Note: Descriptions are shown in the official language in which they were submitted.
1
OPHTHALMIC COMPOSITION COMPRISING ATROPINE OR ATROPINE SULPHATE
[0001]
BACKGROUND OF THE DISCLOSURE
[0002] Pharmaceutical formulations have an expiration date which is based
on the degradation
of the active ingredient.
SUMMARY OF THE DISCLOSURE
[0003] Provided herein are ophthalmic compositions. In some embodiments,
disclosed herein is
an ophthalmic composition, comprising from about 0.001 wt% to about 0.05 wt%
of a muscarinic
antagonist and deuterated water, at a pD of from about 4.2 to about 7.9.
[0004] In some embodiments, the muscarinic antagonist comprises atropine,
atropine sulfate,
noratropine, atropine-N-oxide, tropine, tropic acid, hyoscine, scopolomine,
tropicamide, cyclopentolate,
pirenzapine, homatropine, or a combination thereof. In some embodiments, the
muscarinic antagonist is
atropine. In some embodiments, the muscarinic antagonist is atropine sulfate.
[0005] In some embodiments, the ophthalmic composition has a pD of one of:
less than about
7.3, less than about 7.2, less than about 7.1, less than about 7, less than
about 6.8, less than about 6.5, less
than about 6.4, less than about 6.3, less than about 6.2, less than about 6.1,
less than about 6, less than
about 5.9, less than about 5.8, less than about 5.2, or less than about 4.8
after extended period of time
under storage condition.
[0006] In some embodiments, the ophthalmic composition comprises one of: at
least about 80%,
at least about 85%, at least about 90%, at least about 93%, at least about
95%, at least about 97%, at least
about 98%, or at least about 99% of the muscarinic antagonist based on initial
concentration after
extended period of time under storage condition. As described in this
disclosure, the percentage of the
ophthalmic agent in the composition after storage is based on the amount of
ophthalmic agent that is
initially present in the composition (i.e. prior to the storage condition).
[0007] In some embodiments, the ophthalmic composition further has a
potency of one of: at
least 80%, at least 85%, at least 90%, at least 93%, at least 95%, at least
97%, at least 98%, or at least
99% after extended period of time under storage condition. As described in
this disclosure, the potency of
the ophthalmic agent in the composition after storage is based on the potency
of ophthalmic agent that is
initially present in the composition (i.e. prior to the storage condition).
[0008] In some embodiments, the extended period of time is one of: about 1
week, about 2
weeks, about 3 weeks, about 1 month, about 2 months, about 3 months, about 4
months, about 5 months,
Date recue / Date received 2021-12-03
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
2
about 6 months, about 8 months, about 10 months, about 12 months, about 18
months, about 24 months,
about 36 months, about 4 years, or about 5 years.
[0009] In some embodiments, the storage condition has a storage temperature
of from about 2 C
to about 10 C or from about 16 C to about 26 C. In some embodiments, the
storage condition has a
storage temperature of about 25 C. In some embodiments, the storage condition
has a storage
temperature of about 40 C. In some embodiments, the storage condition has a
storage temperature of
about 60 C.
[0010] In some embodiments, the storage condition has a relative humidity
of about 60%. In
some embodiments, the storage condition has a relative humidity of about 75%.
[0011] In some embodiments, the muscarinic antagonist is present in the
composition at a
concentration of one of: from about 0.001 wt% to about 0.04 wt%, from about
0.001 wt% to about 0.03
wt%, from about 0.001 wt% to about 0.025 wt%, from about 0.001 wt% to about
0.02 wt%, from about
0.001 wt% to about 0.01 wt%, from about 0.001 wt% to about 0.008 wt%, or from
about 0.001 wt% to
about 0.005 wt%.
[0012] In some embodiments, the composition comprises less than 20% of
major degradant
based on the concentration of the ophthalmic agent after extended period of
time under storage condition.
In some embodiments, the composition comprises less than 15% of major
degradant based on the
concentration of the ophthalmic agent after extended period of time under
storage condition.
[0013] In some embodiments, the composition comprises less than 10% of
major degradant
based on the concentration of the ophthalmic agent after extended period of
time under storage condition.
In some embodiments, the composition comprises less than 5% of major degradant
based on the
concentration of the ophthalmic agent after extended period of time under
storage condition. In some
embodiments, the composition comprises less than 2.5% of major degradant based
on the concentration
of the ophthalmic agent after extended period of time under storage condition.
In some embodiments, the
composition comprises less than 2.0% of major degradant based on the
concentration of the ophthalmic
agent after extended period of time under storage condition. In some
embodiments, the composition
comprises less than 1.5% of major degradant based on the concentration of the
ophthalmic agent after
extended period of time under storage condition. In some embodiments, the
composition comprises less
than 1.0% of major degradant based on the concentration of the ophthalmic
agent after extended period
of time under storage condition. In some embodiments, the composition
comprises less than 0.5% of
major degradant based on the concentration of the ophthalmic agent after
extended period of time under
storage condition. In some embodiments, the composition comprises less than
0.4% of major degradant
based on the concentration of the ophthalmic agent after extended period of
time under storage condition.
In some embodiments, the composition comprises less than 0.3% of major
degradant based on the
concentration of the ophthalmic agent after extended period of time under
storage condition. In some
embodiments, the composition comprises less than 0.2% of major degradant based
on the concentration
of the ophthalmic agent after extended period of time under storage condition.
In some embodiments, the
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
3
composition comprises less than 0.1% of major degradant based on the
concentration of the ophthalmic
agent after extended period of time under storage condition. In some
embodiments, the major degradant
is tropic acid. As described in this disclosure, the percentage of the primary
degradant in the composition
after storage is based on the amount of ophthalmic agent that is initially
present in the composition (i.e.
prior to the storage condition).
[0014] In some embodiments, the composition is in a form of an aqueous
solution.
[0015] In some embodiments, the composition further comprises an osmolarity
adjusting agent.
In some embodiments, the osmolarity adjusting agent is sodium chloride.
[0016] In some embodiments, the ophthalmic composition further comprises a
preservative. In
some embodiments, the preservative is selected from benzalkonium chloride,
cetrimonium, sodium
perborate, stabilized oxychloro complex, SofZia, polyquaternium-1,
chlorobutanol, edetate disodium,
polyhexamethylene biguanide, or combinations thereof.
[0017] In some embodiments, the ophthalmic composition further comprises a
buffer agent. In
some embodiments, the buffer agent is selected from borates, borate-polyol
complexes, phosphate
buffering agents, citrate buffering agents, acetate buffering agents,
carbonate buffering agents, organic
buffering agents, amino acid buffering agents, or combinations thereof
[0018] In some embodiments, the ophthalmic composition further comprises a
tonicity adjusting
agent. In some embodiments, the tonicity adjusting agent is selected from
sodium chloride, sodium
nitrate, sodium sulfate, sodium bisulfate, potassium chloride, calcium
chloride, magnesium chloride, zinc
chloride, potassium acetate, sodium acetate, sodium bicarbonate, sodium
carbonate, sodium thiosulfate,
magnesium sulfate, disodium hydrogen phosphate, sodium dihydrogen phosphate,
potassium dihydrogen
phosphate, dextrose, mannitol, sorbitol, dextrose, sucrose, urea, propylene
glycol, glycerin, or a
combination thereof
[0019] In some embodiments, the composition is stored in a plastic
container. In some
embodiments, the material of the plastic container comprises low-density
polyethylene (LDPE).
[0020] In some embodiments, the ophthalmic composition is essentially free
of procaine and
benactyzine, or pharmaceutically acceptable salts thereof
[0021] In some embodiments, the composition has a dose-to-dose ophthalmic
agent
concentration variation of less than 50%. In some embodiments, the composition
has a dose-to-dose
ophthalmic agent concentration variation of less than 40%. In some
embodiments, the composition has a
dose-to-dose ophthalmic agent concentration variation of less than 30%. In
some embodiments, the
composition has a dose-to-dose ophthalmic agent concentration variation of
less than 20%. In some
embodiments, the composition has a dose-to-dose ophthalmic agent concentration
variation of less than
10%. In some embodiments, the composition has a dose-to-dose ophthalmic agent
concentration
variation of less than 5%. In some embodiments, the dose-to-dose ophthalmic
agent concentration
variation is based on 10 consecutive doses. In some embodiments, the dose-to-
dose ophthalmic agent
concentration variation is based on 8 consecutive doses. In some embodiments,
the dose-to-dose
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
4
ophthalmic agent concentration variation is based on 5 consecutive doses. In
some embodiments, the
dose-to-dose ophthalmic agent concentration variation is based on 3
consecutive doses. In some
embodiments, the dose-to-dose ophthalmic agent concentration variation is
based on 2 consecutive doses.
[0022] In some embodiments, the composition further comprises a pD
adjusting agent. In some
embodiments, the pD adjusting agent comprises DCI, Na0D, CD3COOD, or C61)807.
[0023] In some embodiments, the composition further comprises a
pharmaceutically acceptable
carrier. In some embodiments, the ophthalmically acceptable carrier further
comprises at least one
viscosity-enhancing agent. In some embodiments, the viscosity-enhancing agent
is selected from
cellulose-based polymers, polyoxyethylene-polyoxypropylene triblock
copolymers, dextran-based
polymers, polyvinyl alcohol, dextrin, polyvinylpyrrolidone, polyalkylene
glycols, chitosan, collagen,
gelatin, hyaluronic acid, or combinations thereof.
[0024] In some embodiments, the ophthalmic composition comprises one of:
less than 60% of
H20, less than 55% of H20, less than 50% of H20, less than 45% of H20, less
than 40% of H20, less than
35% of H20, less than 30% of H20, less than 25% of H20, less than 20% of H20,
less than 15% of H20,
or less than 10% of H20.
[0025] In some embodiments, the ophthalmic composition comprises one of:
less than 5% of
H20, less than 4% of H20, less than 3% of H20, less than 2% of H20, less than
1% of H20, less than
0.5% of H20, less than 0.1% of H20, or 0% of H20.
[0026] In some embodiments, the ophthalmic composition is stored below room
temperature
prior to first use. In some embodiments, the ophthalmic composition is stored
at between about 2 C to
about 10 C prior to first use. In some embodiments, the ophthalmic
composition is stored at between
about 4 C to about 8 C prior to first use.
[0027] In some embodiments, the ophthalmic composition is stored at room
temperature after
first use. In some embodiments, the ophthalmic composition is stored at
between about 16 C to about 26
C after first use.
[0028] In some embodiments, the ophthalmic composition is not formulated as
an injectable
formulation.
[0029] In some embodiments, the ophthalmic composition is formulated as an
ophthalmic
solution for the treatment of an ophthalmic disorder. In some embodiments, the
ophthalmic disorder or
condition is pre-myopia, myopia, or progression of myopia. In some
embodiments, the ophthalmic
composition is formulated as an ophthalmic solution for the treatment of pre-
myopia, myopia, or
progression of myopia.
[0030] In some embodiments, the ophthalmic composition is a solution.
[0031] In some embodiments, disclosed herein is a method of arresting
myopia development
that comprises administering to an eye of an individual in need thereof an
effective amount of an
ophthalmic composition described herein. Also described herein is a method of
preventing myopia
development that comprises administering to an eye of an individual in need
thereof an effective amount
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
of an ophthalmic composition described herein. In some embodiments, described
herein is a method of
arresting or preventing myopia development, comprising administering to an eye
of an individual in need
thereof an effective amount of an ophthalmic composition comprising from about
0.001 wt% to about
0.05 wt% of a muscarinic antagonist and deuterated water, at a pD of from
about 4.2 to about 7.9. In
some embodiments, the ophthalmic composition is administered at predetermined
time intervals over an
extended period of time. In some embodiments, the ophthalmic composition is
administered once every
day. In some embodiments, the ophthalmic composition is administered every
other day. In some
embodiments, the ophthalmic composition is administered over 1 week, 2 weeks,
1 month, 2 months, 3
months, 6 months, 1 year, 2 years, 3 years, 4 years, 5 years, 6 years, 7
years, 8 years, 9 years, 10 years,
11 years, or 12-15 years. In some embodimemts, the ophthalmic composition is
stored below room
temperature prior to first use. In some embodiments, the ophthalmic
composition is stored at between
about 2 C to about 10 C prior to first use. In some embodiments, the
ophthalmic composition is stored
at between about 4 C to about 8 C prior to first use. In some embodiments,
the ophthalmic composition
is stored at room temperature after first use. In some embodiments, the
ophthalmic composition is stored
at between about 16 C to about 26 C after first use.
[0032] In some embodiments, disclosed herein is an ophthalmic solution that
comprises from
about 0.001 wt% to about 0.05 wt% of a muscarinic antagonist and deuterated
water, at a pD of from
about 4.2 to about 7.9. In some embodiments, the ophthalmic solution has a pD
of one of: less than about
7.3, less than about 7.2, less than about 7.1, less than about 7, less than
about 6.8, less than about 6.5, less
than about 6.4, less than about 6.3, less than about 6.2, less than about 6.1,
less than about 6, less than
about 5.9, less than about 5.8, less than about 5.2, or less than about 4.8
after extended period of time
under storage condition. In some embodiments, the muscarinic antagonist
comprises atropine, atropine
sulfate, noratropine, atropine-N-oxide, tropine, tropic acid, hyoscine,
scopolomine, tropicamide,
cyclopentolate, pirenzapine, homatropine, or a combination thereof. In some
embodiments, the
ophthalmic solution comprises one of: less than 5% of H20, less than 4% of
H20, less than 3% of H20,
less than 2% of H20, less than 1% of F120, less than 0.5% of H20, less than
0.1% of H20, or 0% of H20.
In some embodiments, the ophthalmic composition comprises one of: at least
about 80%, at least about
85%, at least about 90%, at least about 93%, at least about 95%, at least
about 97%, at least about 98%,
or at least about 99% of the muscarinic antagonist based on initial
concentration after extended period of
time under storage condition. In some embodiments, the ophthalmic composition
further has a potency of
one of: at least 80%, at least 85%, at least 90%, at least 93%, at least 95%,
at least 97%, at least 98%, or
at least 99% after extended period of time under storage condition. In some
embodiments, the extended
period of time is one of: about 1 week, about 2 weeks, about 3 weeks, about 1
month, about 2 months,
about 3 months, about 4 months, about 5 months, about 6 months, about 8
months, about 10 months,
about 12 months, about 18 months, about 24 months, about 36 months, about 4
years, or about 5 years. In
some embodiments, the muscarinic antagonist is present in the composition at a
concentration of one of:
from about 0.001 wt% to about 0.04 wt%, from about 0.001 wt% to about 0.03
wt%, from about 0.001
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
6
wt% to about 0.025 wt%, from about 0.001 wt% to about 0.02 wt%, from about
0.001 wt% to about 0.01
wt%, from about 0.001 wt% to about 0.008 wt%, or from about 0.001 wt% to about
0.005 wt%. In some
embodiments, the storage condition has a storage temperature of from about 2 C
to about 10 C or from
about 16 C to about 26 C. In some embodiments, the ophthalmic composition has
a dose-to-dose
muscarinic antagonist concentration variation of one of: less than 50%, less
than 40%, less than 30%, less
than 20%, less than 10%, or less than 5%. In some embodiments, the dose-to-
dose muscarinic antagonist
concentration variation is based on one of: 10 consecutive doses, 8
consecutive doses, 5 consecutive
doses, 3 consecutive doses, or 2 consecutive doses.
[0033] In some embodiments, disclosed herein is an ophthalmic composition,
comprising from
about 0.001 wt% to about 0.05 wt% of a muscarinic antagonist and water, at a
pH of from about 3.8 to
about 7.5.
[0034] In some embodiments, the muscarinic antagonist comprises atropine,
atropine sulfate,
noratropine, atropine-N-oxide, tropine, tropic acid, hyoscine, scopolomine,
tropicamide, cyclopentolate,
pirenzapine, homatropine, or a combination thereof. In some embodiments, the
muscarinic antagonist is
atropine or atropine sulfate.
[0035] In some embodiments, the ophthalmic composition comprises one of: at
least about 80%,
at least about 85%, at least about 90%, at least about 93%, at least about
95%, at least about 97%, at least
about 98%, or at least about 99% of the muscarinic antagonist based on initial
concentration after
extended period of time under storage condition.
[0036] In some embodiments, the ophthalmic composition has a pH of one of:
less than about
7.3, less than about 7.2, less than about 7.1, less than about 7, less than
about 6.8, less than about 6.5, less
than about 6.4, less than about 6.3, less than about 6.2, less than about 6.1,
less than about 6, less than
about 5.9, less than about 5.8, less than about 5.2, less than about 4.8, or
less than about 4.2 after
extended period of time under storage condition.
[0037] In some embodiments, the ophthalmic composition further has a
potency of one of: at
least 80%, at least 85%, at least 90%, at least 93%, at least 95%, at least
97%, at least 98%, or at least
99% after extended period of time under storage condition.
[0038] In some embodiments, the extended period of time is one of: about 1
week, about 2
weeks, about 3 weeks, about 1 month, about 2 months, about 3 months, about 4
months, about 5 months,
about 6 months, about 8 months, about 10 months, about 12 months, about 18
months, about 24 months,
about 36 months, about 4 years, or about 5 years.
[0039] In some embodiments, the storage condition has a storage temperature
of one of: about
25 C, about 40 C, or about 60 C. In some embodiments, the storage condition
has a storage temperature
of from about 2 C to about 10 C or from about 16 C to about 26 C.
[0040] In some embodiments, the storage condition has a relative humidity
of about 60% or
about 75%.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
7
[0041] In some embodiments, the muscarinic antagonist is present in the
composition at a
concentration of one of: from about 0.001 wt% to about 0.04 wt%, from about
0.001 wt% to about 0.03
wt%, from about 0.001 wt% to about 0.025 wt%, from about 0.001 wt% to about
0.02 wt%, from about
0.001 wt% to about 0.01 wt%, from about 0.001 wt% to about 0.008 wt%, or from
about 0.001 wt% to
about 0.005 wt%.
[0042] In some embodiments, the ophthalmic composition further comprises an
osmolarity
adjusting agent. In some embodiments, the osmolarity adjusting agent is sodium
chloride.
[0043] In some embodiments, the ophthalmic composition further comprises a
preservative. In
some embodiments, the preservative is selected from benzalkonium chloride,
cetrimonium, sodium
perborate, stabilized oxychloro complex, SofZia, polyquaternium-1,
chlorobutanol, edetate disodium,
polyhexamethylene biguanide, or combinations thereof
[0044] In some embodiments, the ophthalmic composition further comprises a
buffer agent. In
some embodiments, the buffer agent is selected from borates, borate-polyol
complexes, phosphate
buffering agents, citrate buffering agents, acetate buffering agents,
carbonate buffering agents, organic
buffering agents, amino acid buffering agents, or combinations thereof.
[0045] In sonic embodiments, the ophthalmic composition further comprises a
tonicity adjusting
agent. In some embodiments, the tonicity adjusting agent is selected from
sodium chloride, sodium
nitrate, sodium sulfate, sodium bisulfate, potassium chloride, calcium
chloride, magnesium chloride, zinc
chloride, potassium acetate, sodium acetate, sodium bicarbonate, sodium
carbonate, sodium thiosulfate,
magnesium sulfate, disodium hydrogen phosphate, sodium dihydrogen phosphate,
potassium dihydrogen
phosphate, dextrose, mannitol, sorbitol, dextrose, sucrose, urea, propylene
glycol, glycerin, or a
combination thereof.
[0046] In some embodiments, the ophthalmic composition is stored in a
plastic container. In
some embodiments, the material of the plastic container comprises low-density
polyethylene (LDPE).
[0047] In some embodiments, the ophthalmic composition has a dose-to-dose
muscarinic
antagonist concentration variation of one of: less than 50%, less than 40%,
less than 30%, less than 20%,
less than 10%, or less than 5%.
[0048] In some embodiments, the dose-to-dose muscarinic antagonist
concentration variation is
based on one of: 10 consecutive doses, 8 consecutive doses, 5 consecutive
doses, 3 consecutive doses, or
2 consecutive doses.
[0049] In some embodiments, the ophthalmic composition has a pH of one of:
from about 3.8 to
about 7.5, from about 4.2 to about 7.5, from about 4.8 to about 7.3, from
about 5.2 to about 7.2, from
about 5.8 to about 7.1, from about 6.0 to about 7.0, or from about 6.2 to
about 6.8.
[0050] In some embodiments, the ophthalmic composition further comprises a
pH adjusting
agent. In some embodiments, the pH adjusting agent comprises HC1, NaOH,
CH3COOH, or C6H807.
[0051] In some embodiments, the ophthalmic composition comprises one of:
less than 60% of
D20, less than 55% of D20, less than 50% of D20, less than 45% of D20, less
than 40% of D20, less than
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
8
35% of D20, less than 30% of D20, less than 25% of D20, less than 20% of D20,
less than 15% of D20,
or less than 10% of D20.
[0052] In some embodiments, the ophthalmic composition comprises one of:
less than 5% of
D20, less than 4% of D20, less than 3% of D20, less than 2% of D20, less than
1% of D20, less than
0.5% of D20, less than 0.1% of D20, or 0% of D20. In some embodiments,
ophthalmic composition is
essentially free of D20.
[0053] In some embodiments, the composition further comprises a
pharmaceutically acceptable
carrier.
[0054] In some embodiments, the ophthalmic composition is formulated as an
ophthalmic
solution for the treatment of an ophthalmic disorder. In some embodiments, the
ophthalmic disorder or
condition is pre-myopia, myopia, or progression of myopia.
[0055] In some embodiments, the ophthalmic composition is not formulated as
an injectable
formulation.
[0056] Other features and technical effects of the methods and compositions
described herein
will become apparent from the following detailed description. It should be
understood, however, that the
detailed description and the specific examples, while indicating specific
embodiments, are given by way
of illustration only.
BRIEF DESCRIPTION OF THE DRAWINGS
[0057] The novel features of the disclosure are set forth with
particularity in the appended claims.
A better understanding of the features and advantages of the present
disclosure will be obtained by
reference to the following detailed description that sets forth illustrative
embodiments, in which the
principles of the disclosure are utilized, and the accompanying drawings of
which:
[0058] Fig. 1A-Fig. 1C show the shelf life prediction of 0.01% atropine
sulfate solution with a
primary degradant RRT 0.87-0.89, and a n.m.t. of 0.5% area, based on data
obtained from samples stored
at 25 C and 40 C. The pH range of the atropine sulfate solution is from 5.9-
6.2.
[0059] Fig. 2A-Fig. 2C show the shelf life prediction of 0.01% atropine
sulfate solution with a
primary dcgradant RRT 0.87-0.89, and a n.m.t. of 0.5% area, based on data
obtained from samples stored
at 25 C and 60 C. The pH range of the atropine sulfate solution is from 5.9-
6.2.
[0060] Fig. 3 illustrates mass balance at 4 weeks and at 60 C condition for
atropine sulfate
formulations disclosed in Example 9.
[0061] Fig. 4 illustrates atropine sulfate (0.010%) formulation stability
in acetic acid. The
atropine sulfate formulation is formulated with acetic acid and either with
H20 (top panel, Formulation 3)
or D20 (bottom panel, Formulation 7). Formulation 3 has a pH of 4.8 and
Formulation 7 has a pD of 5.2.
Both formulations are stored at 60 C for 4 weeks prior to analysis.
[0062] Fig. 5 illustrates atropine sulfate (0.01%) formulation stability in
citric acid. The atropine
sulfate formulation is formulated with citric acid and either with H20 (top
panel, Formulation 5) or D20
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
9
(bottom panel, Formulation 8). Formulation 5 has a pH of 5.8 and Formulation 8
has a pD of 6.2. Both
formulations are stored at 60 C for 4 weeks prior to analysis.
[0063] Fig. 6 illustrates comparison of total RS and tropic acid for
atropine sulfate (0.025%)
formulation (Formulation 4) at pH 4.8 in H20.
[0064] Fig. 7 illustrates comparison of total RS and tropic acid for
atropine sulfate (0.01%)
formulation (Formulation 7) at pD 5.2 in D20.
[0065] Fig. 8 illustrates comparison of total RS and tropic acid for
atropine sulfate (0.01%)
formulation (Formulation 5) at pH 5.8 in H70.
[0066] Fig. 9 illustrates comparison of total RS and tropic acid for
atropine sulfate (0.025%)
formulation (Formulation 6) at pH 5.8 in 1+0.
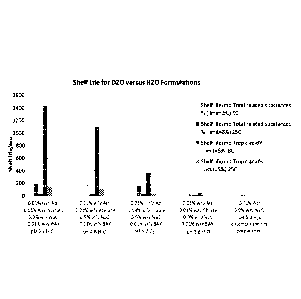
[0067] Fig. 10 illustrates estimated shelf lifes for D20 and H20
formulations disclosed in
Examples 11 and 12.
DETAILED DESCRIPTION OF THE DISCLOSURE
[0068] The present disclosure recognizes that there is a need for a
stabilized ophthalmic
composition with extended shelf life upon storage. The present disclosure also
recognizes that there is a
need for stabilizing an ophthalmic composition through arresting or reducing
hydrolysis of at least some
of its active agents. The present disclosure further recognizes that there is
a need for an ophthalmic
composition that provides convenient and effective delivery of a muscarinic
antagonist such as atropine
in the eye of a patient.
[0069] The present disclosure recognizes that muscarinic antagonist (e.g.
atropine or its
pharmaceutically acceptable salts) prevents or arrests the development of
myopia in humans, for example
as evidenced by reduction of the rate of increase of myopia in young people.
The present disclosure also
recognizes the effects of muscarinic antagonist (e.g. atropine or its
pharmaceutically acceptable salts) on
reduction of axial elongation and myopia in visually impaired chick eyes, and
on ocular growth and
muscarinic cholinergic receptors in young rhesus monkeys.
[0070] In addition, the present disclosure recognizes that systemic
absorption of muscarinic
antagonist (e.g. atropine) sometimes leads to undesirable side effect, and
that localized delivery of
muscarinic antagonist (e.g. atropine or its pharmaceutically acceptable salts)
reduces or prevents the
aforementioned systemic exposure.
[0071] Further, the present disclosure recognizes that some liquid
muscarinic antagonist (e.g.
atropine) compositions are formulated at a relatively lower pH range (e.g.
less than 4.5) for stability of
muscarinic antagonist (e.g. atropine or its pharmaceutically acceptable
salts). For some individuals, the
lower pH range in some instances causes discomfort or other side effects such
as pain or burning
sensation in the eye, which is prevented or alleviated by formulating
muscarinic antagonist (e.g. atropine)
compositions at higher pH ranges. For some individuals, the lower pH in some
instances elicits a tear
response which reduces the absorption of the drug in the eye and therefore the
effectiveness.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
[0072] Still further, the present disclosure recognizes that some
muscarinic antagonist (e.g.
atropine) liquid compositions formulated at lower concentrations (e.g. 0.001%
to 0.05%) present stability
challenges that are less so in higher concentrations (e.g. 0.1-1%). Without
wishing to be bound by any
particular theory, it is contemplated that the some muscarinic antagonist
(e.g. atropine) contributes to the
stability of an ophthalmic composition, such as an aqueous solution. For
example, the concentration of
the muscarinic antagonist (e.g. atropine) in some embodiments affects the pH
or pD of the ophthalmic
composition, such as with the muscarinic antagonist acting as a buffering
agent. Furthermore, the
concentration of the muscarinic antagonist (e.g. atropine) in some embodiments
affects the interaction
between the muscarinic antagonist and other ingredients of the ophthalmic
composition, which in turn
affects the stability of the ophthalmic composition.
[0073] Finally, the present disclosure recognizes that deuterated water
stabilizes ophthalmic
compositions. In some cases, the deuteratcd water is a weak acid as compared
to H20, as such deuterated
water comprises a lower concentration of the reactive species (e.g., -OD)
which in some instances leads
to base catalyzed hydrolysis of an active agent in the ophthalmic composition.
As such, in some
instances compositions comprising deuterated water leads to reduced base
catalyzed hydrolysis when
compared to compositions comprising H20. In some instances, deuterated water
further lowers the
buffering capacity of an ophthalmic composition, leading to less tear reflex
in the eye.
[0074] Myopia, axial elongation of the eye, affects a large proportion of
the population. The
onset of myopia is generally during the grade school years and progresses
until growth of the eye is
completed. The present disclosure recognizes the importance of compositions
and treatments for
preventing or arresting the development of myopia, especially compositions and
treatments that allow
convenient administration, reduce potential side effects, has suitable
stability, and/or provide relatively
consistent therapeutic effects.
[0075] Ophthalmic Muscarinic Antagonist Composition
[0076] Provided herein is an ophthalmic composition containing low
concentrations of an
ophthalmic agent. In some embodiments, the ophthalmic composition includes
from about 0.001 wt% to
about 0.05 wt% of an ophthalmic agent for treatment of an ophthalmic disorder
or condition; and an
ophthalmically acceptable carrier, wherein the ophthalmic agent is distributed
with substantial uniformity
throughout the ophthalmically acceptable carrier. In some instances, the
ophthalmic agent is a muscarinic
antagonist.
[0077] Provided herein is an ophthalmic composition containing low
concentrations of a
muscarinic antagonist. In some embodiments, the ophthalmic composition
includes from about 0.001
wt% to about 0.05 wt% of a muscarinic antagonist for treatment of an
ophthalmic disorder or condition;
and an ophthalmically acceptable carrier, wherein the muscarinic antagonist is
distributed with
substantial uniformity throughout the ophthalmically acceptable carrier.
[0078] In some instances, the muscarinic antagonist includes atropine,
atropine sulfate,
noratropine, atropine-N-oxide, tropine, tropic acid, atropine methonitrate,
diphenhydramine,
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
11
dimenhydrinate, dicyclomine, flavoxate, oxybutynin, tiotropium, hyoscine,
scopolomine (L-hyoscine),
hydroxyzine, ipratropium, tropicamide, cyclopentolate, pirenzapine,
homatropine, solifenacin,
darifenacin, benzatropine, mebeverine, procyclidine, aclidinium bromide,
trihexyphenidyl/benzhexol,
tolterodine, or a combination thereof In some instances, the muscarinic
antagonist includes atropine,
atropine sulfate, noratropine, atropine-N-oxide, tropine, tropic acid,
hyoscinc, scopolominc, tropicamide,
cyclopentolate, pirenzapine, homatropine, or a combination thereof In some
embodiments, the
muscarinic antagonist is atropine, or a pharmaceutically acceptable salt or
prodrug thereof In some
embodiments, the muscarinic antagonist is atropine sulfate.
[0079] In some embodiments, the ophthalmic composition comprise a
muscarinic antagonist
selected from atropine, atropine sulfate, noratropine, atropine-N-oxide,
tropine, tropic acid, atropine
methonitrate, diphenhydramine, dimenhydrinate, dicyclomine, flavoxate,
oxybutynin, tiotropium,
hyoscine, scopolomine (L-hyoscine), hydroxyzine, ipratropium, tropicamide,
cyclopentolate, pirenzapine,
homatropine, solifenacin, darifenacin, benzatropine, mebeverine, procyclidine,
aclidinium bromide,
trihexyphenidylibenzhexol, tolterodine, or a combination thereof In some
instances, the muscarinic
antagonist includes atropine, atropine sulfate, noratropine, atropine-N-oxide,
tropine, tropic acid,
hyoscine, scopolomine, tropicamide, cyclopentolate, pirenzapine, or
homatropine.
[0080] In some embodiments, the ophthalmic composition comprise two or more
muscarinic
antagonists in which the two or more muscarinic antagonists comprises
atropine, atropine sulfate,
noratropine, atropine-N-oxide, tropine, tropic acid, atropine methonitrate,
diphenhydramine,
dimenhydrinate, dicyclomine, flavoxate, oxybutynin, tiotropium, hyoscine,
scopolomine (L-hyoscine),
hydroxyzine, ipratropium, tropicamide, cyclopentolate, pirenzapine,
homatropine, solifenacin,
darifenacin, benzatropine, mebeverine, procyclidine, aclidinium bromide,
trihexyphenidyl/benzhexol,
tolterodine, or a combination thereof In some instances, the muscarinic
antagonist includes atropine,
atropine sulfate, noratropine, atropine-N-oxide, tropine, tropic acid,
hyoscine, scopolomine, tropicamide,
cyclopentolate, pirenzapine, homatropine, or any combination thereof
[0081] In some embodiments, the ophthalmic composition comprises one or
more muscarinic
antagonist in combination with one or more sympathetic agonists. In some
embodiments, the sympathetic
agonist is selected from phenylephrine or hydroxyamphetamine. In some
embodiments, the ophthalmic
composition comprises one or more of muscarinic antagonist: atropine, atropine
sulfate, noratropine,
atropine-N-oxide, tropine, tropic acid, atropine methonitrate,
diphenhydramine, dimenhydrinate,
dicyclomine, flavoxate, oxybutynin, tiotropium, hyoscine, scopolomine (L-
hyoscine), hydroxyzine,
ipratropium, tropicamide, cyclopentolate, pirenzapine, homatropine,
solifenacin, darifenacin,
benzatropine, mebeverine, procyclidine, aclidinium bromide,
trihexyphenidyl/benzhexol, or tolterodine;
in combination with one or more of sympathetic agonists: phenylephirine or
hydroxyamphetamine.
[0082] Provided herein is an ophthalmic composition containing low
concentrations of atropine
or its pharmaceutically acceptable salts. In some embodiments, the ophthalmic
composition includes
from about 0.001 wt% to about 0.05 wt% of atropine or its pharmaceutically
acceptable salts for
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
12
treatment of an ophthalmic disorder or condition; and an ophthalmically
acceptable carrier, wherein the
ophthalmic agent is distributed with substantial uniformity throughout the
ophthalmically acceptable
carrier.
[0083] Provided herein is an ophthalmic composition containing low
concentrations of atropine
sulfate. In some embodiments, the ophthalmic composition includes from about
0.001 wt% to about 0.05
wt% of atropine sulfate for treatment of an ophthalmic disorder or condition;
and an ophthalmically
acceptable carrier, wherein the ophthalmic agent is distributed with
substantial uniformity throughout the
ophthalmically acceptable carrier.
[0084] In some embodiments, the ophthalmic disorder or condition is pre-
myopia, myopia or
progression of myopia.
[0085] The present disclosure further recognizes that the clinical use of
atropine as a therapy has
been limited due to its ocular side effects including glare from pupillary
dilation and blurred vision due to
loss of accommodation. Without wishing to be bound by any particular theory,
it is contemplated that
the limited use of atropine against myopia development, include its ocular
side effects, is attributable to
the concentration of atropine used in known ophthalmic formulations (e.g. lwt%
or higher).
[0086] The present disclosure further recognizes the challenges present in
formulation of
compositions that contain low concentrations, especially very low
concentrations (e.g. from about 0.001
wt% to about 0.5 wt%), of ophthalmic agents, such as muscarinic antagonist
(e.g. atropine or its
pharmaceutically acceptable salts). In particular, pharmaceutical compositions
with ophthalmic agent at
such low concentrations are difficult to maintain dose-to-dose uniformity in
term of ophthalmic agent
content and/or distribution.
[0087] In some aspects, described herein are formulations or solutions of
muscarinic antagonist
(e.g., atropine) formulated in deuterated water. In some aspects, formulations
or solutions of muscarinic
antagonist (e.g., atropine) formulated in deuterated water are stable at
different temperatures, at different
relative humidity, with an acidic pD, and with a potency of at least 80%
relative to the ophthalmic agent.
In additional aspects, formulations or solutions of muscarinic antagonist
(e.g., atropine) formulated in
deuterated water has a lowered buffering capacity. In such instances, the
lowered buffering capacity of
the ophthalmic formulations or solutions when administered into the eye allows
the ophthalmic
formulation or solution to reach physiological pH at a faster rate than
compared to an equivalent
ophthalmic formulation or solution formulated in H20.
[0088] In some aspects, described herein are formulations of muscarinic
antagonist (e.g.
atropine) at low concentrations that does not have a dose-to-dose variation.
In some aspects, described
herein are formulations of muscarinic antagonist (e.g. atropine) at low
concentrations that are stable at
different temperatures, at different relative humidity, with an acidic pD, and
with a potency of at least
80% relative to the ophthalmic agent.
[0089] In other aspects, described herein include formulating the
ophthalmic composition as an
ophthalmic gel or an ophthalmic ointment. For example, some ophthalmic gel or
an ophthalmic ointment
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
13
described herein allows desirable dose-to-dose uniformity, reduced or limited
systemic exposure, or
combinations thereof.
[0090] Ophthalmic Solution Muscarinic Anta2onist Composition
[00911 Disclosed herein, in certain embodiments, is an ophthalmic
composition formulated as
an aqueous solution. In some embodiments, the ophthalmic composition comprises
from about 0.001
wt% to about 0.05 wt% of a muscarinic antagonist and deuterated water. As used
herein, deuterated
water refers to D20, DHO, heavy water, and/or deuterium oxide.
[0092] In some embodiments, the composition comprises at least about 80% of
the ophthalmic
agent (e.g. muscarinic antagonist) for an extended period of time under
storage condition. In some
embodiments, the composition comprises at least about 81% of the ophthalmic
agent (e.g. muscarinic
antagonist) for an extended period of time under storage condition. In some
embodiments, the
composition comprises at least about 82% of the ophthalmic agent (e.g.
muscarinic antagonist) for an
extended period of time under storage condition. In some embodiments, the
composition comprises at
least about 83% of the ophthalmic agent (e.g. muscarinic antagonist) for an
extended period of time
under storage condition. In some embodiments, the composition comprises at
least about 84% of the
ophthalmic agent (e.g. muscarinic antagonist) for an extended period of time
under storage condition. In
some embodiments, the composition comprises at least about 85% of the
ophthalmic agent (e.g.
muscarinic antagonist) for an extended period of time under storage condition.
In some embodiments, the
composition comprises at least about 86% of the ophthalmic agent (e.g.
muscarinic antagonist) for an
extended period of time under storage condition. In some embodiments, the
composition comprises at
least about 87% of the ophthalmic agent (e.g. muscarinic antagonist) for an
extended period of time
under storage condition. In some embodiments, the composition comprises at
least about 88% of the
ophthalmic agent (e.g. muscarinic antagonist) for an extended period of time
under storage condition. In
some embodiments, the composition comprises at least about 89% of the
ophthalmic agent (e.g.
muscarinic antagonist) for an extended period of time under storage condition.
In some embodiments, the
composition comprises at least about 90% of the ophthalmic agent (e.g.
muscarinic antagonist) for an
extended period of time under storage condition. In some embodiments, the
composition comprises at
least about 91% of the ophthalmic agent (e.g. muscarinic antagonist) for an
extended period of time
under storage condition. In some embodiments, the composition comprises at
least about 92% of the
ophthalmic agent (e.g. muscarinic antagonist) for an extended period of time
under storage condition. In
some embodiments, the composition comprises at least about 93% of the
ophthalmic agent (e.g.
muscarinic antagonist) for an extended period of time under storage condition.
In some embodiments, the
composition comprises at least about 94% of the ophthalmic agent (e.g.
muscarinic antagonist) for an
extended period of time under storage condition. In some embodiments, the
composition comprises at
least about 95% of the ophthalmic agent (e.g. muscarinic antagonist) for an
extended period of time
under storage condition. In some embodiments, the composition comprises at
least about 96% of the
ophthalmic agent (e.g. muscarinic antagonist) for an extended period of time
under storage condition. In
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
14
some embodiments, the composition comprises at least about 97% of the
ophthalmic agent (e.g.
muscarinic antagonist) for an extended period of time under storage condition.
In some embodiments, the
composition comprises at least about 98% of the ophthalmic agent (e.g.
muscarinic antagonist) for an
extended period of time under storage condition. In some embodiments, the
composition comprises at
least about 99% of the ophthalmic agent (e.g. muscarinic antagonist) for an
extended period of time
under storage condition.
[0093] In some embodiments, the composition has a potency of at least about
80% after
extended period of time under storage condition. In some embodiments, the
composition has a potency of
at least about 81% after extended period of time under storage condition. In
some embodiments, the
composition has a potency of at least about 82% after extended period of time
under storage condition.
In some embodiments, the composition has a potency of at least about 83% after
extended period of time
under storage condition. In some embodiments, the composition has a potency of
at least about 84%
after extended period of time under storage condition. In some embodiments,
the composition has a
potency of at least about 85% after extended period of time under storage
condition. In some
embodiments, the composition has a potency of at least about 86% after
extended period of time under
storage condition. In some embodiments, the composition has a potency of at
least about 87% after
extended period of time under storage condition. In some embodiments, the
composition has a potency
of at least about 88% after extended period of time under storage condition.
In some embodiments, the
composition has a potency of at least about 89% after extended period of time
under storage condition.
In some embodiments, the composition has a potency of at least 90% after
extended period of time under
storage condition. In some embodiments, the composition has a potency of at
least 91% after extended
period of time under storage condition. In some embodiments, the composition
has a potency of at least
92% after extended period of time under storage condition. In some
embodiments, the composition has a
potency of at least 93% after extended period of time under storage condition.
In some embodiments, the
composition has a potency of at least 94% after extended period of time under
storage condition. In
some embodiments, the composition has a potency of at least 95% after extended
period of time under
storage condition. In some embodiments, the composition has a potency of at
least 96% after extended
period of time under storage condition. In some embodiments, the composition
has a potency of at least
97% after extended period of time under storage condition. In some
embodiments, the composition has a
potency of at least 98% after extended period of time under storage condition.
In some embodiments, the
composition has a potency of at least 99% after extended period of time under
storage condition.
[0094] In some embodiments, the extended period of time is at least 1 week.
In some
embodiments, the extended period of time is at least 2 weeks. In some
embodiments, the extended period
of time is at least 3 weeks. In some embodiments, the extended period of time
is at least 1 month. In
some embodiments, the extended period of time is at least 2 months. In some
embodiments, the extended
period of time is at least 3 months. In some embodiments, the extended period
of time is at least 4
months. In some embodiments, the extended period of time is at least 5 months.
In some embodiments,
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
the extended period of time is at least 6 months. In some embodiments, the
extended period of time is at
least 7 months. In some embodiments, the extended period of time is at least 8
months. In some
embodiments, the extended period of time is at least 9 months. In some
embodiments, the extended
period of time is at least 10 months. In some embodiments, the extended period
of time is at least 11
months. In some embodiments, the extended period of time is at least 12 months
(i.e. 1 year). In some
embodiments, the extended period of time is at least 18 months (i.e. 1.5
years). In some embodiments,
the extended period of time is at least 24 months (i.e. 2 years). In some
embodiments, the extended
period of time is at least 36 months (i.e. 3 years). In some embodiments, the
extended period of time is at
least 3 years. In some embodiments, the extended period of time is at least 5
years, or more.
[0095] In some embodiments, the temperature of the storage condition is
between about 20 C
and about 70 C. In some embodiments, the temperature of the storage condition
is between about 25 C
and about 65 C, about 30 C and about 60 C, about 35 C and about 55 C, or about
40 C and about 50 C.
In some embodiments, the temperature of the storage condition is about 25 C.
In some embodiments, the
temperature of the storage condition is about 40 C. In some embodiments, the
temperature of the storage
condition is about 60 C.
[0096] In some embodiments, the relative humidity of the storage condition
is between about
50% and about 80%, or between about 60% and about 75%. In some embodiments,
the relative humidity
of the storage condition is about 60%. In some embodiments, the relative
humidity of the storage
condition is about 75%.
[0097] In some embodiments, the composition comprises less than 60% of H20.
In some
embodiments, the composition comprises less than 55% of H20. In some
embodiments, the composition
comprises less than 50% of H20. In some embodiments, the composition comprises
less than 45% of
H20. In some embodiments, the composition comprises less than 40% of H20. In
some embodiments,
the composition comprises less than 35% of H20. In some embodiments, the
composition comprises less
than 30% of H20. In some embodiments, the composition comprises less than 25%
of H20. In some
embodiments, the composition comprises less than 20% of H20. In some
embodiments, the composition
comprises less than 15% of H20. In some embodiments, the composition comprises
less than 10% of
H20.
[0098] In some embodiments, the composition comprises from less than 5% of
H20 to 0% of
H2O. In some embodiments, the composition comprises less than 5% of H20. In
some embodiments, the
composition comprises less than 4.5% of H20. In some embodiments, the
composition comprises less
than 4% of H20. In some embodiments, the composition comprises less than 3.5%
of H20. In some
embodiments, the composition comprises less than 3% of H20. In some
embodiments, the composition
comprises less than 2.5% of H20. In some embodiments, the composition
comprises less than 2% of
H20. In some embodiments, the composition comprises less than 1.5% of H20. In
some embodiments,
the composition comprises less than 1% of H20. In some embodiments, the
composition comprises less
than 0.5% of H20. In some embodiments, the composition comprises less than
0.4% of H20. In some
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
16
embodiments, the composition comprises less than 0.3% of H20. In some
embodiments, the composition
comprises less than 0.2% of 1120. In some embodiments, the composition
comprises less than 0.1% of
H20. In some embodiments, the composition comprises 0% of H20.
[0099] In some embodiments, the composition has a pD of between about 4 and
about 8, about
4.5 and about 7.8, about 5 and about 7.5, or about 5.5 and about 7. In some
embodiments, the
composition has a pD of less than about 7.5. In some embodiments, the
composition has a pD of less
than about 7.4. In some embodiments, the composition has a pD of less than
about 7.3. In some
embodiments, the composition has a pD of less than about 7.2. In some
embodiments, the composition
has a pD of less than about 7.1. In some embodiments, the composition has a pD
of less than about 7. In
some embodiments, the composition has a pD of less than about 6.9. In some
embodiments, the
composition has a pD of less than about 6.8. In some embodiments, the
composition has a pD of less
than about 6.7. In some embodiments, the composition has a pD of less than
about 6.6. In some
embodiments, the composition has a pD of less than about 6.5. In some
embodiments, the composition
has a pD of less than about 6.4. In some embodiments, the composition has a pD
of less than about 6.3.
In some embodiments, the composition has a pD of less than about 6.2. In some
embodiments, the
composition has a pD of less than about 6.1. In some embodiments, the
composition has a pD of less
than about 6. In some embodiments, the composition has a pD of less than about
5.9. In some
embodiments, the composition has a pD of less than about 5.8. In some
embodiments, the composition
has a pD of less than about 5.7. In some embodiments, the composition has a pD
of less than about 5.6.
In some embodiments, the composition has a pD of less than about 5.5. In some
embodiments, the
composition has a pD of less than about 5.4. In some embodiments, the
composition has a pD of less than
about 5.3. in some embodiments, the composition has a pD of less than about
5.2. in some embodiments,
the composition has a pD of less than about 5.1. In some embodiments, the
composition has a pD of less
than about 5. In some embodiments, the composition has a pD of less than about
4.9. In some
embodiments, the composition has a pD of less than about 4.8. In some
embodiments, the composition
has a pD of less than about 4.7. In some embodiments, the composition has a pD
of less than about 4.6.
In some embodiments, the composition has a pD of less than about 4.5. In some
embodiments, the
composition has a pD of less than about 4.4. In some embodiments, the
composition has a pD of less than
about 4.3. In some embodiments, the composition has a pD of less than about
4.2. In some embodiments,
the composition has a pD of less than about 4.1. In some embodiments, the
composition has a pD of less
than about 4.
[00100] In some embodiments, the composition comprising deuterated water
has a lowered
buffering capacity than an equivalent composition comprising FLO. As described
elsewhere herein, in
some embodiments, the lowered buffering capacity allows the composition
comprising deuterated water
to normalize to physiological pH at a faster rate than a composition
comprising H20. In some
embodiments, the lowered buffering capacity allows the composition to induce
less tear reflex than an
equivalent composition comprising H20.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
17
[00101] In some instances, the composition comprising deuterated water
stabilizes muscarinic
antagonist (e.g., atropine). In some embodiments, this is due to a lower
concentration of the reactive
species (e.g., -OD) in the D20 aqueous system compared to the concentration of
the reactive species
(e.g., -OH) in an equivalent F120 aqueous system. In some cases, base
catalyzed hydrolysis leads to the
presence of tropine degradant from atropine. In some cases, with a lower
concentration of the reactive
species that causes tropine degradant formation, atropine solution is more
stable in a D20 aqueous system
than compared to an equivalent H20 aqueous system. In some embodiments, the
ophthalmic
composition formulated with deuterated water allows for a more stable
ophthalmic composition relative
to the ophthalmic composition formulated with H20.
[00102] In some embodiments, the composition comprises less than 20% of
major degradant
based on the concentration of the ophthalmic agent after extended period of
time under storage condition.
In some embodiments, the composition comprises less than 15% of major
degradant based on the
concentration of the ophthalmic agent after extended period of time under
storage condition.
[00103] In some embodiments, the composition comprises less than 10% of
major degradant
based on the concentration of the ophthalmic agent after extended period of
time under storage condition.
In some embodiments, the composition comprises less than 5% of major degradant
based on the
concentration of the ophthalmic agent after extended period of time under
storage condition. In some
embodiments, the composition comprises less than 2.0% of major degradant based
on the concentration
of the ophthalmic agent after extended period of time under storage condition.
In some embodiments, the
composition comprises less than 1.5% of major degradant based on the
concentration of the ophthalmic
agent after extended period of time under storage condition. In some
embodiments, the composition
comprises less than 1.0% of major degradant based on the concentration of the
ophthalmic agent after
extended period of time under storage condition. In some embodiments, the
composition comprises less
than 0.5% of major degradant based on the concentration of the ophthalmic
agent after extended period
of time under storage condition. In some embodiments, the composition
comprises less than 0.4% of
major degradant based on the concentration of the ophthalmic agent after
extended period of time under
storage condition. In some embodiments, the composition comprises less than
0.3% of major degradant
based on the concentration of the ophthalmic agent after extended period of
time under storage condition.
In some embodiments, the composition comprises less than 0.2% of major
degradant based on the
concentration of the ophthalmic agent after extended period of time under
storage condition. In some
embodiments, the composition comprises less than 0.1% of major degradant based
on the concentration
of the ophthalmic agent after extended period of time under storage condition.
In some embodiments, the
major degradant is tropic acid.
[00104] In some embodiments, the primary degradant is an early eluting
related substance at
RRT of 0.87-0.89 according to the UPLC method described herein (Table 10). In
some instances, the
early eluting related substance is referred to as RRT 0.87-0.89. In some
embodiments, the primary
degradant is RRT 0.87-0.89.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
18
[00105] Ophthalmic Muscarinic Antagonist Concentration
[00106] In some embodiments, the compositions described herein have a
concentration of
ophthalmic agent between about 0.001% to about 0.050%, between about 0.005% to
about 0.050%,
between about 0.010% to about 0.050%, between about 0.015% to about 0.050%,
between about 0.020%
to about 0.050%, between about 0.025% to about 0.050%, between about 0.030% to
about 0.050%,
between about 0.035% to about 0.050%, between about 0.040% to about 0.050%, or
between about
0.045% to about 0.050% of the ophthalmic agent, or pharmaceutically acceptable
prodrug or salt thereof,
by weight of the composition. In some instances, the prodrug of the ophthalmic
agent (e.g. muscarinic
antagonist) is chemically converted into the ophthalmic agent (e.g. muscarinic
antagonist) after the
administration of the ophthalmic composition. In a non-limiting example, the
muscarinic antagonist
prodrug has a chemical bond that is cleavable by one or more enzymes in tears.
In some embodiments,
the ophthalmic agent is a muscarinic antagonist. In some embodiments, the
muscarinic antagonist
comprises atropine, atropine sulfate, noratropine, atropine-N-oxide, tropine,
tropic acid, hyoscine,
scopolomine, tropicamide, cyclopentolate, pirenzapine, homatropine, or a
combination thereof. In some
embodiments, the muscarinic antagonist is atropine, or a pharmaceutically
acceptable salt thereof. In
some embodiments, the muscarinic antagonist is atropine sulfate. As described
herein, the ophthalmic
agent includes optically pure stereoisomers, optically enriched stereoisomers,
and a racemic mixture of
stereoisomers. For example, some ophthalmic compositions disclosed herein
includes atropine or
atropine sulfate in which the atropine is a racemic mixture of D- and L-
isomers; and some ophthalmic
compositions disclosed herein includes atropine or atropine sulfate in which
the atropine is a optically
enriched in favor of the more ophthalmically active L-isomer.
[00107] In some embodiments, the compositions described herein have a
concentration of
ophthalmic agent between about 0.001% to about 0.045%, between about 0.005% to
about 0.045%,
between about 0.010% to about 0.045%, between about 0.015% to about 0.045%,
between about 0.020%
to about 0.045%, between about 0.025% to about 0.045%, between about 0.030% to
about 0.045%,
between about 0.035% to about 0.045%, or between about 0.040% to about 0.045%
of the ophthalmic
agent, or pharmaceutically acceptable prodrug or salt thereof, by weight of
the composition. In some
embodiments, the ophthalmic agent is a muscarinic antagonist. In some
embodiments, the muscarinic
antagonist comprises atropine, atropine sulfate, noratropine, atropine-N-
oxide, tropine, tropic acid,
hyoscine, scopolomine, tropicamide, cyclopentolate, pirenzapine, homatropine,
or a combination thereof
In some embodiments, the muscarinic antagonist is atropine, or a
pharmaceutically acceptable salt
thereof. In some embodiments, the muscarinic antagonist is atropine sulfate.
[00108] In some embodiments, the compositions described herein have a
concentration of
ophthalmic agent between about 0.001% to about 0.040%, between about 0.005% to
about 0.040%,
between about 0.010% to about 0.040%, between about 0.015% to about 0.040%,
between about 0.020%
to about 0.040%, between about 0.025% to about 0.040%, between about 0.030% to
about 0.040%,
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
19
between about 0.035% to about 0.040% of the active ingredient, or
pharmaceutically acceptable prodrug
or salt thereof, by weight of the composition. In some embodiments, the
ophthalmic agent is a
muscarinic antagonist. In some embodiments, the muscarinic antagonist
comprises atropine, atropine
sulfate, noratropine, atropine-N-oxide, tropine, tropic acid, hyoscine,
scopolomine, tropicamide,
cyclopentolate, pirenzapine, homatropine, or a combination thereof In some
embodiments, the
muscarinic antagonist is atropine, or a pharmaceutically acceptable salt
thereof. In some embodiments,
the muscarinic antagonist is atropine sulfate.
[00109] In some embodiments, the compositions described herein have a
concentration of
ophthalmic agent between about 0.001% to about 0.035%, between about 0.005% to
about 0.035%,
between about 0.010% to about 0.035%, between about 0.015% to about 0.035%,
between about 0.020%
to about 0.035%, between about 0.025% to about 0.035%, or between about 0.030%
to about 0.035% of
the ophthalmic agent, or pharmaceutically acceptable prodrug or salt thereof,
by weight of the
composition. In some embodiments, the ophthalmic agent is a muscarinic
antagonist. In some
embodiments, the muscarinic antagonist comprises atropine, atropine sulfate,
noratropine, atropinc-N-
oxide, tropine, tropic acid, hyoscine, scopolomine, tropicamide,
cyclopentolate, pirenzapine,
homatropine, or a combination thereof. In some embodiments, the muscarinic
antagonist is atropine, or a
pharmaceutically acceptable salt thereof In some embodiments, the muscarinic
antagonist is atropine
sulfate.
[00110] In some embodiments, the compositions described herein have a
concentration of
ophthalmic agent between about 0.001% to about 0.030%, between about 0.005% to
about 0.030%,
between about 0.010% to about 0.030%, between about 0.015% to about 0.030%,
between about 0.020%
to about 0.030%, or between about 0.025% to about 0.030% of the active
ingredient, or phallnaceutically
acceptable prodrug or salt thereof, by weight of the composition. In some
embodiments, the ophthalmic
agent is a muscarinic antagonist. In some embodiments, the muscarinic
antagonist comprises atropine,
atropine sulfate, noratropine, atropine-N-oxide, tropine, tropic acid,
hyoscine, scopolomine, tropicamide,
cyclopentolate, pirenzapine, homatropine, or a combination thereof In some
embodiments, the
muscarinic antagonist is atropine, or a pharmaceutically acceptable salt
thereof. In some embodiments,
the muscarinic antagonist is atropine sulfate.
[00111] In some embodiments, the compositions described herein have a
concentration of
ophthalmic agent between about 0.001% to about 0.025%, between about 0.005% to
about 0.025%,
between about 0.010% to about 0.025%, between about 0.015% to about 0.025%, or
between about
0.020% to about 0.025% of the ophthalmic agent, or pharmaceutically acceptable
prodrug or salt thereof,
by weight of the composition. In some embodiments, the ophthalmic agent is a
muscarinic antagonist. In
some embodiments, the muscarinic antagonist comprises atropine, atropine
sulfate, noratropine, atropine-
N-oxide, tropine, tropic acid, hyoscine, scopolomine, tropicamide,
cyclopentolate, pirenzapine,
homatropine, or a combination thereof In some embodiments, the muscarinic
antagonist is atropine, or a
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
pharmaceutically acceptable salt thereof. In some embodiments, the muscarinic
antagonist is atropine
sulfate.
[00112] In some embodiments, the compositions described herein have a
concentration of
ophthalmic agent between about 0.001% to about 0.020%, between about 0.005% to
about 0.020%,
between about 0.010% to about 0.020%, or between about 0.015% to about 0.020%
of the active
ingredient, or pharmaceutically acceptable prodrug or salt thereof, by weight
of the composition. In
some embodiments, the ophthalmic agent is a muscarinic antagonist. In some
embodiments, the
muscarinic antagonist comprises atropine, atropine sulfate, noratropine,
atropine -N-oxide, tropine, tropic
acid, hyoscine, scopolomine, tropicamide, cyclopentolate, pirenzapine,
homatropine, or a combination
thereof. In some embodiments, the muscarinic antagonist is atropine, or a
pharmaceutically acceptable
salt thereof. In some embodiments, the muscarinic antagonist is atropine
sulfate.
[00113] In some embodiments, the compositions described herein have a
concentration of
ophthalmic agent between about 0.001% to about 0.015%, between about 0.005% to
about 0.015%, or
between about 0.010% to about 0.015% of the ophthalmic agent, or
pharmaceutically acceptable prodrug
or salt thereof, by weight of the composition. In some embodiments, the
ophthalmic agent is a muscarinic
antagonist. In some embodiments, the muscarinic antagonist comprises atropine,
atropine sulfate,
noratropine, atropine-N-oxide, tropine, tropic acid, hyoscine, scopolomine,
tropicamide, cyclopentolate,
pirenzapine, homatropine, or a combination thereof In some embodiments, the
muscarinic antagonist is
atropine, or a pharmaceutically acceptable salt thereof. In some embodiments,
the muscarinic antagonist
is atropine sulfate.
[00114] In some embodiments, the compositions described herein have a
concentration of
ophthalmic agent between about 0.001% to about 0.010%, between about 0.005% to
about 0.010%, or
between about 0.008% to about 0.010% of the ophthalmic agent, or
pharmaceutically acceptable prodrug
or salt thereof, by weight of the composition. In some embodiments, the
ophthalmic agent is a
muscarinic antagonist. In some embodiments, the muscarinic antagonist
comprises atropine, atropine
sulfate, noratropine, atropine-N-oxide, tropine, tropic acid, hyoscine,
scopolomine, tropicamide,
cyclopentolate, pirenzapine, homatropine, or a combination thereof In some
embodiments, the
muscarinic antagonist is atropine, or a pharmaceutically acceptable salt
thereof. In some embodiments,
the muscarinic antagonist is atropine sulfate.
[00115] In some embodiments, the compositions described herein have a
concentration of
ophthalmic agent about 0.001%, 0.005%, 0.010%, 0.015%, 0.020%, 0.025%, 0.030%,
0.035%, 0.040%,
0.045%, or 0.050% of the ophthalmic agent, or pharmaceutically acceptable
prodrug or salt thereof, by
weight of the composition. In some embodiments, the ophthalmic agent is a
muscarinic antagonist. In
some embodiments, the muscarinic antagonist comprises atropine, atropine
sulfate, noratropine, atropine-
N-oxide, tropine, tropic acid, hyoscine, scopolomine, tropicamide,
cyclopentolate, pirenzapine,
homatropine, or a combination thereof In some embodiments, the muscarinic
antagonist is atropine, or a
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
21
pharmaceutically acceptable salt thereof. In some embodiments, the muscarinic
antagonist is atropine
sulfate.
[00116] Without wishing to be bound by any particular theory, it is
contemplated herein that the
low concentration of the ophthalmic agent (e.g. muscarinic antagonist such as
atropine or atropine
sulfate) in the disclosed ophthalmic composition provides sufficient and
consistent therapeutic benefits to
an individual in need thereof, while reducing or avoiding the ocular side
effects including glare from
pupillary dilation and blurred vision due to loss of accommodation that are
associated with ophthalmic
formulations containing higher concentrations of the ophthalmic agent (e.g.
muscarinic antagonist such
as atropine or atropine sulfate).
[00117] Aqueous Solution Stability
[00118] In some embodiments, the composition described herein comprises a
buffer. In some
embodiments, a buffer is selected from borates, borate-polyol complexes,
phosphate buffering agents,
citrate buffering agents, acetate buffering agents, carbonate buffering
agents, organic buffering agents,
amino acid buffering agents, or combinations thereof. In some embodiments, the
composition described
herein comprises buffer comprising deuterated water. In some embodiments, a
deuterated buffer is
selected from borates, borate-polyol complexes, phosphate buffering agents,
citrate buffering agents,
acetate buffering agents, carbonate buffering agents, organic buffering
agents, amino acid buffering
agents, or combinations thereof, formulated in deuterated water.
[00119] In some instances, borates include boric acid, salts of boric acid,
other pharmaceutically
acceptable borates, and combinations thereof. in some cases, borates include
boric acid, sodium borate,
potassium borate, calcium borate, magnesium borate, manganese borate, and
other such borate salts.
[00120] As used herein, the term polyol includes any compound having at
least one hydroxyl
group on each of two adjacent carbon atoms that are not in trans configuration
relative to each other. In
some embodiments, the polyols is linear or cyclic, substituted or
unsubstituted, or mixtures thereof, so
long as the resultant complex is water soluble and pharmaceutically
acceptable. In some instances,
examples of polyol include: sugars, sugar alcohols, sugar acids and uronic
acids. In some cases, polyols
include, but are not limited to: mannitol, glycerin, xylitol and sorbitol.
[00121] In some embodiments, phosphate buffering agents include phosphoric
acid; alkali metal
phosphates such as disodiurn hydrogen phosphate, sodium dihydrogen phosphate,
trisodium phosphate,
dipotassium hydrogen phosphate, potassium dihydrogen phosphate, and
tripotassium phosphate; alkaline
earth metal phosphates such as calcium phosphate, calcium hydrogen phosphate,
calcium dihydrogen
phosphate, monomagnesium phosphate, dimagnesium phosphate (magnesium hydrogen
phosphate), and
trimagnesium phosphate; ammonium phosphates such as diammonium hydrogen
phosphate and
ammonium dihydrogen phosphate; or a combination thereof. In some instances,
the phosphate buffering
agent is an anhydride. In some instances, the phosphate buffering agent is a
hydrate.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
22
[00122] In some embodiments, borate-polyol complexes include those
described in U.S. Pat. No.
6,503,497. In some instances, the borate-polyol complexes comprise borates in
an amount of from about
0.01 to about 2.0% wiv, and one or more polyols in an amount of from about
0.01% to about 5.0% w/v.
[00123] In some cases, citrate buffering agents include citric acid and
sodium citrate.
[00124] In some instances, acetate buffering agents include acetic acid,
potassium acetate, and
sodium acetate.
[00125] In some instances, carbonate buffering agents include sodium
bicarbonate and sodium
carbonate.
[00126] In some cases, organic buffering agents include Good's Buffer, such
as for example 2-
(N-morpholino)ethanesulfonic acid (MES), N-(2-Acetamido)iminodiacetic acid, N-
(Garb amoylmethyDiminodiac ctic acid (ADA), piperazinc-N,N'-bis(2-
ethanesulfonic acid (PIPES), N-(2-
acetamido)-2-aminoethanesulfonic acid (ACES), 3-Hydroxy-4-
morpholinepropanesulfonic acid, 3-
Morpholino-2-hydroxypropanesulfonic acid (MOPS0), cholamine chloride, 3-(N-
morpholino)propansulfonic acid (MOPS), N,N-bis(2-hydroxyethyl)-2-
aminoethanesulfonic acid (BES),
2-[(2-Hydroxy-1,1-bis(hydroxymethyl)ethyDamino]ethanesulfonic acid (TES), 4-(2-
hydroxyethyl)-1-
piperazineethanesulfonic acid (HEPES), 3-(N,N-Bis[2-hydroxyetbyl]amino)-2-
hydroxypropanesulfonic
acid (DIPSO), acetamidoglycine, 3- {[1,3-Dihydroxy-2-(hydroxymethyl)-2-
propanyl]aminol -2-hydroxy-
1 -propanesulfonic acid (TAPSO), piperazine-1,4,-bis (2-
hydroxypropanesulphonic acid) (POPSO), 4-(2-
hydroxyethyppiperazine-1-(2-hydroxypropanesulfonic acid) hydrate (HEPPSO), 344-
(2-hydroxyethyl)-
1-piperazinyl]propanesulfonic acid (HEPPS), tricine, glycinamide, bicine or N-
tris(hydroxymethyl)methy1-3-aminopropanesulfonic acid sodium (TAPS); glycine;
and diethanolamine
(DEA).
[00127] In some cases, amino acid buffering agents include taurinc,
aspartic acid and its salts
(e.g., potassium salts, etc), E-aminocaproic acid, and the like.
[00128] In some instances, the composition described herein further
comprises a tonicity
adjusting agent. Tonicity adjusting agent is an agent introduced into a
preparation such as an ophthalmic
composition to reduce local irritation by preventing osmotic shock at the site
of application. In some
instances, buffer solution and/or a pD adjusting agent that broadly maintains
the ophthalmic solution at a
particular ion concentration and pD are considered as tonicity adjusting
agents. In some cases, tonicity
adjusting agents include various salts, such as halide salts of a monovalent
cation. In some cases, tonicity
adjusting agents include mannitol, sorbitol, dextrose, sucrose, urea, and
glycerin. In some instances,
suitable tonicity adjustors comprise sodium chloride, sodium nitrate, sodium
sulfate, sodium bisulfate,
potassium chloride, calcium chloride, magnesium chloride, zinc chloride,
potassium acetate, sodium
acetate, sodium bicarbonate, sodium carbonate, sodium thiosulfate, magnesium
sulfate, disodium
hydrogen phosphate, sodium dihydrogcn phosphate, potassium dihydrogen
phosphate, dextrose,
mannitol, sorbitol, dextrose, sucrose, urea, propylene glycol, glycerin, or a
combination thereof.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
23
[00129] In some instances, the concentration of the tonicity adjusting
agent in a composition
described herein is between about 0.5% and about 2.0%. In some instances, the
concentration of the
tonicity adjusting agent in a composition described herein is between about
0.7% and about 1.8%, about
0.8% and about 1.5%, or about 1% and about 1.3%. In some instances, the
concentration of the tonicity
adjusting agent is about 0.6%, 0.7%, 0.8%, 0.9%, 1.0%, 1.1%, 1.2%, 1.3%, 1.4%,
1.5%, 1.6%, 1.7%,
1.8%, or 1.9%. In some cases, the percentage is a weight percentage.
[00130] In some cases, the composition described herein further comprises a
pD adjusting agent.
In some embodiments, the pD adjusting agent used is an acid or a base. In some
embodiments, the base
is oxides, hydroxides, carbonates, bicarbonates and the likes. In some
instances, the oxides are metal
oxides such as calcium oxide, magnesium oxide and the likes; hydroxides are of
alkali metals and
alkaline earth metals such as sodium hydroxide, potassium hydroxide, calcium
hydroxide and the likes or
their deuterated equivalents, and carbonates are sodium carbonate, sodium
bicarbonates, potassium
bicarbonates and the likes. In some instances, the acid is mineral acid and
organic acids such as
hydrochloric acid, nitric acid, phosphoric acid, acetic acid, citric acid,
fumaric acid, malic acid tartaric
acid and the likes or their deuterated equivalents. In some instances, the pD
adjusting agent includes, but
is not limited to, acetate, bicarbonate, ammonium chloride, citrate,
phosphate, pharmaceutically
acceptable salts thereof and combinations or mixtures thereof. In some
embodiments, the pD adjusting
agent comprises DC1 and Na0D.
[00131] In some instances, the composition has a pD of between about 4 and
about 8, about 4.5
and about 7.8, about 5 and about 7.5, or about 5.5 and about 7. In some
embodiments, the composition
has a pD of less than about 7.5. In some embodiments, the composition has a pD
of less than about 7.4.
In some embodiments, the composition has a pD of less than about 7.3. In some
embodiments, the
composition has a pD of less than about 7.2. In some embodiments, the
composition has a pD of less than
about 7.1. In some embodiments, the composition has a pD of less than about 7.
In some embodiments,
the composition has a pD of less than about 6.9. In some embodiments, the
composition has a pD of less
than about 6.8. In some embodiments, the composition has a pD of less than
about 6.7. Tn some
embodiments, the composition has a pD of less than about 6.6. In some
embodiments, the composition
has a pD of less than about 6.5. In some embodiments, the composition has a pD
of less than about 6.4.
In some embodiments, the composition has a pD of less than about 6.3. In some
embodiments, the
composition has a pD of less than about 6.2. In some embodiments, the
composition has a pD of less
than about 6.1. In some embodiments, the composition has a pD of less than
about 6. In some
embodiments, the composition has a pD of less than about 5.9. In some
embodiments, the composition
has a pD of less than about 5.8. In some embodiments, the composition has a pD
of less than about 5.7.
In some embodiments, the composition has a pD of less than about 5.6. In some
embodiments, the
composition has a pD of less than about 5.5. In some embodiments, the
composition has a pD of less than
about 5.4. In some embodiments, the composition has a pD of less than about
5.3. In some embodiments,
the composition has a pD of less than about 5.2. In some embodiments, the
composition has a pD of less
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
24
than about 5.1. In some embodiments, the composition has a pD of less than
about 5. In some
embodiments, the composition has a pD of less than about 4.9. In some
embodiments, the composition
has a pD of less than about 4.8. In some embodiments, the composition has a pD
of less than about 4.7.
In some embodiments, the composition has a pD of less than about 4.6. In some
embodiments, the
composition has a pD of less than about 4.5. In some embodiments, the
composition has a pD of less than
about 4.4. In some embodiments, the composition has a pD of less than about
4.3. In some embodiments,
the composition has a pD of less than about 4.2. In some embodiments, the
composition has a pD of less
than about 4.1. In some embodiments, the composition has a pD of less than
about 4. In some
embodiments, the pD is the pD of the composition after extended period of time
under storage condition.
[00132] In some instances, the composition has an initial pD of between
about 4 and about 8,
about 4.5 and about 7.8, about 5 and about 7.5, or about 5.5 and about 7. In
some embodiments, the
composition has an initial pD of about 7.5. In some embodiments, the
composition has an initial pD of
about 7.4. In some embodiments, the composition has an initial pD of about
7.3. In some embodiments,
the composition has an initial pD of about 7.2. In some embodiments, the
composition has an initial pD
of about 7.1. In some embodiments, the composition has an initial pD of about
7. In some embodiments,
the composition has an initial pD of about 6.9. In some embodiments, the
composition has an initial pD
of about 6.8. In some embodiments, the composition has an initial pD of about
6.7. In some
embodiments, the composition has an initial pD of about 6.6. In some
embodiments, the composition has
an initial pD of about 6.5. In some embodiments, the composition has an
initial pD of about 6.4. In
some embodiments, the composition has an initial pD of about 6.3. In some
embodiments, the
composition has an initial pD of about 6.2. In some embodiments, the
composition has an initial pD of
about 6.1. in some embodiments, the composition has an initial pD of about 6.
in some embodiments,
the composition has an initial pD of about 5.9. In some embodiments, the
composition has an initial pD
of about 5.8. In some embodiments, the composition has an initial pD of about
5.7. In some
embodiments, the composition has an initial pD of about 5.6. In some
embodiments, the composition has
an initial pD of about 5.5. In some embodiments, the composition has an
initial pD of about 5.4. In some
embodiments, the composition has an initial pD of about 5.3. In some
embodiments, the composition has
an initial pD of about 5.2. In some embodiments, the composition has an
initial pD of about 5.1. In some
embodiments, the composition has an initial pD of about 5. In some
embodiments, the composition has
an initial pD of about 4.9. In some embodiments, the composition has an
initial pD of about 4.8. In some
embodiments, the composition has an initial pD of about 4.7. In some
embodiments, the composition has
an initial pD of about 4.6. In some embodiments, the composition has an
initial pD of about 4.5. In some
embodiments, the composition has an initial pD of about 4.4. In some
embodiments, the composition has
an initial pD of about 4.3. In some embodiments, the composition has an
initial pD of about 4.2. In some
embodiments, the composition has an initial pD of about 4.1. In some
embodiments, the composition has
an initial pD of about 4.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
[00133] In some embodiments, the pD of the composition described herein is
associated with the
stability of the composition. In some embodiments, a stable composition
comprises a pD of between
about 4 and about 8, about 4.5 and about 7.8, about 5 and about 7.5, or about
5.5 and about 7. hi some
embodiments, a stable composition comprises a pD of less than about 7.5. In
some embodiments, a
stable composition comprises a pD of less than about 7.4. In some embodiments,
a stable composition
comprises a pD of less than about 7.3. In some embodiments, a stable
composition comprises a pD of
less than about 7.2. In some embodiments, a stable composition comprises a pD
of less than about 7.1. In
some embodiments, a stable composition comprises a pD of less than about 7. In
some embodiments, a
stable composition comprises a pD of less than about 6.9. In some embodiments,
a stable composition
comprises a pD of less than about 6.8. In some embodiments, a stable
composition comprises a pD of
less than about 6.7. In some embodiments, a stable composition comprises a pD
of less than about 6.6.
In some embodiments, a stable composition comprises a pD of less than about
6.5. In some
embodiments, a stable composition comprises a pD of less than about 6.4. In
some embodiments, a
stable composition comprises a pD of less than about 6.3. In some embodiments,
a stable composition
comprises a pD of less than about 6.2. In some embodiments, a stable
composition comprises a pD of
less than about 6.1. In some embodiments, a stable composition comprises a pD
of less than about 6. In
some embodiments, a stable composition comprises a pD of less than about 5.9.
In some embodiments, a
stable composition comprises a pD of less than about 5.8. In some embodiments,
a stable composition
comprises a pD of less than about 5.7. In some embodiments, a stable
composition comprises a pD of
less than about 5.6. In some embodiments, a stable composition comprises a pD
of less than about 5.5. In
some embodiments, a stable composition comprises a pD of less than about 5.4.
In some embodiments, a
stable composition comprises a pD of less than about 5.3. In some embodiments,
a stable composition
comprises a pD of less than about 5.2. In some embodiments, a stable
composition comprises a pD of
less than about 5.1. In some embodiments, a stable composition comprises a pD
of less than about 5. In
some embodiments, a stable composition comprises a pD of less than about 4.9.
In some embodiments, a
stable composition comprises a pD of less than about 4.8. In some embodiments,
a stable composition
comprises a pD of less than about 4.7. In some embodiments, a stable
composition comprises a pD of
less than about 4.6. In some embodiments, a stable composition comprises a pD
of less than about 4.5. In
some embodiments, a stable composition comprises a pD of less than about 4.4.
In some embodiments, a
stable composition comprises a pD of less than about 4.3. In some embodiments,
a stable composition
comprises a pD of less than about 4.2. In some embodiments, a stable
composition comprises a pD of
less than about 4.1. In some embodiments, a stable composition comprises a pD
of less than about 4.
[00134] As described elsewhere herein, in some instances, the D20 aqueous
system stabilizes a
muscarinic antagonist (e.g., atropine). In some embodiments, this is due to a
lower concentration of the
reactive species (e.g., -OD) in the D20 aqueous system compared to the
concentration of the reactive
species (e.g., -OH) in an equivalent H20 aqueous system. In some instances,
the concentration of the
reactive species (e.g., -OD) in the DA) aqueous system is about one third less
than the concentration of
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
26
the reactive species (e.g., -OH) in the equivalent H20 aqueous system. In some
cases, this is due to a
lower or smaller dissociation constant of D20 than H20. For example, the
Ka(H2O) is 1x10-14, whereas
the Ka(D20) is 1x10-15. As such, D20 is a weaker acid than H20. In some cases,
base catalyzed hydrolysis
leads to the presence of tropine degradant from atropine. In some cases, with
a lower concentration of the
reactive species that causes tropinc dcgradant formation, atropine solution is
more stable in a D20
aqueous system than compared to an equivalent H20 aqueous system. In some
embodiments, the
ophthalmic composition formulated with deuterated water allows for a more
stable ophthalmic
composition relative to the ophthalmic composition formulated with H2O.
[00135] In some embodiments, the presence of deuterated water shifts the
pKa of the buffer. In
some embodiments, the presence of deuterated water allows for the ophthalmic
composition to simulate
the stability of a lower pH system. In some instances, the buffer capacity of
the ophthalmic composition
is lowered, thereby allowing a faster shift in pH. In some instances, the
lowered buffering capacity of the
ophthalmic composition when administered into the eye allows the ophthalmic
composition to reach
physiological pH at a faster rate than compared to an ophthalmic composition
formulated in H2O. In
some instances, the ophthalmic composition formulated with deuterated water
allows for a lower tear
production, or less tear reflex in the eye, in comparison with an ophthalmic
composition formulated with
H20.
[00136] In some instances, the composition described herein further
comprises a disinfecting
agent. In some cases, disinfecting agents include polymeric biguanides,
polymeric quarternary
ammonium compounds, chlorites, bisbiguanides, chlorite compounds (e.g.
potassium chlorite, sodium
chlorite, calcium chlorite, magnesium chlorite, or mixtures thereof), and a
combination thereof.
[00137] In some instances, the composition described herein further
comprises a preservative. In
some cases, a preservative is added at a concentration to a composition
described herein to prevent the
growth of or to destroy a microorganism introduced into the composition. In
some instances,
microorganisms refer to bacteria (e.g. Proteus mirabilis, Serratia marcesens),
virus (e.g. Herpes simplex
virus, herpes zoster virus), fungus (e.g. fungi from the genus Fusarium),
yeast (e.g. Candida albicans),
parasites (e.g. Plasmodium spp., Gnathostoma spp.), protozoan (e.g. Giardia
larnblia), nematodes (e.g.
Onchocercus volvulus), worm (e.g. Dirofilaria immitis), and/or amoeba (e.g.
Acanthameoba).
[00138] In some instances, the concentration of the preservative is between
about 0.0001% and
about 1%, about 0.001% and about 0.8%, about 0.004% and about 0.5%, about
0.008 % and about 0.1%,
and about 0.01% and about 0.08%. In some cases, the concentration of the
preservatives is about 0.001%,
0.002%, 0.003%, 0.004%, 0.005%, 0.006%, 0.008%, 0.009%, 0.009%, 0.01%, 0.015%,
0.02%, 0.025%,
0.03%, 0.04%, 0.05%, 0.06%, 0.07%, 0.08%, 0.09%, 0.1%, 0.2%, 0.3%, 0.4%, 0.5%,
0.6%, 0.7%, 0.8%,
0.9% or 1.0%.
[00139] In some embodiments, the preservative is selected from benzalkonium
chloride,
cetrimonium, sodium perborate, stabilized oxychloro complex, SofZia (Alcon),
polyquatemium-1,
chlorobutanol, edetate disodium, and polyhexamethylene biguanide.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
27
[00140] In some embodiments, the composition described herein is stored in
a plastic container.
In some embodiments, the material of the plastic container comprises high
density polyethylene (HDPE),
low density polyethylene (LDF'E), polyethylene terephthalate (PET), polyvinyl
chloride (PVC),
polyprolyene (PP), polystyrene (PS), fluorine treated HDPE, post-consumer
resin (PCR), K-resine
(SBC), or bioplastic. In some embodiments, the material of the plastic
container comprises LDPE.
[00141] In some embodiments, the composition described herein is stored in
a plastic container.
In some embodiments, the composition stored in a plastic container has a pD of
between about 4 and
about 8, about 4.5 and about 7.9, or about 4.9 and about 7.5. In some
embodiments, the composition
stored in a plastic container has a pD of less than about 7.4. In some
embodiments, the composition
stored in a plastic container has a pD of less than about 7.3. In some
embodiments, the composition
stored in a plastic container has a pD of less than about 7.2. In some
embodiments, the composition
stored in a plastic container has a pD of less than about 7.1. In some
embodiments, the composition
stored in a plastic container has a pD of less than about 7. In some
embodiments, the composition stored
in a plastic container has a pD of less than about 6.9. In some embodiments,
the composition stored in a
plastic container has a pD of less than about 6.8. In some embodiments, the
composition stored in a
plastic container has a pD of less than about 6.7. In some embodiments, the
composition stored in a
plastic container has a pD of less than about 6.6. In some embodiments, the
composition stored in a
plastic container has a pD of less than about 6.5. In some embodiments, the
composition stored in a
plastic container has a pD of less than about 6.4. In some embodiments, the
composition stored in a
plastic container has a pD of less than about 6.3. In some embodiments, the
composition stored in a
plastic container has a pD of less than about 6.2. In some embodiments, the
composition stored in a
plastic container has a pD of less than about 6.1. In some embodiments, the
composition stored in a
plastic container has a pD of less than about 6. In some embodiments, the
composition stored in a plastic
container has a pD of less than about 5.9. In some embodiments, the
composition stored in a plastic
container has a pD of less than about 5.8. In some embodiments, the
composition stored in a plastic
container has a pD of less than about 5.7. In some embodiments, the
composition stored in a plastic
container has a pD of less than about 5.6. In some embodiments, the
composition stored in a plastic
container has a pD of less than about 5.5. In some embodiments, the
composition stored in a plastic
container has a pD of less than about 5.4. In some embodiments, the
composition stored in a plastic
container has a pD of less than about 5.3. In some embodiments, the
composition stored in a plastic
container has a pD of less than about 5.2. In some embodiments, the
composition stored in a plastic
container has a pD of less than about 5.1. In some embodiments, the
composition stored in a plastic
container has a pD of less than about 5. In some embodiments, the composition
stored in a plastic
container has a pD of less than about 4.9. In some embodiments, the
composition stored in a plastic
container has a pD of less than about 4.8. In some embodiments, the
composition stored in a plastic
container has a pD of less than about 4.7. In some embodiments, the
composition stored in a plastic
container has a pD of less than about 4.6. In some embodiments, the
composition stored in a plastic
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
28
container has a pD of less than about 4.5. In some embodiments, the
composition stored in a plastic
container has a pD of less than about 4.4. In some embodiments, the
composition stored in a plastic
container has a pD of less than about 4.3. In some embodiments, the
composition stored in a plastic
container has a pD of less than about 4.2. In some embodiments, the
composition stored in a plastic
container has a pD of less than about 4.1. In some embodiments, the
composition stored in a plastic
container has a pD of less than about 4.
[00142] In some embodiments, the composition stored in a plastic container
has a potency of at
least 80% after extended period of time under storage condition. In some
embodiments, the composition
stored in a plastic container has a potency of at least 85% after extended
period of time under storage
condition. In some embodiments, the composition stored in a plastic container
has a potency of at least
90% after extended period of time under storage condition. In some
embodiments, the composition stored
in a plastic container has a potency of at least 93% after extended period of
time under storage condition.
In some embodiments, the composition stored in a plastic container has a
potency of at least 95% after
extended period of time under storage condition. In some embodiments, the
composition stored in a
plastic container has a potency of at least 97% after extended period of time
under storage condition. In
some embodiments, the composition stored in a plastic container has a potency
of at least 98% after
extended period of time under storage condition. In some embodiments, the
composition stored in a
plastic container has a potency of at least 99% after extended period of time
under storage condition. In
some instances, the storage condition comprises a temperature of about 25 C,
about 40 C, or about 60 C.
In some instances, the extended period of time is at least 1 week, at least 2
weeks, at least 3 weeks, at
least 1 month, at least 2 months, at least 3 months, at least 4 months, at
least 5 months, at least 6 months,
at least 8 months, at least 10 months, at least 12 months, at least 18 months,
or at least 24 months.
[00143] In some embodiments, the composition stored in a plastic container
has a potency of at
least 80% at a temperature of about 25 C, about 40 C, or about 60 C. In some
embodiments, the
composition stored in a plastic container has a potency of at least 85% at a
temperature of about 25 C,
about 40 C, or about 60 C. In some embodiments, the composition stored in a
plastic container has a
potency of at least 90% at a temperature of about 25 C, about 40 C, or about
60 C. In some
embodiments, the composition stored in a plastic container has a potency of at
least 93% at a temperature
of about 25 C, about 40 C, or about 60 C. In some embodiments, the composition
stored in a plastic
container has a potency of at least 95% at a temperature of about 25 C, about
40 C, or about 60 C. In
some embodiments, the composition stored in a plastic container has a potency
of at least 97% at a
temperature of about 25 C, about 40 C, or about 60 C. In some embodiments, the
composition stored in
a plastic container has a potency of at least 98% at a temperature of about 25
C, about 40 C, or about
60 C. In some embodiments, the composition stored in a plastic container has a
potency of at least 99%
at a temperature of about 25 C, about 40 C, or about 60 C.
[00144] In some embodiments, the composition stored in a plastic container
has a potency of at
least 80% for a period of at least 1 week, at least 2 weeks, at least 3 weeks,
at least 1 month, at least 2
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
29
months, at least 3 months, at least 4 months, at least 5 months, at least 6
months, at least 8 months, at
least 10 months, at least 12 months, at least 18 months, or at least 24
months. In some embodiments, the
composition stored in a plastic container has a potency of at least 85% for a
period of at least 1 week, at
least 2 weeks, at least 3 weeks, at least 1 month, at least 2 months, at least
3 months, at least 4 months, at
least 5 months, at least 6 months, at least 8 months, at least 10 months, at
least 12 months, at least 18
months, or at least 24 months. In some embodiments, the composition stored in
a plastic container has a
potency of at least 90% for a period of at least 1 week, at least 2 weeks, at
least 3 weeks, at least 1 month,
at least 2 months, at least 3 months, at least 4 months, at least 5 months, at
least 6 months, at least 8
months, at least 10 months, at least 12 months, at least 18 months, or at
least 24 months. In some
embodiments, the composition stored in a plastic container has a potency of at
least 93% for a period of
at least 1 week, at least 2 weeks, at least 3 weeks, at least 1 month, at
least 2 months, at least 3 months, at
least 4 months, at least 5 months, at least 6 months, at least 8 months, at
least 10 months, at least 12
months, at least 18 months, or at least 24 months. In some embodiments, the
composition stored in a
plastic container has a potency of at least 95% for a period of at least 1
week, at least 2 weeks, at least 3
weeks, at least 1 month, at least 2 months, at least 3 months, at least 4
months, at least 5 months, at least
6 months, at least 8 months, at least 10 months, at least 12 months, at least
18 months, or at least 24
months. In some embodiments, the composition stored in a plastic container has
a potency of at least
97% for a period of at least 1 week, at least 2 weeks, at least 3 weeks, at
least 1 month, at least 2 months,
at least 3 months, at least 4 months, at least 5 months, at least 6 months, at
least 8 months, at least 10
months, at least 12 months, at least 18 months, or at least 24 months. In some
embodiments, the
composition stored in a plastic container has a potency of at least 98% for a
period of at least 1 week, at
least 2 weeks, at least 3 weeks, at least 1 month, at least 2 months, at least
3 months, at least 4 months, at
least 5 months, at least 6 months, at least 8 months, at least 10 months, at
least 12 months, at least 18
months, or at least 24 months. In some embodiments, the composition stored in
a plastic container has a
potency of at least 99% for a period of at least 1 week, at least 2 weeks, at
least 3 weeks, at least 1 month,
at least 2 months, at least 3 months, at least 4 months, at least 5 months, at
least 6 months, at least 8
months, at least 10 months, at least 12 months, at least 18 months, or at
least 24 months.
[00145] In some embodiments, the composition stored in a plastic container
comprises less than
20% of primary degradant based on the concentration of the ophthalmic agent
after extended period of
time under storage condition. In some embodiments, the composition stored in a
plastic container
comprises less than 15% of primary degradant based on the concentration of the
ophthalmic agent after
extended period of time under storage condition. In some embodiments, the
composition stored in a
plastic container comprises less than 10% of primary degradant based on the
concentration of the
ophthalmic agent after extended period of time under storage condition. In
some embodiments, the
composition stored in a plastic container comprises less than 5% of primary
degradant based on the
concentration of the ophthalmic agent after extended period of time under
storage condition.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
[00146] In some embodiments, the composition stored in a plastic container
comprises from less
than 2.5% of primary degradant to less than 0.1% of primary degradant based on
the concentration of the
ophthalmic agent after extended period of time under storage condition. In
some embodiments, the
composition stored in a plastic container comprises less than 2.5% of primary
degradant based on the
concentration of the ophthalmic agent after extended period of time under
storage condition. In some
embodiments, the composition stored in a plastic container comprises less than
2.0% of primary
degradant based on the concentration of the ophthalmic agent after extended
period of time under storage
condition. In some embodiments, the composition stored in a plastic container
comprises less than 1.5%
of primary degradant based on the concentration of the ophthalmic agent after
extended period of time
under storage condition. In some embodiments, the composition stored in a
plastic container comprises
less than 1.0% of primary degradant based on the concentration of the
ophthalmic agent after extended
period of time under storage condition. In some embodiments, the composition
stored in a plastic
container comprises less than 0.5% of primary degradant based on the
concentration of the ophthalmic
agent after extended period of time under storage condition. In some
embodiments, the composition
stored in a plastic container comprises less than 0.4% of primary degradant
based on the concentration of
the ophthalmic agent after extended period of time under storage condition. In
some embodiments, the
composition stored in a plastic container comprises less than 0.3% of primary
degradant based on the
concentration of the ophthalmic agent after extended period of time under
storage condition. In some
embodiments, the composition stored in a plastic container comprises less than
0.2% of primary
degradant based on the concentration of the ophthalmic agent after extended
period of time under storage
condition. In some embodiments, the composition stored in a plastic container
comprises less than 0.1%
of primary degradant based on the concentration of the ophthalmic agent after
extended period of time
under storage condition. In some instances, the storage condition comprises a
temperature of about 25 C,
about 40 C, or about 60 C. In some instances, the extended period of time is
at least 1 week, at least 2
weeks, at least 3 weeks, at least 1 month, at least 2 months, at least 3
months, at least 4 months, at least 5
months, at least 6 months, at least 8 months, at least 10 months, at least 12
months, at least 18 months, or
at least 24 months.
[00147] In some embodiments, the composition stored in a plastic container
comprises less than
20% of primary degradant based on the concentration of the ophthalmic agent at
a temperature of about
25 C, about 40 C, or about 60 C. In some embodiments, the composition stored
in a plastic container
comprises less than 15% of primary degradant based on the concentration of the
ophthalmic agent at a
temperature of about 25 C, about 40 C, or about 60 C. In some embodiments, the
composition stored in a
plastic container comprises less than 10% of primary degradant based on the
concentration of the
ophthalmic agent at a temperature of about 25 C, about 40 C, or about 60 C. In
some embodiments, the
composition stored in a plastic container comprises less than 5% of primary
degradant based on the
concentration of the ophthalmic agent at a temperature of about 25 C, about 40
C, or about 60 C.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
31
[00148] In some embodiments, the composition stored in a plastic container
comprises from less
than 2.5% of primary degradant to less than 0.1% of primary degradant based on
the concentration of the
ophthalmic agent at a temperature of about 25 C, about 40 C, or about 60 C. In
some embodiments, the
composition stored in a plastic container comprises less than 2.5% of primary
degradant based on the
concentration of the ophthalmic agent at a temperature of about 25 C, about 40
C, or about 60 C. In some
embodiments, the composition stored in a plastic container comprises less than
2.0% of primary
degradant based on the concentration of the ophthalmic agent at a temperature
of about 25 C, about 40 C,
or about 60 C. in some embodiments, the composition stored in a plastic
container comprises less than
1.5% of primary degradant based on the concentration of the ophthalmic agent
at a temperature of about
25 C, about 40 C, or about 60 C. In some embodiments, the composition stored
in a plastic container
comprises less than 1.0% of primary degradant based on the concentration of
the ophthalmic agent at a
temperature of about 25 C, about 40 C, or about 60 C. In some embodiments, the
composition stored in a
plastic container comprises less than 0.5% of primary degradant based on the
concentration of the
ophthalmic agent at a temperature of about 25 C, about 40 C, or about 60 C. In
some embodiments, the
composition stored in a plastic container comprises less than 0.4% of primary
degradant based on the
concentration of the ophthalmic agent at a temperature of about 25 C, about 40
C, or about 60 C. In some
embodiments, the composition stored in a plastic container comprises less than
0.3% of primary
degradant based on the concentration of the ophthalmic agent at a temperature
of about 25 C, about 40 C,
or about 60 C. In some embodiments, the composition stored in a plastic
container comprises less than
0.2% of primary degradant based on the concentration of the ophthalmic agent
at a temperature of about
25 C, about 40 C, or about 60 C. In some embodiments, the composition stored
in a plastic container
comprises less than 0.1% of primary degradant based on the concentration of
the ophthalmic agent at a
temperature of about 25 C, about 40 C, or about 60 C.
[00149] In some embodiments, the composition stored in a plastic container
comprises less than
20% of primary degradant based on the concentration of the ophthalmic agent
for a period of at least 1
week, at least 2 weeks, at least 3 weeks, at least 1 month, at least 2 months,
at least 3 months, at least 4
months, at least 5 months, at least 6 months, at least 8 months, at least 10
months, at least 12 months, at
least 18 months, or at least 24 months. In some embodiments, the composition
stored in a plastic
container comprises less than 15% of primary degradant based on the
concentration of the ophthalmic
agent for a period of at least 1 week, at least 2 weeks, at least 3 weeks, at
least 1 month, at least 2 months,
at least 3 months, at least 4 months, at least 5 months, at least 6 months, at
least 8 months, at least 10
months, at least 12 months, at least 18 months, or at least 24 months. In some
embodiments, the
composition stored in a plastic container comprises less than 10% of primary
degradant based on the
concentration of the ophthalmic agent for a period of at least 1 week, at
least 2 weeks, at least 3 weeks, at
least 1 month, at least 2 months, at least 3 months, at least 4 months, at
least 5 months, at least 6 months,
at least 8 months, at least 10 months, at least 12 months, at least 18 months,
or at least 24 months. In
some embodiments, the composition stored in a plastic container comprises less
than 5% of primary
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
32
degradant based on the concentration of the ophthalmic agent for a period of
at least 1 week, at least 2
weeks, at least 3 weeks, at least 1 month, at least 2 months, at least 3
months, at least 4 months, at least 5
months, at least 6 months, at least 8 months, at least 10 months, at least 12
months, at least 18 months, or
at least 24 months.
[00150] In some embodiments, the composition stored in a plastic container
comprises from less
than 2.5% of primary degradant to less than 0.1% of primary degradant based on
the concentration of the
ophthalmic agent for a period of at least 1 week, at least 2 weeks, at least 3
weeks, at least 1 month, at
least 2 months, at least 3 months, at least 4 months, at least 5 months, at
least 6 months, at least 8 months,
at least 10 months, at least 12 months, at least 18 months, or at least 24
months. In some embodiments,
the composition stored in a plastic container comprises less than 2.5% of
primary degradant based on the
concentration of the ophthalmic agent for a period of at least 1 week, at
least 2 weeks, at least 3 weeks, at
least 1 month, at least 2 months, at least 3 months, at least 4 months, at
least 5 months, at least 6 months,
at least 8 months, at least 10 months, at least 12 months, at least 18 months,
or at least 24 months. In
some embodiments, the composition stored in a plastic container comprises less
than 2.0% of primary
degradant based on the concentration of the ophthalmic agent for a period of
at least 1 week, at least 2
weeks, at least 3 weeks, at least 1 month, at least 2 months, at least 3
months, at least 4 months, at least 5
months, at least 6 months, at least 8 months, at least 10 months, at least 12
months, at least 18 months, or
at least 24 months. In some embodiments, the composition stored in a plastic
container comprises less
than 1.5% of primary degradant based on the concentration of the ophthalmic
agent for a period of at
least 1 week, at least 2 weeks, at least 3 weeks, at least 1 month, at least 2
months, at least 3 months, at
least 4 months, at least 5 months, at least 6 months, at least 8 months, at
least 10 months, at least 12
months, at least 18 months, or at least 24 months. In some embodiments, the
composition stored in a
plastic container comprises less than 1.0% of primary degradant based on the
concentration of the
ophthalmic agent for a period of at least 1 week, at least 2 weeks, at least 3
weeks, at least 1 month, at
least 2 months, at least 3 months, at least 4 months, at least 5 months, at
least 6 months, at least 8 months,
at least 10 months, at least 12 months, at least 18 months, or at least 24
months. In some embodiments,
the composition stored in a plastic container comprises less than 0.5% of
primary degradant based on the
concentration of the ophthalmic agent for a period of at least 1 week, at
least 2 weeks, at least 3 weeks, at
least 1 month, at least 2 months, at least 3 months, at least 4 months, at
least 5 months, at least 6 months,
at least 8 months, at least 10 months, at least 12 months, at least 18 months,
or at least 24 months. In
some embodiments, the composition stored in a plastic container comprises less
than 0.4% of primary
degradant based on the concentration of the ophthalmic agent for a period of
at least 1 week, at least 2
weeks, at least 3 weeks, at least 1 month, at least 2 months, at least 3
months, at least 4 months, at least 5
months, at least 6 months, at least 8 months, at least 10 months, at least 12
months, at least 18 months, or
at least 24 months. In some embodiments, the composition stored in a plastic
container comprises less
than 0.3% of primary degradant based on the concentration of the ophthalmic
agent for a period of at
least 1 week, at least 2 weeks, at least 3 weeks, at least 1 month, at least 2
months, at least 3 months, at
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
33
least 4 months, at least 5 months, at least 6 months, at least 8 months, at
least 10 months, at least 12
months, at least 18 months, or at least 24 months. In some embodiments, the
composition stored in a
plastic container comprises less than 0.2% of primary degradant based on the
concentration of the
ophthalmic agent for a period of at least 1 week, at least 2 weeks, at least 3
weeks, at least 1 month, at
least 2 months, at least 3 months, at least 4 months, at least 5 months, at
least 6 months, at least 8 months,
at least 10 months, at least 12 months, at least 18 months, or at least 24
months. In some embodiments,
the composition stored in a plastic container comprises less than 0.1% of
primary degradant based on the
concentration of the ophthalmic agent for a period of at least 1 week, at
least 2 weeks, at least 3 weeks, at
least 1 month, at least 2 months, at least 3 months, at least 4 months, at
least 5 months, at least 6 months,
at least 8 months, at least 10 months, at least 12 months, at least 18 months,
or at least 24 months.
[00151] In some embodiments, the composition described herein is stored in
a glass container. In
some embodiments, the glass container is a glass vial, such as for example, a
type I, type II or type III
glass vial. In some embodiments, the glass container is a type I glass vial.
In some embodiments, the type
I glass vial is a borasilicate glass vial.
[00152] In some embodiments, the composition stored in a glass container
has a pD of higher
than about 7. In some embodiments, the composition stored in a glass container
has a pD of higher than
about 7.5. In some embodiments, the composition stored in a glass container
has a pD of higher than
about 8. In some embodiments, the composition stored in a glass container has
a pD of higher than about
8.5. In some embodiments, the composition stored in a glass container has a pD
of higher than about 9.
[00153] In some embodiments, the composition stored in a glass container
has a potency of less
than 60% at a temperature of about 25 C, about 40 C, or about 60 C. In some
embodiments, the
composition stored in a glass container has a potency of less than 60% for a
period of at least 1 week, at
least 2 weeks, at least 3 weeks, at least 1 month, at least 2 months, at least
3 months, at least 4 months, at
least 5 months, at least 6 months, at least 8 months, at least 10 months, at
least 12 months, at least 18
months, or at least 24 months.
[00154] In some embodiments, the composition stored in a glass container is
less stable than a
composition stored in a plastic container.
[00155] In some embodiments, the composition is stored under in the dark.
In some instances, the
composition is stored in the presence of light. In some instances, the light
is indoor light, room light, or
sun light. In some instances, the composition is stable while stored in the
presence of light.
[00156] In some embodiments, the composition described herein is formulated
as an aqueous
solution. In some embodiments, the aqueous solution is a stable aqueous
solution. In some instances, the
aqueous solution is stored in a plastic container as described above. In some
instances, the aqueous
solution is not stored in a glass container. In some instances, the aqueous
solution is stored in the dark. In
some instances, the aqueous solution is stored in the presence of light. In
some instances, the aqueous
solution is stable in the presence of light.
CA 02953363 2016-12-21
WO 2015/200361
PCT/US2015/037249
34
[00157] In a
specific embodiment, the ophthalmically acceptable formulations alternatively
comprise a cyclodextrin. Cyclodextrins are cyclic oligosaccharides containing
6, 7, or 8 glucopyranose
units, referred to as a-cyclodextrin, 3-cyclodextrin, or y-cyclodextrin
respectively. Cyclodextrins have a
hydrophilic exterior, which enhances water-soluble, and a hydrophobic interior
which forms a cavity. In
an aqueous environment, hydrophobic portions of other molecules often enter
the hydrophobic cavity of
cyclodextrin to form inclusion compounds. Additionally, cyclodextrins are also
capable of other types of
nonbonding interactions with molecules that are not inside the hydrophobic
cavity. Cyclodextrins have
three free hydroxyl groups for each glucopyranose unit, or 18 hydroxyl groups
on a-cyclodextrin, 21
hydroxyl groups on 3-cyclodextrin, and 24 hydroxyl groups on -y-cyclodextrin.
In some embodiments,
one or more of these hydroxyl groups are reacted with any of a number of
reagents to form a large variety
of cyclodextrin derivatives, including hydroxypropyl ethers, sulfonates, and
sulfoalkylethers. Shown
below is the structure of 3-cyclodextrin and the hydroxypropyl-3-cyclodextrin
(HPPCD).
RO
RO 0
7A/ CPR RO
0
OR
OR
0 RO
0 R = H
RD 13 cyclodextrin
RO 0
OR ROk R = CH2CH(OH)CH3
0
(21%,. hydroxypropyl 13-cyclodextrin
OR
0 OR R. 0 o
_IR 0 0
0c)
RD
0
OR
[00158] In some
embodiments, the use of cyclodextrins in the pharmaceutical compositions
described herein improves the solubility of the drug. Inclusion compounds are
involved in many cases of
enhanced solubility; however other interactions between cyclodextrins and
insoluble compounds also
improves solubility. Hydroxypropyl-P-cyclodextrin (HPPCD) is commercially
available as a pyrogen free
product. It is a nonhygroscopic white powder that readily dissolves in water.
HIVCD is thermally stable
and does not degrade at neutral pH. Thus, cyclodextrins improve the solubility
of a therapeutic agent in a
composition or formulation. Accordingly, in some embodiments, cyclodextrins
are included to increase
the solubility of the ophthalmically acceptable ophthalmic agents within the
formulations described
herein. In other embodiments, cyclodextrins in addition serve as controlled
release excipients within the
formulations described herein.
[00159] By way of
example only, cyclodextrin derivatives for use include a-cyclodextrin, 3-
cyclodextrin, -y-cyclodextrin, hydroxyethy1-13-cyclodextrin, hydroxypropyl-y-
cyclodextrin, sulfated 3-
cyclodextrin, sulfated a-cyclodextrin, sulfobutyl ether P-cyclodextrin.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
[00160] The concentration of the cyclodextrin used in the compositions and
methods disclosed
herein varies according to the physiochemical properties, pharmacokinetic
properties, side effect or
adverse events, formulation considerations, or other factors associated with
the therapeutically
ophthalmic agent, or a salt or prodrug thereof, or with the properties of
other excipients in the
composition. Thus, in certain circumstances, the concentration or amount of
cyclodextrin used in
accordance with the compositions and methods disclosed herein will vary,
depending on the need. When
used, the amount of cyclodextrins needed to increase solubility of the
ophthalmic agent and/or function
as a controlled release excipient in any of the formulations described herein
is selected using the
principles, examples, and teachings described herein.
[00161] Other stabilizers that are useful in the ophthalmically acceptable
formulations disclosed
herein include, for example, fatty acids, fatty alcohols, alcohols, long chain
fatty acid esters, long chain
ethers, hydrophilic derivatives of fatty acids, polyvinyl pyrrolidoncs,
polyvinyl ethers, polyvinyl
alcohols, hydrocarbons, hydrophobic polymers, moisture-absorbing polymers, and
combinations thereof.
In some embodiments, amide analogues of stabilizers are also used. In further
embodiments, the chosen
stabilizer changes the hydrophobicity of the formulation, improves the mixing
of various components in
the formulation, controls the moisture level in the formula, or controls the
mobility of the phase.
[00162] In other embodiments, stabilizers are present in sufficient amounts
to inhibit the
degradation of the ophthalmic agent. Examples of such stabilizing agents,
include, but are not limited to:
glycerol, methionine, monothioglycerol, EDTA, ascorbic acid, polysorbate 80,
polysorbate 20, arginine,
heparin, dextran sulfate, cyclodextrins, pentosan polysulfate and other
heparinoids, divalent cations such
as magnesium and zinc, or combinations thereof
[00163] Additional useful stabilization agents for ophthalmically
acceptable formulations include
one or more anti-aggregation additives to enhance stability of ophthalmic
formulations by reducing the
rate of protein aggregation. The anti-aggregation additive selected depends
upon the nature of the
conditions to which the ophthalmic agents, for example a muscarinic antagonist
(e.g. atropine or its
pharmaceutically acceptable salts), are exposed. For example, certain
formulations undergoing agitation
and thermal stress require a different anti-aggregation additive than a
formulation undergoing
lyophilization and reconstitution. Useful anti-aggregation additives include,
by way of example only,
urea, guanidinium chloride, simple amino acids such as glycine or arginine,
sugars, polyalcohols,
polysorbates, polymers such as polyethylene glycol and dextrans, alkyl
saccharides, such as alkyl
glycoside, and surfactants.
[00164] Other useful formulations optionally include one or more
ophthalmically acceptable
antioxidants to enhance chemical stability where required. Suitable
antioxidants include, by way of
example only, ascorbic acid, methionine, sodium thiosulfate and sodium
metabisulfite. In one
embodiment, antioxidants are selected from metal chelating agents, thiol
containing compounds and
other general stabilizing agents.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
36
[00165] Still other useful compositions include one or more ophthalmically
acceptable
surfactants to enhance physical stability or for other purposes. Suitable
nonionic surfactants include, but
are not limited to, polyoxyethylene fatty acid glycerides and vegetable oils,
e.g., polyoxyethylene (60)
hydrogenated castor oil; and polyoxyethylene alkylethers and alkylphenyl
ethers, e.g., octoxynol 10,
octoxynol 40.
[00166] In some embodiments, the ophthalmically acceptable pharmaceutical
formulations
described herein are stable with respect to compound degradation (e.g. less
than 30% degradation, less
than 25% degradation, less than 20% degradation, less than 15% degradation,
less than 10%
degradation, less than 8% degradation, less than 5% degradation, less than 3%
degradation, less than
2% degradation, or less than 5% degradation) over a period of any of at least
about 1 day, at least about
2 days, at least about 3 days, at least about 4 days, at least about 5 days,
at least about 6 days, at least
about 1 week, at least about 2 weeks, at least about 3 weeks, at least about 4
weeks, at least about 5
weeks, at least about 6 weeks, at least about 7 weeks, at least about 8 weeks,
at least about 3 months, at
least about 4 months, at least about 5 months, or at least about 6 months
under storage conditions (e.g.
room temperature). In other embodiments, the formulations described herein are
stable with respect to
compound degradation over a period of at least about 1 week. Also described
herein are formulations that
are stable with respect to compound degradation over a period of at least
about 1 month.
[00167] In other embodiments, an additional surfactant (co-surfactant)
and/or buffering agent is
combined with one or more of the pharmaceutically acceptable vehicles
previously described herein so
that the surfactant and/or buffering agent maintains the product at an optimal
pD for stability. Suitable
co-surfactants include, but are not limited to: a) natural and synthetic
lipophilic agents, e.g.,
phospholipids, cholesterol, and cholesterol fatty acid esters and derivatives
thereof; b) nonionic
surfactants, which include for example, polyoxyethylene fatty alcohol esters,
sorbitan fatty acid esters
(Spans), polyoxyethylene sorbitan fatty acid esters (e.g., polyoxyethylene
(20) sorbitan monooleate
(Tween 80), polyoxyethylene (20) sorbitan monostearate (Tween 60),
polyoxyethylene (20) sorbitan
monolaurate (Tween 20) and other Tweens, sorbitan esters, glycerol esters,
e.g., Myrj and glycerol
triacetate (triacetin), polyethylene glycols, cetyl alcohol, cetostearyl
alcohol, stearyl alcohol, polysorbate
80, poloxamers, poloxamines, polyoxyethylene castor oil derivatives (e.g.,
Cremophor RH40, Cremphor
A25, Cremphor A20, Cremophor EL) and other Cremophors, sulfosuccinates, alkyl
sulphates (SLS);
PEG glyceryl fatty acid esters such as PEG-8 glyceryl caprylate/caprate
(Labrasol), PEG-4 glyceryl
caprylate/caprate (Labrafac Hydro WL 1219), PEG-32 glyceryl laurate (Gelucire
444/14), PEG-6
glyceryl mono oleate (Labrafil M 1944 CS), PEG-6 glyceryl linoleate (Labrafil
M 2125 CS); propylene
glycol mono- and di-fatty acid esters, such as propylene glycol laurate,
propylene glycol
caprylate/caprate; Brij 700, ascorby1-6-palmitate, stearylamine, sodium
lauryl sulfate,
polyoxethyleneglycerol triiricinoleate, and any combinations or mixtures
thereof; c) anionic surfactants
include, but are not limited to, calcium carboxymethylcellulose, sodium
carboxymethylcellulose, sodium
sulfosuccinate, dioctyl, sodium alginate, alkyl polyoxyethylene sulfates,
sodium lauryl sulfate,
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
37
triethanolamine stearate, potassium laurate, bile salts, and any combinations
or mixtures thereof; and d)
cationic surfactants such as cetyltrimethylammonium bromide, and
lauryldimethylbenzyl-ammonium
chloride.
[00168] In a further embodiment, when one or more co-surfactants are
utilized in the
ophthalmically acceptable formulations of the present disclosure, they are
combined, e.g., with a
pharmaceutically acceptable vehicle and is present in the final formulation,
e.g., in an amount ranging
from about 0.1% to about 20%, from about 0.5% to about 10%.
[00169] In one embodiment, the surfactant has an HLB value of 0 to 20. In
additional
embodiments, the surfactant has an HLB value of 0 to 3, of 4 to 6, of 7 to 9,
of 8 to 18, of 13 to 15, of 10
to 18.
[00170] np.
[00171] In some embodiments, the pD of a composition described herein is
adjusted (e.g., by use
of a buffer and/or a pD adjusting agent) to an ophthalmically compatible pD
range of from about 4 to
about 8, about 4.5 to about 7.5, or about 5 to about 7. In some embodiments,
the ophthalmic composition
has a pD of from about 5.0 to about 7Ø In some embodiments, the ophthalmic
composition has a pD of
from about 5.5 to about 7Ø In some embodiments, the ophthalmic composition
has a pD of from about
6.0 to about 7Ø
[00172] In some embodiments, useful formulations include one or more pD
adjusting agents or
buffering agents. Suitable pD adjusting agents or buffers include, but are not
limited to acetate,
bicarbonate, ammonium chloride, citrate, phosphate, deuterated forms of
acetate, bicarbonate,
ammonium chloride, citrate, phosphate, pharmaceutically acceptable salts
thereof and combinations or
mixtures thereof. In some embodiments, the pD adjusting agents or buffers
include deuterated
hydrochloric acid (DC1), deuterated sodium hydroxide (Na0D), deuterated acetic
acid (CD3COOD), or
deuterated citric acid (C6D807).
[00173] In one embodiment, when one or more buffers are utilized in the
formulations of the
present disclosure, they are combined, e.g., with a pharmaceutically
acceptable vehicle and are present in
the final formulation, e.g., in an amount ranging from about 0.1% to about
20%, from about 0.5% to
about 10%. In certain embodiments of the present disclosure, the amount of
buffer included in the gel
formulations are an amount such that the pD of the gel formulation does not
interfere with the body's
natural buffering system.
[00174] In one embodiment, diluents are also used to stabilize compounds
because they provide a
more stable environment. In some instances, salts dissolved in buffered
solutions (which also provides
pD control or maintenance) are utilized as diluents in the art, including, but
not limited to a phosphate
buffered saline solution.
[00175] In some embodiments, the pD is calculated according to the formula
disclosed in Glasoe
et al., "Use of glass electrodes to measure acidities in deuterium oxide," J.
Physical Chem. 64(1): 188-
190 (1960). In some embodiment, the pD is calculated as pD = pH* + 0.4, in
which pH* is the measured
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
38
or observed pH of the ophthalmic composition formulated in a solution
comprising deuterated water
(e.g., D20).
[00176] In some embodiments, the ophthalmic aqueous, gel, or ointment
composition described
herein has a pD of between about 4 and about 8, between about 4.5 and about 8,
between about 4.9 and
about 7.9, between about 5.4 and about 7.9, between about 5.9 and about 7.9,
between about 6.4 and
about 7.9, or between about 7.4 and about 7.9. In some embodiments, the
ophthalmic aqueous, gel, or
ointment composition described herein has a pD of between about 4.5-7.5,
between about 5.0 and about
7.5, between about 5.5 and about 7.5, between about 6.0 and about 7.5, or
between about 7.0 and about
7.5. In some embodiments, the ophthalmic aqueous, gel, or ointment composition
described herein has a
pD of between about 4.5-7.0, between about 5.0 and about 7.0, between about
5.5 and about 7.0,
between about 6.0 and about 7.0, or between about 6.5 and about 7Ø In some
embodiments, the
ophthalmic aqueous, gel, or ointment composition described herein has a pD of
between about 4.9-7.4,
between about 5.4 and about 7.4, between about 5.9 and about 7.4, between
about 6.4 and about 7.4, or
between about 6.9 and about 7.4. In some embodiments, the ophthalmic aqueous,
gel, or ointment
composition described herein has a pD of between about 4.5-6.5, between about
5.0 and about 6.5,
between about 5.5 and about 6.5, or between about 6.0 and about 6.5. In some
embodiments, the
ophthalmic aqueous, gel, or ointment composition described herein has a pD of
between about 4.9-6.9,
between about 5.4 and about 6.9, between about 5.9 and about 6.9, or between
about 6.4 and about 6.9.
In some embodiments, the ophthalmic aqueous, gel, or ointment composition
described herein has a pD
of between about 4.5-6.0, between about 5.0 and about 6.0, or between about
5.5 and about 6Ø In some
embodiments, the ophthalmic aqueous, gel, or ointment composition described
herein has a pD of
between about 4.9-6.4, between about 5.4 and about 6.4, or between about 5.9
and about 6.4. In some
embodiments, the ophthalmic aqueous, gel, or ointment composition described
herein has a pD of
between about 4.5-5.5, or between about 5.0 and about 5.5. In some
embodiments, the ophthalmic
aqueous, gel, or ointment composition described herein has a pD of between
about 4.9-5.9, or between
about 5.4 and about 5.9. In some embodiments, the ophthalmic aqueous, gel, or
ointment composition
described herein has a pD of between about 4.5-5Ø In some embodiments, the
ophthalmic aqueous, gel,
or ointment composition described herein has a pD of between about 4.9-5.4.
[00177] In some embodiments, the ophthalmic composition is an ophthalmic
aqueous
composition. In some instances, the ophthalmic aqueous composition has a pD of
between about 4 and
about 8, about 4.5 and about 7.8, about 5 and about 7.5, or about 5.5 and
about 7. In some embodiments,
the ophthalmic aqueous composition has a pD of about 7.5. In some embodiments,
the ophthalmic
aqueous composition has a pD of about 7.4. In some embodiments, the ophthalmic
aqueous composition
has a pD of about 7.3. In some embodiments, the ophthalmic aqueous composition
has a pD of about 7.2.
In some embodiments, the ophthalmic aqueous composition has a pD of about 7.1.
In some
embodiments, the ophthalmic aqueous composition has a pD of about 7. In some
embodiments, the
ophthalmic aqueous composition has a pD of about 6.9. In some embodiments, the
ophthalmic aqueous
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
39
composition has a pD of about 6.8. In some embodiments, the ophthalmic aqueous
composition has a pD
of about 6.7. In some embodiments, the ophthalmic aqueous composition has a pD
of about 6.6. In some
embodiments, the ophthalmic aqueous composition has a pD of about 6.5. In some
embodiments, the
ophthalmic aqueous composition has a pD of about 6.4. In some embodiments, the
ophthalmic aqueous
composition has a pD of about 6.3. In some embodiments, the ophthalmic aqueous
composition has a pD
of about 6.2. In some embodiments, the ophthalmic aqueous composition has a pD
of about 6.1. In some
embodiments, the ophthalmic aqueous composition has a pD of about 6. In some
embodiments, the
ophthalmic aqueous composition has a pD of about 5.9. In some embodiments, the
ophthalmic aqueous
composition has a pD of about 5.8. In some embodiments, the ophthalmic aqueous
composition has a pD
of about 5.7. In some embodiments, the ophthalmic aqueous composition has a pD
of about 5.6. In some
embodiments, the ophthalmic aqueous composition has a pD of about 5.5. In some
embodiments, the
ophthalmic aqueous composition has a pD of about 5.4. In some embodiments, the
ophthalmic aqueous
composition has a pD of about 5.3. In some embodiments, the ophthalmic aqueous
composition has a pD
of about 5.2. In some embodiments, the ophthalmic aqueous composition has a pD
of about 5.1. In some
embodiments, the ophthalmic aqueous composition has a pD of about 5. In some
embodiments, the
ophthalmic aqueous composition has a pD of about 4.9. In some embodiments, the
ophthalmic aqueous
composition has a pD of about 4.8. In some embodiments, the ophthalmic aqueous
composition has a pD
of about 4.7. In some embodiments, the ophthalmic aqueous composition has a pD
of about 4.6. In some
embodiments, the ophthalmic aqueous composition has a pD of about 4.5. In some
embodiments, the
ophthalmic aqueous composition has a pD of about 4.4. In some embodiments, the
ophthalmic aqueous
composition has a pD of about 4.3. In some embodiments, the ophthalmic aqueous
composition has a pD
of about 4.2. In some embodiments, the ophthalmic aqueous composition has a pD
of about 4.1. In some
embodiments, the ophthalmic aqueous composition has a pD of about 4. In some
embodiments, the pD is
an initial pD of the ophthalmic aqueous composition. In some embodiments, the
pD is the pD of the
ophthalmic aqueous composition after extended period of time under storage
condition.
[00178] In some instances, the ophthalmic aqueous composition has an
initial pD of between
about 4 and about 8, about 4.5 and about 7.8, about 5 and about 7.5, or about
5.5 and about 7. In some
embodiments, the ophthalmic aqueous composition has an initial pD of about
7.5. In some embodiments,
the ophthalmic aqueous composition has an initial pD of about 7.4. In some
embodiments, the
ophthalmic aqueous composition has an initial pD of about 7.3. In some
embodiments, the ophthalmic
aqueous composition has an initial pD of about 7.2. In some embodiments, the
ophthalmic aqueous
composition has an initial pD of about 7.1. In some embodiments, the
ophthalmic aqueous composition
has an initial pD of about 7. In some embodiments, the ophthalmic aqueous
composition has an initial pD
of about 6.9. In some embodiments, the ophthalmic aqueous composition has an
initial pD of about 6.8.
In some embodiments, the ophthalmic aqueous composition has an initial pD of
about 6.7. In some
embodiments, the ophthalmic aqueous composition has an initial pD of about
6.6. In some embodiments,
the ophthalmic aqueous composition has an initial pD of about 6.5. In some
embodiments, the
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
ophthalmic aqueous composition has an initial pD of about 6.4. In some
embodiments, the ophthalmic
aqueous composition has an initial pD of about 6.3. In some embodiments, the
ophthalmic aqueous
composition has an initial pD of about 6.2. In some embodiments, the
ophthalmic aqueous composition
has an initial pD of about 6.1. In some embodiments, the ophthalmic aqueous
composition has an initial
pD of about 6. In some embodiments, the ophthalmic aqueous composition has an
initial pD of about 5.9.
In some embodiments, the ophthalmic aqueous composition has an initial pD of
about 5.8. In some
embodiments, the ophthalmic aqueous composition has an initial pD of about
5.7. In some embodiments,
the ophthalmic aqueous composition has an initial pD of about 5.6. In some
embodiments, the
ophthalmic aqueous composition has an initial pD of about 5.5. In some
embodiments, the ophthalmic
aqueous composition has an initial pD of about 5.4. In some embodiments, the
ophthalmic aqueous
composition has an initial pD of about 5.3. In some embodiments, the
ophthalmic aqueous composition
has an initial pD of about 5.2. In some embodiments, the ophthalmic aqueous
composition has an initial
pD of about 5.1. In some embodiments, the ophthalmic aqueous composition has
an initial pD of about 5.
In some embodiments, the ophthalmic aqueous composition has an initial pD of
about 4.9. In some
embodiments, the ophthalmic aqueous composition has an initial pD of about
4.8. In some embodiments,
the ophthalmic aqueous composition has an initial pD of about 4.7. In some
embodiments, the
ophthalmic aqueous composition has an initial pD of about 4.6. In some
embodiments, the ophthalmic
aqueous composition has an initial pD of about 4.5. In some embodiments, the
ophthalmic aqueous
composition has an initial pD of about 4.4. In some embodiments, the
ophthalmic aqueous composition
has an initial pD of about 4.3. In some embodiments, the ophthalmic aqueous
composition has an initial
pD of about 4.2. In some embodiments, the ophthalmic aqueous composition has
an initial pD of about
4.1. In some embodiments, the ophthalmic aqueous composition has an initial pD
of about 4.
[00179] In some instances, the ophthalmic aqueous composition has a pD of
between about 4 and
about 8, about 4.5 and about 7.8, about 5 and about 7.5, or about 5.5 and
about 7. In some embodiments,
the ophthalmic aqueous composition has a pD of less than about 7.5. In some
embodiments, the
ophthalmic aqueous composition has a pD of less than about 7.4. In some
embodiments, the ophthalmic
aqueous composition has a pD of less than about 7.3. In some embodiments, the
ophthalmic aqueous
composition has a pD of less than about 7.2. In some embodiments, the
ophthalmic aqueous composition
has a pD of less than about 7.1. In some embodiments, the ophthalmic aqueous
composition has a pD of
less than about 7. In some embodiments, the ophthalmic aqueous composition has
a pD of less than about
6.9. In some embodiments, the ophthalmic aqueous composition has a pD of less
than about 6.8. In
some embodiments, the ophthalmic aqueous composition has a pD of less than
about 6.7. In some
embodiments, the ophthalmic aqueous composition has a pD of less than about
6.6. In some
embodiments, the ophthalmic aqueous composition has a pD of less than about
6.5. In some
embodiments, the ophthalmic aqueous composition has a pD of less than about
6.4. In some
embodiments, the ophthalmic aqueous composition has a pD of less than about
6.3. In some
embodiments, the ophthalmic aqueous composition has a pD of less than about
6.2. In some
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
41
embodiments, the ophthalmic aqueous composition has a pD of less than about
6.1. In some
embodiments, the ophthalmic aqueous composition has a pD of less than about 6.
In some embodiments,
the ophthalmic aqueous composition has a pD of less than about 5.9. In some
embodiments, the
ophthalmic aqueous composition has a pD of less than about 5.8. In some
embodiments, the ophthalmic
aqueous composition has a pD of less than about 5.7. In some embodiments, the
ophthalmic aqueous
composition has a pD of less than about 5.6. In some embodiments, the
ophthalmic aqueous composition
has a pD of less than about 5.5. In some embodiments, the ophthalmic aqueous
composition has a pD of
less than about 5.4. In some embodiments, the ophthalmic aqueous composition
has a pD of less than
about 5.3. In some embodiments, the ophthalmic aqueous composition has a pD of
less than about 5.2. In
some embodiments, the ophthalmic aqueous composition has a pD of less than
about 5.1. In some
embodiments, the ophthalmic aqueous composition has a pD of less than about 5.
In some embodiments,
the ophthalmic aqueous composition has a pD of less than about 4.9. In some
embodiments, the
ophthalmic aqueous composition has a pD of less than about 4.8. In some
embodiments, the ophthalmic
aqueous composition has a pD of less than about 4.7. In some embodiments, the
ophthalmic aqueous
composition has a pD of less than about 4.6. In some embodiments, the
ophthalmic aqueous composition
has a pD of less than about 4.5. In some embodiments, the ophthalmic aqueous
composition has a pD of
less than about 4.4. In some embodiments, the ophthalmic aqueous composition
has a pD of less than
about 4.3. In some embodiments, the ophthalmic aqueous composition has a pD of
less than about 4.2. In
some embodiments, the ophthalmic aqueous composition has a pD of less than
about 4.1. In some
embodiments, the ophthalmic aqueous composition has a pD of less than about 4.
In some embodiments,
the pD is the pD of the ophthalmic aqueous composition after extended period
of time under storage
condition.
[00180] In some embodiments, the pD of the ophthalmic aqueous composition
described herein
is associated with the stability of the ophthalmic aqueous composition. In
some embodiments, a stable
composition comprises a pD of between about 4 and about 8, about 4.5 and about
7.8, about 5 and about
7.5, or about 5.5 and about 7. In some embodiments, a stable composition
comprises a pD of less than
about 7.5. In some embodiments, a stable composition comprises a pD of less
than about 7.4. In some
embodiments, a stable composition comprises a pD of less than about 7.3. In
some embodiments, a stable
composition comprises a pD of less than about 7.2. In some embodiments, a
stable composition
comprises a pD of less than about 7.1. In some embodiments, a stable
composition comprises a pD of
less than about 7. In some embodiments, a stable composition comprises a pD of
less than about 6.9. In
some embodiments, a stable composition comprises a pD of less than about 6.8.
In some embodiments, a
stable composition comprises a pD of less than about 6.7. In some embodiments,
a stable composition
comprises a pD of less than about 6.6. In some embodiments, a stable
composition comprises a pD of
less than about 6.5. In some embodiments, a stable composition comprises a pD
of less than about 6.4.
In some embodiments, a stable composition comprises a pD of less than about
6.3. In some
embodiments, a stable composition comprises a pD of less than about 6.2. In
some embodiments, a
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
42
stable composition comprises a pD of less than about 6.1. In some embodiments,
a stable composition
comprises a pD of less than about 6. In some embodiments, a stable composition
comprises a pD of less
than about 5.9. In some embodiments, a stable composition comprises a pD of
less than about 5.8. In
some embodiments, a stable composition comprises a pD of less than about 5.7.
In some embodiments, a
stable composition comprises a pD of less than about 5.6. In some embodiments,
a stable composition
comprises a pD of less than about 5.5. In some embodiments, a stable
composition comprises a pD of
less than about 5.4. In some embodiments, a stable composition comprises a pD
of less than about 5.3. In
some embodiments, a stable composition comprises a pD of less than about 5.2.
In some embodiments, a
stable composition comprises a pD of less than about 5.1. In some embodiments,
a stable composition
comprises a pD of less than about 5. In some embodiments, a stable composition
comprises a pD of less
than about 4.9. In some embodiments, a stable composition comprises a pD of
less than about 4.8. In
some embodiments, a stable composition comprises a pD of less than about 4.7.
In some embodiments, a
stable composition comprises a pD of less than about 4.6. In some embodiments,
a stable composition
comprises a pD of less than about 4.5. In some embodiments, a stable
composition comprises a pD of
less than about 4.4. In some embodiments, a stable composition comprises a pD
of less than about 4.3. In
some embodiments, a stable composition comprises a pD of less than about 4.2.
In some embodiments, a
stable composition comprises a pD of less than about 4.1. In some embodiments,
a stable composition
comprises a pD of less than about 4.
[00181] In some embodiments, the D20 aqueous system stabilizes a muscarinic
antagonist (e.g.,
atropine). In some embodiments, this is due to a lower concentration of the
reactive species (e.g., -OD) in
the D20 aqueous system compared to the concentration of the reactive species
(e.g., -OH) in an
equivalent H2O aqueous system. In some instances, the concentration of the
reactive species (e.g., -OD)
in the D20 aqueous system is about one third less than the concentration of
the reactive species (e.g., -
OH) in the equivalent H20 aqueous system. In some cases, this is due to a
lower or smaller dissociation
constant of D20 than H2O. For example, the Ka(H20) is 1x10-14, whereas the
Ka(D20) is 1x10-15. As such,
D20 is a weaker acid than H20. In some cases, base catalyzed hydrolysis leads
to the presence of tropine
degradant from atropine. In some cases, with a lower concentration of the
reactive species that causes
tropine degradant formation, atropine solution is more stable in a D20 aqueous
system than compared to
an equivalent H20 aqueous system. In some embodiments, the ophthalmic
composition formulated with
deuterated water allows for a more stable ophthalmic composition relative to
the ophthalmic composition
formulated with H20.
[00182] In some embodiments, the presence of deuterated water shifts the
pKa of the buffer. In
some embodiments, the presence of deuterated water allows for the ophthalmic
composition to simulate
the stability of a lower pH system. In some instances, the buffer capacity of
the ophthalmic composition
is lowered, thereby allowing a faster shift in pH. In some instances, the
lowered buffering capacity of the
ophthalmic composition when administered into the eye allows the ophthalmic
composition to reach
physiological pH at a faster rate than compared to an ophthalmic composition
formulated in H2O. In
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
43
some instances, the ophthalmic composition formulated with deuterated water
allows for a lower tear
production, or less tear reflex in the eye, in comparison with an ophthalmic
composition formulated with
H20.
[00183] In some embodiment, the ophthalmic gel or ointment composition
described herein has a
pD of about 4, about 4.1, about 4.2, about 4.3, about 4.4, about 4.5, about
4.6, about 4.7, about 4.8, about
4.9, about 5.0, about 5.1, about 5.2, about 5.3, about 5.4, about 5.5, about
5.6, about 5.7, about 5.8, about
5.9, about 6.0, about 6.1, about 6.2, about 6.3, about 6.4, about 6.5, about
6.6, about 6.7, about 6.8, about
6.9, about 7.0, about 7.1, about 7.2, about 7.3, about 7.4, about 7.5, about
7.6, about 7.7, about 7.8, or
about 7.9.
[00184] In some embodiment, the pD of the ophthalmic aqueous, gel, or
ointment composition
described herein is suitable for sterilization (e.g., by filtration or aseptic
mixing or heat treatment and/or
autoclaving (e.g., terminal sterilization)) of ophthalmic formulations
described herein. As used in in the
present disclosure, the term "aqueous composition" includes compositions that
are based on D20.
[00185] In some embodiments, the pharmaceutical formulations described
herein are stable with
respect to pD over a period of any of at least about 1 day, at least about 2
days, at least about 3 days, at
least about 4 days, at least about 5 days, at least about 6 days, at least
about 1 week, at least about 2
weeks, at least about 3 weeks, at least about 4 weeks, at least about 5 weeks,
at least about 6 weeks, at
least about 7 weeks, at least about 8 weeks, at least about 1 month, at least
about 2 months, at least about
3 months, at least about 4 months, at least about 5 months, at least about 6
months, at least about 7
months, at least about 8 months, at least about 9 months, at least about 10
months, at least about 11
months, at least about 12 months, at least about 18 months, at least about 24
months, at least about 3
years, at least about 4 years, at least about 5 years, at least about 6 years,
at least about 7 years, at least
about 8 years, at least about 9 years, at least about 10 years, or more. In
other embodiments, the
formulations described herein are stable with respect to pD over a period of
at least about 1 week. In
other embodiments, the formulations described herein are stable with respect
to pD over a period of at
least about 2 weeks. Tn other embodiments, the fomiulations described herein
are stable with respect to
pD over a period of at least about 3 weeks. In other embodiments, the
formulations described herein are
stable with respect to pD over a period of at least about 1 month. Also
described herein are formulations
that are stable with respect to pD over a period of at least about 2 months,
at least about 3 months, at least
about 4 months, at least about 5 months, at least about 6 months, at least
about 12 months, at least about
18 months, at least about 2 years, or more.
[00186] Aqueous Solution Dose-To-Dose Uniformity
[00187] Typical ophthalmic aqueous solutions are packaged in eye drop
bottles and administered
as drops. For example, a single administration (i.e. a single dose) of an
ophthalmic aqueous solution
includes a single drop, two drops, three drops or more into the eyes of the
patient. In some embodiments,
one dose of the ophthalmic aqueous solution described herein is one drop of
the aqueous solution
composition from the eye drop bottle.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
44
[00188] In some cases, described herein include ophthalmic aqueous
compositions which provide
a dose-to-dose uniform concentrations. In some instances, the dose-to-dose
uniform concentration does
not present significant variations of drug content from one dose to another.
In some instances, the dose-
to-dose uniform concentration does provide consistent drug content from one
dose to another.
[00189] In some embodiments, the composition has a dose-to-dose ophthalmic
agent
concentration variation of less than 50%. In some embodiments, the composition
has a dose-to-dose
ophthalmic agent concentration variation of less than 40%. In some
embodiments, the composition has a
dose-to-dose ophthalmic agent concentration variation of less than 30%. In
some embodiments, the
composition has a dose-to-dose ophthalmic agent concentration variation of
less than 20%. In some
embodiments, the composition has a dose-to-dose ophthalmic agent concentration
variation of less than
10%. In some embodiments, the composition has a dose-to-dose ophthalmic agent
concentration
variation of less than 5%.
[00190] In some embodiments, the dose-to-dose ophthalmic agent
concentration variation is
based on 10 consecutive doses. In some embodiments, the dose-to-dose
ophthalmic agent concentration
variation is based on 8 consecutive doses. In some embodiments, the dose-to-
dose ophthalmic agent
concentration variation is based on 5 consecutive doses. In some embodiments,
the dose-to-dose
ophthalmic agent concentration variation is based on 3 consecutive doses. In
some embodiments, the
dose-to-dose ophthalmic agent concentration variation is based on 2
consecutive doses.
[00191] A nonsettling formulation should not require shaking to disperse
drug uniformly. A "no-
shake" formulation is potentially advantageous over formulations that require
shaking for the simple
reason that patients' shaking behavior is a major source of variability in the
amount of drug dosed. It has
been reported that patients often times do not or forget to shake their
ophthalmic compositions that
requires shaking before administering a dose, despite the instructions to
shake that were clearly marked
on the label. On the other hand, even for those patients who do shake the
product, it is normally not
possible to determine whether the shaking is adequate in intensity and/or
duration to render the product
uniform. In some embodiments, the ophthalmic gel compositions and ophthalmic
ointment compositions
described herein are "no-shake" formulations that maintained the dose-to-dose
uniformity described
herein.
[00192] To evaluate the dose-to-dose uniformity, drop bottles or tubes
containing the ophthalmic
aqueous compositions, the ophthalmic gel compositions, or ophthalmic ointment
compositions are stored
upright for a minimum of 12 hours prior to the start of the test. To simulate
the recommended dosing of
these products, predetermined number of drops or strips are dispensed from
each commercial bottles or
tubes at predetermined time intervals for an extended period of time or until
no product was left in the
bottle or tube. All drops and strips are dispensed into tared glass vials,
capped, and stored at room
temperature until analysis. Concentrations of a muscarinic antagonist such as
atropine in the expressed
drops were determined using a reverse-phase HPLC method.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
[00193] Aqueous Solution Viscosity
[00194] In some embodiments, the composition has a Brookfield RVDV
viscosity of from about
10 to about 50,000 cps at about 20 C and sheer rate of ls1. In some
embodiments, the composition has a
Brookfield RVDV viscosity of from about 100 to about 40,000 cps at about 20 C
and sheer rate of 1s-1.
In some embodiments, the composition has a Brookfield RVDV viscosity of from
about 500 to about
30,000 cps at about 20 C and sheer rate of 1s-1. In some embodiments, the
composition has a Brookfield
RVDV viscosity of from about 1000 to about 20,000 cps at about 20 C and sheer
rate of Isl. In some
embodiments, the composition has a Brookfield RVDV viscosity of from about
2000 to about 10,000 cps
at about 20 C and sheer rate of ls-1. In some embodiments, the composition has
a Brookfield RVDV
viscosity of from about 4000 to about 8000 cps at about 20 C and sheer rate of
ls-1.
[00195] In some embodiments, the ophthalmic aqueous formulation contains a
viscosity
enhancing agent sufficient to provide a viscosity of between about 500 and
50,000 centipoise, between
about 750 and 50,000 centipoise; between about 1000 and 50,000 centipoise;
between about 1000 and
40,000 centipoise; between about 2000 and 30,000 centipoise; between about
3000 and 20,000
centipoise; between about 4000 and 10,000 centipoise, or between about 5000
and 8000 centipoise.
[00196] In some embodiments, the compositions described herein are low
viscosity compositions
at body temperature. In some embodiments, low viscosity compositions contain
from about 1% to about
10% of a viscosity enhancing agent (e.g., gelling components such as
polyoxyethylene-polyoxypropylene
copolymers). In some embodiments, low viscosity compositions contain from
about 2% to about 10% of
a viscosity enhancing agent (e.g., gelling components such as polyoxyethylene-
polyoxypropylene
copolymers). In some embodiments, low viscosity compositions contain from
about 5% to about 10% of
a viscosity enhancing agent (e.g., gelling components such as polyoxyethylene-
polyoxypropylene
copolymers). In some embodiments, low viscosity compositions are substantially
free of a viscosity
enhancing agent (e.g., gelling components such as polyoxyethylene-
polyoxypropylene copolymers). hi
some embodiments, a low viscosity ophthalmic agent composition described
herein provides an apparent
viscosity of from about 100 cP to about 10,000 cP. In some embodiments, a low
viscosity ophthalmic
agent composition described herein provides an apparent viscosity of from
about 500 cP to about 10,000
cP. In some embodiments, a low viscosity ophthalmic agent composition
described herein provides an
apparent viscosity of from about 1000 cP to about 10,000 cP.
[00197] Osmolarity
[00198] In some embodiments, a composition disclosed herein is formulated
in order to not
disrupt the ionic balance of the eye. In some embodiments, a composition
disclosed herein has an ionic
balance that is the same as or substantially the same as the eye. In some
embodiments, a composition
disclosed herein does not does not disrupt the ionic balance of the eye.
[00199] As used herein, -practical osmolarity/osmolality" or -deliverable
osmolarity/osmolality"
means the osmolarity/osmolality of a composition as determined by measuring
the osmolarity/osmolality
of the ophthalmic agent and all excipients except the gelling and/or the
thickening agent (e.g.,
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
46
polyoxyethylene-polyoxypropylene copolymers, carboxymethylcellulose or the
like). The practical
osmolarity of a composition disclosed herein is measured by a suitable method,
e.g., a freezing point
depression method as described in Viegas et. al., Int. J. Pharm., 1998, 160,
157-162. In some instances,
the practical osmolarity of a composition disclosed herein is measured by
vapor pressure osmometry
(e.g., vapor pressure depression method) that allows for determination of the
osmolarity of a composition
at higher temperatures. In some instances, vapor pressure depression method
allows for determination of
the osmolarity of a composition comprising a gelling agent (e.g., a
thermoreversible polymer) at a higher
temperature wherein the gelling agent is in the form of a gel.
[00200] In some embodiments, the osmolarity at a target site of action
(e.g., the eye) is about the
same as the delivered osmolarity of a composition described herein. In some
embodiments, a
composition described herein has a deliverable osmolarity of about 150 mOsm/L
to about 500 mOsm/L,
about 250 mOsm/L to about 500 mOsm/L, about 250 mOsm/L to about 350 mOsm/L,
about 280
mOsm/L to about 370 mOsm/L or about 250 mOsm/L to about 320 mOsm/L.
[00201] The practical osmolality of an ophthalmic composition disclosed
herein is from about
100 mOsm/kg to about 1000 mOsm/kg, from about 200 mOsm/kg to about 800
mOsm/kg, from about
250 mOsm/kg to about 500 mOsm/kg, or from about 250 mOsm/kg to about 320
mOsm/kg, or from
about 250 mOsm/kg to about 350 mOsm/kg or from about 280 mOsm/kg to about 320
mOsm/kg. in
some embodiments, a composition described herein has a practical osmolarity of
about 100 mOsm/L to
about 1000 mOsm/L, about 200 mOsm/L to about 800 mOsmIL, about 250 mOsm/L to
about 500
mOsm/L, about 250 mOsm/L to about 350 mOsm/L, about 250 mOsm/L to about 320
mOsny'L, or about
280 mOsm/L to about 320 mOsm/L.
[00202] In some embodiments, suitable tonicity adjusting agents include,
but are not limited to
any pharmaceutically acceptable sugar, salt or any combinations or mixtures
thereof, such as, but not
limited to dextrose, glycerin, mannitol, sorbitol, sodium chloride, and other
electrolytes. In some
instances, the tonicity adjusting agent is selected from sodium chloride,
sodium nitrate, sodium sulfate,
sodium bisulfate, potassium chloride, calcium chloride, magnesium chloride,
zinc chloride, potassium
acetate, sodium acetate, sodium bicarbonate, sodium carbonate, sodium
thiosulfate, magnesium sulfate,
disodium hydrogen phosphate, sodium dihydrogen phosphate, potassium dihydrogen
phosphate,
dextrose, mannitol, sorbitol, dextrose, sucrose, urea, propylene glycol,
glycerin, or a combination thereof.
[00203] In some embodiment, the ophthalmic compositions described herein
include one or more
salts in an amount required to bring osmolality of the composition into an
acceptable range. Such salts
include those having sodium, potassium or ammonium cations and chloride,
citrate, ascorbate, borate,
phosphate, bicarbonate, sulfate, thiosulfate or bisulfite anions; suitable
salts include sodium chloride,
potassium chloride, sodium thiosulfate, sodium bisulfite and ammonium sulfate.
[00204] Sterility
[00205] In some embodiments, the compositions are sterilized. Included
within the embodiments
disclosed herein are means and processes for sterilization of a pharmaceutical
composition disclosed
47
herein for use in humans. The goal is to provide a safe pharmaceutical
product, relatively free of infection
causing micro-organisms. The U. S. Food and Drug Administration has provided
regulatory guidance in
the publication "Guidance for Industry: Sterile Drug Products Produced by
Aseptic Processing" available
at: http://www.fda.gov/cder/guidance/5882fn1.htm.
[00206] As used herein, sterilization means a process used to destroy or
remove microorganisms
that are present in a product or packaging. Any suitable method available for
sterilization of objects and
compositions is used. Available methods for the inactivation of microorganisms
include, but are not
limited to, the application of extreme heat, lethal chemicals, or gamma
radiation. In some embodiments, a
process for the preparation of an ophthalmic formulation comprises subjecting
the formulation to a
sterilization method selected from heat sterilization, chemical sterilization,
radiation sterilization or
filtration sterilization. The method used depends largely upon the nature of
the device or composition to
be sterilized. Detailed descriptions of many methods of sterilization are
given in Chapter 40 of
Remington: The Science and Practice of Pharmacy published by Lippincott,
Williams & Wilkins.
Filtration
[00207] Filtration sterilization is a method used to remove but not destroy
microorganisms from
solutions. Membrane filters are used to filter heat-sensitive solutions. Such
filters are thin, strong,
homogenous polymers of mixed cellulosic esters (MCE), polyvinylidene fluoride
(PVF; also known as
PVDF), or polytetrafluoroethylene (PTFE) and have pore sizes ranging from 0.1
to 0.22 0 m. Solutions
of various characteristics are optionally filtered using different filter
membranes. For example, PVF and
PTFE membranes are well suited to filtering organic solvents while aqueous
solutions are filtered
through PVF or MCE membranes. Filter apparatus are available for use on many
scales ranging from the
single point-of-use disposable filter attached to a syringe up to commercial
scale filters for use in
manufacturing plants. The membrane filters are sterilized by autoclave or
chemical sterilization.
Validation of membrane filtration systems is performed following standardized
protocols
(Microbiological Evaluation of Filters for Sterilizing Liquids, Vol 4, No. 3.
Washington, D.C: Health
Industry Manufacturers Association, 1981) and involve challenging the membrane
filter with a known
quantity (ca. 107/cm2) of unusually small microorganisms, such as
Brevundimonas diminuta (ATCC
19146).
[00208] Pharmaceutical compositions are optionally sterilized by passing
through membrane
filters. Formulations comprising nanoparticles (U.S. Pat No. 6,139,870) or
multilamellar vesicles
(Richard et al., International Journal of Pharmaceutics (2006), 312(1-2):144-
50) are amenable to
sterilization by filtration through 0.22 0 m filters without destroying their
organized structure.
[00209] In some embodiments, the methods disclosed herein comprise
sterilizing the formulation
(or components thereof) by means of filtration sterilization. In ophthalmic
gel compositions that includes
thermosetting polymers, filtration is carried out below (e.g. about 5 C) the
gel temperature (Tgel) of a
Date recue / Date received 2021-12-03
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
48
formulation described herein and with viscosity that allows for filtration in
a reasonable time using a
peristaltic pump (e.g. below a theoretical value of 100cP ).
[00210] Accordingly, provided herein are methods for sterilization of
ophthalmic formulations
that prevent degradation of polymeric components (e.g., thermosetting and/or
other viscosity enhancing
agents) and/or the ophthalmic agent during the process of sterilization. In
some embodiments,
degradation of the ophthalmic agent (e.g., a muscarinic antagonist such as
atropine or atropine sulfate) is
reduced or eliminated through the use of specific pD ranges for buffer
components and specific
proportions of viscosity enhancing agents in the formulations. in some
embodiments, the choice of an
appropriate viscosity enhancing agents or thermosetting polymer allows for
sterilization of formulations
described herein by filtration. In some embodiments, the use of an appropriate
thermosetting polymer or
other viscosity enhancing agents in combination with a specific pD range for
the formulation allows for
high temperature sterilization of formulations described with substantially no
degradation of the
therapeutic agent or the polymeric excipients. An advantage of the methods of
sterilization provided
herein is that, in certain instances, the formulations arc subjected to
terminal sterilization via autoclaving
without any loss of the ophthalmic agent and/or excipients and/or viscosity
enhancing agents during the
sterilization step and are rendered substantially free of microbes and/or
pyrogens.
Radiation Sterilization
[00211] One advantage of radiation sterilization is the ability to
sterilize many types of products
without heat degradation or other damage. The radiation commonly employed is
beta radiation or
alternatively, gamma radiation from a 6 Co source. The penetrating ability of
gamma radiation allows its
use in the sterilization of many product types, including solutions,
compositions and heterogeneous
mixtures. The germicidal effects of irradiation arise from the interaction of
gamma radiation with
biological macromolecules. This interaction generates charged species and free-
radicals. Subsequent
chemical reactions, such as rearrangements and cross-linking processes, result
in the loss of normal
function for these biological macromolecules. The formulations described
herein are also optionally
sterilized using beta irradiation.
Sterilization by Heat
[00212] Many methods are available for sterilization by the application of
high heat. One method
is through the use of a saturated steam autoclave. In this method, saturated
steam at a temperature of at
least 121 C is allowed to contact the object to be sterilized. The transfer
of heat is either directly to the
microorganism, in the case of an object to be sterilized, or indirectly to the
microorganism by heating the
bulk of an aqueous solution to be sterilized. This method is widely practiced
as it allows flexibility,
safety and economy in the sterilization process.
Microorganisms
[00213] In some embodiments, the compositions are substantially free of
microorganisms.
Acceptable bioburden or sterility levels arc based on applicable standards
that define therapeutically
acceptable compositions, including but not limited to United States
Pharmacopeia Chapters <1111> et
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
49
seq. For example, acceptable sterility (e.g., bioburden) levels include about
10 colony forming units (cfu)
per gram of formulation, about 50 cfu per gram of formulation, about 100 cfu
per gram of formulation,
about 500 cfu per gram of formulation or about 1000 cfu per gram of
formulation. In some embodiments,
acceptable bioburden levels or sterility for formulations include less than 10
cfu/mL, less than 50 cfu/mL,
less than 500 cfu/mL or less than 1000 cfu/mL microbial agents. In addition,
acceptable bioburden levels
or sterility include the exclusion of specified objectionable microbiological
agents. By way of example,
specified objectionable microbiological agents include but are not limited to
Escherichia coil (E. colt),
Salmonella sp., Pseudornonas aeruginosa (P. aeruginosa) and/or other specific
microbial agents.
[00214] An important component of the sterility assurance quality control,
quality assurance and
validation process is the method of sterility testing. Sterility testing, by
way of example only, is
performed by two methods. The first is direct inoculation wherein a sample of
the composition to be
tested is added to growth medium and incubated for a period of time up to 21
days. Turbidity of the
growth medium indicates contamination. Drawbacks to this method include the
small sampling size of
bulk materials which reduces sensitivity, and detection of microorganism
growth based on a visual
observation. An alternative method is membrane filtration sterility testing.
In this method, a volume of
product is passed through a small membrane filter paper. The filter paper is
then placed into media to
promote the growth of microorganisms. This method has the advantage of greater
sensitivity as the entire
bulk product is sampled. The commercially available Millipore Steritest
sterility testing system is
optionally used for determinations by membrane filtration sterility testing.
For the filtration testing of
creams or ointments Steritest filter system No. TLHVSL210 are used. For the
filtration testing of
emulsions or viscous products Steritest filter system No. TLAREM210 or
TDAREM210 are used. For
the filtration testing of pre-filled syringes Steritest filter system No.
TTHASY210 are used. For the
filtration testing of material dispensed as an aerosol or foam Steritest
filter system No. TTHVA210 are
used. For the filtration testing of soluble powders in ampoules or vials
Steritest filter system No.
TTHADA210 or TTHADV210 are used.
[00215] Testing for E. coil and Salmonella includes the use of lactose
broths incubated at 30 ¨ 35
C for 24-72 hours, incubation in MacConkey and/or EMB agars for 18-24 hours,
and/or the use of
Rappaport medium. Testing for the detection of P. aeruginosa includes the use
of NAC agar. United
States Pharmacopeia Chapter <62> further enumerates testing procedures for
specified objectionable
microorganisms.
[00216] In certain embodiments, the ophthalmic formulation described herein
has less than about
60 colony forming units (CFU), less than about 50 colony forming units, less
than about 40 colony
forming units, or less than about 30 colony forming units of microbial agents
per gram of formulation. In
certain embodiments, the ophthalmic formulations described herein are
formulated to be isotonic with the
eye.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
Endotoxins
[00217] An additional aspect of the sterilization process is the removal of
by-products from the
killing of microorganisms (hereinafter, "Product"). The process of
depyrogenation removes pyrogens
from the sample. Pyrogens are endotoxins or exotoxins which induce an immune
response. An example
of an endotoxin is the lipopolysaccharide (LPS) molecule found in the cell
wall of gram-negative
bacteria. While sterilization procedures such as autoclaving or treatment with
ethylene oxide kill the
bacteria, the LPS residue induces a proinflammatory immune response, such as
septic shock. Because the
molecular size of endotoxins varies widely, the presence of endotoxins is
expressed in "endotoxin units"
(EU). One EU is equivalent to 100 picograms of E. colt LPS. In some cases,
humans develop a response
to as little as 5 EU/kg of body weight. The bioburden (e.g., microbial limit)
and/or sterility (e.g.,
endotoxin level) is expressed in any units as recognized in the art. In
certain embodiments, ophthalmic
compositions described herein contain lower endotoxin levels (e.g. <4 EU/kg of
body weight of a
subject) when compared to conventionally acceptable endotoxin levels (e.g., 5
EU/kg of body weight of a
subject). In some embodiments, the ophthalmic formulation has less than about
5 EU/kg of body weight
of a subject. In other embodiments, the ophthalmic formulation has less than
about 4 EU/kg of body
weight of a subject. In additional embodiments, the ophthalmic formulation has
less than about 3 EU/kg
of body weight of a subject. In additional embodiments, the ophthalmic
formulation has less than about 2
EU/kg of body weight of a subject.
[00218] In some embodiments, the ophthalmic formulation has less than about
5 EU/kg of
formulation. in other embodiments, the ophthalmic formulation has less than
about 4 EU/kg of
formulation. In additional embodiments, the ophthalmic formulation has less
than about 3 EU/kg of
formulation. In some embodiments, the ophthalmic formulation has less than
about 5 EU/kg Product. In
other embodiments, the ophthalmic formulation has less than about 1 EU/kg
Product. In additional
embodiments, the ophthalmic formulation has less than about 0.2 EU/kg Product.
In some embodiments,
the ophthalmic formulation has less than about 5 EU/g of unit or Product. In
other embodiments, the
ophthalmic formulation has less than about 4 EU/ g of unit or Product. In
additional embodiments, the
ophthalmic formulation has less than about 3 EU/g of unit or Product. In some
embodiments, the
ophthalmic formulation has less than about 5 EU/mg of unit or Product. In
other embodiments, the
ophthalmic formulation has less than about 4 EU/mg of unit or Product. In
additional embodiments, the
ophthalmic formulation has less than about 3 EU/mg of unit or Product. In
certain embodiments,
ophthalmic formulations described herein contain from about Ito about 5 EU/mL
of formulation. in
certain embodiments, ophthalmic formulations described herein contain from
about 2 to about 5 EU/mL
of formulation, from about 3 to about 5 EU/mL of formulation, or from about 4
to about 5 EU/mL of
formulation.
[00219] In certain embodiments, ophthalmic compositions described herein
contain lower
endotoxin levels (e.g. <0.5 EU/mL of formulation) when compared to
conventionally acceptable
endotoxin levels (e.g., 0.5 EU/mL of formulation). In some embodiments, the
ophthalmic formulation
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
51
has less than about 0.5 EU/mL of formulation. In other embodiments, the
ophthalmic formulation has
less than about 0.4 EU/mL. of formulation. In additional embodiments, the
ophthalmic formulation has
less than about 0.2 EU/mL. of formulation.
[00220] Pyrogen detection, by way of example only, is performed by several
methods. Suitable
tests for sterility include tests described in United States Pharmacopoeia
(USP) <71> Sterility Tests (23rd
edition, 1995). The rabbit pyrogen test and the Limulus amebocyte lysate test
are both specified in the
United States Pharmacopeia Chapters <85> and <151> (U5P23/NF 18, Biological
Tests, The United
States Pharniacopeial Convention, Rockville, MD, 1995). Alternative pyrogen
assays have been
developed based upon the monocyte activation-cytokine assay. Uniform cell
lines suitable for quality
control applications have been developed and have demonstrated the ability to
detect pyrogenicity in
samples that have passed the rabbit pyrogen test and the Limulus amebocyte
lysate test (Taktak et al, J.
Pharm. Pharmacol. (1990), 43:578-82). In an additional embodiment, the
ophthalmic formulation is
subject to depyrogenation. In a further embodiment, the process for the
manufacture of the ophthalmic
formulation comprises testing the formulation for pyrogcnicity. In certain
embodiments, the formulations
described herein are substantially free of pyrogens.
[00221] Ophthalmic Muscarinic Antaaonist-Mucus Penetratinu Particle (MPP)
Composition
[00222] Mucus-penetrating particles (MPPs) are particles that rapidly
traverse mucus (e.g. human
mucus). In some cases, MPPs comprise of a nanoparticle with a particle size of
between about 200nm
and 500nm. In some instances, the nanoparticle is further coated with a mucus
penetrating agent. In some
instances, a composition described herein is formulated with MPPs for mucus
penetration. In some
instances, an ophthalmic agent composition described herein is formulated with
MPPs for mucus
penetration. In some instances, the ophthalmic agent is a muscarinic
antagonist. In some instances, a
muscarinic antagonist composition described herein is formulated with MPPs for
mucus penetration. In
some instances, a muscarinic antagonist comprises atropine, atropine sulfate,
noratropine, atropine-N-
oxide, tropine, tropic acid, atropine methonitrate, diphenhydramine,
dimenhydrinate, dicyclomine,
flavoxate, oxybutynin, tiotropium, hyoscine, scopolomine (L-hyoscine),
hydroxyzine, ipratropium,
tropicamide, cyclopentolate, pirenzapine, homatropine, solifenacin,
darifenacin, benzatropine,
mebeverine, procyclidine, aclidinium bromide, tribexyphenidyl/benzbexol, or
tolterodine. In some
instances, a muscarinic antagonist is atropine or its pharmaceutically
acceptable salt thereof In some
instances, a muscarinic antagonist is atropine sulfate. In some instances, an
atropine composition
described herein is formulated with MPPs for mucus penetration. In some
instances, an atropine sulfate
composition described herein is formulated with MPPs for mucus penetration. In
a non-limiting
example, the MMPs for use in the disclosed composition is obtained from Kala
Pharmaceuticals, Inc.
(100 Beaver Street #201, Waltham, MA 02453).
[00223] In some embodiments, the nanoparticle comprises of any suitable
material, such as an
organic material, an inorganic material, a polymer, or combinations thereof.
In some instances, the
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
52
nanoparticle comprises of inorganic material, such as for example, a metal
(e.g., Ag, Au, Pt, Fe, Cr, Co,
Ni, Cu, Zn, and other transition metals), a semiconductor (e.g., silicon,
silicon compounds and alloys,
cadmium selenide, cadmium sulfide, indium arsenide, and indium phosphide), or
an insulator (e.g.,
ceramics such as silicon oxide). In some instances, the nanoparticle comprises
organic materials such as a
synthetic polymer and/or a natural polymer. Examples of synthetic polymers
include non-degradable
polymers such as polymethacrylate and degradable polymers such as polylactic
acid, polyglycolic acid
and copolymers thereof. Examples of natural polymers include hyaluronic acid,
chitosan, and collagen.
[00224] In some embodiments, the nanoparticle is coated with a mucus
penetrating agent. In
some instances, the mucus penetrating agent comprises any suitable material,
such as a hydrophobic
material, a hydrophilic material, and/or an amphiphilic material. In some
instances, the mucus
penetrating agent is a polymer. In some instances, the polymer a synthetic
polymer (i.e., a polymer not
produced in nature). In other embodiments, the polymer is a natural polymer
(e.g. , a protein,
polysaccharide, rubber). In certain embodiments, the polymer is a surface
active polymer. In certain
embodiments, the polymer is a non-ionic polymer. In certain embodiments, the
polymer is a non-ionic
block copolymer. In some embodiments, the polymer is a diblock copolymer, a
triblock copolymer, e.g.,
e.g., where one block is a hydrophobic polymer and another block is a
hydrophilic polymer. In some
embodiments, the polymer is charged or uncharged.
[00225] Additional examples of suitable polymers include, but are not
limited to, polyamines,
polyethers, polyamides, polyesters, polycarbamates, polyureas, polycarbonates,
polystyrenes, polyimides,
polysulfones, polyurethanes, polyacetylenes, polyethylenes, polyethyeneimines,
polyisocyanates,
polyacrylates, polymethacrylates, polyacrylonitriles, and polyarylates. Non-
limiting examples of specific
polymers include poly(caprolactone) (PCL), ethylene vinyl acetate polymer
(EVA), poly(lactic acid)
(PLA), poly(L-lactic acid) (PLLA), poly(glycolic acid) (PGA), poly(lactic acid-
co-glycolic acid)
(PLGA), poly(L-lactic acid-co-glycolic acid) (PLLGA), poly(D,L-lactide)
(PDLA), poly(L- lactide)
(PLLA), poly(D,L-lactide-co-caprolactone), poly(D,L-lactide-co-caprolactone-co-
glycolide), poly(D,L-
lactide-co-PEO-co-D,L-lactide), poly(D,L-lactide-co-PPO-co-D,L-lactide),
polyalkyl cyanoacrylate,
polyurethane, poly-L-lysine (PLL), hydroxypropyl methacrylate (HPMA),
poly(ethylene glycol), poly-L-
glutamic acid, poly(hydroxy acids), polyanhydrides, polyorthoesters,
poly(ester amides), polyamides,
poly(ester ethers), polycarbonates, polyalkylenes such as polyethylene and
polypropylene, polyalkylene
glycols such as poly(ethylene glycol) (PEG), polyalkylene oxides (PEO),
polyalkylene terephthalates
such as poly(ethylene terephthalate), polyvinyl alcohols (PVA), polyvinyl
ethers, polyvinyl esters such as
poly(vinyl acetate), polyvinyl halides such as poly(vinyl chloride) (PVC),
polyvinylpyrrolidone,
polysiloxanes, polystyrene (PS), polyurethanes, derivatized celluloses such as
alkyl celluloses,
hydroxyalkyl celluloses, cellulose ethers, cellulose esters, nitro celluloses,
hydroxypropylcellulose,
carboxymethylcellulose, polymers of acrylic acids, such as
poly(methyl(meth)acrylate) (PMMA),
poly(ethyl(meth)acrylate), poly(butyl(meth)acrylate),
poly(isobutyl(meth)acrylate),
poly(hexyl(meth)acrylate), poly(isodecyl(meth)acrylate),
poly(lauryl(meth)acrylate),
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
53
poly(phenyl(meth)acrylate), poly(methyl acrylate), poly(isopropyl acrylate),
poly(isobutyl acrylate),
poly(octadecyl acrylate) (jointly referred to herein as "polyacrylic acids"),
and copolymers and mixtures
thereof, polydioxanone and its copolymers, polyhydroxyalkanoates,
polypropylene fumarate),
polyoxymethylene, poloxamers, poly(ortho)esters, poly(butyric acid),
poly(valeric acid), poly(lactide-co-
caprolactone), and trimethylene carbonate, polyvinylpyrrolidonc.
[00226] In some cases, an ophthalmic agent (e.g. a muscarinic antagonist
such as atropine or
atropine sulfate) is present in the MPP formulation at a concentration of
between about 0.001 wt% and
about 0.05 wt%, between about 0.005% to about 0.050%, between about 0.010% to
about 0.050%,
between about 0.015% to about 0.050%, between about 0.020% to about 0.050%,
between about 0.025%
to about 0.050%, between about 0.030% to about 0.050%, between about 0.035% to
about 0.050%,
between about 0.040% to about 0.050%, or between about 0.045% to about 0.050%
of the ophthalmic
agent, or pharmaceutically acceptable prodrug or salt thereof, by weight of
the composition. In some
instances, additional agents such as buffers, pD adjusting agents, and/or
preservatives are formulated in
the MPP formulation.
[00227] In some instances, ophthalmic agent-MPP composition is formulated
using any suitable
method. In some embodiments, a milling process is used to reduce the size of a
solid material to form
particles in the micrometer to nanometer size range. In some cases, dry and
wet milling processes such as
jet milling, cryo-milling, ball milling, media milling, and homogenization are
known and are used in
methods described herein. Generally, in a wet milling process, a suspension of
the material to be used as
the nanoparticle is mixed with milling media with or without excipients to
reduce particle size. Dry
milling is a process wherein the material to be used as the nanoparticle is
mixed with milling media with
or without excipients to reduce particle size. In a cryo-milling process, a
suspension of the material to be
used as the nanoparticle is mixed with milling media with or without
excipients under cooled
temperatures.
[00228] In some embodiments, any suitable grinding medium is used for
milling. In some
embodiments, a ceramic and/or polymeric material and/or a metal is used.
Examples of suitable materials
include zirconium oxide, silicon carbide, silicon oxide, silicon nitride,
zirconium silicate, yttrium oxide,
glass, alumina, alpha- alumina, aluminum oxide, polystyrene, poly(methyl
methacrylate), titanium, steel.
In some cases, a grinding medium has any suitable size. For example, the
grinding medium has an
average diameter of at least about 0.1 mm, at least about 0.2 mm, at least
about 0.5 mm, at least about 0.8
mm, at least about 1 mm, at least about 2 mm, or at least about 5 mm. In some
cases, the grinding
medium has an average diameter of less than or equal to about 5 mm, less than
or equal to about 2 mm,
less than or equal to about 1 mm, less than or equal to about 0.8, less than
or equal to about 0.5 mm, or
less than or equal to about 0.2 mm. Combinations of the above-referenced
ranges are also possible (e.g.,
an average diameter of at least about 0.5 millimeters and less than or equal
to about 1 mm). Other ranges
are also possible.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
54
[00229] In some embodiments, any suitable solvent are used for milling. In
some cases, the
choice of solvent is depend on factors such as the solid material (e.g., a
muscarinic antagonist such as
atropine) being milled, the particular type of stabilizer/mucus penetrating
agent being used (e.g., one that
renders the particle mucus penetrating), the grinding material be used, among
other factors. In some
cases, suitable solvents are ones that do not substantially dissolve the solid
material or the grinding
material, but dissolve the stabilizer/mucus penetrating agent to a suitable
degree. Non-limiting examples
of solvents include, but are not limited to, water, buffered solutions, other
aqueous solutions, alcohols
(e.g. , ethanol, methanol, butanol), and mixtures thereof that optionally
include other components such as
pharmaceutical excipients, polymers, pharmaceutical agents, salts,
preservative agents, viscosity
modifiers, tonicity modifier, taste masking agents, antioxidants, pD modifier,
and other pharmaceutical
excipients. In other embodiments, an organic solvent is used. In some cases, a
pharmaceutical agent (e.g.
a muscarinic antagonist such as atropine) has any suitable solubility in these
or other solvents, such as a
solubility in one or more of the ranges described above for aqueous solubility
or for solubility in a
coating solution.
[00230] In some instances, a MPP is a MPP as described in W02013/166385. In
some instances,
a MPP is a MPP as described in Lai et al., "Rapid transport of large polymeric
nanoparticles in fresh
undiluted human mucus," PNAS 104(5):1482-1487 (2007). In some instances, an
ophthalmic agent-MPP
composition is formulated using a method as described in W02013/166385. In
some instances, an
ophthalmic agent-MPP composition is formulated using a method as described in
Lai et al., "Rapid
transport of large polymeric nanoparticles in fresh undiluted human mucus,"
PNAS104(5):1482-1487
(2007). In some instances, the ophthalmic agent is a muscarinic antagonist
such as atropine or atropine
sulfate.
[00231] Ophthalmic Gel Muscarinic Antagonist Composition
[00232] Gels have been defined in various ways. For example, the United
States Pharmacopoeia
defines gels as semisolid systems consisting of either suspensions made up of
small inorganic particles or
large organic molecules interpenetrated by a liquid. Gels include a single-
phase or a two-phase system. A
single-phase gel consists of organic macromolecules distributed uniformly
throughout a liquid in such a
manner that no apparent boundaries exist between the dispersed macromolecules
and the liquid. Some
single-phase gels are prepared from synthetic macromolecules (e.g., carbomer)
or from natural gums,
(e.g., tragacanth). In some embodiments, single-phase gels are generally
aqueous, but will also be made
using alcohols and oils. Two-phase gels consist of a network of small discrete
particles.
[00233] In some embodiments, gels are also classified as being hydrophobic
or hydrophilic. In
certain embodiments, the base of a non-limiting example of a hydrophobic gel
includes a liquid paraffin
with polyethylene or fatty oils gelled with colloidal silica, or aluminum or
zinc soaps. In contrast, the
base of a non-limiting example of a hydrophilic gel includes water, glycerol,
or propylene glycol gelled
with a suitable gelling agent (e.g., tragacanth, starch, cellulose
derivatives, carboxyvinylpolymers, and
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
magnesium-aluminum silicates). In certain embodiments, the rheology of the
compositions disclosed
herein is pseudo plastic, plastic, thixotropic, or dilatant.
[00234] In some embodiments, the ophthalmic composition is an ophthalmic
gel, and wherein the
ophthalmically acceptable carrier comprises water and at least one viscosity-
enhancing agent. In some
embodiments, the viscosity-enhancing agent is selected from cellulose-based
polymers, polyoxyethylene-
polyoxypropylene friblock copolymers, dextran-based polymers, polyvinyl
alcohol, dextrin,
polyvinylpyrrolidone, polyalkylene glycols, chitosan, collagen, gelatin,
hyaluronic acid, or combinations
thereof.
[00235] In some embodiment, the ophthalmic gel composition described herein
is a semi-solid or
id in a gelled state before it is topically administered (e.g. at room
temperature). For example, suitable
viscosity-enhancing agents for such gels include by way of example only,
gelling agents and suspending
agents. In one embodiment, the enhanced viscosity formulation does not include
a buffer. In other
embodiments, the enhanced viscosity formulation includes a pharmaceutically
acceptable buffer. Sodium
chloride or other tonicity agents are optionally used to adjust tonicity, if
necessary.
[00236] By way of example only, the ophthalmically acceptable viscosity
agent includes
hydroxypropyl methylcellulose, hydroxyethyl cellulose, polyvinylpyrrolidone,
carboxymethyl cellulose,
polyvinyl alcohol, sodium chondroitin sulfate, sodium hyaluronate. Other
viscosity enhancing agents
compatible with the targeted ocular site include, but are not limited to,
acacia (gum arabic), agar,
aluminum magnesium silicate, sodium alginate, sodium stearate, bladderwrack,
bentonite, carbomer,
carrageenan, Carbopol, xanthan, cellulose, microcrystalline cellulose (MCC),
ceratonia, chitin,
carboxymethylated chitosan, chondrus, dextrose, furcellaran, gelatin, Ghatti
gum, guar gum, hectorite,
lactose, sucrose, maltodextrin, mannitol, sorbitol, honey, maize starch, wheat
starch, rice starch, potato
starch, gelatin, sterculia gum, xanthum gum, gum tragacanth, ethyl cellulose,
ethylhydroxyethyl
cellulose, ethylmethyl cellulose, methyl cellulose, hydroxyethyl cellulose,
hydroxyethylmethyl cellulose,
hydroxypropyl cellulose, poly(hydroxyethyl methacrylate), oxypolygelatin,
pectin, polygeline, povidone,
propylene carbonate, methyl vinyl ether/maleic anhydride copolymer (PVM/MA),
poly(methoxyethyl
methacrylate), poly(methoxyethoxyethyl methacrylate), hydroxypropyl cellulose,
hydroxypropylmethyl-
cellulose (HPMC), sodium carboxymethyl-cellulose (CMC), silicon dioxide,
polyvinylpyrrolidone (PVP:
povidone), Splenda0 (dextrose, maltodextrin and sucralose) or combinations
thereof. In specific
embodiments, the viscosity-enhancing excipient is a combination of MCC and
CMC. In another
embodiment, the viscosity-enhancing agent is a combination of
carboxymethylated chitosan, or chitin,
and alginate. The combination of chitin and alginate with the ophthalmic
agents disclosed herein acts as a
controlled release formulation, restricting the diffusion of the ophthalmic
agents from the formulation.
Moreover, the combination of carboxymethylated chitosan and alginate is
optionally used to assist in
increasing the permeability of the ophthalmic agents in the eye.
[00237] In some embodiments is an enhanced viscosity formulation,
comprising from about 0.1
mM and about 100 mM of an ophthalmic agent, a pharmaceutically acceptable
viscosity agent, and water
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
56
for injection, the concentration of the viscosity agent in the water being
sufficient to provide an enhanced
viscosity formulation with a final viscosity from about 100 to about 100,000
cP. In certain embodiments,
the viscosity of the gel is in the range from about 100 to about 50,000 cP,
about 100 cP to about 1,000 cP,
about 500 cP to about 1500 cP, about 1000 cP to about 3000 cP, about 2000 cP
to about 8,000 cP, about
4,000 cP to about 50,000 cP, about 10,000 cP to about 500,000 cP, about 15,000
cP to about 1,000,000
cP. In other embodiments, when an even more viscous medium is desired, the
biocompatible gel
comprises at least about 35%, at least about 45%, at least about 55%, at least
about 65%, at least about
70%, at least about 75%, or even at least about 80% or so by weight of the
ophthalmic agent. In highly
concentrated samples, the biocompatible enhanced viscosity formulation
comprises at least about 25%, at
least about 35%, at least about 45%, at least about 55%, at least about 65%,
at least about 75%, at least
about 85%, at least about 90% or at least about 95% or more by weight of the
ophthalmic agent.
[00238] In one embodiment, the pharmaceutically acceptable enhanced
viscosity ophthalmically
acceptable formulation comprises at least one ophthalmic agent and at least
one gelling agent. Suitable
gelling agents for use in preparation of the gel formulation include, but are
not limited to, celluloses,
cellulose derivatives, cellulose ethers (e.g., carboxymethylcellulose,
ethylcellulose,
hydroxyethylcellulose, hydroxymethylcellulose, hydroxypropylmethylcellulose,
hydroxypropykellulose,
methylcellulose), guar gum, xanthan gum, locust bean gum, alginates (e.g.,
alginic acid), silicates, starch,
tragacanth, carboxyvinyl polymers, carrageenan, paraffin, petrolatum and any
combinations or mixtures
thereof. In some other embodiments, hydroxypropylmethylcellulose (Methoce1C4')
is utilized as the
gelling agent. In certain embodiments, the viscosity enhancing agents
described herein are also utilized as
the gelling agent for the gel formulations presented herein.
[00239] In some embodiments, the ophthalmic gel composition described
herein is an in situ gel
formulation. In some instances, the in situ gel formation is based on
increased pre-corneal residence time
of the ophthalmic composition which improves ocular bioavailability, corneal
mucoadhesion, lysosomal
interaction and ionic gelation, improved corneal absorption, thermal gelation,
or a combination thereof.
In some instances, the in situ gel formulation is activated by pH,
temperature, ion, UV, or solvent
exchange.
[00240] In some instances, the ophthalmic gel composition comprises a
muscarinic antagonist
and one or more gelling agents. In some instances, the gelling agent includes,
but is not limited to,
poloxamer (e.g. Poloxamer 407), tetronics, ethyl (hydroxyethyl) cellulose,
cellulose acetate phthalate
(CAP), carbopol (e.g. Carbopol 1342P NF, Carbopol 980 NF), alginates (e.g. low
acetyl gellan gum
(Gelritek)), gellan, hyaluronic acid, pluronics (e.g. Pluronic F-127),
chitosan, polyvinyl alcohol (PVA),
polyvinylpyrrolidone (PVP), dextran, hydroxy propyl methyl cellulose (HPMC),
hydroxyethylcellulose
(HEC), methylcellulose (MC), thiolated xyloglucan, polymethacrilic acid
(PMMA), polyethylene glycol
(PEG), pseudolatexes, xyloglucans, or combinations thereof
[00241] In some instances, the in situ gel formation further comprises a
permeation enhancer. In
some instances, the permeation enhancer includes surfactants (e.g. non-ionic
surfactants), benzalkonium
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
57
chloride, EDTA, surface-active heteroglycosides, calcium chelators, hydroxyl
propyl beta cyclodextrin
(HP beta CD), bile salts, and the like.
[00242] In some embodiments, other gel formulations are useful depending
upon the particular
ophthalmic agent, other pharmaceutical agent or excipients/additives used, and
as such are considered to
fall within the scope of the present disclosure. For example, other
commercially-available glycerin-based
gels, glycerin-derived compounds, conjugated, or crosslinked gels, matrices,
hydrogels, and polymers, as
well as gelatins and their derivatives, alginates, and alginate-based gels,
and even various native and
synthetic hydrogel and hydrogel-derived compounds are all expected to be
useful in the ophthalmic agent
formulations described herein. In some embodiments, ophthalmically acceptable
gels include, but are not
limited to, alginate hydrogels SAFO-Gel (ConvaTec, Princeton, N.J.), Duoderm
Hydroactive Gel
(ConvaTec), Nu-gel (Johnson & Johnson Medical, Arlington, Tex.); Carrasyng(V)
Acemannan
Hydrogel (Carrington Laboratories, Inc., Irving, Tex.); glycerin gels Elia
Hydrogel (Swiss-American
Products, Inc., Dallas, Tex.) and K-YO. Sterile (Johnson & Johnson). In
further embodiments,
biodegradable biocompatible gels also represent compounds present in
ophthalmically acceptable
formulations disclosed and described herein.
[00243] In some embodiments, the viscosity-enhancing agent is a cellulose-
based polymer
selected from cellulose gum, alkylcellulose, hydroxyl-alkyl cellulose,
hydroxyl-alkyl alkylcellulose,
carboxy-alkyl cellulose, or combinations thereof In some embodiments, the
viscosity-enhancing agent is
hydroxyl-alkyl alkylcellulose. In some embodiment, the viscosity-enhancing
agent is hydroxypropyl
methylcellulose.
[00244] In certain embodiments, the enhanced viscosity formulation is
characterized by a phase
transition between room temperature and body temperature (including an
individual with a serious fever,
e.g., up to about 42 C). In some embodiments, the phase transition occurs at
1 C below body
temperature, at 2 C below body temperature, at 3 C below body temperature,
at 4 C below body
temperature, at 6 C below body temperature, at 8 C below body temperature,
or at 10 C below body
temperature. In some embodiments, the phase transition occurs at about 15 C
below body temperature,
at about 20 C below body temperature or at about 25 C below body
temperature. In specific
embodiments, the gelation temperature (Tgel) of a formulation described herein
is about 20 C, about 25
C, or about 30 C. In certain embodiments, the gelation temperature (Tgel) of
a formulation described
herein is about 35 C, or about 40 C. Included within the definition of body
temperature is the body
temperature of a healthy individual, or an unhealthy individual, including an
individual with a fever (up
to ¨42 C). In some embodiments, the pharmaceutical compositions described
herein are liquids at about
room temperature and are administered at or about room temperature.
[00245] Copolymers polyoxypropylene and polyoxyethylene (e.g.
polyoxyethylene-
polyoxypropylene triblock copolymers) form thermosetting gels when
incorporated into aqueous
solutions. These polymers have the ability to change from the liquid state to
the gel state at temperatures
close to body temperature, therefore allowing useful formulations that are
applied to the targeted ocular
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
58
site. The liquid state-to-gel state phase transition is dependent on the
polymer concentration and the
ingredients in the solution.
[00246] In some embodiments, the amount of thermosetting polymer in any
formulation
described herein is about 10%, about 15%, about 20%, about 25%, about 30%,
about 35% or about 40%
of the total weight of the formulation. In some embodiments, the amount of
thermosetting polymer in any
formulation described herein is about 10%, about 11%, about 12%, about 13%,
about 14%, about 15%,
about 16%, about 17%, about 18%, about 19%, about 20%, about 21%, about 22%,
about 23%, about
24% or about 25% of the total weight of the formulation. In some embodiments,
the amount of
thermosetting polymer (e.g., Poloxamer 407) in any formulation described
herein is about 7.5% of the
total weight of the formulation. In some embodiments, the amount of
thermosetting polymer (e.g.,
Poloxamer 407) in any formulation described herein is about 10% of the total
weight of the formulation.
In some embodiments, the amount of thermosetting polymer (e.g., Poloxamer 407)
in any formulation
described herein is about 11% of the total weight of the foimulation. In some
embodiments, the amount
of thermosetting polymer (e.g., Poloxamer 407) in any formulation described
herein is about 12% of the
total weight of the formulation. In some embodiments, the amount of
thermosetting polymer (e.g.,
Poloxamer 407) in any formulation described herein is about 13% of the total
weight of the formulation.
In some embodiments, the amount of thermosetting polymer (e.g., Poloxamer 407)
in any formulation
described herein is about 14% of the total weight of the formulation. In some
embodiments, the amount
of thermosetting polymer (e.g., Poloxamer 407) in any formulation described
herein is about 15% of the
total weight of the formulation. In some embodiments, the amount of
thermosetting polymer (e.g.,
Poloxamer 407) in any formulation described herein is about 16% of the total
weight of the formulation.
In some embodiments, the amount of thermosetting polymer (e.g., Poloxamer 407)
in any formulation
described herein is about 17% of the total weight of the formulation. In some
embodiments, the amount
of thermosetting polymer (e.g., Poloxamer 407) in any formulation described
herein is about 18% of the
total weight of the formulation. In some embodiments, the amount of
thermosetting polymer (e.g.,
Poloxamer 407) in any formulation described herein is about 19% of the total
weight of the formulation.
In some embodiments, the amount of thermosetting polymer (e.g., Poloxamer 407)
in any formulation
described herein is about 20% of the total weight of the formulation. In some
embodiments, the amount
of thermosetting polymer (e.g., Poloxamer 407) in any formulation described
herein is about 21% of the
total weight of the formulation. In some embodiments, the amount of
thermosetting polymer (e.g.,
Poloxamer 407) in any formulation described herein is about 23% of the total
weight of the formulation.
In some embodiments, the amount of thermosetting polymer (e.g., Poloxamer 407)
in any formulation
described herein is about 25% of the total weight of the formulation. In some
embodiments, the amount
of thickening agent (e.g., a gelling agent) in any formulation described
herein is about 1%, about 5%,
about 10%, or about 15% of the total weight of the formulation. In some
embodiments, the amount of
thickening agent (e.g., a gelling agent) in any formulation described herein
is about 0.5%, about 1%,
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
59
about 1.5%, about 2%, about 2.5%, about 3%, about 3.5%, about 4%, about 4.5%,
or about 5% of the
total weight of the formulation.
[00247] In an alternative embodiment, the thermogel is a PEG-PLGA-PEG
triblock copolymer
(Jeong etal, Nature (1997), 388:860-2; Jeong etal, J. Control. Release (2000),
63:155-63; Jeong etal, Adv.
Drug Delivery Rev. (2002), 54:37-51). The polymer exhibits sol-gel behavior
over a concentration of
about 5% w/w to about 40% w/w. Depending on the properties desired, the
lactide/glycolide molar ratio
in the PLGA copolymer ranges from about 1:1 to about 20:1. The resulting
coploymers are soluble in
water and form a free-flowing liquid at room temperature, but form a hydrogel
at body temperature. A
commercially available PEG-PLGA-PEG triblock copolymer is RESOMER RGP t50106
manufactured
by Boehringer Ingelheim. This material is composed of a PLGA copolymer of
50:50 poly(DL-lactide-co-
glycolide) and is 10% w/w of PEG and has a molecular weight of about 6000.
[00248] Additional biodegradable thermoplastic polyesters include AtriGelg
(provided by Atrix
Laboratories, Inc.) and/or those disclosed, e.g., in U.S. Patent Nos.
5,324,519; 4,938,763; 5,702,716;
5,744,153; and 5,990,194; wherein the suitable biodegradable thermoplastic
polyester is disclosed as a
thermoplastic polymer. Examples of suitable biodegradable thermoplastic
polyesters include
polylactides, polyglycolides, polycaprolactones, copolymers thereof,
terpolymers thereof, and any
combinations thereof. In some such embodiments, the suitable biodegradable
thermoplastic polyester is a
polylactide, a polyglycolide, a copolymer thereof, a terpolymer thereof, or a
combination thereof. In one
embodiment, the biodegradable thermoplastic polyester is 50/50 poly(DL-lactide-
co-glycolide) having a
carboxy terminal group; is present in about 30 wt. % to about 40 wt. % of the
composition; and has an
average molecular weight of about 23,000 to about 45,000. Alternatively, in
another embodiment, the
biodegradable thermoplastic polyester is 75/25 poly (DL-lactide-co-glycolide)
without a carboxy
terminal group; is present in about 40 wt. % to about 50 wt. % of the
composition; and has an average
molecular weight of about 15,000 to about 24,000. In further or alternative
embodiments, the terminal
groups of the poly(DL-lactide-co-glycolide) are either hydroxyl, carboxyl, or
ester depending upon the
method of polymerization. Polycondensation of lactic or glycolic acid provides
a polymer with terminal
hydroxyl and carboxyl groups. Ring-opening polymerization of the cyclic
lactide or glycolide monomers
with water, lactic acid, or glycolic acid provides polymers with the same
terminal groups. However, ring-
opening of the cyclic monomers with a monofunctional alcohol such as methanol,
ethanol, or 1-
dodecanol provides a polymer with one hydroxyl group and one ester terminal
groups. Ring-opening
polymerization of the cyclic monomers with a diol such as 1,6-hexanediol or
polyethylene glycol
provides a polymer with only hydroxyl terminal groups.
[00249] Since the polymer systems of thermosetting gels dissolve more
completely at reduced
temperatures, methods of solubilization include adding the required amount of
polymer to the amount of
water to be used at reduced temperatures. Generally after wetting the polymer
by shaking, the mixture is
capped and placed in a cold chamber or in a thermostatic container at about 0-
10 C in order to dissolve
the polymer. The mixture is stirred or shaken to bring about a more rapid
dissolution of the thermosetting
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
gel polymer. The ophthalmic agent and various additives such as buffers,
salts, and preservatives are
subsequently added and dissolved. In some instances the pharmaceutically agent
is suspended if it is
insoluble in water. The pD is modulated by the addition of appropriate
buffering agents.
[00250] Ophthalmic Ointment Muscarinic Antagonist Composition
[00251] An ointment is a homogeneous, viscous, semi-solid preparation, most
commonly a
greasy, thick oil (e.g. oil 80% - water 20%) with a high viscosity, intended
for external application to the
skin or mucous membranes. Ointments have a water number that defines the
maximum amount of water
that it contains. They are used as emollients or for the application of active
ingredients to the skin for
protective, therapeutic, or prophylactic purposes and where a degree of
occlusion is desired. Ointments
are used topically on a variety of body surfaces. These include the skin and
the mucous membranes of the
eye (an eye ointment), vulva, anus, and nose
[00252] The vehicle of an ointment is known as the ointment base. The
choice of a base depends
upon the clinical indication for the ointment. The different types of ointment
bases are: hydrocarbon
bases, e.g. hard paraffin, soft paraffin, microcrystalline wax and ceresine;
absorption bases, e.g. wool fat,
beeswax; water soluble bases, e.g. macrogols 200, 300, 400; emulsifying bases,
e.g. emulsifying wax,
cetrimide; vegetable oils, e.g. olive oil, coconut oil, sesame oil, almond oil
and peanut oil.
[00253] Ointments are formulated using hydrophobic, hydrophilic, or water-
emulsifying bases to
provide preparations that are immiscible, miscible, or emulsifiable with skin
secretions. In some
embodiments, they are also derived from hydrocarbon (fatty), absorption, water-
removable, or water-
soluble bases. The active agents are dispersed in the base, and later they get
divided after the drug
penetration into the target sites (e.g. membranes, skins, etc.).
[00254] The present disclosure recognizes that it is sometimes difficult to
incorporate into the
ointment a drug of low concentration with sufficient dose-to-dose uniformity
for effectively treating a
disorder or disease. In some embodiments, poly(ethylene-glycols),
polyethoxylated castor oils
(CremophorkEL), alcohols having 12 to 20 carbon atoms or a mixture of two or
more of said
components are effective excipients for dispersing and/or dissolving effective
amounts of ophthalmic
drugs, in particular of ascomycins and staurosporine derivatives, in an
ointment base, in particular in an
ointment base substantially comprising oleaginous and hydrocarbon components,
and that the resulting
ointments are excellently tolerated by the skin and by ocular tissue.
[00255] The present disclosure further recognizes that ophthalmic drugs,
such as a muscarinic
antagonist (e.g. atropine or its pharmaceutically acceptable salts),
incorporated in the ointment
compositions describes herein target the choroid and/or retina in a patient
when the compositions are
topically administered to the ocular surface, in particular to the sclera of
said patient. In some
embodiments, an ophthalmic ointment composition includes an ophthalmic drug,
an ointment base and
an agent for dispersing and/or dissolving said drug in the ointment base,
selected from a poly(ethylene-
glycol), a polyethoxylated castor oil, an alcohol having 12 to 20 carbon atoms
and a mixture of two or
more of said components.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
61
[00256] In some embodiments, the ointment bases include ophthalmically
acceptable oil and fat
bases, such as natural wax e.g. white and yellow bees wax, camauba wax, wool
wax (wool fat), purified
lanolin, anhydrous lanolin; petroleum wax e.g. hard paraffin, microcrystalline
wax; hydrocarbons e.g.
liquid paraffin, white and yellow soft paraffin, white petrolatum, yellow
petrolatum; or combinations
thereof.
[00257] The above mentioned oil and fat bases are described in more detail,
for instance, in the
British Pharmacopoeia, Edition 2001, or the European Pharmacopoeia, 3rd
Edition.
[00258] In some embodiments, the ointment base is present in amounts of
about 50 to about 95,
preferably of 70 to 90% by weight based on the total weight of the
composition.
[00259] A preferred ointment base comprises a combination of one or more of
one or more
natural waxes like those indicated above, preferably wool wax (wool fat), and
one or more hydrocarbons
like those indicated above, preferably a soft paraffin or a petrolatum, more
preferably in combination
with liquid paraffin.
[00260] A special embodiment of the aforementioned ointment base comprises
e.g. 5 to 17 parts
by weight of wool fat, and 50 to 65 parts by weight of white petrolatum as
well as 20 to 30 parts by
weight of liquid paraffin.
[00261] In some embodiments, the agent for dispersing and/or dissolving the
ophthalmic drug in
the ointment base is selected from a poly(ethylene-glycol), a polyethoxylated
castor oil, an alcohol
having 12 to 20 carbon atoms and a mixture of two or more of said components.
The agent is preferably
used in amounts of 1 to 20 percent, more preferably 1 to 10 percent by weight
of the entire semisolid
ophthalmic composition.
[00262] Alcohols having 12 to 20 carbon atoms include particularly stearyl
alcohol
(C18H370H), cetyl alcohol (C16H330H) and mixtures thereof. Preferred are so-
called cetostearyl
alcohols, mixtures of solid alcohols substantially consisting of stearyl and
cetyl alcohol and preferably
comprising not less than 40 percent by weight of stearyl alcohol and a sum of
stearyl alcohol and cetyl
alcohol amounting to at least 90 percent by weight, and compositions
comprising not less than 80 percent
by weight of cetylstearyl alcohol and an emulsifier, in particular sodium
cetostearyl sulfate and/or
sodium lauryl sulfate, preferably in amounts not less than 7 percent by weight
of emulsifier.
[00263] Polyethoxylated castor oils are reaction products of natural or
hydrogenated castor oils
and ethylene glycol. In some instances, such products are obtained in known
manner, e.g. by reaction of a
natural or hydrogenated castor oil or fractions thereof with ethylene oxide,
e.g. in a molar ratio of from
about 1:30 to about 1:60, with optional removal of free polyethylene glycol
components from the
product, e.g. in accordance with the methods disclosed in German
Auslegeschriften 1,182,388 and
1,518,819. Especially suitable and preferred is a product commercially
available under the trade name
Cremophor*EL having a molecular weight (by steam osmometry)=ca. 1630, a
saponification no.=ca. 65-
70, an acid no.--ca. 2, an iodine no.--ca. 28-32 and an nD 25=ca.1.471. Also
suitable for use in this
category is, for instance, NikkoltHCO-60, a reaction product of hydrogenated
castor oil and ethylene
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
62
oxide exhibiting the following characteristics: acid no.=ca. 0.3;
saponification no.=ca. 47.4; hydroxy
value=ca. 42.5. pH (5%)=ca. 4.6; Color APHA=ca. 40; m.p.=ca. 36.0 C.;
Freezing point=ca. 32.4 C.;
H20 content (%, KF)=ca. 0.03.
[00264] Poly(ethylene-glycols) are used in some embodiments as the agent
for dispersing and/or
dissolving the ophthalmic drug in the ointment base according to the present
disclosure. Suitable
poly(ethylene-glycol)s are typically mixtures of polymeric compounds of the
general formula H
(OCH2¨CH2)n0H, wherein the index n typically range from 4 to 230 and the mean
molecular weight
from about 200 to about 10000. Preferably 11 is a number from about 6 to about
22 and the mean
molecular weight between about 300 and about 1000, more preferably n ranges
from about 6 to about 13
and the mean molecular weight from about 300 to about 600, most preferably n
has a value of about 8.5
to about 9 and the relative molecular weight is about 400. Suitable
poly(ethylene-glycols) are readily
available commercially, for example poly(ethylene-glycols) having a mean
molecular weight of about
200, 300, 400, 600, 1000, 1500, 2000, 3000, 4000, 6000, 8000 and 10000.
[00265] The poly(ethylene-glycols), in particular the preferred types
described in the foregoing
paragraph, are preferably used in amounts of 1 to 10, more preferably 1 to 5
percent by weight of the
entire semisolid ophthalmic composition.
[00266] An especially preferred embodiment of the compositions according to
the instant
disclosure comprises an agent for dispersing and/or dissolving of the drug in
the ointment base which is
selected from a poly(ethylene-glycol), a polyethoxylated castor oil and
preferably a mixture of said
components.
[00267] Gel/Ointment Viscosity
[00268] In some embodiments, the composition has a Brookfield RVDV
viscosity of from about
10,000 to about 300,000 cps at about 20 C and sheer rate of 1s'. In some
embodiments, the composition
has a Brookfield RVDV viscosity of from about 15,000 to about 200,000 cps at
about 20 C and sheer
rate of ls-1. In some embodiments, the composition has a Brookfield RVDV
viscosity of from about
50,000 to about 150,000 cps at about 20 C and sheer rate of 1s'. In some
embodiments, the composition
has a Brookfield RVDV viscosity of from about 70,000 to about 130,000 cps at
about 20 C and sheer
rate of ls-1. In some embodiments, the composition has a Brookfield RVDV
viscosity of from about
90,000 to about 110,000 cps at about 20 C and sheer rate of 1s-1.
[00269] In some embodiments, the ophthalmic gel formulation contains a
viscosity enhancing
agent sufficient to provide a viscosity of between about 500 and 1,000,000
centipoise, between about 750
and 1,000,000 centipoise; between about 1000 and 1,000,000 centipoise; between
about 1000 and
400,000 centipoise; between about 2000 and 100,000 centipoise; between about
3000 and 50,000
centipoise; between about 4000 and 25,000 centipoise; between about 5000 and
20,000 centipoise; or
between about 6000 and 15,000 centipoise. In some embodiments, the ophthalmic
gel formulation
contains a viscosity enhancing agent sufficient to provide a viscosity of
between about 50,0000 and
1,000,000 centipoise.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
63
[00270] In some embodiments, the compositions described herein are low
viscosity compositions
at body temperature. In some embodiments, low viscosity compositions contain
from about 1% to about
10% of a viscosity enhancing agent (e.g., gelling components such as
polyoxyethylene-polyoxypropylene
copolymers). In some embodiments, low viscosity compositions contain from
about 2% to about 10% of
a viscosity enhancing agent (e.g., gelling components such as polyoxyethylene-
polyoxypropylene
copolymers). In some embodiments, low viscosity compositions contain from
about 5% to about 10% of
a viscosity enhancing agent (e.g., gelling components such as polyoxyethylene-
polyoxypropylene
copolymers). In some embodiments, low viscosity compositions are substantially
free of a viscosity
enhancing agent (e.g., gelling components such as polyoxyethylene-
polyoxypropylene copolymers). In
some embodiments, a low viscosity ophthalmic agent composition described
herein provides an apparent
viscosity of from about 100 cP to about 10,000 cP. In some embodiments, a low
viscosity ophthalmic
agent composition described herein provides an apparent viscosity of from
about 500 cP to about 10,000
cP. In some embodiments, a low viscosity ophthalmic agent composition
described herein provides an
apparent viscosity of from about 1000 cP to about 10,000 cP.
[00271] In some embodiments, the compositions described herein are viscous
compositions at
body temperature. In some embodiments, viscous compositions contain from about
10% to about 25% of
a viscosity enhancing agent (e.g., gelling components such as polyoxyethylene-
polyoxypropylene
copolymers). In some embodiments, the viscous compositions contain from about
14% to about 22% of a
viscosity enhancing agent (e.g., gelling components such as polyoxyethylene-
polyoxypropylene
copolymers). In some embodiments, the viscous compositions contain from about
15% to about 21% of a
viscosity enhancing agent (e.g., gelling components such as polyoxyethylene-
polyoxypropylene
copolymers). In some embodiments, a viscous ophthalmic composition described
herein provides an
apparent viscosity of from about 100,000 cP to about 1,000,000 cP. In some
embodiments, a viscous
ophthalmic composition described herein provides an apparent viscosity of from
about 150,000 cP to
about 500,000 cP. In some embodiments, a viscous ophthalmic composition
described herein provides an
apparent viscosity of from about 250,000 cP to about 500,000 cP. In some of
such embodiments, a
viscous ophthalmic composition is a liquid at room temperature and gels at
about between room
temperature and body temperature (including an individual with a serious
fever, e.g., up to about 42 C).
In some embodiments, a viscous ophthalmic composition is administered as
monotherapy for treatment
of an ophthalmic disease or condition described herein.
[00272] In some embodiments, the viscosity of the gel formulations
presented herein is measured
by any means described. For example, in some embodiments, an LVDV-II+CP Cone
Plate Viscometer
and a Cone Spindle CPE-40 is used to calculate the viscosity of the gel
formulation described herein. In
other embodiments, a Brookfield (spindle and cup) viscometer is used to
calculate the viscosity of the gel
formulation described herein. In some embodiments, the viscosity ranges
referred to herein are measured
at room temperature. In other embodiments, the viscosity ranges referred to
herein are measured at body
temperature (e.g., at the average body temperature of a healthy human).
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
64
[00273] Gel/Ointment Dose-To-Dose Uniformity
[00274] Typical ophthalmic gels are packaged in eye drop bottles and
administered as drops. For
example, a single administration (i.e. a single dose) of an ophthalmic gel
includes a single drop, two
drops, three drops or more into the eyes of the patient. Furthermore, typical
ophthalmic ointments are
packaged in tubes or other squeezable containers with a dispensing nozzle
through which strips of the
ointment are delivered. For example, a single administration (i.e. a single
dose) of an ophthalmic
ointment includes a single strip, or multiple strips into the eyes of the
patient. In some embodiments, one
dose of the ophthalmic gel described herein is one drop of the gel composition
from the eye drop bottle.
In some embodiments, one dose of the ophthalmic ointment is one strip of the
ointment composition
dispensed through the nozzle of a dispersing tube.
[00275] In some cases, described herein include ophthalmic gel compositions
which provide a
dose-to-dose uniform concentrations. In some instances, the dose-to-dose
uniform concentration does not
present significant variations of drug content from one dose to another. In
some instances, the dose-to-
dose uniform concentration does provide consistent drug content from one dose
to another.
[00276] In some cases, described herein include ophthalmic ointment
compositions which
provide a dose-to-dose uniform concentrations. In some instances, the dose-to-
dose uniform
concentration does not present significant variations of drug content from one
dose to another. In some
instances, the dose-to-dose uniform concentration does provide consistent drug
content from one dose to
another.
[00277] In some embodiments, the composition has a dose-to-dose ophthalmic
agent
concentration variation of less than 50%. In some embodiments, the composition
has a dose-to-dose
ophthalmic agent concentration variation of less than 40%. In some
embodiments, the composition has a
dose-to-dose ophthalmic agent concentration variation of less than 30%. In
some embodiments, the
composition has a dose-to-dose ophthalmic agent concentration variation of
less than 20%. In some
embodiments, the composition has a dose-to-dose ophthalmic agent concentration
variation of less than
10%. In some embodiments, the composition has a dose-to-dose ophthalmic agent
concentration
variation of less than 5%.
[00278] In some embodiments, the dose-to-dose ophthalmic agent
concentration variation is
based on 10 consecutive doses. In some embodiments, the dose-to-dose
ophthalmic agent concentration
variation is based on 8 consecutive doses. In some embodiments, the dose-to-
dose ophthalmic agent
concentration variation is based on 5 consecutive doses. In some embodiments,
the dose-to-dose
ophthalmic agent concentration variation is based on 3 consecutive doses. In
some embodiments, the
dose-to-dose ophthalmic agent concentration variation is based on 2
consecutive doses.
[00279] A nonsettling formulation should not require shaking to disperse
drug uniformly. A "no-
shake" formulation is potentially advantageous over formulations that require
shaking for the simple
reason that patients' shaking behavior is a major source of variability in the
amount of drug dosed. It has
been reported that patients often times do not or forget to shake their
ophthalmic compositions that
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
requires shaking before administering a dose, despite the instructions to
shake that were clearly marked
on the label. On the other hand, even for those patients who do shake the
product, it is normally not
possible to determine whether the shaking is adequate in intensity and/or
duration to render the product
uniform. In some embodiments, the ophthalmic gel compositions and ophthalmic
ointment compositions
described herein arc "no-shake" formulations that maintained the dose-to-dose
uniformity described
herein.
[00280] To evaluate the dose-to-dose uniformity, drop bottles or tubes
containing the ophthalmic
aqueous compositions, the ophthalmic gel compositions, or ophthalmic ointment
compositions are stored
upright for a minimum of 12 hours prior to the start of the test. To simulate
the recommended dosing of
these products, predetermined number of drops or strips are dispensed from
each commercial bottles or
tubes at predetermined time intervals for an extended period of time or until
no product was left in the
bottle or tube. All drops and strips are dispensed into tared glass vials,
capped, and stored at room
temperature until analysis. Concentrations of a muscarinic antagonist such as
atropine in the expressed
drops were determined using a reverse-phase HPLC method.
[00281] Methods of Treatment
[00282] Disclosed herein are methods of arresting myopia development by
administering to an
eye of an individual in need thereof an effective amount of an ophthalmic
composition as described
above. Also disclosed herein are methods of preventing myopia development by
administering to an eye
of an individual in need thereof an effective amount of an ophthalmic
composition as described above.
[00283] In some embodiments, the ophthalmic aqueous formulations described
herein are
packaged in eye drop bottles and administered as drops. For example, a single
administration (i.e. a
single dose) of an ophthalmic aqueous formulation includes a single drop, two
drops, three drops or more
into the eyes of the patient. In some embodiments, the ophthalmic gel
formulations described herein are
packaged in eye drop bottles and administered as drops. For example, a single
administration (i.e. a
single dose) of an ophthalmic gel includes a single drop, two drops, three
drops or more into the eyes of
the patient. In some embodiments, the ophthalmic ointment formulations
described herein are packaged
in tubes or other squeezable containers with a dispensing nozzle through which
strips of the ointment are
delivered. For example, a single administration (i.e. a single dose) of an
ophthalmic ointment includes a
single strip, or multiple strips into the eyes of the patient. In some
embodiments, one dose of the
ophthalmic aqueous formulation described herein is one drop of the aqueous
composition from the eye
drop bottle. In some embodiments, one dose of the ophthalmic gel described
herein is one drop of the gel
composition from the eye drop bottle. In some embodiments, one dose of the
ophthalmic ointment is one
strip of the ointment composition dispensed through the nozzle of a dispersing
tube.
[00284] In some embodiments of the disclosed method, the ophthalmic
composition is stored
below room temperature prior to first use. In some embodiments of the
disclosed method, the ophthalmic
composition is stored at between about 2 C to about 10 C prior to first use.
in some embodiments of the
disclosed method, the ophthalmic composition is stored at about 2 C, about 3
C, about 4 C, about 5 C,
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
66
about 6 C, about 7 C, about 8 C, about 9 C, or about 10 C prior to first
use. In some embodiments of
the disclosed method, the ophthalmic composition is stored at between about 4
C to about 8 C prior to
first use.
[00285] In some embodiments of the disclosed method, the ophthalmic
composition is stored at
room temperature after first use. In some embodiments of the disclosed method,
the ophthalmic
composition is stored at between about 16 C to about 26 C after to first
use. In some embodiments of
the disclosed method, the ophthalmic composition is stored at about 16 C,
about 17 C, about 18 C,
about 19 C, about 20 C, about 21 C, about 22 C, about 23 C, about 24 C,
about 25 C, or about
26 C after to first use.
[00286] In some embodiments, the ophthalmic aqueous formulations are
administered as follows:
the lower lid of the eye to be administered was pulled down and a
predetermined amount of the aqueous
formulation (e.g. 1-3 drops) is applied to the inside of the eyelid. The
ophthalmic tip of the dispensing
mechanism does not touch any surface to avoid contamination and/or injury.
[00287] In some embodiments, the ophthalmic gel formulations are
administered as follows: the
lower lid of the eye to be administered was pulled down and a predetermined
amount of gel (e.g. 1-3
drops) is applied to the inside of the eyelid. The ophthalmic tip of the
dispensing mechanism does not
touch any surface to avoid contamination and/or injury.
[00288] In some embodiments, the ophthalmic ointment formulations are
administered as
follows: the lower lid of the eye to be administered was pulled down and a
small amount of ointment
(approximately 0.25 inches) was applied to the inside of the eyelid. The
ophthalmic tip of the dispensing
mechanism does not touch any surface to avoid contamination and/or injury.
[00289] In some embodiments, the ophthalmic composition is administered at
predetermined
time intervals over an extended period of time. In some embodiments, the
ophthalmic composition is
administered once every day. In some embodiments, the ophthalmic composition
is administered every
other day. In some embodiments, the ophthalmic composition is administered
over 1 week, 2 weeks, 1
month, 2 months, 3 months, 6 moths, 1 year, 2 years, 3 years, 4 years, 5
years, 6 years, 7 years, 8 years, 9
years, 10 years, 11 years, or 12-15 years.
[00290] In some embodiments, the ophthalmic composition is administered in
doses having a
dose-to-dose ophthalmic agent concentration variation of less than 50%, less
than 40%, less than 30%,
less than 20%, less than 10%, or less than 5%.
[00291] The number of times a composition is administered to an individual
in need thereof
depends on the discretion of a medical professional, the disorder, the
severity of the disorder, and the
individual's response to the formulation. In some embodiments, a composition
disclosed herein is
administered once to an individual in need thereof with a mild acute
condition. In some embodiments, a
composition disclosed herein is administered more than once to an individual
in need thereof with a
moderate or severe acute condition. In the case wherein the patient's
condition does not improve, upon
the doctor's discretion the administration of an ophthalmic agent is
administered chronically, that is, for
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
67
an extended period of time, including throughout the duration of the patient's
life in order to ameliorate
or otherwise control or limit the symptoms of the patient's disease or
condition.
[00292] In the case wherein the patient's condition does not improve, upon
the doctor's
discretion the administration of the ophthalmic agent is administered
chronically, that is, for an extended
period of time, including throughout the duration of the patient's life in
order to ameliorate or otherwise
control or limit the symptoms of the patient's disease or condition.
[00293] In the case wherein the patient's status does improve, upon the
doctor's discretion the
administration of the ophthalmic agent is given continuously; alternatively,
the dose of drug being
administered is temporarily reduced or temporarily suspended for a certain
length of time (i.e., a "drug
holiday"). The length of the drug holiday varies between 2 days and 1 year,
including by way of example
only, 2 days, 3 days, 4 days, 5 days, 6 days, 7 days, 10 days, 12 days, 15
days, 20 days, 28 days, 35 days,
50 days, 70 days, 100 days, 120 days, 150 days, 180 days, 200 days, 250 days,
280 days, 300 days, 320
days, 350 days, and 365 days. The dose reduction during a drug holiday is from
10%-100%, including by
way of example only 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%,
65%, 70%, 75%,
80%, 85%, 90%, 95%, and 100%.
[00294] Once improvement of the patient's ophthalmic conditions has
occurred, a maintenance
ophthalmic agent dose is administered if necessary. Subsequently, the dosage
or the frequency of
administration, or both, is optionally reduced, as a function of the symptoms,
to a level at which the
improved disease, disorder or condition is retained. In certain embodiments,
patients require intermittent
treatment on a long-term basis upon any recurrence of symptoms.
[00295] The amount of ophthalmic agent that will correspond to such an
amount will vary
depending upon factors such as the particular compound, disease condition and
its severity, according to
the particular circumstances surrounding the case, including, e.g., the
specific ophthalmic agent being
administered, the route of administration, the condition being treated, the
target area being treated, and
the subject or host being treated. The desired dose is presented in a single
dose or as divided doses
administered simultaneously (or over a short period of time) or at appropriate
intervals.
[00296] In some embodiments, the initial administration is a particular
ophthalmic agent and the
subsequent administration a different formulation or ophthalmic agent.
[00297] Kits/Articles of Manufacture
[00298] The disclosure also provides kits for preventing or arresting
myopia development. Such
kits generally will comprise one or more of the ophthalmic compositions
disclosed herein, and
instructions for using the kit. The disclosure also contemplates the use of
one or more of the ophthalmic
compositions, in the manufacture of medicaments for treating, abating,
reducing, or ameliorating the
symptoms of a disease, dysfunction, or disorder in a mammal, such as a human
that has, is suspected of
having, or at risk for developing myopia.
[00299] In some embodiments, kits include a carrier, package, or container
that is
compartmentalized to receive one or more containers such as vials, tubes, and
the like, each of the
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
68
container(s) including one of the separate elements to be used in a method
described herein. Suitable
containers include, for example, bottles, vials, syringes, and test tubes. In
other embodiments, the
containers are formed from a variety of materials such as glass or plastic.
[00300] The articles of manufacture provided herein contain packaging
materials. Packaging
materials for use in packaging pharmaceutical products are also presented
herein. See, e.g., U.S. Patent
Nos. 5,323,907, 5,052,558 and 5,033,252. Examples of pharmaceutical packaging
materials include, but
are not limited to, drop bottles, tubes, pumps, bags, vials, containers,
syringes, bottles, and any packaging
material suitable for a selected formulation and intended mode of
administration and treatment. A wide
array of ophthalmic compositions provided herein are contemplated as are a
variety of treatments for any
disease, disorder, or condition that benefits by controlled release
administration of an ophthalmic agent to
the eye.
[00301] In some embodiments, a kit includes one or more additional
containers, each with one or
more of various materials (such as rinses, wipes, and/or devices) desirable
from a commercial and user
standpoint for use of a formulation described herein. Such materials also
include labels listing contents
and/or instructions for use and package inserts with instructions for use. A
set of instructions is optionally
included. In a further embodiment, a label is on or associated with the
container. In yet a further
embodiment, a label is on a container when letters, numbers or other
characters forming the label are
attached, molded or etched into the container itself; a label is associated
with a container when it is
present within a receptacle or carrier that also holds the container, e.g., as
a package insert. In other
embodiments a label is used to indicate that the contents are to be used for a
specific therapeutic
application. In yet another embodiment, a label also indicates directions for
use of the contents, such as in
the methods described herein.
[00302] In certain embodiments, the ophthalmic compositions are presented
in a dispenser device
which contains one or more unit dosage forms containing a compound provided
herein. In a further
embodiment, the dispenser device is accompanied by instructions for
administration. In yet a further
embodiment, the dispenser is also accompanied with a notice associated with
the container in form
prescribed by a governmental agency regulating the manufacture, use, or sale
of pharmaceuticals, which
notice is reflective of approval by the agency of the form of the drug for
human or veterinary
administration. In another embodiment, such notice, for example, is the
labeling approved by the U.S.
Food and Drug Administration for prescription drugs, or the approved product
insert. In yet another
embodiment, compositions containing a compound provided herein formulated in a
compatible
pharmaceutical carrier are also prepared, placed in an appropriate container,
and labeled for treatment of
an indicated condition.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
69
EXAMPLES
Example 1 ¨ Ophthalmic Formulations
[00303] Exemplary compositions for preparation of ophthalmic formulations
are described in
Tables 1-8.
Table 1 ¨Aqueous Solution Formulation (Atropine)
, 'In it.redicid, Qua, ntity (nib,
Con.cnLt t t ton vtsi ---
Atropine 0.01-0.5 0.001-0.05 (wt%)
Buffer agent and/or pD adjusting agent (e.g. q.s. for pD=4.2-7.9
, borates and/or DC1)
Preservative (e.g. benzalkonium chloride, q.s.
to prevent the growth of or
cetrimonium sodium perborate, etc.) to destroy microorganism
introduced into the solution
Tonicity and/or Osmolarity adjustor (e.g. q.s. to 0.5-2.0 wt%
NaC1, mannitol, etc)
Deuterated Water q.s. to 100 wt%
Table 2 ______________ Aqueous Solution Formulation (Atropine Sulfate)
Atropine sulfate 0.01-0.5 0.001-0.05 (wt%)
Buffer agent and/or pD adjusting agent (e.g. q.s. for pD=4.2-7.9
, borates and/or DC1)
Preservative (e.g. benzalkonium chloride, q.s.
to prevent the growth of or
cetrimonium sodium perborate, etc.) to destroy microorganism
introduced into the solution
Tonicity and/or Osmolarity adjustor (e.g. q.s. to 0.5-2.0 wt%
NaCl, mannitol, etc)
Deuterated Water q.s. to 100 wt%
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
Table 3 ¨Aqueous Solution Formulation (Atropine Sulfate)
FT:1111T6R.6)10:aiiiiii:(WiTar-7:
Atropine sulfate 0.05-0.15 0.005-0.015 (wt%)
Buffer agent and/or pD adjusting agent (e.g. q.s. for pD=4.2-7.9
, borates and/or DC1)
Preservative (e.g. benzalkonium chloride, q.s.
to prevent the growth of or
cetrimonium sodium perborate, etc.) to destroy microorganism
introduced into the solution
Tonicity and/or Osmolarity adjustor (e.g. q.s. to 0.5-2.0 wt%
NaCl, mannitol, etc)
Deuterated Water q.s. to 100 wt%
Table 4 ¨Mucus Penetrating Particle Formulation (Atropine)
' :]Concentration \ t 01:
Atropine 0.01-0.5 0.001-0.05 (wt%)
Buffer agent and/or pD adjusting agent (e.g. q.s. for pD=4.2-7.9
, borates and/or DC1)
Preservative (e.g. benzalkonium chloride, q.s.
to prevent the growth of or
cetrimonium sodium perborate, etc.) to destroy microorganism
introduced into the solution
Mucus penetrating particles q.s. to formulate atropine
at
0.001-0.05 wt%
Deuterated Water q.s. to 100 wt%
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
71
Table 5 ¨Mucus Penetrating Particle Formulation (Atropine Sulfate)
Atropine sulfate 0.01-0.5 0.001-0.05 (wt%)
Buffer agent and/or pD adjusting agent (e.g. q.s. for pD=4.2-7.9
, borates and/or DC1)
Preservative (e.g. benzalkonium chloride, q.s. to prevent the growth
of or
cetrimonium sodium perborate, etc.) to destroy microorganism
introduced into the solution
Mucus penetrating particles q.s. to formulate
atropine at
0.001-0.05 wt%
Deuterated Water q.s. to 100 wt%
Table 6 ________________ Cellulose Gel Formulation (Atropine Sulfate)
Atropine Sulfate 0.01-0.5 0.001-0.05 (wt%)
Viscosity enhancing agent (e.g. 10-50 1-5 (wt%)
hydroxypropyl methylcellulose)
Buffer agent and/or pD adjusting agent (e.g. q.s. for pD=4.2-7.9
, sodium acetate and/or DC1)
Stabilizer (e.g. EDTA, cyclodextrin, etc.) q.s. for low degradation
of
atropine sulfate (e.g. less than
10%, 5% or 1% degradation)
Osmolarity modifier (e.g. NaCl) q.s. 150-500 mOsm/L
Deuterated Water q.s. to 100 wt%
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
72
Table 7 ¨Thermosetting Gel Formulation (Atropine Sulfate)
Atropine sulfate 0.01-0.5 0.001-0.05 (wt%)
Viscosity enhancing agent (e.g. poloxamer 100-250 10-25 (wt%)
407)
Buffer agent and/or pD adjusting agent (e.g. q.s. for pH=4.2-7.9
, sodium acetate and/or DC1)
Stabilizer (e.g. EDTA, cyclodextrin, etc.) q.s. for low degradation of
atropine sulfate (e.g. less than
10%, 5% or 1% degradation)
Osmolarity modifier (e.g. NaC1) q.s. 150-500 mOsin/L
Deuterated Water q.s. to 100 wt%
Table 8 ¨ Ointment Formulation (Atropine Sulfate)
11L. SOIUtiOfL:: aqueous solution
Atropine sulfate 0.01-0.5 0.001-0.05 (wt%)
Dispersing agent (e.g. polyethyleneglycol, 10-200 1-20 (wt%)
and/or polyethoxylated castor oil and/or C12-
C20 alcohol
Buffering agent pD adjusting agent (e.g. q.s. for pD=4.2-7.9
DC1)
Stabilizer (e.g. EDTA, cyclodextrin, etc.) q.s. for low degradation of
atropine sulfate (e.g. less than
10%, 5% or 1% degradation)
Osmolarity modifier (e.g. NaCl) q.s. 150-500 mOsni/L
Ointment base (e.g. wool wax and/or q.s. to 100 wt%
petrolatum and/or liquid paraffin)
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
73
Example 2 - Preparation of an Aqueous Solution Formulation Containing 0.01%
Atropine in D20
[00304] Stock 1% Solution
[00305] In a 100mL solution, 1 gram of atropine, and 0.77 g of NaCl (and
other
ingredients/components preferably in their dry state) are added along with a
quantity sufficient to equal
100mL sterile deuterated water for injection. The solution is mixed in an
appropriately sized beaker with
a stir bar on a hot plate until all of the solid powders have dissolved and
the solution has become clear
with no visible particles. Next, the stir bar is removed, and the solution is
poured into a filter bottle and
vacuum filtered through a 0.22 micron pothyethersulfone membrane filter into a
sterile bottle. The filter
top is removed from the sterile stock bottle and the stock bottle is capped
for storage with a sterile bottle
cap.
[00306] Diluted 0.01% Solution
[00307] 0.3rnL of the 1% solution was combined with a quantity sufficient
to achieve 30mL total
of sterile 0.9% Sodium Chloride For Injection USP. The solution was thoroughly
mixed. The pH of the
solution was recorded. A 0.22 micron filter was placed on the tip of the
syringe and the solution was
aliquotted into separate sterile containers.
Example 3 - Preparation of an Aqueous Solution Formulation Containing 0.01%
Atropine Sulfate
[00308] Stock 1% Solution
[00309] In a 100mL solution, 1 gram of atropine sulfate, and 0.77 g of NaC1
(and other
ingredients/components preferably in their dry state) were added along with a
quantity sufficient to equal
100mL sterile water for injection. The solution was mixed in an appropriately
sized beaker with a stir bar
on a hot plate until all of the solid powders had dissolved and the solution
became clear with no visible
particles. Next, the stir bar was removed, and the solution was poured into a
filter bottle and vacuum
filtered through a 0.22 micron pothyethersulfone membrane filter into a
sterile bottle. The filter top was
removed from the sterile stock bottle and the stock bottle was capped for
storage with a sterile bottle cap.
[00310] Diluted 0.01% Solution
[00311] 0.3mL of the 1% solution was combined with a quantity sufficient to
achieve 30mL total
of sterile 0.9% Sodium Chloride For Injection USP. The solution was thoroughly
mixed. The pH of the
solution was recorded. A 0.22 micron filter was placed on the tip of the
syringe and the solution was
aliquotted into separate sterile containers.
Example 4 ¨ Stability Analysis
[00312] Five 0.01% atropine sulfate solutions were prepared from the 1%
atropine sulfate stock
solution (preparation as described in Example 2). The pH of the five solutions
was 5.87, 5.97, 5.90, 6.24,
and 6.16 for solutions 1-5, respectively. Each solution was thoroughly mixed.
A 0.22 micron filter was
placed on the tip of the syringe and the solution was aliquotted into separate
sterile containers according
to Table 9.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
74
Table 9. Container Filling Outline
Volume of 0.01% Total
Type of Container Atropine Sulfate Drug Containers
Product in Container Filled
Sterile Eyedroppers 5-mL 12
Sterile Glass Vials 5-mL 12
[00313] The samples were then stored at different conditions for stability
analysis. The samples
were analyzed at different time points up to 2 months. The storage conditions
include: 40 C with 75%
relative humidity (RH) (samples were transferred from 2-8 C condition after 3
days), 25 C with 60% RH,
and 60 C. The time points were 1 week, 2 weeks, 1 month, and 2 months. At each
of the time point, one
plastic eyedropper (LDPE plastic) and one glass vial from each of the stored
condition were removed and
allowed to equilibrate to ambient conditions. Once equilibrated, both the
plastic eyedropper and the glass
vials were inverted 3 times. The solution in the eyedroppers was transferred
to an HPLC vial in a drop
wise fashion through the dropper. The solution in the glass vial was
aliquotted into an HPLC vial using a
glass Pasteur pipette. The samples were then tested for purity and potency
using the UPLC method listed
in Table 10.
Table 10. UPLC Method Parameters
Parameter Condition
Column EMD, Hiber HR PurospherSTAR C-18, 100 x2.1 mm, 2 gm
Mobile Phase/Diluent 87:13, 50 InM Potassium Phosphate: Acetonitrile, pH
3.5
Flow Isocratic
Flow Rate 0.5 mL/min
Detection Wavelength 210 nm
Column Temperature 30 3 C
Autosampler Temperature 5 3 C
Run Time 6.0 minutes
Injection Volume 10 gL*
Needle Wash Solution 90/10 Water: Acetonitrile
* Modified from original method to maintain sensitivity at 100iug/mL nominal.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
[00314] Table 11 lists the stability data for the 0.01% atropine sulfate
solutions.
Table 11. Stability Data for 0.01% Atropine Sulfate Solutions
t=c) t=l week t=2 week.' tel mon&
t=2 month3
Storage
Analyst Container Type
Condition Purity Potency pH Purity Potency
Purity Potency Purity Potency pH Purity Potency pH
25 C/60% RH ND ND 99.1 99.9 ND ND
ND 95.4 97.4 6.3
Eyedropper,
40'C/75% RH 99.5 99.8 5.9 ND ND 96.2 97.3 95.1
95.6 5.2 ND ND ND
LDPE (Plastic)
1 60 C 80.8 83.3 86.2 88.6 88.3
91.5 4.2 ND ND ND
25'C/60% RH ND ND 92.2 93.1 80.7 80.5
7.8 73.0 74.5 7.3
Glass Vial 40'C/75% RH 99.8 100.4 ND ND ND 73.6 74.1
50.1 50.2 7.4 ND ND ND
60 C 43.1 43.9 28.3 28.4 ND ND
ND ND ND ND
25 C/60% RH ND ND 99.1 99.6 ND ND
ND 97.0 99.1 6.1
Eyedropper,
40 C/75% RH 99.7 99.9 6.0 ND ND 96.6 97.2 95.5
95.8 5.6 ND ND ND
LDPE (Plastic)
2 60 C 89.4 92.2 92.2 94.0 90.6
94.4 4.1 ND ND ND
25'C/6036 RH ND ND 92.6 92.9 82.5 82.2
7.6 80.2 81.6 7.3
Glass Vial 40'C/75% RH 99.8 100.2 ND ND ND 74.7 75.1
59.1 59.0 7.2 ND ND ND
60'C 54.2 55.2 37.3 37.4 ND ND
ND ND ND ND
25C/60% RH ND ND 98.7 96.1 ND ND
ND 95.8 94.8 6.3
Eyedropper,
40 C/75% RH 99.3 96.3 5.9 ND ND 96.7 93.1 94.8
91.8 5.5 ND ND ND
LDPE (Plastic)
3 60 C 88.8 89.0 88.0 86.8 88.6
87.7 4.1 ND ND ND
25'C/60% RH ND ND 94.1 91.2 85.0 81.9
7.5 79.3 78.3 7.3
Glass Vial 40'C/75% RH 99.4 9&4 ND ND ND 72.2 74.6
61.3 63.0 7.2 ND ND ND
60'C 48.6 51.1 34.1 34.9 ND ND
ND ND ND ND
edro er 25 C/60% RH ND ND 99.1 98.8 ND ND
ND 96.4 97.6 6.3
Eypp,
LDPE (Plastic) 40 C/75% RH 99.8 99.6 Ea ND ND 96.3
97.0 94.5 94.2 5.6 ND ND ND
4 60 C 90.5 93.0 89.3 90.6 84.2
85.8 4.2 ND ND ND
25'C/60% RH ND ND 90.7 90.0 76.9 75.1
7.6 72.5 71.6 7.4
Glass Vial 40'C/75% RH 99.8 98.8 ND ND ND 71.0 68.7
57.0 56.7 7.2 ND ND ND
60'C 52.4 52.1 29.7 28.6 ND ND
ND ND ND ND
25 C/60% RH ND ND 99.3 100.4 ND ND
ND 97.8 100.5 6.2
dropper Eye,
40 C/75% RH 99.6 100.6 6.2 ND ND 95.9 96.7 96.8
97.6 5.5 ND ND ND
LDPE (Plastic)
5 60 C 91.2 94.6 91.4 93.6 90.3
92.8 4.2 ND ND ND
25'C/60% RH ND ND 90.5 91.3 79.3 79.7
7.8 72.8 74.6 7.3
Glass Vial 40'C/75% RH 99.8 100.7 ND ND ND 71.3 71.9
56.0 56.4 7.3 ND ND ND
60'C 46.3 47.4 29.5 29.6 ND ND
ND ND ND ND
'The 25 T and the 60 Csa mples were psi led at 15 days, the 40 C samples
were pulled at 11 days.
The 25 "C and the 60 "C samples were pulled at 28 days, the 40 C samples were
pulled at 24 days.
The 25 'C and the 60 Csa mples were psi led at 46 days.
[00315] A change
in the pH of the 0.01% Atropine Sulfate solutions was observed over the
course of the stability study. The plastic (LDPE) eyedroppers maintained pH
around 6.2 when stored at
25 C for 2 months. However at the same time point, the pH of the 0.01%
atropine has increased to 7.2
when stored in glass vials. Additionally, when stored at elevated temperatures
(e.g. 40 C and 60 C), the
pH in the plastic (LDPE) eyedroppers dropped to approximately 4-5, while the
pH maintained around 7.2
when stored in the glass vials.
[00316] There was also a significant difference in the rate of degradation
for Atropine Sulfate
(0.01%) when stored in plastic (LDPE) eyedroppers versus Type I glass vials.
However, in both
containers there was an increase of an early eluting related substance at
relative retention time (RRT)
=0.87-0.89. In some cases, this early eluting related substance is referred to
as primary degradant. In
some instances, the primary degradant is referred to as RRT 0.87-0.89. This
related substance is likely to
be the first parameter to fail specification regardless of the container. The
amount of this related
substance was tracked at each time point and is listed in Table 12.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
76
Table 12. Area (%) of the Main Degradation Species for 0.01% Atropine Sulfate
(RRT 0.87-0.89)
Temperature
Analyst t=0 t=1 week t=2 week t=1 month t=2
months
C
25 0.08 NA 0.92 NA 3.98
1 40 NA NA 3.74 4.78 NA
60 NA 17.78 13.49 11.51 NA
25 0.07 NA 0.88 NA 2.46
2 40 NA NA 3.26 4.37 NA
60 NA 9.38 7.67 9.13 NA
25 0.07 NA 1.05 NA 2.88
3 40 NA NA 2.98 4.85 NA
60 NA 9.59 11.57 10.55 NA
25 0.08 NA 0.92 NA 3.09
4 40 NA NA 3.43 5.32 NA
60 NA 8.30 10.46 15.49 NA
25 0.08 NA 0.64 NA 1.66
40 NA NA 3.96 3.07 NA
60 NA 7.61 8.35 9.7 NA
Average 25 C 0.08 NA 0.88 NA 2.81
Average 40 C NA NA 3.47 4.48 NA
Average 60 C NA 10.53 10.31 11.28 NA
[00317] Arrhenius based shelf life predictions were calculated using the
related substance data
from Table 12. These predictions are based on an assumption that the
degradation is first order (linear).
These predictions are illustrated in Figs. 1 and 2. Fig. 1 shows the shelf
life prediction of 0.01% atropine
sulfate solution with a primary degradant RRT 0.87-0.89, and a n.m.t. of 0.5%
area, based on data
obtained from samples stored at 25 C and 40 C. The pH range of the atropine
sulfate solution is from
5.9-6.2. Fig. 2 shows the shelf life prediction of 0.01% atropine sulfate
solution with a primary degradant
RRT 0.87-0.89, and a n.m.t. of 0.5% area, based on data obtained from samples
stored at 25 C and 60 C.
The pH range of the atropine sulfate solution is from 5.9-6.2.
Example 5 -1% Atropine Sulfate (Bausch + Lomb) Sample Analysis
[00318] The 1% atropine sulfate sample was obtained from Bausch + Lomb
(Lot 198421). For
comparison the pH of the 1% Atropine Sulfate drug product was determined in
the neat solution as well
as a sample that was diluted to the current nominal concentration (0.01%
Atropine Sulfate) using the
vehicle. Additionally a sample was diluted to the nominal concentration with
method diluent. Both
samples diluted to the nominal concentration were analyzed using the RP-UPLC
method (Table 10). The
results are listed in Table 13.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
77
Table 13. pH and Purity of the Bausch + Lomb Atropine Sulfate Sample
Purity
Sample pH
(`)/0 area)
1% Atropine Sulfate 4.89 ND
0.01% Atropine Sulfate,
6.16 99.6%
diluted with Vehicle
0.01% Atropine Sulfate,
ND 99.6%
diluted with Diluent
Vehicle 7.94 ND
ND= not determined
Example 6 ¨ Dose Uniformity (10-Dose)
[00319] To evaluate the dose-to-dose uniformity, drop bottles containing
the ophthalmic aqueous
composition are stored upright for a predetermined period of time (e.g. 12
hours) prior to the start of the
test. To simulate the recommended dosing of the product, 10 drops of the
aqueous composition are
dispensed from each bottle at predetermined time intervals (e.g.
consecutively, every 1 minute, every 10
minutes, every hour or every 24 hours). All drops are dispensed into tared
glass vials, capped, and stored
at room temperature until analysis. Concentrations of atropine in the
expressed drops are determined
using a reverse-phase HPLC method.
Example 7 ¨ Dose Uniformity (5-Dose)
[00320] To evaluate the dose-to-dose uniformity, drop bottles containing
the ophthalmic aqueous
composition are stored upright for a predetermined period of time (e.g. 12
hours) prior to the start of the
test. To simulate the recommended dosing of the product, 5 drops of the
aqueous composition are
dispensed from each bottle at predetermined time intervals (e.g.
consecutively, every 1 minute, every 10
minutes, every hour or every 24 hours). All drops are dispensed into tared
glass vials, capped, and stored
at room temperature until analysis. Concentrations of atropine in the
expressed drops are determined
using a reverse-phase HPLC method.
Example 8 ¨ Dose Uniformity (2-Dose)
[00321] To evaluate the dose-to-dose uniformity, drop bottles containing
the ophthalmic aqueous
composition are stored upright for a predetermined period of time (e.g. 12
hours) prior to the start of the
test. To simulate the recommended dosing of the product, 2 drops of the
aqueous composition are
dispensed from each bottle at predetermined time intervals (e.g.
consecutively, every 1 minute, every 10
minutes, every hour or every 24 hours). All drops are dispensed into tared
glass vials, capped, and stored
at room temperature until analysis. Concentrations of atropine in the
expressed drops are determined
using a reverse-phase HPLC method.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
78
Exam])le 9 - Formulation Stability Comnarison
[00322] Atropine sulfate monohydrate (MP Bio; Lot Number 7825K) and tropic
acid (Sigma
Aldrich; Lot Number STBD6457V) were used for this experiment. Eight
formulations illustrated in
Table 14A were analyzed at t=0, 2 weeks, and 4 weeks. A RP-HPLC method was
used to carry out the
analysis.
Table 14A. Atropine sulfate formulations
Formulation Atropine Benzalkonium Sodium Acetic Citric pH/pD Aqueous
Sulfate Chloride Chloride Acid Acid
Monohydrate (BAK)
1 0.010 0.01 0.90 0.01 - 4.2 SWFI
2 0.025 0.01 0.90 0.01 - 4.2 SWFI
3 0.010 0.01 0.90 0.01 - 4.8 SWFI
4 0.025 0.01 0.90 0.01 - 4.8 SWFI
0.010 0.01 0.90 - 0.04 5.8 SWFI
6 0.025 0.01 0.90 - 0.04 5.8 SWFI
7 0.010 0.01 0.90 0.01 - 5.2 D20
(pD)
8 0.010 0.01 0.90 - 0.04 6.2 D20
(pD)
[00323] The values are % w/v. The formulations were prepared at 100mL scale
in volumetric
glassware. The pD of Formulation 7 and Formulation 8 are 5.2 and 6.2,
respectively. In some instances,
the pD is calculated as pD = 0.4+pH*, in which pH* is the measured or observed
pH of the solution
formulated in a solution containing deuterated water.
[00324] Table 14B illustrates analysis time points for the formulations
listed in Table 14A.
Table 14B. Schedule for atropine sulfate formulation testing
Storage Time Point
Condition Initial
2 Week 4 Week
(Horizontal) (t=0)
25 C/60%RH X X
40 C/75%RH X X X
60 C X X
[00325] Table 15 illustrates the atropine sulfate purity data associated
with each of the eight
formulations. Purity is indicated as percentage of area under the curve.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
79
Table 15. Atropine sulfate purity as Area-%
Solvent Condition t=0 t=2 weeks t=4 weeks'
Formulation 1 25/60 97.39 97.76 98.20
pH 4.2 40/75 97.25 97.04
60 C 94.98 93.87
Formulation 2 25/60 98.85 99.03 99.08
pH 4.2 40/75 98.50 98.32
60 C 97.47 96.65
Formulation 3 25/60 98.16 98.16 98.45
pH 4.8 40/75 97.98 97.35
60 C 95.94 94.65
Formulation 4 25/60 98.81 98.75 98.46
pH 4.8 40/75 98.26 98.01
60 C 96.22 94.04
Formulation 5 25/60 98.16 97.92 97.54
pH 5.8 40/75 95.88 93.51
60 C 80.94 66.83
Formulation 6 25/60 99.08 98.91 98.46
pH 5.8 40/75 97.65 96.20
60 C 89.15 80.68
Formulation 7 25/60 98.93 99.07 98.39
pD 5.2 40/75 98.51 97.55
60 C 96.70 94.01
Formulation 8 25/60 98.93 98.95 98.51
pD 6.2 40/75 98.53 97.44
60 C 95.97 92.72
'Some chromatographic interference were observed to occur late in the run (-27-
32 minutes) for many of
the 1=4 week stability samples and in some instances is proposed to be system
related.
[00326] After four weeks of storage at 60 C, in some instances the
atropine sulfate concentration
have an impact on the stability for the formulations containing acetic acid at
pH 4.2. For example,
atropine sulfate concentration at 0.025 % w/v (Formulation 2) showed a 2.8%
increase in % purity at pH
4.2 compared to the atropine sulfate concentration at 0.010 % w/v (Formulation
1). This trend was not
observed for the acetic acid formulations at pH 4.8 (Formulations 3 and 4);
rather a 0.6% decrease in %
purity was observed for the higher doses.
[00327] The dose dependent stability trend that was observed at pH=4.2 was
also seen in the
formulations containing citric acid at pH 5.8 (Formulations 5 and 6). After
four weeks of storage at 60 C
there is approximately 14% less degradation in the higher does than observed
in the lower dose.
[00328] At both the high and the low doses, more degradation is observed in
the formulations
that start at a higher pH. This degradation is predominantly the growth of
tropic acid. In some instances,
buffer species plays a role in the observed degradation between the different
pH values.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
[00329] The percentage of tropic acid observed for each of the formulations
at t=4 weeks and at
60 C are as follow:
[00330] Formulation 1- Tropic acid observed is 0.54%.
[00331] Formulation 2-Tropic acid observed is 0.93%.
[00332] Formulation 3-Tropic acid observed is 1.58%.
[00333] Formulation 4-Tropic acid observed is 3.03%.
[00334] Formulation 5-Tropic acid observed is 29.13%.
[00335] Formulation 6-Tropic acid observed is 16.84%.
[00336] Formulation 7-Tropic acid observed is 1.07%.
[00337] Formulation 8-Tropic acid observed is 4.03%.
[00338] In some embodiments, switching the water source to deuterated water
(D20) has an
impact on stabilizing the growth of the tropic acid peak for the formulation
containing acetic acid at pD
5.2 (Formulation 7), see Fig. 4. In addition, in the formulation containing
citric acid at pD 6.2
(Formulation 8), the deuterated water also stabilizes atropine sulfate, see
Fig. 5.
[00339] Table 16 illustrates tropic acid as area under the curve for each
of the eight formulations.
Tropic acid is a degradant of atropine sulfate. In some instances, LOQ was
previously found to be 0.05%
for the RP-HPLC method.
Table 16. Tropic acid as area-%
Solvent Condition t=0 t=2 weeks t=4 weeks
Formulation 1 25/60 <LOQ 0.08 <LOQ
pH 4.2 40/75 0.10 0.10
60 C 0.37 0.51
Formulation 2 25/60 <LOQ 0.05 <LOQ
pH 4.2 40/75 0.11 0.12
60 C 0.46 0.93
Formulation 3 25/60 <LOQ 0.12 0.05
pH 4.8 40/75 0.19 0.27
60 C 0.90 1.58
Formulation 4 25/60 <LOQ 0.10 0.13
pH 4.8 40/75 0.31 0.53
60 C 1.84 3.03
Formulation 5 25/60 <LOQ 0.40 0.71
pH 5.8 40/75 2.22 4.35
60 C 16.62 29.13
Formulation 6 25/60 <LOQ 0.24 0.42
pH 5.8 40/75 1.30 2.44
60 C 9.32 16.84
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
81
Formulation 7 25/60 <LOQ 0.07 0.08
pD 5.2 40/75 0.14 0.24
60 C 0.71 1.07
Formulation 8 25/60 <LOQ 0.11 0.14
pD 6.2 40/75 0.33 0.65
60 C 2.32 4.03
[00340] Table 17 illustrates percentage of potency of atropine in the eight
formulations.
Table 17. % Potency
Solvent Condition t=0 t=2 weeks t=4 weeks
Formulation 1 25/60 109.4 110.3 112.8
pH 4.2 40/75 111.0 112.4
60 C 112.8 114.8
Formulation 2 25/60 102.9 107.1 109.7
pH 4.2 40/75 108.4 109.6
60 C 109.4 111.0
Formulation 3 25/60 106.3 108.0 109.6
pH 4.8 40/75 108.1 110.0
60 C 108.0 109.9
Formulation 4 25/60 102.5 107.9 109.2
pH 4.8 40/75 107.4 108.9
60 C 107.9 108.8
Formulation 5 25/60 105.0 105.9 107.1
pH 5.8 40/75 103.8 103.5
60 C 90.2 77.7
Formulation 6 25/60 107.2 107.1 109.1
pH 5.8 40/75 106.8 107.1
60 C 99.0 93.7
Formulation 7 25/60 107.3 111.3 112.9
pD 5.2 40/75 111.6 113.5
60 C 111.8 113.5
Formulation 8 25/60 99.0 103.0 105.0
pD 6.2 40/75 104.9 104.7
60 C 101.6 103.0
[00341] After 4 weeks of storage, the observed potency values were elevated
from the t=0 and 2
week time points, with the exception of Formulations 5 and 6 at 60 C where
the potencies dropped due
to degradation. In some instances, these potency values are within the error
of the HPLC method, but
appear to be trending upward. Mass balance was calculated for the 60 C data
and results were consistent
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
82
across the formulations and levels of degradation, although skewed lower due
to the higher than
anticipated potency values at 4 weeks, see Figure 3.
[00342] Table 18 illustrates pH or pD stability of the eight formulations.
Table 18. pH/pD Stability
Solvent Condition t=0 t=2 weeks t=4 weeks
Formulation 1 25/60 4.21 3.93 4.02
(pH) 40/75 3.86 3.96
60 C 3.71 3.86
Formulation 2 25/60 4.26 4.11 4.25
(1:41) 40/75 4.04 4.17
60 C 3.93 4.10
Formulation 3 25/60 4.85 4.44 4.61
(pH) 40/75 4.41 4.54
60 C 4.32 4.40
Formulation 4 25/60 4.98 4.93 5.05
(pH) 40/75 4.89 4.98
60 C 4.77 4.77
Formulation 5 25/60 5.87 5.93 6.03
(pH) 40/75 5.96 5.96
60 C 5.82 5.78
Formulation 6 25/60 5.80 5.69 5.77
(pH) 40/75 5.65 5.67
60 C 5.54 5.50
Formulation 7 25/60 5.31 5.10 5.24
(pD) 40/75 5.08 5.15
60 C 5.00 4.93
Formulation 8 25/60 6.25 5.72 5.88
(pD) 40/75 5.74 5.78
60 C 5.58 5.50
[00343] The italicized values are pD values for a deuterated sample. In
some embodiments, the
pD of the deuterated samples are pD = pHread,õ +0.4 (Glasoe, et al. "Use of
glass electrodes to measure
acidities in deuterium oxide" J. Physical Chem. 64(1): 188-190 (1960)).
[00344] At the two lower temperatures, the pH values at t=4 week are
slightly elevated from the
t=2 week time point. These data were generated using a new glass pH probe. In
some instances, the
observed differences are due to the probe differences or additional variables
such as for example, the age
of the standard buffers or temperature gradients within the laboratory
environment. The downward pH
trend for each formulation with increasing temperatures at t=4 week is
consistent with previous data and
is consistent with the increase in the amount of tropic acid present in the
stability sample.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
83
Example 10-Determination of Shelf Life and Activation Energy
[00345] Activation energy was calculated for the eight formulations
disclosed in Example 9 and
comparison with a reference standard was made with Formulations 4-7.
[00346] Table 19 illustrates the activation energy (Ea) calculation. The Ea
minimum is 17.8
Kcal/mol, the Ea maximum is 21.3 Kcal/mol, and the Ea mean is 19.5 Kcal/mol.
Mean is +/- 3* stdev.
Figs. 6 and 7 illustrate the poor correlation between RS and tropic acid with
Formulation 4 and
Formulation 7, respectively. Figs. 8 and 9 illustrate improved correlation
between RS and tropic acid
with Formulation 5 and Formulation 6, respectively. At a lower pH (e.g. pH 4.8
or lower), there was a
poor correlation observed (Formulation 4 and Formulation 7). This was due to a
slowed hydrolysis and
increased alternative degradation pathways. At a higher pH (e.g., pH 5.8 or
higher), an improved or
better correlation was observed (Formulation 5 and Formulation 6). This was
due to the hydrolysis of
atropine as the primary degradant. It is noted that the activation energy is
for the specific acid catalyzed
degradation to tropic acid- the predominant degradation product and
degradation mechanism operating at
pH 5.8 or higher.
Table 19. Activation energy for total related substance (RS) and tropic acid.
Total Tropic
RS Acid
EgigiguipoogigighwCorr Con
Egiiiigigigigg
2 12.2 Poor
Corr
imprripbsppr li.igcrmoRpla
iildwomoonammtiammoodialiii
.... 4 .. 16.8 18.1
EnianNinaWn =MgV
6 19.2 20.0
8 Poor 18.9
Corr
Ø eimmipimmiimogo*No4mi.;
Mean 16.2 18.4 Kcal/mole
1:S..a6P.M;GUEN4.106Relp6QACZaagiS
RSD 21% 9%
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
84
[00347] Table 20 illustrates the rate of RS or tropic acid formation per
week at 40 C.
Table 20.
Formulation Rate 40 C (total RS Rate 40 C (Tropic
%/wk) acid %/wk)
Formulation 5 0.01% Atr Citrate pH5.8 1.16 1.09
Formulation 6 0.025% Atr Citrate pH5.8 0.72 0.61
Formulation 8 0.01% Atr Citrate pD 6.2 D20 0.163
[00348] Table 21 illustrates the activation energy and predicted shelf life
at 30 C calculated based
on Table 20. It is assumed for the calculation that tropic acid and total RS
is 5% (self-life).
Table 21A.
Rate @30 C (Total RS %/wk) Estimated Shelf life @30 C (mo)
Formulation Ea min Ea mean Ea max Ea min Ea mean Ea max
0.45 0.41 0.38 2.78 3.04 3.33
6 0.28 0.26 0.23 4.47 4.90 5.37
8 -- -- -- -- -- --
Table 21B.
Rate @30 C (Tropic acid %/wk) Estimated Shelf life iii30 C (mo)
Formulation Ea min Ea mean Ea max Ea min Ea mean Ea max
5 , 0.42 0.39 0.35 2.95 3.24 3.54
6 0.24 0.22 0.20 5.28 5.78 6.33
8 0.06 0.06 0.05 19.75 21.64 23.70
[00349] At pD 6.2, the deuterated formulation (Formulation 8) has a
predicted shelf life of close
to 2 years at 30 C.
[00350] Table 22 illustrate the predicted shelf life at temperatures of 40
C, 30 C, 25 C, and 2-8 C
for Formulations 4-8 for total RS and tropic acid, respectively.
Table 22
Stability Prediction RS Tropic Acid
Temperature Temperature
Formulation weeks months weeks months
( C) ( C)
4 40 16.5 4.1 40 7.7 1.9
30 40.2 10.1 30 20.0 5.0
25 64.2 16.0 25 33.0 8.3
2-8 493.4 123.4 2-8 296.8 74.2
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
5 40 2.8 0.7 40 0.9 0.2
30 7.9 2.0 30 2.7 0.7
25 13.7 3.4 25 4.6 1.2
2-8 151.1 37.8 2-8 50.5 12.6
6 40 5.8 1.4 40 1.7 0.4
30 15.9 4.0 30 4.8 1.2
25 27.3 6.8 25 8.4 2.1
2-8 281.6 70.4 2-8 95.9 24.0
7 40 11.5 2.9 40 16.9 4.2
30 23.2 5.8 30 38.4 9.6
25 33.4 8.4 25 59.1 14.8
2-8 165.7 41.4 2-8 388.2 97.1
8 40 - - 40 6.2 1.6
30 - _
30 17.0 4.3
25 - _
25 28.9 7.2
2-8 - - 2-8 287.1 71.8
Example 11-Additional Formulation Stability Comparison
[00351] Atropine sulfate monohydrate (MP Bio; Lot Number 7825K) and tropic
acid (Sigma
Aldrich; Lot Number STBD6457V) were used for this experiment. Thirteen
formulations illustrated in
Table 23A were analyzed. Formulations 1-8 had been analyzed at t=0, 2 weeks, 4
weeks, and 8 weeks.
Formulations 9-13 had been analyzed at t=0, 2 weeks, and 4 weeks. The pH
values reported herein are
the measured pH values obtained using the Thermo Scientific, Orion Dual Star
pH/ISE benchtop pH
meter and the Orion Double Junction Micro pH probe SiN S01-18520 calibrated
with H20 based
standards.
Table 23A. Atropine sulfate Formulations
Formulation Atropine Benzalkonium Sodium Acetic Citric pH/pD Aqueous
Sulfate Chloride Chloride Acid Acid
Monohydrate (BAK)
1 0.010 0.01 0.90 0.01 - 4.2 SWFI
2 0.025 0.01 0.90 0.01 - 4.2 SWFI
3 0.010 0.01 0.90 0.01 - 4.8 SWFI
4 0.025 0.01 0.90 0.01 - 4.8 SWFI
5 0.010 0.01 0.90 - 0.04 5.8 SWFI
6 0.025 0.01 0.90 - 0.04 5.8 SWFI
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
86
7 0.010 0.01 0.90 0.01 - 5.2 D20
(pD)
8 0.010 0.01 0.90 - 0.04 6.2 D20
(pD)
9 0.010 - 0.90 - 0.04 6.8 D20
(pD)
0.010 0.90 0.04 6.4 H20 (control)
11 0.010 - 0.90 - 0.08 6.4 H20
(control)
12 0.010 - 0.90 - 0.04 7.2 D20
(pD)
13 0.010 - 0.90 - 0.04 6.8 H20
(control)
[00352] The
values are % w/v. The formulations were prepared at 100 mL scale in volumetric
glassware and filled into LDPE eye droppers. In some instances, the pD is
calculated as pD = 0.4+pH*,
in which pH* is the measured or observed pH of the solution formulated in a
solution containing
deuterated water.
[00353] Table 23B illustrates analysis time points for the formulations
listed in Table 23A.
Table 23B. Schedule for atropine sulfate formulation testing
Storage Time Point
Condition Initial
2 Week 4 Week
(Horizontal) (t=0)
25 C/60%RH X X
40 C/75%RH X X X
60 C X X
[00354] Table 24A and Table 24B illustrate atropine sulfate purity data
associated with the
atropine sulfate formulations. Purity is indicated as percentage of area under
the curve. The I & ,I,
indicate the high or low concentration of atropine sulfate monohydrate (0.01%
and 0.025%). The A & C
indicate the buffer species used, acetic acid and citric acid respectively.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
87
Table 24A. Atropine Sulfate Purity as Area-% for H20 Formulations
Solvent Condition t=0 t=2 weeks t=4 weeks
Formulation 3 25/60 98.16 98.16 98.45
,I,A H20 pH 4.8 40/75 97.98 97.35
60 C 95.94 94.65
Formulation 5 25/60 98.16 97.92 97.54
IC H20 pH 5.8 40/75 95.88 93.51
60 C 80.94 66.83
Formulation 10 25/60 98.66 96.67 95.81
iC H20 pH 6.4 40/75 91.07 85.27
60 C 59.77 42.87
Formulation 11 25/60 99.47 97.87 96.69
.0 (2x) H20 pH 6.4 40/75 90.97 84.26
60 C 54.96 34.40
Formulation 13 25/60 97.21 95.42 93.24
IC H20 pH 6.8 40/75 83.05 73.00
60 C 43.99 27.50
Table 24B. Atropine Sulfate Purity as Area-% for D20 Formulations
Solvent Condition t=0 t=2 weeks t=4 weeks
Formulation 7 25/60 98.93 99.07 98.39
,[A D20 pD 5.2 40/75 98.51 97.55
60 C 96.70 94.01
Formulation 8 25/60 98.93 98.95 98.51
IC D20 pD 6.2 40/75 98.53 97.44
60 C 95.97 92.72
Formulation 9 25/60 99.29 98.42 98.07
IC D20 pD 6.8 40/75 95.20 93.22
60 C 75.17 65.97
Formulation 12 25/60 98.53 97.17 95.99
IC D20 pD 7.2 40/75 90.75 84.64
60 C 56.78 46.05
[00355] Table 25A and Table 25B illustrate tropic acid formation associated
with the atropine
sulfate formulations. Tropic acid is a degradant of atropine sulfate, and is
indicated as percentage of area
under the curve. LOQ was found to be 0.05% for the RP-HPLC method. The is & 1
indicate the high or
low concentration of atropine sulfate monohydrate (0.01% and 0.025%). The A &
C indicate the buffer
species used, acetic acid and citric acid, respectively.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
88
Table 25A. Tropic Acid as Area-% for H20 Formulations
Solvent Condition t=0 t=2 weeks t=4 weeks
Formulation 3 25/60 <LOQ 0.12 0.05
IA H20 pH 4.8 40/75 0.19 , 0.27
60 C 0.90 1.58
Formulation 5 25/60 <LOQ 0.40 0.71
IC H20 pH 5.8 40/75 2.22 4.35
60 C 16.62 29.13
Formulation 10 25/60 0.74 1.90 3.21
IC H20 pH 6.4 40/75 7.61 13.49
60 C 37.44 54.06
Formulation 11 25/60 0.09 1.31 2.64
.0 (2x) H20 pH 6.4 40/75 7.61 14.68
60 C 42.43 62.23
Formulation 13 25/60 2.21 3.66 6.11
IC H20 pH 6.8 40/75 15.47 25.80
60 C 53.24 69.34
Table 25B. Tropic Acid as Area-% for D20 Formulations
Solvent Condition t=0 t=2 weeks t=4 weeks
Formulation 7 25/60 <LOQ 0.07 0.08
,[A D20 pD 5.2 40/75 0.14 0.24
60 C 0.71 1.07
Formulation 8 25/60 <LOQ 0.11 0.14
,IC D20 pD 6.2 40/75 0.33 0.65
60 C 2.32 4.03
Formulation 9 25/60 0.06 0.55 1.06
,I,C D20 pD 6.8 40/75 3.16 6.29
60 C 21.09 29.25
Formulation 12 25/60 0.42 1.35 2.62
C D20 pD 7.2 40/75 7.27 13.53
60 C 38.58 48.15
[00356] Table 26A and Table 26B illustrate the percentage of potency of
atropine in the
formulations. The I & ,I, indicate the high or low concentration of atropine
sulfate monohydrate (0.01%
and 0.025%). The A & C indicate the buffer species used, acetic acid and
citric acid respectively.
Table 26A. Percentage of potency for H20 Formulations
Solvent Condition t=0 t=2 weeks t=4 weeks
Formulation 3 25/60 106.3 108.0 109.6
JA H20 pH 4.8 40/75 108.1 110.0
60 C 108.0 109.9
Formulation 5 25/60 105.0 105.9 107.1
,I,C H20 pH 5.8 40/75 103.8 103.5
60 C 90.2 77.7
Formulation 10 25/60 101.7 100.0 98.0
IC H20 pH 6.4 40/75 89.4 87.0
60 C 63.7 45.7
CA 02953363 2016-12-21
WO 2015/200361
PCT/US2015/037249
89
Formulation 11 25/60 97.5 96.1 94.3
.0 (2x) H20 pH 6.4 40/75 89.4 82.0
60 C 55.7 35.20
Formulation 13 25/60 99.4 96.9 94.1
IC H20 pH 6.8 40/75 85.0 74.0
60 C 46.4 29.8
Table 26B. Percentage of potency for D20 Formulations
Solvent Condition t=0 t=2 weeks t=4 weeks
Formulation 7 25/60 107.3 111.3 112.9
IA D20 pD 5.2 40/75 111.6 113.5
60 C 111.8 113.5
Formulation 8 25/60 99.0 103.0 105.0
IC D20 pD 6.2 40/75 104.9 104.7
60 C 101.6 103.0
Formulation 9 25/60 101.4 99.9 100.1
IC D20 pD 6.8 40/75 97.4 93.2
60 C 78.7 68.9
Formulation 12 25/60 104.9 103.5 101.6
IC D20 pD 7.2 40/75 96.9 89.1
60 C 62.5 50.9
[00357] Table 27A
and Table 27B illustrate the stability of pH or pD for the atropine sulfate
formulations. The I & I indicate the high or low concentration of atropine
sulfate monohydrate (0.01%
and 0.025%). The A & C indicate the buffer species used, acetic acid and
citric acid respectively.
Table 27A. Stability of pH for H20 Formulations
Solvent Condition t=0 t=2 weeks t=4 weeks
Formulation 3 25/60 4.85 4.44 4.61
IA H20 pH 4.8 40/75 4.41 4.54
60 C 4.32 4.40
Formulation 5 25/60 5.87 5.93 6.03
IC H20 pH 5.8 40/75 5.96 5.96
60 C 5.82 5.78
Formulation 10 25/60 6.43 6.41 6.46
IC H20 pH 6.4 40/75 6.62 6.67
60 C 6.01 5.92
Formulation 11 25/60 6.44 6.47 6.72
IC(2x) H20 pH 6.4 40/75 6.66 6.61
60 C 6.27 6.23
Formulation 13 25/60 6.77 6.91 6.91
IC H20 pH 6.8 40/75 6.65 6.62
60 C 6.30 6.19
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
Table 27B. Stability of pD for D20 Formulations
Solvent Condition t=0 t=2 weeks t=4 weeks
Formulation 7 25/60 5.31 5.10 5.24
JA D20 pD 5.2 40/75 5.08 5.15
60 C 5.00 4.93
Formulation 8 25/60 6.25 5.72 5.88
IC D20 pD 6.2 40/75 5.74 5.78
60 C 5.58 5.50
Formulation 9 25/60 6.76 6.80 6.81
IC D20 pD 6.8 40/75 6.78 6.86
60 C 6.45 6.24
Formulation 12 25/60 7.25 7.18 7.26
[C D20 pD 7.2 40/75 7.14 7.15
60 C 6.52 6.36
Example 12. Determination of shelf life and activation engergy for atropine
sulfate formulations of
Example 11
[00358] Activation energy was calculated for the atropine sulfate
formulations disclosed in
Example 11. Specifically, activation energies were calculated from the total %
of related substances (RS)
at 40 C and 60 C (2 point calculations) and from tropic acid formation at 40 C
and 60 C (2 point
calculations). These values were then averaged. Table 28 illustrates the
activation energy calculation.
Table 29 illustrates estimated shelf-lifes from the 40 C rate of formation of
%RS and tropic acid,
respectively. Figure 10 illustrates estimated shelf lifes for D20 and H20
formulations.
Table 28. Activation Energy
Atropine Total RS Tropic Acid
Formulations
7 14 19
3 16 17
8 20 21
5 14 Poor Corr
6 15 16
Mean 16.3 18.7
Stdev 2.68 1.90
RSD 16% 10%
Poor COM One or more curve had R2 < 0.95
CA 02953363 2016-12-21
WO 2015/200361
PCT/US2015/037249
91
Table 29. Estimated Shelf Life
Estimated Shelf 1ife/mo
Total related substances % Tropic acid %
(limit=8%) (limit=5%)
Formulation 8 C 25 C 8 C 25 C
0.01% w/v Atr
0.01% w/v Acetate
0.9% w/v NaC1 189 26 1427 147
0.01% w/v BAK
pD 5.2 D20 (Formulation 7)
0.01% w/v Atr
0.01% w/v Acetate
0.9% w/v NaC1 211 29 1095 113
0.01% w/v BAK
pH4.8 H20 (Formulation 3)
0.01% w/v Atr
0.04% w/v Citrate
0.9% w/v NaC1 158 22 369.8 38
0.01% w/v BAK
pD 6.2 D20 (Formulation 8)
0.01% w/v Atr
0.04% w/v Citrate
0.9% w/v NaCl 37 5.2 54 5.5
0.01% w/v BAK
pH5.8 1120 (Formulation 5)
0.01% Atr
0.9% w/v NaC1
015.9 H20 13.6 2.6
extemporaneous
preparation
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
92
[00359] Tables 30 illustrate the predicted shelf life at temperatures of 40
C, 30 C, 25 C, and
2-8 C for Formulations 2-8 for total RS and tropic acid, respectively.
Table 30
Stability Prediction RS Tropic Acid
Temperature Temperature
Formulation weeks months weeks months
( C) ( C)
2 40 64.5 16.1 40
30 153.2 38.3 30 - -
25 241.2 60.3 25 - -
2-8 1747.9 437.0 2-8 _ -
3 40 31.1 7.8 40 99.5 24.9
30 73.9 18.5 30 268.3 67.1
25 116.3 29.1 25 451.8 113.0
2-8 842.9 210.7 2-8 4382.0 1095.5
4 40 30.7 7.7 40 42.1 10.5
30 73.0 18.2 30 113.7 28.4
25 114.9 28.7 25 191.5 47.9
2-8 832.6 208.1 2-8 1857.0 464.2
40 5.5 1.4 40 4.9 1.2
30 13.1 3.3 30 13.2 3.3
25 20.6 5.2 25 22.2 5.5
2-8 149.3 37.3 2-8 215.0 53.8
6 40 10.7 2.7 40 8.8 2.2
30 25.5 6.4 30 23.7 5.9
25 40.1 10.0 25 39.8 10.0
2-8 290.5 72.6 2-8 386.5 96.6
7 40 27.9 7.0 40 129.6 32.4
30 66.4 16.6 30 349.6 87.4
25 104.5 26.1 25 588.7 147.2
2-8 757.3 189.3 2-8 5709.4 1427.4
8 40 23.3 5.8 40 33.6 8.4
30 55.3 13.8 30 90.6 22.6
25 87.2 21.8 25 152.5 38.1
2-8 631.6 157.9 2-8 1479.2 369.8
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
93
Example 13 - Effect of pH on Ophthalmic Acceptance in Guinea Pigs
[00360] A cohort of guinea pigs is administered 50 !IL of ophthalmic
formulations having
different pH values described herein. For example, ophthalmic formulations
comprising H20 or
deuterated water (e.g., D20) are administered to the animals. Animal behavior
is recorded at
predetermined time intervals to evaluate the acceptance of the ophthalmic
formulations
Example 14 ¨ In vivo Rabbit Eve Irritation Test
[00361] The exemplary compositions disclosed herein are subjected to rabbit
eye irritation test to
evalaute their safety profile. The test composition are tested for eye
irritation test in New Zealand Rabbits
(see for example Abraham M H, et al., Draize rabbit eye test compatibility
with eye irritation thresholds
in humans: a quantitative structure-activity relationship analysis. Toxicol
Sci. 2003 December;
76(2):384-91. Epub 2003 Sep. 26; see also Gettings SD et al., A comparison of
low volume, Draize and
in vitro eye irritation test data. 111. Suifactant-based formulations. Food
Chem Toxicol. 1998 March;
36(3):209-31). The study involves single ocular administration into the right
eye and the same volume of
its placebo in the left eye of each of the three rabbits. Rabbits are examined
immediately and after
instillation of the compositions for 4, 24, 48 and 72 hours post instillation
to note the signs/symptoms of
eye irritation, if any. The test compositions show no sign of irritancy in
cornea, iris and conjunctivae of
the rabbit eyes.
Example 15 ¨ In vivo Testing of Ophthalmic Aqueous Formulation in Guinea Pigs
[00362] Focus deprivation myopia (FDM) is achieved using a latex shield to
cover one eye. For
defocus-induced myopia, a latex-made facemask was held in place by a rubber-
band around the bead of
animals, leaving both eyes, the nose, mouth and ears freely exposed. A - 4.00
D lens is glued onto a
plastic lens frame. The lens frame is then attached to the facemask around one
eye by a fabric hook-and-
loop fastener after the optical center of the lens was aligned with the pupil
center. The lens is detached
and cleaned on both sides with a water-wetted gauze at least once daily
followed by re-attachment to the
facemask. All the animals are maintained on a cycle of 12-h illumination (500
Lux) and 12-h darkness
during the experimental period
[00363] A cohort of guinea pigs at age of 3 weeks are randomly assigned to
FDM (a facemask
worn monocularly) or defocus-induced myopia (a -4.00 D lens worn monocularly)
and control groups.
The FDM groups were treated with the ophthalmic aqueous formulation, the
ophthalmic carrier (without
the opthalmic agent), or FDM-only. The defocus-induced myopia groups were
treated with the
ophthalmic aqueous formulation, the ophthalmic carrier (without the opthalmic
agent), or defocus-only.
The control groups were treated with the ophthalmic aqueous formulation, the
ophthalmic carrier
(without the opthalmic agent), or no treatment. Ocular biometric parameters
are measured in both eyes
of individual animals before and at 11 days of treatment
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
94
[00364] Biometric parameters (e.g. refraction, corneal curvature, and axial
components of the
eye) are measured by an optometrist, orthoptist, or ophthamologist with help
from an animal care
assistant during the light cycle (daytime) after removal of the faccmask or
lens. The optometrist,
orthoptist, or ophthamologist is masked in regard to the treatment conditions
for each animal.
[00365] Refraction is measured by retinoscopy after the pupil is completely
dilated by topical
administration of 1% cyclopentolate hydrochloride. The results of retinoscopy
are recorded as the mean
value of the horizontal and vertical meridians.
[00366] Corneal curvature is measured with a keratometer modified by
attachment of an +8 D
lens onto the anterior surface of the keratometer. A group of stainless steel
balls with diameters from 5.5
to 11.0 mm are measured by the modified keratometer. Three readings are
recorded for each
measurement to provide a mean result. The radius of corneal curvature is then
deduced from the readings
on the balls with known radii.
[00367] A-scan ultrasonagraph is used to measure axial components of the
eye (lens thickness
and vitreous length and axial length). The conducting velocity was 1,723.3 m/s
for measurement of the
lens thickness and 1,540 m/s for measurement of the vitreous length as
described previously. Each of the
axial components is calculated as the mean of 10 repeated measurements.
Example 16 ¨ Safety and Efficacy Studies of Ophthalmic Aqueous Formulation
[00368] A clinical trial is performed to investigate the efficacy and
safety of ophthalmic aqueous
formulations described herein in patents with myopia. In some instances, the
study is open-label, single
blind, or double blind study. Patient selection criteria include myopic
refraction of at least 1.0D in both
eyes, and additional factors such as astigmatism, a documented myopic
progression, age, sex, and/or
health conditions.
[00369] The patients are randomized to receive 0.05%, 0.01%, or 0.001
atropine aqueous
formulation formulated in either FI20 or deuterated water (e.g., D20) once
nightly in both eyes.
Allocation ratio in some instances is defined based the patient population.
[00370] The patients are evaluated on day 0 (baseline), day 14, day 30, and
then at 2, 3, 4, 5, 6, 8,
10, 12, 18, 20, 24, and 36 months. At each visit, best-coorected distance
logMar visual acuity (BCVA) is
assessed by an optometrist, orthoptist, or ophthamologist using the Early
Treatment Diabetic Retinopathy
study chart. Near visual acuity is assessed using best-corrected distance
spectable correction with a
reduced logMar reading chart placed at 40cm under well-lit conditions. The
near point of accommodation
(NPA) is measured using a RAF rule using best-corrected distance spectable
correction. Patients are
instructed to move the target inwards till the N5 print becomes slightly
blurred and then outwards till it
just becomes clear. Accommodation amplitude is calculated as the inverse of
NPA. Mesopic pupil size is
measured with Procyon 3000 pupillometer. Photopic pupil size is measured using
the Neuroptics
pupillometer.
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
[00371] Cycloplegic autorefraction is determined 30 minutes after 3 drops
of cyclopentolate 1%
are administered at 5 minutes apart using a Canon RK-F1 autorefractor. A Zeiss
IOL Master, a non-
contact partial coherence interfcrometry, is used to measure the ocular axial
length.
[00372] The primary outcome is myopia progression over the time period of
the study. Safety is
assessed by adverse events including allergic reactions, irritation, or
development of blurring of vision in
one or both eyes.
Example 17¨ Preparation of an Ointment Formulation Containing Atropine Sulfate
[00373] Atropine sulfate is mixed with the dispersing agent (e.g.
polyethyleneglyeol) under
heating and sonication and this mixture is thither thoroughly mixed with a
molten ointment base (e.g. a
mixture of wool wax, white petrolatum, and liquid paraffin). The mixture is
placed in a pressure vessel,
and sterilized at 125 C for 30-45 minutes and cooled to room temperature. in
another embodiment,
autoclaving is conducted under nitrogen. The resulting ophthalmic ointment is
aseptically filled into pre-
sterilized containers (e.g. tubes).
Example 18 ¨ Atropine-Mucus Penetrating Particle Composition
[00374] A 0.01% atropine-mucus penetrating particle composition was
prepared utilizing a
milling procedure. An aqueous dispersion containing atropine particles and an
MPP-enabling mucus
penetrating agent was milled with grinding medium until particle size was
reduced to approximately
200nm with a polydispersity index less than 0.15 as measured by dynamic light
scattering. Additional
agents such as preservatives are also added during the milling procedure.
Subsequently, the atropine-
MPP composition are be stored at temperatures of between about 15 C and about
25 C.
Example 19 ¨ Atropine Sulfate-Mucus Penetrating Particle Composition
[00375] A 0.01% atropine sulfate-mucus penetrating particle composition was
prepared utilizing
a milling procedure. An aqueous dispersion containing atropine particles and
an MPP-enabling mucus
penetrating agent was milled with grinding medium until particle size was
reduced to approximately
200nm with a polydispersity index less than 0.15 as measured by dynamic light
scattering. Additional
agents such as preservatives are also be added during the milling procedure.
Subsequently, the atropine-
MPP composition are be stored at temperatures of between about 15 C and about
25 C.
[00376] According to another aspect of the disclosure, described herein is
an ophthalmic
composition that comprises from about 0.001 wt% to about 0.05 wt% of a
muscarinic antagonist and
water, at a pH of from about 3.8 to about 7.5.
[00377] In some instances, the muscarinic antagonist comprises atropine,
atropine sulfate,
noratropine, atropine-N-oxide, tropine, tropic acid, hyoscine, scopolomine,
tropicamide, cyclopentolate,
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
96
pirenzapine, homatropine, or a combination thereof In some cases, the
muscarinic antagonist is atropine.
In some cases, the muscarinic antagonist is atropine sulfate.
[00378] In some instances, the ophthalmic composition comprises one of: at
least about 80%, at
least about 85%, at least about 90%, at least about 93%, at least about 95%,
at least about 97%, at least
about 98%, or at least about 99% of the muscarinic antagonist based on initial
concentration after
extended period of time under storage condition.
[00379] In some instances, the ophthalmic composition has a pH of one of:
less than about 7.3,
less than about 7.2, less than about 7.1, less than about 7, less than about
6.8, less than about 6.5, less
than about 6.4, less than about 6.3, less than about 6.2, less than about 6.1,
less than about 6, less than
about 5.9, less than about 5.8, less than about 5.2, less than about 4.8, or
less than about 4.2 after
extended period of time under storage condition.
[00380] In some instances, the ophthalmic composition further has a potency
of one of: at least
80%, at least 85%, at least 90%, at least 93%, at least 95%, at least 97%, at
least 98%, or at least 99%
after extended period of time under storage condition.
[00381] In some instances, the extended period of time is one of: about 1
week, about 2 weeks,
about 3 weeks, about 1 month, about 2 months, about 3 months, about 4 months,
about 5 months, about 6
months, about 8 months, about 10 months, about 12 months, about 18 months,
about 24 months, about 36
months, about 4 years, or about 5 years.
[00382] In some instances, the storage condition has a storage temperature
of one of: about 25 C,
about 40 C, or about 60 C. In some cases, the storage condition has a storage
temperature of from about
2 C to about 10 C or from about 16 C to about 26 C. In some cases, the storage
condition has a relative
humidity of about 60% or about 75%.
[00383] In some instances, the ophthalmic composition is in the form of an
aqueous solution. In
some cases, the muscarinic antagonist is present in the composition at a
concentration of one of: from
about 0.001 wt% to about 0.04 wt%, from about 0.001 wt% to about 0.03 wt%,
from about 0.001 wt% to
about 0.025 wt%, from about 0.001 wt% to about 0.02 wt%, from about 0.001 wt%
to about 0.01 wt%,
from about 0.001 wt% to about 0.008 wt%, or from about 0.001 wt% to about
0.005 wt%.
[00384] In some instances, the ophthalmic composition further comprises an
osmolarity adjusting
agent. In some cases, the osmolarity adjusting agent is sodium chloride.
[00385] In some instances, the ophthalmic composition further comprises a
preservative. In some
cases, the preservative is selected from benzalkonium chloride, cetrimonium,
sodium perborate,
stabilized oxychloro complex, SofZia, polyquaternium-1, chlorobutanol, edetate
disodium,
polyhexamethylene biguanide, or combinations thereof
[00386] In some instances, the ophthalmic composition further comprises a
buffer agent. In some
cases, the buffer agent is selected from borates, borate-polyol complexes,
phosphate buffering agents,
citrate buffering agents, acetate buffering agents, carbonate buffering
agents, organic buffering agents,
amino acid buffering agents, or combinations thereof
CA 02953363 2016-12-21
WO 2015/200361 PCT/US2015/037249
97
[00387] In some instances, the ophthalmic composition further comprises a
tonicity adjusting
agent. In some cases, the tonicity adjusting agent is selected from sodium
chloride, sodium nitrate,
sodium sulfate, sodium bisulfate, potassium chloride, calcium chloride,
magnesium chloride, zinc
chloride, potassium acetate, sodium acetate, sodium bicarbonate, sodium
carbonate, sodium thiosulfate,
magnesium sulfate, disodium hydrogen phosphate, sodium dihydrogen phosphate,
potassium dihydrogen
phosphate, dextrose, mannitol, sorbitol, dextrose, sucrose, urea, propylene
glycol, glycerin, or a
combination thereof.
[00388] In some instances, the ophthalmic composition is stored in a
plastic container. In some
cases, the material of the plastic container comprises low-density
polyethylene (LDPE).
[00389] In some instances, the ophthalmic composition has a dose-to-dose
muscarinic antagonist
concentration variation of one of: less than 50%, less than 40%, less than
30%, less than 20%, less than
10%, or less than 5%. In some cases, the dose-to-dose muscarinic antagonist
concentration variation is
based on one of: 10 consecutive doses, 8 consecutive doses, 5 consecutive
doses, 3 consecutive doses, or
2 consecutive doses.
[00390] In some instances, the ophthalmic composition has a pH of one of:
from about 3.8 to
about 7.5, from about 4.2 to about 7.5, from about 4.8 to about 7.3, from
about 5.2 to about 7.2, from
about 5.8 to about 7.1, from about 6.0 to about 7.0, or from about 6.2 to
about 6.8.
[00391] In some instances, the ophthalmic composition further comprises a
pH adjusting agent.
In some cases, the pH adjusting agent comprises HC1, NaOH, CH3COOH, or C6H807.
[00392] In some instances, the ophthalmic composition comprises one of:
less than 5% of D90,
less than 4% of D70, less than 3% of D70, less than 2% of D20, less than 1% of
D20, less than 0.5% of
D20, less than 0.1% of D,O, or 0% D20. In some cases, the ophthalmic
composition is essentially free of
D20.
[00393] In some instances, the ophthalmic composition further comprises a
pharmaceutically
acceptable carrier.
[00394] In some instances, the ophthalmic composition is formulated as an
ophthalmic solution
for the treatment of an ophthalmic disorder. In some cases, the ophthalmic
disorder or condition is pre-
myopia, myopia, or progression of myopia.
[00395] In some instances, the ophthalmic composition is not formulated as
an injectable
formulation.
[00396] While preferred embodiments of the present disclosure have been
shown and described
herein, such embodiments are provided by way of example only. Various
alternatives to the embodiments
described herein are optionally employed in practicing the disclosure. It is
intended that the following
claims define the scope of the disclosure and that methods and structures
within the scope of these claims
and their equivalents be covered thereby.