Note: Descriptions are shown in the official language in which they were submitted.
COMPOSITIONS AND METHODS FOR PRODUCING LIPIDS AND OTHER
BIOMATERIALS FROM GRAIN ETHANOL STILLAGE AND STILLAGE
DERIVATIVES
FIELD OF THE INVENTION
The invention is directed to engineered yeasts and methods for converting
stillage
organics into lipids and other biomaterials.
BACKGROUND
Renewable fuels are environmentally desirable, especially fuels with higher
energy
density such as biodiesel. U.S. biodiesel production capacity however, is
underused. From
January of 2014 to July 2016, annual production of biodiesel did not exceeded
60% of plant
capacity. This was due in part to a lack of inexpensive feedstock supply,
which the industry
greatly needs as an alternative source of seed oils (Anon 2016).
Plant triglycerides from soybeans, canola, rapeseed and palm oil can be
converted into
methyl or ethyl esters for use as biodiesel. Biodiesel produced in the U.S. is
mainly derived
from waste cooking oils, the edible oils of soybean and rapeseed (canola)
(Hammond et al.
2005), from the inedible corn oil recovered following fermentation and
distillation of grain to
ethanol, and with lesser amounts generated from animal fats.
If the U.S. were to use all of its domestic soybeans to make biodiesel, it
would result in
about 5.1 billion gallons of biofuel. This approach, however, is not realistic
because these
oilseeds are also key components of the food chain and are used for the
production of many
household and industrial products. Thus, increasing the production of
biodiesel from foodstuffs
would lead to higher prices of commodities derived from them, and economic
hardship for the
consumer.
Biodiesel can be made from the triglycerides generated by oleaginous
(lipogenic) yeast
or algae. In fact, 2 to 3 times more lipid/g dry weight is generated by these
microbial sources
than from seed oils. Certain algae can accumulate lipids when cultivated on
sunlight and CO2,
but fixation of CO2 by photosynthesis requires a great deal of metabolic
energy, so cell growth
and lipid accumulation is relatively slow. Some algae can grow
heterotrophically on simple
1
CA 2982734 2017-10-17
organic compounds dissolved in water, which greatly increases their rates of
lipid accumulation.
Algae, however, do not generally assimilate more complex organic materials
such as starch,
cellulosic or hemicellulosic oligomers. Ascomyceteous and basidiomyceteous
lipogenic yeasts
and filamentous fungi will, however, readily assimilate these compounds.
Moreover, because
these yeasts and fungi are heterotrophic, their growth rates on simple or
complex dissolved
organic materials are much faster than algae cultivated under heterotrophic
conditions.
If it were possible to produce biodiesel from cellulosic residues or other
waste organic
materials in yields similar to what could be achieved with ethanol production,
domestic
biodiesel production could satisfy a significant fraction of the national
transport energy demand
without affecting the food supply. Furthermore, this increased domestic
production would also
decrease dependency on foreign oil.
Agricultural residue (e.g. corn stover) is a potential source of renewable
biomass that
could be converted into liquid transportation fuels such as biodiesel if
recalcitrance of the
biomass to hydrolysis, the presence of inhibitors mixed with the hydrolyzed
sugars, and the
difficulty in obtaining microbial catalysts that will convert the sugars to
lipids in high yield can
be overcome. The potential for biodiesel production from agricultural residues
is significant. If
the residues from U.S. soybean production alone were collected and converted
by a microbial
process with a mass yield of 35% based on the starting sugar, it would be
possible to produce
about 10 million metric tons of lipid annually or about 15% more oil than the
total of what is
presently recovered from the processing of soybeans itself
Cellulosic biodiesel produced by a lipogenic yeast cultivated on agricultural
hydrolysate
would generate an animal feed byproduct similar to that obtained from
processing oil from
soybeans or palm oil. Based on comparisons with existing prices for wholesale
yeast protein
from brewing, biodiesel production by lipogenic yeast would yield residual
yeast protein with
the same or slightly higher market value as soy protein.
While several technologies exist to pretreat and enzymatically saccharify
agricultural
residues for hydrolysis to create fermentable sugars, new microbial
biocatalysts are needed to
convert the resulting mixed sugars into lipids and other higher value
materials.
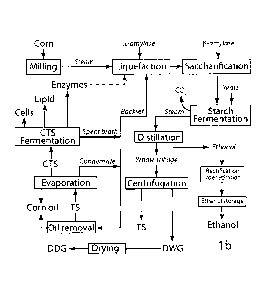
Grain ethanol plants are a potential source of unused, soluble and insoluble
organic
materials suitable for biodiesel production. In wet and dry-mill ethanol
operations, cornstarch
2
CA 2982734 2017-10-17
is enzymatically converted into sugar then fermented to ethanol. The process
leaves behind
significant amounts of corn fiber and generates soluble organics as byproducts
of ethanol
production.
Grain ethanol plants are becoming less economical to operate due to lack of
demand for
ethanol and to low profit margins when grain prices are high and petroleum
prices are low.
Ethanol derived from grain is also criticized for having poor compatibility
with fuel distribution
systems, reducing the food supply, contributing to soil erosion, and releasing
net CO2 emissions
that are only marginally better than gasoline. Reduced operating costs,
increased process
efficiency, better fuel compatibility, and higher product value and diversity
could significantly
improve the economics and environmental acceptability of this process.
In a conventional dry mill process, whole grain is hammer milled, then steam
treated in
a jet cooker as it is sent to the fermentation tank for liquefaction with a
thermostable alpha-
amylase. Following cooling and saccharification, the mash is inoculated for
fermentation.
Variations on this basic process can involve separation of corn hulls (fiber),
starch and germ
gluten (protein) prior to saccharification, use of less steam for cooking, use
of raw starch,
recovery of edible corn oil from the germ and other changes. Following
fermentation, ethanol
is recovered by distillation, and the bulk of the fiber and protein, along
with yeast cells and corn
oil, are separated from the dissolved organics by centrifugation. This yields
wet cake or
distiller's wet grain (DWG) solids and thin stillage (TS) solubles. In a
conventional process,
the distiller's wet grain is dried to make distiller's dried grain (DDG) and
the thin stillage
containing the solubles (S) is evaporated to make a syrup, which is sprayed
back onto the
distiller's dried grain to make DDGS. Evaporation of the thin stillage
separates a fraction of the
inedible corn oil, which can be recovered for biodiesel production (FIG. 1).
Stillages (vinasse) following distillation of ethanol from industrial ethanol
fermentations of grain include corn gluten and yeast protein, residual corn
fiber, yeast cells,
corn oil, and dissolved organics. Thin stillage contains significant
quantities of glycerol (14 to
20 g/1), glucose disaccharides (e.g., cellobiose, trehalose, etc.) (6 to 10
g/l), xylose, lactic acid,
corn oil and various oligosaccharides derived from residual undigested starch,
dextrins,
cellulose and hemicellulose. The total dissolved and suspended organic content
of thin stillage
3
CA 2982734 2017-10-17
is about 10% w/v. Table 1 presents a published summary of stillage components
(Kim et al.
2008).
Table 1. Exemplary Stillage Composition
Stillage Component g/1
Glucose 0.9
Glucan (oligosaccharide) 12.4
Xylose 0.7
Xylan (oligosaccharide) 3.7
Arabinose 0.4
Arabinan (oligosaccharide) 0.5
Lactic acid 16.8
Glycerol 14.4
Acetic acid 0.3
Butanediol 1.9
Ethanol 0.6
The glycerol and oligosaccharide contents of thin stillage retain water during
evaporation and prevent drying. This makes thin stillage evaporation energy-
inefficient.
Removing glycerol and oligosaccharides prior to evaporation is therefore
desirable. Stillages
from other ethanol distillation processes also present disposal problems.
Stillage is difficult and
non-economical to treat in a waste water system because of its high biological
oxygen demand
(BOD), its high organic content and low pH. Stillage can also have relatively
high nitrogen and
phosphorous contents, about 2 g/1 and 130 mg/I respectively (Yen et al. 2012).
The massive
volumes of thin stillage resulting from fuel ethanol production are
particularly difficult to
handle. A significant fraction of the thin stillage is therefore recycled or
"backset" into the
liquefaction stage, which increases the level of dissolved organics in the
fermentation and whole
stillage.
Methods and tools for converting on-site low value soluble organic stillage
byproducts
from ethanol production into biodiesel are needed to increase fuel production
without
4
CA 2982734 2017-10-17
harvesting more grain. Such methods and tools would reduce the organic load in
the backset
while creating higher value products such as yeast oil, enzymes, and animal
feed from
underutilized organic byproducts.
SUMMARY OF THE INVENTION
The invention addresses the aforementioned needs by providing engineered
yeasts and
methods for converting stillage organics into lipids and other biomaterials.
An exemplary method of the invention for converting stillage organics into
yeast oil,
cell protein, and enzymes is shown in FIG. 2. This method is similar to
standard dry mill
operations up through the centrifugation of whole stillage to separate thin
stillage (TS) and
distiller's wet grain (DWG). Instead of sending thin stillage directly to
evaporation, however,
oil (e.g., corn oil) is first removed. Second stage thin film evaporators then
remove 50 to 80%
of the water, and residual protein is separated. The clarified, concentrated
thin stillage (CTS) is
used as a medium to cultivate native or bioengineered lipogenic yeasts that
produce enzymes
that will accelerate liquefaction of starch, rapidly consume glycerol,
cellobiose, trehalose
xylose, lactic acid and residual oligosaccharides while accumulating yeast
lipids and biomass.
An advantage of microbial bioconversion is that a glycerol byproduct generated
during
conversion of triglycerides to biodiesel can be fed back into the bioreactor
to increase both the
rate and yield of lipid production.
An amylolytic, lipogenic yeast specifically bioengineered for rapid lipid and
enzyme
production from dissolved organics in the clarified thin stillage and other
stillage forms is
preferred for use in this process. The yeasts are modified to express,
constitutively express, or
overexpress an acetyl-CoA carboxylase, an alpha-amylase, an ATP citrate lyase,
a
diacylglycerol acyltransferase, a fatty acid synthase, a glycerol kinase, a 6-
phosphogluconate
dehydrogenase, a glycerol-3-phosphate dehydrogenase, a malic enzyme, a fatty
acyl-CoA
reductase, a delta-9 acyl-CoA desaturase, a glycerol-3-phosphate
acyltransferase, a
lysophosphatidate acyltransferase, a glucose-6-phosphate dehydrogenase, a beta-
glucosidase, a
hexose transporter, a glycerol transporter, a glycoside hydrolase enzyme, and
an auxiliary
activity family 9 enzyme, or any combination thereof The yeasts are in some
cases also
modified to reduce or ablate activity of other enzymes. The bioengineered
yeasts of the
5
CA 2982734 2017-10-17
invention produce enzymes that accelerate liquefaction of starch, rapidly
consume glycerol,
cellobiose, trehalose, xylose, lactic acid and residual oligosaccharides while
accumulating yeast
lipids.
The technology described herein can increase profit margins for grain ethanol
producers
by making higher value and more diverse byproducts. The technology also has
the added
benefits of decreasing thin stillage viscosity, increasing protein production,
reducing organic
load from wastewaters, reducing natural gas-based processing expenses, and, in
some cases,
releasing a larger fraction of corn oil from the dried grain solids. Hence,
the technologies would
be of interest to several industries by providing a means to diversify
products and increase the
supply of high energy density renewable fuels while at the same time reducing
environmental
pressures.
The objects and advantages of the invention will appear more fully from the
following
detailed description of the preferred embodiment of the invention made in
conjunction with the
accompanying drawings.
BRIEF DESCRIPTION OF THE DRAWINGS
FIG. 1 shows a schema of a conventional dry mill ethanol process.
FIG. 2 shows a schema of a process of the present invention for converting
stillage
organics into yeast oil, protein, and enzymes.
FIG. 3 shows a schema of intermediary metabolism linked to fatty acid
biosynthesis in
yeast. PDH, pyruvate dehydrogenase; PYC Pyruvate carboxylase; Ac, aconitase;
ME malic
enzyme; MDc malate decarboxylase, cytosolic; MDm malate dehydrogenase,
mitochondrial;
ACL ATP:citrate lyase; ACC, acetyl-CoA carboxylase; ICDH, isocitrate
dehydrogenase; FAS,
fatty acid synthase; DGA, diacylglycerol acyltransferase. a, b, c and d
are transporters.
FIG. 4 shows a graph of the effects of different culture media on basal lipid
content of
Lipomyces starkeyi type strain NRRL Y-11557. The effects of six types of
culture media on L.
starkeyi basal lipid production were evaluated. YPD is yeast peptone dextrose
media. M1 (LN)
is a minimally defined, low nitrogen media containing yeast nitrogen base and
supplemented
with urea, M2 (LN) is a low nitrogen media having the same components as YPD
but containing
6
CA 2982734 2017-10-17
only 3.64% and 1.82% of the yeast extract and peptone contained in YPD. M3
(HN) and M4
(I-IN) are the high nitrogen containing versions of Ml, with M4 HN containing
peptone. mTS
is modified thin stillage, prepared by clarifying and concentrating ethanol
thin stillage. Results
are shown as normalized fluorescence to 0D630.
FIG. 5 is a diagram of a base vector used for creating genomic integrating
cassettes. The
origin of replication (On) and kanamycin resistance gene (Kan R) permit
propagation and
maintenance in E. coli. Two multiple cloning sites (MCSs) enable insertion of
gene target
cassettes adjacent to loxP sites (Xs), which flank an expression cassette for
nourseothricin
resistance (NAT R) driven by the constitutive TDH3 promoter (TDH3p) and
terminator
(TDH3t). Digestion of the vector with AsiS1 enables linearization and
integration into the yeast
genome. The sequence of the base vector is represented by SEQ ID NO:91.
FIG. 6 shows a schema of a screening pipeline used for generating
metabolically
engineered yeast.
FIG. 7 shows growth of various strains of L. starkeyi on glucose as the sole
carbon
source (left panel) or glycerol as the sole carbon source (right panel).
FIGS. 8A and B show Nile Red screening of yeast transformants in YPD or mTS.
Results are shown normalized to the wild-type and 0D600 after two (YPD) or
three (mTS) days
of growth. FIG. 8A shows averages of the top 50% of transformants with each
gene in
preliminary screening. Of 234 transformants cultivated, preliminary screening
revealed three
strains transformed with three genes (cDGA1-1233, cDGA1-1389, and cACC/) that
showed
large standard deviations or higher means than the base vector transformed
strains. Data is
shown for the end of the growth phase in each media (YPD = 2 days, mTS = 3
days). FIG. 8B
shows results of a validation screen of the prime performers of each gene in
mTS evaluated in
triplicate, revealing that the transformants DGA1-1233 6L, GUT]-1602 6L, and
GUT]-1617
2L show superior lipid accumulation over the WT as deemed by Nile Red
fluorescence.
FIG. 9 shows consumption of organics and cell density of wild-type L. starkeyi
(Ls-1)
and a GUT] engineered strain (Ls-11). The presence of glycerol, xylitol,
cellobiose, lactic acid,
and arabinose in 200 mL of mTS solution in shake flasks were monitored by HPLC
analysis
during cultivation of wild-type L. starkeyi (Ls-1) and a strain with an
engineered version of the
glycerol kinase gene (GUT] Ls-11). Cell density (0D600) and direct cell counts
were also
7
CA 2982734 2017-10-17
determined. In this experiment, the Ls-11 engineered strain achieved higher
cell density and
consumed more glycerol than the wild-type strain.
FIGS. 10A and 10B show a difference in the cellular morphologies of wild-type
L.
starkeyi (FIG. 10A) and a strain overexpressing GUT] (FIG. 10B) when cultured
on mTS.
Besides generating larger liposomes in some cells, the strain overexpressing
GUT] also formed
cellular assemblies similar to pseudomycelium. Photos were taken after two
days of growth in
mTS.
FIGS. 11A-11C show a comparison of wild-type L. starkeyi with engineered
strain
DGAI-1233 6L comprising the DGA / -1233 gene. FIG. 11A shows growth rates and
carbon
utilization of wild-type (solid shapes) and DGAI-1233 6L (open shapes) of
glycerol and
cellobiose. Cultures were grown in triplicate on mTS media, error bars denote
the standard
deviation. FIGS. 11B and 11C show cellular morphologies for wild-type (FIG.
11B) and DGAI-
1233 6L (FIG. 11C), showing significantly more liposomes in DGA / -1233 6L.
FIGS. 12A and 12B show Nile Red fluorescence and percent lipid analysis of
wild-type
L. starkeyi and the engineered strain designated DGAI-1233 6L cultured in mTS
using
bioreactors. FIG. 12A shows relative Nile Red fluorescence of the wild-type
strain and the
DGAI-1233 transformant measured at different time points. Spent media was
drawn off and
the bioreactors refilled with thin stillage after 48 hours of growth. FIG. 12B
shows percent lipid
content of cell dry weight of wild-type L. starkeyi and the DGA / -1233
transformant. The
engineered DGA / -1233 6L strain had more than double the Nile Red
fluorescence and percent
lipid content.
FIG. 13 shows a comparison of strains overexpressing DGA1 variants in
synthetic thin
stillage medium (sTS) by Bodipy fluorescence. A) Bodipy fluorescence of the
wild-type Ls-1
(black), gDGA1 163M (dark gray), cDGA-1233 6L (light gray), and cDGA 154L
(white) L.
starkeyi transformants as monitored over the course of 4 days (denoted as D1,
D2, D3, and D4).
The cDGA 154L strain was chosen as the platform strain for further improvement
due to its
similar performance to cDGA 6L and Hygromycin B resistance marker.
FIG. 14 shows dilution-corrected Bodipy fluorescence in mutant and wild-type
Accl-
transformed L. starkeyi. The S1146 point mutation increases fluorescence
values relative to the
wild-type strain, whereas S639A and the double mutation leads to slightly
inferior performance.
8
CA 2982734 2017-10-17
Data is plotted as the average difference in fluorescence from the
untransformed wild-type
strain over the course of four days from 63-188 transformants in each pool.
FIG. 15 shows dilution-corrected Bodipy fluorescence in wild-type and SCT1-
transformed L. starkeyi. Average wild-type fluorescence, average transformant
fluorescence,
SCT1 110L transformant fluorescence, and transformant SCT1 131L fluorescence
are shown.
In FIG. 15, as well as in FIGS. 16, 24, 25, and 27, there are no error bars
included with the data
for the single transformants because each data point represents fluorescence
from one culture
of each transformant.
FIG. 16 shows dilution-corrected Bodipy fluorescence in wild-type and SLC1-
transformed L. starkeyi. Average wild-type fluorescence, average transformant
fluorescence,
and SCT1 30L transformant fluorescence are shown.
FIG. 17 shows dilution-corrected Bodipy fluorescence with dual overexpression
of
DGA1 and the gene for the deregulated protein Accl(S1146A) in L. starkeyi.
Dilution corrected
Bodipy fluorescence of wild-type strain (black), the strain overexpressing
cDGA1 (dark gray),
and the strain in which overexpressed cDGA1 was crossed with Accl(S1146A)
overexpressing
strains was monitored over the course of 4 days (D1, D2, D3, and D4). The
mated strain was
obtained by crossing the top performers from each cDGA1 and Acc 1 (S1146A)
transformant
pool. Overexpression of both the DGA1 gene and the gene for the mutated
protein
Acc 1 (S1146A) synergistically enhanced lipid accumulation compared to
overexpression of
each gene alone.
FIG. 18 shows dilution-corrected Bodipy fluorescence of L. starkeyi strains
with
combinatorial expression of lipogenic cassettes. The strains are shown in the
following order
(from left (black) to right (white): wild-type, cDGA-NAT (the top cDGA1-1233
strain), cDGA-
HPH (a new platform strain), and cDGA-HPH crossed with either cDGA-NAT or a
strain
transformed with engineered SCTI. Fluorescence was monitored over the course
of 4 days (D1,
D2, D3, and D4). The top strains (lightest gray and white) exhibited
improvement in lipid
production through dual overexpression of lipogenic cassettes.
FIG. 19 shows dilution-corrected Bodipy fluorescence of L. starkeyi strains
with
combinatorial expression of lipogenic and auxiliary cassettes. The strains are
shown in the
following order (from left (black) to right (white): wild-type, ATP citrate
lyase a and 13 subunit
9
CA 2982734 2017-10-17
overexpressing strain (designated as cAc11/2), malic enzyme cloned from gDNA
overexpressing strain (designated as gMalic Enzyme), diacylglycerol
transferase (designated as
cDGA 1 -1233) cloned from cDNA overexpressing strain (cDGA 1), combinatorial
cDGA1-1233
and Ac11/2 overexpressing strain (cDGA1 x cAc11/2), and combinatorial cDGA1-
1233 and
genomic malic enzyme overexpressing strain (cDGA1 X gMalic Enzyme).
Combinatorial
expression of lipogenic and auxiliary cassettes improved lipid production.
FIG. 20 shows Bodipy analysis in sTS of a lipogenic diacylglycerol transferase
overexpressing strain (cDGA) and the same strain transformed with a second
base vector. The
introduction of expressing a second resistance marker does not increase lipid
accumulation on
its own, and may even decrease lipid levels.
FIG. 21 shows wild-type L. starkeyi (first column), or transformed cells
overexpressing
the gene for glycerol kinase (GUT], second column) or GUT] and an FAD-
dependent glycerol-
3-phosphate dehydrogenase (GUT1/GUT2, third column) plated onto YP solid media
containing 2% glycerol and incubated for 4-5 days at 30 C. The growth defect
of the GUT]
single transformant was rescued by overexpression of GUT2.
FIG. 22 shows average dilution-corrected Bodipy fluorescence in a parental
wild-type
L. starkeyi strain (black), a strain overexpressing GUT2 (dark gray), a strain
overexpressing
GUT] (light gray), and a GUT1/GUT2 double transformant (white). The GUT1/GUT2
double
transformant accumulates more lipid than parental wild-type or GUT]
overexpressing strains.
Overexpression of GUT2 alone has little effect on lipid accumulation.
FIGS. 23A-23C show cell density (OD), glucose consumption, and glycerol
consumption in a parental wild-type L. starkeyi strain (FIG. 23A), a GUT1
single transformant
(FIG. 23B), and a GUT1/GUT2 double transformant (FIG. 23C). FIG. 23D shows
superimposed curves of glycerol utilization in the wile-type strain and the
GUT1/GUT2 double
transformant. Glycerol was depleted in just 67 hours in the GUT1/GUT2 double
transformant
versus 84 hours in the wild-type strain.
FIG. 24 shows dilution-corrected Bodipy fluorescence of wild-type L. starkeyi
(black)
and a pool of GPD1 transformants (gray). Bodipy fluorescence was monitored in
synthetic thin
stillage over the course of 4 days. Overall, the transformants exhibited
moderately higher
fluorescence over the wild-type, with almost 13% improvement on the fourth
day. The top
CA 2982734 2017-10-17
transformant GPD1 67M (white) is also shown. The data indicate that
overexpression of
glycerol-3-phosphate in L. starkeyi increases lipid production.
FIG. 25 shows dilution-corrected Bodipy fluorescence in wild-type L. starkeyi
and
GND1/ZWF1 transformants. Average wild-type fluorescence, average transformant
fluorescence, transformant GND1 + ZWF1 37L fluorescence, and transformant GND1
+ ZWF1
26L fluorescence are shown.
FIG. 26 shows dilution-corrected Bodipy fluorescence in wild-type L. starkeyi
and
FOX1 and PDX1 knockouts.
FIG. 27 shows dilution-corrected Bodipy fluorescence in wild-type L. starkeyi
and
DGA2 transformants. Average wild-type fluorescence, average transformant
fluorescence,
transformant DGA2 20L fluorescence, and transformant DGA2 70L fluorescence are
shown.
FIG. 28 shows an engineered yeast constitutively secreting a glucoamylase that
retains
activity following incubation for 1 hour at 70 C. A) Supernatant of a wild-
type L. starkeyi
culture conditioned to secrete enzyme incubated on 1% corn starch plates prior
to (top) or after
(bottom) boiling. The presence of a clearing zone indicates starch hydrolytic
activity, which is
lost after boiling. B) Supernatant of wild-type cells display glucoamylase
activity when cultured
in starch containing media, but not YPD (left two boxes). In this case, starch
hydrolysis is
indicated by a zone impervious to iodine staining on a 2% starch plate. The
supernatant of the
engineered strain exhibits activity when cultured on both YPD and starch
containing media,
and is retained following incubation for one hour at 70 C (right two boxes).
DETAILED DESCRIPTION OF THE INVENTION
The elements and method steps described herein can be used in any combination
whether explicitly described or not.
All combinations of method steps as used herein can be performed in any order,
unless
otherwise specified or clearly implied to the contrary by the context in which
the referenced
combination is made.
As used herein, the singular forms "a," "an," and "the" include plural
referents unless
the content clearly dictates otherwise.
11
CA 2982734 2017-10-17
Numerical ranges as used herein are intended to include every number and
subset of
numbers contained within that range, whether specifically disclosed or not.
Further, these
numerical ranges should be construed as providing support for a claim directed
to any number
or subset of numbers in that range. For example, a disclosure of from 1 to 10
should be
construed as supporting a range of from 2 to 8, from 3 to 7, from 5 to 6, from
1 to 9, from 3.6
to 4.6, from 3.5 to 9.9, and so forth.
All patents, patent publications, and peer-reviewed publications (i.e.,
"references") cited
herein are expressly incorporated by reference to the same extent as if each
individual reference
were specifically and individually indicated as being incorporated by
reference. In case of
conflict between the present disclosure and the incorporated references, the
present disclosure
controls.
All protein identification (PID) numbers provided herein refer to proteins in
the database
of the Joint Genome Institute (JGI) of the United States Department of Energy.
See, e.g., Jeffries
2013.
It is understood that the invention is not confined to the particular
construction and
arrangement of parts herein illustrated and described, but embraces such
modified forms thereof
as come within the scope of the claims.
Lipogenic (Oleaginous) Yeasts
An aspect of the invention encompasses bioengineered yeasts. The bioengineered
yeasts
are preferably derived from lipogenic yeasts. The methods of the invention are
preferably
performed with either native, non-bioengineered lipogenic yeasts or
bioengineered lipogenic
yeasts.
Lipogenic yeasts (also known as oleaginous yeasts) have been recognized for
more than
50 years. They are defined as those that accumulate lipids in intracellular
oil bodies to greater
than 20% of their dry mass. In some yeasts, lipids have been reported to
account for up to 71%
of the cell's total biomass (Holdsworth et al. 1988). Out of the 1200 to 1500
known yeast
species, only a fraction qualifies as lipogenic. Lipomyces starkeyi was among
the earliest
lipogenic yeasts studied (Lodder et al. 1952). Other known lipogenic yeasts
include Yarrowia
Upo/ytica (Papanikolaou et al. 2001), and species in the genera of
Rhodotorula, Cryptococcus
12
CA 2982734 2017-10-17
(Ratledge 2002), Candida, Trichosporon (Holdsworth et al. 1988),
Rhodosporidium,
Sporidiobolus, Sporodobolomyces, and various other ascomyceteous and
basidiomycete genera
amounting for a total of about 100 species (Garay et al. 2016).
Lipogenic yeasts belong to the larger taxonomic groups of filamentous
ascornyceteous
and basidiomycetous fungi. Exemplary lipogenic yeasts include yeasts from the
genus
Lipomyces, such as L. starkeyi, L. anomalus, L. arxii, L. chichibuensis, L.
doorenjongii, L.
japonicus, L. kockii, L. kononenkoae, L. lipofer, L. mesembrius, L.
oligophaga, L. orientalis, L.
smithiae, L. spencermartinsiae, L. suomiensis, L. tetrasporus, L. yamadae, L.
yarrowii, and L.
Sp.; yeasts from the genus Yarrowia, such as Y lipolytica, Y. bubula, Y.
deformans, Y. divulgata,
Y. keelungensis, Y porcina, Y. yakushimensis, and Y. Sp.; yeasts from the
genus Candida, such
as C. Sp.; yeasts from the genus Hansenula, such as H. polymorpha; yeasts from
the genus
Cunninghamella, such as S. bigelovii sp nov CGMCC 8094, S. echinulate, S.
blakesleeana
JSK2, and S. Sp. Salicorn 5; yeasts from the genus Mortierella, such as M
alpina, M isabellina,
and M Sp.; yeasts from the genus Rhodosporidium, such as R. torulo ides, R.
babjevae, R.
diobovatum, R. fluviale, R. kratochvilovae, R. paludigenum, R. sphaerocarpum,
R. araucariae,
R. colostri, R. dairenensis, R. gram is, R. lusitaniae, and R. mucilaginosa;
yeasts from the
genus Sporidiobolus, such as S. johnsonii, S. pararoseus, S. ruineniae, S.
ruineniae, and S.
salmonicolor; yeasts from the genus Sporobolomyces, such as S. bannaensis, S.
beijingensis, S.
carnicolor, S. metaroseus, S. odoratus, S. poonsookiae, S. sin gularis, and S.
inositophilus;
yeasts from the genus Occuitifur, such as 0. externus; yeasts from the genus
Rhodotorula, such
as R. bogoriensis, R. hylophila, R. glutinis, and R. rhodochrous; yeasts from
the genus
Trichosporon, such as T fermentans, T. oleaginosus ATCC 20509, and T cutaneum;
and yeasts
from the genus Cryptococcus, such as C. curvatus and C. Sp.
Certain filamentous fungi and unicellular algae can also be lipogenic. These
include
filamentous fungi from the genus Aspergillus, such as A. nidulans, and from
the genus Mucor,
such as M circinelloides and M rouxii. Lipogenic algae include species from
the genus
Scenedesmus, such as S. quadricauda.
Nontraditional lipogenic yeasts have an innate ability to convert poorly
metabolized
wastes from ethanol fermentation into lipids, protein, and enzymes. Some
lipogenic yeasts
13
CA 2982734 2017-10-17
naturally make large amounts of lipids from a wide variety of carbon sources,
and this
prodigious capacity renders them amenable to many bioprocessing applications.
Lipomyces starkeyi is a particularly preferred lipogenic yeast in this regard.
L. starkeyi
can utilize many different substrates, including the oligosaccharides and
sugars found in both
agricultural waste products and the hydrolysates of lignocellulosic material
(CaIvey et al. 2014,
Gong et al. 2012, Zhao et al. 2008).
L. starkeyi genes of notable importance include 24 glycoside hydrolases, three
alpha-
amylases, a highly versatile dextranase to metabolize the lateral starch side
chains, and 15
copies of maltase that convert oligosaccharides into glucose (Riley et al.
2016, Kang et al.
2005). Overproduction and secretion of the thermostable alpha-amylase and
dextranase found
in L. starkeyi are particularly useful for starch saccharification in the
integrated process
described here (FIG. 2).
The L. starkeyi genome also encodes at least two secreted [3-1,4-glucosidases
and four
non-secreted 13-1,4-glucosidases, all of which have cellulosic carbohydrate
binding domains,
and at least two endo-1,4-13-D-glucanases (cellulases) (Chen et al. 2008).
These enzymatic
activities are useful for hydrolyzing corn fiber or other agricultural
residues for additional
ethanol or biodiesel production.
An ensemble of lipogenic genes are found in the genome of L. starkeyi, along
with the
metabolic machinery to supply the acetyl-CoA and NADPH needed to support high
levels of
lipid biosynthesis. For example, L. starkeyi has two complete genes for
pyruvate carboxylase
(LsPYCl, LsPYC2). This enzyme combines CO2 with pyruvate to make oxaloacetate,
which in
turn citrate synthase (LsCIT1, LsC1T2, LsCIT3) combines with acetyl-CoA to
make citrate.
Genes coding for the alpha and beta subunits of ATP :citrate lyase (LsACL1,
LsACL2) are found
in a tandem bidirectional operon. Likewise, the genome of L. starkeyi codes
for two complete
fatty acid synthase complexes (LsFAS1.1, LsFAS1.2; LsFAS2.1, LsFAS2.2) with
each
organized into tandem bidirectional operons. Most yeasts and fungi have only a
single FAS
complex with the alpha and beta-subunits occurring in different parts of the
genome.
Mitochondrial NAD-dependent malate dehydrogenase (MDm, LsMDH2; PID 5229), and
cytosolic NAD-dependent malate dehydrogenase (MDc, LSMDH1; PID_3988) exist as
single
copies in wild type L. starkeyi. Tandem bidirectional operons or clusters of
metabolically
14
CA 2982734 2017-10-17
associated genes indicate close interdependence and coordinated regulation of
genes
comprising a metabolic trait (Jeffries 2013).
L. starkeyi yeast maintains a basal lipid content that increases throughout
fermentation,
which already meets or exceeds that of any other native lipogenic yeast or
alga. The lipid profile
produced by L. starkeyi is remarkably similar to that of palm oil, one of the
most common
biodiesel feedstocks, which indicates that a biodiesel produced from this
species naturally has
desirable fuel properties (CaIvey et al. 2016).
L. starkeyi is generally regarded as safe (GRAS) and is easily propagated,
rendering
residual cells and cell proteins useful for fortifying livestock or other
animal feeds (Collett et
al. 2014). Lastly, its genome has been sequenced, which enables the use of
rational metabolic
engineering methodologies to target specific genes for overexpression or
deletion. By simply
altering the expression or regulation of genes already present in the genome
it is possible to
improve lipid production and accumulation on poorly utilized substrates or
under conditions
when lipogenesis would not normally occur.
Lipomyces starkeyi is among the yeasts that can metabolize glucose, xylose and
cellobiose, which are the main sugars released from the hydrolysis of
lignocellulosic materials
(Pan et al. 2009). With L. starkeyi, optimal lipid production is attained when
growing on a 2:1
mixture of glucose and xylose, the same ratio found in enzymatic hydrolysates.
Some strains of
L. starkeyi also produce lipid from glycerol.
The lipid profile of Lipomyces is similar to that of palm oil (Calvey et al.
2016), which
is important for both food and fuel production. By developing technology that
uses Lipomyces
to produce biofuels from the hydrolysates of agricultural cellulosic residues
as an alternative to
seed-based oils, the present invention provides for generating fuel from a
renewable,
environmentally benign resource that does not compete with food production.
Furthermore,
biodiesel derived from non-food sources such as lignocellulosic hydrolysate is
advantageous
because it meets the criteria delineated under Renewable Fuel Standard 2
(RFS2), which
mandates the increased use of advanced cellulosic biofuels.
In addition to yeasts, a large number of eukaryotic algae also accumulate
lipids,
particularly when cultivated under heterotrophic conditions (US Patent
8,110,670). These
eukaryotic algae can also be used in the methods of the present invention. One
advantage to
CA 2982734 2017-10-17
using lipogenic yeasts over algae, however, is that they can be grown readily
in bioreactors and,
unlike heterotrophically cultivated lipogenic algae, lipogenic yeasts can be
cultivated on a wide
range of organic substrates.
Lipid accumulation typically occurs when a readily assimilated carbon source
is present
in excess and nitrogen is limiting (Wei et al. 2009, Zhu et al. 2012). For
example, when L.
starkeyi transitions into a nitrogen-limited environment the biosynthetic
pathways dependent
on abundant nitrogen shut down and lipogenesis becomes the dominant metabolic
feature of
the cell. The cells continue to assimilate carbon, and in the absence of new
cell growth, they
store it as triacylglycerols. The most readily assimilated lipogenic carbon
source is typically
glucose (Ratledge 2002), and xylose has been reported to increase lipid
accumulation even more
(Gong et al. 2012, Zhao et al. 2008). Other substrates include cellobiose,
glycerol,
oligosaccharides, various industrial organic byproducts and hydrolysate from
non-edible
cellulosic feedstocks (Vicente et al. 2010). The yeasts engineered herein are
capable of
producing lipid when glucose is limited and carbon organics and nitrogen are
in abundance.
Any or all of the genes described above can be driven by promoters that may be
regulated or constitutive, strong, moderately strong or weak in L. starkeyi to
modulate their
activity therein. Alternately these genes may be deleted or inactivated.
Likewise genes for
metabolic pathways competing with the desired product of the genes described
above may be
modulated, deleted, or inactivated in order to improve the desired product or
its formation rate.
Any or all of the genes described above in L. starkeyi can be incorporated in
any other
lipogenic yeast to confer similar benefits therein.
Biochemistry of Lipid Synthesis by Yeasts
The biochemistry of lipid synthesis by yeasts is outlined in FIG. 3. Pyruvate
is
transported from the cytosol into the mitochondrion where pyruvate carboxylase
(PYC)
combines CO2 with pyruvate to make oxaloacetate, and pyruvate dehydrogenase
(PDT')
oxidizes pyruvate to acetyl-CoA and NADH. Citrate synthase (CIT) combines with
acetyl-CoA
to make citrate while mitochondrial malate dehydrogenase (MDm) reduces
oxaloacetate to
malate.
16
CA 2982734 2017-10-17
The first step in response to nitrogen limitation is the activation of AMP
deaminase,
which cleaves adenosine monophosphate (AMP) into inosine monophosphate (IMP)
and
ammonia (NH4). This activation lowers the intracellular concentrations of AMP,
which inhibits
the TCA cycle at the level of isocitrate dehydrogenase (ICDH), whose function
is uniquely
dependent on AMP in lipogenic yeasts (Ratledge 2004).
Significant evidence exists to support this mechanism. Intracellular AMP
concentrations fall 11-fold within 24 hours after a transition to nitrogen
limitation (Boulton et
al. 1983). Also, assays of the Lipomyces ICDH1/2 enzyme show that its activity
decreases about
5-fold when cells are transferred to a nitrogen-limiting medium (Tang et al.
2009). Activation
of AMP deaminase and decreases in ICDH activity result in the accumulation of
isocitrate and
citrate (Boulton et al. 1983, Tang etal. 2009). Furthermore, lipid-
accumulating Lipomyces cells
display higher ACL enzymatic activity than proliferating cells (Naganuma et
al. 1987).
When ICDH is no longer active, but the flux of glycolysis to pyruvate
continues,
isocitrate accumulates in the mitochondria and then equilibrates with citrate
via isomerization.
Citrate is exported into the cytosol via the citrate efflux system, which
transports citrate out of
the mitochondria in exchange for malate. The reduction in NADH formation by
ICDH also
results in less need for oxygen uptake and a lower rate of ATP synthesis.
Next, ATP:citrate lyase (ACL) catalyzes the cleavage of citrate into
oxaloacetate and
acetyl-CoA. This reaction is thought to be the primary source of the acetyl-
CoA used for lipid
synthesis and has been recognized as a key to efficient lipid production in
lipogenic yeasts.
ACL occurs in all lipogenic yeasts, but not in non-oleaginous species, which
suggests a central
role in lipid accumulation (Ratledge 2002, Evans et al. 1985). In the presence
of CoA and ATP
this enzyme cleaves citrate to acetyl-CoA and oxaloacetate, and supplies
acetyl-CoA for lipid
synthesis (Ratledge 1987). ACL is a dimeric protein with alpha and beta-
subunits coded for by
ACL1 and ACL2, which occur in a tandem bidirectional operon in the Lipomyces
starkeyi
genome (Jeffries 2013). The holoenzyme has a high affinity for citrate and ATP
and is inhibited
by ADP, glucose 6-phosphate, palmitoyl-CoA, and oleoyl-CoA. These allosteric
inhibitors
indicate that low ATP levels, high glycolytic flux, and the accumulation of
fatty acid end
products limit activity. The oxaloacetate formed by ACL can be converted to
malate by a
17
CA 2982734 2017-10-17
cytosolic malate dehydrogenase (MDc) and thus facilitate the export of more
citrate from the
mitochondrion.
Malate can also undergo conversion by malic enzyme (ME) into pyruvate and CO2,
while regenerating NADPH in the cytosol. The NADPH produced by the action of
malic
enzyme is thought to be the primary provider of reducing power for both fatty
acid biosynthesis
and desaturation reactions (Wynn et al. 1999). Lipid production may, however,
draw on sources
of NADPH in the cell such as the oxidative pentose phosphate pathway (Ratledge
2014).
Specifically, two molecules of NADPH are required for each acetyl-CoA added to
the growing
fatty acyl chain during the standard fatty acid elongation cycle on the fatty
acid synthase (FAS)
complex (Ratledge 2004).
Malic enzyme (ME) has a high affinity for malate, and is only weakly inhibited
by
citrate, pyruvate, oxaloacetate, and ATP. ME is found in all lipogenic yeasts
and its activity
disappears following the period of active lipid accumulation. In contrast,
acetyl-CoA
carboxylase (ACC), fatty acid synthase (FAS), diacyglycerol acyltransferase
(DGA),
ATP:citrate lyase (ACL) and the NADPH-generating enzymes glucose-6-phosphate
dehydrogenase, 6-phosphogluconate dehydrogenase and NADP(+):isocitrate
dehydrogenase,
do not demonstrate any changes in activities correlating with the accumulation
of storage lipid
(Wynn et al. 1999). ME therefore appears to be a source of NADPH for
lipogenesis and a target
for modification.
Metabolic pathways are often regulated at the first committing step. In this
case,
synthesis of malonyl-CoA, which is catalyzed by acetyl-CoA carboxylase (ACC),
is a limiting
step in attaining high titers of fatty acids and polyketides. Some versions of
acetyl-CoA
carboxylase are deactivated by a serine/threonine protein kinase, which is in
turn activated by
AMP. This relationship causes the acetyl-CoA carboxylase to be inactivated
when glucose is
depleted. To prevent this deactivation, a site-directed mutation is introduced
in the serine
residues of acetyl-CoA carboxylase that are phosphorylated by the AMP-
activated protein
kinase (AMPK). By converting these serines in Acc 1 into an alanine or other
non-serine and
non-threonine residue, phosphorylation and inactivation is avoided. When the
mutated gene is
introduced into a host, acetyl-CoA carboxylase activity and total lipid
accumulation increase.
18
CA 2982734 2017-10-17
Overexpression of the L. starkeyi ACC1 and GPD1 genes increase lipid
production.
Overexpression of ACC1, GUT2, and GUT] also increases lipid production. In the
case of
GUT2 and GUT] overexpression, higher levels of the coded proteins should
increase the uptake
of glycerol from thin stillage. In the case of ACC] (acetyl-CoA carboxylase)
overexpression
and modification, altering the regulation should increase the formation of
malonyl-CoA as a
precursor for lipid synthesis and thereby increase the flux of malonyl-CoA
into malonyl
transferase which is an enzymatic activity of fatty acid synthase (FAS1).
Enzymes Involved in Substrate Assimilation
Lipomyces species and other lipogenic yeasts produce active amylases and other
enzymes that degrade polysaccharides, and so are able to produce lipids from
starch. L. starkeyi
and other lipogenic yeasts also use a wide range of other polysaccharides
(Gallagher et al. 1991,
Punpeng et al. 1992, Steyn et al. 1995, Bignell et al. 2000, Ryu et al. 2000,
Wilkie et al. 2000,
Lee et al. 2003).
The genome of Lipomyces starkeyi contains numerous starch degrading alpha-
amylase
glycoside hydrolases (CAZY family GH13). Genes for several of these enzymes
occur in
clusters along with sugar transporters (e.g. Lipomyces starkeyi [PID_5034, PID
5035,
PID 5036, PID 29016]; [PID 73677, PID 5097]; [PID 5097, PID 73677]; [PID
205534,
PID 205437]; [PID 32360, PID 71673, PID 3625]). At least one of the alpha-
amylases
encoded in the L. starkeyi genome is thermostable (TAM], PID 272826).
Thermostable alpha-
amylases are rarely found in yeasts. A gene for an amylo-alpha-1,6-glycosidase
(dextranase) is
also present (PID_5189) (Jeffries 2013). These features make L. starkeyi
particularly suitable
for lipid production when cultivated on starch (Gallagher et al. 1991, Punpeng
et al. 1992).
Different strains of Lipomyces starkeyi show variable capacities for the
assimilation of
glycerol. The first step in glycerol metabolism is phosphorylation of glycerol
by glycerol kinase
(GUT I, PID_332345) to form glycerol-3-phosphate. This is followed by
oxidation of glycerol-
3-phosphate to dihydroxyacetone phosphate by an FAD-dependent glycerol-3-
phosphate
dehydrogenase (GUT2, PID 3942). Genes for both of these enzymes are present in
Lipomyces
starkeyi NRRL Y-11557 (Jeffries 2013).
19
CA 2982734 2017-10-17
Beta-1,4-endo-glucanase (BGL; GH5) depolymerizes glucan oligosaccharides. The
genome of L. starkeyi encodes at least two GH5 cellulases, EGC1 and EGC2
(PID_72543 and
PID 5513). The gene EGC1 is adjacent to a HXT2 sugar transporter (PID 4247) in
the
Lipomyces starkeyi genome. The HXT2 imports cellobiose (or
cellulooligosaccharides). Beta-
glucosidase (BGL; EC 3.2.1.21; GH3) is involved in the bioconversion of
cellobiose to glucose.
The genome of Lipomyces starkeyi encodes six putative enzymes in this family.
At least one
appears to be highly expressed (PID_69491) and at least two appear to be
secreted (PID_147
and PID 5081). Several of the genes for GH3 proteins are proximally associated
with genes
coding for HXT2 sugar transporters (PID_3714, PID_334883, PID_7343).
Such proximal clustering of genes having associated physiological functions
indicates
that L. starkeyi has evolved for efficient use of beta- and alpha-linked
glucans such as cellulose
and starch.
The genome of L. starkeyi also contains a putative "glycoside hydrolase Family
61"
auxiliary redox enzyme for cellulose hydrolysis (PID_61479) (Jeffries 2013).
This protein
includes a secretion signal and occurs rarely in ascomyceteous yeasts (Riley
et al. 2016). This
enzyme family is now known to not belong to the glycoside hydrolases, but
rather is a "lytic
polysaccharide mono-oxygenase" (LPMO). Enzymes belonging to this class are
important
components of commercial cellulase preparations (Cannella et al. 2014).
Glycerol kinase (GUT]) and glycerol-3-phosphate dehydrogenase (GUT2) are the
first
two genes involved in the assimilation of glycerol. Both of these genes are
present in the
genome of L. starkeyi NRRL- 1 1 557 (Jeffries 2013).
Any or all of the genes described above in L. starkeyi can be constitutively
or
overexpressed in L. starkeyi to enhance their activity therein.
Any or all of the genes described above in L. starkeyi can be incorporated in
any other
lipogenic yeast to confer similar benefits therein.
Genetic Targets for Metabolic Engineering
Various versions of the invention are directed to yeasts genetically modified
to comprise
one or more recombinant nucleic acids configured to express one or more
proteins. The one or
more recombinant nucleic acids are preferably configured to constitutively
express or to
CA 2982734 2017-10-17
overexpress the one or more proteins. The one or more recombinant nucleic
acids preferably
comprise one or more recombinant genes configured to constitutively express or
to overexpress
the one or more proteins. The expressed proteins include enzymes and other
types of proteins
such as transporters. If a cell endogenously expresses a particular protein,
the nucleic acid
expressing that protein may be modified to exchange or optimize promoters,
exchange or
optimize enhancers, or exchange or optimize any other genetic element that
results in increased
or constitutive expression of the proteins. Alternatively or additionally, one
or more additional
copies of a gene or coding sequence thereof may be introduced to the cell for
enhanced
expression of the proteins. If a cell does not endogenously express a
particular protein, one or
more copies of a recombinant nucleic acid configured to express that protein
may be introduced
to the cell for expression of the protein. The recombinant nucleic acid may be
incorporated into
the genome of the cell or may be contained on an extra-chromosomal plasmid.
Techniques for
genetic manipulation are described in further detail below. The genetically
modified yeasts of
the invention are also referred to herein as "recombinant," "engineered," or
"bioengineered"
yeasts, or other designations.
The recombinant yeasts of the invention may comprise one or more recombinant
nucleic
acids configured to express any one or more of the following proteins in any
combination: an
acetyl-CoA carboxylase, an alpha-amylase, an ATP citrate lyase, a
diacylglycerol
acyltransferase, a fatty acid synthase, a glycerol kinase, a 6-
phosphogluconate dehydrogenase,
a glycerol-3-phosphate dehydrogenase, a malic enzyme, a fatty acyl-CoA
reductase, a delta-9
acyl-CoA desaturase, a glycerol-3-phosphate acyltransferase, a
lysophosphatidate
acyltransferase, a glucose-6-phosphate dehydrogenase, a beta-glucosidase, a
hexose
transporter, a glycerol transporter, a glycoside hydrolase enzyme, and an
auxiliary activity
family 9 enzyme. The one or more recombinant nucleic acids preferably comprise
one or more
recombinant genes configured to express the above-referenced proteins.
For example, the recombinant yeasts of the invention may comprise one or more
recombinant genes configured to express an acetyl-CoA carboxylase alone or
with any one or
more of an alpha-amylase, an ATP citrate lyase, a diacylglycerol
acyltransferase, a fatty acid
synthase, a glycerol kinase, a 6-phosphogluconate dehydrogenase, a glycerol-3-
phosphate
dehydrogenase, a malic enzyme, a fatty acyl-CoA reductase, a delta-9 acyl-CoA
desaturase, a
21
CA 2982734 2017-10-17
glycerol-3-phosphate acyltransferase, a lysophosphatidate acyltransferase, a
glucose-6-
phosphate dehydrogenase, a beta-glucosidase, a hexose transporter, a glycerol
transporter, a
glycoside hydrolase enzyme, and an auxiliary activity family 9 enzyme in any
combination.
The recombinant yeasts of the invention may comprise one or more recombinant
genes
configured to express an alpha-amylase alone or with any one or more of an
acetyl-CoA
carboxylase, an ATP citrate lyase, a diacylglycerol acyltransferase, a fatty
acid synthase, a
glycerol kinase, a 6-phosphogluconate dehydrogenase, a glycerol-3-phosphate
dehydrogenase,
a malic enzyme, a fatty acyl-CoA reductase, a delta-9 acyl-CoA desaturase, a
glycerol-3-
phosphate acyltransferase, a lysophosphatidate acyltransferase, a glucose-6-
phosphate
dehydrogenase, a beta-glucosidase, a hexose transporter, a glycerol
transporter, a glycoside
hydrolase enzyme, and an auxiliary activity family 9 enzyme in any
combination.
The recombinant yeasts of the invention may comprise one or more recombinant
genes
configured to express an ATP citrate lyase alone or with any one or more of an
acetyl-CoA
carboxylase, an alpha-amylase, a diacylglycerol acyltransferase, a fatty acid
synthase, a
glycerol kinase, a 6-phosphogluconate dehydrogenase, a glycerol-3-phosphate
dehydrogenase,
a malic enzyme, a fatty acyl-CoA reductase, a delta-9 acyl-CoA desaturase, a
glycerol-3-
phosphate acyltransferase, a lysophosphatidate acyltransferase, a glucose-6-
phosphate
dehydrogenase, a beta-glucosidase, a hexose transporter, a glycerol
transporter, a glycoside
hydrolase enzyme, and an auxiliary activity family 9 enzyme in any
combination.
The recombinant yeasts of the invention may comprise one or more recombinant
genes
configured to express a diacylglycerol acyltransferase alone or with any one
or more of an
acetyl-CoA carboxylase, an alpha-amylase, an ATP citrate lyase, a fatty acid
synthase, a
glycerol kinase, a 6-phosphogluconate dehydrogenase, a glycerol-3-phosphate
dehydrogenase,
a malic enzyme, a fatty acyl-CoA reductase, a delta-9 acyl-CoA desaturase, a
glycerol-3-
phosphate acyltransferase, a lysophosphatidate acyltransferase, a glucose-6-
phosphate
dehydrogenase, a beta-glucosidase, a hexose transporter, a glycerol
transporter, a glycoside
hydrolase enzyme, and an auxiliary activity family 9 enzyme in any
combination.
The recombinant yeasts of the invention may comprise one or more recombinant
genes
configured to express a fatty acid synthase alone or with any one or more of
an acetyl-CoA
carboxylase, an alpha-amylase, an ATP citrate lyase, a diacylglycerol
acyltransferase, a
22
CA 2982734 2017-10-17
glycerol kinase, a 6-phosphogluconate dehydrogenase, a glycerol-3-phosphate
dehydrogenase,
a malic enzyme, a fatty acyl-CoA reductase, a delta-9 acyl-CoA desaturase, a
glycerol-3-
phosphate acyltransferase, a lysophosphatidate acyltransferase, a glucose-6-
phosphate
dehydrogenase, a beta-glucosidase, a hexose transporter, a glycerol
transporter, a glycoside
hydrolase enzyme, and an auxiliary activity family 9 enzyme in any
combination.
The recombinant yeasts of the invention may comprise one or more recombinant
genes
configured to express a glycerol kinase alone or with any one or more of an
acetyl-CoA
carboxylase, an alpha-amylase, an ATP citrate lyase, a diacylglycerol
acyltransferase, a fatty
acid synthase, a 6-phosphogluconate dehydrogenase, a glycerol-3-phosphate
dehydrogenase, a
malic enzyme, a fatty acyl-CoA reductase, a delta-9 acyl-CoA desaturase, a
glycerol-3-
phosphate acyltransferase, a lysophosphatidate acyltransferase, a glucose-6-
phosphate
dehydrogenase, a beta-glucosidase, a hexose transporter, a glycerol
transporter, a glycoside
hydrolase enzyme, and an auxiliary activity family 9 enzyme in any
combination.
The recombinant yeasts of the invention may comprise one or more recombinant
genes
configured to express a 6-phosphogluconate dehydrogenase alone or with any one
or more of
an acetyl-CoA carboxylase, an alpha-amylase, an ATP citrate lyase, a
diacylglycerol
acyltransferase, a fatty acid synthase, a glycerol kinase, a glycerol-3-
phosphate dehydrogenase,
a malic enzyme, a fatty acyl-CoA reductase, a delta-9 acyl-CoA desaturase, a
glycerol-3-
phosphate acyltransferase, a lysophosphatidate acyltransferase, a glucose-6-
phosphate
dehydrogenase, a beta-glucosidase, a hexose transporter, a glycerol
transporter, a glycoside
hydrolase enzyme, and an auxiliary activity family 9 enzyme in any
combination.
The recombinant yeasts of the invention may comprise one or more recombinant
genes
configured to express a glycerol-3-phosphate dehydrogenase alone or with any
one or more of
an acetyl-CoA carboxylase, an alpha-amylase, an ATP citrate lyase, a
diacylglycerol
acyltransferase, a fatty acid synthase, a glycerol kinase, a 6-
phosphogluconate dehydrogenase,
a malic enzyme, a fatty acyl-CoA reductase, a delta-9 acyl-CoA desaturase, a
glycerol-3-
phosphate acyltransferase, a lysophosphatidate acyltransferase, a glucose-6-
phosphate
dehydrogenase, a beta-glucosidase, a hexose transporter, a glycerol
transporter, a glycoside
hydrolase enzyme, and an auxiliary activity family 9 enzyme in any
combination.
23
CA 2982734 2017-10-17
The recombinant yeasts of the invention may comprise one or more recombinant
genes
configured to express a malic enzyme alone or with any one or more of an
acetyl-CoA
carboxylase, an alpha-amylase, an ATP citrate lyase, a diacylglycerol
acyltransferase, a fatty
acid synthase, a glycerol kinase, a 6-phosphogluconate dehydrogenase, a
glycerol-3-phosphate
dehydrogenase, a fatty acyl-CoA reductase, a delta-9 acyl-CoA desaturase, a
glycerol-3-
phosphate acyltransferase, a lysophosphatidate acyltransferase, a glucose-6-
phosphate
dehydrogenase, a beta-glucosidase, a hexose transporter, a glycerol
transporter, a glycoside
hydrolase enzyme, and an auxiliary activity family 9 enzyme in any
combination.
The recombinant yeasts of the invention may comprise one or more recombinant
genes
configured to express a fatty acyl-CoA reductase alone or with any one or more
of an acetyl-
CoA carboxylase, an alpha-amylase, an ATP citrate lyase, a diacylglycerol
acyltransferase, a
fatty acid synthase, a glycerol kinase, a 6-phosphogluconate dehydrogenase, a
glycerol-3-
phosphate dehydrogenase, a malic enzyme, a delta-9 acyl-CoA desaturase, a
glycerol-3-
phosphate acyltransferase, a lysophosphatidate acyltransferase, a glucose-6-
phosphate
dehydrogenase, a beta-glucosidase, a hexose transporter, a glycerol
transporter, a glycoside
hydrolase enzyme, and an auxiliary activity family 9 enzyme in any
combination.
The recombinant yeasts of the invention may comprise one or more recombinant
genes
configured to express a delta-9 acyl-CoA desaturase alone or with any one or
more of an acetyl-
CoA carboxylase, an alpha-amylase, an ATP citrate lyase, a diacylglycerol
acyltransferase, a
fatty acid synthase, a glycerol kinase, a 6-phosphogluconate dehydrogenase, a
glycerol-3-
phosphate dehydrogenase, a malic enzyme, a fatty acyl-CoA reductase, a
glycerol-3-phosphate
acyltransferase, a lysophosphatidate acyltransferase, a glucose-6-phosphate
dehydrogenase, a
beta-glucosidase, a hexose transporter, a glycerol transporter, a glycoside
hydrolase enzyme,
and an auxiliary activity family 9 enzyme in any combination.
The recombinant yeasts of the invention may comprise one or more recombinant
genes
configured to express a glycerol-3-phosphate acyltransferase alone or with any
one or more of
an acetyl-CoA carboxylase, an alpha-amylase, an ATP citrate lyase, a
diacylglycerol
acyltransferase, a fatty acid synthase, a glycerol kinase, a 6-
phosphogluconate dehydrogenase,
a glycerol-3-phosphate dehydrogenase, a malic enzyme, a fatty acyl-CoA
reductase, a delta-9
acyl-CoA desaturase, a lysophosphatidate acyltransferase, a glucose-6-
phosphate
24
CA 2982734 2017-10-17
dehydrogenase, a beta-glucosidase, a hexose transporter, a glycerol
transporter, a glycoside
hydrolase enzyme, and an auxiliary activity family 9 enzyme in any
combination.
The recombinant yeasts of the invention may comprise one or more recombinant
genes
configured to express a lysophosphatidate acyltransferase alone or with any
one or more of an
acetyl-CoA carboxylase, an alpha-amylase, an ATP citrate lyase, a
diacylglycerol
acyltransferase, a fatty acid synthase, a glycerol kinase, a 6-
phosphogluconate dehydrogenase,
a glycerol-3-phosphate dehydrogenase, a malic enzyme, a fatty acyl-CoA
reductase, a delta-9
acyl-CoA desaturase, a glycerol-3-phosphate acyltransferase, a glucose-6-
phosphate
dehydrogenase, a beta-glucosidase, a hexose transporter, a glycerol
transporter, a glycoside
hydrolase enzyme, and an auxiliary activity family 9 enzyme in any
combination.
The recombinant yeasts of the invention may comprise one or more recombinant
genes
configured to express a glucose-6-phosphate dehydrogenase alone or with any
one or more of
an acetyl-CoA carboxylase, an alpha-amylase, an ATP citrate lyase, a
diacylglycerol
acyltransferase, a fatty acid synthase, a glycerol kinase, a 6-
phosphogluconate dehydrogenase,
a glycerol-3-phosphate dehydrogenase, a malic enzyme, a fatty acyl-CoA
reductase, a delta-9
acyl-CoA desaturase, a glycerol-3-phosphate acyltransferase, a
lysophosphatidate
acyltransferase, a beta-glucosidase, a hexose transporter, a glycerol
transporter, a glycoside
hydrolase enzyme, and an auxiliary activity family 9 enzyme in any
combination.
The recombinant yeasts of the invention may comprise one or more recombinant
genes
configured to express a beta-glucosidase alone or with any one or more of an
acetyl-CoA
carboxylase, an alpha-amylase, an ATP citrate lyase, a diacylglycerol
acyltransferase, a fatty
acid synthase, a glycerol kinase, a 6-phosphogluconate dehydrogenase, a
glycerol-3-phosphate
dehydrogenase, a malic enzyme, a fatty acyl-CoA reductase, a delta-9 acyl-CoA
desaturase, a
glycerol-3-phosphate acyltransferase, a lysophosphatidate acyltransferase, a
glucose-6-
phosphate dehydrogenase, a hexose transporter, a glycerol transporter, a
glycoside hydrolase
enzyme, and an auxiliary activity family 9 enzyme in any combination.
The recombinant yeasts of the invention may comprise one or more recombinant
genes
configured to express a hexose transporter alone or with any one or more of an
acetyl-CoA
carboxylase, an alpha-amylase, an ATP citrate lyase, a diacylglycerol
acyltransferase, a fatty
acid synthase, a glycerol kinase, a 6-phosphogluconate dehydrogenase, a
glycerol-3-phosphate
CA 2982734 2017-10-17
dehydrogenase, a malic enzyme, a fatty acyl-CoA reductase, a delta-9 acyl-CoA
desaturase, a
glycerol-3-phosphate acyltransferase, a lysophosphatidate acyltransferase, a
glucose-6-
phosphate dehydrogenase, a beta-glucosidase, a glycerol transporter, a
glycoside hydrolase
enzyme, and an auxiliary activity family 9 enzyme in any combination.
The recombinant yeasts of the invention may comprise one or more recombinant
genes
configured to express a glycerol transporter alone or with any one or more of
an acetyl-CoA
carboxylase, an alpha-amylase, an ATP citrate lyase, a diacylglycerol
acyltransferase, a fatty
acid synthase, a glycerol kinase, a 6-phosphogluconate dehydrogenase, a
glycerol-3-phosphate
dehydrogenase, a malic enzyme, a fatty acyl-CoA reductase, a delta-9 acyl-CoA
desaturase, a
glycerol-3-phosphate acyltransferase, a lysophosphatidate acyltransferase, a
glucose-6-
phosphate dehydrogenase, a beta-glucosidase, a hexose transporter, a glycoside
hydrolase
enzyme, and an auxiliary activity family 9 enzyme in any combination.
The recombinant yeasts of the invention may comprise one or more recombinant
genes
configured to express a glycoside hydrolase enzyme alone or with any one or
more of an acetyl-
CoA carboxylase, an alpha-amylase, an ATP citrate lyase, a diacylglycerol
acyltransferase, a
fatty acid synthase, a glycerol kinase, a 6-phosphogluconate dehydrogenase, a
glycerol-3-
phosphate dehydrogenase, a malic enzyme, a fatty acyl-CoA reductase, a delta-9
acyl-CoA
desaturase, a glycerol-3-phosphate acyltransferase, a lysophosphatidate
acyltransferase, a
glucose-6-phosphate dehydrogenase, a beta-glucosidase, a hexose transporter, a
glycerol
transporter, and an auxiliary activity family 9 enzyme in any combination.
The recombinant yeasts of the invention may comprise one or more recombinant
genes
configured to express an auxiliary activity family 9 enzyme alone or with any
one or more of
an acetyl-CoA carboxylase, an alpha-amylase, an ATP citrate lyase, a
diacylglycerol
acyltransferase, a fatty acid synthase, a glycerol kinase, a 6-
phosphogluconate dehydrogenase,
a glycerol-3-phosphate dehydrogenase, a malic enzyme, a fatty acyl-CoA
reductase, a delta-9
acyl-CoA desaturase, a glycerol-3-phosphate acyltransferase, a
lysophosphatidate
acyltransferase, a glucose-6-phosphate dehydrogenase, a beta-glucosidase, a
hexose
transporter, a glycerol transporter, and a glycoside hydrolase enzyme in any
combination.
Acetyl-CoA carboxylases include enzymes falling under Enzyme Commission (EC)
number 6.4.1.2. An exemplary acetyl-CoA carboxylase that may be expressed
includes Acc 1
26
CA 2982734 2017-10-17
(SEQ ID NO:2) encoded by Accl (SEQ ID NO:1) from L. starkeyi (PID_72701) Other
exemplary acetyl-coA carboxylases include Acc 1 mutants that comprise a
residue other than
serine at a position corresponding to position 1146 of SEQ ID NO:2. The
residue at position
1146 is preferably a residue other than serine and threonine. The residue at
position 1146 may
be any amino acid other than serine or, more preferably any amino acid other
than serine and
threonine. The residue at position 1146 may be alanine. Other exemplary acetyl-
coA
carboxylases include Accl mutants that comprise serine at a position
corresponding to position
639 of SEQ ID NO:2 and a residue other than serine and threonine at a position
corresponding
to position 1146 of SEQ ID NO:2.
Alpha-amylases include enzymes falling under EC number 3.2.1.1. Exemplary
alpha-
amylases that may be expressed include the secreted alpha-amylase designated
by PID_3772
(SEQ ID NO:4) encoded by the corresponding nucleic acid (SEQ ID NO:3) from
Lipomyces
starkeyi, the thermostable a-amylase designated by PID_272826 (SEQ ID NO:6)
encoded by
the corresponding nucleic acid (SEQ ID NO:5) from L. starkeyi, and a chimera
of the secreted
alpha-amylase and the thermostable a-amylase (SEQ ID NO:8) encoded by the
corresponding
nucleic acid (SEQ ID NO:7).
ATP citrate lyases include enzymes falling under EC number 2.3.3.8. An
exemplary
ATP citrate lyase that may be expressed includes the alpha subunit Acll (SEQ
ID NO:10)
encoded by Acll (SEQ ID NO:9) and the beta subunit Ac12 (SEQ ID NO:12) encoded
by Ac12
(SEQ ID NO:11) from L. starkeyi. The alpha and beta subunits are preferably
expressed as a
pair.
Diacylglycerol acyltransferases (DGAs) include enzymes falling under EC number
2.3.1.20. Exemplary diacylglycerol acyltransferases that may be expressed
include a 1233
variant of DGA1 (SEQ ID NO:14) encoded by DGA1-1233 (SEQ ID NO:13), a 1389
variant
of DGA1 (SEQ ID NO:16) encoded by DGA1-1389 (SEQ ID NO:15), and DGA2 (SEQ ID
NO:58) encoded by DGA2 (SEQ ID NO:57), all derived from L. starkeyi. The 1233
variant of
DGA1 is identical to the 1389 variant except that it lacks the first 52
residues of the 1389
variant. The 1233 variant unexpectedly confers enhanced lipogenic properties
compared to the
1389 variant. Accordingly, preferred diacylglycerol acyltransferases of the
invention lack a
sequence corresponding to positions 1-52 of SEQ ID NO:16.
27
CA 2982734 2017-10-17
Fatty acid synthases (FASs) include enzymes falling under EC number 2.3.1.85.
Exemplary fatty acid synthases that may be expressed include any combination
of alpha and
beta fatty acid synthase subunits from L. starkeyi. Alpha fatty acid synthase
subunits from L.
starkeyi include FAS2.1 (SEQ ID NO:18) encoded by FAS2.1 (SEQ ID NO:17) and
FAS2.2
(SEQ ID NO:22) encoded by FAS2.2 (SEQ ID NO:21). Beta fatty acid synthase
subunits from
L. starkeyi include FAS1.1 (SEQ ID NO:20) encoded by FAS1.1 (SEQ ID NO:19) and
FAS1.2
(SEQ ID NO:24) encoded by FAS1.2 (SEQ ID NO:23). FAS2.1 is preferably
expressed with
FAS1.1, and FAS2.2 is preferably expressed with FAS1.2. However, FAS2.1 may be
expressed
with FAS1.2, and FAS2.2 may be expressed with FAS1.1.
Glycerol kinases include enzymes falling under EC number 2.7.1.30. Exemplary
glycerol kinases that may be expressed include a 1602 variant of GUT1 (SEQ ID
NO:26)
encoded by GUT1-1602 (SEQ ID NO:25) and a 1617 variant of GUT1 (SEQ ID NO:28)
encoded by GUT1-1617 (SEQ ID NO:27), both derived from L. starkeyi. The 1602
variant of
DGA1 is identical to the 1617 variant except that it lacks the first 5
residues of the 1617 variant.
Accordingly, some glycerol kinases of the invention lack a sequence
corresponding to positions
1-5 of SEQ ID NO:28.
6-Phosphogluconate dehydrogenases include enzymes falling under EC number
1.1.1.44. An exemplary 6-phosphogluconate dehydrogenase that may be expressed
includes
GND1 (SEQ ID NO:30) encoded by GNDI (SEQ ID NO:29) from L. starkeyi.
Glycerol-3-phosphate dehydrogenases include enzymes falling under EC number
1.1.1.8. Exemplary glycerol-3-phosphate dehydrogenases that may be expressed
include GPD1
(SEQ ID NO:32) encoded by GPD1 (SEQ ID NO:31) and the FAD-dependent glycerol-3-
phosphate dehydrogenase GUT2 (SEQ ID NO:56) encoded by GUT2 (SEQ ID NO:55),
both
from L. starkeyi.
Malic enzymes include enzymes falling under EC numbers 1.1.1.38, 1.1.1.39,
1.1.1.40,
and 83. An exemplary malic enzyme that may be expressed includes ME (SEQ ID
NO:34)
encoded by ME (SEQ ID NO:33) from L. starkeyi.
Fatty acyl-CoA reductases include enzymes falling under EC numbers 1.2.1, such
as
1.2.1.84 and others. These enzymes include the preferred alcohol-forming fatty
acyl-CoA
reductases (EC 1.2.1.84). An exemplary fatty acyl-CoA reductase that may be
expressed
28
CA 2982734 2017-10-17
includes FALDR (SEQ ID NO:36) encoded by FALDR (SEQ ID NO:35) from
Marinobacter
aquaeolei.
Delta-9 acyl-CoA desaturases include enzymes falling under EC number
1.14.19.1. An
exemplary delta-9 acyl-CoA desaturase that may be expressed includes OLE1 (SEQ
ID NO:38)
encoded by OLE1 (SEQ ID NO:37) from L. starkeyi. In addition to or instead of
expressing a
delta-9 acyl-CoA desaturase, other fatty acid desaturases may be expressed,
such as acyl-CoA
(8-3)-desaturases (delta-5 desaturases) (EC 1.14.19.44) and acyl-CoA 6-
desaturases (delta-6
desaturases) (EC 1.14.19.3).
Glycerol-3-phosphate acyltransferases include enzymes falling under EC number
2.3.1.15. An exemplary glycerol-3-phosphate acyltransferase that may be
expressed includes
SCT1 (SEQ ID NO:40) encoded by SCT1 (SEQ ID NO:39) from L. starkeyi.
Lysophosphatidate acyltransferases include enzymes falling under EC number
2.3.1.51.
An exemplary lysophosphatidate acyltransferase that may be expressed includes
SLC1 (SEQ
ID NO:42) encoded by SLC1 (SEQ ID NO:41) from L. starkeyi.
Glucose-6-phosphate dehydrogenases include enzymes falling under EC number
1.1.1.49. An exemplary glucose-6-phosphate dehydrogenase that may be expressed
includes
ZWF1 (SEQ ID NO:44) encoded by ZWF1 (SEQ ID NO:43) from L. starkeyi.
Beta-glucosidases include enzymes falling under EC number 3.2.1.21. An
exemplary
beta-glucosidase that may be expressed includes BGL1. / (SEQ ID NO:46) encoded
by BGL1.1
(SEQ ID NO:45) from L. starkeyi.
Hexose transporters include proteins falling under the Hxt family (e.g., Hxtl
, Hxt2,
Hxt3, Hxt4, Hxt5, Hxt6, Hxt7, Hxt8, Hxt9, Hxtl 1, etc.). An exemplary hexose
transporter that
may be expressed includes Hxt2.2 (SEQ ID NO:48) encoded by Hxt2.2 (SEQ ID
NO:47) from
L. starkeyi.
Glycerol transporters that may be expressed include the glycerol/H+ symporters
STL1
(SEQ ID NO:64) encoded by STL1 (SEQ ID NO:63) and STL2 (SEQ ID NO:66) encoded
by
STL2 (SEQ ID NO:65) from L. starkeyi. Glycerol transporters that may be
expressed also
include the glycerol facilitator FPS1 (SEQ ID NO:68) encoded by FPS] (SEQ ID
NO:67) from
L. starkeyi.
29
CA 2982734 2017-10-17
Glycoside hydrolases include a number of families. Preferred glycoside
hydrolases that
may be expressed include glycoside hydrolase family 5 enzymes. Glycoside
hydrolase family
enzymes include endoglucanases (EC 3.2.1.4), beta-mannanases (EC 3.2.1.78),
exo-1,3-
glucanases (EC 3.2.1.58), endo-1,6-glucanases (EC 3.2.1.75), xylanases (EC
3.2.1.8),
5 endoglycoceramidases (EC 3.2.1.123), and trehalases (EC 3.2.1.28).
Preferred glucosidase
hydrolase family 5 enzymes that may be expressed include endoglucanases.
Exemplary
endoglucanases that may be expressed include EGC1 (SEQ ID NO:50) encoded by
EGG] (SEQ
ID NO:49) from L. starkeyi and EGC2 (SEQ ID NO:52) encoded by EGC2 (SEQ ID
NO:51)
from L. starkeyi. Exemplary trehalases that may be expressed include the
"neutral" trehalase
NTH1 (SEQ ID NO:60) encoded by NTH] (SEQ ID NO:59) and the "acidic" trehalase
ATH1
(SEQ ID NO:62) encoded by ATH1 (SEQ ID NO:61), both from L. starkeyi.
Auxiliary activity family 9 enzymes are copper-dependent lytic polysaccharide
monooxygenases (LPM0s). These enzymes are involved in the cleavage of
cellulose chains
with oxidation of various carbons (C-1, C-4 and C-6). Auxiliary activity
family 9 enzymes were
originally classified as glycoside hydrolases under glycoside hydrolase family
61 (GH61). An
exemplary auxiliary activity family 9 enzyme that may be expressed includes
AAC9 (SEQ ID
NO:54) encoded by AAC9 (SEQ ID NO:53) from L. starkeyi.
Other suitable proteins that may be expressed include those comprising
polypeptide
sequences at least about 80%, at least about 85%, at least about 90%, at least
about 95%, at
least about 97%, or at least about 99% identical to the sequences listed
above. Other suitable
proteins that may be expressed include orthologs and paralogs of the proteins
listed above.
Other suitable proteins that may be expressed include those comprising
polypeptide sequences
at least about 80%, at least about 85%, at least about 90%, at least about
95%, at least about
97%, or at least about 99% identical to orthologs and paralogs of the proteins
listed above. The
orthologs are preferably from lipogenic yeasts, such as any of the lipogenic
yeasts described
herein. The recombinant gene encoding the proteins may include introns or be
devoid of introns
or any or all other non-coding regions in the native gene. Any nucleotide
sequences capable of
expressing the polypeptide sequences encompassed herein are acceptable.
Tremendous
variation from the exemplary nucleotide sequences described herein is possible
due to the
redundancy in the genetic code and codon optimization.
CA 2982734 2017-10-17
Coding sequences of the above-mentioned proteins are preferably operably
linked to a
promoter. The promoter may be a constitutive promoter or an inducible
promoter. Exemplary
promoters that may be operably linked to the coding sequences of the above-
mentioned proteins
include the L. starkeyi ATPase 3900 promoter (SEQ ID NO:75), the L. starkeyi
citrate synthase
(CIT1) promoter (SEQ ID NO:76), the L. starkeyi fructose bisphosphate aldolase
(FBA1)
promoter (SEQ ID NO:77), the L. starkeyi glutamine synthetase (GLN1) promoter
(SEQ ID
NO:78), the L. starkeyi glyceraldehyde 3-phosphate dehydrogenase (TDH3)
promoter (SEQ ID
NO:79), the L. starkeyi pyruvate kinase (PYK1) promoter (SEQ ID NO:80), the L.
starkeyi
translation elongation factor (TEF1) promoter (SEQ ID NO :81), the L. starkeyi
triosephosphate
isomerase (TPI) promoter (SEQ ID NO:82), the L. starkeyi enolase (EN01)
promoter (SEQ ID
NO:83), the copper inducible (CUP]) promoter (SEQ ID NO:84), or sequence
variants at least
about at least about 80%, at least about 85%, at least about 90%, at least
about 95%, at least
about 97%, or at least about 99% identical thereto.
Coding sequences of the above-mentioned proteins are preferably operably
linked to a
terminator. Exemplary terminators that may be operably linked to the coding
sequences of the
above-mentioned proteins include the L. starkeyi ATPase 3900 terminator (SEQ
ID NO:85),
the L. starkeyi fructose bisphosphate aldolase (FBA]) terminator (SEQ ID
NO:86), the L.
starkeyi glutamine synthetase (GLN1) terminator (SEQ ID NO:87), the L.
starkeyi
glyceraldehyde 3-phosphate dehydrogenase (TDH3) terminator (SEQ ID NO:88), the
L.
starkeyi pyruvate kinase (PYK1) terminator (SEQ ID NO: 89), the L. starkeyi
triosephosphate
isomerase (TPI) terminator (SEQ ID NO:90), or sequence variants at least about
at least about
80%, at least about 85%, at least about 90%, at least about 95%, at least
about 97%, or at least
about 99% identical thereto.
In addition to expressing any one or more of the proteins listed above, the
recombinant
yeasts of the invention may be modified to reduce or ablate the activity of
one or more native
or non-native proteins. The recombinant yeasts, for example, may comprise a
modification that
reduces or ablates the activity of one or more of the following native
proteins: a delta-9 acyl-
CoA desaturase, a glycerol-3-phosphate dehydrogenase, an acyl-CoA oxidase, a 3-
hydroxyacyl-CoA dehydrogenase, and an enoyl-CoA hydratase.
31
CA 2982734 2017-10-17
Delta-9 acyl-CoA desaturases include enzymes falling under EC number
1.14.19.1. An
exemplary delta-9 acyl-CoA desaturase whose activity may be reduced or ablated
includes
OLE1 (SEQ ID NO:38) encoded by OLE1 (SEQ ID NO:37) from L. starkeyi. In
addition to or
instead of reducing or ablating the activity of a delta-9 acyl-CoA desaturase,
the activity of
other fatty acid desaturases may be reduced or ablated, such as acyl-CoA (8-3)-
desaturases
(delta-5 desaturases) (EC 1.14.19.44) and acyl-CoA 6-desaturases (delta-6
desaturases) (EC
1.14.19.3).
Glycerol-3-phosphate dehydrogenases include enzymes falling under EC number
1.1.1.8. An exemplary glycerol-3-phosphate dehydrogenase whose activity may be
reduced or
ablated includes the FAD-dependent glycerol-3-phosphate dehydrogenase GUT2
(SEQ ID
NO:56) encoded by GUT2 (SEQ ID NO:55) from L. starkeyi.
Acyl-CoA oxidases include enzymes falling under EC number 1.3.3.6. An
exemplary
acyl-CoA oxidase whose activity may be reduced or ablated includes PDX1 (SEQ
ID NO:70)
encoded by PDX] (SEQ ID NO:69) from L. starkeyi. Acyl-CoA oxidases catalyze
the first step
of beta oxidation.
Enoyl-CoA hydratases include enzymes falling under EC number 4.2.1.17. An
exemplary enoyl-CoA hydratase whose activity may be reduced or ablated
includes FOX1
(SEQ ID NO:72) encoded by FOX1 (SEQ ID NO:71) from L. starkeyi. Enoyl-CoA
hydratases
catalyze the third step of beta oxidation.
3-Hydroxyacyl-CoA dehydrogenases include enzymes falling under EC number
1.1.1.35. An exemplary 3-hydroxyacyl-CoA dehydrogenase whose activity may be
reduced or
ablated includes FOX1 (SEQ ID NO:72) encoded by FOX] (SEQ ID NO:71) from L.
starkeyi.
3-hydroxyacyl-CoA dehydrogenases catalyze the second step of beta oxidation.
Certain enzymes are multifunctional and have more than one enzymatic activity.
Examples include the FOX1 (SEQ ID NO:72) encoded by FOX1 (SEQ ID NO:71) from
L.
starkeyi, which has both 3-hydroxyacyl-CoA dehydrogenase and enoyl-CoA
hydratase
activities, and is thereby considered to be both a 3-hydroxyacyl-CoA
dehydrogenase and an
enoyl-CoA hydratase.
Other suitable proteins whose activity may be reduced or ablated include those
comprising amino acid sequences at least about 80%, at least about 85%, at
least about 90%, at
32
CA 2982734 2017-10-17
least about 95%, at least about 97%, or at least about 99% identical to the
sequences listed
above. Other suitable proteins whose activity may be reduced or ablated
include orthologs and
paralogs of the proteins listed above. Other suitable proteins whose activity
may be reduced or
ablated include those comprising amino acid sequences at least about 80%, at
least about 85%,
at least about 90%, at least about 95%, at least about 97%, or at least about
99% identical to
orthologs and paralogs of the proteins listed above.
A modification that reduces or ablates the activity of a gene product such as
a protein is
referred to herein as a "functional deletion." "Functional deletion" or its
grammatical
equivalents refers to any modification to a microorganism that ablates,
reduces, inhibits, or
otherwise disrupts production of a gene product, renders a produced gene
product non-
functional, or otherwise reduces or ablates a produced gene product's
activity. Accordingly, in
some instances, a gene product that is functionally deleted means that the
gene product is not
produced by the microorganism at all. "Gene product" refers to a protein or
polypeptide
encoded and produced by a particular gene. "Gene" refers to a nucleic acid
sequence capable
of producing a gene product and may include such genetic elements as a coding
sequence
together with any other genetic elements required for transcription and/or
translation of the
coding sequence. Such genetic elements may include a promoter, an enhancer,
and/or a
ribosome binding site (RBS), among others.
One of ordinary skill in the art will appreciate that there are many well-
known ways to
functionally delete a gene product. For example, functional deletion can be
accomplished by
introducing one or more genetic modifications. As used herein, "genetic
modifications" refer
to any differences in the nucleic acid composition of a cell, whether in the
cell's native
chromosome or in endogenous or exogenous non-chromosomal plasmids harbored
within the
cell. Examples of genetic modifications that may result in a functionally
deleted gene product
include but are not limited to substitutions, partial or complete deletions,
insertions, or other
variations to a coding sequence or a sequence controlling the transcription or
translation of a
coding sequence, such as placing a coding sequence under the control of a less
active promoter,
etc. In some versions, a gene or coding sequence can be replaced with a
selection marker or
screenable marker. Various methods for introducing genetic modifications are
well known in
the art and include homologous recombination, among other mechanisms. See,
e.g., Green et
33
CA 2982734 2017-10-17
al., Molecular Cloning: A laboratory manual, 4th ed., Cold Spring Harbor
Laboratory Press
(2012) and Sambrook et al., Molecular Cloning: A Laboratory Manual, 3rd ed.,
Cold Spring
Harbor Laboratory Press (2001). In some versions, functional deletion can be
accomplished by
expressing ribozymes or antisense sequences that target the mRNA of the gene
of interest.
Functional deletion can also be accomplished by inhibiting the activity of the
gene product, for
example, by chemically inhibiting a gene product with a small-molecule
inhibitor, by
expressing a protein that interferes with the activity of the gene product, or
by other means. In
some versions, the functional deletion may comprise an activity-reducing or
activity-ablating
mutation in the endogenous gene. The activity-reducing or activity-ablating
mutation in the
endogenous gene may comprise a nucleotide substitution in the endogenous gene,
a nucleotide
insertion in the endogenous gene, a partial deletion of the endogenous gene,
and/or a complete
deletion of the endogenous gene.
In certain versions of the invention, the functionally deleted gene product
may have less
than about 95%, less than about 90%, less than about 85%, less than about 80%,
less than about
75%, less than about 70%, less than about 65%, less than about 60%, less than
about 55%, less
than about 50%, less than about 45%, less than about 40%, less than about 35%,
less than about
30%, less than about 25%, less than about 20%, less than about 15%, less than
about 10%, less
than about 5%, less than about 1%, or about 0% of the activity of the non-
functionally deleted
gene product.
In certain versions of the invention, a cell with a functionally deleted gene
product may
have less than about 95%, less than about 90%, less than about 85%, less than
about 80%, less
than about 75%, less than about 70%, less than about 65%, less than about 60%,
less than about
55%, less than about 50%, less than about 45%, less than about 40%, less than
about 35%, less
than about 30%, less than about 25%, less than about 20%, less than about 15%,
less than about
10%, less than about 5%, less than about 1%, or about 0% of the activity of
the gene product
compared to a cell with the non-functionally deleted gene product.
In certain versions of the invention, the functionally deleted gene product
may be
expressed at an amount less than about 95%, less than about 90%, less than
about 85%, less
than about 80%, less than about 75%, less than about 70%, less than about 65%,
less than about
60%, less than about 55%, less than about 50%, less than about 45%, less than
about 40%, less
34
CA 2982734 2017-10-17
than about 35%, less than about 30%, less than about 25%, less than about 20%,
less than about
15%, less than about 10%, less than about 5%, less than about 1%, or about 0%
of the amount
of the non-functionally deleted gene product.
In certain versions of the invention, the functionally deleted gene product
may result
from a genetic modification in which at least 1, at least 2, at least 3, at
least 4, at least 5, at least
10, at least 20, at least 30, at least 40, at least 50, or more nonsynonymous
substitutions are
present in the gene or coding sequence of the gene product.
In certain versions of the invention, the functionally deleted gene product
may result
from a genetic modification in which at least 1, at least 2, at least 3, at
least 4, at least 5, at least
10, at least 20, at least 30, at least 40, at least 50, or more bases are
inserted in the gene or
coding sequence of the gene product.
In certain versions of the invention, the functionally deleted gene product
may result
from a genetic modification in which at least about 1%, at least about 5%, at
least about 10%,
at least about 15%, at least about 20%, at least about 25%, at least about
30%, at least about
35%, at least about 40%, at least about 50%, at least about 55%, at least
about 60%, at least
about 65%, at least about 70%, at least about 75%, at least about 80%, at
least about 85%, at
least about 90%, at least about 95%, or about 100% of the gene product's gene
or coding
sequence is deleted or mutated.
In certain versions of the invention, the functionally deleted gene product
may result
from a genetic modification in which at least about 1%, at least about 5%, at
least about 10%,
at least about 15%, at least about 20%, at least about 25%, at least about
30%, at least about
35%, at least about 40%, at least about 50%, at least about 55%, at least
about 60%, at least
about 65%, at least about 70%, at least about 75%, at least about 80%, at
least about 85%, at
least about 90%, at least about 95%, or about 100% of a promoter driving
expression of the
gene product is deleted or mutated.
In certain versions of the invention, the functionally deleted gene product
may result
from a genetic modification in which at least about 1%, at least about 5%, at
least about 10%,
at least about 15%, at least about 20%, at least about 25%, at least about
30%, at least about
35%, at least about 40%, at least about 50%, at least about 55%, at least
about 60%, at least
about 65%, at least about 70%, at least about 75%, at least about 80%, at
least about 85%, at
CA 2982734 2017-10-17
least about 90%, at least about 95%, or about 100% of an enhancer controlling
transcription of
the gene product's gene is deleted or mutated.
In certain versions of the invention, the functionally deleted gene product
may result
from a genetic modification in which at least about 1%, at least about 5%, at
least about 10%,
at least about 15%, at least about 20%, at least about 25%, at least about
30%, at least about
35%, at least about 40%, at least about 50%, at least about 55%, at least
about 60%, at least
about 65%, at least about 70%, at least about 75%, at least about 80%, at
least about 85%, at
least about 90%, at least about 95%, or about 100% of a sequence controlling
translation of the
gene product's mRNA is deleted or mutated.
In certain versions of the invention, the decreased activity or expression of
the
functionally deleted gene product is determined with respect to the activity
or expression of the
gene product in its unaltered state as found in nature. In certain versions of
the invention, the
decreased activity or expression of the functionally deleted gene product is
determined with
respect to the activity or expression of the gene product in its form in a
corresponding
microorganism. In certain versions, the genetic modifications giving rise to a
functionally
deleted gene product are determined with respect to the gene in its unaltered
state as found in
nature. In certain versions, the genetic modifications giving rise to a
functionally deleted gene
product are determined with respect to the gene in its form in a corresponding
microorganism.
As used herein, "corresponding microorganism" refers to a microorganism of the
same species
having the same or substantially same genetic and proteomic composition as a
microorganism
of the invention, with the exception of genetic and proteomic differences
resulting from the
manipulations described herein for the microorganisms of the invention.
The yeasts of the invention with the modifications described herein preferably
exhibit a
property selected from the group consisting of increased lipid production,
increased lipid
secretion, increased lipid production under nitrogen-rich conditions,
increased lipid yield,
increased lipid secretion under nitrogen-rich conditions, increased enzyme
production,
increased enzyme secretion, increased carbohydrase production, increased
carbohydrase
secretion, increased growth rates, and/or increased organic consumption, such
as increased
glycerol consumption and/or and increased disaccharide (cellobiose and/or
trehalose)
36
CA 2982734 2017-10-17
consumption relative to a non-recombinant control. "Carbohydrase" refers to
any enzyme
capable of breaking down a carbohydrate, such as amylases, cellulases,
glucosidases, etc.
Nitrogen-rich conditions exist when the form, the amount, or form and amount
of
nitrogen biologically available to the cell exceeds the amount of carbon
source necessary for
balanced cell growth. The form of nitrogen refers to the chemical form in
which it is supplied
to the cells. An easily assimilated nitrogen source includes amino acids or
extracts of yeast
cells. These nitrogen sources require less metabolic energy for assimilation
and provide carbon
at the same time. A less readily assimilated nitrogen source includes
inorganic salts of
ammonium or nitrate or nitrogen supplied as urea. Elemental nitrogen or
nitrogen bound in
insoluble minerals are not generally considered biologically available to
fungi. Nitrogen-rich
conditions can exist when either readily assimilated or less-readily
assimilated nitrogen sources
are provided to a cell in excess of the amount of carbon required for protein,
nucleic acid and
cell wall synthesis.
Nitrogen-poor conditions can exist when the amount of readily available or
less readily
available nitrogen supplied is substantially less than the amount of available
carbon source that
can be assimilated. A nitrogen-poor or nitrogen-limiting condition could also
exist when an
easily assimilated nitrogen source is supplied slowly or in a slow-release
formulation.
As in the case of the nitrogen supply, the carbon source can be readily
assimilated, less
readily assimilated, poorly assimilated or not assimilated. The cell can use
readily assimilated
carbon source such as glucose to rapidly generate metabolic energy. A carbon
source such as
glycerol, cellobiose or other oligosaccharides might or might not be readily
assimilated
depending on the enzymes available for its metabolism and the conditions of
growth such as
the supply of oxygen.
A nitrogen-rich condition in a native organism can be identified as a
concentration
above a nitrogen-limited concentration, wherein the nitrogen-limited
concentration includes all
concentrations at or below the concentration of nitrogen in which any
decreases thereof increase
the amount of lipid produced and/or accumulated by the organism.
Acetyl-CoA carboxylase and diacylglycerol acyltransferase are both preferred
targets
for increasing lipid production. Overexpression of acetyl-CoA carboxylase
results in increased
rates of fatty acid biosynthesis and fatty acid content. The Acc 1 acetyl-CoA
carboxylase of L.
37
CA 2982734 2017-10-17
starkeyi has serine residues at positions corresponding to 639 and 1146 of SEQ
ID NO2. The
inventors have determined that the serine at position 1146 is responsible for
downregulating
ACC1 activity upon post-translational modification. Certain versions of this
invention modify
the serine at position 1146 to an amino acid other than serine or threonine,
such as alanine or
any other amino acid, to prevent this downregulation.
Overexpression of diacylglycerol acyltransferase leads to an increase in lipid
production. This lipid production is increased even further following
overexpression of acetyl-
CoA carboxylase. The present inventors surmise that a sink for triglyceride
synthesis, such as
through high expression of diacylglycerol acyltransferase, should be present
in order for
overexpression of acetyl-CoA carboxylase to be fully effective.
One of the prerequisites of oleaginous organisms is the ability to produce a
continuous
supply of acetyl-CoA precursors in the cytosol. ATP citrate lyase acts as the
primary source of
cytosolic acetyl-CoA in these species, and is believed to be one of the rate
limiting steps of lipid
biosynthesis. The activity of ATP citrate lyase correlates well with the
specific rate of lipid
biosynthesis in L. starkeyi. In Lipomyees both intracellular and extracellular
citrate levels rise
in response to nitrogen limitation (Holdsworth et al. 1988). Citrate
accumulation may represent
a bottleneck, which could be overcome by increasing ATP citrate lyase activity
in the cytosol
or citrate synthase (CS) in the mitochondria.
NADPH, which is required for lipid biosynthesis, is largely supplied by the
oxidative
pentose phosphate pathway (PPP). Native lipogenic yeasts possess a highly
active oxidative
pentose phosphate pathway along with an enzyme system that exports citric acid
to the cytosol
where it is converted into acetyl-CoA and NADPH that feed into lipid
synthesis. Modification
of lipogenic enzymes or alteration of gene expression that boosts these
systems can further
increase lipid synthesis in native lipogenic yeasts. Overexpression of 6-
phosphogluconate
dehydrogenase (GND1) and/or glucose-6-phosphate dehydrogenase (ZWF1), for
example, are
predicted to increase the supply of NADPH for lipid synthesis, but since ZWF1
and
(particularly) GND1 are subject to strong allosteric regulation by
physiological levels of
NADPH (Barcia-Vieitez etal. 2014, Rippa et al. 1998, Velasco et al. 1995),
modification of the
regulatory controls are predicted to be more effective. For example,
substitution of the tyrosine
at position 99 (i.e., Y99) of the ZWF1 of L. starkeyi represented by SEQ ID
NO:44 can alter
38
CA 2982734 2017-10-17
binding of NADPH and thereby render this enzyme resistant to allosteric
regulation by
NADPH. The tyrosine in ZWF1 can be substituted by any amino acid. The tyrosine
in ZWF1
is preferably substituted with serine, threonine, glutamine asparagine,
cysteine, alanine, proline,
leucine, isoleucine, phenylalanine, valine, histidine, lysine, asparagine,
aspartic, glutamic acid,
or glycine; more preferably substituted with serine or threonine; and most
preferably substituted
with serine. An analogous substitution in GND1 can also or alternatively be
performed to render
GND1 resistant to allosteric regulation by NADPH.
The NADPH supplied by malic enzyme has a strong influence on lipid
accumulation in
lipogenic yeasts. Increasing malic enzyme activity is predicted to provide
lipogenic yeasts with
more NADPH for lipid synthesis.
Another approach to increasing the production of lipid is to reduce activity
of the 0-
oxidation pathway. The f3-oxidation pathway is responsible for consuming lipid
after lipogenic
yeasts exhaust their carbohydrate sources. Eliminating the breakdown of lipids
is therefore
another target for developing yeasts with enhanced lipid production. Exemplary
modifications
in this regard include functional deletion of an acyl-CoA oxidase, a 3-
hydroxyacyl-CoA
dehydrogenase, and/or an enoyl-CoA hydratase. Acyl-CoA oxidases catalyze the
first step in
beta oxidation. Enoyl-CoA hydratases catalyze the second step in beta
oxidation. 3-
Hydroxyacyl-CoA dehydrogenases catalyze the third step in beta oxidation. PDX1
is an
exemplary acyl-CoA oxidase in L. starkeyi. FOX1 is a multifunctional enzyme in
L. starkeyi
that can be considered to be both a 3-hydroxyacyl-CoA dehydrogenase and an
enoyl-CoA
hydratase, as it has both 3-hydroxyacyl-CoA dehydrogenase and enoyl-CoA
hydratase
activities. Other modifications that inhibit lipid oxidation are acceptable.
Disruption of the gene for the regulatory protein CreA/Migl in Y upolytica
increased
the lipid content from 36% to 48.7% of its dry weight while increasing the
C181 content (Wang
et al. 2013). A CreA homolog in the L. starkeyi genome is similar to the CreA
transcriptional
activator of Y. hpo/ytica, and could be a good target to determine whether
disruption of the
CREA/MIG1 homolog in L. starkeyi results in an increase in lipid production.
For Mucor circinelloides (Zhang et al. 2008), Rhodotorula glutinis (Li et al.
2012), and
Yarrowia lipolytica (Tai et al. 2013), the basal lipid production contents of
the parental strains
are all considerably lower than what has been observed in native (non-
engineered) L. starkeyi
39
CA 2982734 2017-10-17
under nitrogen limiting conditions (z, 65%), indicating that there is
substantial room for
improvement in these strains with the modifications described herein.
Genetic Engineering
The cells of the invention may be genetically altered to functionally delete,
express, or
overexpress any of the specific genes or gene products explicitly described
herein or homologs
thereof. Proteins and/or protein sequences are "homologous" when they are
derived, naturally
or artificially, from a common ancestral protein or protein sequence.
Similarly, nucleic acids
and/or nucleic acid sequences are homologous when they are derived, naturally
or artificially,
from a common ancestral nucleic acid or nucleic acid sequence. Nucleic acid or
gene product
(amino acid) sequences of any known gene, including the genes or gene products
described
herein, can be determined by searching any sequence databases known in the art
using the gene
name or accession number as a search term. Common sequence databases include
GenBank
(www.ncbi.nlm.nih.gov), ExPASy (expasy.org), KEGG (www.genomejp), among
others.
Homology is generally inferred from sequence similarity between two or more
nucleic acids or
proteins (or sequences thereof). The precise percentage of similarity between
sequences that is
useful in establishing homology varies with the nucleic acid and protein at
issue, but as little as
25% sequence similarity (e.g., identity) over 50, 100, 150 or more residues
(nucleotides or
amino acids) is routinely used to establish homology (e.g., over the full
length of the two
sequences to be compared). Higher levels of sequence similarity (e.g.,
identity), e.g., 30%, 35%
40%, 45% 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, or 99% or more, can
also
be used to establish homology. Accordingly, homologs of the genes or gene
products described
herein include genes or gene products having at least about 30%, 35%, 40%,
45%, 50%, 55%,
60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, or 99% identity to the genes or gene
products
described herein. Methods for determining sequence similarity percentages
(e.g., BLASTP and
BLASTN using default parameters) are described herein and are generally
available. The
homologous proteins should demonstrate comparable activities and, if an
enzyme, participate
in the same or analogous pathways. Homologs include orthologs and paralogs.
"Orthologs" are
genes and products thereof in different species that evolved from a common
ancestral gene by
speciation. Normally, orthologs retain the same or similar function in the
course of evolution.
CA 2982734 2017-10-17
Paralogs are genes and products thereof related by duplication within a
genome. As used herein,
"orthologs" and "paralogs" are included in the term "homologs."
For sequence comparison and homology determination, one sequence typically
acts as
a reference sequence to which test sequences are compared. When using a
sequence comparison
algorithm, test and reference sequences are input into a computer, subsequence
coordinates are
designated, if necessary, and sequence algorithm program parameters are
designated. The
sequence comparison algorithm then calculates the percent sequence identity
for the test
sequence(s) relative to the reference sequence based on the designated program
parameters. A
typical reference sequence of the invention is a nucleic acid or amino acid
sequence
corresponding to the genes or gene products described herein.
Optimal alignment of sequences for comparison can be conducted, e.g., by the
local
homology algorithm of Smith & Waterman, Adv. Appl. Math. 2:482 (1981), by the
homology
alignment algorithm of Needleman & Wunsch, J. Mol. Biol. 48:443 (1970), by the
search for
similarity method of Pearson & Lipman, Proc. Nat'l. Acad. Sci. USA 85:2444
(1988), by
computerized implementations of these algorithms (GAP, BESTFIT, FASTA, and
TFASTA in
the Wisconsin Genetics Software Package, Genetics Computer Group, 575 Science
Dr.,
Madison, Wis.), or by visual inspection (see Current Protocols in Molecular
Biology, F. M.
Ausubel et al., eds., Current Protocols, a joint venture between Greene
Publishing Associates,
Inc. and John Wiley & Sons, Inc., (supplemented through 2008)).
One example of an algorithm that is suitable for determining percent sequence
identity
and sequence similarity for purposes of defining homologs is the BLAST
algorithm, which is
described in Altschul et al., I Mol. Biol. 215:403-410 (1990). Software for
performing BLAST
analyses is publicly available through the National Center for Biotechnology
Information. This
algorithm involves first identifying high scoring sequence pairs (HSPs) by
identifying short
words of length W in the query sequence, which either match or satisfy some
positive-valued
threshold score T when aligned with a word of the same length in a database
sequence. T is
referred to as the neighborhood word score threshold (Altschul et al., supra).
These initial
neighborhood word hits act as seeds for initiating searches to find longer
HSPs containing them.
The word hits are then extended in both directions along each sequence for as
far as the
cumulative alignment score can be increased. Cumulative scores are calculated
using, for
41
CA 2982734 2017-10-17
nucleotide sequences, the parameters M (reward score for a pair of matching
residues;
always>0) and N (penalty score for mismatching residues; always<0). For amino
acid
sequences, a scoring matrix is used to calculate the cumulative score.
Extension of the word
hits in each direction are halted when: the cumulative alignment score falls
off by the quantity
X from its maximum achieved value; the cumulative score goes to zero or below,
due to the
accumulation of one or more negative-scoring residue alignments; or the end of
either sequence
is reached. The BLAST algorithm parameters W, T, and X determine the
sensitivity and speed
of the alignment. The BLASTN program (for nucleotide sequences) uses as
defaults a
wordlength (W) of 11, an expectation (E) of 10, a cutoff of 100, M=5, N=-4,
and a comparison
of both strands. For amino acid sequences, the BLASTP program uses as defaults
a wordlength
(W) of 3, an expectation (E) of 10, and the BLOSUM62 scoring matrix (see
Henikoff &
Henikoff (1989) Proc. Natl. Acad. Sci. USA 89:10915).
In addition to calculating percent sequence identity, the BLAST algorithm also
performs
a statistical analysis of the similarity between two sequences (see, e.g.,
Karlin & Altschul, Proc.
Natl. Acad. Sci. USA 90:5873-5787 (1993)). One measure of similarity provided
by the BLAST
algorithm is the smallest sum probability (P(N)), which provides an indication
of the probability
by which a match between two nucleotide or amino acid sequences would occur by
chance. For
example, a nucleic acid is considered similar to a reference sequence if the
smallest sum
probability in a comparison of the test nucleic acid to the reference nucleic
acid is less than
about 0.1, more preferably less than about 0.01, and most preferably less than
about 0.001. The
above-described techniques are useful in identifying homologous sequences for
use in the
methods described herein.
The terms "identical" or "percent identity", in the context of two or more
nucleic acid
or polypeptide sequences, refer to two or more sequences or subsequences that
are the same or
have a specified percentage of amino acid residues or nucleotides that are the
same, when
compared and aligned for maximum correspondence, as measured using one of the
sequence
comparison algorithms described above (or other algorithms available to
persons of skill) or by
visual inspection.
The phrase "substantially identical" in the context of two nucleic acids or
polypeptides
refers to two or more sequences or subsequences that have at least about 60%,
about 65%, about
42
CA 2982734 2017-10-17
70%, about 75%, about 80%, about 85%, about 90, about 95%, about 98%, or about
99% or
more nucleotide or amino acid residue identity, when compared and aligned for
maximum
correspondence, as measured using a sequence comparison algorithm or by visual
inspection.
Such "substantially identical" sequences are typically considered to be
"homologous", without
reference to actual ancestry. Preferably, the "substantial identity" exists
over a region of the
sequences that is at least about 50 residues in length, more preferably over a
region of at least
about 100 residues, and most preferably, the sequences are substantially
identical over at least
about 150 residues, at least about 250 residues, or over the full length of
the two sequences to
be compared.
Terms used herein pertaining to genetic manipulation are defined as follows.
Deletion: The removal of one or more nucleotides from a nucleic acid molecule
or one
or more amino acids from a protein, the regions on either side being joined
together.
Derived: When used with reference to a nucleic acid or protein, "derived"
means that
the nucleic acid or polypeptide is isolated from a described source or is at
least 70%, 80%, 90%,
95%, 99%, or more identical to a nucleic acid or polypeptide included in the
described source.
Endogenous: As used herein with reference to a nucleic acid molecule, genetic
element
(e.g., gene, promoter, etc.), or polypeptide in a particular cell,
"endogenous" refers to a nucleic
acid molecule, genetic element, or polypeptide that is in the cell and was not
introduced into
the cell or transferred within the genome of the cell using recombinant
engineering techniques.
For example, an endogenous genetic element is a genetic element that was
present in a cell in
its particular locus in the genome when the cell was originally isolated from
nature. The term
"native' is used herein interchangeably with "endogenous."
Exogenous: As used herein with reference to a nucleic acid molecule, genetic
element
(e.g., gene, promoter, etc.), or polypeptide in a particular cell, "exogenous"
refers to any nucleic
acid molecule, genetic element, or polypeptide that was introduced into the
cell or transferred
within the genome of the cell using recombinant engineering techniques. For
example, an
exogenous genetic element is a genetic element that was not present in its
particular locus in
the genome when the cell was originally isolated from nature. The term
"heterologous" is used
herein interchangeably with "exogenous."
43
CA 2982734 2017-10-17
Expression: The process by which a gene's coded information is converted into
the
structures and functions of a cell, such as a protein, transfer RNA, or
ribosomal RNA. Expressed
genes include those that are transcribed into mRNA and then translated into
protein and those
that are transcribed into RNA but not translated into protein (for example,
transfer and
ribosomal RNAs).
Introduce: When used with reference to genetic material, such as a nucleic
acid, and a
cell, "introduce" refers to the delivery of the genetic material to the cell
in a manner such that
the genetic material is capable of being expressed within the cell.
Introduction of genetic
material includes both transformation and transfection. Transformation
encompasses
techniques by which a nucleic acid molecule can be introduced into cells such
as prokaryotic
cells or non-animal eukaryotic cells. Transfection encompasses techniques by
which a nucleic
acid molecule can be introduced into cells such as animal cells. These
techniques include but
are not limited to introduction of a nucleic acid via conjugation,
electroporation, lipofection,
infection, and particle gun acceleration.
Isolated: An "isolated" biological component (such as a nucleic acid molecule,
polypeptide, or cell) has been substantially separated or purified away from
other biological
components in which the component naturally occurs, such as other chromosomal
and
extrachromosomal DNA and RNA and proteins. Nucleic acid molecules and
polypeptides that
have been "isolated" include nucleic acid molecules and polypeptides purified
by standard
purification methods. The term also includes nucleic acid molecules and
polypeptides prepared
by recombinant expression in a cell as well as chemically synthesized nucleic
acid molecules
and polypeptides. In one example, "isolated" refers to a naturally-occurring
nucleic acid
molecule that is not immediately contiguous with both of the sequences with
which it is
immediately contiguous (one on the 5' end and one on the 3' end) in the
naturally-occurring
genome of the organism from which it is derived.
Nucleic acid: Encompasses both RNA and DNA molecules including, without
limitation, cDNA, genomic DNA, and mRNA. Nucleic acids also include synthetic
nucleic acid
molecules, such as those that are chemically synthesized or recombinantly
produced. The
nucleic acid can be double-stranded or single-stranded. Where single-stranded,
the nucleic acid
44
CA 2982734 2017-10-17
molecule can be the sense strand, the antisense strand, or both. In addition,
the nucleic acid can
be circular or linear.
Operably linked: A first nucleic acid sequence is operably linked with a
second nucleic
acid sequence when the first nucleic acid sequence is placed in a functional
relationship with
the second nucleic acid sequence. For instance, a promoter is operably linked
to a coding
sequence if the promoter affects the transcription or expression of the coding
sequence. An
origin of replication is operably linked to a coding sequence if the origin of
replication controls
the replication or copy number of the nucleic acid in the cell. Operably
linked nucleic acids
may or may not be contiguous.
Operon: Configurations of separate genes that are transcribed in tandem as a
single
messenger RNA are denoted as operons. Thus, a set of in-frame genes in close
proximity under
the transcriptional regulation of a single promoter constitutes an operon.
Operons may be
synthetically generated using the methods described herein.
Overexpress: When a gene is caused to be transcribed at an elevated rate
compared to
the endogenous or basal transcription rate for that gene. In some examples,
overexpression
additionally includes an elevated rate of translation of the gene compared to
the endogenous
translation rate for that gene. Methods of testing for overexpression are well
known in the art,
for example transcribed RNA levels can be assessed using RT-PCR and protein
levels can be
assessed using SDS-PAGE gel analysis.
Recombinant: A recombinant nucleic acid molecule, genetic element (e.g., gene,
promoter, etc.), or polypeptide is one that has a sequence that is not
naturally occurring, is
present in a different locus (e.g., genetic locus or on an extrachromosomal
plasmid) within a
particular cell than in a corresponding native cell, or both. A recombinant
cell or microorganism
is one that contains a recombinant nucleic acid molecule, genetic element, or
polypeptide.
Vector or expression vector: An entity comprising a nucleic acid molecule that
is
capable of introducing the nucleic acid, or being introduced with the nucleic
acid, into a cell for
expression of the nucleic acid. A vector can include nucleic acid sequences
that permit it to
replicate in the cell, such as an origin of replication. A vector can also
include one or more
selectable marker genes and other genetic elements known in the art. Examples
of suitable
vectors are found below.
CA 2982734 2017-10-17
Unless explained otherwise, all technical and scientific terms used herein
have the same
meaning as commonly understood to one of ordinary skill in the art to which
this disclosure
belongs. Although methods and materials similar or equivalent to those
described herein can be
used in the practice or testing of the present disclosure, suitable methods
and materials are
described below.
Exogenous nucleic acids can be introduced stably or transiently into a cell
using
techniques well known in the art, including electroporation, lithium acetate
transformation,
calcium phosphate precipitation, DEAE-dextran mediated transfection, liposome-
mediated
transfection, conjugation, transduction, and the like. For stable
transformation, a nucleic acid
can further include a selectable marker. Suitable selectable markers include
antibiotic resistance
genes that confer, for example, resistance to nourseothricin, G418, hygromycin
B, neomycin,
tetracycline, chloramphenicol, or kanamycin, genes that complement auxotrophic
deficiencies,
and the like. (See below for more detail.)
Various embodiments of the invention use an expression vector that includes a
recombinant nucleic acid encoding a protein involved in a metabolic or
biosynthetic pathway.
Suitable expression vectors include, but are not limited to viral vectors,
phage vectors,
bacteriophage vectors, plasmids, phagemids, cosmids, fosmids, bacterial
artificial
chromosomes, P1-based artificial chromosomes, yeast plasmids, yeast artificial
chromosomes,
and any other vectors specific for cells of interest.
Useful vectors can include one or more selectable marker genes to provide a
phenotypic
trait for selection of transformed cells. The selectable marker gene encodes a
protein necessary
for the survival or growth of transformed cells grown in a selective culture
medium. Cells not
transformed with the vector containing the selectable marker gene will not
survive in the culture
medium. Typical selection genes encode proteins that (a) confer resistance to
antibiotics or
other toxins, e.g., nourseothricin, G418, hygromycin B, ampicillin, neomycin,
methotrexate, or
tetracycline, (b) complement auxotrophic deficiencies, or (c) supply critical
nutrients not
available from complex media. In alternative embodiments, the selectable
marker gene is one
that encodes orotidine 5' -phosphate decarboxylase, dihydrofolate reductase or
confers
neomycin resistance (for use in eukaryotic cell culture).
46
CA 2982734 2017-10-17
The coding sequence in the expression vector is operably linked to an
appropriate
expression control sequence (promoters, enhancers, and the like) to direct
synthesis of the
encoded gene product. Such promoters can be derived from endogenous or
exogenous sources.
Thus, the recombinant genes of the invention can comprise a coding sequence
operably linked
to a heterologous genetic element, such as a promoter, enhancer, ribosome
binding site, etc.
"Heterologous" in this context refers to a genetic element that is not
operably linked to the
coding sequence in nature. Depending on the cell/vector system utilized, any
of a number of
suitable transcription and translation control elements, including
constitutive and inducible
promoters, transcription enhancer elements, transcription terminators, etc.
can be used in the
expression vector (see e.g., Bitter et al. (1987) Methods in Enzymology,
153:516- 544).
Non-limiting examples of suitable promoters for use within a eukaryotic cell
are
typically viral in origin and include the promoter of the mouse
metallothionein I gene (Hamer
et al. (1982) 1 MoI AppL Gen. 1:273); the TK promoter of Herpes virus
(McKnight (1982)
Cell 31:355); the SV40 early promoter (Benoist et al. (1981) Nature (London)
290:304); the
Rous sarcoma virus promoter; the cytomegalovirus promoter (Foecking et al.
(1980) Gene
45:101); and the yeast gal4 gene promoter (Johnston et al. (1982) PNAS (USA)
79:6971; Silver
et al. (1984) PNAS (USA) 81:5951.
Coding sequences can be operably linked to an inducible promoter. Inducible
promoters
are those wherein addition of an effector induces expression. Suitable
effectors include proteins,
metabolites, chemicals, or culture conditions capable of inducing expression.
Alternatively, a coding sequence can be operably linked to a repressible
promoter.
Repressible promoters are those wherein addition of an effector represses
expression.
In some versions, the cell is genetically modified with a recombinant nucleic
acid
encoding a biosynthetic pathway gene product that is operably linked to a
constitutive promoter.
Suitable constitutive promoters are known in the art.
In some versions, the cell is genetically modified with an exogenous nucleic
acid
encoding a single protein. In other embodiments, a modified cell is one that
is genetically
modified with exogenous nucleic acids encoding two or more proteins. Where the
cell is
genetically modified to express two or more proteins, those nucleic acids can
each be contained
in a single or in separate expression vectors. When the nucleic acids are
contained in a single
47
CA 2982734 2017-10-17
expression vector, the nucleotide sequences may be operably linked to a common
control
element (e.g., a promoter), that is, the common control element controls
expression of all of the
coding sequences in the single expression vector.
When the cell is genetically modified with recombinant nucleic acids encoding
two or
more proteins, one of the nucleic acids can be operably linked to an inducible
promoter, and
one or more of the nucleic acids can be operably linked to a constitutive
promoter. Alternatively,
all can be operably linked to inducible promoters or all can be operably
linked to constitutive
promoters.
Nucleic acids encoding proteins desired to be expressed in a cell may be codon-
optimized for that particular type of cell. Codon optimization can be
performed for any nucleic
acid by "OPTIMUMGENE"-brand gene design system by GenScript (Piscataway, NJ).
Methods for transforming yeast cells with recombinant DNA and producing
polypeptides therefrom are disclosed by Clontech Laboratories, Inc., Palo
Alto, Calif., USA (in
the product protocol for the "YEASTMAKER"-brand yeast transformation system
kit); Reeves
et al. (1992) FEMS Microbiology Letters 99:193-198; Manivasakam and Schiestl
(1993)
Nucleic Acids Research 21(18):4414-5; and Ganeva et al. (1994) FEMS
Microbiology Letters
121:159-64. Expression and transformation vectors for transformation into many
yeast strains
are available. For example, expression vectors have been developed for the
following yeasts:
Candida albicans (Kurtz, et al. (1986) Mol. Cell. Biol. 6:142); Candida
maltosa (Kunze et al.
(1985) 1 Basic Microbiol. 25:141); Hansenula polymorpha (Gleeson et al. (1986)
1 Gen.
Microbiol. 132:3459) and Roggenkamp et al. (1986) Mol. Gen. Genet. 202:302);
Kluyveromyces fragilis (Das et al. (1984) 1 BacterioL 158:1165); Kluyveromyces
lactis (De
Louvencourt et al. (1983)1 BacterioL 154:737) and Van den Berg et al. (1990)
Bio/Technology
8:135); Pichia quillerimondii (Kunze et al. (1985) 1 Basic Microbiol. 25:141);
Pichia pastoris
(Cregg et al. (1985) Mol. Cell. Biol. 5:3376; U.S. Pat. No. 4,837,148; and
U.S. Pat. No.
4,929,555); Saccharomyces cerevisiae (Hinnen et al. (1978) Proc. NatL Acad.
Sci. USA
75:1929 and Ito et al. (1983) 1 BacterioL 153:163); Schizosaccharomyces pombe
(Beach et al.
(1981) Nature 300:706); and Yarrowia lipolytica (Davidow et al. (1985) Curr.
Genet. 10:380-
471 and Gaillardin et al. (1985) Curr. Genet. 10:49). Genetic transformation
systems for
metabolic engineering have been developed specifically for a number of
lipogenic yeasts
48
CA 2982734 2017-10-17
including Mucor circinelloides (Zhang et al. 2007), Yarrowia lipolytica (Xuan
et al. 1988),
Rhodotorula glutinis (Li et al. 2012), Rhodosporidium toruloides (Zhu et al.
2012), Lipomyces
starkeyi (CaIvey et al. 2014, Oguro et al. 2015), and Trichosporon oleaginosus
(Gorner et al.
2016).
Organic Substrate Conversion
An aspect of the invention includes methods of processing organics. The
methods can
be performed with the native, non-genetically modified yeasts or genetically
modified yeasts
such as those described herein. If non-genetically modified, the yeasts are
preferably native
lipogenic (oleaginous) yeasts, such as Lipomyces starkeyi.
The methods involve consuming certain organics while producing other organics.
As
used herein, "organic" refers to any organic compound, molecule, or polymer
capable of being
consumed or produced by a microorganism. Exemplary organics include but are
not limited to
carbohydrates (simple sugars, oligosaccharides, polysaccharides), nucleotides,
nucleosides,
nucleic acids, polypeptides, organic acids (including amino acids), and
organic compounds.
Specific examples of organics include glucose, glucan, xylose, xylan,
arabinose, arabinan, lactic
acid, glycerol, acetic acid, butanediol, ethanol, fatty acids, acylglycerols,
enzymes (amylases,
glucosidases, etc.), among others. As used herein, "consume" refers to the
reduction of a certain
component from a medium and can encompass direct uptake of the component for
internal
metabolic processing thereof or external processing of the component
optionally followed by
uptake of resulting products for internal metabolic processing.
The engineered yeasts of the invention and certain lipogenic yeasts are
particularly
effective at consuming certain organics that other microorganisms either
cannot consume or
cannot do so effectively. These organics include glycerol, cellobiose, xylose,
lactic acid,
trehalose, and oligosaccharides. Accordingly, an aspect of the methods of the
invention includes
the consumption of these and other organics from the medium. In certain
versions of the
invention, contacting the medium with the yeast reduces an amount of any one
or more of these
or other organics to less than about 80%, less than about 70%, less than about
60%, less than
about 50%, less than about 45%, less than about 40%, less than about 35%, less
than about
49
CA 2982734 2017-10-17
30%, less than about 25%, less than about 20%, less than about 15%, less than
about 10%, less
than about 5%, less than about 2.5%, or less than about 1% of the initial
amount.
In some aspects of the invention, the medium comprises one or more components
selected from glucose, glucan, trehalose, xylose, xylan, arabinose, arabinan,
lactic acid,
glycerol, acetic acid, butanediol, and ethanol. The medium, for example, may
comprise glucose
in an amount of from about 0.01 to about 100 g/L, from about 0.1 g/L to about
10 g/L, or about
1 g/L; glucan in an amount of from about 0.1 g/L to about 1000 g/L, from about
1 g/L to about
100 g/L, or about 10 g/L; trehalose in an amount of from about 0.01 to about
100 g/L or from
about 0.1 g/L to about 10 g/L; xylose in an amount of from about 0.01 g/L to
about 100 g/L,
from about 0.1 g/L to about 10 g/L, or about 1 g/L; xylan in an amount of from
about 0.05 g/L
to about 500 g/L, from 0.5 g/L to about 50 g/L, or about 5 g/L; arabinose in
an amount of from
about 0.005 g/L to about 50 g/L; from about 0.05 g/L to about 5 g/L, or about
0.5 g/L; arabinan
in an amount of from about 0.005 g/L to about 50 g/L, from about 0.05 g/L to
about 5 g/L, or
about 0.5 g/L; lactic acid in an amount of from about 0.15 g/L to about 1500
g/L, about 1.5 g/L
to about 150 g/L, or about 15 g/L; glycerol in an amount of from about 0.15
g/L to about 1500
g/L, from about 1.5 g/L to about 150 g/L, or about 15 g/L; acetic acid in an
amount of from
about 0.005 g/L to about 50 g/L; from about 0.05 g/L to about 5 g/L, or about
0.5 g/L; butanediol
in an amount of from about 0.02 g/L to about 200 g/L, from about 0.2 g/L to
about 20 g/L, or
about 2 g/L; and/or ethanol in an amount of from about 0.005 g/L to about 50
g/L, from about
0.05 g/L to about 5 g/L, or about 0.5 g/L. Contacting the medium with a yeast
may reduce an
amount of any one or more of these or other organics to less than about 80%,
less than about
70%, less than about 60%, less than about 50%, less than about 45%, less than
about 40%, less
than about 35%, less than about 30%, less than about 25%, less than about 20%,
less than about
15%, less than about 10%, less than about 5%, less than about 2.5%, or less
than about 1% of
the initial amount.
In certain versions of the invention, the medium comprises a grain ethanol
distillation
stillage or a processed grain ethanol distillation stillage. The processed
grain ethanol distillation
stillage may be made by centrifuging distiller's wet grain therefrom, removing
oil,
concentrating, and filtering, or other thin-stillage processing steps
described elsewhere herein
or known in the art. The concentrating may comprise evaporating. In some
versions, the
CA 2982734 2017-10-17
processed grain ethanol distillation stillage comprises thin stillage. The
thin stillage may be
further processed by removing oil and concentrating to generate the medium.
The medium may comprise various amounts of the grain ethanol distillation
stillage or
processed grain ethanol distillation stillage. In some versions, the medium
may comprise at
least about 5%, at least about 15%, at least about 20%, at least about 25%, at
least about 30%,
at least about 35%, at least about 40%, at least about 45%, at least about
50%, at least about
55%, at least about 60%, at least about 65%, at least about 70%, at least
about 75%, at least
about 80%, at least about 85%, at least about 90%, at least about 95%, at
least about 97%, at
least about 99%, or about 100% grain ethanol distillation stillage or
processed grain ethanol
distillation stillage. The grain ethanol distillation stillage or processed
grain ethanol
distillation stillage may be diluted with water or other solvents.
The engineered yeasts of the invention and certain lipogenic yeasts are
particularly
effective at producing certain organics that other microorganisms either
cannot produce or
cannot do so effectively. These organics include lipids (triacylglycerols,
diacylglycerols,
monoacylglycerols, fatty acids, etc.), enzymes (amylases, glucosidases, etc.),
and other
proteins. Particular enzymes include glycoside hydrolases, alpha-amylases
(thermostable and
secreted), dextranases (amylo-alpha-1,6-glycosidase), maltases, beta-1,4-
glucosidases, endo-
1,4-beta-D-glucanases (cellulases) or any other of the enzymes described
elsewhere herein.
Accordingly, an aspect of the methods of the invention includes the production
of these and
other organics.
The organic produced by the yeast may be separated or purified from any other
component of the spent medium for downstream use in other applications. For
example,
enzymes produced by the yeast may be used in liquefaction of starch and/or
saccharification of
liquefaction-processed starch. Such enzymes may include any of the enzymes
produced by the
yeast as described above, such as carbohydrases. Lipid produced by the yeast
may be used for
producing biofuels therefrom or used as a replacement for palm or other oils
in food
applications or other applications. The yeast grown in the medium may
themselves be harvested
and processed to yield a source of protein as a replacement, for example, for
soy protein or as
a source of long-chain polyunsaturated fatty acids for fish grown in
aquaculture.
51
CA 2982734 2017-10-17
The spent medium may also be mixed, either alone or with other liquids (such
as thin
stillage), with starch as a backset for liquefaction of the starch. The
reduction in dissolved
organics by virtue of the yeast consumption reduces the build-up of organics
in grain ethanol
production systems as a whole and in the liquefaction fermentation in
particular.
EXAMPLES
Background and Overview
The existence of an annotated genome for Lipomyces starkeyi greatly
facilitates cloning
and homologous expression, but even with a complete or nearly-complete genome,
various
problems are encountered in cloning and expressing target genes.
One aspect of a suitable cloning strategy involves overexpressing endogenous
genes
involved in lipid production in L. starkeyi under constitutive promoters.
Genomic DNA
(gDNA) can be a preferable source for gene amplification, because all introns
should be
natively excised, and genomic DNA preparation is faster and cheaper than
creating cDNA
libraries. However, certain introns may have regulatory functions or their
presence may impede
or promote the post-transcriptional processing, subsequent protein
translation, or stability of the
transcript when overexpressed. Therefore, some gene targets are preferably
cloned from cDNA
and other targets are cloned from gDNA.
Another issue to consider in cloning involves possible alternate start codons
present in
the sequences of transcribed mRNA of certain gene targets. As an example, both
GUT] and
DGA/ of L. starkeyi have two possible initiator methionine sites differing by
a respective 15
and 156 base pairs upstream from the sequence annotated on the Joint Genome
Institute site.
Detailed sequence alignments with orthologs of these genes in yeast species
with well-
annotated sequences are inconclusive, owing to the large divergence of L.
starkeyi from other
yeast species and the high degree of variability at the N-terminus of these
proteins. Hence, in
such situations, both versions of these genes are cloned. As shown below,
results sometimes
demonstrate a large difference in lipid content, as in the case for the two
DGA1 variants,
whereas others, such as the two GUT] variants, behave similarly.
It is not evident a priori whether genes from cDNA can be amplified under all
conditions. For example, seven out of nine gene targets from cDNA could be
amplified from
52
CA 2982734 2017-10-17
cDNA derived from standard yeast cultures growing in liquid (yeast peptone
dextrose) YPD
media. However, one construct required gDNA as a cloning template. The
exception was
ACC1, which could not be amplified as a product from this cDNA library (as
deemed by agarose
gel electrophoresis) despite many PCR optimization efforts. Amplification of
the gene was
possible with the same primers when gDNA was used as the template. When L.
starkeyi was
cultivated under highly lipogenic conditions and a new cDNA library created
from these cells,
amplification of ACC1 was successful. Such evidence indicates that Acc 1 is
not constitutively
expressed and that expression of ACC1 is inducible under lipogenic conditions.
This finding
emphasizes the importance of ACC1 as an important gene target for enhancing
lipid
biosynthesis.
Generation of vectors involves assembly of 4 to 6 fragments to create the
desired gene
cassettes using the well-established "Gibson" method for in vitro enzymatic
assembly of DNA
fragments (Gibson et al. 2009). Cloning attempts can be unsuccessful in
producing any of the
desired constructs. Gel purification methods to isolate amplified genes can
introduce errors in
DNA terminal regions. Therefore, gel extraction of DNA fragments and column
PCR
purification are preferred to improve the purity and concentration of the PCR
products used in
the assembly reaction.
Correct and complete constructs of the desired cassettes can be identified by
colony
PCR. This can be accomplished through colony PCR by amplifying either parts of
the construct
or the entire construct. During routine screening and sequencing, some of the
construct
candidates can contain partial cassettes (i.e. some will be missing part or
all of one or more of
the assembly components). To avoid selecting these partial constructs, it is
preferable to use
colony PCR for the entire cassette (instead of portions of the cassette). That
way only candidates
containing the appropriately sized insert are then sequenced.
L. starkeyi is cultivated under special conditions and genetic transformation
is optimized
in order to obtain transformants with genes integrated into the genome (Calvey
et al. 2014). L.
starkeyi can be transformed relatively easily with a base vector that contains
only essential
elements as well as a selectable marker. An exemplary base vector is
represented by SEQ ID
NO:91. Success depends upon having cells at an appropriate stage of
development, at an
appropriate cell density and with the correct ratio of cells to DNA.
Transformation conditions
53
CA 2982734 2017-10-17
are systematically evaluated for each method used to determine cell density,
and for the activity
and condition of critical reagents such as salmon sperm DNA.
In native strains of L. starkeyi, each DNA expression cassette integrates
either randomly
via non-homologous end joining (NHEJ) or in a targeted manner via homologous
recombination to create a transformant. In native strains of L. starkeyi, NEHJ
integration occurs
much more frequently than homologous recombination. Even genes that normally
increase lipid
production when overexpressed from most integration sites can generate
transformants with
reduced lipid production relative to the wild-type when integrated randomly
into various sites.
Many strains are therefore screened under controlled conditions in order to
obtain those with
improved performance.
Nile Red fluorescence can be used as a rapid assay for cells or strains of
cells showing
greater relative lipid accumulation. However, the assay is highly variable,
and it is important to
conduct the trials under specific conditions. Selection of media that will
enable identification
of strains with the capacity for elevated lipid production on stillage is
particularly critical. Some
types of rich media used to cultivate L. starkeyi, such as YPD or those based
on modified thin
stillage (mTS), have intrinsic fluorescent properties that interfere with the
proper quantification
of the bona fide Nile Red fluorescent signal from the cells. Moreover,
unmodified thin stillage
contains significant amounts of corn oil and lipids from the hydrolysis of
Saccharomyces
cerevisiae. A partial solution to this problem is to perform a series of
washes to remove the
media from the cells, followed by suspension of the cells in H20. This
treatment can eliminate
interference due to media components that can be removed by washing. As
described elsewhere
herein, other methods can be used to reduce background fluorescence.
When examining cultures for increased lipid accumulation it is important to
distinguish
higher levels of lipid due to higher cell density from higher levels of lipid
due to higher lipid
content per cell. The fluorescence response is therefore normalized to the
cell density.
Effects of Medium Components
The fluorescence assay is intended to identify transformants that overproduce
lipid
when cultivated on an industrial medium composed in part or in whole of thin
stillage. Medium
components can strongly affect the fluorescence assay for intracellular lipid
by increasing or
54
CA 2982734 2017-10-17
decreasing intracellular lipid production. When screening for a transformant
with the capacity
for increased lipid production, it is more difficult to identify improved
mutants when the
medium enables the cells to accumulate high levels of lipid than when native
cells normally
produce relatively little lipid under the growth conditions.
For example, wild-type, untransformed L. starkeyi cultivated in a defined
minimal
medium using an easily assimilated carbon source and a low amount of a poorly
assimilated
nitrogen source would produce a high level of lipid. The same organism
cultivated with the
same carbon source but an amount of readily assimilated nitrogen sufficient
for cell growth
would accumulate less lipid as illustrated by published studies (CaIvey 2016).
It is easier to identify a transformant with increased capacity for lipid
production when
it is cultivated in a medium that allows lower lipid accumulation than in a
medium that enables
higher lipid accumulation.
As taught elsewhere in this disclosure, the relationship between the amount
and the
source of the carbon and the nitrogen provided to the cells can affect growth
and lipid
production in complex ways. Moreover, media used in certain commercial
applications will
include a complex mixture of glycerol, cellobiose, trehalose, xylose, xylitol,
acetic acid residual
corn oil and oligosaccharides derived from residual starch, hemicellulosic and
cellulosic
components from corn along with hydrolysis products from lysed yeast cells. In
practice it is
not reasonable to predict how strains or transformants will perform on a
complex medium of
such composition by screening their performance against simple or complex
media composed
of typical laboratory substrates such as glucose, yeast extract, peptone or
other common
medium constituents. For this reason, empirical testing is important to
identify media that is
representative of the industrial substrate and appropriate for strain
screening. These
considerations were addressed with various media as described below.
Six types of growth media were examined to determine how they influenced the
native
lipid accumulation of L. starkeyi over a 3-day fermentation period. The types
of media included
in the analysis were M1 (LN), M2 (LN), M3 (FIN), M4 (HN), YPD, and mTS. M1
(LN) is a
minimally defined low nitrogen media containing 1.72g/L of yeast nitrogen
base, 0.417g/L
ammonium sulfate 0.179 g/L urea, and 30 g/L of dextrose. The M1 (LN)
formulation is nitrogen
limiting and gives a C:N ratio of approximately 150:1. M2 (LN) is a low
nitrogen media
CA 2982734 2017-10-17
identical to YPD but containing only 3.64% of the amount of yeast extract that
YPD contains
and only 1.82% of the amount of peptone that YPD contains. Converted to
grams/liter, M2
(LN) contains 0.364g/L of yeast extract and 0.364 g/L of peptone. The amount
of dextrose in
M2 (LN) is the same as in traditional YPD (20g/liter). The M2 (LN) formulation
is nitrogen
limiting and gives a C:N ratio of approximately 70:1. M3 (FIN) and M4 (FIN)
are high nitrogen-
containing versions of Ml, with M4 FIN containing peptone. More specifically,
M3 (I-IN)
contains 6.7g/L yeast nitrogen base, 2.145g/L urea, and 30g/L dextrose. M4 FIN
media contains
6.7g/L yeast nitrogen base, 2.145g/L urea, 3.0g/L peptone, and 30g/L dextrose.
YPD is yeast
peptone dextrose media. mTS is modified thin stillage. The mTS was prepared by
clarifying
thin stillage to remove oil by skimming, boiling to concentrate the stillage,
removing
precipitates produced by the boiling, autoclaving, and removing precipitates
generated during
the autoclaving.
As shown in FIG. 4, the type of media greatly influences the basal lipid
production of
L. starkeyi NRRL Y-11557, even if nitrogen is not considered limiting. These
wild-type cells
appear to accumulate a moderate amount of lipids (z, 30%) when cultivated in
mTS during
extended fermentation times, despite its nitrogen content. The mildly
lipogenic property of mTS
with L. starkeyi NRRL Y-11557 is likely due to the presence of glycerol and/or
other
compound(s).
The choice of a screening media should be done carefully. Choosing a media
limiting
in nitrogen could mask small lipogenic effects of certain genes, while media
high in nitrogen
or complexity may suppress those effects. Likewise, even media that is not
nitrogen limiting
can be slightly lipogenic, as seen in M3 (HN) media, because the lack of some
nutrients can
induce lipogenesis.
In light of these considerations, transformants of NRRL Y-11557 are screened
using
mTS medium to identify yeast strains that convert the soluble organics and
sugars to other
products. Evaluation on this medium also eliminates the possibility that some
transformants
perform well during screening on synthetic media but present significantly
dampened or null
effects when grown on a commercially relevant fermentation stream.
Transformants are also
screened using YPD to control for any possible variation in various sources of
mTS.
56
CA 2982734 2017-10-17
Gene Cassette Selections and Primer Design
Gene cassette integration vectors are designed using a base vector as shown in
FIG. 5
and having a sequence of SEQ ID NO:91. The base vector contains an origin of
replication
(On) and a kanamycin resistance cassette that permits maintenance and
propagation in bacteria.
In addition, it contains two multiple cloning sites (MCS) and two modified
LoxP sites (RE and
LE) that flank the strong constitutive LsTDH3 promoter coupled to a codon
optimized
nourseothricin (NAT) resistance gene and its respective transcription
terminator region.
Measures are taken to ensure no antibiotic resistance gene is incorporated in
the final
yeast strain. The bacterial kanamycin resistance gene is not integrated into
the yeast genome if
it is interrupted by restriction digestion prior to transformation. The
presence of the loxP sites
enables excision of the NAT resistance gene after genome integration. The LoxP
sites
themselves are modified (i.e. dead) such that the product after one
recombination event resists
further recombination, thereby protecting the genomic integrity of the
organism. The gene
overexpression cassettes therefore include a constitutive promoter, the
gene(s) of interest, and
one or more transcription terminator regions inserted into one of the MCSs.
Genes are overexpressed to increase lipid production under nutrient-rich
conditions.
These include: acetyl-coenzyme-A carboxylase (LsACC/), delta-9 acyl-CoA
desaturase
(LsOLE1), ATP-citrate lyase alpha and beta subunits (LsACL1/LsACL2), two
variants of
glycerol kinase (LsGUT1-1602) and (LsGUT1-1617), two variants of
diacylglycerol
acyltransferase (LsDGA1-1233) and (LsDGA1-1389) having different start sites,
and malic
enzyme cloned from genomic DNA (LsgME) and cDNA (LscME), among other genes
described herein. Precise promoter, gene, and terminator sequences are
selected based on
Illumina RNAseq data and knowledge of promoter strength and expression
patterns in this
organism. All contain a single promoter, gene, and terminator region, with
exception to the
LsAc11/LsAc12 construct, which involves a bidirectional promoter expressing
the two genes
with different transcription terminator regions. These sequences are then
analyzed for the
presence of specific restriction digest sites to determine into which one of
two MCSs contained
in the base vector they would be cloned. This step facilitates future
subcloning into other
vectors. For example, the diacylglycerol acyltransferase (LsDGA1) construct is
capable of being
inserted upstream of the loxP site using the Sbfl restriction enzyme to
linearize the vector.
57
CA 2982734 2017-10-17
Other constructs are capable of being inserted downstream of the opposing loxP
site, using
RsrII and AvrII to linearize the vector. The desired sequence combinations and
restriction
enzyme sites are entered into the NEBuilder assembly tool
(nebuilder.neb.com/) with a
minimum overlap setting of 20 base pairs to construct the primer sequences for
performing the
Gibson assembly reaction.
Schematic of the Pipeline for Creating and Characterizing Strains
An assembly line of sequential steps to create metabolically engineered yeast
is shown
in FIG. 6. These steps included target gene, promoter, and terminator
selection, molecular
biology techniques (PCR, Gibson assembly), yeast transformations, selection,
cataloging and
storage, Nile Red screening, and further validation assays.
gDNA Extraction and cDNA Library Creation: A MasterpureTM Yeast DNA
Purification Kit (Epicentre, Madison, WI) is used to extract L. starkeyi gDNA.
Nitrogen rich
(YPD) or nitrogen limited (YPD 70:1 (C:N)) cultures are used for RNA
extraction. The YPD
70:1 media contain only 3.64 % and 1.82% percent of the yeast extract and
peptone as YPD,
respectively. A freshly saturated 5 mL culture of L. starkeyi grown under
constant agitation at
30 C is pelleted by centrifugation, washed, suspended in 5 mL of sterile H20,
then used to
inoculate 50 mL of either YPD or YPD 70:1 media in a 125 mL shaker flask 0.8
OD600 and
allowed to incubate overnight at 30 C under 225 rpm. Cells are observed
microscopically to
determine lipid production in the YPD 70:1 media as compared to the YPD media,
and the
0D600 is measured to calculate the quantity of cells to use in the RNA
extraction protocol. RNA
is extracted using an RNeasy Mini Kit (Qiagen), following enzymatic
disruption. cDNA is
synthesized from these RNA preparations using a QuantiTect Reverse
Transcription Kit
(Qiagen), following the instructions of the manufacturer.
PCR of Fragments and Gibson Enzymatic Assembly: The base vector (pXC301 ¨ FIG.
5 ¨ SEQ ID NO:91) linearized with the enzymes listed in Table 2 is used to
clone L. starkeyi
genes in conjunction with the "Gibson method" for in vitro enzymatic assembly
of DNA
fragments. All PCR amplifications are performed using Phusion High Fidelity
Taq polymerase
(NEB) and the manufacturer protocol in either 5X Phusion GC buffer (ACC1) or
5X Phusion
buffer (all others) using the annealing temperatures in Table 2. Annealing
steps are carried out
58
CA 2982734 2017-10-17
using the lowest Tm of the primer pair, or the experimentally optimized
annealing temperature,
where appropriate. The reaction products are analyzed by agarose gel
electrophoresis
containing ethidium bromide and subsequently visualized and photographed.
Successful
reactions of identical fragments are pooled and then all samples (including
the digested base
vector) are subjected to a PCR cleanup column (Qiagen), and then quantified on
a Nanodrop
2000 instrument (Thermo Scientific). The vector and inserts are then added in
equimolar
quantities (0.1 pmoles/fragment) in a separate tube, and the final volume
adjusted to 20 pi, by
the addition of 15 [IL premade Gibson assembly reaction mix and water. All
reactions are
allowed to incubate at 50 C for one hour, and then 5 iL of the assembly
reaction is used to
transform 20 [t1_, of EnduraTM DUO competent cells (Lucigen) using standard
techniques.
Transformants are selected on LB plates containing kanamycin, and positive
candidates are
identified by colony PCR and sent for sequencing confirmation.
59
CA 2982734 2017-10-17
Table 2. Summary of Gene Targets, Promoters, and Terminators with
Cloning Information
Promoter and Cloning Enzymes
for
Target PCR Annealing
Full Name Teminator Digest Yeast
Gene Temperature
Pairings Enzymes
Transformation
,
Acetyl- FBA I Promoter . 55-64 C
Rsrll and
ACC I coenzyme-A ACC1 67 C Avr1I AsiSI
carboxylase FBA1 Terminator 55-64 C
GLN1 Promoter 60 C
Delta-9 acyl- RsrlI and
OLE1 OLE I _____ 63 C AsiS I
CoA desaturase Avr11
GLN1 Terminator 60 C
EN01 Promoter 57 C
ACL1 58 C
ACL1 ATP-citrate
ATPase RsrlI and
and lyase, alpha and AsiS1
Terminator 64 C Avr11
ACL2 beta subunits
ACL2 58 C
TPI Terminator 65 C
ATPase (3900)
55.3-61.2 C
Glycerol Promoter
Glycerol Kinase ________________________________________ Rsrll and
Kinase 1602 Variant AvrIl Glycerol Kinase
63.9-67 C AsiS1
1602 ATPase (3900)
62.75-66.25 C
Terminator
ATPase (3900)
58 C
Promoter
Glycerol Diacylglycerol
Glycerol Kinase Rsrll and
Kinase acyltransferase 65 C Asi SI
1617 AvrII
1617 1617 Variant
ATPase (3900)
65 C
Terminator
Diacylglycerol TEF1 Promoter 60 C
DGA1
1233 acyltransferase DGA I 72 C Sbfl XmaI/Avr11
1233 Variant TDH3 Terminator 70 C
Diacylglycerol TEF1 Promoter 60 C
DGA I
1389 acyltransferase DGA I 72 C Sbll X maI/Avr11
1389 Variant 11)I13 Terminator 70 C
TDH3 Promoter 60 C
gMalic Malic Enzyme ____________________ Rsrll and
gMalic Enzyme 60 C AsiSI
Enzyme (gDNA) Avr11
TDH3 Terminator 60 C
TPI (196787)
62 C
Promoter)
cMalic Mal ic Enzyme __________________ Rsrll and
cMalic Enzyme 58 C AsiS1
Enzyme (cDNA) _________________________________________ Awl(
TPI (196787)
66 C
Terminator
Vectors are linearized so the entire target gene cassette, including the loxP
flanked
region, randomly integrates into the L. starkeyi genome as one fragment Table
2 lists restriction
enzymes for linearization. Linearized DNA is ethanol precipitated and
suspended in TE to
increase its concentration (>160 nM) and purity. To transform the cells, a
procedure based on
CA 2982734 2017-10-17
the lithium acetate method was used (Calvey et al. 2014, Gietz et al. 2002). A
near stationary
phase culture of L. starkeyi is inoculated into 50 ml YPD to between 0.6 and
0.8 0D600 and
grown overnight to reach between 3.0 and 4.0 0D600. The culture is harvested
and washed twice
with 25 mL sterile water, and resuspended to 1.5 mL in sterile water or 0.1M
LiAc. Aliquots of
150 [it are dispensed into 1.5 mL tubes and centrifuged. The remaining cell
pellet is then
suspended in 360 iaL transformation mix containing 240 L 50% w/v PEG 3350, 50
j.tL boiled
ssDNA, 36 j.it 1.0 M LiAc, and 36 [IL of the desired plasmid DNA (added last).
Samples are
incubated at 30 C for 3 hours, heat shocked at 40 C for 5 minutes, and then
centrifuged to
remove the transformation suspension. Cells are allowed to recover in 3 mL YPD
for 4 hours
before being plated onto appropriate selective media. After 6 days of growth,
transformants are
selected, catalogued by size, and streaked onto appropriate antibiotic
selection plates for
creating glycerol stocks until characterization.
Yeast Lipid Screening using Nile Red: Both YPD and mTS are used to screen for
lipid
accumulation in L. starkeyi. Starter cultures are inoculated with the desired
transformants in 5
mL tubes containing YPD-NAT or YPD liquid media at 30 C under constant
agitation. This
lipid screening protocol is based on Nile Red fluorescence adapted from a
previous study
(Sitepu et al. 2012). Samples and appropriate dilutions are then prepared in a
96-well black
clear-bottomed plate to contain 100 [iL in each well. Nile Red is prepared as
a 2 mg/mL stock
solution and then diluted to a 2X working concentration (8 pg/mL). Fluorescent
signals and
OD63o readings are read independently from two plate readers (BioTekTm FLx800
and
BioWhittakerTM ELx808, respectively). Data is analyzed by normalizing the
fluorescent signal
to the 0D630 of the culture. To enable comparisons between different runs,
normalized
fluorescence is standardized relative to the WT culture of each group.
Lipid Extraction Analysis: An extraction protocol is used for crude
gravimetric
assessments of total lipid content based on the classic Bligh and Dyer method
(Bligh et al. 1959)
(20). First, 2 mL of cell culture is centrifuged at 3,000 rpm for 5 minutes in
15 mL falcon tubes,
and the cell pellets frozen at -20 C until lipid extraction analysis. Thawed
cell pellets are
suspended in 1 mL H20 containing 200 lit of concentrated HC1, and the
suspension was heated
to 90 C for 1 hour to lyse cells. Lipids are then extracted by addition of 6
mL of a 2:1 (v:v)
methanol:chloroform solution and 3 mL of 1M NaC1, followed by vortexing for 5
minutes.
61
CA 2982734 2017-10-17
Tubes are then centrifuged at 3,000 rpm for 10 minutes to induce phase
separation. The lipid-
containing lower chloroform layer (z-,' 2 mL) is then carefully removed using
a glass Pasteur
pipette, and transferred into a clean, pre-weighed, 5 mL glass vial. Finally,
the extracted
chloroform layer is completely evaporated by incubation in a 40 C heat block
under a constant
stream of air for 1 hour. Vials are then re-weighed to determine the mass of
extracted total
lipids.
HPLC Analysis of Organics: Concentrations of sugars and mTS metabolites
(including
glucose, xylose, cellobiose, arabinose, xylitol, ethanol, glycerol, lactic
acid, and acetic acid) are
determined by high performance liquid chromatography (HPLC) using an Agilent
1100 Series
auto sampler, pump, and refractive index detector, with a Bio-Rad Aminex HPX-
87H ion
exclusion column (300 x 7.8 mm) held at 65 C. The mobile phase is 0.01N H2SO4
at a flow
rate of 0.6 mL/min. Samples are diluted 1:10 prior to injection.
Selection of a Yeast Platform
The constituents of thin stillage obtained from a supplier were characterized
in order to
better monitor the consumption of organics during fermentation of engineered
yeast strains. It
was discovered that mTS contains between 18.6 to 20.5 g/L glycerol, among
other components.
To establish a yeast platform for bioprocessing of mTS and metabolic
engineering, the growth
performance of several strains of L. starkeyi on different media was
monitored. All of them
grew well on starch but the Y-11558 strain grew slightly faster (Table 3).
When challenged with glycerol as a carbon source, none of the South African
strains
(Y-27493, 27494 and 27495) grew, and Y-11558 was much slower ( = 0.047; Td =
21 h). See
FIG. 7. However, mutagenesis and serial subculturing reduced the doubling time
of Y-11557
to 9.9 h on glycerol. These findings established that L. starkeyi grows on
glycerol, a major
component of mTS, and that adaptation increases growth rates. Based on these
results and given
that the genome of L. starkeyi NRRL Y-11557 has been sequenced (Grigoriev et
al. 2012 and
DOEJGI 2011), this strain was selected as the exemplary platform yeast for
metabolic
engineering.
62
CA 2982734 2017-10-17
Table 3. Doubling Times of Different Lipomyces
starkeyi Strains Grown on starch
Strain Doubling Time (Hrs)
Y-1388 3.56 0.28
Y-11557 3.60 0.28
Y-11558 3.36 0.30
Y-27493 3.52 0.28
Y-27494 3.97 0.25
Y-27495 4.28 0.23
Metabolic Engineering and Screening
The next objective was to modify the physiology of L. starkeyi so that it
would generate
lipids under high nitrogen conditions, such as those found in thin stillage
(TS) and modified
thin stillage (mTS). As described in Table 2, specific genes targets were
selected with
constitutive promoter and terminator pairings based on knowledge of the L.
starkeyi genome,
its lipid biochemistry, and gene expression profile under these conditions.
These were all cloned
into target vectors and transformed into L. starkeyi. Since the vector
integrates at random within
the genome, each transformant might display different lipid biosynthetic
capabilities based
simply upon the site of genomic integration, or on the number of copies
introduced within the
genome. It was hypothesized that although gene cassettes conferring genuine
lipogenic effects
could occasionally integrate into unfavorable regions that could mask their
effect, the average
normalized fluorescence of that particular transformant pool should be
somewhat higher than
the average of the WT and base vector transformed strains. In addition, there
may also be more
variation among the transformants of these genes than those with no effect,
since each
transformant could have different degrees of increased lipid biosynthesis, as
opposed to genes
with null effects. Following this logic, a total of 234 transformants were
screened from the pool
63
CA 2982734 2017-10-17
of metabolically engineered strains for Nile Red fluorescence when cultivated
on YPD (234),
mTS (11), or both (145). The screening was performed over the course of 2
(YPD) or 3 (mTS)
days. The results of this preliminary screening are shown in FIG. 8A, which
displays the top
50% performers of each gene. Three gene targets (DGA1-1233, DGA1-1389, and
ACC1) had
either large standard deviations, higher relative normalized fluorescence (1.5-
2 times higher),
or both when compared to the WT yeast transformed with the base vector. This
behavior was
independent of the screening medium for DGA1-1233 and ACC1. Additionally, DGA1-
1389,
GUT]-1617 and ACL1/ACL2 displayed moderate standard deviations when screened
in mTS
media. The complexity of mTS likely contributed to this phenomenon. In
general, however,
transformants that performed well in YPD media also performed well in mTS
media.
Considering the variability of thin stillage across ethanol plants, this is
encouraging because it
implies that the engineered strains have the flexibility to overproduce lipids
in various types of
media. In fact, a total of 12 transformants were identified that exhibited one
full standard
deviation higher normalized fluorescence compared to the empty vector
transformed yeast at
the end of the growth phase when cultivated on both YPD and mTS. Eight of
these are DGA /-
1233 transformants, while the other 4 are ACC] transformed strains.
In order to obtain a more statistically significant analysis of these results,
the top
performers of each gene were rescreened in triplicate in mTS. The results of
this analysis are
shown in FIG. 8B, and clearly demonstrate increased normalized fluorescent
signals relative to
the WT in DGA1-1233 6L, GUT1-1602 6L, and GUT1-1617 2L. In these
transformants, it was
also confirmed that the entire cassette had integrated into the genome by PCR
amplification
from genomic DNA using primers specific to the cassette. All had the expected
fragment (data
not shown).
Effects of Overexpressing Glycerol Kinase in L. starkeyi
The presence of sugars or oligosaccharides can repress glycerol utilization in
some
yeasts, so overexpressing the glycerol kinase gene (GUT]) in L. starkeyi could
increase glycerol
utilization if that were the rate limiting step. When this hypothesis was
tested, L. starkeyi
transformants showed increased lipid content by Nile Red fluorescence, but
presented lower
growth rates. In screening about twelve GUT]-1602 and GUT]-1617 transformants
for growth
64
CA 2982734 2017-10-17
on thin stillage plates, one GUT]-1617 transformant, Ls-11, appeared to grow
relatively faster
than the others. This strain was selected to use in shake flask and 3-L
bioreactor trials to
compare against the WT strain. Ls-11 showed higher cell growth and glycerol
utilization rates
as compared to WT cells in shake flasks (FIG. 9). The cells from the 200 ml
shake flasks were
then used to inoculate 200 ml of medium in 3-L bioreactors. In the bioreactor
studies glycerol
utilization by the Ls-11 transformant was initially faster. Besides these
characteristics, the Ls-
11 transformant accumulated larger liposomes when examined microscopically,
and formed
pseudomycelia structures (FIG. 10B).
It was hypothesized that in the GUT] transformant, glycerol kinase shunts
glycerol
directly towards triglyceride production via glycerol-3-phosphate, and that
this accounts for
higher lipid accumulation with lower growth rates. To increase the growth rate
on glycerol
glycerol-3-phosphate dehydrogenase (GUT2) can also be overexpressed, as
described in
subsequent sections. Overexpression of GUT] alone has complex and differing
effects on the
growth and morphology of L. starkeyi. Different transformants of GUT] alone
show various
rates of growth and increased lipid accumulation, some of which are superior
to the WT or Ls-
11.
Remarkably, L. starkeyi is capable of using cellobiose, trehalose,
amylodextrins,
cellulo-oligosaccharides, xylose, xylan oligosaccharides and glycerol ¨ the
primary
carbohydrate sources present in thin stillage. The fact that L. starkeyi
effectively consumes all
of the oligosaccharide components and all of the glycerol in thin stillage
highlights the
importance and bioprocessing capabilities of this organism.
Evaluation of Other Cellular Features of DGAJ-Transformed Strain
To gain further insight into the physiology of the metabolically engineered
strains, a
deeper evaluation into the DGA /-transformed strain (DGA1-1233 6L) was
performed by
evaluating its whole crude lipid content, dry cell weight, growth rate,
consumption of organics,
and cellular morphology relative to the WT when cultivated in mTS in a
bioreactor. The DGA1-
1233 6L strain grew slightly slower than the WT, leading to marginally reduced
glycerol and
cellobiose utilization rates (Fig. 11). Nevertheless, both strains consumed
all of the available
oligosaccharides and carbon sources present in mTS within five days, except
for trace amounts
CA 2982734 2017-10-17
of lactic acid (data not shown). When the cells were examined under the
microscope,
significantly more liposomes were observed in the DGA1-1233 transformant than
the WT (Fig.
11). Remarkably, it was found that overexpression of DGA1 increased the final
lipid content
from 8.25 g/L to 22.7 g/L, which is an increase of about 14.5 g/L or 275%.
This also correlates
to an increase in the lipid content (g lipid/g dry cell weight) from
approximately 30% to 85%
(Fig. 12). It is hypothesized that overexpression of DGA1 is seeding liposome
formation, and
this in turn is increasing the lipid carrying capacity of the cells. Taken
together, the present
examples have demonstrated the creation of a DGA /4233 engineered strain of L.
starkeyi capable of efficiently converting mTS into cell mass with nearly 85%
lipid content and
22.7 g/L lipids. This was also accomplished without optimization of culturing
conditions
(aeration, pH, temperature, etc.), which will surely lead to further
improvements in lipid
production and productivity.
Improved Screening by Substituting Bodipy Dye for Nile Red
Nile Red dye is useful in identifying engineered yeast strains that produce
more lipid
when cultivated on modified thin stillage (mTS), but it has several
limitations: First, thin
stillages from different plants vary greatly, and this variance can affect the
response of the Nile
Red assay. Stillages are altered by the method and extent of corn oil
extraction, the extent of
stillage backset, and whether clarification, and/or concentration is employed.
Each of these
factors affect cell growth, lipid production and the response of the Nile Red
assay. Second, the
mTS medium used in the screens can interfere with the Nile Red fluorescent
signal, which
necessitates washing cells prior to dye addition. Third, Nile Red dye itself
is unstable, and signal
loss occurs within several minutes following addition. Finally the number of
handling steps
reduces throughput in sample processing. All of these factors are exacerbated
because random
integration of genes into the chromosome requires examining many transformants
in order to
identify the top performers. An effective screening assay must be rapid, offer
similar responses
on many different stillages and effective in-situ without cell harvest or
washing. We therefore
modified our screening process to accommodate higher sample throughput by
using a different
fluorescent dye, Bodipy, which is superior to Nile Red in many aspects, and by
standardizing
the medium for screening.
66
CA 2982734 2017-10-17
Bodipy (boron-dipyrromethene) fluorescent dyes are much more sensitive and
stable
than Nile Red. They have been used successfully for conducting in vivo lipid
trafficking and
accumulation studies. One Bodipy variant (4,4-Difluoro-1,3,5,7,8-Pentamethy1-4-
Bora-3a,4a-
Diaza-s-Indacene) is ideal for detecting neutral lipids, such as those that
accumulate in the
liposomes of L. starkeyi. We established a new screening methodology in which
Bodipy dye
(0.6 [tg/mL) is added directly to a synthetic thin stillage (sTS) to mimic
cultivation in mTS
under more consistent conditions. Synthetic thin stillage is composed of 6.7
g/L yeast nitrogen
base with ammonium sulfate and amino acids, 20g/L glycerol, and 6 g/L
cellobiose. This
method enables a 6.5 fold higher throughput in screening. It also requires
fewer steps, as
samples from actively growing cultures can be immediately assessed for
fluorescence without
washing off stillage prior to dye addition.
We also developed an improved automated method for ranking transformants based
on
their raw fluorescence with Bodipy. In short, samples are normalized by
dilution to a target
0D630 and then measured for fluorescence once a day for four days. An
algorithm is then
applied to rank the transformants for easy identification of prime performers.
The data can be
presented as bar graphs of either entire transformant pools or replicates of
the same
transformants. These graphs account for the dilution factor of the cells/dye,
which we refer to
as fluorescence in the figures for simplicity, even though it is fluorescence
following correction
for dilution. This approach streamlines the data analysis process with minimal
manipulation.
As proof of principle, many of the same transformants that had been selected
manually in Nile
Red assays were also identified by this approach, along with other high
performing strains that
might have been overlooked based on the previous method.
Investigating DGA1 Cassette Variants on Lipid Accumulation
One of the most lipogenic cassettes discussed above overexpresses
diacylglycerol
acyltransferase (DGA1-1233), which was cloned from cDNA (cDGA1-1233). It
employs a
NAT marker for resistance to the antibiotic nourseothricin to enable selection
of transformed
strains. In order to facilitate overexpression of cDGA/-1233-NAT in
combination with other
genes ¨ either by sequential transformations or by mating ¨ we constructed a
second DGA1-
1233 overexpression cassette using an alternate resistance marker. We then
determined the
67
CA 2982734 2017-10-17
conditions under which L. starkeyi transformants could be recovered on plates
containing an
antibiotic other than nourseothricin.
We next questioned whether overexpression of cDNA or genomic DNA (gDNA) of
DGAI (gDGA1-1233) resulted in better transformants. The principal difference
between the
gDNA and cDNA clones of DGAI-1233 is the presence or absence of introns.
Introns are
known to enable alternative splicing of mRNA so a single gene can create
multiple proteins,
and while introns do not encode protein products, they can be integral to the
regulation of gene
expression.
Little is known about how or whether introns regulate gene expression in L.
starkeyi.
To address this, we created sibling cDGA/-1233 and gDGA/-1233 cassettes cloned
from cDNA
and gDNA into a vector that confers resistance to Hygromycin B. These were
then transformed
into L. starkeyi and the Bodipy fluorescence of the resulting transformant
pools were monitored
in sTS over the course of four days. Although both versions yielded more
fluorescence over the
wild-type strain, the gDNA version did not induce as many liposomes as the
cDNA version,
which indicated that the presence of introns modulated cDGA 1-1233 expression.
Subsequent
screening tests revealed that the top cDNA transformant (cDGA1-1233 154L)
outperformed the
top gDNA transformant (gDGA 163M). Moreover, cDGA 154L behaved similarly to
strain 6L,
the top performer from the Nile Red screen. See FIG. 13. Therefore, we
selected cDGA 1-1233
154L as our platform strain for further development.
Deregulated Accl Mutants
ACC1 expression is induced under lipogenic conditions in L. starkeyi, and when
overexpressed, we identified four transformants that exhibited one full
standard deviation
higher normalized fluorescence when compared to yeast transformed with the
empty vector.
However, these transformants were clearly less prodigious at producing lipids
compared to our
DGAI transformants. This was surprising, since ACC catalyzes the carboxylation
of acetyl-
CoA into malonyl-CoA, which is a regulated commitment step in fatty acid
biosynthesis.
Enzyme activity can be regulated by more factors than gene expression alone.
In fact,
ACC activity in S. cerevisiae and other organisms is regulated by an AMP-
activated protein
68
CA 2982734 2017-10-17
kinase (Snfl), which phosphorylates certain serine residues. Mutation of these
residues can
prevent inhibition of ACC, and increase lipid yields.
To determine if ACC could also be deregulated in L. starkeyi, we identified
two possible
serine phosphorylation sites in the L. starkeyi ACC1 ortholog based on the
AMPK
phosphorylation target motif, and created three mutated versions of the ACC1
gene (S63 9A,
S1146A, and the double mutant S639A/S1146A). These were made because it was
unknown
which mutations and in what combination could deregulate Acc 1 activity.
Following
transformation and fluorescence screening in yeast, we discovered that
constitutive expression
of the single S1146A mutant (but not S639A nor the double mutant cassette),
increased average
fluorescence levels, as shown in FIG. 14.
Overexpression of Acyltransferases
Given the dramatic improvement in lipid accumulation that occurs in DGA1-1233
engineered strains, we hypothesized that expression of additional genes
involved in the
biosynthesis of triacylglycerols might coax cellular metabolism towards
additional lipid
production. The DGA1 gene product carries out the third and final acylation
step in
triacylglycerol synthesis, so we focused our attention on the two
acyltransferases that catalyze
the previous reactions, namely SCT1 and SLC1. The former (SCT1) is a glycerol-
3-phosphate
acyltransferase that carries out the first acylation step, whereas the latter
(SLCI) is a
lysophosphatidate acyltransferase, which catalyzes the second acylation step.
To test these
genes, we constructed SCT1 and SLC1 overexpression cassettes from gDNA under
the control
of the CIT1 promoter/terminator pair (SCTI) or the FBA/ promoter/terminator
pair (SLCI), and
generated over 200 transformants from each cassette to assay for increased
lipid production.
These strains were evaluated on sTS medium using our Bodipy screening
methodology.
Data gathered from an exemplary screen of SCT1 transformants is shown in FIG.
15.
Overexpression of SCT1 caused a moderate increase in lipid accumulation under
these
conditions. SCT1 transformants, on average, yielded 5-10% increased lipid
content as measured
by Bodipy fluorescence. However, some of the top performing SCT1 strains
produced
fluorescent readings approximately 30% higher than the wildtype across all
time points tested.
Taken together, these results demonstrate that SCTI plays a critical role in
initiating
triacylglycerol biosynthesis in L. starkeyi. Additionally, SCT1 is an
attractive target because it
69
CA 2982734 2017-10-17
utilizes glycerol-3-phosphate as a substrate, and could potentially improve
the rate of glycerol
utilization in feedstocks with high glycerol content, such as thin stillage.
SCT1 was also
introduced into other genetic backgrounds by both mating and secondary
transformations, as
described below, to generate strains with further improvements.
In contrast to SCTI, overexpression ofSLCI did not result in any statistically
significant
improvements in lipid accumulation. On average the SLCI strains performed no
better, or
perhaps slightly worse, than the wild-type in terms of Bodipy fluorescence
readings (FIG. 16).
Growth rates of all strains were approximately similar. However, there were a
few strains that
performed better than any wild-type replicates (e.g. SLC1-30L, FIG. 16), which
could occur if
multicopy integration or specific integration sites are required for SLC1 to
have a measurable
impact on lipid production.
Given that overexpression of SLC1 alone does not typically increase lipid
production,
this suggests that the second acylation reaction catalyzed by SLC1 is not a
rate-limiting or
highly regulated step during the accumulation of lipids in Lipomyces. Since we
have
demonstrated that overexpression of SCTI (1st acylation) and DGA1/2 (3rd
acylation) have
such significant effects on improving lipid accumulation, it follows that
these are the more
significant bottlenecks than the SLCI reaction.
However, it remains to be seen if combinatorial expression of SLC1 with other
genes
may synergistically increase lipid production. It is possible that
overexpression of SLC1 in a
SCT1 engineered strain could further improve lipid production if the
bottleneck resolved by
acceleration of the first acylation step (SCT1) subsequently leads to the
creation of a new rate-
limiting step at the second acylation (SLC1). Alternatively, overexpression of
SLC1 in a DGA1
engineered strain could also improve lipid production by introducing a
metabolic "pull" towards
triacylglycerol production.
Combinatorial Expression of Lipogenic Cassettes
In order to evaluate the effects of overexpressing more than one lipogenic
cassette in
the same organism, we pursued two strategies. First, top strains with
different resistance
markers were mated together and subjected to sporulation, and progeny
harboring both
integrated cassettes were selected on double antibiotic plates. Alternatively,
top strains were
CA 2982734 2017-10-17
also used as a platform for a second round of transformations with another
cassette, as long as
the resistance markers were different. The resulting double transformed
strains were then
evaluated in our Bodipy screen against the progenitor transformant strain for
enhanced lipid
production. In this way, we screened over 70 mated strains of top performers
and 200 double
transformed strains.
We first evaluated mated transformants overexpressing DGA1-1233 and ACC1,
ACC1(S639A), and ACC1(S1146A). In some cases, the mated strains performed
better than
the parental strains. The biggest improvement was seen in the DGA1-
1233/ACC1(S1146A)
cross (FIG. 17). The DGA1-1233/ACC1(S639A) cross performed no better than the
DGA1-
1233/ACC1 cross and also no better than the parental DGA1-1233 strain, again
suggesting that
S639 plays no role in regulating ACC1. Hence, the combination of
overexpressing DGA1-1233
and ACC1 in L. starkeyi enhances lipid production compared to independent
overexpression of
these proteins, but is particularly pronounced when ACC1 is mutated at the
S1146
phosphorylation site.
We also evaluated mated transformants constitutively expressing DGA1-1233 and
SCT1, and a strain with two DGA1-1233 overexpression cassettes. In both cases,
each strain
performed better than the parental strains. These are shown in FIG. 18, along
with the wild-
type and our previously identified top performer (cDGA NAT) for comparison
purposes.
Combinatorial Expression of Lipogenic and Auxiliary Cassettes
We previously determined that certain cassettes resulted in no significant
increase in
lipid accumulation relative to control strains as deemed by Nile Red analysis
in Lipomyces
starkeyi. See FIG. 8. These included the dual overexpression of ATP citrate
lyase subunits 1
and 2 (Ac11/Ac12), and the cDNA and gDNA versions of the malic enzyme (gME and
cME).
We re-evaluated these transformants in our sTS screening methodology using
Bodipy and
obtained similar results. We hypothesized that overexpressing these genes has
little effect on
lipid accumulation since they are not directly involved in the regulatory
mechanisms that
govern lipogenesis. However, their activities may synergistically enhance
lipid accumulation
in strains that have been deregulated from one or more of these mechanisms.
71
CA 2982734 2017-10-17
To test this hypothesis, we selected our top DGA1 transformant (cDGA1) to
cross with
select transformants overexpressing the cDNA version of the ATP citrate lyase
cassette
(cAc11/2), or the cDNA and gDNA versions of the malic enzyme cassettes (cMalic
Enzyme,
gMalic Enzyme). After mating, the progeny and parental strains were evaluated
in sTS
containing Bodipy for lipid accumulation. As observed previously, the
Ac11/Ac12 and malic
enzyme sole transformants performed marginally better than the wild-type
strain due to the
presence of resistance marker integration and overexpression (FIG. 19). Sole
overexpression of
a resistance marker cassette can be marginally lipogenic due to a number of
possibilities,
including slower transformant growth and the position in which the cassette
integrated in the
genome. However, when coupled with overexpression of DGA1, the crossed strains
performed
significantly better than either of the parental strains, particularly on the
third and fourth days
of culture growth (FIG. 19). As a control, we also evaluated the parental DGA1
strain
transformed with the empty base vector overexpressing the same resistance
marker as the
lipogenic transformants. The overall performance of these control strains was
slightly worse
than the parental DGA1 strain (FIG. 20), implying that the lipid accumulation
observed in the
mated strains could be synergistic, as the total improvement is the same if
not higher than the
individual improvements combined.
Enhanced glycerol utilization is another feature that is particularly
important, due to the
large amount of glycerol present in thin stillage. Glycerol Kinase (GUT])
catalyzes the
phosphorylation of glycerol to glycerol-3-phosphate, and is the first step in
glycerol utilization.
However, when GUT] was overexpressed in L. starkeyi, the cells presented with
significantly
lower growth rates, presumably due to the introduction of a metabolic
bottleneck. To overcome
this, we transformed a strain overexpressing GUT] with an FAD dependent
glycerol-3-
phosphate dehydrogenase (GUT2), which catalyzes the second reaction in
glycerol utilization
by conversion of glycerol-6-phosphate to dihydroxyacetone phosphate (DHAP),
and screened
the resulting double transformants for growth on glycerol and lipid
accumulation. Dual
overexpression of GUT] and GUT2 rescued the growth rates in approximately half
of the
transformants evaluated (FIG. 21), with some exhibiting superior lipid
accumulation relative to
the parental strains (FIG. 22). Importantly, overexpression of GUT2 alone did
not significantly
affect lipid accumulation (FIG. 22). One of the highly lipogenic strains
(GUT]/GUT2 IL) was
72
CA 2982734 2017-10-17
chosen for scale-up in shake flask experiments to better evaluate its glycerol
utilization rate in
media containing both glucose and glycerol as a carbon source (FIGS. 23A-23D).
The results
of this experiment demonstrate that the GUT1/GUT2 double transformant consumed
glycerol
at a faster rate than the wild-type, once glucose catabolite repression was
alleviated.
The highly lipogenic nature of some of the double GUT1/GUT2 transformants was
not
anticipated, since enhanced glycerol utilization might diminish glycerol pools
needed for
triacylglyceride synthesis. One explanation for this is that only one molecule
of glycerol is
needed for every triacylglceride formed, and even though the wild-type strain
uses glycerol as
a substrate, expression of the assimilation pathway is not optimal.
Biosynthesis of acyl carbon
chains could be enhanced by increased metabolic carbon flux due to faster
glycerol catabolism.
Overexpression of Glycerol-3-phosphate Dehydrogenase (GPD1)
Intracellular production of glycerol involves the reduction of
dihydroxyacetone
phosphate (DHAP) into glycerol-3-phosphate by the NADH dependent enzyme
glycerol-3-
phosphate dehydrogenase (GPD1). Overexpression of GPD1 could alleviate a
potential
bottleneck in glycerol formation and introduce a metabolic "push" towards
triacylglycerol
production. Hence, we constructed a GPD1 overexpression cassette driven by a
pyruvate kinase
(PYK1) promoter and the cognate PYK1 terminator. This was transformed into L.
starkeyi and
several hundred transformants were obtained, of which a subset were selected
for Bodipy
analysis in sTS. The results of this are shown in FIG. 24. The transformant
pool evaluated had
moderately higher fluorescence over the wild-type, which became progressively
more
pronounced throughout the duration of the screen. In fact, by the last day the
average
fluorescence of the transformant pool was almost 13% higher than the wild-
type. This is
counterintuitive, since by this time the cells have consumed all of the
cellobiose and most of
the glycerol present in sTS. Lipogenic effects should be observed when the
cells are consuming
cellobiose and producing DHAP to convert into glycerol, not consuming
glycerol. This could
be explained if GPD1 favors conversion of glycerol-3-phosphate into DHAP
during glycerol
consumption. The DHAP could then enter the glycolytic pathway and be used to
produce
energy, which indirectly effects lipid levels and/or cell robustness. A
similar Bodipy screen
73
CA 2982734 2017-10-17
performed on media lacking glycerol but containing glucose could provide more
insight into
this phenomenon.
Engineering the Pentose Phosphate Pathway
To test our prediction that overexpression of 6-phospholuconate dehydrogenase
(GNDI) and/or glucose-6-phosphate dehydrogenase (ZWFI) could improve lipid
production
via increasing the cytoplasmic pool of NADPH, we constructed a plasmid to
simultaneously
express both genes. The plasmid was transformed into Lipomyces, and several
hundred
transformants were obtained. Of these, 60 colonies were selected for further
screening using
our sTS and Bodipy methodology. The results of this screen are shown in FIG.
25.
We found that dual overexpression of GND1 and ZWF1 leads to moderately
improved
lipid accumulation in Lipomyces (FIG. 25). The average fluorescence readings
of all
transformants outperformed the wild-type by about 10-15% at each time point.
Two exceptional
strains, "GND1+ZWF1 37L" and "GND1+ZWF1 26L" had fluorescent values
approximately
30% and 60% higher than the wild-type, respectively. These results suggest
that when
engineered cells are grown on sTS, overexpression of GND1 and ZWF1 helps to
"pull" glucose
(derived from cellobiose or amylodextrins) towards the pentose phosphate
pathway. Catabolism
of glucose via this pathway generates more NADPH than via glycolysis, which
increases the
availability of reducing power required for lipid biosynthesis.
Generation and Use of An NHEJ-Deficient (lig4A) Strain for Further Metabolic
Engineering
In Lipomyces starkeyi, genetic transformation occurs primarily via random
integration
into the genome, likely by a non-homologous end joining (NHEJ) mechanism.
Previous
attempts to disrupt genes of interest using homologous recombination with
knockout constructs
were unsuccessful. However, it is well known that disruption of the NHEJ
pathway in a wide
range of yeast species greatly increases homologous recombination
efficiencies, and facilitates
targeted deletion or insertion of genes. Knockouts of Ku70, Ku80, and/or Lig4
have all been
commonly used to ablate NHEJ. Therefore, we sought to disrupt the DNA ligase
LIG4
(PID 2300, SEQ ID NO:73 (nucleotide) and SEQ ID NO:74 (protein)), to generate
an NHEJ-
74
CA 2982734 2017-10-17
deficient strain of Lipomyces. A strain with improved homologous recombination
could be used
to knock out members of the beta-oxidation pathway, for example, to eliminate
the ability of
Lipomyces starkeyi to consume accumulated lipids.
A LIG4 knockout construct was compiled, using a LoxP-flanked NAT resistance
construct bordered by 3kb of cloned genomic DNA homologous to the upstream
(promoter)
and downstream (terminator) regions neighboring the LIG4 gene. The
transformation rate was
very low when introduced into Lipomyces, due to the large size of the
construct (z 8.4 kb). A
total of 22 colonies were selected for further examination to determine if
they were truly LIG4
knockouts (lig4A). Following genomic DNA extraction and PCR genotyping, we
found that
only 1/22 transformants (4.5%) harbored a band consistent with replacement of
the LIG4 gene
by the NAT resistance construct (lig4::NAT). Thus, random integration
elsewhere into the
genome was the dominant mode of genetic transformation and that homologous
recombination
occurs only about 5% of the time even when very long flanking regions are
used. Subsequent
amplification and sequencing of the LIG4 locus in the putative knockout strain
confirmed that
gene replacement by homologous recombination occurred exactly as intended.
To determine the utility of this strain as a genetic tool, we designed two
additional
knockout constructs, targeting the acyl-CoA oxidase PDX] and multifunctional
enzyme (3-
hydroxyacyl-CoA dehydrogenase & enoyl-CoA hydratase) FOX] in the lig4A strain.
These
constructs utilized a LoxP-flanked HPH resistance marker, bordered by only 1
kb of cloned
genomic DNA homologous to the upstream/downstream regions neighboring each
respective
gene. Transformation of the lig4A strain with these constructs resulted in
about 70 colonies
each. From these, a total of 18 strains were selected for analysis by PCR
genotyping. Of the
PDX] transformants, we found that 7 out of 9 (78%) were confirmed as true
knockouts. As for
the FOX] transformants, only 2 out of 9 (22%) were confirmed to be true
knockouts.
Regardless, these significantly improved homology recombination rates
demonstrate that gene
deletions are much easier to achieve in the lig4A:. NAT background, and can be
accomplished
with shorter regions of homology.
CA 2982734 2017-10-17
Disruption Of The P-Oxidation Pathway in L. Starkeyi
As described above, we generated a NHEJ-deficient Lipomyces strain by gene
replacement of the DNA ligase LIG4 with a NAT resistance construct
(lig4A::NA7). Using this
strain as a platform, two additional transformations were carried out in order
to subsequently
knock out either the acyl-CoA oxidase PDX] or the multifunctional enzyme FOX]
by
homologous recombination mediated gene replacement with a LoxP-flanked HPH
construct.
The resulting strains were knockouts for both LIG4 and either PDX] or FOX]
(ligA::NAT,
poxIA::HPH), (lig4A : :NAT, foxlA : HPH).
To test our prediction that disruption of the 0-oxidation pathway could
improve lipid
accumulation, we subjected several replicates of validated poxlA or foxIA
knockouts to our
standard Bodipy screening methodology in sTS medium (FIG. 26). We found that
deletion of
either PDX] or FOX] mildly enhances lipid accumulation when cells are
incubated under
aerobic conditions. Average fluorescent readings of these knockout strains
ranged from about
5-15% higher values that the wildtype controls. These results suggest that, in
addition to
overexpressing lipogenic genes, eliminating lipid consumption by the 0-
oxidation pathway is
another viable route for improving lipid accumulation in Lipomyces starkeyi.
Overexpression Of DGA2 ¨ A Second Diacylglycerol Acyltransferase
Overexpression of diacylglycerol acyltransferases, alone or in combination
with other
lipogenic genes, is predicted to increase lipid accumulation. Unlike
Saccharomyces cerevisiae,
some oleaginous yeast species (e.g. Yarrowia hpolytica) possess two or more
protein encoding
genes with diacylglycerol acyltransferase activity. Upon searching the genome
for additional
putative DGA genes, we discovered that Lipomyces starkeyi contains a second,
previously
unknown, diacylglycerol acyltransferase gene (DGA2, PID_6231, SEQ ID NOS: 57
and 58).
The sequences of DGA1 and DGA2 are quite divergent, as DGA2 is a member of the
MBOAT
family (Membrane Bound 0-Acyl Transferase), and possesses several
transmembrane domains.
DGA1 is most likely localized to liposomes, while we predict DGA2 resides in
the endoplasmic
retieulum.
It is unknown whether DGA1 and DGA2 might have differing metabolic roles,
activities, or regulation. We therefore tested whether overexpression of the
DGA2 gene was
76
CA 2982734 2017-10-17
capable of increasing lipid production. A plasmid was designed to overexpress
DGA2 under the
control of the native Lipomyces TPI1 promoter and PGK1 terminator. Hundreds of
transformants were obtained and subjected to the Bodipy screening methodology
described
previously. In an exemplary screen, 87 individual DGA2 transformant strains
were grown and
assessed for lipid accumulation as compared to wild-type replicates. We found
that
overexpression of DGA2 leads to significantly improved lipid accumulation in
Lipomyces (FIG.
27). The average of all DGA2 transformants, which includes poor performers,
still
outperformed the wild-type by 5-10% on each time point. Some of the most
promising strains,
such as "DGA2-20L" and "DGA2-70L," had fluorescent values 20-30% higher than
the
wildtype (FIG. 27). Therefore, we have identified a second diacylglycerol
acyltransferase as an
engineering target for increasing lipid accumulation in Lipomyces starkeyi.
Presence of Trehalose in Thin Stillage and its Utilization in Lipomyces
Starkeyi
Thin stillage is a complex mixture containing a variety of short-chain
oligosaccharides
that are difficult to distinguish from each other using typical analytical
methods (e.g. HPLC).
For example, disaccharides of glucose include cellobiose, maltose, and
trehalose, which differ
only in the location of their glycosidic bonds. For simplicity, our synthetic
thin stillage medium
(sTS) includes cellobiose as the sole analog for all disaccharides which might
be present.
However, trehalose accumulates in Saccharomyces cerevisiae in response to many
environmental stress conditions, particularly as an adaptation mechanism to
ethanol tolerance.
Consequently, in the yeast ethanol industry, trehalose is found among the
residual sugars at the
termination of fermentation. Trehalose makes up a significant portion of the
remaining
disaccharide fraction, referred to as the "DP2 peak" in reference to the
degree of polymerization
of the oligosaccharides eluted together during HPLC analysis.
Therefore, complete conversion of the organics present in thin stillage
requires either
addition of a trehalase enzyme or use of organisms capable of naturally
utilizing trehalose as a
carbon source. We have observed nearly complete disappearance of the "DP2
peak" when
Lipomyces is cultured on thin stillage, which suggests that this species
natively possesses the
ability to consume the residual trehalose. We discovered two trehalases in L.
starkeyi, which
are categorized based on their predicted pH optimum: the "neutral" trehalase
NTH] (PID_4858,
77
CA 2982734 2017-10-17
SEQ ID NOS: 59 and 60), and the "acidic" trehalase ATHI (PID 4583, SEQ ID NOS:
61 and
62). Interestingly, the L. starkeyi ATH1 has a strong secretion signal, which
indicates its
usefulness in engineering strains for hyper-secretion of trehalase, or other
glycoside hydrolases.
If spent Lipomyces broth were recycled back into primary fermentation vessels,
secreted
trehalases would reduce the residual sugars present in the DP2 peak, resulting
in increased
ethanol yields and plant profitability.
Targeting Glycerol Transporters In Lipomyces starkeyi for Enhanced Glycerol
Utilization
There are multiple potential routes for glycerol assimilation in yeast. The
phosphorylative pathway, which involves the glycerol kinase GUT] and the
mitochondrial
FAD-dependent glycerol 3-phosphate dehydrogenase GUT2, ultimately generates
DHAP for
glycolysis. Simultaneous overexpression of GUT] and GUT2 improves the rate of
glycerol
utilization, as described elsewhere. Another strategy for improving glycerol
utilization is the
overexpression of transporters involved in the uptake of glycerol into the
cell. Several glycerol
transporters have been identified in yeast.
The primary route for glycerol uptake in Saccharomyces is through active
transport via
STL1, a glycerol/H+ symporter. STL1 is a member of the major facilitator
superfamily, a broad
group of transporters for many substrates, which can make identification of
specific genes
difficult. We have identified two a glycerol/H+ symporters in Lipomyces
starkeyi, referred to
as STLI (PID 114515, SEQ ID NOS:63 and 64 and STL2 (PID 201837, SEQ ID NOS:65
and
66).
A secondary route for glycerol uptake in Saccharomyces is through passive
transport by
FPS], a glycerol facilitator, which is mainly used for controlled export of
glycerol during
osmoregulation. However, FPS] homologs in other species with superior glycerol
utilization
rates (such as Pachysolentannophilus) have been shown to be the main route of
glycerol import.
We have identified a FPS] homolog in L. starkeyi (PID_ 67294, SEQ ID NOS:67
and 68).
Interestingly, the L. starkeyi FPS1 is more similar to the Pachysolen FPS1
than the
Saccharomyces FPS], so it may function as a passive glycerol transporter by
facilitated
diffusion.
78
CA 2982734 2017-10-17
We have observed that when Lipomyces is grown in the presence of both glucose
and
glycerol, glucose is preferentially utilized first due to the effects of
carbon catabolite repression.
One of the methods for controlling this response arises from transcriptional
regulation of
glycerol transporters, which are not highly expressed while glucose is
present. Transporters
unconstrained by transcriptional regulation could help to improve glycerol
uptake and enable
simultaneous utilization of glucose and glycerol. Therefore, the STL1, STL2,
and FPS1 can be
overexpressed with constitutive promoters to overcome carbon catabolite
repression and
improve glycerol utilization rates in L. starkeyi.
Engineering Constitutive Glucoamylase Secretion
Alpha-amylase and glucoamylases are major expenses in ethanol plants, required
for
the hydrolysis of starch polysaccharides into fermentable glucose monomers.
Interestingly, L.
starkeyi is known to synthesize both dextranases and amylases. We found
evidence of inducible
extracellular glucoamylase activity in the supernatant of cells cultured in
media containing
starch, but not dextrose. This activity was also eliminated by boiling,
indicating that it is the
result of one or more secreted enzymes, induced by the presence of complex
carbohydrates
(FIG. 28 (A)). If one or more of these secreted enzymes could be engineered
for high levels of
secretion in L. starkeyi, then it could serve as a valuable co-product during
the production of
lipid. To demonstrate the feasibility of this strategy, we identified a
secreted a-amylase in L.
starkeyi and engineered it for constitutive expression. The supernatant of the
resulting
transformants exhibited glucoamylase activity independent of the culture
medium, and this
activity was retained following incubation for 1 hour at 70 C (FIG. 28, (B)).
Thus, we have
achieved constitutive secretion of a relatively thermostable a-amylase in L.
starkeyi. Optimally,
this will result in a spent culture broth rich in a collection of starch
degrading enzymes with
exceptional processivity and substrate diversity that could be used in ethanol
plants to reduce
operating costs, or other related industries.
Consolidation of Transformed Traits
Combinations of gene cassettes from the top lipid producing strains described
above
further enhance lipid accumulation, and can be done through yeast mating.
Mating techniques
79
CA 2982734 2017-10-17
for Lipomyces starkeyi and isolation of spores for screening progeny enables
identification of
crossed strains with more than one cassette integrated in the genome without
needing to create
cassettes with alternate resistance markers and repeat the screening process.
Nonetheless,
integration vectors that confer resistance to G418 and Hygromycin B that are
functional in L.
starkeyi can be used. Lastly, a Lipomyces starkeyi strain that constitutively
secretes a native a-
amylase and could be mated to the DGA1-1233 transformant to obtain a strain
that converts
mTS to lipids and a-amylase. Lipids and a-amylase are products of much higher
value than the
mTS substrate, which currently presents a disposal cost.
REFERENCES
Anon, Edn. July 29, 2016 (U.S. Energy Information Administration, 2016).
Athenstaedt, K. YALI0E32769g (DGA1) and YALIOE16797g (LR01) encode major
triacylglycerol synthases of the oleaginous yeast Yarrowia lipolytica.
Biochimica Et
Biophysica Acta-Molecular and Cell Biology of Lipids 1811, 587-596 (2011).
Barcia-Vieitez, R. & Ramos-Martinez, J.I. The Regulation of the Oxidative
Phase of the
Pentose Phosphate Pathway: New Answers to Old Problems. Iubmb Life 66, 775-779
(2014).
Becker, J. et al. Metabolic flux engineering of L-lysine production in
Corynebacterium
glutamicum - over expression and modification of G6P dehydrogenase. Journal of
Biotechnology 132, 99-109 (2007).
Beopoulos, A. et al. Identification and characterization of DGA2, an
acyltransferase of the
DGAT1 acyl-CoA:diacylglycerol acyltransferase family in the oleaginous yeast
Yarrowia hpolytica. New insights into the storage lipid metabolism of
oleaginous
yeasts. Applied Microbiology and Biotechnology 93, 1523-1537 (2012).
Bignell, G.R., Bruce, I.J. & Evans, I.H. Amylolytic enzymes of Lipomyces
starkeyi: purification
and size-determination. Biotechnology Letters 22, 1713-1718 (2000).
Bligh and Dyer. A rapid method of total lipid extrµaction and purification.
Canadian Journal of
Biochemistry and Physiology 37(8):911-7 (1959).
CA 2982734 2017-10-17
Boulton, C.A. & Ratledge, C. Use of transition studies in continuous cultures
of Lipomyces
starkeyi, an oleaginous yeast, to investigate the physiology of lipid
accumulation.
Journal of general microbiology 129, 2871-2876 (1983).
Calvey, C.H., Willis, L.B. & Jeffries, T.W. An optimized transformation
protocol for
Lipomyces starkeyi. Current Genetics 60, 223-230 (2014).
Calvey, C.H., Su, Y.K., Willis, L.B., McGee, M. & Jeffries, T.W. Nitrogen
limitation, oxygen
limitation, and lipid accumulation in Lipomyces starkeyi. Bioresource
Technology 200,
780-788 (2016).
Cannella, D. & Jorgensen, H. Do New Cellulolytic Enzyme Preparations Affect
the Industrial
Strategies for High Solids Lignocellulosic Ethanol Production? Biotechnology
and
Bioengineering 111, 59-68 (2014).
Cardenas, J. & Da Silva, N.A. Engineering cofactor and transport mechanisms in
Saccharomyces cerevisiae for enhanced acetyl-CoA and polyketide biosynthesis.
Metabolic Engineering 36, 80-89 (2016).
Chen, L., Zhou, X.S., Fan, W.M. & Zhang, Y.X. Expression, purification and
characterization
of a recombinant Lipomyces starkeyi dextranase in Pichia pastoris. Protein
Expression
and Purification 58, 87-93 (2008).
Choi, J.W. & Da Silva, N.A. Improving polyketide and fatty acid synthesis by
engineering of
the yeast acetyl-CoA carboxylase. Journal of Biotechnology 187, 56-59 (2014).
Collett, J.R., Meyer, S. & Jones, S. Preliminary economics for hydrocarbon
fuel production
from cellulosic sugars. (2014).
Courchesne, N.M.D., Parisien, A., Wang, B. & Lan, C.Q. Enhancement of lipid
production
using biochemical, genetic and transcription factor engineering approaches. J
Biotechnol 141, 31-41 (2009).
Evans, C.T. & Ratledge, C. Possible regulatory roles of ATP-citrate lyase,
malic enzyme and
AMP deaminate in lipid accumulation by Rhodosporidium toruloides CBS-14.
Canadian Journal of Microbiology 31, 1000-1005 (1985).
Flores, C.L., and Gancedo, C. Yarrowia lipolytica mutants devoid of pyruvate
carboxylase
activity show an unusual growth phenotype, Eukaryot Cell 4 (2005) 356-364.
81
CA 2982734 2017-10-17
Gallagher, A.M., Kelly, C.T. & Fogarty, W.M. A novel extracellular
carbohydrase produced
by Lipomyces tetrasporus. Applied Microbiology and Biotechnology 35, 455-460
(1991).
Garay, L.A. et al. Eighteen new oleaginous yeast species. Journal of
industrial microbiology &
biotechnology 43, 887-900 (2016).
Gibson, D.G. et al. Enzymatic assembly of DNA molecules up to several hundred
kilobases.
Nature Methods 6, 343-U341 (2009).
Gietz, R. D., and R.A. Woods, Transformation of yeast by lithium
acetate/single stranded
carrier DNA/polyethylene glycol method. Methods in Enzymology 350:87-96
(2002).
Gomma, A.E., Lee, S.K., Sun, S.M., Yang, S.H. & Chung, G. Improvement in Oil
Production
by Increasing Malonyl-CoA and Glycerol-3-Phosphate Pools in Scenedesmus
quadricauda. Indian Journal of Microbiology 55, 447-455 (2015).
Gong, Z.W. et al. Co-fermentation of cellobiose and xylose by Lipomyces
starkeyi for lipid
production. Bioresource Technology 117, 20-24 (2012).
Gorner, C. et al. Genetic engineering and production of modified fatty acids
by the non-
conventional oleaginous yeast Trichosporon oleaginosus ATCC 20509. Green
Chemistry 18, 2037-2046 (2016).
Hamid, A.A., Molchtar, N.F., Taha, E.M., Omar, 0. & Yusoff, W.M.W. The role of
ATP citrate
lyase, malic enzyme and fatty acid synthase in the regulation of lipid
accumulation in
Cunninghamella sp 2A1. Annals of Microbiology 61, 463-468 (2011).
Hammond, E.G., Johnson, L.A., Su, C., Wang, T. & White, P.J. Soybean oil.
Bailey's Industrial
Oil and Fat Products (2005).
Holdsworth, J.E. & Ratledge, C. Lipid turnover in oleaginous yeasts. Journal
of General
Microbiology 134, 339-346 (1988).
Holdsworth, J.E., Veenhuis, M. & Ratledge, C. Enzyme activities in oleaginous
yeasts
accumulating and utilizing exogenous or endogenous lipids. Journal of General
Microbiology 134, 2907-2915 (1988).
Jeffries, T.W. Lipomyces starkeyi NRRL Y-11557 Genome Sequencing Project.
European
Nucleotide Archive (2013).
82
CA 2982734 2017-10-17
Kang, H.K. et al. Cloning and characterization of a dextranase gene from
Lipomyces starkeyi
and its expression in Saccharomyces cerevisiae. Yeast 22, 1239-1248 (2005).
Kildegaard, K.R. et al. Engineering and systems-level analysis of
Saccharomyces cerevisiae for
production of 3-hydroxypropionic acid via malonyl-CoA reductase-dependent
pathway.
Microbial Cell Factories 15 (2016).
Kim, Y. et al. Composition of corn dry-grind ethanol by-products: DDGS, wet
cake, and thin
stillage. Bioresource Technology 99, 5165-5176 (2008).
Lee, S.Y. et al. Demonstration of two independent dextranase and amylase
active sites on a
single enzyme elaborated by Lipomyces starkeyi KSM 22. Journal of Microbiology
and
Biotechnology 13, 313-316 (2003).
Li, Z. et al. Overexpression of malic enzyme (ME) of Mucor circinelloides
improved lipid
accumulation in engineered Rhodotorula glutinis. Appl Microbiol Biotechnol
(2012).
Lodder, J., Acomina & Kreger-Van Rij, N.J.W. The yeasts-a taxonomic study.
(1952).
Moritz, B., Striegel, K., De Graaf, A.A. & Sahm, H. Kinetic properties of the
glucose-6-
phosphate and 6-phosphogluconate dehydrogenases from Corynebacterium
glutamicum
and their application for predicting pentose phosphate pathway flux in vivo.
European
journal of biochemistry / FEBS 267, 3442-3452 (2000).
Naganuma, T., Uzuka, Y., Tanaka, K. & Iizuka, H. Differences in enzyme
activities of
Lipomyces starkeyi between cells accumulating lipid and proliferating cells.
Journal of
basic microbiology 27, 35-42 (1987).
Oguro, Y. et al. Multicopy integration and expression of heterologous genes in
the oleaginous
yeast, Lipomyces starkeyi. Bioscience Biotechnology and Biochemistry 79, 512-
515
(2015).
Ohnishi, J., Katahira, R., Mitsuhashi, S., Kakita, S. & Ikeda, M. A novel gnd
mutation leading
to increased L-lysine production in Corynebacterium glutamicum. FEMS
Microbiology
Letters 242, 265-274 (2005).
Pan, L.-X. et al. Isolation of the oleaginous yeasts from the soil and studies
of their lipid-
producing capacities. Food Technol. Biotechnol 47, 215-220 (2009).
Papanikolaou, S., Chevalot, I., Komaitis, M., Aggelis, G. & Marc, I. Kinetic
profile of the
cellular lipid composition in an oleaginous Yarrowia lipolytica capable of
producing a
83
CA 2982734 2017-10-17
cocoa-butter substitute from industrial fats. Antonie Van Leeuwenhoek
International
Journal of General and Molecular Microbiology 80, 215-224 (2001).
Punpeng, B., Nakata, Y., Goto, M., Teramoto, Y. & Hayashida, S. A novel raw-
starch digesting
yeast alpha amylase from Lipomyces starkeyi. Journal of Fermentation and
Bioengineering 73, 108-111 (1992).
Rangasamy, D. & Ratledge, C. Genetic Enhancement of Fatty Acid Synthesis by
Targeting Rat
Liver ATP:Citrate Lyase into Plastids of Tobacco. Plant Physiology 122, 1231-
1238
(2000).
Ratledge, C. Lipid biotechnology - a wonderland for the microbial
physiologist. Journal of the
American Oil Chemists Society 64, 1647-1656 (1987).
Ratledge, C. Regulation of lipid accumulation in oleaginous micro-organisms.
Biochemical
Society Transactions 30, 1047-1050 (2002).
Ratledge, C. Fatty acid biosynthesis in microorganisms being used for Single
Cell Oil
production. Biochimie 86, 807-815 (2004).
Ratledge, C. The role of malic enzyme as the provider of NADPH in oleaginous
microorganisms: a reappraisal and unsolved problems. Biotechnol. Lett. 36,
1557-1568
(2014).
Riley R, et al. (2016) Comparative genomics of biotechnologically important
yeasts.
Proceedings of the National Academy of Sciences 113(35):9882-9887.
Rippa, M., Giovannini, P.P., Barrett, M.P., Dallocchio, F. & Hanau, S. 6-
phosphogluconate
dehydrogenase: the mechanism of action investigated by a comparison of the
enzyme
from different species. Biochhnica Et Biophysica Acta-Protein Structure and
Molecular
Enzymology 1429, 83-92 (1998).
Ruenwai, R., Cheevadhanarak, S. & Laoteng, K. Overexpression of acetyl-CoA
carboxylase
gene of Mucor rouxii enhanced fatty acid content in Hansenula polymorpha. Mol
Biotechnol 42, 327-332 (2009).
Ryu, S.J. et al. Purification and partial characterization of a novel
glucanhydrolase from
Lipomyces starkeyi KSM 22 and its use for inhibition of insoluble glucan
formation.
Bioscience Biotechnology and Biochemistry 64, 223-228 (2000).
84
CA 2982734 2017-10-17
Saenge, C., Cheirsilp, B., Suksaroge, T.T. & Bourtoom, T. Potential use of
oleaginous red yeast
Rhodotorula glutinis for the bioconversion of crude glycerol from biodiesel
plant to
lipids and carotenoids. Process Biochemistry 46, 210-218 (2011).
Sitepu, et al. An improved high-throughput Nile red fluorescence assay for
estimating
intracellular lipids in a variety of yeast species. Journal of Microbiological
Methods
91:321-328 (2012).
Shi, S.B., Chen, Y., Siewers, V. & Nielsen, J. Improving Production of Malonyl
Coenzyme A-
Derived Metabolites by Abolishing Snfl -Dependent Regulation of Accl. Mbio 5
(2014).
Steyn, A.J.C., Marmur, J. & Pretorius, I.S. Cloning, sequence analysis and
expression in yeasts
of a cDNA-containing a Lipomyces kononenkoae alpha-amylase encoding gene. Gene
166, 65-71 (1995).
Tai, M. & Stephanopoulos, G. Engineering the push and pull of lipid
biosynthesis in oleaginous
yeast Yarrowia lipolytica for biofuel production. Metabolic Engineering 15, 1-
9 (2013).
Tang, W., Zhang, S., Wang, Q., Tan, H. & Zhao, Z.K. The isocitrate
dehydrogenase gene of
oleaginous yeast Lipomyces starkeyi is linked to lipid accumulation. Can J
Microbiol
55, 1062-1069 (2009).
Tang, X.L., Feng, H.X. & Chen, W.N. Metabolic engineering for enhanced fatty
acids synthesis
in Saccharomyces cerevisiae. Metabolic Engineering 16, 95-102 (2013).
Tang, X.L. & Chen, W.N. Investigation of fatty acid accumulation in the
engineered
Saccharomyces cerevisiae under nitrogen limited culture condition. Bioresource
Technology 162, 200-206 (2014).
van Rossum, H.M., Kozak, B.U., Pronk, J.T. & van Mans, A.J.A. Engineering
cytosolic acetyl-
coenzyme A supply in Saccharomyces cerevisiae: Pathway stoichiometry, free-
energy
conservation and redox-cofactor balancing. Metabolic Engineering 36, 99-115
(2016).
Velasco, P., Sieiro, A.M., Ibarguren, I., Ramosmartinez, J.I. & Barcia, R. The
Modulation of
the Oxidative Phase of the Pentose-Phosphate Pathway in Mouse Liver.
International
Journal of Biochemistry & Cell Biology 27, 1015-1019 (1995).
Vicente, G. et al. Direct transformation of fungal biomass from submerged
cultures into
biodiesel. Energy & Fuels 24, 3173-3178 (2010).
CA 2982734 2017-10-17
Wang, Z.P., Xu, H.M., Wang, G.Y., Chi, Z. & Chi, Z.M. Disruption of the MIG1
gene enhances
lipid biosynthesis in the oleaginous yeast Yarrowia lipolytica ACA-DC 50109.
Biochimica Et Biophysica Acta-Molecular and Cell Biology of Lipids 1831, 675-
682
(2013).
Wang, J.C., Xu, R.H., Wang, R.L., Hague, M.E. & Liu, A.Z. Overexpression of
ACC gene
from oleaginous yeast Lipomyces starkeyi enhanced the lipid accumulation in
Saccharomyces cerevisiae with increased levels of glycerol 3-phosphate
substrates.
Bioscience Biotechnology and Biochemistry 80, 1214-1222 (2016).
Wei, T., Sufang, Z., Qian, W., Haidong, T. & Zongbao Kent, Z. The isocitrate
dehydrogenase
gene of oleaginous yeast Lipomyces starkeyi is linked to lipid accumulation.
Canadian
Journal of Microbiology 55, 1062-1069 (2009).
Wilkie, A.C., Riedesel, K.J. & Owens, J.M. Stillage characterization and
anaerobic treatment
of ethanol stillage from conventional and cellulosic feedstocks. Biomass &
Bioenergy
19, 63-102 (2000).
Wynn, J.P., bin Abdul Hamid, A. & Ratledge, C. The role of malic enzyme in the
regulation of
lipid accumulation in filamentous fungi. Microbiology 145 ( Pt 8), 1911-1917
(1999).
Wynn, J.P., Hamid, A.B.A. & Ratledge, C. The role of malic enzyme in the
regulation of lipid
accumulation in filamentous fungi. Microbiology-Uk 145, 1911-1917 (1999).
Xuan, J.W., Fournier, P. & Gaillardin, C. Cloning of the Lys5 gene endocing
saccharopine
dehydrogenase from the yeast Yarrowia lipolytica. Current Genetics 14, 15-21
(1988).
Yen, H.-W., Yang, Y.-C. & Yu, Y.-H. Using crude glycerol and thin stillage for
the production
of microbial lipids through the cultivation of Rhodotorula glutinis. Journal
of
Bioscience and Bioengineering 114, 453-456 (2012).
Zha, J., Shen, M.H., Hu, M.L., Song, H. & Yuan, Y.J. Enhanced expression of
genes involved
in initial xylose metabolism and the oxidative pentose phosphate pathway in
the
improved xylose-utilizing Saccharomyces cerevisiae through evolutionary
engineering.
Journal of Industrial Microbiology & Biotechnology 41, 27-39 (2014).
Zhang, Y., Adams, I.P. & Ratledge, C. Malic enzyme: the controlling activity
for lipid
production? Overexpression of malic enzyme in Mucor circinelloides leads to a
2.5-fold
increase in lipid accumulation. Microbiology-Sgm 153, 2013-2025 (2007).
86
CA 2982734 2017-10-17
Zhang, Y., Wang, Z.Y., He, X.P., Liu, N. & Zhang, B.R. New industrial brewing
yeast strains
with ILV2 disruption and LSD1 expression. International Journal of Food
Microbiology 123, 18-24 (2008).
Zhang, M., Galdieri, L. & Vancura, A. The Yeast AMPK Homolog SNF1 Regulates
Acetyl
Coenzyme A Homeostasis and Histone Acetylation. Molecular and Cellular Biology
33, 4701-4717 (2013).
Zhao, X., Kong, X.L., Hua, Y.Y., Feng, B. & Zhao, Z.B. Medium optimization for
lipid
production through co-fermentation of glucose and xylose by the oleaginous
yeast
Lipomyces starkeyi. European Journal of Lipid Science and Technology 110, 405-
412
(2008).
Zhou, Y.J.J. et al. Production of fatty acid-derived oleochemicals and
biofuels by synthetic
yeast cell factories. Nature Communications 7 (2016).
Zhu, Z.W. et al. in Nature Communications, Vol. 3 (2012).
EXEMPLARY EMBODIMENTS OF THE INVENTION
Exemplary embodiments of the invention are as follows:
Embodiment 1. A recombinant yeast comprising one or more recombinant nucleic
acids
configured to express one or more proteins selected from the group consisting
of an acetyl-CoA
carboxylase, an alpha-amylase, an ATP citrate lyase, a diacylglycerol
acyltransferase, a fatty
acid synthase, a glycerol kinase, a 6-phosphogluconate dehydrogenase, a
glycerol-3-phosphate
dehydrogenase, a malic enzyme, a fatty acyl-CoA reductase, a delta-9 acyl-CoA
desaturase, a
glycerol-3-phosphate acyltransferase, a lysophosphatidate acyltransferase, a
glucose-6-
phosphate dehydrogenase, a beta-glucosidase, a hexose transporter, a glycerol
transporter, a
glycoside hydrolase enzyme, and an auxiliary activity family 9 enzyme.
Embodiment 2. The recombinant yeast of embodiment 1, wherein the one or more
recombinant nucleic acids comprises one or more recombinant genes configured
to express the
one or more proteins.
Embodiment 3. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express a glycerol kinase and a glycerol-3-phosphate dehydrogenase.
87
CA 2982734 2017-10-17
Embodiment 4. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express a diacylglycerol acyltransferase and at least one of an ATP citrate
lyase and a malic
enzyme.
Embodiment 5. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express an acetyl-CoA carboxylase comprising a sequence at least about 90%
identical to SEQ
ID NO:2.
Embodiment 6. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express an acetyl-CoA carboxylase comprising a sequence at least about 90%
identical to SEQ
ID NO:2, wherein the sequence comprises a residue other than serine and
threonine at a position
corresponding to position 1146 of SEQ ID NO:2.
Embodiment 7. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express an acetyl-CoA carboxylase comprising a sequence at least about 90%
identical to SEQ
ID NO:2, wherein the sequence comprises and a serine or threonine at a
position corresponding
to position 639 of SEQ ID NO:2 a residue other than serine and threonine at a
position
corresponding to position 1146 of SEQ ID NO:2.
Embodiment 8. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express one or more alpha-amylases comprising a sequence selected from the
group consisting
of a sequence at least about 90% identical to SEQ ID NO:4, a sequence at least
about 90%
identical to SEQ ID NO:6, and a sequence at least about 90% identical to SEQ
ID NO:8.
Embodiment 9. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express an ATP citrate lyase comprising a sequence selected from the group
consisting of a
sequence at least about 90% identical to SEQ ID NO:10 and a sequence at least
about 90%
identical to SEQ ID NO:12.
88
CA 2982734 2017-10-17
Embodiment 10. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express a diacylglycerol acyltransferase comprising a sequence selected from
the group
consisting of a sequence at least about 90% identical to SEQ ID NO:14, a
sequence at least
about 90% identical to SEQ ID NO:16, and a sequence at least about 90%
identical to SEQ ID
NO:58.
Embodiment 11. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express a diacylglycerol acyltransferase comprising a sequence at least about
90% identical to
SEQ ID NO:14 and devoid of a sequence corresponding to positions 1-52 of SEQ
ID NO:16.
Embodiment 12. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express a fatty acid synthase comprising a sequence selected from the group
consisting of a
sequence at least about 90% identical to SEQ ID NO:18, a sequence at least
about 90% identical
to SEQ ID NO:20, a sequence at least about 90% identical to SEQ ID NO:22, and
a sequence
at least about 90% identical to SEQ ID NO:24.
Embodiment 13. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express a glycerol kinase comprising a sequence selected from the group
consisting of a
sequence at least about 90% identical to SEQ ID NO:26 and a sequence at least
about 90%
identical to SEQ ID NO:28.
Embodiment 14. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express a glycerol kinase comprising a sequence at least about 90% identical
to SEQ ID NO:26
and devoid of a sequence corresponding to positions 1-5 of SEQ ID NO:28.
Embodiment 15. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express a 6-phosphogluconate dehydrogenase comprising a sequence at least
about 90%
identical to SEQ ID NO:30.
89
CA 2982734 2017-10-17
Embodiment 16. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express a glycerol-3-phosphate dehydrogenase comprising a sequence selected
from the group
consisting of a sequence at least about 90% identical to SEQ ID NO:32 and a
sequence at least
about 90% identical to SEQ ID NO:56.
Embodiment 17. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express a glycerol kinase comprising a sequence selected from the group
consisting of a
sequence at least about 90% identical to SEQ ID NO:26 and a sequence at least
about 90%
identical to SEQ ID NO:28 and a glycerol-3-phosphate dehydrogenase comprising
a sequence
at least about 90% identical to SEQ ID NO:56.
Embodiment 18. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express a malic enzyme comprising a sequence at least about 90% identical to
SEQ ID NO:34.
Embodiment 19. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express a diacylglycerol acyltransferase comprising a sequence selected from
the group
consisting of a sequence at least about 90% identical to SEQ ID NO:14, a
sequence at least
about 90% identical to SEQ ID NO:16, and a sequence at least about 90%
identical to SEQ ID
NO:58, in combination with at least one of an ATP citrate lyase comprising a
sequence selected
from the group consisting of a sequence at least about 90% identical to SEQ ID
NO:10 and a
sequence at least about 90% identical to SEQ ID NO:12 and a malic enzyme
comprising a
sequence at least about 90% identical to SEQ ID NO:34.
Embodiment 20. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express a fatty acyl-CoA reductase comprising a sequence at least about 90%
identical to SEQ
ID NO:36.
Embodiment 21. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
CA 2982734 2017-10-17
express a delta-9 acyl-CoA desaturase comprising a sequence at least about 90%
identical to
SEQ ID NO:38.
Embodiment 22. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express a glycerol-3-phosphate acyltransferase comprising a sequence at least
about 90%
identical to SEQ ID NO:40.
Embodiment 23. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express a lysophosphatidate acyltransferase comprising a sequence at least
about 90% identical
to SEQ ID NO:42.
Embodiment 24. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express a glucose-6-phosphate dehydrogenase comprising a sequence at least
about 90%
identical to SEQ ID NO:44.
Embodiment 25. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express a beta-glucosidase comprising a sequence at least about 90% identical
to SEQ ID
NO:46.
Embodiment 26. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express a hexose transporter comprising a sequence at least about 90%
identical to SEQ ID
NO:48.
Embodiment 27. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express a glycerol transporter comprising a sequence selected from the group
consisting of a
sequence at least about 90% identical to SEQ ID NO:66, a sequence at least
about 90% identical
to SEQ ID NO:66, and a sequence at least about 90% identical to SEQ ID NO:68.
Embodiment 28. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express one or more glycoside hydrolase family 5 enzymes comprising a sequence
selected
91
CA 2982734 2017-10-17
from the group consisting of a sequence at least about 90% identical to SEQ ID
NO:50 and a
sequence at least about 90% identical to SEQ ID NO:52.
Embodiment 29. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express a trehalase comprising a sequence selected from the group consisting
of a sequence at
least about 90% identical to SEQ ID NO:60 and a sequence at least about 90%
identical to SEQ
ID NO:62.
Embodiment 30. The recombinant yeast of any prior embodiment, wherein the one
or
more recombinant nucleic acids comprises one or more recombinant genes
configured to
express an auxiliary activity family 9 enzyme comprising a sequence at least
about 90%
identical to SEQ ID NO:54.
Embodiment 31. The recombinant yeast of any prior embodiment, wherein at least
one
of the one or more recombinant nucleic acids comprises a recombinant gene
comprising a
constitutive promoter or an inducible promoter.
Embodiment 32. The recombinant yeast of any prior embodiment, wherein at least
one
of the one or more recombinant nucleic acids comprise a recombinant gene
comprising a
promoter operably linked to a coding sequence of at least one of the one or
more enzymes,
wherein the promoter has a sequence selected from the group consisting of a
sequence at least
about 90% identical to any one of SEQ ID NOS:75-84.
Embodiment 33. The recombinant yeast of any prior embodiment, wherein at least
one
of the one or more recombinant nucleic acids comprise a recombinant gene
comprising a
terminator operably linked to a coding sequence of at least one of the one or
more enzymes,
wherein the terminator has a sequence selected from the group consisting of a
sequence at least
about 90% identical to any one of SEQ ID NOS:85-90.
Embodiment 34. The recombinant yeast of any prior embodiment, wherein the
yeast
exhibits increased expression of at least one of the one or more enzymes
relative to a non-
recombinant control.
Embodiment 35. The recombinant yeast of any prior embodiment, wherein the
yeast
comprises a modification that reduces or ablates the activity of one or more
native enzymes in
the yeast selected from the group consisting of a delta-9 acyl-CoA desaturase,
a glycerol-3-
92
CA 2982734 2017-10-17
phosphate dehydrogenase, an acyl-CoA oxidase, a 3-hydroxyacyl-CoA
dehydrogenase, and an
enoyl-CoA hydratase.
Embodiment 36. The recombinant yeast of any prior embodiment, wherein the
yeast
comprises a modification that reduces or ablates the activity of a native
delta-9 acyl-CoA
desaturase comprising a sequence at least about 90% identical to SEQ ID NO:38.
Embodiment 37. The recombinant yeast of any prior embodiment, wherein the
yeast
comprises a modification that reduces or ablates the activity of a native
glycerol-3-phosphate
dehydrogenase comprising a sequence at least about 90% identical to SEQ ID
NO:56.
Embodiment 38. The recombinant yeast of any prior embodiment, wherein the
yeast
comprises a modification that reduces or ablates the activity of a native acyl-
CoA oxidase
comprising a sequence at least about 90% identical to SEQ ID NO:70.
Embodiment 39. The recombinant yeast of any prior embodiment, wherein the
yeast
comprises a modification that reduces or ablates the activity of a native 3-
hydroxyacyl-CoA
dehydrogenase comprising a sequence at least about 90% identical to SEQ ID
NO:72.
Embodiment 40. The recombinant yeast of any prior embodiment, wherein the
yeast
comprises a modification that reduces or ablates the activity of a native
enoyl-CoA hydratase
comprising a sequence at least about 90% identical to SEQ ID NO:72.
Embodiment 41. The recombinant yeast of any prior embodiment, wherein the
yeast
exhibits a property selected from the group consisting of increased lipid
production, increased
lipid secretion, increased carbohydrase production, increased carbohydrase
secretion, increased
growth rate, increased glycerol consumption, increase trehalose consumption,
and increased
cellobiose consumption relative to a non-recombinant control.
Embodiment 42. The recombinant yeast of any prior embodiment, wherein the
yeast is
a recombinant lipogenic yeast.
Embodiment 43. The recombinant yeast of any prior embodiment, wherein the
yeast is
a recombinant Lipomyces starkeyi.
Embodiment 44. A method of processing comprising:
contacting a medium comprising a first organic with a yeast, wherein the yeast
consumes the first organic and produces a second organic.
93
CA 2982734 2017-10-17
Embodiment 45. The method of embodiment 44, wherein the first organic is
selected
from the group consisting of glycerol, cellobiose, xylose, lactic acid,
trehalose, and an
oligosaccharide.
Embodiment 46. The method of any one of embodiments 44-45, wherein the
contacting
reduces an amount of the first organic to less than 25% of an initial amount
in the medium.
Embodiment 47. The method of any one of embodiments 44-46, wherein the second
organic is selected from the group consisting of a lipid and a protein.
Embodiment 48. The method of any one of embodiments 44-47, wherein the second
organic is an enzyme.
Embodiment 49. The method of any one of embodiments 44-48, further comprising,
after the contacting, separating at least a portion of a component selected
from the group
consisting of lipid produced by the yeast, enzymes produced by the yeast, and
the yeast from at
least a portion of one other component of spent medium resulting from the
contacting.
Embodiment 50. The method of any one of embodiments 44-49, further comprising,
after the contacting, conducting a process selected from the group consisting
of liquefaction of
starch and saccharification of liquified starch with enzymes produced by the
yeast.
Embodiment 51. The method of any one of embodiments 44-50, further comprising,
after the contacting, mixing spent medium resulting from the contacting with
starch and
conducting liquefaction of the starch in the presence of the spent medium.
Embodiment 52. The method of any one of embodiments 44-51, wherein the medium
comprises a component selected from the group consisting of glucose, glucan,
trehalose, xylose,
xylan, arabinose, arabinan, lactic acid, glycerol, acetic acid, butanediol,
and ethanol.
Embodiment 53. The method of any one of embodiments 44-52, wherein the medium
comprises a component selected from the group consisting of glucose in an
amount of from
about 0.1 g/L to about 10 g/L, glucan in an amount of from about 1 g/L to
about 100 g/L, xylose
in an amount of from about 0.1 g/L to about 10 g/L, trehalose in an amount of
from about 0.01
g/L to about 100 g/L, xylan in an amount of from 0.5 g/L to about 50 g/L,
arabinose in an
amount of from about 0.05 g/L to about 5 g/L, arabinan in an amount of from
about 0.05 g/L to
about 5 g/L, lactic acid in an amount of from about 1.5 g/L to about 150 g/L,
glycerol in an
amount of from about 1.5 g/L to about 150 g/L, acetic acid in an amount of
from about 0.05
94
CA 2982734 2017-10-17
g/L to about 5 g/L, butanediol in an amount of from about 0.2 g/L to about 20
g/L, and ethanol
in an amount of from about 0.05 g/L to about 5 g/L.
Embodiment 54. The method of any one of embodiments 44-53, wherein the medium
comprises a grain ethanol distillation stillage or a processed grain ethanol
distillation stillage.
Embodiment 55. The method of any one of embodiments 44-45, wherein the medium
comprises a processed grain ethanol distillation stillage made by processing
grain ethanol
distillation stillage with a step selected from the group consisting of
centrifuging, removing oil,
and concentrating.
Embodiment 56. The method of any one of embodiments 44-54, wherein the yeast
is a
lipogenic yeast.
Embodiment 57. The method of any one of embodiments 44-55, wherein the yeast
is a
non-genetically modified lipogenic yeast.
Embodiment 58. The method of any one of embodiments 44-56, wherein the yeast
is
non-genetically modified Lipomyces starkeyi.
Embodiment 59. The method of any one of embodiments 44-57, wherein the yeast
is a
recombinant yeast as recited in any one of embodiments 1-43.
CA 2982734 2017-10-17