Note: Descriptions are shown in the official language in which they were submitted.
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
METHOD OF DECONTAMINATING METAL SURFACES IN A HEAVY WATER
COOLED AND MODERATED NUCLEAR REACTOR
TECHNICAL FIELD OF THE INVENTION
The invention relates to a method of decontaminating a heavy water cooled
and moderated nuclear reactor, and in particular to a method of
decontaminating
metal surfaces in the primary circuit and/or the moderator circuit of a heavy
water
nuclear reactor wherein the metallic surface has a radioactive oxide layer.
BACKGROUND OF THE INVENTION
The piping of a nuclear reactor is usually made of stainless steel, carbon
steel
and/or Co alloys. The steam generator tubes and main surfaces inside the
primary circuit may include Ni alloys. Under operational conditions of a
nuclear
reactor metal ions are leached out of the alloys of the piping and are
dissolved
and transported into the coolant. When passing the reactor core during
operation,
some of the metal ions are activated to form radioisotopes. During operation
of
the reactor these metal ions and radioisotopes are deposited as a radioactive
oxide layer on metal surfaces inside the reactor cooling system. The removal
of
these radioactive metal oxide layers is necessary to reduce the level of
personnel
radiation exposure prior to carrying out inspection, maintenance, repair and
dismantling procedures on the reactor cooling system.
Many procedures are described to remove the oxide layers containing
radioisotopes from metal surfaces of the cooling system in a light water
cooled
nuclear reactor. A commercially successful method comprises the steps of
treating the oxide layer with an oxidant such as permanganate in order to
convert
Cr(III) to Cr(VI), and subsequently dissolving the oxide layer under acidic
conditions using a decontamination solution of an organic acid such as e.g.
oxalic
acid. The organic acid additionally serves to reduce a possible excess of
oxidant
from the preceding oxidation step, and to reduce the dissolved Cr(VI) to
Cr(III) in
the decontamination solution. An additional reducing agent can be added to
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 2 -
remove the oxidant and convert Cr(VI) to Cr(III). The decontamination solution
containing metal ions and radioisotopes originating from the oxide layer, such
as
Fe(ll), Fe(III), Ni(II), Co(II), Co(III) and Cr(III), is then passed through
an ion
exchange resin to remove the radioisotopes and some or all of the metal ions
from the decontamination solution. The organic acid in the decontamination
solution is decomposed by photocatalytic oxidation to form carbon dioxide and
water.
However, this decontamination method is not directly applicable to a heavy
water cooled and moderated reactor.
The operation of a heavy water cooled and moderated nuclear reactor
requires that the concentration of heavy water is maintained at >99.8 percent.
Replacing the heavy water by light water prior to decontamination is too
costly
and would also require large storage capacities for the low level contaminated
heavy water, as well as sophisticated test methods to control the heavy water
concentration for re-starting a power generating operation in the nuclear
reactor.
U.S. Patent 3,737,373 discloses that a heavy water cooled and moderated
reactor can be decontaminated by employing a heavy water solution of
deuterated oxalic acid. The deuterated oxalic acid is produced by dissolving
oxalic acid anhydride in heavy water. The radioactive substances eluted in the
deuterated oxalic acid solution are removed by decomposing the deuterated
oxalic acid by means of irradiation with gamma-rays. The radioactive
substances
are precipitated and removed by filtration. The ions in the filtered heavy
water are
removed by ion exchange resin techniques.
CA 1 062 590 is directed to a method of decontaminating a heavy water
moderated and cooled nuclear reactor or a light water cooled reactor. A
relatively
small quantity of acidic reagent composition is injected into the circulating
coolant
of the reactor, which is shut down but not defueled, so as to provide a dilute
solution of reagent which dissolves radioactive contaminants in the system.
The
coolant is then passed through cationic exchange resin to remove the
contaminant and leave the regenerated reagent which is returned to the cooling
system. When the cationic resin stops removing contaminants it is removed and
discarded. The reagent is finally removed from the system by anionic exchange
resin. Suitable reagents include mixtures of certain organic acids such as
oxalic
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 3 -
acid, acetic acid, and citric acid with or without chelating agents such as
EDTA or
hydrazine. In case of heavy water moderated and cooled reactors, a preferred
cationic resin is in D+ form, and preferred anionic resin is in OD- form so
that the
original coolant composition is restored.
CA 1 136 398 A is an improvement over the method disclosed in
CA 1 062 590 A. A metal surface contaminated with radioactive materials is
decontaminated by circulating an aqueous solution of decontaminating reagents
comprising oxalic acid, formic acid, citric acid and EDTA. The efficacy of the
organic acid decontaminating reagents is prolonged under ionizing radiation by
the inclusion of formic acid therein. Where heavy water coolant or moderator
is
used in the decontamination, the ion exchange resins are converted to the D+
and OD- forms in order to avoid downgrading the deuterium content.
These prior art decontamination methods suffer from the fact that a high
number of treatment cycles are necessary in order to completely remove the
metal oxide layer and to achieve a satisfactory reduction of activity on the
metal
surfaces, thus resulting in a high amount of radioactive waste produced
therewith.
SUMMARY OF THE INVENTION
Therefore, it is an object of the invention to provide an effective
decontamination method for a heavy water cooled and moderated reactor which
prevents the entrainment of light water into the primary heavy water coolant
during the decontamination treatment and which reduces the number of
treatment cycles and minimizes the amount of radioactive waste resulting from
the decontamination treatment.
According to the invention, the object is solved by a method of
decontaminating a metal surface in a heavy water cooled and moderated nuclear
reactor, wherein the metal surface has a coating comprising one or more metal
oxides and radioisotopes, and wherein the metal surface is in contact with a
heavy water coolant or moderator, the method comprising one or more treatment
cycles each comprising:
a) an oxidation step wherein the metal surface is contacted with a
solution of an oxidant in heavy water;
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 4 -
b) a decontamination step wherein the metal surface subjected to the
oxidation step is contacted with a decontamination reagent in heavy
water for dissolving at least part of the one or more metal oxides and
to form a decontamination solution containing the decontamination
reagent, one or more metal ions dissolved from the metal oxides and
the radioisotopes, and wherein the decontamination solution is passed
over an ion exchange resin to immobilize the metal ions and the
radioisotopes; and
c) a decomposition step wherein the decontamination reagent in the
decontamination solution is decomposed;
wherein the oxidant, the decontamination reagent and the ion exchange resin
are provided in a deuterated form and/or are free of active hydrogen.
The decontamination method of the present invention avoids a dilution of the
heavy water coolant and moderator by light water during the decontamination
treatment cycles since all decontamination chemicals are provided in their
deuterated form and no light water is formed as a byproduct.
Therefore, the primary coolant and moderator itself can be used as the
solvent for the decontamination chemicals. No further cleaning of the heavy
water
coolant and moderator after decontamination is necessary to remove light water
impurities. The process also saves the costs involved with replacing the heavy
water in the reactor cooling and moderating system by light water just for
performing a decontamination of the primary coolant and moderator circuit of
the
heavy water reactor.
Heavy water is widely available at the facilities of a heavy water cooled and
moderated reactor, and can be used for preparing a bulk of deuterated ion
exchange resins. The deuterated ion exchange resins can then be used to
produce a stock solution of the deuterated oxidant in heavy water for use in
the
oxidation step, as well as a stock solution of the deuterated decontamination
reagent in heavy water for dissolving the metal oxide coating in the
decontamination step.
The decontamination solution containing metal ions and radioisotopes from
the dissolved metal oxide coating is passed over the deuterated ion exchange
resin to immobilize radioactive components and metal ions dissolved in the
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 5 -
decontamination solution. Thus, the decontamination solution is depleted of
the
radioisotopes and metal ions, while the decontamination reagent is
regenerated.
In the following decomposition step, the decontamination reagent is
decomposed.
The side products of the decontamination treatment are only carbon dioxide
and heavy water so that the heavy water coolant will not be diluted with light
water during the decontamination treatment. Oxidation of the metal surfaces
prior
to the decontamination step is effective to reduce the number of treatment
cycles.
Use of deuterated oxidants and decontamination reagents also results in a
reduction of radioactive waste.
According to a preferred embodiment, the oxidation step is performed at a
temperature of from 20 to 120 C and optionally under higher than atmospheric
pressure to avoid boiling of the heavy water coolant and moderator in low
pressure parts of the primary circuit. Preferably, the temperature is in a
range of
from 80 to 95 C. Oxidation treatment at high temperatures is effective to
facilitate
the formation of pores in the metal oxide layer.
In a further embodiment, the oxidation step may be performed at a
temperature of from 95 to 120 C under elevated pressure.
During the oxidation step, chromium(III) in the metal oxide coating is
converted into soluble chromate (Cr(VI)) and is dissolved in the oxidant
solution.
Additionally, a certain amount of nickel(11) is solubilized by mechanisms not
necessarily involving a change of oxidation state of the nickel.
The dissolution of Cr(VI) and Ni(II) can be shown by analyzing the oxidant
solution during the oxidation step. The oxidation step is terminated as soon
as no
increase of the amount of chromium(VI) in the oxidant solution can be
determined.
Preferably, the oxidant is deuterated permanganic acid, DMnat, which is
preferred over alkali metal permanganate salts because less waste is produced.
More preferably, the deuterated permanganic acid is controlled at a
concentration of from 10 to 800 mg/I during the oxidation step, preferably 100
to
200 mg/I.
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 6 -
Still more preferably, the deuterated permanganic acid is prepared by ion
exchange reaction between an alkali metal permanganate salt and a cationic ion
exchange resin in deuterated form.
The deuterated permanganic acid DMnat can be provided as a stock solution
in heavy water, D20, having a concentration of from 1 to 45 g/I preferably a
concentration of from 30 to 40 g/I.
According to a further preferred embodiment, the decontamination reagent is
selected from the group consisting of deuterated oxalic acid, linear, fully
deuterated alkyl dicarboxylic acids having active acidic deuterium atoms,
alkali
metal salts of oxalic acid, alkali metal salts of fully deuterated straight-
chain alkyl
dicarboxylic acids, and mixtures thereof.
Preferably, the decontamination reagent is a deuterated dicarboxylic acid
selected from at least one of deuterated oxalic acid and a linear alkyl
dicarboxylic
acid having active deuterium atoms, and most preferably deuterated oxalic
acid.
More preferably, the decontamination reagent is provided as a stock solution
in heavy water having a concentration of from 25 to 150 g/I, preferably from
25 g/I
to 120 g/I, and more preferably from 25 g/I to 100 g/I.
Still more preferably, the decontamination reagent is prepared by ion
exchange between an alkali metal salt of the decontamination reagent,
preferably
an alkali metal salt of the dicarboxylic acid, and a cationic ion exchange
resin in
deuterated form.
The decomposition step preferably comprises the step of decomposing the
decontamination reagent to form carbon dioxide and heavy water. More
preferably, the decontamination reagent is decomposed by reaction of the
decontamination reagent with ozone and exposure to UV radiation.
Preferably, the temperature during the decomposition step is between 20 and
95 C.
The chemicals used for the decontamination treatment can be injected into
the primary heavy water coolant and moderator at a dosing station located in
the
low-pressure part of the coolant and moderator circuit of the heavy water
reactor.
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 7 -
Preferably, the decontamination treatment can be applied using external
decontamination equipment to monitor the decontamination treatment and
achieve the decontamination targets. The process temperatures are preferably
kept below the boiling point of heavy water to eliminate the need of using
complex and expensive pressure-proof components for the external
decontamination equipment.
Accordingly, a further aspect of the invention is a heavy water cooled and
moderated reactor adapted to perform the above decontamination method
wherein the reactor comprises a primary coolant circuit having a low-pressure
section and a high pressure section, a moderator circuit and an external
decontamination device connected to the low-pressure circuit of the primary
coolant circuit and/or the moderator circuit, wherein the oxidant and/or the
decontamination reagent are injected into the primary coolant circuit and/or
the
moderator circuit by means of the external decontamination device.
Preferably, the low-pressure section of the primary circuit comprises a high
pressure pump, a volume control system upstream of the high pressure pump,
and a pressure reducing station upstream of the volume control system, wherein
the decontamination device is connected to the primary coolant circuit at a
position upstream of the high pressure pump, preferably upstream of the volume
control system and downstream of the pressure reducing station.
The construction and method of operation of the invention together with
additional objects and advantages thereof will be best understood from the
following description of specific embodiments when read in connection with the
accompanying drawings.
BRIEF DESCRIPTION OF THE DRAWINGS
- Figure 1 is a schematic diagram of a heavy water cooled and moderated
nuclear reactor; and
- Figure 2 is a schematic diagram showing a decontamination device
connected to a primary coolant circuit of the heavy water reactor.
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 8 -
DETAILED DESCRIPTION OF PREFERRED EMBODIMENTS
According to the method of the present invention, a metal oxide coating
containing radioisotopes is effectively removed from metal surfaces in the
cooling
and moderator system of a heavy water nuclear reactor. The cooling and
moderator system is understood as comprising all systems and components
which are in contact with the heavy water coolant and moderator during reactor
operation, including but not limited to the primary cooling and moderator
circuits
including the reactor vessel, reactor coolant and moderator pumps, pipework
and
steam generators, and auxiliary systems such as the volume control system,
pressure reducing station and reactor water clean-up system.
Referring to the embodiment shown in Fig. 1, a heavy water reactor 10
comprises a primary coolant circuit 12 for circulating a primary heavy water
coolant under high pressure through fuel bundles 14 and steam generator 16.
The primary coolant is circulated by means of reactor coolant pump 18 and
pressurized by pressure tank 20.
The fuel bundles 14 are located in a low-pressure vessel or calandria 22
containing the heavy water moderator surrounding the fuel bundles 14. The
moderator circuit 23 includes a moderator pump 24 and heat exchanger 26 for
cooling the heavy water moderator. Adjustment rods 28 are provided for
controlling neutron flux in the fuel bundles 14.
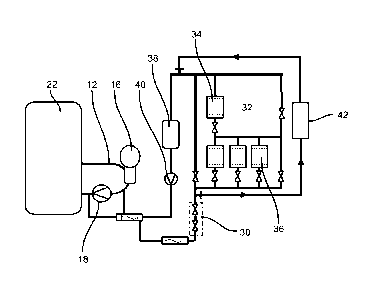
As shown in Fig. 2, the primary coolant circuit 12 further comprises a low-
pressure section including a pressure reducing station 30, a reactor water
clean-
up system 32 including anionic ion exchanger 34 and cationic ion exchangers
36,
a volume control system 38 and a high pressure pump 40 downstream of the
volume control system 38 which are also in contact with the primary heavy
water
coolant during power generating operation of the reactor.
A decontamination circuit 42 including external decontamination device 44 is
coupled to primary coolant circuit 12 by connecting decontamination circuit 44
to
the low-pressure section of the primary coolant circuit 12 downstream of the
pressure reducing station 32 and the intake side of high pressure pump 40,
preferably upstream of volume control system 38. In alternative embodiments,
the decontamination circuit 42 may be connected to other components in the low-
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 9 -
pressure section of the primary coolant circuit 12 at different positions,
depending
on the specific reactor design.
For decontaminating the moderator circuit 23 operated under low pressure,
the external decontamination device 44 can be operated in parallel to a
moderator cleaning system (not shown) and connected to any part of the
moderator circuit 23.
The decontamination device 44 preferably has a modular design and may
comprise an UV reactor and at least one circulation pump, heaters, ion
exchange
columns, filters, sampling modules, monitoring systems, automation and remote
controls and chemical injection equipment (not shown).
The UV reactor 34 is used for photocatalytic decomposition of the
decontamination reagent. The sampling devices will be used during the
treatment
cycles for process control, and mechanical filtration may be performed during
the
decontamination step.
It is understood by those skilled in the art that the reactor design
schematically shown in Figures 1 and 2 may vary and is not limiting to the
present invention.
The decontamination method of the present invention is particularly useful for
decontamination of the cooling and moderator system in a boiling water reactor
or a pressurized water reactor such as CANDU or KWU heavy water reactors,
and preferably a heavy water reactor comprising steam generator piping having
metal surfaces of nickel alloys such as lnconelTM 600, lnconelTM 690 or
I ncoloyTm 800.
The decontamination treatment can be carried out on reactor subsystems or
as full system decontamination, without degrading the heavy water
concentration
by entrainment of light water. During full system decontamination the
contaminated metal oxide coating is removed from all metal surfaces in the
reactor cooling and moderator system that are in contact with the heavy water
coolant and moderator during reactor operation. Typically, full system
decontamination involves all parts of the primary coolant circuit and the
moderator circuit as well as the volume control system, the pressure reducing
station and possibly other systems which are contaminated to a certain extent.
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 10 -
Ion exchange resins and chemicals used in the decontamination method of
the present invention are prepared specifically for use in the heavy-water
reactor.
Since heavy water has limited availability worldwide, the deuterated ion
exchange
resins and decontamination chemicals are preferably prepared directly at the
nuclear power plant facilities where heavy water is widely available.
A) Preparation of deuterated ion exchange resins and decontamination
chemicals
Preparation of deuterated cationic and anionic exchange resins
Deuterated ion exchange resins are required for preparing the
decontamination chemicals as well as for removing and immobilizing of the
radioisotopes from the decontamination solution during the decontamination
step.
Therefore, the decontamination method preferably comprises the step of
providing a bulk of deuterated ion exchange resins.
Cationic and anionic exchange resins are commercially available in
regenerated form. Cationic exchange resins have sulfonic acid groups and
anionic exchange resins have quaternary amine groups. These commercial ion
exchange resins cannot be used directly in the primary coolant circuit of a
heavy
water reactor because both types of ion exchange resins would emit hydrogen
ions into the heavy water coolant thereby causing a dilution of the heavy
water
with light water. This process is shown in following equations 1 and 2 wherein
"Polymer" denotes the inert resin backbone of the ion exchange resins:
Cationic exchange resin
D20 + Polymer ¨ SO2 ¨ OH DHO + Polymer ¨ SO2 ¨ OD
Equation 1
Anionic exchange resin
D20 + Polymer ¨ NR3 OH DHO + Polymer ¨ NR3 OD
Equation 2
For preparing the bulk of deuterated ion exchange resins, the resins are
poured into an ion exchange column filled with heavy water, and are left
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 11 -
overnight. The heavy water is then removed from the ion exchange column, and
the amount of light water in the eluate is determined. The ion exchange column
is
again filled with heavy water, and the process is repeated until the amount of
light
water in the eluate has reached a predetermined threshold. Preferably, the
amount of light water in the eluate is 1 weight percent or less. It has been
found
that a degree of deuteration of > 99% is sufficient for use.
The ion exchange column may be part of the facilities of the nuclear power
plant, such as the reactor water cleaning system or is provided as an external
mobile ion exchange column. Preferably, the ion exchange column is connected
to the external decontamination device 42.
Flushing of the ion exchange resins with heavy water will also exchange light
water bound in the resin skeleton and/or used for swelling the ion exchange
resin.
Preferably, the anion exchange resin has functional groups consisting of
tertiary amino end groups such as trisalkyl ammonium groups so that only one
hydrogen atom is exchanged per functional group, as shown in Equation 2.
Anionic exchange resins having primary or secondary amino end groups bear
an additional one or two hydrogen atoms, and would therefore be able to elute
additional hydrogen atoms into the heavy water, as shown in the following
Equations 3a to 3c.
Secondary amino groups
D20 + Polymer ¨ NHR2 OD DHO +
Polymer ¨ NDR2 OD
Equation 3a
Primary amino groups:
D20 + Polymer ¨ NRH2 OD DHO +
Polymer ¨ NRDH OD
Equation 3b
D20 + Polymer ¨ NRDH OD DHO +
Polymer ¨ NRD 20D
Equation 3c
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 12 -
The reactions according to Equations 3a to 3c would consume additional
amounts of heavy water. Therefore, anionic exchange resins having primary and
secondary amino end groups are less preferred.
The above process is suitable for the removal of light water from anionic
exchange resins and cationic exchange resins and providing a bulk of
deuterated
ion exchange resins.
The bulk of deuterated cationic exchange resins obtained by this process are
also used to prepare the decontamination chemicals such as the deuterated
oxidant and the deuterated dicarboxylic acid. Both of the deuterated anionic
exchange resin and deuterated cationic exchange resins are used in the
decontamination step of the decontamination treatment cycle for immobilizing
the
radioisotopes and metal ions.
Preferably, the bulk of ion exchange resins have a light water content of less
than 1 weight percent. It has been found that a degree of deuteration of > 99%
is
sufficient for use. Therefore, the light water content of the decontamination
chemicals produced from these deuterated ion exchange resins can also be
controlled to be less than 1 percent.
In cases were a degree of deuteration of 99% would exceed the concentration
limit of light water, such as for decontamination of sub-systems with small
volumes, the degree of deuteration of both the anion and cation ion exchange
resin may be adapted accordingly. In order to increase the degree of
deuteration,
the above described preparation process is extended for some additional days.
For any case of the application it is assumed that the total content of light
hydrogen stored on each deuterated ion exchange resin, which will be used
during the chemical decontamination process, is dissolved in the final heavy
water filling of the subsystem. According to this assumption the degree of
deuteration of each ion exchange resin being used is adapted by calculation.
The
above described mechanism for leaching out light water can be easily
controlled
by atomic mass spectroscopy.
Preparation of the oxidant
The preferred oxidant used in the oxidation step is deuterated permanganic
acid. Alkali metal salts of permanganate, free of crystal water, such as
potassium
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 13 -
permanganate can also be used but are less preferred because the alkali metal
ions would increase the mass of the waste ion exchange resin to be disposed.
Preferably, deuterated permanganic acid is prepared from dry potassium
permanganate by ion exchange using a deuterated cationic exchange resin, as
prepared above. An alkali metal salt of permanganate is dried, and the water
free
salt is dissolved in heavy water, as shown in the following Equation 4:
D2 0
KMn04 K+(solv) + Mn04-(solv)
Equation 4
The solution of potassium permanganate in heavy water is passed over the
deuterated cationic exchange resin and transformed into deuterated permanganic
acid to provide a stock solution of deuterated permanganic acid in heavy
water.
This reaction is shown in the following Equation 5 wherein "Polymer" denotes
the
backbone of the ion exchange resin:
K+ + MnO4- + Polymer ¨ SO2 ¨ OD cooling D+ + MnO4- +
Polymer ¨ SO2 ¨ OK
Equation 5
Preferably, the stock solution of deuterated permanganic acid in heavy water
has a concentration of from 1 to 45 g DMnat per liter of heavy water, more
preferably a concentration of from 30 to 40 g/I. Less concentrated solutions
of the
oxidant are inefficient because of the high amount of heavy water to be
transported. Oxidant solutions having a concentration of deuterated
permanganic
acid of greater than 40 g/I are difficult to obtain because of the limited
solubility of
the alkali metal permanganate salt in heavy water. In addition, stock
solutions of
deuterated permanganic acid having a concentration of greater than 40 g/I tend
to decompose autocatalytically even at room temperature, as shown in the
following Equation 6.
Room temperature
4 D30+ +4 Mn04- ____________________________________________________ > 6 D20
+4 Mn02 + 3 02
Equation 6
The concentrated stock solution of deuterated permanganic acid prepared in
accordance with Equation 5 can be used in the oxidation step of the
decontamination treatment cycle without changing or degrading the heavy water
concentration in the primary coolant circuit.
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 14 -
The concentrated stock solutions of deuterated permanganic acid obtained in
accordance with Equation 5 are stable at room temperature for several weeks,
preferably at least four weeks. The stock solution can therefore be prepared
in
sufficient time prior to performing the chemical decontamination treatment.
Preparation of the decontamination reagent
Preferably, the decontamination reagent used in the decontamination step is
a deuterated dicarboxylic acid selected from at least one of deuterated oxalic
acid, DO2C-CO2D, and fully deuterated linear alkyl dicarboxylic acids used for
dissolving the metal oxides and radioisotopes deposited on the metal surfaces
during operation of the nuclear power plant. Examples for the linear alkyl
dicarboxylic acids are deuterated malonic acid and deuterated succinic acid.
The deuterated dicarboxylic acid can also act as a complexing agent or
chelating agent by forming metal complexes with the metal cations from the
metal
oxides and radioisotopes and keeping the metal complexes in solution.
Deuterated oxalic acid and linear, fully deuterated alkyl dicarboxylic acids
which do not have a secondary or tertiary carbon atom are preferred because
these reagents can be decomposed to form water and carbon dioxide without any
intermediary products. Secondary and tertiary carbon atoms cannot be oxidized
to tetravalent carbon and result in intermediary products which have to be
removed from the primary coolant using anionic exchange resins. This would
result in an additional consumption of anionic exchange resin during
decontamination and cause additional costs for preparing the required
deuterated
anionic exchange resin. For example, deuterated malonic acid is suitable for
use
as a decontamination reagent in the process of the present invention, whereas
deuterated 2-trisdeuteromethyl malonic acid will not be useful because it has
a
secondary carbon atom and is not completely decomposed in the cleaning step.
In an alternative embodiment, the decontamination reagent is an alkali metal
salt of oxalic acid such as disodium oxalate, or an alkali metal salt of the
linear,
fully deuterated alkyl dicarboxylic acid. This embodiment is less preferred
because the decontamination process will then introduce alkali metal ions into
the
primary coolant which could lead to corrosion. Moreover, the additional alkali
metal ions may increase the amount of secondary waste because the alkali metal
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 15 -
ions must be removed from the heavy water coolant using additional deuterated
cationic exchange resin.
For preparing the deuterated dicarboxylic acid, an alkali metal salt of the
dicarboxylic acid is dissolved in heavy water and passed over a deuterated
cationic exchange resin to obtain a stock solution of the deuterated
dicarboxylic
acid by ion exchange.
Still more preferably, the ion exchange process is carried out at room
temperature, as illustrated in the following Equation 7 showing the
preparation of
deuterated oxalic acid.
Room Temperature
2 N a+ + C20,21,- + 2 Polymer ¨ SO2 ¨OD ______________ > 2 D+ +
C20,21,- + 2 Polymer ¨ SO2 ¨ ON a
Equation 7
The solubility of sodium oxalate in heavy water at room temperature is about
37 g/I. Preferably, the deuterated oxalic acid used as the decontamination
reagent has a concentration of 25 g/I in heavy water, after the ion exchange
process.
The stock solution of the decontamination reagent in heavy water can be
further concentrated to a concentration of 100 g/I to 200 g/I by evaporating
the
heavy water solvent. Preferably, the solution of the decontamination reagent
is
evaporated in vacuum at temperatures of about 100 C in a rotary evaporator
until the concentration of the decontamination reagent is about 100 g/I or
more.
The concentrated stock solution so obtained can be stored at room temperature
for several weeks, preferably at least four weeks.
A stock solution of the decontamination reagent in heavy water at a
concentration of about 100 g/I or more has been found useful in the
decontamination method of the present invention. For example, the
decontamination of a primary coolant circuit having a volume of about 100 m3
requires about 20001 of the decontamination reagent such as deuterated oxalic
acid having a concentration of 100 g/I in heavy water. This volume of the
decontamination reagent fits well into the technical capacities of a nuclear
power
plant.
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 16 -
If deuterated oxalic acid is used as the decontamination reagent, the
temperature of the evaporation process for preparing the concentrated stock
solution must not exceed 140 C. Otherwise, a thermal decomposition of
deuterated oxalic acid will take place at temperatures above 150 C. Similar
conditions apply to deuterated malonic acid. Deuterated succinic acid was
found
to be stable up to 200 C.
B) Chemical Decontamination Treatment
Metal surfaces in the primary coolant and moderator circuit of an operating
nuclear power plant are coated with metal oxide deposits including radioactive
isotopes such as Co-60 during reactor operation. Chemical decontamination of
the metal surfaces dissolves these metal oxide coatings together with the
radioisotopes incorporated therein. The metal surfaces are cleaned, and
metallic
bright surfaces without oxide deposits are obtained.
The method of the present invention is directed to the decontamination of
metal surfaces in the primary coolant and moderator circuit of a heavy water
reactor. Similar to a pressurized water reactor, the metal surfaces in the
primary
coolant and moderator circuit of the heavy water reactor will have chromium
containing metal oxide deposits. The inventors therefore contemplate that the
decontamination treatment cycle must include an oxidation step.
The decontamination method is suitable for use with any CANDU reactors as
well as other heavy water reactors, but is not limited to these reactor types.
The
inventors also contemplate use of the decontamination method in a boiling
water
reactor operated with heavy water. In this case, the oxidation step could be
omitted if the metal oxide deposits on the metal surfaces in the primary
coolant
circuit include iron oxides having a chromium content of less than 1 weight
percent.
Under operational conditions of a nuclear reactor at temperatures of up to
330 C, metal ions are leached out of the alloys of the piping in the primary
coolant and moderator circuit and are dissolved and transported into the heavy
water coolant and moderator. When passing the reactor core during operation,
some of the metal ions are activated to form radioisotopes. During operation
of
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 17 -
the reactor these metal ions and radioisotopes are deposited as an oxide layer
on
metal surfaces inside the reactor cooling and moderator system.
Depending on the type of alloy used for a component or system, the oxide
layers which are formed contain mixed iron oxides with divalent and trivalent
iron
as well as other metal oxide species including chromium(III) and nickel(11)
spinels.
Especially the oxide deposits formed on the metal surfaces of the steam
generator tubes have a high chromium(III) or Ni(II) content which makes them
very resistant and difficult to remove from the metal surfaces.
Over extended reactor operation periods, the amount of the radioisotopes,
such as Co-60, Co-58, Cr-51, Mn-54 etc., deposited together with the metal
oxides on the inner metal surfaces of the reactor cooling system accumulates.
This results in an increased surface activity or dose rate of the components
of the
reactor cooling and moderator system. The removal of this radioactive matter
results in a measurable reduction of personnel radiation exposure.
In general, one or more decontamination treatment cycles are carried out in
order to achieve a satisfactory reduction of activity on the metal surfaces.
The
reduction of surface activity and/or the dose reduction correlating to surface
activity reduction is referred to as "decontamination factor". The
decontamination
factor is calculated either by the surface activity in Bq/cm2 before
decontamination treatment divided by the surface activity in Bq/cm2 after the
decontamination treatment, or by the dose rate before decontamination
treatment
divided by the dose rate after decontamination treatment.
Preferably, the decontamination factor of a technically satisfying
decontamination treatment is greater than 100.
A decontamination treatment cycle of the present invention comprises an
oxidation step, wherein the metal surface is contacted with a solution of an
oxidant in heavy water; a decontamination step wherein the metal surface
subjected to the oxidation step is contacted with a decontamination reagent in
heavy water for dissolving at least part of the one or more metal oxides and
to
form a decontamination solution containing the decontamination reagent, one or
more metal ions dissolved from the metal oxides and the radioisotopes, and
wherein the decontamination solution is passed over an ion exchange resin to
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 18 -
immobilize the metal ions and the radioisotopes; and a decomposition step
wherein the decontamination reagent in the decontamination solution is
decomposed.
Preferably, the decontamination treatment cycle comprises a reduction step
wherein the oxidant is reacted with the decontamination reagent.
In the decomposition step, the decontamination reagent is decomposed to
form carbon dioxide and heavy water.
More preferably, only deuterated cationic ion exchange resins are used for
cleaning the decontamination solution in the decomposition step and during
decomposition of the decontamination reagent.
As soon as the concentration of the deuterated dicarboxylic acid in the
decontamination solution is less than 50 mg/kg, a cleaning step can be
performed wherein anionic and cationic ion exchange resins are used to further
remove the deuterated dicarboxylic acid and remaining metal ions. Preferably,
the anionic ion exchange resin is operated downstream of the cationic ion
exchange resin. The concentration at which the cleaning step will be started
can
vary according to local waste regulations. Some countries have limits for the
concentration of organic acids on waste resin. Therefore the the decomposition
step can be extended to reach concentrations less than 10 mg/kg. This
extension
is technically possible. It will extend the decomposition time but lower the
concentration of dicarboxylic acid on the resulting waste from spent anionic
exchange resin.
A preferred embodiment of the decontamination method for a heavy water
cooled and moderated reactor may comprise the following treatment cycles:
First decontamination treatment cycle:
a) Oxidation of metal oxide coating using a solution of deuterated
permanganic acid in heavy water;
b) Reducing the deuterated permanganic acid and dissolving metal oxide
coating using a solution of deuterated dicarboxylic acid in heavy water;
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 19 -
c) Photocatalytic decomposition of deuterated dicarboxylic acid including
exposure to UV radiation;
d) Performing an intermediate Cleaning step
Second decontamination treatment cycle:
a) Oxidation of metal oxide coating using a solution of deuterated
permanganic acid in heavy water;
b) Reducing the deuterated permanganic acid and dissolving metal oxide
coating using a solution of deuterated dicarboxylic acid in heavy water;
c) Photocatalytic decomposition of deuterated dicarboxylic acid including
exposure to UV radiation;
d) Performing an intermediate Cleaning step
....
Last decontamination treatment cycle:
a) Oxidation of metal oxide coating using a solution of deuterated
permanganic acid in heavy water;
b) Reducing the deuterated permanganic acid and dissolving metal oxide
coating using a solution of deuterated dicarboxylic acid in heavy water;
c) Photocatalytic decomposition of deuterated dicarboxylic acid including
exposure to UV radiation;
d) Final cleaning step.
Preferably, the decontamination method comprises two to four
decontamination treatment cycles. It has been found that a sufficient
decontamination factor could be achieved with this number of treatment cycles
in
full system decontamination and/or decontamination of subsystems or
components of a heavy water cooled and moderated reactor. However, the
number of decontamination treatment cycles is not limited to the numbers given
above, but may also depend on reactor design and level of radioactive
contamination.
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 20 -
According to the invention, the oxidant, the decontamination reagent and the
ion exchange resin are provided in a deuterated form and/or are free of active
hydrogen, preferably free of any hydrogen. "Active hydrogen" is understood as
acidic hydrogen atoms which are reactive against a Grignard reagent such as
methyl magnesium bromide. Thus, the decontamination method may comprise a
plurality of treatment cycles without dilution of the heavy water coolant and
moderator by light water during the decontamination treatment.
The various steps of the decontamination method are described in greater
detail below
Oxidation step
For carrying out the oxidation step, a stock solution of the deuterated
oxidant
such as deuterated permanganic acid is injected into the primary coolant and
moderator circuit or the subsystem which is to be decontaminated, and the
oxidant solution is circulated through the cooling and moderator system. The
oxidant solution can be introduced into the cooling and moderator system by
means of the external decontamination device 42.
Preferably, the deuterated oxidant is injected into a low-pressure section of
the cooling and moderator system. Examples for suitable injection positions
are
the volume control system, the reactor water cleaning system and/or a residual
heat removal system.
The deuterated permanganic acid reacts with spinell-type metal oxides in the
metal oxide coating which are inert to organic and mineral or acids by
oxidizing
Cr(III) to soluble Cr(VI).
Preferably, the oxidation step is carried out at a temperature of between
about
20 to 120 C, more preferably at a temperature of from 80 to 95 C. The
oxidation
step is faster at higher temperatures.
Accordingly, higher oxidation temperatures are preferred. Moreover, the
boiling point of a solution of deuterated permanganic acid in heavy water
under
atmospheric pressure is higher than 95 C so that the oxidant solution can
easily
be circulated through the cooling and moderator system using external pumps of
the decontamination device.
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 21 -
However, it is also possible to carry out the oxidation step at temperatures
of
up to 120 C at a higher than atmospheric pressure. Thus, the temperature of
the
oxidation step is selected depending on the pressure conditions in the
decontamination device. Generally, the temperature is selected as high as
possible within the temperature range of from 95 to 120 C, but is controlled
to be
at least 5 K below the boiling point of the heavy water solution as calculated
for
the actual hydrostatic pressure within the decontamination system. Thus, the
heavy water solution is prevented from boiling to protect the circulating
pumps
against cavitation. Preferably, boiling graphs for light water can be used to
calculate the boiling point of the heavy water solution due to the negligible
difference between the boiling points of light water and heavy water, as a
function
of the ambient pressure.
Preferably, the concentration of the deuterated oxidant in the cooling and
moderator system is controlled to be in the range of from 10 to 800 mg/kg
during
the oxidation step, and preferably to range from 100 to 200 mg/kg. If the
concentration of the deuterated oxidant in the oxidation solution is lower
than
10 mg/kg, and preferably lower than 100 mg/kg, the reaction rate of the
oxidation
is too low. If the concentration of the oxidant in the oxidant solution
exceeds
800 mg/kg, a large excess of the oxidant will be present at the end of the
oxidation step. Preferably, the amount of the oxidant is controlled to be as
low as
possible at the end of the oxidation step because the deuterated oxidant is
expensive, and removal of excess deuterated oxidant will increase the amount
of
secondary waste.
Preferably, the amount of the deuterated oxidant in the oxidation solution
during the oxidation step is controlled by monitoring the concentration of
Cr(VI) in
the oxidation solution. As long as the oxidation reaction continues and the
oxidation of the metal oxide layer is incomplete, the concentration of Cr(VI)
increases, as shown in Equation 7:
24 D2 0 + 5 Cr3+ + 3 Mn04- 5 Cr0j,- (solv) + 16 D3 0+ + 3 Mn2+
Equation 7
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 22 -
The residence time of the oxidant solution in the cooling and moderator
system during the oxidation step may comprise a plurality of hours, preferably
up
to 30 hours. It is desired that the oxidation of the metal oxide layer is
substantially
complete so that as much as possible of the oxide coating thickness is reacted
during the oxidation step. Preferably, the oxidation step is terminated when
no
further increase in the Cr(VI) concentration can be determined.
Instead of monitoring the concentration of Cr(VI), it is also possible to
monitor
the presence of the radioisotope Cr-51 in the oxidant solution by means of
gamma spectroscopy.
As soon as the oxidation step is terminated, preferably a reduction step is
started.
Reduction step
The oxidation step may be followed by a reduction step which is the shortest
step of the treatment cycle. The reduction step comprises reducing an excess
of
the oxidant remaining in the oxidant solution at the end of the oxidation step
by
reacting the oxidant with the decontamination reagent. Preferably the oxidant
is
deuterated permanganic acid, and the decontamination reagent is a fully
deuterated dicarboxilic acid such as deuterated oxalic acid, as shown in the
following Equation 8:
16 D30' + 2Mn04- + 5 C20,21,- 2 Mn2+ + 24 D20 + 10 CO2
Equation 8
A stock solution of the decontamination reagent is injected into the primary
coolant and moderator circuit or the subsystem which is to be decontaminated,
and the solution containing the decontamination reagent is circulated through
the
cooling and moderator system. The stock solution of the decontamination
reagent
can be introduced into the cooling and moderator system by means of the
external decontamination device 42 at the same position as described above
with
respect to the oxidant solution.
Mangenese cations generated by the reduction of permanganic acid are
dissolved in the decontamination reagent solution as a manganese(II) oxalate
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 23 -
complex. Carbon dioxide is dissolved in the heavy water solution under high
pressure and will be released into the environment in the low-pressure part of
the
cooling and moderator system such as in the volume control system.
All metal ions dissolved from the metal oxide coating during the reduction
step
solution as well as Cr(VI) generated in the oxidation step will be reduced by
the
decontamination reagent to a lower oxidation step. Cr(VI) will be converted to
Cr(III), most of the iron will be present as iron(II), and nickel and
manganese will
be present as nickel(11) and Mn(II).
The duration of the reduction step is dependent on the excess of deuterated
permanganic acid in the oxidant solution. Accordingly, it is desired to
terminate
the oxidation step at a concentration of the deuterated permanganic acid as
low
as possible. The duration of the reduction step is further influenced by the
effectivity of removal of the carbon dioxide dissolved in the heavy water
solution,
because the carbon dioxide must be removed in the low-pressure part of the
cooling and moderator system without damaging the pumps in the
decontamination circuit due to cavitation. Moreover, the duration of the
reduction
step is also influenced by the injection rate of the deuterated oxalic acid
which is
preferably injected into the heavy water coolant circuit at a dosing station
located
in the low-pressure part of the heavy water coolant circuit.
The reduction step is controlled by monitoring the removal of carbon dioxide
and the concentration of permanganic acid in the decontamination solution
solution. As soon as the reaction between deuterated permanganic acid and
deuterated oxalic acid is completed, the decontamination step is started.
However, it is understood that the reduction step may also be considered part
of
the decontamination step.
Decontamination of the metal surfaces
The decontamination step comprises the step of contacting the metal oxide
layer subjected to the oxidation step with the decontamination reagent to
dissolve
metal ions and radioisotopes incorporated in the metal oxide coating and to
form
a decontamination solution containing the decontamination reagent, one or more
metal ions dissolved from the metal oxides and the radioisotopes, and passing
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 24 -
the decontamination solution over an ion exchange resin to immobilize the
metal
ions and the radioisotopes.
Preferably, the decontamination solution is passed over a deuterated cationic
exchange resin which is prepared as described above by flushing with heavy
water. Accordingly, no light water is entrained into the decontamination
solution
during the ion exchange reaction. Equation 9 shows an example of the ion
exchange reaction using nickel ions:
Ni2+ + 2 Polymer ¨ SO2 ¨ OD 2 D+ +
Ni(Polymer ¨ SO2 ¨ 0)2
Equation 9
Similar to nickel ions, all other cations dissolved in the decontamination
solution, including manganese(II) generated from the deuterated permanganic
acid, as well as the radioisotopes dissolved in the decontamination solution
are
removed from the decontamination solution and immobilized on the cationic ion
exchange resin.
The progress of the decontamination step and the ion exchange reaction can
be monitored by measuring the concentration of selected radioisotopes and
metal
ions. Samples can be taken from the decontamination solution and analyzed by
spectroscopic methods such as atom absorption spectroscopy. The amount of
radioisotopes dissolved in the decontamination solution can be determined by
gamma spectroscopy or by using a gamma counter.
The decontamination step is terminated as soon as no substantial increase of
the amount of metal ions removed from the decontamination solution and
immobilized on the cationic ion exchange resin is determined and/or no further
increase of the activity of the radioisotopes immobilized at the ion exchange
resins is determined.
The deuterated dicarboxylic acid in the decontamination solution is
regenerated by release of deuterium ions during the ion exchange reaction as
exemplified above in Equation 9. Therefore, the deuterated dicarboxylic acid
is
not depleted in the decontamination step. Rather, the decontamination of the
metal surfaces is only limited by a decrease of the solubility of metal ions
dissolved from the metal oxide coating. The reason for the decrease of the
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 25 -
solubility of the metal oxides in the decontamination solution is found in the
fact
that the metal oxide layer reacted in the oxidation step is completely removed
at
the end of the decontamination step, and a further oxidation of the remaining
metal oxide coating is required to dissolve further metal ions into the
decontamination solution.
Decomposition step
In order to start a further oxidation step, the decontamination reagent must
be
removed from the decontamination solution. Theoretically, the decontamination
reagent such as deuterated dicarboxylic acid could be reacted with deuterated
permanganic acid as shown in Equation 8 above. For example, this process can
be used for decontamination systems having small volumes, e.g. during the
decontamination of isolated heat exchangers and the like.
However, this reaction would require a substantial amount of permanganic
acid and also generate additional secondary waste in the form of manganese
ions and/or manganese oxide. Therefore, the decontamination method of the
present invention includes a decomposition step comprising a photocatalytic
oxidation of the decontamination reagent. The photocatalytic oxidation of the
decontamination reagent such as fully deuterated dicarboxylic acid does not
generate additional waste but results in the formation of heavy water and
carbon
dioxide.
Preferably, the decontamination reagent is reacted with ozone. Use of oxygen
would also be possible but is less preferred. The byproducts of the
photocatalytic
oxidation of the decontamination reagent are carbon dioxide and heavy water.
No
light water is produced since no hydrogen-containing reagents are used.
The reaction of ozone and deuterated dicarboxylic acid such as deuterated
oxalic acid is shown in the following Equation 10. Use of ozone as the oxidant
in
the photocatalytic oxidation reaction is preferred since six electrons per
molecule
of ozone are available for the oxidation reaction. Thus, three moles of
oxalate can
be reacted with one mole of ozone to form carbon dioxide and heavy water.
UV
03 + 3 C2 0i, - + 6 D30+ 6 CO2 + 9D20
Equation 10
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 26 -
Preferably, the photocatalytic oxidation reaction is conducted at a
temperature
of from 20 to 95 C.
Use of ozone as the oxidant in the photocatalytic oxidation reaction further
has the advantage that no hydrogen atoms are introduced into the reaction
solution, as it would be the case if hydrogen peroxide was used.
Preferably, the ozone is generated using pure oxygen. If air was used for
generating ozone by means of an electrical ozone generator, a small amount of
nitrogen oxides NOx would be generated and converted into nitrate in the
decontamination solution. The nitrates must be removed from the
decontamination solution by passing the solution over an anionic ion exchange
resin, which would increase the amount of secondary waste.
Preferably, a UV reactor is immersed into the decontamination solution, and
the ozone is injected into the decontamination solution by means of a Venturi
mixer upstream of the UV reactor. Thus, the ozone is thoroughly mixed with the
decontamination solution.
The injection of ozone into the decontamination solution is controlled so that
no dissolved ozone is determined downstream of the UV reactor.
Preferably, the ozone concentration in the decontamination solution is
determined by measuring the oxidation potential against a standard electrode
Ag/AgCI, and more preferably by controlling the oxidation potential of the
decontamination solution subjected to ozone treatment to be less than +200 mV
downstream of the UV reactor.
Alternatively or simultaneously, the ozone concentration can be measured
indirectly by monitoring the concentration of iron(II) in the decontamination
solution. If the concentration of iron(II) downstream of the UV reactor is
greater
than 2 mg/kg, ozone is completely eliminated. Otherwise, ozone would
immediately react with iron(II) to form iron(III), as shown in the following
Equation
11.
03+6 Fe2+ + 6 D30+ 6 Fe3+ + 9 D20
Equation 11
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 27 -
Preferably, the concentration of ozone downstream of the UV reactor is
measured continuously, and the rate of the ozone injection is adjusted
continuously.
Preferably, the UV reactor comprises a medium pressure mercury lamp. A
power of 10 kW was found to be sufficient for a volume flow rate of 15 to 50
m3/h
of the decontamination solution. The minimum total amount of iron, including
iron(II) and iron(III), in the decontamination solution should preferably
exceed
mg/kg in order to enable a reliable measurement of the ozone concentration.
In a further preferred embodiment, the concentration of ozone is determined
10 by means of an ion selective electrode sensitive to ozone.
The reaction rate of the photocatalytic oxidation of the fully deuterated
dialkyl
carboxylic acid is a first order reaction, if sufficient ozone is present. The
progress
of the decomposition reaction of the deuterated dicarboxylic acid can
therefore be
determined as shown in the following Equation 12.
F.k
N(t) = No *env
Equation 12
wherein
No denotes the initial concentration of the deuterated dicarboxylic acid
[mg/kg]
N(t) denotes the concentration of the deuterated dicarboxylic acid at time t
t denotes the decomposition time [h]
n denotes the number of UV reactors operated in parallel
F denotes the flow rate per UV reactor [m3/h]
V denotes the volume of the decontamination solution [m3]; and
k denotes the reaction constant specific to the deuterated dicarboxylic acid.
During the photocatalytic decomposition of the decontamination reagent,
dissolved metal ions and radioisotopes are removed from the decontamination
solution and are immobilized on the cationic ion exchange resins. The removal
of
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 28 -
the metal ions and radioisotopes in the decomposition step and/or the
decontamination step may take place in a bypass conduit in the low-pressure
part
of the reactor, most preferably using cationic ion exchange columns present in
the water cleaning system of the heavy water reactor. Alternatively or in
addition,
external ion exchange columns can be operated in parallel to the ion exchange
columns of the reactor water cleaning system.
The decomposition step is terminated if the decontamination solution is
depleted of the decontamination reagent and the concentration of the
decontamination reagent such as deuterated dicarboxylic acid in the
decontamination solution is 50 mg/kg or less.
Intermediate and final cleaning step
After terminating the decomposition step, when the concentration of the
decontamination reagent in the decontamination solution is 50 mg/kg or less,
an
intermediate or final cleaning step is performed wherein the decontamination
solution depleted of the decontamination reagent is cleaned by further removal
of
metal ions and deuterated dicarboxylic acid by means of deuterated cationic
exchange resins and deuterated anionic exchange resins operated downstream
of the cationic exchange resins.
If a further oxidation step is to be carried out, the concentration of the
decontamination reagent in the decontamination solution is preferably
controlled
to be less than 10 mg/kg so that the consumption of deuterated permanganic
acid in the initial phase of the oxidation step is as low as possible.
In a final cleaning step, the conductivity of the heavy water coolant is
controlled to be 10 pS/cm at 20 C. Preferably, the final cleaning step is
conducted at a temperature of 60 C or less, more preferably 30 C or less.
The decontamination method of the present invention is preferably applied to
the decontamination of both, the primary coolant circuit, and the moderator
circuit
of the heavy water reactor. The primary coolant circuit is provided for
cooling of
the reactor core including the fuel bundles and for transferring the hot heavy
water to the steam generator where energy is transferred from the primary
coolant to a secondary light water circuit passing through the steam
generator.
The moderator circuit comprises the reactor vessel filled with heavy water
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 29 -
surrounding the fuel elements and is provided with a separate cooling and
cleaning system.
Since the heavy water moderator circuit is operated at low pressure, a
decontamination device for injection of the decontamination chemicals can be
coupled to the moderator circuit at any suitable position and operated in
parallel
to the moderator circuit.
A decontamination of the primary coolant circuit requires that the
decontamination device is connected to a low-pressure section of the primary
coolant circuit using the volume control system as described above. In the
high-pressure section of the primary coolant circuit, the heavy water is
circulated
under a pressure of 100 bar or higher. Thus, connecting the decontamination
device to the high-pressure section of the primary coolant circuit may damage
the
pumps of the decontamination device and/or requires use of expensive
pressure-proof equipment.
Preferably, the decontamination device is connected to the low-pressure
section of the primary coolant circuit for injecting the decontamination
chemicals
into the primary coolant upstream of the intake side of the high-pressure pump
and downstream of a pressure reducing station for transferring the
decontamination solution out of the primary coolant circuit back into the
decontamination device.
The decontamination method of the present invention avoids a dilution of the
heavy water coolant and moderator by light water during the decontamination
treatment cycles since all decontamination chemicals are provided in their
deuterated form and no light water is formed as a byproduct. No further
cleaning
of the heavy water coolant and moderator after decontamination is necessary to
remove light water impurities. The heavy water primary coolant and moderator
itself can be used as the solvent for the decontamination chemicals. The
process
also saves the costs involved with replacing the heavy water in the reactor
cooling and moderating system by light water just for performing a
decontamination of the primary coolant and moderator cycle of the heavy water
reactor.
CA 03003488 2018-04-27
WO 2017/076431
PCT/EP2015/075591
- 30 -
Oxidation of the metal surfaces prior to the decontamination step is effective
to reduce the number of treatment cycles. Use of deuterated oxidants and
decontamination reagents prepared from bulk deuterated ion exchange resins
also results in a reduction of radioactive and secondary waste.
Although the invention is illustrated and described herein as embodied in a
method for surface decontamination, it is nevertheless not intended to be
limited
to the details shown, since various modifications and structural changes may
be
made therein without departing from the scope of the appended claims.