Note: Descriptions are shown in the official language in which they were submitted.
WO 2021/011675
PCT/US2020/042163
A METHOD FOR EFFICIENT ELECTROCATALYTIC SYNTHESIS OF PURE
LIQUID PRODUCT SOLUTIONS INCLUDING H202, OXYGENATES, AMMONIA,
AND SO ON
CROSS-REFERENCE TO RELATED APPLICATIONS
[001] This Application claims priority from U.S. Provisional Application No.
62/874,176,
which was filed in the United States of America on July 15, 2019.
BACKGROUND
[002] Hydrogen peroxide (H202) is a nexus chemical for a variety of
industries, and it is
currently produced through an indirect, energy-demanding, and waste-intensive
anthraquinone process. This traditional method usually generates H202 mixtures
with
concentrations of 1-2 wt.%, followed with further purifications and
distillations,
where significant costs adds up, to reach concentrated pure 11202 solutions
for
commercial use. However, this process requires centralized infrastructures and
thus
relies heavily on transportation and storage of bulk 11202 solutions, which
are unstable
and hazardous.
[003] The direct synthesis of 11202 from hydrogen (112) and oxygen (02)
mixture (Fig. 1A)
provides an alternative route for small-scale on-site generation. Exciting
progresses
have been made in developing selective catalysts over the past decade, such as
the
palladium-tin catalyst with high selectivity (>95%) and productivity (61 mol
kgcat-
1 h-1) towards H202. However, due to the wide range of It flammability limits,
one
big challenge of this direct synthesis route is the inherent hazard associated
with
mixing high-pressure H2 and 02. Thus, in practice, H2 feedstock needs to be
heavily
diluted using CO2 or N2 carrier gas, significantly lowering the yields of
H202. In
addition, the use of methanol in solvents can lead to extra cost in product
separation
for pure H202 aqueous solutions.
[004] Different from the direct synthesis, where 02 and H2 are mixed and
catalyzed on the
same catalytic surface, the direct electrosynthesis of H202 can de,couple the
H2/02
redox into two half-cell reactions (alkaline conditions for example):
1
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
26-02 reduction reaction (26-ORR): 02+ H20 + 2e- ¨'1102- + OH-; (Eq. 1)
H2 (HOR): H2 + 20H- - 2e- ¨>- 2H20. (Eq. 2)
[005] Advantages of this electrochemical route are obvious, including 1) 02
and H2 can be
completely separated without safety issue, and fed with high purity for high
reaction
rates; 2) different catalysts can be designed separately for 2e-ORR and
HORJOER,
with each half-cell reaction optimized; 3) the synthesis can be operated under
ambient
conditions for renewable and on-site 11202 generation; and 4) the 112/02 redox
couple
could even output electricity during 11202 synthesis. Although there have been
selective catalysts such as noble metals or carbon materials developed for the
2e-
ORR pathway, the generated H202 products were usually mixed with solutes in
traditional liquid electrolytes ranging from acidic to alkaline solutions.
Extra
separation processes to recover pure H202 solutions for use were therefore
required.
Other designs including using deionized water (DI water), or polymer
electrolyte
membrane as ion conducting electrolyte were rarely proposed for obtaining pure
11202
solutions, but they generally suffered from low reaction rates, product
concentrations,
or Faradaic Efficiencies (FEs).
[006] Embodiments herein relate to an alternative and highly efficient
concept that employs
a porous solid electrolyte electrolytic cell comprised of a cathodic catalyst,
an anodic
catalyst, ion exchange membranes, and solid electrolyte wherein a porous solid
electrolyte design is used to realize the direct electrosynthesis of pure H202
as well
as many other liquid product solutions. Depending on the pure liquid product
to be
produced, the cathodic catalyst could be 2e oxygen reduction reaction catalyst
(such
as oxidized carbon) to generate pure H202 solutions, or CO2/C0 reduction
catalyst
for pure oxygenates solutions, or N2/N031NO2- reduction catalyst for pure N
species
solutions, and so on. Solid electrolytes can also be replaced with the
corresponding
liquid products if high ionic conductivity can be maintained.
[007] In one aspect, embodiments disclosed herein generally relate to a
porous solid
electrolyte electrosynthesis cell for direct synthesis of high purity liquid
products
wherein the electrosynthesis cell comprises a cathode compartment including a
cathode electrode comprising a gas diffusion layer loaded with a selective
reduction
reaction electrocatalyst for specific reduction reactions wherein the
reduction
2
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
reactions comprise oxygen reduction reactions, CO2 reduction reactions, CO
reduction reactions, N2 reduction reactions, nitrate reduction reactions and
nitrite
reduction reactions. The electrosynthesis cell further includes an anode
compartment
including an anode electrode comprising a gas diffusion layer loaded with a
catalyst
for oxidation reactions; a solid electrolyte compartment comprising a porous
solid
electrolyte; a cation exchange membrane; and an anion exchange membrane;
wherein
the solid electrolyte compartment is separated from the cathode and the anode
by the
anion exchange membrane and/or the cation exchange membrane.
[008] In another aspect, embodiments disclosed herein generally relate to a
process for
producing high purity and concentrated liquid products through
electrocatalytic
reaction in an electrosynthesis cell comprising a cathode compartment
including a
cathode electrode comprising a gas diffusion layer loaded with a selective
electrocatalyst for reduction reactions; an anode compartment including an
anode
electrode comprising a gas diffusion layer loaded with a catalyst for
oxidation
reactions; a solid electrolyte compartment comprising a porous solid
electrolyte, an
inlet, and an outlet; a cation exchange membrane; and an anion exchange
membrane.
The process further includes supplying hydrogen gas or water solutions to the
anode
to be electrochemically oxidized on the oxidation reaction catalysts; and
supplying
an oxygen, CO2, CO, or N2 containing gas to the cathode to be selectively
reduced by
the selective reduction reaction catalyst; wherein the solid electrolyte
compartment is
separated from the cathode and the anode by the anion exchange membrane and
the
cation exchange membrane. The process further includes supplying deionized
water
or N2 gas to an inlet of the solid electrolyte compartment to flow through the
porous
solid electrolyte to bring out the generated liquid product.
[009] In yet another aspect, embodiments disclosed herein generally relate
to a porous solid
electrolyte electrosynthesis cell for direct synthesis of high purity liquid
products
wherein the porous solid electrolyte electrosynthesis cell includes a cathode
compartment including a cathode electrode including a gas diffusion layer
loaded
with a selective reduction reaction electrocatalyst for specific reduction
reactions.
The specific reduction reactions may include oxygen reduction reactions, CO2
reduction reactions, CO reduction reactions, N2 reduction reactions, nitrate
reduction
reactions and nitrite reduction reactions. The electrosynthesis cell may
further include
3
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
an anode compartment including an anode electrode comprising a gas diffusion
layer
loaded with a catalyst for oxidation reactions_ The electrosynthesis cell may
include
a solid electrolyte compartment comprising a porous solid electrolyte, a first
cation
exchange membrane, and a second cation exchange membrane, where the solid
electrolyte compartment may be separated from the each of the cathode and the
anode
by the first and second cation exchange membranes.
BRIEF DESCRIPTION OF THE DRAWINGS
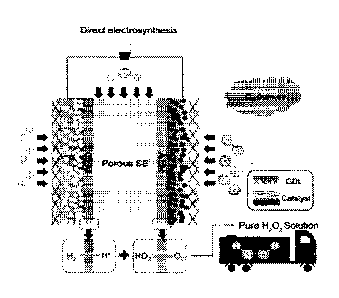
[010] FIGS. 1A-1D show a (Fig. 1A) schematic of direct synthesis of H202
using diluted
H2 and 02 under high pressure. Fig. 1B shows a schematic of direct
electrosynthesis
of H202 using pure 142 and 02 streams separated into anode and cathode,
respectively.
Fig_ 1C shows an I-V curve of ORR on CB-10% catalyst using the standard three-
electrode setup in a traditional flow-cell system with 1.0 M Na2SO4 (pH =7)
and 1.0
M KOH as the electrolyte (pH = 14) and Fig. 1D shows the corresponding FEs of
H202 under different potentials.
[011] FIG. 2 is a schematic design for an electrosynthesis cell for pure
H202.
[012] FIG. 3 is a schematic design for an electrosynthesis cell for CO2
reduction for the
production of a variety of liquid products.
[013] FIGS. 4A-4D show an (Fig. 4A) SEM image and (Fig. 4B) and BET surface
area
analysis of carbon black catalyst with different surface oxygen content. Fig.
4C shows
a representative SEM image of a spray coated CB-10% electrode with a roughly
70
pm thick catalyst layer. Fig. 4D shows an enlarged SEM image of the CB-10%
catalyst electrode demonstrating the high porosity of the catalyst layer on
the GDL to
provide for improved 02 diffusion and catalytic current density.
[014] FIGs. 5A-5B show high-resolution (Fig. 5A) C is and (Fig. 5B) 0 is
XPS spectra.
[015] FIGs. 6A-6C shows (Fig. 6A) XPS survey scans of carbon black
catalysts with
different surface oxygen contents, (Fig. 613) faradaic efficiencies, and (Fig.
6C)
curves of carbon black catalysts with different surface oxygen contents for 26-
ORR
using 02/./SE/M-fr0 cell configuration with solid proton conductor.
[016] FIGs. 7A-H show a comparison of the four types of TM-CNT samples,
including Fe,
Pd, Co, and Mn, which are demonstrated to have similar structures by
transmission
electron microscopy (TEM) (FIGs 7A-D) and aberration-corrected high-angle
annular dark-field scanning TEM (HAADF-STEM) (FIGs 7E-H).
4
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
[On] FIGs. 8A-8G show (Fig. 8A) SEM, (Fig. 8B) TEM and
(Fig. 8C) high-resolution
TEM views of the BOON nanosheet. Fig. 8D shows STEM-EDS elemental mapping
of BOON. Fig. 8E shows TEM and (Fig. 8F) high-resolution TEM images of in-situ
reduced metallic 2D-Bi. Fig. 8G shows in-situ Bi L3-edge XAS spectra of BOON
at
different potentials.
[018] FIGs. 9A-9C show SEM images of solid polymer proton conductors. Fig. 9A
shows
a zoomed out SEM view of sulfonated styrene-divinylbenzene copolymer proton
conductor with successive zoom in views Fig. 9B and Fig. 9C demonstrating a
uniform spherical morphology.
[019] FIG. 10 shows 2e--ORR performance of CB-10% in solid electrolyte for
a three-
electrode cell.
[020] FIGs. 11A-11D show (Fig. 11A) The 1-V curve of CB-10%//SE//Pt-C cell
with H+
conducting porous solid-electrolyte. Fig. 11B shows the corresponding FEs and
production rates of 14202 under different cell voltages. Fig. 11C shows the
dependences of 11202 concentration on the DI water flow-rate under an overall
current
density of 200 mA/cm2. Fig 11D shows the removal of TOC in Houston rainwater
using the generated pure H202 solution under a current density of 200 mA/cm2.
[021] FIGs. 12A-12B show stability tests of continuous generation of pure
H202 solutions
with concentrations over 1,000 and 10,000 ppm, respectively. No degradations
were
overserved in cell voltages and H202 concentrations over the 100-hour
continuous
operation. The cell currents and DI flow-rates are (Fig. 12A) 60 mA under and
27 mL
h-1 and (Fig. 12B) 120 mA and 5.4 inL h-1, respectively.
[022] FIGs. 13A-13B show 11202 productivity of the presently described
02//SEHH2 system
for (Fig. 13A) direct electrosynthesis and (Fig. 13B) direct synthesis
compared with
systems of the previous literature.
[023] FIGs. 14A-14B show online 112 detection during H202 production and
the XPS
analysis of post-stability catalyst. Fig. 14A shows gas chromatography
analysis of
cathode gas flow of CB-IOW/SEM:1-C cell during H202 production using 02 and
H2.
Fig. 14B shows XPS survey scans of CB-10% catalyst after stability test under
a
relatively high current density.
[024] FIGs. 15A-15D show pure H202 generation using 02 and 112 with polymer
anion
conductor and inorganic proton conductor. The current densities over cell
voltages of
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
CB-10%//SE/54-C cell with (Fig. 15A) an inorganic Cs.,143.xPW12040 proton
solid
conductor and (Fig. 1513) anion conducting solid-electrolyte. The
corresponding FEs
and concentration of H202 products under different cell voltages are shown for
(Fig.
15A) an inorganic Csitl3a12040 proton solid conductor and (Fig. 15D) anion
conducting solid-electrolyte. Note that the DI flow-rate is 27 mL/h.
[0251 FIGs. 16A-16B show H202 faradaic efficiencies (FE)s
as a function of DI water flow
rate for (Fig. 16A) 02//SE/M2 and (Fig. 16B) scaled-up unit cell, showing that
the
11202 selectivity was inhibited with increased 11202 concentration.
[026] FIG. 17 shows a long-term operation test of the direct electro-
synthesis of pure H202
solution using 02//SE//H20 cell, showing high selectivity and stability at 60
mA using
this proposed system. The FE of H202 is maintained constant (- 95%) over the
100-
hour continuous operation. The DI flow-rate is 27 mUh.
[027] FIGs. 18A-18B show (Fig. 18A) the I-V curve of an 02//SW/H20 cell
where H20 is
oxidized on the anode side into protons and 02. The 0.5 M H2SO4 in water
solution
was used for improving the ionic conductivity on the anode side, and was not
consumed during electrosynthesis. Fig. 18B shows the corresponding FEs of
H2//SW/H20 cell.
[028] FIGs. 19A-19D show (Fig. 19A) the I-V curve and FEs of Aid/SE/4120
cell for
generating pure H202 solutions. It demonstrated the generation of pure H202
solutions
at a high production rate of 2.3 mmol cm-2 h-1 (2490 mol kgcat-1 h-1) using
only air
and water as cathode and anode feedstock, respectively, when pure H2 and 02
are not
available. Fig. 19B shows the I-V curve of the scaled-up unit cell module (80
cm2
electrode, no iR-compensation), and (Fig. 19C) the corresponding 11202 FEs. It
confirms that the present approach can be scaled up with negligible sacrifice
in
performance. Fig. 19D shows the dependence of H202 concentration (up to - 20
wt.%) on the DI water flow-rate while maintaining an overall current of 8 A.
[029] FIGs. 20A-20E show (Fig. 20A) the current densities over cell
voltages on 2D-Bi
catalyst using the electrosynthesis cell for CO2 reduction with Fr and HC00-
conducting solid-electrolyte. The corresponding faradaic efficiencies for the
reduction products under different cell voltages using (Fig. 20B) 11+ and
(Fig. 20C)
HC00- conducting solid-electrolyte. Fig. 20D shows the dependencies of HCOOH
concentration on the DI flow-rate maintaining an overall current density of
100 mA/
6
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
cm2, indicating that concentrated pure HCOOH solution (up to 6.73 M) can be
continuously produced. Fig. 20E shows the production of electrolyte-free C2+
liquid
fuel solutions using commercial Cu2O catalyst, showing that small molecular
oxygenates liquid fuels can also be efficiently collected.
[030] FIG. 21 shows the long-term operation test of CO2 reduction to pure
HCOOH solution
demonstrating the high selectivity and the stability of the 2D-Bi catalyst at
30 mA/cm2
using this proposed CO2 reduction system. The FE of HCOOH maintains more than
80% over the 100-hour continuous operation.
[031] FIGs. 22A-B displays the current-voltage profile (Fig. 22B) of the
direct
electrocatalytic CO2 hydrogenation cell (Fig. 22A) for HCOOH vapor generation.
[032] FIGs. 23A-E show the ORR performance of M-CNT catalysts cast RRDE in
0.1M
KOH. Fig. 23A shows linear sweep voltanunetry measurements; Fig. 23B shows the
calculated H202 selectivity and electron transfer number during potential
sweep; Fig.
23C shows a stability measurement of Fe-CNT; and Figs 23D-E show a comparison
of LSV and corresponding 14202 selectivity.
[033] FIGs. 24A-B show the effects of Fe atom loading at respective amounts
of 0, 0.05,
0.1, and 0.2 at% on H202 activity and selectivity
[034] FIGs. 25A-B show the bulk electrolysis for H202 generation in a
homemade H-cell
electrolyzer. Fig. 25A shows an SE1VI image of GDL supported catalyst at a
loading
of 0.5 mg cm-2, and Fig. 25B shows a polarization curve of Fe-CNT/GDL catalyst
in
1 M KOH electrolyte
[035] FIGs. 26A-B show the (Fig. 26A) XANES comparison of before, during,
and after 2-
h's continuous ORR electrolysis at 0.55 V vs. RUE and (Fig. 2611) EXAFS of
post-
catalysis Fe-CNT with Fe metal and Fe304 as references.
[036] FIGs. 27A-D show the disinfection performance of Fe-CNT in neutral
pH. Fig. 27A-
B is an LSV of Fe-CNT catalyst on RRDE with corresponding selectivity under
different potentials; Fig 27C shows LSV of Fe-CNT catalyst on GDL electrode in
an
H-cell electrolyzer; Fig 27D shows bulk electrolysis at a constant current
density in
PBS containing E. coli bacteria.
[037] FIG. 28 shows the The disinfection efficiency as a function of
treatment time.
[038] FIGs. 29A-C show the production of pure HCOOH solution and vapor using
optimized CO2 reduction system with solid electrolyte. Fig. 29A shows a
Schematic
7
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
illustration of the proposed four-chamber CO2 reduction cell with solid
electrolyte.
Fig. 29B shows the current densities over cell voltages and the corresponding
HCOOH FEs. Fig. 29C shows the the concentration of pure KOH which is
simultaneously produced using the four-chamber solid cell during CO2
reduction.
[039] FIGs. 30A-C show ORR performance of the catalysts cast RRDE in: Figs.
30A and
30C :0.1 M KOH; Figs, 30B and 30D: 0.1M Na2SO4. Figs. 30A and 30B show linear
sweep voltanunetry (LSV) measurements of H2-annealed carbon black (denoted as
'Pure C') and boron, nitrogen, phosphorous, sulfur-doped carbon (denoted as `B-
C',
'N-C', `P-C', `S-C', respectively). Figs. 30C and 30D show calculated H202
molar
selectivity (left y-axis) and faradaic efficiency (right y-axis) during the
potential
sweep for different catalysts in 0.1M KOH and 0.1M Na2SO4, respectively.
[040] FIGs. 31A-D show the three-electrode flow cell performance of
catalysts. Figs. 31A
and 31B show I-V curves for Pure C, B-C and 0-C in1M KOH and 1M Na2SO4,
respectively.
[041] FIGs. 32A-B show solid-electrolyte cell performance for pure 11202
generation. Fig.
32A shows I-V curve and corresponding 11202 faradaic efficiency, and Fig. 32B
shows H202 partial currents and H202 production rates under different applied
potentials. Fig. 32C shows stability test of B-C fixed at 50tnA cm-2 of
generation of
-1,100 ppm pure H202 solution. The DI water feeding rate is fixed at 54 mL
R0421 FIG. 33 shows a schematic of a H202 production
reactor with two cation exchange
membranes on each of the cathode and anode side.
[043] FIG. 34 shows the stability of Ni-N-C single atom catalyst in the all
CEM solid
reactor.
[044] FIG. 35 shows the electrochemical 26-ORR performance of B-doped carbon.
DETAILED DESCRIPTION OF THE INVENTION
[045] Specific embodiments will now be described in detail with reference
to the
accompanying figures. Like elements in the various figures are denoted by like
reference numerals for consistency.
[046] In the following detailed description of embodiments, numerous
specific details are
set forth in order to provide a more thorough understanding.
8
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
[047] However, it will be apparent to one of ordinary skill in the art that
embodiments may
be practiced without these specific details. In other instances, well-known
features
have not been described in detail to avoid unnecessarily complicating the
description.
[048] In the following description, any component described with regard to
a figure, in
various embodiments of the present disclosure, may be equivalent to one or
more
like-named components described with regard to any other figure.
[049] Throughout the application, ordinal numbers (e.g., first, second,
third, etc.) may be
used as an adjective for an element (i.e., any noun in the application). The
use of
ordinal numbers is not to imply or create any particular ordering of the
elements nor
to limit any element to being only a single element unless expressly
disclosed, such
as by the use of the terms "before," "after," "single," and other such
terminology.
Rather, the use of ordinal numbers is to distinguish between the elements. By
way of
an example, a first element is distinct from a second element, and the first
element
may encompass more than one element and succeed (or precede) the second
element
in an ordering of elements, if an ordering exists.
[050] One or more embodiments of the present disclosure relate to methods
and systems
for the production of high purity concentrated liquid products through
electrocatalytk
reactions.
[051] One or more embodiments of the present disclosure relate to the
production of H202.
In yet another embodiment, the described electrosynthesis cell may be used in
the
production of highly pure formic acid and/ or other liquid fuels through the
elctrocatalytic reduction of CO2 or CO with solid electrolytes. Beyond the
continuous
production of pure H202, other pure liquid products including methanol,
ethanol, n-
propane', formic acid, acetic acid and other organic oxygenates from CO2
reduction
reactions (CO2RR) or CO reductions (CORR) can be realized utilizing the
general
process and electrosynthesis cell described in one or more embodiments of the
present
disclosure.
[052] In accordance with one or more embodiments of the present disclosure,
the produced
liquid product may be produced through superior selectivity of employed
catalyst to
achieve the continuous production of high purity concentrated liquid products
that do
not require any additional separation steps to achieve pure product solutions.
9
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
[053] One or more embodiments of the present disclosure may be directed
towards
processes for the highly efficient and large-scale synthesis of commercial-
level
concentrated H202 via a cost-effective electrocatalytic oxygen reduction route
(ORR).
[054] One or more embodiments of the present disclosure may relate to
systems that may
include a three-compartment electrosynthesis cell for direct pure liquid
product
production without any additional energy-intensive purification steps.
[055] One or more embodiments of the present disclosure may relate to systems
and
methods that may include a four-compartment electrosynthesis cell for the
simultaneously production of up to three kinds of high-purity products, in
which an
alkaline/neutral solution can be used for OER anode catalyst for direct pure
liquid
product production without any additional energy-intensive purification steps.
In one
or more embodiments, systems and methods that may include a four-compartment
electrosynthesis cell, where the solid electrolyte may be split and separated
by bipolar
membrane. To separate the anode and cathode reaction. In such embodiments,
noble
metal catalysts of the anode may be excluded and/or replaced. For example, in
one or
more embodiments, a nickel iron layered double hydroxide (NiFe-LDH) and KOH
may be chosen as the OER catalyst and electrolyte to decrease the catalyst
cost and
anode over potential.
[056] One or more embodiments of the present disclosure are provided to
introduce a highly
efficient process and electrolytic cell capable of achieving high current
efficiency for
the direct and continuous production of pure (-20wt%) hydrogen peroxide (H202)
via
electrocatalytic synthesis.
[057] One or more embodiments of the present disclosure is directed to
processes
(electrosynthesis cell with highly active electrocatalyst) to achieve highly
pure and
concentrated liquid products from electrocatalytic reactions, e.g. 20wt% pure
H202
solution from ORR. Notably, unlike many traditional pure liquid product
synthetic
systems and processes, such as an 1202 synthetic system, the present
disclosure
subverts the need for an energy-intensive purification step, as the immediate
product
of the governing system is an already pure form of 1202 solutions.
[058] One or more embodiments of the present disclosure may include a three-
compartment
electrolytic cell including a (cathode), a catalyst, such as 1r02/C for water
oxidation
or Pt/C for H2 oxidation (anode) and a solid electrolyte. More particularly,
one or
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
more embodiments herein relate to a process for the on-site production of
highly pure
hydrogen peroxide via electrocatalytic oxygen reduction reaction (ORR, 02+ H20
+
2e- ¨> H02- + OW), which can be used for bleaching, medical uses, food
cleaning and
processing, and other applications, together with oxygen at the counter side
by water
oxidation (OER, 2H20 ¨> 02 + 4H+ +
In accordance with one or more
embodiments, a three-compartment porous solid electrolyte electrolytic cell
with
solid electrolyte provided between a cathode and an anode is disclosed for
carrying
out this process.
[059] In one or more embodiments of the present disclosure, the cathode and
anode of the
proposed cell may be catalyst coated gas diffusion layer (GDL) electrodes,
which are
separated by an anion and cation exchange membrane, respectively.
Electrocatalytic
reduction of oxygen (ORR) at the cathode and water oxidation at the anode may
be
used to generate anions (such as HOf ) and cations (such as Ir) respectively,
which
when driven through the appropriate ion exchange membranes ionically recombine
to form pure 11202. 02 generated at the anode can be driven back to the
cathode to
further undergo reduction, thereby contributing to the overall efficiency of
the
presented H202 synthetic system.
[060] In accordance with one or embodiments described, the processes for the
electrosynthesis of highly pure and concentrated liquid products may utilize a
three-
compartment electrolsynthesis cell, i.e. a cell partitioned into an anode
compartment,
an intermediate solid electrolyte compartment, and a cathode compartment
wherein
the cells are partitioned by cation or anion ion-exchange membranes, to
produce liquid
products.
[061] As schematically illustrated in Fig. 1B, the cathode and anode of the
proposed cell
may be catalyst coated GDL electrodes, which were separated by anion and
cation
exchange membranes, respectively.
[062] In one or more emboidments, porous solid ion conductors, e.g. Fr or
H02- conductors,
may be filled in between the membranes or electrodes with close contact. In
accordance with one or more embodiments, a PSMIM anion exchange membrane and
a Nation membrane may be used for anion and cation exchange, respectively.
Other
anion and cation exchange membranes may be used, alternatively. Two Nation
membranes may be used, alternatively. The solid electrolyte, as denoted in
Fig. 1B,
11
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
may be made of ion-conducting polymers with different functional groups, such
as
porous styrene-divinylbenzene copolymer consisting of sulfonic acid functional
groups for cation conduction, or quaternary amino functional groups for anion
conduction. Other forms of solid electrolyte used in batteries, such as
ceramics,
polymer/ceramic hybrids, or solidified gel electrolytes (e.g. 10 wt%
H3PO4/polyviny1pyrrolidone gel), may also be employed. The cathode electrode
where 02 is reduced can be supplied with humidified 02 gas to facilitate 02
mass
transport, whereas the anode side can be circulated with an acid solution,
such as 0.5
M H2SO4, for water oxidation using commercially-available Ir02/C catalyst. The
anode side can also oxidize H2 to generate fr, in one or more embodiments.
[063] In the electrosynthesis cell of one or more embodiments of the
present disclosure,
oxygen gas (or an oxygen-containing gas such as air) may be supplied to the
cathode,
while hydrogen gas or water is supplied to the anode. These gases may be
externally
fed. Alternatively, the two gases produced by water electrolysis can be
rerouted and
directly fed to the electrolytic cell.
[064] CATHODE
[065] As schematically illustrated in Fig. 1B, the cathode of the
electrosynthesis cell may
be catalyst coated gas diffusion layer (GDL) electrodes. The cathode electrode
may
be included in the cathode compartment of the three-compartment
electrosynthesis
cell. The cathode electrode may include a gas diffusion layer that may be
loaded with
a selective reduction reaction electrocatalyst for specific reduction
reactions. The
specific reduction reactions may include one or more of, oxygen reduction
reactions,
CO2 reduction reactions, CO reduction reactions, N2 reduction reactions,
nitrate
reduction reactions and nitrite reduction reactions, or combinations thereof.
[066] In one or more embodiments, the cathode may be selected from an oxygen-
reducing
electrode that includes a gas diffusion layer coated in a product selective
electrocatalyst such as oxidized carbon material including carbon black,
graphene,
carbon nanotubes, or a mixture thereof. In one or more embodiments, the
product
selective electrocatalyst such as carbon material including carbon black,
grapheme,
carbon nanotubes, or a mixture thereof, where the carbon material may include
a non-
metal dopant anchored on the carbon substrate. Non-metal dopants may include
boron, nitrogen, phosphorous, sulfur, or a combination thereof. In another
12
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
embodiment, examples of other electrocatalyst for coating a gas diffusion
layer may
includ N-, P-, S-, B-, Si-, or metal-doped carbon materials, or Bi, Cu, Ni,
Fe, Co, Pd,
In, Pb, Tn, transition metals, single atom catalysts of transition metals
anchored into
carbon nanotubes (CNT), oxides, chalcogenides thereof, or a mixture thereof.
[067] In one or more embodiments, the cathode maybe comprised of a gas
diffusion layer
coated in a carbon black electrocatalyst that may be optionally oxidized. For
example,
in one or more embodiments, the carbon black may be pretreated before coating
the
GDL during the preparation of the cathode electrode. Carbon black may be acid
treated to realize and optimize surface ether and carboxyl functionalization
to
improve selectivity towards the desired 2C-ORR pathway.
[068] In one or more embodiments, the cathode of the electrosynthesis cell
may be catalyst
coated gas diffusion layer (GDL) electrodes where the catalyst may be loaded
on the
GDL electrode in an amount ranging from 0.01 mg/cm2 to 20 mg/cm2. In one or
more
embodiments, the cathode of the electrosynthesis cell may be catalyst coated
gas
diffusion layer (GDL) electrodes where the catalyst may be loaded on the GDL
electrode in an amount ranging from 0.01, 0.1, 0.3, 0.5, 1, 3, 5, 7, and 9
mg/cm2 to
412, 03, 0_4, 0.6, 1, 2, 5, 8, 10, 15, and 20 mg/cm2, where any lower limit
may be
combined with any mathematically feasible upper limit
[069] In one or more embodiments, the specific electrocatalyst for H202
production may be
a low-cost oxidized carbon black, which may be directly synthesized and
treated by
oxidation of commercial carbon black (such as Vulcan XR-72R) in acid solution.
In
one or more embodiments, carbon black may be oxidized by mixing and refluxing
the carbon black in a concentrated acid solution for an amount of time ranging
from
0.5, 1, 2, 3, 4, 6, 8, 10, 12, 16, 20, 24, 30, 36 and 40 hours (his) to 2, 3,
5, 8, 10, 12,
16, 20, 24, 30, 36, 40, and 48 his, where any lower limit may be combined with
any
mathematically feasible upper limit. For example, in one or more embodiments
commercial carbon black may be oxidized in a solution of 12 M HNO3 for 3 hrs.
[070] In one or more embodiments the treated and oxidized carbon black
cathode catalyst
may have a surface oxygen content ranging from 0.1, 1, 2, 3, 5, 7, 10, 15, 20,
and
25% to 2, 3, 7, 8, 11, 13, 15, 18, 20, 25, and 30%, wherein any lower limit
may be
combined with any mathematically feasible upper limit.
13
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
[071] In one or more embodiments, the cathode may be selected from an oxygen-
reducing
electrode comprised of a gas diffusion layer coated in a product selective
electrocatalyst such as transition metal (TM) single atoms including Fe, Co,
Ni, Cu,
Zen, Pt, Pd, Ii, Mn, Cr that may be optionally anchored into carbon nanotube
(TM-
CNT) vacancies. For Example, the cathode may be selected from an oxygen-
reducing
electrode comprised of a gas diffusion layer coated in a product selective
electrocatalyst such as Fe-CNT, Pd-CNT, Co-CNT, and Mn-CNT, or combinations
thereof.
[072] In one or more embodiments, the cathode may be selected from an oxygen-
reducing
electrode comprised of a gas diffusion layer coated in a product selective
electrocatalyst such as transition metal (TM) single atoms and non-metal
dopants that
include B, N, 0, F, S, P, Si, Cl, etc. For example, Fe-C-0 single atom
catalyst is
shown herein to demonstrate an excellent H202 Faradaic efficiency in both
alkaline
and neutral pH (Figure 4), which can be directly used in our solid electrolyte
cell for
pure 11202 solutions.
[073] In one or more embodiments, the single atom TM may anchored into the CNT
in an
amount ranging from 0.01 to 5 at%. In one or more embodiments the TM may
anchored into the CNT in an amount ranging from 0.01, 0-05,0.1, 0.15, 0.2,
0.3, 0.5,
0.8, 1, 1.5,2, 3, and 4 at% to 0.1,0.15. 0.18, 0.2 0.25, 0.3, 0.5, 0.8, 1,
1.5, 2, 3, 4, and
at%, wherein any lower limit may be combined with any mathematically feasible
upper limit. For example, TM single atoms catalysts of one or more embodiments
of
the present disclosure may be prepared from metal cations (-0.1 at%) that may
be
first dispersed onto commercial surface-functionalized CNTs as the carbon
matrix,
and suspended in water. They may then be further treated through steps of
freeze
drying and thermos annealing under inert gas at about 500 to 1000 'C.
[074] In one or more embodiments the product selective catalyst may be an
ultrathin two-
dimensional Bismuth (2D-Bi) catalyst for CO2-to-HCOOH conversion. In one or
more embodiments the product selective catalyst may be an ultrathin two-
dimensional Bi (2D-Bi) catalyst where at least 50% of the Bi sites of the 2D-
Bi were
electrochemically active using cyclic voltammetry. This high percentage may
ensure
high Bi atom efficiency during CO2RR catalysis.
14
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
[075] In one or more embodiments, the electrosynthesis cell including a
cathode of a gas
diffusion layer coated in a product selective electrocatalyst may generate a
liquid
product with a Faradaic selectivity ranging from 10% to 99.9%. In one or more
embodiments, the gas diffusion layer coated in a product selective
electrocatalyst may
generate a liquid product with a Faradaic selectivity ranging from 10, 20, 30,
40, 50,
60, 70, 80, 90 95, and 97% to 50, 60,70, 80, 85, 90, 93, 95, 97, and 99.9%,
wherein
any lower limit may be combined with any mathematically feasible upper limit.
[076] In one or more embodiments the electrosynthesis cell including a
cathode of a gas
diffusion layer coated in a product selective electrocatalyst may deliver a
final liquid
product with a tunable FE that ranges from 10% to 99.9%. In one or more
embodiments the electrosynthesis cell including a cathode of a gas diffusion
layer
coated in a product selective electrocatalyst may deliver a liquid product
with a FE
that ranges from 30, 40, 50, 60,70, 80, 9095, and 97% to 50, 60,70, 80, 85,
90, 93,
95, 97, and 99%, wherein any lower limit may be combined with any
mathematically
feasible upper limit. In one or more embodiments, the FE may be tunes by
controlling
the current density.
[077] In one or more embodiments, the cathode electrode may have an
electrode area that
ranges from 1 cm2 to 10 m2 per unit cell, which can be scaled up by stacking
multiple
cells.
[0781 ANODE
[079] In one or more embodiments, the anode for use in the present
disclosure may be
selected from a gas diffusion electrode, hydrogen-oxidizing electrode, or a
catalyst
coated gas diffusion electrode, according to the electrolysis conditions. In
one or more
embodiments, examples of anode electrocatalyst for coating a gas diffusion
layer
include metal-doped carbon materials, or Ru, It, Pt, Ni, Ce, among other
transition
metals, single atom catalysts, an oxide or a chalcogenide thereof.
[080] For example, the oxygen-generating electrode may be a gas diffusion
layer coated
with a catalyst electrode material consisting mainly of a metal such as
platinum,
iridium, or ruthenium, an oxide of such a metal, or an oxide metal carbon
compound
as a catalyst and is used as such. In other embodiments, the oxygen-generating
electrode may be a gas diffusion layer coated with a catalyst electrode
material
consisting of a nickel iron layered double hydroxide (NiFe-LDH).
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
[081] In one or more embodiments, the anode of the electrosynthesis cell
may be catalyst
coated gas diffusion layer (GDL) electrodes where the catalyst may be loaded
on the
GDL electrode in an amount ranging from 0.01 mg/cm2 to 10 mg/cm2. In one or
more
embodiments, the cathode of the electrosynthesis cell may be catalyst coated
gas
diffusion layer (GDL) electrodes where the catalyst may be loaded on the GDL
electrode in an amount ranging from 0.1, 0.2, 0.3, 0.35, 0.4, 0.5, 0.8, 1,
1.5, 3, 5, and
7 mg/cm2 to 0.2, 0.3, 0.4, 0.5, 0.6, 0.8, 1, 1.5, 3, 5, 8, and 10 mg/cm2,
where any lower
limit may be combined with any mathematically feasible upper limit.
[082] In one or more embodiments, the anode electrode may have an electrode
area that
ranges from 1 cm2 to 10 m2 per unit cell, which can be scaled up by stacking
multiple
cells.
[083] ION EXCHANGE MEMBRANES
[084] In one or more embodiments, the ion exchange membrane may be a cation
and/or
anion exchange membrane. The cation and/or anion exchange membranes of the
present disclosure may not be particularly limited. In one or more
embodiments, the
cation exchange membrane may be a perfluorosulfonic acid (PFSA) membrane and
the anion exchange membrane may be a membrane comprising a co-polymer of
polystyrene cross linked with divinylbenzene and polystyrene methyl
ifnidazolium
chloride (PSM1M). Other AEMs are also feasible, such as polybenzimidazole
membrane (PBI), benzyltrimethylammonium grafted PTFE membrane, vinyl-benzyl
chloride grafted fully fluorinated poly(tetrafluoroethylene-co-
hexafluoropropylene)
membrane and chloromethylated polysulfones membrane.
[085] In one or more embodiments, the cation and anion exchange membranes may
be
interchangeable or selectively used in multiple configurations at either the
cathode
side or the anode side of the electrosynthesis cell.
[086] SOLID ELECTROLYTE
[087] In one or more embodiments, the solid electrolyte material disposed
between the
cathode and anode may include ion-exchange resins and matrixes comprising an
ion-
conducting material.
[088] In one or more embodiments, the porous solid electrolyte may be
selected from a
group of ion conducting polymers including polymers or copolymers of styrene,
acrylic acid, aromatic polymers, or a combination thereof.
16
CA 03150909 2022-3-10
WO 2021/011675
PCT/U52020/042163
[089] In one or more embodiments, the porous solid electrolyte may be
selected from an
inorganic ceramic solid electrolyte, a polymer/ceramic hybrid solid
electrolyte,
solidified gel electrolytes, or ion conducting polymers, or a combination
thereof.
[090] In one or more embodiments, the porous solid electrolyte is a porous
styrene
divinylbenzene copolymer consisting of sulfonic acid functional groups for
cation
conduction, or quaternary amino functional groups for anion conduction.
[091] In one or more embodiments, the solid electrolyte resins may include
hydrocarbon
resins such as styrene polymers, acrylic acid polymers and aromatic polymers.
The
sulfonated inorganic materials, like sulfonated carbon, SiO2, TiO2, W03, Ce02,
TIC,
MoC et al., may also be used as solid electrolyte. The solid proton conductor
may be
prepared by refluxing porous (pore size ranges from 2 nm to 100 nm) or solid
polymer
or inorganic matrix in fuming acid, such as H2S0.4 for about 24-h at an
elevated
temperature of about 80 C.
[092] In one or more embodiments of the present disclosure, the solid
electrolyte comprised
ion-conducting polymers with different functional groups, such as porous
styrene-
divinylbenzene copolymer consisting of sulfonic acid functional groups for IV
conduction, or quaternary amino functional groups for anion conduction. The
solid
electrolyte is not limited and may be an anion polymer conductor for anion or
an
inorganic solid cation conductor for pure generation comprised of Csx1-
13,13W1204o.
The porous styrene-divinylbenzene copolymer may be one of styrene-
divinylbenzene
sulfonated copolymer such as SSE 50 or SSE 300. One or more embodiments may
comprise other forms of solid electrolytes, such as ceramics, polymer/ceramic
hybrids, or solidified gel electrolytes (e.g. 10 wt%
H3PO4/polyvinylpyrrolidone gel).
[093] GAS DIFFUSION LAYER ELECTRODE
[094] In one or more embodiments, the gas diffusion layers of the present
disclosure are not
particularly limited. In one or more embodiments, the gas diffusion layer may
be a
thin carbon-based porous medium that must provide high electrical and thermal
conductivity and chemical and corrosion resistance, in addition to controlling
the
proper flow of reactant gases (hydrogen and air) to ensure uniform
distribution of
reactive gases on the surface of the electrodes. In one or more embodiments,
the gas
diffusion layers may be coated in a catalyst to form either the cathode or
anode of the
electrosynthesis cell.
17
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
[095] GAS FEED
[096] In one or more embodiments, the hydrogen gas and oxygen gas may be
supplied, or
hydrogen and oxygen gases may be generated by water electrolysis and may be
directly supplied to the electrolytic cell.
[097] In one or more embodiments the cathode side may be supplied with a
controlled and
tunable amount of 02, CO2, CO, N2, air, or other reactants and the anode side
may be
supplied with enough of H2, 1120, alkaline solutions, acidic solutions, or
other
reactants.
[098] In one or more embodiments the cathode side may be supplied with a
controlled and
tunable amount of gas at a flow rate ranging from 0.001 to 1000 SLM. In one or
more
embodiments, the gas flow rate may change depending upon the device capacity.
[099] PURE WATER FEED
[0100] In one or more embodiments, pure water may be fed to the solid
electrolyte
compartment at a suitable rate depending on the size of reactor and the need
of
product concentration. In one or more embodiments, the water flow rate in a
unit cell
(one cathode and one anode) may range from 1 uL/h to 10 m3/h. In one or more
embodiments, the specific water flow rate may be tuned relative to the target
product
fluid and its concentration.
[0101] LIQUID PRODUCTS FORMED
[0102] The process and electrosynthesis cell according to one or more
embodiments may be
used to obtain pure liquid products such as H202, methanol, ethanol, n-
propanol,
formic acid, acetic acid, other organic alcohols and acids, or ammonia from
CO2
reduction reactions (CO2RR), CO reduction reactions, N2 reduction reactions,
nitrate
or nitrite reductions, and so on.
[0103] In one or more embodiments, the electrosynthesis cell may capable of
generating a
concentrated liquid product. For example, in one or more embodiments the
electrosynthesis cell may generate a liquid product, such as H202, with a
concentration ranging from 0.01 to 20 wt.%. in one or more embodiments the
electrosynthesis cell may generate a liquid product, such as H2Oz with a
concentration ranging from 0.01, 0.1, 1, 3, 5, 8, 10, 14, 16, and 18 wt.% to
8, 10, 12,
14, 16, 18 and 20 wt.%, wherein any lower limit may be combined with any
mathematically feasible upper limit.
18
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
[0104] In one or more embodiments, the electrosynthesis cell may be tuned to
selectively
operate at a current density ranging from 1 mA/cm2 to 100 A/cm2.
[0105] In one or more embodiments, the electrolysis conditions of the
electrosynthesis cell
may include operating at a liquid temperature ranging from 1 to 95 C.
[0106] ELECTROSYNTHESIS CELL
[0107] In one or embodiments wherein a pure product, such as hydrogen peroxide
(11202),
may be obtained, 02 may be reduced by the H202-selective catalyst, and the
generated
negatively charged H02- may then be driven by the electrical field to travel
through
the AEM towards the middle solid electrolyte channel. At the same time,
protons
generated by water oxidation or hydrogen oxidation on the anode side may move
across the CEM to compensate the charge. Depending on the type of ion-
conducting
polymers in between, pure H202 product can be formed via the ionic
recombination
of crossed ions either at the left (Fr-conducting polymer) or right (H02-
conducting
polymer) interface between the middle channel and membrane. Then, the formed
liquid products may be quickly released by the slow deionized water (DI)
stream or
humidified inert gas flow.
[0108] Pure liquid product solutions with a wide range of concentrations may
be produced
by adjusting the flow rate of the DI and gas as demonstrated in the examples
below.
In one or more embodiments, the DI flow rate may be at least 1 uUhr and may be
dependent on the size and/or capacity of the device.
[0109] In one or more embodiments the cathode electrode, where 02 is reduced,
may be
supplied with humidified 02 gas to facilitate 02 mass transport, whereas the
anode
side may be circulated with a solution such as 0.5 M 142SO4 for water
oxidation using
commercial-available Ir02/C catalyst, or H2 gas using commercial-available PUC
catalyst.
[0110] As illustrated in Fig. 1B, H2 and 02 streams are separated into anode
and cathode,
respectively, avoiding their mixture as in the case of direct synthesis (Fig.
1A).
[0111] On the anode side, 112 can be electrochemically oxidized on a HOR
catalyst, which
may be coated on a gas diffusion layer electrode, into Ir; on the cathode
side, by
designing a 2e--ORR selective catalyst, 02 can be selectively reduced through
the 2e-
pathway into H02- (Eq.. 1), instead of OH- as in traditional H2/02 fuel cells.
Both HOR
and 26-ORR catalysts are in close contact with cation and anion exchange
19
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
membranes (CEM and AEM), respectively, to avoid flooding issues from the
direct
contact with liquid water.
[0112] As shown in Fig. 2, the electrochemically generated anions (H02-) and
cations (Fr)
then move across the corresponding membranes into a thin layer of porous solid
electrolyte, which plays a key role in both ion conduction and pure product
collection.
First, ions can be efficiently conducted through the solid electrolyte with
small ohmic
losses for high cell efficiencies, particularly under large current densities.
Second,
11202 molecules can be formed via the ionic recombination of crossed 1102- and
H+
ions in the solid electrolyte layer, which were dissolved in the DI water
stream and
quickly released as pure H202 solutions with no other impurity ions involved.
By
tuning the H02- generation rate or the DI water flow rate, a wide range of
H202
concentrations (from hundreds of ppm to tens of percentage) can be directly
obtained
for different purposes of use. No further energy-consuming downstream
purifications
are needed in this case, dramatically differentiating the present electro
synthesis
design and process from traditional anthraquinone process or direct synthesis
methods.
[0113] The porous solid electrolyte may be either anion or cation solid
conductor, which can
be made of ion conducting polymers with different functional groups, inorganic
compounds, or other types of solid electrolyte materials such as ceramics,
polymer/ceramic hybrids or solidified gels.
[0114] With different solid electrolyte properties, the electrosynthesis cell
and process can
be further extended to other electrocatalytic synthesis of pure products
beyond H202,
such as CO2 reduction, N2 reduction and so on. For example, Fig_ 3 illustrates
an
electrosynthesis cell for the reduction of CO2 wherein the anode is coated
with a stable
and active HOR or oxygen evolution reaction catalyst (0ER, in acidic
solutions),
which helps release Ir from water to compensate for negative charges of
generated
formic acid ions.
[0115] The electrosynthesis cell and process in accordance with one or more
embodiments
of the present disclosure may be able to achieve high H202 selectivity of 95%,
productivity (at 180 mA/cm2 partial current or 3660 mol/kg cat h), and a
liquid
product concentration of 20 wt.%.
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
[0116] Additionally, a 100-hour continuous and stable generation of - 1.1 wt.%
(- 11,000
ppm) pure H202 solution is demonstrated herein. It is also shown that similar
H202
activity and selectivity can be obtained while using air and water for 2c-ORR
and
oxygen evolution reaction (0ER), respectively, making on-site applications
more
accessible compared to pure H2 and 02. To demonstrate potential applications,
the
total organic carbon (TOC) in Houston rainwater was successfully treated with
a
processing rate up to 2180 L m-2electrode h-1 to meet Texas drinking water
standards, as demonstrated below.
[0117] To deliver efficient energy conversions, electrocatalysts with high
activity and
selectivity for 26-ORR and HORJOER are a prerequisite. It is straightforward
to
employ the state-of-the-art platinum on carbon (Pt/C) catalyst for HOR at the
anode
side with high H2-to-H conversion rates and small over-potentials. On the
other side,
however, electrocatalysts with both high activity and selectivity for 2c-ORR
towards
H202 are much less explored compared to the extensively studied 4c-ORR to H20
in
fuel cell catalysis.
[0118] SELECTIVE ELECTROCATALYSTS FOR 11202
[0119] Commercial carbon black is demonstrated herein as the starting material
due to the
following detailed and demonstrated reasons. First, it is significantly
cheaper than
graphene and/or noble metals, which makes it particularly suitable for large-
scale
applications. Second, it has a high surface area (Figs. 4A-4D) for high mass
activities;
and third, it is different from graphene nanosheets where their layer-by-layer
stacking
can block gas diffusions. The nanoparticulate morphology of carbon black
allows for
effective 02 diffusions from GDL to the surface layer of catalyst (Figs. 4C-
4D). This
ensures efficient operations particularly under large current densities. For
carbon
materials, surface functional groups such as ether (C-O-C) and carboxyl (HO-
C=0)
have been identified to possibly activate the adjacent carbon atomic sites for
selective
2c-ORR. Hence, carbon black nanoparticles may be treated in nitric acid to
realize
surface ether and carboxyl functionalization.
[0120] Example 1-Preparation and treatment of Carbon Black Catalysts
[0121] To demonstrate, three example compositions of carbon black were
prepared by
adding 600 mg of commercial carbon black (XC-72, FuelCellStore) into 600 iriL
of
12.0 M nitric acid. Then, the above solution was refluxed at 85 C for 1, 3
and 12 h,
21
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
respectively, to obtain oxidized carbon black with surface oxygen content of
7.33%,
10.19% and 11.62%, respectively. After natural cooling, the slurry was taken
out,
centrifuged and washed with water and ethanol until the pH was neutral.
Finally, the
sample was dried at 70 C in a vacuum oven. The as-received commercial carbon
black shows a 2.33% surface oxygen content. Otherwise, a comparative 500 mg
sample of commercial carbon black was annealed in a tube furnace at a
temperature
of 500 C for 2 h under a mixed hydrogen (5%)/argon atmosphere to obtain the
surface oxygen-free carbon black. Following the preparation of the
functionalized
carbon black examples, appropriate characterization was conducted as detailed
below.
[0122] No morphological evolution was observed for those carbon black
nanoparticles after
acid treatment (Fig. 4B). The high-resolution X-ray photoelectron spectroscopy
(XPS) spectra of treated carbon black (Figs. 5A-5B) confirmed that the acid
treatment
enriched oxygen-containing functional groups, including C-0-C/C-OH and HO-C=0
as de-convolved from carbon and oxygen is regions. The carbon is spectrum,
shown
in Fig. 5A, of the CB-10% catalyst can be de-convoluted into five
contributions that
are sp2 carbon at 284.6 eV, sp3 carbon at 285.5 eV, C-0 at 286.8 eV, -COOH at
288.9 eV and the characteristic shakeup line of carbon in aromatic compounds
at
291.2 eV ot-rr* transition). The 0 is peaks, shown in Fig. 5B, could be de-
convoluted
into three peaks. The components centered at 531.7 and 532.9 eV were
attributed to
the C-OH/C-O-C and C=0 surface functional groups, respectively. The last
component with B.E. around 535.5 eV was characteristic of adsorbed water.
These
results indicate that the acid treatment induced surface oxygen
functionalization of
carbon black.
[0123] Surface characterization was further conducted to tune the surface
oxygen on carbon
black for optimized ORR performance. Carbon black with different surface
oxygen
contents and Ir02-C was used as cathode and anode catalyst, respectively. The
cathode side was supplied with 50 sccm of humidified 02 gas. The anode was
circulated with 0.5 M H2804 for water oxidation. The surface oxygen strongly
correlates to the 11202 selectivity and activity (Fig. 6A). The maximal 11202
selectivity
of carbon black quickly ramped up to - 98% with relatively low surface oxygen
coverage (2.11%), whereas that of oxygen-free carbon black was only - 80%
(Fig.
22
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
6B). While the 11202 selectivity was not obviously enhanced by further
increasing the
surface oxygen from 2.11 to 11.62%, it was found that the ORR catalytic
activity was
gradually improved (Fig. (C). This improvement can be ascribed to the
increased
concentration of active sites for 2c-ORR. After optimization, carbon black
with ca.
10% surface oxygen coverage (CB-10%) was selected as the cathode catalyst for
efficient 2C-ORR.
[0124] A standard three-electrode setup was used to evaluate the intrinsic
activity of CB-
10%. 11202 can be reliably detected at 0.56 and 0.82 V vs. reversible hydrogen
electrode (RHE) in 1.0 M Na2SO4 and 1.0 M KOH electrolyte, respectively (Fig.
1C).
With a wide potential window to deliver high H202 selectivity (>90%) in both
neutral
and alkaline solutions, the catalyst reached a maximal faradaic efficiencies
(FEs) of
98 and 99%, respectively (Fig. 1D). More importantly, impressive H202 partial
currents of 410 and 300 mA/cm2 were achieve while high FEs were still
maintained
in alkaline and neutral solutions, respectively, which is among the highest 02
-to-
11202 conversion rates achieved so far.
[0125] Example 2- Preparation and Analysis of single atom TM-CNT
[0126] In one or more embodiments, as indicated, the cathode may be selected
from an
oxygen-reducing electrode comprised of a gas diffusion layer coated in a
product
selective electrocatalyst such as transition metal (TM) single atoms including
Fe, Pd,
Co, and Mn that may be optionally anchored into carbon nanotube (TM-CNT)
vacancies.
[0127] In the following example TM-CNT catalysts were prepared by an
impregnation and
reduction method. In the synthesis of Fe-CNT, a 7.5-rrEM iron nitrate stock
solution
was first prepared by dissolving Fe(NO3)3-9H20 (ACS Grade, Alfa Aesar) into
Millipore water (18.2 Mi2-cm). A carbon suspension was prepared by mixing 50
mg
multi-walled carbon nanotubes (Carbon Nanotubes Plus GCM389, used as received)
with 20 mL of Millipore water, and tip sonicated (Branson Digital Sonifier)
for
30 min till a homogeneous dispersion. Then 200 1., of Fe solution, given a
raw
atomic ratio of Fe:C to be ¨0.1 at.%, was dropwise added into CNT solution
under
vigorous stirring, followed by a quickly frozen in liquid nitrogen. The as-
prepared
Fe(NO3)3/CNT powder was heated up in a tube furnace to 600 C at a pressure of
1
23
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
Tor and a gas flow of 100 seem Ar (UHP, Airgas) within 20 min, and kept at
same
temperature for another 40 min before cooling down to room temperature.
[0128] Other Pd-, Co-, and Mn-CNTs were prepared in a similar way to Fe-CNT
except for
various metal salt precursors, i.e., Pd(NO3)2=2H20, Co(NO3)2=6H20, and
Mn(NO3)2=4H20 (Puriss or ACS Grade, Sigma-Aldrich), respectively.
[0129] N doped Fe-N-CNT was prepared by heating up the above-mentioned
Fe(NO3)3/CNT
powder under a same temperature program with Fe-CNT but within a mixed gas
flow
of 50 sccm NI-I3 (anhydrous, Airgas) + 100 seem Ar.
[0130] FIGs 7A-H show a comparison of the four types of TM-CNT samples,
including Fe,
Pd, Co, and Mn, which are demonstrated to have similar structures by
transmission
electron microscopy (TEM) and aberration-corrected high-angle annular dark-
field
scanning TEM (HAADF-STEM). No nanoparticles or clusters were observed in the
bright field TEM images by different scales as shown by FIGs. 7A-D. This
suggests
a good dispersion of TM atoms. Isolated TM atoms can be resolved by HAADF-
STEM due to their high Z-contrast compared to those neighboring light elements
such
as C or 0. While all four isolated metal atoms were observed as the white dots
in
FIG. 7E¨H, Pd-CNT presents the most distinguishable single atoms due to its
heaviest
atomic mass compared to the other three metal elements. In addition, the
oxidation
state of coordinated Fe is lower than simply adsorbed Fe on CNT, suggesting
the
different chemical environment between the adsorption case and coordination
case
[0131] Among the different potential transition metals carbon nanotube
catalysts, Fe-CNT is
further demonstrated herein to provide excellent performance towards
H202 generation in terms of activity and selectivity. Fe-CNT was analyzed as a
representative of other M-CNTs
[0132] An improved onset potential to reach 0.1 mA cm-2 H202 generation
current is
achieved at only 0.822 V versus reversible hydrogen electrode (vs. RHE) in 0.1
M
KOH on rotating ring-disc electrode (RRDE), while a peak H202 selectivity of
more
than 95% is delivered in both alkaline and neutral pH. With the 02 mass
transport
facilitated by a gas diffusion layer (GDL) electrode, the H202 generation rate
by Fe-
CNT can reach to 43 mA cm-2 with a 95.4% selectivity under only 0.76 V. By
switching the neighboring 0 with N coordination (through doping), the 2e- ORR
24
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
pathway can also be successfully shifted towards 4e- of H20, demonstrating a
wide
range of reaction tunability in this materials platform.
[0133] Density functional theory (DFT) calculations were conducted and suggest
that the
catalytically active C and Fe sites in Fe¨C-0 and Fe¨C¨N motifs may be
responsible
for the H202 and H20 pathways, respectively. In a variety of Fe¨C-0 motifs
calculated, the incorporation of Fe atoms significantly improves their
catalytic
activities for H202 generation compared to those with only 0 dopants. As a
prototype
demonstration of potential applications, this high-performance H202 generation
catalyst enables an effective water disinfection of >99.9999% bacteria removal
at a
treating rate of 125 L 111-2e1ectrode
[0134] SELECTIVE ELECTROCATALYST FOR HCOOH IN CO2 REDUCTION
REACTION
[0135] Similarly, other electrocatalyst were explored, in particular towards
specific
selectivity to HCOOH from CO2 reduction. In one or more embodiments, when the
target liquid product is HCOOH a variety of HCOOH-selective electrocatalysts,
such
as Bi, Co, Pd, In, Pb, Sn, and carbonaceous material, could be coupled into
the
electrosynthesis cell for a CO2RR system for pure HCOOH solution generation.
Among them, Bi-based catalysts are demonstrated herein to have achieved peak
faradaic efficiencies (FEs) of over 95% under high current densities (>50
ntA/cm2),
outperforming most of other non-noble metal catalysts. CO2 reduction to
formate was
the most energetically favorable among the competing cathodic processes on Bi
surface. However, large overpotentials were usually required to drive
significant
CO2RR currents, which leads to low energy conversion efficiencies. More
importantly, conventional Bi-based electrocatalysts generally involve multi-
step or
complicated synthesis methods, making it difficult for low-cost and largescale
productions in the future.
[0136] A facile and scalable hydrolysis approach was developed, followed by in-
situ
electrochemical-reduction to synthesize ultrathin two-dimensional Bi (2D-Bi)
catalyst for CO2-to-HCOOH conversion, which thereby presents abundant under-
coordinated active Bi sites for significantly improved catalytic performance.
Due to
the simplicity of the synthesis method, kilogram-scale synthesis of this Bi
catalyst
has been demonstrated using a 1-liter reactor.
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
[0137] Example 3: Synthesis of two-dimensional Bi (2D-Bi) catalyst for CO2-to-
HCOOH
conversion
[0138] Specifically, commercial bismuth nitrate was firstly hydrolyzed to form
layered basic
bismuth nitrates ¨ Bi606(OH)3(NO3)3-1.5H20 (BOON) which was then
topotactically
converted into 2D-Bi by in-situ electro-reduction. During the hydrolysis step,
cetyltrimethylammonium bromide (CTAB) was used as surface capping agent to
obtain ultrathin 2D-Bi. Br- ions have been demonstrated to suppress the
stacking of
monolayers for Bi-compound during bottom-up synthesis system, and the extra
surface repulsion from the hydrophobic long chains of CTA+ ions could further
terminate the stacking of layered basic bismuth nitrates.
[0139] Scanning electron microscopy (SEM) and aberration-corrected
transmission electron
microscopy (TEM) images (Figs. 8A-8B) showed the few-layer-thick BOON
nanosheets with good homogeneity. Notably, the nanosheets were almost
transparent
to the electron beam, indicating their ultrathin feature. The lattice spacing
in the high-
resolution TEM image was measured to be 0.288 nm (Fig. 8C), corresponding to
the
(006) planes of tetragonal BOON. In addition, the corresponding fast Fourier
transform (FFT) analysis of an individual BOON nanosheets indicated its single-
crystalline nature. Scanning transmission electron microscopy-energy
dispersive
spectroscopy (STEM-EDS) elemental mapping (Fig. 8D) showed a uniform
distribution of Bi, 0, N, C and Br of the BOON nanosheet, confirming the CTAB
capping effect. The BOON nanosheets were then electrochemically reduced to 2D-
Bi metal in CO2-saturated 0.5 M KHCO3 solution. The XRD measurement of the
reduced material showed obvious diffraction peaks assignable to metallic Bi,
consistent with the X-ray photoelectron spectroscopy (XPS) analysis. The XPS
and
STEM-EDS studies also demonstrated the absence of Br at the Bi surface,
suggesting
the formation of clean metallic Bi. It is noted that the in-situ formed Bi
metal retains
the original nanosheet morphology of BOON (Fig. 8E). The lattice spacing in
Fig. 8F
is 0.238 nm, agrees well with the interplanar spacing in rhombohedral Bi. The
thicknesses of individual Bi nanosheet determined by atomic force microscopy
(AFM) was only a few nanometers, revealing its 2D nature with maximally
exposed
surface sites. It is also interesting that the present process results in 2D-
Bi with quasi-
single-crystal nature, which could benefit in-plane electron transportation.
In
26
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
addition, it was found that ca. 59.8% Bi sites of 2D-Bi were electrochemically
active
using cyclic voltamrnetry. This high percentage could ensure high Bi atom
efficiency
during CO2RR catalysis.
[0140] In-operando X-ray absorption spectroscopic (XAS) can help to elucidate
the
electronic structure change of the Bi catalyst under reaction conditions. Fig.
8G
further shows the in-situ Bi L3-edge normalized absorption spectra under
different
applied potentials, as well as commercial Bi metal as a reference. A negative
energy
shift of 131 edge was observed from open circuit voltage (OCV) to -0.32 V vs.
reversible hydrogen electrode (RHE), suggesting the reduction of Bi oxidation
states.
With the negative potential further increased to -0.92 V. the Bi L3-edge
spectra
overlapped with metallic Bi reference, which indicated that the active phase
under
CO2RR conditions was metallic.
[0141] ELECTROSYNTHESIS CELL FOR 11202 PRODUCTION
[0142] Example 4: Electrocatalytic Characterization of Carbon Black Catalyst
[0143] The excellent 2e--ORR and HOR performances of CB-10% and Pt-C catalysts
therefore make good preparations for the direct electrosynthesis of pure H202
solutions using the presently described design with solid electrolytes. In one
or more
embodiments styrene-divinylbenzene copolymer microspheres (Figs. 9A-9C),
consisting of sulfonic acid functional groups for cation (r) conductions,
serves as
the SE layer with micron pores in between for water flow and product release.
Other
types of solid electrolytes including anion (H02-) polymer conductors and
cation
inorganic conductors were also demonstrated for pure H202 generation. It was
first
confirmed that there are no obvious negative or positive impacts on H202
selectivity
of CB-10% catalyst when switching from traditional liquid electrolyte to the
solid
electrolyte in a standard three-electrode setup (Fig. 10), which different
from the two-
electrode cell, can also calibrate the potentials in RHE scale. Figure 10A
plots the I-
V curve of CB-10%//SE//Pt-C cell with 02 and H2 gas streams in the cathode and
anode, respectively.
[0144] It is noted that, for all of the two-electrode cell measurements in
this work, the cell
voltages are defined as negative when the device can output electrical energy
during
the production of H202. The positive cell voltages thereby suggest the
external energy
input to this reactor. The DI water flow rate was fixed at 27 mL/h for this 4
cm2
27
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
electrode cell to prevent significant product accumulation particularly under
large
currents. H202 was readily detected starting from a cell voltage of -034 V.
suggesting
an early onset considering the equilibrium voltage of -0.76 V (30). The H202
selectivity was maintained above 90% across the whole cell voltages, reaching
upto
a maximum of 95% (Fig. 11B).
[0145] An H202 generation current of - 30 inA/cm2 (0.53 mmol/cm2 h) can be
obtained
under 0 V (no external energy input), indicating an energy-efficient route
compared
to traditional anthraquinone or direct synthesis methods. In addition, only
0.61 V cell
voltage was required to deliver a significant current density of 200 mA/cm2
with a
high H202 FE of 90%. This large current represents an H202 generation rate of
3.37
mmollcm2 h, or 3660 mol kgcat-1 h-1 considering both cathode and anode
catalyst,
setting up a new productivity benchmark in both direct synthesis and electro
synthesis
of H202 (Table 1 and Figs. 13A-13B). No H2 byproduct (possibly from H2
evolution
due to large ovetpotentials) was detected from the cathode side under such a
high
current density (Fig. 14A), indicating an exclusive ORR. Other types of solid
electrolyte with different material properties, including anion polymer
conductors for
anion conduction and cation inorganic compound conductors (CsxH3-xPW1204.0),
can
also be employed for pure H202 solution generation (Figs. 15A-15D), which
suggests
the wide tunability and versatility of the solid electrolyte design. The
relatively low
H202 FEs for cell using anion conducting solid-electrolyte is probably caused
by the
self-decomposition of H202 in the solid electrolyte layer as significant gas
bubbles
observed, as the anion conducting solid-electrolyte provides a high alkaline
environmental for ion-conduction.
[0146] Under the fixed DI water flow rate of 27 mL/h, the produced H202
concentration from
the electrosynthesis cell can reach up to - 1.7 wt.% with an overall cell
current of 800
mA (4 cm2 electrode). By speeding up or slowing down the DI water flow rate
while
maintaining the H202 generation current, a wide range of product
concentrations may
were obtained which could satisfy different application scenarios (Fig. 11C).
Up to
20 wt.% (200,000 ppm) concentrated pure H202 solutions can be directly and
continuously obtained via electrochemical synthesis.
[0147] It was observed that the H202 selectivity was inhibited with increased
H202
concentration (Fig. 16A). This observed decrease (98% at 0.3 wt.% vs. 70 % at
6.6
28
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
wt.%) was ascribed to the following three reasons: the concentrated 11202
solution in
the solid electrolyte layer 1) may self-decompose into 02 and H20 during the
present
quantification process; 2) may thermodynamically retard the 2c-ORR while the
selectivity of the competing 4e- pathway picks up; and 3) may diffuse across
the CEM
and become oxidized on the anode side as frequently observed in methanol or
formic
acid fuel cells.
[0148] In addition to the activity and selectivity, long-term stability is
another important
metric for evaluating catalysis. The electrosynthesis device demonstrated a
100-hour
continuous and stable production of - 1,200 ppm and - 11,000 pure H202
solutions
with no degradations in H202 activity and selectivity (Fig. 12A and 128). XPS
characterization of post-catalysis CB-10% catalyst reveals that its surface
oxygen was
robust and cannot be electrochemically reduced during the operation of ORR
(Fig.
14B).
[0149] Table 1. Performance metrics of different H202 generation methods.
Max,
IPsoductilit, Productity
9electicirk,
Parity
Stability Concentration
($hg.õõ,-1 fa-I) itemard earl Irl)
ekt iffPni)
Char Method Fuse aose a.at
90 ¨ 95 > 100 h 200-400
&Emote
Up to 4 cyclezt
Direct Spaltesis a ak A 34- a¨
ISO N/A 00.7 ¨ 5,300
4 It
361-
?taw
0.05 ¨ 12
47 ¨ 93,5 2 ¨ 6 h 3,499 ¨ 60A0
(3742)
Electrochemical
_______________________________________________________________________________
________________________________
Sipa:the-al
WA
Puce
265
6 h SOõOtIO
[0150] Possible impurities in collected products, examined by inductively
coupled plasma
atomic emission spectroscopy (ICP-OES), such as sodium (common impurity ions
in
water), iron (from device), sulfur (from SE), and platinum (from anode), were
at ppm
or lower level, demonstrating the ultra-high purity of the generated H202
solutions.
Table 2 shows the concentration of impurities for generated H202 using
02//SE//H20
cell. Note that the reported concentrations are average results acquired from
5
independent tests. Therefore, those electrochemically synthesized pure 11202
are
ready for immediate use out of the cell without any further purification
processes,
reducing a significant portion of cost compared to other methods, and more
29
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
importantly simplifying the setup for the deployment of delocalized generation
in the
future.
[0151] Table 2. Shows the concentration of impurities for generated H202 using
02//SE111120 cell.
Sodium Iron Sulfur Platinum
Lower than
0.872 ppm 0.022 ppm
2.62 ppm
detection limit
[0152] APPLICATION OF PRODUCT FOR WATER PURIFICATION
[0153] Example 5: Water Purification
[0154] This renewable and simple on-site generation of pure H202 solutions
opens great
opportunities in practical applications ranging from drinking water treatment,
disinfection, bleaching and so on. Rainwater is one of the most important
drinking
water supply for much of the world's population, which however may contain
contaminates such as bacteria, or small organic molecules particularly in
industrial
area, such as Houston. Compared to the traditionally used chlorine compounds
which
may produce carcinogens in the processed drinking water, H202 is safe to both
human
health and environments when disinfecting bacteria and decomposing organics.
Specifically, it is capable of removing total organic carbon (TOC)
contaminants in
rainwater for drinking.
[0155] The generated H202 stream (200 mA/cm2, 4 cm2 electrode, 27 mL/h DI
water flow)
was directly mixed with the rainwater stream with a tunable feeding rate to
optimize
the purification efficiency. The TOC of the pristine rainwater collected in
Houston
was detected to be ¨ 5 ppm, which is above the Texas treated water standard of
¨ 2
ppm. As shown in Fig. 11D, the TOC was gradually decreased when the rainwater
feeding rate was slowed down, demonstrating the efficacy of H202 in water
treatment.
A maximal processing rate of 2180 L/(m2 electrode hr) was achieved in bringing
down the TOC level to meet the drinking water standards, making the design
appealing for practical rainwater treatment when scaled-up.
[0156] Example 6: Anode Water Oxidation Coupling with ORR
[0157] It was also demonstrated that the oxidation reaction on the anode side,
to be coupled
with the cathode 2e--ORR, could be flexibly changed for applications where H2
is not
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
available. Water oxidation to 02 with protons released can be more easily
accessed
than HOR. Sulfuric acid (0.5 M H2504) was added in water to reduce the ionic
resistance on the anode side, where H2504 was not consumed during catalysis
and
continuously circulated. Figure 18A exhibits the I-V curve of 0211SE/4120
cell, with
the corresponding H202 FEs and production rates shown in Fig. 18B. Further
analysis
provided that the H202 selectivity under the same current densities is very
close to
that of 02//SEll H2 design (Fig. 11B), ruling out any impacts on the cathode
26-ORR
catalysis when the anode reaction was changed. Similarly, high 11202
productivity of
3.3 mmol cm-2 h-1 (3565 mol kgcat-1 h-1) can be achieved at a cell voltage of
2.08
V. representing an electricity-to-chemical energy conversion efficiency of
22.6% to
deliver this practical production rate. The ultra-high purity of synthesized
H202 was
continued using ICP-OES with negligible amount of impurities. A 100-hour
continuous generation of pure 11202 solutions suggested the high stability of
02
//SE//H20 cell (Fig. 17). To further simplify the design instead of using pure
02, air
was directly pumped into the cathode side as the 02 source for 26-ORR (Fig.
18A).
While higher cell voltages were required to drive the reaction due to
dramatically
decreased 02 concentration/activity, the AirIISE/4120 cell still presented
high H202
selectivity of over 90%. A maximal H202 partial current of - 123 inA/cm2 was
reached at 2.36 V. corresponding to an impressive H202 productivity of 2.3
mmol
cm-2 h-1 (2490 mol kgcat-1 h-1).
[0158] SCALABILITY AND STABILITY
[0159] Example 7: Scalability
[0160] To validate the scalability of the porous solid electrolyte design for
large-scale
synthesis of pure H202 solutions, the electrode area was extended from 4 cm2
used
for performance evaluation to - 80 cm2 in one unit modular cell (Figs. 19B to
D),
which can be further stacked in the future for scaled up capacities. A maximal
cell
current of over 20 A was achieved, with a high H202 selectivity of - 80% and
production rate of - 0.3 mol/h. Under a fixed cell current of 8 A, the scaled-
up device
is also capable of producing highly concentrated pure H202 solutions up to 20
wt.%
under a DI flow rate of 5.4 mL h-1 (Fig. 19D and Fig. 16B).
[0161] As demonstrated above, an electrosynthesis cell according to one or
more
embodiments of the present disclosure may produce highly pure, concentrated
H202
31
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
with high current efficiency (95-95 %). Pure oxygen or oxygen in air can be
directly
reduction into H202 at the cathode using an oxidized carbon material.
Additionally,
water may be oxidized into oxygen at the anode using Ir02/C catalyst. Then,
the anode
02 can be feed back to the cathode to produce H202 in order to enhance the
overall
electricality-to-H202 efficiency of the device.
[0162] High current efficiency towards H202 (- 90%) even at very high current
density (>
200 inA/cm2) can be obtained by the present electrosynthesis cell and
corresponding
process. A pure -1.6wt% 11202 can be continuously produced under a constant DI
flow-rate of 27 mL min-1.
[0163] A 70-hour continuous and stable production of - 0.13wt% pure H202
solution was
demonstrated using the carbon catalyst in this solid electrolyte ORR cell. The
current
density was fixed at 15 mA/cm2 (60 mA cell current) and the DI flow rate of 27
mL
Ii', resulting in a total of 1.89 L 0.13wt% pure H202 product. Over this 80-
hour
course, the cell voltage showed negligible change, and the H202 selectivity
was
maintained above 99%.
[0164] It was also shown that the 4 cm2 device can be easily scaled up to a
100 cm2 unit
module for ultra-concentrate pure H202 production. A maximal 20 A current can
be
achieved using the unit module with high H202 selectivity (>90%) By simply
tuning
the flow-rate of the DI water, concentrated H202 can be obtained.
Specifically, it was
shown that commercial-level 3-20wt% pure H202 can be continuously produced
using the presently disclosed electrosynthesis cell and process.
[0165] Based on the design of porous solid electrolyte layer as well as the
good performance
of 26-ORR catalysts, this demonstrated approach for direct electrosynthesis of
pure
H202 solutions, with high production rates, selectivity, and energy
efficiencies can
applied to a wide variety of electrochemical synthesis techniques of liquid
products
which are in most cases generated and mixed in liquid electrolytes. The
current
process and electrolytic cell can be extended beyond H202 generation to other
applications in electrocatalysis, such as CO2 reduction to pure liquid fuel
solutions
and 142 reduction to pure ammonia solutions. Future improvements on the
intrinsic
activity of 2e--ORR catalysts under neutral pH environments will further boost
the
device energy efficiencies. Earth-abundant catalysts, with similarly high
32
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
performances in HOR, may also be employed as alternative materials to Pt for
large-
scale renewable H202 generation.
[0166] With different solid electrolyte properties, the present design for a
three-compartment
electrolytic cell device can be further extended to other electrocatalytic
synthesis of
pure products beyond H202, such as CO2 reduction, N2 reduction and so on.
[0167] PRODUCTION OF PURE LIQUID FUELS VIA CO2RR
[0168] Example 8: Performance of the 2D-Di catalyst
[0169] The excellent CO2RR performance of the 2D-Bi catalyst as well as its
easy scalability
provide good preparations for the demonstration of producing pure HCOOH
solution
in the presently proposed CO2 reduction cell with solid electrolytes. 1r02-C
on the
anode side was selected as very stable and active OER catalyst in acidic
solutions,
which can help to release Fr from water to compensate for the negative charges
of
generated HC00-.
[0170] Figure 20A plots the CO2RR activity of 2D-Billsolid-electrolyteffIr02-C
cell with
different types of solid ion-conductors. In the case of Hi-conductor, the
overall current
density can reach to over 100 mA/cm2 at a cell voltage of 3.27 V. while the
LICOO
conductor delivers a relatively lower current of 50 mA/cm2 at ca. 347 V. No
other
liquid products were observed except HCOOH by 1H and 13C NMR. In the cell with
Fr conducting solid electrolyte, a peak HCOOH FE of 93.1% with a partial
current
of 32.1 mA/cm2 was achieved under 3.08 V (Fig. 20B), corresponding to 0.112 M
pure HCOOH solution under a DI flow-rate of 12 mL h-1 (with electrode
geometric
area of 4 cm2). The pH of this produced HCOOH solution was measured to be ca.
23-24, which agrees well with the theoretical pH value of 0.112 M pure HCOOH
solution (pH = 2.36). Furthermore, negligible amounts of impurity ions
including
potassium, sodium, iron, bismuth, sulfur (all lower than 100 ppm) and iridium
(lower
than 10 ppb) were detected by inductively coupled plasma atomic emission
spectroscopy (ICP-OES). The pH and ICPOES results demonstrate the ultra-high
purity of the produced HCOOH solution by the electrosynthesis device. Under
this
maximal HCOOH selectivity, an impressive energy conversion efficiency of 42.3%
from electricity to pure HCOOH was delivered.
[0171] In addition, a similar peak HCOOH FE of 90.1% with a HCOOH partial
current of
28 mA/cm2 was obtained at 3.21 V using a HC00- conductor (Fig. 20C),
33
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
demonstrating the generality of the solid-state electrolyte concept for pure
HCOOH
solution production. Importantly, it was shown that the concentration of pure
HCOOH solution can be easily controlled by tuning the flow-rate of DI stream
(Fig.
20D). By slowing down the DI flow, higher HCOOH concentration of 6.73 M (- 29
wt%) was achieved in CO2-to-HCOOH conversion with a FE of 30%. The decreased
HCOOH FEs with increased HCOOH concentrations might be due to two reasons:
the concentrated HCOOH solution in the solid electrolyte channel 1) may
thermodynamically lower the CO2-to- HCOOH conversion rate; and 2) can
crossover
the Nafion film and get oxidized by anode which has been typically observed in
direct
formate fuel cells 49. It was found that if the Nafion 1110(254 pm) CEM was
used
instead of Nafion 117 (183 gm), then a higher HCOOH FE can be achieved.
Specifically, an HCOOH FE of 40.3% was obtained at 100 mAJcm2 under 0.6 mL h-
1 DI rate for Nafion 117, while 51.1% HCOOH FE is achieved for Nation 1110
under
the same condition. It is also worthwhile to note that the improvement of
HCOOH
FE is still limited even when a thicker CEM (254 vs. 183 gm) was employed to
block
the HCOOH crossover. Thus, the concentrated HCOOH solution in the solid
electrolyte layer may also thermodynamically lower the CO2-to-HCOOH conversion
rate.
[0172] A 100-hour continuous and stable production of 0.11 M pure HCOOH
solution was
demonstrated using the 2D-Bi catalyst in this solid electrolyte CO2RR cell
(Fig. 21).
The current density was fixed at 30 mA/cm2 (120 tnA cell current) and the DI
flow
rate of 16.2 mL h-1, resulting in a total of 1.6 L 0.1 M pure HCOOH product.
Over
this 100-hour course the cell voltage showed negligible change, and the HCOOH
selectivity was maintained above 80%. The water contact angle test unveils the
superhydrophobicity of 100-hour aged cathode GDL, demonstrating that no water-
flooding occurred. SEM characterization of post-stability catalyst reveals
that no Bi
particulate agglomerates were observed, further highlighting the advantages of
the
electrosynthesis cell configuration for long-term stability compared with that
in an H-
cell. Moreover, higher concentration HCOOH solutions (e.g. > 1.0 M) can be
stably
and continuously obtained. Analysis of the HCOOH in the generated concentrate
HCOOH solution (150 mA cell current, 2 mL/h DI flow for 20 hours) leads to an
average HCOOH FE of ca. 80.9%, translating to -1.13 M HCOOH. In addition,
34
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
besides the polymer solid electrolyte, it was further shown that an inorganic
solid
proton conductor, like insoluble Csx1-13,PW12040, can also be employed for
pure
HCOOH generation, significantly expanding the application range of solid
electrolyte
design.
[0173] Example 9: Extension to other Products
[0174] In accordance with one or more embodiments of the present disclosure,
other types
of electrolyte-free CO2RR liquid products can be obtained using this porous
solid
electrolyte cell design.
[0175] To demonstrate its wide applicability for other pure liquid fuel
productions beyond
HCOOH, a Cu catalyst was selected, which can generate multiple C2+ oxygenate
fuels. Based on the Cu catalyst derived from commercial Cu2O nanoparticles, it
was
found that electrolyte-free dense C2+ oxygenate fuels, including ethanol,
acetic acid,
and n-propanol can be efficiently collected (Fig. 20E). At 3.45 V, the
electrolyte-free
oxygenates solution was obtained containing 4.6 mM ethanol, 3.4 mlvl n-
propanol
and 1.3 mN1 acetic acid.
[0176] The GC and NMR results present an overall ca. 100% FE, indicating that
all the
generated liquid fuels have been successfully collected by the DI stream. The
above
discussion confirms that the solid electrolyte cell design can be easily
extended to
produce other pure liquid fuels such as pure ethanol solutions once highly
selective
and exclusive CO2RR catalyst is developed.
[0177] Experiment 10: Electrocatalytic Hydrogenation to Pure Vapor
[0178] Here the electrocatalytic CO2 hydrogenation to pure HCOOH vapor under
ambient
conditions is demonstrated based on the solid state electrolyte design, which
excludes
the OER process without any liquid streams involved.
[0179] As illustrated in Fig. 22A, hydrogen was electrochemically oxidized
into proton at
the anode, which is catalyzed by commercial Pt/C, whereas the CO2 gets reduced
into
formate at cathode using the 2D-Bi. The electroreduced FIC00- ions will
recombination with the generated protons, which across from the hydrogen
oxidation
reaction (HOR) side, to form pure HCOOH. Then, the formed HCOOH vapor at the
solid electrolyte surface will be brought out by the continuous humified N2
flow. Of
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
note, no liquid stream was required for entire cell, leading to an all vapor
phase
operation. This solid electrolyte electrochemical cell can offer a 100% atom
utilization without byproduct for HCOOH production using CO2 and H2 as
feedstocks
(CO2 + H2 ¨> HCOOH). Fig. 22B displays the current-voltage profile of the
direct
electrocatalytic CO2 hydrogenation cell for HCOOH vapor generation. A peak
HCOOH FE of 83.3% was obtained at only 1.1 V.
[0180] An HCOOH partial current of 163 rnA cm-2 (HCOOH FE of 73.3%) can be
achieved
at low cell voltage of mere 1.33 V. It is important to mention that the formed
HCOOH
can be detected at as low as 0.45 V. translating to a small cell overpotential
of only
0.26 V.
[0181] Given the wide variety of solid electrolytes, as well as different
liquid fuels from
CO2RR or many other electrocatalytic reactions, we demonstrate a general
approach
using solid electrolyte design in generating pure liquid product solutions or
vapors in
electrocatalysis.
[0182] ELECTROCATALYTIC CHARACTERIZATION OF TIVI-CNT CATALYST
[0183] Example 11: Electrocatalytic Characterization of Single Atom TM-CNT
Catalyst
[0184] The ORR performances of TM-CNT as prepared in Example 2 were further
evaluated
in 0.1 M KOH by casting a thin catalyst layer onto rotation ring disk
electrode
(RRDE), with the collection efficiency pm-calibrated by the redox reaction of
[Fe(CN6)]4-/EFe(CN6)113-.
[0185] The potential of the reference electrode was double confirmed by
purging pure It gas
onto a physically and electrochemically polished polycrystalline Pt wire or Pt
rotation
disc electrode at a reasonable rotation speed. The ORR peak of Fe-CNT was
observed
in the cyclic voltanunetry in 02-saturated electrolyte, in contrast with the
double layer
current when 02 was switched to N2. Figure 23A shows the polarization curves
of M-
CNTs for their performance screening at a constant catalyst loading of 0.1 mg
cm-2,
together with the H202 generation current detected by the Pt ring electrode.
Note that
the possible H202 decomposition on metal oxides compared to the generation
should
be negligible. The corresponding H202 selectivity and electron transfer
numbers were
plotted in Fig. 23B as a function of potential.
[0186] Among the prepared different TMs, Fe-CNT presents the strongest 11202
generation
performance evaluated by RRDE, with a maximal 14202 selectivity of more than
95%,
36
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
and a high potential of 0.822 V vs. RUE to deliver a 0.1 mA cm-2 H202 onset
current,
as showin in Fig. 2313. This early onset is superior to the so-far reported
H202 catalysts such as Pd-Hg, Au-Pd, Pt single atoms, and highly oxidized
CNTs,
representing a facile ORR kinetics with negligible overpotential for 02-to-
H202 conversion.
[0187] By switching the metal dopants from Fe to Pd, Co, and Mn, the H202
selectivity was
changed to 90.3, 74.8, and 39.8%, respectively, suggesting a wide range tuning
of
electron transfer numbers from 2.09 to 3.20. Figs. 24A-E4 show the effects of
Fe atom
loading at respective amounts of 0, 0.05, 0.1, and 0.2 at% on H202 activity
and
selectivity. Compared to bare CNT, the performance was gradually increased
with
the increase of Fe atom loading, but dramatically dropped once Fe clusters was
formed demonstrating the critical role of atomically dispersed Fe.
[0188] Fe-CNT maintains its high 1202 selectivity and activity when applied
onto a GDL
(Fig 25A) electrode with facilitated 02 mass diffusion for large current
densities in
electrolyzer, where the colorimetric quantification of 11202 was employed
instead. In
1 M KOH, the catalyst delivered a steady-state 11202 partial current of 43 mA
cm-2 at
0.76 V with a Faradaic efficiency of 954%, corresponding to a 1202 production
rate
of -1.6 mol g-1 11-1 or 8 mol m-2 fri (Fig. 25B).
[0189] The catalytic activity of Fe-CNT in both RRDE test and bulk
electrolysis presents
significant improvements compared to conventional catalysts. The performance
stability of Fe-CNT single atom catalyst was also demonstrated on RRDE in Fig.
23C,
with a stable H202 selectivity of above 90% over the 8-h continuous operation.
Post-
catalysis XAS analysis of Fe K-edge XANES overlaps well with that of pristine
Fe-
CNT, suggesting that the electronic structure and coordination of Fe single
atoms
remains unchanged as demonstrated in Fig. 26K The corresponding Fourier
transformed EXAFS spectrum (Fig. 26B) of post-catalysis Fe-CNT reveals that Fe
atoms still maintain an atomic dispersion. The reaction pathway can also be
tuned by
maintaining the metal center while switching its neighboring metalloid
coordination,
which combined with the corresponding changes in catalytic performances, could
further reveal the possible active coordination motifs for 11202 generation.
The
H202 selectivity of Fe-CNT was decreased to a maximum of 60% when the catalyst
was annealed in forming gas with Fe-C-0 coordination reduced (Red. Fe-CNT);
the
37
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
4e- ORR pathway was preferred when 0 was replaced with N to form Fe-C-N
coordination, with the electron transfer number boosted from 2.09 of Fe-CNT to
3.71
of Fe-N-CNT, and even to 3.90 when the mass loading was increased to a typical
fuel
cell test condition.
[0190] Example 12: Water Disinfection by FT-CNT Catalyst
[0191] In the following Example, Fe-CNT catalyst were employed in a prototype
Example
to test the catalyst's disinfection effectiveness. Neutral pH was used instead
of
alkaline solutions to mimic the practical applications, therefore the ORR
selectivity
of Fe-CNT was first evaluated in 0.1 M PBS electrolyte using RRDE as shown in
Figs. 27A and 27B. H202 generation started at -0.53 V and maintained a high
selectivity above 90% from 0.5 to 0.3 V. The practical electrolysis was
performed in
an H-cell where Fe-CNT catalyst was casted onto a GDL electrode
(0.5 mg cm-2 catalyst loading), with the catalytic performance plotted in Fig.
27C.
The potential to deliver a 20 mA cm-2 constant current for 11202 generation
remained
unchanged over the course of electrolysis (Fig. 27D). Around 1613 ppm H202 was
generated within 210 min electrolysis as determined by the colorimetric
quantification method, representing an average Faradaic Efficiency of 90.8%.
[0192] With those performance metrics obtained, electrolyte with Escherichia
coil coli)
was then used as a model system at a bacteria concentration of -107 colony
forming
units (c.f.u.) mL-I. The disinfection process was monitored by picking up
several
droplets during the 20 niA cm-2 chronopotentiometric measurement, followed by
serially dilution and spread plating onto LB agar for overnight culture. The
calculated
killing rate is plotted in Fig. 28. Fe-CNT demonstrates a rapid disinfection
efficiency
for E. coil, delivering a 43% bacteria inactivation in 5 mm and more than
99.9999%
in 120 min (equals to a 125 L h-i m2searode processing rate) with no recovery
observed.
[0193] These results highlight that the TM single atom coordination motifs can
effectively
tune the ORR pathways and product selectivity. Among different catalysts
examined,
Fe-C-0 coordination was identified as highly active and selective motif for
02 reduction to H202-
[01194] ANODE CATALYST
38
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
[0195] Example 13: Electrocatalytic synthesis with NiFe-LDH Anode Catalyst
[0196] As discussed, the generation of protons by water oxidation on the anode
side is
provided in order to produce pure formic acid using the above proposed solid
electrocatalytic cell. However, the electrocatalytic water oxidation in acidic
solution
is challenging. Alternative embodiments of the present application may include
a
four-component electrosynthesis cell where the SE is separated by a bipolar
membrane. In such embodiments, the anode may be prepared by coating a GDL
electrode with a nickel iron layered double hydroxide (NiFe-LDH) as the OER
catalyst and KOH electrolyte to decrease the catalyst cost and anode
overpotential.
[0197] As illustrated in Fig. 29A, AEM and CEM were also used to separate
catalyst coated
GDLs and the porous SSE-50 solid ion conductors. A bipolar membrane was
employed to separate the cathode and anode compartments, which dissociates
water
in into H+ and OH- during CO2 reduction.
[0198] The generated Fl+ ions from bipolar membrane can neutralize the
negatively charged
HC00- in the left solid electrolyte layer to produce pure HCOOH. At the same
time,
more concentrated KOH can be obtained in the right solid electrolyte layer via
ionic
recombination of OH- and K. The experimentally measured current-voltage
profile
and the corresponding HCOOH FE of this four-chamber cell is presented in Fig.
29B.
A peak HCOOH partial current of ca. 150 mA cm-2 could be achieved under 3.36
V.
More importantly, we successfully collected the pure KOH solution of
concentration
up to 0.66 M under a DI flow-rate of 16.2 mL 11-1 (Fig. 29C), demonstrating
the
feasibility of our strategy.
[0199] Impressively, the cell performance showed no obvious changes during the
course of
stability test. In future applications, the brine streams can be used as
anolyte to drive
the chlorine evolution at the anode side to replace the OER. Then, three kinds
of
valuable pure products (HCOOH, NaOH and C12) can be simultaneously generated.
Implementation of NaOH, C12 and HCOOH production from brine stream and CO2
using our solid electrolyte concept can offer environmentally sound, economic
strategies for sustainable desalination and carbon-cycling.
[0200] CATALYST INCLUDING NON-METAL DOPANTS
[0201] Example 14: Carbon Catalyst Comprising Non-metal Dopants.
39
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
[0202] In the following Example, the trade-off between high activity and high
selectivity in
carbon materials is tested by introducing non-metal dopants, and to see
demonstrate
how the induced electronic structural changes can enhance the catalysts' 2e-
ORR
activity under large currents while maintaining high selectivity towards H202.
In this
Example, a series of nonmetal dopants, including but not limited to boron,
nitrogen,
phosphorous and sulfur, were anchored on carbon black substrates, and the
result
catalysts were compared together with 142-annealed pristine carbon black (Pure
C) as
the control sample. Samples were prepared in accordance with methods described
above.
[0203] Among all the materials, boron-doped carbon (B-C) showed the best
intrinsic activity
while maintaining high selectivity in both alkaline and neutral conditions
from
rotation ring-disk electrode (RRDE), as show in Figs. 30C-D, with a positive
onset of
0.79V and 0.42V (vs. RHE) in 0.1M KOH and 0.1M Na2SO4, respectively (Figs.
30A-B).
[0204] Figs. 31A-B show I-V curve data plots for Pure C, B-C and O-C in 1M KOH
and 1M
Na2SO4, respectively. Figs. 31C-D further show FE and 11202 partial currents
measured in 1M KOH and 1M Na2SO4. Note that all the I-V curves and faradaic
efficiency were taken average of 2-3 independent tests for each of the
samples. For
the large current performance in a three-electrode flow cell, B-C showed
improved
kinetics compared to oxidized carbon (0-C), while maintaining comparably high
selectivity in contrast to Pure C, in both alkaline and neutral electrolytes.
[0205] Furthermore, as demonstrated in Figs. 32A-C, the B-C sample is shown to
efficiently
generate pure 11202 production using the three-compartment solid-electrolyte
cell
configuration demonstrated above. The boron doped sample can achieve a high
faradaic efficiency (FE) of over 87% within a broad potential window until the
current
density reaches as high as 400mA cm-2, as shown in Fig. 32A. A high production
rate
of 7.36 mrnol cm-2h' was achieved at 500mA cm-2 (Fig. 32B) and the cell is
capable
of operating for 30 hours without performance decay (Fig. 32C).
[0206] CEM-CEM THREE COMPONENT CELL
[0207] Example 15: Dual CEM in Three Component Electrosynthesis Cell
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
[0208] In the following Example, the cathode anion exchange membrane (AEM) as
described above was replaced with a CEM for pure H202 solution generation, as
shown in Fig. 33.
[0209] All other parts, except ORR catalysts, were used, unchanged, compared
with the
previous design. Similarly, independent water and 02, streams were
respectively
delivered to water oxidation and 2e--ORR catalysts coating gas diffusion layer
(GDL)
electrodes.
[0210] The anode and cathode were sandwiched with CEM layers to avoid flooding
by direct
contact with liquid water. In the center, a thin porous solid electrolyte
layer facilitated
ionic conduction of Fr crossing from the anode to cathode with small ohmic
losses
and a flowing DI water stream was confined to this middle layer that could
then
dissolve the pure H202 product with no introduction of ionic impurities. By
tuning
the H202 generation rate or the DI water flow rate, a wide range of H202
concentrations could be directly obtained with no need for further energy-
consuming
downstream purification.
[0211] Similar to above, the 02 from air will be used in the electrochemical
reduction into
11202 at the cathode (Cathode: 02 + 2e- + 211+
11202). And the water will be
electrochemically oxidized into 02, while simultaneously releasing protons
(Anode:
H20 - 4e-
02 + 411.). The protons, as
the electrical carriers, will move across the
CEMs and the porous solid-electrolyte layer to compensate the charge. Since
the
locally generated H202 molecules at the CEM and cathode catalyst interface
have a
relatively high concentration, they will then chemically and/or electro-
osmotically
diffuse into the middle solid electrolyte layer, and be further carried out by
the water
flow as pure 11202 solution streams.
[0212] The CEM provides an extremely acidic environment for ORR. The catalyst
tested
included metal and non-metal doped carbon catalysts to demonstrate this
concept. For
example, a nitrogen doped carbon supported nickel single atom (Ni-N-C) was
used
as the catalyst for 2e-ORR in this CEM//solid electrolyteIICEM device. As
shown in
Fig. 34, the Ni-N-C single atom catalyst can deliver a stable 11202 Faradic
efficiency
(FE) of ca. 30% under 20 mA cm-2 at least for 150 hours. The concentration of
generated H202 stream was -560 ppm under 20 mA cm-2 current density. The
stable
41
CA 03150909 2022-3-10
WO 2021/011675
PCT/US2020/042163
operation of this new all CEM based reactor demonstrates the feasibility of
the three-
compartment design for pure H202 generation. Additionally, other types of
carbon
catalysts, including but not limited to surface functionalized carbon, such as
B-doped
carbon, showed good selectivity in generating pure H202 solutions (Fig. 35).
[0213] While the disclosure has been described with respect to a limited
number of
embodiments, those skilled in the art, having benefit of this disclosure, will
appreciate
that other embodiments can be devised which do not depart from the scope of
the
disclosure as disclosed herein. Accordingly, the scope of the disclosure
should be
limited only by the attached claims.
42
CA 03150909 2022-3-10