Note: Descriptions are shown in the official language in which they were submitted.
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
MONOGENIC OR POLYGENIC DISEASE MODEL ORGANISMS HUMANIZED
WITH TWO OR MORE GENES
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application claims the benefit of U.S. Provisional Patent
Application No.
62/821,377, filed on 20 March 2019, the content of which is incorporated
herein by reference in
its entirety.
[0002] This application claims priority to pending U.S. Ser. No. 16/281,988,
filed on 21
February 2019, and to pending PCT/US19/19027, filed 21 February 2019, the
contents of which
are each incorporated herein by reference in their entirety.
SEQUENCE LISTING
[0003] The instant application contains a Sequence Listing which has been
submitted
electronically in ASCII format via EFS-Web and hereby incorporated by
reference in its entirety.
Said ASCII copy, created on 21 February 2020, is named NEMA013PCT ST25.TXT and
is
2384 bytes in size.
FIELD OF THE DISCLOSURE
[0004] This application pertains generally to transgenic animals comprising
two or more
heterologous polypeptide coding sequences, wherein expression of the
heterologous polypeptide
coding sequence product contributes to the same heterologous phenotype; and
their use in
assessing monogenic or polygenic diseases and gene variants thereof.
BACKGROUND OF THE DISCLOSURE
[0005] Clinical genomics is revealing genetic variation occurs at high
prevalence in the human
population. Accumulated genomic data reveals each person has about 500
sequence variants that
create mis sense or indel mutations in the coding regions of their genome
(Jansen I et al.
Establishing the role of rare coding variants in known Parkinson's disease
risk loci. Neurobiol
Aging. 2017 Nov; 59:220.e11-220.e18). With estimates as high as 30% of the
genes in the
human genome being involved in disease biology (Hegde M et al. Development and
Validation
1
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
of Clinical Whole-Exome and Whole-Genome Sequencing for Detection of Germline
Variants in
Inherited Disease. Arch Pathol Lab Med. 2017 Jun;141(6):798-805.), any one
individual harbors
over 100 codon-changing variations in their important "disease" genes.
Surprisingly,
frameshifting indels with a high likelihood of pathogenicity account for only
7% of these
variants. As a result, there remains a significant number of questionable
alleles that are part of
the background of anyone's personal genome. The challenge to the physician is
to determine if a
suspect allele is contributing to the disease as a pathogenic variant or if
the clinical variant is not
consequential and can be classified as a benign variant. For many of the
genetic differences seen
in a patient's genome, the benign or pathogenic status remains undefined and
the variant is a
Variant of Uncertain Significance (VUS). As a result, variant interpretation
is the major
bottleneck now that large scale sequencing is increasingly being used in
clinical settings.
[0006] Genome wide association studies (GWAS) reveal multiple genes are
involved in many
types of disease. For instance, a study of the polygenic genetic architecture
of schizophrenia
identified more than 10% of genome (2725 candidate genes) may be acting as
risk factors for
disease (Lee et al "Estimating the proportion of variation in susceptibility
to schizophrenia
captured by common SNPs." Nat Genet. 2012 Feb 19;44(3):247-50). Another SNP-
based
GWAS in epilepsy identified 16 genetic regions containing 21 epilepsy target
genes as highly-
associated with adult onset disease (Abou-Khalil et al. "Genome-wide mega-
analysis identifies
16 loci and highlights diverse biological mechanisms in the common epilepsies"
Nat Commun.
2018 Dec 10;9(1):5269). Yet a challenge of GWAS is to identify the molecular
nature of the
polygenic drivers of disease. Most SNPs in an association cluster occur in non-
coding regions.
For the rare GWAS SNP that occurs in coding segments they tend to be in non-
conserved
regions. As a result, they are rarely the molecular cause of the disease risk
factor. Instead it is a
rare minor allele at a nearby SNP located within a low to non-recombination
interval on the same
strand as one of the GWAS high frequency SNPs. Since thousands of rare SNPs
can fall into
this category it becomes challenging and tedious to identify the molecular
cause of a polygenic
contribution to disease. Systems are needed for looking at the additive
effects on gene
disfunction for a set of rare alleles distributed across more than one loci.
[0007] A significant proportion of clinical variants seen in patients with
genetic disease are
caused by missense changes resulting in altered amino acid usage. Unlike the
rarer frameshift
and stop-codon mutations and some intra-/inter-genic variants, the functional
consequence of
2
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
missense amino acid changes can remain elusive. Change of function due to
missense can result
in partial loss of gene activities or gain-of-function changes that are highly
pathogenic. There is
an emergent need for the functional analysis of variant pathogenicity that
occurs as a result of
these amino acid changes.
[0008] A variety of technologies from bioinformatics to biochemical assays can
be deployed to
assess functional consequence of mis sense changes. Yet the most reliable are
the in vivo
systems. Most commonly used are cell culture assays that translate to animal
model studies.
The lack of intact animal biology occurring in cell culture systems renders
this technique
intractable to many transcellular pathogenicities. As a result, transgenic
animal models are
favored for capturing the nuances of intra- and inter- cellular pathogenicity
in native contexts.
[0009] Transgenic mice are the traditional animal model for probing functional
consequence of
genomic variation. Yet their high expense and low throughput leave their use
as intractable to
address the 100,000,000's of coding altering variants predicted to occur in
human populations.
Many groups are now focusing on using alternative model organisms (Zebrafish,
drosophila and
C. elegans) as a more affordable and timely approach to assessing variant
specific effects on
gene function, for example, the Undiagnosed Disease Network). Yet current
design compositions
and features of the transgenics used in these studies are not as efficient or
appropriate as they
could be for accurate assessment of variant function.
[0010] As one of the five classical model organisms for genetic studies (worm,
fly, yeast,
zebrafish and mice) the C. elegans nematode worm has a unique set of
attributes that make it
highly optimal for high-throughput clinical variant phenotyping. At the
genetic level, the C.
elegans nematode rivals the Drosophila fly for having orthologs to 80% of
human disease genes,
wherein 6460 genes detected in ClinVar Miner database as human disease genes
were queried
for homologs using the DIOPT database (Hu Y et al. An integrative approach to
ortholog
prediction for disease-focused and other functional studies. BMC
Bioinformatics. 2011 Aug
31;12:357). Of the multicellular models, the C. elegans animal model has the
fastest life cycle (3
days). It has optical transparency for easy tissue and organ system expression
observation.
Finally, in a unique advantage of interpretability, the C. elegans animals are
easy to breed as
self-fertilizing hermaphrodites, which allow rapid population expansion of
nearly identical
animals with very minimal polymorphism load in the genetic background. This
allows
3
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
transgenesis and subsequent population phenotyping to be performed in a matter
of a few weeks
instead of years.
[0011] Transgenic C. elegans are optimal for drug screening capacity. Of the
five animal
models, only yeast provides higher diversity screening per meter of bench
space in comparison
to C. elegans. Yet, yeast exist in a single cellular context and it becomes
challenging to
accurately model human biology where variant function (or disfunction)
operates in a 3-
dimensional tissue-based architecture. The advent of iPSC (Csobonyeiova, M et
al. Recent
Advances in iPSC Technologies Involving Cardiovascular and Neurodegenerative
Disease
Modeling. General Physiology and Biophysics 35, no. 1 (January 2016): 1-12)
and organoid
(Breslin S and O'Driscoll L. Three-Dimensional Cell Culture: The Missing Link
in Drug
Discovery. Drug Discovery Today 18, no. 5-6 (March 2013): 240-49) technologies
bring more
biological-context relevance, yet they remain undemonstrated for capacity to
deploy in robust
high-throughput formats. The C. elegans animal model, on the other hand, is
robust and fast for
high density screens of biological alterations. For instance, a recent screen
for SKN-1 inhibitors
as anthelmintic therapeutics found promising hits in few weeks screen of
340,000 compounds
(Leung CK et al. An ultra high-throughput, whole-animal screen for small
molecule modulators
of a specific genetic pathway in Caenorhabditis elegans. PLoS One. 2013 Apr
29;8(4):e62166).
Many other groups have used transgenic C. elegans for medium- to high-
throughput drug
discovery (Artal-Sanz M et al. Caenorhabditis elegans: a versatile platform
for drug discovery.
Biotechnol J. 2006 Dec;1(12):1405-18; O'Reilly LP et al. C. elegans in high-
throughput drug
discovery. Adv Drug Deliv Rev. 2014 Apr;69-70:247-53; Xiong H et al. An
enhanced C. elegans
based platform for toxicity assessment. Sci Rep. 2017 Aug 29;7(1):9839; Kim W
et al. An
update on the use of C. elegans for preclinical drug discovery: screening and
identifying anti-
infective drugs. Expert Opin Drug Discov. 2017 Jun;12(6):625-633; and, Kim H
et al. A co-
CRISPR strategy for efficient genome editing in Caenorhabditis elegans.
Genetics. 2014
Aug;197(4):1069-80).
[0012] C. elegans are a microscopic organism, with intact nervous system
capable of learned
behavior, where the animal can pack into 96 well, 384 well and even 1536 well
assays (Leung,
C. K., Deonarine, A., Strange, K. & Choe, K. P. High-throughput Screening and
Biosensing with
Fluorescent C. elegans Strains. J Vis Exp (2011)). It has complex tissue
structure (nervous
system, muscles, germ line, intestine, mouth-like pharynx, periodic excretion
through anal
4
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
sphincter, macrophage-like celomocytes, and a tough skin-like hypodermis). As
a result, the C.
elegans nematode provides complex tissue biology in an intact, easy-to-culture
animal model.
[0013] Zebrafish have developed into a popular animal model platform for drug
discovery with a
fast-growing conference support (Zebrafish Disease Modeling Society) now in
its 13th year.
Advantages of the use of zebrafish as an animal model are its inclusion in the
vertebrate phylum
which results in a high degree of homologous gene structures and organ systems
in relation to
humans. Breeds of zebrafish are available with high transparency (e.g. CASPER)
which enable
direct in vivo monitoring of gene activity and organ variability in live
animals. Like the liquid
format used in C. elegans, animal growth and handling of zebrafish is easily
automated with a
variety of fluidic systems.
[0014] Current variant modeling systems in zebrafish, C. elegans, and other
animals are
predominantly done as site directed mutagenesis to insert a variant at the
native ortholog locus.
Only a few groups have tried expression of human transgenes in these animal
models to varying
levels of success. A simple and robust approach to create ideal transgenic
compositions is
lacking. As a result, there remains a need for a ubiquitous transgenics
platform that can be used
to assess function of broad categories of clinical variants, and their
interaction with expression of
wild-type genes in vivo, and screen for drug discovery in the treatment of
pathogenic clinical
variants. Moreover, there remains a need for looking at the additive effects
on gene disfunction
for a set of rare alleles distributed across more than one loci.
[0015] Herein we provide an animal model transgenic platform wherein the
animal model
configuration frequently has the animal's ortholog replaced by a chimeric
heterologous
transgene, such as human disease exon coding sequences paired with a host
animal (e.g.
nematode) intron sequences, that can be used to increase understanding of
individual variants
(clinical and biological) as well as their interaction or additive effects
with other variants or wild-
type sequences that contribute to a particular disease. Furthermore, the
resulting transgenic
animal systems can be used to provide highly-personalized (variant-specific)
discovery of
therapeutic approaches.
SUMMARY OF THE INVENTION
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
[0016] Herein are provided transgenic non-human animals systems for assessing
a heterologous
polygenic or monogenic phenotype and methods thereof. In embodiments, the non-
human
animal is a nematode or zebrafish. In embodiments, a transgenic nematode
system comprises a
host nematode comprising and expressing a first heterologous polypeptide
coding sequence and a
second heterologous polypeptide coding sequence, wherein the first and second
heterologous
polypeptide coding sequences are integrated into the host nematode genome, and
wherein
expression of the first and second heterologous polypeptide coding sequences
contribute to the
heterologous phenotype. The first and second heterologous polypeptide coding
sequence(s) are
interrelated in that their expression contributes to the same phenotype or
trait. That phenotype
may be a particular disease, such as a neurodegenerative disease.
[0017] In embodiments, the host animal further comprises and expresses one or
more additional
heterologous polypeptide coding sequence that contribute to the heterologous
phenotype. In
embodiments, the host nematode comprises and expresses 2 to 15 heterologous
polypeptide
coding sequences; or 3 to 15 heterologous polypeptide coding sequences. In
certain
embodiments, the one or more additional heterologous polypeptide coding
sequence(s)
comprises one or more mutations in exon coding sequences of the heterologous
polypeptide
coding sequence as compared to a wildtype reference sequence resulting in at
least one amino
acid change when the one or more additional heterologous polypeptide coding
sequence is
expressed.
[0018] In embodiments, the heterologous polypeptide coding sequence replaces
the nematode
ortholog using gene swap techniques involving removing the native coding
sequence of the host
nematode ortholog and replacing with modified cDNA coding sequence from a
heterologous
polypeptide sequence.
[0019] The choice of introduced transgene sequence can vary widely but in one
embodiment the
sequence is a modified cDNA coding sequence from any eukaryotic organism. In
embodiments,
Applicants found that using modified intron sequences from a highly expressed
gene of the host
nematode, paired with or interspersed with the heterologous exon coding
sequences - a chimeric
heterologous polypeptide coding sequence - improved expression of the
heterologous
polypeptide coding sequence in the host nematode. (See USN 16/281,988, filed
21 February
2019, incorporated in its entirety herein by reference). Accordingly, in
certain embodiments, at
least one of the first heterologous polypeptide coding sequence or the second
heterologous
6
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
polypeptide coding sequence is a chimeric heterologous polypeptide coding
sequence comprising
heterologous exon coding sequences interspersed with artificial host nematode
intron sequences
optimized for expression in the host nematode. In further embodiments, each of
the first and
second heterologous polypeptide coding sequence is individually a chimeric
heterologous
polypeptide coding sequence comprising heterologous exon coding sequences
interspersed with
artificial host nematode intron sequences optimized for expression in the host
nematode.
[0020] In embodiments provided herein is a transgenic nematode comprising and
expressing a
first heterologous polypeptide coding sequence and a second heterologous
polypeptide coding
sequence, wherein the host nematode comprises a chimeric heterologous
polypeptide coding
sequence comprising heterologous exon coding sequences interspersed with
artificial host
nematode intron sequences optimized for expression in the host nematode
selected from SEQ ID
NO: 1, 2, 3, 4, 5 or 6. In addition to introduction of artificial host intron
sequences into the
cDNA sequence from the heterologous polypeptide coding sequence, the chimeric
heterologous
polypeptide coding sequence may be optimized for expression in the host
nematode wherein the
heterologous polypeptide coding sequence is codon optimized for the host
nematode and
aberrant splice donor and/or acceptor sites removed.
[0021] In embodiments, at least one of the first heterologous polypeptide
coding sequences or
the second heterologous polypeptide coding sequence replaced an entire host
nematode gene
ortholog at a native locus. In certain embodiments, each of the first and
second heterologous
polypeptide coding sequences individually replaced an entire host nematode
gene ortholog at a
native locus. In certain embodiments, the host nematode ortholog gene of the
first heterologous
polypeptide coding sequence and/or the second heterologous polypeptide coding
sequence has
been knocked-out.
[0022] In embodiments, the first and second heterologous polypeptide coding
sequences
comprise human exon coding sequences. In certain embodiments, the human genes
are selected
from those listed in Table 1, Table 3 or Example 3. In embodiments, the
chimeric heterologous
polypeptide coding sequence is integrated in the nematode genome. In certain
embodiments, the
chimeric heterologous polypeptide coding sequence is inserted into a native
locus of the host
nematode. In alternative embodiments, the chimeric heterologous polypeptide
coding sequence
is inserted into a non-native locus of the host nematode or is inserted into a
random site of the
host nematode genome.
7
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
[0023] In embodiments, at least one of the first heterologous polypeptide
coding sequence or the
second heterologous polypeptide coding sequence comprise one or more mutations
in the
heterologous polypeptide coding sequence exon coding sequences as compared to
a wildtype
reference sequence resulting in at least one amino acid change when the first
heterologous
polypeptide coding sequence or the second heterologous polypeptide coding
sequence is
expressed. In embodiments, the mutation corresponds to a human disease gene
clinical variant.
[0024] In embodiments, the heterologous phenotype is a monogenic human disease
phenotype.
In certain other embodiments, the heterologous phenotype is a polygenic human
disease
phenotype. In embodiments, the heterologous polypeptide coding sequence is a
human gene,
and in certain embodiments, the heterologous polypeptide coding sequence is a
human disease
gene.
[0025] In embodiments provided herein is a transgenic nematode system for
assessing a
heterologous disease phenotype, wherein the system comprises a host nematode
comprising and
expressing a first heterologous polypeptide coding sequence and a second
heterologous
polypeptide coding sequence, wherein the first and second heterologous
polypeptide coding
sequence(s) are integrated into the host nematode genome, wherein at least one
of the first
heterologous polypeptide coding sequence or the second heterologous
polypeptide coding
sequence comprises one or more mutations in the heterologous exon coding
sequence as
compared to a wildtype reference sequence resulting in at least one amino acid
change when the
first heterologous polypeptide coding sequence or the second heterologous
polypeptide coding
sequence is expressed, and wherein expression of the first and second
heterologous polypeptide
coding sequences contribute to the heterologous disease phenotype.
[0026] In certain embodiments provided herein is a humanized transgenic
nematode system for
assessing a monogenic or polygenic human disease phenotype, wherein the system
comprises a
host nematode comprising and expressing a first human polypeptide coding
sequence and a
second human polypeptide coding sequence, wherein the first and second human
polypeptide
coding sequences are integrated into the host nematode genome, wherein at
least one of the first
human polypeptide coding sequence or the second human polypeptide coding
sequence
comprises one or more mutations in the human gene exon coding sequence as
compared to a
wildtype reference sequence resulting in at least one amino acid change when
the first human
polypeptide coding sequence or the second human polypeptide coding sequence is
expressed,
8
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
and wherein expression of the first and second human polypeptide coding
sequences contribute
to the monogenic or polygenic human disease phenotype.
[0027] In embodiments, at least one, or each, heterologous polypeptide coding
sequences (e.g.,
first, second, or additional heterologous polypeptide coding sequence) is
present as a single copy
providing a heterozygote transgenic nematode. In certain embodiments, the
heterozygote is
maintained by labeling each chromosome with a marker.
[0028] In embodiments, the transgenic nematode systems are used to assess
function of the
heterologous phenotype resulting from expression of the first and second
heterologous
polypeptide coding sequence. Those polypeptide coding sequences may be a
wildtype sequence
(e.g. human sequence) or a clinical variant thereof, wherein the system may be
used as a screen
for therapeutic agents to identify drugs that may be used to treat individuals
with those
heterologous phenotype and/or clinical variants. In certain embodiments, the
method comprises
culturing a host transgenic nematode wherein at least one of the first and
second heterologous
polypeptide coding sequence is a human clinical variant; and, performing a
phenotypic screen to
identify a monogenic or polygenic phenotype of the transgenic nematode,
wherein a change in
phenotype as compared to a control transgenic animal (validated transgenic
animal) comprising a
corresponding wildtype human heterologous polypeptide coding sequence(s)
indicates an altered
function of the clinical variant in the transgenic host nematode.
[0029] In embodiments, the phenotypic screen is selected from a measurement of
electrophysiology of pharynx pumping, a food race, lifespan extension and
contraction assay,
movement assay, fecundity assay with egg lay or population expansion,
apoptotic body
formation, chemotaxis, lipid metabolism assay, body morphology changes,
fluorescence
changes, drug sensitivity and resistance assays, oxidative stress assay,
Endoplasmic Reticulum
stress assay, nuclear stress assay, response to vibration, response to
electric shock, or a
combination thereof. In certain embodiments, the identified phenotype is
selected from
electropharyngeogram variant, feeding behavior variant, defecation behavior
variant, lifespan
variant, electrotaxis variant, chemotaxis variant, thermotaxis variant,
mechanosensation variant,
movement variant, locomotion variant, pigmentation variant, embryonic
development variant,
organ system morphology variant, metabolism variant, fertility variant, dauer
formation variant,
stress response variant, or a combination thereof.
9
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
BRIEF DESCRIPTION OF THE DRAWINGS
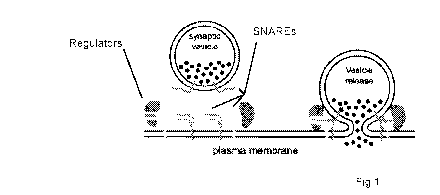
[0030] Figure 1 is an illustration of SNARE genes and their associated
presynaptic proteins.
SNARE proteins act as machinery to cause vesicle fusion (syntaxins, VAMPs and
SNAPs). A
set of additional proteins regulate vesicle fusion to coordinate
neurotransmitter release with
membrane depolarization events.
[0031] Figure 2 shows expected electrophysiology of a wildtype control
nematode (black bar)
and transgenic nematodes comprising humanized synapse genes following
replacement of host
SNARE genes with human SNARE genes (e.g. STX1A, SNAP25 and VAMP2, individually
(hollow box bar) and additive as STX1A, SNAP25 and VAMP2 humanized complex
(grey bar)).
[0032] Figure 3 is an illustration of genes involved in homologous
recombination (HR). Five
events are involved in activation of HR. Recognition recognizes double strand
break (DSB)
damage and recruits other recognition partners, RBBP8, BARD1, BRCA1 and BRIP1.
Resection
is an activity to removed DNA from DSBs by the activity of RAD50, MREllA and
NBN.
Filament is the formation of a primed end via the activity of RPA with RAD51
paralogs. Strand
invasion creates crossovers into sister chromosome by activity of RAD54.
Resolution is an
activity mediated by POLD1 with contribution from BLM, TOP3A and MUS81 to
synthesize
new DNA then ligate back to original chromosome.
[0033] Figure 4 shows expected fluorescence signal from homologous-
recombination-activity-
activated fluorescent reporter. Wildtype control nematode (black bar) and
transgenic nematodes
comprising humanized HR apparatus genes (e.g. ATM, RAD50, RAD51, RAD54, and
POLD1
individually (hollow box bar) and additive as ATM, RAD50, RAD51, RAD54 and
POLD1
humanized complex (grey bar)).
DETAILED DESCRIPTION OF THE INVENTION
[0034] Introduction
[0035] Provided herein is a transgenic non-human animal system, and uses
thereof for assessing
a heterologous phenotype (polygenic or monogenic) wherein a host animal of the
system
comprises (and expresses) a plurality (e.g. at least a first heterologous
polypeptide coding
sequence and a second heterologous polypeptide coding sequence) heterologous
polypeptide
coding sequences, wherein expression of those polypeptide coding sequences
contribute to the
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
heterologous phenotype in the host animal due to their interrelated function
as they relate to an
observable phenotype. In embodiments, the non-human transgenic animal is a
nematode or
zebrafish. The present transgenic non-human animal system provides a model for
assessing both
monogenic and polygenic diseases, wherein a plurality of interrelated
heterologous polypeptide
coding sequences are expressed and, interact in vivo to provide an observable
phenotype. In
embodiments, each of the at least two heterologous polypeptide coding
sequences comprise wild
type coding sequences, for example a common allele of a human gene. In certain
other
embodiments, at least one of the heterologous polypeptide coding sequences
(e.g. a first
heterologous polypeptide coding sequences and a second heterologous
polypeptide coding
sequences) comprise wildtype coding sequence and the remaining heterologous
polypeptide
coding sequences comprise a variant of a wildtype coding sequence resulting in
at least one
amino acid change. In certain embodiments, the plurality of heterologous
polypeptide coding
sequences comprise variant coding sequences. In embodiments, those
heterologous polypeptide
coding sequences comprise clinal variant coding sequences.
[0036] In embodiments, the plurality of heterologous polypeptide coding
sequences in the host
nematode are integrated into the host genome. In certain embodiments, one or
more of the
plurality of heterologous polypeptide coding sequences are integrated at a
native locus and
replace the nematode ortholog. Host nematodes are validated when the
heterologous polypeptide
coding sequences rescues (or at least partially restores) function of the
removed nematode
ortholog. As used herein, this method of replacing the host nematode
ortholog(s) with the
heterologous polypeptide coding sequence(s), may also be referenced as "gene-
swap". USN
16/281,988, incorporated in its entirety by reference, discloses a method of
optimizing a
heterologous polypeptide coding sequences for insertion and expression in a
nematode wherein
host intron sequences from a highly expressed gene are interspersed into the
heterologous exon
sequences, codons are optimized for expression in the nematode and any
aberrant donor or
acceptor sites, which may have been introduced via intron and exon splicing,
are removed. That
method is one way in which the present transgenic nematodes are made. In
embodiments,
heterologous polypeptide coding sequences are introduced in sequence until a
host nematode
comprising a particular number of heterologous polypeptide coding sequences is
made. In other
embodiments, two or more heterologous polypeptide coding sequences may be
introduced into
the host nematode genome simultaneously. In other embodiments, transgenic
nematodes, each
11
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
comprising and expressing a single heterologous polypeptide coding sequences
are crossed
producing progeny with a desired number of unique heterologous polypeptide
coding sequences
integrated into the host nematode genome. See Example 4.
[0037] As used herein, "chimeric heterologous polypeptide coding sequence"
refers to a
sequence comprising heterologous (to the host animal) exon coding sequences
interspersed, or
paired, with artificial (or modified) host animal intron sequences, wherein
the chimeric
heterologous polypeptide coding sequences is optimized for expression in the
host animal (e.g.
nematode) which may include codon optimization and removal of any aberrant
splice donor
and/or acceptor sites that were introduced as a function of the chimeric
sequences. In
embodiments, the heterologous exon coding sequences are "wild type" or from an
allele that is
reflective of a heterogenous population or a common allele in a population. In
certain
embodiments, the heterologous exon coding sequences are from human genes. A
"validated"
transgenic animal system are those animals that have a phenotypic profile that
is deemed to have
demonstrated rescue or partial restoration of function of the swapped genes,
as compared to a
control host animal (e.g., wild type (N2) animal that is genetically identical
to the host animal
prior to the introduction of the heterologous polypeptide coding sequences).
[0038] In embodiments, the validated transgenic animal system may be used for
assessing the
interrelated function of the expressed plurality of heterologous polypeptide
coding sequences in
host organism.
[0039] Provided further is a transgenic animal system for assessing function
of one or more
variant heterologous polypeptide coding sequences, wherein clinical variants
(expressed
heterologous polypeptide coding sequences comprising one or more amino acid
changes as
compared to the wild type heterologous gene) are installed in the heterologous
polypeptide
coding sequences via site directed mutagenesis. In this instance, the host
nematode may
comprise two or more heterologous polypeptide coding sequences that comprise
clinical variant
coding sequences, or the host nematode may comprise one or more heterologous
polypeptide
coding sequences that comprise wildtype coding sequences and one or more
heterologous
polypeptide coding sequences that comprise clinical variant coding sequences.
Clinical variants
are typically classified as pathogenic, likely pathogenic, benign, likely
benign or a variant of
unknown significance (VUS). The system provides a platform that can be used to
test the
12
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
function of those heterologous polypeptide coding sequences (e.g. human
genes), variants of
those heterologous polypeptide coding sequences (e.g. human clinical
variants), or as a drug
screening platform identifying therapeutic agents or drugs that alter the
function of the expressed
heterologous polypeptide coding sequences or for treatment of animals,
including humans (e.g.
drug candidates specific to the clinical variants of the heterologous
polypeptide coding
sequences) in the context of their interaction with other expressed
interrelated heterologous
polypeptide coding sequences in vivo.
[0040] The animals of the invention are "genetically modified" or "transgenic"
at multiple loci,
which means that they have at least two transgenes, or other foreign DNAs,
added or
incorporated, or an endogenous gene modified, including, targeted, recombined,
interrupted,
deleted, disrupted, replaced, suppressed, enhanced, or otherwise altered, to
mediate a genotypic
or phenotypic effect in at least one cell of the animal and typically into at
least one germ line cell
of the animal. In some embodiments, the animal may have each of the plurality
of transgenes
integrated on one allele of its genome (heterozygous transgenic). In other
embodiments, animal
may have each of the plurality of transgenes on two alleles (homozygous
transgenic).
[0041] In certain embodiments, the transgenic animals are model organisms
including, but not
limited to, nematodes, zebrafish, fruit fly, xenopus, or rodents, such as mice
and rats.
[0042] In certain embodiments, the present transgenic animals provide a
plurality of
heterologous polypeptide coding sequences, wherein each is a single gene copy
wherein a
chimeric optimized cDNA of a heterologous polypeptide coding sequence, e.g.
modified human
cDNA, is inserted to replace coding sequences of a C. elegans ortholog. The
humanized
nematode is then compared to a nematode lacking the orthologous C. elegans
genes, to confirm
significant restoration of wild type function. The validated transgenic animal
is then modified by
installation of at least one clinical variant and tested in one or more
phenotyping assays to detect
aberrant function. These transgenic animal models have distinct advantages for
testing and
exploring variant biology. For example, humanized models circumvent
differences in compound
binding between humans and other species.
[0043] In embodiments, the chimeric heterologous polypeptide coding sequences
each comprise
human heterologous exon coding sequences interspersed, or paired, with
artificial host nematode
intron sequences optimized for expression in the host nematode. In
embodiments, the host
13
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
nematode intron coding sequences are from a highly expressed C. elegans gene
and may be
further modified for optimized expression. Provided herein are transgenic
nematodes comprising
and expressing heterologous polypeptide coding sequences, wherein the host
nematode
comprises a plurality of chimeric heterologous polypeptide coding sequences
comprising
heterologous exon coding sequences interspersed with artificial host nematode
intron sequences
optimized for expression in the host nematode and selected from SEQ ID NO: 1
to 6. In
embodiments, the heterologous exon coding sequences are human selected from
the human
genes of Table 1, Table 3 or Example 3.
[0044] Definitions
[0045] As used herein, the terms "a" or "an" are used, as is common in patent
documents, to
include one or more than one, independent of any other instances or usages of
"at least one" or
"one or more."
[0046] As used herein, the term "or" is used to refer to a nonexclusive or,
such that "A or B"
includes "A but not B," "B but not A," and "A and B," unless otherwise
indicated.
[0047] As used herein, the term "about" is used to refer to an amount that is
approximately,
nearly, almost, or in the vicinity of being equal to or is equal to a stated
amount, e.g., the state
amount plus/minus about 5%, about 4%, about 3%, about 2% or about 1%.
[0048] "Clustered Regularly Interspaced Short Palindromic Repeats" and
"CRISPRs", as used
interchangeably herein refers to loci containing multiple short direct repeats
that are found in the
genomes of approximately 40% of sequenced bacteria and 90% of sequenced
archaea.
[0049] "Coding sequence" or "encoding nucleic acid" as used herein means the
nucleic acids
(RNA or DNA molecule) that comprise a nucleotide sequence which encodes a
protein. The
coding sequence can further include initiation and termination signals
operably linked to
regulatory elements including a promoter and polyadenylation signal capable of
directing
expression in the cells of an individual or mammal to which the nucleic acid
is administered. The
coding sequence may be codon optimized. "Polypeptide coding sequence" as used
herein means
the nucleic acid coding sequence that encodes for a specific amino acid
sequence, such as a
heterologous polypeptide.
14
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
[0050] "cDNA" as used herein means the deoxyribonucleic acid sequence that is
derived as a
copy of a mature messenger RNA sequence and represents the entire coding
sequence needed for
creation of a fully functional protein sequence.
[0051] As used herein, the terms "disrupt," "disrupted," and/or "disrupting"
in reference to a
gene mean that the gene is degraded sufficiently such that it is no longer
functional. In
embodiments, the native ortholog gene is replaced with the (chimeric)
heterologous polypeptide
coding sequence effectively disrupting the native host gene.
[0052] "Donor DNA", "donor template" and "repair template" as used
interchangeably herein
refers to a double, or single-stranded DNA fragment or molecule that includes
at least a portion
of the gene of interest. The donor DNA may encode a full-functional protein or
a partially-
functional protein.
[0053] As used herein, the term "donor homology" refers to a sequence at a
target edit site that is
also include in the nucleic acid sequence of a plasmid DNA construct that is
necessary to instruct
endogenous homologous repair machinery of the cell to create in frame
insertion of a transgene
sequence. Typically, a plasmid for instructing transgenesis contains a both a
left-side and right-
side donor homology sequence
[0054] As used herein, the term "gene editing" refers to a type of genetic
engineering in which
DNA is inserted, replaced, or removed from a genome using gene editing tools.
Examples of
gene editing tools include, without limitation, zinc finger nucleases, TALEN
and CRISPR.
[0055] "Genetic disease" as used herein refers to a disease, partially or
completely, directly or
indirectly, caused by one or more abnormalities in the genome, especially a
condition that is
present from birth. The abnormality may be a mutation, an insertion or a
deletion. The
abnormality may affect the coding sequence of the gene or its regulatory
sequence. The genetic
disease may be, but is not limited to epilepsy, DMD, hemophilia, cystic
fibrosis, Huntington's
chorea, familial hypercholesterolemia (LDL receptor defect), hepatoblastoma,
Wilson's disease,
congenital hepatic porphyria, inherited disorders of hepatic metabolism, Lesch
Nyhan syndrome,
sickle cell anemia, thalassaemias, xeroderma pigmentosum, Fanconi's anemia,
retinitis
pigmentosa, ataxia telangiectasia, Bloom's syndrome, retinoblastoma, and Tay-
Sachs disease.
"Clinical variants" are used herein, are those genes that lead to a genetic
disease wherein
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
expression of the gene results in one or more amino acid changes as compared
to benign allele
that does not lead to disease.
[0056] A "heterologous gene" or "heterologous polypeptide coding sequence" as
used herein
refers to a nucleotide sequence not naturally associated with a host animal
into which it is
introduced, including for example, exon coding sequences from a human gene
introduced, as a
(chimeric) heterologous polypeptide coding sequence, into a host nematode. In
embodiments,
the heterologous polypeptide coding sequence may comprise one or more point
mutation(s)
which results in one or more amino acid changes in the expressed product,
wherein any change
as compared to a host wild type sequence is considered a "heterologous
polypeptide coding
sequence" regardless if the entire sequence, or just one nucleic acid change,
was introduced into
the host genome.
[0057] The term "heterologous polygenic or monogenic phenotype" as used
herein, refers to any
measurable phenotype that is different as compared to a host "wild-type"
phenotype.
"Polygenic" and "monogenic" refer to a phenotype that is induced by one
("monogenic"), or
more expressed transgenes.
[0058] The term "human disease phenotype" as used herein, including both
"monogenic" and
polygenic", refers to an observable phenotype induced by expression of one or
more human
disease transgenes. In other words, an observable phenotype seen in the host
animal after
insertion into the genome of a sequence the encodes for a human disease gene,
such as a clinical
variant. The phenotype may not be related to a phenotype seen in a human with
a corresponding
genetic disease, but is any observable phenotype that is different, and or
distinct, from an
observable phenotype of a wild type host animal. The observable human disease
phenotype, in
the instant disclosure, is used as a readout to enable study of human genetic
diseases via a host
animal (e.g. nematodes or zebrafish) expressing the disease gene product.
[0059] The term "homolog" refers to any gene that is related to a reference
gene by descent from
a common ancestral DNA sequence. The term "ortholog" refers to homologs in
different species
that evolved from a common ancestral gene by speciation. Typically, orthologs
retain the same
or similar function despite differences in their primary structure
(mutations).
[0060] As used herein, the term "homology driven recombination" or "homology
direct repair"
or "HDR" is used to refer to a homologous recombination event that is
initiated by the presence
16
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
of double strand breaks (DSBs) in DNA (Liang et al. 1998); and the specificity
of HDR can be
controlled when combined with any genome editing technique known to create
highly efficient
and targeted double strand breaks and allows for precise editing of the genome
of the targeted
cell; e.g. the CRISPR/Cas9 system (Findlay et al. 2014; Mali et al. February
2014; and Ran et al.
2013).
[0061] As used herein, the term "enhanced homology driven insertion or knock-
in" is described
as the insertion of a DNA construct, more specifically a large DNA fragment or
construct
flanked with homology arms or segments of DNA homologous to the double strand
breaks,
utilizing homology driven recombination combined with any genome editing
technique known to
create highly efficient and targeted double strand breaks and allows for
precise editing of the
genome of the targeted cell; e.g. the CRISPR/Cas9 system. (Mali et al. Feb
2013).
[0062] As used herein, the terms "increase," "increased," "increasing,"
"improved," (and
grammatical variations thereof), describe, for example, an increase of at
least about 5%, 10%,
15%, 20%, 25%, 35%, 50%, 75%, 80%, 85%, 90%, 95%, 97%), 98%), 99%), or 100% as
compared to a control. In embodiments, the increase in the context of a
heterogenous gene or
clinical variant thereof, is measured and/or determined via phenotypic assay
to assess function of
the expressed gene.
[0063] As used herein, the term "genomic locus" or "locus" (plural loci) is
the specific location
of a gene or DNA sequence on a chromosome and, can include both intron or exon
sequences of
a particular gene. A "gene" refers to stretches of DNA or RNA that encode a
polypeptide or an
RNA chain that has functional role to play in an organism and hence is the
molecular unit of
heredity in living organisms. For the purpose of this invention it may be
considered that genes
include regions which regulate the production of the gene product, whether or
not such
regulatory sequences are adjacent to coding and/or transcribed sequences.
Accordingly, a gene
includes, but is not necessarily limited to, introns, exons, promoter
sequences, terminators,
translational regulatory sequences such as ribosome binding sites and internal
ribosome entry
sites, enhancers, silencers, insulators, boundary elements, 5' or 3'
regulatory sequences,
replication origins, matrix attachment sites and locus control regions. As
used herein "native
locus" refers to the specific location of a host gene (e.g., ortholog to the
heterologous
polypeptide coding sequence) in a host animal.
17
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
[0064] "Mutant gene" or "mutated gene" as used interchangeably herein refers
to a gene that has
undergone a detectable mutation. A mutant gene has undergone a change, such as
the loss, gain,
or exchange of genetic material, which affects the normal transmission and
expression of the
gene. As used herein, "clinical variant" is a disease gene that comprises one
or more amino acid
changes as compared to wild type and is thus a mutant gene.
[0065] A "normal" or "wild type" nucleic acid, nucleotide sequence,
polypeptide or amino acid
sequence refers to a naturally occurring or endogenous nucleic acid,
nucleotide sequence,
polypeptide or amino acid sequence that has not undergone a change. As used
herein, the wild
type sequence may be a disease gene, but does not comprise a mutation leading
to a pathogenic
phenotype. It is understood there is a distinction between a wild type disease
gene (e.g. those
without a mutation leading to a pathogenic phenotype and may be an allele
reflective of a
"normal" heterogenous population) and clinical variants that comprise one or
more mutations of
those disease genes and that may have a pathogenic phenotype. In embodiments,
the normal
gene or wild type gene may be the most prevalent allele of the gene in a
heterogenous
population. N2 are wild type C. elegans nematodes.
[0066] "Operably linked" as used herein means that expression of a gene is
under the control of
a promoter with which it is spatially connected. A promoter may be positioned
5' (upstream) or 3'
(downstream) of a gene under its control. The distance between the promoter
and a gene may be
approximately the same as the distance between that promoter and the gene it
controls in the
gene from which the promoter is derived. As is known in the art, variation in
this distance may
be accommodated without loss of promoter function.
[0067] "Partially-functional" as used herein describes a protein that is
encoded by a mutant gene
and has less biological activity than a functional protein but more than a non-
functional protein.
In embodiments, function is determined via one or more phenotypic assays
wherein a phenotypic
profile for the mutant (disease) gene may be generated.
[0068] As used herein, the term "percent sequence identity" or "percent
identity" refers to the
percentage of identical nucleotides in a linear polynucleotide of a reference
("query")
polynucleotide molecule (or its complementary strand) as compared to a test
("subject")
polynucleotide molecule (or its complementary strand) when the two sequences
are optimally
18
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
aligned. In some embodiments, "percent identity" can refer to the percentage
of identical amino
acids in an amino acid sequence
[0069] As used herein, the term "percent sequence similarity" or "percent
similarity" refers to the
percentage of near-identical nucleotides in a linear polynucleotide of a
reference ("query")
polynucleotide molecule (or its complementary strand) as compared to a test
("subject")
polynucleotide molecule (or its complementary strand) when the two sequences
are optimally
aligned. In some embodiments, "percent similarity" can refer to the percentage
of near-identical
amino acids in an amino acid sequence. Near-identical amino acids are residues
with similar
biophysical properties (e.g., the hydrophobic leucine and isoleucine, or the
negatively-charged
aspartic acid and glutamic acid).
[0070] As used herein, the term "polynucleotide" refers to a heteropolymer of
nucleotides or the
sequence of these nucleotides from the 5' to 3' end of a nucleic acid molecule
and includes DNA
or RNA molecules, including cDNA, a DNA fragment or portion, genomic DNA,
synthetic (e.g.,
chemically synthesized) DNA, plasmid DNA as DNA construct, mRNA, and anti-
sense RNA,
any of which can be single stranded or double stranded. The terms
"polynucleotide," "nucleotide
sequence" "nucleic acid," "nucleic acid molecule," and "oligonucleotide" are
also used
interchangeably herein to refer to a heteropolymer of nucleotides. Except as
otherwise indicated,
nucleic acid molecules and/or polynucleotides provided herein are presented
herein in the 5' to 3'
direction, from left to right and are represented using the standard code for
representing the
nucleotide characters as set forth in the U.S. sequence rules, 37 CFR 1.821 -
1.825 and the
World Intellectual Property Organization (WIPO) Standard ST.25.
[0071] "Promoter" as used herein means a synthetic or naturally-derived
molecule which is
capable of conferring, activating or enhancing expression of a nucleic acid in
a cell. A promoter
may comprise one or more specific transcriptional regulatory sequences to
further enhance
expression and/or to alter the spatial expression and/or temporal expression
of same. A promoter
may also comprise distal enhancer or repressor elements, which may be located
as much as
several thousand base pairs from the start site of transcription. A promoter
may be derived from
sources including viral, bacterial, fungal, plants, insects, and animals. A
promoter may regulate
the expression of a gene component constitutively, or differentially with
respect to cell, the tissue
or organ in which expression occurs or, with respect to the developmental
stage at which
19
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
expression occurs, or in response to external stimuli such as physiological
stresses, pathogens,
metal ions, or inducing agents.
[0072] As used herein, the terms "reduce," "reduced," "reducing," "reduction,"
"diminish,"
"suppress," and "decrease" (and grammatical variations thereof), describe, for
example, a
decrease of at least about 5%, 10%, 15%, 20%, 25%, 35%, 50%, 75%, 80%, 85%,
90%, 95%,
97%), 98%), 99%), or 100% as compared to a control. In embodiments, the
reduction in the
context of a heterogenous gene or clinical variant thereof, is measured and/or
determined via
phenotypic assay to assess function of the expressed gene.
[0073] The term "safe harbor" locus as used herein refers to a site in the
genome where
transgenic DNA (e.g., a construct) can be added whose expression is insulated
from neighboring
transcriptional elements such that the transgene expression is fully depend on
only the introduced
transgene regulatory elements. In certain embodiments, the present invention
involves
incorporation and expression of transgenic DNA includes transgenes within a
safe harbor locus.
[0074] As used herein "sequence identity" refers to the extent to which two
optimally aligned
polynucleotide or peptide sequences are invariant throughout a window of
alignment of
components, e.g., nucleotides or amino acids. "Identity" can be readily
calculated by known
methods including, but not limited to, those described in: Computational
Molecular Biology
(Lesk, A. M., ed.) Oxford University Press, New York (1988); Biocomputing:
Informatics and
Genome Projects (Smith, D. W., ed.) Academic Press, New York (1993); Computer
Analysis of
Sequence Data, Part I (Griffin, A. M., and Griffin, H. G., eds.) Humana Press,
New Jersey
(1994); Sequence Analysis in Molecular Biology (von Heinje, G., ed.) Academic
Press (1987);
and Sequence Analysis Primer (Gribskov, M. and Devereux, J., eds.) Stockton
Press, New York
(1991).
[0075] As used herein, the phrase "substantially identical," or "substantial
identity" and
grammatical variations thereof in the context of two nucleic acid molecules,
nucleotide
sequences or protein sequences, refers to two or more sequences or
subsequences that have at
least about 70%, 71%, 72%, 73%, 74%, 75%, 76%, 77%, 78%, 79%, 80%, 81%, 82%,
83%,
84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%,
99%,
and/or 100%> nucleotide or amino acid residue identity, when compared and
aligned for
maximum correspondence, as measured using one of the following sequence
comparison
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
algorithms or by visual inspection. In particular embodiments, substantial
identity can refer to
two or more sequences or subsequences that have at least about 70%, at least
about 75%, at least
about 80%, at least about 85%, at least about 90%, at least about 95, 96, 96,
97, 98, or 99%
identity.
[0076] For sequence comparison, typically one sequence acts as a reference
sequence to which
test sequences are compared. When using a sequence comparison algorithm, test
and reference
sequences are entered into a computer, subsequence coordinates are designated
if necessary, and
sequence algorithm program parameters are designated. The sequence comparison
algorithm
then calculates the percent sequence identity for the test sequence(s)
relative to the reference
sequence, based on the designated program parameters.
[0077] Optimal alignment of sequences for aligning a comparison window are
well known to
those skilled in the art and may be conducted by tools such as the local
homology algorithm of
Smith and Waterman, the homology alignment algorithm of Needleman and Wunsch,
the search
for similarity method of Pearson and Lipman, and optionally by computerized
implementations
of these algorithms such as GAP, BESTFIT, FASTA, and TFASTA available as part
of the
GCG Wisconsin Package (Accelrys Inc., San Diego, CA). An "identity fraction"
for aligned
segments of a test sequence and a reference sequence is the number of
identical components
which are shared by the two aligned sequences divided by the total number of
components in the
reference sequence segment, i.e., the entire reference sequence or a smaller
defined part of the
reference sequence. Percent sequence identity is represented as the identity
fraction multiplied by
100. The comparison of one or more polynucleotide sequences may be to a full-
length
polynucleotide sequence or a portion thereof, or to a longer polynucleotide
sequence. For
purposes of this invention "percent identity" may also be determined using
BLASTX version 2.0
for translated nucleotide sequences and BLASTN version 2.0 for polynucleotide
sequences.
[0078] Software for performing BLAST analyses is publicly available through
the National
Center for Biotechnology Information. This algorithm involves first
identifying high scoring
sequence pairs (HSPs) by identifying short words of length W in the query
sequence, which
either match or satisfy some positive-valued threshold score T when aligned
with a word of the
same length in a database sequence. T is referred to as the neighborhood word
score threshold
(Altschul et al, 1990). These initial neighborhood word hits act as seeds for
initiating searches to
21
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
find longer HSPs containing them. The word hits are then extended in both
directions along each
sequence for as far as the cumulative alignment score can be increased.
Cumulative scores are
calculated using, for nucleotide sequences, the parameters M (reward score for
a pair of
matching residues; always > 0) and N (penalty score for mismatching residues;
always <0). For
amino acid sequences, a scoring matrix is used to calculate the cumulative
score. Extension of
the word hits in each direction are halted when the cumulative alignment score
falls off by the
quantity X from its maximum achieved value, the cumulative score goes to zero
or below due to
the accumulation of one or more negative-scoring residue alignments, or the
end of either
sequence is reached. The BLAST algorithm parameters W, T, and X determine the
sensitivity
and speed of the alignment. The BLASTN program (for nucleotide sequences) uses
as defaults a
wordlength (W) of 11, an expectation (E) of 10, a cutoff of 100, M=5, N=-4,
and a comparison
of both strands. For amino acid sequences, the BLASTP program uses as defaults
a wordlength
(W) of 3, an expectation (E) of 10, and the BLOSUM62 scoring matrix (see
Henikoff &
Henikoff, Proc. Natl. Acad. Sci. USA 89: 10915 (1989)).
[0079] In addition to calculating percent sequence identity, the BLAST
algorithm also performs
a statistical analysis of the similarity between two sequences (see, e.g.,
Karlin & Altschul, Proc.
Nat'l. Acad. Sci. USA 90: 5873-5787 (1993)). One measure of similarity
provided by the
BLAST algorithm is the smallest sum probability (P(N)), which provides an
indication of the
probability by which a match between two nucleotide or amino acid sequences
would occur by
chance. For example, a test nucleic acid sequence is considered similar to a
reference sequence if
the smallest sum probability in a comparison of the test nucleotide sequence
to the reference
nucleotide sequence is less than about 0.1 to less than about 0.001. Thus, in
some embodiments
of the invention, the smallest sum probability in a comparison of the test
nucleotide sequence to
the reference nucleotide sequence is less than about 0.001.
[0080] "Subject" and "patient" as used herein interchangeably refers to any
vertebrate, including,
but is not limited to, a mammal (e.g., cow, pig, camel, llama, horse, goat,
rabbit, sheep, hamsters,
guinea pig, cat, dog, rat, and mouse, a non-human primate (for example, a
monkey, such as a
cynomolgus or rhesus monkey, chimpanzee, etc.) and a human). In some
embodiments, the
subject may be a human or a non-human. The subject or patient may be
undergoing other forms
of treatment. In embodiments, the patient is a human wherein a clinical
variant is a sequence of
a disease gene from the patient.
22
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
[0081] "Target gene" as used herein refers to any nucleotide sequence encoding
a known or
putative gene product. As used herein the target gene may be the (chimeric)
heterologous
polypeptide coding sequence, either in normal or wild type form, or as a
clinical variant, or the
host animal ortholog of the heterologous polypeptide coding sequence. The
target gene may be a
mutated gene involved in a genetic disease, also referred to herein as a
clinical variant.
[0082] "Target nucleotide sequence" as used herein refers to the region of the
target gene to
which the Type I CRISPR/Cas system is designed to bind.
[0083] The terms "transformation," "transfection," and "transduction" as used
interchangeably
herein refer to the introduction of a heterologous nucleic acid into a cell.
Such introduction into a
cell may be stable or transient. Thus, in some embodiments, a host cell or
host organism is stably
transformed with a polynucleotide of the invention. In other embodiments, a
host cell or host
organism is transiently transformed with a polynucleotide of the invention.
"Transient
transformation" in the context of a polynucleotide means that a polynucleotide
is introduced into
the cell and does not integrate into the genome of the cell. By "stably
introducing" or "stably
introduced" in the context of a polynucleotide introduced into a cell is
intended that the
introduced polynucleotide is stably incorporated into the genome of the cell,
and thus the cell is
stably transformed with the polynucleotide. "Stable transformation" or "stably
transformed" as
used herein means that a nucleic acid molecule is introduced into a cell and
integrates into the
genome of the cell. As such, the integrated nucleic acid molecule is capable
of being inherited by
the progeny thereof, more particularly, by the progeny of multiple successive
generations.
"Genome" as used herein also includes the nuclear, the plasmid and the plastid
genome, and
therefore includes integration of the nucleic acid construct into, for
example, the chloroplast or
mitochondrial genome. Stable transformation as used herein can also refer to a
transgene that is
maintained extrachromasomally, for example, as a mini-chromosome or a plasmid.
In certain
embodiments, the nucleotide sequences, constructs, expression cassettes can be
expressed
transiently and/or they can be stably incorporated into the genome of the host
organism, such as
in a native, non-native locus or safe harbor location.
[0084] "Transgene" as used herein refers to a gene or genetic material
containing a gene
sequence that has been isolated from one organism and is introduced into a
different organism.
This non-native segment of DNA may retain the ability to produce RNA or
protein in the
23
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
transgenic organism, or it may alter the normal function of the transgenic
organism's genetic
code. The introduction of a transgene has the potential to change the
phenotype of an organism.
[0085] The term "3'untranslated region" or"3'UTR" refers to a nucleotide
sequence downstream
(i.e., 3') of a coding sequence. It generally extends from the first
nucleotide after the stop codon
of a coding sequence to just before the poly(A) tail of the corresponding
transcribed mRNA. The
3' UTR may contain sequences that regulate translation efficiency, mRNA
stability, mRNA
targeting and/or polyadenylation. In embodiments, the 3' UTR may be native, or
non-native in
the context of the (chimeric) heterologous polypeptide coding sequence.
[0086] "Variant" with respect to a peptide or polypeptide that differs in one
or more amino acid
sequence by the insertion, deletion, or conservative substitution of amino
acids as compared to a
normal or wild type sequence. The variant may further exhibit a phenotype that
is quantitatively
distinguished from a phenotype of the normal or wild type expressed gene. In
embodiments,
clinical variant refers to a disease gene with one or more amino acid changes
as compared to the
normal or wild type disease gene.
[0087] Transgenic Nematodes
[0088] The instant transgenic nematode system comprises a host nematode that
comprises and
expresses a first heterologous polypeptide coding sequence and a second
heterologous
polypeptide coding sequence. As used herein, at least a first heterologous
polypeptide coding
sequence and second heterologous polypeptide coding sequence, may be referred
to collectively
as a "plurality" of heterologous polypeptide coding sequences. The present
transgenic
nematodes comprise at least two distinct heterologous polypeptide coding
sequences that are
interrelated as to an observable phenotype, such as a monogenic or polygenic
disease. As used
herein "distinct heterologous polypeptide coding sequences" means a sequence
that codes for a
unique protein wherein each are under control of a separate promotor and/or
other regulatory
elements. In embodiments, the plurality of heterologous polypeptide coding
sequences do not
include a reporter gene or a prokaryotic gene. In embodiments, the first and
second heterologous
polypeptide coding sequences are integrated into the host nematode genome,
wherein expression
of the first and second heterologous polypeptide coding sequences contribute
to the heterologous
phenotype.
24
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
[0089] The present host nematodes comprise at least two ("digenic")
heterologous polypeptide
coding sequences, wherein their expression products, directly or indirectly,
are interrelated such
as in a pathway (e.g. homologous recombination) or a disease phenotype (e.g.
autism, epilepsy or
neurodegenerative disorder). In many instances a variant of pathogenic
consequence occurs at a
protein-protein interaction domain, therefore modeling a pathogenic variant in
a single gene
humanized animal will be insufficient for creating a condition in which
pathogenic behavior can
be detected. At a minimum, at least two human genes need to be installed in
the host animal
genome so the protein-protein interaction variant can be modeled in vivo. In
other conditions, the
pathogenic behavior will only manifest if two genes in a pathway are humanized
so that
polygenic additive effects reaching a pathogenicity threshold can be observed.
As a result,
multiple polypeptide coding sequences need to be installed so that proper
protein complex,
pathway signaling, and/or metabolic processes can be faithful recapitulates as
observed in the
human condition.
[0090] In embodiments, the host nematode comprises and expresses additional
heterologous
polypeptide coding sequences that are also interrelated as to the first and
second heterologous
polypeptide coding sequences. In embodiments, the present host nematodes
comprise and
express from two (2) to about fifteen (15) heterologous polypeptide coding
sequences, optionally
from three (3) to about fifteen (15) polypeptide coding sequences. Those
plurality of
heterologous polypeptide coding sequences may individually code for a wild
type sequence or a
variant thereof including identified clinical variants. It is also an aspect
of the invention that the
host transgenic nematodes, in addition to the plurality of heterologous
polypeptide coding
sequences, comprise and/or express a reporter heterologous polypeptide coding
sequences.
[0091] In embodiments, one or more of the plurality of heterologous
polypeptide coding
sequences is a (chimeric) heterologous polypeptide coding sequence. As used
herein "chimeric
heterologous polypeptide coding sequence" refers to a sequence comprising
heterologous exon
coding sequences and host animal (e.g. nematode) intron sequences interspersed
or paired with
the exon coding sequences. In embodiments, the heterologous polypeptide coding
sequence
corresponds to a nematode ortholog, wherein the chimeric heterologous
polypeptide coding
sequence replaced the entire host nematode ortholog, either prior to or at the
same time the
chimeric heterologous polypeptide coding sequence is installed, and wherein
the chimeric
heterologous polypeptide coding sequence is installed at the host nematode
ortholog native
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
locus. In embodiments, each of the heterologous polypeptide coding sequences
are integrated
into the native locus of the nematode as a chimeric heterologous polypeptide
coding sequence. It
is not an aspect of the invention for partial removal with partial
replacement, of the host animal
ortholog. Further, the plurality of interrelated heterologous polypeptide
coding sequences are
eukaryotic; it is not an aspect of the invention for the plurality of
interrelated heterologous
polypeptide coding sequences to be prokaryotic. In embodiments, the host
nematode is a C.
elegans, C. brig gsae, C remanei, C. tropicalis, or P. pacificus. (Sugi T et
al. Genome Editing in
C. elegans and Other Nematode Species. Int J Mol Sci. 2016 Feb 26;17(3):295).
[0092] In embodiments, the plurality of heterologous polypeptide coding
sequences are selected
from a different species of nematode (e.g. parasitic nematode), an avian,
mammal or fish. In
certain embodiments, the plurality of heterologous polypeptide coding
sequences are human. In
embodiments, the heterologous polypeptide coding sequences replace the entire
nematode
ortholog gene at their respective native loci, accordingly the heterologous
polypeptide coding
sequences must have a homolog as an identified ortholog in the host nematode.
In one
embodiment, the homolog is of substantial quality when sequence identity
between heterologous
source and host exceeds 70%. In one embodiment, the homolog is of high quality
when
sequence identity between heterologous source and host exceeds 50%. In other
embodiments,
the homolog is good when its identity exceeds 35%. In other embodiments, the
homolog is
adequate when its identity exceeds 20%. In other embodiments, the homolog is
poor but
acceptable when its identity is less than 20%. See Example 1 for
identification of host nematode
orthologs; and, Tables 1 and 3 for a pairing of human polypeptide coding
sequences and
nematode orthologs.
[0093] In alternative embodiments, the plurality of heterologous polypeptide
coding sequences
are from a parasitic nematode, which are selected from Trichuris muris,
Ascaris lumbricoides,
Ancylostoma duodenale, Necator americanus, Trichuris trichiura, Enterobius
vermicularis,
Strongyloides stercoralis, Trichinella spiralis, Wuchereria bancrofti, Brugia
malayi, Brugia
timori, Loa loa, Mansonella streptocerca, Onchocerca volvulus, Mansonella
perstans,
Mansonella ozzardi, Cooperia punctata, Cooperia oncophora, Ostertagia
ostertagi,
Haemonchus contortus, Ascaris suum, Aphelenchoides, Dhylenchus, Globodera,
Heterodera,
Longidorus, Meloidogyne ,Nacobbus, Pratylenchus, Trichodorus, Xiphinema,
Bursaphelenchus,
26
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
Dirofilaria immitis, Toxocara canis, Toxocara cati, Ancylostoma braziliense,
Ancylostoma
tuba eforme, Ancylostoma caninum, Dirofilaria repens, and Uncinaria
stenocephala.
[0094] In certain embodiments, the plurality of heterologous polypeptide
coding sequences are
human polypeptide coding sequences. In certain embodiments, the human
polypeptide coding
sequences are wild type polypeptide coding sequences. Provided herein is a
transgenic nematode
system comprising a host nematode comprising a plurality of chimeric
heterologous polypeptide
coding sequences optimized for expression in the host nematode wherein the
heterologous
polypeptide coding sequences replace their respective host nematode gene
ortholog and the
heterologous polypeptide coding sequences rescues, or at least partially
restores, function of the
replaced nematode orthologs. Heterologous polypeptide coding sequences that
rescue function
of the replaced nematode ortholog are referred to herein as "wild type"
heterologous polypeptide
coding sequences.
[0095] In other embodiments, the plurality of heterologous polypeptide coding
sequences are
human disease genes. As used herein, "disease gene" or "disease polypeptide
coding sequence"
refers to a gene or expressed sequence involved in or implicated in a disease.
In certain
embodiments provided herein are transgenic nematodes comprising a plurality of
heterologous
polypeptide coding sequences that are human wild type disease genes that have
replaced the host
nematode orthologs at their native loci. See Examples 1 to 4. Those human
heterologous disease
polypeptide coding sequences represent targets for drug discovery and drugs
that rescue function
of human clinical variants.
[0096] In embodiments, the heterologous polypeptide coding sequences rescue,
or at least
partially restore, function of the removed host nematode orthologs. Rescue or
restoration of
function, which is measured in a phenotypic assay, identifies those transgenic
nematodes that are
validated and may be used as a transgenic control animal. As used herein
"validated transgenic
control nematode" means a transgenic nematode expressing a plurality of
chimeric heterologous
polypeptide coding sequences in place of host nematode orthologs, wherein at
least partial
function is rescued by expression of the heterologous polypeptide coding
sequences. Rescued
function can be from 1% to 100% as compared to a host nematode expressing the
heterologous
"wild-type" polypeptide coding sequence. In other embodiments, rescued
function can be from
1% to 100% as compared to a host nematode with a knock-out of the ortholog.
27
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
[0097] In addition to quantitative rescue effects, rescue can be qualitative
as to essential genes,
wherein rescue with a heterologous transgene provides sufficient lifespan and
fecundity for
establishment of a propagating colony.
[0098] In embodiments, rescue of function is measured by analyzing, observing
or monitoring
the transgenic nematodes in a phenotypic assay as compared to host nematodes
(KO of ortholog
sequence or expressing the heterologous wild type polypeptide coding sequence)
and/or null
variants. In embodiments, the phenotypic assay is selected from a measurement
of
electrophysiology of pharynx pumping, a food race, lifespan extension and
contraction assay,
movement assay, fecundity assay with egg lay or population expansion,
apoptotic body
formation, chemotaxis, lipid metabolism assay, body morphology changes,
fluorescence
changes, drug sensitivity and resistance assays, or a combination thereof.
There is no limitation
as to the phenotypic assay that may be used, including those developed in the
future, provided a
useful phenotype profile can be generated for assessing function of the
installed heterologous
polypeptide coding sequence. The above are representative phenotype assays,
but others may be
used to validate the transgenic nematode, as well as for assessing variants of
the heterologous
polypeptide coding sequences.
[0099] In embodiments, a phenotype profile of the transgenic nematode is
identified from the
assay wherein the identified phenotype is selected from electropharyngeogram
variant, feeding
behavior variant, defecation behavior variant, lifespan variant, electrotaxis
variant, chemotaxis
variant, thermotaxis variant, mechanosensation variant, movement variant,
locomotion variant,
pigmentation variant, embryonic development variant, organ system morphology
variant,
metabolism variant, fertility variant, dauer formation variant, stress
response variant, or a
combination thereof.
[00100] In certain embodiments provided herein are validated transgenic
control
nematodes of the present system, comprising a plurality of heterologous
polypeptide coding
sequences optimized for expression in the host nematode wherein the
heterologous polypeptide
coding sequences replace their respective host nematode gene orthologs and the
heterologous
polypeptide coding sequences rescue function of the replaced nematode
orthologs. In
embodiments, the heterologous polypeptide coding sequences are human disease
genes.
28
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
[00101] In embodiments, the transgenic nematodes further comprise an
inducible reporter
gene operably linked to an inducible promoter. See US Patent No. 8,937,213,
herein incorporated
by reference, which disclose use of inducible and constitutive promoters
operably linked to
reporter genes. Reporter genes are well known in the art and include
luminescent and
fluorescent proteins that can be expressed in living cells. Well known
examples include GFP,
mCherry, mTurquoise and mVenus. In certain embodiments the inducible promoter
is from a
gene induced by the heterologous polypeptide coding sequence, or the variant
heterologous
polypeptide coding sequence. In certain embodiments, the inducible promoter is
from a gene
inhibited by the variant heterologous polypeptide coding sequence.
[00102] The present validated transgenic nematodes are prepared via
homologous
recombination at the native locus of the host nematode ortholog wherein a
plurality of nematode
orthologs are replaced with the heterologous polypeptide coding sequences.
This method is
advantageous in that it provides a platform for further testing and
modifications and provides an
improvement over previously disclosed methods that use amino acid substitution
for generation
of humanized nematodes expressing clinical variants. The use of gene-swap
(i.e. heterologous
polypeptide coding sequence replaces the nematode ortholog at the native
locus) avoids the
expression level issues that are a challenging problem with extrachromosomal
array studies.
Instead, CRISPR techniques are deployed to directly mutate at native loci
(Farboud B and Meyer
BJ. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved
guide RNA
design. Genetics. 2015 Apr;199(4):959-71; Paix A et al. High Efficiency,
Homology-Directed
Genome Editing in Caenorhabditis elegans Using CRISPR-Cas9 Ribonucleoprotein
Complexes.
Genetics. 2015 Sep;201(1):47-54).
[00103] Gene swap involves removal of the native coding sequence of the
host nematode
(e.g. C. elegans) ortholog and replacement with cDNA from the heterologous
polypeptide coding
sequence (e.g., human gene), wherein the exon coding sequences of the
heterologous polypeptide
coding sequence are paired with, or interspersed with, host nematode intron
sequences. The host
intron sequences are derived from a highly expressed host gene and may be
further modified for
expression of the heterologous exon coding sequences. As used herein "chimeric
heterologous
polypeptide coding sequence" refers to a sequence of heterologous (to the host
animal) exon
coding sequences that are paired or interspersed with the host animal intron
sequences.
Representative modified host nematode intron sequences are selected from SEQ
ID NO: 1 to 6.
29
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
In certain embodiments, the present transgenic nematodes comprise a chimeric
heterologous
polypeptide coding sequence comprising one or more of SEQ ID NO: 1 to 6. Those
sequences,
when used with human exon coding sequences have demonstrated good expression
in a host
nematode.
[00104] To execute a gene-swap, the coding sequence from heterologous cDNA
is
optionally adjusted for optimal expression in the host nematode, e.g., C.
elegans. In addition to
the use of host animal intron sequences paired with heterologous exon coding
sequences,
optimization includes codon optimization for the host animal and removal of
any aberrant splice
donor and/or acceptor sites that were generated as a result of the chimeric
sequence.
Accordingly, in embodiments provided herein are transgenic nematodes
comprising a chimeric
heterologous polypeptide coding sequences optimized for expression in the host
nematode
wherein a heterologous polypeptide coding sequence replaces a host nematode
gene ortholog,
wherein the chimeric heterologous polypeptide coding sequence comprises
heterologous exon
coding sequences interspersed with artificial host nematode intron sequences.
[00105] In embodiments, optimization comprises codon optimization (e.g.
removal of rare
codons), introduction of host intron sequences into the heterologous cDNA and
removal of any
aberrant splice sites. For codon optimization, rare codon usage must be
avoided to enable
sufficient levels of protein translation from a mRNA message. For intron
sequences, the
artificial host intron sequences are added to the codon optimized heterologous
cDNA sequence,
which results in improved mRNA stability, and a chimeric sequence. Performing
those
techniques are well known in the art and online tools exist for performing
both. Conveniently,
codon optimization and identification of aberrant splice sites are achieved
with the C elegans
codon adapter that encodes optimal amino acid sequence (Redemann S et al., C.
elegans codon
Adapter ¨ GGA, Nat Methods. 2011 Mar;8(3):250-2) and NextGene2 which adjust
splice donor
and acceptor sites for optimal performance (Hebesgaard SM et al., Nucleic
Acids Res. 1996 Sep
1;24(17):3439-52).
[00106] Those chimeric sequences, heterologous cDNA optimized, and
artificial host
intron sequences added may result in a sequence with highly repetitive
sequences that prevent
gene synthesis by DNA sequence providers. As a result, the sequence may be
hand curated to
minimize repeat sequence formation and enable synthesis to proceed from
suppliers. The need to
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
hand curate sequence content creates a need for removal of aberrant splice
site donor and
acceptor site. Online tools exist for identify unintentional splice site donor
and acceptor sites
(Hebesgaard SM et al., Nucleic Acids Res. 1996 Sep 1;24(17):3439-52).
Additional hand
curated sequence adjustments are made iteratively until on-line software no
longer detects
aberrant splice site donor and acceptor sites. Because a given optimization
may fail to express
properly for unforeseen reasons, three sets of expression-optimized human cDNA
are frequently
made so that at least three attempts at null rescue can be attempted.
[00107] In embodiments, the intron sequences provided by the C. elegans
codon Adapter
are synthetic introns that are not ideal for expression. However, the
synthetic host intron
sequences can be modified to meet certain criteria optimal for expression of
the heterologous
polypeptide coding sequence. Those criteria include intron sequences, for
expression in a host
nematode such as C. elegans, that are: from a gene highly expressed native C.
elegans genes;
small (less than 80bp); do not contain stop codons; are divisible by 3; and,
have a low
hydropathy index. Host intron sequences that do not meet those criteria can be
modified by
deleting or changing bases. Host intron sequences meeting the above criteria
are likely to not
negatively affect gene expression or plasmid building and at the same time,
even if un-spliced in
synthetic DNA, will retain reading frame and code for peptides with low
hydrophobicity content.
As a result, functional protein is likely even if all the intron sequences
fail to splice.
[00108] In some embodiments, the intron position is based on the protein
structure.
Protein structure can be identified by using published data such as X-ray
crystallography. An
alignment of orthologs and paralogs is performed. Un-conserved regions are
mapped to the
structure to find loop regions. The target gene is labeled for loop regions.
Amino acid pairs are
identified in the loop region that can be coded for a good splice donor and
acceptor such as KE,
KD, QE, QD, EE, ED, KV, QV, and EV. The introns as disclosed above are
inserted between
the splice donor and acceptor and the sequence is checked for aberrant
splicing as disclosed
above.
[00109] In certain embodiments, the transgenic control nematodes may be
prepared by
methods other than homologous recombination into the native locus of the
nematode, provided
the cDNA of the plurality of heterologous polypeptide coding sequences are
optimized for
expression in the host nematode by codon optimization, addition of host intron
sequences to the
31
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
cDNA sequence of the heterologous polypeptide coding sequence and removing
aberrant splice
donor and acceptor sites. Those alternative methods comprise inserting the
optimized chimeric
heterologous polypeptide coding sequences via homologous recombination into a
native locus of
the nematode wherein a nematode gene orthologs are removed, wherein the
heterologous
polypeptide coding sequences are rescued, or at least partially restored, for
function of the
removed nematode orthologs; or, inserting the optimized heterologous
polypeptide coding
sequences into a non-native locus of the nematode; or, inserting the optimized
heterologous
polypeptide coding sequences into a random site of the nematode genome; or,
adding the
optimized heterologous polypeptide coding sequences as an expression vector
wherein the
optimized heterologous polypeptide coding sequences are not integrated into
the nematode
genome.
[00110] In embodiments are provided transgenic test nematodes, which are
based on the
validated transgenic control nematode and comprise a variant of a heterologous
polypeptide
coding sequence. As used herein, "variant heterologous polypeptide coding
sequences" refers to
an expressed gene with one or more amino acid changes as compared to a
heterologous
polypeptide coding sequence that was used to prepare the validated transgenic
control nematode.
Accordingly, a transgenic test nematode comprises a transgenic control
nematode that is a
modified validated transgenic nematode, wherein an expressed heterologous
polypeptide coding
sequence comprises one or more amino acid changes providing a variant of the
heterologous
polypeptide coding sequence. The transgenic test nematodes may be used for
assessing function
of the heterologous variant polypeptide coding sequence and drug discovery. In
embodiments, a
transgenic test nematode comprises a purality of (chimeric) variant
heterologous polypeptide
coding sequences, comprising heterologous exon coding sequences interspersed
with artificial
host nematode intron sequences optimized for expression in the host nematode,
wherein the exon
coding sequences comprise one or more mutations resulting in an amino acid
change as
compared to a wildtype reference sequence (wild type heterologous polypeptide
coding sequence
of transgenic control animal), and wherein the (chimeric) variant heterologous
polypeptide
coding sequence replaces the entire host nematode gene ortholog at a native
locus, and wherein
the heterologous polypeptide coding sequences is a eukaryotic gene.
[00111] In embodiments, a variant heterologous polypeptide coding sequence
may be
introduced by amino acid swap of the transgenic control nematode or by gene
swap of a variant
32
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
containing heterologous polypeptide coding sequence in as replacement of the
native coding
sequence. In embodiments, the variant heterologous polypeptide coding
sequences is a human
disease gene comprising one or more amino acid changes as compared to the wild
type disease
gene. In embodiments, the variant comprises a single amino acid change wherein
the change
was installed into the integrated heterologous polypeptide coding sequence of
the transgenic
control animal via a co-CRIPSR method. The resulting transgenic animals are
transgenic test
animals (e.g. nematode or zebrafish). In certain embodiments, the mutations
(of the
heterologous exon coding sequence) are created from a pool of DNA repair
templates each
containing one or more mutations. In other embodiments, the variant comprises
more than one
amino acid change. In certain embodiments, those mutations are created from a
pool of DNA
repair templates each containing two or more mutations. Variants with more
than one amino
acid change, as compared to the wild type gene, may be a known clinical
variant or a
combination of two or more variants of the same gene. The combination of
clinical variants in
one variant heterologous transgenic test animal may be beneficial for
assessing function of
variants as to their synergistic, antagonistic, additive etc. function as
measured in phenotypic
assays.
[00112] Like drosophila studies, electrophysiology measurements in C.
elegans on
functional variants can provide a rich and diverse set of phenotyping data
(Sorkac A et al. In
Vivo Modelling of ATP1A3 G316S-Induced Ataxia in C. elegans Using CRISPR/Cas9-
Mediated Homologous Recombination Reveals Dominant Loss of Function Defects.
PLoS One.
2016 Dec 9;11(12)). These published studies were done by making "humanizing"
mutations at
native loci. A homology alignment is used to determine where conserved
positions occur
between the human gene and its animal model ortholog. Clinical variants are
then mapped to the
sequence alignment and, if they occur at a conserved amino acid, the clinical
variant can be
installed by CRISPR as an amino-acid-swap which substitutes the native amino
acid with the
amino acid change seen in the patient.
[00113] In embodiments, the variant heterologous polypeptide coding
sequences are
human clinical variants. Accordingly, when at least partial rescue of function
is achieved with
expression of a plurality of heterologous polypeptide coding sequences, the
system (comprising
validated transgenic nematodes) becomes valid for installation of clinical
variants (test
transgenic nematodes). Six classes of clinical variants can be installed
(Pathogenic, Likely
33
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
Pathogenic, Uncertain Significance, Likely Benign, Benign, and the
unassessed). On average,
dbSNP data indicates 80% of known variants are unassessed and nearly half
(40%) of the
remaining assessed variants are Variants of Uncertain Significance (VUS).
(NCBI) Variation
Viewer. Installation of known Pathogenic and Benign variants helps determine
how conserved
are the existing assignments when installed into the human cDNA expressing
nematode model.
When most of the pathogenic and benign variants give expected activities
(e.g., phenotype) in
the humanize nematode model the system then is valid for assessment of
pathogenicity of VUS
and unassigned variants.
[00114] In embodiments, methods are provided herein for assessing function
of a human
clinical variant, comprising the steps of culturing a test transgenic
nematode, wherein at least one
of variant heterologous polypeptide coding sequences contains human clinical
variant; and,
performing a phenotypic screen to identify a phenotype of the test transgenic
nematode, wherein
a change in phenotype as compared to a control transgenic nematode comprising
of wildtype
heterologous polypeptide coding sequences (e.g. corresponding validated
transgenic nematode)
indicates an altered function of the clinical variant in the test transgenic
nematode. The
phenotypic screens and identified phenotypes are disclosed above and are the
same as those used
when validating the transgenic control nematode for rescue of function.
[00115] In embodiments, the phenotypic screen is a food race wherein
decreased time to
reach food, as compared to the control transgenic nematode, indicates
pathogenicity of the
human clinical variant. In embodiments, the methods further comprise
classifying the human
clinical variant as pathogenic, likely pathogenic, uncertain significance,
likely benign, or benign
following the phenotypic screen.
[00116] In certain embodiments, the transgenic test nematode comprises an
inducible
promoter operably linked to a reporter gene, wherein the promoter is from a
gene induced by
expression of the human clinical variant gene, wherein the method for
assessing function of a
human clinical variant comprises culturing a test transgenic nematode, wherein
the variant
heterologous polypeptide coding sequence is a human clinical variant and,
observing the
inducible report gene expression, whereby human clinical variant genes with
altered function are
identified as pathogenic or likely pathogenic when the inducible reporter gene
is expressed.
34
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
[00117] In further embodiments provided herein are methods using the
transgenic test
nematode system for drug screening. For humanized platforms exhibiting
pathogenic activity
with a given installed variant, screens of novel and existing compounds can be
performed in
efforts to find drug candidates with capacity to restore function back towards
wild type. In
embodiments, the methods for screening therapeutic agents to treat altered
function of a human
clinical variant, comprises placing a test transgenic nematode in a medium
comprising a test
compound, wherein a variant heterologous polypeptide coding sequence is a
human clinical
variant identified as pathogenic, likely pathogenic, unknown significance or
unassigned;
incubating the test transgenic nematode with the test compound for a period
from 2 minutes to 7
hours, or from 1 to 7 days including 1 day, 2 days, 3 days, 4 days, 5 days, 6
days or 7 days; and,
performing a screening assay, whereby therapeutic agents are identified from
the test compounds
when the outcome of the screening assay is deemed positive. An altered
phenotype back towards
wildtype is considered positive. The screening assays are phenotypic assays
disclosed above,
including fluorescent assay wherein transgenic test nematode further comprises
an inducible
promoter operably linked to a reporter gene wherein the promoter is from a
gene inhibited in
response to expression of the human clinical variant, whereby therapeutic
agents are identified
when the inducible reporter gene is expressed.
[00118] In embodiments provided herein are methods for screening
therapeutic agents to
treat altered function of a human clinical variant. Those methods comprise use
of a present
transgenic test animal. In certain embodiments, those methods comprise placing
a present
transgenic test nematode, with an identified behavioral or molecular phenotype
that is different
from an identified phenotype of a control transgenic nematode expressing a
wildtype
heterologous polypeptide coding sequence, in a medium comprising a test
compound, wherein
the variant heterologous polypeptide coding sequence is a human clinical
variant; incubating the
test transgenic nematode with the test compound for a period from 2 minutes to
seven days,
including 1 day, 2 days, 3 days, 4 days, 5 days, 6 days or 7 days; and,
performing a phenotypic
assay to identify a post-test compound behavioral or molecular phenotype of
the test transgenic
nematode, whereby therapeutic agents are identified from the test compounds
when the post-test
compound phenotype is more similar, as compared to the phenotype of the test
transgenic
nematode, to the phenotype of the control transgenic nematode.
[00119] Specific Embodiments
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
[00120] In certain embodiments, provided herein is a non-human animal
transgenic
system for assessing a heterologous polygenic or monogenic phenotype,
comprising: a host non-
human animal comprising and expressing a first heterologous polypeptide coding
sequence and a
second heterologous polypeptide coding sequence, wherein the first and second
heterologous
coding sequences are integrated into the host animal genome, and wherein
expression of the first
and second heterologous polypeptide coding sequences in the animal contribute
to the
heterologous phenotype. In embodiments, the host non-human animal is a
nematode or a
zebrafish. In certain embodiments, at least one of the first heterologous
polypeptide coding
sequence or the second heterologous polypeptide coding sequence is a chimeric
heterologous
polypeptide coding sequence comprising heterologous exon coding sequences
interspersed with
artificial host intron sequences optimized for expression in the host. In
embodiments, each of the
first and second heterologous polypeptide coding sequences is individually a
chimeric
heterologous polypeptide coding sequence comprising heterologous exon coding
sequences
interspersed with artificial host intron sequences optimized for expression in
the host animal. In
embodiments, at least one of the first heterologous coding sequence or the
second heterologous
coding sequence replaced an entire host gene ortholog at a native locus. In
embodiments, each
of the first and second heterologous coding sequences individually replaced an
entire host gene
ortholog at a native locus. In embodiments, a host ortholog gene sequence
corresponding to the
first heterologous coding sequence and/or the second heterologous coding
sequence has been
knocked-out. In embodiments, the first and second heterologous coding
sequences comprise
human exon coding sequences. In other embodiments, at least one of the first
heterologous
polypeptide coding sequence or the second heterologous polypeptide coding
sequence comprises
one or more mutations in the first and/or second heterologous polypeptide
coding sequence
coding sequences as compared to a wildtype reference sequence resulting in at
least one amino
acid change in the first and/or second polypeptide coding sequences when the
one or more
additional heterologous polypeptide coding sequence is expressed in the host,
optionally wherein
the mutation corresponds to a human disease gene clinical variant. In some
embodiments, the
present system further comprises and expresses one or more additional
heterologous polypeptide
coding sequence that contributes to the heterologous phenotype, optionally
wherein the one or
more additional heterologous polypeptide coding sequences comprises one or
more mutations in
polypeptide coding sequence as compared to a wildtype reference sequence
resulting in at least
36
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
one amino acid change when the one or more additional heterologous polypeptide
coding
sequence is expressed in the host; or optionally wherein the host animal
comprises and expresses
3 to 15 heterologous polypeptide coding sequences, wherein optionally a host
ortholog gene
corresponding to each of the heterologous polypeptide coding sequences has
been knocked-out.
In certain embodiments, the heterologous phenotype is a monogenic human
disease phenotype or
alternatively a polygenic human disease phenotype.
[00121] In certain embodiments provided herein is a non-human animal
transgenic system
for assessing a heterologous disease phenotype, comprising: a host animal
comprising and
expressing a first heterologous polypeptide coding sequence and a second
heterologous
polypeptide coding sequence, wherein the first and second heterologous
polypeptide coding
sequences are integrated into the host genome, wherein at least one of the
first heterologous
polypeptide coding sequence or the second heterologous polypeptide coding
sequence comprises
one or more mutations in the heterologous polypeptide coding sequence as
compared to a
wildtype reference sequence resulting in at least one amino acid change when
the first
heterologous polypeptide coding sequence or the second heterologous
polypeptide coding
sequence is expressed, and wherein expression of the first and second
heterologous polypeptide
coding sequence contribute to the heterologous disease phenotype. In
embodiments, at least one
of the first heterologous polypeptide coding sequence or the second
heterologous polypeptide
coding sequence is a chimeric heterologous polypeptide coding sequence
comprising
heterologous exon coding sequences interspersed with artificial host intron
sequences optimized
for expression in the host. In other embodiments, each of the first and second
heterologous
polypeptide coding sequences is individually a chimeric heterologous
polypeptide coding
sequence comprising heterologous exon coding sequences interspersed with
artificial host intron
sequences optimized for expression in the host. In certain embodiments, at
least one of the first
heterologous polypeptide coding sequence or the second heterologous
polypeptide coding
sequence replaced an entire host gene ortholog at a native locus. In certain
other embodiments,
each of the first and second heterologous polypeptide coding sequences
individually replace an
entire host gene ortholog at a native locus. In embodiments, a host animal
ortholog gene
corresponding to the first heterologous polypeptide coding sequence and/or the
second
heterologous polypeptide coding sequence has been knocked-out. In embodiments,
the first and
second heterologous polypeptide coding sequences of the system comprise human
exon coding
37
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
sequences. In certain embodiments, the one or more mutations corresponds to a
human disease
gene clinical variant. In other embodiments, the system further comprises and
expresses one or
more additional heterologous polypeptide coding sequence that contribute to
the heterologous
disease phenotype, optionally wherein the one or more additional heterologous
polypeptide
coding sequences comprises one or more mutations in exon coding sequences of
the
heterologous polypeptide coding sequence as compared to a wildtype reference
sequence
resulting in at least one amino acid change when the one or more additional
heterologous
polypeptide coding sequence is expressed in the host, or optionally wherein a
host ortholog gene
for each of the heterologous polypeptide coding sequences has been knocked-
out. In
embodiments, the host of the system comprises and expresses 2 to 15, or 3 to
15 heterologous
polypeptide coding sequences. In embodiments, heterologous disease phenotype
of the system is
a monogenic human disease phenotype or alternatively, a polygenic human
disease phenotype.
[00122] Provided herein in certain embodiments is a non-human animal
humanized
transgenic system for assessing a monogenic or polygenic human disease
phenotype, comprising:
a host animal comprising and expressing a first human polypeptide coding
sequence and a
second human polypeptide coding sequence, wherein the first and second human
polypeptide
coding sequences are integrated into the genome of the host animal, wherein at
least one of the
first human polypeptide coding sequence or the second human polypeptide coding
sequence
comprises one or more mutations in the human gene exon coding sequence as
compared to a
wildtype reference sequence resulting in at least one amino acid change when
the first human
gene or the second human gene is expressed in the host animal, and wherein
expression of the
first and second human polypeptide coding sequences contribute to the
monogenic or polygenic
human disease phenotype.
EXAMPLES
[00123] The following examples are put forth so as to provide those of
ordinary skill in
the art with a complete disclosure and description of how to use the
embodiments provided
herein and are not intended to limit the scope of the disclosure nor are they
intended to represent
that the Examples below are all of the experiments or the only experiments
performed. Efforts
have been made to ensure accuracy with respect to numbers used (e.g. amounts,
temperature,
38
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
etc.) but some experimental errors and deviations should be accounted for.
Unless indicated
otherwise, parts are parts by volume, and temperature is in degrees
Centigrade. It should be
understood that variations in the methods as described can be made without
changing the
fundamental aspects that the Examples are meant to illustrate.
[00124] Example 1: Presynaptic terminus activity. In certain embodiments,
the
presynaptic genes involved in neurotransmission in C. elegans are replaced
with human gene
sequences, specifically, the SNARE proteins, their regulators, and other
proteins involved in
neurotransmitter release at the presynaptic terminus. See Figure 1. There are
three SNARE
proteins, syntaxin, VAMP (vesicle-associated-membrane protein) and SNAP
(synaptosome-
associated protein) that act to drive vesicle fusion (Malsam J, Saner TH
(2011). "Organization
of SNAREs within the Golgi stack". Cold Spring Harbor Perspectives in Biology.
3 (10):
a005249.). Of the neurotransmission regulators, there are six key genes acting
to coordinate
neurotransmitter release (STXBP1, NSF, SYT1, UNC13A, CPLX1, RAB3A). There are
31
additional genes that function at presynaptic terminus locations, and that are
also involved in
neurotransmitter release with involvement in human disease. As a result, there
are up to 40
genes identified that may be replaced in the C. elegans with human orthologs,
wherein each of
those genes are useful to humanize due to their known disease associations.
Creation of a
humanized pathway via expression of multiple (e.g. polygenic) genes integrated
into the host
nematode genome creates an improved platform for disease modeling and
discovery because
protein-protein interactions between pairs of human genes are absolutely
maintained.
[00125] In embodiments, a host nematode comprises and expresses all
heterologous
polypeptide coding sequences in a pathway, such as proteins and regulators
(which may not
necessarily be expressed) involved in neurotransmitter release at the
presynaptic terminus. In
other embodiments, the host nematode comprises at least two genes involved in
a pathway, such
as proteins and regulators involved in neurotransmitter release at the
presynaptic terminus. In
embodiments, the nematode may comprise from two (2) to 40 human genes, and
that are
expressed and contribute to the same trait or phenotype (e.g. neurotransmitter
release at the
presynaptic terminus). That phenotype output may be recorded using various
assays known to
one of skill in the art.
39
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
[00126]
Table 1: presynaptic genes with their disease associations, C. elegans
ortholog
and loss-of-function phenotypes. Human genes and their paralogs chosen based
on KEGG
pathway hsa04721 for disease-associated genes:
worm
human gene disease association gene similarity
pheno
ATP6V LB 1 Renal tubular acidosis with deafness vha-12 92,
lethal
ATP6V1B2 congenital deafness with vha-12 91
lethal
onychodystrophy. Zimmermann-
Laband syndrome 2
RAB3A Ependymoma rab-3 81
movement
STX1A Schizophrenia, Autism, Cystic fibrosis unc-64 81
lethal
STX1B Generalized epilepsy with febrile unc-64 81
lethal
seizures 9,
VAMP1 Ataxia, Myasthenia snb-1 81
lethal
VANIP2 Major depressive disorder, -1Thipolar snb-1 78
lethal
depression
DNM1. Early infantile epileptic encephalopathy dyn-1 78
lethal
31
DNM2 Myopathy, Charcot-Marie-Tooth, dyn-1 76
lethal
Lethal congenital contracture syndrome
STXBP1. Early infantile
epileptic encephalopathy unc-18 75 movement
4, West syndrome, Intellectual
disability, Neurodevelopmental
disorders, Schizophrenia
STX3 Microvillus inclusion disease, unc-64 77 lethal
:Intellectual disability
STX2 Male sterility, Male infertility unc-64 72, lethal
NSF Cocaine dependence, Epilepsy, nsf-1 71 lethal
Parkinson disease
SNAP25 Congenital Myasthenic syndrome 18, ric-4 71
movement
ADFID, Bipolar disorder, Depressive
disorder, Diabetes mellitus, Myasthenia
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
SYT1 Baker-Gordon syndrome, Visual snt-1
71 lethal
seizure,
S LC6A2 Orthostatic intolerance, Mental dat-
1 67 movement
depression. Mitral valve prolapse
syndrome. Neurocirculatory asthenia,
irritable heart. Depressive disorder
SLC I7A8 Deafness, autosontal dominant 25
eat-4 66 movement
ATP6V0A4 Distal renal tubular acidosis unc-32 66 lethal
SNAP23 Liver Cirrhosis, Myocardial Ischemia ric-4 63 lethal
CASK FG syndrome 4, Mental retardation and lin-2 61
development
microcephaly, Intellectual disability
ATP6V0A2 Cutis laxa type IIA, Wrinkly skin
unc-32 62, lethal
syndrome
CADPS Glaucoma unc-31 61 movement
SYNI1 Early infantile epileptic unc-26 60 lethal
encephalopathy-63, Parkinson disease
20, Intellectual disability
SLC18A3 Congenital myasthenia 21 Asthma unc-17 60 lethal
DNAJC5 Neuronal ceroid lipofuscinosis 4,
dnj-14 58 movement
Ataxia
CP1_,X1 Early infantile epileptic
encephalopathy cpx-1 56 movement
63,
TORG1 Osteopetrosis I unc-32 56 lethal
UNC13A Amyotrophic lateral sclerosis, unc-
13 54 movement
Intellectual disability
CACNA1A Early infantile epileptic encephalopathy unc-2 52 movement
42, Episodic ataxia Familial hemiplegic
migraine 1 , Spinocerebellar ataxia 6
DIVIXL2 Autosomal dominant deafness 71, rbc-
1 50 n. d.
Polyendocrine-polyneuropathy
syndrome, Intellectual Disability
EPN1 Middle cerebral artery infarction
epti-1 50 lethal
SNAPAP Abnormality of brain morphology
snpri-1 50 development
41
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
SYNGR1 Schizophrenia, Bipolar disorder, Acute sng-1 47
movement
myeloid leukemia, Libman-Sacks
disease, Systemic lupus erythematosus
SYNI X-linked epilepsy, Schizophrenia, snn4 47
movement
Depressive disorder, Autism,
Intellectual disability
APBAI Intelligence lin-10 47
morphology
STXBP6 Autism sec-3 45
lethal
NRXNI Pitt-Hopkins-like syndrome 2, nrx-1 44
development
Schizophrenia.
SYP X-linked mental retardation 96 sph-1 44 n.d.
BIN1 Centronuclear tnyopathy 2 amph-1 44 morphology
RPH3A Tetralogy of .Fallot rbf-1 42
movement
B LOC1S 6 Hermansky-pudlak syndrome 9, glo-2 42,
lethal
SV2A Schizophrenia svop-1 40 morphology
RIMS 1 Cone-rod dystrophy 7 unc-10 37
movement
PCLO Pontocerebellar hypoplasia 3 unc-1.0 33
movement
BSN Heart disease, Epilepsy cIa-i 30 movement
[00132]
Creation of a humanized presynaptic terminus in C. elegans involves creating
clusters of humanized genes starting with the core synaptic-vesicle-fusion
machinery. Genes
selected for core machinery with disease associations include members of the
SNARE complex
(STX1A, STX1B, STX2, STX3, VAMP1, VAMP2, SNAP25 and SNAP23) Although many
combinations of disease-associated SNARE are possible, in this example, the
unc-64 gene in C.
elegans is replaced with human STX1A, the ric-4 gene in C. elegans is replaced
with human
SNAP25, and the snb-1 gene in C. elegans is replaced with human VAMP 1. A
synthetic
sequence is obtained containing the human gene coding sequence codon optimized
for C.
elegans. In addition, at least one but typically 3 artificial introns are
inserted within the coding
sequence as selected from table 2. The artificial intron sequences are derived
from highly
expressed nematode proteins, wherein the gene to be inserted is a chimeric
comprising the
42
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
human or heterologous exon coding sequences interspersed with nematode
artificial intron
sequences. Due to the creation of the chimeric sequence, aberrant donor and
acceptor splice sites
may be introduced and must be removed. The optimized chimeric heterologous
sequence is
inserted into the native locus using published CRISPR-transgenesis techniques
(Dickinson DJ
and Goldstein B "CRISPR-Based Methods for Caenorhabditis elegans Genome
Engineering"
Genetics. 2016 Mar; 202(3): 885-901), wherein the nematode ortholog is
replaced with the
chimeric heterologous polypeptide coding sequence. Each polygenic animal is
made by
consecutively installing each human gene into the previously modified animal.
[00133] Table 2. Six artificial intron sequences derived from nematode
genes
name sequence
Syntronl Gtacttgagatccttaaacgcagtcgaaaattggtaattttacag
(SEQ ID NO: 1)
5yntron2 Gtaagttcctccactagaaatatcaggtgctataattgtgttcag
(SEQ ID NO: 2)
5yntron3 Gtgagttattataattatttgatcacaacgattattttaattttcag
(SEQ ID NO: 3)
5yntron4 Gtgagtgattttaaacattatctgtacttaaattataaattctctattcag
(SEQ ID NO: 4)
5yntron5 Gtaaataattatacattcgatgataaatttatgcgtactatttttcag
(SEQ ID NO: 5)
5yntron6 Gttaaatgtacaaacaactatttgaaagattttctcacccgattttttcag
(SEQ ID NO: 6)
[00134] Further humanization of the presynaptic terminus is performed to
introduce key
regulators of SNARE activity. Building on the SNARE humanized animal, the unc-
18 gene is
replaced with STXBP1. Similar human gene optimization and genomic insertion as
used for
SNARE protein insertion, a consecutive gene swap insertion procedure is used
to insert the
remaining regulators. The nsf-1 gene is replaced with human NSF, the snt-1
gene is replaced
with human SYT1, the unc-13 gene is replaced with human UNC13A, the cpx-1 gene
is replaced
with human CPXL1 and the rab-3 gene is replaced with human RAB3A. The
transgenic
43
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
nematode comprises a humanized presynaptic terminus that uses human genes to
control
neurotransmission activity.
[00135] Successful installation of the humanized presynaptic terminus in
the host
nematode is detected by using a set of functional tests for measuring the
phenotypic consequence
of the polygenic gene-swap. A Screenchip electrophysiology test (US Patent No.
9,723,817) is
used to determine if the heterologous polygenic animal can retain wild type
electrical activity.
See Figure 2. Preparation of an animal with co-expression of human STX1A,
VAMP2, and
SNAP25 is shown to retain electrical activity. As shown in Figure 2, a
nematode comprising
and expressing a single heterologous polypeptide coding sequence (replacing
the nematode
ortholog) can be useful when it rescues activity, but multiple heterologous
polypeptide coding
sequences that are expressed provide a polygenic system that has even greater
capacity to rescue
function. Similar results are expected to occur when the vesicle release
regulators are installed.
[00136] The humanized polygenic pathway may be characterized utilizing
additional
phenotypic behavior assays such as thrashing in liquid, chemotaxis to food
source, and
movement on solid surface.
[00137] Example 2: Homologous recombination activity.
[00138] In certain embodiments provided herein is a host nematode
comprising and
expressing a first heterologous polypeptide coding sequence and a second
heterologous
polypeptide coding sequence. In embodiments, the heterologous polypeptide
coding sequences
are human and involved in the homologous recombination repair pathway. There
are 5
steps/functionalities in executing homologous recombination repair:
recognition, resection,
filament, invasion, and resolution. See Figure 3. Each involves a specific
protein complex
formation. (Lange SS, Takata K, Wood RD Nat Rev Cancer. 2011 Feb;11(2):96-110.
doi:
10.1038/nrc2998). At a dsDNA break, ATM recognizes damage and recruits other
recognition
partners: RBBP8, BARD1, BRCA1 and BRIP1. Next the resection activity is
activated and
executed by RAD50, MREllA and NBN. Filament formation occurs with RPA
associations
RAD51 paralogs. Strand invasion involves RAD54 activity. Resolution utilizes
POLD1 with
contribution from BLM, TOP3A and MUS81
44
CA 03131145 2021-08-20
WO 2020/172587
PCT/US2020/019308
[00139] Table 3: Homologous recombination pathway genes with their disease
associations, C. elegans ortholog and loss-of-function phenotypes. Human genes
and their
paralogs chosen based on KEGG pathway hsa03440 for disease-associated genes
worm
human gene disease aSSOCiatiOTI gene
similarity pbeno
= = =
RAD51 Fanconi anemia complementation
group R, rad-51 74 lethal
Mirror movements 2, Breast cancer
susceptibility
RAD54L Somatic colonic adenocarcinoma, non- rad-54 67
lethal
Hodgkin Lymphoma, non-Hodgkin, Invasive
ductal breast cancer
POLD1 Mandibular hypoplasia, Deafness, F1OC2.4 66 lethal
Progeroid, Lipodystrophy, Colorectal cancer
susceptibility 10
TOP3A Progressive external
ophthalmoplegia with top-3 57 lethal
mitochondrial DNA deletions,
Microcephaly, Growth restriction
MRE 1 1A Ataxia-telangiectasia-like disorder 1 mre-11 54
developmen
RPA1 Chloracne rpa-1 49
developmen
BLM Bloom syndrome him-6 49
developmen
RAD50 Nijmegen breakage syndrome-like
disorder rad-50 46 lethal
RAD51D Breast-ovarian cancer susceptibility 4 rfs-1 46
developmen
Fanconi anemia complementation developmen
group J, Breast cancer early-onset
BRIP1 susceptibility dog-1 45
BRCA1 Fanconi anemia, Familial breast-ovarian
brc-1 39 developmen
cancer 1, Pancreatic cancer 4
NBN Aplastic anemia, Acute lymphoblastic ttn-1 38
lethal
Leukemia, Nijmegen breakage syndrome
MU581 Arterial tortuosity syndrome,
Emphysema, mus-18 37 developmen
Marfan syndrome
CA 03131145 2021-08-20
WO 2020/172587
PCT/US2020/019308
Malignant neoplasm of breast, Breast cancer brd-1 36 developmen
BARD1 susceptibility t
ATM Ataxia-telangiectasia, B-
cell non-Hodgkin atm-1 35 developmen
lymphoma, Mantle cell lymphoma, T-cell t
prolymphocytic leukemia, Breast cancer
susceptibility
RBBP8 Jawad syndrome, Pancreatic carcinoma, com-1 35
lethal
Seckel syndrome 2
RAD52 Malignant neoplasm of lung, Squamous cell D1081.7 34
movement
carcinoma
[00140] Construction of the polygenic animal, comprising at least a first
heterologous
polypeptide coding sequence and a second heterologous polypeptide coding
sequence, wherein
the host animal expresses heterologous polypeptide coding sequences involved
in homologous
recombination repair. Replacing host nematode orthologs with heterologous
polypeptide coding
sequences in the homologous recombination pathway to create a humanized
recognition complex
involves making substitutions of ATM with atm-1, RBBP8 with com-1, BARD1 with
brd-1,
BRCA1 with brc-1, and BRIP1 with dog-1. For the resection system,
substitutions are RAD50
with rad-50, MREllA with mre-11 and NBN with ttn-1. For the filament
formation,
substitutions are RPA1 with rpa-1 and RAD51 with rad-51 and RAD52 with 1081.7
and
RAD51D with rfs-1. For the strand invasion system, substitution is RAD54L with
rad-54. In the
resolution system, substitutions are POLD1 with F 10C2.4, BLM with him-6,
TOP3A with top-3
and MUS81 with mus-81. As disclosed in Example 1, construction involves
creation of human
chimeric gene optimized for expression in a nematode, wherein the chimeric
sequence replaces
the host nematode ortholog using CRISPR techniques.
[00141] Successful humanized homologous recombination activity is measured
using
either an epi-chromosomal or genome integrated fluorescent reporter of HDR
activity as
disclosed in WO patent application PCT/US2019/45374 filed 06 August 2019. As
each
nematode host gene is replaced with human transgene, the fluorescence activity
of the reporter is
measured and quantified relative to the wild type animal. See Figure 4.
[00142] Example 3. Modeling variants that modify the severity of disease
presentation.
46
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
[00143] In one embodiment, the native C. elegans gene mthf-1 is replaced
with the human
coding sequence for MTHFR. Function of the MTHFR in the C. elegans background
is
determined by monitoring the expression of acdh-1 or growth rate. A known risk
factor variant
of A222V is introduced into the MTHFR sequence in the line. This strain is
then used as a
background for other humanizations and variant modeling. Humanizations are for
epilepsy genes
such as STXBP1, SCN1A, KCNQ2, CDKL5, SCN2A, PCDH19, STXBP1, PRRT2, SLC2A1,
MECP2, SCN8A, UBE3, ATSC2, GABRG2, GRIN2A, FOXG1, TPP1, and GABRAL Variants
in these epilepsy genes are assessed with and without the MTHFR risk factor
variant A222V to
see if the epilepsy gene variant has a more severe phenotype with the risk
factor variant present.
[00144] Example 4. Exemplary digenic-humanized nematode
[00145] An exemplary digenic-humanized nematode was made and found to be
functional. First, a monogenic-humanized animal (hSTXBP1) was constructed
STXBP1 coding
sequence as gene replacement of the coding sequence at the unc-18 genetic
locus. This line was
compared with the unc-18 KO line to confirm functional rescue.
[00146] Second, another monogenic-humanized animal (hSTX1A) was
constructed
expressing STX1A coding sequence as gene replacement of the coding sequence at
the unc-64
genetic locus. This line was compared with the unc-64 KO line to confirm
functional rescue.
[00147] Sequential construction was used to create a digenic-humanized
animal
(hSTXBP1; hSTX1A). Examination of the activity of the monogenic vs diagenic
showed no
detectable compromise of activity occurs in digenic humanized animals. This
successful
creation of a digenic humanized animal forecasts that further humanization of
the nematode
nervous system can be pursued to enable creation of a human avatar system for
use in genetic
diagnosis and drug discovery.
[00148] Construction of the hSTXBP1 and comparison with the unc-18 KO line
was
described previously. Construction of the monogenic-humanized hSTXBP1 was
performed as
described in Example 1 of US Serial No. 16/281,988 the contents of that
Example herein is
incorporated by reference.
[00149] The full deletion of unc-64 was created using guide RNAs targeting
Cas9 for
genomic DNA cleavage at the beginning and end of the unc-64 locus (sgRNA
targeting
sequences: ACAACAACATGACTAAGGAC (SEQ ID NO:7) and
GAAACTTTCAGAATGCAGGA (SEQ ID NO: 8)). A gene editing mixture of Cas9 protein,
47
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
guide RNAs and donor homology (5ug Cas9, 50 pmol each sgRNA, and 500 ng donor
homology) was made and microinjected into the gonad of young N2 adult
hermaphrodites. Also
included in the injection mix was the dpy-10 co-CRISPR selection components.
Donor
homology was an oligonucleotide DNA (ODN) sequence containing a right and left
homology
arm sequences of 35bp lengths. In between the homology arms a cargo sequence a
3-frame start,
a sequence for PCR, and a restriction enzyme site. The sequence of the ODN
was:
CGAGACCTGTCAACAGGAACAACAACATGACTAAGTAAATAAATAAACCCCAGAAGTCCTCCAG
TCCCTCGAGGGAAGGGTTCCCATGCACTTGGTCGATTTGCACCT (SEQ ID NO: 9).
[00150] After injection of the gene editing mixture, 39 Fl animals
containing the co-
CRISPR screening phenotype were isolated to new plates. After the F2
population was
established, the Fl animals were harvested and screened by PCR for the
presence of the deletion.
The PCR is specifically designed to distinguish between homozygous mutant,
homozygous wild-
type and heterozygous animals. F2 progeny from Fl animals, PCR positive as
heterozygous for
the deletion were isolated to try and identify homozygous animals. Four rounds
of homozygosing
were attempted, before it was determined that the deletion was homozygous
lethal. The deletion
was confirmed by DNA sequencing.
[00151] Construction of the monogenic-humanized hSTX1A occurred similarly
to the
construction of hSTXBP1. Guide RNAs targeting Cas9 for genomic DNA cleavage at
the
beginning and end of the unc-64 locus were prepared (targeting sequences:
ACAACAACATGACTAAGGAC (SEQ ID NO:7) and TAATCGGCTTCGTTTCTCTG (SEQ
ID NO. 8)). A gene editing mixture of Cas9 plasmid, guide RNA plasmids and
donor homology
plasmids (50 ng/ul Cas9, 25 ng/ul each sgRNA, and 50 ng/ul donor homology)
along with
selection markers was made and microinjected into the gonad of young N2 adult
hermaphrodites.
Donor homology was a plasmid containing a right and left homology arm
sequences of 725bp
and 818bp lengths respectively. In between the homology arms a cargo sequence
encoding a
nematode-codon-biased cDNA sequence for the most abundant isoform of the human
STX1A
gene. Immediately after the hSTX1A cDNA stop codon is a 3'UTR of the eft-3
gene. After the
UTR is a selection marker cassette coding for hygromycin resistance. Three
days after injection
of the gene-editing reagents, Hygromycin B was added to the plates containing
the progeny of
the injected young adults. After 10 days, the plates were examined for
surviving animals which
were singled onto fresh growth plates. After progeny were established, the
founding adult was
48
CA 03131145 2021-08-20
WO 2020/172587
PCT/US2020/019308
harvested for PCR analysis. Allele specific PCR for desired edit was used to
detect presence of
desired edit. Confirmation of homozygosity was confirmed with allele-specific
PCR for wild
type locus. The hSTX1A strain was considered to rescue the function of the
native unc-64 due to
comparison to the KO of the unc-64. No homozygous unc-64 KO were isolated,
which indicated
that the unc-64 KO was lethal. However, homozygous KI strains with the hSTX1A
replacing the
unc-64 gene were isolated, indicating the function of unc-64 could be replaced
by hSTX1A.
[00152] Construction of digenic-humanized animals occurred by injection of
the
hSTXBP1 strain with the components to create the hSTX1A strain. Homozygous
animals were
isolated as described above.
[00153] An alternative method to create the digenic nematode is to perform
a genetic
cross. Heat shock is performed on the hSTX1A plates to create males. The males
of hSTX1A
strain are mated with the hSTXBP1 hermaphrodites. Fl progeny are isolated on
new growth
plates. After F2 progeny are established, the founding Fl adult is harvested
for PCR analysis.
Allele-specific PCR was used to detect the presence of hSTX1A edit. F2 progeny
are isolated on
new growth plates. After F3 progeny are established, the founding F2 adult is
harvested for PCR
analysis. Allele-specific PCR is used to detect presence of hSTXBP1 and hSTX1A
edits and a
second allele-specific PCR is used to detect the presence of wild type (unc-18
and unc-64) at the
hSTXBP1 and hSTX1A edit sites. Animals isolated as positive for hSTXBP1 and
hSTX1A
alleles and negative for wild-type are designated to be the desired digenic-
humanized strains.
[00154] Knock-ins for the digenic-humanized and monogenic humanized
animals were
compared to gene knock-outs for the unc-18 and unc-64 locus (Table 4). Both
the di-genic and
monogenic humanized knock-ins had near wild-type activity, while the gene
knock out for unc-
18 was severely uncoordinated and the gene knock-out for unc-64 was not viable
as homozygote.
[00155] Table 4:
hSTXBP1;
Wild type hSTXBP1 hSTX1A hSTX1A unc-
18 unc-64
(N2) knock-in knock-in knock-in knock-out
knock-out
++++ +++ +++ +++ +
(lethal)
[00156] Example 5. Transgenic nematodes expressing human variants
49
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
[00157] CRISPR, crossing, self-fertilization, and similar techniques are
used to create
animal strains expressing multiple interacting human proteins within the
synaptic bouton. Since
the STXBP1 single-locus humanization line and STX1A single-locus humanization
lines have
already been created and crossed to generate a double-locus humanization line
(as described
above), humanized SNAP25 lines are created.
[00158] To generate the humanized SNAP25 line, the C. elegans ortholog ric-
4 (53%
identity) is replaced on Chromosome V. The human cDNA of 618bp is optimized
for expression
in C. elegans and cloned into a plasmid for CRISPR/Cas9 gene editing. This
plasmid also
contains homology arms for ric-4 and a selection marker. A determination of
whether the
SNAP25 is functional is made by comparing it with the loss of function mutant
which is reported
to be sluggish, small, uncoordinated, and resistant to aldicarb. The donor
homology plasmid is
combined with the human STX1A with plasmids for the sgRNAs, Cas9, and other
injection
markers. All created lines are confirmed with PCR and/or sequencing, and
expression levels
quantified relative to the native gene by qPCR. The humanized SNAP25 line is
then crossed
with the STXBP1/STX1A double insertion line to create a triple insertion line,
confirmed by
PCR assays and sequencing. By these methods, a transgenic animal strain is
created with at least
three interacting human proteins replacing native orthologous proteins.
[00159] Example 6. Molecular Phenotyping
[00160] C. elegans animals with loss of function mutations in ric-4, unc-
18, and unc-64
are characterized for differential expression by RNA-seq relative to the
humanized lines.
Pathway reporter genes common to the three genes being manipulated are
targeted. Candidates
are validated by qPCR assays and those with at least a 2-fold change in
expression will be
selected to create fluorescent biosensors. See US Patent No. 8,937,213, herein
incorporated by
reference, which disclose use of inducible and constitutive promoters operably
linked to reporter
genes. Plasmid constructs are created as promoter-RFP fusions. Promoter
regions for the
candidate reporter genes are selected using ChIP-seq data from the wormbase
database.
Typically a 1000-2000 bp region upstream of a gene's start codon is chosen for
PCR
amplification and then inserted into a red fluorescent protein (RFP)
expression cassette plasmid
("response plasmid"). Promoter-RFP fusion constructs (response plasmid) are co-
injected with a
constitutively expressed reporter plasmid ("control plasmid") to enable
ratiometric analysis. The
CO2F5.3 gene is chosen for control plasmid construction because the gene has a
sufficient
CA 03131145 2021-08-20
WO 2020/172587 PCT/US2020/019308
expression (FPKM: 94) and an interstrain analysis indicates the gene has less
than 6% variance
across all animal types (N2 vs. CL2355 vs. BR5270 vs. UM0001). For the CO2F5.3
control
plasmid, the promoter fusion is made to green fluorescent protein (GFP). The
constitutive GFP
expression acts as internal control allowing ratiometric normalization
(RFP/GFP) for expression
changes observed with each response plasmid. By these methods, at least three
new molecular
phenotypic indicators are identified and validated in knock-out vs. humanized
lines.
51