Note: Descriptions are shown in the official language in which they were submitted.
PHARMACEUTICAL FORMULATIONS FOR TREATING ENDOMETRIOSIS AND
UTERINE FIBROIDS
JOINT RESEARCH AGREEMENT
[0001] Subject matter disclosed in this application was made by or on
behalf of AbbVie
Inc. and/or Neurocrine Biosciences, Inc., whom are parties to a joint research
agreement that was
in effect on or before the effective filing date of this application, and such
subject matter was
made as a result of activities undertaken within the scope of the joint
research agreement.
TECHNICAL FIELD
[0002] The present invention relates to pharmaceutical compositions and
methods of use
of such compositions.
BACKGROUND
[0003] Endometriosis is a disease in which tissue normally found in the
uterine cavity
(i.e., endometrium) is found outside the uterus, usually implanted on the
peritoneal lining of the
pelvis. Endometriosis affects an estimated 1 in 10 women of reproductive age
and can cause
pain, infertility, and sexual dysfunction. Growth of endometrial tissue
outside of the uterine
cavity is believed to be estrogen-dependent. Thus, current therapies for
endometriosis are aimed
at altering estrogen levels.
[0004] Uterine fibroids (leiomyomas) are benign tumors and are highly
prevalent in
women of reproductive age. Symptoms associated with uterine fibroids most
commonly include
heavy or prolonged menstrual bleeding, pelvic pressure and pelvic organ
compression, back
pain, and adverse reproductive outcomes. Heavy menstrual bleeding (HMB;
menorrhagia,
defined as greater than 80 mL per menstrual cycle) (The Menorrhagia Research
Group.
Quantification of menstrual blood loss. The Obstetrician & Gynaecologist.
2004; 6:88-92) is
inconvenient and may lead to iron-deficiency anemia, which is the leading
cause of surgical
interventions that may include hysterectomy. Other symptoms, in particular
pressure symptoms,
are largely dependent on the size, number, and location of the tumors.
[0005] Although the pathogenesis has yet to be fully elucidated, the
growth of uterine
fibroids is known to be highly dependent on both estrogen and progestogen.
Fibroids tend to
shrink after menopause due to a decrease in hormone production. On this basis,
most medical
CA 2977938 2017-09-01
treatments for women with symptomatic uterine fibroids are aimed at either
hormone-blocking or
hormone-modulating strategies.
[0006] Gonadotropin-releasing hormone (GnRH) is a peptide that stimulates
the
secretion of the pituitary hormones that are responsible for sex steroid
production and normal
reproductive function. GnRH agonists are used to treat endometriosis and
uterine fibroids by
suppressing the activity of the pituitary-gonadal axis. However, GnRH agonists
cause an initial
stimulation of gonadotropic and gonadal hormones, such as estrogen.
[0007] Peptide GnRH antagonists competitively bind to GnRH receptors in
the pituitary
gland, blocking the release of gonadotropins, such as luteinizing hormone (LH)
and follicle-
stimulating hormone (FSH) from the pituitary. Such peptide GnRH antagonists
have been
approved for oncology and assisted reproduction. However, administration is
inconvenient, with
peptide GnRH antagonists being delivered as daily subcutaneous injections or
as long-acting
depot formulations.
[0008] Thus, there is a need in the art for new orally administered
treatments for
endometriosis and uterine fibroids and, in particular, management of pain
associated with
endometriosis and heavy menstrual bleeding associated with uterine fibroids.
Moreover, there
remains a need in the art to develop orally bioavailable dosage forms
comprising such treatments
and, in particular, a nonpeptide GnRH antagonist.
SUMMARY OF THE INVENTION
[0009] The disclosure is directed to pharmaceutical compositions
comprising 44(R)-2-
[5-(2-fluoro-3-methox y-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-meth
y1-2,6-dioxo-3,6-
dihydro-2H-pyrimidin-1-y1]-1-phenyl-ethylamino)-butyric acid (Compound A) or a
pharmaceutically acceptable salt thereof; methods of using such compositions;
and methods of
facilitating the release of Compound A from such compositions.
[0010] The present application identifies at least two challenges to
developing
pharmaceutical formulations comprising Compound A or a pharmaceutically
acceptable salt
thereof. One challenge was that Compound A and, in particular, the monosodium
salt of
Compound A has a tendency to form a gel, particularly when present at an
amount greater than
about 10% by weight in the absence of an appropriate anti-gelling agent. Such
gel formation
limits the dissolution of API and, ultimately, can lead to highly variable
inter-and intra patient
2
CA 2977938 2017-09-01
bioavailability. Another challenge was that Compound A can degrade to form a
compound
having a lactam moiety (referred to herein as Compound B). Reducing conversion
of the drug
substance into its lactam-containing degradation product is desirable, for
example, to maintain
safety and efficacy over the life of the product. Thus, it has been determined
in the present
application that a preferred pharmaceutical composition reduces gelling of the
API and/or
reduces generation of the lactam degradation product (i.e., Compound B).
[0011] In one aspect, the disclosed pharmaceutical compositions comprise
Compound A
or a pharmaceutically acceptable salt thereof, and at least one anti-gelling
agent.
[0012] In certain embodiments, the salt of Compound A is the monosodium
salt (sodium
44(R)-2-[5-(2-fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-
methyl-2,6-
dioxo-3,6-dihydro-2H-pyrimidin-1-y11-1-phenyl-ethylamino)butanoate).
[0013] In certain embodiments, the anti-gelling agent facilitates release
of Compound A
or a pharmaceutically acceptable salt thereof from a solid dosage form, such
as a tablet.
[0014] In certain embodiments, the anti-gelling agent also acts as a
stabilizer to, for
example, reduce formation of (R)-5-(2-fluoro-3-methoxypheny1)-1-(2-fluoro-6-
(trifluoromethyl)benzy1)-6-methy1-3-(2-(2-oxopyrrolidin-1-y1)-2-
phenylethyl)pyrimidine-
2,4(1H,3H)-dione (Compound B) in the composition relative to an otherwise
identical
composition without the anti-gelling agent.
[0015] In certain embodiments, the anti-gelling agent acts as a buffer.
[0016] In certain embodiments, the anti-gelling agent is an alkali metal
salt, such as
sodium carbonate. Sodium carbonate may be either sodium carbonate monohydrate
or sodium
carbonate anhydrous.
[0017] In certain embodiments, the weight ratio of Compound A or a
pharmaceutically
acceptable salt thereof to the anti-gelling agent is from about 1:1 to about
4:1.
[0018] In certain embodiments, the weight ratio of Compound A or a
pharmaceutically
acceptable salt thereof to the anti-gelling agent is about 2:1.
[0019] In certain embodiments, the anti-gelling agent is present in the
pharmaceutical
composition in an amount from about 5% to about 35% by weight of the
pharmaceutical
composition.
3
CA 2977938 2017-09-01
[0020] In certain embodiments, the anti-gelling agent is present in the
pharmaceutical
composition in an amount from about 15% to about 25% by weight of the
pharmaceutical
composition.
[0021] In certain embodiments, the pharmaceutical composition further
comprises at
least one additional excipient selected from the group consisting of a binder,
a filler, a lubricant,
a glidant, and a combination thereof.
[0022] In certain embodiments, the binder is polyvinylpyrrolidone.
[0023] In certain embodiments, the filler is a starch and/or mannitol. In
certain
embodiments, the filler is a water soluble filler, such as mannitol or
pregelitanized starch or a
combination thereof. In certain embodiments, the filler is a water insoluble
filler, such as
microcrystalline cellulose. In some such embodiments, the pharmaceutical
composition further
comprises a surfactant, such as sodium lauryl sulfate.
[0024] In certain embodiments, the lubricant is magnesium stearate.
[0025] In certain embodiments, the glidant is colloidal silicon dioxide.
[0026] In certain embodiments, the pharmaceutical composition is an oral
dosage form.
In some such embodiments, the oral dosage form is a tablet.
[0027] In certain embodiments, the pharmaceutical composition comprises
Compound A
or a pharmaceutically acceptable salt thereof in an amount of about 100 mg to
about 600 mg; and
at least about 10% by weight of the anti-gelling agent. In certain
embodiments, the
pharmaceutical composition comprises Compound A or a pharmaceutically
acceptable salt
thereof in an amount of about 100 mg to about 350 mg; and at least about 10%
by weight of the
anti-gelling agent.
[0028] In one aspect, the disclosed pharmaceutical compositions comprise
about 150 mg
of Compound A or a pharmaceutically acceptable salt thereof, at least one anti-
gelling agent,
and, optionally, at least one binder. In certain embodiments, the anti-gelling
agent is sodium
carbonate, such as sodium carbonate monohydrate. In some such embodiments, the
weight ratio
of Compound A or a pharmaceutically acceptable salt thereof to sodium
carbonate is about 2:1.
In certain embodiments, the binder is polyvinylpyrrolidone. In certain
embodiments, the salt of
Compound A is the monosodium salt (sodium 44(R)-2-[5-(2-fluoro-3-methoxy-
pheny1)-3-(2-
fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-l-
y1]-1-phenyl-
ethylamino)butanoate). In some such embodiments, the weight ratio of sodium 4-
((R)-2-[5-(2-
4
CA 2977938 2017-09-01
fluoro-3-methox y-pheny1)-3-(2-fluoro-6-trifluorometh yl-benzy1)-4-meth y1-2,6-
dioxo-3,6-
dihydro-2H-pyrimidin-1-y1]-1-phenyl-ethylamino)butanoate to sodium carbonate
monohydrate is
about 2:1.
[0029] In another aspect, the disclosed pharmaceutical compositions
comprise about 200
mg of Compound A or a pharmaceutically acceptable salt thereof, at least one
anti-gelling agent,
and, optionally, at least one binder. In certain embodiments, the anti-gelling
agent is sodium
carbonate, such as sodium carbonate monohydrate. In some such embodiments, the
weight ratio
of Compound A or a pharmaceutically acceptable salt thereof to sodium
carbonate is about 2:1.
In certain embodiments, the binder is polyvinylpyrrolidone. In certain
embodiments, the salt of
Compound A is the monosodium salt (sodium 44(R)-2-[5-(2-fluoro-3-methoxy-
pheny1)-3-(2-
fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-
y1]-1-phenyl-
ethylamino)butanoate). In some such embodiments, the weight ratio of sodium
44(R)-2-[5-(2-
fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-
dioxo-3,6-
dihydro-2H-pyrimidin-1-y1]-1-phenyl-ethylamino)butanoate to sodium carbonate
monohydrate is
about 2:1.
[0030] In still another aspect, the disclosed pharmaceutical compositions
comprise about
300 mg of Compound A or a pharmaceutically acceptable salt thereof, at least
one anti-gelling
agent, and, optionally, at least one binder. In certain embodiments, the anti-
gelling agent is
sodium carbonate, such as sodium carbonate monohydrate. In some such
embodiments, the
weight ratio of Compound A or a pharmaceutically acceptable salt thereof to
sodium carbonate is
about 2:1. In certain embodiments, the binder is polyvinylpyrrolidone. In
certain embodiments,
the salt of Compound A is the monosodium salt (sodium 44(R)-245-(2-fluoro-3-
methoxy-
pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-dioxo-3,6-dihydro-
2H-pyrimidin-1-
y1]-1-phenyl-ethylamino)butanoate). In some such embodiments, the weight ratio
of sodium 4-
((R)-2-[5-(2-fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-
methyl-2,6-
dioxo-3,6-dihydro-2H-pyrimidin-1-y1]-1-phenyl-ethylamino)butanoate to sodium
carbonate
monohydrate is about 2:1.
[0031] In certain embodiments, the pharmaceutical composition is a tablet
comprising
sodium 44(R)-2-{5-(2-fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-
benzy1)-4-
methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-y11-1-phenyl-ethylamino)butanoate
in an amount
equivalent to about 125 mg to about 175 mg, preferably about 150 mg of
Compound A; and at
CA 2977938 2017-09-01
least about 10%, preferably between about 15% and about 20%, by weight of the
anti-gelling
agent; wherein the tablet when administered as a single dose to a population
of human subjects
provides an average Tmax value that is less than about 3 hours, and preferably
less than about 2
hours, for the population of human subjects. In certain embodiments, the
pharmaceutical
composition is a tablet sodium 44(R)-245-(2-fluoro-3-methoxy-pheny1)-3-(2-
fluoro-6-
trifluoromethyl-benzy1)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-l-y1]-1-
phenyl-
ethylamino)butanoate in an amount equivalent to about 175 mg to about 225 mg,
preferably
about 200 mg of Compound A; and at least about 10%, preferably between about
15% and about
20%, by weight of the anti-gelling agent; wherein the tablet when administered
as a single dose
to a population of human subjects provides an average Tmax value that is less
than about 3 hours,
and preferably less than about 2 hours, for the population of human subjects.
In certain
embodiments, the pharmaceutical composition is a tablet comprising sodium
44(R)-245-(2-
fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-
dioxo-3,6-
dihydro-2H-pyrimidin-1-yI]-1-phenyl-ethylamino)butanoate in an amount
equivalent to about
275 mg to about 325 mg, preferably about 300 mg of Compound A; and at least
about 10%,
preferably between about 15% and about 20%, by weight of the anti-gelling
agent; wherein the
tablet when administered as a single dose to a population of human subjects
provides an average
Tmax value that is less than about 3 hours, and preferably less than about 2
hours, for the
population of human subjects.
[0032] In certain embodiments, the pharmaceutical composition is a tablet
comprising
sodium 44(R)-2-[5-(2-fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-
benzy1)-4-
methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-y1]-1-phenyl-ethylamino)butanoate
in an amount
equivalent to about 125 mg to about 175 mg, preferably about 150 mg of
Compound A; and at
least about 10%, preferably between about 15% and about 20%, by weight of the
anti-gelling
agent; wherein the tablet when administered as a single dose to a population
of human subjects
provides an average Cmax value that is at least about 380 ng/mL for the
population of human
subjects. In certain embodiments, the pharmaceutical composition is a tablet
comprising sodium
44(R)-2-[5-(2-fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-
methyl-2,6-
dioxo-3,6-dihydro-2H-pyrimidin-1-y1]-1-phenyl-ethylamino)butanoate in an
amount equivalent
to about 175 mg to about 225 mg, preferably about 200 mg of Compound A; and at
least about
10%, preferably between about 15% and about 20%, by weight of the anti-gelling
agent; wherein
6
CA 2977938 2017-09-01
the tablet when administered as a single dose to a population of human
subjects provides an
average Cimix value that is at least about 550 ng/mL for the population of
human subjects. In
certain embodiments, the pharmaceutical composition is a tablet comprising
sodium 4-((R)-2-[5-
(2-fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-
dioxo-3,6-
dihydro-2H-pyrimidin-1-y1]-1-phenyl-ethylamino)butanoate in an amount
equivalent to about
275 mg to about 325 mg, preferably about 300 mg of Compound A; and at least
about 10%,
preferably between about 15% and about 20%, by weight of the anti-gelling
agent; wherein the
tablet when administered as a single dose to a population of human subjects
provides an average
CIMIX value that is at least about 1030 ng/mL for the population of human
subjects.
[0033] In certain embodiments, the pharmaceutical composition is a tablet
comprising
sodium 44(R)-215-(2-fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-
benzy1)-4-
methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-y11-1-phenyl-ethylamino)butanoate
in an amount
equivalent to about 125 mg to about 175 mg, preferably about 150 mg of
Compound A; and at
least about 10%, preferably between about 15% and about 20%, by weight of the
anti-gelling
agent; wherein the tablet when administered as a single dose to a population
of human subjects
provides an average AUCt value that is at least about 940 ng=h/mL for the
population of human
subjects. In certain embodiments, the pharmaceutical composition is a tablet
comprising sodium
44(R)-2-[5-(2-fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-
methyl-2,6-
dioxo-3,6-dihydro-2H-pyrimidin-1-y1]-1-phenyl-ethylamino)butanoate in an
amount equivalent
to about 175 mg to about 225 mg, preferably about 200 mg of Compound A; and at
least about
10%, preferably between about 15% and about 20%, by weight of the anti-gelling
agent; wherein
the tablet when administered as a single dose to a population of human
subjects provides an
average AUCt value that is at least about 1410 ng=h/mL for the population of
human subjects. In
certain embodiments, the pharmaceutical composition is a tablet comprising
sodium 44(R)-2-15-
(2-fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-
dioxo-3,6-
dihydro-2H-pyrimidin-1-y1]-1-phenyl-ethylamino)butanoate in an amount
equivalent to about
275 mg to about 325 mg, preferably about 300 mg of Compound A; and at least
about 10%,
preferably between about 15% and about 20%, by weight of the anti-gelling
agent; wherein the
tablet when administered as a single dose to a population of human subjects
provides an average
AUCt value that is at least about 2800 ng=h/mL for the population of human
subjects.
7
CA 2977938 2017-09-01
[0034] In certain embodiments, the pharmaceutical composition is a tablet
comprising
sodium 44(R)-2-[5-(2-fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-
benzy1)-4-
methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-y1]-1-phenyl-ethylamino)butanoate
in an amount
equivalent to about 125 mg to about 175 mg, preferably about 150 mg of
Compound A; and at
least about 10%, preferably between about 15% and about 20%, by weight of the
anti-gelling
agent; wherein the tablet when administered as a single dose to a population
of human subjects
provides an average AUG, value that is at least about 950 ng=h/mL for the
population of human
subjects. In certain embodiments, the pharmaceutical composition is a tablet
comprising sodium
44(R)-2-[5-(2-fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-
methyl-2,6-
dioxo-3,6-dihydro-2H-pyrimidin-1-y11-1-phenyl-ethylamino)butanoate in an
amount equivalent
to about 175 mg to about 225 mg, preferably about 200 mg of Compound A; and at
least about
10%, preferably between about 15% and about 20%, by weight of the anti-gelling
agent; wherein
the tablet when administered as a single dose to a population of human
subjects provides an
average AUG, value that is at least about 1430 ng-h/mL for the population of
human subjects. In
certain embodiments, the pharmaceutical composition is a tablet comprising
sodium 4-((R)-2-[5-
(2-fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluorometh yl-benzy1)-4-methy1-
2,6-dioxo-3,6-
dihydro-2H-pyrimidin-1-y1]-1-phenyl-ethylamino)butanoate in an amount
equivalent to about
275 mg to about 325 mg, preferably about 300 mg of Compound A; and at least
about 10%,
preferably between about 15% and about 20%, by weight of the anti-gelling
agent; wherein the
tablet when administered as a single dose to a population of human subjects
provides an average
AUGDo value that is at least about 2820 ng=h/mL for the population of human
subjects.
[0035] The disclosure is also directed to pharmaceutical compositions
comprising
Compound A or a pharmaceutically acceptable salt thereof and an amount of an
alkali metal salt
sufficient to facilitate release of Compound A or the pharmaceutically
acceptable salt thereof
from the composition.
[0036] In certain embodiments, the release is measured using USP apparatus
II in 900
mL of sodium phosphate, pH 6.8, at 37 C and paddle speed of 50 rpm.
[0037] In certain embodiments, the release is measured using USP Apparatus
II in 900
mL of hydrochloric acid, pH 1.2, at 37 C and paddle speed of 50 rpm.
[0038] In certain embodiments, the alkali metal salt also acts as a
stabilizer.
[0039] In certain embodiments, the alkali metal salt acts as a buffer.
8
CA 2977938 2017-09-01
[0040] In certain embodiments, the weight ratio of Compound A or a
pharmaceutically
acceptable salt thereof to the alkali metal salt is from about 1:1 to about
4:1.
[0041] In certain embodiments, the weight ratio of Compound A or a
pharmaceutically
acceptable salt thereof to the alkali metal salt is about 2:1.
[0042] In certain embodiments, the alkali metal salt is present in the
pharmaceutical
composition in an amount from about 10% to about 30% by weight of the
pharmaceutical
composition.
[0043] In certain embodiments, the alkali metal salt is present in the
pharmaceutical
composition in an amount from about 15% to about 25% by weight of the
pharmaceutical
composition.
[0044] In certain embodiments, the pharmaceutical composition is an oral
dosage form.
[0045] In certain embodiments, the oral dosage form is a tablet.
[0046] The disclosure is also directed to a solid oral dosage form, such
as a tablet,
comprising Compound A or a pharmaceutically acceptable salt thereof and sodium
carbonate.
[0047] In certain embodiments, the salt of Compound A is a sodium salt.
[0048] In certain embodiments, the weight ratio of Compound A or a
pharmaceutically
acceptable salt thereof to sodium carbonate is from about 1:1 to about 4:1.
[0049] In certain embodiments, the weight ratio of Compound A or a
pharmaceutically
acceptable salt thereof to sodium carbonate is about 2:1.
[0050] In certain embodiments, sodium carbonate is present in the
pharmaceutical
composition in an amount from about 10% to about 30% by weight of the
pharmaceutical
composition.
[0051] In certain embodiments, sodium carbonate is present in the
pharmaceutical
composition in an amount from about 15% to about 25% by weight of the
pharmaceutical
composition.
[0052] The disclosure is also directed to methods of facilitating release
of Compound A
or a pharmaceutically acceptable salt thereof from an oral dosage form.
[0053] In certain embodiments, the methods comprise preparing a
pharmaceutical
composition comprising at least one anti-gelling agent and Compound A or a
pharmaceutically
acceptable salt thereof.
9
CA 2977938 2017-09-01
[0054] Compound A has a tendency to form a gel in the presence of water,
further
complicating the development process. Thus, in one aspect this disclosure
provides methods of
manufacturing a pharmaceutical composition comprising Compound A or a
pharmaceutically
acceptable salt thereof in the substantial absence of water. In certain
embodiments, the
pharmaceutical composition is manufactured using a roller compaction process.
[0055] The disclosure is also directed to methods for treating
endometriosis in a subject
in need of such treatment, wherein the method comprises administering to the
subject a
pharmaceutical composition of the present disclosure. In some such
embodiments, the
pharmaceutical composition is administered to the subject once daily (QD). In
some such
embodiments, the pharmaceutical composition is administered to the subject
twice daily (BID).
[0056] The disclosure is also directed to pharmaceutical compositions for
use in treating
endometriosis.
[0057] The disclosure is also directed to methods for treating uterine
fibroids in a subject
in need of such treatment, wherein the method comprises administering to the
subject a
pharmaceutical composition of the present disclosure. In some such
embodiments, the
pharmaceutical composition is administered to the subject once daily (QD). In
some such
embodiments, the pharmaceutical composition is administered to the subject
twice daily (BID).
[0058] The disclosure is also directed to pharmaceutical compositions for
use in treating
uterine fibroids.
[0059] The disclosure is also directed to methods for providing rapid
suppression of
luteinizing hormone (LH) and/or follicle-stimulating hormone (FSH) in a female
patient with
endometriosis or uterine fibroids, wherein the method comprises administering
to the female
patient a pharmaceutical composition of the present disclosure. In some such
embodiments, the
pharmaceutical composition is administered to the subject once daily (QD). In
some such
embodiments, the pharmaceutical composition is administered to the female
patient twice daily
(BID).
[0060] The disclosure is also directed to pharmaceutical compositions for
use in
providing rapid suppression of LH and/or FSH in a female patient with
endometriosis or uterine
fibroids.
[0061] The disclosure is also directed to methods for providing partial to
substantially
full suppression of estradiol in a female patient with endometriosis or
uterine fibroids, wherein
to
CA 2977938 2017-09-01
the method comprises administering to the subject a pharmaceutical composition
of the present
disclosure. In some such embodiments, the pharmaceutical composition is
administered to the
female patient once daily (QD). In some such embodiments, the pharmaceutical
composition is
administered to the female patient twice daily (BID).
[0062] The disclosure is also directed to pharmaceutical compositions for
use in
providing partial to substantially full suppression of estradiol in a female
patient with
endometriosis or uterine fibroids.
[0063] These and other objects of the invention are described in the
following
paragraphs. These objects should not be deemed to narrow the scope of the
invention.
BRIEF DESCRIPTION OF THE DRAWINGS
[0064] Figure 1 is a roller compaction process flow diagram.
[0065] Figure 2 is a plot showing apparent solubility of Compound A in
water.
[0066] Figure 3 is a two-step wet granulation process flow diagram.
[0067] Figure 4 is a graph showing an in vitro dissolution profile for
Formulation F5
after storage for 18 or 24 months.
[0068] Figure 5 is a graph showing an in vitro dissolution profile for
Formulation F6
after storage for 1, 3, 6, or 9 months.
[0069] Figure 6 is a graph showing an in vitro dissolution profile for
Formulation F10
(uncoated and film-coated tablets).
[0070] Figure 7 is a graph showing an in vitro dissolution profile for
compositions
containing varying amounts of sodium carbonate monohydrate.
[0071] Figure 8 is a bar graph showing percentage of a degradation product
(Compound
B) after storage for one week.
[0072] Figure 9 is a bar graph showing formation rate of a degradation
product
(Compound B) under various storage conditions.
DETAILED DESCRIPTION OF THE INVENTION
[0073] This detailed description is intended only to acquaint others
skilled in the art with
the present invention, its principles, and its practical application so that
others skilled in the art
may adapt and apply the invention in its numerous forms, as they may be best
suited to the
11
CA 2977938 2017-09-01
requirements of a particular use. This description and its specific examples
are intended for
purposes of illustration only. This invention, therefore, is not limited to
the embodiments
described in this patent application, and may be variously modified.
[0074] A. DEFINITIONS
[0075] As used in the specification and the appended claims, unless
specified to the
contrary, the following terms have the meaning indicated:
[0076] The term "API" as used herein stands for "active pharmaceutical
ingredient." The
preferred API as disclosed herein is 44(R)-245-(2-fluoro-3-methoxy-pheny1)-3-
(2-fluoro-6-
trifluoromethyl-benzy1)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-l-y11-1-
phenyl-
ethylamino)-butyric acid (Compound A) or a pharmaceutically acceptable salt
thereof and,
preferably is sodium 44(R)-245-(2-fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-
trifluoromethyl-
benzy1)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-y1]-1-phenyl-
ethylamino)butanoate.
[0077] As used herein, the term "pharmaceutical composition" means a
composition
comprising Compound A or a pharmaceutically acceptable salt thereof and,
optionally, one or
more pharmaceutically acceptable excipients.
[0078] The term "pharmaceutically acceptable" is used adjectivally to
mean that the
modified noun is appropriate for use as a pharmaceutical product for human use
or as a part of a
pharmaceutical product for human use.
[0079] The term "subject" includes humans and other primates as well as
other
mammals. The term subject includes, for example, a healthy premenopausal
female as well as a
female patient having, for example, endometriosis or uterine fibroids. In
certain embodiments,
the subject is a human. In certain embodiments, the subject is an adult human
female. In certain
embodiments, the subject is a woman, typically a premenopausal woman, having
endometriosis.
In certain embodiments, the subject is a woman, typically a premenopausal
woman, having
uterine fibroids.
[0080] The term "therapeutically effective amount" means a sufficient
amount of the API
or pharmaceutical composition to treat a condition, disorder, or disease, at a
reasonable
benefit/risk ratio applicable to any medical treatment.
[0081] The terms "treat", "treating" and "treatment" refer to a method of
alleviating or
abrogating a condition, disorder, or disease and/or the attendant symptoms
thereof.
12
CA 2977938 2017-09-01
[0082] B. DRUG SUBSTANCE
[0083] Pharmaceutical compositions disclosed herein comprise at least one
active
pharmaceutical ingredient: 44(R)-245-(2-fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-
trifluoromethyl-benzy1)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-y1]-1-
phenyl-
ethylamino)-butyric acid (Compound A) or a pharmaceutically acceptable salt
thereof.
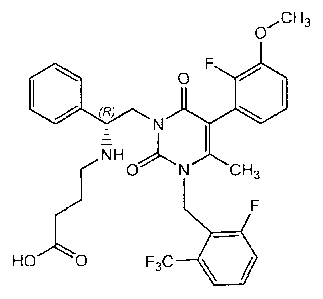
[0084] Compound A has the following formula:
,CH3
0
0
(R)
NH 0 03
LF
HO 0 F3C
[0085] Compound A is an orally active, non-peptide GnRH antagonist and is
unlike other
GnRH agonists and injectable (peptide) GnRH antagonists. Compound A produces a
dose
dependent suppression of pituitary and ovarian hormones in women. Methods of
making
Compound A and a pharmaceutically acceptable salt thereof, as well as similar
compounds, are
described in W02001/055119 and WO 2005/007165, the contents of which are
herein
incorporated by reference.
[0086] In certain embodiments, 44(R)-2-[5-(2-fluoro-3-methoxy-phenyl)-3-
(2-fluoro-6-
trifluoromethyl-benzy1)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-y11-1-
phenyl-
ethylamino)-butyric acid exists in zwitterionic form. For example, both the
carboxylic acid and
the tertiary amine are ionized and, thus, the molecule has no overall charge
but does have charge
separation. Such zwitterionic forms are included within the scope of the term
"Compound A or a
pharmaceutically acceptable salt thereof."
[0087] Compound A may be present in a pharmaceutical composition in the
form of acid
or base addition salts. Acid addition salts of the free amino compounds of the
present invention
may be prepared by methods well known in the art, and may be formed from
organic and
inorganic acids. Suitable organic acids include maleic, fumaric, benzoic,
ascorbic, succinic,
methanesulfonic, acetic, trifluoroacetic, oxalic, propionic, tartaric,
salicylic, citric, gluconic,
13
CA 2977938 2017-09-01
lactic, mandelic, cinnamic, aspartic, stearic, palmitic, glycolic, glutamic,
and benzenesulfonic
acids. Suitable inorganic acids include hydrochloric, hydrobromic, sulfuric,
phosphoric, and
nitric acids. Suitable base addition salts include those salts that form with
the carboxylate anion
and include salts formed with organic and inorganic cations such as those
chosen from the alkali
and alkaline earth metals (for example, lithium, sodium, potassium, magnesium,
barium and
calcium), as well as the ammonium ion and substituted derivatives thereof (for
example,
dibenzylammonium, benzylammonium, 2-hydroxyethylammonium, and the like). Thus,
the term
"pharmaceutically acceptable salt" of Compound A is intended to encompass any
and all
acceptable salt forms.
[0088] In certain embodiments, Compound A is present in a pharmaceutical
composition
in the form of a pharmaceutically acceptable salt. In certain embodiments, a
pharmaceutically
acceptable salt of Compound A is the sodium salt of Compound A. The monosodium
salt of
Compound A has a molecular formula of C32H29F5N305Na, which corresponds to a
molecular
weight of about 653.6 (salt) and about 631.6 (free form). The monosodium salt
of Compound A
has the following formula:
,01-13
0
0
(R)
=NH I
0 N CH3
Na -0
0 F3C
[0089] In certain embodiments, the monosodium salt is in the form of an
amorphous
solid. In certain embodiments, the monosodium salt is in crystalline form,
such as a partially
crystalline form.
[0090] As used herein, and in the absence of a specific reference to a
particular
pharmaceutically acceptable salt of Compound A, any dosages, whether expressed
in milligrams
or as a percentage by weight or as a ratio with another ingredient, should be
taken as referring to
the amount of Compound A free form.
14
CA 2977938 2017-09-01
[0091] In certain embodiments, Compound A or a pharmaceutically acceptable
salt
thereof is present in a pharmaceutical composition in an amount from about 45
mg to about 650
mg of Compound A. In certain embodiments, the amount of Compound A, or
pharmaceutically
acceptable salt thereof, is from about 50 mg to about 400 mg. In certain
embodiments, the
amount of Compound A, or pharmaceutically acceptable salt thereof, is from
about 100 mg to
about 350 mg. In some such embodiments, the amount of Compound A, or
pharmaceutically
acceptable salt thereof, is from about 140 mg to about 160 mg, preferably
about 150 mg. In other
such embodiments, the amount of Compound A, or pharmaceutically acceptable
salt thereof, is
from about 190 mg to about 210 mg, preferably about 200 mg. In still other
embodiments, the
amount of Compound A, or pharmaceutically acceptable salt thereof, is from
about 290 mg to
about 310 mg, preferably about 300 mg.
[0092] C. PHARMACEUTICAL COMPOSITIONS
[0093] Pharmaceutical compositions comprising Compound A or a
pharmaceutically
acceptable salt thereof may be used to treat endometriosis or uterine
fibroids. Pharmaceutical
compositions or dosage forms as described herein may preferably be oral dosage
forms, and, in
particular, solid oral dosage forms, which can be administered to humans. An
oral dosage form
may be in the form of, for example, capsules, granules, granulates, pellets,
pills, powders, caplets
and/or tablets.
[0094] The present disclosure provides pharmaceutical formulations and
functional
excipients to, inter alia, facilitate drug dissolution and/or enhance
stability of the drug product
and/or drug substance (e.g., by controlling the formation of degradation
products).
[0095] In certain embodiments, the pharmaceutical composition is an
immediate release
pharmaceutical composition. The development of an immediate release tablet for
Compound A
was not straightforward. Initial tablets prepared by granulating Compound A
with typical
pharmaceutical excipients showed incomplete dissolution of Compound A into 900
mL of pH
1.2 buffer. If the percent of drug in the tablet exceeded 10%, only 30-40% of
the drug load was
dissolved. The remaining amount of Compound A was present as an insoluble
precipitate at the
top of the dissolution vessels.
[0096] In at least one aspect, the pharmaceutical compositions comprising
Compound A
or a pharmaceutically acceptable salt thereof include an anti-gelling agent.
CA 2977938 2017-09-01
[0097] As referred to herein, an "anti-gelling agent" is an agent that
reduces or prevents
gel formation. In certain embodiments, the anti-gelling agent reduces or
prevents gel formation
relative to an otherwise identical composition without the anti-gelling agent.
In certain
embodiments, the anti-gelling agent reduces or prevents gel formation such
that release of
Compound A or a pharmaceutically acceptable salt thereof from a composition is
facilitated. In
certain embodiments, the anti-gelling agent improves release of Compound A or
a
pharmaceutically acceptable salt thereof from a pharmaceutical composition
relative to that same
pharmaceutical composition without the anti-gelling agent.
[0098] In certain embodiments, the anti-gelling agent is present in the
pharmaceutical
composition in an amount from about 3% to about 50% by weight (w/w) of the
pharmaceutical
composition. In certain embodiments, the anti-gelling agent is present in the
pharmaceutical
composition in an amount from about 5% to about 35% by weight (w/w) of the
pharmaceutical
composition. In certain embodiments, the anti-gelling agent is present in the
pharmaceutical
composition in an amount from about 10% to about 25% by weight (w/w) of the
pharmaceutical
composition. In certain embodiments, the anti-gelling agent is present in the
pharmaceutical
composition in an amount from about 15% to about 20% by weight (w/w) of the
pharmaceutical
composition.
[0099] In certain embodiments, the pharmaceutical composition is a film-
coated tablet. In
some such embodiments, the anti-gelling agent is present in an amount from
about 3% to about
50%, alternatively from about 5% to about 35%, alternatively from about 10% to
about 25%,
alternatively from about 15% to about 20%, by weight of the uncoated tablet.
In some such
embodiments, the anti-gelling agent is present in an amount from about 3% to
about 50%,
alternatively from about 5% to about 35%, alternatively from about 10% to
about 25%,
alternatively from about 15% to about 20%, by weight of the coated tablet.
[00100] In certain embodiments, the weight ratio of Compound A or a
pharmaceutically
acceptable salt thereof to the anti-gelling agent is from about 0.5:1 to about
4:1. In certain
embodiments, the weight ratio of Compound A or a pharmaceutically acceptable
salt thereof to
the anti-gelling agent is from about 1:1 to about 3:1. In certain embodiments,
the weight ratio of
Compound A or a pharmaceutically acceptable salt thereof to the anti-gelling
agent is about 2:1.
[00101] In certain embodiments, the anti-gelling agent comprises a water
soluble salt of a
weak acid, such as a carbonate (e.g., sodium carbonate, sodium hydrogen
carbonate, potassium
16
CA 2977938 2017-09-01
carbonate, potassium hydrogen carbonate), an acetate (e.g., sodium acetate,
potassium acetate,
ammonium acetate), or a phosphate (e.g., mono-, di-, or tri-sodium phosphate),
or a combination
thereof.
[00102] In certain embodiments, the anti-gelling agent comprises a basic
amino acid, such
as arginine, lysine, histidine, or combinations thereof. In certain
embodiments, the buffering
agent comprises poly(meth)acrylate polymers, such as Eudragit E 100, Eudragit
E 12, Eudragit E
5, Eudragit E PO, or combinations thereof.
[00103] In certain embodiments, the anti-gelling agent comprises an alkali
metal salt or a
combination thereof. Exemplary alkali metal salts include sodium carbonate,
sodium hydrogen
carbonate, or sodium phosphate.
[00104] In certain embodiments, the alkali metal salt is present in an
amount sufficient to
provide a microenvironment to reduce or prevent gel formation. In certain
embodiments, the
alkali metal salt is present in an amount sufficient to provide a
microenvironment to facilitate
release of Compound A or the pharmaceutically acceptable salt thereof from the
tablet in an
aqueous medium. In certain embodiments, the microenvironment to facilitate
release of
Compound A or the pharmaceutically acceptable salt thereof from the tablet
comprises a pH of
about 3.5 to about 8Ø
[00105] In certain embodiments, the anti-gelling agent is sodium
carbonate, such as
sodium carbonate monohydrate or sodium carbonate anhydrous.
[00106] In certain embodiments, sodium carbonate is present in the
pharmaceutical
composition in an amount from about 3% to about 50% by weight (w/w) of the
pharmaceutical
composition. In certain embodiments, sodium carbonate is present in the
pharmaceutical
composition in an amount from about 5% to about 35% by weight (w/w) of the
pharmaceutical
composition. In certain embodiments, sodium carbonate is present in the
pharmaceutical
composition in an amount from about 10% to about 25% by weight (w/w) of the
pharmaceutical
composition. In certain embodiments, sodium carbonate is present in the
pharmaceutical
composition in an amount from about 15% to about 20% by weight (w/w) of the
pharmaceutical
composition.
[00107] In certain embodiments, the pharmaceutical composition is a film-
coated tablet. In
some such embodiments, sodium carbonate is present in an amount from about 3%
to about
50%, alternatively from about 5% to about 35%, alternatively from about 10% to
about 25%,
17
CA 2977938 2017-09-01
alternatively from about 15% to about 20%, by weight of the uncoated tablet.
In some such
embodiments, sodium carbonate is present in an amount from about 3% to about
50%,
alternatively from about 5% to about 35%, alternatively from about 10% to
about 25%,
alternatively from about 15% to about 20%, by weight of the coated tablet.
[00108] In certain embodiments, the weight ratio of Compound A or a
pharmaceutically
acceptable salt thereof to sodium carbonate is from about 0.5:1 to about 4:1.
In certain
embodiments, the weight ratio of Compound A or a pharmaceutically acceptable
salt thereof to
sodium carbonate is from about 1:1 to about 3:1. In certain embodiments, the
weight ratio of
Compound A or a pharmaceutically acceptable salt thereof to sodium carbonate
is about 2:1.
[00109] In certain embodiments, the weight ratio of sodium 44(R)-2-[5-(2-
fluoro-3-
methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-dioxo-3,6-
dihydro-2H-
pyrimidin-1-y1]-1-phenyl-ethylamino)butanoate to sodium carbonate monohydrate
is from about
0.5:1 to about 4:1. In certain embodiments, the weight ratio of sodium 44(R)-
245-(2-fluoro-3-
methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-dioxo-3,6-
dihydro-2H-
pyrimidin-1-y1]-1-phenyl-ethylamino)butanoate to sodium carbonate monohydrate
is from about
1:1 to about 3:1. In certain embodiments, the weight ratio of sodium 44(R)-2-
[5-(2-fluoro-3-
methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-dioxo-3,6-
dihydro-2H-
pyrimidin-l-y11-1-phenyl-ethylamino)butanoate to sodium carbonate monohydrate
is about 2:1.
[00110] As used herein, and in the absence of a specific reference to a
particular hydrate
(or anhydrous) form of sodium carbonate, any amounts, whether expressed in
milligrams or as a
percentage by weight or as a ratio with another ingredient, should be taken as
referring to the
amount of sodium carbonate monohydrate.
[00111] Drugs administered via oral solid dosage forms should dissolve in
viva before
systemic absorption can take place. There are number of factors which affect
drug dissolution,
including physicochemical properties of the drug substance. Poorly water
soluble drugs, such as
BCS class II (low solubility and high permeability), often exhibit poor
dissolution and
bioavailability. Even highly soluble drugs, such as BCS class III (high
solubility and low
permeability), may exhibit poor dissolution and bioavailability. Incomplete
dissolution may
result in highly variable inter-and intra patient bioavailability. It has been
determined in the
present application that Compound A is a BCS class III drug. It also has been
determined in the
present application that the monosodium salt of Compound A has a tendency to
form a gel,
18
CA 2977938 2017-09-01
particularly when present at an amount greater than about 10% by weight in the
absence of an
appropriate anti-gelling agent. Thus, it is desirable to provide oral solid
dosage forms that
facilitate drug dissolution.
[00112] In certain embodiments, dissolution is assessed utilizing USP
apparatus II in 900
mL of sodium phosphate, pH 6.8, at 37 C and paddle speed of 50 rpm. In
certain embodiments,
dissolution is assessed utilizing USP apparatus II in 900 mL of hydrochloric
acid, pH 1.2, at 37
C and paddle speed of 50 rpm. The analytical finish may be by a high
performance liquid
chromatography (HPLC) system with ultraviolet (UV) detection
[00113] The solid oral dosage forms described herein will typically be in
the form of a
tablet and, in particular, an immediate release tablet. In certain
embodiments, the immediate
release tablet releases at least 80% of Compound A or a pharmaceutically
acceptable salt thereof
in 30 minutes, measured using USP apparatus II, in 900 mL of sodium phosphate,
pH 6.8, at 37
C and paddle speed of 50 rpm. In certain embodiments, the immediate release
tablet releases at
least 80% of Compound A or a pharmaceutically acceptable salt thereof in 45
minutes, measured
using USP apparatus II, in 900 mL of sodium phosphate, pH 6.8, at 37 C and
paddle speed of 50
rpm.
[00114] In at least one aspect, the pharmaceutical compositions disclosed
herein are
stable during, for example, storage, distribution, and the duration of the
product's shelf-life (e.g.,
up to two years at room temperature/ambient conditions). A stable
pharmaceutical composition
may, for example, exhibit less degradation of the API and/or lower amounts of
degradation
products. Degradation products that arise during storage of the drug substance
and/or drug
product are undesirable and, in extreme cases, might even be harmful to a
patient being treated
with such drug product. Thus, it is desirable to control the formation of
degradation products,
particularly potentially harmful impurities in the drug product.
[00115] Assay and degradation product determination of pharmaceutical
compositions,
particularly solid oral dosage forms, and more particularly tablets, may be
performed using
HPLC with UV detection.
[00116] Pharmaceutical compositions may be assessed for degradation
products following
storage for at least two weeks, at least one month, at least two months, at
least three months, at
least six months, at least twelve months, at least eighteen months, or at
least twenty four months.
In particular, degradation products may be assessed at time intervals of one,
three, six, nine,
19
CA 2977938 2017-09-01
twelve, eighteen, twenty four, thirty six, and/or forty eight months. Storage
conditions may be
long term, intermediate, or accelerated conditions. In particular, storage
conditions may be, for
example, 25 C 2 C/40% relative humidity (RH) 5% RH, 25 C 2 C/60% RH 5%
RH,
30 C 2 C/35% RH 5% RH, 30 C 2 C/65% RH 5% RH, 40 C 2 C/25% RH 5% RH,
40 C 2 C/75% RH 5% RH, 50 C + 2 C/75% RH + 5 A) RH, 60 C 2 C/5% RH 5%
RH,
60 C 2 C/40% RH 5% RH, 70 C 2 C/5% RH 5% RH, 70 C + 2 C/75% RH 5% RH,
and/or 80 C + 2 C/40% RH 5% RH.
[00117] One exemplary degradation product of Compound A is (R)-5-(2-fluoro-
3-
methoxypheny1)-1-(2-fluoro-6-(trifluoromethyl)benzy1)-6-meth y1-3-(2-(2-
oxopyrrolidin-1-y1)-2-
phenylethyl)pyrimidine-2,4(1H,3H)-dione (Compound B), which has the following
structure:
õCH3
0
0
(R)
01\1 CH3
F3C
[00118] It has been identified in the present application that formation of
Compound B
was not adequately controlled in certain formulations. For example, in
formulations without
sodium carbonate monohydrate stored for one week at 65 C/75% RH, Compound B
was present
at greater than 1%. Thus, in certain embodiments, sodium carbonate is included
in the
pharmaceutical composition as a stabilizing agent to decrease degradation
and/or to reduce or
prevent gel formation.
[00119] In certain embodiments, sodium carbonate is present in amount from
about 10%
to about 25%, and preferably from about 15% to about 20%, by weight of the
pharmaceutical
composition. In certain embodiments, the weight ratio of Compound A or a
pharmaceutically
acceptable salt thereof to sodium carbonate is from about 1:1 to about 4:1,
and preferably about
2:1 (drug substance:sodium carbonate).
[00120] In certain embodiments, Compound B is present in a pharmaceutical
composition
in an amount less than about 1.0% by weight after storage for at least one
month, at least two
CA 2977938 2017-09-01
months, at least six months, at least twelve months, at least eighteen months,
or at least twenty-
four months at 25 C and 60% relative humidity. In certain embodiments,
Compound B is present
in a pharmaceutical composition in an amount less than about 0.7% by weight
after storage for at
least one month, at least two months, at least six months, at least twelve
months, at least eighteen
months, or at least twenty-four months at 25 C and 60% relative humidity. In
certain
embodiments, Compound B is present in a pharmaceutical composition in an
amount less than
about 0.5% by weight after storage for at least one month, at least two
months, at least six
months, at least twelve months, at least eighteen months, or at least twenty-
four months at 25 C
and 60% relative humidity. In certain embodiments, Compound B is present in a
pharmaceutical
composition in an amount less than about 0.03% by weight after storage for at
least one month,
at least two months, at least six months, at least twelve months, at least
eighteen months, or at
least twenty-four months at 25 C and 60% relative humidity.
[00121] In one aspect, this disclosure provides a stable pharmaceutical
composition
comprising Compound A or pharmaceutically acceptable salt thereof. A stable
pharmaceutical
composition may, for example, contain less than 1% Compound B following
storage for at least
one, at least three, at least six, at least nine, at least twelve, at least
eighteen, or at least twenty-
four months.
[00122] In certain embodiments, Compound B is present in a pharmaceutical
composition
in an amount less than about 1.0% by weight after storage for at least one
month, at least two
months, at least six months, at least twelve months, at least eighteen months,
or at least twenty-
four months at 40 C and 75% relative humidity. In certain embodiments,
Compound B is present
in a pharmaceutical composition in an amount less than about 0.7% by weight
after storage for at
least one month, at least two months, at least six months, at least twelve
months, at least eighteen
months, or at least twenty-four months at 40 C and 75% relative humidity. In
certain
embodiments, Compound B is present in a pharmaceutical composition in an
amount less than
about 0.5% by weight after storage for at least one month, at least two
months, at least six
months, at least twelve months, at least eighteen months, or at least twenty-
four months at 40 C
and 75% relative humidity. In certain embodiments, Compound B is present in a
pharmaceutical
composition in an amount less than about 0.03% by weight after storage for at
least one month,
at least two months, at least six months, at least twelve months, at least
eighteen months, or at
least twenty-four months at 40 C and 75% relative humidity.
21
CA 2977938 2017-09-01
[00123] The solid oral dosage forms described herein will typically be in
the form of a
tablet. The provision of a tablet with particular pharmacokinetic parameters
is a particular
advantage afforded by the present invention. Pharmacokinetic parameters refer
to any suitable
pharmacokinetic parameters, such as Trnax, Cmax, and AUC. Parameters should be
measured in
accordance with standards and practices which would be acceptable to a
pharmaceutical
regulatory agency such as FDA, EMA, MHLW, or WHO. The values may be based on
measurements taken at appropriate intervals following the time of tablet
ingestion, such as every
hour, or at increasingly sparse sampling intervals, such as 2, 4, 6, 8, 10,
12, 16, and 24 hours
after ingestion. The pharmacokinetic parameters can be assessed either
following a single-dose
of drug or at steady state, preferably following a single-dose. In certain
embodiments,
pharmacokinetic parameters are determined following a single dose of the
pharmaceutical
composition. In certain embodiments, pharmacokinetic parameters are determined
in a multiple
dosing regimen. For example, pharmacokinetic parameters may be determined
after several
dosing intervals, e.g., at steady state. The pharmacokinetic parameters can be
assessed under
fasting or fed conditions, preferably under fasting conditions.
[00124] Cmax refers to the peak concentration and, in particular, the
maximum observed
plasma/serum concentration of drug. T. refers to the time to reach the peak
concentration.
AUCt refers to the area under the plasma concentration-time curve, where t is
the time of the last
measurable plasma concentration in the study. AUC,, refers to the area under
the plasma
concentration-time curve from time zero to infinity following a single dose.
[00125] In certain embodiments, a solid oral dosage form (in particular a
tablet) is
' provided as described herein, wherein the dosage form when administered as a
single dose to a
population of human subjects provides an average T. less than about 3 hours
for the population
of human subjects. In certain embodiments, a solid oral dosage form (in
particular a tablet) is
provided as described herein, wherein the dosage form when administered as a
single dose to a
population of human subjects provides an average Trnax from about 0.5 hours to
about 2.0 hours
for the population of human subjects. In some such embodiments, the solid oral
dosage form is
administered under fasting conditions.
[00126] In certain embodiments, a solid oral dosage form (in particular,
a tablet) is
provided as described herein, wherein the dosage comprises sodium 4-((R)-2-[5-
(2-fluoro-3-
methoxy-phenyl)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-dioxo-3,6-
dihydro-2H-
22
CA 2977938 2017-09-01
pyrimidin-1-y1]-1-phenyl-ethylamino)butanoate in an amount equivalent to about
150 mg of
Compound A and wherein the dosage form when administered as a single dose to a
population of
human subjects provides an average Cmcix from about 400 ng/mL to about 660
ng/mL, an average
AUCt from about 1000 ng=hr/mL to about 1600 ng=hr/mL, and/or an average AUC.
from about
1010 ng=hr/mL to about 1610 ng=hr/mL for the population of human subjects. In
some such
embodiments, the solid oral dosage form is administered under fasting
conditions.
[00127] In certain embodiments, a solid oral dosage form (in particular a
tablet) is
provided as described herein, wherein the dosage comprises sodium 44(R)-2-[5-
(2-fluoro-3-
methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-dioxo-3,6-
dihydro-2H-
pyrimidin-1-y1]-1-phenyl-ethylamino)butanoate in an amount equivalent to about
200 mg of
Compound A and wherein the dosage form when administered as a single dose to a
population of
human subjects provides an average C. from about 590 ng/mL to about 1100
ng/mL,
preferably from about 590 ng/mL to about 930 ng/mL, an average AUCt from about
1510
ng=hr/mL to about 2980 ng-hr/mL, preferably from about 1520 ng=hr/mL to about
2390
ng=hr/mL, and/or an average AUC. from about 1520 ng=hr/mL to about 2990 ng-
hr/mL,
preferably from about 1530 ng=hr/mL to about 2400 ng=hr/mL, for the population
of human
subjects. In some such embodiments, the solid oral dosage form is administered
under fasting
conditions.
[00128] In certain embodiments, a solid oral dosage form (in particular a
tablet) is
provided as described herein, wherein the dosage comprises sodium 44(R)-2-[5-
(2-fluoro-3-
methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-dioxo-3,6-
dihydro-2H-
pyrimidin-1-y1]-1-phenyl-ethylamino)butanoate in an amount equivalent to about
300 mg of
Compound A and wherein the dosage form when administered as a single dose to a
population of
human subjects provides an average Cmax from about 1100 ng/mL to about 1730
ng/mL, an
average AUCt from about 2990 ng=hr/mL to about 4670 ng=hr/mL, and/or an
average AUG-,
from about 3020 ng=hr/mL to about 4720 ng-hr/mL for the population of human
subjects. In
some such embodiments, the solid oral dosage form is administered under
fasting conditions.
[00129] In some embodiments, a solid oral dosage form (in particular a
tablet) as
described herein is provided, for which the 90% confidence interval of log -
transformed C.x,
log-transformed AUCt, and/or log-transformed AUC. for Compound A or a
pharmaceutically
acceptable salt thereof in a population of human subjects falls completely
within the range 80-
23
CA 2977938 2017-09-01
125% of the log -transformed C., log-transformed AUCt, and/or log-transformed
AUC.,
respectively, of a reference tablet, wherein the reference tablet comprises
sodium 44(R)-2-[5-(2-
fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-
dioxo-3,6-
dihydro-2H-pyrimidin-l-y1]-1-phenyl-ethylamino)butanoate in an amount
equivalent to about
150 mg of Compound A; about 150 mg mannitol; about 44 mg pregelatinized
starch; about 14
mg povidone; about 78 mg sodium carbonate monohydrate; about 8 mg magnesium
stearate; and
a film coating consisting of polyvinyl alcohol; titanium dioxide; polyethylene
glycol; talc; and
high tint carmine (such as Opadry 0 II Pink).
[00130] In some embodiments, a solid oral dosage form (in particular a
tablet) as
described herein is provided, for which the 90% confidence interval of log -
transformed Cmax,
log-transformed AUCt, and/or log-transformed AUC. for Compound A or a
pharmaceutically
acceptable salt thereof in a population of human subjects falls completely
within the range 80-
125% of the log -transformed Cmax, log-transformed AUCt, and/or log-
transformed AUC.,
respectively, of a reference tablet, wherein the reference tablet comprises
sodium 44(R)-245-(2-
fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-
dioxo-3,6-
dihydro-2H-pyrimidin-1-y11-1-phenyl-ethylamino)butanoate in an amount
equivalent to about
200 mg of Compound A; about 200 mg mannitol; about 59 mg pregelatinized
starch; about 18
mg povidone; about 104 mg sodium carbonate monohydrate; about 11 mg magnesium
stearate;
and a film coating consisting of polyvinyl alcohol; titanium dioxide;
polyethylene glycol; talc;
and iron oxide red (such as Opadry 8 II Salmon).
[00131] In some embodiments, a solid oral dosage form (in particular a
tablet) as
described herein is provided, for which the 90% confidence interval of log -
transformed C.,
log-transformed AUCt, and/or log-transformed AUC. for Compound A or a
pharmaceutically
acceptable salt thereof in a population of human subjects falls completely
within the range 80-
125% of the log -transformed Cmax, log-transformed AUCt, and/or log-
transformed AUC.,
respectively, of a reference tablet, wherein the reference tablet comprises
sodium 44(R)-2-[5-(2-
fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-
dioxo-3,6-
dihydro-2H-pyrimidin-1-y1]-1-phenyl-ethylamino)butanoate in an amount
equivalent to about
300 mg of Compound A; mannitol; pregelatinized starch; povidone; sodium
carbonate
monohydrate; magnesium stearate; and a film coating.
24
CA 2977938 2017-09-01
[00132] In certain embodiments, administration of a pharmaceutical
composition
comprising Compound A or a pharmaceutically acceptable salt thereof results in
rapid
suppression of luteinizing hormone (LH) and/or follicle-stimulating hormone
(FSH) levels in a
female patient with endometriosis or uterine fibroids. In certain embodiments,
administration of
a pharmaceutical composition comprising Compound A or a pharmaceutically
acceptable salt
thereof results in partial to substantially full suppression of estradiol
levels in a female patient
with endometriosis or uterine fibroids. In some such embodiments, estradiol
levels are less than
about 50 pg/mL. In some such embodiments, estradiol levels are between about
20 pg/mL and
about 50 pg/mL. In some such embodiments, estradiol levels are less than about
20 pg/mL. In
some such embodiments, estradiol levels are less than about 12 pg/mL (e.g.,
below the lowest
limit of quantitation).
[00133] The pharmaceutical compositions may comprise other excipients such
as
excipents that function as fillers, binders, disintegrants, glidants and
lubricants. Thus, a
pharmaceutical composition comprising Compound A or a pharmaceutically
acceptable salt
thereof, further optionally comprises one or more conventional
pharmaceutically acceptable
excipients.
[00134] In certain embodiments, the disclosed pharmaceutical compositions
comprise at
least one excipient that functions as a filler. Fillers may include polyols,
such as dextrose,
isomalt, mannitol, sorbitol, lactose, and sucrose; natural or pre-gelatinized
potato or corn starch;
microcrystalline cellulose (e.g., Avice10); or a combination thereof. Examples
of suitable fillers
include mannitol, such as spray dried mannitol (e.g., Pearlito10 100SD,
Pearlitolg 200SD);
pregelatinized starch, such as Starch 1500e; microcrystalline cellulose, such
as Avice10 ;
lactose monohydrate, such as Foremost 316 Fast Floe; mixtures of isomaltulose
derivatives
such as gaIenIQTM 720; and other suitable fillers and combinations thereof.
[00135] In certain embodiments, the disclosed pharmaceutical compositions
comprise at
least one water soluble filler. In some such embodiments, the water soluble
filler is a polyol,
such as mannitol, sorbitol, lactose, or sucrose; a pregelatinized starch; or a
combination thereof.
In some such embodiments, the water soluble filler is mannitol. In some such
embodiments, the
water soluble filler is mannitol and pregelitanized starch. In certain
embodiments, the disclosed
pharmaceutical compositions comprise at least one water insoluble filler. In
some such
embodiments, the water insoluble filler is a starch, microcrystalline
cellulose (e.g., Avice10), or
CA 2977938 2017-09-01
calcium phosphate. In some such embodiments, the disclosed pharmaceutical
compositions
comprise a water insoluble filler and a surfactant, such as sodium lauryl
sulfate (SLS).
[001361 In certain embodiments, a filler is present in the pharmaceutical
composition in an
amount from about 5% to about 70% by weight (w/w) of the pharmaceutical
composition. In
certain embodiments, the filler is present in the pharmaceutical composition
in an amount from
about 10% to about 60% by weight (w/w) of the pharmaceutical composition. In
certain
embodiments, the filler is present in the pharmaceutical composition in an
amount from about
20% to about 50% by weight (w/w) of the pharmaceutical composition. In certain
embodiments,
the filler is present in the pharmaceutical composition in an amount from
about 30% to about
45% by weight (w/w) of the pharmaceutical composition.
[00137] In certain embodiments, the pharmaceutical composition includes a
first filler in
an amount from about 20% to about 50% by weight and a second filler in an
amount from about
1% to about 20% by weight of the pharmaceutical composition. In certain
embodiments, the
pharmaceutical composition includes a first filler in an amount from about 25%
to about 40% by
weight and a second filler in an amount from about 5% to about 15% by weight
of the
pharmaceutical composition. In certain embodiments, the pharmaceutical
composition includes a
first filler in an amount from about 30% to about 35% by weight and a second
filler in an amount
from about 8% to about 12% by weight of the pharmaceutical composition. In
certain
embodiments, the pharmaceutical composition includes a first filler in an
amount of about 33%
by weight and a second filler in an amount of about 10% by weight of the
pharmaceutical
composition. In certain embodiments, the first filler is mannitol and the
second filler is
pregelitanized starch.
[00138] In certain embodiments, the disclosed pharmaceutical compositions
comprise at
least one excipient that functions as a binder. Binders may include
polyvinylpyrrolidone (e.g.,
povidone), a copolymer of vinylpyrrolidone and vinyl acetate (e.g.,
copovidone); cellulose, such
as hydroxymethylpropylcellulose (HPMC), hydroxypropylethylcellulose, or
microcrystalline
cellulose; sucrose, starches, and combinations thereof. In certain
embodiments, the binder is a
hydrophilic polymer. The hydrophilic polymer may be selected from copolymer of
N-vinyl
lactam, cellulose ester, cellulose ether, polyalkylene glycol, polyacrylate,
polymethacrylate,
polyacrylamide, polyvinyl alcohol, vinyl acetate polymer, oligosaccharide,
polysaccharide, or
combinations thereof. In some such embodiments, the binder is
polyvinylpyrrolidone.
26
CA 2977938 2017-09-01
[00139] In certain embodiments, a binder is present in the pharmaceutical
composition in
an amount from about 0.1% to about 20% by weight (w/w) of the pharmaceutical
composition.
In certain embodiments, a binder is present in the pharmaceutical composition
in an amount from
about 1% to about 10% by weight (w/w) of the pharmaceutical composition. In
certain
embodiments, a binder is present in the pharmaceutical composition in an
amount from about 2%
to about 5% by weight (w/w) of the pharmaceutical composition. In certain
embodiments, the
pharmaceutical composition includes about 3% by weight of a binder. In certain
embodiments,
the binder is polyvinylpyrrolidone.
[00140] In certain embodiments, the disclosed pharmaceutical compositions
comprise at
least one excipient that functions as a glidant. Glidants may include, for
example, colloidal
silicon dioxide, including highly dispersed silica (Aerosile) or any other
suitable glidant such as
animal or vegetable fats or waxes.
[00141] In certain embodiments, a glidant is present in the pharmaceutical
composition in
an amount from about 0.1% to about 5% by weight (w/w) of the pharmaceutical
composition. In
certain embodiments, a glidant is present in the pharmaceutical composition in
an amount from
about 0.3% to about 2% by weight (w/w) of the pharmaceutical composition. In
certain
embodiments, a glidant is present in the pharmaceutical composition in an
amount from about
0.3% to about 1.2% by weight (w/w) of the pharmaceutical composition. In
certain
embodiments, the pharmaceutical composition includes about 0.5% by weight of a
glidant. In
certain embodiments, the glidant is colloidal silicon dioxide.
[00142] In certain embodiments, a glidant is included in an intragranular
portion of the
pharmaceutical composition. In certain embodiments, the intragranular portion
of the
pharmaceutical composition comprises a glidant in an amount from about 0.1% to
about 5% by
weight (w/w), on the basis of the weight of the total pharmaceutical
composition. In certain
embodiments, the intragranular portion of the pharmaceutical composition
comprises a glidant in
an amount from about 0.5% to about 3% by weight (w/w), on the basis of the
weight of the total
pharmaceutical composition.
[00143] In certain embodiments, a glidant is included in an extragranular
portion of the
pharmaceutical composition. In certain embodiments, the extragranular portion
of the
pharmaceutical composition comprises a glidant in an amount from about 0.1% to
about 5% by
weight (w/w), on the basis of the weight of the total pharmaceutical
composition. In certain
27
CA 2977938 2017-09-01
embodiments, the extragranular portion of the pharmaceutical composition
comprises a glidant in
an amount from about 0.5% to about 3% by weight (w/w), on the basis of the
weight of the total
pharmaceutical composition.
[001441 In certain embodiments, a glidant is included in both an
intragranular portion and
an extragranular portion of the pharmaceutical composition. In certain
embodiments, the
intragranular portion of the pharmaceutical composition comprises a glidant in
an amount from
about 0.1% to about 5% by weight (w/w), on the basis of the weight of the
total pharmaceutical
composition and the extragranular portion of the pharmaceutical composition
comprises a glidant
in an amount from about 0.1% to about 5% by weight (w/w), on the basis of the
weight of the
total pharmaceutical composition. In certain embodiments, the intragranular
portion of the
pharmaceutical composition comprises a glidant in an amount from about 0.5% to
about 3% by
weight (w/w), on the basis of the weight of the total pharmaceutical
composition and the
extragranular portion of the pharmaceutical composition comprises a glidant in
an amount from
about 0.5% to about 3% by weight (w/w), on the basis of the weight of the
total pharmaceutical
composition.
[00145] In certain embodiments, the disclosed pharmaceutical compositions
comprise at
least one excipient that functions as a lubricant. Lubricants may include, for
example,
magnesium and calcium stearates, sodium stearyl fumarate, talc, or any other
suitable lubricant.
[00146] In certain embodiments, a lubricant is present in the
pharmaceutical composition
in an amount from about 0.1% to about 10% by weight (w/w) of the
pharmaceutical
composition. In certain embodiments, a lubricant is present in the
pharmaceutical composition in
an amount from about 0.5% to about 5% by weight (w/w) of the pharmaceutical
composition. In
certain embodiments, a lubricant is present in the pharmaceutical composition
in an amount from
about 1% to about 3% by weight (w/w) of the pharmaceutical composition. In
certain
embodiments, the pharmaceutical composition includes about 1.9% by weight of a
lubricant. In
certain embodiments, the lubricant is magnesium stearate.
[00147] In certain embodiments, a lubricant is included in an
intragranular portion of the
pharmaceutical composition. In certain embodiments, the intragranular portion
of the
pharmaceutical composition comprises a lubricant in an amount from about 0.5%
to about 5% by
weight (w/w), on the basis of the weight of the total pharmaceutical
composition. In certain
embodiments, the intragranular portion of the pharmaceutical composition
comprises a lubricant
28
CA 2977938 2017-09-01
in an amount from about 1% to about 3% by weight (w/w), on the basis of the
weight of the total
pharmaceutical composition.
[00148] In certain embodiments, a lubricant is included in an extragranular
portion of the
pharmaceutical composition. In certain embodiments, the extragranular portion
of the
pharmaceutical composition comprises a lubricant in an amount from about 0.5%
to about 5% by
weight (w/w), on the basis of the weight of the total pharmaceutical
composition. In certain
embodiments, the extragranular portion of the pharmaceutical composition
comprises a lubricant
in an amount from about 1% to about 3% by weight (w/w), on the basis of the
weight of the total
pharmaceutical composition. In certain embodiments, magnesium stearate is used
as a lubricant
and the magnesium stearate is in the extragranular portion.
[00149] In certain embodiments, a lubricant is included in both an
intragranular portion
and an extragranular portion of the pharmaceutical composition. In certain
embodiments, the
intragranular portion of the pharmaceutical composition comprises a lubricant
in an amount from
about 0.5% to about 5% by weight (w/w), on the basis of the weight of the
total pharmaceutical
composition and the extragranular portion of the pharmaceutical composition
comprises a
lubricant in an amount from about 0.5% to about 5% by weight (w/w), on the
basis of the weight
of the total pharmaceutical composition. In certain embodiments, the
intragranular portion of the
pharmaceutical composition comprises a lubricant in an amount from about 1% to
about 3% by
weight (w/w), on the basis of the weight of the total pharmaceutical
composition and the
extragranular portion of the pharmaceutical composition comprises a lubricant
in an amount
from about 1% to about 3% by weight (w/w), on the basis of the weight of the
total
pharmaceutical composition. In certain embodiments, the intragranular portion
of the
pharmaceutical composition comprises a lubricant in an amount of about 0.9% by
weight (w/w),
on the basis of the weight of the total pharmaceutical composition and the
extragranular portion
of the pharmaceutical composition comprises a lubricant in an amount of about
1% by weight
(w/w), on the basis of the weight of the total pharmaceutical composition. In
certain
embodiments, magnesium stearate is used as a lubricant at a level of about
1.9% weight/weight
of the formulation with about 0.9% added intragranular and about I% added
extragranular.
[00150] In certain embodiments, the disclosed pharmaceutical compositions
comprise at
least one excipient that functions as a disintegrant. Disintegrants may
include, for example,
sodium starch glycolate (e.g., Explotab), cross-linked polymers such as cross-
linked modified
29
CA 2977938 2017-09-01
starches, cross-linked polyvinylpyrrolidone, also known as crospovidone, and
cross-linked
carboxymethyl cellulose, also known as croscarmellose. In certain embodiments,
a disintegrant is
present in the pharmaceutical composition in an amount from about 0.1% to
about 20% by
weight (w/w) of the pharmaceutical composition.
[00151] In certain embodiments, the pharmaceutical composition is a tablet,
which may be
coated with any suitable coating such as a film coat. A film coat may be used
to, for example,
contribute to the ease with which the tablet can be swallowed. A film coat may
also be employed
to improve taste and provide an elegant appearance. The film coat may comprise
a polyvinyl
alcohol-polyethylene glycol graft copolymer, such as Opadry II and Kollicoat
IR. The film
coat may also comprise talc as an anti-adhesive. The film coat may account for
less than about
5% by weight of the weight of the tablet.
[00152] In at
least one aspect, this disclosure is directed to providing Compound A or a
pharmaceutically acceptable salt thereof in a single, stable solid oral dosage
form that is
pharmacologically efficacious and physically acceptable. The solid oral dosage
forms disclosed
herein are intended for pharmaceutical use in human subjects. Accordingly,
they should be of an
appropriate size and weight for oral human administration (e.g., they should
have a total weight
of less than about 1.5 g), in addition to being therapeutically efficacious.
In order to facilitate the
intake of such a dosage form by a mammal, the dosage form may be shaped into
an appropriate
shape such as a round or elongated shape.
[00153] For
example, as set forth in Table 1, the disclosed pharmaceutical compositions
may include one or more fillers, disintegrants, glidants and/or lubricants in
combination with the
active agent and anti-gelling agent.
[00154] Compound
A referenced in Table 1 below is Compound A sodium salt and the
corresponding weight percent is provided based on that salt form.
[001551 Table 1.
Quantity %a Quantity %a
Ingredient Function
(mg/Tablet) (w/w) (mg/Tablet) (w/w)
Tablet Core
Intragranular
Compound A, sodium salt Drug Substance 155.2 33 207.0 33
Mannitol, USP Filler 150.3 32 200.3 32
CA 2977938 2017-09-01
Pregelatinized Starch, NF Filler/Binder 44.3 9 59.1 9
Povidone K 29/32, USP Binder 13.8 3 18.4 3
Sodium carbonate Anti-gelling
78.0 17 104.0 17
monohydrate, NF Agent
Magnesium stearate, NF Lubricant 3.9 1 5.2 1
Weight subtotal of intragranular
445.5 594.0
components
Extragranular
Magnesium stearate, NF Lubricant 4.5 1 6.0 1
Uncoated tablet weight 450.0 600.0
Film Coating
Film-Coating Powder Film Coat
18.0 4 24.0 4
(Op adry0 II)
Coated tablet weight 468.0 100 624.0 100
[00156]
"Percents given based on the coated tablet weight. Total percentage may not be
100% due to rounding.
[00157] The
amount (mg) of Compound A or pharmaceutically acceptable salt thereof
referenced in the following tables refers to the amount (mg) of Compound A
free form (i.e., in
the case of a pharmaceutically acceptable salt, the free form equivalent
weight).
[00158] In certain
embodiments, the pharmaceutical composition comprises:
Ingredient Amount (mg)
Compound A or pharmaceutically acceptable salt thereof 125-175
Mannitol 125-175
Pregelitanized starch 35-55
Povidone 13-15
Sodium Carbonate 66-90
Magnesium Stearate 7-10
Optional film-coating 16-20
31
CA 2977938 2017-09-01
[00159] In certain
embodiments, the pharmaceutical composition comprises:
Ingredient Amount (mg)
Compound A or pharmaceutically acceptable salt thereof 170-230
Mannitol 170-230
Pregelitanized starch 47-71
Povidone 17-20
Sodium Carbonate 88-120
Magnesium Stearate 9-13
Optional film-coating 21-27
[00160] In certain
embodiments, the pharmaceutical composition comprises:
Ingredient Amount (mg)
Compound A or pharmaceutically acceptable salt thereof 255-345
Mannitol 255-345
Pregelitanized starch 70-107
Povidone 25-30
Sodium Carbonate 132-180
Magnesium Stearate 14-20
Optional film-coating 24-30
[00161] In certain
embodiments, the pharmaceutical composition comprises:
Ingredient Amount (mg)
Compound A or pharmaceutically acceptable salt thereof 150
Mannitol 150
Pregelitanized starch 44.3
32
CA 2977938 2017-09-01
Povidone 13.8
Sodium Carbonate 78
Magnesium Stearate 8.4
Optional film-coating 18
[00162] In certain embodiments, the pharmaceutical composition comprises:
Ingredient Amount (mg)
Compound A or pharmaceutically acceptable salt thereof 200
Mannitol 200
Pregelitanized starch 59.1
Povidone 18.4
Sodium Carbonate 104
Magnesium Stearate 11.2
Optional film-coating 24
[00163] In certain embodiments, the pharmaceutical composition comprises:
Ingredient Amount (mg)
Compound A or pharmaceutically acceptable salt thereof 300
Mannitol 300
Pregelitanized starch 88.7
Povidone 27.7
Sodium Carbonate 156
Magnesium Stearate 16.8
Optional film-coating 27
33
CA 2977938 2017-09-01
[00164] D. METHODS OF MANUFACTURE
[00165] The disclosed pharmaceutical compositions may be prepared by any
suitable
method. Methods such as direct compression, fluid bed granulation, roller
compaction or dry
granulation, and wet granulation may be used to blend Compound A or a
pharmaceutically
acceptable salt thereof with an anti-gelling agent and any other excipients of
the pharmaceutical
composition, including a water soluble filler or a water insoluble filler and
a surfactant.
[00166] In certain embodiments, the disclosed pharmaceutical compositions
are prepared
using a wet granulation process and by compressing the final blend into
tablets.
[00167] In certain embodiments, the disclosed pharmaceutical compositions
are prepared
using a roller compaction process. The roller compaction process may include
any suitable steps.
For example, as illustrated in Figure 1, roller compaction may include steps
such as blending the
active agent with one or more intragranular excipients sized for blending;
feeding the blend into
a roller compactor to densify loose powder into ribbons; milling the resultant
ribbons into
granules; optionally blending the granules with extragranular excipients such
as lubricants;
compressing the granules into tablets; and optionally coating the tablets with
a film-coating.
[00168] E. METHODS OF USE
[00169] In at least one aspect, the present invention includes a method of
treating
endometriosis comprising administering to a patient a pharmaceutical
composition comprising
Compound A or a pharmaceutically acceptable salt thereof. In certain
embodiments, the method
of treating endometriosis comprises administration of a pharmaceutical
composition comprising
Compound A or a pharmaceutically acceptable salt thereof at a dose of about
150 mg. In some
such embodiments, the composition is administered once per day ("QD"). In
certain
embodiments, the method of treating endometriosis comprises administration of
a
pharmaceutical composition comprising Compound A or a pharmaceutically
acceptable salt
thereof at a dose of about 200 mg. In some such embodiments, the composition
is administered
twice per day ("BID"). In certain embodiments, the method of treating
endometriosis comprises
administration of a pharmaceutical composition comprising Compound A or a
pharmaceutically
acceptable salt thereof at a dose of about 300 mg. In some such embodiments,
the composition is
administered twice per day ("BID"). In certain embodiments, the method of
treating
endometriosis comprises administration of a pharmaceutical composition
comprising Compound
34
CA 2977938 2017-09-01
A or a pharmaceutically acceptable salt thereof at a dose of about 600 mg. In
some such
embodiments, the composition is administered once per day ("QD").
[00170] In at least one aspect, the present invention includes a method of
treating uterine
fibroids comprising administering to a patient a pharmaceutical composition
comprising
Compound A or a pharmaceutically acceptable salt thereof. In certain
embodiments, the method
of treating uterine fibroids comprises administration of a pharmaceutical
composition comprising
Compound A or a pharmaceutically acceptable salt thereof at a dose of about
150 mg. In some
such embodiments, the composition is administered QD. In certain embodiments,
the method of
treating uterine fibroids comprises administration of a pharmaceutical
composition comprising
Compound A or a pharmaceutically acceptable salt thereof at a dose of about
200 mg. In some
such embodiments, the composition is administered BID. In certain embodiments,
the method of
treating uterine fibroids comprises administration of a pharmaceutical
composition comprising
Compound A or a pharmaceutically acceptable salt thereof at a dose of about
300 mg. In some
such embodiments, the composition is administered BID. In certain embodiments,
the method of
treating uterine fibroids comprises administration of a pharmaceutical
composition comprising
Compound A or a pharmaceutically acceptable salt thereof at a dose of about
600 mg. In some
such embodiments, the composition is administered QD.
[001711 In certain embodiments, any of the above methods further comprise
administering
to the subject a hormone to reduce or alleviate potential side effects of
Compound A or a
pharmaceutically acceptable salt thereof. For example, the method may comprise
administration
of an estrogen, a progestin, or a combination thereof. Such treatments are
commonly referred to
as "add-back" therapy.
(00172) In some such embodiments, the add-back therapy comprises a
progestogen, such
as a progestin. In some such embodiments, the add-back therapy comprises an
estrogen. In some
such embodiments, the add-back therapy comprises a progestin and an estrogen.
[00173] The estrogen and/or progestogen can be administered orally,
transdermally or
intravaginally. Suitable progestogens for use in the add-back therapy include,
for example,
progesterone, norethindrone, norethindrone acetate, norgestimate,
drospirenone, and
medroxyprogestogen. Suitable estrogens for use in the add-back therapy
include, for example,
estradiol, ethinyl estradiol, and conjugated estrogens. Combined oral
formulations containing an
CA 2977938 2017-09-01
estrogen and a progestogen are known in the art and include, for example,
Activellae,
Angeliqe, FemHRTO, JenteliTM, MimveyTM, PrefestTM, Premphaset, and Premproe.
[00174] In certain embodiments, the estrogen is estradiol, ethinyl
estradiol, or a
conjugated estrogen. In some such embodiments, the estrogen is estradiol. In
some such
embodiments, the estradiol is administered once a day. In some such
embodiments, the dose of
estradiol is 0.5 mg. In other such embodiments, the dose of estradiol is 1.0
mg. In some such
embodiments, the estrogen is ethinyl estradiol. In some such embodiments, the
ethinyl estradiol
is administered once a day. In some such embodiments, the dose of ethinyl
estradiol is 2.5 meg.
In other such embodiments, the dose of ethinyl estradiol is 5.0 meg. In some
such embodiments,
the estrogen is a conjugated estrogen. In some such embodiments, the
conjugated estrogen is
administered once a day. In some such embodiments, the dose of conjugated
estrogen is 0.3 mg.
In other such embodiments, the dose of conjugated estrogen is 0.45 mg or 0.625
mg.
[00175] In certain embodiments, the progestogen is progesterone,
norethindrone,
norethindrone acetate, norgestimate, medroxyprogesterone, or drospirenone. In
some such
embodiments, the progestogen is oral progesterone. In some such embodiments,
the oral
progesterone is used cyclically (for the last 12 days of the 28-30 day cycle).
In some such
embodiments, the dose of the oral progesterone is 100 or 200 mg. In some such
embodiments,
the progestogen is norethindrone or norethindrone acetate. In some such
embodiments, the
norethindrone or norethindrone acetate is administered once a day. In some
such embodiments,
the dose of norethindrone or norethindrone acetate is 0.1 mg. In some such
embodiments, the
dose of norethindrone or norethindrone acetate is 0.5 mg. In some such
embodiments, the dose of
norethindrone or norethindrone acetate is 1.0 mg. In some such embodiments,
the progestogen is
norgestimate. In some such embodiments, the norgestimate is administered once
a day. In some
such embodiments, the dose of norgestimate is 0.09 mg. In some such
embodiments, the
progestogen is medroxyprogesterone. In some such embodiments, the
medroxyprogesterone is
administered once a day. In some such embodiments, the dose of
medroxyprogesterone is 1.5
mg. In some such embodiments, the dose of medroxyprogesterone is 2.5 mg or 5
mg. In some
such embodiments, the progestogen is drospirenone. In some such embodiments,
the
drospirenone is administered once a day. In some such embodiments, the dose of
drospirenone is
0.25 mg. In some such embodiments, the dose of drospirenone is 0.5 mg.
36
CA 2977938 2017-09-01
[00176] In certain embodiments, the add-back therapy comprises a
norethisterone prodrug,
such as norethindrone acetate. In some such embodiments, the add-back therapy
further
comprises estradiol. Thus, in some such embodiments, the add-back therapy
comprises estradiol
and norethindrone acetate. In some such embodiments, estradiol and
norethindrone acetate are
administered orally once per day. In some such embodiments, estradiol is
administered in an
amount of about 0.5 mg and norethindrone acetate is administered in an amount
of about 0.1 mg
per day. In other such embodiments, estradiol is administered in an amount of
about 1.0 mg and
norethindrone acetate is administered in an amount of about 0.5 mg per day.
Alternatively, in
certain embodiments, estradiol is administered continuously and norethindrone
acetate is
administered once per day during the last 12-14 days of a menstrual cycle.
[00177] In certain embodiments, the dose of Compound A or a
pharmaceutically
acceptable salt thereof is administered twice a day. In some such embodiments,
add-back therapy
is administered once a day. The administration of Compound A or a
pharmaceutically acceptable
salt thereof may be prior to, immediately prior to, during, immediately
subsequent to or
subsequent to the administration of the add-back therapy.
[00178] In certain embodiments, a dose of Compound A or pharmaceutically
acceptable
salt thereof (e.g., 300 mg) is administered in the morning with add-back
therapy, such as a
combination of an estrogen and a progestogen (e.g., estradiol and
norethindrone acetate) and a
dose of Compound A or pharmaceutically acceptable salt thereof (e.g., 300 mg)
is administered
in the evening without add-back therapy.
[00179] In certain embodiments, Compound A or a pharmaceutically acceptable
salt
thereof is co-packaged with the add-back therapy. For example, a blister pack
may contain a
dose of Compound A or a pharmaceutically acceptable salt thereof and a dose of
the add-back
therapy.
[00180] In certain embodiments, Compound A or a pharmaceutically acceptable
salt
thereof is present in a fixed dose combination with the add-back therapy. For
example, a capsule
may contain a caplet or tablet comprising Compound A or a pharmaceutically
acceptable salt
thereof and a caplet or tablet comprising the add-back therapy, such as a
combination of an
estrogen and a progestogen (e.g., estradiol and norethindrone acetate). In
some such
embodiments, the capsule comprises 300 mg Compound A or a pharmaceutically
acceptable salt
thereof, 1 mg estradiol, and 0.5 mg norethindrone acetate.
37
CA 2977938 2017-09-01
[00181] The pharmaceutical compositions, methods, and uses described herein
will be
better understood by reference to the following exemplary embodiments and
examples, which
are included as an illustration of and not a limitation upon the scope of the
invention.
[00182] F. EXEMPLARY EMBODIMENTS
[00183] This disclosure provides at least the following enumerated
exemplary
embodiments:
Al. A pharmaceutical composition comprising about 150 mg, about 200 mg,
or about
300 mg 4-((R)-2-[5-(2-fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-
benzy1)-4-
meth y1-2,6-dioxo-3,6-dihydro-2H-pyrimidin-l-y11-1-phenyl-ethylamino)-butyric
acid
(Compound A) or a pharmaceutically acceptable salt thereof; and an anti-
gelling agent; wherein
the pharmaceutical composition comprises at least 10% by weight of Compound A
or the
pharmaceutically acceptable salt thereof.
A2. The pharmaceutical composition of embodiment Al, wherein the anti-
gelling
agent is a water soluble salt of a weak acid, a basic amino acid, or a
poly(meth)acrylate polymer.
A3. The pharmaceutical composition of embodiment Al or embodiment A2,
wherein
the anti-gelling agent further acts as a stabilizer to reduce formation of (R)-
5-(2-fluoro-3-
methox ypheny1)-1-(2-fluoro-6-(trifluoromethyl)benzy1)-6-meth y1-3-(2-(2-
oxopyrrolidin-l-y1)-2-
phenylethyl)pyrimidine-2,4(1H,3H)-dione (Compound B) in the composition
relative to an
otherwise identical composition without the anti-gelling agent.
A4. The pharmaceutical composition of embodiment A3, wherein the anti-
gelling
agent comprises an alkali metal salt.
A5. The pharmaceutical composition of embodiment A4, wherein the alkali
metal salt
is selected from the group consisting of sodium carbonate, sodium hydrogen
carbonate,
potassium carbonate, potassium hydrogen carbonate, sodium phosphate, and
combinations
thereof.
A6. The pharmaceutical composition of embodiment A4, wherein the alkali
metal salt
is sodium carbonate, preferably sodium carbonate monohydrate.
38
CA 2977938 2017-09-01
A7. The pharmaceutical composition of any one of embodiments A3-A6, wherein
the
weight ratio of Compound Aor the pharmaceutically acceptable salt thereof to
the anti-gelling
agent, is from about 1:1 to about 4:1, preferably about 2:1.
A8. The pharmaceutical composition of any one of embodiments A3-A6, wherein
the
anti-gelling agent is present in an amount from about 5% by weight to about
35% by weight of
the pharmaceutical composition, preferably in an amount from about 10% by
weight to about
25% by weight of the pharmaceutical composition.
A9. The pharmaceutical composition of any one of the preceding embodiments,
further comprising (a) at least one water soluble filler or (b) at least one
water insoluble filler and
a surfactant.
A10. The pharmaceutical composition of any one of the preceding embodiments,
wherein the composition releases at least about 80% of Compound A or the
pharmaceutically
acceptable salt thereof in about 45 minutes, preferably at least about 80% of
Compound Aor the
pharmaceutically acceptable salt thereof in about 30 minutes, measured using
USP apparatus II
in 900 mL of sodium phosphate, pH 6.8, at 37 C and paddle speed of 50 rpm.
All. The pharmaceutical composition of any one of the preceding embodiments,
further comprising at least one lubricant.
Al2. The pharmaceutical composition of any one of the preceding embodiments,
wherein the pharmaceutical composition is a solid oral dosage form.
A13. The pharmaceutical composition of embodiment Al2, wherein the solid oral
dosage form is a tablet.
A14. The pharmaceutical composition of any one of the preceding embodiments,
wherein the pharmaceutical composition comprises a salt of Compound A.
A15. The pharmaceutical composition of embodiment A14, wherein the salt of
Compound A is sodium 44(R)-245-(2-fluoro-3-methoxy-phenyl)-3-(2-fluoro-6-
trifluoromethyl-
benzy1)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-l-y11-1-phenyl-
ethylamino)butanoate.
39
CA 2977938 2017-09-01
A16. The pharmaceutical composition of embodiment A14, wherein the salt of
Compound A is amorphous sodium 44(R)-2-[5-(2-fluoro-3-methoxy-pheny1)-3-(2-
fluoro-6-
trifluoromethyl-benzy1)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-y1]-1-
phenyl-
ethylarnino)butanoate.
A17. The pharmaceutical composition of embodiment A15, wherein the composition
when administered as a single dose to a population of human subjects provides
an average Tmax
value that is less than about 3 hours, preferably about 0.5 to about 2.0
hours.
A18. The pharmaceutical composition of embodiment A15, wherein 44(R)-2-[5-(2-
fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-
dioxo-3,6-
dihydro-2H-pyrimidin-1-y11-1-phenyl-ethylamino)butanoate is present in an
amount equivalent
to about 150 mg of Compound A and the composition when administered as a
single dose to a
population of human subjects provides an average Cmax value that is about 400
ng/mL to about
660 ng/mL for the population of human subjects.
A19. The pharmaceutical composition of embodiment A15, wherein 4-((R)-2-[5-(2-
fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-
dioxo-3,6-
dihydro-2H-pyrimidin-1-y11-1-phenyl-ethylamino)butanoate is present in an
amount equivalent
to about 150 mg of Compound A and the composition when administered as a
single dose to a
population of human subjects provides an average AUCt from about 1000 ng=hr/mL
to about
1600 ng=hr/mL for the population of human subjects.
A20. The pharmaceutical composition of embodiment A15, wherein 4-((R)-2-[5-(2-
fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-
dioxo-3,6-
dihydro-2H-pyrimidin-l-y11-1-phenyl-ethylamino)butanoate is present in an
amount equivalent
to about 150 mg of Compound A and the composition when administered as a
single dose to a
population of human subjects provides an average AUC. from about 1010 ng=hr/mL
to about
1610 ng=hr/mt for the population of human subjects.
A21. The pharmaceutical composition of embodiment A15, wherein 4-((R)-215-(2-
fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-
dioxo-3,6-
dihydro-2H-pyrimidin-1-y1]-1-phenyl-ethylamino)butanoate is present in an
amount equivalent
CA 2977938 2017-09-01
to about 200 mg of Compound A and the solid oral dosage form when administered
as a single
dose to a population of human subjects provides an average Cmax from about 590
ng/mL to about
1100 ng/mL for the population of human subjects.
A22. The pharmaceutical composition of embodiment A15, wherein 44(R)-245-(2-
fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-
dioxo-3,6-
dihydro-2H-pyrimidin-1-y1]-1-phenyl-ethylamino)butanoate is present in an
amount equivalent
to about 200 mg of Compound A and the solid oral dosage form when administered
as a single
dose to a population of human subjects provides an average AUCt from about
1510 ng=hr/mL to
about 2980 ng=hr/mL for the population of human subjects.
A23. The pharmaceutical composition of embodiment A15, wherein 44(R)-245-(2-
fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-
dioxo-3,6-
dihydro-2H-pyrimidin-l-y1]-1-phenyl-ethylamino)butanoate is present in an
amount equivalent
to about 200 mg of Compound A and the solid oral dosage form when administered
as a single
dose to a population of human subjects provides an average AUG from about
1520 ng=hr/mL to
about 2990 ng-hr/mL for the population of human subjects.
A24. The pharmaceutical composition of embodiment A15, wherein 44(R)-2-[5-(2-
fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-
dioxo-3,6-
dihydro-2H-pyrimidin-1-y1]-1-phenyl-ethylamino)butanoate is present in an
amount equivalent
to about 300 mg of Compound A and the solid oral dosage form when administered
as a single
dose to a population of human subjects provides an average Cmax from about
1100 ng/mL to
about 1730 ng/mL for the population of human subjects.
A25. The pharmaceutical composition of embodiment A15, wherein Compound A or
the pharmaceutically acceptable salt thereof is present in an amount
equivalent to about 300 mg
of Compound A and the solid oral dosage form when administered as a single
dose to a
population of human subjects provides an average AUCt from about 2990 ng=hr/mL
to about
4670 ng=hr/mL for the population of human subjects.
A26. The pharmaceutical composition of embodiment A15, wherein Compound A or
the pharmaceutically acceptable salt thereof is present in an amount
equivalent to about 300 mg
41
CA 2977938 2017-09-01
of Compound A and the solid oral dosage form when administered as a single
dose to a
population of human subjects provides an average AUC. from about 3020 ng=hr/mL
to about
4720 ng=hr/mL for the population of human subjects.
A27. The pharmaceutical composition of any one of the preceding embodiments,
wherein the composition when administered to a female subject provides rapid
suppression of
luteinizing hormone (LH) and follicle-stimulating hormone (FSH) levels.
A28. The pharmaceutical composition of any one of the preceding embodiments,
wherein the pharmaceutical composition comprises less than about 0.7% (R)-5-(2-
fluoro-3-
methoxypheny1)-1-(2-fluoro-6-(trifluoromethyl)benzy1)-6-methyl-3-(2-(2-
oxopyrrolidin-1-y1)-2-
phenylethyl)pyrimidine-2,4(1H,3H)-dione after storage for at least one month
at 25 C and 60%
relative humidity.
A29. The pharmaceutical composition of any one of embodiments A1-A27, wherein
the pharmaceutical composition comprises less than about 0.7% (R)-5-(2-fluoro-
3-
methoxypheny1)-1-(2-fluoro-6-(trifluoromethyl)benzy1)-6-methyl-3-(2-(2-
oxopyrrolidin-1-y1)-2-
phenylethyl)pyrimidine-2,4(1H,31/)-dione after storage from about one month to
about three
months at 25 C and 60% relative humidity.
B1. A pharmaceutical composition comprising about 150 mg of 44(R)-245-(2-
fluoro-
3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-dioxo-3,6-
dihydro-2H-
pyrimidin-1-y1]-1-phenyl-ethylamino)-butyric acid (Compound A) or a
pharmaceutically
acceptable salt thereof; and an anti-gelling agent; wherein the weight ratio
of Compound A or the
pharmaceutically acceptable salt thereof is from about 1:1 to about 4:1,
preferably about 2:1.
B2. A pharmaceutical composition comprising about 200 mg of 44(R)-245-(2-
fluoro-
3-metboxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-dioxo-3,6-
dihydro-2H-
pyrimidin-1-y1]-1-phenyl-ethylamino)-butyric acid (Compound A) or a
pharmaceutically
acceptable salt thereof; and an anti-gelling agent; wherein the weight ratio
of Compound A or the
pharmaceutically acceptable salt thereof is from about 1:1 to about 4:1,
preferably about 2:1.
B3. A pharmaceutical composition comprising about 300 mg of 44(R)-2-[5-(2-
fluoro-
3-methoxy-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-dioxo-3,6-
dihydro-2H-
42
CA 2977938 2017-09-01
pyrimidin-1-y1]-1-phenyl-ethylamino)-butyric acid (Compound A) or a
pharmaceutically
acceptable salt thereof; and an anti-gelling agent; wherein the weight ratio
of Compound A or the
pharmaceutically acceptable salt thereof is from about 1:1 to about 4:1,
preferably about 2:1.
B4. The pharmaceutical composition of any one of embodiments B1-B3, wherein
the
anti-gelling agent is sodium carbonate.
B5. The pharmaceutical composition of embodiment B4, wherein the weight
ratio of
Compound A or a pharmaceutically acceptable salt thereof to sodium carbonate
is about 2:1.
B6. The pharmaceutical composition of any one of embodiments B1-B5, further
comprising a water soluble filler.
B7. The pharmaceutical composition of any one of embodiments B1-B5, further
comprising a water insoluble filler and a surfactant.
B8. The pharmaceutical composition of any one of embodiments B1-B7, further
comprising a binder.
B9. The pharmaceutical composition of embodiment B8, wherein the binder is
polyvinylpyrrolidone.
B10. The pharmaceutical composition of any one of embodiments B1-B9, wherein
the
pharmaceutical composition comprises a salt of Compound A.
B11. The pharmaceutical composition of embodiment B10, wherein the salt of
Compound A is sodium 4-((R)-2-[5-(2-fluoro-3-methoxy-pheny1)-3-(2-fluoro-6-
trifluoromethyl-
benzy1)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-y1]-1-phenyl-
ethylamino)butanoate.
Cl. A pharmaceutical composition comprising:
(a) about 20 to about 50% by weight of 4-((R)-2-[5-(2-fluoro-3-methoxy-pheny1)-
3-
(2-fluoro-6-trifluoromethyl-benzy1)-4-methy1-2,6-dioxo-3,6-dihydro-2H-
pyrimidin-l-y1]-1-phenyl-ethylamino)-butyric acid (Compound A)or a
pharmaceutically acceptable salt thereof;
(b) a binder;
43
CA 2977938 2017-09-01
(c) an anti-gelling agent; and
(d) a water soluble filler.
C2. The pharmaceutical composition of embodiment Cl, wherein the binder is
selected from the group consisting of polyvinylpyrrolidone,
hydroxymethylpropylcellulose
(HPMC), hydroxypropylethylcellulose, microcrystalline cellulose, starch, and a
combination
thereof.
C3. The pharmaceutical composition of embodiment Cl, wherein the binder
comprises polyvinylpyrrolidone.
C4. The pharmaceutical composition of any one of embodiments C1-C3, wherein
the
binder is present in an amount from about 1% to about 10% by weight.
C5. The pharmaceutical composition of any one of embodiments C1-C4, wherein
the
anti-gelling agent comprises an alkali metal salt.
C6. The pharmaceutical composition of embodiment C5, wherein the alkali
metal salt
is selected from the group consisting of sodium carbonate, sodium hydrogen
carbonate, sodium
phosphate, and combinations thereof.
C7. The pharmaceutical composition of embodiment Cl, wherein the anti-
gelling
agent comprises sodium carbonate, preferably sodium carbonate monohydrate.
C8. The pharmaceutical composition of any one of embodiments C1-C7, wherein
the
anti-gelling agent is present in an amount from about 10% to about 25% by
weight.
C9. The pharmaceutical composition of any one of embodiments C1-C8, wherein
the
water soluble filler comprises mannitol and pregelatinized starch.
C10. The pharmaceutical composition of any one of embodiments C1-C9, wherein
the
water soluble filler is present in an amount from about 30% to about 50% by
weight.
C11. The pharmaceutical composition of any one of embodiments Cl-C10, further
comprising about 1 to about 5% lubricant.
44
CA 2977938 2017-09-01
C12. The pharmaceutical composition of embodiment C11, wherein the lubricant
is
magnesium stearate.
C13. The pharmaceutical composition of any one of embodiments C1-C12, wherein
the
pharmaceutical composition is a solid oral dosage form.
C14. The pharmaceutical composition of embodiment C13, wherein the solid oral
dosage form is a tablet.
C15. The pharmaceutical composition of embodiment C14, wherein the tablet
comprises a film coating.
C16. The pharmaceutical composition of any one of embodiments C1-C15, wherein
the
pharmaceutical composition comprises a salt of Compound A.
C17. The pharmaceutical composition of embodiment C16, wherein the salt of
Compound A is sodium 44(R)-2-[5-(2-fluoro-3-methoxy-phenyl)-3-(2-fluoro-6-
trifluoromethyl-
benzy1)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-y11-1-phenyl-
ethylamino)butanoate.
C18. The pharmaceutical composition of embodiment C16, wherein the salt of
Compound A is amorphous sodium 44(R)-2-[5-(2-fluoro-3-methoxy-phenyl)-3-(2-
fluoro-6-
trifluoromethyl-benzy1)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-y1]-1-
phenyl-
ethylamino)butanoate.
Dl. A solid oral dosage form comprising:
(a) about 33.2% by weight of sodium 4-((R)-2-[5-(2-fluoro-3-methoxy-pheny1)-3-
(2-
fluoro-6-trifluoromethyl-benzy1)-4-methy1-2,6-dioxo-3,6-dihydro-2H-pyrimidin-
1-y1]-1-phenyl-ethylamino)butanoate;
(b) about 16.7% by weight of an alkali metal salt; and
(c) about 51.1% by weight of one or more excipients.
D2. The solid oral dosage form of embodiment D1, wherein the one or
more
excipients comprises a binder, a water soluble filler, a lubricant, and a film-
coating.
El. A solid oral dosage form comprising:
CA 2977938 2017-09-01
(d) about 33.2% by weight of sodium 44(R)-2-[5-(2-fluoro-3-methoxy-phenyl)-3-
(2-
fluoro-6-trifluoromethyl-benzy1)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-
1-y11-1-phenyhethylamino)butanoate;
(e) about 2.9% by weight of a binder;
(f) about 16.7% by weight of an alkali metal salt;
(g) about 41.6% by weight of a water soluble filler;
(h) about 1.8% by weight of a lubricant; and
(i) about 3.8% by weight of a film-coating.
E2. The solid oral dosage form of embodiment El, wherein the binder is
polyvinylpyrrolidone.
E3. The solid oral dosage form of embodiment El or embodiment E2, wherein
the
alkali metal salt is sodium carbonate, preferably sodium carbonate
monohydrate.
E4. The solid oral dosage form of any one of embodiments E1-E3, wherein the
water
soluble filler comprises mannitol and pregelatinized starch.
ES. The solid oral dosage form of embodiment E4, wherein the solid oral
dosage form
comprises about 32% by weight of mannitol and about 9% by weight of
pregelatinized starch.
E6. The solid oral dosage form of any one of embodiments El-ES, wherein
the
lubricant is magnesium stearate.
E7. The solid oral dosage form of any one of embodiments E1-E6, wherein
the dosage
form comprises an intragranular portion and an extragranular portion.
Fl. A pharmaceutical composition comprising Compound A or a
pharmaceutically
acceptable salt thereof and Compound B or a salt thereof; wherein Compound A
is 4-((R)-2-[5-
(2-fluoro-3-methox y-pheny1)-3-(2-fluoro-6-trifluoromethyl-benzy1)-4-methyl-
2,6-dioxo-3,6-
dihydro-2H-pyrimidin-l-y1]-1-phenyl-ethylamino)-butyric acid; Compound B is
(R)-5-(2-fluoro-
3-methox ypheny1)-1-(2-fluoro-6-(trifluoromethyl)benzy1)-6-methyl-3-(2-(2-
oxopyrrolidin-1-y1)-
2-phenylethyl)pyrimidine-2,4(1H,3H)-dione; and Compound B is present in the
composition in
an amount less than about 0.7% by weight.
46
CA 2977938 2017-09-01
F2. The pharmaceutical composition of embodiment Fl, wherein Compound B
is
present in the composition in an amount less than about 0.7% by weight after
storage for at least
one month, at least two months, or preferably at least six months at 25 C and
60% relative
humidity.
G1. A method of treating endometriosis, the method comprising
administering the
pharmaceutical composition of embodiments Al-A29, B1-B11, C1-C18, D1-D2, or Fl-
F2 or the
solid oral dosage form of embodiments El-E7 to a patient in need thereof.
H1. A method of treating uterine fibroids, the method comprising
administering the
pharmaceutical composition of embodiments A1-A29, B1-B11, Cl-C18, Dl-D2, or F1-
F2 or the
solid oral dosage form of embodiments E1-E7 to a patient in need thereof.
J1. A method for providing partial to substantially full suppression of
estradiol in a
female patient with endometriosis or uterine fibroids, the method comprising
administering the
pharmaceutical composition of embodiments Al-A29, Bl-B11, C1-C18, D1-D2, or F1-
F2 or the
solid oral dosage form of embodiments E1-E7to the patient.
J2. The method of embodiment J1, wherein estradiol levels in the female
patient are
less than about 50 pg/mL.
J3. The method of embodiment J1, wherein estradiol levels in the female
patient are
less than about 20 pg/mL.
[00184] G. EXAMPLES
[00185] The following Examples demonstrate certain challenges encountered
during
formulation development and describe formulations that overcome those
challenges.
[00186] Example 1: Gel Formation by Compound A Monosodium Salt
[00187] To estimate the solubility of Compound A in water, various amounts
of
Compound A sodium salt were added to a fixed volume of 1.5 mL and equilibrated
at 37 C;
solutions were assayed for Compound A concentration.
[00188] Table 2 lists the raw data and observations of the experiment, and
Figure 2 shows
the concentration as a function of the amount of Compound A solid added. The
dotted line in
Figure 2 is the theoretical concentration based on the weights of the solids
added and the volume
47
CA 2977938 2017-09-01
of water. As shown in Figure 2, the concentration of Compound A agrees with
the simple
calculation up to 100 mg solid / 1.5 mL. Deviation of the concentrations from
the theoretical line
is due to the volume expansion upon dissolution of large amount of solutes.
Beyond that, the
concentrations deviate from the theoretical line, but the solution is still
clear and no visible
gelling was observed. When more than 500 mg of Compound A solid was added,
visible gelling
was observed, therefore, concentrations were not determined.
[00189] Table 2: Raw Data of Compound A Solubility Experiment in Water at
37 C
Amount
Measured Concentration
(mg) added
(mg/mL) Observation Final pH
to 1.5 mL
0 0 N/A ¨6
12.2 7.45 Clear solution 9.66
28.7 17.3 Clear solution 9.96
49.8 28.0 Clear solution 10.10
100.4 60.8 Clear solution 10.16
202.5 111 Clear solution 10.18
295.6 149 Clear solution 10.20
503.2 170 Clear solution 10.17
700.9 N/A Gel formation N/A
990.0 N/A Gel formation N/A
[00190] Further experiments revealed that if the percent of Compound A or a
salt thereof
in a tablet formulation is greater than 10 percent (and in the absence of an
appropriate anti-
gelling agent), incomplete dissolution occurs ¨ Compound A was present as an
insoluble
precipitate. Therefore, a formulation of Compound A sodium salt was evaluated,
at about 10%
drug loading, where minimal gelling was observed.
[00191] Example 2: In Vitro Release in the Absence of an Anti-Gelling Agent
[00192] An immediate release formulation was prepared without an anti-
gelling agent. All
components, except magnesium stearate, were blended in a high-shear granulator
and granulated
with neat, de-ionized water. The granules were tray-dried at 40 C and passed
through a #20 US
Standard sieve and tubed with magnesium stearate. Compound A referenced in the
table below is
the Compound A sodium salt.
48
CA 2977938 2017-09-01
[00193] Composition of Formulation without Anti-Gelling Agent
Quantity
Ingredient
(mg/Tablet)
Compound A, sodium salt 207.3
Mannitol 304.0
Pregelatinized Starch 59.1
Povidone K 29/32 18.4
Magnesium stearate 11.2
[00194] The dissolution profile for the uncoated tablets in pH 1.2 medium
is shown in
Table 3.
[00195] Table 3:
Time (min) Mean % (Std Dev)
15 15 (0.5)
30 31(0.5)
45 45 (0.6)
60 57 (0.7)
[00196] Example 3: Formulations Having an Anti-Gelling Agent
[00197] Table 4 presents additional non-limiting examples of components of
the disclosed
formulations and their percentage by weight (w/w) of the final coated tablet.
Compound A
referenced in the table below is the Compound A sodium salt and the
corresponding amount
(mg/tablet) and weight percent is provided based on that salt form.
49
CA 2977938 2017-09-01
n
1001981 Table 4. Composition of Exemplary Formulations.
i..)
to
,.1
,.1 Fl (150 mg) F2
(50 mg) F3 (150 mg)
to
w Ingredient Function
co Quantity %a
Quantity c/oa Quantity Voa
i..)
(mg/Tablet) (w/w) (mg/Tablet) (w/w) (mg/Tablet)
(w/w)
0
I-.
,.1 Compound A, sodium salt Drug Substance 155.5 25.2
51.8 33.5 155.5 33.5
,
0
to
1 Mannitol, USP Filler 271.0 43.9
50.2 32.5 150.5 32.5
0
1-.
Corn Starch, NF Filler 68.2 11.0
16.0 10.4 48.0 10.4
Pregelatinized Starch Filler/Binder -- -- --
-- -- --
Povidone K 29/32, USP Binder 21.3 3.4
5.0 3.9 15.0 3.2
Sodium carbonate Anti-gelling
75.0 12.1
25.0 16.2 75.0 16.2
monohydrate, NF Agent/Buffer
Silicon Dioxide, NF Glidant 3.0 0.5 --
-- -- --
Magnesium stearate, NF Lubricant 6.0
1.0 2.0 1.3 6.0 1.3
Uncoated tablet weight 600.0 -- 150.0 -- 450.0
Opadry Film Coat 18.0 2.9
4.5 2.9 13.5 2.9
Opadry II Film Coat -- --
-- -- -- --
Eudragit L 30 D-55 +
Enteric Coat -- -- --
-- -- --
plasticizer + glidant
Coated tablet weight 618.0 100 154.5 100 463.5 100
"Percents given based on the coated tablet weight. Total percentage may not be
100% due to rounding.
n
100199j Table 4, cont. Composition of Exemplary
Formulations.
I)
to
=-.1 F4
(100 mg) F5 (150 mg)
=-.1
to Ingredient Function
w Quantity %a
Quantity %a
co
(mg/Tablet) (w/vv) (mg/Tablet) (w/w)
n.)
0
1-.
=-.1 Compound A, sodium
salt Drug Substance 103.7 33.6 155.5 33.5
1
0
to Mannitol, USP Filler 100.0 32.4
150.0 32.4
1
0
1-. Corn Starch, NF Filler -- -- --
--
Pregelatinized Starch Filler/Binder 29.5 9.5
44.3 9.6
Povidone K 29/32, USP Binder 9.2 3.0
13.8 3.0
Sodium carbonate Anti-gelling
52.0 16.8 78.0 16.8
monohydrate, NF Agent/Buffer
Silicon Dioxide, NF Glidant -- -- --
--
Magnesium stearate, NF Lubricant 5.6 1.8
8.4 1.8
Uncoated tablet weight 300.0 -- 450.0 --
Opadry Film Coat 9.0 2.9
13.5 2.9
Opadry II Film Coat -- -- --
--
Eudragit L 30 D-55 +
Enteric Coat -- -- --
--
plasticizer + glidant
Coated tablet weight 309.0 100 463.5 100
"Percents given based on the coated tablet weight. Total percentage may not be
100% due to rounding.
51
n [00200] Table 4, cont. Composition of Exemplary
Formulations.
I)
to F6 (150 mg) F7
(200 mg) F7DR (200 mg) F8 (300 mg)
=-.1
=-.1
to di Ingreent Function
w Quantity (Yoa
Quantity (3/0" Quantity (Yoa Quantity %a
co
(mg/Tablet) (w/w) (mg/Tablet) (w/w) (mg/Tablet) (w/w) (mg/Tablet) (w/w)
n.)
0
1-.
=-.1 Compound A, sodium
salt Drug Substance 155.5 33.5 207.4 33.6 207.4 32.0
310.9 33.5
1
0
to Mannitol, USP Filler 150.01 32.4
199.9h 32.3 199.91? 30.8 299.91? 32.4
1
0
1-. Corn Starch, NF Filler -- -- --
-- -- -- --
Pregelatinized Starch Filler/Binder 44.3 9.6
59.1 9.6 59.1 9.1 88.7 9.6
Povidone K 29/32, USP Binder 13.8 3.0
18.5 3.0 18.5 2.9 27.7 3.0
Sodium carbonate Anti-gelling
78.0 16.8 104.0 16.8 104.0 16.0 156.0 16.8
monohydrate, NF Agent/Buffer
Silicon Dioxide, NF Glidant -- -- --
-- -- -- -- --
Magnesium stearate, NF Lubricant 8.4 1.8
11.2 1.8 11.2 1.7 16.8 1.8
Uncoated tablet weight 450.0 -- 600.1 600.1 --
900.0 --
Opadry Film Coat -- -- --
-- -- -- -- --
Opadry II Film Coat 13.5 2.9
18.0 2.9 -- -- 27.0 2.9
Eudragit L 30 D-55 +
Enteric Coat -- -- --
-- 48.0 7.4 -- --
plasticizer + glidant
Coated tablet weight 463.5 100 618.1 100 648.1 100
927.0 100
'Percents given based on the coated tablet weight. Total percentage may not be
100% due to rounding.
hMannitol (12.3%) added extragranular.
52
n
100201] Table 4, cont. Composition of Exemplary
Formulations.
I'.)
to
=-.1 F9
(150 mg) F10 (200 mg) Fll (200 mg)
=-.1
to Ingredient Function
w Quantity %a
Quantity %a Quantity %a
co
(mg/Tablet) (w/w) (mg/Tablet) (w/w) (mg/Tablet) (w/w)
i..)
0
1-.
=-.1 Compound A, sodium
salt Drug Substance 155.2 33.2 207.4 33.2 207.4 50.3
1
0
to Mannitol, USP Filler 150.3 32.1
200.3 32.1 74.2 18.0
1
0
1-. Corn Starch, NF Filler -- -- --
-- -- --
Pregelatinized Starch Filler/Binder 44.3 9.5
59.1 9.5 -- --
Povidone K 29/32, USP Binder 13.8 2.9
18.4 2.9 12.0 2.9
Sodium carbonate Anti-gelling
78.0 16.7 104.0 16.7 100.0 24.3
monohydrate, NF Agent/Buffer
Silicon Dioxide, NF Glidant -- -- --
-- -- --
Magnesium stearate, NF Lubricant 8.4 1.8
11.2 1.8 6.4 1.6
Uncoated tablet weight 450.0 -- 600.0 -- 400.0 --
Opadry Film Coat -- -- --
-- -- --
Opadry II Film Coat 18.0 3.8
24.0 3.8 12.0 29
Eudragit L 30 D-55 +
Enteric Coat -- -- --
-- -- --
plasticizer + glidant
Coated tablet weight 468.0 100 624.0 100 412.0 100
'Percents given based on the coated tablet weight. Total percentage may not be
100% due to rounding.
53
Docket No. 025148.8372 (ABV12349USL1)
[00202] 3.1. Preparation.
[00203] Fl was prepared using a two-step granulation process. The
manufacturing process
flow diagram is presented in Figure 3. In this process, the binder and a
portion of the filler were
added to the single pot processor bowl. Sodium carbonate, the remaining
filler, Compound A,
and colloidal silicon dioxide are blended in an IBC. In the first granulation
step, the filler/binder
blend in the SPP bowl was granulated with water. In the second granulation
step, the Compound
A blend was added to the SPP bowl and granulated by mixing for a short time.
The granulation
was then dried in the SPP bowl using vacuum and swing mode. The dried
granulation was milled
using a Comil into another IBC. The lubricant magnesium stearate was added to
the granules and
blended. The granules were compressed into 600 mg tablet for 150 mg dose
strength.
[00204] Formulations F2 and F3 were prepared using blending, fluid bed
granulation,
milling, tableting, and tablet coating.
[00205] Formulations F4-F11 were prepared using blending, roller compaction
and
milling, tableting, and tablet coating, generally as shown in Figure 1.
Formulations F4 and F5
were developed with a target drug loading of ¨35% Compound A to obtain
uncoated tablet
weights of 300 and 450 mg for 100 and 150 mg dose strengths, respectively.
[00206] A subset of the immediate release tablets were coated with an
enteric coating to
provide delayed release tablets (F7DR). The tablets were coated with an
enteric coating
comprising Eudragit L 30 D-55, Plasacryl T20, and triethyl citrate. Typical
coating parameters
were maintained during this process.
[00207] 3.2. In Vitro Dissolution Profile for Formulations Fl & F5 in pH
1.2 Buffer
[00208] Multiple batches of the formulation with 25% drug loading were
manufactured.
The dissolution profile for Fl tablets in pH 1.2 medium is shown in Table 5.
[00209] Table 5.
Mean % Dissolution (SD)
Time
(min)
Batch 1 Batch 2
15 54(3.4) 46(1.7)
30 99 (1.1) 87 (0.3)
45 102 (1) 99 (1.9)
60 104 (0.7) 101 (1.5)
54
CA 2977938 2017-09-01
[00210] The dissolution profile for F5 tablets in pH 1.2 medium is shown in
Table 6.
[00211] Table 6:
Time (min) Mean % Dissolution
15 20
30 54
45 80
60 95
[00212] 3.3. In Vitro Dissolution Profile After Storage for up to 24 months
[00213] Formulations F5, F6, and F10 were tested for dissolution using USP
apparatus II
in 900 mL of sodium phosphate, pH 6.8, at 37 C and paddle speed of 50 rpm.
Formulations F5,
F6, and F10 all showed similar dissolution profiles both tested initially and
upon storage for up
to 24 months. The dissolution profiles of Formulation F5 tablets at 18 and 24
months are shown
in Figure 4. The stability dissolution profile of Formulation F7 tablets is
shown in Figure 5. The
dissolution profile of uncoated and film coated Formulation F10 tablets is
shown in Figure 6.
[00214] 3.4. Pharmacokinetic Profile for 150 mg, 200 mg, & 300 mg Dosage.
[00215] 3.4.1. Pharmacokinetics of F3 and F5 (150 mg)
[00216] A study was conducted to explore the bioayailability of single
doses of 2 IR tablet
formulations (F3 and F5) at a 150 mg dose under fasting conditions. The
pharmacokinetic
parameters are shown in Table 7. Data for pharmacokinetic parameters are
presented as the mean
SD.
CA 2977938 2017-09-01
[00217] Table 7:
Formulations
F3 F5
150 mg IR tablet 150 mg IR tablet
Pharmacokinetic (N = 23) (N = 23)
Parameters (units)
Cmax (ng/mL) 523 + 247 510 + 225
Tmd. (hr) 1.07 + 0.35 1.12 + 0.42
AUCt (ng=hr/mL) 1263 560 1273 520
AUC. (ng=hr/mL) 1271 + 560 1281 + 520
(hr) 2.03 0.41 2.21 0.60
[00218] 3.4.2. Pharmacokinetics of F4 and F7 (200 mg)
[00219] A study was conducted to compare the relative bioavailability of
single doses of
one 200 mg IR tablet of Formulation F7 with that of two 100 mg IR tablets of
Formulation F4
under fasting conditions. Pharmacokinetic assessments showed that Formulation
F7 (200 mg IR
tablet) was bioequivalent to Formulation F4 (2 x 100 mg IR), with respect to
maximum
concentration (Cmax) and area under the curve (AUC), with 90% CI that fell
within the limits of
0.80 to 1.25. The pharmacokinetic parameters are shown in Table 8. Data for
pharmacokinetic
parameters are presented as the mean + SD.
[00220] Table 8:
Formulations
F4 F7
2 x 100 mg IR tablets 200 mg IR tablet
Pharmacokinetic (N = 23) (N = 23)
Parameters (units)
Cmax (ng/mL) 879 + 401 845 +329
TillaX (hr) 1.1 + 0.4 1.1 +0.3
AUC, (ng=hr/mL) 2384 + 916 2211 853
AUC. (ng=hr/mL) 2391 + 917 2217 + 854
t1/2 (hr) 3.86 0.70 3.91 + 0.48
[00221] 3.4.3. Pharmacokinetics of F4 and F10 (200 mg)
[00222] A further study was conducted to explore the bioavailability a
single dose of one
200 mg IR tablet of Formulation F10 with that of two 100 mg IR tablets of
Formulation F4 under
fasting conditions. The pharmacokinetic parameters are shown in Table 9. Data
for
pharmacokinetic parameters are presented as the mean SD.
56
CA 2977938 2017-09-01
[00223] Table 9:
Formulations
F4 F10
2 x 100 mg IR tablets 200 mg IR tablet
Pharmacokinetic (N = 54) (N = 54)
Parameters (units)
Cmt. (ng/mL) 744 + 353 (47) 738 + 419 (57)
Tma. (h) 1.0 (0.5 - 3.0) 1.0 (0.5 - 3.0)
t1/2 (h) 5.91 2.82 6.20 + 2.93
AUCt (ng=h/mL) 1890 + 852 (45) 1910 960 (50)
AUC. (ng=h/mL) 1900 + 853 (45) 1920 961(50)
[00224] 3.4.4. Phartnacokinetics of F5 (300 rng)
[00225] A study was conducted to explore the bioavailability of a single
dose of two 150
mg IR tablets of Formulation F5 under fasting conditions. The pharmacokinetic
parameters are
shown in Table 10. Data for pharmacokinetie parameters are presented as the
mean SD.
[00226] Table 10:
F5
2x 150 mg IR tablet
Pharmacokinetic (N = 10)
Parameters (units)
Cmax (ng/mL) 1378 487
TIMIX (hr) 1.6 0.6
AUCt (ng=hr/mL) 3732 1356
AUC. (ng=hr/mL) 3772 1368
[00227] 3.4.5. Pharmacokinetics of F7 & F7DR (200 mg)
[00228] A clinical study was conducted to compare the in vivo performance
of F7 and
F7DR. Additionally, the study assessed the potential effects of a high-fat
meal on the
pharmacokinetics of F7DR. Adult premenopausal healthy female subjects were
administered a
single dose of F7DR under fasting conditions, a single dose of F7DR 30 minutes
after consuming
a high-fat meal, or a single dose of F7 under fasting conditions.
[00229] A high-fat meal reduced the concentrations of the F7DR tablet
formulation. A
delay in absorption was observed for the F7DR tablet formulation, regardless
of the meal
conditions. Food reduced the C.. and AUC of F7DR. The delay of absorption was
longer for
under fed conditions.
57
CA 2977938 2017-09-01
[00230] .. The pharmacokinetic parameters are shown in Table 11. Data for C.
and AUC
are presented as the mean (% CV); data for Tmax are presented as median (min ¨
max); and data
for t1/2 are presented as harmonic mean (pseudo CV).
[00231] Table 11:
Regimens
F7 - fasted F7DR - fasted F7DR - fed
200 mg IR tablet 200 mg eIR tablet 200 mg eIR tablet
Pharmacokinetic (N = 24) (N = 11) (N = 11)
Parameters (units)
CMdX (ng/mL) 850 (34) 977 (63) 332 (51)
TMdX (hr) 1.0 (0.75 ¨ 1.5) 3.0 (1.5 ¨6.0) 7.0 (4.0 ¨ 12.0)
AUCt (ng=hr/mL) 2106 (43) 2253 (53) 1241 (46)
AUC. (ng=hr/mL) 2115 (43) 2262 (53) 1250 (46)
t1/2 (hr) 4.4 (33) 3.80 (34) 3.01 (26)
[00232] Example 4: Estradiol Concentrations Following Treatment with F4 or
F5
[00233] A further study was conducted in premenopausal women with moderate
to severe
endometriosis-associated pain. The women enrolled in this study were
representative of the
general population of women with moderate to severe endometriosis-associated
pain. Baseline
characteristics, including the subjects' endometriosis-associated pain at
study entry, were
comparable across treatment groups.
[00234] Treatment groups were: (a) one F5 tablet once daily (i.e., 150 mg
QD) and (b) two
F4 tablets twice daily (i.e., 200 mg BID). Blood samples were collected during
the monthly
clinic visits to measure hormone concentrations. Over 800 female subjects
across 151 sites in
North America were randomized into the study in a 3:2:2 ratio to placebo, 150
mg QD, or 200
mg BID, respectively.
[00235] A dose-dependent suppression of estradiol was observed in the
treatment groups,
compared with placebo during the treatment period. For the placebo group, the
median estradiol
levels at their monthly visits were between 70.0 and 91.6 pg/mL, with 2% to 4%
of women with
estradiol concentrations <20 pg/mL. For the 150 mg QD group, the median
estradiol levels at
their monthly visits were between 36.8 and 45.7 pg/mL, with 15% to 24% of
women with
estradiol concentrations < 20 pg/mL. For the 200 mg BID group, the median
estradiol levels at
their monthly visits were at the limit of quantification (12.4 pg/mL), with
71% to 81% of women
with estradiol concentrations <20 pg/mL.
58
CA 2977938 2017-09-01
Docket No. 025148.8372 (ABV12349USL1)
n
[00236] Table 12: Estradiol Serum Concentrations
n.)
to 1
=-.1 Treatment/Parameter Day
Month 1 Month 2 Month 3 Month 4 Month 5 Month 6
=-.1 (Predose)
to
w Placebo
co
n.) N 366 342 329
310 300 288 223
0
1-.
=-.1 Median 48.6 70.0
83.4 88.6 77.7 82.9 91.6
1
0 (Mean SD) (66.5+56.3) (91.8174.8) (104.4179.4)
(110.6+89.7) (107.1+86.8) (107.3182.7) (111.7179.9)
to
1
0 % subjects < 20 pg/mL 6.3 3.8 4.0
1.9 2.7 3.8 2.7
1-.
% subjects 20-50 pg/mL 45.6 33.6 23.4
26.1 25.7 23.6 21.1
% subjects > 50 pg/mL 48.1 62.6 72.6
71.9 71.7 72.6 76.2
150 mg QD
N 244 229 217
213 198 195 165
Median 45.7 36.8 39.2
41.0 41.2 41.5 45.7
(Mean+SD) (70.9+70.0) (56.7+58.5) (60.7+59.0)
(65.8+66.9) (66.7+82.4) (70.1+72.5) (75.4177.6)
% subjects <20 pg/mL 9.4 23.6 19.4
21.1 20.7 15.4 15.2
% subjects 20-50 pg/mL 45.5 43.7 44.7
37.1 38.9 41.5 41.2
% subjects > 50 pg/mL 45.1 32.8 35.9
41.8 40.4 43.1 43.6
200 mg BID
N 246 222 208
197 188 181 150
Median 46.5 12.4 12.4
12.4 12.4 12.4 12.4
(Mean+SD) (77.5+84.9) (25.0+38.2) (25.5+44.7)
(27.4+42.9) (22.8+32.4) (25.3135.3) (31.1+50.8)
% subjects <20 pg/mL 8.1 80.6 80.8
76.1 78.2 75.1 70.7
% subjects 20-50 pg/mL 45.5 8.6 11.1
13.7 14.4 14.9 15.3
% subjects > 50 pg/mL 46.3 10.8 8.2
10.2 7.4 9.9 14.0
59
Docket No. 025148.8372 (ABV12349USL1)
[00237] Example 5: Estradiol Concentrations Following Treatment with F4 or
F5
[00238] Another study was conducted in premenopausal women with moderate to
severe
endometriosis-associated pain. The women enrolled in this study were
representative of the
general population of women with moderate to severe endometriosis-associated
pain. Baseline
characteristics, including the subjects' endometriosis-associated pain at
study entry, were
comparable across treatment groups.
[00239] Treatment groups were: (a) one FS tablet once daily (i.e., 150 mg
QD) and (b) two
F4 tablets twice daily (i.e., 200 mg BID). Blood samples were collected during
the monthly
clinic visits to measure hormone concentrations. Over 800 female subjects
across 187 sites in
North America, South America, Europe, Africa, and Australia were randomized
into the study in
a 3:2:2 ratio to placebo, 150 mg QD, or 200 mg BID, respectively.
[00240] A dose-dependent suppression of estradiol was observed in the
treatment groups,
compared with placebo during the treatment period. For the placebo group, the
median estradiol
levels at their monthly visits were between 70.7 and 105 pg/mL with 4% to 6%
of women with
estradiol concentrations < 20 pg/mL. For the 150 mg QD dose, the median
estradiol levels at
their monthly visits were between 37.2 and 55.8 pg/mL, with 14% to 22% of
women with
estradiol concentrations <20 pg/mL. For the 200 mg BID dose, the median
estradiol levels at
their monthly visits were between 8.43 and 13.1 pg/mL, with 62% to 77% of
women with
estradiol concentrations < 20 pg/mL.
CA 2977938 2017-09-01
Docket No. 025148.8372 (ABV12349USL1)
n
100241i Table 13: Estradiol Serum Concentrations
n.)
to 1
=-.1 Treatment/Parameter Day
Month 1 Month 2 Month 3 Month 4 Month 5 Month 6
=-.1 (Predose)
to
w Placebo
co
n.) N 324 312 293
272 261 256 232
0
1-.
=-.1 Median 58.1 70.7
85.2 84.9 77.8 80.2 105
1
0
to (Mean+SD)
(91.4+81.5) (99.6187.1) (105+79.1)
(106+80.6) (100+89.4) (107+101) (114+79.6)
1
0 % subjects < 20 pg/mL 5.6 4.8 4.1
3.7 6.1 3.5 4.7
1-.
% subjects 20-50 pg/mL 37.4 29.2 25.6
26.8 24.9 26.6 19.0
% subjects > 50 pg/mL 57.1 66.0 70.3
69.5 69.0 69.9 76.3
150 mg QD
N 205 201 185
190 167 175 158
Median 67.0 43.6 37.2
41.7 47.5 46.9 55.8
(Mean+SD)
(87.1169.6) (74.6196.5) (56.6+53.1)
(64.9+63.4) (69.5+82.6) (61.1+52.0) (76.8+64.9)
% subjects <20 pg/mL 5.9 18.4 22.2
20.5 19.2 18.3 13.9
% subjects 20-50 pg/mL 28.3 38.8 41.6
35.3 32.9 36.0 32.9
% subjects > 50 pg/mL 65.9 42.8 36.2
44.2 47.9 45.7 53.2
200 mg BID
N 211 192 185
184 180 177 153
Median 63.2 8.43 8.95
10.9 10.6 13.0 13.1
(Mean+SD)
(83.9+74.5) (22.4152.2) (21.2+32.0)
(28.6+45.6) (24.3+34.9) (35.0162.9) (31.8+46.6)
A subjects <20 pg/mL 5.7 77.1 70.8
67.4 70.0 63.8 62.1
% subjects 20-50 pg/mL 35.1 13.5 17.8
16.3 15.0 17.0 18.3
% subjects > 50 pg/mL 59.2 9.4 11.4
16.3 15.0 19.2 19.6
61
Docket No. 025148.8372 (ABV12349USL1)
[00242] Example 6: Impact of Water-Insoluble Filler & Surfactant An
immediate
release formulation containing sodium carbonate was prepared. All components,
except
magnesium stearate, were blended in a high-shear granulator and granulated
with neat, de-
ionized water. The granules were tray-dried at 40 C and passed through a #20
US Standard sieve
and lubed with magnesium stearate. Compound A referenced in Table 14 below is
the
Compound A sodium salt.
[00243] Table 14: Composition of Formulation F12
Quantity (mg/Tablet)
Ingredient
F12 Fl2A
Compound A, sodium salt 155.5 153.1
Microcrystalline Cellulose 150.5 148.2
Corn Starch 48.0 47.3
Povidone K 29/32 15.0 14.8
Sodium Carbonate, monohydrate 75.0 73.8
Magnesium stearate 6.0 5.9
Sodium dodecyl sulfate 6.8
[00244] Formulation F12A was prepared by combining 6.3 g Formulation F12
and
97.3mg sodium dodecyl sulfate (1.5% w/w) in a bottle and rolling the bottle by
hand to blend.
[00245] An in vitro dissolution study was conducted. The release of
Compound A was
monitored using USP apparatus II in 900 mL of pH 6.8 buffer, at 37 C and
paddle speed of 50
rpm. The dissolution results are presented in Table 15:
[00246] Table 15: Percent Compound A released in pH 6.8 buffer
Mean %
Minutes
F12 Fl2A
15 14.3 27.4
30 39.2 62.1
45 59.6 86.4
62
CA 2977938 2017-09-01
[00247] Example 7: Impact of sodium carbonate on dissolution
[00248] The impact of sodium carbonate monohydrate level on dissolution was
examined.
The amount of sodium carbonate monohydrate was varied by 20% of the nominal
level to
study the impact on dissolution. Mannitol level was adjusted in the
formulation to maintain the
overall tablet weight. A slugging process was used to manufacture tablets to a
target solid
fraction of 0.88 hardness of 125 N. Dissolution profiles for tablets from the
batches are presented
in Figure 7. All results passed the proposed dissolution specification at t=
30 minutes. The results
indicate the 20% change in the level of sodium carbonate monohydrate does
not impact
dissolution.
[00249] Example 8: Impact of sodium carbonate on degradation products,
including
Compound B
[00250] One degradation product of Compound A is Compound B, which has a
lactam
moiety.
[00251] Excipient compatibility studies were conducted using mixtures of
excipients and
Compound A with and without sodium carbonate. The results are shown in Figure
8. All
excipients (corn starch, mannitol, pregelatinized starch (PGS),
microcrystalline cellulose (MCC),
dibasic calcium phosphate (DCP), isomalt, colloidal silicon dioxide (SiO2))
showed much higher
formation of lactam in absence of sodium carbonate. In the presence of sodium
carbonate, the
excipients showed much lower content of lactam, very close to the detectable
limit of about
0.03%.
[00252] Formulations using 2:1, 3:1, and 4: 1 w/w ratio of Compound A to
sodium
carbonate monohydrate were prepared. These formulations contained ¨35%
Compound A,
sodium carbonate monohydrate, mannitol, pregelatinized starch, povidone, and
magnesium
stearate. The tablets were film coated and tested under accelerated stability
protocol conditions
of 50 C/75% RH, 60 C/5% RH, 60 C/40% RH, 70 C/5% RH, 70 C/75% RH, 80 C/40% RH
over a period ranging from 2 to 25 days. The results are shown in Figure 9.
Formulations
prepared with 3:1 and 4:1 w/w ratio of Compound A to sodium carbonate showed
higher
presence of the lactam degradant, whereas formulations with 2:1 w/w ratio
showed relatively less
formation of the lactam degradant.
[00253] Additional stability testing was performed on Formulations F5 and
F7. The tablets
were prepared, placed in clear blister pack with aluminum foil, and stored
under the following
63
CA 2977938 2017-09-01
conditions: 25 C/60% RH or 40 C/75% RH. Tablets were assessed for the presence
of
degradation products, including Compound B, at 0 (initial), 1, 3, 6, 9, 12,
18, and 24 months for
the 25 C/60% RH condition and at 0 (initial), 1, 3, and 6 months for the 40
C/75% RH
condition. The results are presented in Table 16.
64
CA 2977938 2017-09-01
Docket No. 025148.8372 (ABV12349USL1)
n
1002541 Table 16: Stability of F5 and F7 up to 24 months
IS.)
to
=-.1
=-.1
to Storage Degradation
Months
w Formulation
co Condition Product [%] 0 1 3
6 9 12 18 24
n.)
0 Compound B
ND ND ND ND ND ND ND ND
1-. =-.1 25 C F5 Total 0 0 0.10
0.10 0 0.11 0.10 0.11
1
0 60%RH F7 Compound B
ND ND ND ND ND ND ND ND
to
1 Total 0 0 0
0 0 0 0.11 0.22
0
1-.
F5 Compound B ND ND <0.10
<0.10 NT NT NT NT
40 C Total
0 0 0.23 0.35 NT NT NT NT
75%RH F7 Compound B ND ND <0.10
<0.10 NT NT NT NT
Total
0 0 0.10 0.33 NT NT NT NT
1002551 ND = not detected; NT = not tested
Docket No. 025148.8372 (ABV12349USL1)
[00256] It is understood that the foregoing detailed description and
accompanying
examples are merely illustrative and are not to be taken as limitations upon
the scope of the
invention, which is defined solely by the appended claims and their
equivalents. Various changes
and modifications to the disclosed embodiments will be apparent to those
skilled in the art. Such
changes and modifications, including without limitation those relating to the
chemical structures,
substituents, derivatives, intermediates, syntheses, formulations, or methods,
or any combination
of such changes and modifications of use of the invention, may be made without
departing from
the spirit and scope thereof.
[00257] All references (patent and non-patent) cited above are incorporated
by reference
into this patent application. The discussion of those references is intended
merely to summarize
the assertions made by their authors. No admission is made that any reference
(or a portion of
any reference) is relevant prior art (or prior art at all). Applicant reserves
the right to challenge
the accuracy and pertinence of the cited references.
66
CA 2977938 2017-09-01