Note: Descriptions are shown in the official language in which they were submitted.
THERAPEUTIC COMPOSITIONS FOR TREATING PANCREATIC CANCER
BACKGROUND OF THE INVENTION
[0001] Pancreatic cancer arises when cells in the pancreas begin to multiply
out of control and
form a mass. These cancerous cells have the ability to invade other parts of
the body. Pancreatic
cancer is the fourth leading cause of cancer death in Western countries and is
also the tenth
leading cause of cancer death in Taiwan. Approximately 60 percent of
pancreatic cancer arises in
the head of pancreas, and about 21 percent invades to whole pancreas.
[0002] Pancreatic cancer can be divided into two general groups. The vast
majority of cases,
about 99%, occur in the part of the pancreas which produces digestive enzymes,
known as the
exocrine component. There are several sub-types of exocrine pancreatic
cancers, but their
diagnosis and treatment have much in common. These small minority of cancers
arise in the
hormone-producing (endocrine) tissue of the pancreas.
[00031 Surgery with the intention of a cure is only possible in around one-
fifth (20%) of new
cases. Although CT scans help, in practice, it can be difficult to determine
whether the tumor can
be fully removed (its "resectability"), and it may only become apparent during
surgery that it is
not possible to successfully remove the tumor without damaging other vital
tissues. Even when
the operation appears to have been successful, cancerous cells are often found
around the edges
("margins") of the removed tissue. After surgery, adjuvant chemotherapy with
gemcitabine or 5-
FU can be offered if the person is sufficiently fit, after a recovery period
of one to two months. In
people not suitable for curative surgery, chemotherapy may be used to extend
life or improve its
quality of life.
SUMMARY OF THE INVENTION
[0004] In one aspect provided herein are composition for treating pancreatic
cancer in a subject
R3 H3
0 R4
Ri.x
OR
comprising a compound having the structure: R2
and one or more anti-
cancer agents,
wherein each of X and Y independently is oxygen, or sulfur;
R is a hydrogen or C(-0)C1-C8alkyl;
each of RI, R2 and R3 independently is a hydrogen, methyl or (CH2)m¨CH3;
1
CA 3020933 2018-10-15
R4 is H, C1-C8alkyl, C2-C8alkenyl, C2-C8alkynyl, or aryl optionally
substituted with one
or more substituents selected from C1-C8 alkyl, C2-C8 alkenyl, C2-C8 alkynyl,
C3-C8
cycloalkyl, and C1-C8 haloalkyl; n=1-12; or a pharmaceutically acceptable
salt,
metabolite, solvate or prodrug thereof.
[0005I In another aspect provided herein are methods for treating pancreatic
cancer in a subject
comprising administering the subject in need thereof of a compound having the
structure:
R3 CH3
0 R4
R"
X 0 R
Y. R2 and one or more anti-cancer agents,
wherein each of X and Y independently is oxygen, or sulfur;
R is a hydrogen or C(=0)C1-C8alkyl;
each of RI, R2 and R3 independently is a hydrogen, methyl or (CH2)m CH3;
R4 is H, C1-C8alkyl, C2-C8alkenyl, C2-C8alkynyl, or aryl optionally
substituted with one
or more substituents selected from C1-C8 alkyl, C2-C8 alkenyl, C2-C8 alkynyl,
C3-C8
cycloalkyl, and C1-C8 haloalkyl; n=1-12; or a pharmaceutically acceptable
salt,
metabolite, solvate or prodrug thereof.
[0006] In another aspect provided herein are methods for the treatment of a
patient whose
pancreatic cancer is resistant, refractory or non-responsive to gemcitabine,
paclitaxel, or a
combination thereof, comprising administering to a patient in need thereof a
compound having
R3 CH3
0 R4
R,.
X 0 R
Y. the structure: R2 or a pharmaceutically acceptable salt,
metabolite,
solvate or prodrug thereof,
wherein each of X and Y independently is oxygen, or sulfur;
R is a hydrogen or C(=0)C1-C8alkyl;
each of R1, R2 and R3 independently is a hydrogen, methyl or (CH2)m¨CH3;
R4 is 14, C1-C8alkyl, C2-C8alkenyl, C2-C8alkynyl, or aryl optionally
substituted with one
or more substituents selected from C1-C8 alkyl, C2-C8 alkenyl, C2-C8 alkynyl,
C3-C8
cycloalkyl, and C1-C8 haloalkyl;
n=1-12; or a pharmaceutically acceptable salt, metabolite, solvate or prodrug
thereof.
2
CA 3020933 2018-10-15
[0007]
BRIEF DESCRIPTION OF THE DRAWINGS
[0008] The novel features of the invention are set forth with particularity in
the appended claims. A
better understanding of the features and advantages of the present invention
will be obtained by
reference to the following detailed description that sets forth illustrative
embodiments, in which the
principles of the invention are utilized, and the accompanying drawings of
which:
[00091FIG. 1A/13 provide the results of cytotoxic activity in AsPC-1
pancreatic cancer cells treated
with exemplary compound 1 for (1A) 48 hours and (1B) 72 hours. MTT assay was
used to measure
cytotoxic activity.
[0010] FIG. 2A/B provide the results of cytotoxic activity in AsPC-1
pancreatic cancer cells treated
with Erlotinib for (2A) 48 hours and (2B) 72 hours. MTT assay was used to
measure cytotoxic activity.
[0011] FIG. 3A/B provide the results of cytotoxic activity in AsPC-1
pancreatic cancer cells treated
with 5-FU for (3A) 48 hours and (3B) 72 hours. MTT assay was used to measure
cytotoxic activity.
[0012] FIG. 4A/B provide the results of cytotoxic activity in AsPC-1
pancreatic cancer cells treated
with Gemcitabine for (4A) 48 hours and (4B) 72 hours. MTT assay was used to
measure cytotoxic
activity.
[0013] FIG. 5A/B provide the results of cytotoxic activity in AsPC-1
pancreatic cancer cells treated
with Irinotecan for (5A) 48 hours and (5B) 72 hours. MTT assay was used to
measure cytotoxic
activity.
[0014] FIG. 6A/B provide the results of cytotoxic activity in AsPC-1
pancreatic cancer cells treated
with Oxaliplatin for (6A) 48 hours and (6B) 72 hours. MTT assay was used to
measure cytotoxic
activity.
[0015] FIG. 7A/B provide the results of cytotoxic activity in AsPC-1
pancreatic cancer cells treated
with Paclitaxel for (7A) 48 hours and (7B) 72 hours. MTT assay was used to
measure cytotoxic
activity.
[0016] FIG. 8 shows the effect of Compound 1 with chemotherapy drugs,
paclitaxel, gemcitabine, 5-
FU, oxaliplatin, erlotinib or irinotecan on the cytotoxicity of AsPC-1
pancreatic
3
Date Recue/Date Received 2023-10-12
cancer cell line for 48 hours. MTT assay was used to measure cytotoxic
activity. Values are
means of survival rate SEM. Different letters (a-e) denote significant
difference (P < 0.05) for
various treatments.
[0017] FIG. 9 shows the cytotoxic activity of Compound 1 with chemotherapy
drugs, paclitaxel,
gemcitabine, 5-FU, oxaliplatin, erlotinib and irinotecan, respectively. AsPC-1
pancreatic cancer
cells were treated antroquinonol or combination formula for or 72 hours. MTT
assay was used to
measure cytotoxic activity. Values are means of survival rate SEM. Different
letters (a-g)
denote significant difference (P < 0.05) for various treatments.
[0018] FIG. 10A-C show the dose-effect for combination of Compound 1 (also
known as
antroquinonol, Aq) with paclitaxel (Px), gemcitabine (Ge) in pancreatic cancer
cell lines. Plots
indicated the Fa values of the treated of combination of antroquinonol with
paclitaxel,
antroquinonol with gemcitabine, antroquinonol plus paclitaxel and gemcitabine
in AsPC-1 (10A),
CaPan-2 (10B) and Panc-1 (10C) pancreatic cancer cell lines. The x-axis
represents the dose of
drugs in gmo1/1_, and the y-axis represents Fa, the fraction of cell affected
(growth inhibition).
[0019] FIG. 11A-C show the analysis results of the combinations by Compound 1
with
paclitaxel, gemcitabine in pancreatic cancer cell lines for 72 hours. CI plots
for the combination
of antroquinonol (Aq) with paclitaxel (Px), antroquinonol with gemcitabine
(Ge), antroquinonol
plus paclitaxel and gemcitabine in AsPC-1 (11A), CaPan-2 (11B) and Panc-1 (I
IC) pancreatic
cancer cell lines. The x-axis represents Fa (fraction of cell affected) and
the y-axis represents CI
(combination index). Cl = 1, < 1 and > 1 indicates additive effect, synergism
and antagonism,
respectively.
[0020] FIG. 12 shows the results of the mice body weight changes during the
test of antitumor
activity by Compound 1 with chemotherapy drugs on AsPC-1 subcutaneous
xenograft model.
[0021] FIG. 13 shows the effect of antitumor activity of Compound 1 plus
chemotherapy drugs
on AsPC-1 subcutaneous xenograft model. Weekly measurements of tumor size were
made and
the mean relative tumor volume was plotted as a function of time after the
start of treatment.
Each point was presented mean of tumor volume.
[0022] FIG. 14 shows the effect of Compound 1 plus chemotherapy drugs on AsPC-
1
subcutaneous xenograft model. Percentage of tumor volume significantly
decreased in treatment
groups of Compound 1 contained combinations which compared with control group
(Ti, corn
oil).
[0023] FIG. 15 shows the analysis results of the Tumor Growth Inhibition (TGI)
percent of
Compound 1 alone or with chemotherapy drugs, paclitaxel and gemcitabine, on
the growth of
AsPC-1 tumor volume. Each column was presented mean of tumor growth
inhibition. Values are
4
CA 3020933 2018-10-15
means SEM. Different superscripts denote significant difference (P <0.05)
between each
treatment group.
[0024] FIG. 16 illustrates the comparison results of representative photo of
AsPC-1 pancreatic
tumor xenograft before and after treatment with Compound 1 and/or plus
chemotherapy drugs,
respectively.
[0025] FIG. 17 illustrates the comparison results of representative ultrasound
images of AsPC-1
pancreatic tumor xenograft before and after treatment with Compound 1 and/or
plus
chemotherapy drugs, respectively.
[0026] FIG. 18A/B show the results of the images (18A) and figures (18B) of
excised tumors at
the time of sacrifice from the subcutaneous AsPC-1 pancreatic tumor xenograft-
bearing male
nude mice after 28 days of treatments. ANOVA was used to analyze the
statistical significance. *
P < 0.05 is considered to be statistically significant when compared with
control group (Ti, corn
oil group).
[0027] FIG. 19 shows the results of inhibitory effect of Compound 1 alone or
with chemotherapy
drugs, paclitaxel and gemcitabine, on the growth of AsPC-1 solid tumor. Each
column was
presented mean of tumor inhibitory rate (TIR). Values are means of TIR SEM.
Different
superscripts denote significant difference (P < 0.05) between each treatment
group.
DETAILED DESCRIPTION OF THE INVENTION
[0028] Surgery with the intention of a cure is only possible in around one-
fifth (20%) of new
cases. Although curative surgery no longer entails the very high death rates
that occurred until
the 1980s, a high proportion of people (about 30 45%) still have to be
treated for a post-
operative sickness that is not caused by the cancer itself. The most common
complication of
surgery is difficulty in emptying the stomach. After surgery, adjuvant
chemotherapy with
gemcitabine or 5-FU can be offered if the person is sufficiently fit, after a
recovery period of one
to two months. Gemcitabine was approved by the United States Food and Drug
Administration
(FDA) in 1997, after a clinical trial reported improvements in quality of life
and a 5-week
improvement in median survival duration in people with advanced pancreatic
cancer.
Chemotherapy using gemcitabine alone was the standard for about a decade, as a
number of trials
testing it in combination with other drugs failed to demonstrate significantly
better outcomes.
[0029] The FOLFIRINOX chemotherapy regimen using four drugs was found more
effective
than gemcitabine, but with substantial side effects, and is thus only suitable
for people with good
performance status. This is also true of protein-bound paclitaxel (nab-
paclitaxel), which was
licensed by the FDA in 2013 for use with gemcitabine in pancreas cancer.
However, the changes
of the last few years have only increased survival times by a few months.
CA 3020933 2018-10-15
100301 A method for treating pancreatic cancer in a subject comprising
administering the subject
in need thereof a cyclohexanone compound was reported in U.S. patent number
8,236,860. An
exemplary compound, Antroquinonol was studied for treating pancreatic cancer
using the
orthotopic PANC-1 human pancreatic cancer xenograft model. Four groups of mice
were treated
with 30 mg/kg, 60 mg/kg, 90 mg/kg, and vehicle control, respectively.
Treatment with
antroquinonol at 30, 60, and 90 mg/kg produced an effective anti-tumor
activity with statistically
significant smaller mean tumor volumes and tumor weights in all three dosage
levels compared
to vehicle control. Although Antroquinonol was shown to be a good candidate as
a drug to treat
pancreatic cancer, there was no consideration to use it in any combination
therapy.
[0031] However, it is found unexpectedly a synergistic effect existed in a
combination therapy
R3 CH3
0 R4
X OR
Y.
comprising a compound having the structure: R2 , and one or more
anti-cancer agents, such as gemcitabine, paclitaxel, or a combination thereof
provides synergistic
effects compared with the mono therapy; wherein each of X and Y independently
is oxygen, or
sulfur;
R is a hydrogen or C(=0)C1-C8alkyl;
each of RI, R2 and R3 independently is a hydrogen, methyl or (CH2)m¨CH3;
R4 is H, C1-C8alkyl, C2-C8alkenyl, C2-C8alkynyl, or aryl optionally
substituted with one
or more substituents selected from C1-C8 alkyl, C2-C8 alkenyl, C2-C8 alkynyl,
C3-C8
cycloalkyl, and C1-C8 haloalkyl;
n=1-12; or a pharmaceutically acceptable salt, metabolite, solvate or prodrug
thereof. In
some embodiments, such combination therapy further comprises administering an
immunotherapy agent.
[0032] In some embodiments, provided herein are compositions for treating
pancreatic cancer in
R3 CH3
R4
R1 .x
OR
a subject comprising a compound having the structure: IN2 ,
and one or
more anti-cancer agents, (see Examples 1-3). The cyclohexenone compounds, in
some
embodiments, are obtained from extracts of natural products and in some other
embodiments, are
prepared synthetically. See for example, U.S. patent No. 9365481.
6
CA 3020933 2018-10-15
[0033] In some embodiments, the one or more anti-cancer agents comprise
gemcitabine,
paclitaxel, idarubicin/cytarabine, etopside phosphate, gleevec (imatinib),
temozolomide,
bortezomib, letrozole, cetuximab, bevacizumab, nab-paclitaxel, docetaxel,
erlotinib, pemetrexed,
pemetrexed/carboplatin, paxlitaxel/carboplatin, letrozole/cyclophsphamide,
temsirolimus,
bevacizumabitemsirolimus, 1pilimumab, RAD001, Pazopanib, FOLFIRI, BKM120,
GSK1120212, PF-05212384/irinotecan, AZD2171, PF-04691502, cyclophosphamide,
cisplatin,
cytarabine/daunorubcin, tersirolimus, erlotinib/temsirolimus, capecitabine,
tamoxifen,
bortezomib, trastuzumab, docetaxel/capecitabine, trastuzumab/tipifarnib,
tipifamib/gemcitabline,
tootecan, or combinations thereof. In certain embodiments, the one or more
anti-cancer agent is
gemcitabine, paclitaxel, or a combination thereof. In certain embodiments, the
one or more anti-
cancer agent are the combination of gemcitabine and paclitaxel.
[0034] For example, paclitaxel is classified as a "plant alkaloid," a "taxane"
and an
"antimicrotubule agent." It is used to treat a number of types of cancer such
as ovarian cancer,
breast cancer, lung cancer, Kaposi sarcoma, cervical cancer, and pancreatic
cancer. Albumin-
bound paclitaxel (trade name Abraxane, also called nab-paclitaxel) is an
alternative formulation
where paclitaxel is bound to albumin nano-particles. Paclitaxel is known to
have some common
side effects including nausea and vomiting, loss of appetite, change in taste,
thinned or brittle
hair, pain in the joints of the arms or legs lasting two to three days,
changes in the color of the
nails, and tingling in the hands or toes. Paclitaxel is one of several
cytoskeletal drugs that target
tubulin. Paclitaxel-treated cells have defects in mitotic spindle assembly,
chromosome
segregation, and cell division. Unlike other tubulin-targeting drugs such as
colchicine that inhibit
microtubule assembly, paclitaxel stabilizes the microtubule polymer and
protects it from
disassembly.
[0035] Another exemplary anticancer drug, gemcitabine is a nucleoside analog
used as
chemotherapy to treat a number of types of cancer including breast cancer,
ovarian cancer, non-
small cell lung cancer, pancreatic cancer, and bladder cancer. Common side
effects include bone
marrow suppression, liver and kidney problems, nausea, fever, rash, shortness
of breath, and hair
loss. Use during pregnancy will likely result in harm to the baby. Gemcitabine
is in the
nucleoside analog family of medication. It works by blocking the creation of
new DNA, which
results in cell death.
[0036] In some embodiments, there are provided methods for treating pancreatic
cancer in a
subject comprising administering the subject in need thereof the combination
therapy
compositions described herein. In some embodiments provide the use of a
therapeutically
7
CA 3020933 2018-10-15
effective amount of the combination therapy compositions described herein in
the manufacture of
a medicament for treating pancreatic cancer in a subject.
[0037] In some embodiments, there are provided methods for the treatment of a
patient whose
pancreatic cancer is resistant, refractory or non-responsive to gemcitabine,
paclitaxel, or a
combination thereof, comprising administering the patient in need thereof
R3 CH3
0 R4
OR
a compound having the structure: R2
or a pharmaceutically acceptable
salt, metabolite, solvate or prodrug thereof. In some embodiments, the mtheods
further comprise
administering an immunotherapy agent. In some embodiments provide the use of a
R3 CH3
0
R4
R1 .x
OR
therapeutically effective amount of a compound having the structure R2
or a pharmaceutically acceptable salt, metabolite, solvate or prodrug thereof,
in the manufacture
of a medicament for treating a patient whose pancreatic cancer is resistant,
refractory or non-
responsive to gemcitabine, paclitaxel, or a combination thereof.
[0038] In certain embodiments, the cancer is resistant, refractory or non-
responsive to a drug
selected from gemcitabine, paclitaxel, idarubicin/cytarabine, etopside
phosphate, gleevec,
temozolomide, bortezomib, letrozole, cetuximab, bevacizumab, nab-paclitaxel,
docetaxel,
erlotinib, pemetrexed, pemetrexed/carboplatin, paxlitaxel/carboplatin,
letrozole/cyclophsphamide, temsirolimus, bevacizumab/temsirolimus, 1pilimumab,
RAD001,
Pazopanib, FOLFIRI, BKM120, GSK1120212, PF-05212384/irinotecan, AZD2171, PF-
04691502, cyclophosphamide, cisplatin, cytarabine/daunorubcin, tersirolimus,
erlotinib/temsirolimus, capecitabine, tamoxifen, bortezomib, trastuzumab,
docetaxel/capecitabine, trastuzumab/tipifarnib, tipifamib/gemcitabline,
tootecan, or combinations
thereof. In certain embodiments, the cancer is resistant, refractory or non-
responsive to
gemcitabine or paclitaxel. In certain embodiments, the cancer is resistant,
refractory or non-
responsive to a combination of gemcitabine and paclitaxel.
8
CA 3020933 2018-10-15
[0039] In some embodiments, the cyclohexenone compound having the structure
R3 CH3
0 R4
R1.
X OR
Y,R2 is prepared synthetically or semi-synthetically from
any suitable
starting material. In other embodiments, the cyclohexenone compound is
prepared by
fermentation, or the like. For example, Compounds 1, 3 and 4 are isolated from
organic solvent
extracts or prepared synthetically or semi-synthetically. The non-limited
exemplary compounds
are illustrated below.
0 Ail
RIP
0 OH HO0 111111111P OH
0
1 3
0
HO OH
4
[0040] In some embodiments, R is a hydrogen, C(=0)C3H7, C(---0)C2H5, or
C(=0)CH3. In some
embodiments, Ri is a hydrogen or methyl. In certain embodiments, R2 is a
hydrogen, methyl,
ethyl, propyl, butyl, pentyl or hexyl. In some embodiments, R3 is a hydrogen,
methyl, ethyl,
propyl, butyl, pentyl or hexyl. In some embodiments, R4 is hydrogen. In some
embodiments, R4
is C1-C8alkyl, C2-C8alkenyl, C2-C8alkynyl, or aryl, optionally substituted
with one or more
substituents selected from C1-C8 alkyl, C2-C8 alkenyl, C2-C8 alkynyl, C3-C8
cycloalkyl, and C1-C8
haloalkyl. In certain embodiments, R4 is CH2CH=C(CH3)2. In certain
embodiments, the
H3 H3 H3 H3
CH3
H3C.0
--MOH
compound is
[0041] In some embodiments, the methods to treat pancreatic cancer described
herein comprise
administering an immunotherapy agent. The immunotherapy agent includes,
without limitation,
any living immune cell that can be administered to a patient, and/or
antibodies specific for a
target cell (e.g., a tumor cell such as pancreatic cancer cell). Preferably,
the immunotherapy agent
is an NK cell or a T cell, or a modification or derivative thereof.
9
CA 3020933 2018-10-15
Immunotherapy
[0042] 1mmunotherapy is the "treatment of disease by inducing, enhancing, or
suppressing an
immune response". Immunotherapies designed to elicit or amplify an immune
response are
classified as activation immunotherapies, while immunotherapies that reduce or
suppress are
classified as suppression immunotherapies.
[0043] In some embodiments, the immunotherapy agent is an anti-cancer
antibody. Non-limiting
examples include trastuzumab (Herceptin6), bevacizumab (Avastin ), cetuximab
(Erbitux0),
panitumumab (Vectibix0), ipilimumab (Yervoy6), rituximab (Rituxane),
alemtuzumab
(Campath8), ofatumumab (Arzerral ), gemtuzumab ozogamicin (Mylotargo),
brentuximab
vedotin (Adcetrise), 90Y-ibritumomab tiuxetan (ZevalinS), 131I-tositumomab
(Bexxar8), anti-
programmed-death 1 (anti-PD-I) antibody such as Nivolumab, Pembrolizumab, and
the like.
Certain Pharmaceutical and Medical Terminology
[0044] Unless otherwise stated, the following terms used in this application,
including the
specification and claims, have the definitions given below. It must be noted
that, as used in the
specification and the appended claims, the singular forms "a," "an" and "the"
include plural
referents unless the context clearly dictates otherwise. Unless otherwise
indicated, conventional
methods of mass spectroscopy, NMR, HPLC, protein chemistry, biochemistry,
recombinant
DNA techniques and pharmacology are employed. In this application, the use of
"or" or "and"
means "and/or" unless stated otherwise. Furthermore, use of the term
"including" as well as other
forms, such as "include", "includes," and "included," is not limiting. The
section headings used
herein are for organizational purposes only and are not to be construed as
limiting the subject
matter described.
[0045] An "alkyl" group refers to an aliphatic hydrocarbon group. The alkyl
group may be a
saturated alkyl group (which means that it does not contain any carbon-carbon
double bonds or
carbon-carbon triple bonds) or the alkyl group may be an unsaturated alkyl
group (which means
that it contains at least one carbon-carbon double bonds or carbon-carbon
triple bond). The alkyl
moiety, whether saturated or unsaturated, may be branched, or straight chain.
[0046] The "alkyl" group may have 1 to 12 carbon atoms (whenever it appears
herein, a
numerical range such as "1 to 12 refers to each integer in the given range;
e.g., "1 to 12 carbon
atoms" means that the alkyl group may consist of 1 carbon atom, 2 carbon
atoms, 3 carbon
atoms, etc., up to and including 12 carbon atoms, although the present
definition also covers the
occurrence of the term "alkyl" where no numerical range is designated). The
alkyl group of the
compounds described herein may be designated as, "Ci-C12 alkyl", "Ci-C8 alkyl"
or similar
designations. By way of example only, "Ci-Cs alkyl" indicates that there are
one, two, three,
CA 3020933 2018-10-15
four, five, six, seven or eight carbon atoms in the alkyl chain. In one aspect
the alkyl is selected
from the group consisting of methyl, ethyl, propyl, iso-propyl, n-butyl, iso-
butyl, sec-butyl, and t-
butyl. Typical alkyl groups include, but are in no way limited to, methyl,
ethyl, propyl, isopropyl,
butyl, isobutyl, sec-butyl, tertiary butyl, pentyl, neopentyl, hexyl, ally!,
but-2-enyl, but-3-enyl,
cyclopropylmethyl, cyclobutylmethyl, cyclopentylmethyl, cyclohexylmethyl, and
the like. In one
aspect, an alkyl is a CI-C8 alkyl.
100471 The term "alkylene" refers to a divalent alkyl radical. Any of the
above mentioned
monovalent alkyl groups may be an alkylene by abstraction of a second hydrogen
atom from the
alkyl. In one aspect, an alkylene is a CI-Ci2alkylene. In another aspect, an
alkylene is a CI-
C8alky1ene. Typical alkylene groups include, but are not limited to, -CH2-, -
CH(CH3)-, -C(CH3)2-
, -CH2CH2-, -CH2CH(CH3)-, -CH2C(CH3)2-, -CH2CH2CH2-, -CH2CH2CH2CH2-, and the
like.
100481 As used herein, the term "aryl" refers to an aromatic ring wherein each
of the atoms
forming the ring is a carbon atom. Aryl rings are formed by five, six, seven,
eight, nine, or more
than nine carbon atoms. Aryl groups are optionally substituted. In one aspect,
an aryl is a phenyl
or a naphthalenyl. In one aspect, an aryl is a phenyl. In one aspect, an aryl
is a C6-Cioaryl.
Depending on the structure, an aryl group can be a monoradical or a diradical
(i.e., an arylene
group). In one aspect, an arylene is a Co-Cio arylene. Exemplary arylenes
include, but are not
limited to, phenyl-1,2-ene, phenyl-1,3-ene, and phenyl-1,4-ene.
100491 The term "aromatic" refers to a planar ring having a delocalized it-
electron system
containing 4n+2 it electrons, where n is an integer. Aromatic rings can be
formed from five, six,
seven, eight, nine, ten, or more than ten atoms. Aromatics are optionally
substituted. The term
"aromatic" includes both carbocyclic aryl ("aryl", e.g., phenyl) and
heterocyclic aryl (or
"heteroaryl" or "heteroaromatic") groups (e.g., pyridine). The term includes
monocyclic or
fused-ring polycyclic (i.e., rings which share adjacent pairs of carbon atoms)
groups.
[00501 The term "halo" or, alternatively, "halogen" or "halide" means fluoro,
chloro, bromo or
iodo.
[0051] The term "alkenyl" as used herein, means a straight, branched chain, or
cyclic (in which
case, it would also be known as a "cycloalkenyl") hydrocarbon containing from
2-10 carbons and
containing at least one carbon-carbon double bond formed by the removal of two
hydrogens. The
alkenyl group of the compounds described herein may be designated as, "C2-
C10alkenyl", "C2-
C8alkenyl" or similar designations. By way of example only, "C2-C8 alkenyl"
indicates that there
are two, three, four, five, six, seven or eight carbon atoms in the alkenyl
chain. In some
embodiments, depending on the structure, an alkenyl group is a monoradical or
a diradical (i.e.,
an alkenylene group). In some embodiments, alkenyl groups are optionally
substituted.
11
CA 3020933 2018-10-15
Illustrative examples of alkenyl include, but are not limited to, ethenyl, 2-
propenyl, 2-methyl-2-
propenyl, 3-butenyl, 4-pentenyl, 5-hexenyl, 2-heptenyl, 2-methyl-1-heptenyl,
and 3-cecenyl.
100521 The term "alkynyl" as used herein, means a straight, branched chain, or
cyclic (in which
case, it would also be known as a "cycloalkynyl") hydrocarbon containing from
2-10 carbons and
containing at least one carbon-carbon triple bond formed by the removal of
four hydrogens. In
some embodiments, depending on the structure, an alkynyl group is a
monoradical or a diradical
(i.e., an alkynylene group). The alkynyl group of the compounds described
herein may be
designated as, "C2-C10alkynyl", "C2-Csalkynyl" or similar designations. By way
of example only,
"C2-Csalkynyl" indicates that there are two, three, four, five, six, seven or
eight carbon atoms in
the alkynyl chain. In some embodiments, alkynyl groups are optionally
substituted. Illustrative
examples of alkynyl include, but are not limited to, ethynyl, propynyl,
butynyl, pentynyl,
hexynyl, heptynyl, and the like.
100531 The term "cycloalkyl" as used herein, means a monocyclic or polycyclic
radical that
contains only carbon and hydrogen, and includes those that are saturated,
partially unsaturated, or
fully unsaturated. Cycloalkyl groups include groups having from 3 to 10 ring
atoms.
Representative examples of cyclic include but are not limited to, the
following moieties:
> _______________ ) 01 f OICICC)
CI> , im=, hp
ope
. In some embodiments, depending
on the structure, a cycloalkyl group is a monoradical or a diradical (e.g., a
cycloalkylene
group).
[00541 The terms "haloalkyl," "haloalkenyl," "haloalkynyl" and "haloalkoxy" as
used herein,
include alkyl, alkenyl, alkynyl and alkoxy structures in which at least one
hydrogen is replaced
with a halogen atom. In certain embodiments in which two or more hydrogen
atoms are replaced
with halogen atoms, the halogen atoms are all the same as one another. In
other embodiments in
which two or more hydrogen atoms are replaced with halogen atoms, the halogen
atoms are not
12
CA 3020933 2018-10-15
all the same as one another. The terms "fluoroalkyl" and "fluoroalkoxy"
include haloalkyl and
haloalkoxy groups, respectively, in which the halo is fluorine. In certain
embodiments, haloalkyls
are optionally substituted.
[0055] The term "acceptable" with respect to a formulation, composition or
ingredient, as used
herein, means having no persistent detrimental effect on the general health of
the subject being
treated.
[0056] Antrodia is a genus of fungi in the family Meripilaceae. Antrodia
species have fruiting
bodies that typically lie flat or spread out on the growing surface, with the
hymenium exposed to
the outside; the edges may be turned so as to form narrow brackets. Most
species are found in
temperate and boreal forests, and cause brown rot.
[0057] The term "carrier," as used herein, refers to relatively nontoxic
chemical compounds or
agents that facilitate the incorporation of a compound into cells or tissues.
[0058] The terms "co-administration" or the like, as used herein, are meant to
encompass
administration of the selected therapeutic agents to a single patient, and are
intended to include
treatment regimens in which the agents are administered by the same or
different route of
administration or at the same or different time.
[0059] The term "diluent" refers to chemical compounds that are used to dilute
the compound of
interest prior to delivery. Diluents can also be used to stabilize compounds
because they can
provide a more stable environment. Salts dissolved in buffered solutions
(which also can provide
pH control or maintenance) are utilized as diluents in the art, including, but
not limited to a
phosphate buffered saline solution.
[0060] The terms "effective amount" or "therapeutically effective amount," as
used herein, refer
to a sufficient amount of an agent or a compound being administered which will
relieve to some
extent one or more of the symptoms of the disease or condition being treated.
The result can be
reduction and/or alleviation of the signs, symptoms, or causes of a disease,
or any other desired
alteration of a biological system. For example, an "effective amount" for
therapeutic uses is the
amount of the composition comprising a compound as disclosed herein required
to provide a
clinically significant decrease in disease symptoms. An appropriate
"effective" amount in any
individual case may be determined using techniques, such as a dose escalation
study.
[0061] A "metabolite" of a compound disclosed herein is a derivative of that
compound that is
formed when the compound is metabolized. The term "active metabolite" refers
to a biologically
active derivative of a compound that is formed when the compound is
metabolized. The term
"metabolized," as used herein, refers to the sum of the processes (including,
but not limited to,
hydrolysis reactions and reactions catalyzed by enzymes) by which a particular
substance is
13
CA 3020933 2018-10-15
changed by an organism. Thus, enzymes may produce specific structural
alterations to a
compound. For example, cytochrome P450 catalyzes a variety of oxidative and
reductive
reactions while uridine diphosphate glucuronyltransferases catalyze the
transfer of an activated
glucuronic-acid molecule to aromatic alcohols, aliphatic alcohols, carboxylic
acids, amines and
free sulphydryl groups. Metabolites of the compounds disclosed herein are
optionally identified
either by administration of compounds to a host and analysis of tissue samples
from the host, or
by incubation of compounds with hepatic cells in vitro and analysis of the
resulting compounds.
[0062] The term "pharmaceutical combination" as used herein, means a product
that results from
the mixing or combining of more than one active ingredient and includes both
fixed and non-
fixed combinations of the active ingredients. The term "fixed combination"
means that the active
ingredients, e.g. a compound (i.e., a cyclohexenone compound described herein)
and a co-agent,
are both administered to a patient simultaneously in the form of a single
entity or dosage. The
term "non-fixed combination" means that the active ingredients, e.g. a
compound (i.e., a
cyclohexenone compound described herein) and a co-agent, are administered to a
patient as
separate entities either simultaneously, concurrently or sequentially with no
specific intervening
time limits, wherein such administration provides effective levels of the two
compounds in the
body of the patient. The latter also applies to cocktail therapy, e.g. the
administration of three or
more active ingredients.
[0063] The term "pharmaceutical composition" refers to a mixture of a compound
(i.e., a
cyclohexenone compound described herein) with other chemical components, such
as carriers,
stabilizers, diluents, dispersing agents, suspending agents, thickening
agents, and/or excipients.
The pharmaceutical composition facilitates administration of the compound to
an organism.
Multiple techniques of administering a compound exist in the art including,
but not limited to:
intravenous, oral, aerosol, parenteral, ophthalmic, pulmonary and topical
administration.
[0064] The term "subject" or "patient" encompasses mammals. Examples of
mammals include,
but are not limited to, any member of the Mammalian class: humans, non-human
primates such
as chimpanzees, and other apes and monkey species; farm animals such as
cattle, horses, sheep,
goats, swine; domestic animals such as rabbits, dogs, and cats; laboratory
animals including
rodents, such as rats, mice and guinea pigs, and the like. In one embodiment,
the mammal is a
human.
[0065] The terms "treat," "treating" or "treatment," as used herein, include
alleviating, abating or
ameliorating at least one symptom of a disease or condition, preventing
additional symptoms,
inhibiting the disease or condition, e.g., arresting the development of the
disease or condition,
relieving the disease or condition, causing regression of the disease or
condition, relieving a
14
CA 3020933 2018-10-15
condition caused by the disease or condition, or stopping the symptoms of the
disease or
condition either prophylactically and/or therapeutically.
Routes of Administration and Dosage
[0066] Suitable routes of administration include, but are not limited to,
oral, intravenous, rectal,
aerosol, parenteral, ophthalmic, pulmonary, transmucosal, transdermal,
vaginal, otic, nasal, and
topical administration. In addition, by way of example only, parenteral
delivery includes
intramuscular, subcutaneous, intravenous, intramedullary injections, as well
as intrathecal, direct
intraventricular, intraperitoneal, intralymphatic, and intranasal injections.
[0067] In certain embodiments, a compound as described herein is administered
in a local rather
than systemic manner, for example, via injection of the compound directly into
an organ, often in
a depot preparation or sustained release formulation. In specific embodiments,
long acting
formulations are administered by implantation (for example subcutaneously or
intramuscularly)
or by intramuscular injection. Furthermore, in other embodiments, the drug is
delivered in a
targeted drug delivery system, for example, in a liposome coated with organ-
specific antibody. In
such embodiments, the liposomes are targeted to and taken up selectively by
the organ. In yet
other embodiments, the compound as described herein is provided in the form of
a rapid release
formulation, in the form of an extended release formulation, or in the form of
an intermediate
release formulation. In yet other embodiments, the compound described herein
is administered
topically.
[0068] In some embodiments, the cyclohexenone compound, or a pharmaceutically
acceptable
salt, metabolite, solvate or prodrug thereof, is administered parenterally or
intravenously. In
other embodiments, the cyclohexenone compound, or a pharmaceutically
acceptable salt,
metabolite, solvate or prodrug thereof, is administered by injection. In some
embodiments, the
cyclohexenone compound, or a pharmaceutically acceptable salt, metabolite,
solvate or prodrug
thereof, is administered orally.
[0069] In the case wherein the patient's condition does not improve, upon the
doctor's discretion
the administration of the compounds may be administered chronically, that is,
for an extended
period of time, including throughout the duration of the patient's life in
order to ameliorate or
otherwise control or limit the symptoms of the patient's disease or condition.
In the case wherein
the patient's status does improve, upon the doctor's discretion the
administration of the
compounds may be given continuously or temporarily suspended for a certain
length of time (i.e.,
a "drug holiday").
[0070] The foregoing ranges are merely suggestive, as the number of variables
in regard to an
individual treatment regime is large, and considerable excursions from these
recommended
CA 3020933 2018-10-15
values are not uncommon. Such dosages may be altered depending on a number of
variables, not
limited to the activity of the compound used, the disease or condition to be
treated, the mode of
administration, the requirements of the individual subject, the severity of
the disease or condition
being treated, and the judgment of the practitioner.
[0071] Toxicity and therapeutic efficacy of such therapeutic regimens can be
determined by
standard pharmaceutical procedures in cell cultures or experimental animals,
including, but not
limited to, for determining the LD50 (the dose lethal to 50% of the
population) and the ED50 (the
dose therapeutically effective in 50% of the population). The dose ratio
between the toxic and
therapeutic effects is the therapeutic index and it can be expressed as the
ratio between LD50 and
ED5o. Compounds exhibiting high therapeutic indices are preferred. The data
obtained from cell
culture assays and animal studies can be used in formulating a range of dosage
for use in human.
The dosage of such compounds lies preferably within a range of circulating
concentrations that
include the ED50 with minimal toxicity. The dosage may vary within this range
depending upon
the dosage form employed and the route of administration utilized. In certain
embodiments, the
CH3 CH3 CH3 CH3
H3
H3C.
T OH
O.
cyclohexenone compound is CH3
CH3 CH3 H3 CH3 H3 CH3 H3 H3
C H3 3 CH3
H3C.
0 T OH HO OH
S'CH3 0
or
CH3 CH3 OH3 CH3
CH3
H 3C
T OH
OH
[0072] In some embodiments, the compounds described herein are formulated into
pharmaceutical compositions. In specific embodiments, pharmaceutical
compositions are
formulated in a conventional manner using one or more physiologically
acceptable carriers
comprising excipients and auxiliaries which facilitate processing of the
active compounds into
preparations which can be used pharmaceutically. Proper formulation is
dependent upon the
route of administration chosen. Any pharmaceutically acceptable techniques,
carriers, and
excipients are used as suitable to formulate the pharmaceutical compositions
described herein:
16
CA 3020933 2018-10-15
Remington: The Science and Practice of Pharmacy, Nineteenth Ed (Easton, Pa.:
Mack
Publishing Company, 1995); Hoover, John E., Remington 's Pharmaceutical
Sciences, Mack
Publishing Co., Easton, Pennsylvania 1975; Liberman, H.A. and Lachman, L.,
Eds.,
Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y., 1980; and
Pharmaceutical
Dosage Forms and Drug Delivery Systems, Seventh Ed. (Lippincott Williams &
Wilkins1999).
[0073] Provided herein are pharmaceutical compositions comprising a compound
(i.e., a
cyclohexenone compound described herein) and a pharmaceutically acceptable
diluent(s),
excipient(s), or carrier(s). In certain embodiments, the compounds described
are administered as
pharmaceutical compositions in which a compound (i.e., a cyclohexenone
compound described
herein) is mixed with other active ingredients, as in combination therapy.
Encompassed herein
are all combinations of actives set forth in the combination therapies section
below and
throughout this disclosure. In specific embodiments, the pharmaceutical
compositions include
one or more compounds (i.e., a cyclohexenone compound described herein).
[0074] A pharmaceutical composition, as used herein, refers to a mixture of a
compound (i.e., a
cyclohexenone compound described herein) with other chemical components, such
as carriers,
stabilizers, diluents, dispersing agents, suspending agents, thickening
agents, and/or excipients.
In certain embodiments, the pharmaceutical composition facilitates
administration of the
compound to an organism. In some embodiments, practicing the methods of
treatment or use
provided herein, therapeutically effective amounts of compounds (i.e., a
cyclohexenone
compound described herein) are administered in a pharmaceutical composition to
a mammal
having a disease or condition to be treated. In specific embodiments, the
mammal is a human. In
certain embodiments, therapeutically effective amounts vary depending on the
severity of the
disease, the age and relative health of the subject, the potency of the
compound used and other
factors. The compounds described herein are used singly or in combination with
one or more
therapeutic agents as components of mixtures.
[0075] In one embodiment, a compound (i.e., a cyclohexenone compound described
herein) is
formulated in an aqueous solution. In specific embodiments, the aqueous
solution is selected
from, by way of example only, a physiologically compatible buffer, such as
Hank's solution,
Ringer's solution, or physiological saline buffer. In other embodiments, a
compound (i.e., a
cyclohexenone compound described herein) is formulated for transmucosal
administration. In
specific embodiments, transmucosal formulations include penetrants that are
appropriate to the
barrier to be permeated. In still other embodiments wherein the compounds
described herein are
formulated for other parenteral injections, appropriate formulations include
aqueous or
17
CA 3020933 2018-10-15
nonaqueous solutions. In specific embodiments, such solutions include
physiologically
compatible buffers and/or excipients.
[0076] In another embodiment, compounds described herein are formulated for
oral
administration. Compounds described herein, including a compound (i.e., a
cyclohexenone
compound described herein), are formulated by combining the active compounds
with, e.g.,
pharmaceutically acceptable carriers or excipients. In various embodiments,
the compounds
described herein are formulated in oral dosage forms that include, by way of
example only,
tablets, powders, pills, dragees, capsules, liquids, gels, syrups, elixirs,
slurries, suspensions and
the like.
[0077] In certain embodiments, pharmaceutical preparations for oral use are
obtained by mixing
one or more solid excipients with one or more of the compounds described
herein, optionally
grinding the resulting mixture, and processing the mixture of granules, after
adding suitable
auxiliaries, if desired, to obtain tablets or dragee cores. Suitable
excipients are, in particular,
fillers such as sugars, including lactose, sucrose, mannitol, or sorbitol;
cellulose preparations
such as: for example, maize starch, wheat starch, rice starch, potato starch,
gelatin, gum
tragacanth, methylcellulose, microcrystalline cellulose,
hydroxypropylmethylcellulose, sodium
carboxymethylcellulose; or others such as: polyvinylpyrrolidone (PVP or
povidone) or calcium
phosphate. In specific embodiments, disintegrating agents are optionally
added. Disintegrating
agents include, by way of example only, cross-linked croscarmellose sodium,
polyvinylpyrrolidone, agar, or alginic acid or a salt thereof such as sodium
alginate.
[0078] In one embodiment, dosage forms, such as dragee cores and tablets, are
provided with
one or more suitable coating. In specific embodiments, concentrated sugar
solutions are used for
coating the dosage form. The sugar solutions, optionally contain additional
components, such as
by way of example only, gum arabic, talc, polyvinylpyrrolidone, carbopol gel,
polyethylene
glycol, and/or titanium dioxide, lacquer solutions, and suitable organic
solvents or solvent
mixtures. Dyestuffs and/or pigments are also optionally added to the coatings
for identification
purposes. Additionally, the dyestuffs and/or pigments are optionally utilized
to characterize
different combinations of active compound doses.
[0079] In certain embodiments, therapeutically effective amounts of at least
one of the
compounds described herein are formulated into other oral dosage forms. Oral
dosage forms
include push-fit capsules made of gelatin, as well as soft, sealed capsules
made of gelatin and a
plasticizer, such as glycerol or sorbitol. In specific embodiments, push-fit
capsules contain the
active ingredients in admixture with one or more filler. Fillers include, by
way of example only,
lactose, binders such as starches, and/or lubricants such as talc or magnesium
stearate and,
18
CA 3020933 2018-10-15
optionally, stabilizers. In other embodiments, soft capsules, contain one or
more active compound that
is dissolved or suspended in a suitable liquid. Suitable liquids include, by
way of example only, one or
more fatty oil, liquid paraffin, or liquid polyethylene glycol. In addition,
stabilizers are optionally
added.
[0080] In other embodiments, therapeutically effective amounts of at least one
of the compounds
described herein are formulated for buccal or sublingual administration.
Formulations suitable for
buccal or sublingual administration include, by way of example only, tablets,
lozenges, or gels. In still
other embodiments, the compounds described herein are formulated for parental
injection, including
formulations suitable for bolus injection or continuous infusion. In specific
embodiments, formulations
for injection are presented in unit dosage form (e.g., in ampoules) or in
multi-dose containers.
Preservatives are, optionally, added to the injection formulations. In still
other embodiments, the
pharmaceutical compositions of a compound (i.e., a cyclohexenone compound
described herein) are
formulated in a form suitable for parenteral injection as a sterile
suspensions, solutions or emulsions in
oily or aqueous vehicles. Parenteral injection formulations optionally contain
formulatory agents such
as suspending, stabilizing and/or dispersing agents. In specific embodiments,
pharmaceutical
formulations for parenteral administration include aqueous solutions of the
active compounds in water-
soluble form. In additional embodiments, suspensions of the active compounds
are prepared as
appropriate oily injection suspensions. Suitable lipophilic solvents or
vehicles for use in the
pharmaceutical compositions described herein include, by way of example only,
fatty oils such as
sesame oil, or synthetic fatty acid esters, such as ethyl oleate or
triglycerides, or liposomes. In certain
specific embodiments, aqueous injection suspensions contain substances which
increase the viscosity
of the suspension, such as sodium carboxymethyl cellulose, sorbitol, or
dextran. Optionally, the
suspension contains suitable stabilizers or agents which increase the
solubility of the compounds to
allow for the preparation of highly concentrated solutions. Alternatively, in
other embodiments, the
active ingredient is in powder form for constitution with a suitable vehicle,
e.g., sterile pyrogen-free
water, before use.
[0081] In one aspect, compounds (i.e., cyclohexenone compounds described
herein) are prepared as
solutions for parenteral injection as described herein or known in the art and
administered with an
automatic injector. Automatic injectors, such as those disclosed in U.S.
Patent Nos. 4,031,893,
5,358,489; 5,540,664; 5,665,071, 5,695,472 and WO/2005/087297 are known. In
general, all automatic
injectors contain a volume of solution that includes a compound (i.e., a
cyclohexenone compound
described herein) to be injected. In general, automatic injectors include a
reservoir for holding the
19
Date Recue/Date Received 2023-10-12
solution, which is in fluid communication with a needle for delivering the
drug, as well as a
mechanism for automatically deploying the needle, inserting the needle into
the patient and
delivering the dose into the patient. Exemplary injectors provide about 0.3
mL, 0.6mL, 1.0mL or
other suitable volume of solution at about a concentration of 0.5 mg to 50 mg
of a compound
(i.e., a cyclohexenone compound described herein) per 1 mL of solution. Each
injector is capable
of delivering only one dose of the compound.
[0082] In still other embodiments, the compounds (i.e., cyclohexenone
compounds described
herein) are administered topically. The compounds described herein are
formulated into a variety
of topically administrable compositions, such as solutions, suspensions,
lotions, gels, pastes,
medicated sticks, balms, creams or ointments. Such pharmaceutical compositions
optionally
contain solubilizers, stabilizers, tonicity enhancing agents, buffers and
preservatives.
[0083] In still other embodiments, the compounds (i.e., cyclohexenone
compounds described
herein) are formulated in rectal compositions such as enemas, rectal gels,
rectal foams, rectal
aerosols, suppositories, jelly suppositories, or retention enemas, containing
conventional
suppository bases such as cocoa butter or other glycerides, as well as
synthetic polymers such as
polyvinylpyrrolidone, PEG, and the like. In suppository forms of the
compositions, a low-melting
wax such as, but not limited to, a mixture of fatty acid glycerides,
optionally in combination with
cocoa butter is first melted.
[0084] In certain embodiments, pharmaceutical compositions are formulated in
any conventional
manner using one or more physiologically acceptable carriers comprising
excipients and
auxiliaries which facilitate processing of the active compounds into
preparations which can be
used pharmaceutically. Proper formulation is dependent upon the route of
administration chosen.
Any pharmaceutically acceptable techniques, carriers, and excipients is
optionally used as
suitable and as understood in the art. Pharmaceutical compositions comprising
a compound (i.e.,
a cyclohexenone compound described herein) may be manufactured in a
conventional manner,
such as, by way of example only, by means of conventional mixing, dissolving,
granulating,
dragee-making, levigating, emulsifying, encapsulating, entrapping or
compression processes.
[0085] Pharmaceutical compositions include at least one pharmaceutically
acceptable carrier,
diluent or excipient and at least one compound (i.e., cyclohexenone compounds
described herein)
described herein as an active ingredient. The active ingredient is in free-
acid or free-base form, or
in a pharmaceutically acceptable salt form. In addition, the methods and
pharmaceutical
compositions described herein include the use crystalline forms (also known as
polymorphs), as
well as active metabolites of these compounds having the same type of
activity. All tautomers of
the compounds described herein are included within the scope of the compounds
presented
CA 3020933 2018-10-15
herein. Additionally, the compounds described herein encompass unsolvated as
well as solvated
forms with pharmaceutically acceptable solvents such as water, ethanol, and
the like. The
solvated forms of the compounds presented herein are also considered to be
disclosed herein. In
addition, the pharmaceutical compositions optionally include other medicinal
or pharmaceutical
agents, carriers, adjuvants, such as preserving, stabilizing, wetting or
emulsifying agents, solution
promoters, salts for regulating the osmotic pressure, buffers, and/or other
therapeutically valuable
substances.
[0086] Methods for the preparation of compositions comprising the compounds
described herein
include formulating the compounds with one or more inert, pharmaceutically
acceptable
excipients or carriers to form a solid, semi-solid or liquid. Solid
compositions include, but are not
limited to, powders, tablets, dispersible granules, capsules, cachets, and
suppositories. Liquid
compositions include solutions in which a compound is dissolved, emulsions
comprising a
compound, or a solution containing liposomes, micelles, or nanoparticles
comprising a
compound as disclosed herein. Semi-solid compositions include, but are not
limited to, gels,
suspensions and creams. The form of the pharmaceutical compositions described
herein include
liquid solutions or suspensions, solid forms suitable for solution or
suspension in a liquid prior to
use, or as emulsions. These compositions also optionally contain minor amounts
of nontoxic,
auxiliary substances, such as wetting or emulsifying agents, pH buffering
agents, and so forth.
[0087] In some embodiments, pharmaceutical composition comprising at least
compound (i.e.,
cyclohexenone compounds described herein) illustratively takes the form of a
liquid where the
agents are present in solution, in suspension or both. Typically when the
composition is
administered as a solution or suspension a first portion of the agent is
present in solution and a
second portion of the agent is present in particulate form, in suspension in a
liquid matrix. In
some embodiments, a liquid composition includes a gel formulation. In other
embodiments, the
liquid composition is aqueous.
[0088] In certain embodiments, pharmaceutical aqueous suspensions include one
or more
polymers as suspending agents. Polymers include water-soluble polymers such as
cellulosic
polymers, e.g., hydroxypropyl methylcellulose, and water-insoluble polymers
such as cross-
linked carboxyl-containing polymers. Certain pharmaceutical compositions
described herein
include a mucoadhesive polymer, selected from, for example,
carboxymethylcellulose, carbomer
(acrylic acid polymer), poly(methylmethacrylate), polyacrylamide,
polycarbophil, acrylic
acid/butyl acrylate copolymer, sodium alginate and dextran.
[0089] Pharmaceutical compositions also, optionally include solubilizing
agents to aid in the
solubility of a compound (i.e., cyclohexenone compounds described herein). The
term
21
CA 3020933 2018-10-15
"solubilizing agent" generally includes agents that result in formation of a
micellar solution or a
true solution of the agent. Certain acceptable nonionic surfactants, for
example polysorbate 80,
are useful as solubilizing agents, as can ophthalmically acceptable glycols,
polyglycols, e.g.,
polyethylene glycol 400, and glycol ethers.
[0090] Furthermore, pharmaceutical compositions optionally include one or more
pH adjusting
agents or buffering agents, including acids such as acetic, boric, citric,
lactic, phosphoric and
hydrochloric acids; bases such as sodium hydroxide, sodium phosphate, sodium
borate, sodium
citrate, sodium acetate, sodium lactate and tris-hydroxymethylaminomethane;
and buffers such as
citrate/dextrose, sodium bicarbonate and ammonium chloride. Such acids, bases
and buffers are
included in an amount required to maintain pH of the composition in an
acceptable range.
[0091] Additionally, pharmaceutical compositions optionally include one or
more salts in an
amount required to bring osmolality of the composition into an acceptable
range. Such salts
include those having sodium, potassium or ammonium cations and chloride,
citrate, ascorbate,
borate, phosphate, bicarbonate, sulfate, thiosulfate or bisulfite anions;
suitable salts include
sodium chloride, potassium chloride, sodium thiosulfate, sodium bisulfite and
ammonium sulfate.
[0092] Other pharmaceutical compositions optionally include one or more
preservatives to
inhibit microbial activity. Suitable preservatives include mercury-containing
substances such as
merfen and thiomersal; stabilized chlorine dioxide; and quaternary ammonium
compounds such
as benzalkonium chloride, cetyltrimethylammonium bromide and cetylpyridinium
chloride.
[0093] Still other pharmaceutical compositions include one or more surfactants
to enhance
physical stability or for other purposes. Suitable nonionic surfactants
include polyoxyethylene
fatty acid glycerides and vegetable oils, e.g., polyoxyethylene (60)
hydrogenated castor oil; and
polyoxyethylene alkylethers and alkylphenyl ethers, e.g., octoxynol 10,
octoxynol 40.
[0094] Still other pharmaceutical compositions may include one or more
antioxidants to enhance
chemical stability where required. Suitable antioxidants include, by way of
example only,
ascorbic acid and sodium metabisulfite.
[0095] In certain embodiments, pharmaceutical aqueous suspension compositions
are packaged
in single-dose non-reclosable containers. Alternatively, multiple-dose
reclosable containers are
used, in which case it is typical to include a preservative in the
composition.
[0096] In alternative embodiments, other delivery systems for hydrophobic
pharmaceutical
compounds are employed. Liposomes and emulsions are examples of delivery
vehicles or
carriers herein. In certain embodiments, organic solvents such as N-
methylpyrrolidone are also
employed. In additional embodiments, the compounds described herein are
delivered using a
sustained-release system, such as semipermeable matrices of solid hydrophobic
polymers
22
=
CA 3020933 2018-10-15
containing the therapeutic agent. Various sustained-release materials are
useful herein. In some
embodiments, sustained-release capsules release the compounds for a few hours
up to over 24
hours. Depending on the chemical nature and the biological stability of the
therapeutic reagent,
additional strategies for protein stabilization may be employed.
[0097] In certain embodiments, the formulations described herein include one
or more
antioxidants, metal chelating agents, thiol containing compounds and/or other
general stabilizing
agents. Examples of such stabilizing agents, include, but are not limited to:
(a) about 0.5% to
about 2% w/v glycerol, (b) about 0.1% to about 1% w/v methionine, (c) about
0.1% to about 2%
w/v monothioglycerol, (d) about 1 mM to about 10 mM EDTA, (e) about 0.01% to
about 2% w/v
ascorbic acid, (f) 0.003% to about 0.02% w/v polysorbate 80, (g) 0.001% to
about 0.05% w/v.
polysorbate 20, (h) arginine, (i) heparin, (j) dextran sulfate, (k)
cyclodextrins, (1) pentosan
polysulfate and other heparinoids, (m) divalent cations such as magnesium and
zinc; or (n)
combinations thereof.
General Consideration for Combination Treatments
[0098] In general, the compositions described herein and, in embodiments where
combinational
therapy is employed and described herein, other agents do not have to be
administered in the
same pharmaceutical composition, and in some embodiments, because of different
physical and
chemical characteristics, are administered by different routes. In some
embodiments, the initial
administration is made according to established protocols, and then, based
upon the observed
effects, the dosage, modes of administration and times of administration is
modified by the
skilled clinician.
[0099] In some embodiments, therapeutically-effective dosages vary when the
drugs are used in
treatment combinations. Combination treatment further includes periodic
treatments that start and
stop at various times to assist with the clinical management of the patient.
For combination
therapies described herein, dosages of the co-administered compounds vary
depending on the
type of co-drug employed, on the specific drug employed, on the disease,
disorder, or condition
being treated and so forth.
[00100] It is understood that in some embodiments, the dosage regimen
to treat, prevent,
or ameliorate the condition(s) for which relief is sought, is modified in
accordance with a variety
of factors. These factors include the disorder from which the subject suffers,
as well as the age,
weight, sex, diet, and medical condition of the subject. Thus, in other
embodiments, the dosage
regimen actually employed varies widely and therefore deviates from the dosage
regimens set
forth herein.
23
CA 3020933 2018-10-15
Examples
Example 1. Preparation of the exemplary cyclohexenone compounds
[00101] Via a particular herbal purification procedure: the following
procedure is for
illustration purposed only. The other purification methods may be used as
well.
[00102] One hundred grams of mycelia, fruiting bodies or mixture of
both from Antrodia
camphorata were placed into a flask. A proper amount of water and alcohol (70-
100% alcohol
solution) was added into the flask and were stirred at 20-25 C for at least 1
hour. The solution
was filtered through a filter and 0.45 tm membrane and the filtrate was
collected as the extract.
[00103] The filtrate of Antrodia camphorata was subjected to High
Performance Liquid
chromatography (HPLC) analysis. The separation was performed on a RP18 column,
the mobile
phase consisted of methanol (A) and 0.3% acetic acid (B), with the gradient
conditions of 0-10
min in 95% - 20% B, 10-20 min in 20%-10% B, 20-35 min in 10%40% B, 35-40 min
in 10%-
95% B, at the flow rate of 1 ml/min. The column effluent was monitored with a
UV-visible
detector.
[00104] The fractions collected at 25 to 30 min were collected and
concentrated to yield 4-
hydroxy-2,3-dimethoxy-6-methy1-5-(3,7,11-trimethyldodeca-2,6,10-
trienyl)cyclohex-2-enone
(compound 1), a product of pale yellow brown liquid. The analysis of compound
1 showed the
molecular formula of C 24H 3804, molecular weight of 390 with melting point of
48 to 52 C.
NMR spectra showed that 1 H-NMR (CDC13) (ppm)=1.51, 1.67, 1.71, 1.75, 1.94,
2.03, 2.07,
2.22, 2.25, 3.68, 4.05, 5.07, and 5.14; 13C-NMR (CDC13) 8 (ppm)=12.31, 16.1,
16.12, 17.67,
25.67, 26.44, 26.74, 27.00, 39.71, 39.81, 40.27, 43.34, 59.22, 60.59, 120.97,
123.84, 124.30,
131.32, 135.35, 135.92, 138.05, 160.45, and 197.12.
CH3 CH3 CH3 CH3
0
CH3
H3C,
OH
0. CH3 1
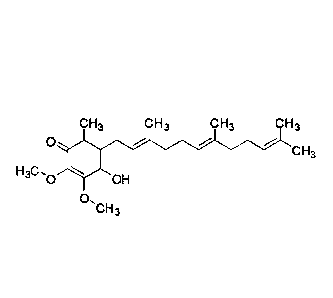
Compound 1: 4-hydroxy-2,3-dimethoxy-6-methy1-5-(3,7,11-trimethyldodeca-2,6,10-
trienyl)cyclohex-2-enone
Other compounds where R4 is H, C1-C8alkyl, C2-C8alkenyl, C2-C8alkynyl, or aryl
optionally
substituted with one or more substituents selected from C1-C8 alkyl, C2-C8
alkenyl, C2-C8
alkynyl, C3-C8 cycloalkyl, and C1-C8 haloalkyl were obtained as well.
[00105] Alternatively, the exemplary compounds may be prepared
synthetically. See for
example, U.S. Patent No. 9365481.
24
CA 3020933 2018-10-15
R3 CH3
0 R4
OR
Y,
Similarly, other cyclohexenone compounds having the structure R2
are
isolated from Antrodia camphorata or prepared synthetically or semi-
synthetically from the
suitable starting materials. An ordinary skilled in the art would readily
utilize appropriate
conditions for such synthesis.
Example 2. Study of Compound 1 with clinical chemotherapy drugs on human
pancreatic
carcinoma in AsPC-1 Cell Culture System and in a Severe Combined
Immunodeficient Mouse
Xenograft Tumor Model
[00106] In order to evaluate the efficacy of a combination therapy
comprising the
exemplary compound 1 with an anticancer agent (e.g., any clinically used
drugs) for the
treatment of pancreatic carcinoma development, an in vitro study and an in
vivo xenografts study
by an exemplary Compound 1 with selected current clinical chemotherapy drugs
were conducted.
[00107] For in vitro study, Compound 1 and each of the selected six (6)
clinical
chemotherapy drugs were used on human pancreatic carcinoma AsPC-1 and/or CaPan-
2, PANC-
1 in cell culture system.
[00108] In in vivo xenograft mouse model, gemcitabine and/or paclitaxel
were selected to
be administered via tail vein twice a week with an oral administration of
Compound 1 once a day
for a total of 28 doses on human pancreatic carcinoma AsPC-1 xenograft model.
[00109] In Vitro Test:
[00110] The human pancreatic carcinoma cell lines, AsPC-1, PANC-1 and
CaPan-2 were
seeded 3 x 104 cells per well and cultured in a forty-eight (48) well cell
culture dish (growth area
0.95 cm2 per well). Sixteen (16) hours later, cells were treated Compound 1
alone or Compound 1
with a clinical chemotherapy drug, respectively. After 48 or 72 hours of the
treatment,
methylthiazoletetrazolium (M'TT) assay (supplemental protocol-1) was performed
to measure the
effect of treatment on the viability of cells during proliferation.
[00111] In vivo Test:
[00112] The human pancreatic carcinoma cell line, AsPC-1, was cultured
and the cells
were injected subcutaneously in a flank of 60 mice. Three days later, the
tumors could be felt in
the mice. When tumors reached approximately 100-200 mg (mm3), total of fifty
one (51) tumor-
bearing mice were randomly selected and sorted into one of six (6) groups.
Vehicle (corn oil)
treated mice were used as negative controls. Compound 1 combined with
gemcitabine or
paclitaxel were administered orally once a day and/or I.V. twice a week for
four (4) weeks.
CA 3020933 2018-10-15
Tumor measurements were recorded once a week using hand-held calipers and
Visualsonic Vevo
2100 image system. Individual mouse weights were taken once a week during
dosing. Mice were
monitored daily for signs of toxicity and morbidity; any abnormal findings
were recorded. The
study was terminated four (4) weeks after initiation of drug administration.
[00113] Mice were euthanized when an individual mouse has a tumor that
is 2,000 mg
(cm3) over one (1) measurement. A mouse was moribund when a tumor impedes the
mouse's
ability to feed, drink or ambulate, or if a mouse loses >20% of its original
body weight. An
individual group was euthanized when tumors reached a mean target size of
1,500 mg per group
over two (2) consecutive measurements. The study coordinators should be
notified of any
unscheduled euthanasia. The study was terminate four (4) weeks after the
initiation of drugs
administration.
Test System:
[00114] A. Cell Lines:
Human pancreatic carcinoma AsPC-1 (ATCC Catalog No. CRL-1682).
Human pancreatic carcinoma PANC-1 (ATCC Catalog No. CRL-1469).
Human pancreatic carcinoma CaPan-2 (ATCC Catalog No. HTB-80).
[00115] B. Cell Preparation
1. Remove and discard culture medium.
2. Briefly rinse the cell layer with 5 mL HBSS. Gently shake and remove the
HBSS.
3. Add 5 mL trypsin-EDTA to 15 cm2 polystyrene dishes. Incubate 5 mins at 37 C
in a humidified incubator with 95% air / 5% CO2 atmosphere.
4. Add 5 mL of complete growth medium (DMEM medium containing 10% FBS)
and aspirate cells by gently pipetting.
5. Count live cells using the trypan blue exclusion assay (supplemental
protocol-
2) with a hemocytometer.
6. For in vitro study, cells will be seeded 3 x 104 cells per well and
cultured in a
forty-eight (48) wells cell culture dish for sixteen (16) hours until
treatment.
7. Incubate cultures at 37 C.
[00116] C. Animal Species/Strain:
a. Nude mice, BALB/cAnN.Cg-Foxn/ nu /CrlNarl
b. Female, athymic nude mice (nu/nu) 4-6 wk of age.
c. Body weight: between 15 and 20 g on the day of inoculation of tumor
cells.
[00117] D. Human pancreatic carcinoma xenograft models:
26
CA 3020933 2018-10-15
1. To create the subcutaneous xenograft model, athymic female nude mice (4-6
weeks old) were subcutaneously inoculated with 1 X 107 AsPC-1 cells in 0.1
mL of serum-free DMEM medium containing 20% Matrigel (BD Biosciences,
Bedford MA).
2. Tumor-bearing animals were divided into the following 6 groups as table 1.
3. The mice were monitored for activity and physical condition every day, and
the
determination of body weight and measurement tumor mass should be done
once a week. Tumor image will be photed by digital camera and Visualsonic
Vevo 2100 image system. Tumor mass is determined by caliper measurement
in two perpendicular diameters of the implant and calculate using the formula
(4/3) X (7cX a X b2), where "a" stands for the long diameter and "b" is the
short
diameter.
[00118] E. Test Articles:
[00119] The clinical chemotherapy drugs were purchased from Sigma-
Aldrich Co. LLC,
and prepared in the following manner:
- Erlotinib (Stock conc. 100 mM in DMSO, Sigma Cat.# E4997).
- 5-Fluorouracil (Stock conc. 100 mM, Sigma Cat.# F6627).
- Gemcitabine (Stock conc. 100 mM in DMSO, Sigma Cat.# G6423).
- 1rinotecan (Stock conc. 100 mM in DMSO, Sigma Cat.# 11406).
- Oxaliplatin (Stock conc. 50 mM in DMSO, Sigma Cat.# 09512).
- Paclitaxel (Stock conc. 10 mM in DMSO, Sigma Cat.# T7402).
[00120] For in vivo test, gemcitabine (G) was purchased from Eli Lilly
(Indianapolis, IN,
USA), and paclitaxel (P) was purchased from Sigma-Aldrich.
[00121] F. in vitro experiment design:
[00122] a. Cytotoxicity of test articles (TC50): AsPC-1 was seeded 3 x
104 cells (counted
by Cell counter, LUNATM Automated Cell Counter or hemocytometer) per well and
cultured in a
48 wells cell culture dish. Cells were treated with single article from 400 or
800 M with serial
dilution to 0 M. After 48 or 72 hours of treated, MCI assay was performed to
measure the
effect of treatment on the viability of AsPC-1 cells during proliferation. The
absorbance at 595
nm was measured using a microplate reader (BioTEK, SynergyTM HO. Article
sensitivity curves
and TC50 values were calculated using GraphPad Prism 4.0 software.
[00123] b. Combination therapy of Compound 1 and chemotherapy drugs:
This study also
contains the cytotoxicity of Compound 1 plus a chemotherapy drug of selections
to investigate
the efficacy of such combination. Compound 1 combined each of paclitaxel,
gemcitabine, 5-FU,
27
CA 3020933 2018-10-15
oxaliplatin, erlotinib, and irinotecan, respectively. After treated 72 hours,
MIT assay was
performed to measure the effect of treatment on the viability of AsPC-1 cells
during
proliferation. The absorbance at 595 nm was measured using a microplate
reader.
[00124] c. Evaluation of drug interactions: MTT assay was also
performed to test the
effect of drug combinations on the viability of AsPC-1, PANC-1 and CaPan-2
cell lines. The
combination index (CI) isobologram method of Chou and Talalay (Chou and
Talalay, 1984;
Chang and Chou, 2000) which is based on the median-effect principle, was used
to calculate
synergism or antagonism for the combined drug effects. Dose-effect curves for
each drug,
separately and in combination, in serially diluted concentrations were plotted
using the median-
effect equation and plot (Chou, 1991) and the Cl equation and plot (Chou et
al., 1994). CI values
at different effect and dose levels and isobolograms were generated
automatically using the
computer software CompuSyn (Chou and Martin, 2005). With this method,
additive, synergistic,
or antagonistic effects are indicated by CI values of 1, <1, and >1,
respectively.
[00125] G. in vivo experiment design:
[00126] a. Cell Injections: Mice were injected subcutaneously into the
right lateral thorax
with 1 x 107 viable human pancreatic carcinoma AsPC-1 Cells. A total of 60
mice were injected,
with approximately 36 tumor-bearing mice were used on this study.
[00127] b. Selections of animals and group Assignment: This study
contained six (6)
groups and each group included eight (8) or nine (9) mice respectively. When
tumors reached a
target window size of approximately 100-200 mg (mm3), 54 tumor-bearing mice
were randomly
selected and sorted into one of 6 groups. Groups 1 is a solvent treated
negative control and
contained mice receiving treatment-1. Group 2, 3, 4, 5 and 6 contained mice
that received
treatment -2, treatment -3, treatment -4, treatment-5 and treatment -6,
respectively. Table 1 lists
test articles concentrations, solvent articles and dosing schedule.
[00128] c. Dose preparation:
c-1: Compound 1 (A)
i. Weighed an appropriate amount of Compound 1 and added into a 50 mL tube.
ii. Added an appropriated amount of corn oil (Sigma-Aldrich) into the 50 mL
tube.
Mixed the formulation with vortex until homogenized completely. Pulled the
formulation up into a disposable syringe and transferred to 4 mL brown tubes,
3.2 mL / tube.
iii. Stored the suspension at frozen conditions (-20 C) until used.
iv. Completely thawed the formulation in a 37 C water bath on day of
intended use.
c-2: Gemcitabine (G)
28
CA 3020933 2018-10-15
i. Stock of gemcitabine was stored as a 50 mg/mL solution in sterile PBS at
-20 C (Ito et al., 2006;
Awasthi et al., 2013).
ii. Before animal studies, weighed an appropriate amount of gemcitabine
into a 4 mL tube.
Added an appropriate volume of vehicle-I (50% Ethanol & 50% Tweeem-80 (v/w))
into the 4
mL tube and shook until the material dissolved completely.
iv. Added an appropriate volume of physiological saline into the 4 mL
tube and moved the tube
side-by-side to completely homogenize the solution.
c-3: Paclitaxel (P)
i. Stock of paclitaxel was dissolved in 100% ethanol to final concentration
10 mg/mL (Shi
et al., 2000; Chang et al., 2006; Awasthi et al., 2013).
ii. Before animal studies, weighed an appropriate amount of paclitaxel into
a 4 mL tube.
iii. Added an appropriate volume of vehicle-Ib into the 4 mL tube and shook
until the
material dissolved completely.
iv. Added an appropriate volume of physiological saline into the 4 mL tube
and moved the
tube side-by-side to completely homogenize the solution.
[00129] Table 1: Treatment groups of AsPC-1 xenograft:
Dose
Group
Treatment label Dose/mouse/day or time Dosing Route volume /
Frequency
Number
mouse
1 Treatment -1 Solvent (corn oil) oral
2 Treatment -2 A (120 mg/kg) oral (A)
A (120 mg/kg)
3 Treatment -3 G (100 mg/kg) oral (A) / IV (G)
100 jiL Orally
once
A (120 mg/kg) for oral, a
day, I.V.
4 Treatment -4 oral (A) / IV (P) 20 ¨ 30
twice
P (5 mg/ kg)
G (100 mg/kg) uL for weekly
for
Treatment -5 N (G & P) I.V. 4 weeks.
P(5 mg/kg)
G(10 mg/kg)
oral (A) IV
6 Treatment -6 P (30 mg)
(G & P)
A(120 mg/kg)
[00130] Observations and Examinations of in vivo system:
29
Date Recue/Date Received 2024-02-23
[00131] A. Clinical Observations: Clinical observations of each study
animal were
performed and at least once daily (including weekends and holidays) for signs
of morbidity,
mortality and Test Article toxicity. Morbidity includes signs of illness such
as, but not limited to,
emaciation, dehydration, lethargy, hunched posture, unkempt appearance,
dyspnea and urine or
fecal staining. All abnormal findings will be recorded.
[00132] B. Animal Weights: Individual animal weights were taken one (1)
time per week
after/before dosing. Weights were recorded first prior to cell injections and
continued for the
duration of the study.
[00133] C. Tumor Measurements: The injection site of each animal was
monitored one (I)
time per week for signs of tumor growth. Throughout the study, the length (a)
and width (b) of
any tumors developed were measured in millimeters (mm) using vernier calipers,
where a is the
longer of the two (2) dimensions. The applicable tumor size in millimeter (mm)
or centimeter
(cm) was calculated by using the formula associated with a prolate ellipsoid:
M = (4/3) X (7E X a
X b2); and these data was recorded.
[00134] D. Evaluation of drug synergistic effect by tumor growth
inhibition (TGI) and
tumor inhibitory ratio (TIR): Volume of tumor growth inhibition (TG1) for each
group was
calculated according to the following formula:
Tumor growth inhibition (%) = (1 - [T ¨ To] / IC ¨ Col) x 100
T and To are the mean tumor volumes on the end day and day 1 of treatment,
respectively,
for the experimental groups. For the control groups, C and Co are the mean
tumor volumes
on a end day and the start of the study, respectively.
For TIR, at the 4th week after treatment, the mice were sacrificed, and tumors
were
ablated carefully and weighed after any remaining blood was washed off with
PBS. The
tumor inhibitory ratio was calculated by the formula:
Tumor inhibitory ratio (%) = t( WControl - WTrea ted) W Control] X 1 0 0%
WTreated and WControl are the average tumor weights of the treated and control
mice,
respectively.
1001351 11 Hematological parameters: Cardiac blood was collected in
EDTA for analyzing
hematological parameters such as white blood cell count (WBC), red blood cell
count (RBC),
hemoglobin concentration (MB), hematocrit, mean corpuscular hemoglobin and
concentration,
platelet distribution width, platelet count, neutrophils, lymphocytes,
monocytes, eosinophils. This
was determined using hematology counter (Abbott Cell-Dyn 3700, IL).
CA 3020933 2018-10-15
[00136] Statistical analysis: All data are presented as mean standard
error of the mean
(SEM). One-way analysis of variance (ANOVA) was used to determine significance
between
groups. For multiple comparisons of more than two groups, the ANOVA analysis
was used and
followed by post-hoc tests with the Bonferroni correction. A value of P < 0.05
was considered to
be significant.
[00137] Illustration of daily schedule of xenograft study:
AsPC-1 human pancreatic carcinoma cells Injection schedule
tll =
I ,1
Start
11011 I tit t 1081 11 it ttItt
1019 (lb
1st week 2nd week 3rd week
4 I4 4 4 I 4 1: AsPC-1 human pancreatic
carcinoma cells
D221 1 11328 029 implanted day
: Administered oral¨Treatment-1, Treatment-2,
Treatment4, Treatment-4 and Treatment4.
14 I : Injection day (IV)¨ Treatment-
3, Treatment-4
and Treatment-6
4th week t : Timor measurement and Body
weight
[00138] Results:
[00139] In vitro cytotoxicity: To evaluate the combinatorial
therapeutic efficiency of the
exemplary compound 1, the viability of treated AsCp-1 cell lines using 3-(4,5-
dimethylthiazol- 2-
yl)-2,5-diphenyltetrazolium bromide (MTT) assay was measured. The toxic
concentration (TC50)
of Compound 1 (i.e., antroquinonol) and chemotherapy drugs on AsPC-1 cell line
was
investigated. FIGs. 1 to 7 and Table 2 show the cytotoxicity of Compound 1 and
chemotherapy
drugs on AsPC-1 cell line. According to these results, it was confirmed that
compound 1 and
combination treatment of chemotherapy drugs on AsPC-1 cell viability following
by drug
concentration on Table3. AsCP-1 cells were individually incubated with
compound 1 or each of
the drug combinations for 48 h and 72 h. In FIGs. 8 and 9, the viability of
AsPC-1 cells incubated
with Compound 1, MTT assays of each chemotherapy drug and their combinations
for 48 h and
72 h respectively were measured. AsPC-1 cells treated with Compound 1 or
chemotherapy drugs
alone showed 75% to 80% viability after incubation for 48 h or 72 h. According
to these results,
31
CA 3020933 2018-10-15
the combination therapies of Compound 1 with the selected chemotherapy drugs
may have a
synergistic effect to control AsPC-1 cell viability. Moreover, these results
also indicated a
combination of the tested clinical drugs and Compound 1 has a more extreme
effect on AsPC-1
human pancreatic cancer cells than the corresponding single drug treatment.
[00140] Table 2. Effect of anticancer drugs on cytotoxicity at 48 and
72 hours
(Antroquinonol = Compound 1)
TCso (pM)a
Drug
48 hrs 72 hrs
Antroquinonol 58.09 24.04
Erlotinib > 800 > 800
5-FU >800 >800
Gemcitabine > 800 > 800
lrinotecan 221.3 68.34
Oxaliplatin 654.7 57.91
Paclitaxel 7.054 3.719
aTC50: the concentration of the compound at which cell viability
was reduced to 50%.
[00141] The combinations of Compound 1 with paclitaxel and/or
gemcitabine, which have
synergistic effect for pancreatic cancer were evaluated. Three pancreatic
cancer cell lines, AsPC-
1, CaPan-2 and Panc-1 were used to test the combinations through median effect
analysis.
100142] Table 3. Drugs concentrations used in the combinations on
cytoxicity of AsPC-1
cells.
Conc. ( M)
Drug
48 hrs 72 hrs
Antroquinonol
20 20
(Compound 1)
Erlotinib 100 100
5-FU 100 100
Gemcitabine 100 100
Irinotecan 200 70
Oxaliplatin 200 60
Paclitaxel 7 3
[00143] Evaluation of the combination therapy effect of Compound 1 with
chemotherapeutic drugs, gemcitabine and paclitaxel
32
CA 3020933 2018-10-15
[00144] The median effect analysis of Chou and Talalay (Chou and
Talalay, 1984) was
used to calculate the CI for each drug combination formula. Therefore, Cl
value was used to
determine if there is a synergy effect between Compound 1 and gemcitabine,
and/or paclitaxel in
this study. In the AsPC-1, CaPan-2 and Pane-1 cell lines, the two or three
compound
combinations of Compound 1 with chemotherapy drugs, paclitaxel and
gemcitabine, were
strongly synergistic (CI < 1, FIG. 11) across the entire range of doses (e.g.,
see FIG. 10 and 11).
These results presented that the exemplary compound (i.e., Compound 1) with a
second or third
currently used chemotherapeutic drugs strongly inhibit pancreatic cancer
growth especially in the
three compound combination.
In vivo antitumor activity on AsPC-1 subcutaneous xenograft model
[00145] The antitumor activity of the combination therapy comprising the
exemplary
Compound 1 was assessed on AsPC-1 tumor bearing nude mice. AsPC-1 cells were
subcutaneously injected 7 days before commencing treatment to the mice (Table
1). These
animals then received daily administration of various drug combinations for 28
days. At day 28,
all test mice were sacrificed and corresponding tumors were excised for
further studies. As
shown in FIGs. 13 and 14, at the end of the experiments, the mice treated with
the combination
of Compound 1 and gemcitabine (T3), or the combination of Compound 1,
gemcitabine and
paclitaxel (T6), showed a highly significant tumor regression with a slow
growing in tumor
volume. Mice in the other four treatment groups showed strict tumor growth
comparing with
groups T3 and T6. The tumor growth inhibition rate increased significantly in
the combination of
Compound 1 and gemcitabine (T3), or the combination of Compound 1, gemcitabine
and
paclitaxel (T6) (FIG. 15) compared with the groups (FIG. 16, T2, T4 and T5).
[00146] FIG. 17 illustrates the treatment results represented by the
ultrasound images of
AsPC-1 pancreatic tumor xenograft during the period of treatments. Photographs
of the tumors
from the sacrificed mice are shown in FIG. 18A. It can be seen that the tumor
volumes in mice
treated with Compound 1 and gemcitabine are significant smaller than those
treated with other
combinations. In particular, FIG. 18B shows that the tumor weight in the mice
treated with
Compound 1 and gemcitabine is much smaller as compared to those in other
groups. All of these
results demonstrate that the combination therapy described herein does have
predominant tumor
growth inhibitory efficacy. The tumor inhibitory rate (TR1) in each group was
also calculated
= based on final tumor weights (FIG. 19). Tumor growth was inhibited
significantly in the groups
of Compound 1 plus gemcitabine (FIG. 19, T3), and Compound 1 plus the other
two drugs (FIG.
19, T6).
33
CA 3020933 2018-10-15
[00147] In order to confirm that therapeutic efficacy of the combination
therapy based on
the exemplary compound 1 is much better than that of mono-chemotherapy drug,
the synergistic
effects of the selected drug combinations were evaluated by tumor volume of
treatment-to-
control ratio (TCR), tumor growth inhibition (TGI) and tumor inhibitory ratio
(TIR). In AsPC-1
xenografts, monotherapy of Compound 1 was able to inhibit tumor growth, with
treatment-to-
control ratios (TCR) of 0.55. The combination of Compound 1 and gemcitabine
(Table 4, Ge +
Aq) produced a dramatically enhanced antitumor activity compared with the
other mono or two-
compound combinations treatments (TCR 0.36; Table 4; Fig. 13). In the
meantime, the
combination with three medicines comprising Compound 1 (Table 4, Px + Ge + Aq)
also induced
strong antitumor activity compared with either mono or other combination
therapies (TCR 0.46;
Table 4; Fig. 13). According to those results, Compound 1 in combinations with
gemcitabine, or
with additional paclitaxel have enhanced antitumor activity, significantly.
[00148] In addition, no significant weight loss was observed in the
tumor-bearing mice
treated with various combinations, indicating negligible side effect of
Compound 1 and other two
chemotherapy drugs for tumor therapy at the employed doses (FIG. 12).
Consistently, Compound
1 and its combinations did not affect number of blood cells except for blood
glucose in AsPC-1
xenograft (Table 5.). According to these data, it suggests that Compound 1,
and/or its
combinations thereof have low safety risk of taking orally.
[00149] Table. 4. Tumor weight of excised tumors at the time of
sacrifice from the
subcutaneous AsPC-1 pancreatic tumor xenograft-bearing male nude mice after 28
days of
treatments.
Treatment Groups"
Ti T2 T3 T4 T5 T6
Tumor
0.42 + 0.11a 0.21 0.07b 0.11 0.05b 0.22 0.16b 0.31 0.21b 0.24 0.09b
weight
"Six treatment groups of the mice administrated corn oil (T1), Compound 1
(T2), combination of
Compound 1 and gemcitabine (T3), combination of Compound 1 and paclitaxel
(T4),
combination of gemcitabine and paclitaxel (T5) and combination of Compound 1,
gemcitabine,
and paclitaxel (T6).
- Tumor weights are means SEM. Values in the same row are followed by a
different
superscript (a-b) are significantly at P <0.05.
34
CA 3020933 2018-10-15
[00150] Table 5. Synergistic antitumor activity by combinations of
Compound 1 (Aq),
paclitaxel (Px) and gemcitabine (Ge) in AsPC-1 xenograft.
Xenograft Treatment Response, TCR TGI (%) TIR (%)
Control 1.00
Aq 0.55 51 45
Ge + Aq 0.36 73 64
AsPC-1
Px + Aq 0.60 49 40
Px + Ge 0.67 40 33
Px + Ge + Aq 0.46 65 54
Abbreviations: Aq, Compound 1; Px, paclitaxel; Ge, gemcitabine; TCR, tumor
volume of
treatment-to-control ratios; TGI, volume of tumor growth inhibition; TIR,
tumor weight inhibitory
ratio.
[00151] Table 6. Hematological values of .each treatment group in AsPC-
1 xenograft
study.
Table 6. Hematological values of .each treatment group in AsPC-1 xenograft
study.
Parameter Unit T1 (control) 12 (A) 13 (G,A) 14 (P,A) 15 (P, G) 16 (P, G,
A) References range of 10-18 week Female ICR mice
WBC 109/ 6.10 t 0.83 7.34
t 0.71 8.92 t 1.11 8.65 t 0.64 7.33 t 1.46 7.65 t 0.92 4.20- 8.86
RBC 1012/1 9.16 t 1.34
9.93 t 1.34 9.12 t 0.94 9.05 t 1.22 9.77 t 1.43 9.26 t 1.57 7.99 - 10.83
HGB gldL 15.5 t 0.9 15.6 t 1.1
15.7 t 0.4 14.6 t 2.9 16.2 t 1.4 16.1 t 1.9 14.3 16.7
HCT % 51.2 t 1.6 50.3 t 1.8
52.1 2.1 54.9 t 2.7 53.9 t 2.1 55.9 t 3.3 49.0- 63.1
ACV fL 54.9 1.7 53.6 t 2.3
57.6 t 4.1 53.1 t 1.4 56.6 t 4.3 53.1 5.3 53.6 - 65.4
MCH pg 15.4 0.5 15.3 t 0.3
15.5 t 0.7 16.1 t 0.6 16.1 t 1.1 16.1 t 1.9 15.8- 16.8
Abbreviations: WBC - white blood count, RBC - red blood cell, HGB -
Hemoglobin, HCT -
Hematocrit, MCV - mean corpuscular volume, MCH - mean corpuscular hemoglobin.
Values are
means SEM (n=3 in each treatment group)
[00152] As shown in the study results, it is clearly shown the superior
unexpected benefits
utilizing the compositions comprising the exemplary Compound 1 and the current
clinical
chemotherapy drugs such as paclitaxel and gemcitabine against pancreatic
cancer. Cell viability
studies showed that the treatments with such combinations resulted in much
higher inhibition
activity than either Compound 1 or the other chemotherapy drugs alone,
suggesting a synergistic
and/or additive effect. Such effect was confirmed by the combinations of
compound 1 with
CA 3020933 2018-10-15
gemcitabine alone, and with additional paclitaxel which exhibit significant
inhibition of tumor
growth on tumor bearing mice.
Example 3. A Phase I study to determine the maximum tolerated dose (MTD) and
to evaluate
safety/tolerability, pharmacokinetics, pharmacodynamics and preliminary
efficacy of Compound
1 in combination with nab+paclitaxel+gemcitabine in first line metastatic
pancreatic cancer
1001531 Introduction: In 2011, the multidrug combination of leucovorin,
fluorouracil,
irinotecan, and oxaliplatin (FOLFIRINOX) was noted to provide an increased
median survival of
4.3 months versus gemcitabine; however, given its side effect profile, it is
available only to a
select group of patients with advanced pancreatic cancer. The patients were
randomly assigned to
receive FOLFIRINOX or gemcitabine. The median overall survival (OS) was 11.1
months in the
FOLFIRINOX group compared with 6.8 months in the gemcitabine group (hazard
ratio [HR] for
death =0.57; 95% confidence interval [CI], 0.45 to 0.73; p<0.001). Median
progression-free
survival (PFS) was 6.4 months in the FOLFIRINOX group and 3.3 months in the
gemcitabine
group (HR for disease progression =0.47; 95% Cl, 0.37 to 0.59; p<0.001).
100154] More recently, the gemcitabine plus nab-paclitaxel (the 130-nm
albumin-bound
nanoparticle formulation of paclitaxel) combination was shown to increase
median survival by
1.8 months, with increased OS at 1 and 2 years; adverse effects were
reasonable and included
cytopenias and peripheral neuropathy. The multi-center, international Phase
III MPACT trial
included 861 patients with previously untreated metastatic pancreatic
adenocarcinoma. The
patients were randomly assigned to receive gemcitabine and nab-paclitaxel or
gemcitabine
monotherapy. The median OS was 8.5 months in the nab-paclitaxel/gemcitabine
group compared
with 6.7 months in the gemcitabine group (FIR for death=0.72; 95% CI, 0.62 to
0.83; p<0.001).
Median PFS was 5.5 months in the nab-paclitaxel/gemcitabine group and 3.7
months in the
gemcitabine group (HR for disease progression=0.69; 95% CI, 0.58 to 0.82,
p<0.001).
1001551 The current National Comprehensive Cancer Network
recommendations suggest
acceptable first-line chemotherapy combinations for patients with good
performance status (i.e.,
Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0 or 1),
based on
patient preference and support system available. These combinations include
FOLFIRINOX for
patients with better/favorable comorbidity profile, or gemcitabine plus nab-
paclitaxel for patients
with adequate/acceptable comorbidity profile, nab-paclitaxel + gemcitabine,
and gemcitabine
plus erlotinib. Gemcitabine alone is recommended for patients with ECOG PS 2
or with a
comorbidity profile that precludes other regimens; the addition of
capecitabine or erlotinib may
be offered. The guidelines for choosing an appropriate treatment regimen for
patients with
metastatic pancreatic cancer thus remain ambiguous, and in the absence of a
randomized trial
36
CA 3020933 2018-10-15
comparing the combination regimens head to head, the most appropriate first-
line therapy for
these patients remains unclear.
[00156] As shown in Example 2, the exemplary compound 1 has shown anti-
tumor activity
and a low toxicity profile in preclinical in vitro and in vivo animal
experiments. In a separate non-
clinical study using the orthotopic PANC-1 human pancreatic cancer xenograft
model with
Compound 1 conducted in 2012, four groups of mice were treated with 30 mg/kg,
60 mg/kg, 90
mg/kg, of Compound 1 and vehicle control, respectively to examine the in vivo
therapeutic
efficacy of Compound 1. Tumor volumes and tumor weights were measured 10 days
after drug
treatment (19 days after tumor implantation). Treatment with Compound 1 at 30,
60, and 90
mg/kg produced an effective anti-tumor activity with statistically significant
smaller mean tumor
volumes and tumor weights in all three dosage levels compared to vehicle
control. All three
doses of antroquinonol were tolerated well by the tumor bearing mice; there
was no severe body
weight loss observed during the study.
[00157] In the early reported clinical trial studies, Antroquinonol
(i.e., Compound 1) at 50,
100, 200, 300, 450, and 600 mg-dose levels, given daily for 4 weeks, was
generally safe and well
tolerated, as no particular safety concerns or DLTs were identified in this
study. The data
generated from the safety and pharmacokinetic (PK) profiles in this study
could support further
studies of antroquinonol in subjects with advanced malignancies to determine
the maximum
tolerated dose (MTD) and efficacy in given patient populations.
STUDY OBJECTIVES
[00158] The primary objectives of this study are: to determine the MTD
or maximum
feasible dose (MFD) of Compound 1 in combination with
nab+paclitaxel+gemcitabine in
subjects with metastatic pancreatic cancer. Although Compound 1 has been
assessed for a cancer
treatment, it is known in the field that many cancer drugs are less effective
in treating metastatic
cancer.
[00159] Cohort expansion: to evaluate the anti-tumor activity of
antroquinonol in
combination with nab+paclitaxel+gemcitabine in subjects with metastatic
pancreatic cancer.
[00160] The secondary objectives of this study are:
[00161] Dose escalation:
= To evaluate the safety and tolerability of the combination of
antroquinonol and
nab+paclitaxel+gemcitabine.
= To characterize the PK of antroquinonol and nab+paclitaxel+gemcitabine.
= To evaluate antroquinonol activity from routine pancreatic cancer
monitoring.
= To explore the preliminary anti-tumor activity of antroquinonol in
combination
37
CA 3020933 2018-10-15
with nab+paclitaxel+gemcitabine in subjects with metastatic pancreatic cancer.
[00162] Cohort expansion: to evaluate the safety and tolerability of
MFD of
antroquinonol and nab+paclitaxel+gemcitabine combination; to characterize the
PK of
antroquinonol and nab-paclitaxel-gemcitabine; to evaluate for antroquinonol
activity from
routine pancreatic cancer monitoring.
STUDY ENDPOINTS
[00163] Primary endpoints
[00164] Dose escalation:
= Occurrence of DLTs in the 1st 28-day cycle of treatment. The DLT
definition
(grading according to NCI Common Terminology Criteria for Adverse Event
[CTCAE]
v.4.03):
= Non-hematological toxicity of Grade 3 or greater excluding: Grade 3
diarrhea,
nausea, or vomiting that resolves within 3 days of onset to Grade 1 or to
baseline
Grade with appropriate treatment
= Grade 4 thrombocytopenia or neutropenia lasting >48 hours
= > Grade 3 febrile neutropenia
= Grade 3 thrombocytopenia with bleeding
= Persisting toxicity of Grade 2 or greater that causes more than a 14 day
delay in
dose administration.
[00165] AEs that are considered by the Investigator to be related to
the underlying disease
condition, concomitant medication, or to new unrelated medical events or
treatment will not be
defined as a DLT.
[00166] Cohort expansion:
[00167] Primary endpoint: To evaluate the anti-tumor activity of
antroquinonol in
combination with nab+paclitaxel+gemcitabine in subjects with metastatic
pancreatic cancer.
[00168] Secondary endpoints:
[00169] Safety: Safety will be evaluated using AEs, physical
examinations, laboratory
findings (including clinical chemistry, hematology, and urinalysis), vital
signs (including blood
pressure and pulse), and electrocardiograms (ECGs).
[00170] Efficacy: Objective response rate (ORR), duration of response
(DoR), disease
control rate (DCR), and PFS using Investigator assessments according to REC1ST
1.1
(assessments to occur every 8 weeks), OS.
[00171] Pharmacokinetics: Plasma concentration of antroquinonol and
nab+paclitaxel+gemcitabine combination and PK parameters will be evaluated.
38
CA 3020933 2018-10-15
[00172] Biomarkers: To evaluate antroquinonol activity from routine
pancreatic cancer
monitoring.
INVESTIGATION PLAN
[00173] Overall Study Design and Plan Description
[00174] This is a single-arm, open-label, multi-center Phase I study of
Compound 1 in
combination with nab-paclitaxel and gemcitabine in first line treatment naïve
subjects with Stage
IV metastatic pancreatic carcinoma.
[00175] The first part of the study will follow a 3+3 dose escalation
design to determine
the MTD or MFD (based on PK and capsules strength) of antroquinonol in
combination with
standard dose regimen of nab-paclitaxel and gemcitabine. Two dose levels of
antroquinonol
given TID are planned based on dose escalation data and capsules strengths.
The final number of
subjects will be based on the number of dose levels investigated and DLTs
occurrence. Up to 12
subjects may be treated in this part of the study. A Safety Monitoring
Committee (SMC) will be
set up for this study to review DLTs and overall safety data and judge the
relevance of events to
the dose escalation scheme.
[00176] After MTD/MFD determination, enrollment will continue at a
fixed dose of
antroquinonol (MTD or MFD). A maximum of 40 evaluable subjects is planned in
this cohort
expansion part of the trial.
[00177] Investigational product, dosage, and mode of administration
= Escalating doses of Compound 1 (200 to 300 mg) per oral,
TID administration, until unacceptable toxicity or disease progression, and in
the absence of discontinuation criteria.
= 125 mg/m2 nab-paclitaxel and 1000 mg/m2 gemcitabine via
intravenous (IV) infusion, both administered on Days 1, 8, and 15 of each 28-
day
cycle (ie, 1 cycle = weekly for 3 weeks, then 1 week off) until unacceptable
toxicity or disease progression.
[00178] DLT definition and evaluation
[00179] Dose limiting toxicity is defined as the occurrence of any of
the toxicities outlined
below occurring during the DLT evaluation period that are determined to be
related or possibly
related to investigational medicinal products (IMP) combination. The DLT
evaluation period will
be the first 28-day treatment cycle. Toxicity grading will be determined using
NCI CTCAE
version 4.03. Dose limiting toxicities will be defined as:
= Non-hematological toxicity of Grade 3 or greater excluding:
¨ Grade 3 diarrhea, nausea, or vomiting that resolves within 3
days of onset to
39
CA 3020933 2018-10-15
Grade 1 or to baseline Grade with appropriate treatment.
= Grade 4 thrombocytopenia or neutropenia lasting 248 hours
= 2 Grade 3 febrile neutropenia
= Grade 3 thrombocytopenia with bleeding
= Persisting toxicity of Grade 2 or greater that causes more than a 14 day
delay in dose administration.
[00180] AEs that are considered by the Investigator to be related to
the underlying disease
condition, concomitant medication, or to new unrelated medical events or
treatment will not be
defined as a DLT.
[00181] Selection of Study Population
[00182] Inclusion Criteria: To be eligible to participate in the study,
subjects must meet
the following criteria:
1. Male and female subjects >18 years or older.
2. Histologically or cytologically confirmed metastatic ductal
adenocarcinoma of the pancreas, measurable according to the RECIST 1.1.
3. Metastatic disease had to have been diagnosed within 6 weeks before
randomization.
4. Treatment-naïve subjects with metastatic pancreatic ductal
adenocarcinoma who have received no previous systemic therapy (except adjuvant
or
neoadjuvant therapy if progression occurred >6 months from last treatment or
surgery,
respectively, and no prior nab-paclitaxel).
5. Adequate hematologic, hepatic, and renal
function, including: - Hemoglobin >9 g/dL
- Absolute neutrophil count >1500/mm3
- Platelet count >100 000/mm3
- Total bilirubin <1.5 x upper limit of normal (ULN) except subjects
with documented Gilbert's syndrome (>3 x ULN)
- Alanine aminotransferase (ALT) and aspartate aminotransferase
(AST) <2.5 x ULN; for subjects with hepatic metastases, ALT and AST <5 x
ULN
- Albumin >3 mg/dL
CA 3020933 2018-10-15
- Serum creatinine <1.5 mg/dL or calculated creatinine clearance 250
mL/min as determined by the Cockcroft-Gault equation
- ECOG of 0 or 1.
6. For women of childbearing potential, a negative serum
pregnancy test result
at Screening and on Day 1.
7. Willing to use two medically accepted and effective methods of
contraception from
the list below during the study (both men and women as appropriate) and for 3
months after the last dose of study drug:
- Established use of oral, injected, or implanted hormonal methods of
contraception
- Placement of an intrauterine device or intrauterine system
- Barrier methods of contraception: Condom or Occlusive cap
(diaphrag or cervical/vault caps) with spermicidal
foarn/gel/film/cream/suppository
- Male sterilization (with the appropriate postvasectomy documentation
of the absence of sperm in the ejaculate)
- True abstinence: When this is in line with the preferred and usual
lifestyle of the subject.
8. Signed ICF.
9. Life expectancy 2:12 weeks.
[00183]
Exclusion Criteria: Subjects who meet any of the following criteria will not
be
eligible to participate in the study:
1. Islet-cell neoplasms or locally advanced disease.
2. Chemo-, hormone-, or immunotherapy or investigational drug within 4 weeks
or five half-lives of the date of first administration of study drug
(whichever is
shorter) and/or persistence of toxicities of prior anti-cancer therapies which
are
deemed to be clinically relevant.
3. Treatment with any drug(s) known to be a strong inhibitor or inducer of
cytochrome P450 (CYP) 2C19, CYP3A4, CYP2C8, and CYP2E1 within 14 days of
the date of first administration of study drug and during study treatment.
4. Other malignancies diagnosed within the past five years (other than
curatively
treated cervical cancer in situ, nonmelanoma skin cancer, superficial bladder
tumors
41
CA 3020933 2018-10-15
Ta [noninvasive tumor] and TIS [carcinoma in situ], or nonmetastatic prostate
cancer
Stage 1 to 2, which has been previously treated with surgery or radiation
therapy, and
serum prostate-specific antigen is within normal limits [test performed within
the
past 12 months prior to the date of first administration of study drug]).
5. Subjects with any serious active infection (ie, requiring an intravenous
antibiotic, antifungal, or antiviral agent).
6. Subjects with known human immunodeficiency virus, active hepatitis B,
or active hepatitis C.
7. Subjects who have any other life-threatening illness or organ system
dysfunction, which in the opinion of the Investigator, would either compromise
subject safety or interfere with the evaluation of the safety of the study
drug.
8. Known or suspected substance abuse or alcohol abuse.
9. Uncontrolled intercurrent illness, including, but not limited to, ongoing
or active
infection, symptomatic congestive heart failure, uncontrolled hypertension,
unstable
angina pectoris, cardiac arrhythmia, interstitial lung disease, or psychiatric
illness/social situations that would limit compliance with study requirement,
substantially increase risk of incurring AEs from study treatment, or
compromise the
ability of the subject to give written informed consent.
10. Inability to swallow oral medications or a recent acute gastrointestinal
disorder with diarrhea, eg, Crohn's disease, malabsorption, or CTCAE Grade >2
diarrhea of any etiology at baseline.
11. Female subjects who are pregnant or breastfeeding, or male or female
subjects of
reproductive potential who are not employing an effective method of birth
contraception.
[00184] Withdrawal of Subjects: Subjects may withdraw their consent to
participate in
the study and investigators may withdraw subjects at any time without
prejudice. Subjects will be
withdrawn from the study or study treatment if any of the conditions set below
has occurred:
1. Documented disease progression according to the response criteria.
2. Unacceptable toxicity.
3. Subject decides to withdraw his/her informed consent.
4. Investigator considers that the subject is no longer physically and/or
psychologically
able to remain in the study.
42
CA 3020933 2018-10-15
[00185] Replacement Procedures: During the dose escalation phase, a
subject who does
not experience DLTs in the first cycle and who meets any of the following
should be replaced
with new subject:
= All planned trial treatment dosing are not completed in the first 28-day
cycle.
= All planned trial treatment dosing are completed with/without delay;
however,
90% of the planned total dose of either drug is not administered in the first
cycle.
[00186] Guidance to Investigators on When to End Study Treatment
Regimen: Subjects
may withdraw their consent to participate in the study and investigators may
withdraw subjects at
any time. Subjects will be withdrawn from the study or study treatment if any
of the following
conditions are met:
I. Lost to follow-up.
2. Any AE or medical condition that the Investigator or Sponsor determines
may jeopardize the subject's safety if he or she continues in the study.
3. Pregnancy or intent to become pregnant.
4. Subject noncompliance that, in the opinion of the Investigator or
Sponsor, warrants withdrawal from study medication (eg, refusal to adhere to
scheduled visits).
5. Initiation of alternative anti-cancer therapy, including another
investigational agent.
6. Confirmed progressive disease and Investigator determination that
the subject is no longer benefiting from study treatment.
7. The Sponsor terminates the study. Reasons for terminating the study may
include, but are not limited to, the following: the incidence or severity of
AEs in this
or other studies indicates a potential health hazard to subjects; subject
enrollment is
unsatisfactory.
[00187] Follow-up of Subjects Prematurely Discontinued From the Study
Treatment
Regimen or Withdrawn From Study:
[00188] All subjects will attend a safety follow-up visit 28 days after
receiving the last
administration of study treatments. Subjects who discontinue investigational
product prematurely
will be asked to return to the clinic for an early termination visit and may
undergo follow-up
assessments. The primary reason for premature investigational product
discontinuation should be
documented on the appropriate electronic case report form (eCRF).
43
CA 3020933 2018-10-15
[00189] Treatment of Subjects
[00190] Treatments Administered: Compound 1 will be administered in 200-
to 300-mg
doses (planned dose levels), TID, given in 100 mg capsules (resulting a daily
dosage of 600 mg to
900 mg). Additional dose levels may be investigated as determined by the study
SMC. The study
drug will be filled in # 2 capsules (100 mg) and then packed in a light-
protected polyethylene
bottle and closed with a piece of polyethylene cap liner fitted in the cap for
each dispensation at
Visit 1 (Day 0), 3 (Day 28), 4 (Day 42), 5 (Day 56), and 6 (Day 84), and
subsequent visits for
subjects entering the Extension Phase. The study drug will be labeled in
accordance with
country-specific requirements. Several doses of antroquinonol are planned for
the first dose
escalation part of the trial. Dose escalation will follow a 3+3 design based
on DLT occurrence.
The starting dose of antroquinonol will be 200 mg TID, which is the dose
currently investigated in
the ongoing Phase II single-agent trial in NSCLC. If there are no DLTs or <1/
6 subjects with
DLT, the dose will be escalated to 300 mg TID. One dose (MTD or MFD) will be
investigated in
the cohort expansion part of the trial.
[00191] Subjects will receive antroquinonol as long as they are
continuing to show clinical
benefit, as judged by the Investigator, and in the absence of discontinuation
criteria.
Antroquinonol should be taken every 8 hours approximately 15 minutes after a
meal or light
snack and not within 1 hour of drinking an ethanol-containing beverage, eg,
an alcoholic drink.
Subjects who forget or are unable to take a dose at the scheduled time should
be instructed to
take the dose as soon as possible. If they do not remember or are unable to
take the dose prior to
the next scheduled dose, they should take the scheduled dose and the missed
dose will not be
made up. The date and time of each study drug administration should be
recorded in the subject
diary.
[001921 The recommended dose of nab-paclitaxel in combination with
gemcitabine is 125
mg/m2 via IV infusion over 30 minutes on Days 1, 8, and 15 of each 28-day
cycle. The
concurrent recommended dose (RD) of gemcitabine is 1000 mg/m2 via IV infusion
over 30
minutes immediately after the completion of nab-paclitaxel administration on
Days 1, 8, and 15
of each 28-day cycle. Treatment with nab-paclitaxel + gemcitabine will
continue until
unacceptable toxicity or disease progression. Per clinical practice and local
regulation, in case of
subject toxicity, the Day 15 treatment administration can be omitted. In this
case, the start of the
subsequent cycle will begin early, on Day 22 of the first cycle administration
(except for Cycle 1,
the DLT period). The tumor assessment schedule will not be altered (ie,
assessments will
continue to be every 8 weeks).
44
CA 3020933 2018-10-15
[00193] Selection and Timing of Dose for Each Subject: In the dose
escalation part of the
study, subjects will be enrolled and treated sequentially following a 3+3 dose
escalation design.
In the cohort expansion part, subjects will be enrolled and treated in
parallel.
[00194] Efficacy and Safety Variables
[00195] Efficacy Assessments: The RECIST 1.1 criteria will be used to
assess subject
response to treatment by determining ORR, DoR, DCR, PFS, PFS 3 months, and PFS
6 months.
The RECIST 1.1 guidelines for measurable, non-measurable, target, and non-
target lesions and
the objective tumor response criteria (complete response [CR], partial
response [PR], stable
disease [SD], or progressive disease) are presented based on revised response
evaluation criteria
in solid tumors guidelines Version 1.1 ¨ adaptation from original publication
with addition of
supplementary explanations. The OS, OS 6 months, and OS 12 months will also be
evaluated.
[00196] The baseline assessments should be performed no more than 28
days before start of
study treatment and ideally should be performed as close as possible to the
start of study
treatment. Efficacy for all subjects will be assessed by objective tumor
assessments every 8 weeks
for the first 12 months and every 12 weeks thereafter until confirmed
objective disease
progression. If an unscheduled assessment is performed and the subject has not
progressed, every
attempt should be made to perform the subsequent assessments at his or her
scheduled visits.
[00197] A confirmatory scan is required following the initial
demonstration of progressive
disease. The confirmatory scan should occur preferably at the next scheduled
visit and no earlier
than 4 weeks after the initial assessment of progressive disease in the
absence of clinically
significant deterioration. Treatment will continue between the initial
assessment of progression
and confirmation for progression. If a subject discontinues treatment (and/or
receives a
subsequent anti-cancer therapy) prior to progression, then the subject should
still continue to be
followed until confirmed objective disease progression.
[00198] Objective tumor response (CR or PR) should be confirmed
preferably at the next
scheduled visit and not less than 4 weeks after the visit when the response
was first observed.
[00199] Following confirmed progression, subjects should continue to be
followed up for
survival every 2 months (8 weeks). In addition, all subjects will be contacted
in the week
following data cutoff to confirm survival status.
[00200] Safety Assessments: Safety will be monitored throughout the
study for all
subjects. The analysis of the safety data will be performed using the Safety
Analysis Set.
[00201] Adverse events:
[00202] Definitions: The term "adverse event," as used by the Sponsor,
is synonymous
with the term "adverse experience," which is used by the FDA.
CA 3020933 2018-10-15
[00203] An AE is any untoward, undesired, unplanned clinical event in
the form of signs,
symptoms, disease, or laboratory or physiological observations occurring in a
human being
participating in a clinical study with a Sponsor test article, regardless of
causal relationship. This
includes the following:
= Any clinically significant worsening of a pre-existing condition
= Note: Emergence of a new pathogen associated with a clinical event during
therapy at a site other than the initial site of infection will be considered
to be an
AE.
= Any recurrence of a pre-existing condition
= An AE occurring from overdose of a Sponsor study drug whether accidental
or
intentional (ie, a dose higher than that prescribed by a health care
professional for
clinical reasons)
= An AE occurring from abuse of a Sponsor study drug (ie, use for
nonclinical
reasons)
= An AE that has been associated with the discontinuation of the use of a
Sponsor
study drug.
[00204] Note: A procedure is not an AE, but the reason for a procedure
may be an AE.
[00205] A pre-existing condition is a clinical condition (including a
condition being
treated) that is diagnosed before the subject signs the ICF and that is
documented as part of the
subject's medical history.
[00206] The questions concerning whether the condition existed before
the start of the
active phase of the study and whether it has increased in severity and/or
frequency will be used to
determine whether an event is a TEAE. An AE is considered to be treatment-
emergent if (1) it is
not present when the active phase of the study begins and is not a chronic
condition that is part of
the subject's medical history, or (2) it is present at the start of the active
phase of the study or as
part of the subject's medical history, but the severity or frequency increases
during the active
phase. The active phase of the study begins at the time of the first dose of
the study drug. The
active phase of the study ends at the follow-up visit.
[00207] Reporting of Adverse Events: At each visit the Investigator, or
delegate, will
determine whether or not any AEs have occurred. The subject will be questioned
in a general
way and no specific symptoms will be suggested. If any AEs have occurred, they
will be
recorded in the AE section of the eCRF and in the subject's medical records.
If known, the
diagnosis should be recorded, in preference to listing the individual signs
and symptoms.
46
CA 3020933 2018-10-15
[00208] Adverse event reporting begins from the time of informed
consent and ends 30
days after the last dose of IMP.
[00209] Assessment of Severity: The AE severity grading scale for the
NCI CTCAE
(version 4.03) will be used for assessing AE severity. For AEs that are not
specifically listed in
the NCI CTCAE, the following definitions will be used:
= Grade 1: Mild; asymptomatic or mild symptoms; clinical or
diagnostic observations only; or intervention not indicated.
= Grade 2: Moderate; minimal, local, or noninvasive intervention
indicated; or limiting age-appropriate instrumental activities of daily
living.
= Grade 3: Severe or medically significant, but not
immediately life-threatening; hospitalization or prolongation of
hospitalization indicated; disabling; or limiting self-care activities of
daily living.
= Grade 4: Life-threatening consequences or urgent intervention
indicated.
= Grade 5: Death related to AE.
[00210] Relationship to Study Treatment: The Investigator will make a
determination of
the relationship of the AE to the study drug using a four-category system
according to the
following guidelines:
= Unrelated: Clinical event with an incompatible time relationship
to drug administration, and that could be explained by underlying disease or
other drugs or chemicals or is incontrovertibly not related to the study drug.
= Unlikely: Clinical event whose time relationship to drug
administration makes a causal connection improbable, but that could plausibly
be explained by underlying disease or other drugs or chemicals.
= Possible: Clinical event with a reasonable time relationship to study
drug administration, but that could also be explained by concurrent disease or
other
drugs or chemicals.
= Definite: Clinical event with plausible time relationship to study
drug administration, and that cannot be explained by concurrent disease or
other
drugs or chemicals.
[00211] Follow-up of Adverse Events: It is important for the
investigators to take
information of underlying diseases, concomitant drugs, and temporal
relationship of the onset of
the event to the time of dosing the study drug, and re-challenging outcomes,
into account when
47
CA 3020933 2018-10-15
making a causal relation decision. It is the Investigators' responsibility to
follow proactively the
outcome of each AE until resolution or stabilization of the condition,
alternative treatment for
pancreatic cancer is started, 6 months after last dose of study drug, or loss
to follow-up,
whichever occurs first. In the event of serious or study drug-related
toxicities, the subject will be
followed until resolution or stabilization. Safety follow-up data may be
collected by telephone
contact every 3 months after the End of Study Visit.
[00212] Serious Adverse Events: A serious adverse event (SAE) is any AE
occurring at
any dose that meets 1 or more of the following criteria:
= Results in death
= Is life-threatening (see below)
= Requires subject hospitalization or prolongation of an existing
hospitalization (see
below)
= Results in a persistent or significant disability or incapacity (see
below)
= Results in a congenital anomaly or birth defect
= Results in an important medical event (see below).
[00213] Additionally, important medical events that may not result in
death, be life-
threatening, or require hospitalization may be considered SAEs when, based on
appropriate
medical judgment, they may jeopardize the subject and may require medical or
surgical
intervention to prevent one of the outcomes listed above. Examples of such
events include allergic
bronchospasm requiring intensive treatment in an emergency room or at home,
blood dyscrasias
or convulsions that do not require hospitalization, or development of drug
dependency or drug
abuse.
[00214] A life-threatening AE is any AE that places the subject at
immediate risk of
death from the event as it occurred. A life-threatening event does not include
an event that might
have caused death had it occurred in a more severe form but that did not
create an immediate
risk of death as it actually occurred. For example, drug-induced hepatitis
that resolved without
evidence of hepatic failure would not be considered life-threatening, even
though drug-induced
hepatitis of a more severe nature can be fatal. Hospitalization is to be
considered only as an
overnight admission.
[00215] Hospitalization or prolongation of a hospitalization is a
criterion for considering
an AE to be serious. In the absence of an AE, the participating Investigator
should not report
hospitalization or prolongation of hospitalization.
[00216] In addition, a hospitalization planned before the start of the
study for a pre-
existing condition that has not worsened does not constitute an SAE (eg,
elective hospitalization
48
CA 3020933 2018-10-15
for a total knee replacement due to a pre-existing condition of osteoarthritis
of the knee that has
not worsened during the study).
[00217] Disability is defined as a substantial disruption in a person's
ability to conduct
normal life functions. If there is any doubt as to whether a case constitutes
an AE or SAE based
on the information available, the case should be treated as an SAE.
[00218] Alternatively, medical and scientific judgment should be
exercised in deciding
whether a case is serious in those situations where important medical events
may not be
immediately life-threatening or result in death, hospitalization, disability,
or incapacity. These
include events that may jeopardize the subject or may require medical
intervention to prevent one
or more outcomes listed in the definition of serious.
[00219] Pharmacokinetic Analysis
[00220] Noncompartmental PK analyses will be performed on individual
plasma
concentration data. The PK analyses will be performed using commercial
software such as
PhoenixTM WinNonline Version 6.4 or higher (Certara USA, Inc.). Maximum plasma
concentration (Cmax), trough (predose) concentration (Ctrough), and the time
of Cmax (Tmax) will be
taken directly from the observed data. The area under the plasma concentration-
time curve
(AUCT) will be calculated using the linear trapezoidal rule. The PK parameters
will be listed for
each individual and summarized by treatment group using descriptive statistics
(sample size [N],
arithmetic mean, standard deviation, coefficient of variation [CV%], median,
minimum,
maximum, and geometric mean). Individual concentration data will be listed and
summarized by
treatment group with descriptive statistics (N, arithmetic mean, standard
deviation, median,
minimum, maximum, geometric mean, and CV%). Mean and individual plasma
concentration-
time profiles will be presented graphically on both linear and semilogarithmic
scales.
[00221] Actual dose administration times and sample collection times
will be used for the
analyses as recorded on the case report form. Plasma concentrations below the
lower limit of
quantification will be set to zero for the analysis. Pharmacokinetic parameter
estimates will be
three or four significant figures for presentation. No attempt will made to
estimate missing data.
Other parameters and data handling procedures may be added as appropriate.
Only subjects who
are given active antroquinonol and have evaluable concentration-time profiles
will be included in
the analysis. All statistical analyses will use nonrounded parameter
estimates.
[00222] A population PK (PopPK) model development and analysis may be
performed
using mixed-effects methods. Dosing, sampling, demographic, and laboratory
data from each
subject will be assembled into a database suitable for a population analysis.
A nonlinear mixed-
effects model will be developed using NONMEM (Globomax, Ellicot City, MD).
Various
49
CA 3020933 2018-10-15
structural models will be evaluated as indicated by the data. Covariate
effects (age, gender, race,
body size, smoking history, etc.) will be evaluated and incorporated into the
model using
univariate or multivariate approaches. Once the optimal model has been
established, it will be
validated using standard methods, such as visual predictive checks and
bootstrap analyses.
[00223] The potential exposure-exposure/concentration relationships
could be evaluated
based on the post hoc estimates of antroquinonol exposure (ie, model predicted
Cmax, Ctrough, and
AUCt) from the above PopPK model or the concentration observations.
[00224] Statistical Methods
[00225] General Consideration: For study reporting purposes the
following aspects will
be considered:
= Descriptive statistics and graphical presentations will be the main
analysis tools.
= Subject disposition will be reported for each dose level using
the All Subjects Set for both Phases of studies, and subject profiles will be
presented for the dose escalation phase of the study only.
= For all analyses, results, and graphical representation of
individual subject data will be presented by dose level. The dose level to
which
a subject was assigned at the first treatment will be indicated on all
outputs.
[00226] Descriptive statistics and graphical representations will be
used to summarize the
data for each dose level. The following summary statistics will be used to
summarize the trial
data per dose level based on their nature, unless otherwise specified:
= Continuous variables: number of nonmissing observations (N), mean,
standard
deviation, median, minimum, and maximum; 95% CI will be presented where
appropriate.
= Categorical variables: frequencies and percentages.
[00227] Details of the statistical analysis will be presented in the
Statistical Analysis Plan.
[00228] Efficacy Analysis
[00229] Primary Outcome Measures:
[00230] Dose escalation phase: The number and proportion of subjects
experiencing
DTLs in each dose level during Cycle I will be assessed on the DLT Analysis
Set.
[00231] Cohort expansion phase: Descriptive summary of subjects with
best overall
response in each category (CR, PR, SD, and progressive disease); the objective
response rate
(CR+PR); the disease control rate (CR+PR+SD [SD of >16 weeks]) with 95% CIs
according to
RECIST 1.1 will be performed on Efficacy Analysis Set.
CA 3020933 2018-10-15
[00232] Criteria in Solid Tumors (RECIST) version 1.1 will be performed
for median OS
time and PFS time with 95% Cl will be evaluated using Kaplan-Meier methods on
Full Analysis
Set. The OS is defined as the time from first dose of study drug to subject
death. The PFS is
defined as the time from first dose of study drug to the start of disease
progression or subject
death, whichever occurred first.
[00233] Analysis of Secondary Outcome Measures: Data of secondary
endpoints will be
listed by dose level, subject, and visit (if applicable). Secondary endpoints
will be descriptive
analyzed by dose level in the relevant analysis set.
[00234] Safety and Tolerability Analysis: The number and proportion of
subjects
experiencing TEAEs; drug exposure; clinically significant changes in
laboratory parameters, vital
signs, physical examinations, weight, ECOG performance status, ECGs, and/or
pulse oximetry,
judged to be related to the trial medication; and number and reasons of deaths
will be
summarized.
[00235] Safety evaluation will include incidence of AEs (or TEAEs),
laboratory test results,
vital signs, ECG results, and physical examination findings. All summaries of
safety data will be
based on the Safety Analysis Set. No formal statistical analysis of the safety
data will be
performed.
[00236] Summary tables will be presented for all Adverse Effects (AEs)
by dose level and
Medical Dictionary for Regulatory Activities SOC and preferred term where
applicable. The
incidence and type of the following AEs will be analyzed.
[00237] An AE will be considered as "treatment-emergent" if it occurred
after the first
study treatment or if it occurred before the first study treatment and
worsened thereafter.
[00238] The TEAE summary tables will include counts of subjects.
Therefore, if a subject
experiences more than one episode of a particular AE, the subject will be
counted only once for
that event. If a subject has more than one AE that is coded to the same
preferred term, the subject
will be counted only once for that preferred term. Similarly, if a subject has
more than one AE
within an SOC, the subject will be counted only once in that SOC.
[00239] All deaths and deaths within 30 days of the last dose of study
treatment as well as
reasons for death will be tabulated by dose level. Drug exposure will be
summarized by dose
level.
[00240] Clinically significant laboratory test variables will be
summarized by dose level
and visit (if applicable) using descriptive statistics (number of subjects,
mean, standard deviation,
minimum, maximum, as well as mean change from baseline, standard deviation for
mean and
standard error for mean change, minimum, median, maximum, and number and
percent of
51
CA 3020933 2018-10-15
subjects within specified categories). Shift tables (ie, cross-tabulations of
below the lower limit
of the normal range, within the limits of the normal range and above the upper
limit of the
normal range at baseline versus scheduled visits) will be presented by
laboratory test (if
applicable). Laboratory tests with categorical results that cannot be analyzed
by change from
baseline or shift table analysis will not be included in these summaries, but
will be listed. Data
obtained from laboratory tests not required by the protocol will not be
summarized, but will be
listed.
[00241] Descriptive statistics of vital signs, weight and ECG results
at each visit will be
presented by treatment group. Physical examination findings will be listed for
each subject. The
overall response rate and the DCR will be estimated along with 95% Cls
calculated using the
method of Clopper and Pearson by dose level.
[00242] Pharmacokinetic Analysis
[00243] Pharmacokinetic Sampling: The PK sampling will be performed on
Day 0 and
Day 28 in all subjects enrolled in the first stage at the following time
points:
= Day 0: (Approximately 5 mL per sample, 60 mL in total) 30
minutes prior to and 0.5, 1, 2, 3, 4, 6, and 8 hours after the first dose.
= Day 28: (Approximately 5 mL per sample, 60 mL in total)
immediately before and 0.5, 1, 2, 3, 4, 6, and 8 hours after the first dose on
Day 28.
1002441 Sparse PK sampling will be performed on Days 28, 42, and 56 in
all subjects
enrolled in Stage 2. At least two samples will be collected on each occasion,
one of which will be
a trough concentration (30 minutes prior to the first dose on Days 28, 42, and
56 and
approximately 8 hours after the last dose on the prior day). At least one
sample per subject will
be timed to coincide with the peak concentration (3 hours after the first
dose). The remainder
may be taken at any time during the dosing interval.
[00245] Each blood sample will be analyzed for antroquinonol plasma
concentration to
determine PK parameters after administration of antroquinonol using a fully
validated
bioanalytical method (a copy of the analytical method validation report and
bioanalytical report
will be included in the clinical study report).
[00246] Pharmacokinetic Sampling Procedures
[00247] Blood Samples: Samples of venous blood will be obtained in 5 mL
sodium heparin
Vacutainere tubes at the pre-scheduled sample times. Immediately after
collection the tube will
be gently inverted 8 to 10 times to mix the anticoagulant with the blood
sample. All samples will
be processed and placed into a freezer within one (1) hour after collection.
The plasma fraction
will be separated by placing the collection tube into a refrigerated
centrifuge (4 C) for 10 minutes
52
CA 3020933 2018-10-15
at 3000 rpm. The plasma fraction will be withdrawn by pipette and divided into
two
polypropylene freezing tubes (with each tube receiving approximately equal
aliquots). All sample
collection and freezing tubes will be clearly labeled with the subject number,
the study period,
and the collection time. Labels will be fixed to freezing tubes in a manner
that will prevent the
label from becoming detached after freezing. All plasma samples will be placed
into a freezer at -
70 C within 1 hour after collection.
[00248] Analytical Methodology: The concentration of study drug will be
determined from
the plasma samples using a validated analytical method. Details of the method
validation and
sample analysis will be included in the final clinical study report.
[00249] Preliminary Results:
[00250] The preliminary results from the patients with 600 mg daily
dosage shows that the
patient tumor size (mm) reduced after the completion of the 3rd cycle (8 weeks
treatment). One
patient's tumor size reduced 7.6% from 91 mm to 84 mm, and the other patient's
tumor size
reduced 22% from 50 mm to 39 mm. The preliminary tumor size reduction further
supports the
superior unexpected benefits of the invention compositions comprising the
exemplary Compound
1 and the current clinical chemotherapy drugs such as paclitaxel and
gemcitabine against
pancreatic cancer as shown in the animal study.
[00251] While preferred embodiments of the present invention have been
shown and
described herein, it will be obvious to those skilled in the art that such
embodiments are provided
by way of example only. Numerous variations, changes, and substitutions will
now occur to
those skilled in the art without departing from the invention. It should be
understood that various
alternatives to the embodiments of the invention described herein may be
employed in practicing
the invention. It is intended that the following claims define the scope of
the invention and that
methods and structures within the scope of these claims and their equivalents
be covered thereby.
53
CA 3020933 2018-10-15