Note: Descriptions are shown in the official language in which they were submitted.
WO 2021/051011
PCT/1JS2020/050574
METHODS AND COMPOSITIONS FOR PROTEIN AND
PEPTIDE SEQUENCING
TECHNICAL FIELD
This disclosure generally relates to methods and compositions for protein and
peptide sequencing,
BACKGROUND
Rapid improvements in DNA sequencing technology in the last decade have
yielded a wealth of molecular information. And while the ability to read
genomes has
revolutionized biological research, a significant amount of phenotypic and
disease-state
information cannot be deduced from the genome. RNA sequencing has provided a
deeper
understanding of the functional elements of the genome and their expression
levels.
However, significant challenges still surround efforts to correlate protein to
mRNA
expression levels (de Sousa (Abreu, Penalva, Marcotte, & Vogel, 2009) (Vogel &
Marcotte, 2012), leading to difficulties in understanding precise protein
quantification,
modification or even sequence, resulting in the loss of information of
cellular state. RNA
analysis falls short in predicting protein presence when evaluating proteins
in serum,
since proteins can be excreted from cells and circulate throughout the blood
system,
resulting in the loss of spatial connection between the RNA sequence and its
translated
target. Additionally, protein sequencing could reveal many unknowns, i.e.
proteins from
other organisms (such as viruses, bacteria etc) present in a host's
bloodstream and
impacting the host organism.
RNA and DNA sequencing gives limited insight into antibody sequences, as the
diversity of antibody repertoire is generated by somatic hypennutation events.
In order to
capture information that occurs alter DNA processing and secretion, such as
post-
translational protein modifications, translational fidelity, protein folding
integrity, etc.,
scientists must be able to sequence proteins (i.e., read their amino acid
sequences) directly
from the sample of interest to infer correlations between protein levels and
its enzymatic
effect. De novo protein sequencing can lead to the discovery of rare and novel
proteins
from any organism (e.g. various tissues, pathogens, mutated cancer cells) from
any
protein-containing sample (e.g. blood, skin, cerebrospinal fluid, soil).
Protein sequencing
1
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
can also serve as a metric for therapeutic efficacy by allowing for extensive
physiological
monitoring through the course of disease treatment. Currently, however, there
exists no
cost and time-effective strategy for the large-scale and high-throughput
sequencing of
proteins and proteomes that spans the entire dynamic range of protein
expression.
Neither is there a robust method to sequence untargeted lowly expressed
proteins. As a
result, sequencing of antibodies and lowly expressed proteins remain wracked
with
obstacles using current technologies and practically inaccessible to all but
the most
specialized research efforts.
SUMMARY
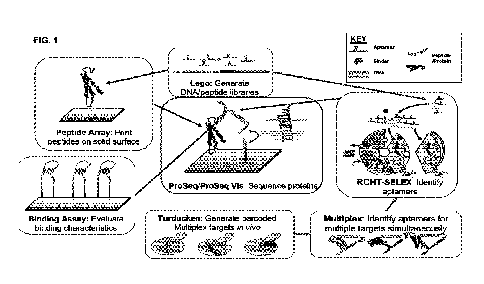
This disclosure describes a collection of methods and compositions that form a
pipeline of developing and using a protein sequencing platform which utilizes
aptamers
that bind specifically to N-terminal amino acids (FIG. 1). Amino acid-specific
aptamers
can be generated using the novel methods described herein (RCHT-SELEX and NTAA-
SELEX). Such amino acid-specific aptamers can be used to recognize, identify
and
convert each amino acid of a protein or peptide into a DNA sequence (PROSEQ)
or such
amino acid-specific aptamers can be used to recognize and identify, based on a
visual
signal, each amino acid of a protein or peptide (PROSEQ-VIS). In addition,
many
different target-specific aptamers can be generated simultaneously, and used
to produce
and screen a large multitude of binders (MULTIPLEX). Simultaneous and specific
aptamer selection relies on robust identification of targets. Nucleic acid
barcoded target
generation can be accomplished in vivo via a non-covalently bond between a
peptide or
protein using an RNA-binding protein and its corresponding recognition
sequence
(TURDUCICEN). Lastly, successful SELEX experiments require that aptamers with
some specific binding preference and affinity for the molecular target be
included in the
original pool of 1014-1015 candidate sequences, which is only a small fraction
of all the
DNA sequences possible. Artificial intelligence (Al), Deep Learning (DL) and
Machine
learning (ML) can optimize experimental seed binders, so, unlike conventional
SELEX
experiments, the most optimal binders do not need to present in the initial
starting library,
but can be generated from features of experimentally discovered binders. The
ability to
construct computationally-derived, customizable DNA libraries to perform SELEX
screens using a controlled input pool can significantly increase the
exploratory space by
systematically assaying aptamer candidates that include sequences with known
binding
properties (LEGO).
2
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
In one aspect, methods of obtaining aptamers having affinity and specificity
to a
target are provided. Such methods typically include (a) providing a plurality
of aptamers;
(b) optionally, performing a negative selection on the plurality of aptamers;
(c) optionally,
spiking the plurality of aptamers with control oligonucleotides prior to PCR
amplification; (d) optionally, amplifying the plurality of aptamers; (e)
incubating the
plurality of aptamers with a plurality of potential targets under conditions
that allow
binding of the plurality of aptamers to the plurality of potential targets;
(f) optionally, for
replicate experiments, incubating the plurality of amplified aptamers with a
plurality of
potential targets or null targets in different reactions under conditions that
allow binding
of the plurality of amplified aptamers to the plurality of potential targets;
(g) removing
unbound aptamers; (h) sequencing target-bound aptamers; and (i) repeating
steps (a) - (10
a plurality of times, thereby obtaining aptamers having affinity and
specificity to the
target.
In some embodiments, the potential targets are polypeptides, amino acids,
nucleic
acids, small molecules, whole proteins or protein complexes, or cells.
In some embodiments, the methods further include amplifying the plurality of
aptamers candidates in the initial random or ML-designed library in a single
bring-up
amplifying step or a double bring-up amplifying step to produce the input pool
into
SELEX containing a plurality of copies of aptamer candidates.
In some embodiments, the same bring-up is assayed against multiple targets, in
replicate experiments, or combinations thereof
In some embodiments, the methods optionally further include introducing a
known amount of a known oligonucleotide into the sample prior to the step of
amplifying
the plurality of aptamers.
In some embodiments, the methods optionally further include introducing a
known amount of a known oligonucleotide into the sample prior to the
sequencing step.
In some embodiments, the sequencing data of the known oligonucleotides spiked
in is observed to detect experimental error.
In some embodiments, the methods further include amplifying a standardized
amount of target-bound aptamers from each sample each time the steps are
repeated.
In some embodiments, the methods further include amplifying the plurality of
aptamers under conditions optimized for maximum amplification with minimal
bias for
the specific primers used.
3
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
In some embodiments, the methods further include digesting the dsDNA post-
PCR into ssDNA such that the desired strand is preserved.
In some embodiments, the methods further include amplifying the plurality of
aptamers in the presence of an abundance of the primer that generates the
desired ssDNA.
In some embodiments, the methods further include performing unit tests before
each dsDNA digestion to determine optimal digestion conditions for each
sample.
In some embodiments, the methods further include changing primer sequences
associated with each member of the plurality of aptamers prior to repeating
the step of
incubating the plurality of aptamers with potential targets a plurality of
times to identify
strong binders independent of the primer region.
In some embodiments, the methods further include alternating targets with
varied
local environment binding regions between each repetition of steps (a) - (h)
for
experiments where the desired aptamers are ones that bind specifically to a
smaller region
of a molecule rather than the whole molecule.
In some embodiments, the methods further include subjecting a small sample of
the aptamer pool prior to step (e) of method 1 through the same PCR reactions
without
assaying against beads or targets to assess the effects of performing SELEX
with the
chosen selection components.
In some embodiments, the methods further include: (a) incubating the plurality
of
aptamers with a plurality of different targets in the same reaction under
conditions that
allow binding of the plurality of aptamers to the plurality of potential
targets; (b)
removing unbound aptamers; (c) amplifying target-bound aptamers; (d)
sequencing target-bound aptamers; (e) repeating steps (a) - (d) a plurality of
times; (1)
incubating the plurality of aptamers with a plurality of a single target in
each experiment
for each different target; (g) repeating steps (b) - (d); thus identifying
aptamer binders to
multiple targets.
In some embodiments, step (e) of claim I is repeated a plurality of times in
separate reactions, each containing a potential target.
The SELEX methods described herein, referred to herein as RCHT SELEX, were
designed with a protocol that is ideal for the integration of machine learning
(ML) into a
SELEX protocol (e.g., prioritizing computational needs). Additional SELEX
methods are
described, referred to herein as N-terminal SELEX (or N-terminal amino acid
(NTAA)
4
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
SELEX), were designed with a protocol that is ideal for small and/or difficult
targets
(e.g., prioritizing experimental needs).
While both of the two SELEX methods can be modified as needed, the differences
between the two methods can include the following:
(a) a bring-up typically is performed in the method referred to as SELEX-RCHT
before the SELEX portion of the method begins, thereby reducing the initial
input pool to
1092 molecules. On the other hand, the method referred to as SELEX-NTAA
typically
does not include a bringup, and, in some instances, works better without one.
Thus, the
SELEX-NTAA method typically starts with a pool of 1094 - 1095 random aptamers.
(b) reactions using the SELEX-RCHT methods typically uses replicates (e.g., 2
to
3 replicate reactions) conducted in parallel; replicate reactions generally
are not necessary
using the SELEX-NTAA methods such that as many experiments as possible can be
done
in parallel.
(c) control reactions using the SELEX-RCHT methods typically are done in
replicate, which takes up 3 of the 12 possible bringup inputs; replicates of
control
reactions are not necessary when using the SELEX-NTAA methods, although it%
suggested to run one target in each experiment to determine global
experimental failure or
contamination. (4) RCHT requires some noise measurement (such as Fake SELEX)
to
determine PCR bias for models; NTAA does not require separate ground truth vs
noise
measurements, but the signal is determined from enrichment curves and testing
top
candidates.
(d) the SELEX-NTAA methods use a switch target step, which allows for the
pursuit of small or difficult targets (or sub-regions of a larger target); the
SELEX-RCHT
methods typically do not utilize this additional step.
(e) the SELEX-NTAA methods incorporate an additional step of counter selection
- especially when pursuing sub-regions of targets - to isolate the best
experimental
binders. The SELEX-RCHT methods do not include a counter selection step,
however, a
counter selection step can be used with the SELEX-RCHT methods, provided such
a
counter selection step is used, alone or in combination with other steps of
the method,
carefully to avoid biasing the results.
In one aspect, methods of sequencing a protein or peptide are provided. Such
methods typically include: (a) incubating the protein or peptide with a
library of DNA
aptamers exhibiting binding specificity toward at least one N-terminal amino
acid under
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
conditions where one or more aptamers bind specifically to at least one N-
terminal amino
acid of the protein or peptide, wherein each aptamer within the library
comprises a
peptide binding ssDNA region and a unique barcode sequence indicative of the
first
sequencing round and the associated peptide binding ssDNA region; (b) ligating
the DNA
aptamer bound to the N-terminal of the protein or peptide onto its proximal
DNA barcode
construct; (c) removing the peptide binding sequence from the DNA aptamer,
thereby
leaving only the barcode of the DNA aptamer and a short consensus sequence for
subsequent ligation covalently attached to the DNA barcode construct, such
that the
identity of the binder and therefore the putative amino acid identity of the N-
terminal of
the peptide is recorded; (d) removing the N-terminal amino acid from the
protein or
peptide to produce an N-terminal amino acid-shortened protein or peptide; (e)
incubating
the N-terminal amino acid-shortened protein or peptide with the library of
aptamers
exhibiting binding specificity toward at least one amino acid under conditions
where one
or more aptamers bind specifically to at least one N-terminal amino acid of
the N-
terminal amino acid-shortened protein or peptide, wherein each aptamer within
the library
comprises a peptide binding ssDNA region and a unique barcode sequence
indicative of
the second sequencing round and the associated peptide binding ssDNA region;
(t)
ligating the DNA aptamer bound to the N-terminal of the protein or peptide
onto its
proximal DNA barcode construct; (g) removing the peptide binding sequence from
the
DNA aptamer, thereby leaving only the barcode of the DNA aptamer and a short
consensus sequence for subsequent ligation covalently attached to the DNA
barcode
construct, such that the identity of the binder and therefore the putative
amino acid
identity of the N-terminal of the peptide is recorded; (h) removing the N-
terminal amino
acid from the N-terminal amino acid-shortened protein or peptide; (i)
repeating steps (a) ¨
(d) a plurality of times to construct a chain of positional barcodes that
correspond to
sequential N-terminal amino acids in the protein or peptide; and (j)
sequencing the chain
of positional barcodes, thereby obtaining the sequence of the protein or
peptide.
In some embodiments, the protein or peptide is from a synthetic sample,
biological sample, or combinations thereof. In some embodiments, the
biological sample
is selected from the group consisting of blood, urine, saliva, tissue biopsy,
sputum, stool,
single cell, environmental samples, bacterial swab, or any sample containing
peptides or
proteins.
6
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
In some embodiments, the protein or peptide is a full-length protein, a
peptide
fragment, or a protein or peptide comprised within a complex. In some
embodiments, the
method further includes, prior to step (a), fragmenting the protein or
peptide. In some
embodiments, the fragmenting step includes fragmenting the protein or peptide
with
ttypsin, Lys-C, another fragmentation enzyme, alternative protein
fragmentation or
degradation methods, or combinations thereof
In some embodiments, the C-terminal end of the protein or peptide is attached
to a
solid support. In some embodiments, the C-terminal end of the protein or
peptide is
attached to an oligonucleotide tail. In some embodiments, removing the aptamer
includes
cleaving the aptamer at the restriction site with a restriction enzyme. In
some
embodiments, the aptamer is attached to the barcode hydrostatically and
removal of the
peptide binding sequence is mediated by hydrogen bond disruption (rather than
DNA
cleavage by restriction enzyme).
In some embodiments, the removing the N-terminal amino acid step comprises
Edman degradation of the protein or peptide, cleaving the protein or peptide
with one or
more aminopeptidases, heat, pH, or combinations thereof. In some embodiments,
the
sequencing step uses a next generation sequencing (NOS) platform. In some
embodiments, the number of sequencing reads associated with amino acid
sequences of
known proteins is analyzed to determine the relative quantity of proteins in a
sample.
In some aspects, methods of identifying novel biomarkers are provided. Such
methods typically include: (a) providing protein samples from biological
samples of
interest and control or comparison biological samples according to the methods
described
herein; (b) optionally removing known proteins of very high concentrations;
(c)
performing the steps (a) - (j) of the methods described herein; (d) removing
high
concentration DNA barcode construct sequences associated with commonly highly
expressed proteins or contaminants such that the ratio of DNA barcode
constructs
associated with lowly expressed proteins to highly expressed proteins
increases, thereby
producing ratio-adjusted DNA barcodes; (e) PCR amplifying the DNA barcode
constructs
post-sup-diff; and (f) comparing the number of sequencing reads associated
with each
lowly expressed proteins from control samples to samples of interest, thereby
identifying
putative biomarkers that have significantly different relative expression
levels between
control samples and samples of interest.
7
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
In some embodiments, methods of using the protein sequencing methods
described herein are provided to evaluate disease state, evaluate response to
treatment,
predict treatment response, or combinations thereof, wherein one or more signs
of those
diseases is aberrant expression levels of known protein biomarkers. Such
methods
typically include: (a) providing protein samples from patient samples
according to the
methods described herein; (b) optionally depleting known proteins of very high
concentrations; (c) performing the steps (a) - (j) of methods described
herein; (d)
removing high concentration DNA barcode construct sequences associated with
commonly highly expressed proteins or contaminants to increase the ratio of
DNA
barcode constructs associated with lowly expressed proteins to highly
expressed proteins,
thereby producing ratio-adjusted DNA barcodes; (e) PCR amplifying the DNA
barcode
constructs post-sup-diff; (f) determining the relative quantity of known
biomarkers by
analyzing the number of sequencing reads associated with known protein
biomarkers; and
(g) determining the presence or absence of expression level deviations of
known
biomarkers from standard values, thereby evaluating disease state, evaluating
response to
treatment, predicting treatment response, or combinations thereof
In some embodiments, the library of aptamers is produced using the RCHT-
SELEX method described herein. In some embodiments, the aptamer exhibits
binding
specificity toward one N-terminal amino acid. In some embodiments, the aptamer
exhibits binding specificity toward two or more N-terminal amino acids. In
some
embodiments, the unique barcode sequence indicative of the aptamer's
associated peptide
binding ssDNA region and sequencing round includes about 6 to about 20
nucleotides. In
some embodiments, a BCS Compatible portion of the aptamer construct may
comprise
one or more complementary DNA sequences hybridized to an aptamer as described
herein. In some embodiments, a proximal DNA barcode foundation contains a
unique
barcode that indicates either the associated protein or peptide (if known) or
the sample
from which the protein or peptide was derived.
In another aspect, articles of manufacture for protein or peptide sequencing
are
provided. Such articles typically include a library of DNA aptamers, wherein
each
member of the library exhibits binding specificity toward at least one N-
terminal amino
acid.
In some embodiments, each member of the library further comprises a common
sequence indicative of the cycle number (e.g., first, second, third, etc.) and
a unique
8
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
barcode sequence. In some embodiments, each member of the library further
comprises a
restriction site. In some embodiments, each member of the library further
comprises at
least one sequence for ligation, annealing, or combinations thereof
The methods described herein also can be used for sequencing MI-length
proteins.
The methods described herein also can be used for sequencing the proteins
within
a protein complex.
The methods described herein also can be used for sequencing the proteins
within
a complex protein pool.
Additional methods are described herein to overcome the difficulties that
result
from the removal of functional P5 adaptors on the surface of Illurnina
sequencing chips as
a result of Edman degradation. The loss of functional P5 adaptors on the
surface of the
sequencing chips prevents the clustering of DNA barcode constructs and,
therefore,
prevents the ability to sequence directly on the same chip.
In some embodiments, after building the DNA barcode construct, which contains
a chain of DNA barcodes that indicates the order of aptamer binding for a
peptide, the
constructs can be amplified on the chip, or cleaved off the chip and amplified
in solution.
Amplification methods used can include, without limitation, PCR, loop mediated
isothermal amplification, nucleic acid sequence based amplification, strand
displacement
amplification, and multiple displacement amplification. Additionally, the
original DNA
barcode constructs can be transcribed on the chip into large amounts of RNA
constructs,
which then can be converted into a cDNA library that includes many copies of
the
original DNA barcode. The amplification products, which are copies of the
original DNA
barcode constructs, can be removed from the microfluidic chamber and sequenced
using
standard DNA sequencing methods including, without limitation, Sanger
sequencing,
NGS, ion semiconductor sequencing, SOLiD technology, cPAS, etc. Numbers of
reads
can be normalized to the number of PCR cycles used to estimate the quantity of
each
protein or peptide sequenced from the initial sample.
In some embodiments, the methods described herein can utilize the empty P7
adaptors available on the chip to perform cluster generation. After DNA
barcode
constructs are built, a second sequencing primer adaptor that contains at
least (a) the
antisense restriction site and (b) the reverse complementary strand to P7
adaptors on the
9
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
chip can be ligated tothe 3' end to the barcode constructs. After bridge
amplification of
the barcode constructs, the reverse strands can be selectively cleaved to
allow for accurate
base-calling in each individual cluster.
In yet another aspect, methods of recording one or more binding events between
a
plurality of putative binders and a plurality of targets are provided (BCS
BINDING
ASSAY). Such methods typically include: (a) incubating known putative binding
partners with a library of DNA barcoded binders of unknown binding affinity
and
specificity, wherein each binder within the library comprises a target binder
and a unique
barcode sequence indicative of the associated binder; (b) ligating the DNA
barcode of the
target binder onto its proximal DNA barcode construct, which itself may
contain a unique
barcode; (c) optionally removing the target binder, thereby leaving only the
barcode of
the target binder and a short consensus sequence for subsequent ligation
covalently
attached to the DNA barcode construct, such that the identity of the binder
and therefore
the putative identity of the bound target is recorded; (d) optionally
repeating steps (b) - (c)
for multiple rounds of validation; (e) optionally, if the binders are
aptamers, not removing
the target binder in step (c), but ligating a sequencing adaptor such that
sequencing will
occur directly through the nucleic acid sequence of the binder; and (f)
ligating appropriate
sequencing adaptors; and (g) sequencing through the foundation and binder
barcodes,
thereby identifying a plurality of targets and their binding partners.
Representative binders include, without limitation, aptamers, antibodies, and
other
small molecule binders. Representative targets include, without limitation,
peptides,
proteins and protein complexes, lipid molecules, viruses, ultramicrobacteria,
and
inorganic molecules.
In some embodiments, the putative binders are immobilized on the solid
substrate,
and the targets are modified with a DNA barcode tail and in solution.
In one aspect, methods of sequencing a protein or peptide with fluorescently-
tagged aptamers are provided. Such methods typically include: (a) providing a
solid
support comprising at least one protein or peptide attached thereto, wherein
the at least
one protein or peptide is attached to the solid support via a nucleic acid
linker, wherein
the nucleic acid linker comprises a sequencing adaptor sequence; (b)
incubating the
protein or peptide with a library of aptamers exhibiting binding specificity
toward at least
one N-terminal amino acid under conditions where one or more aptamers bind
specifically to at least one N-terminal amino acid of the protein or peptide,
wherein each
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
aptamer within the library comprises a unique optical signature; (c) detecting
the unique
optical signature and the position of the unique optical signature; (d)
removing the
aptamer and removing the N-terminal amino acid from the protein or peptide to
produce
an N-terminal amino acid-shortened protein or peptide; (e) incubating the N-
terminal
amino acid-shortened protein or peptide with a library of DNA aptamers
exhibiting
binding specificity toward at least one N-terminal amino acid under conditions
where one
or more aptamers bind specifically to at least one N-terminal amino acid of
the protein or
peptide, wherein each aptamer within the library comprises a peptide binding
ssDNA
region and a unique barcode sequence comprising of individual DNA barcodes
indicative
of the first probe iteration and the associated peptide binding ssDNA region;
(f) detecting
the unique optical signature and the position of the unique optical signature;
(g) removing
the aptamer and removing the N-terminal amino acid from the protein or peptide
to
produce an N-terminal amino acid-shortened protein or peptide; (h) repeating
steps (b) ¨
(g) a plurality of times to construct a positional chain of optical barcodes;
thereby
obtaining the sequence of the protein or peptide.
In another aspect, methods of sequencing a protein or peptide with aptamers
complementary to fluorescently-tagged probes are provided. Such methods
typically
include: (a) providing a solid support comprising at least one protein or
peptide attached
thereto, wherein the at least one protein or peptide is attached to the solid
support via a
nucleic acid linker, wherein the nucleic acid linker comprises a sequencing
adaptor
sequence; (b) incubating the protein or peptide with a library of DNA aptamers
exhibiting
binding specificity toward at least one N-terminal amino acid under conditions
where one
or more aptamers bind specifically to at least one N-terminal amino acid of
the protein or
peptide, wherein each aptamer within the library comprises a series of one or
more
sequences that are complementary to optically labelled nucleic acid probes
that is
indicative of the sequencing round and the associated peptide binding ssDNA
region, and
wherein the probe hybridization regions are hybridized to a protective
complementary
oligo; (c) denaturing and washing off the protective complementary oligo; (d)
incubating
the bound aptamers with fluorescently -tagged oligo probes that are
complementary to
specific regions of the aptamer barcode tail; (e) detecting the unique optical
signature and
the position of the unique optical signature; (f) denaturing and washing off
bound probes;
(g) repeating steps (d) - (f) the required number of iterations; (h) removing
the aptamer
and removing the N-terminal amino acid from the protein or peptide to produce
an N-
11
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
terminal amino acid-shortened protein or peptide; (i) repeating steps (b) ¨
(h) a plurality
of times to construct a positional chain of optical barcodes; thereby
obtaining the
sequence of the protein or peptide.
In still another aspect, methods of using any of the protein sequencing
methods
described herein to identify novel biomarkers. Such methods typically include:
(a)
providing protein samples from biological samples of interest and control or
comparison
biological samples; (b) optionally removing known proteins of very high
concentrations;
(c) performing the steps (a) - (h) of the method of claim 1 or steps (a) - (i)
of claim 2; (d)
comparing the number of optical barcode reads associated with each lowly
expressed
proteins from control samples to samples of interest; thereby identifying
putative
biomarkers that have significantly different relative expression levels
between control
samples and samples of interest.
In another aspect, methods of using the protein sequencing methods described
herein are provided to evaluate disease state, evaluate response to treatment,
predict
treatment response, or combinations thereof, wherein one or more signs of
those diseases
is aberrant expression levels of known protein biomarkers. Such methods
typically
include: (a) providing protein samples from patient samples; (b) optionally
depleting
known proteins of very high concentrations; (c) performing the steps (a) - (h)
of method
in claim 1 or steps (a) - (i) of claim 2; (d) determining the relative
quantity of known
biomarkers by analyzing the number of optical barcode reads associated with
known
protein biomarkers; thereby determining the presence or absence of expression
level
deviations of known biomarkers from standard values.
In still another aspect, methods of using the protein sequencing methods
described
herein are provided to screen for potential antibodies. Such methods typically
include: (a)
providing plasma sample from immunized and naive biological samples; (b)
optionally
depleting known proteins of very high concentrations; (c) optionally isolating
immunoglobulins; (d) performing steps (a) - (h) of the methods described
herein or steps
(a) - (i) of the methods described herein; (e) comparing the number of optical
barcode
reads associated with each peptide from naive samples to immunized samples of
interest,
thereby identifying putative antibodies that have significantly different
relative expression
levels between naive samples and immunized samples of interest.
In some embodiments, the methods further include, prior to step (a),
fragmenting
the protein or peptide. In some embodiments, the fragmenting step comprises
12
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
fragmenting the protein or peptide with trypsin, another fragmentation enzyme,
or
combinations thereof
In some embodiments, the C-terminal end of the protein or peptide is attached
to a
solid support. In some embodiments, the C-terminal end of the protein or
peptide is
attached to an oligonucleofide tail. In some embodiments, the aptamer is
optionally
cross-linked to the N-terminal amino acid after step (b) and prior to step
(c).
In some embodiment, the protein or peptide is from a biological sample. In
some
embodiments, the biological sample is selected from the group consisting of
blood, urine,
saliva, tissue biopsy, sputum, stool, single cell, environmental samples,
bacterial swab, or
any sample containing peptides or proteins. In some embodiments, the protein
or peptide
is a full-length protein, a peptide fragment, or a protein or peptide
comprised within a
complex.
In some embodiments, the unique label is selected from the group consisting of
a
fluorophore, a dye, a nanolanthinade, and quantum dot. In some embodiments,
the
optically-labelled probes are oligos complementary to barcode sequences. In
some
embodiments, one or more oligo probes of one or more colors are hybridized to
the
aptamer barcode tail in the same iteration of probe incubation. In some
embodiments, the
detecting step is performed using optical imaging, total internal reflection
fluorescence
(TIRF), super-resolution microscopy, structured-light microscopy, widefield
microscopy,
or confocal microscopy.
In some embodiments, the aptamer library comprises of aptamers that are
partially
dsDNA in regions that are not related to aptamer binding. In some embodiments,
the
dsDNA is denatured and the protective complementary oligo is washed off. In
some
embodiments, the bound aptamer is cross-linked to the N-terminal amino acid
with PFA.
In some embodiments, the removing the aptamer step comprises cleaving the
aptamer
with a restriction enzyme. In some embodiments, the removing the N-terminal
amino
acid step comprises Edman degradation of the protein or peptide, cleaving the
protein or
peptide with one or more aminopeptidases, heat, pH, or combinations thereof
In some embodiments, the amino acid recognized by the members in the aptamer
library is a natural amino acid, an unmodified amino acid, and a modified
amino acid. In
some embodiments, the library of aptamers is produced using the RCHT-SELEX
method
described herein. In some embodiments, the aptamer exhibits binding
specificity toward
13
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
one N-terminal amino acid. In some embodiments, the aptamer exhibits binding
specificity toward two or more N-terminal amino acids.
In one aspect, methods of screening a library of DNA aptamers for protein or
peptide binding partners is provided. Such methods typically include: (a)
incubating a
plurality of proteins or peptides with a library of DNA aptamer candidates
that may
exhibit binding specificity toward proteins or peptides under conditions where
the
aptamers bind specifically to a protein or peptide within the plurality of
proteins or
peptides, wherein each protein or peptide in the plurality of proteins or
peptides
comprises a DNA bridge annealing sequence and a unique DNA barcode, wherein
each
aptamer within the library comprises a DNA bridge annealing sequence; (b)
incubating
the pool of barcoded proteins or peptides and DNA aptamer candidates with a
short
oligonucleotide bridge, wherein part of the short oligonucleotide bridge is
complementary
to the bridge annealing sequence at the 3' end of the aptamer and wherein an
additional
portion of the short oligonucleotide bridge is complementary to the bridge
annealing
sequence conjugated to the 5' peptide tail; (c) ligating the bridge annealing
portions of
each member of the aptamer library that are specifically bound to a
polypeptide to the
bridge annealing portions of those polypeptides joined by the oligonucleotide
bridge; (d)
amplifying aptamers within the library that are specifically bound to the
protein or
peptide; (e) repeating steps (a) ¨ (d) a plurality of times to identify
aptamers that exhibit
binding specificity toward each protein or peptide; and (t) sequencing the
annealed
aptamer and DNA barcode,; thereby identifying a plurality of polypeptides and
their
aptamer binding partners.
In some embodiments, the amplifying step comprises performing nested PCR. In
some embodiments, the method further includes, optionally, separating the
proteins or
peptides from their specifically-bound aptamers and purifying the aptamers
prior to step
(d). In some embodiments, the sequencing step uses a next generation
sequencing (NGS)
platform.
In one aspect, methods of generating a barcoded polypeptide are provided. Such
methods typically include: transforming an expression construct into
microorganism cells
under conditions in which about one construct is introduced into each cell,
wherein the
expression construct comprises nucleic acids encoding (a) a fusion protein
comprising the
polypeptide, a purification tag, and a nucleic acid-binding protein (naBP);
and (b) nucleic
acid sequence that is recognized by the naBP and a unique nucleic acid
barcode; and
14
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
culturing the microorganism under conditions in which the construct is
expressed, and the
naBP portion of the fusion protein binds to the naBP-recognition sequences,
thereby
producing barcoded polypeptides.
In some embodiments, the microorganism cells are selected from the group
consisting of eukaryotic or prokaryotic cells. In some embodiments, the method
further
includes purifying the barcoded polypeptides. In some embodiments, the
expression
construct comprises any copy number origin of replication compatible with the
host
organism. In some embodiments, expression is driven by any combination of
constitutive, inducible or repressible promoters compatible with the host
organism. In
some embodiments, the components of the system are expressed using distinct
promoters.
In some embodiments, the components of the system are expressed using the same
promoter present at different locations within the expression construct. In
some
embodiments, the components are expressed using Gal 1,10-bidirectional
promoter,
ADH1, GDS, TEF, CMV, EF la, SV40, T7, lac, or any other promoter and promoter
combinations compatible with the host organism.
In some embodiments, the purifying step comprises pulling down the barcoded
polypeptides with a pull-down assay corresponding to the encoded purification
tag. In
some embodiments, the immunoprecipitation step comprises pulling down the
barcoded
polypeptides with protein purification magnetic beads (such as anti-His,
agarose, nickel,
etc). In some embodiments, the method further includes eluting the barcoded
polypeptide
from the beads by using gentle elution buffers such as glycine to release the
fusion
peptide in the absence of denaturing the RNA-protein / peptide binding.
In some embodiments, the polypeptide comprises one or more of any site-
specific
protease cleavage sites such that the barcoded polypeptide is released from
anti-affinity
tag beads using site-specific proteases (e.g., enterokinase, Factor Xa,
Tobacco etch virus
protease, thrombin). In some embodiments, the nucleic acid sequence comprises
restriction enzyme cleavage site such that the barcoded polypeptide is
released from the
beads using restriction endonuclease.
In some embodiments, the nucleic acid sequence that is recognized by the
nucleic
acid binding protein and the nucleic acid binding protein are the MS2 RNA
hairpin or its
variants and MS2 phage coat protein or its mutants. In some embodiments, the
nucleic
acid that is recognized by the nucleic acid binding protein and the nucleic
acid binding
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
protein are the box.B sequence or its variants and the bacteriophage anti-
terminator
protein N (lambdaN).
In some embodiments, the cells are illuminated with UV radiation prior to
barcoded polypeptide purification. In some embodiments, the purified complexes
are
illuminated with UV radiation.
In another aspect, DNA barcoded polypeptides or proteins made by the methods
described herein are provided.
In one aspect, methods of generating dsDNA oligo with high control over
sequence content are provided. Such methods typically include: (a) ligating a
dsDNA
first position LEGO piece with a 5' phosphorylated one nucleotide overhang in
the
direction that the sequence is being extended to a dsDNA second position LEGO
piece
with a 5' phosphorylated one nucleotide overhang at each end, one of which is
complementary to the first position LEGO piece's overhang and one overhang
that is not,
with a dsDNA ligase, leaving one 5' phosphorylated one nucleotide overhang on
the
second position LEGO piece in the direction that sequence is being extended;
(b) ligating
the dsDNA second position LEGO piece to a dsDNA third position LEGO piece with
a 5'
phosphorylated one or more nucleotide overhang at each end, one overhang that
is
complementary to the second position LEGO piece's overhang and one overhang
that is
not, with a dsDNA ligase, leaving one 5' phosphorylated one nucleotide
overhang on the
third position LEGO piece in the direction that the sequence is being
extended; (c)
repeating steps (a) - (b) a multitude of times until sequence construct is one
LEGO piece
short of the desired length; and (d) ligating the sequence construct to a
dsDNA last
position LEGO piece with a 5' phosphorylated one nucleotide overhang in the
opposite
direction that the sequence is being extended.
In some embodiments, the 3' or 5' modification of the LEGO pieces are
compatible with the dsDNA ligase used. In some embodiments, for random library
generation, a heterogenous pool of LEGO pieces is used at particular
position(s) where
diversity is desired. In some embodiments, the double-stranded LEGO pieces are
enzymatically ligated using T4 DNA ligase, or any other dsDNA ligase that is
compatible
with the 3' or 5' end modification utilized by the selected ligase. In some
embodiments,
the ligation reaction is performed in solution, on beads, on a solid support,
in a gel, etc.
In some embodiments, the first position dsDNA LEGO piece is a PCR primer. In
some
embodiments, the last position dsDNA LEGO piece is a PCR primer. In some
16
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
embodiments, the dsDNA products are PCR amplified to generate a library with
replicates. In some embodiments, the post-PCR amplification dsDNA products are
digested to generate an ssDNA library.
In another aspect, methods of generating a ssDNA oligo with high control over
sequence content is provided. Such methods typically include: (a) ligating a
ssDNA first
position LEGO piece 3' end to the 5' end of a second position ssDNA LEGO
piece, with
one of the ends involved in the ligation being phosphorylated; (b) ligating
the ssDNA
second position LEGO piece 3' end to the 5' end of a third position ssDNA LEGO
piece,
with one of the ends involved in the ligation being phosphorylated; (c)
repeating steps (a)
- (b) a multitude of times until sequence construct is one LEGO piece short of
the desired
length; and (d) ligating the sequence construct to a last position LEGO piece.
In some embodiments, the 3' or 5' modification of the LEGO pieces are
compatible with the ssDNA or RNA ligase used. In some embodiments, single
stranded
LEGO pieces are enzymatically ligated using RtcB ssRNA ligase, CircLigase, or
any
other ssDNA or RNA ligase that is compatible with the 3' or 5' end
modification required
by the selected ligase. In one embodiment, the ligation reaction is performed
in solution,
on beads, on a solid support, in a gel, etc. In some embodiments, the first
position ssDNA
LEGO piece is a PCR primer. In some embodiments, the last position ssDNA LEGO
piece is a PCR primer. In some embodiments, the ssDNA products are PCR
amplified to
generate a library of double-stranded replicates. In some embodiments, the
post-PCR
amplification dsDNA products are digested to generate an ssDNA library.
In still another aspect, methods of generating an RNA oligo with high control
over
sequence content are provided. Such methods typically include: (a) ligating an
RNA first
position LEGO piece 3' end to the 5' end of a second position RNA LEGO piece,
with
one of the ends involved in the ligation being phosphorylated; (b) ligating
the RNA
second position LEGO piece 3' end to the 5' end of a third position RNA LEGO
piece,
with one of the ends involved in the ligation being phosphorylated; (c)
repeating steps (a)
- (b) a multitude of times until sequence construct is one LEGO piece short of
the desired
length; and (d) ligating the sequence construct to a last position LEGO piece.
In some embodiments, the 3' or 5' modification of the lego pieces are
compatible
with the RNA ligase used. In some embodiments, RNA LEGO pieces are
enzymatically
ligated using any RNA ligase that is compatible with the 3' or 5' end
modification needed
by the selected ligase. In some embodiments, the ligation reaction is
performed in
17
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
solution, on beads, on a solid support, in a gel, etc. In some embodiments,
the first
position RNA LEGO piece is a PCR primer. In some embodiments, the last
position
RNA LEGO piece is a PCR primer. In some embodiments, to generate an ssDNA
library,
the RNA products are reversed transcribed into cDNA, a second strand
synthesized with a
DNA polymerase, the dsDNA product PCR amplified, and the antisense strand
digested.
In still another aspect, oligo pools made by any of the methods described
herein
are provided.
Definitions
Nucleic acids can be single stranded or double stranded, which usually depends
upon its intended use. As used herein, an "isolated" nucleic acid molecule is
a nucleic
acid molecule that is free of sequences that naturally flank one or both ends
of the nucleic
acid in the genome of the organism from which the isolated nucleic acid
molecule is
derived (e.g., a cDNA or genomic DNA fragment produced by PCR or restriction
endonuclease digestion). Such an isolated nucleic acid molecule is generally
introduced
into a vector (e.g., a cloning vector, or an expression vector) for
convenience of
manipulation or to generate a fusion nucleic acid molecule, discussed in more
detail
below. In addition, an isolated nucleic acid molecule can be an engineered
nucleic acid
molecule such as a recombinant or a synthetic nucleic acid molecule.
Aptamers are single stranded nucleic acid sequences, which can be composed of
RNA, DNA, XNAs such as TNA, modified nucleic acids (such as substituting
natural
DNA nucleotides are substituted alternative functional groups (Chelsea et al.,
2019 and
Pfeiffer et al., 2017)), or other synthetic nucleic acid monologues. Aptamers
are typically
identified with a SELEX assay, which relies heavily on the evolution of a
diverse pool of
sequences amplified from round to round with PCR_ Aptamer sequences are
typically 20 -
45 base pairs (bp) plus additional flanking primer regions (typically 20-23 bp
in length
each for a forward and reverse primer). Capillary electrophoresis SELEX (CE-
SELEX)
does not rely on using aptamers with primer regions, however, CE-SELEX is
limited to
working with volumes in nL, thus limiting the initial starting pool of
sequences from
1014-1016 down to 108-109.
As used herein, a "purified" polypeptide is a polypeptide that has been
separated
or purified from cellular components that naturally accompany it. Typically,
the
polypeptide is considered "purified" when it is at least 70% (e.g., at least
75%, 80%,
18
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
85%, 90%, 95%, 01 99%) by dry weight, free from the polypeptides and naturally
occurring molecules with which it is naturally associated. Since a polypeptide
that is
chemically synthesized is, by nature, separated from the components that
naturally
accompany it, a synthetic polypeptide is "purified."
Nucleic acids can be isolated using techniques routine in the art. For
example,
nucleic acids can be isolated using any method including, without limitation,
recombinant
nucleic acid technology and/or the polymerase chain reaction (PCR). General
PCR
techniques are described, for example in PCR Primer: A Laboratory Manual,
Dieffenbach
& Dveksler, Eds., Cold Spring Harbor Laboratory Press, 1995. Recombinant
nucleic acid
techniques include, for example, restriction enzyme digestion and ligation,
which can be
used to isolate nucleic acids. Isolated nucleic acids also can be chemically
synthesized,
either as a single nucleic acid molecule or as a series of oligonucleotides
via traditional
methods such as bead purification, enzymatic digestion, column purification
etc.
Polypeptides can be purified from natural sources (e.g., a biological sample)
by
known methods such as DEAE ion exchange, gel filtration, HIS-tag bead pull-
down
assays, affinity chromatography, and hydroxyapatite chromatography. A
polypeptide also
can be purified, for example, by expressing a nucleic acid in an expression
vector. In
addition, a purified polypeptide can be obtained by chemical synthesis. The
extent of
purity of a polypeptide can be measured using any appropriate method, e.g.,
column
chromatography, polyaciylamide gel electrophoresis, or HPLC analysis.
A vector containing a nucleic acid (e.g., a nucleic acid that encodes a
polypeptide)
also is provided. Vectors, including expression vectors, are commercially
available or
can be produced by recombinant DNA techniques routine in the art. A vector
containing
a nucleic acid can have expression elements operably linked to such a nucleic
acid, and
further can include sequences such as those encoding a selectable marker
(e.g., an
antibiotic resistance gene). A vector containing a nucleic acid can encode a
chimeric or
fusion polypeptide (e.g., a polypeptide operatively linked to a heterologous
polypeptide,
which can be at either the N-terminus or C-terminus of the polypeptide).
Representative
heterologous polypeptides are those that can be used in purification of the
encoded
polypeptide (e.g., 6xHis tag, glutathione S-transferase (GST)).
Expression elements include nucleic acid sequences that direct and regulate
expression of nucleic acid coding sequences. One example of an expression
element is a
promoter sequence. Expression elements also can include introns, enhancer
sequences,
19
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
response elements, or inducible elements that modulate expression of a nucleic
acid.
Expression elements can be of bacterial, yeast, insect, mammalian, or viral
origin, and
vectors can contain a combination of elements from different origins. As used
herein,
operably linked means that a promoter or other expression element(s) are
positioned in a
vector relative to a nucleic acid in such a way as to direct or regulate
expression of the
nucleic acid.
Vectors as described herein can be introduced into a host cell. As used
herein,
"host cell" refers to the particular cell into which the nucleic acid is
introduced and also
includes the progeny of such a cell that carries the vector. A host cell can
be any
prokaryotic or eulcaryotic cell. For example, nucleic acids can be expressed
in bacterial
cells such as E. coil, or in insect cells, yeast or mammalian cells (such as
Chinese hamster
ovary cells (CHO) or COS cells). Other suitable host cells are known to those
skilled in
the art. Many methods for introducing nucleic acids into host cells, both in
vivo and in
vitro, are well known to those skilled in the art and include, without
limitation,
electroporation, calcium phosphate precipitation, polyethylene glycol (PEG)
transformation, heat shock, lipofection, microinjection, and viral-mediated
nucleic acid
transfer.
As used herein, "specifically" recognizes or "specifically" binds refers to a
molecule that exhibits high substrate specificity for a given target with very
low to no
substrate specificity for anything else within a known operating concentration
range.
As used herein, "semi-specifically" recognizes or "semi-specifically" binds
refers
to a molecule exhibiting high substrate specificity for a known target, and
medium to low
binding specificity to a subset of other targets
As used herein, "prefix" refers to at least the N-terminal amino acid and also
may
include the penultimate N-terminal amino acids at the N-terminal of a protein
or peptide.
As used herein, "suffix" refers to one or more amino acids in the peptide C-
terminal to the "prefix" amino acids as defined previously.
As used herein, "DNA barcode" refers to an oligo sequence with information
indicative of at least one molecule's identity. While barcodes are referred to
as constructs
of "DNA" throughout, the barcode molecules may actually comprise DNA, RNA,
XNAs,
modified nucleic acids or combinations thereof.
As used herein, "DNA barcode construct" refers to the strand of DNA comprising
at least two DNA barcodes.
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
As used herein, "Barcode Sequencing (RCS) compatible" aptamer refers to a
partially double stranded aptamer wherein one or more regions that do not
participate in
target binding can be hybridized with a complementary oligo, and may or may
not
contain an overhang.
As used herein, a "blocked aptamer" refers to a partially double stranded
aptamer
wherein at least the primer region of the aptamer but not the aptamer region
itself can be
hybridized with a protective complementary oligo.
As used herein, "sup-dill" refers to a method of removing DNA barcode
constructs of highly expressed proteins.
As used herein, "optical barcode" or "optical signature" refers to detection
of a
fluorescently-tagged molecule either integrated into the oligo directly or
attached via one
or more binders.
As used herein, "optical barcode" refers to an ordered combination of optical
signatures.
As used herein, "dsDNA lego piece" refers to a 5 or more base-pair-long DNA
oligo with a 5' nucleotide overhang (e.g., of one or more nucleotides) at one
or both ends,
where the 5'-most nucleotide on at least one strand is phosphorylated.
As used herein, "ssDNA lego piece" refers to a 5 or more nucleotide long DNA
oligo with a phosphorylated 3' or 5' end_
As used herein, "RNA lego piece" refers to a 5 or more nucleotide long RNA
oligo with a phosphorylated 3' or 5' end.
Unless otherwise defined, all technical and scientific terms used herein have
the
same meaning as commonly understood by one of ordinary skill in the art to
which the
methods and compositions of matter belong. Although methods and materials
similar or
equivalent to those described herein can be used in the practice or testing of
the methods
and compositions of matter, suitable methods and materials are described
below. In
addition, the materials, methods, and examples are illustrative only and not
intended to be
limiting. All publications, patent applications, patents, and other references
mentioned
herein are incorporated by reference in their entirety.
DESCRIPTION OF DRAWINGS
FIG. 1 is a schematic depicting how all the individual inventions described
herein
make up a pipeline of developing the PROSEQ platform
FIG. 2 is a schematic showing the two amino acid identity redundancy scheme,
21
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
wherein each di-peptide aptamer binding event provides the putative identity
of the two
N-terminal amino acids, while each round of degradation only removes one amino
acid,
thus allowing each amino acid except the original N-terminal amino acid to be
exposed to
two rounds of aptamer binding.
FIG. 3A is a schematic showing the steps in a representative conventional
SELEX
method.
FIG. 3B is a schematic showing the steps in one embodiment of the ML-SELEX
methods described herein.
FIG. 4A is a schematic showing that conventional SELEX methods can
undesirably enrich for aptamers that bind to components of the selection
process ("non-
specific high affinity binders") in addition to aptamers that bind to the
desired target
("specific high affinity binders").
FIG. 4B is a schematic showing that the addition of a negative selection step
in the
SELEX methods described herein can reduce the ultimate enrichment of aptamers
that
bind non-specifically to selection components by first pulling out aptamers
that bind to
the beads, biotin, oligo, or other selection components prior to Bring-Up
amplification or
input into SELEX.
FIG. 5A is a schematic demonstrating the various steps within the RCHT-SELEX
procedure (from FIG. 2) into which the single bring-up experiments, double
bring-up
experiments and/or in-experiment replicas can be incorporated.
FIG. 5B is a schematic demonstrating the single bring-up experiments, double
bring-up experiments, in-experiment replicates, and all-bead control
experiments that can
be used, in parallel or sequentially, during the RCHT-SELEX methods described
herein.
FIG. 6 is a schematic showing a bead-based multiplex version of RCHT-SELEX
that allows for selection of aptamers to multiple targets per experiment.
Aptamers
identified in a bead-based multiplex version of RCHT-SELEX can be de-
multiplexed in
the final round by incubating those aptamers separately with beads that are
conjugated to
only one of the initial targets.
FIG. 7 is a schematic of a method of identifying aptamers that bind
specifically to
an N-terminal amino acid prefix independent of the composition of the
peptide's suffix
tail by assaying aptamers in iterative rounds where the peptide suffix is
changed round to
round while the desired N-terminal amino acid prefix remains the same_ Four
types of
iterations are shown: dipeptide switch (Column 1), wherein the N-terminal
amino acids
22
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
remain the same while the suffix is switched; single amino acid switch (Column
2);
consistent peptide target (Column 3); complete switch or null (Column 4),
wherein
peptide targets differ completely between alternating rounds.
FIG. 8 is a schematic showing how lambda exonuclease can be used to convert
double-stranded (ds) DNA into single-stranded (ss) DNA. Lambda exonuclease
prefers
to degrading targets at an approximate ratio of 20:1 that are phosphorylated
on the 5' end.
Aptamers must be single stranded to fold and bind to peptides, so bound
aptamers are
PCR-amplified with specific protected/phosphorylated primers which produces
dsDNA,
then digested with Lambda exonuclease to convert amplified products such that
the
forward ssDNA aptamer survives.
FIG. 9A ¨ 9C are electropherograms displaying the extent of lambda
endonuclease digestion of the random aptamer library was monitored using Small
RNA
kits on Agilent's Bioanalyzer Chip System. Representative bioanalyzer profiles
are
shown that correspond to (A) dsDNA, (B) partially digested DNA and (C) ssDNA
aptamers. Data is represented on the right of each electropherogram in a gel-
like image,
with the green line representing the RNA marker. Confirmation of complete
conversion
to ssDNA occurred prior to the introduction of each aptamer library into each
new
RCHT-SELEX round.
FIG. 10A ¨ 10C is a schematic showing that oligonucleotide spike-in controls
and
fake experiments can be used in the SELEX methods described herein. Positional
spike-
ins added in specific wells of a 96-well plate can be used to determine local
contamination across wells (A). Different spike-ins are added at different
stages of
SELEX (i.e. prior to the Bring-Up, after each round of incubation before PCR
amplification, and in each NGS sample) to determine PCR bias at each step (B).
In Fake
SELEX, a pull from the bring up is incubated in the absence of beads and
targets and
PCR amplified (C).
FIG. 11A is a schematic showing threshold PCR, wherein similar concentrations
of DNA from different samples of varying concentrations are PCR-amplified to
ensure
similar amounts of input are introduced into each reaction in subsequent
rounds of
SELEX.
FIG. 11B is a graph displaying the expression intensities of every 8mer
combination from the sequencing runs of a DNA pool prior (above) and after
(below)
threshold PCR. The X and Y axes are every 4mer DNA sequence possible.
Comparison
23
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
of the expression intensities between the pools are extremely similar, with a
log variance
of 0.132.
FIG. 11C is a heat map reporting the log ratio of the division of expression
intensities of every 8mer combination from the sequencing runs of a DNA pool
after and
prior to threshold PCR in FIG. 11B. The minimal (black) signal demonstrates
that
threshold PCR can reduce the effects of compounding bias.
FIG. 12 is a schematic showing that primer switching can be used to select for
aptamers with binding affinities independent of the primer region.
FIG. 13 is a schematic showing the peptide sequencing methods described
herein.
Step 0 includes building the foundation consisting of a 5' phosphorylated
barcode
foundation, a forward and reverse colocalization linker, and a protein or
peptide target
(PT) tagged with a C-terminal oligonucleotide sequence oriented with the 3'
end
connected to the protein or peptide and a free, phosphorylated 5' end; Step I
includes the
tethering of the peptide-foundation complex on a solid substrate; Step 2
includes
incubating the bound proteins or peptides with a barcoded aptamer library
under
conditions that allow the appropriate aptamer to bind specifically to the
appropriate N-
terminal amino acid; Step 3 includes ligating the aptamer tail to a second
oligonucleotide
bound to the substrate; and Step 4 includes cleaving off the aptamer, leaving
the DNA
barcode associated with that particular amino acid bound to the second
oligonucleotide.
Upon removal of the N-terminal amino acid from the protein or peptide using
Edman
degradation and/or arninopeptidases, Steps 2-5 are repeated, generating a
chain of DNA
barcodes that can be used to identify each subsequent N-terminal amino acid.
FIG. 14 are schematics showing the construct of the aptamer tail and bridge
oligo.
FIG. 14A is a schematic depicting a Barcode-Specific bridge wherein the bridge
is
entirely complementary to the aptamer tail, including barcode region, except
for the 3'
single stranded overhang region. FIG. 14B is a schematic depicting a Universal
bridge
wherein the bridge is complementary to the restriction site spacer and
consensus sequence
only, both of which are conserved across all aptamers and flank the barcode.
FIG. 15A is a schematic showing the peptide or protein sequencing methods
described herein, where the peptide or protein sequence is determined based on
a DNA
sequence. Step 1 in this embodiment includes attaching the C-terminal end of a
protein or
peptide to a DNA primer oligonucleotide bound to a substrate; Step 2 includes
incubating
the bound proteins or peptides with a barcoded aptamer library under
conditions that
24
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
allow the appropriate aptamer to bind specifically to the appropriate N-
terminal amino
acid; Step 3 includes ligating the aptamer tail to a second oligonucleotide
bound to the
substrate; and Step 4 includes cleaving off the aptamer, leaving the DNA
barcode
associated with that particular amino acid bound to the second
oligonucleotide. Upon
removal of the N-terminal amino acid from the protein or peptide using Edman
degradation and/or aminopeptidases, Steps 1-4 are repeated, generating a chain
of DNA
barcodes that can be used to identify each subsequent N-terminal amino acid.
FIG. 15B is a schematic showing an example of the correlation between
individual amino acids and the corresponding aptamer barcodes.
FIG. 16 is a schematic showing an a priori and a non a priori sup-diff
strategy to
pull out DNA constructs associated with known targets, or unknown but high
concentration DNA constructs.
FIG. 17 shows examples of variations of steps within the PROSEQ platform.
FIG 18. is a heatmap showing the estimated percentage of human proteome
potentially identifiable for each binder library with up to 100 binders that
each bind to up
to 400 different dipeptides on the ProSeq platform wherein proteins are
digested at each
lysine, resulting in peptides of 12mer or less. Details of simulation to get
percent
proteome coverage for hypothetical binder sets are as follows: (a) proteins
are digested by
LysC into fragments, (b) a protein is identified when one of its fragments has
a matching
barcode that is distinct among the proteome, then one of its fragments is
uniquely
identified, (c) the set of dipeptide (pair of amino acids) that a binder has
affinity for is
randomly chosen from the 400 possible, (d) 20 sets of binders is randomly
chosen, (e)
given the binder set and the dipeptides each binder has affmity for, the
barcode read for
each protein fragment is determined and the number of uniquely identified
proteins is
determined, (f) 12 cycles of Edman degradation, binding, and barcoding are
performed on
each fragment. The simulation does not model noise (binders failing to bind
when they
should or binding where they should not). In the real system some noise will
be mitigated
by the redundancy in dipeptide reads and by reading multiple copies of the
same protein.
Additionally, only 20 possible sets were evaluated to obtain a percentage
match, so a
smoother curve would be expected for binder sets of less specificity.
FIG. 19 is a schematic showing the binding validation methods described
herein.
Step 0 includes building the foundation consisting of a 5' phosphorylated
barcode
foundation, a forward and reverse colocalization linker, and a target tagged
with a C-
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
terminal oligonucleotide sequence oriented with the 3' end connected to the
protein or
peptide and a free, phosphorylated 5' end; Step 1 includes the tethering of
the target-
foundation complex on a solid substrate; Step 2 includes incubating the
targets with a
barcoded putative library under conditions that allow putative binders to
targets; Step 3
includes ligating the oligonucleotide barcode tail to a second proximal
foundation
oligonucleotide barcode bound to the substrate; and Step 4 includes cleaving
off the
binder barcode tail, leaving the barcode associated with that particular
putative binder
ligated to the foundation oligonucleotide barcode. Optionally, upon removal of
the
putative binders from the tethered targets, Steps 2-5 are repeated, generating
a chain of
DNA barcodes that can be used to identify multiple binding events. Note that
binding
events are not restricted to N-terminal amino acids or attached target free
end, and can
occur at any exposed region of the target.
FIG. 20 is an overview of the peptide sequencing methods described herein,
where
the peptide or protein sequence is determined using fluorescence and
microscopy.
Peptide is tethered to known adaptor on chip (A). Library of fluorescent dye-
conjugated
aptamers, selected for specific N-terminal amino acid binding properties, is
flowed across
the peptides, incubated with targets and unbound aptamers are washed off the
chip (B).
The optical barcode of bound aptamers are imaged. For each round, a z-stack of
images
are taken in order to generate a spectral signature for the N-terminal amino
acid (C). N-
terminal amino acid on the fixed peptide is removed, the sample is washed and
the same
aptamer pool is flowed on to interrogate the newly exposed N-terminal amino
acid (D).
After repeating this series of steps on the slide, the identity of successive
N-terminal
amino acids at each round can be computationally deduce by comparing the
optical
barcodes for each peptide against the organism proteome (E).
FIG. 21 is a schematic showing one embodiment of the method described herein
in which proteins from cells are isolated and processed prior to tethering the
protein to a
solid substrate.. For example, cells (A) can be lysed and the proteins
isolated (B), and
denatured and digested (C). The side chains and N-terminus of the peptides can
be
protected (D), the C-terminal amino acid modified with an oligo or a linker
(E), and
tethered to a solid substrate. (F). Optically-labeled aptamers can be flowed
onto the
complex (G), an image captured, and the process repeated.
FIG. 22 is a schematic showing the construction of aptamers with regions to
bind
to complementary fluorescently-tagged oligos. The aptamers comprises of (a)
the
26
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
effective binding region, (b) an optional spacer, and (c) a barcode tail of
one or more
combinations of barcode units (BC) indicative of the probing iteration number
and
fluorescent tag, with each BC being complementary to a fluorescently-tagged
oligo.
There are two variations of barcode tail design: (1) BCs are spatially
separate and can
anneal with one or up to all unique complimentary probes at a time and (2) BCs
are
designed such that BC sequences overlap and can only anneal to probes
complementary
to non-overlapping BCs at a time. Note that BCs need not be spatially oriented
in
chronological order of probe incubation iterations (shown in picture) as the
BC sequence
itself contains the probing iteration number information.
FIG. 23 is a schematic showing the peptide sequencing methods described
herein.
Step 1 includes the immobilization of the peptide-oligo target on a solid
substrate; Step 2
includes incubating the bound proteins or peptides with a barcoded aptamer
library under
conditions that allow the appropriate aptamer to bind specifically to the
appropriate N-
terminal amino acid; Step 3 includes removing the protective complementary
oligo,
exposing the barcode region for probe annealing; Step 4 includes incubating
the incubated
with a library of probes that hybridize to barcode regions indicative of probe
iteration 1;
Step 5 includes washing off the unbound probes and imaging the bound probes;
Step 6
includes denaturing the bound probes from the aptamer and washing off the
probes off the
substrate; Step 7 includes repeating steps 4-6 for all the probe iterations
necessary for
aptamer identification. Upon removal of the N-terminal amino acid from the
protein or
peptide using Edman degradation and/or aminopeptidases, Steps 2-8 are
repeated,
generating a series of optical barcodes that can be used to identify each
subsequent N-
terminal amino acid.
FIG. 24 is a schematic depicting the methods for PROSEQ VIS described herein
when the library of aptamer probes consists of high affinity binders that bind
specifically
to a unique N-terminal amino acid prefixes. Single binding events that
indicate the
putative identity of the probed N-terminal amino acid prefix are observed by
detecting
aptamers that are directly conjugated to unique combination of dyes or a
combination of
dye-conjugated oligos hybridized to the aptamer. In Step 1, peptides are
localized to the
sequencing platform, and incubated with aptamers that recognize specific N-
terminal
dipeptides. In Step 2, each aptamer has multiple binding sites for dye-
conjugated binders.
These strong binders can simultaneously hybridize with the aptamer, and remain
bound.
The identity of the aptamer, and by extension that of the N-terminal amino
acid (SEQ ID
27
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
NO:121), is determined by evaluating the combination of colors detected at
each location.
In Step 3, aptamers are washed off and a new N-terminal amino acid exposed via
degradation. The cycle is repeated for the remaining amino acids (SEQ ID
NO:122).
FIG. 25 is a schematic depicting the methods for PROSEQ VIS described herein
when the library of aptamer probes consists of medium affinity binders that
bind non-
specifically to a set of N-terminal amino acid prefixes with variable
probability
distributions for each unique binding pair. Multiple binding events that
indicate the
putative identity of the probed N-terminal amino acid prefix are observed by
detecting
dye-modified aptamers over multiple cycles of incubation and wash off for
each. In Step
1, peptides are localized to the sequencing platform, and incubated with
aptamers that
recognize a set of N-terminal dipeptides. In Step 2, Dye-conjugated binders
hybridize to
a single stranded portion of the aptamers, but because they are 'weak'
binders, they lack
specificity of a stronger binder. The dye-conjugated binders fluorescing at
each peptide
location is tracked over cycles to determine accuracy of call rate of amino
acid. Can be
used with either individual color or optical barcode. In Step 3, the identity
of N-terminal
amino acids at each round is computationally deduced by comparing the observed
combination of fluorescent signals against probability distribution of binding
events for
each aptamer to each N-terminal amino acid prefix (SEQ ID NO:123).
FIG. 26A ¨ 26C are schematics showing the MULTIPLEX methods described
herein. An aptamer library (A) is incubated with a diverse pool of unbound DNA-
barcoded protein or peptide targets (FIG. 188). Upon aptamer binding to a
barcoded
target, the 3' end of the single stranded aptamer is joined to the ssDNA
barcode that is
specific to target identity by an ssDNA bridge that is half complementary to
the 3' end of
the aptamer and half complementary to the 5' end of the ssDNA barcode (C). The
nick
between the aptamer and ssDNA peptide barcode can be ligated and sequenced
through to
obtain the aptamer sequence and peptide barcode, which, in turn, provides the
target to
which the aptamer was bound.
FIG. 26D is a schematic that indicates the steps of the SELEX procedure (from
FIG. 3) into which multiplexing can be incorporated.
FIG. 27 is a schematic of a peptide-oligonucleotide conjugate (POC), which
includes a single-stranded (ss) DNA tail (a) whose 3' end is covalently linked
to the C-
terminus of a peptide or protein target (b). The ssDNA tail (a) includes a 3'
primer region
(c), a unique DNA barcode (d), and a 5' bridge-binding sequence (e). An
aptamer (t)
28
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
includes a 3' bridge-binding sequence (g). A short oligonucleotide bridge (h)
where half
is complementary to the 3' bridge-binding sequence (g) at the 3' end of the
aptamer (t)
and the other half is complementary to the 5' bridge-binding sequence (e) of
the ssDNA
tail (a) can be used to ligate the aptamer (f) to the peptide (b).
FIG. 28 is a schematic of the Nested PCR technique in MULTIPLEX.
FIG. 29 is a schematic showing the barcoded (1)) protein of interest (POI) (A)
complex that is produced in vivo in the TURDUCKEN methods described herein.
This
approach exploits the non-covalent interactions between an RNA-binding protein
(B) and
its corresponding binding site (C).
FIG. 30A - 30C are schematics showing one embodiment of the TURDUCICEN
methods described herein. A pool containing the plasmids of various protein of
interest
(POI)-RNA binding protein (RBP) fusion genes as well as their corresponding
RNA
barcode sequence are transformed into cells at an approximate dilution of 1
plasmid per
cell (A), the POI-RBP fusions are expressed and bind their corresponding RNA
barcodes
(B), which then are purified (C).
FIG. 30D is a schematic that indicates the steps of the SELEX procedure (from
FIG. 3) into which the TURDUCICEN methods can be incorporated.
FIG. 31A - 31B are schematics showing embodiments of LEGO methods
described herein for dsDNA (A) ligation, and ssDNA and RNA ligation (B).
FIG. 32A - 32C are schematics showing one embodiment of the LEGO methods
described herein. Pools of first position, second position, third position,
etc. LEGO pieces
(A) are sequentially ligated (B) and PCR amplified to generate replicates. The
resulting
dsDNA is then digested into ssDNA to form a library of folded aptamers (C).
FIG. 32D is a schematic that indicates the steps of the SELEX procedure (from
FIG. 3) into which the LEGO methods can be incorporated.
FIG. 33 is a schematic of the general workflow of all SELEX (RCHT-SELEX and
NTAA-SELEX) experiments.
FIG. 34A is a schematic that depicts the 400 potential amino acid prefixes
that the
SELEX methods described herein is used to find aptamers for PROSEQ and PROSEQ
VIS.
FIG. 34B is a schematic that depicts how the 400 potential amino acid prefixes
are
organized into 16 blocks.
FIG. 34C is a schematic that depicts how the suffix paired with the 2-mer
prefix
29
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
was alternated between odd and even rounds, with only the 2-mer prefix the
constant
peptide combination through all 4 rounds.
FIG. 34D is a specific example of how the suffixes ("backbone") are switched
in
alternating rounds while the prefix remains the same to find aptamers specific
to the DD
and DC prefix regardless of the suffix (DD/DD, SEQ ID NOs:124-127; DC/DC, SEQ
ID
NOs:128-131; DD/DC, SEQ ID NOs:132-135). The same bring up is also used to
assay
targets with alternating backbones and prefixes that are similar to tease out
aptamers that
are not specific to DD and DX.
FIG. 35 are embodiments of the three variations of SELEX aptamer incubation
(Variant 1-3) with peptides compared to RCS conditions (RCS).
FIG. 36 is a graph displaying the log ratio of expression levels of every 12-
mer
combination from the sequencing runs of DNA pools after bringup divided by
expression
levels prior to the bringup for 96 conditions, two of which failed (two bottom
right
panels). The X and Y axes of each panel are every 6-mer DNA sequence possible.
Panels with high ratios of red or blue demonstrate increased variance from a
Gaussian
distribution, indicating that the experimental conditions perturbed the random
input pool
further from ifs input condition.
FIG. 37 are two tables displaying the sequences and read counts of the top 20
most common sequences from a random sampling of 100,000 reads in the aptamer
pool
after one round of Fake SELEX and SELEX. Sequences derived from Fake SELEX
(SEQ ID NOs:136-155) are all different from the sequences from SELEX (SEQ ID
NOs:156-175), suggesting that aptamers pulled down by peptide targets exhibit
greater
affinity than random sequences.
FIG. 38 is a table exhibiting the counts of replicate sequences between any of
9
experiments, 3 replicates experiments for 3 targets, performed with the same
bringup
pool. All replicates for a rounds were merged together and non-specific
aptamers were
filtered from the counts by bead control subtraction. Counts highlighted in
red are counts
of the same sequences that were found in experiments of differing targets.
BRADY1r5
means target bradykinin, position 1, replicate 1 and SELEX round 5. GNRH4r5 is
target
GnRH, position 4, replicate 1, and SELEX round 5. Sequence contamination
occurs
across nearest neighbor replicates, indicated by the red regions, which was
significantly
reduced after altering automation protocols and target position on the plate.
FIG. 39 are two examples of aptamers selected using RCHT-SELEX methods
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
herein to small peptides: one to vasopressin (SEQ ID NOs:176-179) and one to
bradykinin (SEQ IL) NOs:180-183). Aptamer structures are the lowest Gibbs free
energy
structures obtained by IDT's licensed UNAFold software.
FIG. 40 reports the top 5 aptarner sequences that are serially enriched
specifically
in the presence of peptides with an N-terminal lysine (SEQ ID NOs:184-188) or
N-
terminal cysteine (SEQ ID NOs:189-193) prefix identified in peptide Switch ML-
SELEX
experiments. These results indicate ML-SELEX's capability to find unique
aptamers to
individual amino acids.
FIG 41A. is a schematic diagram of the N-terminal Amino Acid SELEX
experiment strategy of Example 2. 12 selections comprising replicates of each
target
mixtures were run for 5 rounds in parallel. The workflow begins with a
negative
selection against streptavidin beads on an initial pool of ssDNA and split
across 12
random pools. 2 parallel selections were performed on each control reference
target and
3 parallel selections on the target (Proline-Proline) with and without the
switching of
backbones (C and D backbones) in alternating rounds. A representative pool of
ssDNA
from every round of every selection was sequenced and analyzed for round-to-
round
enrichment of sequences.
FIG 41B reports the target compositions and amino acid sequences (SEQ ID
NOs:194-203) in Non-Switch and Switch SELEX.
FIG. 42 reports the sequencing counts of the top 10 most enriched sequences
per
round. X axis is the round of SELEX, Y axis is the number of counts seen
during
sequencing for the 10 sequences. The 10 sequences displayed were chosen
because their
calculated enrichment values.
FIG. 43A is a box plot summarizing the enrichment of the top aptamers for each
target. Specifically, enrichment was calculated from round 2 to round 5. Each
boxplot
shows the summary (minimum, first quartile, median, third quartile, and
maximum) for
the top ten aptamers from each selection performed for the given target. Total
number of
sequences for Backbone, Brady, Beads = 20, Total number of sequences for PP-C
and
PPCD = 30). X axis is in log scale and shows the enrichment. Y axis is the
target of each
selection. The median enrichment for PPCD switch is higher than the negative
control
(Beads), but lower than the positive control (Brady).
FIG. 43B is a categorical scatter plot reporting differences in enrichment
among
the top most enriched sequences for each selection for each target. Two
selections were
31
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
performed for Backbone, Beads and Brady each. Three selections were performed
for
PPC and PPCD. (Total number of sequences for Backbone, Brady, Beads = 20,
Total
number of sequences for PPC and PPCD = 30). Y axis is target, x axis is
enrichment
(pen_growth). For some selections/replicates (rep), higher enrichment was seen
for the
same target. For example, high enrichment ( >3, equivalent to 1000-fold) was
seen for 3
unique sequences in rep 2 while only 1 unique sequence in rep 1 in the
selections
performed for the target Backbone.
FIG. 44 is a confusion matrix of top 10 enriched sequences for each replicate
(rep)
of each target (Backbone, Beads, Brady, PP-C, PP-CD). 0 indicates no sequence
overlap
between two selections, (indicates one sequence overlap, etc. -1 indicates the
same
selections. Within these selections, it is observed that there is some overlap
of sequences
(1-2 sequences) . This information can be incorporated into final candidate
selection.
Candidate aptamers for PP-CD can be chosen to have no overlap with other
control
targets (Backbone, Beads, Brady) but it is permissible to choose candidates
that may
recognize PP-C and PP-CD switch, as these may recognize the PP on the N-
terminal.
FIG. 45 is the results of a single point binding assay for 10 potential
aptamer
candidates. Binding, indicated by fluorescent signal (y axis) was measured for
10
aptamers at 100 nM. Apt 4 shows higher binding than the controls (non-aptamer
and
buffer) for target PP-C. Apt 1,2,3,4,7,8,9 show higher binding than controls
for PP-D.
Data was normalized to the positive control (FAM conjugated directly to
beads).
FIG. 46A and 46B are binding curves for Apt 1 and Apt 4 respectively. Apt 1
(Panel A) shows increasing signal against PP-13, much greater than against PP-
C. It looks
to saturate against PP-C, while not saturating against PP-D, indicative of non-
specific
binding. Apt 4 (Panel B) shows saturation binding against PP-D and no binding
against
PP-C.
FIG. 47 is an example of an electropherograrn from the Agilent Bioanalyzer
assay
with a desirable peak shape at 60 seconds, indicating proper digestion of PCR
products
into ssDNA.
FIG. 48 is an example of an electropherogram from the Agilent Bioanalyzer
assay
with a desirable peak shape indicating most products are of the desired length
(86nt for
the examples described herein).
FIG. 49 is a schematic of the BCS core sequencing unit.
FIG. 50A is a heatmap reporting the counts of reads of barcodes added in each
32
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
cycle, each with an expected position on the barcode construct, at each
position on the
barcode construct for 12 cycles of barcode ligation. In a ideal situation,
barcodes added
in the nth cycle should be in the nth position on the barcode construct. In
the event of x
failed ligations or no aptamer binding event, a barcode would be observed in
the (n-x)th
position. Results confirm it is possible to achieve serial ligation of 12
barcodes in the
expected positions. Note, barcodes used in cycles 1-6 are repeated in the same
order in
cycles 7-12 and results were not de-multiplexed; thus a small fraction of
counts from each
boxed number from Expected Cycles 1-6 may be attributed to the cell five cells
to its
right (marked with *), meaning no barcodes were not ligated until at least
after cycle 6 for
those sequences.
FIG. 50B is an arrow plot depicting successful ligation of 3 barcodes in a row
in 3
cycles of ligation mediated with a universal bridge design, confirming serial
ligation is
possible with universal bridges.
FIG. 51 are heatmaps reporting the instances of each target foundation
sequenced
with the aptamer barcode ligated to it. FIG. 51A reports total counts (SEQ ID
NOs:204-
243), while FIG. 51B reports the normalized percentage (SEQ ID NOs:244-279).
Argipressin aptamers (highlighted in red) identified through RCTH-SELEX show
specificity for argipressin over bradykinin targets and peptide targets with a
DD N-
terminal (DD targets), as their barcodes are ligated on all types of
argipressin foundations,
but to little to no empty controls, bradykinin, and DD target foundations.
FIG. 52 are fluorescent images of a flow cell with bradykinin attached to its
surface prior to Edman Degradation and after 2 cycles of Edman Degradation.
Flow cells
were probed with fluorescent bradykinin antibody and imaged through the 555
channel.
Diminishing but not absent signal indicates decreased antibody binding, which
may
suggest peptides are partially degraded while still remaining attached to the
flow cell
surface.
FIG. 53A is a 100% stacked column chart depicting the distribution of RNA
baits
complementary to 5 different sequences (9, 13, 11, 12, 19) generated from an
original
pool of 0.000125% sequence 9, 0.01% sequence 13, OA% sequence 11, 10% sequence
12,
and 89% sequence 10 by weight with various concentrations of in vitro
transcription
enzyme (IVT). Changes in frequency of RNA bait sequences indicate that
treatment with
varying concentrations of IVT can generate different ratios of RNA bait
sequences.
FIG. 53B is a table reporting the percentage of each RNA bait sequence by
count
33
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
generated with various concentrations of IVT.
FIG. 54 is an image of an electrophoretic mobility shift assay (EMSA) gel
demonstrating that Spot-tag nanobody was conjugated to oligos (VH1-1-oligo).
The first
four gel lanes show electrophoretic mobility of unconjugated Spot-tag nanobody
by itself.
In subsequent lanes, multiple higher molecular weight bands were observed on
the gel,
presumably corresponding to multiple oligos conjugated to a single nanobody.
FIG. 55 is a schematic of the full core sequencing unit constructs of each
target
and their corresponding structures ligated onto the sequencing chip after
ligation and
formamide wash. The DNA targets serve as positive controls. 5'Phos.01 control
is for
noise associated with the full oligo tail that is connected to all peptide
targets, while the
CLR.Null.Block.Br control is for noise associated with sequencing chip
components.
FIG. 56 is a heatmap reporting the instances of each target foundation
sequenced
with the binder barcodes ligated to foundations when Spot-tag nanobodies are
conjugated
to oligos. Controls are run in triplicate with different barcodes associated
for each
replicate, and DNA and Spot-tag experiments are run with 6 experimental
replicates.
DNA controls (Kd pM) bound and tagged complementary oligos with high fidelity
(in
terms of sequencing counts), and the Spot-tag nanobody bound and tagged the
Spot-tag
peptide (Kd 6nM) with strong fidelity. Difference in sequencing counts between
experimental replicates is thought to be due to the difference in barcode used
for each
replicate. The impact of barcode sequence was screened and analyzed to derive
a set of
barcodes used for downstream experimentation. No known variables (GC content,
sequential basepairs, etc.) were found to be related to a barcode's impact on
sequencing
noise outside of target type (DNA vs Nanobody, etc). Experiments were repeated
and
validated, confirming the protocol utilization for a DNA:DNA binding system
and
peptide:nanobody binding system.
FIG. 57 is a heatmap reporting the instances of each target foundation
sequenced
with the binder barcode ligated to foundations when Spot-tag nanobodies were
not
conjugated to oligos. Experiments are run in triplicate with different
barcodes associated
for each replicate. Difference in sequencing counts between experimental
replicates is
thought to be due to the difference in barcode used for each replicate. The
impact of
barcode sequence was screened and analyzed to derive a set of barcodes used
for
downstream experimentation. No known variables (GC content, sequential
basepairs,
etc.) were found to be related to a barcode's impact on sequencing noise
outside of target
34
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
type (DNA vs Nanobody, etc.). For this experiment, only the DNA binder,
AV.84.U2. SA4.2, with its corresponding target (SP9) have high sequencing
counts.
Experiments were repeated and validated, confirming the protocol utilization
for a
DNA: DNA binding system and peptide:nanobody binding system,
FIG. 58 are embodiments of results and computational deconvolution process
from imaging to peptide identification for a single molecule peptide. FIG. 58A
is an
embodiment of a series of images generated by four iterations of probe
incubation for a
single peptide molecule at location (X,Y) on a chip. FIG. 58B is a table
reporting the
fluorescent signal observed by each channel (350, 433, 532, 555, 647) that
reflect the
results of FIG. 58A. Colored regions indicate signal above a noise threshold,
which
together make up the optical signature of the bound aptamer. FIG. 58C is an
embodiment
of a lookup table matching each aptamer identity to the optical signature
observed
through multiple iterations. FIG. 5813 is an embodiment of the series of
aptamers
observed at location (X,Y) on a chip computed from 8 rounds of aptamer
incubation.
Overlapping N-terminal acid amino calls from the two amino acid identity-
redundancy
scheme are indicated in black while disagreeing calls are indicated in red.
FIG. 58E is a
schematic of a sequence calling strategy wherein the computed sequence
generated by the
peptide sequencing methods described herein is matched to a database of known
peptides
or a reference proteome.
FIG. 59 are images of fluorescent bead-streptavidin conjugates on a glass
slide
(single molecule control) and bound to single oligos on a sequencing chip at
20x, 60x,
and 100x magnification. The similarity of sizes of the observed spots between
the
fluorescent beads on the chip and sequencing chip suggests the observed spots
on the
sequencing chip are single molecules.
FIG. 60A are fluorescent images of fluorescent bead-streptavidin conjugates on
a
sequencing chip and the intensity measurement after background subtraction
using a local
threshold. The threshold value is the median intensity for the local
neighborhood (30 by
30 pixel) of pixels.
FIG. 60B are thresholded intensity distribution of all the fluorescent spots
in FIG.
60A.
FIG. 61 is a heat map reporting MULTIPLEX selectivity performance. hi a five
target (GNRH, NC2, NC3, Ti, Vaso) assay, aptamers were first filtered for
abundance (at
least 12 reads) and the top 5 sequences to each target were ranked based on
selectivity
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
(reads to the desired target! reads to all targets). Off-target hits are
shown, with the
selectivity highlighted by the red (low specificity) to blue (greater
specificity) color
gradient. The top 5 target-specific aptamers for each target exhibits 0.500 to
0.923
selectivity, indicating that at least half of the reads of each aptamer was
bound to its
intended target. In comparison, no more than 25.0% of the reads of the same
aptamers
were bound to any individual unintended target.
FIG. 62 illustrates the peptide target sequences used in MULTIPLEX experiments
(SEQ ID NOs:280-285).
FIG. 63 is an image of an SDS-PAGE gel showing that denatured peptides
purified using an Anti-His affinity pull down assay were of the expected size
for dMS-
EmGFP and dMS2, indicating that both dMS-EmGFP and dMS2 were expressed. BSA
was included as a standard.
FIG. 64 is an image of an electrophoretic mobility shift assay (EMSA) gel
demonstrating that dMS2-EmGFP fusion protein bound to 2 nM RNA (protein
concentrations used in binding reaction indicated on the top, nM) containing
the MS2
coat protein binding site.
FIG. 65 is an image of an EMSA gel showing that the dM52 proteins (without
EmGFP) bound to ¨2 nM RNA (protein concentrations used in binding reaction
indicated
on the top, nM) containing the MS2 coat protein binding site, verifying the
identity of
dMS2 proteins.
FIG. 66 is a violin plot displaying the percentage of sequences from each
experiment that were the desired full length constructs using lOmer dsDNA
pieces with 1
base pair overhangs, one of which reached 78.9% efficiency.
FIG. 67 reports the percentages of unique sequences produced in LEGO
experiment 87P from FIG. 66, wherein 78.9% of the constructs were sequences of
the
desired length, order, and orientation of lego pieces.
DETAILED DESCRIPTION
This disclosure describes methods and compositions that form a pipeline of
developing and using a protein sequencing platform which utilizes aptamers
that bind
specifically to N-terminal amino acids (FIG. 1). The protein sequencing
methods
described herein primarily rely upon aptamers having a variety of different
features
depending upon the particular application. For example, amino acid-specific
aptamers
36
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
can be generated using the novel methods described herein (RCHT-SELEX or NTAA-
SELEX). Such amino acid-specific aptamers can be used to recognize, identify
and via a
nucleic barcoded region convert 1-2 amino acid residues of a protein or
peptide into a
DNA sequence (PROSEQ), or such amino acid-specific aptamers can be generated
and
used to recognize and identify, based on a visual signal, each amino acid of a
protein or
peptide (PROSEQ-VIS). In addition, many target-specific aptamers can be
generated
simultaneously, and used to produce and screen a large multitude of binders
(MULTIPLEX). Simultaneous and specific aptamer selection relies on robust
identification of targets. Nucleic acid barcoded target generation can be
accomplished i n
vivo via a non-covalently bond between a peptide or protein using an RNA-
binding
protein and its corresponding recognition sequence (TURDUC10EN). Lastly,
successful
SELEX experiments require that aptamers with some specific binding preference
and
affinity for the molecular target be included in the original pool of 1014-
1015 candidate
sequences, which is only a small fraction of all of the DNA sequences
possible.
Machine-learning (ML) can help to optimize experimental seed binders, so
unlike
conventional SELEX experiments, optimal binders do not need to occur in the
experimental dataset. The ability to construct computationally-derived,
customizable
DNA libraries to perform SELEX screens using a controlled input pool can
drastically
customize the exploratory space by systematically assaying aptamer candidates
that
include sequences with known effective binding properties (LEGO).
Aptamers
Aptamers are short, single-stranded nucleic acid strands, which can be
composed
of RNA, DNA, modified nucleic acids, or other synthetic nucleic acid
monologues, that
fold into unique conformations that allow for binding specificity to
biological targets such
as proteins and peptides (Mckeague & Derosa, 2012), Aptamers are used to
examine
binding interactions involving molecular targets in a number of research areas
including
drug development, diagnostics, imaging, and basic science. Specifically,
aptamers bind
to targets with high specificity and affinity, can be generated and modified
more quickly
and at a lower-cost than antibodies, have a wider range of potential targets
than antibodies
(Thou & Rossi, 2016), and are less likely to provoke immunologic side effects
than
antibodies (Bouchard, Hutabarat, & Thompson, 2010). However, aptamers have not
experienced widespread success in clinical or industrial uses due, in large
part, to the
37
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
laborious nature of discovering and identifying aptamers with desired binding
characteristics (Zhou 8c Rossi, 2016). Additionally, aptamers discovered in
isolated
environments (i.e. selected against purified targets) exhibit high binding
affinity in the
experimental conditions, but fail to bind to its intended target in in vivo
conditions (Chen,
et al., 2016). The present disclosure provides methods of making and using
aptamers
having very specific binding characteristics to amino acid residues at the N-
terminal end
of a peptide chain.
Aptamers with a high peptide binding affinity have an increased chance of
binding
and of generating a binding event record over aptamers with lower binding
affinities.
Aptamers that are specific only bind to a small number of possible peptides
and, as such,
generate records that are informative about which molecules are present. Thus,
aptamers
with high affinity (lcuis <30 nM) and specificity (10x binding preference
desired target
over other targets) are desired for the protein sequencing technologies
herein, however,
sets of aptamers having various affinities can be used to retrieve information
'bits' about
the protein sequence (i.e. PROSEQ AND PROSEQ-VIZ). In end-to-end simulations,
results suggest that aptamers of only moderate binding affinity (1(.4is
nM) and
selectivity will enable us to accurately quantify mixtures of known proteins
with relative
ease. For non de novo applications, PROSEQ and PROSEQ-VIZ technologies can use
a
proteome map to resolve any resolution gaps in the data. Additionally,
subsequent cycles
can be repeated prior to removing the amino acid to allow for additional bits
of
information to be obtained before cleavage. Finally, if PROSEQ and PROSEQ-VIZ
are
restricted to aptamers that selectively bind to N-terminal dipeptide prefixes,
highly
specific aptamers are not necessary even for de novo sequencing. The noise
from the
reduction in specificity is offset by the additional observed binding events
resulting from
the two-amino acid identity-redundancy scheme, since it allows for the
observation of
two binding events per amino acid (except for the first N-terminal amino acid)
to confirm
its identity (FIG. 2). Each dipeptide aptamer binding event provides insight
towards the
identity of the two N-terminal amino acids, while each round of degradation
only
removes one amino acid, thus allowing each amino acid except the original N-
terminal
and C-terminal amino acids (which will only be read once) to be exposed to two
rounds
of aptamer binding. In the event of amino acid identification errors,
downstream
computation algorithms can be used to correct or detect inaccurate readbit
results with a
certain level of confidence.
38
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Robust & Compressed High Throughput - Systematic Evolution of Ligands by
Exponential Enrichment (RCHT-SELEX) and N-terminal amino acid SELEX (NTAA-
SELEX)
Systematic Evolution of Ligands by Exponential Enrichment (SELEX) is a known
high-throughput screening (HTS) process that has been used to identify
aptamers that
bind to a specific target ligand in in vitro selection (Tuerk & Gold, 1990).
Conventional
SELEX protocols typically include screening a diverse and random
oligonucleolide
library against a single peptide or protein target by flowing aptamers onto
bead-bound
targets and eliminating weak binding aptamers through multiple rounds of
selection
where weak binding aptamers and non-binding aptamers are washed away (Blind &
Blank, 2015).
Conventional SELEX methods begin with the synthesis of about 1014-1015 unique
sequences for oligonucleotide libraries, followed by 10-20 iterative rounds of
a) single
target incubation with a random pool of candidate aptamer sequences to promote
aptamer
/ target binding, b) separation of target-bound ofigonucleotides from unbound
sequences,
and c) amplification and characterization of bound aptamers (FIG. 3A). Several
variations of the original SELEX method, such as capillary electrophoresis
SELEX (CE-
SELEX), microfluidic SELEX, and CELL SELEX, have been developed to fulfill
different research needs.
The goal of conventional SELEX methods has been to increase the binding
affinity of aptamers identified through experimental screening. Conventional
SELEX
methods for identifying aptamers suffer from two main problems that prohibit
large-scale
screening:
= Conventional SELEX methods rely on a repeated screening process, in which
experimental error can be compounded in every subsequent round of screening.
For example, in each round, aptamers undergo PCR amplification, DNA
cleanup, and conversion from double-stranded to single-stranded DNA via
separation or enzymatic digestion. Variability in one or more of these
processes across rounds and/or experiments can encourage the biased selection
of an aptamer pool engineered to withstand the selection process of a specific
experimental setup.
39
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
= The lack of parallel selections with the same input library against
controls and
replicates prevents: (a) inter-experimental and intra-experimental comparison
respectively, (b)signal-to-noise ratio analysis, and (c) ground truth
measurements, all of which complicate downstream computational analysis,
data cleaning and the application of predictive modeling such as ML. Models
are attracted to the strongest signal, regardless of source. Often in the case
of
biology experiments, there exists operator error/noise, instrumentation noise,
biological processes noise and noise due to the handling of physical reagents
(i.e. contamination), and the combination of all of these different elements
of
noise can often drown out the experimental signal. As a result, models will
often predict based on a very noisy signal, unless they are trained in advance
to
the different noise elements. To this end, several different features were
designed (bringup, replicates, spike-in controls, Fake SELEX, etc.) to
calculate
and remove the noise during pre-model data processing, or to train the models
on the elements of noise during the prediction stage. Additionally, there are
several classes of models that have limited predictive capabilities outside of
the
linear range, and, often in biology, processes are nonlinear (e.g. such as
PCR).
Linear models have an advantage as they are well-studied, computationally
inexpensive and often give robust predictions. However, when applied to non-
linear datasets, linear models can often give improper predictions. On the
other
end of the spectrum, non-linear modeling approaches can be computationally
more expensive and also are subject to overfilling (ea!, polynomial modeling
on sparse data), but are often required to be utilized when linear models fail
to
describe data sets accurately. As a result, numerous unit tests were run to
calculate the regions of linear and non-linear processes in order to best
determine which type of modeling approaches can be applied.
= Conventional SELEX methods allow for the screening of an aptamer pool
against only one peptide or protein target at once. That is, each protein or
peptide target must be screened in isolation to be able to identify the target
Therefore, screening against 1,000 peptide targets using conventional SELEX
methods would require 1,000 individual SELEX experiments, each involving
multiple rounds of screening.
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
In addition, for a 40-mer ssDNA oligo, for example, there are 1024 possible
oligos
that could be produced, and an exploration of 1012'15 of the total possible
experimental
space can result in difficulty finding a unique aptamer to a target.
Currently, there are
numerous bathers to efficiently screening such a large volume of candidates:
= low hit rate: successful SELEX experiment requires that aptamers with a
high affinity for the molecular target be included in the original pool of
10"4-
10" candidate sequences. For a 10'4 sample, only 8.27 x 104' % of the
experimental space of possible DNA sequences is being explored, such that
in practice, even the most optimized experiment has a high probability of
failure.
= time-prohibitive: generally takes >6 months to a year to identify
specific
aptamer candidates.
= non-specificity: traditional SELEX experiments incubate candidates with
one target at a time, which only demonstrates the candidate aptamers'
relative affinity, but not their specificity in a competitive environment
= inability for direct comparison: since most experiments start with a new
random pool of input oligos, direct comparison across experiments is
impossible.
= difficulty in translating to environments differing from discovery
conditions:
translation of discovered aptamers can also be fraught with difficulties, due
to their sensitive structural nature that is correlated with their discovery
environment. Since structure informs function, aptamers selected in a
particular environment may not fold and bind to their target in the same
manner when conditions differ from experimental ones.
There are two significant gaps in current SELEX protocols. No existing method
is
tailored to accommodate large scale computational analysis for multiple
targets between
every round, for the purpose of using experimental data to supplement
computationally-
derived aptamers. If a working protocol existed, then empirical datasets could
smoothly
integrate with machine learning analysis and prediction pipelines, allowing
for in silico
prediction of aptamers to targets. Computationally predicted aptamers would
allow for
exploration across a wider range of sequences for optimal aptamer targets and
also save
resources and time in aptamer search queries. Also, SELEX protocols lack the
precision
and resolution to discover binders high-resolution for aptamer candidates that
bind to a
41
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
small portion of a larger target and can be used as N-terminal amino acid
binders.
Developed methods addressing both gaps are detailed below. A new SELEX method
(referred to herein as RCHT-SELEX) is provided in Section A that optimizes the
selection of high affinity and specificity aptamers in a time-efficient manner
via an
innovative combination of existing and novel techniques to address the gap in
developing
ML-compatible empirical datasets. In addition, another novel SELEX method
developed
with the priority of discovering N-terminal amino acid specific binders
(referred to herein
as NTAA- SELEX) is provided in Section B.
SECTION A RCHT-SELEX
FIG. 3 is a schematic showing how conventional SELEX methods (FIG. 3A) have
been modified to produce RCHT-SELEX (FIG. 3B). The main differences between
the
two techniques are highlighted below:
= Step #1 of conventional SELEX does not amplify the input pool; RCHT-
SELEX amplifies input pool ("Bringup") after a negative selection step
and spike-in addition such that: (a) there exists approximately 100 copies
of each aptamer binder, and (b) the same input pool is used across
experiments.
= Step #2 of conventional SELEX is a single experiment of incubation of a
single target to a library of aptamers; in contrast, RCHT-SELEX
o splits the Bringup pool across many experiments of several targets
in triplicate (including 3 experimental bead-only controls) to be run
in parallel, and
o assays the aptamers against targets with alternating region(s) in
different rounds such that the only constant region driving the
selection process is the region that the user desires a specific binder
for, regardless of the targets' neighboring regions.
= Step #4 of conventional SELEX sequences the evolved aptamer pool after
8-20 rounds of repeating Steps #2 - #5, whereas Step #4 of RCHT-SELEX
includes sequencing after every round of selection, and multiple
techniques to maximize and standardize the amount of DNA input into the
next rounds in each experiment.
42
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
= Step #5 of conventional SELEX includes obtaining the aptamers that
bound to target proteins in the previous round so that those aptamers can
continue the selection process by repeating Steps #2, #3, #4 and #5 8-20
rounds; RCHT-SELEX can be performed in only 4 rounds and assays the
aptamers against targets again after the primer regions are replaced with
alternative primer sequences.
Since several experiments are run in parallel in RCHT-SELEX, and the goal is
to reduce
experimental bias across each experiment, several additional steps have been
added to the
RCHT-SELEX protocol to support running >36 experiments simultaneously. RCHT-
SELEX can include techniques such as:
= threshotding the same amount of DNA as inputs to subsequent rounds to
reduce PCR bias ("Threshold PCR")
= optimizing the PCR conditions for the specific candidate pool ("PCR
Optimization")
= performing unit tests before each digestion to determine optimal
digestion
conditions for each sample ("dsDNA Digestion").
Additional alterations of RCHT-SELEX can include:
= assaying the same aptamer candidate pool with multiple targets pooled
together in
early rounds and demultiplexing by incubating the aptamers against each target
individually in the final round ("Bead-Based Multiplex-SELEX")
= alternating targets with varied local environment binding regions between
alternating rounds of RCHT-SELEX for experiments where the desired aptamers
are ones that bind specifically to a smaller region of a molecule rather than
the
whole molecule ("Switch")
= switching primers mid-experiment to identify aptamers that are strong
binders
independent of the primer region ("Primer Switch").
Negative SELEX:
One technique that can be used to reduce the enrichment of aptamers to
unwanted
target(s) is to screen the initial pool of aptamer candidates for aptamers
that bind to the
selection components used in SELEX experiments (e.g., beads, streptavidin).
Aptamers
that express binding affinity to selection components are non-specific to the
targets and
can be removed from the candidate pool so that only aptamers that do not bind
the
43
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
selection components would be part of the aptamer candidate pool assayed
against
targets. See, for example, FIG. 4.
Single Bring-Up, Double Bring-Up and In-Experiment Replicates:
For example, a pool of 1012 DNA aptamers are selected from an original pool of
1015 and amplified through 13 cycles of PCR with unmodified primers resulting
in
approximately 2000 copies of each aptamer. Amplification is dependent on
primer
sequences and PCR conditions, and the bringup PCR protocol can be tuned to
each
individual library. The goal is to have at least 100 copies of most sequences
present in
each experiment with a minimum of 30 copies of each aptamer sequence present.
Libraries are sequenced during protocol optimization stages to help
approximate uniform
amplification copy number across sequences.
Post amplification, about 2000 copies of each aptamer is distributed across 12
samples, resulting in approximately 166 copies of each aptamer in each initial
starting
library pool. The process of having multiple copies of the same aptamer
present before
initiating a selection allows for the direct comparison of results of the same
initial bring-
up to each other. Computationally, this feature allows for direct experimental
replicates to
occur side-by-side, and also provides the ability to train models to walk
towards a
particular target and away from another. Since it would take many sequencing
runs to
determine the precise amplification of 1012 sequences, a single NextSeq run of
400
million reads can be performed as an approximation of the amplification
features of the
library across the entire pool. Single Bring-Up stops at this step.
For Double Bring-Up, a second bring-up is conducted by taking about 75 copies
of each aptamer from the first bring up and amplifying it through 6 cycles
with protected
phosphorylated primers, which allows for comparison of results from the same
initial
bring-up across approximately 300 experiments (approximately 2000 copies of
each
aptamer from a single bring-up, 75 aptamers selected yield 26 possible pulls;
each group
of 75 aptamers will yield a double-bringup pool for 12 experiments, so 12*26 =
312 total
experiments; NB there can be some loss in purification, digestion and other
processes and
amplification yield is highly correlative to the properties of primers and
components of
the PCR Mastermix). Amplification of aptamer candidates from each bring-up
also
increases the likelihood that strong and medium binders would carry through
past early
rounds. See, for example, FIG. 5B, which schematically demonstrates the single
and
double bring-ups and experimental replicates described herein, and FIG. 5A,
which
44
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
schematically demonstrates where in the RCHT-SELEX methods the single and
double
bring-ups and experimental replicates can be used.
Bead-Based Multiplex-SELEX
After, for example, four rounds of RCHT-SELEX is performed with multiple
bead-bound targets pooled together, aptamers can be de-multiplexed in round 5
by
incubating pools of amplified aptamers separately with beads that are
conjugated to only
one of the initial targets (see, for example, FIG. 6). Bead-based Multiplex-
SELEX adds a
competitive target environment, and scales the number of targets that can be
explored
within the same experiment.
Peptide switch
When designing binders for protein sequencing, four goals must be
accomplished:
(1) target the specific amino acid, (2) target the specific amino acid in an N-
terminal
location, (3) do not bind to the same amino acid in non-N-terminal locations,
and (4) bind
robustly to the targeted N-terminal amino acid(s) regardless of the
neighboring amino
acids. The rationale for goal #4 is that local biochemical environments (e.g.,
neighboring
amino acids) can influence the binding activity of aptamers, reducing their
effective KA.
Since the goal in protein sequencing is to build binders that can be utilized
in peptide
strings across the entire proteome, binder design must account for local
environmental
impacts. In order to accomplish goal #4, altering changes in local
environments were
introduced during binder selection to develop binders agnostic to neighboring
amino
acids. This was conducted by fixing 1-2 amino acids in a precise location
within a
peptide string (typically the N-terminal position) and varying the connected
or
surrounding amino acids from round to round. FIG. 7 illustrates a method of
identifying
aptamers that bind specifically to an N-terminal amino acid prefix,
independent of the
composition of the peptide's suffix tail. This technique, labelled as 'peptide
switch',
evolves aptamers in iterative rounds where only the peptide suffix is changed
while the
desired N-terminal amino acid prefix remains the same, removing negative
binders.
Peptide switch experiments can include a null, scrambled or 'fake' target as
well to define
promiscuous binders to remove false positives.
PER Optimization
PCR conditions can be optimized to maximize DNA output while minimizing
unwanted products, such as concatemers. PCR optimization must be conducted for
each
individual library. In SELEX experiments, initial library primers must be
replaced often
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
between experiments to prevent PCR contamination in experiments. Mastermix and
PCR
optimization unit tests are conducted for each library after every change in
library
primers, which consist of tuning as many parameters as possible (buffer
conditions, cycle
number, enzyme, primer concentration, number of protected base pairs etc)
before a
SELEX library can be used in experiments. Results are analyzed with
sequencing, Qubit,
TapeStation, Bioanalyzer and digestion unit tests to choose the ideal
optimization settings
for the individual library. For example, amplification can be performed in a
50 piL
reaction volume consisting of 38.49 pL nuclease free water, 0.30 pL 1 InM
forward
primer complementary to the first 6 nucleotides (referred to as 6XP), 030 [IL
linM
phosphorylated reverse primer (referred to as RP04), 0.50 pL Herculase II
Fusion DNA
Polymerase, 10 pL Here Buffer, 0.40 pL 25mM dNTP, and 0.01 pL template. PCR
can
be performed using an Eppendorf Mastercycler nexus eco PCR machine. The
thermal
cycle can be programmed for 5 minutes at 95 C for initial denaturation,
followed by 13
cycles of 30 seconds of 95 C for denaturation, 30 seconds at 55 C for
annealing, 30
seconds at 72 C for extension, and 5 minutes at 72 C for the final extension.
The
conditions for annealing are primer dependent and can be re-optimized for
different
primer sets used.
Digestion of dsDNA
Lambda exonuclease is a highly processive exodeoxyribonuclease that prefers to
digest the 5-phosphorylated strand(s) of dsDNA and has significantly lower
activity on
ssDNA and non-phosphorylated DNA (Little, 1967) (Mitsis & Kwagh, 1999). Lambda
exonuclease can be used to efficiently digest PCR-amplified dsDNA into ssDNA
in the
following three steps: a) unit tests for optimal digestion conditions, b)
segmenting pre-
digested library into thirds, and c) bioanalyzer quality control (QC) assay to
test amount
of ssDNA vs dsDNA. Single-stranded PCR products can be produced by first
performing
PCR with two different primers (e.g., 3'-phosphorothioate protection primer
complementary to the unwanted reverse strand and 5'-phosphorylated primer
complementary to the desired forward strand) followed by PCR amplification,
where the
phosphorylated strand of the PCR product then can be removed by digestion with
lambda
exonuclease. RNA kits of the Bioanalyzer system can be repurposed to quantify
ssDNA
as the dyes in the RNA kits bind ssDNA as well. Although the measurement
outputs are
not calibrated for ssDNA, inferences from the bands and peaks can be made.
See, for
example, FIG. 8. RNA kits of the Bioanalyzer system can be hacked to quantify
the
46
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
amount of ssDNA relative to dsDNA in a sample as the dyes in the RNA kits bind
to both
ssDNA and dsDNA. When a sample with both ssDNA and dsDNA is processed through
capillary electrophoresis on the Bioanalyzer, a unique non-overlapping peak is
generated
for ssDNA and dsDNA, where the relative area under each curve delineates the
percentage of the sample that is attributed to ssDNA and dsDNA. The goal of
utilizing
the RNA Bioanalyzer kit analysis in a digestion assay is to confirm that all
of the dsDNA
has been converted into ssDNA without overdigestion of the ssDNA library.
Although
the measurement outputs are not calibrated for ssDNA, inferences about the
nature of the
DNA mixture can be made from the bands and peaks.
During experimentation, data demonstrated that quality and quantity of PCR
yields influenced the ability to predict the digestion behaviors of lambda
exonuclease.
Libraries with additional concatemers products either digested very slowly or
very
quickly depending on the fraction of protected or phosphorated base pairs that
were
present in the concatemer sequences. Thus, unit tests can be performed when
evaluating
new libraries to prevent complete digestion of the sample. Before conducting
digestion of
all the PCR products, unit tests can be conducted to determine the optimal
reaction time
for efficient ssDNA production for each sample. Time course analysis of lambda
exonuclease digestion can be performed on small samples of the purified PCR
product
following incubation at 37 C for, for example, 2, 5, 10, 15, or 20 minutes, 75
C for 10
minutes, and held at 4 C. An RNA bioanalyzer can be run on each of the samples
to
assess digestion and determine the optimal digestion conditions to apply to
the rest of the
PCR product sample.
Lambda exonuclease digestion of the entire sample can be performed by
incubating at 37 C for the optimal time determined by the time course
analysis, followed
by heat de-activation of the enzyme at 75 C for 10 min and held at 4 C.
Representative samples of the final lambda exonuclease digestion mixture can
be
run on another RNA bioanalyzer chip to ensure sufficient digestion of the PCR
product to
ssDNA prior to the next cycle of RCHT-SELEX (FIG. 9). If digestion is not
complete,
more lambda nuclease and Al? can be spiked in.
Additional controls: bead controls, spike-ins ant/fake SELEX
= spike-in oligonucleotides: small spike-ins of known aptamer mimics can be
added as controls in various steps throughout RCHT-SELEX to detect
experimental error. For example, a mixture of nine oligonucleotides with 3
47
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
representative sequences at three different levels of GC contents (e.g., 40%,
50%, 60%) of known sequences (i.e., "a 9-oligo mix") can be added before
PCR to provide information regarding sample variability relating to PCR
differences. See, for example, FIG. WA. Alternatively or additionally, a
known sequence can be added to each well (e.g., a positional spike-in) to
provide information regarding spatial position on, for example, a 96-well
plate. See, for example, FIG. 10B.
= all bead controls: all bead controls include in-parallel and sequential
controls.
See, for example, FIG. 10B. For in-parallel experiments, an all bead control
(e.g., non-peptide conjugated bead sample) can be run in triplicate alongside
experiments to determine the number of aptamers from the bring-up pool that
bind to only the beads. In addition, these controls can be used to determine
the level of well-to-well contamination or noise from each experiment.
Sequential bead controls can be used after each round of RCHT-SELEX,
where aptamers that bind to the peptide-conjugated beads are incubated with
beads not conjugated to peptides. If desired, aptamers that bind to empty
beads can be sequenced to identify common sequences among aptamers that
are binding to the empty beads.
= fake SELEX: before each round of RCHT-SELEX, a small sample of the
original input can be removed and kept at room temperature as a control to
determine the effects of PCR bias since no target is present. See, for
example,
FIG. 10C.
Threshold PCR
Bound aptamers from bead-based RCHT-SELEX experiments can be amplified
directly on magnetic beads. Thus, aptamers do not need to be denatured from
the beads
prior to running PCR, limiting the number of processing, handling and
potential library
loss steps at a sensitive stage in the SELEX assay (loon, Zhou, Janda,
Brenner, &
Scolnick, 2011). However, PCR reactions can reach a saturation point where
reagents
become limited or concentrations have become too high for uniform replication
to
continue. Since the concentration of aptamers bound are not known a priori to
PCR
amplification, and can only be estimated; it can not be determined precisely
how many
amplification cycles will be needed before amplification saturation will
occur.
Furthermore, PCR amplification can be impacted by some magnetic beads which
are
48
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
coated with bovine serum albumin (BSA), where, if the concentration of BSA is
too high,
then the total product produced by PCR is reduced. Additionally, in-house
experimentation demonstrated that there was a non-uniform distribution of
aptamers
across beads, such that if the aptamer libraries on beads were physically
split into separate
solutions prior to amplification, different end-point amounts and variance in
undesirable
PCR products were seen across splits leading to unknown introduced variance
across
samples. In order to (a) resolve the complexity of introducing unquantifiable
bias across
samples, (b) amplify each library to the same concentration end-point, and (c)
mitigate
issues caused by PCR saturation and the presence of BSA, PCR amplification
occurred in
two stages: (1) PCR on beads and (2) threshold PCR. Conducting PCR
amplification in
two-stages provides the benefit of library redundancy if issues occur with
digestion.
When running many experiments in parallel from the same bringup pool, PCR
reactions can produce mixtures of aptamers with different end point
concentrations based
on the amount of DNA pulled down in each experiment (e.g., low, medium and
high;
FIG. 11). In order to conduct computational comparisons across many
experiments, and
to balance experimental requirements of the minimum amounts of material that
automation can handle (e.g., pipetting volume minimums for magnetic beads),
input
library amounts are normalized prior to a second amplification step. Variance
in input
DNA template amount can impact the effects of PCR bias. The DNA concentration
of
each library after PCR on beads can be measured and the post-PCR library with
the
lowest concentration of DNA, or a standard amount, can be used as a standard
for a
threshold quantity. The rest of the samples are then subjected to the
threshold quantity,
and subsequent rounds of PCR follow before generating inputs to subsequent
RCHT-
SELEX rounds. See, for example, FIG. 11A. Numerous control experiments have
demonstrated that the shape of the sequence distribution is not altered with
this threshold
PCR approach (FIG. 11B and 11C).
Primer switch
Constructs of the aptamer candidates can include a) random sequence of DNA
which participates in or facilitates binding to a target and b) one or more
regions to which
DNA primers can hybridize so that the aptamer sequence can be PCR amplified.
Primer
regions can contribute to aptamer structure and binding affinity to a target
molecule. The
primer regions can be alternated to different primer sequences or removed
entirely, and
49
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
aptamers can be assayed again to isolate aptamers that have high affinity to
the target
molecule independent of the primer region. See, for example, FIG. 12
Sequencing aptamer pool after every round
A representative pull of dsDNA, prior to Threshold PCR, from every round of
every selection were sequenced and analyzed for round-to-round enrichment of
sequences. Unit tests have been conducted of sequencing pre- and post-
Threshold PCR,
which demonstrated that the distribution of sequences did not change during
Threshold
PCR_ Since there wasn't a shift in sequence distribution, and for
computational analysis a
direct comparison point at each stage of SELEX is ideal, the pre-Threshold PCR
stage
was selected to: (1) reduce additional steps at the end of a SELEX experiment,
and (2)
allow for storage of DNA samples at higher concentrations and reduced volumes
without
additional manipulation (i.e. SpeedVac, etc).
As discussed herein, the RCHT-SELEX methods incorporate several novel
modifications: (1) screening of up to 300 different targets simultaneously,
(2)
maintenance of high DNA concentrations between selection rounds with reduced
PCR
bias, (3) additional features for advanced post hoc computational analysis,
including
comparisons across every possible experiment regardless of the day it was
conducted, and
(4) increased binding specificity to small molecule targets, such as small
peptides or
amino acid targets. These capabilities can accelerate the large-scale
identification of
aptamers to biological targets for potential use in diagnostics, therapeutics,
and basic
science research. Novel features of the RCHT-SELEX methods described herein
include,
without limitation:
= single or double bring-up allows for direct comparison of results across
targets, experiments and/or replicates from the same initial bring up;
= analysis of in-experiment replicates strengthens positive signal and
saves time
and money from testing undesirable aptamer candidates;
= threshold PCR generates robust aptamer library inputs for multiple
parallel
experiments with minimized PCR bias, provides an earlier recovery point if
experimental issues with converting post-PCR dsDNA libraries into ssDNA
libraries, and reduces library loss to concatemers;
= switch allows for the detection of aptamers that are specific to a
desired
sequence at a specific location of a target (e.g., small fragments of a larger
molecule);
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
= bead-based multiplex SELEX increases targets within the same experiment,
and reveals aptamer bind capabilities within a competitive environment;
= spike-in concentrations can be used to detect experimental error and PCR
bias;
= Next Generation Sequencing (NGS) at every round combined with sensitive
analysis can: (a) localize binders earlier and (b) generate input data for
machine learning (ML) models. ML models can predict highly specific novel
aptamers with fewer rounds of SELEX and explore a larger DNA input space
than experimentally possible. The use of ML in aptamer prediction can
increase the power of the SELEX methods described herein while saving
precious research funds and time.
RCHT-SELEX methods described herein reduce labor and reagent costs while,
more importantly, improving data quality, downstream analysis and broadening
screening
capabilities. In addition, the multiplex methods described herein can produce
aptamers to
targets that bind specifically in an environment with a multitude of available
targets (e.g.,
cell surfaces, human blood), thus, vastly increasing the discovery to
application pipeline
for aptamers.
The RCHT-SELEX methods described herein can be used to examine substrate
binding beyond DNA: peptide interaction_ For example, binding between a number
of
biological targets can be examined provided both targets include
oligonucleotides that can
be ligated to each other. For example, a similar technique can be employed to
screen for
RNA aptamers that bind small molecule targets or protein complexes.
Additionally, many procedural modifications can be made to adapt this method
to
suit different applications. For example, and without limitation, other
"input" nucleic
acids, such as RNA or modified nucleic bases, can be screened for binding
affinities with
molecular targets of interest, or to screen for aptamers that bind to targets
other than
proteins or peptides (e.g., small molecules, intact proteins, other nucleic
acids, specific
cell lines). Another example of a modification is the replacement of Lambda
exonuclease
dsDNA digestion with asymmetric PCR to produce the ssDNA input into subsequent
rounds of SELEX.
The RCHT-SELEX method described herein can be used to screen for aptamers
with selective binding to specific peptide targets within a competitive multi-
peptide
environment. Like selective antibodies, the resulting aptamers can be used
alone or in
combinations with two or more aptamers to create a complex that exhibits multi-
target
51
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
binding distributions. For example, two aptamers, each highly selective for
different
targets, can be used sequentially, in-tandem, or joined together in order to
create a single
construct that binds to the two separate targets. Alternatively, two aptamers
for the same
primary target but with different off-target binding distributions can be
joined together to
increase the selectivity of binding to their common target through avidity
while
simultaneously decreasing off-target effects.
In addition to being used for measuring binding between aptamers and target,
the
RCHT-SELEX methods described herein can be used for measuring binding between
different mixtures of any of the molecule classes previously described (ag.,
by replacing
the aptamer with a molecule that has been DNA barcoded and has a 3' C overhang
arm),
enabling bi-directional multi-way competitive measurements of any of the
combinations
of molecule classes including, without limitation, peptide vs protein, protein-
protein,
antibody-protein, small molecule-protein, peptide- cell surface marker,
antibody-cell
surface marker, etc. In some embodiments, both binding molecules (e.g., the
binder and
the target) can be drawn from a mixture of molecules from any of the above
classes,
allowing for measurement of cross binding in complex competitive environments.
SECTION B NTAA-SELEX
We have developed a new SELEX method to optimize the selection of high
affinity and specific aptamers in a time-efficient manner via an innovative
combination of
existing and novel techniques:
I) Negative Selection
A common technique to reduce the enrichment of aptamers to unwanted targets
(such as magnetic beads, PEG, reagents in binding buffers (such as BSA, etc)
is to screen
the initial pool of aptamer candidates aptamers that bind to the selection
components used
in the SELEX experiments, in our case, streptavidin beads in SELEX buffer (lx
PBS,
0.025% Tween-20, 0.1 mg/mL BSA, 1 mM MgCl2). Aptamers that express binding
affinity to selection components are non-specific to the targets and are
removed from the
candidate pool so that only aptamers that do not bind the selection components
would be
part of the aptamer candidate pool assayed against targets. A single or
multiple rounds of
negative selection can take place for a library before initiating SELEX
rounds. When
choosing a target library size (e.g., 10" molecules), a larger library needs
to be used for
52
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
negative selection to ensure that the supernatant includes enough molecules
for the
downstream SELEX experiments.
2) Peptide Backbone Switch
During each parallel selection, for each replicate of the target of interest,
a peptide
switch can be performed. Specifically, a "switch" target can be developed with
a
different backbone sequence, e.g., the amino acid sequences of the peptide
target differs
except for, e.g., the two amino acids at the N-terminus. By switching between
at least
two different backbones in rotating rounds, the chances of enriching aptamers
that bound
to anything that was not the dipeptide of interest were lowered.
3) Multiple Parallel Target Screening
In this technique, parallel selections of DNA aptamers for closely related, as
well
as unrelated targets can be used. The following metrics can be used across
targets: 1)
counts of each aptamer in each round, as determined by NGS sequencing and 2)
the
enrichment of each aptamer from round-to-round and 3) enrichment from the
first round
sequenced to the last round sequenced. By comparing these metrics across
different
target selections, one is able to determine what the binding signal looks like
for a 'real
binder', which is binding to a known target which has previously been shown to
be
'aptagenic', and also what the binding signal looks like for a 'non-specific
binder', which
is non specifically binding to the surface on which the targets are
immobilized (e.g.,
beads). These metrics across the parallel targets screening allows tracking
the specificity
of the aptamers and prevent unknown contamination effects.
4) Replicate Target Screening
In this technique, parallel selections of DNA aptamers can be used for the
same
target. Unique random DNA libraries can be used to perform SELEX for the same
target
either 2 or 3 times, at the same time. This allows the experimentalist to have
confidence
in the previously described metrics for each aptamer, especially if they fall
within the
same order of magnitude. In addition, it allows the experimenter to see if
there are
outliers within the aptamer pools. For example, if one random library has
significantly
lower enrichment than the other random library when looking for the final
aptamer
candidate, the experimentalist could choose to work with only the lead aptamer
candidates from the library that showed higher enrichment.
5) Counter SELEX
53
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Counter SELEX is a technique similar to negative selection, except that the
aptamer library is incubated with molecules similar to the desired target on
beads, the
beads are pulled down with a magnet and the resulting supernatant contains the
library of
aptamers that do not bind to the similar targets. The supernatant then can be
used for
downstream experiments to assist with the enrichment of N-terminal binders. A
counter
SELEX can be conducted in parallel or sequentially to a negative selection at
the start of
an experiment, and can be run in single or multiple cycles. Counter SELEX can
be run in
between conventional SELEX rounds, or after the final SELEX round to enhance
the
signal of N-terminal aptamer binders in the library pool.
Many types of molecules can be used during counter SELEX. Counter SELEX
can be used on targets that are similar in nature to the target but with
slight modifications
(e.g., to differentiate a post-translationally modified N-terminal amino acid
from an
unmodified N-terminal amino acid), peptide backbones (or suffices) used during
a peptide
switch or against a large pool of targets representing the proteome to ensure
specific N-
terminal aptamer binders towards the unique goal target.
If multiple backbones are used in a peptide switch experiment, then multiple
peptide suffices can be used sequentially during a counter SELEX experiment.
For
instance, if two different backbones are used for a peptide switch, a parallel
counter
SELEX on a mixture of targets can be run in between SELEX rounds, where the
'target'
pool for counter SELEX consisted of one half of one backbone bound to beads
and one
half of the other backbone bound to beads. Other embodiments could vary
stringencies
and/or introduce a combination of other molecules, such as random peptide
libraries,
various backbone designs, backbones with other N-terminal dipeptide suffixes.
6) PCR and Digestion Techniques
PCR Optimization, Threshold PCR, and Digestions of dsDNA techniques can be
employed in NTAA-SELEX and are described in SECTION A RCHT-SELEX
Novel features of theNTAA-SELEX methods described herein include, without
limitation:
1) This protocol provides a patio discover aptamer binders to N-terminal amino
acids, which can revolutionize approaches to enable high resolution
identification
of protein sequences and high throughput protein sequencing assays. The
stability
and flexibility of nucleic acids make aptamers a versatile tool for multiple
54
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
approaches to protein sequencing and quantification technologies, including
imaging and DNA barcoding methods described herein;
2) Multiple parallel SELEX experiments can allow for scaling aptamer discovery
and the removal of aptamers that are non-specific binders to multiple peptide
targets;
3) Sequencing of counter SELEX experiments can prompt the discovery of N-
terminal binders and the removal of aptamer binders to other regions along the
target;
4) Control targets can be run in each SELEX experiment to allow for the
evaluation
of inter-experiment comparison metrics;
5) Peptide Backbone Switch allows for the detection of aptamers that are
specific to
N-terminal amino acid(s) of a larger peptide, or, if desired, generation of
aptamers
to amino acid sequences internal to a peptide string or modified amino acids.
Protein or Peptide Sequencing (PROSEO)
The PROSEQ methods described herein use barcoded amino acid-specific
aptamers to convert a protein sequence into a readable DNA signal on a next
generation
sequencing (NGS) platform. Mass spectrometry (MS) is one of the common tools
in
identification and quantification of proteins, however the technology lacks
the ability to
cover the wide dynamic range necessary to detect lowly expressed proteins in
complex
samples (Schiess, Wollscheid, & Aebersold, 2008). Other existing specific
protein
quantification assays include antibody or aptamer binding assays where
detectable
antibodies, aptamers, or other small molecule binders bind specifically to
known proteins,
thus incapable of de novo sequencing or measuring proteins for which no
specific binder
has been found. The PROSEQ protein sequencing methods described herein can be
used
on small sample inputs (including single cells or small blood volumes) to
identify the
entire proteome, including low-expression proteins and single amino acid
mutations to
better understand diseases caused by aberrant or degenerative proteins.
Additionally, the
PRO SEQ methods described herein allow for the ability to sequence
heterogeneous
samples or multiple samples in parallel since proteins can be barcoded with
unique DNA
tags, which can be incorporated into the DNA sequences that encode protein
sequence
information. Further, the methods described herein enable significantly deeper
sequencing than existing methods such as mass spectrometry, since DNA
sequences are
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
derived from single peptides, amplified and read off from a sequencer (DR
100409),
which is not subject to the same dynamic range constraints as mass
spectrometry (DR
>10') (Yates, Ruse, & Nakorchevsky, 2009). Additionally, samples can be
processed to
remove reads associated with high abundance proteins within a sample by 1)
removing
highly abundant proteins in the original input pool into PROSEQ or 2)
separating out the
DNA barcodes associated with highly abundant proteins to increase NUS read
count of
DNA sequences associated with low abundance proteins.
The PROSEQ methods described herein can be used in a clinical setting for
quantifying protein expression levels or identifying novel protein fusions or
mutations
that are linked to disease from individual patient samples to assist with
patient diagnosis
and disease onset. In addition, the methods described herein can be broadly
used for
research areas of molecular and cellular biology, and protein engineering such
as:
sequencing proteins, discovering novel biomarkers, analyzing entire proteomes
or
metaproteomes, evaluating mechanisms associated with protein abundance and
more.
1) Aptatners provide the capabilby to perform de novo sequencing.
The methods described herein rely on a library of aptamers specific for unique
combinations of one or two N-terminal amino acids, where each residue or
residue pair
has at least one or multiple possible aptamer binders. The ssDNA aptamers are
designed
to contain a 5' phosphate for ligation, a unique DNA barcode (which indicates
the
identity of the particular aptamer and the corresponding cycle number), a
spacer/consensus region for subsequent barcode ligations (e.g., ligation
consensus
sequence), a restriction enzyme site with spacer, and an amino-acid
recognition sequence
(e.g., a single stranded DNA aptamer sequence). See, for example, FIG. 13.
These
aptamers may be incubated with the peptide targets either with or without a
complementary DNA strand that covers some or all of the barcode sequence, the
ligation
consensus sequence, and the restriction enzyme site with spacer. In the case
where these
regions are uncovered, DNA complementary to the ligation and restriction sites
can be
hybridized after incubation to facilitate ligation and restriction,
respectively.
The aptamers described herein can be used to sequence proteins or peptides in
any
of the following ways:
(A) Peptide fi-agments from proteins processed in solution or on a solid
substrate
Proteins from a sample (e.g., a blood sample, cell lysate or a single cell)
can be
obtained, denatured, conjugated to oligos and digested into peptide fragments.
It would
56
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
be understood that there are multiple methods of obtaining and digesting
proteins, and
conjugating peptide fragments to oligos prior to the sequencing steps. One
such strategy
includes denaturing proteins using a mild surfactant, and reducing and
alkylating the
denatured proteins to protect cysteine side chains. For example, amine groups
on the side
chain of lysine amino acids react with aldehyde-modified oligonucleotide
through
reductive amination reaction using sodium cyanoborohydride. The protein can be
digested with Lys-C, which cleaves proteins on the C-terminal side of lysine.
By using
this approach, each digested peptide has a lysine residue that is attached to
the
oligonucleotide tail. Reductive animation reaction also can happen between the
side
chain of lysines and alkynes with an aldehyde functional group, preparing it
for click
chemistry reaction with azide modified DNA oligos. In another approach, side
chains of
the proteins can be protected, modified with an oligo or click chemistry
linker, and then
cleaved into peptide fragments using, for example, a conventional try psin
approach to cut
at lysines and arginines, and/or other fragmentation enzymes that cleave at
random amino
acid sites (FIG. 13, step 2) or they can be processed in solution (see
modifications below).
At this point, the DNA-conjugated protein fragments can be ligated to DNA
oligos on the
surface of the sequencing substrate, where they will remain tethered
throughout the DNA
barcoding process and removed prior to DNA sequencing.
Aptarners can be taken directly from a SELEX experiment and applied to a BCS
assay via the creation of a BCS Compatible aptamer pool, where one of the
SELEX
primer regions is converted into a BCS handle. The aptamer region of the
binder will be
sequenced and considered the `barcode' of the binder. To generate the BCS
Compatible
aptamer pool, prior to incubating the peptide targets with the aptamers, a
single stranded
aptamer pool is incubated with bridge oligos that are partially complementary
to the
aptamer tail and partially complementary to the ligation region on the barcode
sequence
on the barcode foundation (BF) (single stranded overhang shown in FIG. 14) to
(a)
facilitate binding of the aptamer tail to the barcode sequence and (b) block
the ssDNA
region of the aptamer that is not involved in target binding from affecting
proper aptamer
folding. A DNA-barcoded library of BCS Compatible aptamers hybridized to
bridges can
be flowed across the peptides and incubated, allowing for the appropriate
aptamers to
bind specifically to the N-terminal amino acid residues (FIG. 13, step 3).
After aptamer binding, unbound aptamers are washed away and the tail of the
bound aptamer can be ligated to a second glass-immobilized DNA oligonucleotide
57
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
colocalized with the peptide (FIG. 13, step 4). A restriction enzyme site
included distal to
the aptamer barcode can then be used to cleave the remainder of the aptamer,
leaving the
DNA barcode attached to the nearby oligonucleotide (FIG. 13, step 5). Then,
Edman
degradation and/or aminopeptidases can be used to remove the N-terminal amino
acid
from the fixed peptide. In Edman degradation, once a new N-terminal amino acid
is
exposed, another aptamer pool, with unique DNA barcodes indicating target
recognition
sequence and cycle number, can be introduced and another cycle of DNA barcode
ligation can occur. After repeating this series of steps a plurality of times,
a chain of
DNA barcodes can be built that indicates the order of aptamer binding for a
peptide that
can be read using conventional NGS techniques. Using this information, the
amino acid
sequences of bound peptides can be obtained. In the case of arninopeptidases,
more than
one N-terminal acid amino acid may be cleaved at a time in a less controllable
manner,
which, although is not conducive for de novo sequencing, may reveal insight
for non de
novo sequencing methodologies.
(B) Full length proteins processed in solution
For full length proteins, the protocol is similar to the above, but with some
important differences. The following steps can be conducted: (a) lyse the
cells (if the
proteins are obtained from cells), isolate or purify, denature and protect the
proteins, (b)
protect reactive side chains of amino acid residues (such as *riot, carboxyl
and amine
groups), (c) conjugate a ssDNA oligonucleotide to the C-terminus of the
protein, where
the ssDNA oligonucleotide contains a primer region, a unique barcode and an
initial
ligation region, (d) deprotect all side chain protecting groups, (e) incubate
proteins with
aptamer pools, where the aptamers can contain a tail that includes a 5'
phosphate for
ligation, a unique DNA barcode (which provides information regarding aptamer
binding
sequence plus sequencing round), a spacer / consensus region for subsequent
barcode
ligations (e.g., ligation consensus sequence), a restriction enzyme site with
spacer, and an
N-terminal amino-acid recognition sequence (e.g., the single stranded DNA
aptamer
sequence), (f) ligate the bound aptamer to the DNA tail of the protein, (g)
pull down the
protein / aptamer complexes with a biotinylated reagent that has
complementarity to the
primer region of the protein/DNA conjugate molecule, (h) wash off unbound
aptamer
pool, (i) cleave the binding region of the aptamer off, leaving its DNA
barcode attached
to the protein's DNA tail, (j) cleave off the N-terminal amino acid, (k)
denature the
protein from its biotinylated oligo, (1) collect the supernatant of DNA
barcoded proteins,
58
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
(m) repeat steps (c)-(l) until the entire protein has been converted into a
DNA strand,
followed by PCR amplifying and sequencing the DNA barcode. If binders stay
bound
during the time and disruption during the protein-aptamer complex pull-down,
then step
(g) can also be performed prior to ligating the bound aptamer to the DNA tail
of the
protein [bind, pull-down, wash, ligate] (step 0. It would also be understood
that the
biotinylated reagent that has complementarily to the primer region of the
protein/DNA
conjugate molecule (from step g) can be added during aptamer incubation (step
e) to
prevent aptamers from binding to DNA region of the peptide target instead of
the N-
terminal prefix.
Barcodes, including the overhangs, can be about 8 to about 26 nucleotides (nt)
in
length (e.g., about 9, 10, 12, 15, 16, 18, 20, 21, 22, 23, 24, or 26 nt in
length). NUS
technologies currently are optimized for short reads, or a maximum of about
300-600
cycles. For many proteins, long sequencing experiments (e.g., by PacBio) can
be
performed or the DNA strands can be fragmented into smaller regions and
realigned post-
sequencing.
(C) Protein complexes processed in solution followed by a solid substrate step
For protein complexes, the proteins within protein complexes can be tagged
with
DNA oligonucleotides via an amino acid side chain and proximal side chains can
be
ligated together before the proteins are denatured, before proceeding with the
protocol
outlined above in the absence of peptide fragmentation (e.g., under section
(B)). The
protocol can be optimized such that only proteins in close proximity (e.g.,
bound
complexes) are tagged with oligonucleotides that can be ligated to each other.
The
protein complexes can be pulled down and attached to a solid substrate, which
can have
DNA adaptors specifically placed so that protein complexes can be processed
locally.
The DNA adaptors on the chip can have a unique DNA starting barcode, which,
when
isolated and sequenced, can reveal insight into what the neighboring sequenced
peptides
fragments are, and therefore, of the protein complexes.
The PROSEQ methods described herein do not rely on previous knowledge of
proteins or protein complexes (as is required when using, for example, mass
spectroscopy), and provide an avenue for de novo sequencing. Once the protein
or
peptide molecule(s) have been converted into a DNA molecule, conventional
tools such
as PCR amplification, biotin pull-down assays and/or digestion can be used to
amplify,
enhance and modify the sequences to allow for pooling of many samples or to
ascertain
59
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
lowly expressed molecules within a sample. There are also many novel
biological insights
that can be obtained with the non-tie novo applications of PROSEQ, such as
high
resolution protein quantification, that are not currently possible with
conventional protein
sequencing technologies.
FIG. 15A is another schematic showing an example of the aptamer-based peptide
sequencing method described herein, with conjugating the C-terminal end of the
peptides
to an amine modified oligonucleotide bound to a substrate or using other
strategies such
as click chemistry or SMCC linker (succinimidyl 4-(N-
maleimidomethyl)cyclohexane-1-
carboxylate) to covalently bind the peptide to oligonucleotide (1), incubating
the bound
peptides with the DNA barcoded aptamer library (2), ligating aptamers that
bound to a
peptide to a second oligonucleotide immobilized on the solid substrate (3),
and cleavage
of the aptamer, leaving the DNA barcode attached to the second oligonucleotide
(4).
FIG. 15B is a schematic showing representative aptamers to different amino
acids and the
corresponding aptamer barcode, the sequence of which identifies the specific
amino acid
at that position.
2) The protein sequencing methods described herein overcome the processivity
limits
of Edman degradation
The methods described herein overcome the processivity limits of Edman
degradation. For example, liquid chromatography (LC) typically is used to
identify
terminal amino acids after cleavage by Edman degradation. A putative drawback
in
standard Edman degradation is that, physically, there exists a maximum cycle
number for
accurate degradation and detection of N-terminal amino acids (-10 cycles).
Since the
present methods are not measuring the amino acid that is cleaved, limitations
of detection
of the cleaved amino acid is not an obstacle. Additionally, any processivity
limitation in
the PROSEQ methods described herein can be overcome by rotating between the
use of
Edman degradation and aminopeptidases (e.g., trypsin and pepsin) to cleave
terminal
amino acids. After approximately 30 cycles, for example, the methods described
herein
can use an exopeptidase to cleave the peptide at a specific amino acid site,
which allows
the sequencing to begin again from a new region of the peptide.
3) The protein sequencing methods described herein allow for sequencing of a
heterogeneous protein pool
One of the important features of the PROSEQ methods described herein is the
ability to sequence large pools of proteins, where one or more of the proteins
of interest
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
(e.g., a target protein) are expressed at low levels or very low levels (e.g.,
a protein that is
present in one part per 10 billion; potentially even lower with the "Sup-Diff'
methods
described herein). This is especially useful when processing samples such as
plasma,
which: (1) are easy to obtain from patients, (b) allow for longitudinal
studies, and (c) can
give insight to difficult to study diseases such as neurodegenerative
diseases, due to the
presence of biomarkers in the bloodstream. In plasma, 13 proteins plus albumin
compose
96% of the protein sample, and some of the most interesting molecules, such as
tissue
leakage products and cytokines, make up the last 4% of the sample and found to
be well
under the instrument detection resolution limit for MS (Schiess, Wollscheid, &
Aebersold, 2008). Thus, it can be extremely difficult to identify biomarkers
or new
proteins on plasma samples with MS. Unlike HPLC and MS, identifying amino
acids
based on aptamer binding is not limited to a detection limit of high
individual protein
concentrations within a sample. Since the final product actually being
sequenced is DNA
and not protein, there exist well developed tools to amplify, anneal, and pull
down
specific DNA populations of interest. After the DNA barcode chain is formed,
the DNA
sequencer platform can clonally amplify the sequences (e.g., using bridge
amplification).
Thousands of clusters of each individual DNA sequence produces a larger
readable signal
than its initial input signal from a lowly expressed protein, bypassing single
molecule
techniques. This ability to sequence large, non-uniform pools allows thousands
of
antigens spaiming entire organism proteomes to be sequenced.
For samples that have a large dynamic range, a method referred to as "sup-
diff'
can be used to remove DNA barcode constructs of highly expressed proteins,
leaving an
enhanced ratio of DNA barcode constructs of lowly expressed peptide or protein
clusters
remaining in the pool of oligonucleotides to be sequenced. For example, there
are two
methods for enhancing the ratio of desired or lowly expressed peptides: an a
priori and a
non a priori method. The general strategy is to develop an ssDNA bait pool
containing
biotinylated RNA sequences complementary to certain sequences in the initial
diverse
pool of ssDNA (Diatchenko et al., 1996) (Gnirke et al., 2009). Said RNA bait
pool is
used to capture ssDNA targets via in solution hybridization and subsequent
pulldown on
streptavidin-coated magnetic beads.
The chief difference between the a priori method and the non a priori method
is
that the a priori method pulls out only known sequences, while the non a
priori method
pulls out high abundance sequences in a pool of unknown distribution and
constitution.
61
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
In the a priori method, the diverse pool of ssDNA is first sequenced and then
the user can
design baits specific to what the user wants to pull out of the pool, which
could include
very high concentration sequences that might be contaminants. The a priori
method
enriches for sequences that were not pulled down by the designed baits, thus
reducing
NGS sequencing reads dedicated to the targets that were originally desired to
be pulled
out of the pool. In the non a priori method, the initial diverse pool of ssDNA
is directly
used to generate the RNA bait pool. The RNA bait pool could have the same
fractionational distribution as the original target pool, or a distribution
slightly skewed
toward the initial high abundance sequences. By the assumption that the higher
abundance target sequences will be more likely to find their RNA bait partners
under
optimized conditions of time, temperature, and ratio of overall bait to
target, when the
RNA baits are hybridized with the initial diverse pool of ssDNA, the high
concentration
sequences are more likely to be pulled out. See, for example, FIG. 16.
4) The protein sequencing methods described herein allows for sequencing the
DNA
barcode using a range of DNA sequencing technologies
The methods described herein for sequencing proteins can be performed in
conjunction with any existing DNA sequencing technology. With custom-built
flow cells
that have DNA printed on the glass in a specified manner and an automated
fluidics
system, the barc,odes can be built as described in the preceding sections
without the need
for reprogramming or repurposing an existing DNA sequencing platform. These
DNA
barcodes that represent the protein/peptide sequence may then be sequenced on
any
existing DNA sequencing platform or technology.
5) The protein sequencing methods described herein include strategies to
ensure
robust protein and DNA sequencing capabilities despite the harsh chemistries
of Edman
degradation
The ProSeq methods described herein use barcoded amino acid-specific aptamers
to convert a protein sequence into a readable DNA signal on a next generation
sequencing
(NGS) platform. The methods described herein overcomes the distortion of the
protein
sequencing platform components caused by Edman degradation, which prevents the
clustering of DNA barcode constructs and, therefore, sequencing directly on
the same
chip. Trifluoroacetic acid (TFA) and the pH oscillations that occur during
Edman
degradation result in two main issues: (1) the loss of DNA cluster generation
through the
removal or modification of the P5 and P7 DNA adaptors on the chip, and (2)
62
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
modifications of the constructed DNA barcodes resulting in sequence-
information and
amplification-capability loss.
(A) Off-Chip Sequencing of DNA Barcode
After building the DNA barcode construct containing a chain of DNA barcodes
indicating the order of aptamer binding for a peptide, the constructs are
amplified on the
chip, or cleaved off the chip and amplified in solution. Amplification methods
used
include, without limitation, PCR, loop mediated isothermal amplification,
nucleic acid
sequence based amplification, strand displacement amplification, and multiple
displacement amplification. Additionally, the original DNA barcode constructs
could be
transcribed on the chip into large amounts of RNA constructs, which could then
be
converted into a cDNA library consisting of many copies of the original DNA
barcode.
The amplification products, copies of the original DNA barcode constructs, can
be
removed from the microfluidic chamber and sequenced using standard DNA
sequencing
methods including, without limitation, Sanger sequencing, NGS, ion
semiconductor
sequencing, SOLiD technology, cPAS, etc. Numbers of reads are normalized to
the
number of PCR cycles used to estimate the quantity of each protein or peptide
sequenced
from the initial sample.
(B) INA or modified DNA/RNA Adaptors, Foundations and Barcodes
The methods described herein are a single-chip strategy to overcome the
degradation of DNA components on the BCS platform by utilizing XNAs or
modified
DNA/FtNAs that are (a) resistant to transformations due to Edman degradation
or highly
acidic conditions, (b) are able to be made into chimeras with conventional DNA
nucleotides, and (c) compatible with existing polymerases that can amplify
these non-
natural nucleic acids or convert modified sequences into conventional DNA bps.
Such
modified nucleic acids may include a modification to the 2' carbon of the
ribose sugar
that enhances its hydrolytic stability or to the purine base itself (Watt, et
al. 2009).
Examples include, but are not limited to, 2'-O-methylated RNA, 2'-fluoro
deoxy adenosine, 7-deaza-T-deoxy adenosine, and 7-deaza-8-aza-deoxyguanosine.
= Addition of XNA or modified DNA/RNA adaptors to degraded P7s: the methods
herein can utilize the degraded P7 adaptors available on the chip as bases for
custom XNA or modified DNA/RNA adaptors. After subjecting the P7 and P5
adaptors to acidic conditions, the 135 adaptors are at least partially removed
and
63
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
the P7s are degraded. Two methods of adding new adaptors for ligation and
barcode generation handles, post-barcode cluster generation, are:
o Approach 1: several cycles of Edman degradation can be conducted to
remove P5 and depurinate P7 and XNA or modified DNA/RNA adaptor
can be ligated to the remainder region of P7. A method of XNA adaptor
ligation is to ligate an XNA or modified DNA/RNA adaptor with a
phosphorylated 5' end to the 3' end of the P7. If the modified nucleic acid
analogs lower the ligase efficiency, the adaptor sequence may be a
chimeric XNA or modified DNA/RNA molecule with one or more
standard cytosine or thymine nucleotides at its 5' end.
o Approach 2: Conduct several cycles of Edman degradation to remove P5
and depurinate P7 and use click-chemistry to attach an XNA or modified
DNA/RNA adaptor to the remainder region of P7. Another strategy to add
an XNA adaptor is to chemically attach an XNA or modified DNA/RNA
adaptor on P7s by ligating on the P7's 3' end an oligo linker with a
reactive group at its 3' end. Chemistry reaction can attach a functional
XNA or modified DNA/RNA adaptor to P7, optionally containing a
cleavage site, with the corresponding reactive group at its 5' end to the
oligo linker. Examples of reactive group pairs include, but are not limited
to, NHS ester with amine (wide reaction), azide with alkyne (triazole
reaction), maleimide with thiol (thioether reaction), and tetrazine with
alkene. The P7 and linker can be blocked from unwanted annealing with
an oligo that is partially complementary to both P7 and the extendor oligo
during aptamer incubation.
= XNA or modified DNA/RNA Foundations and Barcodes: the methods herein
foundation pieces, binding regions of aptamers, BCS cassette components,
aptamer barcode regions, or combinations thereof can comprise of XNAs or
modified DNA/RNAs.
The Illtimina sequencing protocol concludes sequencing runs once it no longer
detects P5 adaptors, so additional steps may be needed to prevent premature
sequencing
cessation in embodiments wherein P5s are removed from the sequencing platform
These
steps could include, individually or combined:
64
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
= Enzymatic or chemical addition of multiple P5s to the chip after final
round of
Edman degradation
= Adapt sequencing instrumentation protocol code to continue sequencing run
in the
absence of P5
= Enzymatically or chemically attach a custom primer sequence into the
cleavage
sites of the altered P7 strands and adapt sequencing protocol code to detect
the
custom primer sequence rather than P5 to determine whether to terminate
sequencing run.
6) Exemplary variations to the protein sequencing methods described herein
include,
without limitation (FIG. 17):
= multiple aptamer binding rounds: in some instances (e.g., if issues with
aptamer
specificity binding exist), several rounds of aptamer binding / DNA barcoding
/
aptamer denaturing can be performed before proceeding on to degrading the N-
terminal amino acid for error correction. The additional data collection will
allow for downstream computational analysis to reduce the noise for each
individual measurement.
= aptamers to two amino acids: in some instances (e.g., if aptamers to
single
amino acids do not have high enough affinity or are not specific enough for
the
methods), aptamers to two or more sequential amino acids can be generated
(FIG. 18). The added benefit of aptamers binding and encoding for two amino
acids is that there is improved signal-to-noise since each amino acid (aside
from
N- and C- terminal) will be read twice.
= substrate: this barcode sequencing method also can be performed on glass,
or
quartz substrates with DNA oligos printed or chemically linked in random or
patterned events. Such types of chips can be custom made or purchased; for
example, academic labs make chips with clean rooms and DNA spotters,
Agilent prints microarrays with known oligo sequences patterned in spots on
glass, and Illumina's next generation sequencing chips are glass slides with
randomly distributed DNA adaptors to the P5 and P7 sequence binding sites
linked to a solid surface. In the case of custom glass slides, or substrates,
DNA
oligonucleotides can have specialized patterning to reduce off-target ligation
noise.
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
= different oligo orientation: the protein sequencing methods described
herein
orients the DNA barcode sequence such that the 5' end is attached to the DNA
adaptors on the chip. With alternative or custom chips, the 3' end of the
barcode
sequence can be attached to the chip surface instead.
= in solution: the need for a solid substrate can be eliminated entirely by
ligating
DNA barcodes directly to the C-terminus of the peptide. The peptide C-termini
initially can contain a short oligonucleotide sequence that allows for
ligation
between the aptamer end and the peptide tail bridged by, for example, a 5-mer
oligonucleotide. Following Edman degradation, subsequent DNA barcodes can
be ligated onto the free end of the peptide tail. The resulting barcode
sequence
then can be PCR amplified and sequenced using standard NGS techniques.
* beads in solution: peptides and oligonucleotides can be tethered to beads
(either
magnetic, glass, glass-covered magnetic bead, or other beads coated in acid-
resistant materials), and serial peptide sequencing steps (e.g., aptamer
binding,
barcode incorporation, and peptide degradation) can be performed by immersion
and separation of beads in solution. After the desired number of sequencing
cycles, the DNA barcodes that provide the sequence of the peptide can be PCR
amplified directly off the beads and sequenced using standard NGS techniques
(Moon, Zhou, Janda, Brenner, & Scolnick, 2011).
= different binders: other than aptamers, barcoded-binders such as RNA,
peptides,
proteins, nanobodies, or other small molecules can be used to recognize amino
acids.
= different proteases: when processing protein samples, different proteases
such as
Lys-C as described above, try psin, or a combination of multiple proteases can
be
applied. Additionally, a sample can be divided into multiple samples that are
treated with multiple proteolysis strategies to build different proteome maps.
= single platform versus separation of steps: it is possible for Edman
degradation
of the peptide and DNA barcode generation to occur off the sequencer platform,
or build a complete end-to-end automated single platform. The DNA barcode
chain can be fixed and sequenced in a separate step.
= bridge design: bridges are oligos that are partially complementary to the
aptamer
tail with a 3' single stranded overhang, which anneals to the restriction site
spacer and barcode (FIG. 14). Bridges can be designed such that they can be
(a)
66
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
a Barcode-Specific bridge wherein the bridge is entirely complementary to the
aptamer tail, including barcode region, except for the 3' single stranded
overhang region, such that each unique aptamer has a unique bridge associated
with it (FIG. MA), or (b) a Universal bridge wherein the bridge is
complementary to the restriction site spacer and consensus sequence only, both
of which are conserved across all aptamers and flank the barcode on the
aptamer
tail, such that all unique aptamers share the same bridge oligo (FIG. 14B).
For
the Universal bridge, the region that duplexes with the barcode on the aptamer
tail can consist of (a) a sequence of universal base analogues, such as 5-
nitroindole, 3-nitropyrrole, and 4-nitrobenzitnidazol among others, or (b) a
gap
with no bases such that the Universal bridge consists of two separate oligos
that
anneal to the regions flanking the barcode.
= ligation method: DNA barcodes can be chemically linked rather than
enzymatically ligated together.
= different readout: instead of using a DNA barcode to identify amino acid
binders, one could use fluorescent dyes, beads, nanoparticles, etc. (see,
also, the
PROSEQ-VIS methods described herein).
= sequential amino acid degradation: cleavage of single amino acids in
between
rounds can be performed either enzymatically or chemically, such as via Edman
Degradation.
= sequencing directionality: single amino acids can be cleaved from the N-
terminal end or C-terminal end (Casagranda and Wilshire, 1994) (Cederlund et
al., 2001). Protein sequencing from the N-terminal end is described in detail
here. Based on this disclosure, it would be appreciated that similar methods
can
be applied to protein sequencing from the C-terminal end in conjunction with
aptamers that have been designed to specifically recognize and bind to one or
more C-terminal amino acids. For C-terminal sequencing, methods to remove
the C-terminal amino acid and generate a C-terminal amino acid-shortened
protein or peptide (instead of using, for example, Edman degradation to
generate
a N-terminal amino acid-shortened protein or peptide) are known in the art and
include, without limitation, Bergman et at. (2001, Anal. Biochem., 290(1):74-
82) and Casagranda and Wilshire (1994, Methods Mol. Biol., 32:335-49) can be
used.
67
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
It would be understood that the PROSEQ methods described herein can also serve
as large-scale, high-throughput binding specificity assay to characterize
interactions in
different substrate binding scenarios (BCS BINDING ASSAY). The key advantage
of
this assay is that it allows the recording of one or more binding events
between many
putative binders and many targets in one experiment. Once the desired targets
are
conjugated to co-localization foundations, the foundations can be tethered on
a glass
substrate, or processed in solution. Then, a diverse DNA-barcoded putative
binder library
(PBL) is incubated with the desired and unintended targets for incubation,
allowing for
binding. Each DNA-barcoded putative binder comprises of a binder molecule
conjugated
to a DNA sequence containing at least a a) restriction site, b) ligation site
(e.g., a first
ligation site), c) unique DNA barcode indicative of the identity of the
putative binder and
binding cycle, and d) another ligation site (e.g., a second ligation site).
When a putative
binder binds a tethered target, its DNA barcode tail is ligated to the
proximal, target-
barcoded DNA foundation that is colocalized with the target. The ligated
barcode is cut
with a restriction enzyme, exposing the DNA barcode construct to be ligated to
another
binder barcode in the next round. After repeating this series of steps on the
chip, a chain
of DNA barcodes containing information on the identity of the binder and
target and
order of binding events can be read off with conventional DNA NGS techniques
(FIG.
19). Using this information, the probability distribution of a putative binder
binding to
the desired and unintended targets in various environments can be deduced.
The PROSEQ methods described herein result in a number of advantages,
including, without limitation, the ability to:
= produce a probability distribution of binding events in one mixture by
interrogating the same targets multiple times;
= isolate binding events from unbound binder molecules via washing steps
for the
solid-state method. The separation of binding and ligation events decreases
off-
target ligation events;
= assay a large library of putative binders in various environments (e.g.
in the
presence of unintended targets, other targets of interest, etc.). This is
especially of
importance to binders identified through a selection process wherein the
binders
were selected in isolation of other putative targets, but to be used in
applications
where various targets would be present;
68
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
= detect rare binding events in a high-noise environment (due to high
resolution data
in NUS);
= determine the dynamic range of the binder's functional buffer conditions;
= simplify the process of separating bound and unbound ligands by simply
flowing
on wash buffer, if the reaction is not in solution.
Peptide or Protein Sequencing with Visualization (PROSEQ-VIS)
The PROSEQ-VIS methods described herein convert an amino acid sequence to
an optical barcode. In the PROSEQ-VIS methods described herein, fluorophore-
conjugated aptamers can be used to deconvolve an amino acid sequence, allowing
for de
novo protein sequencing The PROSEQ-VIS methods described herein are capable of
sequencing diverse samples, and particular samples in which one or more of the
proteins
of interest (e.g., target proteins) are present at low or very low
concentrations (e.g., a
protein present in one pail per 10 billion). The PROSEQ-VIS methods described
herein
also provide for computational tools to determine the identity of the N-
terminal amino
acid based on the observed unique spectral signatures of binding events.
The PROSEQ-VIS method described herein uses amino acid-specific aptamer
binding to convert a protein sequence into a series of fluorescent images or
an "optical
barcode," which can be read via microscopy imaging. The optical fluorophores
can be
assigned to their aptarners, revealing the underlying protein sequence. See,
for example,
FIG. 20. This protein sequencing method can be used on small samples
(including single
cells or small blood volumes) to identify the entire proteome of expression,
low-
expression proteins and single amino acid mutations to better understand
complex disease
phenotypes. Additionally, the PROSEQ-VIS methods described herein can be
performed
on intact cells and tissues to visualize, not only the sequence of proteins,
but also the
location within a sample. See, for example, Table 1.
Table I
Biological Source of Proteins Type of Protein
Approach for Peptide Sequencing
Cell Lysate Peptide
Fragmented; Solid Substrate
Blood Full-length protein
Fragmented; In Solution
Saliva Protein complex
Whole Proteins; Solid Substrate
Urine Membrane protein
Full-length proteins; In Solution
Biopsy Post-translational modified
protein Protein-Ligand Complex; Solid
Tissue
Substrate
Single cells
Protein-Ligand Complex;
In Solution
69
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
The PROS EQ-VIS methods described herein can be used in a clinical setting for
identifying novel protein fusions or mutations that are linked to disease from
individual
patient samples, developing a diagnosis or prognosis, evaluating patient
response to
treatment, or predicting the likelihood of possible responses to certain
treatments. In
addition, the methods described herein can be broadly used for characterizing
proteins,
discovering novel biomarkers, analyzing whole proteomes or metaproteomes,
building
cell lines and evaluating mechanisms associated with protein abundance,
sequence or
function.
Aptamers provide the capability to perform de novo sequencing
The PROSEQ-VIS methods described herein use a library of aptamers as
described herein that are specific for unique combinations of one or two N-
terminal
amino acids, where each residue pair has at least one (e.g., or more than one;
e.g.,
multiple) aptamer binders. The ssDNA aptamers are designed to contain a region
that
includes either a fluorophore or a region for annealing short dye-coupled
ssDNA probes,
such that the N-terminal amino acids can be identified by its unique spectral
signature of
binding events between the N-terminal amino acid and its corresponding
aptamer(s).
Proteins from a sample (e.g., a blood sample, cell lysis or a single cell) can
be
obtained, denatured, blocked and cleaved into peptide fragments. While
denatured whole
proteins can be analyzed without cleavage, proteins cleaved into smaller
peptide
fragments are optimal since: (1) rounds of Edman raise the noise-floor in
imaging, and so
fewer rounds of sequencing can be used to determine the sequence of a peptide
fragment,
and (2) certain imaging modalities (like TIRF) have a narrow focus window (10s
- 100s
of nms) and signal detection is highly dependent on samples being fully
contained within
the optimal imaging window. Proteins can be cleaved into peptide fragments
using, for
example, a conventional trypsin approach to cut at lysines and arginines,
and/or other
fragmentation enzymes that cleave at random amino acid sites. The combination
of both
methods can help reduce error in post-sequencing computational alignment. Once
the
proteins are converted into short peptides, the free and unblocked C-terminal
end can be
conjugated to DNA primer oligonucleotides on a glass substrate or conjugated
directly to
the glass (FIG. 21). Then, a library of aptamers can be flowed across the
peptides for
incubation, allowing for aptamers to bind specifically to N-terminal amino
acid residues.
There are many ways to fluorescently label aptamer tails. Two potential
imaging options
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
are that the aptamer tail can have either: (a) an optical barcoded tail for
imaging, or (b) a
region where one or more short fluorescently-tagged DNA probes can anneal to
an
aptamer: amino acid complex.
1.1 Direct Aptainer-Dye Conjugation
After aptamer binding to N-terminal prefixes, the optical signature of the
aptamer
(a) can be imaged by a multi-channel single-molecule epifluorescent or total
internal
reflection fluorescence (TIRF) imaging setup. For each N-terminal prefix read
out
("round"), the unbound aptamers are washed off and a z-stack of images can be
obtained
during the incubation period in order to confirm the spectral signature for
the N-terminal
amino acid(s). The next round then begins by using Edman degradation and/or
aminopeptidases to remove the N terminal amino acid on the fixed peptide. The
same
aptamer pool then can be used to interrogate the newly exposed N-terminal
amino acid
(FIG 20A ¨ 200). After repeating this series of steps, the identity of each N-
terminal
amino acid can be computationally deduced at each round by comparing the
observed
binding events for each peptide against the probability distribution of
binding events for
each an-tamer-amino acid complex. Using this information, the amino acid
sequence of
each peptide can be deduced based on the series of amino acid signatures
obtained in
serial rounds of imaging and degradation_ See, for example, FIG. 20E.
1.2 Oh go-Conjugated Dyes Hybridization to Aptarner
In the case of using aptamers with regions that bind to complementary
fluorescently-tagged oligos, the assay includes multiple "iterations" of probe
incubation
and imaging per "round" of N-terminal prefix read out. The aptamers include 3
regions:
(a) the effective binding region, (b) an optional spacer, and (c) a barcode
tail of one or
more combinations of barcode units indicative of the probing iteration number
and
fluorescent tag, with each barcode being complementary to a fluorescently-
tagged oligo
(FIG. 22). To prevent the barcode regions from affecting the folding of the
aptamer's
binding region, when the library of aptamers is flowed on, the oligo regions
not related to
N-terminal prefix binding can be partially or fully protected by hybridizing a
complementary oligo to form aptamers that are partially double-stranded. The
aptamer:
amino acid complexes can be incubated with a library of probes that hybridize
to barcode
regions indicative of probe iteration 1. The number of unique fluorescent tags
that can be
employed per iteration is dependent on how many channels are in the imaging
set-up,
properties of the fluorescent dyes and emission filters, and sensitivity of
the detector.
71
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
During each iteration, each aptamer can hybridize to one or multiple oligo-
bound probes
for multiplexing as long as the complementary barcode units on the aptamers do
not
overlap for that iteration. The unbound probes then can be washed off and
bound probes
can be imaged to acquire the first section of the optical barcode. Thereafter,
the bound
aptamers can be incubated with the next set of probes that hybridize to
barcode regions
indicative of probe iteration 2. Iterations of probe incubation, imaging, and
washing can
be repeated until full optical barcodes are acquired. Lastly, Edman
degradation can be
performed to remove the N-terminal amino acid and the aptamer it is bound with
to reveal
the next N-terminal amino acid for the next round of sequencing (FIG 23).
It would be understood that procedural modifications, especially to the
imaging
and downstream signal deconvolution strategy, can be made to accommodate the
affinity
and specificity of the aptamers used to probe the N-tenninal amino acids. In
the case of
utilizing highly specific binders, a library of aptamers specific to a unique
N-terminal
amino acid prefix and with low ICA (tight binding) are flowed on, the unbound
aptamers
washed away, and the optical barcodes observed as described above (FIG. 24).
In the
case of aptamers with medium-to-low specificity, a library of fluorophore-
conjugated
aptamers can be flowed across the peptides for incubation, allowing for
aptamers to bind
semi-specifically to a set of N-terminal amino acid residues. Such aptamers
preferentially
bind to a given target, and may also bind to a subset of known N-terminal
amino acids
with a known probability distribution for each binding pair. For each round of
sequencing, images can be taken before (for background), after (for specific
binding) or
during (Kon, Koff measurements) cycles of aptamer incubations in order to
generate a
spectral signature for the N-terminal amino acid prefix composed of multiple
binding
events before the N-terminal amino acid is removed to reveal the next amino
acid to be
probed. Several rounds of incubation and detection can occur before removing
the N-
terminal amino acid via Fdman in order to increase the confidence in the
detected signal.
After repeating multiple rounds of aptamer binding, the identity of N-terminal
amino
acids can be computationally deduced at each round by comparing the observed
binding
events for each peptide against a known probability distribution of binding
events for
each aptamer amino acid prefix, as each unique N-terminal amino acid is
expected to
have its own distinct binding signature given a pool of medium-strength
binders (FIG.
25). Additionally, binders such as RNA or small molecules can be used, in
addition to or
as an alternative to aptamers, to recognize amino acids.
72
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
The methods described herein do not rely on previous knowledge of proteins
(such as with a peptide database required in mass spectroscopy) and provide an
avenue
for de novo sequencing. If a database of proteins is available, however, it is
likely, then,
that only a subset of amino acids need to be identified in order to accurately
map peptide
fragments back to full-length proteins. Additionally, if purification or
selection for (e.g.
by molecular weight, charge, or affinity to a known molecule) proteins were
performed
prior to sequencing, it would further focus the list of candidates based on a
subset amino
acid sequence of a full-length protein identified.
The PROSEQ-VIS methods described herein result in a number of advantages and
applications, including, without limitation, the ability to:
1) sequence peptides irrespective of peptide concentration;
2) convert a protein sequence to an optical sequence, which allows for
isolation of the signal of lowly expressed proteins;
3) conduct de novo protein sequencing (e.g., to allow direct discovery of
sequences in molecules such as cytokines);
4) process small volume samples, down to single cell protein sequencing; and
5) sequence peptides in situ for protein localization data in intact tissues.
Instead of using fluorophore-conjugated aptamers or oligo probes to identify
amino acids, other optical methods such as quantum dots, dye-conjugated
nanoparticles,
or the like could be used. Instead of TIRF, other microscopy means can be used
for
imaging with varying degrees of resolution quality. Lastly, replacing the
aptamer in the
PROSEQ-VIS methods described herein with another type of N-terminal amino acid
binding small molecule that has been barcoded with an optical barcode
similarly allows
for protein sequencing on the PROSEQ-VIS platform.
Concurrent Screening of Multiple Targets (MULTIPLEX)
Attempts by others to screen against multiple targets using SELEX have
successfully multiplexed up to 30 biological similar targets in one SELEX
experiment
(e.g., VENNmultiplex SELEX by BasePair). Although the specific methods that
achieve
this are not known, it is likely that targets are bound to beads with
different spectral
content and incubated with aptamer candidates before being sorted by
fluorescence
activated cell sorting (FACS). This method limits the number of targets that
one can
multiplex at a time due to the optical limitations of the machinery.
73
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
The MULTIPLEX methods described herein allow for screening binders for
multiple peptide or protein targets at once. In addition, the MULTIPLEX
methods
described herein allow for detecting rare binding events in a high-noise
environment;
increasing target specificity; and conducting specificity assays for multiple-
target cross-
validation matrix analysis and machine learning analysis. The MULTIPLEX
methods
described herein can be used to identify interactions between essentially any
two
biological molecules (e.g., two DNA or RNA barcoded molecules such as
oligonucleotides and molecular targets, proteins and antibodies, small
molecules and
barcoded proteins) as long as both targets can be conjugated to
oligonucleotides that can
then be ligated to each other.
The MULTIPLEX methods described herein involve incubating the aptamer
candidates (FIG. 26A) with a diverse pool of unbound DNA-barcoded peptide
targets
(FIG. 26B). Upon aptamer binding, the 3' end of the single stranded aptamer is
ligated to
the peptide ssDNA barcode (FIG_ 26C), and the DNA portion is amplified via
PCR.
Sequencing the aptamer and its covalently attached DNA barcode provides the
aptamer
sequence along with the unique identifier that indicates which target the
aptamer was
bound to, thus eliminating the obstacle of identifying which aptamers are
bound to which
targets. FIG. 26D is a schematic that indicates the steps of the SELEX
procedure (from
FIG. 3) into which multiplexing can be incorporated.
The MULTIPLEX methods described herein can reduce labor and reagent costs
while improving data quality and broadening screening capabilities. In
addition, the
MULTIPLEX methods described herein can produce aptamers that specifically bind
to
their unique targets in an environment with a multitude of available targets
(e.g., cell
surfaces, human blood), thus, vastly increasing the pipeline for aptamer
discovery to
application.
1) Use of a DNA barcode to identifr peptide or protein targets
As described herein, the targets in the MULTIPLEX methods described herein are
peptide-oligonucleotide conjugates (POCs), which, with reference to FIG. 27,
are single-
stranded (ss) DNA tails (a) whose 3' ends are covalently linked to the C-
termini of
peptide or protein targets (b). A ssDNA tail (a) includes a 3' primer region
(c), a unique
DNA barcode (d), and a 5' bridge-binding sequence (e). An aptamer (t) includes
a 3'
bridge-binding sequence (g). After POC-aptamer binding in solution, a short
oligonucleotide bridge (h) can be introduced, where half of the short
oligonucleotide
74
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
bridge (h) is complementary to the 3' bridge-binding sequence (g) at the 3'
end of the
aptamer (f) and the other half is complementary to the 5' bridge-binding
sequence (e) of
the ss DNA tail (a). After the bridge oligonucleotide binds both the aptamer
and peptide
tail, a ligase enzyme can be added to seal the nick, unused bridge
oligonucleotides can be
degraded and/or removed, and the ligase enzyme deactivated. This results in
covalent
linkage of the aptamer (f) to the peptide (b).
Following ligation, bead-bound POC targets can be obtained (e.g., pulled down
using complementarily to biotinylated oligonucleotides), followed by removal
of (e.g.,
washing) unbound aptamers. PCR can be performed on the beads through the ssDNA
tail
and the aptamer, and the resulting DNA construct can be sequenced to obtain
the aptamer
sequence along with the barcode identifier of its protein binding partner
(boxed region in
FIG. 27).
2) Use of proximity-dependent DNA ligation to identify local aptamer binding
events
from global noise
One difficulty encountered in the MULTIPLEX methods disclosed herein is
constraining the assay in a way that favors the ligation of bound partners
over random
available substrates in solution, since peptide tails and aptamers that are
physically close
together are more likely to ligate to each other than to free-floating DNA.
Therefore,
ligation reaction conditions can be developed and optimized to maximize local
signal by
optimizing several experimentally-tested parameters including, without
limitation,
reaction time, substrate concentration, temperature, and reaction solution.
Additionally,
tails of varying lengths and bridge regions of varying lengths can be designed
and
characterized to optimize local interaction in a high-noise environment.
3) Nested PCR for additional rounds of Multiplex-SELEX
To achieve multiple rounds in the MULTIPLEX methods described herein, the
aptamer segment of the ligated aptamer-barcode product can be re-amplified
(e.g., using
nested PCR on the ligated complex with primers flanking the aptamer sequence)
and
processed (e.g., using purification via automated electrophoretic gel
separation), followed
by conversion to ssDNA (e.g., using enzymatic digestion). See FIG. 28.
4) Alternatives and variations on the multiplex methods
Many procedural modifications can be made to adapt the multiplex methods
described herein to suit different applications.
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
The MULTIPLEX methods described herein can be used to examine interactions
in different substrate binding scenarios; for example, and without limitation:
a) DNA -
peptide binding, where the interacting region includes an aptamer bound to a
peptide
target; b) DNA - DNA binding, where the interacting region includes a region
of base
complementarity between two strands of DNA. With DNA - DNA interaction, the
ability
to identify local signals has been demonstrated when binding partners
represent as low as
0001% of the total pool in a 500 nM concentration solution, demonstrating the
sensitivity
of the MULTIPLEX methods described herein.
Additionally, the MULTIPLEX methods described herein can be used to examine
substrate binding beyond DNA - DNA or DNA - peptide interactions. For example,
the
MULTIPLEX methods described herein can be used to examine binding between any
number of biological targets provided both targets can be bound to each other
(e.g., via
ligation of oligonucleotides). For example, a MULTIPLEX method similar to that
described herein can be employed to screen for RNA aptamers that bind small
molecule
targets or protein complexes.
An ssDNA tail can be attached to the C-terminus of a peptide or protein using
any
number of different techniques, including, without limitation, chemical
linkers (e.g., click
chemistry, SMCC linker, EMCS linker, etc.), biological linkers (e.g., biotin-
streptavidin
systems), cross-linking (e.g., using formaldehyde or UV), or the like.
In addition, it would be appreciated that a ssDNA tail can be attached to a
different region of the protein or peptide (i.e., other than the C-terminus).
For example,
the ssDNA tails can be attached to the N-tenninus, to a specific functional
group, amino
acid side chains, etc. Additionally or alternatively, multiple ssDNA tails can
be attached
to a single peptide or protein.
Ligation between the DNA ends can occur in multiple ways. Enzymatic ligation
in aqueous solution can be used, but it is also possible to ligate the DNA
ends chemically.
In some embodiments, alternative ends of the bridge can be used for ligation.
The
overhangs andVor the bridge can also be modified to include base-pairing
mismatches to
introduce a gradient of binding interactions, such that the binding
interaction between the
binder and target takes precedence over the binding interaction of the bridge.
It would be understood that the MULTIPLEX methods described herein can be
conducted in aqueous solutions or they can be tailored for use in a different
system, such
as on a fixed surface, on beads, in vivo, in a gel, or the like.
76
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
The MULTIPLEX methods described herein have been used to identify aptamers
with selective binding to peptide targets in a competitive multi-peptide
environment.
Like selective antibodies, the resulting aptamers are suitable to be used
alone or in
combinations of two or more to create constructs that control their multi-
target binding
distributions. For example, two aptamers, each highly selective for different
targets, can
be joined together in order to create a construct that binds two separate
targets;
alternatively, two aptamers with the same primary target but with different
off target
binding distributions can be added to the pool in parallel or sequentially to
increase the
binding readout to their common target through analysis of regions of
overlapping
distributions.
Replacing the aptamer in the MULTIPLEX methods described herein with a
molecule that has been DNA barcoded and has a 3' C overhang arm allows for
measuring
binding between different mixtures of any of the molecule classes previously
described,
enabling bi-directional multiway competitive measurements of any of the
combinations of
molecule classes: including, peptide vs protein, protein-protein, antibody-
protein, small
molecule-protein, peptide-cell surface marker, antibody-cell surface marker,
etc. In some
embodiments, both the binder and the target molecules can be drawn from any
mixture of
molecules from any of the above classes, allowing for measurement of cross
binding in
complex competitive environments.
The MULTIPLEX methods described herein provide a high-sensitivity tool for
detecting low-level binding events in a large substrate pool. The MULTIPLEX
methods
described herein reduce the need for a large number of rounds of SELEX (e.g.,
8 to 20
rounds) and simultaneously allow for multiplexing several peptide targets in
one solution.
As a result of reduced rounds, the MULTIPLEX methods described herein
minimizes the
number of PCR amplifications that must be performed on the aptamer pool and,
thus,
minimizes the bias introduced with every round of amplification. Increased
specificity
and reduction of off-target binding is an added benefit in the MULTIPLEX
methods
described herein. For example, if a unique aptamer is isolated that binds to
peptide target
#1 in a mixture containing targets #1-10, it also is known that the aptamer,
in addition to
binding to target #1, does not bind to targets #2-10 (under those same
conditions). This
reduces the likelihood of selecting non-specific aptamers that may bind other
targets in
addition to the target of interest.
77
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Target Protein and RNA-Binding Protein Fusion (TURDUCICEN)
The classification of binding interactions is highly desirable in a number of
research areas including in drug development, diagnostics, and basic research.
Protein
and peptide libraries contain a bank of interesting biological targets against
which binders
(e.g., aptamers, small molecules, antibodies, etc.) can be screened.
Presently, screening is
typically performed in individual reactions where the identity of the protein
or peptide
target is known, making large-scale screening, particularly of unknown
targets, cost and
labor prohibitive. Pooling and screening several targets at once allows for
scaling and
greater binding specificity, however, there is currently no available method
for creating
target libraries where the identity of each target in a pool can be easily
deduced.
Biological approaches for creating protein or peptide libraries rely on the
cloning
and purification of each protein individually into a model system such as
yeast or E. colt
(Jia & Jeon, 2016). To create a library of 1,000 unique proteins, researchers
must
perform 1,000 separate transformation reactions, protein purifications, and QC
processes,
before finally pooling the proteins together. Chemical synthesis can reliably
produce
peptide pools, but quickly can become cost-prohibitive and technically
challenging for
larger proteins and protein complexes.
Importantly, existing methods for creating libraries do not enable scientists
to
easily identify individual elements once the components are pooled. Common
techniques
for identifying proteins include mass spectrometry, antibody binding assays,
and affinity
tag binding assays (Miteva, Budayeva, & Cristea, 2012). Concentration
thresholds of
unique elements within a pool of proteins limit the use of mass spectrometry
for the
identification of lowly expressed individual proteins from a large pool;
antibodies are
often inconsistent, non-existent, or cost prohibitive for novel targets; and
the affinity tag
approach limits pool diversity to the number of unique affinity tags
available.
The TURDUCICFN methods described herein allow a mixture of thousands of
unique proteins to be made, tagged, screened and identified in one pool. The
TURDUCICEN methods described herein allow for the production of a diverse
protein
pool and the screening of such a diverse protein pool.
1) Protein Expression
An in vivo system in S. cerevisiae and E. coli is described in which each
transformed cell is engineered to produce a different protein of interest
(POI), which can
be non-covalently linked to a RNA barcode whose sequence can be used to
identify the
78
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
POI; the non-covalent linkage relies on the natural interaction between an RNA
binding
site and its corresponding RNA-binding protein (REP). See, for example, FIG.
29.
Representative RNA binding sites and their corresponding RBPs that can be used
in such
constructs include, without limitation, the MS2 RNA hairpin bound by the MS2
phase
coat protein and the boxE sequence bound by the bacteriophage anti-terminator
protein N
(lambdaN). Each POI ( FIG. 29A) can be expressed as a fusion protein with a
RNA-
binding protein (part B of FIG. 29) in which the POI can be non-covalently
linked to a
specific RNA binding site (part C of FIG. 29), which is recognized by the RNA-
binding
protein and a unique barcode (part D of FIG. 29). Each construct in the pool
typically
contains a POI fused to a REP, a DNA sequence that encodes the RNA sequence
that is
recognized by the RBP, a unique RNA barcode, and a promoter to drive
expression.
Representative promoters include, for example, the Gal 1,10 bidirectional
promoter,
ADH1, GDS, TEF, CMV, EF la, SV40, T7, lac, or any other promoter and promoter
combinations compatible with the host organism. A pool containing the plasmids
of
various POI-REP fusion genes as well as their corresponding RNA barcode
sequence can
be transformed into S. cerevisiae with an approximate dilution of I plasmid
per cell (FIG.
30A). POI fusions made in vivo then bind their corresponding RNA barcodes
inside the
cell (FIG. 30B), which then can be purified (FIG. 30C). FIG. 300 is a
schematic that
demonstrates where, relative to the SELEX method (FIG. 3), the products of
TURDUCKEN as described herein can be used.
2) Protein Purification
POI-RNA complexes can be obtained using any number of methods, resulting in
only complexes containing both the POI fusion protein and the RNA barcode are
collected. Simply by way of example, the complexes can be pulled down from a
cell
lysate via a His-tag or other purification tags, which can be included in the
protein fusion
component of the POI. POIs then can be washed and released from the anti-His
beads or
other pull-down assays compatible with the purification tag used, and further
purified
using a streptavidin-coated bead and a biotinylated oligo that is reverse
complementary to
a sequence in the RNA barcode. After this pull-down step, a mixture of beads
are
obtained that are bound to the POI-RNA complex, biotinylated oligonucleotides
annealed
to random RNA sequences, or nothing. The POI-RNA complex can be released from
the
streptavidin-coated beads and purified by heating and washing the mixture to
denature the
79
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
RNA and biotinylated oligonucleotide or by releasing the complex using
restriction
endonucleases.
3) Protein pool for use in aptainer binding assays
The final product from this method is a diverse pool of proteins, each
identifiable
by an attached RNA barcode. This design allows for the use of this protein
pool in
multiplexed aptamer screening assays. For example, a pool of potential
aptamers that
also contain their own unique nucleic acid barcode can be incubated together
with the
protein pool and aptamers from the pool of potential aptamers are allowed to
bind their
targets. Through controlled enzymatic ligation (e.g., see the MULTIPLEX
methods
described herein), the non-covalently bound aptamer's barcode can be ligated
(e.g.,
covalently) to the POI-RNA complex barcode. By sequencing through the ligated
product, the aptamer sequence can be obtained, which provides the identity of
its target.
The TURDUCICEN methods described herein allow for:
a) labeling of proteins in vivo using a nucleic acid barcode;
b) producing a large, diverse protein pool in a single transformation
reaction;
c) identifying each component of the protein pool using NGS sequencing; and
d) carrying out screening against multiple targets in one pooled reaction.
Other methods of generating DNA-barcoded proteins, such as chemical synthesis,
are unable to operate on a large scale and must be performed in individual
samples or
wells. The TURDUCICEN methods described herein provide the ability to express
and
barcode thousands to millions of different proteins in the same pool ill vivo
with low rates
of mislabeling proteins. This method saves significant time and money.
Additionally, the
TURDUCKEN methods described herein provide the advantage of being able to
screen
many targets at once simultaneously.
It would be understood that procedural modifications can be made to adapt the
TURDUCKEN methods described herein to suit different applications. For
example:
.10 any number of organisms in addition to yeast
(e.g., E colt, mammalian CHO
cells) can be engineered to produce protein-of-interest and nucleic acid (POI-
NA)
complexes.
= the nucleic acids used in the TURDUCIC_EN methods described herein can be
expressed from a variety of different constructs or vectors (e.g., circular
plasrnids,
linear inserts, or chromosomally-integrated DNA).
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
= alternate strategies for linking two substrates in vivo to create the POI-
NA
complexes (e.g., different RNA-binding proteins such as MS2 or Box13/1ambdaN
systems, HUH-endortuclease domains, CRISPR associated protein).
= DNA barcodes can be used instead of RNA barcodes using linker systems
such as
Spycatcher / Spytag, TALE, etc.
There are many potential uses for the in vivo protein labeling provided by the
TURDUCICEN methods described herein. For example, the TURDUCKEN methods
described herein can be used to study interactions between molecular targets
(e.g.,
aptamers, small molecules, etc.) for basic or translational research. For
example,
fluorescent probes hybridized to the POI-DNA complex can be used to visualize
proteins
in vivo as a screening tool for drug discovery applications. For example, the
TURDUCICEN methods described herein can be used to mine for aptamers that then
can
be used as an alternative to antibodies (e.g., as molecular probes, for
targeted drug
delivery, etc.).
Generating Large, Diverse, and Controlled DNA Libraries by Ligation (LEGO)
Systematic Evolution of Ligands by Exponential Enrichment (SELEX) is a
biomolecular technique traditionally used to identify aptamers that is
designed to isolate
strong binders from a large pool of random aptamer candidates since it is
extremely
difficult and expensive to synthesize such a large pool of specific sequences.
However, if
one could generate their own initial SELEX starting aptamer pools, the
landscape of
SELEX experiments could allow for specialized adaptations, such as using ML-
predicted
sequences for a target as the starting aptamer pool. In order to accomplish
the generation
of such large, diverse, yet controlled or known libraries, a protocol referred
to as LEGO
was developed. For a 40-mer ssDNA oligo, there are 1024 possible oligos that
could be
explored, but each SELEX experiment only assays 108-1014 of the total possible
experimental space. This represents only a small fraction of all the DNA
sequences
possible, such that, in practice, even the most optimized experiment has a low
probability
of finding the best aptamers for a particular target. Research has
demonstrated that there
are particular two dimensional structures, or secondary structures such as G-
quadruplexes, that are often seen in aptamers (Tucker, Shunt, & Tanner, 2012),
and it is
hypothesized that these secondary structures increase the aptamer's binding
capabilities.
The ability to generate an initial input library, rather than being restricted
with the use of
81
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
a random library, that biases towards popular secondary structures over
unstructured
aptamers would accelerate binder discoveries. Additionally, as artificial
intelligence
predictive algorithms, such as ML, increases their predictive capabilities; ML-
guided
input libraries for aptamer experiments would significantly increase the
relative ratio of
the potential aptamer candidates to non-candidates in the starling pool, and
potentially
reduce the number of rounds to find equally high affinity aptamers. As a
result, with
fewer SELEX rounds, aptamer candidates could be discovered faster, require
less cost for
discovery and discovered candidates would have reduced impacts from
experimental
noise such as PCR bias. In other words, fewer downstream quality control
assays would
need to be conducted to confirm that top aptamer candidates are true binders
over aptamer
candidates that happen to PCR extremely well and without specifically
preference for the
target of interest. Additionally, one could consider iterating an approach
where a few
rounds of SELEX are conducted from a random library, the library is sequenced,
the
resulting data is fed into an MI. model, the model predicts what the next
initial starting
pool should look like (either features such as secondary structure or GC
content, or direct
sequences), and then a new library is generated for a new, more targeted SELEX
experiment is started.
While random libraries can be synthesized cheaply, there is no current cost-
effective method for generating large pools whose parameters (e.g., GC
content, recurring
motifs, fixed regions, length, etc.) can be easily determined and manipulated.
Current
methods for synthesizing short (>200 bp) DNA pools provide either
a) high diversity with little control over sequence content: random DNA
libraries
with customizable primer regions can be chemically synthesized at low cost
(e.g., under
$300, TriLink Biotech). However, generating 1014 specified sequences by
conventional
microarray synthesis is prohibitively expensive (e.g., Integrated DNA
Technologies:
$2000 for lk sequences 200 bp long; Agilent: $13,000 for 244K sequences 90-bp
max;
Twist Biosciences $46k for 1M sequences).
b) high control over sequence content with limited sequence diversity: groups
have developed methods to construct DNA libraries by stitching together
building blocks
using 12-base fragments in a one-pot reaction (Fujishima et al., 2015) or 8-
base fragments
sequentially on an immobilized system (Horspool et al., 2010). Both of these
methods
possess constraints which restrict their use for aptamer library construction.
82
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
The LEGO methods described herein allows for the construction of
computationally-derived, customizable DNA libraries that allow scientists to
perform
SELEX screens using a controlled input pool at a reasonable cost It makes use
of
commercially available ligase enzymes to assemble a library of random 40-mers
from
sequential ligation of 5-mer or longer DNA LEGO pieces. There are at least two
ways
this can be done: by double-stranded ligation using a dsDNA ligase such as T4
DNA
ligase (FIG. 31A) or by template-independent single-stranded ligation using a
ssDNA or
ssRNA ligase such as RNA ligase RtcB (FIG. 318). In both strategies, ligation
begins
with the linkage of a forward PCR primer to the first LEGO piece, and
continues by
adding one LEGO piece at a time. The final ligation reaction takes place
between the
final LEGO piece and the reverse PCR primer (FIG. 32A ¨ 328). Production of
the
primed 40-mer can be followed by amplification methods such as PCR using a
protected
forward primer and phosphorylated reverse primer. The PCR product can be
cleaned
using any preferred method and products of the correct base pair length can be
selected
using size selection methods such as the automated PippinHT program. The
library can
then be converted from double to single-stranded DNA, for example, using
lambda
exonuclease digestion, and the single-stranded product can be cleaned and
concentrated
(FIG. 32C). FIG. 32D is a schematic that demonstrates where, relative to the
SELEX
method (FIG. 3), the products of LEGO as described herein can be used.
The methods described herein have several several unique features that make it
optimal for creating aptamer libraries:
1) Unique overhang design allows for positional
control for dsDNA ligation
Successful ligation between two fragments of double-stranded DNA requires
complementary single-base overhangs on both fragments. A pair of DNA blocks
possessing compatible overhangs (e.g., A and T, G and C) preferentially ligate
together.
Blocks with incompatible overhangs (e.g., A and C, G and T, etc.) legate
together
significantly less often. By using blocks with different combinations of A, T,
C, and G
overhangs, block positioning can be controlled. For example, blocks can be
encouraged
to assemble in the order 1-2-3 instead of 2-1-3, 3-1-2, etc. by designing them
such that the
overhangs of blocks 1 & 2 are compatible while those of 1 & 3 are not
2) Short building blocks allow the whole DNA space, including sequences which
are difficult to synthesize to be explored
83
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Libraries several magnitudes more diverse than those generated by other
ligation
methods can be created using shorter LEGO pieces. Using a bank of 1,024 5-
mers, the
entire space of 40-met DNA libraries (1024 unique sequences) can be generated.
With the
use of a single 1536 plate, any 40-nrier aptamer or feature-spaced library
that an
experiment demands can be assembled. Additionally, certain sequences (e.g.,
long chains
of G's) are difficult to synthesize accurately by conventional methods.
Stitching together
many shorter blocks provides a useful way to access these sequences.
It is understood that a number of modifications can be made to the methods
described herein. For example:
= Library design: while the methods described herein use 5-mers to
construct
40-mers, libraries of a different length/multiple lengths from building
blocks of a different length/ multiple lengths can be built. During DNA
synthesis, there is a low rate of 5' phosphorylation for oligonucleotides
that are short (i.e. <6 nt) due to steric interactions from the glass
substrate.
Increasing the length of constructs used will increase the percentage of
phosphorylated oligo reagents. However, increasing the lengths of the
oligo pieces will require a larger number of different oligo pieces for
assembling a library of the desired statistical distribution of sequences.
= Building block design: the methods described herein with dsDNA use
blocks that have phosphate group modifications on the 5' ends of both
strands in order to facilitate ligation of the block to the growing strand and
to the next piece in the sequence. Instead, pieces on which there is only
one 5' phosphorylation can be used to reduce the potential for a flipped
DNA block to be integrated/ligated into growing sequences. Alternatively,
ligation-inhibiting modifications could be added onto 5' or 3' strands to
discourage ligation of flipped pieces. For ssDNA ligation, the methods
described herein use pieces that have 3' phosphorylation modifications,
which is required for the RtcB enzyme to facilitate this reaction.
= Starting material: XNAs, RNA, modified RNAs, single-stranded DNA or
modified DNA, instead of unmodified double-stranded DNA, could be
used to construct libraries with compatible ligases.
= Linking method: there are multiple ways to connect strands of DNA
together. The methods described herein uses T4 DNA ligase or RtcB
84
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
ssRNA ligase to enzymatically link DNA building blocks together.
Different ligase enzyme (ex. K con DNA ligase, CircLigase, thermostable
ligases, etc.) or link building blocks chemically (e.g., click chemistry)
could be used.
= Ligation method: instead of doing a one-pot sequential ligation reaction,
several smaller ligation reactions could be performed to create large
blocks, and then pool the products to ligate the large blocks together. This
can increase control over block position.
= Medium: instead of doing the library construction in solution, the
reaction
can be performed on beads, on a solid support, in a gel, etc.
= Size selection: in the ligation of these small pieces of DNA together,
oftentimes the ligation products are not of the desired length. In order to
purify the full-length products, manual and automated size selection
methods such as the PippinHT automated DNA size selection system can
be used.
Additionally, while the methods described herein can be used to generate
random
libraries for SELEX aptamer screens, the methods described herein also can be
used to
generate DNA libraries for different applications, such as:
= building ML-derived DNA libraries for peptide/protein generation via
translation. A priority in the SELEX aptamer screens described herein is
to find aptamers that are specific to their amino acid targets. In order to do
so, the same pool of random aptamers can be incubated with peptides of
different sequences. Obtaining all the different peptide sequences that
may be needed from vendors can be quite expensive, given that,
oftentimes, many different variations of the same sequence need to be
tested. In order to expand the space of random peptides that is available to
use for SELEX, it would be helpful to be able to produce these peptides in-
house. The methods described herein of random DNA library generation
can produce these peptide libraries via cell-free translation kits or
conventional DNA plasmid transformation experiments in cells. Promoter
sequences can be included in the design of the adapter region blocks, or
ligated on post library generation, and peptides could be generated from
these sequences in vivo or in vitro.
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
= building out sequences of DNA barcodes. The key to performing protein
sequencing is the ability to encode and subsequently readout a sequence of
amino acids. In a number of the protein sequencing methods described
herein, DNA barcodes can be used to encode for identified regions of an
amino acid sequence. In these methods, when an aptamer binds to the
portion of the protein or peptide being sequenced, a DNA barcode region
on the aptamer attaches, through any suitable linkage method, to a growing
barcode chain. The enzymatic ligation methods described herein can be
used to link together the barcodes to form the barcode chain or to attach a
barcode to a universal adaptor.
= modifying PROSEQ reagents. In a number of the protein sequencing
methods described herein, functional aptamers and processed peptides
contain regions of DNA such as spacers, barcodes, and ligation consensus
regions. For peptides to be sequenced, a shorter oligo linker (e.g., 6 nt),
can be conjugated to an amino acid residue to increase the rate of reaction
before ligating the rest of the DNA elements in a LEGO-like manner. For
aptamers found in SELEX, DNA tails that include a unique barcode for
aptamer identity, cycle number, restriction site, etc. can be directly ligated
onto the 5' end of the aptamer using a single stranded ligase such as Rtc13.
Additionally, asymmetric PCR can be employed to modify binders found
in SELEX to be used directly on the PROSEQ platform.
The LEGO methods described herein allow for the creation of oligo libraries
that
can be customized to have certain properties (e.g., (IC content, recurring
motifs, etc.).
These libraries are several magnitudes more diverse than those generated by
other ligation
methods and can be assembled at a reasonable cost.
In accordance with the present invention, there may be employed conventional
molecular biology, microbiology, biochemical, and recombinant DNA techniques
within
the skill of the art. Such techniques are explained fully in the literature.
The invention
will be further described in the following examples, which do not limit the
scope of the
methods and compositions of matter described in the claims.
86
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
EXAMPLES
Relevant information for both RCHT and N-terminal amino acid SELEX
Experimentation
The following will be described below:
A. General methods for all SELEX Experimentation
B. RCHT-SELEX Experimentation
B.1 RCHT-SELEX General Experimentation Part I
B.2 RCHT-SELEX Incubation Variations
B.3 RCHT-SELEX General Experimentation Part!!
B.4 RCHT-SELEX Additional Components
C. RCHT-SELEX Results
D. N-terminal Amino Add SELEX Experimentation
E. N-terminal Amino Acid SELEX Results
F. Generalized SELEX protocol
General workflow for all SELEX (RCHT-SELEX and N-terminal Amino Acid
SELEX) experiments is shown in FIG. 33.
Reagents
Aptamer libraries were purchased from TriLink Biotechnologies and IDT, with
all
other oligonucleotides purchased from IDT or synthesized in-house by K&A
LABORGERATE H-8 DNA & RNA Synthesizer. All oligos were purified via HPLC
(either IDT internal system or in-house Agilent 1290 Infinity II). All
automated
procedures were performed on the Agilent Bravo NGS Workstation or Opentrons OT-
2.
All SPRI purifications utilized Mag-Bind TotalPure NGS beads from Omega
Biotek. All
DNA quantifications were obtained using dsDNA and/or ssDNA High Sensitivity
Qubit
Fluorescence Quantification Assay (Thertnofisher). A9932 All water used was
AmbionTm Nuclease-Free water.
Libraries
87
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Single-stranded N40 aptamer libraries consisted of 40 random bases, flanked by
custom primer regions. In order to mitigate contamination by excessively
enriched
aptamers from past experiments, the primers on N40 libraries were switched
every 2-3
months. The initial N40 library
(TAGCiGAAGAGAACGACATATGATNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNTFGACTAGTACATGACCACTI7CiA (SEQ ID NO:1)) was
ordered directly from TriLink Technologies. Subsequent custom primers were
designed
by using random sequence generator tools to generate putative sequences, cross-
validated
against in-house primer sets to avoid sequences that were too similar, and
then using the
IDT Oligo Analyzer to check for melting temperature as well as self and
heterodimers,
The custom primers were also quality checked using an abbreviated SELEX cycle
before
being used for the full SELEX process.
N40 libraries used:
= SELEX N40 Libraryl (also referred to as the TriLink library):
TAGQGAAGAGAAGQACATATGATNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNYFGACTAGTACATGACCACYFGA (SEQ ID
NO:2)
= SELEX N40 1ibraiy2 (also referred to as 0MB63):
CACTFGANNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNCACATCAGACTGGACGACAGAA (SEQ ID
NO:3))
= SELEX N40 Library 3 (also referred to as OMB105 or Wolverine2):
TGATGCTATGCGACTFATFGTACNNNNNNNNNTThJNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNTACYfGGCGYCTUACCACCA (SEQ ID NO:4)
Peptides
Biotinylated peptides were synthesized by Genscript. To facilitate attachment
of
the peptide to biotin, all C-terminal residues were lysines. The construct of
each peptide
was as follows: N-terminus- (2-mer prefix) - (8-mer suffix) - C-terminus-
BIOTIN.
2-mer prefixes: The 20 naturally occurring amino acid prefixes were divided
into
4 groups with 5 amino acids each. 2-rner prefixes were determined by pairing
amino
acids within a block with each other, and with amino acids from other groups.
Each 2-
88
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
mer prefix therefore belonged to one of 16 blocks (with 25 potential 2-mers to
a block).
In total, there are 400 possible 2-mer prefixes. For reference, the 400
potential prefixes
have been depicted in FIG. 34A. The 16 blocks are depicted in FIG. 34B.
8-mer suffixes: For the dipeptide switch experiments, each 2-mer prefix was
associated with 2 suffixes, out of four possible suffixes. Furthermore,
whether there is a K
or C on the end is dependent if the peptide is biotinylated (without DNA oligo
attached)
or made with a DNA oligo attached (PoC) respectively. These suffixes were:
= A' suffix: ADRWADR(K or C) (SEQ ID NO:5)
= B' suffix: MSQPLQP(K or C) (SEQ ID NO:6)
= C' suffix: NHFENEI(K or C) (SEQ ID NO:7)
= D' suffix: TKYVGTG(K or C) (SEQ ID NO:8)
= E' suffix: TAYVETE(K or C) (SEQ ID NO:9)
= F' suffix: QGHSIDN(K or C) (SEQ ID NO:10)
The two suffixes assigned to each 2-mer prefix were chosen to avoid similarity
with the 2-mer prefix. For example, a 2-mer prefix from the AB block would be
associated with the C' and D' suffixes, but not the A' and B' suffixes.
The suffix paired with the 2-mer prefix was alternated between odd and even
rounds, with only the 2-mer prefix the constant peptide combination exerting
selective
pressure on the aptamers through all 4 rounds (FIG. 34C). Examples of suffix
and prefix
combinations for DD and DC prefix experiments are depicted in FIG. 3413.
Section B RCHT-SELEX Experimentation
B.1 RCHT-SELEX General Experimentation Part I
Example 1¨RCHT-SELEX Experimentation
Methods
Pre-SELEX cycle methods:
Bring Up
Depending on experimental needs, bring ups were performed via one of three
variations. All bring ups were performed using 50 microliter PCR reactions,
using
Herculase II Fusion DNA Polymerase (Agilent Technologies). PCRs were SPRI-
purified
89
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
at a 0.6X ratio using Mag-Bind TotalPure NGS beads (Omega-Biotek) with the
addition
of 100% ethanol on a Bravo Automated Liquid Handling Platform (Agilent). The
amplification conditions for this and all subsequent PCR reactions (with the
exception of
NGS preparation) were as follows: an initial denaturation at 95 C for 5
minutes followed
by 13 amplification cycles of 30 seconds of denaturation at 95 C, 30 seconds
annealing at
55T, 30 seconds elongation at 72 C, and a final elongation of 5 minutes at 72
C.
To facilitate regeneration of ssDNA libraries for aptamer incubation (detailed
in
the section on digestion), protected and phosphorylated primers were used. For
the
following primer constructs, * indicates the nucleotide was modified such that
the sulfur
atom in the phosphate backbone was substituted for a phosphorothioate bond
substitutes a
sulfur atom, which renders the sequence more resistant to nuclease digestion.
= SELEX N40 Libraryl (also referred to as the TriLink library):
o Forward primer: 5'- T*A*G*G*G*A*AGAGAAGGACATATGAT -3'
(SEQ ID NO:11)
0 Reverse primer: /5Phos/ - TCAAGTGGTCATGTACTAGTCAA - 3' (SEQ
ID NO:12)
= SELEX N40 1ibrary2 (also referred to as 0MB63):
o Forward primer: 5' - T*T*G*A*C*T*AGTACATGACCACTTGA -3'
(SEQ ID NO:13)
o Reverse primer: /5Phos/ - TTCTGTCGTCCAGTCTGATGTG -3' (SEQ
ID NO:14)
= SELEX N40 Library 3 (also referred to as OMB105 or Wolverine2):
o Forward primer: 5' - T*G*A* T*G*C* TAT GCG ACT TAT TGT AC -3'
(SEQ ID NO:15)
o Reverse primer: /5phos/ -TGG TGG TAA GAACGCCAAGTA -3' (SEQ
ID NO:16)
Bring Up Variations
Option 1 (primarily used):
A sample of 1012 sequences (-48 ng) from the single-stranded N40 library were
amplified across 288 reactions of 50 microliters each. The SPRI-purified
product of all
288 reactions were pooled, to give us a fmal bring up with a diversity of 1012
sequences
with approximately 1200 copies to be split across 12 SELEX reactions. This
method was
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
used to identify aptamers to the biological controls bradykinin, argipressin,
and GnRH, as
well as a subset of the dipeptide switch experiments.
Option 2:
Two samples of 1012 sequences (-48 ng each, ¨96 ng total) from the single-
stranded N40 library were amplified across 576 reactions of 50 microliters
each. The
SPRI-purified product of all 576 reactions were pooled, to give us a final
bring up with a
diversity of 2 x 1012 sequences, to be split across 36 SELEX reactions. This
method
provided the input pools for the majority of the dipeptide switch experiments.
Option 3: Double Bring Up:
A bring up was performed in the style of variation 1, but with unmodified
primers
instead of the protected and phosphotylated versions. Aliquots of the purified
bring up
(with diversity of 1012 sequences) were used as a dsDNA input library for a
second bring
up (of either Variation 1 or 2) with the modified primers. A total of ¨48 ng
of each
dsDNA aliquot was amplified across 288 reactions. The double bring up allows
for the
same input of 1012 sequences to be used across multiple sets of experiments,
far
exceeding the customary 12-18 SELEX reactions to which its distribution is
usually
limited.
Bring Ups: Spike-ins
Depending on experimental needs, N40 constructs with known sequences were
spiked into the bring up and carried through subsequent rounds of SELEX. These
sequences were:
= A6: high_gc_5:
TAGGGAAGAGAAG-GACATATGATCACCGCATCCTGAGG-CCGGTGTGG
AGGGCACGAAGTCTGGITGACTAGTACATGACCACTTGA (SEQ ID
NO:17)
= C2: hig,h_gc_5:
TAGGGAAGAGAAGGACATATGATCTAGCATGGTGCCCTTACCCTCAGA
GCGGAAGTACCTGATTTGACTAGTACATGACCACTTGA (SEQ ID NO:18)
¨5.39 million molecules of each spike-in were present in each 50 ul reaction
during the initial bring up, making each spike-in 53,947 limes more abundant
than the
average random N40 sequence
Refolding
91
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Aptamer libraries were heated to 95 C for 5 minutes and then cooled on ice for
30
minutes to refold the DNA secondary structure into their lowest energy state.
Negative Selection
To remove aptamers that would otherwise bind to reagents consistently present
across samples throughout the assay, oligo libraries underwent negative
selection before
they are used as input for SELEX. 166.62 pmol (4650 ng) of refolded ssDNA
library are
added to 500 ug of streptavidin coated beads (Cl, Ti, M270, or M280 depending
on
experimental needs) and brought to a final volume of 400 ul, at a
concentration of lx
PBS, 0.025% Tween, and 10 ing/inli BSA. The reaction is incubated at room
temperature
(RT) of 22-24 C with rotation for 30 minutes before the supematant is
collected.
When using peptide-oligo conjugates, the oligo-only tail is selected against.
The
oligo tail is incubated with a 5' biotinylated oligo with full length
complementarity to the
oligo tail at a 1:2 tail:complement ratio. Then, a sample containing 1.67 pmol
of the oligo
tail and 3.34 pmol of the complement are added to 166.62 pmol of the refolded
ssDNA
library previously negatively selected against beads. The reaction is
incubated at room
temperature RT with rotation for 30 minutes before adding 200 ug of
streptavidin coated
beads and incubating for a further 30 minutes. The supernatant from this
incubation is
then collected as the final negatively selected input.
Digestion
Amplified libraries were converted to single-stranded DNA (ssDNA) by
enzymatic digestion using lambda exonuclease (New England BioLabs) and SPRI-
purified by automated bead clean up. ssDNA digestion completion was qualified
using
the small RNA kit (Agilent) on the Bioanalyzer 2100 (Agilent), and the
concentration
quantified post-clean via a ssDNA Qubit Assay (Thermofisher).
SELEX cycle methods:
Refolding
Before each SELEX incubation, aptamer libraries were heated to 95 C for 5
minutes and then cooled on ice for 30 minutes to refold the DNA secondary
structure into
their lowest energy state before every SELEX incubation.
B.2 RCHT-SELEX Incubation Variations
SELEX Incubation:
92
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
There are three variations on how the peptide may be incubated with the ssDNA
aptamers. With variant 1, the initial SELEX incubation happens in the presence
of
streptavidin beads (Variation I: SsDNA incubation with peptide-bead
conjugate); with
variant 2, streptavidin beads are added after the majority of the incubation
is complete
(Variation 2: SsDNA incubation with peptide-oligo target followed by bead
pulldown).
With variant 3, the peptide-oligo target is incubated with a biotinylated
primer prior to
addition of a partially double-stranded aptamers (Variation 3: (5') Blocked
Aptamer
incubation with peptide oligo-conjugate, with bead pulldown). See FIG. 35.
In all cases, ssDNA pools were heated to 95c-C for five minutes, then rapidly
cooled on ice prior to incubation. For each reaction, up to 166.62 pmol (4650
ng) of
folded aptamers were added to the peptide or peptide-bead conjugate and
brought up to
400 ul total volume at a final concentration of 1X PBS and 0.025% TWEEN20. The
final
incubation buffer for variant 3 also incorporates BSA at a final concentration
of 10
mg/ml. These buffer conditions can be distinguished as:
= SELEX BUFFER V.1 (also referred to as SELEX buffer): 1X PBS and 0.025%
TWEEN20
= SELEX BUFFER V.2 (also referred to as SELEX buffer with BSA enrichment):
1X PBS, 0.025% TWEEN20, 10 mg/ml BSA
These buffers are prepared from 10X PBS (Sigma-Aldrich), TWEEN20 (Sigma
Aldrich),
and powdered Bovine Serum Albumin (Sigma Aldrich).
Variation I: SsDNA incubation with peptide-bead conjugate
Peptide conjugation with beads
After deciding on a concentration gradient for the SELEX experiment, the
peptide
targets on beads can be made in advance in one large batch to avoid round-to-
round error
caused by multiple conjugations. The beads can be frozen and thawed a single
time
without any experimental defects. Aliquots for each round were made and stored
in
either Eppendorf LoBind or Nunc plates in -20t until taken out to thaw. Unit
tests were
performed on freshly conjugated beads vs frozen beads to ensure similar
properties, and
no discrepancies were found. The amount of target to produce should be based
on the
number of rounds, the starting concentration of the first round and a buffer
stock in case
there are experimental mishaps. In this example, 1:10 starting ratio of
target:DNA
aptamers is used. Using the Bravo Automated Liquid Handling Platform
(Agilent), 18.5
93
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
pmol of peptide was incubated with 87.2 ug (8.72 ul of a 10 mg/ml stock) of
MyOne
Streptavidin Cl Beads (ThermoFisher) for 30 minutes with mixing. After 2
additional
washes with SELEX buffer, each initial mixture of 18.5 pml of peptide and 87.2
ug of
beads was resuspended in 50 iii of SELEX buffer. These numbers were scaled up
proportionately in order to create a large volume bead-conjugate stock that
could be
aliquoted and frozen at the beginning of each experiment. 50 ul of this stock
could be
added to 4650 ng of input ssDNA for a 1:10 target:ssDNA stringency experiment,
and
directly scaled down to a smaller volume for experiments with less than 4650
ng of input
ssDNA. For experiments with the higher stringency of 1:25, the volume of
peptide-bead
conjugate added was further scaled down using a multiplier of 0.6X,
Depending on experimental needs, BSA-blocked M280 or T1 beads were used, or
unblocked M270 or Cl beads. M280 and M270 beads had a diameter of 2.7 tun, and
Cl
and Ti beads had a diameter of 1 urn. Unit tests demonstrated that Cl beads,
which
manufacturers indicated were best for automation, pulled down different
aptamer
sequences from a bringup than M280, M270 and Ti beads. The mechanism for this
result
is unknown. As a result of the unit tests, M280 beads were selected for
experiments
moving forward since BSA-blocking was preferred to prevent for the selection
of
aptamers to the bead surface, and the larger surface area targets could
provide a platform
where individual peptides are placed further apart reducing selection for
aptamers that
prefer peptide dimerization.
Blank bead 'conjugates' were created by putting a mixture of beads and water
through the same automated Bravo protocol, with the full 30 minute incubation
and 2-3
wash cycles. Each initial input of 87.2 ug of beads was also resuspended in 50
ul of
SELEX buffer, and later added to ssDNA at a ratio of 87.2 ug of beads for
every 4650 ng
of ssDNA (for 1:10 stringency reactions) or 34.88 ug of beads for every 4650
ng of
ssDNA (1:25 stringency reactions).
SELEX Incubation
Up to 50 ul of the bead-conjugate was added to 166.62 pmol (4650 ng) of folded
aptamer, and incubated with rotation at RT for 2 hours.
Streptavidin-Biotin Purniown
Streptavidin M280 beads (Invitrogen) were added to the SELEX incubations at
83.33 ug for every 51.02 pmol of peptide present for 30 minutes under
rotation.
94
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Variation 2: SsDNA incubation with peptide-oligo and aptamer incubation
followed by
bead pulldown
Peptide conjugation
No conjugation is required before incubation for this variation. Target is a
peptide-oligo.
SELEX incubation
Amount of added target depends on the desired stringency gradient. Often for
small molecule targets a range of 1:1 to 1:10 (target:ssDNA) stringency
conditions were
used as starting conditions, held through target switch rounds and then the
ratio between
target: DNA was increased in subsequent rounds until sequencing data
demonstrated
enrichment for aptamers. Here, the methods used for an approach for a starting
with a
1:10 target:ssDNA is described. For rounds 1 and 2, 166.62 pmol (4650 ng) of
folded
aptamers were directly added to 18.51 pmol of the peptide-oligo construct, for
a
stringency of 1:10 target:ssDNA. To account for the reduced 1:25 stringency in
rounds 3
and 4, 166.62 pmol (4650 ng) of aptamer was directly added to 7.40 pmol of the
peptide.
The peptides and ssDNA were incubated for 2 hours with rotation at RT.
Streptavidin-Biotin Pulldown
In cases where targets had DNA oligo tails, a biotinylated primer (5' Biotin
TAGGGAAGAGAAGGACATATGAT 3' (SEQ ID NO:19)) that anneals to part of the
oligo tail was added to the SELEX incubations at a 1:2 peptide:biotinylated
oligo ratio for
every 51.02 pmol of peptide present for 30 minutes under rotation. The primer
had two
functions: (1) to prevent aptamers from binding to the DNA oligo tail, and (2)
to allow for
the target to be pulled down via a biotin-streptavidin reaction that would
occur post-
incubation.
Streptavidin M280 beads (Invitrogen) were then added to the SELEX incubations
at
8133 ug for every 51.02 pmol of peptide present for 30 minutes under rotation.
After the
incubation with the beads allowing for the biotin-streptavidin reaction to
come to
completion, the beads were pulled down with a magnet (manually or with
automation),
washed and prepared for PCR.
Variation 3: (5') Blocked Aptamer incubation with peptide oligo-conjugate,
with bead
pulldown
Incubation Solution Preparation (POC and Biotinylated Primer Incubation)
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
In addition to blocking a region of the tail portion of the peptide-oligo
conjugates
(POCs), a portion of the aptamer can also be blocked to prevent unnecessary
binding
between the primer region of the aptamer and the region of the DNA tail on the
POC.
POCs were added to a 5' biotinylated primer complementary to the length of the
ohs tail
at a 1:2 POC:biotinylated primer ratio. 10X PBS, TWEEN-20, BSA, and water were
added to bring each reaction to a final 265 iii solution at 1X PBS, 0.025%
TWEEN-20,
and 0.1509 mg/ml BSA. The entire solution was incubated with rotation for 30
minutes
at RT.
The POC input for each reaction was determined by the anticipated aptamer
input.
An example method is presented below for a 1:10 target:ssDNA stringency round.
For
rounds 1 and 2, 18.5 pmol of POC was prepared for an input of 166.62 pmol
(4650 ng) of
aptamers, culminating in a stringency of 1:10 target:ssDNA. In this particular
gradient,
after two rounds of 1:10 stringency, the next two rounds were accelerated to a
1:25
stringency to increase the signal of the enriched aptamers. It should be noted
that
increasing a stringency too quickly, or starting a stringency too high, will
result in loss or
no true aptamer signal. However, increasing a stringency too slowly, or
starting at a
stringency that does not generate competition between binders will result in
time and
resources lost to additional rounds of SELEX required before enrichment can be
seen. In
this example, to account for the reduced target needed for the 1:25 stringency
in rounds 3
and 4, the amount of POC prepared for a 166.62 pmol (4650 ng) aptamer input
reduced to
7.40 pmol.
SELEX Incubation
The peptides and ssDNA were incubated for 2 hours with rotation at RT. The
final incubation buffer for the 400 ul reaction was 1X PBS, 0.025% TWEEN20,
and
BSA-matched concentration to the Hybridization Buffer used in BCS experiments
(see
below in Example 3¨ProSeq Experimentation and Example 4¨BCS Binding Assay
Experimentation, variations ranged from 0.10 mg/ml - 10mg/m1).
POC Controls
For negative controls for Variation 3 of SELEX, aptamers are incubated with
just
the POC's oligo tail and no peptide.
Possible oligo tails for this purpose are as follows:
= /5phos/cttagatgcacgtggataATCATATGTCCTTCTCTTCCCTA (SEQ ID NO :20)
= /5phos/cttagatgcacgcagcatATC ATATGTCCITCTCTTCCCTA (SEQ ID NO :21)
96
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Streptavidin-Biotin Pulldown
Streptavidin M280 beads (Invitrogen) were added to the SELEX incubations at
83.33 ug for every 51.02 pmol of peptide present for 30 minutes under
rotation.
B.3 RCHT-SELEX General Experimentation Part II
Post-SELEX cycle methods:
Post-incubation Wash (applicable for all variants)
The bead-peptide-aptamer conjugates were collected using an automated wash
protocol on the Bravo. Each SELEX reaction was incubated on a magnetic plate
for 2
minutes. Supernatant containing unbound aptamers was aspirated away and the
beads
were washed two times with SELEX buffer, followed by a final wash with IX PBS.
The
1X PBS was aspirated at the end of the protocol.
PCR on beads
Immediately after the automated wash protocol finished, 50 ul of PCR solution
was added to each well with beads. Unmodified variants of the bring up primers
were
used to amplify the 86 nt construct, except for the Wolverine2 library which
is 84 nt long
(full library constructs previously provided in the description of the
libraries).
NGS Preparation
After PCR amplification on beads, DNA concentrations were measured via Qubit
dsDNA assay and 10 ng samples of SPRI-purified PCRs on beads were taken for
NGS
preparation. Each aptamer identified from sequencing these samples were
associated
with the 6bp barcode of the peptide they putatively bound to in solution. The
P5 and P7
adapters required for Illumina sequencing were incorporated through PCR with
custom
NGS primers (5'-CAAGCAGAAGACGGCATACGAGA
-(Forward
primer)-3' (SEQ ID NO:22) and 5'-
AATGATACGGCGACCACCGAGATCTACACNNNNNN-(Reverse primer)-3') (SEQ
ID NO:23). The forward and reverse primer regions are variable, depending on
which
N40 library was used for SELEX. The amplification conditions for these PCR
reactions
were as follows: an initial denaturation at 95 C for 5 minutes followed by 10
amplification cycles of 30 seconds of denaturation at 95t, 30 seconds
annealing at 65 C,
30 seconds elongation at 72'C, and a final elongation of 5 minutes at 72t. The
final
NGS library was SPRI-purified, pooled, and size-selected for 177 bp constructs
via
97
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
PippinHT (Sage Science).
Threshold PCR
For each SELEX reaction, 4.08 ng of the SPRI-purified product from the PCR on
beads was amplified across twenty-four 50 ul PCR reactions using the custom
modified
primers for each library (sequences provided in the Bring Up section). The
SPRI-purified
dsDNA product of this library is an 86-bp (or 84-bp for Wolverine2 library)
amplicon
with the same construct as the original N40 library, with protected and
phosphorylated
ends that will facilitate enzymatic digestion of the reverse strand. The
regenerated
ssDNA library serves as the input for the next round of SELEX.
SELEX cycles
The protocol steps between aptamer refolding, target selection, aptamer
incubation, unbound separation, washing, amplification, NGS sample pull,
threshold
amplification, ssDNA library generation, and refolding can be repeated as a
`SELEX
round' until enriched aptamers are discovered in the NGS sequencing data.
Bring ups
and initial negative selections are not repeated between rounds.
BA RCHT-SELEX Additional Components
Fake SELEX
During the first 2 hours of Variation 2 of SELEX, negative controls are
incubated
with just water and SELEX buffer. After each round of SELEX, samples from Fake
SELEX were sequenced in order to determine the effects of PCR bias (since no
enrichment should occur due to the lack of a target. Fake SELEX is useful in
computational analysis and ML modeling of aptamers to train models to focus on
the
enrichment signal of the aptamer counts instead of the noise of operator
error,
contamination, PCR bias or other experimental or instrument noise.
BCS Compatible Aptamer Preparation
BCS, or the application of the DNA aptamers in ProSeq, requires a modification
of the primer regions of the aptamers to include the correct ligation,
restriction enzyme
and spacer sequences to facilitate the binding and recording events in BCS. A
unique
barcode, however, is not required since sequencing can proceed through the
entire
aptamer sequence in order to record which aptamer bound to which target on the
BCS
chip. There are a few ways to convert the aptamer library into a BC S-
compatible one,
however the fastest, cheapest and most high-throughput method is to use PCR to
modify
98
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
the primer regions of the aptamers. To this end, ssDNA pools (up to 166.62
pmol for
each reaction) were added to a 23 nt oligo "bridge mimic" complementary to the
forward
primer region of the aptamer at a 1:10 aptamer:bridge mimic ratio. The
solution was
brought up to a 135 ul solution at 1X PBS and 0.25% TWEEN 20. The mixture was
heated to 95 C for 5 minutes, rapidly cooled on ice, then added to the
incubation solution.
For SELEX N40 Library 3 (aka OMB105, Wo1verine2) which has the construct
.5'
TGATGCTATGCGACflAflGTACNNNNNNNNNTThINNNNNNNNNNNNN
NNNNNNNNNNNNNNNNTACTTGQCGTTCTTACCACCA 3' (SEQ ID
NO:24)
And the forward primer
= 5' TGATGCTATGCGACTTATTGTAC 3' (SEQ ID NO:25)
The bridge mimic used was 5' GTACAATAAGTCGCATAGCATCA 3' (SEQ ID
NO:26)
Bead-Based Multiplex SELEX
This assay was almost identical to SELEX, with the exception that multiple
peptides were added to each reaction. Peptides were separately conjugated with
beads at
the beginning of the experiment and aliquoted into individual stocks, to be
mixed in equal
molar proportions at the beginning of the SELEX incubation. The first four
rounds were
processed via the customary bring up/threshold PCR, digestion, incubation,
automated
wash, and PCR on beads cycles. To demultiplex in the final round, N * 4.08 ng
of each
reaction resulting from PCR on beads was amplified across N * 24 reactions,
with N
being the number of peptides that were concurrently incubated with the aptamer
pool.
SsDNA from this reaction was incubated in individual SELEX reactions at a
stringency
of 1:50, with only one peptide present in each reaction.
After using the Bravo's automated wash protocol to wash away unbound
aptamers, 50 ul of PCR solution were added to each demultiplexed well. The
SPR1-
purified product of each of these PCR reactions was barcoded during NGS prep
and
sequenced to reveal the aptamers associated with each peptide in isolation.
Primer Switch
The custom primers flanking the N40 regions are excised and replaced with
alternative primer sequences between rounds. The purpose of this primer switch
is to
99
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
mitigate contamination by excessively enriched aptamers from experiments using
the
same N40 library.
The current primer switch design was designed for the TriLink N40 library. By
amplifying the original N40 construct
TAGQGAAGAGAACGACATATGATNNNNNNNNNNNNNNNNNNNNNNNNNN
TTGACTAGTACATGACCACTTGA (SEQ ID NO:27)) with
primers TriLinkFwd_FokI (5'TAGGGAAGAGGGATGAAGGACATATGAT (SEQ ID
NO:28)) and TriLinkRev_FokI (5'TCAAGTGGTCGGATGATGTACTAGTCAA (SEQ
ID NO:29)), a Fokl restriction site is introduced to create the new full
length construct
TAGGGAAGAGGGATGAAGGACATATGATNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNTFGACTAGTACATC
(SEQ
ID NO:30)).
By digesting this altered PCR product with Fokl (NEB), a nuclease that cleaves
9
bp and 13 bp downstream of its restriction site
(5'...GGATG(N)9/3'...CCTAC(N)13 (SEQ
ID NO:31)), we cleaved off (5'TAGGGAAGAGGGATGAAGGACATA (SEQ ID
NO:32) and 5TTGACTAGTACATCATCCGACCACTTGA (SEQ ID NO:33)), leaving
sticky ends. End-filling this construct with Klenow fragment (NEB) leads to
the creation
of blunt ends. Incubating this blunt-ended double-stranded library with new
double-
stranded primers and ligase completes the protocol, leaving us with our
original N40
library with a new primer set swapped in. The success of each digestion and
ligation
event was analyzed via the Bioanalyzer Small RNA kit (Agilent).
Plate Layouts
In order to minimize the effects of local contamination between proximate
wells,
technical replicates (3 per experimental condition) were spatially randomized
across
different rows and/or different plates_ For the dipeptide switch experiments,
none of the
technical replicates were adjacent to each other. This allowed computational
filtering of
noise during post-sequencing analysis.
SECTION C RCHT-SELEX Results
Bring Up
For the bringup, 96 unit tests were conducted to determine optimal bringup
conditions for the each library, defined as the condition that introduces the
least bias or
variation in expression levels of all combinations of 6-mers possible after
the bringup was
100
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
performed. The expression intensities of every combination of 6-mer possible
from the
sequencing runs of DNA pools after the bringup divided by the expression
intensities
prior to the bringup. The best conditions for the 0MB63 library resulting in
the least
variation in expression levels of every combination of 6-mers was 11 PCR
amplification
cycles, using Herculase II Fusion DNA Polymerase and 0% DMSA, with input of
1010
DNA molecules (FIG. 36).
Fake SELEX
Top 20 sequences from a random sampling of 100,000 sequences from Fake
SELEX samples and real SELEX rounds were confirmed to be different, suggesting
that
DNA pools post-SELEX incubation were altered by the presence of bead-
conjugated
targets rather than a result of pulling down random sequences (FIG. 37). Fake
SELEX
analysis can be used to determine PCR bias elements during a SELEX experiment,
and
also be used to train models towards the ground truth of a positive aptamer
signal.
Digestion
Bioanalyzer Small RNA kit traces show single clear peaks after digestion
process
at approximately 75 nt, which, considering the error of measurement in the
technique,
correlates to ssDNA product size desired (86 bp for most SELEX libraries)
(FIG. 9C).
Confirmation of complete conversion of dsDNA PCR product to ssDNA occurred
prior to
the introduction of each aptamer libraiy into each new round of SELEX.
Threshold PCR
Unit tests have shown that threshold PCR introduced minimal bias. Comparing
the sequencing data of the DNA prior to and after a threshold PCR run
indicated that
threshold PCR results in low variance (0.132 variance of log ratio) in the
distribution of
sequences between the pool prior to and after threshold PCR (FIG. 11B and FIG.
11C).
Replicate Experiments
Aptamer sequences from the same bringup replicated across experiments of the
same targets up to round 5, giving greater confidence in identified aptamers.
Wells in
which bradykinin and GNRH experiments were conducted were physically adjacent
on
the same plate. Within a biocontrols SELEX experiment, significant
bleedthrough
between targets bradykinin and GNRH were detected, allowing for detection of
spatial
contamination (FIG. 38). As a result, randomization of sample placement
occurred on
each plate, where different targets were positioned on the same row with no
spaces
inbetween each experiment and replicates of the same target were positioned
with a
101
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
distance of 2 columns between each replicate to reduce contamination. After
significant
evaluation, it was found that the contamination observed was a result of
reagent carryover
from automation.
Aptamers
Biocontrols
As proof-of-concept of the RCHT-SELEX process, DNA aptamers to argipressin
(peptide sequence: CYFQNCPRG{LYS(BIOTIN)} (SEQ ID NO:34) and bradykinin
(peptide sequence: RPPGESPER{LYS(BIOTIN)} (SEQ ID NO:35)) were identified to
have high binding affinity with an estimated equilibrium dissociation constant
(1(.40 value
of 45 nM based on the experimental conditions of SELEX incubation (FIG. 39).
Further
characterization of the aptamers can be performed to determine the Kit with
and without
the primers. The N40 binding region sequences of the identified aptamers for
each target
are:
= argipressin: 5'-
ATATTCTAGGTTGGTAGGGAAGGCATGTATCTAATTCCTG-3' (SEQ ID
NO:36)
= bradykinin: 5'-
CAAATCGGTGCCGGCCGGGAAGGGGCAAAAACAGTGCAAC-3' (SEQ ID
NO:37)
Both aptamers were flanked by the following primers during RCH1'-SELEX:
= Forward primer: TAGGGAAGAGAAGGACATATGAT (SEQ ID NO:38)
= Reverse primer reverse complement: TTGACTAGTACATGACCACTTGA (SEQ
ID NO:39)
The same bringup was assayed against argipressin and bradykinin in 3 replicate
experiments for each target; the identified sequences replicated in
experiments of the
same target, and did not replicate in experiments with different targets. The
findings
suggested that these aptamers may be specific aptamers for argipressin and
bradykinin
peptides, and useful for the detection of these targets in samples.
Peptide Switch
Within Block A peptide switch experiments, sequences serially enriched for
specific N-terminal amino acids. Representative top aptamers for lysine and
cysteine,
defined as aptamers with the highest sequence counts after filtering for
noise, are reported
102
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
in FIG. 40. Both sets of aptamers were flanked by the following primers during
RCHT-
SELEX:
ID Forward primer: TAGGGAAGAGAAGGACATATGAT (SEQ ID NO:40)
= Reverse primer reverse complement: TTGACTAGTACATGACCACTTGA (SEQ
ID NO:41)
Future experiments can be conducted to characterize and validate identified
aptamers for
protein sequencing.
SECTION D N-terminal Amino Acid SELEX Experimentation
Example 2¨N-terminal Amino Acid SELEX
Reagents
DNA libraries were purchased from TriLink Biotechnologies and all DNA
primers were purchased from Integrated DNA Technologies with HPLC
purification. All
peptides were purchased from Genscript. 10X PBS and Tween-20 were purchased
from
Sigma-Aldrich. Lambda Exonuclease and buffer were purchased from New England
Biolabs. Mag-Bind Total Pure NUS beads were purchased from Omega-Biotek. The
bioanalyzer and all reagents, the Bravo liquid handler, and Herculase II
Phusion
polymerase and buffer were purchased from Agilent. Tubes, plates, and
thermocyclers
were purchased from Eppendorf Nunc plates were purchased from VWR. Both 70%
and 200 proof ethanol was purchased from Fisher Scientific. Nuclease-free
water,
MgCl2, Bovine Serum Albumin, dNTP mix, Dynabeads M280 Streptavidin, and QuBit
reagents were purchased from Thermo Scientific.
Methods
In this example, aptamers specific to the dipeptide Proline-Proline (PP) were
isolated using the N-terminal Amino Acid SELEX method (FIG. 41). 12 selections
were
run in parallel, against 5 total targets: 2 targets of 'interest and 3 control
targets. 3
selections were run against each target of interest and 2 selections against
each control
target. All rounds of positive selection were sequenced and used for analysis
of
enrichment across rounds and targets. Additionally, automation was used in
several steps
to ensure minimization of potential errors across samples and to facilitate
running parallel
selections. For this experiment, the dipeptide, PP, was chosen as the N-
terminal dipeptide
of interest because it's bulky cyclic side chain allows multiple potential
binding sites. PP
103
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
targets were 10-mer peptides with two prolines at the N-terminal and an 8
additional
amino acid region ("backbone"), followed by a C-terminal conjugated biotin
(biotinylated
targets) or DNA tail (PoC targets). To increase the chances of isolating an
aptamer
specific to the N-terminal PP dipeptide , both "switch" and "non-switch"
targets were
utilized, with multiple selections for each. Targets are referred to as PP-C
for PP targets
with the C backbone ("non-switch") or PP-D for PP targets with the D backbone
("non-
switch"). If both targets were used in the selection ("switch"), they are
referred to as
PPCD
Target-bead conjugation
Target-bead conjugations were performed fresh before each round of incubation.
Biotinylated peptide targets were conjugated to M280 streptavidin beads using
the
Agilent Bravo liquid handling platform. Beads were vortexed to homogeneity
before 25
uL beads were added to the appropriate volume for 75 ng peptide target for
each
conjugation reaction. The beads and target incubated on a chilled plate (4 C)
for 2
minutes to allow the biotin and streptavidin to interact and form a tight bond
before the
beads were washed several times with SELEX buffer (lx PBS, 0.025% Tween-20,
0.1
mg/mL BSA, 1 m1VI MgC12). The final product of the bead conjugation reaction
was
resuspended in 50 tiL of SELEX buffer.
Negative SELEX
DNA aptamer generation was carried out with a protocol involving aptamers in
solution and biotinylated targets conjugated to streptavidin beads. The
initial library of
1015 aptamers was pulled from the library stock and underwent 30 minutes of
negative
selection against 50 ul 10mg/mLstreptavidin beads in SELEX buffer. The
supernatant
was kept and put directly into a positive selection against the peptide
targets. This
positive selection was the first step of 5 rounds of SELEX with the following
workflow:
selection, amplification (small-scale PCR and large-scale PCR), and single
strand
generation.
Positive SELEX
Prior to every selection step, aptamers were annealed in Refold Buffer (lx
PBS,
0.025% Tween-20, 1 inM MgCl2) for 5 minutes at 95 C and at least 30 minutes at
room
temperature (RT) of 22-24 C. Selections were carried out in SELEX Buffer for
30
104
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
minutes (negative selection) or 1 hour (positive selections) with rotation.
Stringencies for
each round for "Switch" and "Non-Switch" incubations are reported in Table
2.1.
Table 2.1 Stringencies by Round and Target Type
Round "Non-Switch" Stringency "Switch"
Stringency "Switch" Backbone
1 1:1
1:1 C
2 1:2
1:1 D
3 1:5
1:2 C
4 1:10
1:2 D
1:25 1:5 C
Amplification was performed in two steps: small scale PCR and large scale PCR.
After washing off non-binders, the remaining target-aptamer conjugates were
put directly
into a small-scale PCR reaction of 1 reaction (50 uL) per sample. PCR reaction
conditions consist of all of the DNA retained from the wash steps, 3 uM
forward primer,
3 uM reverse primer, Herculase buffer, 0.2 mM DNTP, 0Ø5 units/ L Herculase
polymerase in a final volume of 50 uL.
After this PCR reaction was cleaned, an aliquot of the products was placed
into a
large-scale PCR with 24 reactions of 50 uL each. The purpose of this large-
scale PCR
was to amplify the DNA as much as possible without introducing excess PCR
bias. PCR
reaction conditions consist of 0.17 ng DNA, 6 uM forward primer, 6 uM reverse
primer,
lx Herculase buffer, 0.2 tnIVI DNTP, 0.5 units/uL Herculase polymerase in a
final
volume of 50 uL.
Both small scale and large scale PCR was performed using a Mastercycler Nexus
with conditions as follows: 5 min at 95 C, 13 cycles of 95 C for 30 seconds,
55 C for 30
seconds, 72 C for 30 seconds, and 72 C for 5 minutes. PCR reactions were
purified
using Mag-Bind TotalPure NGS beads from Omega Bio-Tek and were performed
using
the Agilent Bravo liquid handling platform. ssDNA and Mag-Bind TotalPure NGS
beads were incubated at a 3:5 ratio and washed with 70% ethanol,
To generate single stranded DNA from the large scale PCR products, digestion
with lambda exonucl ease was performed at optimized times. Digestion was
tracked
qualitatively using a bioanalyzer. Cleaned digestions were quantified and used
as input
into the next selection.
105
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
NOS preparation and Sequencing
Samples after the SELEX rounds were prepared for sequencing. The samples
were normalised to a concentration of 10 ng/ul. A 50 ul PCR reaction (2 ul of
6.25 uM
forward and reverse primers, 10 ul of 10 ng/ul DNA sample, 36 ul Master Mix)
was set
up for each sample to amplify the DNA and the reaction was performed using the
Mastercycler Nexus (PCR condition: 98 C for 5 minutes, 10 cycles of 98 C for
30
seconds, 65 C for 30 seconds, 72 C for 30 seconds and 72 C for 5 min). After
the
reaction, the PCR product was cleaned (Agilent Bravo liquid handling
platform). The
Tapestation was then used to quantify the size of the PCR product to determine
if the
PCR reaction was successful. The samples should have DNA size of 170-190 bp.
The
concentration of the PCR product was determined using the qubit dsDNA assay.
The
PCR products were then pooled in a tube according to the concentrations of
each product.
The concentration of the pooled products were determined using the qubit dsDNA
assay.
PCR product was purified by selecting DNA size 177 bp (Pippin Prep system,
Sage
Science). The concentration of the purified product was determined using the
qubit
dsDNA assay. After purification, 10 al, of the purified product was finally
sent for NUS
sequencing.
Analysis
Rapid increase in enrichment for all targets was observed from round 2 to 3
and
plateaued over rounds 3 to 5 (FIG. 42). Additionally log enrichment values
around 3.5,
3.2, and 3.0 for aptamers bound to Brady, PP-C, and PP-CD targets respectively
were
observed, indicating that these targets had putative binders (FIG. 43A). To
examine these
binders further, the top 10 binders by enrichment per replicate for each
target was pulled
out (FIG. 43B). Enrichment for binders to each target clustered among
experimental
replicates, indicating that selections for these targets were isolating
binders of interest.
Further analysis of experimental replicates of binders to targets indicates
that overall there
is little overlap between binders in different replicates (FIG. 44). Due to
the size of the
initial random pools, there is little chance that identical sequences would be
found in
different experimental replicates or to different targets, suggesting that
these are instead
contaminant sequences, allowing for the filtering of these likely contaminant
sequences
prior to testing. These candidates were further filtered down to a short list
of candidates
to test binding characteristics in vitro.
106
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
To identify the final aptamer sequences to fully characterize, two filtering
steps
were performed. Candidate aptamers from PP-CD targets that had high enrichment
(greater than 2, which correlates to at least 100-fold increase from R2 to R5)
and which
bound selectively to PP-CD (binders that did not bind other targets) were
chosen.
Filtering candidate sequences resulted in 26 candidates of which 10 were
selected for
final testing. These final ten candidates were chosen based on a variety of
factors: highest
enrichment ratio, total sequencing counts, representation within each
selection replicate
and zero sequence contamination in selection replicates.
Enrichment Calculation (Formulas defining growth and pen_growth:)
The number of times a given aptamer sequence appeared in the sequencing data
set is the aptamer count. Two rounds of SELEX are defined, "before" and "after
", as the
subset of sequencing data to track the unique aptamer sequences. "Before" is
the subset
from round 2 and "after" is the subset from round 5. A logarithmic scaling
factor was
applied to each aptamer count to accommodate the wide range of aptamer counts,
from 0
to 105
before = log io(before ct + 1)
after = log io(after ct + 1)
Growth is defined as the enrichment of a given aptamer between the "before"
round,
round 2, and the "after" round, round 5.
growth = after¨ before =log id(before ct + 1)/(after a + 1)]
A raw_penalty value was calculated that penalizes sequences that have low
count
numbers in both round 2 and round 5, multiplied it by a factor y and applied
it to the
growth factor by subtracting the product of y and raw_penalty.
raw_penalty = j10-afterin after +10-before in before
y= 1.26
pen_growth = growth ¨ y = raw_penalty
Technicality: If before Cc ,c can be used in the formulas instead, where:
c 2Iog 10(¨ /og(10)) ¨ log 10(n
before)
2
Kif Measurement
200 pmol peptide (PP-C, PP-D) was conjugated to 100 tiL L}ynabeadsTM M-280
Streptavidin (Thermo Scientific) following manufacturer's protocol and
resuspended to
107
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
original concentration in SELEX buffer. 5 mg fluorescein biotin (Biotinium,
#80019) was
resuspended in DMSO. 650 pmol fluorescein biotin was conjugated to 100 uL
DynabeadsTM M-280 Streptavidin (Thermo Scientific) following manufacturer's
protocol,
as a positive control, and resuspended to original concentration. 5' end FAM
labelled
aptamer candidates #1-10 were purchased from IDT. Aptamers were synthesized
with
forward primer and reverse primer complements and tested with the full length.
The full
sequence of each aptamer is as follows: 5'-TTGACTAGTACATGACCACTTGA-N40-
TTCTGTCGTCCAGTCTGATGTG-3' (SEQ ID NO:42). N40 sequences of aptamers
tested is reported in Table 21
Table 12 Aptamer candidate sequences tested.
Random Region N40 only
GACGGTACAGCTTAGTGAA'TTGCCCCCCGACGCAGGGGTT (SEQ ID
Apt 1 NO:43)
TITGCCGCTGTCTGACGCAAGACCACATCAACTTIATTTC (SEQ ID
Apt 2 NO:44)
CGCTCGTGTTGCTCGATCAAGGGTCTGTGCGTCTAGCTGG (SEQ ID
Apt 3 NO:45)
ACACCCAGACACCGCTGTCCGACGCAGGACTGACTGGGGC (SEQ ID
Apt 4 NO:46)
AACGACCGGTTAGACTGTGACCGCTTATCGCCGCAGATAT (SEQ ID
Apt 5 NO:47)
CGCATCCGGCGCAGGATTCAAGCGGGATTGTAAGGTAAGA (SEQ ID
Apt 6 NO:48)
GACATTGCCCTTCGCCGCAGAAGTGATGAAAGGGTTTGTG (SEQ ID
Apt 7 NO:49)
CGCTCGTGTTGCTCGATCAAGTGGACTAGAATTTGCTTCT (SEQ ID
Apt 8 NO:50)
CCACGGAATAATGATGGTGGTTGCTCCCCGACGCAGGGGC (SEQ ID
Apt 9 NO:51)
ACGCACCGATCGCAGGTTCACGTGGTATAACACTTTGTAA (SEQ ID
Apt 10 NO:52)
Peptide conjugated beads were diluted to 0.03 mg/mL, or 1:320 of original
concentration for the binding assay. 100 uL diluted peptide conjugated beads
or
fluorescein conjugated beads were aliquoted into individual wells of a 96 well
plate.
Plate was placed on a magnetic rack for 2 minutes and the supernatant was
removed. 100
uL of 5' end FAM labelled aptamer candidates at varying concentrations (0, 100
nM, 250
nM, 500 nM,750 nM, 1 uM, 2.5 uM, 5 uM, 10 uM, 20 uM), diluted in SELEX buffer,
108
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
was added to the appropriate well. Plate was sealed with plate seal (AB 0558
Adhesive
PCR film, ThermoFisher) and rotated in the dark at room temperature for 1
hour. After
incubation, seal was removed and beads were washed 3 times with 100 uL SELEX
buffer
and resuspended in 100 uL SELEX buffer. Beads were transferred to a black
plate and
single endpoint fluorescent readout was measured using a plate reader
(Biotek).
Note, this is one method of performing a binding assay to measure Ka. Other
methods, which will produce even more accurate measurements include:
microscale
thermophoresis, biolayer interferometiy, flow cytometry and surface plasmon
resonance.
SECTION E N-terminal SELEX Results
Aptamers were tested via plate-based Kd measurement method described above.
At a single concentration (100 nM), 7 aptamers showed higher fluorescent
signal than the
controls (non-aptamer and buffer only) towards the target PP-D. One aptamer
showed
higher fluorescent signal than controls towards the target PP-C (FIG. 45). Two
aptamers
were chosen for further testing, Apt 1 and 4. Apt 1 showed potential
saturation binding
towards PP-C but non specific binding towards PP-D (FIG. 46A). Apt 4 showed
saturation binding towards PP-13 but no binding towards PP-C (FIG. 46B).
SECTION F Generalized SELEX protocol
Above are listed a wide variety of methods that were used, optimized and
utilized
in order to achieve aptamer binders from SELEX results, however for each
application of
SELEX described here: (1) RCHT-SELEX for ML-analysis or (2) N-terminal binder
aptamers with NTAA-SELEX, there were different combinations of methods
employed.
Below is a template protocol that can be used to decipher the combination of
methods
required.
Overall Workflow:
1. Negative Selection
2. Bead conjugation
3. Aniplification
4. Sin2ie Strand CienerationtAntisena,µ Dittestion of Brin2un
5. Incubation
6. Amplification off Incubation Beads
7. Threshold Amplification
8. Simle Strand Generation/Antisense Dietestion of Threshold
109
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
9. Counter Selection
Equipment Protocols:
I. Qubit: Qubit was used to measure DNA concentration according to
manufacturer
protocols.
2. Bravo: Three types of protocols were run on the Bravo liquid handler: (I)
PCR
clean ups ("large volume" and "variable volume"), (2) Bead conjugations ("bead
conjugation") and (3) Bead washing ("Wash no elute post SELEX"). For PCR
clean ups, the Bravo was programmed to follow the manufacturer's guidelines
for
using Mag-Bind TotalPureNGS. For Bead conjugations, the Bravo was
programmed to follow the manufacturer's guidelines for using Dynabeadsrm M-
280 Streptavidin. Incubation time and buffer was optimized for the peptide
being
conjugated. For bead washing, the Bravo was programmed to perform 3 washes
of the peptide beads (after incubation with aptamer). The plate was incubated
on
a magnet for 2 minutes. The first two washes were performed with SELEX
buffer, and the last wash was performed with lx PBS. After the last wash, the
beads were not resuspended but left in the plate for the next step of the
SELEX
protocol.
3. BioAnalyzer: Two types of protocols were run on the Agilent 2100
Bioanalyzer
with 2100 Expert software. Library quality checks and post-PCR quality checks
were performed using high sensitivity DNA chips with the high sensitivity DNA
protocol according to the manufacturer's instructions. Post-digestion/single-
strand generation quality checks were performed using small RNA chips with the
Small RNA Series II protocol according to the manufacturer's instructions.
Table 2.3 SELEX Stringency Gradients:
RI R2
R3 R4 ItS
Gradient 1 1:10 1:10
1:25 1:25 n/a
Gradient 2 1:5 1:10
1:25 1:50 1:100
SELEX Buffer 1X PBS, 0.025% tween-20, 1mM MgCl2, 0.1 mg/mL BSA, Nuclease-free
H20
110
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Technical Terms:
Fwd RC: forward reverse complement of the 5' end of the aptamer. This is a
mimic of
the bridge used in BCS because it makes the 5' end of the aptamer double
stranded.
POC: Peptide-Oligo-Conjugate: this is the target of SELEX, i.e. what we are
finding
aptamer binders to. The POC is created from a 10-mer peptide and a41 nt ssDNA
tail.
bt peptide oligo comp: also known as peptide primer. biotinylated primer. DNA
tail
complement. blocking piece. This piece is the complement of the ssDNA "Tail"
region
of the Peptide-Oligo-Conjugate (POC). This piece has a biotin on the 3' side
to bind to
streptavidin beads, and is a full "block" of the oligo tail of the POC. It is
incubated with
the POC at a 2:1 ratio prior to incubating this target with aptamers.
Tail: Refers to the DNA tail that is conjugated to a peptide in the PoC (but
may be used
alone without peptide attached).
Backbone: also known as the suffix. This is the 8-mer region on dipeptide
targets (both
biotinylated and PoC) that is between the N-terminal dipeptide and the C-
terminal
conjugated biotin (biotinylated targets) or DNA tail (PoC targets). Backbones
are named
by the following convention: [let-tell' (example: C' or D').
Stringency: this corresponds to the ratio of targetaptamer. For example: 1:10
stringency
means there are 10 aptamers sequences for every 1 target, and vice versa 10:1
stringency
means that there are 10 targets for every 1 aptamer. 10:1 is not very
stringent, whereas
1:100 is extremely stringent.
Positive Selection: A selection where the aptamers are incubated with their
targets,
pulled down, and the supernatant is discarded (contains non-binders).
Negative Selection: A selection where the aptamers are incubated against
random
surfaces (tube sides, beads, etc), and the supernatant is kept (contains
sequences that do
not bind to random surfaces).
Counter Selection: A selection where the aptamers are incubated against things
that
closely resemble the target (example: different dipeptide or a backbone only),
and the
supernatant is kept
111
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Workflow
Negative Selection (Beads Only or Beads-I-Tail)
Purpose: to eliminate aptamers from the library that have a high binding
affinity for the
beads.
1. Dilute input ssDNA (1015 molecules) into refolding solution (1X PBS, 0.025%
Tween-20, 1 inM MgCl2, NF H20). Total volume is 150 uL.
2. Annealing (refolding aptamer): Heat to 95 C for 5 minutes and cool on bench
for
30 minutes.
3. Wash 55 uL of 10 mg/mL M280 beads in 500 uL of SELEX buffer 3 times.
Resuspend in 55 uL of SELEX buffer
4. Incubate 50 uL of washed M280 beads in 200 uL of modified SELEX buffer (1X
PBS, 0.025% Tween-20, 1 In.M MgCl2, 0.16 mg/mL BSA, NF H20) with cooled
annealed library solution (150 uL) for 30 minutes with rotation in 1.5 nit lo-
bind
tube.
5. Place tube in magnetic rack and wait 1 minute for beads to fully aggregate
next to
the magnet.
6. Take supernatant (-200 uL) and transfer to new tube.
7. Measure DNA concentration using Qubit ssDNA kit. Typical expectation
concentrations are in the range of 8-20 ng/uL.
Bead conjugation
Purpose: Biotinylated peptide targets are conjugated to streptavidin beads
that
magnetically pull down aptamer binders during incubation.
Note: Peptide-bead conjugates can be made ahead of time and aliquoted in 96-
well
eppendorf plates for freezing (1 freeze/thaw cycle maximum), or made before
each
incubation to be used fresh.
I. Dilute stock peptide to appropriate concentration so that peptides and
beads can be
combined at the ratio of 200 pmol peptide target to 1 mg of Dynabeads M280
beads (according to the manufacturer's protocol).
112
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
2. Pipette in corresponding amounts of peptide and water to a volume of 50 uL
per
well into a 96-well eppendorf plate.
3. Pipette in corresponding amount of M280 Streptavidin beads into a NUNC
plate,
only filling wells that will be used.
4. Using a liquid handler, run protocol "bead conjugations". This performs the
incubation, mixing and washing steps as defined by the manufacturer.
5. Dilute peptide beads to appropriate stringency, aliquot, and store at -20
C.
Amplification (Bring Up)
Purpose: create more copies of each aptamer of the negatively selected
library.
1. Prepare a master mix using a 50 mL conical tube. Master Mix: 3 uM forward
primer, 3 uNI reverse primer, Herculase buffer, 0.2 mM dNTP, 0.5 units/uL
Herculase polymerase in a final volume of 16000 uL (This is a total of 320
reactions of 50 tit per reaction). Each 50 id, reaction should have 0.17 ng
DNA.
2. Aliquot master mix across 3 96 well plates, with 50 uL per reaction.
a Seal 96 well plate and place in thennocycler using the following PCR
protocol: 95
It for 5 min, (95 C for 30 sec, 55 C for 30 sec, 72 C for 30 sec) x 13
cycles, 72
CC for 5 min, 4 012 Hold.
4. Pool 3 plates into one plate of 150 uL reactions.
5. Clean on liquid handler using protocol "large volume". This uses the
manufacturer's protocol for Mag-Bind TotalPure NGS beads.
6. Pool bring-up into one 5 mL eppendorf lo-bind tube.
7. Measure the concentration of double stranded DNA using QuBit dsDNA kit to
check concentration. Typically, concentrations are in the range: 40-90 ng/tiL.
Single Strand Generation (Digestion of a Bringup)
Purpose: lambda exonuclease is used to digest the antisense strand of the
double stranded
DNA. ssDNA must be generated so that the aptamer can bind to the target_
1. Set up single strand generation reaction according to Lambda Exonuclease
(M0262, NEB) manufacturer's specifications (For a 50 uL reaction, use up to 5
ug
DNA, 5 uL 10x Reaction buffer, 1 id, lambda exonuclease and up to 50 uL H20).
113
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Add 10x reaction buffer to DNA first, vortex to mix. Add lambda next, pipet to
mix.
2. Incubate reaction at 37 C for 10-20 minutes depending on DNA input
concentration.
3. Heat inactivate the exonuclease by incubating at 72 C for 10 minutes, hold
at 4 C.
4. Check the quality of DNA after digestion by running the DNA product on the
Bioanalyzer small RNA kit according to manufacturer's protocol. If the traces
show that there is still a double stranded product, add the same amount of
lambda
exonuclease as the original reaction and extend the incubation at 37 C for 5-
10
minutes. Check quality again.
5. Pool the DNA onto one plate and clean up on liquid handler using protocol
"variable volume". This uses Mag-Bind TotalPure NGS beads according to the
manufacturer protocol.
6. Check DNA concentration using the QuBit ssDNA kit. Normally the
concentration is around or above 30 ng/ul.
PoC Target Incubation - No Bead Conjugation
Purpose: to incubate the aptamer library with the targets to see which
aptamers bind to the
targets.
This incubation is used for PoC targets ONLY, where the PoC is exposed to the
aptamers
prior to introduction of beads and pulldown. For any protocols using bead
conjugation,
use the biotinylated target incubation.
1. Dilute input ssDNA (10's molecules), with FWD RC/Bridge if using, into
refolding solution (1X PBS, 0.025% Tween-20, 1 inM MgCl2, NF H20). Total
volume is 150 uL.
2. Annealing (refolding aptamer): Heat to 95 C for 5 minutes and cool on bench
for
30 minutes.
3. TARGET TAIL BLOCKING INCUBATION: Incubate target with bt peptide
oligo comp primer at a ratio of 1:2 in modified SELEX buffer (1X PBS, 0.025%
Tween-20, 1 inM MgC12, 0.16 mg/mL BSA, NF H20) at a total volume of 250 uL
for 30 minutes with rotation in a sealed NUNC plate. Target concentration will
vary depending on stringency gradient.
114
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
4. SELECTION INCUBATION: Combine 150 uL of cooled ssDNA in refolding
solution with 250 uL of target and annealed biotinylated peptide oligo comp in
modified SELEX buffer for a total volume of 400 uL in a sealed NUNe plate with
rotation for I hour.
5. SEPARATION/PULL DOWN INCUBATION: Wash M280 beads beforehand 3x
in SELEX buffer and resuspend in SELEX buffer at original concentration. Add
beads to 400 tiL selection incubation reaction after it has finished and
incubate for
30 minutes.
6. Wash away non specifically binding or non binding DNA from target beads
using
liquid handler (Protocol: "wash no elute").
Biotinylated Target Incubation - Using Bead Conjugation
Purpose: to incubate our aptamer library with the targets to see which
aptamers bind to
the targets.
This incubation protocol should be used for any targets (biotinylated or PoC)
that were
conjugated to beads prior to the start of SELEX. Note that in this protocol,
the aptamers
are being exposed to targets with beads, as opposed to the "PoC Target
Incubation"
protocol where the Poe is exposed to the aptamers prior to introduction of
beads and
pulldown.
1. Dilute input ssDNA GO molecules) in refolding solution (1X PBS, 0.025%
Tween-20, 1 inM MgCl2, NF H20). Total volume is 150 uL.
2. Annealing (refolding aptamer): Heat to 95 C for 5 minutes and cool on bench
for
30 minutes.
3. Thaw frozen bead conjugation plate, add modified SELEX buffer (IX PBS,
0.025% Tween-20, 1 mM MgCl2, 0.16 mg/mL BSA, NF H20) to a total volume of
250 uL.
4. Combine 150 uL of cooled ssDNA in refolding solution with 250 uL of bead
target conjugation in modified SELEX buffer for a total volume of 400 uL and
incubate in a sealed NUNC plate with rotation for 1 hour.
5. Wash away non specifically binding or non binding DNA from target beads
using
liquid handler (Protocol: "wash no elute").
115
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Amplification (PCR Off Beads [Poll])
Purpose: to amplify aptamers bound to target using PCR. Currently, the
aptarners are still
bound to the target and all non specific DNA has been washed away.
1. Add Master Mix (3 itM forward primer, 3 uM reverse primer, Herculase
buffer,
0.2 mM DNTP, 0.5 units/ pL Herculase polymerase in a final volume of 50 uL)
to wells immediately after washing protocol ends to avoid beads drying out.
2. Transfer to Eppendorf lo-bind 96 well plate, seal, and place in
thermocycler using
the following PCR protocol: 95 'V for 5 min, (95 "V for 30 sec, 55 'DC for 30
sec,
72 C for 30 sec) x 13 cycles, 72 C for 5 min, 4 C Hold.
a Cleanup on liquid handler using protocol "variable volume".
4. Measure the concentration of double stranded DNA using QuBit dsDNA kit on a
plate reader to check concentration. Typical concentrations are in the range:
4-20
ng/ii.L.
Threshold PCR
Purpose: to amplify aptamer library with protected primer and (Forward primer
has 6
thiol sulfates, reverse primer has 5' Phosphate).
1. Prepare a master mix using a 50 mL conical tube. Master Mix: 3 M forward
primer, 3 uM reverse primer, Herculase buffer, 0.2 inM dNTP, 0.5 units/ pt
Herculase polymerase in a final volume of 16000 uL (This is a total of 320
reactions of 50 uL per reaction).
2. Make 1:10 dilution of PoB DNA and normalize input concentrations by
pipetting
0.17 ng dsDNA per 50 uL reaction. Prepare a stock solution per sample by
adding
4.3 ng dsDNA, 300 uL H20, and 954 uL of master mix to each well. Aliquot
each sample stock solution into 50 uL per reaction.
3. Seal plates and place in thermocycler using the following PCR protocol: 95
C for
min, (95 C for 30 sec, 55 C for 30 sec, 72 C for 30 sec) x 13 cycles, 72 C for
5
min, 4 C Hold.
4. Cleanup DNA on liquid handler using protocol "large volume".
5. Measure the concentration of double stranded DNA using QuBit dsDNA kit on a
plate reader to check concentration. Concentrations are typically in the
range: 30-
90 ng/uL.
116
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Single Stranded Regeneration (Digestion of Threshold)
Purpose: to generate ssDNA for the next round of SELEX. This needs to be
performed as
multiple reactions because there are different concentrations of DNA for each
selection.
1. Set up single strand generation reaction according to Lambda Exonuclease
(M0262, NEB) manufacturer's specifications (For a 50 uL reaction, use up to 5
ug
DNA, 5 uL 10x Reaction buffer, 1 uL lambda exonuclease and up to 50 uL H20).
Add 10x reaction buffer to DNA first, vortex to mix. Add lambda next, pipet to
fliL
2. Incubate reaction at 37 C for 10-20 minutes depending on DNA input
concentration. Group reactions on different plates depending on reaction
times.
3. Heat inactivate the exonuclease by incubating at 72 C for 10 minutes, hold
at 4 C.
4. Check the quality of DNA after digestion by running the DNA product on the
Bioanalyzer small RNA kit according to manufacturer's protocol. If the traces
show that there is still a double stranded product, add the same amount of
lambda
exonuclease as the original reaction and extend the incubation at 37 C for 5-
10
minutes. Check quality again.
5. Pool the DNA onto one plate and clean up using protocol "variable volume".
This
uses Mag-Bind TotalPure NOS beads according to the manufacturer protocol.
6. Check DNA concentration using the QuBit ssDNA kit. Normally the
concentration is around or above 30 ng,/uL.
Counter Selection
Purpose: to incubate targets against other targets that closely resemble one
or more
aspects of the target, in order to ensure aptamers being enriched are specific
and actually
binding to the target itself. This is very similar to a positive selection,
except the targets
are different and there is no "wash no elute" step.
1. Depending on the experiment, refold aptamers and set up incubation
according to
either the PoC or Biotinylated Incubation steps listed above.
2. After incubation, place the plate on the magnet for 2 minutes to allow all
beads to
aggregate by the magnet.
117
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
3. Remove the supernatant from each well and store into a clean eppendorf 96
well
PCR plate.
4. Measure DNA concentration using Qubit ssDNA kit.
NGS Preparation
PoB DNA from round 2 onward is sequenced. Samples are prepared using NextSeq
protocol (NGS preparation).
Additional protocols:
Post-digestion Bioanalyzer check (small RNA kit):
The purpose of the Bioanalyzer test is to verify that dsDNA from the
bringup/threshold
has been effectively digested to ssDNA by lambda exonucleaseµ The Small RNA
kit is
used according to manufacturer's instructions.
To analyze results of the bioanalyzer assay, look for the locations of the
ssDNA
and dsDNA peaks. The ssDNA peak is at 60 seconds, dsDNA peak is at 40 seconds.
If
there are concatemers, they are observed at 55-65 seconds (wide, uneven peak).
Digestions are complete when a sharp peak is seen at 60 seconds. See FIG. 47
for an
example electropherogram.
dsDNA Bioanalyzer check
The purpose of this Bioanalyzer test is to evaluate the quality of post-
PCR/post-
bringup+clean dsDNA in terms of size (basepairs). We use the High Sensitivity
DNA kit
according to manufacturer's instructions.
To analyze the results of this assay, look for the lower marker at 35 bp,
upper
marker at 10380 bp. Check that the aptamer length matches up with the expected
library
length, in this example 86 bp. See FIG. 48 for an example of electropherogram.
Example 3¨PROSEQ Experimentation
Below the following will be described:
SECTION A ProSeq Experimentation Methods
SECTION B ProSeq Results
SECTION C Generalized ProSeq protocol
SECTION A ProSeq Experimentation Methods
118
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Reagents
Aptamers and foundation oligos were either purchased from IDT, or synthesized
in-house by K&A LABORGERATE 11-8 DNA & RNA Synthesizer and purified via
HPLC (Agilent 1290 Infinity Peptide-
oligonucleotide constructs bradykinin,
argipressin, and GNRH were commercially obtained from Genscript. Aptamer
incubation
and later DNA barcode sequencing was performed on NextSeq or MiSeq Reagent
Kits,
supplemented with PhiX Control v3, and sequenced on a MiSeq500 (IIlumina).
Bound
aptamers were ligated to the barcode foundations using T4 ligase (blunt/TA
Master mix
formulation) and cleaved with EcoRI in Cutsmart Buffer, all purchased from
New
England Biolabs. Excess aptamers and hybridization buffer were washed away
with
Cutsmart buffer. For Edman degradation, peptides were coupled with phenyl
isothiocyanate (PITC) in coupling buffer (0.4 M dimethyl allylamine in 3:2
(v/v)
pyridine:water, pH 9.5), cleaved in trifluoroacetic acid (TFA), and dried
under a stream of
nitrogen gas. MI reagents for Edman degradation were purchased from Sigma-
Aldrich.
All buffers were diluted with AmbionTm Nuclease-Free water. Analysis of NUS-
data was
accomplished with a custom analysis pipeline running on Colaboratory notebook
environment.
Methods
Protein Sequencing
Build and Tether Foundations to Solid Substrate
The core sequencing unit consists of four individual pieces of DNA: a 5'
phosphorylated barcode foundation (BF), a forward and reverse colocalization
linker (FC
and RC), and a protein or peptide target (PT) tagged with a C-terminal
oligonucleotide
sequence oriented with the 3' end connected to the protein or peptide and a
free,
phosphorylated 5' end. The 5' end of the BF sequence is complementary to the
5' end of
the FC to allow for hybridization, while the BF 3' end contains a unique
barcode (for
either sample multiplexing or associated PT identification) and a short
consensus
sequence complementary to a bridge sequence to facilitate aptamer ligation to
the BE
The FC consists of the BF-complementary region at the 5' end, followed by
sequence
complementary to the glass-bound oligo, followed by a flexible T-spacer, with
a short,
high GC-content sequence at the 3' end complementary to the RC. In turn, the
3' end of
the RC is complementary to the 3' end of the FC, followed by a long T-spacer,
followed
119
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
by a sequence complementary to the glass-bound oligo, followed by a sequence
complementary to the PT-bound oligo. The 5' end of the PT oligo is similarly
complementary to the 5' end of the RC, followed by a spacer before attachment
of the PT
at the 3' end (FIG. 49).
These four pieces were then combined and hybridized in solution such that PTs
were connected to a unique BF via the FC and RC, which allows for either PT
identification (in the case of validation and spike-in controls) or sample
demultiplexing
(in the case where multiple peptide pools are sequenced simultaneously). After
hybridization, the four component complex was incubated on the oligo-seeded
glass
substrate. The FC and RC hybridized to the glass-bound oligo, and, with the
addition of a
DNA ligase, the BF and PT oligos were covalently connected to the glass bound
oligos
via ligation (in this case, a 'nick repair' ligation). In this way, BF-PT
pairs were co-
localized and spatially separated from all other BF-PT pairs to ensure that
binding events
on a given PT were confined to a single BF. Furthermore, the covalent
attachment of the
BF and PT to the glass promotes remaining colocalization of the BF and PT over
multiple
rounds of PT sequencing despite the harsh reagents required for PT
degradation. Once
the BF and PT are covalently attached to the glass bound oligos, the forward
and reverse
colocalization linker annealed to the BF and PT is washed away with
formainide.
Aptamer Incubation
After the BF and PT are covalently attached to the substrate the sequencing
process begins by incubating the first BCS Compatible aptamer pool, followed
by
washout of unbound aptamers and addition of a ligase to covalently connect the
aptamer
to the BF. This cycle of incubation and ligation is performed multiple times,
where
ligation is performed after each incubation or after all aptamer pools have
been
introduced. Prior to incubating the peptide targets with the aptamers, the
single stranded
aptamer pool is incubated with bridge oligos to form the library of BCS
Compatible
aptamers. It should be noted that only a single barcode is recorded between
cycles of
restriction digestion (described below). Following ligation, a restriction
enzyme is
introduced (along with an excess of the complementary sequence to the
restriction site
and spacers) to cleave the peptide-binding sequence of the aptamer from the
aptamer
barcode on the 5' end, leaving only the aptamer barcode and the short
consensus
sequence for subsequent ligation attached to the BF. After restriction, the PT
is degraded
processessively from the N-terminal using Edrnan degradation, aminopeptidases,
or any
120
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
other processessive degradation process. Significantly, the technique of
building the
sequence of aptamer-encoded barcodes can be applied equally to C-terminal to N-
terminal peptide or protein sequencing, as the barcode sequence synthesis
process is
agnostic to PT orientation on its oligo tether. Furthermore, multiple cycles
of aptamer
incubation, ligation and restriction can be used to interrogate the same N-
terminal amino
acid sequence multiple times prior to PT degradation to more accurately
identify the N-
terminal composition.
Following degradation, another aptamer pool is incubated and the process is
repeated. The aptamers in each round contain unique barcodes (even when the
peptide
binding sequences are the same), such that missed incorporation events (e.g.,
apparent
deletions) may be easily identified and accounted for in subsequent data
analysis steps.
DNA Barcode Construct Sequencing
The final step in the sequencing process is the addition of a PCR or next-
generation sequencing (NGS) adapter. Using the same consensus and bridge
sequences,
the adapter is ligated to the 3' end of the sequence of aptamer barcodes that
represent the
series of aptamer binding events, which in turn is used to determine the
sequence of the
PT. Using the glass-bound oligo sequence and/or the BF 5' sequence as one
primer and
the PCR/NGS adapter as the other, the barcode construct is amplified off the
chip and
sequenced using standard NGS techniques, or, in the case of an NGS sequencing
flow cell
serving as the PT sequencing platform and the NGS adapter having the proper
design, the
barcode construct is amplified and sequenced directly on the NGS flow cell
without
further processing.
Sup-Dfif
A Priori Sup-Di
Biotinylated RNA bait generation
A priori Sup-Dill is performed on a pool of BCS barcode constructs. A
preliminary NGS datacet reveals sequences of high readcount to be targets for
depletion
by Sup-Dill. The target is made in isolation of the other pool constituents by
IDT or an
in-house K&A H8 DNA Synthesizer. PCR is performed on the target sequence using
a
standard forward primer and a reverse primer containing a T7 RNA polymerase
promoter
sequence. The PCR product is cleaned on an automated Bravo wash protocol (-1-2
ug)
and then used as a template to generate complementary biotinylated RNA bait
via in vitro
121
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
transcription in a 20 ul TranscriptAid Ti High Yield Transcription Kit (Thermo
Scientific) reaction containing 10 rtiN1 ATP, CTP, and GIP, 7.5 mM UTP and 2.5
mM
Biotin-16-UTP (Roche). After 4-6 hours at 37 C, the DNA template and
unincorporated
nucleotides are removed by DNase I (NEB) treatment and RNeasy Mini Kit column
filtration (Qiagen).
In-solution Hybridization and Bead Pulldown
A mix containing the target pool and nuclease-free water is heated for 5
minutes at
95 C, cooled on ice for 2 min and then mixed with biotinylated RNA bait with
SUPERase In RNase Inhibitor (Invitrogen) in prewarmed (65 C) 2X hybridization
buffer
(10X SSPE, 10X Denhardt's, 10 mM EDTA and 0.2% SDS). After 16 hours at 65 C,
the
hybridization mix is added to MyOne Cl streptavidin Dynabeads (Invitrogen),
that are
washed 3 times and resuspended in 2X B&W buffer (10 inNI Tris-HC1 (pH 7.5), 1
mM
EDTA, 2 M NaC1). After 30 minutes at RT, the beads are pulled down and the
supernatant retained.
"Soup" Processing and Sequencing
The supernatant ("soup") is treated with a mixture of two RNases, RNase H
(NEB) and RNase A (Zymo), for 30 minutes at 37 C. The treated ssDNA is then
amplified for 18 or more cycles. Initial denaturation is 5 min at 95 C. Each
cycle is 30
seconds at 95 C, 30 s at 55 C and 30 s at 72 C. Final extension is 5 min at 72
C. Bravo-
washed PCR product is then NGS-prepped for sequencing with custom primers on
an
Illumina Miseq.
Non a Priori Sup-Diti
There also may be circumstances in which a non a priori version of Sup-Diff
may
be necessary. In such a case, a sample of the target pool may be used as a
template for in
vitro transcription (IVT). As a proof of concept, PIT optimizations were
conducted in
order to skew the representation of baits in the RNA bait pool toward the high
abundance
species.
RNA bait pool generation
A gradient of SELEX spike-in sequences was created (% by mass): sequence 9
(0.000125%), sequence 13 (0.01%), sequence 11(1%), sequence 12(10%), sequence
10
(88.98%). This ssDNA gradient pool was used as a template in a 20 ul
TranscriptAid Ti
High Yield Transcription Kit (Thermo Scientific) reaction containing 0.1 mM,
0.25 mM,
122
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
1 mM, 2.5 mM, or 10 mM rNTPs (no biotinylated UTP). After 4-6 hours at 37 C,
the
DNA template and unincorporated nucleotides are removed by DNase I (NEB)
treatment
and RNeasy Mini Kit column filtration (Qiagen).
Reverse Transcription
The purified RNA bait pool was then reverse transcribed into cDNA using the
Maxima Reverse Transcriptase kit (Thermo Fisher). A 28 ul initial reaction
containing
500 ng of the RNA bait pool, 15-20 pmol of TriLink Forward primer, 0.5 inIVI
of
equimolar dNTP mix, and nuclease free water was incubated at 65 C for 5 min.
Then, 8
ul of 5X Reverse Transcriptase Buffer, 25 of SUPERase In RNase Inhibitor
(Invitrogen), and 2 ill of Maxima Reverse Transcriptase enzyme were added and
the
reaction was incubated at 50 C for 30 min followed by heat inactivation at 85
C for 5
min. The resultant cDNA pool was treated with a mixture of two RNases, RNase H
(NEB) and RNase A (Zymo), for 30 min at 37 C.
Amplification and Sequencing
The treated ssDNA was then amplified for 13 or more cycles. Initial
denaturation
was 5 minutes at 95 C. Each cycle was 30 seconds at 95 C, 30 seconds at 55 C
and 30 s
at 72 C. Final extension was 5 min at 72 C. Bravo-washed PCR product was then
NUS-
prepped for sequencing with custom primers on an Illumina Miseq. A 41x8x6 read
was
conducted using a Miseq V2 Nano kit
SECTION B ProSeq Results
Results - Barcode Sequence Synthesis Proof of Concept
As a proof-of-concept for synthesizing the DNA barcode representing the series
of
binding events that, in turn, represents the putative amino acid sequence of
the protein or
peptide being sequenced, the barcode synthesis process was performed using a
'simulated
aptamer' DNA-DNA binding (e.g., hybridization) system. In this way, the
uncertainty of
the binding kinetics and binder-target specificity was reduced to create an
'ideal' binder-
target system in which to demonstrate the serial barcode addition strategy. In
addition,
these DNA-DNA binders can be used as internal controls in future experiments
to
evaluate overall run quality.
Using this idealized platform with Barcode-Specific bridges, up to 12 cycles
of
aptamer barcode ligation and restriction have been performed with as high as
63.8%
efficiency based on the number of perfect 12/12 reads, with a per-cycle
efficiency up to
123
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
75.5% for 3 cycles of barcode incorporation (FIG. 50A). This is consistent
with current
estimates of efficiency for each step, where the assumed efficiency of correct
binder-
target interaction is ¨90%, the efficiency of ligation of the target-bound
aptamer to its
associated sequence of barcodes is > 99%, and the efficiency of the
restriction is
estimated (conservatively) at 85%. In idealized conditions on the platform,
the restriction
enzyme efficiency has been measured at > 95%, which means that given a
moderate
increase in binder specificity (to 95% accuracy) and idealization of
restriction conditions
in the context of the full sequencing cycle, it should be possible to achieve
a per-cycle
barcode incorporation efficiency above 90%.
With the Universal bridge design
5'CTGCGCCTATACGAATTCGTTATC<figref></figref><figref></figref>##4I#CTCTCCGTTATC (SEQ ID
NO:53), wherein each # is a 5-Nitroindole, three out of three serial barcode
ligations of
the correct order and orientation was achieved with an estimated per-round
efficiency of
71% (FIG. 50B). In the same experiment, >36% of the reads associated with a
unique
foundation (Foundation 11) contained all three aptamer barcodes in the correct
order,
confirming that serial ligation and restriction is possible with Universal
bridges.
Results - Peptide Target Identification Proof of Concept
Preliminary results using aptamers with binding sequences derived from RCHT-
SELEX experiments against biologically relevant 10-mer peptides have shown
that,
within a given pool of SELEX-derived sequences, there are binders with
affinities in the
sub-nanomolar range.
Initial evidence of specific aptamer binding to 10-mer argipressin biopeptide
has
been shown in a combination of RCIT-SELEX and PROSEQ conditions. When a
library
of prospective aptamers for argipressin was incubated with foundations
attached to either
argipressin, bradykinin, DD, DNA, or no target (null control) in solution,
barcodes of
prospective bradykinin aptamer were ligated to all types of argipressin-linked
barcode
foundations and to no DD-linked barcode foundations (FIG. 51). The sequences
for the
top specific argipressin aptamers with its DNA barcode tail are:
= /5Phos/GAGAGTAAAGCCGATAGGATAACGAATTCGTATAGGCGCAGGA
TGGACTTGATAACCITCTGCTGCGTGCCTTGATGTGCTTACTTGGCGTT
CTTACCACCA (SEQ ID NO:54)
124
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
= /5Phos/GAGAGTTAGTCAGCAGGGATAACGAATTCGTATAGGCGCAGCA
TTTGATTCTGCTGCGTGCATACCCCTGTGTGTTATCCCTACTTGGCGTT
CTTACCACCA (SEQ ID NO:55)
= /5Phos/GAGAGTCCAC GT6CACAGATAACGAATTCGTATAGGC GC MICA
TACATCGGACATACATCCTGCGTGCATCCACCTTTGCATACTTGGCGTT
CTTACCACCA (SEQ ID NO:56)
The barcodes of all three aptamers above have over 100 hits on all different
argipressin
foundations and no off-target hits. This data suggests that argipressin
aptamers derived
from the RCHT-SELEX methods preferentially bind to argipressin over DD
peptides and
bradykinin. They also do not bind to the oligo that is attached to all targets
as shown by
the lack of counts of argipressin aptamer barcodes to null foundations.
Additionally,
although the aptamers were isolated in RCHT-SELEX without the aptamer barcode
necessary for compatibility with PROSEQ, specificity is still preserved after
the aptamer
tail sequences were added to the 5' end.
Results - Degradation
Preliminary studies of Edinan degradation on a biologically relevant peptide
(Bradykinin) tethered to a glass substrate via an oligonucleotide suggest that
the
oligonucleotide tether is stable (e.g., antibody staining shows a strong
signal both pre- and
post-degradation). Furthermore, after multiple cycles of Edman degradation,
the signal
from the antibody staining is diminished but not entirely absent, suggesting
that the
peptide is in place post TFA exposure, and the degradation in signal is due to
the loss of
antibody binding due to the cleavage of amino acids (FIG. 52).
Results - Sup-Duff
Preliminary data on PIT optimization is promising for the method of non a
priori
Sup-Diff Using the standard 10 inM rNTP IVT protocol to generate a pool of RNA
baits
from a target pool of the following distribution: 89% sequence 10, 10%
sequence 12, 1%
sequence 11, 0.01% sequence 13, and 0.000125% sequence 9, an RNA pool with the
following composition was generated: 81% sequence 10, 18.5% sequence 12, 0.6%
sequence 11, 0.008% sequence 13, and 0.0055% sequence 9. As the final
concentration
of each rNTP was reduced, a shift in the RNA bait distribution was achieved
such that
there is an increase in frequency of RNA baits to high abundance targets. From
10 mM
125
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
final rNTP concentration to 0.25 mM final rNTP concentration there was an 8.5%
average increase in frequency of RNA bait to the highest concentration target,
sequence
(FIG. 53). It demonstrated that the distribution of an RNA bait pool generated
from
the target pool may be skewed toward a high abundance sequence, allowing for
preferential pull-down of the high abundance species when the RNA bait pool is
hybridized to the target sequence pool.
SECTION C Generalized ProSeg protocol
Below is a template protocol used in developmental experiments.
Technical Terms
PoC (protein-oligo conjugate): Protein or peptide conjugated to the 3' end of
an oligo
containing a linker region, a region sequence complementary to 5' end of
reverse
cololinker and a 5' phosphate group.
RC (reverse cololinker): 3' end of the RC is complementary to the 3' end of
the forward
cololinker, followed by a flexible T-spacer, followed by a sequence
complementary to the
glass-bound oligo adaptor, followed by a sequence complementary to the oligo
on the
PoC.
FC (forward cololinker): The FC consists of the foundation-complementary
region at
the 5' end, followed by sequence complementary to the glass-bound oligo
adaptor,
followed by a flexible T-spacer, with a short, high GC-content sequence at the
3' end
complementary to the RC.
Foundation: An oligo containing a barcode specific to a target and on which
DNA
barcodes bound to the target is built upon. 5' end of the foundation sequence
is
complementary to the 5' end of the FC to allow for hybridization, while the 3'
end
contains a unique barcode (for either sample multiplexing or associated PT
identification)
and a short consensus sequence complementary to a bridge sequence to
facilitate binder
DNA barcode ligation to the foundation.
Colocalized constructs: Complete core sequencing unit consisting of a PoC, RC,
FC,
and foundation piece hybridized together.
Restriction/Consensus Bridge: An oligo that is complementary to the
restriction digest
sequence in the BCS cassette. This sequence is added during the restriction
digestion step
to hybridize to the 5' end of aptamers that were ligated to the 3' end of the
126
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
foundation/previous aptamer barcode in case the universal bridge has been
washed away
so that digestion can still occur. Improves efficiency of the digestion
reaction.
Table 3.1 Buffer solutions
Buffer
Formulation
Hybridization Buffer
0.025% TWEEN20 in lx PBS
Blocking Buffer
0.025% TWEEN20 in lx PBS + 10mg/m1
BSA
Chip Blocking Buffer 10
uM of P5 Complementary oligo (5'-
TCTCGGTGGTCGCCGTATCATT-3'
(SEQ ID NO:57))/P7 Complementary oligo
(5'-ATCTCGTATGCCGTCTTCTGCTTG-
3' (SEQ ID NO:58)) sequences + 10 uM
POC Tail blocking sequence (5'-
TAGGGAAGAGAAGGACATATGATTA
TCCACGTGCATCTAAG-3' (SEQ ID
NO:59))
Aptamer Incubation Buffer
0.025% TWEEN20 in lx PBS + 0.1mg/m1
BSA
Foundation Hybridization and Flow Cell Preparation
Foundation Hybridization
Purpose: to hybridize cololinkers, foundations, and targets at the correct
ratios to form
colocalized constructs.
Goal is to get final concentration of 120 pM total foundation concentration,
aim for a
lower concentration if risk of sequencing failure of off-target ligation is
high, i.e. first
time using a new pool/set of aptamers
1. Thaw sequencing unit components on ice (FC/RC stock, foundations, and
targets)
2. Hybridize sequencing unit components at 10 tiM Forward Cololinker
concentration (foundation, target, reverse cololinker in excess). In a 96 well
plate
combine sequencing unit components (1 well per target) in the order of:
a 91 uL Hybridization Buffer
b. 1 uL Cololinker at 1 uM (1 uM stock has FC:RC 1:2)
c. 5 uL Foundation at luM stock (Multichannel from 96 well plate stock)
d. 3 uL Target at 10 uM stock (Minimum final concentration of at least 50
nM)
127
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
e. 100 uL Total
3. Denature/anneal foundations, cololinlcers, and targets using the following
cycling
parameters on the thermocycler:
a 5 minutes at 95 C
b. 1 minutes at 85 C
c. 2 minutes at 75 C
d. 3 minutes at 65 C
e. 5 minutes at 55 C
f 5 minutes at 45 C
g. 5 minutes at 35 C
h. 40 minutes at 25 C ¨> 5 minutes in start step 5
4. Yields 10 nM colocalized constructs
5. With approximately 35 minutes remaining in denature/anneal therrnocycling,
start
refolding for aptamers in round 1 (see below for dilutions)
Foundation Ligation
Purpose: to ligate the colocalized constructs to the flow cell to ensure
targets and
foundations are available for aptamer incubation.
1. Dilute 10 nM colocalized constructs 1:20 to get 500 pM working solution in
Hybridization Buffer
a 95 uL Hybridization buffer + 5 ul of 10 nM colocalized constructs mixture
2. In single Foundation Ligation Tube combine:
a Equal amounts of each target foundation (Final concentration of all
foundations is 120 pM, i.e. 12 uL of 1 nM foundations - may need to dilute
1:20 in order to avoid small volume pipetting)
b. 10 uL 2xBlunt/TA MM (T4) Ligase
c. Dilute in Hybridization Buffer for total volume of 100 uL
3. NOTE. Adjust Foundation Volume and NE .1120 Volume as needed to reduce
loading concentration to avoid overclustering
4. Pipette mix GENTLY at least 15 seconds or until glycerol from Ligase is
completely homogenized
5. Wash chip with 30 uL of Foundation Ligation mix
6. Add 30 it, Foundation mix to chip twice
128
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
7. Incubate for 15 minutes at 28 C
8. Wash chip with 100 uL of 100% Formamide
9. Incubate for 90 seconds at 40 C
Start of Barcoding Cycles (Repeated for Each Cycle)
01/go Tail Block + BSA Block
Purpose: to reduce availability of flow cell surfaces and ssDNA ligated to the
flow cell
for non-specific binding of aptamers during aptamer incubation.
1. Wash chip with 500 uL of Binding Buffer
2, Wash chip with 30 uL of Chip Blocking Solution
a To prepare 100 uL of Chip Blocking Buffer:
i. 60 uL of Blocking Buffer
(0.025% TWEEN-20 + 10 ing/mL BSA
in lx PBS)
uL of 100 uM P.5 Complement (final conc. 10 uM) (sequence in
Table 3.1)
10 uL of 100 uM P7 Complement (final conc. 10 uM) (sequence in
Table 3.1)
iv. 10 uL of 100 uM POC TailBlock (final conc. 10 tiM) (sequence in
Table 3.1)
v. 10 uL of 100 uM Foundation Base Block (final conc. 10 uM)
3, Add 30 uL of Chip Blocking Buffer to chip twice
4. Incubate 15 minutes at 37 C
Aptamer Incubation
Purpose: to expose targets on the flow cell to aptamers to initiate binding
between (1)
target and binding region of aptamer and (2) foundation and BCS cassette of
aptamer.
1. Aptamer Incubation Solution Prep:
Aptamers + Bridge at 1:2 Ratio in Hybridization Buffer
b. Heat aptamer mix to 95 C for 5 minutes in PCR tube (keep in middle of
strip to prevent melt compression of PCR tube)
c. Cool aptamer tube at RT on benchtop for 1 hour
129
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
d. Immediately prior to incubation of aptarners and bridges on chip, add 10
mg/mL BSA to achieve final BSA concentration of 100 ug/mL
i. Example: Add 1 uL of 10 mg/mL
BSA to 99 uL of aptamer mix
2. After 15 minutes Chip Blocking Buffer incubation, wash chip with 100 uL of
Hybridization buffer for 60 seconds
3. Repeat 60 second Hybridization buffer wash
4. Wash lx with 30 uL Aptamer Incubation Solution
5. Load 30 uL of Aptamer Incubation Solution to chip
6. Incubate for 30 minutes at 25 C
Aptamer Ligation
Purpose: to ligate aptamers bound to targets to the colocalized foundations so
the aptamer
barcodes can be sequenced.
1. Wash 3x 90 seconds with 100 uL Aptamer Incubation Buffer
2. Prepare Ligation Solution:
a 63 uL NF H20 + 7 uL 2x Blunt/TA MM Ligase
3. Wash lx with 30 uL Ligation solution
4. Load 30 uL Ligation solution
5. Incubate for 3 minutes at 28 C
Aptamer Restriction Digest
Purpose: To prepare the 3' end of the aptamer barcode ligated to the
foundation for NGS
ligation so it can be sequenced.
1. Wash 3x for 90 seconds in lx Cutsmart buffer
2. Prepare Restriction Solution:
a 77 uL NF H20
b. 10 uL 10x Cutsmart
c. 3 uL of 10 uM Restriction/Consensus Bridge
d. 10 uL EcoRI HF (100,000 U/ml)
3. Wash lx with 30 uL Restriction Solution
4. Load 30 uL Restriction Solution
5. Incubate for 30 minutes at 40 C
6. Wash chip with 100 uL of 100% Formamide
130
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
7. Incubate for 90 seconds at 40 C
8. Wash chip with 500 uL SELEX Buffer
[Repeat for Each Cycle]
End of Final Barcoding Cycle
NGS Adapter Ligation
Purpose: to ligate the P5 complement sequence to the 3' end of the barcode
constructs so
it will be read during sequencing.
1. Prepare NGS Ligation Mix:
a 615 uL NF H20
b. 1.5 uL NGS Adapter + Bridge (1 uM NGS Adapter, 2 uM Bridge)
c. 10 uL 10x Cutsmart buffer
d. 25 uL Blunt/TA MM Ligase
2. Pipette mix solution until ligase is fully incorporated
3. Load 2x 30 uL of NGS Ligation Mix
4. Incubate 165 seconds at 40 C
5. Wash 2x with 500 uL NF H20, incubate each wash for 90 seconds
Load chip on Sequencer
Purpose: to prepare the flow cell and MiSeq for the sequencing run.
1. Change Sample Sheet to reflect read length, experiment/sample name
2. Load 20 uL of 20 pM denatured PhiX 111 580 uL HT1 Buffer (supplied with
sequencing cartridge) into the Sample port on the Miseq cartridge
3. Start Sequencer
a If a flow error arises during the pre-run check, exchange the plastic
hinged
piece that contains the gasket on the flow cell with the same piece from an
old flow cell (after thoroughly rinsing with 70% Ethanol and NF H20)
Example 4¨BCS Binding Assay
Reagents
131
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Aptamers foundation oligos, and DNA targets were HPLC- or PAGE-purified by
and purchased from IDT. Spot-Tag and bradykinin peptide-oligonucleotide
constructs
were commercially obtained from Genscript. The Spot-tag nanobody was purchased
from Chromotek. Spot-tag nanobody-oligo conjugates were prepared using
SoluLINK
Protein¨Oligonucleotide Conjugation Kit. Aptamer incubation and DNA barcode
sequencing was performed on MiSeq Reagent Nano v2 Kits, supplemented with PhiX
Control v3, and sequenced on a MiSeq500 (I1lumina). Bound aptamers were
ligated to
the barcode foundations using T4 ligase (Blunt/TA Master mix formulation) and
cleaved
with EcoRI in CutSmart Buffer, all purchased from New England Biolabs. Excess
aptamers and hybridization buffer were washed away with the 100% formamide
purchased from Millipore Sigma Analysis of NGS data was accomplished with a
custom
analysis pipeline running on a Colaboratory notebook environment.
Methods
Conjugate Spot-Tag nanobody to DNA tail
The commercially obtained Spot-tag nanobodies (Chromotek) were conjugated to
the 3' end of a 5' phosphorylated oligo
(3'ATCCCTTCTCTICCTGTATACTAATAGGTGCACGTAGATTC/5Phos/ (SEQ ID
NO:60)) in anon-site directed manner using the SoluLINK Protein-
Oligonucleotide
Conjugation Kit according to manufacturer instructions.
Success of Spot-tag nanobody-oligo conjugation was confirmed by PAGE
electrophoresis (FIG. 54). Labeling of the protein was not site-directed but
could be
achieved using the sortase-enzyme method. Multiple higher molecular weight
bands
were observed on the gel, presumably corresponding to multiple oligos
conjugated to a
single nanobody. Importantly, for BCS experiments these constructs are less of
a concern
because they will either 1) be non-functional, in which case they will not
bind Spot-Tag
and be washed away, or 2) will bind to the Spot-Tag, following which either of
the
multiple tails can then become ligated to the nearby foundation.
Build and Tether Foundations to Solid Substrate
As a proof-of-concept experiment to validate the ability of the BCS platform
to
record specific binding events in a complex environment, the Spot-Tag-oligo
conjugates
(Spot-Tag.01) and 6 other control targets were seeded onto a MiSeq Nano v2
sequencing
132
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
chip. The other peptide target was Bradykinin conjugated to a 5'
phosphorylated DNA
tail (Brady.01). 2 null targets (oligo tails without target) comprised a 5'
phosphorylated
oligo (5'Phos.01), and an oligo lacking a 5' phosphate, which therefore can
not be
attached to the chip (CLR.Null.Block), 2 DNA controls (SP6.01 and SP4.01),
continuous oligo sequences that contained both a 5' phosphorylated linking
region to
tether to the P7 primers and a binding region to hybridize to a complementary
strand,
served as positive controls (FIG. 55). The binding region and DNA tail
sequences of
each target is reported in Table 4. L
Table 4.1. Sequences of targets and oligo tail
Target Target Name
Sequence
Type
Peptide Spot-Tag* Spot-Tag.01 (N-
terminus)-PDRVRAVSHWSSGGG-Cys (SEQ
target ID
NO:61)
(C-terminus)-
31ATCCCTICTCTIVCTGTATACTAATAGGT
GCACGTAGA'TTC/5Phos/ (SEQ ID NO:62)
Peptide Bradykinin Brady.01 (N-
terminus)-RPPGFSPER-Cys (SEQ ID NO:63)
target (C-
terminus)-
control for
3'ATCCCTTCTCTTCCTGTATACTAATAGGT
non-specific
GCACGTAGATTC/5Phos/ (SEQ ID NO:64)
binding
Null control DNA** CLR.Null.Block
CTTAGATGCACGTGGATAAT (SEQ ID
NO:65)
DNA** 5'Phos.01
/5Phos/CTTAGATGCACGTGGATA (SEQ ID
NO:66)
Positive DNA** SP6.01 /5Phos/CTTAGATGCACGTGGATAATCATAT
control
GTCCTTCTCTTCCCTAATGAAGTACTAACC
TGA (SEQ ID NO:67)
DNA** SP4.01 /5Phos/CTTAGATGCACGTGGATAATCATAT
GTCCTICTCTICCCTAATAGGATTCC (SEQ
ID NO:68)
*The C-terminal of the peptide targets is directly conjugated to the 3' end of
one DNA
tail via a cysteine
**Binding sequences and DNA tails of DNA targets are continuous oligos rather
than
conjugated through another chemical conjugation method.
133
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
To tether a target-oligo conjugate and a DNA barcode foundation containing a
sequence indicative of its associated target in proximity to each other to a
solid substrate,
it must be further assembled into a core sequencing unit. The core sequencing
unit of the
BCS platform consists of four individual pieces of DNA or oligo-conjugated
molecules: a
5' phosphorylated barcode foundation (BF), a forward and reverse
colocalization linker
(FC and RC), and a target tagged with a C-terminal oligonucleotide sequence
oriented
with the 3' end connected to the target and a free phosphorylated 5' end. The
5' end of
the BF sequence is complementary to the 5' end of the FC to allow for
hybridization,
while the BF 3' end contains a unique barcode (for either sample multiplexing
or
associated target identification) and a short consensus sequence complementary
to a
bridge sequence to facilitate aptamer ligation to the BF, The FC consists of
the BF-
complementary region at the 5' end, followed by sequence complementary to the
glass-
bound oligo, followed by a flexible T-spacer, with a short, high GC-content
sequence at
the 3' end complementary to the RC. In turn, the 3' end of the RC is
complementary to
the 3' end of the FC, followed by a long T-spacer, followed by a sequence
complementary to the glass-bound oligo, followed by a sequence complementary
to the
target-conjugated oligo. The 5' end of the target oligo is similarly
complementary to the
5' end of the RC, followed by a spacer before attachment of the target at the
3' end (FIG.
49).
Each control target was tested in triplicates and Spot-Tag in sextuplicate.
Their
respective FC, RC, and BF were thawed on ice before each set of sequencing
units were
combined in 91 uL of Hybridization Buffer (0,025% TWEEN20 in lx PBS) in
separate
wells to generate solutions of 10 nM FC, with RCs, BFs and targets in excess.
In this
experiment, all targets employed the same FC sequence
(5'CATCAGCTCGCAGTCGATCTCGTATGCCGTCITCTG1T1111-1-1-1-1-1-1-1-1-1-1-1
IT11-1-1-1-11=1-11-14-11-1-1-1-1-ritr1-riCCAGCCACCGCCAACCATCC-3' (SEQ ID
NO:69)) and RC sequence
(5'ATTATCCACGTGCATCTAAGATCTCGTATGCCGTCTTCTG
__________________________________________________________ 1 11 1 IT! IT1 1 1
1-1-11-ITI1I1I1I1ITITITITITITIGGATGGTMGCGGTGGCTGG-3' (SEQ ID
NO:70)). FCs and RCs were kept in a stock solution with a ratio of 3:1 FC:RC
in
Hybridization Buffer. The components were added in the order of Hybridization
Buffer,
FC and RC stock, and BFs. Targets were added to the mixtures immediately prior
to
134
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
hybridization. Sequences and concentrations of each set of targets, FCs, RCs,
and BFs
are reported in Table 4.2. The final ratios of individual pieces are:
= 5:1 BF:FC
= 3:1 FC:RC
= 10:1 Target:RC
To assemble the sequencing units, the complete mixtures were mixed thoroughly,
spun down for 30 seconds, sealed, and heated in a thermocycler with the
following
conditions: 5 minutes at 95 C, 1 minute at 85 C, 2 minutes at 75 C, 3 minutes
at 65 C, 5
minutes at 55 C, 5 minutes at 4.5 C, 5 minutes at 3.5 C, 40 minutes at 25 C.
Table 4,2, Foundation sequence of each target replicate
Target* Foundation Name
Foundation (5' - 3')
/5Phos/CGACTGCGAGCTGATGTGGCATCTGATAACG
Spot-Tag.01 Fd31 (SEQ ID NO:71)
/5Phos/CGACTGCGAGCTGATGAGGTACCAGATAACG
Spot-Tag.01 Fd19 (SEQ ID NO:72)
/5Phos/CGACTGCGAGCTGATGCACTTACGGATAACG
Spot-Tag.01 Fd20 (SEQ ID NO:73)
/5Phos/CGACTGCGAGCTGATGTCATGTGGGATAACG
Spot-Tag.01 Fd27 (SEQ ID NO:74)
/5Phos/CGACTGCGAGCTGATGCACC AAACGATAACG
Spot-Tag.01 Fd28 (SEQ ID NO:75)
/5Phos/CGACTGCGAGCTGATGATTGTCCCGATAACG
Spot-Tag.01 Fd29 (SEQ ID NO:76)
/5 Phos/C GACTGCGAGCTGATGCGTTTGC AGATAACG
Brady.01 Fd12 (SEQ ID NO:77)
/5Phos/CGACTGCGAGCTGATGTCTI1CCGGATAACG
Brady_01 Fd13 (SEQ ID NO:78)
/5Phos/CGACTGCGAGCTGATGTTGCTCACGATAACG
Brady .01 Fd14 (SEQ ID NO:79)
/5Phos/CGACTGCGAGCTGATGAGGAGCAAGATAAC
CLR.Null.Blk Fd24 G (SEQ ID
1,40:80)
/5Phos/CGACTGCGAGCTGATGITCCCTTCGATAACG
CLR.Null.Blk Fd25 (SEQ ID NO:81)
/5Phos/CGACTGCGAGCTGATGTCTGAGGTGATAACG
CLR.Null.Filk Fd26 (SEQ ID NO:82)
135
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
/5Phos/CGACTGCGAGCTGATGGCCTTGATGATAACG
5Phos.01 Fd7 (SEQ ID NO:83)
/5Phos/CGACTGCGAGCTGATGCGTACTAGGATAACG
5Phos.01 Fd8 (SEQ ID NO:84)
/5Phos/CGACTGCGAGCTGATGTGTACGCAGATAACG
5Phos.01 Fd11 (SEQ ID NO:85)
/5Phos/CGACTGCGAGCTGATGAGTACTGCGATAACG
SP6.01 Fd21 (SEQ ID NO:86)
/5Phos/CGACTGCGAGCTGATGTTGGGCAAGATAACG
SP6.01 Fd22 (SEQ ID NO:87)
/5Phos/CGACTGCGAGCTGATGTTCCACGTGATAACG
SP6.01 Fd23 (SEQ ID NO:88)
/5Phos/CGACTGCGAGCTGATGGAGTTACGGATAACG
SP4.01 Fd15 (SEQ ID NO:89)
/5Phos/CGACTGCGAGCTGATGTGATATAGGATAACG
SP4.01 Fd16 (SEQ ID NO:90)
/5Phos/CGACTGCGAGCTGATGACCTTAGAGATAACG
SP4.01 Fd17 (SEQ ID NO:91)
*See Table 4.1 for target sequences
Prior to seeding the colocalized constructs, the sequencing chip was washed
with
100 uL Hybridization Buffer twice. Each mixture of colocalized constructs were
diluted
to 0.5 nIVI and and 1.14 uL of each mixture was combined with 10 la, of 2x Bhp-
it/TA
MM Ligase Master Mix and 44 uL of Hybridization Buffer, and gently mixed for a
final
concentration of 120 pM of colocalized constructs. To ligate the colocalized
constructs
onto the chip, the sequencing chip was washed with 30 uL of Foundation Mix
twice and
heated at 28 C for 15 minutes on a hotplate. Then it was washed once with 100
uL of
100% formamide to remove tmligated colocalized constructs. The chip was heated
again
at 40 C for 90 seconds on a hotplate, washed with 500 uL of Blocking Buffer
(0.025%
TWEEN20 in lx PBS + 10 mg/nil BSA) once, washed with 30 uL of Chip Blocking
Solution twice (10 uM of P5 Complementary oligo (5%
TCTCGGTGGTCGCCGTATCATT-3' (SEQ ID NO:92))/P7 Complementary oligo (5%
ATCTCGTATGCCGTCTTCTGCTTG-3' (SEQ ID NO:93)) sequences + 10 uM POC
Tail blocking sequence (5'-
TAGGGAAGAGAAGGACATATGATTATCCACGTGCATCTAAG-3' (SEQ ID
NO:94))), incubated for 37 C for 15 minutes on a hotplate, and washed with 100
uL
136
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Hybridization Buffer twice for 60 seconds one immediately before loading the
prepared
binder library (see Barcoded-Binder Library Preparation section below).
Bareoded-Binder Library Preparation
4 DNA barcoded "binders" were incubated with the targets, each consisting of a
binder region, a DNA spacer region, a restriction site, DNA barcode indicative
of the
binder region identity, and ligation site. 2 DNA binders, U4.SA1.3 and
U4,SA2.3,
contained a binder region consisting of DNA that were complementary to SP4.01
and
SP6.01 respectively. These binders were positive controls that should bind to
SP4..01
and SP6.01 with high affinity and specificity. Another DNA binder, U4.SA4.2,
contained a binder region consisting of a scramble DNA sequence that should
bind to
none of the targets present, serving as a negative control to measure noise.
The last
binder was the Spot-tag nanobody-oligo conjugate.
Prior to incubation each binder was hybridized to a universal bridge (5"-
CTGCGCCTATAGGAATTCGTTATC/i5NitInd//i5NitInd//i5NitInd//i5NitIndlli5NitInd/
/i5NitIndlli5NitIndlli5NitIndlli5NitInd//i5NitIndlii5NitIndlli5NitInd/GGACACCIG
CCGT
TATC-3' (SEQ ID NO:95)), an oligo that was partially complementary to the
restriction
site spacer and partially complementary to the consensus sequence (FIG. 14B).
Each
/i5NitInd/ is a 5-Nitroindole, a universal base analogue that exhibits high
duplex stability
and hybridizes indiscriminately with each of the four natural bases (Loakes
and Brown,
1994). The DNA binders and the Spot-tag nanobody target were hybridized with
their
respective bridges in separate reactions. The DNA binders were added to 2x
excess
bridge oligos per DNA binder in Hybridization Buffer to generate a 50 uL
solution with
an end concentration of 200 nM of each DNA binder (600 nM of all DNA binders
combined). The solution was heated to 95 C for 5 minutes at room temperature
(RT)
(22-24 C) for an hour.
To hybridize the Spot-tag nanobody target to the universal bridge, it was
added to
5x excess bridge per Spot-tag nanobody target in Hybridization Buffer to
generate a 49
uL solution with an end concentration of approximately 400 nM Spot-tag
nanobody
target In the preparations of nanobody-oligo conjugates, the DNA tails are
added in
excess and are not purified away. It is possible that the excess of
unconjugated DNA tails
present in the solution hybridize to the Spot-tag-oligo conjugates, preventing
hybridization of the universal bridge needed for the subsequent ligation of
the Spot-tag
137
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
nanobody barcode to the nearby foundation. A ratio of 5:1 bridge:Spot-tag
nanobody
target was used such that any excess DNA tail that were in the solution but
not conjugated
to Spot-tag nanobody target from the protein-oligo conjugation reaction were
hybridized
to a bridge, promoting bridge hybridization with all oligo tails conjugated to
Spot-tag
nanobody targets. This solution was heated to 37 C for 30 minutes and cooled
at RT for
30 minutes. After cooling, the solutions containing the DNA binders and Spot-
tag
nanobody targets, both hybridized to universal bridges were combined and 1 uL
of
Blocking Buffer (0.025% TWEEN20 in lx PBS + 10mg/m1 BSA) was added. The final
binder library solution had a concentration of 100 nM of each DNA binder (300
nM of all
DNA binders combined) and 200 nM of Spot-tag nanobody target.
Barcoded-Binder Library Incubation, Binder Barcode Ligation, and Restriction
Digest
After the step of washing the sequencing chip with 100 uL Hybridization Buffer
twice for 60 seconds (see Build and Tether Foundations to Solid Substrate
section above),
the chip was washed with Aptamer Incubation Buffer (0.025% TWEEN20 in lx PBS +
0.1mg/m1 BSA) for 60 second& The binder library was gently mixed and the
sequencing
chip was slowly loaded with 30 uL binder library solution twice. The
sequencing chip
was incubated with the binder library solution on a hotplate at 25 C for 30
minutes. After
incubation, the chip was washed with 100 uL of Aptainer Incubation Buffer for
90
seconds three times to wash away unbound and weakly bound binders.
To prepare the ligation reaction, 7 uL of 2x Blunt/TA MM Ligase solution was
diluted in 63 uL of Hybridization buffer arid gently mixed, 30 uL of the
diluted ligase
solution was loaded onto the chip twice before the chip was incubated for 5
minutes in a
hotplate at 28 C to ligate the DNA tail of the binders to its bound target's
respective
foundation oligo. The ligation reaction was terminated by washing the plate
with 100 uL
of lx CutSmart solution for 60 seconds three times.
The rest of the binder besides the consensus region and binder barcode was
removed from the barcode-foundation construct with a restriction digestion
reaction. The
restriction enzyme mix was prepared by adding 10 uL of 20 units/uL EcoR1 to 30
uL 10
uM Restriction bridge (5'-CTGCGCCTATACGAATTCGTTATC-3' (SEQ ID NO:96)),
uL of 10x CutSmart solution, and 77 uL of Nuclease-Free H20 before the
contents
were gently mixed. 30 uL of the restriction enzyme mix was loaded onto the
chip twice
and incubated at 40 C on a hotplate for 30 minutes. To terminate the ligation
reaction
138
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
and wash off any hybridized DNA, the chip was loaded with 100 uL of 100%
fonnamide,
incubated at 40 C on a hotplate for 90 seconds, and washed with 500 uL of
Hybridization
Buffer.
Sequencing
The final step in the sequencing process was the addition of Next Generation
Sequencing (NGS) adapters. 1.5 uL of 2:1 luM Universal NGS Adapter
(/5Phos/AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTC
(iCCGTATCATT (SEQ ID NO:97)) + Universal NGS Adapter Bridge9/5 (5'-
TTCCGATCTCGTTA-3' (SEQ ID NO:98)) was added to 10 uL of 10x CutSrnart, 25 uL
of 2x Blunt/TA MM Ligase, and diluted in 63.5 uL of Nuclease-Free H20. 30 uL
of the
NGS ligation mix was loaded onto the sequencing chip twice and the chip was
incubated
at 40 C on a hotplate for 2 minutes and 45 seconds. The chip was washed with
500 uL of
Nuclease-Free H20 twice with 90 seconds in between the washes. 20 uL of 20 pM
denatured PhiX (Illtunina) was diluted in 580 uL of HT1 buffer (Illumina) and
loaded into
the sample well of the sequencing cartridge. A 45 cycle read was conducted
using MiSeq
V2 chemistry.
Results
Conjugate Spot-Tag nanobody to DNA tail
Labeling of the protein was not site-directed as it was with the sortase-
mediated
method. Multiple higher molecular weight bands were observed on the gel,
presumably
corresponding to multiple oligos conjugated to a single nanobody. Importantly,
for BCS
experiments these constructs are less of a concern because they will either 1)
be non-
functional, in which case they will not bind Spot-Tag and be washed away, or
2) will bind
to the Spot-Tag, following which either of the multiple tails can then become
ligated to
the nearby foundation.
Results - BCS Binding Assay Proof of Concept
Preliminary results adapting a nanobody against its known peptide target on
the
BCS platform have shown that, within a complex environment, specific binding
events
with binders in the sub-micromolar range can be recorded into a DNA signal and
deconvoluted. When a library of prospective binders was incubated with
foundations
139
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
attached to either bradykinin (Brady.01), no target (CLR.Null.Blk and
5'Phos.01 as null
controls), DNA targets (SP4.01 and 5P6 as positive controls), or Spot-Tag
protein (Spot-
Tag.01), barcodes of the Spot-Tag binder were ligated to all foundations
associated with
Spot-tag targets at a significantly higher rate than foundations corresponding
to other
targets. Sequencing counts of a Spot-tag binder barcode ligated to Spot-tag
target
foundations compared to other foundations was 3383-10630 vs 0-1617 counts
(FIG. 56).
Sequencing counts showed that 32-73% of Spot-tag target foundations were
ligated to
Spot-tag binder barcodes, while 0.3-10.7% of other foundations were ligated to
Spot-tag
binder barcodes. For positive target controls, SP4.01 and SP6.01, sequencing
counts
report a high number of DNA binder barcodes were ligated to its intended
target
foundation compared to foundations of other targets. Foundations of null
control targets
and the peptide target control for non-specific binding (Brady.01) ligated to
any binder
barcodes were at or below the noise floor. No sequencing counts were observed
for the
negative control binder, AV.B4.U2.SA4.2.
To confirm that true signal was observed, in experiments where only
unconjugated Spot nanobodies and oligos were loaded onto the sequencing chip,
no Spot-
tag nanobody barcodes were observed on respective foundations (FIG. 57). For
further
optimization experiments, it would be important to work with carefully
purified protein-
oligo conjugates, validate BCS process for oligo tails comprised of two parts
to enable
modularity of design, validate the BCS platform for protein-based binders with
low
affinity such as Myc-tag, and characterize BCS performance with binders across
a
different range of affinities and concentrations.
Example 5¨PROSEQ-VIS Experimentation
Methods
Peptide Tethering
Proteins from cells are isolated, digested and processed prior to tethering
peptide
fragments to a solid substrate. Cells are first lysed and then proteins are
isolated by
precipitation. Isolated proteins are denatured using a surfactant, and then
reduced and
allcylated to protect Cysteine side chains_ In order to attach oligo strands
to the amino
side chain of Lysines, the proteins are incubated in a reaction mixture of
sodium
phosphate buffer (pH 4-5), sodium cyanoborohydride, deionized water, and
oligos
modified with an aldehyde on their 3' end and a phosphate group at us 5' end.
140
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Afterwards. proteins are digested with Lys-C, resulting in peptide fragments
with an
oligo-modified lysine at each C-terminal. Then the 5' ends of the oligos are
covalently
attached to the 3' adaptor on a flow cell with a DNA ligase, tethering the
peptide-oligo
constructs to a solid substrate.
Aptarner Incubation and Imaging
After the oligo-peptide constructs are covalently attached to the substrate
the
sequencing process begins by incubating the first aptamer pool, followed by
washout of
unbound aptamers. On a single chip, 25 million to 5 billion peptide fragments
can be
immobilized across multiple fields of view. After target immobilization, a
library of
unique, aptamers with a unique tail of barcodes hybridized to a protective
complementary
oligo are incubated with the chip to allow for target binding. The unbound
aptamers are
washed off. The bound aptamers are treated with paraformaldehyde (PFA) before
the
dsDNA portion is denatured and the protective complementary oligo washed away
to
expose the barcode-containing region for probe hybridization. The aptamer:
amino acid
complexes are incubated with a library of probes that hybridize to barcode
regions
indicative of probe iteration 1. The unbound probes are then washed off and
bound
probes are imaged to acquire the first section of the optical barcode. After
imaging, the
bound probes are denatured from the aptamer barcode tail and washed off the
chip.
Thereafter, the bound aptamers are incubated with the next set of probes that
hybridize to
barcode regions indicative of probe iteration 2. Iterations of probe
incubation, imaging,
and washing are repeated until full optical barcodes are acquired. The
peptides, along
with the covalently bound aptamer, is degraded processessively from the N-
terminal
using Edman degradation, aminopeptidases, or any other processessive
degradation
process. Then, the cycle of aptamer incubation, iterations of probe incubation
and single
molecule imaging, and amino acid cleavage repeats for multiple rounds to
obtain the
sequence of the peptide molecule (FIG. 23).
As proof-of-concept that single molecule imaging can be achieved without TIRF
microscopy, forward and reverse colocalization linkers (FC and RC) were tagged
with
fluorescent Streptavidin beads and imaged on a flow cell. The FC consisted of
the
barcode foundation-complementary region at the 5' end, followed by sequence
complementary to the glass-bound oligo, followed by a flexible T-spacer, with
a short,
high GC-content sequence at the 3' end complementary to the RC. In turn, the
3' end of
the RC was complementary to the 3' end of the FC, followed by a long T-spacer,
141
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
followed by a sequence complementary to the glass-bound oligo, followed by a
sequence
complementary to another oligo. The FC and RC was biotinylated at the 5' end.
The FC,
LC, and Streptavidin beads, and flow cell surface were blocked separately with
a BSA
buffer (lx PBS, .05% Tween, 10mg/m1 BSA) for 1 hour at RT. In two separate
reactions,
the FC was incubated with FluoSpheresTM Streptavidin-Labeled Microspheres,
0.04 gm,
yellow-green fluorescent (505/515), and the RC with TransFluoSpheresTm
Streptavidin-
Labeled Microspheres, 0.04 p.m (488/645) in a 1 : 4 oligo to beads ratio such
that each
biotinylated oligo likely binding to at least one bead for 30 minutes at RT.
The FC and
RC were combined in a 1 : 2 ratio for 1 hour at RT. The solution was loaded
onto a
Illumina MiSeq v2 (50-cycles) chip and incubated for 30 minutes at 37 C to
allow for the
FC and RCs to hybridize to the P7 adaptors in the chip. The imaging system is
a wide-
field upright fluorescence microscope with a 20X Nikon objective (NA = 0.75).
Glass
piece of the chip was taken out from the MiSeq cassette and imaging was
performed on
the external top surface of the chip. The beads inside the chip were excited
at 488 nm
with SPECTRA X LED light engine and the emitted fluorescence signal was
collected at
515 nm (with a 520/35 bandpass emission filter) and 645 nm (with a 676/29 nm
bandpass
emission filter). Images were acquired with an Andor EMCCD camera with 16
micron
pixel size and 2 second exposure time.
Optical Bareode Decor:volution
After repeating this series of steps on the slide, the identity of successive
N-terminal
amino acids at each round is computationally deduced by colocalizing the
optical
barcodes and generating a peptide sequence. Once peptide sequences are
generated they
will be compared against the organism proteome for protein identification and
quantification.
Results
Imaging Single Molecules
In each iteration of probe incubation and imaging, single peptide molecules at
known locations on the chip (i.e. assigned coordinates (X,Y), generates
spatially
overlapping fluorescent signals (FIG. 58A) that can be detected by separate
channels
(FIG. 5813).
Preliminary data has shown that single oligonucleotide imaging can be achieved
with widefield fluorescence microscopy. Since each biotinylated oligo is
binding to at
142
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
least one streptavidin bead, each fluorescent spot represents at least one
bead (FIG. 59).
In the case where each biotinylated oligo is binding to a cluster of beads,
spots will
appear bigger, or brighter compared to spots with the same size. Streptavidin
beads not
bound to oligos were imaged on a glass as a control (FIG. 58). The similarity
of sizes of
the observed spots between the fluorescent beads on the chip and sequencing
chip
suggests the observed spots on the sequencing chip are single molecules. FIG.
608
shows the intensity distribution of all the fluorescent spots in an image
snapshot. The
local maxima of every 10,000 grayscale count (in the case of channel one: 488
nm
excitation and 645 nm emission, FIG. 60B ) can be used to distinguish spots
with various
peak intensities. For example, the first interval (grayscale count from 0 -
10,000
grayscale count) in FIG. 6013 indicates only one streptavidin bead bound to
one
biotinylated oligo. The second or third interval suggests a cluster of (two or
three)
streptavidin beads were binding to one biotinylated oligo. Data from size
comparison
analysis and intensity distribution suggests that single oligo molecules were
detected.
Fluorescent Signal Deconvolution Into Aptamer Identity
The fluorescent signature that combines fluorescent signal in each channel for
each iteration of a round is compared against the known optical barcodes of
each unique
aptamer, thus deducing the likely identity of the bound N-terminal prefix
based on
probability distributions of binding events for each aptamer against each
prefix (FIG.
58C).
Aptamer Identity to Protein Sequence
For each single peptide molecule at a known location on the chip, the N-
terminal
prefix calls from each round is used to computationally deduce the likely
amino acid
sequence of the peptide tethered at (X,Y). If the N-terminal prefix associated
with the
ssDNA binding regions of the recorded aptamers overlap such that the second
amino acid
of a round is the same as the first amino acid of the subsequent round, there
is greater
confidence in the computationally derived peptide sequence (FIG. 58D).
Protein Sequencing for Full Proteins
Contiguous peptide sequences are linked together in a series of non-contiguous
assay-derived peptide sequences into a scaffold by stitching overlapping
sequences to
143
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
generate the sequence of the full-length protein. The sequences are mapped
against a
proteome map to identify known proteins in the sample, for example argipressin
(FIG.
58E). Relative quantification of a unique protein/peptide in the sample is
calculated from
the number of derived peptide sequences associated with that protein/peptide.
Example 6¨Multiplex Experimentation
Reagents
Aptamer libraries were purchased from TriLink Biotechnologies, and all other
oligonucleotides were purchased from IDT. Peptide oligo conjugates were
ordered from
Gen.script. All automated procedures were performed on the Agilent Bravo NGS
Workstation. All DNA quantifications were obtained using dsDNA and/or ssDNA
High
Sensitivity Qubit Fluorescence Quantification Assay (Thermofisher). All water
used was
Ambionni Nuclease-Free water.
Methods
Bring Up
N40 aptamer libraries consisted of 40 random bases, flanked by custom primer
regions. Aliquots of these initial libraries
(TTGACTAGTACATGACCACTTG
NNNNNNNNNNNNNCACATCAGACTGGACGACAGAA (SEQ ID NO:99)) were
ordered from TriLink. A sample of 1012 sequences (-48 ng) from this initial
library were
amplified across 288 reactions of 50 microliters each using Herculase II
Fusion DNA
Polymerase (Agilent Technologies) and SPRI-purified using Mag-Bind TotalPure
NGS
beads on a Bravo Automated Liquid Handling Platform (Agilent). The
amplification
conditions for this and all subsequent PCR reactions (with the exception of
NGS
preparation) were as follows: an initial denaturation at 95 C for 5 minutes
followed by 13
amplification cycles of 30 seconds of denaturation at 95 C, 30 seconds
annealing at 55eC,
30 seconds elongation at 72'C, and a final elongation of 5 minutes at 72"C.
Digestion
Amplified libraries were converted to single-stranded DNA (ssDNA) by
enzymatic digestion using lambda exonuclease (NEB) and purified by automated
bead
clean up. ssDNA digestion completion was qualified using the small RNA kit
(Agilent)
on the Bioanalyzer 2100 (Agilent), and the concentration quantified post-clean
via a
ssDNA Qubit Assay (Thermofisher).
144
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Peptide-oligo constructs
Peptide-oligo constructs were synthesized by Genscript (full construct: (N-
terminus)- -Cys (SEQ ID NO:100) (C-
terminus)-
3'ATCCCITCTCTTCCTGTATACTANNNNNNGCACGTAGATTC 5' phosphate
(SEQ ID NO:101)). The C-terminus of a 10-mer peptide (with the exception of
(mnRH,
which was an 11-mer, and argipressin, which was a 9-mer) was attached to the
3' end of a
41-nucleotide oligo. All but the final amino acid residue of the peptides were
derived
from naturally occurring peptides (such as (inRH, bradykinin, and argipressin)
or
synthetic peptide designs, with the N-terminal residue reserved for a cysteine
that
facilitated peptide attachment to the oligo. The 41-nucleotide (nt) oligo
featured a 9-
nucleotide bridge-binding region at the 3' end, a 3 nt spacer, a 6 nt DNA
barcode uniquely
associated with the peptide, and a 23 nt primer region at the 5' end.
Incubation
SsDNA pools were heated to 95t for five minutes, then rapidly cooled on ice
prior to incubation with peptide. For the ideal experimental condition in the
first and
second rounds of MULTIPLEX, 166.62 pmol (4650 ng) of folded aptamers were
added to
18.51 pmol of the peptide-oligo construct (for a final stringency of 1:10
target: DNA).
These numbers were scaled according to the amount of ssDNA available for
incubation in
each individual experiment. For rounds 3 and 4, the stringency was increased
to 1:25. A
final buffer solution was prepared from 10X PBS (Sigma-Aldrich), TWEEN20
(Sigma
Aldrich), and HiFi Tag Ligase buffer (NEB) to bring the final incubation
solution to 400
ul total volume, at a concentration of 1X PBS, 1X HiFi Tag Ligase Buffer, and
0.025%
TWEEN20. The peptide-oligo constructs and aptamers were allowed to bind for 2
hours
at RT under rotation.
Ligation
HiFi Tag Ligase (NEB) and a 18-mer DNA bridge
(GCAUCUAAGUUCUGUCGU (SEQ ID NO:102)) were added to the 400 ul mixture of
aptamers and peptide-oligo constructs, with 1 ul of HiFi Tag for every 50 ul
of incubation
solution and the I8-mer bridge at a final concentration of 100 nmol. Ligation
happened at
25 C for 30 minutes. The bridge was subsequently degraded by adding USER
enzyme
(NEB) and 10X cutsmart, and incubating the solution at 37 C for 15 minutes.
Incubation with Biotin
145
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
A biotinylated oligo (/5Biosg/TAGGGAAGAGAAGGACATATGAT-3' (SEQ ID
NO:103)) that hybridizes to the 5*-ATCATATGTCCTTCTCTICCCTA-3' (SEQ ID
NO:104) region of the peptide oligo construct was added to the reaction at an
equimolar
ratio to the peptide-oligo construct. The reaction was incubated for 30
minutes under
rotation.
Streptavidin-Biotin Pulldown
Streptavidin Cl beads (Invitrogen) were incubated with the solution at 83.33
ug
for every 51.02 pmol of peptide present for 30 minutes. Bead-bound peptide
aptamer
constructs were collected using an automated wash protocol on the Bravo. The
MULTIPLEX reactions were incubated on a magnetic plate for 2 minutes. The
supernatant containing unbound aptamers was aspirated away and the beads were
washed
two times with SELEX buffer, followed by a final wash with IX PBS. The 1X PBS
was
aspirated at the end of the protocol.
PCR on beads
Immediately after the automated wash protocol finishes, 50 ul of PCR Mastennix
solution was added to the beads. The primers 51-TAGGGAAGAGAAGGACATATGAT-
3 (SEQ ID NO:105) and TTGACTAGTACATGACCAC'TTGA-3' (SEQ ID NO:106)
were used to amplify the 126 nt construct (5'
ACTFGANNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNCACATCAGACTGGACGACAGAACTTAGATCCACGNNNNN
NATCATATGTCCTTCTCTTCCCTA 3' (SEQ ID NO:107)).
NGS Preparation
ng samples of SPRI-purified PCRs on beads were taken for NGS preparation.
Each aptamer identified from sequencing these samples were associated with the
6bp
barcode of the peptide they putatively bound to in solution. The P5 and P7
adapters
required for Illumina sequencing were incorporated through PCR with custom NGS
primers (5'-
CAAGCAGAAGACGGCATACGAGATNNNNNNNNGTGCGTGCGTGCTTCTGTCG
TCCAGTCTGATGTG-3' (SEQ ID NO:108) and 5'-
AATGATACGGCGACCACCGAGATCTACACNNNNNNGCATGCAGCCGGTTGAC
TAGTACATGACCACTTGA-3' (SEQ ID NO:109)). The amplification conditions for
these PCR reactions were as follows: an initial denaturation at 95"C for 5
minutes
followed by 10 amplification cycles of 30 seconds of denaturation at 95t, 30
seconds
146
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
annealing at 65t, 30 seconds elongation at 72rt, and a final elongation of 5
minutes at
72t. The final NGS library was SPRI-purified, pooled, and cleaned via PippinHT
(Sage
Science).
Threshold PCR/Nested PCR
For each MULTIPLEX reaction, 4.08 ng of the SPRI-purified product was
amplified
across twenty-four 50 ul PCR reactions using 5'-
T*A*G*G*G*A*AGAGAAGGACATATGAT-3' (SEQ ID NO:110) and /5Phos'/-
TTGACTAGTACATGACCACTTGA-3' (SEQ ID NO: 111)), wherein *indicates the
nucleotide was modified such that the sulfur atom in the phosphate backbone
was
substituted for a phosphorothioate bond substitutes a sulfur atom, which
renders the
sequence more resistant to nuclease digestion. The end product of this nested
PCR is a
86-bp amplicon that matches the original N40 library. It can be converted to
ssDNA via
enzymatic digestion, and used for another round of MULTIPLEX.
Results
The resulting data provided information about how aptamers preferentially bind
to
alternative targets in the same experiment. Presently, up to 6 targets have
been
concurrently evaluated via MULTIPLEX.
Within a given MULTIPLEX experiment, target-specific sequences showed
selective binding behavior towards their associated targets (FIG. 61).
Analysis measured
selectivity as reads to the desired target divided by reads to all targets at
round 4. The top
sequences of each target (GNRH, NC2, NC3, Ti, Vaso) showed selectivity of asoo
to
0.924 to their intended targets, and no more than 0.250 to any individual
unintended
target.
Within a MULTIPLEX experiment, there is significant bleedthrough between
targets, with no aptamers that are exclusively identified with a single target
(though there
are round 4 aptamers identified with argipressin up to 58.3% of the time, GnRH
50% of
the time, and Targetl NC2 up to 83.3% of the time). As three of the six
targets had
peptides of similar sequences (Target 1: (N-terminus)-YQNTSQNTS-Cys (C-
terminus)
(SEQ ID NO:112); Targetl NC2: (N-terminus)-KQNTYQNTS-Cys (C-terminus) (SEQ
ID NO:113); Targetl NC3: (N-terminus)-QNTSYQNTS-Cys (C-terminus) (SEQ ID
NO:114)), it is not surprising that they may pull down the same aptamer (FIG.
62).
147
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Example 7¨Turducken Experimentation
Reagents
Constructs for expression of RNA-binding proteins and RNA sequences were
assembled using the standard tools and methods of molecular biology, such as
PCR
amplification, restriction digest, infusion assembly or ligation. Genes of
interest or the
DNA sequences encoding RNA hairpins were ordered as geneblocs or assembled by
PCR_ All regions amplified by PCR were verified in the final bacterial clones
by Sanger
sequencing. Cloning of the expression construct for both RNA-binding protein
and RNA
was performed sequentially, with the gene encoding the RNA binding proteins
inserted
first, followed by restriction digest of these vectors and insertion of the
DNA fragment
encoding the RNA hairpin to produce vectors for expression of both the RBP and
the
RNA. Experiments were performed with a tandem fusion of the MS2 coat protein
(dMS2) tagged with a 9xHis motif for affinity purification, with or without a
molecular
fusion to Emerald GFP (EmGFP). MS2 binding site contains a U to C mutation,
which
improves the affinity of the RNA-protein interaction. For bacterial
expression, dM52-
EmGFP or dMS2 were cloned into pRSFDuet1 vector under the control of 17
promoter
using Infusion (Takata) cloning, and transformed into NEB Turbo cells for
plasmid
amplification. Plasmids were purified from NEB Turbo cells using standard
miniprep
kits (Zymo or Thermo) and sequence verified. All water used was AmbionTh
Nuclease-
Free water.
Methods
Transformation
For overexpression of proteins in bacteria, plasmids carrying dMS2-EmGFP or
dM52 were transformed into T7 Express lysY/Iq Competent E. coil from NEB, and
plated on kanamycin antibiotic selection plates (50 ug/ml) overnight at 37 C.
Protein Expression
Single colonies were resuspended in 5 ml of LB liquid culture media with 50
ug/ml kanamycin and incubated with shaking at 37 C until 0D600 reached 0.4-0.8
to
produce a starter culture. 50-500 ul of starter culture was used to inoculate
5 ml of fresh
LB media with 50 tag/ml kanamycin, and protein production was induced by the
addition
of 0.1 -1 mM IPTG, shaking either overnight at 22-27 C or for 3-5 hours at 37
C.
Protein Isolation
148
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Following protein induction, cells were pelleted by centrifugation at 3,000-
5,000 g
for 5 minutes, washed once with 1 ml of ice-cold PBS, pelleted again and re-
suspended in
200-1000 ul of Y-PER Plus Dialyzable Yeast Protein Extraction Reagent
supplemented
with Halt Protease Inhibitor Cocktail. The weight of the cell pellet
determined the
volume of Y-PER reagent added per manufacturer's recommendation. The mixture
was
gently agitated at room temperature for 20 minutes, and soluble proteins were
isolated
from cell debris by centrifuging at 14,000 x g for 10 minutes.
Supernatant containing soluble cell proteins was removed, analyzed by SOS-
PAGE and Coomassie staining or BCA assays. dMS2 or dMS2-EmGFP were further
isolated by DynabeadsTm His-Tag Isolation and Pulldown using manufacturer's
protocol.
Briefly, lysates from 5-ml liquid cultures were incubated with 100 ul of
Dynabeads in
final volume of 700-1400 ul, with the lysate volume adjusted using
Binding/Wash buffer
(50 niM Sodium Phosphate, pH 8.0, 300 niM NaCI, 0.01% Tween-20). After 5-10
minute incubation, the beads were washed 4x with 300-600 ul of Binding/Wash
buffer,
with the supernatant discarded after each wash and beads resuspended fully in-
between.
To elute the protein, following the final wash beads were incubated for 10
minutes
on a roller with 100-200 ul Binding/Wash buffer containing 300 mM imida.zole.
Eluted
protein was exchanged into PBS and concentrated to ¨1 mg/ml using 10 kDa
Amicon
Ultra-0.5 Devices. Purified protein was quantified using Pierce BCA Protein
Assay Kit
or SOS-PAGE gels stained with SimplyBlue SafeStain.
Binding Verification
Binding of dMS2-EmGFP and dM52 to MS2 RNA was verified by
electrophoretic mobility shift assays (EMSA).
Product Quantification
¨ 350-nt long RNA containing MS2 binding site was produced by in vitro
transcription using TranscriptAid T7 High Yield Transcription Kit, purified
with Qiagen
RNeasy Mini Kit and quantified using Nanodrop.
Product Identity Verification
The presence of the correct product was verified by agarose gel
electrophoresis
following purification. RNA was diluted in TE buffer to 1-10 uM final
concentration and
stored at -80C. Prior to binding experiments, RNA was heated to 70-80 C for 5
minutes
and snap cooled on ice for 5 minutes. Electrophoretic mobility shift assays
were
performed by incubating 1-3 nM RNA with increasing protein concentrations (0 -
200
149
CA 03149852 2022-3-1
WO 2021/051011
PCT/U52020/050574
nM) in 80 mM KC1, 10 mM MgCl2, 100 mM Hepes, pH 7.5 (20 ul final volume) for
30-
60 min at room temperature. SUPERase RNase Inhibitor was added to all binding
reactions. RNA and RNA-protein complexes were resolved by non-denaturing PAGE
using Novex 4-12% Tris-Glycine Gels in Novex Tris-Glycine Native Running
Buffer.
RNA was stained using SYBR Green nucleic acid stain and gels imaged using E-
Gel
imager.
Results
Expression Verification
SDS-PAGE demonstrated that denatured peptides or proteins purified using an
Anti-His affinity pull-down assay were of the expected size for dMS-EinGFP and
dMS2,
indicating that both dMS-EmGFP and dMS2 were expressed. BSA was included as a
standard (FIG. 63).
Binding Verification
EMSA demonstrated dMS2-EmGFP fusion protein bound to ¨2 nM RNA
containing the MS2 coat protein binding site (FIG. 64).
Product Verification
EMSA demonstrated that the c1M52 proteins (without EinGFP) bound to ¨2 nM
RNA containing the MS2 coat protein binding site, verifying the identity of
the protein.
(FIG. 651
Example 8¨LEGO Experimentation
Reagents
Double-stranded DNA primers (TriLink Forward:
TAGGGAAGAGAAGGACATATGAT (SEQ ID NO:115); TriLink Reverse with Lego
4: GCTCTACAGTATTGACTAGTACATGACCACTTGA (SEQ ID NO:116)) and
LEGO pieces (10-mers with 5' phosphorylated single base-pair overhangs) were
obtained
from IDT. The LEGO sequences were:
= Legol: AATGCTGAGC (SEQ ID NO:117)
= Lego2: CACTACAGCC (SEQ ID NO:118)
= Lego3: TAGCACTGAG (SEQ ID NO:119)
= Lego4 with TriLink Reverse:
GCTCTACAGTATTGACTAGTACATGACCACTTGA (SEQ ID NO:120)
150
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Methods
Ligation Reaction
An initial ligation reaction was performed at 25 C (on a thermocycler) for 15
minutes using 2 ul of 2.5 uM TriLinIc Forward dsDNA primer, 2 ul of 2.5 uM
initial
dsDNA LEGO piece (LEG01), 2 ul 10X CutSmart Buffer (NEB), 5 ul BlunvTA Ligase
Master Mix (NEB), 1 uL 2 inM ATP, and 10 uL of water. A subsequent LEGO piece
was ligated to the extending product by adding 2 uL of 2.5 uM LEGO2 and 5 ul
of
Blunt/TA Ligase MM to the initial reaction and allowing it to incubate for 15
min at
2.5 C. This process was repeated two more times until the TriLink Reverse
dsDNA
primer with LEGO4 was added.
Post-Processing & Sequencing
Ligation product was collected with a cleanup assay run on a Bravo Automated
Liquid Handling Platform (Agilent), PCR amplified, and then cleaned again with
the
same Bravo protocol. The cleaned PCR product was NGS-prepped for sequencing
with
custom primers. The NGS-prepped sample was size-selected for a 177-bp-long
product
using a PippinHT automated gel extraction system. A 40x8x6x38 (Read
lxi7xi5xRead2)
read was conducted using NextSeq V2.5 chemistry.
Results
Sequencing results demonstrated that with sequential ligations and unique
single-
base overhangs, 10-mers can be directed to assemble into a goal 40-mer
sequence (with
one 23 bp primer on each end) with ¨80% efficiency (FIGs. 66 and 67). These
results
indicate that generating diverse pools with discrete sequences in various
positions is
feasible.
References
Blind, M., & Blank, M. (2015). Aptamer Selection Technology and Recent
Advances. Molecular Therapy - Nucleic Acids, 4. doi:10.1038/mtna.2014.74
Bergman, T., Cederlund, E., & JOmvall, H. (2001). Chemical C-Terminal
Protein Sequence Analysis: Improved Sensitivity, Length of Degradation,
Proline
Passage, and Combination with Edman Degradation. Analytical Biochemistry,
290(1), 74-82. doi:10.1006/abio.2000.4922
151
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Bouchard, P., Hutabarat, R,, & Thompson, K. (2010). Discovery and
Development of Therapeutic Aptamers. Annual Review of Pharmacology and
Toxicology, 50(1), 237-257. doi:10.1146/atuturev.pharmtox.010909.105547
Casagranda, F., & Wilshire, J. F. (1994). C-Terminal Sequencing of
Peptides: The Thiocyanate Degradation Method. Basic Protein and Peptide
Protocols, 335-350. doi:10.1385/0-89603-268-x:335
Chelsea K. L. Gordon, Diana Wu, Trevor A. Feagin, Anusha Pusuluri, Andrew T.
Csordas, Michael Eisenstein, Craig J. Hawker, ha Niu, H. Tom Soh. (2019).
Click-PD: A Quantitative Method for Base-Modified Aptamer Discovery. bioRxiv
626572; doi: https://doi.org/10.1101/626572
Chen, M., Yu, Y., Jiang, F., Zhou, J., Li, Y,, Liang, C.,. . Zhang, G. (2016),
Development of Cell-SELEX Technology and Its Application in Cancer
Diagnosis and Therapy. International Journal of Molecular Sciences,17(12),
2079. doi:10.3390/ijms17122079
de Sousa Abreu, R. D., Penalva, L. 0., Marcotte, E. M., & Vogel, C. (2009).
Global signatures of protein and mRNA expression levels. Molecular BioSystems.
doi:10.1039/b908315d
Diatchenko, L., Lau, Y. F., Campbell, A. P., Chenchik, A., Moqadam, F., Huang,
B., . . . Siebert, P. D. (1996). Suppression subtractive hybridization: A
method for
generating differentially regulated or tissue-specific cDNA probes and
libraries.
Proceedings of the National Academy of Sciences, 93(12), 6025-6030.
doi :10. 1073/pnas ,93, 12.6025
Fujishima, K., Venter, C., Wang, K., Ferreira, R., & Rothschild, L. J. (2015).
An
overhang-based DNA block shuffling method for creating a customized random
library. Scientific Reports, 5(1). doi:10.1038/srep09740
Moon, S., Zhou, B., Janda, K., Brenner, S., & Scolnick, J. (2011). Aptamer
selection by high-throughput sequencing and informatic analysis.
BioTechniques,
51(6). doi:10.2144/000113786
Horspool, D. R., Coope, R. J., & Holt, K A. (2010). Efficient assembly of very
short oligonucleotides using T4 DNA Ligase. BMC Research Notes, 3(1), 291.
doi:10.1186/1756-0500-3-291
152
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Jia, B., & Jeon, C. O. (2016). High-throughput recombinant protein expression
in
Escherichia coli: Current status and future perspectives. Open Biology, 6(8),
160196. doi:10.1098/rsob.160196
Little, J. W. (1967). An exonuclease induced by bacteriophage lambda. IL
Nature
of the enzymatic reaction. J. Biol. Chem. 1967; 242:679-686.
Loakes, D., & Brown, D. M. (1994). 5-Nitroindole as an universal base
analogue.
Nucleic acids research, 22(20), 4039-4043.
https://doi.org/10.1093/nar/22.20.4039
Mckeague, M., & Derosa, M. C. (2012). Challenges and Opportunities for Small
Molecule Aptamer Development. Journal of Nucleic Acids, 2012, 1-20.
doi:10.1155/2012/748913
Miteva, Y. V., Budayeva, H. G., & Cristea, I. M. (2012). Proteomics-Based
Methods for Discovery, Quantification, and Validation of Protein¨Protein
Interactions. Analytical Chemist-1y, 85(2), 749-768. doi:10.1021/ac3033257
Mitsis, P. G., & Kwagh, J. G (1999). Characterization of the interaction of
lambda exonuclease with the ends of DNA. Nucleic acids research, 27(15), 3057-
3063. https://doi. org/10.1093/nar/27. 15.3057
Pfeiffer, F., Rosenthal, M., Siegl, J., Ewers, J., & Mayer, G. (2017).
Customised
nucleic acid libraries for enhanced aptamer selection and performance. Current
opinion in biotechnology, 48, 111-118
Schiess, R., Wollscheid, B., & Aebersold, R. (2008). Targeted proteomic
strategy
for clinical biomarker discovery. Molecular Oncology, 3(1), 33-44.
doi: 10. 1016/j . molonc.2008. 12.001
Tucker, W. 0., Shunt, K. T., & Tanner, J. A. (2012). G-quadruplex DNA
Aptamers and their Ligands: Structure, Function and Application. Current
Pharmaceutical Design, 18(14), 2014-2026. doi:10.2174/138161212799958477
Tuerk, C., & Gold, L. (1990). Systematic evolution of ligands by exponential
enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science,
249(4968), 505-510. doi:10.1126/science.2200121
Vogel, C., & Marcotte, E. M. (2012). Insights into the regulation of protein
abundance from proteomic and transcriptomic analyses. Nature Reviews Genetics,
13(4), 227-232. doi:10.1038/nrg3185
153
CA 03149852 2022-3-1
WO 2021/051011
PCT/US2020/050574
Yates, J. R., Ruse, C. I., & Nakorchevsky, A. (2009). Proteomics by Mass
Spectrometry: Approaches, Advances, and Applications. Annual Review of
Biomedical Engineering, 11(1), 49-79. doi:10.1146/ifimurev-bioeng-061008-1249
Zhou, J., & Rossi, J. (2016). Aptamers as targeted therapeutics: Current
potential
and challenges. Nature Reviews Drug Discovery, 16(3), 181-202.
doi: 10. 1038/nrd.2016.199
It is to be understood that, while the methods and compositions of matter have
been described herein in conjunction with a number of different aspects, the
foregoing
description of the various aspects is intended to illustrate and not limit the
scope of the
methods and compositions of matter. Other aspects, advantages, and
modifications are
within the scope of the following claims.
Disclosed are methods and compositions that can be used for, can be used in
conjunction with, can be used in preparation for, or are products of the
disclosed methods
and compositions. These and other materials are disclosed herein, and it is
understood
that combinations, subsets, interactions, groups, etc. of these methods and
compositions
are disclosed. That is, while specific reference to each various individual
and collective
combinations and permutations of these compositions and methods may not be
explicitly
disclosed, each is specifically contemplated and described herein. For
example, if a
particular composition of matter or a particular method is disclosed and
discussed and a
number of compositions or methods are discussed, each and every combination
and
permutation of the compositions and the methods are specifically contemplated
unless
specifically indicated to the contrary. Likewise, any subset or combination of
these is
also specifically contemplated and disclosed.
154
CA 03149852 2022-3-1