Note: Descriptions are shown in the official language in which they were submitted.
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
BISPECIFIC HER2 AND CD3 BINDING MOLECULES
[0001] This application is a continuation-in-part of International Patent
Application No.
PCT/US2015/041989, filed July 24, 2015, which claims the benefit of U.S.
Provisional
Application No. 62/029,342, filed on July 25, 2014, each of which is
incorporated by reference
herein in its entirety.
[0002] This application incorporates by reference a Sequence Listing
submitted with this
application as text file entitled "Sequence Listing 13542-039-228.txt" created
on January 27,
2017 and having a size of 184 kbytes.
1. FIELD
[0003] Provided herein are compositions, methods, and uses involving
bispecific binding
molecules that specifically bind to HER2, a receptor tyrosine kinase, and to
CD3, a T cell
receptor, and mediate T cell cytotoxicity for managing and treating disorders,
such as cancer.
2. BACKGROUND
[0004] HER2 is a receptor tyrosine kinase of the epidermal growth factor
receptor family.
Amplification or overexpression of HER2 has been demonstrated in the
development and
progression of cancers. Hercepting (trastuzumab) is an anti-HER2 monoclonal
antibody
approved for treating HER2-positive metastatic breast cancer and HER2-positive
gastric cancer
(Trastuzumab [Highlights of Prescribing Information]. South San Francisco, CA:
Genentech,
Inc.; 2014). Ertumaxomab is a tri-specific HER2-CD3 antibody with intact Fc-
receptor binding
(see, for example, Kiewe et al. 2006, Clin Cancer Res, 12(10): 3085-3091).
Ertumaxomab is a
rat-mouse antibody; therefore, upon administration to humans, a human anti-
mouse antibody
response and a human anti-rat antibody response are expected. 2502A, the
parental antibody of
ertumaxomab, has low affinity for HER2 and low avidity (Diermeier-Daucher et
at., MAbs,
2012, 4(5): 614-622). There is a need for therapies capable of mediating T
cell cytotoxicity in
HER2-positive cancers.
3. SUMMARY
[0005] In a specific embodiment, provided herein is a method of treating a
HER2-positive
cancer in a subject in need thereof, comprising administering to the subject a
therapeutically
effective amount of a bispecific binding molecule comprising an aglycosylated
monoclonal
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
antibody that is an immunoglobulin that binds to HER2, said immunoglobulin
comprising two
identical heavy chains and two identical light chains, said light chains being
a first light chain
and a second light chain, wherein the first light chain is fused to a first
single chain variable
fragment (scFv), via a peptide linker, to create a first light chain fusion
polypeptide, and wherein
the second light chain is fused to a second scFv, via a peptide linker, to
create a second light
chain fusion polypeptide, wherein the first and second scFv (i) are identical,
and (ii) bind to
CD3, and wherein the first and second light chain fusion polypeptides are
identical, and wherein
the cancer expresses a low level of HER2, and preferably wherein the cancer is
not a head and
neck cancer.
[0006] In a specific embodiment, the cancer is deemed to express a low
level of HER2 when
the cancer expresses a lower level of HER2 than the level of HER2 expressed by
cancers that are
indicated for treatment with trastuzumab and are of the same tissue type as
the HER2-positive
cancer.
[0007] In a specific embodiment, the cancer is deemed to express a low
level of HER2 when
the cancer has been determined not to overexpress HER2 based on the following
characterization
of the cancer: (a) a first determination of a level of HER2 in a test specimen
comprising cells of
the cancer is reported as negative, or (b) a first determination of a level of
HER2 in a test
specimen comprising cells of the cancer is reported as equivocal, and a second
determination of a
level of HER2 in a test specimen comprising cells of the cancer is reported as
equivocal or
negative. In a specific embodiment, the determination of the level of HER2 in
the test specimen
is reported as negative when the level of HER2 in the test specimen is
characterized as (i) (1)
immunohistochemistry (IHC) 1+, wherein the level of HER2 in the test specimen
is
characterized as IHC 1+ when the test specimen exhibits an incomplete HER2
membrane
staining that is faint/barely perceptible and within greater than 10% of the
invasive tumor cells,
wherein the staining is readily appreciated using a low-power objective; (2)
IHC 0, wherein the
level of HER2 in the test specimen is characterized as IHC 0 when the test
specimen exhibits no
HER2 staining observed, wherein the lack of staining is readily appreciated
using a low-power
objective, or a HER2 membrane staining that is incomplete and is faint/barely
perceptible and
within less than or equal to 10% of the invasive tumor cells, wherein the
staining is readily
appreciated using a low-power objective; or (ii) in situ hybridization (ISH)
negative, wherein the
level of HER2 in the test specimen is characterized as ISH negative when the
test specimen
-2-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
exhibits (1) a single-probe average HER2 copy number of less than 4.0 signals
per cell; or (2) a
dual-probe HER2/CEP17 ratio of less than 2.0 with an average HER2 copy number
of less than
4.0 signals per cell. In a specific embodiment, the determination of the level
of HER2 in the test
specimen is reported as equivocal when the level of HER2 in the test specimen
is characterized
as: (i) IHC 2+, wherein the level of HER2 in the test specimen is
characterized as IHC 2+ when
the test specimen exhibits (1) a circumferential HER2 membrane staining that
is incomplete
and/or weak/moderate and within greater than 10% of invasive tumor cells,
wherein the staining
is observed in a homogenous and contiguous population, and wherein the
staining is readily
appreciated using a low-power objective; or (2) a complete and circumferential
HER2 membrane
staining that is intense and within less than or equal to 10% of invasive
tumor cells, wherein the
staining is readily appreciated using a low-power objective; or (ii) ISH
equivocal, wherein the
level of HER2 in the test specimen is characterized as ISH equivocal when the
test specimen
exhibits (1) a single-probe ISH average HER2 copy number of greater than or
equal to 4.0 and
less than 6.0 signals/cell, wherein the copy number is determined by counting
at least 20 cells
within the area and is observed in a homogenous and contiguous population; or
(2) a dual-probe
HER2/CEP17 ratio of less than 2.0 with an average HER2 copy number of greater
than or equal
to 4.0 and less than 6.0 signals per cell, wherein the copy number is
determined by counting at
least 20 cells within the area and is observed in a homogenous and contiguous
population.
[0008] In a specific embodiment, the cancer is deemed to express a low
level of HER2 when
a level of HER2 in a test specimen comprising cells of the cancer is
characterized as IHC 2+ or
less according to applicable American Society of Clinical Oncology/College of
American
Pathologists guideline recommendations for human epidermal growth factor
receptor 2 testing in
cancer. In a preferred embodiment, the cancer is deemed to express a low level
of HER2 when a
level of HER2 in a test specimen comprising cells of the cancer is
characterized as IHC 2+ or
less according to applicable American Society of Clinical Oncology/College of
American
Pathologists guideline recommendations for human epidermal growth factor
receptor 2 testing in
breast cancer. In a specific embodiment, a level of HER2 in a test specimen
comprising cells of
the cancer is characterized as IHC 2+. In a specific embodiment, the level of
HER2 in the test
specimen is characterized as IHC 2+ when the test specimen exhibits (1) a
circumferential HER2
membrane staining that is incomplete and/or weak/moderate and within greater
than 10% of
invasive tumor cells, wherein the staining is observed in a homogenous and
contiguous
-3-
CA 03051484 2019-07-24
WO 2018/140026
PCT/US2017/015278
population, and wherein the staining is readily appreciated using a low-power
objective; or (2) a
complete and circumferential HER2 membrane staining that is intense and within
less than or
equal to 10% of invasive tumor cells, wherein the staining is readily
appreciated using a low-
power objective. In a specific embodiment, a level of HER2 in a test specimen
comprising cells
of the cancer is characterized as IHC 1+. In a specific embodiment, the level
of HER2 in the test
specimen is characterized as IHC 1+ when the test specimen exhibits an
incomplete HER2
membrane staining that is faint/barely perceptible and within greater than 10%
of the invasive
tumor cells, wherein the staining is readily appreciated using a low-power
objective. In a
specific embodiment, a level of HER2 in a test specimen comprising cells of
the cancer is
characterized as IHC 0. In a specific embodiment, the level of HER2 in the
test specimen is
characterized as IHC 0 when the test specimen exhibits no HER2 staining
observed, wherein the
lack of staining is readily appreciated using a low-power objective, or a HER2
membrane
staining that is incomplete and is faint/barely perceptible and within less
than or equal to 10% of
the invasive tumor cells, wherein the staining is readily appreciated using a
low-power objective.
[0009] In a
specific embodiment, the HER2-positive cancer that expresses a low level of
HER2 is a programmed death-ligand 1 (PDL1)-positive cancer. In a specific
embodiment, the
HER2-positive cancer overexpresses PDL1 relative to expression of PDL1 in
analogous
noncancerous cells of the same tissue type as the cancer. In a specific
embodiment, the HER2-
positive cancer is deemed to overexpress PDL1 when a test specimen comprising
cells of the
cancer expresses a detectable level of PDL1 above background. In a specific
embodiment, the
cancer is resistant to PDL1 blockade with an anti-PDL1 therapy. In a specific
embodiment, the
anti-PDL1 therapy is an anti-PDL1 antibody. In a specific embodiment, the anti-
PDL1 antibody
is atezolizumab. In a specific embodiment, the cancer is resistant to
programmed cell death
protein 1 (PD1) blockade with an anti-PD1 therapy. In a specific embodiment,
the anti-PD1
therapy is an anti-PD1 antibody. In a specific embodiment, the anti-PD1
antibody is
pembrolizumab.
[0010] In a
specific embodiment, the HER2-positive cancer that expresses a low level of
HER2 is breast cancer, gastric cancer, an osteosarcoma, desmoplastic small
round cell cancer,
ovarian cancer, prostate cancer, pancreatic cancer, glioblastoma multiforme,
gastric junction
adenocarcinoma, gastroesophageal junction adenocarcinoma, cervical cancer,
salivary gland
cancer, soft tissue sarcoma, leukemia, melanoma, Ewing's sarcoma,
rhabdomyosarcoma, or
-4-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
neuroblastoma. In a specific embodiment, the cancer is gastric cancer or
breast cancer. In a
specific embodiment of a method described herein, the HER2-positive cancer
that expresses a
low level of HER2 is a metastatic tumor. In a specific embodiment, the
metastatic tumor is a
peritoneal metastasis. In a specific embodiment, the cancer is resistant to
treatment with
trastuzumab, cetuximab, lapatinib, erlotinib, or any other small molecule or
antibody that targets
the HER family of receptors.
[0011] In a specific embodiment, also provided herein is a method of
treating a HER2-
positive cancer in a subject in need thereof, comprising administering to the
subject a
therapeutically effective amount of a bispecific binding molecule comprising
an aglycosylated
monoclonal antibody that is an immunoglobulin that binds to HER2, said
immunoglobulin
comprising two identical heavy chains and two identical light chains, said
light chains being a
first light chain and a second light chain, wherein the first light chain is
fused to a first single
chain variable fragment (scFv), via a peptide linker, to create a first light
chain fusion
polypeptide, and wherein the second light chain is fused to a second scFv, via
a peptide linker, to
create a second light chain fusion polypeptide, wherein the first and second
scFv (i) are identical,
and (ii) bind to CD3, and wherein the first and second light chain fusion
polypeptides are
identical, and wherein the cancer is not indicated for treatment with
trastuzumab, and preferably
wherein the cancer is not a head and neck cancer. In a specific embodiment,
the cancer is
determined not to be indicated for treatment with trastuzumab based on the
following
characterization of the cancer: (a) a first determination of a level of HER2
in a test specimen
comprising cells of the cancer is reported as negative, or (b) a first
determination of a level of
HER2 in a test specimen comprising cells of the cancer is reported as
equivocal, and a second
determination of a level of HER2 in a test specimen comprising cells of the
cancer is reported as
equivocal or negative. In a specific embodiment, the determination of the
level of HER2 in the
test specimen is reported as negative when the level of HER2 in the test
specimen is
characterized as (i) (1) immunohistochemistry (IHC) 1+, wherein the level of
HER2 in the test
specimen is characterized as IHC 1+ when the test specimen exhibits an
incomplete HER2
membrane staining that is faint/barely perceptible and within greater than 10%
of the invasive
tumor cells, wherein the staining is readily appreciated using a low-power
objective; (2) IHC 0,
wherein the level of HER2 in the test specimen is characterized as IHC 0 when
the test specimen
exhibits no HER2 staining observed, wherein the lack of staining is readily
appreciated using a
-5-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
low-power objective, or a HER2 membrane staining that is incomplete and is
faint/barely
perceptible and within less than or equal to 10% of the invasive tumor cells,
wherein the staining
is readily appreciated using a low-power objective; or (ii) in situ
hybridization (ISH) negative,
wherein the level of HER2 in the test specimen is characterized as ISH
negative when the test
specimen exhibits (1) a single-probe average HER2 copy number of less than 4.0
signals per cell;
or (2) a dual-probe HER2/CEP17 ratio of less than 2.0 with an average HER2
copy number of
less than 4.0 signals per cell. In a specific embodiment, the determination of
the level of HER2
in the test specimen is reported as equivocal when the level of HER2 in the
test specimen is
characterized as: (i) IHC 2+, wherein the level of HER2 in the test specimen
is characterized as
IHC 2+ when the test specimen exhibits (1) a circumferential HER2 membrane
staining that is
incomplete and/or weak/moderate and within greater than 10% of invasive tumor
cells, wherein
the staining is observed in a homogenous and contiguous population, and
wherein the staining is
readily appreciated using a low-power objective; or (2) a complete and
circumferential HER2
membrane staining that is intense and within less than or equal to 10% of
invasive tumor cells,
wherein the staining is readily appreciated using a low-power objective; or
(ii) ISH equivocal,
wherein the level of HER2 in the test specimen is characterized as ISH
equivocal when the test
specimen exhibits (1) a single-probe ISH average HER2 copy number of greater
than or equal to
4.0 and less than 6.0 signals/cell, wherein the copy number is determined by
counting at least 20
cells within the area and is observed in a homogenous and contiguous
population; or (2) a dual-
probe HER2/CEP17 ratio of less than 2.0 with an average HER2 copy number of
greater than or
equal to 4.0 and less than 6.0 signals per cell, wherein the copy number is
determined by
counting at least 20 cells within the area and is observed in a homogenous and
contiguous
population. In a specific embodiment, the HER2-positive cancer is a programmed
death-ligand 1
(PDL1)-positive cancer. In a specific embodiment, the HER2-positive cancer
overexpresses
PDL1 relative to expression of PDL1 in analogous noncancerous cells of the
same tissue type as
the cancer. In a specific embodiment, the HER2-positive cancer is deemed to
overexpress PDL1
when a test specimen comprising cells of the cancer expresses a detectable
level of PDL1 above
background. In a specific embodiment, the cancer is resistant to PDL1 blockade
with an anti-
PDL1 therapy. In a specific embodiment, the anti-PDL1 therapy is an anti-PDL1
antibody. In a
specific embodiment, the anti-PDL1 antibody is atezolizumab. In a specific
embodiment, the
cancer is resistant to programmed cell death protein 1 (PD1) blockade with an
anti-PD1 therapy.
-6-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
In a specific embodiment, the anti-PD1 therapy is an anti-PD1 antibody. In a
specific
embodiment, the anti-PD1 antibody is pembrolizumab. In a specific embodiment,
the HER2-
positive that is not indicated for treatment with trastuzumab is breast
cancer, gastric cancer, an
osteosarcoma, desmoplastic small round cell cancer, ovarian cancer, prostate
cancer, pancreatic
cancer, glioblastoma multiforme, gastric junction adenocarcinoma,
gastroesophageal junction
adenocarcinoma, cervical cancer, salivary gland cancer, soft tissue sarcoma,
leukemia,
melanoma, Ewing's sarcoma, rhabdomyosarcoma, or neuroblastoma. In a specific
embodiment,
the HER2-positive that is not indicated for treatment with trastuzumab is
gastric cancer or breast
cancer. In a specific embodiment, the HER2-positive that is not indicated for
treatment with
trastuzumab is a metastatic tumor. In a specific embodiment, the metastatic
tumor is a peritoneal
metastasis. In a specific embodiment, the cancer is resistant to treatment
with trastuzumab,
cetuximab, lapatinib, erlotinib, or any other small molecule or antibody that
targets the HER
family of receptors.
[0012] Also provided herein is a method of treating a HER2-positive, PDL1-
positive cancer
in a subject in need thereof, comprising administering to the subject a
therapeutically effective
amount of a bispecific binding molecule comprising an aglycosylated monoclonal
antibody that
is an immunoglobulin that binds to HER2, said immunoglobulin comprising two
identical heavy
chains and two identical light chains, said light chains being a first light
chain and a second light
chain, wherein the first light chain is fused to a first single chain variable
fragment (scFv), via a
peptide linker, to create a first light chain fusion polypeptide, and wherein
the second light chain
is fused to a second scFv, via a peptide linker, to create a second light
chain fusion polypeptide,
wherein the first and second scFv (i) are identical, and (ii) bind to CD3, and
wherein the first and
second light chain fusion polypeptides are identical, wherein the cancer is
resistant to PDL1
blockade with an anti-PDL1 therapy and/or is resistant to PD1 blockade with an
anti-PD1
therapy. In a specific embodiment, the HER2-positive cancer overexpresses PDL1
relative to
expression of PDL1 in analogous noncancerous cells of the same tissue type as
the cancer. In a
specific embodiment, the HER2-positive cancer is deemed to overexpress PDL1
when a test
specimen comprising cells of the cancer expresses a detectable level of PDL1
above background.
In a specific embodiment, the anti-PDL1 therapy is an anti-PDL1 antibody. In a
specific
embodiment, the anti-PDL1 antibody is atezolizumab. In a specific embodiment,
the anti-PD1
therapy is an anti-PD1 antibody. In a specific embodiment, the anti-PD1
antibody is
-7-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
pembrolizumab. In a specific embodiment, the HER2-positive cancer is breast
cancer, gastric
cancer, an osteosarcoma, desmoplastic small round cell cancer, squamous cell
carcinoma of head
and neck cancer, ovarian cancer, prostate cancer, pancreatic cancer,
glioblastoma multiforme,
gastric junction adenocarcinoma, gastroesophageal junction adenocarcinoma,
cervical cancer,
salivary gland cancer, soft tissue sarcoma, leukemia, melanoma, Ewing's
sarcoma,
rhabdomyosarcoma, or neuroblastoma. In a specific embodiment, the HER2-
positive cancer is a
metastatic tumor. In a specific embodiment, the metastatic tumor is a
peritoneal metastasis. In a
specific embodiment, the cancer is resistant to treatment with trastuzumab,
cetuximab, lapatinib,
erlotinib, or any other small molecule or antibody that targets the HER family
of receptors.
[0013] In a specific embodiment of a method described herein, the sequence
of each heavy
chain is any of SEQ ID NOs: 23, 27, 62 or 63. In a specific embodiment, the
sequence of each
light chain is SEQ ID NO: 25. In a specific embodiment, the sequence of the
peptide linker is
any of SEQ ID NOs: 14 or 35-41. In a specific embodiment, the sequence of a VH
domain in the
first scEv is any of SEQ ID NOs: 15, 17 or 64. In a specific embodiment, the
sequence of an
intra-scEv peptide linker between a VH domain and a VL domain in the first
scEv is any of SEQ
ID NOs: 14 or 35-41. In a specific embodiment, the sequence of a VL domain in
the first scEv is
any of SEQ ID NOs: 16 or 65. In a specific embodiment, the sequence of the
scEv is any of SEQ
ID NOs: 19 or 48-59. In a specific embodiment, the sequence of the first light
chain fusion
polypeptide is any of SEQ ID NOs: 29, 34, 42-47, or 60. In a specific
embodiment, the sequence
of each heavy chain is SEQ ID NO: 27 and the sequence of each light chain is
SEQ ID NO: 25.
In a specific embodiment, the sequence of the scEv is SEQ ID NO: 19. In a
specific
embodiment, the peptide linker is 5-30, 5-25, 5-15, 10-30, 10-20, 10-15, 15-
30, or 15-25 amino
acids in length. In a specific embodiment, the sequence of the peptide linker
is SEQ ID NO: 14.
In a specific embodiment, the sequence of the first light chain fusion
polypeptide is SEQ ID NO:
60. In a specific embodiment, the sequence of the heavy chain is SEQ ID NO: 62
and the
sequence of each light chain fusion polypeptide is SEQ ID NO: 60. In a
specific embodiment,
the sequence of the first light chain fusion polypeptide is SEQ ID NO: 47. In
a specific
embodiment, the sequence of the heavy chain is SEQ ID NO: 27 and the sequence
of each light
chain fusion polypeptide is SEQ ID NO: 47. In a specific embodiment, the
sequence of the first
light chain fusion polypeptide is SEQ ID NO: 29. In a specific embodiment, the
sequence of the
heavy chain is SEQ ID NO: 27 and the sequence of each light chain fusion
polypeptide is SEQ
-8-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
ID NO: 29. In a specific embodiment, the KD of the bispecific binding molecule
is between
70nM and 1 i.tM for CD3. In a specific embodiment, the bispecific binding
molecule does not
bind an Fc receptor in its soluble or cell-bound form. In a specific
embodiment, the heavy chain
has been mutated to destroy an N-linked glycosylation site. In a specific
embodiment, the heavy
chain has an amino acid substitution to replace an asparagine that is an N-
linked glycosylation
site, with an amino acid that does not function as a glycosylation site. In a
specific embodiment,
the heavy chain has been mutated to destroy a Clq binding site. In a specific
embodiment, the
bispecific binding molecule does not activate complement. In a specific
embodiment, the scFv is
disulfide stabilized.
[0014] In a specific embodiment of a method described herein, the
administering is
intravenous. In a specific embodiment of a method described herein, the
administering is
intraperitoneal, intrathecal, intraventricular in the brain, or
intraparenchymal in the brain. In a
specific embodiment of a method described herein, the administering is
performed in
combination with multi-modality anthracycline-based therapy.
[0015] In a specific embodiment of a method described herein, the method
further comprises
administering to the subject doxorubicin, cyclophosphamide, paclitaxel,
docetaxel, and/or
carboplatin. In a specific embodiment of a method described herein, the method
further
comprises administering to the subject radiotherapy. In a specific embodiment
of a method
described herein, the method further comprises administering to the subject an
agent that
increases cellular HER2 expression.
[0016] In a specific embodiment of a method described herein, the
bispecific binding
molecule is not bound to a T cell during said administering step.
[0017] In a specific embodiment of a method described herein, the method
further comprises
administering T cells to the subject. In a specific embodiment, the T cells
are bound to
molecules identical to said bispecific binding molecule.
[0018] In a specific embodiment of a method described herein, the subject
is a human. In a
specific embodiment, the subject is a canine.
[0019] In a specific embodiment of a method described herein, the
bispecific binding
molecule is contained in a pharmaceutical composition, which pharmaceutical
composition
further comprises a pharmaceutically acceptable carrier.
-9-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
[0020] Also provided herein is a method of treating a HER2-positive cancer
in a subject in
need thereof, comprising administering to the subject a therapeutically
effective amount of a cell
expressing a bispecific binding molecule of the invention, wherein the cancer
expresses a low
level of HER2, and preferably wherein the cancer is not a head and neck
cancer. In a specific
embodiment, the sequence of the heavy chain of the bispecific binding molecule
is SEQ ID NO:
27. In a specific embodiment, the nucleotide sequence encoding the heavy chain
of the
bispecific binding molecule is SEQ ID NO: 26.
[0021] Also provided herein is a method of treating a HER2-positive cancer
in a subject in
need thereof, comprising administering to the subject (a) a therapeutically
effective amount of an
ex vivo cell comprising a vector comprising (i) a first polynucleotide
comprising nucleotide
sequences encoding a light chain fusion polypeptide comprising an
immunoglobulin light chain
fused to a scFv, via a peptide linker, operably linked to a first promoter,
and (ii) a a second
polynucleotide encoding an immunoglobulin heavy chain that binds to HER2
operably linked to
a second promoter, wherein the light chain binds to HER2 and wherein the scFv
binds to CD3, or
(b) a therapeutically effective amount of an ex vivo cell comprising a mixture
of polynucleotides
comprising (i) a first polynucleotide comprising nucleotide sequences encoding
a light chain
fusion polypeptide comprising an immunoglobulin light chain fused to a scFv,
via a peptide
linker, operably linked to a first promoter, and (ii) a second polynucleotide
encoding an
immunoglobulin heavy chain that binds to HER2 operably linked to a second
promoter; and
wherein the cancer expresses a low level of HER2, and wherein the cancer is
not a head and neck
cancer. In a specific embodiment, the sequence of the heavy chain is SEQ ID
NO: 27. In a
specific embodiment, the nucleotide sequence encoding the heavy chain is SEQ
ID NO: 26.
[0022] Also provided herein is a method of treating a HER2-positive cancer
in a subject in
need thereof, comprising administering to the subject a therapeutically
effective amount of a cell
expressing a bispecific binding molecule of the invention, wherein the cancer
is not indicated for
treatment with trastuzumab, and preferably wherein the cancer is not a head
and neck cancer. In
a specific embodiment, the sequence of the heavy chain of the bispecific
binding molecule is
SEQ ID NO: 27. In a specific embodiment, the nucleotide sequence encoding the
heavy chain of
the bispecific binding molecule is SEQ ID NO: 26.
[0023] Also provided herein is a method of treating a HER2-positive cancer
in a subject in
need thereof, comprising administering to the subject (a) a therapeutically
effective amount of an
-10-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
ex vivo cell comprising a vector comprising (i) a first polynucleotide
comprising nucleotide
sequences encoding a light chain fusion polypeptide comprising an
immunoglobulin light chain
fused to a scFv, via a peptide linker, operably linked to a first promoter,
and (ii) a a second
polynucleotide encoding an immunoglobulin heavy chain that binds to HER2
operably linked to
a second promoter, wherein the light chain binds to HER2 and wherein the scFv
binds to CD3, or
(b) a therapeutically effective amount of an ex vivo cell comprising a mixture
of polynucleotides
comprising (i) a first polynucleotide comprising nucleotide sequences encoding
a light chain
fusion polypeptide comprising an immunoglobulin light chain fused to a scFv,
via a peptide
linker, operably linked to a first promoter, and (ii) a second polynucleotide
encoding an
immunoglobulin heavy chain that binds to HER2 operably linked to a second
promoter; and
wherein the cancer is not indicated for treatment with trastuzumab, and
wherein the cancer is not
a head and neck cancer.
[0024] In certain embodiments, provided herein is a bispecific binding
molecule comprising
an aglycosylated monoclonal antibody that is an immunoglobulin that binds to
HER2,
comprising two identical heavy chains and two identical light chains, said
light chains being a
first light chain and a second light chain, wherein the first light chain is
fused to a first single
chain variable fragment (scFv), via a peptide linker, to create a first light
chain fusion
polypeptide, and wherein the second light chain is fused to a second scFv, via
a peptide linker, to
create a second light chain fusion polypeptide, wherein the first and second
scFv (i) are identical,
and (ii) bind to CD3, and wherein the first and second light chain fusion
polypeptides are
identical.
[0025] In certain embodiments of the bispecific binding molecule, the
sequence of each
heavy chain is any of SEQ ID NOs: 23 or 27. In certain embodiments of the
bispecific binding
molecule, the sequence of each light chain is SEQ ID NO: 25. In certain
embodiments of the
bispecific binding molecule, the sequence of the peptide linker is SEQ ID NO:
14. In certain
embodiments of the bispecific binding molecule, the sequence of a VH domain in
the first scFv is
any of SEQ ID NOs: 15 or 17. In certain embodiments of the bispecific binding
molecule, the
sequence of an intra-scFv peptide linker between a VH domain and a VL domain
in the first scFv
is of SEQ ID NO: 14. In certain embodiments of the bispecific binding
molecule, the sequence
of a VL domain in the first scFv is of SEQ ID NO: 16. In certain embodiments
of the bispecific
binding molecule, the sequence of the scFv is SEQ ID NO: 19. In certain
embodiments of the
-11-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
bispecific binding molecule, the sequence of the first light chain fusion
polypeptide is SEQ ID
NO: 29.
[0026] In certain embodiments of the bispecific binding molecule, the
sequence of each
heavy chain is any of SEQ ID NOs: 23, 27, 62 or 63. In certain embodiments of
the bispecific
binding molecule, the sequence of each light chain is SEQ ID NO: 25. In
certain embodiments
of the bispecific binding molecule, the sequence of the peptide linker is any
of SEQ ID NOs: 14
or 35-41. In certain embodiments of the bispecific binding molecule, the
sequence of a VH
domain in the first scFv is any of SEQ ID NOs: 15, 17 or 64. In certain
embodiments of the
bispecific binding molecule, the sequence of an intra-scFv peptide linker
between a VH domain
and a VL domain in the first scFv is any of SEQ ID NOs: 14 or 35-41. In
certain embodiments of
the bispecific binding molecule, the sequence of a VL domain in the first scFv
is any of SEQ ID
NOs: 16 or 65. In certain embodiments of the bispecific binding molecule, the
sequence of the
scFv is any of SEQ ID NOs: 19 or 48-59. In certain embodiments of the
bispecific binding
molecule, the sequence of the first light chain fusion polypeptide is any of
SEQ ID NOs: 29, 34,
42-47, or 60.
[0027] In certain embodiments of the bispecific binding molecule, the
sequence of each
heavy chain is SEQ ID NO: 27 and the sequence of each light chain is SEQ ID
NO: 25. In
certain embodiments of the bispecific binding molecule, the sequence of the
scFv is SEQ ID NO:
19. In certain embodiments of the bispecific binding molecule, the sequence of
the heavy chain
is SEQ ID NO: 27, the sequence of each light chain is SEQ ID NO: 25 and the
sequence of the
scFv is SEQ ID NO: 19. In certain embodiments of the bispecific binding
molecule, the peptide
linker is 5-30, 5-25, 5-15, 10-30, 10-20, 10-15, 15-30, or 15-25 amino acids
in length. In certain
embodiments, the sequence of the peptide linker is SEQ ID NO: 14.
[0028] In certain embodiments, the sequence of the first light chain fusion
polypeptide is
SEQ ID NO: 60. In certain embodiments, the sequence of the heavy chain is SEQ
ID NO: 62
and the sequence of each light chain fusion polypeptide is SEQ ID NO: 60.
[0029] In certain embodiments, the sequence of the first light chain fusion
polypeptide is
SEQ ID NO: 47. In certain embodiments, the sequence of the heavy chain is SEQ
ID NO: 27
and the sequence of each light chain fusion polypeptide is SEQ ID NO: 47.
-12-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
[0030] In certain embodiments, the sequence of the first light chain fusion
polypeptide is
SEQ ID NO: 29. In certain embodiments, the sequence of the heavy chain is SEQ
ID NO: 27
and the sequence of each light chain fusion polypeptide is SEQ ID NO: 29.
[0031] In certain embodiments of the bispecific binding molecule, the KD is
between 70 nM
and 1 i.tM for CD3.
[0032] In certain embodiments of the bispecific binding molecule, the scFy
of the bispecific
binding molecule comprises one or more mutations to stabilize disulfide
binding. In certain
embodiments of the bispecific binding molecule, the stabilization of disulfide
binding prevents
aggregation of the bispecific binding molecule. In certain embodiments of the
bispecific binding
molecule, the stabilization of disulfide binding reduces aggregation of the
bispecific binding
molecule as compared to aggregation of the bispecific binding molecule without
the stabilization
of disulfide binding. In certain embodiments of the bispecific binding
molecule, the one or more
mutations to stabilize disulfide binding comprise a VH G44C mutation and a VL
Q100C mutation
(e.g., as present in SEQ ID NOS: 54-59). In certain embodiments of the
bispecific binding
molecule, the one or more mutations to stabilize disulfide binding are the
replacement of the
amino acid residue at VH44(according to the Kabat numbering system) with a
cysteine and the
replacement of the amino acid residue at VL100 (according to the Kabat
numbering system) with
a cysteine so as to introduce a disulfide bond between VH44 and VL100 (e.g.,
as present in SEQ
ID NOS: 54-59).
[0033] In certain embodiments of the bispecific binding molecule, the
bispecific binding
molecule does not bind an Fc receptor in its soluble or cell-bound form. In
certain embodiments
of the bispecific binding molecule, the heavy chain has been mutated to
destroy an N-linked
glycosylation site. In certain embodiments of the bispecific binding molecule,
the heavy chain
has an amino acid substitution to replace an asparagine that is an N-linked
glycosylation site,
with an amino acid that does not function as a glycosylation site. In certain
embodiments of the
bispecific binding molecule, the heavy chain has been mutated to destroy a Clq
binding site. In
certain embodiments, the bispecific binding molecule does not activate
complement.
[0034] In certain embodiments, provided herein is a bispecific binding
molecule comprising
an aglycosylated monoclonal antibody that is an immunoglobulin that binds to
HER2,
comprising two identical heavy chains and two identical light chains, said
light chains being a
first light chain and a second light chain, wherein the first light chain is
fused to a first single
-13-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
chain variable fragment (scFv), via a peptide linker, to create a first light
chain fusion
polypeptide, and wherein the second light chain is fused to a second scFv, via
a peptide linker, to
create a second light chain fusion polypeptide, wherein the first and second
scFv (i) are identical,
and (ii) bind to CD3, wherein the first and second light chain fusion
polypeptides are identical,
and wherein (a) the sequence of each heavy chain is SEQ ID NO: 62; and (b) the
sequence of
each light chain fusion polypeptide is SEQ ID NO: 60.
[0035] In certain embodiments, provided herein is a bispecific binding
molecule comprising
an aglycosylated monoclonal antibody that is an immunoglobulin that binds to
HER2,
comprising two identical heavy chains and two identical light chains, said
light chains being a
first light chain and a second light chain, wherein the first light chain is
fused to a first single
chain variable fragment (scFv), via a peptide linker, to create a first light
chain fusion
polypeptide, and wherein the second light chain is fused to a second scFv, via
a peptide linker, to
create a second light chain fusion polypeptide, wherein the first and second
scFv (i) are identical,
and (ii) bind to CD3, wherein the first and second light chain fusion
polypeptides are identical,
and wherein (a) the sequence of each heavy chain is SEQ ID NO: 27; and (b) the
sequence of
each light chain fusion polypeptide is SEQ ID NO: 47.
[0036] In certain embodiments, provided herein is a bispecific binding
molecule comprising
an aglycosylated monoclonal antibody that is an immunoglobulin that binds to
HER2,
comprising two identical heavy chains and two identical light chains, said
light chains being a
first light chain and a second light chain, wherein the first light chain is
fused to a first single
chain variable fragment (scFv), via a peptide linker, to create a first light
chain fusion
polypeptide, and wherein the second light chain is fused to a second scFv, via
a peptide linker, to
create a second light chain fusion polypeptide, wherein the first and second
scFv (i) are identical,
and (ii) bind to CD3, wherein the first and second light chain fusion
polypeptides are identical,
and wherein (a) the sequence of each heavy chain is SEQ ID NO: 27; and (b) the
sequence of
each light chain fusion polypeptide is SEQ ID NO: 29.
[0037] In certain embodiments, provided herein is a polynucleotide
comprising nucleotide
sequences encoding a light chain fusion polypeptide comprising an
immunoglobulin light chain
fused to a scFv, via a peptide linker, wherein the light chain binds to HER2
and wherein the scFv
binds to CD3. In certain embodiments of the polynucleotide, the sequence of
the light chain is
SEQ ID NO: 25. In certain embodiments of the polynucleotide, the nucleotide
sequence
-14-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
encoding the light chain is SEQ ID NO: 24. In certain embodiments of the
polynucleotide, the
sequence of the scFv is SEQ ID NO: 19. In certain embodiments of the
polynucleotide, the
nucleotide sequence encoding the scFv is SEQ ID NO: 18. In certain embodiments
of the
polynucleotide, the sequence of the light chain is SEQ ID NO: 25 and the
sequence of the scFv is
SEQ ID NO: 19. In certain embodiments of the polynucleotide, the nucleotide
sequence
encoding the light chain is SEQ ID NO: 24 and the nucleotide sequence encoding
the scFv is
SEQ ID NO: 18. In certain embodiments of the polynucleotide, the peptide
linker is 5-30, 5-25,
5-15, 10-30, 10-20, 10-15, 15-30, or 15-25 amino acids in length. In certain
embodiments of the
polynucleotide, the sequence of the peptide linker is SEQ ID NO: 14. In
certain embodiments of
the polynucleotide, the nucleotide sequence encoding the peptide linker is SEQ
ID NO: 13.
[0038] In certain embodiments, provided herein is a vector comprising a
polynucleotide
encoding nucleotide sequences encoding a light chain fusion polypeptide
comprising an
immunoglobulin light chain fused to a scFv, via a peptide linker, wherein the
light chain binds to
HER2 and wherein the scFv binds to CD3, operably linked to a promoter. In
certain
embodiments, provided herein is an ex vivo cell comprising the polynucleotide
provided herein
operably linked to a promoter. In certain embodiments, provided herein is an
ex vivo cell
comprising the vector.
[0039] In certain embodiments, provided herein is a vector comprising (i) a
first
polynucleotide comprising nucleotide sequences encoding a light chain fusion
polypeptide
comprising an immunoglobulin light chain fused to a scFv, via a peptide
linker, wherein the light
chain binds to HER2 and wherein the scFv binds to CD3 operably linked to a
first promoter, and
(ii) a second polynucleotide encoding an immunoglobulin heavy chain that binds
to HER2
operably linked to a second promoter. In certain embodiments, provided herein
is an ex vivo cell
comprising the vector.
[0040] In certain embodiments, provided herein is a method of producing a
bispecific
binding molecule comprising (a) culturing the cell comprising the vector
comprising (i) a first
polynucleotide comprising nucleotide sequences encoding a light chain fusion
polypeptide
comprising an immunoglobulin light chain fused to a scFv, via a peptide
linker, wherein the light
chain binds to HER2 and wherein the scFv binds to CD3 operably linked to a
first promoter, and
(ii) a second polynucleotide encoding an immunoglobulin heavy chain that binds
to HER2
operably linked to a second promoter, to express the first and second
polynucleotides such that a
-15-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
bispecific binding molecule comprising said light chain fusion polypeptide and
said
immunoglobulin heavy chain is expressed, and (b) recovering the bispecific
binding molecule.
[0041] In certain embodiments, provided herein is a pharmaceutical
composition comprising
a therapeutically effective amount of (i) the first polynucleotide operably
linked to the first
promoter, and (ii) the second polynucleotide encoding an immunoglobulin heavy
chain that
binds to HER2 operably linked to the second promoter. In certain embodiments,
provided herein
is a pharmaceutical composition comprising a therapeutically effective amount
of a vector
comprising (i) the first polynucleotide operably linked to the first promoter,
and (ii) the second
polynucleotide encoding an immunoglobulin heavy chain that binds to HER2
operably linked to
the second promoter. In certain embodiments, the vector is a viral vector.
[0042] In certain embodiments, provided herein is a mixture of
polynucleotides comprising
(i) a polynucleotide comprising nucleotide sequences encoding a light chain
fusion polypeptide
comprising an immunoglobulin light chain fused to a scFv, via a peptide
linker, wherein the light
chain binds to HER2 and wherein the scFy binds to CD3 operably linked to a
first promoter, and
(ii) a second polynucleotide encoding an immunoglobulin heavy chain that binds
to HER2
operably linked to a second promoter. In certain embodiments of the mixture of
polypeptides,
the sequence of the heavy chain is SEQ ID NO: 27. In certain embodiments of
the mixture of
polypeptides, the nucleotide sequence encoding the heavy chain is SEQ ID NO:
26. In certain
embodiments, provided herein is an ex vivo cell comprising the mixture of
polynucleotides
provided herein.
[0043] In certain embodiments, provided herein is a method of producing a
bispecific
binding molecule, comprising (i) culturing the cell comprising the mixture of
polynucleotides to
express the first and second polynucleotides such that a bispecific binding
molecule comprising
said light chain fusion polypeptide and said immunoglobulin heavy chain is
produced, and (ii)
recovering the bispecific binding molecule.
[0044] In certain embodiments, provided herein is a method of producing a
bispecific
binding molecule, comprising (i) expressing the mixture of polynucleotides
such that a bispecific
binding molecule comprising said first light chain fusion polypeptide and said
immunoglobulin
heavy chain is produced, and (ii) recovering the bispecific binding molecule.
[0045] In certain embodiments, provided herein is a method of making a
therapeutic T cell
comprising binding a bispecific binding molecule described herein to a T cell.
In certain
-16-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
embodiments, the T cell is a human T cell. In certain embodiments, the binding
is
noncovalently.
[0046] In certain embodiments, provided herein is a pharmaceutical
composition comprising
a therapeutically effective amount of the bispecific binding molecule and a
pharmaceutically
acceptable carrier.
[0047] In certain embodiments, provided herein is a pharmaceutical
composition comprising
a therapeutically effective amount of the bispecific binding molecule, a
pharmaceutically
acceptable carrier, and T cells. In certain embodiments, the T cells are bound
to the bispecific
binding molecule. In certain embodiments, the binding of the T cells to the
bispecific binding
molecule is noncovalently. In certain embodiments, the T cells are
administered to a subject for
treatment of a HER2-positive cancer in the subject. In certain embodiments,
the T cells are
autologous to the subject to whom they are administered. In certain
embodiments, the T cells are
allogeneic to the subject to whom they are administered. In certain
embodiments, the T cells are
human T cells.
[0048] In certain embodiments, provided herein is a method of treating a
HER2-positive
cancer in a subject in need thereof comprising administering a pharmaceutical
composition
provided herein. In certain embodiments, provided herein is a method of
treating a HER2-
positive cancer in a subject in need thereof comprising administering a
therapeutically effective
amount of a bispecific binding molecule provided herein. In certain
embodiments, the HER2-
positive cancer is breast cancer, gastric cancer, an osteosarcoma,
desmoplastic small round cell
cancer, squamous cell carcinoma of head and neck cancer, ovarian cancer,
prostate cancer,
pancreatic cancer, glioblastoma multiforme, gastric junction adenocarcinoma,
gastroesophageal
junction adenocarcinoma, cervical cancer, salivary gland cancer, soft tissue
sarcoma, leukemia,
melanoma, Ewing's sarcoma, rhamdomyosarcoma, neuroblastoma, small cell lung
cancer, or any
other neoplastic tissue that expresses the HER2 receptor. In certain
embodiments, the HER2-
positive cancer is a primary tumor or a metastatic tumor, e.g., a brain or
peritoneal metastases.
[0049] In certain embodiments of the method of treating, the administering
is intravenous.
In certain embodiments of the method of treating, the administering is
intraperitoneal,
intrathecal, intraventricular, or intraparenchymal. In certain embodiments of
the method of
treating, the method further comprises administering to the subject
doxorubicin,
cyclophosphamide, paclitaxel, docetaxel, and/or carboplatin. In certain
embodiments of the
-17-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
method of treating, the method further comprises administering to the subject
radiotherapy. In
certain embodiments of the method of treating, the administering is performed
in combination
with multi-modality anthracycline-based therapy. In certain embodiments of the
method of
treating, the administering is performed in combination with cytoreductive
chemotherapy. In a
specific embodiment, the administering is performed after treating the subject
with cytoreductive
chemotherapy. In certain embodiments of the method of treating, the bispecific
binding
molecule is not bound to a T cell. In certain embodiments of the method of
treating, the
bispecific binding molecule is bound to a T cell. In certain embodiments of
the method of
treating, the binding of the bispecific binding molecule to the T cell is non-
covalently. In certain
embodiments of the method of treating, the administering is performed in
combination with T
cell infusion. In a specific embodiment, the administering is performed after
treating the patient
with T cell infusion. In certain embodiments, the T cell infusion is performed
with T cells that
are autologous to the patient to whom the T cells are administered. In certain
embodiments, the
T cell infusion is performed with T cells that are allogeneic to the patient
to whom the T cells are
administered. In certain embodiments, the T cells can be bound to molecules
identical to a
bispecific binding molecule as described herein. In certain embodiments, the
binding of the T
cells to the molecules identical to a bispecific binding molecule is
noncovalently. In certain
embodiments, the T cells are human T cells.
[0050] In certain embodiments of the method of treating, the method further
comprises
administering to the subject an agent that increases cellular HER2 expression.
In certain
embodiments of the method of treating, the HER2-positive cancer is resistant
to treatment with
trastuzumab, cetuximab, lapatinib, erlotinib, or any other small molecule or
antibody that targets
the HER family of receptors. In certain embodiments of the method of treating,
the subject is a
human. In certain embodiments of the method of treating, the subject is a
canine.
4. BRIEF DESCRIPTION OF THE DRAWINGS
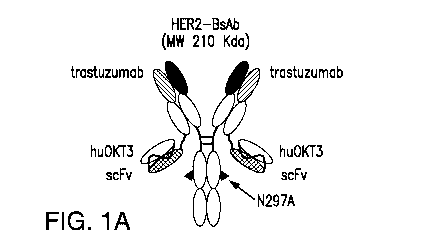
[0051] Fig. 1A, Fig. 1B, Fig. 1C, Fig. 1D, and Fig. 1E describe HER2-BsAb.
Fig. 1A
depicts a schematic of the HER2-BsAb. The arrow points to the N297A mutation
introduced
into the heavy chain to remove glycosylation. Fig. 1B depicts the purity of
HER2-BsAb as
demonstrated under reducing SDS-PAGE conditions. Fig. 1C depicts the purity of
HER2-BsAb
as demonstrated by SEC-HPLC. Fig. 1D demonstrates that the N297A mutation in
the human
-18-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
IgGl-Fc inhibits binding to the CD16A Fe receptor. Fig. 1E demonstrates that
the N297A
mutation in the human IgGl-Fc inhibits binding to the CD32A Fe receptor.
[0052] Fig. 2A and Fig. 2B demonstrate that HER2-BsAb binds to a breast
cancer cell line
and to T cells. Fig. 2A depicts the staining of AU565 breast cancer cells with
trastuzumab (left)
or with HER2-BsAb (right). Fig. 2B depicts the staining of CD3+ T cells with
huOKT3 (left) or
with HER2-BsAb (right).
[0053] Fig. 3 demonstrates that HER2-BsAb displays potent cytotoxic T
lymphocyte activity
in a 4 hour 51Cr release assay. For a description of trastuzumab-mOKT3, see,
Thakur et at.,
2010, Curr Opin Mol Ther, 12: 340.
[0054] Fig. 4 compares the HER2 expression against HER2-BsAb T cell
cytotoxicity in a
panel of cancer cell lines.
[0055] Fig. 5A and Fig. 5B demonstrate that HER2-BsAb-redirected T cell
cytotoxicity is
antigen specific. Fig. 5A demonstrates that HER2-BsAb mediates T cell
cytotoxicity against the
HER2-positive cell line, UM SCC 47, but not the HER2-negative cell line HTB-
132. Fig. 5B
demonstrates that huOKT3 and trastuzumab can block the ability of HER2-BsAb to
mediate T
cell cytotoxicity.
[0056] Fig. 6 demonstrates that HER2-BsAb detects low levels of HER2 by
comparing the
HER2-BsAb mediated T cell cytotoxicity to the HER2 threshold of detection by
flow cytometry.
[0057] Fig. 7A, Fig. 7B, and Fig. 7C provide the specificity, affinity, and
antiproliferative
action of HER2-BsAb. Fig. 7A demonstrates that pre-incubation of the HER2-
positive SKOV3
ovarian carcinoma cell line blocks binding of HER2-BsAb. Fig. 7B demonstrates
that SKOV3
cells labeled with dilutions of trastuzumab or with HER2-BsAb display similar
curves when
mean fluorescence intensity (MFI) is plotted against antibody concentration.
Fib. 7C
demonstrates the antiproliferative action of HER2-BsAb compared against
trastuzumab in the
trastuzumab sensitive breast cancer cell line SKBR3.
[0058] Fig. 8 demonstrates that HER2-BsAb is effective against squamous
cell carcinoma of
the head and neck (SCCHN) cell lines. A panel of SCCHN cells were analyzed for
HER2-
BsAb-mediated cytotoxicity and EC50 and compared to the expression level of
HER2 in each
cell line as determined by flow cytometry and by qRT-PCR.
[0059] Fig. 9A, Fig. 9B, and Fig. 9C. HER2-BsAb mediates T cell
cytotoxicity against
SCCHN resistant to other HER targeted therapies. Fig. 9A demonstrates that the
SCCHN cell
-19-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
line PCI-30 expresses EGFR and HER2. Fig. 9B demonstrates that PCI-30 cells
are resistant to
HER-targeted therapies lapatinib, erlotinib, neratinib, trastuzumab, and
cetuximab. Fig. 9C
demonstrates that PCI-30 cells are sensitive to T cells in the presence of
HER2-BsAb. Data
represents the average of three different cytotoxicity assays.
[0060] Fig. 10 demonstrates that HER2-BsAb is effective against
osteosarcoma cell lines. A
panel of osteosarcoma cell lines were analyzed for HER2-BsAb-mediated
cytotoxicity and EC50
and compared to the expression level of HER2 in each cell line as determined
by flow cytometry
and by qRT-PCR
[0061] Fig. 11A, Fig. 11B, and Fig. 11C demonstrate that HER2-BsAb is
effective against
osteosarcoma cell lines resistant to other targeted therapies. Fig. 11A
demonstrates that the
osteosarcoma cell line U2OS expresses EGFR and HER2. Fig. 11B demonstrates
that USOS
cells are resistant to HER-targeted therapies lapatinib, erlotinib, neratinib,
trastuzumab, and
cetuximab. Fig. 11C demonstrates that USOS cells are sensitive to T cells in
the presence of
HER2-BsAb. Data represents the average of three different cytotoxicity assays.
[0062] Fig. 12A, Fig. 12B, Fig. 12C and Fig. 12D demonstrate that HER2-BsAb
is effective
against the HeLa cervical carcinoma cell line resistant to other targeted
therapies. Fig. 12A
demonstrates that HeLa cells express EGFR and HER2. Fig. 12B demonstrates that
HeLa cells
are resistant to HER-targeted therapies lapatinib, erlotinib, neratinib,
trastuzumab, and
cetuximab. Fig. 12C demonstrates that HeLa cells are sensitive to T cells in
the presence of
HER2-BsAb. Data represents the average of three different cytotoxicity assays.
Fig. 12D
demonstrates that pre-treatment with lapatinib enhances HeLa sensitivity to
HER2-BsAb.
[0063] Fig. 13 demonstrates that HER2-BsAb reduces tumor growth in vivo.
Fig. 13
demonstrates that HER2-BsAb protects against tumor progression in implanted
MCF7 breast
cancer cells mixed with PBMCs.
[0064] Fig. 14 demonstrates that HER2-BsAb protects against tumor
progression in
implanted HCC1954 breast cancer mixed with peripheral blood mononuclear cells
(PBMC) in
vivo.
[0065] Fig. 15 demonstrates that HER2-BsAb protects against a metastatic
model of tumor
progression induced by intravenous introduction of luciferase-tagged MCF7
cells in vivo.
[0066] Fig. 16A, Fig. 16B, Fig. 16C, and Fig. 16D demonstrate that HER2-
BsAb blocks the
metastatic tumor growth of luciferase-tagged MCF7 cells in vivo. Fig. 16A
represents mice
-20-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
without treatment. Fig. 16B represents mice treated with PBMC and HER2-C825.
Fig. 16C
represents mice treated with HER2-BsAb. Fig. 16D represents mice treated with
PBMC and
HER2-BsAb.
[0067] Fig. 17A, Fig. 17B, and Fig. 17C describe HER2-BsAb. Fig. 17A
depicts a
schematic of the HER2-BsAb. The arrow points to the N297A mutation introduced
into the
heavy chain to remove glycosylation. Fig. 17B depicts the purity of HER2-BsAb
as
demonstrated under reducing SDS-PAGE conditions. Fig. 17C depicts the purity
of HER2-BsAb
as demonstrated by size exclusion chromatography high performance liquid
chromatography
(SEC-HPLC).
[0068] Fig. 18A, Fig. 18B, and Fig. 18C demonstrate that HER2-BsAb has the
same
specificity, similar affinity, and antiproliferative effects as trastuzumab.
[0069] Fig. 19A and Fig. 19B demonstrate that HER2-BsAb redirected T cell
cytotoxicity is
HER2-specific and dependent on CD3.
[0070] Fig. 20 depicts HER2 expression and half maximal effective
concentration (EC50) in
the presence of ATC and HER2-BsAb in 35 different cell lines from different
tumor systems.
[0071] Fig. 21A, Fig. 21B, Fig. 21C, Fig. 21D, Fig. 21E, Fig. 21F, Fig.
21G, Fig. 2111, and
Fig. 211 demonstrate that HER2-BsAb mediates cytotoxic responses against
carcinoma cell lines
resistant to other HER-targeted therapies.
[0072] Fig. 22 demonstrates that the EC50 of HER2-BsAb correlates with the
HER2 level of
expression determined by flow-cytometry. pM=picomolar; MFI=mean fluorescence
intensity.
[0073] Fig. 23A, Fig. 23B, and Fig. 23C demonstrates that HER2-BsAb
mediates T cell
cytotoxicity against PD-Li-positive HCC1954 targets in a manner that is
relatively insensitive to
PD-1 blockade by pembrolizumab, even with PD-1 expression on effector T cells.
[0074] Fig. 24A and Fig. 24B demonstrates that HER2-BsAb mediates T cell
cytotoxicity
against PD-Li-positive HEK-293 targets in a manner that is relatively
insensitive to PD-1
expression on effector T cells. The cytotoxicity is an average of 6
experiments.
[0075] Fig. 25A, Fig. 25B, Fig. 25C, and Fig. 25D demonstrate that HER2-
BsAb is
effective against HER2-positive xenografts.
[0076] Fig. 26A, Fig. 26B, Fig. 26C, Fig. 26D, and Fig. 26E demonstrate in
vitro
characterization of HER2-BsAb. Fig. 26A: HER2-BsAb has the same specificity as
trastuzumab. Pre-Incubation of the HER2(+)high SKOV3 cells with trastuzumab
prevents
-21-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
HER2-BsAb binding. Fig. 26B: HER2-BsAb and trastuzumab have similar avidity
for SKOV3
cells. Mean fluorescence intensity ("MFI") was plotted against the antibody
concentration. Fig.
26C: HER2-BsAb maintained same anti-proliferative effects as trastuzumab
against the
trastuzumab-sensitive SKBR3 cells. Fig. 26D: HER2-BsAb mediates T-cell
cytotoxicity against
the HER2(+) MCF-7 cells but not the HER2(-) HTB-132 cells. Fig. 26E: Blocking
of HER2 or
CD3 by trastuzumab or huOKT3, abrogates HER2-BsAb T-cell cytotoxicity. HER2(+)
SCCHN
PCI-13 cells were used in the cytotoxicity assay. For this experiment 0.1
pg/mL of HER2-BsAb
with 10 pg/mL of the blocking antibodies were used.
[0077] Fig. 27A and Fig. 27B demonstrate HER2-BsAb binding to T cells and
redirecting
T-cell killing. Fig. 27A: FACS histograms of HER2-BsAb binding to naive T
cells purified from
fresh PBMC (left panel) or ATCs (right panel). Concentrations of BsAbs (m/106
cells) were
recorded on the top of the left histogram, and Rituxan was used as negative
control (mean
fluorescence intensity set at 5). Fig. 27B: HER2-BsAb redirected T-cell
killing of HER2(+)
AU565 breast cancer cells by 4-hour 51Cr release assay. BsAb was either mixed
directly with T
cells and AU565 together (mixing), or pre-incubated with T cells/target first
(T cells pre-armed
or AU565 pre-targeted), and unbound BsAb washed off before adding the other
cells. ATC-to-
target ratio was 10:1. Data points are shown as Mean SEM.
[0078] Fig. 28A, Fig. 28B, Fig. 28C, and Fig. 28D demonstrate that HER2-
BsAb mediates
cytotoxic responses against carcinoma cell lines resistant to other HER
targeted therapies. Fig.
28A, Fig. 28B, and Fig. 28C: Three representative cell lines were used for
FACS assay (upper
panel), proliferation assay (middle panel), and HER2-BsAb mediated CTL assay
(lower panel):
(Fig. 28A) SCCHN PCI-30, (Fig. 28B) breast carcinoma HCC-1954, and (Fig. 28C)
osteosarcoma U20S. Fig. 28D: HER2-BsAb EC50 inversely correlates with level of
HER2
expression. Each of the cell lines used in a cytotoxicity assay (Table 9) was
assayed at least
twice. The EC50 was determined each time and averaged. These values (except
those beyond
assay limit 5 nM) were compared to HER2 expression (MFI).
[0079] Fig. 29A and Fig. 29B demonstrate that HER2-BsAb-mediated in vitro T-
cell
cytotoxicity was relatively insensitive to PD-Li expression on the tumor
targets or PD-1
expression on T cells. Fig. 29A: FACS analysis of PD-Li expression in HCC1954
cells (left
panel), of induced PD-1 expression in ATCs (middle panel), and HER2-BsAb-
mediated
cytotoxicity (right panel). Fig. 29B: FACS analysis of PD-Li expression in HEK-
293 cells (left
-22-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
panel), and HER2-BsAb mediated cytotoxicity using the ATCs as in Fig. 29A
(middle panel).
Mean + SEM (n=6).
[0080] Fig. 30A, Fig. 30B, Fig. 30C, Fig. 30D, and Fig. 30E demonstrate
that HER2-BsAb
is effective against HER2(+) breast cancer cell line xenografts. Treatment
schedules were
marked on the figures, and doses of BsAbs and effector cells were detailed in
the Results of
Section 6.3.3. Data shown as mean + SEM (n = 5). Fig. 30A: intravenous
("i.v.") tumor plus
i.v. effector cells model: Bioluminescence changes of MCF7 breast cancers
during treatment.
Fig. 30B and Fig. 30C: subcutaneous ("s.c." tumor plus s.c. effector cells
(mixing) model: %
tumor growth of MCF7 (Fig. 30B), and tumor volume changes of HCC1954 (Fig.
30C). Fig.
30D: s.c. tumor plus i.v. effector cells model: tumor volume changes of
HCC1954. Fig. 30E:
HCC1954 s.c. tumor model as in (Fig. 30D), with treatments of one dose of PBMC
(2x107 cells
i.v.) at day 14, and two doses of BsAbs (100 i.v.) at day 12 and 15.
Representative images
(200X magnifications) of IHC staining of tumor sections collected 5 days after
i.v. PBMC were
shown.
[0081] Fig. 31A and Fig. 31B demonstrate that HER2-BsAb is effective
against HER2(+)
ovarian cancer cell line xenografts. Treatment schedules were marked on the
figures, and doses
of BsAbs and effector cells were detailed in the Results of Section 6.3.3.
Data shown as mean +
SEM (n = 4). Fig. 31A: intraperitoneal ("i.p.") tumor plus i.p./i.v. effector
cells model:
Bioluminescence changes of SKOV3-luc ovarian cancers during treatment. Fig.
31B:
Representative bioluminescence images at the beginning (Day 13) and ending
(Day 34) of the
treatment were shown.
[0082] Fig. 32A, Fig. 32B, Fig. 32C, Fig. 32D, and Fig. 32E demonstrate
that HER2-BsAb
is effective against HER2(+) PDXs. s.c. tumor plus i.v. effector cells model
was used for PDXs.
Treatment schedules were marked on the figures, and doses of BsAbs and
effector cells were
detailed in the Results of Section 6.3.3. Data shown as mean + SEM (n = 5).
Fig. 32A: Tumor
volume changes of EK gastric cancer PDX. Fig. 32B: IHC images of CD3 staining
from another
experiment with similar setting as in Fig. 32A. Representative images (200X
magnifications) of
IHC staining of tumor sections collected 36 days after i.v. PBMC were shown.
Fig. 32C: IHC
images (200X magnifications) of HER2 staining of control treated tumor
sections. Fig. 32D and
Fig. 32E: Average tumor volume changes of M37 breast cancer PDX (Fig. 32D),
and tumor
-23-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
growth of 5 individual mouse (black thin line) and averages (black thick line)
in each group (Fig.
32E).
[0083] Fig. 33A and Fig. 33B demonstrate that HER2-BsAb binding to CD3 on T
cells was
functionally monovalent. Fig. 33A: Cytokine release from naive T cells induced
by 16.7 nM
HER2-BsAb when compared to bivalent huOKT3 lgG and monovalent huOKT3 Fab, in
the
absence (left panel) or presence (right panel) of HER2(+) NCI-N87 gastric
tumor cells.
Cytokine release level below detection level was assigned as 1 pg/ml. Fig.
33B: T cell
proliferation stimulated by 67 nM of the related antibodies, in the absence of
tumor targets. T
cells only (Control) as the negative control. OD reading at 450 nm (AU) was
shown. All data
points are shown as Mean+ SD.
[0084] Fig. 34 demonstrates that HER2-BsAb is effective against HER2(+)
breast cancer cell
line xenografts that express PDL1 but are resistant to PD1 or PDL1 treatment.
s.c. tumor plus
i.v. effector cell model: tumor volume changes of HCC1954. Data shown as mean
+ SEM (n =
5). Treatment schedules are marked on the figures. s.c. 5 x 106 HCC1954
xenografts were
treated with i.v. PBMC (7.5 x 106, once per week for 2 weeks), and i.v. HER2-
BsAb, anti-PD1
Pembrolizumab, or anti-PDL1 Atezolizumab (100 ug each, twice per week for 4
weeks).
Tumors were completely eradicated with HER2-BsAb treatment, in contrast to no
effect for
treatment with PD1/PDL1 blockade (i.e., treatment with anti-PD1 Pembrolizumab
or anti-PDL1
Atezolizumab).
5. DETAILED DESCRIPTION
[0085] Provided herein are bispecific binding molecules that bind to both
HER2 and CD3.
Also provided herein are isolated nucleic acids (polynucleotides), such as
complementary DNA
(cDNA), encoding such bispecific binding molecules or fragments thereof.
Further provided are
vectors (e.g., expression vectors) and cells (e.g., ex vivo cells) comprising
nucleic acids
(polynucleotides) or vectors (e.g., expression vectors) encoding such
bispecific binding
molecules or fragments thereof. Also provided herein are methods of making
such bispecific
binding molecules, cells, and vectors. Also provided herein are T cells bound
to bispecific
binding molecules provided herein. Also provided herein are methods of binding
such bispecific
binding molecules to T cells. In other embodiments, provided herein are
methods and uses for
treating HER2-positive cancers using the bispecific binding molecules, nucleic
acids, vectors,
-24-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
and/or T cells described herein. Additionally, related compositions (e.g.,
pharmaceutical
compositions), kits, and diagnostic methods are also provided herein.
[0086] In certain embodiments, provided herein are bispecific binding
molecules that
specifically bind to HER2 and to CD3, and invoke T cell cytotoxicity for
treating cancer.
Without being bound by any theory, it is believed that the bispecific binding
molecules described
herein not only bind tumors to T cells, they also cross-link CD3 on T cells
and initiate the
activation cascade, and, this way, T cell receptor (TCR)-based cytotoxicity is
redirected to
desired tumor targets, bypassing major histocompatibility complex (MHC)
restrictions.
5.1 BISPECIFIC BINDING MOLECULES
[0087] Provided herein are bispecific binding molecules that bind to HER2
and CD3. A
binding molecule, which can be used within the methods provided herein, is a
bispecific binding
molecule comprising an aglycosylated monoclonal antibody that is an
immunoglobulin that
binds to HER2, comprising two identical heavy chains and two identical light
chains, said light
chains being a first light chain and a second light chain, wherein the first
light chain is fused to a
first single chain variable fragment (scFv), via a peptide linker, to create a
first fusion
polypeptide, and wherein the second light chain is fused to a second scFv, via
a peptide linker, to
create a second fusion polypeptide, wherein the first and second scFv (i) are
identical, and (ii)
bind to CD3, and wherein the first and second fusion polypeptides are
identical.
[0088] HER2 is a member of the epidermal growth factor receptor (EGFR)
family of
receptor tyrosine kinases. In a specific embodiment, HER2 is human HER2.
GenBankTM
accession number NM 004448.3 (SEQ ID NO: 1) provides an exemplary human HER2
nucleic
acid sequence. GenBankTM accession number NP 004439.2 (SEQ ID NO: 2) provides
an
exemplary human HER2 amino acid sequence. In another specific embodiment, HER2
is canine
HER2. GenBankTM accession number NM 001003217.1 (SEQ ID NO: 3) provides an
exemplary canine HER2 nucleic acid sequence. GenBankTM accession number NP
001003217.1
(SEQ ID NO: 4) provides an exemplary canine HER2 amino acid sequence.
[0089] CD3 is a T cell co-receptor comprised of a gamma chain, a delta
chain, and two
epsilon chains. In a specific embodiment, CD3 is a human CD3. GenBankTM
accession number
NM 000073.2 (SEQ ID NO: 5) provides an exemplary human CD3 gamma nucleic acid
sequence. GenBankTM accession number NP 000064.1 (SEQ ID NO: 6) provides an
exemplary
human CD3 gamma amino acid sequence. GenBankTM accession number NM 000732.4
(SEQ
-25-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
ID NO: 7) provides an exemplary human CD3 delta nucleic acid sequence.
GenBankTM
accession number NP 000723.1 (SEQ ID NO: 8) provides an exemplary human CD3
delta
amino acid sequence. GenBankTM accession number NM 000733.3 (SEQ ID NO: 9)
provides
an exemplary human CD3 epsilon nucleic acid sequence. GenBankTM accession
number
NP 000724.1 (SEQ ID NO: 10) provides an exemplary human CD3 epsilon amino acid
sequence. In another specific embodiment, CD3 is a canine CD3. GenBankTM
accession number
NM 001003379.1 (SEQ ID NO: 11) provides an exemplary canine CD3 epsilon
nucleic acid
sequence. GenBankTM accession number NP 001003379.1 (SEQ ID NO: 12) provides
an
exemplary canine CD3 epsilon amino acid sequence.
[0090] The immunoglobulin in the bispecific binding molecules of the
invention can be, as
non-limiting examples, a monoclonal antibody, a naked antibody, a chimeric
antibody, a
humanized antibody, or a human antibody. As used herein, the term
"immunoglobulin" is used
consistent with its well known meaning in the art, and comprises two heavy
chains and two light
chains. Methods for making antibodies are described in Section 5.3.
[0091] A chimeric antibody is a recombinant protein that contains the
variable domains
including the complementarity-determining regions (CDRs) of an antibody
derived from one
species, preferably a rodent antibody, while the constant domains of the
antibody molecule is
derived from those of a human antibody. For veterinary applications, the
constant domains of
the chimeric antibody may be derived from that of other species, such as, for
example, horse,
monkey, cow, pig, cat, or dog.
[0092] A humanized antibody is an antibody produced by recombinant DNA
technology, in
which some or all of the amino acids of a human immunoglobulin light or heavy
chain that are
not required for antigen binding (e.g., the constant regions and the framework
regions of the
variable domains) are used to substitute for the corresponding amino acids
from the light or
heavy chain of the cognate, nonhuman antibody. By way of example, a humanized
version of a
murine antibody to a given antigen has on both of its heavy and light chains
(1) constant regions
of a human antibody; (2) framework regions from the variable domains of a
human antibody;
and (3) CDRs from the murine antibody. When necessary, one or more residues in
the human
framework regions can be changed to residues at the corresponding positions in
the murine
antibody so as to preserve the binding affinity of the humanized antibody to
the antigen. This
change is sometimes called "back mutation." Similarly, forward mutations may
be made to
-26-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
revert back to murine sequence for a desired reason, e.g., stability or
affinity to antigen. Without
being bound by any theory, humanized antibodies generally are less likely to
elicit an immune
response in humans as compared to chimeric human antibodies because the former
contain
considerably fewer non-human components.
[0093] The term "epitope" is art-recognized and is generally understood by
those of skill in
the art to refer to the region of an antigen that interacts with an antibody.
An epitope of a protein
antigen can be linear or conformational, or can be formed by contiguous or
noncontiguous amino
acid sequences of the antigen.
[0094] A scFv is an art-recognized term. An scFv comprises a fusion protein
of the variable
regions of the heavy (VH) and light (VL) chains of an immunoglobulin, wherein
the fusion
protein retains the same antigen specificity as the whole immunoglobulin. The
VH is fused to the
VL via a peptide linker (such a peptide linker is sometimes referred to herein
as an "intra-scFv
peptide linker").
[0095] In certain embodiments of the invention, the scFv has a peptide
linker that is between
5-30, 5-25, 5-15, 10-30, 10-20, 10-15, 15-30, or 15-25 amino acid residues in
length. In certain
embodiments, the scFv peptide linker displays one or more characteristics
suitable for a peptide
linker known to one of ordinary skill in the art. In certain embodiments, the
scFv peptide linker
comprises amino acids that allow for scFv peptide linker solubility, such as,
for example, serine
and threonine. In certain embodiments, the scFv peptide linker comprises amino
acids that allow
for scFv peptide linker flexibility, such as, for example, glycine. In certain
embodiments, the
scFv peptide linker connects the N-terminus of the VH to the C-terminus of the
VL. In certain
embodiments, the scFv peptide linker can connect the C-terminus of the VH to
the N-terminus of
the VL. In certain embodiments, the scFv peptide linker is a linker as
described in Table 1,
below (e.g., any one of SEQ ID NOs: 14, or 35-41). In a preferred embodiment,
the peptide
linker is SEQ ID NO: 14.
[0096] In certain embodiments of the bispecific binding molecules of the
invention, the scFv
that binds to CD3 comprises the VH and the VL of a CD3-specific antibody known
in the art,
such as, for example, huOKT3 (see, for example, Adair et at., 1994, Hum
Antibodies
Hybridomas 5 :41-47), YTH12.5 (see, for example Routledge et at., 1991, Eur J
Immunol, 21:
2717-2725), HUM291 (see, for example, Norman et al., 2000, Clinical
Transplantation, 70(12):
1707-1712), teplizumab (see, for example, Herold et at., 2009, Clin Immunol,
132: 166-173),
-27-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
huCLB-T3/4 (see, for example, Labrijn et at., 2013, Proceedings of the
National Academy of
Sciences, 110(13): 5145-5150), otelixizumab (see, for example, Keymeulen et
al., 2010,
Diabetologia, 53: 614-623), blinatumomab (see, for example, Cheadle, 2006,
Curr Opin Mol
Ther, 8(1): 62-68), MT110 (see, for example, Silke and Gires, 2011, MAbs,
3(1): 31-37),
catumaxomab (see, for example, Heiss and Murawa, 2010, Int J Cancer, 127(9):
2209-2221),
28F11 (see, for example, Canadian Patent Application CA 2569509 Al), 27H5
(see, for
example, Canadian Patent Application CA 2569509 Al), 23F10 (see, for example,
Canadian
Patent Application CA 2569509 Al), 15C3 (see, for example, Canadian Patent
Application CA
2569509 Al), visilizumab (see, for example, Dean et at., 2012, Swiss Med Wkly,
142: w13711),
and Hum291 (see, for example, Dean et at., 2012, Swiss Med Wkly, 142: w13711).
[0097] In certain embodiments, the scFv in a bispecific binding molecule of
the invention
binds to the same epitope as a CD3-specific antibody known in the art. In a
specific
embodiment, the scFv in a bispecific binding molecule of the invention binds
to the same epitope
as the CD3-specific antibody huOKT3. Binding to the same epitope can be
determined by
assays known to one skilled in the art, such as, for example, mutational
analyses or
crystallographic studies. In certain embodiments, the scFv competes for
binding to CD3 with an
antibody known in the art. In a specific embodiment, the scFv in a bispecific
binding molecule
of the invention competes for binding to CD3 with the CD3-specific antibody
huOKT3.
Competition for binding to CD3 can be determined by assays known to one
skilled in the art,
such as, for example, flow cytometry. See, for example, Section 6.1.2.4. In
certain
embodiments, the scFv comprises a VH with at least 85%, 90%, 95%, 98%, or at
least 99%
similarity to the VH of a CD3-specific antibody known in the art. In certain
embodiments, the
scFv comprises the VH of a CD3-specific antibody known in the art, comprising
between 1 and 5
conservative amino acid substitutions. In certain embodiments, the scFv
comprises a VL with at
least 85%, 90%, 95%, 98%, or at least 99% similarity to the VL of a CD3-
specific antibody
known in the art. In certain embodiments, the scFv comprises the VL of a CD3-
specific antibody
known in the art, comprising between 1 and 5 conservative amino acid
substitutions.
[0098] Conservative amino acid substitutions are amino acid substitutions
that occur within a
family of amino acids, wherein the amino acids are related in their side
chains. Generally,
genetically encoded amino acids are divided into families: (1) acidic,
comprising aspartate and
glutamate; (2) basic, comprising arginine, lysine, and histidine; (3) non-
polar, comprising
-28-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
isoleucine, alanine, valine, proline, methionine, leucine, phenylalanine,
tryptophan; and (4)
uncharged polar, comprising cysteine, threonine, glutamine, glycine,
asparagine, serine, and
tyrosine. In addition, an aliphatic-hydroxy family comprises serine and
threonine. In addition,
an amide-containing family comprises asparagine and glutamine. In addition, an
aliphatic family
comprises alanine, valine, leucine and isoleucine. In addition, an aromatic
family comprises
phenylalanine, tryptophan, and tyrosine. Finally, a sulfur-containing side
chain family comprises
cysteine and methionine. As an example, one skilled in the art would
reasonably expect an
isolated replacement of a leucine with an isoleucine or valine, an aspartate
with a glutamate, a
threonine with a serine, or a similar replacement of an amino acid with a
structurally related
amino acid will not have a major effect on the binding or properties of the
resulting molecule,
especially if the replacement does not involve an amino acid within a
framework site. Preferred
conservative amino acid substitution groups include: lysine-arginine, alanine-
valine,
phenylalanine-tyrosine, glutamic acid-aspartic acid, valine-leucine-
isoleucine, cysteine-
methionine, and asparagine-glutamine.
[0099] In a preferred embodiment, the scFv is derived from the huOKT3
antibody, and thus
contains the VH and VL of huOKT3 monoclonal antibody (SEQ ID NOS: 15 and 16,
respectively). See, for example, Van Wauwe et at., 1991, nature, 349: 293-299.
In specific
embodiments of the bispecific binding molecule, the scFv is derived from the
huOKT3
monoclonal antibody and has no more than 5 amino acid mutations relative to
native huOKT3
VH and VL sequences. In certain embodiments of the bispecific binding
molecule, the scFv is
derived from the huOKT3 monoclonal antibody and comprises one or more
mutations, relative to
native huOKT3 VH and VL sequences, to stabilize disulfide binding. In certain
embodiments of
the bispecific binding molecule, the stabilization of disulfide binding
prevents aggregation of the
bispecific binding molecule. In certain embodiments of the bispecific binding
molecule, the
stabilization of disulfide binding reduces aggregation of the bispecific
binding molecule as
compared to aggregation of the bispecific binding molecule without the
stabilization of disulfide
binding. In certain embodiments of the bispecific binding molecule, the one or
more mutations
to stabilize disulfide binding comprise a VH G44C mutation and a VL Q100C
mutation (e.g., as
present in SEQ ID NOS: 54-59). In certain embodiments of the bispecific
binding molecule, the
one or more mutations to stabilize disulfide binding are the replacement of
the amino acid
residue at VH44(according to the Kabat numbering system) with a cysteine and
the replacement
-29-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
of the amino acid residue at VL100 (according to the Kabat numbering system)
with a cysteine so
as to introduce a disulfide bond between VH44 and VL100 (e.g., as present in
SEQ ID NOS: 54-
59). In an especially preferred embodiment, the scFv comprises the VH of
huOKT3 comprising
the amino acid substitution at numbered position 105, wherein the cysteine is
substituted with a
serine (SEQ ID NO: 17). In certain embodiments, the sequence of the VH of the
scFv is as
described in Table 4, below (e.g., any one of SEQ ID NOs: 15, 17, or 64). In
certain
embodiments, the sequence of the VL of the scFv is as described in Table 5,
below (e.g., any one
of SEQ ID NOs: 16 or 65). In certain embodiments, the sequence of the scFv is
as described in
Table 6, below (e.g., any one of SEQ ID NOs: 19 or 48-59). In a preferred
embodiment, the
sequence of the scFv is SEQ ID NO: 19. In a specific embodiment, the scFv
comprises a variant
of the VH of huOKT3 that has no more than 5 amino acid mutations relative to
the native
sequence of huOKT3 VH. In a specific embodiment, the scFv comprises a variant
of the VL of
huOKT3 that has no more than 5 amino acid mutations relative to the native
sequence of
huOKT3 VL.
[00100] The sequences of the variable regions of an anti-CD3 scFv may be
modified by
insertions, substitutions and deletions to the extent that the resulting scFv
maintains the ability to
bind to CD3, as determined by, for example, ELISA, flow cytometry, and
BiaCoreTM. The
ordinarily skilled artisan can ascertain the maintenance of this activity by
performing the
functional assays as described herein below, such as, for example, binding
analyses and
cytotoxicity analyses.
[00101] In certain embodiments, the peptide linker conjugating the
immunoglobulin light
chain and the scFv is between 5-30, 5-25, 5-15, 10-30, 10-20, 10-15, 15-30, or
15-25 amino
acids in length. In certain embodiments, the peptide linker displays one or
more characteristics
suitable for a peptide linker known to one of ordinary skill in the art. In
certain embodiments,
the peptide linker comprises amino acids that allow for peptide linker
solubility, such as, for
example, serine and threonine. In certain embodiments, the peptide linker
comprises amino
acids that allow for peptide linker flexibility, such as, for example,
glycine. In certain
embodiments, the sequence of the peptide linker conjugating the immunoglobulin
light chain and
the scFv is as described in Table 1, below (e.g., any one of SEQ ID NOs: 14 or
35-41). In
preferred embodiments, the peptide linker is SEQ ID NO: 14.
-30-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
[00102] In certain embodiments of the bispecific binding molecules of the
invention, the
immunoglobulin that binds to HER2 comprises the heavy chain and/or the light
chain of a
HER2-specific antibody known in the art, such as, for example, trastuzumab
(see, for example,
Baselga et al. 1998, Cancer Res 58(13): 2825-2831), M-111 (see, for example,
Higgins et at.,
2011, J Clin Oncol, 29(Suppl): Abstract TPS119), pertuzumab (see, for example,
Franklin et at.,
2004, Cancer Cell, 5: 317-328), ertumaxomab (see, for example, Kiewe and
Thiel, 2008, Expert
Opin Investig Drugs, 17(10): 1553-1558), MDXH210 (see, for example, Schwaab et
al., 2001,
Journal of Immunotherapy, 24(1): 79-87), 2B1 (see, for example, Borghaei et
al., 2007, J
Immunother, 30: 455-467), and MM-302 (see, for example, Wickham and Futch,
2012, Cancer
Research, 72(24): Supplement 3). In certain embodiments of the bispecific
binding molecules of
the invention, the immunoglobulin that binds to HER2 comprises the heavy chain
of
trastuzumab. In certain embodiments of the bispecific binding molecules of the
invention, the
immunoglobulin that binds to HER2 comprises the sequence as set forth in SEQ
ID NO: 23. In
certain embodiments of the bispecific binding molecules of the invention, the
immunoglobulin
that binds to HER2 comprises a variant of the heavy chain of trastuzumab (see,
e.g., Table 2,
below). In a specific embodiment of the bispecific binding molecules of the
invention, the
immunoglobulin that binds to HER2 comprises a variant of the light chain of
trastuzumab that
has no more than 5 amino acid mutations relative to the native sequence of
trastuzumab. In
certain embodiments of the bispecific binding molecules of the invention, the
immunoglobulin
that binds to HER2 comprises the light chain of trastuzumab (SEQ ID NO: 25).
In certain
embodiments of the bispecific binding molecules of the invention, the
immunoglobulin that
binds to HER2 comprises a variant of the light chain of trastuzumab. In a
specific embodiment
of the bispecific binding molecules of the invention, the immunoglobulin that
binds to HER2
comprises a variant of the light chain of trastuzumab that has no more than 5
amino acid
mutations relative to the native sequence of trastuzumab.
[00103] In certain embodiments of the bispecific binding molecules of the
invention, the
immunoglobulin that binds to HER2 binds to the same epitope as a HER2-specific
antibody
known in the art. In a specific embodiment, the immunoglobulin in a bispecific
binding
molecule of the invention binds to the same epitope as trastuzumab. Binding to
the same epitope
can be determined by assays known to one skilled in the art, such as, for
example, mutational
analyses or crystallographic studies. In certain embodiments, the
immunoglobulin that binds to
-31-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
HER2 competes for binding to HER2 with an antibody known in the art. In a
specific
embodiment, the immunoglobulin in a bispecific binding molecule of the
invention competes for
binding to HER2 with trastuzumab. Competition for binding to HER2 can be
determined by
assays known to one skilled in the art, such as, for example, flow cytometry.
See, for example,
Section 6.1.2.4. In certain embodiments, the immunoglobulin comprises a VH
with at least 85%,
90%, 95%, 98%, or at least 99% similarity to the VH of a HER2-specific
antibody known in the
art. In certain embodiments, the immunoglobulin comprises the VH of a HER2-
specific antibody
known in the art, comprising between 1 and 5 conservative amino acid
substitutions. In certain
embodiments, the immunoglobulin comprises a VL with at least 85%, 90%, 95%,
98%, or at least
99% similarity to the VL of a HER2-specific antibody known in the art. In
certain embodiments,
the immunoglobulin comprises the VL of a HER2-specific antibody known in the
art, comprising
between 1 and 5 conservative amino acid substitutions. In certain embodiments,
the
immunoglobulin comprises a VH of a heavy chain described in Table 2, below
(e.g., the VH of
any one of SEQ ID NOs: 23, 27, 62, or 63). In certain embodiments, the
immunoglobulin
comprises a VL of a light chain described in Table 3, below (e.g., the VL of
SEQ ID NO: 25).
[00104] The sequences of the variable regions of an anti-HER2 antibody may be
modified by
insertions, substitutions and deletions to the extent that the resulting
antibody maintains the
ability to bind to HER2, as determined by, for example, ELISA, flow cytometry,
and BiaCoreTM.
The ordinarily skilled artisan can ascertain the maintenance of this activity
by performing the
functional assays as described herein below, such as, for example, binding
analyses and
cytotoxicity analyses.
[00105] In certain embodiments of the bispecific binding molecules of the
invention, the
immunoglobulin that binds to HER2 is an IgG1 immunoglobulin.
[00106] Methods of producing human antibodies are known to one skilled in the
art, such as,
for example, phage display methods described above using antibody libraries
derived from
human immunoglobulin sequences. See also, U.S. Pat. Nos. 4,444,887 and
4,716,111; and PCT
publications WO 98/46645, WO 98/60433, WO 98/24893, WO 98/16664, WO 96/34096,
WO
96/33735, and WO 91/10741; each of which is incorporated herein by reference
in its entirety.
The techniques of Cole et at., and Boerder et at., are also available for the
preparation of human
monoclonal antibodies (Cole et at., Monoclonal Antibodies and Cancer Therapy,
Alan R. Riss,
(1985); and Boerner et al., J. Immunol., 147(1):86-95, (1991)).
-32-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
[00107] In certain embodiments, human antibodies are produced using transgenic
mice, which
are incapable of expressing functional endogenous mouse immunoglobulins, but
which can
express human immunoglobulin genes. For example, the human heavy and light
chain
immunoglobulin gene complexes may be introduced randomly or by homologous
recombination
into mouse embryonic stem cells. Alternatively, the human variable region,
constant region, and
diversity region may be introduced into mouse embryonic stem cells in addition
to the human
heavy and light chain genes. The mouse heavy and light chain immunoglobulin
genes may be
rendered non-functional separately or simultaneously with the introduction of
human
immunoglobulin loci by homologous recombination. In particular, homozygous
deletion of the
JH region prevents endogenous antibody production. The modified embryonic stem
cells are
expanded and microinjected into blastocysts to produce chimeric mice. The
chimeric mice are
then bred to produce homozygous offspring which express human antibodies. The
transgenic
mice are immunized in the normal fashion with a selected antigen, for example,
all or a portion
of a polypeptide provided herein. Monoclonal antibodies directed against the
antigen can be
obtained from the immunized, transgenic mice using conventional hybridoma
technology. The
human immunoglobulin transgenes harbored by the transgenic mice rearrange
during B cell
differentiation, and subsequently undergo class switching and somatic
mutation. Thus, using
such a technique, it is possible to produce therapeutically useful IgG, IgA,
IgM and IgE
antibodies. For an overview of this technology for producing human antibodies,
see Lonberg
and Huszar, Int. Rev. Immunol. 13:65-93 (1995). For a detailed discussion of
this technology for
producing human antibodies and human monoclonal antibodies and protocols for
producing such
antibodies, see, for example, PCT publications WO 98/24893; WO 92/01047; WO
96/34096;
WO 96/33735; European Patent No. 0 598 877; U.S. Pat. Nos. 5,413,923;
5,625,126; 5,633,425;
5,569,825; 5,661,016; 5,545,806; 5,814,318; 5,886,793; 5,916,771; and
5,939,598, which are
incorporated by reference herein in their entirety. In addition, companies
such as Abgenix, Inc.
(Freemont, Calif), Genpharm (San Jose, Calif.), and Medarex, Inc. (Princeton,
N.J.) can be
engaged to provide human antibodies directed against a selected antigen using
technology
similar to that described above.
[00108] Human monoclonal antibodies can also be made by immunizing mice
transplanted
with human peripheral blood leukocytes, splenocytes or bone marrows (e.g.,
Trioma techniques
of XTL). Completely human antibodies which recognize a selected epitope can be
generated
-33-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
using a technique referred to as "guided selection." In this approach a
selected non-human
monoclonal antibody, for example, a mouse antibody, is used to guide the
selection of a
completely human antibody recognizing the same epitope. See, for example,
Jespers et at.,
Bio/technology 12:899-903 (1988). Human antibodies may also be generated by in
vitro
activated B cells. See U.S. Pat. Nos. 5,567,610 and 5,229,275, which are
incorporated in their
entirety by reference.
[00109] Methods for making humanized antibodies are known to one skilled in
the art. See,
for example, Winter EP 0 239 400; Jones et at., Nature 321:522-525 (1986);
Riechmann et at.,
Nature 332:323-327 (1988); Verhoeyen et at., Science 239: 1534-1536 (1988);
Queen et at.,
Proc. Nat. Acad. ScL USA 86:10029 (1989); U.S. Pat. No. 6,180,370; and Orlandi
et al., Proc.
Natl. Acad. Sd. USA 86:3833 (1989); the disclosures of all of which are
incorporated by
reference herein in their entireties. Generally, the transplantation of murine
(or other non-
human) CDRs onto a human antibody is achieved as follows. The cDNAs encoding
heavy and
light chain variable domains are isolated from a hybridoma. The DNA sequences
of the variable
domains, including the CDRs, are determined by sequencing. The DNAs, encoding
the CDRs
are inserted into the corresponding regions of a human antibody heavy or light
chain variable
domain coding sequences, attached to human constant region gene segments of a
desired isotype
(e.g., gamma-1 for CH and K for CL), are gene synthesized. The humanized heavy
and light
chain genes are co-expressed in mammalian host cells (e.g., CHO or NSO cells)
to produce
soluble humanized antibody. To facilitate large scale production of
antibodies, it is often
desirable select for high expressor using a DHFR gene or GS gene in the
producer line. These
producer cell lines are cultured in bioreactors, or hollow fiber culture
system, or WAVE
technology, to produce bulk cultures of soluble antibody, or to produce
transgenic mammals
(e.g., goats, cows, or sheep) that express the antibody in milk (see, e.g.,
U.S. Pat. No. 5,827,690).
[00110] Antibody fragments can be produced by enzymatic cleavage, synthetic or
recombinant techniques, as known in the art and/or as described herein.
Antibodies can also be
produced in a variety of truncated forms using antibody genes in which one or
more stop codons
have been introduced upstream of the natural stop site. For example, a
combination gene
encoding a F(ab')2 heavy chain portion can be designed to include DNA
sequences encoding the
CH, domain and/or hinge region of the heavy chain. The various portions of
antibodies can be
joined together chemically by conventional techniques, or can be prepared as a
contiguous
-34-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
protein using genetic engineering techniques. In certain embodiments, elements
of a human
heavy and light chain locus are introduced into strains of mice derived from
embryonic stem cell
lines that contain targeted disruptions of the endogenous heavy chain and
light chain loci. The
transgenic mice can synthesize human antibodies specific for human antigens,
and the mice can
be used to produce human antibody-secreting hybridomas. Methods for obtaining
human
antibodies from transgenic mice are described by Green et at., Nature Genet.
7:13 (1994),
Lonberg et at., Nature 368:856 (1994), and Taylor et at., Int. Immun. 6:579
(1994). A fully
human antibody also can be constructed by genetic or chromosomal transfection
methods, as
well as phage display technology, all of which are known in the art. See, for
example,
McCafferty et al., Nature 348:552-553 (1990) for the production of human
antibodies and
fragments thereof in vitro, from immunoglobulin variable domain gene
repertoires from
unimmunized donors. In this technique, antibody variable domain genes are
cloned in-frame into
either a major or minor coat protein gene of a filamentous bacteriophage, and
displayed as
functional antibody fragments on the surface of the phage particle. Because
the filamentous
particle contains a single-stranded DNA copy of the phage genome, selections
based on the
functional properties of the antibody also result in selection of the gene
encoding the antibody
exhibiting those properties. In this way, the phage mimics some of the
properties of the B cell.
Phage display can be performed in a variety of formats, for their review, see
e.g. Johnson and
Chiswell, Current Opinion in Structural Biology 3:5564-571 (1993).
[00111] Antibody humanization can also be performed by, for example,
synthesizing a
combinatorial library comprising the six CDRs of a non-human target monoclonal
antibody
fused in frame to a pool of individual human frameworks. A human framework
library that
contains genes representative of all known heavy and light chain human
germline genes can be
utilized. The resulting combinatorial libraries can then be screened for
binding to antigens of
interest. This approach can allow for the selection of the most favorable
combinations of fully
human frameworks in terms of maintaining the binding activity to the parental
antibody.
Humanized antibodies can then be further optimized by a variety of techniques.
[00112] Antibody humanization can be used to evolve mouse or other non-human
antibodies
into "fully human" antibodies. The resulting antibody contains only human
sequence and no
mouse or non-human antibody sequence, while maintaining similar binding
affinity and
specificity as the starting antibody.
-35-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
[00113] For full length antibody molecules, the immunoglobulin genes can be
obtained from
genomic DNA or mRNA of hybridoma cell lines. Antibody heavy and light chains
are cloned in
a mammalian vector system. Assembly is documented with double strand sequence
analysis.
The antibody construct can be expressed in other human or mammalian host cell
lines. The
construct can then be validated by transient transfection assays and Western
blot analysis of the
expressed antibody of interest. Stable cell lines with the highest
productivity can be isolated and
screened using rapid assay methods.
[00114] In one approach, a hybridoma is produced by fusing a suitable immortal
cell line
(e.g., a myeloma cell line such as, but not limited to, 5p2/0, 5p2/0-AG14,
NSO, NS1, N52, AE-
1, L.5, >243, P3X63Ag8.653, 5p2 5A3, 5p2 MAT, 5p2 SS1, 5p2 5A5, U937, MLA 144,
ACT
IV, MOLT4, DA-1, JURKAT, WEHI, K-562, COS, RAJI, NIH 3T3, HL-60, MLA 144,
NAMAIWA, NEURO 2A), or the like, or heteromylomas, fusion products thereof, or
any cell or
fusion cell derived therefrom, or any other suitable cell line as known in the
art. See, for
example, the ATCC or LifeTech website, and the like, with antibody producing
cells, such as,
but not limited to, isolated or cloned spleen, peripheral blood, lymph,
tonsil, or other immune or
B cell containing cells, or any other cells expressing heavy or light chain
constant or variable or
framework or CDR sequences, either as endogenous or heterologous nucleic acid,
as
recombinant or endogenous, viral, bacterial, algal, prokaryotic, amphibian,
avian, insect,
reptilian, fish, mammalian, rodent, equine, ovine, goat, sheep, primate,
eukaryotic, genomic
DNA, cDNA, rDNA, mitochondrial DNA or RNA, chloroplast DNA or RNA, hnRNA,
mRNA,
tRNA, single, double or triple stranded, hybridized, and the like or any
combination thereof.
See, for example, Ausubel, supra, and Colligan, Immunology, supra, chapter 2,
entirely
incorporated herein by reference. The fused cells (hybridomas) or recombinant
cells can be
isolated using selective culture conditions or other suitable known methods,
and cloned by
limiting dilution or cell sorting, or other known methods. Cells which produce
antibodies with
the desired specificity can be selected by a suitable assay (e.g., ELISA).
[00115] In a preferred specific embodiment, the bispecific binding molecule
comprises a
variant Fc region, wherein said variant Fc region comprises at least one amino
acid modification
relative to a wild-type Fc region, such that said molecule does not bind or
has reduced binding to
an Fc receptor (FcR), in soluble form or cell-bound form (including on immune-
effector cells,
such as, for example, NK cells, monocytes, and neutrophils). These FcRs
include, but are not
-36-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
limited to, FcR1 (CD64), FcRII (CD32), and FcRIII (CD16). The affinity to
FcR(n), the
neonatal Fe receptor, is not affected, and thus maintained in the bispecific
binding molecule. For
example, if the immunoglobulin is an IgG, preferably, the IgG has reduced or
no affinity for an
Fe gamma receptor. In certain embodiments, one or more positions within the Fe
region that
makes a direct contact with Fe gamma receptor, such as, for example, amino
acids 234-239
(hinge region), amino acids 265-269 (B/C loop), amino acids 297-299 (C'/E
loop), and amino
acids 327-332 (F/G) loop, are mutated such that the bispecific binding
molecule has a decreased
or no affinity for an Fe gamma receptor. See, for example, Sondermann et at.,
2000, Nature,
406: 267-273, which is incorporated herein by reference in its entirety.
Preferably, for an IgG,
the mutation N297A is made to destroy Fe receptor binding. In certain
embodiments, affinity of
the bispecific binding molecule or fragment thereof for an Fe gamma receptor
is determined by,
for example, BiaCoreTM assay, as described, for example, in Okazaki et at.,
2004. J Mol Biol,
336(5):1239-49. See also, Section 6. In certain embodiments, the bispecific
binding molecule
comprising such a variant Fe region binds an Fe receptor on a FcR-bearing
immune-effector cell
with less than 25%, 20%, 15%, 10%, or 5% binding as compared to a reference Fe
region.
Without being bound by any particular theory, a bispecific binding molecule
comprising such a
variant Fe region will have a decreased ability to induce a cytokine storm. In
preferred
embodiments, the bispecific binding molecule comprising such a variant Fe
region does not bind
an Fe receptor in soluble form or as a cell-bound form.
[00116] In certain embodiments, the bispecific binding molecule comprises a
variant Fe
region, such as, for example, an Fe region with additions, deletions, and/or
substitutions to one or
more amino acids in the Fe region of an antibody provided herein in order to
alter effector
function, or enhance or diminish affinity of antibody to FcR. In a preferred
embodiment, the
affinity of the antibody to FcR is diminished. Reduction or elimination of
effector function is
desirable in certain cases, such as, for example, in the case of antibodies
whose mechanism of
action involves blocking or antagonism but not killing of the cells bearing a
target antigen. In
certain embodiments, the Fe variants provided herein may be combined with
other Fe
modifications, including but not limited to modifications that alter effector
function. In certain
embodiments, such modifications provide additive, synergistic, or novel
properties in antibodies
or Fe fusions. Preferably, the Fe variants provided herein enhance the
phenotype of the
modification with which they are combined.
-37-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
[00117] In preferred embodiments, the bispecific binding molecule of the
invention is
aglycosylated. Preferably, this is achieved by mutating the anti-HER2
immunoglobulin portion
of the bispecific binding molecule in its Fc receptor to destroy a
glycosylation site, preferably an
N-linked glycosylation site. In another specific embodiment, an immunoglobulin
is mutated to
destroy an N-linked glycosylation site. In certain preferred embodiments, the
bispecific binding
molecule has been mutated to destroy an N-linked glycosylation site. In
certain embodiments,
the heavy chain of the bispecific binding molecule has an amino acid
substitution to replace an
asparagine that is an N-linked glycosylation site, with an amino acid that
does not function as a
glycosylation site. In a preferred embodiment, the method encompasses deleting
the
glycosylation site of the Fc region of a bispecific binding molecule, by
modifying position 297
from asparagine to alanine (N297A). For example, in certain embodiments, the
bispecific
binding molecule comprises a heavy chain with the sequence of SEQ ID NO: 20.
As used
herein, "glycosylation sites" include any specific amino acid sequence in an
antibody to which
an oligosaccharide (i.e., carbohydrates containing two or more simple sugars
linked together)
will specifically and covalently attach. Oligosaccharide side chains are
typically linked to the
backbone of an antibody via either N- or 0-linkages. N-linked glycosylation
refers to the
attachment of an oligosaccharide moiety to the side chain of an asparagine
residue. 0-linked
glycosylation refers to the attachment of an oligosaccharide moiety to a
hydroxyamino acid, e.g.,
serine, threonine. Methods for modifying the glycosylation content of
antibodies are well known
in the art, see, for example, U.S. Pat. No. 6,218,149; EP 0 359 096 B 1; U. S
. Publication No. US
2002/0028486; WO 03/035835; U.S. Publication No. 2003/0115614; U.S. Pat. No.
6,218,149;
U.S. Pat. No. 6,472,511; all of which are incorporated herein by reference in
their entirety. In
another embodiment, aglycosylation of the bispecific binding molecules of the
invention can be
achieved by recombinantly producing the bispecific binding molecule in a cell
or expression
system incapable of glycosylation, such as, for example, bacteria. In another
embodiment,
aglycosylation of the bispecific binding molecules of the invention can be
achieved by
enzymatically removing the carbohydrate moieties of the glycosylation site.
[00118] In preferred embodiments, the bispecific binding molecule of the
invention does not
bind or has reduced binding affinity (relative to a reference or wild type
immunoglobulin) to the
complement component Clq. Preferably, this is achieved by mutating the anti-
HER2
immunoglobulin portion of the bispecific binding molecule to destroy a Clq
binding site. In
-38-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
certain preferred embodiments, the method encompasses deleting the Cl q
binding site of the Fc
region of an antibody, by modifying position 322 from lysine to alanine
(K322A). For example,
in certain embodiments, the bispecific binding molecule comprises a heavy
chain with the
sequence of SEQ ID NO: 21. In certain embodiments, affinity of the bispecific
binding molecule
or fragment thereof for the complement component Clq is determined by, for
example,
BiaCoreTM assay, as described, for example, in Okazaki et at., 2004. J Mol
Biol, 336(5):1239-49.
See also, Section 6. In certain embodiments, the bispecific binding comprising
an anti-HER2-
immunoglobulin comprising a destroyed Clq binding site binds the complement
component Clq
with less than 25%, 20%, 15%, 10%, or 5% binding compared to a reference or
wild type
immunoglobulin. In certain embodiments, the bispecific binding molecule does
not activate
complement.
[00119] In preferred embodiments, the bispecific binding molecule of the
invention comprises
an immunoglobulin, wherein the immunoglobulin (i) comprises at least one amino
acid
modification relative to a wild-type Fc region, such that said molecule does
not bind or has
reduced binding to an Fc receptor in soluble form or as cell-bound form; (ii)
comprises one or
more mutations in the Fc region to destroy an N-linked glycosylation site; and
(iii) does not or
has reduced binding to the complement component Clq. For example, in certain
embodiments,
the bispecific binding molecule comprises an IgG comprising a first mutation,
N297A, in the Fc
region to (i) abolish or reduce binding to an Fc receptor in soluble form or
as cell-bound form;
and (ii) destroy an N-linked glycosylation site in the Fc region; and a second
mutation, K322A,
in the Fc region to (iii) abolish or reduce binding to the complement
component Clq. See, for
example, SEQ ID NO: 27.
[00120] In a preferred embodiment, the immunoglobulin that binds to HER2
comprises the
variable regions of trastuzumab (see, e.g., Tables 2 and 3), and preferably a
human IgG1 constant
region. In a preferred embodiment, the immunoglobulin that binds to HER2
comprises the
variable regions of trastuzumab wherein the sequence of the heavy chain is SEQ
ID NO: 27 and
wherein the sequence of the light chain is SEQ ID NO: 25. In a preferred
embodiment, the
immunoglobulin that binds to HER2 is a variant of trastuzumab, wherein the
heavy chain does
not bind or has reduced binding to an Fc receptor in soluble form or as cell-
bound form. In a
preferred embodiment, the heavy chain that does not bind an Fc receptor in
soluble form or as a
cell-bound form comprises a mutation in the Fc region to destroy an N-linked
glycosylation site.
-39-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
In a preferred embodiment, the heavy chain has an amino acid substitution to
replace an
asparagine that is an N-linked glycosylation site, with an amino acid that
does not function as a
glycosylation site. In a preferred embodiment, the mutation to destroy an N-
linked glycosylation
site is N297A in the Fc region (SEQ ID NO: 20). In a preferred embodiment, the
immunoglobulin that binds to HER2 comprises the variable regions of
trastuzumab, wherein the
sequence of the heavy chain comprises a mutation in the Fc region to destroy a
Clq binding site.
In a preferred embodiment, the immunoglobulin does not activate complement. In
a preferred
embodiment, the mutation to destroy a Clq binding site is K322A in the Fc
region (SEQ ID NO:
21). In an especially preferred embodiment, the immunoglobulin that binds to
HER2 comprises
the variable regions of trastuzumab, wherein the immunoglobulin heavy chain
comprises a
mutation in the Fc region to destroy an N-linked glycosylation site and a
mutation in the Fc
region to destroy a Clq binding site (see, for example, SEQ ID NO: 27). In an
especially
preferred embodiment, the immunoglobulin that binds to HER2 comprises the
variable regions
of trastuzumab wherein the sequence of the heavy chain of the immunoglobulin
has been
mutated in the Fc region and is SEQ ID NO: 27 and wherein the sequence of the
light chain is
SEQ ID NO: 25. In an especially preferred embodiment, the sequence of the
light chain fusion
polypeptide is SEQ ID NO: 29. In certain embodiments, the heavy chain
comprises the constant
region of trastuzumab. In certain embodiments, the heavy chain comprises the
constant region of
a heavy chain described in Table 2, below (e.g., the constant region of any
one of SEQ ID NOs:
23, 27, 62, or 63). In certain embodiments, the sequence of the heavy chain is
as described in
Table 2, below (e.g., any one of SEQ ID NOs: 23, 27, 62, or 63). In certain
embodiments, the
light chain comprises the constant region of a light chain described in Table
3, below (e.g., the
constant region of SEQ ID NO: 25). In certain embodiments, the sequence of the
light chain is
as described in Table 3, below (e.g., SEQ ID NO: 25).
[00121] In certain embodiments, the bispecific binding molecule has a
trastuzumab-derived
sequence that contains one or more of the modifications in the trastuzumab
immunoglobulin, and
has a huOKT3-derived sequence that contains one or more of the modifications
in the huOKT3
VH and VL sequences, as described in Table 8, below. Bispecific binding
molecules having other
immunoglobulin or scEv sequences can contain analogous mutations at
corresponding positions
in these other immunoglobulin or scEv sequences. In certain embodiments, the
bispecific
binding molecule is (a) derived from trastuzumab and huOKT3; and (b) contains
one or more of
-40-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
the modifications as described in Table 8, below. In certain embodiments, the
sequence of the
peptide linker conjugating the immunoglobulin light chain and the scFv is as
described in Table
1, below (e.g., any one of SEQ ID NOs: 14 or 35-41). In certain embodiments,
the sequence of
the heavy chain is as described in Table 2, below (e.g., any one of SEQ ID
NOs: 23, 27, 62, or
63). In certain embodiments, the sequence of the light chain is as described
in Table 3, below
(e.g., SEQ ID NO: 25). In certain embodiments, the sequence of the VH of the
scFv is as
described in Table 4, below (e.g., any one of SEQ ID NOs: 15, 17, or 64). In
certain
embodiments, the sequence of the VL of the scFv is as described in Table 5,
below (e.g., any one
of SEQ ID NOs: 16 or 65). In certain embodiments, the sequence of the scFv
peptide linker is as
described in Table 1, below (e.g., any one of SEQ ID NOs: 14 or 35-41). In
certain
embodiments, the sequence of the scFv is as described in Table 6, below (e.g.,
any one of SEQ
ID NOs: 19 48-59, or 66). In certain embodiments, the sequence of the light
chain fusion
polypeptide is as described in Table 7, below (e.g., any one of SEQ ID NOs:
29, 34, 42-47, or
60).
[00122] In certain embodiments, the bispecific binding molecule comprises a
glycosylated
monoclonal antibody that is an immunoglobulin that binds to HER2, comprising
two identical
heavy chains and two identical light chains, said light chains being a first
light chain and a
second light chain, wherein the first light chain is fused to a first single
chain variable fragment
(scFv), via a peptide linker, to create a first light chain fusion
polypeptide, and wherein the
second light chain is fused to a second scFv, via a peptide linker, to create
a second light chain
fusion polypeptide, wherein the first and second scFv (i) are identical, and
(ii) bind to CD3,
wherein the first and second light chain fusion polypeptides are identical,
wherein the sequence
of each heavy chain is SEQ ID NO: 62, and wherein the sequence of each light
chain fusion
polypeptide is SEQ ID NO: 60.
[00123] In certain embodiments, the bispecific binding molecule comprises a
glycosylated
monoclonal antibody that is an immunoglobulin that binds to HER2, comprising
two identical
heavy chains and two identical light chains, said light chains being a first
light chain and a
second light chain, wherein the first light chain is fused to a first single
chain variable fragment
(scFv), via a peptide linker, to create a first light chain fusion
polypeptide, and wherein the
second light chain is fused to a second scFv, via a peptide linker, to create
a second light chain
fusion polypeptide, wherein the first and second scFv (i) are identical, and
(ii) bind to CD3,
-41-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
wherein the first and second light chain fusion polypeptides are identical,
wherein the sequence
of each heavy chain is SEQ ID NO: 27, and wherein the sequence of each light
chain fusion
polypeptide is SEQ ID NO: 47.
[00124] In certain embodiments, the bispecific binding molecule comprises a
glycosylated
monoclonal antibody that is an immunoglobulin that binds to HER2, comprising
two identical
heavy chains and two identical light chains, said light chains being a first
light chain and a
second light chain, wherein the first light chain is fused to a first single
chain variable fragment
(scFv), via a peptide linker, to create a first light chain fusion
polypeptide, and wherein the
second light chain is fused to a second scFv, via a peptide linker, to create
a second light chain
fusion polypeptide, wherein the first and second scFv (i) are identical, and
(ii) bind to CD3,
wherein the first and second light chain fusion polypeptides are identical,
wherein the sequence
of each heavy chain is SEQ ID NO: 27, and wherein the sequence of each light
chain fusion
polypeptide is SEQ ID NO: 29.
[00125] In certain embodiments, the bispecific binding molecule has low
immunogenicity.
Low or acceptable immunogenicity and/or high affinity, as well as other
suitable properties, can
contribute to the therapeutic results achieved. "Low immunogenicity" is
defined herein as
raising significant HAHA, HACA or HAMA responses in less than about 75%, or
preferably less
than about 50% of the patients treated and/or raising low titres in the
patient treated (Elliott et at.,
Lancet 344:1125-1127 (1994), entirely incorporated herein by reference).
[00126] The bispecific binding molecules provided herein can bind HER2 and CD3
with a
wide range of affinities. The affinity or avidity of an antibody for an
antigen can be determined
experimentally using any suitable method. See, for example, Berzofsky, et at.,
"Antibody-
Antigen Interactions," In Fundamental Immunology, Paul, W. E., Ed., Raven
Press: New York,
N.Y. (1984); Kuby, Janis Immunology, W.H. Freeman and Company: New York, N.Y.
(1992);
and methods described herein. The measured affinity of a particular antibody-
antigen interaction
can vary if measured under different conditions (e.g., salt concentration,
pH). Thus,
measurements of affinity and other antigen-binding parameters are preferably
made with
standardized solutions of antibody and antigen, and a standardized buffer,
such as the buffer
described herein. The affinity, KD is a ratio of koilkoff. Generally, a KD in
the micromolar range
is considered low affinity. Generally, a KD in the picomolar range is
considered high affinity. In
another specific embodiment, the bispecific binding molecule has high affinity
for HER2 and
-42-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
low affinity for CD3. In another specific embodiment, the bispecific binding
molecule has high
affinity for HER2 and average affinity for CD3. In a specific embodiment, the
bispecific binding
molecule has a KD of between 70 nM and 1 M for CD3. In a specific embodiment,
the
bispecific binding molecule has a KD of between 70 nM and 500 nM for CD3. In a
specific
embodiment, the bispecific binding molecule has a KD of between 500 nM and 1
M for CD3.
[00127] In certain embodiments, the bispecific binding molecule binds to one
or more HER2-
positive carcinoma cell lines, as determined by assays known to one skilled in
the art, such as,
for example, ELISA, BiaCoreTM, and flow cytometry. In certain embodiments, the
carcinoma
cell line is a breast carcinoma cell line, such as, for example, MDA-MB-361,
MDA-MB-468,
AU565, SKBR3, HTB27, HTB26, HCC1954, and/or MCF7. In certain embodiments, the
carcinoma cell line is an ovarian carcinoma cell line, such as, for example,
OVCAR3 and/or
SKOV3. In certain embodiments, the carcinoma cell line is a gastric carcinoma
cell line, such
as, for example, NCI-N87, KATO III, AGS, and/or SNU-16. In certain
embodiments, the
carcinoma cell line is a melanoma cell line, such as, for example, HT144,
SKMEL28, M14,
and/or HTB63. In certain embodiments, the carcinoma cell line is an
osteosarcoma cell line,
such as, for example, RG160, RG164, CRL1427, and/or U20S. In certain
embodiments, the
carcinoma cell line is a Ewings sarcoma cell line, such as, for example, SKEAW
and/or SKES-1.
In certain embodiments, the carcinoma cell line is a rhabdomyosarcoma cell
line, such as, for
example, HTB82. In certain embodiments, the carcinoma cell line is a
neuroblastoma cell line,
such as, for example, NMB7, SKNBE(2)C, IMR32, SKNBE(2)S, SKNBE(1)N, and/or
NB5. In
certain embodiments, the carcinoma cell line is a squamous cell carcinoma head
and neck
(SCCHN) cell lines, such as, for example, 15B, 93-VU-147T, PCI-30, UD-SCC2,
PCI-15B,
SCC90, and/or UMSCC47. In certain embodiments, the carcinoma cell line is a
cervical cancer
cell line, such as, for example, HeLa. In certain embodiments, the carcinoma
cell line is a small
cell lung cancer cell line, such as, for example, NCI-H524, NCI-H69, and/or
NCI-H345. In
certain embodiments, the bispecific binding molecule binds to the HER2-
positive carcinoma cell
line with an EC50 in the picomolar range. See, for example, Section 6.1.3.4
and Section 6.1.3.6.
[00128] In certain embodiments, the bispecific binding molecule binds to CD3+
T cells, as
determined by assays known to one skilled in the art, such as, for example,
ELISA, BiaCoreTM,
and flow cytometry. In certain preferred embodiments, the bispecific binding
molecule binds to
-43-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
CD3+ T cells with greater than 15-fold less binding than huOKT3 binding to
CD3+ T cells. See,
for example, Section 6.1.3.1. In certain embodiments, the CD3+ T cells are
human T cells.
[00129] In certain embodiments, the bispecific binding molecule the bispecific
binding
molecule mediates T cell cytotoxicity against HER2-positive cells, as
determined by assays
known to one skilled in the art, such as, for example, cytotoxicity assays. In
preferred
embodiments, the bispecific binding molecule mediates T cell cytotoxicity
against HER2-
positive cell lines with an EC50 in the picomolar range. In certain
embodiments, the HER2-
positive cells are breast carcinoma cell lines, such as, for example, MDA-MB-
361, MDA-MB-
468, AU565, SKBR3, HTB27, HTB26, and/or MCF7. In certain embodiments, the HER2-
positive cells are of an ovarian carcinoma cell line, such as, for example,
OVCAR3 and/or
SKOV3. In certain embodiments, the HER-2 positive cells are of a gastric
carcinoma cell line,
such as, for example, NCI-N87, KATO III, AGS, and/or SNU-16. In certain
embodiments, the
HER2-positive cells are of a melanoma cell line, such as, for example, HT144,
SKMEL28, M14,
and/or HTB63. In certain embodiments, the HER2-positive cells are of an
osteosarcoma cell
line, such as, for example, RG160, RG164, CRL1427, and/or U205. In certain
embodiments,
the HER2-positive cells are of an Ewings sarcoma cell line, such as, for
example, SKEAW
and/or SKES-1. In certain embodiments, the HER2-positive cells are of a
rhabdomyosarcoma
cell line, such as, for example, HTB82. In certain embodiments, the HER2-
positive cells are of a
neuroblastoma cell line, such as, for example, NMB7, SKNBE(2)C, IMR32,
SKNBE(2)S,
SKNBE(1)N, and/or NB5. In certain embodiments, the HER2-positive cells are of
a squamous
cell carcinoma head and neck (SCCHN) cell line, such as, for example, 15B, 93-
VU-147T, PCI-
30, UD-SCC2, PCI-15B, SCC90, and/or UMSCC47. In certain embodiments, the HER2-
positive cells are of a cervical cancer cell line, such as, for example, HeLa.
In certain
embodiments, the HER2-positive cells are of a small cell lung cancer cell
line, such as, for
example, NCI-H524, NCI-H69, and/or NCI-H345. See, for example, Section 6.1.3.4
and
Section 6.1.3.6.
[00130] In certain embodiments, preincubation of HER2-positive cells with
huOKT3 blocks
the ability of the bispecific binding molecule to induce T cell cytotoxicity.
In certain
embodiments, preincubation of HER2-positive cells with trastuzumab blocks the
ability of the
bispecific binding molecule to induce T cell cytotoxicity. See, for example,
Section 6.1.3.3.
-44-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
[00131] In certain embodiments, the bispecific binding molecule mediates T
cell cytotoxicity
against HER2-positive cells, wherein the level of HER2-expression in said
cells is below the
threshold of detection by flow cytometry performed with the bispecific binding
molecule. See,
for example, Section 6.1.3.4.
[00132] In certain embodiments, the bispecific binding molecule mediates T
cell cytotoxicity
against HER2-positive cells resistant to other HER-targeted therapies, such
as, for example,
trastuzumab, cetuximab, lapatinib, erlotinib, neratinib, or any other small
molecule or antibody
that targets the HER family of receptors. In a specific embodiment, the tumor
that is resistant to
HER-targeted therapies, such as, for example, trastuzumab, cetuximab,
lapatinib, erlotinib,
neratinib, or any other small molecule or antibody that targets the HER family
of receptors is
responsive to treatment with a bispecific binding molecule to the invention.
See, for example,
Section 6.1.3.7, Section 6.1.3.8, Section 6.1.3.9, and Section 6.1.3.10.
[00133] In certain embodiments, the bispecific binding molecule reduces HER2-
positive
tumor progression, metastasis, and/or tumor size. See, for example, Section
6.1.3.11.
[00134] In certain embodiments, the bispecific binding molecule is bound to a
T cell. In
certain embodiments, the binding of the bispecific binding molecule to a T
cell is noncovalently.
In certain embodiments, the T cell is administered to a subject. In certain
embodiments, the T
cell is autologous to the subject to whom the T cell is to be administered. In
certain
embodiments, the T cell is allogeneic to the subject to whom the T cell is to
be administered. In
certain embodiments, the T cell is a human T cell.
[00135] In certain embodiments, the bispecific binding molecule is not bound
to a T cell.
[00136] In certain embodiments, the bispecific binding molecule is conjugated
to an organic
moiety, a detectable marker, and/or isotope as described in Section 5.2.
[00137] In certain embodiments, the bispecific binding molecule or fragment
thereof is
produced as described in Section 5.3. In certain embodiments, the bispecific
binding molecule
or fragment thereof is encoded by a polynucleotide as described in Section
5.3.1. In certain
embodiments, the bispecific binding molecule or fragment thereof is encoded by
a vector (e.g.,
expression vector) as described in Section 5.3.2. In certain embodiments, the
bispecific binding
molecule or fragment thereof is produced from a cell as described in Section
5.3.2.
-45-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
[00138] In certain embodiments, the bispecific binding molecule is a component
of a
composition (e.g., pharmaceutical composition) and/or as part of a kit as
described in
Section 5.5.
[00139] In certain embodiments, the bispecific binding molecule is used
according to the
methods provided in Section 5.6. In certain embodiments, the bispecific
binding molecule is
used as a diagnostic tool according to the methods provided in Section 5.6.2.
In certain
embodiments, the bispecific binding molecule is used as a therapeutic
according to the methods
provided in Section 5.6.1. In certain embodiments, the bispecific binding
molecule is
administered to a subject, such as a subject described in Section 5.7, for use
according to the
methods provided in Section 5.6. In certain embodiments, the bispecific
binding molecule is
administered to a subject as part of a combination therapy as described in
Section 5.9, for use
according to the methods provided in Section 5.6.
[00140] Table. 1. Linker Sequence
DESCRIPTION SEQUENCE (SEQ ID NO:)
(G45)3 GGGGSGGGGSGGGGS (SEQ ID NO: 14)
TS(G45)3Linker TSGGGGSGGGGSGGGGS (SEQ ID NO: 35)
G45 Linker GGGGS (SEQ ID NO: 36)
(G45)2 Linker GGGGSGGGGS (SEQ ID NO: 37)
(G45)3 Linker GGGGSGGGGSGGGGS (SEQ ID NO: 38)
(G45)4 Linker GGGGSGGGGSGGGGSGGGGS (SEQ ID NO: 39)
(G45)5 Linker GGGGSGGGGSGGGGSGGGGSGGGGS (SEQ ID NO: 40)
(G45)6Linker GGGGSGGGGSGGGGSGGGGSGGGGSGGGGS (SEQ ID NO: 41)
[00141] Table 2. Heavy Chain Sequence. The non-italicized, non-underlined
sequence
represents the VH domain. The italicized sequence represents the constant
region. The
underlined, italicized, and bold sequences represent the mutations described
in the
"DESCRIPTION" column.
DESCRIPTION SEQUENCE (SEQ ID NO:)
Trastuzumab VH EVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKG
domain with human LEWVARIYPTNGYTRYADSVKGRFTISADTSKNTAYLQMNSLRA
IgG1 constant region EDTAVYYCSRWGGDGFYAMDYWGQGTLVTVSSASTKGPSVFPL
APSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQS
SGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHT
CPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEV
KFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEY
KCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTC
LVKGFYPSDIAVEWESNGQPEN1VYKTTPPVLDSDGSFFLYSKLTVDKS
-46-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
RWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK (SEQ ID NO: 23)
Trastuzumab VH EVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKG
domain with human LEWVARIYPTNGYTRYADSVKGRFTISADTSKNTAYLQMNSLRA
IgG1 constant ED T AVYYC SRWGGDGFYAMDYWGQ GTL VT VS SA STKGPSVFPL
region; N297A; APSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQS
K3 22A SGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHT
CPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEV
KFNWYVDGVEVHNAKTKPREEQYASTYRVVSVLTVLHQDWLNGKEY
KCAVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTC
LVKGFYPSDIAVEWESNGQPEN1VYKTTPPVLDSDGSFFLYSKLTVDKS
RWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK (SEQ ID NO: 27)
Trastuzumab VH EVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKG
domain with human LEWVARIYPTNGYTRYADSVKGRFTISADTSKNTAYLQMNSLRA
IgG1 constant ED T AVYYC SRWGGDGFYAMDYWGQ GTL VT V S SA STKGPSVFPL
region; N297A APSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQS
SGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHT
CPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEV
KFNWYVDGVEVHNAKTKPREEQYASTYRVVSVLTVLHQDWLNGKEY
KCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTC
LVKGFYPSDIAVEWESNGQPEN1VYKTTPPVLDSDGSFFLYSKLTVDKS
RWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK (SEQ ID NO: 62)
Trastuzumab VH EVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKG
domain with human LEWVARIYPTNGYTRYADSVKGRFTISADTSKNTAYLQMNSLRA
IgG1 constant ED T AVYYC SRWGGDGFYAMDYWGQ GTL VT VS SA STKGPSVFPL
region; K322A APSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQS
SGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHT
CPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEV
KFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEY
KCAVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTC
LVKGFYPSDIAVEWESNGQPEN1VYKTTPPVLDSDGSFFLYSKLTVDKS
RWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK (SEQ ID NO :63)
[00142] Table 3. Light Chain Sequence. The non-italicized sequence represents
the VL
domain. The italicized sequence represents the constant region.
DESCRIPTION SEQUENCE (SEQ ID NO:)
Trastuzumab light DIQMTQSPSSLSASVGDRVTITCRASQDVNTAVAWYQQKPGKAP
chain KLLIYSASFLYSGVPSRF SGSRSGTDFTLTIS SLQPEDFATYYCQQH
YTTPPTFGQGTKVEIKRTVAAPSVHFPPSDEQLKSGTASVFCLLNNF
YPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYE
KHKVYACEVTHQGLSSPVTKSFNRGECTS (SEQ ID NO: 25)
[00143] Table 4. scFv VH Sequence. The underlined, italicized, and bold
sequences
represent the mutations described in the "DESCRIPTION" column.
DESCRIPTION SEQUENCE (SEQ ID NO:)
-47-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
huOKT3 VH QVQLVQSGGGVVQPGRSLRLSCKASGYTFTRYTMEIWVRQAPGK
GLEWIGYINPSRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRP
EDTGVYFCARYYDDHYCLDYWGQGTPVTVSS (SEQ ID NO: 15)
huOKT3 VH; Cl 05S QVQLVQSGGGVVQPGRSLRLSCKASGYTFTRYTMHWVRQAPGK
GLEWIGYINPSRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRP
EDTGVYFCARYYDDHYSLDYWGQGTPVTVSS (SEQ ID NO: 17)
huOKT3 VH; Cl 05S QVQLVQSGGGVVQPGRSLRLSCKASGYTFTRYTMHWVRQAPGK
+ VH-G44C CLEWIGYINP SRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRP
EDTGVYFCARYYDDHYSLDYWGQGTPVTVSS (SEQ ID NO: 64)
[00144] Table 5. scFv VL Sequence. The underlined, italicized, and bold
sequence represent
the mutations described in the "DESCRIPTION" column.
DESCRIPTION SEQUENCE (SEQ ID NO:)
huOKT3 VL DIQMTQ SP S SL SASVGDRVTITC SAS S SVSYMNWYQQTPGKAPKR
WIYDT SKLASGVP SRF SGSGSGTDYTFTIS SLQPEDIATYYCQQWS
SNPFTFGQGTKLQITR (SEQ ID NO: 16)
huOKT3 VL; DIQMTQSPSSLSASVGDRVTITCSASSSVSYMNWYQQTPGKAPKR
Q100C WIYDT SKLASGVP SRF SGSGSGTDYTFTIS SLQPEDIATYYCQQWS
SNPFTFGCGTKLQITR (SEQ ID NO:65)
[00145] Table 6. scFv Sequence. The uppercase, non-italicized, non-bold, non-
underlined
sequence represents the VH domain. The uppercase, italicized sequence
represents the VL
domain. The uppercase, underlined, italicized, and bold sequences represent
the mutations
described in the "DESCRIPTION" column. The lowercase bold sequences represent
the intra-
scFv linker.
DESCRIPTION SEQUENCE (SEQ ID NO:)
huOKT3 scFv QVQLVQSGGGVVQPGRSLRLSCKASGYTFTRYTMEIWVRQAPGK
C105S; 15 amino GLEWIGYINPSRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRP
acid intra-scFv EDTGVYFCARYYDDHYSLDYWGQGTPVTVS SggggsggggsggggsD/
linker QMTQSPSSLSASVGDRVTITCSASSSVSYMNWYQQTPGKAPKRWIYDT
SKLASGVP SRFSGSGSGTDYTFTISSLQPEDIATYYCQQWSSNPFTFGQ
GTKLQITR (SEQ ID NO: 19)
huOKT3 scFv QVQLVQSGGGVVQPGRSLRLSCKASGYTFTRYTMEIWVRQAPGK
C105 S ; 5 amino acid GLEWIGYINPSRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRP
intra-scFv linker ED T GVYF CARYYDDHYSLDYWGQ GTPVTV S SggggsDIQMTQSPSS
LSASVGDRVTITCSASSSVSYMNWYQQTPGKAPKRWIYDTSKLASGVP S
RFSGSGSGTDYTFTISSLQPEDIATYYCQQWSSNPFTFGQGTKLQITR
(SEQ ID NO: 48)
huOKT3 scFv QVQLVQSGGGVVQPGRSLRLSCKASGYTFTRYTMEIWVRQAPGK
C105S; 10 amino GLEWIGYINPSRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRP
acid intra-scFv ED T GVYF CARYYDDHYSLDYWGQ GTPVTV S SggggsggggsD/QMT
linker QSPSSLSASVGDRVTITCSASSSVSYMNWYQQTPGKAPKRWIYD TSKLA
-48-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
SGVPSRFSGSGSGTDYTFTISSLQPEDIATYYCQQWSSNPFTFGQGTKL
QITR (SEQ ID NO: 49)
huOKT3 scFv QVQLVQSGGGVVQPGRSLRLSCKASGYTFTRYTIVIEIWVRQAPGK
C105 S ; 20 amino GLEWIGYINPSRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRP
acid intra-scFv EDTGVYF CARYYDDHYSLDYWGQGTPVT VS Sggggsggggsggggsgg
linker ggsDIQMTQSPSSLSASVGDRVTITCSASSSVSYMNWYQQTPGKAPKRW
IYDTSKLASGVPSRFSGSGSGTDYTFTISSLQPEDIATYYCQQWSSNPFT
FGQGTKLQITR (SEQ ID NO: 50)
huOKT3 scFv QVQLVQSGGGVVQPGRSLRLSCKASGYTFTRYTIVIEIWVRQAPGK
C105 S ; 25 amino GLEWIGYINPSRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRP
acid intra-scFv EDTGVYF CARYYDDHYSLDYWGQGTPVT VS Sggggsggggsggggsgg
linker ggsggggsDIQMTQSPSSLSASVGDRVTITCSASSSVSYMNWYQQTPGKA
PKRWIYDTSKLASGVPSRFSGSGSGTDYTFTISSLQPEDIATYYCQQWS
SNPFTFGQGTKLQITR (SEQ ID NO: 51)
huOKT3 scFv QVQLVQSGGGVVQPGRSLRLSCKASGYTFTRYTIVIEIWVRQAPGK
C105 S ; 30 amino GLEWIGYINPSRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRP
acid intra-scFv EDTGVYF CARYYDDHYSLDYWGQGTPVT VS Sggggsggggsggggsgg
linker ggsggggsggggsDIQMTQSPSSLSASVGDRVTITCSASSSVSYMNWYQQTP
GKAPKRWIYDTSKLASGVPSRFSGSGSGTDYTFTISSLQPEDIATYYCQ
QWSSNPFTFGQGTKLQITR (SEQ ID NO: 52)
huOKT3 scFv QVQLVQSGGGVVQPGRSLRLSCKASGYTFTRYTIVIEIWVRQAPGK
C105 S ; VL-Q100C; CLEWIGYINP SRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRP
VH-G44C; 5 amino EDTGVYFCARYYDDHYSLDYWGQGTPVTVSSggggsD/QMTQSPSS
acid intra-scFv LSASVGDRVTITCSASSSVSYMNWYQQTPGKAPKRWIYDTSKLASGVPS
linker RFSGSGSGTDYTFTISSLQPEDIATYYCQQWSSNPFTFGCGTKLQITR
(SEQ ID NO: 53)
huOKT3 scFv QVQLVQSGGGVVQPGRSLRLSCKASGYTFTRYTIVIEIWVRQAPGK
C105 S ; VL-Q100C; CLEWIGYINP SRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRP
VH-G44C; 10 amino EDTGVYFCARYYDDHYSLDYWGQGTPVTVSSggggsggggsD/QMT
acid intra-scFv QSPSSLSASVGDRVTITCSASSSVSYMNWYQQTPGKAPKRWIYDTSKLA
linker SGVPSRFSGSGSGTDYTFTISSLQPEDIATYYCQQWSSNPFTFGCGTKL
QITR (SEQ ID NO: 54)
huOKT3 scFv QVQLVQSGGGVVQPGRSLRLSCKASGYTFTRYTIVIEIWVRQAPGK
C105 S ; VL-Q100C; CLEWIGYINP SRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRP
VH-G44C; 15 amino EDTGVYFCARYYDDHYSLDYWGQGTPVTVSSggggsggggsggggsD/
acid intra-scFv QMTQSPSSLSASVGDRVTITCSASSSVSYMNWYQQTPGKAPKRWIYDT
linker SKLASGVPSRFSGSGSGTDYTFTISSLQPEDIATYYCQQWSSNPFTFGC
GTKLQITR (SEQ ID NO: 55)
huOKT3 scFv QVQLVQSGGGVVQPGRSLRLSCKASGYTFTRYTIVIEIWVRQAPGK
C105 S ; VL-Q100C; CLEWIGYINP SRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRP
VH-G44C; 20 amino EDTGVYFCARYYDDHYSLDYWGQGTPVTVSSggggsggggsggggsgg
acid intra-scFv ggsDIQMTQSPSSLSASVGDRVTITCSASSSVSYMNWYQQTPGKAPKRW
linker IYDTSKLASGVPSRFSGSGSGTDYTFTISSLQPEDIATYYCQQWSSNPFT
FGCGTKLQITR (SEQ ID NO: 56)
huOKT3 scFv QVQLVQSGGGVVQPGRSLRLSCKASGYTFTRYTIVIEIWVRQAPGK
C105 S ; VL-Q100C; CLEWIGYINP SRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRP
-49-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
VH-G44C; 25 amino EDTGVYFCARYYDDHYSLDYWGQGTPVTVSSggggsggggsggggsgg
acid intra-scFv ggsggggsDIQMTQSPSSLSASVGDR VTITCSASSSVSYMNWYQQTPGKA
linker PKRWIYDTSKLASGVP SRFSGSGSGTDYTFTISSLQPEDIATYYCQQWS
SNPFTFGCGTKLQITR (SEQ ID NO: 57)
huOKT3 scFv QVQLVQ SGGGVVQPGRSLRLSCKASGYTFTRYTMHWVRQAPGK
C105 S ; VL-Q100C; CLEWIGYINP SRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRP
VH-G44C; 30 amino EDTGVYFCARYYDDHYSLDYWGQGTPVTVSSggggsggggsggggsgg
acid intra-scFv ggsggggsggggsDIQMTQSPSSLSASVGDRVTITCSASSSVSYMNWYQQTP
linker GKAPKRWIYDTSKLASGVP SRFSGSGSGTDYTFTISSLQPEDIATYYCQ
QWSSNPFTFGCGTKLQITR (SEQ ID NO: 58)
huOKT3; 15 amino QVQLVQ SGGGVVQPGRSLRLSCKASGYTFTRYTMHWVRQAPGK
acid intra-scFv GLEWIGYINPSRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRP
linker EDTGVYFCARYYDDHYCLDYWGQGTPVTVS SggggsggggsggggsD/
QMTQSPSSLSASVGDRVTITCSASSSVSYMNWYQQTPGKAPKRWIYDT
SKLASGVP SRFSGSGSGTDYTFTISSLQPEDIATYYCQQWSSNPFTFGQ
GTKLQITR (SEQ ID NO: 59)
[00146] Table 7. Light Chain Fusion Polypeptide Sequence. The uppercase, non-
italicized, non-bold, non-underlined sequence represents the VL domain of the
trastuzumab light
chain. The uppercase, italicized sequence represents the constant region of
the trastuzumab light
chain. The lowercase, non-italicized, non-bold, non-underlined sequence
represents the linker
conjugating the light chain to the scFv. The uppercase, underlined sequence
represents the VH
domain of the scFv. The uppercase, bold sequence represents the VL domain of
the scFv. The
uppercase, underlined, italicized, and bold sequences represent the mutations
described in the
"DESCRIPTION" column. The lowercase bold sequences represent the intra-scFv
linker.
DESCRIPTION SEQUENCE (SEQ ID NO:)
Trastuzumab light DIQMTQ SP S SL S A S VGDRVTIT CRA S QDVNTAVAWYQ QKP GKAP
chain; C105 S ; 17 KLLIYSASFLYSGVPSRF SGSRSGTDFTLTIS SLQPEDFATYYCQQH
amino acid linker YTTPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNF
conjugating the light YPREAKVQWKVDNALQSGNSQESVTEQDSKDSTY SLSSTLTLSKADYE
chain to the scFv; 15 KHKVY ACEVTHQGLSSPVTKSFNRGECtsggggsggggsggggsQVQLVQS
amino acid intra- GGGVVQPGRSLRLSCKASGYTFTRYTMEIWVRQAPGKGLEWIGY
scFv peptide linker INPSRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRPEDTGVYF
CARYYDDHYSLDYWGQGTPVTVSSggggsggggsggggsDIQMTQSP
SSLSASVGDRVTITCSASSSVSYMNWYQQTPGKAPKRWIYDTS
KLASGVPSRFSGSGSGTDYTFTISSLQPEDIATYYCQQWSSNPF
TFGQGTKLQITR (SEQ ID NO: 29)
Trastuzumab light DIQMTQ SP S SL S A S VGDRVTIT CRA S QDVNTAVAWYQ QKP GKAP
chain; C105 S ; 17 KLLIYSASFLYSGVPSRF SGSRSGTDFTLTIS SLQPEDFATYYCQQH
amino acid linker YTTPPTF GQGTKVEIKRTVAAP SVFIFPP SDEQLKSGTASVVCLLNNF
conjugating the light YPREAKVQWKVDNALQSGNSQESVTEQDSKDSTY SLSSTLTLSKADYE
chain to the scFv; 5 KHKVYACEVTHQGLSSPVTKSFNRGECtsggggsggggsggggsQVQLV QS
-50-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
amino acid intra- GGGVVQPGRSLRLSCKASGYTFTRYTMHWVRQAPGKGLEWIGY
scFv peptide linker INPSRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRPEDTGVYF
CARYYDDHYSLDYWGQGTPVTVSSggggsDIQMTQSPSSLSASVG
DRVTITCSASSSVSYMNWYQQTPGKAPKRWIYDTSKLASGVP
SRFSGSGSGTDYTFTISSLQPEDIATYYCQQWSSNPFTFGQGTK
LQITR (SEQ ID NO: 30)
Trastuzumab light DIQMTQ SP S SL S A S VGDRVTIT CRA S QDVNTAVAWYQ QKP GKAP
chain; C105 S ; 17 KLLIYSASFLYSGVPSRF SGSRSGTDFTLTIS SLQPEDFATYYCQQH
amino acid linker YTTPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNF
conjugating the light YPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSTSSTLTLSKADYE
chain to the scFv; 10 KHKVYACEVTHQGLSSPVTKSFNRGECtsggggsggggsggggsQVQLVQS
amino acid intra- GGGVVQPGRSLRLSCKASGYTFTRYTMHWVRQAPGKGLEWIGY
scFv peptide linker INPSRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRPEDTGVYF
CARYYDDHYSLDYWGQGTPVTVSSggggsggggsDIQMTQSPSSLS
ASVGDRVTITCSASSSVSYMNWYQQTPGKAPKRWIYDTSKLA
SGVPSRFSGSGSGTDYTFTISSLQPEDIATYYCQQWSSNPFTFG
QGTKLQITR (SEQ ID NO: 31)
Trastuzumab light DIQMTQ SP S SL S A S VGDRVTIT CRA S QDVNTAVAWYQ QKP GKAP
chain; C105 S ; 17 KLLIYSASFLYSGVPSRF SGSRSGTDFTLTIS SLQPEDFATYYCQQH
amino acid linker YTTPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNF
conjugating the light YPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSTSSTLTLSKADYE
chain to the scFv; 20 KHKVYACEVTHQGLSSPVTKSFNRGECtsggggsggggsggggsQVQLVQS
amino acid intra- GGGVVQPGRSLRLSCKASGYTFTRYTMHWVRQAPGKGLEWIGY
scFv peptide linker INPSRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRPEDTGVYF
CARYYDDHYSLDYWGQGTPVTVSSggggsggggsggggsggggsDIQMT
QSPSSLSASVGDRVTITCSASSSVSYMNWYQQTPGKAPKRWIY
DTSKLASGVPSRFSGSGSGTDYTFTISSLQPEDIATYYCQQWSS
NPFTFGQGTKLQITR (SEQ ID NO: 32)
Trastuzumab light DIQMTQ SP S SL S A S VGDRVTIT CRA S QDVNTAVAWYQ QKP GKAP
chain; C105 S ; 17 KLLIYSASFLYSGVPSRF SGSRSGTDFTLTIS SLQPEDFATYYCQQH
amino acid linker YTTPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNF
conjugating the light YPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSTSSTLTLSKADYE
chain to the scFv; 25 KHKVYACEVTHQGLSSPVTKSFNRGECtsggggsggggsggggsQVQLVQS
amino acid intra- GGGVVQPGRSLRLSCKASGYTFTRYTMHWVRQAPGKGLEWIGY
scFv peptide linker INPSRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRPEDTGVYF
CARYYDDHYSLDYWGQGTPVTVSSggggsggggsggggsggggsggggsDI
QMTQSPSSLSASVGDRVTITCSASSSVSYMNWYQQTPGKAPK
RWIYDTSKLASGVPSRFSGSGSGTDYTFTISSLQPEDIATYYCQ
QWSSNPFTFGQGTKLQITR (SEQ ID NO: 33)
Trastuzumab light DIQMTQ SP S SL S A S VGDRVTIT CRA S QDVNTAVAWYQ QKP GKAP
chain; C105 S ; 17 KLLIYSASFLYSGVPSRF SGSRSGTDFTLTIS SLQPEDFATYYCQQH
amino acid linker YTTPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNF
conjugating the light YPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSTSSTLTLSKADYE
chain to the scFv; 30 KHKVYACEVTHQGLSSPVTKSFNRGECtsggggsggggsggggsQVQLVQS
amino acid intra- GGGVVQPGRSLRLSCKASGYTFTRYTMHWVRQAPGKGLEWIGY
scFv peptide linker INPSRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRPEDTGVYF
-51-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
CARYYDDHYSLDYWGQGTPVTVSSggggsggggsggggsggggsggggsgg
ggsDIQMTQSPSSLSASVGDRVTITCSASSSVSYMNWYQQTPGK
APKRWIYDTSKLASGVPSRFSGSGSGTDYTFTISSLQPEDIATY
YCQQWSSNPFTFGQGTKLQITR (SEQ ID NO: 34)
Trastuzumab light DIQMTQ SP S SL S A S VGDRVTIT CRA S QDVNTAVAWYQ QKP GKAP
chain; C105 S ; 17 KLLIYSASFLYSGVPSRFSGSRSGTDFTLTIS SLQPEDFATYYCQQH
amino acid linker YTTPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNF
conjugating the light YPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYE
chain to the huOKT3 KHKVYACEVTHQGLSSPVTKSFNRGECtsggggsggggsggggsQVQLVQS
scFv; 5 amino acid GGGVVQPGRSLRLSCKASGYTFTRYTMHWVRQAPGKCLEWIGYI
intra-scFv peptide NPSRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRPEDTGVYF
linker; VL-Q100C; CARYYDDHYSLDYWGQGTPVTVS SggggsDIQMTQSPSSLSASVG
VH-G44C DRVTITCSASSSVSYMNWYQQTPGKAPKRWIYDTSKLASGVP
SRFSGSGSGTDYTFTISSLQPEDIATYYCQQWSSNPFTFGCGTK
LQITR (SEQ ID NO: 42)
Trastuzumab light DIQMTQ SP S SL S A S VGDRVTIT CRA S QDVNTAVAWYQ QKP GKAP
chain; C105 S ; 17 KLLIYSASFLYSGVPSRFSGSRSGTDFTLTIS SLQPEDFATYYCQQH
amino acid linker YTTPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNF
conjugating the light YPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSTSSTLTLSKADYE
chain to the huOKT3 KHKVYACEVTHQGLSSPVTKSFNRGECtsggggsggggsggggsQVQLVQS
scFv; 10 amino acid GGGVVQPGRSLRLSCKASGYTFTRYTMHWVRQAPGKCLEWIGYI
intra-scFv peptide NPSRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRPEDTGVYF
linker; VL-Q100C; CARYYDDHYSLDYWGQGTPVTVS SggggsggggsDIQMTQSPSSLS
VH-G44C ASVGDRVTITCSASSSVSYMNWYQQTPGKAPKRWIYDTSKLA
SGVPSRFSGSGSGTDYTFTISSLQPEDIATYYCQQWSSNPFTFG
CGTKLQITR (SEQ ID NO: 43)
Trastuzumab light DIQMTQ SP S SL S A S VGDRVTIT CRA S QDVNTAVAWYQ QKP GKAP
chain; C105 S ; 17 KLLIYSASFLYSGVPSRFSGSRSGTDFTLTIS SLQPEDFATYYCQQH
amino acid linker YTTPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNF
conjugating the light YPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSTSSTLTLSKADYE
chain to the huOKT3 KHKVYACEVTHQGLSSPVTKSFNRGECtsggggsggggsggggsQVQLVQS
scFv; 15 amino acid GGGVVQPGRSLRLSCKASGYTFTRYTMHWVRQAPGKCLEWIGYI
intra-scFv peptide NPSRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRPEDTGVYF
linker; VL-Q100C; CARYYDDHYSLDYWGQGTPVTVS SggggsggggsggggsDIQMTQSP
VH-G44C SSLSASVGDRVTITCSASSSVSYMNWYQQTPGKAPKRWIYDTS
KLASGVPSRFSGSGSGTDYTFTISSLQPEDIATYYCQQWSSNPF
TFGCGTKLQITR (SEQ ID NO: 44)
Trastuzumab light DIQMTQ SP S SL S A S VGDRVTIT CRA S QDVNTAVAWYQ QKP GKAP
chain; C105 S ; 17 KLLIYSASFLYSGVPSRFSGSRSGTDFTLTIS SLQPEDFATYYCQQH
amino acid linker YTTPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNF
conjugating the light YPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSTSSTLTLSKADYE
chain to the huOKT3 KHKVYACEVTHQGLSSPVTKSFNRGECtsggggsggggsggggsQVQLVQS
scFv; 20 amino acid GGGVVQPGRSLRLSCKASGYTFTRYTMHWVRQAPGKCLEWIGYI
intra-scFv peptide NPSRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRPEDTGVYF
linker; VL-Q100C; CARYYDDHYSLDYWGQGTPVTVS SggggsggggsggggsggggsDIQMT
VH-G44C QSPSSLSASVGDRVTITCSASSSVSYMNWYQQTPGKAPKRWIY
-52-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
DTSKLASGVPSRFSGSGSGTDYTFTISSLQPEDIATYYCQQWSS
NPFTFGCGTKLQITR (SEQ ID NO: 45)
Trastuzumab light DIQMTQ SP S SL S A S VGDRVTIT CRA S QDVNTAVAWYQ QKP GKAP
chain; C105 S ; 17 KLLIYSASFLYSGVPSRF SGSRSGTDFTLTIS SLQPEDFATYYCQQH
amino acid linker YTTPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNF
conjugating the light YPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSTSSTLTLSKADYE
chain to the huOKT3 KHKVYACEVTHQGLSSPVTKSFNRGECtsggggsggggsggggsQVQLVQS
scFv; 25 amino acid GGGVVQPGRSLRLSCKASGYTFTRYTMEIWVRQAPGKCLEWIGYI
intra-scFv peptide NPSRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRPEDTGVYF
linker; VL-Q100C; CARYYDDHYSLDYWGQGTPVTVS SggggsggggsggggsggggsggggsDI
VH-G44C QMTQSPSSLSASVGDRVTITCSASSSVSYMNWYQQTPGKAPK
RWIYDTSKLASGVPSRFSGSGSGTDYTFTISSLQPEDIATYYCQ
QWSSNPFTFGQCGTKLQITR (SEQ ID NO: 46)
Trastuzumab light DIQMTQ SP S SL S A S VGDRVTIT CRA S QDVNTAVAWYQ QKP GKAP
chain; C105 S ; 17 KLLIYSASFLYSGVPSRF SGSRSGTDFTLTIS SLQPEDFATYYCQQH
amino acid linker YTTPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNF
conjugating the light YPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSTSSTLTLSKADYE
chain to the huOKT3 KHKVYACEVTHQGLSSPVTKSFNRGECtsggggsggggsggggsQVQLVQS
scFv; 30 amino acid GGGVVQPGRSLRLSCKASGYTFTRYTMEIWVRQAPGKCLEWIGYI
intra-scFv peptide NPSRGYTNYNQKFKDRFTISRDNSKNTAFLQMDSLRPEDTGVYF
linker; VL-Q100C; CARYYDDHYSLDYWGQGTPVTVS Sggggsggggsggggsggggsggggsgg
VH-G44C ggsDIQMTQSPSSLSASVGDRVTITCSASSSVSYMNWYQQTPGK
APKRWIYDTSKLASGVPSRFSGSGSGTDYTFTISSLQPEDIATY
YCQQWSSNPFTFGCGTKLQITR (SEQ ID NO: 47)
Trastuzumab light DIQMTQ SP S SL S A S VGDRVTIT CRA S QDVNTAVAWYQ QKP GKAP
chain; 17 amino acid KLLIYSASFLYSGVPSRFSGSRSGTDFTLTISSLQPEDFATYYCQQH
linker conjugating YTTPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNF
the light chain to the YPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSTSSTLTLSKADYE
scFv; 15 amino acid KHKVYACEVTHQGLSSPVTKSFNRGECtsggggsggggsggggsQVQLVQS
intra-scFv peptide GGGVVQPGRSLRLSCKASGYTFTRYTMEIWVRQAPGKGLEWIGY
linker INP SRGYTNYNQKFKDRFTISRDNSKNTAFLQMD SLRPEDTGVYF
CARYYDDHYCLDYWGQGTPVTVS SggggsggggsggggsDIQMTQSP
SSLSASVGDRVTITCSASSSVSYMNWYQQTPGKAPKRWIYDTS
KLASGVPSRFSGSGSGTDYTFTISSLQPEDIATYYCQQWSSNPF
TFGQGTKLQITR (SEQ ID NO: 60)
[00147] Table 8. Modifications to bispecific binding molecules
LOCATION OF DESCRIPTION
MODIFICATION
Heavy chain Mutation to reduce binding to the Fc receptor (as an
example, N297A
mutation)
Mutation to destroy a glycosylation site (as an example, N297A
mutation)
Mutation to reduce Clq binding (as an example, K322A mutation)
Linker conjugating Increase or decrease the length of the linker
-53-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
the light chain to the
huOKT3 scFv
huOKT3 scFv VH Mutation to increase stabilization and/or reduce aggregation
(as an
example, introduce disulfide binding between VH40 and VL100
(according to Kabat numbering), as an example, VH G44C and VL
Q100C)
Reduce aggregation (as an example, C105S mutation)
huOKT3 scFv VL Mutation to increase stabilization and/or reduce aggregation
(as an
exampleõ introduce disulfide binding between VH40 and VL100
(according to Kabat numbering), as an example, VH G44C and VL
Q100C)
huOKT3 intra-scFv Increase or decrease the length of the linker
linker
5.2 BISPECIFIC BINDING MOLECULE CONJUGATES
[00148] In preferred embodiments, a bispecific binding molecule provided
herein is not
conjugated to any other molecule, such as an organic moiety, a detectable
label, or an isotope. In
alternative embodiments, a bispecific binding molecule provided herein is
conjugated to one or
more organic moieties. In alternative embodiments, a bispecific binding
molecule provided
herein is conjugated to one or more detectable labels. In alternative
embodiments, a bispecific
binding molecule provided herein is conjugated to one or more isotopes.
5.2.1 DETECTABLE LABELS AND ISOTOPES
[00149] In certain embodiments, a bispecific binding molecule provided herein
is conjugated
to one or more detectable labels or isotopes, e.g., for imaging purposes. In
certain embodiments,
a bispecific binding molecule is detectably labeled by covalent or non-
covalent attachment of a
chromogenic, enzymatic, radioisotopic, isotopic, fluorescent, toxic,
chemiluminescent, nuclear
magnetic resonance contrast agent or other label.
[00150] Non-limiting examples of suitable chromogenic labels include
diaminobenzidine and
4-hydroxyazo-benzene-2-carboxylic acid.
[00151] Non-limiting examples of suitable enzyme labels include malate
dehydrogenase,
staphylococcal nuclease, delta-5-steroid isomerase, yeast-alcohol
dehydrogenase, alpha-glycerol
phosphate dehydrogenase, triose phosphate isomerase, peroxidase, alkaline
phosphatase,
asparaginase, glucose oxidase, beta-galactosidase, ribonuclease, urease,
catalase, glucose-6-
phosphate dehydrogenase, glucoamylase, and acetylcholine esterase.
-54-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
[00152] Non-limiting examples of suitable radioisotopic labels include 3H,
"In, 1251, 1311, 32p,
35s, 14C, 51 -r,
U 57TO, 58CO, 59Fe, 75Se, 152Eu, 90y, 67cti, 2170, 211At, 212pb, 47se, 223Ra,
223R- "Zr,
177
Lu, and 1 9Pd. In certain embodiments, "In is a preferred isotope for in vivo
imaging as it
avoids the problem of dehalogenation of 1251 or 131I-labeled bispecific
binding molecules in the
liver. In addition, "In has a more favorable gamma emission energy for imaging
(Perkins et at.,
Eur. J. Nucl. Med. 70:296-301 (1985); Carasquillo et ah, J. Nucl. Med. 25:281-
287 (1987)). For
example, "In coupled to monoclonal antibodies with 1-(P-isothiocyanatobenzy1)-
DPTA has
shown little uptake in non-tumorous tissues, particularly the liver, and
therefore enhances
specificity of tumor localization (Esteban et at., J. Nucl. Med. 28:861-870
(1987)).
[00153] Non-limiting examples of suitable non-radioactive isotopic labels
include 157Gd,
55Mn, 162Dy, 52Tr, and 56Fe.
[00154] Non-limiting examples of suitable fluorescent labels include a 152Eu
label, a
fluorescein label, an isothiocyanate label, a rhodamine label, a phycoerythrin
label, a
phycocyanin label, an allophycocyanin label, a Green Fluorescent Protein (GFP)
label, an o-
phthaldehyde label, and a fluorescamine label.
[00155] Non-limiting examples of chemiluminescent labels include a luminol
label, an
isoluminol label, an aromatic acridinium ester label, an imidazole label, an
acridinium salt label,
an oxalate ester label, a luciferin label, a luciferase label, and an aequorin
label.
[00156] Non-limiting examples of nuclear magnetic resonance contrasting agents
include
heavy metal nuclei such as Gd, Mn, and iron.
[00157] Techniques known to one of ordinary skill in the art for binding the
above-described
labels to a bispecific binding molecule provided herein are described in, for
example, Kennedy et
at., Clin. CMm. Acta 70:1-31 (1976), and Schurs et al., Clin. CMm. Acta 81:1-
40 (1977).
Coupling techniques mentioned in the latter are the glutaraldehyde method, the
periodate
method, the dimaleimide method, the m-maleimidobenzyl-N-hydroxy-succinimide
ester method,
all of which methods are incorporated by reference herein.
[00158] In certain embodiments, the bispecific binding molecule is conjugated
to a diagnostic
agent. A diagnostic agent is an agent useful in diagnosing or detecting a
disease by locating the
cells containing the antigen. Useful diagnostic agents include, but are not
limited to,
radioisotopes, dyes (such as with the biotin-streptavidin complex), contrast
agents, fluorescent
compounds or molecules and enhancing agents (e.g., paramagnetic ions) for
magnetic resonance
-55-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
imaging (MRI). U.S. Pat. No. 6,331,175 describes MM technique and the
preparation of
antibodies conjugated to a MRI enhancing agent and is incorporated in its
entirety by reference.
Preferably, the diagnostic agents are selected from the group consisting of
radioisotopes,
enhancing agents for use in magnetic resonance imaging, and fluorescent
compounds. In order
to load an antibody component with radioactive metals or paramagnetic ions, it
may be necessary
to react it with a reagent having a long tail to which are attached a
multiplicity of chelating
groups for binding the ions. Such a tail can be a polymer such as a
polylysine, polysaccharide,
or other derivatized or derivatizable chain having pendant groups to which can
be bound
chelating groups such as, for example, ethylenediaminetetraacetic acid (EDTA),
diethylenetriaminepentaacetic acid (DTPA), porphyrins, polyamines, crown
ethers, bis-
thiosemicarbazones, polyoximes, and like groups known to be useful for this
purpose. Chelates
are coupled to the antibodies using standard chemistries. The chelate is
normally linked to the
antibody by a group which enables formation of a bond to the molecule with
minimal loss of
immunoreactivity and minimal aggregation and/or internal cross-linking other,
more unusual,
methods and reagents for conjugating chelates to antibodies are disclosed in
U.S. Pat. No.
4,824,659 to Hawthorne, entitled "Antibody Conjugates," issued Apr. 25, 1989,
the disclosure of
which is incorporated herein in its entirety by reference. Particularly useful
metal-chelate
combinations include 2-benzyl-DTPA and its monomethyl and cyclohexyl analogs,
used with
diagnostic isotopes for radio-imaging. The same chelates, when complexed with
non-radioactive
metals, such as manganese, iron and gadolinium are useful for MM, when used
along bispecific
binding molecules provided herein. Macrocyclic chelates such as NOTA, DOTA,
and TETA are
of use with a variety of metals and radiometals, most particularly with
radionuclides of gallium,
yttrium and copper, respectively. Such metal-chelate complexes can be made
very stable by
tailoring the ring size to the metal of interest. Other ring-type chelates
such as macrocyclic
polyethers, which are of interest for stably binding nuclides, such as 223Ra
for RAIT are
encompassed herein.
5.2.2 ORGANIC CONJUGATES
[00159] In certain embodiments, the bispecific binding molecules provided
herein comprise
one or more organic moieties that are covalently bonded, directly or
indirectly, to the bispecific
binding molecule. Such modification can produce an antibody or antigen-binding
fragment with
improved pharmacokinetic properties (e.g., increased in vivo serum half-life).
The organic
-56-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
moiety can be a hydrophilic polymeric group, fatty acid group, or fatty acid
ester group. As used
herein, the term "fatty acid" encompasses mono-carboxylic acids and di-
carboxylic acids. As
used herein, a "hydrophilic polymeric group" refers to an organic polymer that
is more soluble in
water than in octane, e.g., polylysine. Hydrophilic polymers suitable for
modifying a bispecific
binding molecule provided herein can be linear or branched and include, for
example, polyalkane
glycols (e.g., polyethylene glycol, (PEG), monomethoxy-polyethylene glycol,
and polypropylene
glycol), carbohydrates (e.g., dextran, cellulose, oligosaccharides, and
polysaccharides), polymers
of hydrophilic amino acids (e.g., polylysine, polyarginine, and
polyaspartate), polyalkane oxides
(e.g., polyethylene oxide and polypropylene oxide) and polyvinyl pyrolidone.
In certain
embodiments, the hydrophilic polymer that modifies a bispecific binding
molecule provided
herein has a molecular weight of about 800 to about 150,000 Daltons as a
separate molecular
entity. For example PEG-5000 and PEG20,000, wherein the subscript is the
average molecular weight
of the polymer in Daltons, can be used. The hydrophilic polymeric group can be
substituted with
one to about six alkyl, fatty acid or fatty acid ester groups. Hydrophilic
polymers that are
substituted with a fatty acid or fatty acid ester group can be prepared by
employing suitable
methods. For example, a polymer comprising an amine group can be coupled to a
carboxylate of
the fatty acid or fatty acid ester, and an activated carboxylate (e.g.,
activated with N,N-carbonyl
diimidazole) on a fatty acid or fatty acid ester can be coupled to a hydroxyl
group on a polymer.
[00160] Fatty acids and fatty acid esters suitable for modifying bispecific
binding molecules
provided herein can be saturated or can contain one or more units of
unsaturation. Fatty acids
that are suitable for modifying bispecific binding molecules provided herein
include, for
example, n-dodecanoate, n-tetradecanoate, n-octadecanoate, n-eicosanoate, n-
docosanoate, n-
triacontanoate, n-tetracontanoate, cis-delta-9-octadecanoate, all cis-delta-
5,8,11,14-
eicosatetraenoate, octanedioic acid, tetradecanedioic acid, octadecanedioic
acid, docosanedioic
acid, and the like. Suitable fatty acid esters include mono-esters of
dicarboxylic acids that
comprise a linear or branched lower alkyl group. The lower alkyl group can
comprise from one
to about twelve, preferably one to about six, carbon atoms.
[00161] The bispecific binding molecule conjugates provided herein can be
prepared using
suitable methods, such as by reaction with one or more modifying agents. As
used herein, an
"activating group" is a chemical moiety or functional group that can, under
appropriate
conditions, react with a second chemical group thereby forming a covalent bond
between the
-57-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
modifying agent and the second chemical group. For example, amine-reactive
activating groups
include electrophilic groups such as, for example, tosylate, mesylate, halo
(chloro, bromo, fluor ,
iodo), N-hydroxysuccinimidyl esters (NHS), and the like. Activating groups
that can react with
thiols include, for example, maleimide, iodoacetyl, acrylolyl, pyridyl
disulfides, 5-thio1-2-
nitrobenzoic acid thiol (TNB-thiol), and the like. An aldehyde functional
group can be coupled
to amine- or hydrazide-containing molecules, and an azide group can react with
a trivalent
phosphorous group to form phosphoramidate or phosphorimide linkages. Suitable
methods to
introduce activating groups into molecules are known in the art (see, for
example, Hernanson, G.
T., Bioconjugate Techniques, Academic Press: San Diego, Calif (1996)). An
activating group
can be bonded directly to the organic group (e.g., hydrophilic polymer, fatty
acid, fatty acid
ester), or through a linker moiety, for example a divalent C1-C12 group,
wherein one or more
carbon atoms can be replaced by a heteroatom such as oxygen, nitrogen or
sulfur. Suitable linker
moieties include, for example, tetraethylene glycol, (CH2)3, and NH. Modifying
agents that
comprise a linker moiety can be produced, for example, by reacting a mono-Boc-
alkyldiamine
(e.g., mono-Boc-ethylenediamine or mono-Boc-diaminohexane) with a fatty acid
in the presence
of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) to form an amide bond
between the
free amine and the fatty acid carboxylate. The Boc protecting group can be
removed from the
product by treatment with trifluoroacetic acid (TFA) to expose a primary amine
that can be
coupled to another carboxylate as described, or can be reacted with maleic
anhydride and the
resulting product cyclized to produce an activated maleimido derivative of the
fatty acid. (See,
for example, Thompson, et at., WO 92/16221 the entire teachings of which are
incorporated
herein by reference.)
[00162] As used herein, a "modifying agent" refers to a suitable organic group
(e.g.,
hydrophilic polymer, a fatty acid, and a fatty acid ester) that comprises an
activating group. For
example, the organic moieties can be bonded to the bispecific binding molecule
in a non-site
specific manner by employing an amine-reactive modifying agent, for example,
an N-
hydroxysuccinimide ester of PEG. Modified bispecific binding molecules can
also be prepared
by reducing disulfide bonds (e.g., intra-chain disulfide bonds) of bispecific
binding molecule.
The reduced bispecific binding molecule can then be reacted with a thiol-
reactive modifying
agent to produce the modified bispecific binding molecule provided herein.
Modified bispecific
binding molecules comprising an organic moiety that is bonded to specific
sites of a bispecific
-58-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
binding molecule provided herein can be prepared using suitable methods, such
as reverse
proteolysis (Fisch et al., Bioconjugate Chem., 3:147-153 (1992); Werlen et
al., Bioconjugate
Chem., 5:411-417 (1994); Kumaran et al., Protein Sci. 6(10):2233-2241 (1997);
Itoh et al.,
Bioorg. Chem., 24(1): 59-68 (1996); Capellas et al., Biotechnol. Bioeng.,
56(4):456-463 (1997)),
and the methods described in Hermanson, G. T., Bioconjugate Techniques,
Academic Press: San
Diego, Calif. (1996).
5.3 BISPECIFIC BINDING MOLECULE PRODUCTION
[00163] Provided herein are methods for producing bispecific binding molecules
as described
in Section 5.1 and Section 5.2. In certain embodiments, provided herein are
methods for
producing a bispecific binding molecule comprising an aglycosylated monoclonal
antibody that
is an immunoglobulin that binds to HER2, comprising two identical heavy chains
and two
identical light chains, said light chains being a first light chain and a
second light chain, wherein
the first light chain is fused to a first single chain variable fragment
(scFV), via a peptide linker,
to create a first fusion polypeptide, and wherein the second light chain is
fused to a second scFv,
via a peptide linker, to create a second fusion polypeptide, wherein the first
and second scFv (i)
are identical, and (ii) bind to CD3, and wherein the first and second fusion
polypeptides are
identical.
[00164] Methods to produce bispecific binding molecules described herein are
known to one
of ordinary skill in the art, for example, by chemical synthesis, by
purification from biological
sources, or by recombinant expression techniques, including, for example, from
mammalian cell
or transgenic preparations. The methods described herein employs, unless
otherwise indicated,
conventional techniques in molecular biology, microbiology, genetic analysis,
recombinant
DNA, organic chemistry, biochemistry, PCR, oligonucleotide synthesis and
modification,
nucleic acid hybridization, and related fields within the skill of the art.
These techniques are
described, for example, in the references cited herein and are fully explained
in the literature.
See, for example, Maniatis et al. (1982) Molecular Cloning: A Laboratory
Manual, Cold Spring
Harbor Laboratory Press; Sambrook et al. (1989), Molecular Cloning: A
Laboratory Manual,
Second Edition, Cold Spring Harbor Laboratory Press; Sambrook et al. (2001)
Molecular
Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring
Harbor, NY;
Ausubel et at., Current Protocols in Molecular Biology, John Wiley & Sons
(1987 and annual
updates); Current Protocols in Immunology, John Wiley & Sons (1987 and annual
updates) Gait
-59-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
(ed.) (1984) Oligonucleotide Synthesis: A Practical Approach, IRL Press;
Eckstein (ed.) (1991)
Oligonucleotides and Analogues: A Practical Approach, IRL Press; Birren et al.
(eds.) (1999)
Genome Analysis: A Laboratory Manual, Cold Spring Harbor Laboratory Press.
[00165] A variety of methods exist in the art for the production of bispecific
binding
molecules. For example, the bispecific binding molecule may be made by
recombinant DNA
methods, such as those described in U.S. Pat. No. 4,816,567. The one or more
DNAs encoding a
bispecific binding molecule provided herein can be readily isolated and
sequenced using
conventional procedures (e.g., by using oligonucleotide probes that are
capable of binding
specifically to genes encoding the heavy and light chains of murine
antibodies, or such chains
from human, humanized, or other sources). Once isolated, the DNA may be placed
into
expression vectors, which are then transformed into host cells such as NSO
cells, Simian COS
cells, Chinese hamster ovary (CHO) cells, yeast cells, algae cells, eggs, or
myeloma cells that do
not otherwise produce immunoglobulin protein, to obtain the synthesis of the
bispecific binding
molecules in the recombinant host cells. The DNA also may be modified, for
example, by
substituting the coding sequence for human heavy and light chain constant
domains of a desired
species in place of the homologous human sequences (U.S. Pat. No. 4,816,567;
Morrison et al.,
supra) or by covalently joining to the immunoglobulin coding sequence all or
part of the coding
sequence for a non-immunoglobulin polypeptide. Such a non-immunoglobulin
polypeptide can
be substituted for the constant domains of a bispecific binding molecule
provided herein. In
certain embodiments, the DNA is as described in Section 5.3.1.
[00166] Bispecific binding molecules provided herein can also be prepared
using at least one
bispecific binding molecule-encoding polynucleotide to provide transgenic
animals or mammals,
such as goats, cows, horses, sheep, and the like, that produce such antibodies
in their milk. Such
animals can be provided using known methods. See, for example, but not limited
to, U.S. Pat.
Nos. 5,827,690; 5,849,992; 4,873,316; 5,849,992; 5,994,616, 5,565,362;
5,304,489, and the like,
each of which is entirely incorporated herein by reference.
[00167] In certain embodiments, bispecific binding molecules provided herein
can
additionally be prepared using at least one bispecific binding molecule-
encoding polynucleotide
provided herein to provide transgenic plants and cultured plant cells (for
example, but not limited
to tobacco and maize) that produce such antibodies, specified portions or
variants in the plant
parts or in cells cultured therefrom. As a non-limiting example, transgenic
tobacco leaves
-60-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
expressing recombinant proteins have been successfully used to provide large
amounts of
recombinant proteins, for example, using an inducible promoter. See, for
example, Cramer et
at., Curr. Top. Microbol. Immunol. 240:95-118 (1999) and references cited
therein. Also,
transgenic maize have been used to express mammalian proteins at commercial
production
levels, with biological activities equivalent to those produced in other
recombinant systems or
purified from natural sources. See, for example, Hood et at., Adv. Exp. Med.
Biol. 464:127-147
(1999) and references cited therein. Antibodies have also been produced in
large amounts from
transgenic plant seeds including antibody fragments, such as scFvs, including
tobacco seeds and
potato tubers. See, for example, Conrad et at., Plant Mol. Biol. 38:101-109
(1998) and
references cited therein. Thus, bispecific binding molecules can also be
produced using
transgenic plants, according to known methods. See also, for example, Fischer
et at., Biotechnol.
Appl. Biochem. 30:99-108 (October, 1999), Ma et al., Trends Biotechnol. 13:522-
7 (1995); Ma
et al., Plant Physiol. 109:341-6 (1995); Whitelam et al., Biochem Soc. Trans.
22:940-944 (1994);
and references cited therein. Each of the above references is entirely
incorporated herein by
reference.
[00168] In certain embodiments, bispecific binding molecules provided herein
can be
prepared using at least one bispecific binding molecule-encoding
polynucleotide provided herein
to provide bacteria that produce such bispecific binding molecules. As a non-
limiting example,
E. coli expressing recombinant proteins has been successfully used to provide
large amounts of
recombinant proteins. See, for example, Verma et at., 1998, 216(1-2): 165-181
and references
cited therein.
[00169] See, also, Section 6.1.2.1 for a detailed example for the design and
production of a
bispecific binding molecule described herein.
[00170] In certain embodiments, the bispecific binding molecules can be
recovered and
purified from recombinant cell cultures by well-known methods including, but
not limited to,
protein A purification, protein G purification, ammonium sulfate or ethanol
precipitation, acid
extraction, anion or cation exchange chromatography, phosphocellulose
chromatography,
hydrophobic interaction chromatography, affinity chromatography,
hydroxylapatite
chromatography and lectin chromatography. High performance liquid
chromatography
("HPLC") can also be employed for purification. See, for example, Colligan,
Current Protocols
-61-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
in Immunology, or Current Protocols in Protein Science, John Wiley & Sons, NY,
N.Y., (1997-
2001), e.g., chapters 1, 4, 6, 8, 9, and 10, each entirely incorporated herein
by reference.
[00171] In certain embodiments, the bispecific binding molecules provided
herein include
naturally purified products, products of chemical synthetic procedures, and
products produced by
recombinant techniques from a eukaryotic host, including, for example, yeast,
higher plant,
insect and mammalian cells. In preferred embodiments, the bispecific binding
molecule is
generated in a host such that the bispecific binding molecule is
aglycosylated. In another
preferred embodiment, the bispecific binding molecule is generated in a
bacterial cell such that
the bispecific binding molecule is aglycosylated. Such methods are described
in many standard
laboratory manuals, such as Sambrook, supra, Sections 17.37-17.42; Ausubel,
supra, Chapters
10, 12, 13, 16, 18 and 20, Colligan, Protein Science, supra, Chapters 12-14,
all entirely
incorporated herein by reference.
[00172] Purified antibodies can be characterized by, for example, ELISA,
ELISPOT, flow
cytometry, immunocytology, BiacoreTM analysis, Sapidyne KinExATM kinetic
exclusion assay,
SDS-PAGE and Western blot, or by HPLC analysis as well as by a number of other
functional
assays disclosed herein.
5.3.1 POLYNUCLEOTIDES
[00173] In certain embodiments, provided herein are polynucleotides comprising
a nucleotide
sequence encoding a bispecific binding molecule described herein or a fragment
thereof (e.g., a
heavy chain and/or a light chain fusion polypeptide) that immunospecifically
binds to HER2 and
CD3, as described in Section 5.1 and Section 5.2. Also provided herein are
vectors comprising
such polynucleotides. See, Section 5.3.2. Also provided herein are
polynucleotides encoding
antigens of the bispecific binding molecules provided herein. Also provided
herein are
polynucleotides that hybridize under stringent or lower stringency
hybridization conditions to
polynucleotides that encode a bispecific binding molecule or fragment thereof
provided herein.
[00174] The language "purified" includes preparations of polynucleotide or
nucleic acid
molecule having less than about 15%, 10%, 5%, 2%, 1%, 0.5%, or 0.1% (in
particular less than
about 10%) of other material, e.g., cellular material, culture medium, other
nucleic acid
molecules, chemical precursors and/or other chemicals. In a specific
embodiment, a nucleic acid
molecule(s) encoding a bispecific binding molecule described herein is
isolated or purified.
-62-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
[00175] Nucleic acid molecules provided herein can be in the form of RNA, such
as mRNA,
hnRNA, tRNA or any other form, or in the form of DNA, including, but not
limited to, cDNA
and genomic DNA obtained by cloning or produced synthetically, or any
combinations thereof.
The DNA can be triple-stranded, double-stranded or single-stranded, or any
combination thereof
Any portion of at least one strand of the DNA or RNA can be the coding strand,
also known as
the sense strand, or it can be the non-coding strand, also referred to as the
anti-sense strand.
[00176] In certain embodiments, provided herein is a polynucleotide comprising
nucleotide
sequences encoding a bispecific binding molecule or fragment thereof as
described in Section 5.1
and Section 5.2, wherein the bispecific binding molecule comprises an
aglycosylated monoclonal
antibody that is an immunoglobulin that binds to HER2, comprising two
identical heavy chains
and two identical light chains, said light chains being a first light chain
and a second light chain,
wherein the first light chain is fused to a first scFv, via a peptide linker,
to create a first light
chain fusion polypeptide, and wherein the second light chain is fused to a
second scFv, via a
peptide linker, to create a second light chain fusion polypeptide, wherein the
first and second
scFv (i) are identical, and (ii) bind to CD3, and wherein the first and second
light chain fusion
polypeptides are identical.
[00177] For a detailed example for the generation of a bispecific binding
molecule as
described herein, see, Section 6.1.2.1 for a detailed example for the design
and production of a
bispecific binding molecule described herein.
[00178] In certain embodiments, provided herein is a polynucleotide comprising
nucleotide
sequences encoding a light chain fusion polypeptide comprising a light chain
fused to a scFv, via
a peptide linker, wherein the light chain binds to HER2 and wherein the scFv
binds to CD3. In
certain embodiments, the light chain is the light chain of a HER2-specific
antibody known in the
art, such as, for example, trastuzumab, M-111, pertuzumab, ertumaxomab,
MDX11210, 2B1, and
MM-302. In certain embodiments, the scFv comprises the VH and VL of an anti-
CD3 antibody
known in the art, such as, for example, huOKT3, YTH12.5, HUM291, teplizumab,
huCLB-T3/4,
otelixizumab, blinatumomab, MT110, catumaxomab, 28F11, 27H5, 23F10, 15C3,
visilizumab,
and Hum291. In a preferred embodiment, the anti-CD3 antibody is huOKT3. In an
especially
preferred embodiment, the scFv comprises the VH of huOKT3, further comprising
the amino
acid substitution at numbered position 105, wherein the cysteine is
substituted with a serine. See,
for example, Kipriyanov et al. 1997, Protein Eng. 445-453. In certain
embodiments, the scFv is
-63-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
derived from the huOKT3 monoclonal antibody and comprises one or more
mutations, relative to
the native huOKT3 VH and VL, to stabilize disulfide binding. In certain
embodiments, the
stabilization of disulfide binding prevents aggregation of the bispecific
binding molecule. In
certain embodiments, the stabilization of disulfide binding reduces
aggregation of the bispecific
binding molecule as compared to aggregation of the bispecific binding molecule
without the
stabilization of disulfide binding. In certain embodiments of the bispecific
binding molecule, the
one or more mutations to stabilize disulfide binding comprise a VH G44C
mutation and a VL
Q100C mutation (e.g., as present in SEQ ID NOS: 54-59). In certain embodiments
of the
bispecific binding molecule, the one or more mutations to stabilize disulfide
binding are the
replacement of the amino acid residue at VH44(according to the Kabat numbering
system) with a
cysteine and the replacement of the amino acid residue at VL100 (according to
the Kabat
numbering system) with a cysteine so as to introduce a disulfide bond between
VH44 and VL100
(e.g., as present in SEQ ID NOS: 54-59). In certain embodiments, the peptide
linker is between
5-30, 5-25, 5-15, 10-30, 10-20, 10-15, 15-30, or 15-25 amino acid residues in
length. In certain
embodiments, the sequence of the peptide linker is as described in Table 1,
above (e.g., any one
of SEQ ID NOs: 14 or 35-41). In a particularly preferred embodiment, the
sequence of the
peptide linker is SEQ ID NO: 14. In certain embodiments, the sequence to the
scFy comprises
one or more modifications as described in Table 8, above.
[00179] In particular aspects, provided herein are polynucleotides comprising
nucleotide
sequences encoding bispecific binding molecules or fragments thereof, which
specifically bind to
HER2 and CD3, and comprise an amino acid sequence as described herein, as well
as antibodies
which compete with such bispecific binding molecules for binding to HER2
and/or CD3, or
which binds to the same epitope as that of such antibodies.
[00180] In a preferred embodiment, the sequence of the light chain is SEQ ID
NO: 25. In a
preferred embodiment, the nucleotide sequence encoding the light chain is SEQ
ID NO: 24. In a
preferred embodiment, the sequence of the scFy SEQ ID NO: 19. In a preferred
embodiment,
the nucleotide sequence encoding the scFy SEQ ID NO: 18. In a preferred
embodiment, the
sequence of the light chain is SEQ ID NO: 25 and the sequence of the scFy is
SEQ ID NO: 19.
In a preferred embodiment, the nucleotide sequence encoding the light chain is
SEQ ID NO: 24
and the nucleotide sequence encoding the scFy is SEQ ID NO: 18. In a preferred
embodiment,
the sequence of the light chain fusion polypeptide is SEQ ID NO: 29. In a
preferred
-64-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
embodiment, the nucleotide sequence encoding the light chain fusion
polypeptide is SEQ ID NO:
28.
[00181] In certain embodiments, the bispecific binding molecule has a
trastuzumab-derived
sequence that contains one or more of the modifications in the trastuzumab
immunoglobulin, and
has a huOKT3-derived sequence that contains one or more of the modifications
in the huOKT3
VH and VL sequences, as described in Table 8, below. Bispecific binding
molecules having other
immunoglobulin or scEv sequences can contain analogous mutations at
corresponding positions
in these other immunoglobulin or scEv sequences. In certain embodiments, the
bispecific
binding molecule is (a) derived from trastuzumab and huOKT3; and (b) contains
one or more of
the modifications as described in Table 8, above. In certain embodiments, the
sequence of the
peptide linker conjugating the immunoglobulin light chain and the scEv is as
described in Table
1, above (e.g., any one of SEQ ID NOs: 14 or 35-41). In certain embodiments,
the sequence of
the heavy chain is as described in Table 2, above (e.g., any one of SEQ ID
NOs: 23, 27, 62, or
63). In certain embodiments, the sequence of the light chain is as described
in Table 3, above
(e.g., SEQ ID NO: 25). In certain embodiments, the sequence of the VH of the
scEv is as
described in Table 4, above (e.g., any one of SEQ ID NOs: 15, 17, or 64). In
certain
embodiments, the sequence of the VL of the scEv is as described in Table 5,
above (e.g., any one
of SEQ ID NOs: 16 or 65). In certain embodiments, the sequence of the scEv
peptide linker is as
described in Table 1, above (e.g., any one of SEQ ID NOs: 14 or 35-41). In
certain
embodiments, the sequence of the scEv is as described in Table 6, above (e.g.,
any one of SEQ
ID NOs: 19 or 48-59). In certain embodiments, the sequence of the light chain
fusion
polypeptide is as described in Table 7, above (e.g., any one of SEQ ID NOs:
29, 34, 42-47, or
60).
[00182] In certain embodiments, provided herein is a polynucleotide comprising
nucleotide
sequences encoding the heavy chain of a HER2-specific antibody described in
Section 5.2. In
certain embodiments, the heavy chain is the heavy chain a HER2-specific
antibody known in the
art, such as, for example, trastuzumab, M-111, pertuzumab, ertumaxomab,
MD)<H210, 2B1, and
MM-302. In a preferred embodiment, the antibody comprises the VH of
trastuzumab, wherein
the sequence of the heavy chain is SEQ ID NO: 27. In a preferred embodiment,
the antibody
comprises the VH of trastuzumab, wherein the nucleotide sequence encoding the
heavy chain is
SEQ ID NO: 26. In a preferred embodiment, the sequence of the heavy chain is
comprises the
-65-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
VH of trastuzumab and comprises the amino acid substitution N297A in the Fc
region (SEQ ID
NO: 26). In a preferred embodiment, the nucleotide sequence encoding the heavy
chain
comprises the nucleotide sequence encoding the trastuzumab VH and comprises
the amino acid
substitution N297A in the Fc region (SEQ ID NO: 26). In preferred embodiment,
the sequence
of the heavy chain comprises the sequence of the trastuzumab VH and comprises
the amino acid
substitution K322A in the Fc region (SEQ ID NO: 27). In a preferred
embodiment, the
nucleotide sequence encoding the heavy chain comprises the nucleotide sequence
encoding the
trastuzumab VH and comprises the amino acid substitution K322A in the Fc
region (SEQ ID NO:
26). In an especially preferred embodiment, the sequence of the heavy chain
comprises the
sequence of the trastuzumab VH and comprises the amino acid substitutions
N297A and K322A
in the Fc region (SEQ ID NO: 27). In an especially preferred embodiment, the
nucleotide
sequence encoding the heavy chain comprises the nucleotide sequence encoding
the trastuzumab
VH and comprises the amino acid substitutions N297A and K322A in the Fc region
(SEQ ID
NO: 26).
[00183] The polynucleotides provided herein can be obtained by any method
known in the art.
For example, if the nucleotide sequence encoding a bispecific binding molecule
or fragment
thereof described herein is known, a polynucleotide encoding the bispecific
binding molecule or
fragment thereof can be may be assembled from chemically synthesized
oligonucleotides (e.g.,
as described in Kutmeier et at., BioTechniques 17:242 (1994)), which, briefly,
involves the
synthesis of overlapping oligonucleotides containing portions of the sequence
encoding the
antibody, annealing and ligating of those oligonucleotides, and then
amplification of the ligated
oligonucleotides by PCR.
[00184] Alternatively, a polynucleotide encoding a bispecific binding molecule
or fragment
thereof may be generated from nucleic acid from a suitable source. If a clone
containing a
nucleic acid encoding a particular bispecific binding molecule or fragment
thereof is not
available, but the sequence of the bispecific binding molecule or fragment
thereof is known, a
nucleic acid encoding the bispecific binding molecule or fragment thereof may
be chemically
synthesized or obtained from a suitable source (e.g., an antibody cDNA
library, or a cDNA
library generated from, or nucleic acid, preferably poly A+ RNA, isolated
from, any tissue or
cells expressing the antibody, such as hybridoma cells selected to express an
antibody provided
herein) by PCR amplification using synthetic primers that hybridize to the 3'
and 5' ends of the
-66-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
sequence or by cloning using an oligonucleotide probe specific for the
particular gene sequence
to identify, for example, a cDNA clone from a cDNA library that encodes the
antibody.
Amplified nucleic acids generated by PCR may then be cloned into replicable
cloning vectors
using any method well known in the art. See, for example, Section 5.3.2.
[00185] In certain embodiments, the amino acid sequence of the antibody of the
bispecific
binding molecule is known in the art. In such embodiments, a polypeptide
encoding such an
antibody may be manipulated using methods well known in the art for the
manipulation of
nucleotide sequences, e.g., recombinant DNA techniques, site directed
mutagenesis, PCR, etc.
(see, for example, the techniques described in Sambrook et at., 1990,
Molecular Cloning, A
Laboratory Manual, 2d Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor,
N.Y. and
Ausubel et at., eds., 1998, Current Protocols in Molecular Biology, John Wiley
& Sons, N.Y.,
which are both incorporated by reference herein in their entireties), to
generate bispecific binding
molecules having a different amino acid sequence, for example, to create amino
acid
substitutions, deletions, and/or insertions. For example, such manipulations
can be performed to
render the encoded amino acid aglycosylated, or to destroy the antibody's
ability to bind to Clq,
Fc receptor, or to activate the complement system.
[00186] Isolated nucleic acid molecules provided herein can include nucleic
acid molecules
comprising an open reading frame (ORF), optionally with one or more introns,
for example, but
not limited to, at least one specified portion of at least one complementarity
determining region
(CDR), as CDR1, CDR2 and/or CDR3 of at least one heavy chain or light chain;
nucleic acid
molecules comprising the coding sequence for an anti-HER2 antibody or variable
region, an anti-
CD3 scFv, or a single chain fusion polypeptide; and nucleic acid molecules
which comprise a
nucleotide sequence substantially different from those described above but
which, due to the
degeneracy of the genetic code, still encode at least one bispecific binding
molecule as described
herein and/or as known in the art.
[00187] Also provided herein are isolated nucleic acids that hybridize
under selective
hybridization conditions to a polynucleotide disclosed herein. Thus, the
polynucleotides of this
embodiment can be used for isolating, detecting, and/or quantifying nucleic
acids comprising
such polynucleotides. For example, polynucleotides provided herein can be used
to identify,
isolate, or amplify partial or full-length clones in a deposited library. In
some embodiments, the
-67-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
polynucleotides are genomic or cDNA sequences isolated, or otherwise
complementary to, a
cDNA from a human or mammalian nucleic acid library.
[00188] The nucleic acids can conveniently comprise sequences in addition to a
polynucleotide provided herein. For example, a multi-cloning site comprising
one or more
endonuclease restriction sites can be inserted into the nucleic acid to aid in
isolation of the
polynucleotide. In addition, translatable sequences can be inserted to aid in
the isolation of the
translated polynucleotide provided herein. For example, a hexa-histidine
marker sequence
provides a convenient means to purify the polypeptides provided herein. The
nucleic acid
provided herein¨excluding the coding sequence¨is optionally a vector, adapter,
or linker for
cloning and/or expression of a polynucleotide provided herein.
[00189] Additional sequences can also be added to such cloning and/or
expression sequences
to optimize their function in cloning and/or expression, to aid in isolation
of the polynucleotide,
or to improve the introduction of the polynucleotide into a cell. Use of
cloning vectors,
expression vectors, adapters, and linkers is well known in the art. (See,
e.g., Ausubel, supra; or
Sambrook, supra).
[00190] In a specific embodiment, using routine recombinant DNA techniques,
one or more of
the CDRs of an antibody described herein may be inserted within framework
regions. The
framework regions may be naturally occurring or consensus framework regions,
and preferably
human framework regions (see, e.g., Chothia et at., J. Mol. Biol. 278: 457-479
(1998) for a
listing of human framework regions). Preferably, the polynucleotide generated
by the
combination of the framework regions and CDRs encodes an antibody that
specifically binds
HER2. One or more amino acid substitutions may be made within the framework
regions, and,
preferably, the amino acid substitutions improve binding of the antibody to
its antigen.
Additionally, such methods may be used to make amino acid substitutions or
deletions of one or
more variable region cysteine residues participating in an intrachain
disulfide bond to generate
antibody molecules lacking one or more intrachain disulfide bonds. Other
alterations to the
polynucleotide are provided herein and within the skill of the art.
[00191] In certain embodiments, the isolated or purified nucleic acid
molecule, or fragment
thereof, upon linkage with another nucleic acid molecule, can encode a fusion
protein. The
generation of fusion proteins is within the ordinary skill in the art and can
involve the use of
-68-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
restriction enzyme or recombinational cloning techniques (see, for example,
Gateway.TM..
(Invitrogen)). See, also, U.S. Pat. No. 5,314,995.
[00192] In certain embodiments, a polynucleotide provided herein is in the
form of a vector
(e.g., expression vector) as described in Section 5.3.2.
5.3.2 CELLS AND VECTORS
[00193] In certain embodiments, provided herein are cells (e.g., ex vivo
cells) expressing (e.g.,
recombinantly) bispecific binding molecules as described herein. Also provided
herein are
vectors (e.g., expression vectors) comprising nucleotide sequences (see, for
example,
Section 5.3.1) encoding a bispecific binding molecule or fragment thereof
described herein for
recombinant expression in host cells, preferably in mammalian cells. Also
provided herein are
cells (e.g., ex vivo cells) comprising such vectors or nucleotide sequences
for recombinantly
expressing a bispecific binding molecule described here. Also provided herein
are methods for
producing a bispecific binding molecule described herein, comprising
expressing such bispecific
binding molecule from a cell (e.g., ex vivo cell). In a preferred embodiment,
the cell is an ex vivo
cell.
[00194] A vector (e.g., expression vector) is a DNA molecule comprising a gene
that is
expressed in a cell (e.g., ex vivo cell). Typically, gene expression is placed
under the control of
certain regulatory elements, including constitutive or inducible promoters,
tissue-specific
regulatory elements and enhancers. Such a gene is said to be "operably linked
to" the regulatory
elements, e.g., a promoter. A recombinant host may be any prokaryotic or
eukaryotic cell that
contains either a cloning vector or expression vector. This term also includes
those prokaryotic
or eukaryotic cells, as well as a transgenic animal, that have been
genetically engineered to
contain the cloned gene(s) in the chromosome or genome of the host cell or
cells of the host cells
(e.g., ex vivo cells).
[00195] In a preferred embodiment, the promoter is the CMV promoter.
[00196] In certain embodiments, provided herein is a vector comprising one or
more
polynucleotide as described in Section 5.3.1.
[00197] In certain embodiments, a polynucleotide as described in Section 5.3.1
can be cloned
into a suitable vector and can be used to transform or transfect any suitable
host. Vectors and
methods to construct such vectors are known to one of ordinary skill in the
art and are described
in general technical references (see, in general, "Recombinant DNA Part D,"
Methods in
-69-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
Enzymology, Vol. 153, Wu and Grossman, eds., Academic Press (1987)). In
certain
embodiments, the vector comprises regulatory sequences, such as transcription
and translation
initiation and termination codons, which are specific to the type of host
(e.g., bacterium, fungus,
plant, insect, or mammal) into which the vector is to be introduced, as
appropriate and taking
into consideration whether the vector is DNA or RNA. In certain embodiments,
the vector
comprises regulatory sequences that are specific to the genus of the host. In
certain
embodiments, the vector comprises regulatory sequences that are specific to
the species of the
host.
[00198] In certain embodiments, the vector comprises one or more marker genes,
which allow
for selection of transformed or transfected hosts. Non-limiting examples of
marker genes
include biocide resistance, e.g., resistance to antibiotics, heavy metals,
etc., complementation in
an auxotrophic host to provide prototrophy, and the like. In a preferred
embodiment, the vector
comprises ampicillin and hygromycin selectable markers.
[00199] In certain embodiments, an expression vector can comprise a native or
normative
promoter operably linked to a polynucleotide as described in Section 5.3.1.
The selection of
promoters, for example, strong, weak, inducible, tissue-specific and
developmental-specific, is
within the skill in the art. Similarly, the combining of a nucleic acid
molecule, or fragment
thereof, as described above with a promoter is also within the skill in the
art.
[00200] Non-limiting examples of suitable vectors include those designed for
propagation and
expansion or for expression or both. For example, a cloning vector can be
selected from the
group consisting of the pUC series, the pBluescript series (Stratagene,
LaJolla, Calif.), the pET
series (Novagen, Madison, Wis.), the pGEX series (Pharmacia Biotech, Uppsala,
Sweden), and
the pEX series (Clontech, Palo Alto, Calif.). Bacteriophage vectors, such as
lamda-GT10,
lamda-GT11, lamda-ZapII (Stratagene), lamda-EMBL4, and lamda-NM1149, can also
be used.
Non-limiting examples of plant expression vectors include pBI110, pBI101.2,
pBI101.3, pBI121
and pBIN19 (Clontech). Non-limiting examples of animal expression vectors
include pEUK-C1,
pMAM and pMAMneo (Clontech). The TOPO cloning system (Invitrogen, Carlsbad,
Calif.) can
also be used in accordance with the manufacturer's recommendations.
[00201] In certain embodiments, the vector is a mammalian vector. In certain
embodiments,
the mammalian vector contains at least one promoter element, which mediates
the initiation of
transcription of mRNA, the bispecific binding molecule coding sequence, and
signals required
-70-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
for the termination of transcription and polyadenylation of the transcript. In
certain
embodiments, the mammalian vector contains additional elements, such as, for
example,
enhancers, Kozak sequences and intervening sequences flanked by donor and
acceptor sites for
RNA splicing. In certain embodiments, highly efficient transcription can be
achieved with, for
example, the early and late promoters from SV40, the long terminal repeats
(LTRS) from
retroviruses, for example, RSV, HTLVI, HIVI and the early promoter of the
cytomegalovirus
(CMV). However, cellular elements can also be used (e.g., the human actin
promoter). Non-
limiting examples of mammalian expression vectors include, vectors such as
pIRES1neo, pRetro-
Off, pRetro-On, PLXSN, or pLNCX (Clonetech Labs, Palo Alto, Calif.), pcDNA3.1
(+/-),
pcDNA/Zeo (+/-) or pcDNA3.1/Hygro (+/-) (Invitrogen), PSVL and PMSG
(Pharmacia,
Uppsala, Sweden), pRSVcat (ATCC 37152), pSV2dhfr (ATCC 37146) and pBC12MI
(ATCC
67109). Non-limiting example of mammalian host cells that can be used in
combination with
such mammalian vectors include human Hela 293, H9 and Jurkat cells, mouse
NIH3T3 and
C127 cells, Cos 1, Cos 7 and CV 1, quail QC1-3 cells, mouse L cells and
Chinese hamster ovary
(CHO) cells.
[00202] In certain embodiments, the vector is a viral vector, for example,
retroviral vectors,
parvovirus-based vectors, e.g., adeno-associated virus (AAV)-based vectors,
AAV-adenoviral
chimeric vectors, and adenovirus-based vectors, and lentiviral vectors, such
as Herpes simplex
(HSV)-based vectors. In certain embodiments, the viral vector is manipulated
to render the virus
replication deficient. In certain embodiments, the viral vector is manipulated
to eliminate
toxicity to the host. These viral vectors can be prepared using standard
recombinant DNA
techniques described in, for example, Sambrook et at., Molecular Cloning, a
Laboratory Manual,
2d edition, Cold Spring Harbor Press, Cold Spring Harbor, N.Y. (1989); and
Ausubel et at.,
Current Protocols in Molecular Biology, Greene Publishing Associates and John
Wiley & Sons,
New York, N.Y. (1994).
[00203] In certain embodiments, a vector or polynucleotide described herein
can be
transferred to a cell (e.g., an ex vivo cell) by conventional techniques and
the resulting cell can be
cultured by conventional techniques to produce a bispecific binding molecule
described herein.
Accordingly, provided herein are cells comprising a polynucleotide encoding a
bispecific
binding molecule or fragment thereof, a heavy or light chain thereof, or a
light chain fusion
polypeptide thereof, operably linked to a promoter for expression of such
sequences in the host
-71-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
cell. In certain embodiments, a vector encoding the heavy chain operably
linked to a promoter
and a vector encoding the light chain fusion polypeptide operably linked to a
promoter can be co-
expressed in the cell for expression of the entire bispecific binding
molecule, as described below.
In certain embodiments, a cell comprises a vector comprising a polynucleotide
encoding both the
heavy chain and the light chain fusion polypeptide of a bispecific binding
molecule described
herein operably linked to a promoter. In certain embodiments, a cell comprises
two different
vectors, a first vector comprising a polynucleotide encoding a heavy chain
operably linked to a
promoter, and a second vector comprising a polynucleotide encoding a light
chain fusion
polypeptide operably linked to a promoter. In certain embodiments, a first
cell comprises a first
vector comprising a polynucleotide encoding a heavy chain of a bispecific
binding molecule
described herein, and a second cell comprises a second vector comprising a
polynucleotide
encoding a light chain fusion polypeptide of a bispecific binding molecule
described herein. In
certain embodiments, provided herein is a mixture of cells comprising such
first cell and such
second cell. In a preferred embodiment, the cell expresses the vector or
vectors such that the
oligonucleotide is both transcribed and translated efficiently by the cell.
[00204] In embodiment, the cell expresses the vector, such that the
oligonucleotide, or
fragment thereof, is both transcribed and translated efficiently by the cell.
[00205] In certain embodiments, the cell is present in a host, which can be an
animal, such as
a mammal. Examples of cells include, but are not limited to, a human cell, a
human cell line, E.
coli (e.g., E. coli TB-1, TG-2, DH5a, XL-Blue MRF' (Stratagene), SA2821 and
Y1090), B.
subtilis, P. aerugenosa, S. cerevisiae, N. crassa, insect cells (e.g., Sf9,
Ea4) and others set forth
herein below. In a preferred embodiment, the cell is a CHO cell. In an
especially preferred
embodiment, the cell is a CHO-S cell.
[00206] In certain embodiments, a polynucleotide described herein can be
expressed in a
stable cell line that comprises the polynucleotide integrated into a
chromosome by introducing
the polynucleotide into the cell. In certain embodiments, the polynucleotide
is introduced into
the cell by, for example, electroporation. In certain embodiments, the
polynucleotide is
introduced into the cell by, for example, transfection of a vector comprising
the polynucleotide
into the cell. In certain embodiments, the vector is co-transfected with a
selectable marker such
as DHFR, GPT, neomycin, or hygromycin to allow for the identification and
isolation of the
transfected cells. In certain embodiments, the transfected polynucleotide can
also be amplified
-72-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
to express large amounts of the encoded bispecific binding molecule. For
example, the DHFR
(dihydrofolate reductase) marker can be utilized to develop cell lines that
carry several hundred
or even several thousand copies of the polynucleotide of interest. Another
example of a selection
marker is the enzyme glutamine synthase (GS) (Murphy, et at., Biochem. J.
227:277-279 (1991);
Bebbington, et at., Bio/Technology 10:169-175 (1992)). Using these markers,
the cells are
grown in selective medium and the cells with the highest resistance are
selected. These cell lines
contain the amplified gene(s) integrated into a chromosome. Chinese hamster
ovary (CHO) and
NSO cells are often used for the production of bispecific binding molecules.
[00207] In a preferred embodiment, the vector comprises (i) a first
polynucleotide sequence
encoding a light chain fusion polypeptide comprising an immunoglobulin light
chain fused to a
scFv, via a peptide linker, wherein the light chain binds to HER2 and wherein
the scFv binds to
CD3, operably linked to a first promoter and (ii) a second polynucleotide
encoding an
immunoglobulin heavy chain that binds to HER2 operably linked to a second
promoter. In
certain embodiments, the vector is a viral vector.
5.4 T CELLS BOUND TO BISPECIFIC BINDING MOLECULES
[00208] Without being bound by any theory, it is believed that when the
bispecific binding
molecules provided herein are bound to T cells, by, for example, procedures
such as those
described herein, an anti-CD3 scFv of the bispecific binding molecule binds to
CD3 on the
surface of the T cell. Without being bound by any theory, it is believed that
binding of the
bispecific binding molecule to the T cell (i.e., binding of an anti-CD3 scFv
to CD3 expressed on
the T cell) activates the T cell, and consequently, allows for the T cell
receptor-based
cytotoxicity to be redirected to desired tumor targets, bypassing MHC
restrictions.
[00209] Thus, the invention also provides T cells which are bound to a
bispecific binding
molecule of the invention (e.g., as described in Section 5.1 and Section 5.2).
In specific
embodiments, the T cells are bound to the bispecific binding molecule
noncovalently. In specific
embodiments, the T cells are autologous to a subject to whom the T cells are
to be administered.
In specific embodiments, the T cells are allogeneic to a subject to whom the T
cells are to be
administered. In specific embodiments, the T cells are human T cells.
[00210] In specific embodiments, the T cells which are bound to bispecific
binding molecules
of the invention are used in accordance with the methods described in Section
5.6. In specific
-73-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
embodiments, the T cells which are bound to bispecific binding molecules of
the invention are
used as part of a combination therapy as described in Section 5.9.
5.5 PHARMACEUTICAL COMPOSITIONS AND KITS
[00211] In certain embodiments, provided herein are compositions (e.g.,
pharmaceutical
compositions) and kits comprising a pharmaceutically effective amount of one
or more bispecific
binding molecule as described in Section 5.1 or Section 5.2. Compositions may
be used in the
preparation of individual, single unit dosage forms. Compositions provided
herein can be
formulated for parenteral, subcutaneous, intramuscular, intravenous,
intrarticular, intrabronchial,
intraabdominal, intracapsular, intracartilaginous, intracavitary, intracelial,
intracerebellar,
intracerebroventricular, intra-Ommaya, intraocular, intravitreous, intracolic,
intracervical,
intragastric, intrahepatic, intramyocardial, intraosteal, intrapelvic,
intrapericardiac,
intraperitoneal, intrapleural, intraprostatic, intrapulmonary, intrarectal,
intrarenal, intraretinal,
intraspinal, intrasynovial, intrathoracic, intrauterine, intravesical, bolus,
vaginal, rectal, buccal,
sublingual, intranasal, intrathecal, intraventricular in the brain,
intraparenchymal in the brain, or
transdermal administration. In a preferred embodiment, the composition is
formulated for
parenteral administration. In an especially preferred embodiment, the
composition is formulated
for intravenous administration. In a preferred embodiment, the composition is
formulated for
intraperitoneal administration. In a specific embodiment, the composition is
formulated for
intraperitoneal administration to treat peritoneal metastases. In a preferred
embodiment, the
composition is formulated for intrathecal administration. In a specific
embodiment, the
composition is formulated for intrathecal administration to treat brain
metastases. See, for
example, Kramer et at., 2010, 97: 409-418. In a preferred embodiment, the
composition is
formulated for intraventricular administration in the brain. In a specific
embodiment, the
composition is formulated for intraventricular administration to treat brain
metastases. See, for
example, Kramer et at., 2010, 97: 409-418. In a preferred embodiment, the
composition is
formulated for intraparenchymal administration in the brain. In a specific
embodiment, the
composition is formulated for intraparenchymal administration to treat a brain
tumor or brain
tumor metastases. See, for example, Luther et at., 2014, Neuro Oncol, 16: 800-
806, and Clinical
Trial ID NO NCT01502917.
[00212] In a specific embodiment, the composition is formulated for
intraperitoneal
administration for peritoneal metastases.
-74-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
[00213] In certain embodiments, provided herein is a composition comprising
one or more
polynucleotide comprising nucleotide sequences encoding a bispecific binding
molecule as
described herein. In certain embodiments, provided herein is a composition
comprising a cell,
wherein the cell comprises one or more polynucleotide comprising nucleotide
sequences
encoding a bispecific binding molecule as described herein. In certain
embodiments, provided
herein is a composition comprising a vector, wherein the vector comprises one
or more
polynucleotide comprising nucleotide sequences encoding a bispecific binding
molecule as
described herein. In certain embodiments, provided herein is a composition
comprising a cell,
wherein the cell comprises a vector, wherein the vector comprises one or more
polynucleotide
comprising nucleotide sequences encoding a bispecific binding molecule as
described herein.
[00214] In certain embodiments, a composition described herein is a stable or
preserved
formulation. In certain embodiments, the stable formulation comprises a
phosphate buffer with
saline or a chosen salt. In certain embodiments, a composition described is a
multi-use preserved
formulation, suitable for pharmaceutical or veterinary use. In certain
embodiments, a
composition described herein comprises a preservative. Preservatives are known
to one of
ordinary skill in the art. Non-limiting examples of preservatives include
phenol, m-cresol, p-
cresol, o-cresol, chlorocresol, benzyl alcohol, phenylmercuric nitrite,
phenoxyethanol,
formaldehyde, chlorobutanol, magnesium chloride (e.g., hexahydrate),
alkylparaben (methyl,
ethyl, propyl, butyl and the like), benzalkonium chloride, benzethonium
chloride, and sodium
dehydroacetate and thimerosal, or mixtures thereof in an aqueous diluent. Any
suitable
concentration or mixture can be used as known in the art, such as 0.001-5%, or
any range or
value therein, such as, but not limited to 0.001, 0.003, 0.005, 0.009, 0.01,
0.02, 0.03, 0.05, 0.09,
0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0, 1.1, 1.2, 1.3, 1.4, 1.5,
1.6, 1.7, 1.8, 1.9, 2.0, 2.1, 2.2,
2.3, 2.4, 2.5, 2.6, 2.7, 2.8, 2.9, 3.0, 3.1, 3.2, 3.3, 3.4, 3.5, 3.6, 3.7,
3.8, 3.9, 4.0, 4.3, 4.5, 4.6, 4.7,
4.8, 4.9, or any range or value therein. Non-limiting examples include, no
preservative, 0.1-2%
m-cresol (e.g., 0.2, 0.3. 0.4, 0.5, 0.9, 1.0%), 0.1-3% benzyl alcohol (e.g.,
0.5, 0.9, 1.1, 1.5, 1.9,
2.0, 2.5%), 0.001-0.5% thimerosal (e.g., 0.005, 0.01), 0.001-2.0% phenol
(e.g., 0.05, 0.25, 0.28,
0.5, 0.9, 1.0%), 0.0005-1.0% alkylparaben(s) (e.g., 0.00075, 0.0009, 0.001,
0.002, 0.005, 0.0075,
0.009, 0.01, 0.02, 0.05, 0.075, 0.09, 0.1, 0.2, 0.3, 0.5, 0.75, 0.9, 1.0%),
and the like.
[00215] It can be sometimes desirable to deliver the compositions provided
herein to a subject
over prolonged periods of time, for example, for periods of one week to one
year or more from a
-75-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
single administration. Various slow release, depot or implant dosage forms can
be utilized. For
example, a dosage form can contain a pharmaceutically acceptable non-toxic
salt of the
compounds that has a low degree of solubility in body fluids, for example, (a)
an acid addition
salt with a polybasic acid such as phosphoric acid, sulfuric acid, citric
acid, tartaric acid, tannic
acid, pamoic acid, alginic acid, polyglutamic acid, naphthalene mono- or di-
sulfonic acids,
polygalacturonic acid, and the like; (b) a salt with a polyvalent metal cation
such as zinc,
calcium, bismuth, barium, magnesium, aluminum, copper, cobalt, nickel, cadmium
and the like,
or with an organic cation formed from e.g., N,N'-dibenzyl-ethylenediamine or
ethylenediamine;
or (c) combinations of (a) and (b) e.g., a zinc tannate salt. Additionally, a
composition provided
herein, preferably, a relatively insoluble salt such as those just described,
can be formulated in a
gel, for example, an aluminum monostearate gel with, e.g., sesame oil,
suitable for injection.
Particularly preferred salts are zinc salts, zinc tannate salts, pamoate
salts, and the like. Another
type of slow release depot formulation for injection would contain the
compound or salt
dispersed for encapsulated in a slow degrading, non-toxic, non-antigenic
polymer such as a
polylactic acid/polyglycolic acid polymer, for example, as described in U.S.
Pat. No. 3,773,919.
The compounds or, preferably, relatively insoluble salts such as those
described above can also
be formulated in cholesterol matrix silastic pellets, particularly for use in
animals. Additional
slow release, depot or implant compositons, e.g., gas or liquid liposomes are
known in the
literature (U.S. Pat. No. 5,770,222 and "Sustained and Controlled Release Drug
Delivery
Systems", J. R. Robinson ed., Marcel Dekker, Inc., N.Y., 1978).
[00216] The range of at least one bispecific binding molecule composition
provided herein
includes amounts yielding upon reconstitution, if in a wet/dry system,
concentrations from about
1.0 microgram/ml to about 1000 mg/ml, although lower and higher concentrations
are operable
and are dependent on the intended delivery vehicle, e.g., solution
formulations will differ from
transdermal patch, pulmonary, transmucosal, or osmotic or micro pump methods.
[00217] In certain embodiments, compositions provided herein comprise at least
one of any
suitable auxiliary, such as, but not limited to, diluent, binder, stabilizer,
buffers, salts, lipophilic
solvents, preservative, adjuvant or the like. In certain embodiments,
pharmaceutically acceptable
auxiliaries are preferred. Non-limiting examples of, and methods of preparing
such sterile
solutions are well known in the art, such as, but not limited to, Gennaro,
Ed., Remington's
Pharmaceutical Sciences, 18th Edition, Mack Publishing Co. (Easton, Pa.) 1990.
-76-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
Pharmaceutically acceptable carriers can be routinely selected that are
suitable for the mode of
administration, solubility and/or stability of the bispecific binding molecule
as described herein.
[00218] In certain embodiments, compositions provided herein contain one or
more
pharmaceutical excipient and/or additive. Non-limiting examples of
pharmaceutical excipients
and additives are proteins, peptides, amino acids, lipids, and carbohydrates
(e.g., sugars,
including monosaccharides, di-, tri-, tetra-, and oligosaccharides;
derivatized sugars such as
alditols, aldonic acids, esterified sugars and the like; and polysaccharides
or sugar polymers),
which can be present singly or in combination, comprising alone or in
combination 1-99.99% by
weight or volume. Non-limiting examples of protein excipients include serum
albumin such as
human serum albumin (HSA), recombinant human albumin (rHA), gelatin, casein,
and the like.
Non-limiting examples of amino acid/antibody components, which can also
function in a
buffering capacity, include alanine, glycine, arginine, betaine, histidine,
glutamic acid, aspartic
acid, cysteine, lysine, leucine, isoleucine, valine, methionine,
phenylalanine, aspartame, and the
like. In certain embodiments, the amino acid is glycine. Non-limiting examples
of carbohydrate
excipients include monosaccharides such as fructose, maltose, galactose,
glucose, D-mannose,
sorbose, and the like; disaccharides, such as lactose, sucrose, trehalose,
cellobiose, and the like;
polysaccharides, such as raffinose, melezitose, maltodextrins, dextrans,
starches, and the like;
and alditols, such as mannitol, xylitol, maltitol, lactitol, xylitol sorbitol
(glucitol), myoinositol
and the like. In certain embodiments, the carbohydrate excipient is mannitol,
trehalose, or
raffinose.
[00219] In certain embodiments, a composition provided herein includes one or
more buffer
or a pH adjusting agent; typically, the buffer is a salt prepared from an
organic acid or base.
Non-limiting examples of buffers include organic acid salts such as salts of
citric acid, ascorbic
acid, gluconic acid, carbonic acid, tartaric acid, succinic acid, acetic acid,
or phthalic acid; Tris,
tromethamine hydrochloride, or phosphate buffers. In certain embodiments, the
buffer is an
organic acid salts such as citrate. Other excipients, e.g., isotonicity
agents, buffers, antioxidants,
preservative enhancers, can be optionally and preferably added to the diluent.
An isotonicity
agent, such as glycerin, is commonly used at known concentrations. A
physiologically tolerated
buffer is preferably added to provide improved pH control. The compositions
can cover a wide
range of pHs, such as from about pH 4 to about pH 10, and preferred ranges
from about pH 5 to
about pH 9, and a most preferred range of about 6.0 to about 8Ø Preferably,
the compositions
-77-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
provided herein have pH between about 6.8 and about 7.8. Preferred buffers
include phosphate
buffers, most preferably sodium phosphate, particularly phosphate buffered
saline (PBS).
[00220] In certain embodiments, a composition provided herein includes one or
more
polymeric excipient/additive such as, for example, polyvinylpyrrolidones,
ficolls (a polymeric
sugar), dextrates (e.g., cyclodextrins, such as 2-hydroxypropyl-.beta.-
cyclodextrin), polyethylene
glycols, flavoring agents, antimicrobial agents, sweeteners, antioxidants,
antistatic agents,
surfactants (e.g., polysorbates such as "TWEEN 20" and "TWEEN 80"), lipids
(e.g.,
phospholipids, fatty acids), steroids (e.g., cholesterol), and/or chelating
agents (e.g., EDTA).
[00221] Other additives, such as a pharmaceutically acceptable solubilizers
like Tween 20
(polyoxyethylene (20) sorbitan monolaurate), Tween 40 (polyoxyethylene (20)
sorbitan
monopalmitate), Tween 80 (polyoxyethylene (20) sorbitan monooleate), Pluronic
F68
(polyoxyethylene polyoxypropylene block copolymers), and PEG (polyethylene
glycol) or non-
ionic surfactants such as polysorbate 20 or 80 or poloxamer 184 or 188,
Pluronic® polyls,
other block co-polymers, and chelators such as EDTA and EGTA can optionally be
added to the
compositions to reduce aggregation. These additives are particularly useful if
a pump or plastic
container is used to administer the composition. The presence of
pharmaceutically acceptable
surfactant mitigates the propensity for the protein to aggregate.
[00222] Additional pharmaceutical excipients and/or additives suitable for use
in a
composition provided herein are known to one of skill in the art and are
referenced in, for
example, "Remington: The Science & Practice of Pharmacy", 19<sup>th</sup> ed.,
Williams &
Williams, (1995), and in the "Physician's Desk Reference", 52nd ed., Medical
Economics,
Montvale, N.J. (1998), which are entirely incorporated herein by reference. In
certain preferred
embodiments, the carrier or excipient materials are carbohydrates (e.g.,
saccharides and alditols)
and buffers (e.g., citrate) or polymeric agents.
[00223] Preferably, the aqueous diluent optionally further comprises a
pharmaceutically
acceptable preservative. Preferred preservatives include those selected from
the group consisting
of phenol, m-cresol, p-cresol, o-cresol, chlorocresol, benzyl alcohol,
alkylparaben (methyl, ethyl,
propyl, butyl and the like), benzalkonium chloride, benzethonium chloride,
sodium
dehydroacetate and thimerosal, or mixtures thereof. The concentration of
preservative used in
the composition is a concentration sufficient to yield an anti-microbial
effect. Such
-78-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
concentrations are dependent on the preservative selected and are readily
determined by the
skilled artisan.
[00224] The compositions provided herein can be prepared by a process which
comprises
mixing at least one bispecific binding molecule as described herein and a
preservative selected
from the group consisting of phenol, m-cresol, p-cresol, o-cresol,
chlorocresol, benzyl alcohol,
alkylparaben, (methyl, ethyl, propyl, butyl and the like), benzalkonium
chloride, benzethonium
chloride, sodium dehydroacetate and thimerosal or mixtures thereof in an
aqueous diluent.
Mixing the at least one bispecific binding molecule and preservative in an
aqueous diluent is
carried out using conventional dissolution and mixing procedures. To prepare a
suitable
composition, for example, a measured amount of at least one bispecific binding
molecule in
buffered solution is combined with the desired preservative in a buffered
solution in quantities
sufficient to provide the bispecific binding molecule and preservative at the
desired
concentrations. The compositions provided herein can be prepared by a process
that comprises
mixing at least one bispecific binding molecule as described herein and a
selected buffer,
preferably a phosphate buffer containing saline or a chosen salt. Mixing the
at least one
bispecific binding molecule and buffer in an aqueous diluent is carried out
using conventional
dissolution and mixing procedures. To prepare a suitable composition, for
example, a measured
amount of at least one bispecific binding molecule in water or buffer is
combined with the
desired buffering agent in water in quantities sufficient to provide the
protein and buffer at the
desired concentrations. Variations of these processes would be recognized by
one of ordinary
skill in the art. For example, the order the components are added, whether
additional additives
are used, the temperature and pH at which the composition is prepared, are all
factors that can be
optimized for the concentration and means of administration used.
[00225] In specific embodiments involving combination therapy with infusion of
T cells,
provided herein is a pharmaceutical composition comprising (a) a bispecific
binding molecule
described herein (see, e.g., Section 5.1 or 5.2); (b) T cells; and/or (c) a
pharmaceutically effective
carrier. In specific embodiments, the T cells are autologous to the subject to
whom the T cells
are administered. In certain embodiments, the T cells are allogeneic to the
subject to whom the
T cells are administered. In specific embodiments, the T cells are bound to
the bispecific binding
molecule. In specific embodiments, the binding of the T cells to the
bispecific binding molecule
is noncovalently. In specific embodiments, the T cells are human T cells.
Methods that can be
-79-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
used to bind bispecific binding molecules to T cells are known in the art.
See, e.g., Lum et al.,
2013, Biol Blood Marrow Transplant, 19:925-33, Janeway et at., Immunobiology:
The Immune
System in Health and Disease, 5th edition, New York: Garland Science;
Vaishampayan et at.,
2015, Prostate Cancer, 2015:285193, and Stromnes et al., 2014, Immunol Rev.
257(1):145-164.
See, also, Vaishampayan et al., 2015, Prostate Cancer, 2015:285193, which
describes the
following exemplary, non-limiting method for binding bispecific binding
molecules to T cells:
Peripheral blood mononuclear cells (PBMCs) are collected to obtain lymphocytes
for activated T cell expansion from 1 or 2 leukopheresis. PBMCs are activated
with, for example, 20 ng/mL of OKT3 and expanded in 100 IU/mL of IL-2 to
generate 40-320 billion activated T cells during a maximum of 14 days of
culture
under cGMP conditions as described in Ueda et al., 1993, Transplantation,
56(2):351-356 and Uberti et al., 1994, Clinical Immunology and
Immunopathology, 70(3):234-240. Cells are grown in breathable flasks (FEP Bag
Type 750-C1, American Fluoroseal Corporation, Gaithersburg, MD) in RPMI
1640 medium (Lonza) supplemented with 2% pooled heat inactivated human
serum. Activated T cells are split approximately every 2-3 days based on cell
counts. After 14 days, activated T cells are cultured with 50 ng of a
bispecific
binding molecule described herein per 106 activated T cells. The mixture is
then
washed and cryopreserved.
[00226] In certain embodiments, a pharmaceutical composition described herein
is to be used
in accordance with the methods provided herein (see, e.g., Section 5.6).
5.5.1 PARENTERAL FORMULATIONS
[00227] In certain embodiments, a composition provided herein is formulated
for parenteral
injectable administration. As used herein, the term "parenteral" includes
intravenous,
intravascular, intramuscular, intradermal, subcutaneous, and intraocular. For
parenteral
administration, the composition can be formulated as a solution, suspension,
emulsion or
lyophilized powder in association, or separately provided, with a
pharmaceutically acceptable
parenteral vehicle. Non-limiting examples of such vehicles are water, saline,
Ringer's solution,
dextrose solution, glycerol, ethanol, and 1-10% human serum albumin. Liposomes
and
nonaqueous vehicles such as fixed oils can also be used. The vehicle or
lyophilized powder can
contain additives that maintain isotonicity (e.g., sodium chloride, mannitol)
and chemical
-80-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
stability (e.g., buffers and preservatives). The formulation is sterilized by
known or suitable
techniques.
[00228] Suitable pharmaceutical carriers are described in the most recent
edition of
Remington's Pharmaceutical Sciences, A. Osol, a standard reference text in
this field.
[00229] Formulations for parenteral administration can contain as common
excipients sterile
water or saline, polyalkylene glycols such as polyethylene glycol, oils of
vegetable origin,
hydrogenated naphthalenes and the like. Aqueous or oily suspensions for
injection can be
prepared by using an appropriate emulsifier or humidifier and a suspending
agent, according to
known methods. Agents for injection can be a non-toxic, non-orally
administrable diluting agent
such as aqueous solution or a sterile injectable solution or suspension in a
solvent. As the usable
vehicle or solvent, water, Ringer's solution, isotonic saline, etc. are
allowed; as an ordinary
solvent, or suspending solvent, sterile involatile oil can be used. For these
purposes, any kind of
involatile oil and fatty acid can be used, including natural or synthetic or
semisynthetic fatty oils
or fatty acids; natural or synthetic or semisynthetic mono- or di- or tri-
glycerides. Parental
administration is known in the art and includes, but is not limited to,
conventional means of
injections, a gas pressured needle-less injection device as described in U.S.
Pat. No. 5,851,198,
and a laser perforator device as described in U.S. Pat. No. 5,839,446 entirely
incorporated herein
by reference.
5.5.2 PULMONARY FORMULATIONS
[00230] In certain embodiments, a composition comprising a bispecific binding
molecule
described herein is formulated for pulmonary administration. For pulmonary
administration, the
composition is delivered in a particle size effective for reaching the lower
airways of the lung or
sinuses. Compositions for pulmonary administration can be delivered by any of
a variety of
inhalation or nasal devices known in the art for administration of a
therapeutic agent by
inhalation. These devices capable of depositing aerosolized formulations in
the sinus cavity or
alveoli of a patient include metered dose inhalers, nebulizers, dry powder
generators, sprayers,
and the like. Other devices suitable for directing the pulmonary or nasal
administration of
bispecific binding molecules described herein are also known in the art. All
such devices use
formulations suitable for the administration for the dispensing of a
bispecific binding molecule
described herein in an aerosol. Such aerosols can be comprised of either
solutions (both aqueous
and non aqueous) or solid particles. Metered dose inhalers like the Ventoling
metered dose
-81-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
inhaler, typically use a propellent gas and require actuation during
inspiration (See, e.g., WO
94/16970, WO 98/35888). Dry powder inhalers like TurbuhalerTm (Astra),
Rotahaler . (Glaxo),
Diskus (Glaxo), devices marketed by Inhale Therapeutics, to name a few, use
breath-actuation
of a mixed powder (U.S. Pat. No. 4,668,218 Astra, EP 237507 Astra, WO 97/25086
Glaxo, WO
94/08552 Dura, U.S. Pat. No. 5,458,135 Inhale, WO 94/06498 Fisons, entirely
incorporated
herein by reference). Nebulizers like the Ultravent nebulizer (Mallinckrodt),
and the Acorn
II nebulizer (Marquest Medical Products) (U.S. Pat. No. 5,404,871 Aradigm, WO
97/22376),
the above references entirely incorporated herein by reference, produce
aerosols from solutions,
while metered dose inhalers, dry powder inhalers, etc. generate small particle
aerosols. Such
examples of commercially available inhalation devices are non-limiting
examples are not
intended to be limiting in scope.
[00231] In certain embodiments, a spray comprising a bispecific binding
molecule as
described herein can be produced by forcing a suspension or solution of at
least one bispecific
binding molecule as described herein through a nozzle under pressure. The
nozzle size and
configuration, the applied pressure, and the liquid feed rate can be chosen to
achieve the desired
output and particle size. An electrospray can be produced, for example, by an
electric field in
connection with a capillary or nozzle feed. Advantageously, particles of a
composition
comprising at least one bispecific binding molecule described herein delivered
by a sprayer have
a particle size less than about 10 um, preferably in the range of about 1 um
to about 5 um, and
most preferably about 2 um to about 3 um.
[00232] Formulations of a composition comprising at least one bispecific
binding molecule
described herein suitable for use with a sprayer typically include the at
least one bispecific
binding molecule in an aqueous solution at a concentration of about 0.1 mg to
about 100 mg per
ml of solution or mg/gm, or any range or value therein, e.g., but not limited
to, 0.1, 0.2, 0.3, 0.4,
0.5, 0.6, 0.7, 0.8, 0.9, 1, 2, 3, 4, 5, 6, 7, 8,9, 10, 11, 12, 13, 14, 15, 16,
17, 18, 19, 20, 21, 22, 23,
24, 25, 26, 27, 28, 29, 30, 40, 45, 50, 60, 70, 80, 90 or 100 mg/ml or mg/gm.
The formulation
can include agents such as an excipient, a buffer, an isotonicity agent, a
preservative, a
surfactant, and, preferably, zinc. The formulation can also include an
excipient or agent for
stabilization of the bispecific binding molecule composition, such as a
buffer, a reducing agent, a
bulk protein, or a carbohydrate. Bulk proteins useful in formulating such a
composition include
albumin, protamine, or the like. Typical carbohydrates useful in formulating
antibody
-82-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
composition proteins include sucrose, mannitol, lactose, trehalose, glucose,
or the like. The
composition can also include a surfactant, which can reduce or prevent surface-
induced
aggregation of the composition caused by atomization of the solution in
forming an aerosol.
Various conventional surfactants can be employed, such as polyoxyethylene
fatty acid esters and
alcohols, and polyoxy ethylene sorbitol fatty acid esters. Amounts will
generally range between
0.001 and 14% by weight of the formulation. Preferred surfactants are
polyoxyethylene sorbitan
monooleate, polysorbate 80, polysorbate 20, or the like.
[00233] In certain embodiments, the composition is administered via a
nebulizer, such as jet
nebulizer or an ultrasonic nebulizer. Typically, in a jet nebulizer, a
compressed air source is
used to create a high-velocity air jet through an orifice. As the gas expands
beyond the nozzle, a
low-pressure region is created, which draws a solution of antibody composition
protein through a
capillary tube connected to a liquid reservoir. The liquid stream from the
capillary tube is
sheared into unstable filaments and droplets as it exits the tube, creating
the aerosol. A range of
configurations, flow rates, and baffle types can be employed to achieve the
desired performance
characteristics from a given jet nebulizer. In an ultrasonic nebulizer, high-
frequency electrical
energy is used to create vibrational, mechanical energy, typically employing a
piezoelectric
transducer. This energy is transmitted to the formulation of antibody
composition protein either
directly or through a coupling fluid, creating an aerosol including the
antibody composition
protein. Advantageously, particles of antibody composition protein delivered
by a nebulizer have
a particle size less than about 10 um, preferably in the range of about 1 um
to about 5 um, and
most preferably about 2 um to about 3 um.
[00234] In certain embodiments, the composition is administered via a metered
dose inhaler
(MDI), wherein a propellant, at least one bispecific binding molecule
described herein, and any
excipients or other additives are contained in a canister as a mixture
including a liquefied
compressed gas. Actuation of the metering valve releases die mixture as an
aerosol, preferably
containing particles in the size range of less than about 10 um, preferably
about 1 um to about 5
um, and most preferably about 2 um to about 3 um. The desired aerosol particle
size can be
obtained by employing a formulation of antibody composition protein produced
by various
methods known to those of skill in the art, including jet-milling, spray
drying, critical point
condensation, or the like. Preferred metered dose inhalers include those
manufactured by 3M or
Glaxo and employing a hydrofluorocarbon propellant.
-83-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
[00235] Formulations of a bispecific binding molecule described herein for use
with a
metered-dose inhaler device will generally include a finely divided powder
containing at least
one Anti-IL-6 antibody as a suspension in a non-aqueous medium, for example,
suspended in a
propellant with the aid of a surfactant. The propellant can be any
conventional material
employed for this purpose, such as chlorofluorocarbon, a
hydrochlorofluorocarbon, a
hydrofluorocarbon, or a hydrocarbon, including trichlorofluoromethane,
dichlorodifluoromethane, dichlorotetrafluoroethanol and 1,1,1,2-
tetrafluoroethane, HFA-134a
(hydrofluoroalkane-134a), HFA-227 (hydrofluoroalkane-227), or the like.
Preferably the
propellant is a hydrofluorocarbon. The surfactant can be chosen to stabilize
the at least one
bispecific binding molecule as a suspension in the propellant, to protect the
active agent against
chemical degradation, and the like. Suitable surfactants include sorbitan
trioleate, soya lecithin,
oleic acid, or the like. In some cases solution aerosols are preferred using
solvents such as
ethanol. Additional agents known in the art for formulation of a protein can
also be included in
the formulation.
5.5.3 ORAL FORMULATIONS
[00236] In certain embodiments, a composition provided herein is formulated
for oral
administration. In certain embodiments, for oral administration, compositions
and methods of
administering at least one bispecific binding molecule described herein rely
on the co-
administration of adjuvants such as, for example, resorcinols and nonionic
surfactants such as
polyoxyethylene oleyl ether and n-hexadecylpolyethylene ether, to artificially
increase the
permeability of the intestinal walls, as well as the co-administration of
enzymatic inhibitors such
as, for example, pancreatic trypsin inhibitors, diisopropylfluorophosphate
(DFF) and trasylol, to
inhibit enzymatic degradation. The active constituent compound of the solid-
type dosage form
for oral administration can be mixed with at least one additive, including
sucrose, lactose,
cellulose, mannitol, trehalose, raffinose, maltitol, dextran, starches, agar,
arginates, chitins,
chitosans, pectins, gum tragacanth, gum arabic, gelatin, collagen, casein,
albumin, synthetic or
semisynthetic polymer, and glyceride. These dosage forms can also contain
other type(s) of
additives, such as, for example, inactive diluting agent, lubricant such as
magnesium stearate,
paraben, preserving agent such as sorbic acid, ascorbic acid, alpha.-
tocopherol, antioxidant such
as cysteine, disintegrator, binder, thickener, buffering agent, sweetening
agent, flavoring agent,
perfuming agent, etc.
-84-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
[00237] In certain embodiments, tablets and pills for oral administration can
be further
processed into enteric-coated preparations. In certain embodiments, liquid
preparations for oral
administration include, for example, emulsion, syrup, elixir, suspension and
solution
preparations allowable for medical use. These preparations can contain
inactive diluting agents
ordinarily used in said field, for example, water. Liposome preparations can
be utilized for oral
administration preparations, for example, as described for insulin and heparin
(U.S. Pat. No.
4,239,754). Additionally, microspheres of artificial polymers of mixed amino
acids (proteinoids)
can be utilized to in oral administration of pharmaceuticals, for example, as
described in U.S.
Pat. No. 4,925,673. Furthermore, carrier compounds, such as those described in
U.S. Pat. No.
5,879,681 and U.S. Pat. No. 5,871,753, are used in oral administration of
biologically active
agents.
5.5.4 MUCOSAL FORMULATIONS
[00238] In certain embodiments, a composition provided herein is formulated
for absorption
through mucosal surfaces. In certain embodiments, for absorption through
mucosal surfaces,
compositions and methods of administering at least one bispecific binding
molecule described
herein include an emulsion comprising a plurality of submicron particles, a
mucoadhesive
macromolecule, a bioactive peptide, and an aqueous continuous phase, which
promotes
absorption through mucosal surfaces by achieving mucoadhesion of the emulsion
particles (U.S.
Pat. No. 5,514,670). Mucous surfaces suitable for application of the emulsions
provided herein
can include, for example, corneal, conjunctival, buccal, sublingual, nasal,
vaginal, pulmonary,
stomachic, intestinal, and rectal routes of administration. Formulations for
vaginal or rectal
administration, for example, suppositories, can contain as excipients, for
example,
polyalkyleneglycols, vaseline, cocoa butter, and the like. Formulations for
intranasal
administration can be solid and contain as excipients, for example, lactose or
can be aqueous or
oily solutions of nasal drops. For buccal administration excipients include,
for example, sugars,
calcium stearate, magnesium stearate, pregelinatined starch, and the like
(U.S. Pat. No.
5,849,695).
5.5.5 TRANSDER1VIAL FORMULATIONS
[00239] In certain embodiments, a composition provided herein is formulated
for transdermal
administration. In certain embodiments, for transdermal administration, the
composition
-85-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
comprises at least one bispecific binding molecule described herein
encapsulated in a delivery
device such as, for example, a liposome or polymeric nanoparticles,
microparticle, microcapsule,
or microspheres (referred to collectively as microparticles unless otherwise
stated). A number of
suitable devices are known for transdermal administration, including
microparticles made of
synthetic polymers such as polyhydroxy acids such as polylactic acid,
polyglycolic acid and
copolymers thereof, polyorthoesters, polyanhydrides, and polyphosphazenes, and
natural
polymers such as collagen, polyamino acids, albumin and other proteins,
alginate and other
polysaccharides, and combinations thereof (U.S. Pat. No. 5,814,599).
5.5.6 KITS
[00240] Provided herein are kits comprising one or more bispecific binding
molecule as
described herein, or one or more composition as described herein. In certain
embodiments, the
kit comprises packaging material and at least one vial comprising a
composition comprising a
bispecific binding molecule or composition described herein. In certain
embodiments, the vial
comprises a solution of at least one bispecific binding molecule or
composition as described
herein with the prescribed buffers and/or preservatives, optionally in an
aqueous diluents. In
certain embodiments, the packaging material comprises a label that indicates
that such solution
can be held over a period of 1, 2, 3, 4, 5, 6, 9, 12, 18, 20, 24, 30, 36, 40,
48, 54, 60, 66, 72 hours
or greater. In certain embodiments, the kit comprises two vials. In certain
embodiments, the
first vial comprises at least one lyophilized bispecific binding molecule or
composition as
described herein and the second vial comprises aqueous diluents of prescribed
buffer or
preservative. In certain embodiments, the packaging material comprises a label
that instructs a
subject to reconstitute the at least one lyophilized bispecific binding
molecule in the aqueous
diluents to form a solution that can be held over a period of twenty-four
hours or greater. In
certain embodiments, the packaging material comprises a label that indicates
that such solution
can be held over a period of 1, 2, 3, 4, 5, 6, 9, 12, 18, 20, 24, 30, 36, 40,
48, 54, 60, 66, 72 hours
or greater.
[00241] In certain embodiments, the compositions provided herein can be
provided to a
subject as solutions or as dual vials comprising a vial of lyophilized at
least one bispecific
binding molecule or composition that is reconstituted with a second vial
containing water, a
preservative and/or excipients, preferably a phosphate buffer and/or saline
and a chosen salt, in
an aqueous diluent. Either a single solution vial or dual vial requiring
reconstitution can be
-86-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
reused multiple times and can suffice for a single or multiple cycles of
subject treatment and thus
can provide a more convenient treatment regimen than currently available.
[00242] In certain embodiments, a kit comprising a bispecific binding molecule
or
composition described herein is useful for administration over a period of
immediately to
twenty-four hours or greater. Accordingly, the kit offers significant
advantages to the patient. In
certain embodiments, a kit comprising a bispecific binding molecule or
composition described
herein can optionally be safely stored at temperatures of from about 2 C to
about 40 C and
retain the biologically activity of the protein for extended periods of time,
thus, allowing a
package label indicating that the solution can be held and/or used over a
period of 6, 12, 18, 24,
36, 48, 72, or 96 hours or greater. In certain embodiments, the kit comprises
a
[00243] If preserved diluent is used, such label can include use up to 1-12
months, one-half,
one and a half, and/or two years.
[00244] The kits can be provided indirectly to a subject, such as a subject
as described in
Section 5.7, by providing to pharmacies, clinics, or other such institutions
and facilities, solutions
or dual vials comprising a vial of lyophilized at least one bispecific binding
molecule or
composition that is reconstituted with a second vial containing the aqueous
diluent. The solution
in this case can be up to one liter or even larger in size, providing a large
reservoir from which
smaller portions of the at least one antibody solution can be retrieved one or
multiple times for
transfer into smaller vials and provided by the pharmacy or clinic to their
customers and/or
patients.
[00245] Recognized devices comprising these single vial systems include those
pen-injector
devices for delivery of a solution such as BD Pens, BD Autojectorg, Humajectg,
e.g., as made
or developed by Becton Dickensen (Franklin Lakes, N.J.,), Disetronic
(Burgdorf, Switzerland;
Bioject, Portland, Oreg.; National Medical Products, Weston Medical
(Peterborough, UK),
Medi-Ject Corp (Minneapolis, Minn.). Recognized devices comprising a dual vial
system
include those pen-injector systems for reconstituting a lyophilized drug in a
cartridge for delivery
of the reconstituted solution such as the HumatroPeng.
[00246] In certain embodiments, the kits comprise packaging material. In
certain
embodiments, the packaging material provides, in addition to the information
required by a
regulatory agencies, the conditions under which the product can be used. In
certain
embodiments, the packaging material provides instructions to the subject to
reconstitute the at
-87-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
least one bispecific binding molecule in the aqueous diluent to form a
solution and to use the
solution over a period of 2-24 hours or greater for the two vial, wet/dry,
product. For the single
vial, solution product, the label indicates that such solution can be used
over a period of 2-24
hours or greater. In a preferred embodiment, the kit is useful for human
pharmaceutical product
use. In certain embodiments, the kit is useful for veterinarian pharmaceutical
use. In a preferred
embodiment, the kit is useful for canine pharmaceutical product use. In a
preferred embodiment,
the kit is useful for intravenous administration. In another preferred
embodiment, the kit is
useful for intraperitoneal, intrathecal, intraventricular in the brain, or
intraparenchymal in the
brain administration.
5.6 USES AND METHODS
5.6.1 THERAPEUTIC USES
[00247] In certain embodiments, provided herein are methods for treating a
HER2-positive
cancer in a subject comprising administering to the subject in need thereof a
therapeutically
effective amount of a bispecific binding molecule as described in Section 5.1
or in Section 5.2, a
therapeutically effective amount of a cell, polynucleotide, or vector encoding
such a bispecific
binding molecule as described in Section 5.3, or a therapeutically effective
amount of a
pharmaceutical composition as described in Section 5.5, or a therapeutically
effective amount of
T cells bound to a bispecific binding molecule as described in Section 5.4. In
a specific
embodiment, the subject is a subject as described in Section 5.7. In a
specific embodiment, the
bispecific binding molecule is administered at a dose as described in Section
5.8. In a specific
embodiment, the bispecific binding molecule is administered according to the
methods as
described in Section 5.5. In a preferred embodiment, the bispecific binding
molecule is
administered intravenously. In another preferred embodiment, the bispecific
binding molecule is
administered intrathecally, intraventricularly in the brain,
intraparenchymally in the brain, or
intraperitoneally. In a specific embodiment, the bispecific binding molecule
is administered in
combination with one or more additional pharmaceutically active agents as
described in
Section 5.9.
[00248] In certain embodiments, provided herein are methods for treating a
HER2-positive
cancer in a subject comprising administering to the subject in need thereof a
pharmaceutical
composition as described in Section 5.1 or in Section 5.2. In a specific
embodiment, the
-88-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
pharmaceutical composition is a composition as described in Section 5.5. In a
specific
embodiment, the subject is a subject as described in Section 5.7. In a
specific embodiment, the
pharmaceutical composition is administered at a dose as described in Section
5.8. In a specific
embodiment, the pharmaceutical composition is administered according to the
methods as
described in Section 5.5. In a preferred embodiment, the pharmaceutical
composition is
administered intravenously. In another preferred embodiment, the bispecific
binding molecule is
administered intrathecally, intraventricularly in the brain,
intraparenchymally in the brain, or
intraperitoneally. In a specific embodiment, the pharmaceutical composition is
administered in
combination with one or more additional pharmaceutically active agents as
described in
Section 5.9.
[00249] For use of a bispecific binding molecule in a subject of a particular
species, a
bispecific binding molecule is used that binds to the HER2 and the CD3 of that
particular
species. For example, to treat a human, the bispecific binding molecule
comprises an
aglycosylated monoclonal antibody that is an immunoglobulin that binds to
human HER2,
comprising two identical heavy chains and two identical light chains, said
light chains being a
first light chain and a second light chain, wherein the first light chain is
fused to a first single
chain variable fragment (scFv), via a peptide linker, to create a first light
chain fusion
polypeptide, and wherein the second light chain is fused to a second scFv, via
a peptide linker, to
create a second light chain fusion polypeptide, wherein the first and second
scFv (i) are identical,
and (ii) bind to human CD3, and wherein the first and second light chain
fusion polypeptides are
identical. In another example, to treat a canine, the bispecific binding
molecule comprises an
aglycosylated monoclonal antibody that is an immunoglobulin that binds to
canine HER2,
comprising two identical heavy chains and two identical light chains, said
light chains being a
first light chain and a second light chain, wherein the first light chain is
fused to a first single
chain variable fragment (scFv), via a peptide linker, to create a first light
chain fusion
polypeptide, and wherein the second light chain is fused to a second scFv, via
a peptide linker, to
create a second light chain fusion polypeptide, wherein the first and second
scFv (i) are identical,
and (ii) bind to canine CD3, and wherein the first and second light chain
fusion polypeptides are
identical. Bispecific binding molecules that are cross-reactive with HER2
and/or CD3 of various
species can be used to treat subjects in those species. For example,
trastuzumab is expected to
bind both human and canine HER2 due to the relative conservation of the
epitope in HER2
-89-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
recognized by trastuzumab. See, also, for example, Singer et at., 2012, Mol
Immunol, 50: 200-
209.
[00250] In addition, for use of a bispecific binding molecule in a subject
of a particular
species, the bispecific binding molecule, preferably, the constant region of
the immunoglobulin
portion, is derived from that particular species. For example, to treat a
human, the bispecific
binding molecule can comprise an aglycosylated monoclonal antibody that is an
immunoglobulin, wherein the immunoglobulin comprises a human constant region.
In another
example, to treat a canine, the bispecific binding molecule can comprise an
aglycosylated
monoclonal antibody that is an immunoglobulin, wherein the immunoglobulin
comprises a
canine constant region. In a specific embodiment, when treating a human, the
immunoglobulin
is humanized. In a specific embodiment, the subject is a human. In a specific
embodiment, the
subject is a canine.
[00251] In a specific embodiment, the HER2-positive cancer is breast cancer,
gastric cancer,
an osteosarcoma, desmoplastic small round cell cancer, squamous cell carcinoma
of head and
neck cancer, ovarian cancer, prostate cancer, pancreatic cancer, glioblastoma
multiforme, gastric
junction adenocarcinoma, gastroesophageal junction adenocarcinoma, cervical
cancer, salivary
gland cancer, soft tissue sarcoma, leukemia, melanoma, Ewing's sarcoma,
rhabdomyosarcoma,
neuroblastoma, or any other neoplastic tissue that expresses the HER2
receptor.
[00252] In a specific embodiment, the HER2-positive cancer is resistant to
treatment with
trastuzumab, cetuximab, lapatinib, erlotinib, or any other small molecule or
antibody that targets
the HER family of receptors. In a specific embodiment, the tumor that is
resistant to treatment
with trastuzumab, cetuximab, lapatinib, erlotinib, or any other small molecule
or antibody that
targets the HER family of receptors is responsive to treatment with a
bispecific binding molecule
of the invention. In a specific embodiment, the HER2-positive cancer is
resistant to treatment
with necitumumab, pantitumumab, pertuzumab, or ado-trastuzumab emtansine. In a
specific
embodiment, the HER2-positive cancer that is resistant to treatment with
necitumumab,
pantitumumab, pertuzumab, or ado-trastuzumab emtansine is responsive to
treatment with a
bispecific binding molecule of the invention.
[00253] In a specific embodiment, the HER2-positive cancer is a cancer that
expresses
programmed death-ligand 1 ("PDL 1" or "PDL- 1") (i.e., a HER2-positive, PDL 1-
positive
cancer). Thus, in a specific embodiment, provided herein is a method of
treating a HER2-
-90-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
positive, PDL1-positive cancer in a subject in need thereof, comprising
administering to the
subject a therapeutically effective amount of a bispecific binding molecule as
described in
Section 5.1 or in Section 5.2, a therapeutically effective amount of a cell,
polynucleotide, or
vector encoding such a bispecific binding molecule as described in Section
5.3, or a
therapeutically effective amount of a pharmaceutical composition as described
in Section 5.5, or
a therapeutically effective amount of T cells bound to a bispecific binding
molecule as described
in Section 5.4. In a specific embodiment, the HER2-positive, PDL1-positive
cancer
overexpresses PDL1. In a specific embodiment, the HER2-positive, PDL1-positive
cancer
overexpresses PDL1 in cancerous cells relative to expression of PDL1 in
analogous
noncancerous cells of the same tissue type as the HER2-positive, PDL1-positive
cancer. The
noncancerous cells are analogous to the cancerous cells by virtue of the fact
that they, for
example, are from the same tissue or organ type or are otherwise suitable for
comparison of
PDL1 expression. The level of PDL1 expression in analogous noncancerous cells
can be a
known, standard level for a population or for particular individual(s) or for
the subject having
cancer, or can be newly measured. The overexpression can be shown, for
example, by detecting
increased PDL1 expression in a test specimen comprising cancerous cells
relative to expression
in a control specimen comprising analogous noncancerous cells. In contrast to
the test specimen,
the control specimen does not contain a significant amount of cancerous cells.
In a specific
embodiment, a HER2-positive, PDL1-positive cancer is deemed to overexpress
PDL1 when the
test specimen expresses a detectable level of PDL1 above background (i.e.,
experimental noise),
preferably as measured by immunohistochemistry ("IHC") since most normal
tissue should be
PDL1-negative. In a specific embodiment, the detectable level of PDL1 above
background is
1% to 5%, or is at least 1%, at least 2%, at least 3%, at least 4%, or at
least 5% above
background. In a specific embodiment in which the HER2-positive, PDL1-positive
cancer is a
melanoma, in a particular embodiment, the melanoma is deemed to overexpress
PDL1 when the
test specimen expresses a detectable level of PDL1 that is at least 5% above
background. In a
specific embodiment in which the HER2-positive, PDL1-positive cancer is a non-
small cell lung
carcinoma, in a particular embodiment, the non-small cell lung carcinoma is
deemed to
overexpress PDL1 if the test specimen expresses a detectable level of PDL1
that is at least 5%
above background. In a specific embodiment where binding to an anti-PDL1
antibody is used to
measure the level of PDL1 expression in the test specimen and the control
specimen, in a
-91-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
particular embodiment, the background level is measured by measuring
nonspecific signal, for
example, arising from binding to an antibody that recognizes an antigen known
not to be
expressed by the test or control specimen, e.g., an anti-IgG antibody. In a
specific embodiment,
PDL1 expression is measured by measuring PDL1 protein levels. In a specific
embodiment,
PDL1 expression is measured by measuring PDL1 nucleic acid levels (e.g., cDNA
or RNA
encoding PDL1). In a specific embodiment, PDL1 protein level is measured
according to any
assay known in the art, such as, e.g., IHC, western blot, enzyme-linked
immunosorbent assay, or
fluorescence-activated cell sorting. In a specific embodiment, PDL1 nucleic
acid level is
measured according to any assay known in the art, such as, e.g., in situ
hybridization ("ISH"),
southern blot, northern blot, quantitative reverse transcriptase polymerase
chain reaction, or deep
sequencing. The test specimen comprises cancer cells from the subject having
cancer, and may
be in the form of various biological specimens known in the art, e.g., from a
biopsy or surgical
resection. In a specific embodiment, a test specimen comprises cancerous cells
from a primary
tumor from the subject having cancer. In a specific embodiment, the test
specimen comprises
cancerous cells from a metastatic tumor from the subject having cancer. In a
specific
embodiment, the control specimen comprising analogous noncancerous cells
(analogous to the
cancerous cells in the test specimen) is a specimen obtained or derived from
the subject who has
cancer. Alternatively, a control specimen may be a specimen obtained or
derived from a healthy
subject or a subject who does not have cancer. In a specific embodiment, the
control specimen
does not comprise cancerous cells. In a specific embodiment, the test specimen
and control
specimen are from the same subject. In a specific embodiment, the test
specimen and the control
specimen are from different subjects. In a specific embodiment, the test
specimen contains
cancerous cells from breast tissue and the control specimen contains
noncancerous cells from
breast tissue.
[00254] Nonlimiting examples of HER2-positive cancers that express PDL1 and
thus can be
treated according to the methods described herein include breast cancer,
gastric cancer, an
osteosarcoma, desmoplastic small round cell cancer, ovarian cancer, prostate
cancer, pancreatic
cancer, glioblastoma multiforme, gastric junction adenocarcinoma,
gastroesophageal junction
adenocarcinoma, cervical cancer, salivary gland cancer, soft tissue sarcoma,
leukemia,
melanoma, Ewing's sarcoma, rhabdomyosarcoma, or neuroblastoma. In a specific
embodiment,
the HER2-positive, PDL1-positive cancer is not a head and neck cancer.
-92-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
[00255] In a specific embodiment, a HER2-positive, PDL1-positive cancer
treated according
to the methods described herein is resistant to PDL1 blockade with an anti-
PDL1 therapy. In a
specific embodiment, the HER2-positive, PDL1-positive cancer is resistant to
programmed cell
death 1 ("PD1" or "PD-1") blockade with an anti-PD1 therapy. In a specific
embodiment, the
HER2-positive, PDL1-positive cancer is resistant to (i) PDL1 blockade with an
anti-PDL1
therapy, and (ii) PD1 blockade with an anti-PD1 therapy.
[00256] In a specific embodiment, PDL1 blockade refers to (i) inhibition of
PDL1-dependent
PD1 activation, or (ii) blocking of PDL1 binding to PD1. In a specific
embodiment, the
inhibition or blocking is partial. In another specific embodiment, the
inhibition or blocking is
complete. In a specific embodiment, PDL1 blockade refers to least 5%, 10%,
15%, 20%, 25%,
30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 98%, or
99%
inhibition of PDL1-dependent PD1 activation as assessed by any method known to
one of skill in
the art, such as, e.g., a phosphorylation assay, as compared to PDL1-dependent
PD1 activation in
the presence of a negative control therapy (e.g., an anti-IgG antibody). For
example, in a
specific embodiment, PDL1 blockade that is inhibition of activation is
assessed by (a) contacting
a PDL1-expressing cell and a PD 1-expressing activated T cell with an anti-
PDL1 therapy (e.g.,
an anti-PDL1 antibody) or a negative control therapy (e.g., an anti-IgG
antibody), and (b)
measuring the phosphorylation of PD1 or dephosphorylation of a downstream
signaling
molecule, such as, e.g., Lck or Zap-70, as assessed by, for e.g., ELISA or
western blot, in the
presence of the anti-PDL1 therapy as compared to the phosphorylation of PD1 or
dephosphorylation of a downstream signaling molecule, such as, e.g., Lck or
Zap-70, as assessed
by, for e.g., ELISA or western blot, in the presence of the negative control
therapy. In a specific
embodiment, PDL1 blockade that is blocking of PDL1 binding to PD1 is assessed
by (a)
contacting a PDL1-expressing cell and a PD 1-expressing activated T cell with
an anti-PDL1
therapy (e.g., an anti-PDL1 antibody) or a negative control therapy (e.g., an
anti-IgG antibody),
and (b) measuring the interaction between PDL1 and PD1 by, for example, co-
localization (as
assessed by, e.g., immunohistochemistry) or co-immunoprecipitation (as
assessed by, e.g.,
western blot) of PDL1 and PD1, in the presence of the anti-PDL1 therapy as
compared to the
interaction between PDL1 and PD1 by, for example, co-localization (as assessed
by, e.g.,
immunohistochemistry) or co-immunoprecipitation (as assessed by, e.g., western
blot), in the
presence of the negative control therapy.
-93-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
[00257] In a specific embodiment, PD1 blockade refers to (i) inhibition of
ligand-dependent
PD1 activation, or (ii) blocking of ligand binding to PD1. In a specific
embodiment, the
inhibition or blocking is partial. In another specific embodiment, the
inhibition or blocking is
complete. In a specific embodiment, PD1 blockade refers to least 5%, 10%, 15%,
20%, 25%,
30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 98%, or
99%
inhibition of ligand-dependent PD1 activation as assessed by any method known
to one of skill
in the art, such as, e.g., a phosphorylation assay, as compared to ligand-
dependent PD1 activation
in the presence of a negative control therapy (e.g., an anti-IgG antibody).
For example, in a
specific embodiment, PD1 blockade that is inhibition of activation is assessed
by (a) contacting a
PD1 ligand-expressing cell and a PD1-expressing activated T cell with an anti-
PD1 therapy (e.g.,
an anti-PD1 antibody) or a negative control therapy (e.g., an anti-IgG
antibody), and (b)
measuring the phosphorylation of PD1 or dephosphorylation of a downstream
signaling
molecule, such as, e.g., Lck or Zap-70, as assessed by, for e.g., ELISA or
western blot, in the
presence of the anti-PD1 therapy as compared to the phosphorylation of PD1 or
dephosphorylation of a downstream signaling molecule, such as, e.g., Lck or
Zap-70, as assessed
by, for e.g., ELISA or western blot, in the presence of the negative control
therapy. In a specific
embodiment, PD1 blockade that is blocking of ligand binding to PD1 is assessed
by (a)
contacting a ligand-expressing cell and a PD1-expressing activated T cell with
an anti-PD1
therapy (e.g., an anti-PD1 antibody) or a negative control therapy (e.g., an
anti-IgG antibody),
and (b) measuring the interaction between ligand and PD1 by, for example, co-
localization (as
assessed by, e.g., immunohistochemistry) or co-immunoprecipitation (as
assessed by, e.g.,
western blot) of ligand and PD1, in the presence of the anti-PD1 therapy as
compared to the
interaction between ligand and PD1 by, for example, co-localization (as
assessed by, e.g.,
immunohistochemistry) or co-immunoprecipitation (as assessed by, e.g., western
blot), in the
presence of the negative control therapy.
[00258] In a specific embodiment, an anti-PDL1 therapy is a PDL1-targeted
therapy that is
effective in the treatment of one or more cancers expressing PDLl. In a
specific embodiment,
the anti-PDL1 therapy comprises an antibody or antigen-binding fragment
thereof (e.g., a Fab
fragment, a F(ab')2 fragment, or a disulfide-linked Fv) or antigen-binding
derivative thereof
(e.g., a bispecific antibody, an scFv, an intrabody, or a camelized antibody),
a polypeptide, a
RNAi-inducing nucleic acid (e.g., an antisense oligonucleotide, a small
interfering RNA, a
-94-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
microRNA, or a short hairpin RNA), or a small molecule that targets PDLl.
Nonlimiting
examples of an anti-PDL1 therapy include mpd13280A (see, e.g., Herbst et al.,
J Clin Oncol.
2013;31(suppl):abstr 3000), durvalumab (e.g., for bladder cancer) (also
referred to as "medi-
4736"; see, e.g., Lutzky et al., J Clin Oncol. 2014;32(suppl 5S):abstr 3001),
avelumab (e.g., for
Merkel cell carcinoma) (also referred to as "MSB0010718C"; see, e.g., Heery et
al. J Clin Oncol.
2014;32(suppl 5S):abstr 3064), and bms-936559 (see, e.g., Brahmer et al. N.
Engl. J. Med.
2012;366, 2455-2465), and atezolizumab (see, e.g., McDermott et al., J Clin
Oncol. 2016;
34(8):833-842). In a preferred embodiment, the anti-PDL1 therapy is an anti-
PDL1 antibody. In
a preferred embodiment, the anti-PDL1 antibody is atezolizumab. In a specific
embodiment, the
anti-PDL1 therapy is a therapy approved by the U.S. Food and Drug
Administration ("FDA") for
treatment of one or more cancers. A nonlimiting example of an anti-PDL1
therapy approved by
the U.S. Food and Drug Administration for treatment of cancer is atezolizumab.
In a specific
embodiment, the anti-PDL1 therapy is a PDL1-targeted therapy approved by the
European
Medicines Agency ("EMA") for treatment of one or more cancers. A nonlimiting
example of an
anti-PDL1 therapy approved by the EMA for treatment of a PDL1-expressing
cancer is
atezolizumab.
[00259] In a specific embodiment, an anti-PD1 therapy is a PD1-targeted
therapy that is
effective in the treatment of one or more cancers expressing PDLl. In a
specific embodiment,
the anti-PD1 therapy comprises an antibody or antigen-binding fragment thereof
(e.g., a Fab
fragment, a F(ab')2 fragment, or a disulfide-linked Fv) or antigen-binding
derivative thereof
(e.g., a bispecific antibody, an scFv, an intrabody, or a camelized antibody),
a polypeptide, a
RNAi-inducing nucleic acid (e.g., an antisense oligonucleotide, a small
interfering RNA, a
microRNA, or a short hairpin RNA), or a small molecule that targets PD1.
Nonlimiting
examples of an anti-PD1 therapy include nivolumab (see, e.g., Topalian et al.,
N Engl J Med.
2012;366:2443-54), pidilizumab (see, e.g., Atkins et al., J Clin Oncol.
2014;32(suppl 55):abstr
9001), AMP-224 (see, e.g., Infante et al., J Clin Oncol. 2013;31(suppl):abstr
3044), MEDI0680
(also referred to as "AMP-514"; see, e.g., Hamid et al., Ann Oncol.
2016;27(supp1 6):1050PD),
and pembrolizumab (see, e.g., Hamid et al., N Engl J Med. 2013;369:134-44). In
a preferred
embodiment, the anti-PD1 therapy is an anti-PD1 antibody. In a preferred
embodiment, the anti-
PD1 antibody is pembrolizumab. In a specific embodiment, the anti-PD1 therapy
is a therapy
approved by the U.S. FDA for treatment of one or more cancers. Nonlimiting
examples of an
-95-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
anti-PD1 therapy approved by the U.S. FDA for treatment of cancer include
pembrolizumab and
nivolumab. In a specific embodiment, the anti-PD1 therapy is a therapy
approved by the EMA
for treatment of one or more cancers. Nonlimiting examples of an anti-PD1
therapy approved by
the EMA for treatment of cancer include pembrolizumab and nivolumab.
[00260] In contrast to trastuzumab (which is indicated for treatment of HER2-
overexpressing
breast cancer, metastatic gastric cancer, and gastroesophageal junction
adenocarcinoma (see
Trastuzumab [Highlights of Prescribing Information], South San Francisco, CA:
Genentech, Inc.;
2014)), the bispecific binding molecules described herein are therapeutically
effective against
HER2-positive cancers that express low levels of HER2. See, e.g., the working
example of
Section 6.3, in particular, Figure 32, which demonstrates that tumor growth
was completely
suppressed in a gastric cancer patient-derived xenograft model with low HER2
expression when
treated with a bispecific binding molecule described herein; in contrast,
treatment of the gastric
cancer patient-derived xenograft model with trastuzumab did not suppress tumor
growth. Thus,
also provided herein is a method of treating a HER2-positive cancer in a
subject in need thereof,
comprising administering to the subject a therapeutically effective amount of
a bispecific binding
molecule as described in Section 5.1 or in Section 5.2, a therapeutically
effective amount of a
cell, polynucleotide, or vector encoding such a bispecific binding molecule as
described in
Section 5.3, or a therapeutically effective amount of a pharmaceutical
composition as described
in Section 5.5, or a therapeutically effective amount of T cells bound to a
bispecific binding
molecule as described in Section 5.4, wherein the cancer is not indicated for
treatment with
trastuzumab, and wherein the cancer is not a head and neck cancer. In a
specific embodiment,
the cancer is breast cancer. In a specific embodiment, the cancer is gastric
cancer. In a specific
embodiment, the cancer is gastroesophageal junction adenocarcinoma.
[00261] In a specific embodiment, the HER2-positive cancer is determined not
to be indicated
for treatment with trastuzumab according to applicable American Society of
Clinical
Oncology/College of American Pathologists ("ASCO/CAP") guideline
recommendations for
HER2 testing in cancer ("ASCO HER2 Testing Guidelines") (see, e.g., Wolff et
al., Journal of
Clinical Oncology, 2013, 31(31):3997-4013 and Bartley et al., Journal of
Clinical Oncology,
2016, 146(6):647-669). The applicable ASCO HER2 Testing Guideline will be
known to one of
skill in the art. In a specific embodiment, the applicable ASCO HER2 Testing
Guideline is the
current (i.e.., most recently published and updated) ASCO HER2 Testing
Guideline as of the
-96-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
date of using the ASCO HER2 Testing Guideline to determine that the cancer is
not indicated for
treatment with trastuzumab. In an alternative, preferred embodiment, the
applicable ASCO
HER2 Testing Guideline is the current (e.g., most recently published and
updated) ASCO/CAP
guideline recommendations for HER2 testing in breast cancer ("ASCO HER2 Breast
Cancer
Testing Guideline") (see, e.g., Wolff et al., Journal of Clinical Oncology,
2013, 31(31):3997-
4013) as of the date of determining that the cancer is not indicated for
treatment with
trastuzumab, regardless of the type of cancer that is determined not to be
indicated for treatment
with trastuzumab (e.g., the type of cancer may be breast cancer or any other
HER2-positive
cancer). In another embodiment, the applicable ASCO HER2 Testing Guideline is
the current
(e.g., most recently published and updated) ASCO HER2 Testing Guideline as of
the date of
determining that the cancer is not indicated for treatment with trastuzumab
and the applicable
ASCO HER2 Testing Guideline is for the same type of cancer (e.g., same tissue
type, for
example, both being breast cancers, or both being gastric cancers) as the
cancer that is
determined not to be indicated for treatment with trastuzumab.
[00262] In a specific embodiment, the HER2-positive cancer is determined not
to be indicated
for treatment with trastuzumab based on the following characterization of the
cancer (see, e.g.,
the 2013 ASCO HER2 Breast Cancer Testing Guideline (e.g., as set forth in
Wolff et al., Journal
of Clinical Oncology, 2013, 31(31):3997-4013)): (a) a first determination of a
level of HER2 in a
test specimen comprising cells of the cancer is reported as negative, or (b) a
first determination
of a level of HER2 in a test specimen comprising cells of the cancer is
reported as equivocal, and
a second determination of a level of HER2 in a test specimen comprising cells
of the cancer is
reported as equivocal or negative. In a specific embodiment, the HER2-positive
cancer is
determined not to be indicated for treatment with trastuzumab when a first
determination of a
level of HER2 in a test specimen comprising cells of the cancer is reported as
negative according
to the applicable ASCO HER2 Testing Guideline (e.g., the 2013 ASCO HER2 Breast
Cancer
Testing Guideline (e.g., as set forth in Wolff et al., Journal of Clinical
Oncology, 2013,
31(31):3997-4013)). In a specific embodiment, the HER2-positive cancer is
determined not to
be indicated for treatment with trastuzumab when a first determination of a
level of HER2 in a
test specimen comprising cells of the cancer is reported as equivocal
according to the applicable
ASCO HER2 Testing Guideline (e.g., the 2013 ASCO HER2 Breast Cancer Testing
Guideline
(e.g., as set forth in Wolff et al., Journal of Clinical Oncology, 2013,
31(31):3997-4013)) and a
-97-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
second determination of a level of HER2 in a test specimen comprising cells of
the cancer is
reported as equivocal or negative according to the applicable ASCO HER2
Testing Guideline
(e.g., the 2013 ASCO HER2 Breast Cancer Testing Guideline (e.g., as set forth
in Wolff et al.,
Journal of Clinical Oncology, 2013, 31(31):3997-4013)). The test specimen can
be from a
primary tumor or a metastatic tumor.
[00263] In a specific embodiment, the determination of the level of HER2 in
the test specimen
is reported as negative when the level of HER2 in the test specimen is
characterized as (see, e.g.,
the 2013 ASCO HER2 Breast Cancer Testing Guideline (e.g., as set forth in
Wolff et al., Journal
of Clinical Oncology, 2013, 31(31):3997-4013)): (i) (1) IHC 1+, wherein the
level of HER2 in
the test specimen is characterized as IHC 1+ when the test specimen exhibits
an incomplete
HER2 membrane staining that is faint/barely perceptible and within greater
than 10% of the
invasive tumor cells, wherein the staining is readily appreciated using a low-
power objective; (2)
IHC 0, wherein the level of HER2 in the test specimen is characterized as IHC
0 when the test
specimen exhibits no staining observed, wherein the lack of staining is
readily appreciated using
a low-power objective, or a HER2 membrane staining that is incomplete and is
faint/barely
perceptible and within less than or equal to 10% of the invasive tumor cells,
wherein the staining
is readily appreciated using a low-power objective; or (ii) ISH negative,
wherein the level of
HER2 in the test specimen is characterized as ISH negative when the test
specimen exhibits (1) a
single-probe average HER2 copy number of less than 4.0 signals per cell; or
(2) a dual-probe
HER2/CEP17 ratio of less than 2.0 with an average HER2 copy number of less
than 4.0 signals
per cell.
[00264] In a specific embodiment, the determination of the level of HER2 in
the test specimen
is reported as equivocal when the level of HER2 in the test specimen is
characterized as (see,
e.g., the 2013 ASCO HER2 Breast Cancer Testing Guideline (e.g., as set forth
in Wolff et al.,
Journal of Clinical Oncology, 2013, 31(31):3997-4013)): (i) IHC 2+, wherein
the level of HER2
in the test specimen is characterized as IHC 2+ when the test specimen
exhibits(1) a
circumferential HER2 membrane staining that is incomplete and/or weak/moderate
and within
greater than 10% of invasive tumor cells, wherein the staining is observed in
a homogenous and
contiguous population, and wherein the staining is readily appreciated using a
low-power
objective; or (2) a complete and circumferential HER2 membrane staining that
is intense and
within less than or equal to 10% of invasive tumor cells, wherein the staining
is readily
-98-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
appreciated using a low-power objective; or (ii) ISH equivocal, wherein the
level of HER2 in the
test specimen is characterized as ISH equivocal when the test specimen
exhibits which
comprises: (1) a single-probe ISH average HER2 copy number of greater than or
equal to 4.0 and
less than 6.0 signals/cell, wherein the copy number is determined by counting
at least 20 cells
within the area and is observed in a homogenous and contiguous population; or
(2) a dual-probe
HER2/CEP17 ratio of less than 2.0 with an average HER2 copy number of greater
than or equal
to 4.0 and less than 6.0 signals per cell, wherein the copy number is
determined by counting at
least 20 cells within the area and is observed in a homogenous and contiguous
population. In a
specific embodiment, when two determinations of the level of HER2 in a subject
are made to
determine that a cancer is not indicated for treatment with trastuzumab (e.g.,
a first determination
is reported as equivalent and a second determination is reported as equivalent
or negative), the
two determinations are either: (1) based on the same test specimen using
different assays; or (2)
based on different test specimens using the same assay. For example, if the
first determination is
based on a first test specimen using ISH, the second determination is based on
the first test
specimen using IHC. In an alternative example, if the first determination is
made based on a first
test specimen using ISH, the second determination is based on a second test
specimen using ISH.
[00265] In a specific embodiment, the level of HER2 in the test specimen is
determined
according to one or more assays approved by the U.S. Food and Drug
Administration ("FDA")
for determining the level of HER2. Nonlimiting examples of U.S. Food and Drug
Administration-approved assays for determining a level of HER2 include
HercepTestTm
(manufactured by DAKO), PATHWAY (manufactured by Ventana Medical Systems
Inc.),
InSite (manufactured by Biogenex Laboratories Inc.), Bond OracleTM
(manufactured by Leica
Biosystems), PathVysion (manufactured by Abbott Molecular Inc.), PharmDxTM
Kit
(manufactured by DAKO), SPoT-Light (manufactured by Life Technologies Inc.),
INFORM
HER2 dual IDS DNA probe cocktail (manufactured by Ventana Medical Systems
Inc.), and
PharmDxTM (manufactured by DAKO). In another specific embodiment, the level of
HER2 in
the test specimen is determined according to a laboratory-developed test
performed in a Clinical
Laboratory Improvement Amendments-certified laboratory.
[00266] Also provided herein is a method of treating a HER2-positive cancer in
a subject in
need thereof, comprising administering to the subject a therapeutically
effective amount of a
bispecific binding molecule as described in Section 5.1 or in Section 5.2, a
therapeutically
-99-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
effective amount of a cell, polynucleotide, or vector encoding such a
bispecific binding molecule
as described in Section 5.3, or a therapeutically effective amount of a
pharmaceutical
composition as described in Section 5.5, or a therapeutically effective amount
of T cells bound to
a bispecific binding molecule as described in Section 5.4, wherein the cancer
expresses a low
level of HER2, and wherein the cancer is not a head and neck cancer. In a
specific embodiment,
the cancer is breast cancer. In another specific embodiment, the cancer is
gastric cancer. In a
specific embodiment, the cancer is gastroesophageal junction adenocarcinoma.
[00267] In a preferred embodiment, the HER2-positive cancer is deemed to
express a low
level of HER2 when a level of HER2 in a test specimen comprising cells of the
cancer is
characterized as IHC 2+ or less (e.g., IHC 1+ or IHC 0) according to the
applicable ASCO HER2
Testing Guideline. In a specific embodiment, the HER2-positive cancer is
deemed to express a
low level of HER2 when a level of HER2 in a test specimen comprising cells of
the cancer is
characterized as IHC 2+ according to the applicable ASCO HER2 Testing
Guideline. In a
specific embodiment, the HER2-positive cancer is deemed to express a low level
of HER2 when
a level of HER2 in a test specimen comprising cells of the cancer is
characterized as IHC 1+
according to the applicable ASCO HER2 Testing Guideline. In a specific
embodiment, the
HER2-positive cancer is deemed to express a low level of HER2 when a level of
HER2 in a test
specimen comprising cells of the cancer is characterized as IHC 0 according to
the applicable
ASCO HER2 Testing Guideline. The applicable ASCO HER2 Testing Guideline will
be known
to one of skill in the art. In a specific embodiment, the applicable ASCO HER2
Testing
Guideline is the current (e.g., most recently published and updated) ASCO HER2
Testing
Guideline as of the date of characterizing the level of HER2 in the test
specimen comprising cells
of the cancer as IHC 2+ or less (e.g., IHC 1+ or IHC 0). In a preferred
embodiment, the
applicable ASCO HER2 Testing Guideline is the current (e.g., most recently
published and
updated) ASCO HER2 Breast Cancer Testing Guideline (see, e.g., Wolff et al.,
Journal of
Clinical Oncology, 2013, 31(31):3997-4013) as of the date of characterizing
the level of HER2
in the test specimen, regardless of the type of cancer of the test specimen
(e.g., the test specimen
may be of breast cancer or any other HER2-positive cancer). In another
embodiment, the
applicable ASCO HER2 Testing Guideline is for the same type of cancer (e.g.,
same tissue type,
for example, both being breast cancers, or both being gastric cancers) as the
cancer of the test
specimen. In another embodiment, the applicable ASCO HER2 Testing Guideline is
the current
-100-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
(e.g., most recently published and updated) ASCO HER2 Testing Guideline as of
the date of
characterizing the level of HER2 in the test specimen and the applicable ASCO
HER2 Testing
Guideline is for the same type of cancer (e.g., same tissue type, for example,
both being breast
cancers, or both being gastric cancers) as the test specimen. In a specific
embodiment, the level
of HER2 in the test specimen comprising cells of the cancer is characterized
as IHC 2+ when the
test specimen exhibits (see, e.g., Wolff et al., Journal of Clinical Oncology,
2013, 31(31):3997-
4013) (1) a circumferential HER2 membrane staining that is incomplete and/or
weak/moderate
and within greater than 10% of invasive tumor cells, wherein the staining is
observed in a
homogenous and contiguous population, and wherein the staining is readily
appreciated using a
low-power objective; or (2) a complete and circumferential HER2 membrane
staining that is
intense and within less than or equal to 10% of invasive tumor cells, wherein
the staining is
readily appreciated using a low-power objective. In a specific embodiment, the
level of HER2 in
a test specimen comprising cells of the cancer is characterized as IHC 1+ when
the test specimen
exhibits (see, e.g., Wolff et al., Journal of Clinical Oncology, 2013,
31(31):3997-4013) an
incomplete HER2 membrane staining that is faint/barely perceptible and within
greater than 10%
of the invasive tumor cells, wherein the staining is readily appreciated using
a low-power
objective. In a specific embodiment, the level of HER2 in the test specimen
comprising cells of
the cancer is characterized as IHC when the test specimen exhibits (see, e.g.,
Wolff et al., Journal
of Clinical Oncology, 2013, 31(31):3997-4013) no HER2 staining observed,
wherein the lack of
staining is readily appreciated using a low-power objective, or a HER2
membrane staining that is
incomplete and is faint/barely perceptible and within less than or equal to
10% of the invasive
tumor cells, wherein the staining is readily appreciated using a low-power
objective.
[00268] In another embodiment, the HER2-positive cancer is deemed to express a
low level of
HER2 when the cancer expresses a lower level of HER2 than the level of HER2
expressed by
cancers that are indicated for treatment with trastuzumab and are of the same
type (e.g., same
tissue type, for example, both being breast cancers, or both being gastric
cancers) as the HER2-
positive cancer. In a specific embodiment, the HER2 positive cancer is deemed
to express a low
level of HER2 when the HER2-positive cancer expresses a level of HER2 that is
at least 10%,
20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, or 100% lower than the level of HER2
expressed
by cancers that are indicated for treatment with trastuzumab and are of the
same type (e.g., same
tissue type, for example, both being breast cancers, or both being gastric
cancers) as the HER2-
-101-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
positive cancer. In a specific embodiment, HER2 expression is measured by
measuring HER2
protein levels. In a specific embodiment, HER2 expression is measured by
measuring HER2
nucleic acid levels (e.g, genomic DNA, cDNA, or RNA encoding HER2). In a
specific
embodiment, HER2 protein level is measured according to any assay known in the
art, such as,
e.g., IHC, western blot, enzyme-linked immunosorbent assay, or fluorescence-
activated cell
sorting. In a preferred embodiment, HER2 protein level is measured according
to IHC. In a
specific embodiment, HER2 nucleic acid level is measured according to any
assay known in the
art, such as, e.g., ISH, southern blot, northern blot, quantitative reverse
transcriptase polymerase
chain reaction, or deep sequencing. In a preferred embodiment, HER2 nucleic
acid level is
measured according to ISH.
[00269] In a specific embodiment, the level of HER2 in the test specimen is
determined
according to one or more assays approved by the U.S. Food and Drug
Administration ("FDA")
for determining the level of HER2. Nonlimiting examples of U.S. Food and Drug
Administration-approved assays for determining a level of HER2 include
HercepTestTm
(manufactured by DAKO), PATHWAY (manufactured by Ventana Medical Systems
Inc.),
InSite (manufactured by Biogenex Laboratories Inc.), Bond OracleTM
(manufactured by Leica
Biosystems), PathVysion (manufactured by Abbott Molecular Inc.), PharmDxTM
Kit
(manufactured by DAKO), SPoT-Light (manufactured by Life Technologies Inc.),
INFORM
HER2 dual IDS DNA probe cocktail (manufactured by Ventana Medical Systems
Inc.), and
PharmDxTM (manufactured by DAKO). In another specific embodiment, the level of
HER2 in
the test specimen is determined according to a laboratory-developed test
performed in a Clinical
Laboratory Improvement Amendments-certified laboratory.
[00270] In a specific embodiment, the HER2-positive cancer that expresses a
low level of
HER2 is a breast cancer, a gastric cancer, an osteosarcoma, desmoplastic small
round cell cancer,
an ovarian cancer, a prostate cancer, a pancreatic cancer, glioblastoma
multiforme, gastric
junction adenocarcinoma, gastroesophageal junction adenocarcinoma, a cervical
cancer, a
salivary gland cancer, a soft tissue sarcoma, a leukemia, a melanoma, a
Ewing's sarcoma, a
rhabdomyosarcoma, a brain tumor, or neuroblastoma. In a preferred embodiment,
the HER2-
positive cancer that expresses a low level of HER2 is breast cancer. In a
specific embodiment,
the HER2-positive cancer that expresses a low level of HER2 is gastric cancer.
In a specific
embodiment, the HER2-positive cancer that expresses a low level of HER2 is
ovarian cancer,
-102-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
pancreatic cancer, a desmoplastic small round cell tumor, an osteosarcoma, a
melanoma, a brain
tumor, a cervical cancer, a prostate cancer, or a salivary gland cancer. In a
specific embodiment,
the HER2-positive cancer that expresses a low level of HER2 is not a head and
neck cancer.
[00271] In a specific embodiment, the HER2-positive cancer that expresses a
low level of
HER2 is resistant to treatment with trastuzumab, cetuximab, lapatinib,
erlotinib, or any other
small molecule or antibody that targets the HER family of receptors, and is
responsive to
treatment with a bispecific binding molecule of the invention. In a specific
embodiment, the
HER2-positive cancer that expresses a low level of HER2 is resistant to
treatment with
necitumumab, pantitumumab, pertuzumab, or ado-trastuzumab emtansine, and is
responsive to
treatment with a bispecific binding molecule of the invention.
[00272] In a specific embodiment, a cancer is considered resistant to a
therapy (e.g., an anti-
PDL1 therapy, an anti-PD1 therapy, trastuzumab, cetuximab, necitumumab,
panitumumab,
pertuzumab, ado-trastuzumab emtansine, lapatinib, erlotinib, or any small
molecule that targets
the HER family of receptors) if it has no response, or has an incomplete
response (a response that
is less than a complete remission), or progresses, or relapses after the
therapy.
[00273] In specific embodiments, treatment can be to achieve beneficial or
desired clinical
results including, but not limited to, alleviation of a symptom, diminishment
of extent of a
disease, stabilizing (i.e., not worsening) of state of a disease, delay or
slowing of disease
progression, amelioration or palliation of the disease state, and remission
(whether partial or
total), whether detectable or undetectable. In a specific embodiment,
"treatment" can also be to
prolong survival as compared to expected survival if not receiving treatment.
5.6.2 DIAGNOSTIC USES
[00274] In certain embodiments, bispecific binding molecules described herein
can be used
for diagnostic purposes to detect, diagnose, or monitor a condition described
herein (e.g., a
condition involving HER2-positive cancer cells). In certain embodiments,
bispecific binding
molecules for use in diagnostic purposes are labeled as described in Section
5.2.
[00275] In certain embodiments, provided herein are methods for the detection
of a condition
described herein comprising (a) assaying the expression of HER2 in cells or a
tissue sample of a
subject using one or more bispecific binding molecules described herein; and
(b) comparing the
level of HER2 expression with a control level, for example, levels in normal
tissue samples (e.g.,
from a subject not having a condition described herein, or from the same
patient before onset of
-103-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
the condition), whereby an increase or decrease in the assayed level of HER2
expression
compared to the control level of HER2 expression is indicative of a condition
described herein.
[00276] Antibodies described herein can be used to assay HER2 levels in a
biological sample
using classical immunohistological methods as described herein or as known to
those of skill in
the art (e.g., see Jalkanen et al., 1985, J. Cell. Biol. 101:976-985; and
Jalkanen et al., 1987, J.
Cell . Biol. 105:3087-3096). Other antibody-based methods useful for detecting
protein gene
expression include immunoassays, such as the enzyme linked immunosorbent assay
(ELISA) and
the radioimmunoassay (MA). Suitable antibody assay labels are known in the art
and include
enzyme labels, such as, glucose oxidase; radioisotopes, such as iodine (1251,
121,,1) ,
carbon (14C),
sulfur (35S), tritium (3H), indium (121In), and technetium (99Tc); luminescent
labels, such as
luminol; and fluorescent labels, such as fluorescein and rhodamine, and
biotin.
[00277] In certain embodiments, monitoring of a condition described herein
(e.g., a HER2-
positive cancer), is carried out by repeating the method for diagnosing for a
period of time after
initial diagnosis.
[00278] Presence of the labeled molecule can be detected in the subject using
methods known
in the art for in vivo scanning. Skilled artisans will be able to determine
the appropriate method
for detecting a particular label. Methods and devices that may be used in the
diagnostic methods
of the invention include, but are not limited to, computed tomography (CT),
whole body scan
such as position emission tomography (PET), magnetic resonance imaging (MM),
and
sonography.
5.7 PATIENT POPULATION
[00279] A subject treated in accordance with the methods provided herein can
be any
mammal, such as a rodent, a cat, a canine, a horse, a cow, a pig, a monkey, a
primate, or a
human, etc. In a preferred embodiment, the subject is a human. In another
preferred
embodiment, the subject is a canine.
[00280] In certain embodiments, a subject treated in accordance with the
methods provided
herein has been diagnosed with a HER2-positive cancer, including but not
limited to, breast
cancer, gastric cancer, an osteosarcoma, desmoplastic small round cell cancer,
squamous cell
carcinoma of head and neck cancer, ovarian cancer, prostate cancer, pancreatic
cancer,
glioblastoma multiforme, gastric junction adenocarcinoma, gastroesophageal
junction
adenocarcinoma, cervical cancer, salivary gland cancer, soft tissue sarcoma,
leukemia,
-104-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
melanoma, Ewing's sarcoma, rhabdomyosarcoma, neuroblastoma, or any other
neoplastic tissue
that expresses the HER2 receptor.
[00281] In a specific embodiment, a subject treated in accordance with the
methods provided
herein has not been diagnosed with HER2-positive squamous cell carcinoma of
head and neck
cancer.
[00282] In certain embodiments, the subject is resistant to treatment with
trastuzumab,
cetuximab, lapatinib, erlotinib, or any other small molecule or antibody that
targets the HER
family of receptors. In a specific embodiment, the tumor that is resistant to
treatment with
trastuzumab, cetuximab, lapatinib, erlotinib, or any other small molecule or
antibody that targets
the HER family of receptors is responsive to treatment with a bispecific
binding molecule to the
invention.
[00283] In certain embodiments, a subject treated in accordance with the
methods provided
herein has a HER2-positive cancer that is resistant to treatment with
trastuzumab, cetuximab,
lapatinib, erlotinib, or any other small molecule or antibody that targets the
HER family of
receptors. In certain embodiments, a subject treated in accordance with the
methods provided
herein has a HER2-positive cancer that is responsive to treatment with a
bispecific binding
molecule to the invention.
[00284] In certain embodiments, the subject treated in accordance with the
methods provided
herein has previously received one or more chemotherapy regimens for
metastatic disease, e.g.,
brain or peritoneal metastases. In certain embodiments, the subject has not
previously received
treatment for metastatic disease.
5.8 DOSES AND REGIMENS
[00285] In certain embodiments, the dose of a bispecific binding molecule as
described in
Section 5.1 administered to a subject according to the methods provided herein
is a dose
determined by the needs of the subject. In certain embodiments, the dose is
determined by a
physician according to the needs of the subject.
[00286] In a specific embodiment, the dose of a bispecific binding molecule
provided herein
is less than the dose of trastuzumab. See, for example, Trastuzumab
[Highlights of Prescribing
Information]. South San Francisco, CA: Genentech, Inc.; 2014. In a specific
embodiment, the
dose of a bispecific binding molecule provided herein is approximately between
20 and 40 fold
less than an FDA-approved dose of trastuzumab.
-105-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
[00287] In certain embodiments, the dose of a bispecific binding molecule as
described in
Section 5.1 administered to a subject according to the methods provided herein
is between 0.01
mg/kg and 0. 025 mg/kg, is between 0.025 mg/kg and 0.05 mg/kg, is between 0.05
mg/kg and
0.1 mg/kg, is between 0.1 mg/kg and 0.5 mg/kg, between 0.1 mg/kg and 0.6
mg/kg, between 0.2
mg/kg and 0.7 mg/kg, between 0.3 mg/kg and 0.8 mg/kg, between 0.4 mg/kg and
0.8 mg/kg,
between 0.5 mg/kg and 0.9 mg/kg, or between 0.6 mg/kg and 1.
[00288] In certain embodiments, the dose of a bispecific binding molecule as
described in
Section 5.1 administered to a subject according to the methods provided herein
is an initial dose
followed by an adjusted dose that is the maintenance dose. In certain
embodiments, the initial
dose is administered once. In certain embodiments, the initial dose is between
0.01 mg/kg and
0.025 mg/kg, is between 0.025 mg/kg and 0.05 mg/kg, is between 0.05 mg/kg and
0.1 mg/kg, is
between 0.1 mg/kg and 0.5 mg/kg, between 0.1 mg/kg and 0.6 mg/kg, between 0.2
mg/kg and
0.7 mg/kg, between 0.3 mg/kg and 0.8 mg/kg, between 0.4 mg/kg and 0.8 mg/kg,
between 0.5
mg/kg and 0.9 mg/kg, or between 0.6 mg/kg and 1. In certain embodiments, the
initial dose is
administered via intravenous infusion over 90 minutes. In certain embodiments,
the adjusted
dose is administered once every about 4 weeks. In certain embodiments, the
adjusted dose is
administered for at least 13, at least 26, or at most 52 weeks. In certain
embodiments, the
adjusted dose is administered for 52 weeks. In certain embodiments, the
adjusted dose is between
0.01 mg/kg and 0.025 mg/kg, is between 0.025 mg/kg and 0.05 mg/kg, is between
0.05 mg/kg
and 0.1 mg/kg, is between 0.1 mg/kg and 0.5 mg/kg, between 0.1 mg/kg and 0.6
mg/kg, between
0.2 mg/kg and 0.7 mg/kg, between 0.3 mg/kg and 0.8 mg/kg, between 0.4 mg/kg
and 0.8 mg/kg,
between 0.5 mg/kg and 0.9 mg/kg, or between 0.6 mg/kg and 1. In certain
embodiments, the
adjusted dose is administered via intravenous infusion over 30 minutes. In
certain embodiments,
the adjusted dose is administered via intravenous infusion over 30 to 90
minutes.
[00289] In another specific embodiment, a bispecific binding molecule as
described in
Section 5.1 for use with the methods provided herein is administered 1, 2, or
3 times a week,
every 1, 2, 3, or 4 weeks. In certain embodiments, the bispecific binding
molecule is
administered according to the following regimen: (i) 1, 2, or 3
administrations in a first week; (ii)
1, 2, 3, or 4 administrations a week after the first week; followed by (iii)
1, 2, or 3
administrations in one week each month for a total of 1, 2, 3, 4, 5, 6, 7, 8,
9, 10, 11, or 12
months. In certain embodiments, the bispecific binding molecule is
administered according to
-106-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
the following regimen: (i) 3 administrations in a first week; (ii) 3
administrations a week after the
first week; followed by (iii) 3 administrations in one week each month for a
total of 1, 2, 3, 4, 5,
6, 7, 8, 9, 10, 11, or 12 months. In certain embodiments, the bispecific
binding molecule is
administered according to the following regimen: (i) 3 administrations in a
first week; (ii) 2
administrations a week after the first week; followed by (iii) 2
administrations in one week each
month for a total of 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, or 12 months. In
certain embodiments, the
bispecific binding molecule is administered according to the following
regimen: (i) 3
administrations in a first week; (ii) 1 administrations a week after the first
week; followed by (iii)
1 administrations in one week each month for a total of 1, 2, 3, 4, 5, 6, 7,
8, 9, 10, 11, or 12
months. In certain embodiments, the bispecific binding molecule is
administered according to
the following regimen: (i) 2 administrations in a first week; (ii) 2
administrations a week after the
first week; followed by (iii) 2 administrations in one week each month for a
total of 1, 2, 3, 4, 5,
6, 7, 8, 9, 10, 11, or 12 months. In certain embodiments, the bispecific
binding molecule is
administered according to the following regimen: (i) 2 administrations in a
first week; (ii) 1
administrations a week after the first week; followed by (iii) 1
administrations in one week each
month for a total of 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, or 12 months. In
certain embodiments, the
bispecific binding molecule is administered according to the following
regimen: (i) 1
administrations in a first week; (ii) 1 administrations a week after the first
week; followed by (iii)
1 administrations in one week each month for a total of 1, 2, 3, 4, 5, 6, 7,
8, 9, 10, 11, or 12
months.
[00290] In certain embodiments, a bispecific binding molecule as described
in Section 5.1 is
administered to a subject according to the methods provided herein in
combination with a second
pharmaceutically active agent as described in Section 5.9.
[00291] In another preferred embodiment, the bispecific binding molecule is
administered
intrathecally, intraventricularly in the brain, intraparenchymally in the
brain, or intraperitoneally.
5.9 COMBINATION THERAPY
[00292] In certain embodiments, a bispecific binding molecule provided herein,
or
polynucleotide, vector, or cell encoding the bispecific binding molecule, may
be administered in
combination with one or more additional pharmaceutically active agents, e.g.,
a cancer
chemotherapeutic agent. In certain embodiments, such combination therapy may
be achieved by
way of simultaneous, sequential, or separate dosing of the individual
components of the
-107-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
treatment. In certain embodiments, the bispecific binding molecule or
polynucleotide, vector, or
cell encoding the bispecific binding molecule, and one or more additional
pharmaceutically
active agents may be synergistic, such that the dose of either or of both of
the components may
be reduced as compared to the dose of either component that would be given as
a monotherapy.
Alternatively, In certain embodiments, the bispecific binding molecule or
polynucleotide, vector,
or cell encoding the bispecific binding molecule and the one or more
additional pharmaceutically
active agents may be additive, such that the dose of the bispecific binding
molecule and of the
one or more additional pharmaceutically active agents is similar or the same
as the dose of either
component that would be given as a monotherapy.
[00293] In certain embodiments, a bispecific binding molecule provided herein,
or
polynucleotide, vector, or cell encoding the bispecific binding molecule is
administered on the
same day as one or more additional pharmaceutically active agents. In certain
embodiments, the
bispecific binding molecule or polynucleotide, vector, or cell encoding the
bispecific binding
molecule is administered 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, or 12 hours before
the one or more
additional pharmaceutically active agents. In certain embodiments, the
bispecific binding
molecule or polynucleotide, vector, or cell encoding the bispecific binding
molecule is
administered 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, or 12 hours after the one or
more additional
pharmaceutically active agents. In certain embodiments, the bispecific binding
molecule or
polynucleotide, vector, or cell encoding the bispecific binding molecule is
administered 1, 2, 3,
or more days before the one or more additional pharmaceutically active agents.
In certain
embodiments, the bispecific binding molecule or polynucleotide, vector, or
cell encoding the
bispecific binding molecule is administered 1, 2, 3 or more days after the one
or more additional
pharmaceutically active agents. In certain embodiments, the bispecific binding
molecule or
polynucleotide, vector, or cell encoding the bispecific binding molecule is
administered 1, 2, 3,
4, 5, or 6 weeks before the one or more additional pharmaceutically active
agents. In certain
embodiments, the bispecific binding molecule or polynucleotide, vector, or
cell encoding the
bispecific binding molecule is administered 1, 2, 3, 4, 5, or 6 weeks after
the one or more
additional pharmaceutically active agents.
[00294] In certain embodiments, the additional pharmaceutically active agent
is doxorubicin.
In certain embodiments, the additional pharmaceutically active agent is
cyclophosphamide. In
certain embodiments, the additional pharmaceutically active agent is
paclitaxel. In certain
-108-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
embodiments, the additional pharmaceutically active agent is docetaxel. In
certain embodiments,
the one or more additional pharmaceutically active agents is carboplatin.
[00295] In certain embodiments, the additional pharmaceutically active agent
is a cytokine,
such as, for example, IL15, IL15R/IL15 complex, IL2, or GMCSF.
[00296] In certain embodiments, the additional pharmaceutically active agent
is an agent that
increases cellular HER2 expression, such as, for example, external beam or
radioimmunotherapy. See, for example, Wattenberg et at., 2014, British Journal
of Cancer, 110:
1472.
[00297] In certain embodiments, the additional pharmaceutically active agent
is a
radiotherapeutic agent.
[00298] In certain embodiments, the additional pharmaceutically active agent
is an agent that
directly controls the HER2 signaling pathway, e.g., lapatinib. See, for
example, Scaltiri et at.,
2012, 28(6): 803-814.
[00299] In certain embodiments, the additional pharmaceutically active agent
is an agent that
increases cell death, apoptosis, autophagy, or necrosis of tumor cells.
[00300] In certain embodiments, a bispecific binding molecule provided herein,
or
polynucleotide, vector, or cell encoding the bispecific binding molecule is
administered in
combination with two additional pharmaceutically active agents, e.g., those
used in combination
with trastuzumab (see, Trastuzumab [Highlights of Prescribing Information].
South San
Francisco, CA: Genentech, Inc.; 2014). In certain embodiments, the two
additional
pharmaceutically active agents are doxorubicin and paclitaxel. In certain
embodiments, the two
additional pharmaceutically active agents are doxorubicin and docetaxel. In
certain
embodiments, the two additional pharmaceutically active agents are
cyclophosphamid and
paclitaxel. In certain embodiments, the two additional pharmaceutically active
agents are
cyclophosphamide and docetaxel. In certain embodiments, the two additional
pharmaceutically
active agents are docetaxel and carboplatin. In certain embodiments, the two
additional
pharmaceutically active agents are cisplatin and capecitabine. In certain
embodiments, the two
additional pharmaceutically active agents are cisplatin and 5-fluorouracil.
[00301] In certain embodiments, a bispecific binding molecule provided herein,
or
polynucleotide, vector, or cell encoding the bispecific binding molecule is
administered as a
single agent following multi-modality anthracycline based therapy.
-109-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
[00302] In certain embodiments, a bispecific binding molecule provided herein,
or
polynucleotide, vector, or cell encoding the bispecific binding molecule is
administered after one
or more chemotherapy regimens for metastatic disease, e.g., brain or
peritoneal metastases.
In specific embodiments, a bispecific binding molecule provided herein, or
polynucleotide,
vector, or cell encoding the bispecific binding molecule is administered in
combination with
cytoreductive chemotherapy. In a specific embodiment, the administering is
performed after
treating the subject with cytoreductive chemotherapy.
[00303] In specific embodiments, a bispecific binding molecule provided
herein,
polynucleotide, vector, or cell encoding the bispecific binding molecule, or a
pharmaceutical
composition comprising the bispecific binding molecule, is administered in
combination with T
cell infusion. In specific embodiments, the bispecific binding molecule is not
bound to a T cell.
In specific embodiments, the bispecific binding molecule is bound to a T cell.
In specific
embodiments, the binding of the bispecific binding molecule to the T cell is
noncovalently. In a
specific embodiment, the administering of a bispecific binding molecule
provided herein,
polynucleotide, vector, or cell encoding the bispecific binding molecule, or a
pharmaceutical
composition comprising the bispecific binding molecule is performed after
treating the patient
with T cell infusion. In specific embodiments the T cell infusion is performed
with T cells that
are autologous to the subject to whom the T cells are administered. In
specific embodiments, the
T cell infusion is performed with T cells that are allogeneic to the subject
to whom the T cells are
administered. In specific embodiments, the T cells can be bound to molecules
identical to a
bispecific binding molecule as described herein. In specific embodiments, the
binding of the T
cells to molecules identical to the bispecific binding molecule is
noncovalently. In specific
embodiments, the T cells are human T cells. Methods that can be used to bind
bispecific binding
molecules to T cells are known in the art. See, e.g., Lum et al., 2013, Biol
Blood Marrow
Transplant, 19:925-33, Janeway et at., Immunobiology: The Immune System in
Health and
Disease, 5th edition, New York: Garland Science; Vaishampayan et at., 2015,
Prostate Cancer,
2015:285193, and Stromnes et at., 2014, Immunol Rev. 257(1):145-164. See,
also,
Vaishampayan et al., 2015, Prostate Cancer, 2015:285193, which describes the
following
exemplary, non-limiting method for binding bispecific binding molecules to T
cells:
Peripheral blood mononuclear cells (PBMCs) can be collected to obtain
lymphocytes for activated T cell expansion from 1 or 2 leukopheresis. PBMCs
-110-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
can be activated with, for example, 20 ng/mL of OKT3 and expanded in 100
IU/mL of IL-2 to generate 40-320 billion activated T cells during a maximum of
14 days of culture under cGMP conditions as described in Ueda et at., 1993,
Transplantation, 56(2):351-356 and Uberti et at., 1994, Clinical Immunology
and
Immunopathology, 70(3):234-240. Cells are grown in breathable flasks (FEP Bag
Type 750-C1, American Fluoroseal Corporation, Gaithersburg, MD) in RPMI
1640 medium (Lonza) supplemented with 2% pooled heat inactivated human
serum. Activated T cells are split approximately every 2-3 days based on cell
counts. After 14 days, activated T cells are cultured with 50 ng of a
bispecific
binding molecule described herein per 106 activated T cells. The mixture is
then
washed and cryopreserved.
6. EXAMPLES
6.1 EXAMPLE 1
6.1.1 INTRODUCTION
[00304] This example describes a HER2 /CD3 bi-specific binding molecule
(herein referred to
as "HER2-BsAb") based on an IgG1 platform. This platform was utilized to allow
for: (1) an
optimal size to maximize tumor uptake, (2) bivalency towards the tumor target
to maintain
avidity, (3) a scaffold that is naturally assembled like any IgG (heavy chain
and light chain) in
CHO cells, purifiable by standard protein A affinity chromatography, (4)
structural arrangement
to render the anti-CD3 component functionally monovalent, hence reducing
spontaneous
activation of T cells, and (5) a platform with proven tumor targeting
efficiency in animal models.
This bispecific binding molecule has the same specificity as trastuzumab; but
also recruits and
activates CD3(+) T cells redirecting them against HER2 expressing tumor cells,
generating
robust anti-tumor responses. Without being bound by any theory, the
effectiveness of this BsAb
centers on the exploitation of the cytotoxic potential of polyclonal T cells,
and its unique
capacity to target tumor cells that express even low levels of HER2,
independent of the
activation status of the HER2 pathway.
6.1.2 MATERIALS AND METHODS
-111-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
6.1.2.1 HER2-BsAb Design, Production, and Purification Analyses
[00305] The HER2-BsAb format was designed as a huOKT3 scFv fusion to the C-
terminus of
the light chain of a human IgG1 . The VH was identical to that of Trastuzumab
IgGl, except
N297A mutation in a standard human IgG1 Fc region for aglycosylated form (SEQ
ID NO: 62),
while the light chain is constructed as VL-C-K-(G45)3-scFv (SEQ ID NO: 60).
Nucleotide
sequences encoding VH and VL domains from Trastuzumab, and the huOKT3 scFv
were
synthesized by GenScript with appropriate flanking restriction enzyme sites,
and were subcloned
into a standard mammalian expression vector. HER2-C825 control BsAb (C825 is a
murine
scFv antibody with high affinity for 1,4,7,10-tetraazacyclododecane-1,4,7,10-
tetraacetic acid
(DOTA)-metal complexes with lanthanides including lutetium and yttrium) was
constructed in a
similar way.
[00306] Linearized plasmid DNA was used to transfect CHO-S cells (Invitrogen)
for stable
production of BsAb. 2 x 106 cells were transfected with 5 [tg of plasmid DNA
by Nucleofection
(Lonza) and then recovered in CD OptiCHO medium supplemented with 8 mM L-
glutamine
(Invitrogen) for 2 d at 37 C in 6-well culture plates. Stable pools were
selected with 500 g/mL
hygromycin for approximately two weeks and single clones were then selected
out with limited
dilution. HER2-BsAb titer was determined by HER2(+) AU565 cell and CD3(+)
Jurket cell
ELISA, respectively, and stable clones with highest expression were selected.
[00307] The BsAb producer line was cultured in OptiCHO medium and the mature
supernatant harvested. A protein A affinity column (GE Healthcare) was pre-
equilibrated with
25 mM sodium citrate buffer with 0.15 M NaCl, pH 8.2. Bound BsAb was eluted
with 0.1 M
citric acid/sodium citrate buffer, pH 3.9 and neutralized with 25 mM sodium
citrate, pH 8.5 (1:10
v/v ratio). For storage, BsAb was dialyzed into 25 mM sodium citrate, 0.15 M
NaC1, pH 8.2 and
frozen in aliquots at -80 C. Two micrograms of the protein was analyzed by SDS-
PAGE under
reducing conditions using 4-15% Tris-Glycine Ready Gel System (Bio-Rad).
Invitrogen
SeeBlue Plus2 Pre-Stained Standard was used as the protein MW marker. After
electrophoresis,
the gel was stained using Coomassie G-250 (GelCode Blue Stain Reagent;
Pierce). The purity of
HER2-BsAb was also evaluated by size-exclusion high-performance liquid
chromatography (SE-
HPLC). Approximately 20 [tg of protein was injected into a TSK-GEL G3000SWXL
7.8 mm x
30 cm, 5 p.m column (TOSOH Bioscience) with 0.4 M NaC104, 0.05 M NaH2PO4, pH
6.0 buffer
-112-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
at flow rate of 0.5 mL/min, and UV detection at 280 nm. Ten microliters of gel-
filtration
standard (Bio-Rad) was analyzed in parallel for MW markers.
6.1.2.2 FACS Analyses
[00308] Cells were incubated with 5 pg/mL of primary antibody (trastuzumab,
HER2-BsAb,
or cetuximab) for thirty minutes at 4 C in PBS, and a secondary phycoerythrin-
labeled antibody
specific for human Fc was used after wash of excess primary antibody. Cells
were fixed with
1% paraformaldehyde (PFA) prior to analysis on FACSCalibur cytometer (BD
biosciences).
Controls were cells with secondary antibody only, for which the mean
fluorescent intensity
(MFI) was set to 5. FACS data display the MFI in the upper right panel of each
plot.
6.1.2.3 51Cr Release Assay
[00309] The 51Cr release assay was performed with effector T cells cultured in
vitro in the
presence of anti-CD3 and anti-CD28 for about 14 days. All target tumor cells
were harvested
with 2 mM EDTA in PBS, labeled with 51Cr (Amersham, Arlington Height, IL) at
100 Ci/106
cells at 37 C for 1 h. 5000 target cells/well were mixed with 50,000 effector
cells (E:T=10:1)
and BsAb antibodies in 96-well polystyrene round-bottom plates (BD
Biosciences) to a final
volume of 250 l/well. The plates were incubated at 37 C for 4 h. The
released 51Cr in
supernatant was counted in a y-counter (Packed Instrument, Downers Grove, IL).
Percentage of
specific release was calculated using the formula: (experimental cpm -
background cpm)/(total
cpm - background cpm) x 100%, where cpm represented counts per minute of 51Cr
released.
Total release was assessed by lysis with 10% SDS (Sigma, St Louis, Mo), and
background
release was measured in the absence of effector cells. EC50 was calculated
using SigmaPlot
software.
6.1.2.4 Competition Assay
[00310] To assess the ability of trastuzumab and/or huOKT3 to interfere with
HER2-BsAb
binding, the HER2-positive SKOV3 cell line was incubated for thirty minutes a
4 C with PBS
or with 10 pg/mL of trastuzumab or huOKT3. Cells were subsequently stained
with 10 pg/mL
of Alexa-Fluor 488-conjugated HER2-BsAb and analyzed by flow cytometry. Alexa-
Fluor 488-
conjugated HER2-BsAb was generated with the Zenon Alexa Fluor 488 Human IgG
Labeling Kit (Life Technologies) according to the manufacturer's instructions.
-113-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
6.1.2.5 Binding Assay
[00311] Binding assays were performed by Surface Plasmon Resonance using
Biacore T100
similar as described in Okazaki et at., 2004, J Mol Biol; 336(5): 1239-1249.
6.1.2.6 Avidity Assay
[00312] To compare the avidity of HER2-BsAb and trastuzumab, HER2-positive
SKOV3
cells were incubated with 10 fold dilutions (from 10 to 1x10-5 g/mL) of
trastuzumab or HER2-
BsAb and analyzed by flow cytometry with FITC-labeled human Fc-specific
antibody as the
secondary antibody. MFI was plotted against the antibody concentration and the
curves were
compared.
6.1.2.7 Proliferation Assay
[00313] To determine anti-proliferative effects, cells were treated with
isotype control
monoclonal antibody, 10 nM lapatinib (as a positive control), 10 g/mL HER2-
BsAb, 10 g/mL
Trastuzumab, 10 nM lapatinib, 10 nM erlotinib, 10 nM neratinib, or 10 g/mL
cetuximab for 72
hours and cell proliferation assayed. Cell proliferation was determined using
an ELISA plate
reader and the WST-8 kit (Dojindo technologies) following the manufacturer's
instructions and
using the formula: % survival rate = (Sample-Background)/(Negative control-
Background).
Lapatinib (MSKCC pharmacy) was ground using a mortar and pestle and suspended
in DMSO as
previously described. To determine statistical significance, the results were
analyzed using one-
way ANOVA using Prism 6Ø
6.1.2.8 qRT-PCR
[00314] RNA was extracted when cells were at 70% confluence and cDNA was
analyzed in a
prism 7700 sequence detection system using the HER2 specific, commercially
available kit
Hs01001580 ml from Applied Biosciences.
6.1.2.9 Animals and In Vivo Assays
[00315] For in vivo studies, BALB-Rag2-KO-IL-2R-yc-KO (DKO) mice (derived from
colony of Dr. Mamoru Ito, CIEA, Kawasaki, Japan). See, for example, Koo et
at., 2009, Expert
Rev Vaccines, 8: 113-120 and Andrade et al., 2011, Arthritis Rheum, 2011, 63:
2764-2773.
MCF7 cells or HCC1954 were mixed at a 1:1 ratio with PMBCs (unactivated, from
buffy coat)
and implanted in DKO mice subcutaneously. Four days post implantation, mice
were treated
-114-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
with PBS, 10 [tg of trastuzumab, or 10 [tg of HER2-BsAb twice a week for two
weeks. Tumor
size was measured at the indicated days post implantation. Tumor size was
determined by either
calipers with the formula V= 0.5 (length x width x width), or by using the
Peira TM900 optical
system.
[00316] For the metastatic model, MCF7 cells expressing luciferase were
administered to
DKO mice intravenously. Four days post administration, mice were treated with
100 ug of
HER2-BsAb, 20 ug or HER2-BsAb, or 20 ug of a HER2-BsAb lacking CD3 targeting
(HER2-
C825) twice a week for three weeks, with or without intravenous administration
of 5x106 PBMC.
Tumor size was quantified at the indicated timepoints using IVIS 200 (Xenogen)
to quantify
luciferin bioluminescence.
6.1.3 RESULTS
6.1.3.1 HER2-BsAb binds to both tumor cells and T cells.
[00317] The HER2-BsAb was generated utilizing a trastuzumab variant comprising
a N297A
mutation in the human IgG1 Fc region to remove glycosylation (SEQ ID NO: 62).
The BsAb
light chain fusion polypeptide was generated by attaching the anti-CD3
humanized OKT3
(huOKT3) single chain Fv fragment (ScFv) to the carboxyl end of the
trastuzumab IgG1 light
chain via a C-terminal (G45)3 linker (Fig. 1A and SEQ ID NO: 60). To avoid
aggregation, a
cysteine at position 105 of the variable heavy chain of huOKT3 was substituted
with serine. A
N297A mutation was also introduced into the HER2-BsAb Fc region to eliminate
binding of
HER2-BsAb to Fc receptors. This mutation has previously been shown to
eliminate the capacity
of human IgGl-Fc binding to CD16A (Fig. 1D) and CD32A Fc receptors (Fig. 1E).
[00318] To produce the HER2-BsAb, a mammalian expression vector encoding both
the
heavy chain and the light chain fusion polypeptide was transfected into CHO-S
cells, stable
clones were selected, supernatants collected, and the HER2-BsAb was purified
by protein A
affinity chromatography. Biochemical purity analysis of the BsAb is depicted
in Fig. 1B and
Fig. 1C. Under reducing SDS-PAGE conditions, HER2-BsAb gave rise to two bands
at around
50 KDa, since the huOKT3 scFv fusion to trastuzumab light chain increased the
MW to ¨50
KDa. SEC-HPLC showed a major peak (97% by UV analysis) with an approximate MW
of 210
KDa, as well as a minor peak of multimers removable by gel filtration. The
HER2-BsAb was
stable by SDS-PAGE and SEC-HPLC after multiple freeze and thaw cycles.
-115-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
[00319] FACS and immunostaining were performed to assess the binding of HER2-
BsAb to
both target cells and effector cells. Trastuzumab and HER2-BsAb displayed
comparable binding
to the HER2-positive breast carcinoma cell line, AU565 (Fig. 2A). In contrast,
HER2-BsAb
demonstrated more than 20-fold less binding to CD3+ T cells than huOKT3 (Fig.
2B). This is
consistent with the observation that light chain-anchored scFv had lower
avidity for T cells than
regular huOKT3 IgGl, purposely designed to minimize cytokine release in the
absence of target
tumor cells.
[00320] The lower avidity of HER2-BsAb for T cells was further confirmed by
the binding
affinity analysis by Biacore as described in Cheung et al. 2012,
OncoImmunology, 1:477-486.
For antigen CD3, HER2-BsAb had a km, at 4.53x105 M's', a koff at 8.68x10-2 s-
1, and overall KID
at 192 nM; comparable to parental huOKT3 IgG1-aGlyco at koff (1.09x101 s'1),
but less at kon
(1.68x106 M's') and overall KD (64.6 nM). In summary, HER2-BsAb had much lower
icon than
its parental huOKT3-aGlyco, suggesting less chance of BsAb binding to and
activating T cells
under the same condition, hence less cytokine release.
6.1.3.2 HER2-BsAb redirected T cell killing of human tumor cell lines.
[00321] To evaluate whether HER2-BsAb could redirect T cells to kill tumor
cells, T cell
cytotoxicity on HER2(+) breast cancer AU565 cells was tested in a standard 4-
hour 51Cr release
assay. Substantial killing of tumor cells was observed n the presence of HER2-
BsAb, with an
EC50 at 300 fM (Fig. 3). Moreover, the killing was effective for an extensive
panel of human
tumor cell lines including breast carcinoma, ovarian carcinoma, melanoma,
osteosarcoma,
Ewing's sarcoma, rhabdomyosarcoma, and neuroblastoma, wherein the killing
potency
correlated with the HER2 expression level in the cells by FACS (Fig. 4).
6.1.3.3 HER2-BsAb mediates tumor antigen specific T cell cytotoxicity.
[00322] To investigate the tumor antigen specificity of HER2-BsAb in T cell
cytotoxicity, a
cytotoxicity assay was performed in the HER2-positive UM SCC 47 cells (a model
for head and
neck cancer) and in the HER2-negative HTB-132 cells (a model for breast
cancer). HER2-BsAb
mediated T cell cytotoxicity against the HER2-positive UM-5CC47 cells (EC50 of
14.5 pM), but
not against the HER2-negative HTB-132 cells (Fig. 5A).
[00323] To investigate the specificity of HER2-BsAb in the T cell
cytotoxicity, HER2-
positive cells were first blocked with huOKT3 or with trastuzumab. In the
absence of HER2-
-116-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
BsAb, the T cells displayed minimal cytotoxicity, reassuring that T cells on
their own have
minimum non-specific cytotoxicity. Both huOKT3 and trastuzumab blocked the
ability of
HER2-BsAb to induce T cell cytotoxicity.
6.1.3.4 HER2-BsAb mediates T cell cytotoxicity against HER2-positive cells
below the
HER2 threshold of detection by flow cytometry.
[00324] The HER2+ ovarian carcinoma cell line SKOV3 was used in a 51Cr
cytotoxicity assay
with 10 fold dilutions of HER2-BsAb in the presence of T cells. These same
cells were stained
using HER2-BsAb at the same concentrations and analyzed by flow cytometry, MFI
was plotted
over the same x-axis as cytotoxicity, and EC50 was calculated for both curves.
HER2-BsAb
mediated T cell cytotoxicity against HER2-positive cells even when HER2-BsAb
binding was
not detected by flow cytometry (Fig. 6). Comparing the EC50 for the
cytotoxicity assay (2pM)
vs EC50 for flow cytometry curve (3.5 nM) suggests that T cells in the
presence of HER2-BsAb
were 2500x more effective in detecting HER2-positive cells than flow
cytometry.
6.1.3.5 HER2-BsAb has the same specificity, affinity and antiproliferative
effects as
trastuzumab.
[00325] Prior to treatment with HER2-BsAb, HER2-positive cells were pre-
incubated with
trastuzumab to determine if HER2-BsAb shares the same antigen specificity as
trastuzumab.
Pre-incubation with trastuzumab blocked HER2-BsAb binding to the cells,
demonstrating a
shared specificity (Fig. 7A). To compare the affinity of HER2-BsAb to
trastuzumab, HER2-
positive cells were incubated with dilutions of trastuzumab or HER2-BsAb and
analyzed by flow
cytometry for cellular binding. Plotting of MFI against the antibody
concentration revealed
similar curves for trastuzumab and HER2-BsAb, demonstrating a similar binding
affinity (Fig.
7B). Further, trastuzumab and HER2-BsAb demonstrated similar anti-
proliferative effects
against HER2-positive cells (Fig. 7C).
6.1.3.6 HER2-BsAb mediated T cell cytotoxicity against SCCHN with an EC50
in the
picomolar range.
[00326] The level and frequency of HER2 in the previously characterized head
and neck
cancer cell lines 93-VU-147T, PCI-30, UD-SCC2, SCC90, UMSCC47 and PCI-15B were
assessed via flow cytometry with trastuzumab. The cells were also tested for
HER2 expression
-117-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
by qRT-PCR (Fig. 8). HER2 was comparably expressed in the panel of head and
neck cancer
cell lines. Finally, the level of cytotoxicity in the presence of T cells and
HER2-BsAb was
correlated with the level of HER2 in the cells, revealing HER2-BsAb displays
an EC50 in the
picomolar range for these head and neck cell lines (Fig. 8).
6.1.3.7 HER2-BsAb mediates T cell cytotoxicity against SCCHN resistant to
other HER
targeted therapies.
[00327] To determine the EGFR and HER2 status of the SCCHN cell line PCI-30,
cells were
stained with trastuzumab or cetuximab and analyzed by flow cytometry as
previously described
(Fig. 9A). A proliferation assay demonstrated that these cells are resistant
to the HER-specific
targeted therapies, trastuzumab, cetuximab, lapatinib, erlotinib and pan-HER
inhibitor neratinib
(Fig. 9B). However, PCI-30 cells were sensitive to treatment with HER2-BsAb
utilizing three
different cytotoxicity assays (Fig. 9C). HER2-BsAb generated potent cytotoxic
responses
against PCI-30 independent of their sensitivity to other HER targeted
therapies, even when these
drugs target more than one of these receptors. These assays suggest that HER2-
BsAb was able
to generate powerful cytotoxic responses, regardless of target cell
sensitivity to EGFR or HER2
targeted therapies.
6.1.3.8 HER2-BsAb mediated T cell cytotoxicity against osteosarcoma cell
lines with an
EC50 in the picomolar range.
[00328] The previously characterized osteosarcoma cell lines, RG-160, CRL 1427
and U2OS,
were assessed for their HER2 expression by flow cytometry with trastuzumab
(Fig. 10) and by
qRT-PCR, and the levels of HER2 were correlated with cytotoxicity in the
presence of T cells
and HER2-BsAb (Fig. 10). All tested cell lines were positive for HER2,
although the expression
level varied. Further, all HER2-positive cells were sensitive to T cell
cytotoxicity mediated by
HER2 BsAb, with an EC50 ranging from 11-25 pM.
6.1.3.9 HER2-BsAb mediates T cell cytotoxicity against HER-therapy
resistant
osteosarcoma cell lines.
[00329] U2OS cells are a HER2-positive, EGFR-positive osteosarcoma cell line
(Fig. 11A).
U2OS cells were analyzed for their sensitivity to trastuzumab, cetuximab,
lapatinib and the pan-
HER inhibitor Neratinib by proliferation assay in the presence of each of the
inhibitors. These
-118-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
cells were resistant to cetuximab and trastuzumab with minimal sensitivity to
Lapatinib, erlotinib
and neratinib (Fig. 11B). These same cells were tested for sensitivity for T
cell cytotoxic
responses mediated by HER2-BsAb. HER2-BsAb generated potent cytotoxic
responses against
U2OS cells using three different cytotoxicity assays, independent of its
sensitivity to other HER
targeted therapies (Fig. 11C).
6.1.3.10 HER2-BsAb mediates T cell cytotoxicity against HER-therapy resistant
cervical
cancer HeLa cells.
[00330] HeLa cells are a HER2-positive, EGFR-positive cervical carcinoma cell
line (Fig.
12A). HeLa cells were analyzed for their sensitivity to HER family tyrosine
kinase inhibitors,
Erlotinib, Lapatinib or Neratinib, or to the HER specific antibodies,
Cetuximab or trastuzumab.
These results demonstrated that HeLa cells are pan-resistant to these
therapies (Fig. 12B).
However, these same cells were tested for sensitivity for T cell cytotoxic
responses mediated by
HER2-BsAb. HER2-BsAb generated potent cytotoxic responses against HeLa cells
using three
different cytotoxicity assays, independent of its sensitivity to other HER
targeted therapies (Fig.
12C). Interestingly, pretreatment with lapatinib increased sensitivity to HER2-
BsAb mediated
cytotoxicity, even when lapatinib alone had no effect on cell proliferation.
6.1.3.11 HER2-BsAb is effective against human breast cancer in humanized mice.
[00331] For in vivo therapy studies, BALB-Rag2-KO-IL-2R-yc-KO (DKO) mice
(derived
from colony of Dr. Mamoru Ito, CIEA, Kawasaki, Japan) were used. See, for
example, Koo et
al. 2009, Expert Rev Vaccines 8: 113-120 and Andrade et al. 2011, Arthritis
Rhem 63: 2764-
2773. MCF7-Luciferase breast cancer cells were mixed with peripheral blood
mononuclear cells
(PBMC) and planted subcutaneously. Four days post cell implantation, the mice
were treated
with HER2-BsAb or with trastuzumab and the tumor size was analyzed over time
(Fig. 13).
HER2-BsAb demonstrated a significant suppression of tumor progression. HER2-
BsAb was
also effective against tumor progression when the trastuzumab resistant
HCC1954 breast cancer
cells (See, for example, Huang et at., 2011, Breast Cancer Research, 13: R84)
were planted
subcutaneously with PBMCs (Fig. 14).
[00332] To assess a metastatic tumor model, MCF7-Luciferace cells were
inoculated
intravenously. HER2-BsAb was administered and subsequently in combination with
PBMC.
Tumor luciferin bioluminescence signal demonstrated HER2-BsAb plus PBMC showed
-119-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
complete suppression of tumor progression (Fig. 15, Fig. 16A, Fig. 16B, Fig.
16C, and Fig.
16D).
6.1.4 CONCLUSIONS
[00333] The aglycosylated HER2-BsAb allowed for minimized Fe functions and
avoidance of
a cytokine storm and elimination of all complement activation, complement
mediated and
complement receptor mediated immune adherence. In addition, despite bivalency
of huOKT3 in
the IgG-scFv platform, binding to CD3 was functionally monovalent; hence there
was no
spontaneous activation of T cells in the absence of tumor target. HER2-BsAb
displayed potent
cytotoxicity against HER2-positive tumor cells in vitro, even against cells
with low antigen
expression, or cells that are resistant to trastuzumab, cetuximab, lapatinib,
erlotinib or the pan-
HER inhibitor neratinib. HER2-BsAb also displayed potent cytotoxicity against
breast cancer,
ovarian cancer, SCCHN, osteosarcomas, and sarcomas. Finally, HER2-BsAb
displayed strong in
vivo efficacy against tumor xenografts, substantially better than the
trastuzumab hIgG1
counterpart.
6.2 EXAMPLE 2
[00334] This example provides (a) a more detailed description of certain of
the experiments
described in Example 1 (Section 6.1); and (b) additional experiments as
compared to Example 1
(Section 6.1).
6.2.1 INTRODUCTION
[00335] Trastuzumab has significantly improved patient outcomes in breast
cancer and has
also been key in the design and implementation of other targeted therapies
(Singh et at., 2014, Br
J Cancer 111:1888-98). However, HER2 expression does not guarantee a clinical
response to
trastuzumab or other HER2 targeted therapies (Gajria et at., 2011, Expert
Review of Anticancer
Therapy, 11(2):263-75; Lipton et at., 2013, Breast Cancer Research and
Treatment, 141(1):43-
53). Less than 35% of patients with HER2 positive breast cancer initially
respond to
trastuzumab and 70% of the initial responders will ultimately progress with
metastatic disease
within a year (Vu and Claret., 2011, Frontiers in Oncology 2:62). In
osteosarcoma and Ewing's
sarcoma, where high levels of HER2 expression are associated with decreased
survival (Gorlick
et al., 1999, Journal of Clinical Oncology: Official Journal of The American
Society of Clinical
-120-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
Oncology 17:2781-2788), trastuzumab has not shown any benefit even when used
in conjunction
with cytotoxic chemotherapy (Ebb et at., 2012, Journal of Clinical Oncology:
Official Journal of
the American Society of Clinical Oncology 30:2545-2551). Furthermore,
trastuzumab, like other
HER targeted therapies, has shown modest or no benefit against HER2-positive
head and neck
cancer (Pollock et at., 2014, Clinical Cancer Research, 21(3):526-33).
[00336] The reasons for these failures are complex and only partially
understood. The
genomic diversity and constant evolution of malignancies make them less prone
to oncogene
addiction, a requirement for the success of targeted therapy. Furthermore,
even when oncogene
addiction is present, resistance can emerge from selection pressure induced by
the use of targeted
therapies (Lipton et at., 2013, Breast Cancer Research and Treatment,
141(1):43-53). In fact,
despite the initial enthusiasm received, the majority of targeted therapies
have not produced a
significant benefit in the overall cure of patients receiving it (Nathanson et
at., 2014, Science,
343:72-76). A different approach, one that selectively targets malignant cells
that overexpress
HER family receptors, and that can generate cytotoxic anti-tumor responses
independently of the
receptor activation status can be beneficial.
[00337] Blinatumomab ¨ a CD19/CD3 BsAb was approved in 2014 for treating Acute
Lymphoplastic Leukemia (Sanford, 2015, Drugs 75:321-7). However, despite its
promising
results, the unfavorable PK of these small size molecules necessitates
prolonged infusions,
complicating their administration (Shalaby et at., 1995, Clin Immunol
Immunopathol 74:185-92,
1995; Portell et at., 2013, Clin Pharmacol 5:5-11). Furthermore, the resulting
cytokine release
syndrome (CRS) still poses costly and often life-threatening complications.
Importantly, despite
the ability of bispecific antibodies to activate T cells, the same inhibitory
pathways that regulate
classic T cell function might still limit their effectiveness. For example,
the heterodimeric design
of a monovalent binding HER2/CD3 bispecific antibody was inhibited by the PD-
1/PD-L1
inhibitory axis (Junttila et at., 2014, Cancer Res 74:5561-71).
[00338] The present example provides a bispecific binding molecule (herein
referred to as
"HER2-BsAb") that offers two distinct advantages over the existing
technologies: (1) it is based
on the fully humanized HER2 specific IgG1 mAb Trastuzumab, preserving its
pharmacologic
advantages (Wittrup et at., 2012, Methods Enzymol 503:255-68) and bivalent
binding to HER2;
maximizing tumor avidity; and (2) its binding to CD3 is functionally
monovalent through the
scFv derived from the humanized huOKT3 mAb sequence. Thus, HER2-BsAb is built
on two
-121-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
mAbs with extensive records of clinical safety. Furthermore, this is a
platform with its Fc
function deleted to eliminate all antibody-dependent cell-mediated
cytotoxicity (ADCC) and
CMC activities in order to reduce the cytokine release syndrome.
[00339] The data presented in this example demonstrate the ability of HER2-
BsAb to produce
potent anti-tumor responses, both in vitro and in vivo, against tumor cells
that are resistant to
HER2 targeted therapy or trastuzumab.
6.2.2 MATERIALS AND METHODS
6.2.2.1 Cell Lines
[00340] All cell lines were purchased from ATCC (Manassas Va) except: UM-
SCC47,
obtained from Dr. Carey at the University of Michigan; SCC-90, PCI-30 and PCI-
15B, obtained
from Dr. Robert Ferris at the University of Pittsburgh; HCC1954, obtained from
Dr. Sarat
Chandarlapaty at Memorial Sloan Kettering Cancer Center; 93-VU-147T and HeLa,
obtained
from Dr. Luc Morris; and UD-SCC2, obtained from Henning Bier at Hals-Nasen-
Ohrenklinik
und Poliklinik. All cells were authenticated by short tandem repeat profiling
using PowerPlex
1.2 System (Promega), and periodically tested for mycoplasma using a
commercial kit (Lonza).
The luciferase-labeled tumor cell lines MCF7-Luc were generated by retroviral
infection with a
SFG-GFLuc vector.
6.2.2.2 HER2-BsAb design and expression in CHO-S cells
[00341] In the HER2-BsAb IgG-scFv format (Fig. 17A, "HER2-BsAb"), the VH was
identical
to that of the trastuzumab IgG1 VH, except that an N297A mutation in the Fc
region was
introduced into the HER2-BsAb to remove glycosylation, thereby depleting Fc
function (SEQ ID
NO: 62). The light chain fusion polypeptide was constructed by extending the
trastuzumab IgG1
light chain with a C-terminal (G45)3 linker followed by huOKT3 scFv (SEQ ID
NO: 60). The
DNA encoding both the heavy chain and the light chain was inserted into a
mammalian
expression vector, transfected into CHO-S cells, and stable clones of the
highest expression were
selected. Supernatants were collected from shaker flasks and the HER2-BsAb was
purified by
protein A affinity chromatography. The control BsAb, HER2-C825 (composed of
SEQ ID NOS:
71 and 72), was generated as previously described (Xu et al., 2015, Cancer
Immunol Res 3:266-
77; Cheal et al., 2014, Mol Cancer Ther 13:1803-12).
-122-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
6.2.2.3 Other Antibodies and Small molecules
[00342] Fluorophore-labeled HER2-BsAb was generated with the Zenon Alexa
Fluor 488
Human IgG Labeling Kit from Life Technologies following the manufacturer's
instructions.
Pembrolizumab, cetuximab, trastuzumab, Erlotinib, Lapatinib and Neratinib were
purchased
from the Memorial Sloan Kettering Cancer Center pharmacy. Small molecules were
re-
suspended in DMSO. The CD4, CD8, CD16 and CD56 antibodies were purchased from
BD
Biosciences (San Jose CA). The commercially available PE labeled PD-Li
specific mAb
10F.9G2 was purchased from BioLegend.
6.2.2.4 Cell proliferation assays
[00343] For cell proliferation assays, 5,000 tumor cells were plated using
RPMI-1640
supplemented with 10% FBS in a 96 well plate for 36 hours before being treated
with lapatinib
or the antibodies at the specified concentrations. Cell proliferation was
determined using an
ELISA plate reader and the WST-8 kit (Dojindo technologies) following the
manufacturer's
instructions and using the formula: % survival rate = (Sample-
Background)/(Negative control-
Background). Lapatinib (Memorial Sloan Kettering Cancer Center pharmacy) was
ground using
a mortar and pestle and suspended in DMSO as previously described (Chen et
at., 2012,
Molecular cancer therapeutics 11:660-669). To determine statistical
significance, the results
were analyzed using one-way ANOVA using Prism 6Ø
6.2.2.5 Cytotoxicity Assays (51chromium release assay)
[00344] Cell cytotoxicity was assayed by 51Cr release as previously
described (Xu et at., 2015,
Cancer Immunol Res 3:266-77), and EC50 was calculated using SigmaPlot
software. Effector T
cells were purified from human PBMC using Pan T cell isolation kit (Miltenyi
Biotec), and then
activated and expanded with CD3/CD28 Dynabeads (Invitrogen) according to the
manufacturer's
protocol.
6.2.2.6 PD-1/PD-L1 expression
[00345] To overexpress PD-Li in HEK293 cells, cells were cultured in DMEM
(Cellgro)
supplemented with 10% heat-inactivated FBS and Penicillin (100 IU/ml) and
streptomycin (100
[tg/m1). On Day(-1), HEK293 cells were trypsinized, counted and plated into 6
well plates at 0.5
M cells/well and kept in 2 mL of fresh media. On the day of transfection,
Day(0), the media was
-123-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
exchanged with 2 mL of fresh media. Transfection reagents were prepared as
follows for both
hPD-L1 and control plasmids: 2.5 tg of DNA was diluted into 250 11.1 of
unsupplemented
DMEM (no serum). 511.1 of Lipofectamine 2000 (Invitrogen) was diluted into a
separate 250 11.1
of DMEM (no serum), and incubated for 5 minutes at room temperature. After 5
minutes, the
diluted DNA was combined with the diluted Lipofectamine 2000 (Invitrogen) and
incubated for
another 30 minutes at room temperature. After 30 minutes, the entire 500 11.1
reaction was added,
dropwise, onto a single well of HEK293 cells. The plate was rocked back and
forth briefly to
help mix the reagents. For the untransfected control, 500 11.1 of
unsupplemented DMEM without
DNA or Lipofectamine 2000 was added to one well. Cells were incubated at 37 C
for 24-48
hours before harvesting. On Day(1) or Day(2), cells were lifted from the plate
using 2 mM
EDTA in PBS, and counted. 100,000-200,000 cells were used for FACS analysis
and the rest
were used for the killing assays.
[00346] To induce PD-1 expression of activated T cells (ATCs), effector cells
were incubated
in a 3:1 ratio for 24 hours with the HER2-high Breast Carcinoma Cell line
HCC1954 after these
target cells were incubated with HER2-BsAb at a concentration of 10 pg/mL for
30 minutes and
antibody excess was removed. Cells were harvested and used in cytotoxicity
assays as
previously described against the HEK293 cells transfected with PD-Li.
6.2.2.7 In vivo experiments
[00347] For in vivo therapy studies, BALB-Rag2-/-IL-2R-yc-KO ("DKO") mice
(derived
from colony of Dr. Mamoru Ito, CIEA, Kawasaki, Japan; see, e.g., Koo et at.,
2009, Expert Rev
Vaccines 8:113-20 and Andrade et at., 2011, Arthritis Rheum 63:2764-73) were
used. Three
humanized mouse xenograft models were used: (1) intravenous tumor plus
intravenous effector
cells; (2) subcutaneous tumor plus subcutaneous effector cells; and (3)
subcutaneous tumor plus
intravenous effector cells. Subcutaneous xenografts were created with 5x106
cells suspended in
Matrigel (Corning Corp, Tewksbury MA) and implanted in the flank of DKO mice.
Effector
peripheral blood mononuclear cell (PBMC) cells were purified from buffy coats
purchased from
the New York Blood Center. Prior to every experimental procedure, PBMCs were
analyzed for
their percentage of CD3, CD4, CD8 and CD56 cells to ensure consistency. HER2-
BsAb was
injected intravenously twice a week at 100 pg/injection, beginning two days
before effectors
cells for three weeks, given as 5-10x106PBMC per injection, once a week for 2
weeks. Tumor
size was measured using (1) hand-held TM900 scanner (Pieira, Brussels, BE);
(2) Calipers; or
-124-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
(3) bioluminescence. Bioluminescence imaging was conducted using the Xenogen
In Vivo
Imaging System (IVIS) 200 (Caliper LifeSciences). Briefly, mice were injected
intravenously
with 0.1 mL solution of D-luciferin (Gold Biotechnology; 30 mg/mL stock in
PBS). Images
were collected 1 to 2 minutes after injection using the following parameters:
a 10- to 60-second
exposure time, medium binning, and an 8 f/stop. Bioluminescence image analysis
was performed
using Living Image 2.6 (Caliper Life Sciences).
6.2.3 RESULTS
6.2.3.1 HER2-BsAb
[00348] HER2-BsAb was designed using an IgG-scFv format (Fig. 17A). The VH was
identical to that of trastuzumab IgGl, except for the N297A mutation in the Fc
region of HER2-
BsAb to remove glycosylation (SEQ ID NO: 62). The light chain fusion
polypeptide was
constructed by extending the trastuzumab IgG1 light chain with a C-terminal
(G45)3 linker
followed by huOKT3 scFv (Xu et at., 2015, Cancer Immunol Res 3:266-77) (SEQ ID
NO: 60).
The DNAs encoding both heavy chain and light chain were inserted into a
mammalian
expression vector, transfected into CHO-S cells, and stable clones of highest
expression were
selected. Supernatants were collected from shaker flasks and purified on
protein A affinity
chromatography.
[00349] SEC-HPLC and SDS-PAGE of the HER2-BsAb is shown in Fig. 17B and Fig.
17C,
respectively. Under reducing SDS-PAGE conditions, HER2-BsAb gave rise to two
bands at
around 50 kDa, since the huOKT3 scFv fusion to trastuzumab light chain
increased the
molecular weight to approximately 50 kDa. SEC-HPLC showed a major peak (97% by
UV
analysis) with an approximate molecular weight of 200 KDa, as well as a minor
peak of
multimers removable by gel filtration. The BsAb remained stable by SDS-PAGE
and SEC-
HPLC after multiple freeze and thaw cycles.
6.2.3.2 HER2-BsAb retained specificity, affinity and anti-proliferative
effects of
trastuzumab
[00350] To determine if HER2-BsAb retained the specificity and anti-
proliferative effects of
trastuzumab, the HER2-positive-high SKOV3 ovarian carcinoma cell line was pre-
incubated
with 10 pg/mL of trastuzumab for 30 minutes and then immunostained using HER2-
BsAb
labeled with Alexa 488 (Fig. 18A). Incubation with trastuzumab prevented HER2-
BsAb binding
-125-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
to SKOV3 cells, demonstrating that these antibodies shared the same
specificity. To compare
the avidity of HER2-BsAb to trastuzumab, the same cell line was incubated with
10-fold
downward dilutions (from 10 pg/m1 to 1x10-5 pg/mL) of trastuzumab or HER2-BsAb
and
analyzed by flow cytometry. The mean fluorescence intensity (MFI) was plotted
against the
antibody concentration in M. The similarity in the binding curves confirmed
that trastuzumab
and HER2-BsAb had similar binding avidities for their common HER2 target (Fig.
18B).
[00351] Finally, the trastuzumab-sensitive breast carcinoma cell line SKBR3
was treated with
isotype control mAb, 10 mM Lapatinib (as a positive control), 10 pg/mL HER2-
BsAb, or 10
pg/mL trastuzumab for 72 hours and cell proliferation was assayed. As shown in
Fig. 18C,
trastuzumab and HER2-BsAb had similar anti-proliferative effects that were
significant as
compared to the negative control. As expected, lapatinib showed the strongest
inhibition of cell
proliferation.
6.2.3.3 HER2-BsAb redirected T cell cytotoxicity was HER2-specific and
dependent on
CD3
[00352] To establish the specificity of cytotoxic responses by T cells in
the presence of
HER2-BsAb; HER2-negative and HER2-positive cell lines were assayed in a
cytotoxicity assays
using ATCs (effector:T cell ("E:T") ratio of 10:1) and HER2-BsAb at decreasing
concentrations
(Fig. 19A and Fig. 20). Cytotoxicity was absent for HER2-negative cell lines.
To demonstrate
the dependency of cytotoxicity on CD3, HER2-BsAb cytotoxicity was tested in
the presence of
the CD3 specific blocking mAb OKT3 (Fig. 19B). Pre-incubation with either
trastuzumab or
OKT3 prevented HER2-BsAb T cell mediated cytotoxicity.
6.2.3.4 HER2-BsAb mediated Cytotoxicity against HER2-positive cell lines
that were
resistant to other HER2 targeted therapies.
[00353] Several cell lines from different tumor systems (e.g., head and
neck, breast, and
sarcoma) were characterized for their HER2 level of expression by flow
cytometry (Fig. 20). In
this panel, 75% of these cells tested positive for HER2 expression by flow
cytometry.
Representative cell lines were assayed for their sensitivity to tyrosine
kinase inhibitors (e.g.,
erlotinib, lapatinib, and neratinib), or HER antibodies (e.g., trastuzumab and
cetuximab), as well
as HER2-BsAb mediated T cell cytotoxicity. Fig. 21 shows representative
examples of these
experiments from three different lines from three different tumor systems. As
shown, HER2
-126-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
expression¨even in low quantities¨was sufficient to mediate T cell
cytotoxicity in the presence
of ATC and HER2-BsAb in cell lines otherwise resistant in vitro to HER-
targeted therapies.
When these cell lines were tested for cytotoxicity in the presence of ATC and
HER2-BsAb,
sensitivity to HER2-BsAb, expressed as EC50, strongly correlated with surface
HER2
expression (Fig. 22)
6.2.3.5 HER2-BsAb mediated T cell cytotoxicity was relatively insensitive
to PD-Li
expression on the tumor target or PD-1 expression on T cells.
[00354] Activation of tumor-specific CTL in the tumor microenvironment is
known to
promote expression of PD-1/PD-L1, leading to T cell exhaustion or suppression,
a phenomenon
termed "adaptive immune resistance" (Tumeh et al., 2014, Nature 515:568-71).
The presence of
the PD-1/PD-L1 pathway has also been reported to limit the anti-tumor effects
of T cell engaging
bispecific antibodies (Junttila et at., 2014, Cancer Res 74:5561-71). To
determine if HER2-
BsAb had the same limitations, PD-1-positive ATCs were used against the HER2-
positive, PD-
Li-positive breast carcinoma cell line HCC1954, with or without the PD-1-
specific mAb
pembrolizumab. As shown in Fig. 23A, Fig. 23B, and Fig. 23C, PD-1-positive T
cells generated
similar cytotoxic responses in the presence of HER2-BsAb, independently of the
presence of
pembrolizumab. When HER2-positive human embryonic kidney cells (HEK-293) were
transfected with the full sequence of PD-Li and used as targets, cytotoxicity
against cells
expressing PD-Li was not significantly different to the cytotoxicity observed
in non-transfected
HEK-293 cells (although maximal cytotoxicity was slightly less with PD-Li-
positive HEK-293
versus PD-Li-negative HEK-293) (Fig. 24A and Fig. 24B shows the average of six
experiments,
and error bars represent standard error).
6.2.3.6 HER2-BsAb was effective against HER2-positive xenografts
[00355] To determine the in vivo efficacy of HER2-BsAb, the breast carcinoma
cell lines
HCC1954 (HER2-high) and MCF-7 (HER2-low) were used in xenograft models in DKO
mice.
Three tumor models differing in tumor locations and effector routes were used:
(1) intravenous
tumor cells and intravenous effector PBMCs; (2) subcutaneous tumor cells and
SC PBMCs; and
(3) subcutaneous tumor cells and intravenous PBMCs. Fig. 25 summarizes the
results of these
experiments. The HER2-low MCF-7-luc (carrying luciferase reporter) cells were
inoculated via
tail vein injection into DKO. When tumor presence was confirmed by
bioluminescence, mice
-127-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
were treated with six doses of intravenous HER2-BsAb or control BsAb twice a
week for 3
weeks. Intravenous effector PBMCs were administered 48 hours after the first
dose of HER2-
BsAb, and again (one week later). Mice were evaluated for tumor burden using
luciferin
bioluminescence every week. In this hematogenous disease model, MCF-7 cells
were
completely eradicated without disease progression (Fig. 25B). This same cell
line was implanted
subcutaneously mixed with effector PBMCs subcutaneously and treated with four
injections of
HER2-BsAb twice a week for 2 weeks (totaling 4 injections in the first
experiment) or twice a
week for 3 weeks (totaling 6 injections in 2nd experiment). In both
experiments, HER2-BsAb
caused a significant delay in tumor progression while PBMC+trastuzumab or PBMC
alone were
ineffective (Fig. 25A). In two other separate experiments, subcutaneous HER2-
positive breast
carcinoma cell line HCC1954 was mixed with subcutaneous PBMCs. Again, both 4
or 6
injections of HER2-BsAb resulted in a complete suppression of tumor growth,
while
trastuzumab or control BsAb HER2-C825 had no effect (Fig. 25C). In the third
model, where
subcutaneous HCC1954 xenografts were treated with intravenous PBMC (once a
week for 3
weeks), and intravenous HER2-BsAb twice a week for 3 weeks, tumor growth was
substantially
delayed (in 2 separate experiments), in contrast to only modest effects for
trastuzumab +
huOKT3 + PBMC, control antibody (HER2-C825) + PBMC, huOKT3 + PBMC, or HER2-
BsAb
alone without PBMC (Fig. 25D). The following observation were made: when
effector PBMCs
were mixed with tumor cells subcutaneously, complete tumor regression without
recurrence was
seen for mice over 90 days post-tumor implantation. When effector PBMCs were
administered
intravenously, there was significant reduction in the size of the tumors, but
complete regression
was only observed in a subset of animals.
6.2.4 CONCLUSIONS
[00356] This example describes a HER2-specific BsAb that has been shown to
have potent T
cell-mediated anti-tumor activity in vitro and in vivo, ablating tumors or
delaying tumor growth
in 3 separate tumor models in the presence of human PBMCs. Unlike monovalent
bispecific
antibodies, this HER2-BsAb had identical anti-proliferative capacity as
trastuzumab. In addition,
the serum half-life and area under the curve of HER2-BsAb were similar to IgG.
Unlike other
bispecific antibodies, which tended to aggregate, HER2-BsAb was stable at -20
C and at 37 C,
despite long term storage. Most importantly, the T cell-mediated cytotoxicity
it induced was
relatively insensitive to inhibition by the PD-1/PD-L1 pathway.
-128-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
[00357] When compared to the existing platforms that target HER2, HER2-BsAb
offers
advantages. The F(ab) x F(ab) format, though effective in vitro, was similar
in size to
Blinatumomab (Sanford, 2015, Drugs, 75:321-7) and was expected to share
similar
pharmacokinetic and toxicity profiles (Shalaby et at., 1995, Clin Immunol
Immunopathol
74:185-92, 1995), having a short half-life, thus requiring daily infusions,
potential leakage into
the central nervous system (CNS), potential CNS toxicity, and potential
significant cytokine
release syndrome. In addition, the anti-proliferative capacity of this F(ab) x
F(ab) univalent
system was 10-fold lower than trastuzumab. The IgG x IgG chemical conjugate
between
trastuzumab and OKT3 was useful for arming T cells ex vivo, but was not useful
as an injectable,
likely due to impurities associated with chemical conjugates (Lum and Thakur,
2011, BioDrugs
25:365-79; Lum et al., Clin Cancer Res 21:2305, 2015); in contrast, the HER2-
BsAb provided
herein is tolerated as an injectable. A heterodimer format was recently
described using a
monovalent system (Junttila et at., 2014, Cancer Res 74:5561-71) that does not
preserve
trastuzumab's anti-proliferative effects retained in HER2-BsAb.
[00358] There are other design features that distinguish HER2-BsAb from other
known
candidates of this class. Unlike most bispecific antibodies, HER2-BsAb's
bivalent binding to the
HER2 target was preserved, providing anti-proliferative activity similar to
that of trastuzumab
IgGl. Unlike F(ab) x F(ab) (Shalaby et al., 1995, Clin Immunol Immunopathol
74:185-92) or
tandem scFv constructs (Sanford, 2015, Drugs, 75:321-7), HER2-BsAb had a
molecular weight
high enough to behave in pharmacokinetic analyses like a wild-type IgG. Unlike
other bivalent
bispecifics (Reusch et al., MAbs, 7:584, 2015), HER2-BsAb's reaction with CD3
was
functionally monovalent. HER2-BsAb also differed from man heterodimeric
bispecifics in its
modified Fc, where aglycosylation removed both ADCC and CMC functions, thereby
reducing
cytokine release syndrome without affecting serum pharmacokinetics or
compromising T cell
activation. The other advantage is manufacturability; HER2-BsAb was produced
in CHO cells
and purified using procedures standard for IgG, without significant
aggregation despite
prolonged incubation at 37 C. HER2-BsAb is an important salvage option for
patients who
progress on standard HER2-based therapies, or a replacement for trastuzumab
given its dual anti-
proliferative and T cell retargeting properties.
-129-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
6.3 EXAMPLE 3
[00359] This example provides (a) a more detailed description of certain of
the experiments
described in Example 1 (Section 6.1); and (b) additional experiments as
compared to Example 1
(Section 6.1).
[00360] T-cell based therapies have emerged as one of the most clinically
effective ways to
target solid and non-solid tumors. HER2 is responsible for the oncogenesis and
treatment
resistance of several human solid tumors. As a member of the HER family of
tyrosine kinase
receptors, its over-activity confers unfavorable clinical outcome. Targeted
therapies directed at
this receptor have achieved responses, although development of resistance is
common. This
example explores a novel HER2/CD3 bispecific antibody (HER2-BsAb) platform
that, while
preserving the anti-proliferative effects of trastuzumab, recruits and
activates non-specific
circulating T-cells, promoting T cell tumor infiltration and ablating HER2-
positive ("HER2(+)")
tumors, even when these are resistant to standard HER2 targeted therapies. Its
in vitro tumor
cytotoxicity, when expressed as EC50, correlated with the surface HER2
expression in a large
panel of human tumor cell lines, irrespective of lineage or tumor type. HER2-
BsAb-mediated
cytotoxicity was relatively insensitive to PD-1/PD-L1 immune checkpoint
inhibition. In four
separate humanized mouse models of human breast cancer and ovarian cancer cell
line
xenografts, as well as in human breast cancer and gastric cancer patient-
derived xenografts
("PDXs"), HER2-BsAb was highly effective in promoting T cell infiltration and
suppressing
tumor growth when used in the presence of human peripheral blood mononuclear
cells
("PBMC") or activated T cells ("ATC"). The in vivo and in vitro antitumor
properties of this
BsAb support its further clinical development as a cancer immunotherapeutic.
6.3.1 INTRODUCTION
[00361] Trastuzumab has significantly improved patient outcomes in breast
cancer, and has
also been key in the design and implementation of other targeted therapies
(Singh et al., Br J
Cancer 2014; 111:1888-98). However, HER2 expression does not guarantee a
clinical response
to trastuzumab or other HER2 targeted therapies (Devika & Sarat, Expert Review
Of Anticancer
Therapy 2011; 11(2):263-75; Lipton et al., Breast Cancer Research and
Treatment 2013;
141(1):43-53). HER2-positive breast cancer patients with metastatic disease
initially respond to
trastuzumab and/or other HER2 targeted therapies, but almost all eventually
will develop
-130-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
resistance and relapse (Montemurro & Scaltriti, J Pathol 2014; 232:219-29). In
osteosarcoma
and Ewing's sarcoma, where high levels of HER2 expression was associated with
decreased
survival (Gorlick et al., J Clin Oncol 1999; 17:2781-8), trastuzumab has not
shown any benefit
even when used in conjunction with cytotoxic chemotherapy (Ebb et al., J Clin
Oncol 2012;
30:2545-51). Furthermore, trastuzumab, like other HER targeted therapies, has
shown modest or
no benefit against HER2(+) positive head and neck cancer (Pollock & Grandis,
Clinical Cancer
Research 2014; 21(3):526-33).
[00362] The reasons for these failures are complex and only partially
understood. The
genomic diversity and constant evolution of malignancies make them less prone
to oncogene
addiction, a requirement for the success of targeted therapy. Furthermore,
even when oncogene
addiction is present, resistance can emerge from selection pressure induced by
the use of targeted
therapies (Lipton et al., Breast Cancer Research and Treatment 2013; 141(1):43-
53). In fact,
despite the initial enthusiasm received, the majority of targeted therapies
have not produced a
significant benefit in the overall cure of patients receiving them (Nathanson
et al., Science 2014;
343:72-6). A different approach, one that selectively targets malignant cells
that overexpress
HER family receptors, and that can generate cytotoxic anti-tumor responses
independently of the
receptor activation status could be beneficial.
[00363] Redirecting the immune system against tumor cells has gained
acceptance as an
effective strategy to overcome resistance to cytotoxic chemotherapy and
targeted therapy. In the
forefront of these treatments, T-cell based therapies constitute the most
promising approach.
Both T-cell engaging bispecific antibodies and immune checkpoint antibody
blockade have
received accelerated approval from the FDA based on their outstanding clinical
performance
(Asher, Nature Reviews Drug Discovery 2015). The clinical success of chimeric
antigen
receptor (CAR) gene modified T-cells against non-solid tumors has further
added to the
enthusiasm among scientists, clinicians and the pharmaceutical industry.
[00364] The outstanding clinical responses seen with these therapies have
consolidated T-cells
as the most powerful effector cells within the immune system to eradicate
tumor cells (Kershew
et al., Clinical & Translational Immunology 2014; 3(5):e16). Thus, a number of
approaches that
redirect them against tumor cells have been proposed and tested by many
investigators. In this
regard, Bispecific antibodies ("BsAb") with specificity for T-cells and for
tumor antigens have
attracted the attention of researchers and big pharma. BsAb, in opposition to
other antibody
-131-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
based therapies, only requires expression of its target of interest to be
effective. By recruiting
polyclonal T-cells through the CD3 surface receptor, BsAb activate T-cells
irrespective of their
lineage, antigen specificity, maturation, HLA restriction or co-stimulatory
receptors. The direct
activation of T-cells, bypassing the classic T cell receptor ("TCR"), removes
the limitations
imposed by HLA restriction and its level of expression (Brischwein et al.,
Journal of
Immunotherapy 2006; 30:798-807), a well-established immune resistance
mechanism (Sabbatino
et al., Clinical transplants 2013:453-63).
[00365] Blinatumomab ¨ a CD19/CD3 BsAb was approved in 2014 for treating Acute
Lymphoplastic Leukemia (Sanford, Drugs 2015; 75:321-7). However, despite its
promising
results, the unfavorable pharmacokinetics of these small size molecules
necessitate prolonged
infusions, complicating their administration (Shalaby et al., J Exp Med 1992;
175:217-25; Portell
et al., Clinical Pharmacology : Advances And Applications 2013; 5:5-11).
Furthermore, the
resulting cytokine release syndrome ("CRS") still poses costly and often life-
threatening
complications. Importantly, despite the ability of bispecific antibodies to
activate T-cells, the
same inhibitory pathways that regulate classic T-cell function might still
limit their effectiveness.
For example, the heterodimeric design of a monovalent binding HER2/CD3
bispecific antibody
was inhibited by the PD-1/PD-L1 inhibitory axis (Junttila et al., Cancer
Research 2014; 74:5561-
71).
[00366] This example reports a BsAb against the HER2 tumor antigen that offers
two distinct
advantages over the existing technologies: (1) it is based on the fully
humanized HER2-specific
IgG1 trastuzumab, preserving its pharmacologic advantages (Wittrup et al.,
Methods Enzymol
2012; 503:255-68) and bivalent binding to HER2, maximizing tumor avidity; (2)
its binding to
CD3 is functionally monovalent through the scFv derived from the humanized
huOKT3 IgG1
sequence. Thus, HER2-BsAb is built on two mAbs with an extensive record of
clinical safety.
Previous studies have also shown that scFv linked to the carboxyl end of the
light chain did not
affect the targeting ability of these IgG forms (Cheal et al., Mol Cancer Ther
2014; 13:1803-12;
Orcutt et al., Protein Eng Des Sel 2010; 23:221-8). Furthermore, this is a
platform with its Fc
function silenced to reduce the cytokine release syndrome. This example
presents data to show
that this HER2-BsAb has potent anti-tumor properties both in vitro and in
vivo, against tumor
cells that are resistant to HER2 targeted therapy or to trastuzumab.
6.3.2 MATERIALS AND METHODS
-132-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
6.3.2.1 Cell Lines
[00367] All cell lines were purchased from ATCC (Manassas Va) except: UM-SCC47
obtained from Dr. Carey at the University of Michigan, SCC-90, PCI-30 and PCI-
15B from Dr.
Robert Ferris at the University of Pittsburgh, SKOV3-luc from Dr. Dmitry
Pankov at MSK, 93-
VU-147T and HeLa from Dr. Luc Morris and UD-SCC2 from Henning Bier at Hals-
Nasen-
Ohrenklinik und Poliklinik. All cells were authenticated by short tandem
repeat profiling using
PowerPlex 1.2 System (Promega), and periodically tested for mycoplasma using a
commercial
kit (Lonza). The luciferase-labeled tumor cell lines MCF7-Luc were generated
by retroviral
infection with a SFG-GFLuc vector.
6.3.2.2 HER2-BsAb Design and Expression in CHO-S Cells
[00368] In the HER2-BsAb IgG-scFv format, VH was identical to that of
trastuzumab IgGl,
except N297A mutation in the Fc region was introduced to remove glycosylation,
thereby
depleting Fc function. The sequence of the heavy chain is set forth in SEQ ID
NO: 62. The light
chain fusion polypeptide (SEQ ID NO: 60) was constructed by extending the
trastuzumab IgG1
light chain with a C-terminal (G45)3 linker followed by huOKT3 scFv. The DNA
encoding both
heavy chain and light chain was inserted into a mammalian expression vector,
transfected into
CHO-S cells, and stable clones of highest expression were selected.
Supernatants were collected
from shaker flasks and the HER2-BsAb was purified by protein A affinity
chromatography. The
other control BsAb, HER2-C825, was generated as previously described (Cheal et
al., Mol
Cancer Ther 2014; 13:1803-12). HuOKT3 IgG1 was made using the same variable
sequences
as in huOKT3 scFv, and huOKT3 Fab was prepared from huOKT3 IgG1 using the
Pierce Fab
Preparation Kit (Thermo Scientific).
6.3.2.3 Other Antibodies And Small Molecules
[00369] Fluorophore-labeled HER2-BsAb was generated with the Zenon Alexa
Fluor 488
Human IgG Labeling Kit from Life Technologies following the manufacturer's
instructions.
Pembrolizumab, cetuximab, trastuzumab, Erlotinib, Lapatinib and Neratinib were
purchased
from the Memorial Sloan Kettering Cancer Center pharmacy. Small molecules were
re-
suspended in dimethylsulfoxide ("DMSO"). The CD3, CD4, CD8 and CD56 antibodies
were
purchased from BD Biosciences (San Jose CA). The commercially available PE
labeled PD-Li
specific mAb 10F.9G2 was purchased from BioLegend.
-133-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
6.3.2.4 Cell Proliferation Assays
[00370] For tumor cell proliferation, 5,000 tumor cells were plated using RPMI-
1640
supplemented with 10% fetal bovine serum ("FBS") in a 96 well plate for 36
hours before being
treated with - kinase inhibitors or the antibodies at the specified
concentration. Cell proliferation
was determined using the cell counting WST-8 kit (Dojindo technologies)
following the
manufacturer's instructions and using the formula: % survival rate = (Sample-
Background)/(Negative control-Background). Lapatinib was ground using a mortar
and pestle
and suspended in DMSO as previously described (Chen et al., Molecular cancer
therapeutics
2012; 11:660-9). To determine statistical significance, the results were
analyzed using one-way
ANOVA using Prism 6Ø
[00371] For T cell proliferation, naive T cells were purified from human
PBMC using Pan T
cell isolation kit (Miltenyi Biotec). 2x105 purified T cells were mixed with
different antibodies
in 96-well cell culture plate to a final volume of 25011.1/well. T cells were
cultured and
maintained in RPMI-1640 supplemented with 10% FBS in 37 C for 6 days. T cell
proliferation
was quantitated using the WST-8 kit as described above.
6.3.2.5 Cytotoxicity Assays (51chromium Release Assay)
[00372] Cell cytotoxicity was assayed by 51Cr release as previously
described (Xu et al.,
Cancer immunology research 2015; 3:266-77), and EC50 was calculated using
SigmaPlot
software. Effector PBMC cells were purified from buffy coats purchased from
the New York
Blood Center. ATCs were first purified from human PBMC using Pan T cell
isolation kit,
and then activated and expanded for approximately 14 days with CD3/CD28
Dynabeads
(Invitrogen) according to manufacturer's protocol. For pre-incubation
experiment, HER2-BsAb
was pre-incubated with either ATCs (T cells pre-armed) or chromium-labeled
tumor target cells
(AU565 pre-targeted) for 30 minutes at room temperature, and unbound BsAb was
washed off
for two times before adding the other cells.
6.3.2.6 Cytokine Release Assay
[00373] Cytokine release was assayed as previously described (Ahmed et al.,
OncoImmunology 2015; 4:e989776), using naive T cells prepared as described
above. T cells
(200,000/well) were cultured with or without NCI-N87 tumor cells (10,000/well)
for 24 hours
before supernatants being harvested for ELISA-based cytokine assay.
-134-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
6.3.2.7 PD-1/PD-L1 Expression
[00374] To overexpress PD-Li in HEK293 cells, cells were cultured in DMEM
(Cellgro)
supplemented with 10% heat-inactivated FBS and Penicillin (100 IU/ml) and
streptomycin (100
ps/m1). HEK293 cells were plated into 6 well plates at 0.5 million cells/well
with 2 ml fresh
media the day before transfection. Transfection was done with 2.5 [tg hPD-L1
plasmid DNA
using Lipofectamine 2000 (Invitrogen) according to manufacturer's protocol.
Cells were
incubated at 37 C for 48 hours before harvesting with 2mM EDTA in PBS. 100,000-
200,000
cells were used for FACS analysis and the rest were used for the killing
assays.
[00375] To induce PD-1 expression in ATCs, effector cells were incubated in a
3:1 ratio for
24 hours with the HER2(+) breast carcinoma cell line HCC1954, after these
target cells were
incubated with HER2-BsAb at a concentration of 10 g/mL for 30 minutes and
excessive
antibody was removed. Cells were harvested and used in cytotoxicity assays as
previously
described against the HEK293 cells or HCC1954 cells. For PD-1 blockade, PD-1-
induced ATCs
were pre-incubated with 10 g/mL pembrolizumab for 30 minutes before adding to
the well.
6.3.2.8 In Vivo Experiments
[00376] All animal procedures were performed in compliance with Institutional
Animal Care
and Use Committee (IACUC) guidelines. For in vivo therapy studies, BALB-Rag2-/-
IL-2R-yc-
KO (DKO) mice (derived from colony of Dr. Mamoru Ito, CIEA, Kawasaki, Japan)(
Koo et al.,
Expert Rev Vaccines 2009; 8:113-20; Andrade et al., Arthritis Rheum 2011;
63:2764-73) were
used. Four humanized mouse xenograft models were used: (1) intravenous
("i.v.") tumor plus
i.v. effector cells, (2) subcutaneous ("sc") tumor plus sc effector cells, (3)
sc tumor plus i.v.
effector cells, and (4) intraperitoneal ("i.p.") tumor plus i.p./i.v. effector
cells. Patient derived
xenografts ("PDXs") were established from fresh surgical specimens with
Memorial Sloan
Kettering Cancer Center Institutional Review Board approval. Effector PBMC
cells and ATCs
were prepared as described above. Prior to every experimental procedure, PBMCs
and ATCs
were analyzed by FACS for their percentage of CD3, CD4, CD8 and CD56 cells to
ensure
consistency. Antibodies were injected i.v. or i.p. twice a week started two
days before effectors
cells for 3-6 weeks, given as i.v. 5-10x106 PBMC/ATC per injection, once a
week for 2-3
weeks. s.c. xenografts were created with tumor cells suspended in Matrigel
(Corning Corp,
Tewksbury MA) and implanted in the flank of DKO mice. Tumor size was measured
using 1)
-135-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
hand-held TM900 scanner (Pieira, Brussels, BE), 2) Calipers, or 3)
Bioluminescence.
Bioluminescence imaging was conducted using the Xenogen In Vivo Imaging System
(IVIS)
200 (Caliper LifeSciences). Briefly, mice were injected i.v. with 0.1 mL
solution of D-luciferin
(Gold Biotechnology; 30 mg/mL stock in PBS). Images were collected 1 to 2
minutes after
injection using the following parameters: a 10- to 60-second exposure time,
medium binning,
and an 8 f/stop. Bioluminescence image analysis was performed using Living
Image 2.6
(Caliper LifeSciences).
6.3.2.9 Immunohistochemistry Staining
[00377] The immunohistochemical detection was performed at Molecular Cytology
Core
Facility of Memorial Sloan Kettering using Discovery XT processor (Ventana
Medical Systems).
Paraffin-embedded tumor sections were deparaffinized with EZPrep buffer
(Ventana Medical
Systems), antigen retrieval was performed with CC1 buffer (Ventana Medical
Systems) and
sections were blocked for 30 minutes with Background Buster solution
(Innovex). Anti-CD3
(DAKO, cat# A0452, 1.2 [tg/m1), anti-HER2 (Enzo, cat# ALX-810-227, 5 [tg/m1),
and anti-PD-1
(Ventana, cat# 760-4895, 3.1ug/m1) antibodies were applied and sections were
incubated for 5
hours, followed by 60 minutes incubation with biotinylated goat anti-rabbit
IgG (Vector labs,
cat# PK6101) for CD3 and HER2 antibodies, or biotinylated horse anti-mouse IgG
(Vector Labs,
cat# MKB-22258) for PD-1 antibodies at 1:200 dilution. The detection was
performed with
DAB detection kit (Ventana Medical Systems) according to manufacturer's
instruction. Slides
were counterstained with hematoxylin and coverslipped with Permount (Fisher
Scientific). For
PD-L1 staining, the sections were pre-treated with Leica Bond ER2 Buffer
(Leica Biosystems)
for 20 minutes at 100 C. The staining was done on Leica Bond RX (Leica
Biosystems) with
PD-L1 mouse monoclonal antibody (Cell Signaling, cat# 29122, 2.5 [tg/m1) for 1
hour on Leica
Protocol F. All images were captured from tumor sections using Nikon ECLIPSE
Ni-U
microscope and NIS-Elements 4.0 imaging software.
6.3.2.10 Statistics
[00378] Differences between samples indicated in the figures were tested for
significance by
one-way ANOVA using Prism 6.0, and p < 0.05 was considered statistically
significant.
6.3.3 RESULTS
-136-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
6.3.3.1 HER2-BsAb
[00379] HER2-BsAb heavy chain was constructed using the standard human IgGl,
except for
the N297A mutation in the Fc region to remove glycosylation. The light chain
was constructed
by extending the trastuzumab IgG1 light chain with a C-terminal (G4S)3 linker
followed by
huOKT3 scFv (Xu et al., Cancer Immunology Research 2015; 3:266-77). The DNAs
encoding
both heavy chain and light chain were inserted into a mammalian expression
vector, transfected
into CHO-S cells, and stable clones of highest expression were selected.
Supernatants were
collected from shaker flasks and purified on protein A affinity chromatography
(Xu et al., Cancer
Immunology Research 2015; 3:266-77).
[00380] The SEC-HPLC and SDS-PAGE of the HER2-BsAb was analyzed. Under
reducing
SDS-PAGE conditions, HER2-BsAb gave rise to two bands at around 50 KDa, since
the
huOKT3 scFv fusion to trastuzumab light chain increased the MW to ¨50 KDa
(data not shown).
SEC-HPLC showed a major peak (97% by UV analysis) with an approximate MW of
210 KDa,
as well as a minor peak of multimers (data not shown). The BsAb remained
stable by SDS-
PAGE and SEC-HPLC after multiple freeze and thaw cycles (data not shown).
6.3.3.2 HER2-BsAb retained specificity, affinity and anti-proliferative
effects of
trastuzumab
[00381] To determine if HER2-BsAb retained the specificity of trastuzumab, the
HER2(+)high SKOV3 ovarian carcinoma cell line was pre-incubated with 10
i.tg/mL of
trastuzumab for 30 minutes and then immunostained using 1 i.tg/mL HER2-BsAb
labeled with
Alexa 488 (Fig. 26A). Pre-incubation with trastuzumab prevented HER2-BsAb from
binding to
SKOV3 cells, demonstrating that these antibodies shared the same specificity.
To compare the
avidity of HER2-BsAb to trastuzumab, the same cell line was incubated with 10
fold downward
dilutions (from 10 tg/m1 to 1x10-5 i.tg/mL) of trastuzumab or HER2-BsAb and
analyzed by flow
cytometry. The mean fluorescence intensity ("MFI") was plotted against the
antibody
concentration in M. Again the similarity in the binding curves confirmed that
trastuzumab and
HER2-BsAb had similar binding avidities for their common HER2 target (Fig.
26B).
[00382] Finally, the trastuzumab-sensitive breast carcinoma cell line SKBR3
was treated with
Isotype control mAb, 10 nM lapatinib (as a positive control), 10 i.tg/mL HER2-
BsAb or 10 i.tg
/mL trastuzumab for 72 hours and cell proliferation was assayed. As shown in
Fig. 26C,
-137-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
trastuzumab and HER2-BsAb had similar anti-proliferative effects that were
significant
compared to the negative control. As expected, lapatinib showed the strongest
inhibition of cell
proliferation.
6.3.3.3 HER2-BsAb redirected T cell cytotoxicity was HER2 specific and
dependent on
CD3
[00383] Prior to the cytotoxicity assay, HER2-BsAb was shown capable of
binding different T
cells at the similar level (MFI around 450 with the BsAb concentration of 1
ug/106 cells), no
matter whether they were naïve T cells purified from fresh PBMC or activated T
cells (ATCs)
(Fig. 27A). To establish the specificity of cytotoxic responses by T cells in
the presence of
HER2-BsAb, HER2-negative ("HER2(-)") breast carcinoma HTB-132 cells and
HER2(+) MCF-
7 cells were tested in a cytotoxicity assays using ATCs (E:T ratio of 10:1)
and HER2-BsAb at
decreasing concentrations (Fig. 26D). Cytotoxicity was robust for HER2(+)
cells but absent for
HER2(-) cells. In fact, HER2-BsAb was able to redirect efficient T cell
killing no matter
whether BsAb was present throughout the 4 hour assay (mixing), or used to pre-
arm T cells and
then washed off, or to pre-target AU565 tumor cells and then washed off.
Although pre-targeted
AU565 cells were killed as well as mixing all three together, pre-armed T
cells were less potent
due to the low avidity of BsAb binding to CD3 on T cells (Fig. 27B). To
demonstrate the
dependency of cytotoxicity on both HER2 and CD3, HER2-BsAb cytotoxicity
against HER2(+)
SCCHN cell line PCI-13 was tested in the presence of trastuzumab, or the CD3
specific blocking
huOKT3 IgG1 (Fig. 26E). Pre-incubation with either trastuzumab or huOKT3
prevented HER2-
BsAb mediated T-cell cytotoxicity.
6.3.3.4 HER2-BsAb mediated Cytotoxicity against HER2(+) cell lines that
were
resistant to other HER2 targeted therapies.
[00384] A panel of a total of 39 cell lines from different tumor systems
(breast, ovarian,
gastric, head and neck, sarcoma, etc.) was characterized for their HER2
expression levels by
flow cytometry and CTL activity (Table 9). In this panel, 75% of these cells
were tested positive
for HER2 expression. Representative cell lines were assayed for their
sensitivity to tyrosine
kinase inhibitors (10 nM each of Erlotinib, Lapatinib, Neratinib), or HER
antibodies (10 g/mL
each of trastuzumab and cetuximab), as well as HER2-BsAb mediated T-cell
cytotoxicity. Fig.
28A, Fig. 28B and Fig. 28C showed three representative lines from three
different tumor
-138-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
systems. As shown, HER2 expression, even in low quantities, was sufficient to
mediate T-cell
cytotoxicity in the presence of ATC and HER2-BsAb in cell lines otherwise
resistant in vitro to
HER-targeted therapies. When these cell lines were tested for cytotoxicity in
the presence of
ATC and HER2-BsAb, sensitivity to HER2-BsAb expressed as EC50 inversely
correlated with
surface HER2 expression in general (Fig. 28D, Table 9).
[00385] Table 9.
Tumor Type Cell Line HER2 Expression (MFI)* EC50 (pM)**
Breast carcinoma AU565 1175 0.3
Gastric Carcinoma NCI-N87 4900 1.1
Ovarian Carcinoma OVCAR3 183 1.8
Breast Carcinoma MDA-MB-361 777 2.5
Ovarian Carcinoma SKOV3 1577 2.8
Melanoma SKMEL28 190 3
Breast Carcinoma SKBR3 2506 4.1
Breast Carcinoma HCC1954 1597 5.5
Head and Neck Cancer SCC90 274 5.7
Ewings SKEAW 246 10
Osteosarcoma CRL1427 108 10
Rhabdomyosarcoma HTB82 204 10
Osteosarcoma RG 160 563 11
Head and Neck Cancer PCI-30 359 12.2
Gastric Carcinoma KATO III 201 13.5
Melanoma HT-144 156 15
Neuroblastoma NB5 66 15.5
Osteosarcoma RG 164 439 17.7
Head and Neck Cancer UM SCC47 302 19.8
Osteosarcoma U2OS 90 22.5
Gastric Adenocarcinoma AGS 172 23
Head and Neck Cancer UDSCC2 178 26.9
Gastric Carcinoma SNU-16 29 30.5
Head and Neck Cancer 93VU147T 127 32.4
Ewings SKES-1 146 50
Breast Carcinoma MDA-MB-231 76 50.2
Head and Neck Cancer 15B 305 62.8
Breast Carcinoma MCF7 398 64.9
Cervical Cancer HeLa 104 120.7
Melanoma M14 57 130
Breast Carcinoma MDA-MB-468 6 >5000
Neuroblastoma NMB7 12 >5000
Neuroblastoma SKNBE(2)C 8 >5000
Neuroblastoma IMR32 6 >5000
Neuroblastoma SKNBE(2)S 4 >5000
-139-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
Neuroblastoma SKNBE(1)N 3 >5000
Small Cell lung Cancer NCI-H524 14 >5000
Small Cell lung Cancer NCI-H69 10 >5000
Small Cell lung Cancer NCI-H345 6 >5000
6.3.3.5 HER2-BsAb mediated in vitro T-cell cytotoxicity was relatively
insensitive to
PD-Li expression on the tumor targets or PD-1 expression on T cells.
[00386] Activation of tumor-specific CTL in the tumor microenvironment is
known to
promote expression of PD-1/PD-L1, leading to T-cell exhaustion or suppression,
a phenomenon
termed "adaptive immune resistance" (Tumeh et al., Nature 2014; 515:568-71).
The presence of
PD-1/PD-L1 pathway has also been reported to limit the anti-tumor effects of T-
cell engaging
bispecific antibodies (Junttila et al., Cancer Research 2014; 74:5561-71). To
determine if HER2-
BsAb had this same limitation, PD-1-positive ("PD-1(+)") ATCs were used
against the HER2(+)
PD-Li-positive ("PD-L1(+)") breast carcinoma cell line HCC1954 with or without
the PD-1-
specific antibody pembrolizumab. As shown in Fig. 29A, PD-1(+) T cells
generated similar
cytotoxic responses in the presence of HER2-BsAb no matter whether
pembrolizumab was
present or not. When HER2(+) human embryonic kidney cells (HEK-293) were
transfected with
the full sequence of PD-Li and used as targets, cytotoxicity against cells
expressing PD-Li was
not significantly different to the cytotoxicity observed in non-transfected
HEK-293 cells,
although maximal cytotoxicity was slightly less with PD-L1(+) HEK-293 versus
PD-Li-negative
("PD-L1(-)") parental HEK-293 (Fig. 29B).
6.3.3.6 HER2-BsAb was effective against HER2(+) xenografts
[00387] To determine the in vivo efficacy of HER2-BsAb, the breast carcinoma
cell lines
HCC1954 (HER2high) and MCF-7 (HER2low), ovarian carcinoma cell line SKOV3, and
HER2(+) patient-derived breast cancer and gastric cancer xenografts ("PDXs")
were used in
DKO mice xenograft models. Four tumor models differing in tumor locations and
effector
routes were used, with the first three described before (Xu et al., Cancer
Immunology Research
2015; 3:266-77) to simulate different clinical situations: (1) intravenous
("i.v.") tumor cells/i.v.
effector PBMC; (2) subcutaneous ("s.c." tumor cells/s.c. PBMC; (3) s.c. tumor
cells/i.v. PBMC;
and (4) intraperitoneal ("i.p." tumor cells plus i.p. or i.v. effector T cells
to simulate ovarian
cancer metastasizing to the peritoneal cavity. Fig. 30 and Fig. 31 summarize
the results of these
-140-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
experiments using cell lines, and Fig. 32 summarizes the results of these
experiments using
PDXs (M37 breast cancer and EK gastric cancer).
[00388] 3 x 106 HER2low MCF-7-luc (carrying luciferase reporter) cells were
inoculated via
tail vein i.v. injection into DKO mice. When tumor presence was confirmed by
bioluminescence, mice were treated with HER2-BsAb or control BsAb (20 i.tg
i.v., 2 times per
week for 3 weeks), in combination with PBMC (5 x 106 i.v., once per week for 2
weeks). Mice
were evaluated for tumor burden using luciferin bioluminescence every week. In
this
hematogenous disease model, MCF-7 cells were completely eradicated without
disease
progression (Fig. 30A). This same cell line was implanted s.c. mixed with
effector PBMC (1:1,
7 x 106 each), and treated with HER2-BsAb (10 i.tg i.v., 2 times per week for
2 weeks in the 1st
experiment; or 20 i.tg i.v. 2 times per week for 3 weeks in the 2nd
experiment). In both
experiments, HER2-BsAb caused a significant delay in tumor progression, while
PBMC +
trastuzumab or PBMC alone was ineffective (Fig. 30B). In two other separate
experiments, 5 x
106 HER2(+) breast carcinoma HCC1954 cells were implanted s.c. mixed with 2.5
x 106 PBMC
(2:1). Again, after either 4 or 6 injections of HER2-BsAb (20 i.tg i.v. per
dose), there was
complete suppression of tumor growth, while trastuzumab or control BsAb HER2-
C825 almost
had no effect (Fig. 30C). In the third model, where s.c. 5 x 106 HCC1954
xenografts were
treated with i.v. PBMC (5 x 106, once per week for 3 weeks) and i.v. HER2-BsAb
(100 i.tg, twice
per week for 3 weeks), tumor growth was substantially delayed (2 separate
experiments), in
contrast to only modest effects for PBMC + trastuzumab + huOKT3, PBMC +
trastuzumab, or
PBMC + huOKT3 (Fig. 30D). For HCC1954 xenografts, the following observations
were made:
(1) when effector PBMCs were mixed with tumor cells s.c., complete tumor
regression without
recurrence was seen past 90 days from tumor implantation; and (2) when
effector PBMCs were
administered i.v., there was significant reduction in the size of the tumors,
but complete
regression was only observed in a subset of animals (data not shown).
[00389] Since T cell homing into tumor is critical for anti-tumor response
in cancer
immunotherapy (Tang et al., Cancer Cell 2016; 29:285-96), T-cell tumor
infiltration was studied
using the s.c. tumor model described in Fig. 30D. Tumors were collected 5 days
after i.v. PBMC
and immunohistochemistry ("IHC") was performed (Fig. 30E). T-cell tumor
infiltration by
CD3(+) staining was detected only in PBMC + HER2-BsAb-treated group, but not
in control
group (PBMC+Trastuzumab+huOKT3). These infiltrated T cells also had PD-1
expression,
-141-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
although it was very weak. Interestingly, PD-Li expression in the tumor cells
was strongly
upregulated in the HER2-BsAb-treated mice, presumably induced by the cytokines
released by
the infiltrated T cells in the vicinity. But still, HER2-BsAb treatment
eradicated these tumors.
This was consistent with the in vitro data (Fig. 29) showing HER2-BsAb-
mediated T-cell
cytotoxicity was relatively insensitive to or sufficient to overcome the PD-
1/PD-L1 immune
checkpoint inhibition.
[00390] To simulate ovarian cancer that metastasized into the peritoneal
cavity, 1 x 105
ovarian cancer SKOV3-luc cells were injected peritoneally in DKO mice, and
treatments were
started after confirming tumor growth by bioluminescence. Besides i.v.ATC,
i.p.ATC was also
tested as a source of effectors. As shown in Fig. 31A and Fig. 31B, after
treatment with ATC
(7.5 x 106 i.v. or i.p., once a week for 2 weeks), and i.p. HER2-BsAb (100 g,
twice per week for
3 weeks), tumors were completely eradicated without evidence of recurrence at
followup. Both
i.v.ATC and i.p.ATC were equally effective in this fourth model.
[00391] HER2-BsAb was next tested using PDXs, since they could approximate the
tumor
heterogeneity and microenvironment typically found in fresh human tumor
specimens. To
determine whether HER2-BsAb is effective against PDXs, two HER2(+) PDXs
(gastric cancer
PDX (EK) and breast cancer PDX (M37)) were tested using the s.c. tumor
cells/i.v. PBMC
model similar to the one described in Fig. 30D. The PDXs were recently
characterized by IHC
using the PATHWAY anti-HER2/neu (4B5) Rabbit Monoclonal Primiary Antibody
(VENTANA) and scored (according to the 2013 ASCO HER2 Breast Cancer Testing
Guidelines
(see, e.g., Wolff et al., Journal of Clinical Oncology, 2013, 31(31):3997-
4013)) as IHC 3+ for
breast cancer PDX M37 and IHC 2+ for gastric cancer PDX EK. When the gastric
PDX (EK)
was passaged s.c. in DKO mice and treated with i.v. PBMC (1 x 107, once a week
for 3 weeks)
and i.v. HER2-BsAb (100 g, twice per week for 5 weeks), tumors were
completely eradicated
without disease progression, (Fig. 32A), accompanied by substantial amount of
T cell tumor
infiltration (Fig. 32B), even though the HER2 expression level was relatively
low compared to
the M37 breast cancer PDX (Fig. 32C). In this next experiment, M37 PDX was
passaged s.c. in
DKO mice, and treated with i.v. PBMC (7.5 x 106, once a week for 3 weeks) and
i.v. HER2-
BsAb (100 g, twice per week for 6 weeks). Tumor growth was completely
suppressed in the
group treated with HER2-BsAb and PBMC (Fig. 32D). Interestingly, despite
characterization of
the M37 PDX as IHC 3+, which is a HER2 level indicative of suitability for
treatment with
-142-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
trastuzumab (see, e.g., Wolff et al., Journal of Clinical Oncology, 2013,
31(31):3997-4013),
tumor growth was not suppressed in the M37 PDX group treated with PBMC +
trastuzumab +
huOKT3 (Fig. 32E). Taken together, these experiments showed that HER2-BsAb was
effective
against early passaged HER2(+) human tumor specimens.
6.3.4 CONCLUSIONS
[00392] This example described a HER2-specific BsAb with potent T cell
mediated anti-
tumor activity in vitro and in vivo, ablating tumors or delaying tumor growth
in four separate
tumor-human PBMC compartment models. Unlike monovalent bispecifics, this HER2-
BsAb
had identical anti-proliferative capacity to its parental trastuzumab. Its
serum half-life and area
under the curve were similar to IgG (data not shown). Most importantly, the T
cell-mediated
cytotoxicity it induced was relatively insensitive to inhibition by the PD-
1/PD-L1 pathway, not
previously described for this IgG-scFv platform (Xu et al., Cancer Immunology
Research 2015;
3:266-77). To date, other than the anti-GD2 hu3F8-BsAb (Xu et al., Cancer
Immunology
Research 2015; 3:266-77), no published T-cell redirecting bispecific
antibodies have used this
format. The ability of this IgG-scFv antibody platform to recruit circulating
lymphocytes into
the tumor stroma is critical, given the importance of tumor-infiltrating
lymphocyte (TIL) cells for
a successful anti-tumor effect in most checkpoint blockade studies to date
(Tumeh et al., Nature
2014; 515:568-71), distinguishing responders from nonresponders (Gajewski et
al., Semin Oncol
2015; 42:663-71).
[00393] Schreiber has proposed that tumor cells evolve to evade the immune
system through a
process termed "immuno-editing". Broadly speaking, this process occurs at two
levels: by
changes within the (1) tumor cells or (2) the tumor microenvironment. Tumor
cells can evade T-
cell responses by down-regulating MHC/peptide complexes or by decreasing tumor-
antigen
expression or through the loss of antigen presenting machinery components. On
the other hand,
suppression of the immune response in the tumor microenvironment is the result
of T-regulatory
cells, Myeloid-derived suppressor cells, M2 macrophages (Diaz-Montero et al.,
Semin Oncol
2014; 41:174-84; Laoui et al., Frontiers in immunology 2014; 5:489; Nishikawa
& Sakaguchi,
Curr Opin Immunol 2014; 27:1-7), immuno-suppressive cytokines (including DO)
(Munn &
Mellor, Trends Immunol 2013; 34:137-43), immune checkpoint molecules (Callahan
et al.,
Frontiers in Oncology 2014; 4:385; Postow et al., J Clin Oncol 2015;
33(17):1974-82) and the
consumption of IL-2 (Schreiber et al., Science 2011; 331:1565-70).
-143-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
[00394] Immune checkpoint antibodies that target the CTLA-4 and PD-1/PD-L1
inhibitory
pathways are capable of reversing the inhibitory tumor-microenvironment and
producing
significant and long-lasting clinical responses (Farolfi et al., Melanoma
research 2012; 22:263-
70). However, these strategies are not effective against all tumor types and
their success is
limited to a subset of patients. Durable clinical responses to the CTLA-4
blockade were recently
correlated with tumor mutational load and the expression of antigenic tetra-
peptides that
resembled those found in viral and bacterial pathogens (Snyder et al., N Engl
J Med 2014;
371:2189-99). Clonal neoantigens were shown to elicit T cell immunoreactivity
and sensitivity
to blockade of the PD-1/PD-L1 axis (McGranahan et al., Science 2016;
351(6280):1463-9).
Based on these data, the pre-existence of CD8(+) T-cells in the tumor (TILs)
would be critical.
More importantly, IHC evidence of negative regulation of tumor infiltrating
lymphocytes (TIL)
by the PD-1/PD-L1 axis, was correlated with clinical response to checkpoint
blockade (Tumeh et
al., Nature 2014; 515:568-71).
[00395] As data continues to accumulate, a consensus is emerging that these
immune
modulations would likely be ineffective against tumors with low immunogenicity
because the
presence of tumor specific lymphocytes is required for their clinical
activity. Indeed, HER2 has
been linked to immune resistance (Seliger & Kiessling, Trends in Molecular
Medicine 2013;
19:677-84). This subset of patients, with "T-cell resistant" HER2(+) tumor
cells and/or
insufficient clonal frequency of tumor-specific T-cells, would likely not
benefit from immune
checkpoint blockade alone. The unique property of the HER2-BsAb described in
this example to
recruit T-cells of any specificity and direct them against established tumors
with relative
insensitivity to the PD-1 immune checkpoint pathway is of interest, as it
directly addresses the
known limitations of immune checkpoint blockade. In fact, preliminary in vivo
data showed no
additional benefit of PD-1 blockade to the HER2-BsAb therapeutic efficacy
(data not shown),
even though tumor PD-Li expression was up-regulated substantially following T
cell infiltration
(Fig. 30E).
[00396] When compared to the existing platforms that target HER2, HER2-BsAb
offers
advantages. Shalaby and colleagues described the development of a bispecific
(Fab')2 antibody
(anti-HER2 Fab' x anti-CD3 Fab') through expressing each Fab' separately and
ligating the two
together by chemical conjugation (Shalaby et al., J Exp Med 1992; 175:217-25).
More recently,
Junttila and colleagues developed a heterodimeric bispecific IgG (anti-HER2 x
anti-CD3) using
-144-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
"knob-and-hole" format (Junttila et al., Cancer Research 2014; 74:5561-71).
Both formats have
monovalent binding to either HER2 or CD3, and are substantially different,
both structurally and
functionally, when compared to the HER2-BsAb described herein for the
following reasons.
First, the bivalent binding to HER2 is critical for the anti-proliferation
capability, which is
preserved in the HER2-BsAb construct described herein (Fig. 26C) but not in
those two
monovalent systems, as demonstrated by Juntala et al. Juntala et al. showed
that the anti-
proliferation capability of monovalent binding to HER2 (either heterodimeric
bispecific IgG or
trastuzumab-Fab) was 10-fold lower than bivalent trastuzumab (Junttila et al.,
Cancer Research
2014; 74:5561-71). Without being bound by any particular theory, it is
hypothesized that the
dual mechanism (anti-proliferation plus T cell cytotoxicity) may create
synergism and partly
explains the potent efficacy of the HER2-BsAb in vivo. This may provide a
salvage option for
patients who progress on standard HER2-based therapies, or a replacement for
trastuzumab
given its dual anti-proliferative and T cell retargeting properties. Second,
the bivalent binding to
HER2 in our BsAb maintains high avidity (Fig. 26B) so as to maximize tumor
binding, while the
monovalent binding to HER2 (either heterodimeric bispecific IgG or trastuzumab-
Fab) is 10-fold
lower than trastuzumab (Junttila et al., Cancer Research 2014; 74:5561-71).
Higher avidity
results in higher T cell dependent cell cytotoxicity, a phenomenon that has
been demonstrated in
T cell engaging bispecific antibodies (Ahmed et al., OncoImmunology 2015;
4:e989776).
Additionally, without being bound by any particular theory, it is hypothesized
that the high
avidity of the HER2-BsAb contributes to overcoming PD-1/PD-L1 checkpoints
(Fig. 29),
whereas the monovalent system by Juntala was shown to be inhibited by the PD-
1/PD-L1 axis.
Third, the BsAb described in this example has the trastuzumab IgG backbone,
preserving its
pharmacologic advantages, while Shalaby's construct doesn't have FcR(n)
affinity, should have
much shorter serum half-life, and probably needs to be administered as a
continuous infusion (as
for Blinatumomab) to be effective in vivo. Fourth, the other advantage is
manufacturability:
once a CHO stable line established, the HER2-BsAb can be produced in large
scale and purified
like normal IgG without significant aggregation despite prolonged incubation
at 37 C, while
chemical conjugates require more complicated syntheses and downstream
processing ¨ each
Fab' expressed and purified separately, chemically modified, and then the two
chemically
conjugated and repurified. To ensure a final product that is pure and
chemically stable for direct
clinical infusion is technically challenging and costly. Such chemically
crosslinked reagents
-145-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
have only been feasible for ex vivo arming of T cells, but not for direct
parenteral injections in
the clinic (Lum & Thakur A, BioDrugs 2011; 25:365-79).
[00397] A primary goal was to build a BsAb that has the bivalent binding to
tumor targets (to
preserve high avidity and/or anti-proliferation capability) and the monovalent
binding to CD3 on
effector T cells (to minimize spontaneous T cell activation in the absence of
tumor targets). A
number of uniquely different bivalent formats were surveyed, including
chemical conjugation
(Yankelevich et al., Pediatr Blood Cancer 2012; 59:1198-205), dual-variable-
domain (DVD), or
attaching huOKT3 scFv to different positions in the IgG backbone (C-terminal
of heavy chain or
C-terminal of light chain) (Kontermann, MAbs 2012; 4), and it was found that
the last option
gave the best functionality. Although the HER2-BsAb has anti-CD3 scFv attached
to both light
chains, its reaction with CD3 on T cells was considered as functionally
monovalent primarily for
the following reasons. First, although the HER2-BsAb format contains two anti-
CD3 scFvs
positioned at the end of the light chains, these scFvs are oriented in
geometrically opposed
directions which restrict their ability to cooperatively bind to neighboring
CD3 on T cells. This
restricts the BsAb from binding bivalently and hence results in functional
monovalency to CD3.
It has previously been shown in a different BsAb format that geometrical
restriction of two anti-
CD3 scFv can result in functionally monovalent binding to T cell and lower
cytokine release
(Ahmed et al., OncoImmunology 2015; 4:e989776). Second, the functional
consequence of
bivalent binding to CD3 on T cells is the triggering of spontaneous T cell
activation, hence
strong cytokine release in the absence of tumor targets. As shown in Fig. 33A,
HER2-BsAb only
stimulated background cytokine release similar to that of the monovalent
huOKT3 Fab, while
bivalent huOKT3 IgG induced substantially more cytokines in the absence of
tumor targets (left
panel). However, in the presence of HER2(+) NCI-N87 tumor target, anti-tumor
TH1 cytokines
(TNFa and IFNy) were released but only in the presence of BsAb (right panel),
a format
previously shown to induce immunologic synapse formation between the T cells
and tumor
targets (Xu et al., Cancer Immunology Research 2015; 3:266-77). Furthermore,
only bivalent
huOKT3 IgG induced robust T cell proliferation, while HER2-BsAb and monovalent
huOKT3
Fab had negligible effects comparable to the T cells only Control (Fig. 33B).
In addition, the
aglycosylation of the Fc removed both ADCC and most CMC functions, thereby
further reducing
cytokine release without affecting serum pharmacokinetics or compromising T
cell activation.
-146-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
[00398] Chimeric antigen receptor technology has rapidly accelerated
investigations into
HER2-directed gene modified T cells in several clinical trials: NCT00902044
(for sarcoma),
NCT00889954 (all HER2(+) cancers), NCT01109095 (GBM), NCT00924287 (metastatic
cancer), and NCT01935843 (HER2(+) solid tumors). Toxicities from off target
effects were
initially concerning (Morgan et al., Mol Ther 2010; 18:843-51), although
subsequent patients
have been safely managed pharmacologically. There, the ability of T cells to
overcome low
levels of antigen expression was again observed. Osteosarcoma was a good
example where the
expression level has been controversial (Thomas et al., Clin Cancer Res 2002;
8:788-93), and
where CAR-modified T cells were highly efficient against locoregional and
metastatic
xenografts (Ahmed et al., Mol Ther 2009; 17:1779-87), and against osteosarcoma
tumor
initiating cells (Rainusso et al., Cancer Gene Ther 2012; 19:212-7). Although
the successful
clinical application of CAR T cells has reassured many skeptics, there remain
obstacles,
including the necessity of cytoreductive chemotherapy prior to T cell infusion
for meaningful
clinical responses, logistics of cell harvest, processing, storage, transport
and product release, T
cell exhaustion (Long et al., Nat Med 2015; 21:581-90) and inadequate T cell
persistence after
infusion.
[00399] In summary, this example demonstrates a successful IgG-scFv platform
to engage T
cells for HER2-directed immunotherapy. This BsAb for retargeting T cells was
built with
structural considerations for bivalency towards the target, and functionally
monovalency towards
CD3 on effector T cells, plus Fc aglycosylation for minimal spontaneous
cytokine release. Its
relative insensitivity to the PD-1/PD-L1 axis was novel. It has excellent anti-
tumor activity both
in vitro and in vivo, which is superior to trastuzumab.
6.4 EXAMPLE 4
[00400] This example demonstrates that HER2-BsAb described in section 6.3.3.1
above
(comprising a heavy chain consisting of the amino acid sequence set forth in
SEQ ID NO: 62 and
a light chain fusion polypeptide consisting of the amino acid sequence set
forth in SEQ ID NO:
60) is effective against HER2(+) breast cancer cell line xenografts that
express PDL1 but are
resistant to PD1 or PDL1 treatment (Fig. 34). See Section 6.3.2.8 for
materials and methods. In
particular, 5 x 106 HCC1954 xenografts were implanted subcutaneously in mice
and mice were
treated intravenously with 7.5 x 106 PBMC once per week for two weeks and
intravenously with
HER2-BsAb, anti-PD1 antibody Pembrolizumab, or anti-PDL1 antibody
Atezolizumab. HER2-
-147-
CA 03051484 2019-07-24
WO 2018/140026 PCT/US2017/015278
BsAb, anti-PD1 antibody, and anti-PDL1 antibody treatments were performed with
100 [tg each,
twice per week for 4 weeks. HER2-BsAb-treated tumors were completely
eradicated. In
contrast, there was no effect on tumors treated with PD1/PDL1 blockade (i.e.,
treatment with
anti-PD1 antibody Pembrolizumab or anti-PDL1 antibody Atezolizumab).
7. EQUIVALENTS
[00401] The invention is not to be limited in scope by the specific
embodiments described
herein. Indeed, various modifications of the invention in addition to those
described will become
apparent to those skilled in the art from the foregoing description and
accompanying figures.
Such modifications are intended to fall within the scope of the appended
claims.
[00402] All references cited herein are incorporated herein by reference in
their entirety and
for all purposes to the same extent as if each individual publication or
patent or patent
application was specifically and individually indicated to be incorporated by
reference in its
entirety for all purposes.
-148-