Note: Descriptions are shown in the official language in which they were submitted.
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
DUAL ATM AND DNA-PK INHIBITORS FOR USE IN ANTI-TUMOR THERAPY
FIELD OF THE INVENTION
The invention relates to compounds and pharmaceutically acceptable salts
thereof and methods of their
use for the treatment of cancer as a monotherapy or in combination with
radiotherapy, chemotherapy,
and/or immunotherapy.
BACKGROUND OF THE INVENTION
Several members of the PIKK (PI-3K-like Kinase) family of serine-threonine
kinases are known mediators
of DNA damage signaling.
Radiation therapy (RD is used to treat >50% of all cancer patients at some
point during their illness.
Despite significant effort, previous approaches to develop clinical
radiosensitizers have not been highly
effective, primarily as a result of targeting non-specific pathways which are
not direct regulators of the
cellular response to radiation.
There is a need for new therapies for oncological diseases.
SUMMARY OF THE INVENTION
In general, the invention provides a compound of formula (I):
h0
R1 N
N¨ R4
HN
(R3) I
0
(I)
or a pharmaceutically acceptable salt thereof,
where
Z is CH, CR3, or N;
Y is CHR5 or NR6;
n is 0, 1,2, 0r3;
R1 is ¨0¨L¨N(R7)2 or optionally substituted, four-memberred, saturated N-
heterocyclyl;
R2 is C1-3 alkyl;
each R3 is independently halogen;
R4 is optionally substituted alkyl;
R5 is hydrogen, optionally substituted C1_3 alkyl, or benzyloxy;
R6 is optionally substituted C1_3 alkyl;
each R7 is independently H or optionally substituted C1_3 alkyl; and
L is optionally substituted ethylene.
1
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
In some embodiments, n is 1. In certain embodiments, the compound is a
compound of formula (IA):
0
N
N-R
HN 4
R2- %=:--s0 R3
0
(IA)
or a pharmaceutically acceptable salt thereof.
In particular embodiments, R3 is halogen (e.g., fluorine). In further
embodiments, n is 0.
In yet further embodiments, the compound is a compound of formula (16):
R1 N iliP
-R4
HN
0
(IB)
or a pharmaceutically acceptable salt thereof.
In still further embodiments, R1 is -0-L-N(R7)2. In some embodiments, one R7
is H, and the remaining
R7 is optionally C1_3 alkyl. In certain embodiments, at least one R7 is
isopropyl. In particular
embodiments, R2 is methyl, ethyl, or isopropyl. In further embodiments, R2 is
methyl. In yet further
embodiments, R4 is methyl. In yet further embodiments, Y is CHR5. In still
further embodiments, R5 is
hydrogen. In other embodiments, R5 is optionally substituted C1_3 alkyl. In
yet other embodiments, R5 is
benzyloxy. In still other embodiments, Y is NR6. In some embodiments, R6 is
optionally substituted C3
alkyl. In certain embodiments, R6 is isopropyl.
In particular embodiments, the compound is selected from the group consisting
of:
)NO 0
0
NH
0==0 NH
F 0==0
0
0
N¨
N¨
NH
0==0 NH
F 0==0
2
CA 03147111 2022-01-11
WO 2021/022078 PCT/US2020/044322
BnO,õ BnO,õ
,....j..., NON ....., 0 1
....--õ,,,õ0 .......N 0
.,,i,/(
I I
H N.¨ H N.¨
HN HN
I I
0=S=0 0=S=0
F N F N
I I
0
N ....--..........õ.0 õ....N )NC.1 iq 1
I N-- 1 H N.¨
H
NH NH \
1 1
0=S=0 0=S=0
I F N I F N
I
Nr.....1
I N¨.
NH
0=S=0
I N
and pharmaceutically acceptable salts thereof.
In further embodiments, the compound is of the following structure:
0
)NO )si
I N¨.
H \
NH \
0==0
F N
or a pharmaceutically acceptable salt thereof.
In yet further embodiments, the compound is of the following structure:
0
)NO )q
I N¨
H
NH \
0==0
I F N
or a pharmaceutically acceptable salt thereof.
In still further embodiments, the compound is of the following structure:
0
,...../. .....--.........õ.0 .....,N
N
I N¨
H
NH \
0=S=0
F N
or a pharmaceutically acceptable salt thereof.
3
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
In another aspect, the invention provides a pharmaceutical composition
including the compound of the
invention or a pharmaceutically acceptable salt thereof and a pharmaceutically
acceptable excipient.
In yet another aspect, the invention provides a method of treating an
oncological disease (e.g., cancer,
e.g., those cancers described herein) by administering a therapeutically
effective amount of the
compound of the invention, or a pharmaceutically acceptable salt thereof, or
the pharmaceutical
composition of the invention to a patient in need thereof. In still another
aspect, the invention provides
pharmaceutical compositions for use in the treatment of an oncological disease
(e.g., cancer, e.g., those
cancers described herein). The pharmaceutical compositions include the
compound of the invention. In
a further aspect, the invention provides use of the compound of the invention
in the manufacture of a
medicament for the treatment of an oncological disease (e.g., cancer, e.g.,
those cancers described
herein). In some embodiments, the oncological disease is a brain cancer,
bladder cancer, breast cancer,
central nervous system cancer, cervical cancer, colon cancer, endometrial
cancer, esophageal cancer,
gastrointestinal stromal tumor, gastric cancer, head and neck cancer, buccal
cancer, cancer of the
mouth, hepatocellular cancer, lung cancer, melanoma, Merkel cell carcinoma,
mesothelioma,
nasopharyngeal cancer, neuroblastoma, osteosarcoma, ovarian cancer, pancreatic
cancer, prostate
cancer, renal cancer, salivary gland cancer, sarcomas, testicular cancer,
urothelial cancer, vulvar cancer,
or Wilm's tumor. In further embodiments, the oncological disease is a breast
cancer, lung cancer, head
and neck cancer, pancreatic cancer, rectal cancer, glioblastoma,
hepatocellular carcinoma,
cholangiocarcinoma, metastic liver lesions, melanoma, bone sarcoma, soft
tissue sarcoma, endometrial
cancer, cervical cancer, prostate cancer, or Merkel cell carcinoma.
In some embodiments, the patient is receiving radiotherapy. In certain
embodiments, the compound or
the pharmaceutical composition is administered to the patient concomitantly
with the radiotherapy. In
particular embodiments, the compound or the pharmaceutical composition is
administered to the patient
before radiotherapy. In further embodiments, the compound or the
pharmaceutical composition is
administered to the patient after radiotherapy. In yet further embodiments,
the radiotherapy comprises
external, internal, brachytherapy, or systemic exposure, e.g., with a
radionuclide (e.g., a [3-emitting
radionuclide (e.g., 32Phosphorus, 87Copper, 7713romine, 89Strontium,
90Yttrium, 105Rhodium, 131I0dine,
137Cesium, 149Prometheum, 153Samarium, 1881-lolmium, 177Lutetium, 188Rhenium,
188Rhenium, or 199Gold),
a-emitting radionuclide (e.g., 211Astatine, 21313ismuth, 223Radium,
225Actinium, or 227Thorium), y-ray
emitting radionuclide (e.g., 192Iridium), or electron capturing radionuclides
(e.g., 87Gallium, 103Palladium, or
125I0dine)), antibody radionuclide conjugate (e.g., 90Y-ibritumomab tiuxetane,
1311-tositumomab, 225AC-
lintuzumab satetraxetan, 227Th-anetumab corixetan, 90Y-epitumomab cituxetan,
90Y-clivatuzumab
.. tetraxetan, 177Ludilotomab satetraxetan, 90Y-rosopatamab tetraxetan, 90Y-
tabituximab barzuxetan, or 9 Y-
tacatuzumab tetraxetan), or another targeted radionuclide conjugate (e.g.,
131I-PSMA, 90Y-PSMA, 177Lu-
PSMA, or 177Lu-satoreotide tetraxetan). Preferably, the radiotherapy comprises
administering an antibody
radionuclide conjugate. In still further embodiments, the patient is receiving
an anti-tumor agent. In other
embodiments, the anti-tumor agent is one or more of cisplatin, oxaliplatin,
carboplatin, valrubicin,
idarubicin, calicheamicin, or a PARP inhibitor. In yet other embodiments, the
anti-tumor agent is an anti-
tumor biological agent and/or anti-tumor immunotherapeutic agent. In still
other embodiments, the
compound or the pharmaceutical composition is administered to the patient
concomitantly with the anti-
4
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
tumor agent. In some embodiments, the compound or the pharmaceutical
composition is administered to
the patient before the anti-tumor agent. In certain embodiments, the compound
or the pharmaceutical
composition is administered to the patient after the anti-tumor agent.
DETAILED DESCRIPTION
DEFINITIONS
It is to be understood that the terminology employed herein is for the purpose
of describing particular
embodiments, and is not intended to be limiting. Further, although any
methods, devices and materials
similar or equivalent to those described herein can be used in the practice or
testing of the invention, the
preferred methods, devices and materials are now described. In addition to the
foregoing, as used in the
specification and appended claims, unless specified to the contrary, the
following terms have the meaning
indicated:
"Amino" refers to the -NH2 radical.
"Cyano" refers to the -CN radical.
"Hydroxyl" refers to the -OH radical.
"Imino" refers to the = NH substituent.
"Nitro" refers to the -NO2 radical.
"Oxo" refers to the = 0 substituent.
"Thioxo" refers to the = S substituent.
"Trifluoromethyl" refers to the -CF3 radical.
"Alkyl" refers to a linear, saturated, acyclic, monovalent hydrocarbon radical
or branched, saturated,
acyclic, monovalent hydrocarbon radical, having from one to twelve carbon
atoms, preferably one to eight
carbon atoms or one to six carbon atoms, and which is attached to the rest of
the molecule by a single
bond, e.g., methyl, ethyl, n-propyl, 1-methylethyl (iso-propyl), n-butyl, n-
pentyl, 1,1-dimethylethyl (t-butyl),
3-methylhexyl, 2-methylhexyl and the like. An optionally substituted alkyl
radical is an alkyl radical that is
optionally substituted, valence permitting, by one, two, three, four, or five
substituents independently
selected from the group consisting of halo, cyano, nitro, aryl, cycloalkyl,
heterocyclyl, heteroaryl, oxo,
trimethylsilanyl, -0R14, -0C(0)-R147 _N(R14)27 _ C(0)R157 -C(0)0R147 -
C(0)N(R14)27
-N(R14)C(0)0R16, -N(R14)C(0)R16, -N(R14)S(0)tR16 (where t is 1 or 2), -
S(0)tOR16 (where t is 1 or 2), -
S(0)R16 (where p is 0, 1, or 2) and -S(0)IN(R14)2 (where t is 1 or 2), where
each R14 is independently
hydrogen, alkyl, haloalkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl,
heterocyclyl, or heteroaryl; each R15 is
independently hydrogen, cycloalkyl, aryl, heterocyclyl, or heteroaryl; and
each R16 is independently alkyl,
haloalkyl, cycloalkyl, aryl, heterocyclyl, or heteroaryl.
"Alkenyl" refers to a linear, acyclic, monovalent hydrocarbon radical or
branched, acyclic, monovalent
hydrocarbon radical, containing one, two, or three carbon-carbon double bonds,
having from two to
twelve carbon atoms, preferably two to eight carbon atoms and which is
attached to the rest of the
molecule by a single bond, e.g., ethenyl, prop-1-enyl, but-1-enyl, pent-1-
enyl, penta-1,4-dienyl and the
like. An optionally substituted alkenyl radical is an alkenyl radical that is
optionally substituted, valence
permitting, by one, two, three, four, or five substituents independently
selected from the group consisting
5
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
of: halo, cyano, nitro, aryl, cycloalkyl, heterocyclyl, heteroaryl, oxo,
trimethylsilanyl, -0R14, -0C(0)-R14, -
N(R14)2, -C(0)R15, -C(0)0R14, -C(0)N(R14)2, -N(R14)C(0)0R16, -N(R14)C(0)R16, -
N(R14)S(0)tR16 (where t
is 1 or 2), -S(0)tOR16 (where t is 1 or 2), -S(0)R16 (where p is 0, 1, or 2)
and -S(0)IN(R14)2 (where t is 1
or 2), where each R14 is independently hydrogen, alkyl, haloalkyl, cycloalkyl,
aryl, heterocyclyl, or
.. heteroaryl; each R15 is independently hydrogen, cycloalkyl, aryl,
heterocyclyl, or heteroaryl; and each R16
is independently alkyl, haloalkyl, cycloalkyl, cycloalkylalkyl, aryl,
heterocyclyl, or heteroaryl.
"Alkynyl" refers to a linear, acyclic, monovalent hydrocarbon radical or
branched, acyclic, monovalent
hydrocarbon radical, containing one or two carbon-carbon triple bonds and,
optionally, one, two, or three
carbon-carbon double bonds, and having from two to twelve carbon atoms,
preferably two to eight carbon
atoms and which is attached to the rest of the molecule by a single bond,
e.g., ethynyl, prop-1-ynyl,
but-1-ynyl, pent-1-ynyl, penta-1-en-4-ynyl and the like. An optionally
substituted alkynyl radical is an
alkynyl radical that is optionally substituted by one, two, three, four, or
five substituents independently
selected from the group consisting of: halo, cyano, nitro, aryl, cycloalkyl,
heterocyclyl, heteroaryl, oxo,
trimethylsilanyl, -0R14, -0C(0)-R14, -N(R14)2, -C(0)R15, -C(0)0R14, -
C(0)N(R14)2, -N(R14)C(0)0R16,
-N(R14)C(0)R16, -N(R14)S(0)tR16 (where t is 1 or 2), -S(0)tOR16 (where t is 1
or 2), -S(0)R16 (where p is
0, 1, or 2) and -S(0)IN(R14)2 (where t is 1 or 2) where each R14 is
independently hydrogen, alkyl,
haloalkyl, cycloalkyl, aryl, heterocyclyl, or heteroaryl; each R15 is
independently hydrogen, cycloalkyl, aryl,
heterocyclyl, or heteroaryl; and each R16 is independently alkyl, haloalkyl,
cycloalkyl, aryl, heterocyclyl, or
heteroaryl.
"Alkylene" or "alkylene chain" refers to a linear, acyclic, saturated,
divalent hydrocarbon chain or
branched, acyclic, saturated, divalent hydrocarbon chain, having from one to
twelve carbon atoms, e.g.,
methylene, ethylene, propylene, n-butylene, and the like. The alkylene chain
is attached through single
bonds. The points of attachment of the alkylene chain may be on the same
carbon atom or on different
carbon atoms within the alkylene chain. An optionally substituted alkylene
chain is an alkylene chain that
is optionally substituted, valence permitting, by one, two, three, four, or
five substituents independently
selected from the group consisting of: halo, cyano, nitro, aryl, cycloalkyl,
heterocyclyl, heteroaryl, oxo,
trimethylsilanyl, -0R14, -0C(0)-R14, -N(R14)2, -C(0)R15, -C(0)0R14, -
C(0)N(R14)2, -N(R14)C(0)0R16,
-N(R14)C(0)R16, -N(R14)S(0)tR16 (where t is 1 or 2), -S(0)tOR16 (where t is 1
or 2), -S(0)R16 (where p is
0, 1, or 2) and -S(0)IN(R14)2 (where t is 1 or 2) where each R14 is
independently hydrogen, alkyl,
haloalkyl, cycloalkyl, aryl, heterocyclyl, or heteroaryl; each R15 is
independently hydrogen, cycloalkyl, aryl,
heterocyclyl, or heteroaryl; and each R16 is independently alkyl, haloalkyl,
cycloalkyl, aryl, heterocyclyl, or
heteroaryl. In some embodiments, alkylene is ethylene.
"Alkenylene" or "alkenylene chain" refers to a linear, acyclic, divalent
hydrocarbon chain or branched,
acyclic, divalent hydrocarbon chain, containing one, two, or three carbon-
carbon double bonds and
having from two to twelve carbon atoms, e.g., ethenylene, propenylene, n-
butenylene and the like. The
alkenylene chain is attached through single bonds. The points of attachment of
the alkenylene chain may
be on the same carbon atom or on different carbon atoms within the alkenylene
chain. An optionally
substituted alkenylene chain is an alkenylene chain that is optionally
substituted, valence permitting, by
one, two, three, four, or five substituents independently selected from the
group consisting of: halo,
6
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
cyano, nitro, aryl, cycloalkyl, heterocyclyl, heteroaryl, oxo,
trimethylsilanyl, -0R14, -0C(0)-R14, -N(R14)2,
-C(0)R15, -C(0)0R14, -C(0)N(R14)2, -N(R14)C(0)0R16, -N(R14)C(0)R16, -
N(R14)S(0)tR16 (where t is 1 or 2),
-S(0)tOR16 (where t is 1 or 2), -S(0)R16 (where p is 0, 1, or 2) and -
S(0)IN(R14)2 (where t is 1 or 2) where
each R14 is independently hydrogen, alkyl, haloalkyl, cycloalkyl, aryl,
heterocyclyl, or heteroaryl; each R15
is independently hydrogen, cycloalkyl, aryl, heterocyclyl, or heteroaryl; and
each R16 is independently
alkyl, haloalkyl, cycloalkyl, aryl, heterocyclyl, or heteroaryl.
"Alkynylene" or "alkynylene chain" refers to a linear, acyclic, divalent,
hydrocarbon chain or branched,
acyclic, divalent hydrocarbon chain, containing one or two carbon-carbon
triple bonds and, optionally,
one, two, or three carbon-carbon double bonds, and having from two to twelve
carbon atoms, e.g.,
propynylene, n-butynylene and the like. The alkynylene chain is attached
through single bonds. The
points of attachment of the alkynylene may be on the same carbon atom or on
different carbon atoms
within the alkynylene chain. An optionally substituted alkynylene chain is an
alkynelene chain that is
optionally substituted by one, two, three, four, or five substituents
independently selected from the group
consisting of: halo, cyano, nitro, aryl, cycloalkyl, heterocyclyl, heteroaryl,
oxo, trimethylsilanyl, -0R14,
-0C(0)-R14, -N(R14)2, -C(0)R15, -C(0)0R14, -C(0)N(R14)2, -N(R14)C(0)0R16, -
N(R14)C(0)R16,
-N(R14)S(0)tR16 (where t is 1 or 2), -S(0)tOR16 (where t is 1 or 2), -S(0)R16
(where p is 0, 1, or 2) and
-S(0)IN(R14)2 (where t is 1 to 2) where each R14 is independently hydrogen,
alkyl, haloalkyl, cycloalkyl,
aryl, heterocyclyl, or heteroaryl; each R15 is independently hydrogen,
cycloalkyl, aryl, heterocyclyl, or
heteroaryl; and each R16 is independently alkyl, haloalkyl, cycloalkyl, aryl,
heterocyclyl, or heteroaryl.
"Alkoxy" refers to a radical of the formula -0Ra where Ra is an alkyl radical
as defined above containing
one to twelve carbon atoms. The alkyl part of the optionally substituted
alkoxy radical is optionally
substituted as defined above for an alkyl radical.
"Alkoxyalkyl" refers to a radical of the formula -Ra-O-Rb where Ra is alkylene
and Rb is alkyl as defined
above. Alkyl and alkylene parts of the optionally substituted alkoxyalkyl
radical are optionally substituted
as defined above for an alkyl radical and alkylene chain, respectively.
"Aralkyl" refers to a radical of the formula -Ra-Rb, where Ra is alkylene and
Rb is aryl as described herein.
Alkylene and aryl portions of optionally substituted aralkyl are optionally
substituted as described herein
for alkylene and aryl, respectively.
"Aryl" refers to an aromatic monocyclic or multicyclic hydrocarbon ring system
radical containing from 6 to
18 carbon atoms, where the multicyclic aryl ring system is a bicyclic,
tricyclic, or tetracyclic ring system.
Aryl radicals include, but are not limited to, groups such as fluorenyl,
phenyl and naphthyl. An optionally
substituted aryl is an aryl radical that is optionally substituted by one,
two, three, four, or five substituents
independently selected from the group consisting of alkyl, akenyl, halo,
haloalkyl, haloalkenyl, cyano,
nitro, aryl, heteroaryl, heteroarylalkyl, -R15-0R14, -R15-0C(0)-R14, -R15-
N(R14)2,
-R15-C(0)R14, -R15-C(0)0R14, -R15-C(0)N(R14)2, -R15-N(R14)C(0)0R16, -R15-
N(R14)C(0)R16,
-R15-N(R14)S(0)tR16 (where t is 1 or 2), -R15-S(0)tOR16 (where t is 1 or 2), -
R15-S(0)R16 (where p is 0, 1,
or 2), and -R15-S(0)IN(R14)2 (where t is 1 or 2), where each R14 is
independently hydrogen, alkyl,
7
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
haloalkyl, cycloalkyl, aryl, heterocyclyl, or heteroaryl; each R15 is
independently a direct bond or a linear
or branched alkylene or alkenylene chain; and each R16 is independently alkyl,
haloalkyl, cycloalkyl,
cycloalkylalkyl, aryl, heterocyclyl, or heteroaryl.
"Arylalkoxy" refers to a group of formula ¨0-R, where R is aralkyl. An
optionally substituted arylalkoxy is
an arylalkoxy that is optionally substituted as described herein for aralkyl.
In some embodiments,
arylalkoxy is benzyloxy.
"Cycloalkyl" refers to a stable non-aromatic monocyclic or polycyclic
hydrocarbon radical having from
three to fifteen carbon atoms, preferably having from three to ten carbon
atoms, and which is saturated or
unsaturated, and which attaches to the rest of the molecule by a single bond.
A polycyclic hydrocarbon
radical is bicyclic, tricyclic, or tetracyclic ring system. An unsaturated
cycloalkyl contains one, two, or
three carbon-carbon double bonds and/or one carbon-carbon triple bond.
Monocyclic cycloalkyl radicals
include, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl,
cycloheptyl, and cyclooctyl.
Polycyclic cycloalkyl radicals include, for example, adamantyl, norbornyl,
decalinyl, and the like. An
optionally substituted cycloalkyl is a cycloalkyl radical that is optionally
substituted by one, two, three,
four, or five substituents independently selected from the group consisting of
alkyl, alkenyl, halo,
haloalkyl, haloalkenyl, cyano, nitro, oxo, aryl, aralkyl, cycloalkyl,
heterocyclyl, heteroaryl, -R15-0R14,
-R15-0C(0)-R147 -R15_N(R14)27 -R15-C(0)R14, -R15-C(0)0R147 _R15-C(0)N(R14)27
-R15-N(R14)C(0)0R16,
-R15_N(R14)c(o)R167 -R15_N fr,krc14,
)S(0)tR16 (where t is 1 or 2), -R15-S(0)tOR16 (where t is 1 or 2),
-R15-S(0)pR16 (where p is 0, 1, or 2) and -R15-S(0)IN(R14)2 (where t is 1 or
2) where each R14 is
independently hydrogen, alkyl, haloalkyl, cycloalkyl, aryl, heterocyclyl, or
heteroaryl; each R15 is
independently a direct bond or a linear or branched alkylene or alkenylene
chain; and each R16 is
independently alkyl, haloalkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl,
heterocyclyl, or heteroaryl.
"Fused" refers to any ring system described herein which is fused to an
existing ring structure in the
compounds of the invention. When the fused ring system is a heterocyclyl or a
heteroaryl, any carbon
atom on the existing ring structure which becomes part of the fused ring
system may be replaced with a
nitrogen atom.
"Halo" refers to the halogen substituents: bromo, chloro, fluor , and iodo.
"Haloalkyl" refers to an alkyl radical, as defined above, that is further
substituted by one or more halogen
substituents. The number of halo substituents included in haloalkyl is from
one and up to the total
number of the hydrogen atoms available for replacement with the halo
substituents (e.g., perfluoroalkyl).
Non-limiting examples of haloalkyl include trifluoromethyl, difluoromethyl,
trichloromethyl,
2,2,2-trifluoroethyl, 1-fluoromethy1-2-fluoroethyl, 3-bromo-2-fluoropropyl, 1-
bromomethy1-2-bromoethyl
and the like. For an optionally substituted haloalkyl, the hydrogen atoms
bonded to the carbon atoms of
the alkyl part of the haloalkyl radical may be optionally replaced with
substituents as defined above for an
optionally substituted alkyl.
8
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
"Haloalkenyl" refers to an alkenyl radical, as defined above, that is further
substituted by one or more halo
substituents. The number of halo substituents included in haloalkenyl is from
one and up to the total
number of the hydrogen atoms available for replacement with the halo
substituents (e.g.,
perfluoroalkenyl). Non-limiting examples of haloalkenyl include 2,2-
difluoroethenyl, 3-chloroprop-1-enyl,
and the like. For an optionally substituted haloalkenyl, the hydrogen atoms
bonded to the carbon atoms
of the alkenyl part of the haloalkenyl radical may be optionally replaced with
substitutents as defined
above for an optionally substituted alkenyl group.
"Haloalkynyl" refers to an alkynyl radical, as defined above, that is further
substituted by one or more halo
substituents. The number of halo substituents included in haloalkynyl is from
one and up to the total
number of the hydrogen atoms available for replacement with the halo
substituents (e.g.,
perfluoroalkynyl). Non-limiting examples of haloalkynyl include 3-chloroprop-1-
ynyl and the like. The
alkynyl part of the haloalkynyl radical may be additionally optionally
substituted as defined above for an
alkynyl group.
"Heteroarylalkyl" refers to a radical of the formula -Ra-Rb, where Ra is
alkylene and Rb is heteroaryl as
described herein. Alkylene and heteroaryl portions of optionally substituted
heteroarylalkyl are optionally
substituted as described herein for alkylene and heteroaryl, respectively.
"Heterocycly1" refers to a stable 3- to 18-membered non-aromatic ring system
radical having the carbon
count of two to twelve and containing a total of one to six heteroatoms
independently selected from the
group consisting of nitrogen, oxygen, phosphorus, and sulfur. A heterocyclyl
radical is a monocyclic,
bicyclic, tricyclic, or tetracyclic ring system. A bicyclic, tricyclic, or
tetracyclic heterocyclyl is a fused, spiro,
and/or bridged ring system. The heterocyclyl radical may be saturated or
unsaturated. An unsaturated
heterocyclyl contains one, two, or three carbon-carbon double bonds and/or one
carbon-carbon triple
bond. An optionally substituted heterocyclyl is a heterocyclyl radical that is
optionally substituted by one,
two, three, four, or five substituents independently selected from the group
consisting of alkyl, alkenyl,
halo, haloalkyl, haloalkenyl, cyano, oxo, thioxo, nitro, aryl, aralkyl,
cycloalkyl, heterocyclyl, heteroaryl, -
R15-0R14, -R15-0C(0)-R14, -R15-N(R14)2, -R15-C(0)R14,
-R15-C(0)0R14, -R15-C(0)N(R14)2, -R15-N(R14)C(0)0R16, -R15-N(R14)C(0)R16, -R15-
N(R14)S(0)tR16 (where t
is 1 or 2), -R15-S(0)tOR16 (where t is 1 or 2), -R15-S(0)R16 (where p is 0, 1,
or 2), and -R15-S(0)IN(R14)2
(where t is 1 or 2), where each R14 is independently hydrogen, alkyl, alkenyl,
haloalkyl, cycloalkyl, aryl,
heterocyclyl, or heteroaryl; each R15 is independently a direct bond or a
linear or branched alkylene or
alkenylene chain; and each R16 is independently alkyl, alkenyl, haloalkyl,
cycloalkyl, aryl, heterocyclyl, or
heteroaryl. The nitrogen, carbon, or sulfur atoms in the heterocyclyl radical
may be optionally oxidized
(when the substituent is oxo and is present on the heteroatom); the nitrogen
atom may be optionally
quaternized (when the substituent is alkyl, alkenyl, aryl, aralkyl,
cycloalkyl, heterocyclyl, heteroaryl, -
R15-0R14, -R15-0C(0)-R14, -R15-N(R14)2, -R15-C(0)R14, -R15-C(0)0R14, -R15-
C(0)N(R14)2, -
R15-N(R14)C(0)0R16, -R15-N(R14)C(0)R16, -R15-N(R14)S(0)tR16 (where t is 1 or
2), -R15-S(0)tOR16 (where t
is 1 or 2), -R15-S(0)R16 (where p is 0, 1, or 2), and -R15-S(0)IN(R14)2 (where
t is 1 or 2), where R15 is a
linear or branched alkylene or alkenylene chain, and R14 and R16 are as
defined above). Examples of
optionally substituted heterocyclyl radicals include, but are not limited to,
azetidinyl, dioxolanyl,
9
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
thienyl[1,3]dithianyl, decahydroisoquinolyl, imidazolinyl, imidazolidinyl,
isothiazolidinyl, isoxazolidinyl,
morpholinyl, octahydroindolyl, octahydroisoindolyl, 2-oxopiperazinyl, 2-
oxopiperidinyl, 2-oxopyrrolidinyl,
oxazolidinyl, piperidinyl, piperazinyl, 4-piperidonyl, pyrrolidinyl,
pyrazolidinyl, thiazolidinyl, tetrahydrofuryl,
trithianyl, tetrahydropyranyl, thiomorpholinyl, thiamorpholinyl, 1-oxo-
thiomorpholinyl, and
1,1-dioxo-thiomorpholinyl.
"Heterocyclylene" refers to a heterocyclyl in which one hydrogen atom is
replaced with a valency. An
optionally substituted heterocyclylene is optionally substituted as described
herein for heterocyclyl.
"Heteroaryl" refers to a 5-to 18-membered ring system radical containing at
least one aromatic ring,
having the carbon count of one to seventeen carbon atoms, and containing a
total of one to ten
heteroatoms independently selected from the group consisting of nitrogen,
oxygen, and sulfur. The
heteroaryl radical is a monocyclic, bicyclic, tricyclic, or tetracyclic ring
system. The bicyclic, tricyclic, or
tetracyclic heteroaryl radical is a fused and/or bridged ring system. An
optionally substituted heteroaryl is
a heteroaryl radical that is optionally substituted by one, two, three, four,
or five substituents
independently selected from the group consisting of alkyl, alkenyl, alkoxy,
halo, haloalkyl, haloalkenyl,
cyano, oxo, thioxo, nitro, oxo, aryl, aralkyl, cycloalkyl, heterocyclyl,
heteroaryl, or heteroarylalkyl, -
R15-0R14.7 -R15-0C(0)-R147 _R15_N(R14)27 -R15-C(0)R14, -R15-C(0)0R147 -R15-
C(0)N(R14)2, -
R15-N(R14)C(0)0R167 _R15_N(R14)
C(0)R16, -R15_Nkrcfr-,14,
)S(0)tR16 (where t is 1 or 2), -R15-S(0)tOR16 (where t
is 1 or 2), -R15-S(0)tR16 (where p is 0, 1, or 2), and -R15-S(0)IN(R14)2
(where t is 1 or 2), where each R14 is
independently hydrogen, alkyl, alkenyl, haloalkyl, cycloalkyl, aryl,
heterocyclyl, or heteroaryl; each R15 is
independently a direct bond or a linear or branched alkylene or alkenylene
chain; and each R16 is alkyl,
alkenyl, haloalkyl, cycloalkyl, aryl, heterocyclyl, or heteroaryl. The
nitrogen, carbon, or sulfur atoms in the
heterocyclyl radical may be optionally oxidized (when the substituent is oxo
and is present on the
.. heteroatom), provided that at least one ring in heteroaryl remains
aromatic; the nitrogen atom may be
optionally quaternized (when the substituent is alkyl, alkenyl, aryl, aralkyl,
cycloalkyl, heterocyclyl,
heteroaryl, -R15-0R14.7 _R15-0C(0)-R147 _R15_N(R14)27 -R15-C(0)R14, _R15-
C(0)0R14.7 _R15-C(0)N(R14)2,
-R15-N(R14)C(0)0R167 _R15_N(R14)c(o)R167 -R15_N fr,krc14,
)S(0)tR16 (where t is 1 or 2), -R15-S(0)tOR16 (where
t is 1 or 2), -R15-S(0)R16 (where p is 0, 1, or 2), and -R15-S(0)IN(R14)2
(where t is 1 or 2), where R15 is a
linear or branched alkylene or alkenylene chain, and R14 and R16 are as
defined above), provided that at
least one ring in heteroaryl remains aromatic. Examples of optionally
substituted heteroaryl radicals
include, but are not limited to, azepinyl, acridinyl, benzimidazolyl,
benzthiazolyl, benzindolyl,
benzodioxolyl, benzofuranyl, benzooxazolyl, benzothiazolyl, benzothiadiazolyl,
benzo[b][1,4]dioxepinyl,
1,4-benzodioxanyl, benzonaphthofuranyl, benzoxazolyl, benzodioxolyl,
benzodioxinyl, benzopyranyl,
benzopyranonyl, benzofuranyl, benzofuranonyl, benzothienyl (benzothiophenyl),
benzotriazolyl,
benzo[4,6]imidazo[1,2-a]pyridinyl, carbazolyl, cinnolinyl, dibenzofuranyl,
dibenzothiophenyl, furanyl,
furanonyl, isothiazolyl, imidazolyl, indazolyl, indolyl, indazolyl,
isoindolyl, indolinyl, isoindolinyl, isoquinolyl,
indolizinyl, isoxazolyl, naphthyl, naphthyridinyl, oxadiazolyl, 2-oxoazepinyl,
oxazolyl, oxiranyl,
1-phenyl-1H-pyrrolyl, phenazinyl, phenothiazinyl, phenoxazinyl, phthalazinyl,
pteridinyl, purinyl, pyrrolyl,
pyrazolyl, pyridinyl, pyrazinyl, pyrimidinyl, pyridazinyl, pyrrolyl,
quinazolinyl, quinoxalinyl, quinolinyl,
quinuclidinyl, isoquinolinyl, tetrahydroquinolinyl, thiazolyl, thiadiazolyl,
triazolyl, tetrazolyl, triazinyl and
thiophenyl (i.e. thienyl).
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
The invention disclosed herein is also meant to encompass all pharmaceutically
acceptable compounds
of formula (I) being isotopically-labelled by having one or more atoms
replaced by an atom having a
different atomic mass or mass number. Examples of isotopes that can be
incorporated into the disclosed
.. compounds include isotopes of hydrogen, carbon, nitrogen, oxygen,
phosphorous, fluorine, chlorine, and
iodine, such as 2H7 3H7 11C, 13C, 14C7 13N7 15N7 1507 1707 1807 311D7 32P7
35s7 18F7 36C1, 1231, and 1251,
respectively. These radiolabelled compounds could be useful to help determine
or measure the
effectiveness of the compounds, by characterizing, for example, the site or
mode of action on ATM and
DNA-PK enzymes, or binding affinity to pharmacologically important site of
action on ATM and DNA-PK
enzymes. Certain isotopically-labelled compounds of formula (I), for example,
those incorporating a
radioactive isotope, are useful in drug and/or substrate tissue distribution
studies. The radioactive
isotopes tritium, i.e. 3H, and carbon-14, i.e. 14C, are particularly useful
for this purpose in view of their
ease of incorporation and ready means of detection.
Substitution with heavier isotopes such as deuterium, i.e. 2H, may afford
certain therapeutic advantages
resulting from greater metabolic stability, for example, increased in vivo
half-life or reduced dosage
requirements, and hence may be preferred in some circumstances.
Substitution with positron emitting isotopes, such as iic7 18F7 150 and 13N,
can be useful in Positron
Emission Topography (PET) studies for examining substrate receptor occupancy.
Isotopically-labeled
compounds of formula (I) can generally be prepared by conventional techniques
known to those skilled in
the art or by processes analogous to those described in the Examples and
Preparations as set out below
using an appropriate isotopically-labeled reagent in place of the non-labeled
reagent previously
employed.
The invention disclosed herein is also meant to encompass the in vivo
metabolic products of the
disclosed compounds. Such products may result from, for example, the
oxidation, reduction, hydrolysis,
amidation, esterification, and the like of the administered compound,
primarily due to enzymatic
processes. Accordingly, the invention includes compounds produced by a process
comprising contacting
.. a compound of this invention with a mammal for a period of time sufficient
to yield a metabolic product
thereof. Such products are typically identified by administering a
radiolabelled compound of the invention
in a detectable dose to an animal, such as rat, mouse, guinea pig, canine,
monkey, or to human, allowing
sufficient time for metabolism to occur, and isolating its conversion products
from the urine, blood or other
biological samples.
"Stable compound" and "stable structure" are meant to indicate a compound that
is sufficiently robust to
survive isolation to a useful degree of purity from a reaction mixture, and
formulation into an efficacious
therapeutic agent.
"Mammal" includes humans and both domestic animals such as laboratory animals
and household pets,
(e.g. cats, dogs, swine, cattle, sheep, goats, horses, rabbits), and non-
domestic animals such as wildlife
and the like.
11
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
"Optional" or "optionally" means that the subsequently described event of
circumstances may or may not
occur, and that the description includes instances where said event or
circumstance occurs and instances
in which it does not. For example, "optionally substituted aryl" means that
the aryl radical may or may not
be substituted and that the description includes both substituted aryl
radicals and aryl radicals having no
substitution.
"Patient" means a human or non-human animal (e.g., a mammal) that is suffering
from a disease or
condition, as determined by a qualified professional (e.g., a doctor, nurse
practitioner, or veterinarian)
with or without known in the art laboratory test(s) of sample(s) from the
patient.
"Pharmaceutically acceptable carrier, diluent or excipient" includes without
limitation any adjuvant, carrier,
excipient, glidant, sweetening agent, diluent, preservative, dye/colorant,
flavor enhancer, surfactant,
wetting agent, dispersing agent, suspending agent, stabilizer, isotonic agent,
solvent, or emulsifier which
has been approved by the United States Food and Drug Administration as being
acceptable for use in
humans or domestic animals.
"Pharmaceutically acceptable salt," as used herein, represents those salts
which are, within the scope of
sound medical judgment, suitable for use in contact with the tissues of humans
and animals without
undue toxicity, irritation, allergic response and the like and are
commensurate with a reasonable
benefit/risk ratio. Pharmaceutically acceptable salts are well known in the
art. For example,
pharmaceutically acceptable salts are described in: Berge et al., J.
Pharmaceutical Sciences 66:1-19,
1977 and in Pharmaceutical Salts: Properties, Selection, and Use, (Eds. P.H.
Stahl and C.G. Wermuth),
Wiley-VCH, 2008. Pharmaceutically acceptable salts include acid and base
addition salts.
"Pharmaceutically acceptable acid addition salt" refers to those salts which
retain the biological
effectiveness and properties of the free bases, which are not biologically or
otherwise undesirable, and
which are formed with inorganic acids such as, but are not limited to,
hydrochloric acid, hydrobromic acid,
sulfuric acid, nitric acid, phosphoric acid and the like, and organic acids
such as, but not limited to, acetic
acid, 2,2-dichloroacetic acid, adipic acid, alginic acid, ascorbic acid,
aspartic acid, benzenesulfonic acid,
benzoic acid, 4-acetamidobenzoic acid, camphoric acid, camphor-10-sulfonic
acid, capric acid, caproic
acid, caprylic acid, carbonic acid, cinnamic acid, citric acid, cyclamic acid,
dodecylsulfuric acid, ethane-
1,2-disulfonic acid, ethanesulfonic acid, 2-hydroxyethanesulfonic acid, formic
acid, fumaric acid,
galactaric acid, gentisic acid, glucoheptonic acid, gluconic acid, glucuronic
acid, glutamic acid, glutaric
acid, 2-oxo-glutaric acid, glycerophosphoric acid, glycolic acid, hippuric
acid, isobutyric acid, lactic acid,
lactobionic acid, lauric acid, maleic acid, malic acid, malonic acid, mandelic
acid, methanesulfonic acid,
mucic acid, naphthalene-1,5-disulfonic acid, naphthalene-2-sulfonic acid, 1-
hydroxy-2-naphthoic acid,
nicotinic acid, oleic acid, orotic acid, oxalic acid, palmitic acid, pamoic
acid, propionic acid, pyroglutamic
acid, pyruvic acid, salicylic acid, 4-aminosalicylic acid, sebacic acid,
stearic acid, succinic acid, tartaric
acid, thiocyanic acid, p-toluenesulfonic acid, trifluoroacetic acid,
undecylenic acid and the like.
12
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
"Pharmaceutically acceptable base addition salt" refers to those salts which
retain the biological
effectiveness and properties of the free acids, which are not biologically or
otherwise undesirable. These
salts are prepared from addition of an inorganic base or an organic base to
the free acid. Salts derived
from inorganic bases include, but are not limited to, the sodium, potassium,
lithium, ammonium, calcium,
magnesium, iron, zinc, copper, manganese, aluminum salts and the like.
Preferred inorganic salts are
the ammonium, sodium, potassium, calcium, and magnesium salts. Salts derived
from organic bases
include, but are not limited to, salts of primary, secondary, and tertiary
amines, substituted amines
including naturally occurring substituted amines, cyclic amines and basic ion
exchange resins, such as
ammonia, isopropylamine, trimethylamine, diethylamine, triethylamine,
tripropylamine, diethanolamine,
ethanolamine, deanol, 2-dimethylaminoethanol, 2-diethylaminoethanol,
dicyclohexylamine, lysine,
arginine, histidine, caffeine, procaine, hydrabamine, choline, betaine,
benethamine, benzathine,
ethylenediamine, glucosamine, methylglucamine, theobromine, triethanolamine,
tromethamine, purines,
piperazine, piperidine, N-ethylpiperidine, polyamine resins and the like.
Particularly preferred organic
bases are isopropylamine, diethylamine, ethanolamine, trimethylamine,
dicyclohexylamine, choline and
caffeine.
Often crystallizations produce a solvate of the compound of the invention. As
used herein, the term
"solvate" refers to an aggregate that comprises one or more molecules of a
compound of the invention
with one or more molecules of solvent. The solvent may be water, in which case
the solvate may be a
hydrate. Alternatively, the solvent may be an organic solvent. Thus, the
compounds of the present
invention may exist as a hydrate, including a monohydrate, dihydrate,
hemihydrate, sesquihydrate,
trihydrate, tetrahydrate and the like, as well as the corresponding solvated
forms. The compound of the
invention may be true solvates, while in other cases, the compound of the
invention may merely retain
adventitious water or be a mixture of water plus some adventitious solvent.
A "pharmaceutical composition" refers to a formulation of a compound of the
invention and a medium
generally accepted in the art for the delivery of the biologically active
compound to mammals, e.g.,
humans. Such a medium includes all pharmaceutically acceptable carriers,
diluents, or excipients
therefor.
"Therapeutically effective amount" refers to that amount of a compound of the
invention which, when
administered to a mammal, preferably a human, is sufficient to effect
treatment, as defined below, in the
mammal, preferably a human or canine. The amount of a compound of the
invention, or another
pharmaceutical agent (e.g., an anti-tumor agent), which constitutes a
"therapeutically effective amount"
will vary depending on the compound, the condition and its severity, the
manner of administration, and
the age of the mammal to be treated, but can be determined routinely by one of
ordinary skill in the art
having regard to his own knowledge and to this disclosure.
"Treating" or "treatment" as used herein covers the treatment of the disease
or condition of interest in a
mammal, preferably a human, having the disease or condition of interest, and
includes:
(i) preventing the disease or condition from occurring in a mammal, in
particular, when such
mammal is predisposed to the condition but has not yet been diagnosed as
having it;
13
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
(ii) inhibiting the disease or condition, i.e., arresting its development;
(iii) relieving the disease or condition, i.e., causing regression of the
disease or condition; or
(iv) relieving the symptoms resulting from the disease or condition, i.e.,
relieving pain without
addressing the underlying disease or condition. As used herein, the terms
"disease" and "condition" may
be used interchangeably or may be different in that the particular malady or
condition may not have a
known causative agent (so that etiology has not yet been worked out) and it is
therefore not yet
recognized as a disease but only as an undesirable condition or syndrome,
wherein a more or less
specific set of symptoms have been identified by clinicians.
The compounds of the invention, or their pharmaceutically acceptable salts may
contain one or more
asymmetric centres and may thus give rise to enantiomers, diastereomers, and
other stereoisomeric
forms that may be defined, in terms of absolute stereochemistry, as (R)- or
(S)- or, as (D)- or (*for
amino acids. The present invention is meant to include all such possible
isomers, as well as their racemic
and optically pure forms. Optically active (+) and (-), (R)- and (S)-, or (D)-
and (*isomers may be
prepared using chiral synthons or chiral reagents, or resolved using
conventional techniques, for
example, chromatography and fractional crystallisation. Conventional
techniques for the
preparation/isolation of individual enantiomers include chiral synthesis from
a suitable optically pure
precursor or resolution of the racemate (or the racemate of a salt or
derivative) using, for example, chiral
high-pressure liquid chromatography (HPLC). When the compounds described
herein contain olefinic
double bonds or other centres of geometric asymmetry, and unless specified
otherwise, it is intended that
the compounds include both E and Z geometric isomers. Likewise, all tautomeric
forms are also intended
to be included.
A "stereoisomer" refers to a compound made up of the same atoms bonded by the
same bonds but
having different three-dimensional structures, which are not interchangeable.
The present invention
contemplates various stereoisomers and mixtures thereof and includes
"enantiomers", which refers to two
stereoisomers whose molecules are nonsuperimposeable mirror images of one
another.
A "tautomer" refers to a proton shift from one atom of a molecule to another
atom of the same molecule.
The present invention includes tautomers of any said compounds.
Also within the scope of the invention are intermediate compounds of formula
(I) and all polymorphs of
the aforementioned species and crystal habits thereof.
BRIEF DESCRIPTION OF THE DRAWINGS
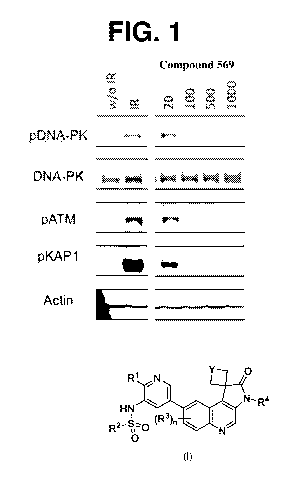
FIG. 1 is an image of an immunoblot from a representative experiment assessing
the effect of compound
569 on MCF7 cells with or without radiation.
FIG. 2 is a graph showing the results of a clonogenic survival assay examining
the MCF7 cell viability
after exposure to vehicle (DMSO) or compound 569 with or without radiation.
14
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
FIG. 3 is an image of an immunoblot showing the induction of phosphorylation
of TBK1 by compound
569, a selective ATM inhibitor (ATMi), a selective DNA-PK inhibitor (DNA-PKi),
and a combination of the
selective ATM inhibitor and the selective DNA-PK inhibitor (Ai+Di) in HCT116
cells expressing wild-type
p53 or HCT116 cells that were negative for p53 expression. In FIG. 3, p.ATM,
p.DNA-PK, p.TBK1, and
p.STING indicate phosphorylated forms of ATM, DNA-PK, TBK1, and STING,
respectively.
FIG. 4 is an immunoblot showing the inhibition of radiation-induced
autophosphorylation of DNA-PK
kinase and radiation-induced phosphorylation of KAP1, an ATM substrate, by
compound 569 in FADU
head and neck squamous cell carcinoma (HNSCC) human tumor xenografts. In FIG.
4, pDNA-PK and
pKAP1 indicate phosphorylated forms of DNA-PK and KAP1, respectively.
FIG. 5 is an immunoblot showing the inhibition of radiation-induced
autophosphorylation of DNA-PK
kinase and radiation-induced phosphorylation of KAP1, an ATM substrate, by
compound 569 in MDA-MB-
231 breast carcinoma human tumor xenografts. In FIG. 5, pDNA-PK and pKAP1
indicate phosphorylated
forms of DNA-PK and KAP1, respectively.
FIG. 6 illustrates the dosing of a FADU subcutaneous human xenograft mouse
model with compound 569
and/or IR, qd x 3. The median relative tumor volume over time for each group
in the study are shown. In
FIG. 6, "569" means compound 569, "Veh." means vehicle, and "Rad." means
radiation.
FIG. 7 represents the Kaplan-Meier quintupling-free survival for each group
dosed in the FADU
subcutaneous human xenograft mouse model with compound 569 and/or IR, qd x 3.
In FIG. 7, "569"
means compound 569, "Veh." means vehicle, and "Rad." means radiation.
FIG.8 illustrates the dosing of a MDA-MB-231 subcutaneous human xenograft
mouse model with
compound 569 and/or IR, qd x 3. The median relative tumor volume over time for
each group in the
study are shown. In FIG. 8, "569" means compound 569, "Veh." means vehicle,
and "Rad." means
radiation.
FIG. 9 represents the Kaplan-Meier quintupling-free survival data for each
group dosed in the MDA-MB-
231 subcutaneous human xenograft mouse model with compound 569 and/or IR, qd x
3. In FIG. 9, "569"
means compound 569, "Veh." means vehicle, and "Rad." means radiation.
COMPOUNDS, COMPOSITIONS, AND METHODS
The invention provides compounds and compositions that may be useful in the
treatment of oncological
diseases (e.g., cancer, e.g., those cancers described herein), e.g., alone or
in combination with
radiotherapy and/or anti-tumor therapy. A compound may be a compound of
formula (I):
R1 N
N¨ R4
HN
(R3) I
0
(I)
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
or a pharmaceutically acceptable salt thereof,
wherein
Z is CH, CR3, or N;
Y is CHR5 or NR6;
n is 0, 1, 2, or 3;
R1 is ¨0¨L¨N(R7)2 or optionally substituted, four-memberred, saturated N-
heterocyclyl;
R2 is C1-3 alkyl;
each R3 is independently halogen or optionally substituted C1-3 alkyl;
R4 is optionally substituted alkyl;
R5 is hydrogen, optionally substituted C1_3 alkyl, or benzyloxy;
R6 is optionally substituted C1_3 alkyl;
each R7 is independently H or optionally substituted C1_3 alkyl; and
L is optionally substituted ethylene.
Advantageously, compounds of the invention (e.g., compound 568, 569, or 570)
may exhibit superior
inhibitory activity for ATM and DNA-PK. Advantageously, compounds of the
invention (e.g., compound
568, 569, or 570) may exhibit superior selectivity as measured by reduced off-
target activity (e.g., mTOR
inhibition, PI3K ato inhibition, and/or hERG inhibition). For example, a
compound of the invention (e.g.,
compound 568, 569, or 570) may have an mTOR IC50 of at least 10 times (e.g.,
at least 20 times) greater
than the ATM IC50 or DNA-PK IC50. A compound of the invention (e.g., compound
568, 569, or 570) may
have an mTOR IC50 of 10 nM or greater (e.g., > 100 nM). Additionally or
alternatively, a compound of
the invention (e.g., compound 568, 569, or 570) may have an hERG IC50 of at
least 100 times (e.g., at
least 500 times, at least 1000 times, or at least 3000 times) greater than the
ATM IC50 or DNA-PK IC50,
when measured at the same compound concentration. A compound of the invention
(e.g., compound
568, 569, or 570) may have an hERG IC50 of 3 pM or greater (e.g., 10 pM or
greater).
Advantageously, compounds of the invention (e.g., compound 568, 569, or 570)
may exhibit superior
pharmacokinetic properties (e.g., Cmax, AUC, and/or t112) .
In some embodiments, the compound is selected from the group consisting of:
trans-N-(5-(3-Ethyl-7'-fluoro-3'-methyl-2'-oxo-2',3'-dihydrospiro[cyclobutane-
1,1'-pyrrolo[2,3-c]quinolin]-
8'-y1)-2-(2-(isopropylamino)ethoxy)pyridin-3-yl)methanesulfonamide
trans-N-(5-(3-(Benzyloxy)-7'-fluoro-3'-methyl-2'-oxo-2',3'-
dihydrospiro[cyclobutane-1,1'-pyrrolo[2,3-
c]quinolin]-8'-y1)-2-(2-(isopropylamino)ethoxy)pyridin-3-yl)methanesulfonamide
N-(5-(7'-Fluoro-3'-methyl-2'-oxo-2',3'-dihydrospiro[cyclobutane-1,1'-
pyrrolo[2,3-c]quinolin]-8'-y1)-2-(2-
(isopropylamino)ethoxy)pyridin-3-yl)propane-2-sulfonamide
N-(5-(7'-Fluoro-3'-methyl-2'-oxo-2',3'-dihydrospiro[cyclobutane-1,1'-
pyrrolo[2,3-c]quinolin]-8'-y1)-2-(2-
(isopropylamino)ethoxy)pyridin-3-yl)methanesulfonamide hydrochloride
N-(5-(7'-Fluoro-3'-methyl-2'-oxo-2',3'-dihydrospiro[cyclobutane-1,1'-
pyrrolo[2,3-c]quinolin]-8'-y1)-2-(2-
(isopropylamino)ethoxy)pyridin-3-yDethanesulfonamide hydrochloride
N-(5-(7'-Fluoro-1-isopropyl-3'-methyl-2'-oxo-2',3'-dihydrospiro[azetidine-3,1'-
pyrrolo[2,3-c]quinolin]-8'-
y1)-2-(2-(isopropylamino)ethoxy)pyridin-3-yl)methanesulfonamide
cis-N-(5-(3-(Benzyloxy)-7'-fluoro-3'-methyl-2'-oxo-2',3'-
dihydrospiro[cyclobutane-1,1'-pyrrolo[2,3-
c]quinolin]-8'-y1)-2-(2-(isopropylamino)ethoxy)pyridin-3-yl)methanesulfonamide
cis-N-(5-(3-Ethyl-7'-fluoro-3'-methyl-2'-oxo-2',3'-dihydrospiro[cyclobutane-
1,1'-pyrrolo[2,3-c]quinolin]-8'-
y1)-2-(2-(isopropylamino)ethoxy)pyridin-3-yl)methanesulfonamide
16
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
N-(2-(3-(Dimethylamino)azetidin-1-y1)-5-(3'-methyl-2'-oxo-2',3'-
dihydrospiro[cyclobutane-1,1'-
pyrrolo[2,3-c]quinolin]-8'-yl)pyridin-3-yOmethanesulfonamide hydrochloride
The compounds of the invention are advantageous in that they can inhibit ATM
(ataxia-telangiectasia,
mutated) and DNA-PK kinases. The ATM (ataxia-telangiectasia, mutated) and DNA-
PK kinases, in
particular, are important modulators of cellular responses to DNA breakage and
inhibition of either of
these molecules markedly increases the sensitivity of cells to ionizing
radiation. Thus, the compounds of
the invention can be effective inhibitors of the actions of ATM and DNA-PK
with or without radiation and
with or without chemotherapy or immunotherapy to provide effective therapy for
the treatment of
oncological diseases (e.g., cancer, e.g., those cancers described herein). The
treatment of a patient with
a compound of the invention can delay or eliminate the repair of DNA damage by
radiation therapy. As a
result, patients receiving a compound of the invention may respond better to
anti-tumor therapies.
Advantageously, patients receiving a compound of the invention may derive
therapeutic benefit by
increasing tumor control from standard doses of radiation therapy or by
achieving similar levels of tumor
control from lower doses of ionizing radiation than routinely used in patients
not receiving a compound of
the invention. Advantageously, lower doses of ionizing radiation may be less
damaging to non-cancerous
tissues than the doses necessary for patients not receiving a compound of the
invention.
Humans and mice having loss-of-function mutations in the ATM or PRKDC genes,
which encode Ataxia
Telangiectasia Mutated (ATM) kinase and DNA-dependent Protein Kinase (DNA-PK),
respectively, are
hypersensitive to ionizing radiation. Inhibition of ATM and DNA-PK kinases
together can be effective in
sensitizing tumor cells to radiation or DNA damaging agents (e.g., anti-tumor
agents). The efficacy of
dual inhibition of ATM and DNA-PK kinases may be superior to inhibition of
either kinase by itself.
In addition, compounds of the invention may advantageously exhibit reduced
inhibition of other kinases
(ATR and mTOR) and thus may exhibit reduced toxicity.
Compounds of the invention may sensitize tumor cells to radiation and/or anti-
tumor agents.
Methods
In another aspect, the invention provides methods for the treatment of an
oncological disease (e.g.,
.. cancer) in a mammal, preferably human or canine, wherein the methods
comprise administering to the
mammal in need thereof a therapeutically effective amount of a compound of the
invention. In some
embodiments, the compound is administered to the mammal receiving
radiotherapy.
In another aspect, the invention provides methods for the treatment of an
oncological disease (e.g.,
cancer) in a mammal, wherein the methods comprise administering to the mammal
in need thereof a
therapeutically effective amount of a compound of the invention. In some
embodiments, the compound is
administered to the mammal in combination with a DNA-damaging agent. Non-
limiting examples of DNA-
damaging agents include cisplatin, oxaliplatin, carboplatin, valrubicin,
idarubicin, calicheamicin, PARP
inhibitors.
17
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
In another aspect, the invention provides pharmaceutical compositions
comprising the compounds of the
invention and pharmaceutically acceptable excipients. In one embodiment, the
pharmaceutical
composition comprises a compound of the invention in a pharmaceutically
acceptable carrier and in an
amount effective to treat an oncoligcal disease in an animal, preferably a
mammal.
A compound of the invention, when used in a combination therapy, may increase
the potency of the other
radiation or drug therapy if it allows the dose of the other treatment to be
reduced, which may reduce the
frequency and/or severity of adverse events associated with the other drug
therapy. For example, side
effects of radiation (e.g., oral or gastrointestinal mucositis, dermatitis,
pneumonitis, or fatigue) may be
reduced in patients receiving a combination therapy including a compound of
the invention and reduced
dose radiotherapy (e.g., incidence of the adverse events may be reduced by at
least 1%, 5%, 10%, or
20%) relative to patients receiving standard full dose radiotherapy without a
compound of the
invention. Additionally, other adverse events that may be reduced in patients
receiving a combination
therapy including a compound of the invention and reduced dose radiotherapy
(e.g., incidence of the
adverse events may be reduced by at least 1%, 5%, 10%, or 20%) relative to
patients receiving standard
full dose radiotherapy without a compound of the invention may be late effects
of radiation, e.g., radiation-
induced lung fibrosis, cardiac injury, bowel obstruction, nerve injury,
vascular injury, lymphedema, brain
necrosis, or radiation-induced cancer. Similarly, when the compound is
administered in a combination
therapy with another anti-cancer drug (e.g., those described herein), the
combined therapy may cause
the same or even increased tumor cell death, even when the dose of the other
anti-cancer drug is
lowered. Reduced dosages of other anti-cancer drugs thus may reduce the
severity of adverse events
caused by the other anti-cancer drugs.
In another aspect, this invention is directed to the use of the compounds of
the invention, as set forth
above, as a stereoisomer, enantiomer, tautomer thereof or mixtures thereof, or
a pharmaceutically
acceptable salt or solvate thereof, or the use of a pharmaceutical composition
comprising a
pharmaceutically acceptable excipient and a compound of the invention, as set
forth above, as a
stereoisomer, enantiomer, tautomer thereof or mixtures thereof, or a
pharmaceutically acceptable salt or
solvate thereof, in the preparation of a medicament for use in the treatment
of a disease. In some
embodiments, the compound of the invention is administered in combination with
radiotherapy. In other
embodiments, the compound of the invention is administered in combination with
a DNA damaging agent.
In further embodiments, the compound of the invention is administered in
combination with an anti-tumor
immunotherapeutic agent (e.g., ipilimumab, ofatumumab, nivolumab,
pembrolizumab, atezolizumab,
avelumab, durvalumab, cemiplimab, obinutuzumab, ocaratuzumab, tremelimumab,
spartalizumab,
camrelizumab, sintilimab, tislelizumab, toripalimab, dostarlimab, veltuzumab,
INCMGA00012, AMP-224,
AMP-514, KN035, CK-301, AUNP12, CA-170, or BMS-986189). In other embodiments,
the anti-tumor
immunotherapeutic agent is ofatumumab, obinutuzumab, ocaratuzumab, or
veltuzumab. In yet other
embodiments, the anti-tumor immunotherapeutic agent is nivolumab,
pembrolizumab, cemiplimab,
spartalizumab, camrelizumab, sintilimab, tislelizumab, toripalimab,
dostarlimab, INCMGA00012, AMP-
224, or AMP-514. In still other embodiments, the anti-tumor immunotherapeutic
agent is atezolizumab,
avelumab, durvalumab, KN035, CK-301, AUNP12, CA-170, or BMS-986189. In certain
preferred
combination therapy embodiments, the compound of the invention is administered
in combination with an
18
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
anti-tumor immunotherapeutic agent.
Methods of the invention may be used in the treatment of an oncological
disease as described herein. An
oncological disease may be, e.g., a premalignant tumor or a malignant tumor
(e.g., a solid tumor or a
liquid tumor). Malignant tumors are typically referred to as cancers. In
certain embodiments, the
oncological disease is cancer.
In further embodiments, examples of cancer to be treated using methods and
uses disclosed herein
include but are not limited to hematologic cancers, e.g., leukemias and
lymphomas. Non-limiting
examples of cancers include acute myelogenous leukemia, acute lymphoblastic
leukemia, acute
megakaryocytic leukemia, promyelocytic leukemia, erythroleukemia,
lymphoblastic T cell leukemia,
chronic myelogenous leukemias, chronic lymphocytic leukemia, hairy-cell
leukemia, chronic neutrophilic
leukemia, plasmacytoma, immunoblastic large cell leukemia, mantle cell
leukemia, multiple myelomas,
malignant lymphoma, diffuse large B-cell lymphoma, Hodgkin's lymphoma, non-
Hodgkin's lymphoma,
lymphoblastic T cell lymphoma, Burkitt's lymphoma, and follicular lymphoma.
In yet further embodiments, examples of cancer to be treated using methods and
uses disclosed herein
include but are not limited to solid tumors. Non-limiting examples of solid
tumors include brain cancers
(e.g., astrocytoma, glioma, glioblastoma, medulloblastoma, or ependymoma),
bladder cancer, breast
cancer, central nervous system cancers, cervical cancer, colon cancer,
endometrial cancer, esophageal
cancer, gastrointestinal stromal tumor, gastric cancer, head and neck cancers,
buccal cancer, cancer of
the mouth, hepatocellular cancer, lung cancer, melanoma, Merkel cell
carcinoma, mesothelioma,
nasopharyngeal cancer, neuroblastoma, osteosarcoma, ovarian cancer, pancreatic
cancer, prostate
cancer, renal cancer, salivary gland cancer, sarcomas, testicular cancer,
urothelial cancer, vulvar cancer,
and Wilm's tumor. Preferably, the methods of the invention are used in the
treatment of lung cancer,
head and neck cancer, pancreatic cancer, rectal cancer, glioblastoma,
hepatocellular carcinoma,
cholangiocarcinoma, metastic liver lesions, melanoma, bone sarcoma, soft
tissue sarcoma, endometrial
cancer, cervical cancer, prostate cancer, or Merkel cell carcinoma.
In still further embodiments, examples of cancer to be treated using methods
and uses disclosed herein
but are not limited to metastases and metastatic cancer. For example, the
methods and uses disclosed
herein for treating cancer may involve treatment of both primary tumors and
metastases.
In some embodiments, methods of the invention may reduce the tumor size in a
subject, e.g., at least by
10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, or 90%, or may eliminate the tumor
(e.g., relative to the
tumor size at the time of the commencement of the therapy or relative to a
reference subject that receives
placebo instead of the compound of the invention). In some embodiments,
methods of the invention may
reduce the tumor burden in a subject, e.g., at least by 10%, 20%, 30%, 40%,
50%, 60%, 70%, 80%, or
90%, or may eliminate the tumor (e.g., relative to the tumor burden at the
time of the commencement of
.. the therapy or relative to a reference subject that receives placebo
instead of the compound of the
invention). In some embodiments, methods of the invention may increase mean
survival time of the
subject, e.g., by at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100%,
150%, or 200% (e.g.,
19
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
relative to a reference subject that receives placebo instead of the compound
of the invention). In some
embodiments, methods of the invention may increase the ability of radiation
therapy or drug therapy to
palliate pain or other symtoms for a longer mean time for the subject, e.g.,
by at least 10%, 20%, 30%,
40%, 50%, 60%, 70%, 80%, 90%, 100%, 150%, or 200% (e.g., relative to a
reference subject that
receives placebo instead of the compound of the invention).
In some embodiments, the methods and uses disclosed herein comprise the pre-
treatment of a patient
with a dual an ATM and DNA-PK inhibitor prior to administration of radiation
therapy or a DNA damaging
agent. Pre-treatment of the patient with a dual ATM and DNA-PK inhibitor may
delay or eliminate the
repair of DNA damage following radiation therapy.
Radiation therapy includes, but is not limited to, external beam radiation
therapy with X-rays (photons),
gamma rays from 80Cobalt or other radioactive isotopes, neutrons, electrons,
protons, carbon ions, helium
ions, and other charged particles. Radiation therapy also includes
brachytherapy and radio-
pharmaceuticals that emits gamma rays, alpha particles, beta particles, Auger
electrons, or other types of
radioactive particles from isotopes including 32Phosphorus, 87Copper,
77Bromine, 89Strontium, 90Yttrium,
105Rhodium, 131 Iodine, 137Cesium, 149Prometheum, 153Samarium, 1881-lolmium,
177Lutetium, 188Rhenium,
188Rhenium, 199Gold, 211Astatine, 213Bismuth, 223Radium, 225Actinium, or
227Thorium, 192Iridium, 87Gallium,
103Palladium, 125I0dine, and other radioactive isotopes (e.g., 192Iridium,
125I0dine, 137Cesium, 103Palladium,
32Phosphorus, 90Yttrium, 87Gallium, 211Astatine, or 223Radium). Radiation
therapy also includes
radioimmunotherapy (RIT) with antibodies or small molecules that are
conjugated to radioactive isotopes
including 131 Iodine, 90Yttrium, 225Actinium, 211Astatine, 87Gallium,
177Lutetium, 227Thorium, and other
radioactive isotopes.
In some embodiments, the combination therapy comprises administration to a
patient of an ATM and
DNA-PK inhibitor and an anti-tumor agent, e.g., cisplatin, oxaliplatin,
carboplatin, topoisomerase I
inhibitors, topoisomerase ll inhibitors, anthracyclines, valrubicin,
idarubicin, calicheamicin, PARP
inhibitors (e.g., olaparib, rucaparib, niraparib, veliparib, or talazoparib),
as well as other anti-cancer
agents known to those skilled in the art.
In certain embodiments, the combination therapy comprises administration to a
patient of an ATM and
DNA-PK inhibitor and an anti-tumor immunotherapeutic agents including by not
limited to ipilimumab,
ofatumumab, nivolumab, pembrolizumab, atezolizumab, avelumab, durvalumab, etc.
In the combination therapies described herein, an ATM and DNA-PK inhibitor may
be administered to the
patient simultaneously or sequentially (e.g., before or after) the other drug.
PREPARATION OF THE COMPOUNDS OF THE INVENTION
The compounds of the present invention can be prepared using methods and
techniques known in the
art. Suitable processes for synthesizing these compounds are provided in the
Examples. Generally,
compounds of Formula (I) can be prepared according to the Schemes described
below. The sources of
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
the starting materials for these reactions are also described.
Protecting groups may be added or removed in the preparation of the compounds
of the invention in
accordance with standard techniques, which are known to one skilled in the art
and as described herein.
The use of protecting groups is described in detail in Greene, T.W. and P.G.M.
Wuts, Greene's Protective
Groups in Organic Synthesis (2006), 41h Ed., Wiley. The protecting group may
also be a polymer resin
such as a Wang resin or a 2-chlorotrityl-chloride resin.
It will also be appreciated by those skilled in the art, although such
protected derivatives of compounds of
this invention may not possess pharmacological activity as such, they may be
administered to a mammal
and thereafter metabolized in the body to form compounds of the invention
which are pharmacologically
active.
All of the compounds described below as being prepared which may exist in free
base or acid form may
be converted to their pharmaceutically acceptable salts by treatment with the
appropriate inorganic or
organic base or acid. Salts of the compounds prepared below may be converted
to their free base or acid
form by standard techniques. It is understood that all polymorphs, amorphous
forms, anhydrates,
hydrates, solvates and salts of the compounds of the invention are intended to
be within the scope of the
invention. Furthermore, all compounds of the invention which contain an acid
or an ester group can be
converted to the corresponding ester or acid, respectively, by methods known
to one skilled in the art or
by methods described herein.
A general representation of preparation of many of these compounds is shown
below in Scheme 1.
Compounds are prepared through the coupling of various components of the
molecule: Suzuki coupling
of halo substituted compound 3 (or 2') with a boronic acid or borate compound
2 (3'). Further reactions
may or may not be needed to furnish the synthesis of the compounds of this
invention. Preparations of
specific compounds of this invention are shown in the following Schemes.
Y
In M Y
n M
X N¨u _______ X
N¨u
A m W boronic acid halo
(V)q (VNZ ( V)
or ester A I
Z
2 (V P 3
Formula I
Y
n M Y
n M
X N¨u _______ X
WO
u
A W halo boronic acid
N¨
(V) Z (V) or ester A
I
q (V NZ
r 3.
Formula I
21
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
Scheme 1
In aryl-aryl coupling reactions, halogen may be iodo, bromo, or chloro,
preferable bromo or iodo. In this
method, halogen substitutions may be transformed to aryl substitutions using
Suzuki coupling reaction
conditions. The conditions of this method are disclosed in many publications
which have been reviewed
by A. Suzuki in an article entitled "The Suzuki reaction with arylboron
compounds in arene chemistry" in
Modern Arene Chemistry 2002, 53-106. In carrying out this reaction any of the
suitable conditions
conventional in a Suzuki reaction can be utilized. Generally, Suzuki coupling
reactions are carried out in
the presence of a transition metal catalyst such as a palladium catalyst
utilizing any conventional organic
solvent for this reaction and a weak inorganic or organic base. Among the
preferred organic solvents are
the polar aprotic solvents. Any conventional polar aprotic solvents can be
utilized in preparing
compounds of the invention. Suitable solvents are customary, especially higher-
boiling solvents, e.g.
dimethoxyethane. The weak inorganic base can be a carbonate or bicarbonate,
such as potassium
carbonate or cesium carbonate. The organic base can be an amine such as
triethylamine.
o
( Y )n o
) n 0
CI ye 0 E5t OEt
m s.
u
haloi1/NO2 halo m NO 2
halo halo IN-
A I em NH
A I
(V (V (V
(V
4
6
1 5 7 8
Scheme 2
Specifically, the other spiro oxindole intermediate 7 is synthesized as shown
in Scheme 2. The cyclyl or
heterocyclyl substituted ester 5 is treated with a strong base such as, but
not limited to, lithium
diisopropylamide at low temperature in anhydrous solvent such as, but not
limited to, tetrahydrofurn to
react with starting material 4, which is either commercially available or
prepared by those skilled in the art
following the literature described methods to provide intermediate 6.
Intermediate 6 is reduced by a
reducing reagent such as, but not limited to, iron to give the corresponding
amino intermediate which
cyclizes to provide the oxindole compound 7 in situ. Thus, the compound 7 is
then N-akylated with an
alkylating reagent in the presence of a base such as, but not limited to,
potassium carbonate or sodium
hydride in a polar solvent such as, but not limited to, N,N-dimethylformamide
or tetrahydrofuran thereby to
generate the spiro oxindole intermediate 8.
22
CA 03147111 2022-01-11
WO 2021/022078 PCT/US2020/044322
Y 1 v ,
(in
m
N-
CI N X N X N
XH "Pd"
0 NBT halo u.. .< N
8
02NBr 02NBr (v
0 P .
9 11 12
"Pd"
0õ0
Y )n 0 Y 1
in 0
R16 ul
X N ( X N (
I m
N-u I m
N-u 15
. Formula I
02N A I H2N A I
(V
P N (V
P N
14
13
Scheme 3
Specifically, the compounds of Formula (I) in this invention can be
synthesized as shown in Scheme 3.
Commercially available 5-bromo-2-chloro-3-nitro-pyridine (9) reacts with a
nucleophile XH (10) in the
presence of a strong base such as, but not limited to, sodium hydride to
provide intermediate 11. Under
palladium catalyzed conditions, borate 12 can be prepared, which then reacts
with the spiro intermediate
8 to provide the cross coupled product 13. The nitro group in compound 13 is
reduced to amino group
using a reducing reagent such as, but not limited to, iron to provide
intermediate 14. Reaction of 14 with
different sulphonyl chlorides (15) furnishes the synthesis of compounds of
Formula (I).
I Y )n 0
0õ0 m
02N Br H2N Br R
16S X N
CI halo ....... N¨u
X N A I
X,N X N 15 Pd" n 0 1.;.
N
''. ¨" . µ-=-=;=W,-- (v
8
_______________________________________________________________________________
_ Formula I
11 16 17 18
Scheme 4
Specifically, the compounds of Formula (I) in this invention can also be
synthesized as shown in Scheme
4. The nitro group in compound 11 is reduced to amino group using a reducing
reagent such as, but not
limited to, iron to provide intermediate 16. Reaction of 16 with different
sulphonyl chlorides (15) provides
the sulphonamide intermediate 17, which is converted to its corresponding
borate 18 under palladium
catalysis. Borate 18 can couple with the halo compound 8 under Suzuki reaction
conditions to provide the
compounds of Formula (I).
In Scheme 3 and Scheme 4, the cross coupled compounds are also synthesizable
using Suzuki coupling
chemistry with components having reversed the halogen and boronate/boronic
acid substitution patterns,
for example, as shown in Scheme 5
23
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
X N
Y r.
n Y
0 ( R.. H
¨u Pd
Br
halo N >17.-13 N¨u 17
A 0
__________________________________________ Formula I
A I
(V
(V
8 19
Scheme 5
Specifically, the compounds of Formula (I) in this invention can also be
synthesized as shown in Scheme
5. The halo compound 8 can be converted to its corresponding borate 19 under
palladium catalysis.
Borate 19 can couple with the halo compound 17 under Suzuki reaction
conditions to provide the
compounds of Formula (I).
In the practice of the method of the present invention, an effective amount of
any one of the compounds
of this invention or a combination of any of the compounds of this invention
or a pharmaceutically
acceptable salt thereof, is administered via any of the usual and acceptable
methods known in the art,
either singly or in combination. The compounds or compositions can thus be
administered orally (e.g.,
buccal cavity), sublingually, parenterally (e.g., intramuscularly,
intravenously, or subcutaneously), rectally
(e.g., by suppositories or washings), transdermally (e.g., skin
electroporation) or by inhalation (e.g., by
aerosol), and in the form or solid, liquid or gaseous dosages, including
tablets and suspensions. The
administration can be conducted in a single unit dosage form with continuous
therapy or in a single dose
therapy ad lithium. The therapeutic composition can also be in the form of an
oil emulsion or dispersion
in conjunction with a lipophilic salt such as pamoic acid, or in the form of a
biodegradable sustained-
release composition for subcutaneous or intramuscular administration.
Useful pharmaceutical carriers for the preparation of the compositions
thereof, can be solids, liquids or
gases; thus, the compositions can take the form of tablets, pills, capsules,
suppositories, powders,
enterically coated or other protected formulations (e.g. binding on ion-
exchange resins or packaging in
lipid-protein vesicles), sustained release formulations, solutions,
suspensions, elixirs, aerosols, and the
like. The carrier can be selected from the various oils including those of
petroleum, animal, vegetable or
synthetic origin, e.g., peanut oil, soybean oil, mineral oil, sesame oil, and
the like. Water, saline, aqueous
dextrose, and glycols are preferred liquid carriers, particularly (when
isotonic with the blood) for injectable
solutions. For example, formulations for intravenous administration comprise
sterile aqueous solutions of
the active ingredient(s) which are prepared by dissolving solid active
ingredient(s) in water to produce an
aqueous solution, and rendering the solution sterile. Suitable pharmaceutical
excipients include starch,
cellulose, talc, glucose, lactose, gelatin, malt, rice, flour, chalk, silica,
magnesium stearate, sodium
stearate, glycerol monostearate, sodium chloride, dried skim milk, glycerol,
propylene glycol, water,
ethanol, and the like. The compositions may be subjected to conventional
pharmaceutical additives such
as preservatives, stabilizing agents, wetting or emulsifying agents, salts for
adjusting osmotic pressure,
buffers and the like. Suitable pharmaceutical carriers and their formulation
are described in Remington's
Pharmaceutical Sciences by E. W. Martin. Such compositions will, in any event,
contain an effective
24
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
amount of the active compound together with a suitable carrier so as to
prepare the proper dosage form
for proper administration to the recipient.
The dose of a compound of the present invention depends on a number of
factors, such as, for example,
the manner of administration, the age and the body weight of the patient, and
the condition of the patient
to be treated, and ultimately will be decided by the attending physician or
veterinarian. Such an amount of
the active compound as determined by the attending physician or veterinarian
is referred to herein, and in
the claims, as an "effective amount".
The invention will now be further described in the Examples below, which are
intended as an illustration
only and do not limit the scope of the invention.
EXAMPLES
Reagents were purchased from Aldrich, Sigma, TCI (Shanghai) Development,
Chembon Pharmaceutical
.. Co., Ltd, Zhangjiagang Aimate Huaxue Youxiangongsi, Changzhou Qinuo BioTech
Co. Ltd, and Shanghai
Weiyuan Fine Fluorine Technology Development Co., Ltd or other suppliers as
indicated below and used
without further purification. Reactions using microwave irradiation for
heating were conducted using a
Biotage Initiator+. The purification of multi-milligram to multi-gram scale
was conducted by methods known
to those skilled in the art such as elution of silica gel flash column
chromatography; preparative flash column
chromatography purifications were also affected in some cases by use of
disposal pre-packed silica gel
columns (Welch/Agela) eluted with a Biotage CombiFlash system.
For the purpose of judging compound identity and purity, typically, the
analytical LC-MS (liquid
chromatography/mass spectroscopy) system was used consisted of a Waters ZQTM
platform with
electrospray ionization in positive ion detection mode with an Agilent 1100
series HPLC with autosampler.
The column was usually a Water Xterra MS C18, 3.0 x 50 mm, 5 pm. The flow rate
was 1 mUmin, and the
injection volume was 10 pL. UV detection was in the range 210-400 nm. The
mobile phase consisted of
solvent A (water plus 0.06% TFA) and solvent B (acetonitrile plus 0.05% TFA)
with a gradient of 100%
solvent A for 0.7 min changing to 100% solvent B over 3.75 min, maintained for
1.1 min, then reverting to
100% solvent A over 0.2 min.
For some separations, the use of super critical fluid chromatography may also
be useful. Super critical fluid
chromatography separations were performed using a Mettler-Toledo Minigram
system with the following
typical conditions: 100 bar, 30 C, 2.0 mUmin eluting a 12 mm AD column with
40% Me0H in super critical
fluid CO2. In the case of analytes with basic amino groups, 0.2% isopropyl
amine was added to the
methanol modifier.
Many compounds of Formula (I) were also purified by reverse phase HPLC, using
methods well known to
those skilled in the art. In some cases, preparative HPLC purification was
conducted using PE Sciex 150
EX Mass Spec controlling a Gilson 215 collector attached to a Shimadzu
preparative HPLC system and a
Leap auto-injector. Compounds were collected from the elution stream using MS
detection in the positive
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
ion detection: The elution of compounds from C-18 columns (2.0 X 10 cm eluting
at 20 mUmin) was
affected using appropriate linear gradation mode over 10 minutes of Solvent
(A) 0.05% TFA/H20 and
Solvent (B) 0.035% TFA/acetyl nitrile. For injection on to HPLC systems, the
crude samples were
dissolved in mixtures of methanol, acetyl nitrile and DMSO.
Compounds were characterized either by 1H-NMR using a Bruker ADVANCE III HD
400MHz
Spectrometer or Bruker AVANCE 300MHz Spectrometer.
LIST OF ABBREVIATIONS
DCE 1,2-dichloroethane
DCM dichloromethane
DIPEA diisopropylethylamine
DMF N, N-dimethylformamide
DMSO dimethylsulfoxide
Et0Ac ethyl acetate
HOAc Acetic acid
HPLC high pressure liquid chromatography
Mel methyl iodide
Me0H methyl alcohol
MW microwave
NMP 1-methyl-2-pyrrolidinone
it ambient temperature
TBDMStert-butyl-dimethylsily1
TEA triethylamine
TFA tifluoroacetic acid
TEMP02,2,6,6-tetramethy1-1-piperidinyloxy
THF tetrahydrofuran
PREPARATION OF COMPOUNDS
0
Br NO 2 Br NO2
LDA, THF, -78 C-0 C, 2 h F
Methyl 1-(6-bromo-7-fluoro-3-nitroquinolin-4-yl)cyclobutane-1-carboxylate:
A solution of methyl
cyclobutanecarboxylate (0.73 g, 6.38 mmol) in tetrahydrofuran (5.00 mL) was
treated with freshly prepared
lithium diisopropylamide (6.38 mmol) in tetrahydrofuran (45.0 mL) for 1 hour
at -78 C under nitrogen
atmosphere followed by the addition of 6-bromo-4-chloro-7-fluoro-3-
nitroquinoline (1.50 g, 4.91 mmol) in
portions over 2 min. After stirring for additional 1 hour at ambient
temperature, the reaction was quenched
by saturated aqueous ammonium chloride (60.0 mL) and diluted with water (120
mL). The resulting mixture
was extracted with ethyl acetate (3 x 60.0 mL). The combined organic layers
was washed with brine (2 x
50.0 mL) and dried over anhydrous sodium sulfate. After filtration, the
filtrate was concentrated under
reduced pressure. The residue was purified by silica gel column
chromatography, eluted with 1%-2% ethyl
26
CA 03147111 2022-01-11
WO 2021/022078 PCT/US2020/044322
acetate in petroleum ether to afford the title compound as a colorless solid
(240 mg, 13%): 1H NMR (400
MHz, CDCI3) 6 9.14 (s, 1H), 8.20 (d, J= 7.2 Hz, 1H), 7.90 (d, J= 8.8 Hz, 1H),
3.84 (s, 3H), 3.12-2.99 (m,
1H), 2.58-2.48 (m, 3H), 1.91-1.83 (m, 1H), 1.45-1.27 (m, 1H); MS: [(M + 1)]+ =
383.17, 385.17.
Br .O2 2 Fe Br NH
HOAc, rt, 18 h
8'-Bromo-7'-fluorospiro[cyclobutane-1,1'-pyrrolo[2,3-c]quinolin]-2'(37-1)-one:
A mixture of methyl 1-(6-
bromo-7-fluoro-3-nitroquinolin-4-yl)cyclobutane-1-carboxylate (240 mg, 0.63
mmol) and iron powder (350
mg, 6.26 mmol) in acetic acid (10.0 mL) was stirred for 18 hours at ambient
temperature. The resulting
mixture was filtered and the filter cake was washed with ethyl acetate (5 x
100 mL). The filtrate was
concentrated under reduced pressure. The residue was purified by silica gel
column chromatography,
eluted with 1%-2% methanol in dichloromethane to afford the title compound as
a light yellow solid (100
mg, 50%): 1H NMR (400 MHz, DMSO-d6) 6 10.75 (s, 1H), 8.68 (s, 1H), 8.52 (d, J=
7.5 Hz, 1H), 7.98 (d, J
= 10.1 Hz, 1H), 2.90-2.75 (m, 2H), 2.50-2.37 (m, 4H); MS: [(M + 1)]+ = 321.15,
323.15.
0 0
Br NH
1) NaH, DMF, 0 C, 30 min Br
2) Mel, 0 C-rt, 40 min
8'-Bromo-7'-fluoro-3'-methylspiro[cyclobutane-1,1'-pyrrolo[2,3-c]quinolin]-
2'(37-1)-one: A solution of 8-
bromo-7-fluoro-2,3-dihydrospiro[cyclobutane-1,1-pyrrolo[2,3-c]quinoline]-2-one
(100 mg, 0.31 mmol) in
N,N-dimethylformamide (10.0 mL) was treated with sodium hydride (19.9 mg, 0.50
mmol, 60% dispersed
in mineral oil) at 0 C for 30 min under nitrogen atmosphere followed by the
addition of iodomethane (66.3
mg, 0.47 mmol). After stirring for additional 40 min at ambient temperature,
the reaction was quenched by
saturated aqueous ammonium chloride (10.0 mL). The resulting mixture was
diluted with water (100 mL)
and extracted with ethyl acetate (3 x 30.0 mL). The combined organic layers
was washed with brine (2 x
20.0 mL) and dried over anhydrous sodium sulfate. After filtration, the
filtrate was concentrated under
reduced pressure. The residue was purified by Prep-TLC (DCM/Me0H = 20/1, v:v)
to afford the title
compound as a colorless solid (102 mg, 98%): 1H NMR (400 MHz, CD30D) 6 8.78
(s, 1H), 8.61 (d, J= 7.4
Hz, 1H), 7.85 (d, J = 9.8 Hz, 1H), 3.36 (s, 3H), 2.94-2.85 (m, 2H), 2.72-2.61
(m, 3H), 2.56-2.48 (m, 1H);
MS: [(M + 1)]+ = 335.00, 337.00.
Boc20
)N OH ________________________________________________ N OH
Me0H, 0 C¨ rt, 2 h Bioc
tert-Butyl N-(2-hydroxyethyl)-N-(propan-2-yl)carbamate: To a solution of 2-
[(propan-2-yDamino]ethan-1-ol
(40.0 g, 388 mmol) in methanol (300 mL) was added di-tert-butyl dicarbonate
(127 g, 586 mmol) dropwise
at 0 C. The resulting mixture was stirred for 2 hours at ambient temperature
and concentrated under
reduced pressure. The residue was purified by silica gel column
chromatography, eluted with 0%-4% ethyl
acetate in petroleum ether. The desired fractions were collected and
concentrated under reduced pressure
27
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
to afford the title compound as a colorless oil (65.0 g, 82%): 1H NMR (400
MHz, CDCI3) 6 4.17 (s, 1H), 3.71
(t, J = 5.4 Hz, 2H), 3.30 (t, J = 5.4 Hz, 2H), 1.47 (s, 9H), 1.12 (d, J = 6.8
Hz, 6H).
CI
I
) NOBr
0 N
N
Boc NaH, THF, 0 C-it, 2 h .. Boc NoBr
tert-Butyl N42-[(5-bromo-3-nitropyridin-2-yDoxy]ethylFA/-(propan-2-
y1)carbamate: A solution of tert-butyl N-
(2-hydroxyethyl)-N-(propan-2-yl)carbamate (15.4 g, 75.8 mmol) in anhydrous
tetrahydrofuran (250 mL) was
treated with sodium hydride (3.30 g, 82.1 mmol, 60% w/w dispersed in mineral
oil) for 1 hour at 0 C under
nitrogen atmosphere followed by the addition of 5-bromo-2-chloro-3-
nitropyridine (15.0 g, 63.2 mmol) over
2 min at 0 C. After additional 2 hours at 25 C, the reaction was quenched by
saturated aqueous
ammonium chloride (50.0 mL) and diluted with water (500 mL). The aqueous layer
was extracted with ethyl
acetate (3 x 150 mL). The combined organic layers was washed with brine (2 x
100 mL) and dried over
anhydrous sodium sulfate. After filtration, the filtrate was concentrated
under reduced pressure. The residue
was purified by silica gel column chromatography, eluted with 1%-18% ethyl
acetate in petroleum ether.
The desired fractions were collected and concentrated under reduced pressure
to afford the title compound
as a light yellow oil (18.0 g, 71%): 1H NMR (400 MHz, CDCI3) 6 8.42 (d, J =
2.4 Hz, 1H), 8.37 (d, J = 2.4
Hz, 1H), 4.57 (t, J = 6.3 Hz, 2H), 4.32 (s, 1H), 3.51 (t, J = 6.3 Hz, 2H),
1.47 (s, 9H), 1.15 (d, J = 6.9 Hz, 6H);
MS: [(M + 1)]+ = 404.00, 406.00.
)NO
Fe
gloc I3oc
NO2 Br HOAc, rt, 1 h NH2 Br
tert-Butyl N42-[(3-amino-5-bromopyridin-2-yl)oxy]ethylFA/-(propan-2-
y1)carbamate: To a solution of
tert-butyl N42-[(5-bromo-3-nitropyridin-2-yl)oxy]ethylFA/-(propan-2-
y1)carbamate (15.0 g, 37.1 mmol) in
acetic acid (150 mL) was added iron powder (20.7 g, 371 mmol) at ambient
temperature. After stirring for
additional 1 hour at ambient temperature, the resulting mixture was filtered
and the filtered cake was
washed with tetrahydrofuran (4 x 100 mL). The filtrate was concentrated under
reduced pressure. The
residue was purified by silica gel column chromatography, eluted with 20%
ethyl acetate in petroleum ether.
The desired fractions were collected and concentrated under reduced pressure
to afford the title compound
as a colorless solid (12.0 g, 86%): 1H NMR (400 MHz, CD30D) 6 7.40 (d, J= 2.2
Hz, 1H), 7.04 (d, J= 2.2
Hz, 1H), 4.40 (t, J= 6.3 Hz, 2H), 4.25-3.99 (m, 1H), 3.52 (t, J= 6.3 Hz, 2H),
1.46 (s, 9H), 1.17 (d, J= 6.8
Hz, 6H); MS: [(M + 1)]+ = 374.10, 376.10.
)N
NON
BI I MsCI, Py, rt, 6 h Boc
NH2Br NHB
oc r
0==0
tert-Butyl (2-((5-bromo-3-(methylsulfonamido)pyridin-2-
yl)oxy)ethyl)(isopropyl)carbamate: To a solution of
tert-butyl N42-[(3-amino-5-bromopyridin-2-yDoxy]ethylFA/-(propan-2-
y1)carbamate (18.8 g, 50.3 mmol) in
pyridine (400 mL) was added dropwse methanesulfonyl chloride (8.63 g, 75.4
mmol) at ambient
28
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
temperature. After stirirng for 6 hours at ambient temperature, the resulting
mixture was concentrated under
reduced pressure. The residue was purified by silica gel column
chromatography, eluted with 3%-25%
ethyl acetate in petroleum ether. The desired fractions were collected and
concentrated under reduced
pressure to afford the title compound as an off-colorless solid (16.3 g, 72%):
1H NMR (400 MHz, DMS0-
d6) 6 9.41 (s, 1H), 8.05 (d, J = 2.2 Hz, 1H), 7.79 (d, J = 2.2 Hz, 1H), 4.35
(t, J = 6.3 Hz, 2H), 4.15 (s, 1H),
3.43 (t, J = 6.3 Hz, 2H), 3.11(s, 3H), 1.39 (s, 9H), 1.09 (d, J = 6.8 Hz, 6H);
MS: [(M + 1)]+ = 452.00, 454.00.
'-S-CI
/...
I
8 BIoc
Bloc NHBr
NH21Etr Py, rt, 6 h
0=S=0
tert-Butyl (2-((5-bromo-3-(ethylsulfonamido)pyridin-2-
yl)oxy)ethyl)(isopropyl)carbamate: To a stirred
solution of tert-butyl (2-((3-amino-5-bromopyridin-2-
yl)oxy)ethyl)(isopropyl)carbamate (5.00 g, 13.4 mmol)
in pyridine (120 mL) was added ethanesulfonyl chloride (5.15 g, 40.1 mmol)
dropwise at ambient
temperature under nitrogen atmosphere. After stirring for 6 hours at ambient
temperature under nitrogen
atmosphere, the resulting mixture was concentrated under reduced pressure. The
residue was purified by
silica gel column chromatography, eluted with 3%-25% ethyl acetate in
petroleum ether. The desired
fractions were collected and concentrated under reduced pressure to afford the
title compound as a light
yellow solid (3.78 g, 61%): 1H NMR (400 MHz, CD30D) 6 7.92 (s, 1H), 7.87 (s,
1H), 4.48 (t, J = 6.6 Hz, 2H),
4.25-4.20 (m, 1H), 3.54 (t, J= 6.6 Hz, 2H), 3.13 (t, J= 7.3 Hz, 2H), 1.49 (s,
9H), 1.35 (t, J= 7.4 Hz, 3H),
1.19 (d, J = 6.8 Hz, 6H); MS: [(M + 1)]+ = 466.10, 468.10.
)N ot3H
B
OH N oc
IBr I
Bl I
PPh3, DIAD, THF, 0 C-rt, 16 h oc
tert-Butyl N42-[(5-bromo-3-iodopyridin-2-yDoxy]ethy1FN-isopropylcarbamate:
To a solution of 5-
bromo-3-iodopyridin-2-ol (10.0 g, 33.3 mmol) in anhydrous tetrahydrofuran (300
mL) were added
triphenylphosphine (11.4 g, 43.3 mmol), tert-butyl N-(2-hydroxyethyl)-N-
isopropylcarbamate (8.80 g, 43.3
mmol) and diisopropyl azodiformate (8.80 g, 43.4 mmol) dropwise at 0 C. The
resulting mixture was stirred
for additional 16 hours at ambient temperature under nitrogen atmosphere. The
resulting mixture was
concentrated under reduced pressure. The residue was purified by silica gel
column chromatography,
eluted with 2%-10% ethyl acetate in petroleum ether. The desired fractions
were collected and
concentrated under reduced pressure to afford the title compound as a
colorless oil (14.0 g, 87%): 1H NMR
(400 MHz, CDCI3) 6 8.14 (s, 2H), 4.43 (t, J= 6.4 Hz, 2H), 4.38-3.96 (m, 1H),
3.50 (t, J= 6.4 Hz, 2H), 1.49
(s, 9H), 1.19 (d, J = 6.8 Hz, 6H); MS: [(M + 1)]+ = 484.95, 486.95.
A-NH2
)N 0
)N K3PO4, Pd2(dba)3.CHCI3 Bac NH
Br
Boc 0==0
I Br Xantphos, toluene,100 C, 48 h
29
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
tert-Butyl (2-((5-bromo-3-((1-methylethyl)sulfonamido)pyridin-2-
yl)oxy)ethyl)(isopropyl)carbamate: To a
mixture of tert-butyl (2-((5-bromo-3-iodopyridin-2-
yl)oxy)ethyl)(isopropyl)carbamate (5.00 g, 10.3 mmol),
4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (1.80 g, 3.10 mmol) and
propane-2-sulfonamide (1.50 g,
12.4 mmol) in toluene (125 mL) were added tripotassium phosphate (10.9 g, 51.5
mmol) and
tris(dibenzylideneacetone)dipalladium-chloroform adduct (1.10 g, 1.10 mmol) at
ambient temperature. The
resulting mixture was stirred for 48 hours at 100 C under argon atmosphere.
After cooling down to ambient
temperature, the resulting mixture was filtered. The filtered cake was washed
with ethyl acetate (3 x 20 mL).
The filtrate was concentrated under reduced pressure. The residue was purified
by silica gel column
chromatography, eluted with 1%-20% ethyl acetate in petroleum ether. The
desired fractions were
collected and concentrated under reduced pressure to afford the title compound
as a colorless solid (1.90
g, 39%): 1H NMR (400 MHz, CDCI3) 6 7.94 (d, J = 2.1 Hz, 1H), 7.89 (d, J = 2.2
Hz, 1H), 4.44 (t, J = 6.3 Hz,
2H), 4.11 (s, 1H), 3.48 (t, J= 6.3 Hz, 2H), 3.24-3.30 (m, 1H), 1.48 (s, 9H),
1.41 (d, J= 6.8 Hz, 6H), 1.14 (d,
J = 6.8 Hz, 6H); MS: [(M + 1)]+ = 480.20, 482.20.
N
I 4 M HCI (g)/dioxane
Boc
NHBr
DCM, 0 C-rt, 40 min
0=S=0 0=S=0
N-(5-Bromo-242-[(propan-2-yDamino]ethoxy]pyridin-3-yOmethanesulfonamide: To a
solution of tert-butyl
N42-[(5-bromo-3-methanesulfonamidopyridin-2-yl)oxy]ethy1FN-(propan-2-
yl)carbamate (3.00 g, 6.63 mmol)
in dichloromethane (5.00 mL) was treated with hydrogen chloride (20.0 mL, 4 M
in 1,4-dioxane) for 40 min
at ambient temperature. The resulting mixture was concentrated under reduced
pressure. The residue was
basified to pH = 8 with saturated aqueous sodium bicarbonate (30.0 mL). The
resulting mixture was
extracted with ethyl acetate (6 x 200 mL). The combined organic layers was
dried over anhydrous sodium
sulfate and filtered. The filtrate was concentrated under reduced pressure.
The residue was purified by
silica gel column chromatography, eluted with 1%-10% methanol in
dichloromethane. The desired fractions
were collected and concentrated under reduced pressure to afford the title
compound as a colorless solid
(1.20 g, 50%): 1H NMR (400 MHz, DMSO-d6) 6 7.68 (d, J = 2.3 Hz, 1H), 7.61 (d,
J = 2.3 Hz, 1H), 5.75 (s,
1H), 4.36 (t, J= 5.2 Hz, 2H), 3.19-3.12 (m, 1H), 3.07 (t, J= 5.1 Hz, 2H), 2.84
(s, 3H), 1.15 (d, J= 6.4 Hz,
6H); MS: [(M + 1)]+ = 352.10, 354.10.
The following intermediates were prepared according to the procedure described
above:
MS:
Intermediates Structure Name 1H NMR
[(M + 1)]+
1H NMR (400 MHz,
N-(5-B ro mos-2- (2-
CDCI3) 6 7.92 (s, 2H),
ON
CC108 NH (isopropylamino)ethoxy)p 366.10
4.50 (t, J = 5.3 Hz, 2H),
- -Br
o==o yridin-3- 368.10
3.15 (q, J = 7.4 Hz, 2H),
yl)ethanesulfonamide
3.07 (t, J = 5.3 Hz, 2H),
3.01-2.93 (m, 1H), 1.40
CA 03147111 2022-01-11
WO 2021/022078 PCT/US2020/044322
(t, J = 7.4 Hz, 3H), 1.16
(d, J = 6.3 Hz, 6H).
1H NMR (400 MHz,
CDCI3) 6 7.95 (d, J =
2.2 Hz, 1H), 7.91 (d, J =
N-(5-Bromo-2-(2- 2.3
Hz, 1H), 4.48 (t, J=
NHBr (isopropylamino)ethoxy)p 380.15 5.4
Hz, 2H), 3.31-3.23
CC110 0=S=o yridin-3-yl)propane-2- 382.15 (m,
1H), 3.02 (t, J= 5.4
sulfonamide Hz, 2H), 2.95-2.87 (m,
1H), 1.40 (d, J = 6.8 Hz,
6H), 1.11 (d, J= 6.2 Hz,
6H).
31
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
\--R p--._(
)NON
______________________________________ B-13, __
I
H H
1:::
_______________________________________________ ...-
'T
NHBr
I Pd(dppf)C12-DCM, KOAc, NIH
B
0=S=00=S=0 0
1,4-dioxane, 90 C, 2 h
II
N-(2-(2-(lsopropylamino)ethoxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-
y0pyridin-3-
yl)methanesulfonamide: To a solution of tert-butyl N42-[(5-bromo-3-
methanesulfonamidopyridin-2-
yDoxy]ethylFA/-(propan-2-yl)carbamate (2.00 g, 5.68 mmol) and
bis(pinacolato)diboron (2.88 g, 11.4 mmol)
in 1,4-dioxane (50.0 mL) were added potassium acetate (2.23 g, 22.7 mmol) and
bis(diphenylphosphino)ferrocene]dichloro palladium (II) dichloromethane adduct
(0.46 g, 0.57 mmol) at
ambient temperature. After stirring for 2 hours at 90 C under nitrogen
atmosphere, the resulting mixture
was concentrated under reduced pressure. The resulting mixture was diluted
with dichloromethane (100
mL). After filtration, the filtered cake was washed with dichloromethane (3 x
10.0 mL). The filtrate was
concentrated under reduced pressure. The residue was purified by reversed
phase flash chromatography
with the following conditions: Column: Spherical C18, 20-40 pm, 330 g; Mobile
Phase A: Water (plus 10
mM NI-141-1CO3), Mobile Phase B: acetonitrile; Flow rate: 65 mUmin; Gradient
(B%): 5%-22%, 6 min;
22%-40%, 20 min; 40%-95%; 2 min; 95%, 5 min; Detector: UV 254 nm; Rt: 15 min.
The fractions containing
desired product were collected and concentrated under reduced pressure to
afford the title compound as
an off-white solid (1.57 g, 87%): 1H NMR (400 MHz, CDCI3) 6 8.27 (d, J= 1.7
Hz, 1H), 8.06 (d, J= 1.7 Hz,
1H), 4.51 (t, J= 5.2 Hz, 2H), 3.05-2.99 (m, 5H), 2.95-2.84 (m, 1H), 1.33 (s,
12H), 1.11 (d, J= 6.3 Hz, 6H);
MS: [(M + 1)]+ = 400.30.
COMPOUND 568
HCI
0
....õ..--.... N -----....õ..- 0 õ....NI
H I NI,
NH
0==0
F N
0
H
Br N----
\
_____________________ B_Bp...
,,L,N....-.,,Oy
HBO..
NH Br __________________ NH 0 0
Pd(dppf)C12.1DCM, KOAc ..... NH
I I 1 1,4-dioxane, H20, N2, 85 C, 6
h =S=0 =S=0 00==0
1,4-dioxane, 90 C, 3 h Pd(PPh3)4, Na2CO3,
.....õc F N
CC110 CC110-1
568
N-(5-(7'-Fluoro-3'-methyl-2'-oxo-2',3'-dihydrospiro[cyclobutane-1,1'-
pyrrolo[2,3-c]quinolin]-8'-y1)-2-(2-
(isopropylamino)ethoxy)pyridin-3-yl)propane-2-sulfonamide: To a solution of
N45-bromo-242-
(isopropylamino)ethoxy]pyridin-3-yl]propane-2-sulfonamide (2.00 g, 5.26 mmol)
in 1,4-dioxane (50.0 mL)
were added bis(pinacolato)diboron (4.01 g, 15.8 mmol), potassium acetate (2.06
g, 21.1 mmol) and
bis(diphenylphosphino)ferrocene]dichloro palladium (II) dichloromethane adduct
(0.34 g, 0.42 mmol) at
ambient temperature. The resulting mixture was stirred for 3 hours at 90 C
under nitrogen atmosphere.
32
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
The resulting mixture was cooled down to ambient temperature followed by the
additions of 8-bromo-7-
fluoro-3-methylspiro[cyclobutane-1,1-pyrrolo[2,3-c]quinolin]-2-one (1.26 g,
3.76 mmol), water (12.5 mL),
sodium carbonate (0.80 g, 7.55 mmol) and tetrakis(triphenylphosphine)palladium
(0) (0.43 g, 0.38 mmol).
The resulting mixture was stirred for 6 hours at 85 C under nitrogen
atmosphere. After cooling down to
ambient temperature, the resulting mixture was concentrated under reduced
pressure. The residue was
purified by reversed phase flash chromatography with the following conditions:
Column: Spherical C18,
20-40 pm, 120 g; Mobile phase A: water (plus 10 mM NI-141-1CO3); Mobile phase
B: acetonitrile; Flow rate:
45 mUmin; Gradient (B%): 5%, 2 min; 5%-25%, 8 min; 25%-39%, 9 min; 39%, 10
min; 39%-95%; 3 min;
95%, 2 min; Detector: UV 254 nm; Rt: 21 min. The fractions containing desired
product were collected and
concentrated under reduced pressure to afford the title compound as a
colorless solid (1.66 g, 80%): 1H
NMR (400 MHz, DMSO-d6) 6 8.88 (s, 1H), 8.35-8.31 (m, 2H), 8.07 (s, 1H), 7.99
(s, 1H), 7.96 (s, 1H), 4.55
(br, 1H), 4.44 (t, J= 5.2 Hz, 2H), 3.35-3.30 (m, 4H), 2.93-2.79 (m, 5H), 2.55-
2.38 (m, 4H), 1.29 (d, J= 7.2
Hz, 6H), 1.02 (d, J = 6.0 Hz, 6H); MS: [(M + 1)]+ = 556.30.
0
0
)NO )NO
N-- dil. HCI (aq.)
N--
NH
HCI NH
0==0 0==0
F F
568 568-HCI
N-(5-(7'-Fluoro-3'-methy1-2'-oxo-2',3'-dihydrospiro[cyclobutane-1,1'-
pyrrolo[2,3-c]quinolin]-8'-y1)-2-(2-
(isopropylamino)ethoxy)pyridin-3-yl)propane-2-sulfonamide hydrogen chloride. A
solution of N-(547-
Fluoro-3-methy1-2-oxospiro[cyclobutane-1,1-pyrrolo[2,3-c]qu inolin]-8-yI]-2-[2-
(isopropylamino)ethoxy]pyridin-3-yl)propane-2-sulfonamide (1.66 g, 2.99 mmol)
in diluted aqueous
hydrochloric acid solution (392 mL, 3.14 mmol, 0.008 M) and acetonitrile (79.0
mL) was lyophilized to afford
the title compound as a yellow solid (1.76 g, 100%): 1H NMR (400 MHz, DMSO-d6)
6 9.45 (s, 1H), 9.02 (br,
2H), 8.90 (s, 1H), 8.40 (s, 1H), 8.32 (d, J = 8.4 Hz, 1H), 8.16 (s, 1H), 8.00
(d, J = 8.4 Hz, 1H), 4.65 (t, J =
4.4 Hz, 2H), 3.54-3.40 (m, 3H), 3.31 (s, 3H), 2.91 (t, J = 11.2 Hz, 2H), 2.55-
2.45 (m, 5H), 1.34-1.30 (m,
12H); MS: [(M + 1)]+ = 556.30.
In a similar manner or palladium (II) couples were affected with Intermediates
CC108 and CC110 to afford
the following compounds example 569 and example 570.
MS:
Comp. # Structure Name 1H NMR
[(M+1)]+
N-(5-(7'-Fluoro-3'- 1H NMR (400
MHz,
0 methyl-2'-oxo-2',3'- DM50-d6) 6
8.88 (s,
)µI
1
H N dihydrospiro[cyclob 1H), 8.31 (d,
J = 8.7 Hz,
¨
569 NH
o=o utane-1 ,1'- 528.30 2H), 8.01-
7.94 (m, 2H),
=e
F N pyrrolo[2,3- 4.44 (t, J =
5.4 Hz, 2H),
c]quinolin]-8'-yI)-2- 3.31 (s, 3H),
3.05 (s,
(2- 3H), 2.97 (t,
J = 5.7 Hz,
33
CA 03147111 2022-01-11
WO 2021/022078 PCT/US2020/044322
(isopropylamino)eth 2H), 2.95-2.85 (m,
3H),
oxy)pyridin-3- 2.60-2.51 (m, 2H),
yl)methanesulfonam 2.49-2.41 (m, 2H),
1.07
ide (d, J = 6.3 Hz, 6H).
1H NMR (400 MHz,
N-(5-(7'-Fluoro-3'-
CD30D) 6 8.77 (s, 1H),
methy1-2'-oxo-2',3'-
8.42 (d, J= 8.1 Hz, 1H),
dihydrospiro[cyclob
8.18 (t, J = 2.2 Hz, 1H),
utane-1,1'-
8.13 (t, J = 1.8 Hz, 1H),
569 0 pyrrolo[2,3-
7.86 (d, J = 12.1 Hz,
(HC1
HCI NH I
N-- c]quinolin]-8'-y1)-2- 528.30
tr.)==c) 1H), 4.63 (t, J = 5.2
Hz,
salt) I F N (2-
2H), 3.40-3.32 (m, 6H),
(isopropylamino)eth
3.04 (s, 3H), 3.00-2.90
oxy)pyridin-3-
(m, 2H), 2.74-2.51 (m,
yl)methanesulfonam
4H), 1.31 (d, J = 5.2 Hz,
ide hydrochloride
6H).
1H NMR (400 MHz,
DMSO-d6) 6 8.88 (s,
N-(5-(7'-Fluoro-3'-
1H), 8.35-8.28 (m, 2H),
methy1-2'-oxo-2',3'-
8.02 (t, J = 2.0 Hz, 1H),
dihydrospiro[cyclob
7.97 (d, J = 12.2 Hz,
N 1H), 4.43 (t, J = 5.4
Hz,
N pyrrolo[2,3-
H
N H 2H), 3.31 (s, 3H), 3.14
570 c]quinolin]-8'-y1)-2- 542.30
=
(2- (q, J = 7.2 Hz, 2H),
2.96
o= o F
(t, J = 5.5 Hz, 2H), 2.93-
(isopropylamino)eth
2.82 (m, 3H), 2.60-2.53
oxy)pyridin-3-
(m, 2H), 2.49-2.40 (m,
yl)ethanesulfonamid
2H), 1.28 (t, J = 7.3 Hz,
3H), 1.05 (d, J= 6.2 Hz,
6H).
1H NMR (400 MHz,
N-(5-(7'-Fluoro-3'-
CD30D) 6 8.82 (s, 1H),
methy1-2'-oxo-2',3'-
8.43 (d, J = 8.0 Hz, 1H),
dihydrospiro[cyclob
8.35 (d, J= 2.1 Hz, 1H),
utane-1,1'-
8.22 (t, J = 1.7 Hz, 1H),
570 0 pyrrolo[2,3-
1 7.88 (d, J = 11.8 Hz,
N--
(HC1 HCI NH c]quinolin]-8'-y1)-2- 542.30
1H), 4.78 (t, J = 5.1 Hz,
salt) F N (2-
2H), 3.60-3.50 (m, 3H),
(isopropylamino)eth
3.39 (s, 3H), 3.27 (t, J=
oxy)pyridin-3-
7.3 Hz, 2H), 3.02-2.89
yl)ethanesulfonamid
(m, 2H), 2.76-2.49 (m,
e hydrochloride
4H), 1.45-1.37 (m, 9H).
34
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
COMPOUND 571
Cl
BrJ NO2 0
Boc ,
Boc,
0 ____________________________________________________ Br NO
2
O LDA, THF, -78 C-0 C, 2 h
1-(tert-Butyl) 3-methyl 3-(6-bromo-7-fluoro-3-nitroquinolin-4-yl)azetidine-1,3-
dicarboxylate: To a
solution of freshly prepared lithium diisopropylamide (137 mmol) in anhydrous
tetrahydrofuran (110 mL)
was added a solution of 1-tert-butyl 3-methyl azetidine-1,3-dicarboxylate
(29.3 g, 137 mmol) in
tetrahydrofuran (100 mL) at -78 C. After stirring for 1 hour, a solution of 6-
bromo-4-chloro-7-fluoro-3-
nitroquinoline (32.0 g, 105 mmol) in tetrahydrofuran (100 mL) was added to the
reaction mixture over 20
min. The resulting mixture was slowly warmed to 0 C for 2 hours. The reaction
was quenched by saturated
aqueous ammonium chloride (20.0 mL) and diluted with water (800 mL). The
resulting mixture was
extracted with ethyl acetate (3 x 150 mL). The combined organic layers was
washed with brine (2 x 100
mL) and dried over anhydrous sodium sulfate. After filtration, the filtrate
was concentrated under reduced
pressure. The residue was purified by silica gel column chromatography, eluted
with 5%-20% ethyl acetate
in petroleum ether to afford the title compound as a light yellow solid (25.0
g, 49%): 1H NMR (400 MHz,
DMSO-d6) 6 9.35 (s, 1H), 8.21 (d, J= 9.3 Hz, 1H), 8.14 (d, J= 7.1 Hz, 1H),
4.18-4.11 (m, 2H), 3.87-3.74
(m, 2H), 3.66 (s, 3H), 1.37 (s, 9H); MS: [(M + 1)]+ = 484.20, 486.20.
Elm\
Boc,N 0 0
0¨ Fe NH
Br NO2 ______________ Br
HOAc, rt, 3 h FN
tert-Butyl 8'-bromo-7'-fluoro-2'-oxo-2',3'-dihydrospiro[azetidine-3,1'-
pyrrolo[2,3-c]quinoline]-1-carboxylate:
To a solution of 1-tert-butyl 3-methyl 3-(6-bromo-7-fluoro-3-nitroquinolin-4-
yl)azetidine-1,3-
dicarboxylate (12.0 g, 24.8 mmol) in acetic acid (300 mL) was added iron
powder (9.69 g, 174 mmol) at
ambient temperature. After stirring for 3 hours at ambient temperature, the
resulting mixture was
concentrated under reduced pressure. The residue was taken up with water (200
mL) and extracted with
.. ethyl acetate (4 x 100 mL). The combined organic layers was washed with
brine (2 x 100 mL) and dried
over anhydrous sodium sulfate. After filtration, the filtrate was concentrated
under reduced pressure. The
residue was purified by silica gel column chromatography, eluted with 1%-5%
methanol in dichloromethane
to afford the title compound as an off-colorless solid (10.4 g, 99%): 1H NMR
(400 MHz, DMSO-d6) 6 11.02
(br, 1H), 8.71 (s, 1H), 8.24 (d, J= 7.3 Hz, 1H), 8.03 (d, J= 10.1 Hz, 1H),
4.29 (d, J= 9.0 Hz, 2H), 4.21 (d,
J = 9.0 Hz, 2H), 1.49 (s, 9H); MS: [(M + 1)]+ = 422.20, 424.20.
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
Boc, Boc,
0 0
Br NH NaH, Mel Br
DMF, 0 C-rt, 1 h
tert-Butyl
8'-bromo-7'-fluoro-3'-methyl-2'-oxo-2',3'-dihydrospiro[azetidine-3,1'-
pyrrolo[2,3-c]quinoline]-1-
carboxylate:
To a stirred solution of tert-butyl 8-bromo-7-fluoro-2-oxo-2,3-
dihydrospiro[azetidine-3,1-
pyrrolo[2,3-c]quinoline]-1-carboxylate (4.22 g, 9.99 mmol) in N,N-
dimethylformamide (100 mL) was added
sodium hydride (0.52 g, 13.0 mmol, 60% dispersed in mineral oil) at 0 C under
nitrogen atmosphere. The
resulting mixture was stirred for 1 hour at ambient temperature followed by
the addition of iodomethane
(1.70 g, 12.0 mmol). After stirring for additional 1 hour at ambient
temperature, the reaction was quenched
by saturated aqueous ammonium chloride (20.0 mL) and diluted with water (1.00
L). The precipitated solids
was collected by filtration, washed with water (3 x 30.0 mL) and hexane (2 x
30.0 mL). The resulting solid
was dried under infrared light to afford the title compound as a light yellow
solid (3.93 g, 90%): 1H NMR
(400 MHz, CDCI3) 6 8.71 (s, 1H), 8.47 (d, J = 7.0 Hz, 1H), 7.94 (d, J = 9.1
Hz, 1H), 4.54 (d, J = 9.1 Hz, 2H),
4.31 (d, J = 9.0 Hz, 2H), 3.40 (s, 3H), 1.56 (s, 9H); MS: [(M + 1)]+ = 436.15,
438.15.
Boc,
0 HN 0
Br N- TFA N-
Br
DCM, rt, 5 h
8-Bromo-7-fluoro-3-methyl-2,3-dihydrospiro[azetidine-3,1-pyrrolo[2,3-
c]quinolin]-2-one: A solution of
tert-butyl
8-bromo-7-fluoro-3-methyl-2-oxo-2,3-dihydrospiro[azetidine-3,1-pyrrolo[2,3-
c]quinoline]-1-
carboxylate (3.93 g, 9.01 mmol) and trifluoroacetic acid (20.0 mL) in
dichloromethane (100 mL) was stirred
for 5 hours at ambient temperature. The resulting mixture was concentrated
under reduced pressure. The
residue was basified to pH = 8 with saturated aqueous sodium bicarbonate
solution. The resulting mixture
was extracted with ethyl acetate (6 x 300 mL). The combined organic layers was
washed with brine (2 x
300 mL) and dried over anhydrous sodium sulfate. After filtration, the
filtrate was concentrated under
reduced pressure to afford the title compound as a light yellow solid (3.00 g,
99%): 1H NMR (400 MHz,
DMSO-d6) 6 9.56 (d, J = 7.7 Hz, 1H), 8.92 (s, 1H), 8.02 (d, J = 10.2 Hz, 1H),
4.18 (d, J = 7.5 Hz, 2H), 3.59
(d, J = 7.5 Hz, 2H), 3.29 (s, 3H); MS: [(M + 1)]+ = 335.95, 337.95.
HN 0
N 0
Br acetone, 3CN NaBH
Br N-
Et0H,50 h
8'-Bromo-7'-fluoro-1-isopropyl-3'-methylspiro[azetidine-3,1'-pyrrolo[2,3-
c]quinolin]-2'(37-1)-one: To a
stirred solution of 8'-bromo-7'-fluoro-3'-methylspiro[azetidine-3,1'-
pyrrolo[2,3-c]quinolin]-2'(37-1)-one (100
36
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
mg, 0.30 mmol) and acetone (4.00 mL) in ethanol (8.00 mL) was added sodium
cyanoborohydride (94.0
mg, 1.50 mmol) at ambient temperature. The resulting mixture was stirred for 3
hours at 50 C under
nitrogen atmosphere. The resulting mixture was purified by reversed phase
flash chromatography with the
following conditions: Column: Spherical C18, 20-40 pm, 120 g; Mobile Phase A:
water (plus 10 mM
NI-141-1CO3); Mobile Phase B: acetonitrile; Flow rate: 50 mUmin; Gradient (B):
5%-20%, 6 min; 20%-50%,
30 min; 50%-95%, 5 min; 95%, 5min; Detector: UV 254 nm. The desired fractions
were collected and
concentrated under reduced pressure to afford the title compound as a
colorless solid (85.0 mg, 77%): 1H
NMR: 1H NMR (400 MHz, DMSO-d6) 6 9.84 (d, J = 8.0 Hz, 1H), 8.93 (s, 1H), 8.00
(d, J = 10.2 Hz, 1H), 3.69
(d, J= 7.6 Hz, 2H), 3.46 (d, J= 7.3 Hz, 2H), 3.30 (s, 3H), 2.63-2.56 (m, 1H),
1.01 (d, J= 6.1 Hz, 6H); MS:
[(M + 1)]+ = 378.10, 380.10.
NH
NQ 0==0 ON
)N
Br
1,4-dioxane, H20, 80 C, 2 h NH
0==0
Pd(PPh3)4, Na2CO3
N-[5[7-Fluoro-3-methyl-2-oxo-1-(propan-2-y1)-2,3-dihydrospiro[azetidine-3,1-
pyrrolo[2,3-c]quinolin]-8-y1F
242-[(propan-2-yDamino]ethoxy]pyridin-3-yl]methanesulfonamide:
To a solution of 8-bromo-7-
fluoro-3-methyl-1-(propan-2-yI)-2,3-di hydrospiro[azetidine-3,1-pyrrolo[2,3-
c]quinolin]-2-
one (80.0 mg, 0.21 mmol) and N-(242-[(propan-2-yDamino]ethoxy]-5-(4,4,5,5-
tetramethyl-1,3,2-
dioxaborolan-2-yl)pyridin-3-yl)methanesulfonamide (169 mg, 0.42 mmol)
in water (2.00 mL) and 1,4-
dioxane (10.00 mL) were added sodium carbonate (33.6 mg, 0.32 mmol) and
tetrakis (triphenylphosphine)
palladium (0) (24.3 mg, 0.021 mmol). After stirring for 2 hours at 80 C under
nitrogen atmosphere, the
resulting mixture was concentrated under reduced pressure. The residue was
purified by Prep-TLC
(DCM/Me0H = 10/1, v/v) to give a crude product which was further purified by
reversed phase flash
chromatography with the following conditions: Column: Spherical C18, 20-40 pm,
120 g; Mobile Phase A:
Water (plus 10 mM NI-141-1CO3 ), Mobile Phase B: acetonitrile; Flow rate: 45
mL/min; Gradient (B%): 5%, 2
min; 5%-24%, 5 min; 24%-34%, 9 min; 34%, 8 min; 34%-95%; 3 min; 95%, 2 min;
Detector: UV 254 nm;
Rt: 21 min. The desired fractions were collected and concentrated under
reduced pressure to afford the
title compound as a colorless solid (65.0 mg, 54%): 1H NMR (400 MHz, DMSO-d6)
6 9.66 (d, J = 8.9 Hz,
1H), 8.89 (s, 1H), 8.25 (t, J= 1.9 Hz, 1H), 7.99-7.91 (m, 2H), 4.42 (t, J= 5.4
Hz, 2H), 3.76 (d, J= 7.2 Hz,
2H), 3.47 (d, J= 7.3 Hz, 2H), 3.31 (s, 3H), 3.02 (s, 3H), 2.96 (t, J= 5.4 Hz,
2H), 2.92-2.84 (m, 1H), 2.62-
2.53 (m, 1H), 1.05 (d, J= 6.3 Hz, 6H), 0.97 (d, J= 6.1 Hz, 6H); MS: [(M + 1)]+
= 571.25.
COMPOUND 572
CI
Br NO2 oI 0
--0
0¨
._ Br NO2R
0 LDA, TI-IF, -78 C-0 C, 1 h
37
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
Methyl 1-(6-bromo-7-fluoro-3-nitroquinolin-4-yI)-3-methoxycyclobutane-1-
carboxylate: To a solution of
freshly prepared lithium diisopropylamide (10.6 mmol) in anhydrous
tetrahydrofuran (100 mL) was added
methyl 3-methoxycyclobutane-1-carboxylate (1.53 g, 10.6 mmol) at -78 C. After
stirring for additional 1
hour, a solution of 6-bromo-4-chloro-7-fluoro-3-nitroquinoline (2.50 g, 8.18
mmol) in tetrahydrofuran (5.00
mL) was added over 5 min. The resulting mixture was slowly warmed to 0 C and
then quenched by
saturated aqueous ammonium chloride (100 mL). The resulting mixture was
diluted with water (1.00 L) and
the organic layer was separated. The aqueous layer was extracted with ethyl
acetate (3 x 200 mL). The
combined organic layers was washed with brine (2 x 200 mL) and dried over
anhydrous sodium sulfate.
After filtration, the filtrate was concentrated under reduced pressure. The
residue was purified by silica gel
column chromatography, eluted with 5%-50% ethyl acetate in petroleum ether to
afford the title compound
as a brown syrup (720 mg, 21%): 1H NMR (400 MHz, DMSO-d6) 6 9.25 (s, 1H), 8.31
(d, J= 7.3 Hz, 1H),
8.17 (d, J= 9.3 Hz, 1H), 4.16-4.13 (m, 1H), 3.71 (s, 3 H), 3.11 (s, 3H), 2.47-
2.38 (m, 2H), 2.00-1.89 (m,
2H); MS: [(M + 1)]+ = 413.20, 415.20.
0
o 0 0
0- Br 2 Fe NO Br NH
HOAc, rt, 3 h
8'-Bromo-7-fluoro-3-methoxyspiro[cyclobutane-1,1'-pyrrolo[2,3-c]quinolin]-
2'(37-1)-one: To a
solution of methyl 1-(6-bromo-7-fluoro-3-nitroquinolin-4-yI)-3-
methoxycyclobutane-1-carboxylate (720
mg, 1.74 mmol) in acetic acid (10.0 mL) was added iron powder (681 mg, 12.2
mmol). The resulting
mixture was stirred for 3 hours at ambient temperature. The resulting mixture
was diluted with water
(150 mL) and extracted with ethyl acetate (4 x 50.0 mL). The combined organic
layers was washed
with brine (2 x 50.0 mL) and dried over anhydrous sodium sulfate. After
filtration, the filtrate was
concentrated under reduced pressure. The residue was purified by silica gel
column chromatography,
eluted with 5%-30% ethyl acetate in petroleum ether to afford the title
compound as a light yellow solid
(550 mg, 89%): 1H NMR (400 MHz, CDCI3) 6 8.93 (s, 0.45 H), 8.88 (d, J= 7.2 Hz,
0.55H), 8.84 (s, 1H),
8.78 (s, 0.55H), 8.27 (d, J= 7.0 Hz, 0.45H) 7.92 (d, J= 9.3 Hz, 1H), 4.71-4.63
(m, 0.45H), 4.57-4.49
(m, 0.55H), 3.48 (d, J= 6.6 Hz, 3H), 3.07-2.83 (m, 4H); MS: [(M + 1)]+ =
351.00, 353.00.
0 0
0 0
Br NH NaH, Mel
N--
Br
DMF, 0 C-rt, 1 h
8'-Bromo-7-fluoro-3-methoxy-3-methylspiro[cyclobutane-1,1'-pyrrolo[2,3-
c]quinolin]-2'(37-1)-one: A
solution of 8-bromo-7-fluoro-3-methoxy-2,3-dihydrospiro[cyclobutane-1,1-
pyrrolo[2,3-c]quinolin]-2-one
(550 mg, 1.56 mmol) in N, N-dimethylformamide (10.0 mL) was treated with
sodium hydride (81.4 mg, 2.04
mmol, 60% dispersed in mineral oil) for 0.5 hours at 0 C followed by the
addition of iodomethane (265 mg,
1.87 mmol) dropwise over 2 min at 0 C. After stirring for additional 1 hour
at ambient temperature, the
38
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
reaction was quenched by saturated aqueous ammonium chloride (20.0 mL) and
diluted with water (150
mL). The resulting mixture was extracted with ethyl acetate (3 x 50.0 mL). The
combined organic layers
was washed with brine (2 x 30.0 mL) and dried over anhydrous sodium sulfate.
After filtration, the filtrate
was concentrated under reduced pressure. The residue was purified by silica
gel column chromatography,
eluted with 5%-10% ethyl acetate in petroleum ether to afford the title
compound as a yellow solid (500
mg, 87%): MS: [(M + 1)]+ = 365.10, 367.10.
-0 HO
0 0
BBr3 in DCM
Br N--. Br N-_
DCM, -78 C-rt, 2 h
8'-Bromo-7'-fluoro-3-hydroxy-3'-methylspiro[cyclobutane-1,1'-pyrrolo[2,3-
c]quinolin]-2'(37-1)-one: .. To .. a
stirred solution of 8'-bromo-7'-fluoro-3-methoxy-3'-methylspiro[cyclobutane-
1,1'-pyrrolo[2,3-c]quinolin]-
2'(37-1)-one (900 mg, 2.46 mmol) in dichloromethane (20.0 mL) was added boron
tribromide (24.6 mL, 24.6
mmol, 1M in dichloromethane) dropwise at -78 C under nitrogen atmosphere. The
resulting mixture was
warmed to ambient temperature slowly. The resulting mixture was stirred for 2
hours at ambient
temperature under nitrogen atmosphere. The mixture was neutralized with
saturated aqueous sodium
bicarbonate solution. The aqueous layer was extracted with ethyl acetate (3 x
100 mL). The combined
organic layers was washed with brine (3 x 100 mL) and dried over anhydrous
sodium sulfate. After filtration,
the filtrate was concentrated under reduced pressure. The residue was purified
by silica gel column
chromatography, eluted with 1%-5% methanol in dichloromethane. The desired
fractions were collected
and concentrated under reduced pressure to afford the title compound as a
colorless solid (530 mg, 62%):
1H NMR (400 MHz, DMSO-d6) 6 8.96-8.89 (m, 1.6H), 8.32 (d, J= 7.4 Hz, 0.4H),
8.05-7.98 (m, 1H), 5.99 (d,
J = 6.5 Hz, 0.6H), 5.73 (d, J = 5.7 Hz, 0.4H), 4.97-4.87 (m, 0.4H), 4.75-4.65
(m, 0.6H), 3.31 (s, 1.2 H), 3.29
(s, 1.8H), 2.98-2.87 (m, 0.8H), 2.81-2.67 (m, 2.4H), 2.65-2.55 (m, 0.8H); MS:
[(M + 1)]+ = 351.00, 353.00.
HO Bn0
0 0
NaH
Br N,
Br
DMF, 0 C-rt, 1 h
3-(Benzyloxy)-8'-bromo-7'-fluoro-3'-methylspiro[cyclobutane-1,1'-pyrrolo[2,3-
c]quinolin]-2'(37-1)-one:
To a solution of 8'-bromo-7'-fluoro-3-hydroxy-3'-methylspiro[cyclobutane-1,1'-
pyrrolo[2,3-
c]quinolin]-2'(37-1)-one (100 mg, 0.29 mmol) in N,N-dimethylformamide (5.00
mL) was added sodium
hydride (13.7 mg, 0.35 mmol, 60% dispersed in mineral oil) at 0 C under
nitrogen atmosphere. The
resulting mixture was stirred for 1 hour at 25 C followed by the addition of
benzyl
bromide (97.4 mg, 0.57 mmol) at 0 C. After stirring for additional 1 hour at
25 C, the reaction was
quenched by saturated aqueous ammonium chloride solution (10.0 mL). The
resulting mixture was diluted
with water (100 mL) and extracted with ethyl acetate (3 x 50.0 mL). The
combined organic layers were
washed with brine (2 x 50.0 mL) and dried over anhydrous sodium sulfate. After
filtration, the filtrate was
concentrated under reduced pressure. The residue was purified by Prep-TLC
(DCM/Me0H = 25/1, v/v) to
afford the title compound as a yellow solid (64.0 mg, 51%): 1H NMR (400 MHz,
DMSO-d6) 6 8.92 (d, J =
3.6 Hz, 1H), 8.79 (d, J = 7.6 Hz, 0.5H), 8.25 (d, J = 7.2 Hz, 0.5H), 8.03 (d,
J = 10.1 Hz, 1H), 7.50 (d, J = 7.0
39
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
Hz, 1H), 7.46-7.37 (m, 3H), 7.37-7.30 (m, 1H), 4.87-4.79 (m, 0.5H), 4.62-4.50
(m, 2.5H), 3.30 (d, J= 5.7
Hz, 3H), 2.98-2.90 (m, 1H), 2.80 (d, J = 6.7 Hz, 2H), 2.72-2.64 (m, 1H); MS:
[(M + 1)]+ = 441.00, 443.00.
)N
Bn0 HNIE3-(< Bn0
0
0
0=s=0
Br
1,4-dioxane, H20, 80 C, 2h HN
FN S
Pd(PPh3)4, Na2CO3 0==0
N-(5-(3-(Benzyloxy)-7'-fluoro-3'-methyl-2'-oxo-2',3'-dihydrospiro[cyclobutane-
1,1'-pyrrolo[2,3-c]quinolin]-8'-
yI)-2-(2-(isopropylamino)ethoxy)pyridin-3-yl)methanesulfonamide: To a
solution of 3-(benzyloxy)-8'-
bromo-7'-fluoro-3'-methylspiro[cyclobutane-1,1'-pyrrolo[2,3-c]quinolin]-2'(37-
1)-one (64.0 mg, 0.15 mmol)
and
N-[242-(isopropylamino)ethoxy]-5-(4,4,5,5-tetramethy1-1,3,2-dioxaborolan-2-
yl)pyridin-3-
yl]methanesulfonamide (69.5 mg, 0.17 mmol) in water (1.00 mL) and 1,4-dioxane
(4.00 mL) were added
sodium carbonate (15.4 mg, 0.15 mmol) and tetrakis (triphenylphosphine)
palladium (0) (33.5 mg, 0.029
mmol). After stirring for 2 hours at 80 C under nitrogen atmosphere, the
resulting mixture was concentrated
under reduced pressure. The residue was purified by Prep-TLC (DCM/Me0H = 10/1,
v/v) to afford the title
compound as a light yellow solid (55.0 mg, 60%): MS: [(M + 1)]+ = 634.55.
Bn0,,
BnO,
Bn0
0
0 .40
N I I I Prep-Chiral-
HPLC +
HN HN
N--
HN
0=S=0
0=S=0
0=5=0
F 572
567
cis-N-(5-(3-(Benzyloxy)-7'-fluoro-3'-methyl-2'-oxo-2',3'-
dihydrospiro[cyclobutane-1,1'-pyrrolo[2,3-
c]quinolin]-8'-y1)-2-(2-(isopropylamino)ethoxy)pyridin-3-yl)methanesulfonamide
(EXAMPLE 572) and trans-
N-(5-(3-(Benzyloxy)-7'-fluoro-3'-methy1-2'-oxo-2',3'-dihydrospiro[cyclobutane-
1,1'-pyrrolo[2,3-c]quinolin]-8'-
y1)-2-(2-(isopropylamino)ethoxy)pyridin-3-yDmethanesulfonamide (EXAMPLE 567).
The above N-(5-(3-
(benzyloxy)-7'-fluoro-3'-methyl-2'-oxo-2',3'-dihydrospiro[cyclobutane-1,1'-
pyrrolo[2,3-c]quinolin]-8'-y1)-2-(2-
(isopropylamino)ethoxy)pyridin-3-yl)methanesulfonamide (55.0 mg, 0.087 mmol)
was seperated by Prep-
Chiral-HPLC with the following conditions: (Column: CHIRALPAK ID, 2 x 25 cm (5
pm); Mobile Phase A:
methyl tert-butyl ether (plus 0.2% isopropylamine), Mobile Phase B: Et0H; Flow
rate: 17 mL/min; Gradient:
10% B in 13 min; Detector: UV 220/254 nm) to afford cis-N-(5-(3-(benzyloxy)-7'-
fluoro-3'-methyl-2'-oxo-
2',3'-dihydrospiro[cyclobutane-1,1'-pyrrolo[2,3-c]quinolin]-8'-y1)-2-(2-
(isopropylamino)ethoxy)pyridin-3-
yl)methanesulfonamide (EXAMPLE 572, RT1: 8.70 min) as a light yellow solid
(15.3 mg, 28%): 1H NMR
(400 MHz, DMSO-d6) 6 8.90 (s, 1H), 8.29 (s, 1H), 8.09 (d, J = 8.3 Hz, 1H),
8.02 (d, J = 1.9 Hz, 1H), 7.98
(d, J= 12.1 Hz, 1H), 7.42-7.34 (m, 4H), 7.32-7.27 (m, 1H), 4.90-4.82 (m, 1H),
4.53 (s, 2H), 4.47 (t, J= 5.4
Hz, 2H), 3.32 (s, 3H), 3.06 (s, 3H), 3.05-2.88 (m, 5H), 2.68 (dd, J= 13.9, 6.2
Hz, 2H), 1.08 (d, J= 6.2 Hz,
6H); MS: [(M + 1)]+ = 634.50 and trans-N-(5-(3-(benzyloxy)-7'-fluoro-3'-methyl-
2'-oxo-2',3'-
dihydrospiro[cyclobutane-1,1'-pyrrolo[2,3-c]quinolin]-8'-y1)-2-(2-
(isopropylamino)ethoxy)pyridin-3-
yl)methanesulfonamide (EXAMPLE 567, RT2:11.2 min) as a colorless solid (12.6
mg, 23%): 1H NMR (400
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
MHz, DMSO-d6) 6 8.88 (s, 1H), 8.59 (d, J= 8.3 Hz, 1H), 8.33 (s, 1H), 8.02-7.94
(m, 2H), 7.36-7.21 (m, 5H),
4.58 (q, J= 6.3 Hz, 1H), 4.44 (t, J= 5.4 Hz, 2H), 4.41-4.33 (m, 1H), 3.28 (s,
3H), 3.05 (s, 3H), 2.00-3.94
(m, 3H), 2.92-2.84 (m, 1H), 2.83-2.74 (m, 1H), 2.69 (dd, J= 13.1, 5.8 Hz, 2H),
2.59-2.54 (m, 1H), 1.35 (d,
J = 6.4 Hz, 3H), 1.05 (d, J = 6.3 Hz, 6H); MS: [(M + 1)]+ = 634.55.
COMPOUND 573
0 0.____7
EDCI, DMAP
0
0
HO DCM, rt, 16 h Me(H2C)90
Decy13-oxocyclobutane-1-carboxylate: To a stirred solution of 3-
methylcyclobutane-1-carboxylic acid
(3.00 g, 26.3 mmol) and 1-decanol (4.16 g, 26.3 mmol) in dichloromethane (90.0
mL) were added 1-ethyl-
3-(3-dimethylaminopropyl)carbodiimide hydrochloride (7.56 g, 39.4 mmol) and 4-
dimethylaminopyridine
(0.32 g, 2.62 mmol) at ambient temperature. The resulting mixture was stirred
for 16 hours at 25 C and
quenched by water (30.0 mL). The resulting mixture was extracted with
dichloromethane (3 x 100 mL). The
combined organic layers was washed with brine (2 x 50.0 mL) and dried over
anhydrous sodium sulfate.
After filtration, the filtrate was concentrated under reduced pressure. The
residue was purified by silica gel
column chromatography, eluted with 1%-8% ethyl acetate in petroleum ether. The
desired fractions were
collected and concentrated under reduced pressure to afford the title compound
as a light yellow oil (3.10
g, 47%): 1H NMR (400 MHz, CDC13) 6 4.15 (t, J= 6.7 Hz, 2H), 3.47-3.35 (m, 2H),
3.35-3.15 (m, 3H), 1.69-
1.61 (m, 2H), 1.39-1.22 (m, 14H), 0.88 (t, J= 6.7 Hz, 3H); MS: [(M + 1)]+ =
252.20.
0....._7
o + _
Ph3PEtBr
1) DMSO, NaH, RT, 0.5 h
Me(H2C)90 2)rt,4h ______ Ac
0
me(H20)90
Decy13-ethylidenecyclobutane-1-carboxylate:
To a solution of ethyltriphenylphosphonium bromide
(17.3 g, 46.5 mmol) in dimethyl sulfoxide (300 mL) was added potassium t-
butoxide (4.94 g, 44.1 mmol) in
portions at 14 C. The resulting mixture was stirred for 0.5 hours at 25 C
under nitrogen atmosphere
followed by the addition of decyl 3-oxocyclobutane-1-carboxylate (8.00 g, 31.5
mmol) dropwise over 2 min
at 14 C. After stirring for 4 hours at 25 C, the reaction was quenched by
saturated aqueous ammonium
chloride solution (20.0 mL) at 0 C. The resulting mixture was diluted with
water (1.00 L) and extracted with
ethyl acetate (3 x 200 mL). The combined organic layers was washed with brine
(2 x 100 mL) and dried
over anhydrous sodium sulfate. After filtration, the filtrate was concentrated
under reduced pressure. The
residue was purified by silica gel column chromatography, eluted with 1%-5%
ethyl acetate in petroleum
ether. The desired fractions were collected and concentrated under reduced
pressure to afford the title
compound as a colorless oil (1.70 g, 21%): 1H NMR (400 MHz, CDC13) 6 5.22-5.14
(m, 1H), 4.09 (t, J= 6.7
Hz, 2H), 3.10 (tt, J= 9.2, 7.2 Hz, 1H), 2.96-2.79 (m, 4H), 1.67-1.58 (m, 2H),
1.49 (dq, J= 6.0, 2.0 Hz, 3H),
1.39-1.20 (m, 14H), 0.88 (t, J= 6.7 Hz, 3H).
41
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
Pd/C, H2
0 Me0H, rt, 16 h
Me(H2C)90 Me(H2C)90
Decyl 3-ethylcyclobutane-1-carboxylate: To a stirred solution of decyl 3-
ethylidenecyclobutane-1-
carboxylate (700 mg, 4.94 mmol) in methanol (10.0 mL) was added anhydrous Pd/C
(70.0 mg, 10%
palladium on charcoal) at ambient temperature. After stirring for 16 hours at
ambient temperature under
hydrogen atmosphere (2 atm.), the resulting mixture was filtered. The filter
cake was washed with methanol
(3 x 20.0 mL). The filtrate was concentrated under reduced pressure to afford
the title compound as a light
yellow oil (666 mg, 95%): 1H NMR (400 MHz, CDC13) 6 4.10-4.02 (m, 2H), 3.10-
2.87(m, 1H), 2.40-2.22 (m,
2H), 2.16-2.03 (m, 1H), 1.91-1.76 (m, 2H), 1.67-1.56 (m, 2H), 1.48-1.21 (m,
16H), 0.92-0.75 (m, 6H).
ci
0
Br NO2
0(CH2)9Me
Br NO
2
-==c0 ________________________________________
Me(H2C)90 LDA, THF, -78 C-0 C, 2 h
F
3 4
Decyl 1-(6-bromo-7-fluoro-3-nitroquinolin-4-y1)-3-ethylcyclobutane-1-
carboxylate: To a solution of freshly
prepared lithium diisopropylamide (2.45 mmol) in anhydrous tetrahydrofuran
(20.0 mL) was added decyl 3-
ethylcyclobutane-1-carboxylate (657 mg, 2.45 mmol) at -78 C under nitrogen
atmosphere. The resulting
mixture was stirred for 1 hour followed by the addition of 6-bromo-4-chloro-7-
fluoro-3-nitroquinoline (575
mg, 1.88 mmol) at -78 C. After stirring for additional 1 hour at 25 C, the
reaction was quenched by
saturated aqueous ammonium chloride solution (10.0 mL). The resulting mixture
was diluted with water
(20.0 mL) and separated. The aqueous layer was extracted with ethyl acetate (5
x 30 mL). The combined
organic layers was washed with brine (4 x 30.0 mL) and dried over anhydrous
sodium sulfate. After filtration,
the filtrate was concentrated under reduced pressure. The residue was purified
by silica gel column
chromatography, eluted with 3%-9% ethyl acetate in petroleum ether. The
desired fractions were collected
and concentrated under reduced pressure to afford the title compound as a
yellow oil (450 mg, crude): MS:
[(M + 1)]+ = 537.25, 539.25.
0
0
0(CH2)9Me
Br NO2 Fe Br NH
HOAc, rt, 16 h
4 5
8'-Bromo-3-ethyl-7'-fluorospiro[cyclobutane-1,1'-pyrrolo[2,3-c]quinolin]-2'(37-
1)-one: A mixture of
crude decyl 1-(6-bromo-7-fluoro-3-nitroquinolin-4-y1)-3-ethylcyclobutane-1-
carboxylate (450 mg, 0.84 mmol)
and iron power (327 mg, 5.86 mmol) in acetic acid (8.00 mL) was stirred for 16
h at ambient temperature .
The resulting mixture was filtered, the filtered cake was washed with
tetrahydrofuran (5 x 300 mL). The
filtrate was concentrated under reduced pressure. The residue was basified to
pH = 8 with saturated
aqueous sodium bicarbonate solution. The resulting mixture was extracted with
ethyl acetate (5 x 100 mL).
The combined organic layers was washed with brine (50.0 mL) and dried over
anhydrous sodium sulfate.
After filtration, the filtrate was concentrated under reduced pressure. The
residue was purified by Prep-TLC
42
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
(DCM/Me0H = 15/1, v/v) to afford the title compound as a yellow solid (40.0
mg, 14%): 1H NMR (400 MHz,
DMSO-d6) 6 10.76 (d, J= 5.3 Hz, 1H), 8.67 (d, J= 1.7 Hz, 1H), 8.45 (d, J = 7.2
Hz, 0.4H), 8.39 (d, J = 7.4
Hz, 0.6H), 7.98 (dd, J= 10.1, 3.4 Hz, 1H), 2.88-2.73 (m, 2H), 2.55-2.51 (m,
2H), 2.29-2.19 (m, 1H), 1.78-
1.64 (m, 2H), 0.99-0.88 (m, 3H); MS: [(M + 1)]+ = 349.05, 351.05.
0
Br NH 1) NaH, DMF, 0 C, 30 min
______________________________________________________ Br N¨_
2) Mel, 0 C-rt, 40 min
5 6
8'-Bromo-3-ethyl-7'-fluoro-3'-methylspiro[cyclobutane-1,1'-pyrrolo[2,3-
c]quinolin]-2'(37-1)-one : To a
solution of 8'-bromo-3-ethyl-7'-fluorospiro[cyclobutane-1,1'-pyrrolo[2,3-
c]quinolin]-2'(37-1)-one (35.0 mg,
0.10 mmol) in N,N-dimethylformamide (3.00 mL) was added sodium hydride (5.21
mg, 0.13 mmol, 60%
dispersed in mineral oil) at 0 C under nitrogen atmosphere. The resulting
mixture was stirred for 30 minutes
at 25 C followed by the addition of iodomethane (21.4 mg, 0.15 mmol) at 0 C.
After stirring for additional
40 minutes at 25 C, the reaction was quenched by saturated aqueous ammonium
chloride solution (20.0
mL). The resulting mixture was diluted by water (100 mL) and extracted with
ethyl acetate (4 x 100 mL).
The combined organic layers was washed with brine (4 x 100 mL) and dried over
anhydrous sodium sulfate.
After filtration, the filtrate was concentrated under reduced pressure. The
residue was purified by Prep-TLC
(DCM/Me0H = 20/1, v/v) to afford the title compound as a yellow solid (30.0
mg, 83%): 1H NMR (400 MHz,
DMSO-d6) 6 8.90 (d, J = 2.9 Hz, 1H), 8.49 (d, J = 7.4 Hz, 0.4H), 8.42 (d, J =
7.4 Hz, 0.6H), 8.02 (dd, J =
10.1, 3.1 Hz, 1H), 3.29 (d, J= 4.5 Hz, 3H), 2.93-2.75 (m, 2H), 2.61-2.52 (m,
2H), 2.29-2.21 (m, 1H), 1.82-
1.67 (m, 2H), 0.98-0.89 (m, 3H); MS: [(M + 1)]+ = 363.05, 365.05.
)NC)
H O. ,0
0
0 N
Br
1,4-dioxane, H20, 80 C, 2 h 0=5=0
I F
Pd(PPh3)4, Na2CO3
6 7
.. N-(5-(3-Ethyl-7'-fluoro-3'-methyl-2'-oxo-2',3'-dihydrospiro[cyclobutane-
1,1'-pyrrolo[2,3-c]quinolin]-8'-yI)-2-
(2-(isopropylamino)ethoxy)pyridin-3-yl)methanesulfonamide:
To a solution of 8'-bromo-3-ethyl-7'-
fluoro-3'-methylspiro[cyclobutane-1,1'-pyrrolo[2,3-c]quinolin]-2'(37-1)-one
(50.0 mg, 0.14 mmol) and N-[2-[2-
(isopropylamino)ethoxy]-5-(4,4,5,5-tetramethy1-1,3,2-dioxaborolan-2-yl)pyridin-
3-yl]methanesulfonamide
(165 mg, 0.41 mmol) in water (0.75 mL) and 1,4-dioxane (3.00 mL) were added
sodium carbonate (17.5
mg, 0.17 mmol) and tetrakis (triphenylphosphine) palladium (0) (31.8 mg, 0.028
mmol). After stirring for 2
hours at 80 C under nitrogen atmosphere, the resulting mixture was
concentrated under reduced pressure.
The residue was purified by Prep-TLC (DCM/Me0H = 10/1, v/v) to give a crude
product which was further
purified by reversed phase flash chromatography with the following conditions:
Column: Spherical C18,
20-40 pm, 120 g; Mobile Phase A: Water (plus 10 mM NI-141-1CO3), Mobile Phase
B: acetonitrile; Flow rate:
45 mL/min; Gradient (B%): 5%-25%, 7 min; 25%-45%, 25 min; 45%-65%, 8 min; 95%,
5 min; Detector:
UV 254 nm. The fractions containing desired product was collected at 17 min
and concentrated under
reduced pressure to afford the title compound as a yellow solid (30.0 mg,
40%): MS: [(M + 1)]+ = 556.20.
43
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
¨õ.
N 0
N
0
N 0 N I I N
-- Prep-Chiral-HPLC
0NH NH
N
FIN
= 0=
0S==0 F N H I
F
I F
573
566
cis-N-(5-(3-Ethyl-7'-fluoro-3'-methyl-2'-oxo-2',3'-dihydrospiro[cyclobutane-
1,1'-pyrrolo[2,3-c]quinolin]-8'-y1)-
2-(2-(isopropylamino)ethoxy)pyridin-3-yOmethanesulfonamide (EXAMPLE 573) and
trans-N-(5-(3-Ethyl-
7'-fluoro-3'-methyl-2'-oxo-2',3'-di hydrospiro[cyclobutane-1,1'-pyrrolo[2,3-
c]quinolin]-8'-yI)-2-(2-
(isopropylamino)ethoxy)pyridin-3-yl)methanesulfonamide (EXAMPLE 566): The
above N-(5-(3-
ethyl-7'-fluoro-3'-methyl-2'-oxo-2',3'-dihydrospiro[cyclobutane-1,1'-
pyrrolo[2,3-c]quinolin]-8'-y1)-2-(2-
(isopropylamino)ethoxy)pyridin-3-yOmethanesulfonamide (30.0 mg) was separated
by Prep-Chiral-HPLC
with the following conditions: Column: Chiralpak IC, 2 x 25 cm, 5 pm; Mobile
Phase A:Hexane/DCM=3/1
(plus 0.2% isopropylamine), Mobile Phase B:Et0H; Flow rate: 20 mL/min;
Gradient: 30% B in 15 min;
Detector: UV 220/254 nm. The desired fractions were collected and concentrated
under reduced pressure
to afford cis-N-(5-(3-ethyl-7'41 uoro-3'-methyl-2'-oxo-2',3'-d ihyd rospi
ro[cyclobutane-1 ,1'-pyrrolo[2,3-
c]quinolin]-8'-yI)-2-(2-(isopropylamino)ethoxy)pyridin-3-yl)methanesulfonamide
(EXAMPLE 573, RT1:
10.94 min) as a colorless solid (12.8 mg, 43%): 1H NMR (400 MHz, DMSO-d6) 6
8.87 (s, 1H), 8.32-8.22
(m, 2H), 7.98 (d, J= 12.2 Hz, 2H), 4.44 (t, J= 5.3 Hz, 2H), 3.30 (s, 3H), 3.04
(s, 3H), 2.99 (t, J= 5.3 Hz,
2H), 2.94-2.78 (m, 4H), 2.26 (dd, J= 12.2, 5.7 Hz, 2H), 1.75 (t, J= 7.2 Hz,
2H), 1.07 (d, J= 6.3 Hz, 6H),
0.89 (t, J = 7.3 Hz, 3H); MS: [(M + 1)]+ = 556.25
and
trans-N-(5-(3-ethyl-7'-fl uoro-3'-methyl-2'-oxo-2',3'-di hyd rospi ro
[cyclobutane-1 ,1'-pyrrolo[2,3-
c]quinolin]-8'-yI)-2-(2-(isopropylamino)ethoxy)pyridin-3-yl)methanesulfonamide
(EXAMPLE 566,
RT2:13.63 min) as a colorless solid (8.6 mg, 29%): 1H NMR (400 MHz, DMSO-d6) 6
8.88 (s, 1H), 8.34-8.26
(m, 2H), 8.01-7.93 (m, 2H), 4.44 (t, J = 5.4 Hz, 2H), 3.31 (s, 3H), 3.05 (s,
3H), 3.00 (d, J = 5.4 Hz, 2H),
2.96-2.83 (m, 2H), 2.70- 2.60 (m, 2H), 1.66 (q, J = 7.2, 6.7 Hz, 2H), 2.56-
2.52 (m, 2H), 1.07 (d, J = 6.2 Hz,
6H), 0.89 (t, J = 7.3 Hz, 3H); MS: [(M + 1)]+ = 556.20.
COMPOUND 574
CI N
NI
02N Br
\-NH HCI ______________________________________________ X;
DIPEA, THF, rt, 3 h
02N Br
1-(5-Bromo-3-nitropyridin-2-yI)-N,N-dimethylazetidin-3-amine: To a stirred
solution of N,N-
dimethylazetidin-3-amine hydrochloride (0.27 g, 2.08 mmol) and 5-bromo-2-
chloro-3-nitropyridine (0.49 g,
2.08 mmol) in tetrahydrofuran (40.0 mL) was added diisopropylethylamine (0.67
g, 5.19 mmol) at ambient
temperature. The resulting mixture was stirred for 3 hours and concentrated
under reduced pressure. The
residue was purified by silica gel column chromatography, eluted with 3%-9%
ethyl acetate in petroleum
ether. The desired fractions were collected and concentrated under reduced
pressure to afford the title
compound as a yellow solid (0.50 g, 59%): 1H NMR (400 MHz, CDCI3) 6 8.37 (d, J
= 2.2 Hz, 1H), 8.29 (d,
J = 2.2 Hz, 1H), 4.20 (ddd, J = 10.0, 7.0, 1.2 Hz, 2H), 3.93 (ddd, J = 9.9,
5.1, 1.2 Hz, 2H), 3.16 (tt, J = 7.0,
5.1 Hz, 1H), 2.20 (s, 6H); MS: [(M + 1)]+ = 301.00, 303.00.
44
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
1 1
Fe N
:rL; AcOH, rt, 2 h
02N Br H2N Br
1-(5-Bromo-3-nitropyridin-2-yI)-N,N-dimethylazetidin-3-amine. To a solution of
1-(5-bromo-3-
nitropyridin-2-y1)-N,N-dimethylpiperidin-4-amine (6.20 g, 20.7 mmol) in acetic
acid (90.0 mL) was added
iron powder (11.5 g, 206 mmol) at ambient temperature. The resulting mixture
was stirred for 2 hours at
ambient temperature. The resulting mixture was filtered and the filtered cake
was washed with
tetrahydrofurane (3 x 100 mL). The filtrate was concentrated under reduced
pressure. The residue was
taken up with saturated aqueous sodium carbonate (100 mL) and extracted with
ethyl acetate (3 x 100 mL).
The combined organic layers was washed with brine (3 x 100 mL) and dried over
anhydrous sodium sulfate.
After filtration, the filtrate was concentrated under reduced pressure. The
residue was purified by silica gel
column chromatography, eluted with 2%-4% methanol in dichloromethane to afford
the title compound as
a grey solid (5.20 g, 92%): 1H NMR (400 MHz, CDCI3) 6 7.74 (d, J = 2.0 Hz,
1H), 6.93 (d, J = 2.0 Hz, 1H),
4.19-4.04 (m, 2H), 3.93-3.88 (m, 2H), 3.21-3.14 (m, 1H), 2.24 (s, 6H); MS: [(M
+ 1)]+ = 271.00, 273.00.
1
1
N N
MsCI, DMAP
I
I Py, rt, 2 h HNBr
H2NBr 0==0
N-(5-Bromo-2-(3-(dimethylamino)azetidin-1-yl)pyridin-3-yl)methanesulfonamide:
To a stirred solution of 5-
bromo-2[3-(dimethylamino)azetidin-1-yl]pyridin-3-amine (2.10 g, 7.77 mmol) and
N,N-4-
dimethylaminopyridine (77.0 mg, 0.63 mmol) in pyridine (70.0 mL) was added
methanesulfonyl chloride
(1.77 g, 15.5 mmol) dropwise at ambient temperature. After stirring for 3
hours at ambient temperature
under nitrogen atmosphere, the resulting mixture was concentrated under
reduced pressure. The residue
was purified by reversed phase flash chromatography with the following
conditions: Column: Spherical C18,
20-40 pm, 330 g; Mobile Phase A: Water (plus 10 mM NI-141-1CO3), Mobile Phase
B: acetonitrile; Flow rate:
65 mL/min; Gradient (B%): 5%-20%, 8 min; 20%-40%, 20 min; 40%-95%, 2 min; 95%,
5 min; Detector:
UV 254 nm. The desired fractions were collected at 19 min and concentrated
under reduced pressure to
afford the title compound as a colorless solid (1.40 g, 52%): 1H NMR (400 MHz,
CD30D) 6 7.92 (d, J = 2.2
Hz, 1H), 7.61 (d, J = 2.2 Hz, 1H), 4.27 (dd, J = 8.8, 7.2 Hz, 2H), 3.96 (dd, J
= 9.0, 5.6 Hz, 2H), 3.24-3.16
(m, 1H), 3.00 (s, 3H), 2.20 (s, 6H); MS: [(M + 1)]+ = 349.00, 351.00.
1
r0 0¨
I I
HNBr pd(dppf)C12.DCM, KOAc
0=S=0 0=S=0 0 __
1,4-dioxane, 90 C, 2 h 1
N-(2-(3-(Dimethylamino)azetidin-1-y1)-5-(4,4,5,5-tetramethyl-1,3,2-
dioxaborolan-2-yl)pyridin-3-
yl)methanesulfonamide: To a solution
of N45-bromo-243-(dimethylamino)azetidin-1-yl]pyridin-3-
yl]methanesulfonamide (1.00 g, 2.86 mmol) and 4 bis(pinacolato)diboron (2.18
g, 8.59 mmol) in 1,4-dioxane
(30.0 mL) were added potassium acetate (1.12 g, 11.5 mmol) and
bis(diphenylphosphino)ferrocene]dichloro palladium (II) dichloromethane adduct
(351 mg, 0.43 mmol) at
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
ambient temperature. The resulting mixture was stirred for 2 hours at 85 C
under nitrogen atmosphere.
The resulting mixture was concentrated under reduced pressure. The residue was
purified by reversed
flash chromatography with the following conditions: Column: Spherical C18, 20-
40 pm, 330 g; Mobile
Phase A: Water (plus 10 mM NI-141-1CO3), Mobile Phase B: acetonitrile; Flow
rate: 65 mL/min; Gradient (B%):
5%-20%, 7 min; 20%-40%, 12 min; 40%-95%; 2 min; 95%, 5 min; Detector: UV 254
nm. The desired
fractions were collected at 20 min and concentrated under reduced pressure to
afford the title compound
as a yellow solid (800 mg, 71%): 1H NMR (400 MHz, DMSO-d6) 6 8.78 (s, 1H),
8.18 (d, J = 1.5 Hz, 1H),
7.50 (d, J= 1.6 Hz, 1H), 4.19 (t, J= 8.0 Hz, 2H), 3.93 (dd, J= 8.9, 5.0 Hz,
2H), 3.12-3.04 (m, 1H), 2.99 (s,
3H), 2.09 (s, 6H), 1.27 (s, 12H); MS: [(M + 1)]+ = 397.20.
NI
Br N¨.
N N 0
N¨.
NH
0 0 1,4-dioxane, H20, 85 C, 2h
=S= 00=5=0
Pd(PPh3)4, Na2CO3
N-(2-(3-(Dimethylamino)azetidin-1-yI)-5-(3'-methyl-2'-oxo-2',3'-
dihydrospiro[cyclobutane-1,1'-pyrrolo[2,3-
c]quinolin]-8'-yl)pyridin-3-yl)methanesulfonamide:
To a solution of 8-bromo-3-methyl-2,3-
dihydrospiro[cyclobutane-1,1-pyrrolo[2,3-c]quinolin]-2-one (320 mg, 1.01 mmol)
and N-[2-[3-
(dimethylamino)azetidin-1-y1]-5-(4,4,5,5-tetramethy1-1,3,2-dioxaborolan-2-
yl)pyridin-3-
yl]methanesulfonamide (600 mg, 1.51 mmol) in 1,4-dioxane (13.0 mL) and water
(2.00 mL) were added
sodium carbonate (160 mg, 1.51 mmol) and tetrakis (triphenylphosphine)
palladium (0) (175 mg, 0.15
mmol). After stirring for 2 hours at 85 C under a nitrogen atmosphere, the
resulting mixture was
concentrated under reduced pressure. The residue was purified by Prep-TLC
(DCM/Me0H = 8/1, v/v) to
afford the title compound as a light yellow solid (350 mg, 69%): 1H NMR (400
MHz, DMSO-d6) 6 9.07 (s,
1H), 8.80 (s, 1H), 8.58 (s, 1H), 8.33 (s, 1H), 8.13 (d, J = 8.9 Hz, 1H), 7.95
(d, J = 9.2 Hz, 1H), 7.91 (d, J =
2.1 Hz, 1H), 4.28-4.21 (m, 2H), 4.02-3.95 (m, 2H), 3.30 (s, 3H), 3.14 (s, 4H),
2.98-2.90 (m, 2H), 2.60-2.52
(m, 4H), 2.13 (s, 6H); MS: [(M + 1)]+ = 507.20.
111 HCI
N 0
N 0
0.006 M, HCI (aq.) N--
NH NH
0=S=0 0=S=0
N-(2-(3-(Dimethylamino)azetidin-1-yI)-5-(3'-methyl-2'-oxo-2',3'-
dihydrospiro[cyclobutane-1,1'-pyrrolo[2,3-
c]quinolin]-8'-yl)pyridin-3-yl)methanesulfonamide hydrochloride: A solution of
N-(2-(3-
(dimethylamino)azetidin-1-y1)-5-(3'-methyl-2'-oxo-2',3'-
dihydrospiro[cyclobutane-1,1'-pyrrolo[2,3-
c]quinolin]-8'-yl)pyridin-3-yl)methanesulfonamide (350 mg, 0.69 mmol) in
diluted aqueous hydrochloric
acid solution (115 mL, 0.69 mmol, 0.006 M) and acetonitrile (20.0 mL) was
lyophilized directly to afford
.. the title compound as an orange solid (375 mg, 100%): 1H NMR (400 MHz, DMSO-
d6) 6 10.77 (s, 1H),
9.24 (s, 1H), 8.83 (s, 1H), 8.64 (d, J= 2.1 Hz, 1H), 8.35 (s, 1H), 8.17 (d, J=
8.9 Hz, 1H), 8.02-7.94 (m,
2H), 4.50-4.41 (m, 2H), 4.39-4.32 (m, 2H), 4.27-4.16 (m, 1H), 3.31 (s, 3H),
3.18 (s, 3H), 2.99-2.88 (m,
2H), 2.82 (d, J = 4.1 Hz, 6H), 2.63-2.52 (m, 4H); MS: [(M + 1)]+ = 507.20.
46
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
TARGET AND OFF-TARGET INHIBITORY ACTIVITY
ATM In Cell Western Assay:
In the morning, MCF-7 breast cancer cells were placed in a 384 well plate at
the density of 10,000
cells/well (Corning, #356663), 25 pL cells per well. The following day,
compounds were added to the
plate using a pin tool (Echo 550) to a final concentration of 1 pM through a 3-
fold serial dilution (a total of
doses). Then, etoposide (Sigma, #E1383) was added to a final concentration of
100 pM. The plate
was incubated at 37 C for 1 h, and the cells were fixed by the addition of 25
pL of fixing solution (8%
paraformaldehyde) for 20 minutes at ambient temperature. Cells were
permeabilized by 5 washes with
1X PBS (phosphate buffered saline) containing 0.1% Triton X-100; each wash was
5 minutes long. Cells
10 were blocked by adding 50 pL of Odyssey Blocking Buffer (LI-COR, #927-
40000) in 384 well plates for
1.5 hours with shaking at ambient temperature. Blocking buffer was then
removed, and 20 pL of anti-
pKAP1 antibody (Bethyl Laboratories, #A300-767A) (1/2000) solution were added
to each well of 384-well
plate. The plate was incubated overnight at 4 C and then washed 5 times with
1X PBST (1X PBS
containing 0.1% Tween-20). A secondary antibody (IRDye 800CW Goat anti-Rabbit
IgG, LI-COR, #926-
32211) (1/5,000) solution containing DNA stain DRAQ5 (CST, #4084L) (1/5,000)
(20 pL) was added to
each well in the plate, and the plate was incubated 1 hour with gentle shaking
in the dark. Cells were
washed 5 times with 1X PBST (1X PBS containing 0.1% Tween-20) at ambient
temperature with gentle
shaking in the dark. After the last wash, the wash solution was removed, the
plate was inverted upside
down onto a thin paper towel and centrifuged at 1000 rpm for 1 min to absorb
all wash buffer. The
bottom of the plate was cleaned with a moist, lint-free paper. The plate was
immediately scanned using
ODYSSEY CLx (LI-COR).
DNA-PK Enzyme-linked immunosorbent Assay:
On day one, a 96-well plate (ThermoFisher, Cat#: 442404) was coated with GST-
p53 (1-101) peptide
(purified by Pharmaron, BCS department) by diluting 3 pg of GST-p53 in each
well with 0.1 M
Na2CO3/NaHCO3 (pH 9.6). The plate was incubated overnight at 4 C. On the
second day, the coating
buffer was removed, and the plate was washed twice with PBST (1X PBS
containing 0.1% Tween-20).
The DNA-PK enzyme solution (Invitrogen, #PR9107A; the final DNA-PK
concentration: 0.1 pg/mL) was
then added. The compounds were serially diluted to the final maximal
concentration of 100 nM (3 fold
series dilution, a total of 10 doses), and an ATP solution (the final ATP
concentration: 20 pM) was added
to the plate. Incubate the plate at 25 C for 1 hour. The plate was washed
three times with PBST (1X
PBS containing 0.1% Tween-20) and blocked with a solution of PBST and 1% BSA
at 4 C overnight.
The third day, the plate was washed four times with PBST (1X PBS containing
0.1% Tween-20). Anti-
phospho-p53 primary antibody (cell signaling Technology, #9286, Phospho-p53
(5er15) (16G8) Mouse
mAb) (1/1000) was added to each well. The plate was sealed, incubated 1 h at
37 C, and washed four
times with PBST (1X PBS containing 0.1% Tween-20). An HRP-linked secondary
antibody (Cell
signaling Technology, #7076, Anti-mouse IgG, HRP-linked Antibody) (1/1000)
(100 pL) was added to
each well. The plate was sealed with tape, incubated 30 min at 37 C, and
washed four times with PBST
(1X PBS containing 0.1% Tween-20). At this time, 100 pL of TMB (Cell signaling
Technology, #7004)
substrate were added to each well. The plate was sealed with tape and
incubated the plate 10 min at
37 C. Stop solution (Cell signaling Technology, #7002) (100 pL) was added to
each well, and the the
plate was subjected to the absorption detection at 450 nm.
47
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
mTOR Biochemical Assay:
mTOR Kinase reactions were performed in a 10 pL volume in low-volume 384-well
plates. Typically,
PerkinElmer model 6008260 plates were used. The composition of the lx kinase
reaction buffer was: 50
mM HEPES pH 7.5, 0.01% Tween 20, 1 mM EGTA, 10 mM MnCl2, and 2 mM DTT. The
solution of
mTOR enzyme (ThermoFisher, # PR8683B; the final mTOR concentration: 0.5 pg/mL)
was added, and
the compounds were serially diluted to the final maximal concentration of 100
nM (3 fold series dilution, a
total of 10 doses). GFP-4E-BP1 (the final concentration: 0.4 pM) and the ATP
solution (the final ATP
concentration: 3 pM) were added to the 384-well plate. The plate was incubated
at 25 C for 1 hour, and
10 pL of the EDTA solution (20 mM) and Tb-labeled anti-p4E-BP1 antibody (4 nM)
in TR-FRET dilution
buffer were added to each well. The plate was sealed, incubated 30 min at 25
C, and read on a plate
reader configured for LanthaScreen TM TRFRET.
PI3Ka and PI3K5 Biochemical Assay:
P13KE and P13KE Kinase reactions were performed in a 5 pL volume in low-volume
384-well plates.
Typically, PerkinElmer model 6008280 plates were used. The lx kinase reaction
buffer consisted of 50
mM HEPES pH 7.5, 3mM MgCl2, 0.03% CHAPS, 1 mM EGTA, 100 mM NaCI, and 2 mM DTT.
PI3KE
(ThermoFisher, # PV4788; the final PI3KE concentration: 120 ng/mL) or PI3KE
enzyme solution
(ThermoFisher, # PV6451; the final PI3KE concentration: 250 ng/mL) was added
to the plate, compounds
were serially diluted to the final maximal concentration of 100 nM (3 fold
series dilution, a total of 10
doses), and the PIP2:3P5 (the final concentration: 10 Eg/mL) and ATP solution
(the final ATP
concentration: 10 pM) was added to the 384-well plate. The plate was incubated
at 25 C for 1 hour.
ADP-Glo reagent buffer (5 pL) was added to each well. The plate was sealed and
incubated for 40 min at
C. ADP-Glo detection buffer (10 pL) was added to each well, and the plate was
incubated for 40 min
25 at 25 C and read on a plate reader configured for Luminescence.
In-vitro hERG Inhibition Assay:
The hERG-T-REx TM HEK 293 cells (Invitrogen, K1236) were generated by
transfecting the hERG coding
sequence in the Tet-regulated expression vector pT-Rex-DEST30 into cells
expressing the Tet-repressor
(T-Rex TM HEK293), thereby producing cells that can be induced to express high
level of hERG channels.
The cells were cultured in a medium containing of 85% DMEM, 10% dialyzed FBS,
0.1 mM NEAA, 25
mM HEPES, 100 U/mL Penicillin-Streptomycin, 5 pg/mL Blasticidin, and 400 pg/mL
Geneticin. The cells
were split using TrypLETm Express (Gibco, 12604) about three times a week and
maintained between
¨40% to ¨80% confluence. Before the assay, the cells were induced with
doxycycline (Sigma, D9891) at
1 pg/mL for 48 hours. On the experiment day, the induced cells were
resuspended and plated onto the
coverslips at 5 X 105 cells /per 6 cm cell culture dish prior to use. The hERG
channel-mediated current
was acquired by manual patch clamp recording systems equipped with amplifiers
(HEKA, EPC10 and
Molecular Devices, multiclamp 700B) and the inverted phase contrast microscope
(Olympus, IX51/71/73).
Glass pipettes were prepared by micropipette puller (Sutter, P97 and
Narishige, PC-10) and qualified by
the pipette resistance in the range of 2-4 ME. The internal pipette solution
was 140 mM KCI, 2 mM
MgCl2, 10 mM EGTA, 5 mM MgATP, and 10 mM HEPES (pH adjusted to 7.35 with KOH),
and the
external buffer was 132 mM NaCI, 4 mM KCI, 3 mM CaCl2, 0.5 mM MgCl2, 11.1 mM
glucose, and 10 mM
48
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
HEPES (pH adjusted to 7.35 with NaOH). The whole-cell configuration was
maintained with access
resistance continuously monitored (< 15 ME). The hERG current was elicited by
depolarizing membrane
to +30 mV for 4.8 sec, and the voltage was set back to -50 mV for 5.2 sec to
remove the inactivation and
measure the deactivating tail current. The maximum amount of tail current size
was used to determine
hERG current amplitude. To evaluate the hERG inhibition, the blank vehicle and
test articles were
perfused to cells under whole-cell recording configuration through the liquid
perfusion system (ALA, VM8
gravity-flow delivery system). For dose response assay, test article was
applied to the cells
accumulatively from low to high concentrations. A positive control
(Dofetilide) was used in the
experiments to ensure the performance of the cells and operations as major
part of method validation.
The percentage hERG current inhibition was fitted against dose concentrations
to build the dose-
response curve and determine ICso.
In some embodiments, the compound of the invention is selected from the group
consisting of
compounds listed in the table below.
TABLE 1 Assay results
ATM ICso DNA-PK mTOR
hERG
Pl3K-a/o Compound nomenclature (nM) ICso (nM)
IC50 I L, 50
,
IC50 (nM)
(cell) (nM)
(PM)
trans-N-(5-(3-Ethyl-7-fluoro-3'-
methyl-2-oxo-2',3'-
566 6.72 1.03 >100 >100/14 2.10
dihydrospiro[cyclobutane-1,1'-
pyrrolo[2,3-c]quinolin]-8'-yI)-2-(2-
(isopropylamino)ethoxy)pyridin-3-
yl)methanesulfonamide
trans-N-(5-(3-(Benzyloxy)-7'-
fluoro-3-methyl-2-oxo-2',3-
567 11 2.70
>100 >100/>100 5.50
dihydrospiro[cyclobutane-1,1'-
pyrrolo[2,3-c]quinolin]-8'-yI)-2-(2-
(isopropylamino)ethoxy)pyridin-3-
yl)methanesulfonamide
N-(5-(7'-Fluoro-3'-methy1-2'-oxo-
2',3'-dihydrospiro[cyclobutane-
568 1,1'-pyrrolo[2,3-c]quinolin]-8'-yI)-2- 2.88 0.79 >100
18/54 14
(2-(isopropylamino)ethoxy)pyridin-
3-yl)propane-2-sulfonamide
N-(5-(7'-Fluoro-3'-methy1-2'-oxo-
2',3'-dihydrospiro[cyclobutane-
1,1'-pyrrolo[2,3-c]quinolin]-8'-yI)-2-
569 2.43 0.63 >100 16/73 >30
(2-(isopropylamino)ethoxy)pyridin-
3-yl)methanesulfonamide
hydrochloride
N-(5-(7'-Fluoro-3'-methy1-2'-oxo-
2',3'-dihydrospiro[cyclobutane-
1,1'-pyrrolo[2,3-c]quinolin]-8'-yI)-2-
570 3.60 0.63 >100 21/55 10.5
(2-(isopropylamino)ethoxy)pyridin-
3-yl)ethanesulfonamide
hydrochloride
N-(5-(7'-Fluoro-1-isopropy1-3'-
methy1-2'-oxo-2',3'-
dihydrospiro[azetidine-3,1'-
571 pyrrolo[2,3-c]quinolin]-8-yI)-2-(2-
5.76/5.51 0.67/0.94 >100 62/100 5.60
'
(isopropylamino)ethoxy)pyridin-3-
yl)methanesulfonamide
49
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
ATM IC50 DNA-PK mTOR
hERG
Pl3K-a/o Compound nomenclature (nM) IC50 (nM)
IC50 I L, 50
,
IC50 (nM)
(cell) (nM)
(PM)
cis-N-(5-(3-(Benzyloxy)-7-fluoro-
3'-methy1-2'-oxo-2',3'-
dihydrospiro[cyclobutane-1,1'-
572 1.05 0.32 >100 23/15
7.5
pyrrolo[2,3-c]quinolin]-8'-yI)-2-(2-
(isopropylamino)ethoxy)pyridin-3-
yl)methanesulfonamide
cis-N-(5-(3-Ethyl-7'-fluoro-3'-
methyl-2'-oxo-2',3'-
dihydrospiro[cyclobutane-1,1'-
573 0.92 0.30 >100 18/12
3
pyrrolo[2,3-c]quinolin]-8'-yI)-2-(2-
(isopropylamino)ethoxy)pyridin-3-
yl)methanesulfonamide
N-(2-(3-(Dimethylamino)azetidin-
1-y1)-5-(3'-methy1-2'-oxo-2',3'-
dihydrospiro[cyclobutane-1,1'-
574 pyrrolo[2,3-c]quinolin]-8'- 1.52 1.03 19.9
0.12/3 >30
yl)pyridin-3-
yl)methanesulfonamide
hydrochloride
CLONOGENIC AND IN VIVO EFFICACY ASSAYS
Methods
Cell Culture. The MCF-7 human breast carcinoma cell line and HCT116 p53 wild-
type and HCT116 p53
-/- cell lines were obtained from ATCC. MCF7 and HCT116 cells were cultured in
Dulbecco's modified
Eagle's medium (DMEM (Gibco #11995-065). MDA-MB-231 human triple negative
breast cancer cell line
and FADU, human head and neck squamous cell carcinoma line, were obtained from
Charles River
Laboratories (Morrisville, NC). MDA-MB-231 cells were cultured in RPM! 1640
medium (Gibco, 11875-
093). FADU cells were cultured in Minimum Essential Medium a (MEM a) (Gibco,
12571-063)
supplemented with 1mM Sodium Pyruvate (Gibco, 11360-070), 1X MEM Non-Essential
Amino Acids
(Gibco, 11140-050). All cell lines were supplemented with 10% fetal bovine
serum (FBS) (Corning, 35-
010-CV) and 1X Antibiotic-Antimycotic (Gibco #15240-062) and were grown at 37
C in 5% CO2. All cell
lines were authenticated by short tandem repeat profiling and tested negative
for mycoplasma.
Antibodies. Antibodies recognizing phosphorylated/activated ATM (S1981,
#ab81292) and DNA-PKcs
(S2056, #ab18192) were purchased from Abcam Biotechnology (Cambridge, MA).
Antibodies to ATM
(#A1106) and DNA-PKcs (#ab1832) were from Sigma and Abcam, respectively.
Phospho-KAP1 (S824,
#A300-767A) and KAP1 (A700-014) antibodies were from Bethyl Labs (Montgomery,
TX). Phospho-TBK1
(#5483), cGAS (#15102), and pSTING (S366, #19781S) antibodies were from Cell
Signaling Technology
(CST) (Danvers, MA). STING (PA5-23381) antibodies were from ThermoFisher
Scientific (Waltham, MA),
and actin (#A5316) antibodies were from Sigma. All antibodies were diluted
1:1000 with the exception of
actin which was diluted 1:3000 in 1X TBST (1X TBS -10X TBS, (Corning, 46-012-
CM), 0.05% Tween-20
(P7949) Sigma and 1% BSA. Secondary antibodies used were (a) Li-Cor method-
Goat anti-Rabbit (926-
3221) and Goat anti-Mouse (926-68020) Li-Cor (b) ECL method: Anti-rabbit IgG,
HRP-linked (#7074S),
CST.
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
lmmunoblotting. Cells were pretreated with Compound 569 at 20, 100, 500, or
1000 nM or DMSO for
30 min and then irradiated with 0 or 10 Gy (XRAD 160, Precision X Ray) as
shown in FIG. 1. In FIG. 3,
cells were pretreated with Compound 569 at 1 pM, ATMi (AZD0156) at 1 pM, DNA-
PKi (peposertib) at 1
pM as single agents or DMSO or with ATMi and DNA-PKi in combination at 0.5
micromolar each or at 1
micromolar each for 30 min and then irradiated with 0 or 10 Gy (XRAD 160,
Precision X Ray) as shown in
FIG. 1. One hour after IR, cells were lysed in RIPA buffer (50nM Tris, pH 8.0,
150mM NaCI, 0.1% SDS,
0.5% sodium deoxycholate, 1% IGEPAL CA-630 (Sigma, 18896) for 20 min at 4 C
in the presence of
protease and phosphatase inhibitor cocktails (Roche, #04693159001 &
#04906837001). Cell lysates
were centrifuged at 13,200g for 10 min at 4 C and supernatants were analyzed
for protein content using
BCA assay (Pierce TM BCA Protein Assay Kit, cat# 23227, ThermoFisher Waltham,
MA 02451) and equal
amounts of protein/lane were used for subsequent immunoblot analyses. Proteins
were denatured by
adding NuPAGE LSD Sample Buffer [4X] (Invitrogen, NP0007) containing [3-
mercaptoethanol at a final
concentration of 2.5% and the samples were boiled for 5 minutes. Samples were
loaded into NuPAGE 3-
8% Tris-Acetate Protein Gels (Invitrogen, EA03785B0X) and electrophoresed with
1X Tris-Acetate SDS
Running Buffer (Invitrogen, LA0041), or were loaded into NuPAGE 4-12% Bis-Tris
Protein Gels
(Invitrogen, NP0336BOX) and electrophoresed with 1X MOPS SDS Running Buffer
(Invitrogen, NP0001).
Proteins were transferred overnight to nitrocellulose membranes (Amersham,
10600003). Membranes
were blocked with 5% non-fat dry milk in lx TBST for 1 hour and incubated with
primary antibodies
.. overnight at 4 C. Membranes were washed 4 x 10 min with 1X TBST at RT, and
then incubated with
secondary antibodies for 1 hr at RT, followed by washing 3 x 10 min with 1X
TBST at RT. Membranes
were visualized with ODYSSEY CLX system, or with ECL (ECL Blotting substrate,
Pierce, 32209, 34075,
34095).
.. Clonogenic survival assay. This assay was performed with compound 569.
Cells were plated in 6-well
plates at different densities: 250 cells for no IR control, 5000 cells for 2
Gy of IR and 10000 cells for 4 Gy
of IR and cultured overnight. The next day, cells were preincubated with
compound 569 at 100, 250, 500,
or 1000 nM or DMSO for 30 min before being exposed to increasing doses of IR
(0, 2, or 4 Gy). After IR,
cells were continuously incubated with DMSO or compound 569 for 5 h before the
culture medium was
removed and cells were washed with PBS. Cells were cultured in complete medium
in the absence of
inhibitor for 9 days. Then cells were stained (PBS, 0.0037% v/v formaldehyde,
0,1% crystal violet) rinsed
with water and dried.
Tumor Studies. Female NCr nu/nu mice (Crl:NU(NCr)-Foxn1nu, Strain 490) were
obtained from Charles
River at 5-6 weeks of age. All animal procedures used in these studies were
approved by the Institutional
Animal Care and Use Committee (IACUC) at Duke University.
FADU or MDA-MB-231 cells were harvested during log phase growth for in vivo
implantation. The
resuspended cells were washed three times in phosphate buffered saline (PBS)
before preparing the
working dilution in PBS. FADU cells were diluted to 1 x 107 cells/mL and MDA-
MB-231 cells were diluted
to 5 x 107 cells/mL. Each mouse was injected subcutaneously in the right flank
with 100pL of the cellular
51
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
suspension. Tumor growth was monitored until the tumors reached a target
volume of at least 100 mm3,
at which point mice were randomly stratified into four treatment groups.
Following implantation of the tumor cell lines, mice were monitored once
weekly for tumor development.
Upon detection, tumors were measured in two dimensions using digital calipers.
Tumors were treated
after reaching at least 100 mm3 and were measured two to three times weekly
following enrollment in one
of the treatment groups. The study endpoint was defined as tumor quintupling
from the volume at time of
treatment.
The vehicle used in this study was 0.5% (Hydroxypropyl)methyl cellulose (HPMC)
and 0.2% Tween80
dissolved in deionized water. The pH of the vehicle was adjusted to 7.0-8.0
and the vehicle was stored at
4 C. On each day of in vivo treatment an appropriate mass of test compound was
added to the vehicle at
room temperature. The mixture was vortexed as necessary to generate a final
concentration of 1.6
mg/mL.
Pharmacodynamic (PD) assays. Tumor studies were performed as follows. Mice
bearing FADU or
MDA-231 tumors were pre-dosed with vehicle alone or compound 569 alone at 3,
6, and 10 mg/kg.
Tumors were then irradiated with 10 Gy 45 min after vehicle or compound 569
administration or left
unirradiated (vehicle and compound 569 at 10 mg/kg). Tumors were harvested 1 h
post radiation.
Tissue homogenates for pharmacodynamic analyses were collected and processed
as follows. Tumor
tissues from FADU or MDA-MB-231 tumors were flash frozen in liquid nitrogen
and pulverized on dry ice
using a tissue pulverizer. A portion of the tumor powder was transferred into
a microtube, followed by
addition of 500 pL of RIPA buffer [50 nM Tris, pH 8.0, 150 mM NaCI, 0.1% SDS,
0.5% sodium
deoxycholate, 1% IGEPAL CA-630 (Sigma, 18896)]. To 10 ml of RIPA buffer, 2
tablets each of Protease
inhibitor and Phosphatase inhibitor (Roche, #04693159001 & #04906837001), 200
pL of 0.1 M PMSF
and 160 pL of Aprotinin, (Sigma, A6279) were added. The suspension was
homogenized on ice with a
pellet pestle motor (KONTES) and incubated on ice for 30 minutes. Cell lysates
were centrifuged at
13,200 rpm for 15 min at 4 C and supernatants were transferred to a new tube
and analyzed for protein
concentration with a BCA kit (Pierce, 23227), followed by immunoblotting.
In vivo tumor growth delay study was performed as follows. Following
implantation of the tumor cell lines,
mice were monitored once weekly for tumor development. Upon development of a
tumor measuring at least
100 mm3, tumor-bearing mice were stratified to one of 4 groups, utilizing 5
mice per group. Tumor volumes
.. were between 100 and 500 mm3. Vehicle or compound 569 (10 mg/kg) were
administered by oral gavage
once daily for three consecutive days (qd x 3), alone or in combination with
focal radiation. The delivered
volume of vehicle or compound 569 was adjusted based on body weight of the
individual animals. Mice
stratified to the radiation therapy groups received 3 Gy per day for three
consecutive days (qd x 3) 45
minutes following administration of the vehicle or compound 569. The mice were
anesthetized with
isoflurane during the radiation treatment, which was performed with the X-RAD
225Cx small animal image-
guided irradiator (Precision X-Ray). The 40mm x 40mm irradiation field was
centered on the tumor-bearing
52
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
leg via fluoroscopy. Mice were irradiated with parallel-opposed anterior and
posterior fields of 225 kVp,
13mA X-rays using a 0.3mm Cu filter at an average dose rate of 300 cGy/min.
Group 1 mice received vehicle qd x 3.
Group 2 mice received 10 mg/kg compound 569 qd x 3.
Group 3 mice received vehicle 45 minutes prior to 3 Gy focal irradiation qd x
3.
Group 4 mice received 10 mg/kg compound 569 45 minutes prior to 3 Gy focal
irradiation qd x 3.
Statistical and Graphical Analysis of the in vivo data. Prism (GraphPad) was
used for graphical
presentations. SAS 9.4 and R 3.6.1 were used for statistical analyses. Tumor
growth rates among four
treatment groups were compared. In order to adjust for the differing tumor
sizes at the start of treatment,
the relative tumor volume was calculated by dividing the tumor volume at each
time point by the volume
at baseline. Line plots were used to display median relative tumor volume over
time (FIGS. 6 and 8).
When an animal exited the study due to tumor quintupling, the final tumor
volume recorded for the animal
was included with the data used to calculate the median volume at subsequent
time points. Kaplan-Meier
plots were used to show the percentage of mice in each treatment group
remaining in the study over time
(FIGS. 7 and 9). Mice that were found dead prior to reaching the quintupling
endpoint were subjected to
right-censoring. Differences in quintupling-free survival between the four
treatment groups were evaluated
with a log-rank test. A P-value less than 0.05 was considered statistically
significant.
Results
FIG. 1 shows an image of an immunoblot from an in vitro experiment assessing
the effect of compound
569 on MCF7 cells with or without radiation. The immunoblot shows the
inhibition of radiation-induced
autophosphorylation of ATM and DNA-PK kinases and radiation-induced
phosphorylation of KAP1, an
ATM substrate, by compound 569 in tumor cells. MCF7, a human breast carcinoma
cell line, was pre-
treated for 30 min with compound 569 at concentrations of 20, 100, 500, and
1000 nM. DMSO was used
as a negative control. The cells were then exposed to 0 or 10 Gy of ionizing
radiation (IR) and one hour
later, cells were harvested for immunoblot analyses. Compound 569 showed
potent dose-dependent
inhibition of radiation-induced phosphorylation of DNA-PKcs at 5er2056 and ATM
at 5er1981 (both
autophosphorylation events) and the ATM substrate KAP1 (5er824) demonstrating
compound 569
inhibited both DNA-PK and ATM kinases in a cell.
FIG. 2 demonstrates the radiosensitizing properties of compound 569 in a
clonogenic survival assay in
vitro. MCF7 cells were pre-treated with vehicle or compound 569 at 100, 250,
500, and 1000 nM for 30
min before irradiation with increasing doses of IR (0, 2, and 4 Gy). After 5h,
the medium was removed
and fresh medium without inhibitors was added to cells, and they were cultured
for 9 days before being
fixed, stained and colonies counted. The 5-h exposure of cells to compound 569
induced a significant
degree of radiosensitization without evidence of cellular toxicity in the
absence of IR in MCF7 cells (FIG.
2, Table 1).
FIG. 3 is an immunoblot showing the induction of phosphorylation of TBK1 by
compound 569, AZD0156
(a selective ATM inhibitor; ATMi), peposertib (a selective DNA-PK inhibitor;
DNA-PKi), and a combination
of AZD0156 and peposertib (Ai+Di) in HCT116 cells expressing wild-type p53 or
HCT116 cells that were
53
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
negative for p53 expression. Phosphorylation of TBK1 is a marker of activation
of the type I interferon
response and has been linked to enhanced tumor response to immune checkpoint
blockade therapy.
Dual inhibition of ATM and DNA-PKcs by combining the two selective chemical
inhibitors (AZD0156 and
peposertib) together resulted in more profound activation of TBK1 than either
of the selective inhibitors
alone. Similarly, exposure of cells to compound 569, a dual inhibitor of ATM
and DNA-PKcs, was a more
potent activator of TBK1 phosphorylation than either of the single target
kinase inhibitors and was
independent of p53 status. The TBK1 activation by 569 and dual inhibition of
ATM and DNA-PKcs
appeared to be independent of the cGAS and phospho-STING induction.
c,
bo OH
N
, N
0 N /
\ N
AZD0156 peposertib
To assess the pharmacodynamic activity of compound 569 in vivo, FADU and MDA-
MB-231 cells were
grown as tumor xenografts in nude mice. Vehicle or compound 569 were
administered by oral gavage
alone or in combination with focal radiation. Mice were dosed with vehicle or
compound 569 at 3, 6, and
10 mg/kg. Tumors then received 10 Gy of focal irradiation 45 min later or were
left unirradiated (0 Gy,
vehicle and 0 Gy + compound 569 at 10mg/kg). Tumors were harvested 1 h post
radiation.
lmmunoblotting of lysates from these tumors shows radiation induced
autophosphorylation of pDNA-PK
and radiation-induced phosphorylation of KAP1, a downstream target of ATM and
dose-dependent
inhibition of those phosphorylation events in FADU and MDA231 tumors treated
with radiation plus
compound 569. Tumor levels of radiation-induced autophosphorylation of DNA-PK
and radiation-induced
phosphorylation of KAP1 were significantly inhibited by ¨55% and >80%,
respectively at 10mg/kg (FIGS.
4 and 5, Tables 2-5). Basal phosphorylation of DNA-PK was also inhibited by
compound 569 in tumors.
TABLE 1 Compound 569 Clonogenic Assay in MCF7 cells (see FIG. 2)
Compound Clonogenic Clonogenic Clonogenic `)/0
survival @ 0
(concentration) survival ratio survival ratio Fold
survival ratio Fold Gy w/out IR
0 Gy 2 Gy 2 Gy 4 Gy 4 Gy
100%
DMSO 1.000 0.662 1.0 0.317 1.0 97%
569 (100 nM) 1.000 0.0882 7.5 0.0146 21.8 92%
569 (250 nM) 1.000 0.0610 10.8 0.00588 54.0 97%
569 (500 nM) 1.000 0.0451 14.7 0.00347 91.5 105%
569 (1000 nM) 1.000 0.0301 22.0 0.00160 198 100%
54
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
TABLE 2 Quantification of pDNA-PK/ DNA-PK in FADU tumors
Treatment pDNA-PK/DNA-PK Standard
deviation
No radiation, vehicle 1.14 0.263
No radiation, 10 mg/kg compound 569 0.343 NA
Gy, vehicle 1 0.0565
10 Gy, 3 mg/kg compound 569 0.938 0.181
10 Gy, 6 mg/kg compound 569 0.658 0.126
10 Gy, 10 mg/kg compound 569 0.476 0.188
TABLE 3 Quantification of pKAP1/KAP1 in FADU tumors
Treatment pKAP1/KAP1 Standard
deviation
No radiation, vehicle 0.00505 0.000265
No radiation, 10 mg/kg compound 569 0.00595 NA
10 Gy, vehicle 1 0.0616
10 Gy, 3 mg/kg compound 569 0.0903 0.005
10 Gy, 6 mg/kg compound 569 0.0982 0.0741
10 Gy, 10mg/kg compound 569 0.156 0.171
5 TABLE 4 Quantification of pDNA-PK/DNA-PK in MDA-231 tumors
Treatment pDNA-PK/DNA-PK Standard
deviation
No radiation, vehicle 0.648 0.0948
No radiation, 10mg/kg compound 569 0.223 NA
10 Gy, vehicle 1 0.231
10 Gy, 3 mg/kg compound 569 0.889 0.414
10 Gy, 6 mg/kg compound 569 0.670 0.278
10 Gy, 10mg/kg compound 569 0.454 0.116
TABLE 5 Quantification of pKAP1/KAP1 in MDA-231 tumors
Treatment pKAP1/KAP1 Standard
deviation
No radiation, vehicle 0.00357 0.000826
No radiation, 10mg/kg compound 569 -0.00319 NA
10 Gy, vehicle 1 0.114
10 Gy, 3 mg/kg compound 569 0.327 0.158
10 Gy, 6 mg/kg compound 569 0.0130 0.00879
10 Gy, 10 mg/kg compound 569 0.00901 0.00242
FADU Tumors. Mice in Group 1 received vehicle, qd x 3. Aggregate tumor growth
in this group was
10 progressive. The median time to endpoint was 14 days with a range of 11-
14 days (FIGS. 6 and 7). Mice
in Group 2 received compound 569 at a dose of 10 mg/kg, qd x 3. The median
time to endpoint was 14
CA 03147111 2022-01-11
WO 2021/022078
PCT/US2020/044322
days with a range of 11 to 17 days. The tumor growth curves and quintupling-
free survival for Group 2
animals are nearly identical to those for Group 1 (FIGS. 6 and 7). Mice in
Group 3 received vehicle and a
focal radiation dose of 3 Gy, which was delivered 45 minutes after
administration of the vehicle. Both the
vehicle and radiation were qd x 3. One animal was found dead on Day 16 due to
unknown causes. The
median time to endpoint was 21 days with a range of 19 to 25 days. Overall
survival was significantly
improved compared to Group 1 and Group 2 (P < 0.01, logrank). The delay in
aggregate tumor growth
and increase in quintupling-free survival for Group 3 relative to Groups 1 and
2 is shown in FIGS. 6 and 7.
Mice in Group 4 received compound 569 and a focal radiation dose of 3 Gy,
which was delivered 45
minutes after administration of the compound. Both compound 569 and radiation
were administered qd x
3. The median time to endpoint was 42 days with a range of 31 to 51 days.
Overall survival was
significantly improved compared to Group 1, Group 2, and Group 3 (P < 0.01,
logrank). The delay in
aggregate tumor growth and increase in quintupling-free survival for Group 4
relative to all other groups is
shown in FIGS. 6 and 7. All treatments were well-tolerated.
MDA-MB-231 Tumors. Mice in Group 1 received vehicle, qd x 3. The median time
to endpoint was 12
days with a range of 12 to 17 days. Mice in Group 2 received compound 569 at a
dose of 10 mg/kg, qd x
3. The median time to endpoint was 17 days with a range of 12 to 22 days. The
tumor growth curves and
quintupling-free survival for Group 2 animals are nearly identical to those
for Group 1 (FIGS. 8 and 9).
Mice in Group 3 received vehicle and a focal radiation dose of 3 Gy, which was
delivered 45 minutes after
__ administration of the vehicle. Both the vehicle and radiation were qd x 3.
The median time to endpoint
was 22 days with a range of 16 to 22 days. Overall survival was significantly
improved compared to
Group 1 animals treated with vehicle alone (P < 0.01, logrank). The increase
in quintupling-free survival
for Group 3 relative to Groups 1 and 2 is shown in FIG. 9. Mice in Group 4
received 10 mg/kg compound
569 and a focal radiation dose of 3 Gy, which was delivered 45 minutes after
administration of compound.
Both compound 569 and radiation were qd x 3. One animal was found dead on Day
35 due to unknown
causes. The median time to endpoint was 43 days with a range of 37 to 58 days.
Overall survival was
significantly improved compared to Group 1, Group 2, and Group 3 (P < 0.01,
logrank). The delay in
aggregate tumor growth and increase in quintupling-free survival for Group 4
relative to all other groups is
shown in FIGS. 8 and 9. All treatments were well-tolerated.
OTHER EMBODIMENTS
Various modifications and variations of the described invention will be
apparent to those skilled in the art
without departing from the scope and spirit of the invention. Although the
invention has been described in
connection with specific embodiments, it should be understood that the
invention as claimed should not
be unduly limited to such specific embodiments. Indeed, various modifications
of the described modes
for carrying out the invention that are obvious to those skilled in the art
are intended to be within the
scope of the invention.
Other embodiments are in the claims.
56