Note: Descriptions are shown in the official language in which they were submitted.
1
DRUG DELIVERY DEVICE FOR PHARMACEUTICAL COMPOSITIONS
Field of the Invention
The present invention relates to a dual-chamber pack with a first chamber
comprising a container; and a second chamber comprising a reservoir, a
biphasic
connector, a plunger, and a plug with a breakable polymeric membrane. The
container of
the first chamber is prefilled with a pharmaceutically acceptable vehicle and
the reservoir
of the second chamber is prefilled with a solid composition of an active
ingredient,
wherein the solid composition of the active ingredient is mixed with the
pharmaceutically
acceptable vehicle to form a liquid pharmaceutical composition upon activation
of the
dual-chamber pack.
Background of the Invention
Liquid pharmaceutical compositions are convenient dosage forms for oral
administration particularly for geriatric and pediatric patients in comparison
to solid
dosage forms such as tablets and capsules. They are easy to administer which
leads to
enhanced patient compliance. Additionally, liquid pharmaceutical compositions
provide a
unique advantage of having a flexible dosing regimen. Liquid pharmaceutical
compositions are also preferred over solid dosage forms in case of high-dose
drugs
considering the size and shape requirements imposed by various regulatory
authorities
worldwide and the swallowability of the dosage form. Liquid pharmaceutical
compositions are generally in the form of a solution, emulsion or a
suspension, wherein
the active ingredient remains in the dissolved or dispersed form in a
pharmaceutically
acceptable vehicle such as water.
However, some of the active ingredients remain unstable in the presence of
pharmaceutically acceptable vehicle such as water when stored for a prolonged
period of
time. To overcome this, the active ingredients are mostly formulated as a dry
powder
which is to be reconstituted with the pharmaceutically acceptable vehicle at
the time of
administration. The reconstitution is done by the end user, wherein the dry
powder is
dissolved or suspended in a household pre-boiled and cooled water to form a
liquid
pharmaceutical composition. Alternatively, the pharmaceutically acceptable
vehicle or
purified water is supplied separately along with the bottle having the dry
powder. This
CA 2989925 2017-12-22
2
conventional pack lacks patient compliance and may lead to contamination due
to
improper quality of water. Further, there remains a possibility of dosing
errors if the
pharmaceutically acceptable vehicle or water is not added to the marked level.
U.S. Patent No. 3,156,369; U.S. Patent No. 3,603,469; U.S. Patent No.
3,840,136;
and U.S. Patent No. 4,982,875 disclose the use of dual-chamber packs for
separately
storing two compositions in two compartments which can be admixed at the time
of use.
The two compartments are separated by a breakable membrane which is ruptured
by the
depression of a plunger so that the one composition gets released into another
and is
mixed. However, there remains a possibility that the membrane fragments may
get
detached and fall into the final product. This may lead to undesirable
contamination and
can pose serious health hazards. Furthermore, the dual-chamber packs disclosed
in the
prior art have a limited capacity for the compartments which may not be
suitable for high-
dose drugs or for drugs which require chronic administration. Also, the liquid
composition
may get permeated into the solid composition across the membrane during
storage which
can lead to the agglomeration of the solid composition. This may result in
poor flow of the
solid composition, thus affecting the content uniformity of the final product.
Also, the
liquid composition on permeation can affect the stability of moisture-
sensitive drugs.
The present invention provides a patient compliant dual-chamber pack with a
significant improvement over the prior art and which fulfills the unmet need
of
incorporating variety of drugs. The present dual-chamber pack can be suitable
for any
class of drugs including the high-dose drugs, drugs requiring chronic
administration, or
moisture-sensitive drugs. Multi-dose liquid compositions can be conveniently
administered using this pack. Further, the plunger used in the pack of the
instant invention
is designed in a way such that the breakable membrane remains adhered to the
plug at the
time of activation and membrane fragments do not fall into the final product.
During
activation, the pack ensures that the final product remains safe for the use
of patients. The
pack also ensures that the solid composition is completely released into the
liquid
composition thereby maintaining the content uniformity of the final product.
Further, the
pack also ensures that there is no permeation of moisture into the chamber
having solid
composition comprising the active ingredient, and the stability of the active
ingredient
remains unaffected during storage.
CA 2989925 2017-12-22
3
Summary of the Invention
The present invention relates to a dual-chamber pack with a first chamber
comprising a container; and a second chamber comprising a reservoir, a
biphasic
connector, a plunger, and a plug with a breakable polymeric membrane. The
container of
the first chamber is prefilled with a pharmaceutically acceptable vehicle and
the reservoir
of the second chamber is prefilled with a solid composition of an active
ingredient,
wherein the solid composition of the active ingredient is mixed with the
pharmaceutically
acceptable vehicle to form a liquid pharmaceutical composition upon activation
of the
dual-chamber pack. The pack allows the end-users ease of dispensing with only
a few
simple steps required for reconstitution. The pack is suitable for drugs
required for chronic
administration, high-dose drugs, and moisture-sensitive drugs. The pack
ensures that the
solid composition falls completely into the pharmaceutically acceptable
vehicle thereby
maintaining the content uniformity. The pack also ensures that final product
remains free
of any contamination from the pack components and is safe to the end-users.
Further, the
pack ensures the stability of the active ingredient during storage.
Brief Description of the Drawings
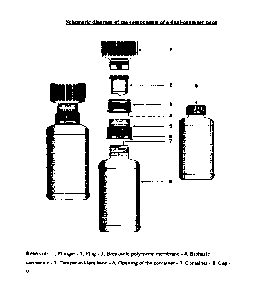
Figure 1: Schematic diagram of the components of a dual-chamber pack
Figure 2: Schematic diagram for the biphasic connector ¨ top view and front
view
Figure 3: Schematic diagram representing the assembly of a dual-chamber pack
Figure 4: Schematic diagram representing the functioning of a dual-chamber
pack
Figure 5: Schematic diagram of the components of a drug delivery device
Figure 6: Schematic diagram of the components of a dual-chamber pack with a
powder for suspension filled in the reservoir
Detailed Description of the Invention
CA 2989925 2017-12-22
4
A first aspect of the invention provides a dual-chamber pack comprising:
(a) a first chamber comprising a container; and
(b) a second chamber comprising a reservoir, a biphasic connector, a
plunger,
and a plug with a breakable polymeric membrane.
According to one embodiment of the above aspect, the container of the first
chamber is prefilled with a pharmaceutically acceptable vehicle and the
reservoir of the
second chamber is prefilled with a solid composition of an active ingredient.
Alternatively,
the reservoir of the second chamber is prefilled with a liquid concentrate
composition of
an active ingredient.
According to another embodiment of the above aspect, the solid composition is
mixed with the pharmaceutically acceptable vehicle to form a liquid
pharmaceutical
composition upon activation of the dual-chamber pack.
According to another embodiment of the above aspect, the liquid pharmaceutical
composition is a solution or a suspension.
According to another embodiment of the above aspect, the dual chamber pack is
used for multi-dose administration of the liquid pharmaceutical composition.
According to another embodiment of the above aspect, the reservoir of the
second
chamber is prefilled with the solid composition in a volume greater than about
40 cc. In a
preferred embodiment of above aspect, the reservoir of the second chamber is
prefi lied
with the solid composition in a volume ranging from about 40 cc to about 500
cc.
According to another embodiment of the above aspect, the biphasic connector of
the second chamber connects the reservoir to the container of the first
chamber.
According to another embodiment of the above aspect, the plunger ensures the
breakable polymeric membrane remains attached to the plug during activation.
According to another embodiment of the above aspect, the plunger comprise of
one
or more sharp projections with an essential continuous blunt area. In a
preferred
embodiment, the plunger comprise of one sharp projection with an essential
continuous
blunt area. The plunger can further have one or more grooves. The body of the
plunger can
be in the form of a cylinder or a funnel.
CA 2989925 2017-12-22
5
According to another embodiment of the above aspect, the plug is made up of
polymeric materials selected from the group comprising polyolefin,
polyethylene,
polypropylene, polyvinyl chloride, cyclic olefin polymer, cyclic olefin co-
polymer,
polyethylene terephthalate, polyethylene terephthalate - G, polypropylene, and
polycarbonate. In a preferred embodiment, the plug is made up of polyethylene.
According to another embodiment of the above aspect, the plug additionally
includes one or more moisture barrier additives.
According to another embodiment of the above aspect, the moisture barrier
additives are selected from the plastic additive group comprising of monomers
and co-
polymers that get activated through polymerization process to form an
effective organic
chemical.
According to another embodiment of the above aspect, the moisture barrier
additives improve the moisture barrier properties by up to 50%. In particular,
the moisture
barrier additives improve the moisture barrier properties by up to 30%.
According to another embodiment of the above aspect, the plug with the
breakable
polymeric membrane prevents moisture permeation from the first chamber into
the second
chamber.
According to another embodiment of the above aspect, the liquid pharmaceutical
composition is a stable composition.
According to another embodiment of the above aspect, the liquid pharmaceutical
composition is a taste-masked composition.
A second aspect of the present invention provides a dual-chamber pack
comprising:
a) a first chamber in the form of a container (8) prefilled with a
pharmaceutically acceptable vehicle provided with an opening (7) at an
upper end;
b) a second chamber comprising:
CA 2989925 2017-12-22
6
(i) a reservoir (I) adapted to fit into a plunger (2) prefilled with a
solid
composition of an active ingredient; the plunger (2) is further
adapted to fit into a plug (3) having a top flat surface,
(ii) the plug (3), with a breakable polymeric membrane (4), adapted to
fit into the biphasic connector (5) optionally having a tamper
evident band (6) which is further connected from the lower end to
the opening (7) of the container (8);
wherein the reservoir (1) at the top of the second chamber has a means to
exert pressure
onto the plunger (2) so as to partially rupture the breakable polymeric
membrane (4) of the
plug and deliver the solid composition into the pharmaceutically acceptable
vehicle of the
container (8); the second chamber is replaced with a cap (9), and wherein the
solid
composition is mixed with the pharmaceutically acceptable vehicle to form a
liquid
pharmaceutical composition.
According to one embodiment of the above aspect, the reservoir of the second
chamber is prefilled with the solid composition in a volume greater than about
40 cc. In a
preferred embodiment of above aspect, the reservoir of the second chamber is
prefilled
with the solid composition in a volume ranging from about 40 cc to about 500
cc.
According to another embodiment of the above aspect, the cap is a conventional
cap or a child-resistant cap.
According to another embodiment of the above aspect, the biphasic connector
has
a tamper evident band on the side connected to the container of the first
chamber and
grooves on another side for locking with the reservoir of the second chamber.
According to another embodiment of the above aspect, the plunger is opened at
both the ends.
According to another embodiment of the above aspect, the reservoir exerts
pressure onto the plunger when it is screwed during the activation of the dual-
chamber
pack.
A third aspect of the present invention provides a method of providing a
liquid
pharmaceutical composition stored in a dual-chamber pack, comprising the steps
of:
CA 2989925 2017-12-22
7
(a) providing a first chamber comprising a container (8), a second chamber
comprising a reservoir (1), a plunger (2), a plug (3) with a breakable
polymeric membrane (4), and a biphasic connector (5);
(b) prefilling the container (8) of the first chamber with a
pharmaceutically
acceptable vehicle to form a first chamber;
(c) prefilling a reservoir (1) of the second chamber with a solid
composition;
(d) fixing the biphasic connector (5) into the reservoir (1);
(e) fixing the plunger (2) in the biphasic connector (5);
(f) mounting the plug (3) onto the plunger of the biphasic connector (5) to
form the second chamber;
(g) mounting the second chamber onto the opening (7) of the container (8)
of
the first chamber;
(h) activating the dual-chamber pack by screwing the reservoir (1) of the
second chamber so that the plunger partially ruptures the circumference of
a breakable polymeric membrane; and
(i) removing the second chamber and replacing it with a cap (9); and
shaking the container (8) to allow the mixing of the solid composition with
the pharmaceutically acceptable vehicle to obtain the liquid pharmaceutical
composition.
According to one embodiment of the above aspect, the reservoir of the second
chamber is prefilled with the solid composition in a volume greater than about
40 cc. In a
preferred embodiment of above aspect, the reservoir of the second chamber is
prefilled
with the solid composition in a volume ranging from about 40 cc to about 500
cc.
According to another embodiment of above aspect, the biphasic connector has a
tamper evident band on the side connected to the container of the first
chamber and
grooves on another side for locking with the reservoir of the second chamber.
The tamper
evident band is removed first to start the activation process.
CA 2989925 2017-12-22
8
The active ingredient used to form a solid composition of the present
invention
may be present in a form to provide an immediate release, delayed release or
an extended
release. The solid composition may comprise of an active ingredient directly
mixed with
one or more pharmaceutically acceptable excipients. Alternatively, the solid
composition
may comprise of cores of an active ingredient, optionally admixed with one or
more
pharmaceutically acceptable excipients. The cores may be coated with an
immediate
release or an extended release coating. The immediate release coating may
comprise a
film-forming agent to mask the taste of bitter active ingredients or to
improve the stability.
Said coating remains insoluble in the reconstituted liquid pharmaceutical
composition
during storage and releases the active ingredient only once ingested. The film-
forming
agent can be a water-soluble polymer in which the release of active ingredient
is prevented
by using a high molar concentration of the solutes in the reconstituted
composition,
wherein the solutes have a higher affinity towards water. The high molar
concentration of
the solutes generates hypertonic conditions leading to high osmolality and
thus prevents
the leaching of the active ingredient from the coated cores. This would help
to mask the
taste of the bitter active ingredients or to improve the stability of active
ingredients.
Further, the film-forming agent can be having a pH-dependent solubility in
which the
release of active ingredient is prevented by using a pre-adjusted pH of the
reconstituted
composition such that the film-forming agent does not get dissolved in the
reconstituted
composition but get dissolved when exposed to the physiological conditions.
Alternatively, the solid composition comprises of active ingredient in a
complexed form
such as ion-exchange resin complex or a cyclodextrin complex, optionally
admixed with
one or more pharmaceutically acceptable excipients. In this case, the active
ingredient is
released when exposed to the physiological conditions upon ingestion. The
extended
release coating may comprise of a pH-dependent release-controlling agent, a pH-
independent release-controlling agent, or mixtures thereof.
Suitable examples of pH-dependent release-controlling agents are selected from
the group comprising acrylic copolymers such as methacrylic acid and methyl
methacrylate copolymers, e.g., Eudragit L 100 and Eudragit S 100,
methacrylic acid and
ethyl acrylate copolymers, e.g., Eudragit L 100-55 and Eudragit L 30 D-55,
dimethylaminoethyl methacrylate and butyl methacrylate and methyl methacrylate
copolymers e.g., Eudragit E 100, Eudragit E PO, methyl acrylate and
methacrylic acid
CA 2989925 2017-12-22
9
and octyl acrylate copolymers, styrene and acrylic acid copolymers, butyl
acrylate and
styrene and acrylic acid copolymers, and ethylacrylate-methacrylic acid
copolymer;
cellulose acetate phthalate; cellulose acetate succinates; hydroxyalkyl
cellulose phthalates
such as hydroxypropylmethyl cellulose phthalate; hydroxyalkyl cellulose
acetate
succinates such as hydroxypropylmethyl cellulose acetate succinate; vinyl
acetate
phthalates; vinyl acetate succinate; cellulose acetate trimelliate; polyvinyl
derivatives such
as polyvinyl acetate phthalate, polyvinyl alcohol phthalate, polyvinyl
butylate phthalate,
and polyvinyl acetoacetal phthalate; zein; shellac; and mixtures thereof.
Suitable examples of pH-independent release-controlling agents are selected
from
the group comprising cellulosic polymers such as ethyl cellulose, methyl
cellulose,
hydroxyethyl cellulose, hydroxypropyl cellulose, hydroxyethylmethyl cellulose,
hydroxypropylmethyl cellulose, and carboxy methylcellulose; acrylic copolymers
such as
methacrylic acid copolymers, e.g., Eudragit RS, Eudragit RL, Eudragit NE 30
D;
cellulose acetate; polyethylene derivatives e.g., polyethylene glycol and
polyethylene
oxide; polyvinyl alcohol; polyvinyl acetate; gums e.g., guar gum, locust bean
gum,
tragacanth, carrageenan, alginic acid, gum acacia, gum arabic, gellan gum, and
xanthan
gum; triglycerides; waxes, e.g., Compritol , Lubritab , and Gelucires ;
lipids; fatty acids
or their salts/derivatives; a mixture of polyvinyl acetate and polyvinyl
pyrrolidone, e.g.,
Kollidon SR; and mixtures thereof.
The term "liquid concentrate composition," as used herein refers to a
concentrated
liquid composition comprising an active ingredient which upon reconstitution
gives the
desired strength.
According to another embodiment of the above aspects, the core is in the form
of a
bead, a pellet, a granule, a spheroid, or the like.
According to another embodiment of the above aspects, the active ingredient is
layered onto an inert particle to form the core.
A fourth aspect of the present invention provides a drug delivery device for
the in situ
preparation of an extended release oral suspension upon activation of the
device, the
device comprising:
a) a first chamber comprising a suspension base;
CA 2989925 2017-12-22
10
b) a second chamber comprising a solid composition comprising cores of active
ingredient coated with a release controlling agent to form coated cores; and
c) a breakable substantially impermeable polymeric membrane separating first
and
second chamber
wherein, the solid composition remains stable when stored at 40 C/75% RH for
at least
three months.
According to one embodiment of the above aspect, the breakable substantially
impermeable polymeric membrane has moisture vapor transmission rate less than
about
5.0 g/m2/day. More preferably, the breakable substantially impermeable
polymeric
membrane has moisture vapor transmission rate less than about 1.0 g/m2/day.
According to another embodiment of the above aspect, the breakable
substantially
impermeable polymeric membrane prevents intimate contact between the contents
of first
and second chamber before activation.
According to another embodiment of the above aspect, the device ensures free
flow
of the coated cores from second chamber to first chamber upon activation of
the device.
According to another embodiment of the above aspect, the breakable
substantially
impermeable polymeric membrane is made up of a polymeric material selected
from the
group consisting of polyethylene (PE), low density polyethylene (LDPE), linear
low
density polyethylene (LLDPE), high density polyethylene (HDPE) and high
barrier grade
PE.
According to another embodiment of the above aspect, the breakable
substantially
impermeable polymeric membrane has a thickness not less than 0.10 mm.
According to another embodiment of the above aspect, the second chamber
comprises a plunger for storing the solid composition and a plug fitted with a
breakable
substantially impermeable polymeric membrane.
According to another embodiment of the above aspect, the solid composition
prefilled in the second chamber is present in a volume ranging from about 0.5
cc to about
40 cc.
CA 2989925 2017-12-22
11
According to another embodiment of the above aspect, the first chamber
comprises
a container and the second chamber comprises an overcap, a plunger, and a plug
with a
breakable substantially impermeable polymeric membrane. The plunger is
prefilled with
the solid composition in a volume ranging from about 0.5 cc to about 40 cc.
According to another embodiment of the above aspect, after activation of the
device, not more than about 50% of the circumference of the breakable
substantially
impermeable polymeric membrane remains attached to the plug to allow free flow
of the
coated cores from second chamber to first chamber. Preferably, after
activation of the
device, not more than about 30% of the circumference of the breakable
substantially
impermeable polymeric membrane remains attached to the plug to allow free flow
of the
coated cores from second chamber to first chamber. More preferably, after
activation of
the device, not more than about 15% of the circumference of the breakable
substantially
impermeable polymeric membrane remains attached to the plug to allow free flow
of the
coated cores from second chamber to first chamber.
According to another embodiment of the above aspect, the plunger comprises one
or more sharp projections with an essential continuous blunt area at an angle
of not more
than about 60 .
According to another embodiment of the above aspect, the drug delivery device
comprises:
(a) a first chamber in the form of a container (7) provided with an opening
(6) at an upper
end, comprising a suspension base containing one or more pharmaceutically
acceptable
inert excipients;
(b) a second chamber comprising:
(i) a plunger (3) adapted to fit into a plug (4) having a top flat surface,
containing a solid
composition comprising cores of active ingredient coated with a release
controlling agent
to form coated cores; and
(ii) the plug (4), with a breakable substantially impermeable polymeric
membrane (5),
adapted to fit into the opening (6) from a lower end and into a cap (I) from
the upper end;
and
CA 2989925 2017-12-22
12
(c) the cap (1) over the second chamber comprising a means to exert pressure
onto the
plunger (3) so as to partially rupture the breakable substantially impermeable
polymeric
membrane (5) of the plug (4) and deliver the solid composition into the
container (7)
wherein the compositions of both chambers are mixed at the time of first
administration by
applying pressure on the cap (1) to in situ form an extended release oral
liquid suspension.
A fifth aspect of the present invention provides an extended release
reconstituted
powder for suspension composition comprising:
a) cores comprising an active ingredient selected from group consisting of a
high-
dose, a low-dose, a water-soluble and a water-insoluble active ingredient; and
b) a coating layer over the core comprising not more than one functional
coating layer
comprising a pH-independent release-controlling agent to form the coated cores
wherein the composition after reconstitution does not settle and exhibit a
sedimentation
volume of about 1 after about at least twelve hours after reconstitution.
According to one embodiment of the above aspects, the composition after
reconstitution does not settle after about at least one month.
According to another embodiment of the above aspects, the coated cores exhibit
an
angle of repose less than about 400.
According to another embodiment of the above aspects, the coated cores exhibit
desired flowability.
According to another embodiment of the above aspects, the coated cores exhibit
a
sphericity (SPHT3) value more than about 0.7 when measured using CamSizer
particle
analyzer from Retsch Technology.
According to another embodiment of the above aspects, the composition is
characterized by having water activity of suspension base sufficiently low to
prevent
growth of Burkholderia cepacia complex.
According to another embodiment of the above aspects, the composition is
characterized by having water activity of suspension base of less than about
0.9.
Preferably, the composition is characterized by having water activity of
suspension base of
about 0.88. Water activity was determined by Rotronic hygropalm.
CA 2989925 2017-12-22
13
According to another embodiment of the above aspects, the composition provides
uniform dose of the active ingredient and has a viscosity ranging from about
500 cps to
about 15,000 cps. Preferably, the viscosity of the composition ranges from
about 1,000 cps
to about 13,000 cps. More preferably, the viscosity of the composition ranges
from about
1300 cps to about 12,000 cps. The viscosity of the composition of the present
invention is
measured by using a Brookfield Viscometer.
A sixth aspect of the present invention provides an extended release powder
for
suspension composition of active ingredient comprising cores of active
ingredient coated
with a release-controlling agent to form coated cores wherein, the coated
cores upon
reconstitution with the suspension base form a suspension which is
characterized by
having no significant leaching of active ingredients from the extended release
coated cores
when placed in a medium having a pH ranging from about 1.5 to about 10.
A seventh aspect of the present invention provides a drug delivery device for
the in
situ preparation of an immediate release oral liquid composition upon
activation of the
device, the device comprising:
a) a first chamber comprising a vehicle;
b) a second chamber comprising a solid composition; and
c) a breakable substantially impermeable polymeric membrane separating first
and
second chamber
wherein solid composition is in the form of an immediate release powder.
The dual chamber pack of the present invention is suitable for multi-dose
administration of the active ingredient. The liquid pharmaceutical composition
ofthe
present invention is in the form of a suspension or a solution.
The pharmaceutically acceptable vehicle of the instant invention may comprise
of
purified water, one or more suitable organic solvents, and mixtures thereof.
The organic
solvents may be selected from the group consisting of ethanol, glycerin,
propylene glycol,
polyethylene glycol, and mixtures thereof. The pharmaceutically acceptable
vehicle may
optionally have one or more pharmaceutically acceptable excipients.
CA 2989925 2017-12-22
14
The term "activation," as used herein means a process which reconstitutes the
solid
composition with the pharmaceutically acceptable vehicle to form a liquid
pharmaceutical
composition. The activation can be done by the end-users such as patients or
pharmacists
or caregiver. The activation process starts by screwing the reservoir.
The term "multi-dose" as used herein, means the liquid pharmaceutical
composition is to be administered in multiple doses after reconstitution, over
a period of
time e.g., for more than seven days, or more than a month, or more than three
months.
The term "about" as used herein, refers to any value which lies within the
range
defined by a variation of up to +10% of the value.
The term "stable," as used herein, refers to chemical stability, wherein not
more
than 5% w/w of total related substances are formed on storage at 40 C and 75%
relative
humidity (R.H.) or at 25 C and 60% R.H. for a period of at least three months
to the extent
necessary for the sale and use of the composition.
The term "pharmaceutically acceptable excipients," as used herein, refers to
excipients that are routinely used in pharmaceutical compositions. The
pharmaceutically
acceptable excipients may comprise glidants, sweeteners, suspending agents,
anti-caking
agents, wetting agents, preservatives, buffering agents, flavoring agents,
anti-oxidants,
chelating agents, solutes, and combinations thereof.
The term "extended release," as used herein, refers to a release profile of
active
ingredient over an extended period of time, e.g., over a period of 0.5, 2, 4,
6, 8, 12, 24
hours, or more.
The term "substantial," as used herein refers to any value which lies within
the
range as defined by a variation of up to +15 from the average value.
The term "suspension base," as used herein, refers to a medium which is used
to
suspend the coated cores of the active ingredient. The suspension base
comprises a
pharmaceutically acceptable vehicle, one or more osmogents, and
pharmaceutically
acceptable excipients. The powder for suspension having coated cores of active
ingredient
of the present invention may be reconstituted with the suspension base having
osmogents,
pharmaceutically acceptable excipients, and a pharmaceutically acceptable
vehicle.
Alternatively, osmogents and pharmaceutically acceptable excipients may be
mixed with
CA 2989925 2017-12-22
15
the coated cores of active ingredient which may then be reconstituted with a
pharmaceutically acceptable vehicle. The suspension base of the present
invention does
not include a saturated solution of active ingredient.
The term "inert particle," as used herein, refers to a particle made from a
sugar
sphere also known as a non-pareil seed, a microcrystalline cellulose sphere, a
dibasic
calcium phosphate bead, a mannitol bead, a silica bead, a tartaric acid
pellet, a wax based
pellet, and the like.
The term "Sphericity," as used herein, refers to the closeness of the shape of
an
object to that of a mathematically perfect sphere. A perfectly spherical
particle has a
sphericity (SPHT3) value of 1. The cores of active ingredient coated with a
release-
controlling agent have sphericity (SPHT3) value more than about 0.7 when
measured using
CamSizer particle analyzer from Retsch Technology.
The term "Angle of repose (AoR)," as used herein, refers to the angle assumed
by
a cone-like pile of the material relative to a horizontal base upon which it
has been poured.
The cores of active ingredient coated with a release-controlling agent exhibit
an angle of
repose less than about 400.
The term "Hausner Ratio (HR)," as used herein, refers to the unsettled volume
divided by the tapped volume (that is the volume after tapping produces no
further change
in volume), or alternatively the tapped density divided by the bulk density.
The cores of
active ingredient coated with a release-controlling agent exhibit Hausner
ratio less than
about 1.25.
The term "Carr's Compressibility Index (CI)," as used herein, can be
calculated
from the Hausner ratio (HR) as Cl = 100 x [1-(1/HR)]. The cores of active
ingredient
coated with a release-controlling agent exhibit Can's Compressibility Index
less than
about 20.
The term "Desired flowability," as used herein, refers to the uniformity of
fill
weight of the coated cores. In other words, second chamber after being filled
coated cores
of the present invention, exhibit weight variation within the range of about +
7.5%.
The term "Sedimentation volume (Suspendibility, F)," as used herein, refers to
the
ratio of the final or ultimate volume (or height) of the sediment, Vu (or Hu)
to the original
CA 2989925 2017-12-22
16
volume (or height) of the suspension, Vo, (or Ho), before settling. Thus,
F=Vu/V0 (or
Hu/Ho].
The term "Water activity (aw)," as used herein, refers to the measurement of
water
vapor pressure generated by the free or non-chemically bound water.
Compositions with
high water activity support growth of microorganisms. One of ways to control
microbial
contamination is to formulate a low water activity composition. The
reconstituted
composition of the present invention is characterized by having water activity
of
suspension base sufficiently low to prevent growth of Burkholderia cepacia
complex.
The term "High-dose" as used herein, refers to an active ingredient having
dose
more than or equal to about 250 mg.
The term "Low-dose" as used herein, refers to an active ingredient having dose
less than about 250 mg.
The term "Water-soluble" as used herein, refers to an active ingredient which
requires less than about 1,000 parts of solvent for dissolution of one part of
solute.
The term "Water-insoluble" as used herein, refers to an active ingredient
which
requires? about 1,000 parts of solvent for dissolution of one part of solute.
The term "Substantially impermeable polymeric membrane" as used herein, refers
to a polymeric membrane having moisture vapor transmission rate less than
about 5.0
g/m2/day.
According to one embodiment of the above aspect, the breakable substantially
impermeable polymeric membrane has moisture vapor transmission rate in the
range of
about 0.8 to about 0.9 g/m2/day.
The average diameter (Dso) of the coated cores ranges from about 10 i_tm to
about
2000 um, particularly from about 100 pm to about 1000 tim, and more
particularly from
about 100 [tm to about 500 um when measured using CamSizer particle analyzer
from
Retsch Technology. The finer sizes of the cores help in avoiding grittiness in
the mouth
and are therefore more acceptable.
This dual-chamber pack can be used for a soluble, a water-insoluble, or a
poorly-
soluble active ingredient. The active ingredient may have a stability problem
due to which
CA 2989925 2017-12-22
'7
the active ingredient is reconstituted using a pharmaceutically acceptable
vehicle at the
time of administration. This dual-chamber pack can be used for active
ingredients such as
valacyclovir, metformin, azithromycin, cloxacillin, clarithromycin,
erythromycin,
amoxicillin alone or in combination with clavulanic acid, cefdinir, cefuroxime
axetil,
cefixime, cefadroxil, cefpodoxime, cefaclor, cefprozil, fluconazole,
voriconazole,
acarbose, miglitol, voglibose, repaglinide, nateglinide, glibenclamide,
glimepride,
glipizide, gliclazide, chloropropamide, tolbutamide, phenformin, alogliptin,
sitagliptin,
linagliptin, saxagliptin, rosiglitazone, pioglitazone, troglitazone,
faraglitazar, englitazone,
darglitazone, isaglitazone, zorglitazone, liraglutide, muraglitazar,
peliglitazar, tesaglitazar,
canagliflozin, dapagliflozin, remogliflozin, sergliflozin, verapamil,
albuterol, salmeterol,
acebutolol, sotalol, penicillamine, norfloxacin, ciprofloxacin, ofloxacin,
levofloxacin,
moxifloxacin, trovafloxacin, gatifloxacin, tetracycline, demeclocycline
hydrochloride,
losartan, irbesartan, eprosartan, valsartan, diltiazem, isosorbide
mononitrate, ranolazine,
propafenone, hydroxyurea, hydrocodone, delavirdine, pentosan polysulfate,
abacavir,
amantadine, acyclovir, ganciclovir, valganciclovir, saquinavir, indinavir,
nelfinavir,
lamivudine, didanosine, zidovudine, nabumetone, celecoxib, mefenamic acid,
naproxen,
propoxyphene, cimetidine, ranitidine, albendazole, mebendazole, thiobendazole,
pyrazinamide, praziquantel, chlorpromazine, sumatriptan, bupropion,
aminobenzoate,
pyridostigmine bromide, potassium chloride, niacin, tocainide, quetiapine,
fexofenadine,
sertraline, chlorpheniramine, rifampin, methenamine, nefazodone, modafinil,
metaxalone,
morphine, sevelamer, lithium carbonate, flecainide acetate, simethicone,
methyldopa,
chlorthiazide, metyrosine, procainamide, entacapone, metoprolol, propanolol
hydrochloride, chlorzoxazone, tolmetin, tramadol, bepridil, phenytoin,
gabapentin,
terbinafine, atorvastatin, doxepine, rifabutin, mesalamine, etidronate,
nitrofurantoin,
choline magnesium trisalicy late, theophylline, nizatidine, methocarbamol,
mycophenolate
mofetil, tolcapone, ticlopidine, capecitabine, orlistat, colsevelam,
meperidine,
hydroxychloroquine, guaifenesin, guanfacine, amiodarone, quinidine,
atomoxetine,
felbamate, pseudoephedrine, carisoprodol, venlafaxine, etodolac, chondroitin,
lansoprazole, pantoprazole, esomeprazole, dexlansoprazole, dexmethylphenidate,
methylphenidate, sodium oxybate, valproic acid or its salts, divalproex,
topiramate,
carbamazepine, oxcarbazepine, isotretinoin, oseltamivir, cholestyramine,
nystatin,
artemether, lumefantrine, or combination thereof.
CA 2989925 2017-12-22
18
The liquid pharmaceutical composition of the present invention may comprise of
two or more different active ingredients or incompatible active ingredients.
Suitable film-forming agents include, but not limited to cellulosic polymers
e.g.,
hydroxypropylmethyl cellulose, hydroxypropyl cellulose, polyvinyl acetate,
polyvinyl
pyrrolidone, acrylic polymers such as these commercially available under the
trade mark
Eudragit E and Eudragit EPO, lipid coating substances such as stearic acid,
palmitic
acid, and glycerol monostearate; hydrophilic colloids such as alginate,
chitosan,
carboxymethylcellulose, xanthan gum, carboxy vinyl polymers e.g., Carbomer
94,
polylysine, gelatin; and mixtures thereof.
The ion-exchange resins such as cation-and anion-exchange matrices are well-
known in the art. Few exemplary resin particles that can be used according to
the
invention include, but are not limited to, Dowex resins and others made by
Dow
Chemical; Amberlite , Amberlyst and other resins made by Rohm and Haas;
Indion
resins made by Ion Exchange, Ltd. (India), Diaion resins by Mitsubishi; Type
AG and
other resins by BioRad; Sephadex and Sepharose made by Amersham; resins by
Lewatit, sold by Fluka; Toyopearl resins by Toyo Soda; IONAC and Whatman
resins
sold by VWR; and BakerBond resins sold by J T Baker; resins having polymer
backbones comprising styrene-divinyl benzene copolymers and having pendant
ammonium or tetraalkyl ammonium functional groups, available from Rohm and
Haas,
Philadelphia, and sold under the tradename DUOLITETm AP143.
Suitable suspending agents are selected from the group comprising cellulose
derivatives such as co-processed spray dried forms of microcrystalline
cellulose and
carboxymethyl cellulose sodium, hydroxypropyl cellulose, hydroxyethyl
cellulose,
hydroxypropylmethyl cellulose, methylcellulose, carboxymethyl cellulose and
its
salts/derivatives, and microcrystalline cellulose; carbomers; gums such as
locust bean
gum, xanthan gum, tragacanth gum, arabinogalactan gum, agar gum, gellan gum,
guar
gum, apricot gum, karaya gum, sterculia gum, acacia gum, gum arabic, and
carrageenan;
pectin; dextran; gelatin; polyethylene glycols; polyvinyl compounds such as
polyvinyl
acetate, polyvinyl alcohol, and polyvinyl pyrrolidone; sugar alcohols such as
xylitol and
mannitol; colloidal silica; and mixtures thereof. Co-processed spray dried
forms of
CA 2989925 2017-12-22
19
microcrystalline cellulose and carboxymethyl cellulose sodium have been
marketed under
the trade names Avicel RC-501, Avicel RC-581, Avicel RC-591, and Avicel CL-
611.
Suitable glidants are selected from the group comprising silica, calcium
silicate,
magnesium silicate, colloidal silicon dioxide, cornstarch, talc, stearic acid,
magnesium
stearate, calcium stearate, sodium stearyl fumarate, hydrogenated vegetable
oil, and
mixtures thereof.
Suitable sweeteners are selected from the group comprising saccharine or its
salts
such as sodium, potassium, or calcium, cyclamate or its salt, aspartame,
alitame,
acesulfame or its salt, stevioside, glycyrrhizin or its derivatives,
sucralose, and mixtures
thereof.
Suitable anti-caking agents are selected from the group comprising colloidal
silicon dioxide, tribasic calcium phosphate, powdered cellulose, magnesium
trisilicate,
starch, and mixtures thereof.
Suitable wetting agents are selected from the group comprising anionic,
cationic,
nonionic, or zwitterionic surfactants, or combinations thereof. Suitable
examples of
wetting agents are sodium lauryl sulphate; cetrimide; polyethylene glycols;
polyoxyethylene-polyoxypropylene block copolymers such as poloxamers;
polyglycerin
fatty acid esters such as decaglyceryl monolaurate and decaglyceryl
monomyristate;
sorbitan fatty acid esters such as sorbitan monostearate; polyoxyethylene
sorbitan fatty
acid esters such as polyoxyethylene sorbitan monooleate; polyethylene glycol
fatty acid
esters such as polyoxyethylene monostearate; polyoxyethylene alkyl ethers such
as
polyoxyethylene lauryl ether; polyoxyethylene castor oil; and mixtures
thereof.
Suitable preservatives are selected from the group comprising parabens such as
methyl paraben and propyl paraben; sodium benzoate; and mixtures thereof.
Suitable buffering agents are selected from the group comprising citric acid,
sodium citrate, sodium phosphate, potassium citrate, acetate buffer, and
mixtures thereof.
Suitable flavoring agents are selected from the group consisting of
peppermint,
grapefruit, orange, lime, lemon, mandarin, pineapple, strawberry, raspberry,
mango,
passion fruit, kiwi, apple, pear, peach, apricot, cherry, grape, banana,
cranberry, blueberry,
black currant, red currant, gooseberry, lingon berries, cumin, thyme, basil,
camille,
CA 2989925 2017-12-22
20
valerian, fennel, parsley, chamomile, tarragon, lavender, dill, bargamot,
salvia, aloe vera
balsam, spearmint, eucalyptus, and combinations thereof.
Suitable anti-oxidants are selected from the group comprising butylated
hydroxytoluene (BHT), butylated hydroxyani sole (BHA), sodium metabisulfite,
ascorbic
acid, propyl gallate, thiourea, tocopherols, beta-carotene, and mixtures
thereof.
Suitable chelating agents are selected from the group comprising
ethylenediamine
tetraacetic acid or derivatives/salts thereof, e.g., disodium edetate;
dihydroxyethyl glycine;
glucamine; acids, e.g., citric acid, tartaric acid, gluconic acid, and
phosphoric acid; and
mixtures thereof.
The term "solute," as used herein, refers to pharmaceutically acceptable inert
agents that have high affinity for the pharmaceutically acceptable vehicle.
The solutes
generates hypertonic conditions leading to high osmolality and thus prevents
the leaching
of the active ingredient from the coated cores. The solutes can be present in
the
pharmaceutically acceptable vehicle or in the solid composition or both.
Suitable solutes
are selected from the group comprising carbohydrates such as xylitol,
mannitol, sorbitol,
arabinose, ribose, xylose, glucose, fructose, mannose, galactose, sucrose,
maltose, lactose,
dextrose and raffinose; water-soluble salts of inorganic acids such as
magnesium chloride,
magnesium sulfate, potassium sulfate, lithium chloride, sodium chloride,
potassium
chloride, lithium hydrogen phosphate, sodium hydrogen phosphate, potassium
hydrogen
phosphate, lithium dihydrogen phosphate, sodium dihydrogen phosphate,
potassium
dihydrogen phosphate, and sodium phosphate tribasic; water-soluble salts of
organic acids
such as sodium acetate, potassium acetate, magnesium succinate, sodium
benzoate,
sodium citrate, and sodium ascorbate; water-soluble amino acids such as
glycine, leucine,
alanine, methionine; urea or its derivatives; propylene glycol; glycerin;
polyethylene
oxide; xanthan gum; hydroxypropylmethyl cellulose; and mixtures thereof.
Particularly,
the solutes used are xylitol, mannitol, glucose, lactose, sucrose, and sodium
chloride.
The cores of the present invention comprising the active ingredient can be
prepared
by any method known in the art, e.g., extrusion-spheronoization, wet
granulation, dry
granulation, hot-melt extrusion granulation, spray drying, and spray
congealing.
Alternatively, the active ingredient can be layered onto an inert particle to
form the core.
Further, the active ingredient particles can be directly coated with a film
forming layer to
CA 2989925 2017-12-22
21
form the microparticles or microcapsules. The microparticles or microcapsules
can be
prepared by a process of homogenization, solvent evaporation, coacervation
phase
separation, spray drying, spray congealing, polymer precipitation, or
supercritical fluid
extraction. The ion-exchange resins comprise loading a plurality of the resin
particles with
the active ingredient to form drug-resin cores. Methods of loading active
ingredients onto
the resin particles are generally known in the art.
The first chamber includes a container which is in the form of a glass or a
plastic or
a metallic bottle. The reservoir of the second chamber can be made of a
plastic, a metal or
a glass; particularly the reservoir is a plastic bottle. The reservoir of the
second chamber
may additionally have a slippery coating or mold polishing. This coating or
polishing will
help to improve the flow characteristics of the solid composition during
activation.
The dual-chamber pack is suitable for incorporating solid composition in a
volume
of greater than about 40 cc. In the dual-chamber pack, the plunger is opened
at both the
ends. The biphasic connector comprises of cross bridges to give the strength.
The bridges
can be tapered at the edges to avoid any powder deposit. Further, the
reservoir can have
serrations to have better grip for the end-users. The biphasic connector have
a tamper-
evident band on the side connected to the container of the first chamber which
is removed
first to start the activation process. The biphasic connector is having
grooves on other side
for locking with the reservoir. On this side, there would be instructions for
the end-users
regarding direction of the rotation such as clockwise rotation for activating
the pack.
The term "tamper-evident band," as used herein, refers to a band attached co-
axially to the biphasic connector. The band breaks easily on pulling apart.
The tamper-
evident band ensures the overall integrity of the product until activation.
The plunger of the instant invention can comprise of one or more sharp
projections
with an essential continuous blunt area. In particular, the plunger comprise
of one sharp
projection with an essential continuous blunt area. Alternatively, the plunger
can have a
single continuous projection with a remaining continuous blunt area which can
be called
as a flute shaped plunger. The plunger can further have one or more grooves.
The body of
the plunger can be in the form of a cylinder or a funnel. The funnel shaped
plunger
provides additional capacity for storing high-dose active ingredients or
active ingredients
required for chronic administration.
CA 2989925 2017-12-22
22
The plunger used in the instant invention ensures that the breakable polymeric
membrane remains attached to the plug during activation. The plug and the
plunger may
be made up of a polymeric material selected from the group comprising
polyolefin,
polyethylene, polypropylene, polyvinyl chloride, cyclic olefin polymer, cyclic
olefin co-
polymer, polyethylene terephthalate, polyethylene terephthalate - G,
polypropylene, and
polycarbonate. Particularly, the plug and the plunger are made up of
polyethylene. More
particularly, the plug and the plunger are made up of linear low density
polyethylene
(LLDPE).
The compositions of the first and second chambers of the container are
separated
by a polymeric breakable membrane of the plug. The plunger used in the instant
invention
helps to rupture the breakable polymeric membrane upon the application of
pressure by a
screw-based mechanism. When pressure is applied on the reservoir, the
breakable
polymeric membrane is ruptured by the plunger. The intact polymeric membrane
remains
attached to the circumference of the plug. In cases, where a bottle liner
exists between the
first and the second chambers, the plunger would break the bottle liner in the
same manner
as it ruptures the breakable polymeric membrane. The unabridged part of the
bottle liner
remains attached to the opening of the container. The plug with the breakable
polymeric
membrane prevents moisture permeation from the first chamber into the second
chamber.
The material used for making the plug may also include moisture barrier
additives
selected from the plastic additive group comprising of monomers and co-
polymers that get
activated through polymerization process to form an effective organic
chemical. The
moisture barrier additives used in the present invention may include any
material that
prevent moisture permeation. The moisture barrier additives may be present in
the form of
a layer inside the plug. The moisture barrier additives may be present in an
amount of
0.1% to 10% w/w, in particularly, 0.5% to 5% w/w based on total weight of the
material
used for making plug.
The material used for making the reservoir may also include the moisture
barrier
additives. The moisture barrier additives may be present in the form of a
layer inside the
reservoir.
The moisture permeation test was carried out on dual chamber packs with
moisture
barrier additives and without moisture barrier additives as per USP (37)¨ 671
Containers
CA 2989925 2017-12-22
23
Performance Testing. The moisture barrier additives used in the present
invention improve
the moisture barrier properties by up to 50%. In particular, the moisture
barrier additives
improves the moisture barrier properties by up to 30%.
The use of moisture barrier additives thus help to prevent the moisture
permeation
from the pharmaceutically acceptable vehicle into the solid composition
comprising the
active ingredient during storage. The active ingredient, particularly moisture-
sensitive
active ingredients thus remains stable during storage.
The invention may be further illustrated by the following example, which is
for
illustrative purposes only and should not be construed as limiting the scope
of the
invention in any way.
CA 2989925 2017-12-22
24
EXAMPLES
Example 1
Ingredients Quantity (mg/mL)
Core
Metformin hydrochloride 80.00
Microcrystalline cellulose spheres 56.00
Hydroxypropylmethyl cellulose 4.00
Purified water q.s.
Extended Release Coating
Ethyl cellulose 68.31
Dibutyl sebacate 1.69
Acetone q.s.
Purified water q.s.
Total Weight of Extended Release
210.00 mg
Beads
Suspension Base
Metformin hydrochloride 20.00
Xylitol 450.00
Microcrystalline cellulose - sodium
carboxymethyl cellulose (Avicel CL- 20.00
611)
Xanthan gum 1.50
Methyl paraben 1.80
Propyl paraben 0.20
Strawberry flavor 2.00
Sucralose 0.50
Colloidal silicon dioxide 3.50
Purified water 472.00 mg
Procedure:
1. Metformin hydrochloride and hydroxypropylmethyl cellulose were dissolved in
purified water.
2. Microcrystalline cellulose spheres were coated with the solution of step
1.
3. Ethyl cellulose and dibutyl sebacate were dispersed in a mixture of
acetone and
purified water.
4. The beads of step 2 were coated with the coating dispersion of step 3
and dried to form
a powder for suspension.
5. Purified water was heated to dissolve methyl paraben and propyl paraben.
CA 2989925 2017-12-22
25
6. Metformin hydrochloride, xylitol, microcrystalline cellulose - sodium
carboxymethyl
cellulose, xanthan gum, strawberry flavor, sucralose, and colloidal silicon
dioxide
were mixed in the solution of step 5 to form a suspension base.
7. The powder for suspension of step 4 was filled in the second chamber of
a drug
delivery device.
8. The suspension base of step 6 was filled in a container of a first
chamber of a drug
delivery device.
9. The two chambers were assembled and the drug delivery device was activated
to form
the extended release liquid composition when required.
In-Vitro Studies of Extended Release Reconstituted Powder for Suspension
The extended release reconstituted powder for suspension prepared as per
Example
1 (for a dose equivalent to 750 mg of metformin hydrochloride) was stored at
room
temperature for 120 days. The in-vitro dissolution was determined at 0, 45,
90, and 120
days using USP type II apparatus at 100 rpm, in 1000 mL of phosphate buffer
with pH 6.8
at 37 C. The results of the release studies are represented in Table I.
Table 1: Percentage (%) of the In-Vitro Metformin Release in USP Type II
Apparatus (Media: Phosphate Buffer, pH 6.8, 1000 mL, and 100 rpm)
Number of Days 0 45 90 120
Time (hours) Percentage of Metformin Release
0.5 20 21 20 21
1 27 25 27 25
2 55 52 55 52
3 74 72 74 72
4 83 81 83 81
85 86 85 86
6 87 90 87 90
8 91 94 91 94
93 96 93 96
12 94 97 94 97
CA 2989925 2017-12-22
26
From the above in-vitro release data, it is evident that the extended release
reconstituted powder for suspension prepared according to Example 1 provides
the
substantially similar in-vitro metformin release for 120 days.
The drug delivery device was kept for 1 month at accelerated conditions i.e.,
40 C/75% R.H. After 1 month, the drug delivery device was activated to form an
extended
release reconstituted powder for suspension which was kept for 120 days at
room
temperature. The in-vitro dissolution was determined at 0, 45, 90, and 120
days using
USP type II apparatus at 100 rpm, in 1000 mL of phosphate buffer with 6.8
at 37 C.
The results of the release studies are represented in Table 2.
Table 2: Percentage (%) of the In-Vitro Metformin Release in USP Type II
Apparatus (Media: Phosphate Buffer, pH 6.8, 1000 mL, and 100 rpm)
Number of Days 0 45 90 120
Time (hours) Percentage of Metformin Release
0.5 21 21 21 20
1 27 25 26 26
2 56 55 52 54
3 74 74 76 72
4 83 81 82 81
96 96 97 94
The drug delivery device was kept for 3 months at accelerated conditions i.e.,
40 C/75% R.H. After 3 months, the drug delivery device was activated to form
an
extended release reconstituted powder for suspension which was kept for 120
days at room
temperature. The in-vitro dissolution was determined at 0, 45, 90, and 120
days using
USP type II apparatus at 100 rpm, in 1000 mL of phosphate buffer with pH 6.8
at 37 C.
The results of the release studies are represented in Table 3.
CA 2989925 2017-12-22
27
Table 3: Percentage (/o) of the In-Vitro Metformin Release in USP Type II
Apparatus (Media: Phosphate Buffer, pH 6.8, 1000 mL, and 100 rpm)
Number of 0 45 90 120
Days
Time (hours) Percentage of Metformin Release
0.5 21 21 21 20
1 26 25 25 26
2 55 53 53 60
3 75 72 72 73
4 80 80 79 82
95 92 96 97
The drug delivery device was kept for 6 months at accelerated conditions i.e.,
40 C/75% R.H. After 6 months, the drug delivery device was activated to form
an
extended release reconstituted powder for suspension which was kept for 120
days at room
temperature. The in-vitro dissolution was determined at 0, 45, 90, and 120
days using
USP type 11 apparatus at 100 rpm, in 1000 mL of phosphate buffer with pH 6.8
at 37 C.
The results of the release studies are represented in Table 4.
Table 4: Percentage CYO of the In-Vitro Metformin Release in USP Type II
Apparatus (Media: Phosphate Buffer, pH 6.8, 1000 mL, and 100 rpm)
Number of 0 45 90 120
Days
Time (hours) Percentage of Metformin Release
0.5 18 19 19 20
1 23 24 25 28
2 50 56 54 57
3 70 71 74 73
4 78 80 79 81
10 95 95 94 94
From the above data, it is clear that the powder for suspension and suspension
base stored
in the drug delivery device of the instant invention at accelerated conditions
for 1 month, 3
CA 2989925 2017-12-22
28
months and 6 months, upon activation of the drug delivery device forms
extended release
reconstituted powder for suspension which when stored for 120 days at room
temperature
provides substantially similar in-vitro metformin release.
Stability Data of Extended Release Reconstituted Powder for Suspension
The related substances for the extended release reconstituted powder for
suspension prepared as per Example 1 were determined at 0 day and after
storage at room
temperature for 45 and 120 days. The powder for suspension and suspension base
was
stored in the drug delivery device for one month and for three months at 40
C/75% R.H.
After one month or three months, the drug delivery device was activated to
form an
extended release reconstituted powder for suspension and then related
substances were
determined at 0 day and after storage at room temperature for 45 days and 120
days.
The related substances of metformin was determined by HPLC method. The results
are shown in Table 5.
Table 5: Stability Data for Metformin in the Drug Delivery Device
Related Initial 1 month 3 month
Substances (40 C/75% R.H) (40 C/75%
(/ow/w) R.H)
0 day 45 120 0 day 45 120 0 day 45 days
days days days days
Cyanoguainidine BLQ 0.001 0.00072 0.001 0.001 0.001 0.001 0.001
Highest unknown 0.05 0.05 0.04 0.05 0.04 0.04 0.05 0.04
impurity
Total impurities 0.05 0.05 0.04 0.05 0.04 0.04 0.09
0.04
*BLQ: Below limit of Quantification
It is evident from the above data that the extended release reconstituted
powder for
suspension prepared as per Example 1 remains stable even after storing at
accelerated
conditions for 3 months.
CA 2989925 2017-12-22
29
Antimicrobial Efficacy Test of Extended Release Reconstituted Powder for
Suspension
The powder for suspension and suspension base prepared as per Example I was
stored in the drug delivery device for twenty four months at 25 C/60% R.H.
After twenty
four months, the drug delivery device was activated to form an extended
release
reconstituted suspension which was tested for antimicrobial efficacy as per
USP and also
by inoculating with Burkholderia cepacia complex. Extended release
reconstituted
suspension prepared after activation of the drug delivery device was found to
comply with
the Antimicrobial Efficacy Test as per USP. Growth of Burkholderia cepacia
complex
was found to be inhibited in the reconstituted suspension.
It is evident from the above test that microbial growth was not promoted in
the
extended release reconstituted powder for suspension composition prepared
after
activation of the drug delivery device.
In-Vitro Studies of Extended Release Coated Cores
Extended release coated cores of Example 1 (step 4) were stored in the drug
delivery
device and kept for 1, 3 and 6 months at accelerated conditions i.e., 40 C/75%
RH. Coated
cores were then subjected to determination of water content after I, 3 and 6
months of
storage at 40 C/75% RH using Karl Fischer Apparatus. The results of the water
content
determination are represented in Table 5A.
Table 5A: Water Content of Extended Release Coated Cores
Time Period Water Content
Initial 1.08
1 month (40 C/75% RH) 1.56
3 month (40 C/75% RH) 0.73
6 month (40 C/75% RH) 1.79
From the above data, it is evident that there was no change in water content
of the
extended release coated cores prepared according to Example l (step 4) after
six months
of storage at 40 C/75% RH.
CA 2989925 2017-12-22
30
Example 2
Ingredients Quantity (mg/mL)
Core
Metformin hydrochloride 80.00
Microcrystalline cellulose spheres 56.00
Hydroxypropylmethyl cellulose 4.00
Purified water q.s.
Extended Release Coating
Ethyl cellulose 75.14
Dibutyl sebacate 1.856
Acetone q.s.
Purified water q.s.
Total Weight of Extended Release Beads 217.00 mg
Lubrication
Magnesium stearate 1.500
Suspension Base
Metformin hydrochloride 20.00
Xylitol 450.00
Microcrystalline cellulose - sodium 20.00
carboxymethyl cellulose (Avicel CL-611)
Xanthan gum 1.50
Methyl paraben 1.80
Propyl paraben 0.20
Strawberry flavor 1.50
Sucralose 0.50
Colloidal silicon dioxide 3.50
Purified water 465.5
Procedure:
1. Metformin hydrochloride and hydroxypropylmethyl cellulose were dissolved in
purified water.
2. Microcrystalline cellulose spheres were coated with the solution of step
1.
3. Ethyl cellulose and dibutyl sebacate were dispersed in a mixture of
acetone and
purified water.
4. The beads of step 2 were coated with the coating dispersion of step 3 and
dried to form
extended release beads.
5. The extended release beads of step 4 were lubricated with Magnesium
stearate to form
powder for suspension.
6. Purified water was heated to dissolve methyl paraben and propyl paraben.
CA 2989925 2017-12-22
31
7. Metformin hydrochloride, xylitol, microcrystalline cellulose - sodium
carboxymethyl
cellulose, xanthan gum, strawberry flavor, sucralose, and colloidal silicon
dioxide
were mixed in the solution of step 6 to form a suspension base.
8. The powder for suspension of step 5 was filled in the second chamber of
a drug
delivery device.
9. The suspension base of step 7 was filled in a container of a first
chamber of a drug
delivery device.
10. The two chambers were assembled and activated to form the extended release
liquid
composition when required.
Flow Properties of Extended Release Coated Cores
Extended release coated cores of Example 2 (step 5) were evaluated for the
following parameters:
Angle of Repose (AoR) - Angle of Repose was determined by passing the extended
release coated cores through Enar Reposograph to make the cone. Then, height
of the cone
(h) thus formed and the radius (r) of the base of the cone were measured.
Angle of repose
(0) was calculated as follows:
= tan-1(h/r)
Hausner ratio (HR) - Hausner ratio was determined by dividing the tapped
density (ptap)
by the bulk density (pbulk).
Carr's Compressibility Index (CI) - Carr's Compressibility Index was
determined from
the Hausner ratio (HR) as Cl = 100 x [1-( UHR)]
Table 6: Flow Property of Extended Release Coated Cores of Metformin
Hydrochloride
Flow Property Parameter Observed Value Flow Character
Angle of Repose ( ) 25.87 Excellent
Carr's Compressibility Index (/0) 7.32 Excellent
Hausner ratio 1.08 Excellent
The powder for suspension was found to have desired flowability.
CA 2989925 2017-12-22
32
In-Vitro Studies of Extended Release Coated Cores
Extended release coated cores of Example 2 (step 5) were stored in the drug
delivery
device and kept for 6 month at accelerated conditions i.e., 40 C/75% RH Coated
cores
were then subjected to in-vitro dissolution testing after six months of
storage at 40 C/75%
RH using USP type II apparatus at 100 rpm, in 1000 mL of phosphate buffer with
pH 6.8
at 37 C. The results of the release studies are represented in Table 7.
Table 7: Percentage (/o) of the In-Vitro Metformin Release from Extended
Release
Coated Cores in USP Type II Apparatus (Media: Phosphate Buffer, pH 6.8, 1000
mL, 100 rpm)
Initial After six months
Time Period at 40 C/75% RH
Time (hours) Percentage of
Metformin Release
. 0.5 1 0
2 46 43
12 95 93
From the above in-vitro release data, it is evident that the extended release
coated
cores prepared according to Example 2 (step 5) provide substantially similar
in-vitro
metformin release after six months of storage at 40 C/75% RH. Thus, the
extended release
coated cores prepared as per the present invention are stable when stored in
the second
chamber of the drug delivery device for at least six months under accelerated
conditions.
Uniformity of Fill Weight of Extended Release Coated Cores
Extended release coated cores prepared according to Example 2 (step 5) were
filled into
the second chamber of drug delivery devices. A total of 960 drug delivery
devices were
filled. Target fill weight was I06.995g. Average weight was found to be
107.52g,
minimum observed fill weight was 106.14g, maximum fill weight was 107.95g and
%
RSD was found to be 0.25. Entire batch was filled within 7.5% of the target
weight.
CA 2989925 2017-12-22
33
Thus, % weight variation with respect to target fill weight of extended
release coated cores
was found to be within the range of about 7.5%.
In-Vitro Studies of Extended Release Reconstituted Powder for Suspension
The extended release reconstituted powder for suspension prepared as per
Example
2 (was stored at room temperature for 100 days. The in-vitro dissolution was
determined at
0, 45, and 100 days using USP type II apparatus at 100 rpm, in 1000 mL of
phosphate
buffer with pH 6.8 at 37 C. The results of the release studies are represented
in Table 8.
Table 8: Percentage (%) of the In-Vitro Metformin Release in USP Type II
Apparatus (Media: Phosphate Buffer, pH 6.8, 1000 mL, and 100 rpm)
Number of Days 0 45 100
Time (hours) Percentage of Metformin Release
0.5 20 21 21
1 24 26 27
2 58 60 61
3 78 75 79
4 86 82 86
12 99 94 100
From the above in-vitro release data, it is evident that the extended release
reconstituted powder for suspension prepared according to Example 2 provides
the
substantially similar in-vitro metformin release for 100 days.
The drug delivery device was kept for 1 month at accelerated conditions i.e.,
40 C/75% R.H. After 1 month, the drug delivery device was activated to form an
extended
release reconstituted powder for suspension which was kept for 100 days at
room
temperature. The in-vitro dissolution was determined at 0, 45, and 100 days
using USP
type 11 apparatus at 100 rpm, in 1000 mL of phosphate buffer with pH 6.8 at 37
C. The
results of the release studies are represented in Table 9.
CA 2989925 2017-12-22
34
Table 9: Percentage CYO of the In-Vitro Metformin Release in USP Type II
Apparatus (Media: Phosphate Buffer, pH 6.8, 1000 mL, and 100 rpm)
Number of Days 0 45 100
Time (hours) Percentage of Metformin Release
0.5 19 19 19
2 55 54 58
12 91 92 97
The drug delivery device was kept for 3 months at accelerated conditions i.e.,
40 C/75% R.H. After 3 months, the drug delivery device was activated to form
an
extended release reconstituted powder for suspension which was kept for 100
days at room
temperature. The in-vitro dissolution was determined at 0 and 100 days using
USP type 11
apparatus at 100 rpm, in 1000 mL of phosphate buffer with pH 6.8 at 37 C. The
results of
the release studies are represented in Table 10.
Table 10: Percentage (%) of the In-Vitro Metformin Release in USP Type II
Apparatus (Media: Phosphate Buffer, pH 6.8, 1000 mL, and 100 rpm)
Number of Days 0 100
Time (hours) Percentage of Metformin Release
0.5 19 19
2 57 60
12 97 95
The drug delivery device was kept for 6 months at accelerated conditions i.e.,
40 C/75% R.H. After 6 months, the drug delivery device was activated to form
an
extended release reconstituted powder for suspension which was kept for 100
days at room
temperature. The in-vitro dissolution was determined at 0 and 100 days using
USP type II
apparatus at 100 rpm, in 1000 mL of phosphate buffer with pH 6.8 at 37 C. The
results of
the release studies are represented in Table 11.
CA 2989925 2017-12-22
35
Table 11: Percentage MO of the In-Vitro Metformin Release in USP Type II
Apparatus (Media: Phosphate Buffer, pH 6.8, 1000 mL, and 100 rpm)
Number of Days 0 100
Time (hours) Percentage of Metformin Release
0.5 19 19
2 59 60
12 96 94
From the above data, it is clear that the powder for suspension and suspension
base stored
in the drug delivery device of the instant invention at accelerated conditions
for 1 month, 3
months and 6 months, upon activation of the drug delivery device forms
extended release
reconstituted powder for suspension which when stored for at least 100 days at
room
temperature provides substantially similar in-vitro metformin release.
Example 3
Ingredients Quantity (mg/ 5mL)
For 228.5 mg strength For 457 mg strength
Solid composition
Amoxicillin Trihydrate 238.15 476.30
Potassium Clavulanate + Silicon dioxide (1:1) 77.63 155.25
Colloidal anhydrous silica 10.00 10.00
Silicon dioxide 126.22 10.45
Xanthan gum 10.00 10.00
Monosodium citrate 6.00 6.00
Sodium citrate 2.00 2.00
Aspartame 10.00 10.00
Strawberry flavor 20.00 20.00
Total Fill Weight 500.00 700.00
Suspension Base
Sodium Benzoate 10.00 10.00
Purified water q.s. to 5 mL q.s. to 5 int.
Procedure:
CA 2989925 2017-12-22
36
1. Amoxicillin was dried at 45-55 C in a tray drier.
2. Xanthan gum, Strawberry flavor, Sodium Citrate, Monosodium citrate,
Aspartame
and Colloidal anhydrous silica were dried at 75-80 C.
3. Dried Monosodium citrate and Sodium citrate were sifted through a suitable
sieve.
4. Strawberry flavor, Xanthan gum, Aspartame and Silicon dioxide were sifted
along
with blend from step 1 using a suitable sieve.
5. Amoxicillin Trihydrate, Potassium Clavulanate and Colloidal anhydrous
silica were
sifted along with blend from step 2 using a suitable sieve.
6. The material from step 3 was blended in low shear blender for 30 to 45
minutes.
7. The blend from step 4 was filled in the second chamber of the drug
delivery device.
8. Sodium Benzoate was dissolved in Purified water to form the vehicle.
9. Vehicle of step 6 was filled in the first chamber of the drug delivery
device.
10. The two chambers were assembled and the device was activated to form the
immediate release liquid composition when required.
Stability Data of Immediate Release Reconstituted Powder for Suspension
The related substances for the immediate release reconstituted powder for
suspension prepared as per Example 3 were determined at 0 day and the powder
for
suspension was stored in the second chamber and suspension base was stored in
the first
chamber of the drug delivery device for one month and for three months at 40
C/75%
R.H. After one month or three months, the device was activated to form an
immediate
release reconstituted powder for suspension and then related substances were
determined.
The related substances were determined by HPLC method. The results are shown
in Table 12.
CA 2989925 2017-12-22
37
Table 12: Stability Data for Amoxicillin in the Amoxicillin and Clavulanic
Acid
Immediate Release Reconstituted Powder for Suspension in the Drug Delivery
Device
228.5 mg/5 mL 457 mg/5 mL
Related Substances 1 month 3 month 1 month 3 month
(%w/w) Initial (40 C/75% (40 C/75% Initial (40 C/75% (40
C/75%
R.H) R.H) R.H) R.H)
Amoxicilloic acid-1 0.007 0.009 0.03 0.01 0.01 0.02
Amoxicilloic acid-2 0.06 0.07 0.08 0.04 0.05 0.05
Amoxilloic acid-1 0.008 0.022 0.05 ND ND 0.03
Amoxilloic acid-2 0.004 0.018 0.03 0.004 0.021 0.02
Diketopiperazine-1 0.01 0.01 0.03 0.01 0.03 ND
Diketopiperazine-2 ND ND ND ND 0.01 ND
2-hydroxy-3-(4-
hydroxypheny1)- ND ND ND ND 0.03 ND
pyrazine
Amoxicillin dimer 0.20 0.27 0.25 0.08 0.15 0.12
Amoxicillin trimer 0.006 ND 0.01 0.01 ND ND
Highest unknown
0.05 0.23 0.03 0.04 0.10 0.08
impurity
Total Unknown impurity 0.32 0.37 0.15 0.17 0.26 0.19
Total Related
0.62 0.77 0.63 0.33 0.56 0.44
Substances
* ND: Not Detectable
CA 2989925 2017-12-22
38
It is evident from the above data that the immediate release reconstituted
powder
for suspension prepared as per Example 3 remains stable even after storing at
accelerated
conditions for 3 months.
CA 2989925 2017-12-22