Note: Descriptions are shown in the official language in which they were submitted.
CATALYST, PREPARING METHOD AND USE THEREOF, AND
SULFUR RECOVERING METHOD
Field of the Invention
The present invention relates to a catalyst, preparing method and use thereof,
and sulfur
recovering method using the catalyst.
Background of the Invention
The main function of the sulfur recovery process is to process the hydrogen
sulfide generated
during the processing procedure of such as petroleum, natural gas and coal-
coking and thus to
recover sulfur resources. At present, as the laws and rules of environmental
protection become
stricter and stricter worldwide, the quality of the crude oil continuously
worsens, and the natural
gas and coal chemical industries develop rapidly, the importance of the sulfur
recovery process
has been increasingly emerging.
Regarding the sulfur recovery catalyst, as one of the key factors influencing
the operation effect
of a sulfur recovery device, the operation effect thereof directly relates to
the sulfur recovery rate
of the entire sulfur recovery device, and finally influences the discharge of
sulfur dioxide in the
flue gas from the device. In April 2015, China issued Emission Standard of
Pollutants for
Petroleum Refining Industry, which regulates: the limited value of the
discharging concentration
of the sulfur dioxide from the sulfur recovery device is 400mg/m3, specific
area executes a
specific limited value of 100mg/m3, which will be executed by the existing
companies from 1
July 2017, and by the newly founded companies from 1 July 2015. Such standard
is the strictest
discharging standard in the world so far. Therefore, a higher requirement on
the performance of
the sulfur recovery catalyst is required. An excellent sulfur recovery
catalyst must have good
activity stability, and higher organo-sulfur hydrolysis activity and Claus
activity. In addition, as
the natural gas and coal chemical industry rise, the properties of the raw
materials for the sulfur
device become more complex, which also requires the sulfur recovery catalyst
to have good
activity stability and organo-sulfur hydrolysis activity.
The sulfur recovery catalyst substantially underwent three developing phases:
a natural bauxite
catalyst phase, an active aluminum oxide catalyst phase and a phase of mutual
development of
1
CA 2969445 2019-01-22
various catalysts. Earlier industrial devices use a natural bauxite catalyst,
the sulfur recovery rate
is only 80-85%, various sulfide not converted is burned and then discharged
into the atmosphere
in the form of SO2, which seriously pollute the environment. Later, the
aluminum oxide based
sulfur recover catalyst is developed, and the total sulfur recovery rate is
remarkably increased.
The sulfur recovery catalysts currently used on the industrial devices mainly
are the active
aluminum oxide catalyst, titaniferous aluminum oxide catalyst and Ti-based
catalyst. Each sulfur
recovery catalyst has its own advantages and disadvantages. The most wildly
used active
aluminum oxide based catalyst has good activity in the initial period, has a
certain extent organo-
sulfur hydrolysis activity, but the activity is reduced rapidly as the using
time increases, which is
mainly caused by catalyst sulfated poisoning. The titaniferous aluminum oxide
based catalyst has
improved organo-sulfur hydrolysis activity, but it still has the disadvantage
of easily being
sulfated poisoned. For example, CN100503034C discloses a titanium dioxide
loading method
when preparing a catalyst, and a bifunctional sulfur recovery catalyst
prepared by the method.
The catalyst, based on the weight ratio relates to: TiO2 is 5-30%, MgO is 3-
7%, and r-A1203 is
63-92%. It overcomes the hydrogen chloride pollution and corrosion generated
by the prior
titanium tetrachloride loading method. However, the main body of the catalyst
carrier thereof is
still aluminum oxide, and it has the disadvantage of being easily sulfated.
The Ti-based sulfur recovery catalyst has received increasing attention due to
its outstanding
organo-sulfur hydrolysis performance. The titanium precursor of the Ti-based
sulfur recovery
catalyst generally is the metatitanic acid generated by sulfuric acid method,
which generally
contains 3-8 wt% of sulfate radical. In order to further improve the organo-
sulfur hydrolysis
performance and activity stability of the Ti-based sulfur recovery catalyst,
those skilled in the art
still conduct a large amount of researches.
For example, CN103111305B discloses a catalyst for a Claus sulfur recovery
process,
characterized in that the catalyst carrier, according to the weight
components, zirconium oxide
20-30, titanium oxide 20-30, and silicon oxide 30-50, are mixed and pressed
into a ball shaped or
a block shaped initial blank. Then an additive, based on the weight
components, two or more of
zinc oxide 10-30, manganese oxide 10-35, chromic oxide 1-5, and iron oxide 1-
3, are pulped. The
catalyst carrier is poured into the pulp, and the additive thereof has a
proportion of 10-35% in the
catalyst carrier. It is calcined in a furnace at 700-1100 C for 1-2 hours and
then cooled. Palladium
2
CA 2969445 2019-01-22
or platinum is added to 40% ammonium nitrate solution to prepare a solution
with a concentration
of 0.5-3.0mol/L. Nickel is added to 30% the ammonium nitrate solution to
prepare a solution with
a concentration of 1.0-4.0mol/L. The aforementioned two solutions are mixed to
obtain a mixed
liquid. The calcined catalyst carrier containing the additive is poured into
the mixed liquid for
immersion, and after dried in the air, the catalyst is obtained. The preparing
process of the catalyst
is complex and the cost for the catalyst is high.
Summary of the Invention
The purpose of the present invention is to provide a novel catalyst and
preparing method thereof.
When applied to the sulfur recovery process, the catalyst has better activity
stability, and better
organo-sulfur hydrolysis activity and Claus activity, and can improve the
sulfur recovery rate of
the sulfur recovery device, and reduce discharging of sulfur dioxide in the
flue gas from the sulfur
recovery device. Preparation of the catalyst is easy to be carried out.
The inventor of the present invention found that, when the catalyst obtained
through using
titanium dioxide as a carrier, together with calcium oxide alkaline regulator,
and lutetium oxide
and/or cerium oxide active components at specific contents is used for sulfur
recovery process,
the activity stability, organo-sulfur hydrolysis activity and Claus activity
of the catalyst are
obviously increased. Moreover, preparation of the catalyst is easy to be
carried out.
A first aspect of the present invention provides a catalyst, comprising a
titanium dioxide as carrier,
lutetium oxide and/or cerium oxide, and calcium oxide, wherein based on 100%
weight of the
catalyst, the content of the titanium dioxide is 80-96 wt%, the content of
calcium oxide is 2-10
wt%, and the content of lutetium oxide and/or cerium oxide is 2-10 wt%.
A second aspect of the present invention provides a method for preparing a
catalyst, comprising:
extrusion moulding, drying and calcining a titanium precursor, a calcium
precursor, soluble salt
of lutetium and/or cerium, an extrusion aid and a binder after homogeneous
mixing; wherein the
amounts of titanium precursor, the calcium precursor, and the soluble salt of
lutetium and/or
cerium are enabled so that based on 100% weight of the obtained catalyst, the
content of titanium
dioxide is 80-96 wt%, preferably 85-95 wt%, the content of calcium oxide is 2-
10 wt%, preferably
2-8 wt%, more preferably 2-5 wt%, and the content of lutetium oxide and/or
cerium oxide is 2-
wt%, preferably 2-8 wt%, more preferably 2-5 wt%.
3
CA 2969445 2019-01-22
A third aspect of the present invention further provides a use of the
aforementioned catalyst in
sulfur recovery.
A fourth aspect of the present invention further provides a method for
recovering sulfur,
comprising: under Claus reaction conditions and in the presence of the
aforementioned catalyst,
contacting acid gas and oxygen-containing gas, to obtain sulfur and Claus tail
gas.
The catalyst of the present invention, taking lutetium and/or cerium as active
ingredients, titanium
dioxide as a carrier, and calcium oxide as an alkaline regulator, with
specific contents for
cooperation, when used in the sulfur recovery process, has better activity
stability, better organo-
sulfur hydrolysis activity and Claus activity, with organo-sulfur hydrolysis
activity>99%, and
Claus activity>80%.
The method for preparing the catalyst provided in the present invention
obtains the catalyst
through kneading, extruding, drying and calcining the metatitanic acid,
calcium precursor, soluble
salt of lutetium and/or cerium, extrusion aid and binder after homogeneous
mixing. As compared
with an impregnation method, the kneading extruding method may ensure the
upper amount of
the active ingredient and the physical stability of the catalyst. According to
a preferred
embodiment of the present invention, taking the metatitanic acid prepared by
using a chlorination
method as the titanium precursor can further improve the organo-sulfur
hydrolysis activity and
Claus activity of the catalyst and further improve the activity stability of
the catalyst. The inventor
of the present invention found that, regarding the existing Ti-based sulfur
recovery catalyst, since
the sulfate radical is attached on the catalyst, the catalyst would be
sulfated easily, thereby
influencing the catalyst activity; on the other hand, a large amount of
sulfate radicals of the sulfur
recovery catalyst will remarkably influence the activity stability thereof.
The catalyst provided in the present invention can be easily prepared, and the
preparing procedure
has no secondary pollution. Using the catalyst can remarkably improve the
sulfur recovery rate
of the device, facilitate reducing the discharge of sulfur dioxide in the flue
gas from the sulfur
recovery device, and have remarkable economic benefits and social benefits
with the
environmental protection standards being gradually stricter and stricter.
4
CA 2969445 2019-01-22
Brief Description of Drawings
The accompanying drawings are provided here to facilitate further
understanding on the present
invention, and constitute a part of this document. They are used in
conjunction with the following
embodiments to explain the present invention, but shall not be comprehended as
constituting any
limitation to the present invention. Among the figures:
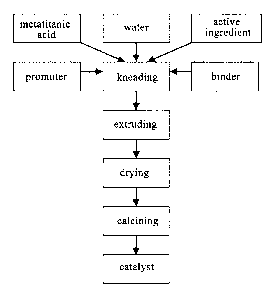
FIG. 1 is a flow chart of preparing the catalyst provide by the present
invention.
FIG. 2 is a flow chart of an evaluation device for catalyst activity.
Detailed Description of the Embodiments
Hereunder some embodiments of the present invention will be detailed. It
should be appreciated
that the embodiments described here are only provided to describe and explain
the present
invention, but shall not be deemed as constituting any limitation to the
present invention.
Limits of scope or any value revealed herein are not limited this specific
scope or value, but rather
these scope or value should be considered as values including those close to
such scope or value.
For numeric ranges, the end points of the ranges, the end points of the ranges
and the discrete
point values, and the discrete point values can be combined to obtain one or
more new numeric
ranges, which shall be deemed as having been disclosed specifically in this
document.
A first aspect of the present invention provides a catalyst, comprising a
titanium dioxide as carrier,
lutetium oxide and/or cerium oxide, and calcium oxide, wherein based on 100%
weight of the
catalyst, the content of the titanium dioxide is 80-96 wt%, the content of
calcium oxide is 2-10
wt%, and the content of lutetium oxide and/or cerium oxide is 2-10 wt%.
In the catalyst provided in the present invention, alkaline earth calcium may
increase the number
of basic sites of the catalyst which facilitates the reaction of the organo-
sulfur hydrolysis.
Based on 100% weight of the catalyst, the content of calcium oxide is 2-8 wt%,
preferably 2-5
According to an embodiment of the present invention, based on 100% weight of
the catalyst, the
content of titanium dioxide is 85-95 wt%, the content of calcium oxide is 2-8
wt%, preferably 2-5
wt%, and the content of lutetium oxide and/or cerium oxide is 2-8 wt%,
preferably 2-5 wt%.
CA 2969445 2019-01-22
In the present invention, the content of lutetium oxide and/or cerium oxide
means (1) the total
content of lutetium oxide and cerium oxide when both present; (2) the content
of lutetium oxide
when cerium oxide not present; (3) the content of cerium oxide when lutetium
oxide not present.
According to a preferred embodiment of the present invention, the catalyst
further contains
promoter. Based on 100% weight of the catalyst, the content of titanium
dioxide is 85-95%, the
content of calcium oxide is 2-5%, preferably 2.5-4%, the content of lutetium
oxide and/or cerium
oxide is 2-5%, preferably 2-4%, and the content of the promoter is 0-5%,
preferably 1-5%, more
preferably 2-4%. The promoter may be one or more of a Y-typed molecular sieve,
silicon dioxide,
and pseudo-boehmite.
In some examples, the specific surface area of the catalyst may be from 210-
250 m2/g; the pore
volume may be not less than 0.25 mL/g; and the lateral pressure strength may
be from 140-170
INI=cm-I. In specific examples, the specific surface area may be from 210-230
m2/g; the pore
volume may be 0.25-0.4 mL/g, and the lateral pressure strength may be 150-165
N. cm-1.
In the present invention, the metal content in the catalyst is measured by
using an X ray
fluorescence spectrometry (XRF) method, which using a ZSX-100e type X ray
fluorescence
spectrograph, using an Rh target, and measuring under the condition of a
current of 50 mA, and
a voltage of 50 kV.
In the present invention, the titanium dioxide preferably is anatase type
titanium dioxide. As
compared with rutile type titanium dioxide, using anatase type titanium
dioxide as a carrier can
ensure the catalyst to have higher organo-sulfur hydrolysis activity, Claus
activity, and activity
stability, and the mechanical strength of the catalyst is higher.
In the present invention, the promoter is used to improve the specific surface
area and pore volume
of the catalyst to increase the Claus activity of the catalyst. The promoter,
for example, can be
one or more of Y-typed molecular sieve, silicon dioxide, and aluminum oxide.
According to a preferred embodiment of the present invention, the content of
the sulfate ions in
the catalyst is less than 1000 ppm, preferably free from sulfate ions.
The metatitanic acid through the chlorination method can be used as a carrier
precursor to obtain
the aforementioned lower sulfate ions content.
The present invention further provides a method for preparing a catalyst,
comprising: kneading,
extruding, drying and calcining titanium precursor, calcium precursor, soluble
salt of lutetium
6
CA 2969445 2019-01-22
and/or cerium, extrusion aid and binder after homogeneous mixing, wherein the
amounts of the
metatitanic acid, calcium precursor, soluble salt of lutetium and/or cerium
are enabled, so that
based on 100% weight of the obtained catalyst, the content of titanium dioxide
is 80-96 wt%,
preferably 85-95 wt%, the content of calcium oxide is 2-10 wt%, preferably 2-8
wt%, more
preferably 2-5 wt%, and the content of lutetium oxide and/or cerium oxide is 2-
10 wt%, preferably
2-8 wt%, more preferably 2-5 wt%.
According to the present invention, the aforementioned titanium precursor may
be various
substances that can obtain the titanium dioxide after calcining, such as
metatitanic acid.
Preferably, the metatitanic acid is made from the chlorination method, and is
free from sulfate
radical. Further preferably the specific surface of the metatitanic acid is
not less than 210 m2/g,
and the pore volume is not less than 0.25 ml/g. Further preferably the
specific surface of the
metatitanic acid is not less than 220 m2/g, for example 220-260 m2/g, and the
pore volume is not
less than 0.28m1/g, for example, 0.28-0.35m1/g. Further preferably the
specific surface is not less
than 230 m2/g, and the pore volume is not less than 0.30 mug. Larger specific
surface area and
pore volume facilitate the catalyst to have a higher Claus activity. The
metatitanic acid meeting
the aforementioned conditions, for example, may be purchased from Shanghai
Yifu Industry Co.,
Ltd.
The catalyst of the present invention is prepared using the extrusion moulding
method. As
compared with the impregnation method, the catalyst obtained using the
extruding method has
higher mechanical strength, more even active ingredients, and larger specific
surface area and
pore volume, so that the catalyst has higher organo-sulfur hydrolysis
activity, Claus activity and
activity stability and a long service life.
The calcium precursor may be one or more of Ca(NO3)2, CaCO3, and calcium
oxalate.
The soluble lutetium salt is preferably one or more of lutetium carbonate,
lutetium nitrate, and
lutetium acetate.
The soluble cerium salt may be one or more of cerium carbonate, cerium
nitrate, and cerium
acetate.
The binder is one of acetic acid, nitric acid, citric acid, soluble glass, and
silica sol, preferably
citric acid. Based on 100% weight of the catalyst, an adding amount of the
binder may be 1-5%,
preferably 2.5-3.5%.
7
CA 2969445 2019-01-22
The extrusion aid may be one or more of sesbania gum, polyvinyl alcohol, Y-
typed molecular
sieve, starch, and citric acid, preferably sesbania gum.
Preferably, the use amounts of the extrusion aid and binder are 1-5% of the
weight of the titanium
precursor, respectively.
The amounts of the extrusion aid and binder after calcining correspond to the
amount of the
promoter in the aforementioned catalyst.
The drying temperature may be 100-150 C, preferably 120-130 C; and the drying
time may be
4-12 hours, preferably 6-10 hours.
The calcining temperature may be 340-500 C, preferably 390-460 C; and the
calcining time may
be 3-8 hours, preferably 4-6 hours. Under the aforementioned calcining
condition, the anatase
type titanium dioxide can be obtained.
According to a preferred embodiment of the present invention, as shown in FIG.
1, the method
for preparing the catalyst of the present invention includes the following
steps:
(1) selecting metatitanic acid prepared through the chlorination method as
a raw material for
preparing a carrier of the catalyst;
(2) according to the proportion of the catalyst weight, respectively
weighing and taking soluble
salt(s) of lutetium and/or cerate, calcium salt, binder and extrusion aid;
dissolving soluble
components using deionized water; evenly stifling to prepare a solution A; and
fully and
homogeneously mixing insoluble components and the metatitanic acid, to obtain
a solid material;
(3) pouring the solution A into the solid material, and fully mixing;
(4) placing the mixed materials into an extruding machine for fully
kneading, until the materials
are homogeneously mixed;
(5) extruding the materials after kneading in the extruding machine to
obtain a catalyst strip;
(6) drying the catalyst strip;
(7) calcining the dried catalyst strip to prepare the catalyst.
The amount of water is based on ensuring the soluble components and subsequent
kneading and
extruding steps to be smoothly carried out. Generally, it is 0.3-0.7 fold of
the weight of the
titanium precursor.
The standard of the catalyst strip may be selected according to requirements.
For the sulfur
recovery process, the standard of the catalyst strip preferably is 4:04 x3-
10mm.
8
CA 2969445 2019-01-22
The present invention further provides a catalyst obtained using the
aforementioned method and
a use thereof in the sulfur recovery. Based on 100% weight of the obtained
catalyst, the content
of titanium dioxide is 80-96 wt%, preferably 85-95 wt%, the content of calcium
oxide is 2-10
wt%, preferably 2-8 wt%, more preferably 2-5 wt%, and the content of lutetium
oxide and/or
cerium oxide is 2-10 wt%, preferably 2-8 wt%, more preferably 2-5 wt%.
The catalyst provided in the present invention is free from sulfate radical,
has strong sulfation
resistance, good activity stability, and good organo-sulfur hydrolysis
activity and Claus activity.
According to a preferred embodiment of the present invention, the catalyst
prepared in the present
invention is free from sulfate radical, the specific surface is greater than
200m2/g, the pore volume
is greater than 0.25 ml/g, the outer shape is a long strip, and the standard
is (1)4x3-10mm. The
catalyst has organo-sulfur hydrolysis activity>99%, and Claus activity>80%.
The catalyst provided in the present invention can be used to process acid gas
generated in
industries such as petroleum refining, natural gas purification, and coal
chemical industry, to
increase the sulfur recovery rate of the sulfur recovery device. The acid gas
generally contains
ingredients such as hydrogen sulfide, carbon dioxide, traces of light
hydrocarbons, ammonia, and
water, which is well known to those skilled in the art.
The present invention further provides a method for recovering sulfur,
comprising: under Claus
reaction conditions and in the presence of a catalyst, contacting acid gas and
oxygen-containing
gas, to obtain sulfur and Claus tail gas. The catalyst contains a titanium
dioxide as carrier, rare-
earth oxide, and alkaline earth oxide. Based on 100% weight of the catalyst,
the content of the
titanium dioxide is 80-96 wt%, the content of the alkaline earth oxide is 2-10
wt%, and the content
of the rare-earth oxide is 2-10 wt%.
The rare-earth oxide is preferably one or more of lanthanum oxide, lutetium
oxide and cerium
oxide, more preferably lutetium oxide and/or cerium oxide.
The alkaline earth oxide is preferably one or more of barium oxide, calcium
oxide and magnesium
oxide, more preferably calcium oxide and/or magnesium oxide, in particular,
preferably calcium
oxide.
According to a preferred embodiment of the present invention, the catalyst is
the catalyst provided
by the first aspect of the present invention. That is, the catalyst contains
titanium dioxide carrier,
lutetium oxide and/or cerium oxide, and calcium oxide. Based on 100% weight of
the catalyst,
9
CA 2969445 2019-01-22
the content of the titanium dioxide is 80-96 wt%, the content of calcium oxide
is 2-10 wt%, and
the content of lutetium oxide and/or cerium oxide is 2-10 wt%.
Preferably, in the acid gas, the content of hydrogen sulfide is 45-95 volume%.
The Claus reaction refers to a chemical reaction of enabling the hydrogen
sulfide to be
incompletely combusted, and then enabling the generated sulfur dioxide and
hydrogen sulfide to
be subjected to the reverse disproportionation reaction for generating sulfur
and water. The Claus
reaction conditions are conventional options in the art. The present invention
has no particular
requirements. For example, it may be the Claus reaction condition recited in
the documents
(Gengliang CHEN, et al, Claus Method in Sulfur Recovery Process Technique,
Petroleum
Industry Press, 2007).
Typically, the Claus reaction conditions include: during the stage in which
the hydrogen sulfide
is incompletely combusted, the temperature is 1000-1400 C, preferably 1100-
1350 , the
pressure is 0.010-0.040 MPa, preferably 0.020-0.030 MPa, and the retention
time is 2-8 seconds,
preferably 3-6 seconds.
During the stage in which the reverse disproportionation reaction occurs to
sulfur dioxide and
hydrogen sulfide, the temperature is 200-350 C, preferably 220-250 C, the
pressure is 0.001-
0.020 MPa, preferably 0.002-0.003 MPa, and the gaseous hourly space velocity
is 600-1200
hours-1.
In the present invention, pressure means gage pressure.
The materials obtained through the reverse disproportionation reaction can be
cooled to 130-150
, and then are subjected to gas-liquid separation to obtain liquid sulfur and
remaining gas (the
Claus tail gas).
The remaining gas can be further contacted with the catalyst under the Claus
reaction conditions
for the next stage of Claus reaction, thereby improving the conversion rate
for converting
hydrogen sulfide in the acid gas to sulfur. That is, multiple stages Claus
reactions can be carried
out. Generally, 2-4 stages Claus reaction, preferably 2 stages Claus reaction
can be carried out.
The conditions of the multiple stage Claus reactions can be the same or
different as long as the
Claus reaction can occur.
The following embodiments will further explain the present invention.
CA 2969445 2019-01-22
In the following examples, the constitution of the catalyst is measured by
using the X ray
fluorescence spectrometry (XRF) method. The X ray fluorescence spectrometry
(XRF) method
includes using a ZSX-100e type X ray fluorescence spectrograph, using an Rh
target, and
measuring under the condition of a current of 50 mA, and a voltage of 50 kV.
The pore volume and specific surface area of the catalyst and the carrier are
measured using a
low-temperature nitrogen adsorption method (see Petroleum and Chemical
Analysis Method
(RIPP experimental method)), edited by Cuiding YANG et al, Science Press,
published in 1990).
The lateral pressure strength of the catalyst is measured using HG/T2783-1996.
The titanium dioxide in the catalyst is detected whether to be the anatase
type titanium dioxide
by X Ray Diffraction (XRD) method. The results are that the Examples 1-23 are
all anatase type
titanium dioxide.
The SiO2 content in the silica sol used in the examples is 25 wt%, and is
manufactured by Qingdao
Ocean Chemical Industry Co., Ltd. The Y-typed molecular sieve is the NaY
molecular sieve
manufactured by Zibo Xinhong Chemical Trade Co., Ltd.
Example 1
Weigh and take 2304g of metatitanic acid prepared by the chlorination method
(purchased from
Shanghai Yifu Industrial Co., Ltd., the following is the same), as a raw
material for preparing the
catalyst carrier. Respectively weigh and take 71g of lutetium nitrate, 76g of
cerium nitrate, and
175g of calcium nitrate. Weigh and take 60g of citric acid as a binder, 60g of
sesbania gum as an
extrusion aid. Add a proper amount of deionized water (50g of deionized water
every 100g of
metatitanic acid) into lutetium nitrate, cerium nitrate, calcium nitrate, and
citric acid for
dissolving; evenly stir to prepare a solution A. Homogeneously mix the
sesbania gum and
metatitanic acid. Slowly pour the solution A into the mixed solid material,
and fully mix. Then
place the resulted material after mixed into an extruding machine for fully
kneading, until the
material is homogeneously mixed. Place the material after kneading into the
extruding machine
for extruding, to obtain a long strip with the standard of 04x3-10mm. Dry the
long strip of 04 x3_
lOmm under the temperature of 125 C for 8 hours. Calcine the dried long strip
of (1)4x3-10mni
under the temperature of 400 C for 5 hours to obtain a catalyst a. The
constitution and physical
and chemical properties of the catalyst are shown in TABLE 2.
11
CA 2969445 2019-01-22
Examples 2-20
Prepare the catalyst according to the method in Example 1, except that types
and ratio of the
materials and the drying and calcining conditions are shown in TABLE 1 as
follows to
respectively obtain a catalyst b to catalyst t. The constitution and physical
and chemical properties
of the catalyst are shown in TABLE 2.
Example 21
Prepare the catalyst according to the method in Example 1, except that the
metatitanic acid
prepared by the chlorination method is replaced by the metatitanic acid
prepared by the sulfuric
acid method (the content of sulfate ions is 3 wt%) with the same weight to
obtain a catalyst u.
The constitution and physical and chemical properties of the catalyst are
shown in TABLE 2.
Comparative examples 1-3
Prepare the catalyst according to the method in Exampe 1, except that types
and ratio of the
materials and the drying and calcining conditions are shown in TABLE 1 as
follows to
respectively obtain catalysts Dl-D3.
Example 22
Using an isovolumetric impregnation method to prepare a catalyst, the
constitution of the catalyst
is the same as that of Example 1, and the detail operations are as follows:
Weigh and take 1880g of titanium dioxide powder (purchased from Jinan Yuxing
Chemical
Industry Co., Ltd., free from sulfate radical, being the anatase type) for
extrusion moulding and
calcining, and then obtain a long strip of (1)4 x3-10mm as a catalyst carrier.
Weigh and take 71g
of lutetium nitrate, 76g of cerium nitrate, and 175g of calcium nitrate,
respectively. Add a proper
amount of deionized water (30g deionized water for 100g of the catalyst
carrier) into lutetium
nitrate, cerium nitrate, calcium nitrate, and citric acid for dissolving, and
evenly stir, to prepare a
solution A. Use the isovolumetric impregnation method to immerse the titanium
dioxide carrier
in the solution A, and then dry at the temperature of 125 C for 8 hours.
Calcine the dried
12
CA 2969445 2019-01-22
substance at the temperature of 400 C for 5 hours, to prepare a catalyst v.
The constitution and
physical and chemical properties of the catalyst are shown in TABLE 2.
Example 23
Prepare the catalyst according to the method in Example 22, except that
titanium dioxide is
replaced by the metatitanic acid of the same weight based on the titanium
dioxide to prepare a
catalyst w. The constitution and physical and chemical properties of the
catalyst are shown in
TABLE 2.
Example 24
Prepare the catalyst according to the method in Example 22, except that the
anatase type titanium
dioxide is replaced by the rutile type titanium dioxide of the same weight, to
prepare a catalyst x.
The constitution and physical and chemical properties of the catalyst are
shown in TABLE 2.
TABLE 1
13
CA 2969445 2019-01-22
Alkaline Dry Calcine
Carrier raw Active ingredient regulator Extrusion
Ex. No. Binder
material raw material raw aid Temperature Time Temperature
Time
material
cerium citric
metatitanic lutetium calcium sesbania
Ex. 1 nitrate acid 125V 8h 400C 5h
acid 2304g nitrate 71g nitrate 175g gum 60g
76g 60g
cerium citric
metatitanic lutetium calcium sesbania
Ex. 2 nitrate acid 125 V 8h 400 V 5h
acid 2353g nitrate 47g nitrate 117g gum 25g
50g 40g
lutetium cerium citric
metatitanic calcium sesbania
Ex. 3 nitrate nitrate acid 125V 8h 400 V 5h
acid 2206g nitrate 293g gum 100g
118g 126g 80g
lutetium cerium citric
metatitanic calcium sesbania
Ex. 4 nitrate nitrate acid 125V 8h 400 V 5h
acid 2084g nitrate 468g gum 50g
189g 151g 70g
lutetium cerium citric
metatitanic calcium sesbania
Ex. 5 nitrate nitrate acid 125V 8h 400 V 5h
acid 2157g nitrate 234g gum 80g
189g 202g 50g
lutetium cerium citric
metatitanic calcium sesbania
Ex. 6 nitrate nitrate acid 125 V 8h 400V 5h
acid 1961g nitrate 585g gum 70g
236g 252g 60g
lutetium 'citric
metatitanic calcium sesbania
Ex. 7 nitrate / acid 125V 8h 400V 5h
acid 2280g nitrate 205g gum 60g
165g 60g
cerium citric
metatitanic calcium sesbania
Ex. 8 / nitrate acid 125V 8h 400 V 5h
acid 2255g nitrate 263g gum 40g
177g 50g
cerium citric
metatitanic lutetium calcium sesbania
Ex. 9 nitrate acid 125V 8h 400r 5h
acid 2206g nitrate 47g nitrate 351g gum 100g
151g 100g
cerium citric
metatitanic lutetium calcium sesbania
Ex. 10 nitrate acid 125 V 8h 400r 5h
acid 2304g nitrate 71g nitrate 175g gum 60g
76g 60g
14
CA 2969445 2019-01-22
cerium citric
metatitanic lutetium calcium sesbania
Ex. 11 nitrate acid 125 C 8h 400 C 5h
acid 2353g nitrate 47g nitrate 117g gum 25g
50g 40g
lutetium cerium citric
metatitanic calcium sesbania
Ex. 12 nitrate nitrate acid 125 V 8h 400t 5h
acid 2206g nitrate 293g gum 100g
118g 126g 80g .
lutetium cerium citric
metatitanic calcium sesbania
Ex. 13 nitrate nitrate acid 125 V 8h 400 V 5h
acid 2084g nitrate 468g gum 50g
189g 151g 70g
,
lutetium cerium citric
metatitanic calcium sesbania
Ex. 14 nitrate nitrate acid 125 C 8h 400V 5h
acid 2157g nitrate 234g gum 80g
189g 202g 50g
lutetium cerium citric
metatitanic calcium sesbania
Ex. 15 nitrate nitrate acid 125 C 8h 400 V 5h
acid 1961g nitrate 585g gum 70g
236g 252g 60g
lutetium citric
metatitanic calcium sesbania
Ex. 16 nitrate / acid 125 V 8h 400V 5h
acid 2280g nitrate 205g gum 60g
165g 60g
' cerium citric
metatitanic calcium sesbania
Ex. 17 / nitrate acid 125 t 8h 400V 5h
acid 2255g nitrate 263g gum 40g
177g 50g
cerium citric
metatitanic lutetium calcium sesbania
Ex. 18 nitrate acid 125 V 8h 400V 5h
acid 2206g nitrate 47g nitrate 351g gum 100g
151g 100g
lutetium cerium calcium acetic Y-typed
metatitanic
Ex. 19 acetate acetate carbonate acid molecular 150 V 4h 340 V
8h
acid 2280g
90g 90g 100g 25g sieve 20g
lutetium acetic acid silica
metatitanic citric acid
Ex. 20 nitrate / calcium sol 140 V 6h 360 V
6h
acid 2280g 20g
100g 110g 400g
metatitanic
same same
acid (sulfuric same as same as same as Ex. same as same as
Ex. same as Ex. same
Ex. 21 as Ex. as
acid Ex. 1 Ex. 1 1 Ex. 1 1 1 as
1 Ex. 1
method)2304g Ex. I
CA 2969445 2019-01-22
,
,
metatitanic
citric
acid (sulfuric calcium sesbania
CEx. 1 / / acid 125 C 8h
400 C 5h
acid nitrate 175g gum 80g
60g
method)2304g
acetic acid same same
same
same as same as same as same as Ex. same
as Ex.
CEx. 2 lanthanum / as Ex. as as
Example 1 Example 1 Ex. 1 1 1
150g 1 Ex. 1
Ex. 1
same as same same
same
same as same as magnesium same as
same as Ex. same as Ex.
CEx. 3 Example as Ex. as as
Example 1 Example I nitrate 175g Ex. 1 1 1
1 1 Ex. 1
Ex. I
TABLE 2
Pore Specific Active Alkaline
Catalyst Lateral pressure
Carrier Promoter
Example No. volume surface ingredient
regulator
No. strength iN=cm-I
(wt%) (wt%)
/mL=g-1 area, m2/g (wt%) (wt%)
Example 1 a 0.35 225 161 94 3 3 /
,
Example 2 b 0.33 222 153 96 2 2 /
Example 3 c 0.33 231 159 90 5 5 /
Example 4 d 0.32 226 158 85 7 8 /
Example 5 e 0.31 223 156 88 8 4 /
,
Example 6 f 0.28 218 157 80 10 10 /
Example 7 g 0.28 217 158 93 3.5 3.5 /
Example 8 h 0.3 220 156 92 3.5 4.5 /
Example 9 i 0.32 230 163 90 4 6 /
Example 10 j 0.33 224 160 93.3 3.6 3.1 /
Example 11 k 0.32 223 155 95.4 2.5 2.1 /
Example 12 I 0.3 222 156 88.6 6.2 5.2 /
Example 13 m 0.31 221 157 91.6 4.8 3.6 /
Example 14 n 0.32 225 156 91.1 4.8 4.1 /
Example 15 o 0.32 224 156 91.3 6.1 2.6 /
Example 16 p 0.29 220 163 92.3 4.1 3.6 /
16
CA 2969445 2019-01-22
Example 17 a 0.32 225 159 91.1 4.4 4.6 /
Example 18 r 0.33 222 158 94.1 3.8 2.1 /
Example 19 s 0.35 228 165 91 5 3 1
Example 20 t 0.32 222 159 90 3 2 5
Example 21 u 0.31 226 160 91 3 3 /
' D 1 3.6 CEx. 1 0.3 225 159 93 / I
CEx. 2 D2 0.28 202 150 92.8 3.8 3.4 /
,
CEx. 3 D3 0.27 205 156 94.6 3 2.4 /
Example 22 v 0.24 185 135 94 3 3 /
Example 23 w 0.25 188 140 94 3 3 /
Example 24 x 0.23 182 142 94 3 3 /
It can be seen from the data in Table 2 that, the catalyst prepared according
to the preferred
embodiment of the present invention has higher specific surface area, pore
volume and
mechanical strength.
Performance Test
The evaluation test of the activity of the sulfur recovery catalyst is carried
out on a 10m1 sulfur
micro reactor device, the reactor is made of a stainless steel tube with an
inner diameter of 20mm,
the reactor is placed in a thermostat container, and the detail process
procedure is shown in FIG.
2. Send hydrogen, oxygen, hydrogen sulfide, sulfur dioxide, nitrogen and
carbon disulfide in a
required proportion by a mass flow meter MFC into a buffer tank. Then the
above kinds of gas
were sent together with water into the reactor for the Claus reaction. The
sulfur collector collects
sulfur. The out gas is sent to a cold trap for cooling, and then enters an
alkaline cleaning system
for alkaline cleaning. The tail gas is unloaded. The filling amount of the
catalyst is 10m1, and
quartz sands with the same granularity are filled in an upper part for mixing
and preheating. The
contents of H2S, SO2, COS, and CS2 in gas at the entrance and exit of the
reactor are analyzed on
line using the Shimadu GC-2014 gas chromatograph, the sulfide is analyzed by
using the GDX-
301 supporter, the 02 content is analyzed by using the 5A molecular sieve, the
column
temperature is 120 C, a thermal conductivity detector is used, hydrogen is
used as carrier gas, and
post-column velocity is 25m1/min.
17
CA 2969445 2019-01-22
3
Take 2H2S Sx
+2H20 as an index reaction, observe the Claus activity of the catalyst,
the volume constitution of the entrance gas is H2S 2%, SO2 1%, 02 3000 ppm,
and H20 30%, and
the remaining is N2. The gaseous hourly space velocity is 2500h1, the reaction
temperature is 230
C, and the Claus Conversion Rate of the catalyst is calculated according to
the following formula:
Mo ¨ MI
11112s+so2= Mo x100%
Wherein: Mo represents the sum of the volume concentrations of H2S and SO2 at
the entrance and
MI represents the sum of the volume concentrations of H2S and SO2 at the exit.
Sample and
analyze once every hour, and the analyzed result is an average value of 10
hours.
Take CS2+ 2H20--4CO2 2H2S as an index reaction, observe the organo-sulfur
hydrolysis
activity of the catalyst, the volume constitution of entrance gas is H2S 2%,
CS2 0.6%, SO2 1%,
02 3000 ppm, and H20 30%, and the remaining is N2, the gaseous hourly space
velocity is
2500h-I, the reaction temperature is 280 C, and the CS2 hydrolysis rate of the
catalyst is calculated
according to the following formula:
Co ¨ CI
1cs2= Co x100%
Wherein: Co and Ci are respectively volume concentrations of CS2 at the
entrance and the exit.
Sample and analyze once every hour, and the analyzed result is an average
value of 10 hours.
The activity of the fresh catalyst after reacting for 5 hours and the activity
of the catalyst after
strict aging, which indicate the activity stability of the catalyst, are
evaluated by using the
aforementioned method.
The evaluation process for the sulfur recovery catalyst activity normally
always is carried out for
hours. For a fresh catalyst, 10 hours of continuous operation has no great
influence on the
using performance of the catalyst. In order to observe the influence of the
operating time on the
using performance of the catalyst, and evaluate the stability of the catalyst,
a method of man-made
strict aging is generally used to process the catalyst so that in a short
period of time, the condition
of the catalyst after using for a long period of time can be simulated.
Regarding the catalyst aged
according to the following strict aging test, the test results are equivalent
to the performance
conditions of the catalyst after used for 3 years.
18
CA 2969445 2019-01-22
The strict aging test: calcine the catalyst at 550 C for 2 hours, then contact
with mixed gas of
SO2: air: water vapor=1: 2.5: 6.5(volume ratio) at the temperature of 260 C
for 2 hours, and the
gaseous hourly space velocity is 100011-1.
The activities of the catalysts prepared according to the aforementioned
method in the
aforementioned examples and comparative examples are evaluated, and the
results are shown in
the following TABLE 3.
TABLE 3
Claus activity, % Organo-sulfur hydrolysis
activity, %
Example No. Catalyst
React for 5 hours After strict aging React
for 5 hours After strict aging
Example 1 a 81.6 66.9 99.6 90.8
Example 2 b 81.5 66.7 99.5 90.6
Example 3 c 81.5 66.8 99.4 90.7
Example 4 d 81.4 66.5 99.2 90.3
Example 5 e 81.5 66.6 99.4 90.3
Example 6 f 81.2 66.3 99.3 90.2
Example 7 g 81.3 66.4 99.4 90.4
Example 8 h 81.4 66.4 99.2 90.3
Example 9 i 81.5 66.6 99.3 90.3
Example 10 j 81.5 66.8 99.4 90.7
Example 11 k 81.4 66.6 99.2 90.3
Example 12 1 81.2 66.3 99.4 90.3
Example 13 m 81.5 66.7 99.4 90.4
Example 14 n 81.4 66.5 99.5 90.3
Example 15 o 81.5 66.7 99.4 90.3
19
CA 2969445 2019-01-22
Example 16 p 81.3 66.4 99.3 90.2
Example 17 q 81.5 66.7 99.4 90.3
Example 18 r 81.4 66.6 99.4 90.4
Example 19 s 81.8 67.3 99.7 91.7
Example 20 t 81.5 66.8 99.6 90.6
Example 21 u 80.5 65.1 98.2 88.5
CEx. 1 D1 76.5 60.2 87.6 76.8
CEx. 2 D2 76.8 60.4 88.5 77.2
CEx, 3 D3 76.7 60.5 88.8 77.0
Example 22 v 80.2 63.5 97.9 88.1
Example 23 w 80.3 63.8 98.4 88.8
Example 24 x 78.2 62.6 92.1 85.3
Note: in above Table 1 to Table 3, Ex. means example, CEx. means comparative
example.
It can be seen from the data in Tables 1-3 that, the catalyst prepared by the
method of the present
invention has higher Claus activity and organo-sulfur hydrolysis activity, and
has higher activity
stability.
While some preferred embodiments of the present invention are described above,
the present
invention is not limited to the details in those embodiments. Those skilled in
the art can make
modifications and variations to the technical scheme of the present invention,
without departing
from the spirit of the present invention. However, all these modifications and
variations shall be
deemed as falling into the protected scope of the present invention.
In addition, it should be appreciated that the technical features described in
the above
embodiments can be combined in any appropriate manner, provided that there is
no conflict
among the technical features in the combination. To avoid unnecessary
iteration, such possible
combinations are not described here in the present invention.
CA 2969445 2019-01-22
=
Moreover, different embodiments of the present invention can be combined
freely as required, as
long as the combinations don't deviate from the ideal and spirit of the
present invention. However,
such combinations shall also be deemed as falling into the scope disclosed in
the present
invention.
21
CA 2969445 2019-01-22