Note: Descriptions are shown in the official language in which they were submitted.
DESCRIPTION
ORAL DOSAGE FORM COMPRISING A CYCLOPROPANECARBOXAMIDE DERIVATIVE FOR
USE IN TREATING INSOMNIA
FIELD OF THE INVENTION
[0001] The present invention is directed to compositions and methods for
treating insomnia. The present
application claims priority on the basis of US Patent Application No.
62/067,443, filed in the United
States on October 23, 2014.
BACKGROUND OF THE INVENTION
[0002] Orexin receptors are G-protein coupled receptors found predominately in
the brain. Their
endogenous ligands, orexin-A and orexin-B, are expressed by neurons localized
in the hypothalamus.
Orodn-A is a 33 amino acid peptide; orexin-B consists of 28 amino acids
(Sakurai T. et al., Cell, 1998,
92 573-585). There are two subtypes of orexin receptors, orexin receptor 1
(hereinafter referred to as
OX I) and orexin receptor 2 (hereinafter referred to as 0X2); OX1 binds orexin-
A preferentially, while
0X2 binds both orexin-A and -B. Orexins stimulate food consumption in rats,
and it has been suggested
that orexin signaling could play a role in a central feedback mechanism for
regulating feeding behavior
(Sakurai et al., supra). It has also been observed that orexins control wake-
sleep conditions (Chemelli
R.M. et at., Cell, 1999, 98, 437-451). Orexins may also play roles in brain
changes associated with opioid
and nicotine dependence (S.L. Borgland et al., Neuron, 2006, 49, 598-601; C.J.
Winrow et al.,
Neuropharmacology, 2010, 58, 185-194), and ethanol dependence (J.R. Shoblock
et al.,
Psychopharmacology, 2011, 215, 191-203). Orexins have additionally been
suggested to play a role in
some stress reactions (T. Ida et al., Biochem. Biophys, Res. Commun., 2000,
270, 318-323). Compound
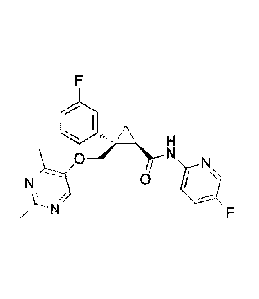
such as
(1R,2S)-2-(((2,4-dimethylpyrimidin-5-yBoxy)methyl)-2-(3-fluorophenyl)-N-(5-
fluoropyridin-2-y1)
cyclopropanecarboxamide (hereinafter referred to as Compound A) have been
found to be potent orexin
receptor antagonists, and may be useful in the treatment of sleep disorders
such as insomnia, as well as
for other therapeutic uses.
[0003] The Formula of Compound A
1
Date recue/date received 2021-10-19
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
.1026-- / N
0
[0004] Regarding the hypnotic agent, when an active pharmaceutical ingredient
(hereinafter referred to as
API) in a pharmaceutical formulation to be taken a once-a-night dosing is too
high a dose, it has the
potential to cause the next-day residual sleepiness, while the single
insufficient dose may cause the
patient to wake up during normal sleep period even if the patients are able to
fall sleep with the hypnotic.
Therefore, it is difficult to set the proper dose with considering the
sensitive balance between easy of
sleep onset and the avoidance of the residual sleepiness, as compared with the
considering only the
balance between side effects and efficacy. Furthermore, even if the dose of a
certain drug for insomnia,
the physiochemical properties of the API and the pharmacokinetic (hereinafter
referred to as PK) profile
after administration of the drug were known, such information would not be
applicable to other APIs for
insomnia because it would be likely effected by a number of factors, including
the mechanism of action,
the route of administration, the rate of absorption, the physiochemical
property such as the solubility and
the stability in plasma or other factors of each API. Indeed, the relationship
between the residual
sleepiness and the characteristics of the hypnotic agents is not always
consistent (CNS Drugs 2004; 18
(5): 297-328). The relation between PK profile and the sleepiness effect such
as the sleep onset or the
residual sleepiness has been unknown yet for compound A.
[0005] There exists a need in the art for more effective methods of treating
insomnia to achieve rapid
sleep onset as well as sleep maintenance, throughout the sleep period, but
avoid residual sleepiness and/or
the next-day impairment, comprising administrating orally a solid dosage form
of a hypnotic agent.
Further, there exists a need in the art for a pharmaceutical composition
comprising a hypnotic agent and
at least one pharmaceutically acceptable excipient for the treatment of
insomnia to achieve rapid sleep
onset as well as sleep maintenance, throughout the sleep period, but avoid
residual sleepiness and/or
4
next-day impairment.
SUMMARY OF THE INVENTION
[0006] It is an object of the present invention to provide methods of treating
insomnia comprising
administrating orally a solid dosage form of the drug compound A.
2
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
[0007] It is further an object of the present invention to provide a
pharmaceutical composition,
comprising a therapeutically effective amount of compound A
[0008] In certain embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single daily dose ranging from about 1 mg
to about 15 mg.
[0009] In certain embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single daily dose ranging from about 2 mg
to about 15 mg.
[0010] In certain embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single daily dose ranging from about 2 mg
to about 10 mg.
[0011] In certain embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single daily dose chosen from about 2,
2.5, 4, 5, 8, 10, or 15 mg.
[0012] In certain embodiment, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single daily dose providing a mean maximum
plasma concentration
(Cmax) of from about 3.0 ng/ml to about 7.2 ng/ml for each 1 mg of compound A,
after single dose
administration to human subjects.
[0013] In certain embodiment, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single daily dose ranging from about 1 mg
to about 15 mg, and
wherein said single daily dose achieves a mean maximum plasma concentration
(Cmax) of from about 3.0
ng/ml to about 7.2 ng/ml for each 1 mg of compound A, after single dose
administration to human
subjects.
[0014] In certain embodiment, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single 1 mg daily dose, and wherein said
single dose achieves a mean
3
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
maximum plasma concentration (Cmax) within the range of about 80% to about
125% of 5.3 ng/ml, after
single dose administration to human subjects.
[0015] In certain embodiment, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single 2.5 mg daily dose, and wherein said
single daily dose achieves a
mean maximum plasma concentration (Cmax) within the range of about 80% to
about 125% of 16 ng/ml,
after single dose administration to human subjects.
[0016] In certain embodiment, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single 5 mg daily dose, and wherein said
single daily dose achieves a
mean maximum plasma concentration (Cmax) of within the range of about 80% to
about 125% of 23
ng/ml, after single dose administration to human subjects.
[0017] In certain embodiment, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single 10 mg daily dose, and wherein said
single daily dose achieves a
mean maximum plasma concentration (Cmax) within the range of about 80% to
about 125% of 36 ng/ml,
after single dose administration to human subjects.
[0018] In further embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single daily dose to achieve a mean AUC(0-
24) of from about 15.9
ng*11r/m1 to about 23.8 ng*hr/m1 for each 1 mg of compound A, after single
dose administration to human
subjects.
[0019] In further embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single 1 mg daily dose, and wherein said
single daily dose achieves a
mean AUC(0-24) within the range of about 80% to about 125% of 17 ng*hr/ml,
after single dose
administration to human subjects.
[0020] In further embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single 2.5 mg daily dose, and wherein said
single daily dose achieves a
4
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
mean AUC(0-24) within the range of about 80% to about 125% of 57 ng*hr/ml,
after single dose
administration to human subjects.
[0021] In further embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single 5 mg daily dose, and wherein said
single daily dose achieves a
mean AUC(0-24) within the range of about 80% to about 125% of 95 ng*hr/ml,
after single dose
administration to human subjects.
[00221 In further embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single 10 mg daily dose, and wherein said
single daily dose achieves a
mean AUC(0-24) within the range of about 80% to about 125% of 159 ng*hr/ml,
after single dose
administration to human subjects.
[0023] In further embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single daily dose to achieve a mean AUC(0-
t) of from about 19.1
ng*hr/ml to about 51.1 ng*hr/ml for each 1 mg of compound A, after single dose
administration to human
subjects.
[0024] In further embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single 1 mg daily dose, and wherein said
single daily dose achieves a
mean AUC(0-t) within the range of about 80% to about 125% of 19 ng*hr/ml,
after single dose
administration to human subjects.
[0025] In further embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single 2.5 mg daily dose, and wherein said
single daily dose achieves a
mean AUC(0-t) within the range of about 80% to about 125% of 80 ng*hr/ml,
after single dose
administration to human subjects.
[0026] In further embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single 5 mg daily dose, and wherein said
single daily dose achieves a
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
mean AUC(0-t) within the range of about 80% to about 125% of 128 ng*hr/ml,
after single dose
administration to human subjects.
[0027] In further embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single 10 mg daily dose, and wherein said
single daily dose achieves a
mean AUC(0-t) within the range of about 80% to about 125% of 284 ng*hr/ml,
after single dose
administration to human subjects.
[0028] In further embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single daily dose to achieve a mean AUC(0-
inf) of from about 19.8
ng*hr/ml to about 53.1 ng*hr/ml for each 1 mg of compound A, after single dose
administration to human
subjects.
[0029] In further embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single 1 mg daily dose, and wherein said
single daily dose achieves a
mean AUC(0-inf) within the range of about 80% to about 125% of 20 ng*hr/ml,
after single dose
administration to human subjects.
[0030] In further embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single 2.5 mg daily dose, and wherein said
single daily dose achieves a
mean AUC(0-inf) within the range of about 80% to about 125% of 80 ng*hr/ml,
after single dose
administration to human subjects.
[0031] In further embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single 5 mg daily dose, and wherein said
single daily dose achieves a
mean AUC(0-inf) within the range of about 80% to about 125% of 149 ng*hr/ml,
after single dose
administration to human subjects.
[0032] In further embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single 10 mg daily dose, and wherein said
single daily dose achieves a
6
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
mean AUC(0-inf) within the range of about 80% to about 125% of 311 ng*hr/ml,
after single dose
administration to human subjects.
[0033] In certain embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single daily dose ranging from about 1 mg
to about 15 mg, and
wherein said single daily dose provides a mean plasma compound A concentration
of about 20 ng/ml or
less at from 8 to 10 hours after single dose administration to human subjects.
[0034] In another embodiment, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
daily dose ranging from
about 1 mg to about 15 mg.
[0035] In another embodiment, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
daily dose to achieve a mean
maximum plasma concentration (Cmax) of from about 3.0 ng/ml to about 7.2 ng/ml
for each 1 mg of the
drug, after single dose administration to human subjects.
[0036] In another embodiment, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
1 mg daily dose, and wherein
said single daily dose achieves a mean maximum plasma concentration (Cmax)
within the range of about
80% to about 125% of 5.3 ng/ml, after single dose administration to human
subjects.
[0037] In another embodiment, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
2.5 mg daily dose, and
wherein said single daily dose achieves a mean maximum plasma concentration
(Cmax) within the range
of about 80% to about 125% of 16 ng/ml, after single dose administration to
human subjects.
[0038] In another embodiment, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
5 mg daily dose, and wherein
said single daily dose achieves a mean maximum plasma concentration (Cmax)
within the range of about
7
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
80% to about 125% of 23 ng/ml, after single dose administration to human
subjects.
[0039] In another embodiment, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
10 mg daily dose, and
wherein said single daily dose achieves a mean maximum plasma concentration
(Cmax) within the range
of about 80% to about 125% of 36 ng/ml, after single dose administration to
human subjects.
[0040] In further embodiments, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
daily dose providing a mean
AUC(0-24) of from about 15.9 ng*hr/ml to about 23.8 ng*hr/ml for each 1 mg of
the drug, after single
dose administration to human subjects.
[0041] In further embodiments, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
1 mg daily dose, and wherein
said single daily dose achieves a mean AUC(0-24) within the range of about 80%
to about 125% of 17
ng*hr/ml, after single dose administration to human subjects.
[0042] In further embodiments, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
2.5 mg daily dose, and
wherein said single daily dose achieves a mean AUC(0-24) within the range of
about 80% to about 125% ,
of 57 ng*hr/ml, after single dose administration to human subjects.
[0043] In further embodiments, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
5 mg daily dose, and wherein
said single daily dose achieves a mean AUC(0-24) within the range of about 80%
to about 125% of 95
ng*hr/ml, after single dose administration to human subjects.
[0044] In further embodiments, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
10 mg daily dose, and
wherein said single daily dose achieves a mean AUC(0-24) within the range of
about 80% to about 125%
8
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
of 159ng*hr/ml, after single dose administration to human subjects.
[0045] In further embodiments, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
daily dose providing a mean
AUC(0-t) of from about 19.1 ng*hr/ml to about 51.1 ng*hr/ml for each 1 mg of
the drug, after single dose
administration to human subjects.
[0046] In further embodiments, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
1 mg daily dose, and wherein
said single daily dose achieves a mean AUC(0-t) within the range of about 80%
to about 125% of 19
ng*hr/ml, after single dose administration to human subjects.
[0047] In further embodiments, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
2.5 mg daily dose, and
wherein said single daily dose achieves a mean AUC(0-t) within the range of
about 80% to about 125%
of 80 ng*hr/ml, after single dose administration to human subjects.
[0048] In further embodiments, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
5 mg daily dose, and wherein
said single daily dose achieves a mean AUC(0-t) within the range of about 80%
to about 125% of 128
ng*hr/ml, after single dose administration to human subjects.
[0049] In further embodiments, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
10 mg daily dose, and
wherein said single daily dose achieves a mean AUC(0-t) within the range of
about 80% to about 125%
of 284 ng*hr/ml, after single dose administration to human subjects.
[0050] In further embodiments, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
daily dose providing a mean
AUC(0-inf) of from about 19.8 ng*hr/ml to about 53.1 ng*hr/ml for each 1 mg of
the drug, after single
9
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
dose administration to human subjects.
[0051] In further embodiments, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
1 mg daily dose, and wherein
said single daily dose achieves a mean AUC(0-inf) within the range of about
80% to about 125% of 20
ng*hr/ml, after single dose administration to human subjects.
[0052] In further embodiments, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
2.5 mg daily dose, and
wherein said single daily dose achieves a mean AUC(0-inf) within the range of
about 80% to about 125%
of 80 ng*hr/ml, after single dose administration to human subjects.
[0053] In further embodiments, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
5 mg daily dose, and wherein
said single daily dose achieves a mean AUC(0-inf) within the range of about
80% to about 125% of 149
ng*hr/ml, after single dose administration to human subjects.
[0054] In further embodiments, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
10 mg daily dose, and
wherein said single daily dose achieves a mean AUC(0-inf) within the range of
about 80% to about 125%
of 311 ng*hr/ml, after single dose administration to human subjects.
[0055] In certain embodiments, the present invention to provide an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
daily dose ranging from
about 1 mg to about 15 mg, and wherein said single daily dose provides a mean
plasma compound A
concentration of about 20 ng/m1 or less at from 8 to 10 hours after single
dose administration to human
subjects.
[0056] In certain embodiments, the present invention is directed to an oral
pharmaceutical dosage form
comprising a pharmaceutically acceptable excipient and an effective amount of
compound A for treating
insomnia, the dosage form providing an dissolution rate of 85 % or more in
dissolution medium (0.1
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
mol/L hydrochloric acid containing 0.5% polysorbate 80, 900 mL, 37 0.5 C)
within 30 minutes from
the onset of dissolution study using the Apparatus 2 (Paddle Apparatus, paddle
speed; 75 rpm) according
to the procedure for immediate-release dosage form in 6.10 Dissolution test of
JP16 or <711> Dissolution
of USP37.
[0057] In certain embodiments, the present invention is directed to an oral
pharmaceutical dosage form
comprising a pharmaceutically acceptable excipient and an effective amount of
compound A for treating
insomnia, the dosage form providing an dissolution rate of 85 % or more in
dissolution medium (0.1
mol/L hydrochloric acid, 900 mL, 37 0.5 C) within 15 minutes from the onset
of dissolution study
using the Apparatus 2 (Paddle Apparatus, paddle speed; 50 rpm) according to
the procedure for
immediate-release dosage form in 6.10 Dissolution test of JP16 or <711>
Dissolution of USP37.
[0058] In certain embodiments, the present invention is directed to an oral
pharmaceutical dosage form
comprising lactose as pharmaceutically acceptable excipient.
[0059] In certain embodiments, the present invention is directed to an oral
pharmaceutical dosage form
comprising low-substituted hydroxypropyl cellulose as pharmaceutically
acceptable excipient.
[0060] In certain embodiments, the present invention is directed to an oral
pharmaceutical dosage form
comprising lactose and low-substituted hydroxypropyl cellulose as
pharmaceutically acceptable excipient.
[0061] The method according to the present invention has a potential use of
the treatment of insomnia
with easy of sleepiness onset, but the avoidance of residual sleepiness and/or
the next-day impairment.
[0062] The pharmaceutical composition according to the present invention has a
potential use of an oral
solid dosage for the treatment of insomnia.
DETAILED DESCRIPTION
I. Definitions
[0063] In order the invention described herein may be more fully understood,
the following definitions
are provided for the purposes of the disclosure:
[0064] The term "effective amount" means an amount of drug of compound A that
is capable of
achieving a therapeutic effect in a human subjective in need thereof.
[0065]The term "drug of compound A" shall mean (1R, 25)-2(((2,4-
dimetylpyrimidin-5-ypoxy)
11
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
methyl)-2-(3-fluoropheny1)-N-(5-fluoropyridin-2-y0cyclopropanecarboxamide or a
pharmaceutically
acceptable salt, hydrate, solvate, polyinorph, free-base or any combination
thereof.
[0066] The term "human subject" shall mean a normal healthy male or female
volunteers and/or any
individual that presents with clinical signs and symptoms of insomnia or any
disease or disorder that
causes insomnia.
[0067] The term "insomnia" as used herein shall mean all of the description as
delineated in the
Diagnostic and Statistical Manual of Mental Disorders, 5th Edition (2013)
(hereafter referred to as
DSM-V), published by the American Psychiatric Association. The DSM-V lists the
diagnostic criteria for
insomnia as follows:
A. A predominant complaint is dissatisfaction with sleep quantity or quality,
associated with one (or
more) of the following symptoms:
1. Difficulty initiating sleep. (In children, this may manifest as difficulty
initiating sleep without
caregiver intervention.)
2. Difficulty maintaining sleep, characterized by frequent awakenings or
problems returning to sleep
after awakenings. (In children, this may manifest as difficulty returning to
sleep without caregiver
intervention.)
3. Early-morning awakening with inability to return to sleep.
B. The sleep disturbance causes clinically significant distress or impairment
in social, occupational,
educational, academic, behavioral, or other important areas of functioning.
C. The sleep difficulty occurs at least 3 nights per week
D. The sleep difficulty is present for at least 3 months.
E. The sleep difficulty occurs despite adequate opportunity for sleep.
F. The insomnia is not better explained by and does not occur exclusively
during the course of another
sleep-wake disorder (e.g, narcolepsy, breathing-related sleep disorder,
circadian rhythm sleep-wake
disorder, a parasomnia.).
G. The insomnia is not attributable to the physiological effects of a
substance (e.g., a drug of abuse, a
medication).
H. Coexisting mental disorders and medical conditions do not adequately
explain the predominant
complaint of insomnia.
[0068] Insomnia shall mean a sleep disorder characterized by symptoms
including, without limitation,
difficulty in falling asleep, difficulty in staying asleep, intermittent
wakefulness, and/or waking up too
early. The term also encompasses daytime symptoms such as sleepiness, anxiety,
impaired concentration,
impaired memory, and irritability. Types of insomnia suitable for treatment
with the compositions of the
12
CA 02964504 2017-04-13
WO 2016/063995 PC
T/JP2015/080304
present invention include, without limitation, short-term, and chronic
insomnia. The term "short-term
insomnia" refers to insomnia lasting for about two to about four weeks. The
term "chronic insomnia"
refers to insomnia lasting for at least one month or longer.
[0069] The expression "bioequivalent" or "bioequivalence" is a term of art and
is intended to be defined
in accordance with Approved Drug Products with Therapeutic Equivalence
Evaluations, 34th Edition,
which is published by the U.S Department of Health and Human Services, and is
commonly known as the
"Orange Book". Bioequivalence of different formulation of the same drug
substance involves equivalence
with respect to the rate and extent of drug absorption. The extent and rate of
absorption of the test
formulation is compared to a reference formulation in order to determine
whether the two formulations
are bioequivalent. The standard bioequivalence study is conducted in crossover
fashion by extensive
testing which includes administering single doses of the test and reference
drugs to a number of
volunteers, usually 12 to 24 healthy normal adults, and then measuring the
blood or plasma levels of the
drug over time. Detailed guidelines for establishing the bioequivalence of a
formulation with a reference
formulation have been published by the FDA Office of Generic Drugs, Division
of Bioequivalence.
[0070] Two formulations whose PK parameters such as Cmax, AUC, or Tmax differ
by -20%/+25% or
less are generally considered to be "bioequivalent". Another approach for
average bioequivalence
involves the calculation of a 90% confidence interval for the ratio of the
averages (population geometric
means) of the measures for the test and reference products. To establish BE,
the calculated confidence
interval should fall within usually 80-125% for the ratio of the product
averages. In addition to this
general approach, the others approach, including (1) logarithmic
transformation of pharmacokinetic data,
(2) methods to evaluate sequence effects and (3) methods to evaluate outlier
data, may be useful for the
establishment of bioequivalence. For example, in the above (1) the confidence
interval should fall within
usually 80-125% for the difference in the mean value of the logarithmic
converted PK parameter.
[0071] The term "sleep time" refers to the time that a subject spends
sleeping. Sleep time can be
continuous or discontinuous.
[0072] "Sleep efficiency" refers to the total sleep time a subject receives
during their time in bed. Sleep
efficiency is measured by the following equation: 100*(total sleep time
(TST)/total time in bed).
[0073] The phrase "residual sleepiness" refers to a patient's subjective
feeling of sleepiness or sedation
upon awakening, usually in the next morning after administration the hypnotic
on the evening before.
"The next-day impairment" refers to a patient's behavior to impair activities
that require alertness,
including driving, which occurs when they are awake in the next morning, but
levels of the insomnia
13
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
medicine in their blood remain high enough. The Karolinska sleepiness scale
(KSS) is one of a number of
tools used for evaluating subjective sleepiness. The KSS was originally
developed to constitute a
one-dimensional scale of sleepiness and was validated against alpha and theta
electroencephalographic
(EEG) activity as well as slow eye movement electrooculographic (El:3G)
activity (Akerstedt and Gillberg,
1990). Other subjective tests for evaluating residual sleepiness or the next
day impairment effect include a
Epworth Sleepiness Scale (ESS), a Stanford Sleepiness Scale (SSS), and a Sleep-
Wake Activity Inventory
(SWAI). Their effects also can be evaluated using one or more of a number of
tests to human subjects by
those of skill in the art to explore their memory, their attention,
information processing and psychomotor
performance, including, for example, a Digit Symbol Substitution Test (DSST),
a Psychomotor Vigilance
Test (PVT), a Choice Reaction Time test (CRT), a Sleep Latency Test (SLT), a
Visual Analog Test
(VAT), a Symbol Copying Test (SCT), a Critical Flicker Fusion threshold test
(CFF), a Simple Reaction
time test (visual or auditory; SRT), a Word Learning Test (WLT), a Critical
Tracking Test (CTT), a
Divided Attention Test (DAT), a digit or letter cancellation test, sleep
staging through polysomnographic
(PSG) measurements, Continuous Performance Task test (CPT), Multiple Sleep
Latency Test (MSLT), a
Rapid Visual Information Processing test (RVIP) and others.
[0074] The term "dosage form(s)" or "pharmaceutical dosage form(s)" shall mean
the means to administer
the drug substance (active pharmaceutical ingredient (API)), or to facilitate
dosing, administration, and
delivery of the medicine to the patient and other mammals. Dosage forms are
classified in terms of
administration routes and application sites, including, for example, oral,
topical, rectal, vaginal,
intravenous, subcutaneous, intramuscular, ophthalmic, nasal, otic and
inhalation administration.
Alternatively, dosage forms are classified in terms of physical form such as
solid, semi-solid or liquid.
Furthermore, dosage forms are subdivided based on their form, functions and
characteristics, including,
without limited, tablet, capsule or injection as described in monograph of
Japanese Pharmacopoeia 16
edition (JP16) or General Chapter <1151> Pharmaceutical Dosage Forms of U.S.
Pharmacopoeia-NF
(37)(USP37).
[0075] The terms "excipient" shall mean a typically inactive ingredient used
as a vehicle (for example,
water, capsule shell etc.), a diluent, or a component to constitute a dosage
form or pharmaceutical
composition comprising a drug such as a therapeutic agent. The term also
encompasses a typically
inactive ingredient that imparts cohesive function (i.e. binder),
disintegrating function (i.e. disintegrator),
lubricant function (lubricating agent), and/or the other function (i.e.
solvent, surfactant etc.) to the
composition.
[0076] The term "a mean" refers to a geometric mean. The pharmacokinetic
parameters such as "a mean
Cmax" or "a mean AUC" refers to the geometric mean value of a Cmax or an AUC.
14
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
[0077] The list of the abbreviations and definitions of the terms used in this
application is presented the
following.
AUC: Area under the plasma concentration-time curve
AUC(0-x): Area under the plasma concentration-time curve from time zero to x
hours after dosing
AUC(0-t): Area under the plasma concentration-time curve from time zero to
time of last quantifiable
concentration
AUC(0-inf): Area under the plasma concentration-time curve from time zero to
infinity
ANCOVA: Analysis of covariance
CI: Confidence interval
Cmax: Maximum drug concentration
Cx: plasma concentration at x hours after dosing
CV: Coefficient of variation
DSST: Digit Symbol Substitution Test
ECG: Electrocardiogram
EEG: Electroencephalogram
EMG: Electromyogram
EOG: Electrooculogram
KSS: Karolinska Sleepiness Scale
LC-MS/MS: Lipid chromatography-mass spectrometry/mass spectrometry
LPS: Latency to persistent sleep, Duration of time measured from lights off to
the first 30 seconds of PSG
recording (epoch) of 20 consecutive epochs of non-wake
LS: Least square
MAD: Multiple ascending dose
MTD: Maximum tolerated dose
PD: Pharmacodynamics
PK: Pharmacokinetic(s)
PSG: Polysomnogram, polysomnography
PVT: Psychomotor Vigilance Test
REM: Rapid eye movement
RT: Reaction time
SE: Sleep efficiency, TST divided by the time in bed (mm) multiplied by 100
SAD: Single ascending dose
SD: Standard deviation
t1/2: Terminal elimination half-life
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
tmax: Time to reach maximum (peak) concentration following drug administration
TST: Total sleep time, Duration of rapid eye movement (REM) + non-REM (NREM)
sleep during Time
in Bed (TIB)
WASO: Wake after sleep onset, Duration of wakefulness from onset of persistent
sleep (LPS) to lights-on
BRIEF DESCRIPTION OF THE FIGURES
[0078] Fig.1 shows Dissolution Profiles of compound A 1 mg and 50 mg Capsules
[0079] Fig.2 shows Dissolution Profiles of compound A 1 mg, 2.5 mg, 5 mg, 10
mg and 25 mg Tablets
[0080] Fig.3 shows Comparative Dissolution Profiles between Compound A
Capsules and Tablets
Obtained in Condition I
II. Description of the Embodiments
[0081] In certain embodiments, the present invention is directed to a method
of treating insomnia,
comprising orally administering a single daily dose of compound A in an amount
from about 1 mg to
about 15 mg, and wherein said single dose provides easy sleep onset, but
avoids residual sleepiness
and/or the next-day impairment.
[0082] In certain embodiments, the present invention is directed to a method
of treating insomnia,
comprising orally administering a single daily dose of compound A in an amount
from about 1 mg to
about 15 mg to achieve a mean maximum plasma concentration (Cmax) of from
about 3.0 ng/ml to about
108 ng/ml. Administration of the single daily dose achieves a mean AUC(0-24)
of from about 15.9
ng*hr/m1 to about 356.4 ng*hr/m1; a mean AUC(0-t) of from about 19.1 ng*hr/m1
to about 766.5
ng*Iir/m1; a mean AUC(0-inf) of from about 19.8 ng*hr/m1 to about 796.5
ng*hr/m1; a mean t1/2 of from
about 12.7 to about 60 hours; and a mean time to reach maximum plasma
concentration (tmax) from
about I to about 3.25 hours are achieved.
[0083] In another embodiment, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single daily dose to achieve a mean
maximum plasma concentration
(Cmax) of from about 3.0 ng/ml to about 7.2 ng/ml for each 1 mg of compound A.
Administration of the
single daily dose achieves a mean AUC(0-24) of from about 15.9 ng*hr/m1 to
about 23.8 ng*hr/m1 for
each 1 mg of compound A; a mean AUC(0-t) of from about 19.1 ng*hr/m1 to about
51.1 ng*hr/m1 for
each 1 mg of compound A; a mean AUC(0-inf) of from about 19.8 ng*Iir/m1 to
about 53.1 ng*hr/m1 for
each 1 mg of compound A are achieved.
16
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
[0084] When a single 1 mg daily dose of compound A is administered to a human
subject, a mean
maximum plasma concentration (Cmax) of about 5.3 ng/ml; a mean AUC(0-24) of
about 17.2 ng*hr/m1; a
mean AUC(0-t) of about 19.1 ng*hr/m1; a mean AUC(0-inf) of about 19.8 ng*hr/m1
; a mean t1/2 of
about 12.7 hours; and a mean time to maximum plasma concentration (tmax) of
about 1 hours are
achieved.
[0085] When a single 2.5 mg daily dose of compound A is administered to a
human subject, a mean
maximum plasma concentration (Cmax) of from about 10 ng/ml to about 18 ng/ml;
a mean AUC(0-24) of
about 57 ng*hr/m1 to about 60 ng*hr/m1; a mean AUC(0-t) of from about 80
nehr/m1 to about 95
ng*hr/m1; a mean AUC(0-inf) of from about 80 ng*hr/m1 to about 103 ng*hr/m1; a
mean t1/2 of from
about 30 to about 37 hours; and a mean time to maximum plasma concentration
(tmax) of from about 1 to
2 hours are achieved.
[0086] When a single 5 mg daily dose of compound A is administered to a human
subject, a mean
maximum plasma concentration (Cmax) of from about 19 ng/ml to about 23 ng/ml;
a mean AUC(0-24) of
from about 95 ng*Iir/m1 to about 110 ng*hr/m1; a mean AUC(0-0 of about 128
ng*hr/m1; a mean
AUC(0-int) of about 150 ng*hr/m1; a mean t1/2 of about 31 hours; and a mean
time to maximum plasma
concentration (tmax) of from about 1 to about 2 are achieved.
[0087] When a single 10 mg daily dose of compound A is administered to a human
subject, a mean
maximum plasma concentration (Cmax) of from about 30 ng/ml to about 58 ng/ml,;
a mean AUC(0-24)
of from about 160 ng*hr/m1 to about 190 ng*hr/m1; a mean AUC(0-t) of from
about 280ng*hr/m1 to
about 510 ng*Iir/m1; a mean AUC(0-inf) of from about 310 nehr/m1 to about 530
ng*hr/m1; a mean t1/2
of from about 56 to about 60 hours; and a mean time to maximum plasma
concentration (tmax) of from
about Ito about 3.25 hours are achieved.
[0088] In certain embodiment, when a single 1 mg daily dose of compound A is
administered to a human
subject, (1) a mean maximum plasma concentration (Cmax) within the range of
about 80% to about 125%
of 5.3 ng/ml; (2) a mean AUC(0-24) within the range of about 80% to about 125%
of 17 ng*hr/m1;(3) a
mean AUC(0-t) within the range of about 80% to about 125% of 19 ng*hr/m1; (4)
a mean AUC(0-inf)
within the range of about 80% to about 125% of 19 nehr/m1 ; and/or (5) a mean
time to maximum
plasma concentration (tmax) within the range of about 80% to about 125% of 1.0
hour is/are achieved.
[0089] In another embodiment, when a single 2.5 mg daily dose of compound A is
administered to a
human subject, (1) a mean maximum plasma concentration (Cmax) within the range
of about 80% to
17
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
about 125% of 16 ng/ml; (2) a mean AUC(0-24) within the range of about 80% to
about 125% of 57
ng*hr/m1; (3) a mean AUC(0-t) within the range of about 80% to about 125% of
80 ng*hr/m1; (4) a mean
AUC(0-inf) within the range of about 80% to about 125% of 80 ng*hr/m1 ; and/or
(5) a mean time to
maximum plasma concentration (tmax) within the range of about 80% to about
125% of 1.0 hour is/are
achieved.
[0090] In another embodiment, when a single 5 mg daily dose of compound A is
administered to a human
subject, (1) a mean maximum plasma concentration (Cmax) within the range of
about 80% to about 125%
of 23 ng/ml; (2) a mean AUC(0-24) within the range of about 80% to about 125%
of 95 ng*hr/m1; (3) a
mean AUC(0-t) within the range of about 80% to about 125% of 128 ng*hr/m1; (4)
a mean AUC(0-inf)
within the range of about 80% to about 125% of 149 ng*hr/m1 ; and/or (5) a
mean time to maximum
plasma concentration (tmax) within the range of about 80% to about 125% of 1.6
hours is/are achieved.
[0091] In another embodiment, when a single 10 mg daily dose of compound A is
administered to a
human subject, (1) a mean maximum plasma concentration (Cmax) within the range
of about 80% to
about 125% of 36 ng/ml; (2) a mean AUC(0-24) within the range of about 80% to
about 125% of 159
ng*hr/m1; (3) a mean AUC(0-t) within the range of about 80% to about 125% of
284 ng*hr/m1; (4) a
mean AUC(0-inf) within the range of about 80% to about 125% of 311 ng*hr/m1 ;
and/or (5) a mean time
to maximum plasma concentration (tmax) within the range of about 80% to about
125% of 1.0 hour is/are
achieved.
[0092] In certain embodiments, when a single 2.5 mg daily dose is administered
over a period of about 14
days, a mean maximum plasma concentration (Cmax) of about 15 ng/ml; a mean
AUC(0-24) of about 120
ng*hr/m1; a mean t1/2 of about 44 hours; and a mean time to maximum plasma
concentration (tmax) of
about 2 hours are achieved.
[0093] In certain embodiments, when a single 5 mg daily dose is administered
over a period of about 14
days, a mean maximum plasma concentration (Cmax) of about 24 ng/ml; a mean
AUC(0-24) of about 190
ng*hr/m1; a mean t1/2 of about 46 hours; and a mean time to maximum plasma
concentration (tmax) of
about 1 hour are achieved.
[0094] In certain embodiments, when a single 10 mg daily dose is administered
over a period of about 14
days, a mean maximum plasma concentration (Cmax) of about 47 ng/ml; a mean
AUC(0-24) of about 360
ng*hr/m1; a mean t1/2 of about 55 hours; and a mean time to maximum plasma
concentration (tmax) of
about 2 hours are achieved.
18 =
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
[0095] In certain embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single daily dose to provide a mean plasma
compound A concentration
of about 20 ng/ml or less, at from 8 to 10 hours after single dose
administration to human subjects.
[0096] In certain embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single daily dose to provide a mean plasma
compound A concentration
of about 18 ng/ml or less, at from 8 to 10 hours after single dose
administration to human subjects.
[0097] In certain embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single daily dose to provide a mean plasma
compound A concentration
of about 15 ng/ml or less, at from 8 to 10 hours after single dose
administration to human subjects.
[0098] In certain embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single daily dose to provide a mean plasma
compound A concentration
of about 9.0 ng/ml or less, at from 8 to 10 hours after single dose
administration to human subjects.
[0099] In certain embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single daily dose to achieve a mean plasma
compound A concentration
of from about 0.4 ng/ml to about 9.0 ng/ml, at from 8 to 10 hours after single
dose administration to
human subjects.
[0100] In certain embodiments, the present invention to provide methods of
treating insomnia, comprises
administrating orally a dosage form with a therapeutically effective amount of
compound A, wherein said
therapeutically effective amount is single daily dose ranging from about 2.5
mg to about 10 mg, and
wherein said single daily dose achieves a mean plasma compound A concentration
of from about 1.8 ng
/ml to about 9.0 ng /ml at 8 hours, or from about 1.5 ng /m1 to about 5.0 ng
/m1 at 9 hours, or from about
2.0 ng /ml to about 8.0 ng /ml at 10 hours, after single dose administration
to human subjects.
[0101] In certain embodiments, the present invention provides an oral dosage
form for treating insomnia,
comprising therapeutically effective amount of compound A and at least one
pharmaceutically acceptable
excipient, wherein said therapeutically effective amount is a single dose
ranging from about 1 mg to about
19
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
15 mg, and wherein said single dose provides easy sleep onset, but avoids
residual sleepiness and/or the
next-day impairment.
[0102] The dosage form of the present invention achieves: 1) a mean maximum
plasma concentration
(Cmax) of from about 3.0 ng/ml to about 108 ng/ml ; 2) a mean AUC(0-24) of
from about 15.9 ng*hr/m1
to about 356.4 ng*hr/m1; 3) a mean t1/2 of from about 12.7 to about 60 hours;
and 4) a mean time to
maximum plasma concentration (tmax) of from about 1 to about 3.25 hours, after
single dose
administration to a human subjects.
[0103] In certain embodiments, the dosage form provides a mean maximum plasma
concentration (Cmax)
of from about 3.0 ng/ml to about 7.2 ng/ml for each 1 mg of compound A, after
single dose
administration to human subjects.
[0104] In certain embodiments, the dosage form comprises 1 mg of compound A
and provides a mean
maximum plasma concentration (Cmax) of about 5.3 ng/ml after single dose
administration to human
subject.
[0105] In certain embodiments, the dosage form comprises 2.5 mg of compound A
and provides a mean
maximum plasma concentration (Cmax) of from about 10 ng/ml to about 18 ng/ml
after single dose
administration to human subject.
[0106] In certain embodiments, the dosage form comprises 5 mg of compound A
and provides a mean
maximum plasma concentration (Cmax) of from about 19 ng/ml to about 23 ng/ml
after single dose
administration to human subject.
[0107] In certain embodiments, the dosage form comprises 10 mg of compound A
and provides a mean
maximum plasma concentration (Cmax) of from about 30 ng/ml to about 58 ng/ml
after single dose
administration to human subject.
[0108] In certain embodiments, the dosage form provides a mean AUC(0-24) of
from about 15.9
ng*hr/m1 to about 23.8 ng*hr/m1 for each 1 mg of compound A, after single dose
administration to human
subjects.
[0109] In certain embodiments, the dosage form comprises 1 mg of compound A
and provides a mean
AUC(0-24) of about 17 ng*Iir/ml, after single dose administration to human
subjects.
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
[0110] In certain embodiments, the dosage form comprises 2.5 mg of compound A
and provides a mean
AUC(0-24) of about 57 ng*hr/ml to about 60 ng*hr/ml, after single dose
administration to human
subjects.
[0111] In certain embodiments, the dosage form comprises 5 mg of compound A
and provides a mean
AUC(0-24) of about 95 ng*hr/ml to about 110 ng*hr/ml, after single dose
administration to human
subjects.
[0112] In certain embodiments, the dosage form comprises 10 mg of compound A
and provides a mean
AUC(0-24) of about 160 ng*hr/ml to about 190 ng*hr/ml, after single dose
administration to human
subjects.
[0113] In further embodiments, the dosage form provides a mean AUC(0-t) of
from about 19.1 ng*hr/ml
to about 766.5 ng*hr/ml, after single dose administration to human subjects.
[0114] In further embodiments, the dosage form comprises from 1 mg to 15 mg of
compound A and
provides a mean AUC(0-t) of from about 19.1 ng*hr/ml to about 766.5 ng*hr/ml,
after single dose
administration to human subjects.
[0115] In certain embodiments, the dosage form provides a mean AUC(0-t) of
from about 19.1 ng*hr/ml
to about 51.1 ng*hr/ml for each 1 mg of compound A, after single dose
administration to human subjects.
[0116] In certain embodiments, the dosage form comprises 1 mg of compound A
and provides a mean
AUC(0-t) of about 19 ng*hr/ml, after single dose administration to human
subjects.
[0117] In certain embodiments, the dosage form comprises 2.5 mg of compound A
and provides a mean
AUC(0-t) of from about 80 ng*hr/ml to about 95 ng*hr/ml, after single dose
administration to human
subjects.
[0118] In certain embodiments, the dosage form comprises 5 mg of compound A
and provides a mean
AUC(0-t) of about 128 ng*hr/ml, after single dose administration to human
subjects.
[0119] In certain embodiments, the dosage form comprises 10 mg of compound A
and provides a mean
AUC(0-t) of from about 280 ng*hr/ml to about 510 ng*hr/ml, after single dose
administration to human
subjects.
21
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
[0120] In further embodiments, the dosage form provides a mean AUC(0-inf) of
from about 19.8
ng*/m1 to about 796.5 ng*hr/ml, after single dose administration to human
subjects.
[0121] In certain embodiments, the dosage form comprises from about 1 mg to
about 15 mg of compound
A and provides a mean AUC(0-inf) of from about 19.8 ng*hr/ml to about 796.5
ng*hr/ml, after single
dose administration to human subjects.
[0122] In certain embodiments, the dosage form provides a mean AUC(0-inf) of
from about 19.8
ng*hr/m1 to about 53.1 ng*hr/ml for each 1 mg of compound A, after single dose
administration to human
subjects.
[0123] In certain embodiments, the dosage form comprises 1 mg of compound A
and provides a mean
AUC(0-inf) of about 19.8 ng*hr/ml, after single dose administration to human
subjects.
[0124] In certain embodiments, the dosage form comprises 2.5 mg of compound A
and provides a mean
AUC(0-inf) of from about 80 ng*hr/ml to about 103 ng*hr/ml, after single dose
administration to human
subjects.
[0125] In ceratin embodiments, the dosage form comprises 5 mg of compound A
and provides a mean
AUC(0-inf) of about 150 ng*hr/ml, after single dose administration to human
subjects.
[0126] In certain embodiments, the dosage form comprises 10 mg of compound A
and provides a mean
AUC(0-inf) of from about 310 ng*hr/ml to about 530 ng*hr/ml, after single dose
administration to human
subjects.
[0127] In certain embodiments, the dosage form provides a mean plasma compound
A concentration of
about 20 rig /ml or less, at from 8 to 10 hours after single dose
administration to human subjects.
[0128] In certain embodiments, the dosage form provides a mean plasma compound
A concentration of
about 18 ng /m1 or less, at from 8 to 10 hours after single dose
administration to human subjects.
[0129] In certain embodiments, the dosage form provides a mean plasma compound
A concentration of
about 15 ng /m1 or less, at from 8 to 10 hours after single dose
administration to human subjects.
[0130] In certain embodiments, the dosage form provides a mean plasma compound
A concentration of
about 9.0 ng /m1 or less, at from 8 to 10 hours after single dose
administration to human subjects.
22
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
[0131] In certain embodiments, the dosage form provides a mean plasma compound
A concentration of
from about 0.4 ng /ml to about 9.0 ng /ml, at from 8 to 10 hours after single
dose administration to human
subjects.
[0132] In further embodiments, the dosage form comprises a therapeutically
effective amount of
compound A and at least one pharmaceutically acceptable carrier or excipient,
wherein said
therapeutically effective amount is single daily dose ranging from about 2.5
mg to about 10 mg, and
wherein said single dose achieves a mean plasma compound A concentration of
from about 1.8 ng /m1 to
about 9.0 ng /m1 at 8 hours, or from about 1.5 ng /m1 to about 5.0 ng /m1 at 9
hours, or from about 2.0 ng
/m1 to about 8.0 ng /m1 at 10 hours, after single dose administration to human
subjects.
[0133] In certain embodiments, the dosage form comprises 1 mg of compound A
and provides an
elimination half-life (t1/2) of about 12.7 hours after single dose
administration to human subjects.
[0134] In certain embodiments, the dosage form comprises 2.5 mg of compound A
and provides an
elimination half-life (t1/2) of from about 30 to 37 hours after single dose
administration to human
subjects.
[0135] In certain embodiments, the dosage form comprises 5 mg of compound A
and provides an
elimination half-life (t1/2) of about 31 hours after single dose
administration to human subjects.
[0136] In certain embodiments, the dosage form comprises 10 mg of compound A
and provides an
elimination half-life (t1/2) of from about 56 to 60 hours after single dose
administration to human
subjects.
[0137] In certain embodiments, the dosage form comprises 1 mg of compound A
and provides a mean
time to maximum plasma concentration (tmax) of about 1 hour after single dose
administration to human
subjects.
[0138] In certain embodiments, the dosage form comprises 2.5 mg of compound A
and provides a mean
time to maximum plasma concentration (tmax) of from about 1 to about 2 hours
after single dose
administration to human subjects.
[0139] In certain embodiments, the dosage form comprises 5 mg of compound A
and provides a mean
time to maximum plasma concentration (tmax) of from about 1 to about 2 hours
after single dose
23
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
administration to human subjects.
[0140] In certain embodiments, the dosage form comprises 10 mg of compound A
and provides a mean
time to maximum plasma concentration (tmax) of from about 1 to about 3.25
hours after single dose
administration to human subjects.
[0141] In certain embodiment, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
1 mg daily dose, and wherein
said single daily dose achieves (1) a mean maximum plasma concentration (Cmax)
within the range of
about 80% to about 125% of 5.3 ng,/m1; (2) a mean AUC(0-24) within the range
of about 80% to about
125% of 17 ng*hr/m1; (3) a mean AUC(0-t) within the range of about 80% to
about 125% of 19 nehr/m1;
(4) a mean AUC(0-inf) within the range of about 80% to about 125% of 19
ng*hr/m1 ; and/or (5) a mean
time to maximum plasma concentration (tmax) within the range of about 80% to
about 125% of 1.0 hour.
[0142] In another embodiment, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
2.5 mg daily dose, and
wherein said single daily dose achieves (1) a mean maximum plasma
concentration (Cmax) within the
range of about 80% to about 125% of 16 ng/ml; (2) a mean AUC(0-24) within the
range of about 80% to
about 125% of 57 nehr/m1; (3) a mean AUC(0-t) within the range of about 80% to
about 125% of 80
ng*hr/m1; (4) a mean AUC(0-inf) within the range of about 80% to about 125% of
80 ng*hr/m1 ; and/or
(5) a mean time to maximum plasma concentration (tmax) within the range of
about 80% to about 125%
of 1.0 hour.
[0143] In another embodiment, the present invention provides an oral dosage
form for treating insomnia
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
5 mg daily dose, and wherein
said single daily dose achieves (1) a mean maximum plasma concentration (Cmax)
within the range of
about 80% to about 125% of 23 ng/ml; (2) a mean AUC(0-24) within the range of
about 80% to about
125% of 95 nehr/m1; (3) a mean AUC(0-t) within the range of about 80% to about
125% of 128
ng*hr/m1; (4) a mean AUC(0-inf) within the range of about 80% to about 125% of
149 ng*Iir/m1 ; and/or
(5) a mean time to maximum plasma concentration (tmax) within the range of
about 80% to about 125%
of 1.6 hours.
[0144] In another embodiment, the present invention provides an oral dosage
form for treating insomnia
24
CA 02964504 2017-04-13
WO 2016/063995 PC T/J
P2015/080304
comprising a therapeutically effective amount of compound A and at least one
pharmaceutically
acceptable excipient, wherein said therapeutically effective amount is single
10 mg daily dose, and
wherein said single daily dose achieves (1) a mean maximum plasma
concentration (Cmax) within the
range of about 80% to about 125% of 36 ng/ml; (2) a mean AUC(0-24) within the
range of about 80% to
about 125% of 159 ng*hr/m1; (3) a mean AUC(0-t) within the range of about 80%
to about 125% of 284
ng*hr/m1; (4) a mean AUC(0-inf) within the range of about 80% to about 125% of
311 ng*hr/m1 ; and/or
(5) a mean time to maximum plasma concentration (tmax) within the range of
about 80% to about 125%
of 1.0 hour.
[0145] In certain embodiments, the present invention is directed to an oral
pharmaceutical dosage form
comprising a pharmaceutically acceptable excipient and an effective amount of
compound A for treating
insomnia, wherein the dosage form provides an dissolution rate of 85 % or more
within 45 minutes from
the onset of dissolution study using the Apparatus 2 (Paddle Apparatus)
according to the procedure for
immediate-release dosage form in 6.10 Dissolution test of JP16 or <711>
Dissolution of USP37. The
dissolution medium (900 mL, 37 0.5 C) is chosen from 0.1 mol/L hydrochloric
acid or 0.1 mol/L
hydrochloric acid containing 0.5% polysorbate 80. The paddle speed is chosen
from 50 rpm or 75 rpm.
[0146] In certain embodiments, the present invention is directed to an oral
pharmaceutical dosage form
comprising a pharmaceutically acceptable excipient and an effective amount of
compound A for treating
insomnia, wherein the dosage form provides an dissolution rate of 85 % or more
within 30 minutes from
the onset of dissolution study using the Apparatus 2 (Paddle Apparatus)
according to the procedure for
immediate-release dosage form in 6.10 Dissolution test of 1P16 or <711>
Dissolution of USP37. The
dissolution medium (900 mL, 37 0.5 C) is chosen from 0.1 mon hydrochloric
acid or 0.1 mol/L
hydrochloric acid containing 0.5% polysorbate 80. The paddle speed is chosen
from 50 rpm or 75 rpm.
[0147] In certain embodiments, the present invention is directed to an oral
pharmaceutical dosage form
comprising a pharmaceutically acceptable excipient and an effective amount of
compound A for treating
insomnia, wherein the dosage form provides an dissolution rate of 85 % or more
within 15 minutes from
the onset of dissolution study using the Apparatus 2 (Paddle Apparatus)
according to the procedure for
immediate-release dosage form in 6.10 Dissolution test of JP16 or <711>
Dissolution of USP37. The
dissolution medium (900 mL, 37 0.5 C) is chosen from 0.1 mol/L hydrochloric
acid or 0.1 mol/L
hydrochloric acid containing 0.5% polysorbate 80. The paddle speed is chosen
from 50 rpm or 75 rpm.
[0148] In certain embodiments, the present invention is directed to an oral
pharmaceutical dosage form
comprising a pharmaceutically acceptable excipient and an effective amount of
compound A for treating
insomnia, wherein the dosage form provides an dissolution rate of 85 % or more
in dissolution medium
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
(pH1 .2, 900 mL, 37 0.5 C) within 15 minutes from the onset of dissolution
study using the Apparatus 2
(Paddle Apparatus, paddle speed;50 rpm) according to the procedure for
immediate-release dosage form
in 6.10 Dissolution test ofJP16 or <711> Dissolution of USP37.
[0149] In certain embodiments, the present invention is directed to an oral
pharmaceutical dosage form
comprising lactose as pharmaceutically acceptable excipient.
[0150] In certain embodiments, the present invention is directed to an oral
pharmaceutical dosage form
comprising low-substituted hydroxypropyl cellulose as pharmaceutically
acceptable excipient.
[0151] In certain embodiments, the present invention is directed to an oral
pharmaceutical dosage form
comprising lactose and low-substituted hydroxypropyl cellulose as
pharmaceutically acceptable excipient.
[0152] In the present invention, compound A may be in the form of free base, a
pharmaceutically
acceptable salt, hydrate, solvate, polymorph or any combination of the
foregoing.
[0153] Pharmaceutically acceptable salts may include, but are not limited to,
inorganic acid salts (for example, a sulfate, a nitrate, a perchlorate, a
phosphate, a carbonate, a
bicarbonate, a hydrofluoride, a hydrochloride, a hydrobromide, a hydroiodide);
organic carboxylates (for
example, an acetate, an oxalate, a maleate, a tartrate, a fumarate, a
citrate); organic sulfonates (for
example, a methanesulfonate, a trifluoromethanesulfonate, an ethanesulfonate,
a benzenesulfonate,
a toluenesulfonate, a camphorsulfonate); amino acid salts (for example, an
aspartate, a glutamate);
quaternary amine salts; alkaline metal salts (for example, a sodium salt, a
potassium salt); and
alkaline-earth metal salts (for example, a magnesium salt, a calcium salt).
[0154] Methods of treating insomnia of the present invention contain compound
A in a therapeutically
effective amount for treatment of insomnia when administered in accordance
with the 'teachings of the
present invention. The effective amount is single daily dose, ranging from 0.5
mg to 100 mg, from 1 mg
to 15 mg, from 2 mg to 15 mg, or from 2 mg to 10 mg.
Formulations
[0155] Dosage forms of the present invention contain compound A in a
therapeutically effective amount
for treatment of insomnia when administered in accordance with the teachings
of the present invention.
Unit dose of the effective amount in a dosage form is from 0.5 mg to 100 mg,
from 1 mg to 15 mg, from
2 mg to 15mg, or chosen from 2 mg, 2.5 mg, 4 mg, 5 mg, 8 mg, 10 mg, or 15 mg.
Unit dose is not limited
by the type of the dosage form or the number of dosage forms for single dose.
26
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
[0156] A dosage form in the present invention may constitute one or more
pharmaceutical composition
comprising compound A together with pharmaceutically acceptable excipients.
[0157] The term "composition" used herein includes a product comprising a
particular ingredient in a
particular amount and any product directly or indirectly brought about by the
combination of particular
ingredients in particular amounts. Such a term related to the pharmaceutical
composition is intended to
include a product comprising an active ingredient and an inert ingredient
constituting a carrier and include
every product directly or indirectly brought about by the combination,
complexation or aggregation of
any two or more ingredients or the dissociation, other kinds of reactions or
interaction of one or more
ingredients. Thus, the pharmaceutical composition of the present invention
includes every composition
prepared by mixing the compound of the present invention with a
pharmaceutically acceptable carrier.
The term "pharmaceutically acceptable" is used to mean that a carrier, a
diluent or a vehicle must be
compatible with other ingredients of a preparation and must be nontoxic to a
taker.
[0158] A dosage form is not limited to as previous said, preferably a solid
dosage form; more preferably
an oral solid dosage form; furthermore preferably an immediate release oral
dosage form.
[0159] The dissolution rate of compound A from the dosage form is over 85%
within 45 minutes,
preferably within 30 minutes, more preferably within 15 minutes, from the
onset of dissolution study
using the Apparatus 2 (Paddle Apparatus, paddle speed) according to the
procedure for immediate-release
dosage form in 6.10 Dissolution test of JP16 or <711> Dissolution of USP37.
The dissolution medium
(900 mL, 37 0.5 C) is chosen from 0.1 mol/L hydrochloric acid or 0.1 mol/L
hydrochloric acid
containing 0.5% polysorbate 80. The paddle speed is chose from 50 rpm or 75
rpm.
[0160] The term "immediate release" in this invention shall mean a dissolution
profile that the dissolution
rate of compound A from the dosage form is over 80%, preferably over 85% in
dissolution medium
(pH1.2, 900 mL, 37 0.5 C) within 15 minutes from the onset of dissolution
study using the Apparatus 2
(Paddle Apparatus, paddle speed; 50 rpm) according to the procedure for
immediate-release dosage form
in 6.10 Dissolution test of JP16 or <711> Dissolution of USP37.
[0161] Solid dosage forms include capsules, granules, lozenges, pellets,
pills, powders, suspensions,
tablets, preferably capsules, granules, pellets, pills, tablets.
[0162] The pharmaceutical composition of the invention may be prepared, using
standard techniques and
manufacturing processes generally known in the art. See, e.g. the monograph of
Japanese Pharmacopoeia
27
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
16 edition or General Chapter <1151> Pharmaceutical Dosage Forms of U.S.
Pharmacopoeia-NF (37).
[0163] The pharmaceutical composition for solid dosage form of the invention
may be prepared, for
example, powders is prepared by dry blending the components. For example,
Compound A, one or more
diluents, one or more optional excipients (e.g., binders and/or disintegrants,
as well as other additional
optional excipients) are blended together. The components of the blend prior
to blending, or the blend
itself, may be passed through a mesh screen, for example a 400-700 pm mesh
screen. A lubricant, which
may also be screened, is then added to the blend and blending is continued
until a homogeneous mixture
is obtained as granules. The mixture is then compressed into tablets.
Alternatively, a wet granulation
technique can be employed. For example, the active agent and excipient(s) are
blended together, for
example by using a granulator, and the powder blend is granulated with a small
volume of purified water.
The resultant wet granule is dried and passed through a mill to obtain as
granules. Furthermore, a
disintegrator and a lubricant are added to the milled granules and after
blending the resultant
homogeneous mixture is compressed into tablets. Alternatively, a vehicle such
as capsule shells is filled
with powders or granules to obtain as capsules. It will be appreciated that
modifications of the dry
blending and wet granulation techniques, including the order of addition of
the components and their
screening and blending prior to compression into tablets, may be carried out
according to principles well
known in the art.
[0164] In the case of production of tablets or granules, it may be coated with
a water-based film, for
example by spray-coating, if necessary.
[0165] Examples of diluents used herein include lactose, corn starch and
crystalline cellulose etc.
Examples of binders used herein include hydroxypropyl cellulose, hypromellose
etc. Examples of
disintegrators used herein include low-substituted hydroxypropyl cellulose,
calcium carboxymethyl
cellulose, sodium croscarmellose etc. Examples of lubricants used herein
include magnesium stearate,
calcium stearate etc. Examples of coloring agents used herein include titanium
oxide etc. Examples of
coating agents used herein include hydroxypropyl cellulose, hypromellose,
methyl cellulose etc. However,
needless to say, examples of above agents are not limited thereto.
Detailed Description of the Preferred Embodiments
EXAMPLES
[0166] The following examples illustrate various aspects of the present
invention. They are not to be
construed to limit the claims in any manner whatsoever.
Example 1
28
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
Single Dose Study (001 Study)
[0167] This was a randomized, double-blind, placebo-and active-controlled,
sequential, single-dose study.
The study consisted of two parts, Part A (healthy subjects) and Part B
(otherwise healthy subjects with
primary insomnia).
[0168] The primary objective in this study was to evaluate the safety and
tolerability of single oral doses
of compound A administered in the morning to healthy subjects, and to evaluate
selected
pharmacodynamic (PD) parameters (e.g., polysomnographically defined sleep
measures) with regard to
dose response in subjects with primary insomnia following single oral dosing
of compound A in the
evening approximately 30 minutes prior to the sleep period, compared with 10
mg zolpidem and placebo.
[0169] The secondary objective was to evaluate the safety and tolerability of
single oral doses of
compound A in otherwise healthy subjects with primary insomnia, and to assess
the pharmacokinetics
(PK) of compound A following administration of single oral doses in healthy
subjects and subjects with
primary insomnia.
[0170] Both parts of the study had two phases, the Prerandomization Phase and
the Randomization Phase.
The Prerandomization Phase lasted up to 21 days and consisted of a screening
period (Day-21 to Day-
3) and a baseline period (Day ¨2 to Day ¨1) during which each subject's study
eligibility was
determined and baseline assessments were conducted on Day-2. In the
Randomization Phase, subjects
were randomized to receive a single oral dose of either compound A or compound
A matching placebo
(Part A), and/or zolpidem, or zolpidem-matched placebo (Part B).
[0171] It was planned to screen approximately 160 healthy subjects and 250
otherwise healthy subjects
with primary insomnia in order to enroll 64 and 60 subjects specifically for
Part A and Part B,
respectively. 160 healthy subjects and 281 otherwise healthy subjects with
primary insomnia were
actually screened to enroll 64 and 58 subjects into Parts A and B,
respectively.
[0172] For Part A, 64 healthy subjects were enrolled into cohorts sequentially
in a gradual dose escalation
manner, to receive either compound A or placebo, and stratified by gender.
Each cohort comprised six
compound A- and two placebo-treated subjects. All study drugs were
administered as single doses using
one or more compound A-capsules or compound A-matched placebo capsules due to
the test dose. After
screening, subjects underwent baseline procedures and randomization on Day ¨2.
Subjects were dosed on
Day 1 in the morning after an overnight fast, 1 hour after lights-on. PK blood
samples were collected at
prespecified timepoints, and PD assessments were performed. Subjects were
administered assessments on
Day 1 predose and every 2 hours from 2 to 12 hours postdose, and each morning
on Days 2 to 6.
29
CA 02964504 2017-04-13
WO 2016/063995 PC T/J
P2015/080304
[0173] These assessments, included the Karolinska Sleepiness Scale (KSS),
Digit Symbol Substitution
Test (DSST), and Psychomotor Vigilance Test (PVT) in order to assess daytime
sleepiness, level of
alertness, and ability to concentrate. Waketime Questionnaires were
administered after PD assessments
each morning on Days 1 to Day 6. Time and duration of naps was recorded on
Days 1 and 2. Safety was
monitored throughout the study.
[0174] Doses for Part A were 1 mg, 2.5 mg, 5 mg, 10 mg, 25 mg, 50 mg, 100 mg,
and 200 mg of
compound A. Escalation to the next higher dose level did not occur until: 1)
the safety, tolerability
(including laboratory and electrocardiogram [ECG]), and available PK data from
the latest completed
cohort were reviewed in a blinded manner and 2) the available data supported
the increase to the next
dose.
[0175] For Part B, 58 otherwise healthy subjects with primary insomnia were
randomized across three
cohorts, and stratified by gender. Part B also included an active control
(zolpidem) and matching placebo.
In each cohort, there were approximately 12 subjects in the compound
A/zolpidem-matched placebo
group, approximately four subjects in the zolpidem/compound A-matched placebo
group, and
approximately four subjects in the compound A-matched placebo/zolpidem-matched
placebo group. Part
B dosing occurred in the evening, 30 minutes prior to the sleep period. The
starting dose for Part B was 3
dose levels that determined to be safe and well tolerated in Part A.
Subsequent dose levels in Part B were
determined based on the PD results of the first cohort in Part B and PD and
safety results from at least 3
completed higher dose cohorts in Part A. Each cohort in Part B was divided
into at least two groups, with
dosing of each group staggered by a minimum of 2 days.
[0176] After the initial Screening visit, eligible subjects were scheduled to
return to the clinic for 2 days
during the Screening Period to conduct screening/baseline PSGs. These two days
occurred at least 3 days
after the initial Screening visit and within a window from Day -7 to Day -6 (
2 days). The first PSG was
used to screen for sleep apnea and periodic limb movements in sleep (PLMS) and
serve as the first
baseline PSG. The second PSG was used as the second baseline PSG. Specific PSG
variables were used
to determine whether subjects met PSG inclusion criteria, and the average of
PSG variables from these
two PSGs was used as Baseline for this PD measure. Subjects who had met PSG
inclusion criteria
returned to the clinic on Day ¨1 for additional baseline procedures and
randomization. Subjects were not
allowed to nap on Day 1. On the evening of Day 1, after fasting a minimum of 3
hours, study drug was=
administered 30 minutes prior to the subject's habitual bedtime (lights-out),
as calculated from the sleep
diary for the first PSG during the Screening Period. PK blood samples were
collected at prespecified
timepoints, and PD assessments were performed. PSG was recorded on Day 1,
postdose. Subjects were
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
administered additional PD assessments each morning on Day 1 through Day 6.
These assessments
included the KSS, DSST, and PVT, in order to assess daytime sleepiness, level
of alertness, and ability to
concentrate. The KSS and DSST were also administered 5 minutes predose and 25
minutes postdose on
Day 1, just prior to lights-out. Waketime Questionnaires were administered
after the PD assessments
within 15 minutes of lights-on, on Day 1 to Day 6. Time and duration of naps
was recorded on Day 2.
Pharmacokinetic:
[0177] For subjects in Part A, Blood samples for determination of plasma
concentrations of compound A
were collected on Day 1 predose and at 0.25 (15 minutes), 0.5 (30 minutes), 1,
2, 3, 4, 5, 6, 9, 12, 24, 48,
72, 96, 120, 168, and 240 hours after oral administration of compound A, but
collected up to 72 hours in
the first three cohorts (1, 2.5 and 5 mg compound A groups). PK samples were
collected preferentially via
an indwelling venous catheter for the first 12 hours and by direct
venipuncture thereafter.
[0178] For subjects in Part B, blood samples for measurements of plasma
concentrations of compound A
were obtained by direct venipuncture on Day 1 predose and at 0.5, 9, 12, 24,
36, 60, 84, 108, 156, and 228
hours postdose.
[0179] The noncompartmental plasma PK parameters that were calculated for
compound A (as data
permitted) included, but were not limited to: Cmax (maximum drug
concentration); tmax (time to reach
maximum (peak) concentration following drug administration); AUC(0-24h) (area
under the
concentration x time curve from time zero to time 24 hours); AUC(0-t) (area
under the concentration x
time curve from time zero to time of last measurable concentration); AUC(0-
inf) (area under the
concentration x time curve from time zero to infinity); t1/2 (terminal
elimination half-life); CL/F
(apparent total body clearance of the drug from after extravascular
administration); and V/F (apparent
volume of distribution).
[0180] Plasma concentrations of compound A were measured using a validated
liquid
chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) assay.
[0181] PK parameters of compound A for subjects in Part A were summarized in
Table 1. In Part B, the
concentration-time profiles were approximately similar to their corresponding
dose groups in Part A.
[0182] Table 1 PK parameters of compound A for subjects in Part A
31
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
lmg 2.5mg 5mg 10mg 25mg 50mg 100mg 200mg
AUC(0-24) N 6 6 6 6 6 6 6 6
(ng*hImL) Mean(SD) 17.2(3.06) 56.8(21.1) 94.6(188)
159(61,9) - 654(97.6) 1110(321) 1930(588) 4080(1040)
N 6 6 6 6 6 6 6 6
Cmax(ng/mL)
Mean(SD) 5.29(1.25) 15.9(5.73) 22.7(4.39) 36.0(18,7) 108(22.0) 168(487)
264(128) 431(51.1)
AUC(0-t) N 6 6 6 6 6 6 6 6
(ng*h/inL) Mean(SD) 19.1(6.18) 80.2(32.2) 128(26.5) 284(80.7) 1450(455)
2080(775) 4490(1300) 9840(3510)
AUC(0-inf) N 5 4 5 6 6 6 6 6
(ng*h/mL) Mean(SD) 19.8(4.01) 79.7(42.0) 149(34.3) 311(90.1) 1540(518)
2150(834) 4740(1420) 10500(3690)
6 6 6 6 6 6 6 6
tmax (h)
Median 1.00 1.01 1.55 1.00 2.01 2.53 3.00 3.00
1/2
6 6 6 6 6 6 6 6
t (h)
Median 12.70 30.10 31.35 56.15 65.50 51.85 59.75
65.20
C9 ng/mL)
N 6 6 6 6 6 6 6 6
(
Mean(SD) 0.384(0.0726) 1.54(0.711) 2,68(0.942) 4.60(1.39) 21.9(4.19)
39.3(14.6) 86.0(47.5) 199(119)
Pharmacodynamic:
[0183] Part A Subjects were administered the KSS, DSST, and PVT starting 30
minutes predose on Day
1, then every 2 hours for 12 hours postdose and on Days 2 to 6 starting 30
minutes after lights-on.
Waketime Questionnaires were administered on the mornings of Days 1 to 6
following the PD
assessments.
[0184] In Part A, measures of sleepiness (KSS, DSST, PVT) indicated a general
dose-response
relationship. Pharmacodynamic response on these measures was generally maximal
at 2 hours postdose,
coinciding with Cmax. Duration of effect correlated with dose, i.e., the
effects were longer with higher
doses.
[0185] Part B subjects were administered the KSS, DSST, and PVT within 15
minutes after lights-on
specifically in that order, on each morning from Day 1 through Day 6, followed
by the Waketime
Questionnaire. The KSS and DSST were also administered 5 minutes predose and
25 minutes postdose on
Day 1 (just prior to lights-out).
[0186] In addition, PSG was performed during the Screening Period at Days -7
and -6 ( 2 days), and
postdose on Day 1. An 8-hour diagnostic PSG consisting of electroencephalogram
(EEG),
electrooculogram (EGG), electromyogram (EMG), ECUs, leg electrodes, and
measures of respiratory
function (airflow, respiratory effort, and oxygen saturation) were performed
starting at the subject's
habitual bedtime as determined from the sleep diary for the 3 nights
immediately prior to the first PSG on
Day ¨7 ( 2 days). PSG variables from this night were used to screen for sleep
apnea and PLMS. On Day
-6 ( 2) and postdose on Dayl, standard PSGs (i.e., not including leg
electrodes or measures of respiratory
function other than oxygen saturation) were performed. Specified PSG variables
from these two PSGs
conducted during the Screening Period were used to determine whether subjects
met PSG inclusion
criteria. The average of PSG variables from these two PSGs conducted during
the Screening Period were
32
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
used as baseline PSG values. The key PD parameters such as PSG LPS, TST, SE,
and WASO have been
obtained from all PSG recordings.
[0187] Polysomnography results indicated preliminary efficacy of compound A.
At 2.5 and 10 mg doses,
LPS was reduced by almost 30 minutes and at 25 mg, LPS was reduced by
approximately 45 minutes
relative to Baseline. At 2.5- and 10-mg doses, WASO was reduced by
approximately 30 minutes and at
25 mg, WASO was reduced by more than 45 minutes relative to Baseline. Relative
to zolpidem, the 2.5-
and 10- mg doses of compound A showed a similar magnitude of effect on LPS and
WASO. Relative to
Baseline, SE was improved by 11% for the 2.5-mg dose of compound A, by 13% for
the 10-mg dose, and
by 18% for the 25-mg dose. This compared to a change from Baseline in SE of 3%
for placebo treatment
and 13% for zolpidem. After a single dose of 25 mg of compound A, SE was
increased to approximately
90%. However, PD assessments indicated that some individuals at this dose
exhibited increases from
baseline on measures of next-day residual sleepiness.
[0188] There were no clinically significant next day effects of any dose of
compound A, zolpidem, or
placebo on KSS, DSST, or PVT.
Example 2
Multiple Ascending Dose Study (002 Study)
[0189] This was a single-center, randomized, double-blind, placebo-controlled,
sequential, multiple -dose
study.
[0190] Primary objective of this study was to evaluate the safety,
tolerability, and pharmacokinetics (PK)
of compound A after multiple doses administered orally, once daily in the
evening for 14 days in healthy
adult subjects. In addition, the objective of this study is to identify the
maximum tolerated dose (MTD) or
a sufficiently high tolerated dose of compound A to provide a safety margin
relative to anticipated
therapeutic dose.
[0191] A total of 48 healthy adult subjects (18 to 55 years) were to be
enrolled into 1 of 6 cohorts
sequentially in a gradual dose escalation manner, and randomized to receive
either compound A or
compound A-matched placebo in the evening 30 minutes before habitual bedtime,
and after 3 hours
fasting, for 14 days. Each cohort was to comprise 6 compound A- treated
subjects and 2 placebo-treated
subjects. Blood samples were collected for PK analysis at prespecified
timepoints, and PD assessments
were conducted.
[0192] The study had two phases, the Prerandomization Phase and the
Randomization Phase. The
33
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
Prerandomization Phase lasted up to 21 days and consisted of a screening
period (Day-21 to Day-3)
and a baseline period (Day -2 to Day -1) during which each subject' s study
eligibility was determined
and baseline assessments were conducted on Day-2. The Randomization Phase
(Days 1-28) consisted of
3 periods: Treatment (Days 1-14) during which subjects were randomized and
received daily oral doses
of either compound A or compound A-matched placebo, Inpatient Follow-up (Days
15-19), and
Outpatient Follow-up (Days 20-28) during which PK and safety assessments were
conducted.
[0193] All subjects were administered PD assessments, including the Karolinska
Sleepiness Scale (KSS),
Digit Symbol Substitution Test (DSST), and Psychomotor Vigilance Test (PVT) in
order to assess acute
sleepiness in the interval between dosing and bedtime, as well as next-day
residual sleepiness and the
level of alertness and ability to concentrate. In addition, a Waketime
Questionnaire was administered
daily in order to assess quality of sleep on the previous night.
[0194] The starting dose for this study was based on the results in the study
of Example 1. Escalation to
the next higher dose level did not occur until the safety, tolerability
(including laboratory and
electrocardiogram [ECG]), and available PK data from the latest completed
cohort and if the available
data supported the increase to the next dose.
[0195] Blood samples for determination of plasma concentrations of compound A
were collected at Day 1
predose and postdose 0.5 (30 minutes), 1, 1.5, 2, 3, 4, 5, 6, 8, 10, and 12
hours; Days 2 to 13: predose;
Day 14: predose and postdose 0.5 (30 minutes), 1, 1.5, 2, 3, 4, 5, 6, 8, 10,
and 12 hours; Day 15: 24 hours
after Day 14 dose; Days 16 to 19: 36, 60, 84, and 108 hours after Day 14 dose;
Day 21, 24, 26, 28: As
close as possible to 156, 228, 276, and 324 hours after Day 14 dose. Plasma
concentrations of compound
A were measured in the same manner as described in Example 1 and the above
noncompartmental plasma
PK parameters were calculated for compound A.
[0196] PD effects were assessed by evaluating postdose and next-day
functioning on the KSS, DSST, and
the PVT, and by self-report of sleep quality on the Waketime Questionnaire.
The KSS, DSST, and PVT
were performed starting Day ¨1 at 15 minutes before habitual bedtime; on Days
1 to 15 within 15 minutes
after habitual waketime, and at 1, 2, 4, 8, and 12 hours after habitual
waketirne, and on Days Ito 14 at 15
minutes predose and 15 minutes postdose. 24-hour Holter recordings were
started 30 minutes before
bedtime on Day ¨2 and just prior to dosing on Day 14. Extractions from these
recordings were used to
conduct ECG analyses, including the HPQT analysis. Waketime Questionnaires
were administered on
Day 1 to Day 19.
[0197] The KSS, PVT, and DSST were administered on each treatment day in the
evening predose and
34
CA 02964504 2017-04-13
WO 2016/063995 PCT/JP2015/080304
postdose to assess the acute effect of compound A on sleepiness. For these
timepoints, the daily 15
minutes predose values on the KSS, DSST, and PVT served as a baseline for that
day's 15 minutes
postdose value (daily baseline). The KSS, PVT, and DSST were also administered
throughout the
daytime hours subsequent to each dosing evening to assess the effect of
compound A on next-day residual
sleepiness. For these timepoints, the assessments taken at Day 1, 15 minutes
and 1, 2, 4, 8, and 12 hours
after habitual waketime served as the baseline for the assessments taken at
the corresponding times after
habitual waketime on Day 2 to Day 15 (time-matched baseline). Waketime
Questionnaires were
administered on Day 1 through Day 19 in the morning hours. The predose value
on Day 1 was used as the
baseline. The difference between placebo and each dose of compound A in change
from baseline at each
timepoint was calculated along with 95% confidence intervals (CIs). Potential
dose-response and time
trend were explored as data allowed.
[0198] Compound A capsules and compound A-matched placebo capsules were
available in strengths of
2.5 mg, 10 mg, and 50 mg. All study drugs were administered as daily doses
using one or more
compound A-capsules or compound A-matched placebo capsules due to the test
dose.
Pharmacokinetic
[0199] PK parameters on Dayl and Day14 were summarized in Table 2 and Table 3
respectively.
[0200] Table 2 PK parameters on Dayl
2.5mg 5mg 10mg 25mg 50mg 75mg
AUC(0-24) N 6 6 6 6 5 6
(ng*h/mL) Mean(SD) 59.4(17.5) 108(34.9) 187(47.9)
549(104) 931(253) 1260(301)
Cmax N 6 6 6 6 5 6
(ng/mL) Mean(SD) 10.1(4.26) 19.4(7.91) 30.4(13.1) 92.0(24.0) 199(81.2)
223(103)
6 6 6 6 5 6
tmax (h)
Median 2.015 1.250 3.250 1.500 2.000 3.000
6 6 6 6 5 6
C8 (ng/mL)
Mean(SD) 2.33(0.967) 4.40(1.73) 8.51(2.89) 24.1(9.26)
38.1(11.0) 54.5(21.7)
6 6 6 6 5 6
C10 (ng/mL)
Mean(SD) 2.04(0.769) 3.63(1.23) 7.72(4.13) 19.8(7.11)
32.9(10.2) 44.2(13.4)
CA 02964504 2017-04-13
WO 2016/063995 PCT/JP2015/080304
[0201] Table 3 PK parameters on Day14
2.5mg 5mg 10mg 25mg 50mg 75mg
AUC(0-24) N 6 6 6 5 5 5
(ng*h/mL) Mean(SD) 120(38.0) 186(87.5) 357(193) 1100(387) 2300(758) 3790(857)
Cmax N 6 6 6 5 5 5
(ng/mL) Mean(SD) 15.4(4.73) 24.0(10.7) 46.9(14.5) 107(38.9) 220(33.5) 420(140)
6 6 6 5 5 5
tmax (h)
Median 2.000 1.000 1.750 3.000 2.020 2.000
6 6 6 5 5 5
t1/2 (h)
Mean(SD) 43.8(13.1) 45.6(16.6) 55.0(23.8) 50.6(10.7)
55.5(21.3) 56.2(20.1)
[0202] Based on graphical assessment of dose-normalized data, Cmax increased
slightly less than in
proportion to dose for both Day 1 and Day 14 assessments. Based on dose-
normalized data, AUC(0-24h)
increased slightly less than in proportion to dose on Day 1 but increased in
approximate proportion to
dose based on Day 14 assessments. The terminal half-life for the 2.5-and 5-mg
doses were similar,
averaging approximately 45 hours. At doses of 10 mg and higher, the mean
terminal half-life was
approximately 55 hours following the last day of Day 14 dosing. Accumulation
was lower than predicted
by the terminal half-life. Based on accumulation, the effective half-life was
ranged from 16.9 to 24.7
hours for doses ranging from 2.5 to 25 mg, and 28.0 and 39.3 hours for the 50-
and 75-mg doses,
respectively.
Pharmacodynamics
[0203] Next-day residual sleepiness effect: There were dose-related increases
in both the magnitude and
duration of next day residual sleepiness as measured by the KSS, PVT, and
DSST. In the 2.5-mg and
5-mg dose groups, there was no meaningful difference from placebo indicative
of an increase in next-day
residual sleepiness groups on any assessment at any time relative to waketime
on any treatment day.
Slight differences from placebo were observed in the 10-mg dose group at
timepoints within 2 hours after
waketime on the KSS on Day 2 through Day 4. In the 25-mg dose group, the
increase in next-day residual
sleepiness was more consistent and slightly larger than in the 10-mg dose
group. The effect was again,
limited to timepoints within 2 hours after waketime, but was observed on the
KSS and to some extent on
PVT Lapses and PVT Mean RRT. The differences from placebo were most consistent
and larger on Day
2 and Day 15, compared with all other treatment days. In the 50-mg and 75-mg
dose groups, there were
consistent and relatively large differences from placebo on all assessments of
sleepiness. These
differences were of greater magnitude at timepoints within 2 hours after
waketime, but were still observed
at 4 hours and 8 hours after waketime, particularly on the PVT Mean RRT. By 12
hours after waketime,
there were no differences from placebo on any measures of sleepiness in any
dose group on any day. For
36
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
dose groups in which next-day residual sleepiness was observed (ie, 10 mg and
higher), sleepiness was
relatively greater on Days 2 to 4 vs Days 5 to 15. This pattern of lessening
sleepiness across treatment
days was generally observed, despite accumulation of compound A in plasma.
[0204] None of the items on the Waketime Questionnaire indicated a systematic
pattern of changes in
nighttime sleep in any dose group or the placebo group, with the exception
that the Quality of Sleep scale
showed a trend for more subjects in both the placebo and compound A groups to
report "restless" or
"very restless" sleep on Day 2 and especially on Day 15, relative to other
days.
Pharmacokinetic-Pharmacodynamics
[0205] For the PK-PD exploratory analysis of next-day residual sleepiness,
population PK model-derived
compound A plasma concentrations at 8, 9, and 10 hours following
administration on the evening of Day
1 were related to change from baseline on the KSS, PVT Lapses, and DSST at 15
minutes, 1 hour, and 2
hours after waketime on the morning of Day 2, respectively. At all timepoints,
both KSS and the PVT
Lapses were observed to increase more from baseline with increasing compound A
concentrations.
Concentrations below 30 ng/mL (which occur at doses below 25 mg) after 9-10
hours postdose, which
correspond to the 1-2 hours after morning awakening, were associated with
minimal or no change from
baseline on the KSS, PVT Lapses, or DSST.
Example 3
Crossover Study of Ralative Bioavailability of Tablet versus Capsule
Formulation (005 Study)
[0206] Single-center, open-label, randomized crossover study was conducted to
evaluate, in healthy adult
subjects, the bioavailability of single solid oral doses of compound A in
tablet formulation relative to
single oral doses of compound A in capsule formulation at 2.5, 10, and 25 mg.
Another objective of the
study was to evaluate the safety and tolerability of tablet formulations of
compound A at 2.5, 10, and 25
mg in healthy adult subjects. Approximately 36 subjects were randomly assigned
to one of three cohorts
(approximately 12 subjects per cohort) and received both a single dose of
compound A as a capsule
formulation and a single dose of compound A as a tablet formulation, in random
sequence, in a 1:1 ratio.
The doses were 2.5 mg, 10 mg, and 25 mg.
[0207] The study had two phases: Prerandomization and Randomization. The
Prerandomization Phase
lasted for up to 21 days and included a Screening Period and Baseline Period
A, during which eligibility
was established and baseline assessments before dosing of the first
formulation occurred. The
Randomization Phase consisted of two Treatment Periods (A and B), separated by
Baseline Period B. On
the first day of Treatment Period A, subjects received a single oral dose of
the first formulation. After the
37
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
first formulation dose, pharmacokinetic (PK) and safety assessments were
obtained throughout Treatment
Period A, and subjects completed a 20-day washout. Before dosing of the second
formulation, subjects
completed Baseline Period B assessments. Subjects then proceeded to Treatment
Period B and received a
single oral dose of the second formulation. Pharmacokinetic and safety
assessments were obtained
throughout Treatment Period B.
[0208] Blood samples for the determination of plasma concentrations of
compound A were collected at
the following times: Treatment Period A (Day 1 to Day 15): before and after
dosing at 0.5 (30 minutes), 1,
1.5, 2, 3, 4, 5, 6, 8, 12, 24, 48, 72, 120, 168, 240, and 336 hours; Treatment
Period B (Day 22 to Day 36):
before and after dosing at 0.5 (30 minutes), 1, 1.5, 2, 3, 4, 5, 6, 8, 12, 24,
48, 72, 120, 168, 240, and 336
hours.
[0209] Plasma concentrations of compound A were measured using a validated
liquid
chromatography-tandem mass spectrometry (LC-MS/MS) assay.
[0210] Noncompartmental methods were used to calculate the following plasma PK
parameters for
compound A: area under the plasma concentration-time curve from time zero to 8
hours after dosing
(AUC(0-8)), area under the plasma concentration-time curve from time zero to
72 hours after dosing
(AUC(0-72)), area under the plasma concentration-time curve from time zero to
time of the last
quantifiable concentration (AUC(0-t)), area under the plasma concentration-
time curve from time zero
extrapolated to time infmity (AUC(0-inf)), maximum observed plasma drug
concentration (Cmax),
terminal elimination half-life (t1/2), absorption lag time (tlag), and time to
reach the maximum (peak)
plasma concentration after drug administration (tmax). The primary PK
parameters were AUC(0-inf) and
Cmax. The individual PK parameters of compound A were presented in the data
listings by formulation
(tablet or capsule) and dose (2.5, 10, or 25 mg). The PK parameters except
tmax and tlag were
summarized by formulation and dose using descriptive statistics: number of
subjects, mean, SD,
coefficient of variation, geometric mean, median, minimum, and maximum. The
parameters tmax and
tlag were summarized by formulation and dose using the following descriptive
statistics: median,
minimum, maximum, and the 90% confidence interval (Cl) of the median point
estimate. The natural log
(ln)-transformed PK parameters for compound A (AUC(0-inf), Cmax, AUC(0-8),
AUC(0-72), and
AUC(04)) were compared separately by dose with a mixed-effects model with
sequence, treatment period,
and formulation as fixed effects and subjects nested within sequence as a
random effect. The ratio of
geometric least squares (LS) means (tablet formulation as test/capsule
formulation as reference) and
corresponding 90% CI were computed by exponentiation of the LS mean difference
and corresponding
90% CI.
38
CA 02964504 2017-04-13
WO 2016/063995 PCT/JP2015/080304
[0211] PK parameters of compound A were summarized in Table 4.
[0212] Table 4 PK parameters of compound A (Tablet versus Capsule)
2.5mg 10mg ' 25mg
Tablet Capsule Tablet Capsule Tablet
Capsule
12 12 12 12 15 13
AUC(0-8) (ng*h/mL)
47.1(16.3) 45.5(14.8) 178(62.2) 164(44.3)
386(67.0) 358(88.7)
Mean(SD)
AUC(0-72) (ng*h/mL)
84.3(30.8) 85.1(27.4) 341(135) 372(114)
795(199) 803(234)
Mean(SD)
AUC(0-t) (ng*h/mL)
93.2(40.0) 94.9(34.6) 455(214) 511(222)
1070(350) 1080(368)
Mean(SD)
AUC(0-inf) (ng*h/mL)
101(42.9) 103(39.3) 472(222) 531(234) 1100(366)
1110(379)
Mean(SD)
Cmax (ng/mL)
18.0(7.50) 15.9(5.93) 58.1(24.0) 49.0(16.5)
120(26.7) 105(33.0)
Mean(SD)
tmax (h) (median) 1.00 1.00 1.00 1.50 1.00 1.50
t1/2 (h)
35.1(14.6) 36.8(17.2) 59.5(19.0) 57.0(22.0)
57.6(18.7) 57.4(20.5)
Mean(SD)
C8 (ng/mL)
1.75(0.610) 1.84(0.618) 7.44(3.23) 8.90(2.70)
17.6(6.11) 19.9(7.62)
Mean(SD)
[0213] Differences between formulations (Treatment A [tablet] compared to
Treatment B [capsule]) in
AUC(0-8), AUC(0-72), AUC(0-t), and AUC(0-inf) across all dose levels were each
less than 13%.
Differences between the tablet and capsule formulations in Cmax across all
dose levels were each less
than 16%. The median tmax was observed at 1 to 1.5 hours after administration
of both the tablet and
capsule formulations across all dose levels. A trend of a 30-minute delay in
median tmax for the capsule
formulation (Treatment B) compared to the tablet formulation (Treatment A) was
observed at higher
doses. There was no observed absorption lag in either formulation at any dose
level.
[0214] Overall, the results indicate that both the rate and extent of compound
A absorption after tablet
administration are comparable to the reference capsule for all strengths
tested. Variability in the derived
PK parameters was also similar for the tablet compared to capsule treatments.
These results support the
conclusion that the relative bioavailability of the tablet at strengths of
2.5, 10, and 25 mg is similar to
corresponding strengths of the capsule. Thus, clinical transition to the
tablet formulation can be made
39
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
without dose adjustment relative to the capsule.
Example 4 (201 Study)
[0215] This was a multi-center, randomized, double-blind, adaptive design,
dose-response study in
subjects with insomnia. Subjects were randomized to 1 of 6 doses of compound A
(1 mg, 2.5 mg, 5 mg,
mg, 15 mg and 25 mg) or placebo.
[0216] The primary objectives of the study were to:
1. Identify a dose or doses of compound A that maximize efficacy and minimize
next-day residual
sleepiness in subjects with chronic insomnia at the beginning of treatment by
comparing the effect of 6
doses of compound A with placebo using a composite utility function
incorporating change from baseline
on sleep efficiency (SE) and change from baseline on the Karolinska Sleepiness
Scale (KSS) at 1 hour
after morning waketime after dosing on Day 2 and Day 3.
2. Compare the effect of 6 doses of compound A with placebo on the KSS at 1
hour after morning
waketime on Day 15 and Day 16 in subjects with chronic insomnia, in order to
confirm that the dose or
doses that maximize efficacy and minimize next-day residual sleepiness at the
beginning of treatment are
not associated with treatment unacceptable levels of next-day residual
sleepiness at the end of treatment.
[0217] The additional objectives of the study were to evaluate:
1. Efficacy at beginning of treatment:
Overall: Compare each dose level of compound A with placebo on change from
mean SE at baseline to
mean SE after dosing on Day 1 and Day 2
Sleep induction: Compare each dose level of compound A with placebo on change
from mean latency to
persistent sleep (LPS) at baseline to mean LPS after dosing on Day 1 and Day 2
Sleep maintenance: Compare each dose level of compound A with placebo on
change from mean
wakefulness after sleep onset (WASO) at baseline to mean WASO after dosing on
Day 1 and Day 2
2. Efficacy at end of treatment: Overall: Compare each dose level of compound
A with placebo on change
from mean SE at baseline to mean SE after dosing on Day 14 and Day 15
Sleep induction: Compare each dose level of compound A with placebo on change
from mean LPS at
baseline to mean LPS after dosing on Day 14 and Day 15
Sleep maintenance: Compare each dose level of compound A with placebo on
change from mean WASO
at baseline to mean WASO after dosing on Day 14 and Day 15
3. Potential habituation of efficacy from beginning to end of treatment:
Overall: Compare each dose level of compound A with placebo on change from
mean SE at baseline to
mean SE after dosing on Day 1 and Day 2 versus change from mean SE at baseline
to mean SE after
dosing on Day 14 and Day 15
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
Sleep induction: Compare each dose level of compound A with placebo on change
from mean LPS at
baseline to mean LPS after dosing on Day 1 and Day 2 versus change from mean
LPS at baseline to mean
LPS after dosing on Day 14 and Day 15
Sleep maintenance: Compare each dose level of compound A with placebo on
change from mean WASO
at baseline to mean WASO after dosing on Day 1 and Day 2 versus change from
mean WASO at baseline
to mean WASO after dosing on Day 14 and Day 15
[0218] A total of 616 subjects were screened, and 291 of these subjects were
randomized to the study; 56 to
placebo, 32 to 1 mg, 27 to 2.5 mg, 38 to 5 mg, 32 to 10 mg, 56 to 15 mg and 50
to 25 mg. 291 subjects were
contained in the Full Analysis Set, Safety Analysis Set, and PD Analysis Set.
There were 222 subjects in the
active dose groups (roughly equal over all doses, 90 to 100%) and 51 (91.1%)
in the placebo group who
completed the planned treatment regimen.
[0219] The study had 2 phases: Prerandomization and Randomization. The
Prerandomization Phase lasted up
to 21 days and consisted of a Screening Period (Days ¨21 to ¨2) and a Baseline
Period (Day ¨1). After the
Baseline Period, all eligible subjects were randomized in a double-blind
manner to receive compound A or
placebo for 15 nights during the Treatment Period (Days 1 to 15). All subjects
then received placebo in a
single-blind manner, for 2 nights (Days 16 to 17) during the Rebound Insomnia
Assessment Period (Days 16
to 18). Subjects did not receive study drug during the Follow-up Period (Days
19 to 30). All subjects came to
the clinic for screening procedures. During the Screening Period, subjects
completed the Sleep Diary each day.
Polysomnographic sleep was measured during the Screening Period on 2
consecutive nights between Day ¨9
and Day ¨3. The 8-hour polysorrmograms (PSGs) were started at the median
habitual bedtime calculated from
responses on the Sleep Diary, which were completed for 7 days before the first
PSG night. These recordings
served as both eligibility screening PSGs and as Baseline PSGs. Subjects could
leave the clinic between the
screening/Baseline PSG nights.
[0220] All subjects returned to the clinic on Day ¨1 for Baseline Period
procedures. They remained in the
clinic until Day 3. Morning assessments on Day 1 provided the Baseline values
for the KSS, the Digit
Symbol Substitution Test (DSST), and the Reaction Time Index (RTI).
Assessments at 6 hours after
waketime provided the Baseline values for the Waking Function Battery (WFB),
and the Profile of Mood
States-Brief (POMS-B). Subjects were then randomized to receive 1 of 6 doses
of compound A or
placebo for the next 15 days. Study drugs were tablets containing compound A-
matched placebo or
compound A of 1 mg, 2.5 mg, 5 mg, or 10 mg and to be ingested 30 minutes
before the median habitual
bedtime calculated from their Sleep Diary responses during the Screening
Period. An 8-hour PSG,
starting at the same bedtime as used for the screening and Baseline PSG
nights, was recorded on the first
2 treatment nights (Days 1 and 2). The Sleep Diary continued to be completed
each day in the clinic, and
41
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
assessments of insomnia severity (ISI), next-day residual effects (KSS, DSST,
and RTI) were conducted
while subjects were in the clinic. On specified study days, plasma
concentrations of compound A were
assessed while subjects were in the clinic in the morning after awakening and
at trough just before dosing.
[0221] Subjects continued to take compound A or placebo 30 minutes before
their anticipated,
self-selected bedtime and continued to complete the Sleep Diary each day while
at home during the
Treatment Period. On Day 14 of the Treatment Period, subjects returned to the
clinic. They remained in
the clinic for 4 nights and the intervening days until Day 18. Eight-hour PSGs
were recorded each night in
the clinic, to start at the median habitual bedtime calculated from responses
on the Sleep Diary completed
on Days 3 to 13. The Sleep Diary continued to be completed each day in the
clinic, and the ISI, KSS,
DSST, RTI, were administered at prespecified time points during the daytime
hours.
[0222] After the Treatment Period ended, all subjects received placebo in a
single-blind manner on the
final 2 nights spent in the clinic (Days 16 and 17). On these 2 nights, 8-hour
PSGs starting on the same
bedtime as Days 14 and 15 were recorded to assess for rebound insomnia
(Rebound Insomnia Assessment
Period).
[0223] During the Treatment period, blood samples for plasma concentrations of
compound A were
obtained within 30 minutes predose each night (except on Day 1) in the clinic
and within 1 hour of
morning waketime following each night spent in the clinic. Plasma
concentrations of compound A were
measured using a validated liquid chromatography-tandem mass spectrometry (LC-
MS/MS) assay.
[0224] The KSS was used to measure next-day residual effects at prespecified
timepoints. In this test,
subjects rate their sleepiness using the KSS, a 9-point verbally anchored
scale. Categories and scores
range from "extremely alert" (score = 1), "alert" (3), "neither alert nor
sleepy" (5), "sleepy-but no
difficulty remaining awake" (7), to "extremely sleepy-fighting sleep" (9). The
key outcome parameter for
the KSS was the score from 1 to 9.
[0225] All statistical tests were based on the 5% level of significance,
except for the Bayesian methods
used for the primary endpoint. Details of statistical methods and analyses
were specified in the Statistical
Analysis Plan (SAP) and body of the clinical study report.
[0226] The Safety Analysis Set was the group of subjects who received at least
1 dose of study drug and
had at least 1 postdose safety assessment. The Full Analysis Set (FAS) was the
group of randomized
subjects who received at least 1 dose of study drug and had at least 1
postdose primary efficacy
measurement. The PK Analysis Set was the group of randomized subjects who
received at least 1 dose of
42
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
compound A and had at least 1 quantifiable compound A concentration. The PD
Analysis Set was the
group of subjects who had sufficient PD data to derive at least 1 PD
parameter. The PK/PD Analysis Set
was the group of randomized subjects who received at least 1 dose of compound
A or placebo, and had at
least 1 quantifiable concentration of compound A concentration (active
subjects), and at least 1 postdose
PD assessment.
[0227] A difference from placebo of at least 6% in the change from baseline of
mean SE at Day 1 and
Day 2 was considered the minimum clinically significant difference (CSD).
[0228] Each dose was assessed for next-day residual sleepiness using the KSS.
A mean difference of
change from baseline in KSS at 1 hour after waketime on Day 2 and Day 3 of
less than 4 units was
incorporated into the utility function. A dose of compound A was considered to
have an acceptable KSS
at Day 15 and Day 16 if the mean difference of change from baseline in KSS at
1 hour after waketime on
Day 15 and Day 16 at this dose relative to placebo was less than 4 units.
Operationally, acceptable KSS
for Day15 and Day 16 was defmed as the lower boundary of a 90% confidence
interval (CI) being less
than 4 units (of the mean difference of change from baseline in KSS at 1 hour
after waketime at this dose
relative to placebo).
[0229] Utility Function: The utility at a dose was a function of both SE and
KSS, constructed by
specifying the 1-dimensional component for each endpoint and then combining
them multiplicatively.
Sufficient utility was defined as a Pr(Utility >1).
[0230] Maximum Utility Dose (dUmax): The dose that produced the maximum
utility score, ie, the best
combination of efficacy and residual sleepiness as judged by the utility
above.
[0231] The PK/PD Analysis Set was used to evaluate relationships between
compound A concentrations
and selected PD parameters. The relationships between exposure to compound A
and selected PD
endpoints (eg. KSS, DSST, RTI) were explored graphically and could be followed
by population PK/PD
modeling. The relationship between plasma concentrations of compound A at
predose (trough), and
within 1 hour after morning waketime, and selected PD parameters, was analyzed
using Nonmem version
7.2 or later.
Results
[0232] The summary statistics for change from baseline in SE are presented in
Table 5. All compound All
doses were statistically significant against placebo for the change from
baseline of Mean of Days 1/2.
Doses compound A 2.5 mg and above were statistically significant against
placebo for the change from
43
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
baseline of Mean of Days 14/15. There was no statistical evidence of an
increase or decrease in SE for the
change from baseline of the mean of Days 1/2 compared to Days 14/15,
indicating no loss of treatment
effect.
[0233] In Table 5, "Baseline" was defined as the mean of the screening PSG 1
and 2, within -9 to -3 days
of randomization. "LS Means Diff' refers to the differences between LS Means
of Placebo and each
compound A dose, "95% CI" means to 95% CI of LS Means Diff. "p-value" was
analyzed using analysis
of covariance (ANCOVA) with baseline as a covariate.
[0234] Table 5 Summary Statistics for Change from Baseline in SE (%)
44
CA 02964504 2017-04-13
WO 2016/063995 PCT/JP2015/080304
Compound A
Placebo
1 mg 2.5 mg 5 mg 10 mg 15 mg 25 mg
1 ,
Baseline
_
N 56 32 27 38 32 56 50
Mean 66.5 61.7 61.3 63.1 65.1 65.1 66.6
SD 9.25 12.30 14.7 12.48 11.75 ,
12.19 10.94
(A) Change from Baseline of Mean of Days 1 & 2
N 56 32 27 38 32 56 50
Mean 12.6 21.1 21.3 21.2 21.9 23.8 22.7
SD 12.18 11.21 14.1 13.20 11.92 12.22 10.98
LS Means Diff 4.57 4.44 5.74 8.09 10.06 10.13
95% CI 0.86, 2.54, 4.73, 7.20, 7.18,
1.19, 7.94
8.01 8.93 11.45 12.93 13.08 _
p-value
I 0.0083 0.0151 0.0005 <0.0001 <0.0001
<0.0001
(B) Change from Baseline of Mean of Days 14 & 15
N 52 31 27 37 31 54 46
Mean 12.3 17.5 20.7 21.0 21.7 21.2 21.4
SD 10.53 13.62 14.66 15.43 = 13.37 12.92
9.96
LS Means Diff 0.34 3.94 5.76 7.78 7.89 8.87
95% CI -3.22, 0.22, 2.40, 4.24, 4.86, 5.72,
3.90 7.66 9.12 11.32 10.92 12.02
p-value 0.8505 0.038 0.0008 <0.0001 <0.0001
<0.0001
(B)-(A) (Potential Habituation Effect)
N 52 31 27 37 31 54 46
Mean -0.1 -4.0 -0.5 0.1 -0.7 -2.3 -2.1
SD 7.19 8.19 10.51 6.89 6.88 6.60 5.39
LS Means Diff -3.3 0.3 1.0 0.3 -1.3 -1.1
95% CI -6.5, 0.0 -3.1, 3.7 -2.1, 4.0 -3.0, 3.5
-4.1, 1.4 -4.0, 1.8
1 I _____________________________________________________
p-value 0.0506 0.8790 0.5441 0.8651 0.3438
0.4493
[0235] The summary statistics for change from baseline in KSS at 1 hour post
walcetime are presented in
Table 6. The LS mean differences between placebo and compound A 1 mg to 15 mg
were not statistically
significant on the Mean of Days 2/3. Only the LS mean difference between
placebo and compound A 25
mg was statistically significant on the Mean of Days 2/3 (LS mean difference
0.47; P = 0.0393),
indicating that subjects rated themselves worse than placebo subjects. This
result was similar for the
CA 02964504 2017-04-13
WO 2016/063995 PCT/JP2015/080304
mean of Days 15/16 at 1 hour post waketime. The 2 hour post waketime
assessments for both the mean of
Days 2/3 showed statistical significance for compound A 15 mg and 25 mg, while
the mean of Days
15/16 was statistically significant for compound A 25 mg. No statistically
significant differences were
seen at the 15 min post waketime timepoints.
[0236] In Table 6, "Baseline" was defined as the time-matched value on Day I.
"LS Means Diff' refers
to the differences between LS Means of Placebo and each compound A dose, "95%
Cl" means to 95% CI
of LS Means Diff. "p-value" was analyzed using analysis of covariance (ANCOVA)
with baseline as a
covariate.
[0237] Table 6 Summary Statistics for Change from Baseline in KSS, 1 hour post
waketime
Compound A
Placebo
1 mg 2.5 mg 5 mg 10 mg 15 mg 25 mg
Baseline
N 55 32 27 38 32 56 50
Mean 4.0 4.0 4.0 4.3 3.8 4.1 3.7
SD 2.01 1.82 1.59 1.61 1.77 1.91 1.76
Median 4.0 4.0 4.0 4.0 3.5 4.0 3.0
Min, Max 1.0, 9.0 1.0, 8.0 1.0, 8.0 2.0, 8.0 1.0, 8.0
10, 8.0 1.0, 8.0
(A) Change from Baseline of Mean of Days 2 & 3
N 55 32 27 38 32 56 50
Mean -0.2 -0.2 -0.3 -0.1 0.0 0.1 0.4
SD 1.27 0.97 1.00 1.54 1.51 1.59 1.36
LS Means Diff 0.02 -0.06 0.20 0.16 0.32 0.47
95% CI -0.49, -0.61, -0.29, -0.36, -0.11,
0.02,
0.54 0.48 0.68 0.67 0.76 0.93
p-value 0.9290 0.8177 0.4304 0.5485 0.1471
0.0393
(B) Change from Baseline of Mean of Days 15 & 16
N 51 31 27 37 31 54 46
Mean -0.2 0.3 -0.1 0.1 -0.1 0.1 0.6
SD 1.43 1.22 1.12 1.39 1.57 1.72 1.51
LS Means Diff 0.51 0.13 0.43 0.01 0.38 0.68
95% Cl -0.03, -0.44, -0.09, -0.54, -0.08,
0.19,
1.06 0.70 0.94 0.55 0.85 1.17
p-value 0.0651 0.6490 0.1059 0.9818 0.1071
0.0063
46
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
[0238] The summary statistics for change from baseline in LPS are presented in
Table 7. As a result of a
non-normal distribution, the data were log¨transformed and analyzed using ANC
OVA as pre-specified.
The geometric mean ratio between placebo and compound A 1 mg was not
statistically significant for the
change from baseline of Mean of Days 1/2. The geometric mean ratios between
placebo and all other
active compound A showed evidence of statistical significance for the change
from baseline of Mean of
Days 1/2. Similar results were shown for the change from baseline of Mean of
Days 14/15.
Compound A 10 mg showed a statistical difference of change from baseline of
the mean of Days 1/2
compared to Days 14/15, showing further improvement in LPS over time. On all
other doses except
compound A 10 mg, there was no other statistical evidence of an increase or
decrease in LPS for the
change from baseline of the mean of Days 1/2 compared to Days 14/15.
[0239] In Table 7, "Baseline" was defined as the mean of the screening PSG 1
and 2, within -9 to -3 days
of randomization. "p-value" was analyzed using analysis of covariance (ANCOVA)
with baseline as a
covariate.
47
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
[0240] Table 7 Summary Statistics for Change from Baseline in LPS (min)
Placebo Compound A
I mg 2.5 mg 5 mg 10 mg 15 mg 25 mg
,
Baseline
N 56 32 27 38 32 56 50 .
Mean 58.8 69.9 73.0 70.4 67.9 72.5 64.3 ,
SD 30.58 39.09 50.94 42.66 52.43 36.12 45.91
_
Median 55.8 68.60 68.5 61.4 52.3 68.9 52.1
Min, Max 7.3, 17.0, 3.3, 5.8, 11.3, 5.5, 2.8,
150.3 160.8 187.8 164.3 218.0 188.3 217.3
(A) Change from Baseline of Mean of Dal. s 1 & 2
N 56 32 27 38 32 56 50
Mean -22.9 -42.9 -52.7 -47.7 -46.8 -51.6 -50.2
SD 44.46 41.86 50.15 39.39 46.11 36.73 43.14
Median -29.9 -42.0 -47.8 -37.4 -26.5 -49.1 -42.6
MM, Max -126.3, -136.0, -173.3, -140.5, -200.5, -
176.0, -199.0,
174.8 71.0 18.3 13.3 3.0 26.8 18.8
Geometric Mean Ratio
0.77 0.55 0.60 0.54 0.52 0.39
(compound A/placebo)
95% CI 0.54, 1.09 0.38, 0.80 0.43, 0.83 0.38, 0.76
0.38, 0.70 0.29, 0.54
p-value 0.1407 0.0018 0.0025 0,0006 <0.0001
<0.0001
(B) Change from Baseline of Mean of Da s 14 & 15
N 52 31 , 27 37 31 54 46
Mean -22.4 -41.2 -54.2 -51.7 -56.1 -51.6 -50.8
SD 29.04 34.62 44.92 41.99 45.55 36.73 40.16
Median -23.9 -34.3 -48.5 -47.3 -39.5 -49.1 -41.6
MM, Max -79.0, -107.0, -165.0, -130.8, -178.5, -
176.0, -200.0,
75.3 27.8 -1.0 28.5 2.3 26.8 1.3
Geometric Mean Ratio
0.73 0.49 0.47 0.32 0.41 0.34
(compound A /placebo)
95% Cl 0.49, 1.08 0.33, 0.75 0.32, 0.69 0.21,
0.47 0.29, 0.57 0.24, 0.48
p-value 0.1158 0.001 0.0001 <0.0001 <0.0001
<0.0001
(B)-(A) (Potential Habituation Effect)
N 52 31 27 37 31 54 46
Mean 0.57 2.7 -1.5 -4.6 -7.9 -1.1 1.4
SD , 40.85 24.33 22.12 22.34 17.41 24.57
12.05
Median 3.6 4.0 -0.8 -3.8 -5.5 -3.60 -0.5
Min, Max -234.0, -43.3, -41.3, -90.0, -75.8, -
49.5, -32.0,
66.8 68.0 62.5 38.8 22.0 74.8 35.5
Geometric Mean Ratio
0.93 0.90 0.77 0.60 0.76 0.89
(compound A/placebo)
95% CI , 0.60, 1.44 0.57, 1.41 0.51, 1.17 0.39,
0.93 0.52, 1.10 0.60, 1.31
1
p-value 0.7491 0.6354 0.2245 0.0216 0.1450
0.5493
48
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
[0241] The summary statistics for change from baseline in WASO are presented
in Table 8. All
compound A doses of 10 mg and above were statistically significant against
placebo for the change from
baseline of the mean of Days 1/2. Doses compound A 15 mg and above were
statistically significant
against placebo for the change from baseline of the mean of Days 14/15. There
was no statistical evidence
of an increase or decrease in WASO between the change from baseline of the
mean of Days 1/2 compared
to Days 14/15.
(0242] In Table 8, "Baseline" was defined as the mean of the screening PSG 1
and 2, within -9 to -3 days
of randomization. "LS Means Diff' refers to the differences between LS Means
of Placebo and each
compound A dose, "95% CI" means to 95% CI of LS Means Diff "p-value" was
analyzed using analysis
of covariance (ANCOVA) with baseline as a covariate.
49
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
[0243] Table 8 Summary Statistics for Change from Baseline in WASO (min)
Compound A
Placebo i
1 mg 2.5 mg 5 mg 10 mg 15 mg 25 mg
, ,
Baseline
, , , _______________________
N 56 32 27 38 32 56 50
J-
Mean 108.9 121.2 119.8 113.7 103.5 103.3 103.0
SD 37.52 49.59 51.18 47.96 34.35 42.90 42.55
(A) Change from Baseline of Mean of Days 1 & 2
N 56 32 27 38 32 56 50
Mean -40.8 -60.9 -51.1 -55.6 -56.7 -66.1 -62.3
SD 46.18 36.69 46.37 52.28 35.45 44.25 41.18
_
LS Means Diff -11.1 -2.3 -11.3 -19.8 -29.3 -25.8
95% CI -24.5, -16.5, -23.9, -33.2,
-40.8, -17.9 -37.6, -14.1
2.3 11.9 1.4 -6.4
p-value 0.1050 0.7501 0.0818 0.0038 <0.0001
<0.0001
(B) Change from Baseline of Mean of Days 14 & 15
N 52 31 27 37 31 54 46
Mean -38.2 -43.6 -48.9 -52.2 -48.5 -53.4 -53.9
SD 45.35 54.27 55.86 59.22 40.17 46.97 42.51
LS Means Diff 5.7 -2.3 -10.7 -14.7 -20.8 -21.5
95% CI -9.6, -18.3, -25.1, -30.0, 0.5 -33.9,-7.7
-35.1, -7.9
21.0 13.6 3.8
p-value 0.4642 0.7754 0.1461 0.0581 0.0019
0.0020
(B)-(A) (Potential Habituation Effect)
N 52 31 27 37 31 54 46
Mean 6.0 18.0 2.2 2.7 9.3 11.8 9.3
SD 31.42 34.30 44.05 29.73 25.89 26.19 23.12
LS Means' Diff 13.2 -2.9 -2.9 2.8 5.2 2.7
95% CI -0.3, -17.0, -15.7, -10.6, -6.4, 16.7
-9.3, 14.7
26.7 11.2 9.8 16.3
p-value 0.0547 0.6852 0.6498 0.6796 0.3792
0.6635
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
[0244] The PK of compound A was best described by a 2-compartment model with
elimination from the
central compartment. Apparent clearance of compound A was independent of dose
and time, indicating
linearity in PK. Measures of the PD effects of compound A included KSS, RTI,
DSST, the WFB (RTI,
Rapid Visual Processing [RVP], and Spatial Span [SSP]), POMS, melatonin
levels, and the DLMO. Due
to high variability and non-normal distribution in change from baseline of
LPS, it was not possible to
reliably model the concentration-response relationship between compound A PK
parameters and LPS.
Nonetheless, higher plasma concentrations of compound A were associated with
larger decreases in LPS,
up to approximately 10 ng/mL. This finding was consistent with the efficacy
results, where LPS was
decreased at doses of 2.5 mg and higher. Above this concentration, the
relationship appeared to reach an
asymptote, suggesting that there was no apparent additional benefit of higher
compound A concentrations
with regard to sleep onset . When modeled, WASO data were best described by
log-linear relationships
with the maximum observed concentration (Cmax). The exposure-response
relationship for WASO
showed a log-linear relationship with Cmax, such that higher concentrations of
compound A at Cmax
were associated with larger decreases in WASO. PK/PD analyses for next-day
residual sleepiness
assessments (KSS, DSST, and RTI) did not show any apparent relationship with
time-matched compound
A plasma concentrations. However, subjects whose compound A plasma
concentrations were greater than
20 ng/mL at 1 hour after waking had slightly greater increases on the KSS and
a higher incidence of AEs
of somnolence. This concentration is predicted to be achieved by most subjects
receiving doses greater
than 10 mg.
Example 5 (Formulation)
[0245] Capsules used for Example 1, 2, and 3 consist of size 2 hypromellose
capsules containing 1 mg,
2.5 mg, 10mg, or 50mg each of compound A drug substance. Compound A 25 mg
capsules which consist
of size 2 hypromellose capsules containing 25 mg compound A drug substance are
also prepared only for
the dissolution evaluation. The placebo consists of size 2 hypromellose
capsules containing 10 mg of
microcrystalline cellulose.
[0246] The components and compositions of tablets used for Examples 3 and 4
are shown in Table 9
51
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
[0247] Table 9 Components and Compositions of Compound A Tablets
Strength
Component
lmg 2.5mg 5mg 10mg 25mg
Core tablet
(Internal phase)
compound A 1.0 2.5 5.0 10.0 25.0
Lactose monohydrate 97.88 96.38 93.88 88.88 222.2
Low-substituted hydroxypropyl cellulose 10.8 10.8 10.8 10.8
27.0
Hydroxypropyl cellulose 3.6 3.6 3.6 3.6 9.0
(External phase)
Low-substituted hydroxypropyl cellulose 6.0 6.0 6.0 6.0 15.0
Magnesium stearate 0.72 0.72 0.72 0.72 1.8
Film-coated tablet
Opadry RED 9.0 9.0 9.0 9.0 15.0
Total Weight (mg) 129 129 129 129 315
[0248] Conventional wet granulation method was used for the manufacturing for
compound A
film-coated tablets. The compound A film-coated tablets were manufactured
through mixing,
wet-granulation, drying, sizing, lubrication, tableting and film-coating
process. Compound A, lactose
monohydrate and low-substituted hydroxypropyl cellulose were mixed using
mixer. The mixture was
wet-granulated using mixer with gradually adding appropriate amount of the
aqueous solution of
hydroxypropyl cellulose. The wet granules were dried using a dryer. The dried
granules are passed
through a 1.0 mm screen using a screening mill. Low-substituted hydroxypropyl
cellulose and magnesium
stearate are weighed depending on the yield of granules. The granules, low-
substituted hydroxypropyl
cellulose and magnesium stearate are lubricated together in a mixer. The
lubricated granules equivalent to
one tablet were compressed into bi-convex tablets using a tablet press. The
core tablets were coated using
a coating machine with spraying the aqueous suspension of Opadry RED.
Test Example
[0249] The dissolution test for the compound A capsules and tablets prepared
in the Example 5 was
executed using Apparatus 2 (paddle apparatus) according to JP 6.10, USP <711>,
and Ph.Eur. 2.9.3. The
capsules and tablets were tested in 900 triL of 0.1 mon hydrochloric acid
containing 0.5% polysorbate
80 at the paddle rotation speed of 75 rpm (Condition I). In addition, the
tablets were tested in 900 mL of
0.1 mon hydrochloric acid at the paddle rotation speed of 50 rpm (Condition
II). A helical wire sinker
was used in the tests for capsules. Aliquots of media were withdrawn through a
filter (pore size: 0.45 p.m)
at the prescribed time point to make sample solutions. The standard solutions
were prepared to have
52
CA 02964504 2017-04-13
WO 2016/063995
PCT/JP2015/080304
compound A concentrations corresponding to those of the sample solutions at
nominal concentration level.
The amount of compound A released was determined chromatographically compared
to the standard
solution. The dissolution conditions and HPLC conditions are provided in
Table10. Testing was carried
out on 6 capsules/tablets and their average value was indicated in each case.
Dissolution profiles of
compound A 1 mg and 50 mg capsules obtained in Condition I are presented in
Fig.1 and Table 11.
Dissolution profiles of compound A 1 mg, 2.5 mg, 5 mg, 10 mg and 25 mg tablets
obtained in Condition
II are presented in Fig.2 and Table 12. Comparative dissolution profiles
between compound A capsules
and tablets obtained in Condition I are presented in Fig 3 and Table 13. The
difference between capsules
and tablets was observed in the dissolution profiles, which was caused by the
lag time for the
disintegration of capsules.
[0250] Table 10 Dissolution Conditions and HPLC Conditions
Dissolution Conditions
Parameter Condition I Condition II
A aratus Apparatus 2 (paddle apparatus) in accordance with JP
6.10, USP <711>, and
pp
Ph. Eur. 2.9.3
Paddle rotation speed 75 rpm 50 rpm
0.1 mol/L hydrochloric acid
Dissolution medium 0.1 mol/L hydrochloric acid
containing 0.5% polysorbate 80
Medium volume 900 mL
Medium temperature 37 C
Compound A capsules: helical wire sinkers
Sinker
Compound A tablets: N/A
Compound A capsules:5, 10, 15, 30, 45 and 60 minutes
Sampling time Compound A tablets: "5, 10, 15, and 30 minutes" or "5, 10,
15, 30, and 45
minutes"
HPLC Conditions
Parameter Condition I Condition II
Detection Wavelength 283 nm
4.6-mm x 7.5-cm column that 4.6-mm x 7.5-cm column that
contains
Column
contains 3.5-um packing Li (USP) , 3- m packing Li
(USP)
Column temperature A constant temperature of about 40 C
A: Water/70% perchloric acid
(1000:1, v/v)
Water/acetonitrile/70% perchloric acid
Mobile phase B: Acetonitrile/70% perchloric acid
(1000:1, v/v) (550:450:1, v/v/v)
Isocratic flow: A=60%13-40%
Flow rate 1.0 mL/min 1.2 mL/min
Injection volume 10 p,L 504
Sample cooler temperature 25 C
Measurement time 5 minutes after injection
53
CA 02964504 2017-04-13
WO 2016/063995 PCT/JP2015/080304
[0251] Table 11 Dissolution Rates of Compound A 1 mg and 50 mg Capsules
Obtained in Condition I
Dissolution rate (%)
Samples
min 10 min 15 min 30 min 45 min 60 min
Compound Al mg capsules 0.0 50.0 86.0 96.4 96.0 95.9
Compound A 50 mg capsules 0.0 1.4 10.6 69.2 94.3 96.9
[0252] Table 12 Dissolution Rates of Compound A 1 mg, 2.5 mg, 5 mg, 10 mg, and
25 mg Tablets
Obtained in Condition II
Dissolution rate (%)
Samples
5 min 10 min 15 min 30 min 45 min 60
min
Compound A 1 mg tablets 77.0 99.7 100.5 100.6
100.4 ______--------'
Compound A 2.5 mg tablets 66.1 100.2 101.7 101.9
102.0 ______--------
Compound A 5 mg tablets 58.0 96.1 99.7 100.3 100.2
______--------
Compound A 10 mg tablets 52.2 96.2 100.6 100.9 100.9
_____---------
Compound A25 mg tablets 51.7 89.0 97.5 100.4 100.5
_____,..---------
[0253] Table 13 Dissolution Rates of Compound A Capsules and Tablets Obtained
in Condition I
Dissolution rate (%)
Samples
5 min 10 min 15 min 30 min 45 min 60
min
Compound A 1 mg capsules 0.0 50.0 86.0 96.4 96.0 95.9
Compound A 2.5 mg capsules 0.0 54.6 88.5 98.4 98.8 98.5
Compound A 10 mg capsules 0.0 41.4 80.7 96.2 96.6 96.5
Compound A 25 mg capsules 0.1 50.7 87.7 99.0 99.2 99.3
,
Compound A 50 mg capsules 0.0 1.4 10.6 69.2 943 96.9
Compound A 1 mg tablets 87.7 97.7 99.7 99.9
Compound A 25 mg tablets 83.3 100.0 102.0 102.3
54