Note: Descriptions are shown in the official language in which they were submitted.
CA 02926693 2016-04-06
WO 2015/078831 PCT/EP2014/075460
METHODS FOR THE ENRICHMENT OF MUTATED NUCLEIC ACID
FROM A MIXTURE
FIELD OF THE INVENTION
The invention pertains to the fields of nucleic acid chemistry and nucleic
acid
amplification. In particular, the invention pertains to the enrichment of low
abundance
mutant target nucleic acids using compounds and methods that can detect base
pair
mismatches in nucleic acids.
BACKGROUND OF THE INVENTION
Most human inherited diseases and cancers are known to be caused by mutations
in
nuclear genes. In general, a mutation is considered to be particular
polymorphic
variants at a genetic locus. The mutation can be a single nucleotide
difference, often
referred to as a point mutation. At the cellular and tissue level,
polymorphisms at a
specific genetic locus may give rise to significantly altered cellular
behavior. However,
because even relatively small cell or tissue samples can contain millions or
billions of
DNA molecules containing the particular genetic locus, a representation of the
range
and frequencies of polymorphic variants at a genetic locus, requires detecting
alleles that
are potentially present at a very low frequency. In most cases, the detection
of the
presence of rare mutations from a biological sample presents tremendous
challenges
due to the simultaneous presence of a vast excess of wild-type DNA.
Thus there exists a need in the art for a method to selectively and accurately
enrich low-
copy mutant DNA such that their presence can be detectable following the
performance
of amplification reactions such as PCR.
CA 02926693 2016-04-06
WO 2015/078831 PCT/EP2014/075460
2
SUMMARY OF THE INVENTION
The present invention is directed to methods for enriching low abundance
alleles (e.g.
mutant DNA) in a sample that allows subsequent detection of such alleles. In a
first
aspect, the invention relates to a method of enriching a variant of a target
nucleic acid in
a mixture of nucleic acids from a sample, the target nucleic acid existing in
the form of
two variant sequences, wherein said variants differ at a single nucleotide
position, the
method comprising, providing the sample that includes the target nucleic acid
wherein
the variant to be enriched is present in the sample in low abundance amongst a
large
excess of the other variant; providing an oligonucleotide that is
complementary to one
strand of the target nucleic acid at a concentration that is in molar excess
to the target
nucleic acid, wherein the oligonucleotide is attached with an affinity label
and is
perfectly matched at the single nucleotide position with the variant to be
enriched and
has a mismatch at the single nucleotide position with the other variant;
providing
conditions suitable for hybridization of the oligonucleotide to the target
nucleic acid to
generate duplex polynucleotides consisting of the oligonucleotide and one
strand of
either variant of the target nucleic acid; contacting the duplex
polynucleotides with a
mismatch intercalating compound that preferentially binds to only the duplex
polynucleotides that contain a mismatch wherein said compound is further
capable of
catalyzing cleavage of one strand of the duplex polynucleotide at the mismatch
site with
light; subjecting the duplex polynucleotides to light resulting in both
cleaved and
uncleaved duplex polynucleotides; applying both cleaved and uncleaved duplex
polynucleotides to an affinity matrix that recognizes and binds to the
affinity label on
the oligonucleotide; providing conditions whereby only the cleaved duplex
polynucleotide is denatured and removing the denatured single strand from the
affinity
matrix; and providing a buffer under conditions to denature the uncleaved
polynucleotide duplex; and collecting the buffer which contains one strand of
the
enriched variant of the target nucleic acid. In one embodiment, the mismatch
CA 02926693 2016-04-06
WO 2015/078831 PCT/EP2014/075460
3
intercalating compound is Rh(bpy)2(chrysi)3 ' or Rh(bpy)2(phzi)3'' or their
respective
analogs. In another embodiment, the invention relates to a further step of
amplifying
and detecting the enriched variant of the target nucleic acid.
In a second aspect, the invention relates to a method for detecting a mutant
allele of a
-- target nucleic acid in a mixture of nucleic acids from a sample wherein the
mutant allele
differs from a wild-type allele at a single nucleotide position and is present
in the sample
in low abundance amongst a large excess of the wild-type allele, the method
comprising
enriching the mutant allele in the sample wherein the enrichment is performed
by
providing an oligonucleotide that is complementary to one strand of the target
nucleic
-- acid at a concentration that is in molar excess to the target nucleic acid,
wherein the
oligonucleotide is attached with an affinity label and is perfectly matched at
the single
nucleotide position with the mutant allele and has a mismatch at the single
nucleotide
position with the wild-type allele; providing conditions suitable for
hybridization of the
oligonucleotide to the target nucleic acid to generate duplex polynucleotides
consisting
-- of the oligonucleotide and one strand of either the mutant allele or the
wild-type allele;
contacting the duplex polynucleotides with a mismatch intercalating compound
that
preferentially binds to only the duplex polynucleotides that contain a
mismatch wherein
said compound is further capable of catalyzing cleavage of one strand of the
duplex
polynucleotide at the mismatch site with light; subjecting the duplex
polynucleotides to
-- light resulting in both cleaved and uncleaved duplex polynucleotides;
applying both
cleaved and uncleaved duplex polynucleotides to an affinity matrix that
recognizes and
binds to the affinity label on the oligonucleotide; providing conditions
whereby only the
cleaved duplex polynucleotide is denatured and removing the denatured single
strand of
the wild-type allele from the affinity matrix; providing a buffer under
conditions to
-- denature the uncleaved polynucleotide duplex; and collecting the buffer
which contains
one strand of the enriched mutant allele of the target nucleic acid;
amplifying the
enriched mutant allele; and detecting the product of the enriched amplified
mutant
CA 02926693 2016-04-06
WO 2015/078831 PCT/EP2014/075460
4
allele or the signal generated from the enriched amplified mutant allele. In
one
embodiment, the mismatch intercalating compound is Rh(bpy)2(chrysi)3F or
Rh(bpy)2(phzi)3F or their respective analogs.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 shows the structures of the rhodium-based intercalators,
Rh(bpy)2(chrysi)3F
(left) and Rh(bpy)2(phzi)3F (right), where N represents nitrogen, Rh
represents rhodium,
and R1, R2 and R3 are independently selected from the group consisting of
hydrogen,
alkyl, aryl, a solid support, and a linker attached with an affinity label.
Figure 2 shows a graphical representation of the method of the present
invention for the
enrichment of a single strand of the mutant allele.
DETAILED DESCRIPTION OF THE INVENTION
Definitions
Unless defined otherwise, all technical and scientific terms used herein have
the
meaning commonly understood by a person skilled in the art to which this
invention
belongs. The following references provide one of skill with a general
definition of many
of the terms used in this invention: Singleton et al., Dictionary of
Microbiology and
Molecular Biology (2nd ed. 1994); The Cambridge Dictionary of Science and
Technology
(Walker ed., 1988); The Glossary of Genetics, 5th Ed., R. Rieger et al.
(eds.), Springer
Verlag (1991); and Hale & Marham, The Harper Collins Dictionary of Biology
(1991). As
used herein, the following terms have the meanings ascribed to them unless
specified
otherwise.
CA 02926693 2016-04-06
WO 2015/078831 PCT/EP2014/075460
The term "nucleic acid" refers to polymers of nucleotides (e.g.,
ribonucleotides,
deoxyribonucleotides, nucleotide analogs etc.) and comprising deoxyribonucleic
acids
(DNA), ribonucleic acids (RNA), DNA-RNA hybrids, oligonucleotides,
polynucleotides,
aptamers, peptide nucleic acids (PNAs), PNA-DNA conjugates, PNA-RNA
conjugates,
5 etc., that comprise nucleotides covalently linked together, either in a
linear or branched
fashion. A nucleic acid is typically single-stranded or double-stranded and
will generally
contain phosphodiester bonds, although in some cases, nucleic acid analogs are
included that may have alternate backbones, including, for example,
phosphoramide
(Beaucage et al. (1993), Tetrahedron 49(10):1925); phosphorothioate (Mag et
al. (1991),
Nucleic Acids Res. 19:1437; and U.S. Pat. No. 5,644,048), phosphorodithioate
(Briu et al.
(1989), J. Am. Chem. Soc. 111:2321), 0-methylphophoroamidite linkages (see
Eckstein,
Oligonucleotides and Analogues: A Practical Approach, Oxford University Press
(1992)), and peptide nucleic acid backbones and linkages (see, Egholm (1992),
J. Am.
Chem. Soc. 114:1895). Other analog nucleic acids include those with positively
charged
backbones (Denpcy et al. (1995), Proc. Natl. Acad. Sci. USA 92: 6097); non-
ionic
backbones (U.S. Pat. Nos. 5,386,023, 5,637,684, 5,602,240, 5,216,141 and
4,469,863) and
non-ribose backbones, including those described in U.S. Pat. Nos. 5,235,033
and
5,034,506. Nucleic acids containing one or more carbocyclic sugars are also
included
within the definition of nucleic acids (see Jenkins et al. (1995), Chem. Soc.
Rev. pp. 169-
176), and analogs are also described in, e.g., Rawls, C & E News Jun. 2, 1997,
page 35.
These modifications of the ribose-phosphate backbone may be done to facilitate
the
addition of additional moieties such as labels, or to alter the stability and
half-life of such
molecules in physiological environments.
In addition to the naturally occurring heterocyclic bases that are typically
found in
nucleic acids (e.g., adenine, guanine, thymine, cytosine, and uracil),
nucleotide analogs
also may include non-naturally occurring heterocyclic bases, such as those
described in,
e.g., Seela et al. (1999), Hely. Chim. Acta 82:1640. Certain bases used in
nucleotide
CA 02926693 2016-04-06
WO 2015/078831 PCT/EP2014/075460
6
analogs act as melting temperature (Tm) modifiers. For example, some of these
include
7-deazapurines (e.g., 7-deazaguanine, 7-deazaadenine, etc.), pyrazolo[3,4-
d] pyrimidines, propynyl-dN (e.g., propynyl-dU, propynyl-dC, etc.), and the
like. See,
e.g., U.S. Pat. No. 5,990,303. Other representative heterocyclic bases
include, e.g.,
hypoxanthine, inosine, xanthine; 8-aza derivatives of 2-aminopurine, 2,6-
diaminopurine, 2-amino-6-chloropurine, hypoxanthine, inosine and xanthine; 7-
deaza-
8-aza derivatives of adenine, guanine, 2-aminopurine, 2,6-diaminopurine, 2-
amino-6-
chloropurine, hypoxanthine, inosine and xanthine; 6-azacytidine; 5-
fluorocytidine; 5-
chloro cytidine; 5 - iodocytidine; 5 -b romocytidine; 5 -methylcytidine; 5 -
propynylcytidine;
5 -b romovinyluracil; 5 -fluorouracil; 5 - chlorouracil; 5 - io douracil; 5 -b
romouracil; 5 -
trifluoromethyluracil; 5-methoxymethyluracil; 5-ethynyluracil; 5-
propynyluracil, and
the like.
A "nucleoside" refers to a nucleic acid component that comprises a base or
basic group
(comprising at least one homocyclic ring, at least one heterocyclic ring, at
least one aryl
group, and/or the like) covalently linked to a sugar moiety (a ribose sugar or
a
deoxyribose sugar), a derivative of a sugar moiety, or a functional equivalent
of a sugar
moiety (e.g. a carbocyclic ring). For example, when a nucleoside includes a
sugar
moiety, the base is typically linked to a l'-position of that sugar moiety. As
described
above, a base can be a naturally occurring base or a non-naturally occurring
base.
Exemplary nucleosides include ribonucleosides, deoxyribonucleosides,
dideoxyribonucleosides and carbocyclic nucleosides.
A "nucleotide" refers to an ester of a nucleoside, e.g., a phosphate ester of
a nucleoside,
having one, two, three or more phosphate groups covalently linked to a 5'
position of a
sugar moiety of the nucleoside.
The terms "polynucleotide" and "oligonucleotide" are used interchangeably.
"Oligonucleotide" is a term sometimes used to describe a shorter
polynucleotide. An
CA 02926693 2016-04-06
WO 2015/078831 PCT/EP2014/075460
7
oligonucleotide may be comprised of at least 6 nucleotides, for example at
least about
10-12 nucleotides, or at least about 15-30 nucleotides corresponding to a
region of the
designated nucleotide sequence.
The term "enriching a variant of a target nucleic acid sequence" refers to
increasing the
amount of the desired variant of the target nucleic acid sequence and
increasing the
ratio of the desired variant relative to the undesired variant in a sample.
Generally, the
desired variant to be enriched is less prevalent in a nucleic acid sample than
the
undesired variant, and makes up less than 50% of the total amount of all the
variants of
the target nucleic acid sequence. In many cases, the desired variant refers to
a mutant
allele and the undesired variant refers to a wild-type allele.
The term "wild-type" as used herein refers to a gene or allele which has the
characteristics of that gene or allele when isolated from a naturally
occurring source. A
wild-type gene or a wild-type allele is that which is most frequently observed
in a
population and is arbitrarily designated as the "normal" or "wild-type" form
of the gene
or allele.
In contrast, the term "mutant" or "mutated" refers to a gene or allele which
displays
modifications in sequence when compared to the wild-type gene or allele. The
term
"mutation" refers to a change in the sequence of nucleotides of a normally
conserved
nucleic acid sequence resulting in the formation of a mutant as differentiated
from the
normal (unaltered) or wild type sequence. Mutations can generally be divided
into two
general classes, namely, base-pair substitutions (e.g. single nucleotide
substitutions) and
frame-shift mutations. The latter entail the insertion or deletion of one to
several
nucleotide pairs.
The term "allele" refers to two sequences which are different by only one or a
few bases.
CA 02926693 2016-04-06
WO 2015/078831 PCT/EP2014/075460
8
The term "mismatch" DNA or "heteroduplex" DNA refers to DNA which includes one
or more mismatch base pairings. A mismatch base pairing refers to a specific
pair of
opposing bases, in the context of a DNA duplex, which cannot form one of the
hydrogen-bonded base pairs, T with A or G with C. Heteroduplex DNA includes
double-stranded DNA in which one or more bases in one strand does or do not
complement the base or bases in the opposing strand, as well as double-
stranded DNA
in which one or more bases of either strand does or do not have an opposing
base, due
to an insertion or deletion in one strand as compared to the opposing strand.
In
contrast, homoduplex DNA refers to double-stranded DNA in which each strand is
a
complete complement of the other strand, and each base forms a hydrogen-bonded
base
pair with an opposing base.
The terms "molecular binding partners" and "specific binding partners" refer
to pairs of
molecules, typically pairs of biomolecules, that exhibit specific binding. Non-
limiting
examples are receptor and ligand, antibody and antigen, biotin and avidin, and
biotin
and streptavidin. Molecular binding partners can also be represented by
binding that
occurs between an "affinity label" and an "affinity matrix" as defined below.
An "affinity" label is a molecule that can specifically bind to its molecular
binding
partner. The binding can be through covalent or non-covalent (e.g., ionic,
hydrogen,
etc.) bonds. As used herein, an affinity label, such as biotin, can
selectively bind to an
affinity matrix, such as streptavidin-coated beads or particles. An affinity
label can be
attached to an oligonucleotide on its 3' terminus, 5' terminus or on an
internal position
of the oligonucleotide.
An "affinity matrix" as used herein refers to a molecule that is attached to
the surface of
a solid support or solid matrix (e.g. magnetic latex particles, glass beads)
that can
specifically bind to its molecular binding partner. The binding can be through
covalent
CA 02926693 2016-04-06
WO 2015/078831 PCT/EP2014/075460
9
or non-covalent bonds. As used herein, an affinity matrix, such as
streptavidin-coated
magnetic latex particles can selectively bind to an affinity label, such as
biotin.
An "alkyl group" refers to a linear, branched, or cyclic saturated hydrocarbon
moiety
and includes all positional isomers, e.g., methyl, ethyl, propyl, butyl, 1-
methylpropyl, 2-
-- methylpropyl, 1,1-dimethylethyl, pentyl, 1-methylbutyl, 2-methylbutyl, 3-
methylbutyl,
2,2-dimethylpropyl, 1-ethylpropyl, hexyl, 1,1-dimethylpropyl, 1,2-
dimethylpropyl, 1-
methylpentyl, 2-methylpentyl, 3-methylpentyl, 4-methylpentyl, 1,1-
dimethylbutyl, 1,2-
dimethylbutyl, 1,3-dimethylbutyl, 2,2-dimethylbutyl, 2,3-dimethylbutyl, 3,3-
dimethylbutyl, 1-ethylbutyl, 2-ethylbutyl, 1,1,2-trimethylpropyl, 1,2,2-
trimethylpropyl,
-- 1-ethyl- 1-methylpropyl and 1-ethyl-2-methylpropyl, n-hexyl, cyclohexyl, n-
heptyl, n-
octyl, 2-ethylhexyl, n-nonyl, n-decyl and the like. An alkyl group typically
comprises
about 1-20 carbon atoms and more typically comprises about 2-15 carbon atoms.
Alkyl
groups can be substituted or unsubstituted.
An "aryl group" refers to a substituent group of atoms or moiety that is
derived from an
-- aromatic compound. Exemplary aryl groups include, e.g., phenyl groups, or
the like.
Aryl groups optionally include multiple aromatic rings (e.g., diphenyl groups,
etc.). In
addition, an aryl group can be substituted or unsubstituted.
"PCR amplification" or simply "PCR" refers to the polymerase chain reaction
that
involves the use of a nucleic acid sequence as a template for producing a
large number of
-- complements to that sequence. The template may be hybridized to a primer
having a
sequence complementary to a portion of the template sequence and contacted
with a
suitable reaction mixture including dNTPs and a polymerase enzyme. The primer
is
elongated by the polymerase enzyme producing a nucleic acid complementary to
the
original template. For the amplification of both strands of a double stranded
nucleic
-- acid molecule, two primers are used, each of which may have a sequence
which is
complementary to a portion of one of the nucleic acid strands. The strands of
the
CA 02926693 2016-04-06
WO 2015/078831 PCT/EP2014/075460
nucleic acid molecules are denatured, for example by heating, and the process
is
repeated, this time with the newly synthesized strands of the preceding step
serving as
templates in the subsequent steps. A PCR amplification protocol may involve a
few to
many cycles of denaturation, hybridization and elongation reactions to produce
5 sufficient amounts of the target nucleic acid.
The term "allele-specific primer" or "AS primer" refers to a primer that
hybridizes to
more than one variant of the target sequence, but is capable of discriminating
between
the variants of the target sequence in that only with one of the variants, the
primer is
efficiently extended by the nucleic acid polymerase under suitable conditions.
With
10 other variants of the target sequence, the extension is less efficient,
inefficient or
undetectable.
The term "common primer" refers to the second primer in the pair of primers
that
includes an allele-specific primer. The common primer is not allele-specific,
i.e. does not
discriminate between the variants of the target sequence between which the
allele-
specific primer discriminates.
The terms "complementary" or "complementarity" are used in reference to
antiparallel
strands of polynucleotides related by the Watson-Crick base-pairing rules. The
terms
"perfectly complementary" or "100% complementary" refer to complementary
sequences that have Watson-Crick pairing of all the bases between the
antiparallel
strands, i.e. there are no mismatches between any two bases in the
polynucleotide
duplex. However, duplexes are formed between antiparallel strands even in the
absence
of perfect complementarity. The terms "partially complementary" or
"incompletely
complementary" refer to any alignment of bases between antiparallel
polynucleotide
strands that is less than 100% perfect (e.g., there exists at least one
mismatch or
unmatched base in the polynucleotide duplex). The duplexes between partially
CA 02926693 2016-04-06
WO 2015/078831 PCT/EP2014/075460
11
complementary strands are generally less stable than the duplexes between
perfectly
complementary strands.
The term "sample" refers to any composition containing or presumed to contain
nucleic
acid. This includes a sample of tissue or fluid isolated from an individual
for example,
skin, plasma, serum, spinal fluid, lymph fluid, synovial fluid, urine, tears,
blood cells,
organs and tumors, and also to samples of in vitro cultures established from
cells taken
from an individual, including the formalin-fixed paraffin embedded tissues
(FFPET)
and nucleic acids isolated therefrom.
The term "primary sequence" refers to the sequence of nucleotides in a
polynucleotide
or oligonucleotide. Nucleotide modifications such as nitrogenous base
modifications,
sugar modifications or other backbone modifications are not a part of the
primary
sequence. Labels, such as chromophores conjugated to the oligonucleotides are
also not
a part of the primary sequence. Thus two oligonucleotides can share the same
primary
sequence but differ with respect to the modifications and labels.
The term "primer" refers to an oligonucleotide which hybridizes with a
sequence in the
target nucleic acid and is capable of acting as a point of initiation of
synthesis along a
complementary strand of nucleic acid under conditions suitable for such
synthesis. As
used herein, the term "probe" refers to an oligonucleotide which hybridizes
with a
sequence in the target nucleic acid and is usually detectably labeled. The
probe can have
modifications, such as a 3'-terminus modification that makes the probe non-
extendable
by nucleic acid polymerases, and one or more chromophores. An oligonucleotide
with
the same sequence may serve as a primer in one assay and a probe in a
different assay.
As used herein, the term "target sequence", "target nucleic acid" or "target"
refers to a
portion of the nucleic acid sequence which is to be either amplified, detected
or both.
CA 02926693 2016-04-06
WO 2015/078831 PCT/EP2014/075460
12
The terms "hybridized" and "hybridization" refer to the base-pairing
interaction of
between two nucleic acids which results in formation of a duplex. It is not a
requirement
that two nucleic acids have 100% complementarity over their full length to
achieve
hybridization.
The terms "selective hybridization" and "specific hybridization" refer to the
hybridization of a nucleic acid predominantly (50% or more of the hybridizing
molecule) or nearly exclusively (90% or more of the hybridizing molecule) to a
particular nucleic acid present in a complex mixture where other nucleic acids
are also
present. For example, under typical PCR conditions, primers specifically
hybridize to
the target nucleic acids to the exclusion of non-target nucleic acids also
present in the
solution. The specifically hybridized primers drive amplification of the
target nucleic
acid to produce an amplification product of the target nucleic acid that is at
least the
most predominant amplification product and is preferably the nearly exclusive
(e.g.,
representing 90% or more of all amplification products in the sample)
amplification
product. Preferably, the non-specific amplification product is present in such
small
amounts that it is either non-detectable or is detected in such small amounts
as to be
easily distinguishable from the specific amplification product. Similarly,
probes
specifically hybridize to the target nucleic acids to the exclusion of non-
target nucleic
acids also present in the reaction mixture. The specifically hybridized probes
allow
specific detection of the target nucleic acid to generate a detectable signal
that is at least
the most predominant signal and is preferably the nearly exclusive (e.g.,
representing
90% or more of all amplification products in the sample) signal.
There is a continuing need for developing new methods that can detect rare
somatic
mutations associated with various types of cancer with increased accuracy and
sensitivity. A particular need for more sensitive detection methods exists in
the field of
cancer biomarker detection from peripheral fluids such as blood, sputum, and
urine.
During the past several years, many studies have been published, that have
clearly
CA 02926693 2016-04-06
WO 2015/078831 PCT/EP2014/075460
13
established the value of blood-based cancer biomarker detection for therapy
prediction,
therapeutic monitoring of drug resistance development, and tumor dynamics and
cancer recurrence by monitoring defined mutations in blood. Furthermore, a
highly
sensitive method for mutation detection from peripheral biological fluids may
someday
deliver on the promise of a "liquid biopsy" approach for cancer screening and
early stage
cancer detection.
Many methods have been developed over the years to increase the sensitivity of
rare
mutation detection. Most of the methods have generally focused on capitalizing
on the
sequence based differences between mutant and wild-type, by employing primer-
or
probe-based discrimination during PCR, commonly referred as allele-specific
PCR (AS-
PCR). These methods are successful down to a level of 0.1-1% mutant levels,
but then
further improvements are limited by enzyme based limitations or PCR errors.
Digital
PCR, pioneered by Bert Vogelstein, to date has been the most promising
technique to
successfully enhance the sensitivity of rare allele detection. This is
accomplished by
dividing the sample into thousands of smaller amplification reactions. This
method, in
effect, dilutes out the wild-type DNA, and enriches the ratio of mutant to
wild-type. An
alternative approach proposed presently is to use an upfront sample
preparation
method that enriches the mutant DNA, and thereby reduces the difficulty of
detection
in the downstream assay.
PCR, in its many different modalities, is a powerful technique that can
readily detect
literally a single copy of a specific sequence in the presence of a large
amount of
background DNA, provided that the nature of the desired sequence is
sufficiently
different from the background DNA. This is the case for example, when trying
to detect
the presence of an exogenous pathogenic sequence from a biological specimen
also
containing an excess of human genomic DNA. However, the problem becomes
increasingly challenging when the sequence of interest becomes increasingly
similar to
the sequences present in the background DNA, as is the case in the detection
of rare
CA 02926693 2016-04-06
WO 2015/078831 PCT/EP2014/075460
14
somatic mutations. Generally, the mutation status must be determined in a
sample that
also contains a large excess of the wild type sequence. This is challenging
because
although the currently available methods for mutation detection are selective
for the
mutant sequence, they are not absolute in specificity, and as the ratio of
wild-type to
mutant increases, it becomes increasingly difficult to distinguish mutant from
wild-type
DNA.
The methods described in the present invention are based on the use of bulky
rhodium
(III) complexes, as disclosed in U.S. Patent No. 6,031,098, U.S. Patent No.
6,306,601,
Nature Protocols 2: 357-371, 2007, where Barton et al. describe the synthesis
and
function of two families of mismatch-specific rhodium-based intercalators
based on a
pair of bulky intercalating ligands, 5,6-chrysenequinone diimine (chrysi) and
3,4-
benzojal phenazine quinone diimine (phzi) to generate, respectively,
Rh(bpy)2(chrysi)3F
or Rh(bpy)2(phzi)3F. These compounds are known for their ability to insert
themselves
selectively into the bulge created by a nucleotide mismatch within a DNA
duplex. The
mechanism of binding has been evaluated by multiple NMR and crystallography
based
investigations, and the results have been remarkable. Unlike the classical
intercalative
binding mode where the binder enters the DNA duplex from the major groove, and
intercalates between base pairs, these novel compounds enter the minor groove
of the
DNA, insert the bulky aromatic ligand, and eject the mismatched bases into the
major
groove. Upon photoactivation, the complex promotes direct strand scission at
single-
base mismatch sites within the duplex. Site-specific cleavage is evident at
nanomolar
concentrations. The main focus so far on the use of these rhodium compounds
has been
in the detection of single-nucleotide
polymorphisms (SNPs), and novel chemotherapeutics (Boon, EM et al., Methods in
Enzymology, 353:506-522, 2002, U.S. Patent No. 6,444,661, U.S. Patent No.
6,777,405).
CA 02926693 2016-04-06
WO 2015/078831 PCT/EP2014/075460
In the present invention, these rhodium compounds have been applied for the
purpose
of the enrichment of rare alleles (e.g. rare mutant alleles). The structures
of the
compounds chosen for this study are shown in Figure 1, where R1, R2, R3 can be
H,
alkyl, aryl, or a solid phase, or linker with an affinity label (e.g. biotin).
Although it has
5 previously been shown that the described compounds can bind to mismatched
DNA
duplexes and catalyze photocleavage, it has also been shown that the
photocleavage only
results in the cleavage of only one of the two strands. This is of little
utility in the
enrichment application of mutant DNA because an uncleaved wild-type strand
would
still be present and can function as a template for amplification.
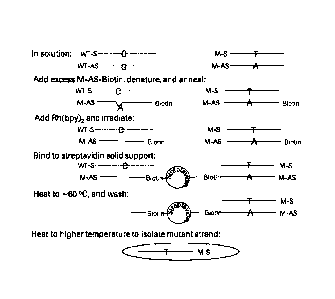
iu The overall strategy of the present invention is graphically represented
on Figure 2 and
takes advantage of the ability of these rhodium complex compounds to bind to
base
mismatched region of interest, and cause the cleavage of a specific
phosphodiester
linkage upon photoactivation. According to the methods described here, a
sample is
provided in which the target nucleic acid contains both the wild-type allele
(shown in
15 Figure 2 as having a "C" nucleotide on the sense strand, "WT-S" and "G"
nucleotide on
the antisense strand "WT-AS" and the mutant allele (shown as having a "T"
nucleotide
on the sense strand "M-S" and a "A" nucleotide on the antisense strand, "M-
AS").
Next, a single-stranded oligonucleotide (represented in Figure 2 as M-AS-
Biotin)
corresponding to one strand of the mutant allele desired to be enriched and
comprising
an affinity ligand (e.g. biotin) is offered in excess relative to amount of
the target nucleic
acid in the sample. (Although Figure 2 shows the oligonucleotide corresponding
to the
antisense strand with the biotin label on its 3' terminus, the method could
also be
practiced with an oligonucleotide having the sequence of the sense strand of
the mutant
allele with the biotin label attached at any position other than mutation
site.) Then the
mixture is first heated in order to denature all double-stranded sample DNA
and then
cooled to allow annealing of complementary single strands. Because the M-AS-
Biotin
oligonucleotide is present in excess, almost all of the sense strand of the
target nucleic
CA 02926693 2016-04-06
WO 2015/078831 PCT/EP2014/075460
16
acid (both the wild-type allele, WT-S and the mutant allele, M-S)will be
hybridized to
the oligonucleotide. While the mutant sense strand duplexes will be perfectly
matched,
the wild-type sense strand will have a single base mismatch at the position of
mutation.
The rhodium complex compound (represented in Figure 2 as "Rh(bpy)2") is then
added
to the mixture. Upon photoactivation, the mismatch-containing duplex will be
cleaved,
either on the M-AS-Biotin oligonucleotide (as shown in Figure 2) or on the
wild-type
sense strand, WT-S, while the duplexes with the M-AS-Biotin oligonucleotide
bound to
the mutant sense strand, M-S, will not be cleaved due to a perfect match at
the mutation
position. Using the affinity portion (shown as Biotin in Figure 2) of the
oligonucleotide,
all the oligonucleotide bound sequences (i.e. wild-type and mutant sense
strands) are
captured on a solid phase (shown in Figure 2 as a streptavidin-coated solid
support),
and all the excess wild-type and mutant antisense strands are washed away. In
the next
step, the solid phase is placed in the appropriate buffer and the temperature
is raised
until only the captured wild-type sense strand is released due to the lower
melting
temperature of the cleavage-containing duplex. This is then washed away,
leaving only
the mutant sense strand on the support. Finally the mutant sense strand is
recovered in
buffer by either a temperature or alkaline pH elution step.
While Figure 2 shows the use of an antisense strand oligonucleotide for the
enrichment
of the mutant sense strand, similarly a sense strand oligonucleotide can be
used for the
enrichment of the mutant antisense strand. Generally, the choice for which
strand to use
for the oligonucleotide depends on the binding affinity of the rhodium complex
compound to the mismatch position, with the most thermodynamically
destabilized
mismatch sites having the highest binding affinities (for further details, see
Jackson, B.A.
and Barton, J.K., Biochemistry 39: 6176-6182, 2000).
The following examples and figures are provided to aid the understanding of
the present
invention, the true scope of which is set forth in the appended claims. It is
understood
CA 02926693 2016-04-06
WO 2015/078831 PCT/EP2014/075460
17
that modifications can be made in the procedures set forth without departing
from the
spirit of the invention.
EXAMPLES
Example 1 Control Experiment using Single Stranded Oligonudeotides
The following experiment is used to enrich a sense-strand mutant (MU-S)
oligonucleotide in the presence of a sense-strand wild-type (WT-S)
oligonucleotide by
using an affinity-labeled anti-sense strand (AL-AS) oligonucleotide that is
perfectly
matched to the mutant oligonucleotide and has a one-base mismatch with the
wild-type
oligonucleotide. The sequences of the oligonucleotides are as follows
(mismatch site
bolded):
WT-S: 5' -CGTGCAGCTCATCACGCAGCTCATGCCCTT-3' (SEQ ID NO: 1)
MU-S: 5' -CGTGCAGCTCATCATGCAGCTCATGCCCTT-3' (SEQ ID NO: 2)
AL-AS: 5' -AAGGGCATGAGCTGCATGATGAGCTGCACG-Biotin-3' (SEQ ID NO: 3)
A reaction mixture is prepared with 10 1 150 mM Glycine, pH 9.5, 2 1 5M NaC1
and
55 1 water. To this solution is added 141 of 1004 WT-S, 141 of 1004 MU-S, and
10 1
of 5004 AL-AS and 3 1 of 100 M Rh(bpy)2(phzi)3F . The solution is vortexed and
incubated at room temperature for 5 minutes. (Final concentrations: 15 0/1
Glycine pH
9.5, 100 0/1 NaC1, 1 0/1 WT-S, 1 0/1 MU-S, 5 0/1 AL-AS, and 3 0/1
Rh(bpy)2phzi3F).
The reaction mixture is then irradiated in a Stratagene UV Stratalinker 1800
using 365
nm bulbs for 30 minutes to cleave one strand of the sense-antisense duplex
that contains
the mismatch.
A separate solution of 25 L of 10 mg/mL Solulink streptavidin magnetic beads
is
washed with 1 mL of 15 0/1 Glycine pH 9.5 buffer and the beads are separated
from the
CA 02926693 2016-04-06
WO 2015/078831 PCT/EP2014/075460
18
supernatant using a magnet. The reaction mixture with the cleaved and
uncleaved
oligonucleotide duplexes is added to the magnetic bead pellet and mixed. The
resulting
mixture is incubated at room temperature for 30 minutes. Afterwards, the
solution is
heated to 60 C or to the determined melting temperature of the uncleaved WT-S
strand
bound to the cleaved AL-AS strand (or of the cleaved WT-S strand bound to the
uncleaved AL-AS strand), magnetically separated, and the supernatant is
removed, such
that only the uncleaved MU-S strand is still bound to the uncleaved AL-AS
strand on
the magnetic bead. The MU-S strand is removed by treating the magnetic beads
with
100 L of 20 M NaOH, magnetically separating the solution, and decanting the
supernatant into a test tube for further analysis.
Example 2 Control Experiment using Double Stranded Oligonudeotides
The following experiment is used to enrich a sense-strand mutant (MU-S)
oligonucleotide in the presence of a sense-strand wild-type (WT-S)
oligonucleotide, an
antisense-strand wild-type (WT-AS) oligonucleotide and an antisense-strand
mutant
(MU-AS) oligonucleotide by using an affinity-labeled anti-sense strand (AL-
AS)oligonucleotide that has the identical sequence to the MU-AS
oligonucleotide and
has a one-base mismatch with the WT-AS oligonucleotide. The sequences of the
oligonucleotides are as follows (mismatch site bolded):
WT-S: 5'- CGTGCAGCTCATCACGCAGCTCATGCCCTT-3' (SEQ ID NO: 1)
WT-AS: 5' -AAGGGCATGAGCTGCGTGATGAGCTGCACG-3' (SEQ ID NO: 4)
MU-S: 5' -CGTGCAGCTCATCATGCAGCTCATGCCCTT-3' (SEQ ID NO: 2)
MU-AS: 5' -AAGGGCATGAGCTGCATGATGAGCTGCACG-3' (SEQ ID NO: 5)
AL-AS: 5'- AAGGGCATGAGCTGCATGATGAGCTGCACG-Biotin-3' (SEQ ID NO: 3)
CA 02926693 2016-04-06
WO 2015/078831 PCT/EP2014/075460
19
A reaction mixture is prepared with 10 1 150 mM Glycine, pH 9.5 and 57 1
water. To
this solution is added 10 1 of a mixture of 1004 WT-S and WT-AS, 141 of a
mixture of
1004 MU-S and MU-AS, and 10 1 of 10004 AL-AS. The resulting solution is
incubated at 95 C for 5 minutes to dissociate the double stranded
oligonucleotides to
single strands, and then the solution is cooled to room temperature to allow
the single
stranded oligonucleotides to re-anneal. 3 !IL of 100 IIM Rh(bpy)2phzi3F is
added to the
solution, the solution is vortexed, and incubated at room temperature for 5
minutes.
(Final concentrations: 15 IIM Glycine pH 9.5, 1 IIM WT-S, 1 IIM WT-AS, 1 IIM
MU-S,
1 IIM MU-AS, 10 IIM AL-AS, and 3 IIM Rh(bpy)2phzi3+). The reaction mixture is
then
irradiated in a Stratagene UV Stratalinker 1800 using 365 nm bulbs for 15
minutes to
cleave one strand of the sense-antisense duplexes that contains the mismatch.
The 10-
fold concentration excess of the AL-AS oligonucleotide serves to increase the
probability
that most of the MU-S strand would bind to AL-AS rather than to the MU-AS
strand.
A separate solution of 50 !IL of 10 mg/mL Solulink streptavidin magnetic beads
is
washed with 1 mL of 15 IIM Glycine pH 9.5 buffer and the beads are separated
from the
supernatant by using a magnet. The reaction mixture with cleaved and uncleaved
oligonucleotide duplexes is added to the magnetic bead pellet and mixed. The
resulting
mixture is incubated at room temperature for 30 minutes. Afterwards, the
solution is
heated to 60 C or to the determined melting temperature of the uncleaved WT-S
strand
bound to the cleaved AL-AS strand (or of the cleaved WT-S strand bound to the
uncleaved AL-AS strand), magnetically separated. All WT-AS, MU-AS and WT-S
strands are removed with the supernatant, such that only the uncleaved MU-S
strand is
still bound to the uncleaved AL-AS strand on the magnetic bead. The MU-S
strand is
removed by treating the magnetic beads with 100 !IL of 20 IIM NaOH,
magnetically
separating the solution, and decanting the supernatant into a test tube for
further
analysis.
CA 02926693 2016-04-06
WO 2015/078831 PCT/EP2014/075460
Example 3 Enrichment and Detection of EGFR Mutant DNA
A sample is provided from which a mixture of nucleic acids, for example, human
genomic DNA, can be extracted. The sample can be from a tissue such as skin,
organs,
and tumors or from fluid such as blood, plasma, serum, urine, or from any
composition
5 containing or presumed to contain nucleic acid. From this mixture of
nucleic acids, a
target gene of interest, for example, the human EGFR gene, may contain a
certain
variation such as a point mutation that is present in low abundance amongst a
large
excess of the other variant of the gene, which would be the non-mutant or wild-
type
gene. An example of an EGFR gene mutation that has clinical relevance to the
10 development of cancer is the T790M mutation.
To enrich for the low-abundance T790M mutant allele of the EGFR gene, an
excess of a
biotin labeled antisense strand oligonucleotide (BL-AS) that is complementary
to and
perfectly matched with the sense strand of the T790M mutant allele is added to
a
solution containing the extracted genomic DNA. The solution is then heated at
90 C or
15 higher temperature to denature the double-stranded genomic DNA and then
gradually
cooled to a temperature to allow reannealing of the single DNA strands to
occur. During
the annealing step, the BL-AS strand can form duplexes with both the T790M
mutant
sense strand with which it is perfectly matched and also with the wild-type
sense strand
which will have a mismatch at the position of the point mutation.
20 The rhodium chelator, Rh(bpy)2(phzi)3F , is then added to the solution
and allowed to
incubate such that the chelator can bind only to the BL-AS: wild-type sense
strand
duplexes at the position of the mismatch. The reaction mixture is then
irradiated in a
Stratagene UV Stratalinker 1800 using 365 nm bulbs for 15 minutes to cleave
one strand
of BL-AS: wild-type sense strand duplexes. Next, a solid matrix coated with
streptavidin
is added. Examples of such solid matrices would be streptavidin coated
magnetic
particles such as Streptavidin-coupled Dynabeads from Invitrogen,
Streptavidin
CA 02926693 2016-04-06
WO 2015/078831 PCT/EP2014/075460
21
MagneSphere Paramagnetic Particles from Promega, and NanoLink- and MagnaLink-
Streptavidin Magnetic Beads from Solulink. Following incubation (e.g. 40 C for
1 hour),
a magnet is used to separate the particles and wash away all the nucleic acid
that is not
bound to the particles, which includes both mutant and wild-type antisense
strands and
any excess BL-AS. The wild-type sense strand (which is either cleaved at the
mismatch
site or is bound to a cleaved BL-AS oligonucleotide is then eluted from the
magnetic
particles using an appropriate elution buffer at a temperature that
corresponds to the
melting temperature of the mismatch duplex. As a result, only the T790M sense
strand
remains attached to the magnetic particles by being hybridized to the
uncleaved biotin-
labeled T790M antisense oligonucleotide. By then subjecting the particles to
either high
temperature or to alkaline pH conditions, the T790M sense strand can
dissociate from
the BL-AS oligonucleotide and collected for use in an amplification reaction
for
detection.
While the invention has been described in detail with reference to specific
examples, it
will be apparent to one skilled in the art that various modifications can be
made within
the scope of this invention. Thus the scope of the invention should not be
limited by the
examples described herein, but by the claims presented below.