Note: Descriptions are shown in the official language in which they were submitted.
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
COMPOSITION FOR REDUCING FREQUENCY OF URINATION,
METHOD OF MAKING AND USE THEREOF
[0001] This application claims priority from U.S. Continuation Patent
Application Serial
No. 14/975,332, filed on December 18, 2015. The entirety of the aforementioned
application is
incorporated herein by reference.
FIELD
[0002] The present application generally relates to methods and compositions
for
inhibiting the smooth muscles of the urinary bladder and, in particular, to
methods and
compositions for reducing the frequency of urination.
BACKGROUND
[0003] The detrusor muscle is a layer of the urinary bladder wall made of
smooth muscle
fibers arranged in spiral, longitudinal, and circular bundles. When the
bladder is stretched, this
signals the parasympathetic nervous system to contract the detrusor muscle.
This encourages the
bladder to expel urine through the urethra.
[0004] For the urine to exit the bladder, both the autonomically controlled
internal
sphincter and the voluntarily controlled external sphincter must be opened.
Problems with these
muscles can lead to incontinence. If the amount of urine reaches 100% of the
urinary bladder's
absolute capacity, the voluntary sphincter becomes involuntary and the urine
will be ejected
instantly.
[0005] The human adult urinary bladder usually holds about 300-350 ml of urine
(the
working volume), but a full adult bladder may hold up to about 1000 ml (the
absolute volume),
varying among individuals. As urine accumulates, the ridges produced by
folding of the wall of
the bladder (rugae) flatten and the wall of the bladder thins as it stretches,
allowing the bladder to
store larger amounts of urine without a significant rise in internal pressure.
[0006] In most individuals, the desire to urinate usually starts when the
volume of urine in
the bladder reaches around 200 ml. At this stage it is easy for the subject,
if desired, to resist
the urge to urinate. As the bladder continues to fill, the desire to urinate
becomes stronger and
harder to ignore. Eventually, the bladder will fill to the point where the
urge to urinate becomes
overwhelming, and the subject will no longer be able to ignore it.
[0007] In some individuals, this desire to urinate starts when the bladder is
less than 100%
full in relation to its working volume. Such increased desire to urinate may
interfere with normal
activities, including the ability to sleep for sufficient uninterrupted
periods of rest. In some cases,
this increased desire to urinate may be associated with medical conditions
such as benign prostate
hyperplasia or prostate cancer in men, or pregnancy in women. However,
increased desire to
1
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
urinate also occurs in individuals, both male and female, who are not affected
by another medical
condition.
[0008] In some individuals, such as in children, involuntary urination (e.g.,
bed wetting)
may occur as a result of lack of control to the bladder muscle. In other
individuals, involuntary
urination (e.g., urinary incontinence) may occur as a result from an
underlying medical condition.
[0009] Accordingly, there exists a need for compositions and methods for the
treatment of
male and female subjects who suffer from an undesired frequency of urination.
SUMMARY
[0010] One aspect of the present application relates to a method for
manufacturing a
pharmaceutical composition for reducing the frequency of urination. In some
embodiments, the
method comprises the steps of forming a first mixture comprising a first
active ingredient
formulated for immediate release and a second active ingredient formulated for
extended release;
coating the first mixture with a delayed release coating to form a core
structure; coating the core
structure with a second mixture comprising a third active ingredient
formulated for immediate
release and a fourth active ingredient formulated for extended release,
wherein at least one of the
first, second, third and fourth active ingredients comprises a prostaglandin
pathway inhibitor. In
some embodiments, the prostaglandin pathway inhibitor is a prostaglandin (PG)
inhibitor, or a
prostaglandin transporter (PGT) inhibitor, or a prostaglandin receptor (PGR)
inhibitor.
[0011] In other embodiments, the method comprises the steps of forming a core
structure
comprising a first active ingredient formulated for immediate release and a
second active
ingredient formulated for extended release; coating the core structure with a
delayed release
coating to form a coated core structure; mixing the coated core structure with
a third active
ingredient formulated for immediate release and a fourth active ingredient
formulated for
extended release to form a final mixture; and preparing a dosage form with the
final mixture,
wherein at least one of the first, second, third and fourth active ingredients
comprises a
prostaglandin pathway inhibitor.
[0012] In other embodiments, the method comprises the steps of forming a core
structure
comprising a first active ingredient formulated for immediate release and a
second active
ingredient formulated for extended release; coating the core structure with a
delayed release
coating to form a coated core structure; coating the coated core structure
with a third active
ingredient formulated for extended release to form an extended-release layer
coated core
structure; and coating the extended-release layer coated core structure with a
fourth active
ingredient, wherein at least one of the first, second, third and fourth active
ingredients comprises a
prostaglandin pathway inhibitor.
[0013] Another aspect of the present application relates to a pharmaceutical
composition
for treating a condition that results in undesired frequency of urination. In
some embodiments,
2
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
the pharmaceutical composition comprises: a first component comprising an
immediate-release
subcomponent and an extended-release subcomponent, wherein the first component
is formulated
to release the subcomponents immediately after administration; and a second
component
comprising an immediate-release subcomponent and an extended-release
subcomponent, wherein
the second component is formulated for a delayed-release of the subcomponents,
wherein at least
one of subcomponents in the first component and the second component comprises
an active
ingredient comprising a prostaglandin pathway inhibitor.
[0014] In other embodiments, the pharmaceutical composition comprises: a first
component comprising an immediate-release subcomponent, wherein the immediate-
release
subcomponent comprises an active ingredient comprising one or more agents
selected from the
group consisting of analgesic agents and prostaglandin pathway inhibitors,
wherein the first
component is formulated to release its subcomponent immediately after oral
administration; and a
second component comprising an immediate-release subcomponent and an extended-
release
subcomponent, wherein the second component is formulated to release its
subcomponent after
gastric emptying of the second component, wherein at least one of the
subcomponents in the first
and the second components comprises an active ingredient comprising one or
more agents
selected from the group consisting of analgesic agents and prostaglandin
pathway inhibitors.
[0015] In other embodiments, the pharmaceutical composition comprises: an
immediate-
release component comprising acetaminophen and an NSAID, each in an amount of
5-2000 mg;
and an extended-release component comprising acetaminophen and an NSAID, each
in an amount
of 5-2000 mg, wherein the immediate-release component, or the extended-release
component, or
both, further comprise a prostaglandin pathway inhibitor.
BRIEF DESCRIPTION OF DRAWINGS
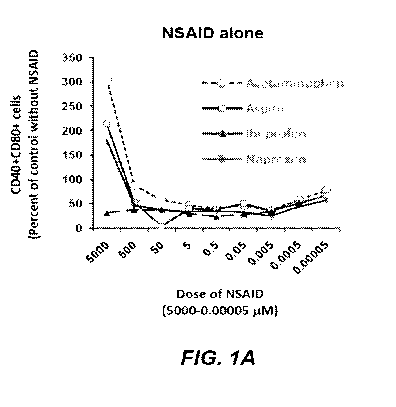
[0016] Figures 1A and 1B are diagrams showing that analgesics regulate
expression of co-
stimulatory molecules by Raw 264 macrophage cells in the absence (Figure 1A)
or presence
(Figure 1B) of LPS. Cells were cultured for 24 hrs in the presence of
analgesic alone or together
with Salmonella typhimurium LPS (0.05 g/m1). Results are mean relative % of
CD4O+CD80+
cells.
DETAILED DESCRIPTION
[0017] The following detailed description is presented to enable any person
skilled in the
art to make and use the invention. For purposes of explanation, specific
nomenclature is set forth
to provide a thorough understanding of the present invention. However, it will
be apparent to
one skilled in the art that these specific details are not required to
practice the invention.
Descriptions of specific applications are provided only as representative
examples. The present
invention is not intended to be limited to the embodiments shown, but is to be
accorded the
broadest possible scope consistent with the principles and features disclosed
herein.
3
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
[0018] As used herein, the term "prostaglandin (PG)" refers to a group of
lipid compounds
that are derived enzymatically from fatty acids and have a variety of
physiological effects, such as
regulating the contraction and relaxation of smooth muscle tissue, in the
animal body. Every
prostaglandin contains 20 carbon atoms, including a 5-carbon ring. Examples of
prostaglandin
include, prostaglandin E1 (PGE1), prostaglandin E2 (PGE2), Prostaglandin D2,
prostaglandin 12
(PGI2, prostacyclin), and prostaglandin F2a (PGF2a).
[0019] As used herein, the term "prostaglandin (PG) pathway inhibitor" refers
to agents
that interact directly or indirectly with one or more components involved in
the synthesis or action
of PG on a target tissue, and interfere with either the levels or the ultimate
actions of
prostaglandin on the target tissue. PG pathway inhibitors include, but are not
limited to, PG
inhibitors, prostaglandin transporter (PGT) inhibitors and prostaglandin
receptor (PGR) inhibitors.
The term "PG pathway inhibitor," however, does not include the analgesics
defined below.
[0020] The term "PG inhibitors," as used herein, include but are not limited
to, inhibitors
of PG synthesis and inhibitors of PG activity. The term "inhibitors of PG
synthesis," as used
herein, refers to agents that inhibit the production of prostaglandin, such as
agents that inhibit the
expression or activity of phospholipase A2, the prostaglandin synthases and
the tissue specific
isomerases and synthase enzymes such as: Thromboxane synthase, PGF synthase,
cytosolic PG
synthase (cPGES), prostaglandin I synthase (PGIS) and the microsomal PGES
enzymes
(mPGES). Examples of PG synthesis inhibitors include flunixin meglumine. As
used herein, the
terms "inhibitors of PG synthesis" and "PG synthesis inhibitors" do not
include the analgesics
defined below.
[0021] The term "inhibitors of PG activity," as used herein, refers to agents
that
antagonize the action of prostaglandin itself by any means. Agents that
interfere solely with the
synthesis of prostaglandins, such as by interfering with the action of
prostaglandin synthases, but
which do not interfere with the action of prostaglandins are not included
within the definition of
inhibitors of PG activity as used in this specification.
[0022] The term "PGT inhibitors," as used herein, refers to agents that
inhibits the
expression or the activity of PG transporters, such as ATP dependent multi-
drug resistance
(MDR) transporter-4, or other MDR channels such as ABCC1, ABCC2, ABCC3, ABCC6,
ABCG2 and ABCB11. Examples of PGT inhibitors that inhibit PGT activity
include, but are not
limited to, compounds that inhibit MDR membrane pumps, such as triazine
compounds,
verapamil, and calcium channel blockers; channels include quinidines,
ketoconazole, itraconazole,
azithromycin, valspodar, cyclosporine, elacridar, fumitremorgin-C, gefitinib
and erythromycin.
Examples of PGT inhibitors that inhibit PGT expression include, but are not
limited to, agents
which control the transcription of the MDR genes by targeting the promoter
region and/or
transcription factors which bind to the promoter or other gene control
regions. The term "PGR
4
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
inhibitors," as used herein, refers to agents that inhibits the activity or
expression of PGRs. In
some embodiments, the PGRs comprise E prostanoid receptor EP1, EP2, EP3, and
EP4 subtypes
of the PGE receptor; PGD receptor (DP1); PGF receptor (FP); PGI receptor (IP);
and
thromboxane receptor (TP). Two additional isoforms of the human TP (TPa and
T113) and FP
(FPA and FPB) and eight EP3 variants are generated through alternative
splicing, which differ
only in their C-terminal tails. In some embodiments, the PGRs further comprise
a G protein-
coupled receptor termed chemo-attractant receptor-homologous molecule (CRHME).
In other
embodiments, the PGRs include all of the receptors that activate rhodopsin-
like 7-transmembrane-
spanning G protein-coupled receptors.
[0023] Examples of PGR activity inhibitors include, but are not limited to,
anti-PGR
antibodies and any agent that inhibits the G-protein coupled receptor
signaling pathway. PGR
expression inhibitors include agents that inhibit PGR expression at the
transcriptional level,
translational level or post transcriptional level. Examples of PGR expression
inhibitors include,
but are not limited to, anti-PGR siRNA and mi RNAs.
[0024] As used herein, the term "an effective amount" means an amount
necessary to
achieve a selected result.
[0025] As used herein, the term "analgesic" refers to agents, compounds or
drugs used to
relieve pain and inclusive of anti-inflammatory compounds. Exemplary analgesic
and/or anti-
inflammatory agents, compounds or drugs include, but are not limited to, non-
steroidal anti-
inflammatory drugs (NSAIDs), salicylates, aspirin, salicylic acid, methyl
salicylate, diflunisal,
sal salate, olsalazine, sulfasalazine, para-aminophenol derivatives,
acetanilide, acetaminophen,
phenacetin, fenamates, mefenamic acid, meclofenamate, sodium meclofenamate,
heteroaryl acetic
acid derivatives, tolmetin, ketorolac, diclofenac, propionic acid derivatives,
ibuprofen, naproxen
sodium, naproxen, fenoprofen, ketoprofen, flurbiprofen, oxaprozin; enolic
acids, oxicam
derivatives, piroxicam, meloxicam, tenoxicam, ampiroxicam, droxicam,
pivoxicam, pyrazolon
derivatives, phenylbutazone, oxyphenbutazone, antipyrine, aminopyrine,
dipyrone, coxibs,
celecoxib, rofecoxib, nabumetone, apazone, indomethacin, sulindac, etodolac,
isobutylphenyl
propionic acid, lumiracoxib, etoricoxib, parecoxib, valdecoxib, tiracoxib,
etodolac, darbufelone,
dexketoprofen, aceclofenac, licofelone, bromfenac, loxoprofen, pranoprofen,
piroxicam,
nimesulide, cizolirine, 3-formylamino-7-methylsulfonylamino-6-phenoxy-4H-1-
benzopyran-4-
one, meloxicam, lornoxicam, d-indobufen, mofezolac, amtolmetin, pranoprofen,
tolfenamic acid,
flurbiprofen, suprofen, oxaprozin, zaltoprofen, alminoprofen, tiaprofenic
acid, pharmacological
salts thereof, hydrates thereof, and solvates thereof
[0026] As used herein, the term "coxib" refers to a compound or composition of
compounds that is capable of inhibiting the activity or expression of COX1 and
COX2 enzymes.
[0027] As used herein, the term "derivative" refers to a chemically modified
compound
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
wherein the modification is considered routine by the ordinary skilled
chemist, such as an ester or
an amide of an acid, or protecting groups such as a benzyl group for an
alcohol or thiol, or a tert-
butoxycarbonyl group for an amine.
[0028] As used herein, the term "analogue" refers to a compound which
comprises a
chemically modified form of a specific compound or class thereof and which
maintains the
pharmaceutical and/or pharmacological activities characteristic of said
compound or class.
[0029] As used herein, the term "pharmaceutically acceptable salts" refer to
derivatives of
the disclosed compounds wherein the parent compound is modified by making acid
or base salts
thereof. Examples of pharmaceutically acceptable salts include, but are not
limited to, mineral or
organic acid salts of basic residues such as amines, alkali or organic salts
of acidic residues such
as carboxylic acids, and the like. The pharmaceutically acceptable salts
include the conventional
non-toxic salts or the quaternary ammonium salts of the parent compound
formed, for example,
from non-toxic inorganic or organic acids. For example, such conventional non-
toxic salts
include those derived from inorganic acids such as hydrochloric acid,
hydrobromic acid, sulfuric
acid, sulfamic acid, phosphoric acid, nitric acid, and the like and the salts
prepared from organic
acids such as acetic acid, propionic acid, succinic acid, glycolic acid,
stearic acid, lactic acid,
malic acid, tartaric acid, citric acid, ascorbic acid, pamoic acid, maleic
acid, hydroxymaleic acid,
phenylacetic acid, glutamic acid, benzoic acid, salicylic acid, sulfanilic
acid, 2-acetoxybenzoic
acid, fumaric acid, toluensulfonic acid, methanesulfonic acid, ethane
dislfonic acid, oxalic acid,
isethionic acid, and the like.
[0030] As used herein, the phrase "pharmaceutically acceptable" is used with
reference to
compounds, materials, compositions and/or dosage forms which are, within the
scope of sound
medical judgment, suitable for use in contact with the tissues of human beings
and animals
without excessive toxicity, irritation, allergic response, or other problems
or complications
commensurate with a reasonable benefit/risk ratio.
[0031] The term "immediate-release" is used herein with reference to a drug
formulation
that does not contain a dissolution rate controlling material. There is
substantially no delay in the
release of the active agents following administration of an immediate-release
formulation. An
immediate-release coating may include suitable materials immediately
dissolving following
administration so as to release the drug contents therein. In some
embodiments, the term
"immediate-release" is used with reference to a drug formulation that releases
the active
ingredient in less than 10 min, 20 min, 30 min, 40 min 50 min, 60 min, 90 min
or 120 min after
administration into a patient.
[0032] As used herein, the term "extended-release," also known as sustained-
release (SR),
sustained-action (SA), time-release (TR), controlled-release (CR), modified
release (MR), or
continuous-release (CR), refers to a mechanism used in medicine tablets or
capsules to dissolve
6
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
slowly and release the active ingredient over time. The advantages of extended-
release tablets or
capsules are that they can often be taken less frequently than immediate-
release formulations of the
same drug and that they keep steadier levels of the drug in the bloodstream,
thus extending the
duration of the drug action and lowering the peak amount of drug in the
bloodstream. In some
embodiments, the term "extended-release" refers to a release profile that the
active ingredient in a
tablet or capsule is released over a period of 2, 3, 4, 5, 6, 7, 8, 9, 10, 12,
14, 16, 18, 20, 22 or 24
hour, either continuously or in pulses after administration into a patient.
[0033] As used herein, the term "delayed-release" refers to a drug release
profile that the
release of the active ingredient(s) of a pharmaceutical composition is delayed
or postponed for a
given period of time (e.g., 1, 2, 3, 4 or 5 hours, or after stomach) after
administration of the
pharmaceutical composition.
[0034] As used herein, the term "delayed-extended-release" refers to a drug
release profile
that the release of the active ingredient(s) of a pharmaceutical composition
is delayed or
postponed for a given period of time (e.g., the lag period of 1, 2, 3, 4 or 5
hours, or after stomach)
after administration of the pharmaceutical composition. Once the release
starts, the active
ingredient(s) is released slowly over time (e.g., over a period of 2, 3, 4, 5,
6, 7, 8, 9, 10, 12, 14, 16,
18, 20, 22 or 24 hour), either continuously or in pulses.
Method for reducing frequency of urination
[0035] One aspect of the present application relates to a method for reducing
frequency of
urination by administering to a subject having a condition that results in an
undesired frequency of
urination an effective amount of a pharmaceutical composition. The
pharmaceutical composition
comprises one or more PG pathway inhibitors and a pharmaceutically acceptable
carrier.
Conditions that result in an undesired frequency of urination include, but are
not limited to,
nocturia, overactive bladder, urinary incontinence and bed wetting.
[0036] In some embodiments, the PG inhibitor is an inhibitor of PG synthetase.
Examples of inhibitors of PG synthesis include, but are not limited to,
inhibitors of PG
synthetase.( this is redundant) In other embodiments, the PG inhibitor is an
inhibitor of PG
activity. Examples of inhibitors of PG activity include, but are not limited
to agents which block
the binding of PG to any of its receptors: EP1, EP2, EP3, EP4. DP1, DP2, FP2,
IP and TP.
Examples of these types of inhibitors include, but are not limited to, the IP
receptor inhibitor
developed by Roche: R03244019, ONO-85-39 which is an EP1 receptor antagonist,
the dual EP1
and EP2 receptor antagonist AH 6809, and the EP4 antagonist. RQ-15986. In
certain
embodiments, the one or more PG pathway inhibitors comprise a PGT inhibitor.
In some
embodiments, the PGT inhibitor is a PGT activity inhibitor. Examples of PGT
activity inhibitor
include, but are not limited to, anti-PGT antibodies, and any known compound
which can inhibit
the ATP-dependent Multi drug resistance transporter-4 or related MDR pumps
that are shown to
7
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
transport PGs. In other embodiments, the PGT inhibitor is a PGT expression
inhibitor. Examples
of PGT expression inhibitor include, but are not limited to, anti-PGT siRNA,
antisense RNAs that
target PGT mRNA, and agents which control the transcription of the gene by
influencing DNA
methylation and or chromatin modification.
[0037] In some embodiments, the one or more PG pathway inhibitors comprise an
inhibitor that targets both the COX active site and the PDX active site which
are contained in both
COX1 and COX2. In other embodiments, the one or more PG pathway inhibitors
comprise an
inhibitor that inhibits the PGE2 pathway.
[0038] In certain embodiments, the one or more PG pathway inhibitors comprise
a PGR
inhibitor. PGRs are G-protein-coupled receptors containing seven transmembrane
domains.
Examples of PGR include, EP1, EP2, EP3, EP4, DP1, DP2, FP, IP1, 1P2, CRTH2 and
TP
receptors. In some embodiments, the one or more PG pathway inhibitors comprise
an inhibitor
that inhibits any of the PG receptors listed above. In some embodiments, the
PGR inhibitor is a
PGR activity inhibitor. Examples of PGR activity inhibitor include, but are
not limited to, anti-
PGR antibodies. In some embodiments, the PGR inhibitor is an inhibitor of PGE2
receptor
activity, such as EP1 activity inhibitor, EP2 activity inhibitor, EP3 activity
inhibitor or EP4
activity inhibitor.
[0039] In other embodiments, the PGR inhibitor is a PGR expression inhibitor.
Examples
of PGR expression inhibitor include, but are not limited to, anti-PGR siRNA,
antisense RNAs that
target PGR mRNA, or agents which control the transcription of the gene by
influencing DNA
methylation and or chromatin modification. In some embodiments, the PGR
expression inhibitor
is an inhibitor of PGE2 receptor expression, such as EP1 expression inhibitor,
EP2 expression
inhibitor, EP3 expression inhibitor or EP4 expression inhibitor. In some
embodiments, the one or
more PG pathway inhibitors comprise a small molecule inhibitor. As used
herein, the term "small
molecule inhibitor" refers to inhibitors having a molecular weight of 1000
dalton or less.
[0040] In some embodiments, the PG pathway inhibitor comprises a short
interfering
RNA (siRNA). An siRNA is a double-stranded RNA that can be engineered to
induce sequence-
specific post-transcriptional gene silencing of mRNAs corresponding to a
component of the PG
pathway. siRNAs exploit the mechanism of RNA interference (RNAi) for the
purpose of
"silencing" gene expression of e.g., targeted PGE2 receptor genes. This
"silencing" was originally
observed in the context of transfecting double stranded RNA (dsRNA) into
cells. Upon entry
therein, the dsRNA was found to be cleaved by an RNase III-like enzyme, Dicer,
into double
stranded small interfering RNAs (siRNAs) 21-23 nucleotides in length
containing 2 nucleotide
overhangs on their 3' ends. In an ATP dependent step, the siRNAs become
integrated into a
multi-subunit RNAi induced silencing complex (RISC) which presents a signal
for AG02-
mediated cleavage of the complementary mRNA sequence, which then leads to its
subsequent
8
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
degradation by cellular exonucleases.
[0041] In some embodiments, the PG pathway inhibitor comprises a synthetic
siRNA or
other class of small RNA targeting a PG synthase RNA, a PGT RNA or a PGR RNA
in the target
cell/tissue. Synthetically produced siRNAs structurally mimic the types of
siRNAs normally
processed in cells by the enzyme Dicer. Synthetically produced siRNAs may
incorporate any
chemical modifications to the RNA structure that are known to enhance siRNA
stability and
functionality. For example, in some cases, the siRNAs may be synthesized as a
locked nucleic
acid (LNA)-modified siRNA. An LNA is a nucleotide analogue that contains a
methylene bridge
connecting the 2'-oxygen of the ribose with the 4' carbon. The bicyclic
structure locks the
furanose ring of the LNA molecule in a 3'-endo conformation, thereby
structurally mimicking the
standard RNA monomers.
[0042] In other embodiments, the PG pathway inhibitor comprises an expression
vector
engineered to transcribe a short double-stranded hairpin-like RNA (shRNA) that
is processed into
a targeted siRNA inside the cell. The shRNAs can be cloned in suitable
expression vectors using
kits, such as Ambion's SILENCER siRNA Construction Kit, Imgenex's
GENESUPPRESSORTM
Construction Kits and Invitrogen's BLOCK-ITTm inducible RNAi plasmid and
lentivirus vectors.
Synthetic siRNAs and shRNAs may be designed using well known algorithms and
synthesized
using a conventional DNA/RNA synthesizer.
[0043] In some embodiments, the secondary active agent comprises an antisense
oligonucleotide or polynucleotide capable of inhibiting the expression of a
component of the PG
pathway. The antisense oligonucleotide or polynucleotide may comprise a DNA
backbone, RNA
backbone or chemical derivative thereof In one embodiment, the antisense
oligonucleotide or
polynucleotide comprises a single stranded antisense oligonucleotide or
polynucleotide targeting
for degradation. In certain embodiments, the anti-inflammatory agent comprises
a single stranded
antisense oligonucleotide complementary to the mRNA sequence of a component of
the PG
pathway. The single stranded antisense oligonucleotide or polynucleotide may
be synthetically
produced or it may be expressed from a suitable expression vector. The
antisense nucleic acid is
designed to bind via complementary binding to the mRNA sense strand so as to
promote RNase H
activity, which leads to degradation of the mRNA. Preferably, the antisense
oligonucleotide is
chemically or structurally modified to promote nuclease stability and/or
increased binding.
[0044] In some embodiments, the antisense oligonucleotides are modified to
produce
oligonucleotides with nonconventional chemical or backbone additions or
substitutions, including,
but not limited to, peptide nucleic acids (PNAs), locked nucleic acids (LNAs),
morpholino
backboned nucleic acids, methylphosphonates, duplex stabilizing stilbene or
pyrenyl caps,
phosphorothioates, phosphoroamidates, phosphotriesters and the like. By way of
example, the
modified oligonucleotides may incorporate or substitute one or more of the
naturally occurring
9
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
nucleotides with an analog; internucleotide modifications incorporating, for
example, uncharged
linkages (e.g., methyl phosphonates, phosphotriesters, phosphoamidates,
carbamates, etc.) or
charged linkages (e.g., phosphorothioates, phosphorodithioates, etc.);
modifications incorporating
intercalators (e.g., acridine, psoralen, etc.), chelators (e.g., metals,
radioactive metals, boron,
oxidative metals, etc.) or alkylators and/or modified linkages (e.g., alpha
anomeric nucleic acids,
etc.).
[0045] In some embodiments, the single stranded oligonucleotides are
internally modified
to include at least one neutral charge in its backbone. For example, the
oligonucleotide may
include a methylphosphonate backbone or peptide nucleic acid (PNA)
complementary to the
target-specific sequence. These modifications have been found to prevent or
reduce helicase-
mediated unwinding. The use of uncharged probes may further increase the rate
of hybridization
to polynucleotide targets in a sample by alleviating the repulsion of
negatively-charges nucleic
acid strands in classical hybridization.
[0046] PNA oligonucleotides are uncharged nucleic acid analogs for which the
phosphodiester backbone has been replaced by a polyamide, which makes PNAs a
polymer of 2-
aminoethyl-glycine units bound together by an amide linkage. PNAs are
synthesized using the
same Boc or Fmoc chemistry as are use in standard peptide synthesis. Bases
(adenine, guanine,
cytosine and thymine) are linked to the backbone by a methylene carboxyl
linkage. Thus, PNAs
are acyclic, achiral and neutral. Other properties of PNAs are increased
specificity and melting
temperature as compared to nucleic acids, capacity to form triple helices,
stability at acid pH, non-
recognition by cellular enzymes like nucleases, polymerases, etc.
[0047] Methylphosphonate-containing oligonucleotides are neutral DNA analogs
containing a methyl group in place of one of the non-bonding phosphoryl
oxygens.
Oligonucleotides with methylphosphonate linkages were among the first reported
to inhibit
protein synthesis via anti-sense blockade of translation.
[0048] In some embodiments, the phosphate backbone in the oligonucleotides may
contain phosphorothioate linkages or phosphoroamidates. Combinations of such
oligonucleotide
linkages are also within the scope of the present invention.
[0049] In other embodiments, the oligonucleotide may contain a backbone of
modified
sugars joined by phosphodiester internucleotide linkages. The modified sugars
may include
furanose analogs, including but not limited to 2-deoxyribofuranosides, a-D-
arabinofuranosides, a-
2'-deoxyribofuranosides and 2',3'-dideoxy-3'-aminoribofuranosides. In
alternative embodiments,
the 2-deoxy-3-D-ribofuranose groups may be replaced with other sugars, for
example, 13-D-
ribofuranose. In addition, P-D-ribofuranose may be present wherein the 2-0H of
the ribose
moiety is alkylated with a C1-6 alkyl group (2-(0--C1-6 alkyl) ribose) or with
a C2-6 alkenyl
group (2-(0--C2-6 alkenyl) ribose) or is replaced by a fluor group (2-
fluororibose).
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
[0050] Related oligomer-forming sugars include those used in locked nucleic
acids (LNA)
as described above. Exemplary LNA oligonucleotides include modified bicyclic
monomeric units
with a 2'-0-4'-C methylene bridge, such as those described in U.S. Patent No.
6,268,490.
[0051] Chemically modified oligonucleotides may also include, singly or in any
combination, 2'-position sugar modifications, 5-position pyrimidine
modifications (e.g., 5-(N-
benzylcarboxyamide)-2'-deoxyuridine, 5-(N-isobutylcarboxyamide)-2'-
deoxyuridine, 5-(N42-
(1H-indole-3y1)ethyl]carboxyamide)-2'-deoxyuridine, 5-(N41-(3-
trimethylammonium)
propyl]carboxyamide)-2'-deoxyuridine chloride, 5-(N-napthylcarboxyamide)-2'-
deoxyuridine, 5-
(N-E1-(2,3-dihydroxypropyl)]carboxyamide)-2'-deoxyuridine), 8-position purine
modifications,
modifications at exocyclic amines, substitution of 4-thiouridine, substitution
of 5-bromo- or 5-
iodo-uracil, methylations, unusual base-pairing combinations, such as the
isobases isocytidine and
isoguanidine and the like.
[0052] In some embodiments, the one or more PG pathway inhibitors comprise a
ribozyme capable of inhibiting the expression of a component of the PG
pathway. Ribozymes are
nucleic acid molecules that are capable of catalyzing a chemical reaction,
either intramolecularly
or intermolecularly. Ribozymes are thus catalytic nucleic acid. It is
preferred that the ribozymes
catalyze intermolecular reactions. There are a number of different types of
ribozymes that
catalyze nuclease or nucleic acid polymerase type reactions which are based on
ribozymes found
in natural systems, such as hammerhead ribozymes, hairpin ribozymes and
tetrahymena
ribozymes. There are also a number of ribozymes that are not found in natural
systems, but which
have been engineered to catalyze specific reactions de novo. Preferred
ribozymes cleave RNA or
DNA substrates and more preferably cleave RNA substrates, such as mRNAs of
components of
the PG pathway. Ribozymes typically cleave nucleic acid substrates through
recognition and
binding of the target substrate with subsequent cleavage. This recognition is
often based mostly
on canonical or non-canonical base pair interactions. This property makes
ribozymes particularly
good candidates for target specific cleavage of nucleic acids because
recognition of the target
substrate is based on the target substrates sequence.
[0053] In some embodiments, the one or more PG pathway inhibitors comprise
triplex
forming oligonucleotide capable of inhibiting the expression of a component of
the PG pathway.
Triplex forming oligonucleotides (TFOs) are molecules that can interact with
either double-
stranded and/or single-stranded nucleic acids, including both coding and non-
coding regions in
genomic DNA targets. When TFOs interact with a target region, a structure
called a triplex is
formed, in which there are three strands of DNA forming a complex dependant on
both Watson-
Crick and Hoogsteen base-pairing. TFOs can bind target regions with high
affinity and
specificity. In preferred embodiments, the triplex forming molecules bind the
target molecule
with a Kd less than 10-6, 10-8, 10-10 or 10-12. Exemplary TFOs for use in the
present invention
11
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
include PNAs, LNAs and LNA modified PNAs, such as Zorro-LNAs.
[0054] In some embodiments, the one or more PG pathway inhibitors comprise an
external guide sequences (EGS). External guide sequences (EGSs) are molecules
that bind a
target nucleic acid molecule forming a complex. This complex is recognized by
RNase P, which
cleaves the target molecule. EGSs can be designed to specifically target an
mRNA molecule of
choice. RNAse P aids in processing transfer RNA (tRNA) within a cell.
Bacterial RNAse P can
be recruited to cleave virtually any RNA sequence by using an EGS that causes
the target
RNA:EGS complex to mimic the natural tRNA substrate. Similarly, eukaryotic
EGS/RNAse P-
directed cleavage of RNA can be utilized to cleave desired targets within
eukaryotic cells.
[0055] In other embodiments, the one or more PG pathway inhibitors comprise a
biomolecule. As used herein, the term "biomolecule" is any molecule that is
produced by a living
organism, including large macromolecules such as proteins, polysaccharides,
lipids, and nucleic
acids, as well as small molecules such as primary metabolites, secondary
metabolites, and natural
products.
[0056] In other embodiments, the one or more PG pathway inhibitors comprise
target
neutralization agents. As used herein, the term "target neutralization agent"
refers to antibodies,
fragments of antibodies, or any other non-antibody peptide or synthetic
binding molecule, such as
an aptamer or synbody, which is capable of specifically binding directly or
indirectly to a
component of the PG pathway so as to interfere with the ultimate actions of
prostaglandin on the
target tissue.
[0057] The target neutralization agents may be produced by any conventional
method for
generating high-affinity binding ligands, including SELEX, phage display and
other
methodologies, including combinatorial chemistry and/or high throughput
methods known to
those of skill in the art.
[0058] An aptamer is a nucleic acid version of an antibody that comprises a
class of
oligonucleotides that can form specific three dimensional structures
exhibiting high affinity
binding to a wide variety of cell surface molecules, proteins and/or
macromolecular structures.
Aptamers are commonly identified by an in vitro method of selection sometimes
referred to as
Systematic Evolution of Ligands by EXponential enrichment or "SELEX." SELEX
typically
begins with a very large pool of randomized polynucleotides which is generally
narrowed to one
aptamer ligand per molecular target. Typically, aptamers are small nucleic
acids ranging from 15-
50 bases in length that fold into defined secondary and tertiary structures,
such as stem-loops or
G-quartets.
[0059] An aptamer can be chemically linked or conjugated to the above
described nucleic
acid inhibitors to form targeted nucleic acid inhibitors, such as aptamer-
siRNA chimeras. An
aptamer-siRNA chimera contains a targeting moiety in the form of an aptamer
which is linked to
12
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
an siRNA. When using an aptamer-siRNA chimera, it is preferable to use a cell
internalizing
aptamer. Upon binding to specific cell surface molecules, the aptamer can
facilitate
internalization into the cell where the nucleic acid inhibitor acts. In one
embodiment both the
aptamer and the siRNA comprises RNA. The aptamer and the siRNA may comprise
any
nucleotide modifications as further described herein. Preferably, the aptamer
comprises a
targeting moiety specifically directed to binding cells expressing the
chemokine-, cytokine- and/or
receptor target genes, such as lymphoid, epithelial cell and/or endothelial
cells.
[0060] Synbodies are synthetic antibodies produced from libraries comprised of
strings of
random peptides screened for binding to target proteins of interest.
[0061] Target neutralization agents, including aptamers and synbodies, can be
engineered
to bind target molecules very tightly with Kds between 10-10 to 10-12 M. In
some embodiments,
the target neutralization agent binds the target molecule with a Kd less than
10-6, less than 10-8,
less than 10-9, less than 10-10 or less than 10-12 M.
[0062] In certain embodiments, the one or more PG pathway inhibitors comprise
a
polynucleotide that encodes and is adapted to express a PGT inhibitor and/or a
PGR inhibitor. In
other embodiments, the one or more PG pathway inhibitors comprise an
expression vector that
encodes and is adapted to express a PGT inhibitor and/or a PGR inhibitor.
[0063] In some embodiments, the PG pathway inhibitor is an engineered protein
containing a TALE sequence or and engineered zinc finger directed at the gene
encoding any
component of the PG pathway. This TALE or zinc finger could be designed to
bind directly to
the gene and inhibit its expression by cleaving the gene, altering its
nucleotide sequence, or
tethering a repressor protein to the gene which serves to silence the gene.
[0064] In some embodiments, the PG pathway inhibitor is produced using the
CRISPR/CAS system. In this strategy a guide molecule specific for the gene
sequence of each pg
pathway gene is designed and introduced into the cell or tissue using delivery
systems described
above (viruses, plasmids, etc). The action of the CRISPR/CAS system would
modify the DNA
sequence of the gene such that the PG pathway gene is deleted or inhibited in
ability to express
the RNA.
[0065] In some embodiments, the PG pathway inhibitor is capable of turning off
the
transcription of one or more PG pathway genes by targeting the chromatin
associated enzymes
which post translationally modify histones in chromatin. Examples of such
enzymes are (but not
limited to) histone deacetylases, histone demethylases, histone
acetyltransferases, histone
methyltransferases, and helicases.
[0066] In some embodiments, the PG pathway inhibitor targets the gene encoding
each
component by altering the DNA methylation status of the gene. Compounds which
target the
TET family of DNA demethylases and the DNA methyltransferases (DNMT1, DNMTa
and
13
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
DNMTb) could change the expression of RNA from the any of the genes in the PG
pathway.
[0067] The expression vector of the present application comprises a
polynucleotide
encoding a PG pathway inhibitor or a portion thereof. The expression vectors
also include one or
more regulatory sequences operably linked to the polynucleotide being
expressed. These
regulatory sequences are selected based on the type of host cells. It will be
appreciated by those
skilled in the art that the design of the expression vector depends on such
factors as the choice of
the host cells and the desired expression levels.
[0068] In some embodiments, the expression vector is a plasmid vector. In
other
embodiments, the expression vector is a viral vector. Examples of viral
vectors include, but are
not limited to, retrovirus, lentivirus, adenovirus, adeno-associated virus
(AAV), herpes virus, or
alphavirus vectors. The viral vectors can also be astrovirus, coronavirus,
orthomyxovirus,
papovavirus, paramyxovirus, parvovirus, picornavirus, poxvirus, or togavirus
vectors. When used
in mammalian cells, the expression vector's control functions are often
provided by viral
regulatory elements. For example, commonly used promoters are derived from
polyoma,
adenovirus 2, cytomegalovirus and Simian Virus 40. In some embodiments, the
expression vector
contains tissue-specific regulatory elements. Delivery of the expression
vector include, but are
not limited to, direct infection with viral vectors, exposing target tissue to
polycationic condensed
DNA linked or unlinked to killed virus, ligand linked DNA, gene guns, ionizing
radiation, nucleic
charge neutralization, or fusion with cell membranes. Naked plasmid or viral
DNA can also be
employed. Uptake efficiency may be improved using biodegradable latex beads.
This method
can be further improved by treating the beads to increase their
hydrophobicity. Liposome-based
methods can also be used to introduce plasmid or viral vector into the target
tissue.
[0069] In some embodiments, the pharmaceutical composition further comprises
one or
more active ingredients selected from the group consisting of analgesic
agents, antimuscarinic
agents, antidiuretics, spasmolytics, inhibitors of phosphodiesterase type 5
(PDE 5 inhibitors) and
zolpedim.
[0070] Examples of antimuscarinic agents include, but are not limited to,
oxybutynin,
solifenacin, darifenacin, fesoterodine, tolterodine, trospium, atropine, and
tricyclic
antidepressants. Examples of antidiuretics include, but are not limited to,
antidiuretic hormone
(ADH), angiotensin II, aldosterone, vasopressin, vasopressin analogs (e.g.,
desmopressin
argipressin, lypressin, felypressin, ornipressin, terlipressin), vasopressin
receptor agonists, atrial
natriuretic peptide (ANP) and C-type natriuretic peptide (CNP) receptor (i.e.,
NPR1, NPR2, and
NPR3) antagonists (e.g., HS-142-1, isatin, [Asu7,23']b-ANP-(7-28)], anantin, a
cyclic peptide
from Streptomyces coerulescens, and 3G12 monoclonal antibody), somatostatin
type 2 receptor
antagonists (e.g., somatostatin), pharmaceutically-acceptable derivatives, and
analogs, salts,
hydrates, and solvates thereof Examples of spasmolytics include, but are not
limited to,
14
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
carisoprodol, benzodiazepines, baclofen, cyclobenzaprine, metaxalone,
methocarbamol, clonidine,
clonidine analog, and dantrolene. Examples of PDE 5 inhibitors include, but
are not limited to,
tadalafil, sildenafil and vardenafil.
[0071] The pharmaceutical composition may be formulated for immediate-release,
extended-release, delayed-release, or combinations thereof
[0072] In some embodiments, the pharmaceutical composition is formulated for
immediate-release.
[0073] In other embodiments, the pharmaceutical composition is formulated for
extended-
release by embedding the active ingredient in a matrix of insoluble
substance(s) such as acrylics or
chitin. The extended-release form is designed to release the active ingredient
at a predetermined
rate by maintaining a constant drug level for a specific period of time. This
can be achieved
through a variety of formulations, including, but not limited to, liposomes
and drug-polymer
conjugates, such as hydrogels.
[0074] An extended-release formulation can be designed to release the active
ingredient at
a predetermined rate so as to maintain a constant drug level for a specified,
extended period of
time, such as up to about 24 hours, about 22 hours, about 20 hours, about 18
hours, about 16
hours, about 14 hours, about 12 hours, about 10 hours, about 9 hours, about 8
hours, about 7
hours, about 6 hours, about 5 hours, about 4 hours, about 3 hours or about 2
hours following
administration or following a lag period associated with delayed-release of
the active ingredient.
The constant active ingredient level may be maintained by a continuous release
of the active
ingredient or pulsed-release of the active ingredient.
[0075] In certain embodiments, the active ingredient in an extended-release
formulation is
released over a time interval of between about 1 to about 24 hours, or between
2 to about 12
hours. Alternatively, the active ingredient may be released over about 3,
about 4, about 5, about
6, about 7, about 8, about 9, about 10 hours, about 11 hours, about 12 hours,
about 14 hours,
about 16 hours, about 18 hours, about 20 hours, about 22 hours or about 24
hours. In yet other
embodiments, the active ingredient in an extended-release formulation is
released over a time
period between about 5 to about 8 hours following administration.
[0076] In some embodiments, the extended-release formulation comprises an
active core
comprised of one or more inert particles, each in the form of a bead, pellet,
pill, granular particle,
microcapsule, microsphere, microgranule, nanocapsule, or nanosphere coated on
its surfaces with
drugs in the form of a drug-containing coating or film-forming composition
using, for example,
fluid bed techniques or other methodologies known to those of skill in the
art. The inert particle
can be of various sizes, so long as it is large enough to remain poorly
dissolved. Alternatively, the
active core may be prepared by granulating and milling and/or by extrusion and
spheronization of
a polymer composition containing the drug substance. As used herein, the term
"drug" refers to
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
the active ingredient of a pharmaceutical composition.
[0077] The active ingredient may be introduced to the inert carrier by
techniques known to
one skilled in the art, such as drug layering, powder coating,
extrusion/spheronization, roller
compaction or granulation. The amount of active ingredient in the core will
depend on the dose
that is required and typically varies from about 1 to 100 weight %, about 5 to
100 weight %, about
to 100 weight %, about 20 to 100 weight %, about 30 to 100 weight %, about 40
to 100 weight
%, about 50 to 100 weight %, about 60 to 100 weight %, about 70 to 100 weight
%, or about 80 to
100 weight %.
[0078] Generally, the polymeric coating on the active core will be from about
1 to 50%
based on the weight of the coated particle, depending on the lag time required
and/or the polymers
and coating solvents chosen. Those skilled in the art will be able to select
an appropriate amount
of drug for coating onto or incorporating into the core to achieve the desired
dosage. In one
embodiment, the inactive core may be a sugar sphere or a buffer crystal or an
encapsulated buffer
crystal such as calcium carbonate, sodium bicarbonate, fumaric acid, tartaric
acid, etc. which
alters the microenvironment of the drug to facilitate its release.
[0079] Extended-release formulations may utilize a variety of extended-release
coatings or
mechanisms facilitating the gradual release of active agents over time. In
some embodiments, the
extended-release agent comprises a polymer controlling release by dissolution
controlled release.
In a particular embodiment, the active agent(s) is incorporated in a matrix
comprising an insoluble
polymer and drug particles or granules coated with polymeric materials of
varying thickness. The
polymeric material may comprise a lipid barrier comprising a waxy material,
such as carnauba
wax, beeswax, spermaceti wax, candellila wax, shallac wax, cocoa butter,
cetostearyl alcohol,
partially hydrogenated vegetable oils, ceresin, paraffin wax, ceresine,
myristyl alcohol, stearyl
alcohol, cetyl alcohol, and stearic acid, along with surfactants, such as
polyoxyethylene sorbitan
monooleate. When contacted with an aqueous medium, such as biological fluids,
the polymer
coating emulsifies or erodes after a predetermined lag-time depending on the
thickness of the
polymer coating. The lag time is independent of gastrointestinal motility, pH,
or gastric
residence.
[0080] In other embodiments, the extended-release agent comprises a polymeric
matrix
effecting diffusion controlled release. The matrix may comprise one or more
hydrophilic and/or
water-swellable, matrix forming polymers, pH-dependent polymers and/or pH-
independent
polymers.
[0081] In one embodiment, the extended-release formulation comprises a water
soluble or
water-swellable matrix-forming polymer, optionally containing one or more
solubility-enhancing
agents and/or release-promoting agents. Upon solubilization of the water
soluble polymer, the
active agent(s) dissolves (if soluble) and gradually diffuses through the
hydrated portion of the
16
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
matrix. The gel layer grows with time as more water permeates into the core of
the matrix,
increasing the thickness of the gel layer and providing a diffusion barrier to
drug release. As the
outer layer becomes fully hydrated, the polymer chains become completely
relaxed and can no
longer maintain the integrity of the gel layer, leading to disentanglement and
erosion of the outer
hydrated polymer on the surface of the matrix. Water continues to penetrate
towards the core
through the gel layer, until it has been completely eroded. Whereas soluble
drugs are released by
this combination of diffusion and erosion mechanisms, erosion is the
predominant mechanism for
insoluble drugs, regardless of dose.
[0082] Similarly, water-swellable polymers typically hydrate and swell in
biological fluids
forming a homogenous matrix structure that maintains its shape during drug
release and serves as
a carrier for the drug, solubility enhancers and/or release promoters. The
initial matrix polymer
hydration phase results in slow-release of the drug (lag phase). Once the
water swellable polymer
is fully hydrated and swollen, water within the matrix can similarly dissolve
the drug substance
and allow for its diffusion out through the matrix coating.
[0083] Additionally, the porosity of the matrix can be increased due to the
leaching out of
pH-dependent release promoters so as to release the drug at a faster rate. The
rate of the drug
release then becomes constant and is a function of drug diffusion through the
hydrated polymer
gel. The release rate from the matrix is dependent upon various factors,
including polymer type
and level, drug solubility and dose, polymer to drug ratio, filler type and
level, polymer to filler
ratio, particle size of drug and polymer, and porosity and shape of the
matrix.
[0084] Exemplary hydrophilic and/or water-swellable, matrix forming polymers
include,
but are not limited to, cellulosic polymers including hydroxyalkyl celluloses
and carboxyalkyl
celluloses such as hydroxypropylmethylcellulose (HPMC), hydroxypropylcellulose
(HPC),
hydroxyethylcellulose (HEC), methylcellulose (MC), carboxymethylcellulose
(CMC); powdered
cellulose such as microcrystalline cellulose, cellulose acetate,
ethylcellulose, salts thereof, and
combinations thereof; alginates; gums including heteropolysaccharide gums and
homopolysaccharide gums such as xanthan, tragacanth, pectin, acacia, karaya,
alginates, agar,
guar, hydroxypropyl guar, veegum, carrageenan, locust bean gum, gellan gum,
and derivatives
therefrom; acrylic resins including polymers and copolymers of acrylic acid,
methacrylic acid,
methyl acrylate, and methyl methacrylate; and cross-linked polyacrylic acid
derivatives such as
Carbomers (e.g., CARBOPOL , including CARBOPOL 71G NE, which is available in
various
molecular weight grades from Noveon, Inc., Cincinnati, OH), carageenan;
polyvinyl acetate (e.g.,
KOLLIDON SR); and polyvinyl pyrrolidone and its derivatives such as
crospovidone,
polyethylene oxides, and polyvinyl alcohol. Preferred hydrophilic and water-
swellable polymers
include the cellulosic polymers, especially HPMC.
[0085] The extended-release formulation may further comprise at least one
binder that is
17
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
capable of cross-linking the hydrophilic compound to form a hydrophilic
polymer matrix (e.g., a
gel matrix) in an aqueous medium, including biological fluids.
[0086] Exemplary binders include homopolysaccharides such as galactomannan
gums,
guar gum, hydroxypropyl guar gum, hydroxypropylcellulose (HPC; e.g., Klucel
EXF), and locust
bean gum. In other embodiments, the binder is an alginic acid derivative, HPC
or
microcrystallized cellulose (MCC). Other binders include, but are not limited
to, starches,
microcrystalline cellulose, hydroxypropyl cellulose, hydroxyethyl cellulose,
hydroxypropylmethyl
cellulose, and polyvinylpyrrolidone.
[0087] In one embodiment, the introduction method is drug layering by spraying
a
suspension of active agent(s) and a binder onto the inert carrier.
[0088] The binder may be present in the bead formulation in an amount of about
0.1% to
about 15% by weight and preferably of from about 0.2% to about 10% by weight.
[0089] In some embodiments, the hydrophilic polymer matrix may further include
an
ionic polymer, a non-ionic polymer, or water-insoluble hydrophobic polymer to
provide a
stronger gel layer and/or reduce pore quantity and dimensions in the matrix so
as to slow diffusion
and erosion rates and concomitant release of the active agent(s). This may
additionally suppress
the initial burst effect and produce a more steady "zero order release" of
active agent(s).
[0090] Exemplary ionic polymers for slowing dissolution rate include both
anionic and
cationic polymers. Exemplary anionic polymers include, for example, sodium
carboxymethylcellulose (Na CMC); sodium alginate, polymers of acrylic acid or
carbomers (e.g.,
CARBOPOL 934, 940, 974P NF); enteric polymers such as polyvinyl acetate
phthalate (PVAP),
methacrylic acid copolymers (e.g., EUIDRAGIT L100, L 30D 55, A, and FS 30D),
and
hypromellose acetate succinate (AQUAT HPMCAS); and xanthan gum. Exemplary
cationic
polymers include, for example, dimethylaminoethyl methacrylate copolymer
(e.g., EUIDRAGIT
E 100). Incorporation of anionic polymers, particularly enteric polymers, is
useful for developing
a pH-independent release profile for weakly basic drugs as compared to
hydrophilic polymer
alone.
[0091] Exemplary non-ionic polymers for slowing dissolution rate, include, for
example,
hydroxypropylcellulose (HPC) and polyethylene oxide (PEO) (e.g., POLYOXTm).
[0092] Exemplary hydrophobic polymers include ethylcellulose (e.g., ETHOCELTm,
SURELEASE ), cellulose acetate, methacrylic acid copolymers (e.g., EUIDRAGIT
NE 30D),
ammonio-methacrylate copolymers (e.g., EUIDRAGIT RL 100 or PO RS100),
polyvinyl acetate,
glyceryl monostearate, fatty acids such as acetyl tributyl citrate, and
combinations and derivatives
thereof.
[0093] The swellable polymer can be incorporated in the formulation in
proportion from
1% to 50% by weight, preferably from 5% to 40% by weight, most preferably from
5% to 20% by
18
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
weight. The swellable polymers and binders may be incorporated in the
formulation either prior
to or after granulation. The polymers can also be dispersed in organic
solvents or hydro-alcohols
and sprayed during granulation.
[0094] Exemplary release-promoting agents include pH-dependent enteric
polymers that
remain intact at a pH value lower than about 4.0 and dissolve at pH values
higher than 4.0,
preferably higher than 5.0, most preferably about 6.0, and are considered
useful as release-
promoting agents for this invention. Exemplary pH-dependent polymers include,
but are not
limited to, methacarylic acid copolymers; methacrylic acid-methyl methacrylate
copolymers (e.g.,
EUDRAGIT L100 (Type A), EUDRAGIT S100 (Type B), Rohm GmbH, Germany),
methacrylic acid-ethyl acrylate copolymers (e.g., EUDRAGIT L100-55 (Type C)
and
EUDRAGIT L30D-55 copolymer dispersion, Rohm GmbH, Germany); copolymers of
methacrylic acid-methyl methacrylate and methyl methacrylate (EUIDRAGIT FS);
terpolymers
of methacrylic acid, methacrylate, and ethyl acrylate, cellulose acetate
phthalates (CAP);
hydroxypropyl methylcellulose phthalate (HPMCP) (e.g., HP-55, HP-50, HP-55S,
Shinetsu
Chemical, Japan); polyvinyl acetate phthalates (PVAP) (e.g., COATERIC , OPADRY
enteric
white OY-P-7171); polyvinylbutyrate acetate, cellulose acetate succinates
(CAS); hydroxypropyl
methylcellulose acetate succinate (HPMCAS) (e.g., HPMCAS LF Grade, MF Grade,
and HF
Grade, including AQOAT LF and AQOAT MF, Shin-Etsu Chemical, Japan), shellac
(e.g.,
MARCOATTm 125 and MARCOATTm 125N); vinyl acetate-maleic anhydride copolymer,
styrene-maleic monoester copolymer, carboxymethyl ethylcellulose (CMEC, Freund
Corporation,
Japan); cellulose acetate phthalates (CAP) (e.g., AQUATERIC ), cellulose
acetate trimellitates
(CAT), and mixtures of two or more thereof at weight ratios between about 2:1
to about 5:1, such
as a mixture of EUIDRAGIT L 100-55 and EUIDRAGIT S 100 at a weight ratio of
about 3:1 to
about 2:1, or a mixture of EUIDRAGIT L 30 D-55 and EUIDRAGIT FS at a weight
ratio of
about 3:1 to about 5:1.
[0095] These polymers may be used either alone or in combination, or together
with
polymers other than those mentioned above. Preferred enteric pH-dependent
polymers are the
pharmaceutically acceptable methacrylic acid copolymers. These copolymers are
anionic
polymers based on methacrylic acid and methyl methacrylate and, preferably,
have a mean
molecular weight of about 50,000 to 200,000, preferably about 135,000. A ratio
of free carboxyl
groups to methyl-esterified carboxyl groups in these copolymers may range, for
example, from
1:1 to 1:3, e.g. around 1:1 or 1:2. The release promoters are not limited to
pH dependent
polymers. Other hydrophilic molecules that dissolve rapidly and leach out of
the dosage form
quickly leaving a porous structure can be also be used for the same purpose.
[0096] In some embodiments, the matrix may include a combination of release
promoters
and solubility enhancing agents. The solubility enhancing agents can be ionic
and non-ionic
19
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
surfactants, complexing agents, hydrophilic polymers, and pH modifiers such as
acidifying agents
and alkalinizing agents, as well as molecules that increase the solubility of
poorly soluble drug
through molecular entrapment. Several solubility enhancing agents can be
utilized
simultaneously.
[0097] Solubility enhancing agents may include surface active agents, such as
sodium
docusate; sodium lauryl sulfate; sodium stearyl fumarate; Tweens and Spans
(PEO modified
sorbitan monoesters and fatty acid sorbitan esters); poly(ethylene oxide)-
polypropylene oxide-
poly(ethylene oxide) block copolymers (aka PLURONICSTm); complexing agents
such as low
molecular weight polyvinyl pyrrolidone and low molecular weight hydroxypropyl
methyl
cellulose; molecules that aid solubility by molecular entrapment such as
cyclodextrins and pH
modifying agents, including acidifying agents such as citric acid, fumaric
acid, tartaric acid, and
hydrochloric acid, and alkalizing agents such as meglumine and sodium
hydroxide.
[0098] Solubility enhancing agents typically constitute from 1% to 80% by
weight, from
1% to 60% by weight, from 1% to 50% by weight, from 1% to 40% by weight and
from 1% to
30% by weight, of the dosage form and can be incorporated in a variety of
ways. They can be
incorporated in the formulation prior to granulation in dry or wet form. They
can also be added to
the formulation after the rest of the materials are granulated or otherwise
processed. During
granulation, solubility enhancing agents can be sprayed as solutions with or
without a binder.
[0099] In one embodiment, the extended-release formulation comprises a water-
insoluble
water-permeable polymeric coating or matrix comprising one or more water-
insoluble water-
permeable film-forming over the active core. The coating may additionally
include one or more
water soluble polymers and/or one or more plasticizers. The water-insoluble
polymer coating
comprises a barrier coating for release of active agents in the core, wherein
lower molecular
weight (viscosity) grades exhibit faster release rates as compared to higher
viscosity grades.
[0100] In some embodiments, the water-insoluble film-forming polymers include
one or
more alkyl cellulose ethers, such as ethyl celluloses and mixtures thereof,
(e.g., ethyl cellulose
grades PR100, PR45, PR20, PR10, and PR7; ETHOCEL , Dow).
[0101] In some embodiments, the water-insoluble polymer provides suitable
properties
(e.g., extended-release characteristics, mechanical properties, and coating
properties) without the
need for a plasticizer. For example, coatings comprising polyvinyl acetate
(PVA), neutral
copolymers of acrylate/methacrylate esters such as commercially available
Eudragit NE3OD from
Evonik Industries, ethyl cellulose in combination with hydroxypropylcellulose,
waxes, etc. can be
applied without plasticizers.
[0102] In yet another embodiment, the water-insoluble polymer matrix may
further
include a plasticizer. The amount of plasticizer required depends upon the
plasticizer, the
properties of the water-insoluble polymer and the ultimate desired properties
of the coating.
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
Suitable levels of plasticizer range from about 1% to about 20%, from about 3
A to about 20%,
about 3 A to about 5%, about '7 A to about 10%, about 12% to about 15%, about
17% to about
200 o, or about 100, about 2%, about 300, about 40, about 50, about 6%, about
70, about 8%,
about 90, about 10%, about 150o, or about 2000 by weight relative to the total
weight of the
coating, inclusive of all ranges and sub-ranges therebetween.
[0103] Exemplary plasticizers include, but are not limited to, triacetin,
acetylated
monoglyceride, oils (castor oil, hydrogenated castor oil, grape seed oil,
sesame oil, olive oil, and
etc.), citrate esters, triethyl citrate, acetyltriethyl citrate acetyltributyl
citrate, tributyl citrate, acetyl
tri-n-butyl citrate, diethyl phthalate, dibutyl phthalate, dioctyl phthalate,
methyl paraben, propyl
paraben, propyl paraben, butyl paraben, diethyl sebacate, dibutyl sebacate,
glyceroltributyrate,
substituted triglycerides and glycerides, monoacetylated and diacetylated
glycerides (e.g.,
MYVACET 9-45), glyceryl monostearate, glycerol tributyrate, polysorbate 80,
polyethyleneglycol (such as PEG-4000 and PEG-400), propyleneglycol, 1,2-
propyleneglycol,
glycerin, sorbitol, diethyl oxalate, diethyl malate, diethyl fumarate,
diethylmalonate, dibutyl
succinate, fatty acids, glycerin, sorbitol, diethyl oxalate, diethyl malate,
diethyl maleate, diethyl
fumarate, diethyl succinate, diethyl malonate, dioctyl phthalate, dibutyl
sebacate, and mixtures
thereof. The plasticizer can have surfactant properties, such that it can act
as a release modifier.
For example, non-ionic detergents such as Brij 58 (polyoxyethylene (20) cetyl
ether), and the like,
can be used.
[0104] Plasticizers can be high boiling point organic solvents used to impart
flexibility to
otherwise hard or brittle polymeric materials and can affect the release
profile for the active
agent(s). Plasticizers generally cause a reduction in the cohesive
intermolecular forces along the
polymer chains resulting in various changes in polymer properties. These
changes include, but are
not limited to, a reduction in tensile strength and increase in elongation and
a reduction in the
glass transition or softening temperature of the polymer. The amount and
choice of the plasticizer
can affect the hardness of a tablet, for example, and can even affect its
dissolution or
disintegration characteristics, as well as its physical and chemical
stability. Certain plasticizers
can increase the elasticity and/or pliability of a coat, thereby decreasing
the coat's brittleness.
[0105] In another embodiment, the extended-release formulation comprises a
combination
of at least two gel-forming polymers, including at least one non-ionic gel-
forming polymer and/or
at least one anionic gel-forming polymer. The gel formed by the combination of
gel-forming
polymers provides controlled release, such that when the formulation is
ingested and comes into
contact with the gastrointestinal fluids, the polymers nearest the surface
hydrate to form a viscous
gel layer. Due to the high viscosity, the viscous layer dissolves away only
gradually, exposing the
material below to the same process. The mass thus dissolves away slowly,
thereby slowly
releasing the active ingredient into the gastrointestinal fluids. The
combination of at least two
21
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
gel-forming polymers enables properties of the resultant gel, such as
viscosity, to be manipulated
in order to provide the desired release profile.
[0106] In a particular embodiment, the formulation comprises at least one non-
ionic gel-
forming polymer and at least one anionic gel-forming polymer. In another
embodiment, the
formulation comprises two different non-ionic gel-forming polymers. In yet
another embodiment,
the formulation comprises a combination of non-ionic gel-forming polymers with
the same
chemistry, but different solubilities, viscosities, and/or molecular weights
(for example, a
combination of hydroxyproplyl methylcellulose of different viscosity grades,
such as HPMC
K100 and HPMC K15M or HPMC KlOOM).
[0107] Exemplary anionic gel forming polymers include, but are not limited to,
sodium
carboxymethylcellulose (Na CMC), carboxymethyl cellulose (CMC), anionic
polysaccharides
such as sodium alginate, alginic acid, pectin, polyglucuronic acid (poly-a-
and -0-1,4-glucuronic
acid), polygalacturonic acid (pectic acid), chondroitin sulfate, carrageenan,
furcellaran, anionic
gums such as xanthan gum, polymers of acrylic acid or carbomers (Carbopol
934, 940, 974P
NF), Carbopol copolymers, a Pemulen polymer, polycarbophil, and others.
[0108] Exemplary non-ionic gel-forming polymers include, but are not limited
to,
Povidone (PVP: polyvinyl pyrrolidone), polyvinyl alcohol, copolymer of PVP and
polyvinyl
acetate, HPC (hydroxypropyl cellulose), HPMC (hydroxypropyl methylcellulose),
hydroxyethyl
cellulose, hydroxymethyl cellulose, gelatin, polyethylene oxide, acacia,
dextrin, starch,
polyhydroxyethylmethacrylate (PHEMA), water soluble nonionic polymethacrylates
and their
copolymers, modified cellulose, modified polysaccharides, nonionic gums,
nonionic
polysaccharides, and/or mixtures thereof.
[0109] The formulation may optionally comprise an enteric polymer as described
above
and/or at least one excipient, such as a filler, a binder (as described
above), a disintegrant and/or a
flow aid or glidant.
[0110] Exemplary fillers include, but are not limited to, lactose, glucose,
fructose, sucrose,
dicalcium phosphate, sugar alcohols also known as "sugar polyol" such as
sorbitol, manitol,
lactitol, xylitol, isomalt, erythritol, and hydrogenated starch hydrolysates
(a blend of several sugar
alcohols), corn starch, potato starch, sodium carboxymethycellulose,
ethylcellulose and cellulose
acetate, enteric polymers, or a mixture thereof.
[0111] Exemplary binders include, but are not limited to, water-soluble
hydrophilic
polymers such as Povidone (PVP: polyvinyl pyrrolidone), copovidone (a
copolymer of polyvinyl
pyrrolidone and polyvinyl acetate), low molecular weight HPC (hydroxypropyl
cellulose), low
molecular weight HPMC (hydroxypropyl methylcellulose), low molecular weight
carboxy methyl
cellulose, ethylcellulose, gelatin, polyethylene oxide, acacia, dextrin,
magnesium aluminum
22
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
silicate, and starch and polymethacrylates such as Eudragit NE 30D, Eudragit
RL, Eudragit RS,
Eudragit E, polyvinyl acetate, enteric polymers, or mixtures thereof.
[0112] Exemplary disintegrants include, but are not limited to, low-
substituted
carboxymethyl cellulose sodium, crospovidone (cross-linked polyvinyl
pyrrolidone), sodium
carboxymethyl starch (sodium starch glycolate), cross-linked sodium
carboxymethyl cellulose
(Croscarmellose), pregelatinized starch (starch 1500), microcrystalline
cellulose, water insoluble
starch, calcium carboxymethyl cellulose, low substituted hydroxypropyl
cellulose, and
magnesium or aluminum silicate.
[0113] Exemplary glidants include but are not limited to, magnesium, silicon
dioxide, talc,
starch, titanium dioxide, and the like.
[0114] In yet another embodiment, the extended-release formulation is formed
by coating
a water soluble/dispersible drug-containing particle, such as a bead or bead
population therein (as
described above), with a coating material and, optionally, a pore former and
other excipients. The
coating material is preferably selected from a group comprising cellulosic
polymers such as
ethylcellulose (e.g., SURELEASEA methylcellulose, hydroxypropyl cellulose,
hydroxypropylmethyl cellulose, cellulose acetate, and cellulose acetate
phthalate; polyvinyl
alcohol; acrylic polymers such as polyacrylates, polymethacrylates, and
copolymers thereof; and
other water-based or solvent-based coating materials. The release-controlling
coating for a given
bead population may be controlled by at least one parameter of the release
controlling coating,
such as the nature of the coating, coating level, type and concentration of a
pore former, process
parameters, and combinations thereof. Thus, changing a parameter, such as a
pore former
concentration, or the conditions of the curing, allows for changes in the
release of active agent(s)
from any given bead population, thereby allowing for selective adjustment of
the formulation to a
pre-determined release profile.
[0115] Pore formers suitable for use in the release controlling coating herein
can be
organic or inorganic agents and include materials that can be dissolved,
extracted or leached from
the coating in the environment of use. Exemplary pore forming agents include,
but are not limited
to, organic compounds such as mono-, oligo-, and polysaccharides including
sucrose, glucose,
fructose, mannitol, mannose, galactose, sorbitol, pullulan, and dextran;
polymers soluble in the
environment of use such as water-soluble hydrophilic polymers,
hydroxyalkylcelluloses,
carboxyalkylcelluloses, hydroxypropylmethylcellulose, cellulose ethers,
acrylic resins,
polyvinylpyrrolidone, cross-linked polyvinylpyrrolidone, polyethylene oxide,
Carbowaxes,
Carbopol, and the like, diols, polyols, polyhydric alcohols, polyalkylene
glycols, polyethylene
glycols, polypropylene glycols, or block polymers thereof, polyglycols, and
poly(a-
S2)alkylenediols; and inorganic compounds such as alkali metal salts, lithium
carbonate, sodium
23
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
chloride, sodium bromide, potassium chloride, potassium sulfate, potassium
phosphate, sodium
acetate, sodium citrate, suitable calcium salts, combination thereof, and the
like.
[0116] The release controlling coating can further comprise other additives
known in the
art, such as plasticizers, anti-adherents, glidants (or flow aids) and
antifoams. In some
embodiments, the coated particles or beads may additionally include an
"overcoat," to provide,
for example, moisture protection, static charge reduction, taste-masking,
flavoring, coloring,
and/or polish or other cosmetic appeal to the beads. Suitable coating
materials for such an
overcoat are known in the art and include, but are not limited to, cellulosic
polymers such as
hydroxypropylmethylcellulose, hydroxypropylcellulose, and microcrystalline
cellulose or
combinations thereof (for example, various OPADRY coating materials).
[0117] The coated particles or beads may additionally contain enhancers that
may be
exemplified by, but not limited to, solubility enhancers, dissolution
enhancers, absorption
enhancers, permeability enhancers, stabilizers, complexing agents, enzyme
inhibitors, p-
glycoprotein inhibitors, and multidrug resistance protein inhibitors.
Alternatively, the formulation
can also contain enhancers that are separated from the coated particles, for
example, in a separate
population of beads or as a powder. In yet another embodiment, the enhancer(s)
may be
contained in a separate layer on coated particles either under or above the
release controlling
coating.
[0118] In other embodiments, the extended-release formulation is formulated to
release
the active agent(s) by an osmotic mechanism. By way of example, a capsule may
be formulated
with a single osmotic unit or it may incorporate 2, 3, 4, 5, or 6 push-pull
units encapsulated within
a hard gelatin capsule, whereby each bilayer push pull unit contains an
osmotic push layer and a
drug layer, both surrounded by a semi-permeable membrane. One or more orifices
are drilled
through the membrane next to the drug layer. This membrane may be additionally
covered with a
pH-dependent enteric coating to prevent release until after gastric emptying.
The gelatin capsule
dissolves immediately after ingestion. As the push pull unit(s) enters the
small intestine, the
enteric coating breaks down, which then allows fluid to flow through the semi-
permeable
membrane, swelling the osmotic push compartment to force drugs out through the
orifice(s) at a
rate precisely controlled by the rate of water transport through the semi-
permeable membrane.
Release of drugs can occur over a constant rate for up to 24 hours or more.
[0119] The osmotic push layer comprises one or more osmotic agents creating
the driving
force for transport of water through the semi-permeable membrane into the core
of the delivery
vehicle. One class of osmotic agents includes water-swellable hydrophilic
polymers, also referred
to as "osmopolymers" and "hydrogels," including, but not limited to,
hydrophilic vinyl and
acrylic polymers, polysaccharides such as calcium alginate, polyethylene oxide
(PEO),
polyethylene glycol (PEG), polypropylene glycol (PPG), poly(2-hydroxyethyl
methacrylate),
24
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
poly(acrylic) acid, poly(methacrylic) acid, polyvinylpyrrolidone (PVP),
crosslinked PVP,
polyvinyl alcohol (PVA), PVA/PVP copolymers, PVA/PVP copolymers with
hydrophobic
monomers such as methyl methacrylate and vinyl acetate, hydrophilic
polyurethanes containing
large PEO blocks, sodium croscarmellose, carrageenan, hydroxyethyl cellulose
(HEC),
hydroxypropyl cellulose (HPC), hydroxypropyl methyl cellulose (HPMC),
carboxymethyl
cellulose (CMC) and carboxyethyl, cellulose (CEC), sodium alginate,
polycarbophil, gelatin,
xanthan gum, and sodium starch glycolate.
[0120] Another class of osmotic agents includes osmogens, which are capable of
imbibing
water to effect an osmotic pressure gradient across the semi-permeable
membrane. Exemplary
osmogens include, but are not limited to, inorganic salts such as magnesium
sulfate, magnesium
chloride, calcium chloride, sodium chloride, lithium chloride, potassium
sulfate, potassium
phosphates, sodium carbonate, sodium sulfite, lithium sulfate, potassium
chloride, and sodium
sulfate; sugars such as dextrose, fructose, glucose, inositol, lactose,
maltose, mannitol, raffinose,
sorbitol, sucrose, trehalose, and xylitol; organic acids such as ascorbic
acid, benzoic acid, fumaric
acid, citric acid, maleic acid, sebacic acid, sorbic acid, adipic acid, edetic
acid, glutamic acid, p-
toluenesulfonic acid, succinic acid, and tartaric acid; urea; and mixtures
thereof
[0121] Materials useful in forming the semipermeable membrane include various
grades
of acrylics, vinyls, ethers, polyamides, polyesters, and cellulosic
derivatives that are water-
permeable and water-insoluble at physiologically relevant pHs, or are
susceptible to being
rendered water-insoluble by chemical alteration, such as crosslinking.
[0122] In some embodiments, the extended-release formulation comprises a
polysaccharide coating that is resistant to erosion in both the stomach and
intestine. Such
polymers can be only degraded in the colon, which contains a large microflora
containing
biodegradable enzymes breaking down, for example, the polysaccharide coatings
to release the
drug contents in a controlled, time-dependent manner. Exemplary polysaccharide
coatings may
include, for example, amylose, arabinogalactan, chitosan, chondroitin sulfate,
cyclodextrin,
dextran, guar gum, pectin, xylan, and combinations or derivatives therefrom.
[0123] In some embodiments, the pharmaceutical composition is formulated for
delayed-
release or delayed-extended-release. In some embodiments, the delayed extended-
release
formulation includes an extended-release formulation coated with an enteric
coating, which is a
barrier applied to oral medication that prevents release of medication before
it reaches the small
intestine. Delayed-release formulations, such as enteric coatings, prevent
drugs having an irritant
effect on the stomach, such as aspirin, from dissolving in the stomach. As
used herein, the term
"enteric coating" is a coating comprising of one or more polymers having a pH
dependent or pH-
independent release profile. An enteric coated pill will not dissolve in the
acidic juices of the
stomach (pH ¨3), but they will in the alkaline (pH 7-9) environment present in
the small intestine
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
or colon. An enteric polymer coating typically resists releases of the active
agents until sometime
after a gastric emptying lag period of about 3-4 hours after administration.
Accordingly, a
formulation that releases it component "after gastric emptying" refers to a
delayed formulation
that releases the active ingredient(s) after the formulation is emptied from
the stomach and enters
intestine.
[0124] Such coatings are also used to protect acid-unstable drugs from the
stomach's
acidic exposure, delivering them instead to a basic pH environment
(intestine's pH 5.5 and above)
where they do not degrade and give their desired action. The term "pulsatile-
release" is a type of
delayed-release, which is used herein with reference to a drug formulation
that provides rapid and
transient release of the drug within a short time period immediately after a
predetermined lag
period, thereby producing a "pulsed" plasma profile of the drug after drug
administration.
Formulations may be designed to provide a single pulsatile release or multiple
pulsatile releases at
predetermined time intervals following administration, or a pulsatile release
(e.g., 20-60% of the
active ingredient) followed with extended release over a period of time (e.g.,
a continuous release
of the remainder of the active ingredient). A delayed-release or pulsatile
release formulation
generally comprises one or more elements covered with a barrier coating, which
dissolves, erodes
or ruptures following a specified lag phase.
[0125] A barrier coating for delayed-release may consist of a variety of
different
materials, depending on the objective. In addition, a formulation may comprise
a plurality of
barrier coatings to facilitate release in a temporal manner. The coating may
be a sugar coating, a
film coating (e.g., based on hydroxypropyl methylcellulose, methylcellulose,
methyl
hydroxyethylcellulose, hydroxypropylcellulose, carboxymethylcellulose,
acrylate copolymers,
polyethylene glycols, and/or polyvinylpyrrolidone) or a coating based on
methacrylic acid
copolymer, cellulose acetate phthalate, hydroxypropyl methylcellulose
phthalate, hydroxypropyl
methylcellulose acetate succinate, polyvinyl acetate phthalate, shellac,
and/or ethylcellulose.
Furthermore, the formulation may additionally include a time delay material
such as glyceryl
monostearate or glyceryl distearate.
[0126] In some embodiments, the delayed-extended-release formulation includes
an
enteric coating comprising one or more polymers facilitating release of active
agents in proximal
or distal regions of the gastrointestinal tract. pH dependent enteric coatings
comprise one or more
pH-dependent or pH-sensitive polymers that maintain their structural integrity
at low pH, as in the
stomach, but dissolve in higher pH environments in more distal regions of the
gastrointestinal
tract, such as the small intestine, where the drug contents are released. For
purposes of the
present invention, "pH dependent" is defined as having characteristics (e.g.,
dissolution) which
vary according to environmental pH. Exemplary pH-dependent polymers have been
described
earlier, pH-dependent polymers typically exhibit a characteristic pH optimum
for dissolution. In
26
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
some embodiments, the pH-dependent polymer exhibits a pH optimum between about
5.0 and 5.5,
between about 5.5 and 6.0, between about 6.0 and 6.5, or between about 6.5 and
7Ø In other
embodiments, the pH-dependent polymer exhibits a pH optimum of >5.0, of >5.5,
of >6.0, of
>6.5, or of >7Ø
[0127] In certain embodiments, the coating methodology employs the blending of
one or
more pH-dependent and one or more pH-independent polymers. The blending of pH-
dependent
and pH-independent polymers can reduce the release rate of active ingredients
once the soluble
polymer has reached its optimum pH of solubilization.
[0128] In some embodiments, a "delayed-release" or "delayed-extended-release"
profile
can be obtained using a water insoluble capsule body containing one or more
active agents,
wherein the capsule body closed at one end with an insoluble, but permeable
and swellable
hydrogel plug. Upon contact with gastrointestinal fluid or dissolution medium,
the plug swells,
pushing itself out of the capsule and releasing the drugs after a pre-
determined lag time, which
can be controlled by, for example, the position and dimensions of the plug.
The capsule body
may be further coated with an outer pH-dependent enteric coating keeping the
capsule intact until
it reaches the small intestine. Suitable plug materials include, for example,
polymethacrylates,
erodible compressed polymers (e.g., HPMC, polyvinyl alcohol), congealed melted
polymer (e.g.,
glyceryl mono oleate), and enzymatically controlled erodible polymers (e.g.,
polysaccharides,
such as amylose, arabinogalactan, chitosan, chondroitin sulfate, cyclodextrin,
dextran, guar gum,
pectin and xylan).
[0129] In other embodiments, capsules or bilayered tablets may be formulated
to contain a
drug-containing core, covered by a swelling layer and an outer insoluble, but
semi-permeable
polymer coating or membrane. The lag time prior to rupture can be controlled
by the permeation
and mechanical properties of the polymer coating and the swelling behavior of
the swelling layer.
Typically, the swelling layer comprises one or more swelling agents, such as
swellable
hydrophilic polymers that swell and retain water in their structures.
[0130] Exemplary water swellable materials to be used in the delayed-release
coating
include, but are not limited to, polyethylene oxides (having e.g., an average
molecular weight
between 1,000,000 and 7,000,000, such as POLY0X(D); methylcellulose;
hydroxypropyl
cellulose; hydroxypropyl methylcellulose; polyalkylene oxides having a weight
average molecular
weight of 100,000 to 6,000,000, including, but not limited to, poly(methylene
oxide),
poly(butylene oxide), poly(hydroxy alkyl methacrylate) having a molecular
weight of 25,000 to
5,000,000, poly(vinyl)alcohol having a low acetal residue, which is cross-
linked with glyoxal,
formaldehyde, or glutaraldehyde, and having a degree of polymerization from
200 to 30,000;
mixtures of methyl cellulose, cross-linked agar, and carboxymethyl cellulose;
hydrogel forming
copolymers produced by forming a dispersion of a finely divided copolymer of
maleic anhydride
27
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
with styrene, ethylene, propylene, butylene, or isobutylene cross-linked with
from 0.001 to 0.5
moles of saturated cross-linking agent per mole of maleic anyhydride in the
copolymer;
CARBOPOL acidic carboxy polymers having a molecular weight of 450,000 to
4,000,000;
CYANAMER polyacrylamides; cross-linked water swellable indenemaleicanhydride
polymers;
GOODRITE polyacrylic acid having a molecular weight of 80,000 to 200,000;
starch graft
copolymers; AQUAKEEPS acrylate polymer polysaccharides composed of condensed
glucose
units such as diester cross-linked polyglucan; carbomers having a viscosity of
3,000 to 60,000
mPas as a 0.5%-1% w/v aqueous solution; cellulose ethers such as
hydroxypropylcellulose having
a viscosity of about 1000-7000 mPa s as a 1% w/v aqueous solution (25 C);
hydroxypropyl
methylcellulose having a viscosity of about 1000 or higher, preferably 2,500
or higher to a
maximum of 25,000 mPas as a 2% w/v aqueous solution; polyvinylpyrrolidone
having a viscosity
of about 300-700 mPas as a 10% w/v aqueous solution at 20 C; and combinations
thereof.
[0131] Alternatively, the release time of the drugs can be controlled by a
disintegration lag
time depending on the balance between the tolerability and thickness of a
water insoluble polymer
membrane (such as ethyl cellulose, EC) containing predefined micropores at the
bottom of the
body and the amount of a swellable excipient, such as low substituted
hydroxypropyl cellulose (L-
HPC) and sodium glycolate. After oral administration, GI fluids permeate
through the
micropores, causing swelling of the swellable excipients, which produces an
inner pressure
disengaging the capsular components, including a first capsule body containing
the swellable
materials, a second capsule body containing the drugs, and an outer cap
attached to the first
capsule body.
[0132] The delayed-release coating layer may further comprise anti-tackiness
agents, such
as talc and glyceryl monostearate. The delayed-release coating layer may
further comprise one or
more plasticizers including, but not limited to, triethyl citrate, acetyl
triethyl citrate, acetyltributyl
citrate, polyethylene glycol acetylated monoglycerides, glycerin, triacetin,
propylene glycol,
phthalate esters (e.g., diethyl phthalate, dibutyl phthalate), titanium
dioxide, ferric oxides, castor
oil, sorbitol, and dibutyl sebacate.
[0133] In another embodiment, the delayed-release formulation employs a water-
permeable but insoluble film coating to enclose the active ingredient and an
osmotic agent. As
water from the gut slowly diffuses through the film into the core, the core
swells until the film
bursts, thereby releasing the active ingredients. The film coating may be
adjusted to permit
various rates of water permeation or release time.
[0134] In another embodiment, the delayed release formulation employs a water-
impermeable tablet coating whereby water enters through a controlled aperture
in the coating until
the core bursts. When the tablet bursts, the drug contents are released
immediately or over a
28
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
longer period of time. These and other techniques may be modified to allow for
a pre-determined
lag period before release of drugs is initiated.
[0135] In another embodiment, the active agents are delivered in a formulation
to provide
both delayed-release and extended-release (delayed-extended-release). The term
"delayed-
extended-release" is used herein with reference to a drug formulation
providing pulsatile release
of active agents at a pre-determined time or lag period following
administration, which is then
followed by extended-release of the active agents thereafter.
[0136] In some embodiments, immediate-release, extended-release, delayed-
release, or
delayed-extended-release formulations comprises an active core comprised of
one or more inert
particles, each in the form of a bead, pellet, pill, granular particle,
microcapsule, microsphere,
microgranule, nanocapsule, or nanosphere coated on its surfaces with drugs in
the form of e.g., a
drug-containing film-forming composition using, for example, fluid bed
techniques or other
methodologies known to those of skill in the art. The inert particle can be of
various sizes, so
long as it is large enough to remain poorly dissolved. Alternatively, the
active core may be
prepared by granulating and milling and/or by extrusion and spheronization of
a polymer
composition containing the drug substance.
[0137] The amount of drug in the core will depend on the dose that is required
and
typically varies from about 1 to 100 weight %, about 5 to 100 weight %, about
10 to 100 weight
%, about 20 to 100 weight %, about 30 to 100 weight %, about 40 to 100 weight
%, about 50 to
100 weight %, about 60 to 100 weight %õ about 70 to 100 weight %, or about 80
to 100 weight
%. Generally, the polymeric coating on the active core will be from about 1 to
50% based on the
weight of the coated particle, depending on the lag time and type of release
profile required and/or
the polymers and coating solvents chosen. Those skilled in the art will be
able to select an
appropriate amount of drug for coating onto or incorporating into the core to
achieve the desired
dosage. In one embodiment, the inactive core may be a sugar sphere or a buffer
crystal or an
encapsulated buffer crystal such as calcium carbonate, sodium bicarbonate,
fumaric acid, tartaric
acid, etc. which alters the microenvironment of the drug to facilitate its
release.
[0138] In some embodiments, for example, delayed-release or delayed-extended-
release
compositions may formed by coating a water soluble/dispersible drug-containing
particle, such as
a bead, with a mixture of a water insoluble polymer and an enteric polymer,
wherein the water
insoluble polymer and the enteric polymer may be present at a weight ratio of
from 4:1 to 1:1, and
the total weight of the coatings is 10 to 60 weight % based on the total
weight of the coated beads.
The drug layered beads may optionally include an inner dissolution rate
controlling membrane of
ethylcellulose. The composition of the outer layer, as well as the individual
weights of the inner
and outer layers of the polymeric membrane are optimized for achieving desired
circadian rhythm
release profiles for a given active, which are predicted based on in vitro/in
vivo correlations.
29
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
[0139] In other embodiments the formulations may comprise a mixture of
immediate-
release drug-containing particles without a dissolution rate controlling
polymer membrane and
delayed-extended-release beads exhibiting, for example, a lag time of 2-4
hours following oral
administration, thus providing a two-pulse release profile.
[0140] In some embodiments, the active core is coated with one or more layers
of
dissolution rate-controlling polymers to obtain desired release profiles with
or without a lag time.
An inner layer membrane can largely control the rate of drug release following
imbibition of
water or body fluids into the core, while the outer layer membrane can provide
for a desired lag
time (the period of no or little drug release following imbibition of water or
body fluids into the
core). The inner layer membrane may comprise a water insoluble polymer, or a
mixture of water
insoluble and water soluble polymers.
[0141] The polymers suitable for the outer membrane, which largely controls
the lag time
of up to 6 hours may comprise an enteric polymer, as described above, and a
water insoluble
polymer at 10 to 50 weight %. The ratio of water insoluble polymer to enteric
polymer may vary
from 4:1 to 1:2, preferably the polymers are present at a ratio of about 1:1.
The water insoluble
polymer typically used is ethylcellulose.
[0142] Exemplary water insoluble polymers include ethylcellulose, polyvinyl
acetate
(Kollicoat SR#OD from BASF), neutral copolymers based on ethyl acrylate and
methylmethacrylate, copolymers of acrylic and methacrylic acid esters with
quaternary
ammonium groups such as EUDRAGIT NE, RS and RS30D, RL or RL30D, and the like.
Exemplary water soluble polymers include low molecular weight HPMC, HPC,
methylcellulose,
polyethylene glycol (PEG of molecular weight>3000) at a thickness ranging from
1 weight % up
to 10 weight % depending on the solubility of the active in water and the
solvent or latex
suspension based coating formulation used. The water insoluble polymer to
water soluble
polymer may typically vary from 95:5 to 60:40, preferably from 80:20 to 65:35.
In some
embodiments, AMBERLITETm IRP69 resin is used as an extended-release carrier.
AMBERLITETm IRP69 is an insoluble, strongly acidic, sodium form cation
exchange resin that is
suitable as carrier for cationic (basic) substances. In other embodiments,
DUOLITETm
AP143/1093 resin is used as an extended-release carrier. DUOLITETm AP143/1093
is an
insoluble, strongly basic, anion exchange resin that is suitable as a carrier
for anionic (acidic)
substances. When used as a drug carrier, AMBERLITETm IRP69 or/and DUOLITETm
AP143/1093 resin provides a means for binding medicinal agents onto an
insoluble polymeric
matrix. Extended-release is achieved through the formation of resin-drug
complexes (drug
resinates). The drug is released from the resin in vivo as the drug reaches
equilibrium with the
high electrolyte concentrations, which are typical of the gastrointestinal
tract. More hydrophobic
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
drugs will usually elute from the resin at a lower rate, owing to hydrophobic
interactions with the
aromatic structure of the cation exchange system.
[0143] In some embodiments, the pharmaceutical composition is formulated for
oral
administration. Oral dosage forms include, for example, tablets, capsules, and
caplets and may
also comprise a plurality of granules, beads, powders, or pellets that may or
may not be
encapsulated. Tablets and capsules represent the most convenient oral dosage
forms, in which
case solid pharmaceutical carriers are employed.
[0144] In a delayed-release formulation, one or more barrier coatings may be
applied to
pellets, tablets, or capsules to facilitate slow dissolution and concomitant
release of drugs into the
intestine. Typically, the barrier coating contains one or more polymers
encasing, surrounding, or
forming a layer, or membrane around the therapeutic composition or active
core. In some
embodiments, the active agents are delivered in a formulation to provide
delayed-release at a pre-
determined time following administration. The delay may be up to about 10
minutes, about 20
minutes, about 30 minutes, about 1 hour, about 2 hours, about 3 hours, about 4
hours, about 5
hours, about 6 hours, or longer.
[0145] Various coating techniques may be applied to granules, beads, powders
or pellets,
tablets, capsules or combinations thereof containing active agents to produce
different and distinct
release profiles. In some embodiments, the pharmaceutical composition is in a
tablet or capsule
form containing a single coating layer. In other embodiments, the
pharmaceutical composition is
in a tablet or capsule form containing multiple coating layers. In some
embodiments, the
pharmaceutical composition of the present application is formulated for
extended-release or
delayed extended-release of up to 100% of the active ingredient.
[0146] In other embodiments, the pharmaceutical composition of the present
application is
formulated for a two-phase extended-release or delayed two-phase extended-
release characterized
by an "immediate-release" component that is released within two hours of
administration and an
"extended-release" component which is released over a period of 2-12 hours. In
some
embodiments, the "immediate-release" component provides about 1-90% of the
total dosage of
the active agent(s) and the "extended-release" component provides 10-99% of
the total dosage of
the active agent(s) to be delivered by the pharmaceutical formulation. For
example, the
immediate-release component may provide about 10-90%, or about 10%, 20%, 25%,
30%, 35%,
40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85% or 90% of the total dosage of
the active
agent(s) to be delivered by the pharmaceutical formulation. The extended-
release component
provides about 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%,
70%, 75%,
80%, 85%, 90%, 95% or 99% of the total dosage of the active agent(s) to be
delivered by the
formulation. In some embodiments, the immediate-release component and the
extended-release
component contain the same active ingredient. In other embodiments, the
immediate-release
31
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
component and the extended-release component contain different active
ingredients (e.g., one PG
pathway inhibitor in one component and another PG pathway inhibitor in another
component). In
some embodiments, the immediate-release component and the extended-release
component each
contains a PG pathway inhibitor and an analgesic selected from the group
consisting of aspirin,
ibuprofen, naproxen sodium, indomethacin, nabumetone, and acetaminophen. In
other
embodiments, the immediate-release component and/or the extended-release
component further
comprises one or more additional active agents selected from the groups
consisting of an
antimuscarinic agent, an antidiuretic, a spasmolytic, an inhibitor of
phosphodiesterase type (PDE
inhibitor) and zolpidem.
[0147] In some embodiments, the pharmaceutical composition comprises a
plurality of
active ingredients selected from the group consisting of PG pathway
inhibitors, analgesics,
antimuscarinic agents, antidiuretics, spasmolytics, PDE 5 inhibitors and
zolpidem. Examples of
antimuscarinic agents include, but are not limited to, oxybutynin,
solifenacin, darifenacin,
fesoterodine, tolterodine, trospium, atropine, and tricyclic antidepressants.
Examples of
antidiuretics include, but are not limited to, antidiuretic hormone (ADH),
angiotensin II,
aldosterone, vasopressin, vasopressin analogs (e.g., desmopressin argipressin,
lypressin,
felypressin, ornipressin, terlipressin); vasopressin receptor agonists, atrial
natriuretic peptide
(ANP) and C-type natriuretic peptide (CNP) receptor (i.e., NPR1, NPR2, and
NPR3) antagonists
(e.g., HS-142-1, isatin, [Asu7,23']b-ANP-(7-28)], anantin, a cyclic peptide
from Streptomyces
coerulescens, and 3G12 monoclonal antibody); somatostatin type 2 receptor
antagonists (e.g.,
somatostatin), pharmaceutically-acceptable derivatives, and analogs, salts,
hydrates, and solvates
thereof. Examples of spasmolytics include, but are not limited to,
carisoprodol, benzodiazepines,
baclofen, cyclobenzaprine, metaxalone, methocarbamol, clonidine, clonidine
analog, and
dantrolene. Examples of PDE 5 inhibitors include, but are not limited to,
tadalafil, sildenafil and
vardenafil.
[0148] In some embodiments, the pharmaceutical composition comprises a
plurality of
active ingredients comprising (1) one or more PG pathway inhibitors and (2)
one or more other
active ingredients selected from the group consisting of analgesics,
antimuscarinic agents,
antidiuretics, spasmolytics, PDE 5 inhibitors and zolpidem. In some
embodiments, the plurality
of active ingredients are formulated for immediate-release. In other
embodiments, the plurality of
active ingredients are formulated for extended-release. In other embodiments,
the plurality of
active ingredients are formulated for delayed-release. In other embodiments,
the plurality of
active ingredients are formulated for both immediate-release and extended-
release (e.g., a first
portion of each active ingredient is formulated for immediate-release and a
second portion of each
active ingredient is formulated for extended-release). In yet other
embodiments, some of the
plurality of active ingredients are formulated for immediate-release and some
of the plurality of
32
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
active ingredients are formulated for extended-release (e.g., active
ingredients A, B, C are
formulated for immediate-release and active ingredients C and D are formulated
for extended-
release). In some other embodiments, the plurality of active ingredients are
formulated for
delayed-extended-release.
[0149] In certain embodiments, the pharmaceutical composition comprises an
immediate-
release component and an extended-release component. The immediate-release
component may
comprise one or more active ingredients selected from the group consisting of
PG pathway
inhibitors, analgesics, antimuscarinic agents, antidiuretics, spasmolytics,
PDE 5 inhibitors and
zolpidem. The extended-release component may comprise one or more active
ingredients
selected from the group consisting of PG pathway inhibitors, analgesics,
antimuscarinic agents,
antidiuretics, spasmolytics, PDE 5 inhibitors and zolpidem. In some
embodiments, the
immediate-release component and the extended-release component have exactly
the same active
ingredients. In other embodiments, the immediate-release component and the
extended-release
component have different active ingredients. In yet other embodiments, the
immediate-release
component and the extended-release component have one or more common active
ingredients. In
some other embodiments, the immediate-release component and/or the extended-
release
component is further coated with a delayed-release coating, such as an enteric
coating. In other
embodiments, the pharmaceutical composition comprises two or more active
ingredients
formulated as two extended-release components, each providing a different
extended-release
profile. For example, a first extended-release component releases a first
active ingredient at a first
release rate and a second extended-release component releases a second active
ingredient at a
second release rate.
[0150] In some embodiments, the pharmaceutical composition comprises an
immediate-
release component and a delayed-release component. In other embodiments, the
pharmaceutical
composition comprises two or more active ingredients formulated as two delayed-
release
components, each providing a different delayed-release profile. For example, a
first delayed-
release component releases a first active ingredient at a first time point,
and a second delayed-
release component releases a second active ingredient at a second time point.
[0151] The components in a combined release profile formulation (e.g.,
formulations with
a combination of an immediate-release component and an extended-release
component, a
combination of an immediate-release component and a delayed-release component,
a combination
of an immediate-release component, a delayed-release component, and an
extended-release
component, a combination of two or more delayed-release components, or a
combination of two
or more extended-release components) may contain the same active ingredient(s)
or different
active ingredient(s). In some embodiments, the immediate-release component may
provide about
1% to about 80% of the total dosage of the active agent(s) to be delivered by
the pharmaceutical
33
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
formulation. In some embodiments, the combined release profile formulation
contains an
immediate-release component and the immediate-release component provide about
100 to about
90%, about 10% to about 90%, about 20 A to about 90%, about 30 A to about 90%,
about 40 A to
about 90%, about 50 A to about 90%, about 60 A to about 90%, about 70 A to
about 90% or about
80 A to about 90% of the total dosage of each active ingredient to be
delivered by the formulation.
In alternate embodiments, the immediate-release component provides up to about
10%, 20%,
30%, 40%, 500o, 60%, 70%, 80% or 90% of the total dosage of each ingredient to
be delivered by
the formulation.
[0152] In some embodiments, the pharmaceutical formulation comprises an active
core
comprised of one or more inert particles, each in the form of a bead, pellet,
pill, granular particle,
microcapsule, microsphere, microgranule, nanocapsule, or nanosphere coated on
its surfaces with
drugs in the form of e.g., a drug-containing film-forming composition using,
for example, fluid
bed techniques or other methodologies known to those of skill in the art. The
inert particle can be
of various sizes, so long as it is large enough to remain poorly dissolved.
Alternatively, the active
core may be prepared by granulating and milling and/or by extrusion and
spheronization of a
polymer composition containing the drug substance.
[0153] The amount of drug in the core will depend on the dose that is required
and
typically varies from about 5 to 90 weight %. Generally, the polymeric coating
on the active core
will be from about 1 to 50% based on the weight of the coated particle,
depending on the lag time
and type of release profile required and/or the polymers and coating solvents
chosen. Those
skilled in the art will be able to select an appropriate amount of drug for
coating onto or
incorporating into the core to achieve the desired dosage. In one embodiment,
the inactive core
may be a sugar sphere or a buffer crystal or an encapsulated buffer crystal
such as calcium
carbonate, sodium bicarbonate, fumaric acid, tartaric acid, which alters the
microenvironment of
the drug to facilitate its release.
[0154] In some embodiments, the pharmaceutical composition comprises a delayed-
release component formed by coating a water soluble/dispersible drug-
containing particle, such as
a bead, with a mixture of a water insoluble polymer and an enteric polymer,
wherein the water
insoluble polymer and the enteric polymer may be present at a weight ratio of
4:1 to 1:1, and the
total weight of the coatings is 10 to 60 weight % based on the total weight of
the coated beads.
The drug layered beads may optionally include an inner dissolution rate
controlling membrane of
ethylcellulose. The composition of the outer layer, as well as the individual
weights of the inner
and outer layers of the polymeric membrane are optimized for achieving desired
circadian rhythm
release profiles for a given active, which are predicted based on in vitro in
vivo correlations.
[0155] In other embodiments, the formulations comprise a mixture of immediate-
release
drug-containing particles without a dissolution rate controlling polymer
membrane and delayed
34
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
release beads exhibiting, for example, a lag time of 2-4 hours following oral
administration, thus
providing a two-pulse release profile. In yet other embodiments, the
formulations comprise a
mixture of two types of delayed-release beads: a first type that exhibits a
lag time of 1-3 hours and
a second type that exhibits a lag time of 4-6 hours. In yet other embodiments,
the formulations
comprise a mixture of two types of release beads: a first type that exhibits
immediate-release and
a second type that exhibits a lag time of 1-4 hours followed with extended-
release.
[0156] In some embodiments, the formulations are designed with a release
profile such
that a fraction of the active ingredient(s) (e.g., 10-80%) is released
immediately or within two
hours of administration, and the rest is released over an extended period of
time (e.g., over a
period of 2-24 hours). In other embodiments, the formulations are designed
with a release profile
such that one active ingredient (e.g., an analgesic) are released immediately
or within two hours
of administration, and one or more other active ingredients (e.g., a PG
pathway inhibitor) are
released over an extended period of time (e.g., over a period of 2-24 hours).
[0157] The pharmaceutical composition may be administered daily or
administered on an
as needed basis. The pharmaceutical composition may be administered orally,
intravenously, or
intramuscularly. In preferred embodiments, the pharmaceutical composition is
administered
orally. In other embodiments, the pharmaceutical composition is administered
by retrograde
perfusion through the urinary tract. In other embodiments, the pharmaceutical
composition is
administered by direct injection into bladder muscle.
[0158] In some embodiments, the pharmaceutical composition is administered
daily, twice
a day or three times a day. In other embodiments, the pharmaceutical
composition is
administered every other day, every 3 days, every 4 days, every 4 days, every
5 days, every 6
days, every week, every 2 weeks, every 3 weeks, every month, every 2 months or
every 3 months.
[0159] In some embodiments, the pharmaceutical composition is administered at
bedtime.
In some embodiments, the pharmaceutical composition is administered within
about two hours
before bedtime, preferably within about one hour before bedtime. In another
embodiment, the
pharmaceutical composition is administered about 2-4 hours before bedtime. In
a further
embodiment, the pharmaceutical composition is administered at least 4 hours
before bedtime.
[0160] The appropriate dosage ("therapeutically effective amount") of the
active
ingredient(s) in the immediate-release component, the extended-release
component, the delayed-
release component or delayed-extended-release component will depend, for
example, on the
severity and course of the condition, the mode of administration, the
bioavailability of the
particular ingredient(s), the age and weight of the patient, the patient's
clinical history and
response to the active agent(s), discretion of the physician, etc.
[0161] As a general proposition, the therapeutically effective amount of the
PG pathway
inhibitor(s) in the immediate-release component, the delayed-release
component, the extended-
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
release component or the delayed-extended-release component is administered in
the range of
about 1 ng/kg body weight/day to about 100 mg/kg body weight/day whether by
one or more
administrations. In some embodiments, the range of each active agent
administered daily in a
single dose or in multiple does is from about 1 ng/kg body weight/day to about
100 mg/kg body
weight/day, 1 ng/kg body weight/day to about 30 mg/kg body weight/day, 1 ng/kg
body
weight/day to about 10 mg/kg body weight/day, 1 ng/kg body weight/day to about
3 mg/kg body
weight/day, 1 ng/kg body weight/day to about 1 mg/kg body weight/day, 1 ng/kg
body
weight/day to about 300 ng/kg body weight/day, 1 ng/kg body weight/day to
about 100 ng/kg
body weight/day, 1 ng/kg body weight/day to about 30 ng/kg body weight/day, 1
ng/kg body
weight/day to about 10 ng/kg body weight/day, 1 ng/kg body weight/day to about
3 ng/kg body
weight/day, 10 ng/kg body weight/day to about 100 mg/kg body weight/day, 10
ng/kg body
weight/day to about 30 mg/kg body weight/day, 10 ng/kg body weight/day to
about 10 mg/kg
body weight/day, 10 ng/kg body weight/day to about 3 mg/kg body weight/day, 10
ng/kg body
weight/day to about 1 mg/kg body weight/day, 10 ng/kg body weight/day to about
300 ng/kg
body weight/day, 10 ng/kg body weight/day to about 100 ng/kg body weight/day,
10 ng/kg body
weight/day to about 30 ng/kg body weight/day, 30 ng/kg body weight/day to
about 100 mg/kg
body weight/day, 30 ng/kg body weight/day to about 30 mg/kg body weight/day,
30 ng/kg body
weight/day to about 10 mg/kg body weight/day, 30 ng/kg body weight/day to
about 3 mg/kg body
weight/day, 30 ng/kg body weight/day to about 1 mg/kg body weight/day, 30
ng/kg body
weight/day to about 300 ng/kg body weight/day, 30 ng/kg body weight/day to
about 100 ng/kg
body weight/day, 100 ng/kg body weight/day to about 100 mg/kg body weight/day,
100 ng/kg
body weight/day to about 30 mg/kg body weight/day, 100 ng/kg body weight/day
to about 10
mg/kg body weight/day, 100 ng/kg body weight/day to about 3 mg/kg body
weight/day, 100
ng/kg body weight/day to about 1 mg/kg body weight/day, 100 ng/kg body
weight/day to about
300 ng/kg body weight/day, 300 ng/kg body weight/day to about 100 mg/kg body
weight/day,
300 ng/kg body weight/day to about 30 mg/kg body weight/day, 300 ng/kg body
weight/day to
about 10 mg/kg body weight/day, 300 ng/kg body weight/day to about 3 mg/kg
body weight/day,
300 ng/kg body weight/day to about 1 mg/kg body weight/day, 1 mg/kg body
weight/day to about
100 mg/kg body weight/day, 1 mg/kg body weight/day to about 30 mg/kg body
weight/day, 1
mg/kg body weight/day to about 10 mg/kg body weight/day, 1 mg/kg body
weight/day to about 3
mg/kg body weight/day, 3 mg/kg body weight/day to about 100 mg/kg body
weight/day, 3 mg/kg
body weight/day to about 30 mg/kg body weight/day, 3 mg/kg body weight/day to
about 10
mg/kg body weight/day, 10 mg/kg body weight/day to about 100 mg/kg body
weight/day, 10
mg/kg body weight/day to about 30 mg/kg body weight/day or 30 mg/kg body
weight/day to
about 100 mg/kg body weight/day.
36
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
[0162] As a general proposition, the therapeutically effective amount of the
analgesic
agent(s) in the immediate-release component, the delayed-release component,
the extended-
release component or the delayed-extended-release component is administered in
the range of
about 10 [tg/kg body weight/day to about 100 mg/kg body weight/day whether by
one or more
administrations. In some embodiments, the range of each active agent
administered daily in a
single dose or in multiple does is from about 10 [tg/kg body weight/day to
about 100 mg/kg body
weight/day, 10 [tg/kg body weight/day to about 30 mg/kg body weight/day, 10
[tg/kg body
weight/day to about 10 mg/kg body weight/day, 10 [tg/kg body weight/day to
about 3 mg/kg body
weight/day, 10 [tg/kg body weight/day to about 1 mg/kg body weight/day, 10
[tg/kg body
weight/day to about 300 [tg/kg body weight/day, 10 [tg/kg body weight/day to
about 100 [tg/kg
body weight/day, 10 [tg/kg body weight/day to about 30 [tg/kg body weight/day,
30 [tg/kg body
weight/day to about 100 mg/kg body weight/day, 30 [tg/kg body weight/day to
about 30 mg/kg
body weight/day, 30 [tg/kg body weight/day to about 10 mg/kg body weight/day,
30 [tg/kg body
weight/day to about 3 mg/kg body weight/day, 30 [tg/kg body weight/day to
about 1 mg/kg body
weight/day, 30 [tg/kg body weight/day to about 300 [tg/kg body weight/day, 30
[tg/kg body
weight/day to about 100 [tg/kg body weight/day, 100 [tg/kg body weight/day to
about 100 mg/kg
body weight/day, 100 [tg/kg body weight/day to about 30 mg/kg body weight/day,
100 [tg/kg
body weight/day to about 10 mg/kg body weight/day, 100 [tg/kg body weight/day
to about 3
mg/kg body weight/day, 100 [tg/kg body weight/day to about 1 mg/kg body
weight/day, 100
[tg/kg body weight/day to about 300 [tg/kg body weight/day, 300 [tg/kg body
weight/day to about
100 mg/kg body weight/day, 300 [tg/kg body weight/day to about 30 mg/kg body
weight/day,
300 [tg/kg body weight/day to about 10 mg/kg body weight/day, 300 [tg/kg body
weight/day to
about 3 mg/kg body weight/day, 300 [tg/kg body weight/day to about 1 mg/kg
body weight/day, 1
mg/kg body weight/day to about 100 mg/kg body weight/day, 1 mg/kg body
weight/day to about
30 mg/kg body weight/day, 1 mg/kg body weight/day to about 10 mg/kg body
weight/day, 1
mg/kg body weight/day to about 3 mg/kg body weight/day, 3 mg/kg body
weight/day to about
100 mg/kg body weight/day, 3 mg/kg body weight/day to about 30 mg/kg body
weight/day, 3
mg/kg body weight/day to about 10 mg/kg body weight/day, 10 mg/kg body
weight/day to about
100 mg/kg body weight/day, 10 mg/kg body weight/day to about 30 mg/kg body
weight/day or
30 mg/kg body weight/day to about 100 mg/kg body weight/day.
[0163] The analgesic agent(s) described herein may be included in an immediate-
release
component or an extended-release component, a delayed-release component, a
delayed-extended-
release component or combinations thereof for daily oral administration at a
single dose or
combined dose range of 1 mg to 2000 mg, 1 mg to 1000 mg, 1 mg to 300 mg, 1 mg
to 100 mg, 1
mg to 30 mg, 1 mg to 10 mg, 1 mg to 3 mg, 3 mg to 2000 mg, 3 mg to 1000 mg, 3
mg to 300 mg,
3 mg to 100 mg, 3 mg to 30 mg, 3 mg to 10 mg, 10 mg to 2000 mg, 10 mg to 1000
mg, 10 mg to
37
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
300 mg, 10 mg to 100 mg, 10 mg to 30 mg, 30 mg to 2000 mg, 30 mg to 1000 mg,
30 mg to 300
mg, 30 mg to 100 mg, 100 mg to 2000 mg, 100 mg to 1000 mg, 100 mg to 300 mg,
300 mg to
2000 mg, 300 mg to 1000 mg or 1000 mg to 2000 mg. As expected, the dosage will
be dependent
on the condition, size, age, and condition of the patient.
[0164] In some embodiments, the pharmaceutical composition comprises a single
analgesic agent. In one embodiment, the single analgesic agent is aspirin. In
another
embodiment, the single analgesic agent is ibuprofen. In another embodiment,
the single analgesic
agent is naproxen or naproxen sodium. In another embodiment, the single
analgesic agent is
indomethacin. In another embodiment, the single analgesic agent is nabumetone.
In another
embodiment, the single analgesic agent is acetaminophen.
[0165] In other embodiments, the pharmaceutical composition comprises a pair
of
analgesic agents. Examples of such paired analgesic agents include, but are
not limited to,
acetylsalicylic acid and ibuprofen, acetylsalicylic acid and naproxen sodium,
acetylsalicylic acid
and nabumetone, acetylsalicylic acid and acetaminophen, acetylsalicylic acid
and indomethancin,
ibuprofen and naproxen sodium, ibuprofen and nabumetone, ibuprofen and
acetaminophen,
ibuprofen and indomethancin, naproxen, naproxen sodium and nabumetone,
naproxen sodium and
acetaminophen, naproxen sodium and indomethancin, nabumetone and
acetaminophen,
nabumetone and indomethancin, and acetaminophen and indomethancin. The paired
analgesic
agents are mixed at a weight ratio in the range of 0.1:1 to 10:1, 0.2:1 to 5:1
or 0.3:1 to 3:1. In one
embodiment, the paired analgesic agents are mixed at a weight ratio of 1:1.
[0166] In some other embodiments, the pharmaceutical composition of the
present
application further comprises one or more antimuscarinic agents. Examples of
the antimuscarinic
agents include, but are not limited to, oxybutynin, solifenacin, darifenacin,
fesoterodine,
tolterodine, trospium, atropine, and tricyclic antidepressants. The daily dose
of antimuscarinic
agent is in the range of 1 [tg to 300 mg, 1 [tg to 100 mg, 1 [tg to 30 mg; 1
[tg to 10 mg, 1 [tg to 3
mg, 1 [tg to 1 mg, 1 [tg to 300 [tg, 1 [tg to 100 [tg, 1 [tg to 30 [tg, 1 [tg
to 10 [tg, 1 [tg to 3 [tg, 3 [tg
to 100 mg, 3 [tg to 100 mg, 3 [tg to 30 mg; 3 [tg to 10 mg, 3 [tg to 3 mg, 3
[tg to 1 mg, 3 [tg to 300
[tg, 3 [tg to 100 [tg, 3 [tg to 30 [tg, 3 [tg to 10 [tg, 10 [tg to 300 mg, 10
[tg to 100 mg, 10 [tg to 30
mg; 10 [tg to 10 mg, 10 [tg to 3 mg, 10 [tg to 1 mg, 10 [tg to 300 [tg, 10 [tg
to 100 [tg, 10 [tg to 30
[tg, 30 [tg to 300 mg, 30 [tg to 100 mg, 30 [tg to 30 mg; 30 [tg to 10 mg, 30
[tg to 3 mg, 30 [tg to 1
mg, 30 [tg to 300 [tg, 30 [tg to 100 [tg, 100 [tg to 300 mg, 100 [tg to 100
mg, 100 [tg to 30 mg;
100 [tg to 10 mg, 100 [tg to 3 mg, 100 [tg to 1 mg, 100 [tg to 300 [tg, 300
[tg to 300 mg, 300 [tg to
100 mg, 300 [tg to 30 mg; 300 [tg to 10 mg, 300 [tg to 3 mg, 300 [tg to 1 mg,
1 mg to 300 mg, 1
mg to 100 mg, 1 mg to 30 mg, 1 mg to 3 mg, 3 mg to 300 mg, 3 mg to 100 mg, 3
mg to 30 mg, 3
mg to 10 mg, 10 mg to 300 mg, 10 mg to 100 mg, 10 mg to 30 mg, 30 mg to 300
mg, 30 mg to
100 mg or 100 mg to 300 mg.
38
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
[0167] In some other embodiments, the pharmaceutical composition of the
present
application further comprises one or more antidiuretics. Examples of the
antidiuretics include, but
are not limited to, antidiuretic hormone (ADH), angiotensin II, aldosterone,
vasopressin,
vasopressin analogs (e.g., desmopressin argipressin, lypressin, felypressin,
ornipressin, and
terlipressin), vasopressin receptor agonists, atrial natriuretic peptide (ANP)
and C-type natriuretic
peptide (CNP) receptor (i.e., NPR1, NPR2, and NPR3) antagonists (e.g., HS-142-
1, isatin,
[Asu7,23']b-ANP-(7-28)], anantin, a cyclic peptide from Streptomyces
coerulescens, and 3G12
monoclonal antibody), somatostatin type 2 receptor antagonists (e.g.,
somatostatin),
pharmaceutically-acceptable derivatives, and analogs, salts, hydrates, and
solvates thereof In
some embodiments, the one or more antidiuretics comprise desmopressin. In
other embodiments,
the one or more antidiuretics is desmopressin. The daily dose of antidiuretic
is in the range of 1
[tg to 300 mg, 1 [tg to 100 mg, 1 [tg to 30 mg; 1 [tg to 10 mg, 1 [tg to 3 mg,
1 [tg to 1 mg, 1 [tg to
300 [tg, 1 [tg to 100 [tg, 1 [tg to 30 [tg, 1 [tg to 10 [tg, 1 [tg to 3 [tg, 3
[tg to 100 mg, 3 [tg to 100
mg, 3 [tg to 30 mg; 3 [tg to 10 mg, 3 [tg to 3 mg, 3 [tg to 1 mg, 3 [tg to 300
[tg, 3 [tg to 100 [tg, 3
[tg to 30 [tg, 3 [tg to 10 [tg, 10 [tg to 300 mg, 10 [tg to 100 mg, 10 [tg to
30 mg; 10 [tg to 10 mg,
[tg to 3 mg, 10 [tg to 1 mg, 10 [tg to 300 [tg, 10 [tg to 100 [tg, 10 [tg to
30 [tg, 30 [tg to 300
mg, 30 [tg to 100 mg, 30 [tg to 30 mg; 30 [tg to 10 mg, 30 [tg to 3 mg, 30 [tg
to 1 mg, 30 [tg to
300 [tg, 30 [tg to 100 [tg, 100 [tg to 300 mg, 100 [tg to 100 mg, 100 [tg to
30 mg; 100 [tg to 10
mg, 100 [tg to 3 mg, 100 [tg to 1 mg, 100 [tg to 300 [tg, 300 [tg to 300 mg,
300 [tg to 100 mg, 300
[tg to 30 mg; 300 [tg to 10 mg, 300 [tg to 3 mg, 300 [tg to 1 mg, 1 mg to 300
mg, 1 mg to 100 mg,
1 mg to 30 mg, 1 mg to 3 mg, 3 mg to 300 mg, 3 mg to 100 mg, 3 mg to 30 mg, 3
mg to 10 mg,
10 mg to 300 mg, 10 mg to 100 mg, 10 mg to 30 mg, 30 mg to 300 mg, 30 mg to
100 mg or 100
mg to 300 mg.
[0168] In other embodiments, the pharmaceutical composition of the present
application
further comprises one or more spasmolytics. Examples of spasmolytics include,
but are not
limited to, carisoprodol, benzodiazepines, baclofen, cyclobenzaprine,
metaxalone,
methocarbamol, clonidine, clonidine analog, and dantrolene. In some
embodiments, the
spasmolytics is used at a daily dose of 0.1 mg to 1000 mg, 0.1 mg to 300 mg,
0.1 mg to 100 mg,
0.1 mg to 30 mg, 0.1 mg to 10 mg, 0.1 mg to 3 mg, 0.1 mg to 1 mg, 0.1 mg to
0.3 mg, 0.3 mg to
1000 mg, 0.3 mg to 300 mg, 0.3 mg to 100 mg, 0.3 mg to 30 mg, 0.3 mg to 10 mg,
0.3 mg to 3
mg, 0.3 mg to 1 mg, 1 mg to 1000 mg, 1 mg to 300 mg, 1 mg to 100 mg, 1 mg to
30 mg, 1 mg to
10 mg, 1 mg to 3 mg, 3 mg to 1000 mg, 3 mg to 300 mg, 3 mg to 100 mg, 3 mg to
30 mg, 3 mg to
10 mg, 10 mg to 1000 mg, 10 mg to 300 mg, 10 mg to 100 mg, 10 mg to 30 mg, 30
mg to 1000
mg, 30 mg to 300 mg, 30 mg to 100 mg, 100 mg to 1000 mg, 100 mg to 300 mg, or
300 mg to
1000 mg.
39
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
[0169] In other embodiments, the pharmaceutical composition of the present
application
further comprises one or more PDE 5 inhibitors. Examples of PDE 5 inhibitors
include, but are
not limited to, tadalafil, sildenafil and vardenafil. In some embodiments, the
one or more PDE 5
inhibitors comprise tadalafil. In other embodiments, the one or more PDE 5
inhibitors is tadalafil.
In some embodiments, the PDE 5 inhibitor is used at a daily dose of 0.1 mg to
1000 mg, 0.1 mg to
300 mg, 0.1 mg to 100 mg, 0.1 mg to 30 mg, 0.1 mg to 10 mg, 0.1 mg to 3 mg,
0.1 mg to 1 mg,
0.1 mg to 0.3 mg, 0.3 mg to 1000 mg, 0.3 mg to 300 mg, 0.3 mg to 100 mg, 0.3
mg to 30 mg, 0.3
mg to 10 mg, 0.3 mg to 3 mg, 0.3 mg to 1 mg, 1 mg to 1000 mg, 1 mg to 300 mg,
1 mg to 100
mg, 1 mg to 30 mg, 1 mg to 10 mg, 1 mg to 3 mg, 3 mg to 1000 mg, 3 mg to 300
mg, 3 mg to 100
mg, 3 mg to 30 mg, 3 mg to 10 mg, 10 mg to 1000 mg, 10 mg to 300 mg, 10 mg to
100 mg, 10
mg to 30 mg, 30 mg to 1000 mg, 30 mg to 300 mg, 30 mg to 100 mg, 100 mg to
1000 mg, 100
mg to 300 mg, or 300 mg to 1000 mg.
[0170] In some other embodiments, the pharmaceutical composition of the
present
application further comprises zolpidem. The daily dose of zolpidem is in the
range of 100 [tg to
100 mg, 100 [tg to 30 mg, 100 [tg to 10 mg, 100 [tg to 3 mg, 100 [tg to 1 mg,
100 [tg to 300 [tg,
300 [tg to 100 mg, 300 [tg to 30 mg, 300 [tg to 10 mg, 300 [tg to 3 mg, 300
[tg to 1 mg, 1 mg to
100 mg, 1 mg to 30 mg, 1 mg to 10 mg, 1 mg to 3 mg, 10 mg to 100 mg, 10 mg to
30 mg, or 30
mg to 100 mg.
[0171] The antimuscarinic agents, antidiuretics, spasmolytics, zolpidem and/or
PDE 5
inhibitors may be formulated, alone or together with other active
ingredient(s) in the
pharmaceutical composition, for immediate-release, extended-release, delayed
release, delayed-
extended-release or combinations thereof
[0172] In certain embodiments, the pharmaceutical composition is formulated
for
extended release and comprises (1) an analgesic agent selected from the group
consisting of
cetylsalicylic acid, ibuprofen, naproxen, naproxen sodium, nabumetone,
acetaminophen, and
indomethancin and (2) a PDE 5 inhibitor, such as tadalafil.
[0173] The pharmaceutical composition may be formulated into a tablet, an
orally
disintegrating tablet, capsule, dragee, powder, granulate, liquid, gel or
emulsion form. Said
liquid, gel or emulsion may be ingested by the subject in naked form or
contained within a
capsule.
[0174] In some embodiments, the pharmaceutical composition comprises a single
analgesic agent and a single PDE 5 inhibitor. In one embodiment, the single
analgesic agent is
aspirin. In another embodiment, the single analgesic agent is ibuprofen. In
another embodiment,
the single analgesic agent is naproxen or naproxen sodium. In another
embodiment, the single
analgesic agent is indomethacin. In another embodiment, the single analgesic
agent is
nabumetone. In another embodiment, the single analgesic agent is
acetaminophen. In another
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
embodiment, the single PDE 5 inhibitor is tadalafil. The analgesic agent and
PDE 5 inhibitor may
be given at doses in the ranges described above.
[0175] In some embodiments, the pharmaceutical composition comprises one or
more
analgesic agents, individually or in combination, in an amount between 10-1000
mg, 10-800 mg,
10-600 mg, 10-500 mg, 10-400 mg, 10-300 mg, 10-250 mg, 10-200 mg, 10-150 mg,
10-100 mg
30-1000 mg, 30-800 mg, 30-600 mg, 30-500 mg, 30-400 mg, 30-300 mg, 30-250 mg,
30-200 mg,
30-150 mg, 30-100 mg, 100-1000 mg, 100-800 mg, 100-600 mg, 100-400 mg, 100-250
mg, 300-
1000 mg, 300-800 mg, 300-600 mg, 300-400 mg, 400-1000 mg, 400-800 mg, 400-600
mg, 600-
1000 mg, 600-800 mg or 800-1000 mg, wherein the composition is formulated for
extended
release with a release profile in which the one or more analgesic agents are
released continuously
over a period of 2-12 hours or 5-8 hours.
[0176] In some embodiments, the composition is formulated for extended-release
with a
release profile in which at least 90% of the one or more analgesic agents are
released continuously
over a period of 2-12 hours or 5-8 hours.
[0177] In some embodiments, the composition is formulated for extended release
with a
release profile in which the one or more analgesic agents are released
continuously over a period
of 5, 6, 7, 8, 10 or 12 hours. In some embodiments, the pharmaceutical
composition further
comprises an antimuscarinic agent, an antidiuretic, a spasmolytic, zolpidem or
a PDE 5 inhibitor.
[0178] In other embodiments, the composition is formulated for extended-
release with a
release profile in which the analgesic agent is released at a steady rate over
a period of 2-12 hours
or 5-8 hours. In other embodiments, the composition is formulated for extended
release with a
release profile in which the analgesic agent is released at a steady rate over
a period of 5, 6, 7, 8,
or 12 hours. As used herein, "a steady rate over a period of time" is defined
as a release profile
in which the release rate at any point during a given period of time is within
30% - 300% of the
average release rate over that given period of time. For example, if 80 mg of
aspirin is released at
a steady rate over a period of 8 hours, the average release rate is 10 mg/hr
during this period of
time and the actual release rate at any time during this period is within the
range of 3 mg/hr to 30
mg/hr (i.e., within 30% - 300% of the average release rate of 10 mg/hr during
the 8 hour period).
In some embodiments, the pharmaceutical composition further comprises an
antimuscarinic agent,
an antidiuretic a spasmolytic, zolpidem or a PDE 5 inhibitor.
[0179] In some embodiments, the analgesic agent is selected from the group
consisting of
aspirin, ibuprofen, naproxen sodium, naproxen, indomethacin, nabumetone and
acetaminophen. In
one embodiment, the analgesic agent is acetaminophen. The pharmaceutical
composition is
formulated to provide a steady release of small amount of the analgesic agent
to maintain an
effective drug concentration in the blood such that the overall amount of the
drug in a single
dosage is reduced compared to the immediate release formulation.
41
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
[0180] In some other embodiments, the pharmaceutical composition comprises one
or
more analgesic agent(s), individually or in combination, in an amount between
10-1000 mg, 10-
800 mg, 10-600 mg, 10-500 mg, 10-400 mg, 10-300 mg, 10-250 mg, 10-200 mg, 10-
150 mg, 10-
100 mg 30-1000 mg, 30-800 mg, 30-600 mg, 30-500 mg, 30-400 mg, 30-300 mg, 30-
250 mg, 30-
200 mg, 30-150 mg, 30-100 mg, 100-1000 mg, 100-800 mg, 100-600 mg, 100-400 mg,
100-250
mg, 300-1000 mg, 300-800 mg, 300-600 mg, 300-400 mg, 400-1000 mg, 400-800 mg,
400-600
mg, 600-1000 mg, 600-800 mg or 800-1000 mg, wherein the analgesic agent(s) are
formulated for
extended release, characterized by a two-phase release profile in which 20-80%
of the analgesic
agent(s) are released within 2 hours of administration and the remainder are
released
continuously, or at a steady rate, over a period of 2-12 hours or 5-8 hours.
In yet another
embodiment, the analgesic agent(s) is formulated for extended-release with a
two-phase release
profile in which 20, 30, 40, 50 or 60% of the analgesic agent(s) are released
within 2 hours of
administration and the remainder are released continuously, or at a steady
rate, over a period of 2-
12 hours or 5-8 hours. In one embodiment, the analgesic agent(s) are selected
from the group
consisting of aspirin, ibuprofen, naproxen sodium, naproxen, indomethacin,
nabumetone, and
acetaminophen. In one embodiment, the analgesic agent is acetaminophen. In
another
embodiment, the analgesic agent is acetaminophen. In some embodiments, the
pharmaceutical
composition further comprises an antimuscarinic agent, an antidiuretic, a
spasmolytic, zolpidem
and/or a PDE 5 inhibitor. In some embodiments, the antimuscarinic agent,
antidiuretic,
spasmolytic, zolpidem and/or PDE 5 inhibitor is/are formulated for immediate-
release.
[0181] Another aspect of the present application relates to a method for
reducing
frequency of urination by administering to a subject in need thereof, two or
more PG pathway
inhibitors alternatively to prevent the development of drug resistance. In one
embodiment, the
method comprises administering a first PG pathway inhibitor for a first period
of time and then
administering a second PG pathway inhibitors for a second period of time. In
another
embodiment, the method further comprises administering a third PG pathway
inhibitor for a third
period of time. The first, second, and third PG pathway inhibitors are
different from each other
and may be formulated for immediate-release, extended-release, delayed-release
or combinations
thereof.
[0182] Another aspect of the present application relates to a method for
treating nocturia
by administering to a person in need thereof a diuretic, followed with the
pharmaceutical
composition of the present application. The diuretic is dosed and formulated
to have a diuretic
effect within 6 hours of administration and is administered at least 8 or 7
hours prior to bedtime.
The pharmaceutical composition of the present application is formulated for
extended-release or
delayed, extended-release, and is administered within 2 hours prior to
bedtime.
42
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
[0183] Examples of diuretics include, but are not limited to, acidifying
salts, such as
CaC12 and NH4C1; arginine vasopressin receptor 2 antagonists, such as
amphotericin B and
lithium citrate; aquaretics, such as Goldenrod and Juniper; Na-H exchanger
antagonists, such as
dopamine; carbonic anhydrase inhibitors, such as acetazolamide and
dorzolamide; loop diuretics,
such as bumetanide, ethacrynic acid, furosemide and torsemide; osmotic
diuretics, such as glucose
and mannitol; potassium-sparing diuretics, such as amiloride, spironolactone,
triamterene,
potassium canrenoate; thiazides, such as bendroflumethiazide and
hydrochlorothiazide; and
xanthines, such as caffeine, theophylline and theobromine.
[0184] Another aspect of the present application relates to a method for
reducing the
frequency of urination, comprising administering to a subject in need thereof
an effective amount
of the pharmaceutical composition of the present application and an effective
amount of
botulinum toxin.
[0185] In some embodiments, the botulinum toxin is administered by injection
into a
bladder muscle; and orally administering to the subject the pharmaceutical
composition of the
present application. In some embodiments, the injecting step comprises
injection of 10-200 units
of botulinum toxin at 5-20 sites in bladder muscle with an injection dose of 2-
10 units per site. In
one embodiment, the injecting step comprises injection of botulinum toxin at 5
sites in bladder
muscle with an injection dose of 2-10 units per site. In another embodiment,
the injecting step
comprises injection of botulinum toxin at 10 sites in bladder muscle at an
injection dose of 2-10
units per site. In another embodiment, the injecting step comprises injection
of botulinum toxin at
15 sites in bladder muscle at an injection dose of 2-10 units per site. In yet
another embodiment,
the injecting step comprises injection of botulinum toxin at 20 sites in
bladder muscle at an
injection dose of 2-10 units per site. In some embodiments, the injecting step
is repeated every 3,
4, 6, 8, 10 or 12 months.
[0186] In some embodiments, the pharmaceutical composition comprises one or
more
analgesic agents selected from the group consisting of aspirin, ibuprofen,
naproxen, naproxen
sodium, indomethacin, nabumetone and acetaminophen in an amount of 5-2000 mg
per agent, and
one or more spasmolytics selected from the group consisting of carisoprodol,
benzodiazepines,
baclofen, cyclobenzaprine, metaxalone, methocarbamol, clonidine, clonidine
analog, and
dantrolene in a total amount of 50-500 mg, wherein the pharmaceutical
composition is formulated
for extended release with a two-phase release profile in which 20-60% of the
active ingredients
are released within 2 hours of administration, and the remainder are released
continuously, or at a
steady rate, in a period of 5-24 hours, 5-8 hours, 8-16 hours or 16-24 hours.
[0187] Another aspect of the present application relates to a pharmaceutical
composition
that comprises a first component having an immediate-release subcomponent and
an extended-
release subcomponent, wherein the first component is formulated to release the
subcomponents
43
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
immediately after administration; and a second component comprising an
immediate-release
subcomponent and an extended-release subcomponent, wherein the second
component is
formulated for a delayed-release of the subcomponents. In some embodiments, at
least one of the
subcomponents in the first component or the second component comprises an
active ingredient
comprising one or more analgesic agents, and at least one of the subcomponents
in the first
component or the second component comprises an active ingredient comprising a
PG pathway
inhibitor, such as a PG inhibitor, a PGT inhibitor or a PGR inhibitor. In some
embodiments, the
PG pathway inhibitor is selected from the groups consisting of inhibitors of
PG activity, inhibitors
of PG synthesis, inhibitors of PGT activity, inhibitors of PGT expression,
inhibitors of PGR
activity, and inhibitors of PGR expression.
[0188] In some embodiments, each of the subcomponents in the first component
or the
second component comprises an active ingredient comprising one or more
analgesic agents and/or
a PG pathway inhibitor, such as a PG inhibitor, a PGT inhibitor or a PGR
inhibitor.
[0189] In some embodiments, the one or more analgesic agents are selected from
the
group consisting of aspirin, ibuprofen, naproxen, naproxen sodium,
indomethacin, nabumetone,
and acetaminophen.
[0190] In some related embodiments, the immediate-release subcomponent and the
extended-release subcomponent in the first component each comprises an active
ingredient
comprising one or more analgesic agents, and/or a PG pathway inhibitor, such
as a PG inhibitor, a
PGT inhibitor or a PGR inhibitor. In other embodiments, the immediate-release
subcomponent
and the extended-release subcomponent in the second component each comprises
an active
ingredient comprising one or more analgesic agents, and/or a PG pathway
inhibitor, such as a PG
inhibitor, a PGT inhibitor or a PGR inhibitor.
[0191] In some embodiments, the one or more analgesic agents comprise
acetaminophen.
In yet other embodiments, at least one of the subcomponents in the first
component or the second
component comprises an active ingredient comprising one or more analgesic
agents and a PG
pathway inhibitor, such as a PG inhibitor, a PGT inhibitor or a PGR inhibitor.
[0192] In some related embodiments, the second component is coated with an
enteric
coating.
[0193] In some related embodiments, the second component is formulated to
release the
subcomponents after a lag time of 1-4 or 2-4 hours or 4-8 hours following oral
administration.
[0194] In some related embodiments, the extended-release subcomponent in the
first
component is formulated to release its active ingredient over a time interval
of about 2-10 hours.
[0195] In some related embodiments, the extended-release subcomponent in the
second
component is formulated to release its active ingredient over a time interval
of about 2-10 hours.
44
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
[0196] In some related embodiments, the active ingredient in the immediate-
release
subcomponent and the extended-release subcomponent in the first component
further comprises
an antimuscarinic agent. In some embodiments, the active ingredient in the
immediate-release
subcomponent and the extended-release subcomponent in the second component
further
comprises an antimuscarinic agent. In some embodiments, the active ingredient
in the immediate-
release subcomponent and the extended-release subcomponent in both the first
and the second
component further comprises an antimuscarinic agent.
[0197] In some related embodiments, the active ingredient in the immediate-
release
subcomponent and the extended-release subcomponent in the first component
further comprises
an antidiuretic agent. In some embodiments, the active ingredient in the
immediate-release
subcomponent and the extended-release subcomponent in the second component
further
comprises an antidiuretic agent. In some embodiments, the active ingredient in
the immediate-
release subcomponent and the extended-release subcomponent in both the first
and the second
component further comprises an antidiuretic agent.
[0198] In some related embodiments, the active ingredient in the immediate-
release
subcomponent and the extended-release subcomponent in the first component
further comprises a
spasmolytic. In some embodiments, the active ingredient in the immediate-
release subcomponent
and the extended-release subcomponent in the second component further
comprises a
spasmolytic. In some embodiments, the active ingredient in the immediate-
release subcomponent
and the extended-release subcomponent in both the first and the second
component further
comprises a spasmolytic.
[0199] In some related embodiments, the immediate-release subcomponent and the
extended-release subcomponent in the first component each comprises an
analgesic agent, such as
acetaminophen, in an amount of 5-2000 mg. In some embodiments, the immediate-
release
subcomponent and the extended-release subcomponent in the second component
each comprises
an analgesic agent, such as acetaminophen, in an amount of 5-2000 mg. In some
embodiments,
the active ingredient in the immediate-release subcomponent and the extended-
release
subcomponent in both the first and the second component each comprises an
analgesic agent, such
as acetaminophen, in an amount of 5-2000 mg.
[0200] In some related embodiments, the active ingredient in the immediate-
release
subcomponent of the first component and the active ingredient in the immediate-
release
subcomponent of the second component both comprise an analgesic agent, such as
acetaminophen. In some embodiments, the active ingredient in the immediate-
release
subcomponent of the first component and the active ingredient in the immediate-
release
subcomponent of the second component comprise different analgesic agents.
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
[0201] Another aspect of the present application relates to a pharmaceutical
composition
that comprises a first component comprising an immediate-release subcomponent,
wherein the
immediate-release subcomponent comprises an active ingredient comprising one
or more agents
selected from the group consisting of analgesic agents and a PG pathway
inhibitor, such as a PG
inhibitor, a PGT inhibitor or a PGR inhibitor, wherein the first component is
formulated to release
its subcomponent immediately after oral administration; and a second component
comprising an
immediate-release subcomponent and an extended-release subcomponent, wherein
the second
component is formulated to release its subcomponent after gastric emptying,
wherein the
subcomponents in the second component each comprises an active ingredient
comprising one or
more agents selected from the group consisting of analgesic agents and a PG
pathway inhibitor,
such as a PG inhibitor, a PGT inhibitor or a PGR inhibitor. In some
embodiments, the PG
pathway inhibitor is selected from the groups consisting of inhibitors of PG
activity, inhibitors of
PG synthesis, inhibitors of PGT activity, inhibitors of PGT expression,
inhibitors of PGR activity,
and inhibitors of PGR expression.
[0202] In some embodiments, the one or more analgesic agents are selected from
the
group consisting of aspirin, ibuprofen, naproxen, naproxen sodium,
indomethacin, nabumetone,
and acetaminophen.
[0203] In some related embodiments, the second component is formulated to
release the
subcomponents after a lag time of 2-12 hours, 2-4 hours, 2-6 hours, 2-8 hours,
or 4-8 hours
following oral administration.
[0204] In some related embodiments, the active ingredient in the immediate-
release
subcomponent and the extended-release subcomponent of the second component
comprises one or
more analgesic agents.
[0205] In some related embodiments, the first component further comprises an
extended-
release subcomponent, wherein the extended-release subcomponent comprises an
active
ingredient comprising one or more agents selected from the group consisting of
analgesic agents
and a PG pathway inhibitor, such as a PG inhibitor, a PGT inhibitor or a PGR
inhibitor. In some
embodiments, the one or more agents comprises an analgesic agent selected from
the group
consisting of aspirin, ibuprofen, naproxen, naproxen sodium, indomethacin,
nabumetone, and
acetaminophen.
[0206] In some embodiments, the immediate-release subcomponent and the
extended-
release subcomponent in the second component each comprises a PG pathway
inhibitor, such as a
PG inhibitor, a PGT inhibitor or a PGR inhibitor.
[0207] In some related embodiments, at least one of the active ingredients in
the
immediate-release subcomponent and/or the extended-release subcomponent of the
first and the
46
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
second components further comprises an agent selected from the group
consisting of
antimuscarinic agents, antidiuretic agents and spasmolytics.
[0208] In some related embodiments, the active ingredient in the immediate-
release
subcomponent and/or the extended-release subcomponent of the first component
further
comprises an agent selected from the group consisting of antimuscarinic
agents, antidiuretic
agents and spasmolytics.
[0209] In some related embodiments, the active ingredient in the immediate-
release
subcomponent and/or the extended-release subcomponent of the second component
further
comprises an agent selected from the group consisting of antimuscarinic
agents, antidiuretic
agents and spasmolytics.
[0210] Another aspect of the present application relates to a pharmaceutical
composition
that comprises a first component comprising an immediate-release subcomponent
and an
extended-release subcomponent, wherein the first component is formulated to
release the
subcomponents immediately after administration; and a second component
comprising an
immediate-release subcomponent and an extended-release subcomponent, wherein
the second
component is formulated for a delayed-release of the subcomponents, wherein
the immediate-
release subcomponent and the extended-release subcomponent in the first
component each
comprises an active ingredient comprising one or more analgesic agents and a
PG pathway
inhibitor, such as a PG inhibitor, a PGT inhibitor or a PGR inhibitor, and
wherein the immediate-
release subcomponent and the extended-release subcomponent in the second
component each
comprises an active ingredient comprising one or more analgesic agents and a
PG pathway
inhibitor, such as a PG inhibitor, a PGT inhibitor or a PGR inhibitor, wherein
the pharmaceutical
composition reduces the frequency of urination in patients in need thereof In
some embodiments,
the PG pathway inhibitor is selected from the groups consisting of inhibitors
of PG activity,
inhibitors of PG synthesis, inhibitors of PGT activity, inhibitors of PGT
expression, inhibitors of
PGR activity, and inhibitors of PGR expression.
[0211] In some embodiments, the one or more analgesic agents are selected from
the
group consisting of aspirin, ibuprofen, naproxen, naproxen sodium,
indomethacin, nabumetone,
and acetaminophen. In some embodiments, the one or more analgesic agents
comprise
acetaminophen.
[0212] In other embodiments, the pharmaceutical composition comprises a pair
of
analgesic agents. Examples of such paired analgesic agents include, but are
not limited to,
acetaminophen and an NSAID, acetylsalicylic acid and ibuprofen,
acetylsalicylic acid and
naproxen sodium, acetylsalicylic acid and nabumetone, acetylsalicylic acid and
acetaminophen,
acetylsalicylic acid and indomethancin, ibuprofen and naproxen sodium,
ibuprofen and
nabumetone, ibuprofen and acetaminophen, ibuprofen and indomethancin, naproxen
sodium and
47
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
nabumetone, naproxen sodium and acetaminophen, naproxen sodium and
indomethancin,
nabumetone and acetaminophen, nabumetone and indomethancin, and acetaminophen
and
indomethancin. The paired analgesic agents are mixed at a weight ratio in the
range of 0.1:1 to
10:1, 0.2:1 to 5:1 or 0.3:1 to 3:1 with a combined dose or single dose (i.e.,
the dose for each
analgesic) in the range of 5 mg to 2000 mg, 20 mg to 2000 mg, 100 mg to 2000
mg, 200 mg to
2000 mg, 500 mg to 2000 mg, 5 mg to 1500 mg, 20 mg to 1500 mg, 100 mg to 1500
mg, 200 mg
to 1500 mg, 500 mg to 1500 mg, 5 mg to 1000 mg, 20 mg to 1000 mg, 100 mg to
1000 mg, 250
mg to 500 mg, 250 mg to 1000 mg, 250 mg to 1500 mg, 500 mg to 1000 mg, 500 mg
to 1500
mg, 1000 mg to 1500 mg, and 1000 mg to 2000 mg. In one embodiment, the paired
analgesic
agents are mixed at a weight ratio of 1:1.
[0213] Another aspect of the present application relates to a pharmaceutical
composition
that comprises an immediate-release component and an extended-release
component. Each
component comprises a pair of analgesic agents as described above and a PG
pathway inhibitor,
such as a PG inhibitor, a PGT inhibitor or a PGR inhibitor. In some
embodiments, the immediate-
release component and the extended-release component comprise different pairs
of analgesic
agents. In some embodiments, the immediate-release component and the extended-
release
component comprise the same pair of analgesic agents. In some embodiments, the
immediate-
release component and the extended-release component each comprises
acetaminophen and an
NSAID. In some embodiments, the immediate-release component and the extended-
release
component each comprises acetaminophen and ibuprofen. In some embodiments, the
immediate-
release component and the extended-release component each consists of
acetaminophen,
ibuprofen and a PG pathway inhibitor, such as a PG inhibitor, a PGT inhibitor
or a PGR inhibitor.
In some embodiments, the PG pathway inhibitor is selected from the groups
consisting of
inhibitors of PG activity, inhibitors of PG synthesis, inhibitors of PGT
activity, inhibitors of PGT
expression, inhibitors of PGR activity, and inhibitors of PGR expression.
[0214] In some embodiments, the extended-release component is formulated for
extended
release over a period of 0.5-24, 2-6, 6-10, 10-14, or 14-24 hours. In some
embodiments, the
extended-release component is formulated for extended release over a period of
about 8 hours. In
some embodiments, the extended-release component is coated with a delayed-
release coating. In
some embodiments, the delayed-release coating delays the release of the
extended-release
component for a period of 0.1-12, 0.5-12, 1-12, 2-12, 1-4, 2-4, 4-8 or 8-12
hours. In some
embodiments, the delayed-release coating is an enteric coating. In some
embodiments, the
pharmaceutical composition with an immediate-release component and an extended-
release
component is formulated into an orally disintegrating tablet.
[0215] As used herein, the term "orally disintegrating tablet" or "orally
disintegrating
formulation" refers to drug tablet or formulation that rapidly disintegrates
or dissolves in the oral
48
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
cavity. Orally disintegrating formulations differ from traditional tablets in
that they are designed
to be dissolved on the tongue rather than swallowed whole. In some
embodiments, the orally
disintegrating formulations are designed to completely disintegrate or
dissolve in the oral cavity
without the aid of additional water (i.e., in saliva only) in 5, 10, 20, 30,
60, 90, 120, 180, 240 or
300 seconds.
[0216] In some embodiments, the pharmaceutical composition with an immediate-
release
component and an extended-release component is formulated into a liquid form
for oral
administration. Examples of the liquid form formulation include, but are not
limited to, gels,
emulsions and particle suspensions. For example, the extended-release
component may be
formulated into a gel form that solidifies in the stomach. In some
embodiments, the
pharmaceutical composition with an immediate-release component and an extended-
release
component is formulated into a pixie pack of powder that can quickly melt on
the tongue. In
some embodiments, the immediate-release component or the extended release
component or both
further comprise one or more additional agents selected from the group
consisting of
antimuscarinic agents, spasmolytics and antidiuretic agents.
Method of manufacture
[0217] Another aspect of the present application relates to methods of
manufacturing
extended-release pharmaceutical compositions for reducing the frequency of
urination. In some
embodiments, the method comprises the steps of forming a first mixture having
a first active
ingredient formulated for immediate release and a second active ingredient
formulated for
extended release; coating the first mixture with a delayed release coating to
form a core structure;
and then coating the core structure with a second mixture comprising a third
active ingredient
formulated for immediate release and a fourth active ingredient formulated for
extended release.
In one embodiment, at least one of the first, second, third and fourth active
ingredients comprises
an analgesic agent and at least one of the first, second, third and fourth
active ingredients
comprises a PG pathway inhibitor, such as a PG inhibitor, a PGT inhibitor or a
PGR inhibitor. In
some embodiments, the PG pathway inhibitor is selected from the groups
consisting of inhibitors
of PG activity, inhibitors of PG synthesis, inhibitors of PGT activity,
inhibitors of PGT
expression, inhibitors of PGR activity, and inhibitors of PGR expression.
[0218] In some embodiments, the analgesic agent is selected from the group
consisting of
aspirin, ibuprofen, naproxen, naproxen sodium, indomethacin, nabumetone and
acetaminophen,
and wherein at least one of the first, second, third and fourth active
ingredients comprises 5 mg to
2000 mg of the analgesic agent.
[0219] In some embodiments, at least one of the first, second, third and
fourth active
ingredients comprises (1) an analgesic agent selected from the group
consisting of aspirin,
49
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
ibuprofen, naproxen, naproxen sodium, indomethacin, nabumetone, and
acetaminophen, and (2) a
PG pathway inhibitor, such as a PG inhibitor, a PGT inhibitor or a PGR
inhibitor.
[0220] In some embodiments, the at least one of the first, second, third and
fourth active
ingredients comprises (1) acetaminophen, and (2) a PG pathway inhibitor, such
as a PG inhibitor,
a PGT inhibitor or a PGR inhibitor.
[0221] In some embodiments, the at least one of the first, second, third and
fourth active
ingredients comprises an agent selected from the group consisting
antimuscarinic agents,
antidiuretic agents and spasmolytics.
[0222] In some embodiments, the delayed release coating is an enteric coating.
In some
embodiments, the enteric coating comprises a pH-dependent polymer. In some
embodiments, the
delayed release coating comprises a swelling layer covered by an outer semi-
permeable polymer
layer. In some embodiments, the delayed release coating is formulated to
release the coated
material after a lag time of 0.1-12 hours, 0.5-12 hours, 1-12 hours, 2-12
hours, 1-4 hours, 2-4
hours, 2-6 hours, 2-8 hours, 4-6 hours or 4-8 hours after oral administration.
[0223] In some embodiments, the second active ingredient, or the fourth active
ingredient
or both comprise an active core comprising an extended-release coating or a
polymeric matrix
effecting diffusion controlled release.
[0224] In some embodiments, the first mixture is prepared by mixing the first
active
ingredient in liquid or powder form with the second active ingredient, which
is formulated for
extended release. As described above, the second active ingredient may be
formulated in an
extended release formulation having an active core comprised of one or more
inert particles, each
in the form of a bead, pellet, pill, granular particle, microcapsule,
microsphere, microgranule,
nanocapsule, or nanosphere coated on its surfaces with drugs in the form of
e.g., a drug-
containing coating or film-forming composition using, for example, fluid bed
techniques or other
methodologies known to those of skill in the art. The inert particle can be of
various sizes, so
long as it is large enough to remain undissolved. Alternatively, the active
core may be prepared
by granulating and milling and/or by extrusion and spheronization of a polymer
composition
containing the drug substance. In some embodiments, the active core comprises
an extended-
release coating or a polymeric matrix effecting diffusion controlled release,
as described in more
detail earlier. In some embodiments, the polymeric matrix is a water soluble
or water-swellable
matrix. In some embodiments, the second active ingredient is simply mixed with
the first active
ingredient. Either ingredient or both ingredients may be in the form of bead,
pellet, granular
particle, pill, microcapsule, microsphere, microgranule, nanocapsule or
nanosphere as a powder or
as a liquid suspension. In other embodiments, the second active ingredient
form an active core
that is coated with the first active ingredient. In some embodiments, the
second active ingredient
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
in the first mixture is formulated to release the active ingredient over a
period of 2-4 hours, 2-6
hours, 2-8 hours or 2-10 hours.
[0225] In some embodiments, the second active ingredient is kept in a
compartment
partially or completely separate from the first active ingredient. In other
embodiments, the first
mixture is formed by keeping the second active ingredient in a compartment
partially or
completely separated from the first active ingredient.
[0226] The first mixture is then coated with a delayed release coating to form
a core
structure. In some embodiments, the delayed release coating is an enteric
coating. In some
embodiments, the enteric coating comprises a pH-dependent polymer that
maintains its structure
integrity at low pH, such as the pH in the stomach (normally in the range of
1.5-3.5). In some
embodiments, the term "low pH" refers to a pH value of 4.0, 3.5, 3.0, 2.5,
2.0, 1.5, 1.0 or lower.
In some embodiments, the enteric coating comprises one or more pH-dependent
polymers and one
or more polysaccharides that are resistant to erosion in both the stomach and
intestine, thus
allowing the release of the first mixture only in the colon. In some
embodiments, the delayed
release coating comprises two or more layers of coating. In some embodiments,
the delayed
release coating comprises a swelling layer and an outer semi-permeable polymer
layer that covers
the swelling layer.
[0227] In the next step, the coated core structure is re-coated with a second
mixture that
comprises a third active ingredient formulated for immediate release and a
fourth active ingredient
formulated for extended release. In some embodiments, the second mixture is
prepared by mixing
the third active ingredient in liquid or powder form with the fourth active
ingredient, which is
formulated for extended release. The fourth active ingredient may be
formulated in an extended
release formulation having an active core comprised of one or more inert
particles, each in the
form of a bead, pellet, pill, granular particle, microcapsule, microsphere,
microgranule,
nanocapsule, or nanosphere coated on its surfaces with drugs in the form of
e.g., a drug-
containing coating or film-forming composition using, for example, fluid bed
techniques or other
methodologies known to those of skill in the art. The inert particle can be of
various sizes, so
long as it is large enough to remain poorly dissolved. Alternatively, the
active core may be
prepared by granulating and milling and/or by extrusion and spheronization of
a polymer
composition containing the drug substance. In some embodiments, the active
core comprises an
extended-release coating or a polymeric matrix effecting diffusion controlled
release, as described
in more detail earlier. In some embodiments, the polymeric matrix is a water
soluble or water-
swellable matrix. In some embodiments, the fourth active ingredient is simply
mixed with the
third active ingredient. Either ingredient or both ingredients may be in the
form of bead, pellet,
granular particle, pill, microcapsule, microsphere, microgranule, nanocapsule
or nanosphere as a
powder or as a liquid suspension.
51
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
[0228] In other embodiments, the coated core structure is re-coated first with
the fourth
active ingredient, and then coated with the third active ingredient. In some
embodiments, the
fourth active ingredient is formulated to release the active ingredient over a
period of 2-4 hours, 2-
6 hours, 2-8 hours or 2-10 hours.
[0229] In some embodiments, the fourth active ingredient is kept in a
compartment
partially or completely separated from the third active ingredient. In other
embodiments, the
second mixture is formed by keeping the fourth active ingredient in a
compartment partially or
completely separated from the third active ingredient.
[0230] In other embodiments, the method comprises the steps of forming a core
structure
comprising a first active ingredient formulated for immediate release and a
second active
ingredient formulated for extended release, coating the core structure with a
delayed release
coating to form a coated core structure, and mixing the coated core structure
with a third active
ingredient formulated for immediate release and a fourth active ingredient
formulated for
extended release. The first, second, third and fourth active ingredients can
be the active
ingredients described above. In one embodiment, the first, second, third and
fourth active
ingredients each comprises an analgesic agent and/or a PG pathway inhibitor,
such as a PG
inhibitor, a PGT inhibitor or a PGR inhibitor. In some embodiments, the PG
pathway inhibitor is
selected from the groups consisting of inhibitors of PG activity, inhibitors
of PG synthesis,
inhibitors of PGT activity, inhibitors of PGT expression, inhibitors of PGR
activity, and inhibitors
of PGR expression. In some embodiments, the analgesic agent is selected from
the group
consisting of aspirin, ibuprofen, naproxen, naproxen sodium, indomethacin,
nabumetone and
acetaminophen. In some embodiments, the method further comprises the step of
preparing a
dosage form with the final mixture. In some embodiments, the dosage form is in
a tablet form. In
some embodiments, the dosage form is in an orally disintegrating form, e.g.,
orally disintegrating
tablet form. In some embodiments, the dosage form is in a beads-in-a-capsule
form. In some
embodiments, the dosage form is in a liquid (e.g., emulsion) form.
[0231] In other embodiments, the method comprises the steps of forming a core
structure
comprising a first active ingredient formulated for immediate release and a
second active
ingredient formulated for extended release, coating the core structure with a
delayed release
coating to form a coated core structure, mixing the coated core structure with
a third ingredient
formulated for immediate release and a fourth ingredient formulated for
extended release.
[0232] Another aspect of the present application relates to a method for
manufacturing a
pharmaceutical composition for reducing the frequency of urination. The method
comprises the
step of forming a core structure comprising a first active ingredient
formulated for immediate
release and a second active ingredient formulated for extended release;
coating the core structure
with a delayed release coating to form a coated core structure; mixing the
coated core structure
52
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
with a third active ingredient formulated for immediate release and a fourth
active ingredient
formulated for extended release to form a final mixture, and compressing the
final mixture into a
tablet. In some embodiments, at least one of the first, second, third and
fourth active ingredients
comprises an analgesic agent and at least one of the first, second, third and
fourth active
ingredients comprises a PG pathway inhibitor, such as a PG inhibitor, a PGT
inhibitor or a PGR
inhibitor. In some embodiments, the PG pathway inhibitor is selected from the
groups consisting
of inhibitors of PG activity, inhibitors of PG synthesis, inhibitors of PGT
activity, inhibitors of
PGT expression, inhibitors of PGR activity, and inhibitors of PGR expression.
[0233] In some embodiments, the analgesic agent is selected from the group
consisting of
aspirin, ibuprofen, naproxen, naproxen sodium, indomethacin, nabumetone and
acetaminophen
and wherein at least one of the first, second, third and fourth active
ingredients comprises 5-2000
mg of the analgesic agent.
[0234] In some embodiments, the at least one of the first, second, third, and
fourth active
ingredients comprises: (1) acetaminophen; and (2) a PG pathway inhibitor, such
as a PG inhibitor,
a PGT inhibitor or a PGR inhibitor.
[0235] In some embodiments, the at least one of the first, second, third and
fourth active
ingredients comprises an agent selected from the group consisting of
antimuscarinic agents,
antidiuretic agents and spasmolytics.
[0236] Another aspect of the present application relates to a method for
manufacturing a
pharmaceutical composition for reducing the frequency of urination. The method
comprises the
steps of forming a core structure comprising a first active ingredient
formulated for immediate
release and a second active ingredient formulated for extended release;
coating the core structure
with a delayed release coating to form a coated core structure; coating the
coated core structure
with a third active ingredient formulated for immediate release to form a
double-coated core
structure. In some embodiments, wherein at least one of the first, second and
third active
ingredients comprises an analgesic agent and at least one of the first, second
and third active
ingredients comprises a PG pathway inhibitor, such as a PG inhibitor, a PGT
inhibitor or a PGR
inhibitor. In some embodiments, the PG pathway inhibitor is selected from the
groups consisting
of inhibitors of PG activity, inhibitors of PG synthesis, inhibitors of PGT
activity, inhibitors of
PGT expression, inhibitors of PGR activity, and inhibitors of PGR expression.
[0237] In some embodiments, the analgesic agent is selected from the group
consisting of
aspirin, ibuprofen, naproxen, naproxen sodium, indomethacin, nabumetone and
acetaminophen
and wherein at least one of the first, second and third active ingredients
comprises 5-2000 mg of
the analgesic agent.
53
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
[0238] In some embodiments, at least one of the first, second and third active
ingredients
comprises: (1) acetaminophen; and (2) a PG pathway inhibitor, such as a PG
inhibitor, a PGT
inhibitor or a PGR inhibitor.
[0239] In some embodiments, the at least one of the first, second and third
active
ingredients comprises an agent selected from the group consisting
antimuscarinic agents,
antidiuretic agents and spasmolytics.
[0240] Another aspect of the present application relates to a method for
manufacturing a
pharmaceutical composition for reducing the frequency of urination. The method
comprises the
steps of forming a core structure comprising a first pair of analgesic agents
formulated for
extended-release, and coating the core structure with a coating layer
comprising a second pair of
analgesics, wherein the second pair of analgesics is formulated for immediate
release and wherein
either the core structure or the coating layer or both further comprise a PG
pathway inhibitor, such
as a PG inhibitor, a PGT inhibitor or a PGR inhibitor. In some embodiments,
the PG pathway
inhibitor is selected from the groups consisting of inhibitors of PG activity,
inhibitors of PG
synthesis, inhibitors of PGT activity, inhibitors of PGT expression,
inhibitors of PGR activity, and
inhibitors of PGR expression.
[0241] In some embodiments, the core structure is first coated with a delayed-
release
coating and then coated with a coating layer comprising a second pair of
analgesics, wherein the
second pair of analgesics is formulated for immediate release.
[0242] In some embodiments, the method comprises the steps of forming a first
mixture
comprising a first pair of analgesic agents formulated for extended-release,
forming a second
mixture comprising a second pair of analgesic agents formulated for immediate-
release, and
combining the first mixture and the second mixture to form a final mixture,
wherein either the
first mixture or the second mixture or both further comprise a PG pathway
inhibitor, such as a PG
inhibitor, a PGT inhibitor or a PGR inhibitor.
[0243] In some embodiments, the first mixture, the second mixture and the
final mixture
are mixtures of solid materials. In some embodiments, the final mixture is in
powder or granulate
form. In some embodiments, the method further comprises the step of pressing
the final mixture
into a tablet form. In some embodiments, the final mixture is in a liquid, gel
or emulsion form.
[0244] Examples of paired analgesic agents include, but are not limited to,
acetaminophen
and an NSAID, acetylsalicylic acid and ibuprofen, acetylsalicylic acid and
naproxen sodium,
acetylsalicylic acid and nabumetone, acetylsalicylic acid and acetaminophen,
acetylsalicylic acid
and indomethancin, ibuprofen and naproxen sodium, ibuprofen and nabumetone,
ibuprofen and
acetaminophen, ibuprofen and indomethancin, naproxen sodium and nabumetone,
naproxen
sodium and acetaminophen, naproxen sodium and indomethancin, nabumetone and
acetaminophen, nabumetone and indomethancin, and acetaminophen and
indomethancin. In some
54
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
embodiments, the first pair of analgesic agents is different from the second
pair of analgesic
agents. In other embodiments, the first pair of analgesic agents is the same
as the second pair of
analgesic agents. In one embodiment, the first pair of analgesic agents and
the second pair of
analgesic agents are both acetaminophen and ibuprofen.
[0245] For example, the extended-release component may be formulated into a
gel form
that solidifies in the stomach. In some embodiments, the pharmaceutical
composition with an
immediate-release component and an extended-release component is formulated
into a pixie pack
of powder that can quickly melt on the tongue. In some embodiments, the
pharmaceutical
composition with an immediate-release component and an extended-release
component is
formulated into an orally disintegrating tablet using loose compression
tableting. In loose
compression, orally disintegrating formulation is compressed at much lower
forces (4 ¨ 20 kN)
than traditional tablets. In some embodiments, the orally disintegrating
formulation contains some
form of sugar, such as mannitol, to improve mouth feel. In some embodiments,
the orally
disintegrating tablet is produced using lyophilized orally disintegrating
formulation.
[0246] The present invention is further illustrated by the following example
which should
not be construed as limiting. The contents of all references, patents, and
published patent
applications cited throughout this application are incorporated herein by
reference.
EXAMPLE 1: INHIBITION OF THE URGE TO URINATE WITH IBUPROFEN
[0247] Twenty volunteer subjects, both male and female were enrolled, each of
which
experienced a premature urge or desire to urinate, interfering with their
ability to sleep for a
sufficient period of time to feel adequately rested. Each subject ingested 400-
800 mg of
ibuprofen as a single dose prior to bedtime. At least 14 subjects reported
that they were able to
rest better because they were not being awakened as frequently by the urge to
urinate.
[0248] Several subjects reported that after several weeks of nightly use of
ibuprofen, the
benefit of less frequent urges to urinate was no longer being realized.
However, all of these
subjects further reported the return of the benefit after several days of
abstaining from taking the
dosages. More recent testing has confirmed similar results can be achieved at
much lower dosages
without any subsequent diminution of benefits.
EXAMPLE 2: EFFECT OF ANALGESIC AGENTS, BOTULINUM NEUROTOXIN AND
ANTIMUSCARINIC AGENTS ON MACROPHAGE RESPONSES TO INFLAMMATORY
AND NON-INFLAMMATORY STIMULI
Experimental Design
[0249] This study is designed to determine the dose and in vitro efficacy of
analgesics
and antimuscarinic agents in controlling macrophage response to inflammatory
and non-
inflammatory stimuli mediated by COX2 and prostaglandins (PGE, PGH, etc.). It
establishes
baseline (dose and kinetic) responses to inflammatory and non-inflammatory
effectors in bladder
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
cells. Briefly, cultured cells are exposed to analgesic agents and/or
antimuscarinic agents in the
absence or presence of various effectors.
[0250] The effectors include: lipopolysaccharide (LPS), an inflammatory agent,
and Cox2
inducer as inflammatory stimuli; carbachol or acetylcholine, stimulators of
smooth muscle
contraction as non-inflammatory stimuli; botulinum neurotoxin A, a known
inhibitor of
acetylcholine release, as positive control; and arachidonic acid (AA), gamma
linolenic acid
(DGLA), or eicosapentaenoic acid (EPA) as precursors of prostaglandins, which
are produced
following the sequential oxidation of AA, DGLA, or EPA inside the cell by
cyclooxygenases
(COX1 and COX2) and terminal prostaglandin synthases.
[0251] The analgesic agents include: Salicylates such as aspirin; iso-butyl-
propanoic-
phenolic acid derivative (ibuprofen) such as Advil, Motrin, Nuprin, and
Medipren; naproxen
sodium such as Aleve, Anaprox, Antalgin, Feminax Ultra, Flanax, Inza, Midol
Extended Relief,
Nalgesin, Naposin, Naprelan, Naprogesic, Naprosyn, Naprosyn suspension, EC-
Naprosyn,
Narocin, Proxen, Synflex and Xenobid; acetic acid derivative such as
indomethacin (Indocin);1-
naphthaleneacetic acid derivative such as nabumetone or relafen; N-acetyl-para-
aminophenol
(APAP) derivative such as acetaminophen or paracetamol (Tylenol); and
Celecoxib.
[0252] The antimuscarinic agents include oxybutynin, solifenacin, darifenacin,
and
atropine.
[0253] Macrophages are subjected to short term (1-2 hrs) or long term (24-48
hrs)
stimulation with:
(1) Each analgesic agent alone at various doses.
(2) Each analgesic agent at various doses in the presence of LPS.
(3) Each analgesic agent at various doses in the presence of carbachol or
acetylcholine.
(4) Each analgesic agent at various doses in the presence of AA, DGLA, or EPA.
(5) Botulinum neurotoxin A alone at various doses.
(6) Botulinum neurotoxin A at various doses in the presence of LPS.
(7) Botulinum neurotoxin A at various doses in the presence of carbachol or
acetylcholine.
(8) Botulinum neurotoxin A at various doses in the presence of AA, DGLA, or
EPA.
(9) Each antimuscarinic agent alone at various doses.
(10) Each antimuscarinic agent at various doses in the presence of LPS.
(11) Each antimuscarinic agent at various doses in the presence of carbachol
or
acetylcholine.
(12) Each antimuscarinic agent at various doses in the presence of AA, DGLA,
or EPA.
[0254] The cells are then analyzed for the release of PGH2; PGE; PGE2;
Prostacydin;
Thromboxane; IL-10; IL-6; TNF-a; the COX2 activity; the production of cAMP and
cGMP; the
56
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
production of IL-113, IL-6, TNF-a, and COX2 mRNA; and surface expression of
CD80, CD86,
and MHC class II molecules.
Materials and Methods
Macrophage cells
[0255] Murine RAW264.7 or J774 macrophage cells (obtained from ATCC) were used
in
this study. Cells were maintained in a culture medium containing RPMI 1640
supplemented with
% fetal bovine serum (FBS), 15 mM HEPES, 2 mM L-glutamine, 100 U/ml
penicillin, and 100
ig / ml of streptomycin. Cells were cultured at 37 C in a 5 % CO2 atmosphere
and split
(passages) once a week.
In vitro treatment of macrophage cells with analgesics
[0256] RAW264.7 macrophage cells were seeded in 96-well plates at a cell
density of
1.5x105 cells per well in 100 IA of the culture medium. The cells were treated
with (1) various
concentrations of analgesic (acetaminophen, aspirin, ibuprofen or naproxen),
(2) various
concentrations of lipopolysaccharide (LPS), which is an effector of
inflammatory stimuli to
macrophage cells, (3) various concentrations of carbachol or acetylcholine,
which are effectors of
non-inflammatory stimuli, (4) analgesic and LPS or (5) analgesic and carbachol
or acetylcholine.
Briefly, the analgesics were dissolved in FBS-free culture medium (i.e., RPMI
1640
supplemented with 15 mM HEPES, 2 mM L-glutamine, 100 U / ml penicillin, and
100 ,g / ml of
streptomycin) and diluted to desired concentrations by serial dilution with
the same medium. For
cells treated with analgesic in the absence of LPS, 50 IA of analgesic
solution and 50 IA of FBS-
free culture medium were added to each well. For cells treated with analgesic
in the presence of
LPS, 50 IA of analgesic solution and 50 IA of LPS (from Salmonella
typhimurium) in FBS-free
culture medium were added to each well. All conditions were tested in
duplicates.
[0257] After 24 or 48 hours of culture, 150 IA of culture supernatants were
collected, spun
down for 2 min at 8,000 rpm at 4 C to remove cells and debris and stored at -
70 C for analysis of
cytokine responses by ELISA. The cells were collected and washed by
centrifugation (5 min at
1,500 rpm at 4 C) in 500 IA of Phosphate buffer (PBS). Half of the cells were
then snapped frozen
in liquid nitrogen and stored at -70 C. The remaining cells were stained with
fluorescent
monoclonal antibodies and analyzed by flow cytometry.
Flow cytometry analysis of co-stimulatory molecule expression
[0258] For flow cytometry analysis, macrophages were diluted in 100 IA of FACS
buffer
(phosphate buffered saline (PBS) with 2% bovine serum albumin (BSA) and 0.01%
NaN3) and
stained 30 min at 4 C by addition of FITC-conjugated anti-CD40, PE-conjugated
anti-CD80, PE-
conjugated anti-CD86 antibody, anti MHC class II (I-Ad) PE (BD Bioscience).
Cells were then
washed by centrifugation (5 min at 1,500 rpm at 4 C) in 300 IA of FACS buffer.
After a second
wash, cells were re-suspended in 200 IA of FACS buffer and the percentage of
cells expressing a
57
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
given marker (single positive), or a combination of markers (double positive)
were analyzed with
the aid of an Accuri C6 flow cytometer (BD Biosciences).
Analysis of cytokine responses by ELISA
[0259] Culture supernatants were subjected to cytokine-specific ELISA to
determine IL-
113, IL-6, and TNF-a responses in cultures of macrophages treated with
analgesic, LPS alone or a
combination of LPS and analgesic. The assays were performed on Nunc MaxiSorp
Immunoplates
(Nunc) coated overnight with 100 ?al of anti-mouse IL-6, TNF-a mAbs (BD
Biosciences) or IL-
113 mAb (R&D Systems) in 0.1 M sodium bicarbonate buffer (pH 9.5). After two
washes with
PBS (200 ill per well), 200 ill of PBS 3% BSA were added in each well
(blocking) and the plates
incubated for 2 hours at room temperature. Plates were washed again two times
by addition of
200 ill per well, 100 ill of cytokine standards and serial dilutions of
culture supernatants were
added in duplicate, and the plates were incubated overnight at 4 C. Finally,
the plates were
washed twice and incubated with 100 ?al of secondary biotinylated anti-mouse
IL-6, TNFa mAbs
(BD Biosciences), or IL-113 (R&D Systems) followed by peroxidase-labeled goat
anti-biotin mAb
(Vector Laboratories). The colorimetric reaction was developed by the addition
of 2,2'-azino-bis
(3)-ethylbenzylthiazoline-6-sulfonic acid (ABTS) substrate and 14702 (Sigma)
and the absorbance
measured at 415 nm with a Victor V multilabel plate reader (PerkinElmer).
Determination of COX2 activity and the production of cAMP and cGMP
[0260] The COX2 activity in the cultured macrophages is determined by
sequential
competitive ELISA (R&D Systems). The production of cAMP and cGMP is determined
by the
cAMP assay and cGMP assay. These assays are performed routinely in the art.
[0261] Table 1 summarizes the experiments performed with Raw 264 macrophage
cell
line and main findings in terms of the effects of analgesics on cell surface
expression of
costimulatory molecules CD40 and CD80. Expression of these molecules is
stimulated by COX2
and inflammatory signals and, thus, was evaluated to determine functional
consequences of
inhibition of COX2.
[0262] As shown in Table 2, acetaminophen, aspirin, ibuprofen, and naproxen
inhibit
basal expression of co-stimulatory molecules CD40 and CD80 by macrophages at
all the tested
doses (i.e., 5x 105 nM, 5x 104 nM, 5x 103 nM, 5x 102 nM, 50 nM, and 5 nM),
except for the
highest dose (i.e., 5x 106 nM), which appears to enhance, rather than inhibit,
expression of the co-
stimulatory molecules. As shown in Figures 1A and 1B, such inhibitory effect
on CD40 and
CD50 expression was observed at analgesic doses as low as 0.05 nM (i.e.,
0.00005 M). This
finding supports the notion that a controlled release of small doses of
analgesic may be preferable
to acute delivery of large doses. The experiment also revealed that
acetaminophen, aspirin,
58
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
ibuprofen, and naproxen have a similar inhibitory effect on LPS induced
expression of CD40 and
CD80.
Table 1. Summary of experiments
LPS
Control Salmonella
typhimurium Acetaminophen Aspirin Ibuprofen Naproxen
TESTS
1 X
2 X Dose responses
(0, 5, 50, 1000)
ng/mL
3 X Dose responses
(0, 5, 50, 500, 5x103, 5x104, 5x105, 5x106) nM
4 X X (5 ng/mL) Dose responses
X (50 ng/mL (0, 5 , 50, 500, 5x103, 5x104, 5x105, 5x106)
nM
X (1000 ng/mL)
ANALYSIS
a Characterization of activation/stimulatory status: Flow cytometry
analysis of CD40, CD80,
CD86, and MHC class II
Mediators of inflammatory responses: ELISA analysis of IL-113, IL-6, TNF-a
Table 2. Summary of main findings
Effectors % Positive Negative LPS Dose analgesic (nM)
Control 5
ng/ml
5x106 5x105 5x104 5x103 500 50 5
CD40+CD80+ 20.6 77.8
Acetaminophen CD40+CD80+ 63 18 12 9.8 8.3 9.5 7.5
Aspirin CD40+CD80+ 44 11 10.3 8.3 8
10.5 7.5
Ibuprofen CD40+CD80+ ND* 6.4 7.7 7.9 6.0 4.9 5.8
Naproxen CD40+CD80+ 37 9.6 7.7 6.9 7.2 6.8 5.2
Analgesic plus LPS
Acetaminophen CD40+CD80+ 95.1 82.7 72.4 68.8 66.8 66.2 62.1
Aspirin CD40+CD80+ 84.5 80 78.7 74.7 75.8 70.1 65.7
Ibuprofen CD40+CD80+ ND 67 77.9 72.9 71.1 63.7 60.3
Naproxen CD40+CD80+ 66.0 74.1 77.1 71.0 68.8 72 73
* ND: not done (toxicity)
59
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
[0263] Table 3 summarizes the results of several studies that measured serum
levels of
analgesic after oral therapeutic doses in adult humans. As shown in Table 3,
the maximum serum
levels of analgesic after an oral therapeutic dose are in the range of iO4
to105 nM. Therefore, the
doses of analgesic tested in vitro in Table 2 cover the range of
concentrations achievable in vivo in
humans.
Table 3. Serum levels of analgesic in human blood after oral therapeutic doses
Maximum serum
Analgesic drug Molecular levels after oral References
weight therapeutic doses
mg/L nM
Acetaminophen 151.16 11-18 7.2x104- * BMC Clinical
Pharmacology.2010, 10:10
(Tylenol) 1.19x105 * Anaesth Intensive Care. 2011,
39:242
Aspirin 181.66 30-100 1.65x105- * Disposition of Toxic Drugs
and Chemicals in
(Acetylsalicylic acid) 5.5x105 Man, 8th Edition, Biomedical
Public, Foster City,
CA, 2008, pp. 22-25
* J Lab Clin Med. 1984 Jun;103:869
Ibuprofen 206.29 24-32 1.16x105- * BMC Clinical
Pharmacology2010, 10:10
(Advil, Motrin) 1.55 x105 * J Clin Pharmacol. 2001,
41:330
Naproxen 230.26 Up to Up to * J Clin Pharmacol. 2001, 41:330
(Aleve) 60 2.6x105
EXAMPLE 3: EFFECT OF ANALGESIC AGENTS, BOTULINUM NEUROTOXIN AND
ANTIMUSCARINIC AGENTS ON MOUSE BLADDER SMOOTH MUSCLE CELL
RESPONSES TO INFLAMMATORY AND NON-INFLAMMATORY STIMULI
Experimental Design
[0264] This study is designed to characterize how the optimal doses of
analgesics
determined in Example 2 affect bladder smooth muscle cells in cell culture or
tissue cultures, and
to address whether different classes of analgesics can synergize to more
efficiently inhibit COX2
and PGE2 responses.
[0265] The effectors, analgesic agents and antimuscarinic agents are described
in Example
2.
[0266] Primary culture of mouse bladder smooth muscle cells are subjected to
short term
(1-2 hrs) or long term (24-48 hrs) stimulation with:
(1) Each analgesic agent alone at various doses.
(2) Each analgesic agent at various doses in the presence of LPS.
(3) Each analgesic agent at various doses in the presence of carbachol or
acetylcholine.
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
(4) Each analgesic agent at various doses in the presence of AA, DGLA, or EPA.
(5) Botulinum neurotoxin A alone at various doses.
(6) Botulinum neurotoxin A at various doses in the presence of LPS.
(7) Botulinum neurotoxin A at various doses in the presence of carbachol or
acetylcholine.
(8) Botulinum neurotoxin A at various doses in the presence of AA, DGLA, or
EPA.
(9) Each antimuscarinic agent alone at various doses.
(10) Each antimuscarinic agent at various doses in the presence of LPS.
(11) Each antimuscarinic agent at various doses in the presence of carbachol
or
acetylcholine.
(12) Each antimuscarinic agent at various doses in the presence of AA, DGLA,
or
EPA.
[0267] The cells are then analyzed for the release of PGH2; PGE; PGE2;
Prostacydin;
Thromboxane; IL-1(3; IL-6; TNF-a; the COX2 activity; the production of cAMP
and cGMP; the
production of IL-113, IL-6, TNF-a, and COX2 mRNA; and surface expression of
CD80, CD86,
and MHC class II molecules.
Materials and Methods
Isolation and purification of mouse bladder cells
[0268] Bladder cells were removed from euthanized animals C57BL/6 mice (8-12
weeks
old), and cells were isolated by enzymatic digestion followed by purification
on a Percoll
gradient. Briefly, bladders from 10 mice were minced with scissors to fine
slurry in 10 ml of
digestion buffer (RPMI 1640, 2% fetal bovine serum, 0.5 mg/ml collagenase,
301.tg/m1DNase).
Bladder slurries were enzymatically digested for 30 minutes at 37 C.
Undigested fragments were
further dispersed through a cell-trainer. The cell suspension was pelleted and
added to a
discontinue 20%, 40%. and 75% Percoll gradient for purification on mononuclear
cells. Each
experiment used 50-60 bladders.
[0269] After washes in RPMI 1640, bladder cells were resuspended RPMI 1640
supplemented with 10 % fetal bovine serum, 15 mM HEPES, 2 mM L-glutamine, 100
U/ml
penicillin, and 100 ,g / ml of streptomycin and seeded in clear-bottom black
96-well cell culture
microculture plates at a cell density of 3x104 cells per well in 100 jil.
Cells were cultured at 37 C
in a 5 % CO2 atmosphere.
In vitro treatment of cells with analgesics
[0270] Bladder cells were treated with analgesic solutions (50 pl/ well)
either alone or
together with carbachol (10-Molar, 50 pl/ well), as an example of non-
inflammatory stimuli, or
lipopolysaccharide (LPS) of Salmonella typhimurium (1 jig/ml, 50 pl/ well), as
an example of
61
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
non-inflammatory stimuli. When no other effectors were added to the cells, 50
.1 of RPMI 1640
without fetal bovine serum were added to the wells to adjust the final volume
to 200 .1.
[0271] After 24 hours of culture, 150 .1 of culture supernatants were
collected, spun down
for 2 min at 8,000 rpm at 4 C to remove cells and debris, and stored at -70 C
for analysis of
Prostaglandin E2 (PGE2) responses by ELISA. Cells were fixed, permeabilized,
and blocked for
detection of Cyclooxygenase-2 (COX2) using a fluorogenic substrate. In
selected experiment
cells were stimulated 12 hours in vitro for analysis of COX2 responses.
Analysis of COX2 responses
[0272] COX2 responses were analyzed by a Cell-Based ELISA using human/mouse
total
COX2 immunoassay (R&D Systems), following the instructions of the
manufacturer. Briefly,
after cells fixation and permeabilization, a mouse anti-total COX2 and a
rabbit anti-total GAPDH
were added to the wells of the clear-bottom black 96-well cell culture
microculture plates. After
incubation and washes, an HRP-conjugated anti-mouse IgG and an AP-conjugated
anti-rabbit IgG
were added to the wells. Following another incubation and set of washes, the
HRP- and AP-
fluorogenic substrates were added. Finally, a Victor V multilabel plate
reader (PerkinElmer) was
used to read the fluorescence emitted at 600 nm (COX2 fluorescence) and 450 nm
(GAPDH
fluorescence). Results are expressed as relative levels of total COX2 as
determined by relative
fluorescence unit (RFUs) and normalized to the housekeeping protein GAPDH.
Analysis of PGE2 Responses
[0273] Prostaglandin E2 responses were analyzed by a sequential competitive
ELISA
(R&D Systems). More specifically, culture supernatants or PGE2 standards were
added to the
wells of a 96-well polystyrene microplate coated with a goat anti-mouse
polyclonal antibody.
After one hour incubation on a microplate shaker, an HRP-conjugated PGE2 was
added and the
plates were incubated for an additional two hours at room temperature. The
plates were then
washed and HRP substrate solution added to each well. The color was allowed to
develop for 30
minutes, and the reaction stopped by the addition of sulfuric acid before
reading the plate at 450
nm with wavelength correction at 570 nm. Results are expressed as mean pg/ml
of PGE2.
Other Assays
[0274] The release of PGH2; PGE, Prostacydin; Thromboxane; IL-113; IL-6; and
TNF-a;
the production of cAMP and cGMP; the production of IL-1(3, IL-6, TNF-a, and
COX2 mRNA;
and surface expression of CD80, CD86, and MHC class II molecules are
determined as described
in Example 2.
Results
Analgesics inhibit COX2 responses of mouse bladder cells to an inflammatory
stimulus
[0002] Several analgesics (acetaminophen, aspirin, ibuprofen, and naproxen)
were tested
on mouse bladder cells at the concentration of 5 [tM or 50 [tM to determine
whether the
62
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
analgesics could induce COX2 responses. Analysis of 24-hour cultures showed
that none of the
analgesics tested induced COX2 responses in mouse bladder cells in vitro.
[0003] The effect of these analgesics on the COX2 responses of mouse bladder
cells to
carbachol or LPS stimulation in vitro was also tested. As indicated in Table
1, the dose of
carbachol tested has no significant effect on COX2 levels in mouse bladder
cells. On the other
hand, LPS significantly increased total COX2 levels. Interestingly,
acetaminophen, aspirin,
ibuprofen, and naproxen could all suppress the effect of LPS on COX2 levels.
The suppressive
effect of the analgesic was seen when these drugs were tested at either 5 i.tM
or 50 i.tM (Table 4).
Table 4. COX2 expression by mouse bladder cells after in vitro stimulation and
treatment with
analgesic
Stimulus Analgesic Total COX2 levels (Normalized RFUs)
None None 158 18
Carbachol (mM) None 149 21
LPS (1 gimp None 420 26
LPS (1 gimp Acetaminophen (5 iaM) 275 12
LPS (1 gimp Aspirin (5 iaM) 240 17
LPS (1 gimp Ibuprofen (5 aM)) 253 32
LPS (1 gimp Naproxen (5 iaM) 284 11
LPS (1 gimp Acetaminophen (50 iaM) 243 15
LPS (1 gimp Aspirin (50 iaM) 258 21
LPS (1 gimp Ibuprofen (50 iaM) 266 19
LPS (1 gimp Naproxen (50 iaM) 279 23
Analgesics inhibit PGE2 responses of mouse bladder cells to an inflammatory
stimulus
[0275] The secretion of PGE2 in culture supernatants of mouse bladder cells
was
measured to determine the biological significance of the alteration of mouse
bladder cell COX2
levels by analgesics. As shown in Table 5, PGE2 was not detected in the
culture supernatants of
unstimulated bladder cells or bladder cells cultured in the presence of
carbachol. Consistent with
COX2 responses described above, stimulation of mouse bladder cells with LPS
induced the
secretion of high levels of PGE2. Addition of the analgesics acetaminophen,
aspirin, ibuprofen,
and naproxen suppressed the effect of LPS on PGE2 secretion, and no difference
was seen
between the responses of cells treated with the 5 or 50 i.tM dose of
analgesic.
63
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
Table 5. PGE2 secretion by mouse bladder cells after in vitro stimulation and
treatment with
analgesic.
Stimulus Analgesic PGE2 levels (pg/ml)
None None <20.5
Carbachol (mM) None <20.5
LPS (1[1g/m1) None 925 55
LPS (1[1g/m1) Acetaminophen (5 [IM) 619 32
LPS (1[1g/m1) Aspirin (5 [IM) 588 21
LPS (1[1g/m1) Ibuprofen (5 [IM)) 593 46
LPS (1[1g/m1) Naproxen (5 [IM) 597 19
LPS (1[1g/m1) Acetaminophen (50 [IM) 600 45
LPS (1[1g/m1) Aspirin (50 [IM) 571 53
LPS (1[1g/m1) Ibuprofen (50 [IM) 568 32
LPS (1[1g/m1) Naproxen (50 [IM) 588 37
[0276] In summary, these data show that the analgesics alone at 5 tM or 50 tM
do not
induce COX2 and PGE2 responses in mouse bladder cells. The analgesics at 5 tM
or 50
however, significantly inhibit COX2 and PGE2 responses of mouse bladder cells
stimulated in
vitro with LPS (1 g/ml). No significant effect of analgesics was observed on
COX2 and PGE2
responses of mouse bladder cells stimulated with carbachol (1 mM).
EXAMPLE 4: EFFECT OF ANALGESIC AGENTS, BOTULINUM NEUROTOXIN AND
ANTIMUSCARINIC AGENTS ON MOUSE BLADDER SMOOTH MUSCLE CELL
CONTRACTION
Experimental Design
[0277] Cultured mouse or rat bladder smooth muscle cells and mouse or rat
bladder
smooth muscle tissue are exposed to inflammatory stimuli and non-inflammatory
stimuli in the
presence of analgesic agent and/or antimuscarinic agent at various
concentrations. The stimulus-
induced muscle contraction is measured to evaluate the inhibitory effect of
the analgesic agent
and/or antimuscarinic agent.
[0278] The effectors, analgesic agents, and antimuscarinic agents are
described in
Example 2.
[0279] Primary cultures of mouse bladder smooth muscle cells are subjected to
short term
(1-2 hrs) or long term (24-48 hrs) stimulation with:
(1) Each analgesic agent alone at various doses.
(2) Each analgesic agent at various doses in the presence of LPS.
(3) Each analgesic agent at various doses in the presence of carbachol or
acetylcholine.
64
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
(4) Each analgesic agent at various doses in the presence of AA, DGLA, or EPA.
(5) Botulinum neurotoxin A alone at various doses.
(6) Botulinum neurotoxin A at various doses in the presence of LPS.
(7) Botulinum neurotoxin A at various doses in the presence of carbachol or
acetylcholine.
(8) Botulinum neurotoxin A at various doses in the presence of AA, DGLA, or
EPA.
(9) Each antimuscarinic agent alone at various doses.
(10) Each antimuscarinic agent at various doses in the presence of LPS.
(11) Each antimuscarinic agent at various doses in the presence of carbachol
or
acetylcholine.
(12) Each antimuscarinic agent at various doses in the presence of AA, DGLA,
or
EPA.
Materials and Methods
[0280] Primary mouse bladder cells are isolated as described in Example 3. In
selected
experiments, cultures of bladder tissue are used. Bladder smooth muscle cell
contractions are
recorded with a Grass polygraph (Quincy Mass, USA).
EXAMPLE 5: EFFECT OF ORAL ANALGESIC AGENTS AND ANTIMUSCARINIC
AGENTS ON COX2 AND PGE2 RESPONSES OF MOUSE BLADDER SMOOTH MUSCLE
CELLS.
Experimental Design
[0281] Normal mice and mice with over active bladder syndrome are given oral
doses of
aspirin, naproxen sodium, ibuprofen, Indocin, nabumetone, Tylenol, Celecoxib,
oxybutynin,
solifenacin, darifenacin, atropine, and combinations thereof. Control groups
include untreated
normal mice and untreated OAB mice with over active bladder syndrome. Thirty
(30) minutes
after last doses, the bladders are collected and stimulated ex vivo with
carbachol or acetylcholine.
In selected experiments, the bladders are treated with botulinum neurotoxin A
before stimulation
with carbachol. Animals are maintained in metabolic cages and frequency (and
volume) of
urination are evaluated. Bladder outputs are determined by monitoring water
intake and cage
litter weight. Serum PGH2, PGE, PGE2, Prostacydin, Thromboxane, IL-113, IL-6,
TNF-a, cAMP,
and cGMP levels are determined by ELISA. CD80, CD86, and MHC class II
expression in whole
blood cells are determined by flow cytometry.
[0282] At the end of the experiment, animals are euthanized, and ex vivo
bladder
contractions are recorded with a Grass polygraph. Portions of bladders are
fixed in formalin, and
COX2 responses are analyzed by immunohistochemistry.
EXAMPLE 6: EFFECT OF ANALGESIC AGENTS, BOTULINUM NEUROTOXIN AND
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
ANTIMUSCARINIC AGENTS ON HUMAN BLADDER SMOOTH MUSCLE CELL
RESPONSES TO INFLAMMATORY AND NON-INFLAMMATORY STIMULI
Experimental Design
[0283] This study is designed to characterize how the optimal doses of
analgesic
determined in Examples 1-5 affect human bladder smooth muscle cells in cell
culture or tissue
cultures and to address whether different classes of analgesics can synergize
to more efficiently
inhibit COX2 and PGE2 responses.
[0284] The effectors, analgesic agents, and antimuscarinic agents are
described in
Example 2.
[0285] Human bladder smooth muscle cells are subjected to short term (1-2 hrs)
or long
term (24-48 hrs) stimulation with:
(1) Each analgesic agent alone at various doses.
(2) Each analgesic agent at various doses in the presence of LPS.
(3) Each analgesic agent at various doses in the presence of carbachol or
acetylcholine.
(4) Each analgesic agent at various doses in the presence of AA, DGLA, or EPA.
(5) Botulinum neurotoxin A alone at various doses.
(6) Botulinum neurotoxin A at various doses in the presence of LPS.
(7) Botulinum neurotoxin A at various doses in the presence of carbachol or
acetylcholine.
(8) Botulinum neurotoxin A at various doses in the presence of AA, DGLA, or
EPA.
(9) Each antimuscarinic agent alone at various doses.
(10) Each antimuscarinic agent at various doses in the presence of LPS.
(11) Each antimuscarinic agent at various doses in the presence of carbachol
or
acetylcholine.
(12) Each antimuscarinic agent at various doses in the presence of AA, DGLA,
or
EPA.
[0286] The cells are then analyzed for the release of PGH2; PGE; PGE2;
Prostacydin;
Thromboxane; IL-1(3; IL-6; TNFa; the COX2 activity; the production of cAMP and
cGMP; the
production of IL-113, IL-6, TNFa, and COX2 mRNA; and surface expression of
CD80, CD86, and
MHC class II molecules.
EXAMPLE 7: EFFECT OF ANALGESIC AGENTS, BOTULINUM NEUROTOXIN AND
ANTIMUSCARINIC AGENTS ON HUMAN BLADDER SMOOTH MUSCLE CELL
CONTRACTION
Experimental Design
66
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
[0287] Cultured human bladder smooth muscle cells are exposed to inflammatory
stimuli
and non-inflammatory stimuli in the presence of an analgesic agent and/or
antimuscarinic agent at
various concentrations. The stimuli-induced muscle contraction is measured to
evaluate the
inhibitory effect of the analgesic agent and/or antimuscarinic agent.
[0288] The effectors, analgesic agents, and antimuscarinic agents are
described in
Example 2.
[0289] Human bladder smooth muscle cells are subjected to short term (1-2 hrs)
or long
term (24-48 hrs) stimulation with:
(1) Each analgesic agent alone at various doses.
(2) Each analgesic agent at various doses in the presence of LPS.
(3) Each analgesic agent at various doses in the presence of carbachol or
acetylcholine.
(4) Each analgesic agent at various doses in the presence of AA, DGLA, or EPA.
(5) Botulinum neurotoxin A alone at various doses.
(6) Botulinum neurotoxin A at various doses in the presence of LPS.
(7) Botulinum neurotoxin A at various doses in the presence of carbachol or
acetylcholine.
(8) Botulinum neurotoxin A at various doses in the presence of AA, DGLA, or
EPA.
(9) Each antimuscarinic agent alone at various doses.
(10) Each antimuscarinic agent at various doses in the presence of LPS.
(11) Each antimuscarinic agent at various doses in the presence of carbachol
or
acetylcholine.
(12) Each antimuscarinic agent at various doses in the presence of AA, DGLA,
or
EPA.
[0290] Bladder smooth muscle cell contractions are recorded with a Grass
polygraph
(Quincy Mass, USA).
EXAMPLE 8: EFFECT OF ANALGESIC AGENTS ON NORMAL HUMAN BLADDER
SMOOTH MUSCLE CELL RESPONSES TO INFLAMMATORY AND NON
INFLAMMATORY SIGNALS
Experimental Design
Culture of normal human bladder smooth muscle cells
[0291] Normal human bladder smooth muscle cells were isolated by enzymatic
digestion
from macroscopically normal pieces of human bladder. Cells were expended in
vitro by culture at
37 C in a 5 % CO2 atmosphere in RPMI 1640 supplemented with 10 % fetal bovine
serum, 15
mM HEPES, 2 mM L-glutamine, 100 U/ml penicillin, and 100 mg / ml of
streptomycin and
67
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
passage once a week by treatment with trypsin to detach cells followed by
reseeding in a new
culture flask. The first week of culture, the culture medium was supplemented
with 0.5 ng/ml
epidermal growth factor, 2 ng/ml fibroblast growth factor, and 5 g/m1
insulin.
Treatment of normal human bladder smooth muscle cells with analgesics in vitro
[0292] Bladder smooth muscle cells trypsinized and seeded in microculture
plates at a
cell density of 3x104 cells per well in 100 .1 were treated with analgesic
solutions (50 .1/ well)
either alone or together carbachol (10-Molar, 50 .1/ well), as an example of
non-inflammatory
stimuli, or lipopolysaccharide (LPS) of Salmonella typhimurium (1 g/ml, 50
.1/ well), as an
example of non-inflammatory stimuli. When no other effectors were added to the
cells, 50 .1 of
RPMI 1640 without fetal bovine serum were added to the wells to adjust the
final volume to
200 1.
[0293] After 24 hours of culture, 150 .1 of culture supernatants were
collected, spun down
for 2 min at 8,000 rpm at 4 C to remove cells and debris, and stored at -70 C
for analysis of
Prostaglandin E2 (PGE2) responses by ELISA. Cells were fixed, permeabilized,
and blocked for
detection of COX2 using a fluorogenic substrate. In selected experiments,
cells were stimulated
12 hours in vitro for analysis of COX2, PGE2, and cytokine responses.
Analysis of COX2, PGE2, and cytokine responses
[0294] COX2 and PGE2 responses were analyzed as described in Example 3.
Cytokine
responses were analyzed as described in Example 2.
[0295] RESULTS
[0296] Analgesics inhibit COX2 responses of normal human bladder smooth muscle
cells
to inflammatory and non- inflammatory stimuli - Analysis of cells and culture
supernatants after
24 hours of cultures showed that none of the analgesics tested alone induced
COX2 responses in
normal human bladder smooth muscle cells. However, as summarized in Table 6,
carbachol
induced low, but significant COX2 responses in normal human bladder smooth
muscle cells. On
the other hand, LPS treatment resulted in higher levels of COX2 responses in
normal human
bladder smooth muscle cells. Acetaminophen, aspirin, ibuprofen, and naproxen
could all suppress
the effect of carbachol and LPS on COX2 levels. The suppressive effect of the
analgesics was
seen on LPS-induced responses when these drugs were tested at either 5 M or
50 M.
68
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
Table 6. COX2 expression by normal human bladder smooth muscle cells after in
vitro
stimulation with inflammatory and non- inflammatory stimuli and treatment with
analgesic
Total COX2 levels Total COX2 levels
Stimulus Analgesic (Normalized RFUs) (Normalized RFUs)
subject 1 subject 2
None None 230 199
Carbachol 10-3M None (50 M) 437 462
Carbachol 10-3 M Acetaminophen (50 M) 298 310
Carbachol 10-3M Aspirin (50 M) 312 297
Carbachol 10-3M Ibuprofen (50 M) 309 330
Carbachol 10-3M Naproxen (50 M) 296 354
LPS (10 gimp None 672 633
LPS (10 gimp Acetaminophen (5 M) 428 457
LPS (10 gimp Aspirin (5 M) 472 491
LPS (10 gimp Ibuprofen (5 M) 417 456
LPS (10 gimp Naproxen (5 M 458 501
LPS (10 gimp Acetaminophen (50 M) 399 509
LPS (10 gimp Aspirin (50 M) 413 484
LPS (10 gimp Ibuprofen (50 M) 427 466
LPS (10 gimp Naproxen (50 M) 409 458
#13ata are expressed as mean of duplicates
[0297] Analgesics inhibit PGE2 responses of normal human bladder smooth muscle
cells
to inflammatory and non- inflammatory stimuli - Consistent with the induction
of COX2
responses described above, both carbachol and LPS induced production of PGE2
by normal
human bladder smooth muscle cells. Acetaminophen, aspirin, ibuprofen, and
naproxen were also
found to suppress the LPS-induced PGE2 responses at either 5 .1\4 or 50 .1\4
(Table 7).
69
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
Table 7. PGE2 secretion by normal human bladder smooth muscle cells after in
vitro stimulation
with inflammatory and non- inflammatory stimuli and treatment with analgesic
Stimulus Analgesic PGE2 levels (pg/ml) PGE2 levels
(pg/ml)
Subject 1 Subject 2
None None <20.5 <20.5
Carbachol 10-3M None 129 104
Carbachol 10-3 M Acetaminophen (50 M) 76 62
Carbachol 10-3M Aspirin (50 M) 89 59
Carbachol 10-3M Ibuprofen (50 M) 84 73
Carbachol 10-3M Naproxen (50 M) 77 66
LPS (10 gimp None 1125 998
LPS (10 gimp Acetaminophen (5 M) 817 542
LPS (10 gimp Aspirin (5 M) 838 598
LPS (10 gimp Ibuprofen (5 M) 824 527
LPS (10 gimp Naproxen (5 M 859 506
LPS (10 gimp Acetaminophen (50 M) 803 540
LPS (10 gimp Aspirin (50 M) 812 534
LPS (10 gimp Ibuprofen (50 M) 821 501
LPS (10 gimp Naproxen (50 M) 819 523
#Data are expressed as mean of duplicates
[0298] Analgesics inhibit cytokine responses of normal human bladder cells to
inflammatory stimuli - Analysis of cells and culture supernatants after 24
hours of culture showed
that none of the analgesics tested alone induced IL-6 or TNFa secretion in
normal human bladder
smooth muscle cells. As shown in Tables 8 and 9, the doses of carbachol tested
induced low, but
significant TNFa and IL-6 responses in normal human bladder smooth muscle
cells. On the other
hand, LPS treatment resulted in massive induction of these proinflammatory
cytokines.
Acetaminophen, aspirin, ibuprofen, and naproxen suppress the effect of
carbachol and LPS on
TNFa and IL-6 responses. The suppressive effect of the analgesics on LPS-
induced responses
was seen when these drugs were tested at either 5 M or 50 M.
Table 8. TNFa secretion by normal human bladder smooth muscle cells after in
vitro stimulation
with inflammatory and non- inflammatory stimuli and treatment with analgesic
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
Stimuli Analgesic TNFoc (pg/ml) # TNFoc (pg/ml)
Subject 1 Subject 2
None None <5 <5
Carbachol 10-3M None 350 286
Carbachol 10-3 M Acetaminophen (50 M) 138 164
Carbachol 10-3M Aspirin (50 M) 110 142
Carbachol 10-3M Ibuprofen (50 M) 146 121
Carbachol 10-3M Naproxen (50 M) 129 137
LPS (10 g/m1) None 5725 4107
LPS (10 g/m1) Acetaminophen (5 M) 2338 2267
LPS (10 g/m1) Aspirin (5 M) 2479 2187
LPS (10 g/m1) Ibuprofen (5 M) 2733 2288
LPS (10 g/m1) Naproxen (5 M 2591 2215
LPS (10 g/m1) Acetaminophen (50 M) 2184 2056
LPS (10 g/m1) Aspirin (50 M) 2266 2089
LPS (10 g/m1) Ibuprofen (50 M) 2603 1997
LPS (10 g/m1) Naproxen (50 M) 2427 2192
#Data are expressed as mean of duplicates.
Table 9. IL-6 secretion by normal human bladder smooth muscle cells after in
vitro stimulation
with inflammatory and non- inflammatory stimuli and treatment with analgesic
Stimulus Analgesic IL-6 (pg/ml) # Subject 1 IL-6 (pg/ml)
Subject 2
None None <5 <5
Carbachol 10-3M None 232 278
Carbachol 10-3 M Acetaminophen (50 M) 119 135
Carbachol 10-3M Aspirin (50 M) 95 146
Carbachol 10-3M Ibuprofen (50 M) 107 118
Carbachol 10-3M Naproxen (50 M) 114 127
LPS (10 g/m1) None 4838 4383
LPS (10 g/m1) Acetaminophen (5 M) 2012 2308
LPS (10 g/m1) Aspirin (5 M) 2199 2089
LPS (10 g/m1) Ibuprofen (5 M) 2063 2173
LPS (10 g/m1) Naproxen (5 M 2077 2229
LPS (10 g/m1) Acetaminophen (50 M) 2018 1983
LPS (10 g/m1) Aspirin (50 M) 1987 2010
LPS (10 g/m1) Ibuprofen (50 M) 2021 1991
LPS (10 g/m1) Naproxen (50 M) 2102 2028
Data are expressed as mean of duplicates
71
CA 03008628 2018-06-14
WO 2017/106032 PCT/US2016/065775
[0299] Primary normal human bladder smooth muscle cells were isolated,
cultured and
evaluated for their responses to analgesics in the presence of non-
inflammatory (carbachol) and
inflammatory (LPS) stimuli. The goal of this study was to determine whether or
not normal
human bladder smooth muscle cells recapitulate the observations previously
made with murine
bladder cells.
[0300] The above-described experiment will be repeated with analgesic agents
and/or
antimuscarinic agents in delayed-release, or extended-release formulation or
delayed-and-
extended-release formulations.
[0301] The above description is for the purpose of teaching a person of
ordinary skill in
the art how to practice the present invention, and it is not intended to
detail all those obvious
modifications and variations of it which will become apparent to the skilled
worker upon reading
the description. It is intended, however, that all such obvious modifications
and variations be
included within the scope of the present invention, which is defined by the
following claims. The
claims are intended to cover the claimed components and steps in any sequence
which is effective
to meet the objectives there intended, unless the context specifically
indicates the contrary.
72