Note: Descriptions are shown in the official language in which they were submitted.
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
Apparatus, Kits and Methods for the Prediction of Onset of Sepsis
The present invention is concerned with kits, methods and apparatus for
analysing a biological
sample from an animal to predict and monitor the development of sepsis
utilising biomarker
signatures/lists of biomarkers to predict whether an animal is likely to
develop the symptoms of
sepsis, and especially biomarker signatures capable of providing a mean
predictive accuracy of at
least 92% to differentiate development of sepsis from non-sepsis, and of at
least 95% to
differentiate development of sepsis from SIRS.
Following exposure to a biological agent there is often a lag phase before
symptoms of sepsis
present. After the onset of clinical symptoms, the effectiveness of treatment
often decreases as the
disease progresses, so the time taken to make any diagnosis is critical. It is
likely that a detection or
diagnostic assay will be the first confirmed indicator of sepsis. The
availability, rapidity and
predictive accuracy of such an assay will therefore be crucial in determining
the outcome. Any time
saved will speed up the implementation of medical countermeasures and will
have a significant
impact on recovery.
The development of technologies to facilitate rapid detection of biological
agent infection is a key
concern for all at risk. During the initial stages of infection many
biological agents are either absent
from, or present at very low concentrations in, typical clinical samples (e.g.
blood). It is therefore
likely that agent-specific assays would have limited utility in detecting
infection before clinical
symptoms arise. Previous studies have shown that infection elicits a pattern
of immune response
involving changes in the expression of a variety of biomarkers that is
indicative of the type of agent.
Such patterns of biomarker expression have proven to be diagnostic for a
variety of infectious
agents. It is now possible to distinguish patterns of gene expression in blood
leukocytes from
symptomatic patients with acute infections caused by four common human
pathogens (Influenza A,
Staphylococcus aureus, Streptococcus pneumoniae and Escherichia coil) using
whole transcriptome
analysis. More recently, researchers have been able to reduce the number of
host biomarkers
1
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
.
required to make a diagnosis through use of appropriate bioinformatic analysis
techniques to select
key biomarkers for the diagnosis of infectious disease.
While host biomarker signatures represent an attractive solution for the pre-
symptomatic detection .. '
of biological agent infection, their discovery relies on the exploitation of
laboratory models of
infection whose fidelity to the pathogenesis of disease in humans varies. An
alternative approach for
pre-symptomatic biomarker discovery in humans is to exploit a common sequela
of biological agent
infection; the life-threatening condition sepsis. Sepsis is traditionally
defined as a systemic
inflammatory response syndrome (SIRS) in response to infection which, when
associated with acute
organ dysfunction, may ultimately cause severe life-threatening complications.
This broad definition
relies on observation of overt symptoms of systemic illness (temperature,
blood pressure, heart rate,
etc.) as well as the indication of the presence of an infectious organism
through microbial culture
from clinical samples. It has been described in animal (primarily murine and
NHP) models of anthrax
(Bacillus anthracis), tularemia (Francisella tularensis), plague (Yersinia
pestis), glanders (Burkholderia
melioidosis (B. pseudomallei), haemorrhagic filovirus and alphavirus
infection. More
importantly, sepsis is directly caused by the same biological agents in
humans.
The incidence of natural biological agent infection is generally extremely
low, making prospective
studies of the onset of disease in a human population non-viable. However, the
development of
severe sepsis, associated with organ dysfunction, hypoperfusion or
hypotension, is a major cause of
morbidity and mortality in intensive care units (ICU). In the UK, severe
sepsis is responsible for 27%
of all ICU admissions. Across Europe the average incidence of severe sepsis in
the ICU is 30%, with a
mortality rate of 27%. In the USA, hospital-associated mortality from sepsis
ranges between 18 to
30%; an estimated 9.3% of all deaths occurred in patients with sepsis. Clearly
there is a very
accessible patient population that could be used to study predictive markers
for the onset of sepsis.
Despite greatly improved diagnosis, treatment and support, serious infection
and sepsis remain
significant causes of death and often result in chronic ill-health or
disability in those who survive
2
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
acute episodes. Although sudden, overwhelming infection is comparatively rare
amongst otherwise
healthy adults, it constitutes an increased risk in immunocompromised
individuals, seriously ill
patients in intensive care, burns patients and young children. In a proportion
of cases, an apparently
treatable infection leads to the development of sepsis; a dysregulated,
inappropriate response to
infection characterised by progressive circulatory collapse leading to renal
and respiratory failure,
abnormalities in coagulation, profound and unresponsive hypotension and, in
about 30% of cases
death. The incidence of sepsis in the population of North America is about
0.3% of the population
annually (about 750,000 cases) with mortality rising to 40% in the elderly and
to 50% in cases of the
most severe form, septic shock.
It should be noted that clinical sepsis may also result from infection with
some viruses (for example
Venezuelan Equine Encephalitis Virus, VEEV) and fungi, and that other
mechanisms are likely to be
involved in such cases.
The ability to detect potentially serious infections as early as possible and,
especially, to predict the
onset of sepsis in susceptible individuals is clearly advantageous, A
considerable effort has been
expended over many years in attempts to establish clear criteria defining
clinical entities such as
shock, sepsis, septic shock, toxic shock and systemic inflammatory response
syndrome (SIRS).
Similarly, many attempts have been made to design robust predictive models
based on measuring a
range of clinical, chemical, biochemical, immunological and cytometric
parameters and a number of
scoring systems, of varying prognostic success and sophistication, proposed.
According to the 1991 Consensus Conference of the American College of Chest
Physicians (ACCP)
and Society of Critical Care Medicine (SCCM) "SIRS" is considered to be
present when patients have
more than one of the following: a body temperature of greater than 38 C or
less than 36 C, a heart
rate of greater than 90/min, hyperventilation involving a respiratory rate
higher than 20/min or
PaCO2 lower than 32mm Hg, a white blood cell count of greater than 12000 cells
/1.11 or less than
4000 cells Jul,
3
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
"Sepsis" has been defined as SIRS caused by infection. It is accepted that
SIRS can occur in the
absence of infection in, for example, burns, pancreatitis and other disease
states. "Infection" was
defined as a pathological process caused by invasion of a normally sterile
tissue, fluid or body cavity
by pathogenic or potentially pathogenic micro-organisms.
"Severe sepsis" is defined as sepsis complicated by organ dysfunction.
"Septic shock" refers (in adults) to sepsis plus a state of acute circulatory
failure characterised by a
persistent arterial hypotension unexplained by other causes.
The correlation of sepsis and a number of specific serum markers has been
extensively studied with
a view to developing specific diagnostic and prognostic tests.
However, although many of these markers correlate with sepsis and some give an
indication of the
seriousness of the condition, no single marker or combination of markers has
yet been shown to be
a reliable diagnostic test, much less a predictor of the development of
sepsis.
Extracting reliable diagnostic patterns and robust prognostic indications from
changes over time in
complex sets of variables including traditional clinical observations,
clinical chemistry, biochemical,
immunological and cytometric data requires sophisticated methods of analysis.
The use of expert
systems and artificial intelligence, including neural networks, for medical
diagnostic applications has
been being developed for some time.
Neural networks are non-linear functions that are capable of identifying
patterns in complex data ,
systems. This is achieved by using a number of mathematical functions that
make it possible for the
network to identify structure within a noisy data set. This is because data
from a system may
produce patterns based upon the relationships between the variables within the
data. If a neural
network sees sufficient examples of such data points during a period known as
"training", it is
capable of "learning" this structure and then identifying these patterns in
future data points or test
CA 02938761 2016-08-04
WO 2015/121605 PCT/GB2015/000004
data. In this way, neural networks are able to predict or classify future
examples by modelling the
patterns present within the data it has seen. The performance of the network
is then assessed by its
ability to correctly predict or classify test data, with high accuracy scores,
indicating the network has
successfully identified true patterns within the data. The parallel processing
ability of neural
networks is dependent on the architecture of its processing elements, which
are arranged to interact
according to the model of biological neurones. One or more inputs are
regulated by the connection
weights to change the stimulation level within the processing element. The
output of the processing
element is related to its activation level and this output may be non-linear
or discontinuous.
Training of a neural network therefore comprises an adjustment of
interconnected weights
depending on the transfer function of the elements, the details of the
interconnected structure and
the rules of learning that the system follows. Such systems have been applied
to a number of clinical
situations, including health outcomes models of trauma patients.
US patent application 2002/0052557 describes a method of predicting the onset
of a number of
catastrophic illnesses based on the variability of the heart-rate of the
patient. A neural network is
among the possible methods of modelling and analysing the data.
International patent application WO 00/52472 describes a rapid assay method
for use in small
children based on the serum or neutrophil surface levels of CD1lb or 'CD11b
complex' (Mac-1, CR3).
The method uses only a single marker, and one which is, arguably, a well-known
marker of
neutrophil activation in response to inflammation.
The alternative approach to analysing such complex data sets where the data
are often qualitative
and discrete, rather than quantitative and continuous, is to use sophisticated
statistical analysis
techniques such as logistic regression. Where logistic regression using
qualitative binary dependent
variables is insufficiently discriminating in terms of selecting significant
variables, multivariate
techniques may be used. The outputs from both multiple logistic regression
models and neural
networks are continuously variable quantities but the likelihoods calculated
by neural network
81790465
models usually fall at one extreme or the other, with few values in the middle
range. In a clinical
situation this is often helpful and can give clearer decisions.
The ability to detect the earliest signs of infection and / or sepsis has
clear benefits in terms of
allowing treatment as soon as possible. Indications of the severity of the
condition and likely
outcome if untreated inform decisions about treatment options. This is
relevant both in
vulnerable hospital populations, such as those in intensive care, or who are
burned or
immunocompromised, and in other groups in which there is an increased risk of
serious infection
and subsequent sepsis. The use or suspected use of biological weapons in both
battlefield and
civilian settings is an example where a rapid and reliable means of testing
for the earliest signs of
infection in individuals exposed would be advantageous.
However, until now neither a test nor a list of biomarkers has been
identified/produced which
can detect or predict sepsis pre-symptomatically with a high predictive
accuracy (for example
> 75%, but preferably > 90%).
The present invention thus aims to provide a biomarker signature (list of
biomarkers), and
methods for classifying biological samples using the biomarker signature, to
pre-symptomatically
predict/detect the development of sepsis with a high predictive accuracy, and
especially a
biomarker signature that could differentiate between sepsis and SIRS with an
accuracy of at least
95%, and/or differentiate between sepsis and non-sepsis with an accuracy of at
least 92%.
In an embodiment, there is provided a diagnostic kit for predicting the
development of sepsis
prior to the onset of symptoms (pre-symptomatic), said kit consisting of means
specific for
detecting in a sample levels of a gene product of each member of a biomarker
signature to pre-
symptomatically predict/detect the development of sepsis, wherein the
biomarker signature
consists of one of the following: all the 266 genes of Table 1, the 44 gene
biomarker signature in
Table 3, the 45 gene biomarker signature of Table 4, the down-selected 25 gene
biomarker
signature of Table 3 or Table 4, or one of the biomarker signatures consisting
of 44 genes selected
from Table 5.
In an embodiment, there is provided a method for analysis of a biological
sample from an animal
to predict or monitor the development of sepsis in said animal prior to the
onset of symptoms,
the method comprising: monitoring, measuring and/or detecting the expression
of a gene
product of each member of a biomarker signature, wherein the biomarker
signature consists of
one of the following: all the 266 genes of Table 1, the 44 gene biomarker
signature of Table 3, the
6
Date Recue/Date Received 2021-06-01
81790465
45 gene biomarker signature of Table 4, the down-selected 25 gene biomarker
signatures of Table 3 or
Table 4, or one of the biomarker signatures consisting of 44 genes selected
from Table 5 and assessing
data produced from the monitoring, measuring and/or detecting to predict or
monitor the development
of sepsis, wherein the monitoring, measuring and/or detecting comprises
producing quantitative, and
optionally qualitative, data for all biomarkers in the biomarker signature,
and the assessing data comprises
inputting said data into an analytical process on a computer, comparing the
data with reference data, and
producing an output from the analytical process which predicts or monitors the
likelihood of
developing sepsis.
In an embodiment, there is provided an apparatus for analysis of a biological
sample from an animal to
predict or monitor the development of sepsis prior to the onset of symptoms
comprising a means specific
for monitoring, measuring or detecting the expression levels of a gene product
of each member of a
biomarker signature, a means for analysis of data produced from the means for
monitoring, measuring or
detecting, and means for providing an output from the analysis which output
provides a prediction for an
animal developing sepsis, or an output suitable for monitoring of sepsis,
wherein the biomarker signature
consists of one of the following: all the 266 genes of Table 1, the 44 gene
biomarker signature of Table 3,
the 45 gene biomarker signature of Table 4, the down-selected 25 gene
biomarker signatures of Table 3
or Table 4, or one of the biomarker signatures consisting of 44 genes selected
from Table 5.
In an embodiment, there is provided use of a biomarker signature consisting of
one of the following: all
the 266 genes of Table 1, the 44 gene biomarker signature of Table 3, the 45
gene biomarker signature of
Table 4, the down-selected 25 gene biomarker signatures of Table 3 or Table 4,
or one of the biomarker
signatures consisting of 44 genes selected from Table 5 to differentiate
development of sepsis from non-
sepsis to predict the development of sepsis prior to the onset of symptoms of
sepsis (pre-symptomatic).
With this in mind, the applicants have determined a biomarker signature (list
of biomarkers)
predictive of the development of sepsis prior to the onset of symptoms (pre-
symptomatic)
and capable of a mean predictive accuracy of at least 75% to differentiate
development of
sepsis from non-sepsis, and sepsis from SIRS, wherein the biomarker signature
comprises
at least 25 genes, or the products expressed by those genes, selected from the
list of genes consisting of
the 266 genes listed in Table 1.The Applicant has identified through a
comprehensive analysis of the host
6a
Date Recue/Date Received 2022-06-02
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
transcriptome, sourced from blood samples from human patients collected prior
to the clinical onset
of sepsis, a panel of 266 genes (Table 1) highly significant to the onset of
symptoms of sepsis. The
full panel and subsets thereof were used in a number of statistical models to
determine
discrimination between sepsis and non-sepsis patients, and between patients
with sepsis and SIRS.
In order to achieve a mean predictive accuracy of greater than 75%, the
Applicant has shown that a
signature of at least 25 gene biomarkers can be randomly selected from the 266
genes listed in Table
1.
The Applicant has in particular shown through an analysis of 44,014
combinations/biomarker
signatures of 44 biomarkers, randomly selected from the list of 266, that all
combinations have a
mean predictive accuracy of greater than 75%. These results are illustrated by
the 15 specific
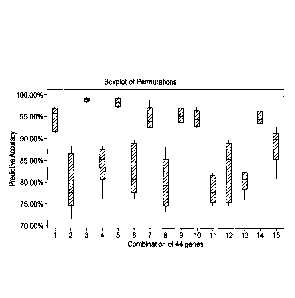
combinations listed in Table 24, which have the accuracies shown in Figure 3.
Thus in one
embodiment the biomarker signature comprises at least 44 genes selected from
the list of genes
consisting of the 266 genes listed in Table 1.
The Applicant has also identified biomarker signatures, comprising at least
25, at least 44, and
comprising all 266 gene biomarkers, which is capable of differentiating
development of sepsis from
non-sepsis with a mean predictive accuracy of at least 92%, and development of
sepsis from SIRS
with a mean predictive accuracy of at least 95%.
The Applicant has produced and trained an artificial neural network (ANN)
which can provide a
predictive accuracy for any selection of biomarkers from the 266 to
differentiate between sepsis and
non-sepsis and/or sepsis from SIRS predict, and thereby provide a likelihood
of whether a patient is
to develop sepsis or not throUgh inputting the patient data set into the ANN.
A patient data set, for example that comprising gene expression levels for the
266 biomarkers in a
patient blood sample, is inputted into the ANN, having selected a biomarker
signature (list of
biomarkers), and will thereby output the predictive accuracy of the selected
biomarker signature,
7
CA 02938761 2016-08-04
WO 2015/121605 PCT/GB2015/000004
and also indicate whether the specific patient data set is indicative of the
development of sepsis,
versus non-sepsis'and/or SIRS. The R script for the trained ANN is detailed in
Table 2.
The Applicant has shown that a biomarker signature (list of biomarkers)
comprising at least 25
genes, but preferably about 44 genes, or the products expressed by those
genes, selected from the
list of genes consisting of the 266 genes listed in Table 1, as inputted into
a mathematical model
such as the ANN detailed in Table 2, can be predictive of the development of
sepsis prior to the
onset of symptoms (pre-symptomatic) and be capable of a mean predictive
accuracy of at least 92%
to differentiate development of sepsis from non-sepsis,.
The Applicant has also shown that a biomarker signature (list of biomarkers)
comprising at least 25
genes, but preferably about 44 genes, or the products expressed by those
genes, selected from the
list of genes consisting of the 266 genes listed in Table 1, as inputted into
a mathematical model
such as the ANN detailed in Table 2 can be predictive of the development of
sepsis prior to the onset
of symptoms (pre-symptomatic) and capable of a mean predictie accuracy of at
least 95% to
differentiate development of sepsis from SIRS. Biomarker signatures providing
such high predictive
accwracies have not until now been identified, and clearly the use of such
signatures could greatly
improve the power of kits, apparatuses and methods to be able to identify
patients likely to develop
sepsis, i.e. presymptomatically, and also to monitor patients with sepsis, and
potentially inform
patient treatment.
Table 1. The 266 gene biomarkers predictive of pre-symptomatic development of
sepsis, as down-
selected from the whole transcriptome using a multitude of mathematical
methods.
ACTR6 EBI2 CXORF42 SORBS3 RPL11 SLC26A8 ATP2A2
B1N1 GAS7 CLASP1 T1MM9 PPP2R2B WDR37 ZNF608
C160RF7 HIS12H4B CD2 TST NOL11 ZNF17 TBC1D8
CD247 ILiRl C140RF112 CCDC65 GZMK ANKS1A RRBP1
8
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
CLNS1A LGALS2 BCL6 NCOA3 ZNF32 CD59 RPL26
CYB561 LTA MRPL24 PDCD4 TMEM42 ElF3D PHCA
FCER1A EEF1B2 L00646483 RASGRP1 TCEA3 GYG1 NSUN7
GRB10 CTSS KLRG1 RPL18A SLC2A11 KIF1B LETMD1
,
HS.445036 CD7 HLA-DRA RPS14 SERTAD2 MMP9 IRAK3
LARP5 CACNA1E GRAMD4 RPS6 RPS20 PAG1 FAM160A2
L00646766 C120RF57 MRPS6 SIVA RPL38 RPL19 CTDP1
MRPL50 A0C2 OLFML2B SS18L2 RPL12 RPS15 ATP8B4
ADRB2 LY6E PTPRCAP TMC6 PRKCQ SLC36A1 RPS3A
BOAT L0C285176 RPL13 TTLL3 OLFM1 WWP1 TDRD9
C210RF7 IL1R2 RPL7A CD01 HLA-DRB3 ARG1 RUNX1
CD3D HLA-DMA RPS27 RPSA ZNF430 CKAP4 RPL27A
CPA3 GBP1 SH2D1A RPS15A TOMM7 EMILIN2 PHTF1
DHRS3 EOMES SMAD2 RPL30 TCTN1 HIBADH NT5DC2
FLT3LG CUTL1 THBS3 RCN2 SLC38A10 MUC1 L0C153561
GTPBP8 CD96 TP53BP2 PECI ACVR1B PFKFB2 ITGAM
1CAM2 CCL5 ZNHIT3 NDST2 C130RF23 RPL22 FBX034
LDHA C120RF62 LEPROTL1 EFCBP1 DACH1 RPS25 CYP1B1
L00652071 ASNSD1 MS4A4A ZFAND1 FBXW2 SLC41A3 ATXN7L3
MRPS27 MAFG P117 TMEM150 ITGAX ZC3H3 TRPM2
AKR1B1 L00644096 PYHIN1 SSBP2 L00647099 NAPB RPL4
BTBD11 IL32 RPL13A SLBP OPLAH LARP4B PLAC8
C50RF39 HLA-DMB RPL9 RTP4 PTPN1 HIPK2
CD3E GBP4 RPS29 RPS17 RPL5 EXOC7
CR1 EXOSC5 SIGIRR RPL32 SIL1 CMTM4
D1P2A CXORF20 SMPDL3A RPL10A UPP1 ARID5B
GALM CDKN2AIP THNSL1 POP5 TFB1M ZDHHC19
9
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
HDC CD177 TRAT1 NMT2 AMD1 SORT1
ICOS C120RF65 OSTALPHA FAM26F C220RF9 RPS8
LDOC1 ATP9A MYBPC3 ZNF195 DNAJCS RPL24
LSG1 METTL7B P2RY5 TMEM204 GOLGA1 PGD
AMPH L00646200 RARRES3 TBCC KIAA1881 NLRC4
C110RF1 I1M2A RPL18 SLC26A6 MACF1 LDLR
C90RF103 HLA-DPA1 RPS10 SELM P4HB HK3
CD6 GPR107 RPS5 RP518 RPL15 EXT1 =
CRIP2 FAM69A SIRPG RPL36 RPS13 CSGALNACT2
Table 2. The R script for a trained artificial neural network (ANN) for
calculating the predictive
accuracy for a biomarker signature selected from the 266 biomarkers to
differentiate development
of sepsis versus non-sepsis and/or SIRS, and thereby indicate the likelihood
that a patient data set
inputted into the ANN is indicative of the development of sepsis or not.
# DATA PROCESSING:
rawdata <- read.csv("Data/44 top performing genes.csv")
transposed <- data.frame(t(rawdata[,-1 ]))
names(transposed) <- c("Diagnosis", "Day",
as.character(rawdata$SAMPLE_ID[3:nrow(rawdata)]))
transposed$Diagnosis <- factor(transposed$Diagnosis, levels=c(0,1),
labels=c("No
Sepsis", "Sepsis"))
for.normalising <- transposed[ ,3:ncol(transposed)]
not.for.normalising <- transposed[ ,1:2]
medians <- apply(for.normalising, 2, median)
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
normalised.genes <- sweep(data.matrix(for.normalising), 2, medians)
normalised.data <- data.frame(not.for.normalising, normalised.genes)
input <- normalised.data[,-2]
# TRAINING/TEST SPLIT:
cases <- nrow(input)
cases.train <- sample(1:cases, round((0.7*cases), digits =0))
training <- input[cases.train, ]
test <- input[-cases.train, ]
# NEURAL NETWORK:
library(nnet)
nntraining <- nnet(Diagnosis ., data = training, size = 1, rang = 1,
decay = 0.01, maxit = 1000, Hess = FALSE, MaxNWts = 1000,
abstol = 1.0e-4, reltol = 1.0e-8, trace = TRUE,
skip = FALSE, lineout = FALSE, softmax = FALSE, censored =
FALSE,
entropy = TRUE)
#Unu.sed nnet arguments: weights = 1, Wts = 1, mask = all,
entropy = FALSE
Outcome <- test$Diagnosis
nn_Prediction <- predict(nntraining, test, type = "class")
dfAll <- data.frame(Outcome, nn_Prediction)
prediction.table <- xtabs(-Outcome+nn_Prediction, data=dfAll)
c(prediction.table[1,1] prediction.table[2,2] prediction.table[1,2),
prediction.table[2,1])/nrow(test)
11
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
Preferred biomarker signatures for use in the present invention are those that
result in a mean
predictive accuracy of at least 92% to differentiate development of sepsis
from non-sepsis, or a
mean predictive accuracy of at least 95% to differentiate development of
sepsis from SIRS which can
be identified by a simple iterative approach, inputting biomarker signatures
into a mathematical
model, such as the trained ANN detailed in table 2. The Applicant has in
particular used this
approach to identify a key biomarker signature of 44 biomarkers which can
differentiate sepsis from
SIRS with 100% predictive accuracy, and sepsis from SIRS with 97% predictive
accuracy
Accordingly, in a first aspect, the present invention provides a diagnostic
kit for predicting the
development of sepsis prior to the onset of symptoms (pre-symptomatic), said
kit comprising means
for detecting levels of a gene or gene product of each member of a biomarker
signature in a sample,
wherein the biomarker signature comprises at least 25 genes, or the products
expressed by those
genes, selected from the list of genes consisting of the 266 genes listed in
Table 1.
The biomarker signature may be capable of a mean predictive accuracy of at
least 75% to
differentiate development of sepsis from non-sepsis, and sepsis from SIRS,
though particularly
= advantageously the biomarker signature is capable of a mean predictive
accuracy of at least 92% to
differentiate development of sepsis from non-sepsis, and/or a mean predictive
accuracy of at least
95% to differentiate development of sepsis from non-sepsis.
Microarray technology was used to obtain gene expression data of samples
derived from pre-
symptomatic sepsis patients and control non-sepsis patient samples. An
unsupervised bioinformatic
approach was used to identify prognostic transcriptomic expression patterns
that characterize sepsis
before the onset of clinical symptoms. These characteristic biomarker patterns
were further
analysed and validated using quantitative RT-PCR.
12
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
The Applicant has shown that use of all 266 biomarkers provides a predictive
accuracy of more than
95% to differentiate both the development of sepsis from non-sepsis and sepsis
from SIRS. A
selection of 44 biomarkers from the 266 can potentially provide a predictive
accuracy up to 100% to
differentiate the development of sepsis from SIRS, and a predictive accuracy
of at least 97% to
differentiate the development of sepsis from non-sepsis.
The Applicant has in particular identified a biomarker signature containing 44
biomarkers, the list
consisting of those biomarkers in Table 3, which when all 44 biomarkers are
used for the prediction
is capable of up to 100% predictivity of sepsis versus SIRS. Use of a specific
list of 25 biomarkers
down-selected from these 45, as listed in Table 3, is capable of a predictive
accuracy of at least 92%
to differentiate development of sepsis from non-sepsis, and at least 95% to
differentiate
development of sepsis from SIRS. These predictive accuracies are in particular
obtainable using the
artificial neural network detailed in Table 2, though such accuracies may be
obtained using other
mathematical models, and other artificial neural networks.
Table 3. Specific (first) biomarker signature consisting of 44 biomarkers
selected from the 266 gene
biomarkers, and a further down-selected list of 25 biomarkers.
44 Gene Biomarker Signature Down-selected 25 Gene Biomarker Signature
ACTR6, B1N1, C160RF7, CD247, CLNS1A, ACTR6, BIN1, C160RF7, CD247, CLNS1A,
CYB561, FCER1A, GRB10, HS.445036, LARP5, CYB561, FCER1A, GRB10, HS.445036,
LARP5,
L00646766, MRPL.50, ADRB2, BOAT, C210RF7 L00646766, MRPL50, ADRB2, BOAT,
C210RF7
CD3D, CPA3, DHRS3, FLT3LG, GTPBP8, 1CAM2, CD3D, CPA3, DHRS3, FLT3LG, GTPBP8,
ICAM2,
LDHA, L00652071, MRPS27, AKR1B1, BTBD11, LDHA, L00652071, MRPS27, AKR1B1
C5ORF39, CD3E, CR1, 0IP2A, GALM, HOC,
ICOS, LDOC1, LSG1, AMPH, C110RF1,
C9ORF103, CD6, CRIP2, EBI2, GAS7, HIST2H4B,
ILiRl
13
CA 02938761 2016-08-04
WO 2015/121605 PCT/GB2015/000004
A further list of 45 gene biomarkers selected from the list of 266 as detailed
in Table 4, was also
shown to have a predictivity of higher than 92% to differentiate sepsis from
non-sepsis, especially
with a specific down-selected list of 25 biomarkers.
Table 4. Further (second) specific biomarker signature consisting of 45
biomarkers selected from the
266 gene biomarkers, and a further down-selected list of 25 biomarkers.
45 Gene Biomarker Signature Down-selected 25 Gene Biomarker Signature
ATP9A, C160RF7, C50RF39, C9ORF103, C160RF7, C50RF39, C90 RF103, CD177,
CACNA1E, CD177, DHRS3, EEF1B2, FCER1A, FCER1A, GAS7, L0C285176, MYBPC3, NDST2,
FLT3LG, GAS7, GRB10, HLA.DMA, HS.445036, EBI2, RPL13A, RPL18A, RPL32, RPL36,
RPL9,
IL1R1, IL1R2, L0C285176, MYBPC3, NCOA3, RPS20, RPS29, RPS6, SIGIRR, TCEA3,
TCTN1,
NDST2, RPL10A, EBI2, L00646483, RPL13A, TIMM9, TOMM7, ZFAND1, ZNHIT3
RPL18, RPL18A, RPL32, RPL36, RPL9, RPS20,
RPS29, RPS6, SIG IRR, SLBP, SLC26A6,
SMPDL3A, SORBS3, TCEA3, TCTN1, THBS3,
THNSL1, TIMM9, TOMM7, ZFAND1, ZNHIT3
These further (second) two biomarker signatures of 45 and 25 have 11 and 6
biomarkers,
respectively, in common with the first 44 gene biomarker signature. The
Applicant has also
evaluated 14 further combinations of 44 biomarkers in detail, of which all
combinations have a mean
predictive accuracy of at least 75%, but of which 6 combinations have a mean
predictive accuracy of
at least 92%. These signatures are listed in Table 5. These six signatures
have at least 5 genes in
common with the first 44 gene biomarker signature in Table 3, and thus in one
embodiment any
combination of 44 biomarkers or 25 biomarkers selected from the 266 may
comprise at least 5
biomarkers from the first 44 in order to provide a mean predictive accuracy of
at least 92%.
In another embodiment, the at least 25 genes comprises at least 11 genes
selected from the first 44
gene biomarker signature. in a third embodiment the at least 25 genes
comprises at least the
complete first 25 gene biomarker signature (listed in Table 3). In a fourth
embodiment, the
biomarker signature of the present invention comprises the complete first 44
gene biomarker
signature (listed in Table 3).
14
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
=
Table 5. Six combinations of 44 biamarkers selected from the list of 266
biomarkers which have a
mean predictive accuracy of sepsis vs non-sepsis of at least 92% by using the
artificial neural network
detailed in Table 2
1 2 3 4 5 6
CYB561 ACTR6 C160RF7 CD6 BCL6 EBI2
GRB10 ' BIN1 LARP5 CD247 CLNS1A CD247 ,
BTBD11 L00646766 C210RF7 CLNS1A CYB561 FCER1A
CD3E ICAM2 GTPBP8 C50RF39 FCER1A ICAM2
EBI2 L00652071 LDHA GALM C210RF7 C50RF39
CD7 ICOS MRPS27 ICOS FLT3LG EEF1B2
L0C285176 CD7 BTBD11 A0C2 CTSS C120RF57
HLA-DMA IL1R2 HDC IL1R2 CD96 A0C2
C120RF62 ASNSD1 CRIP2 CUTL1 CCL5 EOMES
' ASNSD1 MAFG URI. CDKN2AIP HLA-DMB i IL32
I
GPR107 GBP4 CACNA1E I1M2A CDKN2AIP ' GBP4
BCL6 CXORF20 L0C285176 CLASP1 GPR107 ; CD177
MRPL24 HLA-DPA1 I HLA-DMA =C140RF112 CXORF42 HLA-DPA1
RPL7A CXORF42 1 ASNSD1 BCL6 CLASP1 , C140RF112
RPL13A ,
MRPL24 L00644096 L00646483 RPS27 1 BCL6 .
RPS5 PTPRCAP = IL32 RPS27 SMAD2 MRPL24
CCDC65 RPL7A ; F= AM69A P117 ZNHIT3 RPS27
NCOA3 PYHIN1 MRPL24 RPL9 RPL13A P117
;
RASGRP1 RASGRP1 ' H= LA-DRA = RPS10 SMPDL3A NCOA3
I
RPS6 RPL30 MRPS6 SORBS3 TMEM150 , RASGRP1
,
NMT2 EFCBP1 RPS27 TST FAM26F RPS6
ZNF32 TMEM150 ; PYHIN1 RPL18A TBCC ; PECI
SERTAD2 RPL32 ' S= IGIRR 5S18L2 TCEA3 ' EFCBP1
' RPL38 ZNF195 SMPDL3A CD01 ITGAX TMEM150
SLC38A10 TMEM 204 ' P= 2RY5 RPS15A PTPN1 NMT2 .
ACVR1B SELM RARRES3 TMEM150 TFB1M SLC26A6
;
P4HB RPS18 TTLL3 RPL32 AMD1 RPL11
SLC26A8 PPP2R2B 1 RPS15A SLC26A6 KIAA1881 PPP2R2B
WDR37 OLFM1 I RPL36 RPS20 CD59 ZNF32
PAG1 TCTN1 TMEM42 HLA-DRB3 KIF1B ACVR1B
RPL19 DACH1 HLA-DRB3 TCTN1 RPL19 TFB1M
SLC41A3 ITGAX FBXW2 P4HB NAPB P4HB .
LARP4B TFB1M L00647099 RPL15 ZDHHC19 RPL15
ZDHHC19 GYG1 ZNF17 RPS13 EXT1 ZNF17
SORT1 MMP9 CD59 WDR37 ZNF608 E1F3D
NLRC4 PAG1 KIF1B ANKS1A TBC1D8 MMP9
EXT1 RPS15 SLC36A1 KIF1B RRBP1 SLC36A1
ATP2A2 CKAP4 PFKFB2 MMP9 ATP8B4 NAPB
,
CA 02938761 2016-08-04
WO 2015/121605 PCT/GB2015/000004
ZNF608 RPL22 SLC41A3 EXOC7 RPS3A ARID5B
RkBP1 ZDHHC19 EX0C7 CMTM4 RPL27A HK3
TDRD9 SORT1 HK3 RPL24 PHTF1 CSGALNACT2
RUNX1 NLRC4 ATP2A2 CSGALNACT2 FBX034 FAM160A2
L0C153561 CSGALNACT2 LETMD1 ATP2A2 CYP1B1 RPS3A
ITGAM RRBP1 ITGAM FAM160A2 RPL4 RPL27A
In a second aspect, the present invention provides a method for analysis of a
biological sample from
an animal to predict and monitor the development of sepsis, especially prior
to onset of symptoms,
comprising monitoring, measuring and/or detecting the expression of all
biomarkers in the selected
biomarker signature (list of biomarkers), and evaluating/assessing data
produced from the
monitoring, measuring and/or detecting to predict and monitor the development
of sepsis.
The method is preferably capable of differentiating sepsis from non-sepsis,
with high levels of
accuracy, such as > 75%, but preferably >90% accuracy, or as high as >92%, and
also potentially
sepsis from SIRS with the same predictivities.
The animal may be a human, and the biological sample is most likely a blood or
serum sample.
The diagnostic kit of the invention provides the means for detecting levels of
a gene or gene product
of the genes comprising the biomarker signatures described above. Although
gene expression may
be determined by detecting the presence of gene products including proteins
and peptides, such
processes may be complex. In a particular embodiment, the means comprises
means for detecting a
nucleic acid and in particular DNA, or a gene product which is RNA such as
mRNA.
The monitoring, measuring or detecting may use any suitable technique,
including use of recognition
elements, or microarray based methods Thus in a particular embodiment, the kit
of the invention
comprises microarray on which are immobilised probes suitable for binding to
RNA expressed by
each gene of the biomarker signature.
16
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
In an alternative embodiment, the kit comprises at least some of the reagents
suitable for carrying
out amplification of genes or regions thereof, of the biomarker signature.
In one embodiment the monitoring, measuring or detecting the.expression of
biomarkers uses real-
time (RT) polymerase chain reaction (PCR). In such cases, the means may
comprise primers for
amplification of said genes or regions thereof. The kits may further
comprise labels in particular
fluorescent labels and/or oligonucleotide probes to allow the PCR to be
monitored in real-time using
any of the known assays, such as TaqMan, LUX, etc. The kits may also contain
reagents such as
buffers, enzymes, salts such as MgCl etc. required for carrying out a nucleic
acid amplification
reaction.
The method of the second aspect is advantageously computer-implemented to
handle the
complexity in monitoring and analysis of the numerous biomarkers, and their
respective
relationships to each other. Such a computer implemented invention could
enable a yes/no answer
as to whether sepsis is likely to develop, or at least provide an indication
of how likely the
development of sepsis is.
The method preferably uses mathematical modelling tools and/or algorithms to
monitor and assess
expression of the biomarkers both qualitatively and quantitatively. The tools
could in particular
include support vector machine (SVM) algorithms, decision trees, random
forests, artificial neural
networks, quadratic discriminant analysis, and Bayes classifiers. In a
preferred embodiment the data
from monitoring all biomarkers in the biomarker signature is assessed by means
of an artificial
neural network, for example the trained artificial neural network detailed in
Table 2.
=
In one embodiment of the second aspect the method is a computer-implemented
method wherein
the monitoring, measuring and/or detecting comprises producing quantitative,
and optionally
qualitative, data for all biomarkers, inputting said data into an analytical
process on the computer, -
using at least one mathematical method, that compares the data with reference
data, and producing
17
CA 02938761 2016-08-04
WO 2015/121605 PCT/GB2015/000004
an output from the analytical process which provides a prediction for the
likelihood of developing .
sepsis, or enables monitoring of the sepsis condition. The reference data may
include data from
healthy subjects, subjects diagnosed with sepsis, and subjects with SIRS, but
no infection.
The output from the analytical process may enable the time to onset of
symptoms to be predicted,
such as 1, 2, or 3 days prior to onset of symptoms, and consequently may be
particularly valuable
and useful to a medical practitioner in suggesting a course of treatment,
especially when the choice
of course of treatment is dependent on the progression of the disease. The
method may also enable
=
monitoring of the success of any treatment, assessing whether the likelihood
of onset of symptoms
decreases over the course of treatment.
In a third aspect, the present invention provides an apparatus for analysis of
a biological sample
from an animal td predict and monitor the development of sepsis comprising
means for monitoring,
measuring or detecting the expression of all biomarkers in the biomarker
signature as described
above, such as RT-PCR using reagents specific to the biomarkers in the
biomarker signature, and
means for analysis of data produced from the means for monitoring, measuring
or detecting, such as
a computer comprising an appropriate mathematical model to analyses the data,
such as an
artificial neural network, and means for providing an output from the analysis
which output provides
a prediction of the likelihood of an animal having sepsis, or an output to
enable monitoring of sepsis,
which output could also be provided by an appropriately programmed computer.
The present invention will now be described with reference to the following
non-limiting examples
and drawings in which
Figure 1 is a display of Bioanalyzer results for a randomised selection of RNA
sample preparations;
Figure 2 is an illustration depicting the rationale for sample selection, and
especially the selection of
control samples, and the matching with sepsis patient samples;
18
CA 02938761 2016-08-04
WO 2015/121605 PCT/GB2015/000004
Figure 3 is a graph detailing the predictive accuracies for sepsis versus non-
sepsis of the 15
combinations detailed in Table 24; and
Figure 4 is a graph and table indicating the predictive accuracies for
different subsets of biomarkers
selected from the 266 biomarkers in Table 1.
Example ¨ Development of a predictive panel of pre-symptomatic biomarkers for
sepsis
The aim of this program of work was to develop a predictive panel of pre-
symptomatic biomarkers
for sepsis, through comprehensive analysis of the host transcriptome, sourced
from blood samples
from human patients collected prior to the clinical onset of sepsis, and to
develop biomarker
signatures that may indicate whether and.when clinical symptoms will arise
following infection. In so
doing it would yield a suitably powered bioinformatic model for
differentiating sepsis patients from
control patients based on transcriptomic biomarker signatures. In turn, this
will assist in the
development of RT-PCR methods for sepsis prediction, where this capability
should provide timely
diagnosis and treatment of infection when medical countermeasures are most
effective.
We used microarray technology to obtain gene expression data of samples
derived from pre.-
symptomatic sepsis patients and control non-sepsis patient samples. An
unsupervised bioinformatic
approach was used to identify prognostic transcriptomic expression patterns
that characterize sepsis
before the onset of clinical symptoms. These characteristic biomarker patterns
were further
analysed and validated using quantitative RT-PCR on the Fluidigm BioMarkTm
real-time PCR array
platform.
Through significance testing a final panel of 266 biomarkers was derived. The
full panel and subsets
of this was then used in a number of statistical models to determine
discrimination between sepsis
and non-sepsis patients. The artificial neural network gave the highest
predictive accuracy, with 44
.biomarkers being the optimal subset.
19
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
Technical Summary
Acquisition and Storage of patient samples ¨Patients were admitted to the
study if they gave
informed consent, were between 18 and 80 years of age and undergoing a
procedure that, in the
clinician's opinion, had a risk of causing infection and ultimately sepsis.
Typically these were
abdominal and thoracic surgeries. However, other surgical procedures were
permitted and included,
with one extensive maxillofacial procedure resulting in sepsis in one case.
Patients were excluded if
they were either pregnant, infected with a known pathogen (HIV, Hepatitis A, B
or C),
immunosuppressed or withdrew consent to take part in the study at any time
during their stay. All
patients received the normal standard of care once enrolled.
Blood samples were collected according to a protocol. Briefly, two 4 ml
aliquots of patient blood
were collected into sterile EDTA vacutainers and then immediately transferred
into RNAse-free vials
containing 10.5 ml of RNAlater (a RNA stabilization media) (Life
Technologies, USA). These were
then stored at -20 C and eventually transported on dry ice. In addition 4m1
of patient blood was
, collected into a serum separation tube, spun, separated and stored at -20 C.
Blood collection
occurred once between 1 and 7 days before surgery and then once daily on each
day post-surgery.
Post-operative blood collection was stopped after the patient was discharged
from hospital, or after
7 days post-surgery, or once sepsis had been confirmed by the clinician.
Additional patient
information (e.g. daily patient metrics, type of surgery and microbiology
results) was captured using
a bespoke database provided by ItemTracker, UK.
We recruited 2273 elective surgery patients into the study with 1842 patient
time courses in
storage; 72 of these patients went on to develop sepsis. The incidence of
sepsis in our patient cohort
is therefore 3.91%. Over 600 of the remaining patients met the criteria set
for SIRS (2 out of the
following four symptoms: increased/decreased temperature; increased heart
rate; increased
ventilation rate, increased/decreased white blood cell count). However, many
of these "SIRS"
patients had very transient changes in symptomology. We suspect that the 438
patients, as
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
identified by the clinical staff at the centres, are more reflective of the
number of patients with
prolonged SIRS.
This patient recruitment was sufficient to satisfy the requirement for 30
sepsis patient time courses
(plus matched non-sepsis patient controls) to be used for biomarker discovery
during 2011 as well as
a further 40 sepsis patients time courses (plus matched non-sepsis patient
controls) for the
validation of biomarkers during 2012.
An initial batch of 61 SIRS patient blood samples was analysed. Of these
samples, 2 were identified
as having microbial DNA present in the blood (one patient had E. coil and the
other had S. aureus).
These patients were re-classified as belonging to the patient cohort that goes
on to develop sepsis.
The remaining 59 patients had undetectable levels of microbial DNA present in
their blood. This
indicated that these patients truly belonged to the SIRS patient group. The
biomarker signatures
from both groups of patients were then used in a biomarker discovery analysis
that provided a
biomarker signature for the pre-symptomatic diagnosis of sepsis in elective
surgery patients. A
second batch of 190 patient samples containing samples from patients who
developed either SIRS or
sepsis, as well as samples from patients who did not develop any post-surgical
symptoms (post-
operative controls) were again sent for analysis using the Sepsitest. All post-
operative control
patient samples were confirmed as negative by the Sepsitest. Additionally all
the patient samples
isolated from sepsis patients with blood borne infections were also identified
correctly. All of the
SIRS patients were confirmed as not septic.
RNA extraction from stabilization media ¨ The RNA from all patient samples
selected for further
microarray and Fluidigm array analysis was extracted using the RiboPureTM -
Blood kit (Life
Technologies, USA), followed by treatment with TURBO DNA-freeTM (Life
Technologies, USA). In order
to give confidence in the quality of sample preparation the quality of all RNA
products were assessed
on the Agilent 2100 BioAnalyser (Agilent USA) using the Agilent BioAnalyser
RNA 6000 Nano kit
(Agilent USA). Having regard to Figure 1 a qualitative indication of the 100s
of RNA samples using 12
21
CA 02938761 2016-08-04
WO 2015/121605 PCT/GB2015/000004
randomly selected samples is shown using the Agilent 2100 BioAnalyser (Agilent
USA). The double
banding in each lane indicates good quality RNA with little degradation.
Further quantitative
measures of the quality and quantity of RNA preparation, like the RNA
integrity number (RI N), and
concentration of RNA in each preparation indicated that RNA isolation
protocols were fit for purpose
(Table 6).
Table 6. Quantification and integrity of typical RNA samples.
Results
Did the sample
Patient RIN Concentration pass QC
Total concentration
sam pie Result (kg/m I) (RIN>7.0/RNA
>2.0)?
1 8.0 49 4.41 Yes
2 7.0 23 2.07 Yes
3 7.0 36 3.24 Yes
4 7.5 34 3.06 Yes
8.5 28 2.52 Yes
6 8.9 30 2.70 Yes
7 8.4 50 4.50 Yes
8 7.3 30 2.70 Yes
9 7.3 45 4.05 Yes
7.9 48 4.32 Yes
11 7.5 38 3.42 Yes
12 - 7.8 47 4.23 Yes
Over 99% of RNA samples achieved a RIN of 7 or above with a yield of 2 kg or
above. This was
sufficient quality and quantity to undertake microarray and quantitative RT-
PCR analyses on these
samples. On the rare occasions when the sample preparations gave an
unsatisfactory yield, the
22
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
process was repeated four times and the product sent for quantitative RT-PCR
only (i.e. there was
sufficient RNA to produce cDNA and subsequently undertake PCR).
The selection of those patients who went on to develop sepsis and those that
did not was the
responsibility of the Principal Investigators (Pis) at each centre. They were
all consultant intensive
care clinicians with many years experience in the clinic with over 265 peer
review publications
between them. Two of the four Pis from the four centres hold prominent
advisory roles to journals
and funding bodies across Europe and the USA. Selections by the clinicians
were double-checked by
the project team to ensure that all patients met the previously agreed
criteria for the definition of
sepsis. Pen-operative antibiotic use was minimal, with only one dose of a
broad-spectrum antibiotic
given in 85.7% of sepsis patient cases, prior to sepsis diagnosis. The
remaining patients received
daily doses of antibiotic but still developed clinical evidence of sepsis.
Under clinical guidance we
have included these patients in the study as they developed sepsis in spite of
treatment, although it
is possible that such treatment may have influenced microbial culture results.
The range of
infectious agents that resulted in sepsis in the study was quite broad and is
listed in Table 7.
Table 7. Infectious agents isolated from sepsis patients in phase I and II of
the study.
Phase 1 - Discovery Phase ll - Validation
Escherichia coil Blood Serratia Haemophilus Enterobacter species
= influenzae
Pseudomonas Candida species Escherichia col/ Stenotrophomonas
aeruginosa maltophilia
Klebsiella species Proteus species Klebsiella species
Gram negative bacilli
coliforms Clostridium difficile Pseudomonas Candida
species
= aeruginosa
Streptococcus Streptococcus co I 'forms
pneumoniae pneumoniae
Staphylococcus aureus Staphylococcus aureus Moraxella
catarrhalis
23
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
Unidentified Gram Streptococcus species Coagulase-
negative
negative bacteria Staphylococcus (CNS)
Stenotroph omon as Enterococcus species CDT
maltophilla
Once patients were confirmed as septic, a comparator group was selected that
matched each sepsis
patient's age, sex and procedure. These patients did not develop SIRS as a
result of their surgery.
Having regard to Figure 2, the rationale for comparator selection is
illustrated as well as which
patient samples were analysed and how the time frames for patient samples that
are taken at
different days post-surgery were standardized. It should be noted that the
main analytical effort
' was focused on the 3 days prior to the diagnosis of sepsis as these are
most likely to yield useful pre-
symptomatic biomarker signatures. The time course of the development of sepsis
in a patient is
indicated by the Sepsis patient #1 bar. From the large number of patients who
do not go on to
develop sepsis following surgery a suitable age/sex/procedure matched control
is identified and
used as a comparator. In this example the day of diagnosis of sepsis is day 7
post-infection.
Therefore the 3 days before sepsis diagnosis are days 4, 5 and 6 post-surgery.
In terms of pre-
symptomatic diagnosis this may also be noted as Days -3, -2 and -1. In order
to provide a robust and
. relevant post-operative comparison for each of the 3 days before sepsis
diagnosis, the equivalent
post-operative blood sample was used. In this case the blood samples taken
from days 4, 5 and 6
post surgery were used for comparison, acting as Day -3, -2 and -1 controls.
The process of matching
the pre-symptomatic blood samples of patients who went on to develop sepsis
with their most
appropriate post-operative comparators was then repeated in Phase I and II of
the study so that the
time courses of 30 and 40 patients who go on to develop sepsis were compared
to 30 and 40 post-
.-
operative comparator patients, respectively.
In addition to the non-sepsis comparator group, further controls were provided
through exploitation
of each patient's pre-operative sample as well as samples from pgients that
developed SIRS and not
24
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
sepsis. This ensured that any changes observed in the transcriptomes of sepsis
patients were a direct
result of infection acquired during surgery. A summary of patients used in
both phase I and II of the
study is given in table 8. It should be noted that antibiotic use was dictated
on a case-by case basis
and under the discretion of the clinician. The study protocol did not
influence patient management;
ethically we were unable to dictate medical countermeasure use during this
study.
Table 8. Summary of patient ages, gender, delay for sepsis and types of
surgery used in phase I and II
Phase I Phaseil
(Discovery) (Validation)
Sepsis I Controls Sepsis Controls
= I n=430 riF30 n=-40 m-40
Age 63148-811 61 152-791
64128-791 644-80
Gender 14/16 14/16 11/20 11/29
(female/male)
Delay for ! 3.511-81 NA 4.7511-91 NA
sepsis I
Surgery Type inc"dc" Ilimrd tic Ir 'Thoracic, lhoradc,
abdxnnaI bdominal abdominal or
abdominal or
maxiloracsal imndlotacial
Microarray analysis (Phase I Biomarker discovery) - Illumine Human HT12y4
Beadarrays were run
on the samples from the 60 phase I patients (30 x sepsis & 30 x comparator),
80 phase II patients (40
x sepsis & 40 x comparator) and 40 Phase II SIRS patients. This corresponded
to 192 transcriptomes
analysed during Phase I and 433 transcriptomes analysed during phase II of the
study. Data were
= collected for 30 sepsis patients and 30 age, sex and surgery matched
controls (or baselines).
Microarray data were collected from 192 blood samples. These represented 4
different time points
corresponding to pre-operation and 1, 2 and 3 days prior to the onset of
sepsis. Samples were taken
for each paired baseliner based on the corresponding day of onset for the
sepsis 'sample,
summarized in table 9.
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
Table 9. The number of samples used during Phase I of the study.
Comparator Sepsis
Pre-op 30 30
Onset Day -1 30 30
Onset Day -2 21 21
Onset Day -3 15 15
The Illumina Human v4 chip contains 48,804 probes mapping to over 27,000
reference sequence
numbers. Each probe is 50 base pairs long providing a high degree of
specificity for each gene. For
each sample globin-reduced RNA (GlobinClearTM, Life Technologies, USA) was
prepared from total
RNA. RNA integrity was measured using a Bioanalyzer 2100 (Agilent, USA) and
concentration was
assessed using a NanoQuantTM (Tecan, USA). cRNA was prepared by amplification
and labelling using
the Illumina TotalPrep" RNA Amplification Kit (Life Technologies) and
hybridized to Human HT-12
v4 Beadarrays (lllumina , USA). The Illumina HighScanHO.TM then imaged each
chip with resulting
intensities indicating the expression level of each probe's corresponding
gene. Background
subtracted data was then generated using GenomeStudioTM Software (Illumina ,
USA).
A variety of preliminary or exploratory analyses on the micrcarray data for
Phase I were undertaken
= to determine whether:
1. There were any batch processing effects on the data.
2. There was a difference between pre- and post-surgical transcriptomes.
3. There was a gross difference between the transcriptomes of patients who
went on to
develop sepsis and their baseliner comparators.
= Batch Effects
26
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
3D Principal Component Analyses (PCA) was used to examine whether the day of
hybridization of
sample had an impact on the transcriptomes of patients in the study.
The data indicated that samples hybridized on different days did not segregate
into distinct groups.
This suggested that there was no batch effect amongst the samples according to
day of
hybridization.
Pre- and Post-Surgical Transcriptomes
3D PCA was also used to indicate whether there were any differences in the
transcriptomes of pre-
and post-surgery patients. The analysis indicated that the transcriptomes of
pre-surgery patients
cluster together. This suggests that they are more similar to each other than
to the transcriptomes
of post-surgery patients. Additionally, the transcriptomes of the entire post-
surgery patient samples
cluster away from the pre-surgery transcriptomes, suggesting that they too
have more in common
with each other than with the transcriptomes of pre-surgery patients.
Differences between patients who go on to develop sepsis and their comparators
Like PCA, Hierarchical Clustering is a tool used for unsupervised analysis of
data sets. It was used to
describe the transcriptomes of both patient groups through use of a heat map.
Hierarchical
clustering involves the
re-ordering of genes in the dataset so that similar transcriptome
patterns (expression profiles) are put next to each other. In effect it is a
tool that helps identify
samples that are related to each other.
Preliminary inspection of the heat map indicated that the pre-surgery samples
as well as the
transcriptomes of baseliner patients on comparative Days -1, -2 and -3 are
clustered near each
other, generally at the top half of the heat map. In contrast the
transcriptomes of patients who go
on to develop on Days -1, -2 and -3 seem to cluster near each other near the
bottom of the heat
map. This suggests that there is a difference in the transcriptomes of
patients who go on to develop
sepsis and their baseliner comparators.
27
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
Following the collection of transcriptomic data from 192 samples, further
analysis was required to
elucidate key biomarkers whose expression was significantly different between
the two patient
groups. These host response genes would form the basis of a biomarker
signature that could be used
to indicate an individual who was likely to deverep the symptoms associated
with life-threatening
disease.
Biomarker Discovery ¨ Microarray (Phase I) ¨
Data Pre-Processing
There were three main steps in the data pre-processing:
1. Log transform - a loge transform was performed on the transcriptomic
data to comply with
assumptions of normality required for further analysis
2. Pre-surgery subtraction ¨ in order to obtain the log expression for each
sample due to the
response to surgery, all samples were normalised to the difference compared
with pre-
surgery expression levels.
3. Median subtraction - This was important within each gene probe to
account for systematic
variation.
Multiple Hypothesis Testing for Determination of Genes of Interest
We used multiple t-tests to discern evidence for significant differences in
gene expression (below
the threshold p-value assigned), for the 3 days before sepsis diagnosis. The
analyses indicated that
452 genes were significantly different between the two patient groups on all 3
days before sepsis
diagnosis. We also determined that there was evidence for significant
differences between the two
groups on each day before sepsis diagnosis. The expression of 91, 1022 and 938
genes had evidence
for significant differential on Days 3, 2 and 1 before sepsis diagnosis,
respectively.
28
CA 02938761 2016-08-04
WO 2015/121605 PCT/GB2015/000004
We then took a similar approach implementing the significance analysis of
microarray (SAM) analysis
method (Tusher VG , Tibshirani R, Chu, X. 2001. Significance analysis of
microarrays applied to the
ionizing radiation response. Proc Nat Am Sci 98:5116-5121) as published by R.
Tibshiriani at Stanford
University. This method is commonly used for microarray analysis. We felt this
alternative was
worth exploring since they were likely to provide an independent validation of
the first findings and
therefore confidence in the eventual selection of biomarkers for pre-
symptomatic diagnosis.
Expression analysis and subsequent SAM
Expression analysis was used as a test for the difference in gene expression
between groups of
subjects based on a known response variable, such as the onset of sepsis.
Response variables were
generated for 4 different tests, defined using the patient groups in Table 10.
Table 10. Patient categories used for expression analysis
Comparator Sepsis
Onset Day -1 B1 Si
Onset Day -2 B2 52
Onset Day -3 B3 53
The four tests were:
1. - S1+52+S3 vs. B1+B2+B3
2. S1 vs. B1+132+83
3. S2 vs. B1+62+63
4. S3 vs. B1+B2+B3
The SAM package in the R statistical language software was used to perform the
expression analysis
for each of the 4 tests described above. For each gene i an expression
statistic d is calculated from
29
CA 02938761 2016-08-04
WO 2015/121605 PCT/GB2015/000004
=
the average difference in the expression between the two response groups. This
average different r
is scaled by the standard deviation s, according to the following equation:
---; ¨ 1, 2, p
This statistic has a natural ordering based on magnitude as it measures the
strength of the
relationship between gene expression and the response variable.
In order to determine which genes are significantly expressed, SAM uses
permutation analysis to
estimate the local false discovery rate (FDR) at a variety of different test
statistic thresholds (delta).
The FDR is fixed at 1% for each test to ensure a consistent risk of falsely
identifying significant genes.
However, the change in FDR as the threshold changes is dependent on the
distribution of expression
statistics, and there is often a minimum FDR for any given range of Delta.
For example, Table 11 shows the estimated false discovery rate for the
diagnosis of sepsis 2 days
prior to onset of symptoms.
Table 11. 90th Percentile for the estimated false discovery rate for range of
delta values for sepsis at
Day -2.
delta number of 90th % FDR
genes called
1.4 158 0.015927
1.41 145 0.016795
1.42 139 0.01752
1.43 129 0.013215
1.44 123 0.0132
1.45 118 0.013759
CA 02938761 2016-08-04
WO 2015/121605 PCT/GB2015/000004
1.46 114 0.007833
1.47 109 0.007448
1.48 86 0.009439
1.49 75 0.010824
1.5 72 0.011275
The 90th percentile is used as an upper bound on the likely false discovery
rate (FDR). A FDR of 1%
(0.01) was deemed an acceptable risk but it is clear from the above table that
this increases again as
we increase the delta beyond 1.47. Since this also satisfies the FDR<1% delta
of 1.47 was chosen to
identify 109 significant genes in total for this diagnosis.
As a consequence of this approach we identified 458 genes whose expression was
different between
the 2 patient groups for all 3 days prior to the onset of sepsis. In addition
the expression of 167, 179
and 226 genes was found to be specifically differentially expressed between
the patient groups on
Days -3, -2 and -1, respectively. Unique to this test, were 163 of the total
number of genes, 18 for
Day-1, 12 for Day-2, and 51 for Day-3.
Models
Any biomarkers selected for further validation must be mathematically modelled
so that their
performance can be assessed both qualitatively and quantitatively. It is
however important to
determine a useful model by:
= ensuring any assumptions are fit for the purpose of the analysis,
= determining precedent for the choice of model, unless the analysis is a
new approach,
= undertaking an appropriate sensitivity analysis to determine the
limitations of the model,
31
CA 02938761 2016-08-04
WO 2015/121605 PCT/GB2015/000004
= correlating the model itself with scientific rationale.
Within the field of biostatistics and bioinformatics, there are many analysis
pathways and algorithms
(or models) available. It would be impossible to use all of these approaches
to help select and
validate the most appropriate biomarkers for pre-symptomatic diagnosis of
sepsis. In the context of
this project the criteria for the analyses used is described in Table 12,
where a number of
approaches are gradually discounted due to likely model requirements.
Table 12. Down-selection of models used for biomarker selection and analysis.
Model Requirements Potential Models
Data are non linear Kernel based PCA, Support Vector Machines
(shown in Lukaszewski et al. 2008) (SVM), Quadratic regression, Decision
Trees,
Random Forests, Artificial Neural Networks
(ANN), Quadratic Discriminant Analysis (QDA),
Naive Bayes classifier, K-Nearest Neighbour
Analysis (KNN) and Factor analysis.
Solution needs to be resolved quickly Kernal based PCA, SVM, Quadratic
regression,
Random Forests, ANN, QDA, Bayes classifier,
KNN, factor analysis.
Due to potential use, model needs to learn and From list above, quadratic
regression, factor
adapt to new variation in data. analysis and KNN will not fit this
criterion.
Provide a classification. PCA will not provide a classification
algorithm,
generally used for exploratory analysis.
What's left? SVM, Decision Tress, Random Forests, ANN,
QDA
and Bayes classifier
Several models were generated to determine the best fit.
Analysis 1
Support vector machines (SVMs), Random Forests and Differential analysis were
used to identify
genes for down selection for targeted ciRT-PCR on the Fluidigm array. Survival
analysis, which makes
use of longitudinal information, was also used. All analysis was carried via R
2.14.0 and relevant R
packages.
32
CA 02938761 2016-08-04
WO 2015/121605 PCT/GB2015/000004
SVMs and random forests are supervised machine learning algorithms commonly
used as
bioinformatic tools. The ease of variable (gene) selection provided by these
methods was a key
factor in adoption of the methods. A SVM uses observations to find a hyper-
plane that best
separates two labelled groups. The Random Forest algorithm is an ensemble
classifier, which uses
bagging to create many independent classification trees. Each tree has its own
training dataset, a
subset of original observations is approximately 66% of the samples, with the
remaining samples .
used to determine that tree's accuracy. Each classification tree was created
using a random subset
of variables allowing genes to be ranked based on a measure of how strongly
they influence tree
accuracies called the mean Gini coefficient. Random forests are probabilistic
classifiers yielding a
value between 0 and 1 indicating the probability that a given sample belongs
to a particular class.
Survival analysis was also employed to find probes that play a role in the
development of sepsis. The
method's main attraction is that it allows microarray data from different days
to be incorporated in
to the model whereas the machine learning approaches use only one point to
find important genes.
However, the technique was not developed for prediction and creates a separate
model for each
gene. Similar to the t-statistic from standard differential expression, a test
statistic is computed for
each gene that is then used to rank genes.
The SVM, Random Forest, differential expression, and survival analysis
approaches showed
significant overlap in gene selection when analysing Phase I microarray data,
as detailed in Table 13.
The top 531 genes prior to sepsis found by random forest and SVM- and survival
analysis using all
post operation time samples overlapped greatly with genes found by
differentially expressed genes.
All overlaps were highly significant (p-value < 0.001) and the numbers of
overlapping genes are given
in Table 13.
33
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
Table 13. Differential expression analysis of expressed genes ¨ overlapping
genes between different
models.
Method SVM Survival Differential Expression
Analysis
Random Forest 154 140 59
SVM 255 98
Survival Analysis 91
Prediction rates using this data were then calculated through Random forests
and support vector
machines (SVMs). Individual days (pre-op, day-1, etc.) were split into sepsis
and control. For day-1,
day-2, and day-3 predictions were made with pre-op normalization by division,
by subtraction, and
without normalization. Averages across days were also considered, for example
day-1 and day-2
averaged for each patient. In order to maintain the assumption that samples
were independent, no
days were grouped together into a meta group (in either sepsis or control).
For survival analysis all
data were used under the false assumption that each time point was equally
spaced (time between
pre-op and day-3 was variable).
Random Forest prediction of Sepsis
Random forests are composed of many simple tree classifiers, each based on a
different random
subset of samples for training and testing each tree (70% vs. 30%) thus
allowing for accurate
estimates of error rates. Below are the sensitivity and specificity for
predicting sepsis in each time
grouping. Note that Day ¨2 and Day ¨3 (D-2 and D-3) have smaller sample sizes.
Normalizing by pre-
op (by division of unlogged data) and combining days showed that averaging
Days ¨1 and 2 yields
the most accurate results. Normalization by subtraction (not shown) performed
no better than
normalization by division.
Table 14. Performance of identified genes using Random Forests
34
CA 02938761 2016-08-04
WO 2015/121605 PCT/GB2015/000004
Filtered Sensitivity Specificity Error Rate
Pre-op 0.667 0.643 0.345
D-3 0.786 0.8 0.207
D-2 0.95 0.857 0.0976
D-1 0.778 0.786 0.218
To provide a comparison to random forests a Support Vector Machine (SVM) with,
a Wilcoxon test to
allow for non-normally distributed probes was employed (Table 15). We
concentrated on the Day-1
and Day-2 average given that this performed the best and used 5 fold cross
validation using 20% for
testing. Standard errors are shown in parentheses.
Table 15. Performance of identified genes using Support Vector Machines.
Sensitivity Specificity Error Rate
D-1n2 ave 0.8 (0.082) 0.69 (0.027) 0.253 (0.044)
D-1n2 ave Filtered 0.853 (0,037) 0.807 (0.11) 0.164 (0.06)
Both approaches demonstrated acceptable, but not outstanding, differentiations
between the two
patient groups. This suggested that other techniques may be useful when trying
to model these
datasets.
Analysis 2
Artificial Neural networks (ANNs) provide the ability to predict classes of
.data given an unknown
pattern in a set of example data, and have been used successfully in a pilot
study. The neural
network analysis is described by the following process. This was performed
separately 5 times to
show possible changes in predictive ability.
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
1. Gene expression data was identified based on SAM analysis for each separate
test, all
sepsis, sepsis Day -1, -2 and -3. This data was normalized by subtracting the
median and
scaling by the standard deviation for each gene.
2. Normalized data was split into 70% subset used for training and 30% used
for validating
the neural network
3. The neural network was trained and weights for each hidden unit are used to
form a
predictor for new data.
4. The 30% subset was passed through the predictor and a probability is given
for
assignment to each of the two groups. (Sepsis, Non-sepsis)
5. The predictive ability of the neural network was based on specificity and
sensitivity
= estimated from the 30% unknown data set.
An average'specificity and sensitivity was then gained from the five separate
neural networks and
the results are summarized in Table 16.
Table 16. Summary results for prediction of sepsis on different days with
intervals based on standard
error of the five repeated predictors, as based on the genes identified by SAM
analysis.
Test Sensitivity Standard Specificity
Standard
Deviation Deviation
(+/-)
sepsis on any day 89,7% 7.4% 89.4% 7.2%
sepsis on Day -1 70.8% 11.8% 91.6% 3.3%
sepsis on Day -2 73.4% 12.7% 93.5% 5.4%
sepsis on Day -3 72.8% 13.0% 94.7% 6.5%
Neural network analysis is restricted by the number of samples which can be
used to estimate the
sensitivity/specificity since new data must be passed through the predictor.
We fully accept that
with this data set, other classification techniques may be able to provide
more accurate results
36
CA 02938761 2016-08-04
WO 2015/121605 PCT/GB2015/000004
based on larger number of patients. However, it did perform better than the
techniques used in
Analysis 1.
Having regard to the 458 genes identified in the SAM analysis, and due to the
natural ordering of the
magnitude of the test statistic, it was possible to select genes from the top
of this list in order to find
a smaller subset with a similar predictive ability (data not shown).
As a consequence of the different analyses conducted in Analysis 1 and
Analysis 2, a down select of
biomarkers was conducted. Those biomarkers identified as most predictive by
any of the techniques
outlined above were selected to be taken forward for further analysis using
the Fluidigm q RT-PCR
array system. In total 270 genes were selected along with 6 housekeeping genes
(BRD7, PWWP2A,
RANBP3, TERF2, SCM H1, FAM105B) selected based on consistent expression across
all samples.
Fluidigm Confirmation and Quantitation of Microarray Biomarkers - The Fluidigm
BioMarkHD was
used to profile 270 genes in the 60 phase I and 80 phase II samples taken at
all time points. The
BloMarkHary is a qPCR assay that runs 96 primer-probe pairs in 96 samples.
Specifically, globin-
reduced RNA (GlobinClear/m, Life Technologies, USA) was converted to cDNA
(High Capacity RT kit,
Life Technologies, USA) and preamplified by limited PCR (PreAmp"' Master Mix,
Life Technologies,
USA) with a pool of primers (DeltaGene, Fluidigm, USA) for all assays of
interest (in this case, a pool
of 276 assay primer pairs). Preamplified cDNAs were treated with Exonuclease I
(New England
Biolabs, USA) and diluted to remove unused primers and dNTPs and to prepare
samples for qPCR.
Preamplified samples were combined with 2x SsoFast EvaGreen Supermix with Low
ROX, (Bio-Rad,
USA) and 20x DNA Binding Dye Sample Loading Reagent (Fluidigm, USA) and assays
(primer pairs)
were combined with 2x Assay Loading Reagent (Fluidigm, USA). Samples and Assay
mixes were
loaded onto a 96 by 96 Dynamic Array IFC for real-time PCR analysis on a
BioMarkHD' (Fluidigm,
USA). Pre-processing was performed, using Fluidigm's Real Time PCR Analysis
Software to
37
CA 02938761 2016-08-04
WO 2015/121605 PCT/GB2015/000004
determine cycle threshold (Ct) values, using linear (derivative) baseline
correction and auto-
detected, assay-specific threshold determination. Reference or housekeeping
genes are used to
normalize each assay and produce delta Ct values. Reference samples are then
used to normalize all
samples on the plate with resulting values referred to as a delta-delta Ct's.
Three 96 by 96 plates
were used to profile all 270 genes plus .6 housekeeping genes on each plate.
A total of 269 (the original Phase I 192 + extra samples from the comparator
group) samples from
the 60 phase I patients and 439 samples from the 80 Phase II patients (+ 40
SIRS patient samples)
were profiled via the Fluidigm BioMark'. It should be noted that data from
Phase I of the study was
first analysed. The initial analysis used SVM to assess the performance of the
down-selected
biomarker list. The array was very good at identifying non-sepsis comparator
patients, as detailed in
Table 17. However, its performance for positive identification of sepsis
patients diminished with
time from sepsis diagnosis. Furthermore, when the data for the three days
prior to sepsis is pooled,_
the array gave an overall predictive accuracy of 78.8%.
Table 17. Performance (%) of down-selected biomarkers in prediction of pre-
symptomatic sepsis
patients and their comparators on Phase I data using the Fluidigm array
Comparator Sepsis
No. of patients % Predictive Accuracy No. of
patients % Predictive Accuracy =
DAY -1 29 100 30 90
DAY -2 25 100 21 71.4
DAY -3 22 100 15 66.67
DAY -4 20 95 11 54.5
Optimising the Biomarker Signature
38
CA 02938761 2016-08-04
WO 2015/121605 PCT/GB2015/000004
Due to the performance of the classifiers within Phase 1, it was decided that
the biomarker list
required updating in the light of new knowledge. The results from Phase 1
enabled down selection
of the gene list based on differential analysis.
Biomarker Validation with Phase II samples - Blind Testing with Independent
Data Sets. A fresh set
of patient samples were obtained over the course of the study and used to
validate the down-
selected genes from Phase I. All RNA samples were prepared, blinded and sent
for analysis using
microarray and Fluidigm array analysis, 266 genes were determined through
several methods such
as using SAM analysis, as mentioned above. This gene set was then further
reduced through the use
of measures taken from the classifying algorithms used, such as the Gini
coefficient in the random
forest classifier.
Two groups of classifiers were then selected, one with 45 genes, and the other
25 genes. These are
indicated in Table 18. ,
Table 18. 45 and 25 gene classifiers whose predictive accuracy was tested in
Phase II of the study
using microarray and Fluidigm array analysis of 433 blinded RNA samples.
45 Gene Classifier 25 Gene Classifier
ATP9A, C160RF7, C5ORF39, C9ORF103, C160RF7, C50RF39, C9ORF103, CD177,
CACNA1E, CD177, DHRS3, EEF1B2, FCER1A, FCER1A, GAS7, L0C285176, MYBPC3,
NIDST2,
FLT3LG, GAS7, GRB10, HLA.DMA, HS.445036, EBI2, RPL13A, RPL18A, RPL32, RPL36,
RPL9,
IL1R2, L0C285176, MYBPC3, NCOA3, RPS20, RPS29, RPS6, SIGIRR, TCEA3, TCTN1,
NDST2, RPL10A, EBI2, L00646483, RPL13A, TIMM9, TOMM7, ZFAND1, ZNHIT3
RPL18, RPL18A, RPL32, RPL36, RPL9, RPS20,
RPS29, RPS6, SIG !RR, SLBP, S1C26A6,
SMPDL3A, SORBS3, TCEA3, TCTN1, THBS3,
THNSL1, TIMM9, TOMM7, ZFAND1, ZNHIT3
We made predictions as to the type of patient that each sample had come from
and sent them to be
unblended. The performance of these classifiers is summarised in Table 19.
39
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
Table 19. Performance (%) of blomarker classifiers - (A) predictive accuracies
for samples from
Comparator patients, (8) predictive accuracies for samples from pre-
symptomatic sepsis patients
' A Number Equivalent Microarray Microarray Fluidigm Fluidigm
of Day Pre-
patients SEPSIS SepClass SepClass SepClass SepClass
,
Diagnosis
25Genes 45Genes 25Genes 45Genes
CONTROL 34 DAY -1 91.2 91.2 61.8 82.4
,
CONTROL 30 DAY -2 86.7 86.7 60.0 76.7
CONTROL 27 DAY -3 88.9 88.9 63.0 74.1
CONTROL 22 DAY -4 81.8 81.8 63.6 72.7
,s.
B Number Equivalent Microarray Microarray Fluidigm
Fluidigm .
of Day Pre- I
I
patients SEPSIS SepClass SepClass SepClass SepClass
Diagnosis
' 25Genes 45Genes 25Genes 45Genes
SEPSIS 37 DAY -1 64.9 64.9 91.9 75.7
SEPSIS 31 DAY -2 71.0 71.0 90.3 80.6 .
SEPSIS 28 DAY -3 67.9 67.9 92.9 78.6
SEPSIS 21 DAY -4 81.0 81.0 95.2 76.2
1
Table 19 demonstrates that the analysis undertaken can classify to a given
level between sepsis
patients and non-sepsis patients and their comparators. The tables also show
that the further away
from the day of sepsis diagnosis and as the N reduces, the classifier
performance increases.
Biomarker Assessment- Following the decision to remove some of the biomarkers
from the original
Fluidigm array, we re-constituted 31.5% of them with more candidate biomarkers
identified for
Phase II with more candidates. This final list of biomarkers still held the
180 highly significant genes
identified and used for Fluidigm validation during Phase I. Additional
candidate biomarkers identified
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
by SAM analysis, were added to increase the likelihood of finding key pre-
symptomatic biomarkers.
This then enabled the gene list to keep what was determined as optimum genes
and add in other
genes into the list that the differential analysis showed as significant. This
final list is listed in Table
20.
_
Table 20. Final Down-Selected genes for use on Fluidigm array during Phase II
ACTR6 EBI2 CXORF42 SORBS3 RPI_11 5LC26A8 ATP2A2
BIN1 GAS7 CLASP1 1IMM9 PPP2R2B WDR37 ZNF608
C160RF7 HIST2H4B CD2 TST NOL11 ZNF17 TBC1D8
CD247 'URI. C140RF112 CCDC65 GZMK ANKS1A RRBP1
CLNS1A LGALS2 BCL6 NCOA3 ZNF32 C059 RPL26
CY8561 LTA MRPL24 PDCD4 TMEM42 ElF3D PHCA .
FCER1A EEF1B2 L00646483 RASGRP1 TCEA3 GYG1 NSUN7
GRB10 CTSS KLRG1 RPL18A SLC2A11 KIF1B LETMD1
HS.445036 CD7 1 HLA-DRA ' RPS14 SERTAD2 MMP9 IRAK3
LARP5 CACNA1E GRAMD4 RPS6 RPS20 PAG1 FAM160A2
L00646766 C120RF57 MRPS6 , ' SIVA RPL38 RPL19 CTDP1
i
MRPL50 A0C2 OLFML2B SS18L2 RPL12 RPS15 ATP8B4
ADRB2 LY6E PTPRCAP TMC6 PRKCQ SLC36A1 . RPS3A
' BOAT L0C285176 RPL13 TTLL3 OLFM1 WWP1 l TDRD9
C210RF7 IL1R2 RPL7A , CD01 HLA-DRB3 ARG1 RUNX1
CD3D HLA-DMA ' RPS27 RPSA ZNF430 CKAP4 RPL27A
CPA3 GBP1 SH2D1A RPS15A TOMM7 EMILIN2 PHTF1
DHRS3 EOMES SMAD2 RPL30 TCTN1 HIBADH , NT5DC2
FLT3LG CUTL1 THBS3 RCN2 SLC38A10 MUC1 L0C153561
,
GTPBP8 CD96 .TP533P2 ' PECI ACVR1B PFKFB2 . ITGAM
i
41
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
ICAM2 CCL5 ZNHIT3 NDST2 C130RF23 RPL22 FBX034
LDHA C120RF62 LEPROTL1 EFCBP1 DACH1 RPS25 CYP1B1
,
L00652071 ASNSD1 MS4A4A ZFAND1 FBXW2 SLC41A3 ATXN7L3
MRPS27 MAFG P117 ; TMEM150 ITGAX ZC3H3 TRPM2
AKR1B1 L00644096 PYHIN1 SSBP2 L00647099 NAPB RPL4
I
I
BTBD11 IL32 RPL13A SLBP OPLAH LARP4B PLAC8
C50RF39 HLA-DMB RPL9 RTP4 PTPN1 HIPK2
CD3E GBP4 RPS29 RPS17 RPL5 EXOC7
CR1 EXOSC5 SIGIRR RP132 SIL1 CMTM4
DIP2A CXORF20 SMPDL3A RPL10A UPP1 ARID5B
GALM CDKN2AIP THNSL1 POP5 TFB1M ZDHHC19
HDC CD177 TRAT1 NMT2 AMD1 SORT1
ICOS C120RF65 OSTALPHA FAM26F C220RF9 RPS8
LDOC1 ATP9A MYBPC3 , ZNF195 DNAJC5 RPL24
LSG1 METTL7B P2RY5 TMEM204 GOLGA1 PGD
AMPH L00646200 RARRES3 . TBCC KIAA1881 NLRC4
C110RF1 ' ITM2A RPL18 SLC26A6 MACF1 LDLR
C9ORF103 HLA-DPA1 RPS10 ' SELM P4HB HK3
_ I
CD6 GPR107 RPS5 RPS18 RPL15 EXT1
1
CRIP2 FAM69A SIRPG RPL36 RPS13 CSGALNACT2
I
=
In order to understand why these candidate biomarkers are indicative of sepsis
in the two patient
populations, the pathways and networks affected by changes in the expression
of these down-
selected genes were analysed using GeneGo software. The complement, epithelial
to mesenchymal
and cytoskeletal remodelling pathways had the highest proportion of genes that
were over-
expressed of all host pathways at 1 day prior to sepsis diagnosis. In
contrast, pathways associated
42
CA 02938761 2016-08-04
WO 2015/121605 PCT/GB2015/000004
with immune cell and G protein signalling had the highest proportion of down-
regulated genes of
host response pathways at 1 day prior to sepsis diagnosis. The inflammatory,
apoptosis and cell
adhesion networks were most up-regulated in the healthy comparator patients,
and consequently
most down-regulated in the sepsis patient group. A similar pattern was
observed for the networks
controlling protein translation, antigen presentation and T cell receptor
signalling.
q RT-PCR validation of highlighted biomarkers - Blind Testing with Independent
Data set (Phase II
samples). Given the performance of the first down-selected biomarker
signatures, it was decided
that we would also down-select a second set of biomarkers that would give good
predictive
accuracies with lower numbers of genes. In order to compare with the first set
2 pre-symptomatic
biomarker classifiers consisting of 44 and 25 gene were down-selected by
taking random samples of
the 266 gene list and determining the predictive accuracy of the ANN. The 44
and 25 gene listed in
Table 21 were the gene lists that enabled the highest value for predictive
accuracy.
Table 21. 44 and 25 gene classifiers whose predictive accuracy was tested in
Phase II of the study
using Fluidigm array analysis of 433 blinded RNA samples
r 44 Gene Classifier 25 Gene Classifier
ACTR6, BIN1, C160RF7, CD247, CLNS1A, ACTR6, BIN1, C160RF7, CD247, CLNS1A,
CYB561, FCER1A, GRB10, HS.445036, LARP5, CYB561, FCER1A, GRB10, HS.445036,
LARP5,
L00646766, MRPL50, ADRB2, BOAT, C210RF7 L00646766, MRPL50, ADRB2, BOAT,
C210RF7
CD3D, CPA3, DHRS3, FLT3LG, GTPBP8, ICAM2, CD3D, CPA3, DHRS3, FLT3LG, GTPBP8,
ICAM2,
LDHA, L00652071, MRPS27, AKR1B1, BTBD11, LDHA, L00652071, MRPS27, AKR1B1
C5ORF39, CD3E, CR1, DIP2A, GALM, HDC,
ICOS, LDOC1, LSG1, AMPH, C110RF1,
C9ORF103, CD6, CRIP2, EBI2, GAS7, HIST2H4B,
MIK
=
Exploratory Analysis
Principal component analysis (PCA) was performed for sepsis, SIRS and
comparator patient data
using 266 genes on the validation cohort. The separation of the three groups
of patients allowed
43
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
further analysis to be undertaken as it demonstrated that there was a
separation between the
groups to be found by the classification algorithms. This separation was made
more noticeable
when PCA analysis, using the Dstl 44 gene classifier, was used on the
validation cohort.
ANN Results
The ANN approach undertaken has already been described as part of the Phase I
work. The training
and testing (70:30) was undertaken using data/samples taken from 70 sepsis
patients and 70
comparators (combination of phase 1 and phase 2 patients), at different time
points corresponding
to pre-operation, and 1, 2, and 3 days prior to the onset of sepsis, and thus
using over 600 samples.
Summary results for prediction of sepsis on different days with intervals
based on standard error of
the five repeated predictors are shown in Table 22, using the artificial
neural network detailed in
Table 2.
Table 22. Summary results for prediction of sepsis through use of an ANN.
Test (no. of Predictive Standard I Sensitivity Standard
Specificity Standard
genes) Accuracy Deviation Deviation Deviation
(+/-)
(V-) 1
266 95.02% 3.9303% 4.7124% 5.3014% 5.5714% 3.0000%
44 97.24% 1.4046% 1.7348% 1.0897% 4.4074% 2.4856%
25 92.00% 3.7202% 9.3839% 8.7481% 8.2601% 4.2226%
The results demonstrate that the ANN can classify sepsis and non-sepsis
patients to a high degree of
confidence. This confidence shows little variation when reducing the number of
genes in the
classifier, for example to 25. The results also suggest that there is an
optimal number of genes on
which to classify.
44
CA 02938761 2016-08-04
WO 2015/121605 PCT/GB2015/000004
Neural Network Analysis :SIRS
A potential confounder for these results is the possibility that the biomarker
signature of patients
who develop SIRS but NOT sepsis is similar to or overlaps with those for
sepsis patients. This could
give rise to false positives and undermine the predictive value of the pre-
symptomatic biomarker
signature for sepsis. This would lead to a lack of confidence in the findings.
For this reason, data for
40 SIRS patients were run through the ANN against the sepsis biomarker
signature, the results of
which are shown in Table 23.
Table 23. Summary results for prediction of sepsis vs. SIRS on different days
with intervals, based on
standard error of the five repeated predictors
Test (no. of Predictive Standard Sensitivity Standard
Specificity Standard
genes) I Accuracy Deviation Deviation Deviation
(+/-) (+/-) (+1-) (+/-)
266 95.22% 10.25% 0.2 0.447214 0.002 0.004472
44 100.00% 0.00% 0 0 0 0
25 1 99.84% 0.36% 0 0 0.001961 0.004384
The results in Table 23 show that the ANN effectively classifies between
sepsis and SIRS patient
biomarker signatures. Again as with Table 22, there appears to be an optimum
number of genes for
the classification. Furthermore the difference between results for 263 genes
and 45 genes suggests .
that the 263 gene biomarker list does have a commonality with the SIRS
signature within it.
Through the production of 44,014 combinations/biomarker signatures of 44
biomarkers, randomly
selected from the list of 266, it has been shown that all combinations have a
mean predictive
accuracy of greater than 75% (in fact above 76.1%). The abundance of
individual genes in the top
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
and bottom 1000 subsets is not uniform; the genes which appear more frequently
in the top subsets
of 44, appear less frequently in the bottom subsets, and vice versa.
These results are illustrated by the 15 specific combinations listed in Table
24, which have the .
._
accuracies shown in Figure 3. Thus in one embodiment the biomarker signature
comprises at least
44 genes selected from the list of genes consisting of the 266 genes listed in
Table 1.
Table 24. Fifteen combinations of 44 biomarkers tested for predictive
accuracies. The predictive
accuracies are illustrated in Figure 3.
ii 2 3 4 5 6 7 8
CYB561 BIN1 ACTR6 BOAT A CTR6 BIN1 C160RF7 I CYB561
¨
GRB10 FCER1A BIN1 L00652071 BIN1 ' ADRB2 LARPS
FCER1A
BTBD11 100646766 L00646766 DIP2A C160RF7 BOAT C210RF7 =
MRPL50
i
CD3E MRPL50 ICAM2 C110RF1 CD247 ' FLT3LG GTPBP8 ADRB2
,
EB12 ADRB2 L00652071 CD6 ' CLN51A L00652071 ' LDHA
L00652071
¨
CD7 CD3E ICOS HIST2H4B CYB561 ' CR1 MRPS27 !COS
L0C285176 CACNAlE CD7 EEF1B2 FCER1A DIP2A BTBD11
AMPH
HLA-DMA A0C2 IL1R2 ' C= 120RF57 GRB10 LY6E HOC C9ORF103
C120RF62 LY6E ASNSD1 IL1R2 HS.445036 HLA-DMA CRIP2 '
C120RF57
_
ASNSD1 GBP4 MAFG CXORF20 LARP5 GBP1 1L1R1 A0C2
GPR107 CDKN2AIP GBP4 ' C= 0177 L00646766 MAFG CACNAlE
, HLA-DMA
BCL6 METTL7B CXORF20 CD2 MRPL50 L00644096 L0C285176
CDKN2AIP
MRPL24 1 HA-DPA1 HLA-DPA1 acL6 ADRB2 ATP9A HLA-DMA
C120RF65
RPL7A 100646483 CXORF42 L00646483 BOAT CLASP1 ASNSD1
ATP9A
RPL13A MRPS6 MRPL24 ' H= LA-DFtA C210RF7 BCL6 L00644096
L00646200
RPS5 OLFML2B PTPRCAP ' PTPRCAP CD3D RPL7A IL32
HLA-DPA1 '
¨
CCOC65 - SH2D1A RPL7A TP53BP2 CPA3 TH1353 FAM69A
L00646483
NCOA3 SIRPG PYHIN1 ' S= ORBS3 DHRS3 P2RY5 MRPL24 RPL7A
-
RASGRP1 RPS14 RASGRP1 NCOA3 FLT3LG RPS5 HLA-DRA
TP53BP2
RPS6 RPS15A RPL30 RPS6 GTPBP8 RASGRP1 . MRPS6 ZNHIT3
NMT2 RCN2 EFCBP1 5S18L2 ICAM2 RPS14 RPS27 RPL13A
ZNF32 EFCBP1 TMEM150 RPS15A i LOHA RPSA PYHIN1 TRAT1
SERTAD2 NMT2 RPL32 ZFAND1 LOC652071 RPS17 SIGIRR P2RY5
RPL38 NOL11 ZNF195 RPS18 MRPS27 NMT2 SMPDL3A RARRES3
SLC38A10 ZNF32 TMEM204 NOL11 AKR1B1 SLC26A6 P2RY5
CCDC65
ACVR1B SLC2A11 SELM TMEM42 BTBD11 DACH1 RARRES3 RPS6
,
P41413 OLFM1 RPS18 RPL12 C50RF39 RPL5 TTLL3 RPSA
t
SLC26A8 100647099 PPP2R2B ZNF430 CD3E TFB1M RPS15A
RCN2
WDR37 OPLAH OLFM1 SLC38A10 CR1 AMD1 RPL36 RPL32
,
46
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
,
PAG1 1 ZNF17 TCTN1 ACVR1B DIP2A MACF1 TMEM42 I RPL36
RPL19 KIF1B DACH1 DACH1 GALM KIF1B HLA-DRB3 , RPL12
5LC41A3 RPS15 ITGAX UPP1 HOC SLC36A1 FBXW2 OLFM1
LARP4B I MUC1 TFB1M GOLGA1 ICOS WWP1 L00647099 1 ZNF430
ZDHHC19 PFKFB2 GYG1 MACF1 LDOC1 PFKFB2 ZNF17 : TOMM7
SORT1 NAPB MMP9 P4HB LSG1 . RPS25 CD59 GOLGA1
NLRC4 I LDLR PAG1 SLC36A1 AMPH CMTM4 KIF1B I ZNF17
EXT1 ATP2A2 RPS15 CKAP4 ' C11ORF1 RPS8 SLC36A1
, C059
' ATP2A2 RRBP1 . CKAP4 PFKFB2 C9ORF103 PGD PFKFB2
PAG1
ZNF608 IRAK3 RPL22 ZC3H3 CD6 TBC1D8 SLC41A3 RPL19
RRBP1 ATP8B4 ZDHHC19 CMTM4 CRIP2 LETMD1 EXOC7
I 2C3H3
TDRDS PHTF1 5ORT1 ZDHHC19 EBI2 IFLAK3 HK3 I NAPB
I ,
RUNX1 NTSDC2 NLRC4 NLRC4 GAS7 RPS3A ATP2A2 = HIPK2
LOC153561. LOC153561 CSGALNACT2 PHTF1 HIST2H4B PHTF1
LETMD1 ZNF608
ITGAM FBX034 RRBP1 TRPM2 IL1R1 = NTSDC2 ITGAM
, PHCA
' 9 10 11 12 13 14 15
. CD6 BCL6 ' CUTL1 CDKN2AIP L00652071 EBI2 . A0C2
CD247 CLNS1A GIPBP8 CLNS1A BIN1 CO247 L00646766
' CLNS1A CYB561 LDHA ADRB2 CYB561 FCER1A BOAT
CSORF39 FCER1A BTBD11 FLT3LG GRB10 ICAM2 DiP2A
GALM C210RF7 ICOS GTPBP8 LARP5 C50RF39 CRIP2 . .
ICOS FLT3LG C110RF1 L1R1 . L00646766 ' EEF1B2 CUTL1
A0C2 CTSS CD6 C07 AKR1B1 C120RF57 CD96
IL1R2 CD96 LGALS2 CACNAlE DIP2A A0C2 HLA-DMB
CUTL1 CCL5 GBP1 GBP4 L5G1 EOMES EXOSCS
CDKN2AIP HLA-DMB ' ASNSD1 EXO5C5 EBI2 IL32 ITM2A
I1M2A CDKN2AIP , CDKN2AIP - C= XORF20 EEF1B2 GBP4
HLA-DPA1
CLASP1 GPR107 ITM2A CD177 CD7 CD177 BCL6
C140RF112 ' CXORF42 C140RF112 METTL7B LY6E HIA-DPA1
MRPL24
. BCL6 CLASP' BCL6 HLA-DPA1 C120RF65 C140RF112
100646483
L00646483 RPS27 HLA-DRA CD2 METTL7B BCL6 KLRG1
RPS27 SMAD2 RPS29 - R= PS27 ITM2A MRPL24 GRAMD4
P117 ZNHI13 OSTALPHA TP53I3P2 L00646483 RPS27
PYHIN1
RPL9 RPL13A 1ST * S= IGIRR SIGIRR P117 SMPDL3A
RPS10 SMPDL3A CCDC65 ' O= STALPHA TRAT1 NCOA3 RPS5
SORBS3 TMEM150 NCOA3 RARRES3 OSTALPHA RASGRP1 RASGRP1
1ST FAM26F PDCD4 SIRPG NCOA3 RPS6 RPL18A
RPL18A TBCC SLBP SORBS3 RPL18A PECI RPSA
SS18L2 TCEA3 RPL10A RPL30 RPS15A EFCBP1 , TMEM150
CD01 ITGAX GZMK NDST2 ' RCN2 TMEM150 ZNF195
RPS15A PTPN1 RPL12 SLBP RTP4 NMT2 SLC26A6
TMEM150 TFB1M PRKCQ RTP4 SLC2A11 SLC26A6 SLC2A11
47 .
CA 02938761 2016-08-04
WO 2015/121605
PCT/GB2015/000004
RPL32 AMD1 HLA-DRB3 RPL11 HLA-DRB3 ' RPL11 PRKCQ
SLC26A6 KIAA1881 OPLAH ZNF32 C130RF23 PPP2R2B HLA-DRB3
RP520 CD59 = UPP1 RPS20 L00647099 ZNF32 TCTN1
HLA-DRB3 KIF1B KIAA1881 HLA-DRB3 OPLAH , ACVR1B ITGAX
TCTN1.= RPL19 P4HB ACVR1B ANKS1A TFB1rse1 , AMD1
P4HB NAPB RP513 C130RF23 RPL19 P4HB DNAJC5
RPL15 ZDHHC19 WDR37 DACI-11 . WWP1 RPL15 GOLGA1
RPS13 EXT1 . ZNF17 AMD1 ARG1 ZNF17 CD59
WDR37 ZNF608 ANKS1A SLC26A8 CKAP4 ElF3D RPS15
ANKS1A TBC1D8 . CD59 WDR37 EXOC7 MMP9 ARG1
,
1
KIF1B RRBP1 CKAP4 KIF1B PGD SLC36A1 EMILIN2
MMP9 ATP8B4 PFKFB2 WWP1 HK3 NAPB HIBADH
I
EXOC7 . RPS3A RPL22 RPS25 RRBP1 . ARID5B MUC1
CMTM4 RPL27A , ZC.3H3 RPL24 RPL26 HK3 2C3H3
RPL24 PHTF1 ZDHHC19 ZNIF608 FAM160A2 CSGALNACT2 ZDHHC19
- C.SGALNACT2 FBX034 NLRC4 RRBP1 CTDP1 FAM160A2 HK3
ATP2A2 CYP1B1 - RRBP1 NSUN7 RPL27A RPS3A PHCA
FAM160A2 RPL4 RPL26 TDRD9 NT5DC2 RPL27A NSUN7
,
Having regard to Figure 4, 98 subsets of different numbers of biomarkers were
formed from the
entire panel of 266 genes, wherein each subset was characterised by only
including genes of a
particular level of abundance in the top 1000 subsets selected from the
44,014. The subsets range
from a single gene (LDLR, which was by far the most abundant) to the whole
panel of 266
biomarkers. Where multiple genes have the same abundance they are all included
in a single subset
associated with that threshold, which is why there are less subsets than there
are total genes. The
trend, derived from a more basic ANN than that detailed in Table 2, shows an
increase in predictive
accuracy with increasing subset size, which begins to plateau at a subset size
of 97 genes. Here a
maximum predictive accuracy is reached at 97.0%. When the subset size exceeds
149 genes the
predictive accuracy begins to drop off, falling to 89.6%. This trend does not
match that obtained
using a random forest, although here we examine single defined subsets rather
than many random
ones. Of the three subsets reaching the maximum predictive accuracy, the
smallest one (subset 50)
contains genes that appeared 165 times or more in the top 1000 subsets of the
test.
48
CA 02938761 2016-08-04
WO 2015/121605 PCT/GB2015/000004
Through use of systems-scale profiling technology, this study identified
biomarker signatures
predictive of the development of post-operative sepsis with high accuracy in a
sizable blinded
validation set. The key to analysing complicated data sets is the method of
analysis. Our approach
was predicated on the conclusion that no one biomarker is likely to be a
predictive for sepsis in
humans. Previous studies of pre-symptomatic biomarker expression have shown
that conventional
linear analyses of biomarker expression may fail to reveal differences between
the two patient
groups i.e. sepsis and non-sepsis patients. A variety of Non-linear techniques
were used with varying
degrees of success to differentiate between the transcriptomes of patients who
go on to develop
sepsis and their comparators. Random Forests and SVM demonstrated some use for
the
differentiation of transcriptomes from different patient groups. However, ANN
analysis, using 25 and
44 gene biomarker signatures performed excellently. These gave high predictive
accuracies as well
as high sensitivities and specificities when differentiating between patients
who went on to develop
sepsis and their comparators. Furthermore, the biomarker signatures derived
were very robust
when tested against potentially confounding transcriptomes from patients who
had SIRS but who
did not go on to develop sepsis.
The strong performance of non-linear techniques is perhaps not unexpected,
since immune markers
fluctuate greatly over the entire course of sepsis. It is unlikely that
analysis using simple linear
techniques could be used as easily to pick out key biomarker signatures.
It is also worth noting that the successful testing of patient transcriptomes
through use of a
- multiplexed q RT-PCR indicates the suitability = of this technology for
further development as a
diagnostic assay.
The functional relevance of a subset of transcripts constituting' this
signature, which is broadly
associated with coordinated molecular and cellular chain of events involved
during inflammation
and sepsis, instils confidence in our results. Indeed activation of the
complement pathway has been
shown to play an important role in sepsis and inflammation. Conversely,
dendritic cells and other
49
CA 02938761 2016-08-04
WO 2015/121605 PCT/GB2015/000004
antigen presenting cells have been shown to disappear from the circulation
during septic episodes,
which may account for the observed decrease in abundance of transcripts
associated with MHC gene
expression. It should be noted that the majority of the candidate markers
identified through this
unbiased global profiling approach have not been as well characterized as
these few functionally
enriched "landmark" transcripts.