Note: Descriptions are shown in the official language in which they were submitted.
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
METHODS AND SYSTEMS FOR RAPID DETECTION OF MICROORGANISMS
USING INFECTIOUS AGENTS
RELATED APPLICATIONS
The present application claims priority to U.S. Provisional Patent Application
No.
62/280,043. filed January 18. 2016 and U.S. Provisional Patent Application No.
621280,465.
filed January 19, 2016. This application also claims priority to U.S.
Application No.
15/263.619. filed September 13, 2016.
REFERENCE TO A SEQUENCE LISTING SUBMITTED AS
A TEXT FILE VIA EFS-WEB
The official copy of the sequence listing is submitted electronically via EFS-
Web as
an ASCII formatted sequence listing with a file named 1035526_ST25.txt,
created on January
17, 2017. and having a size of 7 kilobytes and is filed concurrently with the
specification.
The sequence listing contained in this ASCII formatted document is part of the
specification
and is herein incorporated by reference in its entirety.
FIELD OF THE INVENTION
This invention relates to methods and systems for the detection of
microorganisms
using infectious agents.
BACKGROUND
There is a strong interest in improving speed and sensitivity for detection of
bacteria.
viruses, and other microorganisms in biological, food, water, and clinical
samples. Microbial
pathogens can cause substantial morbidity among humans and domestic animals,
as well as
immense economic loss. Also, detection of microorganisms is a high priority
for the Food
and Drug Administration (FDA) and Centers for Disease Control (CDC) given
outbreaks of
life-threatening or fatal illness caused by ingestion of food contaminated
with certain
microorganisms. e.g.. Escherichla col) or Salmonella spp.
Traditional microbiological tests for the detection of bacteria rely on non-
selective
and selective enrichment cultures followed by plating on selective media and
further testing
to confirm suspect colonies. Such procedures can require several days. A
variety of rapid
methods have been investigated and introduced into practice to reduce the time
requirement.
HON\ ever, these methods have drawbacks. For example. techniques involving
direct
immunoassays or gene probes generally require an overnight enrichment step in
order to
1
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
obtain adequate sensitivity. Polymerase chain reaction (PCR) tests also
include an
amplification step and therefore are capable of both very high sensitivity and
selectivity:
however, the sample size that can be economically subjected to PCR testing is
limited. With
dilute bacterial suspensions. most small subsamples will be free of cells and
therefore
purification and/or lengthy enrichment steps are still required.
The time required for traditional biological enrichment is dictated by the
growth rate
of the target bacterial population of the sample, by the effect of the sample
matrix, and by the
required sensitivity. In practice, most high sensitivity methods employ an
overnight
incubation and take about 24 hours overall. Due to the time required for
cultivation, these
methods can take up to three days. depending upon the organism to be
identified and the
source of the sample. This lag time is generally unsuitable as the
contaminated food, water
(or other product) may have already made its way into livestock or humans. In
addition,
increases in antibiotic-resistant bacteria and biodefense considerations make
rapid
identification of bacterial pathogens in water, food and clinical samples
critical priorities
w orldw ide.
Therefore, there is a need for more rapid, simple and sensitive detection and
identification of microorganisms. such as bacteria and other potentially
pathogenic
microorganisms.
SUMMARY
Embodiments of the invention comprise compositions. methods, systems and kits
for
the detection of microorganisms. The invention may be embodied in a variety of
ways.
In some aspects, the invention comprises a recombinant bacteriophage
comprising an
indicator gene inserted into a late gene region of a bacteriophage genome. In
some
embodiments the recombinant bacteriophage is a genetically modified CBA120
genome. In
some embodiments the recombinant bacteriophage is a genetically modified T4-
like or Vi!-
like bacteriophage genome. In some embodiments the recombinant bacteriophage
specifically infects E. coli 0157:H7. In an embodiment. the recombinant
bacteriophage can
distinguish E. coil 0157:H7 in the presence of more than 100 other types of
bacteria.
In some embodiments of recombinant indicator bacteriophage, the indicator gene
can
be codon-optimized and can encode a soluble protein product that generates an
intrinsic
signal or a soluble enzyme that generates signal upon reaction with substrate.
Some
recombinant bacteriophage further comprise an untranslated region upstream of
a codon-
2
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
optimized indicator gene, wherein the untranslated region includes a
bacteriophage late gene
promoter and a ribosomal entry site. In some embodiments, the indicator gene
is a luciferase
gene. The luciferase gene can be a naturally occurring gene, such as
Oplophonis luciferase,
Firefly luciferase. Lucia luciferase. or Renilla luciferase. or it can be a
genetically engineered
gene.
Also disclosed herein are methods for preparing a recombinant indicator
bacteriophage. Some embodiments include selecting a wild-type bacteriophage
that
specifically infects a target pathogenic bacterium: preparing a homologous
recombination
plasmid/vector comprising an indicator gene: transforming the homologous
recombination
plasmid/vector into target pathogenic bacteria: infecting the transformed
target pathogenic
bacteria with the selected wild-type bacteriophage, thereby allowing
homologous
recombination to occur between the plasmid/vector and the bacteriophage
genome: and
isolating a particular clone of recombinant bacteriophage. In some embodiments
the selected
wild-type bacteriophage is CBA120. In some embodiments the selected wild-type
bacteriophage is or Vil-like.
In some embodiments, preparing a homologous recombination plasmid/vector
includes determining the natural nucleotide sequence in the late region of the
genome of the
selected bacteriophage: annotating the genome and identifying the major capsid
protein gene
of the selected bacteriophage: designing a sequence for homologous
recombination
downstream of the major capsid protein gene, wherein the sequence comprises a
codon-
optimized indicator gene: and incorporating the sequence designed for
homologous
recombination into a plasmid/vector. The step of designing a sequence can
include inserting
an untranslated region, including a phage late gene promoter and ribosomal
entry site,
upstream of the codon-optimized indicator gene. Thus in some methods the
homologous
recombination plasmid comprises an untranslated region including a
bacteriophage late gene
promoter and a ribosomal entry site upstream of the codon-optimized indicator
gene.
Some embodiments of the invention are compositions that include a recombinant
indicator bacteriophage as described herein. For example, compositions can
include one or
more wild-type or genetically modified infectious agents (e.g..
bacteriophages) and one or
more indicator genes. In some embodiments, compositions can include cocktails
of different
indicator phages that may encode and express the same or different indicator
proteins.
3
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
In some embodiments. the invention comprises a method for detecting a
microorganism of interest in a sample comprising the steps of incubating the
sample with a
recombinant bacteriophage that infects the microorganism of interest, wherein
the
recombinant bacteriophage comprises an indicator gene inserted into a late
gene region of the
bacteriophage such that expression of the indicator gene during bacteriophage
replication
following infection of host bacteria results in a soluble indicator protein
product, and
detecting the indicator protein product. wherein positive detection of the
indicator protein
product indicates that the microorganism of interest is present in the sample.
In some embodiments of methods for preparing recombinant indicator
bacteriophage,
the wild-type bacteriophage is CBA120 and the target pathogenic bacterium is
E. coil
0157:H7. In some embodiments, isolating a particular clone of recombinant
bacteriophage
comprises a limiting dilution assay for isolating a clone that demonstrates
expression of the
indicator gene.
Other aspects of the invention include methods for detecting bacteria, such as
E. coil
0157:H7. in a sample, including steps of incubating the sample with a
recombinant
bacteriophage derived from CBA120 and detecting an indicator protein product
produced by
the recombinant bacteriophage, wherein positive detection of the indicator
protein product
indicates that E. coil 0157:H7 is present in the sample. The sample can be a
food,
environmental, water. commercial. or clinical sample. In some embodiments, the
sample
comprises beef or vegetables.
In some embodiments of methods for detecting bacteria, the sample is first
incubated
in conditions favoring growth for an enrichment period of 9 hours or less. 8
hours or less. 7
hours or less, 6 hours or less. 5 hours or less, 4 hours or less. 3 hours or
less. or 2 hours or
less. In some embodiments, the total time to results is less than 12 hours,
less than 11 hours,
less than 10 hours, less than 9 hours, less than 8 hours, less than 7 hours,
or less than 6 hours.
In some embodiments, the ratio of signal to background generated by detecting
the indicator
is at least 2.0 or at least 2.5. In some embodiments, the method detects as
few as 1, 2. 3, 4, 5,
6, 7, 8, 9, 10. 15. 20. 30. 40. 50, 60, 70. 80, 90, or 100 of the specific
bacteria in a sample of a
standard size for the food safety industry.
Additional embodiments include systems and kits for detecting E. coil 0157:H7.
wherein the systems or kits include a recombinant bacteriophage derived from
CBA120.
Some embodiments further include a substrate for reacting with an indicator to
detect the
4
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
soluble protein product expressed by the recombinant bacteriophage. These
systems or kits
can include features described for the bacteriophage, compositions, and
methods of the
invention. In still other embodiments, the invention comprises non-transient
computer
readable media for use with methods or systems according to the invention.
BRIEF DESCRIPTION OF THE FIGURES
The present invention may be better understood by referring to the following
non-
limiting figures.
Figure 1 shovvs a portion of the genome of the wild-type CBA120 bacteriophage
and
the annotated late gene region in particular.
Figure 2 shows one embodiment of a plasmid designed for homologous
recombination with the CBA120 bacteriophage genome. Capsid protein gp23
(0RF187) is
believed to represent the major capsid protein. As this virion protein is
expressed at a very
high level, any genes inserted into this region can be expected to have
similar expression
levels, as long as late gene promoters and/or other similar control elements
are used.
Figure 3 sholys an embodiment of homologous recombination of the wild-type
CBA120 genome in Figure 1 with the plasmid illustrated in Figure 2.
Figure 4 depicts the isolation of recombinant bacteriophage from a mixture of
wild-
type and recombinant bacteriophage derived from transforming target bacteria
with a plasmid
carrying a sequence designed to recombine in homologous fashion with the
natural
bacteriophage genome, and then infecting the transformed bacteria with wild-
type
bacteriophage to allow homologous recombination. A series of sequential
infection and
dilution steps allow identification and isolation of recombinant phage that
expresses an
indicator/reporter gene.
Figure 5 is an electron micrograph of one embodiment of a recombinant
indicator
bacteriophage. the CBA120NanoLuc bacteriophage.
Figure 6 depicts the use of indicator bacteriophage encoding a soluble
reporter (e.g..
luciferase) to detect bacterial cells via detection of luciferase generated
from replication of
indicator bacteriophage during infection of the bacterial cells, according to
an embodiment of
the imention.
Figure 7 demonstrates the detection of pathogenic bacteria using different
phage
concentrations of CBA120NanoLuc for infecting samples with known numbers of
cells, with
106 phage/mL yielding the highest signal to background ratio.
5
CA 03011704 2018-07-17
WO 2017/127-134
PCT/US2017/013955
Figure 8 demonstrates that replicates of experiments using 106 phagelmL
CBA120NanoLuc for infecting samples with known numbers of cells show
significant
differences between signal from a single cell and signal from 0 cells. 2
cells, or more.
Figure 9 demonstrates that the signal to background ratio for the experiment
shown in
Figure 8 is greater than 2Ø
Figure 10 shows Relative Light Units (RLU) and signal to background ratios for
detection of E. coil 0157:H7 in a 1 mL concentration sample from 25 g ground
beef when the
assay is conducted after 5, 6, and 7 hours of enrichment.
Figure 11 summarizes detection of E. coli 0157:H7 in a I mL concentration
sample
from 25 g ground beef as shown in Figure 10 with confirmation of the results
using a
secondary method.
Figure 12 shows RLU and signal to background ratios for detection of E. coil
0157:H7 in a 10 mL concentration sample from 25 g ground beef when the assay
is
conducted after 5 hours of enrichment with confirmation of the results using a
secondary
.. method.
Figure 13 shows RLU and signal to background ratios for detection of E. coil
0157:H7 in 1 mL concentration samples from 125 g beef trim when the assay is
conducted
after 7, 8. and 9 hours of enrichment.
Figure 14 shows RLU and signal to background ratios for detection of E. coil
.. 0157:H7 in 10 mL concentration samples from 125 g beef trim when the assay
is conducted
after 7. 8. and 9 hours of enrichment.
Figure 15 summarizes detection of E. coil 0157:H7 in 1 mL concentration
samples
from 125 g beef trim as shown in Figure 13 with confirmation of the results
using a
secondary method.
Figure 16 summarizes detection of E. coil 0157:H7 in 10 mL concentration
samples
from 125 g beef trim as shown in Figure 14 with confirmation of the results
using a
secondary method.
Figure 17 shows RLU and signal to background ratios for detection of E. coil
0157:H7 in 100 mL spinach wash filtered and subjected to a filter assay format
with
.. confirmation of the results using a secondary method.
6
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
DETAILED DESCRIPTION OF THE INVENTION
Disclosed herein are compositions, methods and systems that demonstrate
surprising
sensitivity for detection of a microorganism of interest in test samples
(e.g., biological, food,
water, and clinical samples). Detection can be achieved in a shorter timeframe
than was
previously thought possible using genetically modified infectious agents in
assays performed
without culturing for enrichment, or in some embodiments with minimal
incubation times
during which microorganisms could potentially multiply. Also surprising is the
success of
using a potentially high multiplicity of infection (MOI). or high
concentrations of plaque
forming units (PFU), for incubation with a test sample. Such high phage
concentrations
(PFU/mL) were previously purported to be detrimental in bacterium detection
assays. as they
were purported to cause -lysis from without.- However, a high concentration of
phage can
facilitate finding, binding, and infecting a low number of target cells.
The compositions. methods. systems and kits of the invention may comprise
infectious agents for use in detection of such microorganisms. In certain
embodiments. the
im ention may comprise a composition comprising a recombinant bacteriophage
having an
indicator gene inserted into a late gene region of the bacteriophage. In
certain embodiments,
expression of the indicator gene during bacteriophage replication following
infection of a
host bacterium results in production of a soluble indicator protein product.
In certain
embodiments. the indicator gene may be inserted into a late gene (i.e., class
III) region of the
bacteriophage. The bacteriophage can be derived from T7, T4. T4-like. Vii. ViI-
like (or Vil
virus. per GenBankINCBI). CBA120. or another wild-type or engineered
bacteriophage.
In some aspects. the invention comprises a method for detecting a
microorganism of
interest. The method may use an infectious agent for detection of the
microorganism of
interest. For example. in certain embodiments, the microorganism of interest
is a bacterium
and the infectious agent is a bacteriophage. Thus, in certain embodiments, the
method may
comprise detection of a bacterium of interest in a sample by incubating the
sample with a
recombinant bacteriophage that infects the bacterium of interest. In certain
embodiments, the
recombinant bacteriophage comprises an indicator gene. The indicator gene may,
in certain
embodiments, be inserted into a late gene region of the bacteriophage such
that expression of
the indicator gene during bacteriophage replication following infection of
host bacteria
results in production of an indicator protein product. The method may comprise
detecting the
indicator protein product, wherein positive detection of the indicator protein
product indicates
7
CA 03011704 2018-07-17
WO 2017/127-134
PCT/US2017/013955
that the bacterium of interest is present in the sample. In some embodiment
the indicator
protein is soluble.
In certain embodiments, the invention may comprise a system. The system may
contain at least some of the compositions of the invention. Also, the system
may comprise at
least some of the components for performing the method. In certain
embodiments, the
system is formulated as a kit. Thus, in certain embodiments, the invention may
comprise a
system for rapid detection of a microorganism of interest in a sample,
comprising: a
component for incubating the sample with an infectious agent specific for the
microorganism
of interest, wherein the infectious agent comprises an indicator moiety; and a
component for
detecting the indicator moiety. In vet other embodiments, the invention
comprises software
for use with the methods or systems.
Thus, some embodiments of the present invention solve a need by using
bacteriophage-based methods for amplifying a detectable signal indicating the
presence of
bacteria. In certain embodiments as little as a single bacterium is detected.
The principles
applied herein can be applied to the detection of a variety of microorganisms.
Because of
numerous binding sites for an infectious agent on the surface of a
microorganism. the
capacity to produce one hundred or more agent progeny during infection, and
the potential
for high level expression of an encoded indicator moiety, the infectious agent
or an indicator
moiety can be more readily detectable than the microorganism itself. In this
embodiments of the present invention can achieve tremendous signal
amplification from even
a single infected cell.
Aspects of the present invention utilize the high specificity of binding
agents that can
bind to particular microorganisms, such as the binding component of infectious
agents, as a
means to detect and/or quantify the specific microorganism in a sample. In
some
embodiments, the present invention utilizes the high specificity of infectious
agents such as
bacteriophage.
In some embodiments, detection is achieved through an indicator moiety
associated
NV -ith the binding agent specific for the microorganism of interest. For
example, an infectious
agent may comprise an indicator moiety. such as a gene encoding a soluble
indicator. In
some embodiments the indicator may be encoded by the infectious agent, such as
a
bacteriophage. and the bacteriophage is designated an indicator phage.
8
CA 03011704 2018-07-17
WO 2017/127-134
PCT/US2017/013955
Some embodiments of the invention disclosed and described herein utilize the
discovery that a single microorganism is capable of binding specific
recognition agents, such
as phage. Following infection and replication of the phage, progeny phage may
be detected
via an indicator moiety expressed during phage replication. This principle
allows
amplification of indicator signal from one or a few cells based on specific
recognition of
microorganism surface receptors. For example, by exposing even a single cell
of a bacterium
to a plurality of phage, thereafter allowing amplification of the phage and
high-level
expression of an encoded indicator gene product during replication, the
indicator signal is
amplified such that the single bacterium is detectable.
Embodiments of the methods and systems of the invention can be applied to
detection
and quantification of a variety of microorganisms (e.g., bacteria, fungi.
yeast) in a variety of
circumstances, including but not limited to detection of pathogens from food,
water, clinical
and commercial samples. The methods of the present invention provide high
detection
sensitivity and specificity rapidly and without the need for traditional
biological enrichment
(e.g.. culturing for enrichment), which is a surprising aspect as all
available methods require
culturing. In some embodiments detection is possible within a single
replication cycle of the
bacteriophage, which is unexpected.
Definitions
Unless otherwise defined herein, scientific and technical terms used in
connection
with the present invention shall have the meanings that are commonly
understood by those of
ordinary skill in the art. Further, unless otherwise required by context,
singular terms shall
include pluralities and plural terms shall include the singular. Generally,
nomenclatures used
in connection with, and techniques of, cell and tissue culture, molecular
biology.
immunolo, microbiology, genetics and protein and nucleic acid chemistry and
hybridization described herein are those well known and commonly used in the
art. Known
methods and techniques are generally performed according to conventional
methods well
known in the art and as described in various general and more specific
references that are
discussed throughout the present specification unless othenyise indicated.
Enzymatic
reactions and purification techniques are performed according to
manufacturer's
specifications, as commonly accomplished in the art or as described herein.
The
nomenclatures used in connection with the laboratory procedures and techniques
described
herein are those well-known and commonly used in the art.
9
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
The following terms, unless otherwise indicated, shall be understood to have
the
following meanings:
As used herein, the terms "a", "an", and "the" can refer to one or more unless
specifically noted otherwise.
The use of the term -or" is used to mean "and/or" unless explicitly indicated
to refer
to alternatives only or the alternatives are mutually exclusive, although the
disclosure
supports a definition that refers to only alternatives and "and/or.- As used
herein -another"
can mean at least a second or more.
Throughout this application, the term "about- is used to indicate that a value
includes
the inherent variation of error for the device, the method being employed to
determine the
value, or the variation that exists among samples.
The term "solid support- or "support- means a structure that provides a
substrate
and/or surface onto which biomolecules may be bound. For example, a solid
support may be
an assay well (i.e., such as a microtiter plate or multi-well plate), or the
solid support may be
a location on a filter, an array, or a mobile support, such as a bead or a
membrane (e.g., a
filter plate or lateral flow strip).
The term "binding agent" refers to a molecule that can specifically and
selectively
bind to a second (i.e., different) molecule of interest. The interaction may
be non-covalent,
for example, as a result of hydrogen bonding, van der Waals interactions, or
electrostatic or
hydrophobic interactions, or it may be covalent. The term "soluble binding
agent- refers to a
binding agent that is not associated with (i.e., covalently or non-covalently
bound) to a solid
support.
As used herein, an "analyte- refers to a molecule, compound or cell that is
being
measured. The analyte of interest may, in certain embodiments, interact with a
binding agent.
As described herein, the term "analyte" may refer to a protein or peptide of
interest. An
anaMe may be an agonist, an antagonist, or a modulator. Or, an analyte may not
have a
biological effect. Analytes may include small molecules, sugars,
oligosaccharides, lipids,
peptides, peptidomimetics, organic compounds and the like.
The term "detectable moiety" or "detectable biomolecule" or "reporter" or
"indicator"
or "indicator moiety- refers to a molecule that can be measured in a
quantitative assay. For
example, an indicator moiety may comprise an enzyme that may be used to
convert a
substrate to a product that can be measured. An indicator moiety may be an
enzyme that
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
catalyzes a reaction that generates bioluminescent emissions (e.g..
luciferase). Or, an
indicator moiety may be a radioisotope that can be quantified. Or, an
indicator moiety may
be a fluorophore. Or, other detectable molecules may be used.
As used herein, "bacteriophage- or -phage- includes one or more of a plurality
of
bacterial viruses. In this disclosure, the terms "bacteriophage- and "phage-
include viruses
such as mycobacteriophage (such as for TB and paraTB), mycophage (such as for
fungi),
mycoplasma phage. and any other term that refers to a virus that can invade
living bacteria,
fungi. mycoplasma, protozoa. yeasts, and other microscopic living organisms
and uses them
to replicate itself. Here, "microscopic- means that the largest dimension is
one millimeter or
less. Bacteriophages are viruses that have evolved in nature to use bacteria
as a means of
replicating themselves. A phage does this by attaching itself to a bacterium
and injecting its
DNA (or RNA) into that bacterium, and inducing it to replicate the phage
hundreds or even
thousands of times. This is referred to as phage amplification.
As used herein. "late gene region- refers to a region of a viral genome that
is
transcribed late in the viral life cycle. The late gene region typically
includes the most
abundantly expressed genes (e.g., structural proteins assembled into the
bacteriophage
particle). Late genes are synonymous with class III genes and include genes
with structure
and assembly functions. For example. the late genes (synonymous with class
111.) are
transcribed in phage T7, e.g.. from 8 minutes after infection until lysis.
class I (e.g., RNA
polymerase) is early from 4-8 minutes. and class II from 6-15 minutes, so
there is overlap in
timing of II and III. A late promoter is one that is naturally located and
active in such a late
gene region.
As used herein. "culturing for enrichment- refers to traditional culturing,
such as
incubation in media favorable to propagation of microorganisms. and should not
be confused
with other possible uses of the word "enrichment,- such as enrichment by
removing the
liquid component of a sample to concentrate the microorganism contained
therein, or other
forms of enrichment that do not include traditional facilitation of
microorganism propagation.
Culturing for enrichment for very short periods of time may be employed in
some
embodiments of methods described herein, but is not necessary and is for a
much shorter
period of time than traditional culturing for enrichment, if it is used at
all.
11
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
As used herein "recombinant" refers to genetic (i.e., nucleic acid)
modifications as
usually performed in a laboratory to bring together genetic material that
would not otherwise
be found. This term is used interchangeably with the term "modified" herein.
As used herein "RLU" refers to relative light units as measured by a
luminometer
(e.g., GLOMAX:k. 96) or similar instrument that detects light. For example,
the detection of
the reaction between luciferase and appropriate substrate (e.g.. NANOLUCt with
NANO-
GLOlit) is often reported in RLU detected.
As used herein "time to results" refers to the total amount of time from
beginning of
sample preparation to the collection of data. Time to results does not include
any
confirmatory testing time.
Samples
Each of the embodiments of the methods and systems of the invention can allow
for
the rapid detection and quantification of microbes in a sample. For example,
methods
according to the present invention can be performed in a shortened time period
with superior
results.
Microbes detected by the methods and systems of the present invention include
pathogens that are of natural. commercial, medical or veterinary concern. Such
pathogens
include Gram-negative bacteria, Gram-positive bacteria, mycoplasmas and
viruses. Any
microbe for which an infectious agent that is specific for the particular
microbe has been
identified can be detected by the methods of the present invention. Those
skilled in the art
will appreciate that there is no limit to the application of the present
methods other than the
availability of the necessary specific infectious agent/microbe pairs.
Bacterial cells detectable by the present invention include, but are not
limited to,
bacterial cells that are food or water borne pathogens. Bacterial cells
detectable by the
present invention include, but are not limited to, all species of Salmonella,
all strains of
Kscherichia coli, including, but not limited to E. co/i 0157:H7, all species
of Lisleria,
including, but not limited to L. monocytogenes, and all species of
Campylohacter. Bacterial
cells detectable by the present invention include, but are not limited to,
bacterial cells that are
pathogens of medical or veterinary significance. Such pathogens include, but
are not limited
to, Bacillus spp.. Bordetella pertussis, Cainplvohacterjejuni. Chltunydia
pneurnoniae.
Clostridium per.fringens, Enterobacter spp.. Klehsiella pneumoniae.
114.ycoplasma
12
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
pneumoniac, Salmonella typhi. Shigella sonnet, Staphylococcus attreits., and
Streptococcus.
App.
The sample may be an environmental or food or water sample. Some embodiments
may include medical or veterinary samples. Samples may be liquid, solid, or
semi-solid.
Samples may be swabs of solid surfaces. Samples may include environmental
materials,
such as the water samples, or the filters from air samples or aerosol samples
from cyclone
collectors. Samples may be of meat, poultry, processed foods, milk. cheese, or
other dairy
products. Medical or veterinary samples include, but are not limited to.
blood, sputum.
cerebrospinal fluid, and fecal samples and different types of swabs.
In some embodiments, samples may be used directly in the detection methods of
the
present invention, without preparation, concentration, or dilution. For
example, liquid
samples, including but not limited to, milk and juices, may be assayed
directly. Samples may
be diluted or suspended in solution, which may include, but is not limited to.
a buffered
solution or a bacterial culture medium. A sample that is a solid or semi-solid
may be
suspending in a liquid by mincing, mixing or macerating the solid in the
liquid. A sample
should be maintained within a pH range that promotes bacteriophage attachment
to the host
bacterial cell. A sample should also contain the appropriate concentrations of
divalent and
monovalent cations. including but not limited to Nat. Mg2f, and K. Preferably
a sample is
maintained at a temperature that maintains the viability of any pathogen cells
contained
within the sample.
Preferably throughout detection assays. the sample is maintained at a
temperature that
maintains the viability of any pathogen cell present in the sample. During
steps in which
bacteriophages are attaching to bacterial cells. it is preferable to maintain
the sample at a
temperature that facilitates bacteriophage attachment. During steps in which
bacteriophages
are replicating within an infected bacterial cell or lysing such an infected
cell, it is preferable
to maintain the sample at a temperature that promotes bacteriophage
replication and lysis of
the host. Such temperatures are at least about 25 degrees Celsius (C), more
preferably no
greater than about 45 degrees C. most preferably about 37 degrees C. It is
also preferred that
the samples be subjected to gentle mixing or shaking during bacteriophage
attachment.
replication and cell lysis.
13
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
Assays may include various appropriate control samples. For example. control
samples containing no bacteriophages or control samples containing
bacteriophages without
bacteria may be assayed as controls for background signal levels.
Indicator Bacteriophage
As described in more detail herein, the compositions, methods, systems and
kits of the
invention may comprise infectious agents for use in detection of pathogenic
microorganisms.
In certain embodiments, the invention comprises a recombinant indicator
bacteriophage.
wherein the bacteriophage genome is genetically modified to include an
indicator or reporter
gene. In some embodiments, the invention may include a composition comprising
a
recombinant bacteriophage having an indicator gene incorporated into the
genome of the
bacteriophage.
A recombinant indicator bacteriophage can include a reporter or indicator
gene. In
certain embodiments of the infectious agent, the indicator gene does not
encode a fusion
protein. For example, in certain embodiments, expression of the indicator gene
during
bacteriophage replication following infection of a host bacterium results in a
soluble indicator
protein product. In certain embodiments. the indicator gene may be inserted
into a late gene
region of the bacteriophage. Late genes are generally expressed at higher
levels than other
phage genes, as they code for structural proteins. The late gene region may be
a class III
gene region and may include a gene for a major capsid protein.
Some embodiments include designing (and optionally preparing) a sequence for
homologous recombination downstream of the major capsid protein gene. In some
embodiments, the sequence comprises a codon-optimized reporter gene preceded
by an
untranslated region. The untranslated region may include a phage late gene
promoter and
ribosomal entry site.
In some embodiments, an indicator bacteriophage is derived from T7. T4 or
another
similar phage. An indicator bacteriophage may also be derived from T4-like. T7-
like. Vil.
Vi 1-like. CBA 120, or another bacteriophage having a genome with at least 70,
71, 72, 73, 74.
75. 76. 77. 78, 79, 80, 81, 82, 83, 84. 85. 86. 87, 88, 89, 90. 91, 92. 93.
94. 95. 96. 97, 98. or
99 <!,O homology to T7, T7-like, T4, T4-like, CBA120, Vii. or Vil-like (or Vii
virus-like. per
GenBank,NCBI) bacteriophages. In some embodiments, the indicator phage is
derived
from a bacteriophage that is highly specific for a particular pathogenic
microorganism. The
genetic modifications may avoid deletions of wild-type genes and thus the
modified phage
14
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
may remain more similar to the wild-type infectious agent than many
commercially available
phage. Environmentally derived bacteriophage may be more specific for bacteria
that are
found in the environment and as such. genetically distinct from phage
available
commercially.
Moreover. phage genes thought to be nonessential may have unrecognized
function.
For example, an apparently nonessential gene may have an important function in
elevating
burst size such as subtle cutting. fitting, or trimming functions in assembly.
Therefore,
deleting genes to insert an indicator may be detrimental. Most phages can
package a DNA
that is a few percent larger than their natural genome. With this
consideration, a smaller
indicator gene may be a more appropriate choice for modifying a bacteriophage,
especially
one with a smaller genome. OpLuc and NANOLUC,*: proteins are only about 20 kDa
(approximately 500-600 bp to encode). while FLuc is about 62 kDa
(approximately 1,700 bp
to encode). For comparison, the genome of T7 is around 40 kbp, while the T4
genome is
about 170 kbp. and the genome of CBA120 is about 157 kbp. Moreover, the
reporter gene
should not be expressed endogenously by the bacteria (i.e.. is not part of the
bacterial
genome), should generate a high signal to background ratio, and should be
readily detectable
in a timely manner. Promega's NANOLUCK: is a modified Oplophorus
gracilirostris (deep
sea shrimp) luciferase. In some embodiments. NANOLUC r< combined with
Promega's
NANO-GLOtz), an imidazopyrazinone substrate (furimazine), can provide a robust
signal
with low background.
In some indicator phage embodiments. the indicator gene can be inserted into
an
untranslated region to avoid disruption of functional genes. leaving wild-type
phage genes
intact, which may lead to greater fitness when infecting non-laboratory
strains of bacteria.
Additionally, including stop codons in all three reading frames may help to
increase
expression by reducing read-through, also known as leaky expression. This
strategy may also
eliminate the possibility of a fusion protein being made at low levels, which
would manifest
as background signal (e.g.. luciferase) that cannot be separated from the
phage.
An indicator gene may express a variety of biomolecules. The indicator gene is
a gene
that expresses a detectable product or an enzyme that produces a detectable
product. For
example, in one embodiment the indicator gene encodes a luciferase enzyme.
Various types
of luciferase may be used. In alternate embodiments, and as described in more
detail herein.
the luciferase is one of Oplophoms luciferase, Firefly luciferase. Lucia
luciferase, Renilla
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
luciferase, or an engineered luciferase. In some embodiments, the luciferase
gene is derived
from Oplophorus In some embodiments, the indicator gene is a genetically
modified
luciferase gene, such as NANOLUCk.
Thus, in some embodiments. the present invention comprises a genetically
modified
bacteriophage comprising a non-bacteriophage indicator gene in the late (class
III) gene
region. In some embodiments, the non-native indicator gene is under the
control of a late
promoter. Using a viral late gene promoter insures the reporter gene (e.g.,
luciferase) is not
only expressed at high levels, like viral capsid proteins, but also does not
shut down like
endogenous bacterial genes or even early viral genes.
In some embodiments, the late promoter is a T4-. 17-, or ViI-like promoter. or
another phage promoter similar to that found in the selected wild-type phage,
i.e.. without
genetic modification. The late gene region may be a class III gene region. and
the
bacteriophage may be derived from17. T4, 14-like, Vil, ViI-like, CBA120, or
another
natural bacteriophage having a genome with at least 70, 75, 80. 85, 90 or 95%
homology to
T7, T4, 14-like. Vii, Vil-like. or CBA120 phage.
Genetic modifications to infectious agents may include insertions, deletions,
or
substitutions of a small fragment of nucleic acid, a substantial part of a
gene, or an entire
gene. In some embodiments, inserted or substituted nucleic acids comprise non-
native
sequences. A non-native indicator gene may be inserted into a bacteriophage
genome such
.. that it is under the control of a bacteriophage promoter. In some
embodiments, the non-
native indicator gene is not part of a fusion protein. That is, in some
embodiments, a genetic
modification ma.v be configured such that the indicator protein product does
not comprise
polypeptides of the wild-type bacteriophage. In some embodiments, the
indicator protein
product is soluble. In some embodiments, the invention comprises a method for
detecting a
bacterium of interest comprising the step of incubating a test sample with
such a recombinant
bacteriophage.
In some embodiments, expression of the indicator gene in progeny bacteriophage
following infection of host bacteria results in a free, soluble protein
product. In some
embodiments, the non-native indicator gene is not contiguous Writh a gene
encoding a
.. structural phage protein and therefore does not yield a fusion protein.
Unlike systems that
employ a fusion of a detection moiety to the capsid protein (i.e.. a fusion
protein), some
embodiments of the present invention express a soluble luciferase. This may
greatly increase
16
CA 03011704 2018-07-17
WO 2017/12743-1
PCT/US2017/013955
the sensitivity of the assay (down to a single bacterium), and simplifies the
assay. allowing
the assay to be completed in less than an hour for some embodiments, as
opposed to several
hours due to additional purification steps required with constructs that
produce detectable
fusion proteins. Further. fusion proteins may be less active than soluble
proteins due, e.g.. to
protein folding constraints that may alter the conformation of the enzyme
active site or access
to the substrate.
Moreover, fusion proteins by definition limit the number of the moieties
attached to
subunits of a protein in the bacteriophage. For example, using a commercially
available
system designed to serve as a platform for a fusion protein would result in
about 415 copies
of the fusion moiety. corresponding to the about 415 copies of the gene 10B
capsid protein in
each T7 bacteriophage particle. Without this constraint, infected bacteria can
be expected to
express many more copies of the detection moiety (e.g.. luciferase) than can
fit on the
bacteriophage. Additionally. large fusion proteins, such as a capsid-
luciferase fusion, may
inhibit assembly of the bacteriophage particle, thus yielding fewer
bacteriophage progeny.
Thus a soluble. non-fusion indicator gene product may be preferable.
In some embodiments, the indicator phage encodes a reporter, such as a
detectable
enzyme. The indicator gene product may generate light and/or may be detectable
by a color
change. Various appropriate enzymes are commercially available, such as
alkaline
phosphatase (AP), horseradish peroxidase (HRP). or luciferase (Luc). In some
embodiments,
these enzymes may serve as the indicator moiety. In some embodiments. Firefly
luciferase
is the indicator moiety. In some embodiments, Oplophorus luciferase is the
indicator moiety.
In some embodiments. NANOLUCt is the indicator moiety. Other engineered
luciferases or
other enzymes that generate detectable signals may also be appropriate
indicator moieties.
In some embodiments, the use of a soluble detection moiety eliminates the need
to
remove contaminating parental phage from the lysate of the infected sample
cells. With a
fusion protein system, any bacteriophage used to infect sample cells would
have the detection
moiety attached, and would be indistinguishable from the daughter
bacteriophage also
containing the detection moiety. As detection of sample bacteria relies on the
detection of a
newly created (de novo synthesized) detection moiety, using fusion constructs
requires
additional steps to separate old (parental) moieties from newly created
(daughter
bacteriophage) moieties. This may be accomplished by washing the infected
cells multiple
times, prior to the completion of the bacteriophage life cycle, inactivating
excess parental
17
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
phage after infection by physical or chemical means, and/or chemically
modifying the
parental bacteriophage with a binding moiety (such as biotin), which can then
be bound and
separated (such as by streptavidin-coated sepharose beads). However, even with
all these
attempts at removal, parental phage can remain when a high concentration of
parental phage
is used to assure infection of a low number of sample cells, creating
background signal that
may obscure detection of signal from infected cell progeny phage.
By contrast. with the soluble detection moiety expressed in some embodiments
of the
present invention, purification of the parental phage from the final lysate is
unnecessary, as
the parental phage do not have any detection moiety attached. Thus any
detection moiety
present after infection must have been created de novo, indicating the
presence of an infected
bacterium or bacteria. To take advantage of this benefit, the production and
preparation of
parental phage may include purification of the phage from any free detection
moiety
produced during the production of parental bacteriophage in bacterial culture.
Standard
bacteriophage purification techniques may be employed to purify some
embodiments of
phage according to the present invention, such as sucrose density gradient
centrifugation.
cesium chloride isopycnic density gradient centrifugation, HPLC. size
exclusion
chromatography, and dialysis or derived technologies (such as Amicon brand
concentrators ¨
Millipore., Inc.). Cesium chloride isopycnic ultracentrifugation can be
employed as part of
the preparation of recombinant phage of the invention, to separate parental
phage particles
from contaminating luciferase protein produced upon propagation of the phage
in the
bacterial host. In this way, the parental recombinant bacteriophage of the
invention is
substantially free of any luciferase generated during production in the
bacteria. Removal of
residual luciferase present in the phage stock can substantially reduce
background signal
observed when the recombinant bacteriophage are incubated with a test sample.
In some embodiments of modified bacteriophage, the late promoter (class III
promoter, e.g.. from T7. T4. or Vil) has high affinity for RNA polymerase of
the same
bacteriophage that transcribes genes for structural proteins assembled into
the bacteriophage
particle. These proteins are the most abundant proteins made by the phage, as
each
bacteriophage particle comprises dozens or hundreds of copies of these
molecules. The use
of a viral late promoter can ensure optimally high level of expression of the
luciferase
detection moiety. The use of a late viral promoter derived from, specific to.
or active under
the original wild-type bacteriophage the indicator phage is derived from
(e.g.. a T4. T7, or
18
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
Vii late promoter yy ith a T4-. T7-. or Vii- based system) can further ensure
optimal
expression of the detection moiety. The use of a standard bacterial (non-
viral/non-
bacteriophage) promoter may in some cases be detrimental to expression, as
these promoters
are often down-regulated during bacteriophage infection (in order for the
bacteriophage to
prioritize the bacterial resources for phage protein production). Thus, in
some embodiments,
the phage is preferably engineered to encode and express at high level a
soluble (free)
indicator moiety, using a placement in the genome that does not limit
expression to the
number of subunits of a phage structural component.
Compositions of the invention may comprise one or more wild-type or
genetically
modified infectious agents (e.g.. bacteriophages) and one or more indicator
genes. In some
embodiments, compositions can include cocktails of different indicator phages
that may
encode and express the same or different indicator proteins.
Methods of Preparing Indicator Bacteriophage
Embodiments of methods for making indicator bacteriophage begin with selection
of
a wild-type bacteriophage for genetic modification. Some bacteriophage are
highly specific
for a target bacterium. This presents an opportunity for highly specific
detection.
Thus, the methods of the present invention utilizes the high specificity of
binding
agents, associated with infectious agents, that recognize and bind to a
particular
microorganism of interest as a means to amplify a signal and thereby detect
low levels of a
microorganism (e.g., a single microorganism) present in a sample. For example.
infectious
agents (e.g., bacteriophage) specifically recognize surface receptors of
particular
microorganisms and thus specifically infect those microorganisms. As such,
these infectious
agents may be appropriate binding agents for targeting a microorganism of
interest.
A variety of infectious agents may be used. In alternate embodiments,
bacteriophages, phages, mycobacteriophages (such as for TB and paraTB).
mycophages
(such as for fungi). mycoplasma phages. and any other virus that can invade
living bacteria.
fungi, mycoplasma, protozoa. yeasts. and other microscopic living organisms
can be
employed to target a microorganism of interest. For example, in an embodiment,
where the
microorganism of interest is a bacterium, the infectious agent may comprise a
bacteriophage.
For example, well-studied phages of E. coil include TI. T2, T3, T4, T5, T7.
and lambda:
other E. coil phages available in the ATCC collection, for example, include
phiX174. S13.
0x6, MS2, phiV 1, fd. PR772. and ZIK I. As discussed herein, the bacteriophage
may
19
CA 03011704 2018-07-17
WO 2017/127-134
PCT/US2017/013955
replicate inside of the bacteria to generate hundreds of progeny phage.
Detection of the
product of an indicator gene inserted into the bacteriophage genome can be
used as a measure
of the bacteria in the sample.
Some embodiments of the invention utilize the specificity of binding and high-
level
genetic expression capacity of recombinant bacteriophage for rapid and
sensitive targeting to
infect and facilitate detection of a bacterium of interest. In some
embodiments, CBA120
bacteriophage is genetically modified to include a reporter gene. In some
embodiments the
late gene region of a bacteriophage is genetically modified to include a
reporter gene. In
some embodiments, a reporter gene is positioned downstream of the major capsid
gene. In
other embodiments. a reporter gene is positioned upstream of the major capsid
gene.
Some embodiments of methods for preparing a recombinant indicator
bacteriophage
include selecting a wild-type bacteriophage that specifically infects a target
pathogenic
bacterium; preparing a homologous recombination plasmid/vector that comprises
an indicator
gene: transforming the homologous recombination plasmid/vector into target
pathogenic
bacteria: infecting the transformed target pathogenic bacteria with the
selected wild-type
bacteriophage, thereby allowing homologous recombination to occur between the
plasmid/vector and the bacteriophage genome: and isolating a particular clone
of recombinant
bacteriophage.
Various methods for designing and preparing a homologous recombination plasmid
are known. Various methods for transforming bacteria with a plasmid are known,
including
heat-shock. F pilus mediated bacterial conjugation, electroporation. and other
methods.
Various methods for isolating a particular clone following homologous
recombination are
also known. Some method embodiments described herein utilize particular
strategies.
Thus, some embodiments of methods for preparing indicator bacteriophage
include
the steps of selecting a wild-type bacteriophage that specifically infects a
target pathogenic
bactenum: determining the natural sequence in the late region of the genome of
the selected
bacteriophage: annotating the genome and identifying the major capsid protein
gene of the
selected bacteriophage: designing a sequence for homologous recombination
adjacent to the
major capsid protein gene, wherein the sequence comprises a codon-optimized
reporter gene:
incorporating the sequence designed for homologous recombination into a
plasmid,/vector:
transforming the plasmid/vector into target pathogenic bacteria: selecting for
the transformed
bacteria; infecting the transformed bacteria with the selected wild-type
bacteriophage.
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
thereby allowing homologous recombination to occur between the plasmid and the
bacteriophage genome: determining the titer of the resulting recombinant
bacteriophage
lysate: and performing a limiting dilution assay to enrich and isolate the
recombinant
bacteriophage. Some embodiments comprise further repeating the limiting
dilution and titer
steps, following the first limiting dilution assay, as needed until the
recombinant
bacteriophage represent a detectable fraction of the mixture. For example. in
some
embodiments the limiting dilution and titer steps can be repeated until at
least 1/30 of the
bacteriophage in the mixture are recombinant before isolating a particular
clone of
recombinant bacteriophage. A ratio of 1:30 recombinant:wild-type is expected
to yield an
average of 3.2 transducing units (TV) per 96 plaques (e.g., in a 96-well
plate). By Poisson
distribution, a 1:30 ratio therefore generates a 96% chance of observing at
least one TU
somewhere in the 96 wells.
Figure 1 depicts a schematic representation of the wild-type CBA120
bacteriophage
genome. The late gene cluster 110 was identified, and open reading frames 120
(ORF) in the
late gene region were annotated. The ORF187/gp23 putative gene for the major
capsid
protein 130 (MCP) was identified and its sequence. along with downstream
sequence in the
late gene cluster, was used to prepare a recombinant plasmid carrying the
desired reporter
gene.
Some embodiments of methods of preparing a recombinant indicator bacteriophage
include designing a plasmid that can readily recombine with the wild-type
bacteriophage
genome to generate recombinant genomes. In designing a plasmid, some
embodiments
include addition of a codon-optimized reporter gene, such as a luciferase
gene. Some
embodiments further include addition of elements into the upstream
untranslated region. For
example, in designing a plasmid to recombine with the CBA120 genome, an
upstream
untranslated region can be added between the sequence encoding the C-terminus
of the gp23 I
Major Capsid Protein and the start codon of the NANOLUC:g.:, reporter gene.
The
untranslated region can include a promoter, such as a T4, T4-like, 17, T7-
like, CBA120, Vi!.
or Vu-like promoter. The untranslated region can also include a Ribosomal
Entry / Binding
Site (RBS), also known as a --Shine-Dalgarno Sequence" with bacterial systems.
Either or
both of these elements, or other untranslated elements, can be embedded NN
ithin a short
upstream untranslated region made of random sequences comprising about the
same GC
21
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
content as rest of the phage genome. The random region should not include an
ATG
sequence, as that will act as a start codon.
There are numerous known methods and commercial products for preparing
plasmids.
For example PCR, site-directed mutagenesis. restriction digestion, ligation,
cloning, and other
techniques may be used in combination to prepare plasmids. Synthetic plasmids
can also be
ordered commercially (e.g.. GeneWiz). Cosmids can also be employed, or the
CRISPRiCAS9 system could be used to selectively edit a bacteriophage genome.
Figure 2 shi:ms an embodiment of a plasmid designed to recombine with the
CBA120
bacteriophage genome to generate a recombinant bacteriophage. This particular
plasmid is
designated pUC57.HR.CBA120.NanoLuc. The detection/indicator moiety is encoded
by the
NANOLUCik reporter gene 941-1540. The insert (396-1883) is in the standard
AmpR
version of pUC57. The major capsid protein C-terminal fragment is represented
by 396-895,
ORF187,1gp23. A T4-like phage late promoter consensus sequence (902-912) &
Shine-
Dalgarno Ribosomal Entry/Binding Site (927-934) within the 5' untranslated
region are
represented by 896-940. The codon-optimized NANOLUCX reporter gene is
represented by
941-1540. The untranslated region (UTR) and ORF185 hypothetical protein N-
Terminal
fragment are represented by 1541-1838. The transcriptional terminator (1839-
1883) is only
in the plasmid. and does not become part of the phage genome as a result of
recombination.
The ORF187/gp23 fragment 396-895 is a part of a structural gene that encodes a
virion protein. As these virion proteins are expressed at a very high level,
any genes inserted
into this region can be expected to have similar expression levels, as long as
late gene
promoters and/or other similar control elements are used.
Figure 3 shows a schematic of the homologous recombination expected between
the
plasmid of Figure 2 and bacteriophage genome of Figure Ito create recombinant
bacteriophage that express the luciferase gene. In this embodiment of
homologous
recombination to generate recombinant bacteriophage. the CBA120 phage genome
is 157.304
base pairs. while the synthesized plasmid is 4.117 base pairs. The final
recombinant genome
resulting from recombination is 157.949 base pairs.
In some embodiments, indicator phage according to the invention comprise
CBA120
bacteriophage genetically engineered to comprise a reporter gene such as a
luciferase gene.
For example. an indicator phage can be the CBA120 bacteriophage wherein the
genome
22
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
comprises the sequence of the NANOLUCai gene. A recombinant CBA120
bacteriophage
genome may further comprise a 14, T7, CBA120, Vii, or another late promoter.
Thus, in the embodiment of the recombinant phage generated as a result of the
recombination illustrated in Figure 3, the indicator gene (i.e.. NANOLUCt) is
inserted into
the late gene region, just downstream of the gene encoding the major capsid
protein, and thus
creates recombinant bacteriophage genomes comprising the NANOLUCt gene. The
construct may additionally comprise the consensus 14, T7, CBA120. Vii, or
another late
promoter or another suitable promoter to drive transcription and expression of
the luciferase
gene. The construct may also comprise a composite untranslated region
synthesized from
several UTRs. This construct ensures soluble luciferase is produced such that
expression is
not limited to the number of capsid proteins inherent in the phage display
system.
Figure 4 depicts the isolation of recombinant phage from the mixture of wild-
type
and recombinant bacteriophage resulting from the homologous recombination
illustrated in
Figure 3, using the plasmid construct shown in Figure 2.
In the first step 402. bacteria transformed with the homologous recombination
plasmid are infected with bacteriophage, resulting in progeny phage with a
mixture of
parental and recombinant phage with a ratio of approximately 120 wild-type 432
:1
recombinant phage 434. The resulting recombinant phage mix is diluted 404 into
96-well
plates 406 to give an average of 3 recombinant transducing units (TU) per
plate. which
corresponds to about 3.8 infectious units (IU) of mostly wild-type phage per
well. The 96-
well plate is assayed for luciferase activity to identify wells 436 containing
recombinant
phage as compared to IN ells 440 containing wild-type bacteriophage. Bacteria
438 are added
408: for example, each well may contain about 50 jiL of a turbid E. coil
0157:H7 culture.
This allows the phage to replicate and produce the luciferase enzyme 442.
After 2 hours of
incubation at 37 C shown in 410. wells may be screened for the presence of
luciferase 442.
Any positive wells are likely to have been inoculated with a single
recombinant phage, and at
this stage the mixture may contain a ratio of approximately 3.8 wild-type
phage: 1
recombinant, an enrichment over the original 120:1 ratio. In one embodiment,
soluble
luciferase and phage Vs ere present at an approximate ratio of 16 NN ild-
type:1 recombinant. If
necessary (i.e., if the ratio of recombinant:wild-type is lower than 1:30).
progeny from this
enriched culture 412 may be subjected to additional limiting dilution assay(s)
414 to increase
the ratio and determine the actual concentration of recombinant phage
transducing units. For
23
CA 03011704 2018-07-17
WO 2017/127-134
PCT/US2017/013955
example, about 3 recombinant TU per 96-well plate 416 may be aliquoted 414
from the first
purification stock, leading to an approximate inoculation of ¨20 mostly wild-
type phage per
well of a second dilution assay plate 420. Anv positive luciferase wells are
likely to have
been inoculated with a single recombinant along with ¨20 wild-type phage.
These wells may
be analyzed for presence of luciferase 442.
After addition of bacteria and incubation (e.g., for 2 hours at 37 C) 418,
soluble
luciferase and phage are present at approximately 20 wild-type: 1 recombinant
420. Finally,
a plaque assay may be performed 422 to screen for recombinants that express
luciferase 446.
A small number of individual (e.g., n=48) plaques may be individually picked
and screened
in a third multiwell plate 426 for luciferase activity 436. In an embodiment,
this approach
should insure that about 3 recombinants would be in the mix of plaques being
screened. One
plaque may be removed from the plate to each well of a 96-well plate 424 and a
luciferase
assay performed 426 to determine which vells contained phage exhibiting
luciferase activity
442. Wells 428 demonstrating luciferase activity represent pure recombinant
phage 434,
while wells without luciferase activity 430 represent pure wild-type phage
432.
Individual plaques may then be suspended in buffer (e.g., 100 1.iL TMS) or
media, and
an aliquot (e.g., about 5 p.1_,) added to a well containing a turbid E. coil
0157:H7 culture, and
assayed after incubation (e.g., about 45 minutes to 1 hour at 37 C). Positive
wells are
expected to contain a pure culture of recombinant phage. Certain embodiments
can include
.. additional rounds of plaque purification.
Thus, as illustrated in Figure 4, recombinant phage generated by homologous
recombination of a plasmid designed for recombination with the wild-type phage
genome can
be isolated from a mixture comprising only 0.005% of total phage genomes.
Following
isolation, large scale production may be performed to obtain high titer
recombinant indicator
phage stocks appropriate for use in the E. coil 0157:117 detection assay.
Furthermore,
cesium chloride isopycnic density gradient centrifugation may be used to
separate phage
particles from contaminating luciferase protein to reduce background.
Figure 5 shows an electron micrograph of one embodiment of a recombinant
indicator bacteriophage generated by recombination of the IA pe CBA120
bacteriophage
genome shown in Figure 1 with the plasmid shown in Figure 2, as illustrated in
Figure 3. To
capture the image, the bacteriophage purified on a 5-20% sucrose density
gradient were
adsorbed onto a glow discharge-treated carbon film and stained with 2% uranyl
acetate. The
24
CA 03011704 2018-07-17
WO 2017/127434 PC
T/US2017/013955
sample was viewed in a FE! Tecnai G2 Spirit BioTwin Transmission Electron
Microscope
and the micrograph taken with an Eagleml 2K CCD. This indicator bacteriophage
is
designated "CBA120NanoLuc" (or "CBA120NanoLuc indicator phage") and was
utilized in
the assays described herein. The data presented in Examples and Figures herein
were
obtained using this Indicator Phage for infection of bacteria in the sample
being tested.
In this way, and as described in more detail in the Examples below,
recombinant
bacteriophage having the reporter gene of interest (e.g.. luciferase gene such
as Firefly.
Oplophorus or an engineered luciferase such as NANOLUCt) inserted into a wild-
type
bacteriophage may be generated.
Methods of Using Infectious Agents for Detecting Microorganisms
As noted herein, in certain embodiments, the invention may comprise methods of
using infectious particles for detecting microorganisms. The methods of the
invention may
be embodied in a variety of ways.
In an embodiment, the invention may comprise a method for detecting a
bacterium of
interest in a sample comprising the steps of: incubating the sample with
bacteriophage that
infects the bacterium of interest, wherein the bacteriophage comprises an
indicator gene such
that expression of the indicator gene during bacteriophage replication
following infection of
the bacterium of interest results in production of a soluble indicator protein
product: and
detecting the indicator protein product. Wherein positive detection of the
indicator protein
product indicates that the bacterium of interest is present in the sample.
In certain embodiments. the assay may be performed to utilize a general
concept that
can be modified to accommodate different sample types or sizes and assay
formats.
Embodiments employing recombinant bacteriophage of the invention (i.e.,
indicator
bacteriophage) may allow rapid detection of specific bacterial strains, with
total assay times
under 1.5. 2.0, 2.5. 3.0, 3.5, 4.0, 4.5, 5.0, 5.5, 6.0, 6.5, 7.0, 7.5, 8.0,
8.5, 9.0, 9.5, 10.0, 10.5,
11.0, 11.5. or 12 hours, depending on the sample type, sample size, and assay
format. For
example, the amount of time required may be somewhat shorter or longer
depending on the
strain of bacteriophage and the strain of bacteria to be detected in the
assay, type and size of
the sample to be tested, conditions required for viability of the target,
complexity of the
physical/chemical environment, and the concentration of "endogenous" non-
target bacterial
contaminants.
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
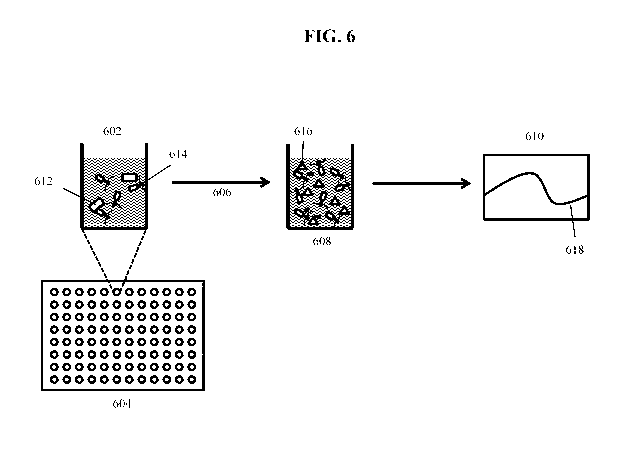
Figure 6 illustrates an embodiment of an assay for detecting a bacterium of
interest
using a modified bacteriophage according to an embodiment of the invention.
Aliquots of
indicator phage 614 are distributed to the individual wells 602 of a multi-
well plate 604, and
then test sample aliquots containing bacteria 612 are added and incubated 606
for a period of
time (e.g., 45 minutes at 37 C) sufficient for phage to replicate and generate
soluble indicator
616 (e.g., luciferase). The plate wells 608 containing soluble indicator and
phage may then
be assayed 610 to measure the indicator activity on the plate 618 (e.g.,
luciferase assay).
Experiments utilizing this method are described herein. In some embodiments,
the test
samples are not concentrated (e.g.. by centrifugation) but are incubated
directly with indicator
.. phage for a period of time and subsequently assayed for luciferase
activity. In other
embodiments, various tools (e.g.. a centrifuge or filter) may be used to
concentrate the
samples before enrichment or before testing. For example. a 10 mL aliquot of a
prepared
sample may be extracted and centrifuged to pellet cells and large debris. The
pellet can be
resuspended in a smaller volume for enrichment or for testing (i.e., before
infecting the
sample with Indicator Bacteriophage).
In some embodiments, the sample may be enriched prior to testing by incubation
in
conditions that encourage growth. In such embodiments, the enrichment period
can be 1, 2,
3. 4, 5, 6. 7, or up to 8 hours or longer, depending on the sample type and
size.
Thus. in some embodiments. the indicator bacteriophage comprises a detectable
.. indicator moiety, and infection of a single pathogenic cell (e.g.,
bacterium) can be detected by
an amplified signal generated via the indicator moiety. Thus the method may
comprise
detecting an indicator moiety produced during phage replication. N\ herein
detection of the
indicator indicates that the bacterium of interest is present in the sample.
In an embodiment, the invention may comprise a method for detecting a
bacterium of
interest in a sample comprising the steps of incubating the sample with a
recombinant
bactenophage that infects the bacterium of interest. wherein the recombinant
bacteriophage
comprises an indicator gene inserted into a late gene region of the
bacteriophage such that
expression of the indicator gene during bacteriophage replication following
infection of host
bacteria results in production of a soluble indicator protein product: and
detecting the
indicator protein product, wherein positive detection of the indicator protein
product indicates
that the bacterium of interest is present in the sample. In some embodiments,
the amount of
26
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
indicator moiety detected corresponds to the amount of the bacterium of
interest present in
the sample.
As described in more detail herein, the methods and systems of the invention
may
utilize a range of concentrations of parental indicator bacteriophage to
infect bacteria present
in the sample. In some embodiments the indicator bacteriophage are added to
the sample at a
concentration sufficient to rapidly find, bind, and infect target bacteria
that are present in very
low numbers in the sample, such as a single cell. In some embodiments, the
phage
concentration can be sufficient to find, bind, and infect the target bacteria
in less than one
hour. In other embodiments, these events can occur in less than two hours, or
less than three
hours, following addition of indicator phage to the sample. For example. in
certain
embodiments, the bacteriophage concentration for the incubating step is
greater than 1 x 105
PFU,'mL, greater than 1 x 106 PFU'inL, or greater than 1 x 107 PFUlmL.
In certain embodiments. the recombinant infectious agent may be purified so as
to be
free of any residual indicator protein that may be generated upon production
of the infectious
agent stock. Thus, in certain embodiments. the recombinant bacteriophage may
be purified
using cesium chloride isopycnic density gradient centrifugation prior to
incubation with the
sample. When the infectious agent is a bacteriophage, this purification may
have the added
benefit of' removing bacteriophage that do not have DNA (i.e., empty phage or -
ghosts-).
In some embodiments of the methods of the invention, the microorganism may be
detected without any isolation or purification of the microorganisms from a
sample. For
example, in certain embodiments, a sample containing one or a few
microorganisms of
interest may be applied directly to an assay container such as a spin column,
a microliter IN ell.
or a filter and the assay is conducted in that assay container. Various
embodiments of such
assays are disclosed herein.
Aliquots of a test sample may be distributed directly into wells of a multi-
well plate.
indicator phage may be added. and after a period of time sufficient for
infection, a lysis buffer
may be added as well as a substrate for the indicator moiety (e.g., luciferase
substrate for a
luciferase indicator) and assayed for detection of the indicator signal. Some
embodiments of
the method can be performed on filter plates. Some embodiments of the method
can be
performed with or without concentration of the sample before infection with
indicator phage.
For example, in many embodiments. multi-well plates are used to conduct the
assays.
The choice of' plates (or any other container in which detecting may be
performed) may affect
27
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013055
the detecting step. For example. some plates may include a colored or white
background,
which may affect the detection of light emissions. Generally speaking, white
plates have
higher sensitivity but also yield a higher background signal. Other colors of
plates may
generate lower background signal but also have a slightly lower sensitivity.
Additionally,
one reason for background signal is the leakage of light from one well to
another, adjacent
well. There are some plates that have white wells but the rest of the plate is
black. This allows
for a high signal inside the well but prevents well-to-well light leakage and
thus may decrease
background. Thus the choice of plate or other assay vessel may influence the
sensitivity and
background signal for the assay.
Methods of the invention may comprise various other steps to increase
sensitivity.
For example. as discussed in more detail herein, the method may comprise a
step for washing
the captured and infected bacterium, after adding the bacteriophage but before
incubating, to
remove excess parental bacteriophage and/or luciferase or other reporter
protein
contaminating the bacteriophage preparation.
In some embodiments, detection of the microorganism of interest may be
completed
NN ithout the need for culturing the sample as a way to increase the
population of the
microorganisms. For example, in certain embodiments the total time required
for detection is
less than 12.0 hours, 11.0 hours, 10.0 hours. 9.0 hours, 8.0 hours, 7.0 hours,
6.0 hours. 5.0
hours, 4.0 hours, 3.0 hours, 2.5 hours, 2.0 hours. 1.5 hours, 1.0 hour, 45
minutes, or less than
.. 30 minutes. Minimizing time to result is critical in food and environmental
testing for
pathogens.
In contrast to assays known in the art, the method of the invention can detect
individual microorganisms. Thus, in certain embodiments, the method may detect
< 10 cells
of the microorganism (i.e., 1. 2, 3, 4, 5, 6, 7, 8, 9 microorganisms) present
in a sample. For
.. example, in certain embodiments, the recombinant bacteriophage is highly
specific for E. coil
0157:H7. In an embodiment, the recombinant bacteriophage can distinguish E.
coil 0157:147
in the presence of more than 100 other apes of bacteria. In certain
embodiments, the
recombinant bacteriophage can be used to detect a single bacterium of the
specific type in the
sample. In certain embodiments. the recombinant bacteriophage detects as few
as 2, 3. 4, 5.
6, 7. 8, 9, 10. 15, 20, 30, 40, 50, 60, 70, 80, 90, or 100 of the specific
bacteria in the sample.
Thus, aspects of the present invention provide methods for detection of
microorganisms in a test sample via an indicator moiety. In some embodiments,
where the
28
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
microorganism of interest is a bacterium, the indicator moiety may be
associated vNith an
infectious agent such as an indicator bacteriophage. The indicator moiety may
react with a
substrate to emit a detectable signal or may emit an intrinsic signal (e.g..
fluorescent protein).
In some embodiments, the detection sensitivity can reveal the presence of as
few as 50, 20,
10. 9. 8, 7, 6, 5, 4. 3, or 2 cells of the microorganism of interest in a test
sample. In some
embodiments, even a single cell of the microorganism of interest may yield a
detectable
signal. In some embodiments, the bacteriophage is a T4-like or Vil-like
bacteriophage. In
some embodiments, the recombinant bacteriophage is derived from CBA120. In
certain
embodiments, a CBA120 recombinant bacteriophage is highly specific for E.coli
0157:H7.
In some embodiments, the indicator moiety encoded by the infectious agent may
be
detectable during or after replication of the infectious agent. Many different
types of
detectable biomolecules suitable for use as indicator moieties are known in
the art, and many
are commercially available. In some embodiments the indicator phage comprises
an
enzyme, which serves as the indicator moiety. In some embodiments, the genome
of the
indicator phage is modified to encode a soluble protein. In some embodiments,
the indicator
phage encodes a detectable enzyme. The indicator may emit light and/or may be
detectable
by a color change. Various appropriate enzymes are commercially available such
as alkaline
phosphatase (AP), horseradish peroxidase (HRP), or luciferase (Luc). In some
embodiments,
these enzymes may serve as the indicator moiety. In some embodiments. Firefly
luciferase
is the indicator moiety. In some embodiments, Oplophorus luciferase is the
indicator moiety.
In some embodiments. NANOLUCt is the indicator moiety. Other engineered
luciferases or
other enzymes that generate detectable signals may also be appropriate
indicator moieties.
Thus, in some embodiments. the recombinant bacteriophage of the methods,
systems
or kits is prepared from wild-type bacteriophage CBA120. In some embodiments,
the
.. indicator gene encodes a protein that emits an intrinsic signal. such as a
fluorescent protein
(e.g.. green fluorescent protein or others). The indicator may emit light
and/or may be
detectable by a color change. In some embodiments, the indicator gene encodes
an enzyme
(e.g., luciferase) that interacts with a substrate to generate signal. In some
embodiments, the
indicator gene is a luciferase gene. In some embodiments, the luciferase gene
is one of
Oplophorus luciferase. Firefly luciferase, Renilla luciferase. External
Gaussia luciferase.
Lucia luciferase, or an engineered luciferase such as NANOLUCK. Rluc8.6-535,
or Orange
Nano-lantern.
29
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
Detecting the indicator may include detecting emissions of light. In some
embodiments, a luminometer may be used to detect the reaction of indicator
(e.g., luciferase)
with a substrate. The detection of RLU can be achieved with a luminometer. or
other
machines or devices may also be used. For example, a spectrophotometer. CCD
camera. or
.. CMOS camera may detect color changes and other light emissions. Absolute
RLU are
important for detection. but the signal to background ratio also needs to be
high (e.g.. > 2Ø>
2.5. or > 3.0) in order for single cells or low numbers of cells to be
detected reliably.
In some embodiments. the indicator phage is genetically engineered to contain
the
gene for an enzyme, such as a luciferase. which is only produced upon
infection of bacteria
that the phage specifically recognizes and infects. In some embodiments, the
indicator
moiety is expressed late in the viral life cycle. In some embodiments, as
described herein, the
indicator is a soluble protein (e.g., soluble luciferase) and is not fused
with a phage structural
protein that limits its copy number.
Thus in some embodiments utilizing indicator phage. the invention comprises a
method for detecting a microorganism of interest comprising the steps of
capturing at least
one sample bacterium; incubating the at least one bacterium with a plurality
of indicator
phage: allowing time for infection and replication to generate progeny phage
and express
soluble indicator moiety: and detecting the progeny phage, or preferably the
indicator,
wherein detection of the indicator demonstrates that the bacterium is present
in the sample.
For example, in some embodiments the test sample bacterium may be captured by
binding to the surface of a plate. or by filtering the sample through a
bacteriological filter
(e.g.. 0.45 um pore size spin filter or plate filter). In an embodiment, the
infectious agent
(e.g., indicator phage) is added in a minimal volume to the captured sample
directly on the
filter. In an embodiment, the microorganism captured on the filter or plate
surface is
subsequently washed one or more times to remove excess unbound infectious
agent. In an
embodiment, a medium (e.g.. Luria-Bertani Broth, also called LB herein, or
Tryptic Soy
Broth or Tryptone Soy Broth, also called TSB herein) may be added for further
incubation
time, to allow replication of bacterial cells and phage and high-level
expression of the gene
encoding the indicator moiety . HONN ever, a surprising aspect of some
embodiments of testing
assays is that the incubation step with indicator phage only needs to be long
enough for a
single phage life cycle. The amplification power of using bacteriophage was
previously
thought to require more time, such that the phage would replicate for several
cycles. A single
CA 03011704 2018-07-17
WO 2017/127-134
PCT/US2017/013955
replication cycle of indicator phage can be sufficient to facilitate sensitive
and rapid detection
according to some embodiments of the present invention.
In some embodiments, aliquots of a test sample comprising bacteria may be
applied to
a spin column and after infection with a recombinant bacteriophage and an
optional washing
to remove any excess bacteriophage, the amount of soluble indicator detected
will be
proportional to the amount of bacteriophage that are produced by infected
bacteria.
Soluble indicator (e.g., luciferase) released into the surrounding liquid upon
lysis of
the bacteria may then be measured and quantified. In an embodiment, the
solution is spun
through the filter, and the filtrate collected for assay in a new receptacle
(e.g., in a
luminometer) following addition of a substrate for the indicator enzyme (e.g.,
luciferase
substrate). Alternatively, the indicator signal may be measured directly on
the filter.
In various embodiments, the purified parental indicator phage does not
comprise the
detectable indicator itself, because the parental phage can be purified before
it is used for
incubation with a test sample. Expression of late (Class III) genes occurs
late in the viral life
cycle. In some embodiments of the present invention, parental phage may be
purified to
exclude any existing indicator protein (e.g.. luciferase). In some
embodiments, expression of
the indicator gene during bacteriophage replication following infection of
host bacteria
results in a soluble indicator protein product. Thus, in many embodiments, it
is not necessary
to separate parental from progeny phage prior to the detecting step. In an
embodiment, the
microorganism is a bacterium and the indicator phage is a bacteriophage. In an
embodiment,
the indicator moiety is soluble luciferase, which is released upon lysis of
the host
microorganism.
Thus, in an alternate embodiment, the indicator substrate (e.g.. luciferase
substrate)
may be incubated with the portion of the sample that remains on a filter or
bound to a plate
surface. Accordingly, in some embodiments the solid support is a 96-well
filter plate (or
regular 96-well plate). and the substrate reaction may be detected by placing
the plate directly
in the luminometer.
For example. in an embodiment, the invention may comprise a method for
detecting
E coil 0157:H7 comprising the steps of infecting cells captured on a 96-well
filter plate
with a plurality of parental indicator phage capable of expressing luciferase
upon infection:
washing excess phage away: adding LB broth and allowing time for phage to
replicate and
lyse the specific E. coil target (e.g., 30-90 minutes). and detecting the
indicator luciferase by
31
CA 03011704 2018-07-17
WO 2017/127-134
PCT/US2017/013955
adding luciferase substrate and measuring luciferase activity directly in the
96-well plate.
wherein detection of luciferase activity indicates that the E. coil 0157:H7 is
present in the
sample.
In another embodiment. the invention may comprise a method for detecting E.
coil
0157:H7 comprising the steps of: infecting cells in liquid solution or
suspension in a 96-well
plate with a plurality of parental indicator phage capable of expressing
luciferase upon
infection: allowing time for phage to replicate and lyse the specific E. coli
target (e.g., 30-120
minutes); and detecting the indicator luciferase by adding luciferase
substrate and measuring
luciferase activity directly in the 96-well plate. wherein detection of
luciferase activity
indicates that the E. coli 0157:H7 is present in the sample. In such an
embodiment no
capturing step is necessary. In some embodiments, the liquid solution or
suspension may be
a consumable test sample, such as a vegetable wash. In some embodiments, the
liquid
solution or suspension may be vegetable wash fortified with concentrated LB
Broth,
Tryptic/Tryptone Soy Broth, Peptone Water or Nutrient Broth. In some
embodiments, the
liquid solution or suspension may be bacteria diluted in LB Broth.
In some embodiments, lysis of the bacterium may occur before, during, or after
the
detection step. Experiments suggest that infected unlysed cells may be
detectable upon
addition of luciferase substrate in some embodiments. Presumably, luciferase
may exit cells
and/or luciferase substrate may enter cells without complete cell lysis. Thus,
for
embodiments utilizing the spin filter system, where only luciferase released
into the lysate
(and not luciferase still inside intact bacteria) is analyzed in the
luminometer, lysis is required
for detection. However. for embodiments utilizing filter plates or 96-well
plates with sample
in solution or suspension, where the original plate full of intact and lysed
cells is directly
assayed in the luminometer, lysis is not necessary for detection.
In some embodiments, the reaction of indicator moiety (e.g., luciferase) with
substrate
may continue for 30 minutes or more, and detection at various time points may
be desirable
for optimizing sensitivity. For example, in embodiments using 96-well filter
plates as the
solid support and luciferase as the indicator, luminometer readings may be
taken initially and
at 10- or 15-minute intervals until the reaction is completed.
Surprisingly, high concentrations of phage utilized for infecting test samples
have
successfully achieved detection of very low numbers of target microorganism in
a very short
timeframe. The incubation of phage with a test sample in some embodiments need
only be
32
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
long enough for a single phage life cycle. In some embodiments, the
bacteriophage
concentration for this incubating step is greater than 7 x 106, 8 x 106, 9 x
106, 1.0 x 107, 1.1 x
107, 1.2 x 107, 1.3 x 107, 1.4 x 107. 1.5 x 107, 1.6 x 107. 1.7 x 107, 1.8 x
107, 1.9x 107, 2.0x
107, 3.0 x 107, 4.0 x 107, 5.0 x 107, 6.0 x 107, 7.0 x 107, 8.0 x 107, 9.0 x
1.07, or 1.0 x 108
PFU/mL.
Success with such high concentrations of phage is surprising because the large
numbers of phage were previously associated with -lysis from without," which
killed target
cells and thereby prevented generation of useful signal from earlier phage
assays. It is
possible that the clean-up of prepared phage stocks described herein helps to
alleviate this
problem (e.g., clean-up by cesium chloride isopycnic density gradient
ultracentrifugation),
because in addition to removing any contaminating luciferase associated with
the phage, this
clean-up may also remove ghost particles (particles that have lost DNA). The
ghost particles
can lyse bacterial cells via "lysis from without,- killing the cells
prematurely and thereby
preventing generation of indicator signal. Electron microscopy demonstrates
that a crude
phage lysate (i.e., before cesium chloride clean-up) may have greater than 50%
ghosts. These
ghost particles may contribute to premature death of the microorganism through
the action of
many phage particles puncturing the cell membrane. Thus ghost particles may
have
contributed to previous problems where high PFU concentrations were reported
to be
detrimental. Moreover, a very clean phage prep allows the assay to be
performed with no
wash steps, which makes the assay possible to perform without an initial
concentration step.
Some embodiments do include an initial concentration step, and in some
embodiments this
concentration step allows a shorter enrichment incubation time.
Some embodiments of testing methods may further include confirmatory assays, A
variety of assays are known in the art for confirming an initial result,
usually at a later point
in time. For example, the samples can be cultured (e.g., CHROMAGARVDYNABEADS*)
assay as described in Example 4), PCR can be utilized to confirm the presence
of the
microbial DNA, or other confirmatory assays can be used to confirm the initial
result.
Figures 7-9 demonstrate data from basic assays (e.g., performed as shown in
Figure
6) on samples derived from E. coil 0157:H7 cultures, using the CBA120NanoLuc
Indicator
Phage. Figure 7 demonstrates three different infecting phage concentrations,
10), 106, and
107 phage/mL. Figure 8 uses 6-10 replicates of each indicated cell number to
demonstrate
significant differences between signals from single cells as compared to zero
cells
33
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
(background) or higher numbers of cells. Figure 9 shows that the signal to
background ratio
for the experiment shown in Figure 8 is greater than 2Ø Example 3 also
describes these
experiments.
Beef Assays
Existing protocols for detection of E coil 0157:H7 in foods are complicated,
expensive, slow, labor-intensive and prone for false positives. Detection with
a recombinant
bacteriophage specific for this pathogen offers an effective, fast and simple
testing
alternative.
Embodiments of beef assays include sample preparation steps. Some embodiments
can include enrichment time. For example, enrichment for I, 2, 3. 4. 5, 6, 7,
or 8 hours may
be needed. depending on sample type and size. Following these sample
preparation steps,
infection with a high concentration of recombinant bacteriophage that
expresses a reporter or
indicator can be performed in a variety of assay formats, such as that shown
in Figure 6.
Embodiments of beef assays can detect a single pathogenic bacterium in sample
sizes
corresponding to industry standards. \ \ ith a reduction in time-to-results of
20-50%. depending
on the sample type and size.
Figures 10-16 show data from beef assay experiments using CBA120NanoLuc
Indicator Bacteriophage, as described in Example 4.
Vegetable Wash Assays
To prepare the vegetable wash, vegetable leaves (e.g., spinach or lettuce) may
be
weighed and added to a clean plastic bag. Liquid can be added to the vegetable
wash. For
example, in some embodiments 5 mL of water are added per each gram (g) of
vegetable.
Other laboratory liquids (e.g.. LB) may also be used. Leaves and solution can
be mixed
manually for a few minutes. Liquid can then be extracted from the plastic bag
and can be
used as the "vegetable wash.- Using this method, - I million -endogenous-
bacterial
contaminants IA ere found to reside on a single spinach leaf (1-2 g).
The assay is quantitative in that the signal detected is proportional to the
amount of
the bacterium of interest in the sample. For example, known numbers of E colt
0157:H7
cells can be added to vegetable wash samples to simulate contamination of
vegetables with
pathogenic bacteria. The experiment using vegetable wash samples described in
Example 5
demonstrates marked differences between the signal from 0 cells. 1 cell, and 7
cells per assay,
demonstrating the ability to detect single-digit cell numbers in vegetable
wash. Using more
34
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
bacterial cells per assay shows increasing signal in a dose-dependent manner.
The vegetable
wash contains about 106 non-target bacteria /mL, corresponding to at least 105
non-target
bacteria per sample in this assay (including the 0 cells E. coil 0157:H7
control). The ability
to discern as few as a single target bacterial cell from 10 non-target
bacteria is surprising and
again demonstrates the specificity and sensitivity of the assay. Figure 17
shoxµs data from a
vegetable wash experiment (Example 5).
In some embodiments, the incubating step of the methods described herein
comprises
a final bacteriophage concentration of greater than 7 x 106. 8 x 106, 9 x 106,
1.0 x 107, 1.1 x
107, 1.2 x 107, 1.3 x 107, 1.4 x 107, 1.5 x 107, 1.6 x 107, 1.7 x 107, 1.8 x
107, 1.9 x 107. 2.0x
107, 3.0 x 107, 4.0 x 107, 5.0 x 107, 6.0 x 107, 7.0 x 107, 8.0 x 107, 9.0 x
107, or 1.0 x 108
PFU/mL. Such high phage concentrations were previously reported to be
detrimental to such
an assay, and therefore successful use of such high concentrations generated
unexpected
results. In some embodiments, the methods of the invention require less than
12, 11, 10, 9, 8,
7, 6, 5, 4, 3, or 2 hours for detection of a microorganism of interest. In
some embodiments,
the methods can detect as few as 100, 50, 20, 10.9. 8, 7, 6. 5.4. 3, or 2
cells of the bacterium
of interest. These are shorter timeframes than were previously thought
possible. In some
embodiments, even a single cell of the bacterium is detectable. In additional
embodiments,
the invention comprises systems (e.g., computer systems, automated systems or
kits)
comprising components for performing the methods disclosed herein, and/or
using the
modified bacteriophage described herein.
Systems and Kits of the Invention
In some embodiments, the invention comprises systems (e.g.. automated systems
or
kits) comprising components for performing the methods disclosed herein. In
some
embodiments, indicator phage are comprised in systems or kits according to the
invention.
Methods described herein may also utilize such indicator phage systems or
kits. Some
embodiments described herein are particularly suitable for automation and/or
kits, given the
minimal amount of reagents and materials required to perform the methods. In
certain
embodiments, each of the components of a kit may comprise a self-contained
unit that is
deliverable from a first site to a second site.
In some embodiments, the invention comprises systems or kits for rapid
detection of a
microorganism of interest in a sample. The systems or kits may in certain
embodiments
comprise a component for incubating the sample with an infectious agent
specific for the
CA 03011704 2018-07-17
WO 2017/127-134
PCT/US2017/013955
microorganism of interest. NN herein the infectious agent comprises an
indicator moiety and a
component for detecting the indicator moiety. In some embodiments of both the
systems and
the kits of the invention, the infectious agent is a recombinant bacteriophage
that infects the
bacterium of interest, and the recombinant bacteriophage comprises an
indicator gene
inserted into a late gene region of the bacteriophage as the indicator moiety
such that
expression of the indicator gene during bacteriophage replication following
infection of host
bacteria results in a soluble indicator protein product. Some systems further
comprise a
component for capturing the microorganism of interest on a solid support.
In other embodiments. the invention comprises a method, system, or kit for
rapid
detection of a microorganism of interest in a sample, comprising an infectious
agent
component that is specific for the microorganism of interest, wherein the
infectious agent
comprises an indicator moiety, and a component for detecting the indicator
moiety. In some
embodiments, the bacteriophage is a T4-like, Vi!. ViI-like, or CBA120
bacteriophage. In one
embodiment. the recombinant bacteriophage is derived from CBA120. In certain
embodiments, the recombinant bacteriophage is highly specific for a particular
bacterium.
For example, in certain embodiments, the recombinant bacteriophage is highly
specific for E
colt 0157:H7. In an embodiment, the recombinant bacteriophage can distinguish
E. coil
0157:H7 in the presence of more than 100 other types of bacteria. In certain
embodiments, a
system or kit detects a single bacterium of the specific type in the sample.
In certain
embodiments, a system or kit detects as few as 2. 3, 4, 5, 6. 7, 8,9. 10, IS,
20. 30, 40, 50, 60.
70. 80. 90. or 100 specific bacteria in the sample.
In certain embodiments, the systems and/or kits may further comprise a
component
for washing the captured microorganism sample. Additionally or alternatively,
the systems
and/or kits may further comprise a component for determining amount of the
indicator
moiety, wherein the amount of indicator moiety detected corresponds to the
amount of
microorganism in the sample. For example. in certain embodiments, the system
or kit may
comprise a luminometer or other device for measuring a luciferase enzyme
activity.
In some systems and/or kits, the same component may be used for multiple
steps. In
some systems and/or kits, the steps are automated or controlled by the user
via computer
input and/or wherein a liquid-handling robot performs at least one step.
Thus in certain embodiments, the invention may comprise a system or kit for
rapid
detection of a microorganism of interest in a sample. comprising: a component
for incubating
36
CA 03011704 2018-07-17
WO 2017/127-134
PCT/US2017/013955
the sample with an infectious agent specific for the microorganism of
interest, wherein the
infectious agent comprises an indicator moiety: a component for capturing the
microorganism
from the sample on a solid support; a component for washing the captured
microorganism
sample to remove unbound infectious agent: and a component for detecting the
indicator
moiet). In some embodiments, the same component may be used for steps of
capturing
and/or incubating and/or washing (e.g., a filter component). Some embodiments
additionally
comprise a component for determining amount of the microorganism of interest
in the
sample, wherein the amount of indicator moiety detected corresponds to the
amount of
microorganism in the sample. Such systems can include various embodiments and
subembodiments analogous to those described above for methods of rapid
detection of
microorganisms. In an embodiment, the microorganism is a bacterium and the
infectious
agent is a bacteriophage. In a computerized system, the system may be fully
automated.
semi-automated, or directed by the user through a computer (or some
combination thereof).
In some embodiments, the system may comprise a component for isolating the
microorganism of interest from the other components in the sample.
In an embodiment, the invention comprises a system or kit comprising
components
for detecting a microorganism of interest comprising: a component for
isolating at least one
microorganism from other components in the sample: a component for infecting
the at least
one microorganism with a plurality of a parental infectious agent: a component
for lysing the
at least one infected microorganism to release progeny infectious agents
present in the
microorganism; and a component for detecting the progeny infectious agents, or
with greater
sensitivity, a soluble protein encoded and expressed by the infectious agent.
wherein
detection of the infectious agent or a soluble protein product of the
infectious agent indicates
that the microorganism is present in the sample. The infectious agent may
comprise
CBA120NanoLuc.
The systems or kits may comprise a variety of components for detection of
progeny
infectious agents. For example, in an embodiment, the progeny infectious agent
(e.g..
bacteriophage) may comprise an indicator moiety. In an embodiment, the
indicator moiety in
the progeny infectious agent (e.g.. bacteriophage) may be a detectable moiety
that is
expressed during replication, such as a soluble luciferase protein.
In other embodiments. the invention may comprise a kit for rapid detection of
a
microorganism of interest in a sample. the system comprising: a component for
incubating
37
CA 03011704 2018-07-17
WO 2017/12743-1
PCT/US2017/013955
the sample NN. i th an infectious agent specific for the microorganism of
interest, wherein the
infectious agent comprises an indicator moiety: a component for capturing the
microorganism
from the sample on a solid support: a component for washing the captured
microorganism
sample to remove unbound infectious agent: and a component for detecting the
indicator
moiety. In some embodiments, the same component may be used for steps of
capturing
and,/or incubating and/or washing. Some embodiments additionally comprise a
component
for determining amount of the microorganism of interest in the sample, wherein
the amount
of indicator moiety detected corresponds to the amount of microorganism in the
sample. Such
kits can include various embodiments and subembodiments analogous to those
described
above for methods of rapid detection of microorganisms. In an embodiment, the
microorganism is a bacterium and the infectious agent is a bacteriophage.
In some embodiments, a kit may comprise a component for isolating the
microorganism of interest from the other components in the sample.
These systems and kits of the invention include various components. As used
herein.
the term -component" is broadly defined and includes any suitable apparatus or
collections of
apparatuses suitable for carrying out the recited method. The components need
not be
integrally connected or situated with respect to each other in any particular
way. The
invention includes any suitable arrangements of the components with respect to
each other.
For example. the components need not be in the same room. But in some
embodiments. the
components are connected to each other in an integral unit. In some
embodiments, the same
components may perform multiple functions.
Computer Systems and Computer Readable Media
The system, as described in the present technique or any of its components.
may be
embodied in the form of a computer system. Typical examples of a computer
system include
a general-purpose computer. a programmed microprocessor, a microcontroller, a
peripheral
integrated circuit element, and other devices or arrangements of devices that
are capable of
implementing the steps that constitute the method of the present technique.
A computer system may comprise a computer, an input device, a display unit,
andlor
the Internet. The computer may further comprise a microprocessor. The
microprocessor may
be connected to a communication bus. The computer may also include a memory.
The
memory may include random access memory (RAM) and read only memory (ROM). The
computer system may further comprise a storage device. The storage device can
be a hard
38
CA 03011704 2018-07-17
WO 2017/127-134
PCT/US2017/013955
disk drive or a removable storage drive such as a floppy disk drive, optical
disk drive. etc.
The storage device can also be other similar means for loading computer
programs or other
instructions into the computer system. The computer system may also include a
communication unit. The communication unit allows the computer to connect to
other
databases and the Internet through an I/O interface. The communication unit
allows the
transfer to, as well as reception of data from, other databases. The
communication unit may
include a modem, an Ethernet card, or any similar device which enables the
computer system
to connect to databases and net orks such as LAN, MAN, WAN and the Internet.
The
computer system thus may facilitate inputs from a user through input device,
accessible to the
system through I/O interface.
A computing device typically will include an operating system that provides
executable program instructions for the general administration and operation
of that
computing device, and typically will include a computer-readable storage
medium (e.g.. a
hard disk, random access memory, read only memory, etc.) storing instructions
that. when
executed by a processor of the server. alloy% the computing device to perform
its intended
functions. Suitable implementations for the operating system and general
functionality of the
computing device are known or commercially available, and are readily
implemented by
persons having ordinary skill in the art, particularly in light of the
disclosure herein.
The computer system executes a set of instructions that are stored in one or
more
storage elements, in order to process input data. The storage elements may
also hold data or
other information as desired. The storage element may be in the form of an
information
source or a physical memory element present in the processing machine.
The environment can include a variety of data stores and other memory and
storage
media as discussed above. These can reside in a variety of locations, such as
on a storage
medium local to (and,'or resident in) one or more of the computers or remote
from any or all
of the computers across the netyv ork. In a particular set of embodiments. the
information may
reside in a storage-area network ("SAN-) familiar to those skilled in the art.
Similarly, any
necessary files for performing the functions attributed to the computers,
servers, or other
network devices may be stored locally and/or remotely , as appropriate. Where
a system
includes computing devices, each such device can include hardware elements
that may be
electrically coupled via a bus, the elements including, for example, at least
one central
processing unit (CPU), at least one input device (e.g., a mouse, keyboard.
controller, touch
39
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
screen, or keypad). and at least one output device (e.g.. a display device,
printer, or speaker).
Such a system may also include one or more storage devices, such as disk
drives, optical
storage devices, and solid-state storage devices such as random access memory
("RAM") or
read-only memory ("ROM"). as well as removable media devices. memory cards,
flash cards.
etc.
Such devices also can include a computer-readable storage media reader, a
communications device (e.g., a modem, a network card (wireless or wired), an
infrared
communication device, etc.). and working memory as described above. The
computer-
readable storage media reader can be connected with, or configured to receive,
a computer-
readable storage medium, representing remote, local, fixed, and/or removable
storage devices
as well as storage media for temporarily and/or more permanently containing,
storing,
transmitting, and retrieving computer-readable information. The system and
various devices
also typically will include a number of software applications, modules,
services, or other
elements located within at least one working memory device including an
operating system
.. and application programs. such as a client application or Web browser. It
should be
appreciated that alternate embodiments may have numerous variations from that
described
above. For example. customized hardware might also be used and/or particular
elements
might be implemented in hardware, software (including portable software, such
as applets).
or both. Further, connection to other computing devices such as network
input/output devices
may be employed.
Non-transient storage media and computer readable media for containing code,
or
portions of code, can include any appropriate media known or used in the art,
including
storage media and communication media, such as but not limited to volatile and
non-volatile,
removable and non-removable media implemented in any method or technology for
storage
and/or transmission of information such as computer readable instructions,
data structures.
program modules, or other data. including RAM. ROM, EEPROM, flash memon or
other
memory technology. CD-ROM, digital versatile disk (DVD) or other optical
storage.
magnetic cassettes, magnetic tape. magnetic disk storage or other magnetic
storage devices.
or any other medium which can be used to store the desired information and Vs,
hich can be
accessed by the a system device. Based on the disclosure and teachings
provided herein, a
person of ordinary skill in the art will appreciate other ways and/or methods
to implement the
various embodiments.
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
A computer-readable medium may comprise. but is not limited to. an electronic.
optical, magnetic, or other storage device capable of providing a processor
with computer-
readable instructions. Other examples include, but are not limited to, a
floppy disk, CD-
ROM. DVD, magnetic disk, memory chip, ROM, RAM, SRAM, DRAM, content-
addressable memory ("CAM"). DDR. flash memory such as NAND flash or NOR flash,
an
ASIC, a configured processor, optical storage. magnetic tape or other magnetic
storage, or
any other medium from which a computer processor can read instructions. In one
embodiment, the computing device may comprise a single type of computer-
readable
medium such as random access memory (RAM). In other embodiments, the computing
device may comprise two or more types of computer-readable medium such as
random
access memory (RAM), a disk drive, and cache. The computing device may be in
communication with one or more external computer-readable mediums such as an
external
hard disk drive or an external DVD or Blu-Ray drive.
As discussed above, the embodiment comprises a processor which is configured
to
execute computer-executable program instructions and/or to access information
stored in
memory. The instructions may comprise processor-specific instructions
generated by a
compiler and/or an interpreter from code written in any suitable computer-
programming
language including, for example, C. C++, C#, Visual Basic. Java, Python. Perl,
JavaScript.
and ActionScript (Adobe Systems, Mountain View, Calif.). In an embodiment, the
computing
device comprises a single processor. In other embodiments, the device
comprises two or
more processors. Such processors may comprise a microprocessor. a digital
signal processor
(DSP), an application-specific integrated circuit (AS1C), field programmable
gate arrays
(FPGAs). and state machines. Such processors may further comprise programmable
electronic devices such as PLCs, programmable interrupt controllers (PICs),
programmable
logic devices (PLDs), programmable read-only memories (PROMs), electronically
programmable read-only memories (EPROMs or EEPROMs), or other similar devices.
The computing device comprises a network interface. In some embodiments, the
network interface is configured for communicating via wired or wireless
communication
links. For example. the network interface may allow for communication over
netw orks via
Ethernet, IEEE 802.11 (Wi-Fi). 802.16 (Wi-Max), Bluetooth. infrared. etc. As
another
example, network interface may allow for communication over networks such as
CDMA.
GSM. UMTS, or other cellular communication networks. In some embodiments, the
network
41
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
interface may allow for point-to-point connections with another device, such
as via the
Universal Serial Bus (USB), 1394 FireWire, serial or parallel connections, or
similar
interfaces. Some embodiments of suitable computing devices may comprise two or
more
network interfaces for communication over one or more networks. In some
embodiments. the
computing device may include a data store in addition to or in place of a net
ork interface.
Some embodiments of suitable computing devices may comprise or be in
communication with a number of external or internal devices such as a mouse, a
CD-ROM.
DVD. a keyboard, a display. audio speakers. one or more microphones, or any
other input or
output devices. For example. the computing device may be in communication with
various
user interface devices and a display. The display may use any suitable
technology including,
but not limited to. LCD. LED, CRT. and the like.
The set of instructions for execution by the computer system may include
various
commands that instruct the processing machine to perform specific tasks such
as the steps
that constitute the method of the present technique. The set of instructions
may be in the
form of a soft w are program. Further, the softw are may be in the form of a
collection of
separate programs. a program module with a larger program or a portion of a
program
module, as in the present technique. The software may also include modular
programming in
the form of object-oriented programming. The processing of input data by the
processing
machine may be in response to user commands, results of previous processing.
or a request
made by another processing machine.
While the present invention has been disclosed with references to certain
embodiments, numerous modifications, alterations and changes to the described
embodiments are possible without departing from the scope and spirit of the
present
invention, as defined in the appended claims. Accordingly, it is intended that
the present
invention not be limited to the described embodiments, but that it have the
full scope defined
by the language of the following claims, and equivalents thereof.
Some of the embodiments of the technology described herein can be defined
according to any of the following numbered paragraphs:
(I) A recombinant bacteriophage comprising an indicator gene inserted into a
late gene
region of the bacteriophage CBA120 genome.
(2) The recombinant bacteriophage of paragraph 1. wherein the recombinant
bacteriophage
specifically infects E. coil 0157:H7.
42
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
(3) The recombinant bacteriophage of paragraph 1 or 2, yherein the indicator
gene is codon-
optimized and encodes a soluble protein product that generates an intrinsic
signal or a soluble
enzyme that generates signal upon reaction with substrate.
(4) The recombinant bacteriophage of any of paragraphs 1-3, further comprising
an
untranslated region upstream of the codon-optimized indicator gene. wherein
the untranslated
region includes a bacteriophage late gene promoter and a ribosomal entry site.
(5) A method of preparing a recombinant indicator bacteriophage comprising:
selecting a
wild-type bacteriophage that specifically infects a target pathogenic
bacterium; preparing a
homologous recombination plasmid/vector comprising an indicator gene;
transforming the
homologous recombination plasmid/yector into target pathogenic bacteria:
infecting the
transformed target pathogenic bacteria with the selected wild-type
bacteriophage, thereby
allowing homologous recombination to occur between the plasmid/vector and the
bacteriophage genome: and isolating a particular clone of recombinant
bacteriophage.
(6) The method of paragraph 5, wherein preparing a homologous recombination
plasmicli'vector comprises: determining the natural nucleotide sequence in the
late region of
the genome of the selected bacteriophage: annotating the genome and
identifying the major
capsid protein gene of the selected bacteriophage: designing a sequence for
homologous
recombination downstream of the major capsid protein gene, wherein the
sequence comprises
a codon-optimized indicator gene; and incorporating the sequence designed for
homologous
recombination into a plasmidivector.
(7) The method of paragraph 5 or 6. wherein designing a sequence further
comprises
inserting an untranslated region including a phage late gene promoter and
ribosomal entry
site upstream of the codon-optimized indicator gene.
(8) The method of any of paragraphs 5-7. wherein the homologous recombination
plasmid
comprises an untranslated region including a bacteriophage late gene promoter
and a
ribosomal entry site upstream of the codon-optimized indicator gene.
(9) The method of any of paragraphs 5-8, wherein the wild-type bacteriophage
is CBA120
and the target pathogenic bacterium is E. colt 0157:H7.
(10) The method of any of paragraphs 5-9. W herein isolating a particular
clone of
recombinant bacteriophage comprises a limiting dilution assay for isolating a
clone that
demonstrates expression of the indicator gene.
43
CA 03011704 2018-07-17
WO 2017/127-134
PCT/US2017/013955
(11) A method for detecting E. coli 0157:H7 in a sample comprising: incubating
the sample
with a recombinant bacteriophage derived from CBA120 and detecting an
indicator protein
product produced bv the recombinant bacteriophage, wherein positive detection
of the
indicator protein product indicates that E. coli 0157:H7 is present in the
sample.
(12) The method of paragraph 11. wherein the sample is a food. environmental,
water.
commercial or clinical sample.
(13) The method of paragraph 11 or 12, wherein the method detects as few as
10, 9, 8, 7. 6,
5, 4, 3, 2. or a single bacterium in a sample of a standard size for the food
safety industry.
(14) The method of any of paragraphs 11-13, wherein the sample comprises beef
or
vegetables.
(15) The method of any of paragraphs 11-14, wherein the sample is first
incubated in
conditions favoring growth for an enrichment period of 9 hours or less. 8
hours or less. 7
hours or less. 6 hours or less. 5 hours or less. 4 hours or less, 3 hours or
less. or 2 hours or
less.
(16) The method of any of paragraphs 11-15, wherein the total time to results
is less than 12
hours, less than I I hours, less than 10 hours, less than 9 hours. less than 8
hours, less than 7
hours, or less than 6 hours.
(17) The method of any of paragraphs 11-16, wherein the ratio of signal to
background
generated by detecting the indicator is at least 2.0 or at least 2.5.
(18) A kit for detecting E. coli 0157:H7 comprising a recombinant
bacteriophage derived
from CBA120.
(19) The kit of paragraph 18. further comprising a substrate for reacting with
an indicator to
detect the soluble protein product expressed by the recombinant bacteriophage.
(20) A system for detecting E. coil 0157:H7 comprising a recombinant
bacteriophage
derived from CBA120.
EXAMPLES
Results depicted in the following examples demonstrate detection of a low
number of
cells, even a single bacterium, in a shortened time to results.
Example 1. Creation of Indicator Phage from C13A120
Indicator Phage CBA120NanoLuc was created through homologous recombination
using the following detailed procedures, as illustrated in Figures 1-3.
44
CA 03011704 2018-07-17
WO 2017/127-134
PCT/US2017/013955
The genomic sequence of the CBA120 bacteriophage was av ailable on the
National
Center for Biotechnology Information's GenBank, filed under -Escherichia phage
Cbal 20,-
ID 12291. The genome was fully annotated, though most of the genes were
labeled as
-hypothetical protein,- denoting that automated Open Reading Frame discover
was used.
Hypothetical proteins need only have a start and stop codon. and may not be
expressed. as
DNA regulation (promoters / enhancers / operators, etc.) are not defined in
the sequence.
The late gene region was determined by comparison with other phage genomes.
CBA120, and all other ViI-like phage, fall under the Vil-like phage group
(genus Vii virus or
Vii virus), which are related to T4-like phages. Bacteriophage T4 being the
most studied
bacteriophage, many of the genes homologs could be found, and were labeled as
such. This
includes the late gene region. which consists of the highly expressed phage
structural
proteins. This region was targeted for insertion of the NANOLUC V: reporter
gene. The
major capsid protein was specifically identified. As the major capsid protein
typically has the
highest expression, inserting the reporter directly downstream of the major
capsid protein can
maximize expression of the reporter.
A sequence was designed to insert a codon-optimized NANOLUCg gene
downstream of the major capsid protein. As illustrated in Figure 2, a
homologous
recombination (HR) plasmid was designed. initially with 500 bp upstream and
downstream of
the insert point. Previous HR plasmids using Firefly Luciferase as a reporter
gave poor
transformation, which was alleviated by using a shorter downstream region.
Presumably.
there was a toxic effect with the full 500 bp region selected against in the
bacteria. As such.
the modified downstream region extends only about 300 bp.
The upstream region consisted of the 3 end of the major capsid protein, with
the
insert occurring immediately after the stop codon (TAA): SEQ ID NO: 1
cificatgctggaagttgaagcgaacggtatcggigttgacacccgtcgtggtaaaggcaaccgtgactgtguctccga
acgtggcat
ccgctctggcgatgtctggcatgctggactatgctccggtictgcaggaaaacactaaactggctgitgacccgactgg
ccagaccttc
gctggtgnctgtccaacggtatgcgcgtctatgttgacccgtatgctgtagcagaatatatcaccctggcatacaaagg
cgcaactgcg
ctggatgccggtatcttatcgcgccgtatgtgccgctggaaatgtaccgcacccagggtgaaaccaccttcgctccgcg
tatggcgtt
caaaacccgttacggcatctgtgctaacccgttcgtacagattccggctaaccaagacccgcaggtnacgtgactgctg
acggtaftg
ctcaagacagcaacccgtatttccgcaaaggtctgatcaaatctctgtictaa
This was followed by an Mlul restriction site, then a T4 late gene promoter
consensus
sequence, which consists of the -10 (57 factor consensus binding sequence
(CTAAATAcCcc
(SEQ ID NO: 2)). This promoter was designed based on compositing known -10
sequences.
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
14 random base pairs later, the ribosomal entry site, the Shine-Dalgarno
consensus sequence
(aaggaggt) was inserted, followed by 6 more random base pairs. The random base
pairs were
chosen to keep a similar GC content to other upstream untranslated regions.
SEQ ID NO: 3
acgcgiCTAAATAcCccaaatactag1aga1aaggaggit11cga
A codon-optimized version of Promega's NANOLUC with excretion signal, from
pNL1.3 was inserted. SEQ ID NO: 4
ATGAATAGCTTTAGC ACC AGCGCCTTTGGCCCTGTTGCCTTTAGCCTGGGCCTGCT
GCTGGTTCTGCCGGCAGC ATTTCCGGCCCCGGTGTTCACCCTGGAAGA ________________ 1 I"I TGTG
GGCGATTGGCGCCAGACCGCCGGTTATAACCTGGATCAGGTGCTGGAACAGGGT
GGIGTGAGCAGCCTGITTCAGAATCTGGGCGTGAGCGTGACCCCGATTCAGCGCA
TTGTGCTGAGCGGCGAGAACGGCCTGAAAATTGATATTCATGTTATTATTCCGTA
TGAGGGTCTGAGCGGC GATCAGATGGGCCAGATTGAAAAAATCTTTAAGGTGGT
GTATCCGGTGGACGACCATCATTTCAAGGTGATCCTGCATTACGGCACACTGGTG
ATTGACGGCGTTACCCCGAACATGATCGACTATTTCGGCCGCCCGTATGAAGGTA
TCGCCGTGTTCGACGGCAAGAAAATTACCGTGACCGGTACCCTGTGGAACGGCA
ACAAGATCATTGACGAGCGCCTGATTAACCCGGATGGTAGCCTGCTGTTTCGCGT
GACCATTAATGGCGTGACCGGCTGGCGTCTGTGTGAACGCATCCTGGCCTAA
This was followed by 298 bp of the downstream HR segment. which includes a
hypothetical gene. SEQ ID NO: 5
gcgacaggtMgataacaaaccccgcttcggcggggttatctttatagggatatgtaagataataaagcctcatttatca
aaggaggtta
awitgtctcatcaattatctggcggtgcagtcgatactctattcgttcttttctggatggacctcgtgaagctggggaa
atacctgctaaatc
tggagaagccgaattggcctccctggggtingtaaacgagttgatgttaaaaacgtaccaaaaggtcgagatacacatc
tgtglgtact
caccgaggaaggttacaaatac
Following this, a consensus transcription terminator was inserted along with
stop
codons, which should only function on the plasmid to reduce any read through
and possible
toxic effects. As homologous recombination occurs only at the HR regions, the
transcriptional terminator shouldn't be included in the recombinant phage. SEQ
ID NO: 6
taaTTTGAT AAC AAACC CC GC TTC GGC GGGGTTTTTC TTTATAGG
The full sequence was synthesized into a plasmid (GeneWiz). The plasmid was
transformed into previously prepared E. col! 0157:H7 electroporation competent
cells using
the protocol included in the Bio-Rad MicroPulser Electroporation Apparatus
Operating
Instructions and Applications Guide (catalog # 165-2100).
46
CA 03011704 2018-07-17
WO 2017/127434 PCT/US2017/013955
Synthesized plasmid DNA (pUC57.CBA.HR.NanoLuc ) (4 tig plasmid DNA) was
dissolved in autoclaved filtered deionized water (40 tiL) to make a 100 ng/tit
stock. This
plasmid (1 tiL) was mixed with 20 tiL thawed (on ice) E. coil 0157:H7
electroporation
competent cells (derived from non-toxic E. coil 0157:H7 bacteria, ATCC 43888).
The
cell+DNA mix was transferred to an ice-cold Bio-Rad 0.1 cm electroporation
cuyette, and
subjected to the MicroPulser Electroporation Apparatus using program Ed. The
mix was
immediately transferred into 1 mL Recovery Medium (Life Technologies), and
incubated for
1 hour at 42 C. 220 rpm.
Aliquots of I titõ 100 tiL, and the remainder of the culture concentrated by
centrifugation (2 min ("a 6800g) and resuspended in 100 1i1_, were plated onto
selective
medium (LB+Amp agar plates from Teknova) and incubated overnight at 37 C.
The next day, 23 colonies (+1 negative control) were screened by inoculating
100 tit
LB+Amp and incubating for 2.5 hours at 37 C, then screened for luciferase
activity. 5 tit of
each culture were subjected to Promega NANO-GLO:izi luciferase assay, and read
on a
.. Promega GLOMAX:k 96 luminometer. All 23 colonies Were positive.
The top 3 wells were mixed and inoculated into 4 mL LB+Amp and grown to
1.8x107
cells/mL. Bacteria were infected with wild-type CBA120 bacteriophage from the
Kutter lab
(see Kutter et al., Virology Journal 2011, 8:430) at an MO! of 0.1. and the
homologous
recombination infection Was incubated for 3 hours à, 37 C.
Bacterial concentration was monitored for 4 hours bacteria doubled by 2 hours,
then
began to drop, indicating a successful phage infection.
Example 2. isolation of CBA120NanoLuc
Following homologous recombination to generate recombinant bacteriophage
genomes, a series of titer and enrichment steps was used to isolate a specific
recombinant
bacteriophage that expresses NANOLUoz.
To reduce background NANOLUCt signal from plasmid expression, the lysate was
washed 3 times with TMS in an Amicon Ultra Concentrator, spun to concentrate
the volume
from 4 mL to 500 TMS was added to bring the volume to 4 mL, and this series
was
repeated.
In order to determine the initial ratio of recombinant to wild-type phage,
limiting
dilution assays based on the TCID50 (tissue culture infectious dose 50%) were
used to both
determine the concentration of infectious units (IU/rriL), akin to number of
virus particles or
47
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
plaque forming units, and to determine the number of luciferase transducing
units (TU/mL).
In these assays, the sample was serially diluted, with each dilution aliquoted
into replicate
wells with E. coil 0157:H7 bacteria. Any wells that showed luciferase activity
must have
been infected with at least one recombinant phage. Any wells that showed cell
lysis had been
infected by at least one phage. Based on the highest dilution where each of
these cases
occurred, the original concentrations were back-calculated. These initial
phage mixtures
from transformed cells typically yielded a ratio of 20,000 wild-type 1U for
each recombinant
phage TU. Steps were then taken to isolate and amplify the recombinant phage.
As illustrated in Figure 4, in some experiments recombinant phage were
isolated
from a mixture comprising 0.83% of total phage. The phage mixtures were
diluted into 96
well plates to give an average of 3 recombinant TU per plate, which
corresponds to about 3.8
infectious units (IU) of mostly wild-type phage per well. Bacteria were added
such that each
well contained 50 1_, of turbid E. coli 0157:H7. After 2 hours of incubation
at 37 C, wells
were sampled and screened for the presence of luciferase. Any positive wells
are likely to
have been inoculated with a single recombinant phage, and at this stage the
mixture contained
an enriched ratio of 1 recombinant phage: 3.8 wild-type phage, which is an
enrichment over
the original 1:120 ratio. Of 96 wells screened, 7 were positive. Further
rounds of limiting
dilution assay were not necessary in this experiment.
A plaque assay was performed, wherein plaques ere individually picked and
screened for luciferase transducing ability, insuring about 3 recombinants
were in the mix of
plaques being screened. Each plaque was suspended in 100 uL TMS, and 5 p.L was
added to
a ell containing a turbid E. co/i 0157:H7 culture, and wells were assayed
after incubation
for 45 minutes to 1 hour at 37 C.
Positive wells were expected to contain a pure culture of recombinant phage,
but an
additional round of plaque purification was performed. Finally, large-scale
production was
performed to obtain high titer stocks appropriate for use in the E, coil
0157:H7 detection
assay. Cesium chloride isopycnic density gradient centrifugation was used to
separate phage
particles from contaminating luciferase protein to reduce background.
Example 3. Bacterial Detection Using CBA1101NanoLuc Indicator Phage
Detection of E. coil 0157:H7 using the CBA120NanoLuc Indicator Phage was
tested
in experiments using the basic assay format depicted in Figure 6. First. cell
numbers ranging
from 1-10,000 were taken from cultures and infected with 105. 106, and 107
phage/mL in
48
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
identical sample volumes of LB for 2 hours. Following the addition of lysis
buffer and
NANO-GLOt reagent, the reaction was read using a GLOMAXt 96 instrument. Figure
7
shows that the highest ratio of signal/background was achieved with 106
phage/mL used for
infecting the sample.
Figure 8 shows the data from 6-10 replicates, each using the same cell numbers
from
cell cultures in LB. A phage concentration of 106 phage/mL was used for
infecting the
sample, and infected cells were incubated for 2 hours at 37 C. Following the
addition of
lysis buffer and NANO-GLOt reagent, the reaction was read using a GLOMAXO; 96
instrument. Figure 8 shows that CBA120NanoLuc can detect a single ( I ) cell
with a signal
that is significantly higher than background.
Figure 9 shows from the data of Figure 8 that CBA120NanoLuc can detect a
single
(1) E. coil 0157:H7 cell with a signal to background ratio of >2Ø
The performance of CBA120NanoLuc indicator phage for detecting E. colt 0157:H7
was also certified August 1, 2016 by the AOAC Research Institute (Certificate
No. 081601).
Example 4. Bacterial Detection in Beef Assays Using CBA1201NanoLuc
CBA120NanoLuc was used to detect E. col/ 0157:H7 in beef assays. For all of
the
beef experiments, 50 RLU was used as the background value. and 3 times
background value
was considered positive (i.e., >150 RLU is positive, or Signal/Background >
3.0). There
were no false positives or negatives when compared to the secondary
confirmation method
described below.
For 25 g beef samples, pre-warmed TSB medium (42 C) was added to the sample to
1:3 sample:medium (25g:75mL). The sample NN as blended with a Stomacher for 30
seconds
on low setting/or equivalent, followed by incubation at 42 C without shaking.
The bag was
closed by folding over the top 2-3 times and clipping closed. After 5 hours
(for 10 mL
aliquots in the next step) or 6 hours (for 1 mL aliquots in the next step) of
enrichment at
42 C. the bag NN as gently massaged to thoroughly mix the contents.
An aliquot of either 1 mL or 10 mL was removed from the bag for testing. These
correspond to the "1 mL concentration" or "10 mL concentration" in the data
presented for
all beef assay experiments in Figures 10-16.
Aliquots of 10 mL were centrifuged at 3400g for 5 minutes. the supernatant was
discarded, and the contents were resuspended in 1 mL pre-warmed TSB. The
CBA120
49
CA 03011704 2018-07-17
WO 2017/127434
PCT/11S2017/013955
Indicator Phage Vs as added to infect any target bacteria in the sample by
adding 10 uL of 1 x
108 phage/mL.
Aliquots of 1 mL were centrifuged for 1 minute at the highest speed in a
microfuge.
the supernatant was discarded, and the contents were resuspended in 200 uL pre-
warmed
TSB. To infect target bacteria. 15 uL of 1.2 x 107 phagelmL of the CBA120
Indicator Phage
was added.
Samples with CBA120 Indicator Phage were incubated for 2 hours at 37 C.
vortexed
briefly, centrifuged for 5-10 seconds to pellet debris. and 150 tit sample was
transferred to a
96-well plate (being careful not to disturb debris pellet). Lysis buffer (10
p.L) was added to
each well and gently mixed by pipetting. Freshly prepared NANO-GLOt reagent
(50 ut)
was added to each well and gently mixed by pipetting (or automatically
injected). (NANO-
GLO:0:' reagent was prepared diluting the NANO-GLO Luciferase Assay Substrate
1:50
into NANO-GLOk Luciferase Assay Buffer, e.g., to make 1 mL of NANO-GU:YR)
reagent.
and 20 uL of NANO-GLO: Luciferase Assay Substrate was added to 1 mL of NANO-
GLOCk, Luciferase Assay Buffer.)
The plate was read on a GLOMAXt 96 instrument 3 minutes after substrate
addition.
Secondary Confirmation Method:
Confirmation of E. coil 0157:H7 was performed on overnight-enriched cultures
using
immuno-magnetic separation (IMS) with particles coated with 0157 antibodies
(DYNABEADS'..k. Life Technologies #71004) and plating onto selective plates
(CHROMAGAR:k plates. BD #214984).
To prepare for the confirmation, the samples were incubated overnight (18-24
hours
total or 13-19 additional hours) at 42 C + 1 . From the overnight culture, 1
mL was removed
and the DYNABEADSik, anti-E. coh 0157 procedure was followed. Briefly. 201.11
of IMS
particles were added to the diluted overnight culture and incubated for 10
minutes at room
temperature. Magnetic particles NN ere isolated for 3 minutes with the magnet,
then washed 3
times with PBS, 1 ml per wash. After the final wash_ particles were plated
onto
CHROMAGARt3; plates (BD #214984) and incubated 18-24 hours at 37 C 1 .
Mauve-colored colonies (presumptive positive) 1\ ere cultured in TSB media
overnight
(18-24 hours) at 37 C + 1 for serological confirmation. Presence of 0157 and
H7 antigens
was determined using an agglutination assay (Remel Wellcolex E. coil 0157:H7
#R30959601). The manufacturer's instructions were followed_ using 40 uL of the
overnight
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
culture. Results confirmed presence or absence of 0157 and/or H7 antigens and
provided
confirmation for E. colt 0157:H7.
Data from 25 g beef samples are shown in Figures 10-12. Figures 10-11
correspond
to the 1 mL concentration and Figure 12 to the 10 mL concentration of enriched
samples. All
positives IN ere detected after 6 hours enrichment for the 1 mL concentration
and after 5 hours
enrichment for the 10 mL concentration. Figures 11-12 show confirmation by
DYNABEADS t/CHROMAGAR plating.
For larger (125 g) beef samples, experiments were performed with both ground
beef
and beef trim. The procedure was similar, except that beef trim samples
required treatment
with the stomacher for at least 120 seconds on high setting/or equivalent.
Enrichment for
either sample type followed for 8 hours at 42 C, and the rest of the procedure
was as
described above.
Data from 125 g beef samples are shown in Figures 13-16. Figures 13 and 15
correspond to the I mL concentration and Figures 14 and 16 correspond to the
10 mL
concentration. Figures 15-16 show confirmation by DYNABEADSk/CHROMAGARO,
plating. All positives were detected after 7 hours of enrichment.
Example 5. Vegetable Wash Assays
Data from spinach wash filter assays are shown in Figure 17, which shows that
the
assay can detect 1 cell of E. coil 0157:H7 in 100 mL of spinach wash following
3 hours of
enrichment. These results were confirmed by using the DYNABEADS(CCHROMAGARg
tests on overnight cultures of each sample according to the manufacturer's
instructions, as
described in the -Secondary Confirmation Method- above.
To prepare the vegetable wash, vegetable leaves (e.g., spinach or lettuce)
were
weighed and added to a clean plastic bag. Five mL of water was added per each
gram (g) of
vegetable. Leaves and solution were mixed manually for a few minutes. Liquid
was then
extracted from the plastic bag and used as the "vegetable wash." Using this
method. - I
million bacteria were found by CFU to reside on a single spinach leaf (1-2 g).
Next. 100 mL of -vegetable wash- was vacuum filtered through a 47 mm 0.45 tiM
filter. The filter was remoNed and placed in a small sealable plastic bag.
Prewarmed (42 C)
TSB medium (600 IA) was added to the bag to cover the filter. The filter was
then incubated
at 42 C for 3 hours with gentle agitation. An aliquot of enriched media (300
ttL) was
removed for confirmation purposes. CBA120NanoLuc Indicator Bacteriophage was
then
51
CA 03011704 2018-07-17
WO 2017/127434
PCT/US2017/013955
added to the remaining medium in the bag to a final concentration of 1 x 106
phage/mL, and
the bag was agitated gently followed by incubation for 2 hours at 37 C.
Finally, 100-150 tiL
of the infection reaction was transferred to a 96-well plate. Lysis buffer (10
L) and prepared
NANO-GLOg, reagent (50 L) were added and the sample was read on a luminometer
(GLOMAXt 96).
Figure 17 shows data from a spinach wash assay, including confirmatory results
from
DYNABEADSX/CHROMAGARX plating. The ability to discern a single target
bacterial
cell from 10 non-target bacteria in vegetable wash is surprising and again
demonstrates the
specificity and sensitivity of the assay.
52