Note: Descriptions are shown in the official language in which they were submitted.
1
CA 02993928 2018-01-26
WO 2017/018820 PCT/KR2016/008270
Description
Title of Invention: PHARMACEUTICAL COMPOSITION WITH
IMPROVED STORAGE STABILITY AND METHOD FOR
PREPARING THE SAME
Technical Field
[11 The present disclosure relates to a pharmaceutical composition with
improved
storage stability and a method for preparing the same, and more specifically,
a pharma-
ceutical composition of poorly water-soluble drug comprising an amphiphilic
block
copolymer wherein the content of a specific related compound is kept within a
specified limit, and a method for preparing the same.
Background Art
[2] Solubilization of a poorly water-soluble drug is a key technology for
delivering the
drug into the body via oral or parenteral administration. Such solubilization
methods
include a method of adding a surfactant to an aqueous solution to form
micelles and
then entrapping a poorly water-soluble drug therein. An amphiphilic block
copolymer
used as a surfactant comprises a hydrophilic polymer block and a hydrophobic
polymer
block. Since the hydrophilic polymer block directly contacts blood proteins
and cell
membranes in vivo, polyethylene glycol or monomethoxypolyethylene glycol, etc.
having biocompatibility has been used. The hydrophobic polymer block improves
affinity to a hydrophobic drug, and polylactide, polyglycolide, poly(lactic-
glycolide),
polycaprolactone, polyamino acid or polyorthoester, etc. having
biodegradability has
been used. In particular, polylactide derivatives have been applied to drug
carriers in
various forms because they have excellent biocompatibility and are hydrolyzed
into
harmless lactic acid in vivo. Polylactide derivatives have various physical
properties
depending on their molecular weights, and have been developed in various forms
such
as microsphere, nanoparticle, polymeric gel and implant agent.
[31 US Patent No. 6,322,805 discloses a composition for delivering a
poorly water-
soluble drug consisting of a polymeric micelle-type drug carrier and a poorly
water-
soluble drug, wherein the polymeric micelle-type drug carrier is formed from a
diblock
or triblock copolymer which is not crosslinked by a crosslinking agent and
consists of
at least one biodegradable hydrophobic polymer selected from the group
consisting of
polylactide, polyglycolide, poly(lactide-glycolide), polycaprolactone and
derivatives
thereof and poly(alkylene oxide) as a hydrophilic polymer, wherein the poorly
water-
soluble drug is physically entrapped in the drug carrier and solubilized, and
wherein
the polymeric micelle-type drug carrier forms a clear aqueous solution in
water and ef-
fectively delivers the poorly water-soluble drug into the body. According to
the above
2
CA 02993928 2018-01-26
WO 2017/018820 PCT/KR2016/008270
US patent, polyethylene glycol-polylactide diblock copolymer is synthesized by
removing moisture from monomethoxypolyethylene glycol, adding stannous octoate
dissolved in toluene thereto and removing toluene under reduced pressure,
adding
D,L-lactide to the resulting mixture and conducting a polymerization reaction,
adding
chloroform to dissolve the produced block copolymer, dropwise adding an excess
amount of diethyl ether in small portions with stiffing to form precipitant
and filtering
the formed precipitant, and washing it several times with diethyl ether.
However, this
method is difficult to employ in mass-scale production and thus is not
commercially
available. In addition, the ether that has been used for purification may
remain in the
final polymeric micelle composition.
[4] US Patent No. 8,853,351 discloses a method for preparing an
amphiphilic block
copolymer, comprising (a) dissolving the amphiphilic block copolymer in a
water-
miscible organic solvent; (b) adding and mixing an aqueous solution of alkali
metal
salt (sodium bicarbonate, sodium carbonate, potassium bicarbonate, potassium
carbonate or lithium carbonate) to the polymeric solution obtained in step
(a); (c)
separating organic and aqueous phases by salting out for the solution obtained
in step
(b); and, (d) isolating the organic phase obtained in step (c) and removing
the organic
solvent therefrom to recover the polymer. However, the method involves
complicated
steps, and requires an additional step for removing the alkali metal salt and
the salt
(sodium chloride or potassium chloride) used for salting out, and may have
residual
metal salts even after the removal thereof.
[51 Impurities of drug must be strictly controlled in various aspects.
Particularly, in case
of impurities derived from active pharmaceutical ingredient (API), each
country de-
termines in its drug approval guideline the upper limit to amount of API-
derived,
known or unknown impurities (related compounds) in a drug product. In
addition,
there are some standards used internationally and ICH guideline Q3A is the
repre-
sentative one. In this guideline, at the time of approving a drug, the amount
of each
related compound in the drug is limited up to 0.1% or 0.2%, etc. and
information such
as toxicity-related data, etc., which should be provided, is discriminately
applied
according to the related compound exceeding the limit. This implies that since
it is
unknown how a related compound of a drug would act in vivo, the amount of the
related compound must be reduced in the procedure of manufacturing the drug.
Therefore, a manufacturing process for reducing the related compounds and
setting of
the upper limit to amount according to the characteristics (structure and
toxicity) of
each related compound are essential factors in quality control of the drug.
Disclosure of Invention
Technical Problem
3
CA 02993928 2018-01-26
WO 2017/018820 PCT/KR2016/008270
[6] One purpose of the present invention is to provide a polymeric micelle-
type pharma-
ceutical composition of poorly water-soluble drug comprising an amphiphilic
block
copolymer, which contains a specific related compound in an amount within a
specified limit.
171 The other purpose of the present invention is to provide a method for
preparing said
pharmaceutical composition.
Solution to Problem
1181 One aspect of the present invention provides a polymeric micelle
pharmaceutical
composition, comprising: a purified amphiphilic block copolymer comprising a
hy-
drophilic block (A) and a hydrophobic block (B), and one or more poorly water-
soluble drugs selected from the group consisting of paclitaxel and docetaxel,
wherein
the pharmaceutical composition contains, when stored at 40 C for 6 months, a
related
compound represented by the following Formula 1 in an amount of less than 0.12
part
by weight, based on 100 parts by weight of the initial amount of the poorly
water-
soluble drug:
1191 [Formula 11
[10] Ri
(7) 0 Hp
0 01-1
/Th
0
t
4 =
R2
RP
0
[11] wherein
[12] R1 is H or COCH3, and R2 is phenyl or 0(CH3)3.
[13] Another aspect of the present invention provides a method for
preparing a polymeric
micelle pharmaceutical composition, comprising: (a) purifying an amphiphilic
block
copolymer comprising a hydrophilic block (A) and a hydrophobic block (B); (b)
dissolving one or more poorly water-soluble drugs selected from the group
consisting
of paclitaxel and docetaxel, and the purified amphiphilic block copolymer in
an
organic solvent; and (c) adding an aqueous solvent to the solution obtained in
step (b)
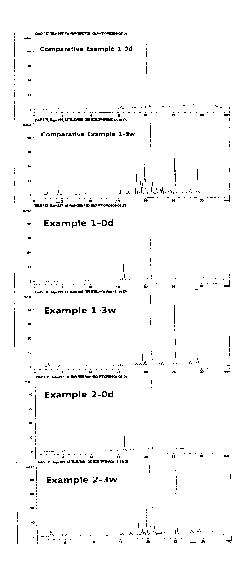
to form polymeric micelles; wherein the pharmaceutical composition contains,
when
stored at 40 C for 6 months, a related compound represented by the above
Formula 1
in an amount of less than 0.12 part by weight, based on 100 parts by weight of
the
initial amount of the poorly water-soluble drug.
Advantageous Effects of Invention
11141 According to the present invention, a pharmaceutical composition of
poorly water-
4
CA 02993928 2018-01-26
WO 2017/018820 PCT/KR2016/008270
soluble drug, which has reduced related compounds and improved storage
stability,
can be obtained.
Brief Description of Drawings
[15] Figure 1 is the resulting chromatogram of HPLC analysis for the
polymeric micelle
composition containing paclitaxel used in Experimental Example 1, which had
been
subjected to the six-month acceleration test.
[16] Figure 2 shows the results of product ion scan in LC/MS/MS analysis
for the related
compound (RRT 1.44 0.05 (1.39-1.49), with which RRT 1.44 is interchangeably
used
hereinafter) obtained in Experimental Example.
[17] Figure 3 shows the results of LC/MS/MS analysis for the material
obtained at RRT
1.44 position in the mixture obtained by thermally decomposing paclitaxel in
Ex-
perimental Example 3.
[18] Figure 4 shows the results of product ion scan in the LC/MS/MS
analysis for the
material obtained at RRT 1.44 position in the mixture obtained by thermally de-
composing paclitaxel in Experimental Example 3, together with the analysis
results of
the six-month acceleration tested sample of the polymeric micelle composition:
[19] (a) Results of analysis of the six-month acceleration tested sample of
the polymeric
micelle pharmaceutical composition
[20] (b) Results of analysis of the material obtained at RRT 1.44 position
in the mixture
obtained by thermally decomposing paclitaxel
[21] Figure 5 shows the results of 1H NMR analysis in the NMR analysis for
the material
obtained at RRT 1.44 position in the mixture obtained by thermally decomposing
pa-
clitaxel in Experimental Example 3.
[22] Figure 6 shows the results of "C NMR analysis in the NMR analysis for
the material
obtained at RRT 1.44 position in the mixture obtained by thermally decomposing
pa-
clitaxel in Experimental Example 3.
[23] Figure 7 shows the results of COSY (Correlation Spectroscopy) analysis
in the NMR
analysis for the material obtained at RRT 1.44 position in the mixture
obtained by
thermally decomposing paclitaxel in Experimental Example 3.
[24] Figure 8 shows the results of HMBC (Heteronuclear Multiple Bond
Correlation
Spectroscopy) analysis in the NMR analysis for the material obtained at RRT
1.44
position in the mixture obtained by thermally decomposing paclitaxel in
Experimental
Example 3.
[25] Figure 9 is the resulting chromatogram of HPLC analysis conducted in
Experimental
Example 6.
Best Mode for Carrying out the Invention
[26] The present invention is explained in more detail below.
5
CA 02993928 2018-01-26
WO 2017/018820 PCT/KR2016/008270
[27] The pharmaceutical composition of an embodiment of the present
invention
comprises a purified amphiphilic block copolymer comprising a hydrophilic
block (A)
and a hydrophobic block (B).
[28] According to one embodiment of the present invention, the amphiphilic
block
copolymer comprises an A-B type diblock copolymer consisting of a hydrophilic
block
(A) and a hydrophobic block (B), or a B-A-B type triblock copolymer.
[29] According to one embodiment of the present invention, the amphiphilic
block
copolymer may comprise the hydrophilic block in an amount of 20 to 95% by
weight,
and more concretely 40 to 95% by weight, based on the total weight of the
copolymer.
In addition, the amphiphilic block copolymer may comprise the hydrophobic
block in
an amount of 5 to 80% by weight, and more concretely 5 to 60% by weight, based
on
the total weight of the copolymer.
[30] According to one embodiment of the present invention, the amphiphilic
block
copolymer may have a number average molecular weight of 1,000 to 50,000
Daltons,
and more concretely 1,500 to 20,000 Daltons.
[31] According to one embodiment of the present invention, the hydrophilic
block is a
polymer having biocompatibility and may comprise one or more selected from the
group consisting of polyethylene glycol or derivatives thereof,
polyvinylpyrrolidone,
polyvinyl alcohol, polyacrylamide and combinations thereof, and more
concretely, it
may comprise one or more selected from the group consisting of polyethylene
glycol,
monomethoxypolyethylene glycol and combinations thereof. The hydrophilic block
may have a number average molecular weight of 200 to 20,000 Daltons, and more
concretely 200 to 10,000 Daltons.
[32] According to one embodiment of the present invention, the hydrophobic
block is a
polymer having biodegradability and may be a polymer of monomers derived from
alpha (a)-hydroxy acid. Concretely, it may comprise one or more selected from
the
group consisting of polylactide, polyglycolide, polymandelic acid,
polycaprolactone,
polydioxan-2-one, polyamino acid, polyorthoester, polyanhydride, polycarbonate
and
combinations thereof, and more concretely, it may comprise one or more
selected from
the group consisting of polylactide, polyglycolide, polycaprolactone,
polydioxan-2-one
and combinations thereof. The hydrophobic block may have a number average
molecular weight of 200 to 20,000 Daltons, and more concretely 200 to 10,000
Daltons.
[33] According to one embodiment of the present invention, an amphiphilic
block
copolymer comprising a hydrophobic polymer block of poly(alpha (a)-hydroxy
acid)
may be synthesized by a known ring-opening polymerization method using a hy-
drophilic polymer having hydroxyl group as an initiator, and a lactone monomer
of
alpha (a)-hydroxy acid. For example, L-lactide or D,L-lactide may be
polymerized
6
CA 02993928 2018-01-26
WO 2017/018820 PCT/KR2016/008270
with hydrophilic polyethylene glycol or monomethoxypolyethylene glycol having
hydroxyl group as an initiator by ring-opening. Synthesis of diblock or
triblock
copolymer is possible according to the number of hydroxyl group existing in
the hy-
drophilic block which is the initiator. In the ring-opening polymerization, an
organometallic catalyst such as tin oxide, lead oxide, tin octoate, antimony
octoate, etc.
may be used, and tin octoate having biocompatibility is preferably used in
preparing
polymer for medical use.
[34] In an embodiment of the present invention, as the amphiphilic block
copolymer, a
purified one is used. According to a preferable embodiment of the present
invention,
the amphiphilic block copolymer is one that has been purified by sublimation.
[35] The purification by sublimation may be conducted at a temperature of
preferably
80 C or higher and lower than 120 C and more preferably 80 to 100 C, and under
a
pressure of a vacuum degree of preferably 10 ton or less, more preferably 5
ton or less
and even more preferably 1 ton or less, for a time of preferably 10 to 74
hours, more
preferably 10 to 48 hours and even more preferably 24 to 48 hours. Conducting
the pu-
rification by sublimation under such conditions can minimize the change in
molecular
weight of the copolymer and remove impurities therefrom.
[36] The pharmaceutical composition of an embodiment of the present
invention
comprises, as active ingredient, one or more poorly water-soluble drugs
selected from
the group consisting of paclitaxel and docetaxel.
[37] According to one embodiment of the present invention, the
pharmaceutical com-
position may further comprise, as additional active ingredient, one or more
poorly
water-soluble drugs other than paclitaxel and docetaxel. As such an additional
active
ingredient, one or more taxane anticancer agents selected from the group
consisting of
7-epipaclitaxel, t-acetylpaclitaxel, 10-desacetylpaclitaxel, 10-desacety1-7-
epipaclitaxel,
7-xylosylpaclitaxel, 10-desacety1-7-glutarylpaclitaxel, 7-N,N-
dimethylglycylpaclitaxel,
7-L-alanylpaclitaxel and cabazitaxel, may be used.
[38] The pharmaceutical composition of an embodiment of the present
invention may
comprise the poorly water-soluble drug in an amount of 0.1 to 50 parts by
weight, and
more concretely 0.5 to 30 parts by weight, based on 100 parts by weight of the
am-
phiphilic block copolymer. If the amount of the poorly water-soluble drug is
too small
as compared with that of the amphiphilic block copolymer, the weight ratio of
the am-
phiphilic copolymer used per drug is high and thus the time for reconstitution
may
increase. On the other hand, if the amount of the poorly water-soluble drug is
too large,
there may be a problem of rapid precipitation of the poorly water-soluble
drug.
[39] As used herein, the "initial" amount of the poorly water-soluble drug
means the
weight of the poorly water-soluble drug incorporated when the pharmaceutical
com-
position was prepared.
7
CA 02993928 2018-01-26
WO 2017/018820 PCT/KR2016/008270
[40] In an embodiment of the present invention, the pharmaceutical
composition contains,
when stored at the accelerated condition (40 C) for 6 months, a related
compound rep-
resented by the following Formula 1 in an amount of less than 0.12 part by
weight,
based on 100 parts by weight of the initial amount of the poorly water-soluble
drug:
[41] [Formula 11
[42]
0013
___________ // 0 Ft3C
j)
0
R2
\o 143C
[43] wherein
[44] R1 is H or COCH3, and R2 is phenyl or 0(CH3)3.
[45] According to one embodiment of the present invention, the poorly water-
soluble drug
is paclitaxel, and the related compound(s) may include the compound
represented by
the following Formula la:
[46] [Formula la]
[47] C
o
___________ \
0
0 HiG
\
0
;'11110,*
[48] The pharmaceutical composition of an embodiment of the present
invention may
contain, when stored at the accelerated condition (40 C) for 6 months, a
related
compound of Formula 1 (particularly, Formula la) in an amount of less than
0.12 part
by weight, preferably 0.1 part by weight or less, more preferably 0.06 part by
weight or
less, even more preferably 0.05 part by weight or less, and most preferably
0.04 part by
weight or less, based on 100 parts by weight of the initial amount of the
poorly water-
soluble drug.
[49] The pharmaceutical composition of an embodiment of the present
invention may
contain, when stored at the severe condition (80 C) for 3 weeks, a related
compound of
8
CA 02993928 2018-01-26
WO 2017/018820 PCT/KR2016/008270
Formula 1 (particularly, Formula la) in an amount of less than 0.93 part by
weight,
preferably 0.8 part by weight or less, more preferably 0.6 part by weight or
less, even
more preferably 0.4 part by weight or less, and most preferably 0.2 part by
weight or
less, based on 100 parts by weight of the initial amount of the poorly water-
soluble
drug.
[501 In an embodiment of the present invention, the pharmaceutical
composition, which
contains a specific related compound in an amount within a specified limit, is
a com-
mercially available composition since it can be produced on a large scale.
[511 In an embodiment, the pharmaceutical composition of the present
invention does not
have ether, for example, diethyl ether, at all.
[52] In an embodiment, the pharmaceutical composition of the present
invention does not
have metal salt, for example, alkali metal salt and/or salt for salting out,
for example,
NaC1 or KC1, at all.
[531 The pharmaceutical composition of an embodiment of the present
invention can be
prepared by a method comprising (a) purifying an amphiphilic block copolymer
comprising a hydrophilic block (A) and a hydrophobic block (B); (b) dissolving
one or
more poorly water-soluble drugs selected from the group consisting of
paclitaxel and
docetaxel, and the purified amphiphilic block copolymer in an organic solvent;
and (c)
adding an aqueous solvent to the solution obtained in step (b) to form
polymeric
micelles.
[541 The purification of the amphiphilic block copolymer is explained
above, and a con-
ventional method can be used for the formation of the polymeric micelles.
[551 In the method for preparing a pharmaceutical composition of an
embodiment of the
present invention, as the organic solvent, a water-miscible organic solvent,
for
example, selected from the group consisting of alcohol (for example, ethanol),
acetone,
tetrahydrofuran, acetic acid, acetonitrile and dioxane and combinations
thereof can be
used, but it is not limited thereto. In addition, as the aqueous solvent, one
selected from
the group consisting of conventional water, distilled water, distilled water
for injection,
physiological saline, 5% glucose, buffer and combinations thereof can be used,
but it is
not limited thereto.
[561 The method for preparing a pharmaceutical composition of an embodiment
of the
present invention may further comprise removing an organic solvent after said
step (a).
[571 In an embodiment, the method may further comprise lyophilizing the
micelle com-
position with addition of a lyophilization aid. The lyophilization aid may be
added for
the lyophilized composition to maintain a cake form. In another embodiment,
the
lyophilization aid may be one or more selected from the group consisting of
sugar and
sugar alcohol. The sugar may be one or more selected from lactose, maltose,
sucrose or
trehalose. The sugar alcohol may be one or more selected from mannitol,
sorbitol,
9
CA 02993928 2018-01-26
WO 2017/018820 PCT/KR2016/008270
maltitol, xylitol and lactitol. The lyophilization aid may also function to
facilitate ho-
mogeneous dissolution of the lyophilized polymeric micelle composition upon
recon-
stitution. The lyophilization aid may be contained at an amount of 1 to 90
weight%,
particularly, 1 to 60 weight%, more particularly 10 to 60 weight%, based in a
total
weight of the lyophilized composition.
[58] The present invention is explained in more detail by the following
examples.
However, these examples seek to illustrate the present invention only, and the
scope of
the present invention is not limited by the examples in any manner.
[59] [EXAMPLES]
[60] Preparation Example 1: Synthesis of diblock copolymer consisting of
monomethoxypolyethylene glycol and D,L-lactide (mPEG-PDLLA) and purification
by sublimation method
[61] 150 g of monomethoxypolyethylene glycol (mPEG, number average
molecular
weight = 2,000) was fed into a 500-ml round-bottom flask equipped with an
agitator,
and agitated at 120 C under vacuum condition for 2 hours to remove moisture.
0.15 g
of tin octoate (Sn(Oct)2) dissolved in 200 [11 of toluene was added in the
reaction flask,
and further agitated under vacuum condition for 1 hour to distill and remove
toluene.
150 g of D,L-lactide was then added and agitated under nitrogen atmosphere for
dis-
solution. After D,L-lactide was dissolved completely, the reactor was tightly
sealed
and the polymerization reaction was conducted at 120 C for 10 hours. After the
reaction was terminated, under agitation with a magnetic bar, the reactor was
connected to a vacuum pump and the product was purified under a pressure of 1
ton or
less by a sublimation method for 7 hours to obtain 262 g of mPEG-PDLLA in
molten
state. The molecular weight (Mn: ¨3740) was calculated by analyzing with 11-I-
NMR
obtaining relative intensities of appropriate peaks with reference to -OCH3
which is the
terminal group of monomethoxypolyethylene glycol.
[62] Preparation Example 2: Purification of diblock copolymer (mPEG-PDLLA)
by
sublimation method
[63] 30 g of mPEG-PDLLA, which was obtained in the polymerization reaction
process
of Preparation Example 1 before conducting the purification process, was fed
into a
one-necked flask and dissolved at 80 C. Under agitation with a magnetic bar,
the
reactor was connected to a vacuum pump and the product was purified under a
pressure of 1 ton or less by a sublimation method for 24 hours and 48 hours.
[64] Preparation Example 3: Purification of diblock copolymer (mPEG-PDLLA)
by
sublimation method
[65] Except that the purification temperature was 100 C, the purification
was conducted
by the same method as in Preparation Example 2.
[66] Preparation Example 4: Purification of diblock copolymer (mPEG-PDLLA)
by
10
CA 02993928 2018-01-26
WO 2017/018820 PCT/KR2016/008270
sublimation method
[67] Except that the purification temperature was 120 C, the purification
was conducted
by the same method as in Preparation Example 2.
[68] Preparation Example 5: Purification of diblock copolymer (mPEG-PDLLA)
by
adsorption method using aluminum oxide (A1203)
[69] 30 g of mPEG-PDLLA, which was obtained in the polymerization reaction
process
of Preparation Example 1 before conducting the purification process, was fed
into a
one-necked flask and dissolved by adding acetone (60 ml). Aluminum oxide (15
g)
was added thereto and completely mixed. The one-necked flask was connected to
a
rotary evaporator, and the contents were mixed at 50 C at 60 rpm for 2 hours.
The
solution was then filtered at room temperature with PTFE filter paper (1 [im)
to
remove aluminum oxide. The filtered acetone solution was distilled using a
rotary
evaporator at 60 C under vacuum to remove acetone, thereby to obtain the
purified
mPEG-PDLLA. The molecular weight (Mn: ¨3690) was calculated by analyzing with'
H-NMR obtaining relative intensities of appropriate peaks with reference to -
OCH3
which is the terminal group of monomethoxypolyethylene glycol.
[70] The molecular weight change of mPEG-PDLLA according to the
purification
conditions in the above Preparation Examples 2 to 5 is shown in the following
Table 1.
[71] [Table 11
[72] Purification
Purification Molecular weight
Temperature ( C) Time (hr) (Mn)
24 3740
Preparation Example 2 80
48 3740
24 3720
Preparation Example 3 100
48 3700
24 3650
Preparation Example 4 120
48 3550
Preparation Example 5 A1203 purification 3690
[73] From the results of Table 1, it can be seen that the reduced amount of
the molecular
weight of mPEG-PDLLA increases as the purification temperature becomes higher.
The purification condition of 80 to 100 C and 24 to 48 hours, particularly 100
C and
24 hours, can be thought of as efficient.
[74] Comparative Example 1: Preparation of polymeric micelle composition
containing paclitaxel
[75] 1 g of paclitaxel and 5 g of mPEG-PDLLA obtained in Preparation
Example 1 were
weighed, and 4 ml of ethanol was added thereto and agitated at 60 C until the
mixture
was completely dissolved to form a clear solution. Ethanol was then removed by
dis-
tillation under reduced pressure using a rotary evaporator equipped with a
round-
bottom flask at 60 C for 3 hours. The temperature was then lowered to 50 C,
and 140
ml of distilled water at room temperature was added and reacted until the
solution
11
CA 02993928 2018-01-26
WO 2017/018820 PCT/KR2016/008270
became clear in blue color to form polymeric micelles. As a lyophilization
aid, 2.5 g of
anhydrous lactose was added thereto and dissolved completely, filtered using a
filter
with a pore size of 200 nm, and freeze-dried to obtain a polymeric micelle
composition
containing paclitaxel in powder form.
[76] Example 1: Preparation of polymeric micelle composition containing
paclitaxel
[77] Except that mPEG-PDLLA purified for 24 hours in Preparation Example 3
was used,
a polymeric micelle composition containing paclitaxel was prepared by the same
method as in Comparative Example 1.
[78] Example 2: Preparation of polymeric micelle composition containing
paclitaxel
[79] Except that mPEG-PDLLA purified in Preparation Example 5 was used, a
polymeric
micelle composition containing paclitaxel was prepared by the same method as
in
Comparative Example 1.
[80] ExperimentalExample 1: Isolation of related compound by liquid chro-
matography
[81] To a vial containing 100 mg of polymeric micelle composition
containing paclitaxel,
which had been subjected to the six-month acceleration test (temperature: 40
C), 16.7
ml of deionized water (DW) was fed and the contents were completely dissolved,
and
the total amount of the liquid was taken and transferred to a 20-ml volumetric
flask,
and the marked line was met to make the total volume 20 ml (5.0 mg/ml). 2 ml
of this
liquid was taken and transferred to a 10-ml volumetric flask, and the marked
line was
met with acetonitrile to make the total volume 10 ml (1 mg/ml). For the above
com-
position, related compound was isolated and fractionally collected using the
following
liquid chromatography.
[82] Conditions for liquid chromatography
[83] 1) Column: Poroshell 120 PFP (4.6 x 150 mm, 2.7 [im, Agilent)
[84] 2) Mobile phase: A: DW / B: Acetonitrile
[85] Time (min) %A %B
0.00 65 35
25.00 45 55
28.00 45 55
30.00 65 35
35.00 65 35
[86] 3) Flow rate: 0.6 ml/min
[87] 4) Injection volume: 10 [11
[88] 5) Detector: UV absorption spectrophotometer (Measurement wavelength:
227 nm)
[89] The resulting chromatogram of HPLC analysis is shown in Figure 1.
[90] ExperimentalExample 2: Qualitative analysis of related compound of RRT
1.44
using LC/MS/MS
[91] The related compound isolated in Experimental Example 1 (RRT: 1.44
0.05
12
CA 02993928 2018-01-26
WO 2017/018820 PCT/KR2016/008270
(1.39-1.49)) was qualitatively analyzed by MS scan of liquid chromatography-
mass
spectrometer (LC/MS/MS). In the following measurement, as the LC/MS/MS, liquid
chromatography 1200 series and electrospray ionization mass spectrometer 6400
series
(Agilent, US) were used. The conditions for analysis were as follows.
[92] Conditions for liquid chromatography
[93] 1) Column: Cadenza HS-C18 (3.0 x 150 mm, 3 [im, Imtakt)
[94] 2) Mobile phase: A: 0.5 mM ammonium acetate with 0.03% acetic acid /
B: Ace-
tonitrile
[95] Time (min) %A %B
0.00 80 20
4.00 55 45
9.00 55 45
9.10 80 20
15.00 80 20
[96] 3) Flow rate: 0.4 ml/min
[97] 4) Injection volume: 2 [11
[98] 5) Detector: UV absorption spectrophotometer (Measurement wavelength:
227 nm)
[99] Conditions for electrospray ionization mass spectrometer
[100] 1) Ionization: Electrospray Ionization, Positive (ESI+)
[101] 2) MS Method: M52 scan / Product ion scan
[102] 3) Ion source: Agilent Jet Stream ESI
[103] 4) Nebulizer gas (pressure): Nitrogen (35 psi)
[104] 5) Ion spray voltage: 3500 V
[105] 6) Drying gas temperature (flow rate): 350 C (7 L/min)
[106] 7) Sheath gas temperature (flow rate): 400 C (10 L/min)
[107] 8) Fragmentor: 135 V
[108] 9) Nozzle voltage: 500 V
[109] 10) Cell accelerator voltage: 7 V
[110] 11) EMV: 0 V
[111] 12) Collision energy: 22 V
[112] 13) Precursor ion: m/z 836.2
[113] 14) Mass scan range: m/z 100-1500
[114] The substance for analysis, which was isolated and came out of the
detection stage,
was set to flow in the mass spectrometer, and at that time the detected ion of
related
compound was qualitatively analyzed selecting the characteristic ion of mass
spectrum
[M+H].
[115] Experimental Example 3: Thermal decomposition test of paclitaxel
[116] In the related compounds which were fractionally collected from the
polymeric
nanoparticle composition containing paclitaxel in Experimental Example 1, many
13
CA 02993928 2018-01-26
WO 2017/018820 PCT/KR2016/008270
polymers existed together and thus direct experiment was very difficult. As a
result of
the qualitative analysis in the preliminary experiment using LC/MS/MS, the
related
compound was presumed as compounds produced by the elimination of water from
pa-
clitaxel. Accordingly, as a method of eliminating water molecule, an
experiment of
heating paclitaxel was carried out to confirm whether the presumed related
compound
was produced. First, 1 g of paclitaxel was vacuum-dried at 170 C for 2-3 hours
and
dissolved completely in 45 ml of acetonitrile, and 5 ml of DW was then added
thereto.
By using this solution, the related compound of RRT 1.44 was isolated and
fractionally
collected on prep-LC.
[117] Experimental Example 4: Analysis of the related compound at RRT 1.44
position
produced after the thermal decomposition reaction of paclitaxel using LC/MS/MS
[118] The related compound fractionally collected in Experimental Example 3
(RRT:
1.44 0.05 (1.39-1.49)) was analyzed by liquid chromatography and mass
spectrometer
(LC/MS/MS). According to the HPLC analysis results, the material fractionally
collected in Experimental Example 3 showed an HPLC peak at the same position
as
that of the related compound of RRT 1.44 in the polymeric micelle composition
(Figure 4). This material was further analyzed by LC/MS/MS. As a result of the
MS
scan first, m/z 836.3 amu which is [M+H1+ was shown (Figure 3). The product
ion scan
was then conducted and the results thereof were shown in Figure 4. The results
of the
related compound of RRT 1.44 formed in the polymeric nanoparticle composition
containing paclitaxel, which had been subjected to the six-month acceleration
test,
were shown together. In conclusion, it could be confirmed that the material
fractionally
collected at RRT 1.44 after thermally decomposing paclitaxel in Experimental
Example 3 was the compound having the same structure as that of the related
compound at RRT 1.44 position after the six-month acceleration test of the
polymeric
micelle composition.
[119] Conditions for liquid chromatography
[120] 1) Column: Poroshell 120 PFP (4.6 x 150 mm, 2.7 [im, Agilent)
[121] 2) Mobile phase: A: DW / B: Acetonitrile
[122] Time (min) %A %B
0.00 65 35
25.00 45 55
28.00 45 55
30.00 65 35
35.00 65 35
[123] 3) Flow rate: 0.6 ml/min
[124] 4) Injection volume: 10 [11
[125] 5) Detector: UV absorption spectrophotometer (Measurement wavelength:
227 nm)
14
CA 02993928 2018-01-26
WO 2017/018820 PCT/KR2016/008270
[126] Conditions for electrospray ionization mass spectrometer
[127] 1) Ionization: Electrospray Ionization, Positive (ESI+)
[128] 2) MS Method: M52 scan / Product ion scan
[129] 3) Ion source: Agilent Jet Stream ESI
[130] 4) Nebulizer gas (pressure): Nitrogen (35 psi)
[131] 5) Ion spray voltage: 3500 V
[132] 6) Drying gas temperature (flow rate): 350 C (7 L/min)
[133] 7) Sheath gas temperature (flow rate): 400 C (10 L/min)
[134] 8) Fragmentor: 135 V
[135] 9) Nozzle voltage: 500 V
[136] 10) Cell accelerator voltage: 7 V
[137] 11) EMV: 0 V
[138] 12) Collision energy: 22 V
[139] 13) Precursor ion: m/z 836.2
[140] 14) Mass scan range: m/z 100-1500
[141] Experimental Example 5: NMR analysis of material obtained at RRT 1.44
position from the mixture obtained by thermally decomposing paclitaxel
[142] The material obtained at RRT 1.44 position from the mixture obtained
by thermally
decomposing paclitaxel in Experimental Example 3 was analyzed by NMR. In the
NMR analysis, the results of 1H NMR analysis are shown in Figure 5, the
results of "C
NMR analysis are shown in Figure 6, the results of COSY (Correlation
Spectroscopy)
analysis are shown in Figure 7, and the results of HMBC (Heteronuclear
Multiple
Bond Correlation Spectroscopy) analysis are shown in Figure 8.
[143] According to the analysis results, it could be confirmed that the
material obtained at
RRT 1.44 position from the mixture obtained by thermally decomposing
paclitaxel
(i.e., the related compound (RRT: 1.44 0.05 (1.39-1.49)) in the polymeric
micelle
composition containing paclitaxel which had been subjected to the six-month ac-
celeration test) was the compound of the following water-eliminated form of pa-
clitaxel.
[144] CH3
Ph2
11). OH
H;C 0
'tp
0
"
\ I/ / 10 9
C11-7
7
3' 2' r 17 16 S 6
011111113 1 H301110,, 15 ¨CH3
3 5
0 14 2 4 \op
0
HO
0 '"20
Phi
100 H3C
15
CA 02993928 2018-01-26
WO 2017/018820 PCT/KR2016/008270
[145] One molecule of water-eliminated form of paclitaxel: C47H49N013
(835.91 g/mol)
[146] Conditions for Nuclear Magnetic Resonance spectroscopy
[147] 1.1H
[148] 1) NMR equipment: Brucker DRX-300 equipped with a temperature
controller
[149] 2) Sample: 1-10mg sample/ 0.6mL chloroform-d in 5mm o.d. NMR tube (In
all
NMR experiments, the same sample was used)
[150] 3) Probe head: Brucker 5mm QNP
[151] 4) Proton 90 pulse: 11[1sec.
[152] 5) Relaxation delay/ Number of scan: 4.0 sec/ 8
[153] 2.13C
[154] 1) Probe head: Brucker 5mm QNP
[155] 2) Carbon 90 pulse, acquisition time: 8[1sec, 4.0 sec
[156] 3) Relaxation delay/ Number of scan: 0.5 sec/ 36,092
[157] 3. COSY
[158] 1) NMR equipment: Brucker DRX-300
[159] 2) Probe head: Brucker 5mm QNP
[160] 3) Pulse sequence: cosyqf45
[161] 4) Proton 90 pulse: 11[1sec.
[162] 5) Relaxation delay/ Number of scan/ Number of experiments for w1:
1.2 sec/ 4/ 256
[163] 4. HMQC
[164] 1) NMR equipment: Brucker DRX-300
[165] 2) Probe head: Brucker 5mm QNP
[166] 3) Pulse sequence: inv4ph
[167] 4) Proton 90 pulse: 11[1sec.
[168] 5) Relaxation delay/ Number of scan/ Number of experiments for w1:
1.2 sec/ 64/
256
[169] 6) Temperature, 1/2(JcH): 300K, 3.5 msec
[170] 5. HMBC
[171] 1) NMR equipment: Brucker DRX-300
[172] 2) Probe head: Brucker 5mm QNP
[173] 3) Pulse sequence: inv41p1rndqf
[174] 4) Proton 90 pulse: 11[1sec.
[175] 5) Relaxation delay/ Number of scan / Number of experiments for al:
1.5 sec/ 256/
256
[176] 6) Temperature/ 1/2(JcH): 300K/ 3.5 msec
[177] 6. DEPT
[178] 1) Pulse sequence: DEPT 135
11791 2) Carbon 90 pulse, acquisition time: 8[1sec, 4.0 sec
16
CA 02993928 2018-01-26
WO 2017/018820
PCT/KR2016/008270
[180] 3) Relaxation delay/ Number of scan/ 1/2(Jc.): 1.2 sec/ 5628/ 3.5
msec
[181] Experimental Example 6: Comparative test of storage stability of
polymeric
micelle containing drug at severe condition (80 C)
[182] The polymeric micelle compositions of paclitaxel prepared in
Comparative Example
1 and Examples 1 and 2 were kept in an oven at 80 C for 3 weeks, and the com-
positions were then analyzed with HPLC to compare the amounts of related
compound. The test solution was prepared by dissolving the micelle composition
in
80% acetonitrile aqueous solution and diluting to 600 ppm concentration of
paclitaxel.
The resulting chromatogram of HPLC analysis is shown in Figure 9 and the
change in
the amount of related compound (%) according to the severe test time is shown
in the
following Table 2.
[183] HPLC conditions
[184] Column: Diameter 2.7 [im, poroshell 120PFP (4.6 x 150 mm, 2.7 [1m)
(Agilent
column)
[185] Mobile phase
[186] Time (min) Water : Acetonitrile
0-25 65:35 45:55
25-28 45:55
28-30 45:55 -4 65:35
30-35 65:35
[187] Detector: UV absorption spectrophotometer (227 nm)
[188] Flow rate: 0.6 ml/min
[189] Amount of each related compound (%) = 100(Ri/Ru)
[190] Ri: Area of each related compound detected in test solution analysis
[191] Ru: Sum of all peak areas detected in test solution analysis
[192] [Table 21
[193]
RRT*
Sample-Storage time 0.87 0.02 0.96 0.02 1.10 0.02
1.12+0.02 1.44 0.05
(0.85-0.89) (0.94 1.00
¨0.98) (1.08-1.12) (1.10-1.14) (1.39-1.49)
Comparative Example 1-0 day(d) 0.04% 0.03% 99.74%
Comparative Example 1-3 weeks(w) 0.76% 1.12% 92.59% 0J8%
0.27% 0.93%
Example 1-0 day(d) 0.03% 0.02% 99.64% 0.02%
Example 1-3 weeks(w) 0.11% 0.15% 95.44% 0.06% 0.08%
0,22%
Example 2-0 day(d) 0.02% 99.72%
Example 2-3 weeks(w) 0.40% 1.04% 93.94% 0.08% 0.05%
0.11%
[194] RRT 0.87 0.02: Paclitaxel, oxetane ring opened compound
[195] RRT 0.96 0.02: Paclitaxel, oxetane ring opened compound
[196] RRT 1.00: Paclitaxel
[1971 RRT 1.10 0.02: Paclitaxel, L-lactide reaction compound
17
CA 02993928 2018-01-26
WO 2017/018820 PCT/KR2016/008270
[198] RRT 1.12 0.02: Paclitaxel, D-lactide reaction compound
[199] RRT 1.44 0.05: Paclitaxel, water eliminated compound
[200]
[201] From Table 2 and Figure 9, it can be known that the stability of the
polymeric
micelle pharmaceutical composition of Example 1 or 2 was improved as compared
with the composition of Comparative Example 1 and the reduction of paclitaxel
amount was relatively smaller, whereby the effect of the drug contained in the
com-
position can be maintained more stably.
[202] Experimental Example 7: Comparative test of storage stability of
polymeric
micelle containing drug at accelerated condition (40 C)
[203] Except that the polymeric micelle composition of paclitaxel prepared
in Comparative
Example 1 and Example 1 respectively was kept in a stability tester at 40 C
for 6
months, the test was conducted by the same method as in Experimental Example
6.
The change in the amount of related compound (%) according to the acceleration
test
time is shown in the following Table 3.
[204] [Table 31
[205]
RRT*
Sample-Storage time 0.8710.02 1.1010.02 1.1210.02
1.4410.05
(0.85-0.89) (1.08-1.12) (1.10-1.14) (1.39-4 .49)
Comparative Example 1-6 months 0.17% 0.22% 0.36% 0.12%
Example 1-6 months 0.04% 0.05% 0.02% 0.04%
[206] The above test result shows an average value of the amounts of each
related
compound and paclitaxel in the test conducted for 3 or more polymeric micelle
com-
positions of different batches.
[207] Through Experimental Example 7, it has been proven that the polymeric
micelle
pharmaceutical composition of Example 1, when stored at the accelerated
storage tem-
perature (40 C) for 6 months, has lower amount of related compound than the
com-
position of Comparative Example 1.