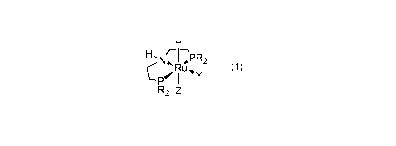Note: Descriptions are shown in the official language in which they were submitted.
CA 02929462 2016-05-03
WO 2015/067899 PCT/FR2014/052839
Procédé de synthèse d'esters et catalyseur de ladite synthèse
Domaine technique
L'invention porte sur un procédé de synthèse d'esters à partir de produits de
départ,
de préférence biosourcés, par couplage déshydrogénant en présence d'un
catalyseur.
Les produits de départ peuvent être des alcools biosourcés tels que des
alcools gras
et/ou des polyols (par exemple le glycérol) issus d'oléagineux, ou encore être
des
alcools produits par fermentation de la biomasse (par exemple l'éthanol et le
butanol).
Art antérieur
De nombreux esters, et notamment l'acétate d'éthyle, sont synthétisés à
l'échelle
industrielle en utilisant des produits de départ d'origine fossile (l'éthylène
dans le cas
de l'acétate d'éthyle) via des procédés multi-étapes. Le marché mondial de
l'acétate
d'éthyle était de 2,5 million de tonnes/an en 2008.
Il est connu d'utiliser un catalyseur à base de ruthénium pour réaliser le
couplage
déshydrogénant de l'éthanol en utilisant par exemple le carbonylchlorohydrido
[bis(2-
diphenylphosphinoethyl)amino]ruthenium(II), de formule A, (CAS : 1295649-40-
9),
Trade Name : Ru-MACHO, ou D (voir ci-dessous) (cf. M. Nielsen, H. Junge, A.
Kammer
and M.Beller, Angew. Chem. Int. Ed., 2012, 51, 5711-5713 et EP2599544A1) ou le
trans-RuCl2(PPh3)[PyCH2NH(CH2)2PPh2], de formule B, (cf. D. Spasyuk and D.
Gusev, Organometallics, 2012, 31, 5239-5242). Ces catalyseurs A et B
requièrent
cependant la présence de tBuOK ou Et0Na, pour être actifs. Le catalyseur D
requière
un long temps de réaction et une charge catalytique élevée pour obtenir des
rendements au maximum de 90% en ester.
H H
Cl CI
N I rrn2
.,,Nõõ I rrn2
Ru_ Ru
CO I 1313h3
Ph2 Cl
A
Un autre catalyseur utilisé pour cette même réaction est le catalyseur
carbonylhydrido(tetrahydroborato)[bis(2-
diphenylphosphinoethyl)amino]ruthenium(1 I),
de formule C (CAS : 1295649-41-0), Trade Name : Ru-MACHO-BH. Cette réaction
est
décrite dans la demande de brevet W02012/144650 où cette synthèse requière la
présence d'un accepteur d'hydrogène telle qu' une cétone, par exemple, la 3-
pentanone. En plus de dissoudre les différentes espèces (catalyseur et
substrat) la 3-
pentanone joue le rôle d'accepteur d'hydrogène. Ainsi une quantité au moins
1
CA 02929462 2016-05-03
WO 2015/067899 PCT/FR2014/052839
stoechiométrique de 3-pentanoneest utilisée dans les exemples et les réactions
décrites ne s'accompagnent donc pas du dégagement d'hydrogène gazeux.
H
H
I /-1-=
Nõõ ,IPPh2
p/
Ph2 HBH3
Il est également connu d'utiliser un catalyseur à base de ruthénium pour
réaliser le
couplage déshydrogénant du butanol en utilisant
par exemple
trans¨RuH2(C0)[HN(C2H4P1Pr2)2], de formule D, (M. Bertoli, A. Choualeb, A. J.
Lough,
B. Moore, D. Spasyuk and D. Gusev, Organometallics, 2011, 30, 3479-3482) ou le
[RuH(PNN)(C0)], de formule E, (cf. J. Zhang, G. Leitus, Y. Ben-David and D.
Milstein,
J. Am. Chem. Soc., 2005, 127,10840-10841) en présence de solvant et en
l'absence
de base et d'accepteur d'hydrogène. Cependant ces catalyseurs nécessitent de
longs
temps de réaction et des charges catalytiques élevées pour obtenir des
rendements au
maximum de 90% en ester.
H H
H
1/-1-=õ
trou2
,rirr2 çe
Rù N-Rù¨CO
,
I CO t.
P NEt2
IPr2 H
D
La mise en place de tels procédés à l'échelle industrielle présente de plus de
nombreux désavantages. Un de ces désavantages est la nécessité d'effectuer
plusieurs étapes de purification pour isoler les produits de la réaction ce
qui complexifie
notablement le procédé. Un autre problème est l'utilisation de produits
organiques, tels
que des solvants, en sus des produits de départs. L'utilisation de tels
produits
augmente sensiblement l'impact environnemental de telles synthèses, ce qui est
bien
sûr à éviter. Il a maintenant été réalisé un procédé qui résout ces problèmes
et qui offre
ainsi une alternative réaliste aux procédés industriels de synthèse d'esters
utilisant des
ressources fossiles. Ce procédé permet en effet d'associer de hauts rendements
à des
étapes de synthèse et de purification simplifiées au niveau de la mise en
oeuvre.
Description de l'invention
L'invention porte sur un procédé de synthèse d'un ester, à partir d'un alcool
qui comprend
l'utilisation d'un catalyseur de formule 1 ci-dessous, en l'absence de
cétones, d'aldéhydes,
d'alcènes, d'alcynes, de soude, d'Et0Na, de Me0Na ou de SuOK.
2
CA 02929462 2016-05-03
WO 2015/067899 PCT/FR2014/052839
Formule 1:
H
Y
P
R2 z
R, identique ou différent, étant un groupement alkyle ou aryle, et plus
particulièrement un groupement phényle, isopropyle ou cyclohexyle et:
- lorsque R est un groupement isopropyle, Z est soit un atome d'hydrogène,
soit un
groupement HBH3, de préférence Z est un groupement HBH3;
- lorsque R est un groupement phényle, Z est, soit un atome d'hydrogène,
soit un
groupement HBH3;
- lorsque R est un groupement cyclohexyle, Z est soit un atome d'hydrogène,
soit un
groupement HBH3;
Y étant un groupement carbonyle CO. Alternativement Y peut-être une phosphine
PR'3, où R' est un groupement alkyle en C1-012 ou aryle en 06-012, plus
particulièrement un groupement méthyle, éthyle, i-propyle ou phényle.
Selon un mode de réalisation préféré de l'invention, ladite synthèse est
effectuée en
l'absence d'un composé accepteur d'hydrogène et en l'absence de base. Cette
synthèse est donc effectuée sans ajout extérieur de ces composés.
Ainsi selon un mode de réalisation particulier l'invention porte sur un
procédé
comprenant la ou les étape(s) suivante(s):
mettre en présence un alcool et un catalyseur tel que défini à la formule 1
sans
ajout de composés accepteurs d'hydrogène et sans ajout de bases.
L'expression accepteur d'hydrogène désigne les composés organiques qui
sont
susceptibles de réagir avec de l'hydrogène moléculaire, H2, pour former un
nouveau
composé dans les conditions réactionnelles de la synthèse d'ester. En
particulier il peut
s'agir de composés tels que les cétones, les aldéhydes, les alcènes, et les
alcynes.
L'absence de composés pouvant réagir avec l'hydrogène moléculaire pour
l'absorber est particulièrement avantageux car il permet notamment la
production
d'hydrogène gazeux H2 qui est facilement isolé du milieu réactionnel et qui
peut ainsi
être utilisé subséquemment. De plus, cela évite la formation stoechiométrique
du
produit d'hydrogénation de l'accepteur d'hydrogène, facilitant les étapes de
purification
en aval.
Ainsi, Selon un mode de réalisation préféré, le procédé selon l'invention
permet
également d'obtenir de l'hydrogène moléculaire gazeux. Celui-ci se sépare du
milieu
réactionnel par simple séparation de phase et peut être évacué et/ou recueilli
directement.
Le terme de base désigne un composé capable de capter un ou plusieurs
3
CA 02929462 2016-05-03
WO 2015/067899 PCT/FR2014/052839
protons. Dans le cadre de l'invention le terme de base désigne tout
particulièrement
des bases telles la soude, ou des sels de métaux alcalins alkoxylés en
particulier
l'Et0Na, Me0Na ou 13u0K.
De manière préférée, la réaction est réalisée en l'absence de toluène ou de
xylène
et en particulier en l'absence de solvant. Le terme solvant désigne une
substance,
liquide à sa température d'utilisation, qui a la propriété de dissoudre, de
diluer ou
d'extraire les alcools et/ou, éventuellement, le catalyseur, sans les modifier
chimiquement dans les conditions réactionnelles de la synthèse d'esters.
Eventuellement un solvant ne se modifie pas lui-même dans les conditions de la
réaction à laquelle il participe. . Il peut s'agir de composés tels que l'eau,
les solvants
inorganiques, et les solvants organiques de type hydrocarboné, oxygéné, et
halogéné.
Ce solvant en l'absence duquel la réaction est réalisée peut évidemment être
également un accepteur d'hydrogène et/ou une base. Ainsi, dans le cadre de
l'invention
le terme solvant désigne notamment les cétones, telles la 3-pentanone,
l'acétone ou la
cyclohexanone. Il désigne également les composés hydrocarbonés aromatiques ou
aliphatiques, éventuellement halogénés, les éthers et les alcools.
L'expression en
l'absence de est utilisée dans son acception normale qui implique l'absence
dans le
mélange réactionnel initial d'une quantité suffisante du composé pour jouer un
rôle
effectif dans la réaction ainsi que l'absence d'ajout extérieur de ce composé
lors de la
réaction. Par exemple la présence dans le milieu réactionnel d'une quantité
minime
(par exemple sous forme de trace) d'un accepteur d'hydrogène, d'une base ou
d'un
solvant n'influencera pas de manière sensible la réaction. Ainsi l'absence de
solvant
implique l'absence d'une quantité suffisante pour dissoudre/diluer/extraire le
ou les
produits de départs (c.à.d. le ou les alcool(s)) ainsi que le catalyseur. Pour
un solvant
cette quantité est généralement supérieure aux nombres de moles de réactifs,
c'est-à-
dire que le solvant est généralement présent dans le milieu réactionnel dans
une
concentration molaire supérieure ou égale à 50%.
De préférence, l'expression absence de implique une concentration molaire
du
dit composé inférieure à 10%, et plus particulièrement inférieure à 5%, voire
son
absence totale c'est-à-dire inférieure à 0,001%.
De préférence, la charge de catalyseur utilisée dans le procédé selon
l'invention est
inférieure à 10000 ppm, plus particulièrement inférieure à 1000 ppm encore
plus
particulièrement inférieure à 500 ppm. Cette charge peut-être par exemple
d'environ
50 10 ppm.
De préférence, la charge de catalyseur utilisée dans le procédé selon
l'invention est
choisie dans une gamme allant de 10000 ppm à 1 ppm, plus particulièrement de
1000
ppm à 10 ppm et encore plus particulièrement de 500 ppm à 50 ppm. Cette charge
peut-être par exemple d'environ 225 10 ppm.
4
CA 02929462 2016-05-03
WO 2015/067899 PCT/FR2014/052839
Les proportions ci-dessus sont données par rapport à la quantité molaire des
produits de départ.
De préférence la température du milieu réactionnel est choisie dans une gamme
de
température allant de 200 C à 15 C, plus particulièrement de 150 C à 40 C et
encore
plus particulièrement de 130 C à 80 C. Cette température peut-être d'environ
130 1 C.
De préférence la réaction est réalisée à une pression allant de 20 bars à 1
bar,
Avantageusement aucune pression particulière n'est appliquée et la réaction
est
réalisée à la pression atmosphérique et/ou dans un système ouvert.
De préférence l'alcool mis en réaction avec le catalyseur de formule 1 est un
alcool
primaire.
De préférence l'alcool mis en réaction avec le catalyseur de formule 1 est un
alcool
primaire, ramifié ou non, en Cl à C30, en particulier en C2 à C6 ou en C8 à
C22 et
plus particulièrement en C16 à C18. Par exemple il peut être choisi dans le
groupe
constitué par l'éthanol, le butanol, l'octanol-l-ol, le 2-éthy1-1-hexanol, le
nonan-1-ol, le
décan-1-ol, l'undécanol, l'alcool laurylique, l'alcool myristylique, l'alcool
cétylique,
l'alcool stéarylique, le docosanol, le policosanol et leurs mélanges.
De préférence l'alcool mis en réaction avec le catalyseur de formule 1 est un
alcool
gras linéaire de formule :
OH
où n est 6, de préférence le nombre de carbone de cet alcool gras est un
nombre
pair et n 12;
ou un alcool issu de la fermentation de la biomasse où n, est compris entre 0
et 5
bornes incluses, préférentiellement où n = 0 ou 2, tel que l'éthanol ou le
butanol.
Dans le procédé selon l'invention, le composé de départ (alcool) peut être
présent
seul ou en mélange avec d'autres réactifs (c'est-à-dire d'autres alcools).
De plus le composé de départ peut être utilisé sous forme purifiée ou sous
forme
brute, notamment non raffinée, ceci en particulier lorsque le composé est
obtenu à
partir d'huiles végétales (par ex. d'oléagineux). Un alcool brut est
généralement une
composition, par exemple un distillat, ne comprenant pas plus de 90%, de
préférence
pas plus de 80 %, par exemple pas plus de 70% en volume d'alcool par rapport
au
volume total de la composition. Ainsi le produit de départ peut être une
composition
comprenant moins de 95%, de préférence moins de 85%, par exemple moins de 75%
en volume d'alcool par rapport au volume total de la composition.
Selon un aspect particulier de l'invention le procédé est réalisé en l'absence
de tout
additif (autre que le catalyseur) pouvant avoir un effet sur la réaction de
couplage de
5
CA 02929462 2016-05-03
WO 2015/067899 PCT/FR2014/052839
l'alcool et/ou sur la production d'hydrogène moléculaire.
Selon un autre aspect préféré de l'invention, le procédé ne comprend pas
d'étape de
séchage et/ou de dégazage des alcools. En effet il a été déterminé de manière
surprenante que le catalyseur (et particulièrement les catalyseurs où Z est
HBH3 tel
que le Ru-MACHO-BH, de formule C)) reste actif en présence d'air et de traces
d'eau.
Il apparait en effet que, de manière inattendue, ces conditions réactionnelles
spécifiques permettent d'obtenir à la fois un temps de réaction plus court et
l'utilisation
d'une charge très réduite de catalyseur tout en préservant un rendement élevé.
Le fait
de pouvoir s'affranchir de l'utilisation de composés chimiques additionnels
tels que
ceux mentionnés ci-dessus permet de réduire les coûts de fabrication, d'éviter
les
problèmes de corrosion liés à leur utilisation et donc de réduire de manière
drastique
l'impact environnemental de tels procédés. Cela permet également de simplifier
grandement les opérations de séparation et/ou de purification des produits de
la
réaction en aval du procédé, opérations telles que l'élimination du solvant de
la réaction
par distillation, la neutralisation du milieu réactionnel utilisant une base,
ou la
séparation des produits de la réaction plus nombreux lors de l'emploi d'un
accepteur
d'hydrogène.
Selon un aspect particulier de l'invention, le catalyseur est de préférence un
catalyseur de formule 1 dans laquelle les quatre radicaux R sont identiques.
Selon un aspect particulier de l'invention, le catalyseur est de préférence un
catalyseur de formule 1, dans laquelle Z est l'HBH3 et/ou Y est un radical CO
et R sont
des radicaux phényles.
Selon un aspect particulièrement préféré de l'invention, le catalyseur utilisé
est un
catalyseur de formule 1 dans laquelle le groupement Z est H et le groupement Y
est
une phosphine PR'3 où R' un groupement alkyle en C1-C12 ou aryle en C6-C12, en
particulier un groupement méthyle, éthyle, isopropyle ou phényle.
Selon une variante préférée de l'invention, le catalyseur est le catalyseur de
Formule
C (R = Ph, Z = HBH3, Y = CO).
Selon une variante préférée de l'invention, le catalyseur est le catalyseur de
Formule
lb (R = i-Pr, Z = HBH3, Y = CO).
Selon une variante préférée de l'invention, le catalyseur est le catalyseur de
Formule
lc (R = Cy, Z = HBH3, Y = CO).
Selon une variante préférée de l'invention, le catalyseur est le catalyseur de
Formule
6a (R = Ph, Z= H, Y = CO).
Selon une variante préférée de l'invention le catalyseur est le catalyseur de
Formule
6c (R = Cy, Z = H, Y = CO).
L'invention porte également sur les catalyseurs décrits dans cette demande en
tant
que tels ainsi que sur leurs procédés de fabrication. En particulier un
complexe de
6
CA 02929462 2016-05-03
WO 2015/067899 PCT/FR2014/052839
formule 1 c, lb, 6a et 6c, ainsi qu'un complexe de même formule où le
substituant
carbonyle est substitué par une phosphine est un objet de l'invention. Leurs
utilisations
dans des procédés de synthèse catalytique ainsi que de tels procédés sont
également
des objets de l'invention. Les procédés de synthèse catalytique peuvent être
des
réactions d'hydrogénation, d'amination d'alcools, de synthèse d'amides ou des
réactions de Guerbet.
L'invention porte également sur un ester obtenu directement par le procédé
décrit ci-
dessus.
Brève description des dessins
L'invention sera mieux comprise à la lecture des figures annexées, qui sont
fournies
à titre d'exemples et ne présentent aucun caractère limitatif, dans
lesquelles:
La FIGURE 1 représente l'évolution du rendement en acétate d'éthyle (Figure
la)
et du TON (Figure lb) en fonction du temps pour le couplage déshydrogénant de
l'éthanol pour différentes charges de catalyseur C selon l'exemple la.
La FIGURE 2 représente l'évolution du rendement de la synthèse selon
l'invention
de l'exemple lb en ester (butyrate de butyle) (FIGURE 2a) et Turn Over
Number
(TON = nombre de mole de substrat convertit par mole de catalyseur) (FIGURE
2b) en
fonction du temps.
La FIGURE 3 représente l'évolution du rendement de la synthèse selon
l'invention
de l'exemple lc en ester (butyrate de butyle) (FIGURE 3a) et du TON (FIGURE
3b) en
fonction du temps.
La FIGURE 4 représente l'évolution du rendement de la synthèse selon
l'invention
de l'exemple 1 d en ester (butyrate de butyle) (FIGURE 4a) et du TON (FIGURE
4b) en
fonction du temps.
La FIGURE 5 représente l'évolution du rendement de la synthèse selon
l'invention
de l'exemple le en ester (butyrate de butyle) (FIGURE 5a) et du TON (FIGURE
5b) en
fonction du temps.
La FIGURE 6 représente l'évolution du TON et rendement en myristyl myristate
en
fonction du temps pour le couplage déshydrogénant du tetradécanol selon
l'invention
décrit à l'exemple 2.
Description détaillée de l'invention
Exemple 1 : Synthèse d'acétate d'éthyle à partir d'éthanol et de butyrate de
butyle à partir de butanol
7
CA 02929462 2016-05-03
WO 2015/067899 PCT/FR2014/052839
Les synthèses d'acétate d'éthyle à partir d'éthanol et de butyrate de butyle à
partir
de butanol peuvent être résumées par les équations suivantes :
0
Ru Cat. ).L
2 + 2 H? (Eq. 1)
éthanol acétate d'éthyle
0
2 0,H/\ Ru Cat.
2H2 (Eq. 2)
butanol butyrate de butyle
Exemple la: Synthèse d'acétate d'éthyle à partir d'éthanol selon un mode de
réalisation de l'invention en utilisant un catalyseur de formule C :
50,2 mg (85,61 p mol) de catalyseur de formule C ([Ru] 500 ppm) est introduit
dans
un tube de Schlenk contenant un barreau d'agitation. 7,8104 g (169,53 mmol) de
d'éthanol est introduit via une seringue sous atmosphère d'argon. Le tube de
Schlenk
est ensuite équipé d'un condenseur surmonté d'un bulleur et d'une entrée
d'argon. Le
système est chauffé à 78 C à l'aide d'un bain d'huile et est agité
magnétiquement
pendant 9 heures. Des échantillons sont prélevés à différents temps à l'aide
d'une
seringue via une entrée latérale du tube de Schlenk. Les échantillons sont
pesés, une
quantité connue de standard interne (cyclohexane) est ajoutée et ils sont
ensuite dilués
par du dichlorométhane.
Les échantillons sont analysés par chromatographie en phase gazeuse équipée
d'un détecteur à ionisation de flamme (GC-FID, Agilent Technologies 7890A, GO
System, colonne Zebron ZB-Bioethanol) pour la détermination de la conversion
et de la
sélectivité de la réaction ainsi que du bilan de matière. Les produits sont
identifiés par
chromatographie en phase gazeuse équipée d'un détecteur spectromètre de masse
(MS: Agilent Technologies 59750 VL MSD) et les résultats comparés avec ceux
des
produits purs. Les résultats obtenus sont compilés dans le tableau I.
Tableau I : TOF (Turn Over Frequency = nombre de mole de substrat convertit
par
mole de catalyseur par heure) obtenues pour le couplage déshydrogénant de
l'éthanol
Catalyseur [Ru] (ppm) TOF0 (h-i)a Rendement (%)b
500 190 (9h) 75(9h)
a : Calculée par régression linéaire de la variation du TON en fonction du
temps du
début de la réaction jusqu'au temps indiqué entre parenthèse.
: Rendement obtenu après le temps indiqué entre parenthèses.
Le couplage de l'éthanol en acétate d'éthyle en présence du catalyseur C
procède à
8
CA 02929462 2016-05-03
WO 2015/067899 PCT/FR2014/052839
une vitesse élevée, avec pour produit l'acétate d'éthyle et des traces
d'acétaldéhyde
(<1%).
Le couplage de l'éthanol en acétate d'éthyle a été étudié pour deux charges
différentes de catalyseur C, soit 50 et 500 ppm (Tableau II, Figure 1). Le
couplage
déshydrogénant de l'éthanol peut être effectué avec une faible charge de
catalyseur,
par ex. 50 ppm. Dans ces conditions, des TOF0 de l'ordre de 500 h-1 sont
obtenus et un
TON supérieur à 6000 après 26 h de réaction est subséquemment observé.
Tableau II : Couplage déshydrogénant de l'éthanol catalysé par
différentes
charges de catalyseur C (T = 78 C)
[C] (PPrn) TOF0 (h-1) TON Ester (`)/0)
500 190(9h) 1507(9h) 75(9h)
6031
(26h)
50 493 (10h) 31 (26h)
Exemple lb : Synthèse de butyrate de butyle en utilisant un catalyseur de
formule C selon un mode de réalisation de l'invention et comparaison avec des
procédés de l'art antérieur:
16,1 mg (27,46 pmol) de catalyseur de formule C ([Ru] ,' 240 ppm) est
introduit dans
un tube de Schlenk contenant un barreau d'agitation. 8,5670 g (115,58 mmol) de
butanol est introduit via une seringue sous atmosphère d'argon. Le tube de
Schlenk est
ensuite équipé d'un condenseur surmonté d'un bulleur et d'une entrée d'argon.
Le
système est chauffé à 130 C à l'aide d'un bain d'huile et est agité
magnétiquement
pendant 5 heures. Des échantillons sont prélevés à différents temps à l'aide
d'une
seringue via une entrée latérale du tube de Schlenk. Les échantillons sont
pesés, une
quantité connue de standard interne (cyclohexane) est ajoutée et ils sont
ensuite dilués
par du dichlorométhane.
Les échantillons sont analysés de la même manière que pour l'exemple la et les
résultats obtenus sont compilés dans le tableau III et représentés aux figures
2a et 2b.
Le couplage du butanol en butyrate de butyle en présence du catalyseur C
procède
à haute vitesse, avec pour produit le butyrate de butyle et des traces de
butyraldéhyde
(<1%). L'expérience a été reproduite dans les mêmes conditions mais cette fois
avec
un catalyseur : Ru-MACHO (ou A). Les résultats sont compilés dans le Tableau
III :
Tableau III: TOF initiales obtenues pour le catalyseur C.
Catalyseur TOF0 (h-l)a
C 2340
A 0
a: Calculée par régression linéaire de la variation du TON en fonction du
temps
9
CA 02929462 2016-05-03
WO 2015/067899 PCT/FR2014/052839
De manière surprenante le couplage du butanol en butyrate de butyle en
présence
du catalyseur C en l'absence de tout additif et notamment en absence de base
et de
solvant procède à haute vitesse, avec pour produit le butyrate de butyle et
des traces
de butyraldéhyde.
Tableau IV:
Comparaison entre les résultats divulgué dans l'exemple 31 de la
demande de brevet W02012/144650 et le procédé décrit ci-dessus pour la
production
du butyrate de butyle
Catalyseur BuOH/Ru en T( C) Solvant Tps Ester
(`)/0)
mol/mol
1000 120 3-pentanone 9 h
100
4150 118 Aucun 3h 96
En plus de dissoudre les différentes espèces (catalyseur et substrat) la 3-
pentanone
joue le rôle d'accepteur d'hydrogène. Ainsi une quantité au moins
stoechiométrique
d'accepteur d'hydrogène (de solvant) est utilisée dans les exemples de la
littérature et
la réaction ne s'accompagne donc pas du dégagement de deux molécules
d'hydrogène
par molécule d'ester produite comme dans le procédé de l'invention où
l'hydrogène
gazeux se dégage du milieu réactionnel.
Outre cette différence importante, les performances des systèmes catalytiques
sont
nettement supérieures selon le procédé de l'invention. Ces conditions
permettent
d'obtenir en des temps 3 fois plus court et avec une charge catalytique 4 fois
plus faible
un rendement similaire en butyrate de butyle.
Exemple lc : Synthèse de butyrate de butyle selon un mode de réalisation de
l'invention et selon l'équation 2 (ci-dessus) en utilisant un catalyseur de
formule
6c:
H
H
Nõõ I 13CY2
PRu,
'CO
CY2
6c
17 mg (28 pmol) de catalyseur de formule 6c ([Ru] 250 ppm) est introduit dans
un
tube de Schlenk contenant un barreau d'agitation. 8,0450 g (108,54 mmol) de
butanol
est introduit via une seringue sous atmosphère d'argon. Le tube de Schlenk est
ensuite
équipé d'un condenseur surmonté d'un bulleur et d'une entrée d'argon. Le
système est
chauffé à 130 C à l'aide d'un bain d'huile et est agité magnétiquement
pendant 5
heures. Des échantillons sont prélevés à différents temps à l'aide d'une
seringue via
CA 02929462 2016-05-03
WO 2015/067899 PCT/FR2014/052839
une entrée latérale du tube de Schlenk. Des échantillons sont prélevés à
différents
temps à l'aide d'une seringue via une entrée latérale du tube de Schlenk. Les
échantillons sont ensuite dilués par du chloroforme deutéré, CDCI3.
Le rendement est déterminé par Spectromètre RMN Bruker Avance 1, 300 MHz,
sonde 5 mm. Les résultats obtenus sont compilés dans le tableau V et
représentés aux
figures 3a et 3b.
Tableau V TOF initiales obtenue
Run TOF0 / h-1 a
1 1005
2 1075
a: Calculée par régression linéaire de la variation du TON en fonction du
temps
En conclusion, le catalyseur 6c est actif pour l'estérification
déshydrogénante du butanol
en l'absence de base et d'accepteur d'hydrogène.
Exemple Id : Synthèse de butyrate de butyle selon un mode de réalisation de
l'invention et selon l'équation 2 (ci-dessus) en utilisant un catalyseur de
formule
lb:
H
H
1/-1¨µõ.õ
N. I n-1-2
'Ru
P
iPr2 HBH3
lb
13 mg (25 pmol) de catalyseur de formule lb ([Ru] 260 ppm) est introduit dans
un
tube de Schlenk contenant un barreau d'agitation. 8,1580 g (110.06 mmol) de
butanol
est introduit via une seringue sous atmosphère d'argon. Le tube de Schlenk est
ensuite
équipé d'un condenseur surmonté d'un bulleur et d'une entrée d'argon. Le
système est
chauffé à 130 C à l'aide d'un bain d'huile et est agité magnétiquement
pendant 5
heures. Des échantillons sont prélevés à différents temps à l'aide d'une
seringue via
une entrée latérale du tube de Schlenk. Les échantillons sont pesés, une
quantité
connue de standard interne (cyclohexane) est ajoutée et ils sont ensuite
dilués par du
dichlorométhane.
Les échantillons sont analysés de la même manière que pour l'exemple la et les
résultats comparés avec ceux des produits purs. Les résultats obtenus sont
compilés
dans le tableau VI et représentés aux figures 4a et 4b.
Tableau VI TOF initiales obtenue
Run TOF) /h a
11
CA 02929462 2016-05-03
WO 2015/067899 PCT/FR2014/052839
1 2465
: Calculée par régression linéaire de la variation du TON en fonction du temps
En conclusion, le catalyseur lb est actif pour l'estérification
déshydrogénante du butanol
en l'absence de solvant, de base et d'accepteur d'hydrogène.
Exemple le: Synthèse de butyrate de butyle selon un mode de réalisation de
l'invention et selon l'équation 2 (ci-dessus) en utilisant un catalyseur de
formule
lc :
H
H
Nõ, I In-,Y2
Ru
Cy2 HBF13
lc
16 mg (26 pmol) de catalyseur de formule 1 c ([Ru] 240 ppm) est introduit dans
un
tube de Schlenk contenant un barreau d'agitation. 8,013 g (108,11 mmol) de
butanol
est introduit via une seringue sous atmosphère d'argon. Le tube de Schlenk est
ensuite
équipé d'un condenseur surmonté d'un bulleur et d'une entrée d'argon. Le
système est
chauffé à 130 C à l'aide d'un bain d'huile et est agité magnétiquement
pendant 5
heures. Des échantillons sont prélevés à différents temps à l'aide d'une
seringue via
une entrée latérale du tube de Schlenk. Des échantillons sont prélevés à
différents
temps à l'aide d'une seringue via une entrée latérale du tube de Schlenk. Les
échantillons sont ensuite dilués par du chloroforme deutéré, CDCI3.
Le rendement est déterminé par Spectromètre RMN Bruker Avance 1, 300 MHz,
sonde 5 mm. Les résultats obtenus sont compilés dans le tableau VII et
représentés
aux figures 5a et 5b.
Tableau VII TOF initiales obtenue
Run TOR) /h a
1 1545
a: Calculée par régression linéaire de la variation du TON en fonction du
temps
En conclusion, le catalyseur 1 c est actif pour l'estérification
déshydrogénante du butanol
en l'absence de solvant, de base et d'accepteur d'hydrogène.
Exemple 2 : Synthèse de myristyl myristate à partir du tetradécanol
12
CA 02929462 2016-05-03
WO 2015/067899 PCT/FR2014/052839
2 OH
H H
rJH,,,s13Ph2
- 2 H2 la Ru
Ph2 H131-13
0 1 a
0
5,4 mg (9,21 pmol) de catalyseur de formule C ([Ru] 225 ppm) est introduit
dans un
tube de Schlenk contenant un barreau d'agitation. 8,7662 g (40,89 mmol) de
tetradécanol est introduit via une seringue sous atmosphère d'argon. Le tube
de
Schlenk est ensuite équipé d'un condenseur surmonté d'un bulleur et d'une
entrée
d'argon. Le système est chauffé à 130 C à l'aide d'un bain d'huile et est
agité
magnétiquement pendant 6 heures. Des échantillons sont prélevés à différents
temps à
l'aide d'une seringue via une entrée latérale du tube de Schlenk. Les
échantillons sont
ensuite dilués par du chloroforme deutéré, CDCI3.
Le rendement est déterminé par Spectromètre RMN Bruker Avance 1, 300 MHz,
sonde 5 mm. Les résultats sont compilés dans le tableau VIII et représenté à
la Figure
6a et 6b.
Tableau VIII : TOF initiale obtenue
Run TOR) / a
1 3070
a: Calculée par régression linéaire de la variation du TON en fonction du
temps.
Exemple 3 : synthèse du
Carbonylchlorohydridolbis(2-di-
isopropylphosphinoethyl)amineuthenium(11) (2b) composé de départ de la
synthèse du composé lb:
Le schéma réactionnel de la réaction est le suivantci :
H Cl
sõPPh3 3a PR
Nõõ 2
Rd ___________________ Y r
Lp/ -CO
Ph3P' 'CO diglyme, 165 C, 2h
H R2 Fi
2b, R = iFr
Synthèse : Dans un tube de Schlenk une
suspension de
carbonylchlorohydrido[tris(triphenylphosphine)]ruthenium(II) (Strem Chemicals,
0,999
13
CA 02929462 2016-05-03
WO 2015/067899 PCT/FR2014/052839
g; 1.04 mmol) et de NH(C2H4P1Pr2)2, 3a (0,357 g; 1.17 mmol) dans du diglyme
(10
mL) est placé dans un bain d'huile préchauffé à 165 C et laissé sous
agitation pendant
deux heures pour donner une solution jaune limpide. La solution est laissée
18h à
température ambiante pour donner un précipité. 10 mL de pentane sont
additionnés et
la suspension refroidie à 0 C pendant 1 heure. Le surnageant est ensuite
éliminé et
les cristaux sont lavés par de l'éther diéthylique (3 x 5 mL) puis séchés sous
pression
réduite pour donner le produit désiré sous forme d'une poudre jaune très pâle.
Rendement : 64% (0,317 g).
RMN 31P-{1H} (CD2Cl2, 121,5 MHz) : O. 74,7 ppm. RMN 1H et 31P en accord avec
les
données spectrales de la littérature. Voir : Bertoli, M.; Choualeb, A.; Lough,
A. J.; Moore
B.; Spasyuk, D.; Gusev D. G. Organometallics, 2011, 30, 3479.
Exemple 4: Synthèse de
Carbonylchlorohydrido[bis(2-
dicyclohexylphosphinoethyl)amineuthenium(11) (2c) composé de départ de la
synthèse des composés lc et 6c:
Le schéma réactionnel de la réaction est le suivant :
y CI
H
/-
Ph313.õ. ,õPPh3 3b N "2
Ru
Ru-
Ph3P- 'CO L
diglyme, 165 C, 19h
Cl R2 H
2c, R = Cy
Synthèse : Dans un tube de Schlenk une suspension
de
carbonylchlorohydrido[tris(triphenylphosphine)]ruthenium(II) (Strem Chemicals,
1,000 g
; 1,05 mmol) et NH(C2H4PCy2)2, 3b (0,498 g; 1,07 mmol) dans du diglyme (10 mL)
est
placé dans un bain d'huile préchauffé à 165 C et laissé sous agitation
pendant 19
heures pour donner une suspension. Le milieu est ensuite refroidi à
température
ambiante, le surnageant est éliminé et le précipité est lavé par de l'éther
diéthylique (3x
5 mL) puis séché sous pression réduite pour donner le produit désiré sous
forme d'une
poudre jaune très pâle. Rendement : 78 % (0,518 g) RMN31P{1H} (CD2Cl2, 121,5
MHz) :
O. 65.6 ppm. RMN 1H et 31P en accord avec les données spectrales de la
littérature.
Voir : Nielsen, M. ; Alberico, E.; Baumann, W.; Drexler H.-J.; Junge, H.;
Gladiali, S.;
Beller, M. Nature, 2013, sous presse (doi:10.1038/nature11891).
Exemple 5 : Synthèse du Carbonylhydrido(tetrahydroboratebis(2-di-i-
propylphosphinoethyl)amineuthenium(11)(1b) :
Le schéma réactionnel de la réaction est le suivant :
14
CA 02929462 2016-05-03
WO 2015/067899 PCT/FR2014/052839
H ci Hi H
õõ PR 7-1----\p
2
p
N2 NaBH4 f '
,CRu ,õ0 i C)
Et0H, PhMe 'eC
L 65 C, 2-3 h
R2 H h2 HBH3
2b, R = 1b, R = iPr
Synthèse : Dans un tube de Schlenk sous flux d'argon, une solution de NaBH4 (5
mg; 0,24 mmol) dans l'éthanol (2 mL) est ajoutée à une suspension de
carbonylchlorohydrido[bis(2-di-i-propylphosphinoethyl)amino]ruthenium(II), 2b,
(50 mg ;
0,08 mmol) dans le toluène (8mL). Le tube de Schlenk est ensuite fermé
hermétiquement, plongé dans un bain d'huile préchauffé à 65 C et laissé sous
agitation pendant 2 h 30 pour donner une solution opalescente. Le solvant est
ensuite
éliminé par distillation sous pression réduite (température ambiante, 1.10-3
mbar). Le
résidu blanc obtenu est extrait par du dichlorométhane (3 x 5 ml) et filtré
sur verre fritté.
Le filtrat est ensuite concentré sous pression réduite (température ambiante,
1.10-3
mbar) pour donner le produit désiré sous la forme d'une poudre blanche (30 mg;
rendement : 62%).
RMN 1H (CD2Cl2, 300 MHz) : O. 3,90 (large ; 1H ; NH) ; 3,30-3,12 (m ; 2H) ;
2.56-
2.44 (m ; 2H) ; 2.30-2.21 (m ; 2H) ; 1.94 ¨ 1.82 (m ; 2H) ; 1,38 (dd ;
JHP=16,2 Hz;
JHH=7,5 Hz; 6H) ; 1,28-1.14 (m ; 18 H) ; -1,92--2,69 (large ; 4H ; RuHBH3) ; -
13,53
(t, JHP=17,7 Hz; 1H; RuH). 31P-{1H} (CD2Cl2, 121,5 MHz) : O. 77,7 ppm.
Exemple 6: Synthèse du carbonylhydrido(tetrahydroboratebis(2-
dicyclohexylphosphinoethyl)amineuthenium(11)(1c):
Le schéma réactionnel de la réaction est le suivant :
Hi Cl H
H
IN 7- --`pp õõ "2 NaBH4 I 7TPR
.e 2
Lpe CO Et0H, PhMe Lpeirh*C0
65 C, 4 h
R2 H R2 HBH3
2c, R = Cy lc, R = Cy
Synthèse : Dans un tube de Schlenk sous flux d'argon, une solution de NaBH4
(48
mg ; 1,37 mmol) dans d'éthanol (12 mL) est ajoutée à une suspension de
carbonylchlorohydrido[bis(2- dicyclohexylphosphinoethyl)amino] ruthenium (II),
2c, (200
mg ; 0,43 mmol) dans le toluène (16 mL). Le tube de Schlenk est ensuite fermé
hermétiquement, plongé dans un bain d'huile préchauffé à 65 C et laissé sous
agitation pendant quatre heures pour donner une solution opalescente. Le
solvant est
ensuite éliminé par distillation sous pression réduite (température ambiante,
1.10'
mbar). Le résidu blanc obtenu est extrait par du dichlorométhane (3 x 5 ml) et
filtré sur
CA 02929462 2016-05-03
WO 2015/067899 PCT/FR2014/052839
verre fritté. Le filtrat est ensuite concentré sous pression réduite
(température
ambiante, 1.10-3 mbar) pour donner le produit désiré sous la forme d'une
poudre
blanche (171 mg ; rendement : 88%).
RMN 1H (CD2Cl2, 300 MHz) : O. 3,85 (large ; 1H; NH) ; 3,27-3,09 (m ; 2H) ;
2,27-2,10
(m; 8H) ; 1,96-1,68 (m; 20H) ; 1,61-1,20 (m; 22H) ; -2,19 - -2,50 (large ; 4H;
RuHBH3) ;-13,60(t,JHp=17, 9Hz ; 1H; RuH). 31P-{1H} (CD2Cl2, 121,5 MHz) : O.
68.9 ppm.
Exemple 7: Synthèse du
carbonyl(dihydridebis(2-di-
cyclohexylphosphinoethyl)amineuthénium(11) (6c)
Le schéma réactionnel de la réaction est le suivant :
H Cl Hi H
17-1¨\ppt 17-1-\pp
-2 NaHBEt3 1 Nõ,
1=11-1.* *C0
THF '
TA, 18 h P
"Ru_0
R2 H R2 H
2c, R = Cy 6c, R = Cy
Synthèse : Dans un tube de Schlenk une suspension blanchâtre de
carbonylhydridoclhoro[bis(2-di-cyclohexylphosphinoethyl)amino]ruthénium(I I)
2c (63
mg; 0,1 mmol) dans du THF (2 ml) est traitée par une solution commerciale de
NaHBEt3 à 1 M dans du toluène (0,12 ml; 0,12 mmol). Puis, le milieu est laissé
sous
agitation à température ambiante sous atmosphère d'argon pendant 18 heures
pour
donner une solution jaune clair opalescente. La solution est ensuite
concentrée sous
pression réduite pour conduire à un résidu solide jaune lequel est dissout
avec du
toluène (2 mL). Cette nouvelle solution est filtrée sur verre fritté et
concentrée sous
pression réduite pour donner une poudre jaune. Rendement 80% (48 mg).
RMN 1H (C6D6, 300 MHz) : O. 2,32 ¨ 2,07 (m ; 6H) ; 1,90¨ 1,60 (m ; 31H) ; 1,34
¨
1,03 (m; 16H) ; -6,18 --6,34 (m ; 2H; RuH2)
RMN 31P{1H} (C6D6, 121,5 MHz) : O. 80,4 ppm
L'invention n'est pas limitée aux modes de réalisations présentées et d'autres
modes
de réalisation apparaîtront clairement à l'homme du métier.
16