Note: Descriptions are shown in the official language in which they were submitted.
CA 03137220 2021-10-18
WO 2019/234248
PCT/EP2019/065040
METAL OXIDE-BASED ELECTRODE COMPOSITIONS
Related Application
This present case is related to, and claims the benefit of, GB 1809467.2 filed
on
08 June 2018 (08.06.2018) and GB 1905218.2 filed on 12 April 2019
(12.04.2019), the
contents of which are hereby incorporated by reference in their entirety.
Field of the Invention
The present invention provides an electrode and an electrochemical cell, such
as a lithium
ion battery, comprising the electrode, together with methods for using the
electrode within
the electrochemical cell.
Background
High-rate lithium ion battery electrode materials can store large quantities
of charge in a few
minutes of charging, rather than hours. Such materials are required to
alleviate the
technological challenges associated with the adoption of electric vehicles and
grid-scale
batteries, and to enable new power-intensive devices.
The most intuitive and commonly used approach to increase the rate performance
of an
electrode material is to create a nanosized or porous (and often hierarchical)
structure. This
minimizes the lithium ion solid-state diffusion distance, enabling more rapid
lithium ion
transport through the electrode and increasing the surface area of electrode
materials in
contact with electrolyte. Carbonaceous hierarchical structures and carbon-
coating are also
frequently employed to improve electronic conductivity, which is another
prerequisite for
high-rate applications.
In practice, despite excellent lithium ion mobility, graphite cannot be used
at high-rates due
to particle fracture and the risk that lithium dendrites form, leading to
short circuits and the
risk of fires and explosions (Zhao, etal.; Downie, etal.). The latter issue
inherently limits the
use of low voltage anodes in high-rate applications, since the electrode
inhomogeneity, or
any source of increased overpotential, can lead to lithium plating potentials
on the surface of
the low-voltage electrode (Downie, etal.).
Li4Ti5012 (lithium titanate; LTO), with an average voltage of 1.55 V against
Li/Li, enables
high-rate (de)intercalation without the risk of lithium dendrites or
substantial solid¨electrolyte
interphase (SEI) formation, albeit with an undesirable but necessary decrease
in full-cell
voltage and thus energy density. However, even in this well-established "high-
rate" anode,
the capacity of 1 pm particles from solid-state synthesis reaches only 60-65
mA=h=g-lat a
rate of 10C (Kim, etal.).
CA 03137220 2021-10-18
WO 2019/234248 PCT/EP2019/065040
2
A number of strategies have been proposed to increase the capacity of LTO
electrodes at
high C-rates, and present carbon-coated nanoparticles of LTO can reach at
least
150 mA=h=g-1 at 10C (Wang etal.; Odziomek, etal.). This corresponds to
approximately
0.5 lithium ions per transition metal (Li/TM).
However, using nanostructured and porous materials for electrochemical energy
storage
applications inherently results in a severe penalty in terms of volumetric
energy density.
Furthermore, these carefully designed porous and nanoarchitectures are time
consuming
and expensive to synthesize, characterize, and manufacture. Synthesis methods
often
result in relatively low yields and/or large quantities of chemical waste
generation (Oszajca,
etal.). Moreover, these porous and nanoarchitectures are also susceptible to
degradation
during electrochemical cycling from processes such as catalytic decomposition
of electrolyte
(Palacin etal.), morphological changes that result in loss of nanostructuring
(Wu etal.), and
higher first cycle capacity loss (Kasnatscheew etal.).
Another problem with the use of nano-LTO electrodes in a full cell is gas
evolution during
repeated charging and discharging cycles, and the associated swelling or
pressure build-up.
This arises from heterogeneous catalysis between the metal oxide surface and
organic
electrolyte (He et al.; Lv, etal.). The small particle sizes required to
compensate for poor
lithium ion and electron diffusion in LTO increase the reactive surface area,
exacerbating this
problem.
Fast charging or high-power delivery from a full cell also requires a cathode
to match the
anode. LiFePat has been used as a promising high-rate cathode (Zaghib, etal.).
However,
both LiFePat and LTO have exceptionally flat voltage profiles. This
combination provides a
constant voltage but presents a serious challenge in terms of battery
management systems
(BMS). Simple and accurate BMS is a crucial factor for battery applications in
electric
vehicles and mobile technology and is even more important at high-rates to
prevent
dangerous and degradative over(dis)charging while maximizing utility. BMS rely
on the
ability to measure state-of-charge, which cannot be done simply by charge
counting alone as
the battery degrades and electron-consuming side reaction occur.
In light of the above challenges, there is a need to provide new electrode
materials for
lithium ion batteries that are capable of operating at high rates.
Summary of the Invention
The invention generally provides an electrode comprising niobium tungsten
oxide, an
electrochemical cell comprising the electrode, and the use of the cell, for
example, in a
lithium ion battery, at high C-rates of 5C or more, such as 10C or more,
during charging
and/or discharging.
CA 03137220 2021-10-18
WO 2019/234248 PCT/EP2019/065040
3
The present inventors have established that extremely high volumetric energy
densities and
impressive charging and discharging rates can be achieved using electrode
materials
comprising niobium tungsten oxides in a high-rate lithium ion cell. The
niobium tungsten
oxides have favorable lithium diffusion properties, and thus exhibit superior
performance.
Above 1.0 V vs. LOLL the formation of SEI is minimal, which means that lithium
will not be
lost into side reactions with the electrolyte.
In addition, a high-rate lithium ion cell using electrode materials comprising
niobium tungsten
oxide can operate above 1.3 V. (For example, the average voltage of Nb16W5055
is 1.57 V
vs Li/Li and the average voltage of Nb18W16093 is 1.67 V vs LOU). Operating in
this voltage
range negates the need to perform an initial formation cycle, simplifying the
cell
manufacturing process. A typical lithium-ion cell comprising a graphite
electrode operates
below 1.3 V and must undergo an initial formation cycle before the cell is
sealed. Typically,
this formation cycle takes place at elevated temperature, for example 60 C,
in order to allow
degassing to occur. This adds signification time and cost to the cell
manufacturing process.
Furthermore, in a full cell against e.g. LiFePat, LiN(CF3S02)2 (LiTFSI) can be
used to
replace the more toxic LiPF6 electrolyte salt commonly used in standard
commercial
electrolytes. Moreover, aluminum can be used as the current collector instead
of the more
expensive copper while avoiding LiAl alloying potentials (0.3 V vs. LOU).
The use of a niobium tungsten oxide as an electrode material, for example in a
high-rate
lithium cell, allows the open-circuit voltage to be used as a measure of state-
of-charge. This
has the potential to provide a simple and reliable BMS, which may prove to be
a significant
advantage for high power/fast charging applications.
In a first aspect of the invention there is provided a method of charging
and/or discharging
an electrochemical cell at a C-rate of at least 5C, such as at least 10C,
wherein the
electrochemical cell has a working electrode comprising niobium tungsten
oxide.
The electrochemical cell may contain a counter electrode and an electrolyte,
and optionally
the electrodes are connectable to or are in connection with a power supply.
The method may be a method of charging and/or discharging an electrochemical
cell at a
current density of at least 750 mkg-1, such as at least 800 mkg-1.
The method may involve a cycle of charging and discharging or discharging and
charging
the electrochemical cell, and the method may comprise 2 cycles or more, 5
cycles or more,
10 cycles or more, 50 cycles or more, 100 cycles or more, 500 cycles or more,
1,000 cycles
or more, or 2,000 cycles or more.
CA 03137220 2021-10-18
WO 2019/234248
PCT/EP2019/065040
4
In one embodiment, the working electrode does not have a porous nor
hierarchical structure.
The working electrode may comprise the niobium tungsten oxide in particulate
form. For
example, the niobium tungsten oxide may have a primary particle size of at
least 1 pm.
The smaller surface area of the particulate niobium tungsten oxides decreases
side
reactions and mitigates the problems of gas evolution and the associated
swelling or
pressure build-up observed with nano-LTO electrodes. Moreover, particulate
niobium
tungsten oxides can be quickly and easily prepared using solid state
synthesis.
The working electrode may have a solid-state lithium diffusion coefficient
(Du) of less than
10-14 m2.s-1 at 298 K, such as less than 10-15 m2.s-1 at 298 K.
The favorable lithium diffusion properties allow micrometer-sized particles of
niobium
tungsten oxide to be used at extremely high rates.
The working electrode may have a capacity of 50 mA=h=g-1 at 200. For example,
75 mA=h=g-1 at 200 or 50 mA=h=g-1 at 600.
The molar ratio of Nb2O5 to W03 in the working electrode may be from 6:1 to
1:15. For
example, from 8:5 to 11:20.
The working electrode may comprise a niobium tungsten oxide selected from the
group
consisting of Nb12W033, Nb26W4077, Nb14W3044, Nb16W5055, Nb18W8069, Nb2W08,
Nb18W16093, Nb22W200115, Nb8W9047, Nb54W820381, Nb20W310143, Nb4W7031, or
Nb2W15050.
For example, Nb16W5055, Nb18W8069, Nb2W08, Nb18W16093 or Nb22W200115.
Additionally, the working electrode may comprise a mixture of niobium tungsten
oxide and
an additional active material. For example, the working electrode may comprise
a mixture of
niobium tungsten oxide and LTO. The ratio of niobium tungsten oxide to LTO may
be from
95:5 to 5:95 by weight. For example, the ratio may be from 90:10 to 10:90 by
weight, from
80:20 to 20:80 by weight, from 70:30 to 30:70 by weight, from 60:40 to 40:60
by weight or
the ratio of niobium tungsten oxide to LTO may be 1:1 by weight.
In a further aspect of the invention, there is provided an electrode, which
may be referred to
as a working electrode, comprising a niobium tungsten oxide, such as wherein
the molar
ratio of Nb2O5 to W03 in the electrode is from 8:5 to 11:20. The working
electrode is suitable
for use in a lithium ion battery.
The working electrode may comprise a niobium tungsten oxide selected from
Nb16W5055,
Nb18W8069, Nb2W08, Nb18W16093 or Nb22W200115. For example Nb16W5055 or
Nb18W16093.
CA 03137220 2021-10-18
WO 2019/234248 PCT/EP2019/065040
In one embodiment, the working electrode does not have a porous nor
hierarchical structure.
The working electrode may comprise a niobium tungsten oxide in particulate
form. For
example, the niobium tungsten oxide may have a primary particle size of at
least 1 pm.
5
Additionally, the working electrode may comprise a mixture of niobium tungsten
oxide and
an additional active material. For example, the working electrode may comprise
a mixture of
niobium tungsten oxide and LTO. The ratio of niobium tungsten oxide to LTO may
be from
95:5 to 5:95 by weight. For example, the ratio may be from 90:10 to 10:90 by
weight, from
80:20 to 20:80 by weight, from 70:30 to 30:70 by weight, from 60:40 to 40:60
by weight or
the ratio of niobium tungsten oxide to LTO may be 1:1 by weight.
In a further aspect of the invention, there is provided an electrochemical
cell comprising the
working electrode of the invention.
In a further aspect of the invention, there is provided a lithium ion battery
comprising one or
more electrochemical cells of the invention. Where there are a plurality of
cells, these may
be provided in series or parallel.
In a further aspect of the invention there is provided the use of a working
electrode
comprising a niobium tungsten oxide in a high-rate electrochemical cell, for
example wherein
the cell is operated at a C-rate of at least 5C, such as at least 10C.
In a further aspect of the invention there is provided a method of charging
and/or discharging
an electrochemical cell at a C-rate of at least 5C, such as at least 10C,
wherein the
electrochemical cell has a working electrode comprising niobium molybdenum
oxide.
The present inventors have found that high energy densities and impressive
rates can be
achieved using electrode materials comprising niobium molybdenum oxides in a
high rate
lithium ion battery. The niobium molybdenum oxides display a higher average
voltage in
comparison to common high-rate anode materials, and so reaction with the
electrolyte is
avoided or minimized.
In a further aspect of the invention, there is provided an electrode, which
may be referred to
as a working electrode, comprising a niobium molybdenum oxide, such as wherein
the molar
ratio of Nb2O5 to Mo03 in the electrode is from 6:1 to 1:3, for example 1:3.
The working
electrode is suitable for use in a lithium ion battery.
The working electrode may comprise a niobium molybdenum oxide selected from
Nb2Mo3014, Nb14Mo3044 or Nb12Mo044. For example Nb2Mo3014.
CA 03137220 2021-10-18
WO 2019/234248 PCT/EP2019/065040
6
These and other aspects and embodiments of the invention are described in more
detail
below.
Summary of the Figures
Figure 1 is a wide-field SEM image of (A) Nb16W5055 and (B) Nb18W16093 at a
low
magnification to show the roughly uniform particle size and presence of
agglomerates. The
background of (A) is a Cu grid and of (B) is adhesive carbon. The scale bars
are 50 pm in
both images.
Figure 2 is an atomic structure and SEM image showing (A¨C) Nb16W5055 and
(D¨F)
Nb18W16093. The scale bars shown in (B) and (E) are 5 pm. The scale bars shown
in (C)
and (F) are 10 pm.
Figure 3 is a diagram showing the bronze-like structure of Nb18W16093 and its
relationship to
the classic tetragonal tungsten bronze (TTB). Partially filled spheres
represent K+ ions,
which are not present in Nb18W16093
Figure 4 is a diagram showing the rate performance of bulk Nb16W5055 and
Nb18W16093. (A)
galvanostatic discharge and charge curves for Nb16W5055 from 60C (leftmost
curve at 1.0 V
and 3.0 V) to 0.2C (rightmost curve at 1.0 V and 3.0 V); (B) dQ/dV plot for
Nb16W5055 from
0.2C (leftmost curve at 50 mA=h=g-1) to 60C (rightmost curve at 50 mA=h=g-1);
(C)
galvanostatic discharge and charge curves for Nb18W16093 from 100C (leftmost
curve at
1.0 V and 3.0 V) to 0.2C (rightmost curve at 1.0 V and 3.0 V); (D) dQ/dV plot
for Nb18W16093
from 0.2C (leftmost curve at 50 mA=h=g-1) to 100C (rightmost curve at 50
mA=h=g-1); (E) a
summary of the rate performance and (F) high-rate cycling of 250 cycles at 10C
followed by
750 cycles at 20C.
Figure 5 shows voltage profiles of Li4Ti5012 (curves with plateau at 1.55 V),
Nb16W5055
(curves crossing at 1.57 V) and Nb18W16093 (curves crossing at 1.65V).
Discharge and
charge voltage profiles normalized to (A) gravimetric capacity and (B) lithium
ions transferred
per transition metal atom. The average voltage of Li4Ti5012, Nb16W5055, and
Nb18W16093 is
1.55 V, 1.57 V, and 1.65 V, respectively. The Li4Ti5012 electrochemistry shown
here
(extracted with WebPlotDigitizer 2017, Ankit Rohatgi) represents an optimal
performance,
consisting of 5-20 nm particles on 15-20 nm carbon nanofibers (CNF) with a
mass loading
of 1 mg=cm-2.
Figure 6 is a comparison of Cu foil to carbon-coated Al foil current
collector. Fig. 6A shows
Nb16W5055 cycled for 1000 cycles under constant current discharge and charge
at 10C (first
200 cycles) and 20C (last 800 cycles); Cu foil at 1.0 V (topmost line); Cu
foil at 1.2 V (second
from top); C-Al at 1.0 V (third from top) and C-Al at 1.2V (bottommost line).
Fig. 6B shows
discharge and charge profiles between 3.0 and 1.0 V for the 100th cycle at
10C; order of
CA 03137220 2021-10-18
WO 2019/234248
PCT/EP2019/065040
7
curves at 100 mA=h=g-1: C-Al charging curve (topmost); Cu foil charging curve
(second from
top); Cu foil discharging curve (third from top) and C-Al discharging curve
(bottommost).
Figure 7 shows longer term cycling as a function of minimum cutoff voltage.
(A) Nip%WsOss
cycled at C/5 (fist 50 cycles) and 1C (second 50 cycles) on Cu foil with a 1 h
potentiostatic
hold at the top of charge; Vi-nin = 1.2 V (topmost curve); Vi-nin = 1.0 V
(bottommost curve). (B)
Nip%WsOss cycled at C/5 on Al foil without a potentiostatic charging step.
Figure 8 shows overpotential in a Li II Li symmetric cell as a function of
current density.
Figure 9 shows the stimulated echo pulsed field gradient sequence used to
measure 7Li
diffusivities showing both radiofrequency (7Li) and magnetic field gradient
(Gz) pulses. Here,
gradient pulse duration (tg) includes the up ramp, time on, and down ramp of
the opt
composite gradient pulses.
Figure 10 shows representative 7Li decay curves showing normalized NMR signal
intensity
as a function of gradient strength for (A) Lilo 2NbisWis0s3 at 453 K, (B) Lis
sNbisWis0s3 at
453 K, (C) Li34Nb18W16093 at 453 K, (D) Lis 3NbisWsOss at 353 K, and (E)
Li84Nb16W5O55 at
383 K. Black circles represent experimental data points and red lines
represent (A¨C; E)
mono- or (D) biexponential fits of the data to the Stejskal¨Tanner equation.
Figure 11 shows lithium diffusion measured via 7Li pulsed field gradient
nuclear magnetic
resonance (PFG NMR) spectroscopy. The lithium diffusion coefficients of Lis
3NbisWsOss
and LixNb18W16093 (x = 3.4, 6.8, 10.2) were measured in the temperature range
333-453 K.
The filled (85% signal contribution) and empty (15% signal contribution)
symbols for
Lis 3NbisWsOss correspond to the observed two-component diffusion.
Figure 12 shows (A) relative changes in lithium diffusion as a function of
open-circuit voltage
(Voc) and (B) open-circuit voltage vs. closed-circuit voltage (Vcc) from
galvanostatic
intermittent titration technique (GITT) measurements showing the
"thermodynamic"
electrochemical profiles at C/20 rate with a 12 h rest period at each point,
reaching a full
discharge in approximately one month.
Figure 13 shows (A) Nip%WsOss and Nb18W16093 from this work as compared to
high-rate
electrode formulations from the literature. Volumetric capacities at 1C and
20C are
determined from reported capacities and reported, measured, or estimated tap
densities.
(B) Ragone (log¨log) plot of energy density and power density on the basis of
anode active
materials vs. a 4.0 V cathode. The mass loading of the niobium tungsten oxides
here was
2.6 mg=cm-2, the mass loading of the other titanium and niobium-based
materials was
typically ca. 1 mg=cm-2. Graphite is included as a reference, though it cannot
be used for
high-rate applications due to lithium plating risks and particle fracture.
Literature values for
the Ragone plot were extracted with WebPlotDigitizer.
CA 03137220 2021-10-18
WO 2019/234248
PCT/EP2019/065040
8
Figure 14 shows theoretical volumetric capacity assuming a reaction of one
electron per
transition metal and crystallographic (single crystal) densities. The capacity
for Li4-1,5012 is
shown for the well-established reaction to Li7Ti5012; the capacity of graphite
is based on the
reaction of one electron per 06 unit.
Figure 15 shows the capacity versus cycle number for Nb2Mo3014 at 0.050 and
from 1.0 to
3.0 V vs. LOLL
Figure 16 shows discharge and charge voltage profiles of Nb2Mo3014 within
three different
voltage windows (1.0 V, 1.2 V and 1.4 V). The discharge and charge curves are
normalized
to gravimetric capacity; order of curves at 200 mA=h=g-1: 1.4 V charging curve
(topmost);
1.2 V charging curve (second from top); 1.0 V charging curve (third from top);
1.2 V
discharging curve (third from bottom); 1.0 V discharging curve and 1.4 V
discharging curve
(overlapping; bottommost).
Figure 17 shows discharge and charge voltage profiles of (A) NM0622
(LiNio60002Mno 202),
(B) LiFePat and (C) LiMn2O4against Li/Li.
Figure 18 compares capacity at varying charge and discharge rates for
electrodes
comprising Nb16W5055 and different separators, binders, binder quantity,
conductive carbon
type and with calendaring of the electrode.
Figure 19 shows discharge and charge voltage profiles of full cells comprising
a Nb16W5055
anode and (A) LiFePat, (B) LiMn204, and (C) NM0622 cathode. Figure 19(D-E)
shows the
rate performance of a cell comprising a Nb16W5055 anode and NM0622 cathode;
order of
cures in (E): capacity (topmost, sloping); coulombic efficiency (lower,
horizontal).
Figure 20 shows (A) long-term cycling at 10 charge and discharge rates of a
cell comprising
a cell comprising a Nb16W5055 anode and NM0622 cathode; capacity (sloping);
columbic
efficiency (horizontal). (B) shows performance of the extracted NM0622
electrode against
LOLL (C) shows performance of the extracted Nb16W5055 electrode against LOLL
Figure 21 shows (A) charge and discharge profiles of Nb16W5055-LTO mixtures in
a ratio of
7:3, 1:1 and 3:7 normalised to gravimetric capacity, order of charging curves
at 3.0 V: 3:7
(leftmost), 5:5 (second from left), 7:3 (rightmost); order of discharging
curves at 1.0 V: 3:7
(leftmost), 5:5 (second from left) and 7:3 (rightmost). (B) rate performance
of the mixtures,
order at 0.20: 7:3 (topmost), 5:5 (second from top), 3:7 (bottommost). (C)
longer term cycle
performance at 100 change and discharge rates, 10 points: 7:3 (topmost), 5:5
(second from
top), 3:7 (bottommost); 100 curves: 3:7 (topmost), 5:5 (second from top), 7:3
(bottommost).
CA 03137220 2021-10-18
WO 2019/234248
PCT/EP2019/065040
9
Figure 22 shows (A) change and discharge profiles of a full cell comprising a
3:7
(Nbi6W5055:LTO) anode and an NM0622 cathode and (B) rate performance of the
cell.
Detailed Description of the Invention
The invention generally provides an electrode comprising a niobium tungsten
oxide, an
electrochemical cell comprising the electrode, and the use of the cell, for
example, in a
lithium ion battery, at high C-rates of 5C during charging and/or discharging.
The preparation of Nb16W5055 has previously been described by, amongst others,
Roth and
Wadsley. The preparation of Nb18W16093 has previously been described by,
amongst
others, Stephensen. However, the electrochemical properties of Nb16W5055 and
Nb18W16093
are not described in these documents.
Electrodes comprising NID8W9047 (Montemayor et al.); Nb26W4077 (Fuentes
etal.); Nb14W3044
(Fuentes etal.); and NbxWi_x03,d2, wherein 0 x 0.25 (Yamada et e/.), have been
previously described. However, the capacity of the materials against the C-
rate was not
measured.
Electrodes comprising Nb8_xW9_x047, wherein 1 x 6, have been described by Cruz
et e/.
However, the electrochemical cell comprising the electrodes is used under
limited
conditions, and there is no disclosure of the cell operating under high-rate
conditions.
Moreover, the authors report that irreversible structural transformations in
the matrix-host
result in loss of capacity after the first cycle.
Electrodes comprising Nb12W033 have been described by Saritha et al. and Yan
et al.
Yan et al. test an electrochemical cell comprising a electrospun Nb12W033
electrode at a
maximum current density of 700 mkg-1 (corresponding to a C-rate of 3.6C).
Saritha et al.
test an electrochemical cell comprising Nb12W033 at a reported C-rate of no
more than 20C.
However, Saritha et al. apparently define the C-rate as reaction (i.e. removal
or insertion) of
one lithium ion in one hour. This corresponds to one electron transfer per
formula unit.
Thus, the 20C rate for Nb12W033 reported by Saritha et al. corresponds to
1.54C using the
convention defined in this work (equivalent to 294 mkg-1).
The present inventors have developed electrodes comprising a niobium tungsten
oxide that
has favorable lithium ion diffusion properties, and thus exhibit superior
performance even
with micron sizes particles. The electrodes exhibit extremely high volumetric
energy
densities and high capacities at high rates of charging and discharging.
The voltage values described herein are made with reference to LOU, as is
common in the
art.
CA 03137220 2021-10-18
WO 2019/234248
PCT/EP2019/065040
The C-rate is a measure of the rate at which a battery is discharged relative
to its maximum
capacity. The C-rate may be defined as the inverse of the number of hours to
reach a
defined theoretical capacity e.g., 10C corresponds to a 6 min discharge or
charge time. In
5 this work, C-rate is defined relative to one electron transfer per
transition metal, e.g., for
Nb16W5055, 1C = 171.3 mA=h=g-1, 20C = 3426 mA=h=g-1. The theoretical capacity
is
calculated by:
2.c,6485.3 [C.
____________________________________________________________________________
¨ _71.3 rnA = h = g-1
= 1.6'1 [.i -]
where n is the number of electrons transferred per formula unit, F is
Faraday's
constant, 3.6 is a conversion factor between Coulombs and the conventional
mA=h=g-1, and
m is the mass per formula unit. Thus, a 1C rate corresponds to the reaction
(i.e. insertion or
removal) of 21 lithium ions per formula unit of Nb16W5055in one hour, as this
material
contains 21 transition metals per formula unit.
The high-rate application may also be described by reference to (gravimetric)
current
density, for example where the current density is at least 800 mkg-1 or 1000
mkg-1.
Current density is related to C-rate by:
' MI g
Thus, for Nb16W5055 a current density of 800 mkg-1 corresponds to a C-rate of
4.67C and
for Nb18W16093 a current density of 800 mkg-1 corresponds to a C-rate of 5.36C
using the
convention defined in this work.
All (gravimetric) capacities are quoted based on the mass of the active
electrode material.
Working Electrode
The invention provides a working electrode comprising a niobium tungsten
oxide. The
working electrode is electrically conductive, and is electrically connectable
to a counter
electrode, for example within an electrochemical cell.
The working electrode may be an anode or cathode during a discharge step, for
example in
a lithium ion battery. Typically, the working electrode is the anode during a
discharge step.
CA 03137220 2021-10-18
WO 2019/234248 PCT/EP2019/065040
11
Typically, the working electrode for use in the method comprises a molar ratio
of Nb2O5 to
W03 from 6:1 to 1:15. Preferably, the molar ratio of Nb2O5 to W03 in the
working electrode
is from 8:5 to 11:20.
Typically, the working electrode for use in the method comprises a niobium
tungsten oxide
selected from Nb12W033, Nb26W4077, Nb14W3044, Nb16W5055, Nb18W8069, Nb2W08,
Nb18W16093, Nb22W200115, NID8W9047, Nb54W820381, Nb20VV310143, Nb4W7031, or
Nb2W15050 or
combinations thereof. Preferably, the working electrode comprises Nb16W5055,
Nb18W8069,
Nb2W08, Nb18W16093 or Nb22W200115 or combinations thereof.
Typically, the molar ratio of Nb2O5 to W03 in the working electrode is from
8:5 to 11:20.
Preferably, the molar ratio of Nb2O5 to W03 in the working electrode is 8:5 or
9:16.
Typically, the working electrode comprises Nb16W5055, Nb18W8069, Nb2W08,
Nb18W16093, or
Nb22W200115, or combinations thereof. Preferably the working electrode
comprises
Nb16W5055 or Nb18W16093, or combinations thereof.
Optionally, the working electrode comprises a mixture of niobium tungsten
oxide and an
additional active material. The additional active material may be an
additional metal oxide.
For example, the working electrode may comprise a mixture of niobium tungsten
oxide and
an additional active material selected from lithium titanate (LTO; Li4Ti5012),
titanium niobium
oxides (for example TiNb207), titanium tantalum oxides (for example TiTa207),
tantalum
molybdenum oxides (for example Ta8W9047) and niobium molybdenum oxides (for
example
Nb2Mo3014)=
Graphite may also be used as an additional active material. A working
electrode comprising
a mixture of niobium tungsten oxide and graphite is cheaper to produce while
maintaining
the beneficial properties outlined above.
Preferably, the working electrode comprises a mixture of niobium tungsten
oxide and LTO.
The ratio of niobium tungsten oxide to LTO may be from 95:5 to 5:95 by weight.
For
example, the ratio may be from 90:10 to 10:90 by weight, from 80:20 to 20:80
by weight,
from 70:30 to 30:70 by weight, from 60:40 to 40:60 by weight or the ratio of
niobium tungsten
oxide to LTO may be 1:1 by weight.
Preferably, the working electrode consists essentially of niobium tungsten
oxide and an
additional active material. For example, the working electrode consists
essential of a
mixture of niobium tungsten oxide and LTO.
Typically, the working electrode does not have a porous nor hierarchical
structure. For
example, the electrode material may have a specific surface area of less than
20 m2.g-1, less
than 10 m2.g-1, less than 5 m2.g-1, less than 3 m2.g-1, less than 2 m2.g-1 or
less than 1 m2.g-1.
CA 03137220 2021-10-18
WO 2019/234248 PCT/EP2019/065040
12
The specific surface area of the electrode material may be known, or it may be
determined
using standard techniques such as N2 adsorption isotherm analysis and
Brunauer¨Emmett¨
Teller (BET) theory.
Alternatively, the working electrode may have a porous structure. For example,
the working
electrode may have a specific surface area of at least 50 m2.g-1, at least 60
m2. 70 m2.g-1,
80 m2.g-1, 90 m2.g-1, 100 m2.g-1, 150 m2.g-1, 200 m2.g-1, 300 m2g. -1, or 400
m2.g-1.
The working electrode may have a pore volume of of at least 0.1 cm3.g-1, at
least 0.2 cm3.g-1,
at least 0.4 cm3.g-1, at least 0.5 cm3.g-1, at least 0.7 cm3.g-1, at least 0.8
cm3.g-1, at least 0.9
cm3.g-1, at least 1.0 cm3.g-1, at least 1.5 cm3.g-1 or at least 2.0 cm3.g-1.
The pore volume of the electrode material may be known, or it may be
determined using
standard techniques such as N2 adsorption isotherm analysis and Barrett-Joyner-
Halenda
(BJH) theory.
The porous working electrode may have an average pore size (largest cross
section) of at
least 1 nm, at least 5 nm, at least, 10 nm, at least 20 nm, at least 30 nm, at
least 40 nm, at
least 50 nm or at least 100 nm.
The porous working electrode may have macroporous structure. Thus, the porous
working
electrode may contain pores having pores having a largest cross section of at
least 200 nm,
at least 500 nm, at least 1 pm, or at least 5 pm.
The pore size of the electrode material may be known, or it may be determined
using
standard techniques such as scanning electron microscopy (SEM).
The working electrode may additionally comprise porous carbon, such as porous
reduced
graphene oxide.
Electrodes comprising porous carbon are generally light and conductive, and
can provide
large pore volumes, which can allow rapid transport of lithium ions and
electrons to the
active materials. They may also increase the electrochemical capacity of the
working
device.
The working electrode may additionally comprise reduced graphene oxide, Ketjen
black or
Super P carbon.
Alternatively, the working electrode may have a hierarchical structure. For
example, the
working electrode may additionally comprise hierarchical reduced graphene
oxide (rG0).
CA 03137220 2021-10-18
WO 2019/234248
PCT/EP2019/065040
13
Typically, the working electrode comprise a niobium tungsten oxide in
particulate form. The
size of the niobium tungsten oxide particles of the working electrode may be
known, or it
may be determined using standard techniques such as SEM.
Typically, the niobium tungsten oxide particles of the working electrode have
primary particle
size of at least 1 pm. The primary particle size is the size of the individual
crystallite. It is
the smallest identifiable subdivision in a particulate system. For example,
the niobium
tungsten oxide particles have a primary particle size of at least 2 pm, 3 pm,
4 pm, 5 pm or
pm.
The individual niobium tungsten oxide particles may agglomerate to form
secondary
particles. Typically, the niobium tungsten oxide particles have an agglomerate
(secondary)
particle size of at least 5 pm. More preferably, the niobium tungsten oxides
have an
agglomerate particle size of at least 10 pm, 15 pm, 20 pm, 25 pm or 30 pm.
Where present, the additional active material may be in particulate form. The
size of the
additional active material particles may be known, or it may be determined
using standard
techniques such as SEM.
Preferably, the additional active material particles have a primary particle
size of 1 pm or
less. For example, the additional active material particles have a primary
particle size of
800 nm or less, 750 nm or less, 700 nm or less, 600 nm or less, 500 nm or
less, 400 nm or
less, 300 nm or less, 200 nm or less or 150 nm or less. Particulate lithium
titanate typically
has a particle size within this range.
Electrodes comprising a mixture of niobium tungsten oxide and an additional
active material
having partial sizes within the ranges described above can be charged and
discharged at
very high C-rates and at very high charge densities.
To improve conductivity at the working electrode, a conductive carbon material
(e.g., carbon
black, graphite, nanoparticulate carbon powder, carbon fiber and/or carbon
nanotubes) is
typically admixed with the working electrode material. Alternatively, the
conductive carbon
material may be coated onto the working electrode material. In one embodiment,
the
working electrode comprises porous carbon, such as porous reduced graphene
oxide, which
may wrap the larger niobium oxides particles.
Typically, the working electrode contains 1-5% by weight of binders.
The electrode may consist essentially of niobium tungsten oxide.
CA 03137220 2021-10-18
WO 2019/234248
PCT/EP2019/065040
14
Alternatively, the working electrode is admixed with a binder or adhesive.
Some examples
of binders or adhesives include PVDF, PTFE, CMC, PAA, PMMA, PEO, SBR and
co-polymers thereof.
.. The working electrode is typically fixed to a current collector, such as a
copper or aluminum
collector, which may be in the form of a plate.
The inventors have assessed a working electrode comprising a particulate
niobium tungsten
oxide using a standard electrode configuration of 8:1:1 active
material/carbon/binder with a
2-3 mg=cm2 loading of active material and a 1.27 cm2 electrode area against a
lithium
counter electrode in a 2032-type coin cell geometry and using 1.0 M LiPF6 in
ethylene
carbonate/dimethyl carbonate as electrolyte.
Under these conditions, the inventors have found that a working electrode
comprising a
niobium tungsten oxide can maintain a capacity of up to 150 mA=h=g-1 at 100
for
1000 cycles, and a capacity of up to 125 mA=h=g-1 at 200 for 750 cycles.
In addition, the inventors have found that a working electrode comprising a
niobium tungsten
oxide has a sloping, rather than flat, voltage profile.
The inventors have assessed solid-state lithium diffusion within niobium
tungsten oxides
using both pulsed field gradient NMR (PFG NMR) and the galvanostatic
intermittent titration
technique (GITT). The inventors have found that the niobium tungsten oxides
have a
solid-state lithium diffusion coefficient (Du) of 10-13 to 10-12 m2.s-1 at 298
K. This corresponds
.. to a characteristic diffusion length of ca. 10 pm for a 1 minute discharge.
The invention also provides a working electrode comprising a niobium
molybdenum oxide.
The working electrode is electrically conductive, and is electrically
connectable to a counter
electrode, for example within an electrochemical cell.
The working electrode may be an anode or cathode during a discharge step, for
example in
a lithium ion battery. Typically, the working electrode is the anode during a
discharge step.
Typically, the working electrode for use in the method comprises a molar ratio
of Nb2O5 to
.. Mo03 of from 6:1 to 1:3. Preferably, the molar ratio of Nb2O5 to Mo03in the
working
electrode is 1:3.
Typically, the working electrode for use in the method comprises a niobium
molybdenum
oxide selected from Nb2Mo3014, Nb14Mo3044 or Nb12Mo044. Preferably, the
working
.. electrode comprises Nb2Mo3014.
CA 03137220 2021-10-18
WO 2019/234248 PCT/EP2019/065040
Typically, the working electrode does not have a porous nor hierarchical
structure. The
working electrode may have a specific surface area, pore volume and average
pore size as
described above.
5 Typically, the working electrode comprise a niobium molybdenum oxide in
particulate form.
The niobium molybdenum oxide particles of the working electrode may have a
primary or
agglomerate particle size as described above.
The working electrode may contain binders and adhesives as described above.
Electrochemical Cell
The present invention also provides an electrochemical cell comprising a
working electrode
of the invention. The working electrode may be an anode or cathode during a
discharge
step, for example in a lithium ion battery. Typically, the working electrode
is the anode
during a discharge step.
The electrochemical cell typically comprises a counter electrode and an
electrolyte. The
electrochemical cell may comprise a current collecting plate. The
electrochemical cell may
be in electrical connection with a power supply. The electrochemical cell may
be in electrical
connection with a measurement device, for example an ammeter or voltmeter.
The counter electrode may be an anode or cathode during a discharge step, for
example in
a lithium ion battery. The counter electrode is typically the cathode during a
discharge step.
Suitable cathode materials include lithium-containing or lithium-intercalated
material, such as
a lithium metal oxide, wherein the metal is typically a transition metal such
as Co, Fe, Ni, V
or Mn, or combination thereof. Some examples of positive electrode materials
include
lithium cobalt oxide (Li0002), lithium nickel manganese cobalt oxide (NMC,
LiNiMnCo02,
e.g. LiNio600o2Mno202), lithium vanadium fluorophosphate (LiVP04F), lithium
nickel cobalt
aluminum oxide (NCA, LiNiCoA102), lithium iron phosphate (LFP, LiFePO4)and
manganese-
based spinels (e.g. LiMn204).
To improve conductivity at the counter electrode, a conductive carbon material
(e.g., carbon
black, graphite, nanoparticulate carbon powder or carbon nanotubes) is
typically admixed
with the counter electrode material. In one embodiment, the counter electrode
comprises
porous carbon, such as porous reduced graphene oxide.
In one embodiment, the counter electrode is substantially free of binders.
CA 03137220 2021-10-18
WO 2019/234248
PCT/EP2019/065040
16
In an alternative embodiment, the counter electrode is admixed with a binder
or adhesive.
Some examples of binders or adhesives include PVDF, PTFE, CMC, PAA, PM MA,
PEO,
SBR and co-polymers thereof.
The counter electrode is typically fixed to a current collecting substrate,
such as an
aluminum plate.
Typically, the electrolyte in the electrochemical cell is suitable for
solubilising lithium ions.
Typically, the electrolyte in a charged and discharged cell contains lithium
ions.
Typically, the electrolyte comprises lithium salts, such as LiTFSI,
(bis(trifluoromethane)sulfonimide lithium salt, LiPF6, LiBF4, LiC104, LiTF
(lithium triflate) or
lithium bis(oxalato) borate (LiBOB).
The electrolyte may be a liquid electrolyte, such as a liquid at ambient
temperature, for
example at 25 C.
The electrolyte may be a non-aqueous electrolyte. The electrolyte may comprise
a polar
aprotic solvent. The electrolyte may comprise an organic solvent. Solvents for
dissolving
lithium ions are well known in the art.
Suitable solvents include carbonate solvents. For example propylene carbonate
(PC),
ethylene carbonate (EC), butylene carbonate (BC), chloroethylene carbonate,
fluorocarbonate solvents (e.g., fluoroethylene carbonate and trifluoromethyl
propylene
carbonate), as well as the dialkylcarbonate solvents, such as dimethyl
carbonate (DMC),
diethyl carbonate (DEC), dipropyl carbonate (DPC), ethyl methyl carbonate
(EMC), methyl
propyl carbonate (MPC), and ethyl propyl carbonate (EPC).
Suitable solvents also include sulfone solvents. For example methyl sulfone,
ethyl methyl
sulfone, methyl phenyl sulfone, methyl isopropyl sulfone (MiPS), propyl
sulfone, butyl
sulfone, tetramethylene sulfone (sulfolane), phenyl vinyl sulfone, allyl
methyl sulfone, methyl
vinyl sulfone, divinyl sulfone (vinyl sulfone), di phenyl sulfone (phenyl
sulfone), dibenzyl
sulfone (benzyl sulfone), vinylene sulfone, butadiene sulfone, 4-methoxyphenyl
methyl
sulfone, 4-chlorophenyl methyl sulfone, 2-chlorophenyl methyl sulfone, 3,4-
dichlorophenyl
methyl sulfone, 4-(methylsulfonyl)toluene, 2-(methylsulfonyl) ethanol, 4-
bromophenyl methyl
sulfone, 2-bromophenyl methyl sulfone, 4-fluorophenyl methyl sulfone, 2-
fluorophenyl methyl
sulfone, 4-aminophenyl methyl sulfone, a sultone (e.g., 1,3-propanesultone),
and sulfone
solvents containing ether groups (e.g., 2-methoxyethyl(methyl)sulfone and 2-
methoxyethoxyethyl(ethyl)sulfone).
Suitable solvents also include silicon-containing solvents such as a siloxane
or silane. For
example hexamethyldisiloxane (HMDS), 1,3-divinyltetramethyldisiloxane, the
polysiloxanes,
CA 03137220 2021-10-18
WO 2019/234248
PCT/EP2019/065040
17
and polysiloxane-polyoxyalkylene derivatives. Some examples of silane solvents
include
methoxytrimethy lsilane, ethoxytrimethy lsilane, dimethoxydimethylsilane,
methyltrimethoxysilane, and 2-(ethoxy)ethoxytrimethylsilane.
Typically, an additive may be included in the electrolyte to improve
performance. For
example vinylene carbonate (VC), vinyl ethylene carbonate, allyl ethyl
carbonate, t-butylene
carbonate, vinyl acetate, divinyl adipate, acrylic acid nitrile, 2-vinyl
pyridine, maleic
anhydride, methyl cinnamate, ethylene carbonate, halogenated ethylene
carbonate, a-
bromo-y-butyrolactone, methyl chloroformate, 1,3-propanesultone, ethylene
sulfite (ES),
propylene sulfite (PS), vinyl ethylene sulfite (VES), fluoroethylene sulfite
(FES), 12-crown-4
ether, carbon dioxide (002), sulfur dioxide (SO2), and sulfur trioxide (SO3).
The electrochemical cell may also include a solid porous membrane positioned
between the
negative and positive electrodes. The solid porous membrane may partially or
completely
replace the liquid electrolyte. The solid porous membrane may comprise a
polymer (e.g.,
polyethylene, polypropylene, or copolymer thereof) or an inorganic material,
such as a
transition metal oxide (e.g., titania, zirconia, yttria, hafnia, or niobia) or
main group metal
oxide, such as silicon oxide, which can be in the form of glass fiber.
The solid non-porous membrane may comprises a lithium-ion conductor. For
example,
LLZO (garnet family), LSPO (LISICON family), LGPS (thio-LISICON family),
LATP/LAGP
(NASICON family), LLTO (perovskite family) and phosphide/sulfide glass
ceramics
The electrochemical cell may be charged or discharged at a C-rate of at least
5C, such as
the electrochemical cell may be charged or discharged at a C-rate of at least
5C with respect
to one electron transfer per transition metal per formula unit of working
electrode material.
Preferably, the electrochemical cell may be charged or discharged at a C-rate
of at least
100, 150, 20C, 25C, 30C, 35C, 40C, 50C, 600 or 800.
The electrochemical cell may be charged or discharged at a current density of
at least 750
mkg-1. Preferably, the electrochemical cell may be charged or discharged at a
current
density of at least 800 mkg-1, 850 mkg-1, 900 mkg-1, 950 mkg-1, 1000 mkg-1,
1050
mkg-1, 1100 mkg-1, 1200 mkg-1 or 1300 mkg-1.
The electrochemical cell may have a volumetric charge density of at least 200,
300, 400,
500, 600 or 700 A=h=L-1 at 1C. Typically, the electrochemical cell has a
volumetric charge
density of up to 100, 200, 300 or 400 A=h=L-1 at 20C.
The electrochemical cell may have a capacity retention of at least 70%, 75%,
80%, 85%,
90%, 95%, 96%, 97%, 98%, 99%, or 100% at 20C maintained over at least 50, 100,
150,
200, 250, 300, 400, 500, 600, 700, 800, 900, 1,000, 1,200, 1,500, 1,800, or
2,000 cycles.
CA 03137220 2021-10-18
WO 2019/234248
PCT/EP2019/065040
18
The electrochemical cell may be regarded as fully charged when the voltage
passes a
threshold value. For example, an electrochemical cell comprising a lithium
metal anode and
a niobium tungsten oxide cathode may be regarded as fully charged when the
voltage rises
above a practicable level, such as where the voltage rises above 2.0 V against
Li/Li, such
as above 2.25 V or above 2.5 V.
The electrochemical cell may be regarded as fully discharged when the voltage
passes a
threshold value. For example, an electrochemical cell comprising a lithium
metal anode and
a niobium tungsten oxide cathode may be regarded as fully discharged when the
voltage
drops below a practicable level, such as where the voltage drops below 1.5 V
against Li/Li,
such as below 1.25 V or below 1.0 V.
The electrochemical cell may be a lithium ion cell.
Methods
The invention provides a method of charging and/or discharging an
electrochemical cell at a
C-rate of at least 5C, such as the electrochemical cell may be charged or
discharged at a C-
rate of at least 5C with respect to one electron transfer per transition metal
per formula unit
of working electrode material. The electrochemical cell comprises a working
electrode
comprising a niobium tungsten oxide and/or niobium molybdenum oxide.
Preferably the
electrochemical cell contains a counter electrode and an electrolyte.
Preferably the method is a method of charging and/or discharging an
electrochemical cell at
a C-rate of at least 10C, 15C, 20C, 25C, 30C, 35C, 40C, 50C, 60C or 80C.
The method may be a method of charging and/or discharging an electrochemical
cell at a
current density of at least 750 mA=g' such as at least 800 mA.V. Preferably
the method is
a method of charging and/or discharging an electrochemical cell at a current
density of at
least 800 mA.V, 850 mA.V, 900 mA.V, 950 mA.V, 1000 mA.V, 1050 mA.V, 1100
mA.V, 1200 mA=g' or 1300 mA=g'.
The method may involve a cycle of charging and discharging or discharging and
charging
the electrochemical cell. The cycle may be repeated more than once. Thus, the
method
comprises 2 cycles or more, 5 cycles or more, 10 cycles or more, 50 cycles or
more, 100
cycles or more, 500 cycles or more, 1,000 cycles or more, or 2,000 cycles or
more.
Battery
The present invention also provides a battery comprising one or more
electrochemical cells
of the invention. The battery may be a lithium ion battery.
CA 03137220 2021-10-18
WO 2019/234248
PCT/EP2019/065040
19
Where there are a plurality of cells, these may be provided in series or
parallel.
A battery of the invention may be provided in a road vehicle, such as an
automobile, moped
or truck. Alternatively, a battery of the invention may be provided in a rail
vehicle, such as a
train or a tram. A battery of the invention may also be provided in an
electric bicycle
(e-bike).
A battery of the invention may be provided in a regenerative braking system.
A battery of the invention may be provided in a portable electronic device,
such as a mobile
phone, laptop or tablet.
A battery of the invention may be provided in a power grid management system.
Uses
The invention generally provides the use of a working electrode comprising a
niobium
tungsten oxide in a high-rate electrochemical cell, such as an electrochemical
cell as
described herein. Typically, the electrochemical cell may be charged or
discharged at a
C-rate of at least 5C, such as the electrochemical cell may be charged or
discharged at a C-
rate of at least 5C with respect to one electron transfer per transition metal
per formula unit
of working electrode material. Preferably, the electrochemical cell may be
charged or
discharged at a C-rate of at least 10C, 15C, 20C, 25C, 30C, 35C, 40C, 50C, 60C
or 80C.
The electrochemical cell may be charged or discharged at a current density of
at least 750
mkg-1. Preferably, the electrochemical cell may be charged or discharged at a
current
density of at least 800 mkg-1, 850 mkg-1, 900 mkg-1, 950 mkg-1, 1000 mkg-1,
1050
mkg-1, 1100 mkg-1, 1200 mkg-1 or 1300 mkg-1.
The working electrode may find use in the methods described herein.
Other Preferences
Each and every compatible combination of the embodiments described above is
explicitly
disclosed herein, as if each and every combination was individually and
explicitly recited.
Various further aspects and embodiments of the present invention will be
apparent to those
skilled in the art in view of the present disclosure.
"and/or" where used herein is to be taken as specific disclosure of each of
the two specified
features or components with or without the other. For example "A and/or B" is
to be taken as
CA 03137220 2021-10-18
WO 2019/234248 PCT/EP2019/065040
specific disclosure of each of (i) A, (ii) B and (iii) A and B, just as if
each is set out individually
herein.
Unless context dictates otherwise, the descriptions and definitions of the
features set out
5 above are not limited to any particular aspect or embodiment of the
invention and apply
equally to all aspects and embodiments which are described.
Certain aspects and embodiments of the invention will now be illustrated by
way of example
and with reference to the figures described above.
Experimental
The following examples are provided solely to illustrate the present invention
and are not
intended to limit the scope of the invention, as described herein.
Synthesis of Nb16W5055 and Nb18W16093
Nb16W 5055 and Nb18W16093 were synthesized by co-thermal oxidation of dark
blue Nb02
(Alfa Aesar, 99+%) and brown W02 (Alfa Aesar, 99.9%) in approximately one to
five gram
batches. The partially reduced oxides were massed to within 0.001 g of the
16:5 or 18:16
ma ratios, ground together by hand with an agate mortar and pestle,
pressed into a pellet
at 10 MPa, and heated in a platinum crucible at a rate of 10 K=min-1 to 1473
K, and naturally
cooled in the furnace over ca. 2 h.
Synthesis of Nb8W9047 and Nb12W033
Nb8W9047 and Nb12W033 were synthesized by co-thermal oxidation of dark blue
Nb02 (Alfa
Aesar, 99+%) and brown W02 (Alfa Aesar, 99.9%) in approximately one to five
gram
batches. The partially reduced oxides were massed to within 0.001 g of the 8:9
or 12:1 -nolar
ratios, ground together by hand with an agate mortar and pestle, pressed into
a pellet at 10
MPa, and heated in a platinum crucible at a rate of 10 K=min-1 to 1473 K, and
naturally
cooled in the furnace over ca. 2 h.
Synthesis of Nb2Mo3014
Nb2Mo3014 synthesized by co-thermal oxidation of dark blue Nb02 (Alfa Aesar,
99+%) and
dark brown Mo02 (Sigma, 99%), or co-thermal oxidation of white Nb2O5 (Sigma,
99.9985%)
and off-white Mo03 (Sigma, 99.5`)/0), in approximately one to five gram
batches. The
partially reduced oxides were massed to within 0.001 g of the 2:3 or 1:3 ..ioi
ratio, ground
together by hand with an agate mortar and pestle, pressed into a pellet at 10
MPa, and
heated in a platinum or alumina crucible at a rate of 10 K=min-1 to 873 K, 923
K or 973 K,
and quenched in air outside the furnace on a metal plate.
CA 03137220 2021-10-18
WO 2019/234248 PCT/EP2019/065040
21
Microscopic Characterisation
Scanning electron microscopy (SEM) images were taken with a Zigma VP
instrument (Zeiss)
at 3.0 kV and a MIRA3 instrument (TESCAN) at 5.0 kV with secondary electron
detection.
Tap density was recorded on an AutoTap (Quantachrome Instruments) instrument
operating
at 257 taps=min-1. Tap densities were measured according to ASTM international
standard
B527-15 modified to accommodate a 5 to 10 cm3 graduated cylinder.
Both Nb16W5055 and Nb18W16093 formed subhedral particles with ca. 3 to 10 pm
primary
particles that appeared in the electron microscope to be intergrown/'cemented'
into larger
secondary particles of ca. 10 to 30 pm (Fig. 1 and 2). The bulk compounds are
a series of
complex "block" (Nb16W5055) or "bronze-like" (Nb18W16093) oxide structures
(Fig. 2) largely
comprised of corner and edge sharing NbO6 and W06 octahedra.
Nb16W5055 has a monoclinic structure comprised of subunits of corner-shared
octahedra
arranged into Re03-like blocks, four octahedra wide by five octahedra long,
and infinite in
the third dimension (Fig. 2A). The block subunits are connected by
crystallographic shear
planes along the edges and by tetrahedra at each corner leading to the
notation (4 x 5)1
where, in (m x n)p, m and n denote block length in units of octahedra and p
relates to the
connectivity of the blocks which may also be joined in pairs (p = 2) or
infinite ribbons (p = 00).
Nb18W16093 is orthorhombic, a 1 x 3 x 1 superstructure of the classic
tetragonal tungsten
bronze (TTB) (Fig. 2D and 3). The TTB structure is sometimes stabilized by
cations (e.g.
K+, partially filled spheres in Fig. 3); alkali cation-free Nb18W16093 is
stabilized by ¨M-0¨
chains partially occupying the tunnels and forming pentagonal bipyramids. The
ionic and
electronic implications for lithium intercalation, respectively, are that the
layers of Nb18W16093
are still fully open and unoccupied by cations (i.e., around the plane (x, y,
1/2)) and all metal
cations are in the desired d electron configuration while in classic TTB
structure neither of
these criteria is met.
Electrochemical Characterization of Nb16W5055 and Nb18W16093
Electrochemical characterisation was conducted using a stainless steel 2032-
coin cell
(Cambridge Energy Solutions) with a conical spring, two 0.5 mm stainless steel
spacer
disks, a plastic gasket, and a glass microfiber separator (Whatman). To form
the niobium
tungsten oxide electrode, the niobium tungsten oxide and conductive carbon
(Super P,
TIMCAL) were ground by hand in an agate mortar and pestle in an 8:1 mass
ratio. This
powder was ground in a 9:1 mass ratio with poly(vinylidene difluoride) (PVDF,
Kynar)
dispersed in N-methyl pyrrolidone (NMP, Sigma-Aldrich, anhydrous, 99.5%). The
slurry was
coated onto aluminium or copper foil with a doctor blade (bar coater). The NMP
was
removed by heating in an oven at 60 C for 24 hours. Though standard, Super P
carbon is a
CA 03137220 2021-10-18
WO 2019/234248 PCT/EP2019/065040
22
nanoparticu late powder and NMP is a hazardous organic solvent so appropriate
nanoparticle
cabinets/fume hoods should be used.
This 80/10/10 metal oxide/carbon/polymer electrode served as the cathode
against a Li
metal disk (LTS Research, 99.95%) anode in a half-cell geometry. In the
electrochemical
tests, the electrolyte was 1 M LiPF6 dissolved in 1:1 v/v ethylene
carbonate/dimethyl
carbonate (EC/DMC; Sigma-Aldrich, battery grade; also known as LP30). No
additives were
used. Electrochemistry was performed in a temperature-controlled room at 293
K. A
Biologic galvanostat/potentiostat with EC-Lab software was used to perform the
electrochemical measurements.
Dense electrodes of large particles with 2 to 3 mg=cm-2 active mass loading
were tested at
current densities corresponding to discharge times of several hours to tens of
seconds.
Nb16W5055 was charged with a 1 h constant voltage step at the top of charge to
ensure a
comparable starting point on discharge; Nb19W16093 was cycled without this
step and stored
over 100 mA=h=g-1 at 600 (i.e., in <60 s). High-rate cycling for 1000 cycles
was performed
on both oxides at 100/200 constant current without any potentiostatic step.
Reaction of Nb16W5055 with lithium (Fig. 4A) proceeds in three regions from
2.5 V to 1.0 V,
with an average voltage of 1.57 V (Fig. 5), comparable to the average voltage
of Li4Ti5012 of
1.55 V. The three regions, more easily observed in the derivative plot (Fig.
4B), are
characterized by their slope and are reminiscent of the three regions observed
in other
crystallographic shear structures (e.g., H-Nb2O5, PNb9025, TiNb207, and
Nb12W033). When
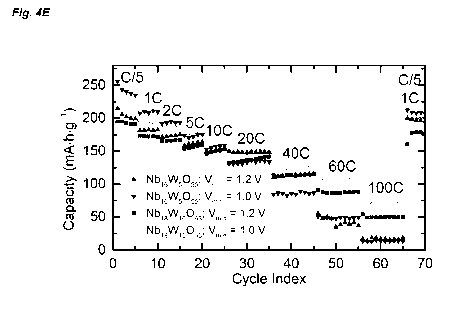
the kinetics were examined over a range of current densities from 0/5 (34.3
mkg-1) up to
600 (10.3 ), Nb16W5055 showed unprecedented bulk rate performance (Fig. 4A
and
40). At 0/5, around 1.3 lithium ions can be reversibly intercalated per
transition metal for a
gravimetric capacity of ca. 225 mA=h=g-1. When the rate is increased by a
factor of 25 to 50,
Nb16W5055 maintains a capacity of 1.0 Li/TM (171 mA=h=g-1). At 200, which
corresponds to
a three minute discharge, it is still possible to exchange 0.86 Li/TM and
access
148 mA=h=g-1. Rate tests on Nb16W5055 were measured with a potentiostatic hold
at the top
of charge to ensure a reliable starting point for discharge. To test the
performance under
more demanding conditions, 1000 cycles were measured with fixed galvanostatic
discharge
and charge conditions of 100 for 250 cycles followed by 200 for 750 cycles
with no hold
(Fig. 4F). Under these conditions, 0.90 Li/TM (avg. 155 mA=h=g-1) were
reversibly
intercalated at 100 with 95% capacity retention after 250 cycles on non-
optimized or
calendared electrodes. At 200, the capacity was 0.75 Li/TM (avg. 128 mA=h=g-
1); the
capacity retention was again 95% over the 750 cycles at 200.
The average voltage of Nb19W16093 is 1.67 V (Fig. 4D-F). In terms of
gravimetric capacity,
Nb19W16093 stores ca. 20 mA=h=g-1 less than Nb16W5055 at 0/5 and 10 due to the
higher
molar mass of the tungsten-rich bronze phase. However, at 200, Nb19W16093 is
still able to
CA 03137220 2021-10-18
WO 2019/234248 PCT/EP2019/065040
23
accommodate a full unit Li+/TM for a capacity of ca. 150 mA=h=g-1. At 600 and
1000
(14.9 A=g-1), the capacity is still 105 and 70 mA=h=g-1, respectively.
Comparison of Cu Foil to Carbon-Coated Al Foil Current Collector
Fig. 6 shows the effect of current collectors, which cannot be ignored at high
rates.
For Nb16W5055cycled for 1000 cycles under constant current discharge and
charge at
100/200, Cu foil current collector displayed moderately higher capacity than
carbon-coated
Al (C@AI) foil (Fig. 6A). Untreated Al foil has been shown to be insufficient
for high current
densities (Griffith et al.); C@AI demonstrated significant improvement over
untreated Al. In
Nb16W5055, the lower voltage region is the source of excess capacity beyond
1.0 Li+/TM at
low rates but is effectively shifted below 1.0 V at moderate rates. The second
discharge
peak (centered at 1.2 V) is only observed at C/5 in the dQ/dV plot (Fig. 4B).
As a result, the
capacity observed when cycling with a minimum voltage limit of 1.2 V vs. 1.0 V
becomes
less significant as rate increases and safety may be further improved by
avoiding lower
voltages. Fig. 6B shows discharge and charge profiles between 3.0 and 1.0 V
for the 100th
cycle at 100.
Longer Term Cycling as a Function of Minimum Cutoff Voltage
Fig. 7 shows the effect of long term cycling at C/5.
Constant current constant voltage charging is a common method to maximize
capacity
without overcharging, as in (Fig. 7A), but the extra time required means that
the cycling
could be considered C/5 and 10 discharge but 0/6 and 0/2 charge. The 2nd to
50th cycle
retention in Fig. 7A is 96% and 93% for a voltage minimum of 1.2 and 1.0 V,
respectively.
The 51st to 100th cycles at 10 show capacity retentions of 99% and 101% for
1.2 and 1.0 V,
respectively. Cycle retention for the 2nd to 50th cycles for C/5 in Fig. 7B is
88%, 90%, and
40% for 1.1, 1.0, and 0.9 V, respectively. Cycling time was approximately one
month.
Overpotential in a Li II Li Symmetric Cell as a Function of Current Density
As a control, Li II Li symmetric cells were cycled at current densities
corresponding to those
in Fig. 4A-E (Fig. 8). Cells were configured identically to those used for
metal oxide testing
with the exception of a second Li disk replacing the composite electrode. Rate
testing was
carried out with 5 cycles at 100 pA (C/5), 500 pA (10), 1 mA (20), 2.5 mA
(5C), 5 mA (100)
and 10 cycles at 10 mA (200), 20 mA (400), 30 mA (600), and 50 mA (1000). The
"rate" in
parentheses indicates the inverse of the amount of time in hours that current
was applied,
simulating the rate test. At low current densities, below 1 mA (20), the
overpotential is
below 100 mV; however, at 5 mA (100) it rises to 200 mV and increases to ca.
700 mV at
1000.
CA 03137220 2021-10-18
WO 2019/234248 PCT/EP2019/065040
24
The overpotentials in the symmetric cell (Fig. 8) closely match those observed
in the
electrochemical cycling curves of Fig. 4A and 4D. This suggests that the
extremely high
rates for a bulk electrode are approaching the limits of Li metal
plating/stripping and/or
lithium ion desolvation and transport in carbonate ester electrolytes at room
temperature,
i.e., a significantfraction of the ohmic drop seen during fast charging
results from the Li
metal and not the complex oxide electrode materials.
'Li Pulsed Field Gradient NMR Spectroscopy
7Li NMR diffusion spectra were recorded on a Bruker Avance III 300 MHz
spectrometer
using a Diff50 probehead equipped with an extended variable temperature
capabilities.
Spectra were recorded with the stimulated echo pulsed field gradient (PFG)
sequence
shown in Fig. 9 to minimize spin-spin relaxation (T2) losses. After the first
90
radiofrequency (rf) pulse, the net magnetization loses coherence due to T2
relaxation; thus,
the time period following this pulse, which includes the first PFG pulse (to
encode spin
position), must be shorter than T2. In the stimulated echo sequence used here,
a second
90 if pulse is applied which stores the net magnetization along the z axis
prior to the
diffusion time, A, allowing the observed species to diffuse for a time
commensurate with the
comparatively longer spin-lattice (Ti) value, as no T2 relaxation occurs.
During A, a short
spoiler gradient (SINE.100) is applied to remove residual transverse
magnetization.
Afterwards, a third 90 if pulse is applied, followed by a PFG pulse to decode
spin position.
Sufficiently long delays between PFG and radiofrequency pulses were used 0.5
ms) to
minimize eddy currents in the diffusion measurements.
During this sequence, the gradient strength, g, was varied from 0.87 to 1800
or 2300 G=cm-1,
and 16 gradient steps were acquired using 'opt' shaped pulses with 1024-4096
transients.
The opt shape is a composite pulse that starts with a quarter of a sine wave,
followed by a
constant gradient, and ends with a ramp down (Fig. 9). The 'opt' gradient
pulses provide the
largest gradient integral for a given time period, maximizing the range of
diffusion coefficients
we could assess in this experiment.
Spectra were analysed in phase-sensitive mode and the response of the NMR
signal
intensity, /, to variation in g, is described by the Stejskal¨Tanner equation:
D) /0 )
where /0 is the intensity in the absence of gradients, y is the gyromagnetic
ratio (y7L, =
103.962x106.s-1.1-- , ) 1,5 is the effective gradient pulse duration, and D is
the diffusion
coefficient. Here, NMR signal intensity and integral gave similar 7Li
diffusivities, but NMR
CA 03137220 2021-10-18
WO 2019/234248
PCT/EP2019/065040
signal intensities gave more reliable data, as evaluated by the standard
deviation of the fit.
Typical 5 values ranged from 0.8 ms to 1.5 ms and .6 values ranged from 50-100
ms for the
bronze and the block phase samples, respectively.
5 Diffusion spectra were recorded at elevated temperatures (333-453 K) due
to the increase
in T2 observed at high temperature (e.g. T2for Li34Nb18W16093 is approximately
700 ps at
room temperature vs. 1.9 ms at 453 K). (N.b. No attempt was made to calibrate
the
temperature for this experimental setup because a single-tuned 7Li coil was
used and no
reliable 7Li reference is routinely used for temperature calibration. The
Bruker manual states
10 that for static measurements, the temperature calibration should be
within 7 degrees of the
set value.) The increase in T2 allowed the use of longer gradient pulses, 5,
that were
necessary to measure diffusion coefficients in the solid oxides.
Representative 7Li diffusion decay curves are shown in Fig. 10. The data
quality is directly
15 correlated with the Li content and subsequent signal-to-noise ratio
(SNR) of the 7Li NMR
spectra, where SNRs of 95, 56, 10, and 12 were observed for Fig. 10A
(Liio2Nbi8W16093),
10B (Lis 8Nbi8W16093), 100 (Li34Nbi8W16093), and 10D (Li63Nbi6W5055),
respectively. We
note that while the SNR of the low Li content Li34Nb18W16093 sample is low and
the signal
decay is only ca. 60%, the observed trend in 7Li diffusivity and activation
energy is consistent
20 with that observed in more lithiated bronze structures.
Lis 3Nbi6W5055 shows two-component behavior with lithium transport as rapid as
4.3x1012
M2' S-1 at 333 K (Fig. 11). Assuming Arrhenius behavior and the measured
activation energy
of 0.23 eV, the room temperature lithium diffusion coefficient is estimated to
be 2.1 x10-12
25 m2=s-1 (Table 1).
LixNb18W16093 (x = 3.4, 6.8, 10.2) exhibited similar diffusion and activation
energies, with
room temperature diffusion coefficients of 1.1 x1013 m2. S-1 and Ea in the
range of 0.27 to
0.30 eV.
CA 03137220 2021-10-18
WO 2019/234248 PCT/EP2019/065040
26
Table 1 - Du and Ea measurements for selected niobium tungsten oxide materials
Compound Du (m2=s-1) at 298 K Du (m2=s-1) at 413 K Ea
(eV)
Li63Nb16W5O55-a (15%) 2.1x10-12 2.7x10-11 0.23
0.04
Li63Nb16W5O55-b (85%) 1.7x10-13 5.2x10-13 0.10
0.04
Li84Nb16W5055 1.6x10-13 6.5x10_13 (403 K) 0.13
0.01
Li34Nb18W16093 1.1x10-13 2.0x10-12 0.27
0.03
Lis sNbi8W16093 1.1x10-13 1.8x10-12 0.30
0.01
Li1o2Nb18W16093 1.1x10-13 2.7x10-12 0.29
0.01
Lithium diffusion in both niobium tungsten oxide structures is markedly faster
than that of
Lia+xTi5012 or LixTiO2 at ca. 10_16_10_15 m2.s-1 and is close to the best
known lithium solid
electrolytes (Table 2).
PFG NMR (Fig. 11) results indicate that initial lithium ion diffusion is of
the order 10-12 to 10-13
m2.s-1 while galvanostatic intermittent titration technique (GITT)
measurements (Fig. 12)
show that the niobium tungsten oxides retain this rapid motion to high
capacities 1.5
Li/TM) where the diffusion then drops by about two orders-of-magnitude toward
2.0 Li/TM.
This suggests that the inherent range of the niobium tungsten oxide electrode
materials for
high-rate multiredox extends to approximately 1.5 Li/TM. The diffusion
coefficients on the
order of 10-12 to 10-13 m2.s-1 measured for these materials are consistent
with the values
required to achieve full lithiation of 6 to 19 pm particles on a 600
timescale.
Table 2 - Du measurements for known materials
0
t..)
o
,-,
o
Compound Structure Type Du (m2=s-1) T (K) Technique
Reference
(...)
Li10GeP2S12 Thio-LISICON 2x10-12 298 PFG NMR
Kuhn, et al. (2013)
t..)
cio
Li10GeP2S12 Thio-LISICON 4x10-11 453 PFG NMR
Kuhn, et al. (2013)
Li7GePS8 Thio-LISICON 2x10-12 298 PFG NMR
Kuhn, et al. (2013)
Li7GePS8 Thio-LISICON 4x10-11 453 PFG NMR
Kuhn, et al. (2013)
Li10SnP2S12 Thio-LISICON 1.4x10-12 298 PFG NMR
Kuhn, et al. (2014)
Li10SnP2S12 Thio-LISICON 4x10-11 453 PFG NMR
Kuhn, et al. (2014)
Li11Si2PS12 Thio-LISICON 3.5x10-12 298 PFG NMR
Kuhn, et al. (2014)
P
Li11Si2PS12 Thio-LISICON 4x10-11 453 PFG NMR
Kuhn, et al. (2014) =,
,
Li7P3S11 Thio-LISICON 1-5x10-12 303 PFG NMR
Hayamizu, et al. (2013)
,
rõ
Li7P3S11 Thio-LISICON 2-11x10' 353 PFG NMR
Hayamizu, et al. (2013) rõ
0
rõ
fl-Li3PS4 Thio-LISICON 5.4)00'3 373 PFG NMR
Gobet, et al.
,
0
,
amorphous-Li3PS4 Amorphous (Thio-LISICON) 6.5x10-13 303 PFG NMR
Hayamizu, et al. (2016) ,
.3
amorphous-Li3PS4 Amorphous (Thio-LISICON) 1.6-3.4x10-12
353 PFG NMR Hayamizu, et al. (2016)
Lio.6[Lio.2Sno.8S2] Layered (01) 2-20x10-12
298 PFG NMR Holzmann, et al.
Lio.6[Lio.2Sno.8S2] Layered (01) 2.6x10-10
407 PFG NMR Holzmann, et al.
Li3.4V0.6Sio.404 LISICON 5x10-15 333 tracer
Ishiyama, et al.
Lit5A10.5Get5(PO4)3 NASICON 2.9x10-13
311 PFG NMR Hayamizu et al. (2017) 1-
d
n
Li12A10.2Ti1.8 (PO4)3 NASICON 1.5x10-12 250
NMR relaxometry
PFG NMR
Arbi, et al.
m
1-d
t..)
o
Li6.6La3Zr1.6Ta0.4012 Garnet 3.5x10-13
353 PFG NMR Hayamizu et al. (2015)
o
O-
Li7La3Zr2012 Garnet 1.8x10-18 298 NMR
relaxometry Kuhn, et al. (2011) o
u,
o
Li7La3Zr2012 Garnet 1.3x10-17 325 NMR
relaxometry Kuhn, et al. (2011)
o
Compound Structure Type Du (m2.s-1) T (K) Technique
Reference
Li7La3Zr2012 Garnet 3.3x10-14 530 NMR
relaxometry Kuhn, et al. (2011) 0
t..)
Graphite (Stage I) Graphite 1-2x10-15 298 NMR
relaxometry Langer, et al. =
,-,
Li metal bcc 5x10-15 298 PFG NMR
(extrapolated) Mali, et al.
(...)
.6.
Li metal bcc 1x10-12 400 PFG NMR
Mali, et al. t..)
.6.
oe
Li4Ti5012 Spinel 3.2x10-15 298 p+-SR
Sugiyama, et al.
Li57Ti5012 Spinel 2.7x10-16 298 NMR
relaxometry Wilkening, et al.
LiTi204 Spinel 3.6x10-15 298 p+-SR
Sugiyama, et al.
/3-Li2TiO3 Li2SnO3 2x10-17 433 NMR
relaxometry Ruprecht, et al.
Lio 12TiO2 micro Anatase component 4.7x10-16 293 NMR
relaxometry Wagemaker, et al. (2001)
Lio 12TiO2 micro Li-titanate component 1.3x10-15 293 NMR
relaxometry Wagemaker, et al.(2001)
P
Lio osTiO2 nano Anatase component 1.9x10-16 293 NMR
relaxometry Wagemaker, et al. (2007) -
,
Lio osTiO2 nano Li-titanate component 5.7x10-16 293 NMR
relaxometry Wagemaker, et al. (2007) ,
oe
.
Lio 12TiO2 nano Anatase component 1.1x10-16 293 NMR
relaxometry Wagemaker, et al. (2007) IV
,
IV
F'
I- i 0 12T i 0 2 nano Li-titanate component 1.8x10-16
293 NMR relaxometry Wagemaker, et
al. (2007) ,A1
,
Lio 12TiO2 nano Li-titanate component 4.9x10-16 413 NMR
relaxometry Wagemaker, et al. (2007) ,
-
Lio55TiO2 nano Li-titanate 1.7x10-16 293 NMR
relaxometry Wagemaker, et al. (2007)
Lio55TiO2 nano Li-titanate 4.6x10-16 413 NMR
relaxometry Wagemaker, et al. (2007)
LiMn204 Spinel 1x10-20 350 NMR
relaxometry Verhoevenm et al.
LiMn204 Spinel 1.8x10-16 623 Tracer
lshiyama, et al. (2016)
LiNb03 LiNb03 7.5x10-15 890 NMR
relaxometry Bork and Heitjans Iv
n
Li3Nb04 Li3Nb04 4x10-21 353 NMR
relaxometry Ruprecht and Heitjans
m
Li3Nb04 Li3Nb04 1x10-16 553 NMR
relaxometry Ruprecht and Heitjans Iv
t..)
o
,-,
Abbreviations: bcc bcc = body-centered cubic, p+-SR = muon spin resonance
o,
u,
o
.6.
o
CA 03137220 2021-10-18
WO 2019/234248 PCT/EP2019/065040
29
Galvanosta tic Intermittent Titration Technique (GITT)
Information on electrode thermodynamics, including phase transitions, and
lithium kinetics
can in principle be extracted from GITT measurements by tracking the voltage
evolution after
a brief current pulse as lithium diffuses and the chemical potential
equilibrates within the
electrode/particles. Reliable quantitative diffusion coefficients, Du, are,
however, difficult to
extract from GITT alone. In order to determine a diffusion coefficient from
GITT
measurements, a diffusion length (L) must be defined but a battery electrode
is a
heterogeneous system. First, it is a composite of active material (here, metal
oxide), porous
carbon, and polymeric binder. Within this composite, there will be a
distribution of particle
sizes (unless single crystals or well-defined particles are employed; even
then the diffusion
varies with lattice direction). Furthermore, different regimes of diffusion
must exist as there
are solid/liquid interfaces and porous electrode structure. Nevertheless, in
an electrode that
does not undergo severe pulverization (e.g. an intercalation electrode), L is
a fixed quantity
throughout the experiment. Variation in L ¨ a parameter required to relate the
rate of
relaxation to the diffusion ¨ causes values of Du to vary significantly
between reports even
for the same material. Thus, while a physically meaningful diffusion
coefficient may not be
extracted, a relative measure of diffusion is readily obtained. For this
reason, we use an
extracted proxy for lithium diffusion (Du=L-2, Fig. 12), which removes the
uncertainty in L and
enables self-consistent analysis of a single electrode and electrodes prepared
under
identical conditions. The addition of quantitative information from another
method, e.g.,
NMR spectroscopy, allows us to calibrate relative changes in Li kinetics to
quantitative
diffusion values throughout a range of lithiation.
As shown in Fig.12, in Nb16W5055, the fastest diffusion is observed from the
dilute limit to
Li45(5)Nbi6W5055, dropping by two orders-of magnitude in the low voltage
window where
more than 1 Li/TM is incorporated. The GITT data indicates that the 2nd
electrochemical
region of Nb16W5055 is broader than typically observed for a two-phase
reaction but the
observed discontinuity in the Du=L-2 values in this region suggests that it is
approaching two-
phase behavior. The average diffusion coefficient in Nb18W16093 is similar to
that of
Nb16W5055. The bronze also displays discontinuities at 2.1, 1.85, and 1.7 V.
In both phases,
the low voltage region ¨ below 1.25 V, well over 1 Li/TM ¨ is characterized by
an increasing
overpotential and suppressed kinetics.
CA 03137220 2021-10-18
WO 2019/234248 PCT/EP2019/065040
Anode Material Ragone Plot
Anode material Ragone plot: the energy density of a cathode material is the
product of
capacity (Q) and voltage (V); however, this product does not work when
comparing anode
materials, where energy and voltage have an inverse relationship. In the
calculation of the
anode material Ragone plot in Fig. 13, the energy (E) is computed based on the
voltage
difference versus a 4.0 V cathode. Thus, normalized to the anode, Eanode =
0/cathodeVanoderQanode=
When compared strictly on the basis of theoretical 1.0 Li+/TM reaction and
crystallographic
density of the active material, titania, niobia, and graphite all display
theoretical charge
densities of greater than 800 A=h=L-1 (Fig. 14). Once experimental capacities
and tap density
are considered (Fig. 13A), the bulk, unoptimized niobium tungsten oxides
presented here
maintain volumetric charge densities of greater than 500 A=h=L-lat 10 and up
to 400 A=h=L-1
at 20 C, volumetric performance that even the most optimised versions of TiO2,
Nb2O5, LTO
cannot achieve. This is not to say that the compounds presented cannot be
improved by
methods such as nanostructuring, calendaring, or carbon-coating as
demonstrated by e.g.
Sun et al. with holey-graphene scaffolds but to prove that large micrometer
particles can be
used for high-rate electrodes and illustrate that nanosizing is not always the
most
appropriate strategy to improve performance. This is evident in a Ragone plot
(Fig. 13B),
which shows the higher energy densities of the new bulk niobium tungsten
oxides, as
compared to state-of-the-art high-rate anode materials and to graphite.
Commercial Materials - Half-Cell Tests
Additionally, in order to test the suitability of the niobium tungsten oxides
as high-rate anode
materials, the commercially-available high-rate cathode materials NM0622
(LiNio6Mno2000202; Targray), lithium iron phosphate (LiFePO4; Johnson Matthey)
and
LiMn204 (MTI Corp) were purchased. These commercial materials were first
characterised
in half-cell geometry against Li metal. Electrochemical measurements were
conducted using
a stainless steel 2032-coin cell and glass microfiber separator in the same
manner as for
Nb16W5055 and Nb18W16093 described above. The commercial cathode material,
conductive
carbon (Li-250 carbon; Deka Chemicals) and PVDF were ground together in the
same
manner as described above to prepare an 80/10/10 electrode (comprising 80 wt%
active
material, 10 wt% carbon and 10wt% polymer) which served as the cathode against
a Li
metal disk anode in half-cell geometry. The electrolyte was 150 pL LP30. No
additives were
used.
NM0622 showed an average voltage of 3.8 V and a practical capacity of 175
mA=h=g-1 under
these conditions (Fig. 17A). LiFePat showed an average voltage of 3.4 V and a
practical
capacity of 165 mA=h=g-1 (Fig. 17B), and LiMn204 showed an average voltage of
4.0 V and a
practical capacity of 120 mA=h=g-1 (Fig. 170).
CA 03137220 2021-10-18
WO 2019/234248 PCT/EP2019/065040
31
Electrode Optimisation - Nbis W5 0 5 5
In order to optimise the performance of the Nb16W5055 electrode, a series of
electrodes were
made to the specifications set out in Table 3, below. Electrochemical
characterisation was
conducting using a stainless steel 2032 coin cell and glass microfiber
separator as described
above. A Li metal disk was used as anode in half-cell geometry. The
electrolyte was 150 pL
LP30. No additives were used.
Table 3 - Electrode Optimisation
Entry Active Separator Carbon Binder
Calendaring
Ref 80% Nb16W5055 Glass microfiber 10% Super P 10% PVDF No
1 80% Nb16W5055 Polypropylenel 10% Super P 10% PVDF No
4% SRB2
2 80% Nb16W5055 Glass microfiber 10% Super P No
1% CMC3
3 80% Nb16W5055 Glass microfiber 15% Super P 5% PVDF No
4 80% Nb16W5055 Glass microfiber 8`)/0 Super P10% PVDF No
2`)/0 CNT4
80% Nb16W5055 Glass microfiber 10% Super P 10% PVDF Yes
1Celgard; 2Styrene-butadiene rubber (Zeon); 3Carboxymethyl cellulose (Sigma
Aldrich); 4Carbon
nanotubes (Sigma Aldrich)
Bulk rate performance was shown to be improved where 5 wt% PVDF or 4 wt% SRB
and
1 w% CMC as binder (Fig. 18). Calandering also consistently improved electrode
performance.
Full Cell Operation with Commercial Electrode Materials
To test the suitability of the niobium tungsten oxides as high-rate anode
materials, full cells
were produced using the commercially-available high-rate cathode materials
NM0622,
LiFePat and LiMn204. Electrochemical measurements were conducted using a
stainless
steel 2032-coin cell and glass microfiber separator in the same manner as for
Nb16W5055
and Nb18W16093 described above. The commercial cathode material, Li-250 carbon
and
PVDF were ground together as described above to prepare an 80/10/10 electrode
(comprising 80 wt% active material, 10 wt% carbon and 10 wt% polymer) which
served as
cathode. The Nb16W5055, Li-250 carbon and PVDF were ground together to prepare
an
80/10/10 electrode (comprising 80 wt% active material, 10 wt% carbon and 10
wt% polymer)
which served as anode. When the cathode comprised NM0622, the anode comprised
Nb16W5055, Li-250 carbon and PVDF in a 80/15/5 ratio. The electrolyte in all
cases was
150 pL LP30. No additives were used. The capacity ratio of the anode and
cathode was
CA 03137220 2021-10-18
WO 2019/234248 PCT/EP2019/065040
32
1:1. For full cell balancing, the theoretical capacity of NM0622 was taken to
be
175 mA=h=g-1.
The full cells were evaluated between 1.0 and 3.0 V. The initial
change/discharge curves of
the full cells with LiFePat and LiMn204 cathodes are shown in Fig. 19A and
19B,
respectively. At a low rate of 0/5, the full cell against LiMn204 showed a
capacity of
120 mA=h=g-1, close to the practical capacity of the cathode. With LiFePat as
cathode, a
capacity of 140 mA=h=g-1 was measured. With NM0622 as cathode, a high capacity
of
165 mA=h=g-1 (Fig. 190). Capacities are quoted based on the mass of the active
cathode
material.
The rate performance of the NMC622 full cell was evaluated in a range of
current densities
from 0/5 to 200. The NM0622 full cell maintained a capacity of 125 mA=h=g-1 at
200,
greater than 75% capacity retention relative to the capacity at 0/5 (Fig 19D).
To test longer
term cycling, the NM0622 full cell was cycled at 100 charge and 100 discharge
rates for
300 cycles. Under these conditions, a capacity of 120 mA=h=g-1 was observed
after
300 cycles, corresponding to greater than 80% capacity retention (Fig. 19E).
At lower rates,
a very high capacity retention of over 98% was achieved for 100 cycles at 10
charge and 10
discharge (Fig 19F).
Investigation of Electrode Degradation
In order to investigate the cause of capacity loss, a full cell using a
Nb16W5055 anode and
NM0622 cathode as described above was cycled for 500 cycles at 10 change and
10
discharge. A 71.1% capacity retention was observed (Fig. 20A). The cell was
disassembled
and the cathode and anode reassembled into a cell against a Li metal electrode
in half-cell
geometry. As shown in Fig. 20B the extracted NM0622 electrode showed a
capacity of
0.48 mAh. The initial capacity of an NM0622 half-cell in this configuration
was 0.72 mAh.
Thus, the NM0622 electrode showed a capacity retention of 67% (a capacity loss
of 33%).
In contrast, the extracted Nb16W5055 electrode showed a capacity of 0.62 mAh
against an
initial capacity of 0.73 mAh in this configuration (Fig. 200). This
corresponds to a capacity
retention of 85% (a capacity loss of 15%). Thus, more than half the capacity
loss observed
in the full cell comprising the Nb16W5055 anode and NM0622 cathode arose from
degradation of the NM0622 cathode. This indicates that the Nb16W5055 material
is
particularly a resilient electrode material.
Mixtures of Nb16W5055 and LTO as anode
Additional experiments were carried out to test the use of a mixture of
Nb16W5055 and LTO
as electrode material. Electrodes comprising three different ratios of
Nb16W5055to LTO were
produced and measured against an Li metal anode in the same manner as for
Nb16W5055
and Nb18W16093 described above. The galvanostatic charge-discharge curves are
shown in
CA 03137220 2021-10-18
WO 2019/234248 PCT/EP2019/065040
33
Fig 21A normalised to gravimetric capacity. The average voltage of 1.55 V is
close to that of
pure LTO (Fig. 5A). In a ratio of 7:3 (Nbi6W5055:LTO), a gravimetric capacity
of
225 mA=h=g-1 was observed at 0/5, while 125 mA=h=g-1 was seen at 200 (Fig.
21B). For 3:7
(Nbi6W5055:LTO), a gravimetric capacity of 190 mA=h=g-1 was observed at 0/5,
while
130 mA=h=g-1 was seen at 200 (Fig 21B). To test longer term cycling, the cell
was cycled at
100 charge and 100 discharge rates for 500 cycles. Under these conditions, a
capacity of
150 mA=h=g-1 was observed, showing greater than 93% capacity retention (Fig.
210).
A full cell comprising a 3:7 (Nbi6W5055:LTO) anode and an NM0622 cathode was
prepared
in a stainless steel 2032-coin cell with glass microfiber separator in the
same manner as for
Nb16W5055 and Nb18W16093 described above. The Nb16W5055, LTO, Super P carbon
and
PVDF were ground together to prepare an 80/15/5 active material/carbon/polymer
anode.
The NM0622, Li250 carbon and PVD were ground together as described above to
prepare
an 80/10/10 active material/carbon/polymer electrode which served as cathode.
The
electrolyte was 150 pL LP30. No additives were used
The full cell was evaluated between 1.0 and 3.25 V, as shown in Fig. 22A. At a
low rate of
0/5 a capacity of 200 mA=h=g-1 was observed. The average voltage was 2.25V.
The rate
performance of the full cell was evaluated in a range of current densities
from 0/5 to 200.
The full cell maintained a capacity of 160 mA=h=g-1 at 200, greater than 78%
capacity
retention relative to the capacity at 0/5 (Fig 22B).
Electrochemical Characterisation of Nb2Mo3014
Electrochemical characterisation of Nb2Mo3014 was conducted in the same manner
as for
Nb16W5055 and Nb18W16093described above, using a stainless steel 2032-coin
cell
(Cambridge Energy Solutions) with a conical spring, two 0.5 mm stainless steel
spacer
disks, a plastic gasket, and a glass microfiber separator (Whatman). The metal
oxide and
conductive carbon (Super P, TIMCAL) were ground by hand in an agate mortar and
pestle in
an 8:1 mass ratio. This powder was ground in a 9:1 mass ratio with
poly(vinylidene
difluoride) (PVDF, Kynar) dispersed in N-methyl pyrrolidone (NMP, Sigma-
Aldrich,
anhydrous, 99.5%). This metal oxide/carbon/polymer electrode served as the
cathode
against a Li metal disk (LTS Research, 99.95%) anode in half-cell geometry.
The electrolyte
was 1 M LiPF6 dissolved in 1:1 v/v ethylene carbonate/dimethyl carbonate
(EC/DMC, Sigma-
Aldrich, battery grade). No additives were used. Electrochemistry was
performed in a
temperature-controlled room at 293 K. A Biologic galvanostat/potentiostat with
EC-Lab
software was used to perform the electrochemical measurements.
At 0/20 it is possible to maintain a gravimetric capacity of ca. 200 mA=h=g-1
(Fig. 15).
Nb2Mo3014 displays an average voltage around 2.0 V. Thus, Nb2Mo3014 may be
used in a
voltage window of, for example 3.0 to 1.4 V, or 3.0 to 1.8 V, and still
provide a high capacity
at a high rate whilst avoiding or minimizing reaction with the electrolyte.
This is
CA 03137220 2021-10-18
WO 2019/234248 PCT/EP2019/065040
34
advantageous as many prospective solid electrolyte materials react with low
voltage anodes
and this could be mitigated using a Nb2Mo3014 anode.
References
All documents mentioned in this specification are incorporated herein by
reference in their
entirety.
Al!press and Roth, J. Solid State Chem. 1971, 3, 209-216.
Arbi, et al., MRS Online Proc. Libr. Arch. 2011, 1313.
Bork and Heitjans, J. Phys. Chem. B 1998, 102, 7303 -7306.
Cruz, at al., Mater. Res. Bull. 2003, 38, 525-531.
Downiem et al., J. Electochem. Soc. 2013, 160, A588-A570.
Fuentes, et al., Solid State Ion. 1996, 92, 103-111.
Fuentes, et al., Solid State Ion. 1997, 93, 245-253.
Gobet, et al., Chem. Mater. 2014, 26, 3558 -3564.
Griffith, et al., lnorg. Chem. 2017, 56, 4002-4010.
Hayamizu, et al., Solid State Ion. 2013, 238, 7-14.
Hayamizu, et al., Solid State Nucl. Magn. Reson. 2015, 70, 21 -27.
Hayamizu, et al., Solid State Ion. 2016, 285, 51 -58.
Hayamizu, et al., Phys. Chem. Chem. Phys. 2017, 19, 23483 -23491.
Holzmann, et al. Energy Environ. Sci. 2016, 9, 2578 -2585.
He at al., Sci. Rep. 2012, 2, 913.
lshiyama, et al., Jpn. J. Appl. Phys. 2014, 53, 110303.
lshiyama, et al., Nucl. lnstrum. Methods Phys. Res. Sect. B Beam Interact.
Mater. At. 2016,
376, 379 -381.
Kasnatscheew, et al., J. Electrochem. Soc. 2017, 164, A2479-A2486.
Kim, et al., Adv. Funct. Mater. 2013, 23, 1214-1222.
Kuhn, et al., Phys. Rev. B 2011, 83, 094302.
Kuhn, et al., Energy Environ. Sci. 2013, 6, 3548-3552.
Kuhn, et al., Phys. Chem. Chem. Phys. 2014, 16, 14669-14674.
Langer et al., Phys. Rev. B 2013, 88, 094304.
Lv, et al., J. Electrochem. Soc. 2017, 164, A2213-A2224.
Mali, et al., J. Phys. F Met. Phys. 1988, 18, 403.
Montemayor, et al., J. Mater. Chem. 1998, 8, 2777 -2781.
Odziomek, et al., Nat. Commun. 2017, 8, 15636.
Oszajca, et al., Chem. Mater. 2014, 26, 5422-5432.
Palacin, et al., Acta Chim. Solv. 2016, 63, 417-423.
Roth and Wadsley, Acta Crystallogr. 1965, 19, 26-32.
Roth and Wadsley, Acta Crystallogr. 1965, 19, 32-38.
Ruprecht, et al., Phys. Chem. Chem. Phys. 2012, 14, 11974 -11980.
Ruprecht and Heitjans, Diffusion Fundamentals 2010, 12,101.
CA 03137220 2021-10-18
WO 2019/234248
PCT/EP2019/065040
Saritha, et al., J. Solid State Chem. 2010, 183, 988-993.
Stephenson, Acta Crystallogr. B 1968, 24, 673-653.
Sugiyama, et al., Phys. Rev. B 2015, 92, 014417.
Sun, et al., Science 2017, 356, 599-604.
Toby and Von Dreele, J. Appl. Crystallogr. 2013, 46, 544 -549.
Verhoevenm, et al. Phys. Rev. Lett. 2001, 86, 4314 -4317.
Wagemaker, et al., J. Am. Chem. Soc. 2001, 123, 11454 -11461.
Wagemaker, et al., Chem. - Eur. J. 2007, 13, 2023 -2028.
Wang, et al., Nano Energy, 2016, 21,133-144.
Waring, et al., J. Res. Nat. Bur. Stand. Sec. A 1966, 70A, 281-303.
Wilkening, et al. Phys. Chem. Chem. Phys. 2007, 9, 6199 -6202.
Wu, at al., Nat. Nanotechnol. 2012, 7, 310-315.
Yamada, et al. Solid State Ion. 2001, 140, 249-255.
Yamada, et al., Mater. Res. Bull. 1999, 34, 955-962.
Yan, et al., J. Mater. Chem. A 2017, 5, 8972 -8980.
Zaghib, et al., Mater. Basel Switz. 2013, 6, 1028-1049.
Zhao, at al., J. Appl. Phys. 2010, 108, 073517.