Note: Descriptions are shown in the official language in which they were submitted.
CA 03058584 2019-09-30
1
METHOD FOR INDUCING EXON SKIPPING BY GENOME EDITING
TECHNICAL FIELD
[0001]
The present invention relates to a gene recombination technique, and relates
to a method of inducing exon skipping by genome editing, which method is
useful in
the field of research and the field of medicine.
BACKGROUND ART
[0002]
Duchenne muscular dystrophy (hereinafter referred to as DMD) is a disease
causing atrophy of muscle fibers due to loss of function of the dystrophin
gene. The
skeletal muscle isoform (Dp427m) of the dystrophin gene is constituted by 79
exons.
In cases where shifting of the reading frame occurs in this gene due to
partial deletion
of the exons or the like, normal production of the dystrophin protein becomes
impossible, leading to development of DMD (Non-patent Document 1).
[0003]
Aiming at radical cure of DMD, various studies have been carried out for
gene therapies in which a functional exogenous dystrophin gene is added to
replace a
dysfunctional endogenous dystrophin gene. Since the full-length cDNA of
dystrophin has a size of as long as 14 kb, attempts are being made to reduce
its size
by elimination of unnecessary domain portions, and to introduce the thus
prepared
minidystrophin or microdystrophin into muscular tissue using various vectors
for
gene transfer (AAV, lentivirus, Sleeping Beauty transposon vectors, and the
like).
However, since introduction of a huge gene is difficult, no effective
therapeutic
method has been established so far.
[0004]
In a study in progress, an antisense oligonucleotide is used to prevent
reading
CA 03058584 2019-09-30
2
of part of a particular exon during splicing of mRNA in order to restore
dystrophin
having the normal function (exon skipping). However, since the antisense
oligonucleotide is only temporarily effective, a method for repairing the gene
itself
has been demanded for the radical cure.
[0005]
As methods for repairing the gene itself, genome editing techniques such as
TALEN and CRISPR-Cas systems have recently been developed. In these
techniques, a particular sequence position is recognized in the genome
sequence, and
then DNA double-strand break is induced to cause local induction of a DNA
repair
mechanism through non-homologous recombination (non-homologous end joining,
NHEJ) or homologous recombination (homology directed repair, HDR), thereby
enabling addition of a base(s) to or deletion of a base(s) from the cleaved
site.
[0006]
For CRISPR-Cas genome editing techniques, the type II and type V CRISPR
systems of bacteria and archaebacteria are widely used. They can bind to a
target
DNA dependently on a spacer sequence contained in a guide RNA (gRNA or sgRNA),
to induce a double-strand DNA break by the action of a Cas nuclease (Cas9 in
cases
of type II, and Cpfl in cases of type V). In the type II CRISPR system, the
guide
RNA is a complex containing crRNA and tracrRNA, or an sgRNA (single guide
RNA) containing crRNA and tracrRNA linked to each other.
[0007]
It has been reported that exon skipping for dystrophin using a genome editing
technique was carried out in myoblasts [Non-patent Documents 2 and 3] or at
the
mdx mouse level [Non-patent Documents 4 to 7].
[0008]
In these studies, both ends of the exon to be skipped are cleaved using two
guide RNAs, to induce a large deletion including the whole exon. However, such
a
CA 03058584 2019-09-30
3
method increases the risk of non-specific cleavage since two gRNAs are
required.
Moreover, cleavage by only one of the gRNAs cannot induce exon skipping, and
the
two gRNA sequences need to act in the same genome. Moreover, at least several
hundred bases need to be deleted, and, in cases where the region contains an
unknown regulatory region or miRNA-coding region, there is a risk of
occurrence of
unexpected side effects.
[0009]
On the other hand, the present inventors have previously reported that, by
using a genome editing technique such as TALEN or CRISPR-Cas9 in iPS cells
derived from DMD patients, a dystrophin gene mutation can be repaired by (1)
exon
skipping, (2) frameshift induction, and (3) knock-in of a deleted exon [Non-
patent
Document 8]. Among these, the method of (1) employs a method in which the
splice acceptor of the exon is deleted. Since exon skipping can be
sufficiently
induced in cases where deletion of several bases to several ten bases can be
induced
with one gRNA, the method is superior to the methods reported by [Non-patent
Documents 2 to 7] in terms of the three facts: a higher efficiency, a smaller
risk of
side-effect mutations, and requirement of only small DNA base deletion.
On the other hand, although a splice acceptor is an attractive target site for
induction of exon skipping, it contains a polypyrimidine sequence (consecutive
T/C's), and similar sequences are contained in a large number of exon
sequences.
Therefore, the method has a problem in that a gRNA having a high specificity
cannot
be easily designed. The double-nicking method, in which the specificity is
increased by combination of two units of nickase-modified CRISPR-Cas
containing a
mutation introduced into the DNA cleavage domain of CRISPR-Cas such that a
single-strand break rather than a DNA double-strand break is induced [Mali P
et al.,
Nat Biotechnol. 2013 Sep; 31(9): 833-8.] [Ran FA et al., Cell. 2013 Sep 12;
154(6):
1380-9.], is known. Since type V AsCpfl is known to have a higher specificity
than
CA 03058584 2019-09-30
4
Type II SpCas9 in human cells [Kleinstiver BP et al., Nat Biotechnol, 2016
Aug;
34(8): 869-74.1 Kim D et al., Nat Biotechnol, 2016 Aug; 34(8): 863-8.], it is
thought
that the problem of the specificity can be avoided by targeting a site near
the splicing
acceptor using the double-nicking method or Cpfl
It is also known that the cleavage activity and the cleavage length vary
depending on the spacer sequence in the guide RNA and the type of the CRISPR-
Cas,
and guide sequences and design methods that enable efficient induction of exon
skipping have been empirically unknown.
PRIOR ART DOCUMENTS
[Non-patent Documents]
[0010]
[Non-patent Document 1] Pichavant et al., Mol Then 2011 May; 19(5): 830-40.
[Non-patent Document 2] Ousterout DG et al., Nat Commun. 2015 Feb 18; 6: 6244
[Non-patent Document 3] Iyombe-Engembe JP Mol Ther Nucleic Acids. 2016 Jan
26; 5: e283.
[Non-patent Document 4] Xu Let al., Mol Then 2016 Mar; 24(3): 564-9.
[Non-patent Document 5] Long C et al., Science, 2016 Jan 22; 351(6271): 400-3.
[Non-patent Document 6] Nelson CE et al., Science. 2016 Jan 22; 351(6271): 403-
7
[Non-patent Document 7] Tabebordbar M et al., Science. 2016 Jan 22; 351(6271):
407-11
[Non-patent Document 8] Li HL et al., Stem Cell Reports. 2015 Jan 13; 4(1):
143-54.
SUMMARY OF THE INVENTION
[0011]
An object of the present invention is to provide an efficient method of exon
skipping. Another object of the present invention is to provide a simple
method of
evaluation of exon skipping.
[0012]
CA 03058584 2019-09-30
The present inventors intensively studied to solve the above problems. The
present inventors discovered that, by using, as a target gene for exon
skipping, a
marker gene containing a sequence which is inserted in a coding region and
which
contains a first intron, an exon to be analyzed, and a second intron, and by
designing
5 the marker gene such that the marker gene functions when the exon to be
analyzed is
skipped, the exon skipping can be efficiently analyzed based on the phenotype
of the
marker gene. As a result, the present inventors discovered that, when targeted
exon
skipping is carried out for a gene of interest in a genome using CRISPR-Cas
and
guide RNA, by arranging the guide RNA such that the site of cleavage by the
CRISPR-Cas is positioned within 80 bases from the splice acceptor site or the
splice
donor site of the target exon, the efficiency of the exon skipping can be
increased,
thereby completing the present invention.
[0013]
More specifically, the present invention provides the followings.
[1] A method of skipping a target exon of a gene of interest in a genome,
comprising
using CRISPR-Cas and guide RNA, wherein the guide RNA contains a spacer
sequence such that the site of cleavage by the CRISPR-Cas is positioned within
80
bases from the splice donor site immediately before the target exon or the
splice
acceptor site immediately after the target exon.
[2] The method according to [1], wherein two or more kinds of the guide RNA
are
used.
[3] The method according to [1], wherein the CRISPR-Cas is a nickase-modified
Cas
containing a substitution in the nuclease activity residue in the RuvC domain,
and
wherein a guide RNA for the sense strand and a guide RNA for the antisense
strand
of the gene of interest are used, the guide RNAs containing spacer sequences
such
that the cleavage site in the sense strand of the gene of interest and the
cleavage site
in the antisense strand of the gene of interest are both positioned within 80
bases
CA 03058584 2019-09-30
6
from the splice donor site immediately before the target exon or the splice
acceptor
site immediately after the target exon.
[4] The method according to any one of [1] to [3], wherein the CRISPR-Cas is
Cas9.
[5] The method according to [4], wherein the Cas9 is derived from
Streptococcus
pyogenes, or derived from Staphylococcus aureus.
[6] The method according to any one of [1] to [3], wherein the CRISPR-Cas is
Cpfl .
[7] The method according to [6], wherein the Cpfl is derived from
Acidaminococcus
sp. BV3L6, or derived from Lachnospiraceae.
[8] The method according to any one of [1] to [7], wherein the gene of
interest is a
human dystrophin gene.
[9] The method according to [8], wherein the target exon is exon 45.
[10] The method according to [9], wherein the guide RNA contains a spacer
sequence
having the base sequence of bases from 17 to 36 in the base sequence of any of
SEQ
ID NOs:17 to 42, the base sequence of bases from 17 to 39 in the base sequence
of
any of SEQ ID NOs:44 to 45, or the base sequence of bases from 24 to 43 in the
base
sequence of any of SEQ ID NOs:50 to 53.
[11] A reagent for skipping a target exon of a gene of interest in a genome,
comprising CRISPR-Cas and guide RNA, wherein the guide RNA contains a spacer
sequence such that the site of cleavage by the CRISPR-Cas is positioned within
80
bases from the splice donor site immediately before the target exon or the
splice
acceptor site immediately after the target exon.
[12] A method of evaluating exon skipping, comprising using a marker gene
containing a sequence which is inserted in a coding region and which contains
a first
intron, an exon to be analyzed, and a second intron, wherein the marker gene
is
designed such that the marker gene functions when the exon to be analyzed is
skipped.
[13] The method according to [12], wherein the exon skipping is exon skipping
using
CA 03058584 2019-09-30
7
CRISPR-Cas and guide RNA.
[14] The method according to [12] or [13], wherein a transposon vector is used
for
inserting the marker gene into the genome of a cell to be analyzed.
[15] The method according to any one of [12] to [14], wherein the marker gene
is a
luciferase gene.
[16] The method according to any one of [12] to [15], wherein the exon to be
analyzed is exon 45 of a human dystrophin gene.
EFFECTS OF THE INVENTION
[0014]
According to the method of the present invention, the efficiency of exon
skipping can be increased in exon skipping utilizing a genome editing
technique, so
that the method is effective for treatment of diseases and the like. Further,
by using,
as a target gene for the exon skipping, a marker gene containing a sequence
which is
inserted in a coding region and which contains a first intron, an exon to be
analyzed,
and a second intron, and by designing the marker gene such that the marker
gene
functions when the exon is skipped, the exon skipping can be efficiently
analyzed
based on the phenotype of the marker gene.
BRIEF DESCRIPTION OF THE DRAWINGS
[0015]
Fig. 1 shows the principle of repair of dystrophin protein by a genome editing
exon skipping method.
Fig. 2 shows that the sequence specificity of the splice acceptor sequence
region is generally low, and that similar sequences are present also in other
regions in
the genome, indicating that designing of a highly specific CRISPR guide RNA
and
the like therefor is difficult. Among arbitrary DNA sequences having lengths
of 10
bases to 16 bases, unique sequences (unique k-mer) present only at one
position in a
human genome sequence (hg19) were used to prepare a database, and the
distribution
CA 03058584 2019-09-30
8
. .
of the unique sequences was plotted in terms of the relative positions in all
exons of
the human RefSeq genes. It can be seen that the inside of human exons
generally
shows accumulation of unique sequences and hence high specificity, but that
the
splice acceptor portion has very low specificity.
Fig. 3 shows part of the sequence of a reporter vector for detection of the
exon
skipping efficiency for the dystrophin gene. The sequence was designed by
finding
"CAGIG", which is the sequence most frequently found as an exon¨exon junction,
in
the middle of cDNA of the firefly luciferase gene, and then placing, at this
position, a
sequence around the splicing donor (SD) immediately after exon 44 of the
dystrophin
gene ("+" symbols), exon 45 (">" symbols) and intron sequences before and
after it,
and a splice acceptor (SA) sequence immediately before exon 46 ("+" symbols),
in
that order. The restriction enzyme sites (Nan, AgeI, and Sall) used for the
construction of the vector are indicated with "#" symbols.
Fig. 4 shows alteration of the Luc sequence by introduction of a mutation,
which alteration was carried out such that splicing occurs only at the
inserted
dystrophin sequence portion. Panel (a) shows the result of observation of the
splicing pattern, which observation was carried out by introducing the exon
skipping
vector of Fig. 3 into 293T cells, extracting mRNA therefrom, and then
performing the
Sanger sequencing method. As a result, it was found that a pseudo-splicing
donor
sequence is present in the middle (at the 31th base in Fig. 3) of the Luc
gene, causing
unexpected splicing (the presence of an extra wave in the sequence
electrogram).
Panel (b) shows the result of an analysis that was carried out by obtaining
the entire
exon sequences of the dystrophin gene from Ensemble BioMart, and then
analyzing
the consensus sequences of the splicing donor portions and the splice acceptor
portions using WebLogo. As a result, trends of base sequences similar to those
of
common human gene base sequences were found, and it could be confirmed that
the
"GT" sequence is conserved with the highest frequency among splicing donors,
and
CA 03058584 2019-09-30
9
that "AG" is conserved with the highest frequency among splice acceptor
sequences.
Panel (c) shows alteration of "G" at the 967th position as counted from the
first base
of the Luc cDNA (the 31st base in Fig. 3) to "A", which alteration was carried
out in
order to prevent the pseudo-splicing donor sequence of (a) from functioning.
By
this, the Val amino acid at the 323rd position of the Luc protein was changed
to Ile.
Fig. 5 shows the result of analysis of the crystal structure of the firefly
luciferase protein (PDB code: 1BA3) for prediction of the influence of the
amino
acid modification site on the activity of the firefly luciferase. Since the
Val residue
at the 323th position (square) was sufficiently distant from the active-center
residue
(circle) [Branchini BR et al., JBC, 1997], it was expected that there may be
no direct
interaction, and that the modification of this amino acid may hardly influence
the
enzyme activity. Since the Val residue is positioned in the middle of the a-
helix, it
was altered to an Ile residue, which is less likely to change the a-helix
structure, and
which has a similar structure and chemical properties.
Fig. 6 shows the results of investigation of the influence of the G967A
(V323I) mutation on splicing and the Luc activity. Panel (a) shows Luc
expression
cassettes used for comparative analysis. All of these were prepared by
insertion into
the piggyBac vector (System Biosciences) pPV-EFla-GW-iP-A. * represents the
G967A (V323I) mutation; each dotted line represents an intron; and each box
represents an exon. Panel (b) shows the results of analysis of the splicing
pattern,
which analysis was carried out by introducing each vector into 293T cells,
extracting
mRNA therefrom, and then performing PCR with primers including an intron
portion.
Lane 2, which is for a case where the intron was inserted into Lu2, showed a
band
indicating remaining of the intron (468 bp) as well as a band (377 bp)
indicating
occurrence of the expected splicing. Lane 4, which is for a case where exon 45
of
dystrophin was inserted, showed the same trend, showing a band of 1166 bp
including the intron as well as the band of 468 bp which indicates occurrence
of the
CA 03058584 2019-09-30
expected splicing. Lane 3 and lane 5, which are for cases where the point
mutation
(G967A (V323I) mutation) was introduced into the Luc2 cDNA, indicated the fact
that the splicing occurred more efficiently in each of them. Panel (c) shows
that the
introduction of the G967A (V323I) mutation into the Luc2 cDNA hardly
influenced
5 the luciferase activity (based on comparison between No. 2 and No. 3). On
the
other hand, in the Luc2 vector in which dystrophin exon 45 is inserted, as the
splicing
efficiency increases to allow higher incorporation of exon 45 into Luc2, the
luciferase
activity is expected to decrease due to occurrence of a frameshift in Luc2.
Since the
background level of the luciferase activity decreased due to the introduction
of the
10 G967A (V323I) mutation, highly sensitive detection of low-frequency exon
skipping
became possible.
Fig. 7 shows vectors having various lengths which were constructed for the
purpose of investigation of the influence of the lengths of the introns before
and after
exon 45 on the splicing. A luciferase reporter is loaded on the piggyBac
vector, and,
by introducing this vector together with a piggyBac transposase expression
vector,
stable incorporation of the luciferase reporter into the chromosome of the
host cell is
possible. Further, a puromycin resistance gene is also loaded following IRES
so
that only cells with the reporter vector introduced can be enriched.
Fig. 8a shows the results of analysis of the splicing pattern carried out by
transfecting 293T cells with the exon skipping vectors (Luc2 G967A) of Fig. 7,
extracting mRNA from the cells two days later, and then performing PCR
analysis.
The unspliced band (1166 bp) tended to become weak as the lengths of the
introns
before and after exon 45 (0.7 to 4.0 kb) increased. However, in all cases, a
band
indicating efficient splicing (468 bp) was found. Further, gRNA1 and an SpCas9-
2 5 expressing plasmid were simultaneously introduced to induce exon
skipping (exon
skipping "+"). As a result, in the reporters with any intron length, a band
indicating
induction of skipping (377 bp) was found.
CA 03058584 2019-09-30
11
Fig. 8b shows the results of measurement of the induction efficiency of exon
skipping, which measurement was carried out by a luciferase assay. Regarding
the
reporter vector in which the G967A point mutation was not introduced (Luc2
+hEx45
(0.7 kb)), the background level was high even without induction of exon
skipping,
and no difference was found between the values before and after the induction.
In
contrast, regarding the exon skipping vectors into which the G967A mutation
was
introduced, exon skipping was induced by introduction of a vector expressing
SpCas9 and sgRNA-DMD1, so that increases in the luciferase activity could be
found.
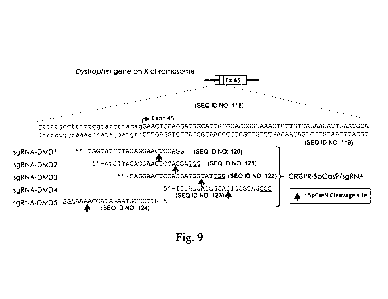
Fig. 9 shows target sequences of CRISPR SpCas9 gRNA for the splice
acceptor site of exon 45 in the human dystrophin gene.
Fig. 10 shows the target DNA cleavage activities obtained using CRISPR-
SpCas9 and CRISPR-SpCas9(D10A) in human 293T cells as measured by an SSA
assay. In the cases of CRISPR-SpCas9, the cleavage activity was found with any
of
the guide RNAs 1 to 5. On the other hand, in the cases where the nickase-
modified
SpCas9(D10A) was used, no cleavage activity, as expected, was found when only
a
single guide RNA was used. A high DNA cleavage activity could be found in the
case where the guide RNA 5 for the sense strand and the guide RNA 4 for the
antisense strand were used in combination.
Fig. 11 shows cleavage patterns as analyzed by cleaved-sequence analysis.
Plasmid DNAs expressing CRISPR-Cas9 and its guide RNA were introduced into
DMD-iPS cells by electroporation, and genomic DNA was extracted. Thereafter,
the genomic DNA cleavage pattern was analyzed using a MiSeq sequencer. The
results are shown as line charts drawn by stacking positions having a base
deletion.
Fig. 12 shows the results of measurement of the exon skipping efficiencies of
sgRNA-DMD 1 to 5 in 293T cells using Luc2 (G967A) +hEx45 (0.7 kb) as an exon
skipping reporter. As a result, increased luciferase activities were found for
CA 03058584 2019-09-30
12
sgRNA-DMD I, sgRNA-DMD4, and sgRNA-DMD2 with significant differences at
P<0.01 compared to a sample without gRNA.
Fig. 13a shows target sequences of gRNAs derived from various kinds of
CRISPR-Cas (SpCas9, SaCas9, and AsCpfl), which were designed for positions
near
the splice acceptor site of dystrophin exon 45. The PAM sequence is indicated
with
an underline.
Fig. 13b shows a schematic view of an AsCpfl gRNA expression cassette. It is
known that G (or A) is desirable as the transcription start site (TSS) of the
H1
promoter. It was thus expected that, by increasing the number of G's from one
base
to three bases, the expression level of the gRNA may be increased, resulting
in an
increased target DNA cleavage efficiency.
Fig. 13c shows the results obtained by introducing AsCpfl and gRNA into
293T cells, and then measuring the target DNA cleavage activity by an SSA
assay.
First, by increasing the number of G's at the transcription start site (TSS)
from one
base to three bases, an increased cleavage activity could be found. Further,
AsCpfl-
gRNA-DMD1 and AsCpfl-gRNA-DMD2, which have TTTT as the PAM sequence,
had very low cleavage activities, but AsCpfl-gRNA-DMD3 and AsCpfl-gRNA-
DMD4, which have TTTG (TTTV) as the PAM, showed cleavage activities
equivalent to or higher than that of SpCas9-gRNA-DMD1.
Fig. 13d shows the exon skipping efficiencies measured using Luc2 (G967A)
+hEx45 (0.7 kb) as an exon skipping reporter in 293T cells, which measurement
was
carried out for cases where the double nicking method, SaCas9/gRNA, or
AsCpfl/gRNA was used. As a result, in the case where sgRNA-DMD4 and
sgRNA-DMD5 were used as a pair in the SpCas9(D10A) double nicking method, a
higher exon skipping activity was induced compared to SpCas9/ sgRNA-DMD1.
Further, also in the cases where sgRNA-DMD3 or sgRNA-DMD4 of AsCpfl was
used, high exon skipping efficiencies could be induced.
CA 03058584 2019-09-30
13
Fig. 14a shows 26 kinds of gRNAs prepared for target sites of SpCas9-gRNA
which can be designed in dystrophin exon 45.
Fig. 14b shows a list of the target sequences and the spacer sequences of
sgRNA-DMD1 to 26 used in Fig. 14a. Each gRNA was introduced into 293T cells
together with an SpCas9 expression vector and a Luc2 (G967A) +hEx45 (0.7 kb)
exon skipping reporter vector, and a T7E1 assay was carried out using the
following
primers: (Luc2-Fwd-Splice: TGCCCACACTATTTAGCTTC (SEQ ID NO:1), Luc2-
Rev-Splice: GTCGATGAGAGCGTTTGTAG) (SEQ ID NO:2)), to measure the
DNA cleavage activity for the target site of each gRNA in the reporter vector
(T7EI
Indel activity [%]). In addition, the exon skipping efficiency was also
measured
(Exon skipping Luc activity [A.U.]).
Fig. 14c shows the results of measurement of the exon skipping efficiencies
of sgRNA-DMD 1 to 26 in 293T cells using Luc2 (G967A) +hEx45 (0.7 kb) as an
exon skipping reporter. As a result, it was found that gRNAs targeting
positions
near the splice acceptor, and gRNAs targeting positions near the splicing
donor, of
exon 45 have high exon skipping efficiencies.
Fig. 15 shows the results of evaluation of the exon skipping efficiencies
using
SpCas9 in the same manner as in Fig. 14b, which evaluation was carried out for
combinations of two kinds of sgRNAs used in Fig. 14b.
EMBODIMENTS FOR CARRYING OUT THE INVENTION
[0016]
The method of the present invention is a method of skipping a target exon of a
gene of interest in a genome, comprising using CRISPR-Cas and guide RNA,
wherein the guide RNA contains a spacer sequence such that the site of
cleavage by
the CRISPR-Cas is positioned within 80 bases from the splice donor site
immediately
before the target exon or the splice acceptor site immediately after the
target exon.
[0017]
CA 03058584 2019-09-30
14
,
<CRISPR-Cas Systems>
As CRISPR systems, class 1, which acts as a complex formed by a plurality
of factors, and class 2, which acts even as a single factor, are known.
Examples of
class 1 include type I, type DI, and type IV, and examples of class 2 include
type II,
type V, and type VI (Makarova KS et al., Nat Rev Microbiol. 2015 1Mohanraju P
et
al., Science, 2016). At present, for use in genome editing in mammalian cells,
class
2 CRISPR-Cas, which acts as a single factor, is mainly used. Representative
examples of the class 2 CRISPR-Cas include type II Cas9 and type V Cpfl.
[0018]
As a CRISPR-Cas9 system, class 2 type 11 Cas9 derived from Streptococcus
pyogenes, which is widely used as a genome editing tool, may be used. Class 2
type
II Cas9 systems derived from other bacteria have also been reported, and, for
example, Cas9 derived from Staphylococcus aureus (Sa), Cas9 derived from
Neisseria meningitidis (Nm), or Cas9 derived from Streptococcus thermophilus
(St)
may also be used.
[I]
An updated evolutionary classification of CRISPR-Cas systems.
Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou
R, Brouns SJ, Charpentier E, Haft DH, Horvath P, Moineau S, Mojica FJ, Terns
RM,
Terns MP, White MF, Yalamin AF, Garrett RA, van der Oost J, Backofen R, Koonin
EV.
Nat Rev Microbiol. 2015 Nov; 13(11): 722-36. doi: 10.1038/nrmicro3569.
[11]
Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas
systems.
Mohanraju P. Makarova KS, Zetsche B, Zhang F, Koonin EV, van der Oost J.
Science. 2016 Aug 5; 353(6299):aad5147. doi: 10.1126/science.aad5147
[illi
CA 03058584 2019-09-30
In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015 Apr 9;
520(7546): 186-91.
[IV]
Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat
Methods.
5 2013 Nov; 10(11): 1116-21.
[V]
Efficient genome engineering in human pluripotent stem cells using Cas9 from
Neisseria meningitidis. Proc Natl Acad Sci U S A. 2013 Sep 24; 110(39): 15644-
9.
[VI]
10 Streptococcus thermophilus CRISPR-Cas9 systems enable specific editing
of the
human genome. Mol Ther. 2016 Mar; 24(3): 636-44.
[0019]
Further, as a class 2 type V CRISPR-Cas system, Cpfl has been identified,
and it is reported that, by using Cpfl derived from Acidaminococcus sp. (As)
or Cpfl
15 derived from Lacluiospiraceae, genome editing in human cells is possible
dependently on the gRNA sequence. Thus, these kinds of CRISPR-Cpfl may also
be used.
[V]
Cpfl Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell.
2015 Oct 22; 163(3): 759-71
[VI]
Multiplex gene editing by CRISPR-Cpfl using a single crRNA array. Nat
Biotechnol. 2017 Jan; 35(1): 31-34.
[0020]
CRISPR-Cas9 has two nuclease domains, the RuvC domain and the HNH
domain, each of which is involved in cleavage of one strand of the double-
stranded
DNA. CRISPR-Cpfl has the RuvC domain and the Nuc domain. In cases where
CA 03058584 2019-09-30
= 16
Asp at the 10th position in the RuvC domain of Cas9 derived from Streptococcus
pyo genes is substituted with Ala (Dl OA), no cleavage occurs in the DNA
strand to
which the gRNA does not bind. In cases where His at the 840th position in the
HNH domain is substituted with Ala (11840A), no cleavage occurs in the DNA
strand
to which the gRNA binds [Jinek M et al., Science. 2012 Aug 17; 337(6096): 816-
21.].
This property was used for development of the double nicking (or paired
nickases)
method, in which nickases each of which cleaves only one strand of a double-
stranded DNA are used, in a state where they are positioned close to each
other, to
cleave the respective separate DNA strands, to induce a DNA double-strand
break in
a target region [Mali P et al., Nat Biotechnol. 2013 Sep; 31(9): 833-8.] [Ran
FA et al.,
Cell. 2013 Sep 12; 154(6): 1380-9.]. By this, genome editing such as targeting
by
insertion of an arbitrary sequence by knock-in became possible while the risk
of
inducing a sequence mutation at a site other than the target site was reduced
[WO
2014204725 Al].
Thus, in the method of the present invention, Dl OA Cas9 nickase may be used
as CRISPR-Cas9, and two kinds of guide RNAs for cleavage of the sense strand
and
the antisense strand, respectively, may be used (double nicking method). A
nickase-
type Cas9 or a nickase-type Cpfl applicable to the double nicking method can
be
obtained also by introducing a mutation to an active amino acid residue of the
RuvC
domain of a system other than the Cas9 derived from Streptococcus pyogenes.
[0021]
A DNA encoding the above CRISPR-Cas can be obtained by performing
cloning based on a sequence encoding CRISPR-Cas deposited in GenBank or the
like.
Further, a commercially available plasmid containing CRISPR-Cas may be
obtained
from Addgene or the like and used; a DNA encoding CRISPR-Cas may be obtained
by PCR using the plasmid as a template, or the DNA may be artificially
prepared
using an artificial gene synthesis technique known to those skilled in the
art. The
CA 03058584 2019-09-30
= 17
method of obtaining the DNA is not limited. A DNA encoding Cas nickase may be
obtained by introducing a mutation to an active amino acid residue of a
nuclease
domain of CRISPR-Cas by a known molecular biological technique, or may be
obtained by cloning from a plasmid or the like containing a CRISPR-Cas gene to
which a mutation was introduced in advance. Further, in order to increase the
expression efficiency of the CRISPR-Cas in the host, codon alteration may be
carried
out.
[0022]
The CRISPR-Cas may be introduced into the cell as mRNA, protein, or DNA.
The guide RNA may be introduced into the cell as RNA or DNA. In cases of
introduction using a vector, examples of the vector include vectors capable of
replicating in eukaryotic cells, vectors capable of maintaining an episome,
and
vectors that can be incorporated into the host cell genome. Examples of virus
vectors therefor include adenovirus vectors, retrovirus vectors, lentivirus
vectors,
Sendai virus vectors, and adeno-associated virus vectors. Examples of
transposon
vectors therefor include piggyBac vectors, piggyBat vectors, Sleeping Beauty
vectors,
Toll vectors, and LINE vectors. For treatment, introduction using a vector
showing
constant expression is not preferred because of an increased risk of side
effects.
Since treatment requires induction of DNA cleavage only immediately after the
administration, introduction as Cas9 mRNA/gRNA or Cas9 protein/gRNA,
introduction as an episomal vector, or the like is preferred.
The vector may contain a selection marker. The "selection marker" means a
genetic element that provides a selectable phenotype to the cell into which
the
selection marker is introduced. The selection marker is generally a gene whose
gene product gives resistance to an agent which inhibits growth of cells or
which kills
cells. Specific examples of the selection marker include the Puro resistance
gene,
Neo resistance gene, Hyg resistance gene, Bls gene, hisD gene, Gpt gene, and
Ble
CA 03058584 2019-09-30
18
gene. Examples of drugs useful for selecting the presence of selection markers
include puromycin for the Puro resistance gene, G418 for the Neo resistance
gene,
hygromycin for the Hyg resistance gene, blasticidin for the Bls gene,
histidinol for
hisD, xanthine for Gpt, and bleomycin for Ble.
[0023]
<Guide RNA>
A guide RNA (gRNA or sgRNA) is a complex containing tracrRNA and
crRNA, or tracrRNA and crRNA artificially linked to each other, in the CRISPR-
Cas9 method. [Jinek M et al., Science. 2012 Aug 17; 337(6096): 816-21.] In the
present invention, the guide RNA means a product prepared by linking a spacer
sequence having a sequence corresponding to the gene of interest to a scaffold
sequence.
As the scaffold sequence of the guide RNA of SpCas9, a known sequence, for
example, the sequence of
5'-GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAA
CTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTT-3' (SEQ ID NO:3)
can be used. Alternatively, an altered scaffold sequence (Chen B et al., Cell,
2013
Dec 19; 155(7): 1479-91)
5'-GTTTTAGAGCTATGCTGGAAACAGCATAGCAAGTTAAAATAAGGCTAGT
CCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTTTT-3' (SE
Q ID NO:4)
may be used.
The guide RNA does not require tracrRNA in CRISPR-Cpfl.
As the scaffold sequence of the guide RNA of AsCpfl, a known sequence, for
example, the sequence of 5'-GTAATTTCTACTCTTGTAGAT-3' (SEQ ID NO:5) or
5'-GGGTAATTTCTACTCTTGTAGAT-3' (SEQ ID NO:6) can be used. The DNA
may be, for example, artificially prepared using an artificial gene synthesis
technique
CA 03058584 2019-09-30
= 19
known to those skilled in the art. The method of obtaining the DNA is not
limited.
[0024]
In the method of the present invention, the spacer sequence of the guide RNA
is arranged such that the site of cleavage by the CRISPR-Cas is positioned
within 80
bases, preferably within 50 bases, more preferably within 30 bases, from the
splice
donor site immediately before the target exon or the splice acceptor site
immediately
after the target exon. It may also be designed for the exonic splicing
enhancer
(ESE) sequence portion.
With such arrangement, cleavage occurs at a position near the splice acceptor
site or the donor site of the target exon, and the splice acceptor site or the
donor site
is disrupted in the repair process, resulting occurrence of skipping of the
target exon
when the splicing reaction occurs in the process of maturation of pre-mRNA
into
mRNA.
[0025]
The splice acceptor site is defined as the two bases immediately before the
target exon, and its examples include the AG sequence.
The splice donor site is defined as the two bases immediately after the target
exon, and its examples include the GT sequence.
The exonic splicing enhancer (ESE) sequence is defined as a binding site of
SR protein (SRSF1 to 12 genes) present in the target exon. The binding site of
SR
protein can be obtained by searching databases, and examples of such databases
include RESCUE-ESE [Fairbrother WG et al., Science, 2002] and ESEfinder
[Cartegni L. et al., NAR, 2003].
[0026]
The spacer sequence of Type II Cas9 can be designed as RNA having a
continuous base sequence of 15 to 30 bases whose 3'-end corresponding to the
base
immediately before the PAM sequence (for example, NGG in the case of S.
pyogenes
CA 03058584 2019-09-30
Cas9, or NNGRRT in the case of Staphylococcus aureus Cas9) in the sequence of
the
sense strand or the antisense strand of the gene of interest (for example,
NNNNNNNNNNNNNNNNNNNNNGG (SEQ NO:7)). (The N's represent the
spacer sequence.)
5 However, since the cleavage occurs even without 100% matching of the
sequence, a mismatch(es) of one or two bases is/are acceptable (especially in
the 5'-
side). For a transcription start site from the human HI Polifi promoter, the
5'-end
of the spacer sequence is preferably not C or T. In cases where the
corresponding
base in the genome is C or T, it is preferably converted to G
10 The spacer sequence of the guide RNA of type V Cfpl can be designed as
RNA having a continuous base sequence of 15 to 30 bases whose 5'-end
corresponds
to the base immediately after the PAM sequence (for example, TTTV in the case
of
Acidaminococcus sp. Cpfl) (for example,
TTTT (SEQ ID
NO:8)). In the case of Cpfl,
15 tracrRNA is not required.
[0027]
Regarding the site of DNA cleavage by S. pyogenes Cas9, the cleavage occurs
between the third base and the fourth base as counted in the 3'¨>5' direction
from the
3'-end base of the spacer sequence, which is regarded as 1. Accordingly, "the
20 cleavage site is positioned within 80 bases from the acceptor site or
donor site of the
target exon" means that the number of bases present between the bases at the
acceptor site or the donor site (for example, GT or AG (in the case of the
antisense
strand, AC or CT)) and the fourth base as counted in the 3'¨>5' direction from
the
base corresponding to the 3'-end base of the spacer sequence is not more than
80
bases.
Regarding the site of DNA cleavage by AsCpfl, the cleavage occurs at the
19th sense-strand base and the 23rd antisense-strand base as counted in the 5'-
6'
CA 03058584 2019-09-30
21
direction from the 5'-end base of the spacer sequence, which is regarded as 1.
[0028]
A description is given with reference to Fig. 3.
Using exon 45 of the human dystrophin (hDMD) gene as a target, the
acceptor site immediately before the exon 45 is disrupted to carry out
skipping of the
exon 45.
In this process, in Sp-sgRNA-DMD1, a spacer sequence corresponding to the
20 bases immediately before the PAM sequence (AGG) (tggtatcttacagGAAC/TCC)
(SEQ ID NO:9) is designed. In this case, there are four bases between the
acceptor
sequence (ag) and the cleavage site (C/T).
Similarly, in Sp-sgRNA-DMD2, a spacer sequence corresponding to the 20
bases immediately before the PAM sequence (TGG) (atcttacagGAACTCCA/GGA)
(SEQ ID NO:10) is designed. In this case, there are eight bases between the
acceptor sequence (ag) and the cleavage site (A/G).
Similarly, in Sp-sgRNA-DMD3, a spacer sequence corresponding to the 20
bases immediately before the PAM sequence (TGG)
(cagGAACTCCAGGATGG/CAT) (SEQ ID NO:11) is designed. In this case, there
are 14 bases between the acceptor sequence (ag) and the cleavage site (G/C).
Similarly, in Sp-sgRNA-DMD4, a spacer sequence corresponding to the 20
bases immediately before the PAM sequence (CGG)
(TCCAGGATGGCATTGGG/CAG) (SEQ ID NO:12) is designed. In this case,
there are 21 bases between the acceptor sequence (ag) and the cleavage site
(G/C).
Sp-sgRNA-DMD5 is arranged for the antisense strand, and a spacer sequence
corresponding to the 20 bases immediately before the PAM sequence (AGG)
(GTTCctgtaagatacca/aaa) (SEQ ID NO:13) is designed.
In this case, there are 11 bases between the acceptor sequence (ct) and the
cleavage site (a/a).
CA 03058584 2019-09-30
22
In each sequence ID number, T is read as U when an RNA sequence is meant.
[0029]
By adding a scaffold sequence to the above spacer sequence, a guide RNA can
be obtained. Plasmids which contains a scaffold sequence therein, and with
which a
desired guide RNA can be expressed by inserting a DNA sequence corresponding
to
an arbitrary spacer sequence, are commercially available (for example, Addgene
plasmid 41824), and they can be simply used for introduction of a guide RNA
into
cells.
[0030]
Two or more kinds of guide RNAs may be used. In such a case, two or more
kinds of guide RNAs satisfying the condition "the site of cleavage by the
CRISPR-
Cas is positioned within 80 bases from the splice donor site immediately
before the
target exon or the splice acceptor site immediately after the target exon" may
be used
for one site to be disrupted (target exon). By using two or more different
kinds of
guide RNAs for the same site to be disrupted, and causing DNA double-strand
breaks
simultaneously at two or more sites, exon skipping can be caused with a very
high
efficiency. The two or more kinds of guide RNAs may be arranged either for one
of
the sense strand and the antisense strand, or for both of these.
[0031]
In cases where Dl OA Cas9 nickase is used, and two kinds of guide RNAs for
cleaving the sense strand and the antisense strand, respectively, are used,
the two
kinds of guide RNAs are designed such that they satisfy the requirement that
the
cleavage site of at least one of them is positioned within 80 bases from the
acceptor
site or the donor site before or after the target exon. In this case, the
distance
between the guide RNA-binding site (spacer sequence) in the sense strand and
the
guide RNA-binding site (spacer sequence) in the antisense strand is preferably
-10 to
200 bases, more preferably 0 to 100 bases. The acceptor site or the donor site
is
CA 03058584 2019-09-30
23
= =
preferably located between the cleavage site in the sense strand and the
cleavage site
in the antisense strand. The spacer sequence of the guide RNA for cleavage of
the
sense strand and the spacer sequence of the guide RNA for cleavage of the
antisense
strand may overlap with each other, but they preferably do not overlap with
each
other.
[0032]
The gene of interest having the target exon is not limited. It is preferably a
mammalian gene, more preferably a human gene, for example, a disease-
associated
gene.
One example of the gene is the dystrophin gene, which is a causative gene of
Duchenne muscular dystrophy. Since it is known that the phenotype of the
mutant
gene can be masked by skipping of exon 45, exon 45 of the human dystrophin
gene
can be suitably used as a target of exon skipping.
The gene of interest may also be an artificially synthesized gene containing
an
exon and an intron.
[0033]
Specific examples of the spacer sequence contained in the guide RNA that
can be used in the method of the present invention include the base sequence
of bases
from 17 to 36 in the base sequence of any of SEQ ID NOs:17 to 42, the base
sequence of bases from 17 to 39 in the base sequence of any of SEQ ID NOs:44
to 45,
and the base sequence of bases from 24 to 43 in the base sequence of any of
SEQ ID
NOs:50 to 53. Their complementary sequences may also be used.
Among these, when two kinds of guide RNAs are used in combination,
preferred examples of the combination of the spacer sequences include the
combinations of sgRNA-DMD1 (the base sequence of bases from 17 to 36 in SEQ ID
NO:17), sgRNA-DMD2 (the base sequence of bases from 17 to 36 in SEQ ID
NO:18), sgRNA-DMD4 (the base sequence of bases from 17 to 36 in SEQ
CA 03058584 2019-09-30
24
NO:20), sgRNA-DMD8 (the base sequence of bases from 17 to 36 in SEQ ID
NO:24), or sgRNA-DMD9 (the base sequence of bases from 17 to 36 in SEQ ID
NO:25) with sgRNA-DMD23 (the base sequence of bases from 17 to 36 in SEQ ID
NO:39).
Further, among these, preferred examples of the combination of the spacer
sequences for use in the double nicking method include sgRNA-DMD4 (the base
sequence of bases from 17 to 36 in SEQ ID NO:20) and sgRNA-DMD5 (the base
sequence of bases from 17 to 36 in SEQ ID NO:21).
[0034]
Transfection of cells with DNA, RNA, or vectors expressing these can be
carried out by using known arbitrary means, and commercially available
transfection
reagents may be used. For example, Lipofectamine 2000 (Thermo Fisher),
StemFect (STEMGEN), FuGENE 6/HD (Promega), jetPRIME Kit (Polyplus-
transfection), DreamFect (OZ Biosciences), GenePorter 3000 (OZ Biosciences),
or
Calcium Phosphate Transfection Kit (OZ Biosciences) can be used.
Electroporation
may be also used. For example, NEPA21 (Nepa Gene), 4D-Nucleofector (Lonza),
Neon (Thermo Fisher), Gene Pulser Xcell (BioRad), or ECM839 (BTX Harvard
Apparatus) can be used. Regarding the transfection of cells, a complex may be
formed with CRISPR-Cas protein and gRNA in advance, and the cells may then be
transfected with the complex. Further, microinjection or electroporation may
be
carried out for introduction of the DNA or RNA into fertilized eggs.
[0035]
The cells are preferably a mammalian cells, more preferably human cells.
The cells may be primary cultured cells isolated from an established cell line
or from
a mammalian tissue, or may be mesenchymal cells or pluripotent stem cells such
as
induced pluripotent stem (iPS) cells. For example, in cases where the
dystrophin
gene is to be targeted, skeletal muscle cells, mesenchymal cells, or iPS cells
derived
CA 03058584 2019-09-30
from a DMD patient may be established, and then guide RNA and CRISPR-Cas, or a
guide RNA pair and Cas nickase, for induction of exon skipping of the present
invention may be simultaneously introduced into the cells, to induce exon
skipping of
the dystrophin gene, thereby enabling recovery of the dystrophin protein. By
5 transplanting such repaired cells or an induced product therefrom to a
patient,
atrophic muscle cells can be complemented.
[0036]
Further, in another mode, guide RNA and CRISPR-Cas, or a guide RNA pair
and Cas nickase, for induction of exon skipping of the present invention are
10 simultaneously introduced into a muscular tissue of a DMD patient, to
induce exon
skipping of the dystrophin gene, thereby enabling recovery of the dystrophin
protein
in the body of the patient.
[0037]
<Method of Evaluation of Exon Skipping>
15 The present invention also provides a method of evaluating exon
skipping
using CRISPR-Cas and guide RNA and the like, comprising use of a marker gene
containing a sequence which is inserted in a coding region and which contains
a first
intron, an exon to be analyzed, and a second intron, wherein the marker gene
is
designed such that the marker gene functions when the exon to be analyzed is
20 skipped (by the action of the guide RNA and the CRISPR-Cas arranged near
the exon
to be analyzed).
Exon skipping can be induced also in antisense nucleic acid or the like, and
the subject to be evaluated is not limited to genome editing.
[0038]
25 Examples of the marker gene include, but are not limited to, genes of
enzymes such as luciferase or LacZ; genes encoding a fluorescent protein such
as
GFP, Ds-Red, or mCherry; and drug resistance genes such as the Puro resistance
gene,
CA 03058584 2019-09-30
26
Neo resistance gene, Hyg resistance gene, Bls gene, hisD gene, Gpt gene, and
Ble
gene. In cases where cDNA of the marker gene contains a sequence which is the
same as or similar to the splicing donor or acceptor sequence, the
corresponding site
is preferably altered such that splicing in the marker does not occur, for the
purpose
of specifically evaluating splicing in the inserted sequence.
[0039]
The position to which the sequence containing the first intron (containing a
donor site and an acceptor site), the exon to be analyzed, and the second
intron
(containing a donor site and an acceptor site) is inserted is a position where
the
marker gene is prevented from functioning (where the activity of the marker
protein
or the phenotype based on the marker gene disappears) when the exon is
inserted.
The fact that the exon insertion prevents the marker gene from functioning may
be
confirmed in advance. As the sequence of the insertion site, the "@" portion
in the
sequence of CAG@G or AAG@G is more preferably selected since, by this, the
splice donor sequence of the first intron and the splice acceptor sequence of
the
second intron become the consensus (most frequent) sequences of the human
splice
acceptor (MAGIGURAG) and the human splice acceptor sequence (NCAGIG).
In the method of the present invention, in cases where the splicing normally
occurs, the marker gene is expressed as a fusion protein (which lost the
original
activity) containing the exon inserted therein, or the marker gene does not
function
since the insertion of the exon prevents normal expression.
On the other hand, in cases where the exon is skipped by genome editing, the
marker gene is expressed as a normal type, so that the activity of the marker
protein
or the phenotype based on the marker gene is found.
Thus, whether or not skipping of the exon occurred can be analyzed based on
the presence or absence of the function of the marker gene. This enables
simple
evaluation of exon skipping, and is also useful for screening for compounds
that
CA 03058584 2019-09-30
27
promote exon skipping.
[0040]
Whether or not exon skipping has occurred in a cell into which the altered
marker gene is introduced can be investigated by introducing guide RNA
corresponding to the inserted sequence together with CRISPR-Cas, and analyzing
the
phenotype of the cell corresponding to the marker gene. Incorporation of the
altered
marker gene into the chromosome is preferably carried out using a transposon
vector
or a virus vector. Examples of the transposon vector include piggyBac vectors,
piggyBat vectors, Sleeping Beauty vectors, Toll vectors, and LINE vectors.
Examples of the virus vector include retrovirus vectors, lentivirus vectors,
adenovirus
vectors, Sendai virus vectors, and adeno-associated virus vectors.
Examples of the type of the exon include, but are not limited to, exon 45 of
the dystrophin gene as described above.
EXAMPLES
[0041]
The present invention is described below concretely by way of Examples.
However, the present invention is not limited to the following modes.
[0042]
<Methods>
<Unique k-mer Database> in relation to Fig. 2
Using a Pen l script, base sequences of 10 to 16-mer (k-mer) with all
combinations were generated. Using the Bowtie program (Langmead et al., 2009),
the k-mer sequences generated were mapped on human genome hgl 9 without
accepting a mismatch. Subsequently, k-mer sequences mapped only once on human
genome hg19 were extracted, and a unique k-mer database was constructed (Li HL
et
al., Stem Cell Reports, 2015). Using the ngs.plot.r program (Shen Let al., BMC
Genomics, 2014), which runs with the R language, the distribution of unique k-
mers
CA 03058584 2019-09-30
= 28
in 200 bp before and after all human exons were investigated and plotted.
[0043]
<Construction of SpCas9 cDNA Expression Vectors>
DNA synthesis (GenScript) was carried out to prepare the pUC57-SphcCas9
vector, which has a Cas9 cDNA derived from Streptococcus pyogenes optimized
for
the human codon frequencies inserted therein, the Cas9 cDNA having an SV40
large
T antigen-derived nuclear localization signal peptide (PICICKRKV) (SEQ ID
NO:217) at the C-terminus. This was cleaved with SalI-XbaI restriction
enzymes,
and then ligated to the SalI-XbaI site of pENTR2B (A10463, Thermo Fisher) to
construct the pENTR-SphcCas9 vector. Subsequently, the SphcCas9 cDNA portion
of the pENTR-SphcCas9 vector was inserted into the pHL-EFla-GW4C-A vector,
the pHL-EFla-GW-iP-A vector, and the pHL-EFla-GW-A vector by the Gateway LR
clonase reaction, to construct the pHL-EFla-SphcCas9-iC-A vector (SpCas9-IRES-
mCheery-polyA), the pHL-EFla-SphcCas94P-A vector (SpCas9-IRES-PuroR-
1 5 polyA) (Addgene, 60599), and the pHL-EFla-SphcCas9-A vector (SpCas9-
polyA).
The EFla promoter is more suitable than virus-derived promoters (the CMV
promoter and the like) since a higher expression level can be obtained in
pluripotent
stem cells.
Further, for preparation of the Dl OA mutant of SpCas9 (nickase), the DNA
sequence SphcCas9-D10A (Ncoi-Sbfl), in which the GaC codon (Asp, D) was
converted to the GcC codon (Ala, A), was synthesized by gBlock (IDT).
The SphcCas9-D10A(NcoI-Sbfl) sequence was cleaved with the NcoI-Sbfl
restriction enzymes, and then inserted into the NcoI-Sbfl site of the pENTR-
SphcCas9 vector using the In-Fusion reaction, to construct the pENTR-SphcCas9-
2 5 Dl OA vector. Subsequently, the SphcCas9-D10A cDNA portion of the pENTR-
SphcCas9-D10A was inserted into the pHL-EFla-GW4C-A vector and the pHL-
EFla-GW-iP-A vector by the Gateway LR clonase reaction, to construct the pHL-
CA 03058584 2019-09-30
. . 29
EFla-SphcCas9-D10A-iC-A vector (SpCas9-IRES-mCheery-polyA) and the pHL-
EFla-SphcCas9-D10A4P-A vector (SpCas9-IRES-PuroR-polyA).
[0044]
SphcCas9-D10A(NcoI-SbfI)
5'-ATTCAGTCGACCATGGATAAGAAATACAGCATTGGACTGGcCATTGGGA
CAAACTCCGTGGGATGGGCCGTGATTACAGACGAATACAAAGTGCCTTCA
AAGAAGTTCAAGGTGCTGGGCAACACCGATAGACACAGCATCAAGAAAA
ATCTGATTGGAGCCCTGCTGTTCGACTCCGGCGAGACAGCTGAAGCAACT
CGGCTGAAAAGAACTGCTCGGAGAAGGTATACCCGCCGAAAGAATAGGAT
CTGCTACCTGCAGGAGATTTTCAGCAA-3' (SEQ ID NO:14)
[0045]
<Construction of SpCas9 gRNA Expression Vectors>
For cloning of gRNA of SpCas9 into an expression vector, 10 pmol each of
the following arbitrary Sp-sgRNA-XXX-fwd primer (one of SEQ ID NOs:17 to 42)
and the Sp-sgRNA-Universal-rev primer were mixed together, and thermal cycling
reaction was performed using KOD Plus Neo DNA polymerase (Toyobo) (heat
denaturation at 98 C: 2 min followed by {94 C: 10 sec, 55 C: 10 sec, 68 C: 10
sec}
x 35 cycles, further followed by incubation at 4 C). The resulting PCR product
was
subjected to electrophoresis in 2% agarose gel, and a DNA band with a size of
about
135 bp was excised and purified. The purified PCR product was inserted, using
the
In-Fusion reaction (Takara-Clontech), into the pHL-H1-ccdB-mEFla-RiH vector
(Addgene 60601) cleaved with BamHI-EcoRI, to construct a pHL-H1-[SpCas9-
gRNA]-mEFla-RiH vector which expresses an arbitrary gRNA.
[0046]
PCR Product Sequence (135 bp)
GAGACCACTTGGATCCRNNNNNNNNNNNNNNNNNNGTTTTAGAGCTAG
AAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGC
CA 03058584 2019-09-30
' . 30
ACCGAGTCGGTGCTTTTTTTGAATTCAAACCCGGGC (SEQ ED NO:15)
[0047]
Sp-sgRNA-XXX-fwd
GAGACCACTTGGATCC
GTTTTAGAGCTAG
AAATAGCA (SEQ ID NO:16)
Sp-sgRNA-DMD1-fwd
GAGACCACTTGGATCCGggtatcttacaggaactccGTTTTAGAGCTAGAAATAGCA
(SEQ ID NO:17)
Sp-sgRNA-DMD2-fwd
GAGACCACTTGGATCCGtcttacaggaactccaggaGTTTTAGAGCTAGAAATAGCA
(SEQ ID NO:18)
Sp-sgRNA-DMD3-fwd
GAGACCACTTGGATCCGaggaactccaggatggcatGTTTTAGAGCTAGAAATAGCA
(SEQ ID NO:19)
Sp-sgRNA-DMD4-fwd
GAGACCACTTGGATCCGCCAGGATGGCATTGGGCAGGTTTTAGAGCTAGA
AATAGCA (SEQ ID NO:20)
Sp-sgRNA-DMD5-fwd
GAGACCACTTGGATCCGTTCCTGTAAGATACCAAAAGTTTTAGAGCTAGAA
ATAGCA (SEQ ID NO:21)
Sp-sgRNA-DMD6-fwd
GAGACCACTTGGATCCGcaTTTTTGTTTTGCCTTTTGTTTTAGAGCTAGAAA
TAGCA (SEQ ID NO:22)
Sp-sgRNA-DMD7-fwd
GAGACCACTTGGATCCGTGCCTTTTTGGTATCTTACGTTTTAGAGCTAGAA
ATAGCA (SEQ ID NO:23)
Sp-sgRNA-DMD8-fwd
CA 03058584 2019-09-30
31
GAGACCACTTGGATCCAGGAACTCCAGGATGGCATTGTTTTAGAGCTAGA
AATAGCA (SEQ ID NO:24)
Sp-sgRNA-DMD9-fwd
GAGACCACTTGGATCCGCCGCTGCCCAATGCCATCCGTTTTAGAGCTAGAA
ATAGCA (SEQ ID NO:25)
Sp-sgRNA-DMD10-fwd
GAGACCACTTGGATCCGTCAGAACATTGAATGCAACGTTTTAGAGCTAGA
AATAGCA (SEQ ID NO:26)
Sp-sgRNA-DMD11-fwd
GAGACCACTTGGATCCGCAGAACATTGAATGCAACTGTTTTAGAGCTAGA
AATAGCA (SEQ ID NO:27)
Sp-sgRNA-DMD12-fwd
GAGACCACTTGGATCCGAGAACATTGAATGCAACTGGTTTTAGAGCTAGA
AATAGCA (SEQ ID NO:28)
Sp-sgR14A-DMD13-fwd
GAGACCACTTGGATCCAATACTGGCATCTGTTTTTGGTTTTAGAGCTAGAA
ATAGCA (SEQ ID NO:29)
Sp-sgRNA-DMD14-fwd
GAGACCACTTGGATCCAACAGATGCCAGTATTCTACGTTTTAGAGCTAGAA
ATAGCA (SEQ ID NO:30)
Sp-sgRNA-DMD15-fwd
GAGACCACTTGGATCCGAATTTTTCCTGTAGAATACGTTTTAGAGCTAGAA
ATAGCA (SEQ ID NO:31)
Sp-sgRNA-DMD16-fwd
GAGACCACTTGGATCCGAGTATTCTACAGGAAAAATGTTTTAGAGCTAGAA
ATAGCA (SEQ ID NO:32)
Sp-sgRNA-DMD17-fwd
CA 03058584 2019-09-30
32
GAGACCACTTGGATCCAGTATTCTACAGGAAAAATTGTTTTAGAGCTAGAA
ATAGCA (SEQ ID NO:33)
Sp-sgRNA-DMD18-fwd
GAGACCACTTGGATCCAATTGGGAAGCCTGAATCTGGTTTTAGAGCTAGA
AATAGCA (SEQ ID NO:34)
Sp-sgRNA-DMD19-fwd
GAGACCACTTGGATCCGGGGAAGCCTGAATCTGCGGGTTTTAGAGCTAGA
AATAGCA (SEQ ID NO:35)
Sp-sgRNA-DMD20-fwd
GAGACCACTTGGATCCAAGCCTGAATCTGCGGTGGCGTTTTAGAGCTAGA
AATAGCA (SEQ ID NO:36)
Sp-sgRNA-DMD21-fwd
GAGACCACTTGGATCCGCTGAATCTGCGGTGGCAGGGTTTTAGAGCTAGA
AATAGCA (SEQ ID NO:37)
Sp-sgRNA-DMD22-fwd
GAGACCACTTGGATCCGCTCCTGCCACCGCAGATTCGTTTTAGAGCTAGA
AATAGCA (SEQ ID NO:38)
Sp-sgRNA-DMD23-fwd
GAGACCACTTGGATCCAGCTGTCAGACAGAAAAAAGGTTTTAGAGCTAGA
AATAGCA (SEQ ID NO:39)
Sp-sgRNA-DMD24-fwd
GAGACCACTTGGATCCGTCAGACAGAAAAAAGAGGTGTTTTAGAGCTAGA
AATAGCA (SEQ ID NO:40)
Sp-sgRNA-DMD25-fwd
GAGACCACTTGGATCCGCAGACAGAAAAAAGAGGTAGTTTTAGAGCTAG
AAATAGCA (SEQ ID NO:41)
Sp-sgRNA-DMD26-fwd
CA 03058584 2019-09-30
= = 33
GAGACCACTTGGATCCGGTAGGGCGACAGATCTAATGTTTTAGAGCTAGA
AATAGCA (SEQ ID NO:42)
[0048]
Sp-sgRNA-Universal-rev
GCCCGGGTTTGAATTCAAAAAAAGCACCGACTCGGTGCCACTTTTTCAAG
TTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAA (SEQ ID
NO:43)
[0049]
<Construction of SaCas9 cDNA Expression Vectors> in relation to Figs. 13a to
13d
DNA synthesis (GenScript) was carried out to prepare the pUC-Kan-
SahcCas9 vector, which has a Cas9 cDNA derived from Staphylococcus aureus
optimized for the human codon frequencies inserted between the Gateway attL1
site
and attL2 site, the Cas9 cDNA having an SV40 large T NLS at the N-terminus,
and a
nucleoplasmin NLS and a 3 xHA tag at the C-terminus. Subsequently, the SaCas9
cDNA portion of the pUC-Kan-SahcCas9 vector was inserted into the pHL-EFla-
GW-A and pHL-EFla-GW-iP-A vectors by the Gateway LR clonase reaction, to
construct the HL-EFla-SaCas9-A and HL-EFla-SaCas9-iC-A vectors, respectively.
[0050]
<Construction of SaCas9 gRNA Expression Vectors>
For cloning of gRNAs of SaCas9 into an expression vector, 10 pmol each of
the following arbitrary sgRNA-DMD-SA-X-fwd primer (one of SEQ ID NOs:44 to
45) and the SA1 -gRNA-Universal-Rev primer were mixed together, and thermal
cycling reaction was performed using KOD Plus Neo DNA polymerase (Toyobo)
(heat denaturation at 98 C: 2 min followed by {94 C: 10 sec, 55 C: 10 sec, 68
C: 10
sec} x 35 cycles, further followed by incubation at 4 C). The resulting PCR
product was subjected to electrophoresis in 2% agarose gel, and a DNA band
with a
size of about 135 bp was excised and purified. The purified PCR product was
CA 03058584 2019-09-30
34
inserted, using the In-Fusion reaction (Takara-Clontech), into the pHL-H1-ccdB-
mEFla-RiH vector (Addgene 60601) cleaved with Bamiff-EcoRI, to construct a
pHL-H1-[SaCas9-gRNA] -mEFla-RiH vector which expresses the arbitrary gRNA.
[0051]
sgRNA-DMD-SA-5
5'-GAGACCACTTGGATCCATTACAGGAACTCCAGGATGGCAGTTTTAGTAC
TCTGGAAACAGAAT-3' (SEQ ID NO:44)
sgRNA-DMD-SA-8
5'-GAGACCACTTGGATCCATTGCCGCTGCCCAATGCCATCCGTTTTAGTACT
CTGGAAACAGAAT-3' (SEQ ID NO:45)
S A I -gRNA-Universal-Rev
5'-GCCCGGGTTTGAATTCAAAAAAATCTCGCCAACAAGTTGACGAGATAA
ACACGGCATTTTGCCTTGTTTTAGTAGATTCTGTTTCCAGAGTACTAA-3' (S
EQ ID NO:46)
[0052]
<Construction of AsCpfl cDNA Expression Vectors> in relation to Figs. 13a to
13d
DNA synthesis (GenScript) was carried out to prepare the pUC57-hcAsCpfl
vector, which has a Cpfl cDNA derived from Acidaminococcus sp. BV3L6
optimized for the human codon frequencies is inserted between the Gateway
attL1
site and attL2 site, the Cpfl cDNA having a nucleoplasmin NLS
(KRPAATICKAGQAKKICK) (SEQ ID NO:218) and a 3xHA tag (YPYDVPDYA
YPYDVPDYAYPYDVPDYA) (SEQ ID NO:219) sequence at the C-terminus.
Subsequently, the hcAsCpfl cDNA portion of the pENTR-hcAsCpfl vector was
inserted into the pHL-EFla-GW-A and pHL-EFla-GW-iP-A vectors by the Gateway
LR clonase reaction, to construct the HL-EFla-hcAsCpfl-A and HL-EFla-
hcAsCpfl 4C-A vectors, respectively.
[0053]
CA 03058584 2019-09-30
= 35
<Construction of AsCpfl gRNA Expression Vectors>
For cloning of gRNAs of AsCpfl into an expression vector, 10 pmol each of
the following arbitrary AsCpfl-gRNA-XXX-rev primer (one of SEQ ID NOs:50 to
53) and the AsCpfl-gRNA-Universal-GGG-fwd primer (or the AsCpfl-gRNA-
Universal-G-fwd primer) were mixed together, and thermal cycling reaction was
performed using KOD Plus Neo DNA polymerase (Toyobo) (heat denaturation at
98 C: 2 min followed by {94 C: 10 sec, 55 C: 10 sec, 68 C: 10 sec} x 35
cycles,
further followed by incubation at 4 C). The resulting PCR product was
subjected to
electrophoresis in 2% agarose gel, and a DNA band with a size of about 80 bp
was
excised and purified. The purified PCR product was inserted, using the In-
Fusion
reaction (Takara-Clontech), into the pHL-H1-ccdB-mEFla-RiH vector cleaved with
BamHI-EcoRI, to construct a pHL-H1-[AsCpfl-gRNA]tnEFla-RiH vector which
expresses the AsCpfl gRNA.
AsCpfl-gRNA-Universal-GGG-fwd (GGG at TSS)
5'-GAGACCACTTGGATCCGGGTAATTTCTACTCTTGTAGAT-3' (SEQ ID
NO:47)
AsCpfl-gRNA-Universal-G-fwd (G at TSS)
5'-GAGACCACTTGGATCCGTAATTTCTACTCTTGTAGAT-3' (SEQ ID NO:48)
AsCpfl -gRNA-XXX-rev
5'-GCCCGGGTTTGAATTCAAAAAAA ATCT
ACAAGAGTAGAAATTA-3' (SEQ ID NO:49)
AsCpfl -gRNA-DMD1-rev
5 '-GCCCGGGTTTGAATTCAAAAAAAGGAGTTCCTGTAAGATACCAATCTAC
AAGAGTAGAAATTA-3' (SEQ ID NO:50)
AsCpfl-gRNA-DMD2-rev
5 '-GCCCGGGTTTGAATTCAAAAAAATGGAGTTCCTGTAAGATACCATCTAC
AAGAGTAGAAATTA-3' (SEQ ID NO:51)
CA 03058584 2019-09-30
' 36
AsCpfl -gRNA-DMD3 -rev
5'-GCCCGGGTTTGAATTCAAAAAAACTGGAGTTCCTGTAAGATACATCTAC
AAGAGTAGA_AATTA-3' (SEQ ID NO:52)
AsCpfl-gRNA-DMD4-rev
5'-GCCCGGGTTTGAATTCAAAAAAAAGGATGGCATTGGGCAGCGGATCTA
CAAGAGTAGAAATTA-3' (SEQ ID NO:53)
[0054]
<Construction of SSA Vector> in relation to Fig. 10
The SSA-DMD-all-ss oligo DNA and the SSA-DMD-all-as oligo DNA,
which have the target sequences of Sp-gRNA-DMD1 to 5, were annealed with each
other, and then ligated into the BsaI site present in firefly Luc2 cDNA of the
pGL4-
SSA vector (Addgene 42962, Ochiai et al., Genes Cells, 2010), to construct the
pGL4-SSA-DMD-all vector. In the pGL4-SSA-DMD-all vector, the firefly Luc2
cDNA is divided, and does not show Luc activity. However, when cleavage of the
target DNA portion in the pGL4-SSA vector is induced by guide RNA, the DNA
cleavage is repaired by the SSA (single strand annealing) pathway, resulting
recovery
of the firefly Luc2 cDNA.
SSA-DMD-all-ss
5 '-gtcgTGCCTTTTTGGTATCTTACAGGAACTCCAGGATGGCATTGGGCAGCG
GCAAACTGTTGTCAGAACATggt-3' (SEQ ID NO:54)
SSA-DMD-all-as
5'-cggtaccATGTTCTGACAACAGTTTGCCGCTGCCCAATGCCATCCTGGAGTT
CCTGTAAGATACCAAAAAGGCA-3' (SEQ ID NO:55)
[0055]
<Target DNA Cleavage Activity by SSA Assay> Fig. 10, Fig. 13c
A mixture of 100 ng of the pGL4-SSA-DMD-All vector, 20 ng of the phRL-
TK vector, which expresses Renilla Luc, 200 ng of the pHL-EF 1a vector, which
CA 03058584 2019-09-30
37
expresses CRISPR-Cas, and 200 ng of the pHL-H1-sgRNA-mEFla-RiH vector,
which expresses sgRNA, was prepared, and then diluted with 25 1 of Opti-MEM.
With 25 ptl of Opti-MEM, 0.7 1 of Lipofectamine 2000 was diluted, and the
resulting dilution was incubated at room temperature for 3 to 5 minutes,
followed by
mixing the dilution with the above DNA solution, and then incubating the
resulting
mixture at room temperature for additional 20 minutes. The cell number of 293T
cells suspended by trypsin-EDTA treatment was counted, and then the cells were
diluted to 60,000 cells/100 ul with a medium, followed by plating the cells on
a 96-
well plate containing the above DNA-Lipofectamine complex at 100 1/well.
After
culturing the cells for 48 hours with 5% CO2 at 37 C, the 96-well plate was
allowed
to cool to room temperature, and then Dual-Glo Reagent was added thereto,
followed
by incubation at room temperature for 30 minutes to lyse the cells to cause
the
luciferase reaction. To a white 96-well plate, 100 1 of the supernatant was
transferred, and the luminescence intensities of Firefly and Renilla were
measured
using Centro LB960 (Berthold Technologies). Since firefly Luc emits light only
when DNA cleavage is induced by CRISPR, the luminescence value of firefly was
normalized against the luminescence value of Renilla to measure the DNA
cleavage
efficiency.
[0056]
<Cell Culture>
293T cells were cultured inDMEM medium supplemented with 5 to 10% FBS.
DMD-iPS cells (clone ID: CiRA00111) were cultured, on SNL feeder cells
whose growth was inhibited by mitomycin C treatment, using Knockout SR medium
{prepared by adding 50 mL of Knockout SR (Thermo Fisher, 10828028), 2.5 mL of
L-glutamine (Thermo Fisher, 25030081), 2.5 mL of non-essential amino acid
mixture
(Thermo Fisher, 11140050), 0.5 mL of 2-mercaptoethanol (Thermo Fisher,
21985023), 1.25 mL of penicillin-streptomycin (Thermo Fisher, 15140122), and 8
CA 03058584 2019-09-30
38
ng/ml human basic FGF (Wako, 6404541) to 200 mL of DMEM/F12 medium
(Thermo Fisher, 10565018)). Alternatively, using StemFit AKO3N (Ajinomoto)
medium, the cells may be cultured on iMatrix-511 (Nippi, 892014) without the
use of
feeder cells.
[0057]
<Analysis of Genome DNA Cleavage Pattern in DMD-iPS Cells> Fig. 11
For iPS cells established from a DMD patient who lacks exon 44, 10 tiM Y-
27632 (Sigma) was added to the medium not less than one hour before
transfection.
Immediately before electroporation, the iPS cells were detached with CTK
solution,
and then dispersed using 0.25% trypsin-EDTA, followed by counting the cells
for
providing 1 x 106 cells for each condition. To these cells, electroporation of
5 [ig of
the pHL-EFla-SphcCas94P-A vector (Addgene, 60599) and 5 lig of the pHL-H1-
[ Sp-gRNA]-mEFla-RiH vector was carried out using a NEPA21 electroporator
(Nepa Gene) with a poration pulse voltage of 125 V, a pulse width of 5
milliseconds,
and a number of pulses of 2. In the cases where double-nicking was carried
out,
electroporation was carried out with 5 pig of the pHL-EFla-SphcCas9-D10A-iP-A
vector and a total of 10 tig, that is, 5 pg each, of two kinds of pHL-H1-[Sp-
gRNA]-
mEFla-RiH vectors. The iPS cells subjected to the electroporation were
cultured
for not less than several days, and genome DNA was extracted therefrom,
followed
by performing primary PCR amplification using the DMD-MiSeq-Rdl-fwd-X and
DMD-MiSeq-Rd2-rev-X primers, and then performing secondary PCR amplification
using the Multiplex P5 fwd primer and the Multiplex P7 rev primer. The
resulting
PCR product was excised from the gel, and then purified, followed by
quantification
using a Qubit 2.0 Fluorometer (Thermo Fisher) and a KAPA Library
Quantification
Kit for Illumina (KAPA Biosystems). The samples were mixed to the same amount,
and the DNA concentration was adjusted to 2 nM, followed by performing alkali
denaturation of the DNA by treatment with 0.2 N NaOH for 5 minutes. Each
CA 03058584 2019-09-30
39
denatured DNA sample was diluted to 12 pM, and then 4 pM PhiX spike-in DNA
was added thereto, followed by performing MiSeq sequencing reaction using a
MiSeq Reagent Kit v2 for 2 x 150 bp (IIlumina). From the FASTQ sequence file
generated as a result of the sequencing, low-quality reads were removed by
using the
fastq_quality filter program in the FASTX-Toolkit. After removing the PhiX
sequence used as the spike-in, the fastx_barcode_splitter program was used to
divide
the samples according to the barcode sequences. The sequences of the samples
were mapped using the BWA program, and the insertion/deletion patterns of the
sequences were extracted from the MD tag information in the CIGAR code.
[0058]
DMD-MiSeq-Rd 1 -fwd-X (N represents the barcode sequence corresponding to each
sample. See below.)
5 '-CTCTTTCCCTACACGACGCTCTTCCGATCTNNNNAATAAAAAGACATG
GGGCTTCA-3' (SEQ ID NO:56)
DMD-MiSeq-Rd2-rev-X (N represents the barcode sequence corresponding to each
sample. See below.)
5 '-CTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNCTGGCATCTGTTTTT
GAGGA-3' (SEQ ID NO:57)
Multiplex P5 fwd
5'-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCT
C-3' (SEQ ID NO:58)
Multiplex P7 rev
5'-CAAGCAGAAGACGGCATACGAGATGTGACTGGAGTTCAGACGTGTGC
TC-3' (SEQ ID NO:59)
[0059]
Specific sequences containing the barcode for the X
DMD-MiSeq-Rdl-fwdl-AGTC
CA 03058584 2019-09-30
= r 40
'-CTCTTTCCCTACACGACGCTCTTCCGATCTagtcAATAAAAAGACATGGG
GCTTCA-3' (SEQ ID NO:60)
DMD-MiSeq-Rd1-fwd2-GTCA
5 '-CTCTTTCCCTACACGACGCTCTTCCGATCTgtcaAATAAAAAGACATGGG
5 GCTTCA-3' (SEQ ID NO:61)
DMD-MiSeq-Rd1-fwd3-TCAG
5 '-CTCTTTCCCTACACGACGCTCTTCCGATCTtcagAATAAAAAGACATGGG
GCTTCA-3' (SEQ ID NO:62)
DMD-MiSeq-Rdl-fwd4-CAGT
5 '-CTCTTTCCCTACACGACGCTCTTCCGATCTcagtAATAAAAAGACATGGG
GCTTCA-3' (SEQ ID NO:63)
DMD-MiSeq-Rd1-fwd5-ATGC
5 '-CTCTTTCCCTACACGACGCTCTTCCGATCTatgcAATAAAAAGACATGGG
GCTTCA-3' (SEQ ID NO:64)
DMD-MiSeq-Rdl-fwd6-TGCA
5 '-CTCTTTCCCTACACGACGCTCTTCCGATCTtgcaAATAAAAAGACATGGG
GCTTCA-3' (SEQ ID NO:65)
DMD-MiSeq-Rd1-fwd7-GCAT
5 '-CTCTTTCCCTACACGACGCTCTTCCGATCTgcatAATAAAAAGACATGGG
GCTTCA-3' (SEQ ID NO:66)
DMD-MiSeq-Rd1-fwd8-CATG
5 '-CTCTTTCCCTACACGACGCTCTTCCGATCTcatgAATAAAAAGACATGGG
GCTTCA-3' (SEQ ID NO:67)
DMD-MiSeq-Rd1-fwd9-AACG
5 '-CTCTTTCCCTACACGACGCTCTTCCGATCTaacgAATAAAAAGACATGGG
GCTTCA-3' (SEQ ID NO:68)
DMD-MiSeq-Rdl-fwd10-ACGA
CA 03058584 2019-09-30
' = 41
'-CTCTTTCCCTACACGACGCTCTTCCGATCTacgaAATAAAAAGACATGGG
GCTTCA-3' (SEQ ID NO:69)
DMD-MiSeq-Rdl-fwdll-CGAA
5 '-CTCTTTCCCTACACGACGCTCTTCCGATCTcgaaAATAAAAAGACATGGG
5 GCTTCA-3' (SEQ ID NO:70)
DMD-MiSeq-Rd1-fwd12-GAAC
5' -CTCTTTCCCTACACGACGCTCTTCCGATCTgaacAATAAAAAGACATGGG
GCTTCA-3' (SEQ ID NO:71)
DMD-MiSeq-Rdl-fwd13-TACC
5 '-CTCTTTCCCTACACGACGCTCTTCCGATCTtaccAATAAAAAGACATGGG
GCTTCA-3' (SEQ ID NO:72)
DMD-MiSeq-Rd 1 -fwd14-ACCT
5 '-CTCTTTCCCTACACGACGCTCTTCCGATCTacctAATAAAAAGACATGGG
GCTTCA-3' (SEQ ID NO:73)
[0060]
DMD-MiSeq-Rd2-revl-AGTC
5 '-CTGGAGTTCAGACGTGTGCTCTTCCGATCTagtcCTGGCATCTGTTTTTGA
GGA-3' (SEQ ID NO:74)
DMD-MiSeq-Rd2-rev2-GTCA
5 '-CTGGAGTTCAGACGTGTGCTCTTCCGATCTgtcaCTGGCATCTGTTTTTGA
GGA-3' (SEQ ID NO:75)
DMD-MiSeq-Rd2-rev3-TCAG
5 '-CTGGAGTTCAGACGTGTGCTCTTCCGATCTtcagCTGGCATCTGTTTTTGA
GGA-3' (SEQ ID NO:76)
DMD-MiSeq-Rd2-rev4-CAGT
5 '-CTGGAGTTCAGACGTGTGCTCTTCCGATCTcagtCTGGCATCTGTTTTTGA
GGA-3' (SEQ ID NO:77)
CA 03058584 2019-09-30
*
' 42
DMD-MiSeq-Rd2-rev5-ATGC
5' -CTGGAGTTCAGACGTGTGCTCTTCCGATCTatgcCTGGCATCTGTTTTTGA
GGA-3' (SEQ ID NO:78)
DMD-MiSeq-Rd2-rev6-TGCA
5 '-CTGGAGTTCAGACGTGTGCTCTTCCGATCTtgcaCTGGCATCTGTTTTTGA
GGA-3' (SEQ ID NO:79)
DMD-MiSeq-Rd2-rev7-GCAT
5 '-CTGGAGTTCAGACGTGTGCTCTTCCGATCTgcatCTGGCATCTGTTTTTGA
GGA-3' (SEQ ID NO:80)
DMD-MiSeq-Rd2-rev8-CATG
5 '-CTGGAGTTCAGACGTGTGCTCTTCCGATCTcatgCTGGCATCTGTTTTTGA
GGA-3' (SEQ ID NO:81)
DMD-MiSeq-Rd2-rev9-AACG
5 '-CTGGAGTTCAGACGTGTGCTCTTCCGATCTaacgCTGGCATCTGTTTTTG
AGGA-3' (SEQ ID NO:82)
DMD-MiSeq-Rd2-rev10-ACGA
5 '-CTGGAGTTCAGACGTGTGCTCTTCCGATCTacgaCTGGCATCTGTTTTTG
AGGA-3' (SEQ ID NO:83)
DMD-MiSeq-Rd2-rev11-CGAA
5 '-CTGGAGTTCAGACGTGTGCTCTTCCGATCTcgaaCTGGCATCTGTTTTTG
AGGA-3' (SEQ ID NO:84)
DMD-MiSeq-Rd2-rev12-GAAC
5 '-CTGGAGTTCAGACGTGTGCTCTTCCGATCTgaacCTGGCATCTGTTTTTG
AGGA-3 ' (SEQ ID NO:85)
DMD-MiSeq-Rd2-rev13-TACC
5 '-CTGGAGTTCAGACGTGTGCTCTTCCGATCTtaccCTGGCATCTGTTTTTGA
GGA-3' (SEQ ID NO:86)
CA 03058584 2019-09-30
v 43
DMD-MiSeq-Rd2-rev14-ACCT
5'-CTGGAGTTCAGACGTGTGCTCTTCCGATCTacctCTGGCATCTGTTTTTGA
GGA-3' (SEQ ID NO:87)
[0061]
<Construction of Exon Skipping Detection Vector Using Luc Reporter>
Luc2 cDNA was amplified by PCR from pGL4-CMV-luc2 (Promega), and
then cloned into the pENTR-D-TOPO vector (Thermo Fisher Scientific Inc.), to
construct the pENTR-D-TOPO-Luc2 vector. The pENTR-D-TOPO-Luc2 was
cleaved with Nan and AgeI, and then the following intron sequence synthesized
by
gBlock (IDT) and the DMD exon 45 sequence were inserted thereto, to construct
the
pENTR-D-TOPO-Luc2-DMD-intron-Ex45[+] vector. Subsequently, the gBlock
sequence was cleaved at the two Sall sites present in both sides of hEx45, and
then
the vector side was re-ligated to construct the pENTR-D-TOPO-Luc2-DMD-intron-
Ex45[-] vector.
[0062]
Nad-AgeI-DMD-Ex45-gBlock (Sequence of Fig. 6)
GCCAGCGGCGGGGCGCCGCTCAGCAAGGAGGTAGGTGAGGCCGTGGCCA
AACGCTTCCACCTACCAGGTAAGTCTTTGATTTGTCGACCGTATCCACGA
TCACTAAGAAACCCAAATACTTTGTTCATGTTTAAATTTTACAACATTTCA
TAGACTATTAAACATGGAACATCCTTGTGGGGACAAGAAATCGAATTTGC
TCTTGAAAAGGTTTCCAACTAATTGATTTGTAGGACATTATAACATCCTCT
AGCTGACAAGCTTACAAAAATAAAAACTGGAGCTAACCGAGAGGGTGCT
TTTTTCCCTGACACATAAAAGGTGTCTTTCTGTCTTGTATCCTTTGGATAT
GGGCATGTCAGTTTCATAGGGAAATTTTCACATGGAGCTTTTGTATTTCTT
TCTTTGCCAGTACAACTGCATGTGGTAGCACACTGTTTAATCTTTTCTCAA
ATAAAAAGACATGGGGCTTCATTTTTGTTTTGCCTTTTTGGTATCTTACAG
GAACTCCAGGATGGCATTGGGCAGCGGCAAACTGTTGTCAGAACATTGA
CA 03058584 2019-09-30
=
44
ATGCAACTGGGGAAGAAATAATTCAGCAATCCTCAAAAACAGATGCCAG
TATTCTACAGGAAAAATTGGGAAGCCTGAATCTGCGGTGGCAGGAGGTC
TGCAAACAGCTGTCAGACAGAAAAAAGAGGTAGGGCGACAGATCTAATA
GGAATGAAAACATTTTAGCAGACTTTTTAAGCTTTCTTTAGAAGAATATT
TCATGAGAGATTATAAGCAGGGTGAAAGGCGTCGACGTTTGCATTAACA
AATAGTTTGAGAACTATGTTGGAAAAAAAAATAACAATTTTATTCTTCTT
TCTCCAGGCATCCGCCAGGGCTACGGCCTGACAGAAACAACCAGCGCCA
TTCTGATCACCCCCGAAGGGGACGACAAGCCTGGCGCAGTAGGCAAGGT
GGTGCCCTTCTTCGAGGCTAAGGTGGTGGACTTGGACACCGGTAAGACAC
TGG (SEQ ID NO:88)
[0063]
The piggyBac 5'TR (Terminal repeat) and 3'TR sequences derived from
Trichoplusia ni were synthesized (DT) as three separate gBlocks sequences
(gBlockll to 13-PV-3'TR-5'TR), and the three fragments were linked to each
other
by PCR, followed by insertion into the Aatil-Pvull site in the pUC 19 vector
by the
In-Fusion reaction, to construct the pPV-synthesized vector.
[0064]
gBlockll-PV-3 ' TR-5 ' TR
GAAAAGTGCCACCTGACGTCATCTGTTAACATTATACGCGTTTAACCCTA
GAAAGATAATCATATTGTGACGTACGTTAAAGATAATCATGCGTAAAATT
GACGCATGTGTTTTATCGGTCTGTATATCGAGGTTTATTTATTAATTTGAA
TAGATATTAAGTTTTATTATATTTACACTTACATACTAATAATAAATTCAA
CAAACAATTTATTTATGTTTATTTATTTATTAAAAAAAAACAAAAACTCA
AAATTTCTTCTATAAAGTAACAAAACTTTTAAACATTCTCTCTTTTACAAA
AATAAACTTATTTTGTACTTTAAAAACAGTCATGTTGTATTATAAAATAA
GTAATTAGCTTAACCTATACATAATAGAAACAAATTATACTTA (SEQ ID
NO:89)
CA 03058584 2019-09-30
' 45
[0065]
gBlock12-PV-3 'TR-5 'TR
CCTATACATAATAGAAACAAATTATACTTATTAGTCAGTCAGAAACAACT
TTGGCACATATCAATATTATGCTCTGCTAGCGATATCTGTAAAACGACGG
CCAGTTCTAGACTTAAGCTTCATGGTCATAGCTGTTTCCTGCTCGAGTTAA
TTAACCAACAAGCTCGTCATCGCTTTGCAGAAGAGCAGAGAGGATATGCT
CATCGTCTAAAGAACTACCCATTTTATTATATATTAGTCACGATATCTATA
ACAAGAAAATATATATATAATAAGTTATCACGTAAGTAGAACATGAAAT
AACAATA (SEQ ID NO:90)
[0066]
gBlock13-PV-3 'TR-5 'TR
ATCACGTAAGTAGAACATGAAATAACAATATAATTATCGTATGAGTTAAA
TCTTAAAAGTCACGTAAAAGATAATCATGCGTCATTTTGACTCACGCGGT
CGTTATAGTTCAAAATCAGTGACACTTACCGCATTGACAAGCACGCCTCA
CGGGAGCTCCAAGCGGCGACTGAGATGTCCTAAATGCACAGCGACGGAT
TCGCGCTATTTAGAAAGAGAGAGCAATATTTCAAGAATGCATGCGTCAAT
TTTACGCAGACTATCTTTCTAGGGTTAATACGTATAATACATATGATTCAG
CTGCATTAATGAATC (SEQ ID NO:91)
[0067]
The PB-EFla-GW-iP vector (Masui H et al., PLOS ONE, 2014 Aug 15; 9(8):
e104957.) was cleaved with NheI-PacI, and then ligated to the NheI-PacI site
of pPV-
synthesized, to construct the pPV-EFla-GW-iP vector. Subsequently, a rabbit-
derived hemoglobin poly A signal was amplified from the pOCLE-EGFP vector
(Okita K et al., Nat Methods, 2011 May; 8(5): 409-12.) using the pHL-PacI-rHBB-
2 5 pA-IF-fw primer (5'-GTATACCTCGAGTTAAATTCACTCCTCAGGTGC-3' (SEQ
ID NO:92)) and the pPV-PacI-rHBB-pA-IF-rev primer (5'-
CGAGCTTGTTGGTTAATTAAGTCGAGGGATCTCCATAA-3' (SEQ ID NO :93)),
CA 03058584 2019-09-30
' ' 46
and then inserted into the Pad site of the pPV-EFla-GW-iP vector using the In-
Fusion reaction, to construct the pPV-EFla-GW-iP-A vector. By the Gateway LR
reaction using the pPV-EFla-GW-iP-A vector and pENTR-D-TOPO-Luc2-DMD-
intron-Ex45 [+], the pPV-EFla-Luc2-hDMD-Ex45[+]-iP-A vector was constructed.
Further, by the Gateway LR reaction using the pPV-EFla-GW-iP-A vector and
pENTR-D-TOPO-Luc2-DMD-intron-Ex45 [-], the pPV-EFla-Luc2-10MD-Ex45-[-]-
iP-A vector was constructed.
[0068]
<Introduction of Luc2 V323I (G867A) Mutation>
Using the piggyBac vector pPV-EFla-Luc2-hDMD-Ex45[-] vector as a
template, PCR was carried out separately using, as primers, the combination of
Luc2-
NcoI-IF-Fwd and Luc2-V3231-fwd, and the combination of Luc2-V3231-rev and
Luc2-SalI-IF-Rev. The two amplified fragments were mixed together, and the
primers at both ends (Luc2-NcoI-IF-Fwd and Luc2-SalI-T-Rev) were used to
prepare
a fragment having a mutation, followed by inserting the fragment into the NcoI-
SalI
cleavage site of the pPV-EFla-Luc2-hDMD-Ex45[-] vector by the In-Fusion
reaction,
to construct the pPV-EFla-Luc2(V3231)-hDMD-Ex45[-] vector.
Luc2-NcoI-IF-Fwd GCCCCCTTCACCATGGAAG (SEQ ID NO:94)
Luc2-V3231-fwd CAGCAAGGAGATAGGTGAGG (SEQ ID NO:95)
Luc2-V3231-rev CCTCACCTATCTCCTTGCTG (SEQ ID NO:96)
Luc2-SalI-IF-Rev TAATGCAAACGTCGACAAATCAAAGAC (SEQ ID
NO:97)
[0069]
<Insertion of 1-kb, 2-kb, 4-kb, or 0.7-kb hDMD Exon 45, and Intron Sequences
in
Vicinity Thereof>
PCR amplification was carried out using pPV-EFla-Luc2-hDMD-Ex45 [+]-
iP-A as a template, and using the DMD-Ex45-SalI-IF-F and DMD-Ex45-SalI-IF-R
CA 03058584 2019-09-30
47
primers. The resulting product was inserted into the Sail cleavage site of the
pPV-
EFla-Luc2(V3231)-hDMD-Ex45[-] vector by the In-Fusion reaction, to construct
the
pPV-EFla-Luc2(V3230-hDMD-Ex45[+] (0.7 kb) vector.
DMD-Ex45-Sa1I-T-F tattgatttGTCGACcgtatc (SEQ ID NO:98)
DMD-Ex45-SalI-IF-R taatgcaaacGTCGACgcc (SEQ ID NO:99)
[0070]
Using, as a template, human genomic DNA prepared from 1383D2 cells, and
using the following primers (HDMD-SR-XkbFrag-fwd & rev), exon 45 and the
introns in the vicinity thereof were amplified. The resulting product was
inserted
into the Sall cleavage site of the pPV-EFla-Luc2(V3231)-hDMD-Ex45[-] vector by
the In-Fusion reaction, to construct the pPV-EFla-Luc2(V3231)-hDMD-Ex45[+] (1
kb), pPV-EFla-Luc2(V3230-hDMD-Ex45[+] (2 kb), and pPV-EFla-Luc2(V3231)-
hDMD-Ex45[+] (4 kb) vectors.
HDMD-SR-1kbFrag-fwd tctttgatttGTCGAGGGATATCTTGATGGGATGCTCC
(SEQ ID NO:100)
HDMD-SR-1kbFrag-rev taatgcaaacGTCGAAAACCACTAACTAGCCACAAG
T (SEQ ID NO:101)
HDMD-SR-2kbFrag-fwd tattgatttGTCGAATTGTGAGGCACCGTGTCAC (SE
Q ID NO:102)
HDMD-SR-2kbFrag-rev taatgcaaacGTCGACTCTTTGGCTCAAGTTCCCCT
(SEQ ID NO:103)
HDMD-SR-4kbFrag-fwd tattgatttGTCGAGCTGCAGCATTAGTTTATAGCA
(SEQ ID NO:104)
HDMD-SR-4kbFrag-rev taatgcaaacGTCGAAACTTTGGCAAGGGGTGTGT
(SEQ ID NO:105)
[0071]
The sequences of the exon skipping reporter cDNA portion constructed are
CA 03058584 2019-09-30
= = 48
shown below.
Luc2(V3231)-hDMD-Ex45 [-]
ATGGAAGATGCCAAAAACATTAAGAAGGGCCCAGCGCCATTCTACCCAC
TCGAAGACGGGACCGCCGGCGAGCAGCTGCACAAAGCCATGAAGCGCTA
CGCCCTGGTGCCCGGCACCATCGCCTTTACCGACGCACATATCGAGGTGG
ACATTACCTACGCCGAGTACTTCGAGATGAGCGTTCGGCTGGCAGAAGCT
ATGAAGCGCTATGGGCTGAATACAAACCATCGGATCGTGGTGTGCAGCG
AGAATAGCTTGCAGTTCTTCATGCCCGTGTTGGGTGCCCTGTTCATCGGTG
TGGCTGTGGCCCCAGCTAACGACATCTACAACGAGCGCGAGCTGCTGAA
CAGCATGGGCATCAGCCAGCCCACCGTCGTATTCGTGAGCAAGAAAGGG
CTGCAAAAGATCCTCAACGTGCAAAAGAAGCTACCGATCATACAAAAGA
TCATCATCATGGATAGCAAGACCGACTACCAGGGCTTCCAAAGCATGTAC
ACCTTCGTGACTTCCCATTTGCCACCCGGCTTCAACGAGTACGACTTCGT
GCCCGAGAGCTTCGACCGGGACAAAACCATCGCCCTGATCATGAACAGT
AGTGGCAGTACCGGATTGCCCAAGGGCGTAGCCCTACCGCACCGCACCG
CTTGTGTCCGATTCAGTCATGCCCGCGACCCCATCTTCGGCAACCAGATC
ATCCCCGACACCGCTATCCTCAGCGTGGTGCCATTTCACCACGGCTTCGG
CATGTTCACCACGCTGGGCTACTTGATCTGCGGCTTTCGGGTCGTGCTCAT
GTACCGCTTCGAGGAGGAGCTATTCTTGCGCAGCTTGCAAGACTATAAGA
TTCAATCTGCCCTGCTGGTGCCCACACTATTTAGCTTCTTCGCTAAGAGCA
CTCTCATCGACAAGTACGACCTAAGCAACTTGCACGAGATCGCCAGCGG
CGGGGCGCCGCTCAGCAAGGAGaTAGGTGAGGCCGTGGCCAAACGCTTC
CACCTACCAGgtaagtetttgatnGTCGACgtttgcattaacaaatagngagaactatgttggaaaa a a a RR
taacaattttattcttctttctecagGCATCCGCCAGGGCTACGGCCTGACAGAAACAACC
AGCGCCATTCTGATCACCCCCGAAGGGGACGACAAGCCTGGCGCAGTAG
GCAAGGTGGTGCCCTTCTTCGAGGCTAAGGTGGTGGACTTGGACACCGGT
AAGACACTGGGTGTGAACCAGCGCGGCGAGCTGTGCGTCCGTGGCCCCA
CA 03058584 2019-09-30
= 49
TGATCATGAGCGGCTACGTTAACAACCCCGAGGCTACAAACGCTCTCATC
GACAAGGACGGCTGGCTGCACAGCGGCGACATCGCCTACTGGGACGAGG
ACGAGCACTTCTTCATCGTGGACCGGCTGAAGAGCCTGATCAAATACAAG
GGCTACCAGGTAGCCCCAGCCGAACTGGAGAGCATCCTGCTGCAACACC
CCAACATCTTCGACGCCGGGGTCGCCGGCCTGCCCGACGACGATGCCGG
CGAGCTGCCCGCCGCAGTCGTCGTGCTGGAACACGGTAAAACCATGACC
GAGAAGGAGATCGTGGACTATGTGGCCAGCCAGGTTACAACCGCCAAGA
AGCTGCGCGGTGGTGTTGTGTTCGTGGACGAGGTGCCTAAAGGACTGACC
GGCAAGTTGGACGCCCGCAAGATCCGCGAGATTCTCATTAAGGCCAAGA
AGGGCGGCAAGATCGCCGTGTAA (SEQ ID NO:106)
Luc2(V323I)-hDMD-Ex45[+]
ATGGAAGATGCCAAAAACATTAAGAAGGGCCCAGCGCCATTCTACCCAC
TCGAAGACGGGACCGCCGGCGAGCAGCTGCACAAAGCCATGAAGCGCTA
CGCCCTGGTGCCCGGCACCATCGCCTTTACCGACGCACATATCGAGGTGG
ACATTACCTACGCCGAGTACTTCGAGATGAGCGTTCGGCTGGCAGAAGCT
ATGAAGCGCTATGGGCTGAATACAAACCATCGGATCGTGGTGTGCAGCG
AGAATAGCTTGCAGTTCTTCATGCCCGTGTTGGGTGCCCTGTTCATCGGTG
TGGCTGTGGCCCCAGCTAACGACATCTACAACGAGCGCGAGCTGCTGAA
CAGCATGGGCATCAGCCAGCCCACCGTCGTATTCGTGAGCAAGAAAGGG
CTGCAAAAGATCCTCAACGTGCAAAAGAAGCTACCGATCATACAAAAGA
TCATCATCATGGATAGCAAGACCGACTACCAGGGCTTCCAAAGCATGTAC
ACCTTCGTGACTTCCCATTTGCCACCCGGCTTCAACGAGTACGACTTCGT
GCCCGAGAGCTTCGACCGGGACAAAACCATCGCCCTGATCATGAACAGT
AGTGGCAGTACCGGATTGCCCAAGGGCGTAGCCCTACCGCACCGCACCG
CTTGTGTCCGATTCAGTCATGCCCGCGACCCCATCTTCGGCAACCAGATC
ATCCCCGACACCGCTATCCTCAGCGTGGTGCCATTTCACCACGGCTTCGG
CA 03058584 2019-09-30
6
CATGTTCACCACGCTGGGCTACTTGATCTGCGGCTTTCGGGTCGTGCTCAT
GTACCGCTTCGAGGAGGAGCTATTCTTGCGCAGCTTGCAAGACTATAAGA
TTCAATCTGCCCTGCTGGTGCCCACACTATTTAGCTTCTTCGCTAAGAGCA
CTCTCATCGACAAGTACGACCTAAGCAACTTGCACGAGATCGCCAGCGG
5 CGGGGCGCCGCTCAGCAAGGAGaTAGGTGAGGCCGTGGCCAAACGCTTC
CACCTACCAGgtaagtctttgatttGTCGACcgtatccacgatcactaagaaacccaaatactttgttcatgttta
aattttacaacatttcatagactattaaacatggaacatccttgtggggacaagaaatcgaatttgctcttgaaaaggt
ttccaa
ctaattgatttgtaggacattataacatcctctagctgacaagcttaca a ana
taaaaactggagctaaccgagagggtgcttt
tttccctgacacataaaaggtgtctttctgtettgtatcctttggatatgggcatgtcagtticatagggaaattttca
catggagc
10
ttttgtatttctttctttgccagtacaactgcatgtggtagcacactgtttaatcttttctcaaatannaagacatggg
gcTTCA
TTtttgttttgcctttttggtatcttacagGAACTCCAGGATGGCATTGGGCAGCGGCAAACT
GTTGTCAGAACATTGAATGCAACTGGGGAAGAAATAATTCAGCAATCCTC
AAAAACAGATGCCAGTATTCTACAGGAAAAATTGGGAAGCCTGAATCTG
CGGTGGCAGGAGGTCTGCAAACAGCTGTCAGACAGAAAAAAGAGgtagggc
15 gacagatctaataggaatga 22a
cattttagcagactttttaagetttctttagaagaatatttcatgagagattataagcagggt
pa aggcGTCGACgtttgcattaacaaatagtttgagaactatgttggaaaaaa
2nataacaattttattcttattctcca
gGCATCCGCCAGGGCTACGGCCTGACAGAAACAACCAGCGCCATTCTGAT
CACCCCCGAAGGGGACGACAAGCCTGGCGCAGTAGGCAAGGTGGTGCCC
TTCTTCGAGGCTAAGGTGGTGGACTTGGACACCGGTAAGACACTGGGTGT
20 GAACCAGCGCGGCGAGCTGTGCGTCCGTGGCCCCATGATCATGAGCGGC
TACGTTAACAACCCCGAGGCTACAAACGCTCTCATCGACAAGGACGGCT
GGCTGCACAGCGGCGACATCGCCTACTGGGACGAGGACGAGCACTTCTT
CATCGTGGACCGGCTGAAGAGCCTGATCAAATACAAGGGCTACCAGGTA
GCCCCAGCCGAACTGGAGAGCATCCTGCTGCAACACCCCAACATCTTCGA
25 CGCCGGGGTCGCCGGCCTGCCCGACGACGATGCCGGCGAGCTGCCCGCC
GCAGTCGTCGTGCTGGAACACGGTAAAACCATGACCGAGAAGGAGATCG
TGGACTATGTGGCCAGCCAGGTTACAACCGCCAAGAAGCTGCGCGGTGG
CA 03058584 2019-09-30
. . 51
TGTTGTGTTCGTGGACGAGGTGCCTAAAGGACTGACCGGCAAGTTGGACG
CCCGCAAGATCCGCGAGATTCTCATTAAGGCCAAGAAGGGCGGCAAGAT
CGCCGTGTAA (SEQ ID NO:107)
Luc2(V323I)-hDMD-Ex45 [+] (1 kb)
ATGGAAGATGCCAAAAACATTAAGAAGGGCCCAGCGCCATTCTACCCAC
TCGAAGACGGGACCGCCGGCGAGCAGCTGCACAAAGCCATGAAGCGCTA
CGCCCTGGTGCCCGGCACCATCGCCTTTACCGACGCACATATCGAGGTGG
ACATTACCTACGCCGAGTACTTCGAGATGAGCGTTCGGCTGGCAGAAGCT
ATGAAGCGCTATGGGCTGAATACAAACCATCGGATCGTGGTGTGCAGCG
AGAATAGCTTGCAGTTCTTCATGCCCGTGTTGGGTGCCCTGTTCATCGGTG
TGGCTGTGGCCCCAGCTAACGACATCTACAACGAGCGCGAGCTGCTGAA
CAGCATGGGCATCAGCCAGCCCACCGTCGTATTCGTGAGCAAGAAAGGG
CTGCAAAAGATCCTCAACGTGCAAAAGAAGCTACCGATCATACAAAAGA
TCATCATCATGGATAGCAAGACCGACTACCAGGGCTTCCAAAGCATGTAC
ACCTTCGTGACTTCCCATTTGCCACCCGGCTTCAACGAGTACGACTTCGT
GCCCGAGAGCTTCGACCGGGACAAAACCATCGCCCTGATCATGAACAGT
AGTGGCAGTACCGGATTGCCCAAGGGCGTAGCCCTACCGCACCGCACCG
CTTGTGTCCGATTCAGTCATGCCCGCGACCCCATCTTCGGCAACCAGATC
ATCCCCGACACCGCTATCCTCAGCGTGGTGCCATTTCACCACGGCTTCGG
CATGTTCACCACGCTGGGCTACTTGATCTGCGGCTTTCGGGTCGTGCTCAT
GTACCGCTTCGAGGAGGAGCTATTCTTGCGCAGCTTGCAAGACTATAAGA
TTCAATCTGCCCTGCTGGTGCCCACACTATTTAGCTTCTTCGCTAAGAGCA
CTCTCATCGACAAGTACGACCTAAGCAACTTGCACGAGATCGCCAGCGG
CGGGGCGCCGCTCAGCAAGGAGaTAGGTGAGGCCGTGGCCAAACGCTTC
CACCTACCAGgtaagtctttgatttGTCGAAGCACGCATTTGGCTTTCTGTGCCTTC
AATACATTCCAAGGGAAATTTAAATGATGATTGAATTTGACAGTAACCTT
CA 03058584 2019-09-30
=
= 52
TTTGAGGTTTTGTTTTCCCCATTAAACTTGTACCTCTTTGGCTCAAGTTCC
CCTTCAAGAATGTATTCACAAATGTGGTGAAACTAGAGGTAAGTGACACT
ATCACTTTTTTTAGCTTCATAGTCATATTCATAGCTATTTTTAAAACTAAG
CAAAGATCTGTCTTTCCTACAAAACAATCATTTATAATTGCTTTCTAAAAT
CTTCTTGAAAAACAACTGAGATTCAGCTTGTTGAAGTTAAAATATATTGA
AGATATTCACCTTTAAGCAATCATGGGTGATTTTTAAAGCAAACTTCAAG
TTTAAAATAGCAGAAAACCACTAACTAGCCACAAGTATATATTTTAGTAT
ATGAAAAAAAGAAATAAAAAATTTCTTTACTGCTGTTGATTAATGGTTGA
TAGGTTCTTTAATGTTAGTGCCTTTCACCCTGCTTATAATCTCTCATGAAA
TATTCTTCTAAAGAAAGCTTAAAAAGTCTGCTAAAATGTTTTCATTCCTAT
TAGATCTGTCGCCCTACCTCTTTTTTCTGTCTGACAGCTGTTTGCAGACCT
CCTGCCACCGCAGATTCAGGCTTCCCAATTTTTCCTGTAGAATACTGGCA
TCTGTTTTTGAGGATTGCTGAATTATTTCTTCCCCAGTTGCATTCAATGTT
CTGACAACAGTTTGCCGCTGCCCAATGCCATCCTGGAGTTCCTGTAAGAT
ACCAAAAAGGCAAAACAAAAATGAAGCCCCATGTCTTTTTATTTGAGAA
AAGATTAAACAGTGTGCTACCACATGCAGTTGTACTGGCAAAGAAAGAA
ATACAAAAGCTCCATGTGAAAATTTCCCTATGAAACTGACATGCCCTCGA
CgtttgcattaacaaatagtttgagaactatgttggaaaaaaaaataacaattttattcttctttctccagGCATCCGC
C
AGGGCTACGGCCTGACAGAAACAACCAGCGCCATTCTGATCACCCCCGA
AGGGGACGACAAGCCTGGCGCAGTAGGCAAGGTGGTGCCCTTCTTCGAG
GCTAAGGTGGTGGACTTGGACACCGGTAAGACACTGGGTGTGAACCAGC
GCGGCGAGCTGTGCGTCCGTGGCCCCATGATCATGAGCGGCTACGTTAAC
AACCCCGAGGCTACAAACGCTCTCATCGACAAGGACGGCTGGCTGCACA
GCGGCGACATCGCCTACTGGGACGAGGACGAGCACTTCTTCATCGTGGA
CCGGCTGAAGAGCCTGATCAAATACAAGGGCTACCAGGTAGCCCCAGCC
GAACTGGAGAGCATCCTGC TGCAACACCCCAACATCTTCGACGCCGGGG
TCGCCGGCCTGCCCGACGACGATGCCGGCGAGCTGCCCGCCGCAGTCGTC
CA 03058584 2019-09-30
53
GTGCTGGAACACGGTAAAACCATGACCGAGAAGGAGATCGTGGACTATG
TGGCCAGCCAGGTTACAACCGCCAAGAAGCTGCGCGGTGGTGTTGTGTTC
GTGGACGAGGTGCCTAAAGGACTGACCGGCAAGTTGGACGCCCGCAAGA
TCCGCGAGATTCTCATTAAGGCCAAGAAGGGCGGCAAGATCGCCGTGTA
A (SEQ ID NO:108)
Luc2(V323I)-hDMD-Ex45[+] (2 kb)
ATGGAAGATGCCAAAAACATTAAGAAGGGCCCAGCGCCATTCTACCCAC
TCGAAGACGGGACCGCCGGCGAGCAGCTGCACAAAGCCATGAAGCGCTA
CGCCCTGGTGCCCGGCACCATCGCCTTTACCGACGCACATATCGAGGTGG
ACATTACCTACGCCGAGTACTTCGAGATGAGCGTTCGGCTGGCAGAAGCT
ATGAAGCGCTATGGGCTGAATACAAACCATCGGATCGTGGTGTGCAGCG
AGAATAGCTTGCAGTTCTTCATGCCCGTGTTGGGTGCCCTGTTCATCGGTG
TGGCTGTGGCCCCAGCTAACGACATCTACAACGAGCGCGAGCTGCTGAA
CAGCATGGGCATCAGCCAGCCCACCGTCGTATTCGTGAGCAAGAAAGGG
CTGCAAAAGATCCTCAACGTGCAAAAGAAGCTACCGATCATACAAAAGA
TCATCATCATGGATAGCAAGACCGACTACCAGGGCTTCCAAAGCATGTAC
ACCTTCGTGACTTCCCATTTGCCACCCGGCTTCAACGAGTACGACTTCGT
GCCCGAGAGCTTCGACCGGGACAAAACCATCGCCCTGATCATGAACAGT
AGTGGCAGTACCGGATTGCCCAAGGGCGTAGCCCTACCGCACCGCACCG
CTTGTGTCCGATTCAGTCATGCCCGCGACCCCATCTTCGGCAACCAGATC
ATCCCCGACACCGCTATCCTCAGCGTGGTGCCATTTCACCACGGCTTCGG
CATGTTCACCACGCTGGGCTACTTGATCTGCGGCTTTCGGGTCGTGCTCAT
GTACCGCTTCGAGGAGGAGCTATTCTTGCGCAGCTTGCAAGACTATAAGA
TTCAATCTGCCCTGCTGGTGCCCACACTATTTAGCTTCTTCGCTAAGAGCA
CTCTCATCGACAAGTACGACCTAAGCAACTTGCACGAGATCGCCAGCGG
CGGGGCGCCGCTCAGCAAGGAGaTAGGTGAGGCCGTGGCCAAACGCTTC
CA 03058584 2019-09-30
I
54
CACCTACCAGgtaagtctttgatttGTCGATCTTTAACTTTGGCAAGGGGTGTGTGT
GTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTT
TAGGTCAACTAATGTGTTTATTTTGTACAAAATATGAATTGTATCTACTTT
CTGAATAATGTAACATGAATAAAGAGGGAAAGAGGAGGTGGGCAAAGA
CAACTGACATAATTCCAAAATCTTCTTTTTAATACATCTTAACGAAAGAT
ATTCATCAATGAGTTGTTCTAGCTTCCTGAATATTAAAATCCACCTATTAT
GTGGATGATGGGTGGGATGCAAGAGCTTGGCAAAAGAACGAAGTTTTCA
TTGTTCATAACAATAGTCTCATTTGGTAAATAAAGGC CAAGTCTTCCTTTA
CGAAACAAGACACATTAACATCAACAACTGGAAGCATAATACAAAATCC
CATTTATAAACTCTCTAGGCTTTCCAACTGCAGCAGCACGCATTTGGCTTT
CTGTGCCTTCAATACATTCCAAGGGAAATTTAAATGATGATTGAATTTGA
CAGTAACCTTTTTGAGGTTTTGTTTTCCCCATTAAACTTGTACCTCTTTGG
CTCAAGTTCCCCTTCAAGAATGTATTCACAAATGTGGTGAAACTAGAGGT
AAGTGACACTATCACTTTTTTTAGCTTCATAGTCATATTCATAGCTATITT
TAAAACTAAGCAAAGATCTGTCTTTCCTACAAAACAATCATTTATAATTG
CTTTCTAAAATCTTCTTGAAAAACAACTGAGATTCAGCTTGTTGAAGTTA
AAATATATTGAAGATATTCACCTTTAAGCAATCATGGGTGATTTTTAAAG
CAAACTTCAAGTTTAAAATAGCAGAAAACCACTAACTAGCCACAAGTAT
ATATTTTAGTATATGAAAAAAAGAAATAAAAAATTTCTTTACTGCTGTTG
ATTAATGGTTGATAGGTTCTTTAATGTTAGTGCCTTTCACCCTGCTTATAA
TCTCTCATGAAATATTCTTCTAAAGAAAGCTTAAAAAGTCTGCTAAAATG
TTTTCATTCCTATTAGATCTGTCGCCCTACCTCTTTTTTCTGTCTGACAGCT
GTTTGCAGACCTCCTGC CAC CGCAGATTCAGGCTTCCCAATTTTTC CTGTA
GAATACTGGCATCTGTTTTTGAGGATTGCTGAATTATTTCTTCCCCAGTTG
CATTCAATGTTCTGACAACAGTTTGCCGCTGCCCAATGCCATCCTGGAGT
TCCTGTAAGATACCAAAAAGGCAAAACAAAAATGAAGCCCCATGTCTTTT
TATTTGAGAAAAGATTAAACAGTGTGCTACCACATGCAGTTGTACTGGCA
CA 03058584 2019-09-30
= 55
AAGAAAGAAATACAAAAGCTCCATGTGAAAATTTCCCTATGAAACTGAC
ATGCCCATATCCAAAGGATACAAGACAGAAAGACACCTTTTATGTGTCAG
GGAAAAAAGCACCCTCTCGGTTAGCTCCAGTTTTTATTTTTGTAAGCTTGT
CAGCTAGAGGATGTTATAATGTCCTACAAATCAATTAGTTGGAAACCTTT
TCAAGAGCAAATTCGATTTCTTGTCCCCACAAGGATGTTCCATGTTTAAT
AGTCTATGAAATGTTGTAAAATTTAAACATGAACAAAGTATTTGGGTTTC
TTAGTGATCGTGGATACGAGAGGTGAAAAAGAACAAACATAGGTTAGTC
ACAGTATTAAAAAAAAACTCTAGAGATATTTAAATAAAATTAATTGCTAT
ATTAGAAGAAAATTCATTTCAAATTCTGTCTGCGTCAATGTATTTTGCATT
AGAAGCCACAAAAAACTGAGAATTAATTGCTTTCAGGAGCATCCCATCA
AGATATCCCTAAGCTACAGTAATAAATTTTAAAATAATCTATAGTCACCA
GAGCATTTTTATGATTGTCATC GACgtttgcattaacaaatagtttgagaactatgttggaaananaa
ataacaattttattcttctttctccagGCATCCGCCAGGGCTACGGCCTGACAGAAACAACC
AGCGCCATTCTGATCACCCCCGAAGGGGACGACAAGCCTGGCGCAGTAG
GCAAGGTGGTGCCCTTCTTCGAGGCTAAGGTGGTGGACTTGGACACCGGT
AAGACACTGGGTGTGAACCAGCGCGGCGAGCTGTGCGTCCGTGGCCCCA
TGATCATGAGCGGCTACGTTAACAACCCCGAGGCTACAAACGCTCTCATC
GACAAGGACGGCTGGCTGCACAGCGGCGACATCGCCTACTGGGACGAGG
ACGAGCACTTCTTCATCGTGGACCGGCTGAAGAGCCTGATCAAATACAAG
GGCTACCAGGTAGCCCCAGCCGAACTGGAGAGCATCCTGCTGCAACACC
CCAACATCTTCGACGCCGGGGTCGCCGGCCTGCCCGACGACGATGCCGG
CGAGCTGCCCGCCGCAGTCGTCGTGCTGGAACACGGTAAAACCATGACC
GAGAAGGAGATCGTGGACTATGTGGCCAGCCAGGTTACAACCGCCAAGA
AGCTGCGCGGTGGTGTTGTGTTCGTGGACGAGGTGCCTAAAGGACTGACC
GGCAAGTTGGACGCCCGCAAGATCCGCGAGATTCTCATTAAGGCCAAGA
AGGGCGGCAAGATCGCCGTGTAA (SEQ ID NO:109)
CA 03058584 2019-09-30
. . 56
Luc2(V3231)-hDMD-Ex45[+] (4 kb)
ATGGAAGATGCCAAAAACATTAAGAAGGGCCCAGCGCCATTCTACCCAC
TCGAAGACGGGACCGCCGGCGAGCAGCTGCACAAAGCCATGAAGCGCTA
CGCCCTGGTGCCCGGCACCATCGCCTTTACCGACGCACATATCGAGGTGG
ACATTACCTACGCCGAGTACTTCGAGATGAGCGTTCGGCTGGCAGAAGCT
ATGAAGCGCTATGGGCTGAATACAAACCATC GGATCGTGGTGTGCAGCG
AGAATAGCTTGCAGTTCTTCATGCCCGTGTTGGGTGCCCTGTTCATCGGTG
TGGCTGTGGCCCCAGCTAACGACATCTACAACGAGCGCGAGCTGCTGAA
CAGCATGGGCATCAGCCAGCCCACCGTCGTATTCGTGAGCAAGAAAGGG
CTGCAAAAGATCCTCAACGTGCAAAAGAAGCTACCGATCATACAAAAGA
TCATCATCATGGATAGCAAGACCGACTACCAGGGCTTCCAAAGCATGTAC
ACCTTCGTGACTTCCCATTTGCCACCCGGCTTCAACGAGTACGACTTCGT
GCCCGAGAGCTTCGACCGGGACAAAACCATCGCCCTGATCATGAACAGT
AGTGGCAGTACCGGATTGCCCAAGGGCGTAGCCCTACCGCACCGCACCG
CTTGTGTCCGATTCAGTCATGCCCGCGACCCCATCTTCGGCAACCAGATC
ATCCCCGACACCGCTATCCTCAGCGTGGTGCCATTTCACCACGGCTTCGG
CATGTTCACCACGCTGGGCTACTTGATCTGCGGCTTTCGGGTCGTGCTCAT
GTACCGCTTCGAGGAGGAGCTATTCTTGCGCAGCTTGCAAGACTATAAGA
TTCAATCTGCCCTGCTGGTGCCCACACTATTTAGCTTCTTCGCTAAGAGCA
CTCTCATCGACAAGTACGACCTAAGCAACTTGCACGAGATCGCCAGCGG
CGGGGCGCCGCTCAGCAAGGAGaTAGGTGAGGCCGTGGCCAAACGCTTC
CACCTACCAGgtaagtctttgatttGTCGATTTGCAACTACAGGGCTCCATATAGAC
ATCTAGCTTGAATTTATACACTTTCTTTCATTGATGTCCCTGGACTAAAAA
ATGTTAAATATTTCTAACCGCTGTACTTAAAGTCCATTACAAACGAAGAC
TACTGTTGTTAAGTTGAATAGGCATCTTATATATTTTTCACCGGTGCAATA
AATAACTTCTATTCCCTTCTAACATCTGCTTGCGTTGCACTGAGAGTACAC
TATTGATTAGCAATAGGTTCGTGATTACAGCCCTTCTATAATTAATTGTTA
CA 03058584 2019-09-30
=
57
GGTTAACATATTATTCATAAAATATTATTTTATTAATTTTTACTTGATTTG
CTACTGGATGCTTAGAAATAGCTATGAGTATATTGGTAGAACCAGTACTT
ATATTTTATTACATTTTTACATTTCATAAAATTTAAGTGATATAAAAATCC
TGAGGAAGTATGCCACAAAAGTGGTCTCAGTGGAAATTTAAATATGTTAA
CATTTATTTTTAAAATGTAGCGTGAAATAGACAACTTTAAAAGCTCAGCT
TAAAAAAAAAACTCAAGGAAGCTGAACTTGACTTTTTAAAGCACTGAAG
TGCAATATTTAATGTAGGTCAACATGTTTAAATGGGAAAA'FITTTTTCCTA
ATTACAGCCAAATCCCTAGCTGTAATTAACTTAAAATTTGTATACTATTTC
ACAACAGAGTCAGCATATACCACTTTCTTATAAAATTAGAAAGATCTAAA
ATTTTAGAGCTTATTTGGTGAAACAGGCATATTGCTACATCTTTGTTTATA
AATTATAATGTGCCTTTAGAGCCCAATAACAGATAACAAGATTTTGAAAA
TTCAGGTGAATTAGAGTTATCAGAGGGAATGTTAATACACTCTATTCAAA
TACTATATGAGTAAGACATTTAAAATAGGAAACAATACTTTATATATTAT
AGAAAAATAATCTTCCAGTCGATTTAATCCACTTTATGAATTCTCTCCGTA
TATATATATTTATAGTATGGTATTCAATTTTTTTAATTTTCTCATTTCTTAC
CATCTTAATTTGGATTAGATTGAGCCTAGTTCAGAAATGACATTATACAG
GTTTATACCTGTTCATAGTATAAGCACATCAGTTATCTAAATAATAAAAT
ACTTGTATGATTAAGAGAAGAATTTCAATCTGGGAAAAAAGTATATGACT
TACCTAAGGAAGTAGTTTAACTACAAAGTTTAGTTCTTTATTTTATCTATC
TATAATCAAGAAGATTTTCAAAACCAAGACTTAATTATTCAAAATATCTT
TTGATGAGGCTATAATTCTTTAACTTTGGCAAGGGGTGTGTGTGTGTGTGT
GTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTTTAGGTCAA
CTAATGTGTTTATTTTGTACAAAATATGAATTGTATC TACTTTCTGAATAA
TGTAACATGAATAAAGAGGGAAAGAGGAGGTGGGCAAAGACAACTGAC
ATAATTCCAAAATCTTCTTTTTAATACATCTTAACGAAAGATATTCATCAA
TGAGTTGTTCTAGCTTCCTGAATATTAAAATCCACCTATTATGTGGATGAT
GGGTGGGATGCAAGAGCTTGGCAAAAGAACGAAGTTTTCATTGTTCATA
CA 03058584 2019-09-30
=
58
ACAATAGTCTCATTTGGTAAATAAAGGCCAAGTCTTCCTTTACGAAACAA
GACACATTAACATCAACAACTGGAAGCATAATACAAAATCCCATTTATAA
ACTCTCTAGGCTTTCCAACTGCAGCAGCACGCATTTGGCTTTCTGTGCCTT
CAATACATTCCAAGGGAAATTTAAATGATGATTGAATTTGACAGTAACCT
TTTTGAGGTTTTGTTTTCCCCATTAAACTTGTACCTCTTTGGCTCAAGTTCC
CCTTCAAGAATGTATTCACAAATGTGGTGAAACTAGAGGTAAGTGACACT
ATCACTTTTTTTAGCTTCATAGTCATATTCATAGCTATTTTTAAAACTAAG
CAAAGATCTGTCTTTCCTACAAAACAATCATTTATAATTGCTTTCTAAAAT
CTTCTTGAAAAACAACTGAGATTCAGCTTGTTGAAGTTAAAATATATTGA
AGATATTCACCTTTAAGCAATCATGGGTGATTTTTAAAGCAAACTTCAAG
TTTAAAATAGCAGAAAACCACTAACTAGCCACAAGTATATATTTTAGTAT
ATGAAAAAAAGAAATAAAAAATTTCTTTACTGCTGTTGATTAATGGTTGA
TAGGTTCTTTAATGTTAGTGCCTTTCACCCTGCTTATAATCTCTCATGAAA
TATTCTTCTAAAGAAAGCTTAAAAAGTCTGCTAAAATGTTTTCATTCCTAT
TAGATCTGTCGCCCTACCTCTTTTTTCTGTCTGACAGCTGTTTGCAGACCT
CCTGCCACCGCAGATTCAGGCTTCCCAATTTTTCCTGTAGAATACTGGCA
TCTGTTTTTGAGGATTGCTGAATTATTTCTTCCCCAGTTGCATTCAATGTT
CTGACAACAGTTTGCCGCTGCCCAATGCCATCCTGGAGTTCCTGTAAGAT
ACCAAAAAGGCAAAACAAAAATGAAGCCCCATGTCTTTTTATTTGAGAA
AAGATTAAACAGTGTGCTACCACATGCAGTTGTACTGGCAAAGAAAGAA
ATACAAAAGCTCCATGTGAAAATTTCCCTATGAAACTGACATGCCCATAT
CCAAAGGATACAAGACAGAAAGACACCTTTTATGTGTCAGGGAAAAAAG
CACCCTCTCGGTTAGCTCCAGTTTTTATTTTTGTAAGCTTGTCAGCTAGAG
GATGTTATAATGTCCTACAAATCAATTAGTTGGAAACCTTTTCAAGAGCA
AATTCGATTTCTTGTCC CCACAAGGATGTTCCATGTTTAATAGTCTATGAA
ATGTTGTAAAATTTAAACATGAACAAAGTATTTGGGTTTCTTAGTGATCG
TGGATACGAGAGGTGAAAAAGAACAAACATAGGTTAGTCACAGTATTAA
CA 03058584 2019-09-30
= 59
AAAAAAACTCTAGAGATATTTAAATAAAATTAATTGCTATATTAGAAGAA
AATTCATTTCAAATTCTGTCTGCGTCAATGTATTTTGCATTAGAAGCCACA
AAAAACTGAGAATTAATTGCTTTCAGGAGCATCCCATCAAGATATCCCTA
AGCTACAGTAATAAATTTTAAAATAATCTATAGTCACCAGAGCATTTTTA
TGATTGTCAAGCTTAAATATTGTTTACTTTTTTCCTGAATGAAATTTTAAG
AGTAAAGTATCAGAAAAATAGCTCAATTGAAAAGGAGAATATTACAACC
AAGTACACACAAAAACAAAAATGCTTTTTACCATTAAATAAAAATGGCA
ATTACGTTCTATTTAACTTTTTAAAAAAGATAATCTAGAATTTGTAAGGCC
ATTAAAATAACATATTAACTAAATACGAACCTTAGAAAATGAAATAATAT
CTGAGAACTTGAGGTACCTACCGTATTTAAATCTGAATGACTCAAATCCT
TATGTCACTGACAGAATAATGTGCGTATGTAGAAAACTCTCCTAATAGAT
GTGATTCATATTCTCTAATATTTTTGTATTCTCCTACTCCTTGACACAATA
GCAAGCTGACAGTAGACCCCAGTACATGCTTCCTAAATGAAGGAAGGAA
TGCATGTTTTCTGAGACTGAGGTAAAGCTCCCTTAGACTCTCGTTTCACAT
ACATTTCTTGGCTTTTTTCTTTTTCTACATTCAAGCAAAATTATTTTCGAAT
ACTGGAAATTTTGGTAGCATACAGTTAGCAATTAAAATACTCTGTAAATC
AGCAAACCGGTGACACGGTGCCTCACAATGAATATAAAACTATGCACAG
TTACTGAACTATTCACAAGCTGTCCTGGCCATACTCTCTTGAATGCCCATG
AGATGTGCTCTAGTAAACATGTGATATTTCCTTGTAACTAGTTGGCTTTGC
TCCATTGCTCGACgtttgcattaacaaatagtttgagaactatgttggaaaannanntaacaattttattcttctttct
ccagGCATCCGCCAGGGCTACGGCCTGACAGAAACAACCAGCGCCATTCTG
ATCACCCCCGAAGGGGACGACAAGCCTGGCGCAGTAGGCAAGGTGGTGC
CCTTCTTCGAGGCTAAGGTGGTGGACTTGGACACCGGTAAGACACTGGGT
GTGAACCAGCGCGGCGAGCTGTGCGTCCGTGGCCCCATGATCATGAGCG
GCTACGTTAACAACCCCGAGGCTACAAACGCTCTCATCGACAAGGACGG
CTGGCTGCACAGCGGCGACATCGCCTACTGGGACGAGGACGAGCACTTC
TTCATCGTGGACCGGCTGAAGAGCCTGATCAAATACAAGGGCTACCAGG
CA 03058584 2019-09-30
= 60
TAGCCCCAGCCGAACTGGAGAGCATCCTGCTGCAACACCCCAACATCTTC
GACGCCGGGGTCGCCGGCCTGCCCGACGACGATGCCGGCGAGCTGCCCG
CCGCAGTCGTCGTGCTGGAACACGGTAAAACCATGACCGAGAAGGAGAT
CGTGGACTATGTGGCCAGCCAGGTTACAACCGCCAAGAAGCTGCGCGGT
GGTGTTGTGTTCGTGGACGAGGTGCCTAAAGGACTGACCGGCAAGTTGG
ACGCCCGCAAGATCCGCGAGATTCTCATTAAGGCCAAGAAGGGCGGCAA
GATCGCCGTGTAA (SEQ ID NO:110)
[0072]
<Exon Skipping Reporter Assay Using Luc Reporter>
A mixture of 40 ng of the pPV-EF1a-Luc2(V323I)11DMD-Ex45[+] vector, 20
ng of the phRL-TK vector, which expresses Renilla Luc, 280 ng of the pHL-EFla
vector (Addgene), which expresses CRISPR-Cas, and 280 ng of the pHL-H1-
sgRNA-mEFla-RiH vector, which expresses a guide RNA, was prepared, and then
diluted with 25 I of Opti-MEM. With 25 1 of Opti-MEM, 0.7 1 of
Lipofectamine 2000 was diluted, and the resulting dilution was incubated at
room
temperature for 3 to 5 minutes, followed by mixing the dilution with the above
DNA
solution, and then incubating the resulting mixture at room temperature for
additional
minutes. The cell number of 293T cells suspended by trypsin-EDTA treatment
20 was counted, and then the cells were diluted to 60,000 cells/100 I with
a medium,
followed by plating the cells on a 96-well plate containing the above DNA-
Lipofectamine complex at 100 l/well. After culturing the cells for 48 hours
with
5% CO2 at 37 C, the 96-well plate was allowed to cool to room temperature, and
then Dual-Glo Reagent was added thereto, followed by incubation at room
temperature for 30 minutes to lyse the cells to cause the luciferase reaction.
To a
white 96-well plate, 100 I of the supernatant was transferred, and the
luminescence
intensities of Firefly and Renilla were measured using Centro LB960 (Berthold
CA 03058584 2019-09-30
61
Technologies). Since firefly Luc emits light only when exon skipping is
induced,
the luminescence value of firefly was normalized against the luminescence
value of
Renilla to measure the exon skipping efficiency.
[0073]
<Results>
In order to develop a therapeutic method for Duchenne muscular dystrophy,
induction of exon skipping in the dystrophin gene was studied. Fig. 1 shows
its
overview.
(1) In the skeletal muscle iso form (Dp427m) of the dystrophin gene in healthy
individuals, 79 exons are linked to each other by splicing, and a dystrophin
protein
composed of 3685 amino acids is encoded.
(2) However, in DMD patients lacking exon 44, the size of exon 44 is not a
multiple
of 3, and therefore the reading frame of the protein shifts to generate a stop
codon in
the following exon 45, resulting in discontinuity of the dystrophin protein.
(3) Here, by disrupting the splice acceptor sequence portion of exon 45 by
genome
editing, recognition of exon 45 can be prevented during splicing, so that,
instead, the
following exon 46 can be linked to exon 43, resulting in recovery of the
reading
frame of the dystrophin protein.
[0074]
The splice acceptor is constituted by a branching sequence, polypyrimidine
(C/U) sequence, and an "AG" acceptor sequence. Since, among these, the "AG"
acceptor sequence is most highly conserved during the splicing reaction,
deletion of
these two bases may enable induction of exon skipping.
[0075]
In designing of gRNAs for CRISPR systems, from the viewpoint of the off-
target risk, that is, recognition and cleavage of sequences other than the
target
sequence in the genome, the unique k-mer method [Li HL et al., Stem Cell
Reports,
CA 03058584 2019-09-30
=
62
2015] was used to investigate the sequence specificity in the region around
the splice
acceptor. As a result, it became clear that the region near the splice
acceptor has an
especially low specificity compared to the exon regions and other intron
regions (Fig.
2).
[0076]
In order to simply and highly sensitively detect the exon skipping efficiency
in
the dystrophin gene, a reporter vector was constructed using the firefly
luciferase
(Luc) gene. A vector in which Luc cDNA was divided into two parts, and a
synthetic intron sequence was inserted thereto (Luc + Int), and a vector in
which a
sequence around human dystrophin exon 45 was further inserted thereto (Luc +
hEx45), were prepared (Fig. 3). Each vector was introduced into 293T cells,
and
mRNA was recovered, followed by performing reverse transcription and then PCR
amplification. As a result, it was found that the first half of the Luc cDNA
contains
a pseudo-splicing donor sequence, causing extra splicing (Fig. 4, Panel (a)).
In
order to disrupt this pseudo-splicing donor sequence, splicing donor sequences
and
acceptor sequences were extracted from all exon sequences contained in the
human
dystrophin gene from the Ensemble Biomart database (http://www.ensembl.org),
and
common bases were analyzed with Weblogo software
(http://weblogo.threeplusone.com/). As a result, they were found to be
matching
well with known splicing donor and acceptor sequences (Fig. 4, Panel (b)).
Thus, it
was expected that, by converting the "G" at the center of the pseudo-splicing
donor
sequence "AGGTA" to a "sequence other than G", the pseudo-splicing donor
sequence can be prevented from functioning as a splicing donor. However, since
this base is the first base of the codon "GTA", which encodes a Val amino
acid,
alteration of this base inevitably changes the amino acid sequence. Alteration
to
"A" results in generation of the codon "ATA", which encodes an Ile amino acid
(Fig.
4, Panel (c)), and alteration to "C" or "T" results in generation of a codon
"CTA" or
CA 03058584 2019-09-30
63
"CTA", which encodes a Leu Amino acid. In order to confirm that this amino
acid
conversion does not affect the Luc activity, the present inventors downloaded
the
spatial structure of luciferase protein (PDB code: 1BA3) from the PDB
database.
By using Chimera software, it could be confirmed that the G967A(V323I) mutant
amino acid site is distant from the active residue [Branchini BR et al., J
Biol Chem.
1997 Aug 1; 272(31): 19359-64.], and hence that there is no direct interaction
(Fig. 5).
[0077]
Subsequently, in order to investigate whether the expected splicing pattern
actually occurs or not, the vectors shown in Fig. 6, Panel (a) were introduced
into
293T cells, and mRNA was extracted therefrom, followed by performing reverse
transcription and then PCR amplification. As a result of investigation of the
cDNA
size by gel electrophoresis, the vectors into which the G967A(V323I) mutation
was
not introduced (lanes 2 and 4) showed a band corresponding to the non-spliced
transcript with high intensity. On the other hand, it could be confirmed that
splicing
with the expected size occurred in almost the entire transcripts from the
vectors into
which the G967A(V323I) mutation was introduced (lanes 3 and 5) (Fig. 6, Panel
(b)).
Further, according to the result of investigation of the luciferase activity
of each
vector, the insertion of the intron sequence or the introduction of the
G967A(V323I)
mutation hardly caused changes. On the other hand, in the cases where the
human
exon 45 sequence was inserted, the vector without the G967A(V323I) mutation
showed some luciferase activity through the pseudo-splicing donor, and
therefore
exhibited a high background level. In contrast, by the introduction of the
G967A(V323I) mutation, the Luc cDNA sequence containing human exon 45
became the majority, and therefore only very low luciferase activity was found
(Fig. 6,
Panel (c)).
[0078]
Subsequently, analysis of the splicing pattern depending on the lengths of the
CA 03058584 2019-09-30
64
intron sequences before and after exon 45 was carried out. Vectors were
constructed by inserting, other than the originally constructed 0.7-kb
sequence, a
sequence with a length of 1.0 kb, 2.0 kb, or 4.0 kb (Fig. 7). According to the
result
of analysis of the splicing patterns of these vectors (Fig. 8a), the vectors
mostly
showed almost expected splicing patterns (the band of 468 bp), but, as the
intron size
increased, the band of the residual intron (1166 bp) tended to disappear. On
the
other hand, as a result of investigation of the luciferase activity, it was
found that, as
the intron size increases, the introduction activity into the cells decreases,
resulting in
a rather high background activity. With any of the vectors, an increase in the
luciferase activity could be found when exon skipping was induced using CRISPR-
sgRNA-DMD1 (Fig. 8b). Thus, the vectors were found to be useful as reporter
vectors that enable simple and sensitive measurement.
[0079]
In order to induce exon skipping of dystrophin while minimizing the off-
target risk, a plurality of gRNAs were designed at the splice acceptor site of
exon 45
(Fig. 9), and an SSA (Single Strand Annealing) assay was carried out for the
cleavage
pattern by an ordinary wild-type SpCas9 and a gRNA, and for cases where a Dl
OA
nickase-type SpCas9 and two gRNAs were used in combination. The DNA
cleavage activities for the target site were measured. As a result, as shown
in Fig.
10, any of five kinds of gRNAs exhibited a high DNA cleavage activity.
Further, it
was found that, in the double nicking method, the cleavage activity is low
when two
gRNAs are overlapping, and that induction of efficient DNA cleavage requires
the
presence of a certain distance.
[0080]
Subsequently, in order to investigate DNA cleavage patterns obtained under
various conditions, the target site was amplified by PCR, and sequence
analysis with
a next-generation sequencer MiSeq was carried out. As a result, when two gRNAs
CA 03058584 2019-09-30
were designed such that they were arranged at an appropriate distance in the
double
nicking method, DNA cleavage patterns with occurrence of deletion between the
nicking induction sites of the gRNAs were frequently observed. Thus, it was
discovered that, in cases where a splice acceptor sequence, especially the
"AG"
5 acceptor sequence, is included in this region, efficient induction of
exon skipping is
possible (Fig. 11).
[0081]
For studying gRNA sequences, types of CRISPR (SpCas9, AsCpfl, and the
like), and genome editing methods (double-nicking) that enable efficient
induction of
10 exon skipping, a study was carried out using the exon skipping reporters
developed
as described above.
As shown in Fig. 12, five kinds of gRNA sequences of SpCas9 were tested,
and, as shown in Figs. 13a to 13d, comparative analysis was carried out for
SpCas9,
SaCas9, AsCpfl, and the SpCas9 double-nicking method. As shown in Figs. 14a to
15 14c, in order to measure the exon skipping efficiency for all sequences
that can be
designed in human exon 45 (sgRNA sequences including the NGG PAM sequence),
26 kinds of gRNAs were designed (Fig. 14a).
The 26 kinds of gRNAs were introduced into human 293T cells, and the
target DNA cleavage activity was measured by a T7E1 assay (Fig. 14b). As a
result,
20 although sgRNA-DMD6 showed a low DNA cleavage activity, other gRNAs
basically showed cleavage activities of not less than 10%. In view of this,
the 26
kinds of gRNAs were subjected to measurement of the exon skipping efficiency
in
293T cells using the Luc2 (G967A) +hEx45 (0.7 kb) reporter. As a result, it
was
found that the gRNA design site and the distance from the splice acceptor or
the
25 splicing donor are important for the exon skipping efficiency (Fig.
14c).
[0082]
Further, seven kinds of gRNAs that individually showed exon skipping
CA 03058584 2019-09-30
=
66
activity (DMD#1, 2, 4, 8, 9, 20, and 23) were selected, and arbitrary
combinations of
two sgRNAs among these were subjected to measurement of the exon skipping
efficiency in 293T cells using the Luc2 (G967A) +hEx45 (0.7 kb) reporter. As a
result, it was found that the exon skipping efficiency can be further
increased by
simultaneous introduction of two kinds of gRNAs (Fig. 15).
[0083]
References
1. Li HL, Fujimoto N, Sasakawa N, Shirai S, Ohkame T, Sakuma T, et al.
Precise correction of the dystrophin gene in duchenne muscular dystrophy
patient
induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Reports.
2015 Jan 13; 4(1): 143-54.
2. Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, et al.
CAS9 transcriptional activators for target specificity screening and paired
nickases
for cooperative genome engineering. Nat Biotechnol. 2013 Sep; 31(9): 833-8.
3. Ran FA, Hsu PD, Lin C-Y, Gootenberg JS, Konermann S, Trevino AE, et al.
Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing
specificity. Cell. 2013 Sep 12; 154(6): 1380-9.
4. Ousterout DG, Kabadi AM, Thakore PI, Majoros WH, Reddy TE, Gersbach
CA. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin
mutations that cause Duchenne muscular dystrophy. Nat Commun. 2015; 6: 6244.
5. Iyombe-Engembe J-P, Ouellet DL, Barbeau X, Rousseau J, Chapdelaine P,
Lague P, et al. Efficient Restoration of the Dystrophin Gene Reading Frame and
Protein Structure in DMD Myoblasts Using the CinDel Method. Mol Ther Nucleic
Acids. 2016; 5:e283.
6. Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, et
al.
Postnatal genome editing partially restores dystrophin expression in a mouse
model
of muscular dystrophy. Science. 2016 Jan 22; 351(6271): 400-3.
CA 03058584 2019-09-30
67
7. Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Castellanos
Rivera RM, et al. In vivo genome editing improves muscle function in a mouse
model of Duchenne muscular dystrophy. Science. 2016 Jan 22; 351(6271): 403-7.
8. Tabebordbar M, Zhu K, Cheng JKW, Chew WL, Widrick JJ, Yan WX, et al.
In vivo gene editing in dystrophic mouse muscle and muscle stem cells.
Science.
2016 Jan 22; 351(6271): 407-11.