an expression vector of the SREBP-1-specific siRNA
(FIG. 5B). The control
cells transfected with the empty vector (neo) grew in the presence of either
serum or IGF1, just as
the parental DU145 cells did. In contrast, the knockdown cells in which the
expression of SREBP-1
is silenced (clones 1 and 2) displayed reduced serum-independent IGF1-driven
growth whereas
their serum-dependent growth had little effects (FIG. 5A).
The requirement of SREBP-1 in the serum-independent growth may be due to the
lack of
external fat sources in the serum-free medium. Without exogenous fatty acids
present in the serum,
cells need to synthesize fatty acids and cholesterol, the building blocks of
membranes, to maintain
the cell growth. To test the importance of fatty acids in the cell growth, the
growth of the SREBP-1
knockdown cells was monitored in a fat-free serum medium (FIG. 5A). The SREBP-
1 silencing
impaired the cell growth in a fat-free medium as much as it did in the serum-
free IGF1-containing
medium. These results indicate that fatostatin A blocks the serum-independent
growth of cancer
cells through the inhibition of SREBP-1.
EXAMPLE 4
Fatostatin A reduces body weight, lowers cholesterol and glucose levels, and
downregulates
lipogenic enzymes in mice
The drug-like chemical structure of fatostatin A prompted the inventors to
investigate its
ability to inhibit SREBP-1 in the liver of whole animals. The effect of
fatostatin A on hepatic SREBP-
1 under lipogenic conditions of prolonged fasting (48 hours) followed by
feeding fat free high
carbohydrate diet for another 48 hours was examined. Mice were
intraperitoneally injected with
fatostatin A at 30 mg/kg/day for 5 days starting one day prior to the 48-hour
fasting period. After 48
hours of fasting, the treated group lost more weight than the control group
did (6.12 0.6 compared
to 4.9 0.3 gram/mouse; p=0.01.) (FIG. 6A). No reduction of food intake or
obvious toxicity were
observed during the treatment (FIG. 6B). Interestingly, after 48 hours of
refeeding with fat free high
carbohydrate diet, there was a trend of lowering glucose levels (110 23
compared to 137 14
mg/di; P= 0.06 and cholesterol (93 20 compared to 120 19 mg/di; P= 0.12)
in the serum of
fatostatin A-treated mice (FIG. 60). Both HDL and LDL decreased in the treated
mice group.
However, the decrease in LDL levels appeared to be more significant (16 5
compared to 30 6
mg/di) (FIG. 60).
33
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
The expression levels of SREBP-1 in the liver extracts were examined by
western blots.
Consistent with the cell culture results, the liver extracts from the mice
treated with fatostatin A
displayed decreased amounts of the 68 KDa mature form of SREBP-1 and increased
amounts of
the 125 KDa precursor form (FIG. 60). The hepatic expression of fatty acid
synthase (FAS), a
representative of SREBP-1-responsive lipogenic enzymes (Boizard et al., 1998),
was also
determined after the treatment. Western blot analysis of liver extracts showed
that expression levels
of FAS was decreased up to 30% by the fatostatin A treatment (FIG. 6E).
Consistent with the
reduction of the expression, its enzymatic activity in the extracts was
similarly decreased. The
activity of acetyl-CoA carboxylase (ACC) (FIG. 6F), which is also regulated by
SREBP-1, decreased
in liver extracts as observed for FAS (FIG. 6G). These results indicate that
fatostatin A blocks the
activation of SREBP-1 in mouse liver just as found in the cultured cells.
Longer treatment (two weeks) of another group of mice fed with normal diet
resulted in 10%
loss of body weight whereas the control group had no change of body weight
(FIGS. 7A-7B). Food
intake was similar between both groups (3.8 and 3.5 g/mouse/day for treated
and control mice,
respectively). Consistent with the results of mice fed under fasting/refeeding
fat free diet, mice fed
with normal diet exhibited significantly lower glucose levels and a trend of
lower triglyceride (TG)
and cholesterol levels in the blood (FIG. 7C). FAS activity and its protein
level were also decreased
about 30% (FIGS. 7D and 7E).
EXAMPLE 5
Significance of the present invention
Bioactive small molecules have proven to be valuable tools for exploring
complex cellular
processes including metabolic pathways. A key regulator of lipid homeostasis
and insulin action is a
family of SREBP transcription factors (Brown and Goldstein, 1997). Small
molecules that modulate
the SREBP functions may find their use in the treatment of metabolic diseases
and may serve as
tools for further molecular understanding of the diseases. The cell-based and
animal data suggest
that fatostatin A impairs the expression of lipogenic genes through
downregulating the amounts of
the mature SREBP-1 form in the nucleus.
Small molecules that activate SREBP-1 and -2 were been reported. These LDL-
lowering
molecules upregulate the expression of LDL receptor by stimulating the
proteolytic activation of
SREBPs. Although the molecular mechanism of the action has not been
elucidated, data suggest
that SOAP is a primary target of the molecules. Unlike these molecules,
fatostain A inhibits the
activation of SREBPs and downregulates the expression of SREBP-responsive
genes including the
gene of LDL receptor (Table 1).
The animal data of fatostatin A are consistent with the cell culture results.
In liver extracts of
mice treated with fatostatin A under refeeding fat free diet, there was a
significantly lower level of
mature SREBP-1 form and a higher level of the precursor form. On the other
hand and as expected,
the level of the mature form was higher than the precursor form in liver
extracts of the control group
(Horton et al., 1998). Interestingly, it seems that the there was no change in
the overall amount of
the combined forms, and the only difference is at the distribution between the
nuclear (mature) and
34
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
the cytosolic forms (precursor). These data indicate that fatostatin A may not
alter the expression
level of SREBP-1, rather enhances the cleavage process of the precursor
resulting in a decrease in
its amount and an increase in the mature and active form. The importance of
the SREBP-1
cleavage in fat synthesis has been shown by the experiments using mice
deficient in SOAP: the
livers of the mice failed to induce the expression of ACC and FAS under
refeeding conditions (Liang
etal., 2002).
In order to assess the physiological significance of the reduced level of the
nuclear SREBP-
1, the levels and activities of ACC and FAS were determined. Their activities
in liver extracts were
downregulated in response to fatostatin A treatment. These results are
consistent with the role of
SREBP-1 as a regulator of the fatty acid synthesis pathway (Shimano, 2000).
Shimano et al.
showed that the levels of FAS and ACC were not induced when SREBP-fl- mice
were fed with high
carbohydrate diet, confirming the role of SREBP-1 in regulating the expression
of the lipogenic
enzymes.
An interesting observation in the fatostatin A treated mice compared to
control is a reduction
of body weight and blood glucose. The reduction in body weight may be due to a
lower lipogenesis
rate, as a result of the downregulation of lipogenic enzymes such as ACC and
FAS. In addition, the
reduction in malonyl-CoA, the product of ACC and a potent inhibitor of
carnitine palmitoyl
transferase, may result in enhanced fatty acid oxidation and fat burning. In a
specific embodiment,
inhibition of SREBP-1 cleavage by fatostatin A downregulates lipogenic
enzymes, enhances fatty
acid oxidation, reduces weight, and increases insulin sensitivity resulting in
lowering glucose.
EXAMPLE 6
Materials
Lipid-depleted serum was prepared as described (Goldstein etal., 1983). Fat-
free FBS was
obtained from Fisher. Rabbit anti-SREBP-1 (sc-8984) and goat anti-actin (sc-
1616) polyclonal
antibody were purchased from Santa Cruz Biotechnology. Mouse anti-SREBP-2
polyclonal antibody
and mouse anti-FAS antibody were obtained from BD Biosciences. Anti-goat IgG
HRP and anti-
rabbit IgG HRP were obtained from Promega. ProLong Gold antifade reagent with
DAPI was
obtained from Molecular Probes Invitrogen Detection Technologies. Anti-rabbit
IgG FITC was
obtained from Chemicon International. Dexamethasone (DEX) and 1-methyl- 3-
isobutylxanthin
(MIX) were obtained from Sigma.
Preparation of fatostatin A
A mixture of 2-bromo-4'-methylacetophenone (1.22 g, 5.70 mmol) and
prothionamide (1.03
g, 5.70 mmol) in ethanol (20 nil) was heated at 70 C with stirring for 0.5
hour, and then cooled to 0
C. A yellow precipitate formed was filtered, washed with cold ethanol, and
dried to give fatostatin A
HBr salt (1.78 g, 83%) as yellow needles: 1H NMR (DMSO-d6, 600 MHz) dH 8.88
(d, J =6.2 Hz, 1H),
8.54(s, 1H), 8.46(d, J =1.4 Hz, 1H), 8.36 (dd, J =1.4, 6.2 Hz, 1H), 7.99(d,
J=7.6 Hz, 2H), 7.31 (d, J
=7.6 Hz, 2H), 3.03 (t, J =7.6 Hz, 2H), 2.35 (s, 3H), 1.80 (m, 2H), 0.96 (t, J
=7.6, 3H); HRMS (FAB)
exact mass calcd for Cl8H18N2S + H requires m/z 295.1269, found m/z 295.1269.
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
Cell culture
DU145 human androgen-independent prostate cancer cells (ATCC) were maintained
in an
Eagle's minimum essential medium containing 2 mM L-glutamine, 1.0 mM sodium
pyruvate, 0.1 mM
nonessential amino acids, and 1.5 g/L sodium biocarbonate with 10% fetal
bovine serum, 100
units/mL penicillin, and 100 pg/mL streptomycin sulfate at 37 C under 5% CO2.
3T3-L1 fibroblasts
cells (ATCC) were maintained in a Dulbecco's modified Eagle's medium
containing 5.5 mM
glucose, 10% fetal bovine serum, 50 pg/mL gentamycin, 0.5 mM glutamine, and
0.5 pg/mL
fungizone at 37 C. Human embryonic kidney 293 cells (ATCC) were maintained in
a Dulbecco's
modified Eagle's medium with 10% fetal bovine serum, 100 units/mL penicillin,
and 100 pg/mL
streptomycin sulfate at 37 C under 5% CO2.
Oligonucleotide microarray analysis
DU145 prostate cancer cells were treated with 5 mM of fatostatin A or DMSO
alone in the
presence of 1 pg /mL of IGF1 for 6 hrs in a serum free medium, total RNA was
extracted in a TRI
reagent (Molecular Research Center) and further isolated by RNeasy Mini Kit
(Qiagen). Purified
mRNA was analyzed in Baylor College of Medicine Microarray Core Facility by
Affymetrix Human
Genome U133 Plus 2.0 Array consisting of almost 45,000 probe sets representing
more than
39,000 transcripts derived from approximately 33,000 well-substantiated human
genes (Affymetrix,
Inc.).
Luciferase reporter assay
On day 0, HEK293 cells were plated out in triplicate at a density of 5 x
103/well onto a 96-
well plate in a Dulbecco's modified Eagle's medium with 10% fetal bovine
serum, 100 units/mL
penicillin, and 100 pg/mL streptomycin sulfate. On day 2, the cells were
transiently co-transfected
with the following plasmids by using Lipofectamine reagent (Invitrogen): 0.4
pig/well pSRE-Luc (an
SRE-1-driven luciferase reporter construct), and 0.1 lug/well a b-gal reporter
in which the expression
of 3-gal is controlled by an actin promoter in a final volume of 150 mL. After
incubation for 5 hrs at
37 C, the cells were washed with phosphate-buffered saline and then incubated
in 100 'al of
Dulbecco's modified Eagle's medium with 10% lipid-depleted serum, 100 units/pt
penicillin, and 100
pg/mL streptomycin sulfate in the absence or presence of fatostatin A. After
20 hrs of incubation, the
cells in each well were lysed with 20 lut of 1 x Reporter Lysis Buffer
(Promega), and aliquots were
used for measurement of luciferase (10 lut) and p-galactosidase (10 pi,L)
activities. For luciferase
assay, photon production was detected as counts per second in a Wallac 1420
ARVOsx multilabel
counter (PerkinElmer). For p-galactosidase assays, hydrolysis of 0-nitrophenyl-
p-D-galactosidase
was measured after incubation for 0.5 h at 37 C by a microplate reader at the
wave length of 405
nm (Tecan). The luciferase activity (counts per second) was normalized by the
acitivity of 3-
galactosidase (OD units). For overexpression of the N-terminal matured form of
SREBP-1c, pCMV-
SREBP-1 c (1-436) was co-transfected with pSRE-Luc. pSRE-Luc and pCMV-SREBP-lc
(1-436)
were provided by J. L. Goldstein (University of Texas Southwestern Medical
Center).
36
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
RT-PCR experiments
Total RNA was extracted from DU145 cells in TRI reagent (Molecular Research
Center) and
isolated with an RNeasy Mini Kit. The RNA sample was subjected to RT-PCR by
using the Access
RT-PCR System. RT-PCR reactions contained total RNA, 1 'LAM of each primer,
0.2 mM dNTP, 1
mM MgSO4, AMV reverse transcriptase (2 units), and Tfl DNA polymerase (2
units) in a final volume
of 25 L. The primer pairs used are as follows: 5'-TCA GAC CGG GAC TGC TTG GAC
GGC TCA
GTC -3 (SEQ ID NO: 1) and 5'-CCA CTT AGG CAG TGG AAC TCG AAG GCC G -3' (SEQ ID
NO:
2) for Low density lipoprotein receptor (LDLR); 5'- GCC TGC TTG ATA ATA TAT
AAA C -3' (SEQ ID
NO: 3) and 5' - CAC TTG AAT TGA GCT TTA G -3' (SEQ ID NO: 4) for stearoyl-CoA
desaturase
(SCD); 5' -AAG AAA AAG TGT CAG ACA GCT GG -3' (SEQ ID NO: 5) and 5' - TGG ACT
GAA
GGG GTG TTA GC -3' (SEQ ID NO: 6) for ATP citrate lyase (ACL); 5'- GCC CGA CAG
TTC TGA
ACT GGA ACA -3' (SEQ ID NO: 7) and 5'- GAA CCT GAG ACC TCT CTG AAA GAG -3'
(SEQ ID
NO: 8) for 3-hydroxy-3-methylglutaryl- CoA reductase (HMG CoA R); 5'- CTG CCT
GAC TGC CTC
AGC -3' (SEQ ID NO: 9) and 5'- ACC TCT CCT GAC ACC TGG G -3' (SEQ ID NO: 10)
for
mevalonate kinase (MVD); 5'-AAG ACT TCA GGG TAA GTC ATC A-3' (SEQ ID NO: 11)
and 5'-
CGT GTA TAA TGG TGT CTA TCA G -3' (SEQ ID NO: 12) for insulin induced gene 1
(INSIG1). The
amplification conditions are as follows: 1 cycle at 94 C for 4 min, then
denatured at 94 C for 40 s,
annealed at 50 C for 40 s, and extended at 68 C for 2 min with 22 cycles for
SCD and HMG CoA R,
annealed at 58 C with 24 cycles for LDLR and INSIG1, or annealed at 60 C with
24 cycles for ATP
citrate lyase (ACL), annealed at 55 C with 30 cycles for MVD. The amplified
DNAs were analyzed
by an agarose gel and quantified with the Scion-image (version 4.02) software.
Western blotting
DU145 prostate cancer cells were seeded on a 6-well plate at a density of 2 x
105 cells/well
in a serum-free MEM incubated at 37 C for overnight. The cells were then
treated with DMSO or
fatostatin A (1 or 5 mM) in presence of IGF1 (1 pg /mL). After 6 hrs of
incubation, the cells were
harvested in PBS and lysated in an SDS buffer. The samples were separated on a
10% SDS-PAGE
gel and blotted by using rabbit anti-SREBP-1 and anti-SREBP-2 antibodies. The
specific bands
were visualized by using enhanced chemiluminescent (ECL) detection reagents
(Amersham).
Immunofluorescence experiments
DU145 prostate cancer cells were seeded on coverslips for overnight in a serum-
free MEM,
and then treated with 5 mM of fatostatin A or DMSO alone in a serum-free MEM
containing IGF1 (1
pg/mL). After 6 hrs of incubation, the cells were fixed for 20 min in methanol
at -20 C and blocked
for 1 hr in a PBS containing 5% milk and 0.1% Tween 20. The samples were
incubated with rabbit
polyclonal anti-SREBP-1 (Santa Cruz: sc-8984) and then fluorescein
isothiocyanate-conjugated
anti-rabbit IgG antibody (Chemicon Inc). The coverslips were visualized under
a Nikon TE200
fluorescence microscope at x400 magnification with appropriate filters for
fluorescence detection.
37
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
siRNA knockdown of SREBPs
Complimentary oligonucleotides derived from the sequence of the SREBP-1 gene
(512-
531), 5'- GAT CCC CGC CAC ATT GAG CTC CTC TCT TCA AGA GAG AGA GGA GCT CAA TGT
GGC TTT TTG GAAA-3 (SEQ ID NO: 13), and 5'-AGC TTT TCC AAA AAG CCA CAT TGA GCT
CCT CTC TCT CTT GAA GGA GGA GCT CAA TGT GGC GGG-3' (SEQ ID NO: 14), were
inserted
into a pSUPER vector (OligoEngine). The resulting plasmid was transfected into
3T3-L1 or DU145
cells with Fugene 6 (Roche). To establish stably transfected clones, neomycin-
derivative G418
(Gibco) was used at a concentration of 500 pg/mL, and stable transformants
were established. The
expression levels of the SREBP-1 were evaluated by western blots. For
adipogenesis experiments,
3T3-L1 cells were seeded onto a 96-well plate in a DMEM with 10% fetal bovine
serum and
incubated for another two days to complete confluence. On day 0, the medium
was switched to the
induction medium: DMEM containing 10% fetal bovine serum, 5 pg/mL of insulin,
0.5 mM 1-methyl-
3-isobutylxanthin (MIX), and 1 pM dexamethazone (DEX). On day 2, the induction
medium was
removed and switched to a DMEM medium containing 10% fetal bovine serum and 5
pg/mL of
insulin. On day 10, adipose oil droplets were stained with Oil-Red 0. For cell
growth experiments,
DU145 cells were seeded onto 96-well plates at density of 2,000 cells/well in
an MEM without serum
or with lpg /mL of IGF1, 2% fat-free fetal bovine serum, or 2% fetal bovine
serum. The cell growth
was estimated by WST-1 assays after 3 days. The experiments were performed in
triplicate.
Animal studies with fatostatin A
Male mice (129Sv background) were housed under controlled conditions (12-hr
light/dark
cycle; 25 C) in the Animal Care Center at Baylor College of Medicine and had
ad libitum access to
standard laboratory chow (Purina Mills) and water. Fatostatin A was
administered intraperitoneally
(30 mg/kg; 150 litL) to 5-6 month old male mice (129Sv background) using two
different protocols.
First protocol involves fasting the mice for 48 hrs, followed by refeeding fat
free diet for another 48
hrs. This treatment induces both activities and levels of lipogenic enzymes
such as ACC and FAS in
addition to SREBPs. The administration of fatostatin A or 10% DMSO in PBS to
control groups
(n=5) started 24 hrs before the fasting and continued daily until the end of
the experiment.
In the second protocol, two groups of male mice (n=5) were treated daily for
two weeks with
either 30 mg/kg fatostatin A or 10% DMSO in PBS. Food intake and body weight
were measured
daily. At the end of the experiments, the mice were briefly fasted for 4-5
hrs, and their blood was
withdrawn for determination of serum constituents. The mice were then
sacrificed, and their livers
were quickly removed and ground to powder in liquid nitrogen. The powdered
tissues were
suspended in 10 ml of PBS containing 0.1 mM PMSF, 5 mM benzamidine, and 5
mg/mL protease
inhibitor cocktail (Roche), homogenized using Polytron (3 x 30 Sec, at a high
speed), and sonicated
briefly to degrade DNA. The extracts were clarified by centrifugation at
16,000xg for 20 min. The
samples were then subjected to western blot analysis using commercially
available antibodies
against FAS and SREBP-1. FAS and ACC activities were determined as described
earlier (Mao et
aL , 2006).
38
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
EXAMPLE 7
Fatostatin A prevents fatty liver, reduces hyperglycemia and induces weight
loss in ob/ob mice
Despite the efforts to unravel the networks that regulate food intake and
energy balance, it
is not fully understood how obesity causes these disease. The effect of
fatostatin A on male ob mice
was investigated, specifically its effect to prevent weight increase by
reducing white adipose size,
diabetic conditions and fatty liver. As mentioned earlier, fatostatin A is an
inhibitor of the master
control of transcription by inhibiting the action of SREBP-1. Normal mice
treated with fatostatin A
lost weight and had lower levels of glucose and cholesterol. Fatostatin A
reduced the active mature
form of SREBP-1 in the liver of treated mice compared to controls.
SREBP-1 and -2 play related but distinct roles in biosynthesis of fatty acids
and cholesterol.
SREBP-1 preferentially activate the genes required for fatty acid synthesis,
and SREBP-2 favors
cholesterogenesis. Since fatostatin A blocks the activation of SREBP-1 and
perhaps SREBP-2,
administration of fatostatin A into obese ob/ob mice transiently modulates the
biosynthesis of both
fatty acids and cholesterol and reveals interesting phenotypes in the obese
mice.
The effect of fatostatin A on body weight and food intake
The study employed 4-5 week old male ob/ob mice of average weight of about 23
g/mouse.
Fatostatin A (30 mg/kg/day) was delivered intraperitoneally daily, and body
weight and food intake
were measured. As shown in FIGS. 8A-8B, the increase in body weight of the
treated mice was
significantly lower than the controls. At the end of first week of treatment
ob control mice injected
with DMSO gained on average of 4.82 g /mouse (from 23.58 0.62 to 28.40
1.45), whereas the
fatostatin treated group gained about 3.37 g/mouse (23.08 1.53 to 26.45
1.2 g/mouse), (p=
0.03). After 28 days of treatment the fatostatin A treated group weighed about
12% less than the
controls weeks (32.1 1.4 compared to 36.2 2.2 g/mouse for the fatostatin)
(P =0.02). The
accumulative food intake was similar in both groups FIG. 80). On average, in
the treated group,
food intake was not significantly different from the controls being 5.4 1.5
compared to 5.9 1.4
g/mouse. day respectively.
Effect of fatostatin A on glucose and lipids profile in the blood
One of the most distinct phenotypes in ob/ob mice is hyperglycemia as a result
of insulin
resistance conditions. To determine the effect of the fatostatin A on blood
glucose and lipids, the
serum levels of glucose, triglycerides, and cholesterol were analyzed in ob/ob
mice fed with
standard diet.
As shown in FIGS. 9A-9H, the glucose levels after over night fasting, in serum
from treated
animals were about 70% lower than the controls; 153.2 30.5 and 429.4 87
mg/di respectively
(P=0.003). The glucose levels in the serum of treated animals became
comparable to that of wt
mice with functional ob gene, whereas the control mice that were given DMSO
were hyperglycemic
as expected. Interestingly, ketone bodies (R-hydroxy butyrate) increased about
seven fold in the
treated animals compared to the controls; 3.62 1.41 and 0.5 0.37 mg/di
respectively (P=0.004).
The high levels of ketone bodies in fatostatin A animals shows a significant
increase in fatty acid
39
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
oxidation in livers in which the main product is ketone bodies that is
secreted in the blood. Also,
blood constituents that increased in the treated mice were none esterified
free fatty acids (NEFA)
measured in the serum, which was about 70% higher than that of the controls;
1.93 0.26 and 0.7
0.2 mEq/I (P = 0.028). This increase in NEFA levels may be due to increased
lipolysis from adipose
tissue due to an increase in demand for fatty acid oxidation. FFA is known to
be associated with
insulin resistance in animals and human. However, despite the elevated levels
of FFA in serum of
fatostatin A treated ob/ob mice, the glucose level was significantly lower
than the controls indicating
an improvement in insulin sensitivity, possibly due to improved insulin
signaling. In addition, it was
recently shown that as a consequence of increased fatty acid oxidation in
mouse tissues (liver,
adipose and muscle) of mutant acetyl-CoA carboxylase mouse (Acc2-/- mutant
mouse), it resulted in
higher ketone bodies in the blood and increased NEFA as a result of increased
lipolysis in
adipocytes. The level of triglycerides (TG) in the serum increased about 30%
in treated mice
compared to controls, 115 11, and 79 12 respectively (P = 0.006),
indicating that fatostatin A
increases secretion and mobilization of TG from the liver. The serum level of
total cholesterol
showed a lower trend in fatostatin treated animals being 183 16 compared to
219 18 mg/di (P =
0.06). However there was a significant decrease of about 35% in [DL (31 3
compared to 48 8; P
= 0.02) and a lesser decrease in HDL of about 22% (144 11 and 183 12; P =
0.02). Since there
was more decrease in LDL level than that of HDL in serum of fatostatin A
treated mice, this shows a
desirable outcome for the treatment with fatostatin A. The level of VLDL,
which transports
triglycerides, phospholipids, and cholesterol and is calculated based on TG
levels, increased about
50% (23.1 2.3 compared to 15.8 2.4 mg/di).
Fatostatin A reduces the size of epididymal fat and ameliorates fatty liver
Because of uncontrolled food intake, ob/ob mice become morbidly obese and
accumulate
excessive levels of fat in fat tissues and in different organs, such as liver-
causing non-alcoholic fatty
liver conditions and insulin resistance. At about 8-9 weeks of age control
untreated mice showed
enlarged liver size and accumulated fat, as evident from pale color, compared
to those treated with
fatostatin A (FIG. 10A). The average weight of livers of fatostatin A treated
mice was about 32%
less than that of the controls (1.59 0.2 compared to 2.34 0.15; P = 0.06)
(FIG. 10D). Liver
sections of control mice stained with oil red for lipid droplets, contained
abundant lipid droplets while
those of the fatostatin A treated mice were devoid of lipid droplets, which
are mainly triglycerides
(FIG. 10B). It has been shown that transgenic mice over expressing SREBP 1
developed fatty liver.
However, in ob/ob mice lacking SREBP-1 (lep blob X Srebp 1-1-), fatty liver
conditions were
significantly improved, suggesting that SREBP 1 is a major player for the
development of fatty liver
in ob/ob mice.
At the end of four weeks of fatostatin A treatment the treated mice weighed
less than the
controls. By examining the epididymal fat pads, which is the major white fat
tissue, it was found that
the fatostatin A treated mice has significantly smaller fat pads (FIG. 10C).
The average weight of the
fat pads was about 20% less than the controls (2.7 0.1 compared to 3.6
0.2; P = 0.02) (FIG.
100). The smaller fat pads may be due to decrease in storage of lipids and/or
decrease in
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
lipogenesis and enhanced fatty acid oxidation in the adipose. Previous studies
with Acc2 mutant
mice showed that the absence of ACC2 also resulted in less fat in livers,
smaller epididymal fat
pads and enhanced oxidation of fatty acids in different tissues including
liver. It was indicated
therein that enhanced fatty acid oxidation, due to lack of inhibition by ACC2-
produced malonyl-CoA,
on carnitine palmitoyltransferase, the mice become highly insulin sensitive
and protected against
diet-inducing obesity and diabetes. In specific aspects, down regulation of
ACC enzymes by
fatostatin A increases fatty acid oxidation and inhibits fatty acid synthesis
in different tissues such as
liver, adipose and muscle, for example. The TG and cholesterol levels were
determined in livers of
fatostatin A treated ob/ob mice and compared to ob/b controls. As shown in
FIG. 11A, the TG levels
in livers of treated mice were reduced by about 65% (14.8 3.7 and 38.7 6.0
mg/gram liver
respectively; P = 0.0004). The cholesterol levels in liver were also reduced
by fatostatin A by more
than 20% (2.8 0.5 and 3.6 0.1; P =0.03) (FIG. 11B). These results further
confirm the Oil Red 0
staining and indicate that fatty liver in ob/ob mice, which is in part caused
by increased hepatic
lipogenesis, can be completely prevented by treatment with fatostatin A. The
reduction of these
lipids in liver of treated ob/ob mice is due to significant inhibition of
lipogenic enzymes needed to
synthesize TG and cholesterol or their precursors. In addition, due to
increased demand for fatty
acid oxidation by different mice tissues, including the liver there is an
increase in lipase liver activity
and also enhanced mobilization of these lipids from liver to the circulation
for utilization by different
fatty acid oxidizing tissues such as heart and muscles. In specific aspects,
this is related to the
higher level of TG in blood of fatostatin A treated ob/ob mice.
Fatostatin A downrequlates lipogenic enzymes in ob/b mice liver
Enzymes in lipogenic pathways are regulated by transcription factors, such as
PPAR and
SREBPs. The effect of fatostatin A on lipogenic enzymes levels and activities
in treated ob/ob mice
was examined. The activity of acetyl-CoA carboxylase (ACC), which carries out
the rate-limiting step
in fatty acid synthesis, was determined. ACC catalyzes the carboxylation of
acetyl-CoA to yield
malonyl-CoA, the building block for fatty acid synthesis, which is carried out
by another
multifunctional enzyme, fatty acid synthase (FAS). In addition to the role of
malonyl-CoA in fatty acid
synthesis, it plays an important role in of fatty acid oxidation by inhibiting
carnitine palmitoyl
transferase 1 (CPT 1). The lipogenic enzymes are significantly induced in
ob/ob mice, partly
explaining the morbidly obese phenotype of these mice. The activity of ACC in
liver extracts of
fatostatin A treated mice decreased by about 40% (3.44 0.44 compared to 5.55
0.57 n
mol/min.mg) (FIG. 12A). Fatty acid synthase activity was also significantly
downregulated in liver
extracts of fatostatin A treated ob/ob mice. FAS activity was reduced by more
than 70% in the
treated mice (8.64 1.91 compare to 22.6 1.37 n mol/min.mg) (FIG. 12B). The
decrease in both
ACC and FAS activities is due to reduction in the expression levels for both
enzymes, as shown by
western blot analysis for both enzymes (FIG. 120). Their product fatty acids,
014:0 and 016:0, are
significantly lower (about 50%) in the livers of fatostatin treated ob/ob mice
than the in the livers of
untreated control mice (Table 2).
41
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
ACC is acutely regulated by phosphorylation/dephosphorylation mechanism,
resulting in
inhibition and activation of the enzyme, respectively. As shown in FIG. 12C,
the level of phosphor-
ACC was higher in the control group, however because the expression level of
ACC is also higher
and to the same level, this suggests that fatostatin A, does not alter the
specific phosphorylation
level (P-ACC/ACC protein). These results indicate that the downregulation of
ACC activity is solely
due to decreased levels of the enzyme and not to a decrease in the
phosphorylation status (FIGS.
12C-12D). It was shown previously that in liver there are two ACC isoforms;
ACC1 (the dominant
isoform in liver) and ACC2 (dominant isoform in muscle) which play distinct
roles in regulating lipid
synthesis and oxidation, respectively. The reduction in ACC and FAS activities
indicate that
lipogenesis is reduced, whereas fat burning is significantly enhanced in liver
of fatostatin A treated
mice, which is consistent with the seven fold increase in ketone bodies in
blood of ob/ob fatostatin A
treated mice as shown in FIGS. 9A-9H. The level of two key enzymes in fatty
acid metabolism,
which are also under transcription regulation of SREBP-1, ATP citrate lyase
(ACL) and Steroyl-CoA
desaturase 1 (SCD1), was determined. The protein level of ACL that converts
cytosolic citrate into
acetyl-CoA, which is the substrate for ACC to yield malonyl-CoA for fatty acid
synthesis, was
reduced about 70% in liver extracts of fatostatin A mice. This downregulation
in ACL level further
amplify the effect of fatostatin A on the reduction in the lipogenic process
in lipogenic tissues such a
liver. SCD1 catalyzes the rate-limiting step in the biosynthesis of
monounsaturated fatty acids, by
introducing a Cis double bond in A9 position of fatty acyl-CoA, such as
palmitoyl-CoA and stearoyl-
CoA. The products, palmitoleoyl-CoA (16:1) and oleoyl-CoA (18:1) are important
components of
triglycerides and cholesterol esters and deletion of SCD1 in mice including
ob/ob mice resulted in
increased metabolic rate, reduced adiposity, preventing fatty liver and
protecting against diabetes
induced by diet. As shown in FIG. 12D, the protein level of SCD1 was reduced
about 50% in liver
extracts of fatostatin A treated mice compared to the controls. This was
confirmed by the reduction
by about 70% of the monounsaturated fatty acids 016:1, 018:1 and 020:1 as well
as the reduction
by about 50% of the desaturation of their elongated products (018:2)N-6,
(018:3)N-6, (C20:2)N-6,
and (020-3)N-6 (Table 2). This effect on the reduction in SCD1 is an important
factor in weight
reduction and decreases TG level in liver and protection against fatty liver,
in specific embodiments
of the invention. Interestingly, the protein level of FADS 1 or A 5 Desaturase
did not change as a
result of fatostatin A treatment.
TABLE 2: Gas Chromatography-Mass Spectrometry (GC-MS) Analyses Of Fatty Acids
in the Livers
of OB/OB Treated Mice and Their Untreated Control
Fatostatin A Control
Fatty Acid pmole/gram liver p mole/gram liver Ratio P
value
treated control
(14:0) 1224.567 + 508.88 2378.586 + 329.454 0.51483 0.047781
(16:0) 37143.23 + 3656.16 85044.75 +5716.175 0.436749
0.001619
(18:0) 15067.14 + 916.48 14129.2 + 272.7029 1.066383 0.260833
(20:0) 164.3537 22.89 192.9773 + 16.46977 0.851674 0.059842
(22:0) 116.1305 + 25.97 91.74762 + 12.60782 1.26576 0.2499
42
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
(24:0) 157.8261 + 7.02 120.6449 + 19.37201 1.308188 0.052305
(16:1) 11429.29 + 227.5 34787.26 + 4482.311 0.328548 0.005542
(18:1) 44471.72 + 8840.37 152218.4 + 12872.57 0.292157
0.001621
(20:1) 593.7212 + 119.68 1829.537 + 230.8927 0.32452
0.00368
(22:1) 94.94588 + 14.91 109.0117 + 11.47413 0.870969 0.379216
(24:1) 397.92 + 216.3 234.2073 + 26.9417 1.699008 0.214302
(18:2)N-6 29673.93 + 2456.22 46513.4 + 2825.032 0.637965
0.00455
(18:3)N-6 237.1671 + 40.5 560.4358 + 61.89641 0.423183 0.00546
(20:2)N-6 426.6694 + 63.52 914.0388 + 90.59441 0.466796 0.001424
(20:3)N-6 1630.394 + 193.18 2589.236 + 154.791 0.629682 0.008568
(20:4)N-6 8747.289 + 781.41 7772.095 + 110.7878 1.125474
0.171108
(22:4)N-6 280.2708 + 46.1 292.2569 + 11.81504 0.958988 0.810355
(22:5)N-6 172.9715 + 64.89 146.5646 + 14.54166 1.180173 0.879433
(18:3)N-3 4416.975 + 643.658 3625.879 + 220.5747 1.21818
0.703923
(20:5)N-3 2145.643 + 265.17 3067.856 + 299.7701
0.699395 0.04467
(22:5)N-3 1720.424 + 221.16 2338.451 + 234.2185
0.735711 0.092988
(22:6)N-3 9223.38 + 700.31 8718.014 + 532.5817 1.057968
0.448218
Liver samples (100 mg) obtained from fatostatin A-treated ob/ob mice and non-
treated
control mice and stored at - 80 C until analyzed for fatty acid contents. The
fatty acids were
extracted according to Folch's protocol and quantitatively analyzed using gas
chromatography-mass
spectrometry (GC-SM). As shown in the above table, there is about 50%
reduction of 014:0 and
016:0, the products of the de novo fatty acid synthesis, and about 70%
reduction of
monounsaturated fatty acids C16:1, C18:1 and C20:1 and their elongation-
desaturation products
(C18:2)N-6, (C18:3)N-6, (020:2)N-6, (C20-3)N-6 and (C20:5)N-3. Myristate
(014:0) and palmitate
(016:0), the products of FAS, were reduced by about 50% (P<0.05). There was no
change in 018
levels, which is derived not only from de novo fatty acid synthesis by FAS,
but also from food and
chain elongation system. Interestingly there was a strong trend in lowering
the C20:0 by about 15%
(P+0.059) and an increase of 30% in 024:0 (P=0.05). In parallel to the
significant decreases in long-
chain saturated fatty acids, results show that there were significant
reductions of about 70% in the
levels of monounsaturated fatty acids 016:1, 018:1 and 020:1 (P<0.004). Also,
the levels of
polyunsaturated long-chain fatty acids (18:2)N-6, (18:3)N-6, (20:2)N-6,
(20:3)N-6 and (20:5)N-3
were reduced 30-60%. These reductions are the result of down regulation of key
enzymes in the
lipogenic pathways (FAS, ACC and SCD, ACL) at transcription and translation.
These results help
explain the effect of fatostatin A in amelioriating fatty liver conditions by
reducing the triglyceride
levels made in the liver.
Downregulation of mRNA levels of lipogenic enzymes
The decrease in protein levels can be attributed to a transcriptional or
translational
regulation. Real time PCR was used to determined the levels of mRNA levels of
representatives of
lipogenic genes ACC1, FAS and SCD1 in addition to the lipogenic transcription
factor PPAR y.
There was about 80% reduction in the mRNA levels of ACC1, FAS and SCD1 (FIG.
13). These
43
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
results are consistent with lower levels of enzyme proteins and activities,
and strongly indicate that
fatostatin A lowers lipogenesis by inhibiting the maturation of SREBP-1. The
down regulation of
lipogenic enzyme involves one of its main transcription factors, PPAR y, in
specific embodiments.
The mRNA level of this transcription factor was reduced by about 40% in
extracts of fatostatin A
treated mice (FIG. 13). Since, ob/ob mice treated with fatostatin A reduced
hyperglycemia and
prevented fatty liver, this indicates that PPAR y in liver is one of several
factors that may affect
these pathological conditions, in specific aspects. In summary, Fatostatin A
through its action on
SREBP-1 ameliorated fatty liver by reducing hepatic TG storage, reduced
adiposity and lowered
hyperglycemia in treated ob/ob mice. These studies indicate that fatostatin A
and its analogs are
useful agents against obesity, fatty liver and diabetes, for example.
Four-to 5-week old homozygous male obese (ob/ob) mice (C57BL/6J, The Jackson
Laboratory, Bar Harbor, ME) were housed under controlled conditions (12-hr
light/dark cycle; 25 C).
The animals were housed 5 per cage and had ad libitum access to standard
laboratory chow
(Purina Mills, Richmond IN) and water for one week after their arrival. On
first day of the experiment
and every day thereafter the weight of the mice and the amount of the food
consumed were
measured. The weight of mice and food remaining were measured daily between 3-
5 p.m. before
the ip injection of fatostatin A (30 mg/kg; 150 tut). The administration of
Fatostatin A or 10% DMSO
in PBS to control groups (n=5) continued daily for four weeks till the end of
the study.
After 28 days of daily injection of fatostatin A mice were fasted overnight
and blood was
withdrawn and Whole blood glucose and P-hydroxybutyrate were measured with a
Glucometer
Precision Xtra (Abbott). For determination of serum constituents. Glucose,
triglyceride and
cholesterol measurements were done by the Comparative Pathology Laboratory
(Baylor College of
Medicine). Serum non-esterified fatty acids (NEFA) were measured by using NEFA
C kit (Wako
Chemicals, Richmond, VA).
Mice were sacrificed and weights of livers, and Epididymal fat pads were
determined.
Frozen sections of Liver slices from individual animals were stained with Oil
Red 0 to visualize the
fat droplets (TG) in liver slices as described earlier (Abu-Elheiga et al.,
2001). The remaining liver
tissues were frozen in liquid nitrogen and kept at ¨ 80 C for further
analysis. Liver triglyceride and
cholesterol contents were carried out as described in the reference (Chandler,
et al., 2003) using
Cholesterol E Kit (Wako) and Infinity Triglyceride Kit (Thermo Electron,
Melbourne, Australia),
adapted for colorimetric analysis in 96-well plate format.
Enzymatic activities and western blot analyses
A portion of the frozen liver was ground to powder in liquid nitrogen. The
powdered tissues
were suspended in 10 ml of PBS containing 0.1 mM PMSF, 5 mM benzamidine, and 5
mg/ml
protease inhibitor cocktail (Roche), and homogenized using Polytron (3 x 30
Sec, at high speed)
and sonicated briefly to degrade DNA. The extracts were clarified by
centrifugation at 16,000 x g for
20 min. Protein concentrations in the supernatant were determined, and
subjected to western blot
analysis using commercially available antibodies against the following
enzymes: FAS (BD
Biosciences), citrate lyase SCD1, FADS1, ACC and phospho-ACC antibodies. The
proteins were
44
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
visualized using Amersham ECL PlusTM Western Blotting Detection Reagents. The
intensity of the
specific bands of proteins of interest were scanned and normalized against
beta-actin for
quantifications. FAS and ACC activities from the liver extracts were
determined as described earlier
(Mao et al., 2006).
Quantitative real time PCR
Total RNA was prepared from mouse tissues using TRIzol reagent (Invitrogen).
Equal
amounts of RNA from 5 mice were pooled and treated with DNase I (Turbo DNA-
free, Ambion, Inc.).
First stranded cDNA was synthesized from 2 pg of DNase I-treated total RNA
with random hexamer
primer using Superscript ll RNase H-reverse transcriptase (Invitrogen). The
real time PCR
contained, in a final volume of 20 pl, 10 ng of reverse transcribed total RNA,
0.5 pM forward and
reverse primers, and 10 pl of 2 x master mix from DyNAmo HS SYBR Green qPCR
kit (Finnzymes).
PCR was carried out in 96-well plate using DNA Engine Opticon System (MJ
Research, Inc). All
reactions were done in triplicate and the relative amounts of mRNAs were
calculated using the
comparative C(t) method. The cycle threshold C(t) was calculated using the
Opticon Monitor
software 2.02 (MJ Research). Mouse [3 -actin mRNA was used as the internal
control. Data were
expressed as the mean SD. Difference between two groups was assessed using
the unpaired
two-tailed Student t-test.
EXAMPLE 8
Identification of target molecules of Fatostatin A and analogs or derivatives
thereof
In certain aspects of the invention, one or more targets of fatostatin A or
its analog or
derivative is identified. Although any suitable method may be employed for
such identification, in
specific embodiments the fatostatin A or analog or derivative thereof, is
labeled. Exemplary labels
include biotin, for example.
EXAMPLE 9
Exemplary compounds and modifications thereof
FIGS. 14A-14F illustrate exemplary compounds of the invention, and their given
names are
provided in Table 3 and Table 4. FIGS. 15-17 demonstrate exemplary luciferase
reporter gene
assays for these exemplary compounds at 20 mM by the same method shown in FIG.
2A. The
adipogenesis assay was performed as decribed (Choi et al., 2003). The
analogues that completely
inhibited the formation of oil droplets in cells were scored to be
adipogenesis-inhibiting analogues.
Table 3: Exemplary Compounds of the Invention
Inhibition of
Name entry Luc/Gal STDEV adipogenesis
none 9.4426 1.0577
2-propy1-4-(4-p-tolylthiazol-2-yl)pyridine 1 6.2297 1.1014
4-(4-(4-bromophenyl)thiazol-2-y1)-2-
propylpyridine 2 4.5130 0.6176
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
4-(4-phenylthiazol-2-y1)-2-propylpyridine 3 7.4643 2.2215
4-(4-(4-chlorophenyl)thiazol-2-y1)-2-
propylpyridine 4 6.3808 2.0425
4-(4-(4-ethylphenyl)thiazol-2-yl)pyridine 5 7.6190 1.6221
4-(4-p-tolylthiazol-2-yl)pyridine 6 13.4689 1.6735
4-(4-(4-methoxyphenyl)thiazol-2-yl)pyridine 7 18.3174 2.9172
4-(2-(2-propylpyridin-4-yl)thiazol-4-yl)phenyl
benzoate 8 7.7585 1.6193
4-(2-(2-propylpyridin-4-yl)thiazol-4-yl)phenol 9 14.5234 2.7276
methyl 2-(4-(2-(2-propylpyridin-4-yl)thiazol-4-
yl)phenoxy)acetate 10 10.7717 0.8662
4-(4-chlorophenyI)-2-(3,4-
dimethoxyphenyl)thiazole 12 5.9214 1.2693
44443 ,4-d ichloro phenyl)thiazol-2-y1)-2-
propylpyridine 13 5.2391 0.4021
444444 uorophe nyl)th iazol-2-y1)-2-
propylpyridine 14 9.6605 0.9824
4-(4-(2,4-difluorophenyl)th iazol-2-y1)-2-
propylpyridine 15 10.8383 1.7661
4-(2-(2-propylpyrid in-4-yl)thiazol-4-
yl)benzenamine 16 10.3338 2.0763
N-isopropy1-4-(2-(2-propylpyridin-4-yl)thiazol-4-
yl)benzenamine 17 4.7079 1.2781
N-(4-(2-(2-propylpyrid in-4-yl)th iazol-4-
yl)phenyl)acetamide 18 11.7685 6.8358
None 15.0759 0.8305
2-propy1-4-(4-p-tolylthiazol-2-yl)pyridine 1 7.5537 0.9784
N-(4-(2-(2-propylpyrid in-4-yl)th iazol-4-
yl)phenyl)methanesulfonamide 19 4.1981 0.4653
N-benzy1-4-(2-(2-propylpyridin-4-yl)thiazol-4-
yl)benzenamine 20 5.6748 0.0613
N-(cyclopropyl methyl)-4-(2-(2-propylpyrid in-4-
yl)thiazol-4-yl)benzenamine 21 6.4378 1.2736
4-(4-bromophe nyI)-2-(2-propyl pyridi n-4-
yl)th iazole-5-carboxyl ic acid 22 21.5911 0.8383
methyl 4-(4-bromophenyI)-2-(2-propylpyridin-4-
yl)thiazole-5-carboxylate 23 8.1137 2.5369
4-(4-(4-methoxyphenyl)thiazol-2-y1)-2-
propylpyridine 24 11.0367 2.1112
4-(4-(3-methoxyphenyl)thiazol-2-y1)-2-
propylpyridine 25 7.8536 1.2799
4-(4-(2-methoxyphenyl)thiazol-2-y1)-2-
propylpyridine 26 8.3046 2.6780
2-phenyl-4-p-tolylthiazole 27 13.8222 1.3938
3-(2-(2-propylpyridin-4-yl)thiazol-4-yl)phenol 28 12.7791 1.1429
2-(2-(2-propylpyridin-4-yl)thiazol-4-yl)phenol 29 8.2379 1.9501
4-(4-bromopheny1)-N-isopropy1-2-(2-
propylpyridin-4-yl)thiazole-5-carboxamide 30 16.0226 2.1917
4-(4-(4-chlorophenyl)thiazol-2-yl)pyridine 31 16.9971 2.6512
4-(4-(4-chlorophenyl)thiazol-2-y1)-2- 32 9.5798 0.8524
46
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
ethylpyridine
4-(4-chlorophenyI)-2-phenylthiazole 33 17.8175 3.7158
2-propy1-4-(4-(thiophen-2-yl)thiazol-2-
yl)pyridine 34 12.1593 1.5587
2-phenyl-4-p-tolylthiazole 27 13.8222 1.3938
None 18 2
2-propy1-4-(4-p-tolylthiazol-2-yl)pyridine 1 9.413845 0.840651
4-(4'-methyl[1,1'-bipheny1]-4-y1)-2-propyl)
pyridine 35 11.35866 0.881475
2-(2-propylpyridin-4-yI)-4-p-tolylthiazole-5-
carboxylic acid 36 18.98889 2.082093
2-ethyl-4-(4-p-tolylthiazol-2-y1)pyridine 37 9.869906 0.71108
4-pheny1-2-(2-propylpyridin-4-yl)thiazole-5-
carboxylic acid 38 22.65811 3.898667
methyl 2-(2-propylpyridin-4-yI)-4-p-tolylthiazole-
5-carboxylate 39 14.92978 2.600443
None 8.2181 0.5097
2-propy1-4-(4-p-tolylthiazol-2-yl)pyridine 1 3.4437 0.2720
tert-butyl 4-(2-(2-propylpyridin-4-yl)thiazol-4-
yl)phenylcarbamate 40 2.4390 0.4730
N-cyclohexy1-4-(2-(2-propylpyridin-4-yl)thiazol-
4-yl)benzenamine 41 6.5229 0.8638
4-(2-(2-propylpyridin-4-yl)thiazol-4-y1)-N-
tosylbenzenamine 42 3.3957 0.3619
N-(4-(2-(2-propylpyridin-4-yl)thiazol-4-
yl)phenyI)-8-quinolinesulfonamide 43 2.8506 0.6396
N-(4-(2-(2-propylpyridin-4-yl)thiazol-4-
yl)phenyI)-2-thiophenesulfonamide 44 1.8538 0.1240
TABLE 4
Name Compound
4-(4-bromophenyI)-2-(pyrrolidin-2-yl)thiazole 45
4-(4-bromophenyI)-2-(1-propylpyrrolidin-2-yl)thiazole 46
tert-butyl 2-(4-(4-bromophenyl)thiazol-2-yl)pyrrolidine-1-carboxylate 47
benzyl 2-(4-(4-bromophenyl)thiazol-2-yl)pyrrolidine-1-carboxylate 48
3-(4-(4-bromophenyl)thiazol-2-y1)-1-propylpiperidine 49
tert-butyl 3-(4-(4-bromophenyl)thiazol-2-yl)piperidine-1-carboxylate 50
benzyl 3-(4-(4-bromophenyl)thiazol-2-yl)piperidine-1-carboxylate 51
benzyl 4-(4-(4-bromophenyl)thiazol-2-yl)piperidine-1-carboxylate 52
benzyl (R)-2-(4-(4-(methylsulfonamido)phenyl)thiazol-2-yl)pyrrolidine-1-
carboxylate 53
benzyl 3-(4-(4-(methylsulfonamido)phenyl)thiazol-2-yl)piperidine-1-carboxylate
54
benzyl 4-(4-(4-(methylsulfonamido)phenyl)thiazol-2-yl)piperidine-1-carboxylate
55
4-(3-(pyridin-2-y1)41,2,4]triazolo[4,3-b]pyridazin-6-y1)-N-tosylbenzenamine
56
3-chloro-4-methyl-N-(6-(4-(3-(trifluoromethyl)benzyl)piperazin-1-yl)pyridin-3-
57
yl)benzenesulfonamide
(4-(5-chloro-2-methylphenyl)piperazin-1-yI)(4-(tosylamino)phenyl) methanone
58
4444(1-methyl-I H-benzo[d]imidazole-2-yl)methyl)piperazin-1-y1)-N- 59
tosylbenzenamine
47
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
4-chloro-N-(4-(4-((1-methy1-1H-benzo[d]imidazol-2-yl)methyl)piperazin-1- 60
yl)phenyl)benzenesulfonamide
(Z)-4-(3-cyano-3-(4-(2,4-dimethylphenyl)thiazol-2-yl)ally1)-N-(thiazol-2-
61
yl)benzenesulfonamide
N-(3-(H-imidazo[1,2-a]pyridine-2-yl)pheny1)-4-methyl-2-phenylthiazole-5- 62
carboxamide
N-(3-(benzo[d]thiazol-2-yl)phenypisonicotinamide 63
3-(4-chloropheny1)-4,5-dihydro-1-pheny1-5-(2-phenylthiazol-4-y1)-1H-pyrazole
64
N-(4-(6-methylbenzo[d]thiazol-2-yl)pheny1)-2-(N-m-tolylmethylsulfonamido)
65
acetamide
N-(4-(6-methylbenzo[d]thiazol-2-yl)pheny1)-2-(N-p-tolylmethylsulfonamido)
66
acetamide
Furthermore, a skilled artisan recognizes that it may be suitable to modify
one or more
aspects of an exemplary compound to assist in identifying other suitable
compounds. For example,
upon determination of suitability for a particular compound for treatment
and/or prevention of one or
more metabolic disorders, the compound may be modified to identify other
related compounds for
use for the same or a different metabolic disorder. Such alterations may occur
in accordance with
exemplary chemical groups as described herein, in specific embodiments.
EXAMPLE 10
Blockage of fat synthesis by inhibiting the activation of SREBP
Upon fat depletion in a cell, sterol regulatory element binding proteins
(SREBPs) are
released proteolytically from the membrane and translocated into the nucleus,
where they activate
transcription of the genes involved in cholesterol and fatty acid
biosynthesis. In the present
invention, it is shown that a small synthetic molecule that blocks
adipogenesis is a selective inhibitor
of the SREBP activation. The diarylthiazole derivative, called fatostatin,
impairs the proteolytic
activation of SREBPs, thereby decreasing the transcription of lipogenic genes
in cells. The
molecular target of fatostatin appears to be SREBP cleavage-activating protein
(SOAP). Fatostatin
blocked increases in body weight, blood glucose, and hepatic fat accumulation
in obese ob/ob mice,
even under uncontrolled food intake.
As described herein, fatostatin inhibits the insulin-induced adipogenesis of
313-L1 cells and
the serum-independent growth of DU145 cells (Choi et al., 2003). Gene
expression profiles of drug-
treated and untreated cells were compared to gain information about specific
molecular pathways
affected by fatostatin. DU145 cells were treated with fatostatin or DMSO
alone, and the extracted
mRNA samples were analyzed by Affymetrix DNA microarrays mapping 33,000 genes.
Of those
genes (all of which are available at the National Center for Biotechnology
Information's GenBank
database on the world wide web), transcription levels of 63 genes decreased at
least 35% in
response to fatostatin treatment (Table 5). Thirty-four of the affected genes
were directly associated
with fat or sterol synthesis, such as genes encoding biosynthetic enzymes, and
18 of the affected
genes have been reported to be controlled by SREBPs (Horton et al., 2003).
Downregulation of the
affected SREBP-responsive genes was confirmed by RT-PCR experiments. The high
occurrence of
the SREBP-responsive genes and fat/cholesterol biosynthesis genes in the list
of the downregulated
48
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
genes implies that fatostatin acts on the SREBP pathway. Table 5 shows the
results of the
microarray analysis. The 18 underlined genes have been reported to be
controlled by SREBPs, the
genes in bold are associated with fat or sterol synthesis.
TABLE 5
P D 0.707107 Hs.17414 NM_00109 ATP citrate lyase/FL=gb:NM
001096.1
P D 0.707107 Hs.11223 NM_00589 Isocitrate dehydrogenase 1
(NADP+),
soluble/FL=gb:AF020038.1 gb:AF113917
P D 0.707107 Hs.268012 D89053.1 Fatty-acid-
Coenzyme A ligase, long-chain
3/FL=gb: NM_004457.2 gbz:D89053.1
gb:AF116690.1
P D 0.707107 Hs.155976 A1670819 Cullin
4B/FL=gb:NM_003588.1 gb:AB014595.1
P D 0.707107 Hs.41693 BG252490 DNAJ (Hsp40) homolog,
subfamily B, member
4/FL=gb:U40992.2 gb:NM_007034.2
P D 0.707107 Hs.14732 AL049699 malic enzyme 1. NADP(+)-
dependent,
cytosolic/FL=gb:NM 002395.2
P D 0.707107 Hs.213289 S70123.1 low density
lipoprotein receptor (familial
hypercholesterolemia)
P D 0.707107 Hs.6986 AL565516 human glucose
transporter pseudogene
P D 0.659754 Hs.75616 NM_01467 seladin-1/FL=gb:AF261758.1
gb:B0004375.1
6 gb:NM_014762.1
P D 0.659754 Hs.77393 NM_00200 farnesyl diphosphate
synthase (farnesyl
pyrophosphate synthetase, dimethylally-
transtransferase geranyltranstransferase
P D 0.659754 Hs.132898 NM_00593 fatty acid desaturase 1
P D 0.659754 Hs.274398 B0002654 Homo sapiens,
similar to tubulin, beta4, clone
MGC:4083, mRNA, complete cds
P D 0.659754 BC005838 Homo sapiens, tubulin,
beta5
P D 0.659754 Hs.115285 BF978872 dihydrolipoamide
S-acetyltransferase (E2
component of pyruvate dehydrogenase
cornplex)
P D 0.659754 Hs.159154 AL565749 Tubulin, beta 4
P D 0.659754 Hs.179817 AF167438 Homo sapiens
androgen-regulated short-
chain dehydrogenasereductase 1 (ARSDR1)
P D 0.615572 Hs.213289 NM_00052 low density lipoprotein
receptor (familial
hypercholesterolemia)/FL=gb:NM 000527.2
P D 0.615572 Hs.75105 NM_00657 emopamil-binding protein
(sterol
isomerase) /FL=gb:NM_006579.1
P D 0.615572 Hs.74304 NM_00270 Homo sapiens periplakin
(PPL)
A D 0.615572 Hs.268515 NM_00243 meningioma (disrupted in balanced
translocation) 1/FL=gb:D63807.1
P D 0.615572 UG=Hs.31 AF096304 Homo sapiens putative
sterol reductanse SR-1
(TM7SF2) transmembrane 7 super family
2/FL=gb:AF096304.1
P D 0.615572 Hs.93199 D63807.1 lanosterol synthase
(2,3-oxidosqualene-
lanosterol cyclase)/FL=gb:D63807.1
P D 0.574349 Hs.119597 AB032261 stearoyl-CoA
desaturase (delta-9-
desaturase)/FL=gb:AF097514.1
gb:NM 005063 gbL/ab932261.1
P D 0.574349 Hs.174140 A1971281 ATP citrate
lyase/FL=gb:NM 001096.1
49
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
P D 0.574349 Hs.171825 NM_00367 basic heliz-loop-helix domain
containing, class
B, 2/FL=gb:AB004066.1 gb:NM_003670.1
P D 0.574349 Hs.79103 AW235051 cytochrome b5 outer
mitochondrial membrane
precursor /FL=gb:BC004373.1
gb:NM_030579.1
P D 0.574349 Hs.11806 NM_00136 dehydrocholesterol reductase
/FL=gb:BC000054.1 gb:AF034544.1
gb:AF067127.1 gb:AF096305
gb:NM_001360.1
P D 0.574349 Hs.3838 NM_00662 serum-inducible
kinase/FL=gb:AF059617.1
gb:NM_006622.1 gb:AF223574.1
P D 0.574349 Hs.213289 A1861942 low density
lipoprotein receptor (familial
hypercholesterolemia)/FL=gb:NM 000527.2
P D 0.574349 Hs.92199 AW084510 lanosterol synthase (2,3-
oxidosqualene-
lanosterol cyclase)/FL=gb:NM_002340.1
gb:NM_U22526.1
P D 0.574349 Hs.5920 NM_00547 UDP-N-acetylglucosamine-2-epimerase-N-
acetrylmannosamine kinase
P D 0.574349 Hs.1524 NM_00381 tumor necrosis factor
(ligand) superfamily,
member9/FL=gb:NM_003811.1 gb:U03308.1
P D 0.574349 Hs.278544 BC000408 acetyl-Coenzyme A acetyltransferase 2
(acetoacetyl Coenzyme A thiolase) /FL=gb:
BC000408.1
P D 0.574349 Hs.174140 U18197.1 ATP citrate lyase
/FL=gb:U18197.1
P D 0.574349 Hs.7232 B3855983 acetyl-Coenzyme A
carboxylase alpha
/FL=gb:NM 000664.1 gb:U19822.1
P D 0.535887 Hs.226213 NM_00078 cytochrome P450, 51 (lanosterol
14-alpha-
demthylase) /FL=gb:U23042.1
gb:NM_000786 gb:D55653.1
A D 0.535887 Hs.268490 NM_00047 nuclear receptor subfamily 0, group
B,
member 1/FL=gb:NM_000475.2
P D 0.535887 Hs.48876 :AA872727 famesyl-diphosphate
farnesyltransferase
/FL=gb:L06070.1 gb:L06105.1
gb:NM_004462.1
P D 0.535887 Hs.65270 NM_00603 lipase,
endothelial/FL=gb:AF118767.1
gb:NM 006033.1
0.535887 Hs.14779 AK000162 acetyl-CoA synthetase
0.535887 Hs.44499 U59479.1 pinin, desmosome associated
protein/DEF=
Human neutrophil protein mRNA, partial cds
P D 0.5 Hs.154654 AU144855 cytochrome P450,
subfamily I (dioxin-
inducible), polypeptide 1 (glaucoma 3, primary
infantile)
P D 0.5 Hs.11899 AL518627 3-hydroxy-3-methylglutaryl-
Coenzyme A
reductase/FL=gb:M11058.1 gb:NM 000859.1
0.5 Hs.2178 NM_00352 H2B histone family, member Q/FL=gb:
NM_003528.1
P D 0.5 Hs.130607 NM_00043 mevalonate kinase (mevalonic
aciduria)
/FL=gb:M88468.1 gbz:NM 000431.1
P D 0.5 Hs.284244 NM_00200 fibroblast growth factor 2 (basic)
/FL=gb:
M27968.1gb:NM_002006.1
P D 0.5 Hs.71465 AF098865 squalene
epoxidase/FL=gb:D78130.1
gb:AF098865.1 gb:NM 003129.2
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
P D 0.5 Hs.288031 D85181.1 sterol-05-desaturase
(fungal ERG3, delta-5-
desaturase)-like/FL=gb:D85181.1
A D 0.5 L32662.1 prostaglandin E2
receptor/DB_XREF=gi:
484163/FL=gb:L32662.1
P D 0.5 Hs.81412 D80010.1 lipin 1
P D 0.466517 Hs.11899 NM_00085 3-hydroxy-3-
methylglutaryl-Coenzyme A
reductase/FL=gb:M11058.1 gb:NM 000859.1
P D 0.466517 Hs.79440 NM_00654 IGF-II mRNA-binding
protein 3/FL=gb:
U07188.1 gb:U76705.1 gb:AF117108.1
gb:NM_006547.1
P D 0.466517 Hs.75318 AL575074 tubulin, alpha 1
(testis specific)
P D 0.466517 Hs.226213 U40053 cytochrome P450,
51 (lanosterol 14-alpha-
demethylase)
P D 0.466517 Hs.187579 NM_01637 hydroxysteroid (17-beta)
dehydrogenase 7
/FL=gb:AF098786.2 gb:NM_016371.1
P D 0.466517 Hs.76038 BC005247 isopentenyl-
diphosphate delta isomerase
/FL=gb:BC005247.1
P D 0.466517 Hs.57698 B0000245 NAD(P) dependent
steroid dehydrogenase-
like; H105e3/FL=gb:BC000245.1 gb:U4710
gb:NM_015922.1
P D 0.353553 Hs.77910 NM_00213 3-hydroxy-3-
methylglutaryl-Coenzyme a
synthase 1 (soluble) /FL=gb:BC000297,1
gb:L25798.1 gb:NM 015922.1
P D 0.329877 Hs.56205 BE300521 insulin induced gene
1/FL=gb:NM 005542.1
P D 0.329877 Hs.154654 NM_00010 cytochrome P450, subfamily
I (dioxin-
inducible), polypeptide 1 (glaucoma3, primary
infantile) /FL=gb:NM_000104.2 gb:UO
P D 0.329877 Hs.3828 A1189359 mevalonate (diphospho)
decarboxylase
/FL=gb:U49260.1 gb:CB000011.1
gb:NM 002461.1
To confirm that fatostatin impairs the function of SREBPs, the ability of
endogenous
SREBPs to activate transcription of an SREBP-responsive reporter gene was
measured in CHO-K1
cells in the presence or absence of fatostatin (FIG. 18A). Fatostatin
decreased activation of the
reporter gene, in which the expression of luciferase is controlled by sterol
regulatory elements.
Fatostatin had limited effect on the ability of an exogenously expressed,
mature form of SREBP-1
(amino acids 1-436) to activate the reporter gene (FIG. 18B), indicating that
fatostatin impairs the
activation process of SREBPs.
To determine if fatostatin affects the ER-Golgi translocation and proteolytic
processing of
SREBPs, a reporter assay developed by Sakai et al. (1998) was used. PLAP-BP2
in transfected
CHO-K1 cells remains membrane-bound unless it is cleaved by S1P in the Golgi
and secreted into
the culture medium. In the assay, a secreted alkaline phosphatase, fused with
an SREBP-2
fragment lacking the NH2-terminal DNA-binding domain (PLAP-BP2513-1141),
permits monitoring
of translocation and processing through changes in the fluorescence of a
fluorogenic phosphatase
substrate (FIG. 18C). When cells were co-transfected with plasmids encoding
PLAP-BP2513-1141
and SCAP, FLAP phosphatase was secreted, generating fluorescence signals.
Secretion was
similarly decreased by the addition of fatostatin or sterols (FIG. 18C). The
fatostatin-mediated
inhibition of SREBP activation was confirmed by western blot analysis of
SREBPs. Treatment of the
51
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
CHO-K1 cells with fatostatin decreased the amount of the 68 KDa mature form of
SREBP-2, and
increased the amount of the 125 KDa precursor form (FIG. 18D). Similar results
were obtained for
SREBP-1 (FIG. 22). These results collectively indicate that fatostatin blocks
the activation process
of both isoforms of SREBP.
The inventors considered that fatostatin impairs either the proteolytic
cleavage of SREBPs
in the Golgi apparatus or the ER-to-Golgi translocation of the SCAP/SREBP
complex. Brefeldin A, a
natural product that blocks anterograde movement of proteins from the ER to
the Golgi, is known to
render SREBPs unresponsive to sterols, and causes SREBPs to be constitutively
processed in the
ER by relocating S1P from the Golgi to the ER (DeBose-Boyd et al., 1999). In
the presence of
brefeldin A, fatostatin had no impact on the SREBP processing (FIG. 19A),
suggesting that
fatostatin does not block the proteolysis itself.
To determine whether fatostatin blocks the ER-to-Golgi translocation of the
SCAP/SREBP
complex, the inventors analyzed the extent of N-linked glycosylation of SCAP
in the Golgi
apparatus. SOAP contains a glycosylated luminal loop that is protected from
proteolysis by trypsin
and recognizes anti-SOAP IgG-9D5. The two oligosaccharides in the loop are
sensitive to
endoglycosidase H when SOAP resides in the ER. As SOAP is transported to the
Golgi, its sugars
become resistant to digestion by endoglycosidase H. The translocated SOAP has
higher levels of
glycosylation, and is more resistant to endoglycosidase H than ER-bound SOAP.
Sterols prevent
SOAP from becoming resistant to endoglycosidase H by inhibiting the ER-Golgi
translocation
(Nohturfft et al., 1998). Cells were grown in the absence or presence of
fatostatin or sterols, and
membrane fractions were treated successively with trypsin and endoglycosidase
H. In cells grown
without fatostatin and sterols, a tryptic fragment of SOAP was more resistant
to endoglycosidase H
and had one or two saccharide chains (FIG. 19B, lane 1). When cells were grown
in the presence of
fatostatin or sterols, the SOAP fragment was less resistant to endoglycosidase
H, and had either
zero or one saccharide chain (FIG. 19B, lanes 2 and 3). Thus fatostatin
appears to inhibit the
translocation of SOAP from the ER to the Golgi.
Studies of the structure-activity relationship of fatostatin indicated that
the molecule retains
or even increases biological activity when its toluene moiety is modified with
a variety of alkyl or aryl
sulfonamide groups. One fluorescent derivative, dansyl fatostatin (FIG. 20A),
retained the ability to
block SREBP activation (FIG. 24) and served as a microscopic probe. Confocal
microscopic
analyses revealed that the localization of dansyl fatostatin overlapped with
that of ER-tracker red, a
specific marker for ER (FIG. 20B). In contrast, the control dansyl molecule,
which lacked fatostatin,
failed to localize to any organelle. The selective ER localization implies
that fatostatin binds to a
protein in the ER; the most likely candidate is SOAP, the target of
cholesterol for the control of
SREBP (Radhakrishnan et al., 2004). To test this hypothesis, proteins bound to
a fatostatin-
polyproline linker-biotin conjugate (FIG. 20A) (Sato et al., 2007) were
purified from cell lysates and
analyzed by western blots with antibodies against SOAP, SREBP-1, SREBP-2, and
ATF6, an
unrelated ER-bound transcription factor (Ye et al., 2000). The results showed
that fatostatin was
bound to SOAP, but not to the other proteins (FIG. 200). The binding was lost
upon addition of
52
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
excess fatostatin, but not excess cholesterol (FIG. 20D), raising the
possibility that fatostatin may
interact with SCAP in a site distinct from that of cholesterol.
Having established a key role of SREBPs in lipogenesis, the pharmacological
effects of
fatostatin on ob/ob mice, a mouse model of obesity with uncontrolled food
intake, was then
examined. Fatostatin was delivered intraperitoneally on a daily basis, and
food intake and body
weight were monitored. The average daily food intake by the treated mice was
not significantly
different from that of the controls (5.4 + 1.5 vs. 5.9 + 1.4 g/mouse/d,
respectively, p > 0.05), and no
obvious toxicity was observed during the treatment. After 28 days of treatment
with fatostatin, the
treated mice weighed about 12% less than the untreated controls (32.1 - 1.4
and 36.22 g/mouse,
respectively, p=0.02). One of the most distinct phenotypes in ob/ob mice is
hyperglycemia resulting
from insulin resistance. Examination of blood constituents revealed that the
average glucose level of
the treated mice was ¨70% lower than that of the untreated mice (153.2 - 30.5
vs. 429.4 - 87 mg/di,
respectively, p=0.003), which is in the range of normal glucose levels. These
results are consistent
with the reported role of SREBP-1 c in the pathogenesis of hepatic insulin
resistance (Ide et al.,
2004).
Another phenotype of ob/ob mice is excessive accumulation of fat in organs,
including non-
alcoholic fatty liver. Enlarged and fatty livers were evident from their pale
color in the untreated
ob/ob mice, while livers of the mice treated with fatostatin appeared normal.
Livers of the treated
mice averaged ¨32% less weight, and fat pads were smaller than those of the
untreated mice. Oil
red staining of the liver sections showed that the livers of the untreated
ob/ob mice contained
abundant lipid droplets, while livers of the treated mice contain lower levels
of lipid accumulation
(FIG. 21). The triglyceride and cholesterol levels in the livers of the
treated mice were also reduced.
The prevention of fatty liver in ob/ob mice by fatostatin is in agreement with
the reported role of
SREBP-1 in developing fatty liver: transgenic mice overexpressing SREBP-1
developed fatty livers,
while ob/ob mice lacking SREBP-1 (/eekb X Srebp 1-) had healthy livers (Yahagi
et al., 2002).
The reduction of hepatic fat levels in the treated mice was thought to be due
to decreased
hepatic expression of SREBP-responsive lipogenic enzymes. Therefore, the
effects of fatostatin
were examined on the hepatic protein levels and enzymatic activities of
representative SREBP-
responsive lipogenic enzymes, including fatty acid synthase (FAS), acetyl-CoA
carboxylase,
stearoyl-CoA desaturase 1 (SCD1), and ATP citrate lyase (ACL). Biochemical
analysis showed that
protein levels and activities of the lipogenic enzymes were reduced in liver
extracts of fatostatin-
treated mice (FIGS. 25A-250). Thus, fatostatin blocks the processing of SREBP-
1 in liver,
downregulates lipogenic enzymes, and reduces hepatic triglyceride storage.
Fatostatin represents
the first non-sterol-like synthetic molecule that inhibits the activation of
SREBPs.
Luciferase reporter assay. On day 0, OHO-K1 cells were plated out onto a 96-
well plate in
medium A (a 1:1 mixture of Ham's F-12 medium and Dulbecco's modified Eagle's
medium, with 5%
fetal bovine serum, 100 units/mL penicillin, and 100 pg/mL streptomycin
sulfate). On day 2, the cells
were transiently co-transfected with pSRE-Luc (an SRE-1-driven luciferase
reporter construct) (Hua
etal., 1995) and pAc-p-gal (n-gal reporter in which the expression of 8-gal is
controlled by an actin
promoter), using Lipofectamine reagent (Invitrogen). After incubation for 5 h,
the cells were washed
53
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
with phosphate-buffered saline (PBS), and then incubated, in the absence or
presence of fatostatin,
in medium B (a 1:1 mixture of Ham's F-12 medium and Dulbecco's modified
Eagle's medium, with
5% lipid-depleted serum, 100 units/mL penicillin, 100 pg/mL streptomycin
sulfate, 50 mM compactin,
and 50 mM sodium mevalonate). After 20 h of incubation, the cells in each well
were lysed, and
aliquots were used to measure luciferase and p-galactosidase activities.
Luciferase activity was
normalized by the activity of P-galactosidase. For overexpression of the N-
terminal matured form of
SREBP-1c, pCMV-SREBP-1c(1-436) was co-transfected with pSRE-Luc and pAc-p-gal.
Western blot analysis of SREBP processing. On day 0, CHO-K1 cells were plated
out onto
a 100 mm dish of medium A. On day 2, the cells were washed with PBS, and then
incubated in
medium B in the absence or presence of fatostatin. On day 3, the cells were
washed once with cold
PBS, and then treated with buffer containing 10 mM Tris-HCI, pH 7.6, 100 mM
NaCI, 1% (w/v) SDS,
and protease inhibitor mixture (1 pg/ml pepstatin A, 10 pg/ml leupeptin, 200
pM
phenylmethylsulfonyl fluoride). The protein concentration of each total cell
extract was measured
(BCA kit; Pierce), after which a 22-33 pg aliquot of cell extract was mixed
with 0.25 volume of buffer
(250 mM Tris-HCI, pH 6.8, 10% SDS, 25% glycerol, 0.2% (w/v) bromophenol blue,
and 5% (v/v) 2-
mercaptoethanol), heated for 7 min at 95 C. The samples were separated on a
10% SDS-PAGE
gel and blotted using mouse monoclonal antibody against SREBP-2 (IgG-7D4)
(Yang et al., 1995).
The specific bands were visualized using enhanced chemiluminescent (ECL)
detection reagents
(Amersham).
Modification of SOAP oligosaccharides. Cell membrane fractions were prepared
as
described elsewhere herein. The membrane pellets were resuspended in 0.1 mL of
buffer
containing 10 mM Hepes=KOH (pH 7.4), 10 mM KCI, 1.5 mM MgC12, 1 mM sodium
EDTA, and 100
mM NaCI. Aliquots of protein were then incubated in the absence or presence of
1 pg of trypsin, in a
total volume of 58 pL, for 30 min at 30 C. Reactions were stopped by addition
of 2 pL (400 units) of
soybean trypsin inhibitor. For subsequent treatment with endoglycosidase H,
individual samples
received 10 pl of solution containing 3.5% (wt/vol) SDS and 7% (vol/vol) 2-
mercaptoethanol. After
heating at 100 C for 10 min, each sample received sequential additions of 9 pl
of 0.5 M sodium
citrate (pH 5.5), 5 pL of solution containing 17' protease inhibitors (a
concentration of lx,
corresponding to 10 pg/mL leupeptin, 5 pg/mL pepstatin A, and 2 pg/mL
aprotinin), followed by 1 pL
(5 units) of endoglycosidase H. The reactions were carried out overnight at 37
C and stopped by
the addition of 20 pL of buffer containing 0.25 M Tris=HCI (pH 6.8), 2% SDS,
10% (vol/vol) glycerol,
0.05% (wt/vol) bromophenol blue, and 4% 2-mercaptoethanol. The mixtures then
were heated at
100 C for 5 min and subjected to SDS/PAGE (12% gels).
Confocal microscopic analyses. CHO-K1 cells on a glass-bottom 96-well plate
(Grainer) at
¨70% confluency were incubated with 0.2 pM ER-tracker Red (lnvitrogen) and 5
mM dansyl
fatostatin for 1 h. Fluorescent images were captured and analyzed with a Carl
Zeiss LSM 510
confocal microscope, equipped with a CSU10 spinning-disk confocal scanner
(Yokogawa Electric
Corporation) and an ORCA-CCD camera (Hamamatsu Photonics). Images were
analysed with
IPLab software (Solution Systems).
54
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
Binding assay. Cell membrane fractions were prepared as described in
Supplementary
Methods. The membrane fraction was extracted with PBS containing 0.1% FOS-
Choline 10
(Hampton Research). The extract was mixed with Neutravidine-agarose beads (10
pL) saturated
with biotinylated fatostatin and incubated for 1 h. The bound proteins were
washed four times with
PBS containing 0.1% FOS-Choline 10, boiled in 25 pL of SDS sample buffer, and
subjected to
western blotting. For the competition assay, saturated amounts of cholesterol
or fatostatin were
added to the membrane extract before incubating with the beads.
Animal studies procedures. Four to five week-old homozygous male obese (ob/ob)
mice
(057BL/6J, The Jackson Laboratory, Bar Harbor, ME) were housed under
controlled conditions (12
h light/dark cycle; 25 C). The animals were housed 5 per cage, and had ad
libitum access to
standard laboratory chow (Purina Mills, Richmond, IN) and water for one week
after their arrival. On
first day of the experiment and every day thereafter, the weight of each mouse
and the amount of
food intake were measured between 3:00 and 5:00 p.m. Following weight
measurements, treated
mice received an ip injection of fatostatin (30 mg/kg; 150 pL), and control
mice received 10% DMSO
in PBS. Daily Injections were continued for four weeks, until the end of the
study.
Blood constituents. After 28 days of daily injection of fatostatin mice were
fasted for 5-6 h,
whole blood glucose and P-hydroxybutyrate were measured with a Glucometer
Precision Xtra
(Abbott). Measurements of the serum constituents, glucose, triglyceride, and
cholesterol, were
performed by the Comparative Pathology Laboratory (Baylor College of
Medicine). Serum non-
esterified fatty acids (NEFA) were measured using a NEFA C kit (Wako
Chemicals, Richmond, VA).
Liver analyses. Mice were sacrificed, and weights of livers and epididymal fat
pads were
determined. Frozen sections of liver slices from individual animals were
stained with Oil Red 0 to
visualize the fat droplets (triglycerides) in liver slices, as described (Abu-
Elheiga et al., 2001). The
remaining liver tissues were frozen in liquid nitrogen and kept at ¨80 C for
further analysis.
Tissue triglyceride and cholesterol contents. Liver triglyceride and
cholesterol contents were
determined as described by Chandler et al. (2003), using a Cholesterol E Kit
(Wako) and an Infinity
Triglyceride Kit (Thermo Electron, Melbourne, Australia).
Synthesis of fatostatin 1, dansyl fatostatin and fatostatin-polyproline linker-
biotin conjugate
FIG. 23 depicts the synthesis of fatostatin 1, dansyl fatostatin, fatostatin-
polyproline linker-
biotin conjugates and the synthetic intermediates.
Synthesis of fatostatin 1
A mixture of prothionamide (1.03 g, 5.70 mmol) and 2-bromo-4'-
methylacetophenone (1.22
g, 5.70 mmol) in ethanol (20 ml) was heated at 70 C with stirring for 0.5 h,
and then cooled to 0 C.
A yellow precipitate formed was filtered, washed with cold ethanol, and dried
to give 2-propy1-4-(4-p-
tolylthiazol-2-yl)pyridine (fatostatin) 1 HBr salt (1.78 g, 83%) as yellow
needles. mp: 190-193 C; 1H
NMR (600 MHz, DMSO-d6): 68.88 (d, J = 6.2 Hz, 1H), 8.54 (s, 1H), 8.46 (d, J =
1.4 Hz, 1H), 8.36
(dd, J = 1.4, 6.2 Hz, 1H), 7.99 (d, J = 7.6 Hz, 2H), 7.31 (d, J = 7.6 Hz, 2H),
3.03 (t, J = 7.6 Hz, 2H),
2.35 (s, 3H), 1.80 (m, 2H), 0.96 (t, J = 7.6 Hz, 3H); 130 NMR (150 MHz, DMSO-
d6): d 161.3, 158.5,
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
156.9, 146.2, 143.2, 138.4, 130.4, 129.5, 126.3, 122.3, 120.3, 119.5, 35.0,
22.4, 20.9, 13.4; HRMS
(m/z): [M4-Hr calcd for C18H19N2S, 295.1269; found, 295.1269.
Synthesis of 4-(2-(2-propylpyridin-4-yl)thiazol-4-yl)benzenamine 16
A pressure tube was charged with 2 (1.08 g, 3.0 mmol), benzophenone imine
(0.57 g, 3.3
mmol), Pd2(dba)3 (86 mg, 0.15 mmol), BINAP (280 mg, 0.45 mmol), sodium tert-
butoxide (1.44 g,
9.0 mmol), and dry toluene (30 mL) and purged with argon gas. The pressure
tube was sealed and
heated in a 100 C bath for 20 h. After being cooled to room temperature, the
reaction mixture was
chromatographed (5i02, 4:1 hexane:Et0Ac) to provide 1.35 g of 8 (98%) as a
yellow oil. Then to a
solution of 8(1.35 g, 2.9 mmol) in THF (20 mL) was added 2 N aqueous NCI
solution (15 mL). After
being stirred at room temperature for 2 h, the reaction mixture was
concentrated under reduced
pressure, then diluted with Et0Ac (100 mL) and washed with saturated Na2CO3
(50 mL) solution.
The aqueous wash was extracted with Et0Ac (3 x 40 mL), and the combined Et0Ac
layers were
dried over Na2504 and concentrated. The crude product was chromatographed
(5i02, 4:1 hexane:
Et0Ac) to provide 0.73 g of 4-(2-(2-propylpyridin-4-yl)thiazol-4-
yl)benzenamine 16 (82%) as a white
crystal. 1H NMR (600 MHz, CDCI3): 6 8.61 (d, J = 4.8 Hz, 1H), 7.80 (d, J = 8.9
Hz, 2H), 7.75 (d, J =
1.4 Hz, 1H), 7.67 (dd, J = 1.4, 4.8 Hz, 1H), 7.36 (s, 1H), 6.75 (d, J = 8.9
Hz, 2H), 3.82 (brs, 1H),
2.85 (t, J= 7.6 Hz, 2H), 1.83 (m, 2H), 1.01 (t, J= 7.6 Hz, 3H); 130 NMR (150
MHz, 0D013): 6 164.9,
163.4, 157.3, 150.0, 146.8, 140.8, 127.7, 124.8, 119.2, 117.8, 115.1, 111.3,
40.4, 23.1, 13.9; HRMS
(m/z): [M+Hr calcd for C17H18N3S, 296.1221; found, 296.1228.
Synthesis of dansyl fatostatin
To a magnetically stirred solution of 16 (50 mg, 0.17 mmol) and pyridine (27
mg, 0.34mmol)
in 0H2012 (5 mL) was added dansyl chloride (50 mg, 0.18 mmol). After being
stirred for 17 h, the
reaction mixture was concentrated under reduced pressure, and the residue was
partitioned
between Et0Ac (50 mL) and saturated NH401 solution (20 mL). The aqueous phase
was extracted
with Et0Ac (2 x 20 mL). The combined extracts were washed with saturated
NaHCO3 solution, dried
over Na2SO4, and concentrated. The crude product was chromatographed (Si02,
2:1 hexane:
Et0Ac) to afford dansylfatostatin (65 mg, 73%) as a yellow crystal. 1H NMR
(600 MHz, CDCI3): 6
8.60 (d, J = 4.8 Hz, 1H), 8.50 (d, J = 8.2 Hz, 1H), 8.36 (d, J = 8.3 Hz, 1H),
8.21 (d, J = 8.3 Hz, 1H),
7.76(d, J = 8.6 Hz, 2H), 7.70(d, J = 1.5 Hz, 1H), 7.62 (dd, J = 1.5, 4.8 Hz,
1H), 7.61 (t, J = 8.3 Hz,
1H) 7.44 (s, 1H), 7.43 (dd, J = 7.5, 8.2 Hz, 1H), 7.19 (d, J = 7.5 Hz, 1H),
7.04 (d, J = 8.6 Hz, 2H),
6.97 (brs, 1H), 2.87 (s, 6H), 2.84(t, J = 7.6 Hz, 2H), 1.81 (m, 2H), 1.00 (t,
J= 7.6 Hz, 3H); 13C NMR
(150 MHz, CDCI3): 6 165.5, 163.5, 156.1, 152.2, 150.0, 140.5, 136.7, 134.0,
131.1, 131.0, 130.5,
129.8, 129.7, 128.7, 127.3, 123.1, 121.6, 119.2, 118.3, 117.8, 115.3, 113.8,
45.4, 40.4, 23.1, 13.9;
HRMS (m/z): [M+H]f calcd for 029H29N402S2, 529.1732; found, 529.1733.
Synthesis of tert-butyl 4-(2-(2-propylpyridin-4-yOthiazol-4-yOphenylcarbamate
40
To a magnetically stirred solution of 16 (0.57 g, 1.92 mmol) and 4-
(dimethylamino)pyridine
(5 mg, 0.4 mmol) in THF (20 mL) was added di(tert-butyl)dicarbonate (0.49 g,
2.21 mmol). After
being stirred for 17 h, the reaction mixture was concentrated under reduced
pressure, and the
56
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
residue was partitioned between Et0Ac (100 mL) and saturated NH401 solution
(30 mL). The
aqueous phase was extracted with Et0Ac (2 x 50 mL). The combined extracts were
washed with
saturated NaHCO3 solution, dried over Na2SO4, and concentrated. The crude
product was
chromatographed (Si02, 2:1 hexane: Et0Ac) to afford tert-butyl 4-(2-(2-
propylpyridin-4-yl)thiazol-4-
yl)phenylcarbamate 40 (0.33 g, 43%) as a yellow foam. 1H NMR (300 MHz, 0D013):
6 8.68 (d, J =
5.1 Hz, 1H), 7.93 (d, J = 8.4 Hz, 2H), 7.79 (s, 1H), 7.72 (d, J = 5.1 Hz, 1H),
7.52 (s, 1H), 7.47 (d, J =
8.4 Hz, 2H), 6.58 (s, 1H), 2.90 (t, J = 7.5 Hz, 2H,), 1.87 (m, 2H), 1.55 (s,
9H), 1.03 (t, J = 7.5 Hz,
3H); 130 NMR (75 MHz, CDCI3): 6 163.1, 156.5, 152.3, 149.6, 140.6, 138.5,
128.7, 128.1, 127.0,
118.3, 117.7,114.3, 112.9, 80.6, 41.0, 28.4, 24.1,13.7; HRMS (m/z): [M+1-1]+
calcd for 022H26N302S,
396.1746; found, 396.1738.
Synthesis of R=N(Boc)CH2CH2CH2000H intermediate
In an N2 atmosphere, 40 (200 mg, 0.51 mmol) was added to a suspension of NaH
(60%
dispersion in mineral oil, 24 mg, 0.6 mmol) in DMF (5 mL) and the mixture was
stirred for 2hr at
room temperature. Then Nal (91 mg, 0.6 mmol) and ethyl 4-bromobutyrate (0.12g,
0.6mmol) in DMF
(2 mL) were added. After being stirred for 18 h, the reaction mixture was
poured into water (20 mL)
and was extracted with Et0Ac (2 x 50 mL). The combined extracts were dried
over Na2504, and
concentrated. The crude product was chromatographed (Si02, 2:1 hexane:AcOEt)
to provide 11(35
mg, 13%) as a yellow oil. Then to a solution of 11(30 mg, 2.9 mmol) in THF (1
mL) and Me0H (0.5
mL) was added 2 N aqueous NaOH solution (0.2 mL). After being stirred at room
temperature for 18
h, the reaction mixture was concentrated under reduced pressure, then diluted
with Et0Ac (10 mL)
and washed with saturated NH401 solution (5 mL). The aqueous wash was
extracted with Et0Ac (2
x 10 mL), and the combined Et0Ac layers were dried over Na2SO4 and
concentrated. The crude
product was chromatographed (Si02, Et0Ac) to provide 14 mg of the
R=N(Boc)0H20H2CH2000H
intermediate (50%) as a white foam. 1H NMR (300 MHz, 00013): 68.69 (d, J= 5.1
Hz, 1H), 7.95 (d,
J = 8.4 Hz, 2H), 7.80(s, 1H), 7.73 (d, J = 5.1 Hz, 1H), 7.53(s, 1H), 7.51 (d,
J= 8.4 Hz, 2H), 6.58 (s,
1H), 2.92 (m, 4H), 2.33 (m, 2H), 1.90 (m, 4H), 1.50 (s, 9H), 1.02 (t, J = 7.5
Hz, 3H); 130 NMR (75
MHz, 0D013): 6 174.2, 162.9, 156.3, 152.9, 149.6, 140.6, 138.5, 129.3, 128.9,
127.2, 119.0, 117.9,
114.8, 113.3, 80.8, 41.0, 40.3, 32.1, 28.4, 24.1, 21.1, 13.7; HRMS (m/z):
[M+H] calcd for
026H32N3045, 482.2114; found, 482.2120.
Synthesis of 4-(4-(2-(2-propylpyridin-4-yl)thiazol-4-y1)phenylamino)-N-
isopropyl butanamide
To a solution of the R=N(Boc)CH2CH2CH2000H intermediate (17 mg, 0.035 mmol),
triethylamine (17 pl, 0.14 mmol) and isopropylamine (4 pL, 0.046 mmol) in DMF
(0.5 mL) was HATU
(16 mg, 0.042 mmol). After being stirred for 18 h, the reaction mixture was
concentrated under
reduced pressure, and the residue was partitioned between Et0Ac (20 mL) and
saturated NH4CI
solution (10 mL). The aqueous phase was extracted with Et0Ac (2 x 10 mL). The
combined extracts
were washed with saturated NaHCO3 solution, dried over Na2SO4, and
concentrated. The crude
product was chromatographed (Si02, 2:1 hexane: Et0Ac) to afford the N-Boc
protected 4-(4-(2-(2-
propylpyridin-4-yl)thiazol-4-yl)phenylamino)-N-isopropyl butanamide (14 mg,
77%) as a yellow oil.
57
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
Then to a solution of 13 (12 mg, 2.9 mmol) in THF (1 mL) was added TFA (0.2
mL). After being
stirred at room temperature for 18 h, the reaction mixture was concentrated
under reduced
pressure, then diluted with Et0Ac (20 mL) and washed with saturated NH4CI
solution (10 mL). The
aqueous wash was extracted with Et0Ac (2 X 5 mL), and the combined Et0Ac
layers were dried
over Na2SO4 and concentrated. The crude product was chromatographed (Si02, 2:1
hexane:
Et0Ac) to provide 6.2 mg of 4-(4-(2-(2-propylpyridin-4-yl)thiazol-4-
yl)phenylamino)-N-isopropyl
butanamide (63%) as a yellow foam. 1H NMR (300 MHz, CDCI3): 6 8.69 (d, J = 5.4
Hz, 1H), 8.04
(1H, s), 7.96 (d, J = 8.7 Hz, 2H), 7.96 (s, 1H), 7.72 (d, J = 5.4 Hz, 1H),
7.51 (s, 1H), 6.65 (d, J = 8.7
Hz, 2H), 3.95(m, 1H), 3.03(m, 4H,), 2.35(m, 2H), 1.92(m, 4H), 1.25(d, J= 6.3
Hz, 6H), 1.03(t, J=
7.5 Hz, 3H); 13C NMR (75 MHz, CDCI3): 6 171.6, 162.9, 156.3, 152.9, 140.6,
138.5, 129.3, 128.9,
127.2, 119.0, 117.9, 114.8, 113.3, 42.2, 41.0, 40.3, 32.1, 24.1, 23.2, 21.1,
13.7; HRMS (m/z):
[M+H] calcd for C24H31N40S, 423.2219; found, 423.2216.
Synthesis of fatostatin-polyproline linker-biotin conjugate
Fatostatin-KPGQFLYELKKPPPPPPPPPKK (SEQ ID NO: 15)-aminocaproic acid-biotin.
Conjugates were synthesized on Rink-Amide MBHA resin by coupling N-a-Fmoc-
protected
amino acids, N-E-Fmoc-e-aminocaproic acid, R=N(Boc)CH2CH2CH2COOH intermediate,
biotin,
purified by a reversed phase HPLC as described (Sato et al., 2007). Conjugate
3: calcd for
C155H238N35029S2+ requires 3119.9. Found (MALDI-TOF-MS) 3119.7 [M +
Plasmids. pSRE-Luc, pCMV-SREBP-1c(1-436), pCMV-PLAP-BP2(513-1141) and pCMV-
SCAP were gifts from J. L. Goldstein and M. S. Brown (University of Texas
Southwestern Medical
Center) (Sakai etal., 1998; Hua etal., 1995).
Antibodies. Monoclonal anti-SREBP-1 IgG (2A4), anti-SCAP IgG (9D5) and anti-
ATF6 IgG
(H-280) were purchased from Santa Cruz Biotechnology. Monoclonal anti-SREBP-2
IgG-7D4 was a
gift from J. L. Goldstein and M. S. Brown (University of Texas Southwestern
Medical Center).
Polyclonal anti-FAS, anti-ACC, anti-SCD1 and anti-ACL IgG were purchased from
BD Biosciences.
Cell culture. Chinese hamster ovary cells K1 (CHO-K1) cells were maintained in
a
Dulbecco's modified Eagle's medium/Ham's F12 medium [1:1] with 5% fetal bovine
serum, 100
units/mL penicillin, and 100 pg/mL streptomycin sulfate at 37 C under 5% CO2.
Human androgen-
independent prostate cancer cells (DU145) were maintained in an Eagle's
minimum essential
medium containing 2 mM L-glutamine, 1.0 mM sodium pyruvate, 0.1 mM
nonessential amino acids,
and 1.5 g/L sodium biocarbonate with 10% fetal bovine serum, 100 units/mL
penicillin, and 100
pg/mL streptomycin sulfate at 37 C under 5% CO2.
Preparation of cell membrane fraction. Cells were harvested, and then
resuspended in
buffer (10 mM Hepes=KOH (pH 7.4), 10 mM KCI, 1.5 mM MgC12 and 1 mM sodium
EDTA), passed
through a 22-gauge needle, and centrifuged at 1,000 x g for 5 min. The
postnuclear supernatants
then were centrifuged at 15,000 x g for 30 min, and the supernatant was
removed.
Oligonucleotide microarray analysis. DU145 prostate cancer cells were treated
with 5 mM of
fatostatin or DMSO alone in the presence of 1 pg /mL of IGF1 for 6 hrs in a
serum free medium,
total RNA was extracted in a TRI reagent (Molecular Research Center) and
further isolated by
58
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
RNeasy Mini Kit (Qiagen). Purified mRNA was analyzed in Baylor College of
Medicine Microarray
Core Facility by Affymetrix Human Genome U133 Plus 2.0 Array consisting of
almost 45,000 probe
sets representing more than 39,000 transcripts derived from approximately
33,000 well-
substantiated human genes (Affymetrix, Inc.).
RT-PCR experiments. Total RNA was extracted from DU145 cells in TRI reagent
(Molecular
Research Center) and isolated with an RNeasy Mini Kit (Qiagen). The RNA sample
was subjected
to RT-PCR by using the Access RT-PCR System (Promega). RT-PCR reactions
contained total
RNA, 1 pM of each primer, 0.2 mM dNTP, 1 mM MgSO4, AMV reverse transcriptase
(2 units), and
Tfl DNA polymerase (2 units) in a final volume of 25 pL. The primer pairs used
are as follows: 5'-
TCA GAC CGG GAC TGC TTG GAC GGC TCA GTC -3 (SEQ ID NO: 16) and 5'-CCA CTT AGG
CAG TGG AAC TCG AAG GCC G -3' (SEQ ID NO: 17) for Low density lipoprotein
receptor (LDLR);
5'- GCC TGC TTG ATA ATA TAT AAA C -3' (SEQ ID NO: 18) and 5' - CAC TTG AAT TGA
GOT
TTA G -3' (SEQ ID NO: 19) for stearoyl-CoA desaturase (SOD); 5' -AAG AAA AAG
TGT CAG ACA
GOT GG -3' (SEQ ID NO: 20) and 5' - TGG ACT GAA GGG GTG TTA GC -3' (SEQ ID NO:
21) for
ATP citrate lyase (ACL); 5'- GCC CGA CAG TTC TGA ACT GGA ACA -3' (SEQ ID NO:
22) and 5'-
GAA CCT GAG ACC TCT CTG AAA GAG -3' (SEQ ID NO: 23) for 3-hydroxy-3-
methylglutaryl- CoA
reductase (HMG-CoAR); 5'- CTG OCT GAC TGC CTC AGO -3' (SEQ ID NO: 24) and 5'-
ACC TOT
COT GAC ACC TGG G -3' (SEQ ID NO: 25) for mevalonate pyrophosphate
decarboxylase (MVD);
5'-AAG ACT TCA GGG TAA GTC ATC A-3' (SEQ ID NO: 26) and 5'- CGT GTA TAA TGG
TGT CTA
TCA G -3' (SEQ ID NO: 27) for insulin induced gene 1 (INSIG1). The
amplification conditions are as
follows: 1 cycle at 94 C for 4 min, then denatured at 94 C for 40 s, annealed
at 50 C for 40 s, and
extended at 68 C for 2 min with 22 cycles for SOD and HMG CoA R, annealed at
58 C with 24
cycles for LDLR and INSIG1, or annealed at 60 C with 24 cycles for ACL,
annealed at 55 C with 30
cycles for MVD. The amplified DNAs were analyzed by an agarose gel and
quantified with the
Scion-image software.
PLAP-BP2 Cleavage. On day 0, OHO-K1 cells were plated out onto a 96-well plate
in
medium A. On day 2, the cells were transiently co-transfected with pCMV-PLAP-
BP2(513-1141),
pCMV-SOAP and pAc-p-gal, using Lipofectamine reagent (Invitrogen). After
incubation for 5 h, the
cells were washed with PBS, and then incubated, in the absence or presence of
fatostatin (20 pM)
or sterols (10 pg/mL cholesterol and 1 pg/mL 25-hydroxycholesterol), in medium
B. After 20 h of
incubation, an aliquot of the medium was assayed for secreted alkaline
phosphatase activity. The
cells in each well were lysed, and used for measurement of P-galactosidase
activities. The alkaline
phosphatase activity was normalized by the acitivity of p-galactosidase.
Enzymatic activities and western blot analyses. A portion of the frozen liver
was ground to
powder in liquid nitrogen. The powdered tissues were suspended in 10 mL of PBS
containing 0.1
mM PMSF, 5 mM benzamidine, and 5 mg/mL protease inhibitor cocktail (Roche),
and homogenized
using Polytron (3 x 30 Sec, at high speed), and sonicated briefly to degrade
DNA. The extracts were
clarified by centrifugation at 16,000 x g for 20 min. Protein concentrations
in the supernatant were
determined, and subjected to western blot analysis using antibodies against
FAS, ACC, SCD1 and
ACL. The intensity of the specific bands of proteins of interest were scanned
and normalized
59
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
against b-actin for quantifications. FAS and ACC activities from the liver
extracts were determined
as described earlier (Mao etal., 2006).
EXAMPLE 11
Synthesis of benzyl (R)-2-(4-(4-(methylsulfonamido)phenyl)thiazol-2-
yl)pyrrolidine-1-carboxylate 53
As shown in FIG. 26, to a solution of L-prolinamide (1.14 g, 10.0 mmol) in
acetonitrile (100
mL) were added benzyl chloroformate (1.55 mL, 11.0 mmol) and triethylamine
(2.79 mL, 20 mmol)
at rt. The mixture was stirred at rt for 12 h and diluted with AcOEt. The
mixture was poured into 0.1
M HCI solution and separated between the organic layer and the aqueous layer.
The organic layer
was washed with sat. aq. NaHCO3 and brine, dried (Na2SO4), filtered, and
concentrated. The
residue was purified by silica gel column chromatography to give 67 (2.26 g,
91%) as colorless oil.
A mixture of 67 (586 mg, 2.36 mmol) and Lawesson's reagent (1.05 g, 2.60 mmol)
in
toluene (30 mL) was stirred at 90 C for 1 h. After cooling to rt, the solvent
was evaporated. The
residue was purified by silica gel column chromatography to give 68 (495 mg,
79%) as a white
amorphous solid.
A mixture of 68 (300 mg, 1.13 mmol) and 4-bromophenacyl bromide (318 mg, 1.13
mmol) in
Et0H (10 mL) was stirred under reflux for 2 h. After evaporated, the residue
was purified by silica
gel column chromatography to give 48 (265 mg, 53%) as a white solid.
A mixture of 48 (230 mg, 0.519 mmol), Pd2(dba)3 (9.50 mg, 2 mor/o), BINAP
(97%, 20.0 mg,
6 mol /0), sodium t-butoxide (69.8 mg, 0.727 mmol), and benzophenone imine
(0.105 mL, 0.623
mmol) in toluene (5.0 mL) under reflux for 8 h. After cooling to rt, the
mixture was filtered through
Celite and the filtrate was evaporated. The residue was purified by silica gel
column
chromatography to give 69 (228 mg, 81%) as a yellow amorphous solid.
To a solution of 69 (175 mg, 0.321 mmol) in THF (3 mL) was added aqueous HCI
solution
(4 M, 0.3 mL) and the mixture was stirred at rt for 1 h. The mixture was
neutralized with sat. aq.
NaHCO3 and extracted with AcOEt. The organic layer was washed with brine,
dired (Na2SO4),
filtered, and concentrated. The residue was purified by silica gel column
chromatography to give 70
(113 mg, 93%) as a colorless oil.
A mixture of 70 (92 mg, 0.24 mmol), methanesulfonyl chloride (0.021 mL, 0.27
mmol), and
pyridine (0.043 mL, 0.53 mmol) in CH2012 (2 mL) was stirred at rt for 8 h.
After addition of Me0H,
the mixture was diluted with AcOEt and washed with sat. aq. NaHCO3 and brine.
The organic layer
was dried (Na2SO4), filtered, and concentrated. The residue was purified by
silica gel column
chromatography to give 53 (91 mg, 83%) as a white solid.
1H NMR (CDCI3, 600 MHz, two rotamers, ratio = 1:0.95) 5 7.80-7.86 (m, 2H),
7.14-7.41 (m,
7H), 6.64 (s, 1H, major rotamer), 6.83 (s, 1H, minor rotamer), 5.07-5.36 (m,
3H), 3.73 (m, 1H), 3.59
(m, 1H), 3.03 (s, 3H, major rotamer), 3.00 (s, 3H, minor rotamer), 2.28-2.39
(m, 2H), 2.00-2.10 (m,
2H); 130 NMR (CDCI3, 150 MHz, two romamers, major peaks are shown) 174.7,
155.4, 154.4,
136.4, 131.9, 128.5, 128.3, 127.9, 127.8, 127.7, 127.7, 120.8, 112.3, 67.1,
59.4, 47.2, 39.4, 32.9,
23.1; HRMS (FAB): Exact mass calcd. for 022H24N304S2 [M+H], 458.1203; Found
458.1200.
60
CA 02922703 2016-02-26
WO 2015/031710 PCT/US2014/053334
EXAMPLE 12
Synthesis of N-(4-(2-(2-propylpyridin-4-yl)thiazol-4-
yl)phenyl)methanesulfonamide (19)
As shown in FIG. 27, in reaction (a) a mixture of prothionamide (9.01 g, 50.0
mmol) and 4-
bromophenacyl bromide 6a (13.9 g, 50.0 mmol) in Et0H (400 mL) was stirred at
70 C for 1 h. After
cooled to room temperature, the precipitation was filtered off with Et0Ac and
washed with Et0Ac
and saturated aqueous NaHCO3. The organic layer was washed with brine, dried
over Na2SO4, and
concentrated to give 4-(4-bromophenyI)-2-(2-propylpyridin-4-yl)thiazole 2
(14.3 g, 79%) as a light-
yellow solid.
In reaction (b) a pressure tube was charged with 2 (1.08 g, 3.0 mmol),
benzophenone imine
(0.579, 3.3 mmol), Pd2dba3 (86 mg, 0.15 mmol), BINAP (280 mg, 0.45 mmol),
sodium tert-butoxide
(1.44 g, 9.0 mmol), and dry toluene (30 mL) and purged with argon gas. The
pressure tube was
sealed and heated in a 100 C bath for 20 hrs. After being cooled to room
temperature, the reaction
mixture was chromatographed (Si02, 4:1 hexane:Et0Ac) to provide 1.35 g of
71(98%) as a yellow
oil.
In reaction (c) then to a solution of 71 (1.35 g, 2.9 mmol) in THF (20 mL) was
added 2 M
aqueous HCI solution (15 mL). After being stirred at room temperature for 2
hrs, the reaction
mixture was concentrated under reduced pressure, then diluted with Et0Ac (100
mL) and washed
with saturated Na2003 (50 mL) solution. The aqueous wash was extracted with
Et0Ac (3 x 40 mL),
and the combined Et0Ac layers were dried over Na2SO4 and concentrated. The
crude product was
chromatographed (5i02, 4:1 hexane:Et0Ac) to provide 0.73 g of 4-(2-(2-
propylpyridin-4-yl)thiazol-4-
yl)benzenamine 16 (82%) as a white crystal.
1H NMR (600 MHz, 0D013): 6 8.61 (d, J = 4.8 Hz, 1H), 7.80 (d, J = 8.9 Hz, 2H),
7.75 (d, J =
1.4 Hz, 1H), 7.67 (dd, J = 1.4, 4.8 Hz, 1H), 7.36 (s, 1H), 6.75 (d, J = 8.9
Hz, 2H), 3.82 (brs, 1H),
2.85(t, J= 7.6 Hz, 2H), 1.83(m, 2H), 1.01 (t, J= 7.6 Hz, 3H); 130 NMR (150
MHz, CDCI3): 5 164.9,
163.4, 157.3, 150.0, 146.8, 140.8, 127.7, 124.8, 119.2, 117.8, 115.1, 111.3,
40.4, 23.1, 13.9; HRMS
(m/z): [M-FH]+ calcd for 017H18N3S, 296.1221; found, 296.1228.
In reaction (d) methanesulfonyl chloride (0.23 mL, 2.97 mmol) was added to a
stirred
solution of 16 (800 mg, 2.71 mmol) and pyridine (0.66 mL, 8.1 mmol) in 0H2Cl2
(20 mL) at 0 C.
After being stirred for 0.5 h, the mixture was poured into 2 M citric acid
solution and extracted with
Et0Ac. The combined extracts were washed with saturated NaHCO3 solution and
brine, dried over
Na2SO4, and concentrated to
produce N-(4-(2-(2-propylpyridin-4-yl)thiazol-4-
yl)phenyl)methanesulfonamide 19 (880 mg, 87%) as a yellow foam.
1H NMR (300 MHz, CD30D): 5 8.55 (d, J = 5.2 Hz, 1H), 8.02 (d, J = 8.8 Hz, 2H),
7.95 (s,
1H), 7.90 (d, J = 1.9 Hz, 1H), 7.84 (dd, J = 1.9, 5.2 Hz, 1H), 7.34 (d, J =
8.8 Hz, 2H), 3.00 (s, 3H),
2.86 (t, J = 7.7 Hz, 2H), 1.80 (m, 2H), 1.01 (t, J = 7.3Hz, 3H). m/z =374
[M+H].
EXAMPLE 13
Synthesis of N-isopropyl-4-(2-(2-propylpyridin-4-yl)thiazol-4-yl)benzamine
(17)
As shown in FIG. 27, the procedure to synthesize compound 17 is utilizes the
same steps a-
c as described for compound 19 in Example 12. For compound 17 at step e,
acetone (2.5 mL, 34.5
61
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
mmol) and acetic acid (2.0 mL, 34.5 mmol) were added to a stirred solution of
16 (1.02 g, 3.45
mmol) in CH2Cl2 (20 mL). After the mixture was stirred for 1 h, Na(0Ac)3BH
(1.5 g, 6.9 mmol) was
added. The mixture was stirred for 20 h. The mixture was poured into saturated
NaHCO3 solution
and extracted with Et0Ac. The combined extracts were dried over Na2SO4 and
concentrated.
Chromatography of the crude product (Si02, 4:1 hexane/Et0Ac) produced 17 (845
mg, 73%) as a
white foam. 1H NMR (300 MHz, CDCI3): 6 8.61 (d, J = 5.2 Hz, 1H), 7.81 (d, J =
8.5 Hz, 2H), 7.76 (d,
J = 1.4 Hz, 1H), 7.68 (dd, J = 1.4, 5.2 Hz, 1H), 7.34 (s, 1H), 6.65 (d, J =
8.5 Hz, 2H), 3.70 (m, 1H),
2.86 (t, J = 7.6 Hz, 2H), 1.83 (m, 2H), 1.25 (d, J = 6.0 Hz, 6H), 1.02 (t, J =
7.4 Hz, 3H). m/z = 338
[M+11]+.
EXAMPLE 14
SREBP activation assays for compounds 45-66
Standard SREBP activation assays were performed on the exemplary compounds
identified
in Table 4 as per the method for Fatostatin A in Example 1. The ability of
endogenous SREBPs to
activate transcription of an SREBP-responsive reporter gene was measured in
the presence or
absence of compounds 45-55 and 19 (FIG. 28), compounds 56-61 and 19 (FIG. 29)
and
compounds 62-66 (FIG. 30) at 5 'LIM in CHOK1 cells. Of these, compound 61
demonstrated
inhibition of SREBP activation at about 25%, with compound 53 at about 30%,
compound 58 at
about 42%, and compound 19 at about 45%. The inhibitory concentrations of
compounds 53 (FIG.
31) and compounds 58 and 61 (FIG. 32) were determined.
EXAMPLE 15
Compound 19 inhibited the growth of the human breast cancer cell lines SUM159
and
downregulated lipogenic pathways
Treating SUM159 cells with different concentrations of compound 19 for 48
hours caused
an inhibition of cell growth and a toxic effect (FIG. 33A). Cells seeded onto
96 well plates at a
density of 5000 cells/well in 100 pl medium containing 2% charcoal treated
serum. After 24 hours
different concentrations of compound 19 were added for an additional 48 hours.
Cell viability was
measured using WST-1 Cell Viability and Proliferation Assay (ScienCell
Research Laboratory,
Carlsbad CA). Changes in expression levels of lipogenic genes mRNA was
determined in cells that
were treated with 10 pM. There was a significant downregulation of the genes
that are under the
control of transcription factors, SREBPs. Insig2 gene which is not known to be
regulated by these
transcription factors was not affected by the treatment (FIG. 33B).
The effect of Compound 19 was also demonstrated on human hepatocellular
carcinoma cell
line (HePG2). As shown in FIGS. 34A-34B, Compound 19 produced a high level of
toxicity as
evidenced by the dramatic change in the morphology and the inhibition of
growth of the treated
cells. When HePG2 cells were treated with 25 pM of Compound 19 there was a
significant decrease
in the level of the mature and active form and an increase in the precursor of
SREBP-1 and
consequently the expression levels of genes that are controlled by SREBP
(FIGS. 34A-34B). These
62
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
results are consistent and supportive of the conceptual action of compound 19
as an inhibitor of
lipogenesis through the inhibition of SREBP activation.
Compound 19 inhibits the growth of human acute lymphoblastic leukemia cell
line MOLT-4
MOLT-4 cells were grown at two different conditions under 5% fetal bovine
serum (FBS)
and under 5% FBS fat free serum (charcoal treated serum). 10,000 Cells were
seeded a 96 well
plate for 24 hours before starting the treatment with different concentrations
of compound 19.
Compound 19 dissolved in DMSO was added to cells for 48 hours before
determining cell growth
using MTT assay. As shown in FIG. 35A, the growth of the cells under 5% FBS
and FBS fat free
was similar. Compound 19 inhibited cell growth about 75-80% at 10 and 20 pM
under both growth
conditions. These results show that at 10 and 20 pM compound 19 was equally
effective even in the
presence of exogenous lipids, which suggest that inhibition of de novo lipid
synthesis result in
growth inhibition in this cancer sell line. Interestingly, under FBS fat free
serum there was 30-50%
growth inhibition at 2 and 5 pM compound 19 (Fig. 35C) and a strong trend of
inhibition at 5 pM in
5% FBS condition (FIG. 35B).
Compound 19 inhibits the growth of human multiple myeloma cell line RPMI8226
RPMI8226 cells were grown under the same conditions as the MOLT-4 cells.
20,000 cells
were seeded into 96 well plates for 24 hrs prior to treatment with increasing
concentrations of
compound 19 as described for MOLT-4 cells. As shown in FIG. 35D, the growth of
the cells under
5% FBS and FBS fat free was similar except that 5 pM compound 19 in 5% FF FBS
inhibited growth
about 1.5 more than the corresponding amount in 5% FBS. Compound 19 was
similarly effective at
10 pM and 20 pM in 5% FBS and 5% FF FBS. There was a strong trend of
inhibition starting at 3
pM in 5% FBS condition (FIG. 35E) and FBS fat free serum at 2 pM (FIG. 35F).
EXAMPLE 16
Compound 17 inhibits the growth of human acute lymphoblastic leukemia cell
line MOLT-4
MOLT-4 cells were grown at two different conditions under 5% fetal bovine
serum (FBS)
and under 5% FBS fat free serum (charcoal treated serum). 20,000 cells were
seeded into a 96
well plate for 24 hours before starting the treatment with different
concentrations of compound 17.
Compound 17 dissolved in DMSO was added to cells for 48 hours before
determining cell growth
using MTT assay. As shown in FIG. 36A, the growth of the cells under 5% FBS
and 5% FBS fat
free was strongly inhibited at 5, 10 and 20 pM. Under 5% FF FBS, 3 pM
exhibited about 2.5 times
the inhibition as under 5% FBS conditions. Compound 17 inhibited cell growth
about 85-100% at 5,
10 and 20 pM under both growth conditions. Under both 5% FBS and 5% FBS fat
free sera there
was a strong trend of inhibition starting at 1 pM (FIGS. 36B-36C).
Compound 17 inhibits the growth of human multiple myeloma cell line RPM!
RPMI8226 cells were grown under the same conditions as the MOLT-4 cells.
20,000 cells
were seeded into 96 well plates for 24 hrs prior to treatment with increasing
concentrations of
63
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
compound 19 as described for MOLT-4 cells. As shown in FIG. 36D, the growth of
the cells under
5% FBS and FBS fat free was initially similar except that compound 17 in 5% FF
FBS inhibited
growth about 1.5 to 6 times more than the corresponding amount in 5% FBS at 5,
10 and 20 pM.
There was a strong inhibition starting at 3 pM in 5% FBS condition (FIG. 35E)
and 5% FBS fat free
serum (FIG. 35F).
EXAMPLE 17
Compound 19 up regulates uncoupling protein 2 (UCP2) and reduces body fat
SD male rats after 3 weeks feeding of Western diet (high fat high carbohydrate
(HFHC)),
which is rich in both carbohydrate (35% kcal) and fat (45% kcal). The results
of the MTD study
suggested that the maximally efficacious dose might be at or below 100 mg/kg.
Hence, 1/40 and
1/10 of 100 mg/kg was chosen for an efficacy study. The rats fed an HFHC diet
were divided into
four groups of 15 animals: (1) a control group that did not receive any
treatment; (2) a control group
that was given 1 ml cottonseed oil; (3) an experimental group treated with 2.5
mg/kg compound 19;
(4) an experimental group treated with 10 mg/kg compound 19. Finally, a group
(n=5) was fed
regular rat chow (RD).
Compound 19 was effective in reducing body weight and total body fat
Feeding the HFHC diet for three weeks increased total body weight of rats
about 12% more
than the rats that were fed a regular diet, 332 + 6.5 vs. 375 + 4.0 gram
respectively (Fig 37A). As
expected, total body fat and % fat in the rats fed the regular diet was
significantly lower than those
fed HFHC diet (44.0 + 2.5 vs. 62.4 + 2.4 g/rat and 13.95 + 0.5 vs. 18.48 +0.48
%, respectively). The
percent lean was also higher in RD fed rats compared to HFHC fed rats (Figs.
37B, 37C and 37D).
Effect of Compound 19 on body weight and composition
Body weight was determined daily and food intake was measured every 2-3 days.
Rats
were given compound 19 in cottonseed oil for six days starting on Sunday until
Friday. The dose
was calculated based on the body weight at the day of the administration of
the drug and the control
vehicle. As shown in Figure 38, the animals gained weight about 200 gram after
three weeks of
feeding the HFHC diet and before the start of the treatment (Fig. 38A). The
control group which was
fed HFHC diet, but not given cottonseed oil and the second control group
(control vehicle) gained
similar weight, suggesting the vehicle does not contribute to changes in body
weight (Fig. 38B).
There was significant reduction in body weight in the 2.5 mg/kg and the 10
mg/kg treated group
after two weeks of the start of the treatment. At the end of the eight weeks
treatment the 2.5 mg/kg
gained about 15% less weight than the control vehicle (357.7 + 10.4 g/rat
compared to 420.9 + 2.4
g/rat respectively) (Fig. 38A). There was more reduction in body weight gain
in the 10 mg/kg treated
rats which was about 24% lower than that of the control vehicle group (321.4 +
10.3 g/rat
respectively). The cumulative food intake was similar between control and 2.5
mg/kg (1370 + 21 vs.
1320 + 18 g/rat respectively) and was slightly decreased in the group of 10
mg/kg (1245 + 21, see
Fig. 2B). When ratio of total body weight gain was calculated relative to food
intake there was about
64
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
13 % and 16 % less body weight gained in 2.5 and 10 mg/kg groups compared to
control (0.326 +
0.011, 0.313 + 0.014 vs. 0.373 + 0.011, respectively). The reduced weight in
the 2.5 mg/kg group in
the absence of decreased food intake could be interpreted to mean that the
drug reduces weight in
the absence of reducing food intake, as observed in the ob/ob mice.
Reduction in weight gain was mainly due to lower body fat
In order to determine the effect of compound 19 on reducing total body fat
body composition
was determined using ECHOMRI method every two weeks. Total body weight of
treated rats with
2.5 and 10 mg/kg was lower than both the control and control vehicle groups
after two weeks of
treatment (Fig. 39A). This reduction was mainly due to lower body fat with no
significant change in
lean mass (Fig. 39B). After eight weeks of treatment, the body weights of the
2.5 and 10 mg/kg
treated rats were 8% and 15% lower than the control groups (545.6 + 9.7, 502.8
+ 7.8 and 592.9 +
5.4 g/rat respectively, see Fig. 39A). The body weight of the 10 mg/kg group
reached similar values
to the rats that were fed a regular diet. More importantly, the difference in
body weight can be
attributed mostly to the difference in fat content between the treated and
control groups (Fig. 39B).
After eight weeks of treatment the total body fat weight was about 25 and 43%
lower in 2.5 and 10
mg/kg compared to control-vehicle (114.7 + 5.2, 86.5 + 3.2 and 150.9 + 4.2
gr/rat respectively). The
total% fat was also significantly lower, with 2.5 mg/kg group had a similar
%fat to the rat group that
was fed the regular diet and 10 mg/kg had a lower percentage than the regular
diet; 26.6 + 0.53
(control); 21.9 + 0.82 (2.5 mg/kg); 18.0 + 0.6 (10 mg/kg ) and 21.2 + 0.82
(regular diet) (Fig. 390).
The lean mass weight was not significantly different between all groups,
although percent lean mass
was higher in 2.5 and 10 mg/kg compared to that of the control groups (Fig.
39D and 39E).
Compound 19 ameliorated fatty liver conditions caused by HFHC diet and
downregulated gene
expression of de novo lipogenesis and potentially by upregulation of
uncoupling proteins and
thermogenesis
Feeding Sprague-Dawley rats with a high fat diet not only induced obesity but
also caused a
fatty liver condition with high levels of triglyceride accumulated in these
livers. The effect of
compound 19 on the livers of SD rats fed HFHC diet was examined after eight
weeks of treatment.
As shown in Figure 40A, the control rats developed larger and fattier livers
compared to the treated
rats. This was further confirmed by staining of frozen section of livers from
five animals of each
group with Oil Red 0 staining where fat accumulation was lower in all treated
groups (n=5)
compared to the controls (Figure 40B). These results were confirmed by
determining the TG and
cholesterol levels in control and treated rats. As shown in figure 40C, the TG
and cholesterol levels
were 40 and 25% lower in both treated animal groups respectively (Fig. 40C).
Using real time PCR
method we found that genes that are under the control of SREBP-1 (ACC, SOD and
ACL) and
genes under control of SREBP-2 MVD, LDLR and INSIG-1) were significantly
downregulated by 40-
80% (Fig. 40D). These results suggest that compound 19 not only reduces total
body fat but also
improve fatty liver conditions and is a promising drug against human fatty
liver disease.
65
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
Upregulation of Uncoupling Protein 2 (UCP2) in Livers of rats treated with
Compound 19
It is well documented that UCPs are involved in the upregulation of
thermogenesis. They
dissipate metabolic energy as heat by lowering the mitochondria membrane
potential. The liver is a
very active metabolic organ, especially in lipid synthesis. The present
invention has shown that food
consumption was not significantly different between control and rats treated
with compound 19.
Despite feeding with lipogenic diet the treated animals accumulated
significantly lower body fat
including the livers. The livers were examined as an example where energy
might have been
uncoupled and determined the levels of UCP2, the major UCP in liver. As shown
in Figure 41, using
real-time PCR method the level of UCP2 was significantly higher and increased
about 1.5 and 3 fold
in the 2.5 and 10 mg/kg treated groups respectively. These results strongly
suggest that the
decrease in lipid contents in liver is the result of decreased synthesis and
uncoupling, where energy
partially is dissipated by uncoupled thermogenesis.
The following references are cited herein.
Abu-Elheiga etal., (2001) Science 291, 2613-6.
Abu-Elheiga, L. et al., (2003) Proc Nat! Acad Sc! U S A 100, 10207-12.
Abu-Elheiga, L. etal., (1997) J Biol Chem 272, 10669-10677.
Abu-Elheiga, L. etal., (2000) Proc. Natl. Acad. Sci. USA 97, 1444-1449.
Abu-Elheiga, L. et al., (1995) Proc. Natl. Acad. Sci. USA 92, 4011-4015.
Boden G, C. X. etal., (1994) J Clin Invest. 93(6):2438-46 93(6), 2438-2446.
Boizard, M. etal., (1998) J Biol Chem 273, 29164-71.
Brown, M. S. etal., (1997) Ce// 89, 331-40.
Chalkley, S. M. etal., (1998) Metabolism 47(9), 1121.
Choi, Y. et al., (2003) J Biol Chem 278, 7320-4.
Cohen, P. et al., (2002) Science 297(5579), 240-243.
Goldstein, J. L. etal., (1983) Methods Enzymol 98, 241-60.
Goldstein, J. L. etal., (2006) Ce// 124, 35-46.
Grand-Perret, T. et al., (2001) Nat Med 7, 1332-8.
Hastings, N. etal., (2001) Proc. Natl. Acad. Sc!. USA 98(25), 14304-14309.
Hookman etal., (2003) American Journal of Gastroenterology 98(9), 2093-2097.
Horton, J. D. etal., (1998) Proc Nat! Acad Sc! USA 95, 5987-92.
Horton, J. D., etal., (2003) Proc Nat! Acad Sc! USA 100, 12027-32.
Horton, J. D. etal., (2003) J. BioL Chem. 278(38), 36652-36660.
Hue, X. etal. etal., (1996) Ce// 87, 415-26.
Hue, X. etal., (1995) J Biol Chem 270, 29422-29427.
Ingalis, A. M. etal., (1950) J. Hered 41, 317-318.
Kim, J. B. etal., (1996) Genes Dev10, 1096-107.
Ktorza A, B. C. etal., (1997) Diabetes Metab. Suppl 2, 38-46.
Kubota N, T. Y., etal. (1999) MoL Cell 4, 597-609.
Liang, G., et al., (2002) J Biol Chem 277, 9520-8.
66
CA 02922703 2016-02-26
WO 2015/031710
PCT/US2014/053334
Mao, J. etal., (2006) Proc Natl Acad Sci USA 103, 8552-7.
Marquardt, A., etal., (2000) Genomics 66(2), 175.
Matsusue, K., etal., (2003) J. Clin. Invest. 111(5), 737-747.
Miyazaki, M., etal., (2000) J. Biol. Chem. 275(39), 30132-30138.
Moller, D. E. etal., (2005) Annual Review of Medicine 56(1), 45-62.
Nakamura, M. T. etal., (2004) Annual Review of Nutrition 24(1), 345-376.
Ntambi, J. M., etal., (2002) Proc Natl Acad Sci USA 99(17), 11482-11486.
Oh, W., etal., (2005) Proc Natl Acad Sci U S A 102,1384-9.
Osborne, T. F. (2000) J Biol Chem 275, 32379-82.
Rader, D. J. (2001) Nat Med 7, 1282-4.
Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990.
Sakai, J. etal., (1996) Cell 85, 1037-46.
Sakai, J. etal. MMol Cell 2, 505-14 (1998).
Sato, S. etal. J Am Chem Soc 129, 873-80 (2007).
Sheng, et al., (1995) Proc Natl Acad Sci U S A 92, 935-938.
Shimano, H. (2000) Trends Cardiovasc Med 10, 275-8.
Shimano, H., etal., (1999) J Biol Chem 274, 35832-9.
Tontonoz, P., etal., (1993) Mo/ Cell Biol 13, 4753-9.
Yahagi, N., etal., (2002) J. Biol. Chem. 277(22), 19353-19357.
Zambrowicz, B. P et al., (2003) Nat Rev Drug Discov. 2(1), 38-51.
67