Note: Descriptions are shown in the official language in which they were submitted.
DEMANDE OU BREVET VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVET COMPREND
PLUS D'UN TOME.
CECI EST LE TOME 1 DE 2
CONTENANT LES PAGES 1 A 245
NOTE : Pour les tomes additionels, veuillez contacter le Bureau canadien des
brevets
JUMBO APPLICATIONS/PATENTS
THIS SECTION OF THE APPLICATION/PATENT CONTAINS MORE THAN ONE
VOLUME
THIS IS VOLUME 1 OF 2
CONTAINING PAGES 1 TO 245
NOTE: For additional volumes, please contact the Canadian Patent Office
NOM DU FICHIER / FILE NAME:
NOTE POUR LE TOME / VOLUME NOTE:
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 1 -
FUSED PENTACYCLIC IMIDAZOLE DERIVATIVES
The present invention relates to classes of fused pentacyclic imidazole
derivatives,
and to their use in therapy. More particularly, this invention is concerned
with
pharmacologically active substituted fused pentacyclic benzimidazo le
derivatives and
analogs thereof In particular, the present invention is concerned with 6,7-
dihydro-7,14-
methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one derivatives and analogs
thereof
These compounds are modulators of the signalling of TNFa, and are accordingly
of
benefit as pharmaceutical agents, especially in the treatment of adverse
inflammatory and
autoimmune disorders, neurological and neurodegenerative disorders, pain and
nociceptive
disorders, cardiovascular disorders, metabolic disorders, ocular disorders,
and onco logical
disorders.
TNFa is the prototypical member of the Tumour Necrosis Factor (TNF)
superfamily of proteins that share a primary function of regulating cell
survival and cell
death. One structural feature common to all known members of the TNF
superfamily is
the formation of trimeric complexes that bind to, and activate, specific TNF
superfamily
receptors. By way of example, TNFa exists in soluble and transmembrane forms
and
signals through two receptors, known as TNFR1 and TNFR2, with distinct
functional
endpoints.
Various products capable of modulating 'TNFa activity are already commercially
available. All are approved for the treatment of inflammatory and autoirnrnune
disorders
such as rheumatoid arthritis and Crohn's disease. All currently approved
products are
macrornolecular and act by inhibiting the binding of human TNFa to its
receptor. Typical
macromolecular TNFa inhibitors include anti-'TNFa antibodies; and soluble TNFa
receptor fusion proteins. Examples of commercially available anti-TNFa
antibodies
include fully human antibodies such as adalimumab (Hum ira*) and golimumab
(Simponio ), chimeric antibodies such as infliximab (Remicade0), and pegylated
Fab'
fragments such as certolizumab pegol (Cimzia0). An example of a commercially
available soluble TNFa receptor fusion protein is etanercept (Enbrelt).
TNF superfamily members, including TNFa itself, are implicated in a variety of
physiological and pathological functions that arc believed to play a part in a
range of
conditions of significant medical importance (see, for example, M.G. Tansey &
D.E.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 2 -
Szymkowski, Drug Discovery Today, 2009, 14, 1082-1088; and F.S. Carneiro et
al., J.
Sexual Medicine, 2010, 7, 3823-3834).
The compounds in accordance with the present invention, being potent
modulators
of human TNFa activity, are therefore beneficial in the treatment and/or
prevention of
various human ailments. These include autoimmune and inflammatory disorders;
neurological and neurodegenerative disorders; pain and nociceptive disorders;
cardiovascular disorders; metabolic disorders; ocular disorders; and
oncological disorders.
In addition, the compounds in accordance with the present invention may be
beneficial as pharmacological standards for use in the development of new
biological tests
and in the search for new pharmacological agents. Thus, in one embodiment, the
compounds of this invention may be useful as radioligands in assays for
detecting
pharmacologically active compounds. In an alternative embodiment, certain
compounds
of this invention may be useful for coupling to a fluorophore to provide
fluorescent
conjugates that can be utilised in assays (e.g. a fluorescence polarisation
assay) for
detecting pharmacologically active compounds.
International patent applications W02013/186229A1, W02014/009295A1 and
W02014/009296A1 relate to fused imidazole derivatives which are modulators of
the
signalling of TNFa.
International patent applications W02015/086525 and W02015/086526 published
June 18th 2015 relate to fused tricyclic imidazole derivatives which are
modulators of the
signalling of TNFa.
None of the prior art available to date, however, discloses or suggests the
precise
structural class of fused pentacyclic imidazole derivatives as provided by the
present
invention.
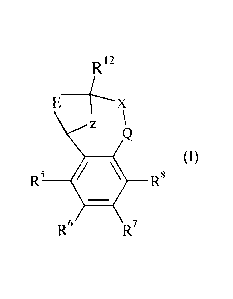
The present invention provides a compound of formula (I) or an N-oxide
thereof,
or a pharmaceutically acceptable salt thereof:
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 3 -
R12
EX
7 \
(1)
R5 R8
R7
wherein
-X-Q- represents -0-, -0-C(0)-, -C(0)-0-, -0-C(CH-CN)-, -S-, -SO-, -SO2-,
-N(R)-CO-, -CO-N(Rf)-, -N(R)-SO2-, -S02-N(10-, -S(0)(NRf)-, -CH2-CH2-, -0-
CH2-, -CH2-0-, -S-CH2-, -SO-CH2-, -502-CH2-, CH2-S-, -CH2-50-, -CH2-S02-, -
N(R8)-
CH2-, -CH2-N(Rg)-, -S(0)(NRf)-CH2-, -CH2-S(0)(NRf)- ,-N(Rf)-C(S)-, -
N=S(0)(CH3)- -
0-C(=CH2)- or -S(=N-CN)-, any of which groups may be optionally substituted by
one or
more substituents.
Z represents methylene;
E represents a fused heteroaromatic ring system selected from the groups of
formula (Ea), (Eb) and (Ec),
R3 R3
R2RN NN
*
R1
R4
R4
R4
(Ea) (Eb) (Ec)
wherein the asterisk (*) represents the site of attachment of E to the
remainder of the molecule;
Rd represents hydrogen, halogen, cyano, trifluoromethyl, trifluoromethoxy,
-SR', -SOW, -SO2Ra, -NRbRc, -NWCORd, -NR`CO2Rd, -NHCONRbRc, -NR`SO2Re,
-CORd, -CO2Rd, -CONRbRc , -SO2NRbRc , or -S(0)(N-Rb)Re; or C1-6 alkyl, C3-7
cycloalkyl, C4_7 cycloalkenyl, aryl, aryl(C1_6)alkyl, C3_7 heterocycloallcyl,
C3-7
heterocycloalkenyl, heteroaryl, heteroaryl(C1_6)alkyl,
(C3_7)heterocycloa1kyl(C1_6)a1ky1-
aryl-, (C3_7)cycloalkyl-heteroaryl-, (C3_7)cycloalkyl(C1_6)alkyl-heteroaryl-,
(C4-
7)cycloalkenyl-heteroaryl-, (C4_9)bicycloalkyl-heteroaryl-,
(C3_7)heterocycloalkyl-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 4 -
heteroaryl-, (C3_7)heterocycloalkyl(Ci_6)a1kyl-heteroary1-,
(C3_7)heterocycloalkenyl-
heteroaryl-, (C4_9)heterobicycloalkyl-heteroaryl- or
(C4_9)spiroheterocycloalky1-
heteroaryl-, any of which groups may be optionally substituted by one or more
substituents; or R1 represents (C3_7)heterocycloalkenyl-aryl-, which group may
be
optionally substituted by one or more substituents;
R2 represents hydrogen, halogen, cyano, nitro, hydroxy, trifluoromethyl,
ttifluoromethoxy or -0Ra ; or C1_6 alkyl optionally substituted by one or more
substituents;
R3 and R4 independently represent hydrogen,halogen or trifluoromethyl; or C1-6
alkyl, optionally substituted by one or more substituents;
R5 and R8 independently represent hydrogen, halogen, hydroxy, cyano,
trifluoromethyl, difluoromethoxy, trifluoromethoxy, -OR', or C1_6
alkylsulphonyl; or C1-6
alkyl optionally substituted by one or more substituents;
R6 and R7 independently represent hydrogen, halogen, trifluoromethyl, C1_6
alkyl
or Ci_6 alkoxy;
R'2 represents hydrogen or C1-6 alkyl;
IV represents Ci_6 alkyl, C3_7 cycloalkyl, C3-7 heterocycloalkyl, aryl,
aryl(Ci-
6)alkyl, heteroaryl or heteroaryl(Ci_6)alkyl, any of which groups may be
optionally
substituted by one or more substituents;
Rb and Re independently represent hydrogen or trifluoromethyl; or C1_6 alkyl,
C3-7
cycloalkyl, C3_7 cycloalkyl(C16)alkyl, aryl, aryl(Ci4alkyl, C3_7
heterocycloalkyl, C3-7
heterocycloalkyl(C1_6)alkyl, heteroaryl or heteroaryl(C1-6)alkyl, any of which
groups may
be optionally substituted by one or more substituents; or
Rh and Re, when taken together with the nitrogen atom to which they are both
attached, represent a heterocyclic moiety selected from azetidin-l-yl,
pyrrolidin-l-yl,
oxazolidin-3-yl, thiazolidin-3-yl, isothiazolidin-2-yl,
piperidin- 1 -yl,
morpholin-4-yl, thiomorpholin-4-yl, piperazin-l-yl, homopiperidin-l-yl,
homomorpholin-
4-yl, homopiperazin-l-yl, (imino)(oxo)thiazinan-4-yl, (oxo)thiazinan-4-y1 and
(dioxo)thiazinan-4-yl, any of which groups may be optionally substituted by
one or more
substituents;
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 5 -
Rd represents hydrogen; or Ci_6 alkyl, C3:7 cycloalkyl, aryl, C3:7
heterocycloalkyl
or heteroaryl, any of which groups may be optionally substituted by one or
more
substituents;
Re represents C1-6 alkyl, aryl or heteroaryl, any of which groups may be
optionally
substituted by one or more substituents;
Rf represents hydrogen; or C1 6 alkyl, C3:7 cycloalkyl, or C3_7
heterocycloalkyl, any
of which groups may be optionally substituted by one or more substituents; and
Rg represents hydrogen; or C1-6 alkyl, C3_7 cycloalkyl, C3-7 heterocycloalkyl,
-CO-
(C16)alkyl, or -S02-(C1_6)alkyl, any of which groups may be optionally
substituted by one
or morc substituents; or Rg represents -CO-(C37)heterocycloalkyl, -S02-
(C3_7)cycloalkyl,
-S02-(C3_7)hetcrocycloalkyl, -S02-aryl or -S02-heteroaryl, any of which groups
may be
optionally substituted by one or more substituents; or Rg represents
heteroaryl or (C2_
6)alkoxycarbonyl, either of which groups may be optionally substituted by one
or more
substituents.
The present invention also provides a compound of formula (I) as defined above
or an N-oxide thereof, or a pharmaceutically acceptable salt thereof, for use
in therapy.
In another aspect, the present invention also provides a compound of formula
(I)
as defined above or an N-oxide thereof, or a pharmaceutically acceptable salt
thereof, for
use in the treatment and/or prevention of disorders for which the
administration of a
modulator of TNFa function is indicated.
In another aspect, the present invention provides a compound of formula (1) as
defined above or an N-oxide thereof, or a pharmaceutically acceptable salt
thereof for use
in the treatment and/or prevention of an inflammatory or autoimmune disorder,
a
neurological or neurodegenerative disorder, pain or a nociceptive disorder, a
cardiovascular disorder, a metabolic disorder, an ocular disorder, or an
oneological
disorder.
In antoher aspect, the present invention provides for the use of a compound of
formula (1) as defined above, or an N-oxide thereof, or a pharmaceutically
acceptable salt
thereof, for the manufacture of a medicament useful for the treatment and/or
prevention
of disorders for which the administration of a modulator of TNFa function is
indicated.
- 6 -
In another aspect, the present invention provides for the use of a compound of
formula (I)
as defined above, or an N-oxide thereof, or a pharmaceutically acceptable salt
thereof, for the
manufacture of a medicament useful for the treatment of an inflammatory or
autoimmune
disorder, a neurological or neurodegenerative disorder, pain or a nociceptive
disorder, a
cardiovascular disorder, a metabolic disorder, an ocular disorder, or an
oncologi cal disorder.
The present invention also provides a method for the treatment and/or
prevention of
disorders for which the administration of a modulator of TNFa function is
indicated which
comprises administering to a patient in need of such treatment an effective
amount of a compound
of formula (I) as defined above, or an N-oxide thereof, or a pharmaceutically
acceptable salt
thereof.
In another aspect, the present invention provides a method for the treatment
and/or
prevention of an inflammatory or autoimmune disorder, a neurological or neuro-
degenerative
disorder, pain or a nociceptive disorder, a cardiovascular disorder, a
metabolic disorder, an ocular
disorder, or an oncological disorder, which comprises administering to a
patient in need of such
treatment an effective amount of a compound of formula (I) as defined above or
an N-oxide
thereof, or a pharmaceutically acceptable salt thereof.
In one aspect, the present invention provides a compound of formula (I) or an
N-oxide
thereof, or a pharmaceutically acceptable salt thereof,
R12
X
z
(I)
R5 R8
R6 R7
wherein
-X-Q- represents -0-, -0-C(0)-, -0-C(=CH-CN)-, -S-, -SO-, -S02-; or is
selected from the group
consisting of -N(R), -N(R)-CO-, -N(R)-SO2-, -0-CH2-, -CH2-S-, -CH2-S0-, -CH2-
S02-,
-N(Rg)-CH2-, -N(Rf)-C(S)-, -N=S(0)(CH3)-, -0-C(=CH2)- and -S(=N-CN)-, any of
which groups
may be optionally substituted by one or more substituents selected from
halogen, (C1-6)alkyl,
carboxy, tri fluorom ethyl, (C2_6)alkylcarbonyl,
(C2_6)alkoxycarbonyl, and
Date Recue/Date Received 2022-02-28
- 6a -
hydroxy(Ci_6)alkyl;
Z represents methylene;
E represents a fused heteroaromatic ring system selected from the groups of
formula (Ea)
and (Eb),
R3 R3
2
R2 R
RI
R4 R4
(Ea) (Eb)
wherein the asterisk (*) represents the site of attachment of E to the
remainder of the molecule;
R1 represents halogen or cyano; or is selected from the group consisting of
aryl, heteroaryl, (C3_
7)cy cl oalkyl -heteroaryl, (C3_7)heterocy cl oalkyl-hetero aryl,
(C4_9)heterobicycloalkyl-heteroaryl,
(C3_7)heterocycloalkyl, (C3_7)heterocycloalkenyl, and (C3_7)heterocycloalkenyl-
aryl, any of which
groups may be optionally substituted by one, two or three substituents
selected from halogen,
cyano, cyano(C1-6)alkyl, C1-6 alkyl, difluoromethyl, trifluoromethyl, hydroxy,
(hydroxy)(C1-
6)alkyl, amino, (amino)(C1_6) alkyl, C1_6 alkoxy, (C1_6) alkoxy(C1_6)alkyl, C2-
6 alkylcarbonyl, C2_6
alkoxycarbonyl, (C2_6) alkoxycarbonyl-amino-C1_6 alkyl, phosphate(C1_6)alkyl,
C1_6 alkylthio, Ci_6
alkylsulphonyl, oxo, (Ci-6)alkylsulphoximinyl, (C1-6)alkylsulphinyl-amino-,
di(Ci_6)alkylamino
(C i_6)alkyl, (C24alkylcarbonylamino(C 1_6)alkyl, di(C
i_o)alkenylamino (C 1_6)alkyl, (C2-
6)alkylcarbonylamino(C1-6)alkyl, C1-6 alkylsulphonyl-amino-Cis alkyl,
tetrahydrofuranyl,
sulphate (C1_6)alkyl, and carboxy-(C1_6)alkyl-carbonyloxy-(Ci_6)alkyl;
R2 represents hydrogen or halogen;
R3 represents hydrogen or trifluoromethyl;
R4 represents hydrogen or trifluoromethyl;
R5 represents halogen, -OR", difluoromethoxy or tifluoromethoxy;
R6 represents hydrogen, halogen or trifluoromethyl;
R7 represents hydrogen or trifluoromethyl;
R8 represents hydrogen, halogen or trifluoromethyl;
R12 represents hydrogen or C1_6 alkyl;
Ra represents Ci_6 alkyl;
Date Recue/Date Received 2022-02-28
- 6b -
Rf represents hydrogen; or C 1-6 alkyl, which group may be optionally
substituted by one or more
substituents selected from trifluoromethyl, carboxy and hydroxy; and
Rg represents hydrogen; or is selected from the group consisting of C1_6
alkyl, -00-(C1_6)alkyl,
-S02-(C 1_6)alkyl, -00-(C3_7)heterocycloalkyl, -S02-(C3_7)cycloalkyl, -S02-
aryl, -S02-heteroaryl,
heteroaryl and (C2-6)alkoxycarbonyl, any of which groups may be optionally
substituted by one or
more substituents selected from halogen, C1-6 alkyl, carboxy, C1_6
alkoxycarbonyl,
trifluoromethyl, C4-9 heterobicycloalkyl, (C1_6 alkyl)sulphonyl, tri(C1_6
alkyl)silyloxy, hydroxy and
(C1-6)alkoxy.
In another aspect, the present invention provides a compound of formula (IIB)
or an N-
oxide thereof, or a pharmaceutically acceptable salt thereof,
R3
R2
Rf
Ri
0
R4
R5
Rs
(IIB)
R6 R7
wherein
RI, R2, R3, R4, R5, R6, R7, R8, and Rf are as defined above.
In another aspect, the present invention provides the above compound or an N-
oxide
thereof, or a pharmaceutically acceptable salt thereof, for use in the
treatment or prevention of a
disorder selected from the group consisting of: an inflammatory disorder, an
autoimmune
disorder, a neurological disorder, a neurodegenerative disorder, a pain
disorder, a nociceptive
disorder, a cardiovascular disorder, a metabolic disorder, an ocular disorder,
and an oncological
disorder.
In another aspect, the present invention provides a use of the above compound
or an N-
oxide thereof, or a pharmaceutically acceptable salt thereof, for the
treatment or prevention of a
disorder selected from the group consisting of: an inflammatory disorder, an
autoimmune
disorder, a neurological disorder, a neurodegenerative disorder, a pain
disorder, or a nociceptive
Date Regue/Date Received 2022-07-11
- 6c -
disorder, a cardiovascular disorder, a metabolic disorder, an ocular disorder,
and an oncological
disorder.
In another aspect, the present invention provides a use of the above compound
or an N-
oxide thereof, or a pharmaceutically acceptable salt thereof, for the
preparation of a medicament
for the treatment or prevention of a disorder selected from the group
consisting of: an
inflammatory disorder, an autoimmune disorder, a neurological disorder, a
neurodegenerative
disorder, a pain disorder, a nociceptive disorder, a cardiovascular disorder,
a metabolic disorder,
an ocular disorder, and an oncological disorder.
In another aspect, the present invention provides a pharmaceutical composition
comprising the above compound or an N-oxide thereof, or a pharmaceutically
acceptable salt
thereof, and a pharmaceutically acceptable carrier.
Where any of the groups in the compounds of formula (I) above is stated to be
optionally
substituted, this group may be unsubstituted, or substituted by one or more
substituents.
Typically, such groups will be unsubstituted, or substituted by one or two
substituents. Suitable
substitutents for each particular groups of compounds formula (I) are further
described here after
in the present specification.
The present invention includes within its scope salts of the compounds of
formula (I)
above. For use in medicine, the salts of the compounds of formula (I) will be
pharmaceutically
acceptable salts. Other salts may, however, be useful in the preparation of
the compounds of use
in the invention or of their pharmaceutically acceptable salts. Standard
principles underlying the
selection and preparation of pharmaceutically acceptable salts are described,
for example, in
Handbook of Pharmaceutical Salts: Properties, Selection and Use, ed. P.H.
Stahl & C.G.
Wermuth, Wiley-VCH, 2002.
The present invention includes within its scope solvates of the compounds of
formula (I)
above. Such solvates may be formed with common organic solvents or water.
Date Recue/Date Received 2022-07-11
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 7 -
The present invention also includes within its scope co-crystals of the
compounds
of formula (1) above. The technical term "co-crystal" is used to describe the
situation
where neutral molecular components are present within a crystalline compound
in a
definite stoichiometric ratio. The preparation of pharmaceutical co-crystals
enables
.. modifications to be made to the crystalline form of an active
pharmaceutical ingredient,
which in turn can alter its physicochemical properties without compromising
its intended
biological activity (see Pharmaceutical Salts and Co-crystals, ed. J. Wouters
& L. Quere,
RSC Publishing, 2012).
The present invention includes within its scope N-oxides of compounds of
formula
(1) above. Particular examples of N-oxides according to the present invention
include
pyrimidine N-oxide and pyridine N-oxide as illustrated in the Examples.The
term "alkyl"
as used herein refers to aliphatic hydrocarbon groups which may be straight or
branched
and may comprise 1 to 20 carbon atoms in the chain, suitably 1 to 15 carbon
atoms in the
chain, more suitably 1 to 10 carbon atoms in the chain. Suitable alkyl groups
which may
be present on the compounds of use in the invention include straight-chained
and branched
C1_6 alkyl groups, for example C1-4 alkyl groups. Illustrative alkyl goups
include methyl
and ethyl groups, and straight-chained or branched propyl, butyl and pentyl
groups.
Suitable alkyl groups include methyl, ethyl, n-propyl, and isopropyl. Derived
expressions
such as "Ci_6 alkoxy", "C1_6 alkylthio", "Ci_6 alkylsulphonyl" and "Ci_6
alkylamino" are to
be construed accordingly.
The term "C3_7 cycloalkyl" as used herein refers to monovalent groups of 3 to
7
carbon atoms derived from a saturated monocyclic hydrocarbon. Suitable C3-7
cycloalkyl
groups may comprise benzo-fused analogues thereof. Illustrative C3-7
cycloalkyl groups
include cyclopropyl, cyclobutyl, benzocyclobutenyl, cyclopentyl, indanyl,
cyclohexyl and
cycloheptyl.
The term "C4_9 bicycloalkyl" as used herein refers to monovalent groups of 4
to 9
carbon atoms derived from a saturated bicyclic hydrocarbon. The term "C4_7
cycloalkenyl"
as used herein refers to monovalent groups of 4 to 7 carbon atoms derived from
a partially
unsaturated monocyclic hydrocarbon. Illustrative C4_7cycloa1kenyl groups
include
cyclobutenyl, cyclopentenyl, cyclohexenyl and cycloheptenyl.
CA 02962826 2017-03-28
WO 2016/050975
PCT/EP2015/072868
- 8 -
The term "aryl" as used herein, refers to an unsaturated aromatic carbocyclic
group
of from 6 to 14 carbon atoms having a single ring (e.g. phenyl) or multiple
condensed
rings (e.g. naphthyl). Illustrative aryl groups include phenyl.
Illustrative aryl(Ci_6)alkyl groups include benzyl, phenylethyl and
phenylpropyl.
The telin "C3.7 heterocycloalkyl" as used herein refers to saturated
monocyclic
rings containing 3 to 7 carbon atoms and at least one heteroatom selected from
oxygen,
sulphur and nitrogen, and may comprise benzo-fused analogues thereof
Illustrative
heterocycloalkyl groups include oxetanyl, azetidinyl, tetrahydrofuranyl,
dihydrobenzo-
furanyl, dihydrobenzothienyl, pyrrolidinyl, indolinyl, dihydroisoindolinyl,
isoindolinyl,
oxazolidinyl, thiazolidinyl, isothiazolidinyl, imidazolidinyl,
tetrahydropyranyl, chromanyl,
tetrahydro-thiopyranyl, piperidinyl, 1,2,3,4-tetrahydroquinolinyl, 1,2,3,4-
tetrahydroisoquinolinyl, piperazinyl, 1,2,3,4-tetrahydroquinoxalinyl,
hexahydro-
[1,2,5]thiadiazolo[2,3-a]pyrazinyl, homopiperazinyl, morpholinyl,
benzoxazinyl,
thiomorpholinyl, azepanyl, oxazepanyl, diazepanyl, thiadiazepanyl, azocanyl,
(imino)(oxo)thiazinanyl, (oxo)thiazinanyl, (dioxo)thiazinanyl,
tetrahydrothiophenyl,
(oxo)tetrahydrothiophenyl, (dioxo)tetrahydrothiophenyl and
(oxo)thiomorpholinyl.
The term "C3_7 heterocycloalkenyl" as used herein refers to monounsaturated or
polyunsaturated monocyclic rings containing 3 to 7 carbon atoms and at least
one
heteroatom selected from oxygen, sulphur and nitrogen, and may comprise benzo-
fused
analogues thereof. Illustrative heterocycloalkenyl groups include thiazolinyl,
imidazolinyl, dihydropyranyl, dihydrothiopyrany1,1,2,3,6-tetrahydropyridinyl,
1,2-
dihydropyridinyl and 1,2-dihydropyrimidinyl.The term "C4_9 heterobicycloalkyl"
as used
herein refers to a C4-9 bicycloalkyl as defined herein, wherein one or more of
the carbon
atoms have been replaced by one or more heteroatoms selected from oxygen,
sulphur and
nitrogen. Illustrative heterobicycloalkyl groups include 3-
azabicyclo[3.1.0]hexanyl, 2-
oxa-5-azabicyclo[2.2.1]heptanyl, 6-azabicyclo[3.2.0]heptanyl, 3-
azabicyclo[3.1.1]heptanyl, 3-azabicyclo[4.1.0]heptanyl, 2-
oxabicyclo[2.2.2]octanyl,
quinuclidinyl, 2-oxa-5-azabicyclo-[2.2.2]octanyl, 3-azabicyclo[3.2.1]octanyl,
8-
azabicyclo [3.2.1]octanyl, 3-oxa-8-azabicyclo[3.2.1]octanyl, 3,8-
.. diazabicyclo[3.2.1]octanyl, 3,6-diazabicyclo[3.2.2]nonanyl, 3-oxa-7-
azabicyclo[3.3.1]nonanyl and 3,9-diazabicyclo[4.2.1]nonanyl. Illustrative
heterobicycloalky groups additionally include 3,7-dioxa-9-azabicyclo[3.3.1]non-
9-yl.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 9 -
The term "C4_9 spiroheterocycloalkyl" as used herein refers to saturated
bicyclic
ring systems containing 4 to 9 carbon atoms and at least one heteroatom
selected from
oxygen, sulphur and nitrogen, in which the two rings are linked by a common
atom.
Illustrative spiroheterocycloalkyl groups include 5-azaspiro[2.3]hexanyl, 5-
azaspiro-
[2.4]heptanyl, 2-azaspiro[3.3]heptanyl, 2-oxa-6-azaspiro[3.3]heptanyl, 2-oxa-6-
azaspiro-
[3.4]octanyl, 2-oxa-6-azaspiro[3.5]nonanyl, 7-oxa-2-azaspiro[3.5]nonanyl, 2-
oxa-7-
azaspiro[3.5]nonanyl and 2,4,8-triazaspiro[4.5]decanyl.
The term "heteroaryl" as used herein represents aromatic carbocyclic groups of
from 5 to 14 carbon atoms having a single ring or multiple condensed rings,
wherein one
or more of the said carbon atoms have been replaced by one or more heteroatoms
selected
from oxygen, sulphur and nitrogen. Illustrative heteroaryl groups include
furyl,
benzofuryl, dibenzofuryl, thienyl, benzothienyl, thieno[2,3-c]pyrazolyl,
thieno[3,4-
b][1,4]dioxinyl, dibenzothienyl, pyrrolyl, indolyl, 2,3-dihydro-1H-isoindolyl,
pyrrolo[2,3-
b]pyridinyl, pyrrolo[3,2-c]pyridinyl, pyrrolo[3,4-b]pyridinyl, pyrazolyl,
pyrazolo[1,5-
a] pyridinyl, pyrazolo[3,4-cl]pyrimidinyl, indazolyl, 4,5,6,7-
tetrahydroindazolyl, oxazolyl,
benzoxazolyl, isoxazolyl, thiazolyl, benzothiazolyl, isothiazolyl, imidazolyl,
benzimidazolyl, imidazo[2,1-b]thiazolyl, imidazo[1,2-alpyridinyl, imidazo[4,5-
b]pyridinyl, purinyl, imidazo[1,2-a]pyrimidinyl, imidazo[1,2-a]pyrazinyl,
oxadiazolyl,
thiadiazolyl, triazolyl, [1,2,4]triazolo[1,5-a]pyrimidinyl, benzotriazolyl,
tetrazolyl,
pyridinyl, quinolinyl, isoquinolinyl, naphthyridinyl, pyridazinyl, cinnolinyl,
phthalazinyl,
pyrimidinyl, quinazolinyl, pyrazinyl, quinoxalinyl, pteridinyl, triazinyl and
chromenyl
groups.
The term "halogen" as used herein is intended to include fluorine, chlorine,
bromine and iodine atoms, typically fluorine, chlorine or bromine atoms.
Where the compounds of formula (1) have one or more asymmetric centres, they
may accordingly exist as enantiomers. Where the compounds of use in the
invention
possess two or more asymmetric centres, they may additionally exist as
diastereorners.
The invention is to be understood to extend to the use of all such enantiomers
and
diastereomers, and to mixtures thereof in any proportion, including racemates.
Formula (I)
and the formulae depicted hereinafter are intended to represent all individual
stereoisomers
and all possible mixtures thereof, unless stated or shown otherwise. In
addition,
compounds of formula (1) may exist as tautomers, for example keto
(CH2C=0)4¨enol
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 10 -
(CH=CHOH) tautomers or amide (NHC=0)4-*hydroxyimine (N=COH) tautomcrs.
Formula (1) and the formulae depicted hereinafter are intended to represent
all individual
tautomers and all possible mixtures thereof, unless stated or shown otherwise.
An illustrative example of a tautomer in accordance with the present
invention, is
2-oxo-(1H)-pyridinyl which is a tautomer of 2-hydroxy-pyridinyl.
Another illustrative example of a tautomer in accordance with the present
invention, is 2-oxo-(1H)-pyrimidinyl which is a tautomer of 2-hydroxy-
pyrimidinyl.
A particular sub-class of compounds in accordance with the present invention
is
the sub-class of compounds of formula (IA), or an N-oxide thereof, or a
pharmaceutically
acceptable salt thereof,
R12
\z
, Q
(1A)
Rs
R8
R6
R7
wherein E, Z, R5, R6, R7 R8 and R12 are as defined above.
It is to be understood that each individual atom present in formula (I), or in
the
formulae depicted hereinafter, may in fact be present in the form of any of
its naturally
occurring isotopes, with the most abundant isotope(s) being preferred. Thus,
by way of
example, each individual hydrogen atom present in formula (I), or in the
formulae depicted
hereinafter, may be present as a 'H, 2H (deuterium) or 3H (tritium) atom,
preferably 'H.
Similarly, by way of example, each individual carbon atom present in formula
(I), or in the
formulae depicted hereinafter, may be present as a 12C, 13C or 14C atom,
preferably 12C.
Generally, -X-Q- represents -0-, -0-C(0)-, -C(0)-0-, -0-C(CH-CN)-, -S-, -SO-,
-N(Rg)-, -N(Rf)-00-, -N(R1')-S02-, -S02-N(Rf)-, -S(0)(NRf)-, -CH2-
CH2-, -0-CH2-, -CH2-0-, -S-CH2-, -S02-CH2-, -CH2-
S02-,
-N(12,8)-CH2-, -CH2-N(R8)-, -S(0)(NR-f)-CH2- , -CH2-S(0)(Nle)- , -N(Rf)-C(S)-,
-
N=S(0)(CH3)_, -0-C(=CH2)- or -S(=N-CN)-, any of which groups may be
substituted by
one or more substituents.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 11 -
More generally, -X-Q- represents -0-, -0-C(0)-, -C(0)-0-, -0-C(CH-CN)-, -S-, -
SO-, -SO2-, -N(Rg)-, -N(R)-CO-, -CO-N(10-, -N(Rf)-S02-, -SO2-N(R)-, -S(0)(NI0-
, -
CH2-CH2-, -0-CH2-, -CH2-0-, -S-CH2-, -SO-CH2-, -S02-CH2-, CH2-S-, -CH2-S0-, -
CH2-
SO2-, -N(Rg)-CH2-, -CH2-N(Rg)-, -S(0)(N10-CH2- , -CH2-S(0)(N10- or -N(Rf)-C(S)-
.
Typically, -X-Q- represents -0-, -0-C(0)-, -C(0)-0-, -0-C(CH-CN)-, -S-, -SO-, -
SO2-, -N(Rg)-, -N(Rf)-00-, -CO-N(10-, -N(R)-SO2-, -SO2-N(R)-, -S(0)(NRf)-, -
CH2-
CH2-, -0-CH2-, -CH2-0-, -S-CH2-, -SO-CH2-, -S02-CH2-, CH2-S-, -CH2-S0-, -CH2-
S02-,
-N(Rg)-CH2-, -CH2-N(Rg)-, -S(0)(NRf)-CH2- or -CH2-S(0)(NI0-.
In a first embodiment, -X-Q- represents -0-. In a second embodiment, -X-Q-
represents -0-C(0)-. In a third embodiment according to the invention, -X-Q-
represents -
C(0)-0-. In a fourth embodiment, -X-Q- represents -0-C(CH-CN)-. In a fifth
embodiment, -X-Q- represents - S-. In a sixth embodiment, -X-Q- represents -SO-
. In a
seventh embodiment, -X-Q- represents -SO2-. In an eighth embodiment, -X-Q-
represents
-N(Rg)-. In a ninth embodiment, -X-Q- represents -N(10-00-. In a tenth
embodiment, -
X-Q- represents -CO-N(Rf)-. In an eleventh embodiment, ¨X-Q- represents -N(10-
S02-.
In a twelfth embodiment, -X-Q- represents -SO2-N(R)-. In a thirteenth
embodiment, -X-
Q- represents -S(0)(N10-. In a fourteenth embodiment, -X-Q- represents
optionally
substituted -CH2-CH2-. In one aspect of that embodiment, -X-Q- represents -CH2-
CH2-.
In a fifteenth embodiment, -X-Q- represents ¨ optionally substituted -0-CH2-.
In one
aspect of that embodiment, -X-Q- represents -0-CH2-. In a sixteenth
embodiment, -X-Q-
represents optionally substituted -CH2-0-. In one aspect of that embodiment, -
X-Q-
represents -CH2-0-. In a seventeenth embodiment, -X-Q- represents optionally
substituted
-S-CH2-. In one aspect of that embodiment, X-Q- represents -S-CH2-. In an
eighteenth
embodiment, -X-Q- represents optionally substituted -SO-CH2-. In one aspect of
that
embodiment, -X-Q- represents -SO-CH2-. In a nineteenth embodiment, -X-Q-
represents
optionally substituted -S02-CH2-. In one aspect of that embodiment, -X-Q-
represents -
S02-CH2-. In a twentieth embodiment, -X-Q- represents optionally substituted -
CH2-S-.
In one aspect of that embodiment, X-Q- represents optionally substituted -CH2-
S-. In a
twenty-first embodiment, -X-Q- represents optionally substituted -CH2-S0-. In
one aspect
of that embodiment, -X-Q- represents -CH2-S0-. In a twenty-second embodiment, -
X-Q-
represents optionally substituted -CH2-S02-. In one aspect of that embodiment,
-X-Q-
represents -CH2-S02-. In a twenty-third embodiment, -X-Q- represents
optionally
substituted -N(Rg)-CH2-. In one aspect of that embodiment, -X-Q- represents -
N(Rg)-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 12 -
CH2-. In a twenty-fourth embodiment, -X-Q- represents optionally substituted -
CH2-
N(Rg)-. In one aspect of that embodiment, -X-Q- represents -CH2-N(R8)-. In a
twenty-
fifth embodiment, -X-Q- represents optionally substituted -S(0)(N111)-CH2-. In
one aspect
of that embodiment, -X-Q- represents -S(0)(NR11)-CH2-. In a twenty-sixth
embodiment, -
X-Q- represents optionally substituted -CH2-S(0)(N10-. In one aspect of that
embodiment, -X-Q- represents -CH2-S(0)(NRf)-. In a twenty-seventh embodiment, -
X-Q-
represents -N(Rf)-C(S)-. In a twenty-eighth embodiment, -X-Q- represents -
N=S(0)(CH3). In a twenty-ninth embodiment, -X-Q- represents -0-C(=CH2)-. In a
thirtieth embodiment ¨X-Q- represents -S(=N-CN)-. Typical substituents on -X-Q-
include halogen, (Ci4alkyl and carboxy. Additional substituents on -X-Q-
include
trifluoromethyl. Further substituents on -X-Q-include (C24a1ky1carbonyl, (C2_
6)alkoxycarbonyl, and hydroxy(Ci_6)alkyl.
Particular examples of substituents on -X-Q- include fluoro, methyl, carboxy,
trifluoromethyl, methylcarbonyl, deuterated methyl, ethoxycarbonyl,
hydroxyisopropyl,
and hydroxymethyl.
Suitable substituents on -X-Q- include fluoro, methyl and carboxy.
Appropriately, -X-Q- represents -0-, -0-C(0)-, -0-C(CH-CN)-, -S-, -SO-, -SO2-;
or -N(Rg)-, -N(Rt)-00-, -N(R)-SO2-, 0-CH2-, -, CH2-S-, -CH2-S0-, -CH2-S02-, -
N(R9-
CH2-, -N(Rf)-C(S)-, -N=S(0)(CH3)_, -0-C(=CH2)- or -S(=N-CN)-, any of which
groups
may be optionally substituted.
Particularly, -X-Q- represents -0-, -0-C(0)-, -0-C(CH-CN)-, -S-, -SO-, -SO2-, -
N(Rg)-, -N(R)-CO-, -N(R8)-CH2-, or -N(Rf)-C(S)-, any of which groups may be
optionally substituted.
Suitably, -X-Q- represents -0-, -0-C(0)-, -0-C(CH-CN)-, -N(R), -N(Rf)-00-, -
N(Rg)-CH2-, or -N(Rf)-C(S)-.
Typically, -X-Q- represents -N(R)-C(0)-, -0-C(0)- or -0-C(CH-CN)-.
Appositely, -X-Q- represents -N(RF)-C(0)-.
Generally, Z represents methylene.
Generally, E represents a fused heteroaromatic ring system of formula (Ea) or
a
fused heteroaromatic ring system of formula (Eb).
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 13 -
In a first embodiment according to the present invention, E represents a fused
heteroaromatic ring system of formula (Ea).
In a second embodiment according to the present invention, E represents a
fused
heteroaromatic ring system of formula (Eb).
In a third embodiment according to the present invention, E represents a fused
heteroaromatic ring system of formula (Ec).
Particular sub-classes of compounds in accordance with the present invention
include compounds of formula (TB), (IC), and (ID).
R3
R RN
2 R 2 12
R12
X
Ri
z R4 z
R4
(IB)
Rs
Rs
(IC) Rs
Rs
6 6
R7
R7
R3
NN R12
X
z
R4
(1D) R5 Rs
R6
R7
wherein ¨X-Q-, Z, R2, R3, R4, R5, R6, R7, Rs and R12 are as defined
above.
Particular sub-classes of compounds in accordance with the present invention
include compounds of formula (IB) and (IC), wherein Z, 111, R2, R3, R4, R5,
R6,
fe,IV and R12 are as defined above.
CA 02962826 2017-03-28
WO 2016/050975
PCT/EP2015/072868
- 14 -
A particular sub-class of compounds in accordance with the present invention
is
the sub-class of compounds of formula (1B), wherein -X-Q-, Z, R1, R25 R3, R4,
R55 R65
R7,R8 and R12 are as defined above.
A further particular sub-class ofcompounds in accordance with the present
invention is the sub-class of compounds of formula (IC), wherein -X-Q-, Z,
R2, R3,
R45 R5, R6, 7 K- R8 and R12 are as defined above.
Generally, re represents hydrogen, halogen, or cyano; or aryl, C3-7
heterocycloalkyl, heteroaryl, (C3_7)cycloa1kyl-heteroaryl,
(C3_7)heterocyc1oalky1-
heteroaryl, (C4_9)bicycloalkyl-heteroaryl, (C4_9)heterobieycloalkyl-heteroaryl-
, (C4-
.. 9)spiroheterocycloalkyl-heteroaryl-, (C3_7)heteroeycloalkenyl, or
(C3_7)heterocycloalkenyl-
aryl-, any of which groups may be optionally substituted by one or more
substituents.
Typically, RI represents hydrogen, halogen, or cyano; or aryl, C3-7
heterocycloalkyl, heteroaryl, (C3_7)cycloalkyl-heteroaryl,
(C37)heterocycloalkyl-
heteroaryl, (C19)bicycloalkyl-heteroaryl, (C19)heterobicycloalkyl-heteroaryl-
or (Ci
9)spiroheterocycloalkyl-heteroaryl-, any of which groups may be optionally
substituted by
one or more substituents.
More typically, R' represents halogen or cyano; or aryl, hcteroaryl, (C;_
7)cycloalkyl-heteroaryl, (C3_7)hcterocycloalkyl-heteroaryl,
(C4_9)heterobicycloalkyl-
heteroaryl, (C37)heterocycloalkyl, (C3_7)heterocycloalkenyl, or
(C3_7)heterocycloalkenyl-
aryl, any of which groups may be optionally substituted by one or more
substituents.
Even more typically, R' represents halogen or cyano; or aryl, heteroaryl, (C3_
7)cycloalkyl-heteroaryl, (C37)heterocycloalkyl-heteroaryl, or
(C4_9)heterobicycloalkyl-
heteroaryl any of which groups may be optionally substituted by one or more
substituents.
Particularly, R1 represents hydrogen, halogen or cyano; or aryl, heteroaryl,
(C3-
7)heterocycloalkyl-heteroaryl, any of which groups may be optionally
substituted by one
or more substituents.
More particularly, le represents halogen or cyano; or aryl, heteroaryl, or
(C3_
7)cycloalkyl-heteroaryl, any of which groups may be optionally substituted by
one or
more substituents.
Suitably, R' represents hydrogen; or IV represents aryl or heteroaryl, either
of
which groups may be optionally substituted by one or more substituents.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 15 -
Appositely, R1 represents aryl or heteroaryl, either of which groups may be
optionally substituted by one or more substituents.
More suitably, R1 represents heteroaryl, either of which groups may be
optionally
substituted by one or more substituents.In a first embodiment, IV represents
hydrogen.
In a second embodiment, R1 represents halogen. In one aspect of that
embodiment, R1 represents bromo. In another aspect of that embodiment, R.1
represents
chloro.
In a third embodiment, IV represents cyano.
In a fourth embodiment, R1 represents optionally substituted aryl. In one
aspect of
that embodiment, R.1 represents optionally substituted phenyl.
In fifth embodiment, R1 represents optionally substituted C3-7
heterocycloalkyl. In
one aspect of that embodiment, R1 represents azetidinyl.
In a sixth embodiment, R1 represents optionally substituted heteroaryl. In one
aspect of that embodiment, R1 represents optionally substituted pyrimidinyl.
In another
aspect of that embodiment, R1 represents optionally substituted pyridinyl.
In a seventh embodiment, R1 represents optionally substituted (C3_7)cycloalkyl-
heteroaryl-. In a first aspect of that embodiment, R1 represents optionally
substituted
cyclohexylpyrazolyl-. In a second aspect of that embodiment, R.1 represents
optionally
substituted cyclohexylpyridinyl-. In a third aspect of that embodiment, R1
represents
optionally substituted cyclopropylpyrimidinyl-. In a fourth aspect of that
embodiment, le
represents optionally substituted cyclobutylpyrimidinyl-. In a fifth aspect of
that
embodiment, R1 represents optionally substituted cyclopentylpyrimidinyl-. In a
sixth
aspect of that embodiment, R1 represents optionally substituted
cyclohexylpyrimidinyl-.
In a seventh aspect of that embodiment, R1 represents optionally substituted
cyclohexyl-
.. pyrazinyl-. In an eighth aspect of that embodiment, R1 represents
optionally substituted
cyclopropylpyridinyl.
In an eighth embodiment, R1 represents optionally substituted (C327)-
heterocycloalkyl-heteroary1-. In a first aspect of that embodiment, R1
represents
optionally substituted pyrrolidinylpyridinyl-. In a second aspect of that
embodiment, R1
represents optionally substituted tetrahydropyranylpyridinyl-. In a third
aspect of that
embodiment, R1 represents optionally substituted piperidinylpyridinyl-. In a
fourth aspect
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 16 -
of that embodiment, R1 represents optionally substituted piperazinylpyridinyl-
. In a fifth
aspect of that embodiment, R1 represents optionally substituted
morpholinylpyridinyl-. In
a sixth aspect of that embodiment, le represents optionally substituted
thiornorpholinyl-
pyridinyl-. In a seventh aspect of that embodiment, le represents optionally
substituted
diazepanylpyridinyl-. In an eighth aspect of that embodiment, R' represents
optionally
substituted oxetanylpyrimidinyl-. In a ninth aspect of that embodiment, R1
represents
optionally substituted azetidinylpyrimidinyl-. In a tenth aspect of that
embodiment, le
represents optionally substituted tetrahydrofuranylpyrimidinyl-. In an
eleventh aspect of
that embodiment, le represents optionally substituted pyrrolidinylpyrimidinyl-
. In a
twelfth aspect of that embodiment, re represents optionally substituted
tetrahydropyranyl-
pyrimidinyl-. In a thirteenth aspect of that embodiment, fe represents
optionally
substituted piperidinylpyrimidinyl-. In a fourteenth aspect of that
embodiment, le
represents optionally substituted piperazinylpyrimidinyl-. In a fifteenth
aspect of that
embodiment, RI represents optionally substituted morpholinylpyrimidinyl-. In a
sixteenth
aspect of that embodiment, R1 represents optionally substituted
thiomotpholinyl-
pyrimidinyl-. In a seventeenth aspect of that embodiment, R' represents
optionally
substituted azepanylpyrimidinyl-. In an eighteenth aspect of that embodiment,
RI
represents optionally substituted oxazepanylpyrimidinyl-. In a nineteenth
aspect of that
embodiment, le represents optionally substituted diazepanylpyrimidinyl-. In a
twentieth
aspect of that embodiment, le represents optionally substituted thiadiazepanyl-
pyrimidinyl-. In a twenty-first aspect of that embodiment, le represents
optionally
substituted oxetanylpyrazinyl-. In a twenty-second aspect of that embodiment,
fe
represents optionally substituted piperidinylpyrazinyl-. In a twenty-third
aspect of that
embodiment, le represents optionally substituted tetrahydropyranylpyridinyl-.
In twenty-
third aspect of that embodiment, RI represents (imino)(oxo)thiazinanyl-
pyrimidinyl-. In
twenty-fourth aspect of that embodiment, le represents (oxo)thiazinanyl-
pyrimidinyl-. In
twenty-fifth aspect of that embodiment, R1 represents (dioxo)thiazinanyl-
pyrimidinyl-. In
a twenty-sixth aspect of that embodiment, le represents substituted
(dioxo)tetrahydrothiophenyl-pyrimidinyl-. In a twenty-seventh aspect of that
embodiment, RI represents substituted tetrahydrothiophenyl-pyrimidinyl-. In a
twenty-
eighth aspect of that embodiment, le represents substituted
(dioxo)thiomorpholinyl-
pyrimidinyl-. In a twenty-ninth aspect of that embodiment, R' represents
substituted
azetidinyl-pyrazolyl. In a thirtieth aspect of that embodiment, le represents
substituted
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 17 -
(oxo)tctrahydrothiophenyl-pyrimidinyl. In a thirty-first aspect, RI represents
substituted
(oxo)thiomorpholinyl.
In a ninth embodiment, RI represents optionally substituted (C4_9)bicycloalkyl-
heteroary1-.
In a tenth embodiment, RI represents optionally substituted (C4-9)-
heterobicycloalkyl-heteroary1-. In one aspect of that embodiment, R1
represents optionally
substituted (3,7-dioxa-9-azabicyclo[3.3.1]non-9-y1)-pyrimidinyl-. In a second
aspect of
this embodiment, R' represents optionally substituted (2-oxa-5-
azabicyclo[2.2.1]heptany1)-pyrimidinyl-. In a third aspect of that embodiment,
RI
represents optionally substituted (3-oxa-8-azabicyclo[3.2.1]oct-8-y1)-
pyrimidinyl-. In a
fourth aspect, RI represents optionally substituted (3,6-
diazabicyclo[3.2.2]nonany1)-
pyrimidinyl-.
In an eleventh embodiment, RI represents optionally substituted (C4-9)-
spiroheterocycloalkyl-heteroary1-.
In a twelfth embodiment, RI represents optionally substituted (C3_
7)heterocycloalkenyl. In one aspect of that embodiment, RI represents
optionally
substituted 1,2-dihydropyridinyl. In a second aspect of that embodiment, RI
represents
optionally substituted 1,2-dihydropyrimidinyl.
In a thirteenth embodiment, RI represents optionally substituted (C3_
7)heterocycloalkenyl-aryl. In one aspect of that embodiment, RI represents
optionally
substituted imidazolyl-phenyl.
Typically, RI represents chloro or cyano; or phenyl, pyridinyl, pyrimidinyl,
cyclopropyl-pyridinyl-, cyclobutyl-pyrimidinyl, cyclobutyl-pyridinyl-,
cyclohexyl-
pyrimidinyl-, (3,7-dioxa-9-azabicyclo[3.3.1]non-9-y1)-pyrimidinyl-, azetidinyl-
pyrimidinyl-, azetidinyl-pyridinyl, pyrrolidinyl-pyridinyl-, pyrrolidinyl-
phenyl-,
piperazinyl-pyridinyl-, piperazinyl-pyrimidinyl-, pyrazolyl-, morpholinyl-
pyrimidinyl-,
thiomorpholinyl-pyrimidinyl-, (dioxo)thiomorpholinyl-pyrimidinyl-,
(oxo)thiomorpholinyl-pyrimidinyl-, oxetanyl-pyridinyl-, oxetanyl-pyrimidinyl-,
imidazolyl-phenyl, diazepanyl-pyrimidinyl-, (oxo)tetrahydrothiophenyl-
pyrimidinyl-,
(dioxo)tetrahydrothiophenyl-pyrimidinyl-, tetrahydrothiophenyl-pyrimidinyl
azetidinyl-
pyrazoly1-, (2-oxa-5-azabicyclo[2.2.1]heptany1)-pyrimidinyl-, (3-oxa-8-
azabicyclo[3.2.11oct-8-y1)-pyrimidinyl-, (3,6-diazabicyclo[3.2.2]nonany1)-
pyrimidinyl-,
CA 02962826 2017-03-28
WO 2016/050975
PCT/EP2015/072868
- 18 -
tetrahydropyranyl-pyrimidinyl, azetidinyl, 1,2-dihydropyridinyl, or 1,2-
dihydropyrimidinyl, any of which groups may be optionally substituted by one
or more
substituents.
Particularly, represents
hydrogen, chloro or cyano; or phenyl, pyridinyl, or
pyrimidinyl, any of which groups may be optionally substituted by one or more
substituents.
Illustratively, R1 represents pyrimidinyl, which may be optionally substituted
by
one or more substituents.
Typical examples of optional substituents on It' include one, two or three
substituents independently selected from halogen, halo(C1_6)alkyl, cyano,
cyano(C1_6)alkyl,
nitro(Ci_6)alkyl, Ci_6 alkyl, phosphate(Ci_6)alkyl, (C3_7)cycloalkyl,
trifluoromethyl,
trifluoroethyl, C2_6 alkenyl, hydroxy, hydroxy(Ci_6)alkyl, C1_6 alkoxy, (C1_6)
alkoxy(Ci_
6)alkyl, trifluoroethoxy, carboxy(C3_7)cycloalkyloxy, C1-6 alkylthio, C1_6
alkylsulphonyl,
(CI 6)alkylsulphonyl(C1 6)alkyl, oxo, amino, amino-(Ci 6)alkyl, CI 6
alkylamino, di(Ci
6)alkylamino, (Ci_6)alkoxy(Ci_6)alkylamino, N-[(C1_6)alky1]-
Nthydroxy(Ci_6)alkyliamino,
(C2_6)alkylcarbonylamino(C1-6)alkyl, Cis alkylsulphonylamino, N-[(C1_6)alkyl]-
N-[(C1-
6)alkylsulphonyllamino, bis[(Ci_6)alkyl-sulphonyl]amino, N-RCi_6)alkyll-N-
[carboxy(Ci_
6)alkyl]amino, carboxy(C3_2)cycloalkyl-amino, carboxy(C3_7)cycloalkyl(C1-
6)alkylamino,
formyl, C2-6 alkylcarbonyl, (C2_6)alkyl-carbony1oxy(C1_6)alkyl, carboxy,
carboxy(Ci-
6)alkyl, C2-6 alkoxycarbonyl, C2-6 alkoxycarbonyl(Cie)alkyl, morpholinyl(Ci-
6)alkoxycarbonyl, C2.6 alkoxycarbonyl-methylidenyl, aminocarbonyl,
aminosulphonyl,
(C1_6)alkylsulphoximinyl and [(C1_6)alkyl][N-(Ci4alkyllsulphoximinyl.
Additional
examples of optional substituents on include C1-6 alkyl phosphate-C1_6
alkyl, sulphate-
Ci_6 alkyl, carboxy(Ci_6)alkyl-carbonyloxy-Ci_6 alkyl, and phosphate- methoxy-
C1_6 alkyl.
A further additional example of optional substituent on It' include (C24
alkoxycarbonyl-
amino-CI-6 alkyl. Additional optional substituents on RI include
difluoromethyl, (C1_
6)alkyl-sulphinyl-amino-, di(C1_6)alkylarnino (C16)alkyl,
di(C1_6)alkenylannino (C14alkyl,
C1_6 alkylsulphonyl-amino-C1 6 alkyl, and tetrahydrofuranyl.
Illustrative examples of optional substituents on R' include one, two or three
substituents independently selected from halogen, cyano, cyano(Ci4alkyl, C1-6
alkyl,
difluoromethyl, trifluoromethyl, hydroxy, (hydroxy)(Ci_6)alkyl, amino,
(amino)(C1-6)
alkyl, Ci_6alkoxy, (C1_6) alkoxy(C1_6)alkyl, C2-6 alkylcarbonyl, C2_6
alkoxycarbonyl, (C24
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 19 -
alkoxycarbonyl-amino-C1_6 alkyl, phosphate(C1_6)alkyl, Ci_6 alkylthio, C1_6
alkylsulphonyl,
oxo, (Ci4alkylsulphoximinyl, (C I _6)alkylsulphinyl-amino-, di(C1_6)alkylamino
(C _
6)alkyl, (C2_6)alkylcarbonylamino(C1_6)alkyl, di(Cl_6)alkenylamino
(C4_6)alkyl, (C2-
6)alkylcarbonylamino(Ci_6)alkyl, C1_6 alkylsulphonyl-amino-Ci_6 alkyl,
tetrahydrofuranyl,
sulphate(Ci_6)alkyl, and carboxy-(Ci_6)allcyl-carbonyloxy-(Ci_6)alkyl.
Particular examples of optional substituents on R1 include one, two or three
substituents independently selected from Ci_6 alkyl, trifluoromethyl, hydroxy,
(hydroxy)(C14 alkyl, (amino)(Ci_6) alkyl, C1_6 alkoxy, (C14 alkoxy(C1-6)alkyl,
(C2-6)
alkoxycarbonyl-amino-Ci_6 alkyl, phosphate(Ci_6)alkyl, Ci_6 alkylsulphonyl,
oxo and (CI_
6)alkylsulphoximinyl.
Suitable examples of optional substituents on R' include one, two or three
substituents independently selected from C1-6 alkyl, hydroxy, (hydroxy)(Ci_6)
alkyl, Ci_6
alkoxy, (C1_6) alkoxy(Ci4alkyl, phosphate(C16)alkyl, C1-6 alkylsulphonyl, oxo
and (C1_
6)alkylsulphoximinyl.
Particular examples of optional substituents on R' include one, two or three
substituents independently selected from (hydroxy)(Ci_6) alkyl and (C1_6)
alkoxy(Ci-
6)alkyl.
Typical examples of particular substituents on RI include one, two or three
substituents independently selected from fluor , chloro, fluoromethyl,
fluoroisopropyl,
cyano, cyanoethyl, nitromethyl, methyl, ethyl, isopropyl, phosphate-isopropyl,
isopropylmethyl, cyclopropyl, cyclobutyl, trifluoromethyl, trifluoroethyl,
ethenyl,
hydroxy, hydroxymethyl, hydroxyisopropyl, methoxy, isopropoxy,
methoxyisopropyl,
trifluoro-ethoxy, carboxycyclobutyloxy, methylthio, methylsulphonyl,
methylsulphonylmethyl, methylsulphonylethyl, oxo, amino, aminomethyl,
aminoisopropyl, methylamino, dimethylamino, methoxyethylamino, N-
(hydroxyethyl)-N-
(methyl)amino, acetylaminomethyl, methylsulphonylarnino, N-methyl-N-
(methylsulphonyl)amino, bis(methylsulphonyl)amino, N-(carboxyethyl)-N-
(methyl)amino,
carboxycyclopentylamino, carboxycyclopropylmethylamino, formyl, acetyl,
acetoxyisopropyl, carboxy, carboxymethyl, carboxyethyl, methoxycarbonyl,
ethoxycarbonyl, n-butoxycarbonyl, tert-butoxycarbonyl, methoxycarbonylmethyl,
ethoxy-
carbonylmethyl, ethoxycarbonylethyl, morpholinylethoxycarbonyl, ethoxycarbonyl-
methylidenyl, methylsulphonylaminocarbonyl, acetylaminosulphonyl, methoxyamino-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 20 -
carbonyl, tetrazolyl, tctrazolylmcthyl, hydroxyoxadiazolyl, aminocarbonyl,
amino-
sulphonyl, methylsulphoximinyl and (methyl)(N-methyl)sulphoximinyl. Additional
typical
examples of optional substituents on R1 include ethyl phosphate-isopropyl,
sulphate-
isopropyl, carboxy-ethyl-carbonyloxy-isopropyl, and phosphate- methoxy-
isopropyl. A
further additional typical example of substituents on R1 include (tert-
butoxycarbonyl)amino-isopropyl. Other additional typical examples of
susbtituents on R'
include (tert-butyl)carbonyl, methoxycarbonylamino-isopropyl,
dimethylaminoisopropyl,
(tert-butyl)sulphinyl-amino, (tert-butypsulphonylamino,
methylsulphonylaminoisopropyl,
methylcarbonylamino-isopropyl, cyanoisopropyl, difluoromethyl,
tetrahydrofuranyl,
di(propenyl)aminoisopropyl and hydroxyisobutyl.
Appropriate examples of optional substituents on 121 include one, two or three
substituents independently selected from methyl, difluoromethyl,
trifluoromethyl,
hydroxy, hydroxyisopropyl, methoxy, methoxyisopropyl, phosphate-isopropyl,
(tert-
butoxycarbonyl)amino-isopropyl, aminoisopropyl, dimethylaminoisopropyl, methyl-
sulphonyl, methylsulphoximinyl, oxo, tert-butoxycarbonyl,
(methoxycarbonyl)amino-
isopropyl, methylthio, (tert-butyl)sulphinyl-amino, amino, (tert-
butyl)sulphonyl-amino,
methylsulphonylamino-isopropyl, methylcarbonylamino-isopropyl, fluoro, cyano,
cyanoisopropyl, tetrahydrofuranyl, di(propenyl)aminoisopropyl, sulphate-
isopropyl,
carboxy-ethyl-carbonyloxy-isopropyl and (hydroxy)isobutyl.
Illustrative examples of optional substituents on R1 include one, two or three
substituents independently selected from methyl, trifluoromethyl, hydroxy,
hydroxyisopropyl, methoxy, methoxyisopropyl, phosphate-isopropyl, (tert-
butoxycarbonyl)amino-isopropyl, amino isopropyl, methyl-sulphonyl and
methylsulphoximinyl.
Suitable examples of optional substituents on R' include one, two or three
substituents independently selected from methyl, hydroxy, hydroxyisopropyl,
methoxy,
methoxyisopropyl, phosphate-isopropyl, methyl-sulphonyl and
methylsulphoximinyl.
Particular examples of substituents on R' include one, two or three
substituents
independently selected from hydroxyisopropyl and methoxyisopropyl.
In a particular embodiment, R' is substituted by hydroxy(C16)alkyl. In one
aspect
of that embodiment, R1 is substituted by hydroxyisopropyl. In a particular
aspect of that
embodiment, R1 is substituted by 2-hydroxyprop-2-yl.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
-21 -
In another particular embodiment, R1 is substituted by (C1_6)
alkoxy(C1_6)alkyl. In
one aspect of that embodiment, RI is substituted by methoxyisopropyl. In a
particular
aspect of this embodiment, R1 is substituted by 2-methoxyprop-2-yl.
Illustrative values of RI include chloro, cyano, methylsulphonyl-phenyl,
methylsulphoximinyl-phenyl, (dihydroxy)(methyDcyclobutylpyrimidinyl,
hydroxyisopropylpyridinyl, hydroxyisopropylpyrimidinyl,
(methyl)(hydroxyisopropyl)pyrimidinyl, phosphate-isopropylpyrimidinyl,
methoxypyridinyl, methoxyisopropylpyrimidinyl, 2-oxo-pyridin-(IH)-yl, (tert-
butoxycarbonyl)aminoisopropyl-pyrimidinyl, aminoisopropylpyrimidinyl, (3,7-
dioxa-9-
azabicyclo[3.3.1]non-9-y1)-pyrimidinyl, (hydroxy)(trifluoromethyl)azetidinyl-
pyrimidinyl,
(methylsulphonyl)(methyl)phenyl, (methyl)(hydroxyisopropyl)pyridinyl, [
(hydroxy)(trifluoromethyl)azetidinyl](methyl)pyrimidinyl
methylsulphonylcyclopropylpyridinyl, (dimethyl)(hydroxyisopropyl)pyrimidinyl,
(hydroxyisopropyl)(trifluoromethyl)pyrimidinyl, (ten-
butoxycarbonyl)(hydroxy)pyrrolidine-pyridinyl, (hydroxy)pyrrolidine-pyridinyl,
(methoxycarbonyl)aminoisopropyl-pyrimidinyl, piperazinylpyridinyl,
(methylsulphonyl)piperazinyl-pyridinyl, (dimethylamino)isopropylpyrimidinyl,
(oxo)piperazinylpyrimidinyl, (N-methyl)pyrazolyl, (methylthio)(methyl)phenyl,
morpholinylpyrimidinyl, ((tert-butyl)sulphinylamino)cyclobutylpyridinyl,
(amino)cyclobutylpyridinyl, ((tert-butyDsulphinylamino)oxetanylpyridinyl,
(amino)oxetanylpyridinyl, ((tert-butyl)sulphonylamino)oxetanylpyridinyl,
pyrrolidinylpyridinyl, (dimethyl)imidazolylphenyl,
(methylsulphonyDaminoisopropylpyrimidinyl,
methylcarbonylaminoisopropylpyrimidinyl,
pyrrolidinyl-phenyl, (oxo)diazepanylpyrimidinyl, (hydroxy)(methyDazetidinyl-
pyrimidinyl, thiomorpholinylpyrimidinyl, (oxo)thiomorpholinylpyrimidinyl,
(dioxo)thiomorpholirtylpyrimidinyl, (difluoro)(hydroxy)cyclohexylpyrimidinyl,
(hydroxy)(oxo)tetrahydrothiophenyl-pyrimidinyl,
(hydroxy)(dioxo)tetrahydrothiophenyl-
pyrimidinyl, (hydroxy)tetrahydrothiophenyl-pyrimidinyl,
(hydroxy)oxetanylpyrimidinyl,
(methylsulphonyDazetidiny1-2,5-pyrazolyl, (oxo)(methyl)-1,2-dihydropyridinyl,
(oxo)-1,2-
dihydropyrimidinyl, (dihydroxy)(methyl)cyclohexylpyrimidinyl,
cyanoisopropylpyrimidinyl, (cyano)(methyl)azetidinylpyrimidinyl, (2-oxa-5-
azabicyclo[2.2.1]heptany1)-pyrimidinyl-, (3-oxa-8-azabicyclo[3.2.1]oct-8-y1)-
pyrimidinyl-,
(oxo)(3,6-diazabicyclo[3.2.2]nonany1)-pyrimidinyl-,
(hydroxyisopropyl)azetidinyl,
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 22 -
(difluoro)azetidinylpyrimidinyl, tetrahydropyranylpyrimidinyl,
methylsulphoxyminylpyridinyl, (thfluoromethyl)(hydroxyisopropyl)pyrimidinyl,
(tetrahydrofuranyl)(hydroxyisopropyl)pyrimidinyl,
di(propenyl)aminoisopropylpyrimidinyl, sulphate-isopropylpyrimidinyl, carboxy-
ethyl-
carbonyloxy-isopropyl-pyrimidinyl and (hydroxy)isobutylpyrimidinyl.Specific
values of
RI include chloro, methylsulphonyl-phenyl, methylsulphoximinyl-phenyl,
(dihydroxy)(methyl)cyclobutylpyrimidinyl, hydroxyisopropylpyridinyl,
hydroxyisopropylpyrimidinyl, (methyl)(hydroxyisopropyl)pyrimidinyl, phosphate-
isopropylpyrimidinyl, methoxypyridinyl, methoxyisopropylpyrimidinyl, 2-oxo-
pyridin-
(1H)-yl, (tert-butoxycarbonyl)aminoisopropyl-pyrimidinyl,
aminoisopropylpyrimidinyl,
(3,7-dioxa-9-azabicyclo[3.3.1]non-9-y1)-pyrimidinyl,
(hydroxy)(trifluoromethyl)azetidinyl-pyrimidinyl,
(methylsulphonyl)(methyl)phenyl and
(methyl)(hydroxyisopropyl)pyridinyl.
Particular values of R1 include methylsulphonyl-phenyl, methylsulphoximinyl-
phenyl, (dihydroxy)(methyl)cyclobutylpyrimidinyl, hydroxyisopropylpyridinyl,
hydroxyisopropylpyrimidinyl, (methyl)(hydroxyisopropyl)pyrimidinyl; phosphate-
isopropylpyrimidinyl, methoxypyridinyl, methoxyisopropylpyrimidinyl and 2-oxo-
pyridin-(1H)-yl.
Selected values of R' include hydroxyisopropylpyrimidinyl, particularly 2-(2-
.. hydroxy-propan-2-y1)-pyrimidin-5-y1; methoxyisopropylpyrimidinyl,
particularly 242-
methoxy-propan-2-y1)-pyrimidin-5-y1; aminoisopropylpyrimidinyl, particularly 2-
(2-
amino-propan-2-y1)-pyrimidin-5-y1; and phosphate-isopropylpyrimidinyl,
particularly 2-
(2-phosphate-propane-2-y1)-pyrimidin-5-yl.
In one embodiment, R1 represents 2-(2-hydroxy-propan-2-y1)-pyrimidin-5-yl. In
another embodiment, R1 represents 2-(2-amino-propan-2-y1)-pyrimidin-5-yl. In a
further
embodiment, R1 represents 2-(2-phosphate-propane-2-y1)-pyrimidin-5-yl.
Illustrative values of R1 include hydroxyisopropylpyrimidinyl, particularly 2-
(2-
hydroxy-propan-2-y1)-pyrimidin-5-yl, and methoxyisopropylpyrimidinyl,
particularly 2-
(2-methoxy-propan-2-y1)-pyrimidin-5-yl.
Typically, R2 represents hydrogen, halogen, trifluoromethyl, trifluoromethoxy
or -
OW; or C1-6 alkyl optionally substituted by one or more substituents.
Suitably, R2 represents hydrogen or halogen.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 23 -
In a first embodiment, R2 represents hydrogen. In a second embodiment, R2
represents halogen. In one aspect of that embodiment, R2 represents fluoro. In
another
aspect of that embodiment, R2 represents chloro. In a third embodiment, R2
represents
cyano. In a fourth embodiment, R2 represents nitro. In a fifth embodiment, R2
represents
hydroxy. In a sixth embodiment, R2 represents trifluoromethyl. In a seventh
embodiment,
R2 represents trifluoromethoxy. In an eighth embodiment, R2 represents -0Ra.
In a ninth
embodiment, R2 represents optionally substituted C1_6 alkyl. In a first aspect
of that
embodiment, R2 represents C1_6 alkyl. In a particular aspect of this
embodiment, R2
represents methyl. In another particular aspect of this embodiment, R2
represents ethyl.
In a second aspect of that embodiment, R2 represents mono substituted methyl
or
monosubstituted ethyl.
Typical examples of optional substituents on R2 include C2_6 alkoxycarbonyl.
Typical examples of particular substituents on R2 include ethoxycarbonyl.
Typical values of R2 include hydrogen, fluoro, chloro, trifluoromethyl,
trifluoromethoxy, -OR', methyl and ethoxycarbonylethyl.
Specific values of R2 include hydrogen, bromo and fluoro.
Particular values of R2 include hydrogen and fluoro.
Generally, R3 represents hydrogen, halogen, trifluoromethyl, or Ci _6 alkyl.
Typically, R3 represents hydrogen, halogen or C1-6 alkyl.
In a first embodiment, R3 represents hydrogen. In a second embodiment, R3
represents halogen. In one aspect of that embodiment, R3 represents fluoro. In
a third
embodiment, R3 represents optionally substituted Ci_6 alkyl. In one aspect of
that
embodiment, R3 represents C1-6 alkyl. In a particular aspect of this
embodiment, R3
represents methyl. In another particular aspect of this embodiment, R3
represents ethyl. In
a fourth embodiment, R3 represents trifluoromethyl.
Illustratively, R3 represents hydrogen or trifluoromethyl.
In a particular embodiment, R3 represents hydrogen.
Generally, R4 represents hydrogen, halogen , trifluoromethyl, or C1-6 alkyl.
Typically, R4 represents hydrogen, halogen or C1-6 alkyl.
CA 02962826 2017-03-28
WO 2016/050975
PCT/EP2015/072868
- 24 -
In a first embodiment, R4 represents hydrogen. In a second embodiment, R4
represents halogen. In one aspect of that embodiment, R4 represents fluoro. In
a third
embodiment, R4 represents optionally substituted C1_6 alkyl. In one aspect of
that
embodiment, R.4 represents CI-6 alkyl. In a particular aspect of this
embodiment, R4
represents methyl. In another particular aspect of this embodiment, R4
represents ethyl. In
a fourth embodiment, R4 represents trifluoromethyl.
Illustratively, R4 represents hydrogen or trifluoromethyl.
In a particular embodiment, le represents hydrogen.
Typically, R5 represents halogen, cyano, difluoromethoxy, trifluoromethoxy, -
ORa, or C1_6 alkylsulphonyl; or C16 alkyl optionally substituted by one or
more
substitucnts.
Generally, R5 represents halogen, -0Ra, difluoromethoxy or trifluoromethoxy.
In a first embodiment, R5 represents hydrogen. In a second embodiment, R5
represents halogen. In one aspect of that embodiment, R5 represents chloro. In
a second
aspect of that embodiment, R5 represents fluoro. In a third embodiment, R5
represents
cyano. In a fourth embodiment, R5 represents hydroxy. In a fifth embodiment,
R5
represents trifluoromethyl. In a sixth embodiment, R5 represents
difluoromethoxy. In a
seventh embodiment, R5 represents trifluoromethoxy. In an eighth embodiment,
R5
represents ¨0Ra. In one aspect of that embodiment, R5 represents methoxy. In a
ninth
embodiment, R5 represents Ci_6 alkylsulphonyl. In one aspect of that
embodiment, R5
represents methylsulphonyl. In a tenth embodiment, R5 represents optionally
substituted
C1_6 alkyl. In one aspect of that embodiment, R5 represents C1_6 alkyl. In a
particular
aspect of this embodiment, R5 represents methyl. In another particular aspect
of this
embodiment, R5 represents ethyl.
Suitably, R5 represents fluoro, methoxy, difluoromethoxy or trifluoromethoxy.
Appositely, R5 represents fluoro, methoxy or difluoromethoxy.
Suitably, R5 represents difluoromethoxy.
Generally, R6 represents hydrogen, halogen or trifluoromethyl.
In a first embodiment, R6 represents hydrogen. In a second embodiment, R6
represents halogen. In one aspect of that embodiment, R6 represents chloro, hi
a second
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 25 -
aspect of that embodiment, R6 represents fluoro. In a third aspect of that
embodiment, R6
represents bromo. In a third embodiment, R6 represents trifluoromethyl. In a
fourth
embodiment, R6 represents C1_6 alkyl. In a first aspect of that embodiment, R6
represents
CIA alkyl. In a second aspect of that embodiment R6 represents Ci_; alkyl. In
a third
aspect of that embodiment, R6 represents C1-2 alkyl. In a particular aspect of
this
embodiment, R6 represents methyl. In another particular aspect of this
embodiment, R6
represents ethyl. In a fifth embodiment, R6 represents C1_6 alkoxy. In a
particular aspect of
that embodiment, R6 represents methoxy.
Particularly, R6 represents hydrogen, bromo or trifluoromethyl.
Illustratively, R6 represents hydrogen or bromo.
Suitably, R6 represents hydrogen.
In a first embodiment, fe represents hydrogen. In a second embodiment, R7
represents halogen. In one aspect of that embodiment, R7 represents chloro. In
a second
aspect of that embodiment, R1 represents fluoro. In a third embodiment, R7
represents
trifluoromethyl. In a fourth embodiment, R7 represents C1_6 alkyl. In a first
aspect of that
embodiment, R7 represents Ci_4 alkyl. In a second aspect of that embodiment R7
represents Ci_3 alkyl. In a third aspect of that embodiment, R7 represents
C1_2 alkyl. In a
particular aspect of this embodiment, R7 represents methyl. In another
particular aspect
of this embodiment, R7 represents ethyl. In a fifth embodiment, R7 represents
Ci_6 alkoxy.
In a particular aspect of that embodiment, R7 represents methoxy.
Illustratively, R7 represents hydrogen or trifluoromethyl.
Suitably, R7 represents hydrogen.
Generally, R8 represents hydrogen, halogen or trifluoromethyl.
In a first embodiment, R8 represents hydrogen. In a second embodiment, R8
represents halogen. In one aspect of that embodiment, R8 represents chloro. In
a second
aspect of that embodiment, R8 represents fluoro. In a third embodiment, R8
represents
cyano. In a fourth embodiment, R8 represents hydroxy. In a fifth embodiment,
R8
represents trifluoromethyl. In a sixth embodiment, R8 represents
difluoromethoxy. In a
seventh embodiment, R8 represents trifluoromethoxy. In an eighth embodiment,
R8
represents -OR'. In one aspect of that embodiment, R8 represents methoxy. In a
ninth
embodiment, R8 represents Ci_6 alkylsulphonyl. In one aspect of that
embodiment, R8
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 26 -
represents mcthylsulphonyl. In a tenth embodiment, R8 represents optionally
substituted
C1_6 alkyl. In one aspect of that embodiment, R8 represents C1_6 alkyl. In a
particular
aspect of this embodiment, R8 represents methyl. In another particular aspect
of this
embodiment, le represents unsubstituted ethyl. In an eleventh embodiment, R8
represents
trifluoromethyl.
Particularly, R8 represents hydrogen, chloro or trifluoromethyl
Illustratively, R8 represents hydrogen or chloro.
Suitably, R8 represents hydrogen.
Generally, Ru represents hydrogen or C1-6 alkyl.
Suitably, R12 represents hydrogen or methyl.
Apositely, R12 represents hydrogen.
Generally, R2 represents C1_6 alkyl, C3-7 cycloalkyl, C37 heterocycloalkyl,
aryl,
aryl(Ci_6)alkyl, heteroaryl or heteroaryl(Ci_6)alkyl, any of which groups may
be optionally
substituted by one or more substituents.
Generally, Rb and Re independently represent hydrogen or trifluoromethyl; or
C1-6
alkyl, C3_7 cycloalkyl, C3_7 cycloalkyl(C16)alkyl, aryl, aryl(Ci_6)alkyl, C3-7
heterocycloalkyl, C3-7 heterocycloalkyl(Ci_6)alkyl, heteroaryl or
heteroaryl(C1_6)alkyl, any
of which groups may be optionally substituted by one or more substituents; or
le and Re, when taken together with the nitrogen atom to which they are both
attached, represent azetidin-l-yl, pyrrolidin-l-yl, oxazolidin-3-yl,
isoxazolidin-2-yl,
thiazolidin-3-yl, isothiazolidin-2-yl, piperidin- I -yl, morpholin-4-yl,
thiomorpholin-4-yl,
piperazin-l-yl, ho mop iperidin-l-yl,
homomorpholin-4-yl, homopiperazin-l-yl,
(imino)(oxo)thiazinan-4-yl, (oxo)thiazinan-4-y1 or (dioxo)thiazinan-4-y1 any
of which
groups may be optionally substituted by one or more substituents.
Generally, Rd represents hydrogen; or C1-6 alkyl, C3-7 cycloalkyl, aryl, C3-7
heterocycloalkyl or heteroaryl, any of which groups may be optionally
substituted by one
or more substituents.
Generally, Re represents Ci_6 alkyl, aryl or heteroaryl, any of which groups
may be
optionally substituted by one or more substituents.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 27 -
Typical examples of suitable substituents which may be present on R2, Rb, Re,
Rd
or Re, or on the heterocyclic moiety -NRbW, include halogen, C1_6 alkyl, C1_6
alkoxy,
difluoromethoxy, trifluoromethoxy, C1_6 alkoxy(Ci_6)alkyl, C1-6 alkylthio, C1-
6
alkylsulphinyl, Ci_6 alkylsulphonyl, hydroxy, hydroxy(C14alkyl,
amino(Ci_6)alkyl, cyano,
trifluoromethyl, oxo, C2-6 alkylcarbonyl, carboxy, C2-6 alkoxycarbonyl, C2 6
alkylcarbonyloxy, amino, C1-6 alkylamino, di(Ci_6)alkylamino, phenylamino,
pyridinylamino, C2-6 alkylcarbonylamino, C2-6 alkylcarbonylamino(C1_6)alkyl,
C2-6
alkoxycarbonylamino, C1_6 alkylsulphonylamino, aminocarbonyl, Ci_6
alkylaminocarbonyl
and di(C1_6)alkylaminocarbonyl.
Typical examples of specific substituents which may be present on R2, Rb, W,
Rd,
or R', or on the heterocyclic moiety -NRbW, include fluoro, chloro, bromo,
methyl, ethyl,
isopropyl, methoxy, isopropoxy, difluoromethoxy, trifluoromethoxy,
methoxymethyl,
methylthio, ethylthio, methylsulphinyl, methylsulphonyl, hydroxy,
hydroxymethyl,
hydroxyethyl, aminomethyl, cyano, trifluoromethyl, oxo, acetyl, carboxy,
methoxycarbonyl, ethoxycarbonyl, tert-butoxycarbonyl, acetoxy, amino,
methylamino,
ethylamino, dimethylamino, phenylamino, pyridinylamino, acetylamino, tert-
butoxycarbonylamino, acetylaminomethyl, methylsulphonylamino, aminocarbonyl,
methylaminocarbonyl and dimethylaminocarbonyl.
Suitably, W represents CI-6 alkyl, aryl(C1_6)alkyl or heteroaryl(C16)alkyl,
any of
which groups may be optionally substituted by one or more substituents.
In a first embodiment, Ra represents optionally substituted CI-6 alkyl. In a
first
aspect of that embodiment, Ra represents C1-6 alkyl. In a particular aspect of
this
embodiment, W represents methyl. In a second aspect of that embodiment, Ra
represents
substituted C1_6 alkyl. In a particular aspect of this embodiment, W
represents
methoxyethyl. In a second embodiment, R2 represents optionally substituted
aryl. In a
first aspect of this embodiment, Ra represents aryl. In a particular aspect of
this
embodiment, Ra represents phenyl. In a second aspect of that embodiment, Ra
represents
monosubstituted aryl. In a particular aspect of this embodiment, Ra represents
methylphenyl. In a third embodiment, Ra represents optionally substituted
aryl(C1_6)a1kyl.
In one aspect of that embodiment, R2 represents aryl(Ci_6)alkyl. In a
particular aspect of
this embodiment, Ra represents benzyl. In a fourth embodiment, Ra represents
optionally
substituted heteroaryl. In one aspect of this embodiment, Ra represents
heteroaryl. In a
fifth embodiment, Ra represents optionally substituted heteroaryl(Ci_6)alkyl.
In a particular
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 28 -
aspect of this embodiment, W represents dioxoisoindolylpropyl. In a sixth
embodiment, le
represents C3-7 cycloalkyl. In a seventh embodiment, Ra represents C3-7
heterocycloalkyl.
Appositely, le represents C1-6alkyl. Illustratively, le represents methyl.
Typically, le represents hydrogen; or C1_6 alkyl, ary1(Ci_6)alkyl, C3-7
heterocycloalkyl or C3-7 heterocycloalkyl(C16)alkyl, any of which groups may
be
optionally substituted by one or more substituents.
Suitably, Rb represents hydrogen or C1_6 alkyl.
In a first embodiment, le represents hydrogen. In a second embodiment, Rh
represents C1-6 alkyl. In a particular aspect of that embodiment, Rh
represents methyl.
Typically RC represents hydrogen; or C1_6 alkyl, Cl_.7 eycloalkyl or C1-7
heterocycloalkyl, any of which groups may be optionally substituted by one or
more
substituents.
Suitably, RC represents hydrogen or C1_6 alkyl.
In a first embodiment, le is hydrogen. In a second embodiment, le represents
C1-6
alkyl. In a one aspect of that embodiment, le represents methyl. In a another
aspect of that
embodiment, 12." represents ethyl.
Appositely, RC represents hydrogen or ethyl.
Alternatively, the moiety -NleItc may suitably represent azetidin-l-yl,
pyrrolidin-
l-yl, oxazolidin-3-yl, isoxazolidin-2-yl, thiazolidin-3-yl, isothiazolidin-2-
yl, piperidin-1-
yl, morpholin-4-yl, thiomorpholin-4-yl, piperazin-l-yl,
homomorpholin-4-y1 or homopiperazin- 1 -yl, (imino)(oxo)thiazinan-4-yl,
(oxo)thiazinan-
4-y1 or (dioxo)thiazinan-4-yl, any of which groups may be optionally
substituted by one
or more substituents.
Specific values of the heterocyclic moiety -NWie include azetidin-l-yl,
hydroxyazetidin-l-yl, hydroxymethylazetidin-l-yl,
(hydroxy)(hydroxymethyl)azetidin-l-
yl, aminomethyl-azetidin-l-yl, cyanoazetidin-l-yl, carboxyazetidin-l-yl,
aminoazetidin-
l-yl, amino carbonylazetidin-l-yl, pyrrolidin-l-yl, amino methy 1pyrrolidin-l-
yl,
oxopyrrolidin-l-yl, acetylaminomethylpyrrolidin-l-yl, tert-
butoxycarbonylaminopyrrolidin-l-yl, oxo-oxazolidin-3-yl, hydroxyisoxazolidin-2-
yl,
thiazolidin-3-yl, oxothiazolidin-3-yl, dioxo-isothiazolidin-2-yl, piperidin-l-
yl,
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 29 -
hydroxypiperidin-l-yl, hydroxymethylpiperidin-1-yl, aminopiperidin-l-yl,
acetylaminopiperidin-l-yl, tert-butoxycarbonylaminopiperidin-l-yl,
methylsulphonylaminopiperidin-l-yl, morpholin-4-yl, piperazin-1 -yl,
methylpiperazin-l-
yl, methylsulphonylpiperazin-l-yl, oxopiperazin-l-yl, acetylpiperazin-l-yl,
.. ethoxycarbonylpiperazin-l-yl, oxohomopiperazin-l-yl, (imino)(oxo)thiazinan-
4-yl,
(oxo)thiazinan-4-yl, and (dioxo)thiazinan-4-yl.
Typically, Rd represents hydrogen; or C1_6 alkyl, aryl or heteroaryl, any of
which
groups may be optionally substituted by one or more substituents.
Selected examples of suitable substituents on Rd include halogen, C1_6 alkyl,
C1-6
.. alkoxy, oxo, C2_6 alkylcarbonyloxy and di(Ci4alkylamino.
Selected examples of particular substituents on Rd include fluor , methyl,
methoxy, oxo, acetoxy and dimethylamino.
Suitably, Rd represents hydrogen or C1_6 alkyl.
In a first embodiment, Rd represents hydrogen. In a second embodiment, Rd
.. represents optionally substituted C1-6 alkyl. In one aspect of that
embodiment, Rd
represents C1-6 alkyl. In a third embodiment, Rd represents optionally
substituted aryl. In
one aspect of that embodiment, Rd represents phenyl. In a fourth embodiment,
Rd
represents optionally substituted heteroaryl.
Appositely, Rd represents hydrogen or methyl.
Typically, R0 represents C1-6 alkyl or aryl, either of which groups may be
optionally substituted by one or more substituents.
Selected examples of suitable substituents on 120 include Cis alkyl,
especially
methyl.
In a first embodiment, RC represents optionally substituted C1-6 alkyl. In a
particular
.. aspect of that embodiment, Re represents C1-6 alkyl. In a particular aspect
of this
embodiment especially methyl. In a second embodiment, RC represents optionally
substituted aryl. In one aspect of that embodiment, RC represents phenyl. In
another aspect
of that embodiment, R0 represents monosubstituted aryl.
Suitably, RC represents methyl, propyl or methylphenyl.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 30 -
Generally, Rf represents hydrogen; or Ci_6 alkyl, C3_6 cycloalkyl, or C4,5
heterocycloalkyl, any of which groups may be optionally substituted by one or
more
substituents.
Suitably, Rf represents hydrogen; or C1_6 alkyl, which group may be optionally
substituted by one or more substituents.
Generally, R5 represents hydrogen; or C1_6 alkyl, -00-(Ci 6)alkyl, -SO2-
(C16)alkyl.
-CO-(C37)heterocycloalkyl, -S02-(C3_7)cycloalkyl, -S02-(C3_7)heterocyc1oalkyl,
-S02-aryl,
-S02-heteroaryl, heteroaryl or (C2_6)alkoxycarbonyl, any of which groups may
be
optionally substituted by one or more substituents.
More generally, R5 represents hydrogen; or C1_6 alkyl, -CO-(C1_6)alkyl, -S02-
(C1_
6)alkyl. -00-(C3-7)hetcrocycloalkyl, -S02-(C37)cycloalkyl, - S02-aryl, -S02-
hctcroaryl,
heteroaryl or (C2_6)alkoxycarbonyl, any of which groups may be optionally
substituted by
one or more substituents.
Typically, R5 represents hydrogen; or C1_6 alkyl, -00-(Ci_6)alkyl,
-CO-(C37)heterocycloalkyl, -S02-(C3_7)cycloalkyl, -S02-(C3_7)heterocycloalkyl,
-S02-aryl
or -S02-heteroaryl, any of which groups may be optionally substituted by one
or more
substitucnts.
Interestingly, R5 represents hydrogen; or C1-6 alkyl, C3-6 cycloalkyl, C4.6
heterocycloalkyl, -00-(Ci4alkyl, or -502-(C i_6)alkyl, any of which groups may
be
optionally substituted by one or more substituents.
Suitably, R5 represents hydrogen, optionally substituted C1_6 alkyl, -00-
(Ci_6)alkyl
or -S02-(C1_6)allcyl.
Typically, substituents on R5 include independently halogen, C1_6 alkyl,
carboxy
and C1-6 alkoxycarbonyl. Additional substituents on Re' include
trifluoromethyl, C4-9
heterobicycloalkyl, (C1_6 alkyl)sulphonyl, tri(Ci_6 alkyl)silyloxy, hydroxy
and (Ci4alkoxy.
Appositely, substituents on Wand R5 include independently halogen and C1_6
alkyl.
Particular examples of substituents on R5 include independently methyl,
trifluoromethyl, ethoxycarbonyl, 3,7-dioxa-9-azabicyclo[3.3.1]non-9-yl,
methylsulphonyl, (tert-butyl)(di-methyl)silyloxyethyl, hydroxy and methoxy.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
-31 -
Particular examples of substituent on Rf include trifluoromethyl, carboxy and
hydroxy.
In a first embodiment, le represents hydrogen. In a second embodiment, Rf
represents optionally substituted C1_6 alkyl. In one aspect of that
embodiment, Rf
represents C1-6 alkyl. In a particular aspect of this embodiment, Rf
represents methyl. In
another particular aspect of this embodiment, Rf represents ethyl. In a
further particular
aspect of this embodiment, Rf represents isopropyl. In another aspect of that
embodiment,
Rf represents deuterated methyl. In a further aspect of that embodiment, Rf
represents
substituted Ci_6 alkyl. In a third embodiment, Rf represents optionally
substituted C3-6
cycloalkyl. In one aspect of that embodiment, R1 represents C3-6 cycloalkyl.
Particular values of Rf include hydrogen, methyl, ethyl, isopropyl,
(carboxy)methyl, (hifluoromethypmethyl, (hydroxyisopropyl)methyl and
deuterated
methyl.
Illustrative values of Rif include hydrogen, methyl, ethyl and isopropyl.
In a first embodiment, Rs represents hydrogen. In a second embodiment, Rs
represents optionally substituted Ci _6 alkyl. In one aspect of that
embodiment, Rs
represents Cis alkyl. In a particular aspect of this embodiment, Rs represents
methyl. In
another particular aspect of this embodiment, Rs represents ethyl. In a
further particular
aspect of this embodiment, Rs represents isopropyl. In a third embodiment, R5
represents
optionally substituted C3 6 cycloalkyl. In one aspect of that embodiment, Rg
represents C3_
6 cycloalkyl. In a fourth embodiment, Rs represents optionally substituted ¨00-
(C1-
6)alkyl. In one aspect of that embodiment, Rs represents ¨00-(Ci 6)alkyl. In a
particular
aspect of this embodiment, Rs represents -CO-CH3. In a fifth embodiment, Rs
represents
optionally substituted -S02-(C1-6)a1kyl. In one aspect of that embodiment, Rs
represents -
S02-(C14alkyl. In a particular aspect of this embodiment, Rs represents -S02-
CH3. In a
sixth embodiment, Rs represents optionally substituted -00-
(C3_7)heterocycloalkyl. In a
particular aspect of that embodiment, Rs represents -CO-azetidinyl. In a
seventh
embodiment, Rs represents optionally substituted -S02-(C37)cycloalkyl. In a
particular
aspect of that embodiment, Its represents -S02-cyclopropyl. In an eighth
embodiment, Rs
represents optionally substituted -S02-(C3-7)heterocycloalkyl. In a ninth
embodiment, Rs
represents optionally substituted -S02-aryl. In a particular aspect of that
embodiment, Rs
represents optionally substituted -S02-phenyl.In a tenth embodiment, Rg
represents
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 32 -
optionally substituted -S02-heteroaryl. In a particular aspect of that
embodiment, R8
represents optionally substituted -S02-pyridinylln an eleventh embodiment, R8
represents optionally substituted heteroaryl. In a particular aspect of that
embodiment, R8
represents optionally substituted pyrimidinyl. In a twelfth embodiment, R.8
represents
optionally substituted (C2_6)allcoxycarbonyl. In a particular aspect of that
embodiment, R8
represents ethoxycarbonyl.
Illustrative values of 118 include hydrogen, methyl, carboxymethyl,
ethoxycarbonylmethyl, methylcarbonyl, methylsulphonyl, (3,7-dioxa-9-
azabi cyclo [33.1 ]n on-9-yl)m ethyl carbonyl , azetidinylcarbonyl,
.. (methylsulphonyl)azetidinylcarbonyl, pyridinylsulphonyl,
cyclopropylsulphonyl, (tert-
butyl)(dimethyl)silyloxyethyl, hydroxyethyl, phenylsulphonyl,
(methoxy)pyridinylsulphonyl, (pyridine-2(1H)-one)sulphonyl, pyrimidinyl and
ethoxycarbonyl.
Selected values of R8 include hydrogen, methyl, carboxymethyl,
ethoxycarbonylmethyl, methylcarbonyl, methylsulphonyl, (3,7-dioxa-9-
azabicyclo[3.3.1]non-9-yl)methylcarbonyl, azetidinylcarbonyl,
(methylsulphonyl)azetidinylcarbonyl, pyridinylsulphonyl, cyclopropylsulphonyl,
(tert-
butyl)(dimethypsilyloxyethyl, hydroxyethyl, phenylsulphonyl and
(methoxy)pyridinylsulphonyl.
Particular values of R8 include hydrogen, methyl, carboxymethyl,
ethoxycarbonylmethyl, methylcarbonyl and methylsulphonyl.
Specific values of R8 include hydrogen and methyl.
Illustrative values of -X-Q- include -0-,-0-00-, -0-C(CH-CN)-, -S-, -SO-, SO2-
,
-NH-, -N(CO-CH3)-, -N(S02-CH3)-, -N(CH2-00-0-CH2-CH3)-, -NRCO-CH2-(3,7-dioxa-
9-azabicyclo[3.3.11non-9-y1)1-, -N[C0-(azetidin-3-y1)]-, -N[C0-
(methylsulphonyl)azetidin-3-ylk, -N(CH2-COOH), -N[(tert-
butyl)(dimethypsilyloxyethyll-, -N(S02-pyridine-3-y1)-, -N-(S02-cyclopropy1)-,
-
N(CH3)-CH2-, -N(CH2-CH2-0H)-, -N(S02-phenyl)-, -N[S02-(6-methoxy-pyridin-3-
y1)]-,
-NH-00-, -N(CH3)-00-, -N(CH2CH3)-00-, -N(CH(CH1)2)-00-, -N(CH2-COOH)-00-, -
N(CH2-CF)-00-, -N(CH2-CH2-0H)-00-, -N(CH2-C(OH)(CH1)2)-00-, -N(CD3)-00-, -
NH-Cf12-- N(CH2-COOH)-CH2-, -NH-CH(CF3)-, -NH-CH(CH3)-, -NH-C(S)-, -N(CO-
CH3)-CH(CH3)-, -N(S02-CH3)-CH2-, -N(CO-CH3)-CH(CH3)-, -N=S(0)(CH3)-, 0-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 33 -
CH(CF3)-, -CH(C00C2H5)-S-, -CH2-S(0)-, -CH2-S(0)2-, -CH(CH(OH)(CH3)2)-S-, -
CH(CH2OH)-S-, -0-C(=CH2)-, -N[S(0)2-(pyridin-1H-2-one)], -NH-S(0)2-, -
N(pyrimidiny1)-, -N(C00C2F15)-, -S(=N-CN)-,¨N(S02-CH3)- and ¨N(C2H5)-00-.
In a particular embodiment, the present invention provides a compound of
formula
.. (I) or an N-oxide thereof, or a pharmaceutically acceptable salt thereof:
Ri2
E
z
(I)
R5 R8
R6 R7
wherein
X-Q- represents -0-, -0-C(0)-, -0-C(CH-CN)-, -S-, -SO-, or -SO2-; or -N(R), -
N(R)-CO-, -N(Rf)-S02-, 0-CH2-, -, CH2-S-, -CH2-S0-, -CH2-S02-, -N(R8)-CH2-, -
N(Rf)-
C(S)-, -N=S(0)(CH3)_, -0-C(=CH2)- or -S(=N-CN)-, any of which groups may be
optionally substituted;
Z represents methylene;
E represents a fused heteroaromatic ring system selected from the groups of
formula (Ea) and (Eb);
R3 R3
R2
N RN
R1 RI N *
R4
R4
(Ea) (Eb)
wherein the asterisk (*) represents the site of attachment of E to the
remainder of the molecule;
R' represents halogen or cyano; or aryl, heteroaryl, (C3_7)cycloalkyl-
heteroaryl, (C3-
7)heterocycloalkyl-heteroaryl, (C4_9)heterobicycloalkyl-heteroaryl,
(C3_7)heterocycloalkyl ,
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 34 -
(C3_7)heterocycloalkenyl, or (C3_7)heterocycloalkenyl-aryl, any of which
groups may be
optionally substituted by one or more substituents;
R2 represents hydrogen or halogen;
R3 represents hydrogen or trifluoromethyl;
R4 represents hydrogen or trifluoromethyl;
R5 represents halogen, -0Ra, difluoromethoxy or trifluoromethoxy;
R6 represents hydrogen, halogen or trifluoromethyl;
R7 represents hydrogen or trifluoromethyl;
R8 represents hydrogen, halogen or trifluoromethyl;
R12 represents hydrogen or C1-6 alkyl;
Ra represents Co alkyl;
Itt' represents hydrogen; or C1-6 alkyl, which group may be optionally
substituted
by one or more substituents; and
Rg represents hydrogen; or Ci_6 alkyl, -00-(Ci_6)alkyl, -S02-(C1_6)alkyl. -00-
(C3-
7)heterocycloalkyl, -S02-(C3_7)cycloalkyl, - S02-aryl, -S02-heteroaryl,
heteroaryl or (C2_
6)alkoxycarbonyl, any of which groups may be optionally substituted by one or
more
substituents.
In a particular aspect of that embodiment, the present invention provides a
compound of formula (I) or an N-oxide thereof, or a pharmaceutically
acceptable salt
thereof,
R12
E
z
(I)
R5 R8
R6
R7
X-Q- include -0-,-0-00-, -0-C(CH-CN)-, -S-, -SO-, SO2-, -NH-, -N(CO-CH3)-, -
N(S02-CH3)-, -N(CH2-00-0-CH2-CH3)-, -NRCO-CH2-(3,7-dioxa-9-
azabicyclo[3.3.1]non-9-y1)]-, -N[C0-(azetidin-3-y1)]-, -N[C0-
(methylsulphonyl)azetidin-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 35 -
3-y1)]-, -N(CH2-COOH), -N[(tert-butyl)(ditnethyl)silyloxyethyd-, -N(S02-
pyridinc-3-y1)-,
-N-(S02-cyclopropy1)-, -N(CH3)-CH2-, -N(CH2-C1-12-0H)-, -N(S02-phenyl)-, -
N[S02-(6-
methoxy-pyridin-3-y1)]-, -NH-00-, -N(CH3)-CO-, -N(CH2CH3)-00-, -N(CH(CH3)2)-
CO-, -N(CH2-COOH)-00-, -N(CH2-CF3)-00-, -N(CH2-CH2-0H)-00-, -N(CH2-
C(OH)(CH3)2)-00-, -N(CD3)-00-, -NH-CH2-, - N(CH2-COOH)-CH2-, -NH-CH(CF3)-, -
NH-CH(CH3)-, -NH-C(S)-, -N(CO-CH3)-CH(CH3)-, -N(S02-CH3)-CH2-, -N(CO-CH3)-
CH(CH3)-, -N=S(0)(CH3)-, 0-CH(CF3)-, -CH(CO0C2H5)-S-, -CH2-S(0)-, -CH2-S(0)2-,
-CH(CH(OH)(CH3)2)-S-, -CH(CH2OH)-S-, -0-C(=CH2)-, -N[S(0)2-(pyridin-1H-2-
one)],
-NH-S(0)2-, -N(pyrimidiny1)-, -N(CO0C2H5)-, -S(-N-CN)-,-N(S02-CH3)- and -
N(C2H5)-00-;
RI represents chloro, cyano, methylsulphonyl-phenyl, methylsulphoximinyl-
phenyl, (dihydroxy)(methyl)cyclobutylpyrimidinyl, hydroxyisopropylpyridinyl,
hydroxyisopropylpyrimidinyl, (methyl)(hydroxyisopropyl)pyrimidinyl, phosphate-
isopropylpyrimidinyl, methoxypyridinyl, methoxyisopropylpyrimidinyl, 2-oxo-
pyridin-
(1H)-yl, (tert-butoxycarbonyl)aminoisopropyl-pyrimidinyl,
aminoisopropylpyrimidinyl,
(3,7-dioxa-9-azabicyclo[3.3.1]non-9-y1)-pyrimidinyl,
(hydroxy)(trifluoromethyl)azetidinyl-pyrimidinyl,
(methylsulphonyl)(methyl)phenyl,
(methyl)(hydroxyisopropyl)pyridinyl, [
(hydroxy)(trifluoromethypazctidinyl](methyl)pyrimidinyl
methylsulphonylcyclopropylpyridinyl, (dimethyl)(hydroxyisopropyl)pyrimidinyl,
(hydroxyisopropyl)(trifluoromethyl)pyrimidinyl, (ten-
butoxycarbonyl)(hydroxy)pyrrolidine-pyri dinyl, (hydroxy)pyrrolidine-
pyridinyl,
(methoxycarbonyl)aminoisopropyl-pyrimidinyl, piperazinylpyridinyl,
(me thy Is ulphonyl)p ip erazinyl-py ridinyl, (dime thy lamino)isopropylpy
rimidinyl,
(oxo)piperazinylpyrimidinyl, (N-methyppyrazolyl, (methylthio)(methyl)phenyl,
morpholinylpyrimidinyl, ((tert-butyl)sulphinylamino)cyclobutylpyridinyl,
(amino)cyclobutylpyridinyl, ((tert-butypsulphinylamino)oxetanylpyridinyl,
(amino)oxetanylpyridinyl, ((tert-butyl)sulphonylamino)oxetanylpyridinyl,
pyrrolidinylpyridinyl, (dimethyl)imidazolylphenyl,
(methylsulphonyl)aminoisopropylpyrimidinyl,
methylcarbonylaminoisopropylpyrimidinyl, pyrrolidinyl-phenyl,
(oxo)diazepanylpyrimidinyl, (hydroxy)(methyl)azetidinyl-pyrimidinyl,
thiomorpholinylpyrimidinyl, (oxo)thiomorpholinylpyrimidinyl,
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 36 -
(dioxo)thiomorpholinylpyrimidinyl, (difluoro)(hydroxy)cyclohcxylpyrimidinyl,
(hydroxy)(oxo)tetrahydrothiophenyl-pyrimidinyl,
(hydroxy)(dioxo)tetrahydrothiophenyl-
pyrimidinyl, (hydroxy)tetrahydrothiophenyl-pyrimidinyl,
(hydroxy)oxetanylpyrimidinyl,
(methylsulphonyl)azetidiny1-2,5-pyrazolyl, (oxo)(methyl)-1,2-dihydropyridinyl,
(oxo)-
1,2-dihydropyrimidinyl, (dihydroxy)(methyl)cyclohexylpyrimidinyl,
cyanoisopropylpyrimidinyl, (cyario)(methyl)azetidinylpyrimidinyl, (2-oxa-5-
azabicyclo[2.2.1]heptany1)-pyrimidinyl-, (3-oxa-8-azabicyclo [3 .2.1]oct-8-y1)-
pyrimidinyl-, (oxo)(3,6-diazabicyclo[3.2.2]nonany1)-pyrimidinyl-,
(hydroxyisopropyl)azetidinyl, (difluoro)azetidinylpyrimidinyl,
tetrahydropyranylpyrimidinyl, methylsulphoxyminylpyridinyl,
(difluoromethyl)(hydroxyisopropyl)pyrimidinyl,
(tetrahydrofuranyl)(hydroxyisopropyl)pyrimidinyl,
di(propenyl)aminoisopropylpyrimidinyl, sulphate-isopropylpyrimidinyl, carboxy-
ethyl-
carbonyloxy-isopropyl-pyrimidinyl and (hydroxy)isobutylpyrimidinyl;
R2 represents hydrogen, bromo or fluoro;
R3 represents hydrogen or trifluoromethy;
R4 represents hydrogen or trifluoromethyl;
R5 represents halogen, methoxy, difluoromethoxy or trifluoromcthoxy;
K6 represents hydrogen, bromo, chloro, or trifluoromethyl;
R7 represents hydrogen or trifluoromethyl;
R8 represents hydrogen, chloro and trifluoromethyl;
Ru represents hydrogen or methyl;
Rf represents hydrogen, methyl, ethyl, isopropyl, (carboxy)methyl,
(trifluoromethyl)methyl, (hydroxyisopropyOmethyl or deuterated methyl;
R8 represents hydrogen, methyl, carboxymethyl, ethoxycarbonylmethyl,
methylcarbonyl, methylsulphonyl, (3,7-dioxa-9-azabicyclo [3.3.1]non-9-
yl)methylcarbonyl, azetidinylcarbonyl, (methylsulphonyl)azetidinylcarbonyl,
pyridinylsulphonyl, cyclopropylsulphonyl, (tert-butyl)(dimethyl)silyloxyethyl,
hydroxyethyl, phenylsulphonyl, (methoxy)pyridinylsulphonyl, (pyridine-2(1H)-
one)sulphonyl, pyrimidinyl or ethoxycarbonyl; and
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 37 -
Z and E are as defined in said particular embodiment.
A particular sub-group of the compounds of formula (IB) above is represented
by
the compounds of formula (IIB) and N-oxides thereof, and pharmaceutically
acceptable
salts thereof:
R3
R2
0
R4
R5 40 R8
(JIB)
R6 R7
wherein RI, R2, R3, R4, R5, R6, R7, Rs and le are as defined above.
A particular sub-group of compounds of formula (JIB) above is represented by
the
compounds of formula (JIB-A) and N-oxides thereof, and pharmaceutically
acceptable
salts thereof:
R2
Rf
Ri
0
(JIB-A)
R5
1 0
wherein wherein RI, R2, R5 and le are as defined above.
A particular sub-group of compounds of formula (JIB-A) above is represented by
the compounds of formula (IIB-AB) and N-oxides thereof, and pharmaceutically
acceptable salts thereof,:
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 38 -
R2
RIO
Rf
N
0
R5
(IIB-AB)
wherein,
W represents N or C-H;
R9 represents hydroxy(C1_6)alkyl, (C i4 alkoxy(Ci_6)alkyl, arnino(Ci_6)alkyl
or
.. phosphate(C16)alkyl;
¨10
K represents hydrogen or C1-45 alkyl; and
R2, 125 and Rf are as defined above.
In one embodiment, W represents N. In another embodiment, W represents C-H.
Suitably, R9 represents hydroxy(Ci_6)alkyl or (Ci_6)alkoxy(Ci4alkyl.
Illustratively, R9 represents hydroxyisopropyl or methoxyisopropyl.
Particular values of R9 include 2-hydroxy-prop-2-y1 and 2-methoxy-prop-2-yl.
In one embodiment, R9 represents hydroxyisopropyl. In a particular aspect of
that
embodiment, R9 represents 2-hydroxy-prop-2-yl.
In another embodiment, R9 represents methoxyisopropyl. In a particular aspect
of
that embodiment, R9 represents 2-methoxy-prop-2-yl.
In a further embodiment, R9 represents amino isopropyl. In a particular aspect
of
that embodiment, R9 represents 2-amino-prop-2-yl.
In another embodiment, R9 represents phosphate(Ci_6)alkyl. . In a particular
aspect
of that embodiment, R9 represents 2-phosphate-prop-2-yl.
In one embodiment, RI represents hydrogen. In another embodiment, Rm
represents C1-6 alkyl. In a particular aspect of this embodiment, Rm
represents methyl.
Illustratively, RI represents hydrogen or methyl.
Particularly, RI represents hydrogen.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 39 -
A particular sub-group of compounds of formula (I1B-AB) above is represented
by
the compounds of formula (IIB-AB-A) and N-oxides thereof, and pharmaceutically
acceptable salts thereof:
R2
RIO
/Rf
'N
N
¨0
R9
R5
(IIB-AB-A)
wherein W, Rf, R2, R5 , R9 and R16 are as defined above.
Another particular sub-group of the compounds of formula (IB) above is
represented by the compounds of formula (TIC) and N-oxides thereof, and
pharmaceutically acceptable salts thereof:
R3
R2
Ri 11101
/ g
R4
R5
Rs
(IIC)
R6
R7
wherein R', R2, R3, IV, R5, R6, R7, R8 and Rg are as defined above.
Specific novel compounds in accordance with the present invention include each
of
the compounds whose preparation is described in the accompanying Examples, and
pharmaceutically acceptable salts and solvates thereof, and co-crystals
thereof.
Therefore, in a particular aspect, the present invention relates to compounds
of
formula (I) which are selected from the group consisting of (7R,14R)-1-
(difluoromethoxy)-10-fluoro-1142-(2-hydroxypropan-2-yl)pyrimidin-5-y1]-6,7-
dihydro-
7,14-methanobenzimidazo[1,2-13][2,5]benzodiazocin-5(14H)-one; (7R,14R)-1-
(difluoromethoxy)-10-fluoro-1142-(2-methoxypropan-2-yl)pyrimidin-5-y11-6-
methy1-6,7-
dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one; (7R,14R)-
1-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 40 -
(difluoromethoxy)-11- [2-(2-hydroxyprop an-2-yl)pyrimidin-5 -y1]-6,7-dihydro -
7,14-
methanob enzimidazo [1,2-b] [2,5]b enzodiazocin-5(14H)-one; (7R,14R)-1-
(di fluorometh oxy)-11- [2-(2-hydroxyprop an-2-yl)pyrim i din-5 -y1]-6-methy1-
6,7-di hydro-
7,14-methanob enzimid azo[1,2-b] [2,5]benzodiazocin-5(14H)-one; (7R,14R)-1-
(difluoromethoxy)-6-ethy1-1142-(2-hydroxypropan-2-yl)pyrimidin-5-y1]-6,7-
dihydro-
7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one; (7R,14R)-1-
(difl uorometho xy)-11- [2-(2-hy droxyprop an-2-y Opyrimidin-5 -y1]-6-(prop an-
2-yI)-6,7-
dihydro-7,14-methanob enzimidazo [1,2-b][2,5]benzodiazocin-5(14H)-one;
(7R,14R)-1-
(difluoromethoxy)-10- fluoro-11-[2-(2-hydroxypropan-2-yl)pyrimidin-5-y1]-7H-
7,14-
methanobenzimidazo [2,1-d] [2,51benzoxazocin-5(14H)-one; (2Z)- [(7R,14R)-1-
(difluoromethoxy)-10- fluoro-1142-(2-hydroxypropan-2-yOpyrimidin-5-y1]-7H-7,14-
methanob enzimidazo [2,1-d] [2 ,5]b enzox azocin-5(14H)-ylidene] acetonitrile;
(2E)-
[(7R,14R)- 1-(difluorometho xy)-10-fluoro -11-[2-(2-hydroxyprop an-2-y
Opyrimidin-5-y1]-
7H-7,14-methanobenzimidazo [2,1-d] [2,5 ]benzoxazocin-5(14H)-ylidene]
acetonitrile;
(7R, 14R) and (7S, 14S)-1142-(2-hydroxypropan-2-Apyrimidin-5-y11-7,14-dihydro-
7,14-
methanopyrido[I',2':1,2]imidazo[4,5-d][2]benzazocin-5(6H)-one; (7R,14R)-11-
chloro-1-
(difluoromethoxy)-1O- fluoro-6,7-dihydro -7,14-methanobenzimidazo [1,2-
b] [2,5]benzodiazo cin-5 (14H)-one ; (7R,14R)-11-chlo ro -1-(difluorometho xy)-
6,7-dihydro-
7,14-methanob enzimidazo [1,2-b] [2 ,5]benzodiazo cin-5(14H)-one; (7R,14R)-11-
chloro-1-
(difluoromethoxy)-5,6,7,14-tetrahydro-7,14-methanobenzimidazo [1,2-
b] [2,5 ]ben zodiazocine; [(7R,14R)-1-(difluoromethoxy)-1142-(2-hydroxypropan-
2-
yl)pyrirnidin-5-y1]-5,14-dihydro-7,14-rnethanobenzimidazo[1,2-
b][2,5]benzodiazoc in-
6(7H)-yl] acet ic acid; (7R,14R)-1-(difluoromethoxy)-1142-(2-hydroxypropan-2-
yl)pyrimidin-5-y1]-6,7-dihydro-7,14-methanobenzimidazo [1,2-b]
[2,5]benzodiazocine-
5(14H)-thione; (7R,14R)-11-chloro-1-(difluoromethoxy)-10-fluoro-5,6,7,14-
tetrahydro-
7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocine; 2- (5-[(7R,14R)-1-
(difluoromethoxy)-10- fluoro-5 ,6,7,14-tetrahydro -7,14-methanob enzimidazo
[1,2-
b] [2,5]benzodiazo cin-11-yllpyrimidin-2-y1) prop an-2-ol; (7R,14R)-10-fluoro-
11-[2-(2-
hydroxypropan-2-yl)pyrimidin-5-y1]-1-methoxy-6,7-dihydro -7,14-
methanobenzimidazo [1,2-b] [2 ,5]b enzodiazoc in-5(14H)-one; (7R,14R)-1,10-
difluoro-11-
[2-(2-hydroxypropan-2-yl)pyrimidin-5-yl] -6,7-dihydro -7,14-methanob
enzimidazo [1,2-
b] [2,5 ]benzodiazo cin-5 (14H)-one ; (6R,12R)-2-chloro-11-(difluoromethoxy)-
7,12-
dihydro-6H-6,12-methanobenzimidazo[2,1-c][1,4]benzodiazepine; 2- {5 -
[(6R,12R)-11-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
-41 -
(difluoromethoxy)-7,12-dihydro-6H-6,12-methanobenzimidazo [2,1-
c] [1 ,4]benzodiazepin-2-yl]pyrimidin-2-yll prop an-2-ol; 1-[(6R,12R)-2-chloro-
11-
(difluoromethoxy)-6H-6,12-methanobenzimidazo[2,1-c][1,4]benzodiazepin-7(1 211)-
ydethanone; 1-[(6R,12R)-11-(d ifluoromethoxy)-2-[2-(2-hydroxypropan-2-
yl)pyrimidin-5-
y1]-6H-6,12-methanobenzimidazo[2,1-c][1,4]benzodiazepin-7(12H)-yl]ethanone;
(6R,12R)-2-chloro-11-(difluoromethoxy)-7-(rnethylsulfony1)-7,12-dihydro-6H-
6,12-
methanobenzimidazo [2,1-c] [1,4]benzodia zepine; 2- {5 - [(6R,12R)-11-
(difluoromethoxy)-
7-(methylsulfony1)-7,12-dihydro-6H-6,12-methanob enzimidazo[2,1 -
c] [1,4]benzodiazepin-2-yllpyrimidin-2-y1} prop an-2-ol; (6R,12R)-2-chloro-11-
(difluoromethoxy)-6H,12H-6,12-methanobenzimidazo [2,1-c] [1,4]b enzo xazepine;
2- {5-
R6R,12R)-11-(difluoromethoxy)-6H,12H-6,12-methanob enzimidazo [2,1-
c] [1,4]benzoxazepin-2-yl]pyrimidin-2-y1} prop an-2-ol; (7R,14R)-1-
(difluoromethoxy)-5-
oxo-5,6,7,14-tetrahydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocine-11-
carbonitrile; (7R,14R)-1-(difluoromethoxy)-11- [4-(rnethylsulfonyl)pheny1]-6,7-
dihydro-
.. 7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(1411)-one; (7R,14R)-1-
(difluoromethoxy)-11-(6-methoxypyridin-3-y1)-6,7-dihydro-7,14-
methanobenzimi dazo [1 ,2-b] [2,5]benzodiazocin-5(14H)-one; (7R,14R)-1-
(di fluoromethoxy)-11-(6-oxo-1,6-di hydropyridin-3-y1)-6,7-d ihydro-7,14-
methanobenzimidazo [1,2-b] [2 ,5]benzodiazocin-5(14H)-one; (7R,14R)-1-
(difluoromethoxy)-10- fluoro-11-[6-(2-hydroxyprop an-2-yl)pyridin-3-y1]-6,7-
dihydro-
7,14-methanob enzimidazo[1,2-b] [2,5]benzodiazocin-5(14H)-one; (7R,14R)-1-
(difluoromethoxy)-11- [2-(2-hy droxyprop an-2-y1)-6-methyl-pyrimidin-5 -y1]-
6,7- dihydro-
7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one; (7R,14R)-1-
(difluorometho xy)-10-fluoro-1142-(2-hydroxypropan-2-y1)-6-methyl-pyrimidin-5-
yl] -
6,7-dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one;
(7R,14R)-
1-(difluoromethoxy)-11-[6-(2-hydroxypropan-2-yl)pyridin-3-y1]-6,7-dihydro-7,14-
methanobenzimidazo [1,2-b] [2 ,5]b enzodiazocin-5(14H)-one; (7R,14R)-1-
(difluoromethoxy)-11- [4-(S-methylsulfonimido yl)pheny1]-6,7-dihydro-7,14-
methanob enzimidazo [1,2-b][2,5]benzodiazocin-5(14H)-one; (1R, 11R)-18-
(difluoromethoxy)-6-fluoro-542-(2-hydroxypropan-2-yOpyrimidin-5-y1]-3,9,12-
triazapentacyclo[9.8.1.02,' 0.03,8.014,'9]icosa-2(10),4,6,8,14(19),15,17-
heptaen-13-one
hydrochloride; (7R,14R)-1-(difluoromethoxy)-1142-(cis-1,3-dihydroxy-3-
methylcyclobutyppyrimidin-5-y1]-6,7-dihydro-7,14-methanobenzimidazo [1,2-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 42 -
b][2,5]benzodiazocin-5(14H)-one; ethyl [(6R,12R)-11-(difluoromethoxy)-3-fluoro-
2-[2-
(2-hydroxyprop an-2-yl)pyrimidin-5-y1]-6H-6,12-methanob enzimidazo [2, 1-
c] [1,4]benzodiazepin-7(12H)-yl]acetate; (7R,14R)-1-(di fluoromethoxy)-10-
fluoro-11-[2-
(2-hydroxyprop an-2-yl)pyrimidin-5-y1]-6-methy1-6,7-dihydro-7,14-
methanobenzimidazo [1,2-b] [2 ,5]benzodiazocin-5(14H)-one; tert-butyl (2- {5-
[(6R,12R)-
11-(difluoromethoxy)-7-(methylsulfony1)-7,12-dihydro-6H-6,12-
methanobenzimidazo [2,1-c] [1,4]benzodiazepin-2-yl]pyrimidin-2-y1} propan-2-
yl)carbamate; 2- {5-[(6R,12R)-11-(difluoromethoxy)-7-(methylsulfony1)-7,12-
dihydro-
6H-6,12-methanobenzimidazo [2,1-c] [1,4]benzodiazepin-2-yllpyrimidin-2-y1}
propan-2-
amine; azetidin-3-y1R6R,12R)-2-chloro-11-(difluoromethoxy)-6H-6,12-
methanobenzimidazo [2,1-c] [1,4]benzodiazepin-7(12H)-yl]methanone; [(6R,12R)-
11-
(difluoromethoxy)-3-fluoro-2-[2-(2-hydroxypropan-2-yl)pyrimidin-5-y1]-6H-6,12-
methanobenzimidazo [2,1-c] [1,4]benzodiazepin-7(12H)-yl][1-
(methylsulfonyl)azetidin-3-
yllmethanone; 2- { 5-K6R,12R)-11-(difluoromethoxy)-3 -fluoro-7,12-dihydro-6H-
6,12-
methanobenzimidazo [2,1-c] [1,4]benzodiazepin-2-yl]pyrimidin-2-yll prop an-2-
ol;
azetidin-3-y1[(6R,12R)-2-chloro-1 1-(difluoromethoxy)-6H-6,12-
methanobenzimi dazo [2,1-c] [1,4]benzodiazepin-7(12H)-yl]methanone; cis-1- { 5-
[(6R,12R)-11 -(difluoromethoxy)-7-(methyl su lfony1)-7, 1 2-dihydro-6H-6, 1 2-
m ethanoben zimidazo [2,1-c] [1,4]benzodiazepin-2-yl]pyrimidin-2-yll -3-
methylcyclobutane-1,3-diol; (6R,12R)-1 1-(difluoromethoxy)-2- {2- [(1s,5s)-3,7-
dioxa-9-
azabicyclo [3.3.1]non-9-yl]pyrimidin-5-y11-7-(methylsulfony1)-7,12-dihydro-6H-
6,12-
methanobenzimidazo [2,1-c] [1,4]benzodiazepine; [(6R,12R)-11-(difluoromethoxy)-
3 -
fluoro-2- [2-(2-hydroxyprop an-2-yl)pyrimidin-5-y1]-6H-6,12-methanob
enzimidazo[2,1-
c] [1,4]benzodiazepin-7(12H)-yl]acetic acid; 2- {5 -[(6R,12R)-11-
(difluoromethoxy)-3-
fluoro-7-(pyridin-3-ylsulfony1)-7,12-dihydro-6H-6,12-methanob enzimidazo [2,1-
c] [1,4]benzodiazepin-2-yl]pyrimidin-2-yllpropan-2-ol; 1- {5 -[(6R,12R)-11-
(difluoromethoxy)-7-(methylsulfony1)-7,12-dihydro-6H-6,12-methanobenzimidazo
[2,1-
c] [1,4]benzodiazepin-2-Apyrimidin-2-y1) -3-(trifluoromethyl)azetidin-3-o1; 2-
{5-
[(6R,12R)-11-(difluorometho xy)-3 -fluoro-7-(methylsulfony1)-7,12-dihydro-6H-
6,12-
methanobenzimidazo [2,1-c] [1,4]benzodiazepin-2-yl]pyrimidin-2-yll prop an-2-
ol; 2- {5 -
[(6R,12R)-7-(cyc lopropylsulfonyl)-11-(difluorometho xy)-3 -fluoro-7,12-
dihydro-6H-6,12-
methanob enzimidazo [2,1-c] [1,4]benzodiazepin-2-yl]pyrimidin-2-yll propan-2-
ol; 2- {5 -
[(6R,12R)-7-(2- [tert-butyl(dimethyl)silyl]oxy} ethyl)-11-(difluoromethoxy)-3-
fluoro-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 43 -
7,12-dihydro-6H-6,12-methanobenzimidazo[2,1-c][1,4]benzodiazepin-2-
yl]pyrimidin-2-
y1) propan-2-ol; 2- {5-[(6R,12R)-11-(difluoromethoxy)-3-fluoro-7-(2-
hydroxyethyl)-7,12-
dihydro-6H-6,12-methanobenzimi dazo[2,1-c][1,4]berizodi azepin-2-yl]pyrimidin-
2-
yl} propan-2-ol; 2- {5- R6R,12R)-11-(difluoromethoxy)-3-fluoro-7-
(phenylsulfony1)-7,12-
dihydro-6H-6,12-methanobenzimidazo[2,1-c][1,4]benzodiazepin-2-yl]pyrimidin-2-
yllpropan-2-ol; 2-(5- { (6R,12R)-11-(difluoromethoxy)-3-fluoro-7- [(6-
methoxypyridin-3-
yl)s ulfony1]-7,12-dihy dro-6H-6,12-methanobenz imidazo [2,1-c]
[1,4]benzodiazepin-2-
yll pyrimidin-2-yl)propan-2-ol; 2- {5 -[(6R,12R)-11-(difluoromethoxy)-3-fluoro-
7-
(phenylsulfony1)-7,12-dihydro-6H-6,12-methanobenzimidazo [2,1-c] [1,4]b
enzodiazepin-
2-yllpyrimidin-2-yl}propan-2-ol; 2-(5- {(6R,12R)-11-(difluoromethoxy)-3-fluoro-
7-[(6-
methoxypyridin-3-yOsulfonyl]-7,12-dihydro-6H-6,12-methanobenzimidazo[2,1-
c] [1,4]benzodiazepin-2-y1 pyrimidin-2-yl)propan-2-ol; [(7R,14R)-1-
(difluoromethoxy)-
1142-(2-hydroxypropan-2-yl)pyrimidin-5-y1]-5-oxo-5,14-dihydro-7,14-
methanobenzimidazo[1,2-b][2,5]benzodiazocin-6(711)-yl]acetic acid; (7R,14R)-1-
(difluorometho xy)- 11- [2-(2-hydroxypropan-2-yOpyrimidin-5 -y1]-6-(2,2,2-
trifluorocthyl)-
6,7-dihydro-7,14-methanob enzimidazo[1,2-h] [2 ,5]benzodiazocin-5(14H)-one;
(7R,14R)-
1-(di fluoromethoxy)-6-(2-hydroxyethyl)-1142-(2-hydroxypropan-2-y1)pyrimi din-
5 -y1]-
6,7-d ihydro-7,14-m eth anobenzimi d azo[1,2-b][2,5]ben7odi a7ocin-5(14H)-one;
(7R,14R)-
1-(di fluorornetho x y)-6-(2-h ydrox y-2-m ethyl propy1)-11 -[2-(2-hydrox
ypropan-2-
yl)pyrimidin-5-y1]-6,7-dihydro-7,14-methanobenzimidazo[1,2-b] [2,5]b
enzodiazoc in-
5(14H)-one; (7R,14R)-1142-(2-aminopropan-2-yl)p yrimidin-5-yl] -1-
(difluoromethoxy)-
6-(trideutero)methy1-6,7-dihydro-7,14-methanob enzimidazo [1,2-b] [2,5
]benzodiazocin-
5(14H)-one; (7R,14R)-1-(difluoromethoxy)-11- {643-hydroxy-3-
(trifluoromethyl)azetidin-l-y1]-4-methylpyridin-3-yll -6,7-dihydro-7,14-
methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one; (6R,12R)-3,10-dibromo-
2-
chloro-11-(difluoromethoxy)-7-(methylsuffony1)-7,12-dihydro-6H-6,12-
methanobenzimidazo [2,1-c] [1,4]benzodiazepine; (6R,12R)-2,8,10-trichloro-11-
(difluoromethoxy)-7,12-dihydro-6H-6,12-methanobenzimidazo [2,1-
c] [1,4]benzodiazcpine; (7R,14R)-1-(difluoromethoxy)-10-fluoro-11-[2-(2-
hydroxypropan-2-y1)-4-methylpyrimidin-5-yl] -7,14-dihydro-7,14-
mcthanopyrido [1',2' :1,2]imidazo [4,5-d] [2]benzazocin-5 (61-0-one; (7R,14R)-
1-
(difluoromethoxy)-11- {243 -hydroxy-3-(trifluoromethyl)azet -6,7-
dihydro-7,14-methanobenzimidazo[l ,2-b] [2,5]ben zodiazocin-5(14H)-one; (7R,
14R)-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 44 -
1-(difluoromcthoxy)-10-fluoro-11- {2-[3-hydroxy-3-(trifluoromethypazetidin-1-
yl]pyrimidin-5-y1}-6,7-dihydro-7,14-methanobenzimidazo[1,2-
b][2,5Thenzodiazocin-
5(14H)-one; (7R,14R)-1-(di fluoromethoxy)-11-12-[(1,5,5s)-3,7-dioxa-9-
azabicyclo [3.3.1]non-9-yl]pyrimidin-5-y11-10-fluoro-6,7-dihydro-7,14-
methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one; (7R,14R)-1-
(difluoromethoxy)-11-[2-methy1-4-(methylsulfonyl)pheny1]-6,7-dihydro-7,14-
methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(1411)-one; (7R,14R)-1-
(difluoromethoxy)-11-[6-(2-hydroxypropan-2-y1)-4-methylpyridin-3-y1]-6,7-
dihydro-
7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(1411)-one; (7R,14R)-11-[2-
(2-
aminopropan-2-yOpyrimidin-5-yl] -1-(difluoromethoxy)-6,7-dihydro-7,14-
methanobenzimidazo [1,2-b] [2 ,5]benzodiazocin-5(14H)-one; (7R,14R)-1142-(2-
aminopropan-2-yl)pyrimidin-5-y1]-1-(difluoromethoxy)-10-fluoro-6,7-dihydro-
7,14-
methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14/1)-one; 2- {5- [(7 R,14R)-1-
(difluoromethoxy)-5 -methyl-5,6,7,14-tetrahydro-7,14-methanobenzimidazo [1,2-
b.] [2,5]benzodiazocin-11-yl]pyrimidin-2-yllpropan-2-ol; 2- {5-[(5R or
5S,7R,14R)-1-
(difluoromethoxy)-5 -methyl-5,6,7,14-tetrahydro-7,14-methanobenzimidazo [1,2-
1)] [2,5]benzodiazocin-11-yl]pyrimidin-2-y11 propan-2-ol ; 2- {5-[(5R or
5S,7R,14R)-1-
(difluoromethoxy)-5-methy1-5,6,7,1 4-tetrahydro-7,1 4-methanoben zimi da7o [ 1
,2-
b][2,5]benzodiazocin-11-yl]pyrimidin-2-yllpropan-2-ol; 1-[(7R,14R)-1 -
(difluoromethoxy)-11-[2-(2-hydroxypropan-2-yl)pyrimidin-5-y1]-5-methy1-5,14-
dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-6(71/)-yl]ethanone; 1-
[(7R,14R)-1 -(difluoromethoxy)-11 42-(2-hydroxypropan-2-yl)pyrimidin-5 -y1]-
5,14-
dihydro-7,14-methanobenzimidazo [1,2-1)] [2,5]benzodiazocin-6(71/)-
yl]ethanone; 2-
{5-[(7R,14R)-1-(difluoromethoxy)-6-methy1-5,6,7,14-tetrahydro-7,14 -
.. methanobenzimidazo [1,2-b] [2,5 ]benzodiazocin-11-yllpyrimidin-2-yllpropan-
2-ol; 2-
{54(7R,14R)-1-(difluoromethoxy)-10-fluoro-6-(methylsulfony1)-5,6,7,14-
tetrahydro-
7,14-methanobenzimidazo [1,2-b] [2,5]benzodiazocin-11-yl]pyrimidin-2-y11
propan-2-
ol ; 2- {5- [(7R,14R)-1-(difluoromethoxy)-5 -(trifluoromethyl)-5,6,7,14-
tetrahydro-7,14-
methanobcnzimidazo [1,2-b] [2,5 ]benzodiazocin-11-yl]pyrimidin-2-yllpropan-2-
ol; 2-
{5- [(5R,7R,14R)-1-(difluoromethoxy)-5 -(trifluoromethyl)-5 ,6,7,14-tetrahydro-
7,14-
methanob cnzimidazo [1,2-b] [2,5 ]benzodiazocin-11-yl] pyrimidin-2-yllpropan-2-
ol ;
2- {5 -[(5S,7R,14R)-1-(di fluoromethoxy)-5 -(trifluorom ethyl)-5,6,7,14-
tetrahydro-7,14-
methanobenzimidazo [1,2-b] [2,5]benzodiazocin-11-yllpyrimidin-2-yllpropan-2-
ol; 2- {5-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 45 -
R5R,7R,14R)-1-(difluoromethoxy)-5-(trifluoromethyl)-5,6,7,14-tetrahydro-7,14-
methanobenzimidazo[1,2-b][2,5Thenzodiazocin-11-ylipyrimidin-2-yllpropan-2-ol;
245 - [(5S,7R,14R)-1-(difluoromethox y)-5-(trifluo romethyl)-5,6,7,14 -
tetrahydro-7,14-
methanobenzimidazo [1,2-b] [2,5 ]benzod iazocin-11 -yl]pyrimidin-2-yllpropan-2-
ol ;
(7R,14R)- 1 -(difluorometho xy)-10-fluoro-11 - [2-(2-hydroxypropan-2 -y1)- 1-
oxidopyrimidin-5 -y1]-6,7-dihydro-7,14-methanobenzimidazo[1,2 -
b] [2,5 ]benzodiazocin-5 (1411)-one; (7 R,14R)-1-(difl uoromethoxy)-11- {243-
hydro xy-3-
(trifluoromethypazetidin-l-yl] -4-methylpyrimidin-5 -y11-6,7-dihydro-7,14-
methanobenzimidazo [1,2-b] [2 ,5]benzodiazocin-5(14H)-one; (7R,14R)-1-
(difluoromethoxy)-10-fluoro-11- {243-hydroxy-3-(trifluoromethyl)azetidin-l-y1]-
4-
methylpyrimidin-5-y1} -6,7-dihydro-7,14-methanobenzimidazo [1,2-b] [2,5
]benzodiazocin-
5(14H)-one; (7R,14R)-1-(difluoromethoxy)-11-16-[3-hydroxy-3-
(trifluoromethyl)azet idin-l-yl] -2-methylpyridin-3 -y1} -6,7-dihydro-7,14-
methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(1411)-one; (6R,12R)-2-chloro-11-
(difluorometho xy)-6H,12H-6,12-methanobenzimidazo [2,1-c]
[1,4]benzothiazepine; 2- {5-
[(6R,12R)-11-(difluoromethoxy)-6H,1211-6,12-methanobenzimidazo [2,1-
c] [1,4]benzothiazepin-2-yl]pyrimidin-2-y11 propan-2-ol ; (6R,7R,12S)-2-chloro-
11-
(difluoromethoxy)-6H,1 211-6,1 2-methanobenzimidazo[2,1-c][1,4]ben7othiazepine
7-
oxide; (6R,7S,125)-2-chloro-11-(difluoromethoxy)-6H,121-1-6,12-
methanobenzimidazo [2,1-c] [1,4]benzothiazepine 7-oxide; 2- {5-[(6R,7R,12S)-11-
(difl uoromethoxy)-7-o xido-6H,12H-6,12-met hanobenzimidazo [2,1-
c] [1,4]benzothiazepin-2-yl]pyrimidin-2-yl}propan-2-ol; 2- {5-[(6R,7S,125)-11-
(difluoromethoxy)-7-oxido-6H,12H-6,12-methanobenzimidazo [2,1-
c] [1,4]benzothiazepin-2-yl]pyrimidin-2-yllpropan-2-ol; (6R,12R)-2-chloro-11-
(difluoromethoxy)-6H,12H-6,12-methanobenzimidazo [2,1-c] [1,4]benzothiazepine
7,7-
dioxide; 2- {5-[(6R,12R)-11-(difluoromethoxy)-7,7-dio xido-6H,12H-6,12-
methanobenzimidazo [2,1-c] [1,4]benzothiazepin-2-yl]pyrimidin-2-y1) propan-2-
ol; 1 -
R5R,7R,14R)-1-(difluoromethoxy)-1142-(2-hydroxypropan-2-yl)pyrimidin-5 -y1]-5 -
methy1-5,14-dihydro-7,14-methanobenzimidazo [1,2-b] [2,5]benzodiazocin-6(7/)-
yl]ethanone; 1-[(5S,7R,14R)-1-(difluoromethoxy)-11-[2-(2-hydroxypropan-2-
yl)pyrimidin-5-yl] -5-methy1-5,14-dihydro-7,14-methanobenzimidazo [1,2-
b] [2,5]benzodiazocin-6(7H)-yl]ethanone; 1 -[(5R,7R,14R)-1-(difluoromethoxy)-
11-[2-(2-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 46 -
hydroxypropan-2-yl)pyrimidin-5-y1]-5-methy1-5,14-dihydro-7,14-
methanobenzimidazo[1,2-b][2,5]benzodiazocin-6(7H)-yl]ethanone;
1-[(5S,7R,14R)-1 -(difluoromethoxy)-1142-(2-hydroxypropan-2-yl)pyrimidin-5-y1]-
5-
methy1-5,14-dihydro-7,14-methanobenzimidazo[1,2-b] [2,5]b enzodiazoc in-6(71)-
yl] ethanone; (7R,14R)-10-fluoro-1-hydroxy-1142-(2-hydroxypropan-2-Apyrimidin-
5-
y1]-6,7-dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one; 2-
{5-
[(7R,14R)-1-(difl uorometho xy)-5-methy1-5 -oxido-7,14-dihy dro-7,14-methano-
5X-4-
benzimidazo [2,1 -d][1,2,5]b enzothiadiazocin-11-yl]pyrimidin-2-y1} propan-2-
ol;
(7R,14R)-1-(difluoromethoxy)-11- {611-(methylsulfonyl)cyclopropyl]pyridin-3 -
y1} -6,7-
dihydro-7,14-methanobenzimidazo[1,2-b] [2,5]b enzodiazoc in-5(1411)-one;
(7R,14R)-1-
(difluoromethoxy)-11-[2-(2-hydroxypropan-2-yppyrimidin-5-y1]-6,7-dimethy1-6,7-
dihydro-7,14-methanobenzimidazo[1,2-b] [2,5]benzodiazocin-5(1410-one; (7R,14R)-
1-
(difluoromethoxy)-11-[2-(2-hydroxypropan-2-yl)pyrimidin-5-y1]-7-methyl-6,7-
dihydro-
7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(141)-one; (7R,14R)-1142-(2-
aminopropan-2-yl)pyrimidin-5-y1]-1-(difluoromethoxy)-6,7-dimethy1-6,7-dihydro-
7,14-
methanobenzimidazo [1,2-h] [2 ,5]b enzodiazocin-5(14H)-one; (7R,14R)-1-
(di fluoromethoxy)-10-fluoro-1142-(2-hydroxypropan-2-y1)-4,6-dimethylpyrimi
din-5 -y1]-
6,7-dihydro-7,14-methanobenzimidazo[l ,2-h][2,5]ben7odia7ocin-5(14H)-one; 2-
{5-
[(5R,7R,14R)-1-(difluoromethoxy)-5-(trifluoromethyl)-5,14-dihydro-7H-7,14-
methanobenzimidazo [2,1-d] [2 ,5]b enzoxazoc in-11-yl]pyrimidin-2-yll propan-2-
ol;
2- {5 4(5S,7R,14R)-1-(difluoromethoxy)-5-(trifluoromethyl)-5,14-dihydro-7H-
7,14-
methanobenzimidazo [2 ,1-d] [2 ,5]benzoxazocin-11-yl]pyrimidin-2-y1} propan-2-
ol;
(7R,14R)-1-(difluoromethoxy)-10-fluoro-1142-(2-hydroxypropan-2-yl)pyrimidin-5-
y1]-
12-(trifluoromethyl)-6,7-dihydro-7,14-methanob enzimidazo [1,2-b] [2,5]b
enzodiazocin-
5(1410-one; (7R,14R)-1-(difluoromethoxy)-10-fluoro-1142-(2-hydroxypropan-2-
yl)pyrimidin-5-y1]-4-(trifluoromethyl)-6,7-dihydro-7,14-methanobenzimidazo
[1,2-
b] [2,5]benzodiazocin-5 (1410-one; (7R,14R)-1-(difluoromethoxy)-10-fluoro-
114242-
hydroxypropan-2-yl)pyrimidin-5-y1]-9-(trifluoromethyl)-6,7-dihydro-7,14-
methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(1410-one; (7R,14R)-1-
(difluoromethoxy)-10-fluoro-1142-(2-hydroxypropan-2-yppyrimidin-5-y1]-2-
(trifluoromethy1)-6,7-dihydro-7,14-methanobenzimidazo [1,2-b] [2,5]benzodiazo
ein-
5(1410-one; (7R,14R)-1-(difluoromethoxy)-10-fluoro-1142-(2-hydroxypropan-2-y1)-
4-
(trifluoromethyppyrimidin-5-yl] -6,7-dihydro-7,14-methanobenzimidazo [1,2-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 47 -
b] [2,5 ]benzodiazo cin-5 (14H)-one ; (7R,14R)-1-(difluoromethoxy)-10-fluoro-
11-[2-(2-
hydroxypropan-2-yl)pyrimidin-5-y1]-3-(trifluoromethyl)-6,7-dihydro-7,14-
methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(1411)-one; 2- {5-[(6R,12R)-11-
(difluoromethoxy)-3-fluoro-7,12-dihydro-6H-6,12-methanopyrido
[1',2':1,2]imidazo [4,5-
c][1]benzazepin-2-yl]pyrimidin-2-y1) prop an-2-ol; (7R,14R)-1-(d
ifluoromethoxy)-11-[2-
(cis-1,3-dihydroxy-3-methylcyclobut yl)pyrimidin-5-y1]-6-tride utero-methy1-
6,7-dihy dro-
7,14-met hanob enzimidazo [1,2-b] [2,5]benzodiazo cin-5(1411)-one ; Ethyl
(7R,145)-11-
chloro-1-(difluoromethoxy)-6,7-dihydro-14H-7,14-methanobenzimidazo [2,1-
d][1,5]benzothiazocine-6-carboxylate ; ethyl (7R,14S)-1-(difluoromethoxy)-11-
[2-(2-
hydroxypropan-2-yl)pyrimidin-5-yl] -6,7-dihydro-14H-7,14-methanobenzimidazo
[2,1-
d][1,5]benzothiazoc ine-6-carboxylate ; 2- 15-[(5R,7R,14R)-1-(difluoromethoxy)-
5-oxido-
6,7-dihydro-14H-7,14-methanobenzimidazo[2,1-d][1,5]benzothiazocin-11-
yl]pyrimidin-
2-y1{ prop an-2-ol; 2-15-[(7R,14R)-1-(difluoromethoxy)-5,5-dioxido-6,7-dihydro-
14H-
7,14-methanobenzimidazo[2,1-d][1,5]benzothiazocin-11-yl]pyrimidin-2-y1 propan-
2-ol
2- {5-[(6R,7R,145)-1-(difluoromethoxy)-6-(2-hydroxypropan-2-y1)-6,7-dihydro-
14H-7,14-methanobenzimidazo[2,1-d][1,5]benzothiazocin-11-yl]pyrimidin-2-y1)
prop an-
2-01; 2- {54(6S,7R,145)-1-(difluoromethoxy)-6-(hydroxymethyl)-6,7-dihydro-14H-
7,14-
methanobenzimidazo [2,1-d] [1,5]benzothiazocin-11-yl]pyrimidin-2-yllpropan-2-
ol; 2- {5-
R6R,7R,145)-1-(difluoromethoxy)-6-(hydroxymethyl)-6,7-dihydro-14H-7,14-
methanobenzimidazo[2,1-d][1,5]benzothiazocin-11-yl]pyrimidin-2-ylIpropan-2-ol;
(7R,14R)-1-(difluoromethoxy)-1146-(2-hydroxypropan-2-yOpyridin-3-y1]-6-
trideuteromethy1-6,7-dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-
5(14H)-one; (7R,14R)- 1-(difluoromethoxy)-1146-(2-hydroxyprop 1-
oxidopyridin-
3-yl]dop
3-yl] -6,7-dihydro-7,14-methanob enzimidazo [1,2-h] [2,5 ]benzodiazo cin-5
(1411)-one;
(7R,14R)-1-(difluoromethoxy)-1146-(2-hydroxypropan-2-y1)-1-oxidopyridin-3-y1]-
6-
trideuteromethy1-6,7-dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-
5(1411)-one; 2-
{54(6R,12R)-11-(difluoromethoxy)-6H,12H-6,12-
methanobenzimidazo [2,1-c] [1,4]benzothiazepin-2-yl]pyrimidin-2-y1) propan-2-
amine,
dihydrochloride salt; tert-Butyl 3- {5 -[(7R,14R)-1-(difluorometho xy)-6-
trideuteromethy1-5
oxo-5,6,7,14-tetrahydro-7,14-methanobenzimidazo [1,2-b] [2 ,5]benzodiazocin-11-
yl]pyridin-2-y1) -hydroxypyrrolidine-l-c arboxylate; (7R,14R)-1-
(difluoromethoxy)-11-
[643-hydroxypyrrolidin-3-yl)pyridin-3-y1]-6-trideuteromethy1-6,7-dihydro-7,14-
methanobenzimidazo [1,2-b] [2 ,5]b enzodiazocin-5(14H)-one, dihydrochloride
salt; methyl
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 48 -
(2- {5-[(7R,14R)-1-(difluoromethoxy)-6-trideuteromethy1-5-oxo-5,6,7,14-
tetrahydro-7,14-
methanobenzimidazo [1,2-b] [2,5]b enzodiazocin-11 -yl]pyrimidin-2-y1) propan-2-
yl)carbamate; (7R,14
R)-1-(difluoromethoxy)-1142-(2-hydroxypropan-2-y1)-4-
methylpyrimidin-5-yll -6- trideutero-methy1-6,7-dihydro-7,14-
methanobenzimidazo [1,2-
b] [2,5]benzodiazocin-5 (1410-one; (7R,14R)-11-chloro-1-(difluoromethoxy)-5
-
methy lidene-5,14-dihydro-7H-7,14-methanobenzimidazo [2,1 -d]
[2,5]benzoxazocine;
(7 R,14R)-1-(difluoromethoxy)-11- {243-hydro xy-3-(trifluoromethyl)azet
methyl pyrimidin-5-y1} -6-trideutero-methy1-6,7-dihydro-7,14-
methanobenzimidazo [1,2-
b] [2,5] benzodiazocin-5(14H)-one; (7R,14R)-1-(difluoromethoxy)-11-[6-
(piperazin-1-
yl)pyridin-3 -yl] -6,7-dihydro-7,14-methanob enzimidazo [1,2-b]
[2,5]benzodiazo cin-5(1411)-
one; (7R,14R)-1-(difluoromethoxy)-6-trideutero-methy1-11-[6-(piperazin-1-
yppyridin-3-
y1]-6,7-dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(1411)-one;
(7R,14R)-1-(difluoromethoxy)-6-trideutero-methy1-11- {644-
(methylsulfonyl)piperazin-1
yllpyridine-3-y1) -6,7-dihydro-7,14-methanob enzimidazo[1,2-b] [2,5
]benzodiazocin-
5(1411)-one; (7R,14R)-1-(difluoromethoxy)-11- {2- [2-(dimethylamino)prop an-
2-
yl]pyrimidin-5-yll -6,7-dihydro-7,14-methanobenzimidazo[1,2-
b][2,5]benzodiazocin-
5(1411)-one; (7R,14R)-1-(difluoromethoxy)-11-[2-(3 -oxopiperazin-l-
yl)pyrimidin-5 -y1]-
6,7-d ihydro-7,1 4-m eth anoben zimi azo[ 1 ,2-b][2,5]ben7odia7ocin-5(1 4H)-
one; 1 -
[(6R,12R)-1 1 -(difluoromethoxy)-2-(1-methy1-1H-pyrazol-4-y1)-6H-6,12-
methanobenzimidazo [2,1-c] [1,4]benzodiazepin-7(12H)-y1]-2-(3,7-dioxa-9-
azabicyclo [3.3.1]non-9-yl)ethanone;
(6R,12R)-2-chloro-11-(difluoromethoxy)-7-[(6-
methoxypyridin-3-ypsulfonyl]-7,12-dihydro-6H-6,12-methanobenzimidazo[2,1-
c] [1,4]benzodiazepine; 5-
{[(6R,12R)-2-chloro-11-(difluoromethoxy)-6H-6,12-
methanobenzimidazo [2,1-c] [1,4]benzodiazepin-7(12H)-yl]sulfonyll pyridin-
2(111)-one; 2-
{5-[(7R,14R)-1-(difluoromethoxy)-5,5-dioxido-6,7-dihydro-14H-7,14-
methanobenzimidazo [2,1-d] [1 ,2,5]b enzothiadiazocin-11-yllpyrimidin-2-yll
prop an-2-ol;
(7R,14R)-1-(difluoromethoxy)-1142-methy1-4-(methylsulfanyl)pheny1]-6,7-dihydro-
7,14-
methanobenzimidazo [1,2-b] [2 ,5]b enzodiazocin-5(141/)-one;
(7R,14R)-1 -
(difluoromethoxy)-11- [2-(morpholin-4-yl)pyrimidin-5 -y1]-6,7-dihydro-7,14-
methanobenzimidazo [1,2-b] [2 ,5]benzodiazocin-5(1411)-one; (6R,12R)-2-
chloro-11-
(difluoromethoxy)-7-(pyrimidin-2-y1)-7,12-dihydro-61/-6,12-methanob enzimidazo
[2,1-
c] [1,4]benzodiazepine; ethyl-
(6R,12R)-2-chloro-11-(difluoromethoxy)-611-6,12-
methanobenzi mi dazo [2,1-c] [1 ,4]benzodiazepine-7(1211)-carboxylate; ethyl-
(6R,12R)-11-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 49 -
(difluoromethoxy)-2-[2-(2-hydroxypropan-2-Apyrimidin-5-y1]-6H-6,12-
methanobenzimidazo [2,1-c] [1,4]benzodiazepine-7(1211)-carboxylate; N-(1- {5-
[(7R,14R)-
1-(di fluoromethoxy)-6-trideuteromethy1-5-oxo-5,6,7,14-tetrahydro-7,14-
methanobenzimidazo [1,2-1)] [2 ,5]benzodiazoc in-11-yl]pyridin-2-y1)
cyclobuty1)-2-
methylpropane-2-sulfinamide; (7R,14R)-
11-[6-(1-aminocyclobutyl)pyridin-3-y1]-1-
(difluoromethoxy)-6-trideuteromethy1-6,7-dihydro-7,14-methanobenzimidazo [1,2-
b] [2,5]benzodiazocin-5(1411)-one; N-(3-
{5-[(7R,14R)-1-(difluorometho xy)-6-
trideuteromethy1-5-oxo-5,6,7,14-tetrahydro-7,14-methanobenzimidazo [1,2-
b] [2,5]benzodiazocin-11-yl]pyridin-2-yll oxetan-3-y1)-2-methylpropane-2-
sulfinamide;
(7R,14R)-1146-(3-aminooxetan-3-yl)pyridin-3-y1]-1-(difluoromethoxy)-6-
trideuteromethy1-6,7-dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-
5(14H)-one; N-(3- {5- [(7R,14R)-1-(difluoromethoxy)-6-methy1-5-oxo-5 ,6,7,14-
tetrahydro-
7,14-met hanob enzimidazo [1,2-b] [2,5]benzodiazocin-11-yl]p yridin-2-y1)
oxetan-3-y1)-2-
methylpropane-2-sulfinamide; N-(3-
{ 5-[(7R,14R)-1-(difluoromethoxy)-5-oxo-5,6,7,14-
tetrahydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-11-y1]-1-
oxidopyridin-2-
ylloxetan-3-y1)-2-methylpropane-2-sulfonamide;
(7R,14R)-1-(difluoromethoxy)-1144-
(2,4-dimethy1-1 ii-imidazol-5-yl)phenyl]-6-trideuteromethy1-6,7-dihydro-7,14-
methanoben zim i dazo [1,2-b][2,5]benzodiazocin-5(14H)-one;
(7R,14R)-1-
(difluoromethoxy)-1144-(2,4-dimethy1-1 H-imidazol-5-yl)phenyl ]-6-
trideuteromethy1-6,7-
dihydro-7,14-methanobenzimidazo[1,2-b] [2,5]benzodiazocin-5(14M-one;
(7R,14R)-1-
uoromethoxy)-11- [4-(2,4-dimethy1-1H-imidazol-5-y 1)phenyl] -6,7-dihydro-7,14-
methanobenzimidazo [1,2-b][2,5]benzodiazocin-5(1411)-one;
[(6R,7E,12R)-2-chloro-11-
(difluoromethoxy)-6H-6,12-methano-7X-4-benzimidazo [2,1-c] [1,4]benzothiazepin-
7(1211)-ylidene]cyanamide; N-(2-
{5-[(7R,14R)-1-(difluoromethoxy)-6-methy1-5 -oxo-
5,6,7,14-tetrahydro-7,14-methanobenzimidazo[1,2-b] [2 ,5]benzodiazocin-11-
yl]pyrimidin-
2-yllpropan-2-yl)methanesulfonamide; N-(2- {5- [(7R,14R)-1-(difluorometho xy)-
6-methyl-
5-oxo-5,6,7,14-tetrahydro-7,14-methanobenzimidazo [1,2-b] [2,5]benzodiazocin-
11-
yl]pyrimidin-2-yl)propan-2-yl)acetamide;
(7R,14R)-1-(difluoromethoxy)-1144-
(pyrrolidin-2-yl)phenyll -6,7-dihydro-7,14-met hanobenzimidazo [1,2-
b][2,5]benzodiazocin-
5(1411)-one; (7R,14R)-1-(difluoromethoxy)-6-trideutero-methy1-1142-(3-
oxopiperazin-l-
y1)pyrimidin-5-y1]-6,7-dihydro-7,14-methanobenzimidazo[1,2-b]
[2,5]benzodiazocin-
5(1410-one;
(7R,14R)-1-(difluoromethoxy)-6-methy1-11-[2-(5-oxo-1,4-diazepan-1-
yl)pyrimidin-5-y1]-6,7-dihydro-7,14-rnethanobenzimidazo [1,2-b]
[2,5]benzodiazocin-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 50 -5(14H)-onc; (7R,14R)-11-[4-(2-
aminopropan-2-yl)phcnyl]-1-(difluoromethoxy)-6,7-
dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one;
(7R,14R)-1-
(di fluoromethoxy)-11- [2-(3-hydroxy-3-meth ylazetidin-l-y1)-4-methylpyrim din-
5-y1]-6,7-
dihydro-7,14-methanobenzimidazo [1,2-b] [2,5]benzodiazocin-5(14H)-one;
(7R,14R)-1-
(difluorometho xy)-10-fluoro-6- trideutero-methyl -114243 -oxopiperazin-l-
yl)pyrimidin-
5-yl] -6,7-dihydro-7,14-methanobenzimidazo [1,2-b] [2,5]benzodiazocin-5 (14H)-
one;
(7R,14R)- 1-(difi uorometho xy)-11-[2-(1-o xidothiomorpholin-4-y Opyrimidin-5 -
y1]-6,7-
dihydro-7,14-methanobenzimidazo [1,2-b][2,5]benzodiazoein-5(14H)-one; (7R,14R)-
11-
[2-(4,4-difluoro-1-hydroxycyc lohexyl)pyrimidin-5 -yl] -1-(dinuoromethoxy)-6,7-
dihydro-
7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one; (7R,14R)-
1-
(difluoromethoxy)-11- [2-(3 -hydroxy-1,1-dioxidotetrahydrothiophen-3-
yl)pyrimidin-5 -y1]-
6,7-dihydro-7,14-methanobenzimidazo[1,2-b] [2 ,5]benzodiazocin-5(14H)-one;
(3R)-3-
[(7R,14R)-1-(difluoromethoxy)-5 -oxo-5 ,6,7,!4-t etrahydro-7,14-
methanobenzimidazo [1,2-
b] [2,5]benzodiazocin-11-yllpyrimidin-2-yll -3-hydroxytetrahydrothiophenium-1-
o late;
(3S)-3- {5 - [(7R,14R)-1-(difluoromethoxy)-5-o xo-5,6,7,14-tetrahydro-7,14-
methanobenzimidazo [1,2-b] [2 ,5]benzodiazocin-11-yl]pyrimidin-2-yl] -3 -
hydroxytetrahydrothiophenium-l-olate;
(7R,14R)-1-(difluoromethoxy)-1142-(3-
hydroxyoxetan-3-yl)pyrimidin-5-y1]-6,7-dihydro-7,1 4-methanobenzimidazo[1 ,2-
b][2,5]ben zodiazocin-5(14H)-one;
(7R,14R)-1-(difluoromethoxy)-11- {1-[1-
(methylsulfonyl)azetidin-3-y1]-1H-pyrazol-4-yl]-6,7-dihydro-7,14-
methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one;
(7R,14R)-1-
(difluoromethoxy)-11-(2-hydroxypyrimidin-5-y1)-6,7-dihydro-7,14-
methanobenzimidazo [1,2-b] [2 ,5]benzodiazocin-5(14H)-one;
(7R,14R)-1-
(difluorometho xy)-11- [2-(1,4-dihydroxy-4-methylcyclo hexyl)pyrimidin-5-y1]-
6,7-
dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one; 2- {5 -
R7R,14R)-1-(difluoromethoxy)-5 -oxo-5 ,6, 7,14-t etrahydro-7,14-
methanobenzimidazo [1,2-
b] [2,5]benzodiazocin-11-yl]pyrimidin-2-y1) -2-methylpropanenitrile; 1- {5 -
[(7R,14R)-1-
(difluoromethoxy)-5-oxo-5,6,7,!4-t etrahydro-7,14-methanobenzimidazo [1,2-
b] [2,5]benzodiazocin-11-yl]pyrimidin-2-yll -3-methylazetidine-3-carbonitrile;
(7R,14R)-1-
(difluoromethoxy)-11-12-[(1S,4S)-2-oxa-5-azabicyclo[2.2.1]hept-5-yl]pyrimidin-
5-yll -
6,7-dihydro-7,14-methanobenzimidazo[1,2-b] [2 ,5]benzodiazocin-5(14H)-one;
(7R,14R)-
1-(difluoromethoxy)-1142-(thiomorpholin-4-yl)pyrimidin-5-y1]-6,7-dihydro-7,14-
methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one;
(7R,14R)-1-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
-51 -
(difluoromethoxy)-11- {2-[3-(2-hydroxypropan-2-yl)azetidin-1-yl]pyrimidin-5-
yll -6,7-
dihydro-7,14-methanobenzimidazo [1,2-b][2,5]benzodiazocin-5(14H)-one; (7R,14R)-
11-
[2-(3,3-difluoroazetidin-1-yl)pyrimidin-5-y1]-1-(difluoromethoxy)-6,7-dihydro-
7,14-
methanobenzimidazo [1,2-b] [2 ,5]benzodiazocin-5(14H)-one;
(7R,14R)-1-
(difluoromethoxy)-11-[2-(3-oxa-8-azabicyclo[3.2.1]oct-8-yl)pyrimidin-5-y1]-6,7-
dihydro-
7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one;
(7R,14R)-1-
(difl uorometho xy)- 1 1 - [2-(7-o xo-3,6-diazabicyc lo [3 .2.2]non-3-yl)p
yrimidin-5 -y1]-6,7-
dihydro-7,14-methanobenzimidazo [1,2-b][2,5]benzodiazocin-5(14H)-one;
(7R,14R)-1-
(difluoromethoxy)-11-[2-(1,1-dioxidothiomorpho lin-4-yl)pyrimidin-5-y1]-6,7-
dihydro-
7,14-met hanob enzimidazo [1,2-b] [2,5]benzodiazocin-5(14H)-one; (7R,14R)-1-
(difluoromethoxy)-11-(1-methy1-6-oxo-1,6-dihydropyridin-3-y1)-6,7-dihydro-7,14-
methanobenzimidazo [1,2-b] [2 ,5]benzodiazocin-5(14H)-one;
(7R,14R)-1-
(difluoromethoxy)-11-[2-(tetrahydro-2H-pyran-4-yl)pyrimidin-5-y1]-6,7-dihydro-
7,14-
methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one;
(7R,14R)-11-[2-(2-
aminopropan-2-yl)pyrimidin-5-y1]-1-(difluoromethoxy)-10-fluoro-6-trideutero
methyl-6,7-
dihydro-7,14-methanobenzimidazo [1,2-h] [2,5]benzodiazocin-5(14H)-one;
(7R,14R)- 10-
fluoro-11- [2-(2-hydroxypropan-2-yl)pyrimidin-5 -y1]-1-(tri fluoromethoxy)-6,7-
dihydro-
7,14-methanoben zimidazo[1,2-b][2,5]benzodia7ocin-5(14H)-one;
(7R,1410-1-
(difluoromethoxy)-10-fluoro-11-(3-(2-hydroxypropan-2-ypazetidin-l-y1)-6,7-
dihydro-
7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one; (7R,14R)-
11-((2-
aminopropan-2-yl)pheny1)-10-fluoro-1-(trifluoromethoxy)-6,7-dihydro-7,14-
methanobenzimidazo [1,2-b] [2 ,5]benzodiazocin-5(14H)-one;
(7R,14R)-11-((2-
aminopropan-2-yl)pheny1)-1-(difluoromethoxy)-10-fluoro-6,7-dihydro-7,14-
methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(1411)-one;
(7R,14R)-1-
(difluoromethoxy)-10-fluoro-1142-(2-hydroxypropan-2-y1)-4-(tetrahydrofuran-3-
y1)pyrimidin-5-y1]-6,7-dihydro-7,14-methanobenzimidazo[1,2-
b][2,5]benzodiazocin-
5(14H)-one; (7R,14R)-1-(difluoromethoxy)-1144-(difluoromethyl)-2-(2-
hydroxypropan-
2-yl)pyrimidin-5 -y1]-10-fluoro-6,7-dihydro-7,14-methanobenzimidazo [1,2-
b] [2,5]benzodiazocin-5 (141/)-one;
(7R,14R)-1-(difluoromethoxy)-6-ethy1-1142-(2-
hydroxypropan-2-yl)pyrimidin-5-y1]-7-methy1-6,7-dihydro-7,14-
methanobenzimidazo [1,2-
b] [2,5]benzodiazocin-5(1411)-one; (7R,14R)-1142-(2-aminopropan-2-yl)pyrimidin-
5 -yl] -
1-(difluoromethoxy)-7-methyl-6,7-dihydro-7,14-methanobenzimidazo [1,2-
b] [2,5]ben zodiazocin-5(1411)-one;
(7R,14R)-1-(difluoromethoxy)-6-ethy1-1142-(2-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 52 -
hydroxypropan-2-yl)pyrimidin-5-y1]-7-incthyl-6,7-dihydro-7,14-
methanobenzimidazo [1,2-
b] [2,5 ]benzodiazo cin-5 (141-1)-one ; (7R,14R)-1142-(2-aminopropan-2-
yl)pyrimidin-5-y1]-
1-(di fluoromethoxy)-7-methyl-6,7-di hydro -7,14-methanobenzimi dazo [1,2-
b] [2,5 ]benzodiazocin-5 (1411)-one; (7R,14R)-1 -(difluoromethoxy)-11 -(2- {2-
[di(prop-2-en-
1 -yl)amino]prop an-2-y1} pyrimidin-5-y1)-6,7-dihydro-7,14-methanobenzimidazo
[1,2-
b] [2,5]benzodiazocin-5(141/)-one; (7R ,14R)-1-(difl uoromethoxy)-6-
trideuteromethy1-11 -
[6-(S-methylsulfo nimido y 1)pyridin-3 -y1]-6,7-dihydro-7,14-methanobenz
imidazo [1,2-
b] [2,5 Thenzodiazocin-5 (1411)-one;
(7R,14R)-1-(difluoromethoxy)-11-[6-(S-
methylsul fonimido yl)pyridin-3 -yl] -6,7-dihydro -7,14 -methanobenzimidazo
[1,2-
b] [2,5 ]benzodiazo cin-5 (1411)-one; (7R,14R)-1-(difluoromethoxy)-11-(2- {2-
[di(prop-2-en-
1 -y0amino]prop an-2-y1} pyrimidin-5-y1)-6,7-dihydro-7,14-methanobenzimidazo
[1 ,2-
b] [2,5 ]benzodiazo cin-5 (1411)-one; (7R,14R)-1-(difluoromethoxy)-10-fluoro-
11-(2-((2R *)-
hydroxybutan-2-yOpyrimidin-5-y1)-6,7-dihydro-7,14-
methanobenzo[f]benzo[4,5]imidazo [1,2-a] [1,4]diazocin-5(1411)-one;
(7R,14R)-1-
(difluoromctho xy)- 10-fluoro- 1 1-(2-((2S*)-hydroxybutan-2-yl)pyrimidin-5-y1)-
6,7-
dihydro-7,14-methanobenzo [I]benzo [4,5 ]imidazo [1,2-a] [1,4]diazocin-5(14H)-
one; 2- {5-
[(7R,14R)-1-(di fluoromethoxy)-10-fluoro-5-oxo-5,6,7,14-tetrahydro-7,14-
methanobenzimidazo[l ,2-h][2,5]benzodiazocin-1 1 -yl]pyrimidin-2-ylIpropan-2-
y1
phosphate, disodium salt; 2- {5-[(7R,14R)-1-(difluoromethoxy)-10- fluoro-6-
methy1-5 -oxo-
5,6,7,14-tetrahydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-11-
yl]pyrimidin-
2-yl}propan-2-y1 phosphate, disodium salt; ammonium 2-(5-((7R,14R)-1-
(difluoromethoxy)-10- fluoro-5-oxo -5,6,7,14-tetrahydro-7,14-
methanob enzo enzo [4,5]imidazo [1,2 -a] [1,4] diazo cin-11-yl)pyrimidin-2-
yl)propan-2-y1
sulphate; and 4-((2-
(5-((7R,14R)-1-(difluoromethoxy)- 10-fluoro-5-oxo-5 ,6,7,14 -
tetrahydro-7,14-methanobenzo [f]b enzo [4 ,5] imi dazo [1,2-a] [1,4]diazocin-
11-yl)pyrimi din-
2-yl)prop an-2-yl)oxy)-4-oxobutano ic acid.
The compounds in accordance with the present invention are beneficial in the
treatment and/or prevention of various human ailments. These include
autoimmune and
inflammatory disorders; neurological and neurodegenerative disorders; pain and
nociceptive disorders; cardiovascular disorders; metabolic disorders; ocular
disorders; and
oncological disorders.
Inflammatory and autoimmune disorders include systemic autoimmune disorders,
autoimmune endocrine disorders and organ-specific autoimmune disorders.
Systemic
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 53 -
autoimmune disorders include systemic lupus erythematosus (SLE), psoriasis,
psoriatic
arthropathy, vasculitis, inflammatory myopathy (including polymyositis,
dermatomyositis,
inclusion body myositis), scleroderma, multiple sclerosis, systemic sclerosis,
ankylosing
spondylit is, rheumatoid arthritis, non-specific inflammatory arthritis,
juvenile
inflammatory arthritis, juvenile idiopathic arthritis (including
oligoarticular and
polyarticular forms thereof), anaemia of chronic disease (ACD), Still's
disease (juvenile
and/or adult onset), Behget's disease and Sjogren's syndrome. Autoimmune
endocrine
disorders include thyroiditis. Organ-specific autoimmune disorders include
Addison's
disease, haemolytic or pernicious anaemia, acute kidney injury (AKI; including
cisplatin-
induced AKI), diabetic nephropathy (DN), obstructive uropathy (including
cisplatin-
induced obstructive uropathy), glomerulonephritis (including Goodpasture's
syndrome,
immune complex-mediated glomerulonephritis and antineutrophil cytoplasmic
antibodies
(ANCA)-associated glomerulonephritis), lupus nephritis (LN), minimal change
disease,
Graves' disease, idiopathic thrombocytopenic purpura, inflammatory bowel
disease
(including Crohn's disease, ulcerative colitis, indeterminate colitis and
pouchitis),
pemphigus, atopic dermatitis, autoimmune hepatitis, primary biliary cirrhosis,
autoimmune
pneumoniti s, autoimmune cardit is, myasthenia gravis, spontaneous
infertility,
osteoporosis, osteopenia, erosive bone disease, chondritis, cartilage
degeneration and/or
destruction, fibrosing disorders (including various forms of hepatic and
pulmonary
fibrosis), asthma, rhinitis, chronic obstructive pulmonary disease (COPD),
respiratory
distress syndrome, sepsis, fever, muscular dystrophy (including Duchenne
muscular
dystrophy) and organ transplant rejection (including kidney allografl
rejection).
Additional inflammatory and autoimmunc disorders include scleritis,Takayasu
arteritis, giant cell arteritis scleritis, hidradenitis suppurativa, pyoderma
gangrenosum,
sarcoidosis, polymyalgia rheumatica, axial spondyloarthritis. Neurological and
neurodegenerative disorders include Alzheimer's disease, Parkinson's disease,
Huntington's disease, ischaemia, stroke, amyotrophic lateral sclerosis, spinal
cord injury,
head trauma, seizures and epilepsy.
Cardiovascular disorders include thrombosis, cardiac hypertrophy,
hypertension,
irregular contractility of the heart (e.g. during heart failure), and sexual
disorders
(including erectile dysfunction and female sexual dysfunction). Modulators of
TNFa
function may also be of use in the treatment and/or prevention of myocardial
infarction
(see J.J. Wu et al.,.1A MA, 2013, 309, 2043-2044).
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 54 -
Metabolic disorders include diabetes (including insulin-dependent diabetes
mellitus
and juvenile diabetes), dyslipidemia and metabolic syndrome.
Ocular disorders include retinopathy (including diabetic retinopathy,
proliferative
retinopathy, non-proliferative retinopathy and retinopathy of prematurity),
macular
.. oedema (including diabetic macular oedema), age-related macular
degeneration (ARMD),
vascularisation (including corneal vascularisation and neovascularisation),
retinal vein
occlusion, and various forms of uveitis (including iritis) and keratitis
Oncological disorders, which may be acute or chronic, include proliferative
disorders, especially cancer, and cancer-associated complications (including
skeletal
complications, cachexia and anaemia). Particular categories of cancer include
haematological malignancy (including leukaemia and lymphoma) and non-
haematological
malignancy (including solid tumour cancer, sarcoma, meningioma, glioblastoma
multiforme, neuroblastoma, melanoma, gastric carcinoma and renal cell
carcinoma).
Chronic leukaemia may be myeloid or lymphoid. Varieties of leukaemia include
lymphoblastic T cell leukaemia, chronic myelogenous leukaemia (CML), chronic
lymphocytic/lymphoid leukaemia (CLL), hairy-cell leukaemia, acute
lymphoblastic
leukaemia (ALL), acute myelogenous leukaemia (AML), myelodysplastic syndrome,
chronic neutrophilic leukaemia, acute lymphoblastic T cell leukaemia,
plasmacytoma,
innmunoblastic large cell leukaemia, mantle cell leukaemia, multiple myeloma,
acute
megakaryoblastic leukaemia, acute megakaryocytic leukaemia, promyelocytic
leukaemia
and erythroleukaemia. Varieties of lymphoma include malignant lymphoma,
Hodgkin's
lymphoma, non-Hodgkin's lymphoma, lymphoblastic T cell lymphoma, Burkitt's
lymphoma, follicular lymphoma, MALT1 lymphoma and marginal zone lymphoma.
Varieties of non-haematological malignancy include cancer of the prostate,
lung, breast,
rectum, colon, lymph node, bladder, kidney, pancreas, liver, ovary, uterus,
cervix, brain,
skin, bone, stomach and muscle. Modulators of TNFa function may also be used
to
increase the safety of the potent anticancer effect of TNF (see F.V.
Hauwermeiren et al.,J.
Clin. Invest., 2013, 123, 2590-2603).
Compounds according to the present invention can be particularly beneficial
for the
treatment of rheumatoid arthritis, psoriasis, psoriatic arthropathy, axial
spondyloarthritis,
juvenile idiopathic arthritis, Crohn's disease, ulcerative colitis, uveitis,
Behyet's disease
and Takayasu arteritis.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 55 -
The present invention also provides a pharmaceutical composition which
comprises a compound in accordance with the invention as described above, or a
pharmaceutically acceptable salt or solvate thereof, in association with one
or more
pharmaceutically acceptable carriers.
Pharmaceutical compositions according to the invention may take a form
suitable
for oral, buccal, parenteral, nasal, topical, ophthalmic or rectal
administration, or a form
suitable for administration by inhalation or insufflation.
For oral administration, the pharmaceutical compositions may take the form of,
for
example, tablets, lozenges or capsules prepared by conventional means with
pharmaceutically acceptable excipients such as binding agents (e.g.
pregelatinised maize
starch, polyvinylpyrrolidone or hydroxypropyl methyl cellulose); fillers (e.g.
lactose,
microcrystalline cellulose or calcium hydrogenphosphate); lubricants (e.g.
magnesium
stearate, talc or silica); disintegrants (e.g. potato starch or sodium
glycollate); or wetting
agents (e.g. sodium lauryl sulphate). The tablets may be coated by methods
well known in
the art. Liquid preparations for oral administration may take the form of, for
example,
solutions, syrups or suspensions, or they may be presented as a dry product
for constitution
with water or other suitable vehicle before use. Such liquid preparations may
be prepared
by conventional means with pharmaceutically acceptable additives such as
suspending
agents, emulsifying agents, non-aqueous vehicles or preservatives. The
preparations may
also contain buffer salts, flavouring agents, colouring agents or sweetening
agents, as
appropriate.
Preparations for oral administration may be suitably formulated to give
controlled
release of the active compound.
For buccal administration, the compositions may take the form of tablets or
lozenges formulated in conventional manner.
The compounds of formula (I) may be formulated for parenteral administration
by
injection, e.g. by bolus injection or infusion. Formulations for injection may
be presented
in unit dosage form, e.g. in glass ampoules or multi-dose containers, e.g.
glass vials. The
compositions for injection may take such forms as suspensions, solutions or
emulsions in
oily or aqueous vehicles, and may contain formulatory agents such as
suspending,
stabilising, preserving and/or dispersing agents. Alternatively, the active
ingredient may
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 56 -
be in powder form for constitution with a suitable vehicle, e.g. sterile
pyrogen-free water,
before use.
In addition to the formulations described above, the compounds of formula (1)
may
also be formulated as a depot preparation. Such long-acting formulations may
be
administered by implantation or by intramuscular injection.
For nasal administration or administration by inhalation, the compounds
according
to the present invention may be conveniently delivered in the form of an
aerosol spray
presentation for pressurised packs or a nebuliser, with the use of a suitable
propellant, e.g.
dichlorodifluoromethane, fluorotric hlo ro methane, dichlorotetrafluoroethane,
carbon
dioxide or other suitable gas or mixture of gases.
The compositions may, if desired, be presented in a pack or dispenser device
which
may contain one or more unit dosage forms containing the active ingredient.
The pack or
dispensing device may be accompanied by instructions for administration.
For topical administration the compounds of use in the present invention may
be
conveniently formulated in a suitable ointment containing the active component
suspended
or dissolved in one or more pharmaceutically acceptable carriers. Particular
carriers
include, for example, mineral oil, liquid petroleum, propylene glycol,
polyoxycthylene,
polyoxypropylene, emulsifying wax and water. Alternatively, the compounds of
use in the
present invention may be formulated in a suitable lotion containing the active
component
suspended or dissolved in one or more pharmaceutically acceptable carriers.
Particular
carriers include, for example, mineral oil, sorbitan monostearate, polysorbate
60, cetyl
esters wax, cetearyl alcohol, benzyl alcohol, 2-octyldodecanol and water.
For ophthalmic administration the compounds of use in the present invention
may
be conveniently formulated as micronized suspensions in isotonic, pH-adjusted
sterile
saline, either with or without a preservative such as a bactericidal or
fungicidal agent, for
example phenylmercuric nitrate, benzylalkonium chloride or chlorhexidine
acetate.
Alternatively, for ophthalmic administration compounds may be formulated in an
ointment
such as petrolatum.
For rectal administration the compounds of use in the present invention may be
conveniently formulated as suppositories. These can be prepared by mixing the
active
component with a suitable non-irritating excipient which is solid at room
temperature but
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 57 -
liquid at rectal temperature and so will melt in the rectum to release the
active component.
Such materials include, for example, cocoa butter, beeswax and polyethylene
glycols.
The quantity of a compound of use in the invention required for the
prophylaxis or
treatment of a particular condition will vary depending on the compound chosen
and the
condition of the patient to be treated. In general, however, daily dosages may
range from
around 10 ng/kg to 1000 mg/kg, typically from 100 ng/kg to 100 mg/kg, e.g.
around 0.01
mg/kg to 40 mg/kg body weight, for oral or buccal administration, from around
10 ng/kg
to 50 mg/kg body weight for parcnteral administration, and from around 0.05 mg
to
around 1000 mg, e.g. from around 0.5 mg to around 1000 mg, for nasal
administration or
administration by inhalation or insufflat ion.
If desired, a compound in accordance with the present invention may be co-
administered with another pharmaceutically active agent, e.g. an anti-
inflammatory
molecule.
It will be apparent to the person skilled in the art that there are various
synthetic
pathways that can lead to the compounds according to the invention. The
following
processes are aimed at illustrating some of these synthetic pathways but
should not be
construed in any way as a limitation on how the compounds according to the
invention
should be made.
It will also be apparent to the person skilled in the art that there may be
variations
in the synthetic pathways depending on the sub-classes of compounds of formula
(1).
Compounds of formula (I) above, may be prepared by a process which comprises
intramolecular cyclisation or includes reaction of an intermediate of formula
(III),
12
X'
Qi
(III)
R¨-- R8
R6 R7
wherein E, Z, R5, R6, R7,R8 and R'2 are as defined above;
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 58 -
X1 represents hydroxy, -SH, CH2-0H, -CO2H, -NHRr, -NHRg, -C(0)-NHItr, Y, or -
CH2-Y;
Q1 represents hydrogen, hydroxy, halogen, amino, -SR', -CO2H, -CH2-Y,
or ¨CH(OH)-CF3
Y represents a suitable leaving group;
Rr and Rg are as defined above,
Ri represents hydrogen, methyl, -CH-C(0)-0-C2H5, or -(CH2)2- C(0)-0-CH2-CH
(CH2CH:3) [(CH2)3CH3]; and
Ri represents hydrogen or methyl.
Suitably, Y represents halogen or (C1õ6 )alkylsulphonate.
Appositely, Y represents bromo or methylsulphonate.
Suitably, R and Rg represent hydrogen.
Compounds of formula (1) wherein R12 represents hydrogen and -X-Q- represents
-0- may be prepared by a process which comprises intramolecular cyclization of
an
intermediate of formula (III) wherein R12 represents hydrogen, X1 represents a
leaving
group Y, e.g. halogen, preferably bromo, and Q1 represents hydroxy, in the
presence of a
base, for example sodium hydride or silver carbonate.
Alternatively, compounds of formula (I) wherein R12 represents hydrogen and -
X-Q- represents -0- may be prepared by a process which comprises
intramolecular
cyclization of an intermediate of formula (111) wherein 102 represents
hydrogen, X1
represents hydroxy and Q1 represents a leaving group Y, e.g. halogen,
preferably bromo,
in the presence of a base, e.g. an inorganic base such as cesium carbonate,
and copper
iodide, at elevated temperature.
Compounds of formula (I) wherein 102 represents hydrogen and -X-Q- represents
-0-C(CH-CN)- may be prepared by a process which comprises intramolecular
cyclization
of an intermediate of formula (III) wherein R12 represents hydrogen,X1
represents hydroxy
and Q1 represents ¨0O2H, in the presence of cyanomethylenetributylphosphorane.
Such intramolecular cyclization is conveniently performed at elevated
temperature
in a suitable solvent, e.g., toluene.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 59 -
Compounds of formula (1) wherein R12 represents hydrogen and ¨X-Q- represents
-0-C(0)- may be prepared by the same process as described above for compounds
of
formula (1) wherein 11.12 represents hydrogen and ¨X-Q- represents ¨0-C(CH-CN)-
followed by treatment with a base, e.g., potassium hydroxide.
Alternatively, compounds of formula (I) wherein RI2 represents hydrogen and ¨
X-Q- represents -0-C(0)- may be prepared prepared by a process which comprises
intramolecular cyclization of an intermediate of formula (III) wherein R'2
represents
hydrogen, X' represents hydroxy and Q1 represents ¨CO2H, in the presence of an
acid,
e.g. a mineral acid, in a suitable solvent.
Compounds of formula (I) wherein R12 represents hydrogen and ¨X-Q- represents
-C(0)-0- may be prepared by a process which comprises intramolecular
cyclization of an
intermediate of formula (III) wherein R12 represents hydrogen, X1 represents
¨CO2H and
Q' represents -OH, in the presence of thionyl chloride, or alternatively, by
using a suitable
coupling reagent, according to methods known to the person skilled in the art.
Compounds of formula (I) wherein R12 represents hydrogen and ¨X-Q- represents
-S-, may be prepared by a process which comprises intramolecular cyclization
of an
intermediate of formula (III) wherein X1 represents -SH and Q' represents
halogen, in the
presence of a transition metal catalyst, according to a method analogous to
that described
in Stambuli J. et al, I Org. Chem., 2009, 74, 4005-4008.
Alternatively, compounds of formula (I) wherein RI2 represents hydrogen and -X-
Q- represents -S-, may be prepared by a process which comprises reacting an
intermediate
of formula (III) wherein R12 represents hydrogen, X' represents -OH and Q'
represents -S-
(CH2)2-C(0)-0-CH2- CH (CH2CH3) [(CH2)3CH3], with methanesulphonylchloride in
the
presence of a base, e.g. N,N-diisopropylethylamine, in a suitable solvent,
e.g.
tetrahydrofuran, to afford the corresponding compound wherein X' represents a
mesylate
moity, followed by reaction with a solution of sodium ethoxide.
Compounds of formula (D wherein R12 represents hydrogen and -X-Q- represents -
N(R), may be prepared by a process which comprises intramolecular cyclization
of an
intermediate of formula (III) wherein R12 represents hydrogen,X1 represents -
NHRg and
Q1 represents halogen, in the presence of a suitable transition metal
catalyst, according to
methods known to the person skilled in the art.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 60 -
The intramolecular cyclization may be conveniently effected in the presence of
palladium(11)acetate and (+/-)-2,2'-bis(diphenylphosphino)1,1'-binaphthyl, in
the presence
of base, e.g., potassium carbonate or cesium carbonate, in a suitable solvent,
e.g., toluene,
at elevated temperature.
Alternatively, compounds of formula (I) wherein R'2 represents hydrogen and -X-
Q- represents -N(R), and Rg represents hydrogen, may be prepared by a process
which
comprises intramolecular cyclization of an intermediate of formula (III)
wherein R'2
represents hydrogen, X' is a leaving group Y, e.g. methylsulphonate, and Q'
represents
amino. The reaction is conveniently effected by first protecting the amino
group of Q1 with
a suitable protecting group, e.g. tert-butoxy carbonyl, according to methods
known to the
person skilled in the art, followed by subsequent addition of a suitable base,
e.g. sodium
hydride in a suitable solvent, e.g. dimethyl foiiiiamide.
Alternatively, compounds of formula (I) wherein R12 represents hydrogen,-X-Q-
represents -N(Rg)-, and Rg represents -00-(C3_7)heterocycloalkyl, may be
prepared by a
process which comprises intramolecular cyclization of an intermediate of
formula (III)
wherein It' represents hydrogen,X1 represents -NHRg , wherein Rg is as defined
above,
and Q1 represents halogen. The reaction is conveniently effected by addition
of a suitable
base, e.g. cesium acetate, and cuprous iodide in a suitable solvent, e.g.
dimethylsulfoxide,
at elevated temperature. Compounds of formula (1) wherein R'2 represents
hydrogen,-X-
Q- represents -N(Rg)-, and Rg represents respectively (C24alkoxycarbonyl,
heteroaryl¨
S02-heteroaryl or ¨S02-aryl may be prepared via an analogous method to the one
described here above for Rg representing -00-(C3_7)heterocycloalkyl.
Compounds of formula (I) wherein R'2 represents hydrogen or methyl, ¨X-Q-
represents -N(Rf)-C(0)-, may be prepared by a process which comprises
intramolecular
cyclization of an intermediate of formula (111) wherein R12 represents
hydrogen or methyl,
X' represents -NFIRf and Q1 is halogen, preferably chloro, in the presence of
carbon
monoxide and a transition metal catalyst.
The reaction is conveniently carried out at elevated temperature, e.g. 100 C
or
150 C, and under an elevated pressure of carbon monoxide, in a suitable sovent
solvent,
e.g., a cyclic ether such as 1,4-dioxane, or dimethylsulfoxide or
dimethylacetamide
The transition metal catalyst of use in the above reaction is suitably
selected from
dichloro [bis(dicyclohexylphosphino)propa,ne] palladium(1I) ,
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 61 -
[bis(diphenylphosphino)xanthenc] palladium(II) and 2,2-dichloro-1,1,3,3-
tetracyclohexy1-
1V, 3X5 palladacyclohexane.
Alternatively, a solution of palladium (II) acetate and 4,5-
bis(diphenylphosphino)-
9,9-dimethylxanthene in a suitable solvent may be used.
An analogous reaction may be performed using molybdenum hexacarbonyl as an
alternative source of carbon monoxide.
Such intramolecular cyclization is conveniently performed in the presence of a
base, e.g. an inorganic base such as sodium carbonate or by activation using
molecular
sieves.
Alternatively, compounds of formula (I) wherein, ¨X-Q- represents -N(Rf)-C(0)-
,
may be prepared by a process which comprises intramolecular cyclization of an
intermediate of formula (III) wherein, X1 represents -NHRf , Rf represents
hydrogen, and
Q1 is -COOH, in the presence of 4-methylmorpholine and (1-cyano-2-cthoxy-2-
oxoethylidenaminooxy) dimethylamino-morpholinocarbenium hexatluorophosphate
(COMU). The reaction is conveniently effected in acetonitrile.
Compounds of formula (I) wherein R12 represents hydrogen and -X-Q- represents
may be prepared by a process which comprises intramolecular cyclization of
an intermediate of formula (III) wherein R12 represents hydrogen , X1 is -C(0)-
NH(Rf)
and Q1 is halogen, preferably bromine, in the presence of a suitable coupling
reagent,
.. according to methods known to the person skilled in the art.
Alternatively, compounds of formula (I) wherein R12 represents hydrogen, -X-Q-
represents -C(0)-N(Rf)-, and Rfrepresents hydrogen, may be prepared by a
process which
comprises intramolecular cyclization of an intermediate of formula (III)
wherein R12
represents hydrogen, X1 represents -CO2H and Q1 represents amino. The reaction
may be
conveniently effected with a suitable coupling agent, e.g., 1-ethyl-3-(3-
dimethylaminopropyl)carbodiimide (EDC), according to methods known to the
person
skilled in the art.
Compounds of foimula (I) wherein R12 represents hydrogen and -X-Q- represents
-N(10-S02- may be prepared by a process which comprises intramolecular
cyclization of
an intermediate of formula (III) wherein R12 represents hydrogen, X1
represents -NHRf
and Q1 represents ¨SH, in the presence of hydrogen peroxide and thionyl
chloride
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 62 -
according to a method analogous to that described by K. Bahrami, M. M.
Khodaci, M.
Soheilizadõ1. Org. Chenz., 2009, 74, 9287-9291.
The reaction is conveniently performed at room temperature in a suitable
solvent,
e.g. an apolar solvent such as acetonitrile and in the presence of an organic
base, e.g.
pyridine.
Compounds of formula (I) wherein R12 represents hydrogen, -X-Q- represents -
S02-N(10-, and represents hydrogen, may be prepared by a process analogous to
the
one described above for ¨X-Q- which represents -N(R)-S02-, from an
intermediate of
formula (III) wherein R'2 represents hydrogen, X' represents -SH and Q'
represents
amino. The reaction is conveniently effected by first protecting the amino
group of Q' with
a suitable protecting group according to methods known to the person skilled
in the art.
Compounds of formula (I) wherein R12 represents hydrogen, -X-Q- represents -
CH2-CH2- may be prepared by a process which comprises intramolecular
cyclization of an
intermediate of formula (III) wherein R12 represents hydrogen , X' represents -
CH2-CO2H
and Q' represents hydrogen, for example applying Friedel Crafts reaction
conditions, in
the presence of polyphosphoric acid. The resulting intermediate, wherein -X-Q-
represents
-CH2-C(0)- is subsequently reduced according to methods known to the person
skilled in
the art.
Compounds of formula (I) wherein -X-Q- represents R12 represents hydrogen and
-0-CH2- may be prepared by a process which comprises intramolecular
cyclization of an
intermediate of formula (III) wherein R12 represents hydrogen, X1 represents
hydroxy and
Q' represents -CH2-Y, wherein Y is a leaving group, e.g. halogen, preferably
bromo, in the
presence of a suitable base, according to methods known to the person skilled
in the art.
Compounds of formula (I) wherein R12 represents hydrogen and -X-Q- represents
-CH2-0- may be prepared by a process which comprises intramolecular
cyclization of an
intermediate of formula (III) wherein R12 represents hydrogen, X' represents -
CH2-0H
and Q' represents halogen, preferably bromo. This reaction is conveniently
effected in the
presence of a suitable transition metal catalyst, e.g. palladium (II) or
copper (II) catalyst,
according to methods known to the person skilled in the art.
Compounds of formula (I) wherein R12 represents hydrogen and -X-Q- represents -
S-CH2 may be prepared by a process analogous to the process of preparation of
compounds of formula (I) wherein -X-Q- represents -0-CH2- starting from
intermediates
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 63 -
of formula (111) wherein R'2 represents hydrogen, X1 represents -SH and Q1
represents -
CH2-Y.
Compounds of formula (I) wherein R12 represents hydrogen and -X-Q- represents -
CH2-S- may be prepared by a process which comprises intramolecular cyclization
of an
intermediate of formula (III) wherein R'2 represents hydrogen, X1 is -CH2-Y, Y
is a
suitable leaving group, e.g. halogen, and Q' represents -SH, in the presence
of suitable
base according to methods known to the person skilled in the art.
Compounds of formula (I) wherein R'2 represents hydrogen, -X-Q- represents -
CH2-N(R5)-, and Rg represents hydrogen, may be prepared by a process which
comprises
intramolecular cyclization of an intermediate of formula (III) wherein IV2
represents
hydrogen , X' is -CH2-Y, wherein Y is a suitable leaving group, e.g.
methylsulphonate,
and Q' represents amino. The reaction is conveniently effected by first
protecting the
amino group of Q' with a suitable protecting group, e.g. tert-butoxy carbonyl,
according to
methods known to the person skilled in the art, followed by subsequent
addition of a
suitable base, e.g. sodium hydride in a suitable solvent, e.g. dimethyl
formamide.
Compounds of formula (I) wherein R12 represents hydrogen, -X-Q- represents -
N(Rg)-CH2- and Rg represents hydrogen may be prepared by a process involving
intramolecular cyclization of the corresponding compound of formula (III)
wherein R'2
represents hydrogen, X1 represents -NH(R) and wherein Q1 represents formyl.
The
reaction is conveniently effected by (i) reaction with an acid, e.g.
trifluoroacetic acid, in a
suitable solvent, e.g. dichloromethane and (ii) reduction of the compound
obtained as a
result of step (i) with an appropriate reducing agent, e.g. polymer supported
cyano
borohydride or borane-dimethylsulphide complex, in a suitable solvent, e.g.
tetrahydrofuran or a mixture of tetrahydrofuran and ethanol.
Compounds of formula (I) wherein 102 represents hydrogen, -X-Q- represents -
N(Rg)-CH(CH3)- and Rg represents hydrogen may be prepared according to a
method
analogous as the one described here above for compounds of formula (I) wherein
-X-Q-
represents -N(Rg)-CH2-, but from intermediate of formula (III) wherein Q1
represents
acetyl.
Compounds of formula (I) wherein R" represents hydrogen, -X-Q- represents -
N(R8)-CH(CF3)- and Rg represents hydrogen may be prepared according to a
method
analogous to the one described here above for compounds of foimula (I) wherein
R'2
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 64 -
represents hydrogen and -X-Q- represents -N(Rg)-CH2-, by reacting compound
obtained as
a result of step (i) with (trifluoromethyptrimethyl silane, in the presence of
trifluoroacetic
acid and potassium hydrogen fluoride, in a suitable solvent, e.g.
dimethylformamide.
Compounds of formula (I) wherein RI' represents hydrogen, -X-Q- represents ¨
N=S(0)(CH3)- may be prepared by a process involving intramolecular cyclization
of the
corresponding compound of formula (III) wherein, X1 represents -NH(Rg), Rg
represents
hydrogen, and wherein Q' represents ¨SCH3. The reaction is conveniently
effected by (i)
adding bromine in dichloromethanc followed by (ii) oxidation e.g. with m-
chloroperbenzoic acid.
Compounds of formula (I) wherein R'' represents hydrogen, -X-Q- represents ¨0-
CH(CF3)- may be prepared by a process involving intramolecular cyclization of
the
corresponding compound of formula (III) wherein, X' represents ¨hydroxy and
wherein
Q' represents ¨CH(OH)CF3. The reaction is conveniently effected using
cyanomethylenetributylphosphorane, in a suitable solvent, e.g.
tetrahydrofuran, at elevated
temperature, e.g. 100 C.
Compounds of formula (I) wherein R12 represents hydrogen, -X-Q- represents ¨0-
C(=CH7)- may be prepared by a process involving intramolecular cyclization of
the
corresponding compound of formula (III) wherein, X' represents halogen, e.g.
bromo, and
wherein Q1 represents ¨CO-Ri and R represents CH3.The reaction is conveniently
effected
in the presence of sodium hydride in a suitable solvent, e.g. tetrahydrofuran,
at low
temperature.
Intermediates of formula (III) wherein E represents (Ea) as defined above, and
X'
represents hydroxy, may be prepared by a process which comprises the
intramolecular
cyclisation and desilylation of an intermediate of formula (IV),
NO2 NC,OSi(CH3)3
Qi
(IV)
R2
R5
R8
R 1
R6
R7
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 65 -
wherein Z, R27 R57 R67 R77
R8 and Q1 are as defined above.
The reaction is suitably performed in the presence of tin(H) chloride at
elevated
temperature in a polar solvent, e.g. ethanol.
Intermediate (IV) as defined above may be prepared by a process comprising
reacting intermediate (V),
NO2
Q2
R2
Rs
Rs
RI
R6 R7
wherein Q2 represents -C(0)-H, and R1, R2, R5, R6, R7, R8 and Q1 are as
defined
above; with zinc iodide and triethylsilyl cyanide in the presence of a base,
e.g.
triethylamine.
Typically, the intermediate of formula (V) wherein Q2 represents -C(0)-H may
be
prepared from the corresponding intermediate wherein Q2 represents -CO2Rh and
Rh
represents Ci_6 alkyl, by reduction with a conventional reducing agent, e.g. a
metal
hydride, such as diisobutyl-aluminium hydride (D1BAL-11).
The intermediate of formula (V) wherein Q2 represents -CO2Rh may be obtained
by
a process which comprises reacting an intermediate of formula (VI) with an
intermediate
of formula (VII),
n2
No2
H2N
Qi
Li
R2 R5
Rs
(VI) (VII)
RI
R6
R7
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 66 -
wherein RI, R2, R5, R6, R7, R8, Z, Qi and Q2 are as defined above; and L1 is a
suitable leaving group, e.g. a halogen atom, for example bromine.
The reaction is conveniently performed in the presence of a base, e.g. an
inorganic
base, such as potassium carbonate, in a suitable solvent, e.g. an apolar
solvent such as
acetonitrile, at elevated temperature.
Intermediates of formula (VII) may be prepared by a multi-step process
starting
from an intermediate of formula (VIII),
0
Q1
R5
R8
(VIII)
R6
R7
wherein R5, R6, R7, R8 and Qi arc as defined above; which process comprises
the
following steps:
(i) Reaction of intermediate (VIII) with (S)-t-butylsulfinamide in the
presence
of K3PO4/1(1-1PO4 in a suitable solvent, e.g. THF;
(ii) Reacting the compound obtained from step (i) with a compound of
formula
L2-Z-Q2 , wherein Z and Q2 are as defined above and L2 is a suitable
leaving group, e.g. halogen, such as bromine, and activated zinc metal dust
prepared according to conditions described in HiIpert, H. et al, Journal of
Medicinal Chemistty, 2013, 56(10), 3980-3995, in the presence of
transition metal salt, e.g. copper chloride at elevated temperature;
(iii) Subsequent reaction with a strong mineral acid, e.g. hydrogen
chloride.
Intermediates of formula (VIII) wherein R5 represents halogen, e.g. chloro,
may be
transformed into the corresponding intermediate of formula (VIII) wherein R5
represents
difluoromethoxy by a process which comprises (i) reaction with potassium
hydroxide, in
water at low temperature and (ii) reaction with
diethyl(bromodifluoromethyl)phosphonate,
at low temperature.
Intermediates of formula (III) wherein E represents (Ea) as defined above, and
-X'
represents -NH(R) and Rg represents hydrogen, may be prepared by a process
which
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 67 -
comprises the reduction, intramolccular cyclization and dcsulfination of an
intermediate
of formula (IVa),
0
H
NO2 NC N _________________________________________ S
Qi
(IVa)
R2
R5 R8
R 1
R6
R7
wherein Z, R', R2, R5, R6, R7, R8 and Q1 are as defined above.
The reaction is suitably performed in the presence of tin(II) chloride,
followed by
addition of a strong acid, e.g. hydrogen chloride, at elevated temperature in
a polar solvent,
e.g. ethanol.
Alternatively, the reduction and cyclization may be performed by a process
involving (i) reduction using hydrogen under pressure, in the presence of zinc
bromine and
of platinum on charcoal and (ii) addition of a strong acid, e.g. hydrogen
chloride, at
elevated temperature in a polar solvent, e.g. ethanol.
Intermediates of formula (IVa), may be prepared by a multi-step process
starting
from corresponding intermediates (IVb),
NO2
V
Qi
(IVb)
R2
R5 R8
R 1
R7 R6
wherein Z, R1, R2, R5, R6, R7, R8 and Q1 are as defined above, and V
represents
CH¨CH2, which process comprises:
(i) Reacting intermediate (IVb) with sodium periodate, in the presence of
potassium
dioxide(dioxo)osmium hydrate and 2,6-dimethyl pyridine, followed by addition
of
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 68 -
sodium thiosulfate, to afford corresponding intermediates of formula (IVb)
wherein V
represents CH=0;
(ii) Reacting intermediates of formula (IVb) wherein V represents CH=0 with
(R)-2-
methylpropane-2-sulfinamide, in the presence of a transition metal catalyst,
eg. titanium
(IV) isopropoxide, in a suitable solvent, e.g. dichloromethane, to afford
corresponding
intermediate of formula (IVb) wherein V represents CH=N-(S0)-tert-butyl;
(iii) Further reaction with sodium cyanide, in the presence of scandium
triflate, in a
suitable solvent, e.g. tetrahydrofuran, to afford intermediates of formula
(IVa).
Intermediates of formula (IVb) wherein Z, Rl, R2, R5, R6, R7, le and 01 are as
defined above, and V represents CH=CH2, may be prepared by a process
comprising
reacting intermediates of formula (Vila) ,
V
F12N
Q1
R5 R8
(V11a)
R6 R7
wherein Z, V, R5, R6, R7, R8 and Q' are as defined above for intermediates of
formula (IVb), with an intermediate of formula (VI) as defined above, wherein
L' is a
halogen, e.g. fluorine, under conditions analogous to those described for the
preparation of
intermediates of formula (V).
Intermediates of formula (VIIa) may be prepared by a process analogous to the
one described for intermediates of formula (VII), but wherein Q2 is replaced
by V.
Intermediates of formula (III), wherein E represents (Eb) or (Ec), as defined
above,
and wherein X1 represents hydroxy may be prepared from intermediates of
formula (IIIA),
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 69 -
0
E z
(IIIA)
R6
R7
wherein R5, R6, R7, R, Z and Q1 are as defined above; by reduction of the
carbonyl
moiety according to methods known to the person skilled in the art.
Intermediates of formula (III) wherein X1 represents ¨N H(Rf), Rf represents
hydrogen and R12 represents methyl may be prepared from intermediate of
formula (IIIA)
using the following sequence of steps:
(i) Reacting intermediate of formula (IIIA) with 2-methyl-2-
propanesulfinamide in the presence of Titanium (IV) isopropoxidc, in a
solvent, e.g. tetrahydrofiwan, at a suitable temperature, e.g. 50 C;
(ii) Adding a solution of methylmagnesium bromide, at low temperature, in a
suitable solvent, e.g. dichloromethane;
(iii) Removing the tert-butyl sulphinyl moiety in the presence of a
strong acid,
e;g. HCI, in a suitable solvent, e.g. 1,4-dioxane.
Intermediates of formula (IIIA) may be prepared by a process which comprises
intramolecular cyclization of an intermediate of formula (IX),
R3
A 0
ORh (IX)
z,, 3
4
R Rs Q1
R6 R8
R7
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 70 -
wherein A is N or C-R2, Q3 is an electron withdrawing group, preferably an
ester moiety,
and R1, R2, R3,1(4, R5, R6, R7, R3, Rh, Z and Q1 are as defined above; in the
presence of a
base, in a suitable solvent at elevated temperature.
Intermediates of formula (IX) may be prepared by a process which includes
reacting an intermediate of formula (X) with an intermediate of formula (XI),
R3
0 0
A
R5 L3
ORh
(XI)
R6
R
z¨Q
R4
Q
(x)
R7
R8
wherein A, RI, R3, R4, R5, R6, R7, R8, Rh, Z, Q1 and Q3 are as defined above;
and L3
is a suitable leaving group, typically a halogen atom, e.g. bromo.
The reaction is conveniently effected at an elevated temperature in a suitable
solvent, e.g. a (CIA)alkanol such as ethanol, or an ether such as 1,4-dioxane
or
dimethoxyethane, and in the presence of magnesium sulphate.
Alternatively, intermediates of formula (IX), wherein Q3 is -CO2H, may be
prepared according to a process which comprises reacting an intermediate of
formula
(XII),
R3
0
A
R1 /L7N---- ORh
R4 (XII)
wherein A, le, R3, R4, and Rh are as defined above; with an intermediate of
formula (VIII) as defined above, in the presence of Meldrum's acid, according
to a method
analogous to the one described in international patent application WO
2009/156091 or by
M. Kerr et al. in J. Org. Chem 2013, 78, 10534.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 71 -
The reaction is conveniently effected in a suitable solvent e.g. acetonitrilc,
in the
the presence of proline and magnesium sulphate, at elevated temperature, e.g.
80 C.
Where they are not commercially available, the starting materials of formula
(VI),
(VIII), (X), (XI) and (XII) may be prepared by methods analogous to those
described in
the accompanying Examples, or by standard methods well known from the art.
Intermediates of formula (III) wherein X1 represents amino, may be prepared
from
intermediates of formula (III) wherein X' is hydroxy, by a process which
comprises (i)
treatment with diphenylphosphorylazide and 1,8-diazabicyclo[5.4.0]undec-7-ene,
in a
suitable solvent, e.g. tetrahydrofuran, at low temperature, e.g. 0 C, and (ii)
subsequent aza-
wittig reaction using triphenylphosphine, in a suitable solvent, e.g. a
mixture of water and
toluene.
Intermediates of formula (Ill), wherein E represents (Eb) or (Ec), as defined
above,
and wherein X' represents amino may be prepared from intermediates of formula
(111A),
wherein R5, R6, R7, R8, Z and Q1 are as defined above; by a process which
comprises
reacting intermediates of formula (IIIA) with a Ci_6 alkylsulfinamide, e.g.
(R)-2-
methylpropane-2-sulfinamide, in the presence of a transition metal catalyst,
e.g. titanium
tetrakis ethanolate, in a suitable solvent, e.g. dichloromethane, followed by
reduction with
a suitable reducing agent, e.g. sodium borohydride, in a suitable solvent,
e.g.
tctrahydrofuran.
Intermediates of formula (111) wherein X1 represents a leaving group Y, e.g.
halogen or (C1_6)alkylsulphonate, may be prepared from intermediates of
founula (111)
wherein X' is hydroxy, according to standard methods known to the person
skilled in the
art.
Intermediates of formula (III) wherein X' represents -SH, may be prepared from
intermediates of formula (III) wherein X' is hydroxy or a leaving group Y,
according to
standard methods known to the person skilled in the art.
Intermediates of formula (III) wherein X' represents -CO2H may be prepared by
hydrolysis of corresponding intermediates of formula (III) wherein X'
represents cyano,
according to standard methods known to the person skilled in the art.
Intermediates of formula (III) wherein X' represents cyano may be prepared by
nucleophilic substitution of intermediates of formula (III) wherein X'
represents a leaving
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 72 -
group Y, and Y represents (C14a1kylsulphonate-, according to standard methods
known to
the person skilled in the art.
Intermediates of formula (III) wherein X' represents -CH2OFI may be prepared
by
reduction of the corresponding intermediate of formula (111) wherein X'
represents -CO2H,
in the presence of a suitable reducing reagent, e.g., BH3.
Intermediates of formula (III) wherein X' represents -CH2-Y may be prepared
from
Intermediates of formula (III) wherein V represents -CH2OH, according to
methods
analogous to those described here above for intermediates of formula (III)
wherein X'
represents a leaving group Y, e.g. halogen or (C1_6)a1kylsulphonate.
Intermediates of formula (III) wherein X1 represents -NH(R) and Rg represents -
CO-(C3_7)heterocycloalkyl may be prepared by reacting compounds of formula
(III)
wherein X' represents -NH2with a (C3_7)heterocycloalkyl-COOH, in the presence
of a
base, e.g. N,N-diisopropylethylamine, and a coupling agent, e.g. HATU (N-
Rdimethylarnino)-1H-1,2,3-triazolo-[4,5-b]pyridin-1-ylmethylene]-N-
ethylmethanaminium hexafluorophosphate N-oxide), in a suitable solvent, e.g.
dimethylformamide.
Intermediates of formula (III) wherein Q' represents formyl may be prepared
from
intermediates of formula (III) wherein Q1 represents halogen, e.g. bromine, by
a process
involving (i) reaction with potassium vinylfluoroborate, in the presence of a
base and a
.. transition metal catalyst and (ii) reaction with sodium periodate and
osmium tetraoxide, in
the presence of a suitable solvent, e.g. a cyclic ether, such as 1,4-dioxane,
at a suitable
temperature, e.g. 0 C.
Suitable bases include inorganic bases, such as cesium carbonate and suitable
transition metal catalysts include 1,1'-bis(diphenylphosphino)ferrocene-
palladium(II)dichloride-dichloromethane complex.
Intermediates of formula (III) wherein Q1 represents acetyl may be prepared
from
intermediates of formula (III) wherein Q1 represents halogen, e.g. bromine, by
a process
involving (i) reaction with tributy1(1-ethoxyvinyl)tin, in the presence of
bis(triphenylphosphine)palladium(II)-dichloride, in a suitable solvent, e.g.
toluene, at
elevated temperature, and (ii) reaction with an acid, e.g. para-
toluenesulphonic acid.
Intermediates of formula (Ill) wherein Q1 represents -S-(CH2)-(CH2)-C(0)-0-CH2-
CH (CH2CH3) [(CH2)3CF13], may be prepared from intermediates of formula (III)
wherein
Q represents halogen, e.g. bromine, by a process involving reaction with 3-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 73 -
mercaptopropionic acid-2-ethylester, in the presence of a suitable transition
metal catalyst,
e.g. tris(benzylideneacetone)dipalladium(0) and 4,5-bis(diphenylphosphino)-9,9-
dimethylxanthene, in a suitable solvent, e.g. 1,4-dioxane, at elevated
temperature.
Similarly, intermediates of formula (III) wherein Q' represents -S-CH2-C(0)-0-
C2H5 [(CH2)3CH3], may be prepared from intermediates of formula (III) wherein
Q1
represents halogen, e.g. bromine, by a process involving reaction with
ethylthioglycolate,
in the presence of a suitable transition metal catalyst, e.g.
tris(benzylideneacetone)dipalladium(0) and 4,5-bis(diphenylphosphino)-9,9-
dimethylxanthene, in a suitable solvent, e.g. 1,4-dioxane, at elevated
temperature.
Intermediates of formula (III) wherein Q1 represents ¨S-CH3 may be prepared
from
intermediates of formula (III) wherein Q1 represents halogen, e.g. bromine, by
a process
which involves treatment with sodium thiomethoxide in a suitable solvent, e.g.
dimethylsulphoxide, at elevated temperature.
Intermediates of formula (III) wherein Q1 represents ¨CH(OH)-CF3 may be
prepared from intermediates of formula (III) wherein Q' represents ¨C(0)-H ,
by a process
which involves reaction with tetrabutylammoniumfluoride, followed by
(trifluoromethyptrimethylsilane, in a suitable solvent, e.g. tetrahydrofuran,
at low
temperature.
It will be understood that any compound of formula (I) initially obtained from
any
of the above processes may, where appropriate, subsequently be elaborated into
a further
compound of formula (I) by techniques known from the art.
As an alternative to the methods described here above, compounds of formula
(I)
wherein ¨X-Q- represents -0-CH2- may be prepared by a process involving
reduction of
compounds of formula (I) wherein -X-Q- represents -0-C(0)- carried out
according to the
method described in Sakai et al, J. Org. Chem. 2007, 72, 5920-5922.
Compounds of formula (1) wherein -X-Q- represents -N(Rg)-CH2- may be prepared
in a similar fashion from compounds of formula (I) wherein -X-Q- represents
represents -
N(R)-00- or under any other conditions used for reduction of a lactam, known
to the
person skilled in the art.
Compounds of formula (I) wherein -X-Q- represents - S-, -CH2-S- or -S-CH2- may
be transformed into compounds of formula (I) wherein -X-Q- represents
respectively -SO-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 74 -
or -S02-; -0-12-S0- or -CH2-S02-; -SO-CH2- or ¨S02-CH2-, by performing
oxidation
according to methods known to the person skilled in the art.
Compounds of formula (I) wherein -X-Q- represents -SO-, -CH2-S0- or -SO-CH2-
may be transformed into compounds of formula (I) wherein -X-Q- represents
respectively
-S(0)(NH)-, -CH2-S(0)(NH)-, or -S(0)(NH)-CH2-, by a method analogous to that
described in Okamura, H. et al, Organic Letters, 2004 , 6(8),1305-1307.
Compounds of formula (I) wherein ¨X-Q- represents ¨S- may be transformed into
compounds of formula (I) wherein ¨X-Q- represents ¨S(=N-CN)- by a process
involving
reaction with iodobenzene diacetate, in the presence of cyanamide. The
reaction is
conveniently effected in acetonitrile at low temperature, e.g. frc.
Compounds of formula (1) wherein -X-Q- represents -N(R)-C(0)- may be
converted into the corresponding compound of formula (1) wherein -X-Q-
represents -
N(le)-C(S)- by treatment with Lawesson's reagent according to methods known to
the
skilled person in the art.
Compounds of formula (I) wherein -X-Q- represents -NH- may be further
transformed into compounds of formula (I) wherein -X-Q- represents -N(R)-
wherein Rg
is an optionally substituted -00-(Ci_6)alkyl, by reaction with ehloracetyl
chloride, in a
suitable solvent, e.g. dichloromethane. Substituents may subsequently be
introduced on
the (CI)alkyl moiety by treatment with a suitable base according to a method
analogous
to the ones described in the accompanying Examples.
Compounds of formula (I) or intermediates of formula (III) wherein le or Rg
represent hydrogen, may further be transformed into the corresponding
compounds of
formula (I) or intermediates of formula (III) wherein le or Rg represent
optionally
substituted C1-6 alkyl, or its deuterated equivalent , by reaction with the
corresponding
optionally substituted C1-6 alkyl halide or deuterated equivalent, e.g. C1_6
alkyl iodide or
its deuterated equivalent, in the presence of a base, e.g. cesium carbonate or
potassium
bis(trimethylsilyl)amide (ICHMDS), in a suitable solvent, e;g.,
dimethylformamide or
THF
Compounds of formula (I) or intermediates of formula (III) wherein Rf or Rg
represent hydrogen, may further be transformed into the corresponding
compounds of
formula (I) or intermediates of formula (III) wherein le or Rg represent
acetyl by reaction
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 75 -
with acetic anhydride, in the presence of base, e.g. pyridine, in a suitable
solvent,
e.g.dichloromethane.
Compounds of formula (I) or intermediates of formula (III) wherein Rf or Rig
represent hydrogen, may further be transformed into the corresponding
compounds of
.. formula (1) or intermediates of formula (III) wherein Rf or R8 represent
methyl, by reaction
with formaldehyde, in a suitable solvent, e.g. 2,2,2-trifluoroethanol,
followed by reaction
with a suitable reducing agent, e.g. sodium borohydride.
Compounds of formula (I) or intermediates of formula (III) wherein Rf or R8
represent hydrogen, may further be transformed into the corresponding
compounds of
formula (I) or intermediates of formula (III) wherein RI or R8 represent
(Ci_6)alkyl-
sulphonyl by treatment with the appropriate (C1-6) alkylsulphonyl halide, e.g.
methane
sulphonyl chloride, in the presence of a suitable base, e.g. N,N-
diisopropylethylamine or
triethylamine, in a suitable solvent e.g. dichloromethane.
A compound of formula (I) or an intermediate of formula (III) which contains a
hydroxy group may be alkylated by treatment with the appropriate alkyl halide
in the
presence of a base, e.g. sodium hydride, or silver oxide.
A compound of formula (I) or an intermediate of formula (111) which contains
hydroxy may be converted into the corresponding fluoro-substituted compound by
treatment with diethylaminosulfur trifluoride (DAST) or bis(2-
methoxyethyl)aminosulfur
trifluoride (BAST). A compound of formula (1) which contains hydroxy may be
converted
into the corresponding difluoro-substituted compound via a two-step procedure
which
comprises: (i) treatment with an oxidising agent, e.g. manganese dioxide; and
(ii)
treatment of the carbonyl-containing compound thereby obtained with DAST.
A compound of formula (I) or an intermediate of formula (III) which contains
an
N-H moiety may be alkylated by treatment with the appropriate alkyl halide,
typically at
an elevated temperature in an organic solvent such as acetonitri le; or at
ambient
temperature in the presence of a base, e.g. potassium hydroxide, in a suitable
solvent, e.g.
THF, in the presence of tetra-butylammoniurn bromide; or at elevated
temperature in the
presence of a base, e.g. sodium hydride, with or without tetra-butylammonium
iodate, in a
suitable solvent, e.g. THF; or at elevated temperature in the presence of a an
alkali metal
carbonate such as potassium carbonate or cesium carbonate, in a suitable
solvent, e.g. a
dipolar aprotic solvent such as N,N-dimethylformamide. A compound of formula
(I)
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 76 -
which contains an N-H moiety may be methylated by treatment with formaldehyde
in the
presence of a reducing agent, e.g. sodium triacetoxyborohydride.
A compound of formula (I) or an intermediate of formula (III) which contains
an
N-H moiety may be acylated by treatment with the appropriate acid chloride,
e.g. acetyl
chloride, or with the appropriate carboxylic acid anhydride, e.g. acetic
anhydride, typically
at ambient temperature in the presence of a base, e.g. an organic base such as
triethylamine.
A compound of formula (I) or an intermediate of formula (III) which contains
an
N-H moiety may be converted into the corresponding compound wherein the
nitrogen
atom is substituted by C1-6 alkyl-sulphonyl, e.g. methylsulphonyl, by
treatment with the
appropriate Ci_is alkylsulphonyl chloride, e.g. methanesulphonyl chloride, or
with the
appropriate Ci_6 alkylsulphonic acid anhydride, e.g. methanesulphonic
anhydride, typically
at ambient temperature in the presence of a base, e.g. an organic base such as
triethylamine
or N,N-diisopropylethyl-amine.
A compound of formula (I) or an intermediate of formula (III) which contains
an
N-H moiety may be converted into the corresponding compound wherein the
nitrogen
atom is substituted by C1_6 alkoxy-carbonyl, e.g. methoxycarbonyl, by
treatment with the
corresponding C1-6 alkoxy-carbonyl halide, in the presence of a base, e.g.
potassium
carbonate, in a suitable solvent, e.g., N, N'-dimethylformamide.
A compound of formula (I) or an intermediate of formula (III) substituted by
amino
(-NH2) may be converted into the corresponding compound substituted by C1-6
alkylsulphonylamino, e.g. methylsulphonyl-amino, or bis[(Ci4alkylsulphonyll
amino, e.g.
bis(methylsulphonyl)amino, by treatment with the appropriate C1_6
alkylsulphonyl halide,
e.g. a C1_6 alkylsulphonyl chloride such as methanesulphonyl chloride, in the
presence of a
suitable base, e.g. N,N-diisopropylethylamine, in a suitable solvent e.g.
dichloromethane
Similarly a compound of formula (I) or an intermediate of formula (Ill)
substituted by amino may be transformed into the corresponding compound of
formula (I)
or intermediate of formula (III) substituted by NH-S02-(C3_7)cycloalkyl, NH -
S02-(C3_
7)heterocycloalkyl, NH-S02-aryl or NH-S02-heteroaryl respectively from the
corresponding (C3 7)cycloalkyl-sulphonylhalide, (C3_7)heterocycloalkyl-
sulphonyl halide,
aryl-sulphonyl-halide or heteroaryl-sulphonyl-halide.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 77 -
Similarly, a compound of formula (1) substituted by hydroxy (-OH) may be
converted into the corresponding compound substituted by Ci_6 alkyl-
sulphonyloxy, e.g.
methylsulphonyloxy, by treatment with the appropriate C1-6 alkyl-sulphonyl
halide, e.g. a
Ci alkylsulphonyl chloride such as methanesulphonyl chloride.
A compound of formula (I) or an intermediate of formula (III) substituted by
amino
(-NH2) may be converted into the corresponding compound substituted by (tert-
butyl)(dimethyl)silyloxyethyl-NH- by treatment with
(bromoethoxy)-tert-
butyldimethylsilane, in the presence of a suitable base, e.g. potassium
carbonate, in a
suitable solvent, e.g. dimethyl formamide, at elevated temperature.
A compound of formula (I) or an intermediate of formula (III) containing the
moiety -S- may be converted into the corresponding compound containing the
moiety -
S(0)- by treatment with 3-chloroperoxy-benzoic acid. Likewise, a compound of
formula
(I) containing the moiety -S(0)- may be converted into the corresponding
compound
containing the moiety -S(0)2- by treatment with 3-chloroperoxybenzoic acid.
Alternatively, a compound of formula (I) containing the moiety -S- may be
converted into
the corresponding compound containing the moiety
-S(0)2- by treatment with Oxone(R) (potassium peroxymonosulfate).
A compound of formula (I) or an intermediate of formula (III) containing an
aromatic nitrogen atom may be converted into the corresponding N-oxide
derivative by
treatment with 3-chloroperoxy-benzoic acid.
A compound of formula (I) or an intermediate of formula (III) which contains a
carbonyl may be converted into the corresponding alcohol by treatment with a
suitable
borohydride, e.g. lithium-tri-sec-butyl-borohydride or sodium borohydride, in
a suitable
solvent e.g. THF.A compound of formula (I) or an intermediate of formula (III)
wherein
le represents halogen, e.g. bromo, may be converted into the corresponding
compound
wherein RI represents an optionally substituted aryl or heteroaryl moiety by
treatment with
the appropriately substituted aryl or heteroaryl boronic acid or a cyclic
ester thereof
formed with an organic diol, e.g. pinacol, 1,3-propanediol or neopentyl
glycol. The
reaction is typically effected at elevated temperature in the presence of a
transition metal
catalyst, e.g. [1,1'-
bis(diphenylphosphino)ferrocene]dichloropalladium(II),
tetrakis(triphenylphosphine)palladium(0), or bis[3-
(diphenylphosphanyl)cyclopenta-2,4-
dien-1-yl]iron-dichloropalladium-dichloromethane complex, and a base, e.g. an
inorganic
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 78 -
base such as sodium carbonate, potassium carbonate or cesium carbonate, or
potassium
phosphate, in a suitable solvent, e.g. 1,4-dioxane or a mixture of 1,4-dioxane
and water.
Alternatively, a compound of formula (I) or an intermediate of formula (III)
wherein R1 represents halogen, e.g. bromo, may be converted into the
corresponding
compound wherein 10 represents an optionally substituted aryl or heteroaryl
moiety by
treatment with the appropriately substituted aryl or heteroaryl boronic acid,
in the presence
of a transition metal catalyst, e.g. tris(dibenzylideneacetone)dipalladium(0),
and
tricyclohexylphosphoniumtetrafluoroborate, in the presence of a base, e.g.
potassium
phosphate, in a suitable solvent, e.g. cyclic ether, such as 1,4-dioxanc. The
reaction is
conveniently effected at elevated temperature and microwave technology may be
used.A
compound of formula (I) wherein R' represents 2-oxo-(1II)-pyridinyl may be
obtained by
treatment of the corresponding compound of formula (I) wherein R1 represents 2-
methoxy-
pyridinyl, with pyridine hydrochloride at elevated temperature, e.g. 160 C.
A compound of formula (I) or an intermediate of formula (III) wherein RI
represents an ester moiety may be obtained by reacting the corresponding
compound of
formula (1) or the intermediate of formula (111) wherein R is halogen, e.g.
chloride, with a
base, e.g. sodium carbonate, and the corresponding alcohol moiety in the
presence of a
transition metal catalyst, typically bis(dicyclohexylphosphino)propane]
palladium(11).
A compound of formula (I) or an intermediate of formula (III) wherein R1
represents cyano may be obtained by reacting the corresponding compound of
formula (I)
or the intermediate of formula (111) wherein R' is halogen, e.g. chloride,
with zinc cyanide,
in the presence of a transition metal catalyst, e.g.
tetrakis(triphenylphosphine)palladium, in
a suitable solvent, e.g., N,N-dimethylformamide. The reaction is conveniently
effected at
elevated temperature, e.g. 180 C, using microwave technology.
In general, a compound of formula (I) containing a -C=C- functionality may be
converted into the corresponding compound containing a -CH-CH- functionality
by
catalytic hydrogenation, typically by treatment with a hydrogenation catalyst,
e.g.
palladium on charcoal, under an atmosphere of hydrogen gas, optionally in the
presence of
a base, e.g. an alkali metal hydroxide such as sodium hydroxide.
A compound of formula (I) containing an ester moiety, e.g. a C2_6
alkoxycarbonyl
group such as methoxycarbonyl or ethoxycarbonyl, may be converted into the
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 79 -
corresponding compound containing a carboxy (-CO2H) moiety by treatment with
an acid,
e.g. a mineral acid such as hydrochloric acid.
A compound of formula (I) containing an ester moiety, e.g. a C2-6
alkoxycarbonyl
group such as methoxycarbonyl or ethoxycarbonyl, may alternatively be
converted into the
.. corresponding compound containing a carboxy (-CO2H) moiety by treatment
with a base,
e.g. an alkali metal hydroxide selected from lithium hydroxide, sodium
hydroxide and
potassium hydroxide; or an organic base such as sodium methoxide or sodium
ethoxide.
A compound of formula (I) containing a carboxy (-CO2H) moiety may be
converted into the corresponding compound containing an amide moiety by
treatment with
the appropriate amine in the presence of a condensing agent such as 1-ethyl-3-
(3-dimethyl-
aminopropyl)carbodiimide.
A compound of formula (I) containing a carbonyl (C=0) moiety may be converted
into the corresponding compound containing a -C(CH3)(OH)- moiety by treatment
with
methylmagnesium bromide. Similarly, a compound of formula (I) containing a
carbonyl
(C=0) moiety may be converted into the corresponding compound containing a
-C(CF3)(OH)- moiety by treatment with (trifluoromethyl)trimethylsilane and
cesium
fluoride. A compound of formula (I) containing a carbonyl (C=0) moiety may be
converted into the corresponding compound containing a -C(CH2NO2)(OH)- moiety
by
treatment with nitromethane.
A compound of formula (1) containing a hydroxymethyl moiety may be converted
into the corresponding compound containing a formyl (-CHO) moiety by treatment
with
an oxidising agent such as Dess-Martin periodinane. A compound of formula (I)
containing a hydroxymethyl moiety may be converted into the corresponding
compound
containing a carboxy moiety by treatment with an oxidising agent such as
tetrapropylammonium perruthenate.
A compound of formula (I) containing an aryl or an heteroaryl moiety may be
transformed into the corresponding compound containing an aryl or heteroaryl
moiety
where a hydrogen atom has been substituted by a chloro or bromo substituent by
reaction
respectively with N-chlorosuccinimide or N-bromosuccinimide, in a suitable
solvent , e.g.
dimethylformamide, according to methods known to the person skilled in the
art.
A compound of formula (1) containing an aryl moiety bearing a difluoromethoxy
group may be transformed into the corresponding compound containing an aryl
moiety
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 80 -
where the difluoromethoxy group has been substituted by a hydroxy group, by
reaction
with sodiumbis(trimethylsilypamide, in a suitable solvent, e.g. THF, at low
temperature.
A compound of formula (I) containing an aryl or an heteroaryl moiety may be
transformed
into the corresponding compound containing an aryl or heteroaryl moiety where
a
hydrogen atom has been substituted by a trifluoromethyl substituent by
reaction
respectively with (i) trifluoroacetic acid in a suitable solvent, e.g.
acetonitrile, (ii) addition
of trifluoromethanesulphonyl chloride, followed by [4,4'-Bis(tert-butyl)-2,2'-
bipyri di ne] bis [3,5-difluoro-245-(trifluoromethyl)-2-pyridinyl]phenyl]
iridium(III)
hexafluorophosphate, according to conditions analogous to those described by
McMillan
ate al. in Nature, 2011, 480, 224.
In one embodiment, the present invention provides compounds of formula (I), in
particular
compounds of formula (IB), (IC) or (ID) as defined above, wherein R'
represents a
substituted heteroaryl selected from the groups represented by formula (i),
(ii), (iii), (iv)
and (v), and their respective corresponding salts represented by formula (ia),
(ib), (iia),
(iiia), (iva), (va), and (vb):
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
-81 -
RI RI
RIO
* .
N''....-"k 1\1------,
N"..-¨.
0 .,k-1 ..' .,,*
I I W II W 0
HO ______ p¨O R11-0¨ P-0 II W
01H HO __ S-0
OH I I
(i) (ii) 0 (iii)
R"
RI
N
0 ,\<1!=,w,:.)
) _______________ 0 0
II
HO ____________________________________ P-0 0
0 (iv)
011 (v)
OH
R" le
,
N . N ---...-
M1 ) 0 ...,,\I__
11 W II W V ,
- 0¨ 13-0 - 0 P-0 1211-0¨ 13-0
0 M2 0 0 -
(ia) (1b) M1 (iia)
N4,
121
R" .
N.-..--"-
N'==='"'- %
DA1 ,.....\<õ
0 __________________________________ (
0
(iiia) 0 - (iva)
/v11
RI RI
* *
0
1 õ w=-i- /il _.., )<I_Lw
õ,!--
-0 _________ P __ 0 0 - 0¨
MI oI - (va) M2 6_
(vb)
kr
wherein
the asterisk () represents the site of attachment of R1 to the remainder of
the
molecule;
W and RI are as defined above;
¨11
tc represents C1-6 alkyl;
MI represents a monovalent cation; and
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 82 -
M2 represents a divalent cation.
In a first aspect of this embodiment, the present invention relates to
compounds of
formula (I) wherein RI is heteroaryl substituted by phosphate-C1_6 alkyl,
e.g., represented
by the group of formula (i), or salts thereof, represented respectively by
groups of
formula (ia) or (ib).
In a second aspect of this embodiment, the present invention relates to
compounds
of formula (I) wherein RI is heteroaryl substituted by CI 6 alkyl phosphate-
C1,5 alkyl e.g.,
represented by the group of formula (ii), or salts thereof, represented by
formula (iia).
In a third aspect of this embodiment, the present invention relates to
compounds
of formula (1) wherein R' is heteroaryl substituted by sulphate-C1_6 alkyl,
e.g. as
represented by the group of formula (iii), or salts thereof represented by
formula (iiia).
In a fourth aspect of this embodiment, the present invention relates to
compounds
of formula (I) wherein R' is heteroaryl substituted by carboxy-(Ci_6)alkyl-
carbonyloxy-
C1_6 alkyl, e.g. as represented by the group of formula (iv), or salts thereof
represented by
formula (iva).
In a fifth aspect of this embodiment, the present invention relates to
compounds of
formula (I) wherein R' is heteroaryl substituted by phosphate- methoxy- C1 -6
alkyl, e.g.,
as represented by the group of formula (v), or salts thereof represented
respectively by the
groups of formula (va) or (vb).
Typical examples of monovalent cation M' in accordance with the present
invention include alkali metal cations or cations represented by formula
+NH(Rk)3
wherein Rk represents hydrogen or C1_6 alkyl.
Suitably, R'' represents ethyl.
In a first embodiment 1141 represents Nat. In a second embodiment, M
represents
K. In a third embodiment, M' represents +NH4. In a fourth embodiment M'
represents
+NH(C1_6 alky1)3. In a particular aspect of this embodiment, M' represents
+NH(CH2CH3)3.
Suitably, M' represents Nat.
Typical examples of divalent cations M2 in accordance with the present
invention
include alkaline earth metal cations.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 83 -
Suitably, M2 represents Ca2+.
In a particular aspect of this embodiment, the present invention relates to
compounds of formula (I), in particular compounds of formula (IB), (IC) or
(ID) as
defined above, wherein R' represents a substituted heteroaryl selected from
the groups
represented by formula (i), (iii), (iv), and their respective salts of formula
(iiia), (va) and
(vb).
A compound of formula (I) wherein R' is heteroaryl substituted by phosphate-
Cr6
alkyl, e.g., as represented above by formula (i) may be prepared from the
corresponding
compound of formula (I) wherein R1 is substituted by hydroxy-Ci_6 alkyl by i)
treatment
with di benzyl N,N-di-isopropylphosphoramidite in a suitable solvent, e.g.
dichloromethane, followed by treatment with hydrogen peroxide at low
temperature and ii)
subsequent hydrogenolysis, e.g. using hydrogen gas under pressure, in the
presence of a
suitable catalyst, e.g. palladium on charcoal, according to a method analogous
to those
described by S. P. Green et al. in Organic Process Research & Development,
2002, 6, 109-
112. A compound of formula (I) wherein R1 is heteroaryl substituted by a salt
of
phosphate-Ci_6 alkyl, e.g. as represented by formula (ia) or formula (ib)
above, wherein M'
and M2 are independently alkali metal cation or alkaline earth metal cation,
may be
prepared by performing the above described hydrogenolysis step (ii) in the
presence of a
suitable alkali metal base or alkaline earth metal base. A compound of formula
(1) wherein
R1 is heteroaryl substituted by Ch6 alkyl phosphate-C1_6 alkyl, e.g. as
represented above by
formula (ii), may be prepared from the corresponding compound of formula (I)
wherein R'
is substituted by hydroxy-C1_6 alkyl by: i) first reacting
cyanoethylphosphoramidite with a
compound of formula R"-OH, in the presence of diisopropylethylamine in a
suitable
solvent e.g. dichloromethane, ii) addition of compound of formula (I) wherein
IV is
substituted by hydroxy-C1_6 alkyl in the presence of a suitable solvent, e.g.
dichbromethane, (iii) followed by oxidation and subsequent treatment with a
suitable
base, according to a method analogous to those described by Nam, N-H. et al.
in Bio-org.
Med Chem., 2004, 12, 6255 or Bennani, L. et al. in international patent
application WO
2012/177707 Al.
A compound of formula (I) wherein R1 is heteroaryl substituted by sulphate-C1-
6
alkyl, e.g. as represented above by formula (iii), may be prepared by
treatment of the
corresponding compound of formula (I) wherein R1 is substituted by hydroxy-
Ci_6 alkyl,
with pyridine:sulphur trioxide complex, according to a method analogous to the
one
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 84 -
described by E. Lacko et al. in Current Medicinal Chemistzy, 2012, 19, 4699,
or
alternatively, by treatment with chloro sulphonic acid in the presence of
triethylamine,
according to a method analogous to the one described in C. Guo et al. in
international
patent application WO 2004/087720 Al.
A compound of formula (I) wherein R' is heteroaryl substituted by carboxy(Ci-
6)alkyl-carbonyloxy-Ci_o alkyl e.g. as represented by the group of formula
(iv), may be
prepared by reacting the corresponding compound of formula (I) wherein R' is
substituted
by hydroxy-C16 alkyl, with an appropriate anhydride, e.g. succinic anhydride,
in the
presence of dimethylaminopyridine, in a suitable solvent, e.g. pyridine, at
elevated
temperature, according to a method analogous to the one described by C. Liu et
al., in
Molecular Pharmaceutics, 2014, 57, 7509 or W.N. Washburn et al. in I Med.
Ghent,
2014, 57, 7509.
A compound of formula (I) wherein R1 is heteroaryl substituted by phosphate-
methoxy-Ci_6 alkyl e.g. as represented by the group of formula (v), may be
prepared by
reacting the corresponding compound of formula (I) wherein R1 is substituted
by hydroxy-
C1_6 alkyl, with a suitable base, e.g. sodium hydride, in a suitable solvent,
e.g.
dimethoxyethane, followed by addition of chloromethyldi-tert-butyl-phosphate,
and
subsequent dealkylation, at high temperature, according to a method analogous
to the one
described in international patent application WO 2012/135082 Al.
Compounds of formula (I) wherein R' is heteroaryl substituted by phosphate-Ci-
o
alkyl, e.g. as represented by formula (i), which can be isolated as a result
of the above
hydrogenolysis step (ii), may be transformed into the corresponding compound
of formula
(1) wherein Rk is heteroaryl substituted by a salt of phosphate-CI -6 alkyl,
e.g. as represented
by formula (ia) wherein 1µ,41 represents an alkali metal cation or a cation of
formula
-11\1H(Rk)3 or may be transformed into the corresponding compound of formula
(1) wherein
121 is heteroaryl substituted by a salt of phosphate-C1_6 alkyl, e.g., as
represented by
formula (ib), wherein M2 represents an alkaline earth metal cation, by
treatment with the
corresponding base, i.e. respectively an alkali metal base or the
corresponding base of
formula N(Rk)3 or an alkaline earth metal base, in a suitable solvent
according to methods
known to the person skilled in the art.Suitable alkali metal bases include
sodium hydroxide
and potassium hydroxide.
Suitable alkaline earth metal bases include calcium hydroxide.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 85 -
Suitable bases of formula N(Rk)3 include ammonia (NH3) and triethylaminc.
Compounds of formula (I) wherein RI is heteroaryl substituted independently by
C1_6 alkyl phosphate-Ci_6 alkyl, e.g. as represented by the group of formula
(ii), or by
sulphate-C1_6 alkyl, e.g. as represented by the group of formula (iii), or by
earboxy(Ci_
6)alkyl-earbonyloxy-CI-6 alkyl, e.g. as represented by the group of formula
(iv), or by
phosphate-methoxy- C1-6 alkyl, e.g. as represented by the group of formula
(v), may be
transformed into their respective corresponding salts represented by formula
(iia), (iiia),
(iva), (va) and (vb), according to a method analogous to the one described
above for
compounds of formula (I) wherein R' is hetcroaryl substituted by phosphate-C16
alkyl.
Illustrative examples of compounds according to the present invention wherein
RI
is a substituted heteroaryl selected from the groups represented by formula
(i), (ii), (iii),
(iv) and (v), and their respective salts of formula (ia), (ib), (iia), (iiia),
(iva), (va) as defined
above, include the following compounds:
F
F
N
b,. b
N,11 N .. ,11
N N
0
OH/
P , \ 0 F¨"-- Na+ _ 0
0,..0
/'=.;,.. F.---
F Na-F -0 0
F
PHD (IB)-(ii)
N N
\ > NH H /
N "N".= N N N N'
I 0 . 0
=
,
/(..N'''
P ________
F--- P
F---<
/ .,=== / ,s
OH 0 Na+ -0 0
F F
(113)-(iii) (M)-(iv) .
N N
"N/'
N N N '''', N
= = , ...- - = ,
, .
N /CN
P ________
F---- F-4
/ s,'= /1)\
F F
(113)-(v) ([B)-(vi)
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 86 -
N N
., 7----
/------
N =--. N '''N IX ``.... N
I 0 0
,
OH No+ 0 0 .
011 0 Na+ - 0 0 F----
T F
(B)-(vii) (IB)-(viii)
N
N
"N
I 0
I 0
'...,
N Na + N
OHõ ,,.-0/C ....". 0 .. -
0
/P.:, F-4 ...õ...-
- or'...o
HO 0 F---C
F F
Na +
(TH)-(ix) (IB)-(x)
F
N F
N
``... Nb I:
N
0 Na + N s
,P
________________________________________ N
OHõ ..,,,-0 0 0 \)
P
F
17----( /
'..../p
OH 0 -o \'0
F F
(B3)-(xi) Na + (IB)-(xii)
1, P
N N
0 ' ,0
_____________________________ CN N ''',.. N CN
, ...-- .
__________ N /CN
0 41 Na+ 0
OH.,.
/P=' F-4
P
.F.---
011 0 Na+ - 0 0
F F
(IB)-(xiii) (IB)-(xiv)
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 87 -
N N
J.,, ,H \ .""Ni'll 'N
N
S I S
0 0
OH., õ....-0 Na+-
/Psz, F---( ..,P, F-----(
OH 0
Na+ -O 0
F F
(IB)-(xv) (IB)-(xvi)
F N F N
b ,Il b.... /11
..
'N
N ''- N N N
OH., ....-OrN
0 '..,_
Na-t-
- 0,, ,õ--OrN---
0 =
P
F-4 P
F"--<
/ /
OH 0 - 0 0
F F
Na+
(IB)-(xvii) (IB)-(xviii)
F F
N N
b ,ll H
b ..... N'
N
I ,
. 0
, ---- .
, .
Na' N
0 0
OH,, 0 _ 0,,
OH 0 -O 0
(IB)-(xix) Na+
(IB)-(7a)
F F
N N
N H \ IIIIJ, ,H
.. "'N
IN N
I .. Na+
P == 0
r OH, ..,,..=0 N -- F .=-
- 0,... ..õ...-0/cN--
F õ
P
/ S'=
OH 0 -O 0
013)-(xxi) Na+ (I13)-(xxii)
0
Ai N 0 N
\>....õ \>õõ_.....,
N 'Nil Na+ I I N ' N ' NH
OH
OH \-.
1 \ N )I\I
P¨ ____________________________________________ y.
0 = Na+ 0 *
F"--4\F F"--1\F
(IB)-(xxiii) (IB)-(xxiv)
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 88 -
N N
0 0
0 ...__.... 0
11 \ N5''N __ KNa
N N N
OH ___ 7------0: I
O PC-- \1 " N 1 \---
.õ
4iiih
OH ___________ N 0
0 = Na+ 0 Mr
F-1\F
F--I\F
(IB)-(xxv) (IB)-(xxvi)
0 N 0 \ z 0 0 N 0 \ z 0
11 xi ------ . \ S /
N .' N'''' '."--- Na+ 11 NV
N
011 __ P--.-- ',' i \.! __ 0 P------- \,-'
I \..)-N I 1 _ \1=1\T õ,
A....L.
OH 0
0 Nis r Na+ 0 fl
F"---(F F----(F
(IB)-(xxvii) (IB)-(xxviii)
N 0
N N
0
........õ,, '....,._
H N 0 Na+
N "' 1 Ni -...' o
P..
I `1\2 ,
õ,
OH N 0 N
0 4. Na+ 0 *
F-I\ F¨JNF
F
(IB)-(xxix) (1B)-(xxx)
F N F N
\ .""N'll
N "=== N N N
I 0 I 0
..., Na+ ..,,
./ ./
0 0
-
F-4 P
- 0 0
/-13:
011:s0 / F-4
F F
(I13)-(xxxi) Na+ (IB)-(xxxii)
N N
b. NN'
NN
,H
N N ..",- N
1
OH. -CP/CN
0 ......
Na+
-o, ..,,,-0 N
/C 0 .....
P,
OH/ \ 0 F--
- 0 0
1; I-
Nal
oBxxxxiii) (m)-(xxxiv)
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 89 -
F F
N N
,I1
/II
""N
N =N'= N
I
..
,
N N
Na+
OH ,.0 /C..'
-. 0 - F-4 0P õ ...õ./0/(...' -
..' 0
./P,:µ,.. /== F----(
OH 0 -o 0
F F
Ns+
(IB)-(xxxv) (1B)-(xxxvi)
N N
,11
b /II
"N .."'N
N ..``-= N
I 0 I 0
Na+
-O
O , ... 0
OH.õ,..õ..0 s0
P
/ -s=
OH 0 -0 0 F----
F F
Ns+
(14)-(xxxvii) (IB)-(xxxviii)
F
N F N
b / b /
N
- .,
OHõ .-0 0 0
./1)\=
F---- Ns+ - 0 ,x;....0 r
Na+ - 0 0
OH 0
F F
(M)-(xxxix) (IB)-(xl)
N
N
N" /--
N.::1
/(kN- ,
' 0
/-0
/P =
OH \ 0 F"---C HO, ..0 17-4
F /P.,= F
OH 0
(M)-(xi) (IB)-(xlii)
N
N
b ..... N'
N
Nb
N `==- N
I 0 I
.. ., 0
F
0 ) __ 0 \ 0
,,
-'S,
----( F-4
OH 0
F 0 F
(IB)-(xliii) OH Oil )-(xliv)
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 90 -
F N
F,...N
\ ..... ,N/
I
N T /
N 0 I , ,
, o
õrN /(..N"" "
./-N. 0 0- .
('),, -0 /-0
P
OH ---( F----(
/ 0 F HO -0 , _
F ,,P,,,, F
H 0
(IB)-(xlv) O (111)-(xlvi)
F N , F N
b =b. ,
I 0 I
0.õ..,, ,0r -= 0 ,_. 0
SF"--- F --"--
/ \S
OH 0
F 0 F
(IB)-(xlvii) OH (TB)-(xlviii)
F N
F
,E1 N
H
.... N
N ..- N
b
1 . 0
,
0
P
F ---( -4
/ HO, ,A20 F
OH 0
F /P\--:, F
OH 0
(113)-(xliz) (113)-(1)
F N F N
b ,Il
N .*.= N
,
' 0 __
) ________________________________________ 0rN 0
F--< F"---
OH 0
F 0 F
(IB)-(10 OH 013)-(Iii)
0 N 0 0
,....\S//
I I
'.. /
0,
*
OH N
0 0 =
F-J\F 1,--(F
(113)-(Iiii) (TB)-(liv)
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 91 -
N 0 0
)...._., s=//
0
N 0 /0 TµV N 'N--- "*....
I \\_ =:...= ,
l', ,,S......
____________________________________________ ''....LN
HO¨S--, N '
\---
1 \,,-I j õµ
o,..., 0 0 I*
OH N
o* ../
F---(,F (30H
(1.13)-(1v) (IB)-Ovi)
0 0 N 0
N
0 11 .1,--, )K
HO ¨P _______________________________________ -==== N ''. - 'N
\---
.,
OH _____________ \----.'N argh...
0 gli OH 'N
0*
F---CF F---(F
(113)-(1vii) (I13)-(lviii)
N 0
s>õ......
11 N / N N=,,N) N 0
N '""
*"....4 N '' )C
'N-
\sr--
.,,..
HO¨ S -----.0 1 1 \----
I A.,,--1,.. , õmi.. 0 0 *
OH N 0
F"-INF
0 glir ..,".
F--.1\ F OH (IB)-(1x) (IB)-(11x)
H ....;-.7-.........-N
H
NN 4111, N-N 4111"."'N/
Nat
0 0
- 0
P
F*----( 0 ...---
'''.11
1,----(
/ =,\ / ..
OH 0 - 0 0
F F
Na I
(IC)-(i) (1C)-(ii)
F ,\,....-i-rN F,õõ_.......s...)-
=õõ........õN
1-1 ,1-1 /
N.õ...--...õ. ......õ, ..,,,,,,,.../..`,, N ip...,,N N .......õ--
=:,....õ, .,.,...,
I 0 0
0 Na+ - 0
P /
F /
-4 F"'"
13
OH 0 Na+ - 0 0
F F
IC)-(ii) IC)-iv)
Where a mixture of products is obtained from any of the processes described
above
for the preparation of compounds according to the invention, the desired
product can be
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 92 -
separated therefrom at an appropriate stage by conventional methods such as
preparative
HPLC; or column chromatography utilising, for example, silica and/or alumina
in
conjunction with an appropriate solvent system.
Where the above-described processes for the preparation of the compounds
according to the invention give rise to mixtures of stereoisomers, these
isomers may be
separated by conventional techniques. In particular, where it is desired to
obtain a
particular enantiomer of a compound of formula (I) this may be produced from a
corresponding mixture of enantiomers using any suitable conventional procedure
for
resolving enantiomers. Thus, for example, diastercomeric derivatives, e.g.
salts, may be
produced by reaction of a mixture of enantiomers of formula (1), e.g. a
racemate, and an
appropriate chiral compound, e.g. a chiral base. The diastereomers may then be
separated
by any convenient means, for example by crystallisation, and the desired
enantiomer
recovered, e.g. by treatment with an acid in the instance where the
diastereomer is a salt.
In another resolution process a racemate of formula (I) may be separated using
chiral
HPLC. Moreover, if desired, a particular enantiomer may be obtained by using
an
appropriate chiral intermediate in one of the processes described above.
Alternatively, a
particular enantiomer may be obtained by performing an enantiomer-specific
enzymatic
biotransformation, e.g. an ester hydrolysis using an esterase, and then
purifying only the
enantiomerically pure hydrolysed acid from the unreacted ester antipode.
Chromatography, recrystallisation and other conventional separation procedures
may also
be used with intermediates or final products where it is desired to obtain a
particular
geometric isomer of the invention. Alternatively the non desired enantiomer
may be
racemized into the desired enantiomer, in the presence of an acid or a base,
according to
methods known to the person skilled in the art, or according to methods
described in the
accompanying Examples.
During any of the above synthetic sequences it may be necessary and/or
desirable
to protect sensitive or reactive groups on any of the molecules concerned.
This may be
achieved by means of conventional protecting groups, such as those described
in
Protective Groups in Organic Chemistry, ed. J.F.W. McOmie, Plenum Press, 1973;
and
T.W. Greene & P.G.M. Wuts, Protective Groups in Organic Synthesis, John Wiley
&
Sons, 3rd edition, 1999. The protecting groups may be removed at any
convenient
subsequent stage utilising methods known from the art.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 93 -
Compounds in accordance with the present invention potently neutralise the
activity of TNFa in a commercially available HEK-293 derived reporter cell
line known
as HEKBIueTM CD4OL. This is a stable HEK-293 transfected cell line expressing
SEAP
(secreted embryonic alkaline phosphatase) under the control of the IFN13
minimal
promoter fused to five NF-KB binding sites. Secretion of SEAP by these cells
is
stimulated in a concentration-dependent manner by TNFa. When tested in the HEK-
293
bioassay, also referred to herein as the reporter gene assay, compounds of the
present
invention exhibit an 1050 value of 50 pM or less, generally of 20 p.M or less,
usually of 5
p.M or less. typically of 1 !AM or less, suitably of 500 nM or less, ideally
of 100 nM or
less, and preferably of 25 nM or less (the skilled person will appreciate that
a lower IC50
figure denotes a more active compound).
Certain compounds in accordance with the present invention potently inhibit
the
binding of a fluorescence conjugate to TNFa when tested in the fluorescence
polarisation
assay described herein. Indeed, when tested in that assay, compounds of the
present
invention exhibit an IC50 value of 50 "AM or less, generally of 20 pM or less,
usually of 5
pM or less, typically of 1 pM or less, suitably of 500 nM or less, ideally of
100 nM or less,
and preferably of 25 nM or less (as before, the skilled person will appreciate
that a lower
IC50 figure denotes a more active compound).
The compounds of the Examples have been tested in one or both of the assays
described below.
Fluorescence Polarisation Assay
Preparation of Compound (A)
1-(2 ,5-Dimethylb enzy1)-6- [4-(piperazin-1 -ylmet hyl)pheny1]-2-(pyridin-4-yl-
methyl)-1H-benzimidazole ¨ hereinafter referred to as "Compound (A)" ¨ can be
prepared
by the procedure described in Example 499 of WO 2013/186229; or by a procedure
analogous thereto.
Preparation offluorescence conjugate
Compound (A) (27.02 mg, 0.0538 mmol) was dissolved in DMSO (2 mL). 5 (-6)
Carboxy-fluorescein succinimyl ester (24.16 mg, 0.0510 mmol) (Invitrogen
catalogue
number: C1311) was dissolved in DMSO (1 mL) to give a bright yellow solution.
The
two solutions were mixed at room temperature, the mixture turning red in
colour. The
- 94 -
mixture was stirred at room temperature. Shortly after mixing a 20 pL aliquot
was removed and
diluted in a 80:20 mixture of AcOH:H20 for LC-MS analysis on the 120ORR-6140
LC-MS
system. The chromatogram showed two closely eluting peaks at retention times
of 1.42 and 1.50
minutes, both with mass (M+H)+ = 860.8 amu, corresponding to the two products
&dined with the
5- and 6-substituted catboxyfluorescein group. A further peak at retention
time 2.21 minutes had a
mass of (M+H)+ = 502.8 amu, corresponding to Compound (A). No peak was
observed for unreacted
5(-6) carboxyfluorescein succinimyl ester. The peak areas were 22.0%, 39.6%
and 31.4% for the
three signals, indicating a 61.6% conversion to the two isomers of the desired
fluorescence conjugate
at that time-point. Further 20 pL aliquots were extracted after several hours
and then after overnight
stirring, diluted as before and subjected to LC-MS analysis. The percentage
conversion was
determined as 79.8% and 88.6% respectively at these time-points. The mixture
was purified on a
UV-directed preparative HPLC system. The pooled purified fractions were freeze-
dried to remove
excess solvent. After freeze-drying, an orange solid (23.3 mg) was recovered,
equivalent to 0.027
mmol of fluorescence conjugate, corresponding to an overall yield of 53% for
the reaction and
preparative HPLC purification.
Inhibition of binding offluorescence conjugate to TNFa
Compounds were tested at 10 concentrations starting from 25 pM in a final
assay
concentration of 5% DMSO, by pre-incubation with TNFa for 60 minutes at
ambient temperature in
mM Tris, 150 mM NaC1, 0.05% TweenTm 20, before addition of the fluorescence
conjugate and a
20 further incubation for 20 hours at ambient temperature. The final
concentrations of TNFu and the
fluorescence conjugate were 10 nM and 10 nM respectively in a total assay
volume of 25 L. Plates
were read on a plate reader capable of detecting fluorescence polarisation
(e.g. an Analyst HT plate
reader; or an Envision plate reader). An 1050 value was calculated using
XLfitTM (4 parameter logistic
model) in ActivityBase.
When tested in the fluorescence polarisation assay, compounds of the
accompanying
Examples were found to exhibit 1050 values of 50 M or better.
When tested in the fluorescence polarisation assay, compounds of the
accompanying
Examples exhibit 1050 values generally in the range of about 0.01 nM to about
50 M, usually in the
range of about 0.01 nM to about 20 pM, typically in the range of about 0.01 nM
to about 5 M,
suitably in the range of about 0.01 nM to about 1
Date Recue/Date Received 2022-02-28
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 95 -
JIM, ideally in the range of about 0.01M to about 500 nM, appositely in the
range of
about 0.01M to about 100 nM, and preferably in the range of about 0.01M to
about 25
nM.
Reporter Gene Assay
Inhibition of TNFa-induced NF-KB activation
Stimulation of HEK-293 cells by TNFa leads to activation of the NF-KB pathway.
The reporter cell line used to determine TNFa activity was purchased from
InvivoGen.
HEKBlueTM CD4OL is a stable HEK-293 transfected cell line expressing SEAP
(secreted
embryonic alkaline phosphatase) under the control of the IFNP minimal promoter
fused to
five NF-KB binding sites. Secretion of SEAP by these cells is stimulated in a
dose-
dependent manner by TNFa, with an EC50 of 0.5 ng/mL for human TNFa. Compounds
were diluted from 10 mM DMS0 stocks (final assay concentration 0.3% DMS0) to
generate a 10-point 3-fold serial dilution curve (30,000 nM to 2 nM final
concentration,
for example). Diluted compound was preincubated with TNFa for 60 minutes prior
to
addition to a 384-well microtitre plate and incubated for 18 h. The final TNFa
concentration in the assay plate was 0.5 ng,/mL. SEAP activity was determined
in the
supernatant using the colorimetric substrates QUANTI-BlueTm or HEKBlueTM
Detection
media (InvivoGen). Percentage inhibitions for compound dilutions were
calculated
between a DMS0 control and maximum inhibition (by excess control compound) and
an
IC50 value calculated using XLfitTM (4 parameter logistic model) in
ActivityBase.
When tested in the reporter gene assay, the compounds of the accompanying
Examples were all found to exhibit IC50 values of 50 p.IVI or better.
When tested in the reporter gene assay, compounds of the accompanying Examples
exhibit IC50 values generally in the range of about 0.01 nM to about 50 p,M,
usually in
the range of about 0.01 nM to about 20 pM, typically in the range of about
0.01 nM to
about 5 p M, usually in the range of about 0.01 nM to about 1 pM, suitably in
the range of
about 0.01M to about 500 nM, ideally in the range of about 0.01M to about 100
nM, and
appositely in the range of about 0.01nM to about 25 nM.
The following Examples illustrate the preparation of compounds according to
the
invention.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 96 -
EXAMPLES
Abbreviations
DCM: Dichloromethane Et0Ac: Ethyl acetate
DMF: N,N-Dimethylfoitnamide MeOH: Methanol
DMSO: Dimethylsulfoxide SiO2: Silica
Et20: Diethyl ether h: Hour
THF: Tetrahydrofuran AcOH: Acetic acid
rt.: Room temperature b s.: Broad singlet
M: Mass
Brine: Saturated aqueous sodium chloride solution
HPLC: High Performance Liquid Chromatography
LCMS: Liquid Chromatography Mass Spectrometry
ES+: Electrospray Positive Ionisation
[LA: Triethylamine
DIPEA: N,N-di-iso-propylethylamine
DIAD: Diisopropyl (E)-1,2-diazenedicarboxylate
RT: retention time
TBAF: tetrabutyl ammonium fluoride
TLC: Thin Layer Chromatography
MeCN: Acetonitrile
D1BAL-H: Diisobutylaluminium hydride
TMSCN: Trimethylsilyl cyanide
DEA: Diethanolamine
pTSA para-toluene sulphonic acid monohydrate
TFA : trifluoroacetic acid
DMA: dimethyl acetamide
HATU: N-RDimethylamino)-1H-1,2,3-triazolo44,5-b]pyridin-l-
ylmethylene]-N-
ethylmethanaminium hexafluorophosphate N-oxide
KHMDS : Potassium bis(trimethylsilyl)amide
COMU: (1-cyano-2-ethoxy-2-oxoethylidenaminooxy) dimethylamino-morpho lino
carbenium hexafluorophosphate
PdC12(dcypp) :dichloro-bis(dicyclohexylphosphino)propane] palladium(II)
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 97 -
ur dF(CF3)ppy12(dtbpy)11)F6 [4,4'-Bis(tert-buty1)-2,2'-bipyridine]bis[3,5-
difluoro-
245-(trifluoromethyl)-2-
pyridinyl]phenyl]iridi urn(III) hexafluorophosphate
Analytical Conditions
All NMRs were obtained either at 300 MHz or 400 MHz.
All reactions involving air-or moisture-sensitive reagents were performed
under a
nitrogen atmosphere using dried solvents and glassware.
All compound LCMS data was determined by using the method below:
Method 1: For Intermediates 1 to 7 and 15 to 17.
Shimadzu 2010- YMC Triart C18, 4.6 x 50mm, 3 pm column
Mobile phase A: 10 mM Ammonium Formate + 0.1 % Ammonia + water
Mobile phase B: 5 % of mobile phase A + 95 % MeCN + 0.1 % Ammonia
Gradient program (Flow Rate 1.4 mUmin, Column Temperature 40 C):
Time A% B%
0.1 70 30
2.5 5 95
3.5 5 95
5.0 70 30
5.5 70 30
Method 2: For Intermediate 8 and 18
Shimadzu 2010- X-bridge C18 Waters 2.1 x 20mm, 2.5 pm column
Mobile phase A: 10 mM Ammonium Formate + 0.1 % Ammonia + water
Mobile phase B: 5 % of mobile phase A + 95 MeCN + 0.1 % Ammonia
Gradient program (Flow Rate 1.0 mUmin, Column Temperature 40 C):
Time A% B%
0.1 95 5
4.0 5 95
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 98 -
5.0 5 95
5.1 95 5
6.5 95 5
Method 3 for all analytical LCMS done in basic conditions: LCMS basic:
A QDA Waters simple quadrupole mass spectrometer is used for LC-MS analysis.
This spectrometer is equipped with an ESI source and an UPLC Acquity Classic
with
diode array detector (210 to 400 nm.)
Data are acquired in a full MS scan from m/z 50 to 1000 in positive mode with
an acidic
elution
The reverse phase separation is carried out at 45 C on
a Waters Acquity UPLC BEH C18 1.7um (2.1x50mm) column for basic elution
Gradient elution is done with:
H20/ACN/Ammonium_formate (95/5/63mg/1) + 50 1 NH4OH (solvent A)
ACN/H20/Ammoniumformate (95/5/63mg/1) + 50 1 NH4OH (solvent B).
Acidic gradient program :
HPLC flow rate: 0.6 ml/min to 0.7 ml/min,
injection volume: 1 ul
Full flow in MS.
Time (min) A (%) B (%) Flow
(ml/min)
0 99 1 0.4
0.3 99 1 0.4
3.2 0 100 0.4
3.25 0 100 0.5
4 0 100 0.5
4.1 99 1 0.4
4.8 90 1 0.4
Method 4 for all analytical LCMS in acid conditions: LCMS acid:
A QDA Waters simple quadrupole mass spectrometer is used for LC-MS analysis.
This spectrometer is equipped with an ES1 source and an UPLC Acquity Hclass
with
diode array detector (210 to 400 nm).
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 99 -
Data arc acquired in a full MS scan from m/z 50 to 1000 in positive mode with
an acidic
elution.The reverse phase separation is carried out at 45 C on a Waters
Acquity UPLC
HSS T3 1.81.1m (2.1x5Ornrn) column for acidic elution
Gradient elution is done with.
Water (solvent A)
Acetonitrile (solvent B)
Water/Acetonitrile/Formic Acid 0.5% (solvent C)
Acidic gradient program :
HPLC flow rate: 0.6 ml/min to 0.7 mUmin, injection volume: 1 p.1
Full flow in MS.
Time (min) A (%) B (%) C (%) Flow
(ml/min)
0 90 0 10 0.6
0.3 90 0 10 0.6
3.2 0 90 10 0.6
3.25 0 90 10 0.7
4 0 90 10 0.7
4.1 90 0 10 0.6
5.4 90 0 10 0.6
Method 5 for all Examples:
Waters Acquity-SQD, Waters Acquity UPLC BEH C18, 2.1 x 50 mm, 1.7 lam column
Mobile phase A: 10 mM Ammonium Formate + 0.1 % Ammonia
Mobile phase B: 95 % MeCN + 5 % H20 + 0.1 % Ammonia
Gradient program (Flow Rate 1.0 mIlmin, Column Temperature 40 C):
Time A% B%
0.00 95 5
0.50 95 5
1.75 5 95
2.00 5 95
2.25 95 5
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 100 -
It will be apparent to the person skilled in the art that different retention
times (RT) may
be obtained for LCMS if different analytical conditions are used.
Additional Analytical HPLC Methods
Method 6
Column: Waters Atlantis dC18 (2.1 x100 mm, 3 jim column)
Flow rate: 0.6 mL/min
Solvent A: 0.1 % Formic acid / water
Solvent B: 0.1 % Formic acid / acetonitrile
Injection Volume: 3 pi,
Column temperature: 40 C
UV Detection wavelength: 215 nm
Eluent: 0 to 5 minutes, constant gradient from 95 % solvent A + 5 % solvent B
to 100 %
solvent B; 5 to 5.4 minutes, 100 % solvent B; 5.4 to 5.42 minutes, constant
gradient from
100 % solvent B to 95 % solvent A + 5 % solvent B; 5.42 to 7.00 minutes, 95 %
solvent
A + 5 % solvent R.
Method 7
Column: Waters Atlantis dC18 (2.1 x 30 mm, 3 mn column)
Flow rate: 1 milmin
Solvent A: 0.1% Formic acid / water
Solvent B: 0.1% Formic acid / acetonitrile
Injection volume: 34
UV Detection wavelength: 215nm
Eluent: 0 to 1.5 minutes, constant gradient from 95 % solvent A + 5 % solvent
B to 100
% solvent B; 1.5 to 1.6 minutes, 100 % solvent B; 1.60 to 1.61 minutes,
constant gradient
from 100 % solvent B to 95 % solvent A + 5 % solvent B; 1.61 to 2.00 minutes,
95 %
solvent A + 5 % solvent B.
Method 8
Column: Phenomenex Gemini CIS (2.0 mm x 100 mm, 3 ium column)
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 101 -
Flow rate: 0.5 rnUmin
Solvent A: 2m1V1 Ammonium bicarbonate / water
Solvent B: Acetonitrile
Injection volume: 3 ittL
Column temperature: 50 C
UV Detection wavelength: 215 nM
Eluent: 0 to 5.5 minutes, constant gradient from 95 % solvent A + 5 % solvent
B to 100
% solvent B; 5.5 to 5.9 minutes, 100 % solvent B; 5.90 to 5.92 minutes,
constant gradient
from 100 % solvent B to 95 % solvent A + 5 % solvent B.
Method 9
Column: Phenomenex Gemini C18 (2.0 mm x 50 mm, 3 gm colurrm)
Flow rate: 1.0 mL/min
Solvent A: 2mM Ammonium bicarbonate / water
Solvent B: Acetonitrile
Injection volume: 3 !IL
Column temperature: 60 C
UV Detection wavelength: 215 nM
Eluent: 0 to 1.8 minutes, constant gradient from 99 % solvent A + 1 % solvent
B to 100
% solvent B; 1.8 to 2.1 minutes, 100 % solvent B; 2.1 to 2.3 minutes, constant
gradient
from 100 % solvent B to 99 % solvent A + 1 % solvent B.
Method 10
Column: Waters XSelect (C18, 50x2.1mm, 3.51.t)
Flow: 0.8 ml/min Column temp; 35 C
Eluent A: 0.1% formic acid in acetonitrile
Eluent B: 0.1% formic acid in water
Lin. Gradient: t=0 mm 5% A, t=3.5 mm 98% A, t=6 mm 98% A
Detection: DAD (220-320 nm)
Detection: MSD (ESI pos/ncg) mass range: 100-800
Method 11
Column: Waters XSelect (C18, 30x2.1mm, 3.5g)
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 102 -
Flow: 1.0 ml/min Column temp; 35 C
Eluent A: 0.1% formic acid in acetonitrile
Eluent B: 0.1% formic acid in water
Lin. Gradient: 1=0 min 5% A, t=1.6 min 98% A,1=3 min 98% A
Detection: DAD (220-320 nm)
Detection: MSD (ESI pos/neg) mass range: 100-800
Method 12
Column: Waters XSelect (C18, 30x2.1mm, 3.50
Flow: 1.0 ml/min Column temp; 35 C
Eluent A: 0.1% formic acid in acetonitrile
Eluent B: 0.1% formic acid in water
Lin. Gradient: 1=0 min 5% A, t=1.6 mm 98% A, t=4 mm 98% A
Detection: DAD (220-320 nm)
Detection: MSD (ESI pos/neg) mass range: 100-800
Method 13
Column: Waters XSelect (C18, 30x2.1mm, 3.5[1)
Flow: 1.0 ml/min Column temp; 35 C
Eluent A: 0.1% formic acid in acetonitrile
Eluent B: 0.1% foitnic acid in water
Lin. Gradient: t=0 min 5% A, t=1.6 min 98% A, 1=3 mm 98% A
Detection: DAD (220-320 nm)
Detection: MSD (ESI pos/neg) mass range: 400-1600Method 14 for the
purification of
Examples 107 to 112
Semi preparative HPLC column: Sunfire prep C18 51Am 10x150 mm
Isocratic elution: 25% of Solvent A (Water/ Acetonitrile/formic acid (v/v/v ;
95/5/0.05))
and 75% Solvent B (acetonitrile/formic acid (v/v ; 100/0.075))
Flow rate: 7 mL/ min
Method 15.* the analysis of Examples 107 to 112
The LCMS control was performed with a QM Waters triple quadrupole mass
spectrometer coupled with an HPLC Alliance Waters 2795 quaternary pump. The
reverse
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 103 -
phase separation is carried out at 45 C on a Waters Sunfire MS C18 column 5m
(4.6 x
15 mm) for acidic elution.
Gradient elution is performed with water (Solvent A), acetonitrile (Solvent B)
and
water/acetonitrile/formic acid (Solvent C v/v/v 50/50/5).
Solution pH 3-4, gradient table as below:
Time A% B% C% Flow
0 90 0 10 1.9
1.5 90 0 10 1.9
7.15 2 88 10 2.4
10.5 2 88 10 2.4
10.6 90 0 10 1.9
13 90 0 10 1.9
Method 16:
Waters UPLC-SQD apparatus; ionization: electrospray in positive and/or
negative mode
(ES+/-); chromatographic conditions: column: Acquity CSH C18 1.7 gm - 1 x 30
mm;
solvents: A: H20 (0.1% formic acid) B: CH3CN (0.1% formic acid); column
temperature:
45 C; flow rate: 0.6 ml/min; gradient (2.0 min): from 5 to 50% of B in 1.0
min; 1.3 min:
100% of B; 1.45 min: 100% off1; 1.75 min: 5% of B; retention time = RT (min).
Method 17:
Waters HPLC-ZQ apparatus; ionization: electrospray in positive and/or negative
mode
(ES+/-); chromatographic conditions: column: XSelect CSH C18 3.5 pm - 3.0 x 75
mm;
solvents: A: H20 (0.1% formic acid) B: CH3CN (0.1% formic acid); column
temperature:
60 C; flow rate: 0.8 ml/min; gradient (6.0 min): 6% of B during 0.8 min; From
6% to
100% of B in 3.9min; 4.8 min: 100% of B; 5.0 min: 6% of B; 6.0 min: 6% of B;
retention
time = RT (min).
Method 18:
Waters HPLC-ZQ apparatus; ionization: electrospray in positive ancUor negative
mode
(ES+/-); chromatographic conditions: column: XSclect CSH C18 3.5 gm - 3.0 x 75
mm;
solvents: A: H20 (0.1% formic acid) B: CH3CN (0.1% formic acid); column
temperature:
60 C; flow rate: 1.0 ml/min; gradient (7.0 min): 10% of B during 0.2 min; From
10 to
100% of B in 4.3 min; 4.85 min: 100% of B; 6.5 min: 10% of B; 7.0 min: 10% of
B;
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 104 -
retention time = RT (min).
Method 19:
Waters UPLC-SQD apparatus; ionization: electrospray in positive and/or
negative mode
(ES+/-); chromatographic conditions: column: Acquity CSH C18 1.7 pm - 1 x 30
mm;
solvents: A: H20 (0.1% formic acid) B: CH3CN (0.1% formic acid); column
temperature:
45 C; flow rate: 0.6 ml/min; gradient (4.0 min): 5% of B during 0.15 min; 1.3
mm: From
5 to 100% of B in 3.15 min; 3.45 mm: 100% of B; 3.85 mm: 5% of B; 4.00 min: 5%
of B;
retention time = RT (min).
Method 20:
Waters UPLC-SQD apparatus; ionization: electrospray in positive and/or
negative mode
(ES+/-); chromatographic conditions: column: Acquity BEH C18 1.7 pm - 2.1 x 50
mm;
solvents: A: H20 (0.1% formic acid) B: CH3CN (0.1% formic acid); column
temperature:
50 C; flow rate: 0.8 ml/min; gradient (2.5 min): from 5 to 100% of B in 1.8
mm; 2.4 min:
100% of B; 2.45 mm: 100% to 5% of B in 0.05 mm; retention time = RT (mm).
INTERMEDIATE 1
2-chloro-6-(difluoromethoxy)benzaldehyde
.. To 2-chloro-6-hydroxy-benzaldehydc (20 g, 128.2 mmol) in McCN (150 mL) was
added
an aqueous solution of potassium hydroxide (71.7 g, 1282 mmol) in water (50
mL) at 0 C
and the reaction mixture was stirred at 0 C for 10 min. Diethyl (bromodifluoro
methyl)
phosphonate (36.4 mL, 205.1 mmol) was added at 0 C. The reaction mixture was
stirred
at 0 C for 30 mm. After completion of reaction (monitored by TLC), the
reaction mixture
was poured into water (500 mL). The aqueous layer was extracted with ethyl
acetate (1 L
X 2). The organic layer was washed with water (500 mL), brine (500 mL) and
dried over
anhydrous sodium sulphate. The organic layer was evaporated under reduced
pressure to
yield the crude product which was purified by column chromatography (SiO2, 5%
Et0Ac
in hexane) yielding the title compound (13.9g, 53% yield) as a yellow oil.
iff NMR (400 MHz, CDCb) 6 10.46 (s, 1H), 7.49 (t, J 8.2 Hz, 1H), 7.37 (dd, J
8.1, 1.1
Hz, 1H), 7.20 (m, 1H), 6.61 (t, 1H).
- 105 -
INTERMEDIATE 2
N4[2-chloro-6-(di fluorom ethoxy)phenyl]methyl en e]-(S)-2-methyl-propan e-2-
sul finamide
To a solution of Intermediate 1 (20 g, 97.08 mmol) in dry THF (100 mL) at 0 C
was added (S)-(-)-t-
butyl sulfinamide (12.92 g, 106.79 mmol), K3PO4 (61.73 g, 291.2 mmol) and
K2HPO4. (50.6 g,
291.2 mmol). Then the reaction mixture was stirred at r.t. for 18h. After
completion of reaction
(monitored by TLC), the reaction mixture was filtered through celiteTM and
washed with ethyl
acetate (1 L). The organic layer was washed with water (500 mL), brine (500
mL) and dried over
anhydrous sodium sulphate. The organic layer was evaporated under reduced
pressure and the
residue was purified chromatography (SiO2, 10 % Et0Ac in hexane) to afford the
title compound
(20g, 87% yield) as a yellow oil.
1H NMR (400 MHz, CDC13) 8 8.90 (s, 1H), 745 __ 732 (m, 2H), 7.29
______________ 7.15 (m, 1H), 6.82
6.34 (m, 111), 1.29 (s, 9H). LCMS (ES+) RT 2.73 mm, 309.90 (M+H)+
INTERMEDIATE 3
Ethyl (3R)-3-U(S)-tert-butylsulfinyl]amino]-342-chloro-6-
(difluoromethoxy)phenyllpropanoate
This procedure used activated zinc and THF dried over sodium and benzophenone
complex.
Activated zinc was prepared using the following procedure: 150 g of zinc
powder was taken in 1N
HCl (500 mL), stirred for 10 mm and decanted. The zinc dust powder was further
washed with
water (3 x 500 mL) and decanted. The powder was further washed with acetone (3
x 500 mL),
decanted and dried under vacuum to afford 105 g of activated zinc.
To activated zinc dust (105 g, 1618 mmol) in dry THF (150 mL) was added CuCl
(19.2 g, 194
mmol) and the reaction mixture was refluxed for 30 min. The reaction mixture
was cooled to room
temperature and ethyl bromoacetate (45 m1,, 404 mmol in THF 100 mL) was added
drop wise. The
reaction mixture was stirred at 50 C for 30 min. The reaction mixture was
cooled to 0 C and
.. Intermediate 2 (50 g, 161 mmol in THF 100 mL) was added. The reaction
mixture was warmed to
r.t. and stirred for 3h. After completion of reaction (monitored by TLC), the
reaction mixture was
filtered through celite and washed with ethyl acetate (700 mL). The organic
layer was washed with
1N citric acid (500 mL), saturated solution of sodium bicarbonate (500 mL),
water (500 mL) and
brine (500 mL).
Date Recue/Date Received 2022-02-28
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 106 -
The organic layer was separated, dried over anhydrous sodium sulphate and
evaporated
under reduced pressure. The residue was purified by chromatography (SiO2, 40 %
Et0Ac
in hexane) to afford the title compound (59 g, 92% yield) as a yellow oil.
NMR (400 MHz, CDCb) 6 7.29 ¨ 7.21 (m, 2H), 7.05 (d, J 7.3 Hz, 1H), 6.82 ¨6.34
(m, 1H), 5.59 (m, 1H), 4.36 (s, 1H), 4.18 ¨4.02 (m, 2H), 3.25 (dd, J 15.6, 7.5
Hz, 1H),
3.01 (dd, J 15.3, 7.5 Hz, 1H), 1.31 ¨ 1.11 (m, 12H).
INTERMEDIATE 4
Ethyl (3R)-3-amino-342-chloro-6-(difluoromethoxy)phenyl]propanoate
hydrochloride
To a solution of Intermediate 3 (32 g, 80.6 mmol) in an Ether: Et0H (75 mL,
2:1)
mixture was added 4M HC1 in 1,4-dioxane (70 mL) and the reaction mixture was
stirred
at r.t. for lh. After completion of reaction (monitored by TLC), the reaction
mixture
was concentrated under reduced pressure and the residue was washed with
diethyl ether
(500 mL) to afford the title compound as a yellow solid (22g, 93% yield).
11-1 NMR (400 MHz, CDC13) 6 8.93 (d, J 6.2 Hz, 2H), 7.32 ¨7.10 (m, 3H), 6.96
(s, 1H),
5.42 (m, 1H), 4.08 (q, J 7.0 Hz, 2H), 3.36 (dd, J 16.5, 7.0 Hz, 1H), 3.14 (dd,
J 16.5, 7.8
Hz, 1H), 1.34 (t, J 7.1 Hz, 3H).
INTERMEDIATE 5
Ethyl (3R)-3-(5-bromo-4-fluoro-2-nitro-anilino)-3-[2-chloro-6-
( di fluoromethoxy)phenyl]propano ate
To a solution of Intermediate 4 (5 g, 17.06 mmol) in MeCN (50 mL) was added
potassium carbonate (7.06 g, 51.18 mmol) and 1-bromo-2,5-difluoro-4-
nitrobenzene (4.86
g, 20.47 mmol). The reaction mixture was stirred at 80 C for 16h. After
completion of
reaction (monitored by TLC), the reaction mixture was diluted with ethyl
acetate (100
mL) and washed with water (100 mL). The organic layer was separated, dried
over
anhydrous sodium sulphate and concentrated under reduced pressure. The residue
was
purified by chromatography (SiO2, 20 % Et0Ac in hexane) to afford the title
compound
(6g, 69% yield) as a yellow viscous liquid.
LCMS (ES+) RT 3.42 min, 510.90/512.90/514.90 (M+H)+
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 107 -
INTERMEDIATE 6
(3R)-3-(5-bromo-4-fluoro-2-nitro-anilino)-342-chloro-6-
(difluoromethoxy)phenyl]propanal
To a solution of Intermediate 5 (6 g, 11.7 mmol) in THF (60 mL) at -78 C was
added
DIBAL-H (23 mL, 23.5 mmol) drop wise. The reaction mixture was stirred for 2h
at -
78 C. After completion of reaction (monitored by TLC), the reaction mixture
was
quenched with an aqueous solution of ammonium chloride (200 mL). The reaction
mixture was diluted with ethyl acetate (200 mL) and filtered through celite.
The filtrate
was washed with water (200 mL) and the organic layer was separated, dried over
sodium
.. sulphate and evaporated under reduced pressure to afford the title compound
(3g, 57%
yield) as a yellow oil, which was used in the next step without purification.
INTERMEDIATE 7
(4R)-4-(5-bromo-4-fluoro-2-nitro -a nilino)-442-chlo ro-6-(d fluor
methoxy)pheny1]-2-
.. trimethylsilylo xy-butanenitrile
To a solution of Intermediate 6 (3 g, 6.42 mmol) in DCM (50 mL) was added ZnI2
(0.2 g,
0.64 mmol), TEA (0.09 mL, 0.64 mmol) and TMSCN (1.6 mL, 12.84 mmol). The
reaction mixture was stirred at Lt. for 3h. After completion of reaction
(monitored by
TLC), the reaction mixture was diluted with water (100 mL) and the organic
layer was
separated. The organic layer was washed with water (100 mL), brine (100 mL),
dried over
anhydrous sodium sulphate and concentrated under reduced pressure to afford
the title
compound (3.25g crude material) which was used for the next step without
purification.
INTERMEDIATE 8
.. (1R)-7-bromo-142-chloro-6-(difluoromethoxy)pheny1]-6-fluoro-23-dihydro-1H-
pyrrolo[1,2-a]benzimidazol-3-ol
CA 02962826 2017-03-28
WO 2016/050975 PCT/FP2015/072868
- 108 -
F N
Si Nb" 0 H
Br
CI I
= 0).......F
F
To a solution of Intermediate 7 (3 g, 5.3 mmol) in Et0H (50 mL) was added
SnC12 (5 g,
26.46 mmol) and the reaction mixture was heated at 80 C for 2h. After
completion of
reaction (monitored by TLC), the reaction mixture was quenched with water (50
mL) and
basified to pH-8 using 1N KOH (100 mL). The reaction mixture was diluted with
ethyl
acetate (100 mL) and filtered through celite. The organic layer was washed
with water
(100 mL), brine (100 mL), dried over anhydrous sodium sulphate and
concentrated under
reduced pressure. The residue was purified by chromatography (SiO2, 0-70%
Et0Ac in
hexane) to afford the title compound (1.1g, 47% yield) as a pale brown solid.
11-1 NMR (400 MHz, CDCI3) 6 7.52 (m, 1H), 7.49 ¨ 7.30 (m, 2H), 7.04 ¨ 6.67 (m,
2H),
6.42 (m, 1H), 6.24 ¨ 5.91 (m, 1H), 5.79 ¨ 5.52 (m, 1H), 3.71 ¨ 3.46 (m, 1H),
3.19 (m,
2H).
LCMS (ES+) RT 2.39 mm, 447.0/449.0/451.0 (M+H)'
INTERMEDIATES 9 AND 10
(1R,3R)-7-bromo-1-12-ch1oro-6-(difluoromethoxy)pheny1]-6-fluoro-23-dihydro-1H-
pyrrolo[1,2-a]benzimidazol-3-oland (1R,3 S)-7-bromo-142-chloro -6-
(difluorometho xy)phenyI]-6-fluoro -2,3-dihydro-1H-p yrro lo [1,2-
a]benzimidazol-3-ol
F F
Br IIN II 6/0N
\ ..." 0 H b. . 0 H
N Br
:
CI s CI I
$
F F
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 109 -
The title compounds were isolated by chiral purification of Intermediate 8
(15g) under
SFC conditions on Chirapak AD (column size: 50*216 mm*mm, flow 360 mL/min,
300mg/injection / frequency: 8.5 minutes, 25 C, CO2 + 20 % Me0H). Chiral
analysis was
done on Chiralpalc AD-H (column size: 250*4.6 mm, 5ittm, flow 1 mL/min at 30 C
using 80 / 20 heptane / ethyl acetate containing 0.1%DEA). Under analytical
conditions
the first eluting diastereoisomers (5.8 and 9.5 minutes) were a mixture of
(1R, 3S) and
(JR. 3R)-7-bromo -142-chloro -6-(difl uoromethoxy)pheny1]-6- fluoro-2,3-
dihydro -1H-
pyrro lo [1,2-a]benzimidazol-3 -el.
(JS, 3R) and (1S, 3S )-7-bromo -142-chloro -6-(di fluoromethoxy)pheny1]-6-
fluoro -2,3-
dihydro- 1H-pyrrolo[1,2-a]benzimidazol-3-ol were isolated at 12.5 minutes and
21.5
minutes.
The mixture of a mixture of (JR. 3S) and (JR. 3R)-7-bromo-1-[2-chloro-6-
(difluoromethoxy)pheny1]-6-fluoro-2,3-dihydro-1H-p yrro lo [1,2-a]b
enzimidazol-3-o1 was
separated by chiral separation under SFC conditions on Chiracel OD (column
size:
50*266 mm*mm, flow 360 mL/min, 80mg/injection / frequency: 4 minutes, 25 C,
CO2+
% Me0H). Chiral analysis was done on Chiralpak AD-H (column size: 250*4.6 mm,
5)1m, flow 1 mL/min at 30 C using 70 / 30 heptane / ethyl acetate containing
0.1%DEA). Under analytical conditions the first eluting diastereoisomer (4.9
minutes)
was the trans isomer, (1R,3 S)-7-bromo-142-chl oro-6-(difluorom ethoxy)pheny1]-
6-
20 fluoro-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazol-3-ol. Combined
fractions were
evaporated to yield Intermediate 10 (12.7 g, 50%). 1HNMR (400 MHz, CDC13) 6
7.41
(m, 3 H), 7.23 (d, J8.0 Hz, 0.4 H), 6.97 (m, 1.2 H), 6.85 (d, J5.8 Hz, 0.4 H),
6.73 (t, J
72.3 Hz, 0.4 H), 6.41 (m, 1 H), 5.95 (dd, J74.2, 70.8 Hz, 0.6 H), 5.71 (m, 0.6
H), 5.62 (d,
J7.4 Hz, 0.4 H), 3.22 (m, 2 H). as a mixture of rotamers 6/4. LCMS basic (ES)
2.50
mm., 446.96/448.95/450.95 (M+H)+.
Under analytical conditions the second eluting diastereoisomer (6.6 minutes)
was the cis
isomer, (1R,3R)-7-bromo-1-[2-chloro-6-(difluoromethoxy)pheny1]-6-fluoro-2,3-
dihydro-
1H-pyrrolo[1,2-a]benzimidazol-3-ol. Combined fractions were evaporated to
yield
Intermediate 9(6.6 g, 26%). Ili NMR (400 MHz, CDC13) 6 7.45 (d, J 8.5 Hz, 1
H), 7.31
(m, 1.8 H), 7.20 (m, 0.6 H), 7.08 (d, J 7.9 Hz, 0.6 H), 6.88 (d, J5.5 Hz, 0.6
H), 6.74 (d, J
5.2 Hz, 0.4 H), 6.61 (t, .172.5 Hz, 0.4 H), 6.15 (t, .172.0 Hz, 0.6 H), 6.08
(m, 1 H), 5.63
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 110 -
(m, 1 Fi), 3.56 (m, 0.6 H), 3.43 (m, 0.4 H), 2.98 (m, 0.4 H), 2.80 (m, 0.6
H),as a mixture
of rotamers 6/4. LCMS acid (ES) 2.20 mm, 446.96/448.95/450.91 (M+H)+.
Under preparative conditions the order of elution was reversed.
INTERMEDIATE 10
(1R,3 S)-7-bromo -142-chloro -6-(difluoromethoxy)pheny11-6-fluoro -2,3 -
dihydro-1H-
pyrro lo [ 1,2-aThenzimidazol-3 -o I
410 Nib/00 H
Br
CI
4* 0
\r-F
The title compound can also be prepared by the following procedure:
Intermediate 9
(3.65 g, 8.146 mmol, 1 eq) and triphenylphosphine (2.62 g, 9.775 mmol, 1.2 eq)
were
solubilized in 8 mL of dry THF, under an inert atmosphere of nitrogen. Acetic
acid (513
pi, 8.960 mmol, 1.1 eq) was added and the mixture cooled to 0 C. A solution of
DIAD
(2.42 mL, 12.220 mmol, 1.5 eq) in 8 ml. of dry THF was added drop wise. The
reaction
was slowly warmed to r.t. and the reaction continued for 2 hours at this
temperature. 20
mL of ethyl acetate were added to the reaction mixture before washing with 3 x
10 mL of
a saturated solution of NaHCO3. The organic layer was dried over anhydrous
magnesium
sulfate and concentrated under vacuum. The residue was purified by
chromatography
(SiO2, 5% McOH in DCM) giving 4.8g (94% yield) of the inverted acetate
intermediate
which was used directly used in the next step. [(1R,3S)-7-bromo-142-chloro-6-
(di fluorometh oxy)ph enyI]-6-fl uoro -2,3-dihydro-1 H-pyrro lo [1,2-a]ben zim
dazol-3-yl]
acetate (4.8 g, 9.800 mmol, 1 eq) was solubilized in 48 mL of methanol.
Potassium
carbonate (1.4 g, 9.800 mmol, 1 eq) was added and the reaction continued for 1
hour at
r.t. The reaction was evaporated and the residue was taken up in ethyl acetate
(50 mL)
and water (20 mL). The organic layer was washed by water (2x 20 mL), dried
over
sodium sulfate, filtered and concentrated under vacuum to give 4.7g of the
crude title
compound as a slightly beige solid.
CA 02962826 2017-03-28
WO 2016/050975 PCT/FP2015/072868
- 111 -
INTERMEDIATE 11
tert-buty1-dimethyl-11-methy1-145-(4.4.5.5-tetramethyl-1.3.2-dioxaborolan-2-
yl)pyrimidin-2-yl] etho xy] si lane
2-(1-hydroxy-1-incthylethyppyrimidinc-5-boronic acid pinacol ester (10 g,
37.8601
mmol), tert-butyldimethylchlorosilane (11.76 g, 75.72 mmol) and imidazo le
(7.890 g,
115.89 mmol) were dissolved in anhydrous DMF (150 mL). The reaction was
stirred at
85 C for 4 days. Et0Ac (100 mL) and water (250 mL) were added, the aqueous
layer was
extracted with 3x20 mL of Et0Ac then the combined organic layers were washed
with
brine (3x20 mL). and dried over MgSO4, filtered and concentrated in vacua. The
residue
was purified by chromatography (SiO2, 0-100% Et0Ac in heptane) to afford the
title
compound as a transparent oil (12.0g, 83.76%).
'H NMR (400 MHz, CDC13) ö 9.04 (s, 2 H), 1.70 (s, 6 H), 1.40 (s, 12 H), 0.94
(s, 9 H),
0.01 (s, 6 H). LCMS acid (ES) RT 3.04min, 297.20 (M+H)+.
INTERMEDIATE 12
(1R,3S)-7-[2-[14tert-butyl(dimethypsilyl]oxy-1-methyl-ethylipyrimidin-5-y1]-
142-
chloro-6-(difluoromethoxy)pheny11-6-fluoro-2,3-dihydro-1H-pyrrolo[1,2-
a]benzimidazol-
3-ol
3,0H
N
I CI
>1õ *
si
Intermediate 10(5.12 g, 11.44 mmol,), Intermediate 11(5.19 g, 13.73 mmol) and
cesium
carbonate (5.59 g, 17.16 mmol, 1.5 eq) were placed in a tube, and filled with
argon.
Degassed 1,4-dioxane (41.2 mL, 3.6 mL/mmol) and degassed water (4.1 mL, 0.36
mL/mmol) were added and the resulting slurry was stirred at r.t. for 5 minutes
before
addition of [1,1'-bis(diphenylphosphino)ferrocene]di chloropalladium(II)
(418.7 mg,
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
-112-
0.572 mmol, 0.05 eq). The reaction mixture was placed on a pre-heated stirring
plate at
90 C and stirred at this temperature for 2 h. Reaction mixture was cooled down
to r.t.
before addition of 50 mL of ethyl acetate and 50 mL of water. The aqueous
layer
extracted with 3 x 20 mL of ethyl acetate. Combined organic layers were dried
over
anhydrous magnesium sulfate, filtered and concentrated under reduced pressure.
The crude was purified by chromatography (SiO2, 30-100% Et0Ae in heptane) to
afford
the title compound (3.8 g, 53% yield).
LCMS basic (ES') RT 3.54min., 619.20 / 621.16 (M+H)+.
INTERMEDIATE 13
(1R,3R)-74241-[tert-b u tyl(d imet hyps ilyl]oxy-l-methyl-e thyll pyrimidin-5-
y1]-112-
chloro-6-(difluoromethoxy)phenyl] -6-fluoro-2,3-dihy dro-1H-pyrro lo
enzimidazo
3-amine
H2
CI
O/
0
Intermediate 12 (3.369 g, 5.441 mmol) was suspended in 11 mL of dry toluene.
At 0 C,
diphenylphosphoryl azide (1.58 mL, 7.071 mmol) was added followed by addition
of 1,8-
diazabicyclo[5.4.0]undec-7-ene (1.139 mL, 7.616 mmol). The reaction was
allowed to
reach r.t. and stirred at this temperature for 2h then heated at 50 C for 18h.
The reaction mixture was diluted by 100 mL of water and 100 mL of ethyl
acetate. The
aqueous layer was extracted by ethyl acetate (3 x 20 mL). Combined organic
layers were
washed with successivelly 20 mL of a saturated solution of NH4C1 and 20 mL of
a
saturated solution of NaHCO3, dried over anhydrous magnesium sulfate and
concentrated
in vacuum.
The crude azide intermediate was solubilized in a solution of tetrahydrofuran
(50 mL, 10
mL / mmol) and water (5 mL, 1 mL / mmol) before addition of 1 M solution of
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 113 -
trimcthylphosphinc in toluene (11 mL, 11 mmol). The reaction mixture was
stirred at r.t.
for 2h. Solvents were evaporated and the residue was purified by
chromatography (SiO2,
5-8% Me0H in DCM) to afford the title compound (2.8 g, 83% yield).
LCMS basic (ES') RT= 3.52min., 618.20/620.20 (M+H)+.
INTERMEDIATE 14
(7R,14R)-1112-(2- qtert-butyl(dimethyl)silyl]oxy) propan-2-yl)pyrimidin-5-yl] -
1-
(di fluoromethoxy)-10- fluoro-6,7-dihydro -7,14-rn eth anobenzi mi dazo [1,2-
b] [2,5]ben zodiazocin-5 (14H)-one
FYy
0N ''''' H
N
0
0 /
Intermediate 13 (2.78 g, 4.50 mmol, 1 eq), sodium carbonate (2.38 g, 22.5
mmol, 5 eq)
and dichloro bis(dicyclohexylphosphino)propane] palladium(II) [Pd-133 from
Johnson
Matthey] (552 mg, 0.899 mmol, 0.2 eq) were solubilized / suspended in degassed
(nitrogen) 1,4-dioxane (54 mL, 12 mL / mmol). The reaction mixture was heated
overnight at 150 C under 5 atmosphere of CO gas. The reaction mixture was
filtered over
celite, which was thoroughly washed with ethanol. Solvents were evaporated and
the
residue was purified by chromatography (SiO2, 80-100% Et0Ac in hcptanc),
giving the
title compound (1.39 g, 50% yield).
LCMS basic (ES) RT 3.47 min., 610.25/611.25 (M+H)"
INTERMEDIATE 15
ethyl (3R)-3-(5-bromo-2-nitro-anilino)-342-chloro-6-
(difluoromethoxy)phenyl]propanoatc
Intermediate 15 was prepared, using the same procedure described for
preparation of
Intermediate 5, from Intermediate 4 (9.3g, 28.3 mmol) and 4-brorno-2-fluoro-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 114 -
nitrobenzene (7.4g, 34 mmol). The reaction was stirred overnight at 80 C and
purified by
chromatography (SiO2, 10% Et0Ac in hexane). Intermediate 15 was obtained as a
yellow oil (12.5g, 90% yield).
LCMS (ES) 495 (M+H)+
INTERMEDIATE 16
(3R)-3(5-bromo-2-nitro-anilino)-342-chloro-6-(difluoromethoxy)phenyl]prop anal
Intermediate /6 was prepared from Intermediate 15 (12.5g, 25.4 mmol) using the
same
procedure described for preparation of Intermediate 6. Following work-up the
crude
Intermediate 16 was purified by chromatography (SiO2, 15% Et0Ac in hexane)
yielding
Intermediate 16 (9g, 80% yield) as a yellow oil.
IHNMR (400 MHz, CDC13) 6 9.80 (d, J 1.3 Hz, 1H), 8.78 (d, J 9.0 Hz, 1H), 7.99
(d, J
9.0 Hz, 1H), 7.27 (d, J3.2 Hz, 2H), 7.21 ¨ 7.08 (m, 1H), 6.81 ¨6.66 (m, 2H),
5.93
(m,1H), 3.56 ¨ 3.38 (m, 2H), 3.12 (dd, J17.9, 5.2 Hz, 1H).
INTERMEDIATE 17
(4R)-445-bromo-2-nitro-anilino)-4-[2-chloro-6-(difluoromethoxy)pheny1]-2-
trimethylsilyloxy-butanenitrile
Intermediate 17 was prepared from Intermediate 16 (9g, 20 mmol) using the same
procedure described for preparation of Intermediate 7. The reaction was
stirred at r.t. for
2h. After completion of reaction (monitored by TLC), water (200 mL) was added
and
extracted with DCM (500 mL). After evaporation of organic layer, the crude
product,
obtained as a yellow oil (9g), was used directly for the next step without any
purification.
INTERMEDIATE 18
(1R)-7-bromo-142-chloro-6-(difluoromethoxy)pheny1}-2,3-dihydro-1H-pyrro lo [1
,2-
a]benzimi dazol-3 -01
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 115 -1411
Br
CI
Intermediate 18 was prepared from Intermediate /7 (9g, 16.4 mmol) using the
same
procedure described for preparation of Intermediate 8. The crude product was
purified by
chromatography (SiO2, 60% Et0Ac in hexane) then triturated with hexane:ethyl
acetate
to yield the title compound (3g, 43% yield) as a yellow solid.
LCMS (ES) 431 (M+H)+
INTERMEDIATE 19 AND 20
(1R,3R)-7-bromo-142-chloro-6-(difluoromethoxy)pheny1]-2,3-dihydro-1H-pyrro to
[1,2-
a]benzirnidazol-3-ol and (1R,3 S)-7-bromo -142-chloro-6-
(difluoromethoxy)pheny11-2,3-
dihydro-1H-pyrro lo [1,2-a]b enzimidazo1-3 -01
ik 0 H 0111 H
Br Br
CI CI
*
The title compounds were isolated by chiral purification of Intermediate 18
(12.5g) by 2
successive chiral separation.
First chiral separation:
Under SFC conditions on Chiracel OD (column size: 50*266 mm*mm, flow 360
mL/min,
20mg/injection / frequency: 4 minutes, 25 C, CO2 + 20 % Me01-1). Chiral
analysis was
done on Chiralcel OD-H (column size: 250*4.6 mm, flow 1 mL/min at 30 C using
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 116 -
100% methanol containing 0.1%DEA). Under analytical conditions the first
eluting
diastereoisomer (3.9 minutes) was either (IS, 3R) or (IS, 3S) 7-bromo-142-
chloro-6-
(difluoromethoxy)pheny1]-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazol-3-ol. The
second
eluting diastereoisomers (4.7 minutes) were a mixture of (1R, 3S) along with
either (1S,
3R) or (1S, 3S)-7-bromo-142-chloro-6-(difluoromethoxy)phenyl] -2,3-dihyd
ro -1H-
pyrrolo[1,2-a]benzimidazol-3-ol and the third eluting diastereoisomer (5.4
minutes) was
(JR. 3R)-7-bromo-142-chloro-6-(difluoromethoxy)p henyll -2,3-dihydro-1H-p
yrrolo [1,2-
a]benzimidazol-3-ol. Combined fractions of the third eluting diastereomer were
evaporated to yield to Intermediate 19 (3.63g, 29%). 1FINMR (400 MHz, DMSO) ö
7.57
(m, 2.3 H), 7.45 (m, 0.8 H), 7.35 (d, J 8.0 Hz, 0.6 H), 7.26 (m, 1 H), 7.17
(m, 0.3 H),
6.83 (t, J72.5 Hz, 1 H), 6.69 (bs, 1 H), 6.15 (m, 1 H), 6.07 (m, 1 H), 5.38
(m, 1 H), 3.38
(m, 1 H), 2.67 (m, 1 H) as a mixture of rotamers 7/3. LCMS acid (ES') RT 4.31
min.,
429.10/431.08/433.05 (M+H)1.
Second chiral separation:
Under SFC conditions on Whelko 01 (R,R) (column size: 50*227 mm*mm, flow 360
mL/min, 690mg/injection / frequency: 5.5 minutes, 25 C, CO2 + 20 % Et0H).
Chiral
analysis was done on Chiralcel OD-H (column size: 250*4.6 mm, flow 1 mL/min at
30 C using 50 / 50 heptane / isopropyl alcohol containing 0.1%DEA).
Under analytical conditions, the first eluting diastereomer (4.1 minutes) was
either (IS,
3R) or (1S,35)-7-bromo-142-chloro-6-(di fluoromethoxy)phenyl] -2,3-d
ihydro -1H-
pyrro lo[1,2-a]benzimidazo 1-3-ol.
Under analytical conditions, the second eluting diastereomer (5.9 minutes) was
the trans
isomer, (1R,3S)-7-bromo-1-[2-chloro-6-(difluoromethoxy)pheny1]-2,3-dihydro-1H-
pyrrolo[1,2-a]benzimidazol-3-ol. Combined fractions were evaporated to yield
Intermediate 20 (4.46g, 36%). 11INMR (400 MHz, DMSO) 8 7.55 (m, 3.4 H), 7.31
(m,
1.4 H), 7.12 (d, J7.8 Hz, 0.6 H), 7.03 (t, J73.0 Hz, 0.6 H), 6.89 (s, 0.6 H),
6.81 (s, 0.4 H),
6.32 (dd, J8.4, 5.9 Hz, 1 H), 6.10 (d, J6.6 Hz, 1 H), 5.32 (m, 0.6 H), 5.26
(t, J6.9 Hz, 0.4
H), 3.13 (m, 1 H), 2.93 (m, 1 H). as a mixture of rotamers 6/4. LCMS acid (ES)
RT 4.40
mm., 429.05/431.08/433.05 (M+H)'.
Under preparative conditions the order of elution was reversed.
CA 02962826 2017-03-28
WO 2016/050975
PCT/FP2015/072868
- 117 -
INTER1VIEDIATE 20
(1R,3S)-7-bromo-142-chloro-6-(difluoromethoxy)phenyll-2,3-dihydro-1H-pyrro lo
[1,2-
a]benzimidazol-3 -01
Nbe0 H
Br
CI
The title compound was prepared from the same procedure as the preparation of
Intermediate 10 starting from Intermediate 19 (3.63 g, 8.450 mmol),
triphenylphosphine
(2,66 g, 10.14 mmol), and acetic acid (0.5 mL, 9.295 mmol) THF (34 mL),DIAD
(2.62
mL, 12.67 mmol) in 5 ml of dry THF giving 3.6 g (91%) of the inverted acetate
intermediate which was used directly in the next step. Using the following
conditions.
[(1R,3S)-7-bromo-142-chloro-6-(difluoromethoxy)pheny11-2,3-dihydro-1H-
pyrrolo[1,2a]benzimidazol-3-yll acetate (4.0 g, 8.480 mmol) was solubilized in
40 mL of
methanol. Potassium carbonate (1.1 g, 8.48 mmol, 1 eq) was added and the
reaction
continued for 1 hour at rt. The methanol was evaporated and the residue was
taken up in
ethyl acetate (50 mL) and water (20 mL). The organic layer was washed by water
(2x 20
mL), dried over anhydrous sodium sulfate, filtered and concentrated under
vacuum to
give 4.9 g of crude title compound as a brown oil use without further
purification.
LCMS basic (ES) RT 2.46 min., 428.94 / 430.96/433.16 (M+H)'.
INTERMEDIATE 21
(1R,3S)-7-[2-[1-[tert-butyl(dimethyl)silyl]oxy-l-methyl-ethyllpyrimidin-5-y11-
142-
chloro-6-(difluoromethoxy)pheny1]-2,3-dihydro-1H-p. olo [
cnzimidazol-3-o I
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 118 -
N N
CI ;
0 /
fit 0 F
Si¨
Intermediate 20 (4.46g, 10.38 mmol), Intermediate //(3.92 g, 10.38 mmol)
following the
protocol described for intermediate 12 using cesium carbonate (5.07 g, 15.57
mmol), 1,4-
dioxane (37.1 mL, 3.6 inUmmol), water (3.7 mL, 0.36 mL/mmol), [1,1'
bis(diphenylphosphino)ferrocene]dichloropalladium(II) (379.8 mg, 0.5191 mmol,
0.05
eq), The crude was purified by chromatography (SiO2, 30-100% Et0Ac in hexane)
to
yield the title compound (5.7g, 92% yield).
LCMS acid (ES) RT 3.64 min., 601.29 / 603.21 (M+H)f
INTERMEDIATE 22
(1R,3R)-74241-[tert-butyl(dimet hyl)silyl]oxy-1-methyl-ethyl]pyrimidin-5-yl] -
142-
chloro-6-(difluoromethoxy)pheny1]-2,3-dihydro -1H-p yrro lo [ 1,2-alb
enzimidazol-3-amine
H2
N
\rL
I
CI ; N
0 /
=.c; *
Intermediate 22 was prepared from Intermediate 21 following the protocol
described for
intermediate 13, using toluene (34 mL), diphcnylphosphoryl azidc (5.0 mL,
24.22 mmol),
1,8-diazabicyclo[5.4.0]undcc-7-cne (3.62 mL, 24.22 mmol) for thc first step
and
tetrahydrofuran (172 mL), water (17 mL), 1 M solution of trimethylphosphine in
toluene
(34.6 mL, 20.8 mmol) for the second step.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 119 -
Thc crude residue was purified by chromatography (SiO2, 0-5% McOH in DCM, 1%
NH4) to afford the title compound (7g, 61% yield).
LCMS basic (ES) RT 3.49min., 600.25 / 602.25 (M+1-1)+
INTERMEDIATE 23
(7R,14R)-11-[2-( 2- { Itert-butyl(dimethypsilyl]oxylpropan-2-yl)pyrimidin-5-
y1]-1-
(difluoromethoxy)-6,7-dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-
5(14H)-one
I 0
0 /
0
Si-
/**.\
Intermediate 23 was prepared from Intermediate 22 (7.00 g, 7.931 mmol)
following the
protocol described for Intermediate 14 using sodium carbonate (6.181 g, 58.31
mmol),
dichloro bis(dicyclohexylphosphino)propane] palladium(I1) [Pd-133 from Johnson
Matthey] (1.43 g, 0.254 mmol), 1,4-dioxane (95 mL, 12 mL / mmol) and 5
atmosphere of
CO gas.
The crude was purified by chromatography (SiO2, 50-100% Et0Ac in heptane) to
afford
the title compound (3.2 g, 62% yield) as a white solid.
1H NMR (400 MHz, DMSO) ö 9.23 (d, J6.8 Hz, 1 H), 9.16 (s, 2 H), 8.32 (dd,
J5.9, 3.5
Hz, 1 H), 7.88 (dd, J51.9, 43.2 Hz, 1 H), 7.84 (s, 1 H), 7.71 (dd, J8.3, 1.8
Hz, 1 H), 7.60
(m, 3 H), 6.47 (d, .17.1 Hz, 1 H), 4.99 (t, J 6.8 Hz, 1 H), 3.58 (m, 1 H),
2.85 (dõ./ 13.4 Hz,
1 H), 1.76 (s, 6 H), 0.95 (s, 9 H), 0.01 (s, 6 H). LCMS basic (ES') RT 3.43
min., 592.27
(M+H).
INTERMEDIATE 24
(7R,14R)-1142-(2- fitert-butyl(dimethyl)silyl]oxy} propan-2-yl)pyrimidin-5-y1]-
1-
(difluoromethoxy)-6-methy1-6,7-dihydro-7,14-methanobenzimidazo [1,2-
b] [2,5]benzodiazocin-5 (14H)-one
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 120 -
N
0
0
Intermediate 23 (1.0 g, 1.690 mmol) was dissolved in dry THF (10 mL/g) and
tetrabutylammonium iodate (0.250 g, 0.676 mmol) was added. At 0 C, sodium
hydride
(60% in mineral oil) (0.081 g, 2.028 mmol) was added and the reaction mixture
was
stirred at r.t. for 35 minutes. lodomethane (0.727 g, 5.070 mmol) was added
and the
reaction mixture was stirred at r.t.for 18h. Distilled water (200 mL) was
added, the
mixture was extracted by of ethyl acetate (3 x 150 mL). Combined organic
layers were
dried over anhydrous magnesium sulfate and concentrated in vacuo . The residue
was
purified by chromatography (SiO2, 50-100% Et0Ac in heptane) to afford the
title
compound (0.868 g, 85% yield).
1H NMR (400 MHz, CDC13) 8.95 (s, 2 H), 8.54 (d, 8.2 Hz, 1 H), 7.93 (d, J 8.5
Hz, 1
H), 7.80 (s, 1 H), 7.59 (d, J 8.5 Hz, 1 H), 7.49 (t, J 8.2 Hz, 1 H), 7.37 (d,
J 8.2 Hz, 1 H),
6.89 (t, J 72.5 Hz, 1 H), 6.42 (d, J 6.9 Hz, 1 H), 5.30 (d, J 6.7 Hz, 1 H),
3.63 (m, 4 H),
2.99 (d, J 13.6 Hz, 1 H), 1.75 (s, 6 H), 0.92 (s, 9 H), 0.00 (s, 6 H). LCMS
basic (ES) RT
3.51 min., 606.25 (M+H)+
INTERMEDIATE 25
(7R,14R)-11-[2-(2- { [tert-butyl(dimet hyl)silyl]oxyl propan-2-yl)pyrimidin-5-
y1]-1-
(difluoromethoxy)-6-ethy1-6,7-dihydro-7,14-methanobenzimidazo[1,2-
b][2,5ibenzodiazocin-5(14H)-one
FQ
0
= Ø
Intermediate 23 (0.075 g, 0.127 mmol, 1 eq) was dissolved in dry THF (10 mL).
At 0 C,
sodium hydride (60% in mineral oil) (0.008 g, 0.190 mmol) was added and the
reaction
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 121 -
mixture was heated at 65 C for 2.5 h, then was allowed to reach r.t. and
iodocthanc (0.059
g, 0.380 mmol) was added. The reaction mixture was then stirred at r.t. for 60
h.
Additional Iodoethane (50 iaL) was added and the reaction mixture was stirred
at r.t.for 2
h. Distilled water (20 mL) was added, the mixture was extracted with 3 x 20 mL
of ethyl
acetate. Combined organic layers were dried over anhydrous magnesium sulfate
and
concentrated in vacuum.The residue was purified by chromatography (SiO2, 50-
100%
Et0Ac in heptane) to afford the title compound as a white solid (0.069 g, 88%
yield).
LCMS basic (ES') RT 2.24 min. 506.23 (M+H).
INTERMEDIATE 26
(7R,14R)-11-[2-(2- {[tert-butyl(dimethyl)silylioxyl propan-2-yl)pyrimidin-5-
y1]-1-
(difluoromethoxy)-6-(propan-2-y1)-6,7-dihydro-7,14-methanobenzimidazo[1,2-
b112,5Thenzodiazocin-5(14H)-one
N
N
0 /
0
sao-
A solution of Intermediate 23 (25 mg, 0.0423 mmoL), potassium hydroxide (2.85
mg,
0.0507 mmoL), tetrabutylammonium bromide (12.26 mg, 0.0380 mmoL) and 2-
iodopropanc (14.36 mg, 0.0845 mmoL) in dry THF (0.8 mL) was stirred at r.t.for
24 h.
The reaction mixture was quenched with water and extracted with ethyl acetate.
The
organic layer was washed with brine, dried over anhydrous magnesium sulphate
and
concentrated to dryness to afford the title compound (10 mg) which was used in
the next
step without further purification.
LCMS basic (ES) RT 3.60 min. 534.30 (M+H)-.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 122 -
INTERMEDIATE 27
(1R,3S)-142-chloro-6-(difluoromethoxy)pheny11-6-fluoro-742-(1-hydroxy-1-methyl-
ethyppyrimidin-5-y11-2,3-dihydro-1H-pyrrolo[1,2-apenzimidazo1-3-ol
r:6=0 H
I
CI
4.1
0 H *
Intermediate 27 was prepared from Intermediate 20 (4.61 g, 10.30 mmol),
following the
protocol described for Intermediate 12, using 2-[5-(4,4,5,5-tetramethy1-1,3,2-
dioxaborolan-2-yl)pyrimidin-2-yl]propan-2-ol (3.00 g, 11.330 mmol), cesium
carbonate
(5.03 g, 15.450 mmol), 1,4-dioxane (37.1 mL), water (3.7 mL), [1,1'-
bis(diphenylphosphino)ferrocene]dichloropalladium(II) (376.8 mg, 0.5150 mmol).
The
crude was purified by chromatography (SiO2, 30-100% Et0Ac in heptane) to
afford the
title compound (3.6 g, 69%).
NMR (400 MHz, CDCb) 6 8.77 (s, 1.2 H), 8.73 (s, 0.8 H), 7.59 (d, J = 11.0 Hz,
1 H),
7.37 (m, 2.8 H), 7.23 (m, 0.6 H), 6.99 (d, J = 8.2 Hz, 0.6 H), 6.84 (d, J =
6.5 Hz, 0.6 H),
6.74 (t, J = 72.5 Hz, 0.6 H), 6.70 (d, J = 6.5 Hz, 0.4 H), 6.51 (m, 1 H), 6.02
(dd, J1 = 74.0
Hz, J2 = 71.0 Hz, 0.4 H), 5.77 (dd, J1 = 7.8 Hz, J2 = 3.3 Hz, 0.6 H), 5.68 (d,
J = 7.1 Hz,
0.4 H), 4.60 (bs, 1 H), 3.27 (m, 2 H), 1.64 (s, 3.60 H), 1.62 (s, 2.40). LCMS
acid (ES)
RT 1.91 mm. 505.15 / 507.15 (M+H)+.
1NTER1V1ED1ATE 28
Butyl 3-(di fluoromethoxy)-2-[(1R,3 S)-6-fluoro -3-hydroxy-742-(1-hydroxy- 1-
methyl-
ethy Opyrimidin-5-y1]-2,3-dihydro -1H-pyrro lo b enzimidazo l-1-yl] b enzo
ate
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 123 -
F
N
I F
0 0
N
0 H F *
A solution of Intermediate 27(900 mg, 1.783 mmoL), sodium carbonate (944 mg,
8.913
mmoL), dichloro [bis(dicyclohexylphosphino)propanc] palladium(11) (54.7 mg,
0.08913
mmol) in 10 mL of 1-butanol was heated for 16 hat 150 C under 4 atm of CO gas.
The reaction mixture was concentrated in vacuo, the residue was taken up in 50
mL of
ethyl acetate and washed with 3x 20 mL of NaOH 0.1 M. The organic layer was
dried
over anhydrous sodium sulphate and concentrated in vacuo. The crude material
was
purified by chromatography (SiO2, 80% Et0Ac in hexane), to afford the title
compound
(490 mg, 48.2 % yield).
LCMS acid (ES) RT 2.66 min. 571.25 (M+H)+.
INTERMEDIATE 29
3-(difluoromethoxy)-2-[(1R,3S)-6-fluoro-3-hydroxy-7-[2-(1-hydroxy-1-methyl-
ethyppyrimidin-5-y1]-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazo1-1-ylThenzoic
acid
FyN
N./.
0 0
0 H F =
OH
Intermediate 28 (470 mg, 0.8237 mmoL) was dissolved in 4.7 mL of methanol. A 5
N
solution of sodium hydroxide (0.3295 mL, 1.647 mmol) was added and the mixture
was
stirred at room temperature for 48h. The reaction mixture was neutralized with
HCI IN
and the solvent was evaporated. The aqueous phase was extracted with ethyl
acetate (3 x
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 124 -
25 mL), the combined organic layers were dried over magnesium sulphate and
concentrated in vacuo to afford crude 511 mg of the title compound used
without further
purification.
LCMS acid (ES) RT 2.05 min., 515.17 (M+H)+.
INTERMEDIATE 30
Ethyl 6-bro mo imidazo [1,2-a] pyrid ine-2-carbo xylate
The title compound was prepared according to the procedure provided in
international patent application WO 2014/009295.
INTERMEDIATE 31
3-(6-bromo-2-ethoxycarbonyl-imidazo[1,2-a]pyridin-3-y1)-3-(2-
chlorophenyl)propanoic acid
NO
BrN 0-\
OH
CI 0
Intermediate 30 (8 g, 29.73 mmol), 2-chlorobenzaldehyde (6.7 ml, 59.58 mmol),
2,2-dirnethy1-1,3-dioxane-4,6-dione (8.6 g, 59.67 mmol), L-proline (170 mg,
1.48 mmol)
and MgSO4 (11 g, 91.39 mmol) in acetonitrile (80 mL) was heated at 90 C for
33 h,
followed by 100 C 15h. The reaction cooled to r.t., and the solid filtered
off, and washed
with methanol (2 x 50 rriL). The filtrate was concentrated in vacua and
triturated with
diethyl ether (50 mL) and sonicated for 10min and resulting gum was filtered
off and
rinsed with diethyl ether (2 x 50 mL) yielding the title compound as beige
solid (10.2g,
76%). Ili NMR (500 MHz, Methanol-d4) 6 8.79 (s, 1H), 7.74 (d, J 7.7 Hz, 1H),
7.67 -
7.55 (m, 2H), 7.43 -7.34 (m, 2H), 7.34 - 7.20 (m, 1H), 5.56 (dd, J 9.5, 6.0
Hz, 1H), 4.35
(qt, J 7.4, 3.7 Hz, 2H), 3.70 (dd, J 16.7, 9.5 Hz, 1H), 3.40 (dd, J 16.7, 6.0
Hz, 1H), 1.34
(t, J 7.1 Hz, 3H). LCMS (ES) RT 1.24 min, 451.0/453.0 (M+H)+.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 125 -
INTERMEDIATE 32
Ethyl 6-bromo-311-(2-chloropheny1)-3-ethoxy-3-oxo-propyl]imidazo[12-alpyridine-
2-
carboxylate
Br \ N 0
0
CI
Thionyl chloride (4 ml, 55.14 mmol) was added to the stirred solution of
Intermediate 31(10.2 g, 20.1 mmol) in Et0H (100 mL) at 0 C. The reaction
mixture
warmed to r.t. and stirred for 20h. The reaction mixture was concentrated in
vacuo and
resulting residue was triturated with Et0Ac (100 mL) and washed with sat.
NaHCO3 (100
mL) and further extracted with Et0Ac (2 x 50 mL). The combined organic layers
were
washed with water (50 mL), brine (50 mL), dried over sodium sulfate, filtered
and
concentrated in vacuo. The residue was purified by chromatography (SiO2, 0-
100%
Et0Ac in heptane) yielding the title compound as an orange gum (8.2g, 85%). 'H
NMR
(500 MHz, CDC13) 6 8.40 (s, 1H), 7.65 (dd, J 7.8, 1.5 Hz, 1H), 7.55 (d, J 9.5
Hz, 1H),
7.34 (dd, J 7.9, 1.4 Hz, 1H), 7.28 (dd, J9.5, 1.8 Hz, 1H), 7.27 ¨ 7.23 (m,
1H), 7.20 (td, J
7.6, 1.6 Hz,1H), 5.44 (dd, J9.8, 5.6 Hz, 1H), 4.42 (q, J 7.1 Hz, 2H), 4.09¨
3.94 (m, 2H),
3.82 (dd, J 16.6, 9.9 Hz, 1H), 3.26 (dd, J 16.6, 5.5 Hz, 1H), 1.40 (t, J 7.1
Hz, 3H), 1.12
(t, J7.1 Hz, 3H). LCMS (ES') RT 1.44 min, 479.0/481.0 (M+H)+.
INTERMEDIATE 33
Ethyl 7-bromo-1-(2-chloropheny1)-3-oxo-2,3 -dihydro-1H-cyclopenta[4,5]imidazo
[1,2-
a1pyridine-2-carboxylate
0
/
Br
'J,0
0
CI sl
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 126 -
Intermediate 32 (4 g, 8.34 mmol) was co-evaporated twice with toluene (50 mL)
and the residue dissolved in dry toluene (400 mL) and degassed with N2(g) for
5 mm. The
mixture was cooled to -10 C (external temp) and a 25% w/w solution of
potassium 2-
methylbutan-2-olate in toluene (7.5 mL, 13.37 mmol) was then added drop wise
and
stirred for 30min at -10 C. The reaction mixture was quenched with acetic acid
(2 mL)
and diluted with water (200 mL), extracted with Et0Ac (2 x 200 mL). The
combined
organic layer was washed with sat. aq. sodium bicarbonate (100 mL), brine (100
mL),
dried over sodium sulfate, filtered and concentrated in vacuo yielding the
title compound
as a pale yellow solid (3.3 g, 82%). 11-1NMR (500 MHz, CDC13) 6 7.77 (s, 1H),
7.65 (d, J
9.8 Hz, 1H), 7.52 (d, J 6.7 Hz, 111), 7.40 (dd, J 9.8, 1.5 Hz, 1H), 7.32 (t, J
8.3 Hz, 1H),
7.25 - 7.15 (m, 1H), 6.67 (s, 1H), 5.59 (s, 1H), 4.30 (q, .17.1 Hz, 2H), 3.89
(s, 1H), 1.33
(t, 7.1 Hz, 3H). LCMS (ES') RT 1.35 min, 433.0/435.0 (M+H)".
INTERMEDIATE 34
7-bromo-1-(2-chloropheny1)-1,2-dihydro-3H-cyc lopenta[4,5] imidazo [1.,2-
alpyridin-3-o no
0
Br
411 CI
Intermediate 33 (3.2 g, 6.64 mmol) was dissolved in DMSO (50 mL) and water
(10 mL) then heated at 100 C for 48h. The reaction was cooled to r.t. and
poured on to
ice and left to stand for lh. The resulting residue was filtered off and
washed with water
yielding the title compound as a pale yellow solid (2.5 g, 99%). NMR
(500 MHz,
CDC13) 6 7.81 (s, 1H), 7.65 (dd, J 9.8, 0.8 Hz, 1H), 7.52 (d, J 8.0 Hz, 1H),
7.38 (dd, J
9.8, 1.8 Hz, 1H), 7.29 (td, J 7.8, 1.6 Hz, 1H), 7.21 (t, J 7.4 Hz, 1H), 6.77
(s, 1H), 5.22
(s,1H), 3.73 (dd, J 18.4, 7.1 Hz, 1H), 2.92 (d, J 19.0 Hz, 1H). LCMS (ES') RT
1.27 min,
361.0/363.0 (M+H)".
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 127 -
INTERMEDIATE 35
1-(2-ehloropheny1)-742-(2-hydroxypropan-2-yl)pyrimidin-5-y1]-12-dihydro-3H-
cyclopenta[4,5]imidazo [1,2-alpyri din-3-one
N
0
N /41111
N ====
OH = ci
Intermediate 34 (1 g, 2.65 mmol), 2-[5-(tetramethy1-1,3,2-dioxaborolan-2-
yl)pyrimidin-2-yl]propan-2-ol (0.84 g, 3.19 mmol) were dissolved in dioxane
(40 mL)
then 2M disodium carbonate (4 mL) was added and the mixture was degassed with
N2
for 5 min. Bis[3-(diphenylphosphanyl)cyclopenta-2,4-dien-1-yl]iron;
dichloromethane;
dichloropalladium (108 mg, 0.13 mmol) was added and the reaction was heated to
80 C
for 1.5 hours. The reaction mixture was then cooled to r.t. and diluted with
water (50 mL)
and extracted with Et0Ac (3 x 50 mL). The combined organic layers were washed
with
brine (50 mL), dried over sodium sulfate, filtered, concentrated in vacuo .
The residue was
purified by chromatography, (SiO2, 10-100% Et0Ac in heptane) yielding the
title
compound as a light brown solid (1.1 g, 96%). 11-1 NMR (500 MHz, CDC13) 6 8.81
(s,
2H), 7.91 (d, J 9.6 Hz, 1H), 7.87 (s, 1H), 7.53 (dd, J 9.5, 1.6 Hz, 2H), 7.29
(td, J 7.8, 1.5
Hz, 1H), 7.21 (t, 7.4 Hz, 1H), 6.84 (s, 1H), 5.31 (s, 1H), 4.45 (s,
1H),3.77 (dd, J 18.3,
7.0 Hz, 1H), 2.99 (d, J 18.9 Hz, 1H), 1.63 (s, 6H). LCMS (ES) RT 1.14 min,
419.0/421.0 (M+1-1)+.
INTERMEDIATE 36
2-15- [(1-(2-chloropheny1)-3-(methoxyimino)-2,3-dihydro-1H-cyclop
enta[4,5Jimidaz0 [1,2-
a]pyridin-7-Apyrimidin-2-yllpropan-2-ol
N
N
0 H C I
- 128 -
Intermediate 35 (500 mg, 1.11 mmol), 0-methylhydroxylamine hydrochloride (185
mg,
2.22 mmol) and sodium acetate (182 mg, 2.22 mmol) in ethanol (20 mL) were
heated at 85 C for
6h. The reaction mixture was cooled to rt., concentrated in vacuo then diluted
with sat. aq. sodium
bicarbonate (50 mL) and extracted with Et0Ac (3 x 50 mL). The combined organic
layers were
washed with brine (25 mL), dried over sodium sulfate, filtered, concentrated
in vacuo yielding the
title compound as beige solid (500 mg, 95%). 1H NMR (500 MHz, CDC13) 8 8.80
(d, J 1.1 Hz, 2H),
7.98 - 7.73 (m, 2H), 7.54 - 7.36 (m, 2H), 7.26 (s, 2H), 6.94 - 6.64 (m, 1H),
5.31 - 5.09 (m, 1H), 4.48
(d, J 9.5 Hz, 1H),4.07 (d, J53.7 Hz, 3H), 3.97(dd, J 18.2, 7.9 Hz, 1H), 3.23 -
3.10 (m, 1H), 1.56 (s,
6H). LCMS (ES) RT 1.17 and 1.29 min, 448.0/450.0 (M+H)+.
INTERMEDIATE 37
2- { 5-[3-amino-1-(2-chloropheny 1)-2,3 -di hy dro-1H-cy c lopenta[4,5] imi
dazo [1,2-al pyri din-7-
yllpyrimidin-2-yl}propan-2-ol
N H2
N N
NyLN
H CI
A solution of Intermediate 36 (500 mg, 1.12 mmol), 7M ammonia in Me0H (0.64
mL) in
methanol (50 mL) was passed over a RaneyTM Nickel cartridge at a flow-rate of
1 ml/min, 60 bar
hydrogen pressure at 80 C in a H-Cube continuous-flow hydrogenation reactor.
This process was
repeated three times and mixture was concentrated in vacuo. The residue was
purified by
preparative HPLC yielding the title compound as an off white solid (300 mg,
64%). A 1.6:1 cis
/trans mixture of distereoisomers was isolated. 1H NMR, cis distereoisomer,
(500 MHz, CDC13) 6
8.78 (s, 2H), 7.74 (d, J20.0 Hz, 211), 7.51 - 7.44 (m, 1H), 7.40 - 7.33 (m,
1H), 7.24 - 7.17 (m, 2H),
7.14 (t, J 7.5 Hz, 1H), 7.00 (dd, J7.6, 1.6 Hz, 1H), 4.96 (dd, J 8.1, 6.0 Hz,
1H), 4.59 (dd, J 7.4, 5.7
Hz, 3H), 3.69 (dt, J 13.5, 8.1 Hz, 1H), 2.11 (dt, J 13.4, 5.6 Hz, 1H), 1.61
(s, 6H). LCMS (ES) RT
1.60min (cis) and 1.65 min (trans), 420.0/422.0 (M+H)+.
Date Recue/Date Received 2022-02-28
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 129 -
INTERMEDIATE 38
\ OH
CI F
r* Br
(1R,35)-7-chloro-142-bromo-6-(difluoromethoxy)pheny1]-2,3-dihydro-1H-
pyrrolo[1,2-
a]benzimidazol-3-ol
The title compound was prepared in a sequence of steps analogous to those
described for
Intermediate 10 starting from 2-bromo-6-hydroxy-benzaldehyde and utilising 4-
chloro-2-
fluoro-nitrobenzene instead of 1-bromo-2,5-difluoro-4-nitrobenzene in the
fifth synthetic
step.
INTERMEDIATE 39
OH
CIF.
F * Br
OR33)-7-chloro-1-[2-bromo-6-(difluoromethoxy)phenyl]-6-fluoro-23-dihydro-1H-
pyrrolo[1,2-a]benzimidazol-3-ol
The title compound was prepared in a sequence of steps analogous to those
described for
Intermediate 10 starting from 2-bromo-6-hydroxy-benzaldehyde and utilising 1-
chloro-
2,5-difluoro-4-nitrobenzene, instead of 1-bromo-2,5-difluoro-4-nitrobenzene in
the fifth
synthetic step.
INTERMEDIATE 40
1411 b.,1\1H2
CIF
Br
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 130 -
(1R,3R)-142-bromo-6-(difluoromethoxy)phenyl]-7-chloro-2,3-dihydro-1H-pyrrolo
[1,2-
aThenzimidazol-3-amine
Intermediate 38 (5 g, 11.64 mmol) was suspended in toluene (22 mL) and cooled
to 0 C
before addition of diphenylphosphoryl azide (3.4 mL, 15 mmol) and 1,8-
diazabicyclo[5.4.0]undec-7-ene (2.5 mL, 16 mmol). The mixture was allowed to
warm up
to r.t and stirred for 2 hours and subsequently at 45 C overnight. The
reaction mixture
was diluted with Et0Ac (150 mL) and the organic phase washed with a saturated
aqueous
solution of ammonium chloride (50 mL) then a saturated solution of aqueous
sodium
bicarbonate (50 mL), and concentrated in vacuo. The crude residue thus
obtained was
solubilized in THF (100 mL) and water (10 mL), trimethylphosphine (17.46 mL,
17.46
mmol) was added and the reaction mixture stirred overnight. The mixture was
concentrated in vacuo, partitioned between Et0Ac (200 mL) and water (150 mL).
The
organic layer was extracted with 0.2M HClaq (3 x 200 mL). The combined acid
layer
was stirred in an ice bath, whilst 10% NaOH solution was added with stirring
until pH
increased to 10. The stirred was continued for further 15 minutes to complete
precipitation. The precipitate was filtered, rinsed with water (20 mL), then
dried under
suction for 10 minutes before drying under high vacuum overnight to afford
3.92 g (78%)
of the title compound as an off white solid. LCMS basic: RT 1.96 rnin. (ES+)
428/430
(M+H)-
INTERMEDIATE 41
CI F 100
H2
41*, Br
(1R,3R)-142-bromo-6-(difluoromethoxy)pheny1]-7-chloro-6-fluoro-2,3-dihydro-1H-
pyrrolo[1,2-aThenzimidazol-3-amine
The title compound was prepared from Intermediate 39 using the experimental
protocol
described for the preparation of Intermediate 40. The crude material was
purified by
column chromatography over silica gel using Et0Ac / Me0H (100/0 to 70/30) as
eluent,
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 131 -
yielding 15g (83%) of the title compound as an amorphous solid. LCMS basic: RT
2.04
min. (ES+) 446/448 (M+H).
INTERMEDIATE 42
to
CI 41:1 411
Fµ _r1
r-
F * r
tert-butyl {(1R,3R)-142-bromo-6-(difluoromethoxy)pheny1]-7-chloro-2,3-dihydro-
1H-
pyrrolo[1,2-albenzimidazol-3-yl}carbamate
To a solution of Intermediate 40 (700 mg, 2 mmol) in DCM (10 mL), at 0 C, was
added
dropwise triethylamine (500 pL, 4 mmol) and di-tert-butyl dicarbamate (400 mg,
2
mmol) portionwise. The reaction was stirred at 0 C for 1 hour and at r.t
overnight. The
reaction mixture was poured into ice-water (20 mL) and the aqueous layer was
extracted
by DCM (3 x 10 mL). The combined organic layers were dried over Na2SO4,
filtered and
concentrated in vacuo. The residue was purified by column chromatography over
silica
gel (heptane / Et0Ac 2 / 8), yielding 626 mg (70 %) of the title compound.
LCMS basic
(ES+) RT 2.92 min., 528.0 / 530.0 (M+H)+
INTERMEDIATE 43
0
H
a F.
tert-butyl {(1R,3R)-7-chloro-142-(difluoromethoxy)-6-ethenylpheny1]-2,3-
dihydro-1H-
pyrrolo[1,2-albenzimidazol-3-ylIcarbamate
Intermediate 42 (250 mg, 0.473 mmol), potassium vinyltrifluoroborate (92.3 mg,
0.662
mmol), cesium carbonate (308 mg, 0.944 mmol), and 1,1'-
bis(diphenylphosphino)ferrocene-palladium(11)dichloride dichloromethane
complex (19.3
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 132 -
mg, 0.0236 mmol) were placed in a tube, and filled with argon. Degassed 1,4
dioxanc (5
mL) and water (0.5 mL) were added and the resulting shiny was stirred at 110 C
overnight. The reaction mixture was cooled to ambient temperature, filtered
and
concentrated in vacuo. The crude material was purified by preparative reverse
phase
HPLC (basic conditions) to afford 175 mg (78%) of the title compound. LCMS
basic
(ES+) RT 2.91 min., 476/478(M+H)+.
INTERMEDIATE 44
0,,13
CI
N H
111111 \40
0 .
tert-butyl {(1R,3R)-7-chloro-142-(difluoromothoxy)-6-formylpheny1]-2,3-dihydro-
1H-
pyrrolo[1,2-aThenzimidazol-3-yll carbamatc
Intermediate 43 (25 mg, 0.0526 mmol) was dissolved in 1,4 dioxane (0.4 mL) and
water
(0.1 mL). At 0 C, sodium periodate (34 mg, 0.158 mmol) followed by osmium
tetroxide
(26 jil, 0.0021 mmol) were added . The reaction mixture was allowed to warm to
r.t and
stirred overnight. The reaction was then diluted with Et0Ac (2 mL) and water
(2 mL).
The aqueous layer was extracted with Et0Ac (2 x 2 mL). The combined organic
layers
were washed with a saturated solution of sodium thiosulfate ( 2 mL), brine,
dried over
magnesium sulphate, filtered and concentrated under vacuum to afford the title
compound
which was used without further purification. LCMS basic (ES+) RT 2.65 min.,
478/480(M+H)+
INTERMEDIATE 45
ci
=
IL/
(7K,14R)-11-chloro-1-(difluoromethoxy)-7,14-dihydro-7,14-methanobenzimidazo [1
,2-
13][2,5]benzodiazocinc
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 133 -
Intermediate 44 (0.0525 mmol) was dissolved in DCM/TFA (1/1). The reaction
mixture
was stirred at r.t for one hour. The reaction mixture was then concentrated
under reduced
pressure to afford the title compound as a TFA salt which was used without
further
purification. LCMS basic (ES+) RT 2.29 min., 360/362 (M+H)+
INTERMEDIATE 46
=
0 *
Ethyl [(7R,14R)-11-ch loro-1-(difluoromethoxy)-5,14-dihydro-7,14-
methanobenzimidazo[1,2-b1[2,5]benzodiazocin-6(7H)-yl]acetate
Example 12 (120 mg, 0.332 mmol) was dissolved in DMF (1 mL). Potassium
carbonate
(2 equiv., 0.663 mmol) and ethyl bromoacetate (1.2 equiv., 0.398 mmol) were
added and
the reaction mixture was stirred at r.t for 1 hour.
The mixture was filtered, rinsed with Et0Ac and the volatiles removed in
vacuo. The
residue was taken up with Et0Ac, washed with water, dried over magnesium
sulphate,
filtered and concentrated in vacuo. The crude compound was purified by reverse
phase
chromatography to afford 34 mg (23%) of the title compound. LCMS (ES+) 448/450
(M+1-1)-
INTERMEDIATE 47
(7R,14R)-11-1-2-(2- (ltert-butyl(dimethyl)silylloxylpropan-2-yl)pyrimidin-5-
y1]-1-
(difluoromethoxy)-6,7-dihydro-7,14-methanobenzimidazo[1,2-
b1[2,5]benzodiazocine-
5(14H)-thione
Intermediate 23 (150 mg, 0.254 mmol) was so lubilized in toluene (6 mL) before
addition
of Lawessons reagent (114 mg, 0.28 mmol). The slurry was heated overnight at
120 C.
The reaction mixture was concentrated in vacuo, and the residue taken up in
DCM and
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 134 -
filtered through a pad of silica gel eluting with DCM / Me0H (1/1) to afford
195 mg of
the title compound as a brown solid used without further purification. LCMS
acidic (ES+)
RT 3.74 min. 608(M+H)t
INTERMEDIATE 49
(S)-N-R1Z)-(2-Chloro-6-methoxyphenyl)methylidene]-2-methylpropane-2-
sulfinamide
To a cooled (0 C) solution of 2-chloro-6-methoxybenzaldehyde (15 g, 87.93
mmol) in
tetrahydrofuran (180 mL) was added sequentially (S)-2-methylpropane-2-
sulfinamide
(11.7 g, 96.7 mmol), tripotassium phosphate (56 g, 264 mmol) and dipotassium
hydrogen
phosphate (46 g, 263.8 mmol). The cooling bath was removed and the resultant
suspension was stirred at r.t. for 18 hours. The reaction mixture was filtered
through a
pad of eelite. The filtrate was diluted with Et0Ac (250 mL), washed with brine
(200
mL), dried over sodium sulfate, filtered and concentrated in vacuo to a crude
residue. The
crude material was purified by flash column chromatography (0-50%
Et0Ae/heptanes) to
afford 22.7 g (94%) of the title compound as a pale yellow solid. . LCMS
Method 6 (ES+)
RT 1.61 min., 274.1 (M+H)+. 'H NMR (500 MHz, Chloroform-d) 6 8.95 (m, 1H),
7.33 (t,
J = 8.3 Hz, 1H), 7.07 (dd, J = 8.1, 0.8 Hz, 11-1), 6.95 ¨ 6.84 (m, 1H), 3.88
(s, 3H), 1.29 (s,
9H)
INTERMEDIATE 50
(S)-N-[(1 R) - 1-(2-chloro-6-methoxyphenyl)but-3-en-1-y1]-2-methylpropane-2-
sulfmamide
To a suspension of zinc powder (27.9 g, 426.5 mmol) in anhydrous THF (100 mL)
was
added 1,2-dibromoethane (620 JAL, 7.19 mmol) and the mixture heated to 70 C.
After 10
minutes at this temperature, the heating was switched off and the reaction
stirred for a
further 30 minutes (internal temperature ca. 50 C) and allowed to cool slowly
to r.t. over
20 minutes. Chloro(trimethyl)silane (910 L, 7.17 mmol) was then added
dropwise.
Effervescence and an exotherrn to ¨ 40 C was observed, along with coagulation
of the
zinc. The reaction was heated to 50 C for 10 min then allowed to cool to
r.t.. 3-
Bromoprop-1-ene (18.5 mL, 213.8 mmol) was then added drop-wise at r.t.. An
exotherm
to ¨ 50 C was observed during addition and the addition rate controlled to
maintain the
exotherm. After completion of addition, the resultant grey suspension was
heated to 70
C for 15 minutes, then cooled first to r.t.. over 30 min, then to -40 C.
Anhydrous THF
(350 ml) was added, then a pre-cooled solution of Intermediate 49(19.5 g, 71.1
mmol) in
dry THF (100 mL) was added dropwise whilst maintaining an internal reaction
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 135 -
temperature of between -35 and -40 C, then the resultant mixture stirred at -
40 C for 1
hour. The reaction was allowed to warm to r.t., decanted and filtered through
a sinter
funnel to remove excess zinc. The solids were washed with THF (2 x 80 mL). The
filtrate
was poured into saturated aqueous ammonium chloride solution (500 mL) and
shaken
well, then extracted with Et0Ac (2 x 500mL). The combined organic layers were
washed
with brine (500 mL), dried over sodium sulfate, filtered and concentrated in
vacua to
afford -56 g crude yellow oil. This material was purified by reverse-phase
flash column
chromatography (elution 0-100% MeCN (+0.1% NH4OH) / H20 (+0.1% NH4OH)). The
clean fractions were extracted with Et0Ac (2 x 3 L). The combined organics
were dried
over sodium sulfate, filtered and concentrated to dryness under vacuum to
yield 16.8 g
(74 %) of the title compound as colourless viscous oil. LCMS Method 6 (ES+) RT
1.63
min., 316.1 (M+H)+. 'FINMR (250 MHz, Chloroform-d) 6 7.14 (t, J = 8.2 Hz, 1H),
6.97
(dd, J = 8.1, 0.9 Hz, 1H), 6.87 - 6.72 (m, 1H), 5.70 (ddt, J = 17.1, 10.1, 7.2
Hz, 1H), 5.26
-4.91 (m, 2H), 4.53 (s, 1H), 3.86 (s, 3H), 2.81 (dtt, J = 21.4, 13.8, 7.6 Hz,
2H), 1.10 (s,
9H).
INTERMEDIATE 51
(1 R)-1-(2-chloro-6-methoxyphenyl)but-3 -en-1 -amine
Intermediate 50 (12.7 g, 40.21 mmol) was dissolved in diethyl ether (40 mT,)
and ethanol
(20 mL) then 4M hydrogen chloride in 1,4-dioxane (31 mL) was added and the
reaction
mixture was stirred for 45 minutes. The reaction mixture was partitioned
between water
(150 mL) and diethyl ether (150 mL). The organic layer was re-extracted with
1M aq HCl
solution (150 mL). The aqueous layers were combined, basified to pH 10 by
addition of
6M aq NaOH solution and extracted with Et0Ac (2 x 200 mL). The combined
organic
layers were dried over Na2SO4 and concentrated to dryness under vacuum to
yield 9.19 g
(97%) of the title compound as a pale yellow viscous oil. LCMS Method 6 (ES+)
RT 1.49
min., 212.3 (M+H)+.11-INMR (500 MHz, Chloroform-d) 6 7.10 (t, J = 8.2 Hz, 1H),
6.96
(dd, J = 8.1, 0.9 Hz, 1H), 6.79 (d, J = 8.2 Hz, 1H), 5.77 (ddt, J = 17.2,
10.2, 7.2 Hz, 1H),
5.11 -4.92 (m, 2H), 4.56 (t, J = 7.6 Hz, 1H), 3.86 (s, 3H), 2.61 (hept, J =
7.3, 6.9 Hz, 2H).
INTERMEDIATE 52
tert-butyl-[145-(2,5-difluoro-4-nitro-phenyl)pyrimidin-2-y1]-1-methyl-ethoxy]-
dimethyl-
silane
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 136 -
The title compound can be prepared from Intermediate 11 and 1-bromo-2,5-
difluoro-4-
nitro-benzene by a palladium catalyzed Suzuki coupling following an analogous
method
to that described for Intermediate 12.
INTERMEDIATE 53
5-(2-{2-[(tert-butyldimethylsilyl)oxy]propan-2-y1}pyrimidin-5-y1)-N-[(1R)-1-(2-
ehloro-
6-methoxyphenyl)but-3-en-1-y1]-4-fluoro-2-nitroaniline
Intermediate 51(2.51 g, 10.67 mmol) and Intermediate 52 7 (4.96 g, 10.9 mmol)
were
dissolved in acetonitrile (40 mL) and K2CO3 (4.4 g, 31.84 mmol) was added. The
reaction
mixture was stirred at 80 C overnight. The reaction mixture was diluted with
Et0Ac
(100 mL) and washed with water (2 x 75 nth), then brine (75 mL), dried
(Na2SO4) and
concentrated to dryness under vacuum to yield 7 g (98%) of the title compound
as an
orange gum. LCMS Method 6 (ES+) RT 2.64 min., 601.1 (M+H)' . 1HNMR (500 MHz,
Chloroform-d) 6 8.94 - 8.74 (m, 3H), 7.99 (d, J = 10.7 Hz, 1H), 7.18 (t, J =
8.2 Hz, 1H),
7.00 (d, J - 7.9 Hz, 2H), 6.83 (d, J = 8.2 Hz, 1H), 5.80 (ddt, J - 17.2, 10.1,
7.1 Hz, 1H),
5.48 - 5.25 (m, 1H), 5.16 (d, J = 16.8 Hz, 11-1), 5.07 (d, J = 10.0 Hz, 1H),
3.88 (s, 3H),
3.04 - 2.86 (m, 1H), 2.79 (dt, J = 13.3, 6.5 Hz, 1H), 1.70 (d, J = 4.4 Hz,
6H), 0.90 (s, 9H),
-0.02 (s, 6H).
INTERMEDIATE 54
(3R)-3-{[5-(2-{2-[(tert-butyldimethylsilypoxy]propan-2-yl}pyrimidin-5-y1)-4-
fluoro-2-
nitrophenyllamino}-3-(2-chloro-6-methoxyphenyl)propanal
Potassium dioxido(dioxo)osmium hydrate (2:1:2) (75 mg, 0.2 mmol) was added in
one
portion to a stirred solution of Intermediate 53 (6.85 g, 10.25 111111101),
sodium periodate
(13.1 g, 61.25 mmol) and 2,6-dimethylpyridine (2.4 mL, 20.67 mmol) in a 3:1
mixture of
1,4-dioxane and water (240 mL). The mixture was stirred overnight then sodium
thiosulfate (11.3 g, 71.47 mmol) was added and the resulting mixture was
stirred for 30
minutes before diluting with DCM (200 mL) and water (200 mL). The biphasic
mixture
was stirred for a further 15 minutes then the two layers were separated and
the aqueous
layer was re-extracted with DCM (2 x 100 mL). The combined organic extracts
were
dried over Na2SO4, filtered and concentrated in vacuo to yield 8.5 g of the
title compound
as an orange gum. LCMS Method 6 (ES+) RT 2.74 min., 603.1 (M+H)'. 1I-1 NMR
(250
MHz, Chloroform-d) 6 9.81 (s, 1H), 8.87 (d, J = 1.6 Hz, 211), 8.24 -8.07 (m,
1H), 7.99 (d,
J = 10.7 Hz, 1H), 7.19 (d, J = 8.2 Hz, 111), 7.02 (dd, J = 8.1, 1.0 Hz, H),
6.86 (d, J = 8.2
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 137 -
Hz, 1H), 6.03 (td, J = 9.3, 5.0 Hz, 1H), 5.62 - 5.45 (m, 1H), 3.94 (s, 3H),
3.53 (ddd, J =
17.6, 8.9, 1.6 Hz, 1H), 3.01 (dd, J = 17.8, 4.8 Hz, 1H), 1.71 (s, 6H), 0.91
(s, 9H), -0.01 (s,
6H).
INTERMEDIATE 55
(R)-N-[(1Z,3R)-3- { [542- {2-[(tert-butyldimethylsilyl)oxy]propan-2-
yllpyrimidin-5-y1)-4-
fluoro-2-nitrophenyliamino}-3-(2-chloro-6-methoxyphenyl)propylidene]-2-
methylpropane-2-sulfinamide
To a solution of Intermediate 54 (8.5 g, 10.29 mmol) and (R)-2-methylpropane-2-
sulfinamide (1.25 g, 10.3 mmol) in DCM (50 mL) was added dropwise titanium(4+)
tetrapropan-2-olate (6.1 mL, 20.6 mmol) and the reaction mixture was stirred
at 40 C
under nitrogen for 3 hours and 20 minutes. The reaction was diluted with DCM
(100 mL)
then quenched by the addition of brine (50 mL). The resultant sticky
suspension was
filtered through celite and the celite washed with further DCM (2 x 100 mL)
and water
(100 mL). The filtrate was separated and the aqueous layer was re-extracted
with DCM
(100 mL). The combined organics were dried (Na2SO4) and concentrated to
dryness undcr
vacuum to yield approximately 8 g of a crude orange gum. The crude product was
purified on slica gel (DCM / Et0Ac 100 / 0 to 95 / 5) to yield 3.61 g (50%) of
the title
compound as an orange gum. T,CMS Method 6 (ES+) RT 2.56 min., 706.1 (M+H)+.
NMR (500 MHz, Chloroform-d) 6 8.96 - 8.79 (m, 3H), 8.10 (dd, J = 5.7, 3.6 Hz,
1H),
7.99 (d, J = 10.6 Hz, 1H), 7.21 (t, J = 8.2 Hz, 1H), 7.11 (s, 1H), 7.02 (d, J
= 8.1 Hz, 1H),
6.85 (d, J = 8.3 Hz, 1H), 5.88 (td, J = 9.3, 4.9 Hz, 1H), 3.92 (s, 3H), 3.63 -
3.46 (m, 1H),
3.11 (d, J = 16.3 Hz, 1H), 1.71 (d, J = 3.8 Hz, 6H), 1.13 (s, 9H), 0.91 (s,
9H), -0.02 (s,
6H).
INTERMEDIATE 56
N-R1R,3R)-3-115-(2-12-[(tert-butyldimethylsily0oxyThropan-2-y1}pyrimidin-5-y1)-
4-
fluoro-2-nitrophenyl]aminol-3-(2-chloro-6-methoxypheny1)-1-cyanopropyl]-2-
methylpropane-2-sulfinamide
Intermediate 55 (2.8 g, 3.96 mmol) was dissolved in anhydrous THF (50 mL)
under
nitrogen and scandium triflatc (400 mg, 0.81 mmol) was added, followed by
sodium
cyanide (220 mg, 4.5 mmol). The reaction mixture was stirred under a flow of
nitrogen
overnight. The reaction mixture was diluted with Et0Ae (100 mL) and washed
with a
saturated solution of NaHCO3 (75 mL). The aqueous layer was re-extracted with
Et0Ac
(75 mL) and the combined organics were washed with saturated brine (75 mL),
dried over
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 138 -
Na2SO4, filtered and concentrated to dryness under vacuum. The crude product
was
purified on silica gel (heptane / Et0Ac 100 / 0 to 60 / 40) to yield to 1.52 g
(44%) of the
title compound as an orange gum. LCMS Method 6 (ES-F) RT 2.41 min., 733.1
(M+H)t
1H NMR (500 MHz, Chloroform-d) 6 8.94 - 8.62 (m, 3H), 8.02 (d, J = 10.5 Hz,
1H), 7.26
-7.14 (m, 2H), 7.03 (d, J - 8.0 Hz, 1H), 6.85 (d, J = 7.9 Hz, 1H), 5.81 -5.61
(m, 1H),
4.49 -4.26 (m, 1H), 3.92 (s, 3H), 3.86 (d, J = 9.4 Hz, 1H), 2.93 (ddd, J =
14.4, 9.7, 4.6
Hz, 1H), 2.48 -2.23 (m, 1H), 1.71 (d, J = 2.4 Hz, 6H), 1.19 (s, 9H), 0.90 (s,
9H), -0.03 (s,
6H).
INTERMEDIATE 57
.01s1 H2
N ,
--O
CI
0 H
2- {5-RIR,3R)-3-amino-1-(2-chloro-6-methoxypheny1)-6-fluoro-2,3-dihydro-1H-
pyrrolo[1,2-a]benzimidazol-7-y1]pyritnidin-2-yllpropan-2-ol
Intermediate 56 (1.52 g, 2.07 mmol) was dissolved in ethanol (16 mL) and tin
(II)
chloride (2.4 g, 12.66 mmol) was added, followed by 12M HC1(1.4 mL) . The
reaction
mixture was stirred at 80 C for 90 minutes. The reaction mixture was cooled
to r.t.,
concentrated in vacuo to approximately 1 mL. The concentrated solution was
dissolved in
DCM (50 mL), basifled with 2M aqueous NaOH solution until pH = 10 and finally
treated with 10% aqueous ICE solution (25 mL). The mixture was filtered and
the solids
were washed with DCM (2 x 20 mL). The filtrate was separated and the aqueous
layer
was re-extracted with DCM (30 mL). The combined organic phases were dried over
Na2SO4, filtered and concentrated to dryness under vacuum to yield the title
compound
(134 mg 14%) as an off-white solid.
The original solids can be washed with further Et0Ac to yield a second crop of
the title
compound after removal of the volatiles in vacuo. If required additional
purification can
be performed by flash chromatography on silica gel (eluted with 0 to 100%
Et0Ac in
heptane, followed by 0 to 20% Me0H in Et0Ac). LCMS Method 6 (ES-I-) RT 3.76
min.,
468.1 (M+H)+. NMR (500 MHz, Chloroform-d) Major atropisomer - 6 8.71 (d, J
= 1.3
Hz, 2H), 7.56 (t, J= 10.2 Hz, 1H), 7.33 -7.27 (m, 1H), 7.16 (d, J = 8.1 Hz,
1H), 6.75 (d, J
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 139 -
= 8.2 Hz, 1H), 6.66 (d, J = 6.6 Hz, 1H), 6.19 (t, J = 7.7 Hz, 1H), 4.74 (s,
1H), 4.60 (s, 2H),
3.61 - 3.52 (m, 1H), 3.38 (s, 3H), 2.67 (dt, J = 14.4, 7.6 Hz, 1H), 1.62 (s,
6H).
INTERMEDIATE 58
b,NH2
>r),, F
OH CI
2- (5- [(1R,3R)-3-amino-1-(2-chloro-6-fluoropheny1)-6-fluoro-2,3-dihydro-lH-
p yrro propan-2-ol
Intermediate 58 was prepared following an analogous 8 steps procedure to that
described
for Intermediate 4/ through to Intermediate 57, starting from 2-chloro-6-
fluoro-
benzaldehyde. to afford 1.1 g of the title compound as a beige solid. LCMS
basic Method
3 (ES+) RT 2.02 min., 456.2 / 458.1 (M+H)+.
INTERMEDIATE 59
NH
CI F
F* Br
N-[(1R,3R)-142-bromo-6-(difluoromethoxy)pheny1]-7-chloro-2,3-dihydro-1H-
pyrrolo[1,2-a]benzimidazD1-3-ylimethanesulfonamide
To a mixture of Intermediate 40 (2.5 g, 5.8 mmol), N,N-diisopropylethylamine
(1.22 mL,
6.97 mmol) in DCM (58.3 mL), methanesulfonyl chloride (0.6 mL, 8 mmol) was
added at
0 C and the mixture was stirred at r.t for 2 hours. Water (30 mL) was added
to the
reaction mixture and extracted with CH2C12 (2 x 30 mL). The organic phase was
washed
with saturated brine (20 mL) and the combined organic phases was dried with
sodium
sulphate, filtered and concentrated in vacua to give a solid. The crude was
triturated in
diethylether, filtered, washed twice with diethylether then hexane and dried
to give the
title compound (2.8 g, 5.53 mmol, 95% yield) as a brown solid. LC/MS Method 3:
RT
2.11 mins (pH 10), m/z 506 and 508.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 140 -
INTERMEDIATE 60
H
CI F
F Br
(1R,3R)-142-bromo-6-(difluoromethoxy)pheny1]-7-chloro-2,3-dihydro-1H-
pyrrolo[1,2-
.. a]benzimidazol-3-ol
Intermediate 38 (5 g, 11.64 mmol) and triphenylphosphine (3.7 g, 14 mmol) were
added
to a round bottom flask followed by acetic acid (0.7 mL, 10 mmol) and THF (12
mL).
The reaction mixture was cooled down to 0 C and DIAD (3.4 mL, 17 mmol) in THF
(12
mL) was added dropwise. The mixture was stirred at 0 C for 2 hours. The
reaction
mixture was warmed to ambient temperature and the crude mixture was extracted
with
Et0Ac (3 x 20 mL). The organic phase was washed with saturated NaHCO1 (20 mL)
and
saturated brine (20 mL), the combined organic phases was dried with sodium
sulphate,
filtered and concentrated in vacuo to give an oil which was purified by flash
chromatography in silica gel (0 to 80% Et0Ac in Hexane) to afford [(1R,3R)-1-
[2-
.. bromo-6-(di fluorometh oxy)ph eny1]-7-chloro-2,3-dihydro-1H-pyrTolo [1,2-
a]berizimidazol-3-yl] acetate. The material was dissolved in Me0H (6.3 mL) and
stirred
with potassium carbonate (1.6 g, 12 mmol) for 45 minutes, the solid was
filtered and
washed with Me0H (30 mL) and water (10 mL) to give the title compound (4.3 g,
10
mmol, 86% yield) as a white solid. LC/MS Method 3: RT 2.07 mins (pH 10), m/z
429 and
.. 431.
INTERMEDIATE 61
244-methy1-5-(4,4.5,5-tetramethyl-1 .3.2-dioxaborolan-2-y1)pyrimidin-2-
yl]propan-2-ol
2-(5-bromo-4-methyl-pyrimidin-2-yl)propan-2-ol (1 g, 4.33 mmol),
bis(pinacolato)diboron (2 equiv., 8.65 mmol), 1,11-
bis(diphenylphosphino)ferrocene-
palladium(II)dichloride dichloromethane complex (0.05 equiv., 0.22 mmol),
potassium
acetate (4 equiv., 17.31 mmol) and 1,4-dioxane (5 mL) were placed in vial and
then
degassed. The mixture was then heated at 105 C for 2 hours.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 141 -
The reaction mixture was cooled and partitioned between Et0Ac and water. The
organic
layer was dried over Na2SO4 and evaporated to give a dark brown solid, stored
at 0 C and
used successfully in ensuing reactions after a period of several days.
Alternatively, intermediate 61 may be prepared by applying the following
procedure:
2-(5-bromo-4-methylpyrimidin-2-yl)propan-2-ol (8 g, 34.6 mmol), BISPIN (9.23
g, 36.3
mmol) and potassium acetate (10.2 g, 104 mmol) were combined in 1,4-dioxane
(300
mL). Argon was bubbled through the mixture over 10 minutes. Then PdC12(dppf)
(0.76 g,
1.04 mmol) was added and the mixture was stirred at 100 C. The reaction
mixture was
cooled to room temperature and filtered over a plug of celite and rinsed with
Et0Ac. The
combined filtrate was concentrated under reduced pressure to give the title
compound as
dark brown oil (16.9 g) which was used as such.
1HNMR (300 MHz, Chloroform-d) 6 8.90 (s, 1H), 5.04 (s, 1H), 2.71 (s, 3H), 1.57
(s,
6H), 1.36 (s, 12H). LC/MS Method 9: 2.15 minutes, [M+H] H 278/279/280.
INTERMEDIATE 62
6-bromo-7-fluoroimidazo[1,2-a]pyridine-2-carboxylate
5-bromo-4-fluoropyridin-2-amine (100 g, 0.52 mol) was dissolved in dioxane
(200 mL)
and added slowly to a solution of ethyl 3-bromo-2-oxopropanoate (70 mL, 0.54
mol) in
1,4-dioxane (800 mL) and stirred at r.t for 1.5 hours. Further 1,4-dioxane
(400 mL) was
added and the mixture heated to 95 C and stirred overnight. The mixture was
cooled to
r.t and concentrated in vacuo. The residue was dissolved in water (700 mL) and
basified
to pH ¨ 9 with saturated aqueous sodium bicarbonate solution. The resulting
solid was
filtered and washed with water (2 x 200 mL) and diethyl ether (300 mL). The
solid was
dried in vacuo at 40 C overnight to give the title compound (133 g, 86%) as a
peach
coloured solid. Method 7 HPLC-MS: MH+ m/z = 288/290, RT = 1.08 min (96 %). 'H
NMR (500 MHz, Chloroform-d) 68.35 (d, J= 6.3 Hz, 1H), 8.11 (s, 1H), 7.39 (d, =
8.5
Hz, 1H), 4.45 (q, J = 7.1 Hz, 2H), 1.43 (t, J = 7.1 Hz, 3H).
INTERMEDIATE 63
{6-bromo-7-fluoroimidazo[1,2-a]pyridin-2-yl}methanol
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 142 -
Intermediate 62 (28.5 g, 95.3 mmol) was dissolved in anhydrous THF (500 mL)
and
cooled to -10 C under nitrogen. 1M diisobutylaluminum hydride in heptane (200
mL) was added dropwise over -30 minutes and the reaction mixture was stirred
at -10 C
under nitrogen for 30 minutes then allowed to warm to 10 C under nitrogen
over 1 hour.
The reaction mixture was cooled to -50 C and quenched by dropwise addition of
saturated aqueous solution of Rochelle's salt (150 mL), stirred at -50 C for
30 minutes,
then allowed to warm to ambient temperature. The mixture was diluted with
water (250
mL) and extracted with Et0Ac (3 x IL). The aqueous layer was filtered through
celite
and extracted with Et0Ac (3 x 500 mL). The combined organics were dried
(Na2SO4) and
concentrated under vacuum to yield the title compound (22.5 g, 93%) as a
yellow solid.
Method 8 HPLC-MS: MH+ m/z = 245/247, RT = 2.93 mm (94 %). 11-1 NMR (500 MHz,
DMSO-d6) 6 9.03 (d, J= 6.7 Hz, 1H), 7.74 (s, 1H), 7.54 (d, J- 9.8 Hz, 1H),
5.23 (t, J-
5.2 Hz, 1H), 4.56 (d, J- 4.4 Hz, 2H).
INTERMEDIATE 64
346-bromo-7-fluoro-2-(hydroxymethypimidazo[1,2-alpyridin-3-y11-342-chloro-6-
(difluoromethoxy)phenyl]propanoic acid
Intermediate 63 (22.4 g, 84.02 mmol), 2-chloro-6-(difluoromethoxy)benzaldehyde
(20.1
g, 92.4 mmol), 2,2-dimethy1-1,3-dioxane-4,6-dione (13.3 g, 92.4 mmol), L-
proline (532
mg, 4.62 mmol) and MgSO4 (15.2 g, 126 mmol) were suspended in anhydrous
acetonitrile (110 mL), wanned to 100 C and stirred under N2(g) overnight. The
mixture
was cooled to RT, diluted with acetonitrile (100 mL) and filtered over a
sintered glass
funnel. The filter cake was further washed with acetonitrile (50 mL) and the
combined
filtrate treated with 6M sodium hydroxide in water (42 ml) and stirred at
ambient
temperature for 1 hour. The mixture was acidified (to pH 4) by treatment with
6M HC1
(aq) then concentrated under vacuum to give a tan solid. The solid thus
obtained was
slurried in water (100 mL) for 2 hours then filtered to give the title
compound (41.8 g,
83%) as a beige powder. Method 7 HPLC-MS: MH+ m/z 493/495, (M-H) m/z 491/493
RT 0.91 min (84%), 'FINMR (500 MHz, DMSO-d6) 6 8.44 (d, J= 6.5 Hz, 1H), 7.65
(d,J
= 9.3 Hz, 1H), 7.41 - 7.37 (n, 2H), 7.19 (t, J= 75.0 Hz, 1H), 7.17 (d, J = 4.9
Hz, 111),
5.37 (dd, Jr8.7, 6.8 Hz, 1H), 4.64 - 4.51 (m, 2H), 3.47 (dd, J= 16.5, 6.7 Hz,
1H), 3.29
(dd, J= 16.5, 8.8 Hz, 1H).
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 143 -
INTERMEDIATE 65
6-bromo-3-{2-carboxy-142-chloro-6-(difluoromethoxy)phenyliethy1}-7-
fluoroimidazo[1,2-a]pyridine-2-carboxylic acid
Chromium trioxide (167 mg, 1.66 mmol) and periodic acid (81.0 g, 355 mmol)
were
suspended in MeCN:H20 (800 mL: 8 mL). The resulting mixture was added to a
stirred
suspension of Intermediate 64 (41.8 g, 71.0 mmol) in MeCN:H20 (312 mL :2.4 n-
iL).
The resulting mixture was stirred at r.t overnight then filtered through a
sintered glass
funnel. The resulting pale green filter cake was washed with acetonitrile (4 x
50 mL) and
the filtrate concentrated under vacuum to give a gummy orange solid. This was
partitioned between Et0Ae (500 mL) and water (250 mL). The layers were
separated and
the aqueous phase further extracted with Et0Ac (2 x 150 mL). The combined
organic
phase was washed with water (3 x 250 mL), dried (MgSO4), filtered and
concentrated
under vacuum to leave an orange paste. This was slurried with water (200 mL)
for 72
hours (weekend period) and filtered. The resulting yellow solid was washed
with water
(100 mL) and dried in vacuo at 40 C to give the title compound (39.1 g, 96%)
as a pale
yellow powder. Method 7 HPLC-MS: MH+ m/z 507/509, RT 1.00 minutes. 'H NMR
(500 MHz, DMSO-do) 6 12.39 (s, 2H), 8.73 (d, .T= 6.6 Hz, 1f1), 7.74 (d, ./=
9.2 Hz, 1H),
7.39 -7.30 (m, 2H), 7.16 - 7.11 (m, 1H), 7.10 (t, J= 75.0 H7, 1H), 5.79 (t, I=
8.4 H7,
1H), 3.46 (dd, J= 17.3, 9.2 Hz, 1H), 3.20 (dd, .1 = 17.3, 7.7 Hz, 1H).
INTERMEDIATE 66
Ethyl 6-bromo-3-{1-[2-chloro-6-(difluoromethoxy)pheny1]-3-ethoxy-3-oxopropyll -
7-
fluoroimidazo[1,2-a]pyridine-2-carboxylate
Potassium carbonate (25.0 g, 180 mmol) and iodoethane (14.6 ml, 181 mmol) were
added
to a stirred solution of Intermediate 65 (34 g, 60.3 mmol) in anhydrous DMF
(350 mL) at
r.t. The mixture was stirred under an atmosphere of nitrogen overnight then
added slowly
to vigorously stirred mixture of ice / water (1.2 L). After stirring at RT for
a further 4
hours, the resulting light beige solid was filtered under vacuum and the
filter cake slurried
with water (50 mL). The solid was dried in vacuo at 40 C to give the title
compound
(31.5 g, 88%) as a light beige powder. Method 7 HPLC-MS: MH+ m/z 564 RT 1.29
mins.
'H NMR (500 MHz, DMSO-d6) 6 8.74 (d, J= 6.5 Hz, 1H), 7.77 (d, J = 9.2 Hz, 1H),
7.40
-7.33 (m, 21-1), 7.30 - 6.96 (m, 211), 5.82 (t, .1 = 8.5 Hz, 111), 4.23 (qq,
.1 = 7.0, 3.8 Hz,
2H), 3.98 (q, J= 7.0 Hz, 2H), 3.49 (dd, J= 17.1, 8.9 Hz, 1H), 3.27 (dd, J =
14.8, 7.7 Hz,
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 144 -
1H), 1.24 (t, J = 7.1 Hz, 3H), 1.03 (t, J = 7.1 Hz, 3H).
INTERMEDIATE 67
rr.N
0
N
Br 0
CI
CH3
Ethyl 11-bromo-312-chloro-6-(difluoromethoxy)pheny11-10-fluoro-5-oxo-1.7-
diazatricyclo[6.4Ø02,6]dodeca-2(6),7,9,11-tetraene-4-carboxylate
Intermediate 67 was prepared from Intermediate 66 according to the method
described
for Intermediate 33. The crude material was purified by flash column
chromatography
(SiO2, 800 g) eluting with a stepwise gradient in 500 mL vessels of 15 - 50 %
Et0Ac in
heptanes then 100% Et0Ac to afford the title compound as a yellow / orange
solid. (13.3
g, 50%). Method 7 HPLC-MS: MH+ m/z 517/519, RT 1.21 min (99%), 1H NMR (500
MHz, DMSO-d6) 8.51 - 8.34 (m, 1H), 7.94 - 7.83 (m, 1H), 7.56 - 7.42 (m, 2H),
7.39 -
6.68 (m, 2H), 5.77 - 5.63 (m, 1H), 4.28 - 4.13 (m, 2H), 4.07 - 4.00 (m, 1H),
1.27 - 1.19
(m, 3H).
INTERMEDIATE 68
0
N
Br
11-bromo-342-chloro-6-(difluoromethoxy)pheny1]-10-fluoro-1,7-
diazatricyclo [6.4 Ø02,6]dodeca-2 (6),7,9,11-tetraen-5 -one
Intermediate 68 was prepared from Intermediate 67 (13.3 g, 25.7 mmol)
according to the
method described for Intermediate 34 to give the title compound (11.3 g, 89%)
as an
orange foam. Method 7 HPLC-MS: MH+ m/z 444/446, RT 1.19 minutes.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 145 -
11-1 NMR (500 MHz, Chloroform-d) 6 7.79 - 7.58 (m, 1H), 7.47 - 7.27 (m, 3H),
7.25 -
7.06 (m, 1H), 6.89 - 5.81 (m, 1H), 5.48 - 5.40 (m, 1H), 3.65 - 3.48 (m, 1H),
3.30 -3.09
(m, 11-1).
INTERMEDIATE 69
F Off:tro
N
Br
(3R)-11-bromo-342-chloro-6-(difluoromethoxy)phcnyl]-10-fluoro-1,7-
diazatricyclo[6.4Ø02,61dodeca-2(6),7,9,11-tetraen-5-one
The title compound was prepared by chiral separation of Intermediate 68 into
the
constituent enantiomers and the isolation of the second eluting peak according
to the
conditions outlined below:
Analytical Method: Liquid Chromatography
Column: Chiralcel OD 250 x 4.6 mm 5 gm
Temperature: 30 C
Eluent: 100% Me0H + 0.1% DEA
Flow rate: 1 ml/min
Enantiomer A: 4.943 min
Enantiomer B (Intermediate 69): 11.887 min
Preperative method, by SFC:
Column: Chiralcel OD 266 x 50 mm
Eluent: CO2 + 20% Me0H
Flow rate: 360 ml/min
Enantiomer A: depending on the amount injected (-5.2 min)
Enantiomer B (Intermediate 69): depending on the amount injected (-8.5 min)
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 146 -
Optical rotation of Enantiomer B Intermediate 69: OD +117.6 (Me0H, conc 0.255
g/100mL, T = 25 C, wavelength 589 nM, cell path 10 cm).
INTERMEDIATE 70
CH3
F 1 ,CH3
yr\,:tfi Ns+)(CH3
Br 0
F
Ff -CI (R)-N-[(3R,5E)-11-bromo-3-[2-chloro-
6-(difluoromethoxy)phenyl] -10- fluoro-1,7-
diazatricyclo[6 .4 Ø02,6]dodeca-2 (6),7,9,11-tetraen-5 -ylidene]-2-
methylpropane-2-
sulfinamide
Ti(0E04 (1.59 mL, 2.48 mmol) was added a stirred solution of Intermediate 69
(0.57 g,
1.22 mmol) in anhydrous THF (12 mL) at r.t then (R)-2-methylpropane-2-
sulfinamide
(0.29 g, 2.43 mmol) was added and mixture was heated at 65 C for 17 hours.
The
mixture was cooled to r.t, diluted with brine (10 mL), Et0Ac (50 mL) and water
(5 mL)
and stirred for 15min. The resulting solids were removed by filtration and the
aqueous
layer was further extracted with Et0Ac (3 x 25 mL). The combined organic
layers were
washed with brine (25 mL), dried over sodium sulfate, filtered and
concentrated in vacuo
to afford the crude product as a beige solid. Purification by column
chromatography (SiO2
Biotage isolera), eluting with 0-100% Et0Ac in heptanes afforded the title
compound
(600 mg, 90%) as a beige solid. Method 7 HPLC-MS: MH+ m/z 548, RT 1.20 min
(99%),
1HNMR (500 MHz, Chloroform-d) 6 7.76 - 7.58 (m, 1H), 7.43 - 7.39 (m, 1H), 7.39
-
7.27 (m, 2H), 7.25 - 7.02 (m, 1H), 6.84 - 5.82 (m, 1H), 5.47 - 5.30 (m, 1H),
4.52 - 4.33
(m, 1H), 3.61 - 3.47 (m, 1H), 1.36 (s, 9H).
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 147 -
INTERMEDIATE 71
HCI
HCI
N /
F\r0 a
(3R,5R)-11-bromo-3- [2-chloro-6-(difluoromethoxy)pheny1-1-10-fluoro-1,7-
diazatricy c lo [6.4 Ø02,6] dodeca-2(6),7,9,11-tetraen-5 -amine dihydro
chloride
Sodium borohydride (90 mg, 2.38 mmol) was added in one portion to a stirred
solution of
Intermediate 70 (600 mg, 1.09 mmol) in THF:water (14.7 mL:0.3 mL) under an
atmosphere of nitrogen at -50 C and the reaction maintained at this
temperature for 30
minutes. The mixture was slowly warmed to 0 C over 2 hours then stirred for a
further 1
hour. The mixture was quenched with Me0H (1 mL) and diluted with water (25
mL). The
intermediate was extracted with Et0Ac (2 x 25 mL) and the combined organic
phases
dried (MgSO4), filtered and concentrated in vacuo to leave an off-white foam
(570 mg).
This was dissolved in dioxane (10 mL) and treated with 4M HC1 in dioxane (1.4
mL, 5.6
mmol). The mixture was stirred at RT for 30 minutes then concentrated to
dryness to
afford the title compound (480 mg, 67%) as an off-white solid. The title
compound was
used directly in the subsequent step without further purification. Method 9
HPLC-MS:
MH+ m/z 446/448, RT 1.42 minutes.
INTERMEDIATE 72
H2
N
N
HO
\ro
H3C CI
CH3
2- (5-R3R,5R)-5-amino-342-chloro-6-(difluoromethoxy)pheny1]-10-fluoro-1,7-
diazatricyclo[6.4Ø02,6]dodeca-2(6),7,9,11-tetraen-11-yl]pyrimidin-2-y1 }
propan-2-ol
An aqueous solution of 2M K2CO3 (1.8 mL) was added to a stirred suspension of
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 148 -
Intermediate 7 I (480 mg, 0.73 mmol) and 245-(tetramethy1-1,3,2-dioxaborolan-2-
yl)pyrimidin-2-ylipropan-2-ol (232 mg, 0.88 mmol) in 1,4-dioxane (10 mL). The
mixture
was degassed under a flow of nitrogen for 10 minutes then treated with bis[3-
(diphenylphosphanyl)cyclopenta-2,4-dien-l-y1] iron; DCM; dichloropalladium (60
mg,
0.07 mmol) and heated to 105 C for 1.5 hours. The mixture was cooled to RT,
diluted
with Et0Ac (30 mL) and filtered through a pad of Celite. The filter cake was
further
washed with Et0Ac (2 x 20 mL). The filtrate was washed with water (25 mL) and
the
aqueous phase further extracted with Et0Ac (2 x 25 mL). The combined organic
phase
was washed with brine (30 mL), dried (MgSO4), filtered and concentrated in
vacuo to
give the crude product (750 mg) as a dark brown oil. Purification by column
chromatography (KP-NH, SiO2, Biotage isolera) eluting with 50-100% Et0Ac in
heptane
followed by 0-50% Me0H in Et0Ac to give the title compound (260mg , 71%) as an
off
white solid. The material was azeotroped twice with toluene prior to use in
the subsequent
step. Method 8 HPLC-MS: MH+ m/z 504, RT 3.86 minutes, NMR (500 MHz,
DMSO-d6) 6 8.90 - 8.78 (m, 2H), 8.18 - 7.90 (m, 1H), 7.68 - 7.54 (m, 1H), 7.45
- 7.36 (m,
1H), 7.37 - 6.66 (m, 3H), 5.14 - 4.92 (m, 2H), 4.48 - 4.32 (m, 1H), 3.57 -
3.34 (m, 1H),
2.95 - 2.54(m, 2H), 2.23 -1.93 (m, 1H), 1.51 - 1.47 (m, 6H).
INTERMEDIATE 73
2,2-Dichloro-3-oxocyclobutyl 2,2-dimethylpropanoate
To a stirred mixture of vinyl pivalate (30 g, 234 mmol) and zinc (31 g, 474
mmol) in
ether (250 mL) was added a solution of 2,2,2-trichloroacetyl chloride (34 mL,
304 mmol)
in ether (250 mL) dropwise over 2.5 hours in a water bath while maintaining
the reaction
temperature between 15 - 30 C. Reaction was filtered through Celite and
washed through
with Et0Ac (200 mL). The filtrate was washed with water (200 mL), brine (200
mL),
dried over sodium sulfate and concentrated under vacuum to afford the title
compound
(68 g, 97 % at 80 % purity) as an orange liquid.
OH (500 MHz, CDC13) ppm 5.40 (dd, J = 8.4, 6.2 Hz, 1H), 3.70 (dd, J = 18.9,
8.4 Hz, 1H),
3.39 (dd, J = 18.9, 6.2 Hz, 1H), 1.28 (s, 9H).
INTERMEDIATE 74
3-0xocyclobutyl 2,2-dimethylpropanoate
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 149 -
Zinc (74 g, 1.1 mol) was added to acetic acid (200 mL) with stirring and the
suspension
was cooled in an ice bath. Intermediate 73 (80 %, 68 g, 228 mmol) in acetic
acid (300
mL) was added dropwise over 2 hours. Upon completion of addition, the reaction
was
warmed to r.t and stirred for 1.5 hours. The reaction was filtered washed with
DCM (100
mL). The filtrate was diluted with Et0Ac (800 mL) and washed sequentially with
water
(3 x 250 mL), saturated aqueous NaHCO3 solution (3 x 250mL) and brine (50 mL).
The
organic phase was dried over sodium sulfate and concentrated under vacuum to
give
crude product as a brown oil (30 g) which was purified by dry flash
chromatography on
silica gel eluting with 0 - 10 % Et0Ac in heptanes to afford the title
compound (11 g, 28
%) as a clear colourless oil
oH (500 MHz, CDC13) 5.26 ¨ 5.19 (m, 1H), 3.51 ¨3.40 (m, 2H), 3.19 ¨ 3.07 (m,
2H),
1.22 (s, 9H).
INTERMEDIATE 75
3-(5-Bromopyrimidin-2-y1)-3-hydroxycyclobutyl 2,2-dimethylpropanoate
5-bromo-2-iodopyrimidine (16.7 g, 58.8 mmol) was dissolved in DCM (200 mL)
with
stirring and cooled to -78 C under N2. 2.5 M n-BuLi in hexane in hexane (23.5
mL) was
added dropwise and stirred for 20 minutes at ¨ 78 C. Intermediate 74 (10 g,
58.8 mmol)
in DCM (50 mL) was cooled in a dry ice bath and added in one portion. The
reaction was
.. stirred at -78 C for 10 minutes. The reaction was quenched by addition of
saturated
aqueous NH4C1 solution (20 mL) and allowed to warm to r.t, saturated aqueous
NH4C1
solution (50 mL) was added and the mixture was extracted with DCM (2 x 100
mL). The
combined organic extracts were dried over sodium sulfate and concentrated
under
vacuum. The cnide product was purified by column chromatography using 0 - 30 %
Et0Ac in heptane to afford the title compound (7.6 g, 35 %) as a yellow solid.
OH (500 MHz, CDC13) 8.78 (s, 2H), 5.22 ¨ 5.14 (m, 1H), 3.03 ¨2.93 (m, 2H),
2.67 ¨ 2.58
(m, 2H), 1.22 (s, 9H).
INTERMEDIATE 76
1-(5-Bromopyrimidin-2-yl)cyclobutane-1,3-dio1
Intermediate 75 (90%, 6 g, 16.4 mmol) was dissolved in Me0H (120 mL) and K2CO3
(11.3 g, 82 mmol) was added and the reaction stirred for 18 hours at r.t. The
reaction was
diluted with DCM (400 mL) and washed with water (150 mL). The aqueous phase
was
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 150 -
extracted with DCM (200 mL). The combined organic extracts were dried over
sodium
sulfate and concentrated under vacuum to afford the title compound (2.94 g, 73
%) as an
off-white solid.
OH (500 MHz, DMSO-d6) 8.98 (s, 2H), 5.63 (s, 1H), 5.08 (d, J = 6.2 Hz, 1H),
4.09 - 3.92
(m, 1H), 2.87 - 2.79 (m, 2H), 2.28 -2.14 (m, 2H).
INTERMEDIATE 77
3-(5-Bromopyrimidin-2-y1)-3-hydroxycyclobutan-1-one
To a stirred solution of Intermediate 76 (2 g, 8.1 mmol) in DCM (200 mL) was
added
Dess-Martin periodinane (4.1 g, 9.8 mmol). The reaction was stirred for 18
hours and the
resulting suspension diluted with DCM (100 mL) and washed with saturated
aqueous
NaHCO3 solution (100 mL). The aqueous layer was re-extracted with DCM (100 mL)
and
the combined organic extracts dried over sodium sulfate and concentrated. The
crude
product was purified by chromatography on silica gel eluting with 0 - 30 %
Et0Ac in
heptanes to afford the title compound (1.37 g, 69 %) as an off white solid.
OH (500 MHz, DMSO-d6) 9.04 (s, 2H), 6.41 (s, 1H), 3.69 - 3.55 (m, 2H), 3.37 -
3.21 (m,
2H).
INTERMEDIATE 78
.. 3-(5-Bromopyrimidin-2-y1)-3-Rtert-butyldimethylsilypoxy]cyclobutan-1-one
Intermediate 77 (1.37 g, 5.64 mmol) was dissolved in dry DMF (20 mL) with
stirring
under N2 and cooled to 0 C. 1H-imidazole (1.9 g, 28.18 mmol) was added
followed by
tert-butyl(chloro)dimethylsilane (2.0 g, 13.5 mmol) and reaction was stirred
at r.t for 20
hours. The reaction was diluted with DCM (150 mL) and washed with water (3 x
50 mL).
The aqueous phase was re-extracted with DCM (50 mL). The combined organic
extracts
were dried over sodium sulfate and concentrated. The crude product was
purified by
chromatography on silica gel eluting with 0 - 20 % Et0Ac in heptanes to afford
the title
compound (1.6 g 79 %) as a pale orange oil.
OH (500 MHz, DMSO-d6) 9.06 (s, 2H), 3.78 - 3.66 (m, 2H), 3.44 - 3.34 (m, 2H),
0.88 (s,
9H), 0.00 (s, 6H).
INTERMEDIATE 79
3-(5-Bromopyrimi din -2-y1)-3 -Rtert-butyl dimethyl si lyl)oxy]-1-m ethyl
cyclobutan-l-ol
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 151 -
Intermediate 78 (1.35 g, 3.78 mmol) was dissolved in dry ether (40 mL) under
N2 with
stirring and cooled to 0 C using an ice bath. 3M MeMgBr in diethylether (2.52
mL) was
added dropwise and reaction stirred for 30 minutes at 0 C. The reaction was
quenched
with saturated aqueous NH4C1 solution (20 mL) and then water (20 mL). The
mixture was
extracted with Et0Ac (2 x 50 mL), dried over sodium sulfate and concentrated
to give a
yellow oil. This was purified by chromatography on silica gel eluting with 0 -
100 %
DCM in heptane followed by 0 - 20 % Et0Ac in DCM to afford the title compound
as a
mixture of separate cis and trans isomers (total yield, 1.19 g, 84 %) as clear
oils.
Major isomer - cis
6H (500 MHz, CDC13) 8.79 (s, 2H), 3.10 - 3.03 (m, 2H), 2.59 - 2.51 (m, 2H),
1.18 (s,
3H), 0.87 (s, 9H), -0.14 (s, 6H).
Minor isomer - trans
OH (500 MHz, CDC13) 8.79 (s, 2H), 2.78 -2.63 (m, 4H), 1.49 (s, 3H), 0.95 (s,
9H), 0.04
(s, 6H).
INTERMEDIATE 80
1-Chloro-2,5-difluoro-4-nitrobenzene
A suspension of 2-chloro-1,4-difluorobenzene (98 g, 660 mmol) in concentrated
sulphuric
acid (250 ml, 4.69 mol) was cooled with an ice/salt mixture after which a
solution of
nitric acid (29.1 ml, 693 mmol) in sulphuric acid (100 ml, 1.88 mol) was added
drop-wise
over 1.5 hours whilst maintaining the temperature between -5 and +2 C. After
30
minutes, the reaction mixture was allowed to warm to -17 C and slowly poured
onto ice
with stirring. The formed solid was isolated by filtration and the residue
washed several
times with water and air dried yielding the title compound (112 g, 88%) as a
pale yellow
powder.
1HNMR (300 MHz, Chloroform-d) 6 7.94 (dd, J = 7.9, 6.5 Hz, 1H), 7.45 (dd, J=
9.8, 5.9
Hz, 1H).
INTERMEDIATE 81
2-(5-(2.5-difluoro-4-nitrophenyppyrimidin-2-yl)propan-2-ol
A mixture of Intermediate 80 (50 g, 258 mmol), 2-(5-(4,4,5,5-tetramethy1-1,3,2-
dioxaborolan-2-yl)pyrimidin-2-yl)propan-2-ol (69.6 g, 264 mmol) and sodium
carbonate
(54.8 g, 517 mmol) in 1,4-dioxane (700 mL) and water (100 mL) was flushed with
argon
3 times. Subsequently, tris(dibenzylideneacetone)dipalladium (5.91 g, 6.46
mmol) and tri-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 152 -
tcrt-butylphosphinc tctrafluoroborate (7.50 g, 25.8 mmol) were added and the
mixture
was stirred at 80 'V overnight. The reaction mixture was cooled to r.t,
filtered over celite
and washed with Et0Ac (1 L). The filtrate was washed with water (100 mL) and
brine (2
x 200 mL) and the combined aqueous layers back-extracted with Et0Ac (200 mL).
The
combined organic layers were dried over Na2SO4, filtered and concentrated in
vacua. The
residue was purified by column chromatography (silica; 30% Et0Ac in heptane).
The
product containing fractions were combined, concentrated in vacuo and
crystallised from
iPrOH to give the title compound as an orange solid (55g) which can be further
purified
if required by trituration with di-isopropyl ether.
LCMS Method 11 RT=1.806 (99.5%); [M+H]= 296.
NMR (300 MHz, Chloroform-d) 6 8.95 (d, J= 1.5 Hz, 2H), 8.02 (dd, J = 9.2, 6.1
Hz,
1H), 7.46 (dd, J = 10.4, 6.0 Hz, 1H), 4.47 (s, 1H), 1.66 (s, 6H).
INTERMEDIATE 82
(S)-N-(2-bromo-6-(difluoromethoxy)benzylidenc)-2-mehylpropanc-2-sulfinamidc.
(S)-2-methylpropane-2-sulfinamide (30 g, 248 mmol), potassium phosphate,
dibasic (129
g, 743 mmol) and phosphoric acid, potassium salt (158 g, 743 mmol) were added
to a
cooled solution of 2-bromo-6-(difluoromethoxy)ben7alrlehyde (68.3 g, 272 mmol)
in
anhydrous THF (500 mL) at 0 C. The reaction mixture was allowed to warm to r.t
and
stirred overnight. The bulk of the THF was removed in vacuo and water and Et20
were
added to the residue. The layers were separated and the aqueous phase
extracted with
Et20. The combined organic layers were washed with brine, dried over Na2SO4,
filtered
and concentrated in vacuo yielding the title compound (90 g) as a dark oil
which was
used as such in the next reaction.
LCMS Method 11: RT=2.084 (97.6%); [M+Hr= 354/356 (Br pattern).
IHNMR (300 MHz, Chloroform-d) 6 8.84 (s, 1H), 7.58 (dd, J= 7.9, 1.2 Hz, 1H),
7.33 (t,
J = 8.1 Hz, 1H), 7.28 - 7.20 (m, 1H), 6.57 (t, J= 73.8 Hz, 1H), 1.30 (s, 9H).
INTERMEDIATE 83
(S)-N-OR)-1-(2-bromo-6-(difluoromethoxy)phcnyl)but-3-en-l-y1)-2-methylpropanc-
2-
sulfinamidc
Under a nitrogen atmosphere, 1,2-dibromoethane (2.19 ml, 25.4 mmol) was added
to a
suspension of zinc (100 g, 1.52 mol) in anhydrous THF (250 mL). The suspension
was
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 153 -
warmed to mild reflux, allowed to cool down and heated to reflux again. This
cycle was
repeated twice more after which TMSC1 (3.24 mL, 25.4 mmol) was added causing
an
exothermic reaction. After 15 minutes, 3-bromoprop-1-ene (55.2 mL, 635 mmol)
was
added dropwise at such a rate that the very light reflux was maintained
without external
heating. The suspension was stirred for an additional 30 minutes while
allowing to cool to
r.t. Stirring was stopped and excess zinc allowed to separate. The grey
supernatant
solution was transferred to a dropping funnel and the flask rinsed twice with
anhydrous
THF. This solution was added relatively fast to a solution of crude
Intermediate 82 (89.9
g, 254 mmol) in anhydrous THF (1000 ml) which was pre-cooled to -60 C (dry
ice/acetone). After the addition was complete, the cooling bath was removed
and the
reaction mixture allowed to slowly warm to r.t overnight. The reaction mixture
was
quenched by adding saturated aqueous NH4C1 solution (20 mL) and some ice. The
bulk of
the THF was removed in vacuo and saturated aqueous NH4C1 solution was added to
the
residue until almost no solids remained. This mixture was extracted with Et20
(3 x
200mL). The combined organic layers were washed with saturated aqueous NH4C1
solution, water and brine, dried over Na2SO4, filtered and concentrated in
vacuo yielding
the title compound (98.9 g) as a yellow-orange oil which was used as such in
the next
reaction.
LCMS Method /0: RT=3.183 (84.1%); [M+H]= 396/398 (Br pattern); d.e.: 94.5%
(the
other diastereomer elutes at 3.43 minutes).
'1-1NMR (300 MHz, Chloroform-d) 6 7.43 (d, J= 7.9 Hz, 1H), 7.14 (t, J= 8.1 Hz,
1H),
7.13 -6.98 (m, 1H), 6.56 (t, J = 73.8 Hz, 1H), 5.82 - 5.62 (m, 1H), 5.19 (q, J
= 8.0 Hz,
1H), 5.10 - 4.95 (m, 2H), 4.40 - 4.03 (m, 1H), 3.00 - 2.65 (m, 2H), 1.14 (s,
9H).
INTERMEDIATE 84
(R) - 1-(2-bromo-6-(difluoromethoxy)phenyl)but-3-en-1-amine
At 0 C, HC1 (1 M in Et20, 666 mL, 666 mmol) was added to a solution of crude
Intermediate 83 (88.0 g, -222 mmol) in ethanol (240 mL). After 3 hours, the
reaction
mixture was diluted with water and the layers were separated. The organic
layer was
extracted twice with aqueous HC1 ((0.2 M). The combined aqueous layers were
made
alkaline (pH = -10) by slowly adding saturated aqueous Na2CO3 solution and the
aqueous
mixture thus obtained was extracted 3 times with Et20. The combined organic
layers
were washed with brine, dried over Na2SO4, filtered and concentrated in vacuo
yielding
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 154 -
the title compound (56.4 g) as a dark orange oil which was used as such in the
next
reaction.
LCMS Method 11: RT=1.360 (93.7%); [M+H]= 292/294 (Br pattern).
1H NMR (300 MHz, Chloroform-d) 6 7.41 (dd, J= 7.6, 1.6 Hz, 1H), 7.14 - 6.98
(m, 2H),
.. 6.56 (t, J= 73.5 Hz, 1H), 5.86 - 5.68 (m, 1H), 5.13 -4.97 (m, 2H), 4.59 (t,
J= 7.6 Hz,
1H), 2.61 (t, J = 7.1 Hz, 2H), 1.85 (br s, 2H).
INTERMEDIATE 85
(R)-2-(5-(5-((1-(2-bromo-6-(difluoromethoxy)phenyl)but-3-en-1-yl)amino)-2-
fluoro-4-
nitrophenyl)pyrimidin-2-yl)propan-2-ol.
Under a N2 atmosphere, Intermediate 81(54.2 g, 183 mmol), Intermediate 84
(56.4 g,
-183 mmol), and potassium carbonate (50.7 g, 367 mmol) were mixed in
acetonitrile
(anhydrous, 500 mL) and stirred at 80 C for 2 days. After cooling to r.t, the
reaction
mixture was filtered through sand and rinsed with Et0Ac. The filtrate was
evaporated to
.. give the title compound (106.1 g) as a dark red oil that was used as such
in the next
reaction.
11-INMR (300 MHz, Chloroform-d) 6 8.83 (s, 2H), 8.70 (hr d, J= 7.0 Hz, 1H),
8.03 (d,./
= 10.7 H7, 1 H), 7.49 (dd, J= 7.0, 2.2 Hz, 1H), 7.24- 7.10 (m, 2H), 6.92 (hr
d, 1= 6.0 Hz,
1H), 6.58 (t, J= 72.3 Hz, 1H), 5.91 -5.73 (m, 1H), 5.36 (q, J= 8.1 Hz, 1H),
5.22 (dd, J =
17.0, 1.4 Hz, 1H), 5.13 (d, J= 10.1 Hz, 1H), 4.67- 4.50 (m, 1H), 3.09 - 2.70
(m, 2H),
1.65 (s, 6H).
INTERMEDIATE 86
(R)-N-(1-(2-bromo-6-(difluoromethoxy)phenyl)but-3-en-1-y1)-5-(2-(2-((tert-
butyldimethylsilyl)oxy)propan-2-yl)pyrimidin-5-y1)-4-fluoro-2-nitroaniline.
Imidazole (60.1 g, 883 mmol) and tert-butyldimethylsily1 chloride (80 g, 531
mmol) were
added to a solution of crude Intermediate 85 (99.8 g, -176 mmol) in anhydrous
N,N-
dimethylformamide (250 mL). The mixture was warmed to 100 C and stirred for
19
hours. After cooling to r.t, the reaction mixture was diluted with brine (1 L)
and extracted
with mixture of heptane and Et0Ac (1:1, 500 + 200 mL). The combined organic
layers
were washed with water (2 x 100 mL) and brine (100 mL), dried over Na2SO4,
filtered
and concentrated in vacuo. The residue was purified by column chromatography
(silica;
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 155 -
5-15% Et0Ac in hcptanc) to give the title compound (107.1 g, 72% (over 5
steps)) as a
red oil.
LCMS Method 12: RT=3.01 (94.9%); [M+H]= 681/683 (Br pattern).
1H NMR (300 MHz, Chloroform-d) 68.78 (s, 2H), 8.65 (br s, 1H), 8.00 (d, J=
10.7 Hz,
1H), 7.44 (dd, J= 6.8, 2.4 Hz, 1H), 7.19 - 7.08 (m, 2H), 6.92 -6.81 (m, 1H),
6.54 (t, J=
72.4 Hz, 1H), 5.88 -5.74 (m, 1H), 5.33 (q, J= 8.0 Hz, 1H), 5.20 (dd, J= 17.0,
1.4 Hz,
1H), 5.11 (d, J= 10.1 Hz, 1H), 3.01 -2.71 (m, 2H), 1.69 (s, 3H), 1.68 (s,
3111), 0.89 (s,
9H), -0.05 (s, 6H).
INTERMEDIATE 87
(4R)-4-(2-bromo-6-(difluoromethoxy)phenyl)-445-(2-(2-((tert-
butyldimethylsilyl)oxy)propan-2-yl)pyrimidin-5-y1)-4-fluoro-2-
nitrophenyl)amino)butane-1,2-diol
Osmium tetroxide (4 wt% in water, 4.80 mL, 0.786 mmol) and 4-methylmorpholine-
4-
oxide (50 wt% in water, 94 mL, 393 mmol) were added to a solution of
Intermediate 86
(107.1 g, 157 mmol) in a mixture of acetone (330 mL) and water (45 mL). The
reaction
mixture was stirred overnight at ambient temperature and concentrated in
vacuo. The
residue was mixed with aqueous Na2S203 solution (10 wt%, 200 mL) and extracted
twice
with Et0Ac. The combined organic layers were washed with brine, dried over
Na2SO4,
filtered and concentrated in vacuo yielding the title compound (120.1 g) as a
dark red
sticky oil that was used as such in the next reaction.
LCMS Method 12: RT=2.58 (95.3%); [M+H]= 715/717 (Br pattern).
INTERMEDIATE 88
(R)-3-(2-bromo-6-(difluoromethoxy)pheny1)-345-(2-(2-((tert-
butyldimethylsilypoxy)propan-2-yppyrimidin-5-y1)-4-fluoro-2-
nitrophenyl)amino)propanal
A solution of crude Intermediate 87(119 g, -155 mmol) in THF (250 mL) was
diluted
with water (200 mL). To the resulting suspension, sodium periodate (68 g, 318
mmol)
was added and another portion of sodium periodate (10.3 g, 48.2 mmol) after 3
hours.
Stirring was continued for another hour after which the reaction mixture was
quenched
with aqueous Na2S203 (10 wt%, 300 mL). The solids were filtered off and washed
with
Et0Ac (500 mL). The organic layers were separated (some NaCl (s) added to
enable layer
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 156 -
separation) and the aqueous phase was extracted with Et0Ac (200 mL). The
combined
organic layers were washed with a mixture of water (100 mL) and brine (50 mL)
and
brine (100 mL), dried over Na2SO4, filtered, and concentrated in vacuo to give
the title
compound (106.6 g) as a dark red thick syrup that was used as such in the next
reaction.
LCMS Method 12: RT=2.73 (72.9%); [M+H]= 683/685 (Br pattern) and RT=2.57
(19.5%); = 701/703 (Br pattern) product as hydrate.
NMR (300 MHz, Chloroform-d) 6 9.81 (s, 1H), 8.86 (d, J = 1.5 Hz, 2H), 8.68 (br
d, J
= 8.6 Hz, 1H), 7.99 (d, J = 10.6 Hz, 1H), 7.45 (dd, J= 6.8, 2.4 Hz, 1H), 7.22 -
7.09 (m,
3H), 6.66 (t, J= 72.2 Hz, 1H), 6.00 (td, J= 9.0, 4.7 Hz, 1H), 3.55 (dd, Jr
17.9, 9.0 Hz,
1H), 3.08 (br d, J= 16.9 Hz, 1H), 1.71 (s, 3H), 1.70 (s, 3H), 0.89 (s, 9H), -
0.03 (s, 6H).
INTERMEDIATE 89
(R)-NAR)-3-(2-bromo-6-(difluoromethoxy)pheny1)-345-(2-(2-((tert-
butyldimethylsilypoxy)propan-2-yl)pyrimidin-5-y1)-4-fluoro-2-
nitrophenyl)amino)propylidene)-2-methylpropanc-2-sultinamidc
Titanium (IV) isopropoxide (87 g, 307 mmol, 91 mL) was added to a solution of
crude
Intermediate 88 (105 g, -154 mmol) and (R)-(+)-2-methyl-2-propanesulfinamide
(18.63
g, 154 mmol) in DCM (180 mT). After stirring overnight, the reaction mixture
was
poured out into a mixture of water (300 mL) and kieselguhr (30 g), stirred for
10 minutes,
and filtered. The yellow solid was washed several times with DCM. The layers
of the
filtrate were separated and the organic layer was dried over Na2SO4, filtered
and
concentrated in vacua yielding the title compound (114.5 g) as a yellow-orange
glass that
was used as such in the next reaction.
LCMS Method 12: RT=2.88 minutes; [M+H]= 786/788 (Br pattern).
INTERMEDIATE 90
(R)-N4R)-3-(2-bromo-6-(difluoromethoxy)phenyl) -34(54242-
((tertbutyldimethylsilyl)oxy)propan-2-y1) pyrimidin-5-y1)-4-fluoro-2-
nitrophenyl)
amino)propylidene) -2-methylpropanc-2-sulfinamide
To a stirred solution of crude Intermediate 89 (103.6 g, -139 mmol) in DCM
(500 mL)
was added yttrium(111) trifluoromethanesulfonate (3.53 g, 6.58 mmol). The
flask was
sealed with a suba seal and to the resulting mixture, trimethylsilyl cyanide
(15.68 g, 158
mmol, 19.77 mL) was added by syringe in a steady stream. After stirring for 5
days, the
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 157 -
reaction mixture was concentrated in vacuo and co-evaporated with Et0Ac
yielding the
title compound (101 g) as a yellow-red foaming oil that was used as such in
the next
reaction.
LCMS Method 13: RT=2.70 minutes; [M+H]+= 813/815 (Br pattern).
INTERMEDIATE 91
(R)-N-R3R)-3-02-amino-5-(2-(2-((tertbutyldimethylsilypoxy)propan-2-
yl)pyrimidin-5-
y1)-4-fluorophenyl)amino)-3-(2-bromo-6-(difluoromethoxy)pheny1)-1-cyanopropy1)-
2-
methylpropane-2-sulfinamide
Under a N2 atmosphere, platinum on charcoal (10 wt%, 13.88 g, 7.11 mmol) was
added to
a mixture of crude Intermediate 90 (106 g, ¨147 mmol) and zinc(II) bromide
(12.46 g,
55.3 mmol) in Et0Ac (1000 mL). Subsequently, the reaction was purged with H2
and
stirred under atmospheric H2 pressure for 3 days. The reaction mixture was
flushed with
N2, filtered over kieselguhr and rinsed with Et0Ac. The filtrate (-1.5 L) was
washed with
water (500 mL) and brine, dried over Na2SO4, filtered and concentrated in
vacuo yielding
the title compound (109 g) as a brown-yellow foaming oil that was used as such
in the
next reaction.
I,CMS Alethod 11: RT=1.360 (76.5%); [M+H]= 783/785 (Br pattern).
INTERMEDIATE 92
N H2
Br
H;)1#1%. *
2-(5-((lR,3R)-3 -amino -1-(2-bromo -6-(difluorometho xy)pheny1)-6- fluoro-2,3 -
dihydro -
1Hbenzo[d]pyrrolo[1,2-alimidazol-7-yl)nyrimidin-2-y1)propan-2-ol.
To a stirred solution of crude Intermediate 91(109 g, ¨147 mmol) in ethanol
(1000 mL)
was added HC1 (4 M in dioxane, 69.5 mL, 278 mmol). The resulting mixture was
stirred
at reflux for 8 hours and allowed to cool to r.t overnight. The reaction
mixture was
concentrated in vacuo and co-evaporated with ethanol. The residue was
triturated in Et20
(1.5 L) and the formed precipitate was isolated by filtration, washed with
Et20, and dried
on the filter. This material was taken up in iPrOH (250 mL) and stirred at
reflux for 15
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 158 -
minutes. The resulting suspension was stirred under N2 while allowing to cool
to r.t. The
resulting thick viscous suspension was diluted with iPrOH (250 mL) and
filtered through
a glass filter. The residue thus obtained was dissolved in water (1.5 L) and
Et20 (500 mL)
while stirring. The layers were separated and the organic layer was extracted
with
aqueous HO (0.1 M, 200 mL). The combined aqueous layers were washed with Et20
(500 mL), made alkaline (pH = 10) with NaOH (s, 40 g), and extracted with DCM
(2 x
500 mL). The combined DCM layers were dried over Na2SO4, filtered and
concentrated
in vacuo. The residue thus obtained was co-evaporated with iPr20 and Et20
yielding the
title compound (40.6 g, 48%) as a green foam.
LCMS Method 10: RT=2.37 minutes; [M+H]= 548/550 (Br pattern).
'H NMR (300 MHz, Chloroform-d): 3:2mixture of rotamers 6 8.74 - 8.70 (m, 2H),
7.61
(d, J= 8.2 Hz, 0.6H), 7.57 (d, J= 11.2 Hz, 1H), 7.45 (dd, J= 6.6, 2.6 Hz,
0.4H), 7.37 -
7.28 (m, 1.4H), 7.04 (d, J= 8.3 Hz, 0.6H), 6.71 (t, J- 72.4 Hz, 0.4H), 6.59
(dd, J= 12.0,
6.6 Hz, 1H), 6.23 -6.13 (m, 1H), 5.92 (dd, J = 74.5, 70.9 Hz, 0.6H), 4.72 (br
s, 1H), 4.61
(br s, 1H), 3.70 -3.41 (m, 1H), 2.77 (dt, J= 13.5, 8.6 Hz, 0.411), 2.63 (dt,
J= 13.7, 7.6
Hz, 0.6H), 2.03 (br s, 2H), 1.62 (s, 3.6H), 1.61 (s, 2.4H).
INTERMEDIATE 93
FN 0
H
H \ r I
0
Br
Ethyl 2-[[(1R,3R)-142-bromo-6-(difluoromethoxy)pheny1]-6-fluoro-7-[2-(1-
hydroxy-1-
methyl-ethyppyrimidin-5-y1]-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazol-3-
yl]aminolacetate
Ethyl bromoacctatc (91.3 mg, 0.54 mmol) was added to a solution of potassium
carbonate
(252 mg, 1.82 mmol), and Intermediate 92 (250 mg, 0.45 mmol) in DMF (1.5 mL).
The
reaction mixture was stirred at 70 C for 2 hours. The reaction mixture was
cooled to
ambient temperature and the crude mixture was quenched with water and
extracted with
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 159 -
Et0Ac (2 x 5 mL). The organic phase was washed with saturated brine (5 mL),
the
combined organic phases were dried with sodium sulphate, filtered and
concentrated in
vacuo to give an oil which was purified by flash chromatography in silica gel
(0 to 100%
Et0Ac in hexanes) to afford the title compound (220 mg, 76% yield) as a brown
solid.
LC/MS Method 3: RT 2.06 mins (pH 10), m/z 634/636.
INTERMEDIATE 94
o3/4(r¨fl j<
I
Br
tert-butyl 3 -[[(1 R ,3R)- 1-[2-bromo-6-(di fluoromethoxy)pheny11-6-fluoro-742-
(1-hydroxy-
1-methyl-ethyppyrimidin-5-y1]-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazol-3-
ylicarbamoyliazetidine-1-carboxylate
Intermediate 92 (550 mg, 1.00 mmol) was added to a solution of 1-tert-
butoxycarbonylazetidine-3-carboxylic acid (222 mg, 1.10 mmol,), HATU (432 mg,
1.10
mmol) and N,N-di-isopropylethylamine (0.38 mL, 2.2 mmol) in DMF (20 mL). The
reaction mixture was stirred at ambient temperature for 3 hours. Water was
added and the
reaction mixture extracted with Et0Ae and the organic phases removed in vacuo
to yield a crude
product.The crude material was purified by column chromatography over silica
gel using
hexane / ethyl acetate (0 to 100%) as eluent, yielding 640 mg (87%) of the
title compound
as an amorphous solid. LCMS basic Method 3: RT 2.25 mm, [M+H-BOC]I= 631.
INTERMEDIATE 95
FIF
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 160 -
tert-Butyl 3- { J(6R,12R)-11-(difluoromethoxy)-242-(2-hydroxypropan-2-
yl)pyrimidin-5-
y1]-6H-6,12-met hanob enzimidazo [2,1-ci[ 1,4]benzodiazepin-7(12H)-
yl]carbonyl}azetidine-1-carboxylate
To Intermediate 94 was added cesium acetate anhydrous (420 mg, 2.18 mmol),
cuprous
iodide (170 mg, 0.87 mmol) and dimethyl sulfoxide (0.9 mL). The mixture was
seal and
purged 3 times with nitrogen. The reaction mixture was stirred for 18 hours at
100 C.
Water and ethyl acetate were added to the reaction mixture and the two layers
were
separated. The aqueous layer was extracted with ethyl acetate and the combined
organics
were filtered through a phase separator and the solvent was evaporated. The
crude
.. material was purified by column chromatography over silica gel using hexane
/ ethyl
acetate (0 to 100%) as eluent, yielding 158 mg (28%) of the title compound as
a brown
solid. LCMS Method 3: RT 2.72 min, [M+Hr= 651
INTERMEDIATE 96
itioj<
olc.C1
H
r
tert-butyl 3-[[(1R,3R)-142-bromo-6-(difluoromethoxy)pheny1]-7-chloro-2,3-
dihydro-1H-
pyrrolo[1.2-a]benzimidazol-3-yl]carbamoyl]azetidine-1-carboxylate
Intermediate 40 (250 mg, 0.58 mmol) was added to a solution of 1-tert-
butoxycarbonylazetidine-3-carboxylic acid (130 mg, 0.65 mmol), HATU (252 mg,
0.643
mmol) and AT,N-diisopropylethylamine (0.18 mL, 1.0 mmol) in DMF (11 mL). The
reaction mixture was stirred at room temperature for 3 hours. Water was added
to the
reaction and the mixture extracted with Et0Ac (x3), dried (sodium sulphate),
filtered and
concentrated in vacuo. The crude material was purified by column
chromatography over
silica gel using hexane / ethyl acetate (0 to 100%) as eluent, yielding 280 mg
(78%) of the
title compound as an amorphous solid.
LCMS Method 3: RT 2.48 minutes, [M+H]= 611/613.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 161 -
INTERMEDIATE 97
0
FIF
tert-butyl 3- {[(6R,12R)-2-chloro-11-(difluoromethoxy)-6H-6,12-
methanobenzimidazo [2.1-c] 1 4 benzodiazepin-7(12H)-Alcarbonyil azetidine-1-
carboxylate
To Intermediate 96 (165 mg, 0.27 mmol) was added cesium acetate (130 mg, 0.67
mmol),
cuprous iodide (52 mg, 0.27 mmol) and dimethyl sulfoxide (0.3 mL). The mixture
was
sealed in a pressure tube and purged 3 times with nitrogen. The reaction
mixture was
stirred at 100 C overnight. Water and ethyl acetate were added to the reaction
mixture
and the two layers were separated. The aqueous layer was extracted with
further ethyl
acetate. The combined organic layer was filtered through a phase separator and
the
solvent was evaporated. The crude material was purified by column
chromatography on
silica gel using hexane / ethyl acetate (0 to 100%) as eluent, yielding 85 mg
(59%) of the
title compound as a brown solid. LCMS Method 3: RT 2.63 min., [M+I-1]'=
531/533.
INTERMEDIATE 98
1
HO
cis-3- { [tert-butyl(dimethypsilyl]oxyl -3- {5 -[(6R,12R )-11-
(difluoromethoxy)-7-
(methylsul fony1)-7,12-dihydro-6H-6,12-methanobenzimidazo[2,1-c] [1
,4]benzodiazepin-
2-yl]pyrimidin-2-y11-1-methyleyclobutanol
The title compound was prepared from 3-[tert-butyhdimethypsilyfloxy-1-methyl-
345-
(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yppyrimidin-2-y1], and Example 23
according
to a method involving the same procedural steps as those described for Example
20, to
give, following purification by column chromatography over silica gel using
hexane /
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 162 -
ethyl acetate (0 to 100%) as cluent the title compound as a white solid (83
mg, 37%
yield).
LC/MS Method 3: RT 2.73 minutes, [M+H]= 684.
INTERMEDIATE 99
5:tof
HC:y311C1:0
tit Br
N-R1R,3 R)-142-bromo-6-(difluoromethoxy)pheny1]-6-fluoro-7-[2-(1-hydroxy-1-
methyl-
ethyppyrimidin-5-y1]-2,3-dihydro-1H-pyrrolo [1,2-a]benzimidazol-3-yl]pyridine-
3-
sulfonamide
Pyridine-3-sulfonyl chloride (1.1 eq, 0.6 mmol) was added to a solution of
Intermediate
92 (300 mg, 0.55 mmol) and /V,N-diisopropylethylamine (0.24 mL, 1.3 mmol) in
dichloromethane (2.8 mL) at room temperature. The mixture was stirred for
lhour before
solvent was partially evaporated. The crude material was purified by column
chromatography on silica gel using hexane / ethyl acetate (0 to 100%) as
eluent, yielding
276 mg (73%) of the title compound. LCMS Method 3: RT 1.91 mm. (pH 10),
[M+H]+=
689.
INTERMEDIATE 100
1-[5-(4,4,5,5-tetramethy1-1,3,2-dioxaborolan-2-yl)pyrimidin-2-y1]-3-
(ttifluoromethypazetidin-3-ol
To a solution of the trifluoroacetate salt of (trifluoromethyl)-3-aziditin-3-
01(5.8g,
22.75mmo1) and 2-chloro-5-(4,4,5,5-tetramethy1-1,3,2-dioxaborolan-2-
yl)pyrimidine (5g,
20.75mmo1) in acetonitrile (80 mL) was added dropwise triethylamine (9.5 mL,
68.5
mmol) and the resultant mixture stirred overnight. LC/MS showed completion of
reaction, concentrated to an off-white solid, ice-water was added, triturated,
filtered,
washed with cold water and dried by suction to give the title compound (6.1g,
50%) as
cream solid. 'H NMR (400 MHz, DMSO-d6): 6 8.53 (s, 2 H), 7.46 (s, 1 H), 4.32
(m, 2
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 163 -
H), 4.10 (d, J = 10.3 Hz, 2 H), 1.29 (s, 12H). LC/MS Method 3:: m/z 346, RT
1.09 min
(pH=10).
INTERMEDIATE 101
11\11,1
I F
N-R1R,3R)-142-bromo-6-(difluoromethoxy)pheny11-6-fluoro-742-(1-hydroxy-l-
methyl-
ethyppyrimidin-5-y11-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazol-3-
ylimethanesulfonamide
To a solution of Intermediate 92 (202 mg, 0.37 mmol) and 4-
dimethylaminopyridine (6
mg, 0.05 mmol) in DCM (4 mL) at 0 C was added N,N-diisopropylethylamine (76
L,
0.43 mmol) followed by methanesulfonyl chloride (30 4, 0.38 mmol). The
reaction
mixture was stirred at 0 C for 10 minutes and then at room temperature for
45 minutes,
after which time the reaction mixture was concentrated in vacuo. The residue
was
purified by column chromatography (SiO2, 25-100% Et0Ac in hexane) to give the
title
compound (193 mg, 83 %) as a yellow solid.
LCMS Method 3: (ES+) 626/628 (M+1-1)+, RT 1.90 minutes.
LCMS Method 4: (ES+) 626/628 (M+H)', RT 1.86 minutes.
INTERMEDIATE 102
NI
Hr
I F
N-R1R,3R)-142-bromo-6-(difluoromethoxy)pheny11-6-fluoro-742-(1-hydroxy-l-
methyl-
ethyl)pyrimidin-5-y1J-2,3-dihydro-1H-pyrrolo[1,2-aThenzimidazol-3-
ylicyclopropanesulfonamide
To a solution of Intermediate 92 (151 mg, 0.28 mmol) and 4-
dimethylaminopyridine (5
mg, 0.04 mmol) in DCM (3 mL) at 0 C was added N,N-diisopropylethylamine (58
1.1L,
0.33 mmol) followed by cyclopropanesulfonyl chloride (172 AL, 1.65 mmol). The
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 164 -
reaction mixture was stirred at 0 C for 10 minutes and then at room
temperature for 18
hours, after which time the reaction mixture was partitioned between DCM (30
mL) and
water (20 mL), the layers separated and the aqueous extracted with DCM (3 x 20
mL).
The combined organics were dried (phase separator), and concentrated in vacuo.
The
residue was purified by column chromatography (SiO2, 25-100% Et0Ac in hexane)
to
give the title compound (110 mg, 61 %) as a purple solid.
LCMS (ES+) Method 3: 652/654 (M+H)+, RT 2.20 minutes.
LCMS (ES+) Method 4: 652/654 (M+H)+, RT 2.01 minutes.
INTERMEDIATE 103
N
H~fN
2-[5-[(1R,3R)-142-bromo-6-(difluoromethoxy)phenyl]-342-[tert-
butyl(dimethypsilylioxyethylamino]-6-fluoro-2,3-dihydro-1H-pyrrolo[1,2-
a]benzimidazol-7-yllpyrimidin-2-yl]propan-2-ol
To a solution of Intermediate 92(251 mg, 0.46 mmol) and potassium carbonate
(252 mg,
1.82 mmol) in DMF (2 mL) was added (2-bromoethoxy)-tert-butyldimethylsilane
(99 ut,
0.46 mmol) and reaction mixture stirred at 70 C for 18 hours. The reaction
mixture was
partitioned between Et0Ac (25 mL) and water (25 mL), layers separated and
aqueous
extracted with Et0Ac (2 x 25 mL), the combined organics dried (phase
separator) and
concentrated in vacuo. The residue was purified by column chromatography
(SiO2, 0-
100% Et0Ac in hexane) to give the title compound (128 mg, 39 %) as a brown
oil.
LCMS (ES+) Method 3: 706/708 (M+H)', RT 3.10 minutes.
INTERMEDIATE 104
F 0
N-R1R,3R)-112-bromo-6-(difluoromethoxy)pheny1]-6-fluoro-742-(1-hydroxy-l-
methyl-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 165 -
ethyppyrimidin-5-y1]-2,3-dihydro-1H-pyrrolo[12-a]benzimidazol-3-
ylibenzenesulfonamide
To a solution of Intermediate 92 (202 mg, 0.37 mmol) and 4-
dimethylaminopyridine (5
mg, 0.04 mmol) in DCM (4 mL) at 0 C was added NAT-diisopropylethylamine (77
0.44 mmol) followed by benzenesulfonyl chloride (49 4, 0.38 mmol). The
reaction
mixture was stirred at 0 C for 5 minutes and then at room temperature for
1.5 hours.
The reaction mixture was concentrated in vacuo and the residue purified by
column
chromatography (SiO2, 0-60% Et0Ac in hexane) to give the title compound (178
mg, 70
%) as a yellow solid.
LCMS (ES+) Method 3: 688/690 (M+H)', RT 2.20 minutes.
LCMS (ES+) Method 4: 688/690 (M+H)+, RT 2.01 minutes.
INTER1V1EDIA1E 105
H =
N I
I F =
r Br
N-R1R,310-142-bromo-6-(difluoromethoxy)pheny1]-6-fluoro-742-(1-hydroxy-l-
methyl-
ethyl)pyrimidin-5-y1]-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazol-3-y1]-6-
methoxy-
pyridine-3-sulfonamide
To a solution of Intermediate 92 (150 mg, 0.27 mmol) and 4-
dimethylaminopyridine (5
mg, 0.04 mmol) in DMF (3 mL) at 0 C was added N,N-diisopropylethylamine (574,
0.33 mmol) followed by 6-methoxypyridine-3-sulfonyl chloride (62 mg, 0.29
mmol).
The reaction mixture was stirred at 0 C for 5 minutes and then at room
temperature 30
minutes. The reaction mixture was concentrated in vacuo. The residue was
purified by
column chromatography (SiO2, 0-100% Et0Ac in hexane) to give the title
compound
(148 mg, 75 %) as a dark beige solid. LCMS (ES+) Method 3: 719/721 (M+H)+, RT
2.30
minutes. LCMS (ES+) Method 4: 719/721 (M+H)+, RT 2.42 minutes. 614 (300 MHz,
DMSO-d6) 8.77-8.97 (m, 1 H), 8.71-8.77 (m, 3 H), 8.19-8.26 (m, 1 H), 7.59-7.68
(m, 1
H), 7.39-7.50 (m, 2 H), 7.38 (t, 1 H, J73.4 Hz), 7.07 (d, 1 H, J8.7 Hz), 6.65-
6.71 (m, 1
H), 6.04-6.21 (m, 1 H), 5.22-5.44 (m, 1 H), 5.05-5.07 (m, 1 H), 3.96 (s, 3 H),
3.05-3.24
(m, 1 H), 2.59-2.79 (m, 1 H), 1.47-1.52 (m, 6 H).
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 166 -
INTERMEDIATE 106
¨Y4NI)1/C(
f 0
.,(Sk
Ethyl [(7R,14R)-11-[2-(2- t[tert-butyl(dimethypsilyl]oxylpropan-2-yppyrimidin-
5-y1]-1-
(difluoromethoxy)-5-oxo-5,14-dihydro-7,14-methanobenzimidazo[1,2-
b][2,5Thenzodiazocin-6(71/)-yl]acetate
To a solution of Intermediate 23 (100 mg, 0.169 mmol) in THF (4 mL) was added
potassium bis(trimethylsilyl)amide (0.2 mL, 0.2 mmol, 1M in THF) at -78 C and
stirred
for 40 minutes before the addition of ethyl bromoacetate (25.0 L, 0.225
mmol). The
reaction mixture was stirred at -78 C for 30 mins before being quenched with
water. The
mixture was extracted with DCM (2 x 20 mL), and the organics were combined,
dried
with Na2SO4, filtered and concentrated in vacuo. The crude product was
columned on
silica eluting with 0-5% Me0H/DCM to give the title compound (88 mg). LC/MS:
Method
3: RT 3.53 minutes.
INTERMEDIATE 107
\ q
.,(Sk
(7R,14R)-11-[2-(2- gtert-butyl(dimethyl)silyfloxy}propan-2-yppyrimidin-5-y1]-1-
(difluoromethoxy)-6-(22.2-trifluoroethyl)-6.7-dihydro-7.14-
methanobenzimidazo[1,2-
b] [2,5]benzodiazocin-5(1410-one
To a solution of Intermediate 23 (90.0 mg, 0.152 mmol) in DMF (3.5 mL) were
added
cesium carbonate (250 mg, 0.767 mmol) and 2-iodo-1,1,1-trifluorocthanc (0.06
mL, 0.60
mmol) at ambient temperature. The reaction mixture was heated at 150 C for 3
hours.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 167 -
The reaction mixture was partitioned between Et0Ac (2 x 10 mL) and water (20
mL),
and the organics were combined, dried with Na2SO4, filtered and concentrated
in vacuo.
The crude product was progressed to the next step without further
purification.
LC/MS: Method 3 MW 674, retention time 2.03 minutes.
INTERMEDIATE 108
=
(7R,14R)-6-(2- fitert-butyl(dimethypsilyl]oxyl ethyl)-11-[2-(2- [_[ ten-
b utyl(dimethyl)sily I] oxy I prop an-2-y Opyrimidin-5-y1]-1-(difl
uoromethoxy)-6,7-dihydro-
7,14-met hanob enzimidazo [1 ,2-bil2 ,5Thenzodiazo cin-5(1411)-one
To a solution of Intennediate 23 (100.0 mg, 0.169 mmol) in THF (4.0 mL) was
added
potassium bis(tritnethylsily0amide (0.20 mL, 0.20 mmol, 1M in THF) at -78 C
followed
by the addition of (2-bromoethoxyl)-tert-butyldimethylsilane (50.0 4, 0.231
mmol). The
reaction mixture was heated in a microwave at 70 C for 24 hours before being
quenched
with aqueous saturated NH4C1 and extracted with Et0Ac (2 x 10 mL). The
organics were
combined, dried with Na2SO4, filtered and concentrated in vacuo. The crude
product was
progressed to the next step without further purification. LC/MS Method 3: ESI
MW 750,
retention time 4.23 minutes.
INTERMEDIATE 109
_\ro joic OH
11
p . 461
(1/4
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 168 -
(7R,14R)-11-[2-(2- lltert-butyl(dimethypsilylloxylpropan-2-y1)pyrimidin-5-y1]-
1-
(difluoromethoxy)-6-(2-hydroxyethyl)-6,7-dihydro-7,14-methanobenzimidazo[1,2-
b][2,5]benzodiazocin-5(141/)-one
To a solution of Intermediate 108 (80.0 mg, 0.107 mmol) in THF (5.0 mL) was
added a
solution of tetrabutylammonium fluoride (0.20 mL, 0.20 mmol, 1M in THF) at
ambient
temperature and the mixture was stirred for 72 hours. The reaction mixture was
partitioned between water (10 mL) and DCM (3 x 10 mL), and the organics were
combined, dried with Na2SO4, filtered and concentrated in vacuo. The crude
product was
progressed to the next step.
LC/MS Method 3: ESI MYI 636, retention time 3.21 minutes.
INTERMEDIATE 110
ct
(7R,14R)-11-chloro-1-(difluoromethoxy)-6-(trideutero)methy1-6,7-dihydro-7,14-
.. methanobenzimidazo[1,2-b112,5]bcnzodiazocin-5(141i)-one
To a solution of Example 11 (90.0 mg, 0.24 mmol) in THF (4 mL) was added a
solution
of KHMDS in THF (1M, 0.25 mL, 0.25 mmol) dropwise at -78 C and the mixture
was
stirred for 30 minutes before the addition of CD3I (30.1 pt, 0.48 mmol). The
reaction
mixture was warmed to 0 C and stirred for 3 hours. The reaction mixture was
quenched
.. with saturated aqueous NH4C1 solution, extracted with Et0Ac, dried with
Na2SO4,
filtered and concentrated in vacuo. The crude product was purified by column
chromatography on silica eluting with 0-10% Me0H/Et0Ac to give the title
compound
(95 mg, 99%). LC/MS Method 3: ES1 MH+ 393, retention time 1.93 minutes.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 169 -
INTERMEDIATE 111
H
Ni<
tert-butyl (2- {5-[(7R,14R)-1-(difluoromethoxy)-6-(trideutero)methy1-5-oxo-
5,6,7,14-
tetrahydro-7,14-methanobenzimidazo[1,2-b1[2,5]benzodiazocin-11-yllpyrimidin-2-
yljpro_pan-2-yl)carbamate
To a solution of tert-butyl N41-methy1-145-(4,4,5,5-tetramethy1-1,3,2-
dioxaborolan-2-
Apyrimidin-2-yl]ethyl]carbamate (260 mg, 0.72 mmol) and Intermediate 110 (94
mg,
0.24 mmol) in 1,4-dioxane (3 mL) were added K3PO4 (294 mg, 1.40 mmol),
tricyclohexyl
phosphonium tetrafluoroborate (15 mg, 0.04 mmol) and
tris(dibenzylideneacetone)
dipalladium(0) (28 mg, 0.03 mmol) were added. The mixture was degassed for 10
minutes with nitrogen before heating at 110 C for 18 hours. The reaction
mixture was
quenched with water and extracted with Et0Ac (2 x 10 mL), dried with Na2SO4,
filtered
and concentrated in vacuo. The crude product was purified by column
chromatography on
silica eluting with 0-10% Me0H/ DCM to give the title compound (40 mg, 28%).
LC/MS
Method 3: ESI MFI' 594, retention time 2.10 minutes.
INTERMEDIATE 112
N H
CI
F-4
rac 2-(5- (1R,3R)-3-amino-1-12-chloro-6-(difluoromethoxy)n heny1]-6-fluoro-2,3-
dihydro-1H-cyclopenta[4,5]imidazo[1,2-a]pyridin-7-y1J -4-methylpyrimidin-2-
yl)propan-
2-ol
Intermediate 61 (0.15 g, 0.6 mmol) and potassium acetate (0.15 g, 1.5 mmol)
were
suspended in anhydrous 1,4-dioaxane (5 mL) in a sealable vessel. The mixture
was stirred
and degassed thoroughly under a stream of N2(g) for 15 min then treated with
bis[3-
(diphenylphosphanyl)cyclopenta-2,4-dien-1-yl]iron; dichloromcthanc;
dichloropalladium
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 170 -
(0.02 g, 0.02 mmol), and a solution of Intermediate 7/ (82%, 0.25 g, 0.4 mmol)
in a
mixture of dioxane (1 mL) and 2M K2CO3 in water (1.24 ml) was added and the
mixture
warmed to 100 C overnight. After cooling to room temperature the mixture was
diluted
with Et0Ac (25 mL) and filtered over a pad of celite. The filtrated was washed
with water
(10 mL), dried (MgSO4), filtered and concentrated in vacuo to give the crude
product (0.4
g) as a brown gum. Column chromatography (C18, biotgae isolera, 60 g) eluting
with 0 to
50% acetonitrile in water spiked with 0.1% NH4OH afforded the title compound
(0.15 g,
58%) as an orange glass. The material was azeotroped twice with toluene prior
to use in
the subsequent step. Method 8 HPLC-MS: MH+ m/z 518, RT 1.43 minutes. 1H NMR
(500 MHz, DMSO-d6) 6 8.56 - 8.51 (m, 1H), 7.99 - 7.77 (m, 1H), 7.63 - 7.53 (m,
1H),
7.43 -7.31 (m, 2H), 7.29 - 7.24 (m, 1H), 7.06- 6.73 (m, 1H), 5.10 -4.90 (m,
2H), 4.47 -
4.31 (m, 1H), 3.52 -3.40 (m, 1H), 2.34- 2.27 (m, 3H), 2.03 - 1.81 (m, 3H),
1.52 - 1.45
(m, 6H).
INTERMEDIATE 113
*sr
(7R,14R)-11-[6-(2- fitert-butyl(dimethypsilyl]oxy}propan-2-y1)-4-methylpyridin-
3-y11-1-
(difluoromethoxy)-6,7-dihydro-7,14-methanobenzimidazo[1.2-b][2,5]benzodiazocin-
5(14H)-one
The title compound was prepared from Example 11(450 mg, 1.20 mmol) and tert-
butyl-
dimethyl-[1-methy1-1-[4-methy1-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-y1)-
2-
pyridyl]ethoxy]si lane (483 mg, 1.86 mmol), by a palladium catalyzed Suzuki
coupling
according to a method involving the same procedural steps as those described
for
Example 20. The crude product was purified by flash chromatography (SiO2, 0 to
100%
Et0Ac in DCM and then 1 to 10% Me0H in Et0Ac) to obtain the title compound
(650
mg, 81%) as a brown solid. LCMS Method 3 (ES+) RT 3.20 mm, 605 (M+H)'
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 171 -
INTERMEDIATE 114
tert-butyl N41-(5-bromopyrimidin-2-y1)-1-methyl-ethyl]carbamate
2-(5-bromopyrimidin-2-yl)propan-2-amine (200 mg, 0.92 mmol) was dissolved in
THF
(5mL) and di-tert-butyl dicarbonate solution (1.0M) (1.3 mL, 1.3 mmol) in THF
added.
After 2 hours the solvents were removed in vacuo to afford the title compound
as an
orange gum (300 mg, 92%). LCMS ,Vethod 3 (ES+) RT 1.88 min, 338.0/340.0
(M+Na)+.
INTERMEDIATE 115
xIz
0
I 0
tert-butyl (2- {5-[(7R,14R)-1-(difluoromethoxy)-5-oxo-5,6,7,14-tetrahydro-7,14-
methanobenzimidazo[1,2-U2,5Thenzodiazocin-11-yl]pyrimidin-2-yl}propan-2-
yl)carbamate
The title compound was prepared from Example 11(300 mg, 0.80 mmol) and
Intermediate 114 (252 mg, 0.80 mmol) in accordance with the Method described
for
Example 70. The crude product was purified by flash chromatography in (SiO2, 0
to
100% Et0Ac in DCM and then 1 to 10% Me0H in Et0Ac) to obtain the title
compound
(154 mg, 33%).
LCMS Method 3 (ES+) RT 1.43 mm, 577.2 (M+H)+
INTERMEDIATE 116
0 H
F
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 172 -
tert-butyl (2- {5- [(7R.14R)-1-(difluoromethoxy)-10-fluoro-5-oxo-5,6,7,14-
tetrahydro-
7,14-met hanob enzimidazo [1,2-bi[2,5]benzodiazo cin-11-ylipyrimidin-2-y1J
propan-2-
yl)carbamate
The title compound was prepared from Example 10 (50 mg, 0.13 mmol) and
Intermediate
114 (100 mg, 0.32 mmol) in accordance with the Method described for Example
70. The
product was purified by flash chromatography (SiO2, 0 to 100% Et0Ac in DCM and
then
1 to 10% Me0H in Et0Ac) to obtain the title compound (55 mg, 71%). LCMS Method
3
(ES+) RT 2.16 mm, 595.2 (M+H)+
INTERMEDIATE 117
o
CFO
io
tert-butyl {(1R,3R)-142-acety1-6-(difluoromethoxy)pheny11-7-chloro-2,3-dihydro-
1H-pyrrolo [1,2 -a]benzimidazol-3 -y1 carbamate
To a degassed solution of Intermediate 42 (1.4 g, 2.6 mmol) in dry toluene (30
mL) under
argon were added tributy1(1-ethoxyvinyl)tin (1.1 mL g, 3.2 mmol) and
.. bis(triphenylphosphine)-palladium(I1)dichloride (100 mg, 0.141 mmol). The
reaction
mixture was heated at 105 C for 48 hours. Further Tributy1(1-ethoxyvinyl)tin
(0.7 g, 2.0
mmol) and bis(triphenylphosphine)palladium(II)-dichloride (64 mg, 0.0912 mmol)
were
added and the reaction mixture was heated for an additional 5 hours at 105 C.
The
reaction mixture was cooled to room temperature and poured onto a saturated
aqueous
.. solution of KF. The mixture was extracted with ethyl acetate, filtered over
celite, dried
over MgSO4, filtered and concentrated in vacuo. The crude compound was re-
dissolved in
THF (50 mL), p-toluene sulfonic acid (200 mg) and water (5 mL) was added and
the
mixture was heated at 45 C for 5 hours. The mixture was poured onto ice,
neutralized
with solid NaHCO3 and extracted with ethyl acetate (x3). The combined organic
layers
.. were dried over MgSO4, filtered and evaporated to dryness. The crude
compound was
purified by flash chromatography with ethyl acetate 50%- heptane 50% to afford
1.27 g of
the title compound as a yellow solid. LCMS Method 3 basic (ES+) RT 2.75 min.
492.1
(M+H)+.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 173 -
INTERMEDIATE 118
= ,A\
(7R,14R)-11-chloro-1-(difluoromethoxy)-5-methy1-7.14-dihydro-7,14-
methanobenzimidazo[1,2-b1[2,5]benzodiazocinc
A solution of Intermediate 117 (0.48 g, 0.976 mmol) in dichloromethane (10 mL)
was
cooled to 0 C and trifluoroacetic acid (10 mL) was added dropwise. The
reaction mixture
was stirred overnight at room temperature. The mixture was poured on ice,
brought to
neutral pH with solid NaHCO3 and extracted with dichloromethane (x3). The
combined
organic layers were dried over MgSO4, filtered and concentrated in vacuo to
afford 0.25 g
(65%) of the title compound. LCMS basic Method 3 (ES+) RT 2.42 mm., 374.1
(M+H)+.
INTERMEDIATE 119
c H
F-<.
(7R,14R)-11-chl oro-1 -(difluoromethoxy)-5-methy1-5,6,7,14-tetrahydro-7,14-
rnethanobenzimidazo[1,2-b][2,5]benzodiazocine
To a solution of Intermediate 118 (350 mg, 0.893 mmol) in a mixture of THF (8
mL)
and Et0H (8 mL) were added macroporous polymer-supported cyanoborohydride
(1.12 g, 4.4 mmol, 4.0 mrnol/g loading) and acetic acid (50 L). The reaction
mixture was stirred overnight at room temperature. The reaction mixture was
filtered
over celite and concentrated in vacuo. The residue was poured onto ice/water
and
solid NaHCO3 was added till pH = 9. The aqueous phase was extracted with DCM
(x3), the combined organic layers dried over MgSO4, filtered and concentrated
to
dryness. The crude compound was purified by normal phase chromatography (DCM
95 % ¨ Me0H 5%) to afford 237 mg (71%) of the title compound as a mixture of
diastereomers. LCMS Method 3 basic (ES+) RT 1.99 mm., 376.1 (M+H) .
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 174 -
INTERMEDIATES 120 and 121
crj0C CI 1.1
^ft.
FFQ
4111
(5R,7R,14R)-11-chloro-1-(difluoromethoxy)-5-methy1-5,6,7,14-tetrahydro-7,14-
methanobenzimidazo[1,2-b][2,5Thenzodiazocine
(5S,7R,14R)-11-chloro-1-(difluoromethoxy)-5-methy1-5,6,7,14-tetrahydro-7,14-
methanobenzimidazo[1,2-b][2,5]benzodiazocine
The title compounds were isolated from Intermediate 119 (0.118 g) by
purification under SFC
conditions on Chiralpak OD-A20 column (50*266, 360 mL/min, 25 C, CO2+20 %
iPrOH, con: 24
g/1), yielding 37 mg (31 %) of Intermediate 120 (RT 3.65 min) and 40.0 mg
(34%) of Intermediate
121 (RT 7.57 min) respectively.
Intermediate 120: LCMS Method 4 (ES+) RT 2.18 min., 376.2 (M+H)+.
Intermediate 121: LCMS Method 4 (ES+) RT 2.14 mm., 376.2 (M+H)+.
INTERMEDIATE 122
t
I F 0
\Y "
tert-butyl {(1R,3R)-7- [2-(2- f[tert-butyl(dimethyl)silyl]oxylpropan-2-
yl)pyrimidin-
5-y1]-1-[2-chloro-6-(difluoromethoxy)pheny11-23-dihydro-lH-pyrrolo[1,2-
a]benzimidazol-3-y1}carbamate
Intermediate 22(5 g, 8,331 mmol) was suspended in DCM (10 mL) and cooled on
an ice bath. Triethylarnine (2.6 mL, 18,33 mmol) and di-tert-butyl dicarbonate
(2.2
g, 10.0 mmol) were added. The reaction was warmed to ambient temperature and
stirred overnight. The reaction mixture was quenched by addition of water (10
mL).
The aqueous layer was extracted by DCM (3 x 10 mL). The combined organic
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 175 -
layers were dried over MgSO4, filtered and concentrated in vacuum. The residue
was purified over silica gel (eluting with heptane / ethyl acetate 7 / 3) to
afford 5.1 g
(87%) of the title compound as a white solid.
LCMS Method 3 (ES+) RT 3.64 min., 700.3 / 702.3 (M+H)+
INTERMEDIATE 123
0 =
=
'WO
>r\
Ethyl 2- {(1R,3R)-3-[(tert-butoxycarbonyl)amino]-742-(2- { [tert-
butyl(dimethyl)silyl]oxylpropan-2-yOpyrimidin-5-y11-2.3-dihydro-1H-pyrrolo[1.2-
a]benzimidazol-1-y1 } -3-(difluoromethoxy)benzoate
Intermediate 122 (5 g, 7.14 mmol), potassium carbonate (1.50 g, 10.71 mmol),
molecular sieve 4 A powder (2 g) and
dichloro[bis(dicyclohexylphosphino)propane]-palladium(H) (350 mg, 0,57 mmol)
were suspended in dry dimethyl sulfoxide (50 mL) and ethanol (1.8 mL, 32
mmol).
The slurry was stirred under 5 bars of CO gas at 100 C, overnight. The slurry
was
filtered through a pad of celite and rinsed with ethyl acetate (30 mL). The
filtrate
was washed successively by a saturated solution of aqueous NH4C1 (20 mL),
brine
(20 mL), dried over MgSO4, filtetred and concentrated in vacuum affording 5.1g
of
the crude title compound as a brown solid, used without further purification.
LCMS
Method 3 (ES+) RT 3.64 min., 738.1 (M+FI)+.
INTERMEDIATE 124
0 t
CYlk
>r\ 0 H
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 176 -
tert-butyl {(1R,3R)-7 -[2-(2- Wert-butyl(dimethyl)silylioxy}propan-2-
y1)pyrimidin-
5-y1]-1-[2-(difluoromethoxy)-6-(hydroxymethyl)pheny1]-2,3-dihydro-1H-
pyrrolo [1,2 a] benzimi dazol-3-ylIcarbamate
Intermediate 123 6 (3 g) was dissolved in ethanol (30 mL). At 0 C, sodium
borohydride
(1.2 g) was added, followed by calcium chloride (1.805 g). The reaction
mixture was
warmed to room temperature and stirred for 4 hours. The reaction mixture was
filtered through a pad of celite, and rinsed with ethyl acetate (2 x 20 mL).
The filtrate
was washed with water (2 x 20 mL) and brine (20 mL), and dried over MgSO4. The
residue was purified by basic reverse phase preparative HPLC yielding 948 mg
(33%) of the title compound as a white solid. LCMS Method 3 (ES+) RT 3.45
minutes
696.3 (M+H)'.
INTERMEDITE 125
LH
$(. 0
tert-butyl 1(1R,3R)-7-[2-(2- 1 Pert-butyl (dimethyl) si lyl] oxy } propan-2-
yl)pyrimi din-
5-y1]- 42-(difluoromethoxy)-6-formylpheny1]-2,3-dihydro-1H-pyrrolo[1,2-
a]benzimidazol-3-yl}carbamate
Intermediate 124 (750 mg, 1.08 mmol) was dissolved in 1,4-dioxane (16 mL)
before
addition of manganese dioxide (2.3 g, 27 mmol) at room temperature. The
reaction was
stirred overnight. The crude reaction mixture was filtered through a pad of
celite and
rinsed with 20 mL of chloroform. The filtrate was concentrated to dryness in
vacuum
yielding to a crude 860 mg of title compound used without further
purification. LCMS
Method 3 basic (ES+) RT 5.85 minutes. 694.4 (M+H)I .
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 177 -
INTERMEDIATE 126
>rõs
(7R,14R)-11 - [2 -(2 -{Ltert-butyl(dimethypsilyl] oxy } propan-2-yOpyrimidin-5-
yl] -1 -
(difluoromethoxy)-7,14-dihydro-7,14-methanobenzimidazo[1,2-
bli2,5]benzodiazocine
Intermediate 125 (860 mg, 1.24 mmol) was treated in accordance with the
procedure
described for the synthesis of Intermediate 118 to afford 710 mg (99%) of the
title
compound as a yellow glass.
LCMS Method 3 basic (ES+) RT 5.76 min., 576.2 (M+H)-.
INTERMEDIATE 127
>rS
(7Rõ14R)-11- [2-(2- { [tert-butyl(dimethyl)silyl] oxy} propan-2-yl)pyrimidin-5
-yl] -1-
(difluoromethoxy)-5.6.7.14-tetrahydro-7.14-methanobenzimidazo[1.2-
.. b][2,5]benzodiazocine
Intermediate 126 (710 mg, 1.23 mmol) was treated in accordance with the
synthetic
method described for Intermediate 119 to afford the title compound 655 mg
(92%) as a
yellow oil.
LCMS Method 3 basic (ES+) RT 5.84 min., 578.7 (M+H) .
INTERMEDIATE 128
0 7
= >04
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 178 -
1-[(7R,14R)-11-[2-(2- { [tert-butyl(dimethyl) oxy}
propan-2-yppyrimidin-5-y1]-
1-(difluoromethoxy)-5,14-dihydro-7,14-methanobenzimidazo[1,2-
b][2,5Thenzodiazocin-6(7H)-yl]ethanone
To a solution of Intermediate 127 (655 mg, 1.13 mmol) in dichloromethane (11
mL) was
added successively pyridine (0.28 mL, 3.4 mmol) and acetic anhydride (0.22 mL,
2.27
mmol). The reaction mixture was stirred at room temperature for 90 minutes.
The
reaction mixture was washed by a saturated aqueous solution of NRIC1 (2 x 20
mL) and
a saturated aqueous solution of NaHCO3 (2 x 10 mL). The aqueous phases were
extracted by DCM (2 x 10 mL). The combined organic layers were dried over
Na2SO4,
filtered and concentrated under vacuum. The residue was purified by
chromatography
over silica gel (eluting with ethyl acetate 100% to ethyl acetate / ethanol
9/1), yielding to
300 mg (43%) of title compound as a yellow glass.
LCMS Method 3 basic (ES+) RT 5.84 mm., 620.3 (M+H)-
INTERMEDIATE 129
\t\IA
>rSi
(7R,14R )-11- [2 -(2 - { [tert-butyl(dimethypsilyl]oxyl propan-2-yppyrimidin-5-
yl] -1 -
(d ifluoromethoxy)-6 -methyl-5 ,6,7,14-tetrahydro-7,14-methanobenzimidazo[1,2-
b][2,5Thenzodiazocine
Intermediate 127 (30 mg, 0.051 mmol) in 2,2,2-trifluoroethanol (3 mL) and
formaldehyde (1 mL, 25.97 mmol) was stirred at room temperature for 30 minutes
before addition of sodium borohydride (20 mg, 0.52 mmol). The reaction mixture
was heated at 70 C for 2 hours. At room temperature, additional sodium
borohydride (20 mg, 0.52 mmol) was added, and the reaction mixture heated at
70 C for addition 1 hour. The reaction mixture was filtered through a pad of
celite,
and the residual solid washed by 2,2,2-trifluoroethanol (2 x 4 mL). The
filtrate was
concentrated in vacuum. The residue was used without further purification.
LCMS Method 3 basic (ES+) RT 5.97 mm., 592.2 (M+H) .
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 179 -
INTERMEDIATE 130
(7R,14R)-11-[2-(2- fftert-butyl(dimethyl)silyl]oxy propan-2-yppyrimidin-5-yll -
1-
(difluoromethoxy)-5-(trifluoromethyl)-5,6,7,14-tetrahydro-7,14-
methanobenzimidazo[1.2-b112.51benzodiazocine
To a solution of Intermediate 126 (0.074 mmol) in acetonitrile (1 mL) and NA
dimethylformamide (17g') was added successively at 0 C, trifluoroacetic acid
(7 lit,
0.092 mmol), potassium hydrogen fluoride (4.4 mg, 0.055 mmol) and
(trifluoromethyptrimethylsilane (16 [iL, 0.11 mmol). The resulting slurry was
allowed to
warm to ambient temperature overnight. The reaction mixture was then
evaporated and
purified by preparative basic reverse phase HPLC. This was followed by a
second
acidic preparative HPLC to afford the TFA salt of the title compound which was
solubilized in Et0Ac (2 mL) and washed with a saturated solution of NaHCO3.
The
aqueous layer was extracted with Et0Ac (2 x 2 mL). The combined organic layers
were dried over magnesium sulphate, filtered and concentrated in vacuo to
afford 7 mg
(15%) of the title compound as a mixture of diastereoisomers.
LCMS Method 3 (ES+) RT 6.08 min., 646.2 (M+H)+.
INTERMEDIATE 131 and INTERMEDIATE 132
and -41--
(5R,7R,14R)-1142-(2-{[tert-butyl(dimethyl)silyl]oxylpropan-2-yppyrimidin-5-y1]-
1-
(difluoromethoxy)-5-(trifluoromethyl)-5 ,6,7, 1 4-tetrahydro-7,1 4-
methanobenzimidazor1,2-13112,51benzodiazocine
and
5S 7R 14R -11- 2- 2- tert-bu 1 dimetlf_,__)_[__(,__( ro an-2- 1 imidin-
5- -1-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 180 -
(difluoromethoxy)-5-(trifluoromethyl)-5,6,7,14-tctrahydro-7,14-
methanobenzimidazo[1,2-h112,51benzodiazocine
To a solution of Intermediate 126 (0.72 mmol) in acetonitrile (5 mL) and DMF
(1664)
was added successively at 0 C, trifluoroacetic acid (69 FL, 0.90 mmol),
potassium
hydrogen fluoride (43 mg, 0.54 mmol) and (trifluoromethyl)trimethylsilane (159
LL, 1.08
mmol). The resulting slurry was allowed to warm to room temperature for 3
hours. The
reaction mixture was then diluted with Et0Ac and a saturated aqueous solution
of
NaHCO3. The two phases were separated and the aqueous layer further extracted
with
Et0Ac (x2). The combined organic extracts were dried over magnesium sulphate,
filtered
and concentrated under reduced pressure.
The crude was purified over silica gel (eluting with dichloromethane-methanol-
aqueous
ammonia / 97:2.7:0.3). This was followed by a second purification by reverse
phase
preparative HPLC to give the following diastereoisomers:
4.8 mg (1%) of Intermediate 131:
LCMS Method 3 (ES+) RT 3.65 min., 646.2 (M+H)'.
NMR (400 MHz, CDCI3) 6 8.72 (m, 2 H), 7.19 (m, 1 H), 7.05 (s, 2 H), 7.00 (m, 1
H),
6.85 (m, 2 H), 6.35 (m, 1 H), 5.33 (m, 1H), 4.52 (d, 1 H, .1= 7.0 Hz), 3.48
(m, 1 H), 2.83
(m, 1 H), 2.42 (m, 1 H), 1.67 (s, 6 H), 0.87 (s, 9 H), -0.07 (d, 6 H, 1= 1.2
Hz).
2.8mg (5%) of Intermediate 132:
LCMS Method 3 (ES+) RT 3.66 min., 646.2 (M+H)+.
NMR (400 MHz, CDCb) 6 8.93 (in, 2 H), 7.94 (m, 1 H), 7.53 (m, 2 H), 7.31 (m, 2
H),
7.23 (m, 1 H), 6.76 (m, 1 H), 6.32 (m, 1 H), 4.88 (m, 1 H), 3.39 (m, 1 H),
3.19 (m, 1 H),
2.67 (m, 1 H), 2.54 (m, 1 H), 1.70 (s, 6 H), 0.89 (s, 9 H), -0.04 (s, 6 H)
INTERMEDIATE 133
144-methy1-5-(4,4,5,5-tetramethy1-1,3,2-dioxaborolan-2-yppyrimidin-2-y1]-3-
(trifluoromethyl)azetidin-3-ol
1-(5-bromo-4-methyl-pyrimidin-2-y1)-3-(trifluoromethyl)azetidin-3-o1 (700 mg,
2.24
mmol), bis(pinacolato)diboron (1.15 g, 4.49 mmol), 1,1'-
bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloro methane
complex (92
mg, 0.112 mmol), potassium acetate (890 mg, 8.97 mmol) and 1,4-dioxane (10 mL)
were
placed in a small RB flask, degassed and placed under nitrogen. The mixture
was then
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 181 -
heated at 105 C for 2 hours. The mixture was cooled and partitioned between
Et0Ac and
water. The organic layer was dried over Na2SO4, filtered and evaporated in
vacuo to give
the title compound as a dark brown solid 1.60g. LC/MS Method 3: RT 1.93
minutes, m/z
360.
INTERMEDIATE 134
1-(5-bromo-6-methy1-2-pyridy1)-3-(trifluoromethypazetidin-3-o1
2,5-dibromo-6-methylpyridine (1.30 g, 5.18 mmol) and 3-
(trifluoromethypazetidin-3-01
hydrochloride (1.00 g, 5.63 mmol) were added to a small RB flask with stirrer
bar. N,N-
diisopropylethylamine (0.90 mL, 5.2 mmol) was added and the mixture heated to
130 C
for 4 hours. The mixture was cooled, diluted with diehloromethane (50mL) and
washed
with sodium bicarbonate solution (50mL). The organic layer was dried (Na2SO4),
filtered and concentrated in vacua. Chromatography (silica, DCM gradient to
15%
Et0Ac in DCM) gave the title compound as a white solid (210 mg, 13.0% yield).
IFT
NMR (400 MHz, DMSO-d6) 6 7.70 (d, J = 8.7 Hz, 1H), 7.32 (s, 1H), 6.32 (dd, J=
8.7,
.. 0.7 Hz, 1H), 4.19 (dd, J = 9.6, 1.0 Hz, 2H), 3.93 (dt, J = 9.4, 1.3 Hz,
2H), 2.42 (s, 3H).
LC/MS Method 3: RT 2.02 minutes, m/z 313/315.
INTERMEDIA'I'E 135
146-methy1-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-y1)-2-pyridy11-3-
(trifluoromethyl)azetidin-3-ol
Intermediate 134 was treated in accordance with the synthetic procedure
described for
Intermediate 133 to afford the title compound as pale brown gum which was used
without
further purification. LC/MS Method 3: RT 2.29 minutes, m/z 359.
INTERMEDIATE 136
2-ethylhexyl 342-[(1R,38)-7-chloro-3-hydroxy-2,3-dihydro-1H-pyrrolo[1,2-
a]benzimidazol-1-y1]-3-(difluoromethoxy)phenyllsulfanylpropanoate
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 182 -
N,N-di-iso-propylethylamine (1.63 mL, 9.31 mmol) was added to a solution of
Intermediate 38 (2.00 g, 4.66 mmol) in 1,4-dioxane (10 mL). The mixture was
evacuated
and refilled with nitrogen. The catalyst,
tris(diberizylideneacetone)dipalladium(0) (213
mg, 0.233 mmol) and 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (275 mg,
0.466
mmol) and 3-mercaptopropionic acid 2-ethylhexyl ester (1.86 mL, 7.93 mmol)
were
added and the mixture was evacuated and filled with nitrogen and heated under
nitrogen
at 105 CC for 18 hours. The mixture was partitioned between Et0Ac (250mL) and
saturated aqueous sodium bicarbonate solution (100 mL). The organic layer was
dried
(sodium sulfate), filtered and concentrated in vacuo to leave a yellow oil,
4.00g.
Purification by chromatography (silica, dichloromethane gradient up to 5%
methanol in
dichloromethane) afforded the title product as a pale yellow foam (2.10 g, 80%
yield). 1H
NMR (300 MHz, DMSO-d6) 6 7.62 (dd, J= 8.6, 1.9 Hz, 1H), 7.54 - 7.41 (m, 1H),
7.36 -
7.21 (m, 1H), 7.21 - 7.09 (m, 1H), 7.06 -6.86 (m, 1H), 6.74 -6.52 (m, 1H),
6.46 - 6.21
(m, 1H), 6.03 (dd, J= 6.7, 3.7 Hz, 1H), 5.36 - 5.07 (m, 1H), 4.05 - 3.80 (m,
2H), 3.35
(td, J- 6.8, 2.6 Hz, 2H), 3.25 - 2.64 (m, 4H), 2.24 (ddt, J= 24.0, 16.6, 8.4
Hz, 1H), 1.50
(dd, .1= 12.5, 6.4 Hz, 1H), 1.37- 1.08 (m, 8H), 0.92 -0.68 (m, 6H). LC/MS
Method 3:
RT 3.00 minutes, tn/z 381/383.
INTERMEDIATE 137
1-(5-bromo-4-methyl-2-pyridy1)-3-(trifluoromethyl)azetidin-3-ol
The title compound was synthesised from 2,5-dibromo-4-methylpyridine and 3-
(trifluoromethyDazetidin-3-ol hydrochloride in accordance with the synthetic
procedure
described for Intermediate 134.
INTERMEDIATE 138
144-methy1-5-(4,4,5,5-tetramethy1-1,3,2-dioxaborolan-2-y1)-2-pyridy1]-3-
(trifluoromethypazetidin-3-ol
Intermediate 137 was treated in accordance with the synthetic procedure
described for
Intermediate 135 to afford the title compound which was used without further
purification.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 183 -
INTERMEDIATE 139
Ethyl 5-bromo-4-methylpyrimidine-2-carboxylate
5-bromo-4-methylpyrimidine-2-carboxylic acid (17.8 g, 82 mmol) dissolved in
ethanol
(185 mL). Sulfuric acid (38.6 g, 394 mmol, 21 mL) was added and the resulting
suspension was placed in a preheated oil bath of 80 C. Additional sulfuric
acid (3.68 g,
37.5 mmol, 2 mL) added and heating continued for a further hour before cooling
to room
temperature. The solid was filtered and residue washed with Et0H. The filtrate
was
evaporated and the residue taken up in Et0Ae (300 mL) and sat. aqueous NaHCO3
solution (300 mL). The layers were separated and the aqueous phase extracted
with
Et0Ac (300 mL). The combined organics were washed with brine (300 mL), dried
(Na2SO4), filtered and evaporated to give a dark solid which was purified by
column
chromatography 300 g silica (20% -> 50% Et0Ac in heptane) to give the title
compound
as a yellow solid (12.24 g, 44%). 1H NMR (300 MHz, Chloroform-d) 6 8.87 (s,
1H), 4.53
(q, J= 7.1 Hz, 2H), 2.76 (s, 3H), 1.46 (t, J= 7.1 Hz, 3H). LC/MS Method 11 RT
1.57
minutes, [M+H]: 245/247 Br-isotope.
INTERMEDIATE 140
2-(5-bromo-4-methylpyrimidin-2-yl)propan-2-ol
Under a nitrogen atmosphere methyl magnesium bromide solution (3M in Et20 109
mmol, 36.4 ml) was added dropwise to a stirred mixture of Intermediate 139
(10.7 g, 43.7
mmol) in diethyl ether (300 mL) while cooled in an ice/water bath. During
addition a
suspension formed. When addition was completed the mixture was stirred at room
temperature. The reaction mixture was carefully quenched with saturated
aqueous NH4C1
solution (300 mL). The resulting organic layer was separated and the aqueous
phase was
extracted with Et20 (300 mL). The combined organics were washed with brine,
dried
(Na2SO4), filtered and evaporated to dryness to give an orange oil.
Purification by flash chromatography (300g silica, 10% -> 50% Et0Ae in
heptane) gave
the title compound as a light yellow oil (7.5 g, 74%). 1H NMR (300 MHz,
Chloroform-d)
6 8.67 (s, 1H), 4.51 (s, 1H), 2.65 (s, 3H), 1.58 (s, 6H). LC/MS Method 9:
[M+H]
231/233 Br-isotope.
INTERMEDIATE 141
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 184 -
N H 2
N
>r\
(1R,3R)-7-[2-(2- utert-butyl(dimethyl)silylioxy I prop an-2-yOpyrimidin-5 -y1]-
112-
(difluoromethoxy)-6-(methylsulfanyl)pheny11-2,3-dihydro-1H-pyrrolo[1,2-
a]benzimidazol-3-amine
To a solution of Intermediate 22 (415 mg, 0.69 mmol) in DMSO (2 mL) was added
sodium thiomethoxide (64 mg, 0.83 mmol). The reaction mixture was stirred for
25
minutes at 100 C. Water (20 mL) and ethyl acetate (40 mL) were added to the
reaction
mixture, and the two layers were separated. The aqueous layer was extracted
with ethyl
acetate (2 x 40 mL) and the combined organics layers were dried over MgSO4,
filtered
and concentrated in vacuo. The crude material was purified over silica gel
using
DCM/Me0H/NH4OH (100% DCM to 90/10/1) as eluent, yielding 310 mg (73%) of the
title compound as a brown solid. LCMS Method 3 (ES+): RT 2.72 min, [M+Fl]+=
612.2
INTERMEDIATE 142
1.1
0
\ =
(7R,14R)-11-[2-(2- {[tert-butyl(dimethyl)silylloxylpropan-2-y1)pyrimidin-5-y1]-
1-
(difluoromethoxy)-5-methy1-7,14-dihydro-7,14-methano-5X-4--benzimidazo[2,1-
d][1,2,5]benzothiadiazocine 5-oxide
To a degassed solution of Intermediate 141 (87 mg, 0.14 mmol) in Me0H (5 mL)
was
added a solution of bromine (19 mg, 0.12 mmol) in Me0H (0.5 mL). The reaction
mixture was evaporated after 1 hour and the residue was solubilised in DCM (5
mL). The
solution was degassed by bubbling of argon through the solution for 5 minutes.
Potassium
carbonate (63 mg, 0.45 mmol) and 3-chloroperbenzoie acid (74 mg, 0.43 mmol)
were
then added to the mixture. The mixture was stirred overnight at ambient
temperature
before dilution with DCM (40 mL) and the organic layer washed with water (2 x
20 mL),
dried by passage through a phase separator cartridge and concentrated in
vacuo. The
crude material was purified over silica gel using DCM/Me0H/NH4OH (100% DCM to
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 185 -
90/10/1) as clucnt, yielding 8 mg (9%) of the title compound as a dark oil.
LCMS Method
3 (ES): RT 3.27 minutes, [M+H]+= 626.2.
INTERMEDIATE 143
5-bromo-2-(1-methylsulfonylcyclopropyl)pyridine
5-bromo-2-[(methylsulfonyl)methyl]pyridine (300 mg, 1.20 mmol), 1,2-
dibromoethane
(0.12 mL, 1.40 mmol), benzyltributylammonium chloride (377 mg, 1.20 mmol) and
sodium hydroxide 50% aqueous solution (7.5 mL, 94 mmol) were mixed in
acetonitrile (8
mL). The reaction mixture was stirred at room temperature for 24 hours.
Aqueous
saturated Nan solution and ethyl acetate were added to the reaction mixture
and the two
layers were separated. The organic layer was dried over MgSO4, filtered and
the solvent
evaporated. The crude was purified by reverse phase basic preparative LCMS to
yield 32
mg (10%) of the title compound as an off-white solid. LCMS Method 3 (ES+): RT
1.86min, [M+H]+= 276.
INTERMEDIATE 144
\
CIF
rit Br
(1R)-142-bromo-6-(difluoromethoxy)pheny1]-7-chloro-1,2-dihydro-3H-pyrrolo[1,2-
a]benzimidazol-3-one
To a solution of Intermediate 19 (3g, 6.98 mmol) in chloroform (60 mL), was
added
manganese dioxide (3.64 g, 42 mmol) and the reaction mixture was stirred at
room
temperature for 3 hours, after which additional manganese dioxide (2 g, 23
mmol) was
added and stirred overnight. The reaction mixture was filtered over celite,
rinsed with
chloroform (2 x 60 mL) and the filtrate concentrated in vacuo to yield 2.91g
(97%) of the
title compound as a beige solid. LCMS Method 3 (ES+): RT 4,88 mm, [M+H]+¨ 427.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 186 -
INTERMEDIATE 145
CIF.
Br
N- {(1R,3E)-142-bromo-6-(difluoromethoxy)pheny1]-7-ehloro-1,2-dihydro-3H-
pyrrolo[1,2-a]benzimidazol-3-ylidene}-2-methylpropane-2-sulfinamide
Titanium(IV) isopropoxide (1,86 mL, 9.52 mmol) was added to a solution of
Intermediate
144 (1.85 g, 4.33 mmol) in dry THF (43 mL). The mixture was stirred at room
temperature for 10 minutes before addition of (R)-(+)-2-methyl-2-
propanesulfinamide
(642 mg, 5.2 mmol). The reaction mixture was stirred at 50 C overnight.
Further (R)-(+)-
2-methy1-2-propanesulfinamide (320 mg, 2.58 mmol) was added and the reaction
mixture
stirred at 50 C overnight. The reaction mixture was cooled to 0 C and methanol
was
added followed by a saturated aqueous solution of NaHCO3until precipitation
was
observed. The slurry was diluted by Et0Ac (50 mL) and filtered over celite.
The filtrate
was washed with brine, dried over Na2SO4, filtered and concentrated in vacuo.
The
residue was purified over silica gel (gradient Et0Ac / heptane 20 to50 %)
yielding 770
.. mg (34%) of the title compound as a brown solid. LCMS Method 3 (ES+): RT
2.82 mm,
[M+H]= 530.
INTERMEDIATE 146
CIF*
r
* Br
N- {(1R,3R)-142-bromo-6-(difluoromethoxy)pheny1]-7-chloro-3-methy1-2,3-dihydro-
1H-
pyrrolo[1,2-a]benzimidazol-3-yll -2-methylpropane-2-sulfinamide
To a solution of Intermediate 145 (8g, 15.1 mmol) in dry DCM (90 mL), cooled
at -70 C,
was added dropwise a solution of methyl magnesium bromide 3 M in diethyl ether
(17.6
mL, 52.8 mmol). The reaction mixture was stirred at -70 C for 10 minutes and
at 0 C for
2 hours before quenching by addition of a saturated solution of NH4C1(100 mL).
The
aqueous layer was extracted with DCM (2 x 100 mL). The combined organic layers
were
washed with saturated brine and dried over MgSO4, filtered and concentrated in
vacuo.
The residue was purified over silica gel (gradient ethyl heptane / acetate 50
to 100%),
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 187 -
yielding to 2.91 g (35%) of the title compound as a beige solid. LCMS Method 3
(ES+):
RT 2.68 min, [M+H]f= 546.10
INTERMEDIATE 147
#
CIF N H
F 411, Br
(1R,3R)-142-bromo-6-(difluoromethoxy)pheny1]-7-chloro-3-methy1-2,3-dihydro-1H-
pyrrolo[1,2-a]benzimidazo1-3-amine
Intermediate 146 (2.91 g, 5.32 mmol) was dissolved in dry 1,4-dioxane (150
mL).
HCVdioxane (4M) (6.65 mL, 27 mmol) was added and the reaction mixture was
stirred at
room temperature for 4 hours. The solvent was evaporated and the residue was
taken up
in Et0Ac (50 mL) and a saturated aqueous solution of NaHCO3 (20 mL) added. The
organic layer was washed with saturated brine, dried over MgSO4, filtered and
concentrated in vacuo. The residue was dissolved in diethyl ether and
evaporated yielding
to 2.2g (93%) of the title compound as a yellow solid. LCMS Method 3 (ES+): RT
2.43
min, [M+H]-= 442.1.
INTERMEDIATE 148
H
Cl
= th,
(7R,14R)-11-chloro-1-(difluoromethoxy)-7-methy1-6,7-dihydro-7,14-
methanobenzimidazo[1,2-h][2,5]benzodiazocin-5(14H)-one
Intermediate 147 (50 mg, 0.11 mmol), potassium carbonate (23 mg, 0.170 mmol),
dichloro[9,9-dimethy1-4,5-bis(diphenylphosphino)xanthene]palladium(Il) (4 mg,
0.006
mmol) were mixed in degassed 1,4-dioxane (2 mL). The mixture was stirred under
CO
gas (3 bar) at 120 C for 4 hours. Additional
bis(diphenylphosphino)xanthene]palladium(l) (4 mg, 0.006 mmol) was introduced
in the
reactor at room temperature and the reaction continued under stirring under CO
(3bar) at
120 C for 16 hours. The crude mixture was purified over silica gel (ethyl
acetate as
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 188 -
eluent), yielding 71 mg (37%) of the title compound as an off-white solid.
LCMS Method
3 (ES+): RT 2.34 min, [114+H]+= 390.
INTERMEDIATE 149
0,1 =
0
= 4111
(7R,14R)-11-chloro-1-(difluoromethoxy)-6,7-dimethy1-6,7-dihydro-7,14-
methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(141/)-one
Intermediate 148 (80 mg, 0.21 mmol) and tetrabutylammonium iodide (30 mg, 0.08
mmol) were mixed in dry THF (2 mL). At 0 C, sodium hydride (60% in mineral
oil) (9
mg, 0.246 mmol) was added. The reaction mixture was stirred at room
temperature for 35
minutes. Iodomethane (0,08 mL, 1.24 mmol) was added and the reaction mixture
was
stirred at ambient temperature overnight. The reaction mixture was quenched by
addition
of water (1 mL). The aqueous layer was extracted by Et0Ac (3 x 2 mL). The
combined
organic layers were dried over MgSO4, filtered and concentrated in vacuo. The
residue
was purified over silica gel (heptane / DCM 50% to 100%), yielding 60 mg (72%)
of the
title compound as a white solid. LCMS Method 4 (ES-f-): RT 2.63 min, [M+H]+
404.
INTERMEDIATE 150
y,
H
tert-butyl (2- {5-[(7R,14R)-1-(difluoromethoxy)-7-methy1-5-oxo-5,6,7,14-
tetrahydro-7,14-
methanobenzimidazo[1,2-b][2,5]benzodiazocin-11-yllpyrimidin-2-yl}propan-2-
ypearbamate
tert-butyl N41-(5-bromopyrimidin-2-y1)-1-methyl-ethylicarbamate (36 mg, 0.11
mmol),
bis(pinacolato)diboron (36 mg, 0.14 mmol), potassium acetate (11 mg, 0.12
mmol), [1,1'-
bis(diphenylphosphino)ferrocene]diehloropalladium(11) (6 mg, 0.008 mmol), were
mixed
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 189 -
in dioxanc (3 mL). The reaction mixture was stirred at 100 C for 2 hours. The
reaction
mixture was filtered through a 45 M filter, and concentrated in vacuo. The
residue was
dissolved in n-butanol (5 mL) and Intermediate 148 (15mg, 0.038 mmol),
tricyclohexylphosphonium tetrafluoroborate (4 mg, 0.009 mmol),
tris(dibenzenylideneacetone)dipaladium(0) (4mg, 0.0038 mmol), potassium
triphosphate
(17 mg, 0.077 mmol) and water (50 L) were added. The reaction mixture was
stirred at
140 C in a microwave for 25 minutes. The reaction mixture was filtered and
purified by
reverse phase basic preparative HPLC-MS to yield 13 mg (57%) of the title
compound as
a beige solid. LCMS Method 3 (ES+): RT 2.42 minutes, [M+H]= 591.
INTERMEDIATE 151
I 111
0,F
(1R,35)-3- atert-butyl(dimethypsilyl]oxy}-712-(2- atert-
butyl(dimethyDsilyl]oxy)propan-2-Apyrimidin-5-y11-1-[2-chloro-6-
(difluoromethoxy)pheny1]-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazole
To a solution of Intermediate 21 (2 g, 3.33 mmol) in DMF (12 mL) was added
imidazole
(283 mg, 4.16 mmol) and tert-butyldimethylchlorosilane (543 mg, 3.49 mmol) and
the
reaction mixture was stirred overnight at room temperature. The reaction was
then diluted
with diethyl ether (30 mL) and water (30 mL). The aqueous layer was extracted
with
.. diethyl ether (2 x 20 mL). The combined organic layers were washed with
brine, dried
over MgSO4, filtered and concentrated under reduced pressure to afford the
title
compound which was used in the next step without further purification. LCMS
Method 3
(ES+): RT 7.09 min.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 190 -
INTERMEDIATE 152
k
Ethyl 2- alR,3R)-3- {[tert-butyl(dimethyl)silylioxy} -7-[2-(2- f[tert-
butyhdimethyl)
silylioxy{ propan-2-yl)pyrimidin-5 -y1]-2,3 -di hydro-1H-pyrro lo[1,2-
albenzimidazol-1-yl -
3-(difluorometboxy)benzoate
Intermediate 151 (2.05 g, 2.87 mmol), potassium carbonate (1.5 equiv., 4.30
mmol),
molecular sieve 4A powder (860 mg) and
dichloro[bis(dicyclohexylphosphino)propane]palladium(II) (0.08 equiv., 0.23
mmol) were
suspended in dry dimethylsulfoxide (20 mL) and ethanol (0.75 mL). The reaction
mixture
was stirred at 100 C under 5 bars of CO gas for 16 hours. After this time,
another portion
of dichloro[bis(dicyclohexylphosphino)propane]palladium(II) was added and the
reaction
stirred overnight at 100 C under 5bars of CO gas to complete the reaction. The
reaction
mixture was allowed to cool to ambient temperature, filtered over celite and
partitioned
between Et0Ac (50 mL) and water (50 mL). The organic layer was washed with
water (2
x 20 mL), dried over MgSO4, filtered and concentrated under reduced pressure.
The
residue was purified over silica gel (DCM : Me0H 99.5%: 0.5%), yielding 824 mg
(38%) of the title compound. LCMS Method 3 (ES+): RT 3.92 mm, 753 (M+H)-.
INTERMEDIATE 153
\4i.k
b
tI F)--49 Wilk OH
J2- {(1R,3S)-3- {{tert-butyl(dimethyl)silylioxyl -7-[2-(2- {[tert-
butyl(dimethypsilynoxy}propan-2-yOpyrimidin-5-y1]-2,3-dihydro-1H-pyrrolo[1,2-
a]benzimidazol- 1-y1}-3-(difluoromethoxy)phenylimethanol
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 191 -
Intermediate 152 (780 mg, 1.04 mmol) was dissolved in dry ethanol (8 mL). At 0
C,
sodium borohydride (317 mg, 8.30 mmol) followed by calcium chloride (460 mg,
4.15
mmol) were added. The reaction was allowed to warm to ambient temperature and
stirred
for 4 hours. The reaction was then diluted with Et0Ac (20 mL) and water (10
mL). The
organic layer was washed with brine, dried over MgSO4, filtered and
concentrated under
reduced pressure. The residue was purified over silica gel (hexane : ethyl
acetate 80 : 20),
yielding 233mg (32%) of the title compound. LCMS Method 3 (ES+): RT 3.69 min,
711
(M+H)4.
INTERMEDIATE 154
I F tr\
\
2- {(1R,3S)-3- {[tert-butyl(dimethyl)silylioxy}-7-[2-(2- ( [ten-
butyl(dimethyl)silyl]oxylpropan-2-yl)pyrimidin-5-y1]-2,3-dihydro-1H-
pyrrolo[1,2-
a]benzimidazo 1-1-yll -3 -(difluoromethoxy)benzaldehyde
Intermediate 153 (233 mg, 0.33 mmol) was dissolved in DCM (5 mL) before the
addition
of Dess-Martin periodinane (157 mg, 0.36 mmol). The slurry was stirred
overnight at
ambient temperature. Additional Dess-Martin periodinane (72 mg, 0.16 mmol) was
added
and the reaction mixture was stirred at room temperature for 3 hours to
complete the
reaction. The slurry was filtered and the filtrate was diluted with DCM (20
mL) and
washed by a saturated aqueous solution of NaHCO1 (10 mL). The aqueous layer
was
extracted with DCM (2 x 10 mL). The combined organic layers were dried over
MgSO4,
filtered and concentrated under reduced pressure. The residue was purified
over silica gel
(hexane : ethyl acetate 80: 20), yielding 190 mg (82%) of the title compound.
LCMS
Method 3 (ES+): RT 3.79 min, 709 (M+H)+.
INTERMEDIATE 155
0 H
I F
0 H
F F
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 192 -
(1R,3S)-112-(difluoromethoxy)-6-(2,2,2-trifluoro-1-hydroxyethyl)pheny1]-7-[2-
(2-
hydroxypropan-2-yl)pyrimidin-5-y1]-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazol-3-
ol
Intermediate 154 (190 mg, 0.27 mmol) was dissolved in tetrahydrofuran (3 mL).
At 0 C,
tetrabutylammonium fluoride (54 ILL, 0.054 mmol) followed by
(trifluoromethyl)trimethylsilane (79 4, 0.54 mmol) were added. The reaction
was
allowed to warm to room temperature and stirred for 2 hours. The reaction was
then
diluted with Et0Ac (10 mL) and water (5 mL). The aqueous layer was extracted
with
further Et0Ac (2 x 5 mL). The combined organic layers were dried over MgSO4,
filtered
and concentrated under reduced pressure. The residue was dissolved in methanol
(1m1)
and p-toluenesulfonic acid monohydrate (255 mg, 1.34 mmol) was added. The
reaction
was stirred overnight at room temperature. Additional p-toluenesulfonic acid
monohydrate (100 mg, 0.53 mmol) was added and the reaction mixture was stirred
at
ambient temperature overnight to complete the reaction. The reaction mixture
was diluted
with Et0Ac (10 mL) and a saturated aqueous solution of NaHCO3 (5 mL). The
aqueous
layer was extracted with Et0Ac (2 x 5 mL). The combined organic layers were
dried over
MgSO4, filtered and concentrated under reduced pressure. The residue was
purified by
reverse phase basic preparative HPLC-MS to afford 97 mg (66%) of the title
compound
as a white solid. I,CMS Method 3 (ES+): RT 2.03 min, 551 (M+H)+.
INTERMEDIATE 156
="*Iµl N H
F I
(1R,3R)-7-[2-(2- {[tert-butyl(dimethypsilyl]oxyl propan-2-yl)pyrimidin-5 -y1]-
112-chloro-
6- difluorometl hm 1 -6- ftoro-2 clo enta 4 5 imidazo 1 2-
cdpyridin-3-amine
To a solution of Intermediate 72 (212 mg, 0.42 mmol) and 4-
dimethylaminopyridine (4
mg, 0.033 mmol) in DCM (4 mL) at 0 C was added NN-di-isopropylethylamine (441
2.52 mmol), followed by tert-butyldimethylsflyl-trifluoromethanesulfonate (395
L,
1.69 mmol). The reaction mixture was stirred at 0 C for 20 minutes before the
cooling
bath was removed and the reaction mixture was allowed to warm up to room
temperature.
After 45 minutes the reaction mixture was diluted with DCM (100 mL) and washed
with
water (2 x 50 mL), brine (50 mL), dried (by passage through a phase separator
cartridge)
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 193 -
and concentrated in vacuo. The crude material was purified by column
chromatography
(SiO2, 60-100 A) Et0Ac in hexane, followed by 0-25% Me0H in Et0Ac) and freeze
dried
from acetonitrile/water to give the title compound (190 mg, 73 %) as an off-
white solid.
LCMS Method 3 (ES+) 618 (M+H)+, RT 3.44 minutes.
LCMS Method 4 (ES+) 618 (M+H)+, RT 3.04 minutes.
INTERMEDIATE 157
>L
Si NI
F.'4F
(6R,12R)-2-[2-(2- {[tert-butyl(dimethypsilyl]oxyl propan-2-yl)pyrimidin-5-y11-
11-
(difluoromethoxy)-3-fluoro-7,12-dihydro-6H-6,12-
methanopyrido[1',21:1,2]im1dazo[4,5-
e][1]benzazepine
To a microwave vial was added tris(dibenzylideneacetone)dipalladium(0) (9.5
mg, 0.01
mmol) and 2-(dicyclohexylphosphino)-2',4',6'-triisopropylbiphenyl (10.2 mg,
0.021
mmol), followed by degassed 1,4-dioxanc (2 mL), and the microwave vial then
scaled
and degassed and stirred at room temperature for 30 minutes. After this time
Intermediate 156 (125 mg, 0.20 mmol) and sodium tert-butoxide (41 mg, 0.40
mmol)
then added and reaction mixture degassed and heated to 110 C for 18 hours.
The
reaction mixture was partitioned between Et0Ac (25 mL) and water (25 mL), the
layers
separated and aqueous extracted with Et0Ac (25 ittL). The combined organics
were
washed with brine (25 mL), dried (MgSO4), filtered and concentrated in vacuo.
The
crude material was purified by column chromatography (SiO2, 0-20% Me0H in DCM)
to
give the title compound (69 mg, 59 %) as a dark brown glass.
1HNMR (300 MHz, Methanol-d4) 6 8.95 (d, J = 1.6 Hz, 2H), 8.37 (d, J = 7.3 Hz,
1H),
7.38 (d, J= 11.2 Hz, 1 H), 6.96 (t, = 75 Hz, 1H), 6.88 (t, õI= 8.1 Hz, 1H),
6.40 ¨ 6.26 (m,
2H), 4.89 ¨4.73 (m, 2H) 3.45 ¨3.32 (m, 1H), 2.21 (d, J= 10.5 Hz, 1H), 1.73 (s,
6H),
0.91 (s, 9H), -0.01 (s, 6H).
LCMS: Method 3 (ES+) 582 (M+H)f, RT 3.39 minutes.
LCMS Method 4 (ES+) 582 (M+H)+, RT 3.48 minutes.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 194 -
INTERMEDIATE 159
N
=
0
(7R,14R)-1-(difluoromethoxy)-6-trideuteromethy1-11-(4,4,5,5-tetramethy1-1,3,2-
dioxaborolan-2-y1)-6,7-dihydro-7,14-methanobenzimidazo[1,2-
b][2,5]benzodiazocin-
5(14H)-one
To a solution of Intermediate 110 (2.01 g, 5.12 mmol) in 1,4-dioxane (18 mL)
was added
bis-(pinacolato)diboron (1.97 g, 7.8 mmol), potassium acetate, (1.5 g, 15.1
mmol),
tricyclohexylphosphonium tetrafluoroborate (197 mg, 0.15 mmol) and
tris(dibenzylideneacetone)dipalladium(0) (242 mg, 0.25 mmol). The reaction
mixture was
degassed for 10 minutes before heating to 140 C in the microwave for 3 hours.
Water and
Et0Ac was added to the reaction mixture and the aqueous phase extracted with
further
Et0Ac. The combined organic layers were evaporated to give a crude residue
which was
purified by column chromatography on silica (eluent Hexane : Et0Ac gradient
from 0 to
100% followed by DCM : Me0H to 10% Me0H) to provide the title compound as a
white solid (2.2 g, 89% yield). LC/MS: Method 3 RT 2.27 mins, [M+H]= 485.
INTERMEDIATE 160
HO
(7R,14R)-11-[2-(cis-1- {[tert-butyl(dimethypsilyl]oxy} -3-hydroxy-3-
methylcyclobutyppyrimidin-5-ylf 1-(difluoromethoxy)-6-trideuteromethy1-6,7-
dihydro-
7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one
Intermediate 159 (401 mg, 0.83 mmol) and Intermediate 79 (403 mg, 1.08 mmol)
in 1,4-
dioxane (10 mL) were degassed, 1, l'-bis(diphenylphospino)ferrocene-
palladium(II)dichloride dichloromethane complex (35 mg, 0.043 mmol) and K3PO4
(282
mg, 1.33 mmol) were added and reaction mixture degassed and then heated at 110
C for
18 hours, or until LCMS analysis showed the reaction to be completed. The
reaction
mixture was allowed to cool to room temperature, partitioned between Et0Ac (20
mL)
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 195 -
and saturated aqueous sodium bicarbonate solution (20 mL), the layers were
separated
and the aqueous phase extracted with further Et0Ac (3 x 25 mL). The combined
organics
were dried over sodium sulphate, filtered and concentrated in vacuo. The crude
material
was purified by column chromatography (SiO2, 0-100% Et0Ac in hexane, followed
by 0-
15 % Me0H in DCM) to give the title compound (221 mg, 41 %) as a yellow foam.
11-1NMR (300 MHz, DMSO-do) 6 9.12 (s, 2H), 8.27 (dd, J= 5.9, 3.6 Hz, 1H), 7.78
(d, J=
8.5 Hz, 2H), 7.69 (t, J= 73.6 Hz, 1H), 7.65 (dd, J= 8.3, 1.8 Hz, 1H), 7.52 -
7.44 (m, 2H),
6.31 (d, J= 7.2 Hz, 1H), 5.25 (d, J= 6.9 Hz, 1H), 5.03 (s, 1H), 3.58 -3.45 (m,
1H), 3.10
- 3.02 (m, 1H), 2.84 (d, J= 13.6 Hz, 1H), 2.56 -2.45 (m, 2H), 2.48 -2.40 (m,
1H), 0.93
(s, 3H), 0.80 (s, 6H), -0.19 (d, J= 1.4 Hz, 9H).
LCMS: Method 3 (ES+) 651 (M+H)+, RT 2.51 minutes.
INTER1V1EDIA1E 161
OH
CI F.
r *
Ethyl 2-[2-[(1R,35)-7-chloro-3-hydroxy-2,3-dihydro-1H-pyrrolo[1,2-
albenzimidazo1-1-
y11-3-(difluoromethoxy)phenyl]sulfanylacetate
The title compound was prepared from Intermediate 38 (2.02 g, 4.7 mmol), N,N-
di-iso-
propylethylamine (1.63 mL, 9.31 mmol),
tris(dibenzylideneacetone)dipalladium(0) (213
mg, 0.23 mmol), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (275 mg, 0.47
mmol)
and ethyl thioglycolate (980 mg, 7.99 mmol) by the method of Intermediate 136.
Crude
material was purified by column chromatography (SiO2, 20-100% Et0Ac in hexane)
to
give the title compound (1.76 g, 80 %) as a yellow solid.
LCMS: Method 3 (ES+) 469 (M+H)', RT 2.08 minutes.
LCMS: Method 4 (ES+) 469 (M+H)', RT 2.05 minutes.
30
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 196 -
INTERMEDIATE 162
01 N
*0
-"ZF F
Ethyl (7S,141?)-11-chloro-1-(difluoromethoxy)-6,7-dihydro-14/1-7,14-
m eth an oben zim i dazo[2,1-d111,5Thenzothiazocin e-6-carboxyl ate 5,5-
dioxide
INTERMEDIATE 163
CI
41 0
F¨(
0 lip
Ethyl (7R,14S)-11-chloro-1-(difluoromethoxy)-6,7-dihydro-1411-7,14-
methanobenzimidazo[2,1-d][1,5]benzothiazocine-6-carboxylate 5-oxide
To a solution of Example 115 (150 mg, 0.33 mmol) in DCM (5 mL) was added 3-
chloroperoxybenzoic acid (149 mg, 0.66 mmol). The reaction mixture was stirred
at
room temperature for 18 hours, after which time the reaction mixture was
partitioned
between DCM (25 mL) and water (25 mL), layers separated and organics washed
with
saturated aqueous sodium bicarbonate solution. The aqueous phase was extracted
with
DCM (20 mL), the combined organics filtered through a phase separator and
concentrated
in vacuo. The residue was purified by column chromatography (SiO2, 0-60% Et0Ac
in
DCM) and freeze dried from acetonitrile/water to give Intermediate 162 (65 mg,
40 %) as
an off-white solid and Intermediate 163 (47 mg, 30 %) as a white solid.
Intermediate 162
LCMS: Method 3 (ES+) 483 (M+H)+, RT 2.12 minutes (minor diastercoisomer) and
2.26
minutes (major diastereoisomer)
LCMS: Method 4 (ES+) 483 (M+H)+, RT 2.09 minutes (minor diastereoisomer) and
2.26
minutes (Major diastereoisorner).
Intermediate 163
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 197 -
LCMS: Method 3 (ES+) 467 (M+H)+, RT 2.22 minutes (major diastercoisomer)
LCMS: Method 4 (ES+) 467 (M+H)+, RT 2.20 minutes (Major diastereoisomer).
INTERMEDIATE 164
H =
C *
=
2-[(7S,14R)-11-chloro-1-(difluoromethoxy)-6,7-dihydro-14H-7,14-
methanobenzimidazo[2,1-d][1,5]benzothiazocin-6-yl]propan-2-ol
To a solution of Example 115 (98 mg, 0.22 mmol) in THF (5 mL) at 0 C was
added
methylmagnesium bromide (3M in diethylether, 0.16 mL, 0.48 mmol). The reaction
mixture was allowed to warm to room temperature and stirred for 18 hours.
After this
time the reaction was quenched with methanol, concentrated in vacuo and the
residue
partitioned between Et0Ac (50 mL) and saturated aqueous sodium bicarbonate
solution
(50 mL), the layers separated and the aqueous phase extracted with Et0Ac (3 x
30 mL),
the combined organics were washed with brine (60 mL), dried (MgSO4), filtered
and
concentrated in vacuo. Purification by column chromatography (SiO2, 0-20% Me0H
in
DCM) and freeze drying from acetonitrile/water gave the title compound (28 mg,
29 %).
LCMS: Method 3 (ES+) 437 (M+H)+, RT 2.44 minutes.
LCMS: Method 4 (ES+) 437 (M+H)+, RT 2.38 minutes.
INTERMEDIATE 165
1111 OH
CI
f(7S,14R)-11-chloro-1-(difluoromethoxy)-6,7-dihydro-14H-7,14-
methanobenzimidazo[2,1-d][1,51benzothiazocin-6-ylimethanol
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 198 -
To a solution of Example 115 (88 mg, 0.15 mmol) in THE (1.5 mL) at -10 C was
added
lithium aluminium hydride (2M solution in THF, 0.1 mL, 0.20 mmol). The
reaction
mixture was stirred below 0 C for 1 hour after which time the reaction was
quenched
with a few drops of 2 M HC1, stirred and then basified with 10 % NaOH (aq) (20
mL) and
extracted with Et0Ac (4 x 25 mL). The combined organic phases were dried
(Na2SO4),
filtered and concentrated in vacuo. Purification by column chromatography
(SiO2, 50-
100% Et0Ac in DCM, followed by 0-20 % Me0H in Et0Ac) and freeze drying from
acetonitrile/water gave the title compound (35 mg, 57 %) as a white solid.
LCMS: Method 3 (ES+) 409 (M+H)+, RT 2.18 minutes (major diastereoisomer) and
2.22
minutes (minor diastereoisomer)
LCMS: Method 4 (ES+) 409 (M+H)+, RT 2.14 minutes.
INTER1V1EDIAIE 166
N H
0
(7R,14R)-1-(difluoromethoxy)-1146-(2-trimethylsilyloxypropan-2-Apyridin-3-y1]-
6,7-
dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(1411)-one
Example 11(750 mg, 2.00 mmol), 6-(2-(trimethylsilyloxy)propan-2-yl)pyridine-3-
boronic acid pinacol ester (1.41 g, 4.00 mmol),
tris(dibenzylideneacetone)dipalladium(0)
(94 mg, 0.1 mmol) and tricyclohexylphosphonium tetrafluoroborate (91 mg, 0.24
mmol,
97 mass%) were added to a round bottom flask, evacuated & refilled with
nitrogen and
1,4-dioxane (10 mL) added followed by potassium phosphate (1.27 g, 6.00 mmol)
in
water (1 mL). The mixture was degassed, placed under nitrogen and heated to
105 C
overnight. The mixture was partitioned between Et0Ac and water, the organic
layer dried
over sodium sulphate, filtered and concentrated in vacuo. The crude material
was purified
by chromatography, (Et0Ac to 15% Me0H gradient). The product fractions were
concentrated in vacuo to give the title compound as an off-white solid, (1.05
g, 96%
yield). LC/MS Method 3: RT 1.61 minutes, nri/z 549.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 199 -
INTERMEDIATE 167
tert-Butyl 3-(5-bromo-2-pyridy1)-3-hydroxy-pyrrolidine-1-carboxylate
2,5-dibromopyridine (2.00 g, 8.27 mmol) was dissolved in toluene (40 mL),
cooled to -
78 C and n-butyllithium (7.1 mL, 9.9 mmol, 1.40 M) solution in n-hexane added
dropwise and stirred for 10 minutes before the addition of N-B0C-3-
pyrrolidinone (1.61
g, 8.69 mmol) in toluene (3 mL). The mixture was stirred at -60 C for 1 hour,
quenched
with methanol (2 mL) and partitioned between Et0Ac and saturated aqueous
ammonium
chloride solution. The organic layer was concentrated in vacuo to yield a
crude residue.
Purification by chromatography (silica, 0 to 60% Et0Ac gradient in iso-hexane)
gave the
title compound as an off-white solid (1.05 g, 3.06 mmol, 37% yield). 1HNMR
(300
MHz, DMSO-d6) 6 8.65 (dd, J= 2.5, 0.7 Hz, 1H), 8.07 (dd, J= 8.5, 2.4 Hz, 1H),
7.66 (d,
J = 8.5 Hz, 1H), 5.76 (s, 1H), 3.63 (t, J = 11.3 Hz, 1H), 3.56 -3.37 (m, 4H),
2.43 - 2.21
(m, 1H), 1.98 - 1.86 (m, 1H), 1.41 (s, 9H). LC/MS Method 3: RT 1.57 minutes,
m/z
341/343 (-ye ion).
INTERMEDIATE 168
Methyl N-[1-(5-bromopyrirn i din-2-y1)-1-methyl-ethyl] c arb am ate
To a cooled (0 C) solution of 2-(5-bronnopyrimidin-2-yl)propan-2-amine (2 g,
9.25
mmol) in dichloromethane (50 mL) was added N,N-diisopropylethylamine (2.2
equiv.,
20.3 mmol) followed by methyl chloroformate (1 equiv., 9.25 mmol) and the
mixture
stirred at room temperature for 4 hours. The dichloromethane solution was
extracted with
2M HC1(x2) washed with saturated aqueous sodium bicarbonate solution, dried
over
sodium sulphate, filtered and the solvents removed in vacua to give 2.6g of a
pale reddish
solid which was used without further purification.
LCMS Method 3 RT = 1.15 minutes
INTERMEDIATE 169
\ B r
CIF* 411
142-(1R,3S)-3-bromo-7-chloro-2,3-dihydro-11-1-pyrrolo[1,2-a]benzimidazol-1-y1]-
3-
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 200 -
(difluoro mahoxy)phenyliethanone
Intermediate 181 (58.0 mg, 0.148 mmol) and triphenylphosphine (43.0 mg, 0.162
mmol)
were dissolved in DCM (2 mL), and at 0 C was added carbon tetrabromide (54.0
mg,
0.163 mmol). The reaction mixture was warmed up to ambient temperature and
stirred for
.. 3 hours, partitioned between water and DCM, and the organics, dried
(MgSO4), filtered
and concentrated in vacua. The crude material was purified by column
chromatography
(0%-5% Me0H in DCM) to afford the title compound (74 mg, 77%). LC/MS: Method 3
ESI MH+ 455/457, retention time 2.18 minutes.
INTERMEDIATE 170
2-(5-bromopyrimidin-2-y1)-N,N-dimethyl-propan-2-amine
To a solution of 1-(5-bromopyrimidin-2-y1)-1-methylethylamine (500 mg, 2.20
mmol) in
THE (15 mL) was added sodium hydride (97 mg, 2.43 mmol) at 0 C. The reaction
mixture was stirred at 0 C for 15 min before the addition of iodomethane (0.17
mL, 2.64
mmol) dropwise. The mixture was stirred at 0 C for 30 mins before warming to
room
temperature and stirred for 16 hours. 1odomethane (0.165 mL, 2.64 mmol) was
added and
stirred for additional hour before being quenched with saturated aqueous NH4C1
solution
and extracted with DCM (3 x 10 ml.). The organics were combined, dried with
Na2SO4,
filtered and concentrated in vacuo. The crude product was purified by column
chromatography (0-5% Me0H in DCM) to give the title compound (120 mg, 22%).
LC/MS: Method 3 retention time 1.23 minutes.
INTERMEDIATE 171
= \
0
0
0
(7R,14R)-1-(difluoromethoxy)-11-(4,4,5,5-tetramethy1-1,3,2-dioxaborolan-2-y1)-
6,7-
dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazoc1n-5(141/)-one
Example 11 (150 mg, 0.40 mmol) in 1,4-dioxane (1.5 mL) was added to
bis(pinacolato)diboron (150 mg, 0.59 mmol), potassium acetate, (117 mg, 1.19
mmol),
tricyclohexylphosphonium tetrafluoroborate (15 mg, 0.040 mmol) and
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 201 -
tris(dibenzylideneaectone)dipalladium(0) (19 mg, 0.02 mmol) were added. The
reaction
mixture was degassed for 10 minutes before heating to 140 C in the microwave
for 3
hours. The reaction mixture was partitioned between Et0Ac and water and
extracted with
further EtOAC (x3). The combined organic phases were filtered through a phase
separator
and the solvents removed in vacuo to give the title compound which was used
without
further purification. LC/MS: Method 3 RT 2.09 mins, [WEI] f= 468.
INTERMEDIATE 172
H
CF
;f0
:r
N-[(1R,3R)-142-bromo-6-(difluoromethoxy)pheny1]-7-ehloro-2,3-dihydro-11/-
pyrrolo11,2-albenzimidazol-3-y11-6-methoxy-pyridine-3-sulfonamide
To a solution of Intermediate 40 (500 mg, 1.17 mmol) and N,N-di-
isopropylethylamine
(0.24 mL, 1.4 mmol, 180 mg) in diehloromethane (12 mL) was added 6-
methoxypyridine-3-sulfonyl chloride (325 mg, 1.52 mmol). The reaction mixture
was
stirred at room temperature for 3 hours before the reaction mixture was
concentrated in
vacuo. The residue was purified by column chromatography (SiO2, 0-100% Et0Ac
in
hexane) to give the title compound (600 mg, 86%) as a dark beige solid. LCMS
(ES+)
Method 3: 600/602 (M+H)f, RT 2.4 minutes
INTERMEDIATE 173
= \ OH
CIFr ?
44ork ti 2
2-[(1R,35)-7-chloro-3-hydroxy-2.3-dihydro-1H-pyrrolo[1,2-a]benzimidazol-1-y1]-
3-
(difluoromethoxy)benzenesulfonamide
To a solution of Intermediate 136 (1.0 g, 1.67 mmol) in dimethyl sulfoxide
(2.8 mL),
sodium ethoxide solution (3M in Et0H, 1.13 mL, 3.3 mmol) was added. The
reaction
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 202 -
mixturc was stirred for 10 minutes before additional sodium ethoxide solution
(3M in
ethanol, 1.13 mL, 3.3 mmol) was added and the mixture stirred for 5 minutes.
Hydroxylarnine-0-sulfonic acid (1.0 g, 8.58 mmol,) and sodium acetate (550 mg,
6.70
mmol) in 2 mL of water were added to the reaction mixture. The reaction
mixture was
stirred overnight at room temperature. Et0Ac and water were added to the
reaction
mixture and the two phases were separated. The aqueous layer was extracted
with Et0Ac
and the combined organic layers were filtered through a phase separator and
the solvent
was evaporated. The crude material was purified by column chromatography over
silica
gel using hexane / ethyl acetate (0 to 100%) then DCM:Me0H (0 to 20%) as
eluent,
yielding 300 mg (42%) of the title compound as a pale yellow solid. LCMS
Method 3: RT
1.57 min, [M-Hr= 430/432.
INTERMEDIATE 174
, CI
CIF 411
r
frN,2
2-[(1R,38)-3,7-dichloro-2,3-dihydro-1H-pyrrolo[1.2-a]benzimidazol-1-y1]-3-
(difluoromethoxy)benzenesulfonamide
To a solution of Intermediate 173 (130 mg, 0.30 mmol) in THF (3 mL), DMAP (4
mg,
0.033 mmol) and N,N-di-isopropylethylamine (0.04 mL, 0.39 mmol) were added.
The
mixture was stirred at 0 C before methanesulfonyl chloride (47 uL, 0.61 mmol)
was
added. The reaction mixture was stirred for lhour 30 minutes. Sodium hydride
(18 mg,
0.45 mmol) was added at 0 C and stirred for 1 hour before the mixture was
heated to
reflux overnight. The reaction was quenched by the addition of water and
extracted with
Et0Ac (x3). The combined organic phases were dried over sodium sulphate,
filtered and
concentrated in vacuo. Purification by column chromatography over silica gel
using
hexane / ethyl acetate (0 to 100%) as eluent, the title compound was obtained
as a mixture
of diasterisomers which were separated by Achiral SFC purification, yielding
14 mg
(11% yield) of the title compound as a white solid. LCMS Method 3: RT 1.63
min, [M-
H]-= 448/450.
INTERMEDIATE 175
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 203 -
H
CI
%0
FF
(7R,14R)-11-ch I oro-1-(di fluoromethoxy)-6,7-dihyd ro-14H-7,14-
methanob enzimid azo [2,1-d] [1 ,2,5]b enzothiad iazocine 5,5-dioxide
To a solution of Intermediate 174 (14 mg, 0.031 mmol) in N,N-dimethylformamide
(0.6
mL) was added sodium hydride (60 mass %, 1.87 mg, 0.047 mmol) and the reaction
mixture was heated at 80 C for 1 hour. Water and Et0Ac were added and the two
phase
were separated. The aqueous layer was extracted with Et0Ac and the combined
organic
layers were washed with brine and filtered through a phase separator. The
solvent was
evaporated, yielding 10 mg (78% yield) of the title compound as a green solid.
LCMS
Method 3: RT 1.70 min, [M-H]= 412/414
INTERMEDIA l'E 176
Br
0
(1R,3R)-142-bromo-6-(difluoromethoxy)pheny1]-7-chloro -N-pyrimidin-2-y1-2,3-
dihydro-
1H-pyrro lo [ 1,2-a]benzimidazol-3 -amine
To a solution of Intermediate 40 (600 mg, 1.40 mmol) and 2-bromopyrimidine
(334 mg,
2.10 mmol) in N,N-dimethylformamide (2.8 mL) was added NN-di-
isopropylethylamine.
The reaction mixture was heated at 80 C for 4 hours and then at 110 C
overnight. Water
and Et0Ac were added and the reaction mixture was extracted with further ethyl
acetate.
The combined organic layer was filtered through a phase separator and the
solvent
evaporated. The crude material was purified by column chromatography over
silica gel
using hexane / ethyl acetate (0 to 100%) then DCM:Me0H (0 to 20%) as eluent,
yielding
205 mg (29% yield) of the title compound as a pale yellow solid. LCMS Method
3: RT
2.24 min, [M-H]= 506/508.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 204 -
INTERMEDIATE 177
or
H
CF
sf.* Br
Ethyl-N-[(1R,3R)-142-bromo-6-(difluoromethoxy)pheny1J-7-chloro-2,3-dihydro-1H-
rovrrolorl,2-albenzimidazol-3-yllcarbamate
To a solution of Intermediate 40 (400 mg, 0.93 mmol,) in dichloromethane (4.6
mL).
N,N-diisopropylethylamine (0.24 mL, 1.4 mmol) followed by ethyl chloroformate
(116
pL, 1.21 mmol) were added and the reaction mixture was stirred for 1 hour. The
solvent
was evaporated and the residue was purified by column chromatography (SiO2, 0-
100%
Et0Ac in hexane) to give the title compound (350 mg, 75% yield) as a pale
yellow solid.
LCMS (ES+) Method 3: 500/502 (M+H)+, RT 2.23 minutes.
INTERMEDIATE 178
141:(415¨(¨
N11-(5-bromo-2-pyridyl)cyclobuty11-2-methyl-propane-2-sulfinamide
2,5-dibromopyridinc (1.4 g, 5.8 mmol) was dissolved in toluene (15 mL) and the
reaction
mixture was cooled to -60 C after which time n-butyllithium (4 mL, 6.4 mmol)
was
added dropwise and the mixture was stirred at this temperature for 10 minutes.
N-
cyclobutylidene-2-methyl-propane-2-sulfinamide (1 g, 5.77 mmol) in 1 mL of
toluene
was added to the reaction mixture and stirred at -60 C for 15 minutes. A
saturated
solution of NH4C1 aq. was added and the reaction mixture was extracted with
Et0Ac and
the combined organic layers was washed with brine and filtered through a phase
separator
and the solvent was evaporated under reduced pressure. The residue was
purified by
column chromatography (SiO2, 0-100% Et0Ac in hexane) to give the title
compound
(1.25 g, 65% yield) as a colourless oil. IFINMR (300 MHz, Chloroform-d) 6 8.64
(dd, J =
2.4, 0.7 Hz, 1H), 7.83 (dd, J = 8.4, 2.4 Hz, 1H), 7.42 (dd, J = 8.5, 0.7 Hz,
1H), 4.31 (s,
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 205 -
1H), 2.72 -2.57 (m, 3H), 2.63 -2.44 (m, 1H), 2.10 (ddt, J = 18.7, 9.0, 7.0 Hz,
1H), 1.96
- 1.75 (m, 1H), 1.22 (s, 9H).
INTERMEDIATE 179
Br
N-[3-(5-bromo-2-pyridyl)oxetan-3-y1]-2-methyl-propane-2-sulfinamide
2,5-dibromopyridine (500 mg, 2.07 mmol) was dissolved in toluene (5 mI,) and
the
reaction mixture was cooled to -60 C before n-butyllithium (1.4 mL, 2.2 mmol)
was
added dropwise and the mixture stirred for 10 minutes. 2-methyl-N-(oxetan-3-
ylidine)propane-2-sulfinamide (420 mg, 2.2 mmol) in 0.5 mL of toluene was
added and
the reaction mixture stirred at -60 C for 15 minutes. A saturated solution of
aqueous
NH4C1 was added and the reaction mixture extracted with Et0Ac and the combined
organic layers washed with brine, filtered through a phase separator and the
solvent
evaporated under reduced pressure. The residue was purified by column
chromatography
(SiO2, 0-100% Et0Ac in hexane) to give the title compound (550 mg, 80% yield)
as a
colourless oil. LC/MS: RT 1.35 mins (pH 10), [M+11]+= 333/335. 1H NMR (400
MHz,
Chloroform-d) 6 8.63 (dd, J = 2.4, 0.7 Hz, 1H), 7.93 (dd, J = 8.5, 2.3 Hz,
1H), 7.71 (dd, J
= 8.5, 0.8 Hz, 1H), 5.41 (s, 1H), 5.33 (d, J = 7.0 Hz, 1H), 5.05 (d, J = 6.6
Hz, 1H), 4.94
(d, J = 6.6 Hz, 1H), 4.84 (dd, J = 7.0, 0.8 Hz, 1H), 1.28 (s, 9H).
INTERMEDIATE 180
5-(4-bromopheny1)-2,4-dimethy1-1H-imidazole
To a solution of 1-(4-bromopheny1)-2-nitropropene (750 mg, 3.09 mmol),
acetamidine
hydrochloride (313 mg, 3.31 mmol) and potassium carbonate (421 mg, 3.015 mmol)
in
ethanol (12 mL) was added indium (III) chloride (33 mg, 0.149 mmol) and the
reaction
mixture was stirred at 70 C overnight. Ethanol was evaporated and the crude
reaction
mixture was diluted with water (2 mL), and extracted with DCM. The organic
layer was
dried over anhydrous sodium sulphate, filtered and evaporated under vacuum to
give a
yellow solid. The residue was purified by column chromatography (hexane/ethyl
acetate)
to afford the title compound (270 mg, 35%) as a pale yellow solid. LC/MS
Method 3: RT
1.62 minutes, [M+H]= 251/253.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 206 -
INTERMEDIATE 181
OH
CIF al
*
1-[2-[(1R,3R)-7-chloro-3-hydroxy-2 ,3-dihydro-1H-pyrro lo [1,2-a]benz imidazol-
1-y1]-3-
(difluoromethoxy)phenyl]ethanone
Intermediate 60 (1.50 g, 3.49 mmol) was dissolved in toluene (20 rriL), and
tributy1(1-
ethoxyvinyl)tin (2.53 mL, 7.67 mmol) and bis(triphenylphosphine)palladium(11)
dichloride (250 mg, 0.36 mmol) were added. The reaction mixture was degassed
and
purged with N2 3 times before heating at 105 C for 18 hours. The reaction
mixture was
diluted with saturated aqueous KF solution and extracted with Et0Ac (x3). The
combined
organics were, dried (sodium sulphate) and concentrated in vacuo. The crude
compound
was purified by column chromatography eluting with 0-10% MeOH:DCM to give the
enol ether intermediate. The intermediate was dissolved in THF/1N HO (1:1, 40
mL) and
stirred at room temperature for 1 hour before neutralisation with saturated
sodium
bicarbonate solution and extraction with Et0Ac (2 x 50 mL). The organics were
combined and concentrated in vacuo to give the desired methyl ketone (1.22 g,
3.10
mmol, 89%). LC/MS: ES1 M1-1+ 393, retention time 1.31 minutes Method 3.
INTERMEDIATE 182
0
-4
0
0
F-
tert-butyl 2- 14-1(7R,14R)-1-(difluoromethoxy)-5-oxo-5,6,7,14-tetrahydro-7,14-
methanobenzimidazo[1,2-N12,5Thenzodiazocin-11-yliphenyl pyrrolidine-l-carboxyl
ate
.. To a solution of Example ii (500 mg, 1.33 mmol) and tert-butyl 2-(4-
(4,4,5,5-
tetramethy1-1,3,2-dioxaborolan-2-yl)phenyl)pyrrolidine-1-carboxylate (768 mg,
2.00
mmol) in 1,4-dioxane (10 mL) was added K3PO4 (566 mg, 2.67 mmol),
tricyclohexylphosphonium tetrafluoroborate (52 mg, 0.14 mmol) and
tris(dibenzylideneacetone)dipalladium(0) (108 mg, 0.11 mmol) with several
drops of
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 207 -
water. The reaction mixture was degassed and flushed with nitrogen and then
heated at
120 degrees in a microwave for 6 hours. The cooled reaction was diluted with
H20
(50rnL) and extracted with Et0Ac x 3. The combined organics were dried over
Na2SO4,
filtered and concentrated in vacuo. Purification by column chromatography,
firstly with
Et0Ac in DCM (0 to 100%) and then with 0-10% Me0H/DCM gave the title compound
(575 mg, 66%). LC/MS Method 3: RT 2.40 minutes, m/z 487.2 (-B0C).
INTERMEDIATE 182(a)
N
NH
H =
>1
tert-Butyl (2-{4-[(7R,I4R)-1-(difluoromethoxy)-5-oxo-5,6,7,14-tetrahydro-7,14-
methanobenzimidazo[1,2-61[2,5Thenzodiazocin-11-yl]phenylJpropan-2-ypcarbamate
The title compound can be prepared from Intermediate 171 (0.35 g, 0.93 mmol,
leq) and
(tert-butyl 2-(4-bromophenyl)propan-2-ylcarbamate (leq) in accordance with the
Method
.. described for Example 137. Purification by flash chromatography on silica
gel (0 to 100%
Et0Ac in DCM followed by 0 to 10% Me0H in DCM) afforded the title compound as
a
brown solid. LC/MS Method 3: RT 2.32 minutes, m/z 575.2
INTERMEDIATE 183
H
Br 41 2
(1R,3R)-142-chloro-6-(difluoromethoxy)phenyl] -7-bromo -6-fluoro-23-dihydro -
1H-
pyrrolo[1,2-a]benzimidazol-3-amine
The title compound may be prepared from Intermediate 10 in accordance with the
Method
described for Intermediate 40.
INTERMEDIATE 184
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 208 -
F
Nib" H I
Br
tert-butyl {(1R,3R)-1-[2-chloro-6-(di fluorometho xy)phenyl] -7-bromo-6-fluoro-
2,3-
dihydro-1H-pyrro lo [1,2-a]b enzimid azol-3 -y1} carbamate
The title compound may be prepared from Intermediate 183 in accordance with
the
Method described for Intermediate 42.
INTERMEDIATE 185
0
F
B a
Fr ,
tert-butyl N-R1R,3R)-7-bromo-1-[2-chloro-6-(difluoromethoxy)pheny1]-6-fluoro-
2,3-
dihydro-1H-pyrrolo[1,2-a]benzimidazol-3-yll-N-(trideuteriomethypearbamate
Intermediate 184 (300 mg, 0.549 mmol) was dissolved in tetrahydrofuran (10
mL).
Potassium bis(trimethylsilyl)amide (0.6 mL, 0.6 mmol) was added dropwise at -
78 degree
and stirred for 30 minutes before the addition of iodomethane-d3 (0.06 mL, 1
mmol). The
reaction mixture was stirred at -78 C for 10 minutes before being left in an
ice-water bath
for 2 hours followed by 1 hour at room temperature. The reaction mixture was
quenched
with saturated ammonium chloride solution and extracted with Et0Ac, the
organics dried
over Na2SO4 and concentrated in vacuo. Purification by column chromatography,
eluting
with 0%-10% Me0H in DCM, to afford the title compound as an off white solid
(320 mg,
99%). LC/MS Method 3: RT 2.75 minutes, m/z 563.0/565.0
25
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 209 -
INTERMEDIATE 186
Hrl
tert-butyl-N-[(1R,3R)-142-chloro-6-(difluoromethoxy)pheny11-6-fluoro-742-(3-
oxoniperazin-l-y1)pyrimidin-5-y1]-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazol-3-
y1]-N-
trideuteriomethyl) carbamate
Intermediate 185 (310 mg, 0.49 mmol), [2-(3-oxopiperazin-1-yl)pyrimidin-5-
yl]boronic
acid (0.55mmo1, 1.1eq), potassium carbonate (115 mg, 0.83 mmol), palladium(II)
acetate (7 mg, 0.03 mmol) and 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene
(19 mg,
0.033 mmol) were dissolved in DMSO (5 mL, 70 mmol) and a drop of water added.
The
mixture was degassed thoroughly and nitrogen flushed. The mixture was heated
for 1
hour in a microwave at 110 C. The mixture was separated between Et0Ac and
brine (25
mL of each) and the aqueous extracted with Et0Ac (25 mL) and the combined
organics
washed with 3 x 20 mL of brine, dried (phase separator) - and evaporated in
vacuo.
Purification by column chromatography on silica using a gradient Et0Ac in DCM
(0-
100%) and then 1 to 15% Me0H in Et0Ac to afford the title compound as an off
white
solid (210 mg, 64%). LC/MS Method 3: RT 2.28 minutes, m/z 661.2.
INTERMEDIATE 187
o, =
F
Pip 41 14
F
rip, 70H
2-[(1R,3R)-3-[tert-butoxycarbonyKtrideuteriomethypamino]-6-fluoro-742-(3-
oxopiperazin-1-yl)pyrimidin-5-y1]-2,3-dihydro-1H-pyrrolo[1.2-a]benzimidazol-1-
y1]-3-
(difluoromethoxy) benzoic acid
Into a 10mL glass vial was placed Intermediate 186 (210 mg, 0.32 mmol),
potassium
carbonate (67 mg, 0.48 mmol), PdC12(dcypp) (15 mg, 0.025 mmol), dimethyl
sulfoxidc (5
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 210 -
mL), water (0.1 mL, 6 mmol) and the vial equipped with a stirring bar was
placed into a
high pressure reactor. The headspace of the reactor was vacuum purge cycled
with CO at
14psi (x 3) and then left with a headspace pressure of 5Bar. The vessel was
heated to
105 C, (heating block temp) for a period of 24 hrs. The reaction mixture was
separated
between ethyl acetate and water (20mL of each) and the organic layer was
extracted with
a further 2 x 20 mL of 10% sodium carbonate solution. The combined aqueous
layers
were then treated with citric acid until no longer basic. The solution was
then extracted
with ethyl acetate (3 x 20 mL) and these organics washed with 4 x 20 mL of
water. The
organics were dried (sodium sulfate), filtered and evaporated in vacuo to
afford the title
compound as a white solid ¨90% pure. (120 mg, 56%). LC/MS Method 3: RT 1.51
minutes, m/z 671.2
INTERMEDIATE 188
CIF J 111111 o,
Ethyl 2424(1R,35)-7-ehloro-3-methylsulfonyloxy-2,3-dihydro-1H-pyrrolo[1,2-
a]benzimidazol-1-y1]-3-(difluoromethoxy)phenyl]sulfanylacetate
To a solution of Intermediate 161 (1.35 g, 2.88 mmol) in DCM (30 mL) at 0 C
was
added 4-dimethylaminopyridine (40 mg, 0.33 mmol), N,N-diisopropylethylamine
(1.01
mL, 5.77 mmol) and methane sulfonyl chloride (335 L, 4.32 mmol) and stirred
for 45
minutes. After this time the reaction mixture was partitioned between DCM (40
mL) and
water (50 mL), layers separated and the aqueous phase extracted with DCM (3 x
50 mL).
Combined organics were washed with saturated aqueous sodium bicarbonate
solution
(100 mL), dried (phase separator) and concentrated in vacuo to give the title
compound as
orange brown solid (1.57 g, quantitative yield). The crude product was
progressed to the
next synthetic step without further purification.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 211 -
INTERMEDIATE 189
tert-Butyl N11-methy1-1-[5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-
yl)pyrimidin-2-
yl]ethyl]carbamate
Bis(pinacolato)diboron (4.55 g, 17.6 mmol), Intermediate 114 (3.70 g, 11.7
mmol),
potassium acetate (4.64 g, 46.8 mmol), 1,1'-bis(diphenylphosphino)ferrocene-
palladium(II)diehloride dichlorornethane complex (1000 mg, 1.22 mmol) and 1,4-
dioxane
(35 mL) were placed in a RBF and then degassed. The mixture was then heated at
105 C
for 1 hour, LCMS showed the completion of the reaction. The reaction mixture
was
diluted with H20 and extracted with Et0Ae (x3), and the organics combined,
dried
(MgSO4), filtered and evaporated in vacuo. The crude material was used in the
subsequent Suzuki coupling (3.6g, 80%). LC/MS Method 3: RT 1.04 minutes, m/z
378.
INTER1VIEDIA l'E 190
F
=
C 411 D
F 0
T
(7R,14R)-11-chloro-1-(difluoromethoxy)-10-fluoro-6-(trideutero)methyl-6,7-
dihydro-
7,14-methanobenzimidazo[1,2-b].[2,5]benzodiazocin-5(14H)-one
Example 10 (650 mg, 1.65 mmol) was dissolved in tctrahydrofuran (15 mL, 184
mmol)
and potassium his(trimethylsilyl)amide (1.8 mL, 1.8 mmol, 1 moFL in TNF) was
added
dropwise at -78 'V and stirred for 30 minutes before the addition of
iodomethane-d3 (0.16
mL, 2.5 mmol). The reaction mixture was stirred at -78 C for 10 minutes
before being
left in an ice-water bath for 1 hour. The reaction mixture was quenched with
aqueous
saturated NH4C1 solution and extracted with Et0Ac (x3), the combined organics
were
dried over Na2SO4, filtered and concentrated in vacuo. The crude material was
purified by
column chromatography eluting with 0%-10% Me0H/DCM to afford the title
compound
(570 mg, 84%). 'HNMR (300 MHz, DMSO-d6) 6 8.27 (dd, J = 5.3, 4.1 Hz, 1H), 7.93
-
7.26 (m, 5H), 6.24 (d, J = 7.1 Hz, 1H), 5.23 (d, J = 7.1 Hz, 1H), 3.49 (dt, J
= 14.3, 7.3 Hz,
1H), 2.81 (d, J = 13.8 Hz, 1H). LC/MS Method 3: RT 2.00 minutes, m/z 411.0
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 212 -
INTERMEDIATE 191
F 0
011,111
>r
tert-Butyl (2- {5-[(7R,14R)-1-(difluoromethoxy)-10-fluoro-6-(trideutero)methy1-
5-oxo-
5,6,7,14-tetrahydro-7,14-methanobenzimidazo[1,2-b112,5Th enzodiazocin-11-
yl]pyrimidin-2-yl}propan-2-yl)carbamate
Intermediate 190 (570 mg, 1.38 mmol), Intermediate 189 (1.8 g, 4.0 mmol),
potassium
phosphate tribasic (1.05 g, 4.85 mmol) and tricyclohexylphosphonium
tetrafluoroborate
(65 mg, 0.17 mmol) were placed in a microwave vial and suspended in 1,4-
dioxane (5
mL). The mixture was degassed and purged with N2 3 times, followed by the
addition of
tris(dibenzylideneacetone) dipalladium(0) (130 mg, 0.13 mmol) and three drops
of water.
The reaction mixture was heated at 140 C for 2 hours in a microwave. The
reaction
cooled to room temperature and quenched with H20 (20 mL) and extracted with
Et0Ac
(3 x 25 mL). The combined organics were dried over (Na2SO4), filtered and the
solvents
removed in vacuo. The residue was purified by column chromatography on silica
eluting
with 0 to 20% Me0H in Et0Ac to afford the title compound (420 mg, 50%). LC/MS
Method 3: RT 2.19 minutes, tn/z 612.2
INTERMEDIATE 192
XX
ciP:0
2- {5-R7R,14R)-1-(difluoromethoxy)-10-fluoro-5-oxo-5,6,7,14-tetrahydro-7,14-
methanobenzimidazo[1,2-b][2,51benzodiazocin-11-yl]pyrimidin-2-yl}propan-2-yl,
di-O-
benzyl phosphate.
Example / (26.0 g, 52.5 mmol) was suspended in dichloromethane (450 nit),
placed
under nitrogen and 5-methy1-1H-tetrazole (8.38 g, 99.7 mmol) was added and the
mixture
cooled to 5 C in an ice bath, evacuated and refilled with nitrogen twice then
stirred for 5
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 213 -
minutes before adding dibenzyl N,N-di-isopropylphosphoramidite (28.2 mL, 83.9
mmol).
The reaction was stirred for 5 minutes before warming to room temperature and
stirred
for one further hour. The mixture was cooled to 0 C in an ice bath then
hydrogen
peroxide solution (5.96 mL, 105 mmol, 50.0% w/w in water) was added. The
mixture was
stirred for 1 hour and monitored by LCMS until oxidation was complete. The
mixture was
washed with 0.25M sodium metabisulphite solution (250 mL), then saturated
aqueous
sodium bicarbonate solution (150 mL) and saturated brine (200 mL). The organic
phase
was dried (sodium sulfate), filtered and concentrated in vacuo to a pale
yellow gum. The
crude product was purified by chromatography on silica (Et0Ac to 15% Me0H in
Et0Ac) to give a crude residue which was azeotroped with toluene (x3) to give
the title
compound as a white solid, (31.0g, 78%). Ili NMR (d6-DMS0 ,300 MHz) 6: 1.87
(s, 6
H), 2.75 (d, 1H, J-13.4 Hz), 3.50 (m, 1H), 4.92 (t, 1H, J-6.8 Hz), 5.02-5.05
(m, 4H), 6.36
(d, 1H, J-7.1 Hz), 7.25 (m, 10 H), 7.55 (m, 3H), 7.69 (d, 1H, J=11.5 Hz), 8.24
(dd, 1H, J=
5.3, 4.2 Hz), 8.99 (s, 2H), 9.16 (d, 1H, J= 6.8 Hz). LC/MS Method 3: RT 2.49
minutes,
in/z756.
INTERMEDIATE 193
tilk I
=
o
2- {5 -R7R,14R)-1-(difluoromethoxy)-10-fluoro-6-methy1-5-oxo-5 ,6,7,144
etrahydro -7,14-
methanobenzimidazo[1,2-b][2,5Thenzodiazocin-11-yllpyrimidin-2-yll propan-2-yl,
di-0-
benzyl phosphate.
To a solution of Intermediate 192 (4.00 g, 5.29 mmol) in THF (70 mL) was added
a
solution of KHMDS in THF (1M, 5.60 mL, 5.60 mmol) dropwisc at -78 C and the
mixture was stirred for 45 minutes before the addition of iodomethane (0.37
mL, 5.90
mmol). The reaction mixture was warmed to 0 C and stirred for 2.5 hours
before the
reaction completed. The reaction mixture was quenched with saturated aqueous
NH4C1
solution and extracted with Et0Ac (x3), the combined organics were dried with
Na2SO4,
filtered and concentrated in vacuo. Purification by column chromatography
eluting with
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 214 -
0-10% Me0H in Et0Ac gave the title compound (3.20 g, 4.2 mmol, 79%). LC/MS
Method 3: ES + (m+H) 770, retention time 2.67 minutes.
INTERMEDIATE 194
(7R ,14R)-11 -chloro-1-(difluoromethoxy)-6-ethy1-7-methy1-6,7-dihydro -7,14-
methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one
Intermediate 148 (100 mg, 0.25 mmol) was dissolved in dry THF (2.5 mL). at the
reaction was cooled to 0 C and sodium hydride (60% in mineral oil) (12 mg,
0.30 mmol)
was added. The reaction mixture was stirred at room temperature for 35
minutes.
lodomethane (0,27 mL, 3.34 mmol) was added and the reaction mixture was
stirred at
50 C overnight. The reaction mixture was quenched by addition of water (2 mL).
The
aqueous layer was extracted by Et0Ac (3 x 150 mL). The combined organic layers
were
dried over MgSO4, filtered and concentrated in vacuo. The residue was purified
over
silica gel (heptane / AcOEt 1/1), yielding to 35 mg (32%) of the title
compound as a white
solid. LCMS (Method 3 ES+): RT 2.60 min, [M+1-1]+ 418.
INTERMEDIATE 195
2-chloro-6-(trifluoromethoxy)benzaldehyde
N,N-Di-isopropylamine (38.4 ml, 271mmo1) was added dropwisc to a solution of n-
butyllithium (1.6 M, 169 ml, 271 mmol) in THF (180 ml) at 0 C and the reaction
mixture
was stirred and allowed to warm to room temperature over 30 mins. The LDA
solution
was then added drop wise over 10minutes to a solution of 1-chloro-3-
(trifluoromethoxy)benzene (50 g, 246 mmol) in THF (500 ml) at -70 C and the
resulting
mixture was stirred at -70 C for 30 minutes. Finally N,N-dimethylformamide (23
ml, 296
mmol) was added dropwise and the resultant mixture stirred at -70 C for 30
minutes.
The reaction was quenched at -70 C by addition of NH4C1(saturated aqueous
solution) to
pH 7-8 and the resulting mixture was extracted with Et0Ac (3 x 75 ml). The
combined
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 215 -
organic phases were washed with water (100 ml), brine (100 ml), dried over
magnesium
sulfate, filtered and concentrated under reduced pressure to provide the title
compound
(55 g, 99% yield).
LCMS (Method 16, ES+) RT 1.32 min., 224 [M+Hr.
INTERMEDIATE 196
(S,E)-N-(2-chloro-6-(trifluoromethoxy)benzylidene)-2-methylpropane-2-
sulfinamide
(S)-2-Methylpropane-2-sullinarnide (49.9g, 411 mmol), potassium hydrogen
phosphate
(196 g, 1.12 mol) and potassium phosphate (238.2 g, 1.12 mot) were added to a
solution
of Intermediate 195 (84 g, 374 mmol) in THF (2000 ml) at room temperature. The
resulting reaction mixture was stirred at room temperature over 20 hours,
filtered and the
solid was washed with Et0Ac (3 x 200 m1). The filtrate was concentrated under
reduced
pressure to isolate the title compound (123 g).
LCMS (Method 16, ES+) RT 1.45 min., 328 [M-I-H]t
INTERMEDIATE 197
Ethyl (R)-3-4(S)-tert-butylsulfinyl)amino)-3-(2-chloro-6-
(trifluoromethoxy)pheny1)-
propanoate
A solution of ethyl bromoacetate (106.1 ml, 938.2 mmol) in THF (100 ml) was
added
dropwise over 2 hours to a solution of zinc (245.5 g, 3.75 mot) and cuprous
chloride (44.6
g, 450 mmol) in THF (800 ml) at 20-25 C (NB: prior to this addition, the
solution of Zn
and CuCl was heated at 70 C over 30mins and cooled down to room temperature).
Once
the addition was complete the reaction mixture was warmed to 50 C over 1 hour.
The
reaction mixture was cooled to 0 C and a solution of Intermediate 196 (123 g,
375.29
mmol) in THF (500 ml) was added dropwise over 60mins. The resulting reaction
mixture
was warmed to room temperature over 20 hours and filtered over charcoal. The
cake was
washed with Et0Ac (3 x 250 ml) and the combined filtrates were washed with
water (500
ml), with 10% citric acid solution (1000 ml) and brine (500 m1). The organic
phase was
dried over magnesium sulfate, filtered and concentrated under reduced pressure
to give
the title compound (155 g, 99% yield).
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 216 -
LCMS (Method 16, ES+) RT 1.36 min., 416 [M+H]t
INTERMEDIATE 198
Ethyl (R)-3-amino-3-(2-chloro-6-(trifluoromethoxy)phenyl)propanoate
hydrochloride
4M Hydrochloric acid solution in 1,4-dioxane (326.1 ml, 1.3 mol) was added
dropwise
over 30 mins to a solution of Intermediate 197 (155 g, 372.7 mmol) in a
mixture of
ethanol (107 ml) and diethyl ether (215 ml) at room temperature. The reaction
mixture
was then stirred for 2 hours and the resulting suspension was filtered. The
cake was
washed with Et20 (3 x 250 ml), with pentane (250 ml) and dried under suction
to yield
49g of the title compound. The filtrate was concentrated under reduced
pressure to
provide a viscous residue. Et20 was added to the residue, and the mixture was
stirred over
hours. The resulting suspension was filtered and the isolated cake was washed
with
Et20 (3 x 250 ml), with pentane (250 ml), and dried to yield 70g of the title
compound as
the hydrochloride salt. LCMS Method 16, ES+) RT 0.59 min., 311 [M+F1]+.
INTERMEDIATE 199
15 Ethyl (R)-3-((5-bromo-4-fluoro-2-nitrophenyl)amino)-3-(2-chloro-6-
(trifluoromethoxy)-
phenyl)propanoate
1-Bromo-2,5-difluoro-4-nitrobenzene (30.2g, 126.7 mmol) was added to a
solution of
ethyl Intermediate 198 (49g, 140.8 mmol) in N,N-dimethylformamide (200 ml) and
N,N-
diisopropylamine (46.76 ml, 281.5 mmol) was added. The reaction mixture was
heated at
20 85 C over two hours and, once cooled to room temperature, was then
concentrated under
reduced pressure. The resulting residue was dissolved in Et0Ac (500 ml),
washed with
water (500 ml), 10% citric acid solution (2 x 500 ml) and brine (500 ml). The
organic
phase was dried over magnesium sulfate, filtered and concentrated under
reduced
pressure. The resulting crude was purified by SiO2 flash chromatography with
Et0Ac/n-
.. Heptane (2/98) as eluent to provide the title compound (45 g, 60% yield).
LCMS (Method 16, ES+) RT 1.60 min., 531 [M+H] .
INTERMEDIATE 200
(R)-34(5-bromo-4-fluoro-2-nitrophenyl)amino)-3-(2-chloro-6-
(trifluoromethoxy)phenyl)-
propanal
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 217 -
Diisobutylaluminium hydride was added dropwisc over 2 hours to a solution of
Intermediate 199 (10 g, 18.9 mmol) in THF (100 ml) at -70 C under argon. The
reaction
mixture was stirred over 2 hours at -70 C. The reaction was quenched at -70 C
by
addition of NH4C1(saturated aqueous solution) to pH = 6 and the resulting
suspension
was filtered over a charcoal pad washing with Et0Ac (75 mL). The filtrate was
extracted
with Et0Ac (3 x75 rriL. The combined organic phases were washed with water
(100 ml) ,
brine (100 ml), dried over MgSO4, filtered and concentrated under reduced
pressure. The
resulting residue was purified by SiO2 flash chromatography with Et0Ac/n-
Heptane (1/4)
as eluent to give the title compound (5.8g, 63% yield).
LCMS (Method 17, ES+) RT 4.83 min., 487 [M+H].
INTERMEDIATE 201
(R)-111-4R,Z)-3-((5-bromo-4-fluoro-2-nitrophenyDarnino)-3-(2-chloro-6-
(trifluoromethoxy)-phenyl)propylidene)-2-methylpropane-2-sulfinamide
(R)-2-Methylpropane-2-sulfinamide (1.47 g, 11.9 mmol) was added to a solution
of
Intermediate 200 (5.8 g, 11.35 mmol) and titanium(IV) isopropoxide (3.36 ml,
11.35
mmol) in dichloromethane (100 ml) at room temperature. The reaction was heated
at
reflux for 20 hours.
After cooling to room temperature the reaction was quenched by addition of
brine (50 ml)
and the resulting mixture was stirred for 30 mins. The suspension was filtered
over a
charcoal pad, washed with dichloromethane until the filtrate is colourless.
The filtrate was
washed with brine (20 mL), dried over magnesium sulfate, filtered and
concentrated
under reduced pressure to afford the title compound (6.6 g, 99% yield).
LCMS (Method 18, ES+) RT 5.31 min., 58 M+Hr.
INTERMEDIATE 202
(R)-N-01R,3R)-345-bromo-4-fluoro-2-nitrophenypamino)-3-(2-chloro-6-
(tri fluorometho x y)phen y1)-1 -cyanopropy1)-2-methylpropane-2-sulfinamide
Scandium(III) trifluoromethanesulfonate (1.30 g, 2.62 mmol) was added to a
solution of
Intermediate 201 (7.7 g, 13.08 mmol) in dichloromethane (75m1) at room
temperature
followed by trimethylsilyl cyanide (3.44 ml, 26.16 mmol). The reaction mixture
was then
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 218 -
stirred over 92 hours at room temperature. The reaction was quenched by
addition of
water (100 ml) and the resulting mixture was extracted with dichloromethane (2
x 50 ml).
Combined organic phases were washed with brine (50 ml), dried over magnesium
sulfate,
filtered and concentrated under reduced pressure to provide the title compound
(7.5 g,
93% yield).
LCMS (Method 18, ES+) RT 4.89 min., 615 [M+Hr.
INTERMEDIATE 203
H2
B 40
=\....F
7-Bromo-1-(2-chloro-6-(trifluoromethoxy)pheny1)-6-fluoro-2,3-dihydro-1H-
benzo[d]pyrrolo[1.2-a]imidazole-3-amine
Stannous chloride (11.78 g, 60.9 mmol) was added to a solution of Intermediate
202 (7.5
g, 12.18 mmol) in ethanol (75 m1). The reaction mixture was heated at reflux
over 2 hours
and water (40 ml) was then added. The resulting solution was heated to reflux
for 16
hours.
Once cooled to room temperature water (100 ml) and 2M Sodium hydroxide aqueous
solution was added until pH = 9.0 while keeping the solution temperature below
30 C.
The resulting suspension was filtered over a charcoal pad and washed through
with
dichloromethane (5x 50 m1). The filtrate was extracted with further
dichloromethane (5x
50 m1). The combined organic phases were washed with brine (20 ml), dried over
magnesium sulfate, filtered and concentrated under reduced pressure to provide
the title
compound as cis/trans mixture of isomers (4/1, 3.00 g, 53% yield).
LCMS (Method 18, ES+) RT 2.83 min., 464 [M+H]t
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 219 -
INTERMEDIATE 204
0
H
e
J.
411110 )CFF
tert-butyl ((1R,3R)-7-bromo-1-(2-chloro-6-(trifluoromethoxy)phenyl)-6-fluoro-
2,3-
dihydro-1H-benzo[d]pyrrolo[1,2-alimidazole-3-y1)carbamate
Di-tert-butyl carbonate (1.99 ml, 9.04 mmol) was added to a solution of
Intermediate 203
(3.00 g, 6.46 mmol) in dichloromethane (20 ml) at room temperature. The
reaction
mixture was then stirred for 20 hours and concentrated under reduced pressure.
The
resulting residue was purified by SiO2 flash chromatography with Et0Ac/n-
Heptane (1/4)
as eluent to give the title compound (1.8g, 49% yield).
LCMS (Method 19, ES+) RT 2.51 min., 464 [M-B0C+H]t
INTERMEDIATE 205
0 ,
nH
H I =
tert-Butyl ((1R,3R)-1-(2-chloro-6-(trifluoromethoxy)pheny1)-6-fluoro-7-(2-(2-
hydroxypropan-2-yl)pyrimidin-5-y1)-2,3-dihydro-1H-benzokilpyrrolo[1,2-
dimidazole-3-
yl)carbamate
2-(5-(4,4,5,5-Tetramethy1-1,3,2-dioxaborolan-2-yppyrimidin-2-yl)propan-2-o1
(0.26 g,
0.97 mmol) was added to a solution of Intermediate 204 (0.50 g, 0.89 mmol) in
1,4-
dioxane (5 ml) under argon. Potassium carbonate (0.370 g, 2.66 mmol) was added
to the
solution and the reaction mixture was purged with argon prior.
bis(diphenylphosphino)ferrocene palladium(I1)dichloride dichloromethane
complex
(0.023 g, 0.026 mmol) was added and the reaction mixture was heated at 90 C
for 2
hours and then cooled to room temperature. The reaction was quenched by
addition of
iced water and the resulting mixture was extracted with Et0Ac (2x 30 m1). The
combined
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 220 -
organic phases were washed with brine ( 2x 20 mL), dried over magnesium
sulfate,
filtered and concentrated under reduced pressure. The resulting residue was
purified by
SiO2 flash chromatography with DCM/Me0H (100/0 to 98/2) followed by SiO2 flash
chromatography with Et0Ac/n-Heptane (3/7 to 1/1) as eluent to give the title
compound
(0.350 g, 64% yield).
LCMS (Method 20, ES+) RT 1.38 min., 622 [M+Hr.
INTERMEDIATE 206
H2
CI
Ft 'F
2-(5-((lR,3R)-3 -amino -1-(2-chloro -6-(trifluoromethoxy)pheny1)-6- fluoro-2,3
-dihydro -
1H-benzo[Apyrrolo[1,2-a]imidazolc-7-yl)pyrimidin-2-yl)propan-2-ol
2N Hydrochloric acid solution in diethyl ether (2.8 ml, 5.63mmo1) was added to
a
solution of Intermediate 205 (0.35 g, 0.56 mmol) in dichloromethane (10 m1).
The
reaction mixture was stirred at room temperature over 72 hours.
Water was added and the aqueous phase was treated with 2N sodium hydroxide
aqueous
solution until pH = 12. The resulting mixture was extracted with
dichloromethane (2x 50
ml) and then with Et0Ac (2x 50 m1). The combined organic phases were dried
over
magnesium sulfate, filtered and concentrated under reduced pressure to afford
the title
compound (300mg).
LCMS (Method 20, ES+) RT 0.67 min., 522 [M+H]t
INTERMEDIATE 207
F
H
MIP
Cl
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 221 -
tert-Butyl ((1R,3R)-1-(2-chloro-6-(difluoromethoxy)pheny1)-6-fluoro-7-(3-(2-
hydroxypropan-2-ypazetidin-1-y1)-2,3-dihydro- 1H-benzo[dipyrrolo[1,2-
a]imidazo1-3-
yl)carbamate
2-(Azetidin-3-yl)propan-2-ol hydrochloride (0.208 g, 1.37 mmol) and cesium
carbonate
(0.890 g, 2.74 mmol) were added to a solution of Intermediate 184 (0.500 g,
0.91 mmol)
in a mixture of toluene/DMF (99/1, 15m1). The resulting mixture was purged
with argon.
(R/S)-(+/-)-2,2'-Bis(diphenylphosphino)-1,1'-binaphthyl (0.018 g, 0.027 mmol)
and
tris(benzylideneacetone)dipalladium(0) (0.009 g, 0.009 mmol) were added. The
reaction
mixture was then heated in a microwave at 100 C over 2 hours. As traces of the
expected
azetidinyl derivative were detected by LCMS, a second addition of all reagents
[2-
(azetidin-3-yl)propan-2-ol hydrochloride (0.208 g, 1.37 mmol), cesium
carbonate (0.890
g, 2.74 mmol), (R/S)-(+/-)-2,2'-Bis(diphenylphosphino)-1,1'-binaphthyl (0.018
g, 0.027
mmol) and tris(benzylideneacetone)-dipalladium(0) (0.009 g, 0.009 mmol)] was
carried
out. The reaction mixture was then heated in microwave at 100 C for a further
2 hours.
Water (50 ml) was added and the resulting mixture was extracted with Et0Ac (3x
50 m1).
The combined organic phases were dried over sodium sulfate, filtered and
concentrated
under reduced pressure. The resulting residue was purified by SiO2 flash
chromatography
with DCM/Me0H (100/0 to 95/5) as eluent to give the title compound (0.249 g,
47%
yield).
LCMS (Method 20, ES+) RT 1.09 min., 581 [MH-H].
INTERMEDIATE 208
H2
2-(1-((lR,3R)-3-amino-1-(2-chloro-6-(difluoromethoxy)pheny1)-6-fluoro-2,3-
dihydro-
1H-benzo rolo 1 2-a imidazol-7- pazetidin-3-Dpropan-2-ol
Trifluoroacetic acid (0.33 nil, 4.29 mmol) was added to a solution of
Intermediate 207
(0.249 g, 0.428 mmol) in dichloromethane (5 m1). The reaction mixture was
stirred at
room temperature over 24 hours.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 222 -
Water (30 ml) was added and the aqueous phase was treated with 2N sodium
hydroxide
aqueous solution until pH = 12. The resulting mixture was extracted with
dichloromethane (2x 30 ml) and Et0Ac (lx 30 m1). The combined organic phases
were
dried over sodium sulfate, filtered and concentrated under reduced pressure to
provide the
title compound (0.157 g, 76% yield).
LCMS (Method 20, ES+) RT 0.64 min., 481 [M+Hr.
INTERMEDIATE 209
0 0,1(0
H
H2 )c..FF
tert-butyl ((lR,3R)-7-(4-(2-aminopropan-2-yl)pheny1)-1-(2-chloro-6-
(trifluoromethoxy)-
phenyl)-6-fluoro-2,3-dihydro-1H-benzo[d]pyrrolo[1,2-alimidazole-3-yl)carbamate
A solution of 2-(4-(4,4,5,5-tetramethy1-1,3,2-dioxaborolan-2-yl)phenyl)propan-
2-amine
in 1,4-dioxane (10 ml) was added to a solution of Intermediate 204 (0.500 g,
0.885 mmol)
in 1,4-dioxane (40 ml) under argon. Sodium carbonate (0.188 g, 1.77 mmol) and
water (1
ml) were added to the solution and purged with argon prior to addition of
tris(dibenzylideneacetone)dipalladium(0) (0.021 g, 0.022 mmol) and tri-tert-
butylphosphonium tretrafluoroborate (0.026 g, 0.088 mmol) were added and the
reaction
mixture was heated at 90 C for 3 hours and then cooled to room temperature
over 15
hours. Water (30 ml added and the resulting mixture extracted with Et0Ac (2x
30 ml).
The combined organic phases were washed with brine ( 2x 20 ml), dried over
sodium
sulfate, filtered and concentrated under reduced pressure. The resulting
residue was
purified by SiO2 flash chromatography with DCM/Me0H (100/0 to 9/1) as cluent
to give
the title compound (0.250 g, 46% yield).
LCMS (Method 20, ES+) RT 0.91 min., 619 [M-l-H]t
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 223 -
INTERMEDIATE 210
H2
H2 FF
(1R,3R)-7-(4-(2-aminopropan-2-yl)pheny1)-1-(2-chloro-6-
(trifluoromethoxy)pheny1)-6-
fluoro-2,3-dihydro-1H-benzo[dipyrrolo[1,2-dmidazole-3-amine
2N Hydrochloric acid solution in diethyl ether (2.0 ml, 4.04 mmol) was added
to a
solution of Intermediate 209 (0.250 g, 0.404 mmol) in dichloromethane (10 m1).
The
reaction mixture was stirred at room temperature for 72 hours. Water (30 ml)
was added
and the aqueous phase was treated with 2N sodium hydroxide aqueous solution
until to
pH = 12. The resulting mixture was extracted with dichloromethane (2x 50 ml)
and then
with Et0Ac (2x 50 m1). The combined organic phases were dried over sodium
sulfate,
filtered and concentrated under reduced pressure to afford the title compound
(0.195 g,
93% yield).
LCMS (Method 20, ES+) RT 0.45 min., 519 [M+H]t
INTERMEDIATE 211
0 0.4..e.
H
Cl
H2
tert-butyl ((1R,3R)-7-(4-(2-aminopropan-2-yl)pheny1)-1-(2-chloro-6-
(difluoromethoxy)phenyl)-6-fluoro-2,3-dihydro-1H-benzo [d]pyrro lo[l,2-
a]imidazo le-3-
Acarbamate
The title compound was prepared from Intermediate 184 (0.500 g, 0.91 mmol) in
accordance with the synthetic procedure described for Intermediate 209 after
purification
by SiO2 flash chromatography with DCM/Me0H (100/0 to 95/5) as eluent to give
(0.324
g, 59% yield).
LCMS (Method 20, ES+) RT 0.86 min., 601 [M+H]t
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 224 -
INTERMEDIATE 212
H2
H2
(1R,3R)-7-(4-(2-aminopropan-2-yl)pheny1)-1-(2-chloro-6-
(difluoromethoxy)pheny1)-6-
fluoro-2,3-dihydro-1H-benzo[d]pyrrolo[1,2-dmidazole-3-amine
The title compound was prepared from Intermediate (0.324 g, 0.54 mmol) in
accordance
with the synthetic procedure described for Intermediate 210 to afford the
title compound
(0.240 g, 89% yield).
LCMS (Method 20, ES+) RT 0.43 min., 501 [M+H] .
INTERMEDIATE 213
073.1:47,sr
N,N-dially1-2-(5-bromopyrimidin-2-yppropan-2-amine
To a mixture of 2-(5-bromopyrimidin-2-yl)propan-2-amine (3.00 g, 13.9 mmol)
and
potassium carbonate (5.81 g, 41.6 mmol) in acetonitrile (50 rnL) was added
ally! bromide
(3.56 g, 29.1 mmol) and the mixture heated to 50 C for 4 hours. A further
portion of
allyl bromide (850 mg, 6.95 mmol) was added and the reaction allowed to stir
at room
temperature for 72 hours. The mixture was partitioned between diethyl ether
(200 mL)
and 2M HC1. The organic phase was further extracted with 2M HC1 (x2) and the
aqueous
phase washed with diethylether (x2). The aqueous phase was cooled on ice and
made
basic with solid sodium hydroxide. The aqueous was then extracted with
dichloromethane
and the organic solvents dried over sodium sulphate, filtered and the
volatiles removed in
vacuo to give the title compound as a red oil, (3.40g, 83% yield). LCMS Method
3 (ES+)
RT 2.63 minutes, 296/298 (M+H)+. NMR (300 MHz, DMSO-d6) 8.96 (s, 2H), 5.80-
5.60 (m, 2H), 5.10-4.85 (m, 4H), 3.20-3.10 (m, 4H), 1.50 (s, 6H).
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 225 -
INTERMEDIATE 214
H
tert-butyl ((1R,3R)-7-(2-(2-((tert-butyldimethylsilyl)oxy)butan-2-yOpyrimidin-
5-y1)-1-
(2-chloro-6-(difluoromethoxy)pheny1)-6-fluoro-2,3-dihydro-1H-
benzo[d]pyrrolo[1,2-
alimidazol-3-Acarbamate
A solution of 2-(2-((tert-butyldimethylsilyl)oxy)butan-2-y1)-5-(4,4,5,5-
tetramethyl-1,3,2-
dioxaborolan-2-y1)pyrimidine [prepared from 5-bromo-2-(2-((tert-
butyldimethylsilyl)oxy)butan-2-yepyrimidine (0.500 g, 1.45mmol),
bis(pinacolato)diboron (0.450 g, 1.74 mmol) potassium acetate (0.426 g, 4.34
mmol),
1,1'-bis(diphenylphosphino)ferrocene palladium(II)dichloride dichloromethane
complex
(0.037 g, 0.043 mmol) in 1,4-dioxane (10 ml) at 95 C during 2 hours] in 1,4-
dioxane (10
ml) was added to a solution of Intermediate 184 (0.650 g, 1.19 mmol) in 1,4-
dioxane (40
ml) under argon. Sodium carbonate (0.253 g, 2.38 mmol), water (1 ml) were
added and
the suspension purged with argon. tris(dibenzylideneacetone)dipalladium(0)
(0.029 g,
0.030 mmol) and tri-tert-butylphosphonium tretrafluoroborate (0.035 g, 0.119
mmol)
were added and the reaction mixture was heated at 95 C for 3 hours before
cooling to
room temperature.
Water (30 ml) was added and the resulting mixture was extracted with Et0Ac (2x
30 m1).
The combined organic phases were washed with brine (30 ml), dried over sodium
sulfate,
filtered and concentrated under reduced pressure. The resulting residue was
purified by
SiO2 flash chromatography with DCM/Me0H (100/0 to 98/2) as eluent to give a
brown
viscous oil. Purification by preparative HPLC provided the title compound
(0.310 g, 36%
yield).
LCMS (Method 20, ES+) RT 1.98 min., 732 [M+H]t
30
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 226 -
INTERMEDIATE 215
H2
I a
,o
(1R,3R)-7-(2-(2-((tert-butyldimethylsidyl)oxy)butan-2-yl)pyrimidin-5-y1)-1-(2-
chloro-6-
(difluoromethoxy)pheny1)-6-fluoro-2,3-dihydro-1H-benzo[d]pyrrolo[1,2-
c]imidazol-3-
amine
2N Hydrochloric acid solution in diethyl ether (1.9 ml, 3.82 mmol) was added
to a
solution of Intermediate 214 (0.280 g, 0.382 mmol) in dichloromethane (10 m1).
The
reaction mixture was stirred at room temperature for 3 hours.
Water (30 ml) was added and the aqueous phase was treated with 2N sodium
hydroxide
aqueous solution until pH = 12. The resulting mixture was extracted with
dichloromethane (2x 30 ml) and with Et0Ac (lx 30 m1). The combined organic
phases
were dried over sodium sulfate, filtered and concentrated under reduced
pressure to
provide the title compound (0.200 g, 78% yield).
LCMS (Method 16, ES+) RT 1.34 min., 632 [M+H].
INTERMEDIATE 216
01
0
0
0:4
(7R,14R)-11-(2-(2-((tert-butyldimethylsilyl)oxy)butan-2-yl)pyrimidin-5-y1)- I-
(difluoromethoxy)-10-fluoro-6,7-dihydro-7,14-
methanobenzoNbenzo[4,5]imidazo[1,2-
a][1,4]diazocin-5(14H)-one
A 0.6M solution of phenol in anhydrous DMSO (0.63 ml, 0.38 mmol) was added to
a
solution of Intermediate 215 (0.200 g, 0.32 mmol) in anhydrous DMSO (6 m1).
Potassium
carbonate (0.066 g, 0.47 mmol), dried 4A molecular sieves (0.240 g), dichloro-
[bis(dicyclohexylphosphino)propane]palladium(II) (0.019 g, 0.032 mmol) were
added.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 227 -
The reaction mixture was heated at 100 C under 3 bars of carbon monoxide over
24
hours.
Et0Ac (100 ml) was added and the resulting mixture was washed with water (3x
100 ml),
brine (100 ml), dried over magnesium sulfate, filtered and concentrated under
reduced
pressure. The resulting crude residue was purified by SiO2 flash
chromatography with
DCM/Me0H (98/2 to 95/5) as eluent to afford the title compound (0.176 g, 89%
yield)
LCMS (Method 20, ES+) RT 1.84 min., 624 [M+H]t
EXAMPLE 1
(7R,14R)-1-(difluoromethoxy)-10-fluoro-1142-(2-hydroxypropan-2-yl)pyrimidin-5-
y11-
6,7-dihydro-7,14-methanobenzimidazo[1,2-b][2,5Thenzodiazocin-5(14H)-one
\> ..
N Nµ "NH
>rL I
0
0
0 H
Intermediate 14 was dissolved with pTSA (2.092, 11.00 mmol, 5 eq) in methanol
(60
mL) and the mixture was stirred overnight at r.t. The reaction mixture was
diluted with
EtOAC (200 mL) and a saturated solution of NaHCO3 (200 mL) was added. The
aqueous
layer was extracted by 3 x 50 mL of Et0Ac and the combined organic layers were
dried
over magnesium sulfate, filtered and concentrated in vacuo. The obtained
residue was
taken up in a minimum of Et0Ac, triturated, and filtered off. The obtained
precipitate was
washed with Et0Ac and dried to afford the title compound (1.6g, 76%).
.. 11-1 NMR (400 MHz, CDC13) 6 8.89 (s, 2 H), 8.46 (d, .1 8.0 Hz, I H), 7.59
(m, 2 H), 7.48
(m, 2 H), 7.38 (m, 1 H), 6.84 (t, J 72.5 Hz, 1 H), 6.37 (d, ./ 7.0 Hz, 1 H),
5.00 (t, .16.4
Hz, 1 H), 4.68 (s, 1 H), 3.51 (dt, .1 13.4, 7.0 Hz, 1 H), 2.90 (d,./ 13.3 Hz,
1 1-1), 1.67 (s, 6
H). LCMS Method 3 (ES') RT 1.28 mm, 496.0 (M+Hr.
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 228 -
EXAMPLE 2
(7R,14R)-1-(difluoromethoxy)-10-fluoro-1112-(2-methoxypropan-2-yl)pyrimidin-5-
y1]-
6-methy1-6,7-dihydro-7,14-methanobenzimidazo[1,2-b][2,5Thenzodiazocin-5(14H)-
one
N .....
(LN I
0
0
0-
Example / (8 mg, 0.01615 mmol) was dissolved in 0.2 mL of dry THF. Sodium
hydride
(60% in mineral oil, 1.6 mg, 0.04037 mmoL) was added and the reaction mixture
was
heated at 65 C for 1.5 h. Methyliodide (2.3 mg, 0.01615 mmoL) was added at
r.t. and the
mixture stirred at r.t for 16h.
An excess of methyliodide was then added to the mixture and stirred for
lh.Water was
added, the mixture was extracted with ethyl acetate, the combined organic
layers were
dried over magnesium sulphate and concentrated in vacuo to the title compound
as an off
white solid( 3 mg, 35.7%).
NMR (400 MHz, DMSO) 6 ppm 8.93 (s, 2H), 8.51 (d, J8.2 Hz, 1H), 7.54 (m, 2H),
7.44 (t, J 8.2 Hz, 1H), 7.32 (d, J 8.1 Hz, 1H), 6.84 (t, J 72.8 Hz, 1H), 6.28
(d, J 7.2 Hz,
1H), 4.98 (d, J 7.1 Hz, 1H), 3.52 (s, 3H), 3.48 (m, 1H), 3.28 (s, 3H), 2.90
(d, J 13.6 Hz,
1H), 1.70 (s, 6H). LCMS Method 3 (ES) RT 1.39 mm, 524.0 (M+H)+.
EXAMPLE 3
(7R,14R)-1-(difluorometho xy)-1142-(2-hydroxypropan-2-yl)pyrimidin-5-yl] -6,7-
dihydro-7,14-methanobenzimidazo [1,2-b11-2,5Thenzodiazocin-5(14H)-onc
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 229 -
)" . ...
"NH
0
0
HO
Intermediate 23 (525 mg, 0.8873 mmoL) was dissolved in 60 mL of methanol and
cooled
to 0 C. pTSA (1g, 0.7031 mmoL) was added and the mixture stirred at room
temperature
for 16h. The mixture was cooled down to 0 C, pTSA (1g, 0.7031 mmoL) was added
and
the reaction mixture was heated at 60 C for 2h.The solvent was evaporated, the
residue
was taken up in dichloromethane and washed with saturated aqueous sodium
hydrogen
carbonate. The aqueous layer was extracted with DCM, the combined layers were
dried
over anhydrous MgSO4, filtered and concentrated in vacuo. The crude compound
was
triturated with Et0Ac filtered, washed with a minimum of cold ethyl acetate
and dried in
vacuo to afford the title compound as a white solid (318 mg, 75%).
11-1NMR (400 MHz, DMSO) 6 ppm 9.14 (d, J 6.9 Hz, 1H), 9.04 (s, 2H), 8.25 (dd,
J 4.6
Hz, J 4.1 Hz, 1H), 7.86-7.50 (m, 6H), 6.38 (dJ 7.0 Hz, 1H), 5.12 (s, 1H), 4.92
(t, J 6.8
Hz, 1H), 3.49 (m, 1H), 2.77 (d, J 13.3 Hz, 1H), 1.54 (s, 6H). LCMS Method 3
(ES) RT
1.26 mm, 478.0 (M+H)'.
EXAMPLE 4
(7 R,14R)-1-(di fluorom ethoxy)-1142-(2-hydroxypropan-2-yl)pyrimidin-5-A-6-
methyl-
6,7-dihydro-7,14-m eth anob en zimi dazo[1,2-b-112,5]benzodiazocin-5(14H)-one
N
I
0
0
0 H
CA 02962826 2017-03-28
WO 2016/050975 PCT/FP2015/072868
- 230 -
Intermediate 24 was dissolved in methanol (20 mL/g), pTSA (0.600 g, 3.152
mmol, 2.2
eq) was added and the reaction mixture was stirred at r.t. overnight. APTS
(0.300 g,
1.176 mmol, 1.1 eq) was added and the reaction mixture was heated at 40 C for
2h, then
45 C for 1 hand following 50 C for 15 minutes until disappearance of starting
material in
LCMS. Et0Ac (600 mL) was added and the mixture was washed successively with a
saturated solution of NaHCO3 (200 mL) and a saturated solution of NaCl (200
mL), dried
over anhydrous magnesium sulfate, filtered and concentrated in vacua, diluted
with
diethylether (150 mL) and concentrated in vacua to afford the title compound
as an off-
white solid (0.701 g, 99%).
NMR (400 MHz, CDC13) ö 8.83 (s, 2 H), 8.43 (d, J = 8.2 Hz, 1 H), 7.84 (d, J =
8.5 Hz,
1 H), 7.70 (s, 1 H), 7.47 (d, J = 8.6 Hz, 1 H), 7.39 (I, J = 8.2 Hz, 1 H),
7.26 (d, J = 8.1 Hz,
1 H), 6.79 (t, J = 72.5 Hz, 1 H), 6.33 (d, J = 7.2 Hz, 1 H), 5.25 (d, J = 7.1
Hz, 1 H), 3.55
(d, J = 7.1 Hz, 1 H), 3.53 (s, 3 H), 3.38 (d, J = 6.7 Hz, 1 H), 2.89 (d, J =
13.7 Hz, 1 H),
1.57 (s, 6 H). LCMS Method 3 (ES') RT 1.30 min, 492.0 (M+H)+.
EXAMPLE 5
(7R,14R)-1 -(di fluoromethoxy)-6-ethy1-1142-(2-hydroxyprop an-2-yOpyrimidin-5-
yli -6,7-
dihydro-7,14-mcthanob cnzimi dazo [1,2-1)] [2,5]b cnzodiazocin-5(14H)-onc
0
F(OL
0 H
Intermediate 25 (69 mg, 0.1113 mmoL) was dissolved in 1 mL of methanol, pTSA
(105.9
mg, 34.0908 mmoL) was added and the mixture was stirred at r.t. for 16 h.
Et0Ac was
added, washed with saturated aqueous sodium hydrogen carbonate and brine. The
organic
layer was dried over magnesium sulphate and concentrated in vacuo. The residue
was
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 231 -
purified by chromatography (SiO2, 75% Et0Ac in heptane)and trituratcd with
diethyl
ether to afford the title compound as an off white solid (23 mg, 40.9%).
'H NMR (400 MHz, CDC13) 6 ppm 8.95 (s, 2 H), 8.56 (d, J = 7.4 Hz, 1 H), 7.93
(m, 1 H),
7.81 (s, 1 H), 7.54 (m, 1 H), 7.48 (m, 1 H), 7.35 (d, J = 7.2 Hz, 1 H), 6.89
(t, J = 72.3 Hz,
1 H), 6.41 (s, 1 H), 5.25 (m, 1 H), 4.17 (m, 1 H), 3.95 (m, 1 H), 3.59 (m, 1
H), 2.93 (m, 1
H), 1.68 (s, 6 H), 1.49 (m, 3 H). LCMS Method 3 (ES') RT 1.36 min, 506.0
(M+H)4.
EXAMPLE 6
(7R, 14R)-1-(difl uorometho xy)-11-[2-(2-hy droxypropan-2-yl)py rimidin-5-y1]-
6-(prop an-
2-y1)-6,7-dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one
>rL I
0
0
0 H
Intermediate 26 (10 mg, 0.0158 mmoL) was dissolved in Me0H (0.2 mL) and pTSA
(6.6
mg, 0.0347 mmoL) was added. The mixture was stirred at r.t.. After 16 h, pTSA
(6.6 mg,
0.0347 mmoL) was again added and the mixture heated at 50 C for 1 h. The
reaction
mixture was concentrated, the residue dissolved in Et0Ac washed with saturated
aqueous
sodium hydrogen carbonate and brine. The organic layer was dried over
anhydrous
magnesium sulphate, concentrated in vacuo and triturated with diethyl ether to
afford the
title compound as a white solid (7 mg, 85.4 % yield).
LCMS (ES) Method 3 RT 1.42 min, 520.0 (MA-1)+,
EXAMPLE 7
(7R, 14R)- I -(difluoromethoxy)-10-fluoro- 142-(2-hydroxypropan-2-
yl)pyrirnidin-5-y1]-
7H-7,14-methanobenzimidazo[2,1-cli[2,5]benzoxazocin-5(14H)-onc
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 232 -
F
yr
N," .41'0
0
0
0 H
To a suspension of Intermediate 29 (50 mg, 0.0972 mmoL) in 1 mL of toluene was
added
cyanomethylenetributylphosphorane (25.9 mg, 0.0282 mmoL) and the reaction
mixture
was heated at 100 C for 16 h. The mixture was cooled to r.t., taken up with
Et0Ac and
washed with IN sodium hydroxide. The organic layer was dried over magnesium
sulphate
and concentrated in vacuo and triturated with diethyl ether to afford the
title compound as
an off-white solid (3 mg, 6.2 % yield).
1H NMR (400 MHz, DMSO) 6 ppm 8.96 (s, 2 H), 8.08 (d, J= 7.9 Hz, 1 H), 7.76 (d,
J =
11.3 Hz, 1 H), 7.65 (t, J = 73.8 Hz, 1 H), 7.61 (m, 2 H), 7.55 (m, 1 H), 6.47
(d, J = 6.8 Hz,
1 H), 6.10 (d, J= 5.3 Hz, 1 H), 5.17 (bs, 1 H), 3.61 (m, 1 H), 3.18 (d, J=
14.7 Hz, 1 H),
1.49 (m, 6 H). LCMS Method 3 (ES ) RT 1.36 mm, 497.0 (M+H)}.
EXAMPLE 8
(2Z)-[(7R,14R)-1-(difluoromethoxy)-10-fluoro-11-[2-(2-hydroxypropan-2-
yl)pyrimidin-
5-y1]-7H-7,14-methanobenzimidazo[2,1-d][2,5]benzoxazoein-5(14H)-
ylidene]acetonitrile
and (2E)-[(7R,14R)-1-(difluoromethoxy)-10-fluoro-1142-(2-hydroxypropan-2-
yl)pyrimidM-5-y1]-7H-7,14-methanobenzinnidazo[2,1-d112,51benzoxazocin-5(14H)-
ylideneiacetonitrile
Frr
0
>rL
CN N
0
0 H
CA 02962826 2017-03-28
WO 2016/050975
PCT/EP2015/072868
- 233 -
To a suspension of Intermediate 29 (50 mg, 0.0972 mmoL) in 1 mL. of toluene
was added
cyanomethylenetributylphosphorane (25.9 mg, 0.0282 mmoL) and the reaction
mixture
was heated at 100 C for 16 h. The reaction was concentrated in vacuo and the
residue was
purified by reverse phase preparative LCMS (basic conditions). The residue was
dissolved in 2 mL of Me0H and passed through an acidic exchange-ion column
(400 mg,
conditioning: Me0H 10mL), followed by washing of the column with 10 mL of
methanol. The compound was discharged from the resin by elution of 10 mL of
ammonia
(1 M in methanol) and concentrated in vacuo. The residue was purified by
preparative
.. TLC with EtOAc - hexane (8/2), to afford the title compound as a colourless
oil (2.5 mg,
4.95 % yield) in a 6/4 mixture of Z/E isomers.
1H NMR (400 MHz, CDC13) 6 ppm 8.93 (s, 1.2 H), 8.90 (s, 0.8 H), 7.69 (t, J =
11.5 Hz, 1
H), 7.60 (d, J = 7.6 Hz, 0.6 H), 7.40 (m, 3 H), 7.25 (d, J = 7.4 Hz, 0.4 H),
6.84 (t, J = 72.5
Hz, 0.4 H), 6.83 (t, J = 72.3 Hz, 0.6 H), 6.28 (m, 1 H), 6.01 (d, J = 4.3 Hz,
0.4 H), 5.87 (d,
J - 4.1 Hz, 0.6 H), 5.34 (s, 0.6 H), 5.07 (s, 0.4 H), 3.32 (m, 1 H), 3.07 (d,
J - 14.1 Hz, 0.4
H), 3.02 (d, J = 13.9 Hz, 0.6 H)õ 1.67 (s, 6 H).
LCMS Method 3 (ES) RT 1.39 min, 520.0 (M+H)+.
EXAMPLE 9
(7R,14R) and (7S, 14S)-1142-(2-hydroxypropan-2-yenyrimidin-5-y1]-7,14-dihydro-
7,14-
methanopyrido[1',2':1,2]imidazo[4,5-d][2]benzazocin-5(6H)-one
/C1
.A_ ,...= ......,N
H
====,)(,,,k,..N./ \ 0 )(Le. 0
IIIP 0 H
and
The title compound was prepared from Intermediate 37 (300 mg, 0.71 mmol),
Na2CO3 (378 mg, 3.57 mmol) and dichloro [bis(dicyclohexylphosphino)propane]
palladium(II) 1Pd-133 from Johnson Matthey] (40 mg, 0.06 mmol), 1,4-dioxane (9
mL)
under CO gas (5 bars) at 150 C for 15h, following the protocol described for
Intermediate 14 (5.5 mg, 2%). 1H NMR (500 MHz, DMSO-d6) 6 9.10 (s, 2H), 8.90
(s,
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 234 -
1H), 8.70 (d, J 6.3 Hz, 1H), 8.28 (d, J 8.0 Hz, 1H), 7.95 (d, J 7.5 Hz, 1H),
7.66 (d, J 9.4
Hz, 1H), 7.61 (d, J 9.4 Hz, 1H), 7.48 (t, ./ 7.4 Hz, 1H), 7.27(t, .1 7.7 Hz,
1H), 5.11 (s,
1H), 4.75 (d, J 6.1 Hz, 11-1), 4.69 (t, ./ 6.3 Hz, 1H), 2.44 (d, 12.6
Hz, 2H), 1.54 (s, 611).
LCMS Method 3 (ES) RT 1.64min, 412.0 (M+H)+.
EXAMPLE 10
C = *II N H
0
=
(7R,14R)-11-chloro-1-(difluoromethoxy)-10-fluoro-6,7-dihydro-7,14-
methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-onc
In a high pressure reactor, Intermediate 41(927 mg, 2.076 mmol) was
solubilized in dry
1,4-dioxane (21 mL). Potassium carbonate (1.4 g, 10.4 mmol) was added. A
solution of
palladium(II) acetate (23.3 mg, 0.1038 mmol) and 4,5-bis(diphenylphosphino)-
9,9-
dimethylxanthene (61.9 mg, 0.1038 mmol) in 1 mL of dry dioxane was then added.
The
reactor was closed and degassed by 3 successive vacuum / nitrogen cycles and
then with
CO by 3 successive vaccuum / CO cycles. The bomb was charged with CO to 8 psi
and
heated at 110 C overnight. The reaction mixture was subsequently filtered
through a pad
of celite and the pad rinsed by 50 mL of Et0Ac. The filtrate was concentrated
in vacuo
and the residue purified over silica gel using AcOEt / Me0H 10/0 to 9/1 to
yield 534 mg
(65%) of the title compound as a pale brown solid. LCMS basic: RT 1.97 min.
(ES+)
394/396 (M-FH)+. 1H NMR (400 MHz, DMS0): 9.13 (d, J= 6.8 Hz, 1 H), 8.23 (dd,
J1 =
6.9 Hz, J2 = 2.6 Hz, 1 H), 7.70 (d, J= 10.1 Hz, 1 H), 7.60 (t, J= 73.2 Hz, 1
H), 7.51 (m,
3 H), 6.30 (d, J= 7.1 Hz, 1 H), 4.88 (t, J= 6.8 Hz, 1 H), 3.45 (m, 1 H), 2.73
(d, J = 13.4
Hz, 1 H).
EXAMPLE 11
010
CI 1H
=
F 110
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 235 -
(7R,14R)-11-chloro-1-(difluoromethoxy)-6,7-dihydro-7,14-methanobenzimidazo[1,2-
b][2,5Thenzodiazocin-5(14H)-one
Intermediate 40 (3.7 g, 8.6 mmol), activated molecular sieve 4A powder (1.2
g),
potassium carbonate (1.5 equiv., 13 mmol) followed by dichloro[9,9-dimethy1-
4,5-
bis(diphenylphosphino)xanthene]palladium(II) (0.04 equiv., 0.35 mmol) were
poured
into the center of the 100 mL Glass Parr reaction vessel. 3 cycles of vacuum (-
20 mmHg)
followed by Argon were applied to the closed reactor.
Anhydrous dimethyl sulfoxide (35 mL) was added, followed by phenol 5M in DMSO
(1.1
equiv., 9.5 mmol). The solution was degassed by 3 vacuum (-20 mmHg) / argon
cycles
followed by 3 cycles of vacuum! CO resulting in a final CO pressure of 1 bar.
The mixture was stirred and heated overnight at 100 C under the CO atmosphere
. The
reaction was cooled to 30 C, the reactor vessel was opened and Et0Ac (40 mL)
was
added. The resulting mixture was filtered on a pad of Celite, evaporated in
vacuo to yield
a green oil.
The residue thus obtained was taken up in Et0Ac (100 mL) and the organic layer
was
washed with water, K2CO3 (saturated aqueous solution) and brine (saturated
aqueous
solution). The aqueous layer was then re-extracted with Et0Ac (1 x 50 mL). The
combined organic layers were dried over MgS0.4, filtered and evaporated to
dryness. The
obtained green solid (3.65 g), was taken up in Et0Ac, the insoluble material
was filtered
and rinsed with Et20 to afford 1.06 g (33.1%) of the title compound as a grey
solid.
The filtrate can be purified by flash chromatography to provide additional
product if
required:
LCMS basic: MH+ m/z = 376, RT 1.90 minutes.
'1-1NMR (300 MHz, DMSO) 69.12 (d, 1 H, J = 6.7 Hz), 8.23 (dd, 1 H, J = 7.0,
2.4 Hz),
7.60 (m, 5 H), 7.20 (dd, 1 H, J = 8.7, 2.1 Hz), 6.29 (d, 1 H, J = 7.1 Hz),
4.87 (dd, 1 H, J =
6.7 Hz, 6.7 Hz), 3.46 (m, 1 H), 2.72 (d, 1 H, J = 13.4 Hz).
EXAMPLE 12
CI
= ip
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 236 -
(7R,14R)-11 -ehloro-1-(difluoromethoxy)-5 ,6,7,14-tetrahydro -7,14-
methanob enzimidazo[1,2-bi[2,5]benzodiazocine
Intermediate 45 (0.0525 mmol) was solubilized in THF (0.5 mL) and ethanol (0.5
mL).
Polymer-supported cyanoborohydride (33 mg, 0.132 mmol, 4 mmol/g) was added.
The
reaction mixture was subjected to an orbital shaker for 2 hours. The reaction
mixture was
then filtered and evaporated under reduced pressure. The crude material was
purified by
preparative reverse phase HPLC (basic condition) to afford 7 mg (37%) of the
title
compound. LCMS basic (ES+) RT 3.5 min., 362/364(M+H)+. LCMS acidic (ES+) RT
2.12 min., 362/364(M+H)+.
'14 NMR (400 MHz, CDC13) 6 7.69 (m, 1 H), 7.20 (m, 3 H), 7.13 (iii 1 H), 6.99
(m, 1 H),
6.73 (m, 1 H), 6.10 (m, 1 H), 4.77 (m, 1 H), 3.66 (m, 1 H), 3.21 (m, 1 H),
3.01 (d, 1 H, J =
15.3 Hz), 2.94 (m, 1 H), 2.50 (m, 1 H).
EXAMPLE 13
OH
I 0
OH j
f(7R,14R)-1-(difluoromethoxy)-11-[2-(2-hydroxypropan-2-yl)pyrimidin-5-y1]-5,14-
dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-6(7H)-yflacetic acid
Intermediate 46 (34 mg, 0.076 mmol) , 2-(1-hydroxy-1-methtylethyl) pyrimidine-
5-
boronic acid pinacol ester (2.5 equiv., 0.190 mmol), K3PO4 (2 equiv., 0.152
mmol),
tris(dibenzylideneacetone)dipalladium(0) (0.05 equiv., 0.0038 mmol), and
tticyclohexylphosphonium tetrafluoroborate (0.12 equiv., 0.0091 mmol) were
dissolved
in a degassed mixture of 1,4 dioxanc (0.9 mL) and water (0.1 mL). The reaction
mixture
was heated overnight at 105 C.
The mixture was cooled to r.t; water was added and the mixture extracted with
Et0Ac.
The aqueous layer was brought to pH 2-3 with HC1 1N, extracted with Et0Ae,
dried over
magnesium sulphate, filtered and concentrated in vacuo. The crude product was
purified
by reverse phase chromatography to afford 3 mg (7.5%) of the title compound as
a white
solid at 90% purity. LCMS (ES+) 522/523 (M+H)+
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 237 -
EXAMPLE 14
H
OH F--e
(7R,14R)-1-(difluoromethoxy)-1142-(2-hydroxypropan-2-yOpyrimidin-5-y1]-6,7-
dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocine-5(14H)-thione
To a solution of Intermediate 47 5 (15 mg, 0.025 mmol) in Me0H (1 mL) was
added p-
toluenesulfonic acid monohydrate (23.5 mg, 0.124 mmol). The slurry was stirred
overnight at r.t. The reaction mixture was quenched with NaHCO3 (10% aqueous
solution) and extracted with Et0Ac (3 x 5 rnL). The organic layer was dried
over MgSO4,
filtered and concentrated in vacuo. The residue was purified by preparative
TLC (DCM /
Me0H 9 / 1) followed by a second purification by flash chromatography on
silica gel
(DCM I Me0H 100 / 0 to 95 / 5) to afford 9 mg (74%) of the title compound.
LCMS
acidic (ES+) RT 2.29 min., 494(M+H)+.
EXAMPLE 15
"===
ci H
=
(7R,14R)-11-ehloro-1-(difluoromethoxy)-10-fluoro-5,6,7,14-tetrahydro-7,14-
methanobenzimidazo[1,2-b][2,5]benzodiazocine
Example 10 (178 mg, 0.452 mmol) was solubilized in dry THF (5 mL). At 0 C,
borane
dimethyl sulfide complex (340 [iL, 2M solution in THF, 0.68 mmol) was added .
The
reaction mixture was allowed to warm to r.t. and stirred overnight. The
reaction mixture
was concentrated in vacuo, the residue was taken up in Me0H, stirred and
heated under
reflux for 48 hours. The reaction mixture was concentrated in vacuo and the
residue was
purified by reverse phase preparative LCMS (basic condition) to afford 65 mg
(38%) of
the title compound. LCMS basic (ES+) RT 3.55 min., 380/382(M+H)+. LCMS acidic
(ES+) RT 3.56 min., 380/382(M+H)+. 11-1NMR (400 MHz, CDC13) 6 ppm 7.52 (d, 1
H, J
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 238 -
= 9.6 Hz), 7.23 (m, 2 H), 7.12 (m, 1 H), 6.97 (d, 1 H, J = 7.4 Hz), 6.71 (m, 1
H), 6.09 (d,
1 H, J = 7.6 Hz), 4.70 (d, 2 H, J = 5.9 Hz), 3.64 (m, 1 H), 3.21 (m, 1 H),
2.98 (d, 1 H, J =
15.3 Hz), 2.48 (d, 1 H, J = 12.6 Hz).
EXAMPLE 16
4.= ,,,,, H
H. 4
__to)
2- {5-[(7R,14R)-1-(difluoromethoxy)-10-fluoro-5,6,7,14-tetrahydro-7,14-
methanobenzimidazo[1,2-b][2,5]benzodiazocin-11-yl]pyrimidin-2-y1} propan-2-ol
Example 15 (47 mg, 0.1237 mmol), 245-(4,4,5,5-tetramethy1-1,3,2-dioxaborolan-2-
yl)pyrimidin-2-yllpropan-2-ol (82 mg, 0.3105 mmol), tricyclohexylphosphonium
tetrafluoroborate (5.524 mg, 0.01485 mmol), tris
(dibenzylideneacetone)dipalladium(0)
(5.6 mg, 0.0061 mmol), and K3PO4(52.5 mg, 0.248 mmol) were placed in a tube,
and
filled with argon. Degassed 1,4 dioxanc (1 mL) and water (100 L) were added
and the
resulting slurry was stirred at 105 C for 2 hours. The reaction mixture was
cooled to r.t.
before addition of Et0Ac (2 mL) and water (2 mL). The aqueous layer extracted
with
Et0Ac (2 x 2 mL). The combined organic layers were dried over magnesium
sulphate,
filtered and concentrated in vacuo. The crude was purified by reverse phase
preparative
LCMS (acidic condition) to afford the TFA salt of the title compound which was
solubilized in Et0Ac (2 mL) and washed with a saturated solution of NaHCO3.
The
aqueous layer was extracted with Et0Ac (2 x 2 mL). The combined organic layers
were
dried over magnesium sulphate, filtered and concentrated in vacuo to afford 33
mg (55%)
of the title compound as a white solid. LCMS basic (ES+) RT 3.69 min.,
482(M+H)+.
LCMS acidic (ES+) RT 1.91 min., 482(M+H)+. 1HNMR (400 MHz, CDC13) 6 ppm 8.87
(m, 2 H), 7.62 (m, 1 H), 7.25 (s, 1 H), 7.20 (m, 1 H), 7.12 (m, 1 H), 6.99 (m,
1 H), 6.72
(m, 1 H), 6.17 (m, 1 H), 4.78 (m, 1 H), 4.66 (m, 1 H), 3.66 (m, 1 H), 3.25 (m,
1 H), 3.05
(m, 1 H), 2.53 (m, 1 H), 1.65 (s, 6 H).
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 239 -
EXAMPLE 17
H
\ 0
0 ip
(7R,14R)-10-fluoro-1142-(2-hydroxypropan-2-yl)pyrimidin-5-y1]-1-methoxy-6,7-
dihydro-7,14-methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one
Intermediate 57(145 mg, 0.31 mmol) was dissolved in anhydrous DMA (3 mL) in
a 25 ml pressure reactor and Na2CO3 (165 mg, 1.56 mmol) was added. The mixture
was
degassed with a stream of nitrogen for 10 minutes then the pressure vessel was
sealed and
subjected to three vacuum/nitrogen flush cycles, before repeating the process
with carbon
monoxide and charging the pressure to 3.0 bar. The mixture was stirred for 5
minutes then
heated to 150 C and stirred at this temperature overnight. The reactor was
allowed to
cool to r.t.. The reaction mixture was diluted with Et0Ac (20 mL) and filtered
through a
pad of celite, washing with excess Et0Ac (20 mL). The filtrate was washed with
water
(15 mL), then brine (15 mL), dried (Na2SO4) and concentrated to dryness under
vacuum
to yield 160 mg of a crude residue. Purification by reverse phase
chromatography (eluting
with 0-100% MeCN (+0.1% NH4OH)/H20 (+0.1% NH4OH)) to yield 2.1 mg (1.5%) of
the title compound as a beige solid. . LCMS Method 6 (ES+) RT 3.52 min., 460.2
(M+H)+. IFINMR (500 MHz, Me0H-d4) 6 8.95 (d, J = 1.5 Hz, 2H), 8.04 (dd, J =
8.0, 1.1
Hz, 1H), 7.64 (d, J = 6.8 Hz, 1H), 7.50 (d, J= 11.2 Hz, 1H), 7.40 (t, J = 8.1
Hz, 1H), 7.37
- 7.29 (m, 1H), 6.62 (d, J = 7.1 Hz, 1H), 4.96 (d, J = 6.7 Hz, 1H), 4.13 (s,
3H), 3.51 (dt, J
= 13.5, 6.9 Hz, 1H), 2.80 (d, J = 13.4 Hz, 1H), 1.65 (s, 6H), 1.63 - 1.57 (m,
1H).
EXAMPLE 18
H
0
HO F *
(7K,14R)-1.10-difluoro-11-[2-(2-hydroxypropan-2-yppyrimidin-5-y11-6,7-dihydro-
7.14-
methanobenzimidazo[1,2-b][2,5]benzodiazocin-5(14H)-one
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 240 -
To a solution of Intermediate 58 (200 mg, 0.44 mmol) in 1,4-dioxane (5 mL) was
added
sodium carbonate (232 mg, 2.19 mmol) dichloropalladium; dicyclohexyl(3-
dicyclohexyl-
phosphanylpropy1)-phosphane (53.8 mg, 0.0877 mmol). The reaction mixture was
stirred
at 160 C under 5 bar of CO pressure overnight. The reaction mixture was
filtered through
.. a celite pad and rinsed with ethanol (10 mL). The filtrate was evaporated
and the crude
was dissolved in DCM (10 mL) and treated with a saturated aqueous solution of
NH4C1(5
mL) The organic layer was concentrated in vacuo, and the residue purified on
silica gel
(DCM/iPrOH/ aq NH3 90:9:1), yielding 32 mg (16%) of the title compound.
LCMS acidic Method 4 (ES+) RT 2.06 min., 448.2 (M+H)+
EXAMPLE 19
CI N
g I
N H
0
F--j= F
.. (6R,12R)-2-chloro-11-(difluoromethoxy)-7,12-dihydro-6H-6,12-
methanobenzimidazo[2,1-cil1,4Thenzod1azepine
A mixture of Intermediate 40 (500 mg, 1.17 mmol), potassium carbonate (322 mg,
2.33
mmol), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (69 mg, 0.12 mmol) and
palladium(H) acetate (26 mg, 0.12 mmol) in 1,4-dioxane (16 mL) was de-gassed
and
stirred at 110 C under nitrogen for 15 hours. The reaction mixture was cooled
to ambient
temperature and 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (69 mg, 0.12
mmol)
and palladium(11) acetate (26 mg, 0.12 mmol) were added to the reaction
mixture. The
solvent was de-gassed and the mixture was stirred at 110 C for 18 hours. The
reaction
mixture was cooled to ambient temperature, filtered through celite and the
latter was
washed with Et0Ac. The combined filtrate and washings were evaporated to
dryness
using an oil pump to give an oil which was purified by flash chromatography on
silica gel
(25 to 100% Et0Ac in hexanes) to afford the title compound (249 mg, 0.71 mmol,
61%
yield) as a brown solid. LC/MS Method 3: RT 2.29 mins (pH 10), m/z 348 and
350.
1HNMR: (CD30D, 300 MHz) 6: 2.48 (d, J = 11.2 Hz, 1H), 2.99 (dt, J = 11.2, 4.5
Hz, 1H),
.. 4.87 (m, 1H (overlap with the residual water), 5.93 (d, J = 4.5 Hz, 1H),
6.40 (d, J = 8.1
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 241 -
Hz, 2H), 6.96 (t, J = 74 Hz, 1H), 7.00 (d, J = 8.1 Hz, 1H), 7.17 (dd, J = 8.6,
2.0 Hz, 1H),
7.46 (d, J = 2.0 Hz, 1H), 7.53 (d, J = 8.6 Hz, 1H).
EXAMPLE 20
H
H CA),.
0
F(F
2- (5- [(6R,12R)-11-(difluoromethoxy)-7,12-dihydro-6H-6,12-
methanobenzimidazo[2,1-
c][1,4]benzodiazepin-2-yl]pyrimidin-2-yllpropan-2-ol
A mixture of 245-(4,4,5,5-tetramethy1-1,3,2-dioxaborolan-2-yOpyrimidin-2-
yl]propan-2-
ol (purchased from commercial sources) (110.5 mg, 0.42 mmol), Example /9 (97
mg,
0.28 mmol), K3PO4 (118.3 mg, 0.56 mmol), and tricyclohexylphosphonium
tetrafluoroborate (15.9 mg, 0.042 mmol) was solubilized in 1,4-dioxane (1.95
mL) and
water (0.19 mL) and the mixture degassed with nitrogen before addition of
tris(dibenzylideneacetone)dipalladium(0) (18.4 mg, 0.019 mmol). The reaction
mixture
was heated at 105 C for 15 hours or until LCMS showed reaction to be
complete. The
reaction mixture was cooled to ambient temperature and the crude mixture was
extracted
with Et0Ac (3 x 10 mL). The combined organic phases were washed with saturated
brine (2 x 10 mL), dried over sodium sulphate, filtered and concentrated in
vacuo to give
an oil which was purified by preparative HPLC (pH = 10) to afford the title
compound
(23 mg, 0.051 mmol, 18% yield) as a white solid. LC/MS Method 3: RT 2.09 mills
(pH
10), miz 450 .
IHNMR: (DMSO-&, 300 MHz) 6: 1.54 (s, 6H), 2.38 (d, J = 11.3 Hz, 1H), 2.94 (dt,
J =
11.3, 4.3 Hz, 1H), 4.91 (t, J = 3.5 Hz, 1H), 5.11 (bs, 1H), 5.91 (d, J = 4.3
Hz, 1H), 6.34 (t,
J = 8.3 Hz, 2H), 6.97 (t, J = 8.3 Hz, 1H), 7.15 (d, J= 3.7 Hz, 1H), 7.38 Hz
(dd, J = 73, 1.7
Hz, 1H), 7.54 (dd, J = 8.4, 1.7 Hz, 1H), 7.68-7.72 (m, 2 H), 9.06 (s, 2H).
CA 02962826 2017-03-28
WO 2016/050975 PCT/EP2015/072868
- 242 -
EXAMPLE 21
0
CI N
0"*
F-J\ F
1-[(6R,12R)-2-chloro-11-(difluoromethoxy)-6H-6,12-methanobenzimidazo [2,1 -
c11-1,41benzodiazepin-7(12H)-yllethanone
Example 19 (245 mg, 0.7044 mmol) was dissolved in pyridine (2 mL) and acetic
anhydride (2 mL). The mixture was stirred at 110 C for 15 hours. The reaction
mixture
was cooled to ambient temperature and quenched with 2N NaOH aq. (2 mL). The
crude
mixture was extracted with Et0Ac (2 x 10 mL). The organic phase was washed
with
saturated brine (10 mL), the combined organic phases was dried with sodium
sulphate,
filtered and concentrated in vacuo to give an oil which was purified by flash
chromatography on silica gel (0 to 100% Et0Ac in hexanes) to afford the title
compound
(177 mg, 65% yield) as a yellow solid. LC/MS Method 3: RT 2.08 mins (pH 10),
m/z 390
and 392.
'HNMR: (CD30D, 300 MHz) 6: 2.66 (d, J = 11.9 Hz, 1H), 2.69 (s, 3H), 3.20 (di,
J =
11.9, 4.5 Hz, 1H), 5.99 (d, J = 3.9 Hz, 1H), 6.10 (d, J = 4.5 Hz, 1H), 6.94
(d, J = 8.3 Hz,
1H), 7.08 (t, J = 73.4 Hz, 1H), 7.20-7.28 (m, 2H), 7.53 (d, J = 8.7 Hz, 1H),
7.59 (d, J = 2.0
Hz, 1H), 8.22 (d, J = 8.7 Hz, 1H).
EXAMPLE 22
0
H
N NI\ 2"N-ic
0 gr
1-[(6R,12R)-11-(difluoromethoxy)-242-(2-hydroxypropan-2-yOpyrimidin-5-y1]-6H-
6,12-
methanobenzimidazo[2,1-c][1,4]benzodiazepin-7(12H)-yl]ethanone
The title compound was prepared from 2-[5-(4,4,5,5-tetramethy1-1,3,2-
dioxaborolan-2-
yl)pyrimidin-2-yl]propan-2-ol (179.9 mg, 0.68 rnmol), and Example 21 according
to a
.. method involving the same procedural steps as those described for Example
20, to give,
DEMANDE OU BREVET VOLUMINEUX
LA PRESENTE PARTIE DE CETTE DEMANDE OU CE BREVET COMPREND
PLUS D'UN TOME.
CECI EST LE TOME 1 DE 2
CONTENANT LES PAGES 1 A 245
NOTE : Pour les tomes additionels, veuillez contacter le Bureau canadien des
brevets
JUMBO APPLICATIONS/PATENTS
THIS SECTION OF THE APPLICATION/PATENT CONTAINS MORE THAN ONE
VOLUME
THIS IS VOLUME 1 OF 2
CONTAINING PAGES 1 TO 245
NOTE: For additional volumes, please contact the Canadian Patent Office
NOM DU FICHIER / FILE NAME:
NOTE POUR LE TOME / VOLUME NOTE: