Note: Descriptions are shown in the official language in which they were submitted.
1 9 CA 03030392 2019-01-09
1
DESCRIPTION
SOLID PREPARATION HAVING IMPROVED LIGHT STABILITY
Technical Field
[0001]
The present invention relates to a solid preparation with improved
photostability, which comprises a 4,5-epoxymorphinan derivative or a
pharmaceutically acceptable acid addition salt thereof.
Background Art
[0002]
Nalfurafine and pharmaceutically acceptable acid addition salts thereof,
which are known to be compounds with remarkable antipruritic effects, are
chemically unstable to, for example, heat, light, and oxygen. Thus, a method
of
improving the chemical stability thereof has been conventionally developed.
[0003]
Specifically, a method in which a substance selected from particular
antioxidants, synergists, saccharides and surfactants is added, and a method
in which
sodium thiosulfate, saccharides, or sugar alcohols and low substituted
hydroxypropyl
cellulose are added have been reported (Patent Documents 1 and 2).
[0004]
The preparation described in Patent Document 1 contains a substance
selected from particular antioxidants, synergists, saccharides and surfactants
to
improve the stability of nalfurafine or a pharmaceutically acceptable acid
addition
salt thereof, and tablets and granules are described as specific dosage forms
for the
solid preparation. Additionally, the solid preparation described in Patent
Document
2 contains sodium thiosulfate, saccharides, or sugar alcohols and low
substituted
hydroxypropyl cellulose as stabilizing agents to improve the stability of
nalfurafine
= CA 03030392 2019-01-09
2
or a pharmaceutically acceptable acid addition salt thereof.
[0005]
In addition, a capsule filler composition prepared by adding a medium-chain
fatty acid triglyceride and propyl gallate to a liquid filler for soft capsule
preparations,
and a soft capsule preparation containing a particular matrix and an
antioxidant are
reported as formulations to stabilize nalfurafine or a pharmaceutically
acceptable
acid addition salt thereof in soft capsule preparations (Patent Document 3 and
4).
[0006]
Specifically, the soft capsule preparations described in Patent Document 3
and 4 contain an antioxidant such as propyl gallate as a stabilizing agent to
improve
the stability of nalfurafine or a pharmaceutically acceptable acid addition
salt thereof.
[0007]
The orally disintegrating tablet described in Patent Document 5 ensures
excellent photostability by applying a coating being composed of a polyvinyl
alcohol-based resin and a particular saccharide and containing a light-
shielding agent
on an orally disintegrating tablet containing nalfurafine or a
pharmaceutically
acceptable acid addition salt thereof.
[0008]
Meanwhile, a light-shielding coating agent containing a polyvinyl
alcohol-based resin and a particular saccharide is reported as a formulation
to
improve the photostability of a drug unstable to light, which contains
nalfurafine or a
pharmaceutically acceptable acid addition salt thereof in solid preparations
(Patent
Document 5).
[0009]
In addition, various methods to stabilize the drug unstable to light are known
in relation to the formulation of those drugs. As methods to improve the
photostability of amlodipine, which is unstable to light, in uncoated tablets,
for
CA 03030392 2019-01-09
3
example, a method in which ferric oxide is added, and a method in which a
dissolved
substance having the similar light absorption behavior to that of the active
ingredient
to be protected from light is added have been reported (Patent Documents 6 and
7).
[0010]
The preparation described in Patent Document 6 is a preparation whose
photostability is improved by combining amlodipine, which is unstable to
light, with
ferric oxide. In the method described in Patent Document 7 to improve
photostability, a dissolved substance having the similar light absorption
behavior to
that of the active ingredient to be protected from light is added to improve
photostability.
Prior Art Documents
Patent Documents
[0011]
Patent Document 1: W099/002158
Patent Document 2: W008/133330
Patent Document 3: JP 2015-168630 A
Patent Document 4: JP 2015-172043 A
Patent Document 5: W010/113841
Patent Document 6: JP 2006-306754 A
Patent Document 7: JP S58-57322 A
Summary of the Invention
Problems to be Solved by the Invention
[0012]
However, no study is conducted in Patent Document 1 on the improvement
of the photostability of nalfurafine or a pharmaceutically acceptable acid
addition salt
thereof, though an improvement in heat and oxidation stability has been
indicated by
data.
1
CA 03030392 2019-01-09
4
[0013]
Also, no study is conducted in Patent Document 2 on the improvement of
the photostability of nalfurafine or a pharmaceutically acceptable acid
addition salt
thereof though an improvement in heat and oxidation stability has been
indicated by
data, and only a commonly used light shielding coating for tablets is applied.
[0014]
In Patent Documents 3 and 4, the stability thereof against heat and oxidation
during storage is indicated by data but there is neither description nor
suggestion on
the improvement of photostability.
[0015]
As seen above, the techniques described in Patent Documents 1 to 4 to
stabilize nalfurafine or a pharmaceutically acceptable acid addition salt
thereof are
methods for stabilization of preparations during production and/or long-term
storage,
and no study has been conducted on the improvement of photostability.
[0016]
When the orally disintegrating tablet described in Patent Document 5 is split
for proper dosing, an uncoated surface appears in each split surface of the
tablet and
deterioration of the tablet, such as degradation or color change, due to
exposure to
light may proceed from the uncoated surfaces. A large amount of coating is
required to ensure sufficient photostability for solid preparations in powder
or
granule form, which may result in decreased stability during production and/or
long-term storage or may cause the production process to be complicated.
[0017]
For the preparation described in Patent Document 6, addition of
butylhydroxyanisole and dibutylhydroxytoluene as stabilizing agents to inhibit
oxidative degradation is described; the Patent Document describes that these
antioxidants reduce the production of degradation products from amlodipine due
to
84981008
its oxidation but do not inhibit color change at all.
[0018]
In the method described in Patent Document 7 to improve photostability, there
is no
description on nalfurafine or a pharmaceutically acceptable acid addition salt
thereof, and different
5 methods to improve photostability are effective for different active
ingredients.
[0019]
Thus, an object of the present invention is to provide a solid preparation
comprising a
4,5-epoxymorphinan derivative or a pharmaceutically acceptable acid addition
salt thereof, which is
stable to light even without light shielding coating.
Means for Solving the Problem
[0020]
The inventors intensively studied to solve the above problems and consequently
arrived at
the following invention. That is, the present invention relates to the
following inventions (1) to (5).
(1) A solid preparation comprising an active ingredient composed of a
4,5-epoxymorphinan
derivative represented by the general formula (I) below or a pharmaceutically
acceptable acid
addition salt thereof, and one or more stabilizing agents selected from the
group consisting of
n-propyl gallate, sodium sulfite, dibutylhydroxytoluene, butylhydroxyanisole,
tocopherol and
D-isoascorbic acid, wherein the solid preparation is in a form other than a
soft capsule, wherein the
active ingredient is formulated as a solid, wherein the amount of the active
ingredient is 0.00001 to
0.01% by weight of the solid preparation, and wherein the amount of the
stabilizing agent is 0.005 to
5% by weight of the solid preparation
Date Regue/Date Received 2022-06-14
CA 03030392 2019-01-09
6
R2
8
R5
=
b (I)
[wherein RI represents cyclopropylmethyl or allyl; R2 represents hydrogen,
hydroxy, acetoxy, or methoxy; R3 represents hydrogen, hydroxy, acetoxy, or
methoxy; A represents -N(R4 )C(---0)- or -N(R4)C(-----0)0-; R4 represents
hydrogen or
a C1_5 linear or branched alkyl; B represents a CI-3 linear alkylene, -CH=CH-,
or
R5 represents hydrogen, phenyl, furyl, or thienyl, provided that a hydrogen(s)
in the above phenyl, the above furyl and the above thienyl is/are optionally
substituted with one or more groups selected from the group consisting of C1_5
alkyl,
C1.5 alkoxy, C1_5 alkanoyloxy, hydroxy, fluorine, chlorine, bromine, iodine,
amino,
nitro, cyano, isothiocyanato, trifluoromethyl, trifluoromethoxy and
methylenedioxy].
(2) The solid preparation according to (1), wherein the stabilizing agent
is
n-propyl gallate.
(3) The solid preparation according to (1) or (2), which contains sodium
thiosulfate.
(4) The solid preparation according to any of (1) to (3), which contains
yellow
ferric oxide, red ferric oxide, or black iron oxide.
(5) The solid preparation according to any of (1) to (4), which contains a
carbohydrate.
(6) The solid preparation according to any of (1) to (5), which is in a
dosage
form selected from the group consisting of tablet, granule, fine granule, hard
capsule,
dry syrup, powder, pill and troche.
Additionally, the present invention relates to the following inventions (7) to
(11).
CA 03030392 2019-01-09
7
(7) A solid preparation comprising an active ingredient composed of a
4,5-epoxymorphinan derivative represented by the general formula (I) below or
a
pharmaceutically acceptable acid addition salt thereof, a carbohydrate, and
one or
more stabilizing agents selected from the group consisting of n-propyl
gallate,
sodium hydrogensulfite, dibutylhydroxytoluene, butylhydroxyanisole, tocopherol
and D-isoascorbic acid, wherein the amount of the above active ingredient is
0.00001
to 0.01% by weight of the solid preparation, and the amount of the above
stabilizing
agent is 0.005 to 5% by weight of the solid preparation
R2
R1 N .
s'
-7
0 (I)
0111] R3
[wherein RI represents cyclopropylmethyl or allyl; R2 represents hydrogen,
hydroxy, acetoxy, or methoxy; R3 represents hydrogen, hydroxy, acetoxy, or
methoxy; A represents -N(R4)C(=0)- or -N(R4)C(=0)0-; R4 represents hydrogen or
a C1-5 linear or branched alkyl; B represents a C1.3 linear alkylene, -0-1=CH-
, or
R5 represents hydrogen, phenyl, furyl, or thienyl, provided that a hydrogen(s)
in the above phenyl, the above furyl and the above thienyl is/are optionally
substituted with one or more groups selected from the group consisting of C
1_5 alkyl,
C1_5 alkoxy, C1_5 alkanoyloxy, hydroxy, fluorine, chlorine, bromine, iodine,
amino,
nitro, cyano, isothiocyanato, trifluoromethyl, trifluoromethoxy and
methylenedioxy].
(8) The solid preparation according to (7), wherein the above stabilizing
agent
is n-propyl gallate.
(9) The solid preparation according to (7) or (8), which contains sodium
thiosulfate.
(10) The solid preparation according to any of (7) to (9), which contains
yellow
= 1
CA 03030392 2019-01-09
8
ferric oxide, red ferric oxide, or black iron oxide.
(11) The solid preparation according to any of (7) to (10),
which is in a dosage
form selected from the group consisting of tablet, granule, fine granule, hard
capsule,
dry syrup, powder, pill and troche.
Effects of the Invention
[0021]
By the present invention, the photostability of a solid preparation containing
a 4,5-epoxymorphinan derivative or a pharmaceutically acceptable acid addition
salt
thereof is improved, which can increase the usefulness of the solid
preparation as a
pharmaceutical product.
Mode for Carrying Out the Invention
[0022]
The mode to practice the present invention will now be described.
However, the present invention is not limited to the following modes. Unless
otherwise clearly indicated, the symbol "%" refers to "% by weight".
[0023]
The solid preparation according to the present invention is a pharmaceutical
product containing an active ingredient which is formulated as a solid, and
examples
of the solid preparation include tablets (including sublingual tablets, orally
disintegrating tablets and mini-tablets), hard capsules, granules, fine
granules,
powders, dry syrups, pills, troches and film preparations. In particular, the
present
invention can conveniently allow tablets to ensure photostability without
forming a
light-shielding coating film layer and thus to disintegrate quickly in oral
cavity when
the present invention is applied to orally disintegrating tablets.
Additionally, the
present invention can also conveniently ensure photostability without
following a
complex production process to form a uniform coating film layer containing a
light-shielding agent on the outer surface of solid preparations in powder
form when
=
CA 03030392 2019-01-09
9
the present invention is applied to granules, fine granules, powders, dry
syrups and
film preparations.
[0024]
The active ingredient according to the present invention is a
4,5-epoxymorphinan derivative represented by the general formula (I) below or
a
pharmaceutically acceptable acid addition salt thereof
R2
'N I
6,
- (I)
R3
[wherein RI represents cyclopropylmethyl or ally!; R2 represents hydrogen,
hydroxy, acetoxy, or methoxy; R3 represents hydrogen, hydroxy, acetoxy, or s
methoxy; A represents -N(R4)C(=0)- or -N(R4)C(=0)0-; R4 represents hydrogen or
a C1..5 linear or branched alkyl; B represents a C _3 linear alkylene, -CH=CH-
, or
-CC-; R5 represents hydrogen, phenyl, fury!, or thienyl, provided that a
hydrogen(s)
in the above phenyl, the above furyl and the above thienyl is/are optionally
substituted with one or more groups selected from the group consisting of C1_5
alkyl,
Ch5 alkoxy, C1_5 alkanoyloxy, hydroxy, fluorine, chlorine, bromine, iodine,
amino,
nitro, cyano, isothiocyanato, trifluoromethyl, trifluorornethoxy and
methylenedioxy).
[0025]
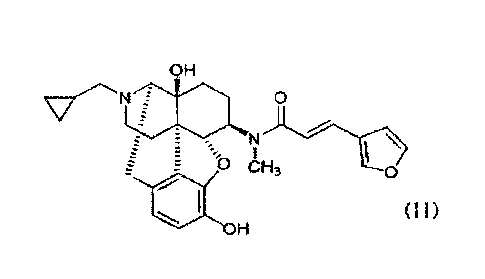
The hydrochloride salt of the compound represented by the general formula
(II) below, which is 17-(cyclopropylmethyI)-3,1413-dihydroxy-4,5a-epoxy-6fl-
[N-methyl-trans-3-(3-furyl)acrylamide]morphinan (hereinafter referred to as
"nalfurafine"), is particularly preferred as the above 4,5-epoxymorphinan
derivative
represented by the general formula (I) or a pharmaceutically acceptable salt
thereof.
CA 03030392 2019-01-09
OH
0
N
\
E 0 CH3 0
411 (ii)
= I-I
[0026]
Examples of the pharmaceutically acceptable acid addition salt include
inorganic acid salts such as hydrochloride, sulfate, nitrate, hydrobromide,
5 hydroiodide, and phosphate; organic carboxylates such as acetate,
lactate, citrate,
oxalate, glutarate, malate, tartrate, fumarate, mandelate, maleate, benzoate,
and
phthalate; organic sulfonates such as methanesulfonate, ethanesulfonate,
benzenesulfonate, p-toluenesulfonate, and camphorsulfonate. Among those, the
hydrochloride, hydrobromide, phosphate, tartrate, maleate, and
methanesulfonate are
10 preferred; the hydrochloride which is commercially available is most
preferred.
[0027]
The amount of the active ingredient of the present invention, which is the
above 4,5-epoxymorphinan derivative represented by the general formula (I) or
a
pharmaceutically acceptable acid addition salt thereof, is preferably from
0.00001 to
0.01% by weight, more preferably from 0.00005 to 0.01% by weight, further
preferably from 0.00025 to 0.01% by weight, of the solid preparation. In cases
where the amount of the active ingredient is more than 0.01% by weight, a
solid
preparation with sufficient photostability is obtained without using a
stabilizing agent.
In cases where the amount of the active ingredient is less than 0.00001% by
weight, a
larger dose of the preparation is required to attain therapeutic effects.
[0028]
The content range of the active ingredient in percent by weight varies
depending on the amount of the solid preparation, and the active ingredient in
an
CA 03030392 2019-01-09
11
amount ranging from 0.01 I.A.g to 50 Itg is typically contained in a daily
dose of the
solid preparation.
[0029]
One or more stabilizing agents selected from the group consisting of
n-propyl gallate, sodium hydrogensulfite, dibutylhydroxytoluene,
butylhydroxyanisole, tocopherol and D-isoascorbic acid are used; preferably
n-propyl gallate and/or tocopherol, more preferably n-propyl gallate, are/is
used as a
stabilizing agent(s) in the present invention.
[0030]
Commercially available products may be used as the stabilizing agents.
n-Propyl gallate is commercially available under various names such as propyl
gallate, gallic acid propyl ester, and 3,4,5-trihydroxybenzoic acid propyl
ester.
Dibutylhydroxytoluene is commercially available under various names such as
BHT,
2,6-di-tert-butyl-p-cresol, 3,5-di-tert-butyl-4-hydroxytoluene, and
2,6-di-tert-butyl-4-methylphenol. Butylhydroxyanisole is commercially
available
under various names such as BHA, 3-t-butyl-4-hydroxyanisole, and
tert-butyl-4-methoxyphenol. Tocopherol is commercially available under various
names such as dl-a-tocopherol, d-a-tocopherol, d-8-tocopherol, and vitamin E.
[0031]
The amount of the stabilizing agent(s) used in the present invention is from
0.005 to 5% by weight, preferably from 0.005 to 1% by weight, of the solid
preparation. In cases where the amount of the stabilizing agent(s) is less
than
0.005% by weight of the solid preparation, a sufficient photostabilizing
effect is not
obtained. In cases where the amount of the stabilizing agent(s) is more than
5% by
weight of the solid preparation, the amount is above the maximum daily dose of
the
stabilizing agent(s) which is confirmed to be safe, which amount is thus not
preferred.
CA 03030392 2019-01-09
12
[0032]
The solid preparation according to the present invention preferably further
contains a carbohydrate. Examples of the carbohydrate include saccharides or
sugar alcohols, and commercially available carbohydrates may be used. Examples
of the carbohydrates include potato starch, sucrose, lactose, mannitol,
erythritol,
maltose, maltitol, trehalose, sorbitol, xylitol, lactitol and glucose, and the
preferred
are lactose, erythritol and mannitol. The use of a carbohydrate can improve
the
storage stability of the above 4,5-epoxymorphinan derivative represented by
the
general formula (I) or a pharmaceutically acceptable acid addition salt
thereof, as
well as the sweetness of the carbohydrate improves drug compliance.
[0033]
The solid preparation according to the present invention may further contain
an antioxidant to prevent degradation of the active ingredient during
production
and/or long-term storage. Examples of such an antioxidant include sodium
thiosulfate.
[0034]
The solid preparation according to the present invention may contain
various additives used in the production of common formulations, in addition
to the
above-described ingredients. Examples of such additives include excipients,
disintegrators, binders, lubricants, coating agents, fluidizing agents, taste
masking
agents, flavoring agents, coloring agents and sweeteners.
[0035]
Examples of the disintegrator include crospovidone, croscarrnellose sodium,
carrnellose calcium, sodium carboxymethyl starch and low substituted
hydroxypropyl cellulose.
[0036]
Examples of the binder include water-soluble polysaccharides, such as
=
CA 03030392 2019-01-09
13
gelatin, pullulan, carrageenan, xanthan gum, tamarind gum, pectin, sodium
alginate
and gum arabic; celluloses, such as hydroxypropyl cellulose, hydroxypropyl
methylcellulose and methyl cellulose; starches, such as alpha starch and
gelatinized
starch; and synthetic polymers, such as polyvinylpyrrolidone, carboxy vinyl
polymer
and polyvinyl alcohol.
[0037]
Examples of the lubricant include magnesium stearate, calcium stearate,
sodium stearyl fumarate, talc, sucrose fatty acid esters, stearic acid,
aluminium
stearate, potassium sodium tartrate, light anhydrous silicic acid, camauba
wax,
carmellose calcium, carmellose sodium, hydrated silicon dioxide, hydrogenated
oil
and hydrogenated rapeseed oil.
[0038]
Examples of the coating agent include hydroxypropyl methylcellulose, ethyl
cellulose, sodium carboxymethyl ethylcellulose and polyvinyl alcohol.
[0039]
Examples of the fluidizing agent include talc, hydrated silicon dioxide and
light anhydrous silicic acid.
[0040]
Examples of the taste masking agent include glutamic acid, fumaric acid,
succinic acid, citric acid, sodium citrate, tartaric acid, malic acid,
ascorbic acid,
sodium chloride and menthol.
[0041]
Examples of the flavoring agent include orange, vanilla, strawberry, or
yogurt flavorings and menthol.
[0042]
Examples of the coloring agent include food colorants, such as titanium
oxide, ferric oxide, yellow ferric oxide, black iron oxide, talc, food red No.
3, food
CA 03030392 2019-01-09
14
yellow No. 5 and food blue No. 1; and riboflavin.
[0043]
Examples of the sweetener include aspartame, saccharin, dipotassium
glycyrrhizinate and stevia.
[0044]
In the present invention, the solid preparation can be manufactured by a wet
granulation process which comprises the steps of dissolving or suspending the
active
ingredient in water or a pharmaceutically acceptable solvent, and adding the
obtained
liquid (solution or suspension) to a carbohydrate. Additionally, the
stabilizing
agent(s) may be added in solid or liquid form at any process steps.
[0045]
The method of adding the stabilizing agent(s) in solid form is not limited,
and examples of the method include methods in which the commercially available
stabilizing agent(s) is/are pulverized as necessary before mixing, and methods
in
which the stabilizing agent(s) suspended in water, an alcohol such as ethanol
or
methanol, or a mixed solution thereof is/are added.
[0046]
The method of adding the stabilizing agent(s) in liquid form is not limited,
and examples of the method include methods in which the stabilizing agent(s)
and
the active ingredient are together dissolved in water or a pharmaceutically
acceptable
solvent and then added to a carbohydrate, and methods in which the active
ingredient
is added to a carbohydrate, the resultant is then subjected to an appropriate
granulation or size selection step, and the stabilizing agent(s) is/are
subsequently
added to the resulting granules. Additionally, the whole amount of the
carbohydrate
may be used at the above-described step of adding the active ingredient, or a
partial
amount of the carbohydrate may be used at the above-described step and the
remaining amount of the carbohydrate may be added at any of the subsequent
steps.
CA 03030392 2019-01-09
[0047]
Commonly used apparatuses are used in the wet granulation process,
including, for example, fluid bed granulator, tumbling fluid bed granulator,
rotating
granulator, cylindrical extrusion granulator, or wet-type extrusion
granulator. In
5 cases where water is used as the solvent to dissolve or suspend the
active ingredient,
a fluid bed granulator or tumbling fluid bed granulator which is capable of
spray
drying is preferred. In cases where a volatile solvent such as ethanol is used
as the
solvent to dissolve or suspend the active ingredient, a fluid bed granulator,
tumbling
fluid bed granulator, or rotating granulator is preferred.
10 [0048]
In cases where the stabilizing agent(s) in solid form is/are added, a
commonly used mixer is used, including, for example, V-type mixer, ribbon
mixer, or
air blender.
[0049]
15 In addition, the addition of yellow ferric oxide, red ferric oxide,
or black
iron oxide as a coloring agent to the solid preparation can further improve
the
photostability of the solid preparation. The method of adding the coloring
agent is
not limited, and the coloring agent can be added in powder form or as a
suspension in
water or a pharmaceutically acceptable solvent.
[0050]
In cases where the solid preparation is in tablet form, commonly used
apparatuses are used for compression molding, including, for example, single
punch
tableting machine or rotary tableting machine. The molding pressure during the
compression process is not limited to a particular pressure as long as the
resulting
tablets are hard enough not to cause problems in handling.
[0051]
The photostability of the active ingredient is improved in the solid
CA 03030392 2019-01-09
16
preparation according to the present invention, which may thus be easily
handled
during drug preparation or drug dosing. In the present invention, the improved
stability means that the above 4,5-epoxymorphinan derivative represented by
the
general formula (1) or a pharmaceutically acceptable acid addition salt
thereof is
maintained in the preparation at a residual rate of not less than 90%, as
described in
W016/052617. The residual rate of the active ingredient is not less than 90%
after
period of at least 24 hours (an overall illumination of 48 thousand lux-hr),
more
preferably not less than 90% after a period of 300 hours (an overall
illumination of
600 thousand lux-hr), when the solid preparation according to the present
invention is
handled at a temperature of 25 C and a relative humidity of 51% RI-I under
white
fluorescent light (with an illumination of 2000 lux) without being packed.
Examples
[0052]
The present invention will be now described by way of examples to
illustrate advantageous effects of this invention. However, the present
invention is
not limited to those examples.
[0053]
(Comparative Example 1)
Into a fluid bed granulator (LAB-I; manufactured by Powrex Corporation),
mannitol (88.8975 parts by weight (hereinafter abbreviated to "parts" unless
otherwise particularly stated) of PEARLITOLO 200SD; manufactured by Roquette
Japan K.K.) was introduced and an aqueous solution of 0.0025 parts of
nalfurafine
hydrochloride was sprayed to produce granules. Then, those granules were mixed
with 10 parts of low substituted hydroxypropyl cellulose (LB-11; manufactured
by
Shin-Etsu Chemical Co., Ltd.) and 1 part of magnesium stearate (manufactured
by
Taihei Chemical Industrial Co., Ltd.) to obtain granules for tablet
compression. The
granules for tablet compression were compressed using a tablet press (Correct
19;
A
CA 03030392 2019-01-09
17
manufactured by Kikusui Seisakusho Ltd.) into tablets with a weight of 99.9 mg
and
a diameter of 7 mm.
[0054]
(Example 1)
Into a fluid bed granulator (LAB-1; manufactured by Powrex Corporation),
mannitol (88.8975 parts of PEARLITOL 200SD; manufactured by Roquette Japan
K.K.) was introduced and an aqueous solution of 0.0025 parts of nalfurafine
hydrochloride was sprayed to produce granules, to which an ethanol solution of
0.1
parts of n-propyl gallate (Wako 1st Grade; manufactured by Wako Pure Chemical
Industries, Ltd.) was added, and the resulting mixture was stirred in a
mortar. After
dried at 40 C for 16 hours in a hot air dryer, the mixture was mixed with 10
parts of
low substituted hydroxypropyl cellulose and 1 part of magnesium stearate to
obtain
granules for tablet compression. The granules for tablet compression were
compressed using a tablet press into tablets with a weight of 100 mg and a
diameter
of 7 mm.
[0055]
(Example 2)
Tablets with a weight of 100 mg and a diameter of 7 mm were obtained in
the same manner as in Example 1, except that 0.1 parts of dl-a-tocopherol
(Guaranteed Reagent; manufactured by Kanto Chemical Co., Inc.) were used
instead
of 0.1 parts of n-propyl gallate in Example I.
[0056]
(Example 3)
Tablets with a weight of 100 mg and a diameter of 7 mm were obtained in
the same manner as in Example 1, except that 0.1 parts of 3-t-butyl-4-
hydroxyanisole
(BHA) (Extra Pure; manufactured by Nacalai Tesque, Inc.) were used instead of
0.1
parts of n-propyl gallate in Example 1.
=
CA 03030392 2019-01-09
18
[0057]
(Example 4)
Tablets with a weight of 100 mg and a diameter of 7 mm were obtained in
the same manner as in Example 1, except that 0.1 parts of 2,6-di-t-butyl-p-
cresol
(BHT) (Extra Pure Reagent; manufactured by Nacalai Tesque, Inc.) were used
instead of 0.1 parts of n-propyl gallate in Example 1.
[0058]
(Example 5)
Tablets with a weight of 100 mg and a diameter of 7 mm were obtained in
the same manner as in Example 1, except that an aqueous solution of 0.1 parts
of
D-isoascorbic acid (Guaranteed Reagent; manufactured by Nacalai Tesque, Inc.)
was
used instead of the ethanol solution of 0.1 parts of n-propyl gallate in
Example 1.
[0059]
(Example 6)
Tablets with a weight of 100 mg and a diameter of 7 mm were obtained in
the same manner as in Example 1, except that an aqueous solution of 0.1 parts
of
sodium sulfite (Guaranteed Reagent; manufactured by Wako Pure Chemical
Industries, Ltd.) was used instead of the ethanol solution of 0.1 parts of n-
propyl
gallate in Example 1.
[0060]
(Comparative Example 2)
Tablets with a weight of 100 mg and a diameter of 7 mm were obtained in
the same manner as in Example 1, except that an aqueous solution of 0.1 parts
of
sodium thiosulfate pentahydrate (Guaranteed Reagent; manufactured by Kokusan
Chemical Co., Ltd.) was used instead of the ethanol solution of 0.1 parts of n-
propyl
gallate in Example 1.
[0061]
=
CA 03030392 2019-01-09
19
(Comparative Example 3)
Tablets with a weight of 100 mg and a diameter of 7 mm were obtained in
the same manner as in Example 1, except that an aqueous solution of 0.1 parts
of
sodium hydrogensulfite (Guaranteed Reagent; manufactured by Wako Pure Chemical
Industries, Ltd.) was used instead of the ethanol solution of 0.1 parts of n-
propyl
gallate in Example 1.
[0062]
(Test Example 1: Photostability Test on Tablets)
The tablets of Examples 1 to 6 and Comparative Examples 1 to 3 were
arranged on glass dishes such that no tablets piled upon each other, and the
glass
dishes were exposed under white fluorescent light (with an illumination of
2000 lux)
to an overall illumination of 48 thousand lux-hr and to an overall
illumination of 600
thousand lux-hr, and the tablets were then collected from the glass dishes.
Addidonally, glass dishes on which the tablets of Example 1 and Comparative
Example 1 were arranged were covered with aluminium foil (for storage in the
dark)
and exposed to an overall illumination of 48 thousand lux-hr, and the tablets
were
then collected from the glass dishes. The tablets were analyzed by the
following
HPLC analysis to calculate the residual rates of the active ingredient.
<Pretreatment Conditions>
A mixed solution of 25 mM phosphate buffer and methanol (40/60; v/v) was
added to the tablets to disintegrate the tablets, and the resulting mixture
was stirred
and then centrifuged to collect the supernatant as a HPLC sample.
<HPLC Conditions>
Mobile phase: 25 mM phosphate buffer (pH 7.0) / acetonitrile = 60/40 (v/v);
Column: "Capcell Pak*" MGII (3.0 x 150 mm in size; manufactured by
Shiseido Co., Ltd.);
Column temperature: 40 C;
CA 03030392 2019-01-09
Detection wavelength: 280 nm;
Injection volume: 100 ?AL.
[0063]
The residual rate of the active ingredient after exposure to light was
5 calculated with the following Formula 1.
Residual rate (%) = (The area value of the HPLC peak of the active
ingredient in the sample after exposure to light / the area value of the HPLC
peak of
the active ingredient in the sample before exposure to light) x 100 ¨
Formula 'I
[0064]
IO The ingredients in the tablets of Examples 1 to 6 and Comparative
Examples
1 to 3, the contents of the respective ingredients in percent by weight of
each solid
preparation, and the residual rates of the active ingredient after exposure to
light
obtained as the results of the HPLC analysis are shown in Table 1.
[0065]
[Table 1]
Comparative Comparative Comparative Example Example Example Example Example
Example .
Example 1 Example 2 Example 3 1 2 3
4 5 6
_
Tablet Tablet Tablet Tablet Tablet
Tablet Tablet Tablet Tablet
Active Nalfurafine
0.0025 0.0025 0.0025 0.0025 0.0025
0.0025 0.0025 0.0025 0.0025
ingredient hydrochloride
Carbohydrate Mannitol 88.9865 88.8975 88.8975 88.8975 88.8975 88.8975
88.8975 88.8975 88.8975
L
Sodium
0.1
thiosulfate ,
,
Sodium
0.1
hydrogensulfite _ . _ _ _
Propyl gallate 0.1
Stabilizing Tocopherol 0.1
.
.
agent BHA 0.1
,.
.
.
.
BHT
0.1
,
Isoascorbic acid 0.1
w
,
,
Sodium sulfite
0.1 .
H
I
0
Low substituted
w
Disintegrator hydroxypropyl 10 10 10 10 10
10 10 10 10
cellulose Magnesium _
Lubricant 1 1 1 I 1 1
1 1 1
stearate .
Total 100 100 100 100 100 100
100 100 100
_
_ _,
The residual rates in samples
79.8 87.6 87.4 99.6 95.7
94.2 91.4 92.3 90.5
exposed to 48 thousand lux=hr (%)
. . _
_
The residual rates in samples
exposed to 600 thousand lux=hr 27.7 27.7 45.3 90.8 76.4
61.1 44.1 71.2 56.4
(%)
CA 03030392 2019-01-09
22
[0066]
As shown in Table 1, the tablets of Examples 1 to 6 in which a particular
stabilizing agent was added maintained the active ingredient at a residual
rate of not
less than 90% in the samples exposed to an overall illumination of 48 thousand
lux=hr,
indicating that the photostability was improved in those samples. The tablets
of
Example 1 in which n-propyl gallate was added as a stabilizing agent further
indicated to maintain the active ingredient at a very high residual rate even
in the
sample exposed to an overall illumination of 600 thousand lux-hr.
Additionally, the
residual rate of the active ingredient stored in the dark was 99.4% in Example
1 and
102.1% in Comparative Example 1, indicating that the residual rate was not
decreased depending on the temperature and humidity conditions.
[0067]
(Comparative Example 4)
Into a fluid bed granulator, 88.5 parts of mannitol was introduced and an
aqueous solution of 0.0025 parts of nalfurafine hydrochloride was sprayed to
produce granules. Then, an ethanol solution of 0.001 parts of n-propyl gallate
was
added to the granules and the resulting mixture was stirred in a mortar. After
dried
at 40 C for 6 hours in a hot air dryer, the mixture was mixed with 10 parts of
low
substituted hydroxypropyl cellulose and 1 part of magnesium stearate to obtain
granules for tablet compression. The granules for tablet compression were
compressed using a tablet press into tablets with a weight of 99.5 mg and a
diameter
of 7 mm.
[0068]
(Example 7)
Tablets with a weight of 99.5 mg and a diameter of 7 mm were obtained in
the same manner as in Comparative Example 4, except that an ethanol solution
of
0.005 parts of n-propyl gallate instead of 0.001 parts of n-propyl gallate was
added to
CA 03030392 2019-01-09
23
the granules of Comparative Example 4.
[0069]
(Example 8)
Tablets with a weight of 99.5 mg and a diameter of 7 mm were obtained in
the same manner as in Comparative Example 4, except that an ethanol solution
of
0.01 parts of n-propyl gallate instead of 0.001 parts of n-propyl gallate was
added to
the granules of Comparative Example 4.
[0070]
(Example 9)
Tablets with a weight of 99.6 mg and a diameter of 7 mm were obtained in
the same manner as in Comparative Example 4, except that an ethanol solution
of 0.1
parts of n-propyl gallate instead of 0.001 parts of n-propyl gallate was added
to the
granules of Comparative Example 4.
[0071]
(Example 10)
Tablets with a weight of 100.5 mg and a diameter of 7 mm were obtained in
the same manner as in Comparative Example 4, except that an ethanol solution
of 1.0
part of n-propyl gallate instead of 0.001 parts of n-propyl gallate was added
to the
granules of Comparative Example 4.
[0072]
(Example 11)
Tablets with a weight of 104.5 mg and a diameter of 7 mm were obtained in
the same manner as in Comparative Example 4, except that an ethanol solution
of 5
parts of n-propyl gallate instead of 0.001 parts of n-propyl gallate was added
to the
granules of Comparative Example 4.
[0073]
The photostability test described in Test Example 1 was performed on the
=
CA 03030392 2019-01-09
24
tablets of Comparative Example 4 and Examples 7 to 11. The ingredients in the
tablets of Comparative Examples 1 and 4 and Examples 7 to 11, the contents of
the
respective ingredients in percent by weight of each solid preparation, and the
residual
rates of the active ingredient after exposure to light obtained as the results
of the
1-1PLC analysis are shown in Table 2.
[0074]
,
[Table 2]
Comparative Comparative
Example 7 Example 8
Example 9 Example 10 Example 11 .
Example 1 Example 4
Tablet Tablet Tablet Tablet
Tablet Tablet Tablet
Active Nalfurafine
0.0025 0.0025 0.0025 0.0025 ,
0.0025 0.0025 0.0024
ingredient hydrochloride
Carbohydrate Mannitol 88.9865 88.9447 88.9447 88.9447
88.8554 88.0597 84.6890
Stabilizing
Propyl gallate - 0.001 0.005 0.01 0.1
1.0 4.8
-
agent
Low substituted
Disintegrator hydroxypropyl 10 10 10 10 10
10 10 0
cellulose
.
,.
Lubricant Magnesium stearate 1 1 1 1 1
1 1 .
.
Total 100 100 100 100 100
100 100 ts.)
,
.
F=I
I The residual rates in samples exposed to
79.8 89.7 96.4 96.4
97.3 99.0 98.2 .
48 thousand lux=hr (%)
.
CA 03030392 2019-01-09
26
[0075]
As shown in Table 2, it is indicated that the stabilizing agent at a dosage of
0.001% (Comparative Example 4) did not provide a sufficient photostabilizing
effect
and, meanwhile, the stabilizing agent at a dosage within the range from 0.005
to 5%
(Examples 7 to 11) contributed to the production of solid preparations with
significantly improved photostability.
[0076]
(Example 12)
To the granules of Comparative Example 4, 1.0 part of n-propyl gallate was
added, and the resulting mixture was mixed with 10 parts of low substituted
hydroxypropyl cellulose and 1 part of magnesium stearate to obtain granules
for
tablet compression. The granules for tablet compression were compressed using
a
tablet press into tablets with a weight of 100.5 mg and a diameter of 7 mm.
[0077]
(Example 13)
Into a fluid bed granulator, 88.5 parts of mannitol was introduced and a
solution containing two solutes, which were 0.0025 parts of nalfurafine
hydrochloride and 0.1 parts of n-propyl gallate, in 30% ethanol was sprayed to
produce granules.
[0078]
(Example 14)
The granules obtained in Example 13 were mixed with 10 parts of low
substituted hydroxypropyl cellulose and I part of magnesium stearate to obtain
granules for tablet compression. The granules for tablet compression were
compressed using a tablet press into tablets with a weight of 99.5 mg and a
diameter
of 7 mm.
[0079]
CA 03030392 2019-01-09
27
(Example 15)
Into a fluid bed granulator, 88.5 parts of mannitol was introduced and an
aqueous solution containing two solutes, which were 0.0025 parts of
nalfurafine
hydrochloride and 0.1 parts of sodium thiosulfate pentahydrate, was sprayed to
produce granules. Then, an ethanol solution of 0.1 parts of n-propyl gallate
was
added to the granules and the resulting mixture was stirred in a mortar. After
dried
at 40 C for 6 hours in a hot air dryer, the mixture was mixed with 10 parts of
low
substituted hydroxypropyl cellulose and 1 part of magnesium stearate to obtain
granules for tablet compression. The granules for tablet compression were
compressed using a tablet press into tablets with a weight of 99.7 mg and a
diameter
of 7 mm.
[0080]
(Example 16)
Tablets with a weight of 100.1 mg and a diameter of 7 mm were obtained in
the same manner as in Example 15, except that an ethanol solution of 0.5 parts
of
n-propyl gallate instead of 0.1 parts of n-propyl gallate was added to the
granules of
Example 15.
[0081]
(Example 17)
Tablets with a weight of 100.6 mg and a diameter of 7 mm were obtained in
the same manner as in Example 15, except that an ethanol solution of 1 part of
n-propyl gallate instead of 0.1 parts of n-propyl gallate was added to the
granules of
Example 15.
[0082]
(Example 18)
Into a fluid bed granulator, 88.5 parts of mannitol was introduced and a
solution containing three solutes, which were 0.0025 parts of nalfurafine
=
CA 03030392 2019-01-09
28
hydrochloride, 0.1 parts of sodium thiosulfate pentahydrate and 0.1 parts of n-
propyl
gallate, in 30% ethanol was sprayed to produce granules.
[0083]
(Example 19)
The granules of Example 18 were mixed with 10 parts of low substituted
hydroxypropyl cellulose and 1 part of magnesium stearate to obtain granules
for
tablet compression. The granules for tablet compression were compressed using
a
tablet press into tablets with a weight of 99.7 mg and a diameter of 7 mm.
[0084]
(Test Example 2: Test to Determine the Ratio of the Generated Main Degradation
Product A to the Active Ingredient during the Production)
The granules of Examples 13 and 18 were further air-dried for 30 minutes,
and the resulting powders were analyzed by the following HPLC analysis to
calculate the ratios of the generated main degradation product A to the active
ingredient.
<Pretreatment Conditions>
Methanol was added to the powder, and the resulting mixture was stirred
and then centrifuged to collect the supernatant. The collected solution was
concentrated to a solid with a rotary evaporator and the resulting solid was
then
redissolved with the mobile phase A to prepare an HPLC sample.
<HPLC Conditions>
Mobile phase A: 50 mM sodium dihydrogen phosphate solution /
acetonitrile = 95/5 (v/v);
Mobile phase B: 50 mM sodium dihydrogen phosphate solution /
acetonitrile = 60/40 (v/v);
Column: YMC-Pack ODS-AM (4. x 250 mm in size; manufactured by YMC
Co., Ltd.);
A
CA 03030392 2019-01-09
29
Column temperature: 40 C;
Detection wavelength: 280 nm;
Flow rate: 1.0 ml/min.
[0085]
The production ratio of the main degradation product A was calculated with
the following Formula 2.
Production ratio (%) = (The area value of the HPLC peak of the main
degradation product A in the sample / the area value of the peak of the active
ingredient in the sample) x 100 Formula 2
[0086]
The production ratios of the main degradation product A in the granules of
Examples 13 and 18 immediately after the production of those granules are
shown in
Table 3.
[0087]
[Table 3]
Example 13 Example 18
Powder Powder
Nalfurafine
Active ingredient 0.0028 0.0028
hydrochloride
Carbohydrate Mannitol 99.8843 99.7717
Stabilizing agent Propyl gallate 0.11 0.11
Antioxidant Sodium thiosulfate 0.11
Total 100 100
The production ratios of the main degradation
1.24 0.38
product A in air-dried samples (%)
[0088]
As shown in Table 3, a significant reduction in the ratio of the main
degradation product during the production was indicated in the granules of
Example
18 to which sodium thiosulfate, which is an antioxidant, was further added, as
compared to those of Example 13.
CA 03030392 2019-01-09
[0089]
The photostability test described in Test Example 1 was performed on the
tablets of Example 12, Examples 14 to 17 and Example 19. The ingredients in
the
tablets of Example 12, Examples 14 to 17 and Example 19, the contents of the
5 respective ingredients in percent by weight of each solid
preparation, and the residual
rates of the active ingredient after exposure to light obtained as the results
of the
HPLC analysis are shown in Table 4.
[0090]
[Table 4]
Example 12 Example 14 Example 15 Example 16
Example 17 Example 19 .
Tablet . Tablet Tablet Tablet
Tablet Tablet
Active Nalfurafine
0.0025 0.0025 0.0025 0.0025
0.0025 0.0025
ingredient hydrochloride
Carbohydrate Mannitol 88.0597 88.9447 88.7663 88.4116
87.9722 88.7663
Stabilizing
Propyl gallate 1.0 0.1 0.1 0.5
1.0 0.1
agent
Antioxidant Sodium thiosulfate - 0.1 0.1
0.1 0.1 .
Low substituted
Disintegrator hydroxypropyl 10 10 10 10
10 10 p
cellulose
LD
,
Lubricant Magnesium stearate 1 1 1 , 1
1 1 .0
.
Total 100 100 1-00 100
100 100
_
.
,...)
.
The residual rates in samples exposed 97.3
98.3 97.7 99.6
100.4 102.7 ,
to 48 thousand lux=hr (%)
.
..
= = CA 03030392 2019-01-09
32
[0091]
As shown in Table 4, the stabilizing agent exhibited an excellent
photostabilizing effect even in case where the stabilizing agent was added in
powder
form (Example 12). Additionally, the improvement of photostability was
indicated
even in cases where the stabilizing agent was sprayed together with the active
ingredient (Example 14). In addition, the photostabilizing effect was
maintained
even in cases where sodium thiosulfate, which is an antioxidant, was used in
combination with the stabilizing agent, indicating that an excellent
stabilizing effect
was obtained irrespective of the way of adding the stabilizing agent and
sodium
thiosulfate.
[0092]
(Comparative Example 5)
Granules described in W099/002158 were produced as follows. In a
mortar, 68.9 parts of lactose (Pharmatoseo 200M) and 31 parts of crystalline
cellulose (CEOLUS PH-101; manufactured by Asahi Kasei Chemicals
Corporation) were placed, and an aqueous solution of 0.1 parts of nalfurafine
hydrochloride was added thereto, and the resulting mixture was stirred. Then,
the
mixture was dried at 40 C for 12 hours to produce granules.
[0093]
(Comparative Example 6)
Granules described in W099/002158 were produced as follows. In a
mortar, 68.8 parts of lactose (Pharmatose 200M) and 31 parts of crystalline
cellulose (CEOLUS PH-101; manufactured by Asahi Kasei Chemicals
Corporation) were placed, and an aqueous solution containing two solutes,
which
were 0.1 parts of nalfurafme hydrochloride and 0.1 parts of sodium
thiosulfate, was
added thereto, and the resulting mixture was stirred. Then, the mixture was
dried at
40 C for 12 hours to produce granules.
CA 03030392 2019-01-09
33
P
[0094]
(Comparative Example 7)
In a mortar, 99.95 parts of mannitol was placed, and an aqueous solution of
0.05 parts of nalfurafine hydrochloride was added thereto, and the resulting
mixture
was stirred. Then, the mixture was dried at 40 C for 12 hours to produce
granules.
[0095]
(Comparative Example 8)
In a mortar, 99.99 parts of mannitol was placed, and an aqueous solution of
0.01 parts of nalfurafine hydrochloride was added thereto, and the resulting
mixture
was stirred. Then, the mixture was dried at 40 C for 12 hours to produce
granules.
[0096]
(Example 20)
In a mortar, 99.89 parts of mannitol was placed, and an aqueous solution of
0.01 parts of nalfurafine hydrochloride was added thereto with stirring, and
an
ethanol solution of 0.1 parts of n-propyl gallate was then added thereto, and
the
resulting mixture was stirred. Then, the mixture was dried at 40 C for 12
hours to
produce granules.
[0097]
(Example 21)
Into a fluid bed granulator, 88.5 parts of mannitol was introduced and a
solution containing three solutes, which were 0.0025 parts of nalfurafine
hydrochloride, 0.1 parts of sodium thiosulfate and 0.1 parts of n-propyl
gallate, in
30% ethanol was sprayed to produce granules. Then, 455.65 parts of mannitol
was
added to 44.35 parts of the granules, and the resulting mixture was mixed
using a
V-type mixer.
[0098]
(Test Example 3: Photostability Test on Powders)
CA 03030392 2019-01-09
34
The powders of Comparative Examples 5 to 8 and Examples 20 and 21 were
spread thinly on glass dishes, and the glass dishes were exposed under white
fluorescent light (with an illumination of 2000 lux) to an overall
illumination of 48
thousand lux=hr, and the powders were then collected from the glass dishes,
and the
powders were analyzed by the following HPLC analysis to calculate the residual
rates of the active ingredient after exposure to light by the same formula as
the
Formula 1.
<Pretreatment Conditions>
Distilled water was added to the powder to suspend or dissolve the powder,
and the resulting mixture was centrifuged to collect the supernatant as an
HPLC
sample.
<HPLC Conditions>
The test was performed under the same HPLC conditions as those in Test
Example 1.
[0099]
The photostability test described in Test Example 2 was performed on the
powders of Comparative Examples 5 to 8 and Examples 20 and 21. The ingredients
in the powders of Comparative Examples 5 to 8 and Examples 20 and 21, the
contents of the respective ingredients in percent by weight of each solid
preparation,
and the residual rates of the active ingredient after exposure to light
obtained as the
results of the HPLC analysis are shown in Table 5.
[0100]
= .
[Table 5] .
Comparative Comparative Comparative Comparative
Example 20
Example 21 "
Example 5 Example 6 Example 7 Example 8
_
Powder Powder Powder Powder
Powder Powder
Active Nalfurafine
0.1 0.1 0.05 0.01
0.01 0.00025
_ ingredient _ hydrochloride
Crystalline
Excipient 31 31
cellulose
Carbohydrate Lactose 68.9000 68.8000
Carbohydrate Mannitol 99.9500 99.9900
99.8900 99.9798
Stabilizing
Propyl gallate
0.1 0.01 0
agent
,s
Sodium
0
.
Antioxidant 0.1 0.01
thiosulfate
Total 100 100 100 100
100 100
,
The residual rates in samples
'7
exposed to 48 thousand lux=hr 102.9 97.6 93.9 75.4
100.8 95.1 2
(%)
CA 03030392 2019-01-09
36
=
[0101]
As shown in Table 5, the solid preparations containing the active ingredient
at a concentration of 0.1% by weight according to W099/002158 (Comparative
Examples 5 and 6) and the solid preparation containing the active ingredient
at a
concentration of 0.05% by weight (Comparative Example 7) ensured
photostability
without any added stabilizing agent, indicating that the problems of the
present
invention are particularly relevant to solid preparations with a low content
of
nalfurafine. Additionally, the effect of the present invention was shown even
in a
solid preparation containing the active ingredient at a concentration of
0.00025% by
weight.
[0102]
(Comparative Example 9)
Vanillin, which is a light absorbing agent described in JP S58-57322 A, was
used. In a mortar, 99.8975 parts of mannitol was placed, and an aqueous
solution of
0.0025 parts of nalfurafine hydrochloride was added thereto and the resultant
was
stirred. Thereafter, an ethanol solution of 0.1 parts of vanillin (Guaranteed
Reagent;
manufactured by Wako Pure Chemical Industries, Ltd.) was added to the
resultant,
and the resulting mixture was stirred. The mixture was then dried at 40 C for
12
hours to produce granules.
[0103]
(Comparative Example 10)
p-Aminobenzoic acid (Guaranteed Reagent; manufactured by Wako Pure
Chemical Industries, Ltd.), which is a light absorbing agent described in JP
S58-57322 A, was used. In a mortar, 99.8975 parts of mannitol was placed, and
an
aqueous solution of 0.0025 parts of nalfurafine hydrochloride was added
thereto and
the resultant was stirred. Thereafter, an ethanol solution of 0.1 parts of
p-aminobenzoic acid was added to the resultant, and the resulting mixture was
stirred.
= CA 03030392 2019-01-09
37
,
The mixture was then dried at 40 C for 12 hours to produce granules.
[0104]
(Example 22)
In a mortar, 99.7975 parts of mannitol was placed, and an aqueous solution
of 0.0025 parts of nalfurafine hydrochloride was added thereto, and the
resultant was
stirred. To the mortar, 0.1 parts of ferric oxide (Sicovit Red 30E172;
manufactured
by Huntsman Corporation) was added, and an ethanol solution of 0.1 parts of
n-propyl gallate was then added thereto, and the resulting mixture was
stirred. Then,
the mixture was dried at 40 C for 12 hours to produce granules.
[0105]
(Example 23)
In a mortar, 100 parts of the granules of Example 21 were placed, and 0.01
parts of ferric oxide and an aqueous ethanol solution were added thereto, and
the
resulting mixture was stirred. Then, the mixture was dried for 12 hours to
produce
granules.
[0106]
The photostability test described in Test Example 2 was performed on the
powders of Comparative Examples 9 and 10 and Examples 22 and 23. The
ingredients in the powders of Comparative Examples 9 and 10 and Examples 22
and
23, the contents of the respective ingredients in percent by weight of each
solid
preparation, and the residual rates of the active ingredient after exposure to
light
obtained as the results of the HPLC analysis are shown in Table 6.
CA 03030392 2019-01-09
38
[0107]
[Table 6]
Comparative Comparative Example Example
Example 9 Example 10 22 23
Powder Powder
Powder Powder
Active Nalfurafine
0.0025 0.0025
0.0025 0.00025
ingredient hydrochloride
Carbohydrate Mannitol 99.8975 99.8975
99.7975 99.9698
Additives Vanillin 0.1
p-aminobenzoic
Additives 0.1
acid
Stabilizing
Propyl gallate 0.1 0.01
agent
Sodium
Antioxidant 0.01
thiosulfate
Coloring
Ferric oxide 0.1 0.01
agent
Total 100 100 100 100
The residual rates in samples
exposed to 48 thousand lux=hr 79.9 89.2 100.0 100.9
(%)
[0108]
As shown in Table 6, vanillin and p-aminobenzoic acid, which are light
absorbing agents described in JP S58-57322 A, provided an insufficient
photostabilizing effect to the active ingredient of the present invention,
which is the
above 4,5--epoxymorphinan derivative represented by the general formula (I) or
a
pharmaceutically acceptable acid addition salt thereof, indicating that
different
stabilization methods are effective for different active ingredients.
Additionally, it
is indicated that the photostabilizing effect was further enhanced by the
addition of
ferric oxide as a coloring agent as well as the stabilizing agent of the
present
invention, as shown in Example 22. In addition, it is indicated that the
photostability of the active ingredient was improved as shown in Example 23
even in
cases where ferric oxide was added at a dosage of 0.01% of the solid
preparation,
4
CA 03030392 2019-01-09
39
which is smaller than the dosage of ferric oxide relative to the composition
according
to JP 2006-306754 A, namely 0.1 %.
Industrial Applicability
[0109]
According to the present invention, the photostability of a solid preparation
containing a 4,5-epoxymorphinan derivative or a pharmaceutically acceptable
acid
addition salt thereof is improved, which greatly improves the convenience in
handling the solid preparation in split tablets or in powder, which has been
difficult
so far, and consequently reduces the risk of phartnacy compounding errors,
improves
the drug compliance in patients, and enhances the therapeutic effect of the
solid
preparation. Moreover, the solid preparation is stable to light even without
light
shielding coating, which contributes to simplifying the production process and
allows
tablets to ensure rapid disintegration properties.