Note: Descriptions are shown in the official language in which they were submitted.
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
VIRAL AND ONCO VIRAL NUCLEASE TREATMENT
Cross-Reference to Related Applications
This application claims benefit of U.S. Provisional Application Serial No.
62/299,792,
filed February 25, 2016, and U.S. Provisional Application Serial No.
62/299,839, filed February
25, 2016, both incorporated by reference.
Technical Field
The invention relates to viral and oncoviral treatment.
Background
Millions of people die each year from cancer. Evidence shows a link between
cancer and
infectious disease. In fact, it is understood that infectious disease
represents the third leading
cause of cancer worldwide. De Flora, 2015, J Prey Med Hyg 56:E15-E20.
Unfortunately, viruses
and cancer are difficult to successfully treat. Some cancer drugs, for
example, may slow the
growth of a tumor yet leave behind affected cells that may proliferate again
after treatment.
Some viruses, including known oncoviruses, exhibit an asymptomatic latent
phase during which
they present no activity or proteins to target with a treatment.
As such, oncoviruses and their resultant tumors may be some of the most
difficult to
treat. Even if a cancer drug successfully removes a tumor, latent viral genes
may later be
expressed if the virus re-enters an active stage of infection. When the virus
re-enters the active
stage of infection, it may trigger cell proliferation resulting in new tumors
such as Burkitt's
lymphoma in the case of Epstein-Barr or a squamous cell carcinoma in the case
of Human
Papilloma Virus.
Even if a viral treatment had good prospects for clearing the infection,
cancerous cells
may still proliferate. That is, even in cases where an oncoviral infection may
have been a causal
factor, tumors may continue to grow once the infection is removed.
1
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
Summary
Compositions and methods for treating tumors include a cancer therapeutic
(i.e., a drug
intended to stop or slow tumor growth or induce cell death in cancer cells)
and a nuclease to
degrade genetic material in the tumor. The nuclease digests nucleic acid from
the tumor genome
or from an oncovirus. The nuclease complements the therapeutic effect of the
cancer drug. For
example, a cancer chemotherapy typically acts to selectively kill tumor cells.
However, no
therapeutic is 100% effective. In combination with the nuclease, the therapy
kills or disables a
greater number of cancer cells. Thus, the nuclease adds a layer of protection
by preventing
proliferation in tumor cells not killed by the cancer drug. Additionally, a
nuclease with a
mechanism of action orthogonal to that of a cancer therapeutic may have
additive or synergistic
effects. Thus, combination of an endonuclease with cancer therapeutics may
facilitate
administering lower dosage of cancer therapeutics, which often have dose-
limiting toxicities
associated in healthy tissues that have higher division rates, e.g. gut
epithelium. Using a
nuclease with a cancer drug may be particularly beneficial where a tumor is
associated with an
infection by an oncovirus, as the cancer drug can cause cell death while the
nuclease can cleave
viral nuclease preventing recurrence of an active oncoviral infection. Thus it
may be preferable
to use a nuclease that preferentially cuts oncoviral nucleic acid over human
genetic material.
Nucleases that are directed to specific targets include transcription-
activator-like effector
nucleases (TALENs), meganucleases, zinc-finger nucleases (ZFNs), and CRISPR-
associated
(Cas) nucleases. Preferred embodiments use a nuclease that may be targeted to
oncoviral DNA
along with a cancer drug. For example, a Cas9 endonuclease and a guide RNA
that matches a
target in a viral genome without having any corresponding match in the human
genome can be
delivered along with an anti-apoptotic inhibitor. For treating an oncovirus
such as Epstein-Barr
virus (EBV), the guide RNA can program Cas9 to degrade key EBV genes while the
chemotherapeutic stops the proliferation of a lymphoma. In another example,
human
papillomavirus (HPV) can be treated using a targetable nuclease to target
genes of the HPV
genome and a chemotherapeutic such as cisplatin to trigger cell death in a
cervical carcinoma. In
another example, merkel cell carcinoma (MCC) associated with merkel cell
polyomavirus
(MCV) may be targeted with MCV-specific guide RNA in combination with
carboplatin and
etoposide. Compositions and methods of the invention attack cancers of
infectious origin at two
defining points: both the causative infecting virus and the cancerous
proliferation of cells.
2
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
The nuclease may be delivered as an active protein¨or ribonucleoprotein in the
case of a
Cas-type nuclease¨or encoded in a vector, such as a plasmid or mRNA, in a
viral vector, such
as adeno-associated virus (AAV), or in a lipid or solid nanoparticle. The
nuclease may be
delivered via a pharmaceutically acceptable composition that also includes the
cancer drug, or
the two may be separately delivered to treat the tumor.
In certain aspects, the invention provides a composition for treating a tumor.
The
composition includes a cancer drug and a nuclease. The cancer drug may be
actinomycin, all-
trans retinoic acid, anthracycline, bleomycin, bortezomib, carboplatin,
carfilzomib, capecitabine,
cisplatin, chlorambucil, cyclophosphamide, cytarabine, daunorubicin,
disulfiram, docetaxel,
doxifluridine, doxorubicin, epirubicin, epothilone, epoxomicin, etoposide,
fluorouracil,
gemcitabine, hydroxyurea, idarubicin, imatinib, interferon alpha, irinotecan,
ixazomib,
lactacystin, mechlorethamine, mercaptopurine, methotrexate, mitoxantrone,
oxaliplatin,
paclitaxel, pemetrexed, salinosporamide A, tenipo side, topotecan, valrubicin,
vinblastine,
vincristine, vindesine, vinorelbine, venetoclax. The cancer drug may be a
biologic. The cancer
drug may be a monoclonal antibody (mAb) that targets cell-specific surface
antigens. A suitable
monoclonal antibody may include, e.g., rituximab, bevacizumab, or
pembrolizumab. Rituximab
(Rituxan) may function as an anti-CD20 to deplete B cells. The cancer drug may
be an immune
checkpoint inhibitors such as, e.g., anti-PD-1 or anti-VEGF. The cancer drug
may be a
recombinant cytokine such as, for example, Interleukin 2 (IL-2), Interleukin
11 (IL-11), or
Interleukin 15 (IL-15). The nuclease may be, for example, an endonuclease, an
exonuclease,
DNase I, a CRISPR-associated nuclease, Cfpl, a transcription-activator-like
effector nuclease, a
meganuclease, and a zinc-finger nuclease.
In certain embodiments, the nuclease preferentially cuts nucleic acid of a an
oncovirus
such as a human papilloma virus (HPV), an Epstein-Barr virus (EBV), Kaposi's
sarcoma-
associated herpesvirus (KSHV), hepatitis B virus (HBV), hepatitis C virus
(HCV), human T-cell
lymphotrophic virus type I (HTLV-I), and Merkel cell polyomavirus (MCV). The
nuclease may
be a CRISPR-associated nuclease, and the composition further include a guide
RNA
complementary to a portion of the nucleic acid.
The cancer drug may be a proteasome inhibitor such as lactacystin, bortezomib,
disulfiram, salinosporamide A, carfilzomib, epoxomicin, and ixazomib. In one
embodiment, the
nuclease is Cas9 and the oncovirus is Epstein-Barr virus. The cancer drug may
be bortezomib.
3
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
The composition may include an antiviral treatment such as ganciclovir or
Gardasil. The
composition may include an epigenetic modifier such as a DNA methyltransferase
(DNMT)
inhibitor or a histone deacetylase inhibitor (HDI or HDACi), such as
vorinostat or panobinostat.
Aspects of the invention provide a composition that includes a cancer drug and
a vector
nucleic acid, such as a plasmid, encoding a nuclease, wherein the cancer drug
and the nuclease
are as described above.
Aspects of the invention provide a method for treating cancer. The method
includes
delivering a cancer drug and a nuclease to a tumor. The cancer drug may be
actinomycin, all-
trans retinoic acid, anthracycline, bleomycin, bortezomib, carboplatin,
carfilzomib, capecitabine,
cisplatin, chlorambucil, cyclophosphamide, cytarabine, daunorubicin,
disulfiram, docetaxel,
doxifluridine, doxorubicin, epirubicin, epothilone, epoxomicin, etoposide,
fluorouracil,
gemcitabine, hydroxyurea, idarubicin, imatinib, interferon alpha, irinotecan,
ixazomib,
lactacystin, mechlorethamine, mercaptopurine, methotrexate, mitoxantrone,
oxaliplatin,
paclitaxel, pemetrexed, salinosporamide A, tenipo side, topotecan, valrubicin,
vinblastine,
vincristine, vindesine, and vinorelbine. The nuclease may be, for example, an
endonuclease, an
exonuclease, DNase I, a CRISPR-associated nuclease, Cfpl, a transcription-
activator-like
effector nuclease, a meganuclease, and a zinc-finger nuclease.
In certain embodiments, the nuclease preferentially cuts nucleic acid of a an
oncovirus
such as a human papilloma virus (HPV), an Epstein-Barr virus (EBV), Kaposi's
sarcoma-
associated herpesvirus (KSHV), hepatitis B virus (HBV), hepatitis C virus
(HCV), and human T-
cell lymphotrophic virus type I (HTLV-I). The nuclease may be a CRISPR-
associated nuclease,
and the composition further include a guide RNA complementary to a portion of
the nucleic acid.
The cancer drug may be a proteasome inhibitor such as lactacystin, bortezomib,
disulfiram, salinosporamide A, carfilzomib, epoxomicin, and ixazomib. In one
embodiment, the
nuclease is Cas9 and the oncovirus is Epstein-Barr virus. The cancer drug may
be bortezomib.
The method may include delivering an antiviral treatment such as ganciclovir
or Gardasil.
The method may include delivering an epigenetic modifier such as a DNA
methyltransferase
(DNMT) inhibitor or a histone deacetylase inhibitor (HDI).
A repair inhibition aspect of the disclosure provides compositions and methods
for
treating a viral infection in a patient by selectively cleaving viral nucleic
acid and preventing
subsequent repair of the viral nucleic acid. A nuclease, such as Cas9, and a
DNA repair inhibitor
4
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
are delivered to infected cells. The nuclease specifically cuts the viral
nucleic acid (e.g., under
the guidance of a guide RNA that does not have any match in a human genome).
The inhibitor
prevents a repair mechanism such as end-joining, synthesis, or ligation. The
combination of gene
editing and inhibition of viral nucleic acid repair act in concert to reduce
or eliminate the effects
of the viral infection. In the case of an oncovirus, this means the reduction
or elimination of the
oncogenic effects of the virus. The combination preferably works without
disrupting host
genomic material (i.e., other than integrated viral sequence). The repair
inhibition embodiment
works on integrated as well as non-integrated virus and is equally effective
on latent and active
virus.
Combination therapies of the repair inhibition embodiment preferably include a
nuclease,
such as Cas9 or a Cas9 variant that is targeted toward oncoviral sequence. It
is recognized
however, that any targeted endonuclease is useful including, but not limited
to, Cas6, Cas5,
Cfpl, a zinc finger nuclease (ZFN), a meganuclease, a transcription activator-
like effector
nuclease (TALEN), or a variant of any of the foregoing. In addition to the
nuclease component,
there is a component that is useful in inhibiting ligation of viral sequence
that has been cleaved.
A ligation inhibitor may be a small molecule that prevents end-joining repair,
an enzyme that
removes a 5' phosphate or 3' hydroxyl, an enzyme that adds blocking groups or
fragments of
DNA that are blocked or that lack an accessible 5' phosphate or 3' hydroxyl,
or other such
moieties. Repair can be inhibited by inhibiting an end-joining repair pathway
or by interfering
with synthesis or ligation, e.g., by preventing function of a synthetase or a
ligase. Additionally or
alternatively, repair may be inhibited by enhancing cell exonuclease activity,
e.g. to increase
degradation of SSB and DSB (single and double strand DNA breaks). For example,
human
exonuclease 1 (hEX01) efficiently repairs DSB. hEX01 is ubiquitinated and
degraded in the
proteasome. Thus, in some embodiments, a combination of a targeted
endonuclease with a
proteasome inhibitor are delivered to enhance hEX0-1 activity and synergize to
kill viral DNA+
cells.
The inhibitor may be co-delivered with the targeted nuclease to suppress
activation of
homologous or non-homologous end repair mechanisms in the resulting fragments.
End repair
mechanisms may be inhibited, for example, by a treatment that includes a small
molecule such as
KU55933, caffeine, or wortmannin.
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
In certain aspects, the repair inhibition embodiment provides a system for
treating cells
with a viral infection, e.g. cells that contain viral nucleic acid. The system
includes a nuclease, a
targeting sequence, and a DNA repair inhibitor. The target nucleic acid is
typically viral nucleic
acid. However, any appropriate nucleic acid may be targeted. In preferred
embodiments, the
system is used to degrade any foreign nucleic acid including, for example,
sequences from
intracellular parasites such as malaria or intracellular bacteria or
mycobacteria such as
tuberculosis. The targeting sequence directs the nuclease to the viral nucleic
acid, the nuclease
cuts the viral nucleic acid into fragments, and the inhibitor prevents repair
of the fragments. The
targeting sequence may include one or more guide RNAs. The nuclease may
include one or more
of a zinc-finger nuclease, a TALENs nuclease, a meganuclease, a Cas9
endonuclease, or others
known in the art. Preferably, the nuclease is Cas9, encoded along with a guide
RNA that
specifically targets the target nucleic acid. In a preferred embodiment, the
nuclease is obtained or
delivered in a ribonucleoprotein (RNP) form, e.g. as a recombinant Cas9
protein duplexed with
sgRNA or with crRNA + tracRNA, or as a recombinant TALEN protein. It may be
found that
delivery as RNP is more effective and less toxic than plasmid DNA, and that
RNP permits
delivery of pre-formed enzymatically active drug (which acts faster), and is
only active in the
cell for a very limited time (<24 hours), thus reducing non-specific toxicity
and off-target
activity. RNP can be directly electroporated into primary tissues, e.g.
peripheral blood
mononuclear cells (PBMCs), for ex vivo transplant indications. RNP, like mRNA
or pDNA, can
also be incorporated into cationic lipid nanoparticles for in vivo delivery
indications, e.g. cancer.
In certain embodiments, the inhibitor prevents homologous or non-homologous
end repair of the
one or more fragments. For example, the treatment may be a small molecule such
as KU55933,
caffeine, VE-821, NU6027, UNC-01, mirin, RI-1, streptonigrin, RI-2, 3-ABA,
olaparib,
NU1025, NSC130813, wortmannin, NU7026, SCR7, or L189. Additionally or
alternatively, the
inhibitor may include an enzyme or protein that functions to inhibit
components essential to end-
repair processes. In one example, the treatment may include the enzyme
Antarctic phosphatase,
which removes the 5' phosphate from DNA and RNA ends. In some embodiments, it
is
recognized that the double-stranded breaks (DSBs) introduced by Cas-type
nuclease are
primarily repaired via non-homologous end joining (NHEJ) and that DNA ligase
IV (LIG4) is
critical for NHEJ. Other LIGs (1-3) are involved in repair of SSB and DSB.
Systems and
methods described herein may include one or more small molecule inhibitors of
LIG4 or other
6
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
LIGs. For example, the compound L82 has been identified as an uncompetitive
inhibitor of DNA
ligase I. L67 is a compound that inhibits LIG1 and LIG3. Other compounds that
have been
identified as inhibitors of a DNA Ligase may be used. Additionally or
alternatively, the inhibitor
may include the delivery of siRNAs that inhibit the function of DNA ligase or
other enzymes
that involved in repair process.
In other embodiments, the inhibitor includes a nucleic acid fragment that is
ligated to an
exposed viral end, wherein the newly-added end (provided by the inhibitor)
lacks either or a 5'
phosphate or a 3' hydroxyl (e.g., it may provide a chain-terminating
nucleotide). In certain
embodiments, the inhibitor includes one or more dideoxy-nucleotides, which
terminate nucleic
acid synthesis when incorporated. The inhibitor may include an enzyme that
removes or that
blocks a 3' hydroxyl or 5' phosphate. Any enzyme or moiety that results in
fragments lacking
fragment ends that are accessible for ligation or polymerization may be used.
Aspects of the repair inhibition embodiment provide methods for treating cells
infected
with a virus. Methods includes obtaining a nuclease that is designed to cut a
target viral nucleic
acid and an inhibitor that prevents repair of the cut viral nucleic acid.
Preferably, the nuclease
cuts the viral nucleic acid without cutting a portion of the human genome
important for normal
cellular function. Suitable targets in viral genomes include, but are not
limited to, a portion of a
genome or gene of a hepatitis virus, a hepatitis B virus (HBV), an Epstein-
Barr virus, a Kaposi's
sarcoma-associated herpesvirus (KSHV), a herpes-simplex virus (HSV), a
cytomegalovirus
(CMV), human papilloma virus (HPV), and Merkel cell polyomavirus. The target
in the viral
genome may lie within one or more of a preC promoter in a hepatitis B virus
(HBV) genome, an
51 promoter in the HBV genome, an S2 promoter in the HBV genome, an X promoter
in the
HBV genome, a viral Cp (C promoter) in an Epstein-Barr virus genome, a minor
transcript
promoter region in a Kaposi's sarcoma-associated herpesvirus (KSHV) genome, a
major
transcript promoter in the KSHV genome, an Egr-1 promoter from a herpes-
simplex virus
(HSV), an ICP 4 promoter from HSV-1, an ICP 10 promoter from HSV-2, a
cytomegalovirus
(CMV) early enhancer element, a cytomegalovirus immediate-early promoter, an
HPV early
promoter, and an HPV late promoter.
In a preferred embodiment of the repair inhibition embodiment, the nuclease is
obtained
or delivered in a ribonucleoprotein (RNP) form, e.g. as a recombinant Cas9
protein duplexed
with sgRNA or with crRNA + tracRNA, or as a recombinant TALEN protein. It may
be found
7
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
that delivery as RNP is more effective and less toxic than plasmid DNA, and
that RNP permits
delivery of pre-formed enzymatically active drug (which acts faster), and is
only active in the
cell for a very limited time (<24 hours), thus reducing non-specific toxicity
and off-target
activity. RNP can be directly electroporated into primary tissues, e.g.
peripheral blood
mononuclear cells (PBMCs), for ex vivo transplant indications. RNP, like mRNA
or pDNA, can
also be incorporated into cationic lipid nanoparticles for in vivo delivery
indications, e.g. cancer.
The repair inhibition embodiment may further involve one or more vectors or
carriers for
delivering the nuclease, targeting sequence, inhibitor, or combination thereof
into cells of a
patient. In certain embodiments, a vector, such as a plasmid, encodes any one
or more of the
nuclease, the targeting sequence, and the inhibitor. In other embodiments, a
first vector encodes
the nuclease and the targeting sequence, and a second vector encodes the
inhibitor. In certain
embodiments, the nuclease and optionally a targeting sequence such as a gRNA
or sgRNA are
encoded on a vector such as a plasmid, and the treatment is a small molecule.
Suitable non-viral
vectors include a plasmid, a nanoparticle, a cationic lipid, a cationic
polymer, a metallic
nanopolymer, a nanorod, a liposome, a micelle, a microbubble, a cell-
penetrating peptide, and a
liposphere. In some instances, the vector may be a viral vector. Suitable
viral vectors include
retrovirus, lentivirus, adenovirus, herpes virus, pox virus, alpha virus, and
adeno-associated
viruses.
In preferred embodiments of the repair inhibition embodiment, a vector that
encodes the
nuclease also encodes the targeting sequence, which then guides the nuclease
to a target on a
genome of a virus. The targeting sequence is typically a guide RNA. The
targeting sequence
preferably matches the target according to a predetermined criteria and does
not match any
portion of a host genome according to the predetermined criteria (e.g., is at
least 60%
complementary within a 20 nucleotide stretch and presence of a protospacer
adjacent motif
adjacent the 20 nucleotide stretch). The guide sequence should not match any
portion of the host
genome (e.g., human genome) according to the predetermined criteria.
Alternatively, compositions of the repair inhibition embodiment may be
delivered via a
liposome, a cell-penetrating peptide, a nanoparticle, polymers, glycopolymers,
transfection,
electroporation, or any other suitable carrier or technique.
In some aspects, the repair inhibition embodiment provides a pharmaceutical
composition
comprising any of the nucleic acids described above. The pharmaceutical
composition may
8
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
include a transfection-facilitating cationic lipid formulation. The
pharmaceutical composition
includes appropriate diluents, adjuvants, and carriers for delivering the
active components to
targeted cells. The carrier may be, for example, a liposome, a nanoparticle, a
peptide, a polymer,
a lipid, or a nanoplex. The formulation may include standard pharmacologic
formulations,
including timed release formulations and other well-known pharmaceutical
formulations.
In related aspects, the repair inhibition embodiment provides for the use of
any of the
compounds or molecules described above in the manufacture of a medicament for
treatment of a
viral infection, preferably a latent viral infection.
Brief Description of the Drawings
FIG. 1 diagrams a cancer treatment method.
FIG. 2 shows a cancer treatment composition.
FIG. 3 shows a composition that includes a nuclease and a chemotherapeutic.
FIG. 4 shows a plasmid that encodes the Cas9 protein.
FIG. 5 shows gRNA targets along a reference genome.
FIG. 6 depicts a proteasome inhibitor.
FIG. 7 illustrates gene delivery with an AAV vector.
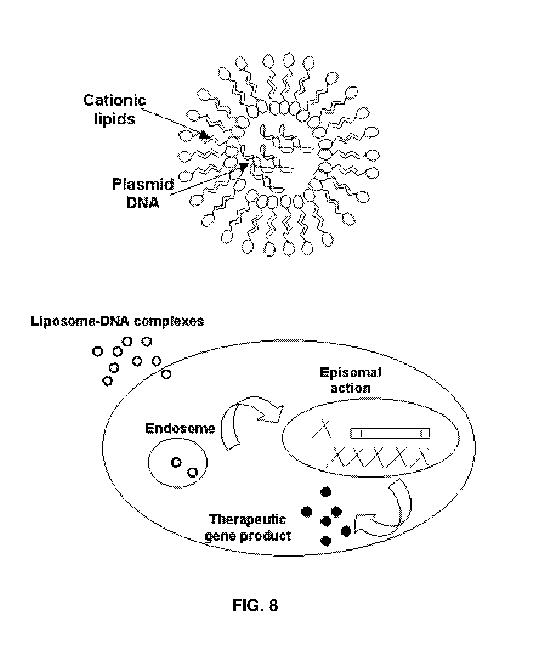
FIG. 8 shows delivery by liposome.
FIG. 9 shows genomic context around guide RNA sgEBV2 and PCR primer locations.
FIG. 10 shows a large deletion induced by sgEBV2.
FIG. 11 gives genome context around guide RNA sgEBV3/4/5 and PCR primer
locations.
FIG. 12 shows large deletions induced by Cas9.
FIG. 13 shows results confirmed by Sanger sequencing.
FIG. 14 shows several cell proliferation curves after different CRISPR
treatments.
FIG. 15 shows nuclear morphology before sgEBV1-7 treatment.
FIG. 16 shows nuclear morphology after sgEBV1-7 treatment.
FIG. 17 shows EBV load after different CRISPR treatments.
FIG. 18 gives a histogram of EBV quantitative PCR Ct values before treatment.
FIG. 19 gives a histogram of EBV quantitative PCR Ct values after treatment.
FIG. 20 diagrams a method of a repair inhibition embodiment.
9
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
FIG. 21 shows a nucleic acid that encodes a nuclease, a targeting sequence and
an
inhibitor of DNA repair.
FIG. 22 shows a plasmid according to certain embodiments.
FIG. 23 shows the results of successfully cleaving the HPV genome using Cas9
endonuclease, a gRNA for E6, and a gRNA for E7.
FIG. 24 shows a gel resulting from an in vitro CRISPR assay against HBV.
FIG. 25 shows three small molecule inhibitors of DNA ligase.
Detailed Description
The invention provides compositions and methods for treating or preventing
oncoviral
infections and tumors. Compositions and methods according to the disclosure
use a nuclease
such as one that may be targeted to viral nucleic acid. For example, a Cas9
nuclease uses a
targeting sequence, or guide RNA, to target the viral nucleic acid. The
targeted cells are treated
with the nuclease and a cancer drug. Each of those treatment modalities are
introduced to the
target cells. The nuclease cuts the viral nucleic acid and the cancer drug
exhibits its
chemotherapeutic effect. Either or both of the nuclease and cancer drug may be
provided in a
pharmaceutically acceptable composition.
i. Oncoviral treatment
FIG. 1 diagrams a cancer treatment method that includes treating cells of a
patient with a
nuclease that cuts nucleic acid of an oncogenic virus and a cancer drug.
Methods include
obtaining a nuclease (e.g., as a protein, ribonucleoprotein, or encoded by a
plasmid). Any
suitable nuclease may be used. In a preferred embodiment, the nuclease is a
CRISPR-associated
nuclease or similar, such as Cas9, Cas6, a modified Cas9, a modified Cas6,
Cfpl, or similar
(collectively, "Cas-type nuclease"). Where a Cas-type nuclease is used, a
targeting sequence is
also used, where a targeting sequencing is an RNA oligomer, which may be about
20 bases long.
In some embodiments, the nuclease comprises Cas9 complexed with a guide RNA
complementary to a portion of the nucleic acid. Methods further include
delivering a cancer
drug. A cancer drug may be selected for any suitable mechanism of action
including, for
example, proteasome inhibition, transcription inhibition, inhibition of
topoisomerase, chromatin
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
remodeling action, inhibition of nucleotide synthesis, causation of DNA cross-
linking, inhibition
of DNA synthesis, affecting tubulin or microtubule binding, or others.
The nuclease (in active form or encoded in nucleic acid) are delivered to the
cells of the
patient. There, the nuclease cleaves nucleic acid of the oncovirus. For
example, the nuclease may
cleave DNA or RNA genome products (e.g., episomal, integrated, or otherwise)
or may cleave
transcripts. Through the use of the targeting sequence, the nuclease leaves
intact important
portions of the host genome necessary for healthy function. The cancer drug
aids in treating or
preventing cell proliferation through its preferred mechanism of action.
Methods of the invention are applicable to in vivo treatment of patients and
may be used
to remove any viral genetic material such as genes of virus associated with a
latent viral
infection. Methods may be used in vitro, e.g., to prepare or treat a cell
culture or cell sample.
When used in vivo, the cell may be any suitable germ line or somatic cell and
compositions of
the invention may be delivered to specific parts of a patient's body or be
delivered systemically.
If delivered systemically, it may be preferable to include within compositions
of the invention
tissue-specific promoters. For example, if a patient has a latent viral
infection that is localized to
the liver, hepatic tissue-specific promoters may be included in a plasmid or
viral vector that
codes for a targeted nuclease.
Any suitable oncovirus may be targeted using methods and compositions of the
invention. For example, a human papilloma virus (HPV), an Epstein-Barr virus
(EBV), Kaposi's
sarcoma-associated herpesvirus (KSHV), hepatitis B virus (HBV), hepatitis C
virus (HCV),
human T-cell lymphotrophic virus type I (HTLV-I), or Merkel cell polyomavirus
(MCV) may be
treated.
FIG. 2 shows a composition for treating a viral infection according to certain
embodiments. The composition preferably includes a vector (which may be a
plasmid, linear
DNA, or a viral vector) that codes for a nuclease and a targeting moiety
(e.g., a gRNA) that
targets the nuclease to viral nucleic acid and a chemotherapeutic such as
etoposide. The vector
may optionally include one or more of a promoter, replication origin, other
elements, or
combinations thereof as described further herein.
In some embodiments, the invention provides a nucleic acid encoding at least
(i) a Cas9
nuclease and (ii) a guide RNA (gRNA) complementary to a portion of the Epstein-
Barr genome
as well as a chemotherapeutic such as etoposide, preferably all of the
components of EPOCH
11
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
with rituximab (rituximab, etoposide, prednisolone, oncovin: vincristine,
cyclophosphamide, and
hydroxydaunorubicin: doxorubicin. In a related embodiment, what is provided
includes (i) a
Cas9 nuclease and (ii) a guide RNA (gRNA) complementary to a portion of the
Epstein-Barr
genome as well as a chemotherapeutic such as etoposide, preferably all of the
components of
EPOCH with rituximab (rituximab, etoposide, prednisolone, oncovin:
vincristine,
cyclophosphamide, and hydroxydaunorubicin: doxorubicin.
ii. Nuclease
Methods of the invention include using a programmable or targetable nuclease
to
specifically target viral nucleic acid for destruction. Any suitable targeting
nuclease can be used
including, for example, zinc-finger nucleases (ZFNs), transcription activator-
like effector
nucleases (TALENs), clustered regularly interspaced short palindromic repeat
(CRISPR)
nucleases, meganucleases, other endo- or exo-nucleases, or combinations
thereof. See Schiffer,
2012, Targeted DNA mutagenesis for the cure of chronic viral infections, J
Virol 88(17):8920-
8936, incorporated by reference.
CRISPR methodologies employ a nuclease, CRISPR-associated (Cas9), that
complexes
with small RNAs as guides (gRNAs) to cleave DNA in a sequence-specific manner
upstream of
the protospacer adjacent motif (PAM) in any genomic location. CRISPR may use
separate guide
RNAs known as the crRNA and tracrRNA. These two separate RNAs have been
combined into a
single RNA to enable site-specific mammalian genome cutting through the design
of a short
guide RNA. Cas9 and guide RNA (gRNA) may be synthesized by known methods.
Cas9/guide-
RNA (gRNA) uses a non-specific DNA cleavage protein Cas9, and an RNA oligo to
hybridize to
target and recruit the Cas9/gRNA complex. See Chang et al., 2013, Genome
editing with RNA-
guided Cas9 nuclease in zebrafish embryos, Cell Res 23:465-472; Hwang et al.,
2013, Efficient
genome editing in zebrafish using a CRISPR-Cas system, Nat. Biotechnol 31:227-
229; Xiao et
al., 2013, Chromosomal deletions and inversions mediated by TALENS and
CRISPR/Cas in
zebrafish, Nucl Acids Res 1-11.
CRISPR(Clustered Regularly Interspaced Short Palindromic Repeats) is found in
bacteria
and is believed to protect the bacteria from phage infection. It has been used
to introduce
insertions or deletions as a way of increasing or decreasing transcription in
the DNA of a
targeted cell or population of cells. See for example, Horvath et al., Science
(2010) 327:167-170;
12
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
Terns et al., Current Opinion in Microbiology (2011) 14:321-327; Bhaya et al.
Ann Rev Genet
(2011) 45:273-297; Wiedenheft et al. Nature (2012) 482:331-338); Jinek Metal.
Science
(2012) 337:816-821; Cong Let al. Science (2013) 339:819-823; Jinek M et al.
(2013) eLife
2:e00471; Mali Pet al. (2013) Science 339:823-826; Qi LS et al. (2013) Cell
152:1173-1183;
Gilbert LA et al. (2013) Cell 154:442-451; Yang H et al. (2013) Cell 154:1370-
1379; and Wang
H et al. (2013) Cell 153:910-918).
FIG. 3 shows a nuclease 201 and a cancer drug 251. Here, the nuclease 201 is
illustrated
as a ribonucleoprotein (RNP) that includes a Cas9/gRNA complex. The Cas9/gRNA
complex
includes a Cas9 endonuclease 225 in a complex with a single guide RNA (sgRNA)
205, bound to
the target 221 oncoviral nucleic acid via the guide sequence 209 of the guide
RNA. The target
221 included to aid in understanding. Compositions of the invention according
to some
embodiments include the RNP (which provides the nuclease 201) and the cancer
drug 251. In
other embodiments, the nuclease may be delivered in the form of a protein or a
nucleic acid (e.g.,
as mRNA or encoded on a plasmid). The cancer drug 251 may be any suitable
agent such as one
of those discussed below.
In an aspect of the invention, the Cas9 endonuclease causes a double strand
break in at
least two locations in oncoviral nucleic acid. These two double strand breaks
cause a fragment to
be deleted. Even if viral repair pathways anneal the two ends, there will
still be a deletion in the
genome. One or more deletions using the mechanism will incapacitate the virus.
The result is
that the host cell will be free of viral infection.
In embodiments of the invention, nucleases cleave the genome of the target
virus. A
nuclease is an enzyme capable of cleaving the phosphodiester bonds between the
nucleotide
subunits of nucleic acids. Endonucleases are enzymes that cleave the
phosphodiester bond within
a polynucleotide chain. Some, such as deoxy-ribonuclease I, cut DNA relatively
nonspecifically
(without regard to sequence), while many, typically called restriction
endonucleases or
restriction enzymes, cleave only at very specific nucleotide sequences. In a
preferred
embodiment of the invention, the Cas9 nuclease is incorporated into the
compositions and
methods of the invention, however, it should be appreciated that any nuclease
may be used.
In preferred embodiments of the invention, the Cas9 nuclease is used to cleave
the
genome. The Cas9 nuclease is capable of creating a double strand break in the
genome. The Cas9
13
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
nuclease has two functional domains: RuvC and HNH, each cutting a different
strand. When
both of these domains are active, the Cas9 causes double strand breaks in the
genome.
In some embodiments of the invention, insertions into the genome can be
designed to
cause incapacitation, or altered genomic expression. Additionally,
insertions/deletions are also
used to introduce a premature stop codon either by creating one at the double
strand break or by
shifting the reading frame to create one downstream of the double strand
break. Any of these
outcomes of the NHEJ repair pathway can be leveraged to disrupt the target
gene. The changes
introduced by the use of the CRISPR/gRNA/Cas9 system are permanent to the
genome.
In some embodiments of the invention, at least one insertion is caused by the
CRISPR/gRNA/Cas9 complex. In a preferred embodiment, numerous insertions are
caused in
the genome, thereby incapacitating the virus. In an aspect of the invention,
the number of
insertions lowers the probability that the genome may be repaired.
In some embodiments of the invention, at least one deletion is caused by Cas9.
In a
preferred embodiment, numerous deletions are caused in the genome, thereby
incapacitating the
virus. In an aspect of the invention, the number of deletions lowers the
probability that the
genome may be repaired. In a highly-preferred embodiment, Cas9 causes
significant genomic
disruption, resulting in effective destruction of the viral genome, while
leaving the host genome
intact. It is noted that in treating a tumor or other oncoviral infection,
repair of cleaved DNA by
host ligases (e.g., by non-homologous end joining) may not be required . The
absence of host-
mediated repair may be an aid in disrupting viral or tumor DNA and may aid in
inducing cell
death.
TALENs uses a nonspecific DNA-cleaving nuclease fused to a DNA-binding domain
that
can be to target essentially any sequence. For TALEN technology, target sites
are identified and
expression vectors are made. Linearized expression vectors (e.g., by Notl) may
be used as
template for mRNA synthesis. A commercially available kit may be use such as
the
mMESSAGE mMACHINE SP6 transcription kit from Life Technologies (Carlsbad, CA).
See
Joung & Sander, 2013, TALENs: a widely applicable technology for targeted
genome editing,
Nat Rev Mol Cell Bio 14:49-55.
TALENs and CRISPR methods provide one-to-one relationship to the target sites,
i.e.
one unit of the tandem repeat in the TALE domain recognizes one nucleotide in
the target site,
and the crRNA, gRNA, or sgRNA of CRISPR/Cas system hybridizes to the
complementary
14
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
sequence in the DNA target. Methods can include using a pair of TALENs or a
Cas9 protein with
one gRNA to generate double-strand breaks in the target. The breaks are then
repaired via non-
homologous end-joining or homologous recombination (HR).
ZFN may be used to cut viral nucleic acid. Briefly, the ZFN method includes
introducing
into the infected host cell at least one vector (e.g., RNA molecule) encoding
a targeted ZFN 305
and, optionally, at least one accessory polynucleotide. See, e.g., U.S. Pub.
2011/0023144 to
Weinstein, incorporated by reference The cell includes target sequence. The
cell is incubated to
allow expression of the ZFN, wherein a double-stranded break is introduced
into the targeted
chromosomal sequence by the ZFN. In some embodiments, a donor polynucleotide
or exchange
polynucleotide is introduced. Swapping a portion of the viral nucleic acid
with irrelevant
sequence can fully interfere transcription or replication of the viral nucleic
acid. Target DNA
along with exchange polynucleotide may be repaired by an error-prone non-
homologous end-
joining DNA repair process or a homology-directed DNA repair process.
Typically, a ZFN comprises a DNA binding domain (i.e., zinc finger) and a
cleavage
domain (i.e., nuclease) and this gene may be introduced as mRNA (e.g., 5'
capped,
polyadenylated, or both). Zinc finger binding domains may be engineered to
recognize and bind
to any nucleic acid sequence of choice. See, e.g., Qu et al., 2013, Zinc-
finger-nucleases mediate
specific and efficient excision of HIV-1 proviral DAN from infected and
latently infected human
T cells, Nucl Ac Res 41(16):7771-7782, incorporated by reference. An
engineered zinc finger
binding domain may have a novel binding specificity compared to a naturally-
occurring zinc
finger protein. Engineering methods include, but are not limited to, rational
design and various
types of selection. A zinc finger binding domain may be designed to recognize
a target DNA
sequence via zinc finger recognition regions (i.e., zinc fingers). See for
example, U.S. Pat. Nos.
6,607,882; 6,534,261 and 6,453,242, incorporated by reference. Exemplary
methods of selecting
a zinc finger recognition region may include phage display and two-hybrid
systems, and are
disclosed in U.S. Pat. 5,789,538; U.S. Pat. 5,925,523; U.S. Pat. 6,007,988;
U.S. Pat. 6,013,453;
U.S. Pat. 6,410,248; U.S. Pat. 6,140,466; U.S. Pat. 6,200,759; and U.S. Pat.
6,242,568, each of
which is incorporated by reference.
A ZFN also includes a cleavage domain. The cleavage domain portion of the ZFNs
may
be obtained from any suitable endonuclease or exonuclease such as restriction
endonucleases and
homing endonucleases. See, for example, Belfort & Roberts, 1997, Homing
endonucleases:
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
keeping the house in order, Nucleic Acids Res 25(17):3379-3388. A cleavage
domain may be
derived from an enzyme that requires dimerization for cleavage activity. Two
ZFNs may be
required for cleavage, as each nuclease comprises a monomer of the active
enzyme dimer.
Alternatively, a single ZFN may comprise both monomers to create an active
enzyme dimer.
Restriction endonucleases present may be capable of sequence-specific binding
and cleavage of
DNA at or near the site of binding. Certain restriction enzymes (e.g., Type
ITS) cleave DNA at
sites removed from the recognition site and have separable binding and
cleavage domains. For
example, the Type ITS enzyme FokI, active as a dimer, catalyzes double-
stranded cleavage of
DNA, at 9 nucleotides from its recognition site on one strand and 13
nucleotides from its
recognition site on the other. The FokI enzyme used in a ZFN may be considered
a cleavage
monomer. Thus, for targeted double-stranded cleavage using a FokI cleavage
domain, two ZFNs,
each comprising a FokI cleavage monomer, may be used to reconstitute an active
enzyme dimer.
See Wah, et al., 1998, Structure of FokI has implications for DNA cleavage,
PNAS 95:10564-
10569; U.S. Pat. 5,356,802; U.S. Pat. 5,436,150; U.S. Pat. 5,487,994; U.S.
Pub. 2005/0064474;
U.S. Pub. 2006/0188987; and U.S. Pub. 2008/0131962, each incorporated by
reference.
Virus targeting using ZFN may include introducing at least one donor
polynucleotide
comprising a sequence into the cell. A donor polynucleotide preferably
includes the sequence to
be introduced flanked by an upstream and downstream sequence that share
sequence similarity
with either side of the site of integration in the chromosome. The upstream
and downstream
sequences in the donor polynucleotide are selected to promote recombination
between the
chromosomal sequence of interest and the donor polynucleotide. Typically, the
donor
polynucleotide will be DNA. The donor polynucleotide may be a DNA plasmid, a
bacterial
artificial chromosome (BAC), a yeast artificial chromosome (YAC), a viral
vector, a linear piece
of DNA, a PCR fragment, a naked nucleic acid, and may employ a delivery
vehicle such as a
liposome. The sequence of the donor polynucleotide may include exons, introns,
regulatory
sequences, or combinations thereof. The double stranded break is repaired via
homologous
recombination with the donor polynucleotide such that the desired sequence is
integrated into the
chromosome. In the ZFN-mediated process, a double stranded break introduced
into the target
sequence by the ZFN is repaired, via homologous recombination with the
exchange
polynucleotide, such that the sequence in the exchange polynucleotide may be
exchanged with a
portion of the target sequence. The presence of the double stranded break
facilitates homologous
16
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
recombination and repair of the break. The exchange polynucleotide may be
physically
integrated or, alternatively, the exchange polynucleotide may be used as a
template for repair of
the break, resulting in the exchange of the sequence information in the
exchange polynucleotide
with the sequence information in that portion of the target sequence. Thus, a
portion of the viral
nucleic acid may be converted to the sequence of the exchange polynucleotide.
ZFN methods
can include using a vector to deliver a nucleic acid molecule encoding a ZFN
and, optionally, at
least one exchange polynucleotide or at least one donor polynucleotide to the
infected cell.
Meganucleases are endo-deoxy-ribonucleases characterized by a large
recognition site
(double-stranded DNA sequences of 12 to 40 base pairs); as a result this site
generally occurs
only once in any given genome. For example, the 18-base pair sequence
recognized by the I-SceI
meganuclease would on average require a genome twenty times the size of the
human genome to
be found once by chance (although sequences with a single mismatch occur about
three times per
human-sized genome). Meganucleases are therefore considered to be the most
specific naturally
occurring restriction enzymes. Meganucleases can be divided into five families
based on
sequence and structure motifs. See, e.g., U.S. Pub. 2010/0086533; U.S. Pub.
2014/0208457; and
Silva et al., 2011, Meganucleases and other tools for targeted genome
engineering, Cur Gene
Ther 11(1):11-27, the contents of each of which are incorporated by reference.
In some embodiments of the invention, a template sequence is inserted into the
genome.
In order to introduce nucleotide modifications to genomic DNA, a DNA repair
template
containing the desired sequence must be present during homology directed
repair (HDR). The
DNA template is normally transfected into the cell along with the gRNA/Cas9.
The length and
binding position of each homology arm is dependent on the size of the change
being introduced.
In the presence of a suitable template, HDR can introduce significant changes
at the Cas9
induced double strand break.
Some embodiments of the invention may utilize modified version of a nuclease.
Modified
versions of the Cas9 enzyme containing a single inactive catalytic domain,
either RuvC- or
HNH-, are called `nickases'. With only one active nuclease domain, the Cas9
nickase cuts only
one strand of the target DNA, creating a single-strand break or 'nick'.
Similar to the inactive
dCas9 (RuvC- and HNH-), a Cas9 nickase is still able to bind DNA based on gRNA
specificity,
though nickases will only cut one of the DNA strands. The majority of CRISPR
plasmids are
17
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
derived from S. pyogenes and the RuvC domain can be inactivated by a DlOA
mutation and the
HNH domain can be inactivated by an H840A mutation.
A single-strand break, or nick, is normally quickly repaired through the HDR
pathway,
using the intact complementary DNA strand as the template. However, two
proximal, opposite
strand nicks introduced by a Cas9 nickase are treated as a double strand
break, in what is often
referred to as a 'double nick' or 'dual nickase' CRISPR system. A double-nick
induced double
strain break can be repaired by either NHEJ or HDR depending on the desired
effect on the gene
target. At these double strain breaks, insertions and deletions are caused by
the CRISPR/Cas9
complex. In an aspect of the invention, a deletion is caused by positioning
two double strand
breaks proximate to one another, thereby causing a fragment of the genome to
be deleted.
iii. Targeting sequence
A nuclease may use the targeting specificity of a guide RNA (gRNA). As
discussed
below, guide RNAs or single guide RNAs are specifically designed to target a
virus genome. As
used herein targeting sequence can mean any combination of gRNA, crRNA,
tracrRNA, sgRNA,
and others. A CRISPR/Cas9 gene editing complex of the invention works
optimally with a guide
RNA that targets the viral genome. Guide RNA (gRNA) (which includes single
guide RNA
(sgRNA), crisprRNA (crRNA), trans-activating RNA (tracrRNA), any other
targeting oligo, or
any combination thereof) leads the CRISPR/Cas9 complex to the viral genome in
order to cause
viral genomic disruption. In an aspect of the invention, CRISPR/Cas9/gRNA
complexes are
designed to target specific viruses within a cell. It should be appreciated
that any virus can be
targeted using the composition of the invention. Identification of specific
regions of the virus
genome aids in development and designing of CRISPR/Cas9/gRNA complexes.
In an aspect of the invention, the CRISPR/Cas9/gRNA complexes are designed to
target
latent viruses within a cell. Once transfected within a cell, the
CRISPR/Cas9/gRNA complexes
cause repeated insertions or deletions to render the genome incapacitated, or
due to number of
insertions or deletions, the probability of repair is significantly reduced.
As an example, the Epstein¨Barr virus (EBV), also called human herpesvirus 4
(HHV-4),
is inactivated in cells using a CRISPR/Cas9/gRNA complex. EBV is a virus of
the herpes family,
and is one of the most common viruses in humans. The virus is approximately
122 nm to 180 nm
in diameter and is composed of a double helix of DNA wrapped in a protein
capsid. In this
18
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
example, the Raji cell line serves as an appropriate in vitro model. The Raji
cell line is the first
continuous human cell line from hematopoietic origin and cell lines produce an
unusual strain of
Epstein-Barr virus while being one of the most extensively studied EBV models.
To target the
EBV genomes in the Raji cells, a CRISPR/Cas9 complex with specificity for EBV
is needed.
FIG. 4 shows a plasmid that includes an EGFP marker fused after the Cas9
protein.
The design of EBV-targeting CRISPR/Cas9 plasmids consisting of a U6 promoter
driven
chimeric guide RNA (sgRNA) and a ubiquitous promoter driven Cas9 that were
obtained from
Addgene, Inc. Commercially available guide RNAs and Cas9 nucleases may be used
with the
present invention. The EGFP marker fused after the Cas9 protein allowed
selection of Cas9-
positive cells.
Preferably guide RNAs are designed, whether or not commercially purchased, to
target a
specific part of an HPV genome. The target area in HPV is identified and guide
RNA to target
selected portions of the HPV genome are developed and incorporated into the
composition of the
invention. In an aspect of the invention, a reference genome of a particular
strain of the virus is
selected for guide RNA design.
In relation to EBV, for example, the reference genome from strain B95-8 was
used as a
design guide. Within a genome of interest, such as EBV, selected regions, or
genes are targeted.
For example, six regions can be targeted with seven guide RNA designs for
different genome
editing purposes.
FIG. 5 shows gRNA targets along a reference genome where # denotes structural
targets,
where * denotes transformation-related targets, and where + denotes latency-
related targets.
In relation to EBV, EBNA1 is the only nuclear Epstein-Barr virus (EBV) protein
expressed in both latent and lytic modes of infection. While EBNA1 is known to
play several
important roles in latent infection, EBNA1 is crucial for many EBV functions
including gene
regulation and latent genome replication. Therefore, guide RNAs sgEBV4 and
sgEBV5 were
selected to target both ends of the EBNA1 coding region in order to excise
this whole region of
the genome. These "structural" targets enable systematic digestion of the EBV
genome into
smaller pieces. EBNA3C and LMP1 are essential for host cell transformation,
and guide RNAs
sgEBV3 and sgEBV7 were designed to target the 5' exons of these two proteins
respectively.
In certain embodiments, the invention uses Cas9 or another Cas-type nuclease
(Cas6,
Cfpl, modified Cas9, modified Cas6, modified Cfpl, etc.) with one or a
plurality of guide RNAs
19
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
with a sequence specific to a target in a genome of Merkel cell polyomavirus
(MCV), delivered
in conjunction with a cancer therapeutic. Merkel cell polyomavirus (MCV),
which can cause
merkel cell carcinoma (MCV). MCV is the fifth polyomavirus that infects humans
to be
discovered. Polyomaviruses are small (-5400 base pair), non-enveloped, double-
stranded DNA
viruses. MCV is one of seven currently known human oncoviruses. It is
suspected to cause the
majority of cases of Merkel cell carcinoma, a rare but aggressive form of skin
cancer.
iv. Cancer drug
Methods and compositions of the invention use one or a plurality of cancer
drug in
conjunction with a nuclease to treat or prevent an oncoviral infection or
resultant condition. The
cancer drug(s) may be selected for its mechanism of action, its clinical
effectiveness, its
suitability to a particular cancer of infectious origin, or any other suitable
trait. When delivered
to a patient, the agent will have an effect according to its mechanism of
action.
It may be preferable to use a proteasome inhibitor. Proteasome inhibitors are
drugs that
block the action of proteasomes, cellular complexes that break down proteins.
Multiple
mechanisms are likely to be involved, but proteasome inhibition may prevent
degradation of pro-
apoptotic factors such as the p53 protein, permitting activation of programmed
cell death in
neoplastic cells dependent upon suppression of pro-apoptotic pathways. For
example,
bortezomib causes a rapid and dramatic change in the levels of intracellular
peptides. Suitable
proteasome inhibitors may include bortezomib, lactacystin, disulfiram,
Salinosporamide A,
bortezomib, carfilzomib, epoxomicin, and ixazomib.
FIG. 6 depicts the proteasome inhibitor bortezomib. A boron atom in bortezomib
binds
the catalytic site of the 26S proteasome with high affinity and specificity.
In normal cells, the
proteasome regulates protein expression and function by degradation of
ubiquitylated proteins,
and also cleanses the cell of abnormal or misfolded proteins. Clinical and
preclinical data support
a role in maintaining the immortal phenotype of myeloma cells, and cell-
culture and xenograft
data support a similar function in solid tumor cancers. While multiple
mechanisms are likely to
be involved, proteasome inhibition may prevent degradation of pro-apoptotic
factors, permitting
activation of programmed cell death in neoplastic cells dependent upon
suppression of pro-
apoptotic pathways.
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
Lactacystin binds and inhibits specific catalytic subunits of the proteasome,
a protein
complex responsible for the bulk of proteolysis in the cell, as well as
proteolytic activation of
certain protein substrates. Lactacystin covalently modifies the amino-terminal
threonine of
specific catalytic subunits of the proteasome
Disulfiram creates complexes with metals (dithiocarbamate complexes) and acts
as a
proteasome inhibitor. A clinical trial of disulfiram with copper gluconate
against liver cancer is
being conducted in Utah and a clinical trial of disulfiram as adjuvant against
lung cancer is
happening in Israel.
Salinosporamide A is a potent proteasome inhibitor and potential anticancer
agent.
Salinosporamide A inhibits proteasome activity by covalently modifying the
active site threonine
residues of the 20S proteasome. In vitro studies using purified 20S
proteasomes showed that
salinosporamide A has lower EC50 for trypsin-like (T-L) activity than does
bortezomib. In vivo
animal model studies show marked inhibition of T-L activity in response to
salinosporamide A,
whereas bortezomib enhances T-L proteasome activity.
Carfilzomib (marketed under the trade name Kyprolis (Onyx Pharmaceuticals,
Inc.) is an
anti-cancer drug acting as a selective proteasome inhibitor. Chemically,
carfilzomib is a
tetrapeptide epoxyketone and an analog of epoxomicin. Carfilzomib irreversibly
binds to and
inhibits the chymotrypsin-like activity of the 20S proteasome, an enzyme that
degrades
unwanted cellular proteins. Inhibition of proteasome-mediated proteolysis
results in a build-up of
poly-ubiquinated proteins, which may cause cell cycle arrest, apoptosis, and
inhibition of tumor
growth.
Epoxomicin is a naturally occurring selective proteasome inhibitor with anti-
inflammatory activity.
Ixazomib is a proteasome inhibitor similar to bortezomib. Ixazomib is
considered to be a
second-generation proteasome inhibitor because it has improved characteristics
and activity over
Velcade.
A cancer drug may be selected for any suitable mechanism of action including,
for
example, transcription inhibition, inhibition of topoisomerase, chromatin
remodeling action,
inhibition of nucleotide synthesis, causation of DNA cross-linking, inhibition
of DNA synthesis,
affecting tubulin or microtubule binding, or others. These categories may
overlap and may not be
mutually exclusive. An exemplary transcription inhibitor includes actinomycin
D. Suitable
21
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
topoisomerase inhibitors include idarubicin, irinotecan, topotecan,
mitoxantrone, and
daunorubicin. In some embodiments, the cancer drug contributes to chromatin
remodeling or to
the breakage of nucleic acid strands. For example, bleomycin and teniposide
are known to cause
breaks in DNA strands. Suitable nucleotide synthesis inhibitors include
capecitabine,
hydroxycarbamide and pemetrexed. Suitable DNA cross-linkers include cisplatin,
mechlorethamine, and oxaliplatin. Cancer drugs that inhibit DNA synthesis
include, e.g.,
chlorambucil, gemcitabine, capecitabine, and cytarabine. Cancer drugs that
affect
tubulin/microtubule binding include, e.g., docetaxel, paclitaxel, vinblastine,
vincristine, and
vinorelbine. By delivery of the cancer drug, cell proliferation is inhibited
and the growth of any
tumor may be suppressed. Thus the adverse effects of infection by an oncovirus
may be
minimized. For example, methods of the invention may be used to treat children
living in areas
associated with a high prevalence of Burkitt's lymphoma. Such a patient may be
treated with a
nuclease that specifically cuts nucleic acid of the Epstein-Barr virus¨without
hindering the
normal, healthy function of the human genome¨and an anti-tumor cancer drug to
prevent or
treat a Burkitt's lymphoma.
Exemplary cancer drugs that may be used include the following.
Actinomycin D is the most significant member of actinomycines, which are a
class of
polypeptide antitumor antibiotics isolated from soil bacteria of the genus
Streptomyces.
Actinomycin D is one of the older anticancer drugs, and has been used for many
years.
Actinomycin D is shown to have the ability to inhibit transcription.
Actinomycin D does this by
binding DNA at the transcription initiation complex and preventing elongation
of RNA chain by
RNA polymerase.
Tretinoin, also known as all-trans retinoic acid, is used to treat at least
one form of cancer
(acute promyelocytic leukemia, also called acute myeloid leukemia subtype M3)
by causing the
immature promyelocytes to differentiate (i.e. mature). The pathology of the
leukemia is due to
the highly proliferative immature cells; retinoic acid drives these cells to
develop into functional
cells, which helps to alleviate the disease.
Anthracyclines (e.g., daunorubicin) are a class of drugs (CCNS or cell-cycle
non-
specific) used in cancer chemotherapy derived from Streptomyces bacterium.
Anthracyclines are
used to treat many cancers, including leukemias, lymphomas, breast, stomach,
uterine, ovarian,
bladder cancer, and lung cancers. Anthracyclines have four mechanisms of
action: inhibition of
22
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
DNA and RNA synthesis; inhibition of topoisomerase II enzyme; iron-mediated
generation of
free oxygen radicals; and induction of histone eviction from chromatin.
Bleomycin acts by induction of DNA strand breaks. Some studies suggest
bleomycin also
inhibits incorporation of thymidine into DNA strands. DNA cleavage by
bleomycin depends on
oxygen and metal ions, at least in vitro. The exact mechanism of DNA strand
scission is
unresolved, but it has been suggested that bleomycin chelates metal ions
(primarily iron),
producing a pseudo-enzyme that reacts with oxygen to produce superoxide and
hydroxide free
radicals that cleave DNA.
Carboplatin is a chemotherapy drug used against some forms of cancer.
Capecitabine is a cancer drug used in the treatment of numerous cancers.
Capecitabine is
metabolized to 5-FU which in turn is a thymidylate synthase inhibitor, hence
inhibiting the
synthesis of thymidine monophosphate (TMP), the active form of thymidine which
is required
for the de novo synthesis of DNA.
Cisplatin is a chemotherapy drug, a member of a class of platinum-containing
anti-cancer
drugs, which now also includes carboplatin and oxaliplatin. These platinum
complexes react in
vivo, binding to and causing crosslinking of DNA, which ultimately triggers
apoptosis
(programmed cell death).
Chlorambucil is a chemotherapy drug that has been mainly used in the treatment
of
chronic lymphocytic leukemia. It is a nitrogen mustard alkylating agent and
can be given orally.
Chlorambucil produces its anti-cancer effects by interfering with DNA
replication and damaging
the DNA in a cell. The DNA damage induces cell cycle arrest and cellular
apoptosis via the
accumulation of cytosolic p53 and subsequent activation of Bax, an apoptosis
promoter.
Chlorambucil alkylates and cross-links DNA during all phases of the cell
cycle, inducing DNA
damage via three different methods of covalent adduct generation with double-
helical DNA
Cyclophosphamide is metabolized to phosphoramide mustard. This metabolite is
only
formed in cells that have low levels of ALDH. Phosphoramide mustard forms DNA
crosslinks
both between and within DNA strands at guanine N-7 positions (known as inter-
strand and intra-
strand cross-linkages, respectively). This is irreversible and leads to cell
apoptosis.
Cyclophosphamide has relatively little typical chemotherapy toxicity as ALDHs
are present in
relatively large concentrations in bone marrow stem cells, liver and
intestinal epithelium.
ALDHs protect these actively proliferating tissues against toxic effects of
phosphoramide
23
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
mustard and acrolein by converting aldophosphamide to carboxycyclophosphamide
that does not
give rise to the toxic metabolites phosphoramide mustard and acrolein. This is
because
carboxycyclophosphamide cannot undergo 13-elimination (the carboxylate acts as
an electron-
donating group, forbidding the transformation), preventing nitrogen mustard
activation and
subsequent alkylation.
Cytarabine is a cancer drug that interferes with the synthesis of DNA. It is
an
antimetabolic agent with the chemical name of 10-arabinofuranosylcytosine. Its
mode of action
is due to its rapid conversion into cytosine arabinoside triphosphate, which
damages DNA when
the cell cycle holds in the S phase (synthesis of DNA). Rapidly dividing
cells, which require
DNA replication for mitosis, are therefore most affected. Cytosine arabinoside
also inhibits both
DNA and RNA polymerases and nucleotide reductase enzymes needed for DNA
synthesis.
Daunorubicin, or daunomycin, is chemotherapeutic of the anthracycline family
that
interacts with DNA by intercalation and inhibition of macromolecular
biosynthesis. This inhibits
the progression of the enzyme topoisomerase II, which relaxes supercoils in
DNA for
transcription. Daunorubicin stabilizes the topoisomerase II complex after it
has broken the DNA
chain for replication, preventing the DNA double helix from being resealed and
thereby stopping
the process of replication. On binding to DNA, daunorubicin intercalates, with
its daunosamine
residue directed toward the minor groove. It can also induce histone eviction
from chromatin
upon intercalation.
Docetaxel is a chemotherapy medication that works by interfering with cell
division.
Docetaxel binds to microtubules reversibly with high affinity and has a
maximum stoichiometry
of 1 mole docetaxel per mole tubulin in microtubules. This binding stabilizes
microtubules and
prevents de-polymerization from calcium ions, decreased temperature and
dilution, preferentially
at the plus end of the microtubule. Docetaxel has been found to accumulate to
higher
concentration in ovarian adenocarcinoma cells than kidney carcinoma cells,
which may
contribute to the more effective treatment of ovarian cancer by docetaxel. It
has also been found
to lead to the phosphorylation of oncoprotein bc1-2, which is apoptosis-
blocking in its
oncoprotein form.
Doxifluridine is a fluoropyrimidine derivative and oral prodrug of the
antineoplastic
agent 5-fluorouracil (5-FU) with antitumor activity. Doxifluridine, designed
to circumvent the
rapid degradation of 5-FU by dihydropyrimidine dehydrogenase in the gut wall,
is converted into
24
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
5-FU in the presence of pyrimidine nucleoside phosphorylase. 5-FU interferes
with DNA
synthesis and subsequent cell division by reducing normal thymidine production
and interferes
with RNA transcription by competing with uridine triphosphate for
incorporation into the RNA
strand.
Doxorubicin is an anthracycline antitumor antibiotic that works by
intercalating DNA.
Epirubicin is an anthracycline drug used for chemotherapy that works by
intercalating
DNA strands. Intercalation results in complex formation which inhibits DNA and
RNA
synthesis. It also triggers DNA cleavage by topoisomerase II, resulting in
mechanisms that lead
to cell death.
The epothilones are a class of potential cancer drugs that prevent cancer
cells from
dividing by interfering with tubulin.
Fluorouracil (5-FU) sold as Adrucil among others, is a drug that is a
pyrimidine analog
which is used in the treatment of cancer. It is a suicide inhibitor and works
through irreversible
inhibition of thymidylate synthase.
Gemcitabine is a nucleoside analog used as chemotherapy. The triphosphate
analogue of
gemcitabine replaces one of the building blocks of nucleic acids, in this case
cytidine, during
DNA replication. The process arrests tumor growth, as only one additional
nucleoside can be
attached to the "faulty" nucleoside, resulting in apoptosis.
Hydroxycarbamide is an antineoplastic drug used in myeloproliferative
disorders.
Hydroxycarbamide decreases the production of deoxyribonucleotides via
inhibition of the
enzyme ribonucleotide reductase by scavenging tyrosyl free radicals as they
are involved in the
reduction NDPs.
Idarubicin is an anthracycline antileukemic drug that inserts itself into DNA
and prevents
DNA unwinding by interfering with the enzyme topoisomerase II.
Imatinib is a tyrosine-kinase inhibitor used in the treatment of multiple
cancers, such as
Philadelphia chromosome-positive (Ph+) chronic myelogenous leukemia (CML).
Imatinib
blocks the BCR-Abl enzyme, and stops it from adding phosphate groups. As a
result, cells stop
growing, and undergo apoptosis. Because the BCR-Abl tyrosine kinase enzyme
exists only in
cancer cells and not in healthy cells, imatinib works as a form of targeted
therapy¨only cancer
cells are killed through the drug's action.
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
Interferon alfa enhances the proliferation of human B cells, as well as being
able to
activate NK cells.
Irinotecan is a chemotherapeutic that prevents DNA from unwinding by
inhibition of
topoisomerase 1.
Mechlorethamine is the prototype of alkylating agents, a group of anticancer
chemotherapeutic drugs. Mechlorethamine works by binding to DNA, crosslinking
two strands
and preventing cell duplication.
Mercaptopurine is an immunosuppressive medication used to treat acute
lymphocytic
leukemia. Mercaptopurine interferes with nucleotide synthesis.
Methotrexate belongs to the class of chemotherapy drugs called
antimetabolites.
Methotrexate exerts its chemotherapeutic effect by being able to counteract
and compete with
folic acid in cancer cells resulting in folic acid deficiency in the cells and
causing their death.
Mitoxantrone is a type II topoisomerase inhibitor that disrupts DNA synthesis
and DNA
repair in both healthy cells and cancer cells by intercalation between the DNA
bases.
Oxaliplatin features a square planar platinum(II) center. In contrast to
cisplatin and
carboplatin, oxaliplatin features the bidentate ligand 1,2-diaminocyclohexane
in place of the two
monodentate ammine ligands. It also features a bidentate oxalate group.
According to in vivo
studies, oxaliplatin fights carcinoma of the colon through non-targeted
cytotoxic effects. Like
other platinum compounds, its cytotoxicity is thought to result from
inhibition of DNA synthesis
in cells. In particular, oxaliplatin forms both inter- and intra-strand cross
links in DNA, which
prevent DNA replication and transcription, causing cell death.
Paclitaxel is a chemotherapeutic that targets tubulin. Paclitaxel-treated
cells have defects
in mitotic spindle assembly, chromosome segregation, and cell division. Unlike
other tubulin-
targeting drugs such as colchicine that inhibit microtubule assembly,
paclitaxel stabilizes the
microtubule polymer and protects it from disassembly. Chromosomes are thus
unable to achieve
a metaphase spindle configuration. This blocks progression of mitosis, and
prolonged activation
of the mitotic checkpoint triggers apoptosis or reversion to the G-phase of
the cell cycle without
cell division.
Pemetrexed is a chemotherapy drug in the class of chemotherapy drugs called
folate
antimetabolites. It works by inhibiting three enzymes used in purine and
pyrimidine synthesis¨
thymidylate synthase (TS), dihydrofolate reductase (DHFR), and glycinamide
ribonucleotide
26
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
formyltransferase (GARFT). By inhibiting the formation of precursor purine and
pyrimidine
nucleotides, pemetrexed prevents the formation of DNA and RNA, which are
required for the
growth and survival of both normal cells and cancer cells.
Teniposide is a chemotherapeutic that causes dose-dependent single- and double-
stranded
breaks in DNA and DNA-protein cross-links. Teniposide has been found to act as
an inhibitor of
topoisomerase II. The cytotoxic effects of teniposide are related to the
relative number of double-
stranded DNA breaks produced in cells, which are a reflection of the
stabilization of a
topoisomerase II-DNA intermediate.
Topotecan is a cancer drug that is a topoisomerase inhibitor.
Valrubicin is a chemotherapy drug used to treat bladder cancer. Valrubicin is
a
semisynthetic analog of the anthracycline doxorubicin, and is administered by
infusion directly
into the bladder.
The cancer drug may be an anti-apoptotic inhibitors, such as venetoclax.
Venetoclax (a
BH3-mimetic) is a small molecule that acts as a Bc1-2 inhibitor. Venetoclax
blocks the anti-
apoptotic B-cell lymphoma-2 (BCL2) protein, leading to programmed cell death
in CLL cells.
Vinblastine is a chemotherapeutic that inhibits mitosis. Vinblastine
suppresses
microtubule dynamics and reduces microtubule polymer mass.
Vincristine is a chemotherapeutic that works partly by binding to the tubulin
protein,
stopping the cell from separating its chromosomes during the metaphase; the
cell then undergoes
apoptosis.
Vindesine is an anti-mitotic vinca alkaloid used in chemotherapy.
Vinorelbine is a chemotherapeutic that inhibits mitosis through interaction
with tubulin.
In some embodiments, the cancer therapeutic is a monoclonal antibody.
Rituximab (trade names Rituxan, MabThera and Zytux) is a chimeric monoclonal
antibody against the protein CD20, which is primarily found on the surface of
immune system B
cells. Rituximab destroys B cells and is therefore used to treat diseases
which are characterized
by excessive numbers of B cells, overactive B cells, or dysfunctional B cells.
This includes many
lymphomas, leukemias, transplant rejection, and autoimmune disorders.
Bevacizumab is a recombinant humanized monoclonal antibody that blocks
angiogenesis
by inhibiting vascular endothelial growth factor A (VEGF-A). VEGF-A is a
chemical signal that
27
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
stimulates angiogenesis in a variety of diseases, especially in cancer.
Bevacizumab was the first
clinically available angiogenesis inhibitor in the United States.
Pembrolizumab (formerly MK-3475 and lambrolizumab, trade name Keytruda[1]) is
a
humanized antibody used in cancer immunotherapy. It targets the programmed
cell death 1 (PD-
1) receptor.
In certain embodiments, the cancer therapeutic comprises an immune checkpoint
inhibitor such as anti-PD-1 or anti-VEGF. A suitable anti-PD-1 may include .
Nivolumab (ONO-
4538, BMS-936558, or MDX1106) is a human IgG4 anti-PD-1 monoclonal antibody
that acts as
an immunomodulator by blocking ligand activation of the programmed cell death
1 (PD-1)
receptor on activated T cells.
In certain embodiments, the cancer therapeutic comprises a recombinant
cytokine such as
Interleukin 2 (IL-2), Interleukin 11 (IL-11), or Interleukin 15 (IL-15). IL2
is a lymphokine that
induces the proliferation of responsive T cells. In addition, it acts on some
B cells, via receptor-
specific binding, as a growth factor and antibody production stimulant.
Interleukin 11 (IL-11) is a secreted protein that stimulates
megakaryocytopoiesis,
resulting in increased production of platelets, as well as activating
osteoclasts, inhibiting
epithelial cell proliferation and apoptosis, and inhibiting macrophage
mediator production.
Interleukin 15 (IL-15) is a cytokine with structural similarity to IL-2. Like
IL-2, IL-15
binds to and signals through a complex composed of IL-2/IL-15 receptor beta
chain (CD122) and
the common gamma chain (gamma-C, CD132). IL-15 is secreted by cells such as
mononuclear
phagocytes after viral infection. This cytokine induces cell proliferation of
natural killer cells;
cells of the innate immune system whose principal role is to kill virally
infected cells.
v. Introduce to cell
Methods of the invention include introducing to cells of a patient a treatment
that
includes: a nuclease or a vector that encodes the nuclease; and a cancer drug.
The nuclease is
targeted to oncoviral nucleic acid by means of the sequence-specific targeting
moiety and it will
cleave the viral nucleic acid without interfering with a host genome. Any
suitable method can be
used to deliver the treatment to the cells. For example, the treatment (or
either part of it) may be
delivered by injection, orally, or by hydrodynamic delivery. The nuclease or
the gene encoding
the nuclease may be delivered to systematic circulation or may be delivered or
otherwise
28
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
localized to a specific tissue type. The nuclease or gene encoding the
nuclease may be modified
or programmed to be active under only certain conditions such as by using a
tissue-specific
promoter so that the encoded nuclease is preferentially or only transcribed in
certain tissue types.
In some embodiments, specific CRISPR/Cas9/gRNA complexes are introduced into a
cell. A guide RNA is designed to target at least one category of sequences of
the viral genome. In
addition to latent infections this invention can also be used to control
actively replicating viruses
by targeting the viral genome before it is packaged or after it is ejected.
In some embodiments, a cocktail of guide RNAs may be introduced into a cell.
The guide
RNAs are designed to target numerous categories of sequences of the viral
genome. By targeting
several areas along the genome, the double strand break at multiple locations
fragments the
genome, lowering the possibility of repair. Even with repair mechanisms, the
large deletions
render the virus incapacitated.
In some embodiments, several guide RNAs are added to create a cocktail to
target
different categories of sequences. For example, two, five, seven or eleven
guide RNAs may be
present in a CRISPR cocktail targeting three different categories of
sequences. However, any
number of gRNAs may be introduced into a cocktail to target categories of
sequences. In
preferred embodiments, the categories of sequences are important for genome
structure, host cell
transformation, and infection latency, respectively.
In some aspects of the invention, in vitro experiments allow for the
determination of the
most essential targets within a viral genome. For example, to understand the
most essential
targets for effective incapacitation of a genome, subsets of guide RNAs are
transfected into
model cells. Assays can determine which guide RNAs or which cocktail is the
most effective at
targeting essential categories of sequences.
For example, in the case of the EBV genome targeting, seven guide RNAs in the
CRISPR
cocktail targeted three different categories of sequences which are identified
as being important
for EBV genome structure, host cell transformation, and infection latency,
respectively. To
understand the most essential targets for effective EBV treatment, Raji cells
were transfected
with subsets of guide RNAs. Although sgEBV4/5 reduced the EBV genome by 85%,
they could
not suppress cell proliferation as effectively as the full cocktail (Fig. 14).
Guide RNAs targeting
the structural sequences (sgEBV1/2/6) could stop cell proliferation
completely, despite not
eliminating the full EBV load (26% decrease). Given the high efficiency of
genome editing and
29
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
the proliferation arrest, it was suspect that the residual EBV genome
signature in sgEBV1/2/6
was not due to intact genomes but to free-floating DNA that has been digested
out of the EBV
genome, i.e. as a false positive.
Additionally, recent data suggest that use of ribonucleoprotein (instead of
delivery as
plasmid DNA) may be preferred for its resulting better DNA cleavage and less
off-target
cytotox. Recent data suggest that EBV may be effectively targeted using only
two EBV guide
RNAs, sgEBV2/6. The data suggest that in mixed cell studies with EBV+ cells
(Raji) and EBV¨
cells (DG-75), compositions and methods described herein may exhibit viral
specificity of
cytotoxicity, preferentially killing infected cells.
Once CRISPR/Cas9/gRNA complexes are constructed, the complexes are introduced
into
a cell. It should be appreciated that complexes can be introduced into cells
in an in vitro model or
an in vivo model. In an aspect of the invention, CRISPR/Cas9/gRNA complexes
are designed to
not leave intact genomes of a virus after transfection and complexes are
designed for efficient
transfection.
Aspects of the invention allow for CRISPR/Cas9/gRNA to be transfected into
cells by
various methods, including viral vectors and non-viral vectors. Viral vectors
may include
retroviruses, lentiviruses, adenoviruses, and adeno-associated viruses. It
should be appreciated
that any viral vector may be incorporated into the present invention to
effectuate delivery of the
CRISPR/Cas9/gRNA complex into a cell. Some viral vectors may be more effective
than others,
depending on the CRISPR/Cas9/gRNA complex designed for digestion or
incapacitation. In an
aspect of the invention, the vectors contain essential components such as
origin of replication,
which is necessary for the replication and maintenance of the vector in the
host cell.
In an aspect of the invention, viral vectors are used as delivery vectors to
deliver the
complexes into a cell. Use of viral vectors as delivery vectors are known in
the art. See for
example U.S. Pub. 2009/0017543 to Wilkes et al., the contents of which are
incorporated by
reference.
A retrovirus is a single-stranded RNA virus that stores its nucleic acid in
the form of an
mRNA genome (including the 5' cap and 3' PolyA tail) and targets a host cell
as an obligate
parasite. In some methods in the art, retroviruses have been used to introduce
nucleic acids into a
cell. Once inside the host cell cytoplasm the virus uses its own reverse
transcriptase enzyme to
produce DNA from its RNA genome, the reverse of the usual pattern, thus retro
(backwards).
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
This new DNA is then incorporated into the host cell genome by an integrase
enzyme, at which
point the retroviral DNA is referred to as a provirus. For example, the
recombinant retroviruses
such as the Moloney murine leukemia virus have the ability to integrate into
the host genome in
a stable fashion. They contain a reverse transcriptase that allows integration
into the host
genome. Retroviral vectors can either be replication-competent or replication-
defective. In some
embodiments of the invention, retroviruses are incorporated to effectuate
transfection into a cell,
however the CRISPR/Cas9/gRNA complexes are designed to target the viral
genome.
In some embodiments of the invention, lentiviruses, which are a subclass of
retroviruses,
are used as viral vectors. Lentiviruses can be adapted as delivery vehicles
(vectors) given their
ability to integrate into the genome of non-dividing cells, which is the
unique feature of
lentiviruses as other retroviruses can infect only dividing cells. The viral
genome in the form of
RNA is reverse-transcribed when the virus enters the cell to produce DNA,
which is then
inserted into the genome at a random position by the viral integrase enzyme.
The vector, now
called a provirus, remains in the genome and is passed on to the progeny of
the cell when it
divides.
As opposed to lentiviruses, adenoviral DNA does not integrate into the genome
and is not
replicated during cell division. Adenovirus and the related AAV may be used as
delivery vectors
since they do not integrate into the host's genome. In some aspects of the
invention, only the
viral genome to be targeted is effected by the CRISPR/Cas9/gRNA complexes, and
not the
host's cells.
Adeno-associated virus (AAV) is a small virus that infects humans and some
other
primate species. AAV can infect both dividing and non-dividing cells and may
incorporate its
genome into that of the host cell. For example, because of its potential use
as a gene therapy
vector, researchers have created an altered AAV called self-complementary
adeno-associated
virus (scAAV). Whereas AAV packages a single strand of DNA and requires the
process of
second-strand synthesis, scAAV packages both strands which anneal together to
form double
stranded DNA. By skipping second strand synthesis scAAV allows for rapid
expression in the
cell. Otherwise, scAAV carries many characteristics of its AAV counterpart.
Addtionally or
alternatively, methods and compositions of the invention may use herpesvirus,
poxvirus,
alphavirus, or vaccinia virus as a means of delivery vectors.
31
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
FIG. 7 illustrates gene delivery with an AAV vector. Using known methods, the
nucleic
acid is packaged in the adenovirus 601. The viral vector 601 fuses with the
cell membrane by
binding to adhesion molecules and becomes an endosome 607 within the lipid bi-
layer. The
vesicle opens in the cytoplasm, releasing the vector and the nucleic acid 101,
which is
transported to and enters the nucleus.
Vectors derived from some AAV serotypes such as AAV9 can cross the blood-brain
barrier and transduce cells of the central nervous system (CNS) following a
single intravenous
injection. In addition to relying on natural diversity, AAV capsids can be
decorated by peptides
or "shuffled" to generate novel capsids that suit specific needs. For example,
a chimeric AAV
capsid "shuffled" from five parental natural AAV capsids was recently found to
efficiently
transduce human liver cells in a humanized mouse model (Lisowski et al., 2014,
Nature
506:382). Similar to AdV vector, rAAV vector can transduce both dividing and
non-dividing
cells, and the recombinant viral genome stays in host nucleus predominantly as
episome.
Interestingly, single or multiple copies of rAAV vector genome can circularize
in a head-to-tail
or head-to-head configuration in host nucleus, thus enhancing stability of the
episomal rAAV
DNA genome and mediating long-term transgene.
An HSV vector may also be used. HSV is a naturally neurotropic virus. After
initial
infection in skin or mucous membranes, HSV is taken up by sensory nerve
terminals, travels
along nerves to neuronal cell bodies, and delivers its DNA genome into nuclei
for replication.
Therefore, HSV vectors are well suited for delivery to neurons.
In certain embodiments of the invention, non-viral vectors may be used to
effectuate
transfection. Methods of non-viral delivery of nucleic acids include
lipofection, nucleofection,
microinjection, biolistics, virosomes, liposomes, immunoliposomes, polycation
or lipid:nucleic
acid conjugates, naked DNA, artificial virions, and agent-enhanced uptake of
DNA. Lipofection
is described in e.g., U.S. Pat. Nos. 5,049,386, 4,946,787; and 4,897,355) and
lipofection reagents
are sold commercially (e.g., Transfectam and Lipofectin). Cationic and neutral
lipids that are
suitable for efficient receptor-recognition lipofection of polynucleotides
include those described
in U.S. Pat. 7,166,298 to Jessee or U.S. Pat. 6,890,554 to Jesse, the contents
of each of which are
incorporated by reference. Delivery can be to cells (e.g. in vitro or ex vivo
administration) or
target tissues (e.g. in vivo administration).
In certain embodiments, non-viral vectors may be used to effectuate
transfection.
32
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
Non-viral vectors and methods for the delivery of nucleic acids and other
moieties
include lipofection, nucleofection, microinjection, biolistics, virosomes,
liposomes,
immunoliposomes, polycation or lipid:nucleic acid conjugates, naked DNA,
artificial virions,
and agent-enhanced uptake of DNA. Lipofection is described in e.g., U.S. Pat.
Nos. 5,049,386,
4,946,787; and 4,897,355) and lipofection reagents are sold commercially
(e.g., Transfectam and
Lipofectin). Preferred embodiments include nanoparticles (e.g., solid lipid
nanoparticles) or
liposomes. Cationic and neutral lipids that are suitable for efficient
receptor-recognition
lipofection of polynucleotides include those described in U.S. Pat. 7,166,298
to Jessee or U.S.
Pat. 6,890,554 to Jesse, the contents of each of which are incorporated by
reference. Delivery can
be to cells (e.g. in vitro or ex vivo administration) or target tissues (e.g.
in vivo administration).
Non-viral vectors may include synthetic vectors based on cationic lipids or
polymers
which can complex with negatively charged nucleic acids to form particles with
a diameter in the
order of 100 nm. The complex protects nucleic acid from degradation by
nuclease. The surfaces
of the cationic non-viral vectors have properties that minimize interaction
with blood
components, reduce reticuloendothelial system uptake, decrease their toxicity
and increase
binding affinity with the target cells.
Synthetic vectors are typically based on cationic lipids or polymers which can
complex
with negatively charged nucleic acids to form particles with a diameter in the
order of 100 nm.
The complex protects nucleic acid from degradation by nuclease. Moreover,
cellular and local
delivery strategies have to deal with the need for internalization, release,
and distribution in the
proper subcellular compartment. Systemic delivery strategies encounter
additional hurdles, for
example, strong interaction of cationic delivery vehicles with blood
components, uptake by the
reticuloendothelial system, kidney filtration, toxicity and targeting ability
of the carriers to the
cells of interest. Modifying the surfaces of the cationic non-virals can
minimize their interaction
with blood components, reduce reticuloendothelial system uptake, decrease
their toxicity and
increase their binding affinity with the target cells. Binding of plasma
proteins (also termed
opsonization) is the primary mechanism for RES to recognize the circulating
nanoparticles. For
example, macrophages, such as the Kupffer cells in the liver, recognize the
opsonized
nanoparticles via the scavenger receptor.
In some embodiments, non-viral vectors are modified to effectuate targeted
delivery and
transfection. PEGylation (i.e. modifying the surface with polyethyleneglycol)
is the predominant
33
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
method used to reduce the opsonization and aggregation of non-viral vectors
and minimize the
clearance by reticuloendothelial system, leading to a prolonged circulation
lifetime after
intravenous administration. PEGylated nanoparticles are therefore often
referred as "stealth"
nanoparticles. The nanoparticles that are not rapidly cleared from the
circulation will have a
chance to encounter infected cells.
However, PEG on the surface can decrease the uptake by target cells and reduce
the
biological activity. Therefore, to attach targeting ligand to the distal end
of the PEGylated
component is necessary; the ligand is projected beyond the PEG "shield" to
allow binding to
receptors on the target cell surface.
FIG. 8 shows a cationic lipid complex and shows the use of cationic lipids to
create a
liposome for delivery (although other lipid complexes and compositions are
within the scope of
the invention) and delivery by liposome. When cationic liposome is used as
gene carrier, the
application of neutral helper lipid is helpful for the release of nucleic
acid, besides promoting
hexagonal phase formation to enable endosomal escape. In some embodiments of
the invention,
neutral or anionic liposomes are developed for systemic delivery of nucleic
acids and obtaining
therapeutic effect in experimental animal model. Designing and synthesizing
novel cationic
lipids and polymers, and covalently or non-covalently binding gene with
peptides, targeting
ligands, polymers, or environmentally sensitive moieties also attract many
attentions for
resolving the problems encountered by non-viral vectors. The application of
inorganic
nanoparticles (for example, metallic nanoparticles, iron oxide, calcium
phosphate, magnesium
phosphate, manganese phosphate, double hydroxides, carbon nanotubes, and
quantum dots) in
delivery vectors can be prepared and surface-functionalized in many different
ways.
In some embodiments, the complexes are conjugated to nano-systems for systemic
therapy, such as solid lipid nanoparticles, liposomes, albumin-based
particles, PEGylated
proteins, biodegradable polymer-drug composites, polymeric micelles,
dendrimers, among
others. See Davis et al., 2008, Nanotherapeutic particles: an emerging
treatment modality for
cancer, Nat Rev Drug Discov. 7(9):771-782, incorporated by reference. Long
circulating
macromolecular carriers such as liposomes, can exploit the enhanced
permeability and retention
effect for preferential extravasation from tumor vessels. In certain
embodiments, the complexes
of the invention are conjugated to or encapsulated into a liposome or
polymerosome for delivery
to a cell. For example, liposomal anthracyclines have achieved highly
efficient encapsulation,
34
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
and include versions with greatly prolonged circulation such as liposomal
daunorubicin and
pegylated liposomal doxorubicin. See Krishna et al., Carboxymethylcellulose-
sodium based
transdermal drug delivery system for propranolol, J Pharm Pharmacol. 1996 Apr;
48(4):367-70.
Liposomal delivery systems provide stable formulation, provide improved
pharmacokinetics, and a degree of 'passive' or 'physiological' targeting to
tissues. Encapsulation
of hydrophilic and hydrophobic materials, such as potential cancer drugs, are
known. See for
example U.S. Pat. No. 5,466,468; U.S. Pat. 5,580,571; U.S. Pat. No. 5,626,869,
the contents of
each of which are incorporated by reference.
Liposomes and polymerosomes can contain a plurality of solutions and
compounds. In
certain embodiments, the complexes of the invention are coupled to or
encapsulated in
polymersomes. As a class of artificial vesicles, polymersomes are tiny hollow
spheres that
enclose a solution, made using amphiphilic synthetic block copolymers to form
the vesicle
membrane. Common polymersomes contain an aqueous solution in their core and
are useful for
encapsulating and protecting sensitive molecules, such as drugs, enzymes,
other proteins and
peptides, and DNA and RNA fragments. The polymersome membrane provides a
physical
barrier that isolates the encapsulated material from external materials, such
as those found in
biological systems. Polymerosomes can be generated from double emulsions by
known
techniques, see Lorenceau et al., 2005, Generation of Polymerosomes from
Double-Emulsions,
Langmuir 21(20):9183-6, incorporated by reference.
Aspects of the invention provide for numerous uses of delivery vectors.
Selection of the
delivery vector is based upon the cell or tissue targeted and the specific
makeup of the
CRISPR/Cas9/gRNA. For example, in the EBV example discussed above, since
lymphocytes are
known for being resistant to lipofection, nucleofection (a combination of
electrical parameters
generated by a device called Nucleofector, with cell-type specific reagents to
transfer a substrate
directly into the cell nucleus and the cytoplasm) was necessitated for DNA
delivery into the Raji
cells. The Lonza pmax promoter drives Cas9 expression as it offered strong
expression within
Raji cells. 24 hours after nucleofection, obvious EGFP signals were observed
from a small
proportion of cells through fluorescent microscopy. The EGFP-positive cell
population
decreased dramatically, however, <10% transfection efficiency 48 hours after
nucleofection was
measured.
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
Aspects of the invention use the CRISPR/Cas9/gRNA complexes and targeted
delivery.
Common known pathways include transdermal, transmucal, nasal, ocular and
pulmonary routes.
Drug delivery systems may include liposomes, proliposomes, microspheres, gels,
prodrugs,
cyclodextrins, etc. Aspects of the invention utilize nanoparticles composed of
biodegradable
polymers to be transferred into an aerosol for targeting of specific sites or
cell populations in the
lung, providing for the release of the drug in a predetermined manner and
degradation within an
acceptable period of time. Controlled-release technology, such as transdermal
and transmucosal
controlled-release delivery systems, nasal and buccal aerosol sprays, drug-
impregnated lozenges,
encapsulated cells, oral soft gels, iontophoretic devices to administer drugs
through skin, and a
variety of programmable, implanted drug-delivery devices are used in
conjunction with the
complexes of the invention of accomplishing targeted and controlled delivery.
In some embodiments, a composition is provided for topical application (e.g.,
in vivo,
directly to skin of a person). The composition may be applied superficially
(e.g., topically). The
composition provides a nuclease or gene therefore and includes a
pharmaceutically acceptable
diluent, adjuvant, or carrier. Preferably, a carrier is approved for animal or
human use by a
competent governmental agency, such as the US Food and Drug Administration
(FDA) or the
like. Examples include, but are not limited to, phosphate buffered saline,
physiological saline,
water, and emulsions, such as oil/water emulsions. The carrier can be a
solvent or dispersing
medium containing, for example, ethanol, polyol (for example, glycerol,
propylene glycol, liquid
polyethylene glycol, and the like), suitable mixtures thereof, and vegetable
oils. These
formulations contain from about 0.01% to about 100%, preferably from about
0.01% to about
90% of the MFB extract, the balance (from about 0% to about 99.99%, preferably
from about
10% to about 99.99% of an acceptable carrier or other excipients. A more
preferred formulation
contains up to about 10% MFB extract and about 90% or more of the carrier or
excipient,
whereas a typical and most preferred composition contains about 5% MFB extract
and about
95% of the carrier or other excipients. Formulations are described in a number
of sources that are
well known and readily available to those skilled in the art.
36
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
vi. Cut nucleic acid
Once inside the cell, the nuclease targets oncoviral nucleic acid sequences.
In some
embodiments, methods and compositions of the invention use a nuclease such as
Cas9 to target
latent oncoviral genomes, thereby reducing the chances of proliferation.
Upon introduction of Cas9 nuclease into target cells, the nuclease forms a
complex with
the gRNA (e.g., crRNA + tracrRNA or sgRNA). The complex cuts the viral nucleic
acid in a
targeted fashion to incapacitate the viral genome. The Cas9 endonuclease
causes a double strand
break in the viral genome. By targeted several locations along the viral
genome and causing not a
single strand break, but a double strand break, the genome is effectively cut
a several locations
along the genome. In a preferred embodiment, the double strand breaks are
designed so that
small deletions are caused, or small fragments are removed from the genome so
that even if
natural repair mechanisms join the genome together, the genome is render
incapacitated.
The nuclease, or a gene encoding the nuclease, may be delivered to cells by
transfection.
For example, the cells may be transfected with DNA that encodes Cas9 and gRNA
(on a single
piece or separate pieces). The gRNAs are designed to localize the Cas9
endonuclease at one or
several locations along the viral genome. The Cas9 endonuclease causes double
strand breaks in
the genome, causing small fragments to be deleted from the viral genome. Even
with repair
mechanisms, the deletions render the viral genome incapacitated.
vii. Host genome
It will be appreciated that method and compositions of the invention can be
used to target
oncoviral nucleic acid without interfering with host genetic material. Methods
and compositions
of the invention employ a targeting moiety such as a guide RNA that has a
sequence that
hybridizes to a target within the viral sequence. Methods and compositions of
the invention may
further use a targeted nuclease such as the cas9 enzyme, or a vector encoding
such a nuclease,
which uses the gRNA to bind exclusively to the viral genome and make double
stranded cuts,
thereby removing the viral sequence from the host.
Where the targeting moiety includes a guide RNA, the sequence for the gRNA, or
the
guide sequence, can be determined by examination of the viral sequence to find
regions of about
20 nucleotides that are adjacent to a protospacer adjacent motif (PAM) and
that do not also
appear in the host genome adjacent to the protospacer motif.
37
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
Preferably a guide sequence that satisfies certain similarity criteria (e.g.,
at least 60%
identical with identity biased toward regions closer to the PAM) so that a
gRNA/cas9 complex
made according to the guide sequence will bind to and digest specified
features or targets in the
viral sequence without interfering with the host genome. Preferably, the guide
RNA corresponds
to a nucleotide string next to a protospacer adjacent motif (PAM) (e.g., NGG,
where N is any
nucleotide) in the viral sequence. Preferably, the host genome lacks any
region that (1) matches
the nucleotide string according to a predetermined similarity criteria and (2)
is also adjacent to
the PAM. The predetermined similarity criteria may include, for example, a
requirement of at
least 12 matching nucleotides within 20 nucleotides 5' to the PAM and may also
include a
requirement of at least 7 matching nucleotides within 10 nucleotides 5' to the
PAM. An
annotated viral genome (e.g., from GenBank) may be used to identify features
of the viral
sequence and finding the nucleotide string next to a protospacer adjacent
motif (PAM) in the
viral sequence within a selected feature (e.g., a viral replication origin, a
terminal repeat, a
replication factor binding site, a promoter, a coding sequence, or a
repetitive region) of the viral
sequence. The viral sequence and the annotations may be obtained from a genome
database.
Where multiple candidate gRNA targets are found in the viral genome, selection
of the
sequence to be the template for the guide RNA may favor the candidate target
closest to, or at the
5' most end of, a targeted feature as the guide sequence. The selection may
preferentially favor
sequences with neutral (e.g., 40% to 60%) GC content. Additional background
regarding the
RNA-directed targeting by endonuclease is discussed in U.S. Pub. 2015/0050699;
U.S. Pub.
20140356958; U.S. Pub. 2014/0349400; U.S. Pub. 2014/0342457; U.S. Pub.
2014/0295556; and
U.S. Pub. 2014/0273037, the contents of each of which are incorporated by
reference for all
purposes. Due to the existence of human genomes background in the infected
cells, a set of steps
are provided to ensure high efficiency against the viral genome and low off-
target effect on the
human genome. Those steps may include (1) target selection within viral
genome, (2) avoiding
PAM+target sequence in host genome, (3) methodologically selecting viral
target that is
conserved across strains, (4) selecting target with appropriate GC content,
(5) control of nuclease
expression in cells, (6) vector design, (7) validation assay, others and
various combinations
thereof. A targeting moiety (such as a guide RNA) preferably binds to targets
within certain
categories such as (i) latency related targets, (ii) infection and symptom
related targets, and (iii)
structure related targets.
38
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
A first category of targets for gRNA includes latency-related targets. The
viral genome
requires certain features in order to maintain the latency. These features
include, but not limited
to, master transcription regulators, latency-specific promoters, signaling
proteins communicating
with the host cells, etc. If the host cells are dividing during latency, the
viral genome requires a
replication system to maintain genome copy level. Viral replication origin,
terminal repeats, and
replication factors binding to the replication origin are great targets. Once
the functions of these
features are disrupted, the viruses may reactivate, which can be treated by
conventional antiviral
therapies.
A second category of targets for gRNA includes infection-related and symptom-
related
targets. Virus produces various molecules to facilitate infection. Once gained
entrance to the host
cells, the virus may start lytic cycle, which can cause cell death and tissue
damage (HBV). In
certain cases, such as HPV-16 or HPV-18, cell products (E6 and E7 proteins)
can transform the
host cells and cause cancers. E6 from HPV-18 is reported as an oncogene
capable of
transforming cells and thus provides a target according to certain
embodiments. Disrupting the
key genome sequences (promoters, coding sequences, etc) producing these
molecules can
prevent further infection, and/or relieve symptoms, if not curing the disease.
A third category of targets for gRNA includes structure-related targets. Viral
genome
may contain repetitive regions to support genome integration, replication, or
other functions.
Targeting repetitive regions can break the viral genome into multiple pieces,
which physically
destroys the genome.
Where the nuclease is a cas protein, the targeting moiety is a guide RNA. Each
cas
protein requires a specific PAM next to the targeted sequence (not in the
guide RNA). This is the
same as for human genome editing. The current understanding the guide
RNA/nuclease complex
binds to PAM first, then searches for homology between guide RNA and target
genome.
Sternberg et al., 2014, DNA interrogation by the CRISPR RNA-guided
endonuclease Cas9,
Nature 507(7490):62-67. Once recognized, the DNA is digested 3-nt upstream of
PAM. These
results suggest that off-target digestion requires PAM in the host DNA, as
well as high affinity
between guide RNA and host genome right before PAM.
It may be preferable to use a targeting moiety that targets portions of the
viral genome
that are highly conserved. Viral genomes are much more variable than human
genomes. In order
to target different strains, the guide RNA will preferably target conserved
regions. As PAM is
39
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
important to initial sequence recognition, it is also essential to have PAM in
the conserved
region.
In a preferred embodiment, methods of the invention are used to deliver a
nucleic acid to
cells. The nucleic acid delivered to the cells may include a gRNA having the
determined guide
sequence or the nucleic acid may include a vector, such as a plasmid, that
encodes an enzyme
that will act against the target genetic material. Expression of that enzyme
allows it to degrade or
otherwise interfere with the target genetic material. The enzyme may be a
nuclease such as the
Cas9 endonuclease and the nucleic acid may also encode one or more gRNA having
the
determined guide sequence.
The gRNA targets the nuclease to the target genetic material. Where the target
genetic
material includes the genome of a virus, gRNAs complementary to parts of that
genome can
guide the degredation of that genome by the nuclease, thereby preventing any
further replication
or even removing any intact viral genome from the cells entirely. By these
means, latent viral
infections can be targeted for eradication.
The host cells may grow at different rate, based on the specific cell type.
High nuclease
expression is necessary for fast replicating cells, whereas low expression
help avoiding off-target
cutting in non-infected cells. Control of nuclease expression can be achieved
through several
aspects. If the nuclease is expressed from a vector, having the viral
replication origin in the
vector can increase the vector copy number dramatically, only in the infected
cells. Each
promoter has different activities in different tissues. Gene transcription can
be tuned by choosing
different promoters. Transcript and protein stability can also be tuned by
incorporating
stabilizing or destabilizing (ubiquitin targeting sequence, etc) motif into
the sequence.
Specific promoters may be used for the gRNA sequence, the nuclease (e.g.,
cas9), other
elements, or combinations thereof. For example, in some embodiments, the gRNA
is driven by a
U6 promoter. A vector may be designed that includes a promoter for protein
expression (e.g.,
using a promoter as described in the vector sold under the trademark
PMAXCLONING by
Lonza Group Ltd (Basel, Switzerland). A vector may be a plasmid (e.g., created
by synthesis
instrument 255 and recombinant DNA lab equipment). In certain embodiments, the
plasmid
includes a U6 promoter driven gRNA or chimeric guide RNA (sgRNA) and a
ubiquitous
promoter-driven cas9. Optionally, the vector may include a marker such as EGFP
fused after the
cas9 protein to allow for later selection of cas9+ cells. It is recognized
that cas9 can use a gRNA
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
(similar to the CRISPR RNA (crRNA) of the original bacterial system) with a
complementary
trans-activating crRNA (tracrRNA) to target viral sequences complementary to
the gRNA. It has
also been shown that cas9 can be programmed with a single RNA molecule, a
chimera of the
gRNA and tracrRNA. The single guide RNA (sgRNA) can be encoded in a plasmid
and
transcription of the sgRNA can provide the programming of cas9 and the
function of the
tracrRNA. See Jinek, 2012, A programmable dual-RNA-guided DNA endonuclease in
adaptive
bacterial immunity, Science 337:816-821 and especially figure 5A therein for
background.
Using the above principles, methods and compositions of the invention may be
used to
target viral nucleic acid in an infected host without adversely influencing
the host genome.
For additional background see Hsu, 2013, DNA targeting specificity of RNA-
guided
Cas9 nucleases, Nature Biotechnology 31(9):827-832; and Jinek, 2012, A
programmable dual-
RNA-guided DNA endonuclease in adaptive bacterial immunity, Science 337:816-
821, the
contents of each of which are incorporated by reference. Since the targeted
locations are selected
to be within certain categories such as (i) latency related targets, (ii)
infection and symptom
related targets, or (iii) structure related targets, cleavage of those
sequences inactivates the virus
and removes it from the host. Since the targeting RNA (the gRNA or sgRNA) is
designed to
satisfy according to similarity criteria that matches the target in the viral
genetic sequence
without any off-target matching the host genome, the latent viral genetic
material is removed
from the host without any interference with the host genome.
viii. Composition
In some embodiments, the invention provides a composition for topical
application (e.g.,
in vivo, directly to skin of a person). The composition may be applied
superficially (e.g.,
topically). The composition provides a nuclease or gene therefore and includes
a
pharmaceutically acceptable diluent, adjuvant, or carrier. Preferably, a
carrier used in accordance
with the subject invention is approved for animal or human use by a competent
governmental
agency, such as the US Food and Drug Administration (FDA) or the like.
Examples include, but
are not limited to, phosphate buffered saline, physiological saline, water,
and emulsions, such as
oil/water emulsions. The carrier can be a solvent or dispersing medium
containing, for example,
ethanol, polyol (for example, glycerol, propylene glycol, liquid polyethylene
glycol, and the
like), suitable mixtures thereof, and vegetable oils. These formulations
contain from about 0.01%
to about 100%, preferably from about 0.01% to about 90% of the MFB extract,
the balance (from
41
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
about 0% to about 99.99%, preferably from about 10% to about 99.99% of an
acceptable carrier
or other excipients. A more preferred formulation contains up to about 10% MFB
extract and
about 90% or more of the carrier or excipient, whereas a typical and most
preferred composition
contains about 5% MFB extract and about 95% of the carrier or other
excipients. Formulations
are described in a number of sources that are well known and readily available
to those skilled in
the art.
ix. Repair inhibition
In a repair inhibition embodiment, an infection is treated by delivering a
nuclease that
cuts viral nucleic acid and an inhibitor that prevents repair of the cut viral
nucleic acid. Any
suitable nuclease can be used. Where a CRISPR-associated (Cas)-type nuclease
(e.g., Cas5,
Cas6, Cas9, Cfpl, or a modified version thereof) is used, the Cas-type
nuclease is delivered
along with an RNA that targets the nuclease to the viral nucleic acid. Any
suitable inhibitor may
be used such as, for example, a small molecule drug, an enzyme, or other
molecular entity. Small
molecules that inhibit enzymes of a DNA repair pathway are known and may be
used.
Additionally or alternatively, the inhibitor may be provided by an enzyme that
modifies a free
end of the cut nucleic acid so that it is not accessible for a repair. The
inhibitor may be a
nucleotide or nucleoside analog or ddNTP that prevents a successful repair.
The nuclease may be initially provided for delivery in any suitable form. For
example,
the nuclease may be delivered as an active enzyme or ribonucleoprotein (RNP)
or the nuclease
may be encoded in a nucleic acid, such as in a DNA vector or as mRNA.
Likewise, where the
inhibitor is a protein, the inhibitor may initially be provided in any
suitable form such as as a
protein or encoded in a nucleic acid. Where the nuclease and the inhibitor are
to be provided in a
nucleic acid form, they may both be encoded on the same nucleic acid (e.g.,
DNA plasmid or
mRNA) with or without a spacer or linker, or they may be separately delivered.
The nuclease and the inhibitor may be delivered to the infected cells together
(e.g., as part
of a single composition) or they may be delivered separately, wholly or
partially simultaneously
or separately. Either or both of the nuclease and inhibitor may be provided
with a
pharmaceutically acceptable carrier or prepared for delivery orally,
intravenously, topically, or
by any suitable method. Either or both of the nuclease and the inhibitor may
be delivered using a
suitable viral or non-viral vector or delivery method or other suitable
format. For example, the
nuclease, targeting sequence, and the treatment may be delivered on the same
vehicle, whether
42
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
nucleic acid, plasmid, or viral vector. Alternatively, the nuclease and
targeting sequence may be
delivered in one manner, and the treatment may be delivered in a separate
manner. For example,
a cocktail may include: (i) a vector encoding the nuclease and the targeting
sequence; and (ii) the
treatment or a vector encoding the treatment. In one embodiment, the delivery
method includes
the use of ribonucleoproteins (RNP). For example, the ribonucleotide may
include Cas9 (as the
protein) and guide RNA as the ribonucleic acid. Delivery as RNP allows control
over dosing and
avoids continuous production of nuclease proteins by the cell. In some
embodiments, mRNA
may be used to deliver the nuclease, to encourage continued production of the
nuclease.
FIG. 20 diagrams a treatment and repair inhibition method 2001 of treating a
viral
infection according to the repair inhibition embodiment. Methods of the repair
inhibition
embodiment are applicable to in vivo treatment of patients and may be used to
remove any viral
genetic material, such as genes of virus associated with a latent viral
infection. Methods may be
used in vitro, or ex vivo, e.g., to prepare or treat a cell culture or cell
sample. When used in vivo,
the cell may be any suitable germ line or somatic cell and compositions of the
repair inhibition
embodiment may be delivered to specific parts of a patient's body or be
delivered systemically.
If delivered systemically, it may be preferable to include tissue-specific
promoters. For example,
if a latent viral infection is localized to the liver, hepatic tissue-specific
promotors may be
included in a plasmid or viral vector that codes for a targeted nuclease.
Methods of the repair inhibition embodiment are suitable for the treatment of
viruses,
including, but not limited to, the following viruses: adenovirus, herpes
simplex virus, varicella-
zoster virus, Epstein-Barr virus, human cytomegalovirus, human herpesvirus
type 8, human
papillomavirus, BK virus, JC virus, smallpox, hepatitis B virus, human
bocavirus, parvovirus,
B19, human astrovirus, Norwalk virus, coxsackievirus, hepatitis A virus,
poliovirus, rhinovirus,
sever acute respiratory syndrome virus, hepatitis C virus, yellow fever virus,
dengue virus, west
nile virus, rubella virus, hepatitis E virus, human immunodeficiency virus,
influenza virus,
guanarito virus, junin virus, las sa virus, machupo virus, sabia virus,
Crimean-Congo hemorrhagic
fever virus, ebola virus, Marburg virus, measles virus, mumps virus,
parainfluenza virus,
respiratory syncytial virus, human metapnemovirus, Hendra virus, nipah virus,
rabies virus,
hepatitis D virus, rotavirus, orbivirus, coltivirus, or banna virus.
Methods of the repair inhibition embodiment involve obtaining a nuclease that
is
designed to cut or cleave a target nucleic acid. Typically, the target nucleic
acid is viral nucleic
43
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
acid. Suitable targets in viral genomes include, for example, a portion of a
genome or gene of a
hepatitis virus, a hepatitis B virus (HBV), an Epstein-Barr virus, a Kaposi's
sarcoma-associated
herpesvirus (KSHV), a herpes-simplex virus (HSV), a cytomegalovirus (CMV), and
a human
papilloma virus (HPV). The target in the viral genome may lie within one or
more of a preC
promoter in a hepatitis B virus (HBV) genome, an 51 promoter in the HBV
genome, an S2
promoter in the HBV genome, an X promoter in the HBV genome, a viral Cp (C
promoter) in an
Epstein-Barr virus genome, a minor transcript promoter region in a Kaposi's
sarcoma-associated
herpesvirus (KSHV) genome, a major transcript promoter in the KSHV genome, an
Egr-1
promoter from a herpes-simplex virus (HSV), an ICP 4 promoter from HSV-1, an
ICP 10
promoter from HSV-2, a cytomegalovirus (CMV) early enhancer element, a
cytomegalovirus
immediate-early promoter, an HPV early promoter, and an HPV late promoter.
While methods of the repair inhibition embodiment may be used to target and
treat
viruses, methods may also be used to directly treat mutated or tumor nucleic
acid. For example,
methods and systems may target gene signatures unique to tumors. The gene
signature unique to
a tumor may include a signature related to proliferation of tumor nucleic acid
or may include a
signature directly related to the responsiveness of the tumor to chemotherapy
or other medicinal
treatments. For example, tumors with Ras mutations have been found less
responsive to
chemotherapy than tumors with normal Ras. In such aspects, methods may target
a nuclease to
Ras-mutated tumor nucleic acid, use the nuclease to cut the Ras-muated tumor
nucleic acid into
fragments, and then use a molecule or moiety to inhibit repair of the ends of
the fragments (e.g.,
by treatment with Antarctic phosphatase, or by ligating fragments with dideoxy
ends to 5' ends
of the fragments). Such a treatment destroys the Ras-mutated tumor nucleic
acid. With the Ras-
mutated tumor nucleic acid destroyed, the tumor may be more receptive to, for
example,
chemotherapy.
Systems and methods may include one or more nucleases, one or more guide or
targeting
sequences, and one or more inhibitor of DNA repair. The nuclease is designed
to cut or cleave
target nucleic acid (such as viral nucleic acid) into fragments, and the guide
sequence targets the
nuclease to a viral genomic target. The inhibitor prevents ligation of the
resulting fragments or
nucleic acid synthesis. In an illustrative embodiment, the nuclease and the
inhibitor are both
provided encoded on a plasmid to be transcribed and translated in the infected
cells.
44
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
FIG. 21 shows nucleic acid 2101 that encodes a nuclease 2105, a guide or
targeting
sequence 2121, and an inhibitor 2109. In the depicted embodiment, the
inhibitor is an enzyme
that prevents repair of cut DNA. For example, the inhibitor may be Antarctic
phosphatase. Other
features may optionally be included in the nucleic acid 2101. For example, the
nucleic acid may
further include a switch 2113 that causes the nuclease to be expressed in the
presence of a viral
nucleic acid (a riboswitch). The nucleic acid 2101 may include one or more
promoter 2117 to aid
in transcription of the included genes. Additionally, the nucleic acid 2101
may include a portion
that codes for a nuclear localization signal 2123 so that the nuclease 105,
the inhibitor, or both,
when expressed by transcription and translation, are tagged for import into
the nucleus of a host
cell so that they can attack the viral DNA there.
FIG. 22 shows a composition for treating a viral infection according to
certain
embodiments. The composition preferably includes a vector (which may be a
plasmid, or a viral
vector) that codes for a nuclease that cuts viral nucleic acid into fragments,
a targeting sequence
(e.g., a gRNA) that targets the nuclease to viral nucleic acid, and a
treatment that prevents DNA
repair or ligation of the fragments. The composition may optionally include
one or more of a
promoter, replication origin, other elements, or combinations thereof as
described further herein.
Systems and methods of the repair inhibition embodiment include using a
programmable
or targetable nuclease to specifically target viral nucleic acid for
destruction. Any suitable
targeting nuclease can be used including, for example, zinc-finger nucleases
(ZFNs),
transcription activator-like effector nucleases (TALENs), clustered regularly
interspaced short
palindromic repeat (CRISPR)-associated (Cas) nucleases, meganucleases, other
endo- or exo-
nucleases, or combinations thereof. See Schiffer, 2012, Targeted DNA
mutagenesis for the cure
of chronic viral infections, J Virol 88(17):8920-8936, incorporated by
reference. Particular
embodiments may employ any nuclease listed in section 'H. Nuclease' above.
A nuclease may use the targeting specificity of a guide RNA (gRNA) or other
such
targeting sequence such as any discussed in section 'Hi. Targeting sequence'
above.
In the repair inhibition embodiments, a nuclease such as a Cas-type nuclease
cleaves
nucleic acid of a virus infecting a cell and an inhibitor of DNA repair
prevents the cleaved
nucleic acid from being repaired.
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
x. Inhibitor of DNA repair
Methods of the repair inhibition embodiments include using one or more
treatments to
inhibit repair of the viral nucleic acid after it is cleaved by the nuclease.
One or more type of
inhibitor may be used. Exemplary types of inhibitors include: inhibitors of
non-homologous end
joining (NHEJ) repair or homologous recombination (HR); enzymes that modify
free ends of
nucleic acid; moieties such as nucleotide/nucleoside analogs that interfere
with DNA synthesis;
ligation of fragments with non-canonical 5' or 3' ends; others; or
combinations thereof.
In preferred embodiments, the inhibitor includes a small molecule drug that
inhibits
NHEJ or HR. By suppressing or destroying those elements essential to end
repair, the end repair
processes are unable to operate and the resulting fragments remain unrepaired
and degraded. The
unrepaired and degraded fragments may promote apoptosis, induce cytotoxicity
or may make the
target nucleic acid more susceptible to other treatments that lead to
apoptosis.
Proteins associated with non-homologous end repair include, but are not
limited to:
Ku80, Ku70, DNA-dependant protein kinase, catalytic subunits (DNA-PKcs),
Artemis, Xrcc4,
and Ligase IV, and non-homologous end repair can be inhibited by suppressing
expression of
those proteins. See, for example, Li, Y.H., Wang, X., Pan, Y.,et al. (2012).
Inhibition of non-
homologous end joining repair impairs pancreatic cancer growth and enhances
radiation
response. PLoS One 7, e39588.; Srivastava, et al. (2012) An inhibitor of
Nonhomologous End-
Joining Abrogates Double Strand Break Repair and Impedes Cancer Progression".
Any enzyme,
chemical or other small molecule that suppresses or inhibits those proteins or
other elements
essential to non-homologous end repair may be used as a treatment to prevent
DNA repair. For
example, Ligase IV has been found as a critical component in the sealing of
double-strand breaks
during non-homologous end joining. SCR7 inhibits expression of Ligase IV,
thereby disrupting
non-homologous end repair of fragmented nucleic acid. Inhibitors of DNA-PK cs
include, for
example, PI-3 Kinases, LY-294002, vanillin (Sigma), and NU-7026 (Valbiochem).
Any suitable
inhibitor of HR may be used. Typical inhibitors of HR will block an enzyme of
a double-
stranded break repair pathway such as ATM, ATR, MRN, RAD51 and paralogs,
BRCA1,
BRCA2, KU70/80, DNA-PKcs, Artemis, Ligase IV, or XRCC4. Suitable HR inhibitors
may
include mirin and caffeine. Specific ATR inhibitors (VE-821 and NU6027) have
been identified
based on cell-based screens and found to be especially toxic to cells
deficient in p53. NU6027
also inhibits RAD51 focus formation (indicative of HR suppression). RI-1
covalently binds to
46
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
the surface of RAD51, thereby reducing its focus formation. HR has also been
targeted by
inhibition of the ATM¨CHK2 or ATR¨CHK1 pathways. The selective ATM inhibitor
KU55933
blocks ionization radiation (IR)-induced, ATM dependent phosphorylation and
sensitized cancer
cells to IR and topoisomerase inhibitors The nonspecific staurosporin analog
UCN-01 is a potent
CHK1 inhibitor. Small-molecule inhibitors of the human RecQ helicases include
BLM (ML216)
and WRN (NSC19630). An ERCC1 inhibitor, N5C130813, has been reported that
synergizes the
effect of cisplatin and mitomycin C. The PARP1 inhibitor olaparib shows
promising results.
Any suitable inhibitor of NHEJ may be used. Genes and proteins associated with
homologous end repair, include but are not limited to, Rad52, Rad51, ATM,
BRAC1, BRAC2,
MRN Complex, ATM, DNA, PK, ATR, and Blm. Homologous repair may be inhibited by
suppressing expression of one or more of those proteins. Any enzyme, chemical
or other
component that negatively affects or suppresses or inhibits those proteins or
other elements
essential to homologous end repair may be used as a treatment to prevent DNA
repair. In one
example, 17-AAG (17-Allylmanio-17-Demethoxygeldanamycin) inhibits homologous
end repair
by causing degrading BRAC2 and altering the behavior of RAD51, which is
critical for
homologous end repair. NHEJ proteins such as the KU70/80 complex, Artemis,
Ligase IV/
XRCC4, Polm, and Poll may be targeted. One of the first inhibitors of DNA-PKcs
was
wortmannin. A derivative of quercetin, LY294002, has also been shown to
possess similar
properties. Recently, NU7026 has been reported to be a very selective and
potent DNA-PK
inhibitor. L189 is a potential Ligase IV inhibitor that blocks the activity of
all three ligases,
Ligase I, Ligase III, and Ligase IV. SCR7 has been identified as a potent
inhibitor of end joining.
For additional background, see Srivastava & Raghavan, 2015, DNA Double-strand
break repair
inhibitors as cancer therapies, Chem & Biol 22:17-29, incorporated by
reference. Thus, the
inhibitor may include small molecule such as KU55933; caffeine; VE-821;
NU6027; UNC-01;
mirin; RI-1; streptonigrin; RI-2; 3-ABA; olaparib; NU1025; N5C130813;
wortmannin; NU7026;
SCR7; or L189 to suppress homologous recombination (HR) or non-homologous end-
joining
(NHER).
An inhibitor may be an enzyme that modify free ends of nucleic acid, such as
Antarctic
phosphatase. Antarctic phosphatase catalyzes the removal of a 5' phosphate
group, rendering that
free end unavailable for repair by end-joining or by template-dependent
extension.
47
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
An inhibitor may include moieties such as nucleotide/nucleoside analogs that
interfere
with DNA synthesis. For example, where DNA repair would require template-
dependent
synthesis, a nucleotide analog may be taken up by a polymerase and arrest it
from further
activity.
Additionally or alternatively, repair may be inhibited by enhancing cell
exonuclease
activity, e.g. to increase degradation of SSB and DSB (single and double
strand DNA breaks).
For example, human exonuclease 1 (hEX01) efficiently repairs DSB. hEX01 is
ubiquitinated
and degraded in the proteasome. Thus, in some embodiments, a combination of a
targeted
endonuclease with a proteasome inhibitor are delivered to enhance hEX0-1
activity and
synergize to kill viral DNA+ cells.
In some embodiments, it is recognized that the double-stranded breaks (DSBs)
introduced
by Cas-type nuclease are primarily repaired via non-homologous end joining
(NHEJ) and that
DNA ligase IV (LIG4) is critical for NHEJ. Other LIGs (1-3) are involved in
repair of SSB and
DSB. Systems and methods described herein may include one or more small
molecule inhibitors
of LIG4 or other LIGs. For example, the compound L82 has been identified as an
uncompetitive
inhibitor of DNA ligase I. L67 is a compound that inhibits LIG1 and LIG3.
Other compounds
that have been identified as inhibitors of a DNA Ligase may be used.
FIG. 25 shows three small molecule inhibitors of DNA ligase, L67, L82, and
L189. L189
inhibits hLigI, hLigIIIP, and hLigIV/XRCC4, L67 inhibits hLigI and hLigIIIP,
and L82 inhibits
hLigI. Additional discussion may be found in Chen et al., 2008, Rational
Design of Human DNA
Ligase Inhibitors that Target Cellular DNA Replication and Repair, Cancer Res
68(9):3169-
3177, incorporated by reference.
In some embodiments, the treated cell is provided with DNA fragments with non-
canonical 5' or 3' ends. Those fragments may include sequence with at least
partial homology to
known, target viral sequences. The end-joining repair mechanisms (HR or NHEJ)
may join those
fragments to the free ends of the cut nucleic acid. In one embodiments, the
fragments have 3'
ends that lack a 3' hydroxyl group and thus present a di-deoxy 3' end, which
is not competent for
further repair.
Additionally or alternatively, the inhibitor may be an enzyme that suppresses
or destroys
components (such as enzymes or proteins) necessary to end repair processes.
Additionally or
alternatively, the treatment may include any added molecule that would inhibit
ligation of the
48
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
resulting fragments after cleavage. For example, the treatment may be a chain
terminator, which
ligates to the fragments before the end repair operations.
An insight is to use gene-editing tools such as Cas9 along with an inhibitor
of DNA
repair, i.e., an inhibitor that would normally prevent successful gene-
editing. By inhibiting end
repair, ligation or both of the resulting fragments, the target nucleic acid
(e.g. viral, mutated, or
cancerous nucleic acid) is disrupted or destroyed. In some embodiments,
leaving the target
nucleic acid fragments unrepaired or non-ligated is enough to destroy the
nucleic acid or treat the
viral infection. In further embodiments, the unrepaired or non-ligated
fragments may make the
target nucleic acid more susceptible or sensitive to other therapies
(including drug, chemical,
radiation, etc.). For example, after the target nucleic acid fragments are
exposed to the treatment
and left unrepaired/non-ligated, methods of the repair inhibition embodiments
provide for
exposing the unrepaired/non-ligated fragments to another therapeutic agent to
further destroy or
degrade the target nucleic acid.
In certain embodiments, an inhibitor is used that prevents formation of a
phosphodiester
bond by, for example, inhibiting a polymerase or functioning as a chain
terminator. For example,
an enzyme or chemical may inhibit bond formation by removing or adding a
phosphate group at
the 5' side or by removing the 3' hydroxyl group to make a dideoxy end.
Inhibitor may include ddNTPs, nucleotides, nucleosides, or analogs thereof
that prevent
ligation or polymerase activity, when contacted and incorporated into a
nucleic acid fragment.
Such a moiety may repress viral reproduction by competing with natural
dNTP/NTP substrates
for incorporation into the nascent viral nucleic acid, thereby leading to
chain termination. In
certain embodiments, the chain terminator is a ddNTP. ddNTPs block
polymerization and
ligation when added to the end of a nucleic acid due to their lack of the 3'
hydroxyl group.
In certain embodiments, antiviral agents (such as nucleoside, nucleotide, and
analogues
thereof) may be used as the treatment. They compete with natural dNTP/NTP
substrates for the
incorporation into the nucleic acid thereby leading to chain termination or
mutagenesis. See,
Clerc and Neyts, Handb Exp Pharmacol. 2009;(189):53-84. doi: 10.1007/978-3-540-
79086-0 3.
For example, nucleoside analogues that possess a 3' hydroxyl may act as a
chain terminator,
where the hydroxyl is conformationally constrained or sterically hindered from
creating a
phosphodiester linkage with incoming nucleotide. In such instance, chain
elongation can be
hampered by, for example, 2'-C-methyl or 4'azido nucleoside inhibitors of HCV
replication.
49
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
Canonical 3'deoxyribonucleotides have also been successfully used as a chain
terminator. See,
Shim et al. Antiviral Res. 2003 May;58(3):243-51. The following nucleosides,
nucleotides, and
analogues may be used as a chain terminator: Lamivudine triphosphate,
Stavudine triphosphate,
Zidoduvine triphosphate, Aciclovir triphosphate, Vidarabine triphosphate,
Ribavirin
triphosphate, 3TC (Lamivudine), d4T (Stavudine), AzT (Zidovudine), ara-A
(Vidarabine),
Aciclovir, Ribavirn, 3TCMP, d4TMP, AzTMP, ara-AMP, Aciclovir monophosphate,
Ribavirin
monophosphate, 3TCTP, d4TTP, AzTTP, ara-ATP, Aciclovir triphosphate, Ribavirin
Triphosphate.
Methods include introducing into a host cell a nuclease and an inhibitor of
DNA repair.
The nuclease may be initially provided for delivery in any suitable form. For
example, the
nuclease may be delivered as an active enzyme or ribonucleoprotein (RNP) or
the nuclease may
be encoded in a nucleic acid, such as in a DNA vector or as mRNA. Likewise,
where the
inhibitor is a protein, the inhibitor may initially be provided in any
suitable form such as as a
protein or encoded in a nucleic acid. Where the nuclease and the inhibitor are
to be provided in a
nucleic acid form, they may both be encoded on the same nucleic acid (e.g.,
DNA plasmid or
mRNA) with or without a spacer or linker, or they may be separately delivered.
In a preferred
embodiment, the nuclease is obtained or delivered in a ribonucleoprotein (RNP)
form, e.g. as a
recombinant Cas9 protein duplexed with sgRNA or with crRNA + tracRNA, or as a
recombinant
TALEN protein. It may be found that delivery as RNP is more effective and less
toxic than
plasmid DNA, and that RNP permits delivery of pre-formed enzymatically active
drug (which
acts faster), and is only active in the cell for a very limited time (<24
hours), thus reducing non-
specific toxicity and off-target activity. RNP can be directly electroporated
into primary tissues,
e.g. peripheral blood mononuclear cells (PBMCs), for ex vivo transplant
indications. RNP, like
mRNA or pDNA, can also be incorporated into cationic lipid nanoparticles for
in vivo delivery
indications, e.g. cancer.
The nuclease and the inhibitor may be delivered to the infected cells together
(e.g., as part
of a single composition) or they may be delivered separately, wholly or
partially simultaneously
or separately. Either or both of the nuclease and inhibitor may be provided
with a
pharmaceutically acceptable carrier or prepared for delivery orally,
intravenously, topically, or
by any suitable method. Either or both of the nuclease and the inhibitor may
be delivered using a
suitable viral or non-viral vector or delivery method or other suitable
format. For example, the
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
nuclease, targeting sequence, and the treatment may be delivered on the same
vehicle, whether
nucleic acid, plasmid, or viral vector. Alternatively, the nuclease and
targeting sequence may be
delivered in one manner, and the treatment may be delivered in a separate
manner. For example,
a cocktail may include: (i) a vector encoding the nuclease and the targeting
sequence; and (ii) the
treatment or a vector encoding the treatment.
In some embodiments, a Cas-type nuclease and inhibitor are introduced into a
cell.
Introduction to the cells may include any of the techniques or materials
discussed in section 'v.
Introduce to cell' above.
xi. Mechanism of repair inhibition
Once inside the cell, the nuclease cuts viral nucleic acid and the inhibitor
prevents repair
of the cut nucleic acid.
Preferably, the nuclease is specifically targeted to viral genomes, e.g., by
the sequence of
ZFN or by a gRNA with a Cas9. The nuclease cuts the viral nucleic acid and the
inhibitor
prevents repair. In some embodiments, methods and compositions of the repair
inhibition
embodiments use a nuclease such as Cas9 to target latent viral genomes,
thereby reducing the
chances of proliferation.
The following describes using Cas9 endonuclease and gRNA for targeted cutting
of the
HPV genome. It is understood that this description is applicable to other
nucleases. FIG. 4 shows
the results of successfully cleaving the HPV genome using Cas9 endonuclease, a
gRNA for E6,
and a gRNA for E7. The nuclease forms a complex with the gRNA (e.g., crRNA +
tracrRNA or
sgRNA). The complex cuts the viral nucleic acid in a targeted fashion to
incapacitate the viral
genome. The Cas9 endonuclease causes a double strand break in the viral
genome. By targeted
several locations along the viral genome and causing not a single strand
break, but a double
strand break, the genome is effectively cut a several locations along the
genome.
The inhibitor prevents repair of the double stranded breaks. In some
embodiments, an
inhibitor such as a small molecule drug prevents homologous or non-homologous
end repair. In
certain embodiments, the inhibitor suppresses or destroys elements (such as
enzymes or proteins)
necessary to end repair processes. Additionally or alternatively, the
inhibitor may otherwise
prevent ligation of the resulting fragments after cleavage. For example, the
treatment may be a
chain terminator, which ligates to the fragments before the end repair
operations. The use of
51
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
terminators is in contrast to targeted nuclease schemes that rely on end
repair mechanisms (e.g.
cleave mutated sequence between fragments and re-ligate fragments) to create a
desired change
within the genomic region of interest, such as altering its expression. By
inhibiting end repair,
ligation or both of the resulting fragments, the target nucleic acid (e.g.
viral, mutated, or
cancerous nucleic acid) is disrupted or destroyed. In some embodiments, the
target nucleic acid
destruction itself may be enough to treat the infection. In further
embodiments, the unrepaired or
non-ligated fragments may make the target nucleic acid more susceptible or
sensitive to other
therapies (including drug, chemical, radiation, etc.)
In preferred embodiments, compositions and methods of the repair inhibition
embodiments are used to treat latent viral infections. Viruses known or
suspected to exhibit a
latency phase include viruses of the Herpesviridae family (e.g., Herpes
simplex virus-1 (HSV-1),
Herpes simplex virus-2 (HSV-2), Varicella zoster virus (VZV), Epstein-Barr
virus (EBV),
Cytomegalovirus (CMV), Roseolovirus, Herpes lymphotropic virus, Kaposi's
sarcoma-
associated herpesvirus (KSHV)) among others (e.g., pseuodrabies virus).
Latency is
distinguished from lytic infection; in lytic infection many Herpes virus
particles are produced
and then burst or lyse the host cell. Lytic infection is sometimes known as
"productive"
infection. Latent cells harbor the virus for long time periods, then
occasionally convert to
productive infection which may lead to a recurrence of symptomatic Herpes
symptoms. During
latency, most of the Herpes DNA is inactive, with the exception of LAT, which
accumulates
within infected cells. Treating a latent viral infection with a targeted
nuclease and a treatment
that prevents DNA repair may be particularly beneficial in preventing any
recurrence of a
productive infection.
After a treatment is used to inhibit end repair, one or more therapeutics may
be applied to
further degrade target nucleic acid or induce apoptosis in the diseased or
infected cell. The
therapeutic may include, for example, application of radiation therapy,
application of
pharmaceuticals, antibiotics, or other chemical compounds, or a combination
thereof. In a
preferred embodiment, a treatment that prevents DNA repair includes an
exonuclease. In one
example, chemotherapy or cytotoxic drugs may be applied after inhibition of
end repair for
further treatment. Suitable chemotherapy drugs include alkylating agents,
antimetabolites,
anthracyclines and other anti-tumor antibiotics, topoisomerase inhibitors, and
miotic inhibitors,
corticosteroids. In another example, hydroxamic acid-based compounds, such as
trichostatin A
52
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
(TSA), can be used to induce cytoxicity or further apoptosis of cells with the
degraded nucleic
acid (i.e. nucleic acid fragments with end repair inhibited).
Incorporation by Reference
References and citations to other documents, such as patents, patent
applications, patent
publications, journals, books, papers, web contents, have been made throughout
this disclosure.
All such documents are hereby incorporated herein by reference in their
entirety for all purposes.
Equivalents
Various modifications of the invention and many further embodiments thereof,
in
addition to those shown and described herein, will become apparent to those
skilled in the art
from the full contents of this document, including references to the
scientific and patent literature
cited herein. The subject matter herein contains important information,
exemplification and
guidance that can be adapted to the practice of this invention in its various
embodiments and
equivalents thereof.
Examples
Example 1: Targeting EBV
Burkitt's lymphoma cell lines Raji, Namalwa, and DG-75 were obtained from ATCC
and
cultured in RPMI 1640 supplemented with 10% FBS and PSA, following ATCC
recommendation. Human primary lung fibroblast IMR-90 was obtained from Coriell
and
cultured in Advanced DMEM/F-12 supplemented with 10% FBS and PSA.
Plasmids consisting of a U6 promoter driven chimeric guide RNA (sgRNA) and a
ubiquitous promoter driven Cas9 were obtained from addgene, as described by
Cong L et al.
(2013) Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 339:819-
823. An
EGFP marker fused after the Cas9 protein allowed selection of Cas9-positive
cells (FIG. 4). We
adapted a modified chimeric guide RNA design for more efficient Pol-III
transcription and more
stable stem-loop structure (Chen B et al. (2013) Dynamic Imaging of Genomic
Loci in Living
Human Cells by an Optimized CRISPR/Cas System. Cell 155:1479-1491).
We obtained pX458 from Addgene, Inc. A modified CMV promoter with a synthetic
intron (pmax) was PCR amplified from Lonza control plasmid pmax-GFP. A
modified guide
53
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
RNA sgRNA(F+E) was ordered from IDT. EBV replication origin oriP was PCR
amplified from
B95-8 transformed lymphoblastoid cell line GM12891. We used standard cloning
protocols to
clone pmax, sgRNA(F+E) and oriP to pX458, to replace the original CAG
promoter, sgRNA and
fl origin. We designed EBV sgRNA based on the B95-8 reference, and ordered DNA
oligos
from IDT. The original sgRNA place holder in pX458 serves as the negative
control.
Lymphocytes are known for being resistant to lipofection, and therefore we
used
nucleofection for DNA delivery into Raji cells. We chose the Lonza pmax
promoter to drive
Cas9 expression as it offered strong expression within Raji cells. We used the
Lonza
Nucleofector II for DNA delivery. 5 million Raji or DG-75 cells were
transfected with 5 ug
plasmids in each 100-ul reaction. Cell line Kit V and program M-013 were used
following Lonza
recommendation. For IMR-90, 1 million cells were transfected with 5 ug
plasmids in 100 ul
Solution V, with program T-030 or X-005. 24 hours after nucleofection, we
observed obvious
EGFP signals from a small proportion of cells through fluorescent microscopy.
The EGFP-
positive cell population decreased dramatically after that, however, and we
measured <10%
transfection efficiency 48 hours after nucleofection. We attributed this
transfection efficiency
decrease to the plasmid dilution with cell division. To actively maintain the
plasmid level within
the host cells, we redesigned the CRISPR plasmid to include the EBV origin of
replication
sequence, oriP. With active plasmid replication inside the cells, the
transfection efficiency rose to
>60%.
To design guide RNA targeting the EBV genome, we relied on the EBV reference
genome from strain B95-8. We targeted six regions with seven guide RNA designs
for different
genome editing purposes. The guide RNAs are listed in Table 51 in Wang and
Quake, 2014,
RNA-guided endonuclease provides a therapeutic strategy to cure latent
herpesviridae infection,
PNAS 111(36):13157-13162 and in the Supporting Information to that article
published online at
the PNAS website, and the contents of both of those documents are incorporated
by reference for
all purposes.
EBNA1 is crucial for many EBV functions including gene regulation and latent
genome
replication. We targeted guide RNA sgEBV4 and sgEBV5 to both ends of the EBNA1
coding
region in order to excise this whole region of the genome. Guide RNAs sgEBV1,
2 and 6 fall in
repeat regions, so that the success rate of at least one CRISPR cut is
multiplied. These
"structural" targets enable systematic digestion of the EBV genome into
smaller pieces.
54
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
EBNA3C and LMP1 are essential for host cell transformation, and we designed
guide RNAs
sgEBV3 and sgEBV7 to target the 5' exons of these two proteins respectively.
EBV Genome Editing.
The double-strand DNA breaks generated by CRISPR are repaired with small
deletions.
These deletions will disrupt the protein coding and hence create knockout
effects. SURVEYOR
assays confirmed efficient editing of individual sites. Beyond the independent
small deletions
induced by each guide RNA, large deletions between targeting sites can
systematically destroy
the EBV genome.
FIG. 9 shows genomic context around guide RNA sgEBV2 and PCR primer locations.
FIG. 10 shows a large deletion induced by sgEBV2, where lane 1-3 are before, 5
days
after, and 7 days after sgEBV2 treatment, respectively. Guide RNA sgEBV2
targets a region
with twelve 125-bp repeat units (FIG. 9). PCR amplicon of the whole repeat
region gave a ¨1.8-
kb band (FIG. 10). After 5 or 7 days of sgEBV2 transfection, we obtained ¨0.4-
kb bands from
the same PCR amplification (FIG. 10). The ¨1.4-kb deletion is the expected
product of repair
ligation between cuts in the first and the last repeat unit (FIG. 9).
DNA sequences flanking sgRNA targets were PCR amplified with Phusion DNA
polymerase. SURVEYOR assays were performed following manufacturer's
instruction. DNA
amplicons with large deletions were TOPO cloned and single colonies were used
for Sanger
sequencing. EBV load was measured with Taqman digital PCR on Fluidigm BioMark.
A
Taqman assay targeting a conserved human locus was used for human DNA
normalization. 1 ng
of single-cell whole-genome amplification products from Fluidigm Cl were used
for EBV
quantitative PCR.
We further demonstrated that it is possible to delete regions between unique
targets.
FIG. 11 shows the region targeted by sgEBV4/5 (e.g., between the forward (4F)
and
reverse (5R) primer binding sites). Six days after sgEBV4-5 transfection, PCR
amplification of
the whole flanking region (with primers EBV4F and 5R) returned a shorter
amplicon, together
with a much fainter band of the expected 2 kb.
FIG. 12 in lane 4 shows the faint band of the expected 2kb. Sanger sequencing
of
amplicon clones confirmed the direct connection of the two expected cutting
sites. A similar
experiment with sgEBV3-5 also returned an even larger deletion, from EBNA3C to
EBNAL
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
Additional information such as primer design is shown in Wang and Quake, 2014,
RNA-
guided endonuclease provides a therapeutic strategy to cure latent
herpesviridae infection, PNAS
111(36):13157-13162 and in the Supporting Information to that article
published online at the
PNAS website, and the contents of both of those documents are incorporated by
reference for all
purposes.
Cell Proliferation Arrest With EBV Genome Destruction.
Two days after CRISPR transfection, we flow sorted EGFP-positive cells for
further
culture and counted the live cells daily. FIG. 11 gives genome context around
guide RNA
sgEBV3/4/5 and PCR primer locations.
FIG. 12 shows large deletions induced by sgEBV3/5 and sgEBV4/5, where lane 1
and 2
are 3F/5R PCR amplicons before and 8 days after sgEBV3/5 treatment; and lane 3
and 4 are
4F/5R PCR amplicons before and 8 days after sgEBV4/5 treatment.
FIG. 13 shows that Sanger sequencing confirmed: genome cleavage and repair
ligation 8
days after sgEBV3/5 treatment (top) and genome cleavage and repair ligation 8
days after
sgEBV4/5 treatment (bottom).
FIG. 14 shows several cell proliferation curves after different CRISPR
treatments.
FIG. 15 shows nuclear morphology before sgEBV1-7 treatment.
FIG. 16 shows nuclear morphology after sgEBV1-7 treatment.
As expected, cells treated with Cas9 plasmids which lacked oriP or sgEBV lost
EGFP
expression within a few days and proliferated with a rate similar rate to the
untreated control
group (FIG. 14). Plasmids with Cas9-oriP and a scrambled guide RNA maintained
EGFP
expression after 8 days, but did not reduce the cell proliferation rate.
Treatment with the mixed
cocktail sgEBV1-7 resulted in no measurable cell proliferation and the total
cell count either
remained constant or decreased (FIG. 14). Flow cytometry scattering signals
clearly revealed
alterations in the cell morphology after sgEBV1-7 treatment, as the majority
of the cells shrank
in size with increasing granulation. Cells in population P3 also demonstrated
compromised
membrane permeability by DAPI staining.
To rule out the possibility of CRISPR cytotoxicity, especially with multiple
guide RNAs,
we performed the same treatment on two other samples: the EBV-negative
Burkitt's lymphoma
cell line DG-75 and primary human lung fibroblast IMR90.
56
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
Eight and nine days after transfection the cell proliferation rates did not
change from the
untreated control groups, suggesting neglectable cytotoxicity.
Previous studies have attributed the EBV tumorigenic ability to its
interruption of host
cell apoptosis (Ruf IK et al. (1999) Epstein-Barr Virus Regulates c-MYC,
Apoptosis, and
Tumorigenicity in Burkitt Lymphoma. Molecular and Cellular Biology 19:1651-
1660).
Suppressing EBV activities may therefore restore the apoptosis process, which
could explain the
cell death observed in our experiment. Annexin V staining revealed a distinct
subpopulation of
cells with intact cell membrane but exposed phosphatidylserine, suggesting
cell death through
apoptosis. Bright field microscopy showed obvious apoptotic cell morphology
and fluorescent
staining demonstrated drastic DNA fragmentation (FIGS. 15-16). Altogether this
evidence
suggests restoration of the normal host cell apoptosis pathway after EBV
genome destruction.
Complete Clearance Of EBV In A Subpopulation.
To study the potential connection between cell proliferation arrest and EBV
genome
editing, we quantified the EBV load in different samples with digital PCR
targeting EBNAL
Another Taqman assay targeting a conserved human somatic locus served as the
internal control
for human DNA normalization.
FIG. 17 shows EBV load after different CRISPR treatments by digital PCR, where
Cas9
and Cas9-oriP had two replicates, and sgEBV1-7 had 5 replicates.
FIG. 18 gives a histogram of EBV quantitative PCR Ct values from single cells
before
treatment.
FIG. 19 gives a histogram of EBV quantitative PCR Ct values from single live
cells 7
days after sgEBV1-7 treatment, where the dash lines in FIGS. 30 & 31 represent
Ct values of one
EBV genome per cell.
On average, each untreated Raji cell has 42 copies of EBV genome. Cells
treated with a
Cas9 plasmid that lacked oriP or sgEBV did not have an obvious difference in
EBV load
difference from the untreated control. Cells treated with a Cas9-plasmid with
oriP but no sgEBV
had an EBV load that was reduced by ¨50%. In conjunction with the prior
observation that cells
from this experiment did not show any difference in proliferation rate, we
interpret this as likely
due to competition for EBNA1 binding during plasmid replication. The addition
of the guide
57
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
RNA cocktail sgEBV1-7 to the transfection dramatically reduced the EBV load.
Both the live
and dead cells have >60% EBV decrease comparing to the untreated control.
Although we provided seven guide RNAs at the same molar ratio, the plasmid
transfection and replication process is likely quite stochastic. Some cells
will inevitably receive
different subsets or mixtures of the guide RNA cocktail, which might affect
the treatment
efficiency. To control for such effects, we measured EBV load at the single
cell level by
employing single-cell whole-genome amplification with an automated
microfluidic system. We
loaded freshly cultured Raji cells onto the microfluidic chip and captured 81
single cells. For the
sgEBV1-7 treated cells, we flow sorted the live cells eight days after
transfection and captured
91 single cells. Following manufacturer's instruction, we obtained ¨150 ng
amplified DNA from
each single cell reaction chamber. For quality control purposes we performed 4-
loci human
somatic DNA quantitative PCR on each single cell amplification product (Wang
et al., 2012,
Genome-wide single-cell analysis of recombination activity and de novo
mutation rates in human
sperm, Cell 150:402-412) and required positive amplification from at least one
locus. 69
untreated single-cell products passed the quality control and displayed a log-
normal distribution
of EBV load with almost every cell displaying significant amounts of EBV
genomic DNA. We
calibrated the quantitative PCR assay with a subclone of Namalwa Burkitt's
lymphoma cells,
which contain a single integrated EBV genome. The single-copy EBV measurements
gave a Ct
of 29.8, which enabled us to determine that the mean Ct of the 69 Raji single
cell samples
corresponded to 42 EBV copies per cells, in concordance with the bulk digital
PCR
measurement. For the sgEBV1-7 treated sample, 71 single-cell products passed
the quality
control and the EBV load distribution was dramatically wider. While 22 cells
had the same EBV
load as the untreated cells, 19 cells had no detectable EBV and the remaining
30 cells displayed
dramatic EBV load decrease from the untreated sample.
Essential Targets For EBV Treatment. The seven guide RNAs in our CRISPR
cocktail
target three different categories of sequences which are important for EBV
genome structure,
host cell transformation, and infection latency, respectively. To understand
the most essential
targets for effective EBV treatment, we transfected Raji cells with subsets of
guide RNAs.
Although sgEBV4/5 reduced the EBV genome by 85%, they could not suppress cell
proliferation
as effectively as the full cocktail (FIG. 14). Guide RNAs targeting the
structural sequences
(sgEBV1/2/6) could stop cell proliferation completely, despite not eliminating
the full EBV load
58
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
(26% decrease). Given the high efficiency of genome editing and the
proliferation arrest, we
suspect that the residual EBV genome signature in sgEBV1/2/6 was not due to
intact genomes
but to free-floating DNA that has been digested out of the EBV genome, i.e. as
a false positive.
We conclude that systematic destruction of EBV genome structure appears to be
more effective
than targeting specific key proteins for EBV treatment.
Example 2: HPV genome and targets
Methods and materials may be used to digest foreign nucleic acid such as a
genome of a
hepatitis B virus (HBV).
It may be preferable to receive annotations for the HB V genome (i.e., that
identify
important features of the genome) and choose a candidate for targeting by
enzymatic degredation
that lies within one of those features, such as a viral replication origin, a
terminal repeat, a
replication factor binding site, a promoter, a coding sequence, and a
repetitive region.
The HPV genome is a double-stranded, circular DNA genome approximately 8 kb in
size
that can be divided, in general, into three major regions (early, late, and a
long control region
(LCR), which regions are separated by two polyadenylation sites. The early
region is over 50%
of the HPV genome from its 5' half and encodes six common open reading frames
(El, E2, E4,
E5, E6 and E7) that translate proteins. The late region is downstream of the
early region and
encodes Ll and L2 ORFs for translation of a major (L1) and a minor (L2) capsid
protein. A
targeting sequence such as a gRNA may be targeted to a capsid protein to
interrupt viral
function. The ¨850 bp LCR region has no protein-coding function, but bears the
origin of
replication as well as transcription factor binding sites for transcription
regulation from viral
early as well as late promoters. See Bernard, 2007, Gene expression of genital
human
papillomaviruses and considerations on potential antiviral approaches.
Antivir.Ther. 7:219-237
incorporated by reference. The HPV-16 genome contains two major promoters. The
P97
promoter lies upstream of the E6 ORF and is responsible for almost all early
gene expression.
The P670 promoter lies within the E7 ORF region and is responsible for late
gene expression.
The HPV-16 P97 promoter, equivalent to P99 in HPV-31 and P105 in HPV-18, is
very potent
and tightly controlled, primarily by upstream cis-elements in the LCR that
interact with cellular
transcription factors and the viral transactivator/repressor E2 and regulate
the transcription of
P97 from undifferentiated basal cells to highly differentiated keratinocytes.
It is believed that E2
59
CA 03015353 2018-08-20
WO 2017/147446 PCT/US2017/019390
functions as a repressor for P97 transcription after TBP or TFIID binding and
its transcriptional
repression only occurs in cells harboring integrated, but not episomal HPV-16
DNA. The HPV-
16 P670 promoter is a late-promoter and its activity can be induced only in
differentiated
keratinocytes. Elements in the E6 and E7 coding regions may regulate late
promoters and both
the late P670 promoter in HPV-16 and P742 in HPV-31 are positioned in the E7
coding region
and transcription from the late promoter has to bypass the early pA site to
allow expression of
the late region. See Zheng & Baker, 2006, Papillomavirus genome structure,
expression, and
post-transcriptional regulation, Front Biosci 11:2286-2302, incorporated by
reference.
The promoters may be used in a vector containing a gene for an antiviral, or
targetable,
endonuclease.
The use of Cas9 may be validated using an in vitro assay. To demonstrate, an
in vitro
assay is performed with cas9 protein and DNA amplicons flanking the target
regions. Here, the
target is amplified and the amplicons are incubated with cas9 and a gRNA
having the selected
nucleotide sequence for targeting. As shown in FIG. 14, DNA electrophoresis
shows strong
digestion at the target sites.
FIG. 24 shows a gel resulting from an in vitro CRISPR assay against HBV. Lanes
1, 3,
and 6: PCR amplicons of HBV genome flanking RT, Hbx-Core, and PreS1. Lane 2,
4, 5, and 7:
PCR amplicons treated with sgHBV-RT, sgHBV-Hbx, sgHBV-Core, sgHBV-PreS1. The
presence of multiple fragments especially visible in lanes 5 and 7 show that
sgHBV-Core and
sgHBV-PreS1 provide especially attractive targets in the context of HBV and
that use of systems
and methods of the disclosure may be shown to be effective by an in vitro
validation assay.
FIG. 24 gives results of digesting foreign nucleic acid. The nuclease forms a
complex
with the gRNA (e.g., crRNA + tracrRNA or sgRNA). The complex cuts the viral
nucleic acid in
a targeted fashion to incapacitate the viral genome. The Cas9 endonuclease
causes a double
strand break in the viral genome. By targeted several locations along the
viral genome and
causing not a single strand break, but a double strand break, the genome is
effectively cut a
several locations along the genome. In a preferred embodiment, the double
strand breaks are
designed so that small deletions are caused, or small fragments are removed
from the genome so
that even with repair mechanisms, the genome is render incapacitated.