Note: Descriptions are shown in the official language in which they were submitted.
WO 2020/023800
PCT/US2019/043525
CLINICAL METHODS AND PHARMACEUTICAL COMPOSITIONS
EMPLOYING AMPA RECEPTOR ANTAGONISTS
TO TREAT GLIOBLASTOMA AND OTHER CANCERS
5 Technical Field
The invention relates to drugs and clinical methods tor treating cancer in
mammalian
subjects. More specifically the invention relates to treating dioblastoma and
other cancers
that are positive for expression of AMPA-receptors.
10 Cross-Reference To Related Applications
This application is related to and claims the priority benefit of prior US
Provisional
Patent Application No. 62/703.952. filed July 27. 2018. and prior US
Provisional Patent
Application No. 62/796.032. tiled January 23. 2019. each incorporated herein
by reference in
its entirety for all purposes.
Background of the Invention
Cancer remains a principal mortality risk in human populations, with available
drugs
and treatment methods falling well short of goals to effectively treat and
manage all forms of
cancer in diverse patients. Today cancer persists as the second leading cause
of death in the
20
United States and in other developed nations. The US
National Cancer Institute (NCI)
reported 8.2 million cancer-related deaths worldwide and 14.1 million new
cases diagnosed
in 2012. New cancer diagnoses are projected to rise globally to roughly 24
million by 2030.
According to current NCI statistics. an estimated 1.735.350 new eases of
cancer will have
been diagnosed and 609.640 cancer deaths tolled in the US for the year 2018.
25
The economic burdens of diagnosing and treating cancer
on healthcare systems
around the world arc enormous. Ns ith estimated national expenses for cancer
care in the
United States in 2017 approaching $150 billion. Cancer health costs will
continue to rise as
mean population age and cancer prevalence increase, and more expensive
treatments are
adopted as standards of care.
30
Conventional treatments for cancer typically involve a
combination of surgery.
chemotherapy, radiation and hormonal therapy. Each of these treatment
modalities imposes
significant morbidity and added risks, tbr example adverse metabolic and
reproductive
impacts on healthy cells. immunosuppression and attendant increased risks of
infection_ and
many other adverse health conditions that attend the rigors and insults
&conventional cancer
35 therapy.
1
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
Despite considerable advances in detection and treatment of cancer over the
past
several decades. conventional treatments like surgery-, chemotherapy and
radiation often
achieve only modest improvements in survival, while imposing significant
adverse impacts
on quality of life¨raising questions about cost-effectiveness and overall
clinical benefits of
such treatments.
In view of the foregoing there persists a dire and compelling need in the
medical arts
for alternative tools and methods to prevent, treat and clinically manage
cancer.
Glioblastoma (GUM) is a high-grade cancer of the central nervous system (CNS)
characterized by a highly invasive and treatment resistant phenotype. Patients
almost always
relapse after an either initially successful surgical resection with or
without cherno- and
radiotherapy (Ishiuchi et al, 2007). GUM tumors often exhibit resistance to
chemotherapy
and/or radiotherapy, which resistance may be acquired during a course of
treatment (Ishiuchi
et al, 2007).
Ternozolomide (TM7.) is a DNA methylating agent that is the current standard
of care
drug for treating GUM. TM/ mediates anti-cancer effects through genotoxie
activity_ and is
etTective in GBM treatment due in part to the drug's ability to bypass the
blood-brain barrier
(BBB) (Prasad et al. 2011). Unfortunately, TM/. shows limited efficacy for
long-term
treatment of GUM, and many patients appear to be refractory to TMZ treatment.
While considerable research has been attempted to elucidate the
pathophysiology of
OBM, a vast majority of drugs tested against GUM are unable to pass the BBB to
yield
sufficient drug levels in the brain to mediate anti-cancer effects (Liu et al.
2015).
Previous reports have suegested a role for glutamate in the proliferation and
migration
of glioma cells_ in a manner similar to glutamate function in neuronal
development (Rzeski et
al, 2001: Ishiuchi et al. 2002). Tumors of the CNS that release glutamate
may cause
eytotoxieity and cell death in neighboring neurons. which is postulated to
facilitate cancer
invasion into neighboring tissues (Ttikano et al_ 2001). Glutamate positive
tumors may also
grow at an enhanced rate. potentially implicating glutamate signaling as an
important
mechanism in the etiology of GUM (Takano et al. 2001).
Among many types of known glutamate receptors ((iRs). the AM PA-type glutamate
receptor (AMPAR) may be overexpressed in certain types of cancer, including
some forms of
CNS cancers (Liu eta!, 2015) and non-CNS cancers (Steptilak et al, 2007:
Herner et al. 2011:
Romeling et al. 2014: Hu et al_ 2014). AMPAR activation may be linked to
increased cancer
cell invasiveness (Pia et al_ 2008). proliferation. andior activation of the
113K/AktlinTOR
signaling axis (Ishiuchi et al. 2007). The 1313K/A ktimIOR signaling axis has
been implicated
2
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
in chemotherapy- resistance in GBMõ and available drugs reported to disrupt
this pathway,
such as raparnyein. do not penetrate the BBB. GUM cancer stern cells
(suspected to be
capable of re-establishing tumors after ablation with surgery, chemotherapy or
radiotherapy)
may express strikingly high amounts of functional AMPARs (Oh eta!, 2012).
In view of the foregoing, a compelling need exists in the art for new drugs
and
therapeutic methods for treating cancer. Related needs are unmet for new drugs
for treating
refractory or treatment-resistant forms of cancer, including cancers of the
CNS. For CNS
cancers, particularly brain cancers such as Cif3M. there is a particularly
urgent need for anti-
cancer drugs capable of transiting the BBB to yield effective drug
concentrations within
protected CNS compartments. most notably within the brain.
Summary of Exemplar\ Embodiments of the Invention
The instant invention satisfies the foregoing needs and fulfills additional
objects and
advantages by providing novel AMPAR antagonist drugs effective to treat AMPAR
positive
cancers and AMPAR dependent cancers_ including AM PAR positive and AMPAR
dependent
cancers of the central nervous system (CNS). In exemplary,' embodiments the
invention
provides compositions and methods employing a novel anti-cancer drug.
Perampanel (PMP)
[2-(2-oxo-1-pheny1-5-pyridin-2-y1-1.2-dihydropyridin-3-y1) benzonitrile],
heretofore reported
for limited clinical use as an antispasmodic drug.
AMPA receptor antagonists have been investigated for antiseizure activity both
preclinically and clinically, with mixed success. The prototypical competitive
AMPA
receptor antagonist 2.3-dihydroxy--6-nitro-7-sulfamoyl-benzoll1 quinoxaline
(NBOX) showed
activity in maximal electroshock (M NS) and pentylenetetnizole (PTl)--induced
seizure
models (Yamaguchi et al.. 1993). but has poor solubility. resulting in
precipitation in the
kidney at therapeutic plasma levels. Derivatives of NBOX with polar
constituents have
shown improved solubility, but these molecules exhibit reduced blood¨brain
barrier (13B13)
penetration (Weiser, 2005). Prototypical noncompetifive AMPA receptor
antagonists. such
as 2.3-benzodiazepine-type compounds. have shown weak in vitro efficacy
compared with
competitive antagonists (Weiser. 2005). Talampanel. a recently developed
noncompetitive
AMPA receptor antagonist. has been evaluated in a number of clinical trials (I
lowes ez. Bell,
2007). but has a relatively short half-life militating- against its potential
clinical utility
(Langan et al., 2003). More recently. Steinhoff et al.. 2013. reported
beneficial activity of
Peramplanelõ a noncompetitive. selective AMPA receptor antagonist, as an
antiepileptic drug
undergoing clinical study for refractory partial-onset seizures.
3
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
According to the surprising discoveries herein, peramplanel (PMP) has now been
identified to possess novel and potent anti-cancer activity. Within the coin
positions and
methods of the invention. PMP. along with its active analogs and derivatives,
and other
selected AMPAR antagonists disclosed herein. potently inhibit AMPAR positive
and
5 AMPAR dependent cancers, including CNS cancers such as GUM.
The invention provides novel compositions and methods for treating cancer
using
AMPAR antagonist compounds such as PMP to reduce or prevent the occurrence.
remission.
growth. severit_v and/or one or more adverse svmptom(s) of AMPA-receptor
positive cancers
in mammalian subjects, including humans. In illustrative embodiments. the
AMPAR
10 antagonist comprises a PMP compound (including anti-cancer effective
chemical analogs,
derivatives, conjugates, solid crystalline forms. solvates and/or different
salt forms of a PMP
compound). which is clinically effective as an anti-cancer agent to treat or
prevent cancer in
mammalian subjects. In exemplary embodiments. PMP is administered to a human
patient
presenting with an AMPAR positive cancer condition in a delivery mode,
formulation and
15 dosage sufficient to alleviate one or more symptoms of the targeted
cancer condition in the
patient
In certain embodiments the PMP compound is perampanel [2-(2-oxo-l-phenyl-5-
pyridin-2-yl-I.2-dihydropyridin-3-y1) benzonitrile] formulated in a
biologically acceptable
composition for administration to a human subject.
20 In related embodiments. novel clinical methods are provided
herein employing a
peramplanel or related compound administered to a mammalian subject, wherein
the
peramplanel compound exerts oncolytie effects against a targeted cancer cell
or tumor
sufficient to kill a targeted cell or tumor. reduce size of a tumor. impair
tumor growth.
prevent or reduce cancer invasiveness, reduce or delay cancer recurrence,
and/or alleviate one
25 or more symptoms associated with the treated cancer condition.
In more detailed embodiments. peramplanel and related compounds are employed
in
effective anti-cancer methods for treating glioblastotna (GUM). The
peramplanel compound
is administered to a mammalian subject with current or prior diagnosis of (IBM
in a dosage
Ibrin. amount and regimen sufficient to prevent or reduce the occurrence.
severity, recurrence
30 and/or related symptoms of GIIM in the subject. In related embodiments,
pharmaceutical
compositions and delivery methods are provided that yield surprisingly high
therapeutic
concentrations of the peramplanel compound in a CNS compartment of the
subject. e.g., in
the brain, yielding potent anti-CNS-cancer therapeutic effects.
4
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
In other detailed embodiments. a peramplanel compound is administered with a
secondary therapeutic agent in combinatorial Ibrinulations or coordinate
treatment methods to
yield desired therapeutic advantages. In exemplary embodiments, a peramplanel
compound
is coordinately administered with a second anti-cancer drug to treat cancer,
whereby anti-
5 cancer efficacy is enhanced and/or adverse side effects are reduced. In
one illustrative
embodiment, peramplanel is coordinately administered with temozolomide (TMZ)
to treat
GBM, pancreatic cancer, or another form of cancer. In another embodiment,
peramplanel is
coordinately administered with eisplatin to treat an AMPAR positive cancer,
for example an
AMPAR positive pancreatic cancer. In other exemplary embodiments, peramplanel
is
10 coordinately administered with hydroxyurea to treat an AMPAR positive
cancer. In other
embodiments, peramplanel is coordinately administered with Carmustine (BCNU)
to treat an
AMPAR positive cancer.
In related treatment methods a peramplanel compound is coordinately
administered
before or after a conventional cancer treatment. Ibr example surgery,
chemotherapy or
15 radiation treatment, with or I.% ithout a secondary anti-cancer agent or
other secondary
therapeutic drug.
In other detailed aspects of the invention, methods and compositions arc
provided
employing a peramplanel compound to reduce oncogenic activity by disrupting a
glutamate-
induced cancer potentiation process (e.g.. glutamate-stimulated cancer cell
proliferation.
20 tumor growth, cancer invasion or other cancer-potentiation activity).
In yet additional embodiments of the invention, AMPAR antagonist compounds are
employed in novel clinical methods and compositions to treat lung cancers,
breast cancers,
pancreatic cancers, liver cancers. colorectal cancers and other forms and
symptoms of cancer
conditions in human subjects.
Brief Description of the Drawing5
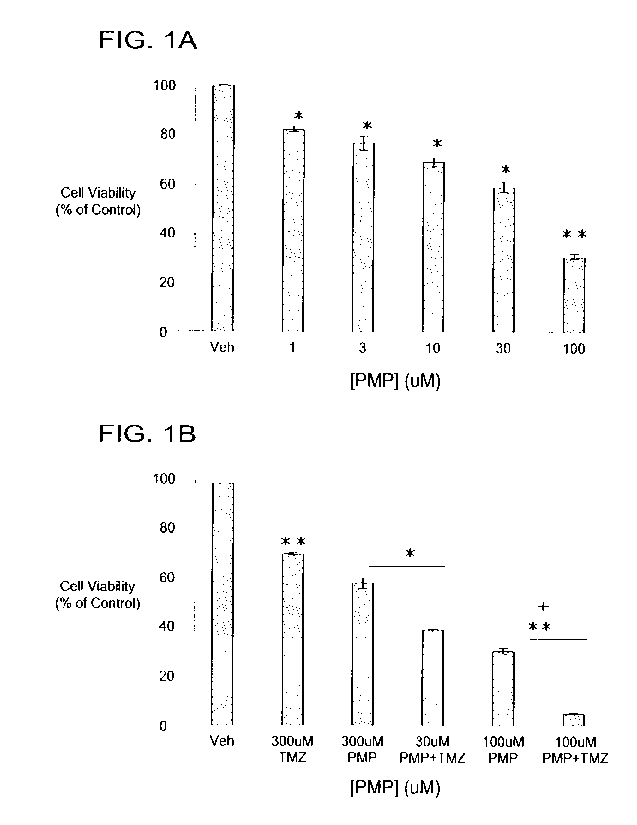
Figure I is a related graph series documenting the effects of PMP on T98G GBM
cell
viability. Dose-response curve of A) PMP on cell viability B) Interactions
with PMP and
TMZ. Data are presented as mean +/- SEM o12 or more independent experiments
performed
30 in quadruplicate. ANOVA, P<0.00 I for PMP suggesting a significant dose-
dependent
response. *p<0.05, **p<0.01 t-test_ compared to vehicle-treated control or
singular drug
treatment. +p<0.05. king's synergy test, demonstrating significant synergistic
interaction.
Figure 2 is a related graph series documenting the effects of PMP on Pane I
cell
viability. Dose-response curve of: A) PMP on cell viability: B) Interactions
with PMP and
5
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
3uM cisplatim C) Effects of glutamate on panel cell viability: and D)
Interactions between
glutamate and PMP. Data are presented as mean 4-/- SEM of 2 or more
independent
experiments performed in quadruplicate. ANOVA. P<0.00 I for PMP and glutamate,
indicating a significant dose-dependent response. *p<0.05. **p<0.0 I. t-test,
compared to
vehicle-treated control or singular drug treatment. -hp<0.05, 4-Fp<0.01,
king's synergy test,
demonstrating significant synergistic interaction. *p<0.01. t-test, compared
to viability of
glutamate- or PMP-treated cells alone.
Haim! 3 is a flow chart illustrating seven candidate anti-cancer chemical
derivatives
(D I 4)7) of peramplanel (PMP), wherein PMP is modified according to known
methods of
conventional rational design chemistry, to yield new candidate compounds for
testing to
determine anti-cancer activity and other beneficial properties.
Detailed Description of Exemplary Embodiments of the Invention
The invention provides AMPAR antagonist.. compounds exemplified by peramplanel
(PMP), shown to be surprisingly effective in treating cancers. including CNS
cancers, in
mammalian subjects. Among the discoveries presented here. peramplanel is shown
to exert
potent. direct oncolytic effects against cancer cells in assays accepted to
predict clinical anti-
cancer activity in human subjects. More specifically the examples below show
that PMP
potently disables viability of CNS and non-CNS cancers. as demonstrated by
direct oncolytic
effects against glioblastorna (613M) and pancreatic cancer cells. Related
studies Further evince
that peramplanel exerts surprisingly potent_ additive or synergistic anti-
cancer effects in
coordinate use with other chemotherapeutic drugs.
The clinical methods and pharmaceutical compositions and formulations of the
invention provide novel tools to treat, prevent and clinically manage a wide
range of cancers
in mammalian subjects, including humans. Any type and form of cancer occurring
in humans
and veterinary subjects may be amenable to treatment according to the
teachings herein,
including, but not limited to: central nervous system (CNS) cancers including
various forms
of brain cancer: lung cancer; prostate cancer: breast cancer: skin cancers_
for example
melanoma: liver cancer: thyroid cancer: esophageal cancer: sarcomas: colon and
rectal
cancers: bladder cancer: gall bladder cancer: stomach cancer: renal cancer:
ovarian cancer:
uterine cancer: cervical cancer: non-I lodgkin's lymphoma: acute myelogenous
leukemia
(AML): acute lymphocytic leukemia: chronic lymphocytic leukemia (CU..):
my,eloma:
mesothelioma: pancreatic cancer. Hodgkin's disease: testicular cancer:
Waldenstrotn's
disease: head/neck cancer: cancer oldie tongue. viral-induced malignancies
(e.g., cancers
6
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
induced by SV40 virus), and other candidate types and forms of cancers that
will be apparent
to skilled artisans.
Subjects amenable to treatment mak have cancer of any stage of development and
etiology, including, but not limited to, cancers marked by rapid increases in
5 cellular/histological abnormalities and/or elevated tumor marker
expression in biopsies or
blood samples_ rapid tumor proliferation and/or growth_ metastasis_ among
other disease
progression indicators, up to and including stage III and stage IV cancers,
even refractory
stage III and IV shown to be -treatment resistant cancers' (e.g., to
effectively subjects with
cancers, such as glioblastoma, persisting or relapsing after ineffective,
conventional anti-
10 cancer treatment(s) (e.g._ surgery. radiation and/or chemotherapy)). In
exemplary
embodiments of the invention an effective AMPAR antagonist drug such as PMP
effectively
prevents or treats (i.e., reduces the severity, progression and/or adverse
side effects of) cancer
in treatment resistant subjects. defined as subjects presenting after one or
more rounds of
conventional oneotherapy (e.g.. chemotherapy. radiation.. surgery and/or
hormonal therapy),
15 with actively progressing or unstable metastatic disease. In other
embodiments the
compositions and methods of the invention are useful for treating other
"refractory- patients
who ma k not otherwise tolerate or be lit for conventional cancer treatments
such as
chemotherapy.
Certain cancer types and disease conditions are contemplated herein to be
particularly
20 amenable to treatment using the AMPAR antagonist drugs and methods of
the invention. In
certain embodiments. the AMPAR antagonist drugs and methods of the invention
are
particularly effective against -AMPAR dependent" cancers. As used herein the
term
-AMPAR dependent refers to cancers that distinctly overexpress AMPA receptors,
or whose
appearance. growth, and/or disease progression may otherwise be determined to
be AMPAR-
25 dependent. In more detailed aspects. "AMPAR-dependent" cancers are not
limited to cancers
whose occurrence, persistence or progression require abnormally elevated AMPAR
receptor
expression or activity, indeed they may include cancers with normal or even
subnormal
expression of A MPARs. which through disease-associated changes in AMPAR
structure or
function_ or any other disease-associated change affecting AMPAR metabolism or
pathology.
30 are particularly susceptible to AMPA receptor interference or blockade
using PMP or other
candidate AMPAR drugs of the invention.
In this context, the use of AMPAR antagonist drugs exemplified by PMP,
according
to the teachings herein. effectively treats or prevents a wide range of
cancers contemplated to
represent -AMPAR-dependent cancersi including but not limited to brain cancer.
breast
7
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
cancer. colorectal cancer. hepatocellular cancer_ leukemia, melanoma, lung
cancer, pancreatic
cancer. renal cancer. and other candidate cancer types or cases determined to
be clinically
susceptible to AMPAR interference or blockade by PMP or another useful AMPAR
antagonist.
5 Each of the anti-cancer methods of the invention involves
administration of a suitable,
effective dosage amount of PMP or another useful AMPAR antagonist to a
subject.
Typically, an effective amount will comprise an amount of the active compound
(e.g.. PMP)
which is therapeutically effective. in a single or multiple unit dosage form.
over a specified
period of therapeutic intervention, to measurably alleviate the targeted
cancer condition.
10 Within exemplary embodiments. PMP is used as the sole or primary active
drug. In other
embodiments. an intermediary or precursor compound to PMP. or a rationally-
designed
analog or derivative of PMP (i.e., a related compound having close structural
and functional
similarity to PMP) is employed. The PMP or other effective AMPAR antagonist is
typically
formulated in a pharmaceutical composition NN ith one or more pharmaceutically
acceptable
15 carriers, excipients, vehicles. emulsifiers, stabilizers_ preservatives,
buffers, and/or other
additives that may enhance stability, delivery. absorption, half-life,
efficacy,
phannacokinetics, and/or pharmacodynamics, reduce adverse side effects, or
provide other
advantages for pharmaceutical use.
Anti-cancer effective dosage amounts of PMP and other effective. anti-cancer
20 AMPAR antagonists of the invention will be readily determined by those
of ordinary skill in
the art, depending on clinical and patient-specific factors. Suitable
effective unit dosage
amounts of the active compounds for administration to mammalian subjects.
including
humans. may range from a minimum daily dose of 1-2 am up to a maximum
prospective dose
between about 200-500 or 300-1.000 mg/day. or greater. In certain embodiments.
the anti-
25 cancer effective dose is between about 2 mg-200 mu/day, in other
embodiments between
about 20-400 mg/day. 50-500 mg/day. 200-600 mg/day. or another anti-cancer
effective dose
or dosage range that can be adjusted based on patient specific factors to
optimize efficacy and
minimize adverse side effects. The PMP or other AMPAR antagonist may be
administered in
a single dose, or in the form of a multiple periodic dosing protocol_ for
example in a dosing
30 regimen comprising from I to 5. or 2-3 doses administered per day. per
week, or per month.
The amount. timing and mode of delivery of the anti-cancer compositions of the
invention will be routinely adjusted on an individual basis, depending on such
factors as
patient weight, age. gender, and condition of the individual, the acuteness of
the subject's
disease and severity symptoms_ whether the administration is prophylactic or
therapeutic,
8
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
prior treatment history (including e.g., any prior history and responsiveness
to chemotherapy
or other cancer treatment treatment) and on the basis of other factors known
to effect drug
delivery, absorption, phannacokinetics and efficacy. An effective dose or
multi-dose
treatment regimen for the instant AM PAR antagonist formulations will
ordinarily be selected
to approximate a minimal dosing regimen necessary and sufficient to
substantially prevent or
alleviate the cancer condition, and/or to substantially prevent or alleviate
one or more
symptoms associated with that condition.
A dosage and administration protocol will often include repeated dosing
therapy over
a course of several days or even one or more weeks, up to several months. or
even a year or
more. An effective treatment regime may also involve prophylactic dosage
administered on a
daily or multi-dose per day basis lasting over a course of days_ weeks, months
or even a year
or more.
Various assays and pre-clinical and clinical model systems can be readily
employed to
determine therapeutic effectiveness of the anti-cancer compositions and
methods invention.
For example. these may detect/monitor a decrease in overt symptoms, such as
pain (e.g., as
measured using any of a variety of pain scales including. but not limited to.
the Visual
Analog Seale. McGill Pain Questionnaire._ Descriptor Differential Scale. Faces
Pain Scale,
Verbal Rating Scale. Simple Descriptive Pain Scale. Numerical Pain Scale (N
PS). or
Dolorimeter Pain Index). More detailed detection/monitoring may document, for
example, a
decrease in circulating tumor cells (CFCs). reduction in tumor size. collapse
or disappearance
of tumors. softening of tumors. liquefaction ofttimors, or a decrease in
cytological or
histochemical cancer markers, among many other conventional diagnostic indicia
of cancer
disease stasis. progression and/or remission.
Effectiveness of the anti-cancer methods and compositions of the invention are
generally demonstrated by a decrease in incidence, severity and/or associated
symptoms of
cancer. which will typically involve a decrease of 5%. 10%. 25%. 30%. 50%,
75%. 90% or
more in comparison to incidence/levels of the same diagnosed indicator/state,
or attendant
symptom(s) in suitable control subjects (or compared to known baseline or
median data for
like, treated or untreated subjects). For example. PMP-treated cancer patients
will often
exhibit a decrease in number or size of targeted tumors, a decrease in
circulating tumor cells
(CTCs) or Cancer Stem Cells (C'S('s) in successive blood assays. and/or a
decrease in one or
more tumor-associated cytological. histoehemical or blood markers, during a
course of
treatment, of from 25%-30%. 50%. 75% or higher. 90% and up to total absence of
the disease
indicator(s) to a limit of detectability associated with the employed
assay(s). Monitoring for
9
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
effective cancer prevention and treatment of the invention can employ any of a
vast array
conventional detection and monitoring tools and indicia. as will be apparent
to those skilled
in the art. For example. CTC monitoring using blood samples of' patients can
utilize flow
cytometry. immunobead capture. fluorescence microscopy, standard and density
5 centrifugation. cell culturing, and immunocytoeltemistry . Similarly,
tumor monitoring can
employ x-ray. MRI_ CT or PET scans. among other methods and tools. For economy
these
and other routine, well-known cancer disease detection and monitoring
technologies will not
be reiterated here.
As noted above, exemplary embodiments of the invention employ peramplanel
(PMP)
10 as an anti-cancer effective AMPAR antagonist compound. Perampanel is
structurally distinct
from other AM PAR antagonists, which as a group show a great deal of
structural diversity
(for example. as illustrated in Table 1 comparing the structure of PMP to two
other AMPAR
antagonists, Telampanel and NRQX.
15 Table I Diverse Chemical Structure of AMP.AR
Antagonists
tIt4(
o
Nit
0=S =0
Fr
N11.,
N
NBQX
CN
<
-1\
(
Perampanet
cr.)
1
Tata m panel
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
According to the discoveries herein. PMP potently reduces or prevents the
occurrence, remission, growth and/or severity of targeted cancers in mammalian
subjects,
including humans. In certain embodiments. PMP is effective to prevent or treat
(including to
reduce one or more adverse symptom(s) on_ an AMPAR positive cancer in a human
cell
5 population, tissue, organ or whole patient. For clinical use, effective
anti-cancer
compositions may comprise a prototypical PMP compound [2-(2-oxo-l-pheny1-5-
pyridin-2-
y1-1.2-dihydropyridin-3-y1) benzonitri le]. or any effective prodrug.
metabolite, analog.
derivative, conjugate, solid crystalline lbrrn, solvate and/or advantageous
salt form of PMP
shown to be clinically effective as an anti-cancer agent.
10 In view of the disclosed potent anti-cancer effects of peramplanel
(PMP). the
invention further provides various chemical analogs and derivatives of PMP
that actively
treat or prevent cancer_ and in certain cases provide additional clinical
advantages, for
example improved solubilit). enhanced blood-brain barrier penetration,
prolonged half-life.
increased AM PAR antagonist activity, among other functional improvements.
15 Figure 3 provides a flow chart illustrating seven candidate anti-
cancer chemical
derivatives (Dl-D7) of peramplanel (PMP) contemplated for anti-cancer testing
within the
clinical methods herein. The illustrated compounds. Dl-D7 can be readily
produced. along
with many additional PMP analogs and derivatives, employing known methods of
conventional rational design chemistry. Such routine design and synthesis
efforts will yield a
20 diverse array of new candidate compounds for testing within the methods
of the invention, to
determine anti-cancer activity and other beneficial properties. In general
terms. each of the
available R groups identified within or attached to each of the aromatic rings
of PMP can be
altered (e.g.. by chemical deletion, substitution or addition) to yield new
candidate PMP
derivative drugs as described. The exemplary derivatives shown in Figure 3 ¨
PMP DI may
25 be designed. tested and selected to have increased solubility. improved
half-life, better
delivery or penetration to desired targets or compartments (e.g.. to deliver
an effective dose
within a tumor mass, to transit the blood brain barrier (BBB) in anti-cancer
effective
amounts. to survive first pass metabolism and transport to a target organ in
effective plasma
concentration. etc.). According to the illustrative embodiments here. PMP
derivatives 1)2,
30 D3 and D4 are designed to have more hydrophilic character. whereby they
will have
improved BBB penetration and accumulate to an effective concentration greater
in the CNS
to mediate clinical benefits of treating and/or preventing CNS related cancers
such as GBM.
PMP D5 is designed to provide increased solubility for improved drug delivery.
bioavailability and clinical efficacy. PMP 1)6 is an exemplar prodrug (a
glycine ester) of
11
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
PMP D5. which will be rapidly hydrolyzed into PMP DS in the blood by plasma
glycine
esterases. thus providing the contemplated prodrug benefits in addition to
increased
solubility. PMP D7 and 138 are likewise more hydrophilic derivatives of PMP
which will
penetrate the BBB and accumulate in effective concentrations for anti-cancer
drug effects in
5 the CNS.
In other exemplary PMP derivative and analog design. each R group may be
modified
to a same. or different. derivative or analog R group identity. Rational
design chemical
alterations to PMP can include alterations wherein an original PMP R group is
altered to a
new structural identity selected from, for example: a substituted or
unsubstituted lower
10 hydrocarbon including an alkyl. alkenyl. alkanoyl. alkynyl. aryl. aroyl.
aralkyl, alkylamino,
aryloxy, hydrogen, carboxyl. nitro, thioalkoxy,-. thioaryloxy. thiol,
cycloalkenyl cycloalkyl,
heterocycloalkyl. heteroaryl. aralkyl, amino acid. peptide_ dye. fitiorophore,
carbohydrate or
polypeptide; a hydrogen. hydroxyl. sulfyhydryl, fluorine, methyl, ethyl,
propyl, benzyl, 2-
bromovinyl amino. hydroxymethyl. methoxy. halogen. pseudohalogen. cyano,
carboxyl,
15 nitro_ thioalkoxy. thioaryloxy, or thiol: a substituted or unsubstituted
lower hydrocarbon
containing 1 to 20 carbons such as alkoxyearbonyl. allkoxycarbonylamino.
amino, amino
acid. aminocarbonyl. arninocarbonyloxy. aralkyl, aryloxy, carboxyl.
cycloalkenyl. alkyl,
cycloalkyl, heterocycloalkyl. aryl. heteroaryl. aralkyl. amino acid, peptide,
dye, tluorophore,
carbohydrate or polypeptide: a heteroatom such as oxygen, sulfur or nitrogen:
and/or an
20 integral member of a new 5. or 6. member exocyclic ring structure, among
other alterations
where feasible to Yield a viable test candidate derivative. In more detailed
embodiments, one
or more R group(s) of PMP can be modified to a hydrogen. hy-droxyl,
sulfyhydryl. benzyl. 2-
bromoyinyl amino, hydroxymethyl. methoxy. halogen. pseutiohalogen, cyano.
carboxyl,
nitro, thioalkyl. thioaryl, thiol, substituted or unsubstituted hydrocarbons
containing 1 to 20
25 carbons. alkoxy.icarbonyl. alkoxycarbonylamino. amino4 amino acid,
aminocarbonyl,
aminoearbonyloxy. aryloxy. carboxyl, cycloalkenyl, substituted or
unsubstituted cycloalkyl,
substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted
aryl. substituted or
unsubstituted heteroaryl. substituted or unsubstituted aralkyl. peptidyl. dye.
fluorophore.
carbohydrate or polypeptidyl. azido, nitrite. substituted benzoyl or hydroxyl
substituted with
30 substituted or unsubstituted hydrocarbon containing 1 to 20 carbons_
and/or an alkanoyl of a
main chain of 1 to 20 carbon atoms, among many other contemplated derivative
changes
obtainable and testable according to the teachings herein without undue
experimentation.
Within additional aspects of the invention, combinatorial formulations and
coordinate
treatment methods are provided that employ an effective amount of PMP or
another anti-
12
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
cancer effective AM PAR antagonist compound or composition, and one or more
secondary
or adjunctive agent(s) combinatorially formulated or coordinately administered
with the
AMPAR antagonist compound or composition to yield an enhanced anti-cancer
composition
or method. Exemplary combinatorial formulations and coordinate treatment
methods in this
5 context employa PMP compound in combination with the one or more
secondary anti-cancer,
anti-viral, and/or immune-stimulatory effective agents or drugs.
Exemplary combinatorial formulations and coordinate treatment methods of the
invention employ PMP or another anti-cancer effective A M PAR antagonist
compound or
composition in combination with one or more secondary or adjunctive anti-
cancer effective
10 agents. for example one or more chemotherapeutic agents. Employing
general terminology
for "chemotherapeutic drugs and adjunctive anti-cancer therapies", these
secondary
agents/therapies for use within the invention may include any anti-cancer or
anti-proliferative
agent. agents that destroy or -'reprograrn- cancer cells, agents that destroy
blood vessels
associated with neoplasms or hyperproliferative conditions and other classes
of drugs
15 harmful to neoplastic cellular targets. In this regard. useful
chemotherapeutics and adjunctive
therapies for use within the invention include. but are not limited to:
(1) Tubulin depolyrnerizing agents like toxoids:
(2) DNA damaging agents and agents that inhibit DNA synthesis:
(3) Anti-metabolics:
20 (4) Anti-angiogenic agents and vascular disrupting agents (VDAs);
(5) Anti-cancer antibodies:
(6) Endocrine cancer therapies:
(7) Immuno-modulators;
(8) Ilistone deacetylase inhibitors:
25 (9) Inhibitors of signal transduction:
( 10) Inhibitors of heat shock proteins:
(11) Retinoids:
(12) Growth Factors and Modulators of growth factor receptors:
( 13) Anti-mitotic compounds:
30 (14) Anti-inflammatory agents such as COX inhibitors: and
(15) Cell cycle regulators (eg, check point regulators and telomerase
inhibitors).
In related embodiments. coordinate anti-cancer treatment methods of the
invention
can include coordinate administration of one or more anti-cancer AMPAR
antagonist
13
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
compounds with a secondary anti-cancer agent selected from az.acitidine.
bevacizumab.
bortezomib. capecitabine, cetuximab. clofarabine. dasatinib, decitabine,
doeetaxel, emend.
erlotinib hydrochloride. exemestane_ fulvestrant. gefitinib. ueincitabine
hydrochloride,
irnati nib mesylate. imiquimod. lenalidomide. letrozole nelarabine.
oxaliplatin, paclitaxel,
5 docetaxel, paliferm in. panitumumab. pegaspargase_ pemetrexed di sodium,
rituximab,
sorafenib tosylate, sunitinib malate, tamoxifen citrate, targretin,
temozolomide, thalidomide,
and/or topotecan hydrochloride. Additional contemplated secondary anti-cancer
effective
agents in this context include. but are not limited to. interleukin-2.
interferon a. filgrasten_ G-
CSF, epoetin alla, erythropoietin. oprelvekin.
trastuzumab, vorinostat. antibiotics,
10 coenzyme q. palladium lipoic complexes including, for example_ poly-
MVAK.
antineoplastins, cartilage_ hydrazine sulfate; milk thistle, electrolytes such
as calcium
carbonate, magnesium carbonate. sodium bicarbonate_ and potassium bicarbonate;
oxidizing
agents. including. but not limited to. cesium chloride, potassium chloride,
potassium orotate
and potassium aspartate: immunoulobulins, colostrum: and vitamin and mineral
supplements,
15 including but not limited to. zinc chloride. magnesium chloride,
pyridoxine, vitamin B- 12, B
complexes_ folic acid, sodium ascorbate_ and probiotics. Additional secondary
therapies may
include conventional chemotherapy, radiation therapy. and/or surgery.
In certain illustrative embodiments directed to treatment of glioblastorna
(GBM),
pancreatic cancer and other AMPAR-dependent types/forms of cancer. the AMP or
other
20 AMPAR antagonist is coordinately administered with temozolomide (TMZ).
In related
embodiments, the PMP or other AMPAR antagonist is coordinately administered
with
eisplatin to treat an AMPAR-dependent cancer, for example an AMPAR-positive
pancreatic
cancer. In other exemplary embodiments. an anti-cancer effective PMP or other
AMPAR
antagonist compound is coordinately administered with hydroxyurea to treat an
AMPAR
25 positive cancer. In other embodiments. the PMP or other AMPAR antagonist
is coordinately
administered with Carmustine (BCNIJ) to treat an AMPAR positive cancer.
In other coordinate methods and compositions, anti-cancer effective AMPAR
antagonist administration is combined with a secondary anti-cancer agent or
therapy, e.g.,
selected from a transcription inhibitor (e.g._ Terameprocol). a telomere
disrupting agent (e.g.,
30 TREI inhibitors such as ETP-4707). an inhibitor of a gene splicing
protein (e.g., a PRMT5
inhibitor), an indoleamine 2_ 3, dioxegenase (ID()) inhibitor. lapatinib
ditosylate enzyme
blocker. anti-cancer antibodies. antibody fragments and related -biologics1
for example
Adavosertib. tumor treating fields, and radiation. alone or in any combination
with other
secondary or adjunctive cancer agents and treatments described herein.
14
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
In related embodiments. coordinate anti-cancer treatment methods of the
invention
can include coordinate administration of one or more anti-cancer AMPAR
antagonist
compounds, such as PMP_ in combination with one or a plurality of any
combination of
secondary therapeutic agent(s) or therapy(ies) selected from: NMDA antagonists
such as
5 memantine for the treatment of various cancer anti PO- I /MI.- I therapy:
CSF I R inhibitors
such as PLX3397 and PLX5622: cannabinoid drugs: anti-rnalarials such as
mefloquine.
primaquine, chloroquine, hydroxychloroquine: Riluzole/troriluzole treatment:
antihistamines
such as clemastine: biguanides such as metforrnin or phenformin: anti-cancer
biologics such
as Pembrolizumab or Nivolumab: selective serotonin reuptake inhibitors
(SSR1s): tricyclic
10 antidepressants (TCAs): AMPA receptor positive allosteric modulators
(Ampakines):
levetiracetam (Keppra). and other agents. therapies and combinations
contemplated herein.
Within exemplary embodiments, typical drug doses or combinatorial drug doses
(median or average doses among a treated patient class) may include, for
example,
peramplanel administered at about 12mg/day (exemplary range 5-50 mg/day),
Memantine at
15 about 20 mg/day. (exemplary ranee 5-75 mg/day). R I tin)) e at about
50mg/day (exemplary
range 10-100 mg/day). PLX3397 at about 1000 mg/day (exemplary range 300 2500
mg/day).
Anti-malarials at about 250 mg every other day (exemplary range 50-200 mg/day
or every
other day), Metformin at about 2 g/day (exemplary range 300 mg-4 g/day).
Pembrolizumab at
about 2 mg/kg every 3 weeks (exemplary range 0.05-10 mtz/kg every 1-4 weeks),
Nivolumab
20 at about 3 me/kg every 2 weeks (exemplary range 1-5 mg/kg, every 1-3
weeks).
Levctiracetain at about 500 mg twice a day. Clemastine lumarate at about 2.5
mg per day.
(exemplary range 1-5 lug per day), Escitalopram at about 20 mg./day (exemplary
range 5-75
mg/day). sertraline at about 200 mg/day (exemplary ranee 50-800 mg/day),
fluoxetine at
about 20 mg/day (exemplary range 5-200 mg/day). Imipramine hydrochloride at
about 25-50
25 mg/day (exemplary range 2-400 mg/day), Ampakines at about 900rng/day,
with high impact
ampakincs at about 100/day (exemplary range 25 me-1.5 g/day): CUD at about 100-
600
mg/day. (exemplary range 20-1000 mg/day.): IIIC at about 5- 1 00 mg/day
(exemplary range 1-
800 mg/day): Ketamine/hydroxynorketamine at about 5-500mg/day (exemplary range
1-1000
mg/day). Disulfiram at about 50-500 me/dak (exemplary range 20-1500 me/day).
or any
30 combination of the foregoing drugs/doses.
In more detailed aspects of the invention employing coordinate treatment with
Ampakines. operable ampakines can be selected from a wide variety of known
ampakine
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
compounds. Ampakines. while structurally diverse as a whole, show many shared
structural
and functional features within classes. Roth between and within known ampakine
classes,
useful drug candidates operable within the anti-cancer methods and
compositions of the
invention can be identified, selected and proven effective according to the
detailed teachings
5 and guidance herein. Following these teachings, anti-cancer active
ampakines can be
selected among positive allosterie A MPA receptor modulators from within a
variety of
known ampakine groups. Among the ampakine classes from which operable ampakine
candidates for use within the invention can be selected include ainpakines
generally classified
as: sulfonamide compounds and derivatives, (bis)stilfonamide compounds and
derivatives, N-
10 substituted sulfonamide compounds and derivatives: heterocyclic
sulfonamide compounds
and derivatives: heterocycly1 sulfonamide compounds and derivatives: alkenyl
sulfonamide
compounds and derivatives; cycloalkenvl sulfonamide compounds and derivatives;
cyclopentyl sulfonamide compounds and derivatives: cycloalkylfluoro
sulfonamide
compounds and; aeetylenic sulfonamide compounds and derivatives; 2-propane-
sulfonamide
15 compounds and derivatives: 2-aminobenzenestilfonamide compounds and
derivatives:
benzoyl piperidinc and benzoyl compounds and derivatives: pyrrolidine
compounds and
derivatives: benzoxazine ring compounds and derivatives: acylbenzoxazine
compounds and
derivatives: carbonvlbenzoxazine compounds and derivatives; substituted 23-
benzodiazepin-
4-one compounds and derivatives: amidophosphate: monotluoralkyl compounds and
20 derivatives; substituted quinazoline compounds and derivatives;
quainoxaline compounds and
derivatives; 2-ethoxy-4.43-(propane-2-sullonylamino)-thiophen-2-y11-biphenyl-4-
carboxylic
and derivatives; p3;rrole and pyrazole compounds and derivatives: thiadiazine
compounds and
derivatives; benzofurazan compounds and derivatives: benzothiazidc compounds
and
derivatives: substituted 5-oxo-5.6.7.8-tetrahydro-4H-l-benzopyran and
benzothiopyran
25 compounds and derivatives; benzoxazepine compounds and derivatives;
among known
classes of compounds comprising AMPA receptor modulator compounds
prospectively
useful within the invention.
According to the teachings and examples presented herein, anti-cancer
effective
ampakines effective within the invention are selected and characterized from
among various
30 structural classes of ampakincs, for example, to demonstrate low impact
convulsant risk and
therapeutically effective anti-cancer activity. In illustrative embodiments
provided herein,
ampakines from the known class of benzoftirazan ampakine compounds and
derivatives (e.g..
as disclosed in U.S. Pat. Nos. 6.110.935: and 6,313.115: and PCT Int'l Pub.
No.
16
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
W09835950) were screened and developed to identify operable drug candidates
within the
compositions and methods of the invention. From these investigations exemplary
anti-cancer
benzolurazan candidates 1-(benzoliirazan-5-ylcarbony1)-4,4-difluoropiperidine,
and 4-
(benzofurazan-5-ylearbonyft and 1-(benzolurazan-5-ylcarbonyl)morpholine.
Within
5 additional compositions and methods of the invention, low impact
ampakines are selected for
combinatorial treatment methods of the invention from another ampakine group
known
collectively as -di-substituted amide ampakines.- These ampakines were first
described by
Cortex (now RespireRx). as detailed in USSN 12/451515, US Publication No.
US2010/0120764, and PCT/1JS/2008/00627 (incorporated herein in their entirety.
for all
10 purposes). Exemplary di-substituted amide ampakines for use within the
invention include
N-Methyl-N-tetrahydro-211-p) ran-4-y1-[2.1.3]-benzoxadiazole-5-carboxamide
("CX1739-),
Trans-4-[(2.1.3-benzoxadiazol-5-ylcarbonyl)(methypaininojcyclohexyl glycinate
hydrochloride (CX1942 ); and. N-(4-trans-hydroxycyclohex)-1)-N-methy142.1,31-
benzoxadiazole-5-carboxamide (CXI763). Within related embodiments of the
invention,
15 useful low impact. anti-cancer ampakines are selected and demonstrated
to be active
according to the teachings herein_ having the exemplary annpakine structure I.
below:
A
20 NA herein:
W is oxygen_ sulfur or C11=01:
X, Y and Z are independently selected from the group consisting of-N. or -CR.
wherein:
R is 1-1, -Br. -0_ -F. -CN. -NO7, -010. -SRI. -NRH, -C1-C6 branched or un-
branched
25 alkyl_ which may be un-substituted or substituted,
wherein:
R' is H, -C1-C6 branched or tin-branched alkyl which_ may be un-substituted or
substituted.
F - 0 or S.
17
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
A is I-1, or -C1-C6 branched or un-branched alkyl, which may be un-substituted
or
substituted_ -C2-C6 branched or un-branched alkenyl. which may be un-
substituted or
substituted, -C2-C6 branched or un-branched alkynyl. which may be un-
substituted or
substituted, -C3-C7 cycloalkyl which may be tin-substituted or substituted, -
C3-C7
5 alkylcycloalkyl which may be un-substituted or substituted, aryl
or heterocycle which
may be un-substituted or substituted. alkylaryl which may be un-substituted or
substituted. alkylheterocycle which may be tin-substituted or substituted
n=0. I. 2. 3_ 4. 5. or 6:
is a -C3-C7 cycloalkyl_ which may be un-substituted or substituted, a -C4-C7
10 azacycloalkyl. which may be tin-substituted or substituted, a C7-
C10 bicycloalkyi
which may be un-substituted or substituted, a -C7-C10 azabicycloalkyl which
may be
tin-substituted or substituted. aryl which may be un-substituted or
substituted or a
heterocycle which may be un-substituted or substituted:
13 is -C=. C-R3, 0, N. S. ('-0. S-0 or SO2.
15 Fr is I-1. a halogen (preferabl) 1--). Oft 0-alkyl, cyano. or a -
CI-C6 alkyl group which
is un-substituted or substituted and which optionally, forms a C3-C.7
cycloalkyl group
with D: and
D is absent when 13 is 0. S. 5-0, C:J:0 or SO2. or if present. is bonded to B
when 13 is
- C. -C-R" or N, and is II. a halogen (preferably F). Rh, a -Ci-C6 branched
or un-
20 branched alkyl. which may he un-substituted or substituted and
which optionally,
Forms a (71-C7 cycloalkyl group with Ra. a -C2-C6 branched or un-branched
alkenyl,
which may be un-substituted or substituted, a -C2-C6 branched or un-branched
alkynyl, which may be tin-substituted or substituted. a -C3-C7 cycloalkyl
which may
be un-substituted or substituted. an aryl which may be tin-substituted or
substituted , a
25 heterocycle which may be un-substituted or substituted. a -C2-C7
carboxyalkyl which
may be un-substituted or substituted. a carboxyaryl which may be un-
substituted or
substituted, a earboxyheteroaryl which may be un-substituted or substituted. a
-Ci-C7
sulfony lalkyl i.shich may be un-substituted or substituted. a sullony-laryl
which may be
un-substituted or substituted or a sullonylheteroaryl which may be un-
substituted or
30 substituted. or when B is -C-Ra. R" and D optionally form a -N-Re
or a =N-OW
group with B. wherein Re is 14 or an unsubstituted or substituted C1-C7 alkyl
group, or
18
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
when B is ¨C-R". R" and D optionally form a ¨14-Re or a ¨N-ORe group with B.
wherein Re is H or an unsubstituted or substituted CI-C7 alkyl group: and
Rb is H, a -CI-C7 alkyl group which ma v he branched or un-branched. un-
substituted
or substituted or a -C2-C7 acvl group which may be un-substituted or
substituted.
Other exemplary ampakines useful within the combinatorial methods herein
include include compounds according to formula II below:
0 0--D
/N, 10
)n
0\
A
11
wherein:
A is -C1-C6 branched or un-branched alkyl, which may be un-substituted or
substituted. a C3-C7 cycloalkyl which may be un-substituted or substituted;
n is O. I _ 1 or 3:
B is C-R". 0 or C-0:
R" is ELF. -OH or alkyl and
D is absent (when B is 0). is H or OH when Ra is H or alkyl, or is F when R is
F, or a
pharmaceutically acceptable salt. solvate. or polyinorph thereof.
Yet additional exemplary ampakines for use within the invention include
compounds according to formula III below:
0
0\
A
I II
w herein:
A is a C1-C6 alkyl which may be substituted or on-substituted:
B is C-ie. 0 or C-0:
Ra is H. F. -011 or alkyl and
D is absent (when B is 0). is 1-1 or OH when R" is II or alkyl. or is F when
R" is F. or a
pharmaceutically acceptable salt, solvate, or polymorph thereof.
19
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
Other exemplary ampakines for use within the invention include compounds
according to formula IV below:
0
lb¨. III
5 A
IV
wherein:
A is a C1-C6 alkyl which may be substituted or tin-substituted.
n is 0. I or 2. or a pharmaceutically acceptable salt. solvate. or polymorph
thereof
100011
Other exemplary embodiments
include compounds according to formula V
below:
2
0
R1
N____4110
0
A
V
15 wherein:
A is a (21-C6 alkyl which may be substituted or un-substituted.
is It F. or CI-C4 alkyl.
R2 is It F. Cl\i. a heterocycle which may be substituted or un-substituted or
OR3.
R3 is H. CI-C6 alkyl which may be substituted or un-substituted, or a
20 pharmaceutically acceptable salt, solvate, or polymorph thereof.
20
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
Other exemplary ampakines for use within the invention include compounds
according to formula VI below:
OH
0 0---R
71, IN
0\
1
A
V I
5 wherein:
A is a C1-C6 alkyl which may he substituted or tin-substituted.
R. is H. or C i-C4 alkyl, or a pharmaceutically acceptable salt, solvate, or
polymorph
thereof.
Other exemplary ampakines for use within the invention include compounds
10 according to formula VII below:
0 Cr-D
/
0
Me
VII
wherein:
B is C-R", 0 or C=0:
15 R" is 11, F. -OH or alkyl and
D is absent (when B is 0). is 11 or 011 when 113 is 11 or alkyl, or is F when
Rd is F. or a
pharmaceutically acceptable salt_ solvate_ or poly morph thereof
Other exemplary ampakines for use within the invention include
compounds according to formula VIII below:
CreD
/ 11101
0
20 Me
VIII
µ herein:
B is C-R". 0 or CO:
R" is H. F. -01-1 or alkyl and
21
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
D is absent (when B is 0). is H or Oil when R" is 11 or alkyl. or is F when Ra
is F. or a
pharmaceutically acceptable salt, solvate. or polyrnorph thereof.
Other exemplary ampakincs for use within the invention include
compounds according to formula IX below:
2
0
3
0 )JR4
N
/
0
A
IX
wherein:
A is a C1-C6 alkyl which may be substituted or un-substituted,
RI is H. or CI-Cr alkyl.
R2 is H. or a C1-C6 alkyl which may be substituted or un-substituted.
R3 is II. or a C -Co alkyl which may he substituted or un-substituted.
R4 is II. or a C1-C6 alkyl which may be substituted or un-substituted, or a
pharmaceutically acceptable salt, solvate or polymorph thereof.
In more detailed embodiments, anti-cancer active compounds are selected
from compounds of Formulas I -IX above that are already isolated and
characterized, selected
from: Al-Cycloheptyl-N-methy1[2.1,31-benzoxadiazole-5-carboxamide:
Dimethylcyclohexyl-N-methy142. I .31-benzoxadiazole-5-carboxamide: N-Methyl-N-
spirol_2.51oct-6-y142. l.3]-benzoxadiazole-5-carboxamide: N-Cyc lohexyl-N-
methy1-[2.1.3]-
benzoxadiazole-5-carboxamide: N-Cyclopentyl-N-methyl-[2,1,3 I-benzoxad iazo le-
5-
carboxamide: N-Cyclobutyl-N-methyl-I2.1.31-benzoxadiazole-5-carboxamide: N-
C)clohexy1-
12.1.3.1-benzoxadiazole-5-carboxarnide: N-Cyclopenty1-1-2.1.31-benzoxadiazole-
5-
carboxamide: N-Cyclobtity1-12, I .31-benzoxadiazole-5-carboxamide: N-(c is-4-
Cyanocy c lohexyl)-N-rinethyl-[2. I .3 I-be nzoxadiazole-5-carboxam ide: N-
(lrans-4-
CyanocyclohexyI)-N-methyl-[2.1,3I-benzoxadiazolc-5-carboxam ide: N-Methyl-N-
tetrahydro-
211-pyran-4-y1-12.1.31-benzoxadiazole-5-carboxamide (CXI739): N-D3-Methyl-
N4etrahydro-
2H-pyran-4-y112. I .31-benzoxadiazole-5-carboxamide: N-(Tetrahydro-2H-pyran-4-
y1)-
12,1.3-1-benzoxadiazole-5-carboxamide: N-(Tetrahydro-211-pyran-3-y1)12,1.3]-
benzoxadiazole-5-carboxamide: N-Methyl-N-(tetrahydro-21/-pyran-3-y1)42,1.31-
berizoxacliazole-5-carboxamide: .11-Fthyl-N-tetrahydro-2H-pyran-4-y142.131-
22
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
be nzoxad azo le-5-ca rboxami de: N-Cy-clohexyl-N-ethy142.1,3J-benzoxadiazole-
5-
carboxam ide:N-(Cyc lohexylmethyl)-N-methy142.1.31-benzoxadiazole-5-
carboxamide; N-
Benzy1-N-methyl-[2,1,311-benzoxad iazolc-5-carboxamide: N-Methy 1-N-
(tetrahydroluran-2-
ylmethy1)12.1,31-benzoxadiazole-5-carboxamide: N-M ethy 1-N-pyrid n-3-
y142.1,3]-
5 benzoxad iazo le-5-earboxami de: N-Methyl-N-pheny1-12,1,31-benzoxadiazole-
5-carboxamide
N-Cyclopropyl-N-tetrahydro-211-pyran-4-y142,1,31-benzoxadiazole-5-earboxamide
N-Tetrahydro-2H-pyran-4-yl-N-(2.2.2-trif1uoroethy1)42.1.31-benzoxadiazole-5-
carboxarnide:
tert-Buty1-44([2_ I .31-benzoxadiazol-5-ylearbonyl)(methyl)amino]pipericline-1-
carboxylate:
N-Methyl-N-piperidin-4-y1-12.1.31-ben7oxadiazole-5-carboxam ide hydrochloride:
N-Methyl-
10 N-(1-rnethylpiperidin-4-y1)-[2.1.31-benzoxadiazole-5-carboxam ide: N-(1-
Acetylpiperid i n-4-
y1)-N-methy112,1.3]-benzoxad azole-5-carboxam ide: N-( -Formylpiperidin-4-y1)-
N-methyl-
[2,1.311-benzoxadiazole-5-carboxamide: N-Methyl-N-I 1-( methylsulfonyl]
piperidin-4-y1)-
[2.1.3 I-benzoxadiazole-5-carboxamide: N-Methy 1-N-(tetrahydro-21-1-pyran-4-
y1)42,13]-
benzothiadiazole-5-carboxam ide: N'-Meth?..:1-N-(tetrahyd ro-211-thiopy ran-4-
y1)42,1.31-
15 benzoxadiazole-5-earboxamide: N-Methyl-N-(1-oxidotetrahydro-2H-thiopyran-
4-y1)-[2.1,31-
benzoxadiazole-5-earboxamide: N-Methyl-N-(1,1-dioxidotetrahydro-2H-thiopyran-4-
5/1)-
[2,1.31-benzoxadiazole-5-carboxamide: N-Methyl-N-tetrahydro-2H-pyran-4-
ylquinoxaline-6-
carboxam ide: N-Methyl-N-(4-oxocyclohexy1)[2.1.31-benzoxadiazole-5-carboxam
ide; N44-
(Hyd roxy im ino)cyclohexyll-N-rnethyl-I 2.1.3[-benzoxadiazole-5-carboxamide:
N
20 (Methoxyimino)eyelohexy1J-N-rnethy112.1.31-benzoxadiazole-5-carboxamide;
N-(4,4-
Dill uoroeyc lohexyl)-N-methy142. I .31-benzoxad iazole-5-carboxam ide: N-(4-
fluoroc ye lohex-
3-en-l-y1)-N-methy142.1.31-bentoxad iazol e-5-ca rboxam idc: N-(4-trans-
H)droxycyclohexy1)42. I _31-benzoxacliazole-5-carboxam ide: N-(irans-4-1-
1ydroxy-4-
methylcyclohexy1)-12.1.3J-bentoxadiazole-5-carboxamide: N-(cis-4-11vdroxy-4-
25 methylcyclohexyl)-N-methyl-[2.1.3]-benzoxadiazole-5-carboxam ide: N-
(trctus-4-1 lydroxy-4-
methyleye lohexyl)-N-m ethy142.1.3]-benzoxadiazolc-5-carboxam ide: N-(cis-4-
Hydroxy-4-
ethylcyc1ohexyl)-N-methy1-12.1.31-benzoxadiazole-5-carboxam ide: N-(irans--4-
Hydroxv-4-
ethylcyclohexy.1)-N-meth y112.1_3 [-benzoxad iazo le-5-carboxam ide: N-(cis-4-
Ethyny1-4-
hydroxy eye lohexyl )-N-methyl-I 2.1.31-benzoxadiazole-5-carboxamide: N-(eis-4-
But-3-en-1-
30 y1-4-hytlroxycyclohexyl)-N-methy1[2.1.31-benzoxadiazole-5-carboxam ide:
N-(trarts-4-But-
3-en-l-y1-4-hvdroxycyc lohexyl )-N-methy142.1.3]-benzoxad iazole-5-carboxa m
ide: N-(4-
trans--1-lyd roxycyc lohexyl)-N-methy142_1_3]-benzoxad iazole-5-earboxam ide
(CX1763): N-
(4-1rans-1-1ydroxycyc1ohexy1)-N-1)3-rnethy142.1.3 I-benzoxadiazole-5-
carboxamide: N-(irans-
4-Metho\vevelohexyl)-AlmetIn142_131-bentoxadiazole-5-carboxamide:
23
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
Methoxycyclohexyl)-N-methy1[2A131-benzoxadiazole-5-carbothioamide; N-(4-cis-
Hydroxycyclohexyl)-N-methyl-12,1.3 I-benzoxadiazole-5-carboxamide; N-Methyl-N-
ftran.s--4-
(211-tetrazol-2-y0cyclohexy1112,1,31-benzoxadiazole-5-carboxarnide: N-(irans-4-
Azidocycbahexyl)-N-Inethyl-[2..1,3]-benzoxadiazole-5-carboxamide: N'-(trans-4-
5 Aminocyclohexyl)-N-methy1-12.1.31-benzoxadiazole-5-carboxamide; N-(cis-3-
Hydroxycyclohexy1)-N-methyl-I2.1.31-benzoxad iazole-5-carboxamide: N-(irans-3-
Hydroxycyclohexyl)-N-met1iy112,1,3 J-benzoxadiazole-5-carboxamide: N-M et hyl-
N-(3-
oxocyclohexy1)12.1.31-benzoxadiazole-5-carboxam ide: N-Methyl-N-(3.3-
d i Iluorocyc lohexyl )-[2.1.31-benzoxad iazole-5-carboxamide: N--(2-H yd
roxycye lohexyl)-N-
10 methyl-42.1.3 J-benzoxadiazole-5-carboxam ide: N-Methyl-N-(2-
oxocyclohexy1)42.1,31-
benzoxadiazolc-5-carboxamide: N-Methyl-N-(2,2-difluorocyclohexyl)-12,1,3)-
benzoxadiazole-5-carboxamide; N-(2-Hydroxytetrahydro-2H-pyran-4-y1)42.1.31-
benzoxadiazole-5-carboxamide: N-(2-oxotetrahydro-2H-pyran-4-y1)12,1,3]-
benzoxadiazole-
5-carboxamide: N-Methyl-N-(2-oxotetrahydro-2H-pyran-4-)=1)-[2. I ,31-
benzoxadiazole-5-
15 carboxamide; N-(2-Hyd roxytetrahydro-2 H-pyran-4-y1)-N-methy142, 1.3]-
benzoxad iazo
carboxamide; trans-4-[(2.1.3-Benzoxadiazo1-5-
ylcarbonyl)(methyl)amino]cyclohexyl N,N-
dimethyl glycinate hydrochloride: tratts-4-[(2.1.3-Benzoxadiazol-5-
ylcarbonyl)(methyl)amino]cyclohexyl L-alaninate hydrochloride: N- (R)-
Tetrahydrofuran-3-
y1-12.1.3-1-benzoxad iazole-5-earboxam ide; N-M ethy 1-N-(R)-tetrahydrofuran-3-
y142,1,3]-
20 benzoxadiazole-5-carboxamide: treurs-4-[(2.13-Benzoxadiazol-5-
ylcarbonyliOnethyDaminolcyclohcxyl glycinate hydrochloride; N-2-(4-
Morpholinyl)ethyl-
[2,1.31-benzoxad iazole-5-carboxam ide: N-Methy1-N-2-(4-morpholiny
pethy142,1,31-
benzoxadiazole-5-carboxamide hydrochloride; N-Methyl-N-tetrahydro-2H-pyran-4-
y1-
1.2,1.3]-benzoxacliazole-5-carbothioamide (CX 1739): 1rans-41(2,1,3-Benzoxad
iazol-5-
25 ylcarbonyl)(methyparninolcyclohexyl L-valinate hydrochloride: irans-4-
[(2,1.3-
Benzoxadiazo1-5-ylcarbonyl)(methyl)aminol-1-methylcyclohexylN.N-dimethyl
glycinate
hydrochloride: N-Methy1-N-tetrahydro-2thpyran-4-ylrnethy142.1.31-
benzoxadiazole-5-
carboxani ide: and trans-4-[(2. 1.3-13enzoxad iazo I-5-y learbony I
)(methyl)amino1-1-
methylcyclohexyl glycinate hydrochloride (C X1942 ).
30 Within additional compositions and methods of the
invention, low impact
ampak Ines are emploNed in the methods and compositions of the invention,
selected from yet
additional ampakine groups. including -"bicyclic amide ampakines.- Among the
many.
bicyclic amide ampakines candidates for use within the invention are the
following
exemplary species: 8-Azabicyclo[3.2.1]oct-8-y1([2.1.31-benzoxadiazol-5-
y1)methanone:
24
CA 03145507 2022-1-24
WO 2020/023800
PCT/1JS2019/043525
([2,1,3]-13enzoxadiazol-5-ylcarbony1)-8-azabicyclo[3.2.1 loctan-3-one: t2, I
.3]-
Benzoxadiazol-5-y1(3.3-difluoro-8-azabicyclo[3.2.1joct-8-yl)methanone:
ench12,1,3]-
Benzoxadiazol-5-y1(3-hydroxy-8-atabicyclo[3.2.1loct-8-y1)inethanone; aro-
12.13k
Bertioxadiazol-5-y,r1(3-hydroxy-8-azabicyclol 3.2.1 loct-8-yflinethanone; 2-
Azabicy-clol 2.2.1 11iept-2-y1(12.13-benzoxadiazol-5-y1)methanone: 1-
Azabicyclo[2.2.11hept-
1-y1(12,1.31-benzoxadiazo I-5-y Dinethanone: 2-Azabicyclo12.2.2]oct-2-y1([2, I
.31-
benzoxadiazol-5-yl)methanone: 12. I .3]-13enzoxadiazol-5-v1(5.6-dichloro-2-
azabicyclo[2.2.11hept-2-yl)nriethanone. Additional bicyclic amide ampakines
for prospective
use within the anti-cancer methods and compositions of the invention include,
but are not
limited to. the following exemplary species: [2.1.31-13enzoxadiazol-5-y1(3-
11uoro-8-
azabicyclo[3.2.1]oct-2-en-8-y1)methanone: 2 -Az_abicyc lo12.2.11hept-5-en-2-
y1([2.1,3]-
benzoxadiazol-5-yl)methanone: R-2-Azabicyclo[2.2.11hept-5-en-2-v1([2.1.31-
benzoxadiazol-
5-yl)methanone: S-2-Azabicyclo[2.2.11hept-5-en-2-y1(12. I 31-benzoxadiazol-5-
yl)methanone; and [2.1.3]-13enzoxadiazo1-5-y1(2-oxa-5azab1cyc10[2.2.11hept-5-
yl)rnethanone.
Yet additional ampakinc compounds for use within the invention will be
selected according to the teachings herein_ using known AMPA receptor
modulator
compounds, reagents, preparative methods and other tools as disclosed in the
following
publications, each of which is incorporated herein for all purposes: PCT Intl
Pub. No. WO
94/02475 and related U.S. Pat. Nos. 5.773.434. 5,488,049, 5,650.409,
5,736,543, 5347.492.
5,773.434, 5,891_876. 6,030,968. 6.274,600, 6.329.368. 6.941159_ and
7.026,475; U.S. Pat.
Pub. No. 20020055508: U.S. Pat. Nos. 6.174.921 6301816. 6358.981, 6.362,230,
6.500.865_ 6,515,026. and 6.552.086: PCI Intl Pub. Nos. WO 0190057. WO
0190056, WO
0168592. WO 0196289. WO 02098846. WO 0006157. WO 9833496_ WO 0006083. WO
0006148. WO 0006149. WO 9943285. and WO 9833496: W00194306: U.S. Pat. No.
6,525.099 and PCT Int'l Pub. No. WO 0006537: U.S. Pat. No. 6.355..655 and PCT
Intl Pub.
Nos. W00214294. W00214275. and W00006159: U.S. Pat. No. 6.358,982 and PCT
Pub. No. W00006158: U.S. Pat. No. 6.387.954 and PCI Int'l Pub. No. W00006539;
PCT
Intl Pub. No. W002098847: U.S. Pat. No. 6.639.107 and PCI Intl Pub. No.
W00142203;
PCT Intl Pub. No. W00232858: PCT Int'l Pub. No. W00218329; U.S. Pat. No.
6.596_716
and PCT Intl Pub. Nos: W02006087169. W02006015827. W02006015828.,
W02006015829. W02007090840. and W02007090841: W002089734: U.S. Pat. Nos.
5.650.409_ 5.747.492. 5.783.587. 5_852.008. and 6.274.600: U.S. Pat. Nos.
5,736.543,
5,962.447. 5.985.871. and PCT Intl Pub. Nos. WO 9736907 and W09933469: U.S.
Pat. No.
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
6,124.278. and PCT Int'l Pub. No. W09951240: PCT Int'l Pub. No. W003045315;
U.S. Pat.
No. 5_891.871: U.S. Pat. Nos. 6.1 10_935 and 6.313.115. and Pcr Int'l Pub. No.
W09835950;
PCT Int'l Pub. No. W09812185: PCT Intl Pub. No. W00075123: U.S. Pat. No.
6,521,605
and PCT Intl Pub. No. W00006176: PCT 1nel Pub, No. WO 0066546; PCT Intl Pub.
No.
WO 9944612; PCT Int'l Pub. No. W02007060144; U.S. Pat. Pub. No. 20060276532;
U.S.
Pat. Pub. No. 20070066573: U.S. Pat. Pub. No. 20070004709: U.S. Pat. Pub. No.
20040171605: PCT Intl Pub. Nos. WO 9942456_ WO 0006156, and WO 0157045_ and
U.S.
Pat. No. 6,617_351.
Additional description and background pertaining also to specific positive
allosteric AMPA receptor modulators, their preparation_ use and selection
within the
compositions and methods of the invention_ is provided in the following
references,
incorporated herein in toto for all purposes: For bervolurazan compounds-PCI
patent
application PCT/US98/02713. United States patent application Serial No.
08/800,108. now
United States Patent 6.110,935. United States patent application Serial No.
09/355,139, now
United States Patent 6,313,115, United States patent application. Serial No.
09/834,349;
United States patent application, Serial No. 09/845_128. now United States
Patent 6,730,677;
For di-substituted amide ampakines-PCT patent application PCT/US2008/00627 I,
United
States patent application Serial No. 12/451_5 15. now United States Patent
8,013,003. United
States patent application. Serial No. 13/226.146_ now issued United States
Patent 8.404482,
and United States patent application_ Serial No. 13/755_210. now issued United
States Patent
8.642.633; For bicyclic amide aunpakines-PCT patent application
PCT/U52008/009508,
United States Provisional patent application. Serial No. 60/964_362: United
States patent
application, Serial No. 12/657.908_ now United States Patent 8_ [19.631 United
States patent
application. Serial No. 12/733.073. now United States Patent 8,263,591. United
States patent
application. Serial No. 13/348,171. now United States Patent 8.507.482. United
States patent
application. Serial No. 13/557.681. United States patent application. Serial
No. 12/657,924.
now United States Patent 8.168.632, PCT patent application PCT/US2010/000255,
and
United States Provisional patent application, Serial No. 61/206.642: For
bicyclic amide
ampakines-PCT patent application PCT/US2010/000254. and United States
Provisional
patent application. Serial No. 61/206_642: For 3-Substituted-I 1,2.3-
113enzotriazinone
ampakines-PCT Patent application PCl/US2007/026415. United States Provisional
patent
application, Serial No. 60/878.626_ United States patent application. Serial
No. 12/448_770,
PCT patent application PCT/US2007/026416_ United States Provisional
application. Serial
No. 60/878_503_ United States Provisional patent application_ Serial No.
60/921,433. and
26
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
United States patent application. Serial No. 12,448.784, now United States
Patent 8,173,644:
For 3-substituted 1.2,3-triazin-4-one and 3-substituted 1.3-pyrimidinone
ampakines-PCT
patent application PCT/US2008/010877. United States Provisional patent
application. Serial
No.60/994,548. and United States patent application. Serial No. 12/733,822;
For benzoxazine
5 ampakines-PCT patent application PC1/U598/27027, and United States patent
application,
Serial No. 08/998.300. now United States Patent 5.985.871: for
Acylbenzoxatines
ampakines- PCT patent application. Serial No. PCT/US99/07325. and United
States patent
application 09/054.916, now United States Patent 6.124.278: for Benzoyl
Piperidinc/Pyrrolidinc ampakines PCI patent application PCT/US96/07607, and
United
10 States patent application Serial No. 08/458367. Filed 2 June 1995. now
United States Patent
5,650.409: For betrzoxazine ampakines-PCT patent application. Serial No,
PCT/US97/05184,
United States patent application. Serial No. 08/624,335. now United States
Patent 5,736,543,
PCT patent application PCT/US93/06916. and United States patent application,
Serial No.
07/919.512. now United States Patent 5_961447; and for carbonylbenzoxazine
ampakines-
15 PCT patent application PCT/U502/37646. United States Provisional patent
application. Serial
No. 60/333334. and United States patent application Serial No. 10/495.049. now
United
States Patent 7,799,913. Each of the foregoing classes and distinct structural
groups of
ampakine compounds disclosed in the above references are suitable for
evaluation to
determine operability within the methods and compositions of the invention.
Persons of
20 ordinary skill in the art will recognize that these various compound
groups, while being
structurally diverse, share common functional characteristics of positive
allosteric AMPA
receptor modulation, as described here, and that because of these common
functional
characteristics, the compounds can be evaluated and determined for their
operability
according to the inventive discoveries and teachings herein. According to the
Examples and
25 other guidance provided here. anti-cancer effective ampakines. for
example, can be selected
and demonstrated for beneficial. clinical use without undue experimentation.
To practice coordinate administration methods of the invention, the anti-
cancer
effective AMPAR antagonist compound is co-administered, simultaneously or
sequentially.
in a coordinate treatment protocol with one or more of the secondary or
adjunctive
30 therapeutic agents contemplated herein. Thus. in certain embodiments the
anti-cancer
effective AMPAR antagonist compound is administered coordinately with a
conventional
cancer chemotherapeutic agent using separate formulations or a combinatorial
formulation.
Coordinate administration may be done simultaneously or sequentially in either
order, and
there may be a time period while only one or both (or all) active therapeutic
agents
27
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
individually and/or collectively exert their therapeutic activities. A
distinguishing aspect of
all such coordinate treatment methods is that the anti-cancer effective AMPAR
antagonist
compound exerts at least some measurably distinct anti-cancer therapeutic
activity, yielding a
distinct clinical response. in addition to any complementary clinical response
provided by the
5 secondary or adjunctive therapeutic agent. Often. the coordinate
administration of the anti-
cancer effective AMPAR antagonist compound with the secondary or adjunctive
therapeutic
agent will yield improved anti-cancer therapeutic or prophylactic results in
the subject
beyond a therapeutic or prophylactic effect elicited by the secondary or
adjunctive therapeutic
agent alone, which benefit contemplates both direct effects, as well as
indirect effects.
10 The anti-cancer effective AM VAR antagonist compounds and
pharmaceutical
compositions of the present invention may be administered by any means that
achieve the
contemplated anti-cancer therapeutic or prophylactic purpose. Suitable routes
of
administration for the compositions of the invention include. but are not
limited to, oral,
buccal, nasal, aerosol, topical. transderrnal. mucosa'. injectable, and
intravenous, as well as
15 all other practicable delivery routes. devices and methods.
The anti-cancer effective AMPAR antagonist compounds of the present invention
may be formulated with a pharmaceutically acceptable carrier appropriate for
the particular
mode of administration employed. Dosage forms of the compositions of the
invention
include excipients recognized in the art of pharmaceutical compounding as
being suitable for
20 the preparation of dosage units as discussed herein. Such excipients
include, without
limitation. solvates. buffers, binders, fillers, lubricants. emulsifiers,
suspending agents,
sweeteners, flavorings. preservatives, wetting agents. disintegrants.
effervescent agents and
other conventional pharmaceutical excipients and additives.
Anti-cancer effective AMPAR antagonist compounds of the invention will often
be
25 formulated and administered in an oral dosage form_ optionally in
combination with a carrier
and/or other additive(s). Suitable carriers for pharmaceutical formulation of
oral dosage
forms include. for example. inicrocrystalline cellulose, lactose, sucrose,
fructose, glucose,
dextrose, or other sugars. di-basic calcium phosphate_ calcium sulfate_
cellulose,
methylcellulose. cellulose derivatives. kaolin_ mannitol. lactitol. maltitol.
xvlitol. sorbitol, or
30 other sugar alcohols, dry starch. dextrin. maltodextrin or other
polysaccharides, inositol_ or
mixtures thereof. Exemplary unit oral dosage forms include ingestible and
sublingual liquids,
tablets. capsules. and films_ among other options, which may be prepared by
any
conventional method known in the art. optionally including additional
ingredients such as
release modifying agents. glidants. cornpression aides. disintegrants.
lubricants. binders.
28
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
flavor enhancers, sweeteners and/or preservatives (e.g.. stearic acid,
magnesium stearate, talc,
calcium stearate. hydrogenated vegetable oils, sodium benzoate. leucine
carbowax.
magnesium lauryl sulfate, colloidal silicon dioxide. glyeeryl monostearate,
colloidal silica,
silicon dioxide, and glyceryl monostearate). Oral dosage forms may further
include an
5 enteric coating that dissolves after passing through the stomach, for
example, a polymer
agent, methacrylate copolymer. cellulose acetate phthalate (CAP).
hydroxypropyl
methylcellulose phthalate (11PMCP).. polyvinyl acetate phthalate (PVAP).
hydroxypropyl
methylcellulose acetate suceinate (HPMCAS), cellulose acetate trimeliitate.
hydroxypropyl
methylcellu lose succinate. cellulose acetate succinate. cellulose acetate
hexahydrophthalate,
10 cellulose propionate phthalate_ cellulose acetate rnaleate_ cellulose
acetate butyrate, cellulose
acetate propionate. copolymer of methylmethacrylic acid and methyl
methacrylate.
copolymer of methyl acrylate. methylmethacrylate and methacrylic acid,
copolymer of
methyl vinyl ether and maleic anhydride (Gantrez ES series). and natural
resins such as zein,
shellac and copal collophorium.
15 If desired. oral, mucosal. gastric. transdermal. topical and
injectable compositions of
the invention can be administered in a controlled release form by use of such
well known
technologies as slow release carriers and controlled release agents.
In certain embodiments the anti-cancer effective AMPAR antagonist compound is
administered to patients in an injectable or intravenous (iv) formulation and
delivery mode.
20 In illustrative aspects a therapeutic unit dosage of PMP is formulated
in a physiological
solution amenable for injection or iv delivery to human subjects. for example
in an aqueous
buffered solution such as saline. Alternative formulations of anti-cancer
effective AMPAR
antagonist compounds for administration to patients intravenously,
intramuscularly,
subcutaneously or intraperitoneally can include nonaqueous sterile injectable
solutions and
25 optionally contain anti-oxidants. buffers. bacteriostats and/or solutes
which render the
formulation isotonic with the blood of the subject, as well as aqueous and non-
aqueous sterile
suspensions which may include suspending agents and/or thickening agents.
Additional
injectable compositions and formulations of the invention may include polymers
and other
controlled deliver) additives or carriers for extended release following
administration.
30 Parenteral preparations may be solutions, dispersions or emulsions
suitable for such
administration. Extemporaneous injection solutions. emulsions and suspensions
may be
prepared from sterile powders. granules and tablets. Preferred unit dosage
formulations are
those containing a daily dose or unit, daily sub-dose. or an appropriate
fraction thereof, of the
anti-cancer effective AMPAR antagonist compound and/or active ingredient(s).
In some
29
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
embodiments, localized delivery of anti-cancer effective AMPAR antagonist
compounds may
be achieved by injecting the parenteral formulation directly into an area
surrounding a
cellular malignancy, directly into a tumor_ into the vasculature supplying a
malignancy itself,
or into a pleural or peritoneal cavity or cerebrospinal compartment proximal
or fluidly
5 connected to a targeted malignancy.
In certain embodiments the methods and compositions of the invention may
employ a
pharmaceutically acceptable salt of an anti-cancer effective AMPAR antagonist
compound,
for example an acid addition or base salt of a PMP compound, derivative or
analog.
Examples of pharmaceutically acceptable addition salts include inorganic and
organic acid
10 addition salts. Suitable acid addition salts are formed from acids which
form non-toxic salts.
for example, hydrochloride. hydrobromide. hydroiodide. sulphate, hydrogen
sulphate, nitrate,
phosphate, and hydrogen phosphate salts. Additional pharmaceutically
acceptable salts
include, but are not limited to. metal salts such as sodium salts, potassium
salts, cesium salts
and the like; alkaline earth metals such as calcium salts. magnesium salts and
the like;
15 organic amine salts such as triethylamine salts, pyridine salts.
picoline salts, ethanolarnine
salts, triethanolamine salts. dicyclohexylamine salts. N1,1\l'-
dibenzylethylenediamine salts and
the like; organic acid salts such as acetate, citrate, lactate, succinate.
tartrate. maleate.
fumarate. mandelate, acetate. dichloroacetate. trilluoroacetate, oxalate, and
formate salts;
sulfonates such as methancsulfbnate. benzenesullonate. and p-toluenesulfonate
salts; and
20 amino acid salts such as arginate, asparginate, glutamate_ tartrate_ and
gluconate salts.
Suitable base salts are formed from bases that form non-toxic salts. for
example aluminum,
calcium_ lithium_ magnesium. potassium, sodium, zinc and diethanolamine salts.
In related
embodiments, optional salt forms of an anti-cancer effective AMPAR antagonist
coin pound
will yield enhanced properties. e.g., improved stability. solubility,
tolerability, etc.
25 In other detailed embodiments. the methods and compositions of
the invention
employ prodrugs of the anti-cancer effective AMPAR antagonist compound, e.g.,
prodrugs of
a PMP compound or derivative, or of an intermediary compound, or precursor
compound of a
PMP compound or derivative. As contemplated herein, prodrugs of anti-cancer
effective
AMPAR antagonist compounds can include the active compound reversibly linked
(e.g.,
30 covalent]) bonded) to any carrier compound or moiety that functions to
release the active
anti-cancer effective AMPAR antagonist compound in vivo (for example to
effectively
mediate delivery, of more active drug. to enhance in vivo half-life of the
drug. or otherwise
enhance phannacokineties or pharmacodynamics of the drug following
administration.
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
Examples of prodrugs useful within the invention include esters or amides with
hydroxyalkyl
or aminoalkyl as a substituent, among many other prodrug constructs known in
the art.
The invention will also be understood to encompass methods and compositions
comprising biologically active metabolites and in vivo conversion products of
the anti-cancer
5 effective AMPAR antagonist compound (either generated in vivo after
administration of the
compound, or directly administered in the form of the metabolite or conversion
product
itself). Such secondary active products may result for example from oxidation,
reduction,
hydrolysis, amidation, esterification and the like. of the administered
compound, primarily
due to enzymatic processes.
10 Example I
Dose-Dependent Anti-Cancer Activity of
Exemplary AMPAR Antagonist Perampanel (PMP)
T9G (Cilioblastoma) and Pane- I (pancreatic adenocarcinoma) were obtained from
ATCC. They were maintained in DM EM media (ATCC) and supplemented with 10% HIS
15 (ATCC) and 1% Penicillin/Streptomycin and maintained in an incubator at
37 C with 95%
air and 5% CO,.
Reagents PMP was purchased from Medkoo and dissolved in DMSO.
Temozolomide, cisplatin and glutamate were purchased from Sigma and dissolved
in
complete media on the day of treatment.
20 Cancer Cell Viability Assays T98G or Panel cells were seeded in
quadruplicate at a
density of 6.000 cells/well in complete DMEM and incubated overnight. T98G
cells were
then treated with increasing concentrations of PMP and temozolomide for 72
hours.
Alternatively. panel cells were treated with PMP. cisplatin or glutamate for
48 hours.
Following this incubation. 15u1_, MIS solution (Promega) was added to each
well and
25 incubated for a further 2 hours. Plates were read at 490nM using the
Fl.x808 microplate
reader. Absorbance values of wells with only media were subtracted out as
background
control. Data were normalized to vehicle-treated cells.
Data analysis Data were analyzed using Microsoft excel using a student's t-
test.
One-way ANOVA were also performed using Statplus. King's Synergy formula was
used to
30 look far synergistic interactions among PMP and chemotherapies. Alpha
value was set at
p=0.05.
Results
The data presented here unexpectedly reveal that AMPAR antagonists.
exemplified
by peramplanel (PMP). induce dose-dependent reductions in cell viability of
T98G cells (Fig
31
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
Ia. ANOVA p<0.0001). The surprisingly potent oneolytic effects of peramplanel
are
mediated in part by antagonistic activity against A MPAR physiology in AM PAR
positive
cancer cell targets. Perampanel exhibited significant inhibitory activities
against GRM
cancer cell viability, even at concentrations of 1-10uM. demonstrating the
clinical utility of
5 this drug at relevant plasma levels for effective cancer chemotherapy.
PMP additively
complements anti-cancer effects of ternozolomide (TMZ) at 30uM, and
synergistically
potentiates TMZ anti-cancer efficacy at I00uM (Fig lb).
Previous work with pancreatic cancer has suggested that ampakines may be able
to
reduce cell viability. Thus. the results provided here demonstrating that PMP
dose-
10 dependently reduces Panel cell viability (ANOVA p=0.0013). with
significant reduction of
cell viability seen at 100uM (Fig 2a). are particularly surprising. Although I
OuM PMP did
not appear to exert potent oncolytic effect alone at this concentration_ it
did significantly
enhance oncoly sis in combination with the drug cisplatin. Accordingly, PMP
will be
clinically effective to improve cancer treatments of this and other
chemotherapeutic agents
15 (Fig 2b). 100uM PMP and 3uM cisplatin also synergistically reduced
pancreatic cancer cell
viability (Fig 2b).
In yet additional working examples provided here, PMP effectively disrupted
the
oneogenic activity of exogenous glutamate. thereby inhibiting glutamate-
potentiated
pancreatic cancer activation (e.g.. as demonstrated by impairment of cancer
cell
20 proliferation). In these studies. glutamate concentrations of 100-1000uM
elicited a dose-
dependent acceleration of pancreatic cancer cell proliferation (Fig 2c). In
test samples panel
cells were exposed to ImM glutamate for 15 minutes prior to the addition of
PMP to cell
culture media. Perampanel addition 15 minutes follow" 11 e. glutamate exposure
was sufficient
to profoundly disrupt oncogenic activities of glutamate (Hg 2d).
Example!!
Dose-Dependent Anti-Cancer Activity of
An Exemplar., AMPAR Antagonist, Perampanel (PMP)
Potent anti-cancer efficacy of PMP compounds and other etTective AM PAR
30 antagonists is readily demonstrated using a range of animal models that
are well known and
wideb. accepted in the art as predictive or anti-cancer activity in humans.
One such model
employs subcutaneous xenografts of tumor cells into useful study animals such
as mice, to
study efficacy of candidate anti-cancer drugs in reducing growth or
proliferation of
xenoffalted tumor cells in test versus control subjects. These studies can
include monitoring
32
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
of a range of indicia of therapeutic efficacy. for example to demonstrate a
dose-dependent
decrease (e.g.. based on average values observed in test versus control
subjects) in
xenografted tumor number, tumor size. tumor metastases, tissue histological
and/or
biochemical cancer markers (e.g.. from biopsy or necropsy-) blood cancer
markers. mortality.
etc. As used herein. cancer "markers- refers to any biomolecule. such as a
growth factor,
genetic regulatory protein. eytokine_ hormone. receptor. etc.. whose presence.
expression,
structure, level or activity is correlated with cancer incidence, severity.
progression, or
another etiologic or therapeutic factor indicative of cancer growth. metabolic
activity,
metastasis. etc.
In useful study protocols relating to central nervous system (CNS) cancers
such as
glioblastoma (GUM). conventional xenograft study designs may be modified to
include
intracranial xenografting, to better capitulate clinical conditions of CUM
(see, for example,
Ozawa et al.. 2010). In one exemplary study protocol employed herein, modified
from
Ozawa et al._ we employ 198G cells. a GUM cell line expressing the enzyme
MGMT, which
functions to repair DNA damage from temozoiomide (Mt). rendering this cell
type
intrinsically resistant to TMZ chemotherapy. These cells are engineered to
express the
bioluminescent enzyme luciferase to allow in vivo xenograft detection and
quantification. A
study total of 24 mice are used, divided into four study groups of 6 members
per group. The
mice are anesthetized using ketamine/zylazine on a warming plate to maintain
core body
temperature. Once anesthetized. the scalp is swabbed with chlorohcxidine and a
sagittal
incision is made over the parieto-occipital bone. about lem long on the left
side. The
exposed skull is cleaned using a cotton swab with 3% hydrogen peroxide.
Xenograft cells are
provided at a concentration of 300.000-500.000 cells in 3u1_, serum-free
media, and this cell
suspension is drawn into a syringe and injected at a depth of 3mm into the
cortical tissue.
The injection is carried out slowly, over a period of one minute. to localize
the xenografted
cells focally to specific brain region and prevent dissemination of the cells
into the ventricles
and spinal cord. After injection., the skull is cleaned with 3% hydrogen
peroxide and sterile
bone wax is to the incised skull defect. The scalp is drawn over the skull and
stapled closed.
Buprenorphine is optionally administered for post-operative pain relief, and
recovery time is
about 30 minutes.
One week after injection the study subjects are divided into 4 groups and
bioluminescent monitoring of the xenografts begins. Group 1 mice receive
placebo saline for
the duration of the experiment. Group 2 receives 20rng/kg/day TMZ. Group 3
receives 5 or
lOmg/kg/day PMP depending on what dose produces a partial effect in
monotherapy
33
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
experiments. Group 4 is a combination group that receives both the TMZ and PMP
treatments. Mice are bioluminescent monitored every 4 days during the study,
for example
using D-luciferin and an in vivo imaging system such as IVIS-200 (PerkinElmer.
Inc,
Norwalk Connecticut) to measure bioluminescent photon release as a
quantitative indicator of
5 tumor growth.
These and related studies will demonstrate that PMP and other anti-cancer
effective
AMPAR antagonists according to the teachings herein potently prevent and treat
AMPAR
positive CNS cancers, including CiBM. in mammalian subjects. Particular
results will
demonstrate a dose-dependent reduction in overall luminescence over an
effective course of
10 AMPAR antagonist treatment, correlated with reduced tumor size, reduced
tumor cell
number and/or reduced xenograft proliferative and/or metastatic capacity
mediated by the
anti-cancer AMPAR antagonist. for example PMP. PMP and other selected AMPAR
antagonist will also significantly decrease tumor cell survival, viability and
proliferation, and
increase correlated indicia including time to tumor doubling and tripling, as
well as subject
15 survival (e.g.. by time and/or numbers of subjects). in addition to
mediating significant
therapeutic benefits corresponding to all other anti-cancer activity
indicators described herein
above
In more detailed in vivo protocols. PMP will exhibit significant inhibitory
activity
against GBM xenograft cell viability, proliferative capacity, tumor growth and
metastases at
20 concentrations of I-10uM or greater, i.e.. at plasma levels that are
safe and effective for
cancer chemotherapy.
In other detailed aspects. PMP will be shown to be combinatorially effective
to
complement anti-GUM effects of secondary anti-cancer drugs and treatments, for
example
temozolomide (TMZ). In certain embodiments. PMP will complement. potentiate or
even
25 synergistically enhance anti-cancer activities of other drugs. for
example to significantly
increase overall anti-cancer effects in combination with Tmz, compared to anti-
cancer
effects mediated by =I'MZ, alone. In these embodiments the combinatorial use
of PMP and
TMZ, e_g.. at therapeutic dosage levels of PMP between about 30uM-100uMõ
provides for
enhanced efficacy of TMZ and lower TMZ dosages with reduced TMZ-associated
side
30 effects. an exemplary model of coordinate treatment that will be
demonstrable across a range
of combinations of AMPAR antagonists and secondary/adjunctive anti-cancer
agents and
therapies.
In related illustrative protocols the efficacy of anti-cancer AMPAR
antagonists such
as PMP is demonstrated in combinatorial usage with a PRMT5 inhibitor, such as
EPZ015666.
34
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
As recently reported by Braun et al (2017). high grade aliomas may be
dependent on deletion
of detained introns of oncogenic transcripts for sustained growth and
survival. PRMT5
ensures proper splicing of these introns to become mature transcripts useful
for production of
various oncogenic proteins. Inhibition of PRMT5 with [PZ015666 reportedly
mediates
5 oncostatic effects against GBM. In one illustrative study here, the
foregoing intraeranial
xenograft study design is adapted to include one individual test group of 6
mice receiving
100mg/kg/day EPZ015666. one group treated with I Omg/kg/day PMP. and a
combinatorial
group treated with both EPZ015666 and PMP. Bioluminescent imaging and other
measures
of anti-cancer efficacy will demonstrate that PMP is anti-cancer effective
alone, and
10 combinatorially effective (e.g.. complementary, additive. potentiating
or synergistic) in
coordinate administration with EPZO I 5666.
In other illustrative protocols of the invention the efficacy of anti-cancer
AMPAR
antagonists such as PMP is demonstrated in combinatorial methods with tumor
treating
fields. Recent studies report that electrical fields using insulated
electrodes applying
15 frequencies of 200kliz can inhibit cell cycle progression in GBM cells
(see. e.g., Kirson et al.
2007; and Stupp et al. 2015). In an exemplary study here. the intracraniai
xenograft protocol
is adapted to include one individual test group of 6 mice receiving an
external insulated
electrode closest to the area of the xenograk applying a 200kIlz current for
the duration of
the study, one group treated with 10mg/kg/day PMP. and a combinatorial group
treated with
20 both therapies. Bioluminescent imaging and other measures of anti-cancer
efficacy will
demonstrate that PMP is anti-cancer etTective alone and combinatorially
effective in
coordinate administration with tumor treating fields.
In other exemplary protocols of the invention the efficacy of anti-cancer
AMPAR
antagonists such as PMP is demonstrated in combinatorial therapies employing
proteins that
25 interfere with telomere function of tumors. for example the TRF I
inhibitor [TP-47037. Due
to rapid proliferation of most tumors. tumor cells are particularly vulnerable
to DNA damage
that can result in cell death. Telomeres are the caps of chromosomes made of
repetitive
DNA. which serve to prevent protein-coding DNA loss or damage during cell
division.
Several proteins are implicated in maintaining telomeres. one of which is a
protein designated
30 TRH. Recent studies report that pharmacological or genetic ablation of
this TRF I reduces
tumor formation and growth in animal models (see, e.g._ Bejarano et al. 2017).
In one report.
75mg/kg of ETP-47037 prevented tumor growth in mice.. In an illustrative
protocol here. the
intracranial xenograft protocol above is adapted to include one test group of
mice receiving a
therapeutic dosage of 75mg/kg of VIP-47037. one group treated with I
Orng/kg/day PMP. and
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
a combinatorial group treated with both therapies. Bioluminescent imaging and
other
measures of anti-cancer efficacy VIII demonstrate that PMP is anti-cancer
effective alone and
combinatorially effective in coordinate administration with ETP-47037.
Additional studies
are contemplated to show that combinatorial treatment with PMP provides for
lower dosing
of the ETP-47037 to achieve the same or greater clinical benefits, with fewer
side effects
(e.g.. wherein a comparable. partial anti-cancer effect as exhibited by
75mg/kg is observed in
combination with PMP at reduced effective dosages of ETP-47037 of 25-50mg/kg
or lower).
In another exemplary combination treatment model of the invention, the
efficacy of
anti-cancer AMPAR antagonists such as PMP is demonstrated in coordinate
protocols with a
transcription inhibitor, such as terameprocol. Terameprocol is a global
transcription inhibitor
that affects proliferation. apoptosis and drug resistance, currently being
clinically evaluated
for treatment of GBM (Grossman et al. 2012). In one representative study the
intracranial
xenograft study includes one test group of mice treated with 20mg/kg/day
Terameprocol. one
group treated with 10mg/kg/day,. PMP. and a combinatorial group treated with
both therapies.
Bioluminescent imaging and other measures of anti-cancer efficacy will
demonstrate that
PMP is anti-cancer effective alone and combinatorially effective in coordinate
administration
with Terameprocol. Additional studies will show that combinatorial treatment
with PMP
provides for lower dosing ofTerameprocol to achieve the same or greater
clinical benefits.
with fewer side effects.
In yet additional combinatorial treatment methods of the invention, the
efficacy of
anti-cancer AMPAR antagonists such as PMP is demonstrated in coordinate
protocols with
NEK2 inhibitors. Recent studies report that EZH2 is vital for maintaining
glioma stem cells,
a subset of glioma cells that are responsible for chemo- and radiotherapy
resistance due to
their ability to regenerate new tumor cells after existing tumor cells are
destroyed. NEK2 is
responsible for guarding EZH2 against premature breakdown, allowing EZH2 to
exert a
longer and more robust oncogenic effect. Recent studies report that an
inhibitor of NEK2,
Cmp3a. exerts anti-cancer effects as demonstrated by prolongation of cancer
survival time in
mice (Wang et al. 2017). According to the modified study protocol here one
group of mice
receives I Oing.kg 'day Cmp3a. one group received IOm/kg/day PMP. and a
combinatorial
group is treated with both therapies. Bioluminescent imaging and other
measures of anti-
cancer efficacy will demonstrate that prvir is anti-cancer effective alone and
combinatorially
effeetke in coordinate administration with NEK2 inhibitors such as Cmp3a.
Additional
studies will show that combinatorial treatment with PMP provides for lower
dosing of NEK3
inhibitors to achieve the same or greater clinical benefits, with fewer side
effects.
36
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
Example III
Anti-Cancer Activity of AMPAR Antagonists In Combination with
Standard Of Care (SOC), Glioma Treatment
5 In exemplary clinical protocols of the invention, anti-cancer
effective AMPAR
antagonist treatment will be combined with secondary anti-cancer therapy
comprising
standard of care (SOC) glioma treatments. In one illustrative example,
patients are initially
treated with SOC maximal safe surgical resection_ followed by an aggressive
SOC
chemoradiation protocol. For the first 6 weeks of treatment, patients receive
75mg/m2/day of
10 Temozolomide (Temodar) starting one hour prior to radiation treatment.
Patients additionally
receive 200cGy focal radiation per day for the First live days of the week
over a 6 week
timespan (30 fractions). for a total of 60Gy radiation. Radiation targets the
tumor area as
well as surrounding edema plus a I em margin. I month after the last radiation
treatment,
patients are administered I 50-200mg/m2/day, temozolomide for the first 5 days
of every
15 month followed by 3 weeks rest as well as antiemetie prophylaxis
treatment as needed.
Treatments are stopped ifplatelet count drops below I00.000/uL. or if there is
evidence of
disease progression or severe treatment-related toxicity (Grossman et at
2009). In
combination with the SOC glioma treatment outlined above, patients receive 8mg
peramplanel orally 1 hour prior to the first radiation session. Perampanel is
further
20 administered once per day, and weekly titrated up 2 mg/day until
patients receive the
maximally tolerated approved dose (mTD) of 12 mg (Gidal et al, 2015) (though
this range
can be adjusted up or down based on patient-specific tolerance and other
clinical factors
determined by the managing physician). Within illustrative methods for
treating GBM,
patients receive the MT[) throughout an initial 6-week treatment period,
during the 1 month
25 SOC rest period, and while patients are taking maintenance Temodar.
Patients are
maintained on perampanel treatment as determined by the managing physician,
unless disease
progression or evidence of perarnplanel-related toxicity is observed_ Subjects
treated
according to this combinatorial protocol will show substantially improved
clinical benefits
over SOC or other conventional anti-glioma therapy.
30 Example IV
Anti-Cancer Activity of AMPAR Antagonists In Combination with
Levetiracetam Co-Treatment
37
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
In additional clinical examples_ patients are treated concomitantly with an
AMPAR
antagonist such as peramplanel and Levetiracetam (Keppra). which has been
shown to
augment temozolonnide efficacy (13obustue et al. 2010) and reduce aggression-
related adverse
events in patients taking perampanel (Kanemura et al, 2019: Kim et al. 2015).
Patients are
administered perampanel as described above along with 500mg levetiracetam 1
hour prior to
the first radiation session. Levetiracetam is administered twice a day. the
second time being
at night before bed. Within illustrative methods for treating GBM, patients
receive 200-
500mg levetiracetam twice a day throughout the first 6-week treatment period,
during the 1
month rest period, and while patients are taking maintenance Temodar. Patients
continue to
receive leveliracetain and perampanel unless disease progression or evidence
of drug-related
toxicity is observed. Subjects treated according to this combinatorial
protocol will show
substantially improved clinical benefits over SOC or other conventional anti-
cancer therapy.
Example V
Anti-Cancer Activity of AMPAR Antagonists In Combination with
NMDA Receptor Antagonist Co-Treatment
In other clinical protocols useful within the invention, patients are treated
concomitantly with an N-methyl-D-aspartate (NMDA) receptor antagonist, such as
memantine. Memantine has been reported to exert anti-cancer effects, possibly
by abrogating
constitutively active growth pathways in cancer (Stepulak et al, 2005: Maraka
et al, 2019).
Patients receive perampanel along with 5-20mg memantine orally prior to the
first radiation
session. Within illustrative methods for treating GBM. perampanel and
memantine are
administered once a day throughout the first 6-week treatment period, during
the 1 month rest
period, and while patients are taking maintenance Temodar. Patients are
maintained on
memantine and perampanel unless disease progression or evidence of drug-
related toxicity is
observed. Subjects treated according to this combinatorial protocol will show
substantially
improved clinical benefits over SOC or other conventional anti-cancer therapy.
In addition to
memantine. ketamine. an NMDA-antagonist used in the setting of
pharrnacoresistant
depression. may also be used. Ketamine is given at doses ranging from 5-
500mg/day and
started prior to the first radiation session. Ketamine and its active
metabolite
hydroxynorketamine (Zanos et al. 2016) may provide anti-cancer benefits (Malsy
et al.
2015). and will be a beneficial adjunct within the methods of the invention,
for example to
complement standard of care + perampanel treatments.
38
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
Example VI
Anti-Cancer Activity of AMPAR Antagonists In Combination with
Riluzole/Troriluzole Co-Treatment
Additional clinical methods of the invention employ an AMPAR antagonist such
as
5 perampanel coordinately administered with Riluzole/ troriluzole (see.
e.g.. Khan et al, 2019).
Patients receive perampanel as above in coordinate treatment with with 20-
50tng
Riluzole/troriluzole orally prior to the first radiation session. Within
illustrative methods for
treating GBM. perampanel and Rilwole/troriluzole are administered once a day
throughout
the first 6-week treatment period, during the I month rest period, and while
patients are
10 taking maintenance Temodar. Patients are maintained on
Riluzole/troriluzole and
perampanel unless disease progression or evidence of drug-related toxicity is
observed.
Subjects treated according to this combinatorial protocol will show
substantially improved
clinical benefits over SOC or other conventional anti-cancer therapy.
Example VII
15 Anti-Cancer Activity of AMPAR Antamonists In Combination
with
CSF1R Inhibitors
Additional clinical methods of the invention employ an AMPAR antagonist such
as
perampanel coordinately administered with a colony stimulating factor 1
receptor (CSF I R)
inhibitor such as PLX3397 (plexidartinib) or PLX5562 (see. e.g., Yan et al,
2017; Butowski
20 et al. 2016). Patients are administered perampanel as above in
coordinate treatment with 100-
1000 mg PLX3397 orally prior to the first radiation session. Within
illustrative methods for
treating GUM. perarnpanel and PLX3397 are administered together or separately
once a day
throughout the first 6-week treatment period, during the 1 month rest period,
and while
patients are taking maintenance Temodar. Patients are maintained on PLX3397
and
25 perampancl unless disease progression or evidence of drug-related
toxicity is observed.
Subjects treated according to this combinatorial protocol .µ ill show
substantially improved
clinical benefits over SOC or other conventional anti-cancer therapy. While
CSH R
inhibition is reported to provide pre-clinical anti-cancer benefitsresults
(Patwardhan et al,
2014: Yan et al. 2017: Quail et al_ 2016). most pre-clinical models of
different types of
30 cancer demonstrate acquired resistance throughout this treatment
(Patwardhan et al.. 2014;
Quail et al. 2016). In particular. for brain cancer, it has been shown that
insulin-like growth
factor I (IGF I ) induces glioma rebound in CSH R inhibitor-treated mice
(Quail et al, 2016).
39
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
Surprisingly. AMPA-glutanaate antagonism according to the methods described
here will
negate oncogenic effects of !GEL and AMPA-glutamate antagonism will complement
with
CSF I R inhibition to lower tumor burden in co-treated subjects. In more
detailed aspects of
the invention, it is noted that P'EX3397 may inhibit PDGFRB signaling in
cancer
5 (Patwardhan et al. 2014). and that PDGF'RB reportedly coordinates an anti-
oxidant program
in cancer through NRF2 transcription (Yang et al. 2018; Nanjaiah et al. 2019).
On this basis,
according to the teachings herein. PLX3397 will be beneficially combined with
glutamate
antagonists like memantine. within AMPAR antagonist methods of the invention,
to bolster
combined efficacy by suppressing an anti-oxidant program, e.g., in glioma
cells.
10 Example VIII
Anti-Cancer Activity of AMPAR Antagonists In Combination with
Anti-Malarial Drugs
Additional clinical methods of the invention employ an AMPAR antagonist such
as
perampanel coordinately administered with one or more anti-malarial drugs such
as
15 chloroquine, hydroxychloroquine. prima/wine and mefloquine (see. e.g.,
Johnson et al, 2015;
Liu et al, 2016: Maraka et al, 2019). In exemplary protocols. patients receive
perampanel as
above along with 250mg mefloquine orally prior to the first radiation session.
Within
illustrative methods for treating GBM. Metloquine is administered once every
two days
throughout the first 6-week treatment period, during the I month rest period,
and while
20 patients are taking maintenance Temodar. Patients are maintained on the
anti-malarial and
perampanel unless disease progression or evidence of drug-related toxicity is
observed.
Subjects treated according to this combinatorial protocol will show
substantially improved
clinical benefits over SOC or other conventional anti-cancer therapy.
Example VIX
25 Anti-Cancer Activity of AMPAR Antagonists In Combination
with
Metformin/Phenformin
Additional clinical methods of the invention employ an AMPAR antagonist such
as
perampanel coordinately administered with metformin (see, e.g.. Benjamin et
al.. 2016:
Maraka et al. 2019). Patients receive perampanel as above along with 500-
2000mg
30 metformin orally prior to the first radiation session. In exemplary
protocols for treating
GBM, peranapanel and metformin are administered once a day throughout the
first 6-week
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
treatment period., during the I month rest period, and while patients are
taking maintenance
Temodar. Patients are maintained on the metform in and perampanel unless
disease
progression or evidence of drug-related toxicity is observed. Subjects treated
according to
this combinatorial protocol will show substantially improved clinical benefits
over SOC or
5 other conventional anti-cancer therapy.
Example X
Anti-Cancer Activity of AMPAR Antagonists In Combination with
PD-I Inhibitor Treatment
Additional clinical methods of the invention employ an AMPAR antagonist such
as
10 perampanel coordinate!) administered with anti-cancer biologics
including programmed cell
death protein I (PD-1) inhibitors, such as pembroliitimab or nivolumab (see,
e.g., Nghiem et
al. 2016: Motzer et al. 2015). Resistance to PD- I antagonism has been
attributed to TNF-a
production in the tumor microenvironment (Neubert et al. 2018). Since Ampa-
glutamate
antagonism has been shown to reduce TNF-a secretion in a model of
intraventricular
15 hemorrhage (Dohare et al. 2016). AMPAR antagonist treatment according to
the invention
will augment the efficacy of PD- I inhibitors. In exemplary protocols,
patients are
administered perampanel as above, in conjunction with I-3mg/kg
pembrolizumabinivolumab
intravenously prior to the first radiation session. Within illustrative
methods for treating
GBM. patients then receive pembrolizumabinivolumab every 2 weeks throughout
the first 6-
20 week treatment period. during the 1 month rest period, and while
patients are taking
maintenance Temodar. Treatment is continued thusly unless disease progression
or evidence
of drug-related toxicity appears. Subjects treated according to this
combinatorial protocol
will show substantially improved clinical benefits over SOC or other
conventional anti-
cancer therapy.
25 Example XI
Anti-Cancer Activity of AMPAR Antagonists In Combination with
PD- I Inhibitor + CSF I R Inhibitor Treatment
Within more detailed examples. AMPAR antagonists such as perampanel are
coordinately administered with PD- I inhibitors, and also with CSF I R
inhibitors. Notably,
30 while PD-I inhibitors reportedl) exhibit robust efficacy in some
patients, they appear to have
little to no therapeutic effects in other patients. Recently. it has been
suggested that CSF I
41
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
and TN F-a secretion by tumor cells may stanch the efficacy of' PD-I therapy
(Neubert et al,
2018). As disclosed herein, patients be treated with a combination of AMPAR
antagonists
PD-1 inhibitors and CSF I R inhibitors (e.g.. with perampancl, pembrolizumab.
and PLX3397)
will benefit by reduced CSF1 signaling in the tumor mieroenvironment, combined
with
5 ampa-glutannate antagonist repression of `INF signaling (see, e.g..
Dohare et al, 2016).
negating PD-1 inhibitor resistance to yield enhanced clinical benefits over
SOC or other
conventional anti-cancer therapy.
,Exaniple XII
Anti-Cancer Activity of AMPAR Antagonists In Combination with
10 Clemastine Fumarate Co-Treatment
Additional clinical methods of the invention employ an AMPAR antagonist such
as
perampanel coordinately administered with clemastine fumarate (see, e.g.,
Diibbeling et al,
2013: Le Joncour et al. 2019). Patients are administered perm-vane' as above
along with 0.5-
2.68mg clemastine fumarate orally prior to the first radiation session. In
illustrative protocols
15 for treating GBM, perampanel and clemastine are taken once a day
throughout the first 6-
week treatment period_ during the I month rest period, and while patients are
taking
maintenance Temodar. Patients are maintained on the clemastine fumarate and
perampanel
unless disease progression or evidence of drug-related toxicity is observed.
Subjects treated
according to this combinatorial protocol will show substantially improved
clinical benefits
20 over SOC or other conventional anti-cancer therapy.
Example XIII
Anti-Cancer Activity of AMPAR Antagonists In Combination with
SSRI Co-Treatment
Additional clinical methods of the invention employ an AMPAR antagonist such
as
25 perampanel coordinately administered with one or more selective
serotonin reuptake
inhibitors (SSR1s) (see. e.g.. Sun et al. 2018: Huang et al. 2011: I,in et al.
2010: Liu et al,
2015: Yuan et al. 2018: Raabe & Gentile. 2008). In exemplary protocols for
treating CiBM,
patients are administered perampanel along with the SSRI(s) orally prior to
the first radiation
session, then once a day throughout the first 6-week treatment period, during
the 1 month rest
30 period, and while patients are taking maintenance Temodar. Patients are
maintained on the
SSRI and perampanel unless disease progression or evidence of drug-related
toxicity is
42
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
observed. Subjects treated according to this combinatorial protocol will show
substantially
improved clinical benefits over SOC or other conventional anti-cancer therapy.
Example XIV
Anti-Cancer Activity of AMPAR Antagonists In Combination with
5 TCA Co-Treatment
Additional clinical methods of the invention employ an AMPAR antagonist such
as
perampanel coordinately administered with one or more tricyclic
antidepressants (TCAs)
(see, e.g._ Jahchan et al, 2013; Jeon et al. 2011: Raabe & Gentile, 2008;
Reynolds & Miller,
1988: Semagor et al_ 1989: Stoll et al. 2007). Patients receive perampanel
along with the
10 TCA(s) orally prior to the first radiation session. In exemplary
protocols for treating GBM,
the perampanel and TCA are then each administered once a day throughout the
first 6-week
treatment period, during the I month rest period_ and while patients are
taking maintenance
Temodar. Patients are maintained on the TCA and perampanel unless disease
progression or
evidence of drug-related toxicity is observed. Subjects treated according to
this
15 combinatorial protocol will show substantially improved clinical
benefits over SOC or other
conventional anti-cancer therapy.
Example XV
Anti-Cancer Activity of AMPAR Antagonists In Combination with
Ampakine Co-Treatment
20 Additional clinical methods of the invention employ an AMPAR
antagonist such as
perampanel coordinately administered with one or more positive allosteric AMPA
receptor
modulators (Ampakines). In exemplary protocols. patients are administered
perampanenl in
combination with one or more ampakines. such as 2.3.6a.7.8.9-hexahydro-1 I H-
l.4-
dioxino[2.3-glpyrrolo[2.1-b][ 1.3Jbenzoxa7ine-I 1-one (-CX614-) (see. e.g.,
Radin et al.
25 2018). Though ampakines are thought to augment AMPA-mediated currents in
neurons, they
have also been reported to induce AMPA receptor desensitization and down
regulation via
endocytosis and degradation after prolonged treatment (Jourdi et al_ 2005),
whereby they may
serve as functional antagonists. Within illustrative methods for treating GBM,
patients are
administered perampanel along with CX614 or another amplakine orally prior to
the first
30 radiation session. then once a day throughout the first 6-week treatment
period, during the 1
month rest period, and while patients are taking maintenance Temodar. Patients
are
43
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
maintained on the ampakine(s) and perampanel unless disease progression or
evidence of
drug-related toxicity is observed. Subjects treated according to this
combinatorial protocol
will show substantially improved clinical benefits over SOC or other
conventional anti-
cancer therapy.
Example XVI
Anti-Cancer Activity of AMPAR Antagonists In Combination with
Cannabinoid Co-Treatment
Additional clinical methods of the invention employ an AMPAR antagonist such
as
perampanel coordinately administered with one or more cannabinoids, for
example
tetrahydroeannabinol (-111C) and/or cannabidiol (CBD) (see. e.g.. Scott et al,
2014; Mareu et
al. 2010; Shrisastava et al. 2011). In exemplary protocols for treating GBM,
patients are
administered perampanel along with I 00-600mg CID and 1-100ing T-IC prior to
the first
radiation session. then once a day throughout the first 6-week treatment
period, during the 1
month rest period, and while patients are taking maintenance Ternodar.
Patients are
maintained on the cannabinoid and perampanel therapy unless disease
progression or
evidence of drug-related toxicit) is observed. Subjects treated according to
this
combinatorial protocol will show substantially improved clinical benefits over
SOC or other
conventional anti-cancer therapy. For treatment of GBM. cannabinoids have been
reported to
potent effects on glioma stem cells (Lepez-Valero et al_ 2018). Considering
that AMPA
receptors are overexpressed on glionia stem cells (Oh et al. 2012). the
combinatorial
treatment methods and compositions described here w ill sensitize resistant
tumor cells to the
DNA-damaging effects of Ternodar and radiation therapy (McLendon et al_ 2006;
Chen et al,
2012) and thereby enhance clinical benefits. Further. cannabinoids reportedly
exert oncolytic
effects through induction of harmful reactive oxygen species (Shrivastava et
al, 2011;
Nanjaiah et al, 2019). whereby the methods and compositions of the invention
combining
cannabinoids with glutamate antagonists will negate antioxidant defenses in
cancer cells and
enhance clinical benefits. particularly in elioma patients.
Example XVI
.Anti-Cancer Activitv of AMPAR Antagonists In Combination with
Distil firam Co-Treatment
44
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
Yet additional clinical methods of the invention employ an AMPAR antagonist
such
as perampanel coordinately administered with disulfirarn (see, e.g., Lun et
al, 2016; Triscon
et al, 2012). Disulfiram targets cancer stem cells and reportedly inhibits
MGMT to boost
efficacy of Temodar (Paranjpe et al. 2014). Within the methods of the
invention, both
5 disulfiram and perampanel augment Temodar's efficacy and refine targeting
of cancer cells,
yielding surprisingly enhanced benefits for treating SOC treatment-resistant
cancers. Within
exemplary methods for treating GBM. patients are administered perampanel along
with 50-
500mu disulfiram orally prior to the first radiation session, then once daily
throughout the
first 6-week treatment period. during the 1 month rest period, and while
patients are taking
10 maintenance Temodar. Patients are maintained on the perampanel and
disulfiram unless
disease progression or evidence of drug-related toxicity is observed. Subjects
treated
according to this combinatorial protocol will show substantially improved
clinical benefits
over SOC or other conventional anti-cancer therapy.
The instant description and examples are provided lor illustration, and those
skilled in
15 the art will realize that the invention extends to additional
embodiments and aspects
following the teachings herein, and is therefore not limited except as by the
appended claims.
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
References
1. Grossman, S. A., Ye, X., Chamberlain. M., Mikkelsen, T.. Batchelor, T.,
Desideri, S.,
... Fine, H. A. (2009). Talampanel with standard radiation and temozolomide in
patients with newly diagnosed glioblastorna: A multicenter phase 11 trial.
Journal of
5 Clinical Oncology, 27(25), 4155-416 I .
2. Gidal. B. E., Laurenza. µA.. Hussein. Z., Yang, H., Fain, It, Edelstein,
J., ... Ferry, J.
(2015). Perampanel efficacy and tolerability with enzyme-inducing AEDs in
patients
with epilepsy. Neurology, 84(19), 1972-1980.
3 Bobustuc, G. C., Baker, C. H.. Limaye. A., Jenkins, W. D., Pearl, G.,
Avgeropoulos.
10 N. G., & Konduri. S. D. (2010). Levetiracetam enhances p53-
mediated MGMT
inhibition and sensitizes glioblastoma cells to temozolomide. Neuro-Oncology,
12(9),
917-927.
4 Kanemura, H., Sano, F., & Aihara. M. (2019).
Usefulness of perampanel with
concomitant levetiracetam for patients with drug-resistant epilepsy. European
Journal
15 q-Paediatric- Neurology, 23(1). 197-203.
5. Kim, Y._ Kim, it Jot), J., Han. J. H.. Kim, Y.
J., Kim. I. A._ . Kim, C. (2015).
Survival benefit of levetiracetam in patients treated with concomitant
chemoradiotherapy and adjuvant chemotherapy with temozolomide for glioblastoma
multiforme. Cancer. 121(17). 2926-2932.
20 6 Stepulak, A.. Sifringer, M. Rzeski. W., Endesfelder, S.. Gratopp, A.,
Pohl, E. E,
lkonomidou. C. (2005). NMDA antagonist inhibits the extracellular signal-
regulated
kinase pathway and suppresses cancer growth. Proceedings of the National
Academy
.Sc.lences the United States of America, 102(43), I 5605- l 5610.
7 Maraka, S., Groves, M. D., Mammoser. A. G., Melguizo-Gavilanes, 1., Conrad,
C. A.,
25 Tremont-Lukats, I. W.,
Penas-Prado, M. (2019). Phase 1 lead-in to a phase 2
factorial study of temozolomide plus memantine, mefloquine, and metformin as
postradiation adjuvant therapy for newly diagnosed glioblastoma. Cancer,
125(3),
424-433.
8 Zanos, P., Moaddel. It. Morris. P. J., Georgiou,
P.. Fischell, J., Elmer, G. 1., .
30 Gould, T. D. (2016). NMDAR inhibition-independent antidepressant
actions of
ketamine metabolites. Nature, 533(7604), 481-486.
9 Malsy, M.. Gebhardt, K.. (lather, M., Wiese. C.. Graf, B.. & Bundscherer, A.
(2015).
Effects of ketamine. s-ketamine. and MK 801 on proliferation, apoptosis. and
necrosis
in pancreatic cancer cells. BAK' Anesthesiologv, 15(1). I 11-118.
35 to Khan, A. J., LaCava, S., Mehta, M., Schiff. D., Thandoni. A.,
Jhawar, S., ... Chen, S.
(2019). The glutamate release inhibitor riluzole increases DNA damage and
enhances
cvtotoxicity in human glioma cells_ in vitro and in vivo. Oncotarget, /0(29),
2824-
2834.
it Van, D., Kovyal, J., Akkari_ L., Schuhmacher_ A. J., Huse. J. T.. West, B.
L., & Joyce,
40 J. A. (2017). Inhibition of colony stimulating factor-1 receptor
abrogates
microenvironment-mediated therapeutic resistance in gliomas. Oncogene, 36(43).
6049-6058.
12 Butowski, N., Colman, H., De Groot, J. F.. Omuro, A. M., Nayak, L., Wen, P.
Y., . .
Prados. M. (2016). Orally administered colony stimulating factor 1 receptor
inhibitor
45 PLX3397 in recurrent glioblastoma: An ivy foundation early phase
clinical trials
consortium phase It study. Nettro-Oncoloff, /8(4). 557-564.
13. Patwardhan. P. P., Surriga, 0.. Beckman, M. J., de Stanehina. E.,
Dematteo, R. P.,
Tap. W. D.. & Schwartz. G. K. (2014). Sustained inhibition of receptor
tyrosine
kinases and macrophage depletion by PLX3397 and rapamycin as a potential new
46
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
approach for the treatment of MPNSTs. Clinical Cancer Research 20(12), 3 I 46-
3158.
14. Yan, D., Kowal, J., Akkari, L., Schuhmacher, A. J., Huse, J. T., West, B.
L., & Joyce,
J. A. (2017). Inhibition of colony stimulating factor-I receptor abrogates
5 microenvironment-mediated therapeutic resistance in gliomas.
Oncogene, 36(43),
6049-6058.
15. Quail, D. F., Bowman, R. L., Akkari, L., Quick, M. L., Schuhmacher, A. J.,
Huse, J.
T.,.. . Joyce, J. A. (2016). The tumor microenvironment underlies acquired
resistance
to CSF-I R inhibition in gliomas. Science. 352(6288).
10 16. Stepulak, A., Sifringer, M., Rzeski, W., Brocke, K., Gratopp, A.,
Pohl, E. F.....
lkonomidou, C. (2007). AMPA antagonists inhibit the extracellular signal
regulated
kinase pathway and suppress lung cancer growth. Cancer Biology & Therapy,
6(12),
1908.
17 Yang, Z.. Yang. Y., Deng. Y., Chen. Y., Chen. X., Zhang. L..... Liu, B.
(2018).
15 Inhibition of PDGFR by CP-67345I induces apoptosis and increases
cisplatin
cytotoxicity in NSCI.0 cells via inhibiting the Nr12-mediated defense
mechanism. 7in/eulogy Letters, 295, 88-98.
ia Nanjaiah, N. D., Ramaswamy, P., Goswarni. K., Hurmath Fathima, K., &
Borkotokey, M. (2019). Survival of glioblastoma cells in response to
endogenous and
20 exogenous oxidative challenges: Possible implication of NMDA
receptor mediated
regulation of redox homeostasis. Cell Biology International, 11193. 1-10.
is. Johnson, C. E., Hunt, D. K., Wiltshire, M., Herbert, T. P., Sampson, J.
R., Errington,
R. J.,.,. . Tee, A. R. (2015). Endoplasmic reticulum stress and cell death in
mTORC1-
overactive cells is induced by nelfinavir and enhanced by chloroquine.
Molecular
25 Oncofoff, 9(3), 675-688.
20 Liu, Y., Chen. S... Xue. R.. Zhao, J.. & Di. M. (2016). Mefloquine
effectively targets
gastric cancer cells through phosphatase-dependent inhibition of
P13K/Akt/inTOR
signaling pathway. Biochemical and Biophysical Research Communications,
470(2),
350-355.
30 21 Benjamin, D., Colombi, M.. Hindupur, S. K.. Betz. C.. Lane* H. A.,
EI-Shemerly, M.
Y. M..... Hall, M. N. (2016). Syrosingopine sensitizes cancer cells to killing
by
metformin. Science Advances. 2(12), el60 I 756-el 601 756.
22 Nghiem. P. T., Bhatia. S., Lipson. E. J.. Kudchadkar, R. It.. Miller, N.
J.., Annamalai,
L..... Cheever, M. A. (2016). PD-1 blockade with pembrolizunaab in advanced
35 merkel-cell carcinoma. The New England Journal of Medicine,
374(26), 2542-2552.
23. Motzer, R. J., Escudier, B., McDermott. D. F., George, S., Hammers, H. J.,
Srinivas,
S., .. CheckMate 025 Investigators. (2015). Nivolumab versus everolimus in
advanced renal-cell carcinoma. The Neu, England Journal of Medicine. 373(19),
1803-1813.
40 24 Neubert. N. J., Schmittnaegel, M., Bordry, N., Nassiri, S., Wald,
N., Martignier, C.,..
. Speiser. D. E. (2018). T cell-induced CSF I promotes melanoma resistance to
PDI
blockade. Science Translational Medicine. 10(436), 1-14.
25 Dohare. P., Zia. M. T.. Ahmed. E.. Ahmed. A.. Yadala. V.. Schober, A. L., .
.
Ballabh, P. (2016). AMPA-kainate receptor inhibition promotes neurologic
recovery
45 in premature rabbits with intraventricular hemorrhage...!
Neuravci. 36(11), 3363-
3377.
26 DObbeling. U.. Waeckerle-Men. Y._ Zabel, F.. Graf. N., Ktindig, T. M., ez.
Johansen,
P. (2013). The antihistamines clemastine and desloratadine inhibit STAT3 and e-
myc
activities and induce apoptosis in cutaneous T-cell lymphoma cell lines.
Evperimental
50 Dennatology, 22(2),, 119.
47
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
2T Le Joncour, V., Filppu, P., Hyvonen, M., Holopainen, M., Turunen, S. P.,
Sihto, H,..
. Laakkonen. P. (2019). Vulnerability of invasive glioblastoma cells to
lysosorrial
membrane destabilization. EMBO Molecular Medicine, 1/(6), e9034.
28 Sun. D., Zhu, L., Zhao, Y., Jiang, Y.. Chen, L., Yu, Y., & Ouyang, L.
(2018).
5 Fluoxetine induces autophagic cell death via eET2K-AMPK-mTOR-ULK
complex
axis in triple negative breast cancer_ Cell Prolijeration, 5/(2). e 12402.
29. Huang, J., Chang, EL, Chou, C., Shu, S., Kuo, C., Tsai, J.,
Jan, C. (2011). The
mechanism of sertraline-induced [Ca2-1-li rise in human PC3 prostate cancer
cells:
Sertraline and human prostate cancer cells. Basic & Clinical Pharmacology &
10 Toxicology. 109(2), 103-110.
30 Lin. C., Robert_ F., Sukarieh, R., Michnick, S.. & Pelletier, J. (2010).
The
antidepressant sertraline inhibits translation initiation by curtailing
mammalian target
of rapamycin signaling. Cancer Research, 70(8), 3199-3208.
31. Yuan, I., Honig, C.. Chen, V. C., Chen, C.. Chen, L., Hsu, T., & Tzang, B.
(2018).
15 Escitalopram oxalate inhibits proliferation and migration and
induces apoptosis in
non-small cell lung cancer cells. Oncology Letters; 15(3), 3376-3382.
32 Sun, D., Zhu. L., Zhao, Y., Jiang, Y., Chen, L., Yu, Y., & Ouyang, L.
(2018).
Fluoxetine induces autophagic cell death via eET2K-AMPK-mTOR-ULK complex
axis in triple negative breast cancer. Cell Praliferation, 51(2), e I 2402.
20 33 Liu, K., Yang, S., Lin. Y., Lin, J.. Lee. Y., Wang, J.... . Shen,
S. (2015). Fluoxetine,
an antidepressant, suppresses glioblastoma by evoking AM PAR-mediated calcium-
dependent apoptosis. Oncotarget, 6(7), 5088-5101.
34. Raabe, R.. & Gentile, L. (2008). Antidepressant interactions with the NMDA
NR1-lb
subunit. Journal of Biophysics, 2008. 474205-8.
25 35. Jahchan, N. S., Dudley. J. -1`., Mazur, P. K., Flores. N., Yang,
D., Palmerton, A.,
Sage, J. (2013). A drug repositioning approach identifies tricyclic
antidepressants as
inhibitors of small cell lung cancer and other neuroendocrine tumors. Cancer
Discovery, 3(12), 1364-1377.
36. icon, S... Kim, Y. S., Kim. Y., Kim, S. H., Lim, Y., Lee, Y. H.. & Shin,
S. Y. (2011).
30 The tricyclic antidepressant imipramine induces autophagic cell
death in 1.1-87MG
glioma cells. Biochemical and Biophysical Research Communications, 4/3(2), 311-
3 I 7.
37. Reynolds, 1. J., & Miller, R. J. (1988). Tricyclic antidepressants block N-
methyl-D-
aspartate receptors: Similarities to the action of zinc. British Journal of
35 Pharmacology, 95(1). 95-102.
38 Sernagor, E., Kuhn, D., Vyklicky, L.. & Mayer, M. L. (1989). Open channel
block of
NMDA receptor responses evoked by tricyclic antidepressants. Neuron, 2(3),
1221-
1227.
39. Stoll, L., Seguin, S., & Gentile, L. (2007). Tricyclic antidepressants,
but not the
40 selective serotonin reuptake inhibitor fluoxetine, bind to the SI
S2 domain of AMPA
receptors. Archives of Biochemistry and Biophysics, 458(2). 213-219.
40 Raclin. D. P.. Purcell. R.. & Lippa. A. S. (2018). Oncolytic properties of
ampakines in
vitro. Anticancer Research. 38(1). 265-269.
41 Jourdi, FL_ Lu, X.. Yanagihara. T., Latitcrborn. J. C.. Hi, X.. Gall, C.
M., & BaucIry,
45 M. (2005). Prolonged positive modulation of alpha-amino-3-hydroxy-
5-methy1-4-
isoxazolepropionic acid (AMPA) receptors induces calpain-mediated PSD-
95/DIWZO-1 protein degradation and AMPA receptor down-regulation in cultured
hippocampal slices. The Journal of Pharmacology and Experimental
Therapeutics, 314(1), 16.
48
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
42 Scott, K. A., Dalgleish, A. G., & Liu, W. M. (2014). The combination of
eannabidiol
and A9-tetrahydrocannabinol enhances the anticancer effects of radiation in an
orthotopic murine glioma model. Molecular Cancer Therapeutics, 13(12), 2955-
2967.
43. Marcus J. P._ Christian. R. T.. Lau, D.. Zielinski. A. J., Horowitz, M.
P.,
5 McAllister. S. D. (2010). Cannabidiol enhances the inhibitory
effects of A9-
tetrahydrocannabinol on human glioblastoma cell proliferation and
survival. Molecular Cancer Therapeutics, 9(1), 180.
44. Shrivastava. A., Kuzontkoski, P. M., Grooptnan, J. E., & Prasad, A.
(2011).
Cannabidiol induces programmed cell death in breast cancer cells by
coordinating the
10 cross-talk between apoptosis and autophagy. Molecular Cancer
Therapeutics, 10(7),
1161-1172.
45. LOpez-Valero, I., Saiz-Ladera. C, Torres, S., Hernandez-Tiedra, S., Garcia-
Taboada,
E., Rodriguez-Fornes.
. Velasco, G. (2018). Targeting
glioma initiating cells
with A combined therapy of eannabinoids and temozolomide.Biochemical
1_5 Pharmacology, 157, 266-274.
46. Oh, M. C., Kim, J. M., Safaee, M., Kaur. a, Sun, M. Z.. Kaur_ R.,
Parsa, A. T.
(2012). Overexpression olcalcium-permeable glutamate receptors in glioblastoma
derived brain tumor initiating cells. PIUS One, 7(10). e47846-e47846.
47. McLendon, R. E., Hjelmeland, A. B., Dewhirst, M. W., Shi, Q., Rich, J. N.,
Wu. Q.,.
20 Hao. Y. (2006). Glioma stem cells promote radioresistance by
preferential
activation of the DNA damage response. Nature, 444(712(1), 756-760.
48 Chen, J., Li_ Y., Yu, T., McKay, R. M., Burns, D. K., Kernie, S. G., &
Parada, L. F.
(2012). A restricted cell population propagates glioblastoma growth after
chemotherapy. Nature, 488(7412), 522-526.
25 49 Lun, X., Wells, J. C., Grinshtein, N., King, J. C., Mao, X., Dang,
N., ... Senger, D. L.
(2016). Disulftram when combined with copper enhances the therapeutic effects
of
temozolomide for the treatment of glioblastoma. Clinical Cancer Research,
22(15),
3860-3875.
50 Triscott, J., Lee.. C., Hu. IC Fotovati, A.. Berns, R., Pambid_ M., ...
Dunn, S. E.
30 (2012). Disulfiram. a drug widely used to control alcoholism,
suppresses the self-
renewal of glioblastoma and over-rides resistance to
temozolomide. maim-gel. 3(10). 1112-1123.
51 Paranjpe, A., Zhang, R., Ali-Osman, F., Bobustuc. G. C., & Srivenugopal, K.
S.
(2014). Disulfiram is a direct and potent inhibitor of human 06-methylguanine-
DNA
35 methyltransferase (MGMT) in brain tumor cells and mouse brain and
markedly
increases the alkylating DNA damage. Carcinogenesis, 35(3), 692-702.
52 Merrier. A.. Sattliunaite. D.. Michalski_ C. W.. Erkan. M., De Oliveira.
T., Abiatari. 1..
. KleelT, J. (2011). Glutamate increases
pancreatic cancer cell invasion and
migration via AMPA receptor activation and Kras-MAPK signaling. in/i Cancer,
40 129(10). 2349-2359.
53 iShiliChi. S.. Tsuzuki, K.. Yoshida, Y._ Yamada. N... Magimura, N., Okado,
H....
07awa, S. (2002). Blockage of Ca(2+)-permeable AMPA receptors suppresses
migration and induces apoptosis in human glioblastoma cells. Nat Med, 8(9),
971-
978.
49
CA 03145507 2022-1-24
WO 2020/023800
PCT/US2019/043525
Ishiuchi, S.. Yoshida. Y.. Sugawara, K.. Aihara, M., Ohtani, T., Watanabe, T.,
. .
Ozawa. S. (2007). Ca2+-permeable AMPA receptors regulate growth of human
glioblastoma via Akt activation. J Neurosci. 27(30), 7987-8001.
55. von Roetneling, C. A., Radisky, D. C., Marlow. L. A.. Cooper, S. J.,
Grebe, S. K.,
5 Anastasiadis, P. Copland. J. A. (2014). Neuronal
pentraxin 2 supports clear
cell renal cell carcinoma by activating the AMPA-selective glutamate receptor-
4.
Cancer Res, 74(17). 4796-4810.
5. Prasad, G., Sottero. T.. Yang. X.., Mueller. S.. James. C. D.. Weiss. W.
A., .. . Haas-
Kogan. D. A. (2011). Inhibition of P13K/mTOR pathways in glioblastoma and
10 implications for combination theram, \ ith ternozolomide. Neut.()
Oncol. /3(4). 384-
_392.
57 Piao. V.. Lu. L.. & de Groot. J. (2009). ANIPA receptors promote
perivascular glioma
invasion via beta I inteurin-dependent adhesion to the extracellular matrix.
Neuro
Oncol, 11(3).260-273.
15 58 Hu. H._ Takano. N.. Xiang. L.. Gilkes. D. M.. Luc,.
Semenza. G. L. (2014).
Hypoxia-inducible factors enhance glutamate signaling in cancer cells.
Oncotarget,
5(19). 8853-8868.
59. Takano. T., Lin, J. H., Areuino. G., Gaff. Q.. Yana, J., & Nedergaard, M.
(2001).
Glutamate release promotes growth of malignant tzliomas. Na! Med, 7(9), 1010-
1015.
20 so Rzeski. W., Turski. L. & lkonomidou. C. (2001). Glutamate
antagonists limit tumor
growth. Prot- Nail Acad Sci US A. 98(11). 6372-6377.
61 Chen. C.. Maw In. Hell_ J. W._ JEL Rogawski. M. A. (2014). Perampanel
inhibition of
AMPA receptor currents in cultured hippocampal neurons. PloS One, 9(9),
e108021.
62 Patsalos. PN. (2015). The clinical pharmacology
profile of the new antiepileptie drug
25 perampanel: A novel noncompetitive AMPA receptor antagonist.
Epilepsia. 56(1).
12-27.
63 Biancur. D. E.. Paulo. J. A... Malachowska. B.. Maria Quiles Del Rey.
Sousa, C. M.,
Wang. X..... Kimmelman. A. C. (2017). Compensatory metabolic networks in
pancreatic cancers upon perturbation of glutamine metabolism. Nature
30 Communications, N. 15965.
CA 03145507 2022-1-24